ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
Данное изобретение касается способа крупномасштабного получения 3-циано-7-алкокси-6-нитро-4-хинолонов, которые являются промежуточными продуктами получения ингибиторов протеинтирозинкиназы (РТК), полезных при лечении рака.
Двумя наиболее часто применяемыми синтетическими способами получения 3-циано-4-хинолонов или 3-карбоалкилоксихинолонов являются внутримолекулярная реакция Фриделя-Крафтса и электроциклическое замыкание кольца N-(2-карбоксивинил)анилиновых производных. Условия реакции Фриделя-Крафтса хорошо работают для богатых электронами анилинов, умеренно для незамещенных анилинов, плохо или совсем не работают для анилинов с дефицитом электронов и совершенно бесполезны для крупномасштабного получения 3-циано-4-хинолонов, использующего анилины с дефицитом электронов. Кстати, электроноакцепторные группы анилина снижают нуклеофильность ароматического кольца таким образом, что побочные взаимодействия конкурируют (если не преобладают) с желательной внутримолекулярной конденсацией. Термические условия электроциклического замыкания кольца N-(2-карбоксивинил)анилиновых производных обычно требуют температур выше 240°С. Однако структуру 3-циано-4-хинолонов получают посредством реакций электроциклического замыкания кольца N-(2-карбоксивинил)анилиновых производных при нагревании до 260°С в дифениловом эфире (патент США №6002008; WO 98/43960). В частности, существует несколько недостатков, связанных с электроциклическим замыканием кольца, для получения количеств материала в технологическом масштабе. Обычно взаимодействия протекают при большом разбавлении (66:1), давая в результате неэффективный крупномасштабный способ вследствие низкой производительности. Кроме того, термическое разложение конечного продукта и/или исходного материала подвергает риску чистоту конечного продукта в результате высокотемпературных условий взаимодействия. К тому же оборудование, необходимое для безопасного проведения высокотемпературных взаимодействий в большом масштабе, является дорогим и недоступно в обычной лаборатории или заводских условиях.
Производство 3-циано-4-хинолонов посредством электроциклического замыкания кольца страдает от всех упомянутых выше проблем, в особенности от термического разложения требуемого конечного продукта или исходного материала. Например, известно, что 7-этокси-4-гидрокси-6-нитрохинолин-3-карбонитрил разлагается при 240°C, тогда как минимальная необходимая для циклизации температура составляет 256°C.
Таким образом, в данной области существует потребность в способе, который направлен на и предпочтительно преодолевает высокотемпературную циклизацию, которая приводит к термическому разложению.
Следующие экспериментальные подробности приведены для облегчения понимания данного изобретения и никоим образом не предназначены и не должны толковаться как ограничения данного изобретения, изложенного в приведенной далее формуле изобретения.
