Изобретение относится к новым солям антагониста AT1-рецептора, представляющего собой (S)-N-(1-карбокси-2-метилпроп-1-ил)-N-пентаноил-N-[2'-(1Н-тетразол-5-ил)бифенил-4-ил-метил]амин(валсартан) формулы
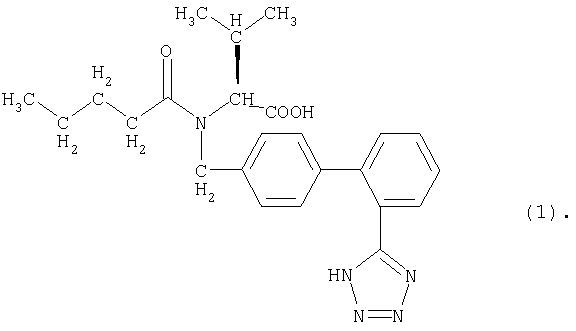
Действующее вещество валсартан представляет собой свободную кислоту, которая описана, в частности, в ЕР 0443983, прежде всего в примере 16; она имеет два кислотных атома водорода: (I) атом водорода (атом Н) карбоксильной группы и (II) атом водорода тетразольного кольца. Следовательно, один кислотный атом Н (прежде всего, атом Н карбоксильной группы) или оба кислотных атома Н могут быть замещены одновалентным или имеющим более высокую валентность, например, двухвалентным, катионом. Возможно также образование смешанных солей.
В ЕР 0443983 не описаны какие-либо конкретные соли валсартана. В заявке не содержится также никаких сведений о специфических свойствах солей. В то же время в ряде стран действующее вещество валсартан применяют в качестве антигипертензивного агента, поступающего в продажу под товарным знаком DIOVAN.
Валсартан в форме свободной кислоты имеет температуру плавления в закрытом тигле 80-95°С, а в открытом тигле 105-110°С, и энтальпию плавления 12 кДж/моль. Оптическое вращение составляет [α]20 D=(-70±2)° для концентрации с=1% (в метаноле).
Плотность кристаллического валсартана и гидратов солей определяли с помощью гелиевого пикнометра (типа Ассирус 1330 фирмы Micromeritics, Норкросс, штат Джоржия, США). Плотность кристаллического валсартана в форме свободной кислоты составляет 1,20±0,02.
Диаграмма дифракции рентгеновских лучей представляет собой в основном очень широкое диффузное отражение рентгеновских лучей; поэтому по данным рентгеновского анализа свободная кислота находится практически в аморфной форме. Данные о температуре плавления наряду с измеренной величиной энтальпии плавления, составляющей 12 кДж/моль, неопровержимо доказывают наличие значительных остаточных структур в виде частиц или структурных доменов в валсартане, находящемся в форме свободной кислоты.
Существует необходимость в создании более стабильных, например кристаллических, форм валсартана, которые легче подвергать процессам сушки или измельчения после заключительной стадии процесса химического получения, а также обрабатывать на стадии приготовления фармацевтических композиций. Было предпринято много тщетных попыток создания улучшенных форм путем получения солей, при этом формы в идеале должны были быть максимально близкими к кристаллическим, а также обладать физической и химической стабильностью. Требуемыми улучшенными свойствами обладают только соли по изобретению, их сольваты и полиморфные формы.
Оказалось, что получение солей валсартана, обладающих требуемыми предпочтительными свойствами, представляет собой трудную задачу. В большинстве случаев получают, например, аморфные соли, обладающие низкой стабильностью (такие как твердые пены, воски или масла). На основе обширных исследований было установлено, что наибольшим преимуществом по сравнению с валсартаном в форме свободной кислоты обладают соли валсартана по настоящему изобретению.
Объектами настоящего изобретения являются соли валсартана, которые выбирают из группы, включающей однонатриевую соль, однокалиевую соль, двукалиевую соль, магниевую соль, кальциевую соль, бисдиэтиламмонийную соль, бисдипропиламмонийную соль, бисдибутиламмонийную соль, моно-L-аргининовую соль, бис-L-аргининовую соль, моно-L-лизиновую соль, бис-L-лизиновую соль, а также смеси солей, или соответственно их аморфные формы, сольваты, прежде всего гидраты, а также их полиморфные формы, соответствующий способ получения и применения и фармацевтические композиции, содержащие такие соли.
Объектами настоящего изобретения являются соли валсартана, которые выбирают из группы, включающей однонатриевую соль, однокалиевую соль, двукалиевую соль, магниевую соль, кальциевую соль, бисдиэтиламмонийную соль, бисдипропиламмонийную соль, бисдибутиламмонийную соль, моно-L-аргининовую соль, бис-L-аргининовую соль, моно-L-лизиновую соль, бис-L-лизиновую соль, или соответственно их аморфные формы, сольваты, прежде всего гидраты, а также их полиморфные формы.
Смеси солей представляют собой (I) формы простых солей, содержащих различные катионы, выбранные из указанной выше группы, или (II) смеси таких форм простых солей, существующих, например, в форме конгломератов.
Предпочтительные соли выбирают, например, из группы, включающей
однонатриевую соль в аморфной форме,
двукалиевую соль валсартана в аморфной или кристаллической форме, прежде всего, в форме ее гидрата,
однокалиевую соль валсартана в аморфной форме,
двукалиевую соль валсартана в аморфной или кристаллической форме, прежде всего в форме ее гидрата,
кальциевую соль валсартана в кристаллической форме, прежде всего в форме ее гидрата, наиболее предпочтительно в форме тетрагидрата,
магниевую соль валсартана в кристаллической форме, прежде всего в форме ее гидрата, наиболее предпочтительно в форме гексагидрата,
смешанную кальциевую/магниевую соль валсартана в кристаллической форме, прежде всего в форме ее гидрата,
бисдиэтиламмонийную соль валсартана в кристаллической форме, прежде всего в форме ее гидрата,
бисдипропиламмонийную соль валсартана в кристаллической форме, прежде всего в форме ее гидрата,
бисдибутиламмонийную соль валсартана в кристаллической форме, прежде всего в форме ее гидрата, наиболее предпочтительно в форме ее полугидрата,
моно-L-аргининовую соль валсартана в аморфной форме,
бис-L-аргининовую соль валсартана в аморфной форме,
моно-L-лизиновую соль валсартана в аморфной форме,
бис-L-лизиновую соль валсартана в аморфной форме.
Соли по изобретению предпочтительно находятся в выделенной и практически очищенной форме, например, имеющей степень чистоты >95%, предпочтительно >98%, наиболее предпочтительно >99%. Степень энантиомерной чистоты солей по изобретению составляет >98%, предпочтительно >99%.
При создании изобретения неожиданно было установлено, что соли по изобретению или их аморфные формы, сольваты, такие как гидраты солей, а также соответствующие полиморфные формы, обладают более предпочтительными свойствами по сравнению со свободной кислотой. В заданных условиях кристаллические соли и гидраты кристаллических солей имеют определенную температуру плавления, которая связана с определенной эндотермической энтальпией плавления. Кристаллические соли по изобретению являются стабильными и по сравнению с валсартаном обладают также более высокими характеристиками при хранении и распределении лекарственных средств. Аморфные или частично аморфные соли имеют ограниченную стабильность, т.е., находясь в твердой форме, они имеют ограниченный диапазон стабильности. Для их стабилизации требуются определенные условия, которые можно достигать, например, путем приготовления галеновых композиций.
Кроме того, как кристаллические, так и аморфные соли по изобретению обладают высокой степенью диссоциации в воде и, следовательно, существенно более высокой растворимостью в воде. Эти свойства являются предпочтительными, поскольку, с одной стороны, процесс растворения происходит более быстро, а, с другой стороны, для таких растворов требуется меньшее количество воды. Кроме того, в случае использования твердых дозируемых форм более высокая растворимость в воде в определенных условиях может приводить также к более высокой биологической доступности солей или гидратов солей. Улучшение свойств, прежде всего, благоприятно сказывается на пациентах. Кроме того, было установлено, что определенные соли по изобретению, прежде всего, соли щелочноземельных металлов, обладают исключительно высокой физической стабильностью. Для различных величин относительной влажности при комнатной температуре, а также при слегка повышенных температурах, гидраты солей по изобретению практически не абсорбируют воду или не теряют воду в широком диапазоне величин относительной влажности в течение нескольких часов, например, четырех часов. Так, например, температура плавления солей по настоящему изобретению не изменяется при хранении в условиях различной относительной влажности.
Улучшение физико-химических свойств определенных солей или гидратов определенных солей имеет большое значение как для их получения в качестве обладающих фармацевтической активностью субстанций, так и для приготовления, хранения и применения галеновых композиций. Таким образом, благодаря более высокому постоянству физических параметров можно обеспечивать еще более высокое качество композиций. Высокая стабильность солей или гидратов солей позволяет также получать экономические преимущества благодаря тому, что процесс обработки состоит из более простых стадий. Высокая степень кристалличности определенных гидратов солей дает возможность применять различные аналитические методы, прежде всего различные рентгеноскопические методы, с помощью которых можно осуществлять точный и простой анализ выхода указанных продуктов. Этот фактор имеет большое значение для обеспечения качества действующего вещества и его галеновых форм в процессе приготовления, хранения и введения пациентам. Кроме того, можно избегать применения сложных мер предосторожности для стабилизации действующего вещества в галеновых композициях.
Таким образом, изобретение относится к кристаллическим, а также частично кристаллическим и аморфным солям валсартана.
Изобретение относится также к сольватам, таким как гидраты, а также к полиморфным формам солей по изобретению.
Сольваты, а также гидраты солей по изобретению могут представлять собой, например, полу-, моно-, ди-, три-, тетра-, пента-, гексасольваты или гидраты соответственно. В кристаллическую решетку могут быть включены применяемые для кристаллизации растворители, такие как спирты, прежде всего метанол, этанол, альдегиды, кетоны, прежде всего ацетон, сложные эфиры, например, этилацетат. Степень сольватации или гидратации при кристаллизации и на последующих стадиях процесса, получаемая с использованием конкретного растворителя или воды, или непосредственное образование свободной кислоты, как правило, являются непредсказуемыми и зависят от комбинации условий, в которых осуществляют процесс, и от различных взаимодействий между валсартаном и конкретным растворителем, прежде всего водой. Соответствующую стабильность образовавшихся кристаллических или аморфных твердых продуктов в форме солей, сольватов и гидратов, а также соответствующих сольватов солей или гидратов солей необходимо определять экспериментальным путем. Таким образом, нельзя ограничиваться только составом химической композиции и стехиометрическим соотношением молекул в образовавшемся твердом продукте, поскольку при таких условиях можно получать как различные кристаллические твердые, так и различные аморфные субстанции.
В качестве соответствующих гидратов могут быть предпочтительными описанные гидраты солей, поскольку молекулы воды в кристаллической структуре связаны большими внутримолекулярными силами и поэтому представляют собой необходимый элемент при формировании структуры таких кристаллов, часть которых обладает очень высокой стабильностью. Однако молекулы воды присутствуют также в определенных кристаллических решетках, которые связаны сравнительно слабыми межмолекулярными силами. Такие молекулы в большей или меньшей степени интегрируются в образующуюся кристаллическую структуру, однако оказывают слабое энергетическое действие. Содержание воды в аморфных твердых веществах, таких как кристаллические гидраты, как правило, можно точно определять, однако оно в значительной степени зависит от сушки и условий окружающей среды. В отличие от этого в случае стабильных гидратов существуют точные стехиометрические соотношения между фармацевтическим действующим веществом и водой. Во многих случаях эти соотношения не полностью совпадают со стехиометрическими величинами, как правило, они меньше теоретических значений вследствие наличия определенных дефектов в кристалле. Соотношение количества органических молекул и молекул воды в том случае, если вода является более слабо связанной, может варьироваться в значительных пределах, например, могут образовываться ди-, три- или тетрагидраты. С другой стороны, в аморфных твердых субстанциях содержание воды в структуре молекулы не является стехиометрическим; это соотношение может оказаться стехиометрическим только случайным образом.
В некоторых случаях оказывается невозможным определить точное стехиометрическое содержание молекул воды, поскольку образуются слоистые структуры, например, в случае солей щелочных металлов, прежде всего калиевой соли, так что нельзя определить точное количество включенных молекул воды.
Для кристаллических твердых субстанций, имеющих идентичный химический состав, существование различных образующихся кристаллических решеток обозначают общим понятием полиморфизм.
Следует иметь в виду, что любые встречающиеся выше и ниже в настоящем описании упоминания солей относятся также к соответствующим сольватам, таким как гидраты, и к полиморфным модификациям, а также к аморфным формам, если это является необходимым и целесообразным.
Наиболее предпочтительными являются тетрагидрат кальциевой соли валсартана и гексагидрат магниевой соли валсартана.
На диаграммах дифракции рентгеновских лучей на порошках этих двух гидратов солей присутствует большое количество дискретных отражений рентгеновских лучей и практически нет признаков наличия некристаллических или аморфных фракций. Таким образом, степень кристаллизации указанных имеющих определенный уровень гидратации солей является неожиданно высокой. Из определенных гидратов солей можно также выращивать относительно большие кристаллы, которые с точки зрения кристаллографии представляют собой монокристаллы. Для таких монокристаллов можно определить структуру твердой субстанции. Это осуществляют на основе компьютеризированного анализа интенсивностей отражения, измеренных с помощью рентгеновского дифрактометра.
Такой метод определения структуры кристалла позволяет в обычных условиях, таких как высокая физическая, химическая и энантиомерная чистота тестируемых кристаллов, осуществлять точное определение структуры на молекулярном или атомном уровне, а именно симметричность и размер элементарных ячеек, положения атомов и температурные коэффициенты, а с учетом оцененного объема ячеек измеренная оптическая плотность фотографического изображения в рентгеновских лучах позволяет оценить молекулярную массу. В то же время определение структуры на основе фотографий в рентгеновских лучах дает сведения о качестве продукта.
