РОДСТВЕННАЯ ЗАЯВКА
Настоящая заявка является родственной заявке на патент США №61/647200, поданной 15 мая 2012 года, полное содержание которой включено в настоящее описание посредством ссылки.
ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится в целом к области терапевтических соединений. В частности, настоящее изобретение относится к соединениям 5-[[4-[[морфолин-2-ил]метиламино]-5-(трифторметил)-2-пиридил]амино]пиразин-2-карбонитрила, (называемым в настоящем документе «TFM-соединения») которые, помимо прочего, ингибируют киназную функцию киназы 1 контрольной точки (CHK1). Настоящее изобретение также относится к фармацевтическим композициям, содержащим такие соединения, и применению таких соединений и композиций как in vitro, так и in vivo для ингибирования киназной функции CHK1, а также для лечения заболеваний и состояний, опосредованных CHK1, поддающихся облегчению путем ингибирования киназной функции CHK1, и т.д., включая пролиферативные заболевания, такие как рак и т.д., необязательно в комбинации с другим агентом, например, (а) ингибитором ДНК-топоизомеразы I или II; (b) ДНК-повреждающим агентом; (с) антиметаболитом или ингибитором тимидилатсинтазы (ТС); (d) нацеленным на микротрубочки агентом; (е) ионизирующим излучением; (f) ингибитором регулятора митоза или регулятора митотической контрольной точки; (g) ингибитором трансдуктора сигнала повреждения ДНК; или (h) ингибитором репаративного фермента повреждения ДНК.
УРОВЕНЬ ТЕХНИКИ
В настоящем документе приведен ряд публикаций с целью более полного описания и раскрытия изобретения и уровня техники в области, к которой относится изобретение. Полное содержание каждой из этих публикаций включено в настоящее описание посредством ссылки в той же степени, как если бы для каждой отдельной публикации конкретно и индивидуально было указано ее включение посредством ссылки.
По всему тексту настоящего описания, включая последующую формулу изобретения, если иное не следует из контекста, слово «содержать» и его вариации, такие как «содержит» и «содержащий», подразумевают включение заявленного целого или стадии или группы целых или стадий, но не исключение любого другого целого или стадии или группы целых или стадий.
Следует отметить, что в настоящем описании и прилагаемой формуле изобретения неопределенная и определенная формы единственного числа включают определяемые объекты во множественном числе, если контекст явно не предписывает иное. Таким образом, например, ссылка на «фармацевтический носитель» включает смеси двух или более таких носителей и т.п.
В настоящем документе диапазоны часто выражены как от «примерно» одного конкретного значения и/или до «примерно» другого конкретного значения. Когда выражен такой диапазон, другой вариант реализации включает величины от одного конкретного значения и/или до другого конкретного значения. Аналогично, когда значения выражены в виде приблизительных величин с использованием ранее упомянутого «примерно», следует понимать, что конкретные значения составляют другой вариант реализации.
Настоящее описание содержит информацию, которая может быть полезной для понимания настоящего изобретения. Это не является допущением того, что любая такая информация представляет собой уровень техники или относится к описываемому в настоящем документе изобретению, или что любая специально или неявно упоминаемая публикация представляет собой уровень техники.
Киназа 1 контрольной точки (CHK1)
Последовательность событий цикла деления клеток представляет собой строго регулируемый процесс и контролируется на некоторых этапах, известных как контрольные точки клеточного цикла (см., например, Weinert и Hartwell, 1989; Bartek and Lukas, 2003). Данные контрольные точки обнаружены на всех четырех стадиях клеточного цикла - G1, S (репликации ДНК), G2 и М (митоза), и они обеспечивают корректное завершение ключевых событий, контролирующих точность репликации ДНК и деления клеток. Контрольные точки клеточного цикла активируются рядом стимулов, включая повреждения ДНК и ошибки в ДНК, вызванные дефектной репликацией. Когда происходит эта активация, клеточный цикл блокируется, что дает время либо для репарации ДНК, либо, если повреждение слишком серьезно, для активации клеточных процессов, ведущих к контролируемой гибели клетки.
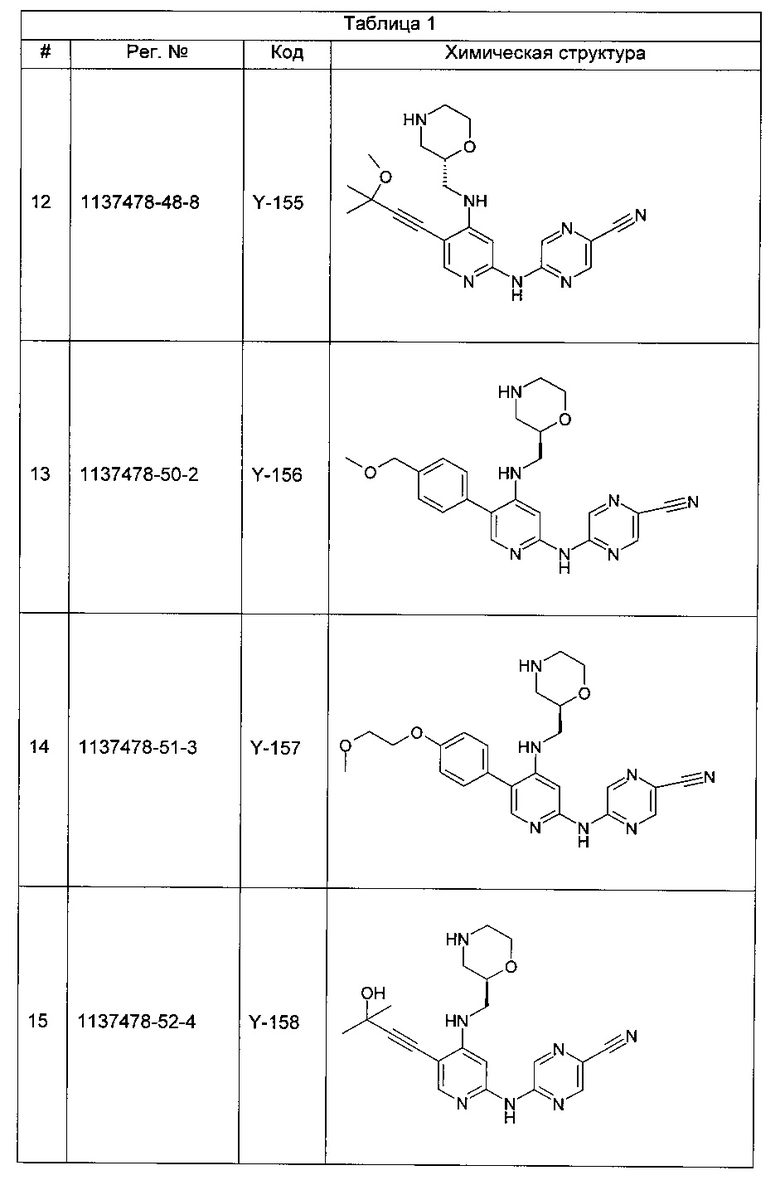
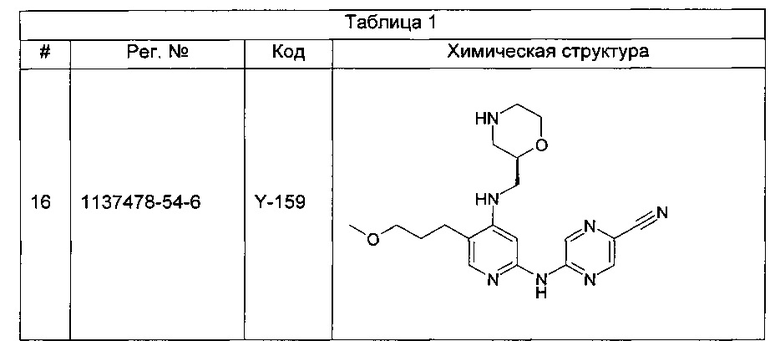
Все виды рака в сущности имеют некоторую аномальную форму цикла деления клеток. Зачастую раковые клетки характеризуются одной или более дефектными контрольными точками клеточного цикла или имеют дефекты конкретного пути репарации ДНК. Следовательно, эти клетки обычно более зависимы от остальных контрольных точек клеточного цикла и путей репарации по сравнению с нераковыми клетками (у которых все контрольные точки и пути репарации ДНК не повреждены). Ответ раковых клеток на повреждение ДНК зачастую является решающим фактором, определяющим, продолжают ли они пролиферировать или активируют процессы клеточной гибели и умирают. Например, опухолевые клетки, которые содержат мутантную форму (формы) опухолевого супрессора р53, являются дефектными в точке контроля повреждения ДНК фазы G1.
Таким образом, ингибиторы контрольных точек фаз G2 или S, как полагают, дополнительно снижают способность опухолевой клетки к репарации поврежденной ДНК.
Многие известные средства для лечения рака вызывают повреждение ДНК посредством либо физической модификации клеточной ДНК, либо нарушения жизненно важных клеточных процессов, которые могут влиять на точность репликации ДНК и деления клеток, таких как метаболизм ДНК, синтез ДНК, транскрипция ДНК и образование веретена деления из микротрубочек. Такие средства лечения включают, например, лучевую терапию, вызывающую разрывы цепей ДНК, и множество химиотерапевтических агентов, включая ингибиторы топоизомеразы, антиметаболиты, алкилирующие ДНК агенты и цитотоксические лекарственные средства, содержащие соединения платины. Существенным ограничением для этих генотоксических способов лечения является лекарственная устойчивость. Один из наиболее важных механизмов, ведущих к этой устойчивости, объясняют активацией контрольных точек клеточного цикла, дающих опухолевой клетке время для репарации поврежденной ДНК. С помощью подавления функции конкретной контрольной точки клеточного цикла или ингибирования конкретного типа репарации ДНК становится, таким образом, возможным преодоление устойчивости опухолевых клеток к генотоксическим агентам и увеличение гибели опухолевых клеток, индуцированной повреждением ДНК, что приводит к повышению терапевтического индекса этих средств для лечения рака.
CHK1 представляет собой серин/треонинкиназу, участвующую в регулировании сигналов контрольных точек клеточного цикла, которые активируются в ответ на повреждение ДНК и возникновение ошибок в последовательности ДНК, вызванное дефектной репликацией (см., например, Bartek и Lukas, 2003). CHK1 передает эти сигналы посредством фосфорилирования субстратов, вовлеченных в ряд клеточных активностей, включая блокирование клеточного цикла и репарацию ДНК. Двумя ключевыми субстратами CHK1 являются фосфатазы Cdc25A и Cdc25C, которые дефосфорилируют циклин-зависимую киназу 1 (CDK1), приводя к активации последней, что является необходимым условием для перехода из фазы G2 в фазу митоза (М-фазу) (см., например, Sanchez et al., 1997). Катализируемое CHK1 фосфорилирование Cdc25C и родственной ей Cdc25A блокирует их способность к активации CDK1, тем самым предотвращая переход клетки из фазы G2 в фазу М. Роль CHK1 в индуцируемой повреждением ДНК контрольной точке фазы G2 клеточного цикла была показана в ряде исследований, в которых функция CHK1 была выключена (см., например, Liu et al., 2000; Zhao et al., 2002; Zachos et al., 2003).
Зависимость индуцируемой повреждением ДНК контрольной точки фазы G2 от CHK1 обеспечивает один пример терапевтической стратегии для лечения рака, включающей направленное ингибирование CHK1. При повреждении ДНК происходят стабилизация и активация белка-супрессора опухолей р53, результатом которых является р53-зависимое блокирование фазы G1, приводящее к апоптозу или репарации ДНК (Balaint и Vousden, 2001). Более чем в половине всех случаев рака обнаружены функциональные дефекты р53, которые могут сделать эти типы рака устойчивыми к генотоксическим средствам лечения, таким как ионизирующее излучение (ИИ) и некоторые виды химиотерапии (см., например, Greenblatt et al., 1994; Carson и Lois, 1995). Эти р53-дефицитные клетки не способны блокировать клеточный цикл в контрольной точке фазы G1 или подвергаться процессам апоптоза или репарации ДНК и, следовательно, могут быть более зависимыми от контрольной точки фазы G2 в отношении жизнеспособности и точности репликации. Соответственно, подавление функции контрольной точки фазы G2 путем ингибирования киназной функции CHK1 может селективно сенсибилизировать р53-дефицитные раковые клетки к генотоксическим средствам лечения рака, что и было продемонстрировано (см., например, Wang et al., 1996; Dixon и Norbury, 2002).
Кроме того, также было показано, что CHK1 участвует в контрольных точках фазы S клеточного цикла и репарации ДНК посредством гомологичной рекомбинации. Таким образом, ингибирование киназы CHK1 в тех случаях рака, которые являются зависимыми от указанных процессов после повреждения ДНК, может обеспечить дополнительные терапевтические стратегии для лечения раковых заболеваний с применением ингибиторов CHK1 (см., например, Sorensen et al., 2005). Кроме того, некоторые виды рака могут демонстрировать репликативный стресс вследствие высоких уровней эндогенного повреждения ДНК (см., например, Cavalier et al., 2009; Brooks et al., 2012) или в результате повышенного уровня репликации, стимулируемой онкогенами, например, амплифицированными или избыточно экспрессируемыми генами МУС (см., например, Di Micco et al., 2006; Cole et al., 2011; Murga et al., 2011). Такие виды рака могут обнаруживать усиленную передачу сигнала через киназу CHK1 (см., например, Höglund et al., 2011). Ингибирование киназы CHK1 в тех случаях рака, которые являются зависимыми от указанных процессов, может обеспечить дополнительные терапевтические стратегии для лечения раковых заболеваний с применением ингибиторов CHK1 (см., например, Cole et al., 2011; Davies et al., 2011; Ferrao et al., 2011).
Последние данные о применении CHK1-специфических миРНК подтверждают целесообразность терапевтического подхода, основанного на селективном ингибировании CHK1, и позволяют предположить, что комбинированное ингибирование с некоторыми другими киназами контрольных точек не обеспечивает дополнительного преимущества и может быть непродуктивным (см., например, Xiao et al., 2006; Guzi et al., 2011). Были описаны низкомолекулярные селективные ингибиторы киназной функции CHK1, относящиеся к различным химическим классам (см., например, Tao et al., 2006).
Известные соединения
Коллинз с соавторами (Collins et al., 2009а) (WO 2009/044162 А1) описывают некоторые соединения следующей формулы, ингибирующие киназную функцию киназы 1 контрольной точки (CHK1) и подходящие для лечения, например, рака:
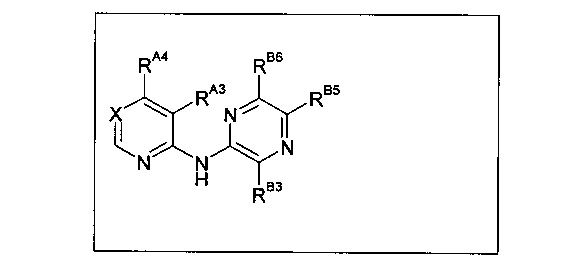
Среди примеров Коллинз с соавторами (Collins et al., 2009а) приводят следующие соединения:
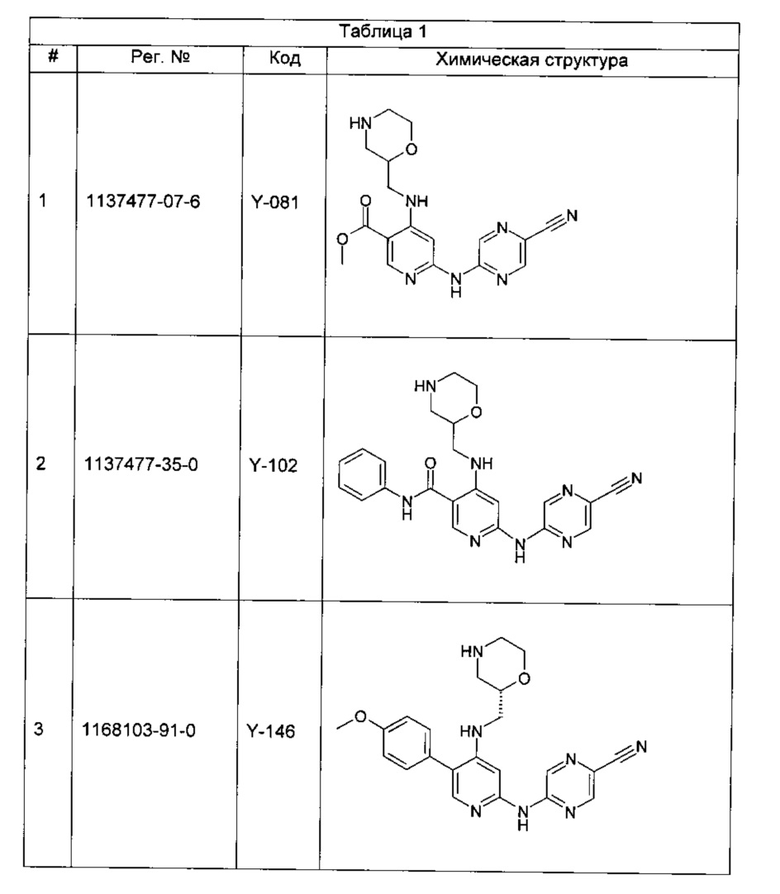
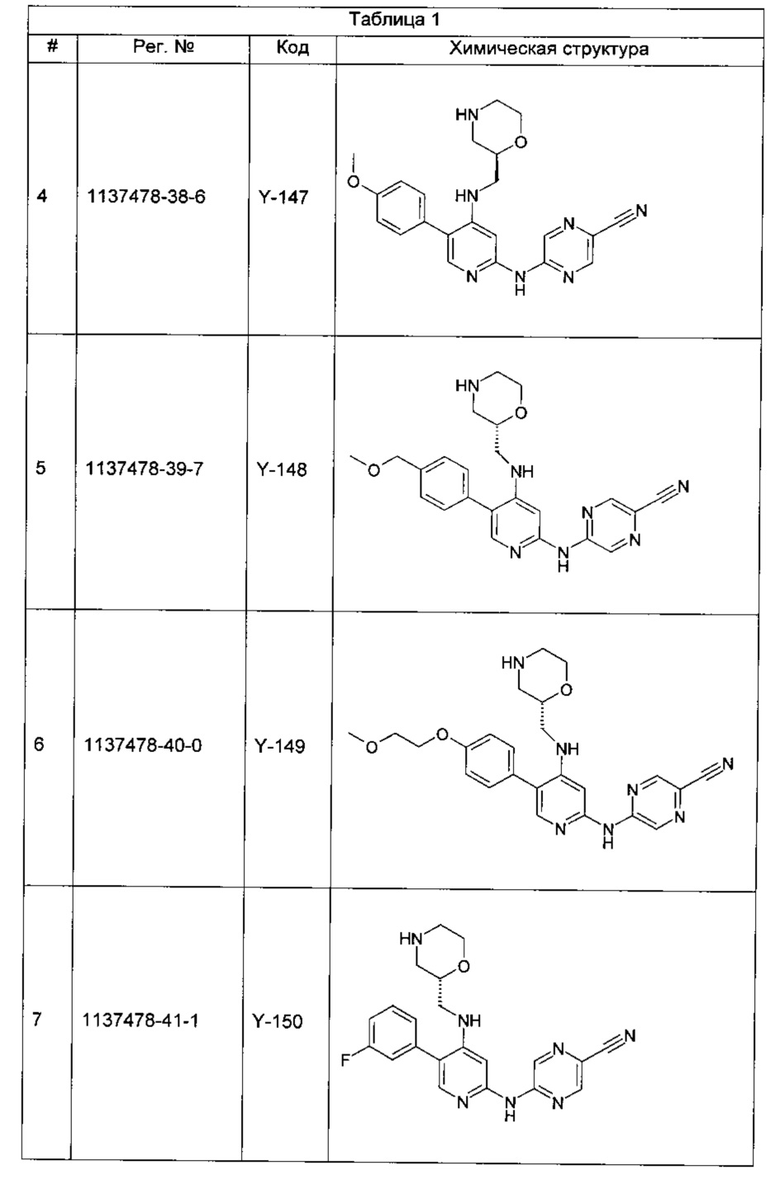
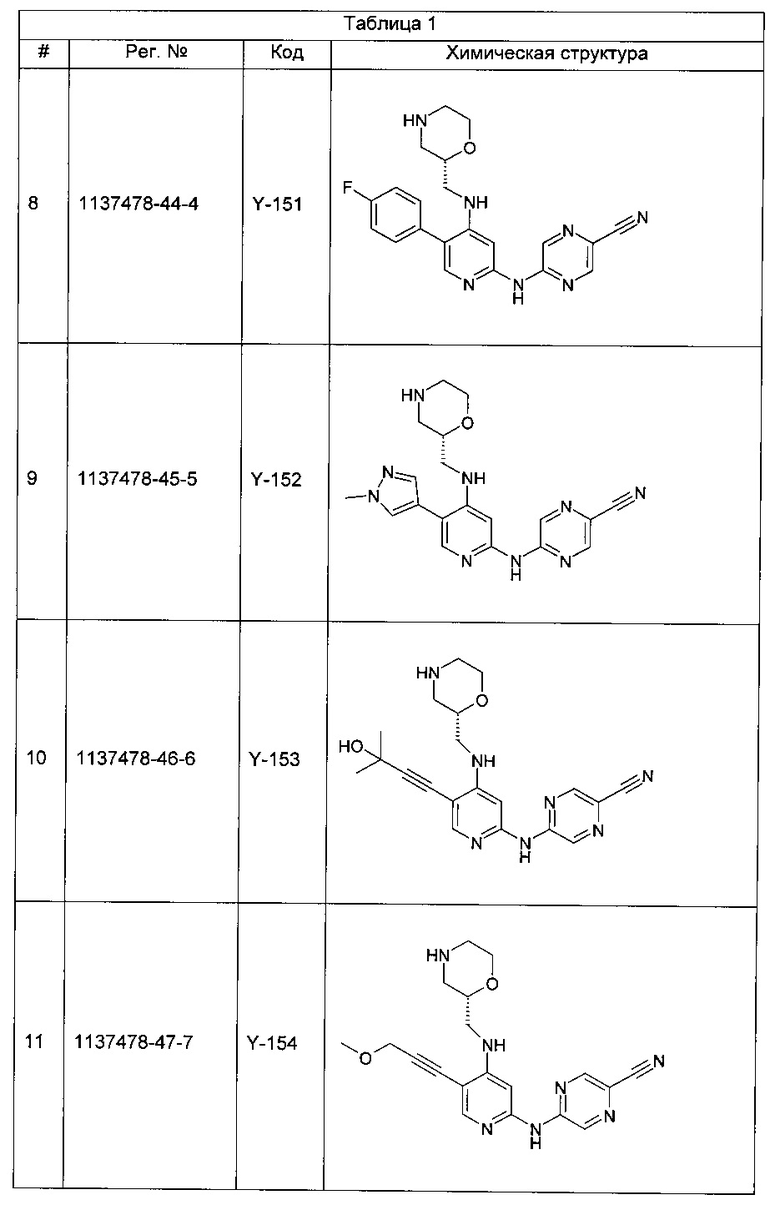
В данном классе, описанном Коллинзом с соавторами, 2009а, X может представлять собой -CRA5- (см., например, с.8, стр. 27 в указанной публикации) и -RA5 может представлять собой -QA5 (см., например, с. 9, стр. 1 в указанной публикации). Группа -QA5 определена широко (см., например, с. 31, стр. 27 - с. 38, стр. 13 в указанной публикации), и может представлять собой, например, -CF3 (см., например, с. 31, стр. 32 и с. 33, стр. 21).
