Область техники, к которой относится изобретение
Настоящее изобретение относится к некоторым дериватизированным ненасыщенными жирными кислотами имеющим лечебное действие глюкокортикоидам и содержащим их фармацевтическим составам. В настоящем описании и формуле изобретения указанные производные именуются «Соединения формулы (I)». Соединения формулы (I) могут быть использованы в терапии раковых и воспалительных заболеваний. Охватывается терапия как рака крови, так и солидного рака и стероид-резистентных типов рака, астмы, а также стероид-резистентных воспалительных заболеваний и хронического обструктивного заболевания легких.
Уровень техники
Глюкокортикоиды широко используются в терапии воспалительных заболеваний, таких как астма, ревматоидный артрит, воспалительные заболевания кишечника и аутоиммунные заболевания. Глюкокортикоиды также являются ключевыми препаратами в терапии детского острого лимфобластного лейкоза (ALL) (1). Хотя их использование в терапии хронических воспалительных заболеваний и рака стало обычным, оптимальная эффективность ограничивается свойственной организму или приобретенной резистентностью к данному препарату. Стероидная резистентность также представляет собой важную проблему при ведении больных с воспалительным заболеванием кишечника (2). Небольшое количество пациентов с астмой не реагируют адекватно или не поддаются лечению кортикостероидами (3). Хотя вопрос кортикостероидной резистентности в случае ревматоидного артрита может быть менее исследован, чем в случае рака крови, этим двум заболеваниям свойственен одинаковый механизм резистентности, и приемы преодоления резистентности после терапии заболевания могут представлять обоюдный интерес (7).
Полагают, что благоприятное воздействие глюкокортикоидов опосредуется подавлением экспрессии воспалительных генов. Они действуют путем связывания с одиночным рецептором (глюкокортикоидным рецептором, GR), расположенным в цитозоле клетки. При активации GR перемещается в ядро, где он либо включает (трансактивация) противовоспалительные гены, либо отключает (трансрепрессия) воспалительные гены (4). Для индукции гена необходима димеризация GR и связывание ДНК со специфичными GRE-элементами, расположенными в промоторных областях генов-мишеней. Глюкокортикоиды могут активировать транскрипцию генов, кодирующих противовоспалительные белки, такие как липокортин-1 и интерлейкин-10. Самым замечательным действием глюкокортикоидов является ингибирование экспрессии полимерных генов (цитокинов, энзимов, рецепторов и адгезивных молекул). Примером является провоспалительный цитокин гранулоцитарно-макрофагальный колониестимулирующий фактор (GM-CSF, ГМКСФ). Глюкокортикоиды опосредуют это ингибирующее действие путем взаимодействия между активированными GR и активированными факторами транскрипции, такими как ядерный фактор-kB (NFkB) и активирующий белок-1 (АР-1), которые регулируют экспрессию воспалительных генов. GM-CSF играет ключевую роль в воспалительных и аутоимунных заболеваниях, а уменьшение количества GM-CSF считается отдельной целью медикаментозной терапии, направленной на подавление симптомов заболевания (8).
Применение глюкокортикоидов имеет ограничения из-за серьезных побочных эффектов (5), обусловленных их воздействием на эндокринную систему и обмен веществ, предположительно, опосредуемым индуцированным транскрипционным регулированием напрямую регулируемых GR генов-мишеней. Известные потенциальные отрицательные действия включают остеопороз и подавление гипоталамо-гипофизарно надпочечниковой системы, после систематического применения - снижение скорости линейного роста детей, потеря костных минералов, симптомы нарушения зрения и изменение кожи после приема ингаляционных кортикостероидов (6). Регулирование трансактивации и трансрепресии как тканеспецифическое, так и линиеспецифическое, может привести к повышению терапевтического индекса и открытию новых высокоселективных кортикостероидов (6) с более высоким терапевтическим эффектом против воспаления или рака, и меньшими побочными эффектами вследствие пониженного транскрипционного регулирования регулируемых GR генов, значимых с точки зрения этих побочных эффектов.
Кроме того, что касается механизма противоракового воздействия глюкокортикоидов, он включает взаимодействие с GR и их генами-мишенями, контролирующими экспрессию белков некроза/апоптоза клеток.
Клеточные линии U937 и ТНР1 являются моноцитарными/макрофаговыми клетками человека и представляют собой модельную систему линии клеток, используемую для испытания продуктов с противовоспалительным и противораковым действием, эти клеточные линии также релевантны в отношении глюкокортикоидной резистентности, поскольку они не подвержены ингибированию роста дексаметазоном и преднизолоном (7).
В принадлежащей тому же патентообладателю публикации WO98/32718 описаны адренокортикостероиды, которые могут образовывать производные с липофильными группами. Среди упомянутых соединений имеются бетаметазон, дексаметазон и беклометазон. Дериватизированные соединения используют для лечения воспаления.
Описаны конъюгаты будесонида с жирными кислотами, более конкретно олеат, пальмитат, линолеат, пальмитолеат и арахидонат будесонида. Эти конъюгаты образуются в ходе лечения воспаления, более конкретно астмы, однако утверждается, что конъюгаты упоминаемых стероидов с жирными кислотами являются фармакологически неактивными конъюгатами.
Описание изобретения
Настоящее изобретение неожиданно показало, что дериватизированные определенными ненасыщенными жирными кислотами глюкокортикоиды обладают неизвестной ранее и необычной терапевтической активностью. Большое число указанных производных составляет новый, ранее не описанный класс химических соединений. Производные соединения настоящего изобретения обладают повышенной активностью и дают меньше побочных эффектов, чем описанные ранее производные глюкокортикоидов, показано, что они активны в стероид-резистентных системах.
Известно, что глюкокортикоиды занимают определенное положение в терапии раковых заболеваний и воспалений. Хорошо известна свойственная организму или приобретенная резистентность к терапии глюкокортикоидными препаратами. Сообщалось, что моноцитарные/макрофаговые клетки U937 и ТНР1 человека представляют собой модельную систему линии клеток, отличающуюся внутренней резистентностью к глюкокортикоидам дексаметазону и преднизолону (7). Эти данные подтверждены, кроме того, показано, что эти клетки обладают перекрестной резистентностью к высокоактивному стероиду - флутиказона пропионату, не проявляющему активность до 100 мкМ.
Как показано на примерах, заявителями неожиданно обнаружено сильное антипролиферативное действие дериватизированных жирными кислотами глюкокортикоидов на эти резистентные клеточные линии. Это открытие делает возможным лечение заболеваний, резистентных к терапии известными глюкокортикоидными препаратами.
Заявителями неожиданно обнаружена повышенная противовоспалительная активность в сочетании с менее выраженной активацией генов, релевантных в отношении побочных эффектов, вызываемых этими дериватизированными жирными кислотами глюкокортикоидами. Для флутиказона, дериватизированного элаидиновой кислотой, наблюдалось в 22 раза более сильное ингибирование секреции цитокина GM-CSF, чем для флутиказона пропионата на активированных клетках легких. В то же время обнаружено 7-кратное ослабление побочных эффектов. Эти побочные эффекты представлены активацией GRE-элементов. Активация генов стероидными производными в этой системе, где активация является мерой побочных эффектов, на 85% меньше. Эти результаты увеличивают относительный терапевтический индекс в 154 раз.