Уникальные свойства этих двух гидратов солей обусловлены их кристаллической структурой, которая при образовании этих солей формируется в результате объединения четырех или шести молекул воды с молекулой валсартана. В результате образуются практически идеальные трехмерные кристаллические решетки. Указанные две соли имеют растворимость в воде, в несколько раз превышающую растворимость валсартана в форме свободной кислоты, что является наиболее неожиданным, принимая во внимание высокие температуры плавления и энтальпии плавления, которые в восемь или в пять раз превышают соответствующие величины, характерные для свободной кислоты. Чрезвычайно высокое качество кристаллических решеток указанных двух гидратов солей является основой химической и физической стабильности этих двух соединений.
В качестве гидрата соли следует особо отметить тетрагидрат кальциевой соли валсартана. В закрытом контейнере для образцов (закрытом тигле) при скорости нагрева Тr=10 К·мин-1 он имеет температуру плавления 205±1,5°С и энтальпию плавления 98±4 кДж·моль-1. При повышенных температурах тетрагидрат кальциевой соли валсартана не обладает стабильностью как в отношении содержания гидратированной воды, так и в отношении структуры молекулы. Указанная температура плавления представляет собой температуру плавления гидрата, которую можно измерять только в закрытом контейнере для образцов. Для этой цели использовали золотые контейнеры, имеющие толщину стенки 0,2 мм; после внесения в них образцов гидрата соли массой от 2 до 4 мг их запечатывали с помощью холодной сварки. Указанные золотые контейнеры имели внутренний свободный объем приблизительно 22 мкл. Количество образца необходимо соответствующим образом адаптировать к объему герметичного контейнера для того, чтобы в процессе измерения температуры плавления не могло происходить значительной дегидратации гидратов солей. Парциальное давление воды при 205°С составляет приблизительно 18 бар, поэтому при измерении температуры плавления в открытом контейнере с помощью ДСК (Дифференциальный Сканирующий Калориметр) происходит превращение в безводное соединение. Если экстраполировать данные, измеренные при нескольких скоростях нагрева (Тr=10, 20, 40 К·мин-1), на бесконечно большую скорость нагрева, то получают температуру плавления 213±2°С и энтальпию плавления 124±5 кДж·моль-1. Как высокая температура плавления, так и величина энтальпии плавления, характеризуют чрезвычайно высокую стабильность кристаллической решетки тетрагидрата кальциевой соли валсартана. Эти две термодинамические характеристики свидетельствуют о более предпочтительных физических свойствах рассматриваемого продукта по сравнению со свободной кислотой, для которой две соответствующие величины составляют: температура плавления в закрытом тигле 90°С, энтальпия плавления 12 кДж·моль-1. Указанные термодинамические данные наряду с рентгеноскопическими данными доказывают высокую стабильность такой кристаллической решетки. Это является причиной высокой физической и химической стабильности тетрагидрата кальциевой соли валсартана.
Измерение инфракрасного спектра абсорбции тетрагидрата кальциевой соли валсартана, включенного в спрессованную таблетку, содержащую бромид калия, позволило выявить наличие перечисленных ниже полос, имеющих значительную интенсивность (данные приведены в виде обратных значений длин волн, т.е. в виде волновых чисел (см-1)): 3750-3000 (st); 3400-2500 (st); 1800-1520 (st); 1500-1380 (st); 1380-1310 (m); 1290-1220 (w); 1220-1190 (w); 1190-1160 (w); 1160-1120 (w); 1120-1050 (w); 1030-990 (m); 989-960 (w), 950-920 (w); 780-715 (m); 710-470 (m). Интенсивности полос абсорбции обозначены следующим образом: (w) = слабая, (m) = средняя и (st) = сильная интенсивность. Измерения инфракрасного спектра осуществляли с помощью ATR-ИК-спектроскопии (Attenuated Total Reflection-Infrared Spectroscopy) с использованием устройства типа Spektrum BX фирмы Perkin-Elmer Corp., Беконсфилд, графство Бакингемшир, Великобритания.
Тетрагидрат кальциевой соли валсартана имеет следующие полосы абсорбции (данные приведены в виде обратных значений длин волн, т.е. в виде волновых чисел (см-1)):
3594 (w); 3306 (w); 3054 (w); 2953 (w); 2870 (w); 1621 (st); 1578 (m); 1458 (m); 1441 (m); 1417 (m); 1364 (m); 1336 (w); 1319 (w); 1274 (w); 1241 (w); 1211 (w); 1180 (w); 1149 (w); 1137 (w); 1106 (w); 1099 (w); 1012 (m); 1002 (w); 974 (w); 966 (w); 955 (w); 941 (w); 863 (w); 855 (w); 844 (w); 824 (w); 791 (w); 784 (w); 758 (m); 738 (m); 696 (m); 666 (m).
Интенсивности полос абсорбции обозначены следующим образом: (w) = слабая, (m) = средняя и (st) = сильная интенсивность.
Наиболее интенсивные полосы абсорбции, полученные с помощью ATR-ИК-спектроскопии, характеризуются следующими значениями (данные приведены в виде обратных значений длин волн, т.е. в виде волновых чисел (см-1)): 3306 (w); 1621 (st); 1578 (m); 1458 (m); 1441 (m); 1417 (m); 1364 (m); 1319 (w); 1274 (w); 1211 (w); 1180 (w); 1137 (w); 1012 (m); 1002 (w); 758 (m); 738 (m); 696 (m); 666 (m).
Погрешность для всех полос абсорбции, измеренных с помощью ATR-ИК, составляет ±2 см-1.
Теоретическое содержание воды для тетрагидрата кальциевой соли валсартана составляет 13,2%. С использованием термошкалы TGS-2 (фирма Perkin-Elmer Corp., Норвалк, штат Коннектикут, США) было установлено, что содержание воды составляет 12,9%. На основе этого была выведена общая формула (С24Н27N5O3)2-Са2+·(3,9±0,1)Н2О.
Потерю массы, т.е. уменьшение содержания воды в тетрагидрате в безводной атмосфере N2 в зависимости от времени, измеряли термогравиметрическим методом при скорости нагрева 10 К·мин-1. Результаты представлены в таблице 1.
В таблице 2 представлены данные, характеризующие растворимость тетрагидрата кальциевой соли валсартана в смесях вода-этанол при 22°С.
В таблице 3 представлено сравнение растворимостей в дистиллированной воде двух имеющих наибольшее значение солей по изобретению и свободной кислоты.
Определение других характеристик тетрагидрата кальциевой соли валсартана осуществляют на основе расстояний между плоскостями кристаллической решетки, которые измеряют с помощью диаграммы дифракции рентгеновских лучей на порошке. Диаграмму дифракции рентгеновских лучей на порошке получали с помощью камеры Гинье (типа FR 552 фирмы Enraf Nonius, Дельфт, Нидерланды) на рентгеновской пленке в проходящих лучах с использованием Cu-Ка1-источника излучения при комнатной температуре. Анализ снимков для вычисления расстояний между плоскостями кристаллической решетки осуществляли как визуально, так и с помощью устройства типа Line-Scanner (фирма Johansson Täby, S), при этом одновременно определяли интенсивности отражения.
Предпочтительно определение характеристик терагидрата кальциевой соли валсартана производят путем измерения расстояний d между плоскостями кристаллической решетки на основе диаграмм дифракции рентгеновских лучей, ниже приведены средние значения и указаны соответствующие пределы погрешности.
d в [Å]: 16,1±0,3, 9,9±0,2, 9,4±0,2, 8,03±0,1, 7,71±0,1, 7,03±0,1, 6,50±0,1, 6,33±0,1, 6,20±0,05, 5,87±0,05, 5,74±0,05, 5,67±0,05, 5,20±0,05, 5,05±0,05, 4,95±0,05, 4,73±0,05, 4,55±0,05, 4,33±0,05, 4,15±0,05, 4,12±0,05, 3,95±0,05, 3,91±0,05, 3,87±0,05, 3,35±0,05.
Ниже приведены расстояния между плоскостями кристаллической решетки для наиболее интенсивных отражений на диаграмме дифракции рентгеновских лучей:
d в [Å]: 16,1±0,3, 9,9±0,2, 9,4±0,2, 7,03±0,1, 6,50±0,1, 5,87±0,05, 5,74±0,05, 4,95±0,05, 4,73±0,05, 4,33±0,05, 4,15±0,05, 4,12±0,05, 3,95±0,05.
Предпочтительный метод проверки указанных выше средних значений расстояний между плоскостями кристаллической решетки и интенсивностей, измеренных на диаграммах дифракции рентгеновских лучей, которые были получены с помощью камеры Гинье для данной субстанции, заключается в вычислении указанных расстояний и соответствующих интенсивностей на основе исчерпывающих данных о структуре монокристалла. Такое определение структуры позволяет получать параметры элементарной ячейки и положения атомов, что позволяет производить обработку диаграммы дифракции рентгеновских лучей для твердой субстанции с помощью компьютерных методов расчета (программа CaRine Crystallography, Universitè de Compiègne, Франция). В таблице 4 представлено сравнение указанных данных, а именно расстояний между плоскостями кристаллической решетки и интенсивностей, представляющих наибольшее значение полос для тетрагидрата кальциевой соли валсартана, измеренных с помощью камеры Гинье и рассчитанных для монокристалла.
Изобретение относится к кристаллическому тетрагидрату кальциевой соли (S)-N-(1-карбокси-2-метилпроп-1-ил)-N-пентаноил-N-[2'-(1Н-тетразол-5-ил)бифенил-4-илметил]амина, т.е. кристаллическому твердому веществу, которое полностью характеризуется экспериментальными данными и параметрами, полученными на основе рентгенографического анализе монокристалла и диаграмм дифракции рентгеновских лучей на порошке. Подробное изложение теории, лежащей в основе методов анализа дифракции рентгеновских лучей на отдельном кристалле, и метода обработки полученных в результате исследования кристаллической структуры данных и параметров приведено в работе Stout и Jensen, X-Ray Structure Determination; A Practical Guide, Mac Millian Co., New York, N.Y. глава 3 (1968).
В таблице 5 представлены данные и параметры, полученные при исследовании с помощью рентгеновских лучей структуры монокристалла тетрагидрата кальциевой соли валсартана.



Элементарная ячейка характеризуется шестью параметрами, а именно постоянными a, b и с кристаллической решетки и углами с оптической осью, т.е. α, β и γ. Эти параметры определяют объем элементарной ячейки Vc. Подробное описание указанных параметров кристаллической решетки приведено в главе 3 работы Stout и Jensen (см. выше). Данные, полученные для тетрагидрата кальциевой соли валсартана на основе измерений, проведенных для монокристалла, прежде всего атомные координаты, изотропические тепловые параметры, координаты атомов водорода, а также соответствующие изотропические тепловые параметры, свидетельствуют о том, что существует элементарная ячейка моноклинного типа, содержащая четыре единицы формулы Са2+ валсартан2- ·4Н2О, которая образуется из двух независимых с кристаллографической точки зрения единиц, каждая из которых находится в двух положениях.
Для рассматриваемой ацентрической пространственной группы P21, которая была установлена на основе определения структуры монокристалла с помощью рентгеноскопического анализа, рацематы не существуют. Это доказывает энантиомерную чистоту S-конфигурации кристаллического тетрагидрата кальциевой соли (S)-N-(1-карбокси-2-метилпроп-1-ил)-N-пентаноил-N-[2'-(1Н-тетразол-5-ил)бифенил-4-илметил]амина.
Важной характеристикой качества чистого действующего вещества как с точки зрения физико-химических процессов, таких как сушка, просеивание, измельчение, так и процессов приготовления галеновых композиций, которые осуществляют с использованием фармацевтических эксципиентов, а именно, процессов смешения, грануляции, сушки распылением, таблетирования, является зависимость поглощения воды или потери воды рассматриваемого действующего вещества от температуры и относительной влажности окружающей среды. Очевидно, что при приготовлении определенных композиций свободная и связанная вода вносится вместе с эксципиентами и/или ее добавляют в обрабатываемую массу в соответствии с условиями соответствующего процесса приготовления композиции. Вследствие этого фармацевтическое действующее вещество подвергается действию свободной воды в течение довольно продолжительных периодов времени, активность которого зависит от температуры (парциальное давление паров).
Точные данные, касающиеся данного вопроса, получают с помощью изотермических измерений, которые проводят в течение предварительно определенных интервалов времени и предварительно определенной относительной влажности на основе динамической сорбции паров (устройство типа DVS-1 фирмы Surface Measurement Systems LTD, Марлоу, графство Бакингемшир, Великобритания). В таблице 6 приведены данные о изменении массы, т.е. об абсорбции или потере воды в течение 4 ч в зависимости от относительной влажности при 25°С для образца тетрагидрата кальциевой соли валсартана массой 9,5 мг. Данные приведены для следующих циклов изменения относительной влажности: 40-90; 90-0; 0-90; 90-0% относительной влажности:
Погрешность измерений указанным сорбционным методом, основанным на термогравиметрии, составляет приблизительно 0,1%. Следовательно можно считать, что в рассматриваемых условиях, которые являются реальными с точки зрения приготовления фармацевтических галеновых форм, для тетрагидрата кальциевой соли валсартана не наблюдается заметной абсорбции или потери воды. Совершенно неожиданным является тот факт, что тетрагидрат, содержащий в кристаллической структуре приблизительно 13% связанной воды, является полностью невосприимчивым к воде даже в условиях очень большой относительной влажности. Это свойство имеет решающее значение на заключительных стадиях химического синтеза, а также для осуществления всех стадий процесса приготовления галеновых композиций для различных дозируемых форм. Такая очень высокая стабильность является также чрезвычайно благоприятной для пациентов, поскольку обеспечивает постоянную биологическую доступность действующего вещества.
