УРОВЕНЬ ТЕХНИКИ
Область техники, к которой относится изобретение
Настоящее изобретение относится к С1-симметричным бисфосфиновым лигандам и соответствующим катализаторам и к их применению в асимметрических синтезах, включая энантиоселективное гидрирование прохиральных олефинов, с целью получения фармацевтически полезных соединений, в том числе (S)-(+)-3-(аминометил)-5-метилгексановой кислоты,
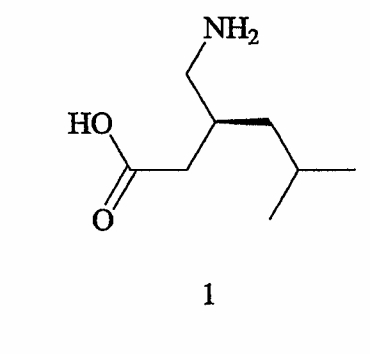
которая обычно известна как прегабалин.
Обсуждение
Хиральные фосфиновые лиганды сыграли важную роль в развитии новых асимметрических реакций, катализируемых переходными металлами, применяемых для обеспечения энантиомерного избытка соединений, обладающих требуемой активностью. Первые успешные попытки асимметрического гидрирования енамидных субстратов были осуществлены в конце 1970 годов с использованием хиральных бисфосфинов в качестве лигандов переходных металлов. См., в частности, B.D. Vineyard et al., J. Am. Chem. Soc. 99(18): 5946-52 (1977); W.S. Knowles et al., J. Am. Chem. Soc. 97(9): 2567-68 (1975). После этих первых опубликованных сообщений последовал всплеск исследований, относящихся к синтезу новых хиральных бисфосфиновых лигандов, которые предназначены для проведения асимметрических гидрирований и осуществления других хиральных каталитических превращений. См. I. Ojima, ed., Catalytic Asymmetrical Synthesis (1993); D.J. Ager, ed., Handbook of Chiral Chemicals (1999).
Некоторые из наиболее эффективных и широко используемых лигандов, разработанных для асимметрического гидрирования, включают BPE лиганды (в частности, (R,R)-Et-BPE, или (+)-1,2-бис((2R,5R)-2,5-диэтилфосфолано)этан); DuPhos лиганды (R,R)-Me-DUPHOS или (-)-1,2-бис((2R,5R)-2,5-диметилфосфолано)бензол); и BisP* лиганд ((S,S)-(-)-1,2-бис(трет-бутилметилфосфино)этан). См., в частности, M.J. Burk, Chemtracts 11(11):787-802 (1998); M.J. Burk et al., Angew Chem., Int. Ed. 37(13/14): 1931-33 (1998); M.J. Burk, et al., J. Org. Chem. 63(18):6084-85 (1998); M.J. Burket al., J. Am. Chem. Soc. 120(18):4345-53 (1998); M.J. Burk et al., J. Am. Chem. Soc. 117(15):4423-24 (1995); M.J. Burk et al., J. Am. Chem. Soc. 115(22): 10125-38 (1993); W.A. Nugent et al., Science 259(5094):479-83 (1993); M.J. Burk et al. Tetrahedron: Asymmetry 2(7):569-92 (1991); M.J. Burk, J. Am. Chem. Soc. 113(22):8518-19 (1991); Т. Imamoto et al., J. Am. Chem. Soc. 120(7):1635-36 (1998); G. Zhu et al., J. Am. Chem. Soc. 119(7): 1799-800 (1997).
Успешное применение BPE, DUPHOS, BisP* и других родственных лигандов в реакциях асимметрического гидрирования относили, помимо других факторов, за счет жесткости их С2-симметричной структуры. Как показано на фиг.1, деление пространства, занимаемого структурой фосфинового лиганда, такого как BisP*, на четыре квадранта дает при присоединении к переходному металлу (в частности, Rh) чередование имеющих и не имеющих пространственные затруднения квадрантов. Указанный структурный фрагмент стимулировал разработку бисфосфиновых лигандов и соответствующих катализаторов для асимметрического гидрирования определенных субстратов, включая енамиды, сложные енольные эфиры и сукцинаты, и, вероятно, мог привести к замедлению разработки бисфосфиновых лигандов, которые не являются С2-симметричными (в частности, С1-симметричных бисфосфиновых лигандов).
Исследователями недавно описаны С1-симметричные бисфосфиновые лиганды и соответствующие катализаторы, которые применимы для осуществления асимметрических превращений, включая реакции энантиоселективного гидрирования. См. совместно поданную заявку на патент США № 2002/0143214 А1, опубликованную 3 октября 2002 года, и совместно поданную заявку на патент США № 2003/0073868 А1, опубликованную 17 апреля 2003 года, полные изложения которых для всех целей включены в данное описание в виде ссылки. Как видно из фиг.2, указанные лиганды, представленные (трет-бутилметилфосфанил)-(ди-трет-бутилфосфанил)этаном, дают при присоединении к переходному металлу, такому как Rh, окружение с тремя стерически затрудненными квадрантами. Тем не менее, способные к образованию связей модели С1-симметричных бисфосфиновых лигандов и соответствующих катализаторов, которые преобразуют их стерические затруднения в энантиоселективность при гидрировании, немногочисленны. См., например, H. Blaser et al., Topics in Catalysis 19: 3 (2002); A. Ohashi et al., European Journal of Organic Chemistry 15: 2535 (2002); K. Matsumura et al., Advanced Synthesis & Catalysis 345: 180 (2003).
Прегабалин, (S)-(+)-3-аминометил-5-метилгексановая кислота, связывается с альфа-2-дельта (α2δ) субъединицей кальциевого канала и имеет отношение к эндогенному ингибирующему нейромедиатору - γ-аминомасляной кислоте (GABA), которая принимает участие в регулировании нейронной активности мозга. Прегабалин обладает антиэпилептической активностью, как описано в патенте США № 5563175, выданном на имя R.B. Silverman et al., и, как полагают, может быть пригоден для лечения, помимо прочих состояний, боли, физиологических состояний, связанных с психомоторными стимуляторами, воспалением, поражением желудочно-кишечного тракта, алкоголизмом, инсомнией и различными психическими расстройствами, включая маниакальный синдром и биполярное расстройство. См., соответственно, патент США № 6242488, выданный на имя L. Bueno et al., патент США № 6326374, выданный на имя L. Magnus & C.A.Segal, и патент США № 6001876, выданный на имя L. Singh; патент США № 6194459, выданный на имя H.C. Akunne et al.; патент США № 6329429, выданный на имя D. Schrier et al.; патент США № 6127418, выданный на имя L. Bueno et al.; патент США № 6426368, выданный на имя L. Bueno et al.; патент США № 6306910, выданный на имя L. Magnus & C.A. Segal; и патент США № 6359005, выданный на имя A.C. Pande, которые для всех целей целиком включены в данное описание в виде ссылки.
Прегабалин получали разными способами. Как правило, получают рацемическую смесь 3-(аминометил)-5-метилгексановой кислоты, а затем разделяют ее на R- и S-энантиомеры. В подобных способах могут быть использованы промежуточный азид (в частности, в патенте США № 5563175, выданном на имя R.B. Silverman et al.), промежуточный малонат (патент США № 6046353, выданный на имя T.M. Grote et al., патент США № 5840956, выданный на имя T.M. Grote et al., и патент США № 5637767, выданный на имя T.M. Grote et al.) или синтез по Гофману (патент США № 5629447, выданный на имя B.K. Huckabee & D.M. Sobieray, и патент США № 5616793, выданный на имя B.K. Huckabee & D.M. Sobieray). В каждом из этих способов рацемат взаимодействует с хиральной кислотой (разделяющим агентом) с образованием пары диастереоизомерных кислот, которые разделяют такими известными методами, как дробная кристаллизация и хроматография. Таким образом, указанные способы включают значительную технологическую обработку, помимо получения рацемата, что вместе с разделяющим агентом приводит к увеличению стоимости процесса. Более того, ненужный R-энантиомер часто отбрасывают, поскольку его нельзя эффективно использовать повторно, и поэтому эффективный выход процесса снижается на 50%.
Кроме того, прегабалин был получен прямым синтезом с использованием вспомогательного хирального вещества (4R,5S)-4-метил-5-фенил-2-оксазолидинона. См., в частности, патенты США №№ 6359169, 6028214, 5847151, 5710304, 5684189, 5608090 и 5599973, все выданные на имя Silverman et al. Несмотря на то что указанные способы позволяют получить прегабалин с высокой энантиомерной чистотой, они являются менее предпочтительными для осуществления синтеза в больших масштабах, поскольку в них используют дорогостоящие реагенты (например, вспомогательное хиральное вещество), которые трудны в обращении, а также используют специальное криогенное оборудование, необходимое для получения требуемых рабочих температур, которые могут быть вплоть до -78°С.
