Это изобретение относится к новому способу получения промежуточного химического соединения, пригодного для получения пропионата флутиказона.
Пропионат флутиказона представляет собой кортикостероид семейства андростана, который обладает мощной противовоспалительной активностью и широко распространен в качестве полезного терапевтического агента для лечения воспалительных и аллергических состояний, таких как ринит и астма. Химическая структура пропионата флутиказона представлена ниже:

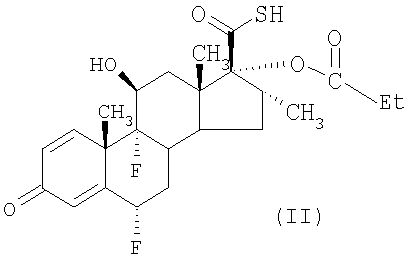
В одном из способов получения пропионата флутиказона соединение формулы (II)

подвергают взаимодействию с соединением формулы LCH2F, где L представляет собой уходящую группу: мезил, тозил или галоген, например Cl, Br или I. Предпочтительно L представляет собой галоген, в особенности Br.
Согласно способам, известным из уровня техники (например, описанным в G.Н.Phillips et al (1994), J. Med. Chem. 37, 3717-3729 и патенте США 4335121 (Glaxo Group Limited)), соединения формулы (II) могут быть получены путем взаимодействия соединения формулы (III)

с активированным производным пропионовой кислоты, например пропионилхлоридом. Активированное производное пропионовой кислоты обычно будут использовать по меньшей мере в двукратном мольном количестве относительно соединения формулы (III), поскольку один моль этого реагента будет взаимодействовать с тиокислотной группировкой, и появится необходимость его удаления, например, путем взаимодействия с амином, таким как диэтиламин.
Однако этот способ получения соединения формулы (II) обладает недостатками, заключающимися в том, что получаемое в результате соединение формулы (II) трудно очистить от загрязняющего его побочного продукта: N,N-диэтилпропанамида. Поэтому авторы изобретения изобрели улучшенный способ осуществления превращения с целью получения соединения формулы (II).
Другие способы получения пропионата флутиказона и родственных соединений описаны в израильской патентной заявке 109656 (Chemagis), WO 01/62722 (Abbott) и Kertesz and Marx (1986), J. Org. Chem., 51, 2315-2328.
Таким образом, согласно данному изобретению предложен способ получения соединения формулы (II)
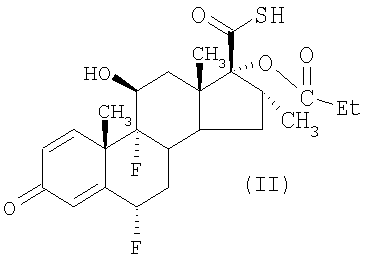
или его соли,
при котором:
(а) соединение формулы (III)
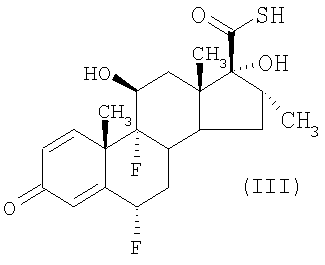
подвергают взаимодействию с активированным производным пропионовой кислоты в количестве по меньшей мере 1,3 моль, подходяще по меньшей мере 2 моль, активированного производного на моль соединения формулы (III) и
(б) удаляют пропионильную группировку, связанную с атомом серы, у любого полученного таким образом соединения формулы (IIA)

путем взаимодействия продукта со стадии (а) с органическим первичным или вторичным, аминным основанием, способным к образованию водорастворимого пропанамида.
На стадии (а) примеры активированных производных пропионовой кислоты включают в себя активированные сложные эсриры или, предпочтительно, галогенангидрид пропионовой кислоты, такой как пропионилхлорид. Эту реакцию традиционно выполняют в присутствии химически инертного органического основания, такого как триC1-4алкиламин, например, три-н-пропиламин, триэтиламин или трибутиламин, особенно триэтиламин, но наиболее предпочтительно три-н-пропиламин. Растворители для этого процесса включают в себя растворители, по существу не смешивающиеся с водой, такие как этилацетат или метилацетат, или растворители, смешивающиеся с водой, такие как ацетон, N,N-диметилформамид или N,N-диметилацетамид, особенно ацетон. По существу несмешивающиеся растворители обеспечивают образование двух фаз при их смешивании и имеют низкий уровень растворимости одного в другом, например, растворимость одного растворителя в другом растворителе составляет менее 30% (мас./мас.), например 10% (мас./мас.), особенно 5% (мас./мас.).
На стадии (б) примеры органического первичного или вторичного аминного основания, способного к образованию водорастворимого пропанамида, включают в себя амины, которые являются более полярными, чем диэтиламин, например аминоспирт, например диэтаноламин, или диамин, например N-метилпиперазин. Предпочтительно используют N-метилпиперазин. Может быть подходящим растворение амина в небольшом количестве органического растворителя, такого как метанол.
Предпочтительно стадии (а) и (б) осуществляют при пониженной температуре, например 0+5°С.
В другом аспекте изобретения предложены способы эффективной очистки соединения формулы (II).
Так, согласно первому такому способу (в1), когда продукт со стадии (б) растворен в по существу несмешивающемся органическом растворителе (таком как метилацетат или этилацетат, или высшем алканоне, таком как пентан-3-он), очистку соединения формулы (II) осуществляют путем вымывания амидного побочного продукта со стадии (б) с использованием водной промывки. Например, к реакционной смеси можно добавить воду, перемешать, оставить фазы разделиться и слить нижний водный слой.
Предпочтительно по существу несмешивающимся растворителем является пентан-3-он.
Остающийся органический слой можно концентрировать перегонкой, возможно при пониженном давлении, а затем можно добавить антирастворитель (например, гексан) для кристаллизации растворенного продукта.
Согласно второму такому способу (в2), когда продукт со стадии (б) растворен в растворителе, смешивающемся с водой (например, ацетоне), очистку соединения формулы (II) осуществляют путем обработки соединения со стадии (б) водной средой для осаждения чистого соединения формулы (II). Соответственно, амидный побочный продукт со стадии (б) будет по существу оставаться в водной фазе.
Водной средой может быть, например, разбавленная водная кислота, такая как разбавленная соляная кислота или уксусная кислота.
Дополнительные общие условия, относящиеся к превращению соединения формулы (III) в соединение формулы (II) и его соли и выделению конечного продукта, будут хорошо известны специалистам в данной области техники.
Однако авторы изобретения обнаружили, что в соответствии с предпочтительным рядом условий, соединение формулы (II) преимущественно можно выделять, следуя способу (в1), в форме твердой кристаллической соли, а не свободного соединения формулы (II). Предпочтительная соль образуется с таким основанием, как диизопропилэтиламин, триэтиламин, 2,6-диметилпиридин, N-этилпиперидин, или с калием. Такие солевые формы соединения формулы (II) более стабильны, их легче фильтровывать и сушить и их можно выделить с более высокой чистотой, чем свободное соединение формулы (II). Наиболее предпочтительной солью является соль, образованная с триэтиламином. Калиевая соль, также представляет интерес.