Таким образом, настоящее изобретение относится к соединениям формулы (I)
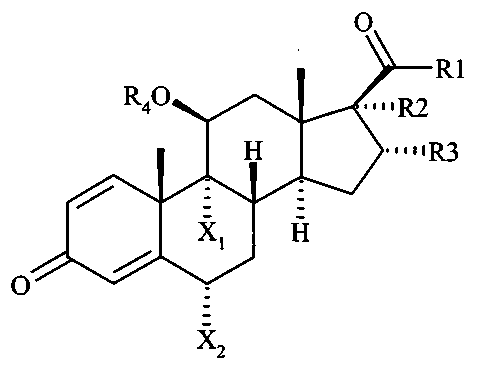
где
Х1 и Х2 независимо друг от друга означают Н или F;
R1 означает ОН, ОСН2СН3, OCH2Cl, SCH2F, СН2ОСОСН(СН3)2, СН2-О-R4 или O-R4;
R2 означает OCOEt, OCOCHCl2, ОСО2СН2СН3 или O-R4;
R3 означает Н, СН3 или ОН; или
R2 и R3, вместе с атомами углерода, с которыми они связаны, образуют 2-R5,R6-1,3-диоксолановое кольцо, где R5 и R6 независимо друг от друга означают Н, алкил С1-С6 или циклоалкил С3-С6;
R4 означает Н или ацильную группу С8-С24 общей формулы
CH3-(CH2)k-(CH=CH-CH2)l-(CH=CH)m-(CH2)n-CO (II)
где k - целое число от 0 до 10, l - целое число от 0 до 6, m - целое число от 0 до 1, n - целое число от 2 до 7;
со следующими условиями:
- когда Х1 является F, Х2 не является Н;
- в соединении имеется, по меньшей мере, одна ацильная группа общей формулы (II); и
- соединение формулы (I) не является одним из следующих соединений: будесонида олеат, будесонида линолеат, будесонида пальмитолеат и будесонида арахидонат;
или их фармацевтически приемлемым солям.
В предпочтительном варианте осуществления изобретения соединение формулы (I) представляет собой соединение, в котором
и Х1, и Х2 означают F;
R1 означает SCH2F;
R2 означает OCOEt или O-R4;
R3 означает СН3;
R4 означает Н или ацильную группу С8-С24 общей формулы
CH3-(CH2)k-(CH=CH-CH2)l-(CH=CH)m-(CH2)n-CO (II)
где k - целое число от 0 до 10, l - целое число от 0 до 6, m - целое число от 0 до 1, n - целое число от 2 до 7;
при условии, что в соединении имеется, по меньшей мере, одна ацильная группа общей формулы (II), или его фармацевтически приемлемая соль.
Другим вариантом осуществления изобретения является соединение формулы (I), в котором
и Х1, и Х2 означают Н;
R1 означает СН2-О-R4;
R2 и R3, вместе с атомами углерода, с которыми они связаны, образуют 2-R5,R6-1,3-диоксолановое кольцо, где R5 и R6 независимо друг от друга означают Н, алкил С1-С6 или циклоалкил С3-С6;
R4 означает Н или ацильную группу С8-С24 общей формулы
CH3-(CH2)k-(CH=CH-CH2)l-(CH=CH)m-(CH2)n-CO (II)
где k - целое число от 0 до 10, l - целое число от 0 до 6, m - целое число от 0 до 1, n - целое число от 2 до 7;
при условии, что в соединении имеется, по меньшей мере, одна ацильная группа общей формулы (II), или его фармацевтически приемлемая соль.
Еще одним вариантом осуществления изобретения является соединение формулы (I), в котором
и Х1, и Х2 означают F;
R1 означает О-R4;
R2 означает О-R4;
R3 означает СН3;
R4 означает Н или ацильную группу С8-С24 общей формулы
CH3-(CH2)k-(CH=CH-CH2)l-(CH=CH)m-(CH2)n-CO (II)
где k - целое число от 0 до 10, l - целое число от 0 до 6, m - целое число от 0 до 1, n - целое число от 2 до 7;
при условии, что в соединении имеется, по меньшей мере, одна ацильная группа общей формулы (II), или его фармацевтически приемлемая соль.
Примерами предпочтительных соединений, соответствующих настоящему изобретению, являются, помимо прочих, следующие соединения:
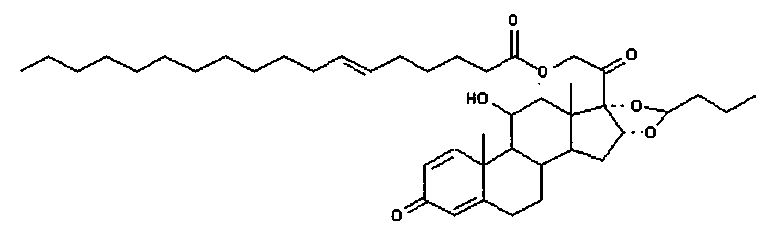
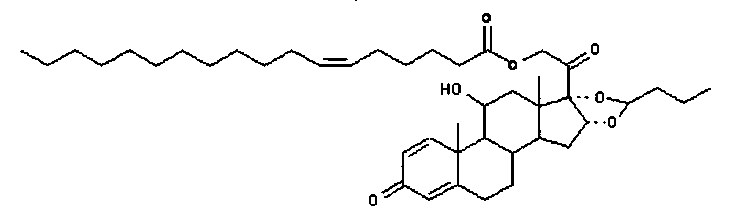
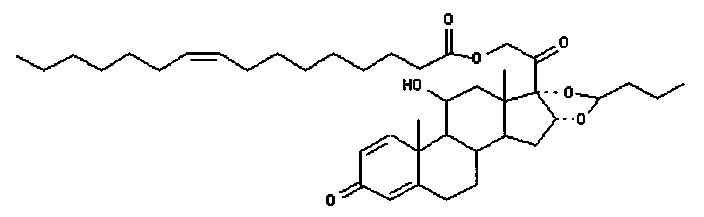
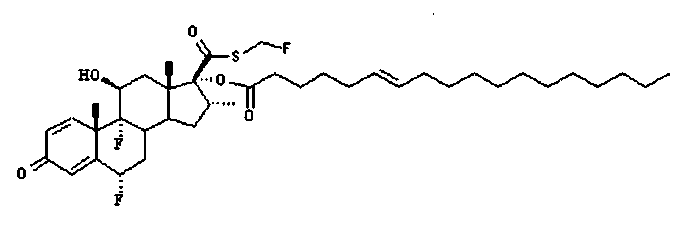
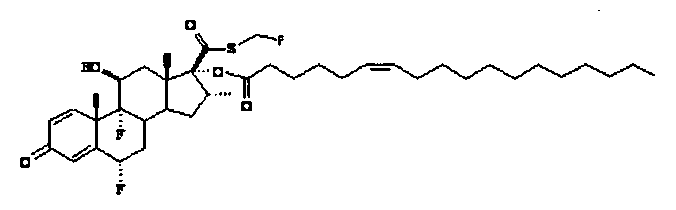
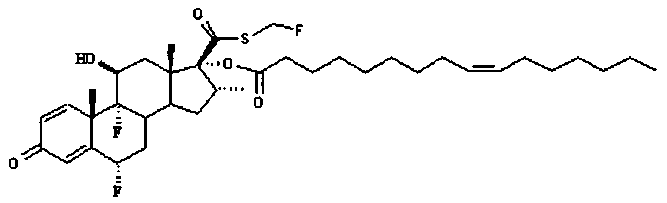
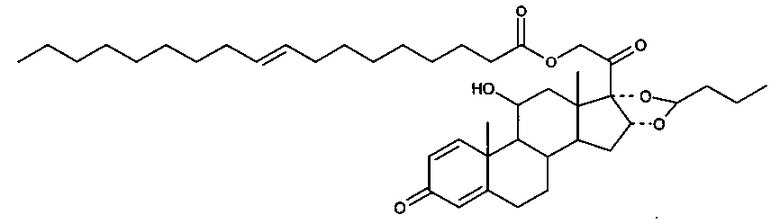
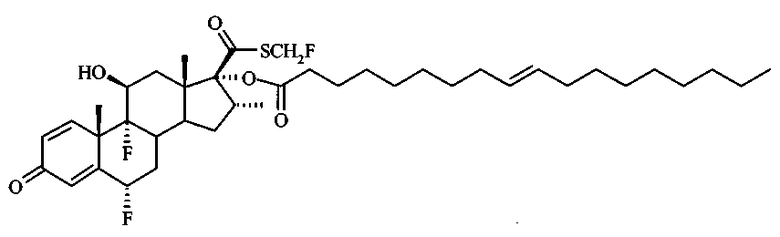
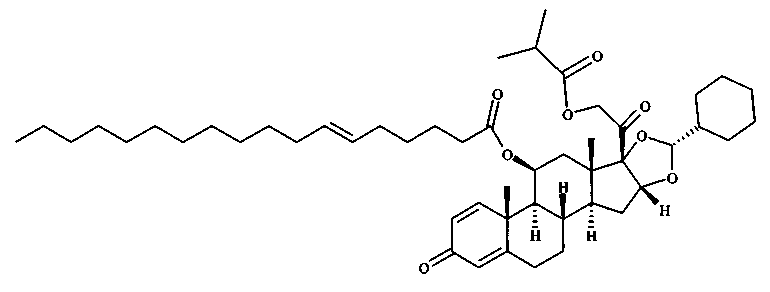
Соединения формулы (I) могут быть использованы для лечения заболеваний. Более конкретно, соединения формулы (I) могут быть использованы для лечения раковых заболеваний. В этой связи раковые заболевания включают рак крови, солидный рак и стероид-резистентный рак. Эти соединения также могут быть использованы для лечения воспаления. Более конкретно, эти соединения хорошо подходят для терапии стероид-резистентных воспалений. Соединения формулы (I) также могут быть использованы для терапии COPD (хронического обструктивного заболевания легких).
Соединения, соответствующие настоящему изобретению, вызывают меньше побочных эффектов, чем базовые стероиды, и обладают более специфичной противовоспалительной активностью.
Соединения настоящего изобретения, обычно, но не обязательно, вводят в состав фармацевтических композиций перед введением пациенту. Следовательно, в другом своем аспекте настоящее изобретение направлено на фармацевтические композиции, содержащие соединение настоящего изобретения и одно или более фармацевтически приемлемое вспомогательное вещество.