Однако в документе Коллинз с соавторами, 2009а ни в одном из примеров X не содержит -CRA5- с -RA5 в виде -CF3.
Коллинз с соавторами (Collins et al., 2009b) описывают некоторые соединения следующей формулы, ингибирующие киназную функцию киназы 1 контрольной точки (CHK1) и подходящие для лечения, например, рака:
Уолтон с соавторами (Walton et al., 2010) описывают доклинические исследования ингибитора CHK1, названного SAR-020106.
Альмейда с соавторами (Almeida et al., 2008) описывают некоторые пиразолиламинозамещенные пиразины, предположительно подходящие для лечения рака.
Иоаннидис с соавторами (Ioannidis et al., 2009) описывают некоторые соединения, ингибирующие Янус-киназу (JAK). См., например, Схему 5 на стр.6526 в указанной публикации.
Лин с соавторами (Lin et al., 2005) описывают некоторые макроциклические соединения на основе мочевины, предположительно подходящие в качестве ингибиторов протеинкиназ. См., например, абзац [0004] на стр. 1 в указанной публикации.
Тао с соавторами (Tao et al., 2005) описывают некоторые макроциклические соединения на основе мочевины, предположительно подходящие в качестве ингибиторов протеинкиназ. См., например, стр. 2 в указанной публикации.
Ли с соавторами (Li et al., 2007) описывают получение и исследование некоторых макроциклических ингибиторов CHK1 на основе мочевины. См., например, таблицу 1 на стр.6502 в указанной публикации.
Тао с соавторами (Тао et al., 2007а) описывают получение и исследование некоторых макроциклических ингибиторов CHK1 на основе мочевины. См., например, таблицу 2 на стр.6596 в указанной публикации.
Тао с соавторами (Тао et al., 2007b) описывают получение и исследование некоторых макроциклических ингибиторов CHK1 на основе мочевины. См., например, таблицу 3 на стр.1517 в указанной публикации.
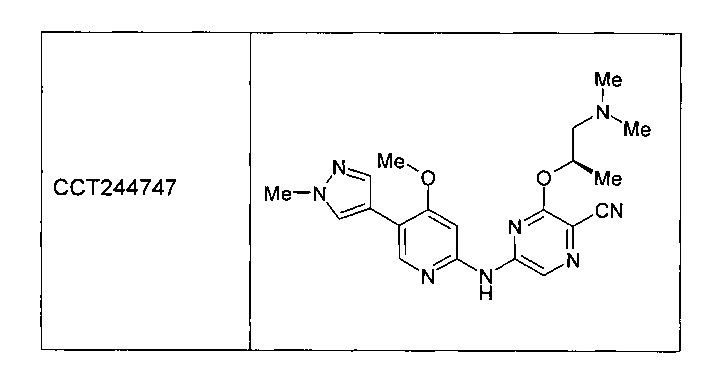
Одним или более из авторов настоящего изобретения внесли вклад в недавние публикации, в которых описан ряд ингибиторов CHK1, включая следующее соединение, названное ССТ244747. См. Lainchbury et al., 2012 (предварительно опубликована в Интернете 19 октября 2012 года) и Walton et al., 2012 (предварительно опубликована 15 октября 2012 года).
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Один аспект изобретения относится к соединениям 5-[[4-[[морфолин-2-ил]метиламино]-5-(трифторметил)-2-пиридил]амино]пиразин-2-карбонитрила (называемым в настоящем документе «TFM-соединения»), описанным в настоящем документе.
Другой аспект изобретения относится к композиции (например, фармацевтической композиции), содержащей описанное в настоящем документе TFM-соединение и фармацевтически приемлемый носитель или разбавитель.
Согласно одному из вариантов реализации композиция (например, фармацевтическая композиция) подходит для перорального введения субъекту.
Согласно одному из вариантов реализации композиция находится в форме таблетки для перорального введения, гранул для перорального введения, порошка для перорального введения, капсулы для перорального введения, саше для перорального введения или пилюли для перорального введения.
Другой аспект изобретения относится к способу получения композиции (например, фармацевтической композиции), включающему стадию смешивания описанного в настоящем документе TFM-соединения и фармацевтически приемлемого носителя или разбавителя.
Другой аспект настоящего изобретения относится к способу ингибирования киназной функции CHK1 в клетке, in vitro или in vivo, включающему приведение клетки в контакт с эффективным количеством TFM-соединения, описанного в настоящем документе.
Согласно одному из вариантов реализации способ дополнительно включает приведение клетки в контакт с одним или более другими агентами, выбранными из: (а) ингибитора ДНК-топоизомеразы I или II; (b) ДНК-повреждающего агента; (с) антиметаболита или ингибитора тимидилатсинтазы (ТС); (d) нацеленного на микротрубочки агента; (е) ионизирующего излучения; (f) ингибитора регулятора митоза или регулятора митотической контрольной точки; (g) ингибитора трансдуктора сигнала повреждения ДНК; и (h) ингибитора репаративного фермента повреждения ДНК.
Другой аспект настоящего изобретения относится к способу регулирования (например, ингибирования) пролиферации клеток (например, пролиферации клетки), ингибирования прогрессии клеточного цикла, стимулирования апоптоза клеток или осуществления комбинации одного или более из вышеперечисленных, in vitro или in vivo, включающему приведение клетки в контакт с эффективным количеством TFM-соединения, описанного в настоящем документе.
Согласно одному из вариантов реализации способ дополнительно включает приведение клетки в контакт с одним или более другими агентами, выбранными из: (а) ингибитора ДНК-топоизомеразы I или II; (b) ДНК-повреждающего агента; (с) антиметаболита или ингибитора тимидилатсинтазы (ТС); (d) нацеленного на микротрубочки агента; (е) ионизирующего излучения; (f) ингибитора регулятора митоза или регулятора митотической контрольной точки; (g) ингибитора трансдуктора сигнала повреждения ДНК; и (h) ингибитора репаративного фермента повреждения ДНК.
Другой аспект настоящего изобретения относится к способу лечения, включающему введение субъекту, нуждающемуся в лечении, терапевтически эффективного количества описанного в настоящем документе TFM-соединения, предпочтительно в форме фармацевтической композиции.
Согласно одному из вариантов реализации указанное введение представляет собой пероральное введение.
Согласно одному из вариантов реализации способ дополнительно включает введение субъекту одного или более других агентов, выбранных из: (а) ингибитора ДНК-топоизомеразы I или II; (b) ДНК-повреждающего агента; (с) антиметаболита или ингибитора тимидилатсинтазы (ТС); (d) нацеленного на микротрубочки агента; (е) ионизирующего излучения; (f) ингибитора регулятора митоза или регулятора митотической контрольной точки; (g) ингибитора трансдуктора сигнала повреждения ДНК; и (h) ингибитора репаративного фермента повреждения ДНК.
Другой аспект настоящего изобретения относится к TFM-соединению, описанному в настоящем документе, для применения в способе лечения тела человека или животного посредством терапии.
Согласно одному из вариантов реализации соединение предназначено для применения в способе лечения тела человека или животного посредством терапии с помощью перорального введения.
Согласно одному из вариантов реализации способ лечения включает лечение с применением как (i) TFM-соединения, так и (ii) одного или более других агентов, выбранных из: (а) ингибитора ДНК-топоизомеразы I или II; (b) ДНК-повреждающего агента; (с) антиметаболита или ингибитора тимидилатсинтазы (ТС); (d) нацеленного на микротрубочки агента; (е) ионизирующего излучения; (f) ингибитора регулятора митоза или регулятора митотической контрольной точки; (g) ингибитора трансдуктора сигнала повреждения ДНК; и (h) ингибитора репаративного фермента повреждения ДНК.
Другой аспект настоящего изобретения относится к применению TFM-соединения, описанного в настоящем документе, для получения лекарственного средства для применения в способе лечения.
Согласно одному из вариантов реализации лекарственное средство представляет собой лекарственное средство для перорального введения. Согласно одному из вариантов реализации лечение включает лечение с применением как (i) лекарственного средства, содержащего TFM-соединение, так и (ii) одного или более других агентов, выбранных из: (а) ингибитора ДНК-топоизомеразы I или II; (b) ДНК-повреждающего агента; (с) антиметаболита или ингибитора тимидилатсинтазы (ТС); (d) нацеленного на микротрубочки агента; (е) ионизирующего излучения; (f) ингибитора регулятора митоза или регулятора митотической контрольной точки; (g) ингибитора трансдуктора сигнала повреждения ДНК; и (h) ингибитора репаративного фермента повреждения ДНК.
Согласно одному из вариантов реализации лечение представляет собой лечение заболевания или состояния, опосредованного CHK1.
Согласно одному из вариантов реализации лечение представляет собой лечение заболевания или состояния, при котором улучшения достигают путем ингибирования киназной функции CHK1.
Согласно одному из вариантов реализации лечение представляет собой лечение пролиферативного заболевания.
Согласно одному из вариантов реализации лечение представляет собой лечение рака.
Согласно одному из вариантов реализации лечение представляет собой лечение рака головы; рака шеи; рака нервной системы; рака мозга; нейробластомы; рака легкого/средостения; рака молочной железы; рака пищевода; рака желудка; рака печени; рака желчевыводящих путей; рака поджелудочной железы; рака тонкой кишки; рака толстой кишки; рака ободочной и/или прямой кишки; рака женских половых органов; рака мочеполовой системы; рака яичника; рака щитовидной железы; рака надпочечника; рака кожи; меланомы; остеосаркомы; саркомы мягких тканей; злокачественных опухолей детского возраста; болезни Ходжкина; неходжкинской лимфомы; миеломы; лейкоза или метастазов из неизвестного первичного очага.
Согласно одному из вариантов реализации лечение представляет собой лечение: рака легкого, рака молочной железы, рака яичника, рака поджелудочной железы, рака ободочной и/или прямой кишки, лимфомы, меланомы, глиомы или нейробластомы.
Согласно одному из вариантов реализации лечение представляет собой лечение р53-дефицитного рака.
Согласно одному из вариантов реализации лечение представляет собой лечение рака с амплифицированным MYC.
Согласно одному из вариантов реализации лечение представляет собой лечение рака с амплифицированным c-MYC.
Согласно одному из вариантов реализации лечение представляет собой лечение рака с амплифицированным MYCN.
Согласно одному из вариантов реализации лечение представляет собой лечение рака, характеризующегося гиперэкспрессией MYC.
Согласно одному из вариантов реализации лечение представляет собой лечение рака, характеризующегося гиперэкспрессией MYCN.
Согласно одному из вариантов реализации лечение представляет собой лечение рака, характеризующегося гиперэкспрессией c-MYC.
Согласно одному из вариантов реализации лечение представляет собой лечение нейробластомы с амплифицированным MYCN.
Согласно одному из вариантов реализации лечение представляет собой лечение В-клеточной лимфомы с амплифицированным c-MYC.
Согласно одному из вариантов реализации лечение представляет собой лечение рака, характеризующегося повышенным эндогенным репликативным стрессом.
Согласно одному из вариантов реализации лечение представляет собой лечение рака, характеризующегося повышенной эндогенной активацией сигнального пути CHK1.
Другой аспект настоящего изобретения относится к набору, содержащему (а) описанное в настоящем документе TFM-соединение, предпочтительно обеспеченное в виде фармацевтической композиции и содержащееся в подходящей емкости и/или с подходящей упаковкой; и (b) инструкции по применению, например, письменные указания относительно введения соединения.
Согласно одному из вариантов реализации набор дополнительно содержит один или более других агентов, выбранных из: (а) ингибитора ДНК-топоизомеразы I или II; (b) ДНК-повреждающего агента; (с) антиметаболита или ингибитора тимидилатсинтазы (ТС); (d) нацеленного на микротрубочки агента; (е) системного радиофармацевтического средства; (f) ингибитора регулятора митоза или регулятора митотической контрольной точки; (g) ингибитора трансдуктора сигнала повреждения ДНК; и (h) ингибитора репаративного фермента повреждения ДНК.
Другой аспект настоящего изобретения относится к TFM-соединению, которое может быть получено посредством способа синтеза, описанного в настоящем документе, или способа, включающего способ синтеза, описанный в настоящем документе.
Другой аспект настоящего изобретения относится к TFM-соединению, полученному посредством способа синтеза, описанного в настоящем документе, или способа, включающего способ синтеза, описанный в настоящем документе.
Другой аспект настоящего изобретения относится к новым промежуточным соединениям, описанным в настоящем документе, которые подходят для применения в описанных в настоящем документе способах синтеза.
Другой аспект настоящего изобретения относится к применению таких новых промежуточных соединений, описанных в настоящем документе, в описанных в настоящем документе способах синтеза.
Специалисту в данной области техники будет понятно, что признаки и предпочтительные варианты реализации одного аспекта изобретения также относятся к другому аспекту изобретения.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Соединения
Один из аспектов настоящего изобретения относится к соединениям следующей формулы, и их фармацевтически приемлемым солям, гидратам и сольватам (для удобства, в целом называемым в настоящем документе соединениями «5-[[4-[[морфолин-2-ил]метиламино]-5-(трифторметил)-2-пиридил]амино]пиразин-2-карбонитрила» или «TFA-соединения»):
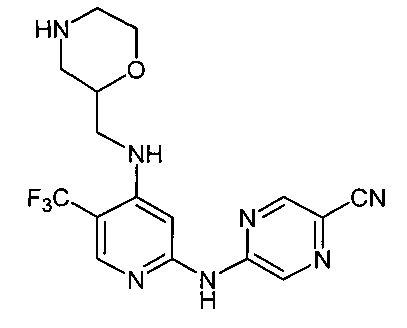
Точка присоединения морфолинильной группы представляет собой хиральный центр, отмеченный звездочкой в следующих формулах, который независимо может иметь (R)-или (S)-конфигурацию. Если не указано иное, подразумевают включение обеих конфигураций.
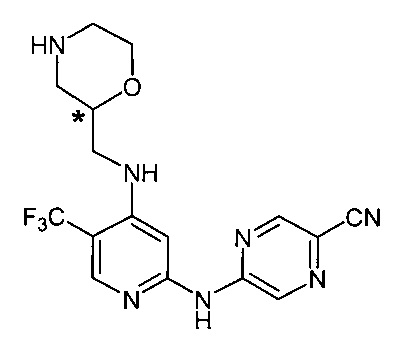
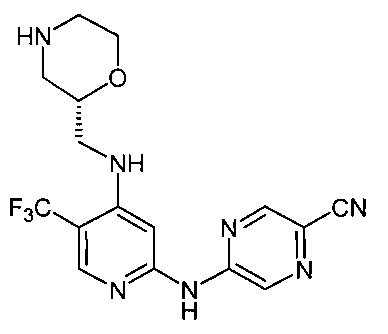
Согласно одному из вариантов реализации соединение представляет собой соединение следующей формулы или его фармацевтически приемлемую соль, гидрат или сольват:
Указанное выше соединение также известно как 5-[[4-[[(2R)-морфолин-2-ил]метиламино]-5-(трифторметил)-2-пиридил]амино]пиразин-2-карбонитрил.
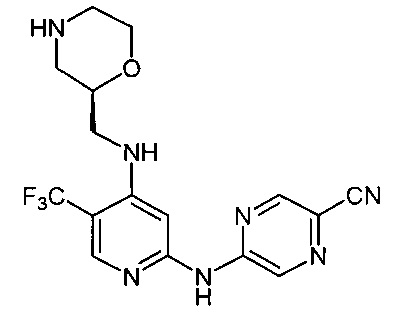
Согласно одному из вариантов реализации соединение представляет собой соединение следующей формулы или его фармацевтически приемлемую соль, гидрат или сольват:
Указанное выше соединение также известно как 5-[[4-[[(2S)-морфолин-2-ил]метиламино]-5-(трифторметил)-2-пиридил]амино]пиразин-2-карбонитрил.