Так, согласно предпочтительному способу, следуя способу (в1), осуществляют обработку органической фазы, содержащей соединение формулы (II), основанием, чтобы соединение формулы (II) выпало в осадок в форме твердой кристаллической соли.
Примеры оснований включают в себя триэтиламин, 2,6-диметилпиридин, N-этилпиперидин или основную калиевую соль, такую как гидрокарбонат калия.
В качестве еще одного аспекта данного изобретения заявлено соединение формулы (II), выделенное в форме твердой кристаллической соли.
В другом аспекте изобретения предложен способ получения пропионата флутиказона, при котором получают соединение формулы (II) или его соль, как описано выше, и превращают соединение формулы (II) или его соль в пропионат флутиказона путем обработки соединением формулы LCH2F, где L представляет собой уходящую группу.
Согласно изобретению соединение формулы (II) преимущественно может быть выделено с более высокой эффективностью, чем способами, известными из уровня техники. Например, способ получения соединения формулы (II), описанный G.H.Phillips et at (J. Med. Chem. 37, 3717-3729 (1994)) включает в себя выделение продукта из системы ацетон/вода. Полученный таким образом продукт чрезвычайно сложно фильтровать. В противоположность этому, соединение формулы (II), когда оно получено в соответствии с настоящим изобретением, фильтровать гораздо легче. Более того, способ по настоящему изобретению также может обеспечить улучшения по чистоте.
Дополнительным преимуществом является получение и применение соединения формулы (III) в виде соли с имидазолом. Известно получение соединения формулы (III) из соединения формулы (IV) (G.H.Phillips et al (1994), J. Med. Chem, 37, 3717-3729). Однако физико-химические свойства соединения формулы (III), полученного таким образом, приводят к образованию продукта с очень низкой скоростью фильтрации. Преимущества, предоставляемые получением соли соединения формулы (III) с имидазолом, включают в себя ее свойства, такие как легкость получения и быстрота фильтрования, легкость обработки и хранения источника соединения формулы (III), которое может быть легко получено из соли путем подкисления, например, соляной кислотой. Кроме того, соединение формулы (III), полученное таким образом, является более чистым. Соль соединения формулы (III) с имидазолом можно получать, выделять и хранить для последующего использования в способе получения соединения формулы (II), как описано в этом документе. Альтернативно, соль соединения формулы (III) с имидазолом можно получать и использовать непосредственно в виде влажного осадка в последующем превращении в соединение формулы (II), избегая таким образом необходимости сушки соли с имидазолом перед дальнейшей реакцией.
Считают, что соль соединения формулы (III) с имидазолом является новой и, следовательно, образует еще один аспект изобретения. Также дополнительно предложен способ получения соли соединения формулы (III) с имидазолом, при котором соединение формулы (IV) подвергают взаимодействию с карбонилдиимидазолом и сероводородом.
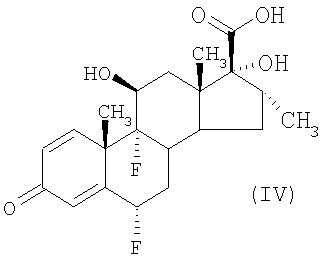
Обычно соединение формулы (IV) и от 1,1 до 2,5 эквивалентов, подходяще 1,8 эквивалента, карбонилдиимидазола перемешивают в подходящем растворителе, например этилацетате, содержащем от 0 до 2 объемов, подходяще 0,5 объема, N,N-диметилформамида, при подходящей температуре, например 18+20°С, в течение подходящего периода времени, например одного часа. Полученную суспензию охлаждают до подходящей температуры, например до температуры от -5°С до 5°С, подходяще от -3°С до 3°С, и за период времени 15-60 минут, подходяще за 20-30 минут, при перемешивании суспензии вводят газообразный сероводород. Реакционную смесь перемешивают в течение дополнительного периода времени приблизительно 30 минут при температуре от -5°С до 5°С, нагревают до приблизительно 10°С за период времени приблизительно 20 минут и перемешивают при 6+12°С в течение 90-120 минут. Затем продукт выделяют фильтрованием при подходящей температуре, подходяще при 5+25°С, предпочтительно при 10+15°С, промывают подходящим растворителем, например этилацетатом, и сушат под вакуумом с получением соли соединения формулы (III) с имидазолом.
Соединение формулы (III) представляет собой одноосновную кислоту и, поэтому, следует ожидать образования соли с имидазолом при стехиометрии имидазольной группировки, приводящей к солеобразованию, к соединению формулы (III) приблизительно 1:1. Однако неожиданно было обнаружено, что стехиометрия имидазольной группировки, приводящей к солеобразованию, к соединению формулы (III) может быть до 4:1 (включительно). Следовательно, во избежание неопределенности, термин "соль с имидазолом" охватывает соли соединения формулы (III) с имидазолом и ассоциированные соединения (association compounds) формулы (III) и имидазола, где стехиометрия имидазольной группировки к соединению формулы (III) до 4:1 (включительно), например от 1:1 до 4:1, подходяще от 1,8:1 до 2,5:1. Пример типичной стехиометрии 2:1. В контексте стехиометрических значений будет понятно, что точные численные значения следует толковать как включающие в себя их незначительные отклонения.
Предпочтительно, соединение формулы (III), используемое в способе, описанном здесь, используют в виде его соли с имидазолом.
Изобретение иллюстрируется со ссылкой на следующие примеры.
ПРИМЕРЫ
Общие методы
1Н-ЯМР спектры регистрировали при 400 МГц и химические сдвиги выражали в ppm (млн-1) относительно тетраметилсилана. Следующие сокращения использовали для описания многообразия сигналов: s (синглет), d (дублет), t (триплет), q (квартет), m (мультиплет), dd (дублет дублетов), ddd (дублет дублета дублетов), dt (дублет триплетов) и b (широкий).
ЖХМС (жидкостная хроматография/масс-спектрометрия) осуществляли на колонке 5 мкм Inertsil ODS-2 25 см × 0,46 см, элюируя 58% {0,1% муравьиной кислоты в 3%-ном метаноле (водном)} (растворитель А) и 42% {0,1% муравьиной кислоты в 3%-ном метаноле (ацетонитрил)} (растворитель Б) с использованием следующего градиента элюирования: 0-40 минут - 42% Б, 40-60 минут - 53% Б, 60-75 минут - 87% Б, 75-85 минут - 42% Б при скорости потока 1 мл/мин. Масс-спектры регистрировали на спектрометре HP LC/MSD с ионизацией (на положительные и отрицательные ионы) методом электрораспыления (ES+ve и ES-ve).