Скорости растворения кальциевой соли валсартана при рН 1, 4,5 и 6,8 превосходят соответствующие значения для валсартана.
Очень высокую стабильность кальциевой соли валсартана, прежде всего его тетрагидрата, в отношении воды можно проиллюстрировать также с помощью тестов по оценке стабильности. Результаты этих тестов свидетельствуют о том, что содержание воды в тетрагидрате кальциевой соли валсартана остается постоянным при хранении в течение четырех недель при 40°С и 75%-ной относительной влажности как открытом контейнере, так и в запечатанной ампуле.
Благодаря тому, что кальциевая соль, прежде всего ее тетрагидрат, обладает лучшими кристаллическими свойствами, указанную соль можно непосредственно подвергать прессованию, получая соответствующие композиции в форме таблетки.
Кроме того, можно обеспечивать улучшенный профиль растворения таблетки. При исследовании профиля растворения было установлено, что из филмтаблетки высвобождение 100% кальциевой соли, прежде всего ее тетрагидрата, происходит в течение 15 мин.
Среди кристаллических твердых соединений нового типа предпочтительным является гидрат магниевой соли валсартана, прежде всего гексагидрат. Термическая характеристика этого гидрата соли в области температуры плавления свидетельствует об определенной химической и физической нестабильности. Следовательно, результаты исследований термических характеристик зависят от условий, при которых проводят измерения. В закрытом золотом контейнере для образцов, имеющем внутренний объем приблизительно 22 мкл, для образца массой 2-4 мг при скорости нагрева Тr=10 К·мин-1, температура плавления гексагидрата магниевой соли валсартана составляет 132±1,5°С, а энтальпия плавления составляет 56±3 кДж·моль-1. Энтальпия плавления, которая приблизительно в 5 раз превышает соответствующую величину для валсартана в форме свободной кислоты, а также более высокая температура плавления гексагидрата магниевой соли валсартана свидетельствуют о стабильности кристаллической решетки нового типа при температуре, близкой к комнатной.
Оптическое вращение для гексагидрата магниевой соли валсартана в 1%-ном растворе в метаноле при 20°С составляет [α]20 D=-14°.
Измерение инфракрасного спектра абсорбции гексагидрата магниевой соли валсартана, включенного в спрессованную таблетку, содержащую бромид калия, позволило выявить наличие перечисленных ниже полос, имеющих значительную интенсивность (данные приведены в виде обратных значений длин волн, т.е. в виде волновых чисел (см-1)): 3800-3000 (st); 3000-2500 (st); 1800-1500 (st); 1500-1440 (m); 1440-1300 (m); 1280-1240 (w); 1240-1190 (w); 1190-1150 (w); 1120-1070 (w); 1050-990 (w); 990-960 (w); 960-920 (w); 920-700 (m); 700-590 (w); 590-550 (w).
Интенсивности полос абсорбции обозначены следующим образом: (w) = слабая, (m) = средняя и (st) = сильная интенсивность.
Измерения инфракрасного спектра осуществляли с помощью ATR-ИК-спектроскопии (Attenuated Total Reflection-Infrared Spectroscopy) с использованием устройства типа Spektrum BX фирмы Perkin-Elmer Corp., Беконсфилд, Бакингемшир, Великобритания.
Гексагидрат магниевой соли валсартана имеет следующие полосы абсорбции (данные приведены в виде обратных значений длин волн, т.е. в виде волновых чисел (см-1)): 3378 (m); 3274 (m); 2956 (m); 2871 (w); 2357 (w); 1684 (w); 1619 (st); 1557 (m); 1464 (m); 1419 (m); 1394 (st); 1374 (m); 1339 (w); 1319 (w); 1300 (w); 1288 (w); 1271 (w) 1255 (w); 1223 (w); 1210 (w); 1175 (m); 1140 (w); 1106 (w); 1047 (w); 1024 (w); 1015 (w); 1005 (w); 989 (w); 975 (w); 955 (w); 941 (w); 888 (w); 856 (w); 836 (m); 820 (w); 766 (st); 751 (m); 741 (st); 732 (st).
Интенсивности полос абсорбции обозначены следующим образом: (w) = слабая, (m) = средняя и (st) = сильная интенсивность.
Наиболее интенсивные полосы абсорбции, полученные с помощью ATR-ИК-спектроскопии, характеризуются следующими значениями (данные приведены в виде обратных значений длин волн, т.е. в виде волновых чисел (см-1)): 3378 (m); 3274 (m); 2956 (m); 1619 (st); 1557 (m); 1464 (m); 1419 (m); 1394 (st); 1271 (w); 1175 (m); 1015 (w); 975 (w); 836 (m); 766 (st); 751 (m); 741 (st); 732 (st).
Погрешность для всех полос абсорбции, измеренных с помощью ATR-ИК, составляет ±2 см-1.
Теоретическое содержание воды в гексагидрате магниевой соли валсартана составляет 19,1%. С помощью объединенного устройства, основанного на термогравиметрическом методе и ИК-спектроскопии с фурье-преобразованием (TG-FTIR, устройство типа IFS 28, выпускается фирмами Netzsch Gerätebau GmbH, Зельб, Bayern and Bruker Optik GmbH, Карлсруэ), которое с помощью инфракрасной спектроскопии позволяет одновременно измерять потерю массы и идентифицировать теряемый при этом компонент (высвобождение воды), было установлено, что содержание воды составляет 18,5%, что хорошо согласуется с теоретическим значением. Для гексагидрата это соответствует молярному соотношению 5,8±0,2 моля Н2О на моль магниевой соли.
В таблице 7 приведены данные о потере воды гексагидратом магниевой соли валсартана в зависимости от температуры, полученные в атмосфере N2 с помощью устройства для термогравиметрического термического анализа при скорости нагрева 10 К·мин-1. На основе измерений TG-FTIR-методом установлено, что потеря массы обусловлена только высвобождением воды.
Гексагидрат магниевой соли валсартана имеет растворимость в дистиллированной воде при 22°С, равную 59 г на литр раствора при значении рН 9.3.
Кристаллическая форма гексагидрата магниевой соли валсартана точно характеризуется расстояниями между плоскостями кристаллической решетки, рассчитанными на основе линий на картине дифракции рентгеновских лучей на порошке. Для этой цели применяют те же методы измерения и анализа, что и для тетрагидрата кальциевой соли валсартана.
Предпочтительно характеристику гексагидрата магниевой соли валсартана получают на основе величин расстояний между плоскостями кристаллической решетки d, средние значения которых представлены ниже наряду с соответствующими пределами погрешностей:
d в [Å]: 19,7±0,3, 10,1±0,2, 9,8±0,2, 7,28±0,1, 6,48±0,1, 6,00±0,1, 5,81±0,1, 5,68±0,1, 5,40±0,05, 5,22±0,05, 5,12±0,05, 5,03±0,05, 4,88±0,05, 4,33±0,05, 4,22±0,05, 4,18±0,5, 4,8±0,5, 3,5±0,5, 3,6±0,5, 3,2±0,5.
Наибольшие интенсивности отражений на диаграмме дифракции рентгеновских лучей соответствуют следующим расстояниям между плоскостями кристаллической решетки:
d в [Å]: 19,7±0,3, 10,11±0,2, 9,8±0,2, 7,28±0,1, 5,81±0,05, 5,68±0,05, 5,03±0,05, 4,88±0,05, 4,18±0,05, 4,08±0,05, 3,46±0,05.
Предпочтительный метод проверки указанных выше средних значений расстояний между плоскостями кристаллической решетки и интенсивностей, измеренных на диаграммах дифракции рентгеновских лучей, которые были получены с помощью камеры Гинье для рассматриваемой субстанции, заключается в вычислении указанных расстояний и соответствующих интенсивностей на основе исчерпывающих данных о структуре монокристалла. Такое определение структуры позволяет получать параметры кристаллической решетки и положения атомов, что дает возможность производить обработку диаграммы дифракции рентгеновских лучей для твердой субстанции с помощью компьютерных методов расчета (программа CaRine Crystallography, Université de Compiégne, Франция). В таблице 8 представлено сравнение указанных данных, а именно расстояний между плоскостями кристаллической решетки и интенсивностей, представляющих наибольшее значение полос для гексагидрата магниевой соли валсартана, полученных с помощью камеры Гинье и на основе расчетов для монокристалла.
Изобретение относится, прежде всего, к кристаллическому гексагидрату магниевой соли (S)-N-(1-карбокси-2-метилпроп-1-ил)-N-пентаноил-N-[2'-(1Н-тетразол-5-ил)бифенил-4-илметил]амина, т.е. кристаллического твердого вещества, которое полностью характеризуется экспериментальными данными и параметрами, полученными на основе рентгенографического анализа монокристалла. Подробное изложение теории, лежащей в основе методов анализа дифракции рентгеновских лучей на отдельном кристалле, и метода обработки полученных в результате исследования кристаллической структуры данных и параметров приведено в работе Stout и Jensen, X-Ray Structure Determination; A Practical Guide, Mac Millian Co., New York, N.Y. глава 3 (1968).
В таблице 9 представлены данные и параметры, полученные при исследовании с помощью рентгеновских лучей структуры монокристалла гексагидрата магниевой соли валсартана.



Элементарная ячейка характеризуется шестью параметрами, а именно постоянными a, b и с кристаллической решетки и углами с оптической осью, т.е. α, β и γ. Эти параметры определяют объем элементарной ячейки Vc. Подробное описание указанных параметров кристаллической решетки приведено в главе 3 работы Stout и Jensen (см. выше).
Данные, полученные для гексагидрата магниевой соли валсартана на основе измерений, проведенных для монокристалла, прежде всего атомные координаты, изотропические тепловые параметры, координаты атомов водорода, а также соответствующие изотропические тепловые параметры, свидетельствуют о том, что существует элементарная ячейка моноклинного типа, содержащая четыре единицы формулы Mg2+ валсартан2- · 6Н2О.
Для рассматриваемой ацентрической пространственной группы C21, которая была установлена на основе определения структуры монокристалла, рацематы не существуют. Это доказывает энантиомерную чистоту S-конфигурации кристаллического гексагидрата магниевой соли валсартана.
В таблице 10 приведены данные о изменении массы, т.е. об абсорбции или потере воды в течение 4 ч в зависимости от относительной влажности при 25°С для образца гексагидрата магниевой соли валсартана массой 9,5 мг. Данные приведены для следующих циклов изменения относительной влажности: 40-90; 90-0; 0-90; 90-0% относительной влажности:
Погрешность измерений указанным сорбционным методом, основанным на термогравиметрии, составляет приблизительно 0,1%. При этом в рассматриваемых условиях, которые являются реальными с точки зрения приготовления фармацевтических галеновых форм, установлено, что для гексагидрата магниевой соли валсартана при относительной влажности 20-80% может иметь место обратимая слабая абсорбция воды или потеря воды. Совершенно неожиданным является тот факт, что гексагидрат, содержащий в кристаллической структуре приблизительно 19% связанной воды, обратимо абсорбирует или высвобождает воду только при очень больших значениях относительной влажности и является сравнительно невосприимчивым при умеренных значениях относительной влажности. Это свойство позволяет разработать несложный физико-химический процесс получения и позволяет создавать дозируемые формы, которые являются наиболее предпочтительными для пациентов.
Очень высокую стабильность магниевой соли валсартана, прежде всего его гексагидрата, в отношении воды можно проиллюстрировать также с помощью тестов по оценке стабильности. Результаты этих тестов свидетельствуют о том, что содержание воды в гексагидрате магниевой соли валсартана остается постоянным при хранении в течение четырех недель при 40°С и 75%-ной относительной влажности как открытом контейнере, так и в запечатанной ампуле.
Благодаря тому, что магниевая соль, прежде всего ее гексагидрат, обладает лучшими кристаллическими свойствами, указанную соль можно непосредственно подвергать прессованию, получая соответствующие композиции в форме таблетки.
Кроме того, это позволяет обеспечивать улучшенный профиль растворения таблетки. При исследовании профиля растворения было установлено, что из филмтаблетки высвобождение 100% магниевой соли, прежде всего, ее гексагидрата, происходит в течение 15 мин.
Кроме того, магниевая соль валсартана, прежде всего ее гексагидрат, обладает предпочтительным профилем сопротивления уплотнению.
Смешанные кальциевые/магниевые соли валсартана также обладают ценными свойствами, например, они могут образовывать однородные кристаллические конгломераты. Их можно предпочтительно применять при приготовлении галеновой композиции.
Скорости растворения, которыми обладает двукалиевая соль валсартана при рН 1, 4,5 и рН 6,8, превышают соответствующие величины, характерные для валсартана.
Следующим объектом изобретения является способ получения солей по изобретению.
Соли по изобретению, включая их аморфные или кристаллические формы, можно получать следующим образом:
Для образования соли процесс осуществляют в системе растворителей, в которой два реагента, а именно валсартан в форме кислоты и соответствующее основание, обладают достаточной растворимостью. Для осуществления кристаллизации или преципитации целесообразно использовать растворитель или смесь растворителей, в котором(ой) образовавшаяся соль слабо растворима или совсем нерастворима. В одном из вариантов получения соли по изобретению применяют растворитель, в котором соль хорошо растворима, и затем к этому раствору добавляют антирастворитель, в результате чего получают растворитель, в котором образовавшаяся соль обладает слабой растворимостью. В другом варианте кристаллизацию соли осуществляют концентрированием раствора соли, например, нагреванием, при необходимости при пониженном давлении, или путем медленного выпаривания растворителя, например, при комнатной температуре, или путем внесения затравочных кристаллов или путем регулирования водной активности, необходимой для образования гидрата.
