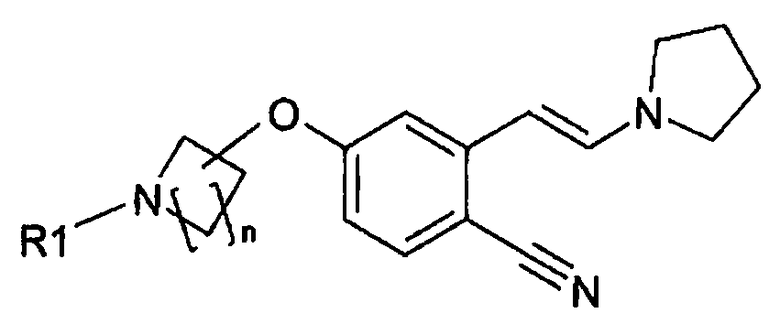
Настоящее изобретение относится к соединению формулы:
 ,
,
в которой
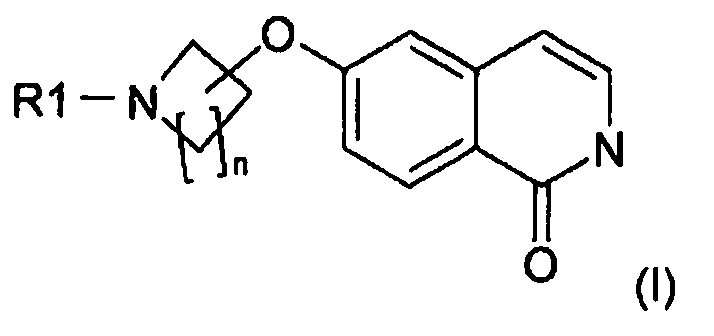
n равно 1, 2, 3 или 4 и R1 означает H или защитную группу. Настоящее изобретение также относится к новому способу получения такого соединения. Оно также относится к применению указанного соединения в качестве промежуточного продукта для получения 6-замещенного производного 1-(2H)-изохинолинона формулы (I):
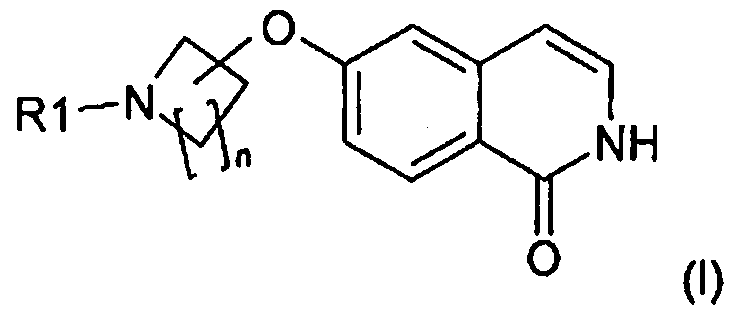 .
.
Соединения формулы (I) являются ингибиторами фермента ро-киназы или их можно использовать в качестве промежуточных продуктов для получения других ингибиторов фермента ро-киназы, которые полезны для лечения, в частности, гипертензии. Такие производные описаны, например, в WO 2007/012421, WO 2007/065916 и WO 2008/077550.
Другой путь синтеза для получения соединения формулы (I) описан в WO 2009/080335. В описанном пути используют производные алкокси-бис(диалкиламино)метана в качестве реагента для получения другого промежуточного продукта, в частности, путем использования трет-бутилокси-бис(диметиламино)метана. Однако эти реагенты являются дорогостоящими, в особенности в случае применения в крупном масштабе, и в случае трет-бутилокси-бис(диметиламино)метана реагент не является хорошо охарактеризованным.
В соответствии с этим задачей настоящего изобретения является разработка альтернативного пути получения соединения (I), который легче и дешевле осуществлять, в особенности в крупном масштабе, и который предпочтительно дает продукт с более высоким выходом. Эта задача решена с помощью настоящего изобретения, и разработан новый путь синтеза, который позволяет получать соединения формулы (I) за небольшое количество стадий химической реакции при описанных условиях проведения реакции с высоким выходом с использованием легко доступных исходных веществ и реагентов. Сами эти производные можно использовать в качестве ингибиторов ро-киназы или можно использовать в качестве промежуточного продукта синтеза других ингибиторов посредством модификации аминогруппы, содержащейся в этих соединениях, путем добавления других заместителей атома N или посредством модификации любого другого положения в изохинолиноновой системе.
Определения
Термин алкильные и соответствующие алкиленовые заместители при использовании в настоящем изобретении означает углеводородный остаток, который может быть линейным, т.е. обладать линейной цепью, или разветвленным и содержит 1, 2, 3, 4, 5 или 6 атомов углерода соответственно, как это указано, например, для (C1-C6)алкила, или (C1-C4)алкила, или (C1-C2)алкила. Это также относится к случаю, когда алкильная группа является заместителем другой группы, например, в алкоксигруппе (O-алкил) или алкоксикарбонильной группе, или арилалкильной группе. Примерами алкильных групп являются метил, этил, пропил, бутил, пентил (амил) или гексил, н-изомеры всех этих групп или разветвленные изомеры изопропил, изобутил, 1-метилбутил, изопентил, неопентил, 2,2-диметилбутил, 2-метилпентил, 3-метилпентил, изогексил, втор-бутил, трет-бутил (1,1-диметилэтил) или трет-пентил (1,1-диметилпропил, трет-амил). Соответствующими алкиленовыми группами являются метилен, этилен, пропилен и т.п.
Галоген означает фтор (F), хлор (Cl), бром (Br) или йод (I).
Арил означает фенил или нафтил, предпочтительно фенил, незамещенный или содержащий 1, 2 или 3, предпочтительно 1, заместителя, независимо выбранные из группы, включающей (C1-C4)алкил, O(C1-C4)алкил или галоген. В алкиленарильной группе, такой как -(C1-C4)алкиленарил или метиленарил, алкилен может быть один, два или три раза замещен арилом по одному и тому же или разным атомам углерода. Алкиленарил включает, например, фенилметилен (также называющийся бензилом), (трифенил)метилен (также называющийся тритилом), (дифенил)метилен (также называющийся бензгидрилом) или (4-метоксифенил)-дифенилметилен.
Подробное описание изобретения
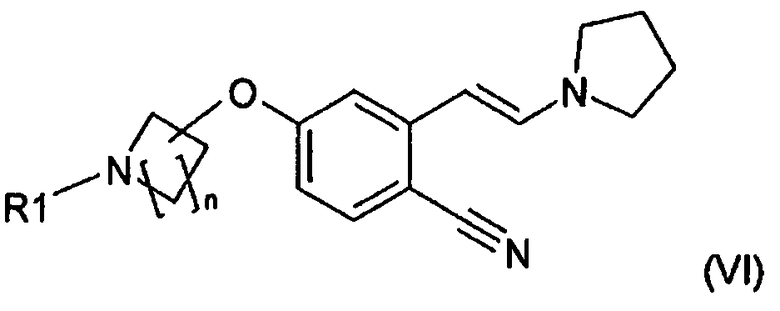
Все стадии способа получения новых соединений и их применение в качестве промежуточных продуктов для получения соединения формулы (I) представлены на схеме 1. В связи с этим соединение (VI) и стадия реакции (B) и также стадия (C) на приведенной ниже схеме являются вариантами осуществления настоящего изобретения.
