Новый способ
Данное изобретение относится к новому способу получения пропионата флутиказона, в особенности пропионата флутиказона в виде кристаллической полиморфной Формы 1.
Пропионат флутиказона представляет собой кортикостероид семейства андростана, который обладает сильным противовоспалительным действием и общеизвестен в качестве полезного терапевтического агента для лечения воспалительных и аллергических состояний, таких как ринит, астма и хроническая обструктивная болезнь легких (COPD). Химическая структура пропионата флутиказона такая, как представлена ниже:

Впервые пропионат флутиказона и способы его получения описаны в патенте Великобритании 2088877 (см. Пример 14). Кристаллическую форму пропионата флутиказона, обозначаемую как полиморфная Форма 1, можно получить растворением неочищенного продукта (полученного, например, как описано в GB 2088877) в этилацетате с последующей перекристаллизацией. Также было показано, что стандартные методы распылительной сушки приводят к образованию Формы 1 пропионата флутиказона. Также описана прямая перекристаллизация пропионата флутиказона из ацетона (Phillips et al (1994), J. Med. Chem. 37, 3717-3729). В международной заявке на патент WO 98/17676 описана новая полиморфная форма пропионата флутиказона, обозначаемая как полиморфная Форма 2, которую можно получить методами сверхкритической жидкости. Модификация сверхкритических условий также смогла привести к образованию полиморфной Формы 1. Примеры условий, описанные в WO 98/17676 для получения Формы 1, включают в себя совместное введение диоксида углерода и раствора пропионата флутиказона, растворенного в ацетоне через коаксиальное сопло в сосуд для образования частиц. Полиморфные Формы 1 и 2 пропионата флутиказона можно различить по их картинам дифракции рентгеновских лучей на порошке (XRPD) и размерам элементарной ячейки. Например, кристаллическая полиморфная Форма 1 обладает моноклинной кристаллической структурой со следующими параметрами:
а=7,722 Å, b=14,176 Å, с=11,290 Å, β=98,458°.
Наоборот, кристаллическая полиморфная модификация Форма 2 имеет орторомбическую кристаллическую структуру со следующими параметрами:
а=23,404 Å, b=14,048 Å, с=7,695 Å, все углы = 90°.
Характеристические пики в картинах XRPD приведены в следующей таблице.
Картины XRPD двух полиморфных модификаций показаны на Фиг.1.
В международной заявке на патент WO 00/38811 (Glaxo Group) раскрыта кристаллизация пропионата флутиказона, растворенного в ацетоне, путем смешивания с водой в присутствии ультразвукового излучения. В международной заявке на патент WO 01/32125 (Glaxo Group) раскрыта кристаллизация пропионата флутиказона при поступлении потока раствора пропионата флутиказона в ацетоне и потока воды в качестве антирастворителя по касательной в цилиндрическую камеру для смешивания, имеющую осевой выпускной канал, так что указанные потоки таким образом тщательно смешиваются вследствие образования "вихря". В настоящее время авторы изобрели новый способ получения пропионата флутиказона в виде кристаллической полиморфной Формы 1.
Таким образом, согласно данному изобретению предложен способ (в дальнейшем называемый "основным способом") получения пропионата флутиказона в виде кристаллической полиморфной Формы 1, при котором раствор пропионата флутиказона в несольватирующем жидком органическом растворителе смешивают с несольватирующим жидким органическим антирастворителем, что приводит к кристаллизации пропионата флутиказона из раствора в виде кристаллической полиморфной Формы 1.
Предпочтительно данный способ осуществляют при таком атмосферном давлении окружающей среды, чтобы при этом атмосферном давлении окружающей среды растворитель и антирастворитель являлись жидкостями.
Несольватирующий жидкий органический растворитель и несольватирующий жидкий органический антирастворитель представляют собой жидкости, которые не имеют тенденции к образованию кристаллических сольватов с пропионатом флутиказона. Примеры несольватирующих жидких органических растворителей включают в себя метилацетат, этилацетат и пентанон, например пентан-3-он, особенно этилацетат, в еще одном особом случае - пентан-3-он. Примеры несольватирующих жидких органических антирастворителей включают в себя толуол, изооктан или гексан, в частности гексан, в более частном случае - толуол. Вода по определению не является органическим жидким антирастворителем.
Способ по изобретению имеет преимущества по сравнению со способами, известными из уровня техники, например: его можно осуществлять при давлении окружающей среды без необходимости применения сложной аппаратуры, например, чтобы сделать возможным использование сверхкритического диоксида углерода, или применения ультразвука, или для создания условий "вихревого" смешивания. Также в одном из аспектов данного изобретения допускается получение пропионата флутиказона в виде кристаллической полиморфной Формы 1 непосредственно из предшественников пропионата флутиказона без необходимости выделения пропионата флутиказона в по существу чистой форме на промежуточной стадии этого способа. Таким образом, данный способ имеет экономические и дополнительные технологические преимущества, поскольку число стадий синтеза может быть уменьшено.
В одном из способов получения пропионата флутиказона (в дальнейшем упоминаемом как "способ (а)") осуществляют взаимодействие соединения формулы (II)

или его соли с соединением формулы LCH2F, где L представляет собой уходящую группу мезил, тозил или галоген, например Cl, Br или I. Предпочтительно L представляет собой галоген, в особенности Br.
Благоприятные условия для осуществления этой стадии, которая является изобретением авторов, включают в себя объединение соединения формулы (II) или его соли, возможно в присутствии катализатора межфазного переноса, с не смешивающимся с водой несольватирующим органическим растворителем и водой. Если применяют соединение формулы (II) в виде свободной кислоты, то для удаления протона в -SH группе также следует добавлять основание. Подходящие основания включают в себя три-н-пропиламин, трибутиламин, бикарбонат натрия, триэтиламин, диизопропилэтиламин и им подобные.
Предпочтительно не смешивающийся с водой несольватирующий органический растворитель представляет собой несольватирующий органический растворитель, выбранный из списка, приведенного выше. Чтобы использовать преимущества способа (а), несольватирующий жидкий органический растворитель, применяемый в основном способе, предпочтительно представляет собой такой же растворитель, т.е. он не смешивается с водой, и наиболее предпочтительно представляет собой этилацетат, еще более предпочтительно - пентан-3-он. Это позволяет избежать необходимости проведения стадии выделения.
При использовании в данном изобретении по существу не смешивающиеся растворители обеспечивают образование двух фаз при смешивании и наличии низкого уровня растворимости одного в другом, например когда растворимость одного растворителя в другом растворителе составляет менее 10% (масс./масс.), в частности менее 5% (масс./масс.). Примеры катализаторов межфазного переноса, которые можно использовать, включают в себя бромид тетрабутиламмония, хлорид тетрабутиламмония, бромид бензилтрибутиламмония, хлорид бензилтрибутиламмония, бромид бензилтриэтиламмония, хлорид метилтрибутиламмония и хлорид метилтриоктиламмония, предпочтительно хлорид бензилтрибутиламмония, бромид бензилтриэтиламмония, особенно хлорид бензилтрибутиламмония.
Предпочтительно, катализатор межфазного переноса применяют в количестве 1-15 молярных % от соединения формулы (II), главным образом 5-12%, в частности около 10%.
К преимуществам данного способа относится тот факт, что присутствие катализатора межфазного переноса в реакционной смеси приводит к значительно более высокой скорости реакции по сравнению со скоростью реакции в его отсутствии. К другим преимуществам изобретения относится тот факт, что пропионат флутиказона в виде кристаллической полиморфной Формы 1 может быть выделен непосредственно из растворителя для алкилирования без необходимости промежуточной стадии выделения.
Еще одним преимуществом является то, что применение метилэтилкетона или добавление метилэтилкетона к этилацетату увеличивает скорость реакции.
Чистоту продукта пропионата флутиказона, присутствующего в органической фазе после осуществления способа (а), предпочтительно увеличивают промывкой сначала водным раствором кислоты, например разведенной HCl, для удаления аминных соединений, таких как триэтиламин и диизопропилэтиламин (которые могут присутствовать после осуществления способа (б), описанного ниже), и затем водным раствором основания, например бикарбоната натрия, для удаления любого непрореагировавшего предшественника соединения формулы (II). Промытый органический слой (который можно отделить от более плотного водного слоя традиционными способами, например путем его отбора) предпочтительно концентрируют перегонкой (возможно при пониженном давлении) перед обработкой органическим антирастворителем согласно основному способу, описанному выше.
