ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к соединениям, обладающим фармакологической активностью, и, более конкретно, к 1-[1-(4-бензилокси-3,5-дифтор-бензоил)-4-фтор-пирролидин-2-карбонил]-пирролидин-2-карбонитрилу, его стереоизомерам и их солям, к способам получения таких соединений, содержащим их фармацевтическим композициям, и их применению в терапии и/или профилактике нейродегенеративных расстройств и/или когнитивных нарушений.
УРОВЕНЬ ТЕХНИКИ
Пролилолигопептидаза (EC 3.4.21.26) (POP), также известная как пролилэндопептидаза (PREP), представляет собой серинпротеазу, которая катализирует гидролиз пептидов на С-концевой стороне остатков L-пролина. Она широко распространена у млекопитающих и может быть выделена из различных органов, включая головной мозг.
Фермент играет важную роль в расщеплении пролинсодержащих нейропептидов, связанных с функциями обучения и памяти (Wilk S et al., Life Sci. 1983; 33:2149-57; OʼLeary RM, OʼConnor B, J. Neurochem. 1995; 65:953-63).
Эффекты ингибирования пролилолигопептидазы были протестированы при лечении когнитивного дефицита, связанного с нейродегенеративными процессами. Болезнь Паркинсона была вызвана у обезьян обработкой 1-метил-4-фенил-1,2,3,6-тетрагидропиридином (MPTP), нейротоксином, вызывающим истощение вещества P. Последующее лечение S-17092, мощным ингибитором POP, повысило выполнение когнитивных задач (Schneider JS et al., Neuropsychopharmacology 2002; 26(2):176-82). Также было обнаружено, что ингибирование POP предотвращает олигомеризацию α-синуклеина ex vivo (Myöhänen TT et al., Br. J. Pharmacol. 2012; 166(3):1097-113). В случае болезни Альцгеймера (AD) несколько экспериментов in vivo на животных моделях показали, что ингибирование POP приводило к нейропротекторным и улучшающим когнитивные функции эффектам (Kato A et al., J. Pharmacol. Exp. Ther. 1997; 283(1):328-35, и Toide K et al., Rev. Neurosci. 1998; 9(1):17-29). Нейропротекторные эффекты первоначально наблюдались группой Кацубе, когда клетки коры и мозжечка были предотвращены от возрастного апоптоза путем обработки ингибитором POP ONO-1603 (Katsube N et al., J. Pharmacol. Exp. Ther. 1999; 288(1):6-13).
Клинические испытания ингибиторов POP при лечении когнитивного дефицита проводились лишь в нескольких случаях. В фазе I клинического исследования группа Морейна (Morain P et al., Br. J. Clin. Pharmacol. 2000; 50(4):350-9) обнаружила, что вышеупомянутый ингибитор пролилэндопептидазы S-17092 обладает свойствами, улучшающими когнитивные функции у здоровых пожилых людей, и имеет четкую зависимость от дозы; кроме того, никаких побочных эффектов обнаружено не было. Более поздние исследования показали дополнительные легкие стабилизирующие настроение свойства этого соединения (Morain P et al., Neuropsychobiology 2007; 55(3-4):176-83).
Сообщалось, что активность пролилолигопептидазы изменяется (посмертно) при нескольких нейродегенеративных заболеваниях, включая болезнь Альцгеймера (AD), болезнь Паркинсона, болезнь Гентингтона и рассеянный склероз (MS) (Mantle D et al., Clin. Chim. Acta 1996; 249(1-2):129-39).
Существует также значительное количество доказательств, указывающих на роль нейровоспаления в патогенезе нейродегенеративных заболеваний, таких как AD, MS и болезнь Паркинсона (Hirsch EC et al., Lancet Neurol. 2009; 8(4):382-97, Philips T et al., Lancet Neurol. 2011; 10(3):253-63). POP считается основным ферментом, участвующим в высвобождении противовоспалительного тетрапептида Ac-SDKP из Tβ4 в головном мозге (Yang F et al., Hypertension 2004; 43(2):229-36, Nolte WM et al., Biochemistry 2009; 48(50):11971-81). Это говорит о том, что ингибирование POP может помочь уменьшить нейровоспаление, и, следовательно, ингибиторы POP могут быть использованы при лечении нейродегенеративных заболеваний с воспалительным компонентом, таких как болезнь Альцгеймера и болезнь Паркинсона, и, в частности, помогают улучшить когнитивные расстройства, связанные с этими заболеваниями.
Старческие бляшки, распространяющиеся на области коры головного мозга, являются типичными нейропатологическими признаками AD. Основным белковым компонентом этих бляшек является β-амилоидный пептид (Аβ). Отложение Aβ вызывает дисфункцию нейронов и гибель в головном мозге. Этот пептид происходит от белка-предшественника β-амилоида (APP). В нормальных условиях АРР расщепляется α-секретазой с образованием растворимого АРРα, который препятствует образованию Аβ.
Интересно, что ингибирование POP увеличивает внутриклеточные уровни IP3, что может способствовать стимуляции продукции APPα, что, в свою очередь, снижает образование Aβ.
Кроме того, Rossner (Rossner S et al., Neurochem. Res. 2005; 30(6-7):695-702) обнаружил меньше иммунореактивных нейронов POP в структурах мозга пациентов с БА, пораженных бляшками Aβ.
Кроме того, представляется, что вещество P может подавлять нейротоксическое действие β-амилоидного белка (Kowall NW et al., Proc. Natl. Acad. Sci. USA 1991; 88(16):7247-51). Ингибиторы пролилолигопептидазы ингибируют метаболизм вещества Р, помогая поддерживать уровни вещества Р, которые могут подавлять нейротоксическое действие β-амилоидного белка.
Ввиду вышеупомянутых эффектов считается, что ингибиторы пролилолигопептидазы могут быть полезными лекарственными средствами для лечения болезни Альцгеймера, помогающими предотвратить повреждение головного мозга и улучшить когнитивные расстройства, связанные с этим заболеванием.
Пролилолигопептидаза также была связана с несколькими факторами, которые могут иметь отношение к рассеянному склерозу (РС). Например, POP участвует в регуляции токсичности микроглии (Klegeris A et al., Glia 2008; 56(6):675-85). Действительно, установлена прямая связь между POP и MS; активность POP в плазме у пациентов с рецидивирующе-ремиттирующим РС (RR-MS) была значительно снижена (Tenorio-Laranga J et al., J Neuroinflammation 2010; 7:23). Интересно, что снижение коррелировало с тяжестью симптомов заболевания, а не с возрастом пациента. Вместо этого у здоровых контролей наблюдалась обратная корреляция между активностью POP и возрастом, а у пожилых контролей уровни были сопоставимы с уровнями, обнаруженными у пациентов с РС.
Нейропатологическим признаком болезни Паркинсона является прогрессирующая дегенерация меланизированных дофаминергических нейронов в компактной части черной субстанции вместе с внутриклеточными включениями, известными как тельца Леви. Основным компонентом телец Леви является белок из 140 аминокислот, α-синуклеин. При определенных условиях мономеры α-синуклеина взаимодействуют с образованием префибриллярных агрегатов или протофибрилл, которые могут создавать цитотоксические нерастворимые фибриллы. Эти фибриллы не могут быть расщеплены протеасомой, и они нарушают функцию этой внутриклеточной протеолитической системы. Это приводит к накоплению протофибрилл α-синуклеина (и других белков, расщепляемых протеасомой) в цитозоле (Bennett MC, Pharmacol. Ther. 2005; 105(3):311-31) и, как следствие, увеличению количества протофибрилл α-синуклеина в головном мозге пациентов с болезнью Паркинсона. Эти фибриллы были связаны с нейротоксичностью в клетках со сверхэкспрессией α-синуклеина и мышиных моделях (Masliah E et al., Science 2000; 287(5456):1265-9; Gosavi N et al., J. Biol. Chem. 2002; 277(50):48984-92). Аномальное накопление неправильно уложенного α-синуклеина может привести к митохондриальным изменениям, которые могут способствовать окислительному стрессу и вызывать гибель клеток (Hsu LJ et al., Am. J. Pathol. 2000; 157(2):401-10). Кроме того, известно, что три точечные мутации (A53T, A30P или E46K) в гене α-синуклеина участвуют в патогенезе семейной формы болезни Паркинсона (Polymeropoulos MH et al., Science 1997; 276(5321):2045-7; Zarranz JJ et al., Ann. Neurol. 2004; 55(2):164-73).
Было показано in vitro, что скорость агрегации α-синуклеина увеличивалась, когда белок инкубировали с клоном POP дикого типа, и это повышение зависело от концентрации POP (Brandt I et al., Peptides 2008; 29(9):1472-8). Более того, мутантный вариант без активности POP (S544A) не увеличивал скорость агрегации.
Повышенную агрегацию также можно было предотвратить добавлением ингибиторов POP, что позволяет предположить, что эффект зависит от ферментативной активности POP. Экспериментальные данные свидетельствуют о том, что ингибиторы POP могут блокировать повышенную агрегацию α-синуклеина, вызванную окислительным стрессом, в клетках нейробластомы SH-SY5Y со сверхэкспрессией α-синуклеина человека (Myöhänen TT et al., Br. J. Pharmacol. 2012; 166(3):1097-113). POP ко-локализуется с α-синуклеином в клетках SH-SY5Y, и эта ко-локализация исчезает после инкубации с ингибиторами POP, указывая на взаимодействие между POP и α-синуклеином. 5-дневное лечение ингибитором POP уменьшало количество растворимого α-синуклеина в мозге трансгенных мышей с α-синуклеином A30P. В связи с этим сообщалось, что ингибиторы POP усиливают клиренс α-синуклеина путем модулирования аутофагии (Myöhänen TT et al., Pharmacological Research 2020; 151:104558).
Таким образом, ингибирование активности POP в головном мозге может предотвращать агрегацию α-синуклеина и, таким образом, предотвращать образование цитотоксических протофибрилл, присутствующих в тельцах Леви. Следовательно, ингибиторы POP потенциально могут иметь терапевтическую ценность при лечении нейродегенеративных заболеваний, при которых была описана ускоренная агрегация α-синуклеина.
Соединения, способные ингибировать POP, эффективны для предотвращения экспериментальных когнитивных нарушений, вызванных скополамином у крыс, что позволяет сделать вывод о том, что ингибиторы POP выполняют функцию облегчения мнемонических дисфункций (Yoshimoto T et al., J. Pharmacobiodyn. 1987; 10:730-5).
Эффект субхронического введения розмариновой кислоты, неконкурентного ингибитора POP (с относительно высоким значением IC50 63,7 мкМ), был протестирован в водном лабиринте Морриса на крысах, и было сообщено об улучшении пространственной памяти (Park DH et al., Fitoterapia 2010; 81(6):644-8).
Было обнаружено, что пациенты с биполярным расстройством имеют высокий уровень активности POP в сыворотке крови. В последние годы POP приобрел значение как мишень для лечения этого заболевания, особенно в связи с его участием в метаболизме инозитол-1,4,5-трифосфата (IP3). IP3 является ключевой молекулой в передаче сигнала в каскаде нейропептидов. Связываясь со специфическими рецепторами, нейропептиды индуцируют увеличение IP3, который связывается со своим рецептором на мембране эндоплазматического ретикулума и индуцирует высвобождение Ca2+, который, как полагают, играет решающую роль в обучении и памяти. Недавние открытия показали, что POP модулирует концентрацию IP3 (Komatsu Y J. Neurosci. 1996; 16:6342-52). Так, известно, что нарушение гена POP у эукариот Dictyostelium discoideum индуцирует устойчивость к литию посредством повышения уровня IP3 (Schulz I et al., Eur. J. Biochem. 2002; 269:5813-20), и также снижала протеолитическую активность POP, что обусловливает высокую концентрацию IP3 в клетках глиомы, антисмысловую для POP человека. Этот эффект также наблюдается, когда эти клетки обрабатывают специфическими ингибиторами POP (Williams RS et al., EMBO J. 1999; 18:2734-45).
Путь передачи сигналов IP3 участвует в действии некоторых лекарственных средств, терапевтических стабилизаторов настроения (литий, карбамазепин и вальпроевая кислота), и дефекты в механизмах, которые регулируют передачу сигналов IP3, могут вызывать биполярное расстройство. Более того, вальпроевая кислота, стабилизатор настроения, который обычно используется для лечения биполярного расстройства, непосредственно ингибирует активность рекомбинантного POP (Cheng L et al., Mol. CeII. Neurosci. 2005; 29: 155-61). Таким образом, имеются убедительные доказательства того, что ингибиторы POP полезны для профилактики и/или лечения биполярного аффективного расстройства у млекопитающих. Таким образом, получение новых ингибиторов POP представляет интерес для терапии этого расстройства или заболевания.
Таким образом, эффекты нескольких ингибиторов POP в различных когнитивных задачах были охарактеризованы, и существует консенсус в отношении того, что ингибиторы POP оказывают положительное влияние на обучение и память (Morain P et al., CNS Drug. Rev. 2002; 8(1):31-52; Shinoda M et al., Eur. J. Pharmacol. 1996; 305(1-3):31-8; Marighetto A et al., Learn. Mem. 2000; 7(3):159-69; Toide K et al., Pharmacol. Biochem. Behav. 1997; 56(3):427-34; Schneider JS et al., Neuropsychopharmacology 2002; 26(2):176-82).
В ряде патентов и патентных заявок описаны ингибиторы POP: WO 2008/077978 A1, WO 2005/027934 A1, JP 2011-037874 A2, WO 2005/002624 A1, WO 2004/060862 A2, WO 03/04468 A1; DE 196 03 510 A1, US 2006/0100253 A1 и US 6159938 A, но только несколько соединений подвергались исследованиям in vivo (JTP-4819, S-17092, Z-321, ONO-1603, Y-29794, ZTTA, Z-Pro-Prolinal и KYP-2047). Из этого списка первые четыре ингибитора в списке прошли клинические испытания, и ни один из них не вышел на рынок.
В WO 2014/072498 A1 описаны ингибиторы POP с высоким сродством к POP и хорошей способностью преодолевать гематоэнцефалический барьер для достижения головного мозга, где проявляется действие ингибитора при лечении когнитивных расстройств. Это важная особенность соединений, позволяющая использовать их для лечения когнитивных расстройств.
В WO 2014/072498 A1 описано, что состав соединений, раскрытых в нем, может быть адаптирован для любого пути введения. Однако только в примерах in vivo, раскрытых в указанной заявке, соединения вводили подкожно.
Было бы преимуществом иметь возможность вводить ингибиторы POP перорально, так как это наиболее удобно с точки зрения пациента.
Таким образом, существует необходимость в поиске новых ингибиторов POP, особенно хорошо адаптированных для перорального введения и эффективно достигающих головного мозга, который является местом действия при использовании указанных ингибиторов POP в терапии когнитивных расстройств. Антагонисты рецепторов серотонина (рецептор 5-HT1A) обычно используются в качестве антидепрессантов. Однако они вызывают значительную сексуальную дисфункцию (Uphouse L, Pharmacol. Biochem. Behav. 2014; 0:31-42), что ограничивает их использование для хронической терапии у взрослого населения. По этой причине ингибитор POP с положительным влиянием на когнитивные функции при аффективных расстройствах и без таких нежелательных явлений окажет значительное влияние на качество жизни пациента.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Авторы изобретения успешно обнаружили, что 1-[1-(4-бензилокси-3,5-дифтор-бензоил)-4-фтор-пирролидин-2-карбонил]-пирролидин-2-карбонитрил, его стереоизомеры и его соли не только способны ингибировать POP с высокой эффективностью, но также демонстрируют высокую проникающую способность в желудочно-кишечном тракте, обеспечивают высокое воздействие на мозг, что позволяет им быть особенно эффективными при пероральном введении и мало связываются с рецептором 5-HT1A, что позволяет избежать негативных побочных эффектов, таких как сексуальная дисфункция.
Таким образом, один аспект изобретения относится к 1-[1-(4-бензилокси-3,5-дифтор-бензоил)-4-фтор-пирролидин-2-карбонил]-пирролидин-2-карбонитрилу, имеющему формулу (I):
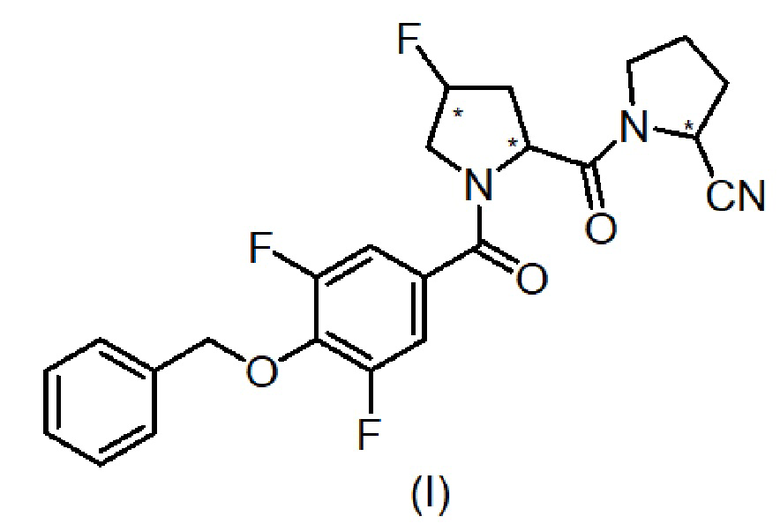
(где звездочка в формуле указывает на наличие хиральных центров), его стереоизомерам и их фармацевтически приемлемым солям.
Другой аспект данного изобретения относится к способам получения соединения формулы (I), определенного выше, его стереоизомеров и его фармацевтически приемлемых солей.
Другой аспект данного изобретения относится к лекарственному препарату или фармацевтической композиции, содержащей по меньшей мере одно соединение формулы (I), определенное выше, его стереоизомеры и их фармацевтически приемлемые соли и фармацевтически приемлемый носитель, адъювант или наполнитель.
Другой аспект данного изобретения относится к соединению формулы (I), определенному выше, его стереоизомерам и его фармацевтически приемлемым солям для применения в качестве лекарственного препарата, в частности, для предупреждения и/или лечения когнитивных расстройств и/или нейродегенеративных расстройств, таких как синуклеинопатии (заболевания, связанные с накоплением синуклеина, такие как болезнь Паркинсона, расстройство поведения во время быстрого сна, деменция с тельцами Леви или множественная системная атрофия). В частности, когнитивные расстройства могут быть связаны с заболеванием, выбранным из группы, включающей шизофрению, большое депрессивное расстройство, биполярное аффективное расстройство, расстройство поведения во время быстрого сна, болезнь Альцгеймера, лобно-височную деменцию, болезнь Паркинсона, деменцию с тельцами Леви, множественную системную атрофию, прогрессирующий надъядерный паралич, кортикобазальную дегенерацию или боковой амиотрофический склероз.
Другой аспект данного изобретения относится к способу лечения или профилактики когнитивных расстройств у млекопитающего, при котором пациенту при необходимости указанного лечения вводят терапевтическое количество соединения формулы (I), определенного выше, его стереоизомеров и его фармацевтически приемлемых солей. В конкретном варианте осуществления изобретения расстройство представляет собой когнитивное расстройство, связанное с заболеванием, выбранным из группы, включающей шизофрению, большое депрессивное расстройство, биполярное аффективное расстройство, расстройство поведения во время быстрого сна, болезнь Альцгеймера, лобно-височную деменцию, болезнь Паркинсона, деменцию с тельцами Леви, множественную системную атрофию, прогрессирующий надъядерный паралич, кортикобазальную дегенерацию или боковой амиотрофический склероз.
Другой аспект данного изобретения относится к применению соединения формулы (I), определенного выше, его стереоизомеров и их фармацевтически приемлемых солей для изготовления лекарственного препарата, в частности, для предупреждения и/или лечения когнитивных расстройств и, более конкретно, когнитивного расстройства, связанного с заболеванием, выбранным из группы, включающей шизофрению, большое депрессивное расстройство, биполярное аффективное расстройство, расстройство поведения во время быстрого сна, болезнь Альцгеймера, лобно-височную деменцию, болезнь Паркинсона, деменцию с тельцами Леви, множественную системную атрофию, прогрессирующий надъядерный паралич, кортикобазальную дегенерацию или боковой амиотрофический склероз. Эти аспекты и их предпочтительные варианты осуществления изобретения дополнительно также определены в формуле изобретения.
