Предпосылки создания изобретения
Валсартан избирательно блокирует связывание ангиотензина II с AT1-рецептором, вызывая расширение кровеносных сосудов, и уменьшает секрецию альдостерона. Современные клинические исследования позволили выявить дополнительные благоприятные действия валсартана у ряда пациентов, перенесших острые сосудистые заболевания. Учитывая, что активация тромбоцитов играет основную роль в патогенезе коронарных и цереброваскулярных окклюзий и что AT1-рецепторы присутствуют на поверхности тромбоцитов, проводили оценку in vitro воздействий валсартана и его основного метаболита, образующегося в печени, валерил-4-гидроксивалсартана, на тромбоциты у пациентов, имеющих множественные факторы риска возникновения сосудистых заболеваний.
В заявке осуществлено реальное сканирование зависимости агрегации тромбоцитов, полученной при исследовании пациента, при исходном уровне и после инкубации с 100 мкМ валсартаном и 1 мкМ валерил-4-гидроксивалсартаном. Высокие концентрации валсартана существенно ингибируют индуцируемую АДФ агрегацию, не оказывая влияния на индуцируемую эпинефрином способность к агрегации, в то время как метаболит блокирует индуцируемую эпинефрином агрегацию в диапазоне терапевтических концентраций.
Краткое изложение сущности изобретения
Одним из объектов настоящего изобретения является способ ингибирования агрегации тромбоцитов, заключающийся в том, что пациенту, который нуждается в этом, вводят терапевтически эффективное количество блокатора рецептора ангиотензина II ("АРБ"), предпочтительно валсартана, или его фармацевтически приемлемой соли необязательно в сочетании с фармацевтически приемлемым носителем.
Другим объектом настоящего изобретения является способ ингибирования агрегации тромбоцитов, заключающийся в том, что пациенту, который нуждается в этом, вводят терапевтически эффективное количество метаболита АРБ, предпочтительно метаболита валсартана, а именно валерил-4-гидроксивалсартана, необязательно в сочетании с фармацевтически приемлемым носителем.
Еще одним объектом настоящего изобретения является способ лечения состояний, связанных с агрегацией тромбоцитов, заключающийся в том, что пациенту, который нуждается в этом, вводят АРБ или метаболит АРБ. Состояния, связанные с агрегацией тромбоцитов, включают острый инфаркт миокарда, ишемический «удар», стенокардию, острые коронарные синдромы, TIA (приступы транзиторной ишемии или острые цереброваскулярные синдромы), сердечную недостаточность, боль в груди, имеющую ишемическую этиологию, синдром X, тромбоэмболию, легочную гипертензию, сахарный, диабет, заболевания периферических сосудов, тромбоз глубоких вен, артериальный тромбоз любого сосуда, связанную с введением катетера тромбозную окклюзию или реокклюзию.
Следующим объектом настоящего изобретения являются фармацевтические композиции, содержащие АРБ или метаболит АРБ и фармацевтически приемлемый носитель.
Подробное описание предпочтительных вариантов осуществления изобретения
Антагонисты AT1-рецептора (также называемые антагонистами рецептора ангиотензина II) представляют собой действующие вещества, которые связываются с подтипом AT1-рецептора рецептора ангиотензина II, но не приводят к активации рецептора. Благодаря тому, что они ингибируют AT1-рецептор, эти антагонисты можно применять, например, для предупреждения агрегации тромбоцитов и лечения, связанных с этим состояний.
Класс антагонистов AT1-рецептора включает соединения, имеющие различные структуры, наиболее предпочтительными являются соединения, не относящиеся к пептидам. АРБ, подпадающие под объем настоящего изобретения, включают валсартан, представляющий собой антагонист рецептора AT1, т.е. (S)-N-(1-карбокси-2-метилпроп-1-ил)-N-пентаноил-N-[2-(1Н-тетразол-5-ил)бифенил-4-ил-метил]амин формулы (I)
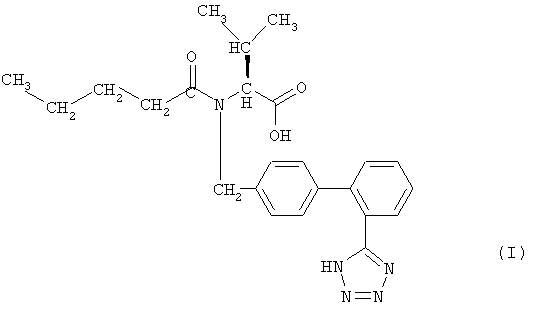
и он описан в ЕР 0443983 А и US 5399578, содержание которых полностью включено в настоящее описание в качестве ссылки. Другие соединения, относящиеся к АРБ, включают (но не ограничиваясь ими) лосартан, кандесартан, эпросартан, ирбесартан, саприсартан, тасосартан, телмисартан, олмесартан, золарсартан, (1-[[3-бром-2-[2-(1Н-тетразол-5-ил)фенил]-5-бензофуранил]метил]-2-бутил-4-хлор-1Н-имидазол-5-карбоновая кислота) и 3-(3-бром-2-[2-(1Н-тетразол-5-ил)-фенил]бензофуран-5-илметил)-2-бутил-5-хлор-3Н-имидазол-4-карбоновая кислота), метил-2-[[4-бутил-2-метил-6-оксо-5-[[2'-(1Н-тетразол-5-ил)[1,1'-бифенил]-4-ил]метил]-1(6Н)-пиримидинил]метил]-3-тиофенкарбоксилат, известный также под названием LR-B/081, и метил-2-[[4-бутил-2-метил-6-оксо-5-[[2'-(1Н-тетразол-5-ил)[1,1'-бифенил]-4-ил]метил]-1-(6Н)-пиримидинил]метил]-3-тиофенкарбоксилат, известный также под названием 3k, т.е. LR-B/081, который несет (карбоксигетероарил)метильный фрагмент в положении 3 (фирма Lusofarmaco), калиевую соль 2,7-диэтил-5-[[2'-(1Н-тетразол-5-ил)бифенил-4-ил]метил]-5Н-пиразоло[1,5-b][1,2,4] триазола, известную также под названием YM 358 (фирма Yamanouchi), которая описана в Biol Pharm Bull, 23(2), февраль 2000, cc.174-181, L-158,809, описанный в Thromb. Res., 15; 105 (6), март 2002, сс.531-536, КТЗ 671, описанный в J. Cardiovasc. Pharmacol.; 25(l), январь 1995, сс.22-29, ТА 606, описанный в J. Cardiovasc. Pharmacol. 31(4), апрель 1998, сс.568-575, ТН 142177, описанный в Fundam. Clin. PharmacoL, 11(5):1997, сс.395-401, UP 269-6, описанный в Br. J. Pharmacol.; 120(3), февраль 1997, сс.488-494, соединение Е-1477, которое имеет следующую формулу
соединение SC-52458, которое имеет следующую формулу:
и соединение ZD-8731, имеющее следующую формулу:
или в каждом случае их фармацевтически приемлемую соль.