В заявке на патент США 2003/0212290 А1 описывается способ приготовления прегабалина асимметрическим гидрированием цианозамещенного олефина с получением хирального цианосодержащего предшественника - (S)-3-(аминометил)-5-метилгексановой кислоты. Цианосодержащий предшественник затем восстанавливают и получают прегабалин. В заявке раскрывается применение различных С2-симметричых лигандов, включая (R,R)-Me-DUPHOS, что приводит к значительному обогащению прегабалина по сравнению с (R)-3-(аминометил)-5-метилгексановой кислотой.
Несмотря на то что способ, раскрытый в заявке на патент США 2003/0212290 А1, представляет собой коммерчески важный метод получения прегабалина, по многим причинам было бы желательно его улучшить. С2-симметричые бисфосфиновые лиганды, включая запатентованный лиганд (R,R)-Me-DUPHOS, часто трудно приготовить, поскольку они содержат два хиральных центра, что увеличивает их стоимость. Кроме того, несмотря на то, что хиральные катализаторы, раскрытые в заявке на патент США 2003/0212290 А1, приводят к получению цианосодержащего предшественника прегабалина с хорошим энантиомерным избытком (в некоторых случаях энантиомерный избыток [ee, ЭИ] составляет около 95% или больше), полезно было бы добиться более высокой энантиоселективности (получить ЭИ, равный приблизительно 98% или больше). Кроме того, были бы полезны хиральные катализаторы, которые можно было бы использовать для достижения большего соотношения субстрат-к-катализатору (s/c), поскольку они позволили бы при заданной загрузке катализатора или заданной концентрации субстрата использовать более значительные концентрации субстрата или меньшие загрузки катализатора. Более значительные концентрации субстрата привели бы к увеличению производительности и, таким образом, снизили производственные затраты на единицу продукции. Аналогично, меньшие загрузки катализатора привели бы к значительному сокращению затрат на единицу продукции.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
В настоящем изобретении предлагаются вещества и способы получения прегабалина (формула 1) и структурно родственных ему соединений. Заявленные способы включают использование новых хиральных катализаторов, каждый из которых включает С1-симметричный бисфосфиновый лиганд, связанный с переходным металлом (в частности, родием) посредством атомов фосфора. Заявляемое изобретение обладает множеством преимуществ перед существующими способами получения прегабалина и аналогичных соединений. Например, С1-симметричные бисфосфиновые лиганды имеют единственный стереогенный центр, что должно сделать приготовление лигандов и соответствующих им хиральных катализаторов относительно недорогим. Кроме того, как показано ниже в примерах, заявленное изобретение позволяет получить хиральный цианосодержащий предшественник прегабалина с большей энантиоселективностью (ЭИ приблизительно 98% или больше), чем в известных способах. Как также показано ниже в примерах, новые хиральные катализаторы могут использоваться с большими соотношениями субстрата к катализатору (s/c), чем известные катализаторы, что должно приводить к значительному снижению затрат на единицу продукции.
В соответствии с одним из аспектов настоящего изобретения предлагается способ получения требуемого энантиомера соединения формулы 2
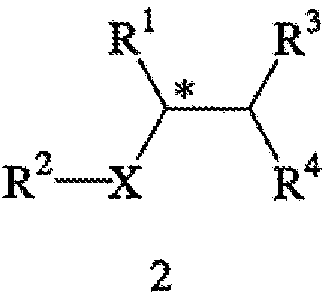
или его фармацевтически приемлемого комплекса, соли, сольвата или гидрата. В формуле 2
R1 обозначает С1-6 алкил, С1-7 алканоиламино, С1-6 алкоксикарбонил,
С1-6 алкоксикарбониламино, амино, амино-С1-6 алкил, С1-6 алкиламино, циано, циано-С1-6 алкил, карбокси или -CO2-Y;
R2 обозначает С1-7 алканоил, С1-6 алкоксикарбонил, карбокси или -CO2-Y;
R3 и R4 независимо обозначают атом водорода, С1-6 алкил, С3-7 циклоалкил, арил или арил-С1-6 алкил, или же R3 и R4 вместе обозначают С2-6 алкандиил;
Х обозначает -NH-, -O-, -CH2- или связь;
Y обозначает катион, а звездочка обозначает стереогенный (хиральный) центр.
Способ включает стадии (а) взаимодействия прохирального субстрата (олефина) формулы 3,
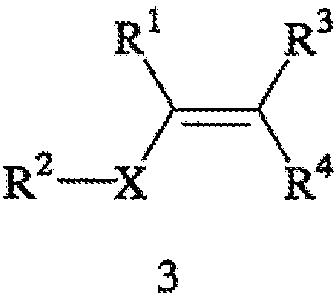
с водородом в присутствии хирального катализатора с образованием соединения формулы 2; и (b) необязательного превращения соединения формулы 2 в фармацевтически приемлемый комплекс, соль, сольват или гидрат. Заместители R1, R2, R3, R4 и Х в формуле 3 те же, что и определенные для формулы 2. Хиральный катализатор включает хиральный лиганд, связанный с переходным металлом посредством атомов фосфора, и имеет структуру, представленную формулой 4,
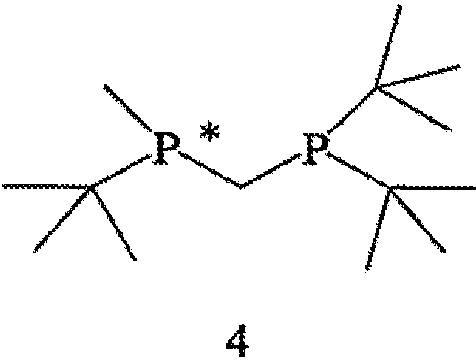
В общем случае способ может применяться для получения требуемого энантиомера соединения формулы 2 с ЭИ, равным приблизительно 95% или больше, а в некоторых случаях с ЭИ, равным приблизительно 99% или больше. Пригодные к использованию прохиральные субстраты включают 3-циано-5-метилгекс-3-еновую кислоту или ее основно-аддитивные соли, такие как 3-циано-5-метилгекс-3-еноат трет-бутиламмониевая соль. Другие пригодные к использованию прохиральные субстраты включают такие субстраты, в которых Y представляет собой ион металла Группы 1 (щелочной), ион металла Группы 2 (щелочно-земельный), ион первичного аммония или ион вторичного аммония.
Наиболее пригодный к использованию хиральный катализатор включает хиральный лиганд формулы 4, который связан с атомом родия посредством атомов фосфора. Другой наиболее пригодный к использованию хиральный катализатор включает энантиомер бисфосфинового лиганда формулы 4, который имеет структуру, представленную формулой 5,
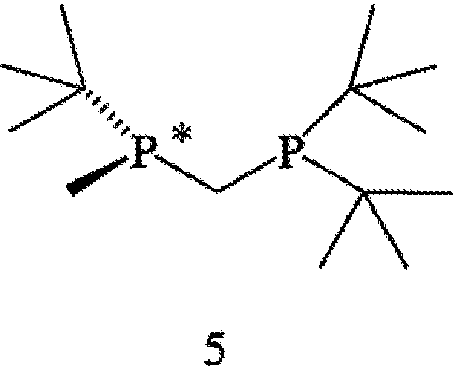
и ЭИ приблизительно 95% или больше. Наиболее пригодный к использованию хиральный катализатор включает энантиомер бисфосфинового лиганда формулы 4, который имеет структуру, представленную формулой 5, и ЭИ приблизительно 99% или больше.