В качестве растворителей можно применять, например, С1-С5алканолы, предпочтительно этанол и изопропанол, а также С1-С5диалкилкетоны, предпочтительно ацетон, и их смеси с водой.
Антирастворители для кристаллизации соли могут представлять собой, например, С3-С7алкилнитрилы, прежде всего ацетонитрил, сложные эфиры, прежде всего С1-С5-алкиловый эфир С2-С7алканкарбоновой кислоты, такой как этил- или изопропилацетат, простые ди(С1-С5алкиловые) эфиры, такие как метил-трет-бутиловый эфир, или тетрагидрофуран, и С5-С8алканы, прежде всего пентан, гексан или гептан.
Для получения гидратов применяют, прежде всего, процесс растворения и кристаллизации или процесс кристаллизации в условиях уравновешивания воды.
Процесс растворения и кристаллизации отличается тем, что
(I) осуществляют взаимодействие валсартана и соответствующего основания предпочтительно в содержащем воду органическом растворителе,
(II) систему растворителей концентрируют, например, нагреванием, при необходимости при пониженном давлении, и путем внесения затравочных кристаллов или медленным выпариванием, например, при комнатной температуре, затем инициируют кристаллизацию или преципитацию и
(III) выделяют образовавшуюся соль.
Применяемая для процесса растворения и кристаллизации содержащая воду система органических растворителей предпочтительно представляет собой смеси спиртов, таких как этанол, и воды, или алкилнитрила, прежде всего ацетонитрила, и воды.
Процесс кристаллизации путем уравновешивания, применяемый для получения гидратов, отличается тем, что
(I) валсартан и соответствующее основание добавляют к содержащему воду органическому растворителю,
(II) растворитель концентрируют, например, путем нагрева, при необходимости при пониженном давлении, или путем медленного выпаривания, например, при комнатной температуре,
(III) остаток, полученный после выпаривания, уравновешивают необходимым количеством воды путем
(а) суспендирования остатка, полученного путем выпаривания, который предпочтительно еще остается теплым и который еще содержит некоторое количество воды, в соответствующем растворителе или
(б) уравновешиванием избытка воды в растворителе;
при этом на стадиях а) и б) имевшаяся в наличии или добавленная вода присутствует в количестве, в котором вода растворяется в органическом растворителе и не образует дополнительную фазу; и
(IV) выделяют образовавшуюся соль.
Система растворителей, которую применяют в качестве содержащего воду органического растворителя, предпочтительно представляет собой смеси соответствующих спиртов, таких как С1-С7алканолы, прежде всего этанол, и воды.
В качестве соответствующего растворителя для уравновешивания применяют, например, сложный эфир, такой как С1-С7алкиловый эфир С1-С7алканкарбоновой кислоты, прежде всего этилацетат, или кетон, такой как диС1-С5алкилкетон, прежде всего ацетон.
Процесс уравновешивания отличается, в частности, тем, что позволяет получать высокие выходы и обладает высокой воспроизводимостью.
При получении одноосновных солей щелочных металлов по настоящему изобретению, как правило, образуются аморфные формы. В то же время можно получать также находящиеся в кристаллической форме и в форме гидратов двухосновные соли щелочных металлов и соли щелочноземельных металлов по настоящему изобретению из соответствующих растворителей, обычно применяемых в процессе производства, таких как сложные эфиры, например, С1-С7алкиловые эфиры С1-С7алканкарбоновой кислоты, прежде всего этилацетат, кетоны, например, диС1-С5алкилкетоны, прежде всего ацетон, С3-С7алкилнитрилы, прежде всего ацетонитрил, или простые эфиры, например, ди(С1-С5алкиловые) эфиры, такие как метил-трет-бутиловый эфир, а также тетрагидрофуран, или смеси растворителей. С помощью процесса растворения и кристаллизации или кристаллизации в условиях уравновешивания воды можно получать воспроизводимым образом определенные гидраты, которые находятся в кристаллической или полиморфной формах.
Получение негидратированных бисдиалкиламмонийных солей по настоящему изобретению предпочтительно осуществляют в одну стадию с использованием соответствующего растворителя, который необязательно смешивают с антирастворителем. Таким путем получают кристаллические соли.
Как правило, аминокислотные соли по настоящему изобретению получают в аморфной форме.
Способы получения солей также являются объектами настоящего изобретения.
Такие соли или гидраты солей по изобретению получают, например, путем нейтрализации валсартана в форме кислоты основанием, несущим соответствующий катион. Такую нейтрализацию целесообразно осуществлять в водной среде, например, в воде или в смеси воды и растворителя, в котором валсартан более растворим, чем в воде. Соли со слабыми основаниями можно превращать в другие соли либо путем обработки более сильными основаниями, либо путем обработки кислотами и последующей нейтрализации другими основаниями.
Кристаллизацию, прежде всего гидратов солей щелочноземельных металлов, проводят в воде или в водной среде, состоящей из воды и по меньшей мере одного растворителя, который может смешиваться или частично смешиваться с водой, т.е. полярность которого не является слишком малой, например, алканола, такого как метанол, этанол, пропанол, изопропанол, бутанол, ацетон, метилэтилкетон, ацетонитрил, ДМФ, ДМСО. Алканольная фракция составляет приблизительно 10-90, или 20-70, предпочтительно 30-50 об.%. В случае применения высших алканолов менее полярный растворитель может присутствовать в более низких концентрациях. Вследствие ограниченной растворимости валсартана в воде процесс часто осуществляют в суспензиях или в растворе, если валсартан растворим в других компонентах растворителя.
Согласно одному из вариантов осуществления, например, для получения кальциевой соли валсартана, водный раствор валсартана нейтрализуют с помощью раствора гидроксида кальция при комнатной температуре и раствор оставляют для кристаллизации. Согласно предпочтительному варианту способа кристаллизацию осуществляют из смеси растворителей вода/этанол, где доля этанола составляет приблизительно 30-50 об.%. Согласно наиболее предпочтительному варианту кристаллизацию осуществляют в закрытой системе с использованием низкого градиента температуры (предпочтительно 1-2°С при 40°С) в растворе, содержащем 30 об.% этанола.
В предпочтительном варианте можно оптимизировать процесс кристаллизации, например, ускорить его, путем добавления по меньшей мере одного затравочного кристалла.
Соли по изобретению можно применять, например, в форме фармацевтических композиций, которые содержат действующее вещество, например, терапевтически эффективное количество действующего вещества, необязательно в сочетании с фармацевтически приемлемым носителем, например, с неорганическим или органическим, твердым или необязательно жидким фармацевтически приемлемым носителем, пригодным для энтерального, например, перорального или парентерального введения.
Изобретение относится в частности к фармацевтической композиции, прежде всего в виде твердой дозируемой формы, предназначенной предпочтительно для перорального введения, необязательно содержащей фармацевтически приемлемый носитель.
Такие фармацевтические композиции можно применять, например, для профилактики и лечения заболеваний или состояний, которые можно облегчать с помощью блокады AT1-рецептора, например,
заболевания или состояния, выбранных из группы, включающей
(а) гипертензию, застойную сердечную недостаточность, почечную недостаточность, прежде всего хроническую почечную недостаточность, рестеноз после чрезкожной внутрипросветной ангиопластики и рестеноз после хирургической операции, связанной с шунтированием коронарной артерии;
(б) атеросклероз, устойчивость к инсулину и синдром X, сахарный диабет типа 2, ожирение, нефропатию, почечную недостаточность, например, хроническую почечную недостаточность, гипотиреоидизм, состояние после инфаркта миокарда (МИ), коронарные заболевания сердца, гипертензию, связанную с возрастом, семейную дислипидемическую гипертензию, повышенное образование коллагена, фиброз и гипертензию, возникающую после ремоделирования (антипролиферативное действие комбинации), при этом все указанные заболевания или состояния связаны с гипертензией или не связаны с ней;
(в) эндотелиальную дисфункцию, сопровождающуюся или не сопровождающуюся гипертензией,
(г) гиперлипедемию, гиперлипопротеинемию, атеросклероз и гиперхолестеролемию и
(д) глаукому.
Композиции по изобретению предпочтительно применяют для лечения повышенного кровяного давления и застойной сердечной недостаточности, а также состояния после инфаркта миокарда.
Специалист в данной области способен выбрать соответствующую стандартную модель для тестирования с использованием животных с целью оценки терапевтических показаний и благоприятных воздействий, приведенных выше и ниже в настоящем описании.
Фармацевтические действия, которые проявляются при введении соединений из числа солей по настоящему изобретению или комбинации действующих веществ по настоящему изобретению, можно продемонстрировать, например, с помощью соответствующих фармакологических моделей, известных в данной области техники. Специалист в данной области способен выбрать соответствующую стандартную модель для тестирования с использованием животных с целью оценки терапевтических показаний и благоприятных воздействий, приведенных выше и ниже в настоящем описании.
Указанные благоприятные действия можно продемонстрировать, например, на модели для тестирования, описанной у G.Jeremic и др., J.Cardovasc. PharmacoL, 27:347-354, 1996.
Например, имеющую большое значение возможность применения солей или композиций по настоящему изобретению для предупреждения и лечения инфаркта миокарда можно выявить с помощью описанной ниже модели для тестирования.
Схема опыта
В проводимом исследовании в качестве модели острого инфаркта миокарда используют хроническую обструкцию коронарной артерии (КАО) у крыс. Эксперименты проводят на 5 группах животных, которые подвергают различным перечисленным ниже обработкам:
- животные, обработанные плацебо
- КАО + носитель
- КАО + соль по настоящему изобретению, необязательно
- КАО + соль по настоящему изобретению + компонент, входящий в состав композиции.
В процессе исследования измеряют следующие параметры:
- размер инфаркта
- объем камеры левого желудочка (ЛЖ)
- плотность интерстициального и периваскулярного коллагена в пораженном миокарде ЛЖ
- содержание протеина COL-I и COL-III в пораженном миокарде ЛЖ (методом вестерн-блотинга)
- площадь поперечного сечения и длину кардиомиоцитов в срезах миокарда ЛЖ
- концентрацию ренина и альдостерона в плазме
- концентрацию натрия, калия и альдостерона в моче
- кровяное давление у находящихся в сознании животных
- кровяное давление в ЛЖ и сонной артерии у анестезированных животных.
Методика
Размер инфаркта: Поперечные гистологические срезы левого желудочка толщиной 6 мкм окрашивают красителем тетразолиевым нитроголубым и регистрируют с помощью CCD-видеокамеры (камера на приборах с зарядовой связью) типа B/W XC-77CE (фирма Sony). Полученное изображение обрабатывают с помощью системы анализа изображений типа KS 300 (фирма Carl Zeiss Vision) с использованием разработанного специально для этой цели программного обеспечения (Porzio и др., 1995). Один оператор, принимающий участие в опыте «вслепую» в интерактивном режиме определяет границы межжелудочковой перегородки и на каждом срезе полуавтоматически идентифицирует область инфаркта, представляющую собой область неокрашенной ткани желудочка. Программа автоматически рассчитывает ряд геометрических параметров для каждого компонента среза желудочка, а именно для камеры, перегородки, области инфаркта, области инфаркта стенки ЛЖ и живой стенки ЛЖ (Porzio и др., 1995).
Гистология: Сердца фиксируют in situ путем направленной вверх перфузии забуференным 4%-ным формальдегидом после остановки сердца в систоле, которую осуществляют с помощью i.v. инъекции 0,5М KCl. После фиксации определяют по отдельности массу левого желудочка (ЛЖ) и свободной стенки правого желудочка; наибольший диаметр ЛЖ измеряют с помощью циркуля. Предназначенные для гистологического исследования срезы ЛЖ окрашивают гематоксилином и эозином для качественного анализа и количественной оценки поперечной области кардиомиоцитов с помощью полуавтоматической процедуры анализа изображений. Накопление интерстициального коллагена в ЛЖ оценивают на окрашенных Сириус красным срезах с помощью полуавтоматической процедуры анализа изображений (Masson и др., 1998).
Содержание коллагена в пораженном миокарде ЛЖ: Ткань пораженного миокарда ЛЖ гомогенизируют, подвергают электрофорезу в ПААГ-ДСН и электроблотингу на нитроцеллюлозной мембране. Блоты обрабатывают первичными антителами, т.е. кроличьей антисывороткой к крысиному коллагену типа I или типа II (фирма Chemicon). Для распознавания первичных антител используют вторичные антитела, конъюгированные с щелочной фосфатазой (для коллагена типа I) или пероксидазой (для коллагена типа II).
Объем камеры левого желудочка: Объем камеры ЛЖ определяют для сердец, остановленных в диастоле (KCl) и фиксированных в формалине при гидростатическом давлении, эквивалентном измеренному конечному диастолическому давлению в ЛЖ. Для измерения длины внутренней полости ЛЖ в ЛЖ погружают измерительный стержень. Поперечные диаметры камеры ЛЖ измеряют на двух поперечных срезах толщиной 1 мм, взятых вблизи основания и верхушки желудочка (Jeremic и др., 1996). Объем камеры вычисляют путем интегрирования уравнения, в которое входят поперечные диаметры и длина внутренней полости.
Системная гемодинамика и гемодинамика левого желудочка: Для измерения систолического и диастолического кровяного давления в правую сонную артерию вводят микродатчик давления (типа Millar SPC-320), соединенный с регистрирующим устройством (типа Windograf, фирма Gould Electronics). Датчик давления продвигают в ЛЖ для измерения систолического (СДЛЖ) и конечного диастолического (КДДЛЖ) давлений в левом желудочке, первой производной давления в ЛЖ по времени (+dP/dt) и частоты сердечных сокращений.
Неинвазивное измерение кровяного давления: Систолическое кровяное давление и частоту сердечных сокращений измеряют у находящихся в сознании крыс с помощью манжетки для хвоста (типа Letica LE 5002).