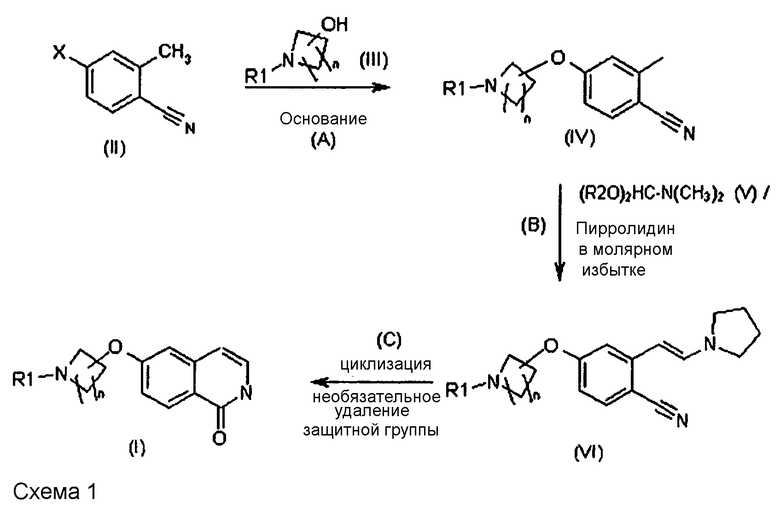
Стадии способа более подробно описаны ниже.
Получение соединения (VI)
В одном варианте осуществления настоящее изобретение относится к способу получения соединения формулы (VI):
,
в которой
n равно 1, 2, 3 или 4 и
R1 означает H или защитную группу,
включающему
(B) реакцию соединения формулы (IV):
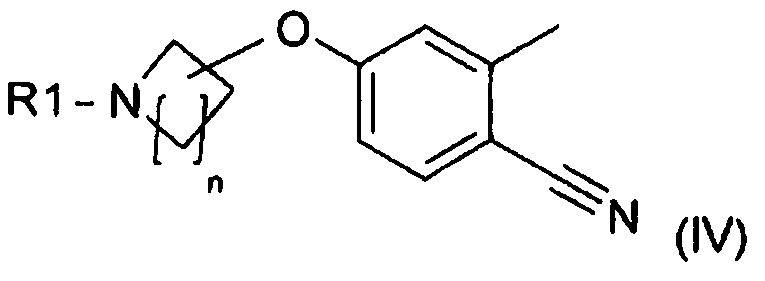 ,
,
в которой
R1 означает защитную группу и
n равно 1, 2, 3 или 4,
с реагентом формулы (R2O)2HC-N(CH3)2 (V), в которой R2 означает (C1-C6)алкил, и пирролидином,
где реагент (V) используют в молярном избытке, составляющем 1,5 или большее количество эквивалентов, и
пирролидин используют в молярном избытке, составляющем 4,0 или большее количество эквивалентов в пересчете на соединение формулы (IV), и
необязательно удаление защитной группы из соединения формулы (VI) с получением соединения формулы (VI), в которой R1 означает H.
В одном варианте осуществления способа R1 означает H. В другом варианте осуществления R1 означает защитную группу.
Формилирование 2-метилнитробензола и его производных ацеталями диметилформамида является известной исходной реакцией для так называемого синтеза индола по Леймгруберу-Батчо (Leimgruber, W.; Batcho, A. D. US Patent № 3732245), в котором сильно электроноакцепторную нитрогруппу используют для подкисления метильной группы, находящейся в орто-положении. Мягкое формилирование диметилацеталем N,N-диметилформамида (DMFDMA, (CH3O)2CH-N(CH3)2) превращает метильную группу в бета-диметиламиностирол, который циклизуется в индол после восстановления нитрогруппы в аминогруппу (схема 2).
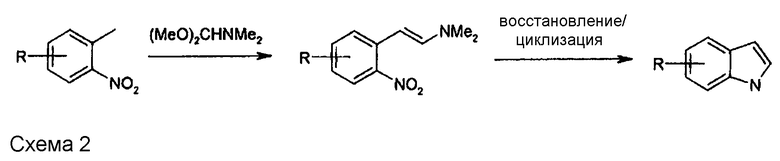
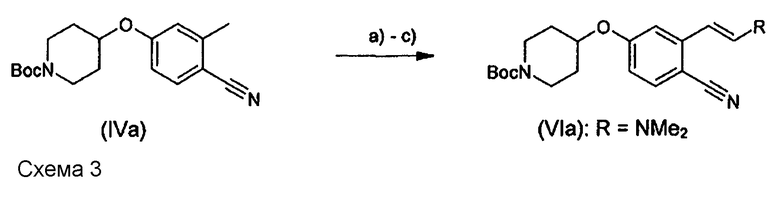
В WO 2009/080335 (схема 2) исследована реакция фенилсодержащих соединений, содержащих группу CN вместо нитрогруппы. На стр. 13 указано, что использование диалкоксиацеталей диметилформамида в соответствии с патентом US № 3732245 приводит к неудаче. Соответствующие неудовлетворительные результаты, полученные по реакции соединения формулы (IVa) в соответствии со схемой 3, в которой R1 означает трет-бутилоксикарбонил и n равно 3, с различными диалкоксиацеталями DMF, представлены ниже в таблице 1.
Другие авторы также сообщали, что эта реакция протекает при добавлении пирролидина или пиперидина к реакционной смеси. Однако не сообщали, что для этой реакции применимы и могут использоваться какие-либо заместители кроме нитрогруппы, находящейся в орто-положении по отношению к метильной группе. Нитрогруппу постоянно используют, поскольку известно, что она сильно активирует метильную группу.
В литературе описана эта реакция, в которой обычно используют примерно стехиометрические количества (1-1,5 экв.) DMFDMA и пирролидина, например, описанная в публикациях
a) Repke в J. Heterocycle Chem. (1992, 19, 845-848), где отношение нитросоединение (40)/DMFDMA/пирролидин составляет 20,6/22,7/24,0 ммоль;
b) Boini в Organic Process Research & Development (2006, 10, 1205-1211), где отношение 2-нитротолуол/DMFDMA/пирролидин составляет 2,19/2,63/2,63 моль;
c) Leonardi в Eur. J. Med. Chem. (1994, 29, 551-559), где отношение нитросоединение 17/DMFDMA/пирролидин составляет 0,077/0,115/0,115 ммоль с использованием до 3 экв., как, например, использует Ohkubo в Tetrahedron (1996, 52, 24, 8099-8112), где отношение нитросоединение 12a/DMFDMA/пирролидин составляет 3,09/9,27/9,27 моль (773 мл).
Кроме того, как отмечено выше, в предшествующем уровне техники не содержится ничего, приводящего к тому, чтобы специалист в данной области техники увеличил количество реагента-ацеталя (V) и/или использовал некоторые количества пирролидина для обеспечения превращения намного менее реакционноспособного замещенного группой CN фенильного субстрата.
В действительности, если пирролидин добавляли в количествах, обычно описанных в литературе, таких как 0,1-3,0 мол.экв. в пересчете на соединение (IVa), реакция получения соединения (VIb) не протекала или давала (VIb) лишь с низким выходом (схема 4; таблица 2 - позиции a), b) и c)).