Указанные вспомогательные вещества подбирают в соответствии с нужной фармацевтической формой и способом введения. Указанные вспомогательные вещества вместе с соединением формулы (I), соответствующим настоящему изобретению, как правило, соединяют с получением лекарственной формы, подходящей для введения пациенту выбранным способом введения. Таким образом, лекарственные формы включают формы, пригодные для: (а) ингаляции, такие как аэрозоли, растворы и сухие порошки; (b) местного применения, такие как кремы, лосьоны, пасты, мази, растворы, спреи и гели; (с) внутривенного введения, такие как стерильные растворы, суспензии и порошки для разведения; (d) орального приема, такие как таблетки, капсулы, драже, порошки, сиропы, эликсиры, суспензии, растворы, эмульсии и саше. Соединения формулы (I) также могут быть введены как глазные капли.
Фармацевтически приемлемые вспомогательные вещества могут быть различными в зависимости от конкретной лекарственной формы. Помимо прочего, фармацевтически приемлемые вспомогательные вещества включают следующие: разбавители, смазывающие вещества, наполнители, дезинтегранты, растворители, увлажняющие агенты, суспендирующие агенты, эмульгаторы, гранулирующие агенты, покровные агенты, связующие, вкусовые добавки, вещества, корректирующие вкус, подсластители, пластификаторы, загустители, антиоксиданты, стабилизаторы, поверхностно-активные вещества и буферные вещества. Некоторые вспомогательные вещества могут выполнять более одной функции.
К пригодным разбавителям и наполнителям относятся лактоза, сахароза, декстроза, маннит, сорбит, картофельный крахмал, кукурузный крахмал, пептизированный крахмал, целлюлоза и их производные. К пригодным связующим относятся крахмал, желатин, триптаника (acaica), альгинат натрия, альгиновая кислота, трагант, гуаровая смола, провидон, целлюлоза и их производные. К пригодным дезинтегрантам относятся натрия крахмалгликолят, альгиновая кислота, кросповидон и натрия карбометилцеллюлоза.
Соединения формулы (I) также могут быть соединены с поддающимися биологическому разложению полимерами с целью управляемого высвобождения препарата.
Фармацевтические композиции, соответствующие настоящему изобретению, могут быть приготовлены с использованием способов и методик, известных специалистам в данной области.
Таким образом, одним из аспектов настоящего изобретения являются соединения формулы (I) для терапии рака, соответствующие пунктам 9-12 формулы изобретения.
Другим аспектом настоящего изобретения является применение соединений формулы (I) для приготовления фармацевтических композиций для терапии указанных заболеваний в соответствии с пунктами 14-21 формулы изобретения.
Соединения формулы (I) могут быть получены, как показано на примерах ниже.
Сложный эфир 11β-элаидиновой кислоты и флутиказона пропионата
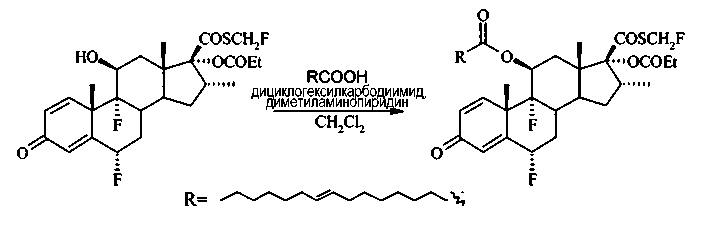
К раствору флутиказона пропионата (1,02 г, 2,04 ммоль) в дихлорметане (35 мл) добавили элаидиновую кислоту (576 мг, 2,04 ммоль), затем 4-(диметиламино)пиридин (249 мг, 2,04 ммоль) и 1,3-дициклогексилкарбодиимид (421 мг, 2,04 ммоль). Полученный раствор перемешивали в течение 24 часов, спустя это время анализ методом тонкослойной хроматографии показал значительное уменьшение количества исходного материала. Смесь концентрировали под вакуумом (~10 мл раствора) и перемешивали еще 140 часов. Смесь дополнительно концентрировали под вакуумом и очистили при помощи флэш-хроматографии на силикагеле, элюируя смесью гексан/EtOAc (3:1) с получением 1,23 г (79%) целевого соединения, имеющего вид бесцветного масла.
6α,9α-дифтор-11β,17α-дигидрокси-16α-метил-3-оксоандроста-1,4-диен-17β-карбоновая кислота
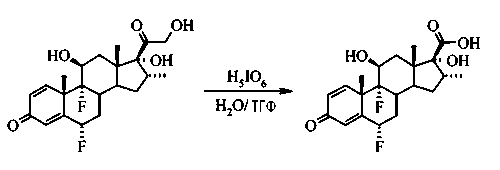
К суспензии флуметазона (1,19 г, 2,9 ммоль) в тетрагидрофуране (11 мл) добавили раствор перйодной кислоты (2,25 г, 9,9 ммоль) в воде (5,3 мл). Полученный раствор перемешивали при комнатной температуре в течение 1 часа. Тетрагидрофуран удалили в вакууме, после чего осталась водная суспензия, которую отфильтровали, твердый материал промыли водой, высушили и получили 1,17 г (100%) соединения, указанного в заголовке, имевшего вид бесцветного твердого вещества.