Содержание электролитов, гормонов в моче: Крыс по отдельности помещают в клетки для исследования метаболизма и собирают в течение 24 ч мочу в 1 мл 6н. HCl. Измеряют поглощение воды. Содержащиеся в моче катехоламины экстрагируют на колонках типа Bondelut С 18 (фирма Varian), разделяют с помощью ЖХВР (Арех-II С 18, 3 мкм, аналитическая колонка 50×4,5 мм, фирма Jones Chromatography) и количественно оценивают с помощью электрохимического детектора (типа Coulochem II, фирма ESA) (Goldstein и др., 1981). Содержание альдостерона в плазме и моче и ангиотензина II в плазме определяют с помощью специфических радиоиммунных анализов (Aldoctk-2, DiaSorin и Angiotensin II, фирма Nichols Diagnostics). Содержание натрия и калия в моче измеряют с помощью пламенной фотометрии.
Размер образца:
Для выявления достоверных биологических различий достаточно иметь в каждой из групп обработки 10 животных, которых подвергают анализу. Для конечного анализа отбирают только тех крыс, у которых размер инфаркта составляет по меньшей мере 10% площади среза ЛЖ.
Известно, что эндотелиальная дисфункция является фактором, имеющим решающее значение для сосудистых заболеваний. Эндотелий играет двойную роль, представляя собой источник различных гормонов или побочных продуктов, обладающих противоположными действиями: вазодилатацией и вазоконстрикцией, ингибированием или стимулированием роста, фибринолизом или тромбогенезом, производством антиоксидантов или окислителей. Генетически предрасположенные к гипертензии животные, имеющие эндотелиальную дисфункцию, представляют собой пригодную модель для оценки эффективности сердечно-сосудистой терапии.
Эндотелиальная дисфункция характеризуется, например, повышенным окислительным стрессом, приводящим к понижению уровня оксида азота, повышенными уровнями факторов, участвующих в коагуляции или фибринолизе, таких как плазминогенактивирующий ингибитор 1 (PAI-1), тканевой фактор (TF), тканевой активатор плазминогена (tPA), повышенными уровнями факторов межклеточной адгезии, таких как ICAM и VCAM, повышенными уровнями факторов роста, таких как bFGF (b-фактор роста фибробластов), TGFb (b-фактор роста Т-клеток), PDGF (тромбоцитарный фактор роста), VEGF (сосудистый эндотелиальный фактор роста), всех факторов, вызывающих связанное с пролиферацией клеток воспаление и фиброз.
Эффективность лечения, например, эндотелиальной дисфункции можно продемонстрировать с помощью следующего фармакологического теста:
Материалы и методы
20-24-недельных самцов крыс линии SHR, полученных от фирмы RCC Ltd. (Фюллингсдорф, Швейцария), содержат в помещении с контролируемым температурным и световым режимом, предоставляя свободный доступ к корму для крыс (Nafag 9331, Госсау, Швейцария) и водопроводной воде. Эксперимент проводят согласно инструкциям NIH (Национальный институт здравоохранения), одобренным ветеринарной службой кантона (Bew 161, Kantonales Veterinäramt, Листаль, Швейцария). Всех крыс обрабатывают ингибитором NO-синтазы L-NAME (фирма Sigma Chemicals), который вводят в питьевой воде (50 мг/л) в течение 12 недель. Средняя суточная доза L-NAME, рассчитанная по количеству поглощенной воды, составляет 2,5 мг/кг/день (диапазон 2,1-2,7).
Крыс можно разделять на 2 или 3 группы: группа 1, контроль (n равно, например, 40); группа 2, обработка солью по настоящему изобретению (n равно, например, 40); группа 3 (для тестирования композиций), обработка компонентом, входящим в композицию (n равно, например, 30). Лекарственные средства вводят в жидкости для питья. Прессорное действие Ang II, применяемого в концентрации 1 мг/кг, полученное для контрольных нормотензивных крыс, может уменьшаться после обработки солью по настоящему изобретению (Gervais и др., 1999).
Каждую неделю измеряют вес тела. За 3 и 2 недели до начала опыта и через 2 недели после введения лекарственного средства регистрируют систолическое кровяное давление и частоту сердечных сокращений с помощью плетисмографа, снабженного манжеткой, охватывающей хвост. У крыс, содержащихся в индивидуальных клетках (для исследования метаболизма), собирают мочу в течение 24 ч перед началом обработки и спустя 4 и 12 недель и стандартными лабораторными методами измеряют объем и проводят анализ на содержание протеина, креатинина, натрия и калия. В эти же моменты времени берут образцы крови из ретроорбитального сплетения (максимум 1 мл) для анализов на содержание креатинина, Na+ и К+.
По истечении 4 недель умерщвляют по десять крыс из каждой группы и выделяют почки и сердце для морфологического анализа. Остальных крыс умерщвляют по истечении 12 недель. Измеряют вес сердца и почек. Последние образцы крови, взятые по истечении 4 (морфометрический анализ) и 12 (завершение исследования) недель, содержащие 5% ЭДТК, используют для оценки уровня альдостерона с помощью радиоиммунного анализа с использованием набора для радиоиммунного анализа альдостерона (РИА) типа DPC coat-a-count (фирма Bühlmann, Швейцария).
Статистический анализ:
Все результаты представлены в виде среднего значения ±СКО. Статистический анализ осуществляют с помощью однонаправленного дисперсионного анализа, после чего применяют анализ с использованием множественного критерия Дункана и критерия Ньюмена-Кеулса для сравнения данных, полученных для различных групп. Статистически достоверными считаются результаты с уровнем вероятности менее 0,05.
Ускорение регресса атеросклероза, не сопровождающееся воздействием на уровни липидов в сыворотке, можно продемонстрировать, например, на модели с использованием животных, согласно методу, описанному у Н.Kano и др., Biochemical and Biophysical Research Communications, 259, 414-419 (1999).
Тот факт, что соли или комбинации по настоящему изобретению можно применять для достижения регресса атеросклероза, индуцированного диетой с высоким содержанием холестерина, можно продемонстрировать с помощью модели для тестирования, описанной, например, у С.Jiang и др. в Br.J.Pharmacol. 104, 1033-1037(1991).
Тот факт, что соли или комбинации по настоящему изобретению можно применять для лечения почечной недостаточности, прежде всего хронической почечной недостаточности, можно продемонстрировать с помощью модели для тестирования, описанной, например, у D.Cohen и др. в Journal of Cardiovascular Pharmacology, 32: 87-95 (1998).
Композиции по настоящему изобретению, которые при необходимости могут содержать другие обладающие фармакологической активностью вещества, получают хорошо известным методом, например с помощью обычных процессов смешения, грануляции, нанесения покрытия, растворения или лиофилизации, и они содержат от приблизительно 0,1 до 100%, предпочтительно от приблизительно 1 до приблизительно 50% лиофилизатов, включающих до 100% действующего вещества.
Изобретение относится также к композициям, содержащим соли по изобретению.
Изобретение относится также к применению солей по изобретению предпочтительно для приготовления фармацевтических композиций, предназначенных, прежде всего, для профилактики и также для лечения заболеваний или состояний, которые можно облегчать путем блокады AT1-рецептора. Их применяют, прежде всего, для лечения повышенного кровяного давления и застойной сердечной недостаточности, а также состояния после инфаркта миокарда.
Изобретение относится также к применению агентов по изобретению для профилактики и лечения заболеваний или состояний, которые можно облегчать путем блокады AT1-рецептора, которое отличается тем, что пациенту, включая человека, нуждающемуся в таком лечении, вводят терапевтически эффективное количество соли по изобретению необязательно в сочетании с по меньшей мере одной композицией для лечения сердечно-сосудистых заболеваний и связанных с ними состояний и заболеваний, перечисленных выше или ниже.
Изобретение относится также к комбинациям, например фармацевтическим комбинациям, содержащим соль по настоящему изобретению или в каждом случае фармацевтически приемлемую соль в сочетании с по меньшей мере одной композицией, предназначенной для лечения сердечно-сосудистых заболеваний и связанных с ними состояний и заболеваний, перечисленных выше или ниже, или в каждом случае ее фармацевтически приемлемую соль. Комбинации агента по изобретению или в каждом случае его фармацевтически приемлемой соли с другими композициями, предназначенные для лечения сердечно-сосудистых заболеваний и связанных с ними состояний и заболеваний, перечисленных выше или ниже, также являются объектами настоящего изобретения.
Комбинация может содержать, например, следующие композиции, выбранные из группы, включающей:
(I) ингибитор HMG-Co-A-редуктазы или его фармацевтически приемлемую соль,
(II) ингибитор ангиотензинпревращающего фермента (АСЕ) или его фармацевтически приемлемую соль,
(III) блокатор кальциевых каналов или его фармацевтически приемлемую соль,
(IV) ингибитор альдостеронсинтазы или его фармацевтически приемлемую соль,
(V) антагонист альдостерона или его фармацевтически приемлемую соль,
(VI) двойной ингибитор ангиотензинпревращающего фермента/нейтральной эндопептидазы (ACE/NEP) или его фармацевтически приемлемую соль,
(VII) антагонист эндотелина или его фармацевтически приемлемую соль,
(VIII) ингибитор ренина или его фармацевтически приемлемую соль, и
(IX) диуретик или его фармацевтически приемлемую соль.
В контексте настоящего описания понятие ингибиторы HMG-Co-A-редуктазы (также называемые ингибиторами β-гидрокси-β-метилглутарил-кофермент-А-редуктазы) относится к таким действующим веществам, которые можно применять для понижения уровней липидов, в том числе холестерина, в крови.
Класс ингибиторов HMG-Co-A-редуктазы включает соединения, обладающие различными структурными особенностями. Следует отметить, например, соединения, выбранные из группы, включающей аторвастатин, церивастатин, компактин, далвастатин, дигидрокомпактин, флуиндостатин, флувастатин, ловастатин, питавастатин, мевастатин, правастатин, ривастатин, симвастатин и велостатин, или в каждом случае их фармацевтически приемлемые соли.
Предпочтительными ингибиторами HMG-Co-A-редуктазы являются поступающие в продажу агенты, наиболее предпочтительными являются флувастатин и питавастатин или в каждом случае их фармацевтически приемлемые соли.
Прекращение ферментативного разложения ангиотензина I, в результате которого образуется ангиотензин II, с помощью так называемых ингибиторов АСЕ (также называемых ингибиторами ангиотензинпревращающего фермента) представляет собой перспективный путь регулирования кровяного давления и позволяет также создать терапевтический метод, пригодный для лечения застойной сердечной недостаточности.
Класс ингибиторов АСЕ включает соединения, обладающие различными структурными особенностями. Следует отметить, например, соединения, выбранные из группы, включающей алацеприл, беназеприл, беназеприлат, каптоприл, церонаприл, цилазаприл, делаприл, эналаприл, энаприлат, фозиноприл, имидаприл, лизиноприл, мовелтоприл, периндоприл, хинаприл, рамиприл, спираприл, темокаприл и трандолаприл, или в каждом случае их фармацевтически приемлемые соли.
Предпочтительными ингибиторами АСЕ являются поступающие в продажу агенты, наиболее предпочтительными являются беназеприл и эналаприл.
Класс ССВ (блокаторы кальциевых каналов) включает, прежде всего, дигидропиридины (ДГП) и соединения, не относящиеся к ДГП, такие как ССВ типа дилтиазема и верапамила.
В качестве ССВ в указанной композиции предпочтительно применяют относящиеся к ДГП соединения, выбранные из группы, включающей амлодипин, фелодипин, риозидин, исрадипин, лацидипин, никардипин, нифедипин, нигулдипин, нилудипин, нимодипин, низолдипин, нитрендипин и нивалдипин, а не относящиеся к ДГП соединения предпочтительно выбирают из группы, включающей флунаризин, прениламин, дилтиазем, фендилин, галлопамил, мибефрадил, анипамил, тиапамил и верапамил, и в каждом случае их фармацевтически приемлемые соли. Все указанные ССВ применяют в терапевтических целях, например, в качестве антигипертензивных лекарственных средств, лекарственных средств против грудной жабы или антиаритмических лекарственных средств.
К предпочтительным ССВ относятся амлодипин, дилтиазем, израдипин, никардипин, нифедипин, нимодипин, низолдипин, нитрендипин и верапамил, или, например, в зависимости от конкретного ССВ, его фармацевтически приемлемая соль. В качестве ДГП наиболее предпочтительным является амлодипин или его фармацевтически приемлемая соль, прежде всего безилат. Наиболее предпочтительным представителем соединений, не относящихся к ДГП, является верапамил или его фармацевтически приемлемая соль, прежде всего гидрохлорид.
Ингибитор альдостеронсинтазы представляет собой фермент, который превращает кортикостерон в альдостерон путем гидроксилирования кортикостерона с образованием 18-ОН-кортикостерона и превращения 18-ОН-кортикостерона в альдостерон. Класс ингибиторов альдостеронсинтазы, которые, как известно, применяют для лечения гипертензии и первичного альдостеронизма, включает как стероидные, так и нестероидные ингибиторы альдостеронсинтазы, при этом последние являются наиболее предпочтительными.
Предпочтительными являются ингибиторы альдостеронсинтазы, которые поступают в продажу, или ингибиторы альдостеронсинтазы, применение которых разрешено органами здравоохранения.
Класс ингибиторов альдостеронсинтазы включает соединения, обладающие различными структурами. Следует отметить, например, соединения, выбранные из группы, включающей нестероидные ингибиторы ароматазы анастрозол, фадрозол (в том числе его (+)-энантиомер), а также стероидный ингибитор ароматазы экземестан, или в каждом случае, если это оказавается возможным, их фармацевтически приемлемые соли.