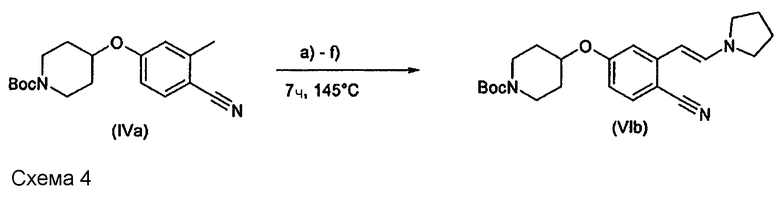
В соответствии с этим предполагается, что на таком субстрате реакция с реагентом (V) протекать не будет. Однако, в отличие от этого предположения, установлено, что в действительности реакцию можно провести при условиях, указанных в настоящем изобретении, и получить продукт с высоким выходом и высокой чистотой (см. схему 4; таблица 3 - позиции d), e) и f)).
Суммарная реакция, протекающая на этой стадии, представлена на схеме 5, где в качестве примера указано соединение формулы (IV), в которой n равно 3.
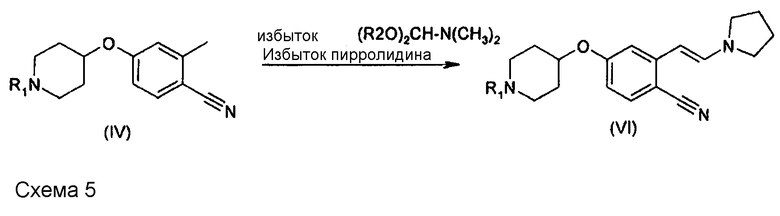
В одном варианте осуществления R2 означает (C1-C4)алкил. В другом варианте осуществления R2 означает метил и реагентом (R2O)2CH-N(CH3)2 является диметилацеталь N,N-диметилформамида (DMFDMA). В другом варианте осуществления реагента (V) R2 означает н-бутил и реагентом является дибутилацеталь N,N-диметилформамида. Другими подходящими ацеталями формамида являются диэтилацеталь N,N-диметилформамида, дипропилацеталь N,N-диметилформамида или диизопропилацеталь N,N-диметилформамида.
Относительные количества реагента (V) и пирролидина, использующиеся в пересчете на соединение (IV), являются частью настоящего изобретения. Хотя для проведения реакции существуют минимальные количества реагентов, в принципе, отсутствует максимальный избыток реагента (V) или пирролидина. Однако по практическим соображениям использующиеся количества реагента (V) и пирролидина не превышают необходимых для проведения реакции.
Поэтому в одном варианте осуществления настоящего изобретения количество реагента (V), такого как DMFDMA, в пересчете на соединение (IV) находится в диапазоне от 2,0 до 7,0 мол.экв. В другом варианте осуществления диапазон составляет 2,0-4,0 мол.экв. В другом варианте осуществления он составляет 2,0-3,0 мол.экв. В еще одном варианте осуществления количество DMFDMA составляет 2,2 мол.экв.
В одном варианте осуществления количество пирролидина находится в диапазоне от 4,0 до 9,0 мол.экв. в пересчете на соединение (IV). В другом варианте осуществления пирролидин используют в количестве, находящемся в диапазоне от 4,0 до 7,0 мол.экв. В еще одном варианте осуществления пирролидин используют в количестве, находящемся в диапазоне от 5,0 до 7,0 мол.экв. в пересчете на соединение (IV). В другом варианте осуществления пирролидин используют в количестве, составляющем примерно 6,6 мол.экв. При использовании этой смеси реагентов обеспечивается практически полное превращение соединения формулы (IV) в (VI).
Не установлено, что в этой реакции применимо другое соединение, кроме пирролидина. Аналогичные соединения, такие как пиперидин, морфолин, диметиламин или третичный амин, такой как триметиламин, описанные в данной области техники (см. обзорную статью Heterocycles, 22, 1, (1984), p. 195-221, в особенности стр. 198-200, и цитированную в ней литературу), неэффективны при использовании для этой цели.
В другом варианте осуществления стадии B), соответствующей настоящему изобретению, к реакционной смеси необязательно можно добавлять DMF (диметилформамид). Установлено, что добавление к реакционной смеси некоторого количества DMF дополнительно катализирует реакцию и дополнительно повышает выход соединения (VI). На добавляемое количество DMF не налагаются ограничения. Однако по практическим соображениям количество DMF, которое можно добавить, ограничено и не превышает необходимого. В одном варианте осуществления количество может меняться в диапазоне примерно 0,1-4,0 мол.экв., предпочтительно примерно 2,0-3,0 мол.экв. в пересчете на соединение (IV). Дополнительное увеличение количества DMF не приводит к повышению суммарного выхода. Другим преимуществом использования DMF является то, что можно уменьшить количество пирролидина и оно находится в диапазоне от 4,0 до 6,0 мол.экв. При использовании DMF длительность проведения реакции и/или температуру, при которой проводят реакцию, можно снизить и таким образом обеспечить более высокий выход и намного более чистый продукт. При отсутствии DMF выход соединения формулы (VI) равен примерно 70-85% (пример 2a), тогда как при использовании DMF выход после обработки превышает 90% (примеры 2b, 2c).
В настоящем изобретении обеспечен выход промежуточного продукта (VI), значительно превышающий суммарный выход промежуточных продуктов, полученных при реакциях, описанных в данной области техники. Настоящее изобретение также относится к более экономичному синтезу нового промежуточного продукта (VI) за счет использования легко доступных реагентов.
Соединение формулы (IV) можно добавлять непосредственно без предварительного разбавления в растворителе к смеси реагента (V) и пирролидина и необязательно DMF. Альтернативно, соединение формулы (IV) можно растворить в подходящем растворителе, таком как DMF, в соответствующем количестве, указанном выше, и затем добавить реагент (V) и пирролидин. В другом альтернативном варианте в качестве растворителя можно использовать низкокипящий простой эфир, такой как MTBE (метил-трет-бутиловый эфир). Этот растворитель предпочтительно непрерывно в значительной степени удалять путем отгонки до прибавления реагента (V) и пирролидина и необязательно DMF. Смесь нагревают и при этом удаляются оставшийся простой эфир, а также продукты, образовавшиеся по реакции соединения (V), такие как метанол или бутанол и диметиламин.
Температура, использующаяся для проведения реакции, находится в диапазоне 80°-200°C, предпочтительно 90°-180°C, более предпочтительно 120-170°C. Путем добавления DMF можно снизить температуру и подходящий диапазон температуры составляет примерно 100°C-120°C. Использующаяся длительность проведения реакции существенна для обеспечения превращения соединения (IV) в соединение (VI) и равна, например, от 2 до 27 часов. При добавлении DMF длительность проведения реакции может быть меньшей, и подходящий диапазон составляет от 2 до 10 ч.
Полученный продукт можно выделить и дополнительно очистить с помощью стандартных синтетических методик. Например, выделение можно провести путем выпаривания реакционной смеси с последующей стандартной обработкой водой и последующей кристаллизацией продукта. Альтернативно, продукт, содержащийся в реакционной смеси, также можно осадить непосредственно из реакционной смеси путем добавления подходящего антирастворителя, такого как вода и/или спирты.
Ацетали DMF общей формулы (R2O)2CH-N(CH3)2 (V), включая DMFDMA, можно получить, как это описал Brederck в Chemische Berichte 1968, 101, 41-50, или можно приобрести у различных поставщиков.
Хотя стереохимическая конфигурация енамина в формуле (VI) изображена в виде E-изомера, он может существовать в виде и E-, и Z-изомера, которые синтетически эквивалентны.
Защитную группу R1 можно выбрать из группы, указанной ниже в разделе "Защитная группа".