По существу очищенные Формы
Один из аспектов настоящего изобретения относится к TFM-соединениям в очищенной форме.
Согласно одному из вариантов реализации соединение присутствует в по существу очищенной форме и/или в форме, по существу не содержащей загрязняющих примесей.
Согласно одному из вариантов реализации соединение присутствует в по существу очищенной форме с чистотой по меньшей мере 50% по массе, например, по меньшей мере 60% по массе, например, по меньшей мере 70% по массе, например, по меньшей мере 80% по массе, например, по меньшей мере 90% по массе, например, по меньшей мере 95% по массе, например, по меньшей мере 97% по массе, например, по меньшей мере 98% по массе, например, по меньшей мере 99% по массе.
Если не указано конкретно, термин «по существу очищенная форма» относится к соединению в любой стереоизомерной или энантиомерной форме. Например, согласно одному из вариантов реализации термин «по существу очищенная форма» относится к смеси энантиомеров, т.е. очищенных от других соединений. Согласно одному из вариантов реализации термин «по существу очищенная форма» относится к эквимолярной смеси энантиомеров (т.е. рацемической смеси, рацемату). Согласно одному из вариантов реализации термин «по существу очищенная форма» относится к одному энантиомеру, например, оптически чистому энантиомеру.
Согласно одному из вариантов реализации соединение находится в форме, по существу не содержащей загрязняющих примесей, при этом загрязняющие примеси составляют не более 50% по массе, например, не более 40% по массе, например, не более 30% по массе, например, не более 20% по массе, например, не более 10% по массе, например, не более 5% по массе, например, не более 3% по массе, например, не более 2% по массе, например, не более 1% по массе.
Если не указано конкретно, термин «загрязняющие примеси» относится к другим соединениям, не являющимся энантиомерами. Согласно одному из вариантов реализации термин «загрязняющие примеси» относится к другим соединениям и другому энантиомеру.
Согласно одному из вариантов реализации соединение находится в по существу очищенной форме с оптической чистотой, составляющей по меньшей мере 60% (т.е. 60% соединения, из расчета на количество молей, представляет собой целевой энантиомер, а 40% является нежелательным энантиомером), например, по меньшей мере 70%, например, по меньшей мере 80%, например, по меньшей мере 90%, например, по меньшей мере 95%, например, по меньшей мере 97%, например, по меньшей мере 98%, например, по меньшей мере, 99%.
Изомеры
Некоторые соединения могут существовать в одной или более конкретных геометрических, оптических, энантиомерных, диастереомерных, эпимерных, атроповых, стереоизомерных, таутомерных, конформационных или аномерных формах, включая, но не ограничиваясь перечисленными, цис- и транс-формы; Е- и Z-формы; с-, t- и r-формы; эндо- и экзо-формы; R-, S- и мезо-формы; D- и L-формы; d- и l-формы; (+) и (-) формы; кето-, енольные и енолятные формы; син- и анти-формы; синклинальные и антиклинальные формы; α- и β-формы; аксиальные и экваториальные формы; формы «ванна», «кресло», «твист», «конверт» и «полукресло» и их комбинации, в дальнейшем совместно именуемые «изомеры» (или «изомерные формы»).
Следует отметить, что кроме таутомерных форм, как указано ниже, в контексте настоящей заявки из термина «изомеры», специально исключены структурные (или конституционные) изомеры (т.е. изомеры, которые различаются по связям между атомами, а не только по положению атомов в пространстве).
Вышеописанное исключение не относится к таутомерным формам, например, кето-, енольной и енолятной формам, как в, например, следующих таутомерных парах: кето/енол (проиллюстрирована ниже), имин/енамин, амид/иминоспирт, амидин/амидин, нитрозо/оксим, тиокетон/ентиол, N-нитрозо/гидроксиазо и нитро/ацинитро.
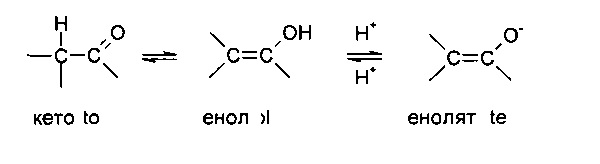
Следует отметить, что в термин «изомеры» конкретно включены соединения с одной или более изотопными заменами. Например, Н может присутствовать в любой изотопной форме, включая 1Н, 2H(D) и 3Н(Т); С может присутствовать в любой изотопной форме, включая 12С, 13С и 14С; О может присутствовать в любой изотопной форме, включая 16O и 18О и т.п.
Если не указано иное, ссылка на конкретное соединение включает все такие изомерные формы, включая их смеси (например, рацемические смеси).
Способы получения (например, асимметрический синтез) и разделения (например, фракционная кристаллизация и хроматографические способы) таких изомерных форм либо известны в данной области техники, либо могут быть легко разработаны путем адаптации предложенных в настоящем документе способов или известных способов известным образом.
Соли
Удобным или целесообразным может оказаться получение, очистка и/или обработка соответствующей соли соединения, например, фармацевтически приемлемой соли. Берг с соавторами (Berge et al., 1977, «Pharmaceutically Acceptable Salts,» J. Pharm. Sci., Vol. 66, pp. 1-19) рассматривают примеры фармацевтически приемлемых солей.
Например, если соединение является анионным или содержит функциональную группу, которая может быть анионной (например, -СООН может быть -СОО-), то может быть образована соль с подходящим катионом. Примеры подходящих неорганических катионов включают, но не ограничиваются ими, ионы щелочных металлов, такие как Na+ и K+, катионы щелочноземельных металлов, такие как Са2+ и Mg2+, и другие катионы, такие как Al3+. Примеры подходящих органических катионов включают, но не ограничиваются ими, ион аммония (т.е. NH4+) и замещенные ионы аммония (например, NH3R+, NH2R2+, NHR3+ NR4+). Примерами некоторых подходящих замещенных ионов аммония являются ионы, полученные из: этиламина, диэтиламина, дициклогексиламина, триэтиламина, бутиламина, этилендиамина, этаноламина, диэтаноламина, пиперазина, бензиламина, фенилбензиламина, холина, меглумина и трометамина, а также аминокислоты, такие как лизин и аргинин. Пример обычного четвертичного аммониевого иона представляет собой N(CH3)4+.
Если соединение является катионным или содержит функциональную группу, которая может быть катионной (например, -NH2 может быть -NH3+), то может быть образована соль с подходящим анионом. Примеры подходящих неорганических анионов включают, но не ограничиваются ими, ионы, полученные из следующих неорганических кислот: соляной, бромоводородной, йодоводородной, серной, сернистой, азотной, азотистой, фосфорной и фосфористой.
Примеры подходящих органических анионов включают, но не ограничиваются ими, ионы, полученные из следующих органических кислот: 2-ацетоксибензойной, уксусной, аскорбиновой, аспарагиновой, бензойной, камфорсульфокислоты, коричной, лимонной, этилендиаминтетрауксусной, этандисульфоновой, этансульфоновой, муравьиной, фумаровой, глюкогептоновой, глюконовой, глутаминовой, гликолевой, гидроксималеиновой, гидроксинафталинкарбоновой, 2-гидроксиэтансульфоновой, молочной, лактобионовой, лауриновой, малеиновой, яблочной, метансульфоновой, слизевой, олеиновой, щавелевой, пальмитиновой, памовой, пантотеновой, фенилуксусной, фенилсульфокислоты, пропионовой, пировиноградной, салициловой, стеариновой, янтарной, сульфаниловой, винной, толуолсульфоновой и валериановой. Примеры подходящих полимерных органических анионов включают, но не ограничиваются ими, ионы, полученные из следующих полимерных кислот: дубильной кислоты, карбоксиметилцеллюлозы.
Если не указано иное, ссылка на конкретное соединение также включает его солевые формы.
Гидраты и сольваты
Удобным или целесообразным может оказаться получение, очистка и/или обработка соответствующего сольвата соединения. В настоящем документе используемый в традиционном смысле термин «сольват» относится к комплексу растворенного вещества (например, соединения, соли соединения) и растворителя. Если растворителем является вода, сольват в целях удобства можно называть гидратом, например, полугидратом, моногидратом, сесквигидратом, дигидратом, тригидратом и т.д.
Если не указано иное, ссылка на конкретное соединение также включает его сольватные и гидратные формы.
Химически защищенные Формы
Удобным или целесообразным может оказаться получение, очистка и/или обработка соединения в химически защищенной форме. В настоящем документе используемый в общепринятом смысле термин «химически защищенная форма» относится к соединению, в котором одна или более реакционно-способных функциональных групп защищены от нежелательных химических реакций при заданных условиях (например, рН, температура, облучение, растворитель и т.п.). На практике применяют хорошо известные химические способы для обратимого перевода в инертное состояние функциональной группы, которая в противном случае была бы реакционно-способной при заданных условиях. В химически защищенной форме одна или более реакционно-способных функциональных групп присутствуют в форме защищенной или защитной группы или (также известной как закрытая или закрывающая группа или блокированная или блокирующая группа). Посредством защиты реакционно-способной функциональной группы можно проводить реакции с участием других незащищенных реакционно-способных функциональных групп без воздействия на защищенную группу; защитная группа может быть удалена, обычно на последующей стадии, причем, удаление по существу не влияет на остальную часть молекулы. См., например, Protective Groups in Organic Synthesis (Т. Greene и P. Wuts; 4th Edition; John Wiley and Sons, 2006).
Большое разнообразие таких способов «защиты», «блокирования» или «закрытия» является широко применяемым и хорошо известным в органическом синтезе. Например, соединение, содержащее две неэквивалентные реакционно-способные функциональные группы, обе из которых обладают реакционной способностью при заданных условиях, может быть превращено в производное с переводом одной из функциональных групп в «защищенное» и, следовательно, инертное состояние при заданных условиях; защищенное таким образом соединение может быть использовано в качестве реагента, который фактически содержит только одну реакционно-способную функциональную группу. После завершения целевой реакции (с участием другой функциональной группы) защищенная группа может быть подвергнута «снятию» защиты с возвращением ее первоначальной функциональности.
Например, аминогруппа может быть защищена, например, превращением в амид (-NRCO-R) или уретан (-NRCO-OR), например, такой как: метиламид (-NHCO-CH3); бензилоксиамид (-NHCO-OCH2C6H5, -NH-Cbz); трет-бутоксиамид (-NHCO-OC(CH3)3, -NH-Boc); 2-бифенил-2-пропоксиамид (-NHCO-OC(CH3)2C6H4C6H5, -NH-Bpoc), 9-флуоренилметоксиамид (-NH-Fmoc), 6-нитровератрилоксиамид (-NH-NVoc), 2-триметилсилилэтилоксиамид (-NH-Teoc), 2,2,2-трихлорэтилоксиамид (-NH-Troc), аллилоксиамид (NH-Alloc), 2-(фенилсульфонил)этилоксиамид (-NH-Psec) или, в соответствующих случаях (например, циклических аминов), превращением в нитроксидный радикал (>N-O•).
Пролекарства
Удобным или целесообразным может оказаться получение, очистка и/или обработка соединения в форме пролекарства. В настоящем документе термин «пролекарство» относится к соединению, которое при метаболизации (например, in vivo) превращается в целевое активное соединение. Как правило, пролекарство является неактивным или менее активным, чем целевое активное соединение, но может обеспечить свойства, полезные с точки зрения обработки, введения или метаболизации.
Композиции
Один из аспектов настоящего изобретения относится к композиции (например, фармацевтической композиции), содержащей TFM-соединение, описанное в настоящем документе, и фармацевтически приемлемые носитель, разбавитель или вспомогательное вещество.
Другой аспект настоящего изобретения относится к способу получения композиции (например, фармацевтической композиции), включающему смешивание TFM-соединения, описанного в настоящем документе, и фармацевтически приемлемого носителя, разбавителя или вспомогательного вещества.
Согласно одному предпочтительному варианту реализации композиция (например, фармацевтическая композиция) подходит для перорального введения субъекту.
Согласно одному предпочтительному варианту реализации композиция находится в форме таблетки для перорального введения, гранул для перорального введения, порошка для перорального введения, капсулы для перорального введения, саше для перорального введения или пилюли для перорального введения.
Применения
TFM-соединения, описанные в настоящем документе, подходят, например, для лечения нарушений (например, заболеваний), при которых улучшения достигают путем ингибирования киназной функции CHK1, описанного в настоящем документе.
Применение в способах ингибирования CHK1
Один из аспектов настоящего изобретения относится к способу ингибирования киназной функции CHK1, in vitro или in vivo, включающему приведение киназы CHK1 в контакт с эффективным количеством TFM-соединения, описанного в настоящем документе.
Один из аспектов настоящего изобретения относится к способу ингибирования киназной функции CHK1 в клетке, in vitro или in vivo, включающему приведение клетки в контакт с эффективным количеством TFM-соединения, описанного в настоящем документе.
Согласно одному из вариантов реализации указанный способ дополнительно включает приведение клетки в контакт с одним или более другими агентами, выбранными из: (а) ингибитора ДНК-топоизомеразы I или II; (b) ДНК-повреждающего агента; (с) антиметаболита или ингибитора тимидилатсинтазы (ТС); (d) нацеленного на микротрубочки агента; (е) ионизирующего излучения; (f) ингибитора регулятора митоза или регулятора митотической контрольной точки; (g) ингибитора трансдуктора сигнала повреждения ДНК; и (n) ингибитора репаративного фермента повреждения ДНК.
Подходящие анализы для определения ингибирования киназной функции CHK1 описаны в настоящем документе и/или известны в данной области техники.
Согласно одному из вариантов реализации способ осуществляют in vitro.
Согласно одному из вариантов реализации способ осуществляют in vivo.
Согласно одному из вариантов реализации TFM-соединение обеспечено в форме фармацевтически приемлемой композиции.
Любой тип клеток может быть подвергнут воздействию, включая, но без ограничения, жировые клетки, клетки легких, желудочно-кишечного тракта (включая, например, клетки кишечника, толстой кишки), груди (молочной железы), яичников, предстательной железы, печени (гепатоциты), почек (почечные), мочевого пузыря, поджелудочной железы, головного мозга и кожи.
Специалист в данной области техники способен легко определить, ингибирует ли соединение-кандидат киназную функцию CHK1. Например, подходящие анализы описаны в настоящем документе.
Применение в способах ингибирования пролиферации клеток и т.д.
Описанные в настоящем документе TFM-соединения, например, (а) регулируют (например, ингибируют) пролиферацию клеток; (b) ингибируют прогрессию клеточного цикла; (с) стимулируют апоптоз клеток; или (d) обеспечивают осуществление комбинации одного или более из вышеперечисленных.
Один из аспектов настоящего изобретения относится к способу регулирования (например, ингибирования) пролиферации клеток (например, пролиферации клетки), ингибирования прогрессии клеточного цикла, стимулирования апоптоза клеток или осуществления комбинации одного или более из вышеперечисленных, in vitro или in vivo, включающему приведение клетки в контакт с эффективным количеством TFM-соединения, описанного в настоящем документе.
Согласно одному из вариантов реализации указанный способ представляет собой способ регулирования (например, ингибирования) пролиферации клеток (например, пролиферации клетки), in vitro или in vivo, включающий приведение клетки в контакт с эффективным количеством TFM-соединения, описанного в настоящем документе.
Согласно одному из вариантов реализации указанный способ дополнительно включает приведение клетки в контакт с одним или более другими агентами, выбранными из: (а) ингибитора ДНК-топоизомеразы I или II; (b) ДНК-повреждающего агента; (с) антиметаболита или ингибитора тимидилатсинтазы (ТС); (d) нацеленного на микротрубочки агента; (е) ионизирующего излучения; (f) ингибитора регулятора митоза или регулятора митотической контрольной точки; (g) ингибитора трансдуктора сигнала повреждения ДНК; и (h) ингибитора репаративного фермента повреждения ДНК.
Согласно одному из вариантов реализации способ осуществляют in vitro.
Согласно одному из вариантов реализации способ осуществляют in vivo.
Согласно одному из вариантов реализации TFM-соединение обеспечено в форме фармацевтически приемлемой композиции.
Любой тип клеток может быть подвергнут воздействию, включая, но без ограничения, клетки легких, желудочно-кишечного тракта (включая, например, клетки кишечника, толстой кишки), груди (молочной железы), яичников, предстательной железы, печени (гепатоциты), почек (почечные), мочевого пузыря, поджелудочной железы, головного мозга и кожи.
Специалист в данной области техники способен легко определить, регулирует ли (например, ингибирует ли) соединение-кандидат пролиферацию клеток и т.д. Например, анализы, которые с удобством могут быть применены для оценки активности, демонстрируемой конкретным соединением, описаны в настоящем документе.
Например, образец клеток (например, из опухоли) может быть выращен in vitro, и соединение введено в контакт с указанными клетками, и проведено наблюдение воздействия соединения на эти клетки. В качестве примера «воздействия» может быть определен морфологический статус клеток (например, живые или мертвые и т.д.). В случае, когда обнаруживают влияние соединения на клетки, этот факт может быть использован в качестве прогностического или диагностического маркера эффективности применения соединения в способах лечения пациента-носителя клеток того же клеточного типа.
Применение в способах терапии
Другой аспект настоящего изобретения относится к TFM-соединению, описанному в настоящем документе, для применения в способе лечения человека или животного посредством терапии.
Другой аспект настоящего изобретения относится к TFM-соединению, описанному в настоящем документе, для применения в способе лечения человека или животного посредством терапии с помощью перорального введения.
Согласно одному из вариантов реализации способ лечения включает лечение с применением как (i) TFM-соединения, описанного в настоящем документе, так и (ii) одного или более других агентов, выбранных из: (а) ингибитора ДНК-топоизомеразы I или II; (b) ДНК-повреждающего агента; (с) антиметаболита или ингибитора тимидилатсинтазы (ТС); (d) нацеленного на микротрубочки агента; (е) ионизирующего излучения; (f) ингибитора регулятора митоза или регулятора митотической контрольной точки; (g) ингибитора трансдуктора сигнала повреждения ДНК; и (h) ингибитора репаративного фермента повреждения ДНК.
Другой аспект настоящего изобретения относится к (а) ингибитору ДНК-топоизомеразы I или II, (b) ДНК-повреждающему агенту, (с) антиметаболиту или ингибитору тимидилатсинтазы (ТС); (d) нацеленному на микротрубочки агенту; (е) системному радиофармацевтическому средству; (f) ингибитору регулятора митоза или регулятора митотической контрольной точки; (g) ингибитору трансдуктора сигнала повреждения ДНК; и (h) ингибитору репаративного фермента повреждения ДНК, описанным в настоящем документе, для применения в способе лечения человека или животного посредством терапии, при этом способ лечения включает лечение с применением как (i) TFM-соединения, описанного в настоящем документе, так и (а) ингибитора ДНК-топоизомеразы I или II, (b) ДНК-повреждающего агента, (с) антиметаболита или ингибитора тимидилатсинтазы (ТС) или (d) нацеленного на микротрубочки агента; (е) системного радиофармацевтического средства; (f) ингибитора регулятора митоза или регулятора митотической контрольной точки; (g) ингибитора трансдуктора сигнала повреждения ДНК; и (h) ингибитора репаративного фермента повреждения ДНК.
Применение для получения лекарственных средств
Другой аспект настоящего изобретения относится к применению TFM-соединения, описанного в настоящем документе, для получения лекарственного средства для применения в способе лечения.