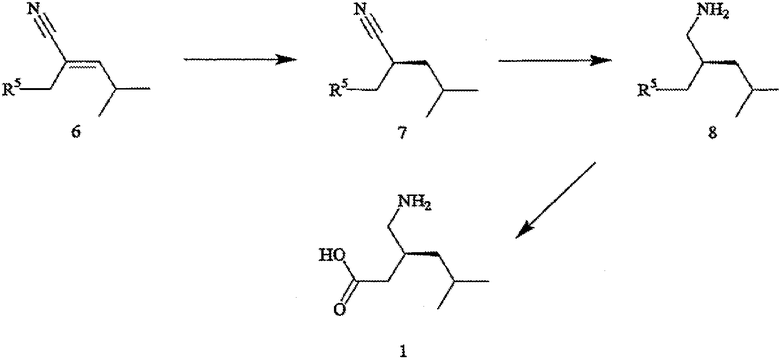
В соответствии с другим аспектом настоящего изобретения предлагается способ получения прегабалина, или (S)-(+)-3-(аминометил)-5-метилгексановой кислоты (формула 1), или его фармацевтически приемлемого комплекса, соли, сольвата или гидрата. Способ включает стадии (а) взаимодействия соединения формулы 6,
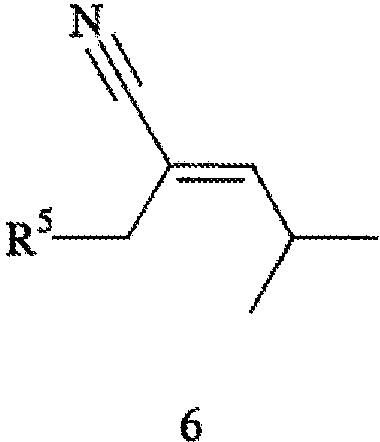
его соответствующего Z-изомера или их смеси, с Н2 (водородом) в присутствии хирального катализатора с получением соединения формулы 7,
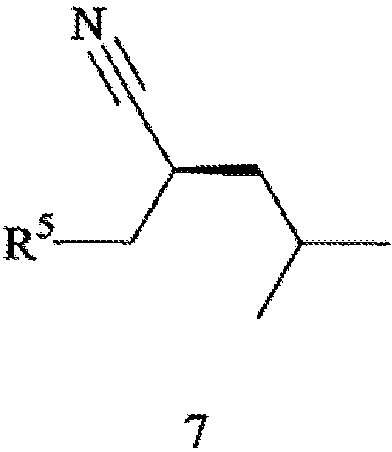
где R5 обозначает карбоксильную группу или -CO2-Y, Y обозначает катион, а хиральный катализатор включает хиральный лиганд (формула 4), связанный с переходным металлом посредством атомов фосфора; (b) восстановления цианового фрагмента соединения формулы 7 с получением соединения формулы 8,
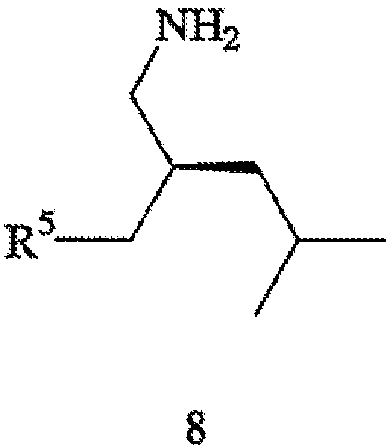
(с) необязательной обработки соединения формулы 8 кислотой с получением прегабалина; и (d) необязательного превращения соединения формулы 8 или формулы 1 в фармацевтически приемлемый комплекс, соль, сольват или гидрат.
Способ может применяться для получения прегабалина, имеющего ЭИ приблизительно 95% или больше или имеющего ЭИ 99% или больше, а в некоторых случаях имеющего ЭИ 99,9% или больше. Пригодные к использованию прохиральные субстраты (формула 6) включают такую основно-аддитивную соль 3-циано-5-метилгекс-3-еновой кислоты, как 3-циано-5-метилгекс-3-еноат трет-бутиламмониевая соль. Другие пригодные к применению прохиральные субстраты включают такие субстраты, в которых Y в формуле 6 обозначает ион металла Группы 1, ион металла Группы 2, ион первичного аммония или ион вторичного аммония. Особенно пригодный к применению хиральный катализатор включает хиральный лиганд формулы 4, который связан с родием посредством атомов фосфора. Другой особенно пригодный к применению хиральный катализатор включает энантиомер бисфосфинового лиганда формулы 4, который имеет структуру, представленную формулой 5 (выше) и имеет ЭИ приблизительно 95% или больше. Наиболее пригодный к применению хиральный катализатор включает энантиомер бисфосфинового лиганда формулы 4, который имеет структуру, представленную формулой 5 и имеет ЭИ приблизительно 99% или больше.
Наконец, в соответствии с еще одним аспектом настоящего изобретения предлагается способ получения требуемого энантиомера соединения формулы 4. Способ включает стадии (а) взаимодействия соединения формулы 9,
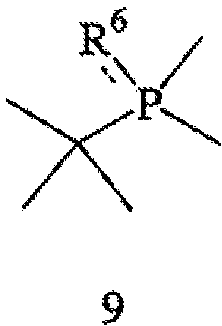
с соединением формулы 10,
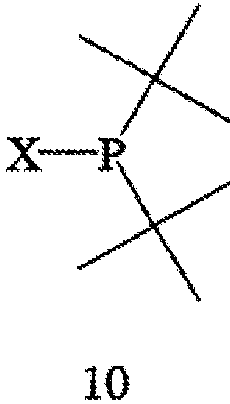
с получением соединения формулы 11,
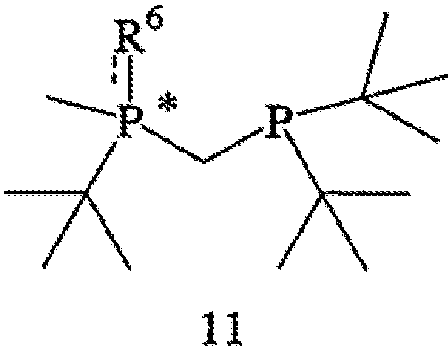
где соединение формулы 9 обрабатывают основанием перед взаимодействием с соединением формулы 10, Х обозначает уходящую группу, а R6 обозначает ВН3, серу или кислород; (b) взаимодействия соединения формулы 11 с бораном, серой или кислородом с образованием соединения формулы 12,
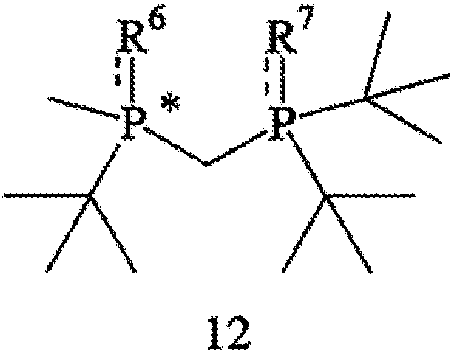
где R7 такой же, как R6, или отличается от R6 и обозначает ВН3, серу или кислород; и (с) удаления R6 и R7 из соединения формулы 12 с образованием соединения формулы 4.
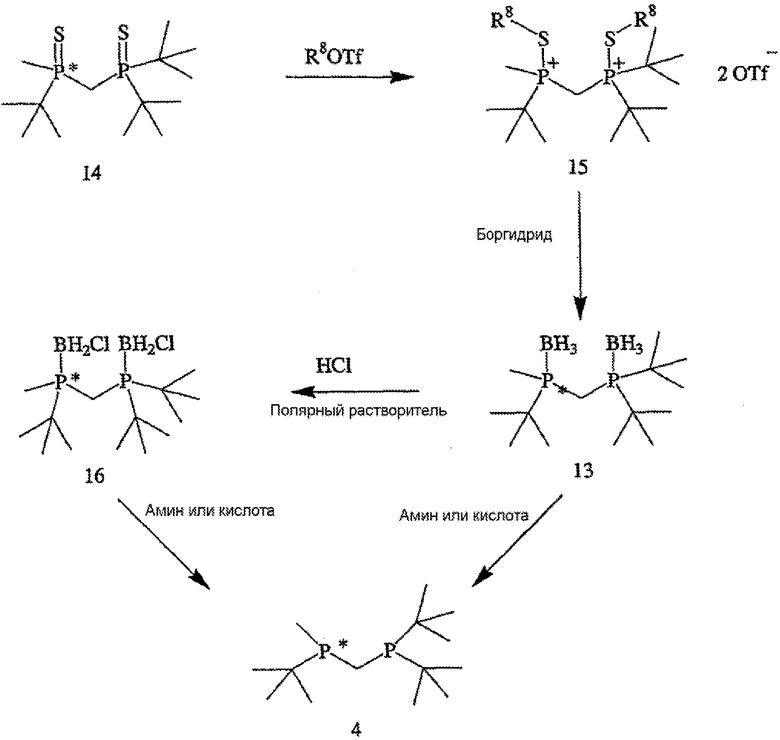
Заявляемый способ особенно пригоден для получения R-энантиомера соединения формулы 5, имеющего ЭИ, равный приблизительно 80%, приблизительно 90%, приблизительно 95% или приблизительно 99% или больше. Как правило, перед удалением R6 и R7 соединение формулы 12 расщепляют на отдельные энантиомеры. Заместители R6 и R7 могут быть удалены различными путями в зависимости от типов конкретных заместителей. Например, в том случае, когда R6 и R7, оба обозначают ВН3, они могут быть удалены путем взаимодействия соединения формулы 13
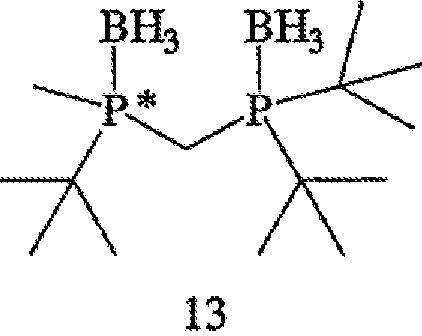
с амином или кислотой с получением соединения формулы 4. Так, например, соединение формулы 13 может вступать во взаимодействие с HBF4•Ме2О с последующим основным гидролизом с образованием соединения формулы 4. Аналогично, с целью удаления R6 и R7 соединение формулы 13 может быть обработано с помощью DABCO, TMEDA, DBU или Et2NH или их комбинацией.