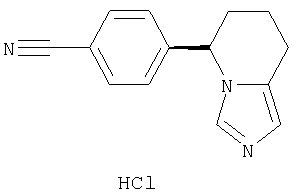
Наиболее предпочтительным нестероидным ингибитором альдостеронсинтазы является (+)-энантиомер гидрохлорида фадрозола (патенты США 4617307 и 4889861) формулы
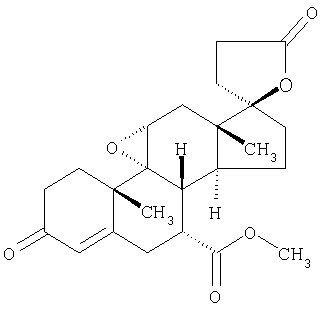
Предпочтительным стероидным антагонистом альдостерона является эплеренон формулы
 или
или
спиронолактон.
Предпочтительным двойным ингибитором ангиотензинпревращающего фермента/нейтральной эндопептидазы (ACE/NEP) является, например, омапатрилат (см. ЕР 629627), фазидоприл или фазидоприлат или, если это возможно, фармацевтически приемлемые соли.
Предпочтительным антагонистом эндотелина является, например, босентан (см. ЕР 526708 А), а также тезозентан (см. WO 96/19459), или в каждом случае их фармацевтически приемлемые соли.
Ингибитором ренина является, например, непептидный ингибитор ренина, такой как соединение формулы
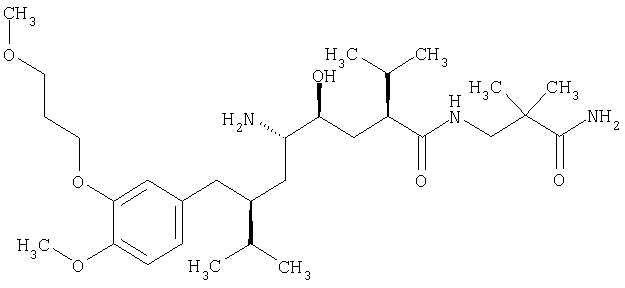
имеющее химическое название 2(S), 4(S), 5(S), 7(S)-N-(3-амино-2,2-диметил-3-оксопропил)-2,7-ди(1-метилэтил)-4-гидрокси-5-амино-8-[4-метокси-3-(3-метоксипропокси)фенил]октаноамид. Данное репрезентативное соединение конкретно описано в ЕР 678503 А. Наиболее предпочтительным является его полуфумарат.
В качестве диуретика можно применять, например, тиазидное производное, выбранное из группы, включающей хлортиазид, гидрохлортиазид, метилхлортиазид и хлорталидон. Наиболее предпочтительным является гидрохлортиазид.
Обладающие в совокупности терапевтической эффективностью количества действующих веществ, входящих в комбинацию по изобретению, можно вводить предпочтительно одновременно или последовательно в произвольном порядке, по отдельности или в виде фиксированной композиции.
Сведения о структуре действующих веществ, обозначенных их родовыми наименованиями или товарными знаками, можно найти в современном издании справочника "The Merck Index" или в базах данных, например Patents International (например, в IMS World Publications). Их содержание, относящееся к данному вопросу, включено в настоящее описание в качестве ссылки. Специалист в данной области может выбирать действующие вещества и на основе этих ссылок также получать их и тестировать на стандартных моделях в опытах in vitro и in vivo их фармацевтические действия и свойства.
Соответствующие действующие вещества или их фармацевтически приемлемые соли можно применять также в форме сольвата, такого как гидрат, или включающего другие растворители, применяемые для кристаллизации.
Соединения, предназначенные для включения в композицию, могут находиться в форме фармацевтически приемлемых солей. Если эти соединения имеют, например, по меньшей мере один основный центр, то они могут образовывать кислотно-аддитивные соли. Если присутствует дополнительный основный центр, то при необходимости можно получать соответствующие кислотно-аддитивные соли. Соединения, несущие кислотную группу (например, СООН) могут образовывать также соли с основаниями.
Один из вариантов настоящего изобретения относится также к «набору компонентов», например, состоящему из компонентов, предназначенных для объединения согласно настоящему изобретению, которые можно вводить независимо или в виде различных фиксированных комбинаций, включающих различные количества компонентов, т.е. одновременно или в различные моменты времени. При этом компоненты набора можно вводить, например, одновременно или через определенные промежутки времени, т.е. в различные моменты времени и с одинаковыми или различными интервалами времени для любого компонента набора. Предпочтительно интервалы времени выбирают таким образом, чтобы воздействие в отношении заболевания или состояния, подлежащего лечению, в случае применения комбинации компонентов превышало воздействие, которое можно получить при использовании только одного любого компонента.
Кроме того, изобретение относится к предназначенной для продажи упаковке, содержащей композицию по настоящему изобретению вместе с инструкцией для одновременного, раздельного или последовательного применения.
Доза может зависеть от различных факторов, таких как путь введения, вид животного, возраст и/или состояние индивидуума. При пероральном введении суточные дозы составляют приблизительно от 0,25 до 10 мг/кг, а для теплокровных животных весом приблизительно 70 кг, предпочтительно приблизительно от 20 до 500 мг, предпочтительно 40, 80, 160 и 320 мг в пересчете на свободную кислоту.
Изобретение проиллюстрировано на примерах и под его объем подпадают также новые соединения, перечисленные в примерах, а также их применение и способы их получения.
Приведенные ниже примеры служат для иллюстрации изобретения и никоим образом не ограничивают объем изобретения.
Например, описан метод получения двукалиевой соли валсартана, прежде всего ее гидрата. Следует особо отметить двукалиевую соль вследствие ее высокой растворимости в воде. Следует отметить также кристаллический тетрагидрат двукалиевой соли валсартана, имеющий температуру плавления 135,0°С. По данным элементарного анализа определенные образцы этого гидрата имеют содержание воды, составляющее 3,72 моля на моль двукалиевой соли. При комнатной температуре при высоких значениях относительной влажности образуется тетрагидрат, а при низких значениях относительной влажности образуется безводная двукалиевая соль.
Аналогичным образом получают магниевую соль валсартана, например, в виде аморфного твердого продукта, содержащего 3,4% H2O. Температура стеклования, представляющая собой среднее значение для стадии стеклования при удельном нагреве 0,85 Дж·[г·0С]-1 составляет 167°С. Не установлено существование температуры плавления. Оба факта, а именно, существование стеклования и отсутствие температуры плавления, в совокупности с измеренным значением изменения удельной теплоемкости свидетельствуют о том, что рассматриваемая магниевая соль валсартана практически на 100% является аморфной. На основе анализа с помощью стереоспецифической хроматографии было установлено, что энантиомерная чистота такой аморфной магниевой соли составляет 83%.
Пример 1:
Получение in situ тетрагидрата кальциевой соли (S)-N-(1-карбокси-2-метилпроп-1-ил)-N-пентаноил-N-[2'-(1Н-тетразол-5-ил)бифенил-4-илметил]амина
21,775 г (S)-N-(1-карбокси-2-метилпроп-1-ил)-N-пентаноил-N-[2'-(1Н-тетразол-5-ил)бифенил-4-илметил]амина растворяют при комнатной температуре в 300 мл этанола. Путем осторожного добавления 300 мл воды концентрацию этанола понижают до 50 об.%. С использованием магнитной мешалки к этому прозрачному слегка кислому раствору (рН 4) медленно добавляют небольшими порциями 3,89 г Са(ОН)2 так, чтобы значение рН в любое время не превышало приблизительно 8. Вследствие абсорбции СО2 из воздуха используемый Са(ОН)2 содержит следовые количества СаСО3; поэтому его добавляют в 5%-ном избыточном количестве. После добавления стехиометрического количества Са(ОН)2, значение рН становится равным приблизительно 6, а после добавления избыточного количества возрастает до 7. Раствор становится мутным вследствие наличия небольшого количества мелкодисперсного СаСО2, который удаляют путем фильтрации через складчатую фильтрующую ткань. После удаления спиртового компонента происходит непрерывная кристаллизация содержащегося в растворе продукта при его выдерживании при комнатной температуре. Процесс можно ускорять, используя плоский поддон в воздушной сушилке с циркуляцией воздуха при 40°С. После концентрирования приблизительно до половины объема содержание спирта понижается до приблизительно 10 об.% и большая часть продукта переходит в кристаллическое состояние. Его фильтруют, промывают в течение небольшого промежутка времени 10 об.% этанола и сушат при 40°С до достижения постоянной массы. В результате получают тетрагидрат кальциевой соли (S)-N-(1-карбокси-2-метилпроп-1-ил)-N-пентаноил-N-[2'-(1Н-тетразол-5-ил)бифенил-4-илметил]амина.
Установлено, что температура плавления тетрагидрата кальциевой соли валсартана, полученного согласно методу, описанному в примере 1, при нагревании со скоростью 10 К·мин-1 в закрытом контейнере для образца, имеющем малый внутренний объем, составляет 205°С, а энтальпия плавления составляет 92 кДж·моль-1.
Плотность кристаллов тетрагидрата кальциевой соли валсартана, полученного согласно методу, описанному в примере 1, по данным измерений с помощью гелиевого пикнометра составляет 1,297 г·см-3. Это значение согласуется со значением 1,298 г·см-3, теоретически рассчитанным для монокристаллической структуры.
Оптическое вращение для тетрагидрата кальциевой соли валсартана, полученной согласно методу, который описан в примере 1, измеренное в 1%-ном растворе метанола, составляет [а]20 D=+1°.
Энантиомерную чистоту гидрата соли, полученного согласно методу, описанному в примере 1, определяют методом стереоспецифической ЖХВР.
Стереоспецифическое разделение осуществляют на хиральной колонке (типа Chiral AGP). Установлено, что энантиомерная чистота составляет ее = 100%.
Ниже приведены расстояния между плоскостями кристаллической решетки, рассчитанные для линий с наибольшей интенсивностью на основе измерений картины дифракции рентгеновских лучей с помощью камеры Гинье для рассматриваемого образца тетрагидрата кальциевой соли валсартана:
d в [Å]: 16,27, 9,90, 9,39, 8,04, 7,71, 7,05, 6,49, 6,34, 6,2, 5,87, 5,75, 5,66, 5,20, 5,05, 4,95, 4,73, 4,55, 4,33, 4,15, 4,12, 3,95, 3,91, 3,87, 3,35.
На основе элементарного анализа получены следующие данные о содержании элементов, присутствующих в тетрагидрате кальциевой соли валсартана, и воды. Определение количества воды осуществляют после ее удаления при 130°С. По данным элементарного анализа в пределах погрешности продукт имеет общую формулу (C24Н27N5О3)-2Са2+·4Н2O.
Пример 2:
Получение in situ гексагидрата магниевой соли (S)-N-(1-карбокси-2-метилпроп-1-ил)-N-пентаноил-N-[2'-(1Н-тетразол-5-ил)бифенил-4-илметил]амина
43,55 г валсартана, т.е. [(S)-N-(1-карбокси-2-метилпроп-1-ил)-N-пентаноил-N-[2'-(1Н-тетразол-5-ил)бифенил-4-илметил]амина], растворяют при комнатной температуре в 600 мл 50 об. %-ного раствора этанола (полученного из абсолютного этанола (см. справочник Merck) и дважды дистиллированной с помощью кварца воды). Слегка мутный раствор становится прозрачным после добавления еще 50 мл 50%-ного этанола. К указанному слабо кислому раствору, имеющему значение рН 4, добавляют с использованием магнитной мешалки небольшими порциями 4,03 г или 0,1М MgO (Merck p.а.). При этом значение рН возрастает приблизительно до 6. Процесс осуществляют с использованием 10%-ного избытка, т.е. добавляют еще 0,40 г MgO. Этот избыток не растворяется полностью и значение рН возрастает приблизительно до 7,5. Небольшое количество остатка фильтруют через складчатую фильтрующую ткань и промывают 50 мл 50%-ного этанола.
Объединенный прозрачный раствор осторожно концентрируют при 40°С при перемешивании с помощью магнитной мешалки в большом поддоне для кристаллизации. По мере приближения к завершению этого процесса раствор начинает густеть, превращаясь в стекловидный гель. На этой фазе путем поскребывания с помощью стеклянной палочки индуцируют кристаллизацию in situ, что можно определить по белому цвету образующегося при этом кристаллического твердого продукта. Продукт сушат при 50°С в воздушной сушилке с циркуляцией воздуха до достижения постоянной массы. Выход гексагидрата магниевой соли валсартана составляет 53,7 г или 95% в пересчете на валсартан, применяемый в форме свободной кислоты.
Измеренная температура плавления гидрата соли, полученного согласно методу, описанному в примере 2, а именно гексагидрата магниевой соли валсартана, при скорости нагрева 10 К·мин-1 в закрытом контейнере небольшого объема для образца массой 2,24 мг, составляет 132°С, а энтальпия плавления составляет 64 кДж·моль-1.
Плотность кристаллов гексагидрата магниевой соли валсартана, полученного согласно методу, который описан в примере 2, измеренная с помощью гелиевого пикнометра, составляет 1,273 г·см-3. Это значение согласуется со значением 1,256 г·см-3, теоретически рассчитанным для монокристаллической структуры.
Оптическое вращение для гексагидрата магниевой соли валсартана, полученного согласно методу, который описан в примере 2, измеренное в 1%-ном растворе метанола, составляет [а]20 D=-14°
Энантиомерную чистоту гидрата соли, полученного согласно методу, описанному в примере 2, определяют с помощью стереоспецифической ЖХВР. Стереоспецифическое разделение осуществляют на хиральной колонке (типа Chiral AGP). Установлено, что энантиомерная чистота составляет ее = 99,6%,
Ниже приведены расстояния между плоскостями кристаллической решетки, рассчитанные для линий с наибольшей интенсивностью на основе измерений картины дифракции рентгеновских лучей с помощью камеры Гинье для рассматриваемого образца гексагидрата магниевой соли валсартана:
d в [Å] 19,78, 10,13, 9,84, 7,28, 6,00, 5,81, 5,67, 5,21, 5,04, 4,88, 4,21, 4,18, 4,08, 3,95, 3,46, 3,42.