При создании изобретения неожиданно было установлено, что метаболиты АРБ также существенно уменьшают агрегацию тромбоцитов. Метаболиты АРБ включают метаболит лосартана, представляющий собой гидрохлорид 2-н-бутил-4-хлор-1-[(2'-(1Н-тетразол-5-ил)бифенил-4-ил) метил]имидазол-5-карбоновой кислоты, также называемый ЕХР-3174, который описан в J. Pharmacol. Exp. Ther.; 255(1), октябрь 1990, c.211-217, и метаболит ЕХР-3174 (II), описанный в J. Chromatogr. 17; 573(2), январь 1992, сс.295-301; метаболиты ирбесартана, представляющие собой 1) конъюгат ирбесартана и тетразол-Н2-бетаглюкуронида, (2) моногидроксилированный метаболит, образующийся в результате омега-1-окисления бутильной боковой цепи, (3, 4) два различных моногидроксилированных метаболита, образующихся в результате окисления спироциклопентанового кольца, (5) диол, образующийся в результате омега-1-окисления в боковой бутильной цепи и окисления спироциклопентанового кольца, (6) кетометаболит, образующийся в результате дополнительного окисления омега-1-моногидроксиметаболита, (7) кетоспирт, образующийся в результате дополнительного омега-1-окисления гидроксила диола, и (8) метаболит карбоновой кислоты, образующийся в результате окисления концевой метильной группы бутильной боковой цепи, который описан в Drug Metab. Dispos.; 26(5), май 1998, сс.408-417; метаболит кандесартанцилексэтил, т.е. кандесартан(2-этокси-1-[[2'-(1Н-тетразол-5-ил)бифенил-4-ил]метил]-1Н-бензимидазол-7-карбоновая кислота), известный также как CV 11974, и CV-15959, описанный в Clin. Pharmacokinet.; 41(1), 2002, сс.7-17, {PRIVATE} J. Chromatogr. В. Biomed. Sci. Appl. 20; 731(2), август 1999, сс.411-417, и {PRIVATE} J. Hum. Hypert.; 11 прил. 2, 1997 сентябрь сс.19-25;
метаболит телмисартана, представляющий собой телмисартан-1-O-ацилглюкуронид, описанный в Drug Metab Dispos; 27(10), октябрь 1999, сс.1143-1149;
метаболит медоксомилолмесартана, представляющий собой олмесартан или CS866, описанный в J. Hypert., Suppl; 19 прил. 1, июнь 2001, с.21-32; метаболит эпросартана, представляющий собой глюкуронид, описанный в Pharmacotherapy; 19(4 Pt 2), апрель 1999, cc.73-78; метаболит тасосартана, представляющий собой энолтасосартан, описанный в J. Pharmacol. Exp. Ther.; 295(2), ноябрь 2000, cc.649-654 и пять других метаболитов, описанных в J. Med. Chem. 22, 41(22), октябрь 1998, cc.4251-4260;
метаболит золасартана, представляющий собой конъюгаты глюкуроновой кислоты и пяти связанных с биотрансформацией продуктов, из которых три гидроксилированы на алифатической боковой цепи, один, кроме того, окислен до кетона и один гидроксилирован на фенильном кольце, описаны в J. Pharm. Biomed. Anal. 12(9), сентябрь 1994, cc.1181-1187 и J. Pharm. Biomed. Anal.; 12(9), сентябрь 1994, cc.1181-1187;
метаболит ZD-8731;
метаболит (5-[(3,5-дибутил-1Н-1,2,4-триазол-1-ил)метил]-2-[2-(1Н-тетразол-5-илфенил)]пиридина, известный также как SC-52458, который описан в J. Cardiovasc. Pharmacol.; 22(4), октябрь 1993, cc.617-625;
и метаболиты следующих АРБ: ZD-8731 (фирма Zeneca), метил-2-[[4-бутил-2-метил-6-оксо-5-[[2'-(1Н-тетразол-5-ил)[1,1'-бифенил]-4-ил]метил]-1(6Н)-пиримидинил]метил]-3-тиофенкарбоксилат, известный также как LR-B/081 и метил-2-[[4-бутил-2-метил-б-оксо-5-[[2'-(1Н-тетразол-5-ил)[1,1'-бифенил]-4-ил]метил]-1-(6Н)-пиримидинил]метил]-3-тиофенкарбоксилат, известный также как 3k, т.е. LR-B/081, в котором в положении 3 введен (карбоксигетероарил)метильный фрагмент (фирма Lusofarmaco), калиевая соль 2,7-диэтил-5-[[2'-(1Н-тетразол-5-ил)бифенил-4-ил]метил]-5Н-пиразол[1,5-b][1,2,4]триазола, известная также как YM 358 (фирма Yamanouchi), описанная в Biol. Pharm. Bull.; 23(2), февраль 2000, cc.174-181, L-158,809, описанный в Thromb. Res. 15; 105(6), март 2002, cc.531-536, КТ3 671, описанный в J. Cardiovasc. Pharmacol.; 25(1), январь 1995, cc.22-29, ТА 606, описанный в J. Cardiovasc. Pharmacol.; 31(4), апрель 1998. cc.568-575, ТН 142177, описанный в Fundam. Clin. Pharmacol.; 11(5), 1997, cc.395-401 и UP 269-6, описанный в Br. J. Pharmacol.; 120(3), февраль 1997, cc.488-494.
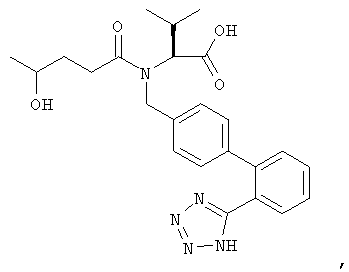
Предпочтительным является метаболит валсартана, представляющий собой валерил-4-гидроксивалсартан, имеющий формулу
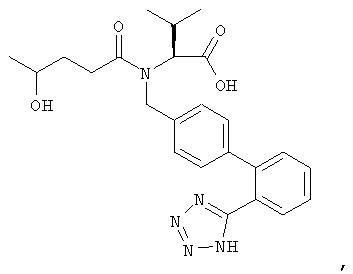
который описан у Waldmeier F. и др., Xenobiotica, том. 27, №.1, 1997, cc.59-71, содержание публикации полностью включено в настоящее описание в качестве ссылки.
В настоящем описании АРБ и их метаболиты называют "соединениями, предлагаемыми в изобретении". В зависимости от природы заместителей соединения, предлагаемые в изобретении, могут иметь один или несколько асимметричных центров. Образующиеся вследствие этого диастереоизомеры, энантиомеры и геометрические изомеры подпадают под объем настоящего изобретения.
В зависимости от выбора исходных продуктов и методов соединения могут находиться в форме одного из возможных изомеров или их смесей, например, в форме практически чистых геометрических (цис- или транс-) изомеров, оптических изомеров (антиподов), рацематов или их смесей. Указанные выше возможные изомеры или их смеси подпадают под объем настоящего изобретения.
Любую образовавшуюся смесь изомеров можно разделять на основе физико-химических различий компонентов на чистые геометрические или оптические изомеры, диастереоизомеры, рацематы, например, с помощью хроматографии и/или фракционированной кристаллизации.
Любые образовавшиеся рацематы конечных продуктов или промежуточных продуктов можно разделять на оптические антиподы известными методами, например, путем разделения их диастереоизомерных солей, полученных с использованием оптически активной кислоты или основания, и выделения производного оптически активной кислоты или основания. Так, промежуточные продукты, представляющие собой карбоновые кислоты, можно разделять на их оптические антиподы, например, с помощью фракционированной кристаллизации D- или L-солей (альфа-метилбензиламина, цинхонидина, цинхонина, хинина, хинидина, эфедрина, дегидроабиэтиламина, бруцина или стрихнина). Рацемические продукты можно разделять также с помощью хиральной хроматографии, например, жидкостной хроматографии высокого давления с использованием хирального адсорбента.
Наконец, соединения, предлагаемые в изобретении, получают либо в свободной форме, либо в форме соли, если присутствуют солеобразующие группы.
Кислотные соединения, предлагаемые в изобретении, можно превращать в соли с использованием фармацевтически приемлемых оснований, например, водного гидроксида щелочного металла, предпочтительно в присутствии растворителя, представляющего собой простой эфир или спирт, такой как (низш.)алканол. Из растворов, содержащих последнее соединение, соли можно осаждать с помощью простых эфиров, например, диэтилового эфира. Образовавшиеся соли можно превращать в свободные соединения путем обработки кислотами. Полученные таким методом соли или другие соли можно также использовать для очистки полученных соединений.