КРАТКОЕ СОДЕРЖАНИЕ ИЗОБРЕТЕНИЯ
Данное изобретение касается способа получения 3-циано-7-алкокси-6-нитро-4-хинолона, включающего
а) взаимодействие замещенного антранилата формулы (I) с диметилацеталем диметилформамида:
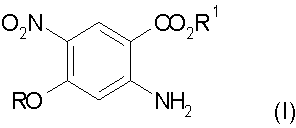
где R и R1 обозначают алкил;
с получением соединения формулы (II):
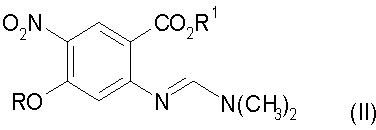
b) конденсацию соединения стадии а) с трет-бутилцианоацетатом с получением соединения формулы (III):
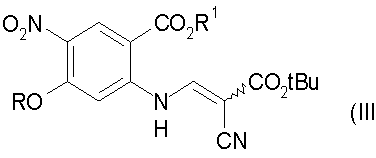
с) гидролиз соединения стадии b) с получением соединения формулы (IV):
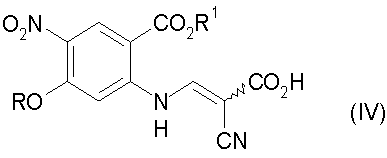
d) декарбоксилирование соединения стадии с) до соединения формулы (V):
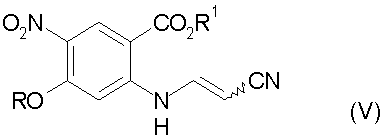
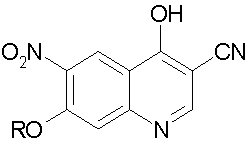
е) циклизацию соединения стадии d) в присутствии основания с получением 3-циано-7-алокси-6-нитро-4-хинолона формулы:
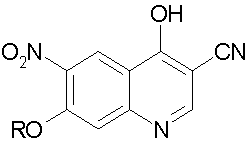
Используемый здесь термин «алкил» обозначает линейную или разветвленную алкильную группу, например, С1-С6 алкильную группу, предпочтительно С1-С4 алкильную группу, более предпочтительно Me, Et, н-Pr, изо-Pr, н-Bu, наиболее предпочтительно Me или Et. R и R1 могут быть одинаковыми или разными. Настоящее изобретение включает все таутомерные формы соединений, а также смеси данных таутомерных форм.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Описанное здесь изобретение для получения 3-циано-7-алкокси-6-нитро-4-хинолонов не требует описанных выше высокой температуры (256°С) и низкопроизводительного сильного разбавления (66:1). Данные условия взаимодействия позволяют проводить реакцию циклизации в стандартном технологическом оборудовании.
Способ данного изобретения показан на схеме I.
Схема I
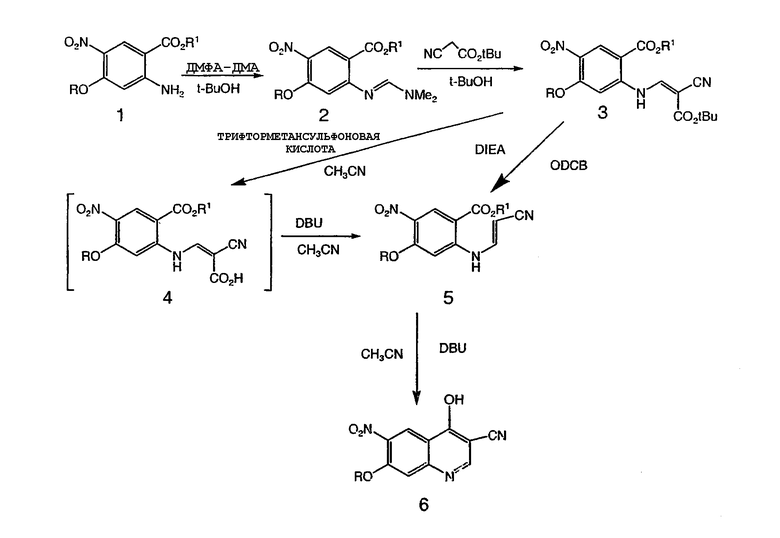
Как описано на схеме I, замещенный антранилат 1, в котором R обозначает алкил, взаимодействует с диметилацеталем диметилформамида (ДМФА-ДМА) или примерно с 1-5 эквивалентами диметилацеталя диметилформамида в спиртовом растворителе с получением N,N-диметиламидина 2. В предпочтительном варианте концентрация (ДМФА-ДМА) составляет от 1 до 2 эквивалентов. Предпочтительные условия для данного взаимодействия используют примерно 1,2 эквивалента диметилацеталя диметилформамида в трет-бутаноле при температуре примерно от 50 до 120°C с предпочтительной температурой 80°C. В предпочтительном варианте данное взаимодействие допускает простое выделение N,N-диметиламидина 2 при охлаждении реакционной смеси с осаждением продукта и сбором осадка фильтрованием. Данная методика обеспечивает почти количественный выход N,N-диметиламидина 2 достаточной степени чистоты для использования его на следующей стадии без дополнительной очистки. В другой предпочтительной методике осуществляют взаимодействие замещенного антранилата 1 с диметилацеталем диметилформамида при кипении (примерно 110°C) и после разбавления водой выделяют N,N-диметиламидин 2 фильтрованием и сушкой собранного продукта.