Таким образом, в данном изобретении предложен способ получения пропионата флутиказона в виде кристаллической полиморфной Формы 1, включающий стадии, на которых
(а) осуществляют взаимодействие соединения формулы (II)

или его соли с соединением формулы LCH2F, где L представляет собой уходящую группу, возможно, в присутствии катализатора межфазного переноса, не смешивающегося с водой несольватирующего органического жидкого растворителя и воды;
(б) возможно повышают чистоту пропионата флутиказона в органическом слое, осуществляя одну или более чем одну стадию промывки водой;
(в) отделяют органический слой от водного слоя и возможно концентрируют пропионат флутиказона в органическом слое;
(г) смешивают раствор пропионата флутиказона в не смешивающемся с водой несольватирующем органическом жидком растворителе, присутствующем в органическом слое, с несольватирующим органическим жидким антирастворителем, тем самым вызывая кристаллизацию пропионата флутиказона из раствора в виде кристаллической полиморфной Формы 1.
В соответствии со способами, известными из уровня техники, например, описанными в G.H.Phillips et al (1994), J. Med. Chem. 37, 3717-3729 и патенте США 4335121 (Glaxo Group Limited), соединения формулы (II) могут быть получены взаимодействием соединения формулы (III)
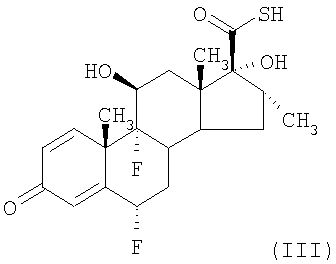
с активированным производным пропионовой кислоты, например пропионилхлоридом. Активированное производное пропионовой кислоты обычно применяют, по меньшей мере в двукратном молярном количестве относительно соединения формулы (III), поскольку один моль реагента взаимодействует с тиокислотной группировкой и требуется, что он был удален, например, взаимодействием с амином, например диэтиламином. Однако этот способ получения соединения формулы (II) обладает недостатками, состоящими в том, что получаемое в результате соединение формулы (II) нелегко очистить от загрязняющего его побочного продукта N,N-диэтилпропанамида. Таким образом, авторы изобрели улучшенный способ осуществления превращения для получения соединения формулы (II). Другие способы получения пропионата флутиказона и родственных соединений описаны в заявке на патент Израиля 109656 (Chemagis), WO 01/62722 (Abbott) и в Kertesz and Marx (1986), J.Org.Chem., 51, 2315-2328).
Таким образом, согласно изобретению также предложен способ получения (в дальнейшем "способ (б)") соединения формулы (II)

или его соли,
при котором:
(а) соединение формулы (III)
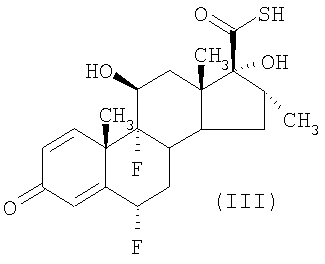
подвергают взаимодействию с активированным производным пропионовой кислоты в количестве по меньшей мере 1,3 моль, приемлемо по меньшей мере 2 моль активированного производного на моль соединения формулы (III); и
(б) удаляют пропионильную группировку, связанную с атомом серы, из любого соединения формулы (IIA),
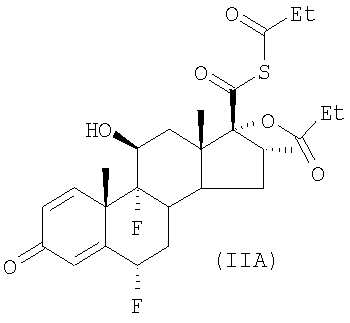
полученного таким образом, путем взаимодействия продукта стадии (а) с органическим основанием, являющимся первичным или вторичным амином, способным к образованию водорастворимого пропанамида, который затем можно удалить при помощи одной или более чем одной стадии промывки водой.
На стадии (а) примеры активированных производных пропионовой кислоты включают в себя активированные сложные эфиры или предпочтительно пропилгалогенид, такой как пропионилхлорид. Эту реакцию традиционно осуществляют в присутствии химически инертного органического основания, такого как триС1-4алкиламин, например триэтиламин, трипропиламин или трибутиламин, особенно триэтиламин, главным образом, три-н-пропиламин. Растворители для этого способа включают в себя по существу не смешивающиеся с водой жидкие органические растворители, такие как этилацетат или метилацетат, либо смешивающиеся с водой жидкие органические растворители, такие как ацетон, N,N-диметилформамид или N,N-диметилацетамид, особенно ацетон.
На стадии (б) примеры органического основания, представляющего собой первичный или вторичный амин, способного к образованию водорастворимого пропанамида, включают в себя амины, которые являются более полярными, чем диэтиламин, например аминоспирт, например диэтаноламин, или диамин, например N-метилпиперазин. Предпочтительно применяют N-метилпиперазин. Может быть удобным растворение амина в небольшом количестве органического растворителя, такого как метанол. Стадии промывки водой удобно осуществлять, используя воду или разведенную кислоту, например разведенную соляную или уксусную кислоту. На этих стадиях более тяжелый водный слой можно отделять от органического слоя традиционными методами, например путем отбора под действием силы тяжести.
Предпочтительно, стадии (а) и (б) осуществляют при пониженной температуре, например 0-5°С.
Соединение формулы (II), полученное способом (б), может быть выделено традиционными методами. Однако предпочтительно, для извлечения максимальной пользы из изобретения, чтобы растворитель, применяемый на стадии (а) этого способа, преимущественно представлял собой по существу не смешивающийся с водой жидкий органический растворитель, более предпочтительно, такой же растворитель, что и применяемый на стадии (а), и еще более предпочтительно, такой же растворитель, который также применяется в основном способе. Таким образом, выделение соединения формулы (II) в твердой форме после осуществления способа (б) не является необходимым, хотя может быть предпочтительным концентрировать соединение формулы (II) в органическом растворителе традиционными методами, например перегонкой, возможно при пониженном давлении. Так, наиболее предпочтительным растворителем, применяемым на стадии (а), является этилацетат, еще более предпочтительным является пентан-3-он.
Дополнительные общие условия, относящиеся к превращению соединения формулы (III) в соединение формулы (II) и его соли и к выделению конечного продукта, хорошо известны специалистам в данной области.
Если планируется выделение соединения формулы (II) (хотя, как отмечено выше, главной особенностью всего изобретения является то, что такое выделение не представляется необходимым), то его можно преимущественно выделять в форме твердой кристаллической соли, а не свободного соединения формулы (II). Предпочтительная соль образована основанием, таким как диизопропилэтиламин, триэтиламин, 2,6-диметилпиридин, N-этилпиперидин, или калием, главным образом триэтиламином. Такие солевые формы соединения формулы (II) более стабильны, их легче отфильтровывать и сушить, и они могут быть выделены с более высокой чистотой, чем свободное соединение формулы (II). Наиболее предпочтительной солью является соль, образованная триэтиламином. Калиевая соль также представляет интерес.
Преимуществом является то, что соединение формулы (II) может быть выделено с более высокой эффективностью, чем посредством известных из уровня техники способов. Например, способ получения соединения формулы (II), описанный G.H.Phillips et al (J.Med.Chem. 37, 3717-3729 (1994)), включает в себя выделение продукта из системы ацетон/вода. Полученный таким образом продукт чрезвычайно сложно фильтровать. Напротив, соединение формулы (II), когда оно получено по настоящему изобретению, фильтровать гораздо легче. Более того, способ по настоящему изобретению также может предложить улучшения по чистоте.
Один из таких способов включает в себя обработку органической фазы, содержащей соединение формулы (II), основанием, с тем, чтобы соединение формулы (II) выпало в осадок в форме твердой кристаллической соли. Примеры оснований включают в себя триэтиламин, 2,6-диметилпиридин, N-этилпиперидин или калиевую соль со свойствами основания, например гидрокарбонат калия. Авторы заявляют соединение формулы (II), выделенное в форме твердой кристаллической соли, в качестве дополнительного аспекта данного изобретения.