6α,9α-дифтор-16α-элаидоилокси-11β-гидрокси-16α-метил-3-оксоандроста-1,4-диен-17β-карбоновая кислота
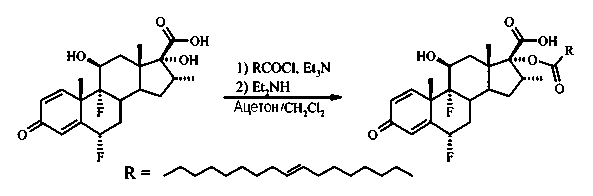
К раствору 6α,9α-дифтор-11β,17α-дигидрокси-16α-метил-3-оксоандроста-1,4-диен-17β-карбоновой кислоты (1,22 г, 3,1 ммоль) и триэтиламина (0,72 г, 7,1 ммоль) в ацетоне (25 мл) добавили элаидоилхлорид в дихлорметане (10 мл). Элаилоилхлорид был получен из элаидиновой кислоты (2,0 г, 7,1 ммоль), оксалилхлорида (2,6 мл, 30 ммоль) и диметилформамида (в каталитическом количестве) в толуоле (45 мл) при перемешивании при комнатной температуре в течение 17 часов, после чего выпарен до сухого состояния. Полученную смесь перемешивали в течение 2 часов при комнатной температуре, обработали диэтиламином (0,97 мл, 9,2 ммоль), полученный раствор перемешивали еще 1,5 часа при комнатной температуре. Затем добавили 1М HCl, смесь экстрагировали дихлорметаном (3×50 мл). Объединенные органические экстракты высушили (Na2SO4), отфильтровали и выделили под вакуумом. Неочищенный продукт растворили в EtOAc, добавили гексан и поместили в холодильник на ночь. Полученное грязно-белое твердое вещество (2,07 г, содержащее некоторое количество N,N-диэтиламида) отделили фильтрованием, высушили и использовали непосредственно на следующей стадии. Некоторое количество указанного в заголовке соединения (170 мг) очистили при помощи флэш-хроматографии на силикагеле, элюируя смесью гексан/EtOAc (4:1), затем смесью гексан/EtOAc/АсОН (100:100:1) и получили 123 мг (72%) белого твердого вещества.
6α,9α-дифтор-17β-(N,N-диметилкарбамоилтио)карбоил-16α-элаидоилокси-11β-гидрокси-16α-метил-3-оксоандроста-1,4-диен
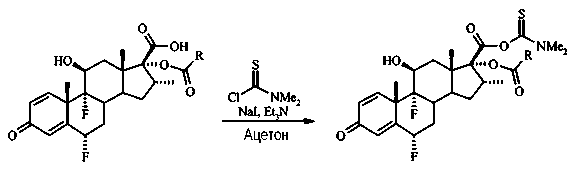
Раствор 6α,9α-дифтор-16α-элаидоилокси-11β-гидрокси-16α-метил-3-оксоандроста-1,4-диен-17β-карбоновой кислоты (1,9 г, 2,9 ммоль) и N,N-диметилтиокарбамоилхлорида (0,71 г, 5,8 ммоль) в ацетоне (40 мл) при комнатной температуре последовательно обработали триэтиламином (0,58 г, 5,8 ммоль), безводным йодидом натрия (0,43 г, 2,9 ммоль) и водой (0,19 мл, 10% вес. со стероидом). Раствор перемешивали 18 часов, добавили воду (100 мл) и EtOAc (100 мл), фазы разделились. Органическую фазу промыли 1М HCl, 5% водным раствором гидрокарбоната натрия, водой, высушили (Na2SO4), отфильтровали и отделили под вакуумом. После флэш-хроматографии на силикагеле с элюированием смесью гексан/EtOAc (3:2) получили 1,55 г (~70%, в две стадии) соединения, указанного в заголовке, в виде желтого твердого вещества.
6α,9α-дифтор-16α-элаидоилокси-11β-гидрокси-16α-метил-3-оксоандроста-1,4-диен-17β-карботионовая кислота
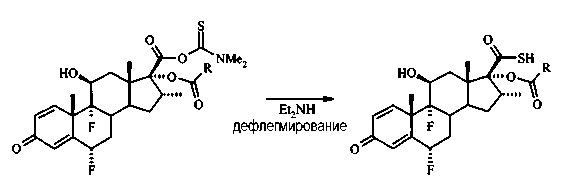
Раствор 6α,9α-дифтор-17β-(N,N-диметилкарбамоилтио)карбоил-16α-элаидоилокси-11β-гидрокси-16α-метил-3-оксоандроста-1,4-диена (1,55 г, 2,1 ммоль) в диэтиламине (16 мл) дефлегмировали в течение 3 часов. Раствор охладили до комнатной температуры, налили в холодную 3М HCl (150 мл) и экстрагировали EtOAc (2×150 мл). Объединенные органические экстракты промыли водой и рассолом перед сушкой (Na2SO4), отфильтровали и выпарили под вакуумом. Неочищенный продукт (1,35 г грязно-белого твердого вещества) использовали непосредственно на следующей стадии.
S-хлорметил 6α,9α-дифтор-16α-элаидоилокси-11β-гидрокси-16α-метил-3-оксоандроста-1,4-диен-17β-карботиоат
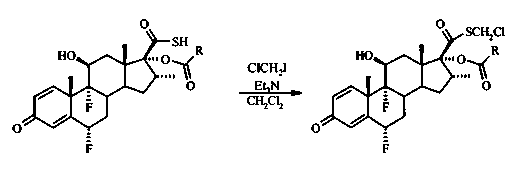
В перемешиваемый раствор 6α,9α-дифтор-16α-элаидоилокси-11β-гидрокси-16α-метил-3-оксоандроста-1,4-диен-17β-карботионовой кислоты (1,35 г, 2 ммоль) в дихлорметане (40 мл) добавили триэтиламин (0,28 мл, 2 ммоль), затем хлорйодметан (0,56 мл, 8 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 70 часов, добавили насыщенный водный раствор хлорида аммония и отделили фазы. Водную фазу экстрагировали дихлорметаном (2×), объединенные органические фазы высушили (Na2SO4), отфильтровали и выпарили под вакуумом. После флэш-хроматографии на силикагеле с элюированием смесью гексан/EtOAc (3:1) получили 1,04 г (72%) соединения, указанного в заголовке, в виде бесцветного твердого вещества.
S-йодметил 6α,9α-дифтор-16α-элаидоилокси-11β-гидрокси-16α-метил-3-оксоандроста-1,4-диен-17β-карботиоат
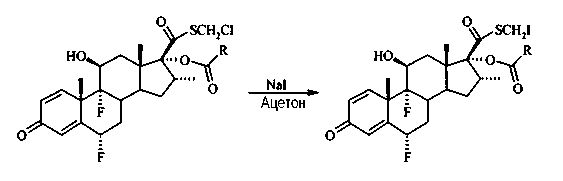
В раствор S-хлорметил 6α,9α-дифтор-16α-элаидоилокси-11β-гидрокси-16α-метил-3-оксоандроста-1,4-диен-17β-карботиоата (1,04 г, 1,4 ммоль) в ацетоне (100 мл) добавили йодид натрия (0,86 г, 5,7 ммоль) и дефлегмировали в течение 22 часов. Растворитель выпарили под вакуумом, остаток растворили в EtOAc и промыли водой, 10% водным раствором гидрокарбоната натрия, водой, высушили (Na2SO4), отфильтровали и выпарили под вакуумом. Неочищенный продукт (1,1 г) использовали непосредственно на следующей стадии.