Реакцию конденсации N,N-диметиламидина 2 с трет-бутилцианоацетатом можно проводить, применяя ацетонитрил, кислоту, толуол или спиртовой растворитель при температуре примерно от 20°C до 110°C, получая N-(2-циано-2-трет-бутоксикарбонилвинил)антранилат 3. В предпочтительном варианте конденсацию проводят, добавляя трет-бутанол при температуре примерно от 25°C до 35°C примерно с 1,5-2,0 экв. трет-бутилцианоацетата, что обеспечивает вещество высокого качества (>98%, жидкостная хроматография при высоком давлении (область ВДЖХ)) с высоким выходом (90-99%).
Гидролиз N-(2-циано-2-трет-бутоксикарбонилвинил)антранилата 3 можно проводить, применяя кислоту в растворителе или непосредственно уксусную кислоту в качестве растворителя при температуре примерно от 20 до 110°C. В предпочтительном варианте гидролиз включает обработку N-(2-циано-2-трет-бутоксикарбонилвинил)антранилата 3 каталитическим количеством трифторметансульфокислоты в ацетонитриле при температуре примерно от 20 до 30°C с получением N-(2-циано-2-карбоксивинил)антранилата 4, как охарактеризовано с помощью ЯМР. N-(2-циано-2-трет-бутоксикарбонилвинил)антранилат 3 можно необязательно нагреть до 180°C в орто-дихлорбензоле (ОДХБ) для удаления трет-бутилового сложного эфира и получения N-(2-циано-2-карбоксивинил)антранилата 4.
Декарбоксилирование N-(2-циано-2-карбоксивинил)антранилата 4 можно проводить в кислотных или основных условиях, получая N- (2-циановинил)антранилат 5. В предпочтительном варианте кислоты включают уксусную кислоту и пара-толуолсульфоновую кислоту, основания включают диизопропилэтиламин, пиридин или диазобицикло[2.2.3]ундецен (DBU) в подходящих растворителях, которые включают ацетонитрил, уксусную кислоту, пиридин и диметилацетамид, при температуре примерно от 80 до 140°С. Если термически индуцированный гидролиз N-(2-циано-2-трет-бутоксикарбонилвинил)антранилата 3 до N-(2-циановинил)антранилата 5 в орто-дихлорбензоле (ОДХБ) проводят в присутствии каталитического количества подходящего основания, которое включает диизопропилэтиламин (ДИЭА), то N-(2-циано-2-трет-бутоксикарбонилвинил)антранилат 3 превращается непосредственно в N-(2-циановинил)антранилат 5. В предпочтительном варианте применяют DBU в ацетонитриле при температуре около 80°С.
Внутримолекулярную анионную циклизацию N-(2-циановинил)антранилата 5 до 3-циано-7-алкокси-6-нитро-4-хинолона 6 можно проводить примерно с 2-13 экв. основания в растворителе. В предпочтительном варианте основание включает DBU, NaH, пиперидин, диметиламинопиридин (DMAP) или трет-бутилат калия (KOtBu). В предпочтительном варианте растворители включают ацетонитрил, простой дифениловый эфир, ОДХБ, смеси ТГФ/ксилол, толуол, N,N-диметилформамид (ДМФА), пропионитрил или изопропанол. В предпочтительном варианте коэффициенты разбавления растворитель : субстрат составляют примерно от 15 до 30:1 при температуре примерно от 60°С до 140°С. Предпочтительной методикой получения 3-циано-7-алкокси-6-нитро-4-хинолона 6 является обработка N-(2-циановинил)антранилата 5 примерно 3-5 экв. DBU в ацетонитриле при температуре около 80°С в течение примерно 4-5 час и гашение водной HCl.