Соединения, предлагаемые в изобретении, имеющие основные группы, можно превращать в кислотно-аддитивные соли, прежде всего, в фармацевтически приемлемые соли. Такие соли образуются, например, с неорганическими кислотами, такими как минеральные кислоты, например, серная кислота, фосфорная или галогенводородная кислота, или с органическими карбоновыми кислотами, такими как С1-С4алканкарбоновые кислоты, которые, например, являются незамещенным или замещенным галогеном, например, уксусная кислота, такими как насыщенные или ненасыщенные дикарбоновые кислоты, например, щавелевая, янтарная, малеиновая или фумаровая кислота, такими как гидроксикарбоновые кислоты, например, гликолевая, молочная, яблочная, винная или лимонная кислота, такими как аминокислоты, например, аспарагиновая или глутаминовая кислота, или с органическими сульфоновыми кислотами, такими как С1-С4алкилсульфоновые кислоты (например, метансульфоновая кислота) или арилсульфоновые кислоты, которые являются незамещенными или замещенными (например, галогеном). Предпочтительными являются соли, образованные с соляной кислотой, метансульфоновой кислотой и малеиновой кислотой.
Учитывая близкие свойства соединений, находящихся в свободной форме, и соединений в форме солей, следует иметь в виду, что если в настоящем описании упоминается соединение, то эта ссылка относится также к соответствующей соли, если это возможно или целесообразно в конкретных условиях.
Соединения, включая их соли, можно получать также в форме гидратов, или они могут содержать другие растворители, применяемые для кристаллизации.
Другим объектом изобретения являются фармацевтические композиции, содержащие терапевтически эффективное количество соединения, предлагаемого в изобретении, и фармацевтически приемлемый носитель. Фармацевтические композиции, предлагаемые в изобретении, можно получать хорошо известным методом и их можно применять для энтерального, такого как оральное или ректальное, введения и парентерального введения млекопитающим (теплокровным животным), включая человека, и они содержат терапевтически эффективное количество фармакологического действующего вещества индивидуально или в сочетании с одним или несколькими фармацевтически приемлемыми носителями, прежде всего пригодными для энтерального или парентерального введения. Типичные оральные композиции представляют собой таблетки, капсулы, сиропы, эликсиры и суспензии. Типичные инъецируемые композиции включают растворы и суспензии. Фармацевтические композиции можно применять для лечения состояний, опосредуемых агрегацией тромбоцитов, в частности, острого инфаркта миокарда, ишемического «удара», стенокардии, острых коронарных синдромов, TIA (приступы транзиторной ишемии или острые цереброваскулярные синдромы), сердечной недостаточности, боли в груди, имеющей ишемическую этиологию, синдрома X, тромбоэмболии, легочной гипертензии, сахарного диабета, заболевания периферических сосудов, тромбоза глубоких вен, артериального тромбоза любого сосуда, вызванного введением катетера тромбозной окклюзии или реокклюзии.
Примерами типичных фармацевтически приемлемых носителей, применяемых в описанных выше композициях, являются: сахара, такие как лактоза, сахароза, маннит и сорбит; крахмалы, такие как кукурузный крахмал, крахмал тапиока и картофельный крахмал; целлюлоза и ее производные, такие как натрийкарбоксиметилцеллюлоза, этилцеллюлоза и метилцеллюлоза; фосфаты кальция, такие как дикальцийфосфат и трикальций фосфат; сульфат натрия; сульфат кальция; поливинилпирролидон; поливиниловый спирт; стеариновая кислота; стеараты щелочно-земельных металлов, такие как стеарат магния и стеарат кальция; растительные масла, такие как арахисовое масло, хлопковое масло, кунжутное масло, оливковое масло и кукурузное масло; неионогенные, катионогенные и анионогенные поверхностно-активные вещества; полимеры этиленгликоля; бетациклодекстрин; жирные спирты и гидролизованные твердые частицы зерновых продуктов, а также другие нетоксичные совместимые наполнители, связующие вещества, разрыхлители, буферы, консерванты, антиоксиданты, замасливатели, корригенты и т.п., которые обычно применяют в фармацевтических композициях.
Такие фармацевтические композиции предназначены для энтерального, такого как оральное, а также для ректального или парентерального введения теплокровным животным, причем композиции содержат фармакологическое действующее вещество либо индивидуально, либо вместе с общепринятыми фармацевтическими вспомогательными веществами. Например, фармацевтические композиции содержат от примерно 0,1 до 90%, предпочтительно от примерно 1 до примерно 80% действующих веществ. Фармацевтические композиции для энтерального или парентерального введения могут находиться, например, в виде стандартных доз, таких как таблетки с покрытием (филм-таблетки), таблетки, капсулы или суппозитории, а также ампулы. Их получают хорошо известным методом, например, использованием стандартных процессов смешения, грануляции, нанесения покрытия, солюбилизации или лиофилизации. Так, фармацевтические композиции для орального введения можно получать путем объединения действующих веществ с твердыми эксципиентами, при необходимости грануляции полученной смеси и, если это является целесообразным или необходимым, обработки смеси или гранул после добавления пригодных вспомогательных веществ с получением таблеток или ядер для филм-таблеток.
АРБ, прежде всего валсартан, и метаболиты АРБ, прежде всего валерил-4-гидроксивалсартан, можно объединять с другими терапевтическими агентами, например, применяя каждый из них в терапевтически эффективной дозе, известной в данной области. Такие терапевтические агенты включают гепарин, варфарин, t-PA, урокиназу, стрептокиназу, аспирин, тиклопидин, клопидогрел, абциксимаб, эптифибатид и тирофибан, гипотнезивные агенты и антидиабетические средства.
Доза действующего вещества может зависеть от различных факторов, таких как путь введения, вид теплокровного животного, возраст и/или индивидуальное состояние.
Предпочтительные дозы действующих веществ, предлагаемых в настоящем изобретении, представляют собой терапевтически эффективные дозы, прежде всего поступающие в продажу дозы валсартана.
Как правило, при оральном введении соединений, предлагаемых в настоящем изобретении, показанная примерная суточная доза составляет от примерно 0,1 до примерно 360 мг для пациента весом примерно 75 кг.
Валсартан поступает в продажу в форме удобных для применения стандартных доз, например, в виде капсулы или таблетки, содержащих терапевтически эффективное количество, например, от примерно 20 до примерно 320 мг валсартана, которые можно вводить пациентам. Введение действующего вещества можно осуществлять до трех раз в день, начиная, например, с суточной дозы 20 или 40 мг валсартана, увеличивая суточную дозу до 80 мг, затем до 160 мг в сутки и далее вплоть до 320 мг в сутки. Предпочтительно пациентам, которым назначена доза 80 или 160 мг, валсартан вводят один раз в день или два раза в день соответственно. Соответствующие дозы можно вводить, например, утром, в полдень или вечером.
В случае валерил-4-гидроксивалсартана предпочтительные стандартные дозы представляют собой, например, таблетки или капсулы, содержащие, например, для млекопитающего весом примерно 50-70 кг от примерно 1 до примерно 1000 мг, предпочтительно от примерно 5 до примерно 500 мг, еще более предпочтительно от примерно 20 до примерно 320 мг, которые вводят один раз в день.
Указанные выше дозы включают терапевтически эффективное количество действующих веществ, предлагаемых в настоящем изобретении.