Жидкостную хроматографию (Метод А) осуществляли на колонке 25 см × 0,46 см (внутренний диаметр), заполненной 5 мкм Inertsil ODS-2, элюируя следующими подкисленными подвижными фазами:
Раствор А: подкисленный ацетонитрил:подкисленный метанол:подкисленная вода (42:3:55)
Раствор Б: подкисленный ацетонитрил:подкисленный метанол:подкисленная вода (53:3:44)
Раствор В: подкисленный ацетонитрил:подкисленный метанол:подкисленная вода (87:3:10)
{где подкисленный ацетонитрил включает в себя 0,05% (об./об.) фосфорной кислоты в ацетонитриле (0,5 мл в 1000 мл), подкисленный метанол включает в себя 0,05% (об./об.) фосфорной кислоты в метаноле (0,5 мл в 1000 мл), и подкисленная вода включает в себя 0,05% (об./об.) фосфорной кислоты в воде (0,5 мл в 1000 мл)}.
Элюирование проводили с использованием следующего градиента: 0-40 минут - раствор А (100%), 40-60 минут - раствор Б (100%), 60-75 минут - раствор В (100%) и 75-90 минут - раствор А (100%) при скорости потока 1,0 мл/мин и температуре термостата 40°С.
Жидкостную хроматографию (Метод Б) осуществляли на колонке из нержавеющей стали (20 см × 0,46 см (внутренний диаметр), 5 мкм октил), элюируя следующими подкисленными подвижными фазами:
Раствор А: ацетонитрил: 0,05 М водный раствор дигидроортофосфата аммония (35:65, об./об.).
Раствор Б: ацетонитрил: 0,05 М водный раствор дигидроортофосфата аммония (70:30, об./об.).
Элюирование проводили с использованием следующего градиента: 0-15 минут - раствор А (100%), 15-40 минут - раствор Б (100%), 40-45 минут - раствор Б (100%) и 45-60 минут - раствор А (100%) при скорости потока 1,5 мл/мин и температуре термостата 30°С.
Температуры плавления получали с использованием прибора для определения температуры плавления Mettler Toledo FP62.
Картины порошковой дифракции рентгеновских лучей (XRPD) получали с использованием порошкового дифрактометра Phillips X'pert MPD.
Пример 1: 6α, 9α-Дифтор-17α-(1-оксопропокси)-11β-гидрокси-16α-метил-3-оксо-андроста-1,4-диен-17β-карботиокислота (с использованием N,N-диметилформамида в качестве растворителя, смешивающегося с водой)
Раствор 6α, 9α-дифтор-11β, 17α-дигидрокси-16α-метил-3-оксо-андроста-1,4-диен-17β-карботиокислоты (полученной в соответствии с методикой, описанной в GB 2088877 В) (7 г) в N,N-диметилформамиде (45 мл) обрабатывают триэтиламином (10,9 мл) при температуре -5°С ÷ -6°С в течение приблизительно 15 минут. Во время добавления и последующей промывки 2,8 мл N,N-диметилформамида раствор перемешивают при температуре от -5°С до 0°С. Полученную в результате суспензию затем охлаждают до -3°С ÷ -7°С и в течение приблизительно 30 минут обрабатывают пропионилхлоридом (6,2 мл), поддерживая температуру от -5°С до +2°С. В качестве поточной промывки добавляют N,N-диметилформамид (2,8 мл). Раствор перемешивают при температуре от -5°С до +2°С в течение дополнительных двух часов. Полученную в результате суспензию затем охлаждают до -3°С + -7°С и в течение приблизительно 30 минут обрабатывают диэтаноламином (23,8 мл) в метаноле (20 мл), поддерживая температуру от -5°С до +2°С. В качестве поточной промывки добавляют N,N-диметилформамид (2,8 мл) и раствор перемешивают при температуре от -5°С до +2°С в течение дополнительных 30 минут. Приблизительно за 30 минут добавляют охлажденную соляную кислоту (содержащую смесь 20 мл концентрированной HCl и 20 мл воды), поддерживая температуру в интервале от -5°С до +5°С, и смесь, приблизительно за 30 минут, гасят охлажденной разбавленной соляной кислотой (содержащей смесь 50 мл концентрированной соляной кислоты и 300 мл воды), поддерживая температуру в интервале от -5°С до +5°С. В качестве промывки емкости добавляют водный N,N-диметилформамид (содержащий смесь 10 мл N,N-диметилформамида и 20 мл воды) и полученную в результате суспензию выдерживают при температуре от -5°С до +5°С в течение по меньшей мере 10 минут. Продукт отфильтровывают, промывают водой и сушат под вакуумом при приблизительно 45°С в течение 24 часов с получением указанного в заголовке соединения в виде твердого вещества от белого цвета до белого цвета с желтоватым оттенком (6,65 г, 83,7%).
ВЭЖХ (высокоэффективная жидкостная хроматография): время удерживания 27,23 мин, m/z 469.2 (положительный молекулярный ион) и m/z 467.2 (отрицательный молекулярный ион).
ЯМР (ДМСОd6 (диметилсульфоксид)) 7.27 (1Н, d, 10 Гц), 6.34 (1Н, d, 10 Гц), 6.14 (1Н, s), 5.31 (1Н, d), 5.17 (1Н, ddd), 4.27 (1Н, m), 2.40 (2Н, q, 7 Гц), 2.00-2.14 (5Н, m), 1.85 (1Н, m), 1.65 (1Н, m), 1.51 (3Н, s), 1.14 (3Н, s), 1.05 (3Н, t, 7 Гц), 0.88 (3Н, d, 7 Гц).
Пример 2: 6α, 9α-Дифтор-17α-(1-оксопропокси)-11β-гидрокси-16α-метил-3-оксо-андроста-1,4-диен-17β-карботиокислота (с использованием ацетона в качестве растворителя, смешивающегося с водой)
Раствор 6α, 9α-дифтор-11β, 17α-дигидрокси-16α-метил-3-оксо-андроста-1,4-диен-17β-карботиокислоты (полученной в соответствии с методикой, описанной в GB 2088877 B) (7 г) в ацетоне (80,6 мл) обрабатывают триэтиламином (10,9 мл) при температуре -5°С + -6°С в течение приблизительно 15 минут. Во время добавления и последующей промывки 2,8 мл ацетона раствор перемешивают при температуре от -5°С до 0°С. Полученную в результате суспензию затем охлаждают до температуры -3°С + -7°С и в течение приблизительно 30 минут обрабатывают пропионилхлоридом (6,2 мл), поддерживая температуру реакционной смеси от -5°С до +2°С. В качестве поточной промывки добавляют ацетон (2,8 мл) и раствор перемешивают при температуре от -5°С до +2°С в течение дополнительных двух часов. Полученную в результате суспензию затем охлаждают до температуры -3°С + -7°С и в течение приблизительно 30 минут обрабатывают диэтаноламином (23,8 мл) в метаноле (20 мл), поддерживая температуру смеси от -5°С до +2°С. В качестве поточной промывки добавляют ацетон (2,8 мл) и раствор перемешивают при температуре от -5°С до +2°С в течение дополнительных 30 минут. Смесь гасят водой (135 мл), поддерживая температуру от -5°С до +5°С. В качестве поточной промывки добавляют ацетон (5,6 мл) и смесь охлаждают до 0+5°С. За период времени от одного часа до двух часов добавляют концентрированную соляную кислоту (65 мл), поддерживая температуру в интервале от 0°С до 5°С, а затем воду (125 мл), поддерживая температуру менее 5°С. Смесь перемешивают при температуре 0°С+5°С в течение 15 минут, продукт отфильтровывают, промывают водой и сушат под вакуумом при приблизительно АУС в течение 18 часов с получением указанного в заголовке соединения в виде твердого вещества от белого цвета до белого цвета с желтоватым оттенком (7,91 г, 99,5%).