Другой аспект настоящего изобретения относится к применению TFM-соединения, описанного в настоящем документе, для получения лекарственного средства для применения в способе лечения с помощью перорального введения.
Согласно одному из вариантов реализации лекарственное средство содержит TFM-соединение.
Согласно одному из вариантов реализации лечение включает лечение с применением как (i) лекарственного средства, содержащего описанное в настоящем документе TFM-соединение, так и (ii) одного или более других агентов, выбранных из: (а) ингибитора ДНК-топоизомеразы I или II; (b) ДНК-повреждающего агента; (с) антиметаболита или ингибитора тимидилатсинтазы (ТС); (d) нацеленного на микротрубочки агента; (е) ионизирующего излучения; (f) ингибитора регулятора митоза или регулятора митотической контрольной точки; (g) ингибитора трансдуктора сигнала повреждения ДНК; и (h) ингибитора репаративного фермента повреждения ДНК.
Другой аспект настоящего изобретения относится к применению (а) ингибитора ДНК-топоизомеразы I или II; (b) ДНК-повреждающего агента; (с) антиметаболита; или ингибитора тимидилатсинтазы (ТС); (d) нацеленного на микротрубочки агента; (е) ионизирующего излучения; (f) ингибитора регулятора митоза или регулятора митотической контрольной точки; (g) ингибитора трансдуктора сигнала повреждения ДНК; и (h) ингибитора репаративного фермента повреждения ДНК, описанных в настоящем документе, для получения лекарственного средства для применения в способе лечения, при этом лечение включает лечение с применением как (i) TFM-соединения, описанного в настоящем документе, так и (а) ингибитора ДНК-топоизомеразы I или II; (b) ДНК-повреждающего агента; (с) антиметаболита или ингибитора тимидилатсинтазы (ТС); (d) нацеленного на микротрубочки агента; (е) ионизирующего излучения; (f) ингибитора регулятора митоза или регулятора митотической контрольной точки; (g) ингибитора трансдуктора сигнала повреждения ДНК; и (h) ингибитора репаративного фермента повреждения ДНК.
Способы лечения
Другой аспект настоящего изобретения относится к способу лечения, включающему введение пациенту, нуждающемуся в лечении, терапевтически эффективного количества TFM-соединения, описанного в настоящем документе, предпочтительно в форме фармацевтической композиции.
Другой аспект настоящего изобретения относится к способу лечения, включающему пероральное введение пациенту, нуждающемуся в лечении, терапевтически эффективного количества TFM-соединения, описанного в настоящем документе, предпочтительно в форме фармацевтической композиции.
Согласно одному из вариантов реализации способ дополнительно включает введение субъекту одного или более других агентов, выбранных из: (а) ингибитора ДНК-топоизомеразы I или II; (b) ДНК-повреждающего агента; (с) антиметаболита или ингибитора тимидилатсинтазы (ТС); (d) нацеленного на микротрубочки агента; (е) ионизирующего излучения; (f) ингибитора регулятора митоза или регулятора митотической контрольной точки; (g) ингибитора трансдуктора сигнала повреждения ДНК; и (h) ингибитора репаративного фермента повреждения ДНК.
Состояния, подлежащие лечению, - состояния, опосредованные CHK1
Согласно одному из вариантов реализации (например, применения в способах лечения, применения для получения лекарственных средств, способов лечения), лечение представляет собой лечение заболевания или состояния, опосредованного CHK1.
Состояния, подлежащие лечению, - состояния, при которых улучшения достигают путем ингибирования киназной функции CHK1
Согласно одному из вариантов реализации (например, применения в способах лечения, применения для получения лекарственных средств, способов лечения), лечение представляет собой лечение заболевания или состояния, при котором улучшения достигают путем ингибирования киназной функции CHK1.
Расстройства, подлежащие лечению, - пролиферативные заболевания
Согласно одному из вариантов реализации (например, применения в способах лечения, применения для получения лекарственных средств, способов лечения), лечение представляет собой лечение пролиферативного заболевания.
В настоящем документе термин «пролиферативное заболевание» относится к нежелательной или неконтролируемой клеточной пролиферации избыточных или аномальных клеток, которая является неблагоприятной, такой как неопластический или гиперпластический рост.
Согласно одному из вариантов реализации лечение представляет собой лечение пролиферативного заболевания, характеризующегося доброкачественной, предзлокачественной или злокачественной пролиферацией клеток, включая, например: новообразования, гиперплазии и опухоли (например, гистоцитому, глиому, астроцитому, остеому), различные виды рака (см. ниже), псориаз, заболевания костей, фибропролиферативные расстройства (например, соединительных тканей), фиброз легких, атеросклероз, пролиферацию гладкомышечных клеток в кровеносных сосудах, такую как стеноз или рестеноз после ангиопластики.
Расстройства, подлежащие лечению, - рак
Согласно одному из вариантов реализации (например, применения в способах лечения, применения для получения лекарственных средств, способов лечения), лечение представляет собой лечение рака.
Согласно одному из вариантов реализации лечение представляет собой лечение пролиферативного заболевания.
Согласно одному из вариантов реализации лечение представляет собой лечение рака.
Согласно одному из вариантов реализации лечение представляет собой лечение рака легкого, мелкоклеточного рака легкого, немелкоклеточного рака легкого, рака желудочно-кишечного тракта, рака желудка, рака кишечника, рака толстой кишки, рака прямой кишки, рака ободочной и/или прямой кишки, рака щитовидной железы, рака молочной железы, рака яичника, рака эндометрия, рака предстательной железы, рака яичка, рака печени, рака почки, почечно-клеточной карциномы, рака мочевого пузыря, рака поджелудочной железы, рака мозга, нейробластомы, глиомы, саркомы, остеосаркомы, рака костей, рака носоглотки (например, рака головы, рака шеи), рака кожи, плоскоклеточного рака, саркомы Капоши, меланомы, злокачественной меланомы, лимфомы или лейкоза.
Согласно одному из вариантов реализации лечение представляет собой лечение:
карциномы, например, карциномы мочевого пузыря, молочной железы, толстой кишки (например, колоректальных карцином, таких как аденокарцинома толстой кишки и аденома толстой кишки), почки, эпидермальной карциномы, печени, легкого (например, аденокарциномы, мелкоклеточного рака легкого и немелкоклеточных карцином легкого), пищевода, желчного пузыря, яичника, поджелудочной железы (например, экзокринной карциномы поджелудочной железы), желудка, шейки матки, щитовидной железы, предстательной железы, кожи (например, плоскоклеточной карциномы);
опухоли кроветворной ткани лимфоидного происхождения, например, лейкоза, острого лимфобластного лейкоза, В-клеточной лимфомы, Т-клеточной лимфомы, лимфомы Ходжкина, неходжкинской лимфомы, волосатоклеточной лимфомы или лимфомы Беркетта;
опухоли кроветворной ткани миелоидного происхождения, например, острых и хронических миелоидных лейкозов, миелодиспластического синдрома или промиелоцитарного лейкоза;
мезенхимальной опухоли, например фибросаркомы или рабдомиосаркомы;
опухоли центральной или периферической нервной системы, например, астроцитомы, нейробластомы, глиомы или шванномы;
меланомы; семиномы; тератокарциномы; остеосаркомы; пигментной ксеродермы; кератоакантомы; фолликулярного рака щитовидной железы или саркомы Капоши.
Согласно одному из вариантов реализации лечение представляет собой лечение рака головы; рака шеи; рака нервной системы; рака мозга; нейробластомы; рака легкого/средостения; рака молочной железы; рака пищевода; рака желудка; рака печени; рака желчевыводящих путей; рака поджелудочной железы; рака тонкой кишки; рака толстой кишки; рака ободочной и/или прямой кишки; рака женских половых органов; рака мочеполовой системы; рака яичника; рака щитовидной железы; рака надпочечника; рака кожи; меланомы; остеосаркомы; саркомы мягких тканей; злокачественных опухолей детского возраста; болезни Ходжкина; неходжкинской лимфомы; миеломы; лейкоза или метастазов из неизвестного первичного очага.
Согласно одному из вариантов реализации лечение представляет собой лечение: рака легкого, рака молочной железы, рака яичника, рака поджелудочной железы, рака ободочной и/или прямой кишки, лимфомы, меланомы, глиомы или нейробластомы.
Согласно одному из вариантов реализации рак характеризуется или дополнительно характеризуется наличием дефицита р53. Согласно одному из вариантов реализации рак представляет собой р53-дефицитный рак.
Согласно одному из вариантов реализации рак характеризуется или дополнительно характеризуется наличием амплификации MYC. Согласно одному из вариантов реализации рак представляет собой рак с амплифицированным MYC.
Согласно одному из вариантов реализации рак характеризуется или дополнительно характеризуется наличием амплификации c-MYC. Согласно одному из вариантов реализации рак представляет собой рак с амплифицированным c-MYC.
Согласно одному из вариантов реализации рак характеризуется или дополнительно характеризуется наличием амплификации MYCN. Согласно одному из вариантов реализации рак представляет собой рак с амплифицированным MYCN.
Согласно одному из вариантов реализации рак характеризуется или дополнительно характеризуется гиперэкспрессией MYC. Согласно одному из вариантов реализации рак представляет собой рак, характеризующийся гиперэкспрессией MYC.
Согласно одному из вариантов реализации рак характеризуется или дополнительно характеризуется гиперэкспрессией MYCN. Согласно одному из вариантов реализации рак представляет собой рак, характеризующийся гиперэкспрессией MYCN.
Согласно одному из вариантов реализации рак характеризуется или дополнительно характеризуется, гиперэкспрессией c-MYC. Согласно одному из вариантов реализации рак представляет собой рак, характеризующийся гиперэкспрессией c-MYC.
Согласно одному из вариантов реализации рак представляет собой нейробластому с амплифицированным MYCN.
Согласно одному из вариантов реализации рак представляет собой В-клеточную лимфому с амплифицированным c-MYC.
Согласно одному из вариантов реализации рак характеризуется или дополнительно характеризуется повышенным эндогенным репликативным стрессом. Согласно одному из вариантов реализации рак представляет собой рак, характеризующийся повышенным эндогенным репликативным стрессом.
Согласно одному из вариантов реализации рак характеризуется или дополнительно характеризуется повышенной эндогенной активацией сигнального пути CHK1. Согласно одному из вариантов реализации рак представляет собой рак, характеризующийся повышенной эндогенной активацией сигнального пути CHK1.
Согласно одному из вариантов реализации лечение представляет собой лечение раковых метастазов.
Согласно одному из вариантов реализации рак характеризуется или дополнительно характеризуется наличием раковых стволовых клеток.
Противораковый эффект может быть опосредован одним или более механизмами, включая, но без ограничения, регуляцию клеточной пролиферации, ингибирование прогрессии клеточного цикла, ингибирование ангиогенеза (образования новых кровеносных сосудов), ингибирование образования метастазов (распространения опухоли от ее первичного очага), ингибирование миграции клеток (распространения раковых клеток в другие части организма), ингибирование опухолевой инвазии (распространения опухолевых клеток в соседние нормальные структуры) или стимулирование апоптоза клеток (программируемой клеточной гибели). Соединения согласно настоящему изобретению могут быть применены для лечения описанных в настоящем документе раковых заболеваний независимо от описанных в настоящем документе механизмов.
Лечение
Термин «лечение», используемый в настоящем документе в контексте лечения расстройства, в целом относится к лечению человека или животного (например, в ветеринарии), при котором достигают некоторого желаемого терапевтического эффекта, например, ингибирования развития заболевания, и включает снижение скорости прогрессирования, прекращение прогрессирования, облегчение симптомов расстройства, уменьшение степени тяжести расстройства и излечение расстройства. Термин также включает лечение в качестве профилактической меры (т.е. профилактику). Например, применение в случае пациентов, у которых еще не произошло развития расстройства, но которые входят в группу риска развития расстройства, охватывается термином «лечение».
Например, лечение включает профилактику рака, снижение заболеваемости раком, облегчение симптомов рака и т.д.
Термин «терапевтически эффективное количество» в настоящем документе относится к такому количеству соединения, или вещества, композиции или лекарственной формы, содержащей соединение, которое эффективно для обеспечения некоторого желаемого терапевтического эффекта, соизмеримого с разумным соотношением польза/риск, при введении в соответствии с требуемой схемой лечения.
Комбинированная терапия
Термин «лечение» включает комбинированное лечение и терапию, при которых сочетают два или более видов лечения или терапии, например, последовательно или одновременно. Например, соединения, описанные в настоящем документе, также могут быть применены в комбинированной терапии, например, в сочетании с другими агентами. Примеры видов лечения и терапии включают, но не ограничиваются ими, химиотерапию (введение активных агентов, включая, например, лекарственные средства, антитела (например, как в иммунотерапии), пролекарства (например, как в фотодинамической терапии, способах GDEPT, ADEPT, и т.д.)); хирургическое лечение; лучевую терапию; фотодинамическую терапию; генную терапию и контролируемый пищевой рацион.
Один из аспектов настоящего изобретения относится к соединению, описанному в настоящем документе, в комбинации с одним или более (например, 1, 2, 3, 4 и т.д.) дополнительными терапевтическими агентами, например, агентами или видами терапии, которые регулируют рост, или выживаемость, или дифференцировку клеток посредством различных механизмов и таким образом воздействуют на некоторые характерные особенности развития рака.
Конкретная комбинация будет составлена по усмотрению лечащего врача, который должен выбирать дозировки, используя свои обычные знания в различных областях, и режимы дозирования, известные квалифицированному практикующему врачу.
Агенты (т.е. соединения, описанные в настоящем документе, а также один или более других агентов) могут быть введены одновременно или последовательно и могут быть введены согласно индивидуальным варьируемым режимам дозирования и посредством различных путей. Например, в случае последовательного введения агенты можно вводить через короткие интервалы времени (например, в течение периода, составляющего 5-10 минут) или более длинные интервалы (например, с промежутком 1, 2, 3, 4 или более часов или даже большим промежутком при необходимости), причем точный режим дозирования должен быть соизмеримым со свойствами терапевтического агента (агентов).
Агенты (т.е. соединения, описанные в настоящем документе, а также один или более других агентов) могут быть получены вместе в виде единой лекарственной формы или, альтернативно, индивидуальные агенты могут быть получены раздельно и представлены вместе в форме набора, необязательно с инструкциями по их применению.
Дополнительные агенты для комбинированной терапии
Как было указано выше в настоящем документе, в некоторых вариантах реализации TFM-соединения применяют в комбинации с (например, в сочетании с) с одним или более другими агентами, выбранными из: (а) ингибитора ДНК-топоизомеразы I или II; (b) ДНК-повреждающего агента; (с) антиметаболита или ингибитора тимидилатсинтазы (ТС); (d) нацеленного на микротрубочки агента; (е) ионизирующего излучения; (f) ингибитора регулятора митоза или регулятора митотической контрольной точки; (g) ингибитора трансдуктора сигнала повреждения ДНК; и (h) ингибитора репаративного фермента повреждения ДНК.
В случае применения как TFM-соединения, так и одного или более других агентов, они могут быть применены (например, путем приведения в контакт, введены и т.д.) в любом порядке. Кроме того, они могут быть применены (например, путем приведения в контакт, введены и т.д.) вместе, как часть единого состава, или раздельно, в виде индивидуальных составов.
Например, касательно способов лечения с применением как TFM-соединения, так и одного или более других агентов, лечение с помощью (например, введение) TFM-соединения может быть осуществлено перед, одновременно с или после лечения с помощью (например, введением) одного или более других агентов или их комбинации.
Согласно одному из вариантов реализации лечение с помощью (например, введение) TFM-соединения осуществляют одновременно с или после лечения с помощью (например, введением) одного или более других агентов.
Согласно одному из вариантов реализации один или более других агентов представляют собой ингибитор ДНК-топоизомеразы I или II; например, этопозид, топотекан, камптотецин, иринотекан, SN-38, доксорубицин, даунорубицин, эпирубицин и митоксантрон.
Согласно одному из вариантов реализации один или более других агентов представляют собой ДНК-повреждающий агент; например, алкилирующий агент, например, темозоломид, дакарбазин, митомицин С, циклофосфамид, ифосфамид, BCNU, CCNU, мелфалан, бусульфан и хлорамбуцил; платинирующий агент, например, цисплатин, карбоплатин, и оксалиплатин; или соединение, которое генерирует свободные радикалы, например, блеомицин.
Согласно одному из вариантов реализации один или более других агентов представляют собой антиметаболит или ингибитор тимидилатсинтазы (ТС); например; 5-фторурацил, гидроксимочевина, гемцитабин, цитарабин, флударабин, капецитабин, неларабин, ралтитрексед, пеметрексед и ZD9331.
Согласно одному из вариантов реализации один или более других агентов представляют собой нацеленный на микротрубочки агент; например, паклитаксел, доцетаксел, кабазитаксел, эрибулин, винкристин, винбластин и винорелбин.
Согласно одному из вариантов реализации один или более других агентов представляют собой ионизирующее излучение (например, как часть лучевой терапии), например, доставленное путем внешнего лучевого облучения или доставленное путем введения системных радиофармацевтических средств, например, 131I- метайодобензилгуанидина, йодида (131I) натрия, тозитумомаб йод (131I) и ибритумомаб (90Y) тиуксетан.
Согласно одному из вариантов реализации один или более других агентов представляют собой ингибитор регулятора митоза или регулятор митотической контрольной точки, например, ингибитор Wee1 киназы, ингибитор Aurora киназы или ингибитор Polo-подобной киназы 1.
Согласно одному из вариантов реализации один или более других агентов представляют собой ингибитор трансдуктора сигнала повреждения ДНК, например, ингибитор ATR киназы, ингибитор ATM киназы, ингибитор CHK2 или ингибитор MK2.
Согласно одному из вариантов реализации один или более других агентов представляют собой ингибитор репаративного фермента повреждения ДНК, например, ингибитор поли(АДФ-рибоза)-полимеразы (PARP), например, олапариб.
Другие применения
TFM-соединения, описанные в настоящем документе, также могут быть применены в качестве добавок при культивировании клеток для ингибирования киназной функции CHK1, например, для ингибирования пролиферации клеток и т.д.
TFM-соединения, описанные в настоящем документе, также могут быть применены как часть исследования in vitro, например, для определения вероятности того, что лечение организма-кандидата с применением рассматриваемого соединения будет иметь положительный результат.
TFM-соединения, описанные в настоящем документе, также могут быть применены в качестве стандартного соединения, например, при проведении анализа, с целью идентификации других соединений, других ингибиторов киназной функции CHK1, других антипролиферативных агентов, других противораковых агентов и т.д.
Наборы
Один из аспектов изобретения относится к набору, содержащему (а) TFM-соединение, описанное в настоящем документе, или композицию, содержащую описанное в настоящем документе TFM-соединение, например, предпочтительно обеспеченное в подходящей емкости и/или с подходящей упаковкой; и (b) инструкции по применению, например, письменные указания относительно введения соединения или композиции.
Согласно одному из вариантов реализации набор дополнительно содержит один или более других агентов, выбранных из:
(а) ингибитора ДНК-топоизомеразы I или II; (b) ДНК-повреждающего агента; (с) антиметаболита или ингибитора тимидилатсинтазы (ТС); (d) нацеленного на микротрубочки агента; (е) системного радиофармацевтического средства; (f) ингибитора регулятора митоза или регулятора митотической контрольной точки; (g) ингибитора трансдуктора сигнала повреждения ДНК; и (h) ингибитора репаративного фермента повреждения ДНК.
Письменные инструкции могут также включать перечень показаний, для которых активный ингредиент представляет собой подходящий вид лечения.