S-фторметил 6α,9α-дифтор-16α-элаидоилокси-11β-гидрокси-16α-метил-3-оксоандроста-1,4-диен-17β-карботиоат
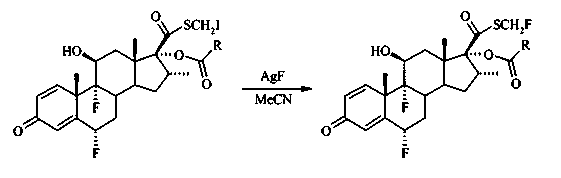
В раствор S-йодметил 6α,9α-дифтор-16α-элаидоилокси-11β-гидрокси-16α-метил-3-оксоандроста-1,4-диен-17β-карботиоата (1,1 г, 1,35 ммоль) в ацетонитриле (65 мл) добавили фторид серебра (1,7 г, 13,5 ммоль) и перемешивали в темноте при комнатной температуре в течение 40 часов. Смесь разбавили EtOAc и отфильтровали на коротком штранге целита и силикагеля. Фильтрат промыли водой, высушили (Na2SO4), отфильтровали и выпарили под вакуумом. Анализ 1Н ЯМР показал наличие некоторого количества непрореагировавшего хлорида, и последние две стадии были повторены для полного превращения хлорида во фторид. Затем продукт очистили методом флэш-хроматографии на силикагеле с элюированием смесью гексан/EtOAc (3:1) и получили 0,65 г (46%, две стадии) соединения, указанного в заголовке, в виде бесцветного твердого вещества.
Примеры
Приведенные ниже примеры поясняют настоящее изобретение. Однако эти примеры не следует рассматривать как ограничивающие объем настоящего изобретения.
Пример 1
Клеточные линии U937 и ТНР-1 высеяли в 96-луночные планшеты по 20000 клеток на лунку. В каждую лунку добавили 50 мкл культуральной среды. В то же время были добавлены исследуемые соединения в 5 разных концентрациях, инкубацию проводили в течение 48 часов. Для изучения цитотоксичности исследуемых соединений в этих клетках использовали набор для анализа пролиферации клеток CellTiter96® Non-Radiactive (Promega). В этом наборе используется колориметрический метод определения количества жизнеспособных пролиферирующих клеток или наборы для анализа химической чувствительности. Он состоит из растворов нового соединения тетразолия (3-(4,5-диметилтиазол-2-ил)-5-(3-карбоксиметоксифенил)-2-(4-сульфофенил)-2Н-тетразолий, внутренняя соль; MTS) и переносчика электронов (феназин метосульфат; PMS). MTS в клетках подвергается биовосстановлению до формазанового продукта, растворимого в среде тканевой культуры. Поглощение излучения 490 нм формазаном можно измерить непосредственно на 96-луночном планшете без дополнительной обработки. Количество формазанового продукта, измеренное по количеству поглощенного излучения 490 нм, прямо пропорционально количеству живых клеток в культуре (величина ИК50). Исследуемыми соединениями были флутиказона пропионат и флутиказон, дериватизированный элаидиновой кислотой. В клеточных линиях, резистентных к глюкокортикоидам, флутиказон, дериватизированный элаидиновой кислотой, оказался высокоактивным, характеризовался величиной ИК50 в микромолярном диапазоне, клетки U937 были наиболее чувствительными.
Пример 2
Клетки COS-1 почки обезьяны (АТСС CRL 1650) были выращены в модифицированной по способу Дульбекко среде Игла (Gibco BRL, Grand Island, Нью-Йорк), как описано в (9). Транзиторная трансфекция клеток COS-1 проведена, как описано в (10). Клетки были высеяны с плотностью 2×105 клеток/лунка. В каждую лунку было помещено 5 мкг исследуемых плазмид, 5 мкг контрольных плазмид β-галактозидаза в качестве внутреннего контроля и либо 2 мкг экспрессионных плазмид рМТ-hGR, либо pGL3-basic в качестве носителя. О плазмидах: мышиный ген PPARα описан в (11). В GL3-LUC векторе (Promega) были конструированы векторы, экспрессирующие ген-репортер LUC под контролем 5'-фланкирующей области и области промотора PPARα. 5'-фланкирующая последовательность PPARα между -28000bp и +100bp была клонирована в расщепляемый NheI pGL3-LUC с образованием плазмиды PPAR(-2800/+100)LUC. Каждую трансфекцию проводили трижды. Клетки трансфектировали 24 часа после посева с добавлением свежей среды, содержащей лиганд. Спустя 72 часа клетки собрали, приготовили цитозольный экстракт и измерили активность LUC в соответствии с протоколом Promega. Результаты нормализовали относительно активности β-галактозидазы, измеренной путем инкубации 100 мкл экстракта с 0,28 о-нитрофенил-D-галактозидой (ONPG) в 50 мМ фосфатного буфера с рН 7,0, 10 мМ KCl, 1 мМ MgCl2 в течение 30 мин при 30ºС и измерения поглощения излучения 420 нм.
Исследуемыми соединениями были флутиказона пропионат, флутиказон, дериватизированный элаидиновой кислотой, и флуметазон, дериватизированный элаидиновой кислотой. Стероидное соединение, флутиказона пропионат, было способно индуцировать 100% активность гена-репортера, тогда как сложные эфиры элаидиновой кислоты и флутиказона и элаидиновой кислоты и флуметазона индуцировали активность гена-репортера на 15% и 2% соответственно, что оказалось неожиданным. Эти стероидные аналоги значительно менее активировали гены, обуславливающие побочные эффекты.
Пример 3
IL-1β-стимулирование вызывает увеличение секреции гранулоцитарно-макрофагального колониестимулирующего фактора (GM-CSF) спустя 24 часа в клетках А549 TRE (АР-1 регуляция). Ингибирование секреции GM-CSF измеряли через 24 часа воздействия исследуемым соединением. Сложноэфирный флутиказон, дериватизированный элаидиновой кислотой, вызывал зависимое от концентрации ингибирование IL-1β-стимулированой секреции GM-CSF в клетках А549 TRE при ИК50=1,4×10-11 М. Флутиказона пропионат вызывал зависимое от концентрации ингибирование секреции GM-CSF в клетках А549 TRE при ИК50=3,1×10-10 М (таблица 3). Флутиказон, дериватизированный элаидиновой кислотой, в 22 раза сильнее ингибировал секрецию GM-CSF, то есть в данном испытании проявил в 22 раза более сильное противовоспалительное действие.
Индуцирование активности GRE-люциферазы наблюдали на стабильно трансфицированных клетках А549 GRE-люциферазы. В клетках А549 GRE-люциферазы не выявлено никакого воздействия на активность GRE-люциферазы по сравнению с зависимой от концентрации индукцией активности гена-репортера GRE-люциферазы в клетках А549 GRE-люциферазы, подвергнутых действию флутиказона пропионата, который вызывает максимальное стимулирование активности люциферазы при ЭК50=5,1×10-10 М.