Получение соединения (I)
В другом варианте осуществления настоящее изобретение относится к способу получения соединения формулы (I):
или его соли, в которой
n равно 1, 2, 3 или 4 и
R1 означает H или защитную группу,
включающему стадии
(B) получения соединения формулы (VI), как показано на схеме 1:
,
в которой
n равно 1, 2, 3 или 4 и
R1 означает защитную группу;
в соответствии со способом, описанным выше,
и
(C) циклизации соединения формулы (VI) в подходящем растворителе и в присутствии галогенводородной кислоты и тем самым необязательно удаления защитной группы с получением соединения формулы (I):
 ,
,
в которой R1 означает H или защитную группу;
(D) необязательно удаления защитной группы из полученного на стадии (C) соединения формулы (I), в которой R1 означает защитную группу, с получением соединения формулы (I), в которой R1 означает H; и
(E) необязательно превращения соединения формулы (I) в его соль.
Превращение 4-гетероциклоалкокси-2-(2'-пирролидинилвинил)бензонитрилов формулы (VI) в 6-гетероциклоалкокси-1-(2H)-изохинолиноны формулы (I) не описано в литературе. В WO 2009/080335 описана циклизация диметиламинопроизводного. Установлены условия и реагенты для циклизации, что давало искомый 6-гетероциклоалкокси-1-(2H)-изохинолинон (I). Эти условия и реагенты для циклизации, применяющиеся в настоящем изобретении, являются частью настоящего изобретения.
В одном варианте осуществления реакцию циклизации 4-гетероциклоалкокси-2-(2'-пирролидинилвинил)бензонитрилов формулы (VI) с получением соединения формулы (I) можно провести по реакции соединения формулы (VI) в присутствии сильной кислоты в качестве циклизирующего реагента, т.е. провести реакцию в кислой среде. Следует понимать, что в кислой среде реакцию циклизации проводят в присутствии галогенводородной кислоты, такой как HCl, HBr или HI, предпочтительно HCl, в подходящем растворителе, таком как спирт, предпочтительно с использованием в качестве растворителя (C1-C6)-алканола, такого как метанол, этанол, пропанол, бутанол или пентанол. Можно использовать н-спирты, а также изомеры. Реакцию предпочтительно проводят в метаноле, этаноле, н-пропаноле или н-бутаноле, и н-бутанол является наиболее предпочтительным.
В качестве источника галогенводородной кислоты можно использовать газообразный HCl, или HBr, или HI и его добавляют к спирту. В качестве альтернативы использованию газообразного HCl также можно использовать другие реагенты, такие как TMSCl или AcCl (ацетилхлорид), которые взаимодействуют со спиртом с образованием безводного спиртового раствора HCl. Предпочтительный набор условий проведения реакции циклизации включает использование газообразного HCl в (C1-C6)-алканоле, таком как н-бутанол, в качестве растворителя.
Реакцию проводят при температуре, находящейся в диапазоне от 40°C до 140°C, более предпочтительный диапазон температуры составляет от 60°C до 120°C в зависимости от температуры кипения использующегося спирта.
Реакцию проводят с использованием от 2 до 30 мол.экв. галогенводородной кислоты, такой как газообразный HCl, более предпочтительно с использованием от 3 до 15 мол.экв. В промышленном масштабе избыток галогенводородной кислоты, такой как HCl, можно легко нейтрализовывать в щелочном скруббере.
При реакции циклизации защитную группу необязательно также можно удалить одновременно с получением соединения формулы (I), в которой R1 означает H. О выборе защитной группы в R1 с получением соединения формулы (I), в которой R1 означает H или защитную группу, см. раздел "Защитная группа".
Получение соединения (IV)
Соединение формулы (IV), применяющееся в связи со способом, предлагаемым в настоящем изобретении, для получения соединения формулы (VI), можно получить с помощью нуклеофильного ароматического замещения. Реакция получения соединения формулы (IV) описана в WO 2009/080335.
Соединение формулы (IV) получают с помощью
(A) реакции соединения формулы (II):
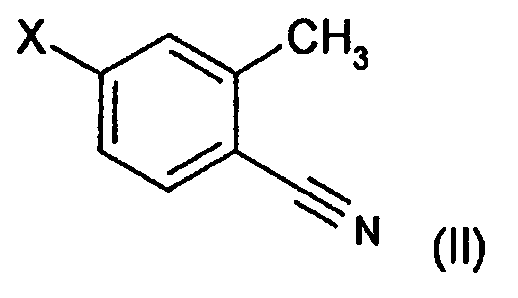 ,
,
в которой X означает галоген,
в подходящем растворителе и в присутствии основания, выбранного из группы, включающей алкоксид щелочного металла, гидрид щелочного металла или щелочной металл, с соединением формулы (III):
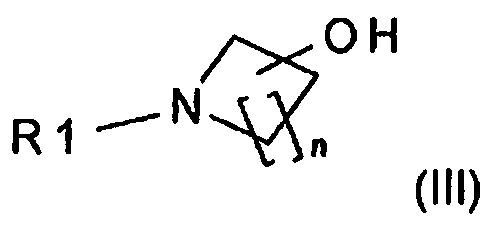 ,
,
в которой
R1 означает H или защитную группу и
n равно 1, 2, 3 или 4,
с получением соединения формулы (IV):
,
и если R1 означает H, то аминогруппу в соединении формулы (IV) защищают с получением соединения формулы (IV), в которой R1 означает защитную группу.
В одном варианте осуществления соединение (III) защищают до его реакции с соединением (II). Подходящим защищенным спиртом формулы (III) является 1-бензил-3-пирролидинол, трет-бутиловый эфир 3-гидроксипиперидин-1-карбоновой кислоты, 1-бензгидрилазетидин-3-ол или трет-бутиловый эфир 4-гидроксипиперидин-1-карбоновой кислоты.
Условия, при которых можно получить соединение (IV), являются следующими.
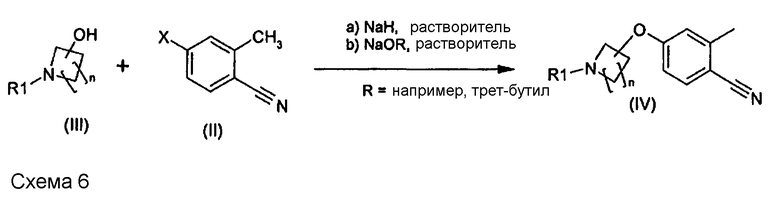
В одном варианте осуществления основание выбрано из группы, включающей трет-бутоксид натрия или калия (KOtBu), трет-амилат натрия или калия. В другом варианте осуществления основанием является NaH. В более предпочтительном варианте осуществления в качестве оснований используют трет-бутоксид калия или трет-амилат калия, наиболее предпочтительно использовать трет-бутоксид калия.
Растворителями, которые можно использовать на этой стадии реакции, включая варианты a) и b), описанные выше на схеме 6, являются простые эфиры, такие как тетрагидрофуран (THF), 2-метил-THF, метил-трет-бутиловый эфир (MTBE), диоксан, диметоксиэтан (DME) или диметоксиметан, а также диполярные апротонные растворители, такие как диметилсульфоксид (DMSO), N-метилпирролидон (NMP), N-этилпирролидон, диметилформамид (DMF) или диметилацетамид. В одном варианте осуществления смесью основание/растворитель является трет-бутоксид калия с MTBE.