Пути введения
TFM-соединение или содержащая TFM-соединение фармацевтическая композиция могут быть введены субъекту посредством любого подходящего пути введения, либо системно/периферически, либо местно (т.е. в место проявления требуемого эффекта).
Пути введения включают, но не ограничиваются ими, пероральный (например, посредством приема внутрь); буккальный; сублингвальный; трансдермальный (включая, например, введение с помощью повязки, пластыря и т.д.); трансмукозальный (включая, например, введение с помощью повязки, пластыря и т.д.); интраназальный (например, с помощью назального аэрозоля); глазной (например, с помощью глазных капель); легочный (например, с помощью ингаляционной или инсуффляционной терапии с применением, например, аэрозоля, например, через полость рта или носа); ректальный (например, с помощью суппозиториев или клизмы); вагинальный (например, с помощью пессария); парентеральный, например, посредством инъекции, включая подкожный, внутрикожный, внутримышечный, внутривенный, внутриартериальный, внутрисердечный, интратекальный, интраспинальный, интракапсулярный, субкапсулярный, интраорбитальный, интраперитонеальный, внутритрахеальный, субкутикулярный, внутрисуставный, субарахноидальный и интрастернальный; имплантацию депо или резервуара, например, подкожно или внутримышечно.
Предпочтительно, путь введения представляет собой пероральный, и TFM-соединение или содержащую TFM-соединение фармацевтическую композицию вводят субъекту перорально.
Субъект/Пациент
Субъектом/пациентом могут являться хордовые, позвоночные, млекопитающие, плацентарные млекопитающие, сумчатые (например, кенгуру, вомбат), грызуны (например, морская свинка, хомяк, крыса, мышь), мышевидные (например, мышь), зайцеобразные (например, кролик), пернатые (например, птица), псовые (например, собака), кошачьи (например, кошка), лошадиные (например, лошадь), свиноподобные (например, свинья), овечьи (например, овца), крупный рогатый скот (например, корова), приматы, обезьяноподобные (например, обезьяна или человекообразная обезьяна), обезьяны (например, мартышка, павиан), человекообразные обезьяны (например, горилла, шимпанзе, орангутан, гиббон) или человек.
Кроме того, субъект/пациент может находиться в любой из его форм развития, например, в форме плода.
Согласно одному предпочтительному варианту реализации субъектом/пациентом является человек.
Составы
Наряду с тем что TFM-соединение можно вводить отдельно, предпочтительно его обеспечение в виде фармацевтического состава (например, композиции, препарата, лекарственного средства), содержащего по меньшей мере одно TFM-соединение, описанное в настоящем документе, вместе с одним или более другими фармацевтически приемлемыми ингредиентами, хорошо известными специалистам в данной области техники, включая, но не ограничиваются ими, фармацевтически приемлемые носители, разбавители, вспомогательные вещества, адъюванты, наполнители, буферы, консерванты, антиоксиданты, смазывающие вещества, стабилизаторы, солюбилизаторы, поверхностно-активные вещества (например, смачивающие агенты), маскирующие агенты, красители, ароматизаторы и подсластители. Состав может дополнительно содержать другие активные агенты, например, другие терапевтические или профилактические агенты.
Таким образом, согласно настоящему изобретению также предложены фармацевтические композиции, определенные выше, и способы получения фармацевтической композиции, включающие смешивание по меньшей мере одного описанного в настоящем документе TFM-соединения вместе с одним или несколькими другими фармацевтически приемлемыми ингредиентами, хорошо известными специалистам в данной области техники, например, носителями, разбавителями, вспомогательными веществами и т.д. При получении композиций в виде отдельных единиц (например, таблеток и т.д.) каждая единица содержит заранее определенное количество (дозу) соединения.
В настоящем документе термин «фармацевтически приемлемый» относится к соединениям, ингредиентам, веществам, композициям, лекарственным формам и т.д., которые, с медицинской точки зрения, подходят для применения в контакте с тканями рассматриваемого субъекта (например, человека), не демонстрируя повышенной токсичности, раздражения, аллергической реакции или других проблем или осложнений, соизмеримых с разумным соотношением польза/риск. Каждый носитель, разбавитель, вспомогательное вещество и т.д., также должен быть «приемлемым» в смысле совместимости с другими ингредиентами состава.
Сведения о подходящих носителях, разбавителях, вспомогательных веществах и т.д. можно найти в стандартной фармацевтической литературе, например, Remington's Pharmaceutical Sciences, 18th edition, Mack Publishing Company, Easton, Pa., 1990 и Handbook of Pharmaceutical Excipients. 5th edition, 2005. Составы могут быть получены посредством любых способов, хорошо известных в области фармации. Такие способы включают стадию объединения соединения с носителем, который содержит один или более вспомогательных ингредиентов. Как правило, составы получают посредством равномерного и однородного объединения соединения с носителями (например, жидкими носителями, мелкодисперсным твердым носителем и т.д.) и далее формования продукта при необходимости.
Состав может быть получен с обеспечением быстрого или медленного высвобождения; немедленного, замедленного, приуроченного к определенному времени или пролонгированного высвобождения или их комбинации.
Составы могут быть надлежащим образом получены в форме жидкостей, растворов (например, водных, неводных), суспензий (например, водных, неводных), эмульсий (например, типа «масло в воде», «вода в масле»), эликсиров, сиропов, электуариев, жидкостей для полоскания рта, капель, таблеток (включая, например, таблетки с покрытием), гранул, порошков, леденцов, пастилок, капсул (включая, например, твердые и мягкие желатиновые капсулы), саше, пилюль, ампул, пилюль больших размеров, суппозиториев, пессариев, настоек, гелей, паст, мазей, кремов, лосьонов, масел, пен, спреев, тонкораспыляемых жидкостей или аэрозолей.
Составы могут быть надлежащим образом обеспечены в виде пластыря, лейкопластыря, бандажа, повязки, и т.п., которые пропитывают одним или более соединениями и необязательно одним или более другими фармацевтически приемлемыми ингредиентами, включая, например, усилители проникновения, проницаемости и всасывания. Составы также могут быть надлежащим образом обеспечены в форме депо или резервуара.
Соединение может быть растворено в, суспендировано в или смешано с одним или более другими фармацевтически приемлемыми ингредиентами. Соединение может быть заключено в липосому или другую микрочастицу, предназначенную для направленной доставки соединения, например, к компонентам крови или одному или более органам.
Составы, подходящие для перорального введения (например, посредством приема внутрь), включают жидкости, растворы (например, водные, неводные), суспензии (например, водные, неводные), эмульсии (например, типа «масло в воде», «вода в масле"), эликсиры, сиропы, электуарии, таблетки, гранулы, порошки, капсулы, саше, пилюли, ампулы, пилюли больших размеров.
Составы, подходящие для буккального введения, включают жидкости для полоскания рта, леденцы, пастилки, а также пластыри, лейкопластыри, депо и резервуары. Леденцы, как правило, содержат соединение в ароматизированной основе, обычно сахарозе и камеди или трагаканте. Пастилки, как правило, содержат соединение в инертной матрице, такой как желатин и глицерин или сахароза и камедь. Жидкости для полоскания рта, как правило, содержат соединение в подходящем жидком носителе.
Составы, подходящие для сублингвального введения, включают таблетки, пастилки, леденцы, капсулы и пилюли.
Составы, подходящие для перорального трансмукозального введения, включают жидкости, растворы (например, водные, неводные), суспензии (например, водные, неводные), эмульсии (например, типа «масло в воде», «вода в масле»), жидкости для полоскания рта, леденцы, пастилки, а также пластыри, лейкопластыри, депо и резервуары.
Составы, подходящие для неперорального трансмукозального введения, включают жидкости, растворы (например, водные, неводные), суспензии (например, водные, неводные), эмульсии (например, типа «масло в воде», «вода в масле»), суппозитории, пессарии, гели, пасты, мази, кремы, лосьоны, масла, а также пластыри, лейкопластыри, депо и резервуары.
Составы, подходящие для трансдермального введения, включают гели, пасты, мази, кремы, лосьоны и масла, а также пластыри, лейкопластыри, бандажи, повязки, депо и резервуары.
Таблетки могут быть получены посредством обычных способов, например, прессованием или формованием, необязательно с одним или более вспомогательными ингредиентами. Прессованные таблетки могут быть получены путем прессования в подходящем таблеточном прессе соединения в легкосыпучей форме, такой как порошок или гранулы, необязательно смешанного с одним или более связующими веществами (например, повидоном, желатином, камедью, сорбитом, трагакантом, гидроксипропилметилцеллюлозой); наполнителями или разбавителями (например, лактозой, микрокристаллической целлюлозой, гидрофосфатом кальция); смазывающими веществами (например, стеаратом магния, тальком, диоксидом кремния); разрыхлителями (например, крахмалгликолятом натрия, поперечно-сшитым повидоном, поперечно-сшитой натрий-карбоксиметилцеллюлозой); поверхностно-активными или диспергирующими или смачивающими агентами (например, лаурилсульфатом натрия); консервантами (например, метил-п-гидроксибензоатом, пропил-п-гидроксибензоатом, сорбиновой кислотой); ароматизаторами, усилителями вкуса и подсластителями. Формованные таблетки могут быть получены путем формования в подходящей формовочной машине смеси порошкообразного соединения, увлажненной инертным жидким разбавителем. Таблетки необязательно могут иметь покрытие или насечку и могут быть приготовлены таким образом, чтобы обеспечивать медленное или контролируемое высвобождение содержащегося в них соединения, с применением, например, гидроксипропилметилцеллюлозы в различных соотношениях для обеспечения желаемого профиля высвобождения. Таблетки необязательно могут быть снабжены покрытием, например, с целью изменения профиля высвобождения, например, кишечнорастворимой оболочкой, с обеспечением высвобождения в отделах кишечника, а не в желудке.
Мази, как правило, получают из соединения и парафиновой или водорастворимой мазевой основы.
Кремы, как правило, получают из соединения и кремовой основы типа «масло в воде». При необходимости водная фаза кремовой основы может содержать, например, по меньшей мере около 30% масс./масс, многоатомного спирта, т.е. спирта, содержащего две или более гидроксильные группы, такого как пропиленгликоль, бутан-1,3-диол, маннит, сорбит, глицерин и полиэтиленгликоль и их смеси. Составы для местного введения могут при необходимости содержать соединение, усиливающее всасывание или проникновение соединения через кожу или другие пораженные участки. Примеры таких усилителей проникновения через кожу включают диметилсульфоксид и родственные аналоги.
Эмульсии, как правило, получают из соединения и масляной фазы, которая необязательно может содержать только эмульгатор (иначе известный как эмульгирующий агент), или она может содержать смесь по меньшей мере одного эмульгатора с жиром или маслом или и жиром, и маслом. Предпочтительно, в эмульсию включают гидрофильный эмульгатор вместе с липофильным эмульгатором, который выступает в качестве стабилизатора. Также предпочтительно включение и масла, и жира. Совместно эмульгатор (эмульгаторы) со стабилизатором (стабилизаторами) или без него образуют так называемый эмульгирующий воск, а воск вместе с маслом и/или жиром составляют так называемую эмульгирующую мазевую основу, которая образует масляную дисперсную фазу в составе крема.
Подходящие эмульгаторы и стабилизаторы эмульсии включают Твин 60 (Tween 60), Спан 80 (Span 80), цетостеариловый спирт, миристиловый спирт, глицерилмоностеарат и лаурилсульфат натрия. Выбор подходящих для состава масел или жиров основан на достижении требуемых косметических свойств, поскольку растворимость соединения в большинстве видов масел, применяемых в фармацевтических эмульсионных составах, может быть очень низкой. Таким образом, крем предпочтительно должен представлять собой нежирный, не оставляющий пятен и смываемый продукт с подходящей консистенцией во избежание утечки из тюбика или других емкостей. Могут быть применены одно- или двухосновные алкиловые сложные эфиры с неразветвленной или разветвленной цепью, такие как диизоадипат, изоцетилстеарат, сложный диэфир пропиленгликоля и жирных кислот кокосового масла, изопропилмиристат, децилолеат, изопропилпальмитат, бутилстеарат, 2-этилгексилпальмитат или смесь сложных эфиров с разветвленной цепью, известная как Кродамол CAP (Crodamol САР), причем, три последних сложных эфира являются предпочтительными. Они могут быть применены отдельно или в комбинации в зависимости от требуемых свойств. Альтернативно, могут быть применены липиды с высокой температурой плавления, такие как белый мягкий парафин и/или жидкий парафин или другие минеральные масла.
Составы, подходящие для интраназального введения, в которых носитель представляет собой жидкость, включают, например, назальный спрей, капли для носа или аэрозоль для введения с помощью распылителя и содержат водные или масляные растворы соединения.
Составы, подходящие для интраназального введения, в которых носитель представляет собой твердое вещество, включают, например, составы в виде крупного порошка с размером частиц, например, в диапазоне от примерно 20 до примерно 500 мкм, которые вводят способом, аналогичным употреблению нюхательного табака, т.е. быстрым вдыханием через носовой ход порошка из емкости, подносимой близко к носу.
Составы, подходящие для легочного введения (например, с помощью ингаляционной или инсуффляционной терапии), включают составы в виде спрей-аэрозоля в упаковке под давлением с применением подходящего пропеллента, такого как дихлордифторметан, трихлорфторметан, дихлортетрафторэтан, диоксид углерода или другие подходящие газы.
Составы, подходящие для глазного введения, включают глазные капли, в которых соединение растворено или суспендировано в подходящем носителе, в частности, водном растворителе для соединения.
Составы, подходящие для ректального введения, могут быть представлены в виде суппозиториев с подходящей основой, включая, например, природные или отвержденные масла, воски, жиры, полужидкие или жидкие полиолы, например, масло какао или салицилат или в виде раствора или суспензии для лечения с помощью клизмы.
Составы, подходящие для вагинального введения, могут быть представлены в виде пессариев, тампонов, кремов, гелей, паст, пен или составов для распыления, содержащие, кроме соединения, подходящие носители, известные в данной области техники.
Составы, подходящие для парентерального введения (например, посредством инъекции), включают водные или неводные изотонические апирогенные стерильные жидкости (например, растворы, суспензии), в котором соединение растворено, суспендировано или обеспечено иным образом (например, заключено в липосому или другую микрочастицу). Такие жидкости могут дополнительно содержать другие фармацевтически приемлемые ингредиенты, такие как антиоксиданты, буферы, консерванты, стабилизаторы, бактериостатические агенты, суспендирующие агенты, загустители и растворенные компоненты, превращающие состав в изотонический крови (или другой соответствующей физиологической жидкости) предполагаемого пациента. Примеры вспомогательных веществ включают, например, воду, спирты, полиолы, глицерин, растительные масла и т.п. Примеры подходящих изотонических носителей для применения в таких составах включают физиологический раствор для инъекций, раствор Рингера, или раствор Рингера с лактатом. Как правило, концентрация соединения в жидкости составляет от около 1 нг/мл до примерно 10 мкг/мл, например, от примерно 10 нг/мл до примерно 1 мкг/мл. Составы могут находиться в герметичных однодозовых или многодозовых емкостях, например, ампулах и флаконах, и могут храниться высушенном сублимацией (лиофилизированном) состоянии, требующем лишь добавления стерильного жидкого носителя, например, воды для инъекций, непосредственно перед применением. Инъекционные экстемпоральные растворы и суспензии могут быть получены из стерильных порошков, гранул и таблеток.
Дозирование
Специалисту в данной области техники будет понятно, что подходящие дозировки TFM-соединений и композиций, содержащих TFM-соединения, могут варьироваться от пациента к пациенту. Определение оптимальной дозы, в общем случае будет включать соизмерение уровней терапевтического эффекта против любого риска или вредных побочных эффектов. Выбранный уровень дозировки будет зависеть от множества факторов, включая, но без ограничения, активность конкретного TFM-соединения, путь введения, время введения, скорость экскреции TFM-соединения, продолжительность лечения, другие лекарственные средства, соединения и/или вещества, применяемые в комбинации, степень тяжести расстройства и вид, пол, возраст, массу тела, состояние, общее состояние здоровья и предшествующий анамнез пациента. Количество TFM-соединения и путь введения в конечном счете будут оставлены на усмотрение лечащего врача, ветеринара или практикующего врача, хотя, как правило, доза будет выбрана с учетом достижения локальных концентраций в месте действия, которые обеспечивают желаемый эффект, по существу не вызывая неблагоприятных или вредных побочных эффектов.
Введение может быть осуществлено в одной дозе, непрерывно или периодически (например, раздельными дозами через подходящие промежутки времени) на протяжении всего курса лечения. Способы определения наиболее эффективных путей и доз для введения хорошо известны специалистам в данной области техники и будет зависеть от применяемого для терапии состава, цели терапии, клетки-мишени (клеток-мишеней), подлежащих лечению, и субъекта, подлежащего лечению. Могут быть осуществлены однократные или многократные введения с уровнем дозы и режимом введения, выбранными лечащим врачом, ветеринаром или практикующим врачом.
В общем, подходящая доза TFM-соединения находится в диапазоне от примерно 10 мкг до примерно 250 мг (чаще от примерно 100 мкг до примерно 25 мг) на килограмм массы тела субъекта в сутки. В случае, когда соединение представляет собой соль, сложный эфир, амид, пролекарство и т.п., вводимое количество рассчитывают на основе исходного соединения и, таким образом, фактическая масса, требующаяся для введения, пропорционально увеличивается.
ПРИМЕРЫ
Химический синтез
Следующие примеры предложены исключительно для иллюстрации настоящего изобретения и не ограничивают объем изобретения, описанного в настоящем документе.
Синтез 1
5-[[4-[[(2R)-морфолин-2-ил]метиламино]-5-(трифторметил)-2-пиридил]амино]пиразин-2-карбонитрил (Соединение 1)
Синтез 1А
(R)-трет-бутил-2-(тозилоксиметил)морфолин-4-карбоксилат

Триэтиламин (15,46 мл, 110 ммоль) добавляли к (R)-трет-бутил 2-(гидроксиметил)-морфолин-4-карбоксилату (21,73 г, 100 ммоль) в дихлорметане (50,0 мл) с получением бесцветного раствора, который охлаждали на ледяной бане. 4-толуолсульфонил хлорид (20,02 г, 105 ммоль) добавляли небольшими порциями при поддержании внутренней температуры ниже 3°С. Суспензию перемешивали в течение 21 часов при комнатной температуре до концентрирования в вакууме. Неочищенный материал растворяли в этилацетате (750 мл), промывали водой (450 мл), солевым раствором (200 мл) и сушили над сульфатом магния. После фильтрования и удаления летучих веществ в вакууме, добавляли гексан (150 мл) и полученный белый осадок фильтровали, промывали гексаном (300 мл) и сушили с получением титульного соединения в виде белого порошка (35,66 г, 96%).
1Н ЯМР (500 МГц, CDCl3) δ 1,46 (9Н, s), 2,46 (3Н, s), 2,62-2,73 (1Н, m), 2,85-2,94 (1Н, m), 3,44-3,49 (1Н, m), 3,58-3,63 (1Н, m), 3,77-3,94 (3Н, m), 3,99-4,06 (2Н, m), 7,36 (2Н, d, J=8,5 Гц), 7,80 (2Н, d, J=8,5 Гц). ЖХ-МС (Agilent 4 мин) Rt 2,90 мин; m/z (ESI) 372 [М+Н+].