Неожиданно обнаружено, что в случае использования флутиказона, дериватизированного элаидиновой кислотой, ингибирование GM-CSF увеличивается более чем в 20 раз по сравнению с отсутствием воздействия на GRE-люциферазу, что в данной испытательной системе также указывает на более высокий терапевтический индекс.
Дополнительные примеры
Сложный эфир будесонида и элаидиновой кислоты
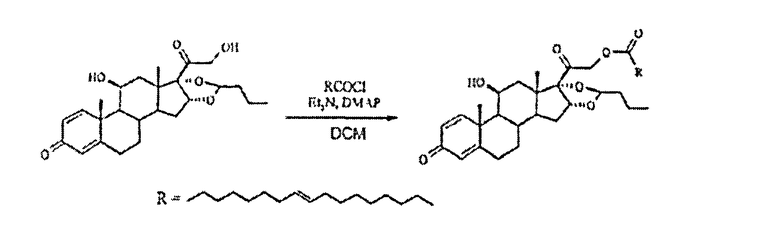
Получали хлорид элаидиновой кислоты из элаидиновой кислоты (0,34 г, 1,1 ммоль), оксалилхлорида (0,21 мл, 2,5 ммоль) и диметилформамида (каталитическое количество) в толуоле (5 мл) путем перемешивания при температуре окружающей среды в течение 1,75 ч, а затем упаривания до сухого состояния. Осадок растворяли в DCM (2 мл) и по каплям добавляли к суспензии будесонида (0,2 г, 0,5 ммоль), TEA (0,07 мл, 0,5 ммоль) и DMAP (каталитическое количество) в DCM (10 мл) в атмосфере N2 при 0°С. Охлаждающую баню удаляли и реакционную смесь перемешивали в темноте в течение 2,5 ч. Затем прозрачный раствор выливали в насыщенный водный раствор NH4Cl, фазы разделяли и водную фазу экстрагировали DCM (2x). Объединенные органические фазы промывали насыщенным водным раствором NaHCO3, рассолом, сушили (Na2SO4), фильтровали и упаривали в вакууме. Флэш-хроматография на силикагеле с элюированием смесью гексан/EtOAc (3:1) дала 309 мг (89%) указанного в заголовке соединения в виде бесцветного масла; 1H-ЯМР (200 МГц; CDCl3) δ 0,93 (м, 6Н), 1,09 (с, 3Н), 1,19 (д, 3Н), 1,34 (м), 1,5-2,6 (м), 2,86 (м), 3,15 (м, 1Н), 4,41 (м, 1Н), 4,92 (с, 2Н), 5,4 (м, 8Н), 6,15 (с, 1Н), 6,38 (дд, 1Н), 7,23 (д, 1Н); MS (электрораспыление); 701 [M+Na]+; ВЭЖХ: 99,4%.
Сложный эфир будесонида и петрозелаидиновой кислоты
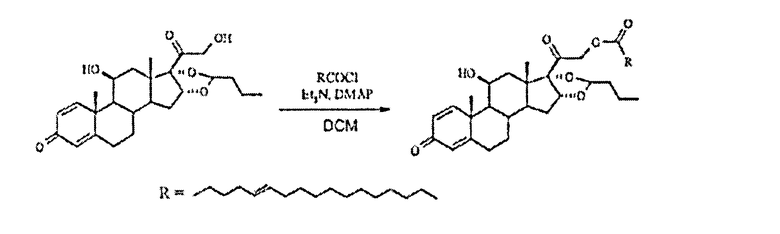
Получали хлорид петрозелаидиновой кислоты из петрозелаидиновой кислоты (0,34 г, 1,1 ммоль), оксалилхлорида (0,21 мл, 2,5 ммоль) и диметилформамида (каталитическое количество) в толуоле (5 мл) путем перемешивания при температуре окружающей среды в течение 1,75 ч, а затем упаривания до сухого состояния. Осадок растворяли в DCM (2 мл) и по каплям добавляли к суспензии будесонида (0,2 г, 0,5 ммоль), TEA (0,07 мл, 0,5 ммоль) и DMAP (каталитическое количество) в DCM (10 мл) в атмосфере N2 при 0°С. Охлаждающую баню удаляли и реакционную смесь перемешивали в темноте в течение 2,5 ч. Затем прозрачный раствор выливали в насыщенный водный раствор NH4Cl, фазы разделяли и водную фазу экстрагировали DCM (2x). Объединенные органические фазы промывали насыщенным водным раствором NaHCO3, рассолом, сушили (Na2SO4), фильтровали и упаривали в вакууме. Флэш-хроматография на силикагеле с элюированием смесью гексан/EtOAc (3:1) дала 309 мг (89%) указанного в заголовке соединения в виде бесцветного масла; 1H-ЯМР (200 МГц; CDCl3) δ 0,93 (м, 6Н), 1,09 (с, 3Н), 1,19 (д, 3Н), 1,34 (м), 1,5-2,6 (м), 2,86 (м), 3,15 (м, 1Н), 4,41 (м, 1Н), 4,92 (с, 2Н), 5,4 (м, 8Н), 6,15 (с, 1Н), 6,38 (дд, 1Н), 7,23 (д, 1Н); MS (электрораспыление); 701 [M+Na]+.
ВЭЖХ: 99,4%
Сложный эфир будесонида и петрозелиновой кислоты
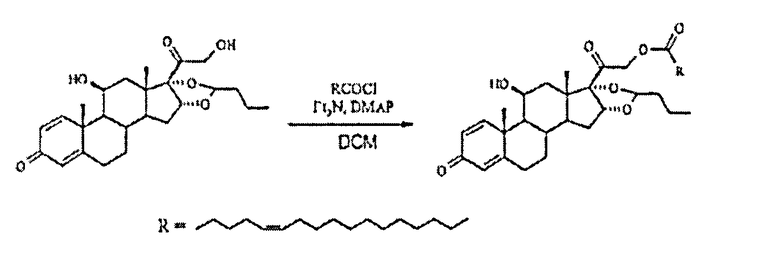
Получали хлорид петрозелиновой кислоты из петрозелиновой кислоты (0,34 г, 1,1 ммоль), оксалилхлорида (0,21 мл, 2,5 ммоль) и диметилфорамида (каталитическое количество) в толуоле (5 мл) путем перемешивания при температуре окружающей среды в течение 1,75 ч, а затем упаривания до сухого состояния. Осадок растворяли в DCM (2 мл) и капельно добавляли к суспензии будесонида (0,2 г, 0,5 ммоль), TEA (0,07 мл, 0,5 ммоль) и DMAP (каталитическое количество) в DCM (10 мл) в атмосфере N2 при 0°С. Охлаждающую баню удаляли и реакционную смесь перемешивали в темноте в течение 2,5 ч. Затем прозрачный раствор выливали в насыщенный водный раствор NH4Cl, фазы разделяли и водную фазу экстрагировали DCM (2x). Объединенные органические фазы промывали насыщенным водным раствором NaHCO3, рассолом, сушили (Na2SO4), фильтровали и упаривали в вакууме. Флэш-хроматография на силикагеле с элюированием смесью гексан/EtOAc (3:1) дала 309 мг (89%) указанного в заголовке соединения в виде бесцветного масла; 1H-ЯМР (200 МГц; CDCl3) δ 0,93 (м, 6Н), 1,09 (с, 3Н), 1,19 (д, 3Н), 1,34 (м), 1,5-2,6 (м), 2,86 (м), 3,15 (м, 1Н), 4,41 (м, 1Н), 4,92 (с, 2Н), 5,4 (м, 8Н), 6,15 (с, 1Н), 6,38 (дд, 1Н), 7,23 (д, 1Н); MS (электрораспыление); 701 [M+Na]+.