ОПИСАНИЕ ФИГУР
На фигуре 1 показаны результаты анализа ингибирования POP для соединений по примеру 1, сравнительному примеру 3 и сравнительному примеру 4.
На фигуре 2 показаны результаты анализа PAMPA для соединений по примеру 1, сравнительному примеру 3 и сравнительному примеру 4.
На фигуре 3 показаны результаты анализа Caco-2 для соединений по примеру 1 и сравнительному примеру 3.
На фигуре 4 показаны результаты анализа воздействия на мозг in vivo для соединений по примеру 1 и сравнительному примеру 3.
На фигурах 5a и 5b показаны результаты анализа задачи NOR для соединений по примеру 1 и сравнительному примеру 3.
На фигуре 6 показаны результаты связывания с рецептором 5-HT1A для соединений по примеру 1 и сравнительному примеру 3.
На фигуре 7 показан ИК-спектр (S)-1-((2S,4R)-1-(4-(бензилокси)-3,5-дифторбензоил)-4-фторпирролидин-2-карбонил)пирролидин-2-карбонитрила.
На фигуре 8 показан 1H-ЯМР спектр (S)-1-((2S,4R)-1-(4-(бензилокси)-3,5-дифторбензоил)-4-фторпирролидин-2-карбонил)пирролидин-2-карбонитрила.
На фигуре 9 показан ИК-спектр (S)-1-((2S,4S)-1-(4-(бензилокси)-3,5-дифторбензоил)-4-фторпирролидин-2-карбонил)пирролидин-2-карбонитрила.
На фигуре 10 показан 1H-ЯМР спектр (S)-1-((2S,4S)-1-(4-(бензилокси)-3,5-дифторбензоил)-4-фторпирролидин-2-карбонил)пирролидин-2-карбонитрила.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В контексте настоящего изобретения следующие термины имеют значения, подробно описанное ниже.
Термин «соль» следует понимать, как любую форму активного соединения, используемого в соответствии с данным изобретением, в которой указанное соединение находится в ионной форме или заряжено и связано с противоионом (катионом или анионом) или находится в растворе. Это определение также включает соли четвертичного аммония и комплексы активной молекулы с другими молекулами и ионами, в частности, комплексы, образованные посредством ионных взаимодействий. Определение включает, в частности, физиологически приемлемые соли; этот термин следует понимать как эквивалент «фармакологически приемлемых солей» или «фармацевтически приемлемых солей».
Термин «фармацевтически приемлемые соли» в контексте настоящего изобретения означает любую соль, которая является физиологически переносимой (обычно это означает, что она не токсична, в частности, из-за противоиона) при использовании соответствующим образом для лечения, применяемым или используемым, в частности, на людях и/или млекопитающих. Эти физиологически приемлемые соли могут быть образованы с катионами или с основаниями, и в контексте настоящего изобретения понимаются, как соли, образованные по крайней мере одним соединением, используемым в соответствии с изобретением, обычно кислотой (депротонированной), такой как анион, особенно при использовании на людях и/или млекопитающих. Эти физиологически приемлемые соли также могут быть образованы с анионами или с кислотами, и в контексте настоящего изобретения под ними понимаются соли, образованные по меньшей мере одним соединением, используемым в соответствии с изобретением, обычно протонированным, например, по азоту, таким как катион, и по меньшей мере с одним физиологически переносимым анионом, особенно при использовании на людях и/или млекопитающих. Это определение конкретно включает в контексте данного изобретения соль, образованную физиологически переносимой кислотой, то есть соли конкретного активного соединения с физиологически переносимой органической или неорганической кислотой, особенно при использовании на людях и/или млекопитающих. Примерами солей этого типа являются хлористоводородная кислота, бромистоводородная кислота, серная кислота, метансульфоновая кислота, муравьиная кислота, уксусная кислота, щавелевая кислота, янтарная кислота, яблочная кислота, винная кислота, миндальная кислота, фумаровая кислота, молочная кислота или лимонная кислота.
Соединения по настоящему изобретению, представленные вышеописанной формулой (I), могут включать энантиомеры, возникающие в результате присутствия в молекуле трех хиральных центров. Отдельные энантиомеры и любая смесь двух или более из них входят в объем настоящего изобретения.
Если не указано иное, соединения по настоящему изобретению также включают меченные изотопами формы, то есть соединения, которые отличаются только наличием одного или нескольких атомов, обогащенных изотопами. Например, в объем настоящего изобретения входят соединения, имеющие настоящую структуру, за исключением замены по крайней мере одного атома водорода на дейтерий или тритий, или замены по крайней мере одного атома углерода на 13C- или 14C-обогащенный углерод, или замены по крайней мере одного атома азота на 15N-обогащенный азот.
Соединения формулы (I) или их соли, предпочтительно. находятся в фармацевтически приемлемой или практически чистой форме. Под фармацевтически приемлемой формой понимается, среди прочего, форма, имеющая фармацевтически приемлемый уровень чистоты, за исключением обычных фармацевтических добавок, таких как разбавители и носители, и не включающая вещества, считающиеся токсичными при нормальных дозировках. Уровни чистоты лекарственного вещества, предпочтительно, выше 50%, более предпочтительно, выше 70%, наиболее предпочтительно, выше 90%. В предпочтительном варианте осуществления изобретения они составляют более 95% соединения формулы (I) или его солей.
Как отмечалось ранее, термин «фармацевтически приемлемые соли» относится к любой соли, которая при введении реципиенту способна (прямо или косвенно) обеспечивать соединение, описанное в настоящем документе. Однако следует понимать, что фармацевтически неприемлемые соли также попадают в объем изобретения, поскольку они могут быть полезны при получении фармацевтически приемлемых солей, сольватов и пролекарств. Получение солей можно осуществлять способами, известными специалистам в данной области техники.
Как используется в настоящем документе, термины «лечить», «лечение» и «излечение» включают устранение, удаление, реверсию, облегчение, модификацию или контроль заболевания или состояния, такого как когнитивное расстройство.
Как используется в настоящем документе, термины «предупреждение», «предотвращение», «предупредительный», «предотвращать» и «профилактика» относятся к способности соединения формулы (I) предотвращать, минимизировать или затруднять возникновение или развитие заболевания или состояния, такого как когнитивное расстройство, до его возникновения.
Следовательно, под «терапией» или «лечением» и/или «предупреждением» или «профилактикой» в целом подразумевается, по меньшей мере, подавление или улучшение симптомов, связанных с состоянием, поражающим субъекта, где подавление и улучшение являются используется в широком смысле для обозначения, по меньшей мере, уменьшения величины параметра, например, симптома, связанного с состоянием, которое лечат, например, когнитивным расстройством. Таким образом, способ по настоящему изобретению также включает ситуации, когда состояние полностью подавлено, например, предотвращено или остановлено, например, прекращено, так, что субъект больше не испытывает состояние. Таким образом, настоящий способ включает как предупреждение, так и лечение когнитивного расстройства.
Термины «когнитивное расстройство» и «умеренные когнитивные нарушения» используются в настоящем документе взаимозаменяемо для обозначения любого состояния, характеризующегося дефицитом умственной деятельности, связанной с мышлением, обучением или памятью. Примеры таких расстройств включают агнозии, амнезии, афазии, апраксии, делирии, деменции и расстройства обучения.
Когнитивное расстройство может быть (и часто бывает) связано (то есть может быть вызвано или возникать при наличии) других состояний, характеризующихся повреждением или потерей нейронов или других структур, участвующих в передаче сигналов между нейронами. Следовательно, когнитивные расстройства могут быть связаны с нейродегенеративными заболеваниями, такими как болезнь Альцгеймера, кортикобазальная дегенерация, болезнь Крейтцфельдта-Якоба, лобно-височная долевая дегенерация, болезнь Хантингтона, рассеянный склероз, гидроцефалия нормального давления, органический хронический мозговой синдром, болезнь Паркинсона, болезнь Пика, сосудистая деменция, деменция с тельцами Леви, множественная системная атрофия, прогрессирующий надъядерный паралич.
Когнитивные расстройства также могут быть связаны с другими состояниями, нарушающими нормальное функционирование центральной нервной системы, включая психические расстройства, такие как тревожные расстройства, диссоциативные расстройства, расстройства настроения, такие как биполярное аффективное расстройство, шизофрения, а также соматоформные и симулятивные расстройства.
Соединения, описанные в настоящем документе, могут быть использованы для лечения агнозии, амнезии, афазии, апраксии, делирия, деменции, расстройств обучения и других когнитивных расстройств.
Примеры деменций, которые можно лечить способами по изобретению, включают комплекс деменции при СПИДе, болезнь Бинсвангера, деменцию с тельцами Леви, лобно-височную деменцию, деменцию с множественными инфарктами, болезнь Пика, семантическую деменцию и сосудистую деменцию.
Примеры расстройств обучения, которые можно лечить способами по изобретению, включают синдром Аспергера, синдром дефицита внимания, синдром дефицита внимания и гиперактивности, аутизм, детское дезинтегративное расстройство, синдром Дауна и синдром Ретта.
Примеры афазии, которые можно лечить способами по изобретению, включают прогрессирующую неплавную афазию.
Соединения, описанные в настоящем документе, могут также применяться для лечения пациентов с нарушениями умственной деятельности, которые являются умеренными или которые иным образом существенно не мешают повседневной жизни. Умеренные когнитивные нарушения являются примером такого состояния: у пациента с умеренными когнитивными нарушениями проявляются симптомы деменции (например, трудности с речью или памятью), но тяжесть этих симптомов такова, что диагноз деменции может быть неподходящим. Соединения, описанные в настоящем документе, могут быть использованы для лечения умеренных когнитивных нарушений и других, менее тяжелых форм когнитивных расстройств.
Таким образом, другим аспектом настоящего изобретения является способ лечения или профилактики когнитивных расстройств у млекопитающего, при котором пациенту, нуждающемуся в указанном лечении, вводят терапевтическое количество соединения по изобретению.
В конкретном варианте осуществления изобретения способ лечения или профилактики осуществляется путем введения пациенту, нуждающемуся в указанном лечении, терапевтического количества соединения по изобретению пероральным путем.
В конкретном варианте осуществления соединения по изобретению вводят перорально в суточной дозе, составляющей от 0,01 до 1,90 мг/кг, например, 0,01, 0,05, 0,1, 0,15, 0,20, 0,25, 0,30, 0,35, 0,40, 0,45, 0,50, 0,55, 0,60, 0,65, 0,70, 0,75, 0,80, 0,85, 0,90, 0,95, 1,0 1,05, 1,10, 1,15, 1,20, 1,25, 1,30, 1,35, 1,40, 1,45, 1,50, 1,55, 1,60, 1,65, 1,70, 1,75, 1,80, 1,50, 1,60, 1,65, 1,70, 1,75, 1,80, 1,55, 1,60, 1,65, 1,70, 1,75, 1, 1,85 или 1,90 мг/кг. Соединения по изобретению можно вводить в любой из указанных выше суточных пероральных доз один раз в день, два раза в день, три раза в день, несколько раз в день, пять раз в день или шесть раз в день.
В конкретном варианте осуществления настоящего изобретения соединения, описанные в настоящем документе, могут применяться для лечения пациентов с когнитивным расстройством, связанным с шизофренией, биполярным аффективным расстройством, болезнью Альцгеймера или болезнью Паркинсона.
В конкретном варианте осуществления настоящего изобретения соединения имеют следующую формулу (Ia):
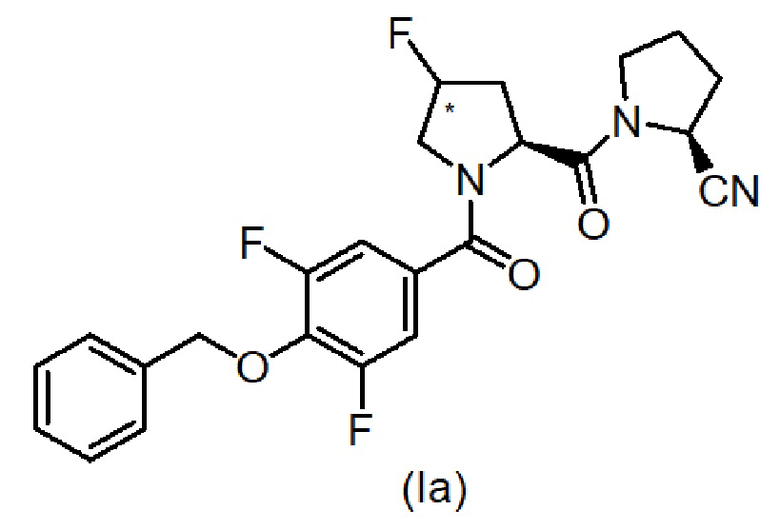
Конкретные индивидуальные соединения по изобретению, подпадающие под формулу (I), включают соединения, перечисленные ниже:
(S)-1-((2S,4R)-1-(4-(бензилокси)-3,5-дифторбензоил)-4-фторпирролидин-2-карбонил)пирролидин-2-карбонитрил
(R)-1-((2S,4R)-1-(4-(бензилокси)-3,5-дифторбензоил)-4-фторпирролидин-2-карбонил)пирролидин-2-карбонитрил
(S)-1-((2R,4R)-1-(4-(бензилокси)-3,5-дифторбензоил)-4-фторпирролидин-2-карбонил)пирролидин-2-карбонитрил
(R)-1-((2R,4R)-1-(4-(бензилокси)-3,5-дифторбензоил)-4-фторпирролидин-2-карбонил)пирролидин-2-карбонитрил
(S)-1-((2S,4S)-1-(4-(бензилокси)-3,5-дифторбензоил)-4-фторпирролидин-2-карбонил)пирролидин-2-карбонитрил
(R)-1-((2S,4S)-1-(4-(бензилокси)-3,5-дифторбензоил)-4-фторпирролидин-2-карбонил)пирролидин-2-карбонитрил
(S)-1-((2R,4S)-1-(4-(бензилокси)-3,5-дифторбензоил)-4-фторпирролидин-2-карбонил)пирролидин-2-карбонитрил
(R)-1-((2R,4S)-1-(4-(бензилокси)-3,5-дифторбензоил)-4-фторпирролидин-2-карбонил)пирролидин-2-карбонитрил
или их фармацевтически приемлемые соли.
Соединения формулы (I), определенные выше, могут быть получены с помощью доступных способов синтеза, как показано на следующих общих схемах:
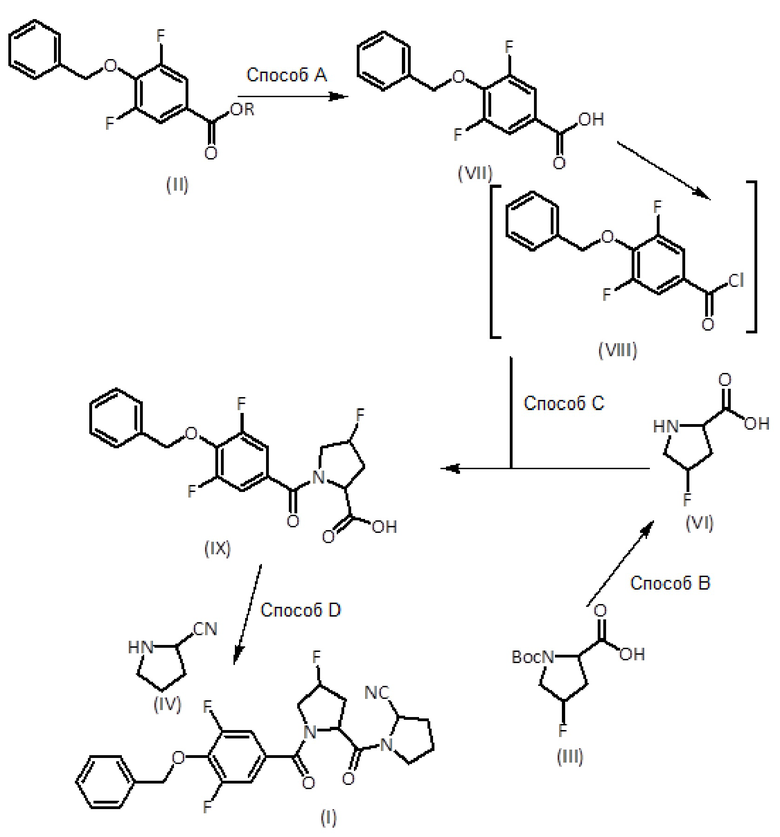
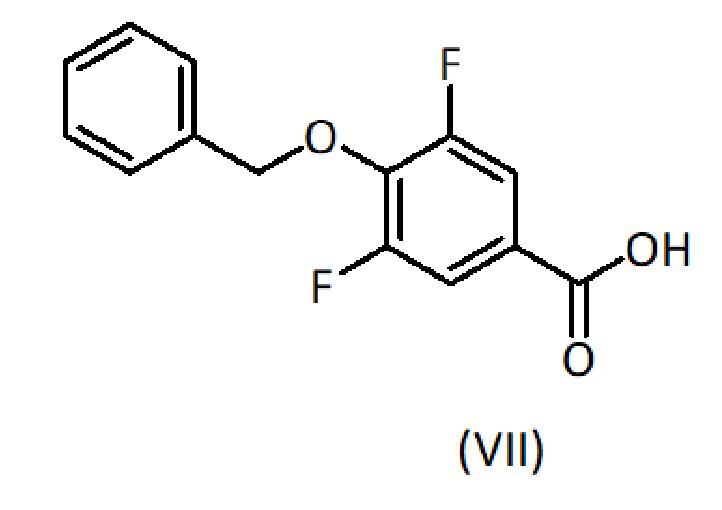
На первой стадии сложный эфир формулы (II) растворяют или суспендируют в полярном органическом растворителе (предпочтительно, протонном полярном органическом растворителе), таком как этанол (EtOH) или метанол, или в смеси полярных органических растворителей. Добавляют водный раствор основания и проводят реакцию гидролиза, выдерживая смесь, как правило, с обратным холодильником, при температуре от комнатной до температуры кипения смеси растворителей до завершения гидролиза, обычно в течение периода от 0,5 до 4 часов, а лучше 1-2 часа. Раствор основания, предпочтительно, имеет неорганическую природу, например разбавленная щелочь, например NaOH. Затем реакционную смесь оставляют для достижения комнатной температуры и, предпочтительно, концентрируют приблизительно до одной пятой реакционного объема. Затем при охлаждении на бане со льдом реакционную смесь медленно добавляют к раствору кислоты, такому как 1М раствор HCl, для осуществления нейтрализации. Если подкисление приводит к осаждению, твердое вещество фильтруют и промывают водой, получая продукт формулы (VII). Если осадка не образуется, полученный раствор несколько раз экстрагируют подходящим органическим растворителем, таким как этилацетат, органическую фазу сушат и выпаривают. Неочищенный продукт формулы (VII) очищают флэш-хроматографией.
Удаление защиты у амина формулы (III) достигается в слабокислых условиях, таких как добавление к раствору хлористого водорода в органическом растворителе, таком как диоксан или смесь ТФУ/ДХМ, при пониженной температуре в диапазоне от 0°C до комнатной температуры. Реакционную смесь перемешивают при комнатной температуре в течение 1-3 часов. Затем растворитель выпаривают досуха с получением гидрохлоридной соли или трифторацетатной соли амина формулы (VI) в зависимости от используемой кислоты.
Соединение формулы (IX) получают из карбоновой кислоты формулы (VII) и амина формулы (VI) в условиях реакции Шоттена-Баумана. Соответственно, к раствору карбоновой кислоты формулы (VII) в органическом растворителе, таком как толуол, добавляют хлорирующий агент, такой как оксалилхлорид. Реакционную смесь перемешивают при температуре от 50°С до 80°С в течение 1-2 часов, чтобы обеспечить образование хлорангидрида карбоновой кислоты формулы (VIII). После выпаривания растворителя полученный сырой продукт солюбилизируют в органическом растворителе, таком как ТГФ, и добавляют к водно-основному раствору амина формулы (VI), обычно водному раствору амина формулы (VI) в NaOH, при пониженной температуре, такой как 0°C. Реакционную смесь перемешивают при пониженной температуре в течение от 1 до 2 часов и при комнатной температуре в течение от 2 до 4 часов. Затем растворитель выпаривают, рН оставшейся водной фракции доводят до кислого (3-4) добавлением раствора HCl и экстрагируют этилацетатом. Органическую фазу промывают насыщенным солевым раствором, сушат, фильтруют и выпаривают. При необходимости сырой продукт очищают флэш-хроматографией.