Кроме того, выгодно получать и применять соединение формулы (III) в виде имидазольной соли. В G.H.Phillips et al (1994), J.Med.Chem. 37, 3717-3729 раскрыто получение соединения формулы (III) из соединения формулы (IV). Однако из-за физико-химических свойств соединения формулы (III), полученного таким образом, продукт имеет очень низкую скорость фильтрации. Преимущества получения имидазольной соли соединения формулы (III) включают в себя ее свойства легко получаемого и быстро фильтруемого, легко обрабатываемого и сохраняемого источника соединения формулы (III), которое может быть без труда получено из этой соли путем подкисления, например, соляной кислотой. Кроме того, соединение формулы (III), полученное таким образом, имеет более высокую чистоту. Имидазольную соль соединения формулы (III) можно получать, выделять и хранить для последующего применения в способе получения соединения формулы (II), описанном в данной заявке. Или же имидазольную соль соединения формулы (III) можно получать и применять в последующем превращении в соединение формулы (II) непосредственно в форме влажного осадка, избегая таким образом необходимости сушить эту имидазольную соль перед дальнейшим взаимодействием.
Считается, что имидазольная соль соединения формулы (III) является новой и, следовательно, образует дополнительный аспект изобретения. Также дополнительно предлагается способ получения имидазольной соли соединения формулы (III), при котором осуществляют взаимодействие соединения формулы (IV) с карбонилдиимидазолом и сероводородом.
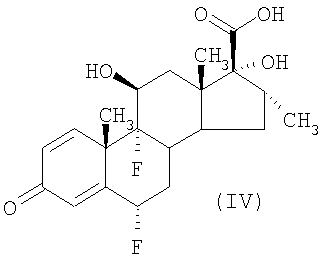
Обычно соединение формулы (IV) и 1,1-2,5 эквивалентов, приемлемо 1,8 эквивалента, карбонилдиимидазола перемешивают в подходящем растворителе, например этилацетате, содержащем от 0 до 2 объемов, приемлемо 0,5 объема, N,N-диметилформамида, при подходящей температуре, например 18-20°С, в течение подходящего периода времени, например одного часа. Полученную суспензию охлаждают до подходящей температуры, например до температуры от -5°С до 5°С, приемлемо от -3°С до 3°С, и при перемешивании суспензии вводят газообразный сероводород за период времени 15-60 минут, приемлемо за 20-30 минут. Реакционную смесь перемешивают в течение еще приблизительно 30 минут при температуре от -5°С до 5°С, нагревают до приблизительно 10°С за период времени приблизительно 20 минут и перемешивают при 6-12°С в течение 90-120 минут. Затем продукт выделяют фильтрацией при подходящей температуре, приемлемо при 5-25°С, предпочтительно при 10-15°С, промывают подходящим растворителем, например этилацетатом, и сушат под вакуумом с образованием имидазольной соли соединения формулы (III).
Соединение формулы (III) представляет собой одноосновную кислоту и поэтому, как ожидается, образует имидазольную соль со стехиометрическим отношением имидазольной солеобразующей группировки к соединению формулы (III) приблизительно 1:1. Однако неожиданно было обнаружено, что стехиометрическое отношение имидазольной солеобразующей группировки к соединению формулы (III) может быть до 4:1 (включительно). Следовательно, во избежание неопределенности, термин "имидазольная соль" охватывает имидазольные соли соединения формулы (III) и ассоциаты соединения формулы (III) и имидазола, где стехиометрическое отношение имидазольной группировки к соединению формулы (III) составляет до 4:1 (включительно), например от 1:1 до 4:1, приемлемо от 1,8:1 до 2,5:1. Примером типичной стехиометрии является отношение 2:1. В контексте стехиометрических значений очевидно, что точные численные значения следует истолковывать как включающие в себя их незначительные отклонения.
Предпочтительно, соединение формулы (III), применяемое в способах, описанных здесь, применяют в виде его имидазольной соли.
Также установлено, что выгодным является получение пропионата флутиказона непосредственно из соединения формулы (IV), т.е. без выделения какого-либо промежуточного соединения. Преимущества такого способа включают в себя большую эффективность, легкость выполнения технологических операций, отсутствие необходимости работы с химическими промежуточными соединениями и их хранения. Этот способ включает в себя получение в одном и том же реакторе соединения формулы (III) из соединения формулы (IV) с последующим in situ превращением в соединение формулы (II) и последующим взаимодействием соединения формулы (II) (без выделения, как описано выше) с образованием пропионата флутиказона.
Следовательно, предлагается способ получения пропионата флутиказона из соединения формулы (IV) без выделения промежуточных соединений, при котором получают соединение формулы (III) или его соль из соединения формулы (IV), затем in situ превращают соединение формулы (III) в соединение формулы (II) и затем in situ превращают соединение формулы (II) в пропионат флутиказона.
Обычно к тщательно перемешанной смеси соединения формулы (IV) и N,N'-карбонилдиимидазола (6,3 г) последовательно добавляют подходящие растворители, например смесь этилацетата и N,N-диметилформамида. Образовавшуюся суспензию перемешивают при подходящей температуре, например при 18-20°С, в течение приблизительно одного часа с образованием бледно-желтого раствора. Затем раствор охлаждают до температуры от -5°С до 25°С, приемлемо от -3°С до 3°С, и в течение 15-60 минут, приемлемо 20-30 минут, через раствор барботируют сероводород, поддерживая температуру содержимого 12±2°С. Полученную суспензию перемешивают при 12±2°С в течение еще 90-120 минут, а затем в условиях умеренного вакуума, медленно выпуская азот, интенсивно перемешивают в течение приблизительно одного часа. Затем сбрасывают вакуум, и сосуд продувают инертным газом, приемлемо азотом. Затем добавляют подходящий растворитель, например 3-пентанон, и суспензию промывают водной минеральной кислотой, например 2М соляной кислотой, и потом водой. Полученный раствор охлаждают до 0-5°С и за 2 минуты добавляют трипропиламин, обеспечивая сохранение температуры реакционной смеси 0-5°С. Раствор перемешивают при 0-5°С и приблизительно за 5 минут добавляют пропионилхлорид, поддерживая температуру реакционной смеси 0-5°С. Затем раствор оставляют нагреваться до приблизительно 10°С и перемешивают при этой температуре в течение 90-120 минут. Затем раствор охлаждают до 0-5°С и добавляют 1-метилпиперазин, поддерживая температуру реакционной смеси 0-5°С. Раствор перемешивают при 0-5°С в течение приблизительно 20 минут, а затем одной порцией добавляют бромфторметан. Раствор оставляют нагреваться до температуры окружающей среды и перемешивают в течение 16 часов. Затем раствор последовательно промывают разбавленной минеральной кислотой, например 1М HCl, водой, водным раствором основания, например 1%-ным NaHCO3, и водой, а потом концентрируют перегонкой при атмосферном давлении до приблизительно 80 мл и затем охлаждают до 90°С. В раствор вносят затравку пропионата флутиказона (Форма 1) и затем добавляют подходящий антирастворитель, например толуол, поддерживая температуру суспензии приблизительно 90°С. Затем суспензию концентрируют перегонкой при атмосферном давлении до приблизительно 8 об. Затем суспензию охлаждают до приблизительно 90°С. Затем за 20 минут добавляют толуол, поддерживая температуру суспензии приблизительно 90°С. Затем за 90 минут суспензию охлаждают до приблизительно 10°С, выдерживают при приблизительно 10°С в течение приблизительно одного часа и потом фильтруют. Осадок на фильтре промывают подходящим растворителем, например смесью 3-пентанона и толуола (1:4), и сушат отсасыванием. Затем твердое вещество сушат традиционными методами.
Изобретение иллюстрируется следующими Примерами.
ПРИМЕРЫ
Общие методы
1H-ЯМР (ядерного магнитного резонанса) спектры регистрировали при 400 МГц и химические сдвиги выражены в ppm (м.д. (миллионных долях)) относительно тетраметилсилана. Для описания мультиплетности сигналов применены следующие сокращения: s (синглет), d (дублет), t (триплет), q (квартет), m (мультиплет), dd (дублет дублетов), ddd (дублет дублета дублетов), dt (дублет триплетов) и b (широкая полоса).