Использующаяся температура обычно находится в диапазоне от 4°C до 220°C, предпочтительно в диапазоне от 80°C до 200°C и более предпочтительно в диапазоне от 40°C до 140°C. При использовании растворителей, обладающих более низкими температурами кипения, реакцию можно провести под давлением в автоклаве. Длительность проведения реакции не является критически важной и может меняться в зависимости от использующихся растворителя и температуры. Реакцию проводят, пока не прореагирует большая часть или все предшественники (II) и (III), что обычно происходит за несколько часов и обычно завершается не более чем за 12 ч.
Получение соединения формулы (IV) также является объектом настоящего изобретения, если затем соединение (IV) превращают в соединение (VI) в соответствии со способом, предлагаемым в настоящем изобретении.
Защитная группа
Защитную группу, применимую на одной из указанных выше стадий реакции A), B) и C) и в соответствующих промежуточных продуктах, можно выбрать из множества групп, например, перечисленных в следующей публикации, но не ограничиваясь только ими: T.W. Greene and P.G.M. Wuts: Protective Groups in Organic Synthesis, Third Edition, John Wiley and Sons, New York, 1999 Chapter 7, page 494. Кроме того, дается ссылка на WO 2009/080335, где описаны группы, подходящие для синтеза соединений формулы (IV), и (VI), и (I).
Защитной группой R1 предпочтительно является такая, которая стабильна при условиях проведения реакции в щелочной среде, использующихся на стадиях A) и B). Подходящие стабильные защитные группы R1, применимые на стадии A) и также на стадиях B) и C) и в промежуточных продуктах (III), (IV) и (VI), можно выбрать из группы, включающей карбаматы, такие как трет-бутилоксикарбонил и бензилоксикарбонил или п-метоксибензилкарбонил, амиды, такие как формил или ацетил, N-алкиленарилы, такие как бензил, (дифенил)метилен, тритил или (4-метоксифенил)дифенилметилен, или N-P и N-сульфонильные защитные группы, такие как диалкилфосфорамидаты и п-толуолсульфонил.
Защитную группу можно ввести по методикам, известным в данной области техники, когда N-гетероциклоалкиловый спирт формулы (III), в которой R1 означает H, взаимодействует с соответствующей защитной группой с образованием реагента для получения защищенного амина. В другом варианте осуществления защитную группу можно ввести в соединение формулы (IV), если R1 означает H, получаемое по указанной выше реакции. Подходящие реагенты, использующиеся для введения защитной группы, известны в данной области техники и имеются в продаже. Например, для введения трет-бутилоксикарбонильной группы можно использовать ди-трет-бутилдикарбонат.
Предпочтительно, если во всем синтезе используют одну и ту же защитную группу. В соответствии с этим на стадиях A), B) и C) предпочтительно использовать защитную группу, стабильную при условиях проведения реакции в щелочной среде. Наиболее подходящими являются стабильные в щелочи, но нестабильные в кислоте защитные группы, которые можно одновременно отщепить на одной и той же стадии реакции, когда протекает реакция циклизации и на стадии C) получают соединение формулы (I), в которой R1 означает H.
В одном варианте осуществления настоящего изобретения нестабильную в кислоте защитную группу используют в качестве защитной группы для R1 в соединении формулы (III), (IV) и (VI). В одном варианте осуществления для R1 используют такую нестабильную в кислоте трет-бутилоксикарбонильную группу, которая также стабильна при реакциях получения соединения (IV) в щелочной среде. При использовании нестабильной в кислоте группы реакция циклизации соединения формулы (VI) с галогенводородной кислотой прямо дает соединение формулы (I), в которой R1 означает H. При использовании нестабильной в кислоте группы можно исключить отдельную стадию (D) удаления защитной группы.
Когда желательно удалить защитную группу после стадии реакции (C), удаление защитной группы можно провести на отдельной стадии (D) с предварительным выделением промежуточного продукта, содержащего защитную группу, или реакционную смесь, полученную после реакции циклизации, можно непосредственно использовать на стадии удаления защитной группы.
В одном варианте осуществления способом, предлагаемым в настоящем изобретении, получают соединение формулы (I), в которой R1 означает H. В другом варианте осуществления соединение формулы (I), в которой R1 означает H, непосредственно получают на стадии (C) путем удаления защитной группы.
Соединение формулы (I), в которой R1 означает H или защитную группу, предпочтительно H, необязательно превращают в его соль. Соединение (I) можно прямо получить в виде соли, если кислоту не удаляют на стадии циклизации для получения свободного основания. Кислоту, использующуюся на стадии циклизации, также можно удалить и заменить другой кислотой по известным методикам и получить соответствующую соль соединения формулы (I).
Соли соединения формулы (I), включая фармацевтически приемлемые соли, можно получить из неорганических кислот, таких как хлористоводородная кислота, бромистоводородная, фосфорная, метафосфорная, азотная и серная кислота, и органических кислот, таких как, например, уксусная кислота, бензолсульфоновая, бензойная, лимонная, этансульфоновая, фумаровая, глюконовая, гликолевая, молочная, малеиновая, яблочная, метансульфоновая, янтарная, п-толуолсульфоновая и винная кислота, по методикам, известным в данной области техники.
В другом варианте осуществления настоящего изобретения соединение формулы (I), в которой R1 означает H, полученное способом, предлагаемым в настоящем изобретении, можно использовать в качестве промежуточного продукта для синтеза других его производных, содержащих заместители R1, не представляющие собой H, для получения ингибиторов ро-киназы. Настоящее изобретение относится к способу получения соединения формулы (I), в которой R1 означает H, и на второй стадии получают соединение формулы (I´), в которой R1 превращается в R7 по реакции подходящего химического эквивалента группы R7 с соединением формулы (I). Например, подходящий альдегид R7-C(O)H, в котором R7 означает, например, (C1-C6)алкил или другую замещенную (C1-C6)алкильную группу, можно ввести в реакцию восстановительного аминирования, как это описано в WO 2007/012421, с соединением формулы (I), в которой R1 означает H, с получением (C1-C6)алкилзамещенного 6-гетероциклоалкокси-1-(2H)-изохинолинона (I´).
В другом варианте осуществления настоящее изобретение относится к способу получения соединения формулы (I):
или его соли,
в которой
n равно 1, 2, 3 или 4 и R1 означает H или защитную группу,
включающему стадии
(C) циклизации соединения формулы (VI):
 ,
,
в которой R1 означает защитную группу и
n равно 1, 2, 3 или 4,
в подходящем растворителе и в присутствии галогенводородной кислоты и тем самым необязательно удаления защитной группы с получением соединения формулы (I):
,
в которой R1 означает H или защитную группу;
(D) необязательно удаления защитной группы из соединения формулы (I), если R1 означает защитную группу, с получением соединения формулы (I), в которой R1 означает H; и
(E) необязательно превращения соединения формулы (I) в его соль.
Этот способ соответствует стадии циклизации (C) в описанном выше синтезе соединения формулы (I). В соответствии с этим формулировки и варианты осуществления, указанные выше в связи со стадией (C), (D) и (E), также применимы к этому варианту осуществления способа.
В другом варианте осуществления настоящее изобретение относится к соединению формулы (VI):
,
в которой R1 означает H или защитную группу и n равно 1, 2, 3 или 4. В одном варианте осуществления R1 означает защитную группу. В другом варианте осуществления R1 означает H.