При создании изобретения неожиданно было установлено, что как АРБ, прежде всего валсартан, так и метаболиты АРБ, прежде всего валерил-4-гидроксивалсартан, обладают выраженной активностью в отношении ингибирования in vitro человеческих тромбоцитов.
Соединения, предлагаемые в настоящем изобретении, ингибируют агрегацию тромбоцитов, и поэтому их можно применять для лечения состояний, опосредуемых агрегацией тромбоцитов, таких, в частности, как острый инфаркт миокарда, ишемический «удар», стенокардия, острые коронарные синдромы, TIA (приступы транзиторной ишемии или острые цереброваскулярные синдромы), сердечная недостаточность, боль в груди, имеющая ишемическую этиологию, синдром X, тромбоэмболия, легочная гипертензия, сахарный диабет, заболевания периферических сосудов, тромбоз глубоких вен, артериальный тромбоз любого сосуда, вызванная введением катетера тромбозная окклюзия или реокклюзия.
Изобретение относится также к соединению, предлагаемому в изобретении, предназначенному для предупреждения, замедления развития, лечения заболевания или состояния опосредуемого агрегацией тромбоцитов, такого, в частности, как острый инфаркт миокарда, ишемический «удар», стенокардия, острые коронарные синдромы, TIA (приступы транзиторной ишемии или острые цереброваскулярные синдромы), сердечная недостаточность, боль в груди, имеющая ишемическую этиологию, синдром X, тромбоэмболия, легочная гипертензия, сахарный диабет, заболевания периферических сосудов, тромбоз глубоких вен, артериальный тромбоз любого сосуда, вызванная введением катера тромбозная окклюзия или реокклюзия.
Изобретение относится также к применению соединения, предлагаемого в изобретении, для приготовления лекарственного средства, предназначенного для предупреждения, замедления развития, лечения заболевания или состояния опосредуемого агрегацией тромбоцитов, такого, в частности, как острый инфаркт миокарда, ишемический «удар», стенокардия, острые коронарные синдромы, TIA (приступы транзиторной ишемии или острые цереброваскулярные синдромы), сердечная недостаточность, боль в груди, имеющая ишемическую этиологию, синдром X, тромбоэмболия, легочная гипертензия, сахарный диабет, заболевания периферических сосудов, тромбоз глубоких вен, артериальный тромбоз любого сосуда, вызванная введением катетера тромбозная окклюзия или реокклюзия.
Указанные выше свойства были продемонстрированы с помощью следующего метода.
У 20 добровольцев, для которых было установлено, что они имеют факторы риска сосудистых заболеваний, брали образцы крови для изучения агрегации тромбоцитов, исследований методом проточной цитометрии и с помощью анализаторов тромбоцитов на основе кассетного метода. Из исследования исключали пациентов, имеющих анамнез геморрагического диатеза, анамнез «удара», «большой» операции или значительной травмы в последние шесть месяцев и гипертензию, характеризующуюся давлением более чем 200/110 мм. Ни один из пациентов не принимал аспирин или какие-либо другие антитромбоцитные агенты. Отбор образцов крови у всех добровольцев осуществляли после того, как они находились в состоянии покоя по меньшей мере в течение 30 мин и не принимали пищу в течение 2 или более часов. Образцы крови брали в период времени от 8 до 10 ч утра для того, чтобы избежать любого влияния дневной деятельности, из расположенной впереди локтя вены с использованием иглы типа «butterfly» 21-размера, содержащей 3,8% цитрата натрия (1:9 объема) после выпускания первых 1,5 мл свободно вытекающей крови. У каждого добровольца, принимавшего участие в исследовании, отбирали в одиннадцать пробирок типа Vacutainer (вместимостью 4,5 мл) в общей сложности 49 мл смеси цельная кровь-цитрат. Одну пробирку сохраняли в качестве внутреннего контроля и инкубировали с буферным раствором. Пять пробирок инкубировали с валсартаном в течение 60 мин при 37°С для получения концентраций соединения 10, 100 нМ, 1, 10, 100 мкМ. Остальные пять пробирок инкубировали аналогичным образом с возрастающими концентрациями валерил-4-гидроксивалсартана, получая концентрации 10, 100 нМ, 1, 10, 100 мкМ. Концентрации валсартана и валерил-4-гидроксивалсартана находились в диапазоне от субтерапевтического уровня до уровня, существенно превышающего терапевтический уровень в плазме, который характерен для пациентов, подвергающихся лечению с использованием валсартана. Пик концентрации валсартана в плазме достигал величины 7,5 мкМ после приема 160 мг, в то время как уровни валерил-4-гидроксивалсартана в плазме составляли лишь 10% от уровней валсартана. Использовали свежеприготовленные растворы валсартана и валерил-4-гидроксивалсартана, которые получали в то же утро, когда проводили исследования тромбоцитов. Для того чтобы избежать субъективной ошибки исследователя, образцы крови кодировали и оценивали «вслепую». Процедуры взятия образцов и исследования тромбоцитов проводили специалисты, не знающие протокола исследования.
Агрегация тромбоцитов
А. Богатая тромбоцитами плазма (тромбоцитемическая плазма)
Смесь цитрата и цельной крови центрифугировали при 1200 г в течение 5 мин для получения богатой тромбоцитами плазмы (БТП), которую выдерживали при комнатной температуре в течение 1 ч. Подсчет тромбоцитов осуществляли для каждого образца БТП с помощью счетчика типа Coulter Counter ZM (фирма Coulter Co., Хилеа, шт. Флорида). Количество тромбоцитов доводили 3,50×108/мл с помощью гомологичной обедненной тромбоцитами плазмы. Агрегацию тромбоцитов (ТА) индуцировали с помощью 5 мкМ АДФ и 5 мкМ эпинефрина. Все агонисты получали от фирмы Chronolog Corporation (Хавертаун, шт. Пенсильвания). Исследование агрегации осуществляли с использованием 4-канального агрегометра типа Chronolog Lumi-Aggregometer (модель 560 - Са). Агрегацию выражали в виде максимального процента изменения прохождения света (% max) по сравнению с исходным уровнем в конце периода регистрации, используя в качестве эталона обедненную тромбоцитами плазму. Зависимости агрегации от времени регистрировали в течение 6 мин и анализировали согласно принятым международным стандартам (Ruggeri ZM, Semin Hemat; 31, 1994, cc.229-239).
Б. Цельная кровь
Методы подробно описаны у Abbate R и др., Amer J Clin Pathol; 86, 1986, cc.91-96. В целом метод заключается в том, что смесь цельной крови и цитрата разбавляли в соотношении 1:1 0,5 мл забуференным трисом изотоническим раствором хлористого натрия (ТБС), затем осторожно перемешивали. Кювету, снабженную стержнем для перемешивания, помещали в лунку для инкубации и давали нагреться до 37°С в течение 5 мин. Затем образец переносили в лунку для анализа. В кювету с образцом помещали электрод. Агрегацию тромбоцитов стимулировали путем добавления 1 мг/мл коллагена. Агрегацию тромбоцитов исследовали с помощью агрегометра для цельной крови типа Chrono-Log Whole Blood Aggregometer с использованием математического обеспечения Aggrolink. Способность тромбоцитов к агрегации оценивали по изменению электрического сопротивления и выражали в омах (Ом).