Затем продукт формулы (IX) связывают с пирролидин-2-карбонитрилом формулы (IV) в присутствии основания, такого как N,N-диизопропилэтиламин (DIEA), и с помощью реагента сочетания, такого как карбодиимид. В частности, соединение формулы (IX) растворяют в апротонном органическом растворителе, таком как дихлорметан, и добавляют к карбодиимиду, например, к карбодиимиду на твердом носителе, такому как N-циклогексилкарбодиимид, Nʼ-метилполистирол, вместе с DIEA. Через 5 мин добавляют пирролидин-2-карбонитрил формулы (IV) и дополнительное количество DIEA. Реакционную смесь перемешивают при комнатной температуре в течение от 8 до 16 часов. Затем реакционную смесь фильтруют и оставшееся твердое вещество промывают апротонным органическим растворителем. Фильтрат упаривают досуха. Сырой продукт затем очищают препаративной ОФ-ВЭЖХ.
Альтернативно, соединения формулы (I) могут быть получены, как показано на следующей схеме и описано ниже:
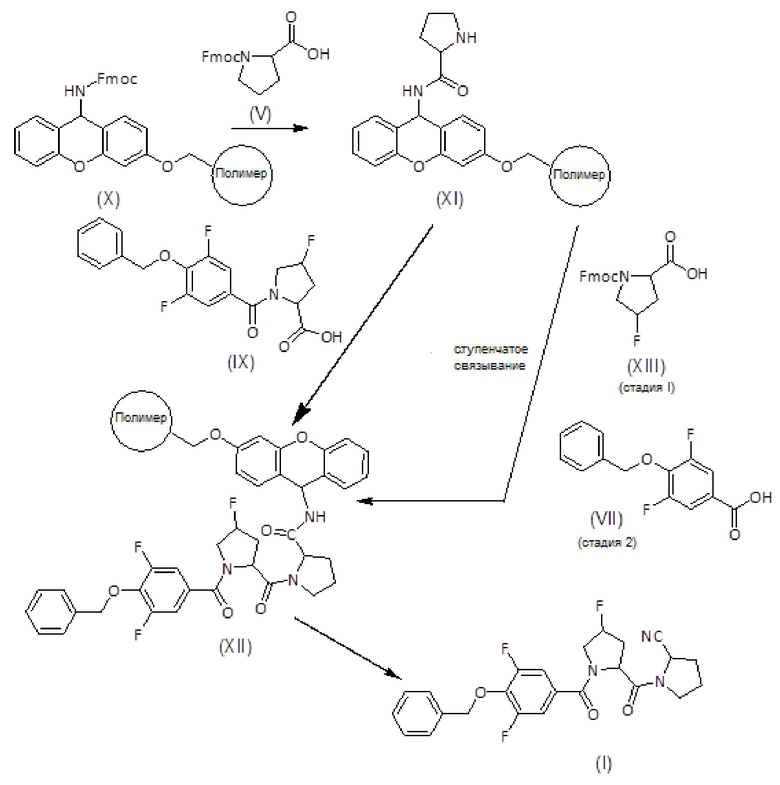
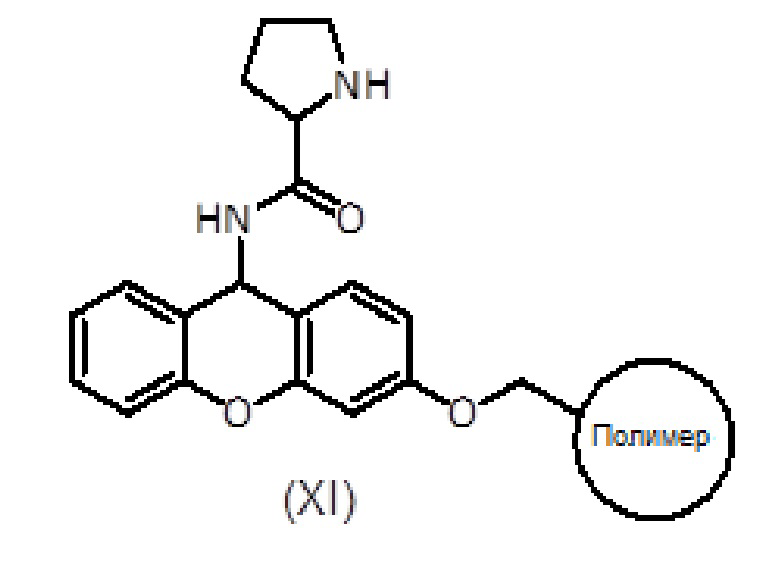
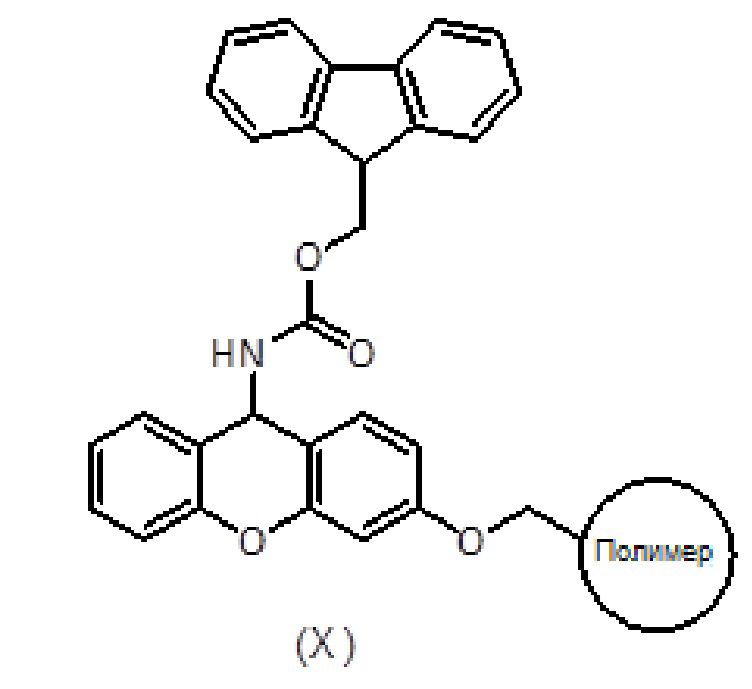
Смолу, функционализированную амином, такую как амидная смола Зибера формулы (X), помещают в шприц, снабженный пористым полиэтиленовым диском. Смола набухает при промывании соответствующими органическими растворителями, такими как дихлорметан (ДХМ) и диметилформамид (ДМФ). Когда аминогруппа смолы защищена (например, в случае амидной смолы Зибера), удаление защитной группы (такой как защитная группа флуоренилметоксикарбонила (Fmoc)) достигается обработкой раствором основания амина, таким как раствор пиперидина в ДМФ.
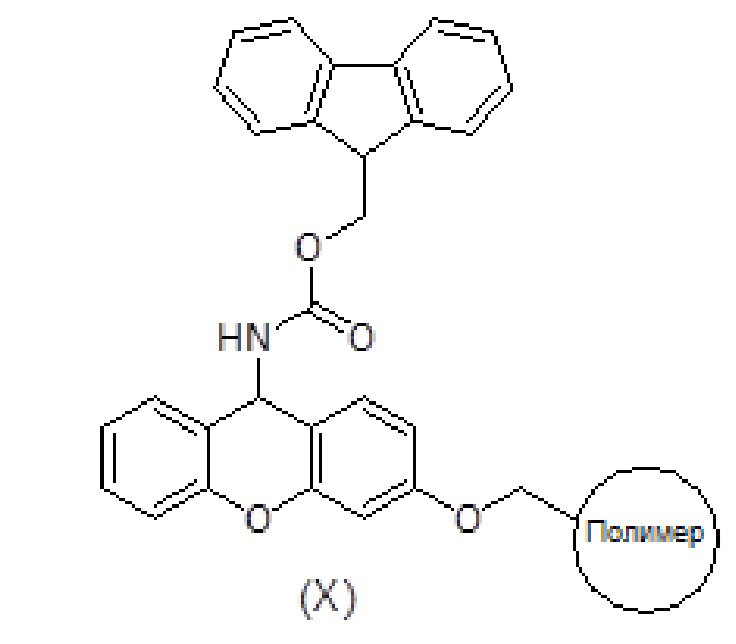
После удаления защитной группы у смолы к смоле присоединяют Fmoc-защищенный пролин формулы (V) с использованием активирующего агента, такого как триазол (то есть TBTU), и основания амина, такого как DIEA, в подходящем органическом растворителе, таком как ДМФ. Смесь перемешивают в течение 1-2 часов. После фильтрации и промывки степень связывания можно контролировать с помощью теста Кайзера, при необходимости проводят повторное связывание. Fmoc удаляют обработкой раствором основания амина, таким как раствор пиперидина в ДМФ и/или смесь пиперидин/DBU/толуол/ДМФ, с получением продукта формулы (XI). Удаление Fmoc можно оценить с помощью теста NF31 с п-нитрофениловым эфиром.

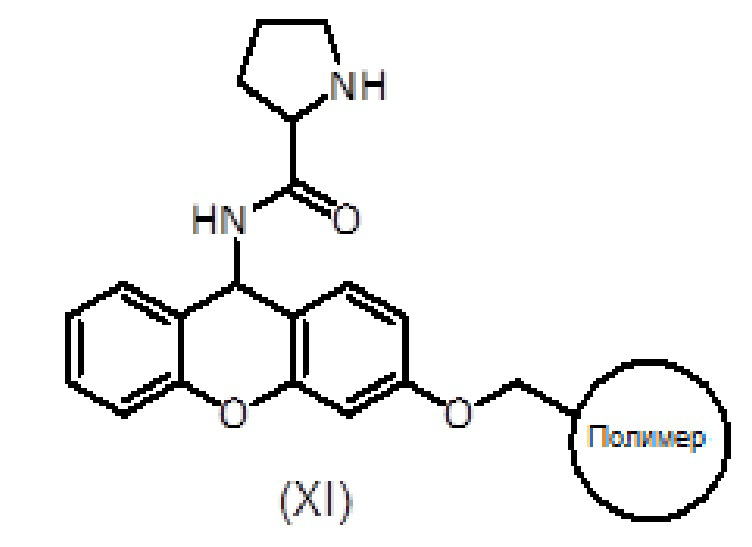
Продукт формулы (XI) связывают с продуктом формулы (IX) с получением продукта формулы (XII) с использованием активирующего агента, такого как PyBOP, в присутствии или в отсутствие добавки, такой как HOAt, и основания амина, такого как DIEA, в подходящем органическом растворителе, таком как ДМФ. Смесь перемешивают вручную в течение общего времени реакции от 1 до 2 часов. Систематическое повторное присоединение осуществляют с использованием тех же количеств и времени. Степень связывания можно контролировать с помощью теста NF31 с п-нитрофениловым эфиром.
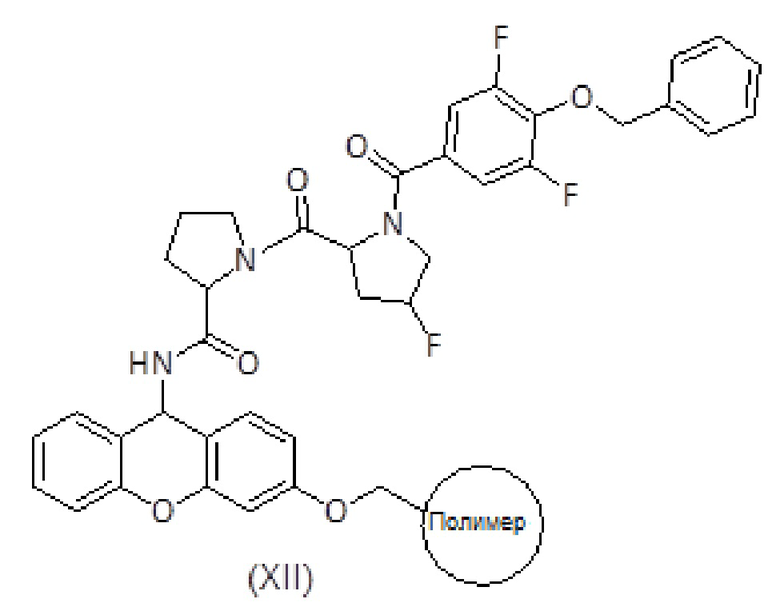
Альтернативно, продукт формулы (XII) также может быть получен путем ступенчатого сочетания продукта (XI) сначала с соединением формулы (XIII), с последующим удалением защитной группы Fmoc и затем сочетанием с соединением формулы (VII).
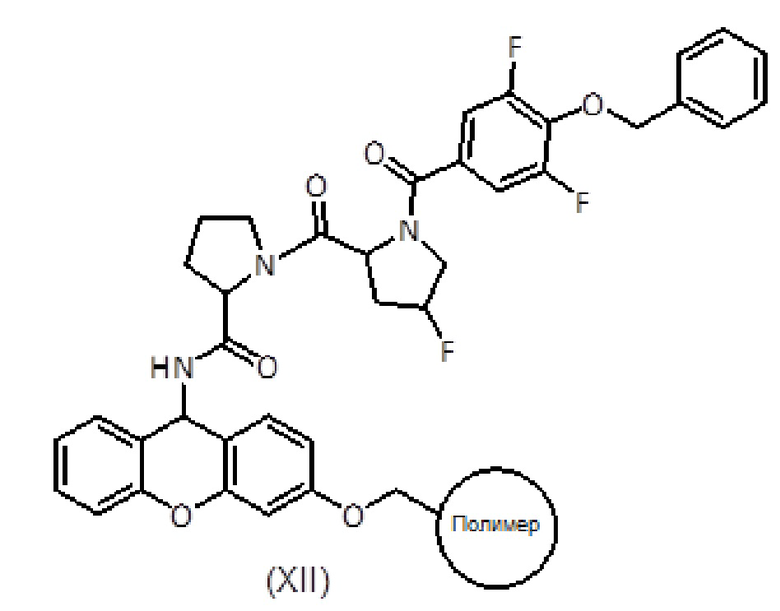
Продукт формулы (XII), тщательно промытый подходящим органическим растворителем, таким как DCM, и высушенный, переносят в колбу, в которую добавляют трифторуксусный ангидрид и пиридин в небольшом количестве органического растворителя. Смесь выдерживают при температуре от 20 до 40°С в течение от 8 до 16 часов. Затем реакционную смесь фильтруют и смолу промывают тем же органическим растворителем Фильтраты собирают и растворитель выпаривают досуха. Полученный сырой продукт растворяют в подходящем растворителе, таком как этилацетат, и промывают насыщенным раствором NaHCO3 и 5% водн. раствором KHSO4. Органическую фазу сушат, фильтруют и упаривают. Сырой продукт растворяют в смеси H2O:CH3CN и лиофилизуют с получением нитрила пептида формулы (I).
Альтернативно, пептидил-смола формулы (XII) может быть обработана смесью ТФУ/H2O/TIS в течение 1-2 часов. Затем смолу фильтруют и промывают ТФУ, фильтраты собирают и растворитель выпаривают досуха. Сырой продукт вновь суспендируют в смеси H2O:CH3CN и лиофилизуют. Полученный сырой амид пептида помещают в соответствующий органический растворитель, такой как ДХМ, и преобразуют в нитрил, например, в присутствии пятиокиси фосфора, тетрахлорида титана, тионилхлорида, трифторуксусного ангидрида/пиридина или трифенилфосфина/четыреххлористого углерода. Смесь выдерживают при комнатной температуре в течение от 8 до 16 часов, растворитель выпаривают и остаток растворяют в этилацетате. Органический раствор затем промывают водн. раствором KHSO4 и водн. раствором NaHCO3. Сушка и выпаривание органической фазы дает нитрил пептида формулы (I).
Сырой продукт очищают ОФ-ВЭЖХ.
Когда описанные выше способы получения соединений по изобретению дают смеси стереоизомеров, эти изомеры могут быть разделены обычными методами, такими как препаративная хроматография. При наличии хиральных центров соединения могут быть получены в рацемической форме или получены отдельные энантиомеры либо путем энантиоспецифического синтеза, либо путем разделения.
Соединения формул (II), (III), (IV) и (V), а также некоторые соединения формулы (VII), используемые в качестве исходных продуктов, являются либо коммерчески доступными, либо также могут быть получены с использованием способов, хорошо известных специалисту в данной области.
Таким образом, в одном аспекте настоящее изобретение относится к способам получения соединения формулы (I) или его фармацевтически приемлемой соли, изомера, пролекарства или сольвата.
В одном варианте осуществления изобретения способ включает стадии:
a) взаимодействие соединения формулы (IX):
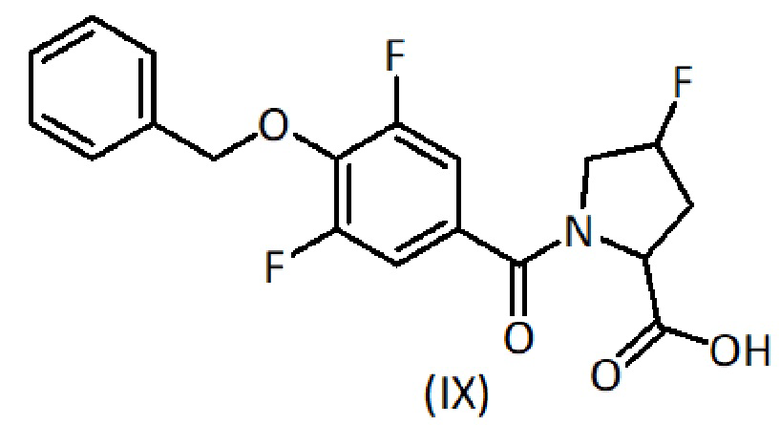
с соединением формулы (XI):
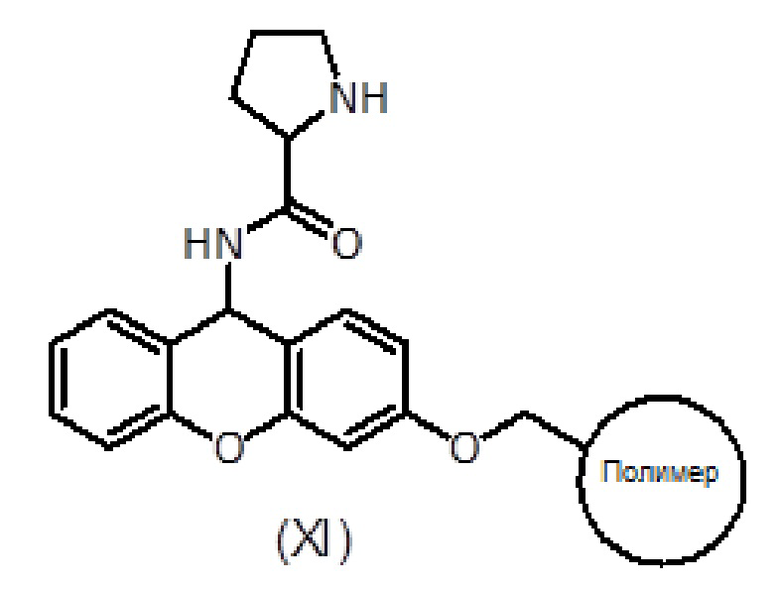
где полимер означает полимер, который является инертным в условиях реакции способа синтеза, описанного в настоящем документе, и нерастворимым, но способным набухать в используемых в данном случае растворителях, таких как малосшитый полистирол и полимеры полистирола с привитым полиэтиленгликолем.