LCMS (сочетание жидкостной хроматографии и масс-спектрометрии) осуществляли на следующей колонке: 25 см ×0,46 см, 5 мкм Inertsil ODS-2, элюируя смесью 58% {0,1% муравьиной кислоты в 3%-ном метаноле (водном)} растворителя А и 42% {0,1% муравьиной кислоты в 3%-ном метаноле (в ацетонитриле)} растворителя Б с использованием следующего градиента элюирования: 0-40 минут - 42% Б, 40-60 минут - 53% Б, 60-75 минут - 87% Б, 75-85 минут - 42% Б, при скорости потока 1 мл/мин. Масс-спектры регистрировали на HP LC/MSD спектрометре с ионизацией на положительные и отрицательные ионы электрораспылением (ES+ve и ES-ve).
Жидкостную хроматографию (Метод А) осуществляли на колонке 25 см × 0,46 см (внутренний диаметр), заполненной 5 мкм Inertsil ODS-2, элюируя следующими подкисленными подвижными фазами:
Раствор А: подкисленный ацетонитрил:подкисленный метанол:подкисленная вода (42:3:55)
Раствор Б: подкисленный ацетонитрил:подкисленный метанол:подкисленная вода (53:3:44)
Раствор В: подкисленный ацетонитрил:подкисленный метанол:подкисленная вода (87:3:10)
{где подкисленный ацетонитрил включает в себя 0,05% (об./об.) фосфорной кислоты в ацетонитриле (0,5 мл в 1000 мл), подкисленный метанол включает в себя 0,05% (об./об.) фосфорной кислоты в метаноле (0,5 мл в 1000 мл) и подкисленная вода включает в себя 0,05% (об./об.) фосфорной кислоты в воде (0,5 мл в 1000 мл)}.
Элюирование проводили с использованием следующего градиента: 0-40 минут - раствор А (100%), 40-60 минут - раствор Б (100%), 60-75 минут - раствор В (100%) и 75-90 минут - раствор А (100%) при скорости потока 1,0 мл/мин и температуре термостата 40°С.
Жидкостную хроматографию (Метод Б) осуществляли на колонке из нержавеющей стали (20 см ×0,46 см внутренний диаметр, 5 мкм Octyl), элюируя следующими подкисленными подвижными фазами:
Раствор А: ацетонитрил: 0,05М водный раствор дигидроортофосфата аммония (35:65, об./об.)
Раствор Б: ацетонитрил: 0,05М водный раствор дигидроортофосфата аммония (70:30, об./об.).
Элюирование проводили с использованием следующего градиента: 0-15 минут - раствор А (100%), 15-40 минут - раствор Б (100%), 40-45 минут - раствор Б (100%) и 45-60 минут - раствор А (100%) при скорости потока 1,5 мл/мин и температуре термостата 30°С.
Точки плавления получали с использованием прибора для определения точки плавления Mettler Toledo FP62.
Картины XRPD получали с использованием порошкового дифрактометра Phillips X'pert MPD.
Промежуточные соединения
Промежуточное соединение 1: 6α,9α-дифтор-17α-(1-оксопропокси)-11β-гидрокси-16α-метил-3-оксо-андроста-1,4-диен-17β-карботионовая кислота
Раствор 6α,9α-дифтор-11β,17α-дигидрокси-16α-метил-3-оксо-андроста-1,4-диен-17β-карботионовой кислоты (полученной в соответствии с методикой, описанной в GB 2088877В) (7 г) в ацетоне (73,6 мл) обрабатывают триэтиламином (10,9 мл) при температуре от -5°С до -6°С в течение приблизительно 15 минут. Во время добавления и последующей промывки ацетоном (2,8 мл) раствор перемешивают при температуре от -5°С до 0°С. Полученную в результате суспензию затем охлаждают до температуры от -3°С до -7°С и в течение приблизительно 30 минут обрабатывают пропионилхлоридом (6,2 мл), поддерживая температуру реакционной смеси от -5°С до +2°С. В качестве промывки добавляют ацетон (2,8 мл) и раствор перемешивают при температуре от -5°С до +2°С в течение еще двух часов. Полученную в результате суспензию затем охлаждают до температуры от -3°С до -7°С и в течение приблизительно 30 минут обрабатывают диэтаноламином (23,8 мл) в метаноле (20 мл), поддерживая температуру от -5°С до +2°С. В качестве промывки добавляют ацетон (2,8 мл) и раствор перемешивают при температуре от -5°С до +2°С в течение дополнительных 30 минут. Смесь гасят в воде (135 мл), поддерживая температуру от -5°С до +5°С. В качестве промывки добавляют ацетон (5,6 мл) и смесь охлаждают до 0-5°С. За период времени от одного часа до двух часов добавляют концентрированную соляную кислоту (65 мл), поддерживая температуру в интервале от 0°С до 5°С, затем добавляют воду (125 мл), поддерживая температуру менее 5°С. Смесь перемешивают при 0-5°С в течение 15 минут, продукт отфильтровывают, промывают водой и сушат под вакуумом при приблизительно 45°С в течение 18 часов с образованием указанного в заголовке соединения в виде твердого вещества от белого до не совсем белого цвета (7,91 г, 99,5%).
ВЭЖХ (высокоэффективная жидкостная хроматография): время удерживания 27,225 мин, m/z 469,2 (положительный молекулярный ион) и m/z 467,2 (отрицательный молекулярный ион).
ЯМР (ДМСОd6 (диметилсульфоксид)) 7.27 (1Н, d, 10 Гц), 6.34 (1Н, d, 10 Гц), 6.14 (1Н, s), 5.31 (1Н, d), 5.17 (1Н, ddd), 4.27 (1Н, m), 2.40 (2Н, q, 7 Гц), 2.00-2.14 (5Н, m), 1.85 (1Н, m), 1.65 (1Н, m), 1.51 (3Н, s), 1.14 (3Н, s), 1.05 (3Н, t, 7 Гц), 0.88 (3Н, d, 7 Гц).
Пример 1: S-Фторметиловый эфир 6α,9α-дифтор-17α-(1-оксопропокси)-11β-гидрокси-16α-метил-3-оксо-андроста-1,4-диен-17β-карботионовой кислоты (с использованием этилацетата в качестве растворителя и гексана в качестве антирастворителя)
Вакуумной перегонкой уменьшают объем раствора S-фторметилового эфира 6α,9α-дифтор-17α-(1-оксопропокси)-11β-гидрокси-16α-метил-3-оксо-андроста-1,4-диен-17β-карботионовой кислоты (5,5 г) (полученного в соответствии с методикой, описанной Philips et al в J.Med.Chem. 37, 3717-3729 (1994)) в этилацетате (300 мл) до приблизительно 32 мл, и в течение по меньшей мере 30 минут этот раствор обрабатывают гексаном (63 мл), поддерживая температуру приблизительно 20°С. Смесь выдерживают при 20°С в течение приблизительно 1 часа, и выпавший осадок собирают фильтрацией, промывают смесью этилацетат/гексан 1:4 (3×5 мл) и сушат при приблизительно 50°С в течение 12 часов с образованием указанного в заголовке соединения в виде белого твердого вещества (5,06 г, 92%).
ЯМР анализ показал отсутствие сольватации продукта и XRPD анализ подтвердил, что продукт находится в полиморфной Форме 1.
ВЭЖХ: время удерживания 35,2 мин.
ЯМР (ДМСОd6) 7.29 (1Н, d, 10 Гц), 6.34 (1Н, d, 10 Гц), 6.17 (1Н, s), 6.00 (2Н, d), 5.68 (1Н, ddd), 4.26 (1Н, m), 3.40 (3Н, s), 2.40 (2Н, q, 7 Гц), 1.80-2.30 (5Н, m), 1.52 (3Н, s), 1.14 (3Н, s), 1.07 (3Н, t, 7 Гц), 1.05 (3Н, s), 0.94 (3Н, d, 7 Гц).