Исследование цельной крови с помощью проточной цитометрии
Экспрессию рецепторов тромбоцитов определяли с использованием следующих моноклональных антител: к CD31 (фактор адгезии тромбоцитов эндотелиальных клеток (РЕСАМ-1)), CD41 (гликопротеин [GP] IIb/IIIa, (IIb (3), CD42b (GP Ib), CD 51/CD61 ((v (3, или рецептор витронектина), CD62p (Р-селектин), CD107a (ассоциированный с лизосомой мембранный протеин-1; LAMP-1), CD 107b (LAMP-2), CD 151 (тромбоцитарный/эндотелиальный тетраспановый антиген-3; РЕТА-3) и РАС-1 к комплексу фибриноген-тромбоцит (фирма PharMingen, Сан-Диего, шт. Калифорния). Взаимодействия тромбоцит-лейкоцит оценивали с использованием двойных антител к пан-тромбоцитарному маркеру (CD 151) и CD 14, маркеру моноцитов/макрофагов. Смесь кровь-цитрат (50 мкл) разбавляли 450 мкл забуференного Трис физиологического раствора (ТБС) (10 ммолей/л Трис, 0,15 моля/л хлорида натрия) и смешивали, осторожно переворачивая пробирку Эппендорфа два раза. Затем к каждому раствору добавляли 5 мкл соответствующих антител и образцы инкубировали в течение 30 мин. После инкубации добавляли 400 мкл 2%-ного забуференного параформальдегида для фиксации. Образцы анализировали с помощью проточного цитометра типа Becton Dicki и др. на FACScan, отрегулированного для измерения рассеяния флуоресцентного света, согласно методу, описанному у Gurbel PA и др., J Am Coil Cardiol. 31, 1998, cc.1466-1473. Данные собирали в виде списочных файлов и затем анализировали. Содержание Р-селектина выражали в виде процента позитивных клеток согласно методу, описанному у Gurbel PA и др. Am Heart J; 139, 2000, cc.320-328. Содержание других антигенов выражали в виде логарифма средней интенсивности флуоресценции.
Кассетные анализаторы тромбоцитов
Анализатор функции тромбоцитов (типа PFA-100', фирма Dade Behring, Деерфилд, шт. Иллинойс) представляет собой стройство, которое моделирует в условиях потока изменения первичного гемостаза после повреждения небольшого сосуда (Kundu SK и др., Semin Thromb Hemost; 21 (приложение 2), 1995, cc.106-112. Осуществляли цифровую регистрацию времени, в течение которого происходила окклюзия просвета, и принимали её в качестве критерия вызываемой сдвигом агрегации тромбоцитов. Оценку времени закрытия осуществляли в двух повторностях.
Применяли быстрый кассетный метод анализа функции тромбоцитов (RPFA-ASA, фирма Ultegra(r) Accumetrics, Inc., Сан-Диего, шт. Калифорния, США) с использованием кассет, с покрытием из полистироловых гранул, которые содержат микрочастицы, с покрытием из лиофилизированного человеческого фибриногена, при этом в качестве агониста применяли пропилгаллат. В кассету добавляли смесь цельной крови и цитрата и регистрировали агглютинацию между тромбоцитами и гранулами с покрытием. Полученные турбидиметрическим методом данные отражали агрегацию тромбоцитов и свидетельствовали о степени блокады тромбоцитов простагаландином (Smith JW и др., Circulation; 99, 1999, cc.620-625). Ultegra(r)-анализы осуществляли в двух повторностях. Каждый день перед отбором образцов для каждого индивидуума для каждого прибора осуществляли электронный контроль качества.
Статистический анализ
Все сравнения осуществляли с использованием t-критерия Стьюдента для выявления конкретных различий в агрегации тромбоцитов, данные о которых были получены с помощью Ultegra(r), Dade-PFA 100(tm), и между исходным уровнем экспрессии рецептора и после инкубации с валсартаном/валерил-4-гидроксивалсартаном. Для анализа непараметрических данных применяли U-тест Манна-Уитни. Распределенные по нормальному закону данные выражали в виде среднего значения ±СКО, а смещенные данные - в виде медианы (с указанием диапазона). При уровнях вероятности р<0,05 данные рассматривали как статистически достоверные. Для статистического анализа данных, распределенных по нормальному закону, полученных для всех участников эксперимента, применяли линейный регрессионный анализ с использованием программы SPSS v9.0 (фирма SPSS Inc. Чикаго, шт. Иллинойс).
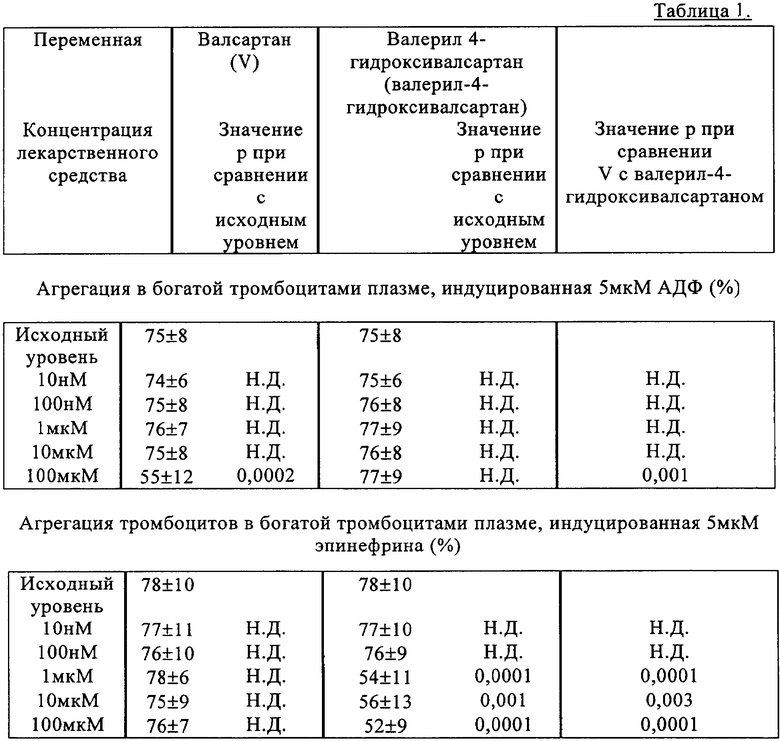
Агрегация тромбоцитов в богатой тромбоцитами плазме
Предварительная инкубация с возрастающими дозами валсартана приводила к ингибированию индуцируемой АДФ агрегации тромбоцитов только при концентрации 100 мкМ, которая превышает терапевтический уровень, и не оказывала влияния на индуцируемую эпинефрином агрегацию тромбоцитов. В отличие от этого инкубация с валерил-4-гидроксивалсартаном приводила к ингибированию индуцируемой эпинефрином агрегации при использовании концентраций 1 и 10 мкМ и не оказывала никакого влияния на индуцируемую АДФ агрегацию. Репрезентативные данные, касающиеся агрегации, стимулированной АДФ в концентрации 5 мкМ и стимулированной эпинефрином в концентрации 5 мкМ, приведены в таблице 1.
Ниже изобретение проиллюстрировано на примерах, которые никоим образом не ограничивают его объем.
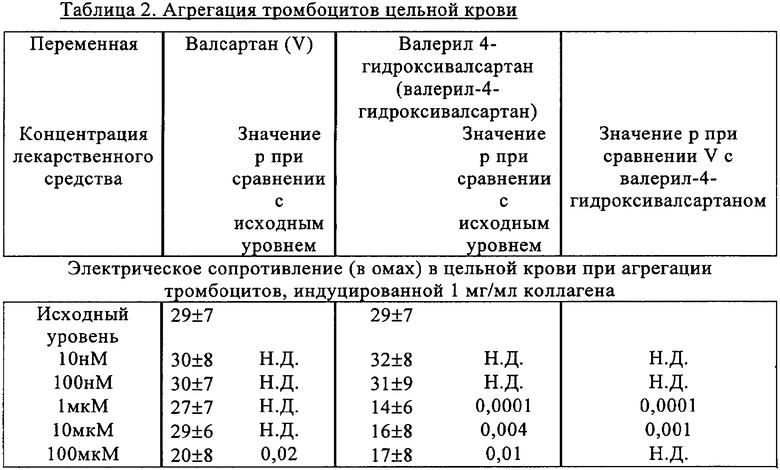
Агрегация тромбоцитов в цельной крови
Валсартан и валерил-4-гидроксивалсартан ингибировали агрегацию человеческих тромбоцитов, индуцируемую 1 мкМ коллагена в цельной крови, в диапазоне концентраций, обладающих терапевтической активностью. Для обоих соединений выявлено зависящее от дозы действие, но предварительная инкубация с валерил-4-гидроксивалсартаном приводила к статистически достоверному уменьшению способности тромбоцитов к агрегации. Результаты, полученные при исследовании агрегации тромбоцитов, вызванной коллагеном, приведены в таблице 2.