с получением соединения формулы (XII):
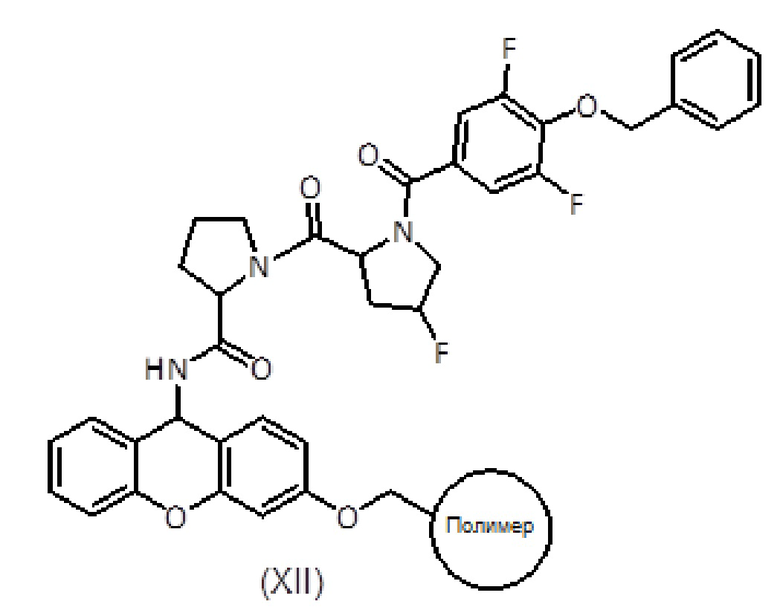
b) гидролиз соединения формулы (XII) с получением соединения формулы (XIV):
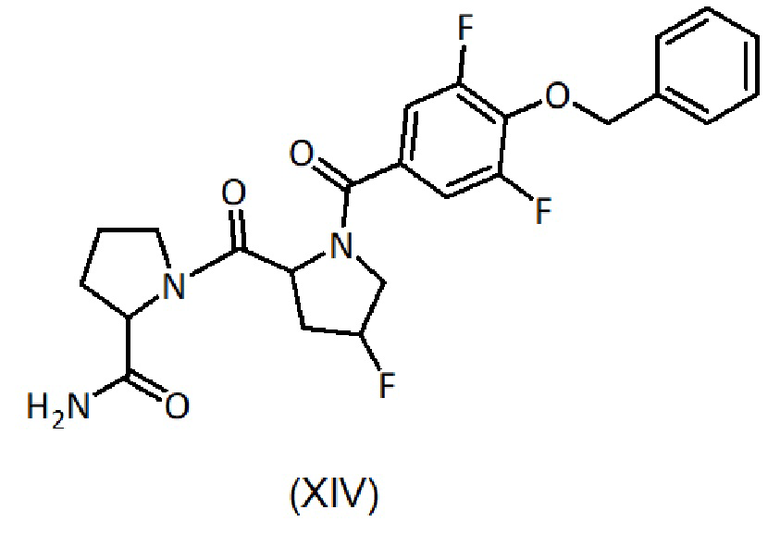
и
c) воздействие на соединение формулы (XIV) условий, способных преобразовать карбоксамидную группу в нитрильную группу с получением соединения формулы (I):
где стадии b) и c) можно проводить отдельно или в одном реакторе.
В другом варианте осуществления настоящего изобретения способ получения соединения формулы (I) или его фармацевтически приемлемой соли, изомера, пролекарства или сольвата включает следующие стадии:
a) взаимодействие соединения формулы (IX):
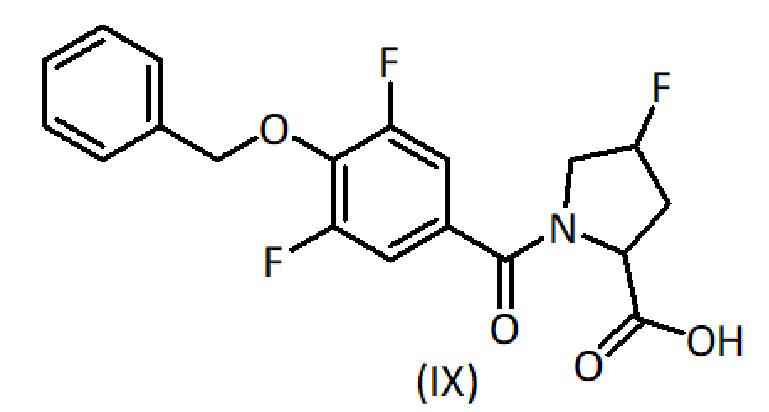
с соединением формулы (IV):
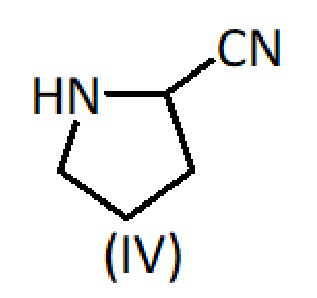
В еще одном варианте осуществления способ включает следующие стадии:
a) взаимодействие соединения формулы (XI):
где полимер означает полимер, который является инертным в условиях реакции описанного здесь синтетического метода и нерастворимым, но набухающим в используемых здесь растворителях, таких как малосшитый полистирол и полистирольные полимеры с привитым полиэтиленгликолем,
с соединением формулы (XIII):
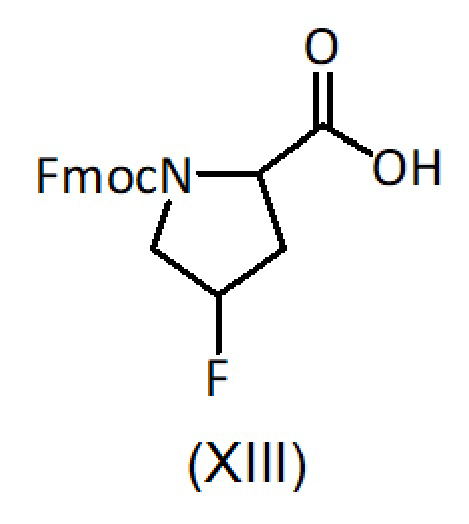
b) удаление защитной группы Fmoc,
c) взаимодействие с соединением формулы (VII):
d) гидролиз полученного продукта из несущего полимера с получением соединения формулы (I):
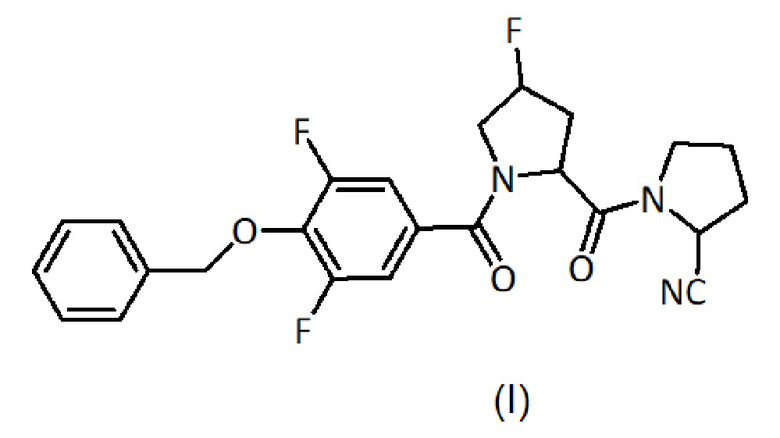
Было обнаружено, что соединения общей формулы (I) могут быть использованы при лечении когнитивных расстройств, в частности, когнитивных расстройств, связанных с другими заболеваниями или состояниями центральной нервной системы.
В конкретном варианте осуществления изобретения когнитивное расстройство представляет собой когнитивное расстройство, связанное с заболеванием, выбранным из группы, включающей шизофрению, биполярное аффективное расстройство, болезнь Альцгеймера и болезнь Паркинсона.
Настоящее изобретение дополнительно относится к лекарственным препаратам или фармацевтическим композициям для введения пациенту, содержащим соединение по данному изобретению или его фармацевтическую соль, производное, пролекарство или стереоизомер вместе с фармацевтически приемлемым носителем, адъювантом или наполнителем.
Вспомогательные материалы или добавки для фармацевтической композиции по настоящему изобретению могут быть выбраны из носителей, эксципиентов, вспомогательных материалов, смазывающих веществ, наполнителей, растворителей, разбавителей, красителей, ароматизаторов, таких как сахара, антиоксидантов, связующих веществ, адгезивов, разрыхлителей, антиадгезивов, глидантов и/или агглютинирующих веществ. В случае c суппозиториями это может подразумевать воски или сложные эфиры жирных кислот, или консерванты, эмульгаторы и/или носители для парентерального применения. Выбор этих вспомогательных материалов и/или добавок и используемых количеств будет зависеть от вида применения фармацевтической композиции.
Лекарственный препарат или фармацевтическая композиция по настоящему изобретению могут быть в любой форме, подходящей для применения к людям и/или животным, предпочтительно, людям, включая младенцев, детей и взрослых, и могут быть получены стандартными способами, известными специалистам в данной области техники. Таким образом, состав в соответствии с изобретением может быть адаптирован для местного или системного применения, в частности, для кожного, трансдермального, подкожного, внутримышечного, внутрисуставного, внутрибрюшинного, внутривенного, внутриартериального, внутрипузырного, внутрикостного, интракавернозного, интраназального, легочного, буккального, сублингвального, глазного, интравитреального, чрескожного, ректального, вагинального, перорального, эпидурального, интратекального, интравентрикулярного, интрацеребрального, интрацеребровентрикулярного, интрацистернального, интраспинального, периспинального, интракраниального, с помощью игл или катетеров с насосными устройствами или без них, или других путей введения.
В предпочтительном варианте осуществления настоящего изобретения фармацевтические композиции находятся в пероральной форме, твердой или жидкой. Подходящие лекарственные формы для перорального введения могут представлять собой таблетки, пилюли, каплеты, желатиновые капсулы, жевательные резинки, капсулы, гранулы, капли, сиропы или растворы и могут содержать обычные эксципиенты, известные в данной области техники, такие как связующие агенты, например, сироп, гуммиарабик, желатин, сорбит, трагакант или поливинилпирролидон; наполнители, например лактоза, сахар, кукурузный крахмал, фосфат кальция, сорбит или глицин; смазывающие вещества для таблетирования, например стеарат магния; разрыхлители, например, крахмал, поливинилпирролидон, крахмалгликолят натрия или микрокристаллическая целлюлоза; или фармацевтически приемлемые смачивающие агенты, такие как лаурилсульфат натрия.
В другом варианте осуществления изобретения фармацевтические композиции находятся в виде продуктов для непарентерального интраназального введения, предпочтительно, в виде продуктов для интраназального введения. Как правило, интраназальное введение осуществляется с использованием назальных спреев, бутылочек и капельниц в качестве средств доставки. Для использования с этими устройствами фармацевтические композиции, предпочтительно, представляют собой жидкие растворы или суспензии соединений по изобретению.
Композиции могут быть приготовлены обычными способами смешивания, заполнения или таблетирования. Повторные операции по смешиванию могут быть использованы для распределения активного агента по композициям, в которых используются большие количества наполнителей. Такие операции являются обычными в данной области техники. Таблетки могут быть получены, например, влажным или сухим гранулированием и, необязательно, покрыты в соответствии со способами, хорошо известными в обычной фармацевтической практике, в частности, энтеросолюбильным покрытием.
Фармацевтические композиции также могут быть адаптированы для парентерального введения, такие как стерильные растворы, суспензии или восстанавливаемые сухие препараты, аэрозоли или спреи в подходящей дозированной форме. Могут быть использованы адекватные эксципиенты, такие как наполнители, буферные агенты или поверхностно-активные вещества.
Композиция по изобретению может быть приготовлена в виде отложений в растворенной форме или в виде пластырей для чрескожного применения.
Кожные аппликации включают мази, гели, кремы, лосьоны, суспензии или эмульсии.
Подходящей формой для ректального применения являются суппозитории.
Упомянутые составы могут быть приготовлены с использованием стандартных способов, таких как описанные или упомянутые в Фармакопеях Испании и США и аналогичной справочной литературе.
В одном варианте осуществлении изобретения является предпочтительным, чтобы соединение формулы (I) использовалось в терапевтически эффективных количествах. Врач определит наиболее подходящую дозировку настоящих терапевтических средств, и она будет варьироваться в зависимости от формы введения и конкретного выбранного соединения, и, кроме того, она будет варьироваться в зависимости от пациента, проходящего лечение, возраста пациента, типа заболевания или состояния, подлежащего лечению. Когда композицию вводят перорально, могут потребоваться большие количества активного агента для получения того же эффекта, что и при введении меньшего количества, вводимого парентерально. Соединения могут быть использованы таким же образом, как и сопоставимые терапевтические агенты, и уровень дозировки имеет тот же порядок величины, что и обычно используемый с такими терапевтическими агентами. Активные соединения обычно вводят один или несколько раз в день, например, 1, 2, 3 или 4 раза в день, с типичными общими дневными дозами в диапазоне от 0,011 до 1,90 мг/кг/день.
Соединения и композиции по данному изобретению могут быть использованы с другими лекарственными средствами для обеспечения комбинированной терапии. Другие лекарственные средства могут составлять часть той же композиции или быть предоставлены в виде отдельной композиции для введения в одно и то же время или в разное время.
В частности, комбинация по меньшей мере одного соединения формулы (I) и по меньшей мере еще одного лекарственного средства может быть составлена для его одновременного, раздельного или последовательного введения по меньшей мере с фармацевтически приемлемым носителем, добавкой, адъювантом или наполнителем. Это подразумевает, что комбинацию соединения формулы (I) и другого лекарственного средства можно вводить следующим образом:
a) В виде комбинации, которая является частью одного и того же лекарственного препарата, оба препарата вводят всегда одновременно.
b) В виде комбинации двух единиц, каждая из которых дает возможность одновременного, последовательного или раздельного введения. В варианте осуществления изобретения соединение формулы (I) вводят независимо от другого лекарственного средства (то есть двумя дозами), но в одно и то же время. В другом конкретном варианте сначала вводят соединение формулы (I), а затем отдельно или последовательно вводят другое лекарственное средство. В еще одном конкретном варианте сначала вводят другое лекарственное средство, а затем вводят соединение формулы (I), отдельно или последовательно, как определено.
В контексте настоящего изобретения использовались следующие акронимы и сокращения, значения которых подробно описаны ниже:
AD
AUC
BBB
Boc
BSA
DBU
DCM
DMEM
DI
DIEA
ДМФ
ДМСО
EtOH
Fmoc
FPLC
FTIR
HBSS
HOAt
hPOP
IP
IP3
IPTG
LB
MALDI-TOF
MK-801
MS
ЯМР
NOR
OD
PAMPA
PBS
PC
PE
pETM10
PI
POP
PS
PyBOP
ОФ-ВЭЖХ
SD
SDS-PAGE
TBTU
TEER
ТФУ
ТГФ
TIS
Tris
Tβ4
УВЭЖХ
Z-G-P-AMC
Болезнь Альцгеймера
Площадь под кривой
Гематоэнцефалический барьер
трет-Бутоксикарбонил
Бычий сывороточный альбумин
1,8-Диазабицикло[5.4.0] ундец-7-ен
Дихлорметан
Модифицированная среда Игла Дульбекко
Индекс дискриминации
N, N’-Диизопропилэтиламин
Диметилформамид
Диметилсульфоксид
Этанол
9-Флуоренилметоксикарбонил
Быстрая белковая жидкостная хроматография
Инфракрасный спектрометр с Фурье-преобразованием
Сбалансированный солевой раствор Хэнкса
1-Гидрокси-7-азабензотриазол
Пролилолигопептидаза человека
Внутрибрюшинно
Инозитолтрифосфат
Изопропил β-D-1- тиогалактопиранозид
Лизогенный бульон
Времяпролетная матрично-активированная лазерная десорбция/ионизация
Дизоцилпин
Рассеянный склероз
Ядерный магнитный резонанс
Распознавания новых объектов
Оптическая плотность
Параллельный анализ проницаемости искусственной мембраны
Фосфатно-солевой буфер
Фосфатидилхолин
Фосфатидилэтаноламин
Плазмида pETM10
Фосфатидилинозитол
Пролилолигопептидаза
Фосфатидилсерин
(Бензотриазол-1-илокси)трипирролидинофосфония гексафторфосфат
Высокоэффективная жидкостная хроматография с обращенной фазой
Стандартное отклонение
Электрофорез в полиакриламидном геле с додецилсульфатом натрия
O-(Бензотриазол-1-ил)-N, N,N',N'-тетраметилурония тетрафторборат
Трансэндотелиальное электрическое сопротивление
Трифторуксусная кислота
Тетрагидрофуран
Триизопропилсилан
Трис(гидроксиметил)аминометан
Белок тимозин бета-4
Ультраэффективная жидкостная хроматография
(N-Бензилоксикарбонил-Gly-Pro-метилкумаринил-7-амид)
Следующие примеры являются просто иллюстрацией некоторых вариантов осуществления изобретения и никоим образом не могут рассматриваться как ограничивающие его.
ПРИМЕРЫ
КОНКРЕТНЫЕ УСЛОВИЯ СИНТЕЗА, ИСПОЛЬЗУЕМЫЕ ПРИ ПОЛУЧЕНИЯХ, ОПИСАННЫХ В ПРИМЕРАХ
Способ A: Гидролиз сложного эфира формулы (II) до карбоновой кислоты формулы (VII)
Сложный эфир формулы (II) (1 ммоль) растворяют в 95% EtOH. Добавляют NaOH (3,7 ммоль) и реакционную смесь кипятят с обратным холодильником в течение примерно 2 часов. Затем оставляют для достижения комнатной температуры. Реакционную смесь концентрируют до прибл. 15-20 мл, затем этот раствор медленно добавляют к 1М раствору HCl при охлаждении на бане со льдом. Белый твердый осадок, который собирают фильтрованием, промывают водой и хорошо сушат перед следующей стадией синтеза. В случае отсутствия осадка полученный раствор экстрагируют AcOEt (3х), органическую фазу сушат и выпаривают. Сырой продукт при необходимости очищают флэш-хроматографией.
Способ B: Удаление защиты у Boc-защищенного амина формулы (III) с получением амина формулы (VI)
Вос-защищенный амин формулы (III) (1 ммоль) медленно добавляют к 4М HCl в диоксане (20 мл) при температуре 0°С. Реакцию перемешивают при комнатной температуре в течение 2 часов. Затем растворитель выпаривают досуха с получением гидрохлоридной соли амина формулы (VI).
Способ C: Связывание амина формулы (VI) с карбоновой кислотой формулы (VII) посредством образования хлорангидрида карбоновой кислоты формулы (VIII).
К раствору карбоновой кислоты формулы (VII) (1 ммоль) в толуоле (5 мл) добавляют оксалилхлорид (1,5 ммоль). Реакционную смесь перемешивают при температуре 50°С в течение 1,5 часов, чтобы обеспечить образование хлорангидрида карбоновой кислоты формулы (VIII). После выпаривания растворителя полученный сырой продукт солюбилизируют в ТГФ и добавляют к водному раствору амина формулы (VI) (1,1 ммоль) в NaOH при температуре 0°С. Реакционную смесь перемешивают при температуре 0°С в течение 1,5 часов и при комнатной температуре в течение 3 часов. Затем ТГФ выпаривают, и оставшуюся водную фракцию доводят до кислого pH (3-4) добавлением 1М раствора HCl и экстрагируют AcOEt. Органическую фазу промывают соляным раствором, сушат, фильтруют и выпаривают. Сырой продукт формулы (IX) при необходимости очищают флэш-хроматографией.
Способ D: Связывание продукта формулы (IX) с пирролидин-2-карбонитрилом формулы (IV) в растворе
Продукт формулы (IX) (1,2 ммоль) растворяют в ДХМ и добавляют к N-циклогексилкарбодиимид-Nʼ-метилполистиролу (3 ммоль) вместе с DIEA (1 ммоль). Через 5 мин добавляют (S)-пирролидин-2-карбонитрил формулы (IV) (1 ммоль) и DIEA (1 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение ночи. Затем реакционную смесь фильтруют и оставшееся твердое вещество промывают ДХМ. Фильтрат упаривают досуха. Затем сырой продукт очищают препаративной ОФ-ВЭЖХ и лиофилизуют, получая продукт формулы (I).
Способ E: Общий способ синтеза на твердой фазе
Набухание/кондиционирование смолы: амидную смолу Зибера формулы (X) (1 экв.) помещают в шприц, снабженный пористым полиэтиленовым диском. Смола набухает от промывок ДХМ и ДМФ. Удаление флуоренилметоксикарбонильной (Fmoc) защитной группы достигается обработкой 20% раствором пиперидина в ДМФ.