В одном варианте осуществления соединения формулы (VI) R1 означает защитную группу, обладающую характеристиками, указанными выше для защитной группы, включая то, что она представляет собой нестабильную в кислоте защитную группу, предпочтительно выбранную из группы, включающей карбамат, такой как трет-бутилоксикарбонильную группу, бензилоксикарбонильную группу или п-метоксибензилкарбонил.
В одном варианте осуществления защитной группой является трет-бутилоксикарбонил и таким образом в другом варианте осуществления соединением формулы (VI) является трет-бутиловый эфир 4-[4-циано-3-((E)-2-пирролидин-1-илвинил)фенокси]пиперидин-1-карбоновой кислоты (VIb), описывающийся формулой
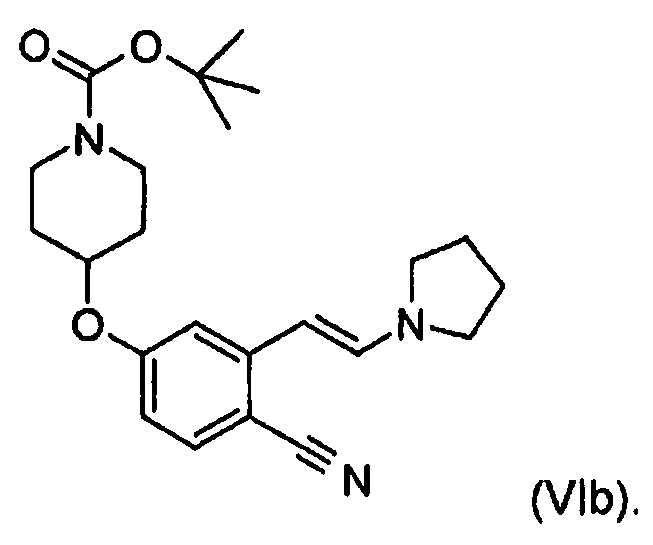
В другом варианте осуществления настоящего изобретения в любом из соединений формулы (I), (III), (IV) или (VI) n равно 2. В другом варианте осуществления этих соединений n равно 3.
Кислород (O) может быть связан с N-содержащим кольцом в любом из соединений формулы (I), (III), (IV) или (VI) в любом положении через кольцевой атом углерода. В одном варианте осуществления n равно 3 и O присоединен в положении 4 получающегося в результате пиперидинового кольца с образованием соединения формулы (Ic):
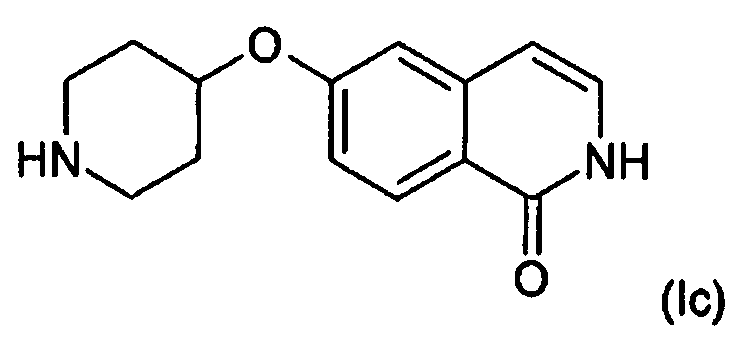 ,
,
или, в другом варианте осуществления, O присоединен в положении 3 пиперидинового кольца с образованием соединения формулы (Id):
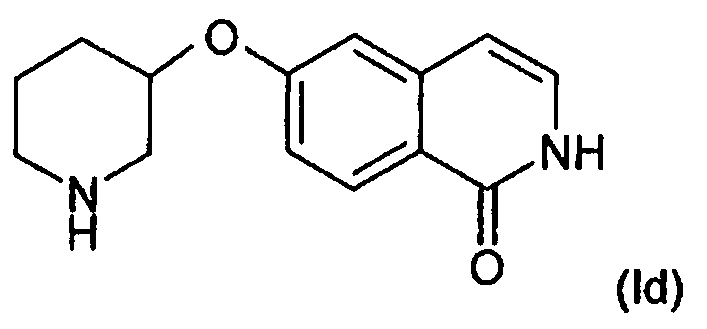 ,
,
во всех его стереоизомерных формах.
В другом варианте осуществления O присоединен в положении 3 пирролидинового кольца с образованием соединения формулы (Ie) во всех его стереоизомерных формах:
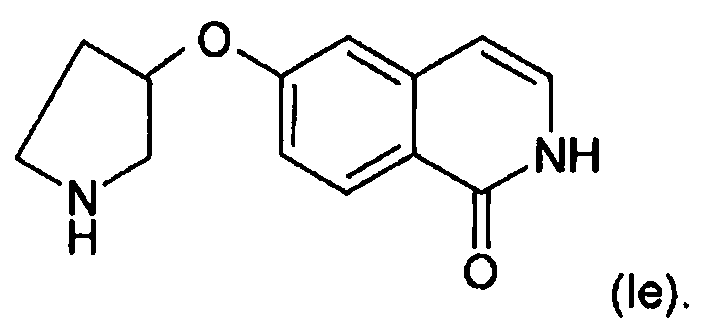
Кроме того, в данной области техники известно, что 2H-изохинолин-1-оны формулы (I), приведенные на схемах, также могут существовать в своей таутомерной форме в виде 1-гидроксиизохинолинов и эти таутомеры входят в объем настоящего изобретения. Кроме того, соединение формул (I), (IV) или (VI) может содержать хиральный атом углерода. Соответственно, эти соединения существуют в стереизомерных формах, включая энантиомеры или диастереоизомеры. Эти стереизомерные формы и смеси стереизомерных форм во всех соотношениях входят в объем настоящего изобретения.
В одном варианте осуществления способа, предлагаемого в настоящем изобретении, получают 6-(пиперидин-4-илокси)-2H-изохинолин-1-он или его соль. В другом варианте осуществления получают 6-(пиперидин-3-илокси)-2H-изохинолин-1-он или его соль. В другом варианте осуществления получают 6-(пирролидин-3-илокси)-2H-изохинолин-1-он или его соль. В одном варианте осуществления солью всех этих соединений является гидрохлорид.
Соединение формулы (VI) можно использовать в качестве промежуточного продукта для получения ингибиторов ро-киназы. Таким образом, настоящее изобретение также относится к применению соединения формулы (VI):
,
в которой R1 означает H или защитную группу, предпочтительно R1 означает защитную группу, и
n равно 1, 2, 3 или 4,
для получения соединения формулы (I):
,
в которой
n равно 1, 2, 3 или 4 и
R1 означает H или защитную группу, предпочтительно R1 означает H.
ПРИМЕРЫ
В приведенных ниже примерах более подробно описаны способы и промежуточные продукты, предлагаемые в настоящем изобретении. Соответственно, приведенные ниже примеры являются частью и вариантами осуществления настоящего изобретения. Кроме того, они предназначены для иллюстрации, а не для ограничения настоящего изобретения.