В том случае, когда оба заместителя являются атомами серы, R6 и R7 могут быть удалены с помощью различных способов. Один из способов включает стадии (а) взаимодействия соединения формулы 14,
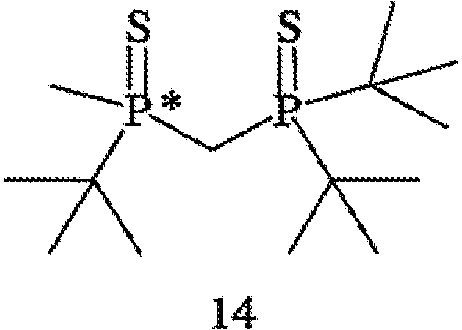
с R8OTf с получением соединения формулы 15,
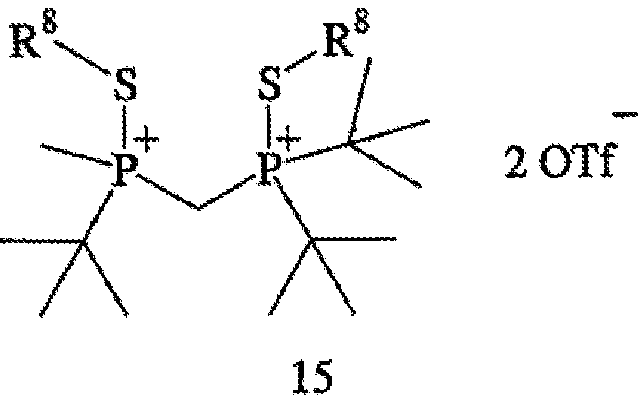
в котором R8 обозначает С1-4 алкил; (b) взаимодействия соединения формулы 15 с боргидридом с образованием соединения формулы 13; и (с) взаимодействия соединения формулы 13 с амином или кислотой с получением соединения формулы 4. Наиболее пригодным заместителем R8 является метил, а наиболее пригодным боргидридом является LiBH4.
Другой способ включает приведенные выше стадии (а) и (b) и далее включает стадии (с) взаимодействия соединения формулы 13 с HCl с образованием соединения формулы 16,
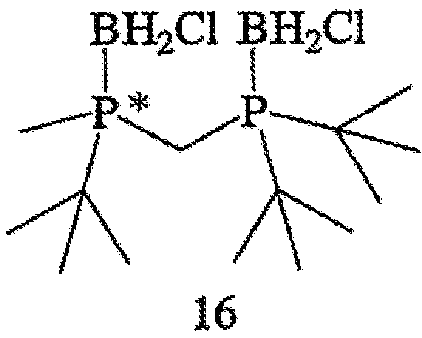
(d) взаимодействия соединения формулы 16 с амином или кислотой с получением соединения формулы 4. В том случае, когда оба заместителя обозначают серу или кислород, R6 и R7 могут быть удалены обработкой соединения формулы 12 восстановителем, включая такой перхлорполисилан, как гексахлордисилан.
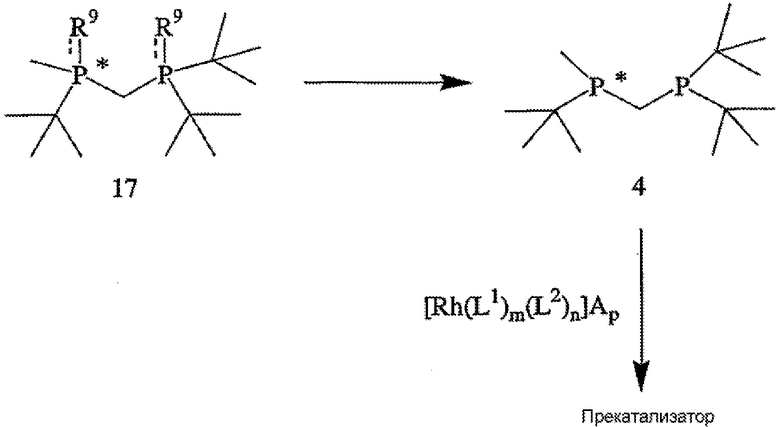
Наконец, в соответствии с еще одним аспектом настоящего изобретения предлагается способ получения катализатора или прекатализатора, образованного хиральным лигандом, который присоединен к переходному металлу посредством атомов фосфора, при этом хиральный лиганд имеет структуру, представленную формулой 4. Способ включает стадии (а) удаления обоих заместителей R9 из соединения формулы 17,
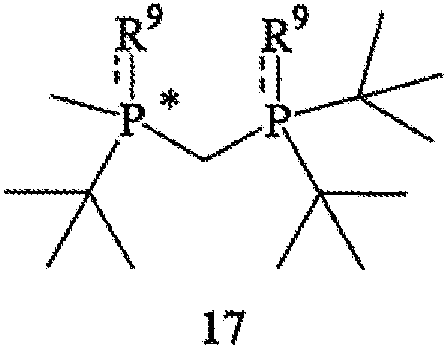
с образованием соединения формулы 4, где R9 обозначает ВН3, серу или кислород; и (b) связывания соединения формулы 4 с переходным металлом (в частности, родием). Стадия (b) может включать взаимодействие соединения формулы 4 с комплексом формулы 18
в котором
L1 обозначает диен, выбранный из COD, норборнадиена или 2,5-диметилгекса-1,5-диена;
L2 обозначает анионный лиганд, выбранный из Cl, Br, I, CN, OR10 или R10, или нейтральный σ-донорный лиганд, выбранный из NR10R11R12, R10OR11, R10SR11, CO или NCR10, в том случае, когда R10, R11 и R12 независимо обозначают Н или С1-6 алкил;
А обозначает анион, выбранный из OTf, PF6, BF4, SbF6или ClO4;
m обозначает целое число от 0 до 2 включительно;
n обозначает целое число от 0 до 4 включительно; и
p обозначает положительное нечетное число, такое, что 4×m+2×n+p=9.
В соответствии с еще одним аспектом настоящего изобретения предлагаются соединения формулы 19,
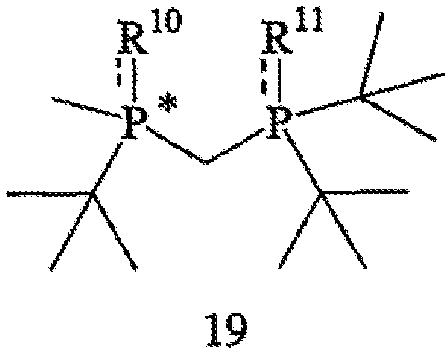
в которых R10 и R11 независимо обозначают ВН3, BH2Cl, серу, кислород, С1-4 алкилтио или же отсутствуют, при условии, что R10 и R11 оба не являются ВН3.
Пригодные соединения формулы 19 включают такие соединения, в которых R10 и R11 отсутствуют, и такие соединения, которые имеют R-абсолютную стереохимическую конфигурацию, а ЭИ составляет приблизительно 95%, или же ЭИ составляет 99% или больше. Другие пригодные соединения формулы 19 включают такие соединения, в которых R10 и R11 совпадают и каждый обозначает кислород, серу или С1-4 алкилтио, и такие, которые имеют R-абсолютную стереохимическую конфигурацию, а ЭИ составляет приблизительно 95% или больше или ЭИ составляет приблизительно 99% или больше. Таким образом, наиболее пригодные соединения, представленные формулой 19, включают:
2-{[(ди-трет-бутилфосфанил)метил]метилфосфанил}-2-метилпропан;
(R)-2-{[(ди-трет-бутилфосфанил)метил]метилфосфанил}-2-метилпропан;
(S)-2-{[(ди-трет-бутилфосфанил)метил]метилфосфанил}-2-метилпропан;
2-[(ди-трет-бутилфосфинотиоилметил)метилфосфинотиоил]-2-метилпропан;
(R)-2-[(ди-трет-бутилфосфинотиоилметил)метилфосфинотиоил]-2-метилпропан;
(S)-2-[(ди-трет-бутилфосфинотиоилметил)метилфосфинотиоил]-2-метилпропан;
2-[(ди-трет-бутилфосфиноилметил)метилфосфиноил]-2-метилпропан;
(R)-2-[(ди-трет-бутилфосфиноилметил)метилфосфиноил]-2-метилпропан;
(S)-2-[(ди-трет-бутилфосфиноилметил)метилфосфиноил]-2-метилпропан;
(ди-трет-бутилметилтиофосфониумилметил)-трет-бутилметилметилтиофосфониум;
(R)-(ди-трет-бутилметилтиофосфониумилметил)-трет-бутилметилметилтиофосфониум; или
(S)-(ди-трет-бутилметилтиофосфониумилметил)-трет-бутилметилметилтиофосфониум.