Затем к смоле присоединяют Fmoc-защищенный пролин формулы (V) (4 экв.) с использованием TBTU (4 экв.) и DIEA (8 экв.) в ДМФ. Смесь периодически перемешивают вручную в течение 90 мин. После фильтрации и промывки степень связывания контролируют с помощью теста Кайзера, при необходимости проводят повторное связывание. Fmoc удаляют с получением продукта формулы (XI) обработкой 20% раствором пиперидина в ДМФ и затем раствором пиперидин/DBU/толуол/ДМФ (20:5:5:70). Удаление Fmoc оценивают с помощью теста NF31 п-нитрофенилового эфира (описанного Madder, A. et al., Eur. J. Org. Chem. 1999; (11):2787-91).
Продукт формулы (IX) (2 экв.) связывают с продуктом формулы (XI) с получением продукта формулы (XII) с использованием PyBOP (2 экв.), HOAt (6 экв.) и DIEA (6 экв.) в ДМФ. Смесь периодически перемешивают вручную в течение всего времени реакции 90 мин. Систематическое повторное соединение осуществляется с использованием тех же количеств и времени. Степень связывания контролируют с помощью теста NF31 с п-нитрофениловым эфиром.
Альтернативно, продукт формулы (XIII) (4 экв.) связывают с продуктом формулы (XI) с использованием PyBOP (4 экв.), HOAt (12 экв.) и DIEA (12 экв.) в ДМФ. Смесь периодически перемешивают вручную в течение всего времени реакции 90 мин. Степень связывания контролируют с помощью теста с п-нитрофениловым эфиром NF31 и при необходимости проводят повторное связывание. Группу Fmoc удаляют обработкой 20% раствором пиперидина в ДМФ и обработкой раствором пипериди/DBU/толуол/ДМФ (20:5:5:70). Затем продукт формулы (VII) (4 экв.) вводят с использованием PyBOP (4 экв.), HOAt (12 экв.) и DIEA (12 экв.) в ДМФ с получением продукта формулы (XII). Смесь периодически перемешивают вручную в течение всего времени реакции 90 мин. Степень связывания контролируют с помощью теста с п-нитрофениловым эфиром NF31 и при необходимости проводят повторное связывание.
Продукт формулы (XII), тщательно промытый ДХМ и высушенный, переносят в круглодонную колбу и добавляют трифторуксусный ангидрид (5 экв.) и пиридин (10 экв.) в ДХМ (приблизительно 2 мл/100 мг). Смесь выдерживают при комнатной температуре в течение ночи. Затем реакционную смесь фильтруют и смолу промывают ДХМ. Фильтраты собирают и растворитель выпаривают досуха. Полученный сырой продукт растворяют в AcOEt и промывают насыщенным раствором NaHCO3 и 5% водн. раствором KHSO4. Органическую фазу сушат, фильтруют и упаривают. Сырой продукт растворяют в H2O:CH3CN (1:1) и лиофилизуют с получением нитрила пептида формулы (I).
Альтернативно, пептидил-смола формулы (XII) может быть обработана смесью ТФУ/H2O/TIS (95:2,5:2,5, прибл. 2-5 мл/100 мг) течение 1-2 часов. Затем смолу фильтруют и промывают ТФУ, фильтраты собирают, растворитель выпаривают досуха. Сырой продукт вновь суспендируют в смеси H2O:CH3CN (1:1) и лиофилизуют. Полученный сырой амид пептида растворяют в ДХМ и добавляют трифторуксусный ангидрид (5 экв.) и пиридин (10 экв.). Смесь выдерживают при комнатной температуре в течение ночи, растворитель выпаривают и остаток растворяют в AcOEt. Органический раствор затем промывают 5% водн. раствором KHSO4 и 10% водн. раствором NaHCO3. Сушка и выпаривание органической фазы дают нитрил пептида формулы (I).
Сырой продукт очищают ОФ-ВЭЖХ.
Пример 1
(S)-1-((2S,4R)-1-(4-(бензилокси)-3,5-дифторбензоил)-4-фторпирролидин-2-карбонил)пирролидин-2-карбонитрил
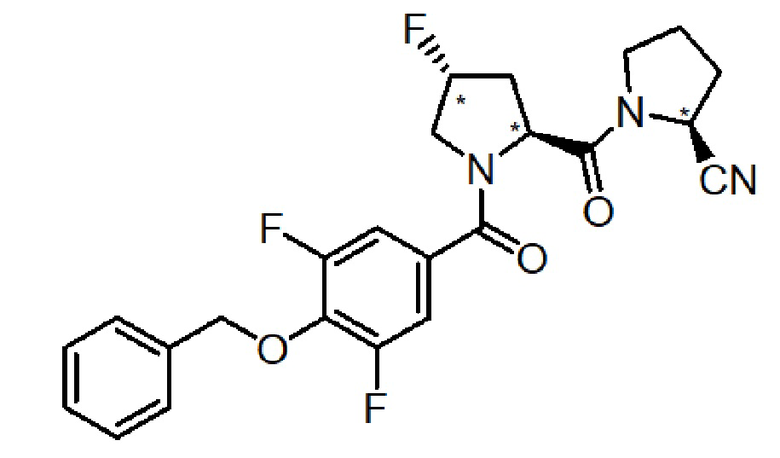
Коммерчески доступный Fmoc-защищенный L-пролин (Fmoc-L-Pro-OH) формулы (V) (249,7 мг, 0,7 ммоль), (2S,4R)-4-фтор-1-Fmoc-пирролодин-2- карбоновую кислоту (533 мг, 1,5 ммоль) формулы (III) и 4-бензилокси-3,5-дифторбензойную кислоту (271 мг, 1 ммоль) формулы (VII) последовательно связывают с коммерчески доступной амидной смолой Зибера (333,3 мг , 0,2 ммоль, 1 экв.), посредством ступенчатого связывания, как описано выше в способе E. Очистка с помощью ОФ-ВЭЖХ дает 156 мг (0,341 ммоль) целевого продукта.
т. пл. 137,6-139,1°C. FTIR: 1642, 1582, 1517, 1414, 1381, 1317, 1230, 1203, 1172, 1064, 1036 см-1. 1H-ЯМР (CDCl3, 400 МГц) δ (м. д.): 7,42 (2H, м), 7,35 (3H, м), 7,12 (2H, м), 5,30 (1H, м), 5,23 (1H, м), 4,93 (1H, дд), 4,82 (1H, м), 3,87 (2H, м), 4,08/3,69 (2H, м), 2,58/2,36 (2H, м), 2,26 (2H, м), 2,24 (2H, м).
Пример 2
(S)-1-((2S,4S)-1-(4-(бензилокси)-3,5-дифторбензоил)-4-фторпирролидин-2-карбонил)пирролидин-2-карбонитрил
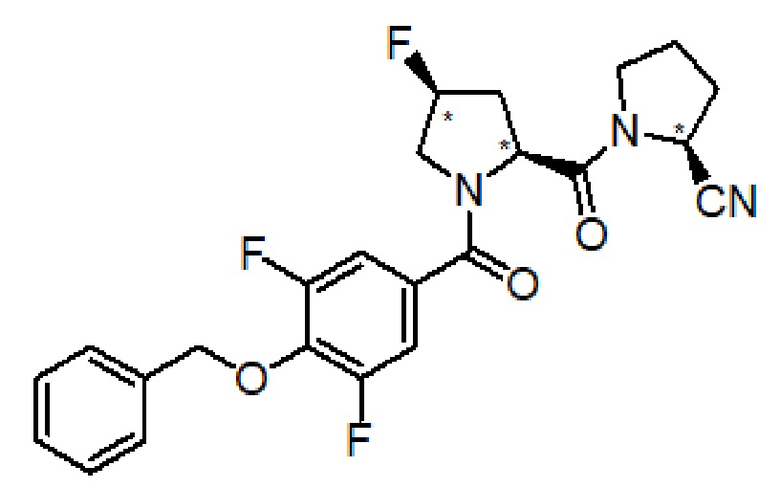
Это соединение было получено по способу, описанному в примере 1, но с заменой (2S,4R)-4-фтор-1-Fmoc-пирролодин-2-карбоновой кислоты на (2S,4S)-4-фтор-1-Fmoc-пирролодин-2-карбоновая кислоту. Очистка с помощью ОФ-ВЭЖХ дает 138 мг (0,302 ммоль) целевого продукта.
т. пл. 135-137°C. FTIR: 3483, 2954, 1653, 1581, 1518, 1437, 1417, 1383, 1356, 1321, 1234, 1203, 1172, 1065, 1039, 1005, 960, 901, 856, 762, 737, 696 см-1.
Сравнительный пример 3:
(S)-1-((2S)-1-(4-(бензилокси)-3,5-диметоксибензоил)-4,4-дифторпирролидин-2-карбонил)пирролидин-2-карбонитрил
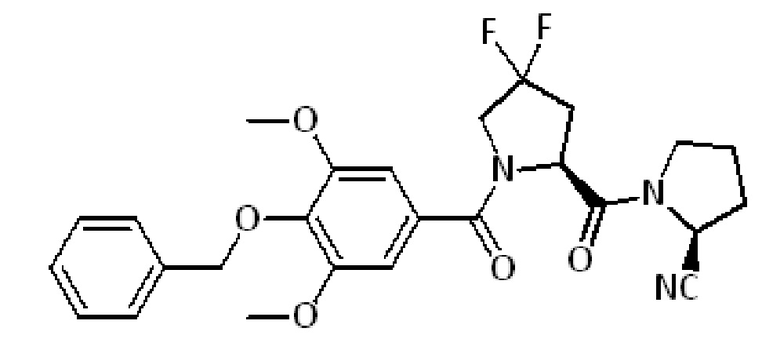
Продукт получают в соответствии с процедурой Е, описанной выше, исходя из коммерчески доступной амидной смолы Зибера (250 мг, 0,19 ммоль, 1 экв.), коммерчески доступного Fmoc-L-пролина (Fmoc-L-Pro-OH) (258 мг, 0,77 ммоль) и (S)-1-(4-(бензилокси)-3,5-диметоксибензоил)-4,4-дифторпирролидин-2-карбоновой кислоты (161 мг, 0,38 ммоль). Очистка с помощью ОФ-ВЭЖХ дает 18 мг (0,036 ммоль) целевого продукта.
Сравнительный пример 4:
(S)-1-((S)-1-(4-(бензилокси)-3-фторбензоил)-4,4-дифторпирролидин-2-карбонил) пирролидин-2-карбонитрил
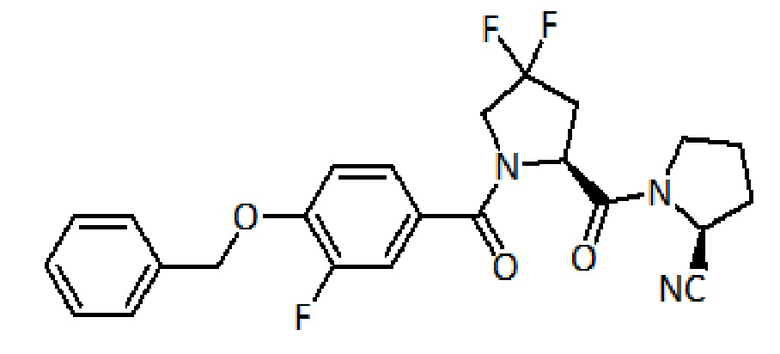
Коммерчески доступный Fmoc-защищенный L-пролин (Fmoc-L-Pro-OH) (150 мг, 0,45 ммоль), Fmoc-4,4-дифтор-L-пролин (166 мг, 0,45 ммоль) и 4-бензилокси-3-фторбензойную кислоту (109 мг, 0,45 ммоль) последовательно связывают с коммерчески доступной амидной смолой Зибера (200 мг, 0,15 ммоль, 1 экв.) посредством ступенчатого связывания, как описано в способе E выше. Очистка с помощью ОФ-ВЭЖХ дает 14 мг (0,030 ммоль) целевого продукта.
ФАРМАКОЛОГИЧЕСКИЕ ДАННЫЕ
ОПРЕДЕЛЕНИЕ ИНГИБИРУЮЩЕГО ДЕЙСТВИЯ НОВЫХ СОЕДИНЕНИЙ НА АКТИВНОСТЬ (ЧЕЛОВЕЧЕСКОЙ) ПРОЛИЛОЛИГОПЕПТИДАЗЫ
Экспрессия и очистка пролилолигопептидазы (POP)
POP была получена экспрессией в E. coli и аффинной очисткой с использованием слияния His-хвоста в соответствии с описанным в литературе способом (Tarragó T et al., ChemBioChem 2006; 7:827-33), кратко изложенным далее:
Экспрессия hPOP: компетентные клетки E. coli BL21 трансформировали pETM10 hPOP. Для индукции экспрессии одну колонию инокулировали прекультурой среды LB (50 мл), содержащей канамицин (50 мкг/мл), и выращивали в течение ночи при температуре 37°C. На следующий день две культуры среды LB (500 мл) инокулировали ночной культурой (10 мл). Инокулированные культуры выращивали при температуре 37°C и 220 об/мин до тех пор, пока OD595 не стала равной 1,2 (2,5-3 часа). Затем добавляли IPTG (конечная концентрация 1 мМ) и проводили индукцию в течение ночи при температуре 25°C. Клетки собирали (3500 g, 15 мин, 4°C) и осадок суспендировали в суспензионном буфере (50 мл) [Трис-HCl pH 8 (50 мМ), NaCl (300 мМ), имидазол (1 мМ)] и обрабатывали ультразвуком с использованием четырех циклов (каждый из которых состоял из 15 секунд обработки ультразвуком и 15 секунд отдыха) при интенсивности 50% и 0,5 импульса, образец хранили на льду. После обработки ультразвуком образец центрифугировали (40000 g, 30 мин, 4°C) и супернатант сразу использовали для очистки POP. Для очистки использовали систему ÅKTA explorer FPLC. Супернатант наносили со скоростью 1 мл/мин на колонку HiTrapQuelating (5 мл), предварительно уравновешенную 5 объемами колонки суспензионного буфера. Колонку промывали суспензионным буфером до тех пор, пока поглощение при 280 нм не возвращалось к исходному уровню. Затем колонку промывали 5 объемами промывочного буфера (50 мМ Трис-HCl, pH 8, 300 мМ NaCl, 30 мМ имидазола). Элюирование проводили 4 объемами элюирующего буфера (50 мМ Трис-HCl, рН 8, 300 мМ NaCl, 500 мМ имидазол). Фракции (4 мл) собирали в течение всего элюирования. Активность POP проверяли во всех фракциях, а положительные фракции анализировали с помощью SDS-PAGE и окрашивали красителем Biosafe Comassie Stain G-250. Положительные фракции собирали и обессоливали с помощью колонки HiPrep 26/10 Desalting с Трис-HCl (50 мМ, pH 8) в качестве буфера. Рекомбинантный hPOP количественно определяли с помощью анализа белка Bio-Rad с BSA в качестве стандарта. Готовили аликвоты рекомбинантного фермента, немедленно замораживали жидким азотом и хранили при температуре -80°C.
Анализы ингибирования POP
Активность POP определяли по методу, описанному Toide et al (Toide K et al., J. Pharmacol. Exp. Ther. 1995; 274:1370-8), с использованием Z-G-P-AMC (N-бензилоксикарбонил-Gly-Pro-метилкумаринил-7-амид) в качестве субстрата для POP. Реакции проводили в 96-луночных титрационных микропланшетах, что позволяло одновременно контролировать несколько реакций. Для каждой реакции буфер активности (134 мкл, 100 мМ фосфатный буфер Na/K, pH 8,0) предварительно инкубировали в течение 15 мин при температуре 37°C с hPOP (в диапазоне от 20 до 60 нМ, в зависимости от активности партии hPOP) и раствором соответствующего нового соединения (3 мкл). Исходный раствор нового соединения готовили в ДМСО (100 мМ), из этого исходного раствора готовили разведения с ДМСО.
После предварительной инкубации добавляли Z-G-P-AMC (10 мкл, 3 мМ в 40% 1,4-диоксане) (3 мкл, 1,5 мМ в 40% 1,4-диоксане, в условиях В) и реакцию инкубировали в течение 1 час при температуре 37°C. Реакцию останавливали добавлением ацетата натрия (150 мкл, 1 М, рН 4) и флуориметрически измеряли образование АМС. Длины волн возбуждения и испускания составляли 360/40 и 485/20 нм, соответственно.
Для каждого соединения измеряли несколько точек концентрации (в диапазоне от 25 пМ до 400 мкМ). Ингибирующую активность в отношении пролилолигопептидазы рассчитывали согласно уравнению 1. Для каждого нового соединения измеряли флуоресценцию в присутствии (a) и в отсутствие hPOP (b). Максимальная флуоресценция (0% ингибирующая активность) была получена от образца hPOP в отсутствие ингибирующих соединений. Для оценки ингибирующей активности нового соединения активность была построена в зависимости от логарифмической концентрации соединения, приведенной к сигмовидной кривой с использованием программного обеспечения GraphPad Prism, и по полученной кривой устанавливали значение IC50, определенное как концентрация соединения, необходимая для ингибирования 50% активности POP.
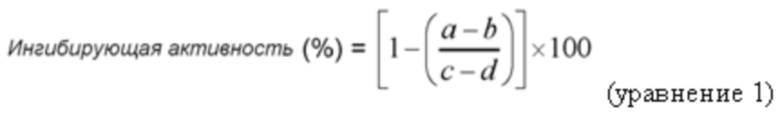
где:
a соответствует интенсивности флуоресценции в присутствии субстрата+исследуемого соединения+hPOP
b соответствует интенсивности флуоресценции в присутствии субстрата+исследуемого соединения
c соответствует интенсивности флуоресценции в присутствии субстрата+hPOP
d соответствует интенсивности флуоресценции в присутствии субстрата.
Новые соединения обладают высокой ингибирующей активностью в отношении пролилолигопептидазы человека. Результаты обобщены в таблице 1.
Данные таблицы 1 представлены на фиг. 1. На указанной фигуре столбцы представляют собой среднее значение±стандартное отклонение (SD).
ОПРЕДЕЛЕНИЕ ПРОНИЦАЕМОСТИ СОЕДИНЕНИЙ
Параллельный анализ проницаемости искусственной мембраны (PAMPA)
Параллельный анализ проницаемости искусственной мембраны (PAMPA), описанный Kansy M et al., J. Med. Chem. 1998; 41(7):1007-10, использовали для определения способности соединений преодолевать гематоэнцефалический барьер (BBB) путем пассивной диффузии (Di L et al., Eur. J. Med. Chem. 2003; 38(3):223-32). Эффективную проницаемость (Pe) соединений измеряли при начальной концентрации 200 мкМ. Буферный раствор готовили из коммерческого концентрированного по инструкции производителя. рН доводили до 7,4 с помощью 0,5 М раствора NaOH. Исходный раствор нового соединения готовили в ДМСО и разбавляли буферным раствором до конечной концентрации 200 мкМ (содержание ДМСО 0,5%). Сэндвич с PAMPA разделяли, и каждую донорную лунку заполняли 200 мкл раствора соединения. Акцепторную пластину помещали в донорскую, следя за тем, чтобы нижняя сторона мембраны находилась в контакте с буфером. В фильтр каждой лунки добавляли по 4 мкл смеси фосфолипидов (20 мг/мл) в додекане, в каждую акцепторную лунку добавляли по 200 мкл буферного раствора. Акцепторную пластину помещали в донорскую, следя за тем, чтобы нижняя сторона мембраны находилась в контакте с буфером. В фильтр каждой лунки добавляли по 4 мкл смеси фосфолипидов (20 мг/мл) в додекане, в каждую акцепторную лунку добавляли по 200 мкл буферного раствора. Планшет накрывали и инкубировали при комнатной температуре в насыщенной влажной атмосфере в течение 4 часов при орбитальном встряхивании со скоростью 100 об/мин. Через 4 часа содержимое акцепторного и донорного отсеков анализировали методом ВЭЖХ: по 150 мкл каждой лунки с донорского планшета и по 150 мкл каждой лунки с акцепторного планшета переносили во флаконы для ВЭЖХ, вводя каждый образец в обращенно-фазовую колонку C18 (150 мм×4,6 мм×5 мкм, 100 Å) (100 мкл/инъекция из акцепторных лунок, 10 мкл/инъекция из донорских лунок и для эталонов t0). Транспорт был также подтвержден спектрометрией MALDI-TOF.