Пример 3: 6α, 9α-Дифтор-17α-(1-оксопропокси)-11β-гидрокси-16α-метил-3-оксо-андроста-1,4-диен-17β-карботиокислота (с использованием N,N-диметилацетамида в качестве растворителя, смешивающегося с водой)
Раствор 6α, 9α-дифтор-11β, 17α-дигидрокси-16α-метил-3-оксо-андроста-1,4-диен-17β-карботиокислоты (полученной в соответствии с методикой, описанной в GB 2088877 В) (7 г) в N,N-диметилацетамиде (40 мл) обрабатывают триэтиламином (10,9 мл) при температуре от -5°С до 0°С в течение приблизительно 15 минут с последующей поточной промывкой 2,8 мл N,N-диметилацетамида. Полученную в результате суспензию затем охлаждают до температуры -3°С - -7°С и в течение приблизительно 30 минут обрабатывают пропионилхлоридом (6,2 мл), поддерживая температуру от -5°С до +2°С. В качестве поточной промывки добавляют N,N-диметилацетамид (2,8 мл) и раствор перемешивают при температуре от -5°С до +2°С в течение дополнительных двух часов. Полученную в результате суспензию затем охлаждают до температуры -3°С + -7°С и в течение приблизительно 30 минут обрабатывают диэтаноламином (23,8 мл) в метаноле (20 мл), поддерживая температуру от -5°С до +2°С. В качестве поточной промывки добавляют N,N-диметилацетамид (2,8 мл) и раствор перемешивают при температуре от -5°С до +2°С в течение дополнительных 30 минут. В течение приблизительно 30 минут добавляют охлажденную соляную кислоту (содержащую смесь 10 мл концентрированной соляной кислоты и 30 мл воды), поддерживая температуру в интервале от -5°С до +5°С. Реакционную смесь, в течение приблизительно 30 минут, гасят разбавленной соляной кислотой (содержащей смесь 55 мл концентрированной соляной кислоты и 300 мл воды), поддерживая температуру от -5°С до +5°С во время переноса. В качестве поточной промывки добавляют водный N,N-диметилацетамид (содержащий смесь 10 мл N,N-диметилформамида и 20 мл воды) и полученную в результате суспензию выдерживают при температуре от -5°С до +5°С в течение по меньшей мере 10 минут. Продукт отфильтровывают, промывают водой и сушат под вакуумом при приблизительно 45°С в течение 24 часов с получением указанного в заголовке соединения в виде твердого вещества от белого цвета до белого цвета с желтоватым оттенком (7,63 г, 96%).
Пример 4: 6α, 9α-Дифтор-17α-(1-оксопропокси)-11β-гидрокси-16α-метил-3-оксо-андроста-1,4-диен-17β-карботиокислота (с использованием этилацетата в качестве растворителя, по существу не смешивающегося с водой)
Раствор 6α, 9α-дифтор-11β, 17α-дигидрокси-16α-метил-3-оксо-андроста-1,4-диен-17β-карботиокислоты (полученной в соответствии с методикой, описанной в GB 2088877B) (7 г) в этилацетате (350 мл) перемешивают при температуре 20-25°С и обрабатывают триэтиламином (10,9 мл) при 0-5°С в течение приблизительно 20 минут, добавляя в качестве поточной промывки дополнительное количество этилацетата (5 мл). Полученную в результате суспензию затем охлаждают до температуры -3°С + -7°С и в течение приблизительно 30 минут обрабатывают пропионилхлоридом (6,2 мл), поддерживая температуру от -5°С до +2°С. В качестве поточной промывки добавляют этилацетат (5 мл) и раствор перемешивают при температуре от -5°С до +2°С в течение дополнительных двух часов. Полученную в результате суспензию затем охлаждают до температуры -3°С + -7°С и в течение приблизительно 30 минут обрабатывают диэтаноламином (23,8 мл) в метаноле (20 мл), поддерживая температуру от -5°С до +2°С. В качестве поточной промывки добавляют этилацетат (5 мл) и раствор перемешивают при температуре от -5°С до +2°С в течение дополнительных 30 минут. В течение приблизительно 10 минут добавляют 25 мл уксусной кислоты, поддерживая температуру в интервале от -5°С до +2°С, и полученную в результате суспензию выдерживают при температуре от -5°С до +5°С в течение по меньшей мере 10 минут. В течение приблизительно 10 минут добавляют 50 мл воды, поддерживая температуру в интервале от -5°С до +2°С, и органическую фазу отделяют и промывают водой (3×50 мл). Водные фазы, возможно, подвергают обратной экстракции этилацетатом (120 мл) при температуре от -5°С до +2°С. Объединенные органические фазы концентрируют до 10 об. вакуумной перегонкой (ниже 30°С) и охлаждают до температуры 0°С + 5°С. Добавляют гексан (70 мл), поддерживая температуру от 0°С до 5°С, и содержимое выдерживают в течение по меньшей мере 30 минут при температуре 0°С+5°С. Продукт отфильтровывают и два раза промывают охлажденной смесью (0+5°С) этилацетата (49 мл) и гексана (49 мл). Продукт сушат под вакуумом при приблизительно 45°С в течение 18 часов с получением указанного в заголовке соединения в виде твердого вещества от белого цвета до белого цвета с желтоватым оттенком (6,37 г, 80,6%).