Синтез 1В
(S)-трет-бутил-2-((2-хлор-5-(трифторметил)пиридин-4-иламино)метил)морфолин-4-карбоксилат
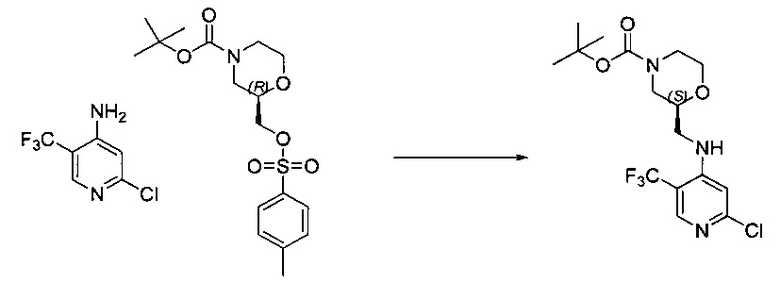
К раствору 2-хлор-5-(трифторметил)пиридин-4-амина (2,9 г, 14,75 ммоль) в диметилформамиде (95 мл) по каплям при комнатной температуре добавляли гидрид натрия (60% по массе в масле; 1,180 г, 29,5 ммоль) и указанную смесь перемешивали в течение 10 минут при 80°С. Добавляли по каплям (R)-трет-бутил-2-(тозилоксиметил)морфолин-4-карбоксилат и реакционную смесь перемешивали при 80°С в течение 2,5 часов. Реакционную смесь охлаждали и выливали в насыщенный водный раствор гидрокарбоната натрия (100 мл), разбавляли водой (250 мл) и экстрагировали этилацетатом (100 мл). После разделения двух слоев водный слой дополнительно экстрагировали этилацетатом (2×100 мл). Объединенные органические слои промывали солевым раствором (4×100 мл), сушили над сульфатом магния, фильтровали, концентрировали и тщательно сушили в вакууме. Неочищенный материал очищали с помощью колоночной хроматографии, элюируя сначала смесью 2,5% диэтиловый эфир / 2,5% этилацетат в дихлорметане, и затем 20% диэтиловым эфиром в дихлорметане до элюирования требуемого продукта из колонки. Соответствующие фракции объединяли и концентрировали с получением титульного соединение в виде беловатого порошка (4,51 г, 77%).
1Н ЯМР (500 МГц, CDCl3) δ 1,49 (9Н, s), 2,70-2,84 (1Н, m), 2,92-3,05 (1Н, m), 3,18-3,23 (1Н, m), 3,33-3,37 (1Н, m), 3,55-3,61 (1Н, m), 3,66-3,71 (1Н, m), 3,80-4,07 (3Н, m), 5,32 (1Н, broad s), 6,61 (1Н, s), 8,24 (1Н, s). ЖХ-МС (Agilent 4 мин) Rt 3,04 мин; m/z (ESI) 396 [МН+].
Синтез 1С
(S)-трет-бутил-2-((2-(5-цианопиразин-2-иламино)-5-(трифторметил)пиридин-4-иламино)метил)морфолин-4-карбоксилат
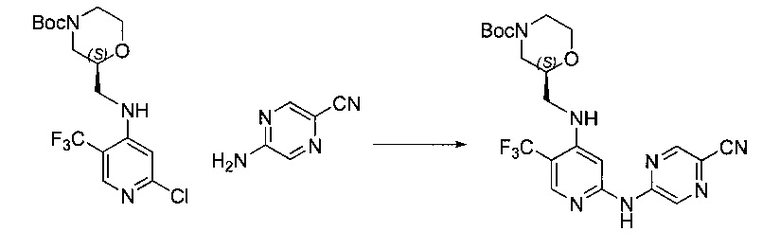
(S)-трет-бутил-2-((2-хлор-5-(трифторметил)пиридин-4-иламино)метил)морфолин-4-карбоксилат (4,67 г, 11,8 ммоль), 2-амино-5-цианопиразин (1,98 г, 16,5 ммоль), трис(дибензилиденацетон)дипалладий (0) (0,86 г, 0,94 ммоль), рац-2,2'-бис(дифенилфосфино)-1,1'-бинафтил (0,54 г, 0,87 ммоль) и карбонат цезия (7,69 г, 23,6 ммоль) суспендировали в безводном диоксане (108 мл) в атмосфере аргона. Аргон барботировали через указанную смесь в течение 30 минут, после чего суспензию нагревали до 100°С в течение 29 часов. Реакционную смесь охлаждали и разбавляли дихлорметаном, затем абсорбировали на силикагеле. Предварительно абсорбированный силикагель добавляли к колонке 340 г KP-Sil SNAP, которую уравновешивали 20% этилацетатом в гексане. В результате колоночной хроматографии, элюируя градиентом 20-35% этилацетатом в гексане, получали частично очищенный материал в виде оранжевой смолы. Его дополнительно очищали с помощью колоночной хроматографии, элюируя 20% этилацетатом в дихлорметане, с получением титульного соединения в виде светло-коричневого порошка (3,28 г, 58%).
1Н ЯМР (500 МГц, CDCl3) δ 1,49 (9Н, s), 2,73-2,86 (1Н, гл), 2,94-3,07 (1Н, m), 3,26-3,31 (1Н, m), 3,38-3,43 (1Н, m), 3,57-3,61 (1Н, m), 3,70-3,75 (1Н, m), 3,83-4,08 (3Н, m), 5,31 (1Н, broads), 7,12 (1Н, s), 8,13 (1Н, s), 8,23 (1Н, s), 8,57 (1Н, s), 8,87 (1Н, s). ЖХ-МС (Agilent 4 мин) Rt 2,90 мин; m/z (ESI) 480 [МН+].
Синтез 1D
5-[[4-[[(2R)-Морфолин-2-ил]метиламино]-5-(трифторметил)-2-пиридил]амино]пиразин-2-карбонитрил (Соединение 1)
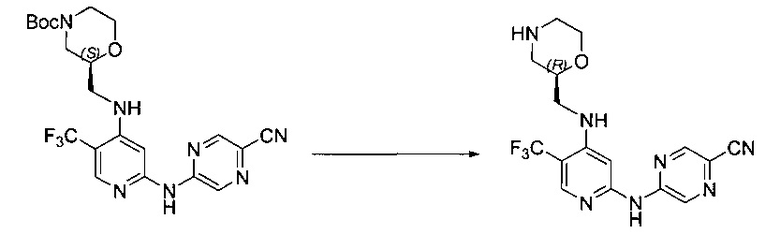
Раствор (S)-трет-бутил-2-((2-(5-цианопиразин-2-иламино)-5-(трифторметил)пиридин-4-иламино)метил)морфолин-4-карбоксилата (1,09 г, 2,273 ммоль) в дихлорметане (8 мл) добавляли по каплям в течение 10 минут к раствору трифторуксусной кислоты (52,7 мл, 709 ммоль) и триизопропилсилана (2,61 мл, 12,73 ммоль) в сухом дихлорметане (227 мл) при комнатной температуре. После перемешивания в течение 30 минут, указанную смесь концентрировали в вакууме. Концентрат повторно суспендировали в дихлорметане (200 мл) и концентрировали в вакууме, затем повторно суспендировали в толуоле (100 мл) и концентрировали.
Описанную выше процедуру проводили трижды (начиная каждый раз с 1,09 г (S)-трет-бутил-2-((2-(5-цианопиразин-2-иламино)-5-(трифторметил)пиридин-4-иламино)метил)морфолин-4-карбоксилата), и три части полученного таким образом неочищенного продукта объединяли для очистки с помощью ионообменной хроматографии на колонках 2×20 г Biotage NH2 Isolute, элюируя метанолом. Элюант концентрировали и добавляли 10% метанол в диэтиловом эфире (25 мл). Полученное твердое вещество фильтровали, промывали диэтиловым эфиром (30 мл) и сушили в вакууме с получением титульного соединения в виде соломенно-желтого окрашенного порошка (2,30 г, 89%).
1Н ЯМР (500 МГц, CD3OD) δ 2,62 (1Н, J=12, 10 Гц), 2,78-2,84 (2Н, m), 2,95 (1Н, dd, J=12, 2 Гц), 3,27-3,38 (2Н, m), 3,63 (1Н, ddd, J=14, 9,5, 3 Гц), 3,73-3,78 (1Н, m), 3,91 (1Н, ddd, J=11, 4, 2 Гц), 7,26 (1Н, s), 8,18 (1Н, s), 8,63 (1Н, s), 9,01 (1Н, s). ЖХ-МС (Agilent 4 мин) R, 1,22 мин; m/z (ESI) 380 [М+Н+]. Оптическое вращение [α]D24=+7,0 (с 1,0, ДМФА).
Синтез 2
5-[[4-[[(2S)-Морфолин-2-ил]метиламино]-5-(трифторметил)-2-пиридил]амино]пиразин-2-карбонитрил (Соединение 2)
Синтез 2А
(S)-трет-бутил-2-(тозилоксиметил)морфолин-4-карбоксилат
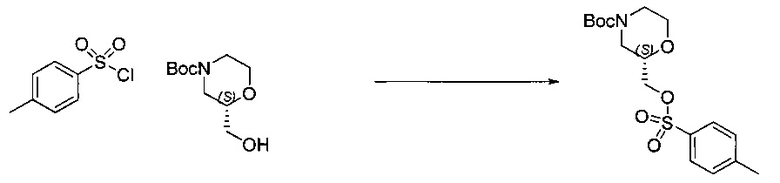
Триэтиламин (6,05 мл, 43,0 ммоль) добавляли к (S)-трет-бутил 2-(гидроксиметил)-морфолин-4-карбоксилату (8,5 г, 39,1 ммоль) в дихлорметане (19,56 мл) с получением бесцветного раствора. 4-толуолсульфонилхлорид (7,83 г, 41,1 ммоль) добавляли небольшими порциями при 0°С. Реакционную смесь перемешивали в течение 18 часов при комнатной температуре, после чего ее концентрировали путем испарения при пониженном давлении. Концентрат растворяли в этилацетате (300 мл) и полученный раствор промывали водой (150 мл), солевым раствором (150 мл), сушили в течение сульфатом магния, фильтровали и концентрировали путем испарения при пониженном давлении. К концентрату добавляли гексан и летучие вещества удаляли в вакууме с получением титульного соединения в виде белого порошка (14,46 г, 99%).
1Н ЯМР (500 МГц, CDCl3) δ 1,46 (9Н, s), 2,46 (3Н, s), 2,61-2,75 (1Н, m), 2,85-2,94 (1Н, m), 3,43-3,49 (1Н, m), 3,58-3,63 (1Н, m), 3,76-3,93 (3Н, m), 3,99-4,06 (2Н, m), 7,35 (2Н, d, J=8,5 Гц), 7,80 (2Н, d, J=8,5 Гц). ЖХ-МС (Agilent 4 мин) R, 2,94 мин; m/z (ESI) 394 [М+Na+].
Синтез 2В
(R)-трет-бутил-2-((2-хлор-5-(трифторметил)пиридин-4-иламино)метил)морфолин-4-карбоксилат
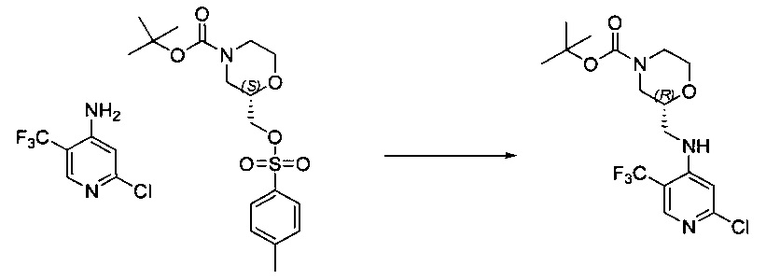
К раствору 2-хлор-5-(трифторметил)пиридин-4-амина (1 г, 5,09 ммоль) в диметилформамиде (32,6 мл) по каплям при комнатной добавляли гидрид натрия (60% по массе в масле; 0,407 г, 10,18 ммоль) температуре, с последующим перемешиванием в течение 10 минут при 80°С. Затем по каплям добавляли (S)-трет-бутил-2-(тозилоксиметил)морфолин-4-карбоксилат (2,268 g, 6,11 ммоль) и реакционную смесь перемешивали при 80°С в течение 2,5 часов. После охлаждения указанную смесь распределяли между насыщенным водным раствором гидрокарбоната натрия (30 мл), водой (100 мл) и этилацетатом (30 мл). Органический слой отделяли и водный слой дополнительно экстрагировали экстрагировали этилацетатом (2×30 мл). Объединенные органические слои промывали солевым раствором (2×70 мл), сушили над сульфатом магния, фильтровали, концентрировали и тщательно сушили в вакууме. Неочищенный материал очищали с помощью колоночной хроматографии на 90 г колонке Thomson SingleStep, элюируя изократической смесью 2,5% диэтиловый эфир/2,5% этилацетат в дихлорметане, с получением титульного соединения в виде прозрачной смолы, которую впоследствии кристаллизовали с получением белого порошка (1,47 г, 73%).
1Н ЯМР (500 МГц, CDCl3) δ 1,48 (9Н, s), 2,71-2,83 (1Н, m), 2,92-3,05 (1Н, m), 3,18-3,23 (1Н, m), 3,33-3,37 (1Н, m), 3,56-3,61 (1Н, m), 3,66-3,71 (1Н, m), 3,80-4,07 (3Н, m), 5,32 (1Н, broads), 6,61 (1Н, s), 8,24 (1Н, s). ЖХ-МС (Agilent 4 мин) Rt 3,04 мин; m/z (ESI) 396 [МН+].
Синтез 2С
(R)-трет-бутил-2-((2-(5-цианопиразин-2-иламино)-5-(трифторметил)пиридин-4-иламино)метил)морфолин-4-карбоксилат
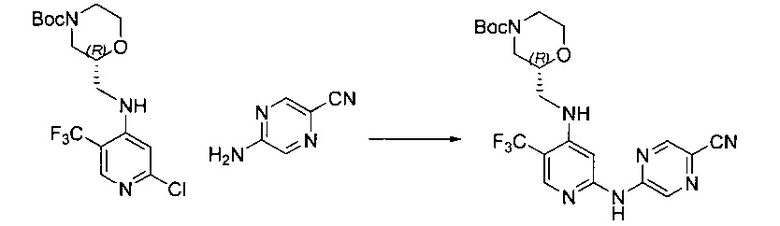
(R)-трет-бутил-2-((2-хлор-5-(трифторметил)пиридин-4-иламино)метил)морфолин-4-карбоксилат (1,44 г, 3,64 ммоль), 2-амино-5-цианопиразин (0,612 г, 5,09 ммоль, 1,4 экв.), трис(дибензилиденацетон)дипалладий(0) (0,267 г, 0,291 ммоль, 0,08 экв.), рац-2,2'-бис(дифенилфосфино)-1,1'-бинафтил (0,362 г, 0,582 ммоль, 0,16 экв.) и карбонат цезия (2,37 г, 7,28 ммоль) суспендировали в безводном диоксане (33 мл) в атмосфере аргона. Аргон барботировали через указанную смесь в течение 30 минут, после чего указанную смесь нагревали до 100°С в течение 22 часов. Реакционную смесь охлаждали и разбавляли дихлорметаном, затем абсорбировали на силикагеле. Предварительно абсорбированный силикагель добавляли к колонке 100 г KP-Sil SNAP, которую элюировали 20-50% этилацетатом в гексанах, с получением частично очищенного продукта в виде оранжевой смолы. Неочищенный продукт растворяли в дихлорметане и очищали с помощью колоночной хроматографии на колонке 90 г SingleStep Thomson, элюируя 20% этилацетатом в дихлорметане, с получением титульного соединения (1,19 г, 68%).
1Н ЯМР (500 МГц, CDCl3) δ 1,50 (9Н, s), 2,71-2,88 (1Н, m), 2,93-3,08 (1Н, m), 3,27-3,32 (1Н, m), 3,40-3,44 (1Н, m), 3,55-3,64 (1Н, m), 3,71-3,77 (1Н, m), 3,82-4,11 (3Н, m), 5,33 (1Н, broad s), 7,19 (1Н, s), 8,23 (1Н, s), 8,58 (1Н, s), 8,84 (1Н, s). ЖХ-МС (Agilent 4 мин) R, 2,93 мин; m/z (ESI) 480 [МН+].
Синтез 2D
5-[[4-[[(2S)-Морфолин-2-ил]метиламино]-5-(трифторметил)-2-пиридил]амино]пиразин-2-карбонитрил (Соединение 2)
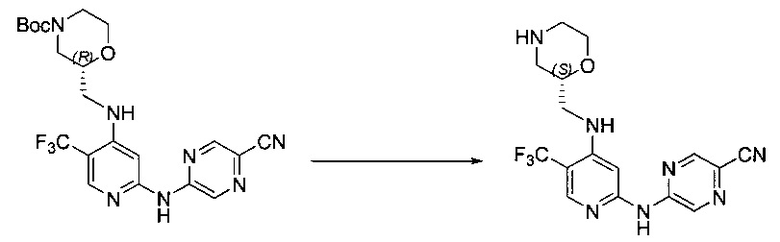
Раствор (R)-трет-бутил-2-((2-(5-цианопиразин-2-иламино)-5-(трифторметил)пиридин-4-иламино)метил)морфолин-4-карбоксилат (1,19 г, 2,48 ммоль) в дихлорметане (8 мл) добавляли по каплям в течение 10 минут к раствору трифторуксусной кислоты (57,5 мл, 774 ммоль) и триизопропилсилан (2,85 мл, 13,90 ммоль) в сухом дихлорметане (248 мл) при комнатной температуре. После перемешивания в течение 30 минут указанную смесь концентрировали в вакууме. Концентрат повторно суспендировали в дихлорметане (200 мл) и концентрировали в вакууме, затем повторно суспендировали в толуоле (100 мл) и концентрировали в вакууме. Неочищенный материал очищали с помощью ионообменной хроматографии на колонке 20 г Biotage NH2 Isolute, элюируя метанолом. Элюант концентрировали и добавляли 10% метанол в диэтиловом эфире (8 мл). Твердое вещество фильтровали, промывали диэтиловым эфиром (20 мл) и сушили в вакууме с получением титульного соединения в виде соломенно-желтого окрашенного порошка (0,604 г, 64% выход).
1Н ЯМР (500 МГц, CD3OD) δ 2,62 (1Н, J=12, 10 Гц), 2,78-2,84 (2Н, m), 2,95 (1Н, dd, J=12, 2 Гц), 3,27-3,38 (2Н, m), 3,63 (1Н, ddd, J=14, 9,5, 3 Гц), 3,73-3,78 (1Н, m), 3,91 (1Н, ddd, J=11, 4, 2 Гц), 7,26 (1Н, s), 8,18 (1Н, s), 8,63 (1Н, s), 9,01 (1Н, s). ЖХ-МС (Agilent 4 мин) Rt 1,26 мин; m/z (ESI) 380 [М+Н+]. Оптическое вращение [α]D24=-6,9 (с 1,0, ДМФА).