Более предпочтительной методикой получения 3-циано-6-алкокси-7-нитро-4-хинолона 6 из N-(2-циано-2-трет-бутоксикарбонилвинил)антранилата 3 является последовательное проведение гидролиза, декарбоксилирования и внутримолекулярной циклизации в одном и том же сосуде без выделения N-(2-циано-2-карбоксивинил)антранилата 4 или N-(2-циановинил)антранилата 5. Более предпочтительный способ получения 3-циано-6-алкокси-7-нитро-4-хинолона 6 включает гидролиз N-(2-циано-2-трет-бутоксикарбонилвинил)антранилата 3 примерно 0,2-0,3 экв. трифторметансульфокислоты в ацетонитриле при температуре примерно от 20 до 30°С в течение примерно 5-60 мин с последующим добавлением примерно 3-5 экв. DBU и кипячением реакционной смеси в течение примерно 4-5 час. 3-Циано-7-алкокси-6-нитро-4-хинолон 6 выделяют, разбавляя реакционную смесь водой и собирая полученный осадок фильтрованием.
Собранный осадок растирают с этилацетатом, получая 3-циано-7-алкокси-6-нитро-4-хинолон 6 в виде бежево-коричневого твердого вещества (выход 70-80%, >98% по данным 1Н ЯМР).
Заявленное здесь изобретение обеспечивает 3-циано-7-алкокси-6-нитро-4-хинолоны при объединении стадий и без необходимости высокотемпературной циклизации. 3-Циано-7-алкокси-6-нитро-4-хинолон 6 получают с хорошим общим выходом (70% для 5 превращений, проводимых за две операции в отдельных реакторах, при чистоте >98% по данным ВЭЖХ и 1H ЯМР).
Для целей данного изобретения кислота представляет собой молекулярный объект или химическое вещество, способное передавать протон или образовывать ковалентную связь с электронной парой. Предпочтительные кислоты включают уксусную кислоту, трифторуксусную кислоту, пара-толуолсульфоновую кислоту, метансульфоновую кислоту и трифторметансульфокислоту.
Для целей данного изобретения «растворитель» является термином, применяемым к исходной жидкой фазе в целом, содержащей экстрагент. Растворитель может содержать только один экстрагент или может представлять собой составную гомогенную смесь экстрагента(ов) с разбавителем(ями). В предпочтительном варианте растворитель включает толуол, ацетонитрил, тетрагидрофуран (ТГФ), диметилацетамид, уксусную кислоту, пиридин, простой дифениловый эфир, ОДХБ, смеси ТГФ/ксилол, толуол, N,N-диметилформамид (ДМФА), пропионитрил или изопропанол.
Для целей данного изобретения основание представляет собой химическое вещество или молекулярный объект, имеющий доступную пару электронов, способную образовывать ковалентную связь с протоном или вакантной орбиталью некоторого другого вещества. В предпочтительном варианте основание включает диизопропилэтиламин, пиридин или диазобицикло[2.2.3]ундецен (DBU), NaH, пиперидин, диметиламинопиридин (DMAP) или трет-бутилат калия (KOtBu).
Для целей данного изобретения термин "алкил" включает линейные и разветвленные алкильные фрагменты, предпочтительно из 1-6 атомов углерода.
Для содействия дальнейшему пониманию данного изобретения способ настоящего изобретения иллюстрируют следующие неограничительные примеры.