В соответствии с дополнительным аспектом настоящего изобретения предлагается катализатор или предшественник катализатора, включающий хиральный лиганд, соединенный с переходным металлом посредством атомов фосфора. Хиральный лиганд имеет структуру, представленную формулой 4.
Особенно пригодный к применению хиральный катализатор или предшественник катализатора содержит родий, связанный с бисфосфиновым лигандом, который имеет структуру, представленную формулой 5. Другие пригодные к применению хиральные катализаторы или предшественники катализаторов включают бисфосфиновый лиганд, который имеет структуру, представленную формулой 5, и ЭИ, равный приблизительно 95% или больше. Наиболее пригодный к применению хиральный катализатор включает бисфосфиновый лиганд, который имеет структуру, представленную формулой 5, и ЭИ, равный приблизительно 99% или больше. Катализатор или предшественник катализатора может дополнительно включать один или несколько диенов (в частности, COD) или анионов галогенов (в частности, Cl), связанных с переходным металлом, и может включать такой противоион, как OTf, PF6, PF4, SbF6, или ClO4, или их смесь.
В соответствии с еще одним аспектом настоящего изобретения предлагается способ получения требуемого энантиомера соединения формулы 32,
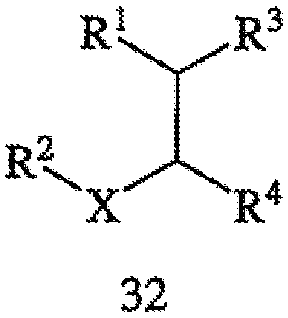
или его фармацевтически приемлемого комплекса, соли, сольвата или гидрата. Способ включает стадии (а) взаимодействия соединения формулы 33,
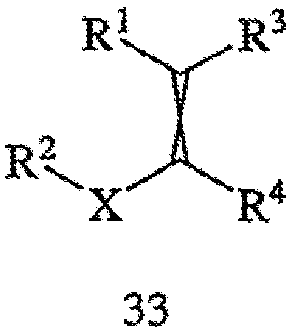
с водородом в присутствии хирального катализатора с получением соединения формулы 32; и (b) необязательно превращения соединения формулы 32 в фармацевтически приемлемый комплекс, соль, сольват или гидрат. Заместители R1, R2, R3, R4 и Х в формуле 32 и формуле 33 те же, что и определенные в формуле 2; хиральный катализатор включает хиральный лиганд, присоединенный к переходному металлу посредством атомов фосфора, при этом хиральный лиганд имеет структуру, представленную выше формулой 4. Пригодные к применению соединения формулы 32 включают оптически активные β-аминокислоты, которые, подобно прегабалину, связываются с α2δ субъединицей кальциевого канала. Указанные соединения, включая их фармацевтически приемлемые комплексы, соли, сольваты или гидраты, пригодны для лечения боли, фибромиалгии и разнообразных психиатрических расстройств и нарушений сна. См., в частности, заявку на патент США № 2003/0195251 А1, выданную на имя Barta et al, полное изложение которой включено в настоящее описание в качестве ссылки.
В объем настоящего изобретения входят все фармацевтически приемлемые комплексы, соли, сольваты, гидраты, полиморфы, сложные эфиры, амиды и пролекарства заявляемых и раскрываемых соединений, включая соединения формул 1, 2, 8 и 32.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Фиг.1 показывает пространственное расположение С2-симметричого бисфосфинового лиганда (например, BisP*), когда он соединен с таким переходным металлом, как Rh.
Фиг.2 показывает пространственное расположение С1-симметричого бисфосфинового лиганда (например, (трет-бутилметилфосфанил)-(ди-трет-бутилфосфанил)этана), когда он соединен с таким переходным металлом, как Rh.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Определения и сокращения
Если не указано иное, в настоящем описании используют приведенные ниже определения. Некоторые определения и формулы могут содержать "-" (тире) для обозначения связи между атомами и места присоединения названного и неназванного атома или группы атомов. Другие определения и формулы могут содержать "=" (знак равенства) или ""(знак идентичности) для обозначения двойной связи или тройной связи соответственно. Некоторые формулы могут также содержать одну или несколько "*" (звездочек) для обозначения стереогенных (хиральных) центров, однако отсутствие звездочек не означает, что в соединении отсутствует один или несколько стереоцентров. Подобные формулы могут относиться к рацематам или к индивидуальным энантиомерам или диастереомерам, которые могут быть или могут не быть практически чистыми. Некоторые формулы могут также включать пересекающуюся двойную связь или удвоение какой-либо связи  с целью обозначения Z-изомера, Е-изомера или смеси Z- и Е-изомеров.
с целью обозначения Z-изомера, Е-изомера или смеси Z- и Е-изомеров.
"Замещенными" группами являются такие группы, в которых один или несколько атомов водорода заменены одним или несколькими отличными от водорода атомами или группами, при условии, что соблюдены требования валентности и в результате замены получается химически устойчивое соединение.
"Алкил" относится к насыщенным углеводородным группам с прямой или разветвленной цепью, которые в общем случае содержат определенное количество атомов углерода (например, С1-6 алкил относится к алкильной группе, имеющей 1, 2, 3, 4, 5 или 6 атомов углерода). Примеры алкильных групп включают, без ограничений, метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, изобутил, трет-бутил, пент-1-ил, пент-2-ил, пент-3-ил, 3-метилбут-1-ил, 3-метилбут-2-ил, 2-метилбут-2-ил, 2,2,2-триметилэт-1-ил, н-гексил и т.п.
"Алкенил" относится к углеводородным группам с прямой или разветвленной цепью, которые имеют одну или несколько ненасыщенных углерод-углеродных связей и в общем случае содержат определенное количество атомов углерода. Примеры алкенильных групп включают, без ограничений, этенил, 1-пропен-1-ил, 1-пропен-2-ил, 2-пропен-1-ил, 1-бутен-1-ил, 1-бутен-2-ил, 3-бутен-1-ил, 3-бутен-2-ил, 2-бутен-1-ил, 2-бутен-2-ил, 2-метил-1-пропен-1-ил, 2-метил-2-пропен-1-ил, 1,3-бутадиен-1-ил, 1,3-бутадиен-2-ил и т.п.
"Алкинил" относится к углеводородным группам с прямой или разветвленной цепью, которые имеют одну или несколько тройных углерод-углеродных связей и в общем случае содержат определенное количество атомов углерода. Примеры алкинильных групп включают, без ограничений, этинил, 1-пропин-1-ил, 2-пропин-1-ил, 1-бутин-1-ил, 3-бутин-1-ил, 3-бутин-2-ил, 2-бутин-1-ил и т.п.
"Алкандиил" относится к дивалентным насыщенным углеводородным группам с прямой или разветвленной цепью, которые в общем случае содержат определенное количество атомов углерода. Примеры включают, без ограничений, метилен, 1,2-этандиил, 1,3-пропандиил, 1,4-бутандиил, 1,5-пентандиил, 1,6-гександиил и т.п.
"Алканоил" и "алканоиламино" относятся, соответственно, к алкил-С(О)- и алкил-С(О)-NH-, где алкил определен выше, и в общем случае содержат определенное количество атомов углерода, включая карбонильный углерод. Примеры алканоильных групп включают, без ограничений, формил, ацетил, пропионил, бутирил, пентаноил, гексаноил и т.п.
"Циклоалкил" относится к насыщенным моноциклическим и бициклическим углеводородным кольцам, которые в общем случае содержат определенное количество атомов углерода, образующих кольцо (например, С3-7 циклоалкил относится к циклоалкильной группе, содержащей 3, 4, 5, 6 или 7 атомов углерода в качестве членов кольца). Циклоалкил может быть присоединен к родственной группе или к субстрату по любому атому кольца, при условии, что подобное присоединение не нарушает требований валентности. Аналогично, циклоалкильные группы могут включать один или несколько отличных от водорода заместителей, при условии, что подобное замещение не нарушает требований валентности. Пригодные заместители включают, без ограничений, алкил, алкокси, алкоксикарбонил и алканоил, как определено ранее, и гидрокси, меркапто, нитро, галоген и амино.