Пример 5: S-Фторметиловый эфир 6α, 9α-дифтор-17α-(1-оксопропокси)-11β-гидрокси-16α-метил-3-оксо-андроста-1,4-диен-17β-карботиокислоты (с использованием единого растворителя для избежания выделения промежуточной 6α, 9α-дифтор-17α-(1-оксопропокси)-11β-гидрокси-16α-метил-3-оксо-андроста-1,4-диен-17β-карботиокислоты)
Раствор 6α, 9α-дифтор-11β, 17α-дигидрокси-16α-метил-3-оксо-андроста-1,4-диен-17β-карботиокислоты (полученной в соответствии с методикой, описанной в GB 2088877 В) (7 г) в этилацетате (350 мл) перемешивают при температуре 20÷25°С и обрабатывают триэтиламином (10,9 мл) при 0÷5°С в течение приблизительно 20 минут, добавляя в качестве поточной промывки дополнительное количество этилацетата (5 мл). Полученную в результате суспензию затем охлаждают до температуры -3°С + -7°С и в течение приблизительно 30 минут обрабатывают пропионилхлоридом (6,2 мл), поддерживая температуру от -5°С до +2°С. В качестве поточной промывки добавляют этилацетат (5 мл) и раствор перемешивают при температуре от -5°С до +2°С в течение дополнительных двух часов. Полученную в результате суспензию затем охлаждают до -3°С + -7°С и в течение приблизительно 30 минут обрабатывают диэтаноламином (23,8 мл) в метаноле (20 мл), поддерживая температуру от -5°С до +2°С. В качестве поточной промывки добавляют этилацетат (5 мл) и раствор перемешивают при температуре от -5°С до +2°С в течение дополнительных 30 минут. В течение приблизительно 10 минут добавляют 25 мл уксусной кислоты, поддерживая температуру в интервале от -5°С до +2°С, и полученную в результате суспензию выдерживают при температуре от -5°С до +5°С в течение по меньшей мере 10 минут. В течение приблизительно 10 минут добавляют 30 мл воды, поддерживая температуру в интервале от -5°С до +2°С, и органическую фазу отделяют и промывают водой (3×50 мл). Водные фазы, возможно, подвергают обратной экстракции этилацетатом (120 мл) при температуре от -5°С до +2°С. Объединенные органические фазы концентрируют приблизительно до 45 об. вакуумной перегонкой (ниже 10°С). Приблизительно половину полученного раствора обрабатывают водой (13,5 мл), хлоридом бензилтрибутиламмония (0,37 г) и триэтиламином (1,3 мл) и смесь охлаждают до 5°С. Добавляют бромфторметан (0,5 мл), поддерживая температуру реакционной смеси приблизительно 5°С. Смесь в течение 2-3 часов нагревают до 20°С и полученную в результате суспензию последовательно промывают 0,5 М соляной кислотой (23 мл), 1%-ным (мас./мас.) водным раствором бикарбоната натрия (3×23 мл) и водой (2×23 мл). Органический слой отделяют и водный слой подвергают обратной экстракции этилацетатом (30 мл). Объединенные органические слои перегоняют до объема, составляющего приблизительно 22 мл, и добавляют дополнительное количество этилацетата (7 мл). Смесь охлаждают до приблизительно 20°С, в течение по меньшей мере 30 минут добавляют гексан (42 мл) и смесь выдерживают при 20°С в течение 15 минут. Выпавший осадок собирают фильтрованием, промывают смесью этилацетата и гексана (1:4, 3×5 мл) и сушат при приблизительно 50°С в течение 18 часов с получением указанного в заголовке соединения в виде белого твердого вещества (3,54 г, 95,7%).
Способ успешно повторили с использованием в качестве растворителя пентан-3-она вместо этилацетата.
Пример 6: Соль 6α, 9α-дифтор-17α-(1-оксопропокси)-11β-гидрокси-16α-метил-3-оксо-андроста-1,4-диен-17β-карботиокислоты с триэтиламином (с использованием этилацетата в качестве растворителя, по существу не смешивающегося с водой)
Раствор 6α, 9α-дифтор-11β, 17α-дигидрокси-16α-метил-3-оксо-андроста-1,4-диен-17β-карботиокислоты (полученной в соответствии с методикой, описанной в GB 2088877 В) (7 г) в этилацетате (350 мл) перемешивают при 20+25°С и обрабатывают триэтиламином (10,9 мл) при 0+5°С в течение приблизительно 20 минут, добавляя в качестве поточной промывки дополнительное количество этилацетата (5 мл). Полученную в результате суспензию затем охлаждают до температуры -3°С + -7°С и в течение приблизительно 30 минут обрабатывают пропионилхлоридом (6,2 мл), поддерживая температуру от -5°С до +2°С. В качестве поточной промывки добавляют этилацетат (5 мл) и раствор перемешивают при температуре от -5°С до +2°С в течение дополнительных двух часов. Полученную в результате суспензию затем охлаждают до температуры -3°С + -7°С и в течение приблизительно 30 минут обрабатывают диэтаноламином (23,8 мл) в метаноле (20 мл), поддерживая температуру от -5°С до +2°С. В качестве поточной промывки добавляют этилацетат (5 мл) и раствор перемешивают при температуре от -5°С до +2°С в течение дополнительных 30 минут. В течение приблизительно 10 минут добавляют 25 мл уксусной кислоты, поддерживая температуру в интервале от -5°С до +2°С, и полученную в результате суспензию выдерживают при температуре от -5°С до +5°С в течение по меньшей мере 10 минут. В течение приблизительно 10 минут добавляют 50 мл воды, поддерживая температуру в интервале от -5°С до +2°С, и органическую фазу отделяют и промывают водой (3×50 мл). Водные фазы, возможно, подвергают обратной экстракции этилацетатом (120 мл) при температуре от -5°С до +2°С. Объединенные органические фазы обрабатывают триэтиламином (15 мл) с последующей азеотропной перегонкой (с этилацетатом) до тех пор, пока содержимое не станет мутным. Затем перегонкой уменьшают объем содержимого до 5 об. или доливают этилацетат, если объем меньше 5 об. Полученный раствор затем охлаждают до 0-5°С. Добавляют гексан (70 мл), поддерживая температуру от 0°С до 5°С, и содержимое выдерживают в течение по меньшей мере 30 минут при 0-5°С. Продукт отфильтровывают и два раза промывают охлажденной смесью (0-5°С) этилацетата (49 мл) и гексана (49 мл). Продукт сушат под вакуумом при приблизительно 45°С в течение 18 часов с получением указанного в заголовке соединения в виде твердого вещества от белого цвета до белого цвета с желтоватым оттенком (7,75 г, 80,6%).
Пример 7: Соль 6α, 9α-дифтор-17α-(1-оксопропокси)-11β-гидрокси-16α-метил-3-оксо-андроста-1,4-диен-17β-карботиокислоты с триэтиламином (альтернативный способ)
Раствор 6α, 9α-дифтор-17α-(1-оксопропокси)-11β-гидрокси-16α-метил-3-оксо-андроста-1,4-диен-17β-карботиокислоты (полученной в соответствии с методикой, описанной в примере 2) (15 г) в ацетоне (90 мл) охлаждают до приблизительно 15°С и обрабатывают триэтиламином (5 мл) в ацетоне (20 мл). Перед охлаждением до 15°С реакционную смесь выдерживают при приблизительно 20°С в течение 0,5 часа. В течение 15 минут добавляют 50 мл гексана и смесь выдерживают в течение 0,5 часа. Продукт отфильтровывают, промывают охлажденными гексаном (120 мл) и этилацетатом (30 мл) и сушат под вакуумом при приблизительно 22°С с получением указанного в заголовке соединения в виде твердого вещества от белого цвета до белого цвета с желтоватым оттенком (17,72 г, 94% от теор.).
Т.пл. (температура плавления) 164,3°С.