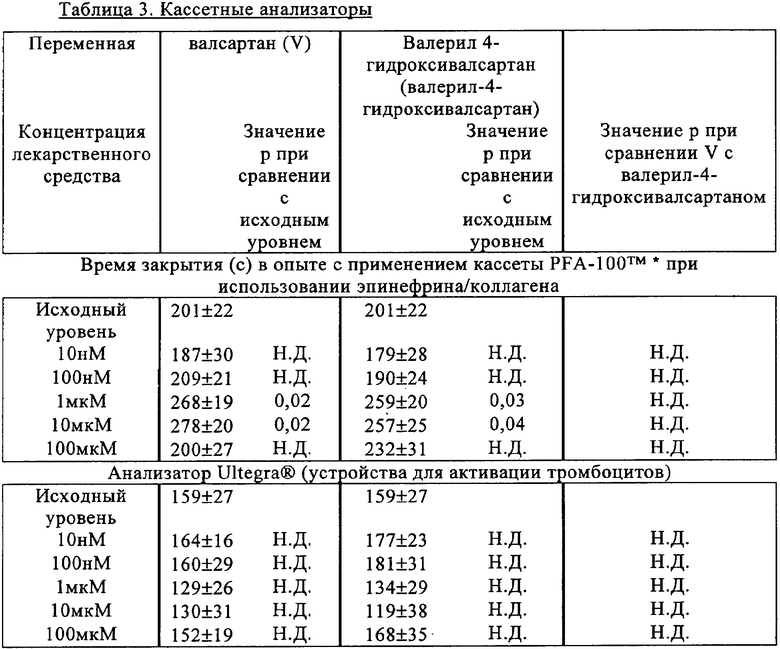
Кассетные анализаторы функции тромбоцитов (PFA-100™ и Ultegra®)
Было выявлено статистически значимое зависящее от дозы увеличение времени закрытия просвета (PFA) при уменьшении количества агрегатов тромбоцитов (Ultegra), свидетельствующее о ингибировании тромбоцитов в условиях с высокими сдвиговыми усилиями. Так же как и при исследовании агрегации в цельной крови и индуцируемой эпинефрином агрегации, валерил-4-гидроксивалсартан обладал более выраженными антитромбоцитными свойствами, чем валсартан. В таблице 3 представлены результаты репрезентативного эксперимента с использованием кассетных анализаторов.
Проточная цитометрия цельной крови
Инкубация с валсартаном и валерил-4-гидроксивалсартаном приводила к небольшому уменьшению экспрессии GP IIb/IIIa (CD 41) и связывания с фибриногеном (РАС-1), причем в случае валерил-4-гидроксивалсартана это действие достигалось при терапевтических концентрациях, в то время как в случае валсартана уменьшение экспрессии CD41 происходило только при использовании высокой дозы. Оба агента существенно понижали концентрацию Р-селектина на поверхности тромбоцитов и в некоторой степени снижали экспрессию рецепторов витронектина, хотя это действие не является статистически достоверным для демонстрации разницы характеристик валсартана и валерил-4-гидроксивалсартана. Инкубация с валсартаном и валерил-4-гидроксивалсартаном не оказывала никакого влияния на РЕСАМ-1 (CD31); GP Ib (CD42) и LAMP-2 (CD107b), PETA-3 (CD 151) и микрочастицы тромбоцит-лейкоцит (CD151+14). Результаты, полученные методом проточной цитометрии, представлены в таблице 4.
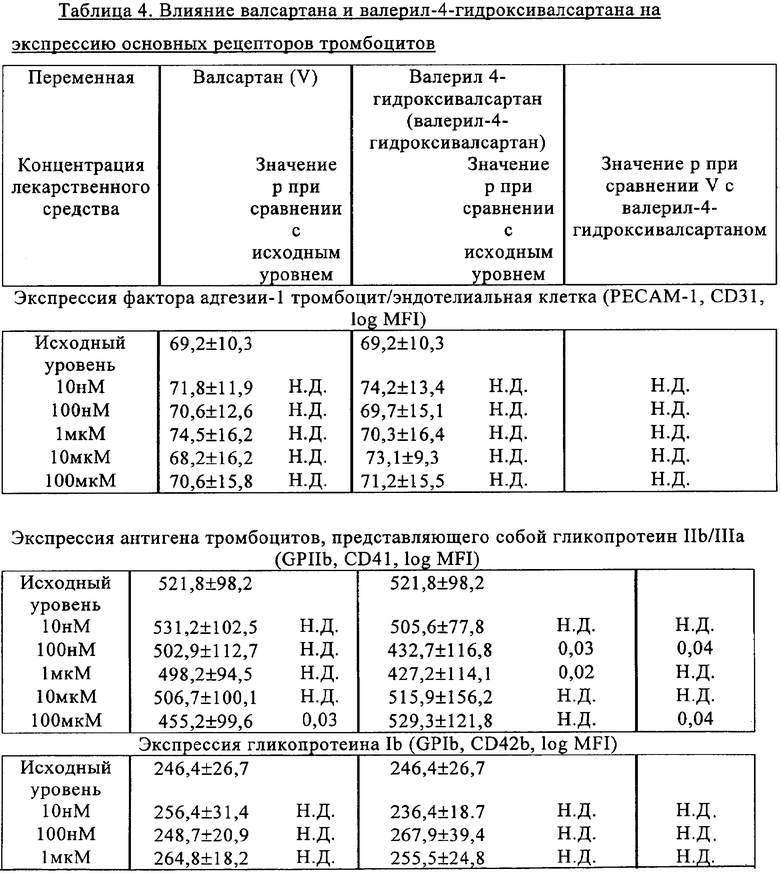
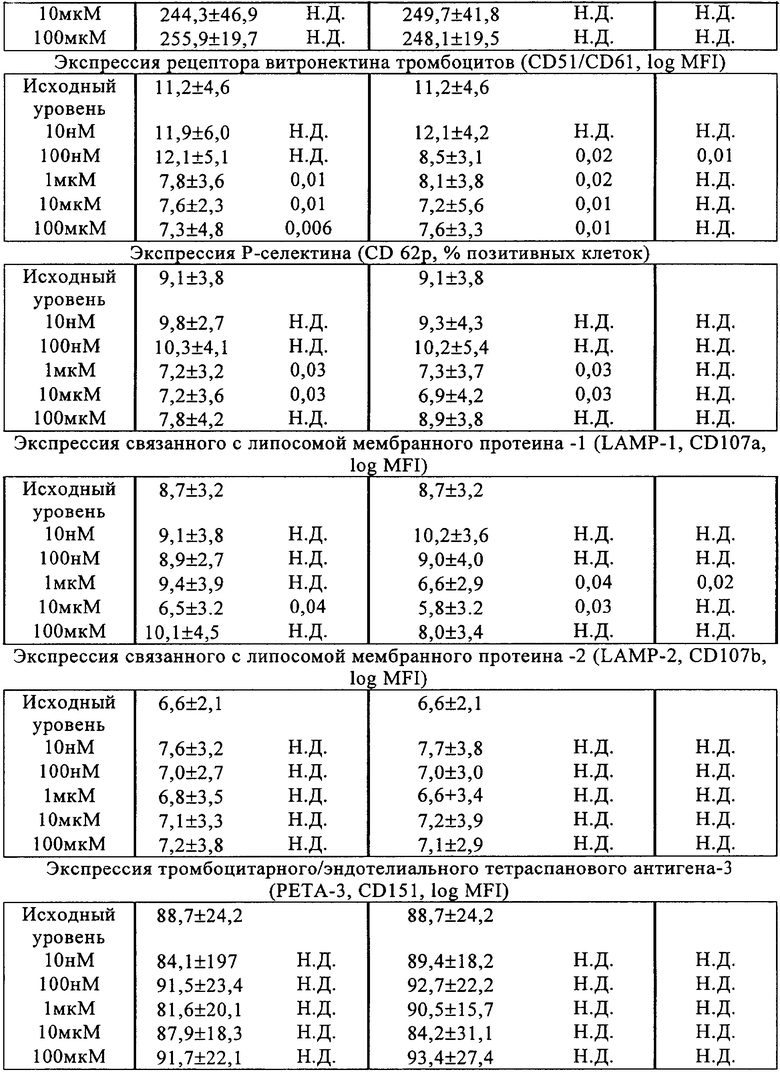
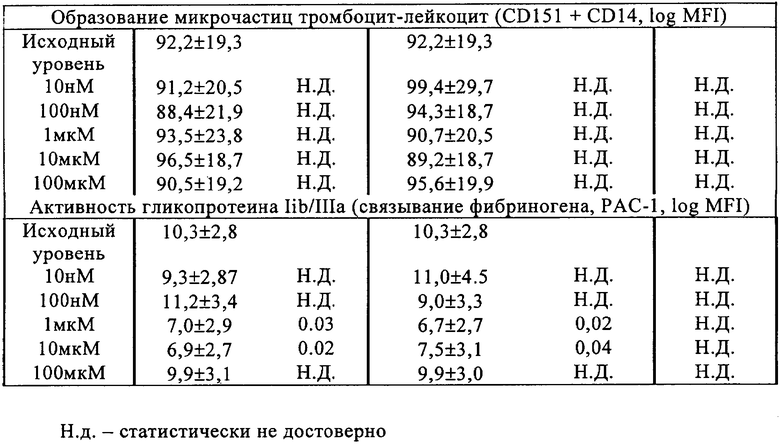
Ниже изобретение проиллюстрировано на примерах, которые никоим образом не ограничивают его объем.
Пример 1
Метод синтеза метаболита валсартана
(S)-2-{(4-гидроксипентаноил)-[2'-(1Н-тетразол-5-ил)-бифенил-4-илметил]амино}-3-метилмасляная кислота
6,7 г (S)-3-метил-2-{(4-оксопентаноил)-[2'-(1Н-тетразол-5-ил)-бифенил-4-илметил]амино} масляной кислоты растворяют в 60 мл метанола и охлаждают до 0°С. Добавляют небольшими порциями 2,25 г борогидрида натрия для того, чтобы поддерживать при перемешивании температуру реакционной смеси ниже 27°С (сильное пенообразование). Смесь перемешивают при комнатной температуре в течение 1 ч, концентрируют в вакууме, растворяют в метиленхлориде и экстрагируют дважды 2н. водным раствором соляной кислоты. Органическую фазу сушат, концентрируют в вакууме и продукт выделяют с помощью хроматографии (колонка для экспресс-хроматографии, 240 г силикагеля 60, KG40-62 мкм, используя смесь растворителей: метиленхлорид, метанол, концентрированный водный раствор аммиака (30:10:1 об./об.). Фракции, содержащие продукт, концентрируют, растворяют в метиленхлориде и экстрагируют 2н. водным раствором соляной кислоты и сушат над сульфатом натрия. После концентрирования остаток сушат в вакууме (60°С) в течение 3 дней, получая (S)-2-{(4-гидроксипентаноил)-[2'-(1Н-тетразол-5-ил)бифенил-4-илметил]амино}-3-метилмасляную кислоту в виде пены белого цвета ([α]D 20=-58° (с=1, метанол). TCX-Rf: 0,18 (толуол/этилацетат/метиленхлорид/муравьиная кислота 16:40:40:4).