Примеры моноциклических циклоалкильных групп включают, без ограничений, циклопропил, циклобутил, циклопентил, циклогексил и т.п. Примеры бициклических циклоалкильных групп включают, без ограничений, бицикло[1.1.0]бутил, бицикло[1.1.1]пентил, бицикло[2.1.0]пентил, бицикло[2.1.1]гексил, бицикло[3.1.0]гексил, бицикло[2.2.1]гептил, бицикло[3.2.0]гептил, бицикло[3.1.1]гептил, бицикло[4.1.0]гептил, бицикло[2.2.2]октил, бицикло[3.2.1]октил, бицикло[4.1.1]октил, бицикло[3.3.0]октил, бицикло[4.2.0]октил, бицикло[3.3.1]нонил, бицикло[4.2.1]нонил, бицикло[4.3.0]нонил, бицикло[3.3.2]децил, бицикло[4.2.2]децил, бицикло[4.3.1]децил, бицикло[4.4.0]децил, бицикло[3.3.3]ундецил, бицикло[4.3.2]ундецил, бицикло[4.3.3]додецил и т.п.
"Циклоаланоил" относится к группе циклоалкил-С(О)-, где циклоалкил определен выше, и в общем случае содержит определенное количество атомов углерода, включая карбонильный углерод. Примеры циклоалканоильных групп включают, без ограничений, циклопропаноил, циклобутаноил, циклопентаноил, циклогексаноил, циклогептаноил и т.п.
"Алкокси", "алкоксикарбонил" и "алкоксикарбониламино" относятся, соответственно, к алкил-О-, алкил-О-С(О)- и алкил-О-С(О)-NH-, где алкил определен выше. Примеры алкоксигрупп включают, без ограничений, метокси, этокси, н-пропокси, изопропокси, н-бутокси, втор-бутокси, трет-бутокси, н-пентокси, втор-пентокси и т.п.
"Алкиламино", "алкиламинокарбонил", "диалкиламинокарбонил", "алкилсульфонил", "сульфониламиноалкил" и "алкилсульфониламинокарбонил" относятся, к алкил-NH-, алкил-NH-С(О)-, алкил2-N-С(О)-, алкил-S(O2)-, HS(O2)-NH-алкил- и алкил-S(O)-NH-С(О)-, где алкил определен выше.
"Аминоалкил" и "цианоалкил" относятся, соответственно, к NH2-алкил и NC-алкил, где алкил определен выше.
"Гало", "галоген" или "галогено" могут использоваться взаимозаменяемо и относятся ко фтору, хлору, брому и йоду.
"Галогеналкил" и "галогеналканоил" относятся, соответственно, к алкильным или алканоильным группам, замещенным одним или несколькими атомами галогена, где алкил и алканоил определены выше. Примеры галогеналкильных или галогеналканоильных групп включают, без ограничений, трифторметил, трихлорметил, пентафторэтил, пентахлорэтил, трифторацетил, трихлорацетил, пентафторпропионил, пентахлорпропионил и т.п.
"Гидроксиалкил" и "оксоалкил" относятся, соответственно, к группам НО-алкил и О=алкил, где алкил определен выше. Примеры гидроксиалкильных и оксоалкильных групп включают, без ограничений, гидроксиметил, гидроксиэтил, 3-гидроксипропил, оксометил, оксоэтил, 3-оксопропил и т.п.
"Арил" и "арилен" относятся к моновалентным и дивалентным ароматическим группам соответственно. Примеры арильных групп включают, без ограничений, фенил, нафтил, бифенил, пиренил, антраценил, флуоренил и т.п., которые могут быть незамещены или содержать от 1 до 4 заместителей. Подобные заместители включают алкил, алкокси, алкоксикарбонил, алканоил и циклоалканоил, как определено выше, и гидрокси, меркапто, нитро, галоген и амино.
"Арилалкил" относится к арил-алкилу, где арил и алкил определены выше. Примеры включают, без ограничений, бензил, флуоренилметил и т.п.
"Арилалканоил" относится к алкил-алканоилу, где арил и алканоил определены выше. Примеры включают, без ограничений, бензоил, фенилэтаноил, фенилпропаноил и т.п.
"Арилалкоксикарбонил" относится к арил-алкоксикарбонилу, где арил и алкоксикарбонил определены выше. Примеры включают, без ограничений, феноксикарбонил, бензилоксикарбонил (CBz) и т.п.
"Гетероцикл" и "гетероциклил" относятся к насыщенным, частично ненасыщенным или ненасыщенным моноциклическим или бициклическим кольцам, содержащим от 5 до 7 или от 7 до 11 членов в кольце соответственно. Указанные группы содержат в кольце члены, образованные атомами углерода и от 1 до 4 гетероатомами, которые независимо представляют собой азот, кислород или серу, и могут включать любые бициклические группы, в которых любые из определенных выше моноциклических гетероциклов конденсированы с бензольным кольцом. Гетероатомы азота и серы необязательно могут быть окислены. Гетероциклическое кольцо может быть присоединено к родственной группе или субстрату по любому из гетероатомов или атомов углерода, при условии, что подобное присоединение не нарушает требований валентности. Аналогично, любые из атомов углерода или азота, являющиеся членами кольца, могут включать отличные от водорода заместители, при условии, что подобное замещение не нарушает требований валентности. Пригодные заместители включают, без ограничений, алкил, алкокси, алкоксикарбонил, алканоил и циклоалканоил, как определено выше, и гидрокси, меркапто, нитро, галоген и амино.
Примеры гетероциклов включают, без ограничений, акридинил, азоцинил, бензимидазолил, бензофуранил, бензотиофуранил, бензотиофенил, бензоксазолил, бензтиазолил, бензтриазолил, бензтетразолил, бензизоксазолил, бензизотиазолил, бензимидазолинил, карбазолил, 4аН-карбазолил, карболинил, хроманил, хроменил, циннолинил, декагидрохинолинил, 2Н,6Н-1,5,2-дитиазинил, дигидрофуро[2,3-b]тетрагидрофуран, фуранил, фуразанил, имидазолидинил, имидазолинил, имидазолил, 1Н-индазолил, индоленил, индолинил, индолизинил, индолил, 3Н-индолил, изобензофуранил, изохроманил, изоиндазолил, изоиндолинил, изоиндолил, изохинолинил, изотиазолил, изоксазолил, морфолинил, нафтиридинил, октагидроизохинолинил, оксадиазолил, 1,2,3-оксадиазолил, 1,2,4-оксадиазолил, 1,2,5-оксадиазолил, 1,3,4-оксадиазолил, оксазолидинил, оксазолил, оксазолидинил, пиримидинил, фенанатридинил, фенантролинил, феназинил, фенотиазинил, феноксатиинил, феноксазинил, фталазинил, пиперазинил, пиперидинил, птеридинил, пуринил, пиранил, пиразинил, пиразолидинил, пиразолинил, пиразолил, пиридазинил, пиридооксазол, пиридоимидазол, пиридотиазол, пиридинил, пиридил, пиримидинил, пирролидинил, пирролинил, 2Н-пирролил, пирролил, хиназолинил, хинолинил, 4Н-хинолизинил, хиноксалинил, хинуклидинил, тетрагидрофуранил, тетрагидроизохинолинил, тетрагидрохинолинил, 6Н-1,2,5-тиадиазинил, 1,2,3-тиадиазолил, 1,2,4-тиадиазолил, 1,2,5-тиадиазолил, 1,3,4-тиадиазолил, тиантренил, тиазолил, тиенил, тиенотиазолил, тиенооксазолил, тиеноимидазолил, тиофенил, триазинил, 1,2,3-триазолил, 1,2,4-триазолил, 1,2,5-триазолил, 1,3,4-триазолил и ксантенил.
"Гетероарил" и "гетероарилен" относятся соответственно к моновалентным и дивалентным гетероциклам или гетероциклильным группам, как определено выше, которые являются ароматическими. Гетероарильные и гетероариленовые группы представляют собой подмножество арильных и ариленовых групп соответственно.