1H ЯМР δ (CD3OD) 7.21 (1Н, d), 6.44 (1Н, s), 6.39 (1Н, d), 5.46 (1Н, ddd), 4.37 (1Н, m), 3.69 (1Н, bs), 3.21 (6Н, m), 2.15-2.55 (7Н, m), 1.54 (3Н, s), 1.49 (1Н, s), 1.38 (9Н, m), 1.15-1.25 (1Н, m), 1.10-1.15 (6Н, m), 0.97 (3Н, d).
Заменяя триэтиламин, используемый в примере 7, на релевантное основание, аналогичным образом получили следующие соли 6α, 9α-дифтор-17α-(1-оксопропокси)-11β-гидрокси-16α-метил-3-оксо-андроста-1,4-диен-17β-карботиокислоты. Высокая чистота всех солей была подтверждена ВЭЖХ:
Калиевая соль 6α, 9α-дифтор-17α-(1-оксопропокси)-11β-гидрокси-16α-метил-3-оксо-андроста-1,4-диен-17β-карботиокислоты
Раствор 6α, 9α-дифтор-17α-(1-оксопропокси)-11β-гидрокси-16α-метил-3-оксо-андроста-1,4-диен-17β-карботиокислоты (2 г) в ацетоне (51 мл) и воде (5 мл) обрабатывают бикарбонатом калия (0,46 г) и смесь перемешивают при температуре окружающей среды до растворения. Вакуумной перегонкой реакционную смесь концентрируют до приблизительно 8 мл перед расслоением с использованием гексана и дихлорметана до начала выпадения в осадок твердых частиц белого цвета. Смесь охлаждают до 15°С в течение ночи. Продукт отфильтровывают, промывают гексаном (16 мл) и этилацетатом (4 мл) и сушат под вакуумом при температуре окружающей среды с получением указанного в заголовке соединения (2,18 г, 99% от теор.).
Т.пл. 290°С (с разложением).
1Н ЯМР δ (СО3OD) 7.27 (1Н, d, J 10 Гц), 6.24 (2Н, dd, J 10 Гц), 6.19 (1Н, s), 5.49 (1Н, ddd), 4.14 (1Н, m), 2.45-2.60 (1Н, m), 2.30-2.40 (3Н, m), 2.24 (q, 2Н, J 7 Гц), 2.05-2.15 (1Н, m), 1.80-1.95 (1Н, m), 1.60 (3Н, s), 1.15 (s, 3Н), 1.05 (t, 3Н, J 7 Гц), 0.88 (d, 3H).
Соль 6α, 9α-дифтор-17α-(1-оксопропокси)-11β-гидрокси-16α-метил-3-оксо-андроста-1,4-диен-17β-карботиокислоты с диизопропилэтиламином
Раствор 6α, 9α-дифтор-17α-(1-оксопропокси)-11β-гидрокси-16α-метил-3-оксо-андроста-1,4-диен-17β-карботиокислоты (15 г) в ацетоне (80 мл) перемешивают при температуре окружающей среды в течение приблизительно 10 минут под слоем азота. Смесь охлаждают до 15°С и добавляют диизопропилэтиламин (5,73 мл) в ацетоне (20 мл). Реакционную смесь выдерживают в течение 0,5 часа при 20°С и, перед охлаждением смеси до 15°С и выдерживанием в течение дополнительных 0,5 часа, в течение 15 минут добавляют 50 мл гексана. Продукт отфильтровывают, промывают охлажденной смесью гексана (120 мл) и этилацетата (30 мл) и сушат под вакуумом при температуре окружающей среды с получением указанного в заголовке соединения (17,56 г, 92%).
Т.пл. 182,1°С.
1H ЯМР δ (ДМСОd6) 7.24 (1Н, d), 6.28 (1Н, d), 6.08 (1Н, s), 5.63 (1Н, add), 5.4 (1Н, s), 5.25 (1Н, bs), 4.06 (1Н, m), 3.62 (2Н, m), 3.25 (2Н, m), 2.40 (2Н, q), 2.15 (1Н, m), 1.85-2.00 (2Н, m), 1.45-1.6 (5Н, s+m с наложением), 1.20-1.35 (17Н, m), 1.15 (1Н, s), 0.95-1.00 (1Н, m), 0.90 (3Н, s) и 0.66 (3Н, d).
Соль 6α, 9α-дифтор-17α-(1-оксопропокси)-11β-гидрокси-16α-метил-3-оксо-андроста-1,4-диен-17β-карботиокислоты с 2.6-диметилпиридином
Раствор 6α, 9α-дифтор-17α-(1-оксопропокси)-11β-гидрокси-16α-метил-3-оксо-андроста-1,4-диен-17β-карботиокислоты (2 г) в дихлорметане (20 мл) перемешивают в атмосфере азота при приблизительно 22°С в течение приблизительно 10 минут. По каплям добавляли 2,6-диметилпиридин (0,46 г) в CH2Cl2 (10 мл) и перемешивали в течение 1 часа. По каплям добавляют гексан (10 мл) и смесь выдерживают в течение по меньшей мере 0,5 часа при 15°С. Выпавшую в осадок соль фильтруют и промывают охлажденной смесью CHzClz (10 мл) и гексана (30 мл) и сушат под вакуумом при температуре окружающей среды с получением указанного в заголовке соединения (1,56 г, 63%).
Т.пл. 111°С.
1Н ЯМР о (CDCl3) 7.57 (1Н, dd), 7.16 (1Н, s), 7.04 (1Н, d), 6.45 (1Н, s), 6.42 (1Н, dd), 5.40 (1Н, ddd), 4.44 (1Н, m), 3.34 (1Н, bs), 2.60 (6Н, s), 2.40 (4Н, m), 2.20-2.35 (2Н, m), 1.65-1.95 (3Н, m), 1.55 (3Н, s), 1.33 (1Н, m), 1.10-1.20 (6Н, m), 1.00 (3Н, d).
Пример 8: 6α, 9α-Дифтор-17α-(1-оксопропокси)-11β-гидрокси-16α-метил-3-оксо-андроста-1,4-диен-17β-ил-S-(1-оксопропокси)-тиоангидрид
Раствор 6α, 9α-дифтор-11β, 17α-дигидрокси-16α-метил-3-оксо-андроста-1,4-диен-17β-карботиокислоты (10 г) в ацетоне (125 мл) охлаждают до приблизительно -5°С и обрабатывают триэтиламином (16 мл) при температуре от 0°С до -5°С в течение приблизительно 15 минут. Суспензию в течение приблизительно 90 минут обрабатывают пропионилхлоридом (8,5 мл), поддерживая температуру от -5°С до 0°С, и раствор перемешивают при температуре от -5°С до 0°С в течение дополнительных двух часов. Реакционную смесь за десять минут вливают в 2 М соляную кислоту (470 мл) и полученную в результате суспензию выдерживают при 5°С в течение 30 минут. Продукт отфильтровывают, промывают водой (3×125 мл) и сушат под вакуумом при приблизительно 40°С в течение 15 часов с получением указанного в заголовке соединения в виде твердого вещества от белого цвета до белого цвета с желтоватым оттенком (12,78 г, 100,6%).