Пример 2: S-Фторметиловый эфир 6α,9α-дифтор-17α-(1-оксопропокси)-11β-гидрокси-16α-метил-3-оксо-андроста-1,4-диен-17β-карботионовой кислоты (с использованием этилацетата в качестве растворителя и толуола в качестве антирастворителя)
Вакуумной перегонкой уменьшают объем раствора S-фторметилового эфира 6α,9α-дифтор-17α-(1-оксопропокси)-11β-гидрокси-16α-метил-3-оксо-андроста-1,4-диен-17β-карботионовой кислоты (5,5 г) (полученного в соответствии с методикой, описанной Philips et al в J.Med.Chem. 37, 3717-3729 (1994)) в этилацетате (300 мл) до 32 мл. Реакционную смесь обрабатывают толуолом (80 мл) и затем перегоняют до общего объема реакционной смеси 32 мл. При 60°С добавляют дополнительную порцию толуола (40 мл) и смесь охлаждают до приблизительно 10°С. Смесь выдерживают при этой температуре в течение приблизительно 1 часа, и выпавший осадок собирают фильтрацией, промывают смесью толуола и этилацетата (в соотношении 5:1) и сушат при приблизительно 50°С в течение 12 часов с образованием указанного в заголовке соединения в виде белого твердого вещества (4,70 г, 85,4%).
ЯМР анализ показал отсутствие сольватации продукта и XRPD анализ подтвердил, что продукт находится в полиморфной Форме 1.
Пример 3: S-Фторметиловый эфир 6α,9α-дифтор-17α-(1-оксопропокси)-11β-гидрокси-16α-метил-3-оксо-андроста-1,4-диен-17β-карботионовой кислоты (с использованием этилацетата в качестве растворителя и изооктана в качестве антирастворителя)
Раствор S-фторметилового эфира 6α,9α-дифтор-17α-(1-оксопропокси)-11β-гидрокси-16α-метил-3-оксо-андроста-1,4-диен-17β-карботионовой кислоты (5,5 г) (полученного в соответствии с методикой, описанной Philips et al в J.Med.Chem. 37, 3717-3729 (1994)) в этилацетате (300 мл) перегоняют до 32 мл, и в течение по меньшей мере 30 минут обрабатывают изооктаном (80 мл), поддерживая температуру смеси приблизительно 20°С. Затем смесь перегоняют до общего объема реакционной смеси приблизительно 32 мл. При 60°С добавляют дополнительную порцию изооктана (40 мл), и смесь охлаждают до приблизительно 10°С и выдерживают при этой температуре в течение приблизительно 60 минут. Выпавший осадок собирают фильтрацией, промывают смесью этилацетата и изооктана (в соотношении 5:1) и сушат при приблизительно 50°С в течение 12 часов с образованием указанного в заголовке соединения в виде белого твердого вещества (5,07 г, 92,2% от теоретического значения).
ЯМР анализ показал отсутствие сольватации продукта и XRPD анализ подтвердил, что продукт находится в полиморфной Форме 1.
Пример 4: S-Фторметиловый эфир 6α,9α-дифтор-17α-(1-оксопропокси)-11β-гидрокси-16α-метил-3-оксо-андроста-1,4-диен-17β-карботионовой кислоты (с использованием метилацетата в качестве растворителя и толуола в качестве антирастворителя)
Раствор S-фторметилового эфира 6α,9α-дифтор-17α-(1-оксопропокси)-11β-гидрокси-16α-метил-3-оксо-андроста-1,4-диен-17β-карботионовой кислоты (5,5 г) (полученного в соответствии с методикой, описанной Philips et al в J.Med.Chem. 37, 3717-3729 (1994)) в метилацетате (300 мл) перегоняют до 32 мл, и в течение по меньшей мере 30 минут обрабатывают изооктаном (80 мл), поддерживая температуру смеси приблизительно 20°С. Затем смесь перегоняют до общего объема реакционной смеси приблизительно 32 мл. При 60°С добавляют дополнительную порцию толуола (40 мл), смесь охлаждают до приблизительно 10°С и выдерживают при этой температуре в течение приблизительно 60 минут. Выпавший осадок собирают фильтрацией, промывают смесью метилацетата и толуола (в соотношении 1:5) (32 мл) и сушат при приблизительно 50°С в течение 12 часов с образованием указанного в заголовке соединения в виде белого твердого вещества (5,06 г, 92% от теоретического значения).
ЯМР анализ показал отсутствие сольватации продукта и XRPD анализ подтвердил, что продукт находится в полиморфной Форме 1.
Пример 5: S-Фторметиловый эфир 6α,9α-дифтор-17α-(1-оксопропокси)-11β-гидрокси-16α-метил-3-оксо-андроста-1,4-диен-17β-карботионовой кислоты (с использованием этилацетата в качестве растворителя и гексана в качестве антирастворителя)
Раствор 6α,9α-дифтор-17α-(1-оксопропокси)-11β-гидрокси-16α-метил-3-оксо-андроста-1,4-диен-17β-карботионовой кислоты (Промежуточное соединение 1) (3,7 г) в этилацетате (167 мл) и воде (13,5 мл) обрабатывают хлоридом бензилтрибутиламмония (0,37 г) и триэтиламином (1,3 мл) и смесь охлаждают до 5°С. Добавляют бромфторметан (0,5 мл), поддерживая температуру реакционной смеси приблизительно 5°С. Смесь за 2-3 часа нагревают до 20°С, и полученную в результате суспензию последовательно промывают 0,5М соляной кислотой (23 мл), 1%-ным (масс.) водным раствором бикарбоната натрия (3×23 мл) и водой (2×23 мл). Органический слой отделяют, и водный слой, возможно, обратно экстрагируют этилацетатом (30 мл). Объединенные органические слои перегоняют до объема приблизительно 22 мл, и добавляют дополнительное количество этилацетата (7 мл). Смесь охлаждают до приблизительно 20°С, в течение по меньшей мере 30 минут добавляют гексан (42 мл), и смесь выдерживают при 20°С в течение 15 минут. Выпавший осадок собирают фильтрацией, промывают смесью этилацетат/гексана 1:4 (3×5 мл) и сушат при приблизительно 50°С в течение 18 часов с образованием указанного в заголовке соединения в виде белого твердого вещества (3,54 г, 95,7%). ЯМР анализ показал отсутствие сольватации продукта и XRPD анализ подтвердил, что продукт находится в полиморфной Форме 1.
Этот пример подтвердил одинаковую эффективность синтеза указанного в заголовке соединения при его повторном воспроизведении с использованием бромида бензилтрибутиламмония в качестве катализатора межфазного переноса или при замене основания диизопропилэтиламином или бикарбонатом натрия.
Пример 6: S-Фторметиловый эфир 6α,9α-дифтор-17α-(1-оксопропокси)-11β-гидрокси-16α-метил-3-оксо-андроста-1,4-диен-17β-карботионовой кислоты (с использованием этилацетата в качестве растворителя и толуола в качестве антирастворителя)
Раствор 6α,9α-дифтор-17α-(1-оксопропокси)-11β-гидрокси-16α-метил-3-оксо-андроста-1,4-диен-17β-карботионовой кислоты (Промежуточное соединение 1) (3,7 г) в этилацетате (167 мл) и воде (13,5 мл) обрабатывают хлоридом бензилтрибутиламмония (0,37 г) и триэтиламином (1,3 мл), и смесь охлаждают до 5°С. Добавляют бромфторметан (0,5 мл), поддерживая температуру реакционной смеси приблизительно 5°С. Смесь за 2-3 часа нагревают до 20°С, и полученную в результате суспензию последовательно промывают 0,5М соляной кислотой (23 мл), 1%-ным (масс.) водным раствором бикарбоната натрия (3×23 мл) и водой (2×23 мл). Органический слой отделяют, и водный слой возможно обратно экстрагируют этилацетатом (30 мл). Объединенные органические слои перегоняют до объема приблизительно 22 мл, и добавляют толуол (54 мл). Затем реакционную смесь перегоняют до общего объема реакционной смеси приблизительно 22 мл. При приблизительно 60°С добавляют дополнительную порцию толуола (27 мл), и смесь охлаждают до приблизительно 10°С и выдерживают при этой температуре в течение приблизительно 1 часа. Выпавший осадок собирают фильтрацией, промывают смесью толуола и этилацетата (в соотношении 5:1) и сушат при приблизительно 50°С в течение 12 часов с образованием указанного в заголовке соединения в виде белого твердого вещества (3,1 г, 83,7%). ЯМР анализ показал отсутствие сольватации продукта, и XRPD анализ подтвердил, что продукт находится в полиморфной Форме 1.