Исходный продукт (S)-3-метил-2-{(4-оксопентаноил)-[2'-(1Н-тетразол-5-ил)бифенил-4-илметил]амино} масляную кислоту можно получать следующим методом.
Бензиловый эфир (S)-2-{(2'-цианбифенил-4-илметил)-[3-(2-метил-[1,3]диоксолан-2-ил)пропионил]амино)-3-метилмасляной кислоты
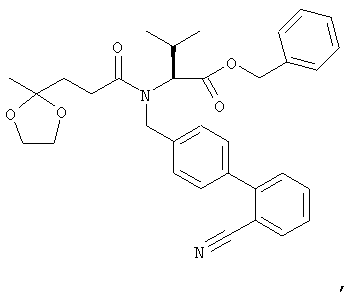
13,8 г гидрохлорида бензилового эфира (S)-2-[(2'-цианбифенил-4-илметил)амино]-3-метилмасляной кислоты (описанного в ЕР 443983) растворяют в 50 мл метиленхлорида, охлаждают до 0°С и обрабатывают 23,8 мл этилдиизопропиламина (основание Хюнига). К этой смеси добавляют при 0°С раствор 3-(2-метил-[1,3]диоксолан-2-ил)пропионилхлорида, полученного из 8,9 г 3-(2-метил-[1,3]диоксолан-2-ил)пропионовой кислоты (Tetrahedron 37, 307, 1981) и 10,31 мл (1-хлор-2-метилпропенил)диметиламина (Tetrahydron 54, 9207, 1998), в 40 мл метиленхлорида. Реакционную смесь перемешивают при комнатной температуре в течение 3-4 дней в зависимости от процесса трансформации. Предпочтительно через 2 дня добавляют 3-(2-метил-[1,3]диоксолан-2-ил)пропионилхлорид в виде 3-4 порций. Реакционную смесь концентрируют в вакууме, растворяют в этилацетате, промывают водой, 1 н. водным раствором соляной кислоты, водой, сушат над сульфатом натрия и концентрируют в вакууме. С помощью экспресс-хроматографии на колонке (240 г силикагеля 60, 40-63 мкм, петролейный эфир/этилацетат от 2:1 до 1:1) получают после сушки продукта в вакууме при 50°С чистый бензиловый эфир (S)-2-{(2'-цианобифенил-4-илметил)-[3-(2-метил-[1,3]диоксолан-2-ил)пропионил]амино}-3-метилмасляной кислоты в виде липкого остатка золотистого цвета. TCX-Rf: 0,23 (петролейный эфир/этилацетат 2:1).
Бензиловый эфир (S)-2-[(2'-цианобифенил-4-илметил')-(4-оксопентаноил)амино]-3-метилмасляной кислоты
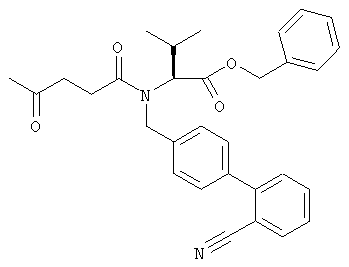
9,8 г бензилового эфира (S)-2-{(2'-цианобифенил-4-илметил)-[3-(2-метил-[1,3]диоксолан-2-ил)пропионил]амино}-3-метилмасляной кислоты растворяют в 100 мл тетрагидрофурана и обрабатывают 50 мл 1 н. водного раствора соляной кислоты. Смесь перемешивают при комнатной температуре в течение 6,5 ч, концентрируют в вакууме и экстрагируют метиленхлоридом. Органическую фазу промывают водой, сушат над сульфатом натрия, концентрируют в вакууме, упаривают и сушат в вакууме при 50°С в течение 1 ч. Получают бензиловый эфир (S)-2-[(2'-цианобифенил-4-илметил)-(4-оксопентаноил)амино]-3-метилмасляной кислоты в виде вязкого масла оранжевого цвета. TCX-Rf: 0,18 (петролейный эфир/этилацетат 2:1).