ВЭЖХ: время удерживания 40,7 мин (чистота 99,5% по площади).
CHN: обнаружено С, 61.8%; Н, 6.7%, S, 6.1%; формула C27H34F2O6S требует С, 61.8%; Н, 6.5%, S, 5.7%.
ЯМР (ДМСОd6) 7.27 (1Н, d, 10 Гц), 6.32 (1Н, d, 10 Гц), 6.12 (1Н, s), 5.81 (1Н, d), 5.65 (1Н, ddd), 4.37 (1Н, m), 2.40 (2Н, q, 7 Гц), 2.00-2.45 (4Н, m), 1.85 (1Н, m), 1.87 (1Н, m), 1.51 (ЗН, s), 1.27 (1Н, m), 1.11-1.22 (4Н, m), 0.90-1.05 (6Н, m), 0.88 (3Н, d, 7 Гц).
Пример 9: Получение соли 6α, 9α-дифтор-11β, 17α-дигидрокси-16α-метил-3-оксо-андроста-1,4-диен-17β-карботиокислоты с имидазолом (1:2)
6α, 9α-Дифтор-11β-гидрокси-16α-метил-3-оксо-17α-гидрокси-андроста-1,4-диен-17β-карбоновую кислоту (35 г, 0,089 моль) и карбонилдиимидазол (25,75 г, 0,16 моль) перемешивают в этилацетате (350 мл) и N,N-диметилформамиде (17,5 мл) при температуре 20±2°С в течение 60 минут. Суспензию охлаждают до 0°С и, пока через погружную трубку (dip pipe) из пористого стекла в течение 32 минут добавляют сероводород (7,7 г, 0,23 моль), содержимое перемешивают при температуре 0±5°С. Всю загрузку перемешивают при 0±3°С в течение 30 минут, за 20 минут нагревают до 9°С и перемешивают при 9±3°С в течение полных 100 минут. Продукт собирают фильтрованием (бумага Whatman 54), осадок промывают этилацетатом (2×105 мл). Продукт сушат под вакуумом при приблизительно 20°С в течение 20 часов с получением указанного в заголовке соединения в виде твердого вещества от белого до бледно-фиолетового цвета (47.7 г, 98,5% от теор).
ЯМР (MeOHd4) 0.86 (3Н) d, J=7.4 Гц; 1.11 (3Н) s; 1.20 (1Н) m; 1.61 (3Н) s; 1.62-1.82 (3Н) m; 2.14-2.25 (2Н) m; 2.33 (1Н) m; 2.54 (1Н) m; 3.19 (1Н) m; 4.26 (1Н) ddd, J=11.2, 4.0, 1.8 Гц; 5.57 (1 H) dddd, J=49.0, 11.6, 6.8, 1.9 Гц; 6.32 (1 Н) m; 6.35 (1Н) dd, J=10.0, 2.0 Гц; 7.35 (4Н) d, J=1.0 Гц (имидазол); 7.41 (1Н) dd, J=10.0, 1.4 Гц; 8.30 (2Н) t, J=1.0 Гц (имидазол).
Т.пл. 120°C (с разложением).
Пример 10: Получение раствора 6α, 9α-дифтор-11β, 17α-дигидрокси-16α-метил-3-оксо-андроста-1,4-диен-17β-карботиокислоты из соли 6α, 9α-дифтор-11β, 17α-дигидрокси-16α-метил-3-оксо-андроста-1,4-диен-17β-карботиокислоты с имидазолом (1:2)
Соль 6α, 9α-дифтор-11β, 17α-дигидрокси-16α-метил-3-оксо-андроста-1,4-диен-17β-карботиокислоты с имидазолом (47,7 г) перемешивают в этилацетате (811 мл) и суспензию охлаждают до температуры 15±3°С. Добавляют 2 М (водную) соляную кислоту (286 мл) и смесь перемешивают в течение приблизительно пяти минут, получая прозрачную двухфазную смесь. Слои разделяют и органический раствор свободной карботиокислоты промывают дополнительным количеством 2 М (водной) соляной кислоты (190 мл).
Альтернативный способ
Соль 6α, 9α-дифтор-11α, 17β-дигидрокси-16α-метил-3-оксо-андроста-1,4-диен-17β-карботиокислоты с имидазолом (47,7 г) перемешивают в пентан-3-оне (954 мл) и суспензию охлаждают до температуры 15±3°С. Добавляют 2 М (водную) соляную кислоту (286 мл) и смесь перемешивают в течение приблизительно пяти минут, получая прозрачную двухфазную смесь. Слои разделяют и органический раствор свободной карботиокислоты промывают водой (190 мл).
В любом случае, влажные осадки соли 6α, 9α-дифтор-11β, 17α-дигидрокси-16α-метил-3-оксо-андроста-1,4-диен-17β-карботиокислоты с имидазолом, содержащие растворитель (а не сухие твердые вещества), можно использовать в качестве исходного материала в упомянутых выше методиках подкисления.
Пример 11: 6α, 9α-Дифтор-17α-(1-оксопропокси)-11β-гидрокси-16α-метил-3-оксо-андроста-1,4-диен-17β-карботиокислота
К тщательно перемешанной смеси 6α, 9α-дифтор-17-α-оксопропокси)-11β-гидрокси-16α-метил-3-оксо-андроста-1,4-диен-17β-карбоновой кислоты (10,0 г) и N,N-карбонилдиимидазола (6,3 г) последовательно добавляли этилацетат (100 мл, 10 об.) и диметилформамид (ДМФ) (5 мл, 0,5 об.). Образовавшуюся суспензию перемешивали при температуре 17±3°С в течение 50 минут с получением бледно-желтого раствора. Раствор охлаждали до 10°С и в течение 25 минут через него барботировали сероводород (2,2 г), поддерживая температуру смеси 12±2°С. Полученную суспензию перемешивали при температуре 12±2°С в течение дополнительных 90 минут и затем фильтровали. Осадок на фильтре промывали этилацетатом (2×30 мл) и сушили под вакуумом. Затем твердое вещество суспендировали в 3-пентаноне (200 мл) и промывали 2 М соляной кислотой (60 мл), а потом водой (60 мл). Полученный раствор охлаждали до 3°С и добавляли за 2 минуты 14,0 мл трипропиламина, обеспечивая сохранение температуры реакционной смеси 3±2°С. Раствор перемешивали при 3±2°С и добавляли за 5 минут 5,3 мл пропионилхлорида, поддерживая температуру реакционной смеси 3±2°С. Затем раствор оставляли нагреваться до 10°С и перемешивали при 12±2°С в течение 90 минут. Затем раствор охлаждали до 3°С и добавляли 1-метилпиперазин (5,1 мл), поддерживая температуру реакционной смеси 3±2°С. Раствор перемешивали при 3±2°С в течение 20 минут, нагревали до 18±3°С и затем последовательно промывали 1 М HCl (60 мл) и водой (60 мл). Затем одну половину раствора (100 мл) в течение 20 минут обрабатывали 2,2,4-триметилпентаном (100 мл). Полученную суспензию перемешивали при 20±3°С в течение по меньшей мере 14 часов и затем фильтровали. Осадок на фильтре промывали смесью 3-пентанона и 2,2,4-триметилпентана (2×20 мл) и сушили под вакуумом. Твердое вещество сушили в вакуумной печи при 40°С в течение 6 часов с получением указанного в заголовке соединения в виде белого твердого вещества (4,1 г, 69% от теор.).