Ниже приведены измеренные на основе элементарного анализа содержания элементов, присутствующих в гексагидрате магниевой соли валсартана, и воды. Определение количества воды осуществляют после ее удаления при 130°С. По данным элементарного анализа в пределах погрешности продукт имеет общую формулу (C24H27N5O3)2-Mg2+·6Н2О.
Пример 3:
Получение гидрата двукалиевой соли (S)-N-(1-карбокси-2-метилпроп-1-ил)-N-пентаноил-N-[2'-(1Н-тетразол-5-ил)бифенил-4-илметил]амина (3,5±1,0 моля Н2O)
5 г (S)-N-(1-карбокси-2-метилпроп-1-ил)-N-пентаноил-N-[2'-(1Н-тетразол-5-ил)бифенил-4-илметил]амина растворяют при осторожном нагревании в 11,5 мл 2н. раствора гидроксида калия и смешивают с 320 мл ацетонитрила. Смесь нагревают в течение 5 мин до температуры дефлегмации (образуется мутный раствор), выдерживают без перемешивания в течение 3 дней при комнатной температуре (образование затравочных кристаллов) и затем выдерживают в течение 24 ч при 0°С. Маточный раствор удаляют декантацией. Кристаллический продукт промывают дважды ацетонитрилом и затем сушат на воздухе в течение 36 ч до достижения постоянной массы. В результате получают гидрат двукалиевой соли (S)-N-(1-карбокси-2-метилпроп-1-ил)-N-пентаноил-N-[2'-(1Н-тетразол-5-ил)бифенил-4-илметил]амина (3,7 моля воды на моль двукалиевой соли). Температура плавления в закрытом контейнере для образцов составляет 135°С.
Элементарный анализ: C24H27N5O3K2·3,72H2O, молекулярная масса 578,72
Диаграмму дифракции рентгеновских лучей измеряют с помощью дифрактометра фирмы Scintag Inc., Купертино, Калифорния, СА 95014, США, с использованием CuKα-источника излучения.
Ниже приведены линии отражения и характеристики интенсивности наиболее значимых линий для гидрата двукалиевой соли валсартана, численные данные приведены в виде 2θ°:
Предпочтительными являются гидраты, характеризующиеся пиками средней и сильной интенсивности.



Пример 4:
Получение двукалиевой соли (S)-N-(1-карбокси-2-метилпроп-1-ил)-N-пентаноил-N-[2'-(1Н-тетразол-5-ил)бифенил-4-илметил]амина
25 г (S)-N-(1-карбокси-2-метилпроп-1-ил)-N-пентаноил-N-[2'-(1Н-тетразол-5-ил)бифенил-4-илметил]амина растворяют в 200 мл этанола. Добавляют 50 мл воды, раствор охлаждают до 0°С и затем смешивают с 57,4 мл 2н. раствора гидроксида калия. Смесь концентрируют путем выпаривания на роторном испарителе, выпаривают снова в присутствии толуола и ацетонитрила и сушат в глубоком вакууме в течение 15 мин при 50°С. Продукт растворяют в 290 мл горячей смеси ацетонитрил/вода (95:5), смешивают с дополнительной порцией ацетонитрила (110 мл), дают остыть и осуществляют затравку приблизительно при 30°С. Смеси дают выстояться в течение 4 дней при комнатной температуре и затем ее подвергают вакуумной фильтрации. Остаток промывают смесью ацетонитрил/вода (95:5) и сушат в глубоком вакууме при 80°С. В результате получают двукалиевую соль (S)-N-(1-карбокси-2-метилпроп-1-ил)-N-пентаноил-N-[2'-(1Н-тетразол-5-ил)бифенил-4-илметил]амина в виде порошка белого цвета. Температура плавления >300°С.
Элементарный анализ: Полученный продукт является гигроскопичным и его можно уравновешивать в воздухе (C24H27N5O3K2, 3,96 молей Н2О).
Пример 5:
Получение двунатриевой соли (S)-N-(1-карбокси-2-метилпроп-1-ил)-N-пентаноил-N-[2'-(1Н-тетразол-5-ил)бифенил-4-илметил]амина
1 г (S)-N-(1-карбокси-2-метилпроп-1-ил)-N-пентаноил-N-[2'-(1Н-тетразол-5-ил)бифенил-4-илметил]амина растворяют в 50 мл этанола, смешивают с 2,3 мл 2н. раствора гидроксида натрия, концентрируют путем выпаривания и остаток упаривают в присутствии этанола и этилацетата. Остаток белого цвета перемешивают с горячим ацетонитрилом и подвергают вакуумной фильтрации при комнатной температуре. После сушки в течение ночи в глубоком вакууме при 80°С получают динатриевую соль (S)-N-(1-карбокси-2-метилпроп-1-ил)-N-пентаноил-N-[2'-(1Н-тетразол-5-ил)бифенил-4-илметил]амина в виде порошка белого цвета. Температура плавления составляет более 260°С, при 295°С происходит окрашивание в коричневый цвет.
Элементарный анализ: Полученный продукт (гигроскопичный) можно уравновешивать в воздухе (C24H27N5O3Na2, 5,36 молей Н2О, молекулярная масса 576,05)
Пример 6:
Получение магниевой соли (S)-N-(1-карбокси-2-метилпроп-1-ил)-N-пентаноил-N-[2'-(1Н-тетразол-5-ил)бифенил-4-илметил]амина
5 г (S)-N-(1-карбокси-2-метилпроп-1-ил)-N-пентаноил-N-[2'-(1Н-тетразол-5-ил)бифенил-4-илметил]амина добавляют к суспензии, содержащей 0,666 г гидроксида магния в 20 мл воды. Добавляют 40 мл метанола, затем смесь перемешивают в течение 2 ч при комнатной температуре и концентрируют. Остаток растворяют в метаноле, фильтруют через фильтр грубой очистки, концентрируют и упаривают в присутствии ацетонитрила. Продукт перемешивают с горячим ацетонитрилом, подвергают вакуумной фильтрации при комнатной температуре и сушат в течение ночи в глубоком вакууме при 90°С. В результате получают магниевую соль (S)-N-(1-карбокси-2-метилпроп-1-ил)-N-пентаноил-N-[2'-(1Н-тетразол-5-ил)бифенил-4-илметил]амин в виде порошка белого цвета. Температура плавления: при нагревании образец приобретает коричневатую окраску и переходит в стекловидное состояние при 300°С.
Элементарный анализ: C24H27N5O3Mg, 0,89 молей Н2О, молекулярная масса: 473,85
Пример 7:
Получение кальциевой соли (S)-N-(1-карбокси-2-метилпроп-1-ил)-N-пентаноил-N-[2'-(1Н-тетразол-5-ил)бифенил-4-илметил]амина
5 г (S)-N-(1-карбокси-2-метилпроп-1-ил)-N-пентаноил-N-[2'-(1Н-тетразол-5-ил)бифенил-4-илметил]амина добавляют к суспензии, содержащей 0,851 г гидроксида кальция в 20 мл воды, и затем смешивают с 200 мл этанола. Смесь перемешивают в течение одного часа при комнатной температуре, концентрируют путем упаривания досуха (повторное упаривание с ацетонитрилом), смешивают с горячим ацетонитрилом (в присутствии следовых количеств этанола и воды) и подвергают вакуумной фильтрации при комнатной температуре.
0,95 г соли нагревают до температуры дефлегмации в 20 мл смеси ацетонитрил/вода (1:1), при этом продукт практически растворяется. Смеси дают остыть до комнатной температуры, смешивают с 20 мл ацетонитрила, подвергают вакуумной фильтрации и дважды промывают смесью ацетонитрил/вода (1:1) и сушат в течение ночи в глубоком вакууме при 80°С. Температура плавления: выше 300°С (разложение).
Элементарный анализ: C24H27N5O3Ca, 1,71 молей Н2О, молекулярная масса 504,39 (определение содержания воды осуществляют после ее удаления при 150°С).
Пример 8:
Получение однокалиевой соли (S)-N-(1-карбокси-2-метилпроп-1-ил)-N-пентаноил-N-[2'-(1Н-тетразол-5-ил)бифенил-4-илметил]амина
2 г (S)-N-(1-карбокси-2-метилпроп-1-ил)-N-пентаноил-N-[2'-(1Н-тетразол-5-ил)бифенил-4-илметил]амина суспендируют в 20 мл воды и смешивают с 2,296 мл 2н. раствора гидроксида калия. Смесь перемешивают в течение 30 мин и смешивают с 50 мл этанола, в результате чего получают бесцветный раствор. Смесь концентрируют путем упаривания, упаривают еще один раз в присутствии ацетонитрила и лиофилизируют из трет-бутанола (в присутствии следовых количеств воды).
Элементарный анализ (после уравновешивания в воздухе). C24H27N5O3Ca, 1,69 молей Н2О, молекулярная масса 504,06 (определение содержания воды осуществляют после ее удаления при 150°С).
Пример 9:
Получение гексагидрата магниевой соли валсартана с помощью процесса уравновешивания воды.
В контейнере для смешения смешивают при комнатной температуре 1600 г валсартана и 6820 г изопропанола до образования суспензии и вносят в стеклянный резервуар объемом 80 л, снабженный мешалкой. Контейнер для смешения промывают порциями изопропанола, общее количество которого составляет 3919 г, и промывной раствор добавляют к основной смеси. После добавления 3800 г деионизированной воды при перемешивании смесь превращается в гомогенный раствор. Затем добавляют 156,3 г оксида магния, суспендированного в 1520 г деионизированной воды, и к суспендии добавляют 1000 г деионизированной воды. При медленном перемешивании при комнатной температуре происходит растворение оксида магния. Значение рН образовавшегося раствора составляет приблизительно 7,2. При добавлении небольшими порциями еще 2,5 г оксида магния значение рН возрастает до приблизительно 8,3. Образовавшаяся смесь является мутной вследствие наличия нерастворившихся частиц неизвестного типа, присутствующих в оксиде магния.
Полученную смесь вносят с использованием фильтра цилиндрической формы из пористого материала в эмалированный испаритель объемом 35 и стеклянный резервуар и передающую трубку промывают 885 г изопропанола и 1122 г деионизированной воды. Для умеренного концентрирования в испарителе создают вакуум, соответствующий первоначальному теоретическому значению 89-100 мбар. При температуре теплоносителя 45-50°С и температуре кипения смеси 37-40°С осуществляют дистилляцию водного раствора изопропанола в общем количестве 13,66 кг. При понижении давления, при котором осуществляют дистилляцию, до конечного значения 10 мбар и при одновременном повышении температуры теплоносителя до 65°С, общее количество дистиллята возрастает до 17,12 кг. К содержимому испарителя при перемешивании добавляют 9300 г этилацетата, а затем 14,9 г гексагидрата магниевой соли валсартана в качестве затравочных кристаллов. В завершении в смеси растворяют еще 6680 г этилацетата и осуществляют охлаждение при комнатной температуре при перемешивании. Процесс перемешивания продолжают в течение по меньшей мере 24 ч. Затем суспензию фильтруют через фильтры Бюхнера. В результате получают влажный осадок на фильтре. Испаритель промывают 1171 г этилацетата и промывочную смесь используют для промывки осадка на фильтре. После сушки определенной части продукта на металлических поддонах в вакуумной сушилке при давлении 50 мбар и температуре печи 40°С в течение 6,5 ч до достижения постоянной массы получают сухую субстанцию.
Физические данные, прежде всего диаграмма дифракции рентгеновских лучей, соответствуют характеристикам гексагидрата магниевой соли, полученной согласно методу, описанному в примере 2.
Пример 10:
Получение тетрагидрата кальциевой соли валсартана.
1600 г валсартана и 7000 г этанола перемешивают до образования суспензии в контейнере для смешения при комнатной температуре и вносят в эмалированный испаритель объемом 35 л, снабженный мешалкой. Контейнер для смешения промывают порциями этанола общей массой 2000 г и промывной раствор добавляют к основной смеси. После добавления при перемешивании 9000 г деионизированной воды смесь превращается в гомогенный раствор. Затем добавляют 272 г гидроксида кальция, суспендированного в 1500 г деионизированной воды, и в суспензию вносят 1300 г деионизированной воды.
При медленном перемешивании при комнатной температуре происходит растворение гидроксида кальция. Значение рН образовавшегося раствора составляет приблизительно 6,9. При добавлении небольшими порциями еще 9,6 г гидроксида кальция значение рН возрастает до приблизительно 10,6. Образовавшаяся смесь является мутной вследствие наличия нерастворившихся частиц, присутствующих в гидроксиде кальция (карбонат кальция). Полученную смесь вносят с использованием фильтра цилиндрической формы из пористого материала в эмалированный испаритель объемом 35 л и стеклянный резервуар и передающую трубку промывают раствором, содержащим 1048 г этанола и 1000 г деионизированной воды. Для умеренного концентрирования в испарителе создают вакуум, соответствующий теоретическому значению 100-120 мбар. При температуре теплоносителя приблизительно 50°С и максимальной температуре кипения 44°С осуществляют дистилляцию водного раствора изопропанола в общем количестве 11,32 кг. В процессе дистилляции происходит спонтанная кристаллизация растворенной соли. Суспензию, оставшуюся после завершения дистилляции, охлаждают при перемешивании до приблизительно 5°С и перемешивают в течение приблизительно 16 ч при 5°С. Затем суспензию фильтруют через фильтры Бюхнера. Испаритель промывают смесью, содержащей 3600 мл деионизированной воды и 400 мл этанола, смесь охлаждают до 5°С и промывочную смесь используют для промывки осадка на фильтре. В результате получают влажный осадок на фильтре. После сушки определенной части продукта на металлических поддонах в вакуумной сушилке при давлении 50 мбар и температуре печи 40°С в течение 24 ч до достижения постоянной массы получают сухую субстанцию.