Пример 1
Метиловый эфир 2-[[(диметиламино)метилен]амино]-4-этокси-5-нитробензойной кислоты
В 3-литровую круглодонную колбу, снабженную верхней мешалкой, холодильником и термопарой, под N2 загружают метиловый эфир 2-амино-4-этокси-5-нитробензойной кислоты (80 г, 333 ммоль) и диметилацеталь N,N-диметилформамида (500 мл). Реакционную смесь нагревают до кипения с обратным холодильником (100°C). Как только густая суспензия становится гомогенной и завершается взаимодействие, реакционную смесь охлаждают до 25-30°C. Реакционную смесь разбавляют водой (3 л) и фильтруют полученную суспензию. Осадок на фильтре промывают водой (3×500 мл) и сушат в вакууме (50 мм рт. ст.) при 55°C, получая указанное в заголовке соединение в виде почти белого твердого вещества (89,6 г, выход 91%, чистота >90% по данным интегрирования ЯМР-спектров). 1H ЯМР (300 МГц, ДМСО-d6): 8,23 (с, 1H), 7,81 (с, 1H), 6,71 (с, 1H), 4,22 (кв., J=7 Гц), 3,68 (с, 3H), 3,09 (с, 3H), 2,97 (с, 3H), 1,40 (т, J=7 Гц, 3H).
Пример 2
Трет-бутиловый эфир 2-циано-3-(5'-этокси-2'-метоксикарбонил-4'-нитрофенил)амино-2-пропеновой кислоты
В 3-литровую круглодонную колбу, снабженную верхней мешалкой, холодильником и термопарой, под N2 загружают метиловый эфир 2-[[(диметиламино)метилен]амино]-4-этокси-5-нитробензойной кислоты (68 г, 230 ммоль), трет-бутанол (500 мл), а затем трет-бутилцианоацетат (65 г, 460 ммоль). Реакционную смесь нагревают до кипения с обратным холодильником. Через 4 час реакционную смесь охлаждают до комнатной температуры и фильтруют суспензию. Осадок на фильтре промывают гептаном (2×100 мл) и сушат в вакууме (50 мм рт.ст.) при 40°С, получая указанное в заголовке соединение в виде бежевого твердого вещества (83 г, выход 91%, чистота >98% по данным ЯМР). 1Н ЯМР (300 МГц, ДМСО-d6): 12,7 (д, J=12,9 Гц, 1Н), 8,77 (д, J=12,9 Гц, 1Н), 8,50 (с, 1Н), 7,47 (с, 1Н), 4,37 (кв., J=7 Гц, 2Н), 3,91 (с, 3Н), 1,52 (с, 9Н), 1,40 (т, J=7 Гц, 3Н).
Пример 3
Трет-бутиловый эфир 2-циано-3-(5'-этокси-2'-метоксикарбонил-4'-нитрофенил)амино-2-пропеновой кислоты
В 3-литровую круглодонную колбу, снабженную верхней мешалкой, холодильником и термопарой, под N2 загружают метиловый эфир 2-амино-4-этокси-5-нитробензойной кислоты (100 г, 0,416 моль), диметилацеталь N,N-диметилформамида (59,5 г, 0,499 моль) и трет-бутанол (800 мл). Реакционную смесь нагревают при кипении с обратным холодильником в течение 1,5 час. Реакционную смесь охлаждают до 22-35°С и добавляют трет-бутилцианоацетат (117 г, 0,832 моль). Реакционную смесь перемешивают при температуре от 20 до 30°С в течение 2 час. Осадок собирают фильтрованием с отсосом, промывают гептаном (500 мл), затем сушат до постоянного веса при пониженном давлении (50 мм рт.ст.)) и 45°С в течение ночи, получая указанное в заголовке соединение в виде не совсем белого твердого вещества (162,9 г, выход 95%, чистота >95% по данным ВЭЖХ). 1H ЯМР (300 МГц, ДМСО-d6): 12,7 (д, J=12,9 Гц, 1Н), 8,77 (д, J=12,9 Гц, 1Н), 8,50 (с, 1Н), 7,47 (с, 1Н), 4,37 (кв., J=7 Гц, 2Н), 3,91 (с, 3Н), 1,52 (с, 9Н), 1,40 (т, J=7 Гц, 3Н).