Аббревиатуры
Примеры
1) Трет-бутиловый эфир 4-(4-циано-3-метилфенокси)пиперидин-1-карбоновой кислоты (ММ (молекулярная масса)=316,40 г/мол.)
a) 1,35 г 4-Фтор-2-метилбензонитрила, растворенного в DMF, добавляли к 2,11 г трет-бутилового эфира 4-гидроксипиперидин-1-карбоновой кислоты и 0,6 г гидрида натрия в 30 мл DMF. Смесь перемешивали при комнатной температуре (КТ) до завершения реакции. Реакцию останавливали водой. Водный слой экстрагировали этилацетатом (AcOEt) или метил-трет-бутиловым эфиром (MTB-эфиром). Объединенные органические слои промывали рассолом, сушили и концентрировали с получением 2,6 г (выход 81%) трет-бутилового эфира 4-(4-циано-3-метилфенокси)пиперидин-1-карбоновой кислоты. Масс-спектр: (C18H24N2O3): рассчитано 316, найдено 261 [M+H-t(C4H9)]+.
b) 22,6 г Трет-бутоксида калия, 33,2 г трет-бутилового эфира 4-гидроксипиперидин-1-карбоновой кислоты и 300 мл MTB-эфира перемешивали в течение 1 ч при кипячении с обратным холодильником. К суспензии за 20 мин добавляли раствор 20,3 г 4-фтор-2-метилбензонитрила в 250 мл MTB-эфира и кипячение с обратным холодильником продолжали в течение 7 ч. Реакцию останавливали водой. Органический слой отделяли и промывали водой и концентрировали с получением 54,8 г (выход 92%) трет-бутилового эфира 4-(4-циано-3-метилфенокси)пиперидин-1-карбоновой кислоты. 1H ЯМР (500 МГц, d6-DMSO) δ 1,40 (с, 9H), 1,47-1,55 (м, 2H), 1,88-1,95 (м, 2H), 2,43 (с, 3H), 3,13-3,22 (м, 2H), 3,63-3,70 (м, 2H), 4,66-4,73 (м, 1H), 6,96 (дд, J=8,6, 2,4 Гц, 1H), 7,07 (д, J=2,3 Гц, 1H), 7,67 (д, J=8,7 Гц, 1H).
2) Трет-бутиловый эфир 4-[4-циано-3-((E)-2-пирролидин-1-илвинил)фенокси]пиперидин-1-карбоновой кислоты (ММ=397,52 г/мол.)
a) 47,3 г (0,15 моль) Трет-бутилового эфира 4-(4-циано-3-метилфенокси)пиперидин-1-карбоновой кислоты, 43,8 мл (0,33 моль) диметилацеталя N,N-диметилформамида и 81,9 мл пирролидина (0,99 моль) смешивали и за 30 мин смесь нагревали до 90°C и выдерживали при этой температуре в течение 2 ч. Все летучие компоненты отгоняли. Температуру повышали до 120°C и выдерживали при этой температуре в течение 27 ч. Нагревание прекращали и темный высоковязкий остаток растворяли в 400 мл MTB-эфира, дважды промывали с помощью 200 мл насыщенного водного раствора NaHCO3 и один раз с помощью 200 мл воды. Органический слой концентрировали и перекристаллизовывали из смеси изопропанол/вода (245 мл/105 мл), собирали и сушили с получением 49,5 г (выход 83%, чистота 96,4%) трет-бутилового эфира 4-[4-циано-3-((E)-2-пирролидин-1-илвинил)фенокси]пиперидин-1-карбоновой кислоты.
b) 94,6 г Трет-бутилового эфира (0,3 моль) 4-(4-циано-3-метилфенокси)пиперидин-1-карбоновой кислоты, 87,6 мл (0,66 моль) диметилацеталя N,N-диметилформамида, 109 мл (1,32 моль) пирролидина и 60,4 мл DMF (0,78 моль) смешивали и смесь за 30 мин нагревали до 90°C и выдерживали при этой температуре в течение 1 ч. Все летучие компоненты отгоняли. Температуру повышали до 110°C и выдерживали при этой температуре в течение 2 ч. Температуру устанавливали равной 120°C и выдерживали при этой температуре в течение 7,5 ч. Нагревание прекращали и смесь охлаждали до температуры окружающей среды. Затем добавляли 200 мл воды и 400 мл изопропанола. Смесь перемешивали в течение 3 ч при температуре окружающей среды и в течение 1 ч при 5°C для осаждения продукта. Твердое вещество собирали, промывали смесью изопропанол/вода (70/30), собирали и сушили с получением 113,7 г (выход 95,5%, чистота 93,3%) трет-бутилового эфира 4-[4-циано-3-((E)-2-пирролидин-1-илвинил)фенокси]пиперидин-1-карбоновой кислоты.
c) 47,3 г (0,15 моль) Трет-бутилового эфира 4-(4-циано-3-метилфенокси)пиперидин-1-карбоновой кислоты, 43,8 мл (0,33 моль) диметилацеталя N,N-диметилформамида, 54,6 мл (0,66 моль) пирролидина и 30,2 мл DMF (0,39 моль) смешивали и смесь за 30 мин нагревали до 90°C и выдерживали при этой температуре в течение 1 ч. Все летучие компоненты отгоняли. Температуру повышали до 110°C и выдерживали при этой температуре в течение 2,5 ч. Температуру устанавливали равной 120°C и выдерживали при этой температуре в течение 5 ч. Нагревание прекращали и смесь охлаждали до температуры окружающей среды, разбавляли с помощью 400 мл MTB-эфира и дважды промывали с помощью 200 мл насыщенного водного раствора NaHCO3 и один раз с помощью 200 мл воды. Органический слой концентрировали и перекристаллизовывали из смеси изопропанол/вода (245 мл/105 мл), собирали и сушили с получением 55,5 г (выход 93%, чистота 100%) трет-бутилового эфира 4-[4-циано-3-((E)-2-пирролидин-1-илвинил)фенокси]пиперидин-1-карбоновой кислоты.
Почти белое кристаллическое твердое вещество, температура плавления 115-117°C, 1H ЯМР (500 МГц, d6-DMSO) δ 1,40 (с, 9H), 1,46-1,55 (м, 2H), 1,85-1,94 (м, 2H), 2,49-2,52 (м, 4H), 3,17-3,26 (м, 2H), 3,26-3,30 (м, 4H), 3,62-3,70 (м, 2H), 4,67-4,76 (м, 1H), 5,09 (д, J=13,6 Гц, 1H), 6,60 (дд, J=8,8, 2,4 Гц, 1H), 7,09 (д, J=2,4 Гц, 1H), 7,46 (д, J=8,9 Гц, 1H), 7,68 (д, J=13,7 Гц, 1H).
3) Гидрохлорид 6-(пиперидин-4-илокси)-2H-изохинолин-1-она (ММ=280,80 г/мол.)
42 г Трет-бутилового эфира 4-[4-циано-3-((E)-2-пирролидин-1-илвинил)фенокси]пиперидин-1-карбоновой кислоты при 15°C порциями добавляли к 135 мл 1-бутанола, насыщенного с помощью 48 г газообразного HCl. Смесь нагревали при 63°C в течение 1 ч и перемешивали при 63-65°C до завершения реакции. Содержащий HCl растворитель заменяли с помощью последующей отгонки и добавления свежего 1-бутанола и осадок собирали, промывали 1-бутанолом и сушили с получением 31,2 г (выход 106%, чистота 96,5%) гидрохлорида 6-(пиперидин-4-илокси)-2H-изохинолин-1-она.