ВЭЖХ: 99,4%
Сложный эфир буденосида и пальмитолеиновой кислоты
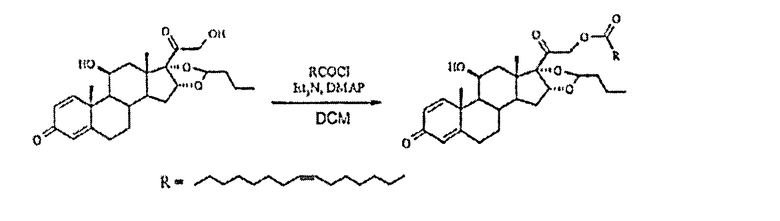
Получали хлорид пальмитолеиновой кислоты из пальмитолеиновой кислоты (0,34 г, 1,1 ммоль), оксалилхлорида (0,21 мл, 2,5 ммоль) и DMF (каталитическое количество) в толуоле (5 мл) путем перемешивания при температуре окружающей среды в течение 1,75 ч, а затем упаривания до сухого состояния. Осадок растворяли в DCM (2 мл) и капельно добавляли к суспензии будесонида (0,2 г, 0,5 ммоль), TEA (0,07 мл, 0,5 ммоль) и DMAP (каталитическое количество) в DCM (10 мл) в атмосфере N2 при 0°С. Охлаждающую баню удаляли и реакционную смесь перемешивали в темноте в течение 2,5 ч. Затем прозрачный раствор выливали в насыщенный водный раствор NH4Cl, фазы разделяли и водную фазу экстрагировали DCM (2x). Объединенные органические фазы промывали насыщенным водным раствором NаНСО3, рассолом, сушили (Na2SO4), фильтровали и упаривали в вакууме. Флэш-хроматография на силикагеле с элюированием смесью гексан/EtOAc (3:1) дала 309 мг (89%) указанного в заголовке соединения в виде бесцветного масла; 1H-ЯМР (200 МГц; CDCl3) δ 0,93 (м, 6Н), 1,09 (с, 3Н), 1,19 (д, 3Н), 1,34 (м), 1,5-2,6 (м), 2,86 (м), 3,15 (м, 1Н), 4,41 (м, 1Н), 4,92 (с, 2Н), 5,4 (м, 8Н), 6,15 (с, 1Н), 6,38 (дд, 1Н), 7,23 (д, 1Н); MS (электрораспыление); 701 [M+Na]+.
ВЭЖХ: 99,4%


Настоящее изобретение относится к некоторым дериватизированным ненасыщенными жирными кислотами имеющим противораковое лечебное действие глюкокортикоидам и содержащим их фармацевтическим составам. 7 н. и 19 з.п. ф-лы, 7 пр., 3 табл.
1. Соединение общей формулы (I)
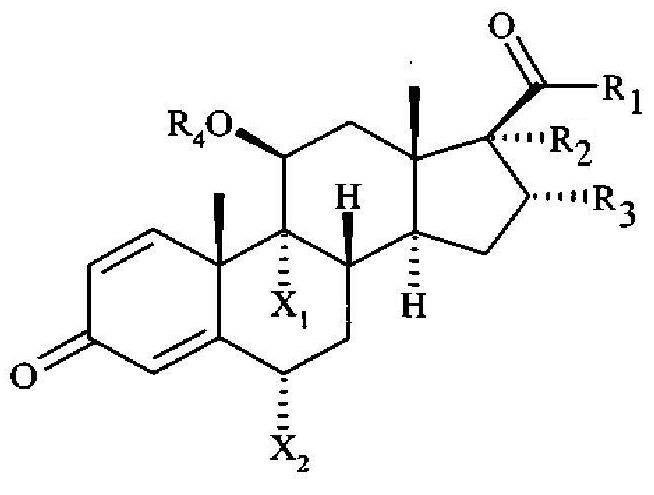
где X1 и Х2 означают F;
R1 означает SCH2F;
R2 означает OCOEt или O-R4;
R3 означает СН3;
R4 означает Н или ацильную группу C8-C24 общей формулы
CH
3
-(CH
2
)
k
-(CH=CH-CH
2
)
l
-(CH=CH)
m
-(CH
2
)
n
-CO, (II)
где k - целое число от 0 до 10; 1 - целое число от 0 до 6; m - целое число от 0 до 1; n - целое число от 2 до 7;
при условии, что в соединении имеется, по меньшей мере, одна ацильная группа формулы (II),
или его фармацевтически приемлемая соль.
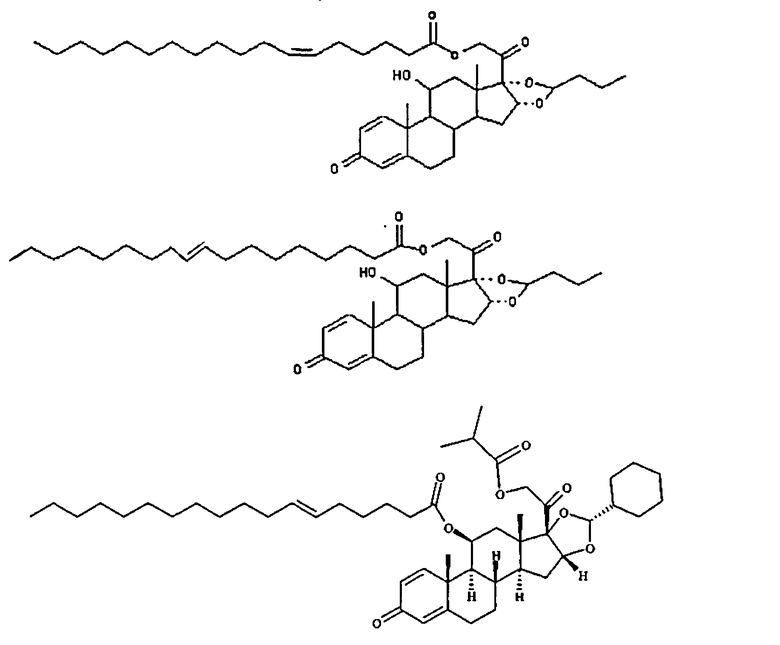
2. Соединение по п.1, определенное как
или
или
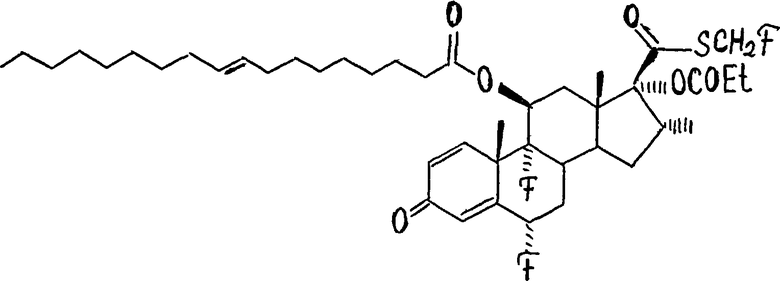
3. Соединение формулы (I) по п.1, в котором:
X1 и X2 означают F;
R1 означает O-R4;
R2 означает O-R4;
R3 означает СН3;
R4 означает Н или ацильную группу C8-C24 общей формулы
CH
3
-(CH
2
)
k
-(CH=CH-CH
2
)
l
-(CH=CH)
m
-(CH
2
)
n
-CO, (II)
где k - целое число от 0 до 10; 1 - целое число от 0 до 6; m - целое число от 0 до 1; n - целое число от 2 до 7,
при условии, что в соединении имеется, по меньшей мере, одна ацильная группа общей формулы (II),
или его фармацевтически приемлемая соль.