Пример 4
N-(2-циановинил)-2-амино-4-этокси-5-нитробензойная кислота
В круглодонную колбу на 500 мл, снабженную мешалкой, холодильником и термопарой, под N2 загружают трет-бутиловый эфир (Z)-2-циано-3-(5'-этокси-2'-метоксикарбонил-4'-нитрофенил)амино-2-пропеновой кислоты (20 г, 51,1 ммоль), N,N-диизопропилэтиламин (1 мл, 5,72 ммоль) и орто-дихлорбензол (200 мл). Реакционную смесь нагревают до кипения с обратным холодильником (180°С). Через 7,5 час взаимодействие завершается, что подтверждают тонкослойной хроматографией. Реакционную смесь охлаждают до комнатной температуры и разбавляют гексаном (500 мл), вызывая осаждение сырого продукта. Твердое вещество выделяют фильтрованием, получая указанное в заголовке соединение в виде бежевого порошка (11,7 г, выход 79% стереоизомеров 65:35, чистота 94% по данным ЯМР). 1H ЯМР (300 МГц, ДМСО-d6): 11,1 (д, J=12,7 Гц, 0,65Н), 10,6 (д, J-12,9 Гц, 0,35Н), 8,47 (с, 0,65Н), 8,37 (с, 0,35Н), 8,29 (дд, J=13,4, 12,9 Гц, 0,35Н), 8,16 (дд, J=12,7, 8,5 Гц, 0,65Н), 7,13 (с, 1Н), 5,49 (д, J=13,4 Гц, 0,35Н), 4,97 (д, J=8,4 Гц, 0,65Н), 4,38-4,28 (м, 2Н), 3,90 (с, 1,95Н), 3,88 (с, 1,05Н), 1,39 (т, J=7 Гц, 3Н).
Пример 5
7-этокси-4-гидрокси-6-нитрохинолин-3-карбонитрил
В круглодонную колбу на 100 мл, снабженную верхней мешалкой, холодильником и термопарой, загружают трет-бутиловый эфир (Z)-2-циано-3-(5'-этокси-2'-метоксикарбонил-4'-нитрофенил)амино-2-пропеновой кислоты (2,5 г, 6,3 ммоль) и ацетонитрил (50 мл). К данной гетерогенной реакционной среде добавляют трифторметансульфокислоту (0,12 мл, 0,21 ммоль). После исчезновения исходного материала, что подтверждают ТСХ (20% EtOAc/гексан), добавляют к реакционной смеси DBU (4,0 мл, 4,25 ммоль). Затем реакционную смесь нагревают до кипения с обратным холодильником и контролируют на завершение процесса (>95% по данным ВЭЖХ - колонка Phenomenex 3 мкм фенил-гексил (150×4,6 мм)). Затем реакционную смесь гасят 10% HCl (100 мл) и разбавляют водой (400 мл). После перемешивания в течение 15 мин при комнатной температуре суспензию фильтруют и собранное твердое вещество оставляют сушиться на воздухе. Собранное твердое вещество суспендируют в этилацетате (25 мл) при комнатной температуре и снова фильтруют и оставляют сушиться на воздухе. Данная методика дает 1,15 г (70%) указанного в заголовке соединения в виде бежевого твердого вещества, которое представляет собой >95% продукта по данным интегрирования спектров ЯМР. 1H ЯМР (300 МГц, ДМСО-d6): 12,9 (с, 1Н), 8,79 (с, 1Н), 8,51 (с, 1Н), 7,24 (с, 1Н), 4,27 (кв., J=7 Гц, 2Н), 1,41 (т, J=7 Гц, 3Н).