1H ЯМР (500 МГц, d6-DMSO) δ 1,85-1,95 (м, 2H), 2,13-2,22 (м, 2H), 3,04-3,14 (м, 2H), 3,20-3,29 (м, 2H), 4,79-4,86 (м, 1H), 6,44 (д, J=7,1 Гц, 1H), 7,10 (дд, J=8,9, 2,5 Гц, 1H), 7,14 (дд, J=7,2, 6,7 Гц, 1H), 7,22 (д, J=2,5 Гц, 1H), 8,09 (д, J=8,6 Гц, 1H), 8,97-9,13 (уш.с, 2H), 11,09 (уш.д, J=5 Гц, 1H).
| название | год | авторы | номер документа |
|---|---|---|---|
| 4-ЗАМЕЩЕННЫЕ-2-ФЕНОКСИФЕНИЛАМИНОВЫЕ МОДУЛЯТОРЫ ДЕЛЬТА-ОПИОИДНЫХ РЕЦЕПТОРОВ | 2010 |
|
RU2553453C2 |
| СПОСОБ ПОЛУЧЕНИЯ СОЕДИНЕНИЯ, ПРИМЕНИМОГО В КАЧЕСТВЕ ИНГИБИТОРА TAFIa | 2010 |
|
RU2538506C2 |
| СПОСОБ ПРОСТОГО И УДОБНОГО ПОЛУЧЕНИЯ ВАБОРБАКТАМА | 2019 |
|
RU2770434C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ ДЛЯ ПОЛУЧЕНИЯ АВИБАКТАМА | 2018 |
|
RU2722625C1 |
| (2S,5R)-5-[(бензилокси)амино]пиперидин-2-карбоксамид | 2012 |
|
RU2610091C2 |
| СПОСОБ ПОЛУЧЕНИЯ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ, ВКЛЮЧАЯ ТРАНС-7-ОКСО-6-(СУЛЬФОКСИ)-1,6-ДИАЗАБИЦИКЛО[3.2.1]ОКТАН-2-КАРБОКСАМИД И ЕГО СОЛИ | 2012 |
|
RU2769076C2 |
| СИНТЕЗ ИНГИБИТОРОВ РЕНИНА С ИСПОЛЬЗОВАНИЕМ РЕАКЦИИ ЦИКЛОПРИСОЕДИНЕНИЯ | 2006 |
|
RU2423348C2 |
| ИНГИБИТОРЫ БЕТА-ЛАКТАМАЗ | 2009 |
|
RU2445314C9 |
| ФЕНОКСИЗАМЕЩЕННЫЕ ПИРИМИДИНЫ ДЛЯ ИСПОЛЬЗОВАНИЯ В КАЧЕСТВЕ МОДУЛЯТОРОВ ОПИОИДНЫХ РЕЦЕПТОРОВ | 2010 |
|
RU2554870C2 |
| ГЕТЕРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ | 2020 |
|
RU2835082C2 |
Изобретение относится к новым замещенным фенилсодержащим соединениям формулы (VI), которые используют в качестве промежуточных продуктов для получения 6-замещенных производных 1-(2H)-изохинолинона. 4 н. и 16 з.п. ф-лы, 3 табл., 1 пр.
1. Способ получения соединения формулы:
,
в которой
n равно 1, 2, 3 или 4 и
R1 означает H или защитную группу,
включающий
(B) реакцию соединения формулы (IV):
,
в которой R1 означает защитную группу,
со смесью реагента формулы (R2O)2HC-N(CH3)2 (V), в которой R2 означает (C1-C6)алкил, и пирролидина,
где реагент (V) используют в молярном избытке, составляющем 1,5 или большее количество эквивалентов, и пирролидин используют в молярном избытке, составляющем 4,0 или большее количество эквивалентов в пересчете на соединение формулы (IV); и
необязательно удаление защитной группы из соединения формулы (VI) с получением соединения формулы (VI), в которой R1 означает H.
2. Способ по п. 1, в котором используют от 2,0 до 7,0 экв. реагента (V) в пересчете на соединение формулы (IV).
3. Способ по п. 1 или 2, в котором реагентом (V) является диметилацеталь N,N-диметилформамида.
4. Способ по п. 1 или 2, в котором используют от 4,0 до 9,0 экв. пирролидина в пересчете на соединение формулы (IV).
5. Способ по п. 1 или 2, в котором добавляют DMF.
6. Способ по п. 1 или 2, в котором n равно 2 или 3.
7. Способ по п. 1 или 2, в котором защитная группа является нестабильной в кислоте.
8. Способ по п. 1 или 2, в котором защитной группой в R1 является трет-бутоксикарбонил.
9. Способ получения соединения формулы (I):
или его соли, в которой
n равно 1, 2, 3 или 4 и
R1 означает H или защитную группу,
включающий стадии
(B) получения соединения формулы (VI):
,
в которой R1 означает защитную группу и
n равно 1, 2, 3 или 4;
(C) циклизации соединения формулы (VI) в присутствии галогенводородной кислоты в подходящем растворителе и тем самым необязательно удаления защитной группы с получением соединения формулы (I), в которой R1 означает H или защитную группу;
(D) необязательно удаления защитной группы из соединения формулы (I), если R1 означает защитную группу, с получением соединения формулы (I), в которой R1 означает H; и
(E) необязательно превращение соединения формулы (I) в его соль.
10. Способ по п. 9, в котором галогенводородной кислотой, использующейся на стадии циклизации, является HCl.
11. Способ по п. 9, в котором n равно 2 или 3.
12. Способ по п. 9, в котором защитная группа является нестабильной в кислоте.
13. Способ по любому из пп. 9-12, в котором защитной группой в R1 является трет-бутоксикарбонил.
14. Способ по п. 1, в котором соединение формулы (IV) получают по реакции соединения формулы (II)
,
в которой X означает галоген,
в подходящем растворителе и в присутствии основания, выбранного из группы, включающей алкоксид щелочного металла, гидрид щелочного металла или щелочной металл, с соединением формулы (III)
,
в которой
R1 означает H или защитную группу и
n равно 1, 2, 3 или 4,
и если R1 означает H, то аминогруппу в соединении формулы (IV) защищают с получением соединения формулы (IV), в которой R1 означает защитную группу.
15. Соединение формулы (VI):
,
в которой R1 означает H или защитную группу и
n равно 1, 2, 3 или 4.
16. Соединение по п. 15, в котором защитная группа является нестабильной в кислоте защитной группой.
17. Соединение по п. 16, в котором нестабильной в кислоте защитной группой является трет-бутилоксикарбонил.
18. Соединение по любому из пп. 15-17, в котором n равно 2 или 3.
19. Соединение по п. 15, которым является трет-бутиловый эфир 4-[4-циано-3-((E)-2-пирролидин-1-илвинил)фенокси]пиперидин-1-карбоновой кислоты.
20. Применение соединения по любому из пп. 15-19 для получения соединения формулы (I):
или его соли,
в которой
n равно 1, 2, 3 или 4 и R1 означает H или защитную группу.
| ПИПЕРИДИНИЛ-ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ ИЗОХИНОЛОНА КАК ИНГИБИТОРЫ Rho-КИНАЗЫ | 2006 |
|
RU2414467C2 |
| WO 2009080335 A1, 02.07.2009 | |||
| US 3732245 A1, 08.05.1973 | |||
| BOINI ET AL., "Development of a Manufacturing Process for 1-(1-Pyridin-2-yl methyl-piperidin-4-yl)-1 H -indole: A Key Intermediate for Protein Kinase C Inhibitor LY317615", ORGANIC PROCESS RESEARCH & DEVELOPMENT, 2006, vol | |||
| Печь-кухня, могущая работать, как самостоятельно, так и в комбинации с разного рода нагревательными приборами | 1921 |
|
SU10A1 |
| Приспособление для точного наложения листов бумаги при снятии оттисков | 1922 |
|
SU6A1 |