Пример 12: Соль 6α, 9α-дифтор-17α-(1-оксопропокси)-11β-гидрокси-16α-метил-3-оксо-андроста-1,4-диен-17β-карботиокислоты с триэтиламином
К тщательно перемешанной смеси 6α, 9α-дифтор-17α-(1-оксопропокси)-11β-гидрокси-16α-метил-3-оксо-андроста-1,4-диен-17β-карбоновой кислоты (10,0 г) и N,N'-карбонилдиимидазола (6,3 г) последовательно добавляли этилацетат (100 мл) и ДМФ (5 мл). Образовавшуюся суспензию перемешивали при 18+20°С в течение 50 минут с получением бледно-желтого раствора. Раствор охлаждали до 10°C и в течение 25 минут через него барботировали сероводород (2,2 г), поддерживая температуру смеси 12±2°С. Полученную суспензию перемешивали при 12±2°С в течение дополнительных 90 минут и затем фильтровали. Осадок на фильтре промывали этилацетатом (2×30 мл) и сушили под вакуумом. Затем твердое вещество суспендировали в 3-пентаноне (200 мл) и промывали 2 М соляной кислотой (60 мл), а потом водой (60 мл). Полученный раствор охлаждали до 3°С и добавляли за 2 минуты 14,0 мл трипропиламина, обеспечивая сохранение температуры реакционной смеси 3±2°С. Раствор перемешивали при 3±2°С и добавляли за 5 минут 5,3 мл пропионилхлорида, поддерживая температуру реакционной смеси 3±2°С. Затем раствор оставляли нагреваться до 10°С и перемешивали при 12±2°С в течение 90 минут. Затем раствор охлаждали до 3°С и добавляли 1-метилпиперазин (5,1 мл), поддерживая температуру реакционной смеси 3±2°С. Раствор перемешивали при 3±2°С в течение 20 минут, нагревали до 18±3°С и затем последовательно промывали 1 М HCI (60 мл) и водой (60 мл). Половину раствора (100 мл) охлаждали до 3°С и обрабатывали триэтиламином (2,1 мл). Раствор перемешивали при 3±2°С в течение 10 минут, нагревали до 20°С и затем добавляли за 20 минут 100 мл 2,2,4-триметилпентана. Полученную суспензию перемешивали при 20±3°С в течение по меньшей мере 14 часов и затем фильтровали. Осадок на фильтре промывали смесью 3-пентанона и 2,2,4-триметилпентана (1:3,2×20 мл) и сушили под вакуумом. Твердое вещество сушили в вакуумной печи при 40°С в течение 6 часов с получением указанного в заголовке соединения в виде белого твердого вещества (6,2 г, 89% от теор.).
Следует понимать, что во всем описании изобретения и формуле, прилагаемой к нему (если в контексте не требуется иначе), под словом "включать в себя" и такими его вариантами, как "включает в себя" и "включающий в себя", подразумевают включение указанных целого либо стадии или группы целых, но и не исключают любые другие целые либо стадию или группу целых или стадий.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ПРОПИОНАТА ФЛУТИКАЗОНА, В ЧАСТНОСТИ ПОЛИМОРФНОЙ ФОРМЫ 1 | 2003 |
|
RU2333218C2 |
| СПЕЦИФИЧЕСКОЕ ГЛЮКОКОРТИКОСТЕРОИДНОЕ СОЕДИНЕНИЕ, ОБЛАДАЮЩЕЕ ПРОТИВОВОСПАЛИТЕЛЬНОЙ АКТИВНОСТЬЮ | 2004 |
|
RU2348645C2 |
| СЛОЖНЫЕ ЭФИРЫ 16 АЛЬФА 17 АЛЬФА - АЦЕТАЛЬЗАМЕЩЕННОЙ АНДРОСТАН-17 БЕТА - КАРБОНОВОЙ КИСЛОТЫ ИЛИ ИХ СТЕРЕОИЗОМЕРЫ | 1991 |
|
RU2081879C1 |
| ПРОИЗВОДНЫЕ АНДРОСТЕНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1997 |
|
RU2194053C2 |
| КОНКРЕТНОЕ ГЛЮКОКОРТИКОСТЕРОИДНОЕ СОЕДИНЕНИЕ, ОБЛАДАЮЩЕЕ ПРОТИВОВОСПАЛИТЕЛЬНОЙ АКТИВНОСТЬЮ | 2004 |
|
RU2359973C2 |
| СПОСОБ ЭТЕРИФИКАЦИИ ТИОКАРБОНОВОЙ КИСЛОТЫ | 2004 |
|
RU2351605C2 |
| СПОСОБ ПОЛУЧЕНИЯ ФЛУМЕТАЗОНА, СОЕДИНЕНИЕ | 2002 |
|
RU2260596C1 |
| ХИМИЧЕСКИЕ СОЕДИНЕНИЯ | 2008 |
|
RU2462472C2 |
| СТЕРОИДНЫЕ ЭФИРЫ ИЛИ ИХ СТЕРЕОИЗОМЕРЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1992 |
|
RU2112775C1 |
| ПРИМЕНЕНИЕ ИНГИБИТОРА Н, К-АТФАЗЫ ДЛЯ ЛЕЧЕНИЯ НОСОВЫХ ПОЛИПОВ | 1997 |
|
RU2197966C2 |
Описан улучшенный способ получения соединения формулы (II) или его соли,

действием на соединение формулы (III)


активированного производного пропионовой кислоты в количестве 1,3 моль на 1 моль соединения III, затем удаляют пропионильную группу при атоме S из соединения формулы IIA действием органического первичного или вторичного аминного основания, способного к образованию водорастворимого пропанамида предпочтительно диэтаноламина или N-метилпиперазина. Способ позволяет получить продукт, не загрязненный N,N-диэтилпропанамидом. 2 н. и 9 з.п. ф-лы.
или его соли,
при котором
(а) соединение формулы (III)

подвергают взаимодействию с активированным производным пропионовой кислоты в количестве по меньшей мере 1,3 моль активированного производного на моль соединения формулы (III) и
(б) удаляют пропионильную группировку, связанную с атомом серы, из любого полученного таким образом соединения формулы (IIA)

путем взаимодействия продукта со стадии (а) с органическим первичным или вторичным аминным основанием, способным к образованию водорастворимого пропанамида.
выделенное в форме твердой кристаллической соли.
| PHILLIPPS G.H | |||
| et al | |||
| J | |||
| of Med | |||
| Chem | |||
| Пишущая машина | 1922 |
|
SU37A1 |
| US 4335121 A, 15.06.1982. | |||