Настоящее изобретение относится к способу получения 3-циано-7-алкокси-6-нитро-4-хинолонов, который включает в себя: а) взаимодействие замещенного антранилата формулы (I) с диметилацеталем диметилформамида, где R обозначает алкил, с получением соединения формулы (II); b) конденсацию соединения формулы (II) с трет-бутилцианоацетатом с получением соединения формулы (III); с) гидролиз соединения формулы (III) с получением соединения формулы (IV); d) декарбоксилирование соединения формулы (IV) до соединения формулы (V); е) циклизацию соединения формулы (V) с получением 3-циано-7-алкокси-6-нитро-4-хинолона. Технический результат: новый способ получения направлен на преодоление высокотемпературной циклизации, которая может приводить к термическому разложению. 33 з.п. ф-лы.
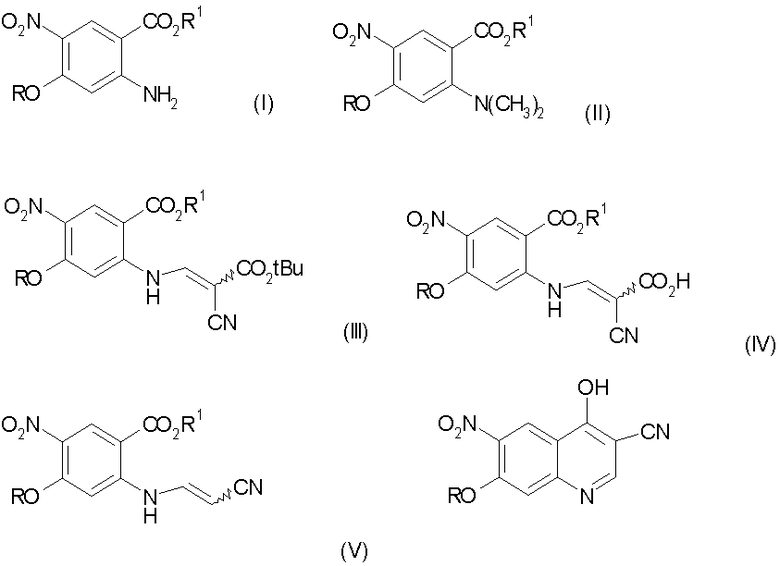
а) взаимодействие замещенного антранилата формулы (I) с диметилацеталем диметилформамида:
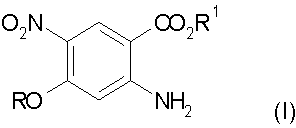
где R обозначает алкил,
с получением соединения формулы (II)
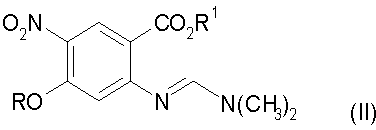
b) конденсацию соединения формулы (II) с трет-бутилцианоацетатом с получением соединения формулы (III)
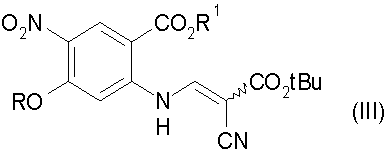
с) гидролиз соединения формулы (III) с получением соединения формулы (IV)
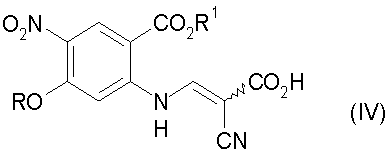
d) декарбоксилирование соединения формулы (IV) до соединения формулы (V)
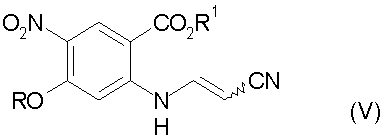
e) циклизацию соединения формулы (V) с получением 3-циано-7-алкокси-6-нитро-4-хинолона формулы
| RU 200119803 А, 10.08.2002 | |||
| Линия для изготовления древесностружечных плит | 1983 |
|
SU1207771A1 |
| WO 9714681 А1, 24.04.1997. | |||