Используемая смесь фосфолипидов представляла собой экстракт полярных липидов головного мозга свиньи, предоставленный компанией Avanti polar lipids, со следующим составом: 12,6% фосфатидилхолина (PC), 33,1% фосфатидилэтаноламина (PE), 18,5% фосфатидилсерина (PS), 4,1% фосфатидилинозита (PI), 0,8% фосфатидной кислоты и 30,9% других соединений.
Эффективную проницаемость (Pe) через 4 часа рассчитывали по уравнению 2, процент переноса (T%) рассчитывали по уравнению 3 и удержание соединения (R%) фосфолипидной мембраны по уравнению 4:
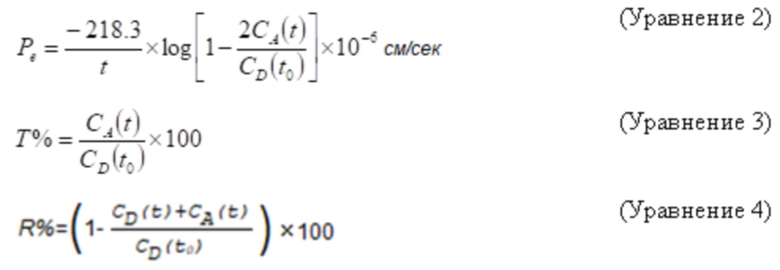
где:
t обозначает время (h)
CA(t) обозначает концентрацию соединения в акцепторной лунке в момент времени t, CD(t) обозначает концентрацию соединения в донорной лунке в момент времени t
и CD(t0) обозначает концентрацию соединения в донорской лунке в момент времени t0.
Основываясь на ориентировочных значениях Pe, показанных в таблице 2, новые соединения демонстрируют хорошую проницаемость через BBB (таблица 3).
Данные таблицы 3 представлены на фиг. 2. На указанной фигуре столбцы представляют собой среднее значение±стандартное отклонение (SD).
Анализ Caco-2
Клетки Caco-2 широко используются в качестве модели in vitro для прогнозирования абсорбции лекарственных средств человеком. Линия клеток Caco-2 происходит от колоректальной карциномы человека, и при культивировании клетки спонтанно дифференцируются в монослои поляризованных энтероцитов.
Было показано, что проницаемость лекарств через монослои клеток Caco-2 хорошо коррелирует с абсорбцией in vivo у человека (Zhao YH et al., 2001, J. Pharmaceut. Sci. 90:749-784) и стала хорошо зарекомендовавшим себя методом in vitro для прогнозирования абсорбции в кишечнике (Kansy M et al., 2001, Pharmcacokinetic optimization in drug research, Ed. Testa B et al., 447-646).
Эксперименты проводились с использованием набора для измерения проницаемости CacoReay (ReadyCell, Barcelona, Spain).
На той же неделе после получения набора транспортную среду клеток разжижали в соответствии с руководством пользователя, прилагаемым к набору, после чего транспортную среду заменяли на свежую среду Игла, модифицированную Дульбекко (DMEM, с низким содержанием глюкозы).
Через три дня, что соответствует 20-му дню культивирования клеток, качество барьерной системы и наличие плотных контактов между клетками проверяли путем измерения трансэндотелиального электрического сопротивления (TEER) клеточного барьера. Значение TEER 200 Ом⋅см2 (или выше) указывает на то, что барьерная система пригодна для анализа поглощения. Электроды стерилизовали, погружая их в этанол (70%) на 30 мин в УФ-свете. После этого электрод уравновешивали в течение 30 мин в среде DMEM, предварительно нагретой до комнатной температуры. Измерения TEER проводились при комнатной температуре, получая среднее значение 1357±168 Ом⋅см2, что подтверждает целостность клеточного барьера.
Перед анализом проницаемости был приготовлен транспортный буфер (HBSS (1×)-Ca2+/Mg2+) и подогрет до температуры 37°C, чтобы избежать температурного стресса клеток во время эксперимента. Параллельно тестируемые соединения растворяли до конечной концентрации 50 мкМ в транспортном буфере (HBSS (1×)-Ca2+/Mg2+). Содержание 0,5% ДМСО, количество, которое хорошо переносится клетками и не влияет на целостность барьера. Соединения тестировали в концентрации 50 мкМ.
Перед анализом проницаемости DMEM из апикального и базального отделов удаляли отсасыванием. В каждом отсеке оставляли по 10 мкл среды для предотвращения высыхания клеток и нарушения их барьерных свойств. Оба отсека промывали транспортным буфером, базальный (нижний) отсек заполняли 300 мкл, а апикальный (верхний) отсек 75 мкл транспортного буфера.
Содержимое базального отсека полностью удаляли отсасыванием и снова заполняли 250 мкл транспортного буфера. Содержимое апикального отсека также удаляли отсасыванием, оставляя 10 мкл транспортного буфера во избежание высыхания клеток. В каждую лунку добавляли 65 мкл раствора тестируемого соединения.
Для экспериментов по транспортировке (апикально-базально) клетки инкубировали с растворами, содержащими соединения, в течение 2 часов при температуре 37°C. После этого содержимое отсеков извлекали и анализировали с помощью ВЭЖХ (колонка C18 Sunfire 100 мм×4,6 мм, 3,5 мм, 100 Å, Waters; скорость потока 1 мл/мин; градиент 0-100% В за 8 мин. А=0,045% трифторуксусной кислоты в H2O и B 0,036% трифторуксусной кислоты в ацетонитриле, обнаружение при 220 нм). Все эксперименты выполняли в трехкратной повторности.
После извлечения образцов из отсеков для проверки целостности клеточных барьеров во время анализов базальные отсеки снова заполняли 250 мкл транспортного буфера, а апикальные отсеки 65 мкл раствора люциферного желтого (LY) в концентрации 20 мкМ в транспортном буфере. Клеточный барьер инкубировали в течение 1 часа при температуре 37°C с раствором LY. LY используется в качестве маркера целостности барьера для анализа проницаемости. После инкубации содержимое отсеков собирали и анализировали с помощью флуориметра.
Для полностью функционального барьера Caco-2 проницаемость LY должна быть менее 0,7% через 1 час. После анализа проницаемость LY была ниже этого среднего транспортного значения, что указывает на целостность барьера во время оценки соединений.
В анализе проницаемости Caco-2 процент транспорта (T%) для соединений рассчитывали в соответствии со следующим уравнением:
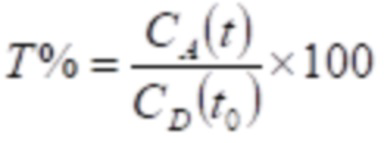
где t обозначает время (час), CA(t) обозначает концентрацию соединения в базальном компартменте (акцептор) в момент времени t, CD(t0) обозначает концентрацию соединения в апикальном компартменте (донор) в момент времени 0.
Как показано выше, проницаемость желудка соединения по примеру 1 намного выше, чем проницаемость сравнительного примера 3. На указанной фигуре столбцы представляют собой среднее значение±стандартное отклонение (SD).
Данные таблицы 4 представлены на фиг. 3.
Воздействие на мозг in vivo
Для каждого соединения использовали двадцать четыре взрослых самца мышей в возрасте 7-8 недель на день эксперимента, которые были случайным образом распределены между группами. Эксперименты проводились на 3 группах животных, как показано в таблице ниже:
(3 животных для каждого времени отбора проб)
(3 животных для каждого времени отбора проб)
После введения соединения брали терминальную кровь в несколько моментов времени после внутрибрюшинного (пример 3) или перорального введения (пример 1).
После транскардиальной перфузии фосфатно-солевым буфером также собирали головной мозг. Все образцы хранились при температуре -80°C до проведения биоанализа.
Образцы плазмы оттаивали при комнатной температуре (КТ) и белки осаждали двукратным объемом ацетонитрила, содержащего 20 нг/мл фенацетина в качестве внутреннего стандарта. Образцы перемешивали, а затем центрифугировали в течение 20 минут при 2200 g. Супернатанты разбавляли 1:1 150 мМ фосфатно-солевым буфером (рН 7,4) и пипеткой переносили на УВЭЖХ 96-луночные планшеты для ожидания анализа.
Образцы мозга взвешивали, оттаивали при комнатной температуре и готовили к анализу путем гомогенизации ткани в бисерном гомогенизаторе с 4-кратным объемом фосфатно-солевого буфера. Гомогенат обрабатывали аналогично образцам плазмы.
Эталонные (калибровочные) образцы готовили так же, как и фактические образцы для исследования, после добавления чистой плазмы и гомогената мозга до и концентрации 0,1, 0,2, 0,5, 1, 2, 5, 10, 20, 50, 100, 200, 500, 1000, 2000, 5000 и 10000 нг/мл соединения по примеру 1 и соед. по пр. 3.
Уровни соединений соед. по пр. 3 и по примеру 1 количественно определяли из плазмы и головного мозга мыши. Образцы анализировали с использованием целевого биоаналитического метода с использованием УВЭЖХ/QE-орбитрап-масс-спектроскопии.
Выстраивали график зависимости концентрации (нг/мл) соединений от времени и рассчитывали площадь под кривой (AUC) в плазме и ткани головного мозга с использованием Graphpad PrismTM 5.0. Отношение плазмы к мозгу рассчитывали следующим образом:
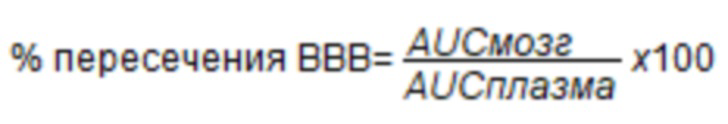
Данные таблицы 5 представлены на фиг. 4.
Как показано выше, при пероральном применении соединения по примеру 1 можно достичь более высокого воздействия на головной мозг, чем воздействие, достигаемое другими соединениями, даже при внутрибрюшинном введении указанных соединений.
Микросомальная стабильность (крыса)
Микросомальная стабильность может быть использована для прогнозирования того, преодолеет ли соединение первый этап метаболизма и достигнет системного воздействия после перорального введения. Также можно прогнозировать период полувыведения соединения после попадания в кровоток.
Стабильность соединений в объединенных микросомах печени крыс (Sprague-Dawley) (номер по каталогу 452501) исследовали в соответствии с инструкциями, предоставленными поставщиком (BD Bioscience). Исходный раствор (500 мкМ) испытуемого соединения готовили в ДМСО. Два мкл соединений инкубировали при конечной концентрации 1 мкМ в следующей смеси:
- 713 мкл H2O
- 200 мкл 0,5 M фосфата калия (pH 7,4)
- 50 мкл регенерирующего системного раствора NADPH A (BD Bioscience, номер по каталогу 451220)
- 10 мкл регенерирующего системного раствора NADPH B (BD Bioscience, номер по каталогу 451200)
Полученную смесь нагревали до температуры 37°C в течение 5 мин. После этого добавляли 25 мкл микросом печени в конечной концентрации 0,5 мг/мл. Смесь инкубировали при температуре 37°C при орбитальном встряхивании (100 об/мин). В выбранные моменты времени аликвоты по 100 мкл экстрагировали и смешивали со 100 мкл ацетонитрила. Полученный осадок встряхивали на вортексе в течение 1 минуты. Образцы выдерживали при температуре 4°C в течение мин, после чего образцы центрифугировали при 20000×g при температуре 4°C в течение 30 мин. Супернатант отфильтровывали и затем анализировали с помощью УВЭЖХ-МС. Все тесты на стабильность выполняли в двух повторностях.
Эти данные предполагают, что соединение по сравнительному примеру 3 и соединение по сравнительному примеру 4 не преодолеют первый этап метаболизма после перорального введения. Следовательно, не достигнет системного кровообращения после перорального введения. Действительно, соединение по сравнительному примеру 3 не показало эффективности после перорального введения, так как обнаруженные уровни лекарственного средства в плазме были очень низкими.
Исследование ФК на мышах
Фармакокинетику соединений после внутривенного болюсного введения исследовали на самцах мышей Swiss Albino. Исследование проводилось в соответствии с рекомендациями Институционального комитета по этике животных (IAEC) и в соответствии с требованиями Комитета по контролю и надзору за экспериментами на животных (CPCSEA), Индия.
Исследование проводилось на 12 мышах на каждое соединение с использованием схемы разреженной выборки (n=3/момент времени) для получения составного профиля. Соединения растворяли в 2% растворе Twee80® в физиологическом растворе (0,9 NaCl) в концентрации 0,5 мг/мл. Мышам вводили однократную дозу 1 мг/кг раствора соединения внутривенно болюсно через латеральную хвостовую вену с использованием шприца BD на 1 мл с иглой 26G в объеме дозы 2 мл/кг.
Образцы крови собирали (n=3 в момент времени) через 0,083, 0,25, 0,5, 1, 2, 4, 6, 8, 24 и 48 ч после введения дозы. В каждый момент времени путем пункции ретроорбитального сплетения собирали 120 мкл крови и переносили в маркированную микроцентрифужную пробирку, содержащую 200 мМ раствор 200 мМ K2EDTA (20 мкл на мл крови). Образцы крови центрифугировали при 5000 g в течение 5 мин при температуре 4±2°C. 20 мкл на мл крови). Образцы крови центрифугировали при 5000 g в течение 5 мин при -60°C до проведения биоанализа.
Образцы плазмы анализировали методом ЖХ-МС/МС с LLOQ 1,06 нг/мл. Фармакокинетические параметры в плазме рассчитывали с использованием инструмента некомпартментного анализа валидированного программного обеспечения Phoenix WinNonlin® (версия 6.3). Площадь под кривой зависимости концентрации от времени (AUClast) рассчитывали по правилу линейных трапеций. Пиковая концентрация в плазме (Cmax) и время достижения пиковой концентрации в плазме (Tmax) были наблюдаемыми значениями. Клиренс (CL) и объем распределения (Vss) были прогнозируемыми значениями.
Соединение по сравнительному примеру 4 демонстрирует очень высокий клиренс после внутривенного (в/в) введения мышам, что указывает на то, что соединение претерпевает высокий метаболизм первого прохождения (в соответствии с данными микросомальной стабильности), предполагая, что соединение не попадет в системный кровоток после перорального введения.
ВЛИЯНИЕ НОВЫХ СОЕДИНЕНИЙ НА ОБУЧЕНИЕ И ПАМЯТЬ В МОДЕЛИ ЖИВОТНЫХ С НАРУШЕНИЕМ КОЗНАНИЯ
Новые соединения оценивали на предмет их эффективности в качестве усилителей когнитивных функций в фармакологической модели когнитивных нарушений. Эффекты новых соединений оценивали на грызунах, не подвергавшихся лечению, и на грызунах, получавших МК-801 (мыши или крысы). MK-801 является неконкурентным антагонистом рецептора N-метил-D-аспартата (NMDA), который ухудшает показатели животных в различных парадигмах обучения и памяти (Castellano C et al., Curr. Drug Targets 2001; 2:273-83.; Riedel G et al., Behav. Brain Res. 2003; 140:1-47). MK-801 также оказывает различные эффекты на поведение грызунов, в том числе дефицит сенсорной обработки, гиперподвижность, стереотипию и атаксию. Поведенческий фенотип, индуцированный лечением MK-801, широко использовался в качестве модели когнитивных нарушений у животных (Bardgett ME et al., Brain Res. Bull. 2003; 60:131-42; Van der Staay FJ et al., Behav. Brain Res. 2011; 220:215-29; Mutlu O et al., Pharmacol. Biochem. Behav. 2011; 99:557-65).
Чтобы определить, действуют ли исследуемые соединения в качестве усилителей когнитивных функций, их способность восстанавливать нормальное когнитивное поведение проверяли с помощью широко используемых тестов, таких как тест распознавания новых объектов (Dere E et al., Neurosci. Biobehav. Rev. 2007; 31:673-704; Boess FG et al., J. Pharmacol. Exp. Ther. 2007; 321:716-25); задача пассивного или тормозящего избегания (Sarter M et al., Psychopharmacology (Berl) 1992; 107:144-59); водный лабиринт Морриса (DʼHooge R et al., Brain Res. Rev. 2001; 36:60-90); и задача с чередованием Т-образного лабиринта (Boess FG et al., Neuropharmacology 2004; 47:1081-92; Spowart-Manning L et al., Behav. Brain Res. 2004; 151:37-46).
В качестве показательного примера для оценки новых ингибиторов POP описывается протокол, которому следовали для каждого из поведенческих тестов, а также результаты, полученные в тесте распознавания объектов и тесте пассивного избегания.
Задача распознавания новых объектов
Задача распознавания новых объектов (NOR) основана на естественном предпочтении грызунов исследовать новые объекты (Ennaceur A et al., Behav. Brain Res. 1988; 31:47-59). Это подходящий тест без вознаграждения для изучения визуального обучения и дефицита памяти. Вкратце, процедура задания NOR состояла из трех испытаний: привыкание, обучение и удержание. Каждое животное приучали к круглой арене диаметром 40 см в течение 10 мин в отсутствие объектов (сеанс привыкания). На следующий день животное помещали на 10 мин на круглую арену для тренировочного испытания, а два одинаковых объекта помещали в симметричное положение. Эту стадию проводили в течение двух дней подряд. На третий день один из объектов заменяли другим объектом. Объект, не использовавшийся в обучающем испытании, был использован в качестве нового объекта в испытании удержания. Затем животным давали возможность свободно исследовать в течение 10 минут, и время, затраченное на изучение каждого объекта, регистрировали. Ожидается, что животное потратит больше времени на изучение нового объекта, что является признаком неповрежденной памяти узнавания. Индекс различения рассчитывали следующим образом: время, затраченное на изучение нового объекта, минус время, затраченное на изучение старого объекта, деленное на общее время, затраченное на изучение обоих объектов, и умноженное на 100. Считалось, что более высокий индекс различения отражает большее сохранение памяти.
Соединения, вводимые перорально
Эксперимент проводили на мышах-самцах линии C57/Bl6 в возрасте 8-9 недель (n=7-9 животных в группе). Соединения растворяли в смеси 5% Tween80® в физиологическом растворе (0,9% NaCl) до получения концентрации раствора 0,5 мг/мл.
Тест был разделен на три части: привыкание, обучение и тест. В первый день животных приучали к манежу (круглому, диаметром 40 см) в течение 10 минут. В следующие два дня животным давали возможность свободно исследовать два одинаковых объекта, размещенных на арене, в течение 10 минут. В тестовых сессиях (четвертый день) один из объектов был заменен новым. Перед этим сеансом за тридцать пять минут до теста животным перорально вводили исследуемые соединения (5% Tween80® в физиологическом растворе, 1 мл на 100 г массы тела). Чтобы вызвать когнитивный дефицит, подкожно вводили дизоцилпин (МК-801) в дозе 0,2 мг/кг в физиологическом растворе за 20 мин до теста в группе отрицательного контроля или в группах тестирования на лекарственное средство. Исходной контрольной группе вводили физиологический раствор.
После введения лекарственного средства каждой мыши давали возможность свободно исследовать объекты в течение 10 минут. В качестве параметра для оценки определяли индекс дискриминации (DI):

Эксперимент записывали с помощью веб-камеры, размещенной на высоте 1,5 м над ареной. Пройденное мышами расстояние как процент времени, проведенного в областях круглой арены, анализировали с использованием программного обеспечения Panlab SMART 2.0.
Соединения, вводимые внутрибрюшинно
Эксперимент проводили на мышах-самцах линии C57/Bl6 в возрасте 8-9 недель (n=7-9 животных в группе). Соединения растворяли в смеси 5% Tween80® в физиологическом растворе (0,9% NaCl) до получения концентрации раствора 0,5 мг/мл.
Тест был разделен на три части: привыкание, обучение и тест. В первый день животных приучали к манежу (круглому, диаметром 40 см) в течение 10 минут. В следующие два дня животным давали возможность свободно исследовать два одинаковых объекта, размещенных на арене, в течение 10 минут. В тестовых сессиях (четвертый день) один из объектов был заменен новым. Перед этим сеансом, за тридцать пять минут до теста, животным внутрибрюшинно вводили исследуемые соединения (5% Tween80® в физиологическом растворе, 5 мг/кг) или (5% Tween80® в физиологическом растворе, 1 мл на 100 г массы тела). Чтобы вызвать когнитивный дефицит, дизоцилпин (МК-801) вводили подкожно в дозе 0,2 мг/кг в физиологическом растворе за 20 мин до теста в группе отрицательного контроля или в группах тестирования на наркотики. Исходной контрольной группе вводили физиологический раствор.