Термин "уходящая группа" относится к любой группе, которая покидает молекулу в процессе фрагментации, включая реакции замещения, реакции элиминирования и реакции добавления-элиминирования. Уходящие группы могут быть нуклеофугивными, при этом группа уходит вместе с парой электронов, которые ранее служили в качестве связи между уходящей группой и молекулой, или могут быть электрофугивными, при этом группа уходит без пары электронов. Способность нуклеофугивной группы уходить зависит от ее силы как основания, при этом наиболее сильные основания являются наихудшими уходящими группами. Обычные нуклеофугивные уходящие группы включают азот (в частности, из солей диазония), сульфонаты (включая тозилаты, брозилаты, нозилаты и мезилаты), трифлаты, нонафлаты, трезилаты, ионы галогенидов, карбоксилатные анионы, фенолятные анионы и алкоксиды. Некоторые такие более сильные основания, как NH2и ОН, могут быть преобразованы в более хорошие уходящие группы обработкой кислотой. Обычные электрофугивные уходящие группы включают протон, СО2 и металлы.
Термин "энантиомерный избыток", или "ЭИ", для данного образца является мерой избытка одного энантиомера по отношению к рацемическому образцу хирального соединения и выражается в процентах. Энантиомерный избыток определяют как 100×(er-1)/(er+1), где "er" обозначает отношение энантиомера с бульшим относительным содержанием к энантиомеру с меньшим относительным содержанием.
Термин "энантиоселективность" относится к конкретной реакции (например, гидрированию), которая приводит к образованию большего количества одного энантиомера, чем другого.
Выражение "высокий уровень энантиоселективности" относится к конкретной реакции, которая дает продукт с ЭИ, по меньшей мере, приблизительно 80%.
Термин "энантиомерно обогащенный" относится к образцу хирального соединения, которое содержит больше одного энантиомера, чем другого. Степень обогащения определяется отношением er или энантиомерным избытком.
Выражение "в значительной степени чистый энантиомер" или "в значительной степени энантиочистый" относится к образцу энантиомера, имеющего ЭИ приблизительно 90% или больше.
Термин "энантимерно чистый" или "энантиочистый" относится к образцу энантиомера, имеющего ЭИ приблизительно 99,9% или больше.
Термин "противоположный энантиомер" относится к молекуле, не являющейся зеркально совмещаемым образом молекулы сравнения, которую можно получить инверсированием всех стереогенных центров молекулы сравнения. Например, если молекула сравнения имеет S-абсолютную стереохимическую конфигурацию, то противоположный энантиомер имеет R-абсолютную стереохимическую конфигурацию. Аналогично, если молекула сравнения имеет S,S-абсолютную стереохимическую конфигурацию, то противоположный энантиомер имеет R,R-абсолютную стереохимическую конфигурацию и т.д.
Термин "прекатализатор" или "предшественник катализатора" относится к соединению или набору соединений, которые превращают в катализатор перед использованием.
Термин "фармацевтически приемлемый" относится к веществам, которые в соответствии с правильным медицинским заключением являются пригодными для применения в контакте с тканями пациентов и не обладают чрезмерной токсичностью, не оказывают раздражающего, аллергического воздействия и т.п., их использование дает разумное отношение пользы к риску, и они эффективны для целей их предполагаемого применения.
Термин "лечение" относится к возвращению к прежнему состоянию, облегчению, ингибированию развития или предотвращения расстройства или состояния, к которому применяют этот термин, или к предотвращению одного или нескольких симптомов подобного расстройства или состояния.
Термин "терапия" относится к акту "лечения", который определен непосредственно выше.
Термин "приблизительно", когда его используют в связи с измеряемой численной переменной, относится к указанному значению переменной и ко всем значениям переменной, которые находятся в пределах экспериментальной ошибки определения для указанного значения (в частности, в пределах 95%-ного доверительного интервала для среднего значения) или в пределах 10% от указанного значения в зависимости от того, какая величина является большей.
Термин "сольват" описывает молекулярный комплекс, включающий прегабалин и стехиометрическое или нестехиометрическое количество молекул одного или нескольких фармацевтически приемлемых растворителей (например, этанола).
Термин "гидрат" описывает сольват, включающий фармацевтически активный ингредиент (в частности, прегабалин) и стехиометрическое или нестехиометрическое количество молекул воды.
Термин "фармацевтически приемлемые сложные эфиры, амиды и пролекарства" относится к кислотно- или основно-аддитивным солям, сложным эфирам, амидам, цвиттерионным формам, если они возможны, и пролекарствам заявляемых и раскрываемых соединений. Примеры фармацевтически приемлемых нетоксичных сложных эфиров включают, без ограничений, С1-6 алкильные сложные эфиры, С5-7 циклоалкильные сложные эфиры и арилалкильные сложные эфиры заявляемых и раскрываемых соединений, где алкил, циклоалкил и арилалкил определены выше. Подобные сложные эфиры могут быть получены обычными способами, как описано, например, M.B. Smith и J. March, March's Advanced Organic Chemistry (5th Ed. 2001).
Примеры фармацевтически приемлемых нетоксичных амидов включают, без ограничений, амиды, полученные из аммиака, первичных С1-6 алкиламинов и вторичных С1-6 диалкиламинов или гетероциклиламинов заявляемых и раскрываемых соединений, где алкил и гетероциклил определены выше. Подобные амиды могут быть получены обычными способами, как описано, например, в March's Advanced Organic Chemistry.
Термин "пролекарство" относится к соединениям, обладающим небольшой фармакологической активностью или не обладающим никакой фармакологической активностью, которые способны в процессе метаболизма в условиях in vivo претерпевать превращения в заявляемые или раскрываемые соединения, обладающие требуемой активностью. Для обсуждения пролекарств см. T. Higuchi and V. Stella, "Pro-drugs as Novel Delivery Systems," ACS Symposium Series 14 (1975), E.B. Roche (ed.), Bioreversible Carriers in Drug Design (1987) è H. Bundgaar, Design of Prodrugs (1985).
В таблице 1 приведен список сокращений, которые используют в данном описании.
Список сокращений
На некоторых приведенных ниже схемах проведения реакций и в примерах определенные соединения могут быть получены с использованием защитных групп, которые препятствуют протеканию нежелательных химических реакций по местам, которые в противном случае были бы активными. Защитные группы могут также использоваться для того, чтобы увеличить растворимость или каким-либо другим образом модифицировать физические свойства соединения. Для обсуждения стратегий использования защитных групп, описания веществ и способов введения и удаления защитных групп, а также компилирования пригодных защитных групп для обычных функциональных групп, включая амины, карбоновые кислоты, спирты, кетоны, альдегиды и т.п., см. T.W. Greene и P.G. Wuts, Protecting Groups in Organic Chemistry (1999) иP. Kocienski, Protecting Groups (2000), которые для всех целей полностью включены в данное описание в качестве ссылки.
Кроме того, на некоторых приведенных ниже схемах и в примерах могут быть опущены детали проведения обычных реакций, включая окисления, восстановления и т.д., которые известны рядовым специалистам в области органической химии. Детали подобных реакций могут быть найдены в ряде научных обзоров, включая Richard Larock, Comprehensive Organic Transformations (1999), и в многотомных выпусках под редакцией Michael B. Smith и других, Compendium of Organic Synthetic Methods (1974-2003). В общем случае исходные вещества и реагенты могут быть получены из коммерческих источников.
В настоящем изобретении предлагаются вещества и способы получения хиральных соединений, представленных выше формулой 2, включая их фармацевтически приемлемые соли, сложные эфиры, амиды и пролекарства. В формуле 2 хиральные соединения содержат, по меньшей мере, один стереогенный центр, как обозначено отметкой *, и включают заместители R1, R2, R3, R4 и Х, которые определены выше. Пригодные соединения, представленные формулой 2, включают такие соединения, в которых R1 обозначает амино, амино-С1-6 алкил, циано или циано-С1-6 алкил; R2 обозначает С1-6 алкоксикарбонил или карбокси; Х обозначает -СН2- или связь; а R3 и R4 независимо обозначают атом водорода или С1-6 алкил. Особенно пригодные соединения включают α-, β- и γ-аминокислоты, в которых R1 обозначает амино или аминометил; R2 обозначает карбокси; Х обозначает связь или -СН2-; а R3 и R4 независимо обозначают атом водорода или С1-6 алкил. Так, наиболее пригодные соединения включают (S)-3-циано-5-метилгексановую кислоту и (S)-(+)-3-(аминометил)-5-метилгексановую кислоту, формула 1, которая известна как прегабалин.