Биологические методы
Анализ 1: Определение ингибиторной активности по отношению к CHK1 в Формате анализа с применением прибора Caliper
Киназную активность CHK1 измеряли посредством анализа с применением микрофлюидного чипа, который позволяет наблюдать отделение фосфорилированного продукта от его субстрата. Анализ проводили на приборе EZ Reader II (Caliper Life Sciences Ltd, Runcorn, UK) с применением буфера для разделения (№по каталогу 760367, Caliper LS), содержащего реагент CR-8 (500 нМ, №по каталогу 760278, Caliper LS). Для построения кривых разведения из 8 точек в двух повторностях применяли акустический дозатор ECHO® 550 (Labcyte Inc™) непосредственно внутри 384-луночных полипропиленовых планшетов для анализа (Greiner Bio-One, Gloucestershire, UK). Для каждого исследуемого соединения использовали стоковый раствор в 100% ДМСО с концентрацией 50 мкМ. Общее количество ДМСО, вносимого в каждую лунку, составляло 250 нл с получением в ходе анализа конечной концентрации ДМСО 2,5% и концентраций исследуемых соединений в диапазоне 0,5-1000 нМ. В лунки этого опытного планшета добавляли 6 мкл CHK1 (конечная концентрация 2 нМ, препарат белка был получен в лаборатории), 2 мкл меченного карбоксифлуоресцеином (FAM) пептида 10 (5-FAM-KKKVSRSGLYRSPSMPENLNRPR-COOH, конечная концентрация 1,5 мкМ, № по каталогу 760354, Caliper LS) и 2 мкл АТФ (конечная концентрация 90 мкМ), разведенных в буфере для определения киназной активности (50 мМ HEPES, 0,02% NaN3, 0,01% БСА, 0,1 мМ ортованадат натрия, 1 мМ DTT, 2 мМ MgCl2, 0,1% Твин 20). Планшет герметично закрывали и центрифугировали (1 минута, 1000 об/мин) перед инкубацией в течение одного часа при комнатной температуре. Реакцию останавливали добавлением буфера для разделения (90 мкл). Сигнал в лунках планшета считывали с помощью прибора EZ Reader II с применением чипа с 12 зондами для отбора образцов из лунок (760137-0372R, Caliper LS) с параметрами настройки: давление -1,5 psi (единиц фунт-силы/квадратный дюйм) и разность потенциалов 1750 В. Процент превращения субстрата в продукт был получен автоматически, а процент ингибирования рассчитывали относительно контрольных лунок (не содержащих фермента и 2,5% ДМСО) и опытных лунок (содержащих все реагенты и 2,5% ДМСО). Величины IC50 для CHK1 рассчитывали с помощью программного обеспечения GraphPad Prism5 с применением нелинейной регрессии для подбора кривой зависимости «логарифм концентрации ингибитора - ответ», описываемой уравнением с переменным угловым коэффициентом.
Анализ 2: Клеточная активность в анализе ингибирования митоза (MIA)
Подавление функции контрольной точки путем применения ингибиторов киназной функции CHK1 в сочетании с генотоксическими агентами оценивали посредством основанного на применении европиевой метки ИФА-анализа, разработанного для количественного определения числа клеток, претерпевающих митоз после обработки генотоксическим агентом (для индукции блокирования фазы G2) с последующей обработкой исследуемым соединением-ингибитором CHK1 в комбинации с нокодазолом с целью прекращения этого блокирования. Клетки линии НТ29 высевали в концентрации 104 клеток/лунка в 96-луночные планшеты в объеме 160 мкл и оставили для прикрепления на 36 часов. Этопозид (10 мМ стоковый раствор в ДМСО) разводили в среде до концентрации 250 мкМ и затем добавляли по 40 мкл в соответствующие лунки с получением конечной концентрации 50 мкМ, и инкубировали в течение 1 часа. Этот вид обработки был ранее оптимизирован до способности индуцировать блоктрование фазы G2 в 80% клеток через 16 часов после обработки. После воздействия генотоксических препаратов среду удаляли и заменяли на свежую среду (160 мкл). Клетки были либо необработанными (необработанный контроль или предварительная обработка исключительно этопозидом), либо подвергнутыми воздействию нокодазола вслед за предварительной обработкой этопозидом или исключительно нокодазола (конечная концентрация 100нг/мл), либо подвергнутыми воздействию возрастающих концентраций исследуемых соединений (конечная концентрация от 0,01 нМ до 200 мкМ) в комбинации с нокодазолом (конечная концентрация 100нг/мл). Исследуемые соединения-ингибиторы CHK1 добавляли в лунки в аликвотах объемом 40 мкл в четырех повторностях для каждой концентрации. После 21-часового периода воздействия среду удаляли и клетки фиксировали с помощью 4% формальдегида в фосфатно-солевом буфере (ФСБ, рН 7,4, предварительно охлажденном до 4°С) в течение 30 минут при 4°С с последующей обработкой 100% метанолом (предварительно охлажденным до -20°С) в течение 10 минут при комнатной температуре. Лунки промывали ФСБ и блокировали 5% сухим молоком (Marvel) в Трис-солевом буфере (ТСБ, рН 7,4) при 37°С в течение 30 минут. Каждую лунку промывали три раза водой, содержащей 0,1% Твин 20. Первые антитела (МРМ-2, № по каталогу 05-368, 1 мкг/мл в 5% растворе молока в ТСБ) добавляли в каждую лунку и инкубировали в течение ночи со встряхиванием при 4°С. Первые антитела удаляли, и лунки промывали водой, содержащей 0,1% Твин 20. Вторые антитела (содержащие европиевую метку антитела против иммуноглобулинов мыши, № по каталогу Perkin Elmer AD0124, 333 нг/мл в буфере для анализа Perkin Elmer, № по каталогу 1244-111) добавляли в каждую лунку и инкубировали при 37°С в течение 1 часа. Каждую лунку промывали водой, содержащей 0,1% Твин 20, и обрабатывали усиливающим сигнал раствором (№ по каталогу Perkin Elmer 1244-105). Интенсивность излучения, опосредованного наличием европиевой метки, подсчитывали с применением счетчика Wallac Victor2 (Perkin Elmer, Bucks, UK). Включали соответствующие контроли, и результаты выражали в виде концентрации исследуемого соединения-ингибитора CHK1, необходимой для протекания митоза в 50% клеток (MIA IC50).
Анализ 3: Определение селективности в клетках по отношению к подавлению функции CHK1-зависимой контрольной точки в зависимости от цитотоксичности
Цитотоксичность соединения оценивали с использованием 96-часового анализа с сульфородамином-В (SRB, Sigma, № по каталогу S9012). Клетки линий НТ29 или SW620 высеивали в концентрации 1,6-3,2×103 клеток на лунку в 96-луночные планшеты в объеме 160 мкл и оставили для прикрепления на 36 часов до обработки. Для проведения анализов цитотоксичности ингибиторов CHK1 (10 мМ стоковый раствор в ДМСО) соединения последовательно разводили в среде, начиная с концентрации 250 мкМ, и затем добавляли по 40 мкл в соответствующие лунки в четырех повторностях с получением конечной концентрации от 50 до 0,1 мкМ (10 концентраций). Для генотоксических агентов, соединения (SN38, LKT laboratories, № по каталогу С0154 и гемцитабин, Lilly "Gemzar", 10 мМ стоковый раствор в ДМСО) последовательно разводили в среде, начиная с концентрации 2 мкМ, и добавляли 40 мкМ в каждую лунку в четырех повторностях с получением конечной концентрации от 200 до 0,39 нМ (10 концентраций). Клетки инкубировали в течение 96 часов (четыре повторности) при 37°C в среде увлажненного 5% CO2 и затем фиксировали и окрашивали SRB. Включали соответствующие контроли, и результаты выражали в виде концентрации исследуемого соединения, необходимой для ингибирования клеточного роста на 50% по сравнению с необработанными контролями (SRB IC50).
Показатель активности (Al), мера селективности исследуемых соединений-ингибиторов CHK1 по подавлению функции CHK1-зависимой контрольной точки в зависимости от цитотоксичности, был рассчитан как отношение IC50 в анализе цитотоксичности ингибитора CHK1 к IC50 в MIA (т.е. Al=SRB IC50 ингибитора CHK1 / MIA IC50), которые оба были измерены на клетках НТ29.
Анализ 4: Клеточная эффективность в линиях клеток рака толстой кишки НТ29 или SW620 в комбинации с SN38 или гемцитабином
Способность соединений-ингибиторов CHK1 повышать цитотоксичность SN38 (активный метаболит ингибитора топоизомеразы I - иринотекана) и гемцитабина (антиметаболит) была оценена с использованием 96 часового анализа с сульфородамином-В (SRB, Sigma, № по каталогу S9012). Клетки линий НТ29 или SW620 высеивали в концентрации 1,6-3,2×103 клеток на лунку в 96-луночные планшеты в объеме среды 160 мкл и оставили для прикрепления на 36 часов до обработки. Анализы потенцирования включали добавление в концентрации IC50 либо гемцитабина, либо SN38 с зафиксированным SRB (определенной с использованием способов Анализы 3, приведенных выше) в объеме 20 мкл среды (10 × конечная концентрация), в каждую лунку четырех повторностях и перемешивание в течение 1 минуты. Исследуемое соединение-ингибитор CHK1 (10 мМ стоковый раствор) последовательно разводили в среде, начиная с концентрации 50 мкМ и добавляли по 20 мкМ в соответствующие лунки в четырех повторностях с получением конечной концентрации от 5 до 0,039 мкМ (8 концентраций) и перемешивали в течение 1 минуты до инкубации при 37°С в увлажненной атмосфере при 37°С в течение 96 часов (четыре повторности) до фиксирования и окрашивания SRB. Включали необработанные контроли и контроли, обработанные только генотоксическими агентами, и результаты выражали в виде концентрации исследуемого ингибитора CHK1, необходимой для ингибирования клеточного роста на 50% (IC50 потенцирования).
Показатель потенцирования (PI) вычисляли как меру способности ингибитора CHK1 увеличивать цитотоксичность SN38 или гемцитабина и его определяли как отношение IC50 цитотоксичности индифидуального ингибитора CHK1 к IC50 потенцирования ингибитора CHK1, объединенного с генотоксическими агентами (т.е., PI=SRB IC50 ингибитора CHK1 / IC50 потенцирования).
Анализ 5: Биодоступность при пероральном введении и фармакокинетика в мыши
Все работы были выполнены в соответствии с законом о животных (научные процедуры) от 1986 года, принятого Министерством внутренних дел, и в соответствии с руководством UKCCCR для экспериментов над животными.
Соединения ингибиторы CHK1 объединяли с 10% ДМСО, 1% Tween 20 и 89% стерильным физиологическим раствором. Соединение ингибитор CHK1 вводили мышам женского пола линии Balb/c (Charles River UK Ltd, Margate, U.K.) внутривенно (iv) и перорально (po) в дозировке 10 мг/кг. Контрольные животные получали только носитель. Группы из 3 мышей инъецировали по расписанию. Через 5, 15 и 30 минут и 1, 2, 4, 6 и 24 часов после введения дозы кровь собирали путем пункции сердца гепаринизированными шприцами под анестезией (смесь галотан/кислород). После центрифугирования (9000 х g, 2 минуты, 4°С) плазму замораживали на сухом льду и хранили при -80°С. Ткани отделяли, быстро замораживали в жидком азоте и хранили при -80°С. Образцы оттаявшей плазмы выделяли путем белкового осаждения с использованием 3 объемов метанол содержащего внутреннего стандарта. Калибровочные стандарты (от 2 до 10000 нМ в плазме) и QC получали путем введения в матрицу чистой плазмы соединения ингибитора CHK1 и экстрагирования в виде тестовых образцов. После центрифугирования супернатант переносили для анализа. Выделенные образцы плазмы анализировали с помощью ЖХ-МС-МС на Agilent 1200 или 1290 LC в тандеме с тройным квадрупольным масс-спектрометром Agilent 6410 для подсчета соединения ингибитора CHK1 и внутреннего стандарта. Соединения отделяли на аналитической колонке Phenomenex Kinetex С18 (50×2,1 мм, 2,6 мкм) при 55°С. Подвижная фаза состояла из 10 мМ ацетата аммония и метанола при расходе 0,4 мл/мин. 7-минутный градиент использовали для отделения анализируемых веществ. Применяли ионизацию электрораспылением в режиме определения положительных ионов и соединения детектировали с помощью MRM с наблюдением адекватного перехода (например, для Соединения 1 он составлял от 380,2 до 320,3, при напряжении на фрагменторе 154 V и энергии столкновения 20 В.
Некомпартментный фармакокинетический анализ (модели 200 и 201) был проведен с помощью программного обеспечения Pharsight WinNonlin (версия 5.2.1).
Биологические данные
Данные для Соединения 1 и Соединения 2, полученные с использованием анализов, описанных выше, приведены в следующей таблице.
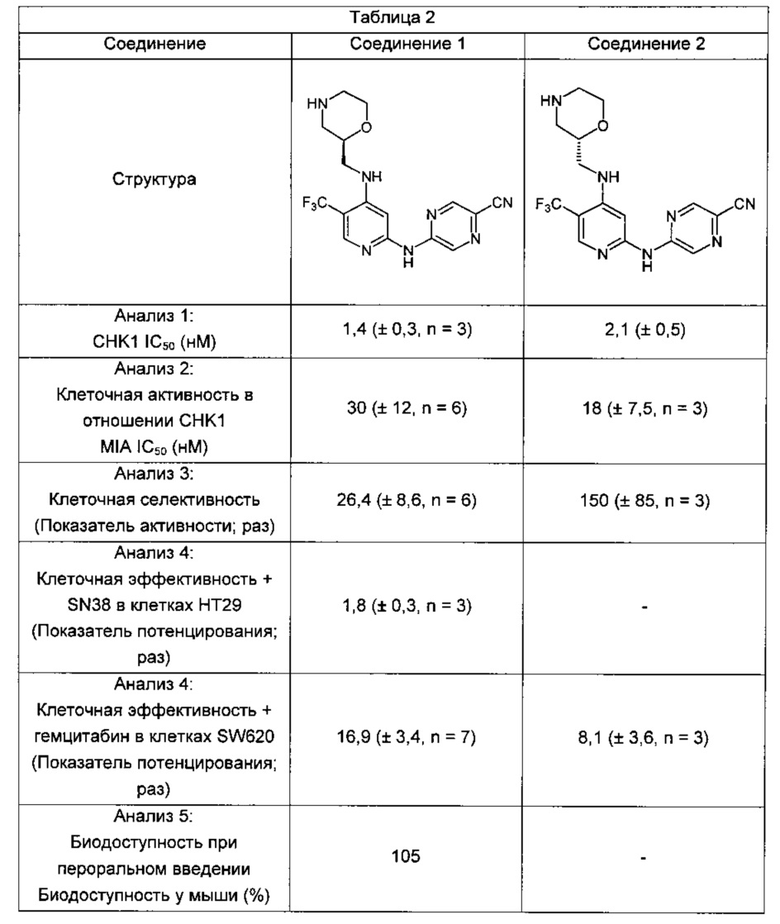
Сравнение данных IC50 для CHK1 (полученные в анализе 1 выше) для Соединений 1 и 2 с соответствующими данными, полученными для 16 аналогичных соединений, показанных у Коллинза с соавт., 2009а (полученными с использованием анализа DELFIA, как описано Коллинзом с соавт., 2009а), приведено в следующей таблице.
Сравнение данных по клеточной активности в отношении CHK1 в анализе MIA (полученных в анализе 2 выше) для Соединений 1 и 2 с соответствующими данными, полученными для 16 аналогичных соединений, показанных у Коллинза с соавт., 2009а, приведено в следующей таблице.
Очевидно, что Соединения 1 и 2 проявляют превосходную клеточную активность в отношении CHK1 (Анализ MIA). Высокая клеточная активность по ингибированию CHK1 является важной с точки зрения разработки ингибитора CHK1. Соединения 1 и 2 проявляли значительно более высокую клеточную активность в отношении эффектов, обусловленных CHK1, чем все 16 соединений, и приблизительно в 3 раза и 5 раз, соответственно, более активны, чем следующее наиболее активное соединение (Y-154).
Сравнение данных по клеточной селективности (полученных в анализе 3 выше) для Соединений 1 и 2 с соответствующими данными, полученными для 16 аналогичных соединений, показанных у Коллинза с соавт., 2009а, приведено в следующей таблице.
Очевидно, что Соединения 1 и 2 проявляют превосходную клеточную селективность, измеренную как отношение клеточной активности по CHK1 (анализ MIA) и неспецифической цитотоксичности в анализе ингибирования роста. Клеточная селективность для эффектов, опосредованных CHK1, по отношению к неспецифической цитотоксичности важна для разработки ингибитора CHK1 для расширения терапевтического окна. Соединения 1 и 2 приблизительно в 26 раз более селективны и 150 раз более селективны, соответственно, в то время как следующее наиболее селективное соединение является только в 11 - более селективным (Y-155).
Сравнение данных по клеточной эффективности (полученных в анализе 4 выше) для Соединений 1 и 2 с соответствующими данными, полученными многих из 16 аналогичных соединений, показанных у Коллинза с соавт., 2009а, приведено в следующих таблицах.
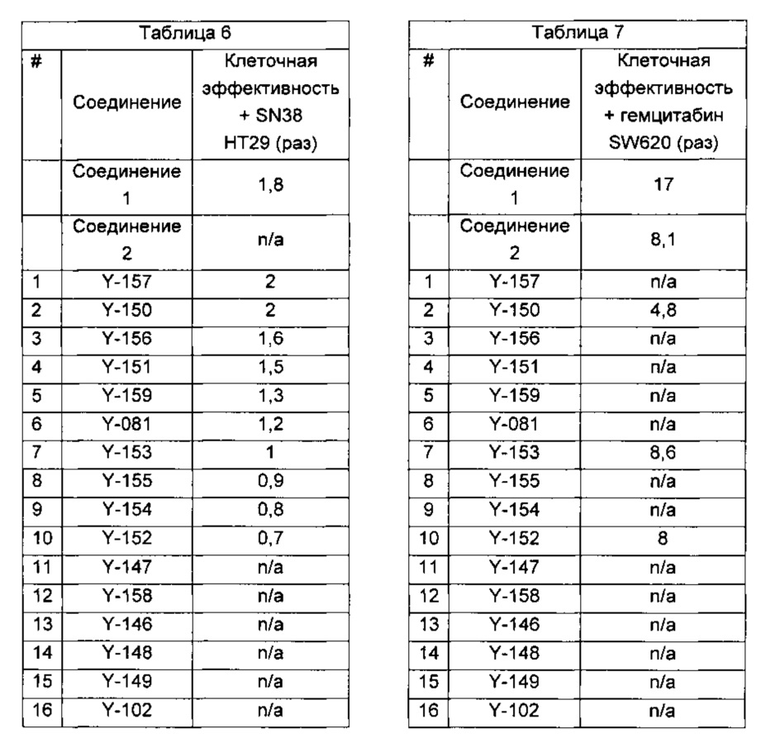
Очевидно, что Соединения 1 и 2 проявляют превосходную клеточную эффективность, определенную как способность к сенсибилизации клеток для двух типичных генотоксических терапевтических агентов: SN38 и гемцитабина. Для разрабатываемого соединения необходимо значение >1 (в ином случае указанное соединение будет снижать эффект генотоксической терапии).
Соединение 1 также проявляет наиболее высокое потенцирование SN38 (приблизительно 1,8-раз) и наиболее высокое потенцирование гемцитабина (приблизительно 17- раз), что свидетельствует о хорошей эффективности. Соединение, проявляющее наиболее высокое потенцирование SN38, представляет собой Y-150 (приблизительно в 2 раза), которое проявляет потенцирование только в 4,8-раза для гемцитабина. Следующее соединение с наиболее высоким показателем потенцирования гемцитабина является Y-153 (приблизительно в 8,6 раз), которое не потенцировало (т.е. приблизительно в 1 раз) SN38.
Кроме того, Соединение 1 имело превосходную для перорального введения биодоступность у мыши: 100% (отмечено как «105%» в таблице выше).
* * *
Выше были описаны принципы, предпочтительные варианты реализации и характер функционирования настоящего изобретения. Однако следует понимать, что изобретение не ограничивается конкретными рассмотренными вариантами реализации. Наоборот, вышеописанные варианты реализации следует считать иллюстративными, а не ограничивающими, и следует понимать, что в этих вариантах реализации специалистами в данной области техники могут быть сделаны изменения без выхода за рамки настоящего изобретения.