После введения препарата каждой мыши давали возможность свободно исследовать объекты в течение 10 минут. В качестве параметра для оценки определяли индекс дискриминации (DI):
Эксперимент записывали с помощью веб-камеры, размещенной на высоте 1,5 м над ареной. Пройденное мышами расстояние как процент времени, проведенного в областях круглой арены, анализировали с использованием программного обеспечения Panlab SMART 2.0.
Результаты, полученные при введении соединения по примеру 1 и соединения по сравнительному примеру 3, представлены в таблице ниже.
Данные таблицы 8 представлены на фиг. 5a (внутрибрюшинное введение) и фиг. 5b. (пероральное введение). На указанных фигурах столбцы представляют среднее значение±стандартная ошибка для n животных. *P <0,05, **P <0,01, ***P <0,001 по сравнению с базовыми (базовый MK 801) условиями когнитивного дефицита (односторонний тест ANOVA с последующим апостериорным тестом Даннета).
Как показано выше, соединение по примеру 1 превосходит соединение по примеру 3 при пероральном введении, что делает его особенно подходящим для перорального введения.
Профиль селективности (связывание с 5-HT1A)
Эксперимент проводился с использованием панели SafetyScreen44® от Cerep/Eurofins. Соединения тестировали в концентрации 100 мкМ (в 1% ДСМО).
Связывание соединения рассчитывали как процент ингибирования связывания радиоактивно меченного лиганда, специфичного для каждой мишени.
Результаты выражены в виде процента ингибирования контрольного специфического связывания.
 ,
,
полученные в присутствии испытуемых соединений.
Считается, что результаты, показывающие ингибирование выше 50%, отражают значительные эффекты испытуемых соединений.
Данные таблицы 9 представлены на фиг. 6.
| название | год | авторы | номер документа |
|---|---|---|---|
| 1-((3S,4R)-4-(3-ФТОРФЕНИЛ)-1-(2-МЕТОКСИЭТИЛ)ПИРРОЛИДИН-3-ИЛ)-3-(4-МЕТИЛ-3-(2-МЕТИЛПИРИМИДИН-5-ИЛ)-1-ФЕНИЛ-1Н-ПИРАЗОЛ-5-ИЛ)МОЧЕВИНА В КАЧЕСТВЕ ИНГИБИТОРА TrkA КИНАЗЫ | 2015 |
|
RU2719489C2 |
| ПРОИЗВОДНЫЕ ПИРРОЛИДИНА ДЛЯ ПРИМЕНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ КАТЕПСИНА | 2011 |
|
RU2548684C2 |
| ИНГИБИТОРЫ КАТЕХОЛ-О-МЕТИЛТРАНСФЕРАЗЫ И ИХ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ ПСИХОТИЧЕСКИХ РАССТРОЙСТВ | 2011 |
|
RU2563634C2 |
| 4-[2-(2-ФТОРФЕНОКСИМЕТИЛ)ФЕНИЛ]ПИПЕРИДИНОВЫЕ СОЕДИНЕНИЯ | 2009 |
|
RU2515612C2 |
| БЕНЗИЛОКСИ-ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ МОНОАМИНОКСИДАЗЫ B | 2005 |
|
RU2378270C2 |
| КРИСТАЛЛИЧЕСКАЯ ФОРМА 4-[2-(2-ФТОРФЕНОКСИМЕТИЛ)ФЕНИЛ]ПИПЕРИДИНОВОГО СОЕДИНЕНИЯ | 2009 |
|
RU2503662C2 |
| СИНТЕЗ И НОВЫЕ СОЛЕВЫЕ ФОРМЫ (R)-5-((E)-2-(ПИРРОЛИДИН-3-ИЛВИНИЛ)ПИРИМИДИНА | 2009 |
|
RU2533819C2 |
| АЛКИЛ [2-(2-{5-[4-(4-{2-[1-(2-МЕТОКСИКАРБОНИЛАМИНО-АЦЕТИЛ)-ПИРРОЛИДИН-2-ИЛ]-3Н-ИМИДАЗОЛ-4-ИЛ}-ФЕНИЛ)-БУТА-1,3-ДИИНИЛ]-1Н-ИМИДАЗОЛ-2-ИЛ}-ПИРРОЛИДИН-1-ИЛ)-2-ОКСО-ЭТИЛ]-КАРБАМАТ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ЛЕКАРСТВЕННОЕ СРЕДСТВО, СПОСОБ ЛЕЧЕНИЯ ВИРУСНЫХ ЗАБОЛЕВАНИЙ | 2013 |
|
RU2518369C1 |
| ПРОИЗВОДНЫЕ ПИРРОЛИДИНА | 2009 |
|
RU2494094C2 |
| Соединения 1-циано-пирролидинов в качестве ингибиторов USP30 | 2016 |
|
RU2717238C2 |
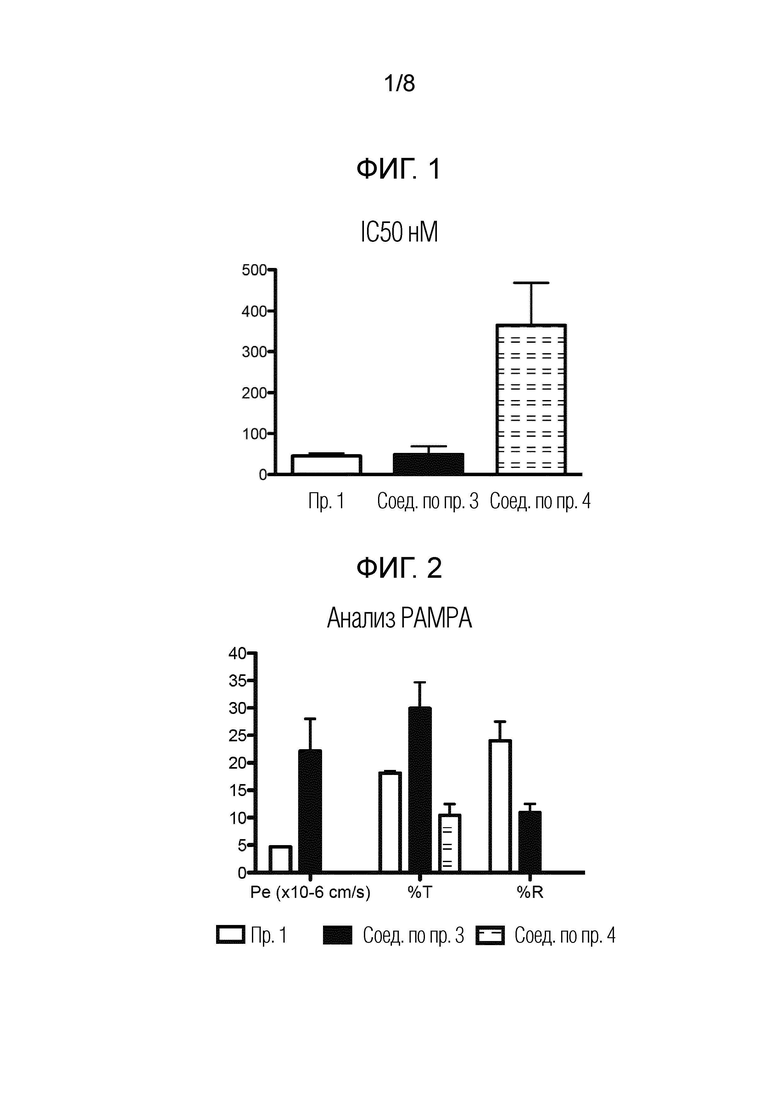
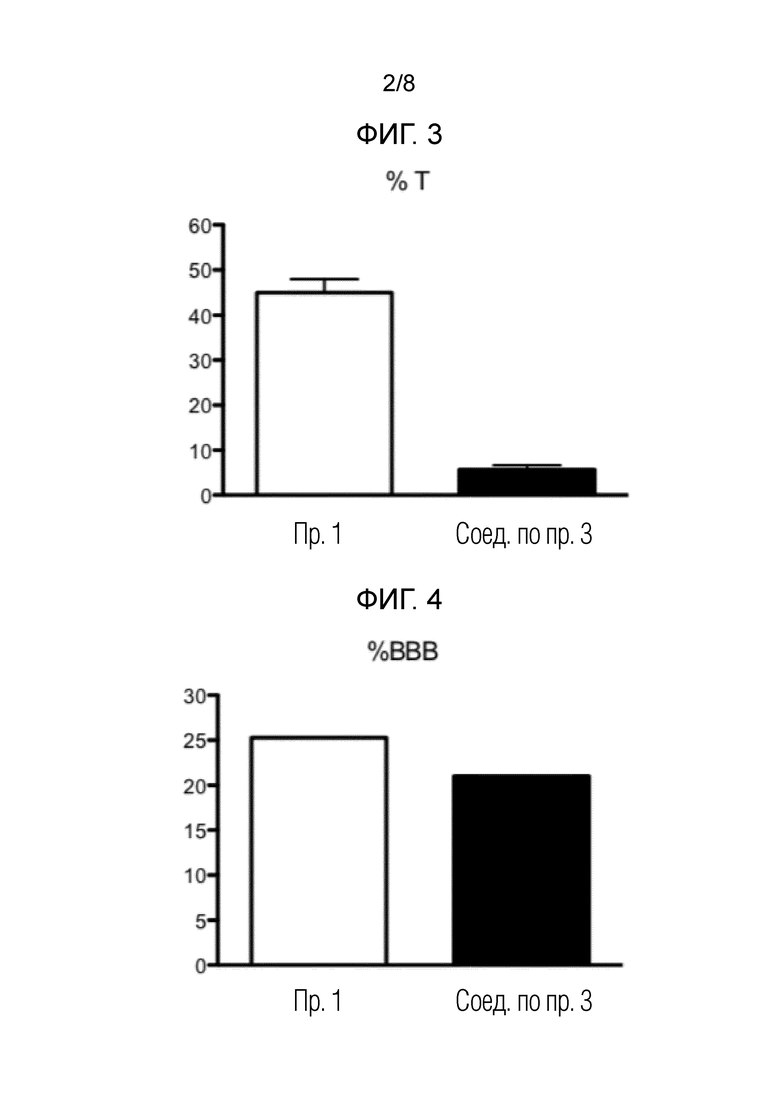
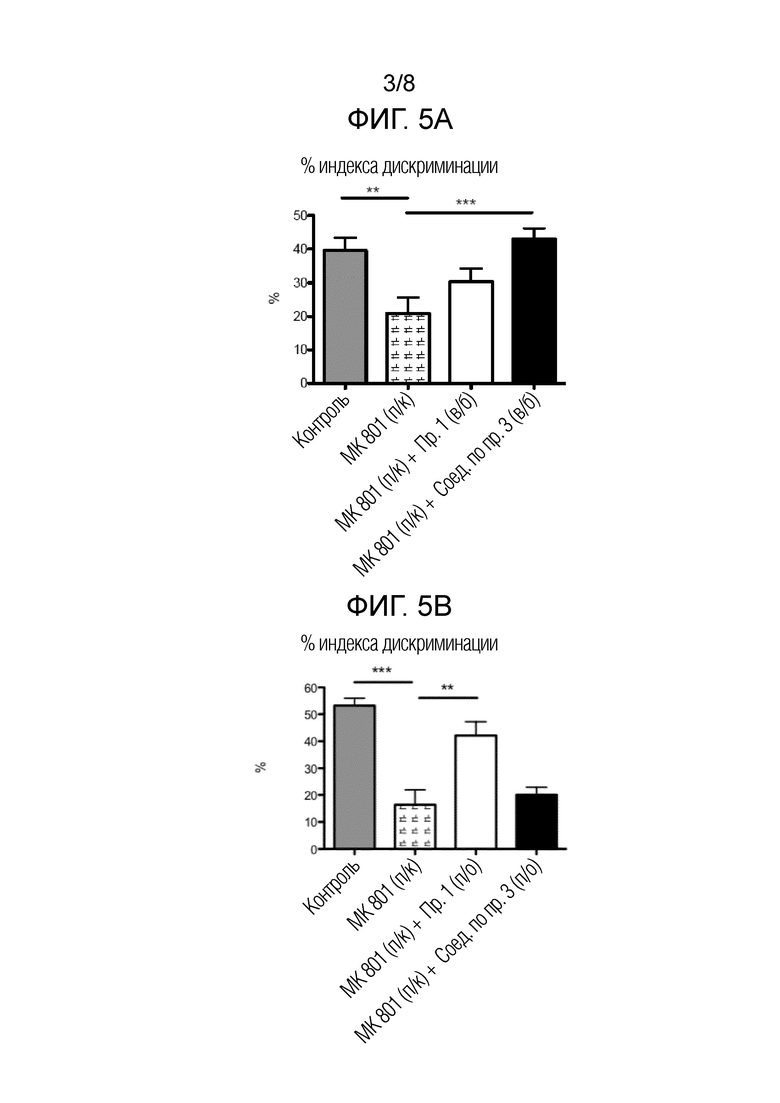
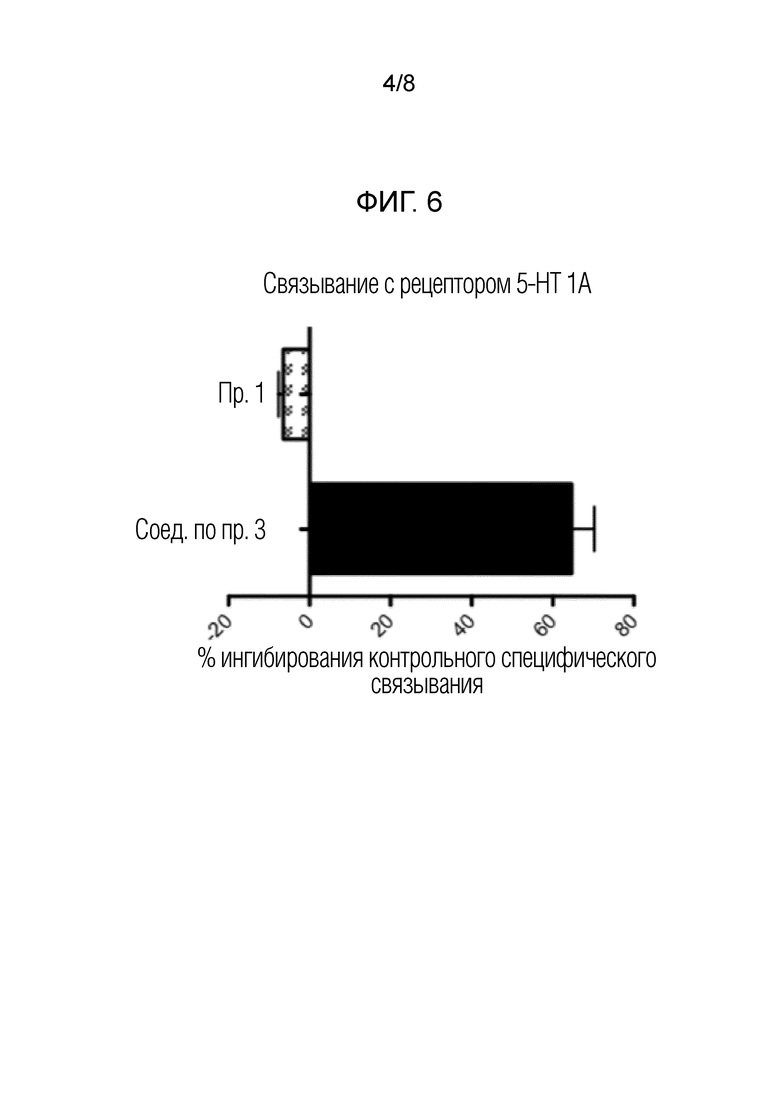
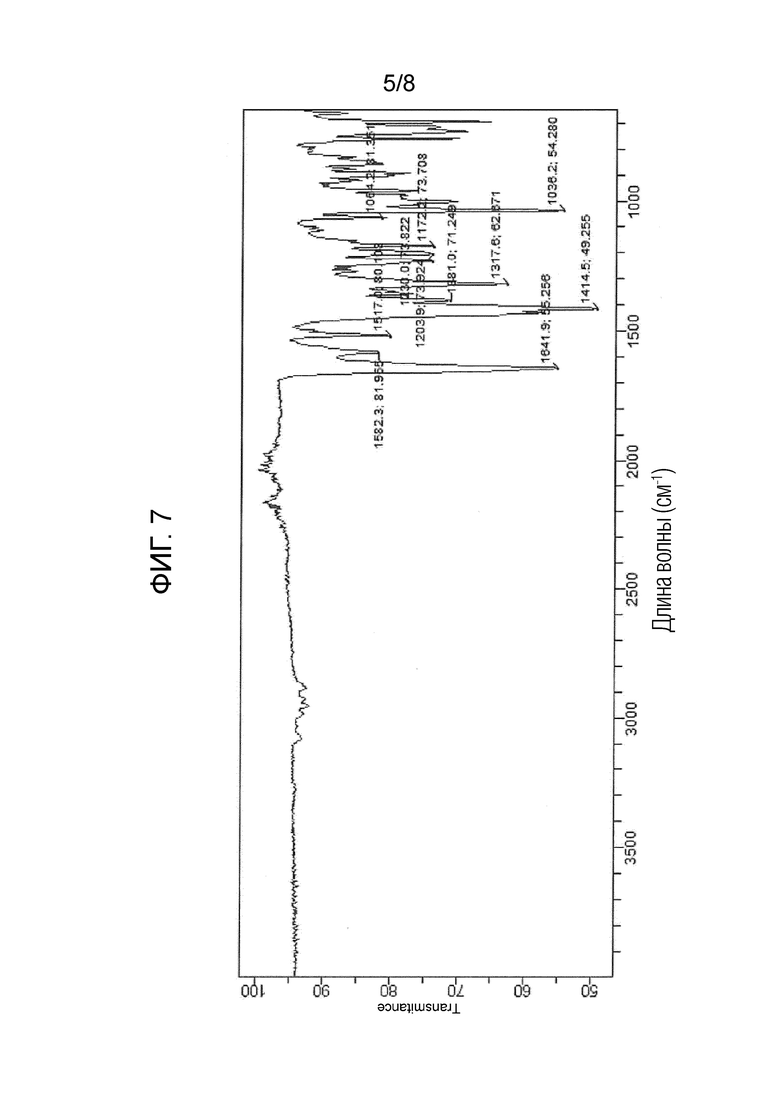
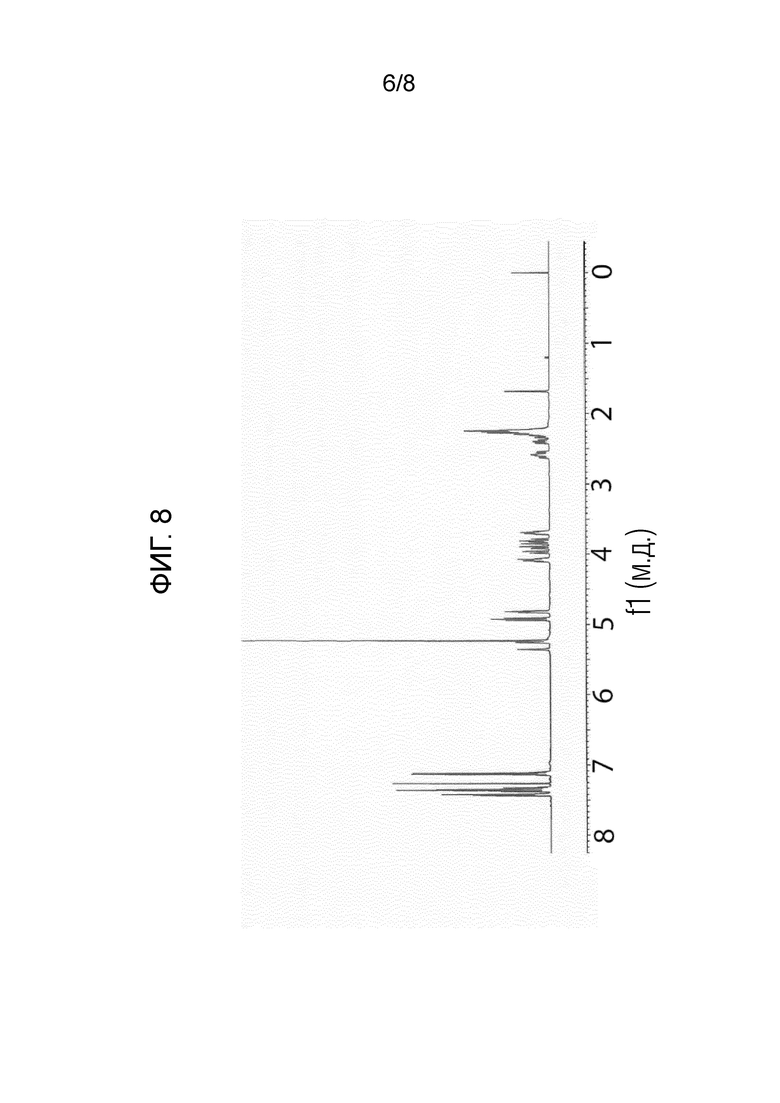
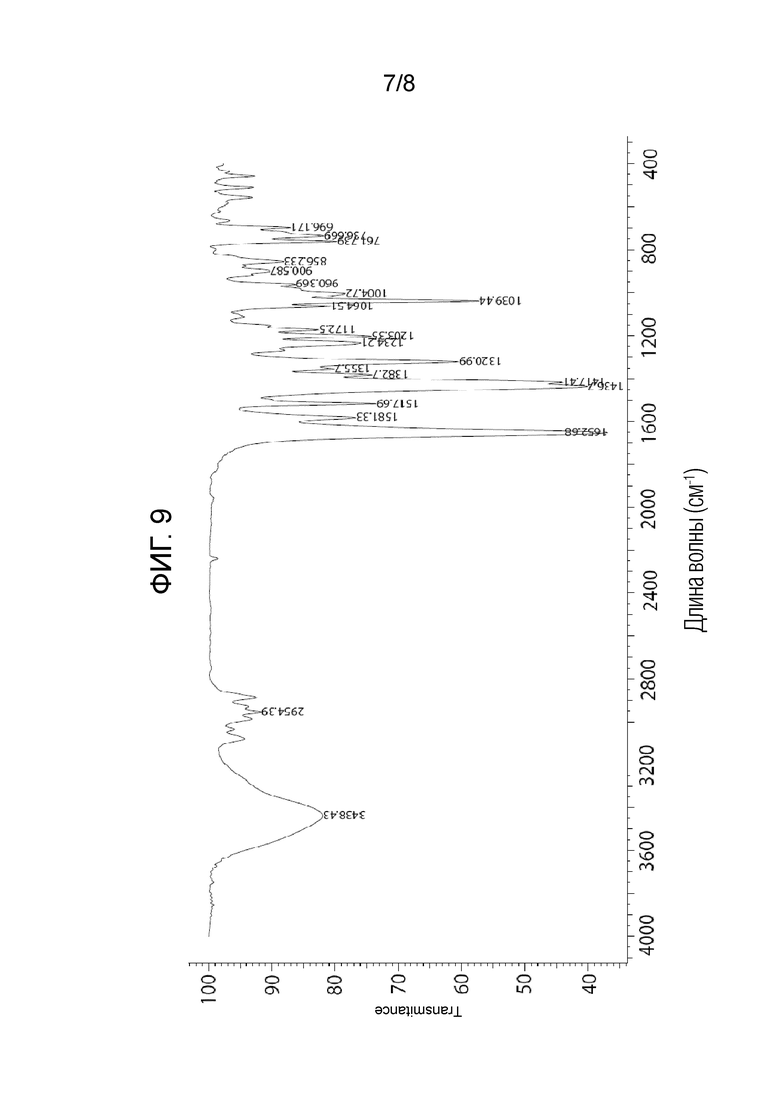
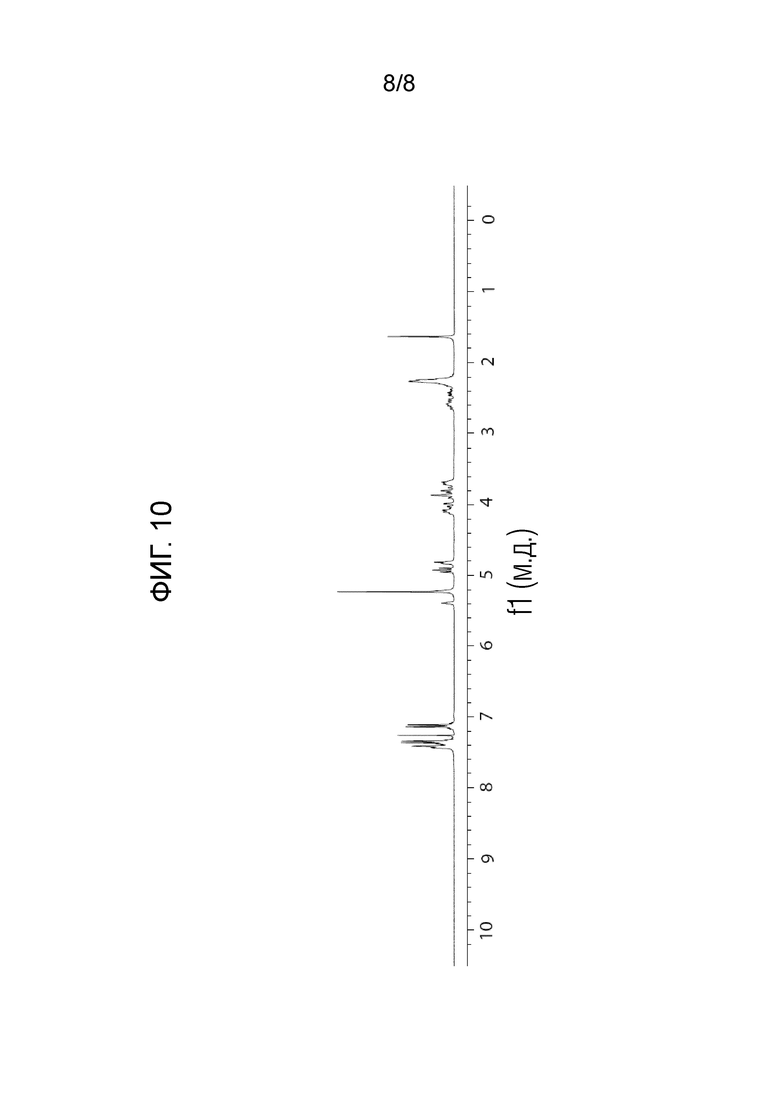
Группа изобретений относится к области фармацевтической химии и включает соединение формулы (I) и его стереоизомеры, его способ получения, фармацевтическую композицию и применения на его основе. Технический результат: соединение формулы (I) и его стереоизомеры, обладающее ингибирующей активностью в отношении пролилолигопептидазы (POP) и используемые для лечения или профилактики когнитивного расстройства. 6 н. и 4 з.п. ф-лы, 11 ил., 9 табл., 4 пр.