Пример 7: S-Фторметиловый эфир 6α,9α-дифтор-17α-(1-оксопропокси)-11β-гидрокси-16α-метил-3-оксо-андроста-1,4-диен-17β-карботионовой кислоты (с использованием этилацетата в качестве растворителя и изооктана в качестве антирастворителя)
Раствор 6α,9α-дифтор-17α-(1-оксопропокси)-11β-гидрокси-16α-метил-3-оксо-андроста-1,4-диен-17β-карботионовой кислоты (3,7 г) (Промежуточное соединение 1) в этилацетате (167 мл) и воде (13,5 мл) обрабатывают хлоридом бензилтрибутиламмония (0,37 г) и триэтиламином (1,3 мл), и смесь охлаждают до 5°С. Добавляют бромфторметан (0,5 мл), поддерживая температуру реакционной смеси приблизительно 5°С. Смесь за 2-3 часа нагревают до 20°С, и полученную в результате суспензию последовательно промывают 0,5М соляной кислотой (23 мл), 1%-ным (масс.) водным раствором бикарбоната натрия (3×23 мл) и водой (2×23 мл). Органический слой отделяют, и водный слой возможно обратно экстрагируют этилацетатом (30 мл). Объединенные органические слои перегоняют до общего объема реакционной смеси приблизительно 22 мл, и в течение по меньшей мере 30 минут обрабатывают изооктаном (54 мл), поддерживая температуру приблизительно 20°С. Затем смесь перегоняют до общего объема реакционной смеси приблизительно 22 мл, охлаждают до приблизительно 10°С и выдерживают при этой температуре в течение по меньшей мере 30 минут. Выпавший осадок собирают фильтрацией, промывают смесью этилацетата и изооктана (в соотношении 1:5) и сушат при приблизительно 50°С в течение 12 часов с образованием указанного в заголовке соединения в виде белого твердого вещества (3,4 г, 92,3%). ЯМР анализ показал отсутствие сольватации продукта, и XRPD анализ подтвердил, что продукт находится в полиморфной Форме 1.
Пример 8: S-Фторметиловый эфир 6α,9α-дифтор-17α-(1-оксопропокси)-11β-гидрокси-16α-метил-3-оксо-андроста-1,4-диен-17β-карботионовой кислоты (с использованием единственного растворителя, чтобы избежать выделения промежуточной 6α,9α-дифтор-17α-(1-оксопропокси)-11β-гидрокси-16α-метил-3-оксо-андроста-1,4-диен-17β-карботионовой кислоты)
Раствор 6α,9α-дифтор-11β,17α-дигидрокси-16α-метил-3-оксо-андроста-1,4-диен-17β-карботионовой кислоты (полученной в соответствии с методикой, описанной в GB 2088877В) (7 г) в этилацетате (350 мл) перемешивают при 20-25°С и обрабатывают триэтиламином (10,9 мл) при 0-5°С в течение приблизительно 20 минут, добавляя в качестве промывки дополнительное количество этилацетата (5 мл). Полученную в результате суспензию затем охлаждают до температуры от -3°С до -7°С и в течение приблизительно 30 минут обрабатывают пропионилхлоридом (6,2 мл), поддерживая температуру от -5°С до +2°С. В качестве промывки добавляют этилацетат (5 мл), и раствор перемешивают при температуре от -5°С до +2°С в течение еще двух часов. Полученную в результате суспензию затем охлаждают до температуры от -3°С до -7°С и в течение приблизительно 30 минут обрабатывают диэтаноламином (23,8 мл) в метаноле (20 мл), поддерживая температуру смеси от -5°С до +2°С. В качестве промывки добавляют этилацетат (5 мл), и раствор перемешивают при температуре от -5°С до +2°С в течение еще 30 минут. Приблизительно за 10 минут добавляют уксусную кислоту (25 мл), поддерживая температуру в интервале от -5°С до +2°С, и полученную в результате суспензию выдерживают при температуре от -5°С до +5°С в течение по меньшей мере 10 минут. Приблизительно за 10 минут добавляют воду (30 мл), поддерживая температуру в интервале от -5°С до +2°С, и органическую фазу отделяют и промывают водой (3×50 мл). Возможно осуществляют обратную экстракцию водных фаз этилацетатом (120 мл) при температуре от -5°С до +2°С, и объединенные органические фазы концентрируют приблизительно до 45 об. вакуумной перегонкой (ниже 10°С). Приблизительно половину полученного раствора обрабатывают водой (13,5 мл), хлоридом бензилтрибутиламмония (0,37 г) и триэтиламином (1,3 мл), и смесь охлаждают до 5°С. Добавляют бромфторметан (0,5 мл), поддерживая температуру реакционной смеси приблизительно 5°С. Смесь за 2-3 часа нагревают до 20°С, и полученную в результате суспензию последовательно промывают 0,5М соляной кислотой (23 мл), 1%-ным (масс.) водным раствором бикарбоната натрия (3×23 мл) и водой (2×23 мл). Органический слой отделяют, и водный слой обратно экстрагируют этилацетатом (30 мл). Объединенные органические слои перегоняют до общего объема реакционной смеси приблизительно 22 мл, и добавляют дополнительное количество этилацетата (7 мл). Смесь охлаждают до приблизительно 20°С, в течение по меньшей мере 30 минут добавляют гексан (42 мл), и смесь выдерживают при 20°С в течение 15 минут. Выпавший осадок собирают фильтрацией, промывают смесью этилацетат/гексан 1:4 (3×5 мл) и сушат при приблизительно 50°С в течение 18 часов с образованием указанного в заголовке соединения в виде белого твердого вещества (3,54 г, 95,7%).
Данный способ был успешно повторно осуществлен с использованием в качестве растворителя пентан-3-она вместо этилацетата.
Пример 9: 6α,9α-Дифтор-17α-(1-оксопропокси)-11β-гидрокси-16α-метил-3-оксо-андроста-1,4-диен-17β-ил S-(1-оксопропокси)тиоангидрид
Раствор 6α,9α-дифтор-11β,17α-дигидрокси-16α-метил-3-оксо-андроста-1,4-диен-17β-карботионовой кислоты (10 г) в ацетоне (125 мл) охлаждают до приблизительно -5°С и обрабатывают триэтиламином (16 мл) при температуре от 0°С до -5°С в течение приблизительно 15 минут. Суспензию в течение приблизительно 90 минут обрабатывают пропионилхлоридом (8,5 мл), поддерживая температуру от -5°С до 0°С, и раствор перемешивают при температуре от -5°С до 0°С в течение дополнительных двух часов. Реакционную смесь за десять минут вливают в 2М соляную кислоту (470 мл), и полученную в результате суспензию выдерживают при 5°С в течение 30 минут. Продукт отфильтровывают, промывают водой (3×125 мл) и сушат под вакуумом при приблизительно 40°С в течение 15 часов с образованием указанного в заголовке соединения в виде твердого вещества от белого до не совсем белого цвета (12,78 г, 100,6%).
ВЭЖХ: время удерживания 40,7 мин (чистота 99,5% по площади).
CHN: обнаружено С 61,8%; Н 6,7%, S 6,1%; для формулы С27Н34F2О6S требуется С 61,8%; Н 6,5%, S 5,7%.
ЯМР (ДМСОd6) 7.27 (1Н, d, 10 Гц), 6.32 (1Н, d, 10 Гц), 6.12 (1Н, s), 5.81 (1Н, d), 5.65 (1Н, ddd), 4.37 (1Н, m), 2.40 (2Н, q, 7 Гц), 2.00-2.45 (4Н, m), 1.85 (1Н, m), 1.87 (1Н, m), 1.51 (3Н, s), 1.27 (1Н, m), 1.11-1.22 (4Н, m), 0.90-1.05 (6Н, m), 0.88 (3Н, d, 7 Гц).