Ссылки
В настоящем документе приведен ряд публикаций с целью более полного описания и раскрытия изобретения и существующего уровня техники, к которому относится изобретение. Ниже представлен полный список публикаций. Полное содержание каждой из этих публикаций включено в настоящее описание посредством ссылки в той же степени, как если бы для каждой отдельной публикации конкретно и индивидуально было указано ее включение посредством ссылки.
Almeida et al., 2008, "Pyrazolyl-amino-substituted pyrazines and their use for the treatment of cancer", international (PCT) patent publication number WO 2008/117050 A1 published 02 October 2008.
Balaint and Vousden, 2001, "Activation and activities of the p53 tumour suppressor protein," Br. J. Cancer. Vol. 85, pp. 1813-1823.
Bartek and Lukas, 2003, "Chk1 and Chk2 kinases in checkpoint control and cancer," Cancer Cell, Vol. 3, pp.421-429.
Brooks et al., 2012, "A potent chk1 inhibitor is selectively toxic in melanomas with high levels of replicative stress," Oncogene, doi:10.1038/onc. 2012.72.
Carson and Lois, 1995, "Cancer progression and p53," Lancet, Vol. 346, pp. 1009-1011.
Cavelier et al., 2009, "Constitutive activation of the DNA damage signaling pathway in acute myeloid leukemia with complex karyotype: Potential importance for checkpoint targeting therapy," Cancer Res., Vol. 69, pp.8652-8661.
Cole et al., 2011 "RNAi screen of the protein kinome identifies checkpoint kinase 1 (chk1) as a therapeutic target in neuroblastoma," Proc. Natl. Acad. Sci. U.S.A., Vol. 108, pp. 3336-3341.
Collins et al., 2009a, "Pyrazin-2-yl-2-yl-amine and pyrazin-2-yl-pyrimidin-4-yl-amine compounds and their use", international (PCT) patent publication number WO 2009/044162 A1 published 09 April 2009.
Collins et al., 2009b, "Bicyclylaryl-aryl-amine compounds and their use", international (PCT) patent publication number WO 2009/103966 A1 published 27 August 2009.
Davies et al., 2011, "Single-agent inhibition of chk1 is antiproliferative in human cancer cell lines in vitro and inhibits tumor xenograft growth in vivo," Oncol. Res., Vol. 19, pp. 349-363.
Di Micco et al., 2006, "Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication," Nature, Vol. 444, pp. 638-642.
Dixon and Norbury, 2002, "Therapeutic exploitation of checkpoint defects in cancer cells lacking p53 function," Cell Cycle. Vol, 1, pp. 362-368.
Ferrao et al., 2011, "Efficacy of chk inhibitors as single agents in myc-driven lymphoma cells," Oncogene, doi:10.1038/onc. 2011.358.
Greenblatt et al., 1994, "Mutations in the p53 tumor suppressor gene: clues to cancer etiology and molecular pathogenesis," Cancer Res., Vol. 54, pp. 4855-4878.
Guzi et al., 2011, "Targeting the replication checkpoint using SCH 900776, a potent and functionally selective CHK1 inhibitor identified via high content screening," Mol. Cancer Ther., Vol. 10, pp. 591-602.
Höglund et al., 2011, "Therapeutic Implications for the Induced Levels of Chk1 in Мус-Expressing Cancer Cells," Clin. Cancer Res., Vol. 17, pp. 7067-7079.
loannidis et al., 2009, "Discovery of pyrazol-3-ylamino pyrazines as novel JAK2 inhibitors", Bioora. Med. Chem. Lett., Vol. 19, pp. 6524-6528.
Lainchbury et al., 2012, "Discovery of 3-alkoxyamino-5-(pyridin-2-ylamino)pyrazine-2-carbonitriles as selective, orally bioavailable CHK1 inhibitors", J. Med. Chem., Vol. 55, No. 22, pp. 10229-10240.
Li et al., 2007, "Synthesis and in-vitro biological activity of macrocyclic urea CHK1 inhibitors", Bioorg. Med. Chem. Lett., Vol.17, pp. 6499-6504.
Lin et al., 2005, "Macrocyclic kinase inhibitors", US patent publication number US 2005/0215556 A1 published 29 September 2005.
Liu et al., 2000, "Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint," Genes Dev., Vol.14, pp. 1448-1459.
Murga et al., 2011, "Exploiting oncogene-induced replicative stress for the selective killing of Мус-driven tumors," Nat. Struct. Mol. Biol., Vol. 18, pp. 1331-1335.
Sanchez et al., 1997, "Conservation of the Chk1 checkpoint pathway in mammals: linkage of DNA damage to Cdk regulation through Cdc25," Science. Vol. 277, pp. 1497-1501. Sorensen et al., 2005, "Cell-cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair," Nat. Cell Biol., Vol 7, pp.195-201.
Tao et al., 2005, "Macrocyclic kinase inhibitors", international (PCT) patent publication number WO 2005/047294 A1 published 26 May 2005.
Tao et al., 2006, "Chk1 inhibitors for novel cancer treatment," Anti-Cancer Agents in Medicinal Chemistry. Vol. 6, pp. 377-388.
Tao et al., 2007a, "Macrocyclic ureas as potent and selective CHK1 inhibitors: an improved synthesis, kinome profiling, structure-activity relationships, and preliminary pharmacokinetics," Bioorg. Med. Chem. Lett., Vol. 17, pp. 6593-6601.
Tao et al., 2007b, "Structure-based design, synthesis, and biological evaluation of potent and selective macrocyclic checkpoint kinase 1 inhibitors," J. Med. Chem.. Vol. 50, pp. 1514-1527.
Walton et al., 2010, "The preclinical pharmacology and therapeutic activity of the novel CHK1 inhibitor SAR-020106," Mol. Cancer Ther., Vol. 9, No. 1, pp. 89-100.
Walton et al., 2012, "CCT244747 is a novel potent and selective CHK1 inhibitor with oral efficacy alone and in combination with genotoxic anticancer drugs", Clin. Cancer Res., Vol. 18, No. 20, pp. 5650-5661.
Wang et al., 1996, "UCN-01: a potent abrogator of G2 checkpoint function in cancer cells with disrupted p53," J. Natl. Cancer Inst., Vol. 8, pp. 956-965.
Weinert and Hartwell, 1989, "Control of G2 delay by the rad9 gene of Saccharomyces cerevisiae," J. Cell Sci. Suppl., Vol. 12, pp. 145-148.
Xiao et al., 2006, "Differential roles of checkpoint kinase 1, checkpoint kinase 2, and mitogen-activated protein kinase-activated protein kinase 2 in mediating DNA damage-induced cell cycle arrest: implications for cancer therapy," Mol. Cancer Ther., Vol. 5, pp. 1935-1943.
Zachos et al., 2003, "Chk1-deficient tumour cells are viable but exhibit multiple checkpoint and survival defects," EMBO J., Vol. 22, pp. 713-723.
Zhao et al., 2002, "Disruption of the checkpoint kinase 1/cell division cycle 25A pathway abrogates ionizing radiation-induced S and G2 checkpoints," Proc. Natl. Acad. Sci. USA, Vol. 99, pp. 14795-14800.
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЕДИНЕНИЯ 5-(ПИРИДИН-2-ИЛАМИНО)-ПИРАЗИН-2-КАРБОНИТРИЛА И ИХ ТЕРАПЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2012 |
|
RU2641693C2 |
| КОМБИНАЦИЯ ИНГИБИТОРОВ ЧЕКПОЙНТ-КИНАЗЫ 1 И ИНГИБИТОРОВ КИНАЗЫ WEE 1 | 2011 |
|
RU2627841C2 |
| КОМБИНИРОВАННАЯ ТЕРАПИЯ (2R,Z)-2-АМИНО-2-ЦИКЛОГЕКСИЛ-N-(5-(1-МЕТИЛ-1Н-ПИРАЗОЛ-4-ИЛ)-1-ОКСО-2,6-ДИГИДРО-1Н-[1,2]ДИАЗЕПИНО[4,5,6-cd]ИНДОЛ-8-ИЛ)АЦЕТАМИДОМ | 2007 |
|
RU2409361C2 |
| ФОСФОРОАМИДАТНЫЕ ПРОИЗВОДНЫЕ 5-ФТОР-2'-ДЕЗОКСИУРИДИНА ДЛЯ ПРИМЕНЕНИЯ ДЛЯ ЛЕЧЕНИЯ РАКА | 2012 |
|
RU2614406C2 |
| ГОМОЛОГИ (ATR) ПОЛИПЕПТИДА rad3, ПОЛИНУКЛЕОТИДЫ И СВЯЗАННЫЕ С НИМИ СПОСОБЫ И МАТЕРИАЛЫ | 1996 |
|
RU2252256C2 |
| СПОСОБ ПОЛУЧЕНИЯ СОЕДИНЕНИЯ | 2014 |
|
RU2672725C2 |
| ИНГИБИТОРЫ 1 КИНАЗЫ КОНТРОЛЬНОЙ ТОЧКИ КЛЕТОЧНОГО ЦИКЛА ДЛЯ УСИЛЕНИЯ ДНК-ПОВРЕЖДАЮЩИХ АГЕНТОВ | 2010 |
|
RU2567044C2 |
| СПОСОБ ЛЕЧЕНИЯ РАКА С ИСПОЛЬЗОВАНИЕМ КОМБИНАЦИИ ИНГИБИТОРОВ СНК1 И ATR | 2015 |
|
RU2736219C2 |
| ХИМИЧЕСКИЕ СОЕДИНЕНИЯ | 2014 |
|
RU2644769C2 |
| ПЕПТИДНАЯ ЗАЩИТА ПРОТИВ ТОКСИЧЕСКОГО ВОЗДЕЙСТВИЯ УЛЬТРАФИОЛЕТОВОГО ИЗЛУЧЕНИЯ | 2010 |
|
RU2565535C2 |
Изобретение относится в целом к соединению следующей формулы
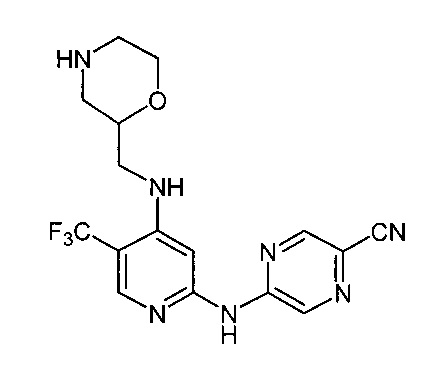 , а также к фармацевтическим композициям, содержащим такие соединения, и к применению таких соединений для ингибирования киназной функции СНK1 и для лечения опосредованных CHK1 заболеваний и состояний. Технический результат: получены новые соединения, пригодные для лечения пролиферативных заболеваний, таких как рак, путем ингибирования киназной функции СНK1. 21 н. и 5 з.п. ф-лы, 7 табл.
, а также к фармацевтическим композициям, содержащим такие соединения, и к применению таких соединений для ингибирования киназной функции СНK1 и для лечения опосредованных CHK1 заболеваний и состояний. Технический результат: получены новые соединения, пригодные для лечения пролиферативных заболеваний, таких как рак, путем ингибирования киназной функции СНK1. 21 н. и 5 з.п. ф-лы, 7 табл.
1. Соединение следующей формулы или его фармацевтически приемлемая соль:
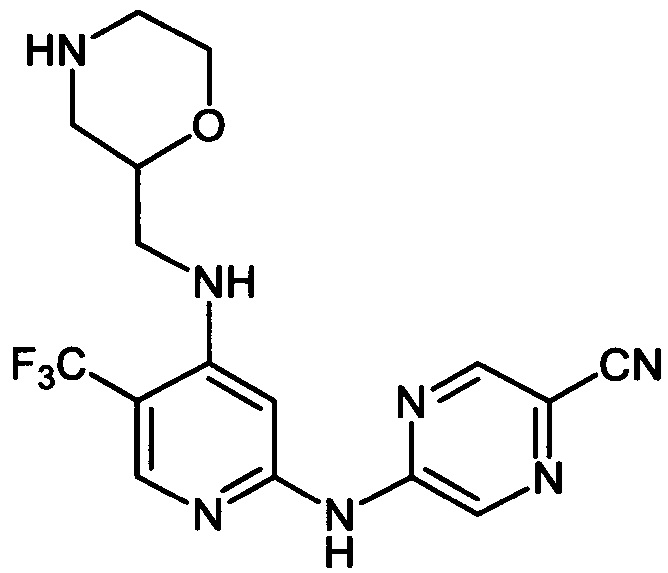
2. Соединение по п. 1, которое представляет собой соединение следующей формулы или его фармацевтически приемлемая соль:
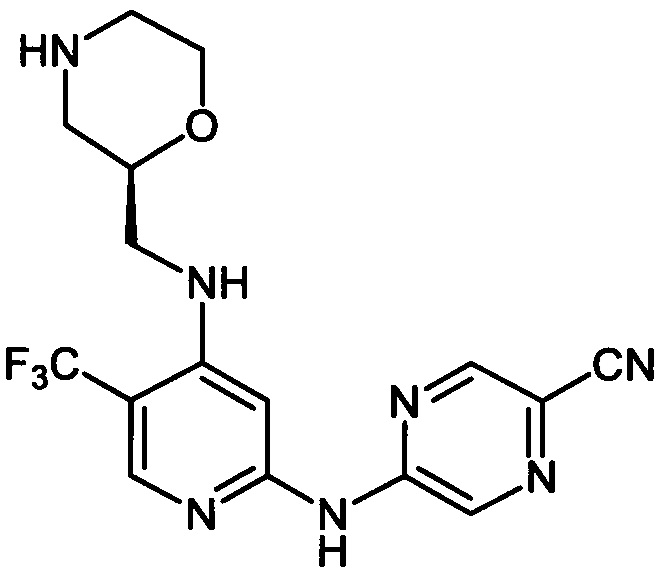
3. Соединение по п. 1, которое представляет собой соединение следующей формулы или его фармацевтически приемлемая соль:
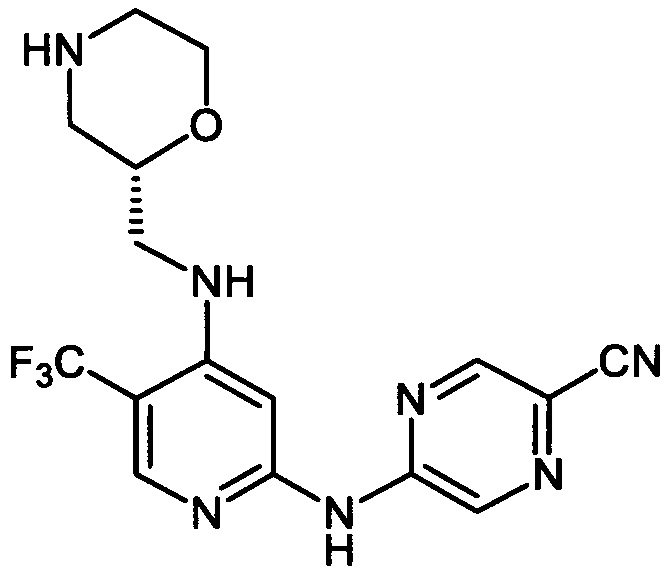
4. Фармацевтическая композиция для применения в способе лечения пролиферативного заболевания, содержащая эффективное количество соединения по любому из пп. 1-3, и фармацевтически приемлемый носитель или разбавитель.
5. Фармацевтическая композиция по п. 4, подходящая для перорального введения субъекту.
6. Способ получения фармацевтической композиции, включающий стадию смешивания соединения по любому из пп. 1-3, и фармацевтически приемлемого носителя или разбавителя.
7. Применение соединения по любому из пп. 1-3, для получения лекарственного средства для лечения пролиферативного заболевания.
8. Применение соединения по любому из пп. 1-3, для получения лекарственного средства для лечения рака.
9. Применение соединения по любому из пп. 1-3, для получения лекарственного средства для лечения рака головы; рака шеи; рака нервной системы; рака мозга; нейробластомы; рака легкого/средостения; рака молочной железы; рака пищевода; рака желудка; рака печени; рака желчевыводящих путей; рака поджелудочной железы; рака тонкой кишки; рака толстой кишки; рака ободочной и/или прямой кишки; рака женских половых органов; рака мочеполовой системы; рака яичника; рака щитовидной железы; рака надпочечника; рака кожи; меланомы; остеосаркомы; саркомы мягких тканей; злокачественных опухолей детского возраста; болезни Ходжкина; неходжкинской лимфомы; миеломы; лейкоза или метастазов из неизвестного первичного очага.
10. Применение соединения по любому из пп. 1-3, для получения лекарственного средства для лечения рака легкого, рака молочной железы, рака яичника, рака поджелудочной железы, рака ободочной и/или прямой кишки, меланомы, глиомы или нейробластомы.
11. Применение соединения по любому из пп. 1-3, для получения лекарственного средства для лечения р53-дефицитного рака.
12. Применение соединения по любому из пп. 1-3, для получения лекарственного средства для лечения рака с амплифицированным MYC.
13. Применение соединения по любому из пп. 1-3, для получения лекарственного средства для лечения рака с амплифицированным c-MYC.
14. Применение соединения по любому из пп. 1-3, для получения лекарственного средства для лечения рака с амплифицированным MYCN.
15. Применение соединения по любому из пп. 1-3, для получения лекарственного средства для лечения рака, характеризующегося гиперэкспрессией MYC.
16. Применение соединения по любому из пп. 1-3, для получения лекарственного средства для лечения рака, характеризующегося гиперэкспрессией MYCN.
17. Применение соединения по любому из пп. 1-3, для получения лекарственного средства для лечения рака, характеризующегося гиперэкспрессией c-MYC.
18. Применение соединения по любому из пп. 1-3, для получения лекарственного средства для лечения нейробластомы с амплифицированным MYCN.
19. Применение соединения по любому из пп. 1-3, для получения лекарственного средства для лечения В-клеточной лимфомы с амплифицированным c-MYC.
20. Применение соединения по любому из пп. 1-3, для получения лекарственного средства для лечения рака, характеризующегося повышенным эндогенным репликативным стрессом.
21. Применение соединения по любому из пп. 1-3, для получения лекарственного средства для лечения рака, характеризующегося повышенной эндогенной активацией сигнального пути CHK1.
22. Применение по п. 10, отличающееся тем, что лекарственное средство представляет собой лекарственное средство для перорального введения.
23. Применение по п. 10, отличающееся тем, что указанное лечение дополнительно включает лечение с применением одного или более других агентов, выбранных из: (а) ингибитора ДНК-топоизомеразы I или II; (b) ДНК-повреждающего агента; (с) антиметаболита или ингибитора тимидилатсинтазы (ТС); (d) нацеленного на микротрубочки агента; (е) ионизирующего излучения; (f) ингибитора регулятора митоза или регулятора митотической контрольной точки; (g) ингибитора трансдуктора сигнала повреждения ДНК; и (h) ингибитора репаративного фермента повреждения ДНК.
24. Способ ингибирования киназной функции CHK1 in vitro, включающий приведение клетки в контакт с эффективным количеством соединения по любому из пп. 1-3.
25. Способ ингибирования киназной функции CHK1 в клетке in vitro, включающий приведение клетки в контакт с эффективным количеством соединения по любому из пп. 1-3.
26. Способ ингибирования пролиферации клеток, ингибирования прогрессии клеточного цикла, стимулирования апоптоза клеток или осуществления комбинации одного или более из вышеперечисленных in vitro, включающий приведение клетки в контакт с эффективным количеством соединения по любому из пп. 1-3.
| WO2009044162 A1, 09.04.2009 | |||
| RU2009110255 A, 27.09.2010. |