Бензиловый эфир (S)-3-метил-2-{(4-оксопентаноил)-[2'-(1Н-тетразол-5-ил)бифенил-4-илметил]амино} масляной кислоты
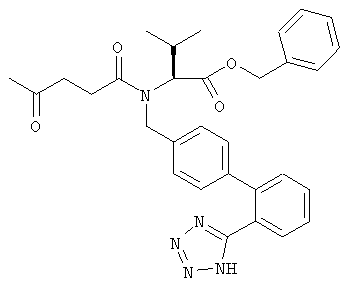
Смесь, содержащую 8,64 г бензилового эфира (S)-2-[(2'-цианбифенил-4-илметил)-(4-оксопентаноил)амино]-3-метилмасляной кислоты и 12,71 г трибутилтиназида (фирма Aldrich), в 20 мл ксилола кипятят с обратным холодильником в течение 28 ч. Смесь обрабатывают 0,5 н. водным раствором гидроксида натрия, водную фазу промывают простым эфиром и эфирную фазу экстрагируют один раз водой. Объединенную водную фазу подкисляют концентрированным водным раствором соляной кислоты, экстрагируют метиленхлоридом, промывают водой, суспендируют с активированным углем, фильтруют, сушат над сульфатом натрия, концентрируют и сушат в вакууме. Продукт, представляющий собой бензиловый эфир (S)-3-метил-2-{(4-оксопентаноил)-[2'-(1Н-тетразол-5-ил)бифенил-4-илметил]амино}масляной кислоты, получают в виде пены коричневого цвета. TCX-Rf: 0,36 (толуол/метиленхлорид/метанол/муравьиная кислота 40:40:40:4).
(S)-3-метил-2-{(4-оксопентаноил)-[2'-(1Н-тетразол-5-ил)бифенил-4-илметил]амино}масляная кислота
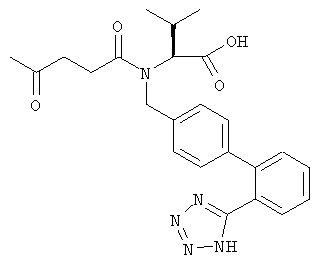
7,9 г бензилового эфира (S)-3-метил-2-{(4-оксопентаноил)-[2'-(1Н-тетразол-5-ил)бифенил-4-илметил]амино}масляной кислоты в 160 мл тетрагидрофурана гидрируют при атмосферном давлении при комнатной температуре в присутствии 1,5 г палладия на угле (10%) до достижения насыщения. Смесь фильтруют и концентрируют в вакууме, получая (S)-3-метил-2-{(4-оксопентаноил)-[2'-(1Н-тетразол-5-ил)бифенил-4-илметил]амино}масляную кислоту в виде пены практически белого цвета. TCX-Rf: 0,1 (толуол/метиленхлорид/метанол/муравьиная кислота 40:40:40:4).
Хотя настоящее изобретение описано достаточно подробно на примере определенных предпочтительных вариантов осуществления изобретения, можно применять другие варианты без отклонения от сущности и объема предпочтительных вариантов, представленных в настоящем описании.
Все упомянутые в настоящем описании публикации и патенты полностью включены в описание в качестве ссылки.
| название | год | авторы | номер документа |
|---|---|---|---|
| ФАРМАЦЕВТИЧЕСКИЕ КОМБИНАЦИИ АНТАГОНИСТА РЕЦЕПТОРА АНГИОТЕНЗИНА И ИНГИБИТОРА NEP | 2006 |
|
RU2459809C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМБИНАЦИИ АНТАГОНИСТА РЕЦЕПТОРА АНГИОТЕНЗИНА И ИНГИБИТОРА NEP | 2006 |
|
RU2503668C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ДВОЙНОГО ДЕЙСТВИЯ НА ОСНОВЕ НАДМОЛЕКУЛЯРНЫХ СТРУКТУР АНТАГОНИСТА/БЛОКАТОРА РЕЦЕПТОРОВ АНГИОТЕНЗИНА (ARB) И ИНГИБИТОРА НЕЙТРАЛЬНОЙ ЭНДОПЕПТИДАЗЫ (NEP) | 2008 |
|
RU2493844C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1997 |
|
RU2183128C2 |
| ИНГИБИТОРЫ NEP ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, ХАРАКТЕРИЗУЮЩИХСЯ УВЕЛИЧЕНИЕМ ИЛИ РЕМОДЕЛИРОВАНИЕМ ПРЕДСЕРДИЯ | 2018 |
|
RU2809222C2 |
| ИНГИБИТОРЫ NEP ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, ХАРАКТЕРИЗУЮЩИХСЯ УВЕЛИЧЕНИЕМ ИЛИ РЕМОДЕЛИРОВАНИЕМ ПРЕДСЕРДИЯ | 2013 |
|
RU2667643C2 |
| ПРИМЕНЕНИЕ ИНГИБИТОРОВ РКС ПРИ ОСЛОЖНЕНИЯХ, ВЫЗВАННЫХ ДИАБЕТОМ | 2007 |
|
RU2447892C2 |
| СОЛИ ВАЛСАРТАНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ПОЛУЧЕНИЯ СОЛЕЙ | 2001 |
|
RU2275363C2 |
| ГАЛЕНОВЫЕ КОМПОЗИЦИИ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ | 2017 |
|
RU2773029C2 |
| ЛЕЧЕНИЕ ГИПЕРТЕНЗИИ И/ИЛИ ПРЕДОТВРАЩЕНИЕ И ЛЕЧЕНИЕ СЕРДЕЧНОЙ НЕДОСТАТОЧНОСТИ У МЛЕКОПИТАЮЩЕГО, ПОЛУЧАЮЩЕГО ТЕРАПИЮ АНТИКОАГУЛЯНТАМИ | 2011 |
|
RU2564941C2 |
Настоящее изобретение относится к области лекарственных средств, в частности к способу ингибирования агрегации тромбоцитов, заключающемуся в том, что пациенту, который нуждается в этом, вводят терапевтически эффективное количество метаболита валсартана, представляющего собой валерил-4-гидроксивалсартан. Кроме того, изобретение также относится к фармацевтической композиции, включающей валерил-4-гидроксивалсартан, и его применению для предупреждения, замедления развития или лечения заболевания и нарушения, опосредуемых агрегацией тромбоцитов. Технический результат заключается в уменьшении способности тромбоцитов к агрегации при применении валерил-4-гидроксивалсартана. 3 н.п. ф-лы, 4 табл.
| BUCZKO W | |||
| et al | |||
| Studies on the antithrombotic action of ATI receptor antagonists | |||
| Medic | |||
| Sci | |||
| moint | |||
| Перекатываемый затвор для водоемов | 1922 |
|
SU2001A1 |
| WALDIMER F | |||
| et al | |||
| Pharmacokinetics, disposition and biotransformation of (14C)-radilabeled valsartan in healthy male volunteers after a singlew oral dose | |||
| Xenobiotica, т.27, N1, 1997, с.59-71 | |||
| MONTON M | |||
| et al. | |||