Пример 10: Получение 6α,9α-дифтор-11β,17α-дигидрокси-16α-метил-3-оксо-андроста-1,4-диен-17β-карботионовой кислоты имидазольной соли (1:2)
6α,9α-дифтор-11β-гидрокси-16α-метил-3-оксо-17α-гидрокси-андроста-1,4-диен-17β-карбоновую кислоту (35 г, 0,089 моль) и карбонилдиимидазол (25,75 г, 0,16 моль) перемешивают в этилацетате (350 мл) и N,N-диметилформамиде (17,5 мл) при 20±2°С в течение 60 минут. Суспензию охлаждают до 0°С и данную партию перемешивают при 0±5°С, пока через погруженную в смесь трубку из плавленого стекла в течение 32 минут добавляют сероводород (7,7 г, 0,23 моль). Данную партию перемешивают при 0±3°С в течение 30 минут, за 20 минут нагревают до 9°С и перемешивают при 9±3°С в течение в общей сложности 100 минут. Продукт собирают фильтрацией (бумага Whatman 54), и осадок на фильтре промывают этилацетатом (2×105 мл). Продукт сушат под вакуумом при приблизительно 20°С в течение 20 часов с образованием указанного в заголовке соединения в виде твердого вещества от белого до бледно-пурпурного цвета (47,7 г, 98,5% от теоретического значения).
ЯМР (MeOHd4) 0.86 (3Н) d, J=7,4 Гц; 1.11 (3Н) s; 1.20 (1Н) m; 1.61 (3Н) s; 1.62-1.82 (3Н) m; 2.14-2.25 (2Н) m; 2.33 (1Н) m; 2.54 (1Н) m; 3.19 (1Н) m; 4.26 (1Н) add, J=11,2, 4,0, 1,8 Гц; 5.57 (1Н) dddd, J=49,0; 11,6; 6,8; 1,9 Гц; 6.32 (1Н) m; 6.35 (1Н) dd, J=10,0, 2,0 Гц; 7.35 (4Н) d, J=1.0 Гц (имидазол); 7.41 (1Н) dd, J=10,0, 1,4 Гц; 8.30 (2Н) t, J=1,0 Гц (имидазол).
Точка плавления 120°С (с разложением).
Пример 11: Получение раствора 6α,9α-дифтор-11β,17α-дигидрокси-16α-метил-3-оксо-андроста-1,4-диен-17β-карботионовой кислоты из имидазольной соли 6α,9α-дифтор-11β,17α-дигидрокси-16α-метил-3-оксо-андроста-1,4-диен-17β-карботионовой кислоты (2:1)
Имидазольную соль 6α,9α-дифтор-11β,17α-дигидрокси-16α-метил-3-оксо-андроста-1,4-диен-17β-карботионовой кислоты (47,7 г) перемешивают в этилацетате (811 мл), и суспензию охлаждают до 15±3°С. Добавляют 2М (водную) соляную кислоту (286 мл), и смесь перемешивают в течение приблизительно пяти минут, получая прозрачную двухфазную смесь. Слои разделяют, и органический раствор свободной карботионовой кислоты промывают дополнительным количеством 2М (водной) соляной кислоты (190 мл).
Альтернативный способ
Имидазольную соль 6α,9α-дифтор-11β,17α-дигидрокси-16α-метил-3-оксо-андроста-1,4-диен-17β-карботионовой кислоты (47,7 г) перемешивают в пентан-3-оне (954 мл) и суспензию охлаждают до 15±3°С. Добавляют 2М соляную кислоту (286 мл), и смесь перемешивают в течение приблизительно пяти минут, получая прозрачную двухфазную смесь. Слои разделяют, и органический раствор свободной карботионовой кислоты промывают водой (190 мл).
В обоих случаях влажные от растворителя осадки имидазольной соли 6α,9α-дифтор-11β,17α-дигидрокси-16α-метил-3-оксо-андроста-1,4-диен-17β-карботионовой кислоты (а не сухие твердые вещества) можно применять в качестве исходного материала в приведенных выше методиках кислотообразования.
Пример 12: S-Фторметиловый эфир 6α,9α-дифтор-17α-(1-оксопропокси)-11β-гидрокси-16α-метил-3-оксоандроста-1,4-диен-17β-карботионовой кислоты
6α,9α-Дифтор-17α-(1-оксопропокси)-11β-гидрокси-16α-метил-3-оксоандроста-1,4-диен-17β-карботионовой кислоты имидазольную соль (1:2) (13,3 г) суспендировали в 3-пентаноне (200 мл) и промывали 2М соляной кислотой (60 мл), а потом водой (60 мл). Полученный раствор охлаждали до 3°С, и за 2 минуты добавляли 14,0 мл трипропиламина, обеспечивая сохранение в реакционной смеси температуры 3±2°С. Раствор перемешивали при 3±2°С, и за 5 минут добавляли 5,3 мл пропионилхлорида, поддерживая температуру реакционной смеси 3±2°С. Затем раствор оставляли нагреваться до 12°С и перемешивали при 12±2°С в течение 90 минут. Затем раствор охлаждали до 3°С и добавляли 1-метилпиперазин (5,1 мл), поддерживая температуру реакционной смеси 3±2°С. Раствор перемешивали при 3±2°С в течение 20 минут, и затем одной порцией добавляли бромфторметан (1,4 мл). Раствор оставляли нагреваться до 18°С и перемешивали при 18±2°С в течение 16 часов. Затем этот раствор последовательно промывали 1М HCl (60 мл), водой (60 мл), 1%-ным NaHCO3 (60 мл) и водой (60 мл), и затем концентрировали перегонкой при атмосферном давлении до приблизительно 80 мл и охлаждали до 90°С. В раствор вносили затравку указанного в заголовке соединения (0,05 г) и затем за 20 минут добавляли толуол (120 мл), поддерживая температуру суспензии 87±3°С. Затем суспензию концентрировали перегонкой при атмосферном давлении до приблизительно 80 мл и после этого охлаждали до 90°С. Затем за 20 минут добавляли толуол (120 мл), поддерживая температуру суспензии 87±3°С. Затем за 90 минут суспензию охлаждали до 10°С, выдерживали при 10±3°С в течение 60 минут, а потом фильтровали. Осадок на фильтре промывали смесью 3-пентанон: толуол (1:4, 2×20 мл) и сушили отсасыванием. Твердое вещество сушили в вакуумном термостате при 50°С в течение 16 часов с образованием указанного в заголовке соединения в виде белого игольчатого твердого вещества (8,7 г, 69% от теоретического значения).
Пример 13: S-Фторметиловый эфир 6α,9α-дифтор-17α-(1-оксопропокси)-11β-гидрокси-16α-метил-3-оксоандроста-1,4-диен-17β-карботионовой кислоты
К тщательно перемешанной смеси 6α,9α-дифтор-17α-(1-оксопропокси)-11β-гидрокси-16α-метил-3-оксоандроста-1,4-диен-17β-карбоновой кислоты (10,0 г) и N,N'-карбонилдимидазола (6,3 г) последовательно добавляли этилацетат (100 мл) и N,N-диметилформамид (5 мл). Образовавшуюся суспензию перемешивали при 18±20°С в течение 50 минут с получением бледно-желтого раствора. Раствор охлаждали до 10°С, и в течение 25 минут через него барботировали сероводород (2,2 г), поддерживая температуру содержимого 12±2°С. Полученную суспензию перемешивали при 12±2°С в течение дополнительных 90 минут, а затем помещали в условия умеренного вакуума (300-350 мбар ((3,0-3,5)×104 Па)), медленно выпуская азот, и интенсивно перемешивали в течение 50 минут. Затем сбрасывали вакуум, и сосуд продували азотом. Затем добавляли 3-пентанон (150 мл), и суспензию промывали 2М соляной кислотой (60 мл), а потом водой (60 мл). Полученный раствор охлаждали до 3°С, и за 2 минуты добавляли трипропиламин (14,0 мл), обеспечивая сохранение температуры реакционной смеси 3±2°С. Раствор перемешивали при 3±2°С, и за 5 минут добавляли 5,3 мл пропионилхлорида, поддерживая температуру реакционной смеси 3±2°С. Затем раствор оставляли нагреваться до 10°С и перемешивали при 12±2°С в течение 90 минут. Затем раствор охлаждали до 3°С и добавляли 1-метилпиперазин (5,1 мл), поддерживая температуру реакционной смеси 3±2°С. Раствор перемешивали при 3±2°С в течение 20 минут, затем одной порцией добавляли бромфторметан (1,4 мл). Раствор оставляли нагреваться до 18°С и перемешивали при 18±2°С в течение 16 часов. Затем раствор последовательно промывали 1М HCl (60 мл), водой (60 мл), 1%-ным NaHCO3 (60 мл) и водой (60 мл), а потом концентрировали перегонкой при атмосферном давлении до приблизительно 80 мл и охлаждали до 90°С. В раствор вносили затравку указанного в заголовке соединения (0,05 г) и затем за 20 минут добавляли толуол (120 мл), поддерживая температуру суспензии 87±3°С. Затем суспензию концентрировали перегонкой при атмосферном давлении до приблизительно 80 мл и потом охлаждали до 90°С. Затем за 20 минут добавляли толуол (120 мл), поддерживая температуру суспензии 87±3°С. Затем за 90 минут суспензию охлаждали до 10°С, выдерживали при 10±3°С в течение 60 минут, а потом фильтровали. Осадок на фильтре промывали смесью 3-пентанон: толуол (1:4, 2×20 мл) и сушили отсасыванием. Твердое вещество сушили в вакуумном термостате при 50°С в течение 16 часов с образованием указанного в заголовке соединения в виде белого игольчатого твердого вещества (9,5 г, 75% от теоретического значения).