Физические данные, прежде всего диаграмма дифракции рентгеновских лучей, соответствуют характеристикам тетрагидрата кальциевой соли, полученного согласно методу, описанному в примере 1.
Пример 11:
Гидрат двунатриевой соли валсартана (2,4±1,0 моля Н2О):
50 мл 2н. раствора гидроксида натрия добавляют по каплям при приблизительно 25°С к раствору, содержащему 21,5 г валсартана в 200 мл изопропанола. Прозрачный раствор (рН приблизительно 7,2) концентрируют в вакууме приблизительно при 40°С. Аморфный остаток двунатриевой соли суспендируют в 100 мл изопропанола и удаляют воду с помощью еще одной стадии концентрирования в вакууме приблизительно при 40°С и дегазации. Аморфный остаток суспендируют в 75 мл ацетона и 2 мл воды приблизительно при 40°С. При температуре приблизительно 25-30°С добавляют 200 мл метил-трет-бутилового эфира, при этом компоненты, которые первоначально представляют собой жирные вещества, постепенно превращаются в кристаллическую суспензию. После перемешивания в течение ночи приблизительно при 25°С суспензию охлаждают до 10°С и спустя приблизительно 1 ч подвергают вакуумной фильтрации для удаления атмосферной влаги. Затем осуществляют промывку с помощью 20 мл метил-трет-бугиловото эфира. Влажный осадок на фильтре сушат в течение ночи приблизительно при 30 мбар и 30°С. В результате получают бесцветный кристаллический порошок, обладающий слабой гигроскопичностью. Элементарный анализ: C24H27N5O3Na2, 2,44 моля Н2О
Ниже приведены данные о диаграмме дифракции рентгеновских лучей (линии отражения и интенсивности наиболее важных линий) кристаллического гидрата двунатриевой соли валсартана, полученные путем измерения с помощью дифрактометра фирмы Scintag Inc. Купертино, Калифорния, СА 95014, US, с использованием в качестве источника излучения CuKα:
Пример 12:
Гидрат двукалиевой соли валсартана (3,4±1,0 моля Н2О):
6,9 г карбоната калия добавляют при приблизительно 25°С к раствору, содержащему 21,7 г валсартана в 150 мл ацетона и 20 мл воды. После перемешивания в течение 2 ч при приблизительно 25°С получают почти прозрачный раствор, который концентрируют в вакууме при температуре бани приблизительно 50°С. К остатку (29,3 г), который содержит остаточное количество воды, добавляют 55 мл ацетона и в течение приблизительно 2 ч при приблизительно 35°С вносят метил-трет-бутиловый эфир в общем количестве 250 мл. После перемешивания при приблизительно 25°С легко поддающуюся перемешиванию суспензию кристаллов охлаждают до 10°С, перемешивают в течение по меньшей мере 1 ч, подвергают вакуумной фильтрации и промывают 20 мл метил-трет-бутилового эфира. Влажный осадок на фильтре сушат в течение ночи при давлении приблизительно 30 мбар и при температуре 30°С. В результате получают бесцветный, обладающий слабой гигроскопичностью кристаллический порошок.
Элементарный анализ: C24H27N5O3K2, 3,42 моля Н2О
Диаграмму дифракции рентгеновских лучей получают путем измерения с помощью дифрактометра фирмы Scintag Inc. Купертино, Калифорния, СА 95014, US, с использованием в качестве источника излучения CuKα.
Ниже приведены линии отражения и характеристики интенсивности наиболее значимых линий для гидрата двукалиевой соли валсартана, численные данные приведены в виде 2θ°:
Предпочтительными являются гидраты, характеризующиеся наличием пиков средней и сильной интенсивности.
Пример 13:
Смешанная кальциевая/магниевая соль валсартана:
21,5 г валсартана в 200 мл изопропанола и 100 мл воды перемешивают в течение приблизительно 3 ч при приблизительно 25°С с 1,5 г гидроксида магния и 1,9 г гидроксида кальция. Практически прозрачный раствор концентрируют в вакууме при приблизительно 50°С. К еще теплому полутвердому остатку, который содержит остаточное количество воды, добавляют при перемешивании в общей сложности 240 мл этилацетата. После перемешивания в течение ночи приблизительно при 25°С компоненты, первоначально находящиеся в вязком состоянии, превращаются в гомогенную суспензию. Суспензию подвергают вакуумной фильтрации и промывают 20 мл этилацетата. Влажный осадок на фильтре сушат в вакууме при 30-40°С. Получают бесцветный кристаллический порошок.
Диаграмма дифракции рентгеновских лучей соответствует конгломерату тетрагидрата кальциевой соли и гексагидрата магниевой соли, которые описаны в примерах 1 и 2.
Пример 14:
Бисдиэтиламмонийная соль валсартана:
1,5 г диэтиламина добавляют по каплям приблизительно при 25°С к раствору, содержащему 4,35 г валсартана в 60 мл ацетона. Через небольшой промежуток времени начинается медленная кристаллизация. После перемешивания в течение ночи кристаллический продукт подвергают вакуумной фильтрации при приблизительно 20°С, промывают холодным ацетоном и сушат в вакууме при приблизительно 50°С. Получают бесцветный кристаллический порошок.
Элементарный анализ: С32Н51N7O3, 0,1 моля H2O
Ниже приведены данные о диаграмме дифракции рентгеновских лучей (линии отражения и интенсивности наиболее значимых линий) для кристаллической бисдиэтиламмонийной соли
Пример 15:
Бисдипропиламмонийная соль валсартана:
2,1 г дипропиламина добавляют по каплям при 25°С к раствору, содержащему 4,35 г валсартана в 60 мл ацетона. После начала кристаллизации температуру повышают в течение короткого промежутка времени до 40°С и затем дают понизиться в течение приблизительно 2 ч до комнатной температуры. После перемешивания в течение ночи кристаллический продукт подвергают вакуумной фильтрации, промывают дважды 15 мл ацетона и сушат в вакууме при приблизительно 40°С. Получают продукт в виде кристаллических гранул.
Элементарный анализ: C36H69N7O3, 0,05 моля H2O
Ниже приведены данные о диаграмме дифракции рентгеновских лучей (линии отражения и интенсивности наиболее значимых линий) для кристаллической бисдипропиламмонийной соли
Пример 16:
Бисдибутиламмонийная соль валсартана:
Раствор, содержащий 2,15 г валсартана в 30 мл ацетона, смешивают с 1,4 г дибутиламина при приблизительно 25°С. Через небольшой промежуток времени начинается кристаллизация и густую суспензию в течение приблизительно 1 ч постепенно разбавляют 20 мл изопропилацетата. После перемешивания в течение 4 ч при приблизительно 25°С кристаллы отделяют вакуумной фильтрацией, промывают дважды 10 мл изопропилацетата и сушат в вакууме при 50°С. В результате получают бесцветный кристаллический порошок, обладающий слабой гигроскопичностью.
Элементарный анализ: C40H67N7O3, 0,5 моля Н2О
Ниже приведены данные о диаграмме дифракции рентгеновских лучей (линии отражения и интенсивности наиболее значимых линий) для кристаллической бисдибутиламмонийной соли
Пример композиции 1:
Таблетка, получаемая путем непосредственного прессования:
Ингредиент №1 просеивают через сито с размером ячеек 0,5 мм и смешивают в течение 15 мин в устройстве типа Turbula с ингредиентами 1-6. Таблетки изготавливают путем прессования с помощью таблетировочного пресса с одним пуансоном, диаметр которого составляет 8 мм.
Пример композиции 2:
Таблетка, получаемая путем валкового уплотнения:
Ингредиенты №1-5 смешивают в течение 50 мин и уплотняют с помощью валкового уплотнителя Фрейнда. Полученную полоску размалывают и после смешения с ингредиентом 6, спрессовывают в таблетки с помощью таблетировочного пресса с одним пуансоном, диаметр которого составляет 8 мм.
| название | год | авторы | номер документа |
|---|---|---|---|
| ФАРМАЦЕВТИЧЕСКИЕ КОМБИНАЦИИ АНТАГОНИСТА РЕЦЕПТОРА АНГИОТЕНЗИНА И ИНГИБИТОРА NEP | 2006 |
|
RU2459809C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМБИНАЦИИ АНТАГОНИСТА РЕЦЕПТОРА АНГИОТЕНЗИНА И ИНГИБИТОРА NEP | 2006 |
|
RU2503668C2 |
| ГИДРАТЫ СОЛЕЙ ЩЕЛОЧНОЗЕМЕЛЬНЫХ МЕТАЛЛОВ И ИРБЕСАРТАНА И ИХ ПОЛУЧЕНИЕ | 2006 |
|
RU2401266C2 |
| СОЛЬ АЛИСКИРЕНА И ОРОТОВОЙ КИСЛОТЫ | 2007 |
|
RU2456267C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ДВОЙНОГО ДЕЙСТВИЯ НА ОСНОВЕ НАДМОЛЕКУЛЯРНЫХ СТРУКТУР АНТАГОНИСТА/БЛОКАТОРА РЕЦЕПТОРОВ АНГИОТЕНЗИНА (ARB) И ИНГИБИТОРА НЕЙТРАЛЬНОЙ ЭНДОПЕПТИДАЗЫ (NEP) | 2008 |
|
RU2493844C2 |
| СОЛЬ АЛИСКИРЕНА И СЕРНОЙ КИСЛОТЫ | 2007 |
|
RU2439054C2 |
| Замещенные производные бисфенилового эфира масляной кислоты в качестве ингибиторов NEP | 2019 |
|
RU2784522C2 |
| ГАЛЕНОВЫЕ КОМПОЗИЦИИ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ | 2017 |
|
RU2773029C2 |
| ТОЗИЛАТНАЯ СОЛЬ ТЕРАПЕВТИЧЕСКОГО СОЕДИНЕНИЯ И ЕЕ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2008 |
|
RU2430099C2 |
| СТРОНЦИЕВАЯ СОЛЬ S-ОМЕПРАЗОЛА ИЛИ ЕЕ ГИДРАТ, СПОСОБ ИХ ПОЛУЧЕНИЯ И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2006 |
|
RU2372345C1 |
Описываются новые соли валсартана, выбранные из группы, включающей однонатриевую, однокалиевую, двунатриевую, двукалиевую, магниевую, кальциевую, бисдиэтил (или дипропил, или дибутил) аммонийные соли или их гидраты, а также смеси указанных солей; способ их получения и фармацевтическая композиция, их содержащая; соли могут находиться в кристаллической форме, частично кристаллической, аморфной или полиморфной форме. Полученные соли имеют высокое качество кристаллических решеток, что является основной химической и физической стабильности новых соединений. 3 н. и 8 з.п., табл.11
(I) имеет диаграмму дифракции рентгеновских лучей на порошке, полученную с помощью камеры Гинье, которая характеризуется следующими расстояниями между плоскостями кристаллической решетки:
d в [Å]: 16,1±0,3, 9,9±0,2, 9,4±0,2, 7,03±0,1, 6,50±0,1, 5,87±0,05, 5,74±0,05, 4,95±0,05, 4,73±0,05, 4,33±0,05, 4,15±0,05, 4,12±0,05, 3,95±0,05; или
(II) имеет ATR-ИК-спектр, содержащий полосы абсорбции, соответствующие следующим обратным волновым числам (см-1):
1621 (st); 1578 (m); 1458 (m); 1441 (m); 1417 (m); 1364 (m); 1012 (m); 758(m);738(m);696(m);666(m).
(I) имеет диаграмму дифракции рентгеновских лучей на порошке, полученную с помощью камеры Гинье, которая характеризуется следующими расстояниями между плоскостями кристаллической решетки:
d в [Å]: 19,7±0,3, 10,11±0,2, 9,8±0,2, 7,28±0,1, 5,81±0,05, 5,68±0,05, 5,03±0,05, 4,88±0,05, 4,18±0,05, 4,08±0,05, 3,46±0,05; или
(II) имеет ATR-ИК-спектр, содержащий полосы абсорбции, соответствующие следующим обратным волновым числам (см-1):
3378 (m); 3274 (m); 2956 (m); 1619 (st); 1557 (m); 1464 (m); 1419 (m); 1394 (st); 1374 (m); 1175 (m); 836 (m); 820 (s); 766 (st); 751 (m); 741 (st); 732 (st).
(I) кристаллическую форму;
(II) частично кристаллическую форму;
(III) аморфную форму и
(IV) полиморфную форму.
| US 6071931 А, 06.06.2000 | |||
| WO 9967231 А, 29.12.1999 | |||
| Устройство для испытания горных пород | 1973 |
|
SU443983A1 |
| СПОСОБ ПОЛУЧЕНИЯ 2-Н.БУТИЛ-3-[[2'-(ТЕТРАЗОЛ-5-ИЛ)БИФЕНИЛ-4-ИЛ]МЕТИЛ]-1,3-ДИАЗАСПИРО[4.4]НОН -1-ЕН-4-ОНА В ФОРМЕ А ИЛИ В ФОРМЕ В, СПОСОБ ПОЛУЧЕНИЯ ФОРМЫ В, ФОРМА В И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ОСНОВЕ ФОРМЫ В | 1995 |
|
RU2144536C1 |
| ПРОИЗВОДНЫЕ ТЕТРАЗОЛА | 1992 |
|
RU2091376C1 |