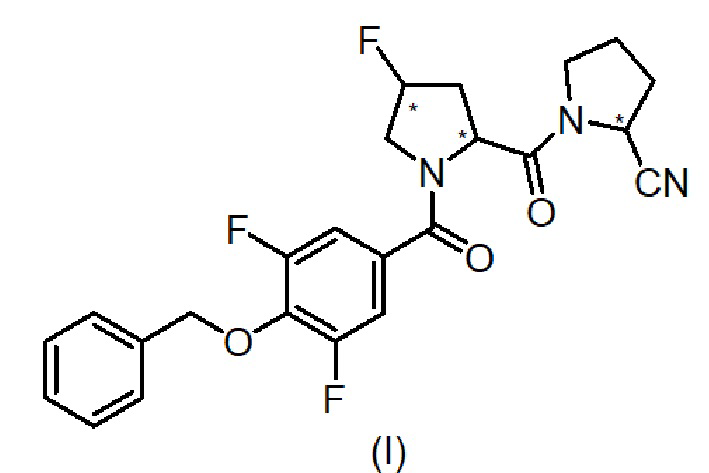
1. Соединение формулы (I)
(где звездочки обозначают хиральные центры) или его стереоизомеры.
2. Соединение по п.1, имеющее формулу (Ia):
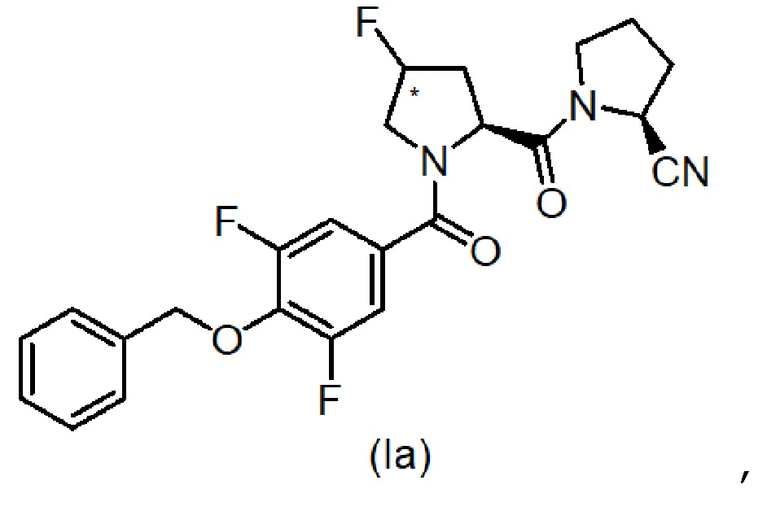
или его стереоизомеры.
3. Соединение по п.1, где указанное соединение выбрано из следующих:
(S)-1-((2S,4R)-1-(4-(бензилокси)-3,5-дифторбензоил)-4-фторпирролидин-2-карбонил)пирролидин-2-карбонитрил,
(R)-1-((2S,4R)-1-(4-(бензилокси)-3,5-дифторбензоил)-4-фторпирролидин-2-карбонил)пирролидин-2-карбонитрил,
(S)-1-((2R,4R)-1-(4-(бензилокси)-3,5-дифторбензоил)-4-фторпирролидин-2-карбонил)пирролидин-2-карбонитрил,
(R)-1-((2R,4R)-1-(4-(бензилокси)-3,5-дифторбензоил)-4-фторпирролидин-2-карбонил)пирролидин-2-карбонитрил,
(S)-1-((2S,4S)-1-(4-(бензилокси)-3,5-дифторбензоил)-4-фторпирролидин-2-карбонил)пирролидин-2-карбонитрил,
(R)-1-((2S,4S)-1-(4-(бензилокси)-3,5-дифторбензоил)-4-фторпирролидин-2-карбонил)пирролидин-2-карбонитрил,
(S)-1-((2R,4S)-1-(4-(бензилокси)-3,5-дифторбензоил)-4-фторпирролидин-2-карбонил)пирролидин-2-карбонитрил,
(R)-1-((2R,4S)-1-(4-(бензилокси)-3,5-дифторбензоил)-4-фторпирролидин-2-карбонил)пирролидин-2-карбонитрил.
4. Способ получения соединения формулы (I), определенного в любом из пп.1-3, который включает
a) взаимодействие соединения формулы (IX):
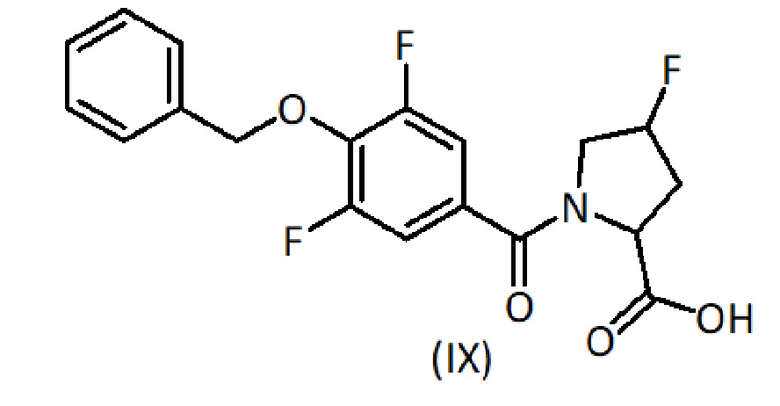
с соединением формулы (XI):
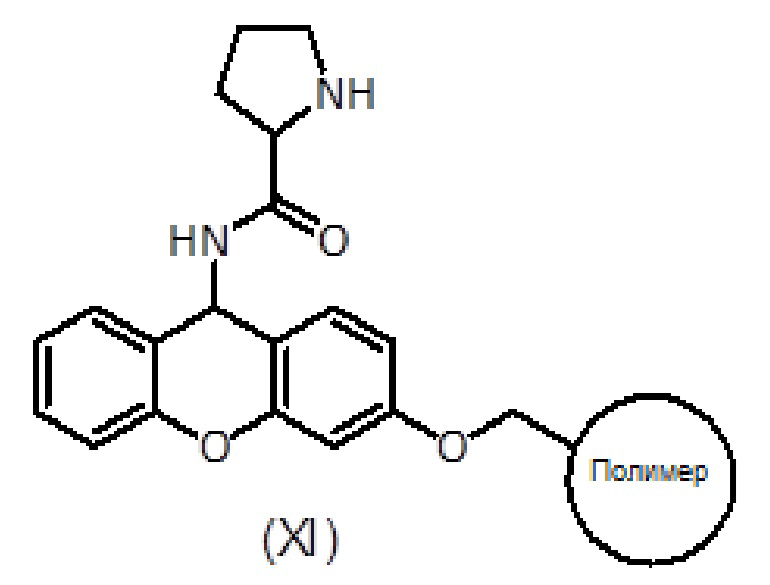
с получением соединения формулы (XII):
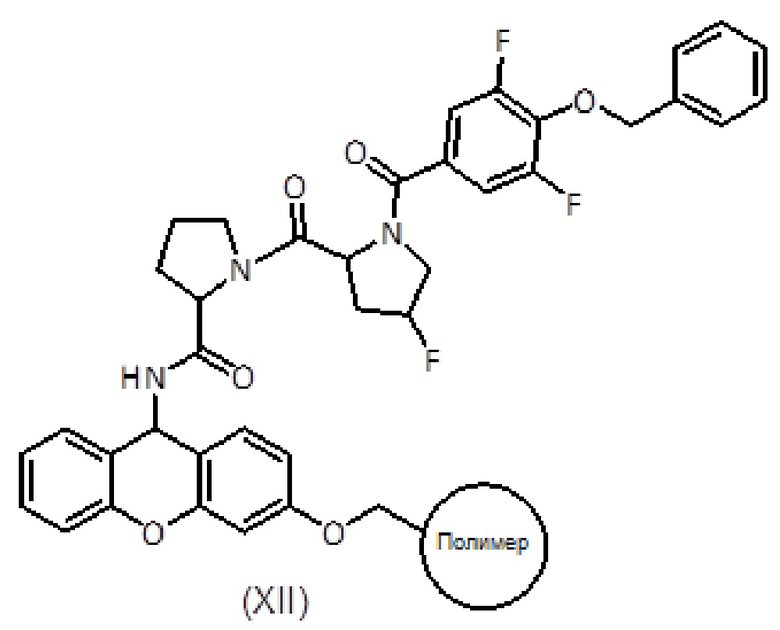
b) гидролиз соединения формулы (XII) с получением соединения формулы (XIV):
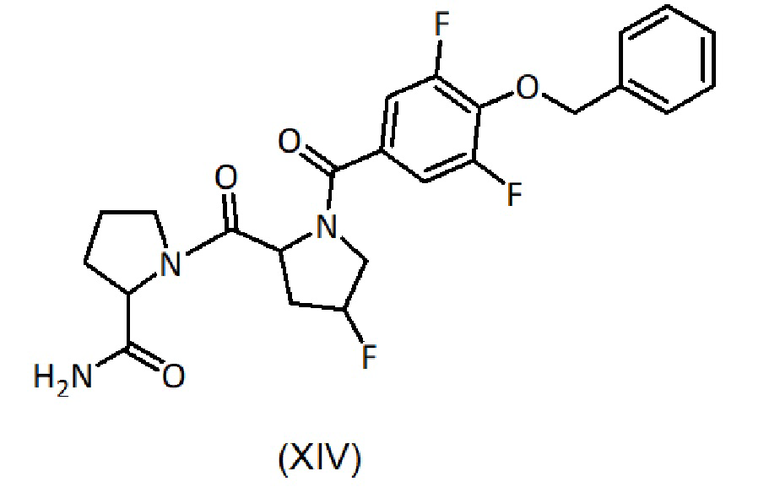
и
c) воздействие на соединение формулы (XIV) условий, способных преобразовать карбоксамидную группу в нитрильную группу с получением соединения формулы (I):
где стадии b) и c) можно проводить отдельно или в одном реакторе.
5. Способ получения соединения формулы (I), определенного в любом из пп.1-3, или его стереоизомеров, который включает взаимодействие соединения формулы (IX):
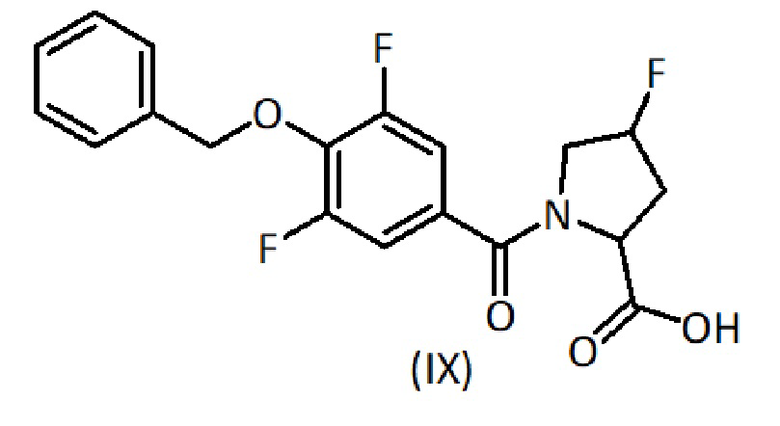
с соединением формулы (IV):
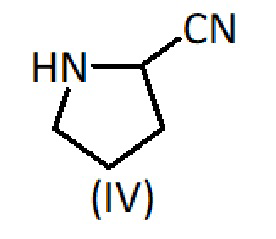
6. Фармацевтическая композиция для лечения или профилактики когнитивного расстройства, содержащая терапевтически эффективное количество соединения формулы (I), определенное в любом из пп.1-3, или его стереоизомера и фармацевтически приемлемый носитель, адъювант или наполнитель.
7. Применение соединения формулы (I), определенного в любом из пп.1-3, для лечения или профилактики когнитивного расстройства.
8. Применение соединения, как определено в п.7, где когнитивное расстройство представляет собой когнитивное расстройство, связанное с заболеванием, выбранным из группы, включающей шизофрению, большое депрессивное расстройство, биполярное аффективное расстройство, расстройство поведения во время быстрого сна, болезнь Альцгеймера, лобно-височную деменцию, болезнь Паркинсона, деменцию с тельцами Леви, множественную системную атрофию, прогрессирующий надъядерный паралич, кортикобазальную дегенерацию или боковой амиотрофический склероз.
9. Применение соединения формулы (I), определенного в любом из пп.1-3, для изготовления лекарственного препарата для лечения или профилактики когнитивного расстройства.
10. Применение, как определено в п.9, где когнитивное расстройство представляет собой когнитивное расстройство, связанное с заболеванием, выбранным из группы, включающей шизофрению, большое депрессивное расстройство, биполярное аффективное расстройство, расстройство поведения во время быстрого сна, болезнь Альцгеймера, лобно-височную деменцию, болезнь Паркинсона, деменцию с тельцами Леви, множественную системную атрофию, прогрессирующий надъядерный паралич, кортикобазальную дегенерацию или боковой амиотрофический склероз.
| WO 2014072498 A1, 15.05.2014 | |||
| RU 2018116882 A, 18.11.2019 | |||
| БД PubChem, PubChem CID: 110198463, create date 18.01.2016. |