Пример 14: S-Фторметиловый эфир 6α,9α-дифтор-17α-(1-оксопропокси)-11β-гидрокси-16α-метил-3-оксо-андроста-1,4-диен-17β-карботионовой кислоты (с использованием пентан-3-она в качестве растворителя и толуола в качестве антирастворителя)
Перемешанную суспензию S-фторметилового эфира 6α,9α-дифтор-17α-(1-оксопропокси)-11β-гидрокси-16α-метил-3-оксоандроста-1,4-диен-17β-карботионовой кислоты (9,1 г) в пентан-3-оне (250 мл) нагревали до кипения с обратным холодильником, получая раствор, который затем концентрировали перегонкой при атмосферном давлении до приблизительно 65 мл. Раствор охлаждали до 85°С, а затем за 20 минут добавляли толуол (120 мл), поддерживая температуру смеси 85±3°С. Образовавшуюся суспензию концентрировали перегонкой при атмосферном давлении до приблизительно 90 мл, охлаждали до 85°С, и затем за 20 минут добавляли толуол (95 мл), поддерживая температуру смеси 85±3°С. Затем суспензию выдерживали при 85±3°С в течение 15 минут и потом за 90 минут охлаждали до 10°С. Суспензию выдерживали при 12±3°С в течение 30 минут и потом фильтровали. Осадок на фильтре промывали смесью пентан-3-он: толуол (1:3, 2×20 мл) и сушили отсасыванием. Твердое вещество сушили в вакуумном термостате при 50°С в течение 4 часов с образованием указанного в заголовке соединения в виде белого игольчатого твердого вещества (9,02 г, 98% от теоретического значения). ЯМР анализ показал отсутствие сольватации продукта, и XRPD анализ подтвердил, что продукт находится в полиморфной Форме 1.
Следует понимать, что во всем описании изобретения и формуле изобретения, следующей за ним, если по контексту не требуется иного, под словом "включать" и такими его вариантами как "включает" и "включающий" подразумевают включение указанных целого, или стадии, или группы целых, но не исключение любого другого целого, или стадии, или группы целых или стадий.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ 6α, 9α-ДИФТОР-17α-(1-ОКСО-ПРОПОКСИ)-11β-ГИДРОКСИ-16α-МЕТИЛ-3-ОКСО-АНДРОСТ-1,4-ДИЕН-17β-КАРБОТИОНОВОЙ КИСЛОТЫ | 2003 |
|
RU2333217C2 |
| СПЕЦИФИЧЕСКОЕ ГЛЮКОКОРТИКОСТЕРОИДНОЕ СОЕДИНЕНИЕ, ОБЛАДАЮЩЕЕ ПРОТИВОВОСПАЛИТЕЛЬНОЙ АКТИВНОСТЬЮ | 2004 |
|
RU2348645C2 |
| СПОСОБ ЭТЕРИФИКАЦИИ ТИОКАРБОНОВОЙ КИСЛОТЫ | 2004 |
|
RU2351605C2 |
| СЛОЖНЫЕ ЭФИРЫ 16 АЛЬФА 17 АЛЬФА - АЦЕТАЛЬЗАМЕЩЕННОЙ АНДРОСТАН-17 БЕТА - КАРБОНОВОЙ КИСЛОТЫ ИЛИ ИХ СТЕРЕОИЗОМЕРЫ | 1991 |
|
RU2081879C1 |
| КОНКРЕТНОЕ ГЛЮКОКОРТИКОСТЕРОИДНОЕ СОЕДИНЕНИЕ, ОБЛАДАЮЩЕЕ ПРОТИВОВОСПАЛИТЕЛЬНОЙ АКТИВНОСТЬЮ | 2004 |
|
RU2359973C2 |
| ПРОИЗВОДНЫЕ АНДРОСТЕНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1997 |
|
RU2194053C2 |
| СПОСОБ ПОЛУЧЕНИЯ ФЛУМЕТАЗОНА, СОЕДИНЕНИЕ | 2002 |
|
RU2260596C1 |
| ХИМИЧЕСКИЕ СОЕДИНЕНИЯ | 2008 |
|
RU2462472C2 |
| ПРИМЕНЕНИЕ ИНГИБИТОРА Н, К-АТФАЗЫ ДЛЯ ЛЕЧЕНИЯ НОСОВЫХ ПОЛИПОВ | 1997 |
|
RU2197966C2 |
| СТЕРОИДНЫЕ ЭФИРЫ ИЛИ ИХ СТЕРЕОИЗОМЕРЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1992 |
|
RU2112775C1 |
Описан улучшенный способ получения пропионата флутиказона в виде кристаллической полиморфной Формы 1, при котором осуществляют взаимодействие соединения формулы (II)

или его соли с соединением формулы LCH2F, где L представляет собой уходящую группу, возможно в присутствии катализатора межфазного переноса, несольватирующего органического жидкого растворителя, не смешивающегося с водой, и воды. Способ позволяет проводить процесс при атмосферном давлении и без выделения промежуточного продукта. 2 н. и 9 з.п. ф-лы.
(а) осуществляют взаимодействие соединения формулы (II)
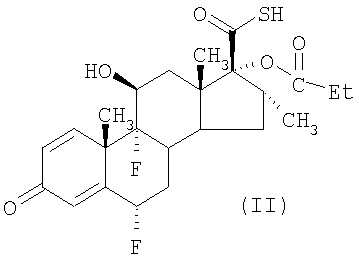
или его соли с соединением формулы LCH2F, где L представляет собой уходящую группу, возможно в присутствии катализатора межфазного переноса, несмешивающегося с водой несольватирующего органического жидкого растворителя, представляющего собой метилацетат, этилацетат или пентанон, и воды;
(б) возможно увеличивают чистоту пропионата флутиказона в органическом слое путем осуществления одной или более чем одной стадии промывки водой;
(в) отделяют органический слой от водного слоя и, возможно, концентрируют пропионат флутиказона в органическом слое;
(г) смешивают раствор пропионата флутиказона в несмешивающемся с водой несольватирующем органическом жидком растворителе, представляющем собой метилацетат, этилацетат или пентанон, присутствующем в органическом слое, с несольватирующим органическим жидким антирастворителем, представляющим собой толуол, изооктан или гексан, что приводит к кристаллизации пропионата флутиказона из раствора в виде кристаллической полиморфной Формы 1.
(а) осуществляют взаимодействие соединения формулы (III)

с активированным производным пропионовой кислоты в количестве по меньшей мере 1,3 моль активированного производного на моль соединения формулы (III); и
(б) удаляют из любого полученного таким образом соединения формулы (IIА) пропионильную группировку, связанную с атомом серы,

путем взаимодействия продукта стадии (а) с органическим основанием: первичным или вторичным амином, способным к образованию водорастворимого пропанамида, который затем можно удалить путем осуществления одной или более чем одной стадии промывки водой.
| PHILLIPS G | |||
| et al | |||
| J | |||
| of Med | |||
| Chem | |||
| Пишущая машина | 1922 |
|
SU37A1 |
| Устройство для укладки слитков в штабель | 1960 |
|
SU132125A1 |
| KARIUKT, BENSON M | |||
| et al | |||
| Chem | |||
| Comm | |||
| Металлический водоудерживающий щит висячей системы | 1922 |
|
SU1999A1 |