4. Соединение формулы (I) по п.1, в котором:
X1 и Х2 означают Н;
R1 означает ОСН2СН3, ОСH2Cl или O-R4;
R2 означает OCO2CH2СН3, ОСОСНСl2 или O-R4;
R3 означает Н;
R4 означает Н или ацильную группу C8-C24 общей формулы
CH
3
-(CH
2
)
k
-(CH=CH-CH
2
)
l
-(CH=CH)
m
-(CH
2
)
n
-CO, (II)
где k - целое число от 0 до 10; 1 - целое число от 0 до 6; m - целое число от 0 до 1; n - целое число от 2 до 7,
при условии, что в соединении имеется, по меньшей мере, одна ацильная группа общей формулы (II),
или его фармацевтически приемлемая соль.
5. Соединение формулы (I) по п.1 или 4, в котором ацильная группа формулы (II) представляет собой остаток элаидиновой кислоты.
6. Соединение, определяемое как
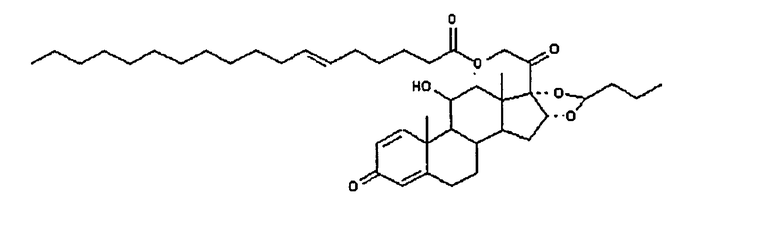
7. Соединение формулы (I)
в которой X1 и Х2 означают Н или F;
R1 означает ОН, ОCH2СН3, ОСН2Сl, SCH2F, СН2OСОСН(СН3)2, CH2-O-R4 или О-R4;
R2 означает OCOEt, OCOCHCl2, ОСO2СН2СН3 или O-R4;
R3 означает СН3 или
R2 и R3, вместе с атомами углерода, с которыми они связаны, образуют 2-R5,R6-1,3-диоксолановое кольцо, где R5 и R6 независимо друг от друга означают Н, алкил C1-С6 или циклоалкил С3-С6;
R4 означает Н или ацильную группу С8-С24 общей формулы
CH
3
-(CH
2
)
k
-(CH=CH-CH
2
)
l
-(CH=CH)
m
-(CH
2
)
n
-CO, (II)
где k - целое число от 0 до 10; 1 - целое число от 0 до 6; m - целое число от 0 до 1; n - целое число от 2 до 7,
со следующими условиями:
- в соединении имеется, по меньшей мере, одна ацильная группа общей формулы (II),
или его фармацевтически приемлемая соль для лечения рака.
8. Соединение по п.7 или его фармацевтически приемлемая соль для лечения рака крови.
9. Соединение по п.7 или его фармацевтически приемлемая соль для лечения стероидрезистентного рака.
10. Соединение по п.7 или его фармацевтически приемлемая соль для применения во вспомогательной и/или паллиативной терапии.
11. Применение соединения формулы (I)
в которой
X1 и Х2 независимо друг от друга означают Н или F;
R1 означает ОН, ОСН2СН3, ОСН2Сl, SCH2F, СН2OСОСН(СН3)2, CH2-O-R4 или O-R4;
R2 означает OCOEt, OCOCHCl2, ОСO2СН2СН3 или O-R4;
R3 означает Н, СН3 или ОН или
R2 и R3 вместе с атомами углерода, с которыми они связаны, образуют 2-R5,R6-1,3-диоксолановое кольцо, где R5 и R6 независимо друг от друга означают Н, алкил C1-С6 или циклоалкил С3-С6;
R4 означает Н или ацильную группу С8-С24 общей формулы
CH
3
-(CH
2
)
k
-(CH=CH-CH
2
)
l
-(CH=CH)
m
-(CH
2
)
n
-CO, (II)
где k - целое число от 0 до 10; 1 - целое число от 0 до 6; m - целое число от 0 до 1; n - целое число от 2 до 7,
при условии, что
- в соединении имеется, по меньшей мере, одна ацильная группа общей
формулы (II),
или его фармацевтически приемлемой соли для изготовления фармацевтической композиции для лечения рака.
12. Применение по п.11 для изготовления фармацевтической композиции для лечения рака крови.
13. Применение по п.11 для изготовления фармацевтической композиции для лечения стероидрезистентного рака.
14. Применение по п.11 для изготовления фармацевтической композиции для использования во вспомогательной и/или паллиативной терапии.
15. Соединение формулы (I) по любому из пп.1-6 или его фармацевтически приемлемая соль для лечения воспаления.
16. Соединение по п.15 или его фармацевтически приемлемая соль для лечения стероидрезистентного воспаления.
17. Применение соединения формулы (I) по любому из пп.1-6 или его фармацевтически приемлемой соли для изготовления фармацевтической композиции для лечения воспаления.
18. Применение по п.17 для изготовления фармацевтической композиции для лечения стероидрезистентного воспаления.
19. Соединение формулы (I) по любому из пп.1-6 или его фармацевтически приемлемой соли для лечения хронического обструктивного заболевания легких (COPD).
20. Применение соединения формулы (I) по любому одному из пп.1-6 или его фармацевтически приемлемой соли для изготовления фармацевтической композиции для лечения хронического обструктивного заболевания легких (COPD).
21. Способ лечения рака у пациента, нуждающегося в таком лечении, включающий введение указанному пациенту терапевтически эффективного количества соединения формулы (I) по п.11 или его фармацевтически приемлемой соли.
22. Способ по п.21, в котором подлежащее лечению заболевание представляет собой рак крови.
23. Способ по п.21, в котором подлежащее лечению заболевание представляет собой стероидрезистентный рак.
24. Способ лечения воспаления у пациента, нуждающегося в таком лечении, включающий введение указанному пациенту терапевтически эффективного количества соединения формулы (I) по любому из пп.1-6 или его фармацевтически приемлемой соли.
25. Способ по п.24, в котором подлежащее лечению заболевание представляет собой стероидрезистентное воспаление.
26. Фармацевтическая композиция для леченияч рака, воспаления и хронического обструктивного заболевания легких, содержащая эффективное количество соединения формулы (I) по любому из пп.1-6 и фармацевтически приемлемые вспомогательные вещества, носители и/или разбавители.
| MILLER-LARSSON et al | |||
| Drug Metabolism and Disposition, v | |||
| Прибор для получения стереоскопических впечатлений от двух изображений различного масштаба | 1917 |
|
SU26A1 |
| Способ и аппарат для получения гидразобензола или его гомологов | 1922 |
|
SU1998A1 |
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| TESHIMA MUGEN et al | |||
| Biological & Pharm | |||
| Bull | |||
| v | |||
| Солесос | 1922 |
|
SU29A1 |
| HASHIGUCHI Т | |||
| et al | |||
| Biological & Pharm | |||
| Bull | |||
| v | |||
| Выбрасывающий ячеистый аппарат для рядовых сеялок | 1922 |
|
SU21A1 |