Схема I поясняет способ получения требуемого энантиомера соединения формулы 2. Энантиоселективный синтез включает стадии (а) взаимодействия прохирального субстрата (олефина) формулы 3 с водородом в присутствии хирального катализатора и органического растворителя с образованием соединения формулы 2; и (b) необязательно превращения соединения формулы 2 в фармацевтически приемлемую соль, сложный эфир, амид или пролекарство. Заместители R1, R2, R3, R4 и Х в формуле 3 определены в формуле 2. В общем случае и если не указано иное, в том случае, когда конкретное определение заместителя (R1, R2, R3 и т.д.) указано впервые в связи с какой-либо формулой, то же определение заместителя, когда его используют в последующих формулах, будет иметь то же самое значение, что и в предыдущих формулах. Так, например, если R20 в первой формуле обозначает водород, галоген или С1-6 алкил, то, если не указано иное или если иное не очевидно из контекста, R20 во второй формуле также обозначает водород, галоген или С1-6 алкил.
Схема I
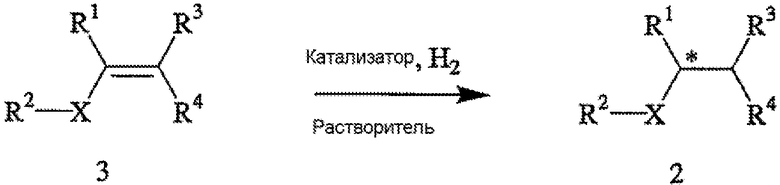
Пригодные к применению прохиральные субстраты формулы 3 включают индивидуальные Z- и Е-изомеры или смесь Z- и Е-изомеров. Пригодные к применению прохиральные субстраты включают также соединения формулы 3, в которых R1 обозначает амино, амино-С1-6 алкил, циано или циано-С1-6 алкил; R2 обозначает С1-6 алкоксикарбонил, карбокси или -CO2-Y; Х обозначает -СН2- или связь; R3 и R4 независимо обозначают атом водорода или С1-6 алкил; а Y обозначает катион. Другие пригодные к применению соединения включают α-, β- и γ-цианокислоты, в которых R1 обозначает циано или цианометил; R2 обозначает карбокси или -CO2-Y; Х обозначает связь или -СН2-; а R3 и R4 независимо обозначают атом водорода или С1-6 алкил; а Y обозначает ион металла Группы 1 (щелочной), металла Группы 2 (щелочно-земельный), ион первичного аммония или ион вторичного аммония. Особенно пригодные к применению соединения формулы 3 включают 3-циано-5-метилгекс-3-еновую кислоту и ее основно-аддитивные соли, такие как 3-циано-5-метилгекс-3-еноат трет-бутиламмониевая соль. Прохиральные субстраты могут быть получены из коммерческих источников или же могут быть приготовлены с помощью известных способов.
Как указано выше, хиральный катализатор включает хиральный лиганд, который связан с переходным металлом (в частности, металлами Группы 3 - Группы 12) посредством атомов фосфора и имеет структуру, представленную формулой 4 или формулой 5 (или ее зеркальным отображением). Наиболее пригодный хиральный катализатор включает бисфосфиновый лиганд формулы 5, который имеет ЭИ приблизительно 95% или больше или предпочтительно имеет ЭИ приблизительно 99% или больше. Пригодные переходные металлы включают родий, рутений и иридий. Из указанных металлов наиболее пригодным является родий.
В приведенных на схеме I реакциях может применяться предшественник хирального катализатора или прекатализатор. Предшественник катализатора или прекатализатор представляет собой соединение или набор соединений, которые перед использованием превращают в хиральный катализатор. Предшественники катализатора обычно включают переходный металл (в частности, родий) в комплексе с бисфосфиновым лигандом (в частности, формулы 4) и диеном (в частности, норборнадиен, COD, (2-метилаллил)2 и т.п.), галогенидом (Cl или Br) или диеном и галогеном, в присутствии противоиона, А, такого как OTf, PF6, BF4, SbF6, ClO4 и т.п. Так, например, предшественник катализатора, составленный из комплекса, [(бисфосфиновый лиганд)Rh(COD)]+A, может быть превращен в хиральный катализатор путем гидрирования диена (COD) в МеОН с образованием соединения [(бисфосфиновый лиганд)Rh(МеОН)2]+A. МеОН затем заменяют прохиральным олефином (формулы 3), который претерпевает энантиоселективное гидрирование в требуемое хиральное соединение (формула 2). Так, например, пригодный к применению предшественник хирального катализатора включает тетрафторборат (S)-(+)-(2-{[(ди-трет-бутилфосфанил)метил]метилфосфанил}-2-метилпропан)-(1,5-циклооктадиен)родия(I).
В зависимости от того, какой из энантиомеров хирального катализатора используют, асимметрическое гидрирование приводит к энантиомерному избытку (ЭИ) (R)-энантиомера или (S)-энантиомера формулы 2. Несмотря на то, что количество получаемого требуемого энантиомера будет зависеть от условий проведения реакции (температуры, давления Н2, загрузки катализатора, растворителя), желателен ЭИ требуемого энантиомера приблизительно 80% или больше; более желателен ЭИ приблизительно 90% или больше; и еще более желателен ЭИ приблизительно 95% или больше. Наиболее пригодными асимметрическими гидрированиями являются такие, при которых ЭИ требуемого энантиомера составляет приблизительно 99% или больше. Для целей настоящего изобретения требуемый энантиомер формулы 2 считается достаточно чистым, если он имеет ЭИ приблизительно 90% или больше.
Для конкретного хирального катализатора и прохирального субстрата молярное отношение субстрата и катализатора (s/c) будет зависеть, среди прочих причин, от давления Н2, температуры реакции и растворителя. Как правило, отношение субстрат/катализатор превышает приблизительно 10:1 или 20:1, а отношения субстрат/катализатор, равные приблизительно 100:1 или 200:1, являются типичными. Хотя хиральный катализатор может повторно использоваться, полезными являются большие отношения субстрат/катализатор. Например, были бы полезны отношения субстрат/катализатор, равные приблизительно 1000:1, 10000:1 и 20000:1 или больше. Асимметрическое гидрирование, как правило, проводят приблизительно при комнатной температуре или выше и под давлением Н2 приблизительно 0,1 МПа (1 атм) или выше. Температура реакционной смеси может составлять в диапазоне от приблизительно 20°С до приблизительно 80°С, а давление Н2 может составлять в диапазоне от приблизительно 0,1 МПа до приблизительно 5 МПа или выше, однако более типично оно составляет в диапазоне от приблизительно 0,3 МПа до приблизительно 3 МПа. Комбинацию температуры, давления Н2 и отношения субстрат/катализатор обычно выбирают таким образом, чтобы обеспечить практически полную конверсию (в частности, приблизительно 95% мас. или больше) прохирального олефина в течение приблизительно 24 часов. В общем случае повышение давления Н2 увеличивает энантиоселективность.
При проведении асимметрического гидрирования могут применяться такие разнообразные растворители, как МеОН, EtOH и i-PrOH. Другие пригодные растворители включают такие апротонные полярные растворители, как ТГФ, MeCl2 и ацетон, или ароматические растворители, такие как толуол, трифтортолуол и хлорбензол. При проведении энантиоселективного гидрирования может использоваться один растворитель или же может использоваться смесь таких растворителей, как МеОН и ТГФ.
Как показано на схеме II, раскрываемое асимметрическое гидрирование пригодно для получения прегабалина, или (S)-(+)-3-(аминометил)-5-метилгексановой кислоты (формула 1). Способ может быть использован для получения прегабалина, имеющего ЭИ приблизительно 95% или больше или имеющего ЭИ приблизительно 99% или больше, а в некоторых случаях имеющего ЭИ приблизительно 99,9% или больше. В способе применяют энантиоселективное гидрирование соединения формулы 6 с использованием хирального катализатора, с целью получения хирального цианового предшественника прегабалина (формула 7). Хиральный циановый предшественник затем восстанавливают и необязательно обрабатывают кислотой, получая прегабалин. В формулах 6-8 заместитель R5 может быть карбоксильной группой или группой -CO2-Y, где Y обозначает катион.
Пригодные прохиральные субстраты (формулы 6) включают основно-аддитивную соль 3-циано-5-метилгекс-3-еновой кислоты, такую как 3-циано-5-метилгекс-3-еноат трет-бутиламмониевая соль. Другие пригодные прохиральные субстраты включают такие субстраты, в которых Y в формуле 6 является ионом металла Группы 1, ионом металла Группы 2, ионом первичного аммония или ионом вторичного аммония. Прохиральные субстраты могут быть получены из коммерческих источников или приготовлены известными способами. Для обсуждения получения пригодных к применению прохиральных субстратов и восстановления хиральных циановых предшественников прегабалина см., например, совместную заявку на патент США № 20003/0212290 А1, опубликованную 13 ноября 2003 года, полное изложение которой для всех целей включено в данное описание в качестве ссылки.
Схема II
На схеме III показан способ получения хирального лиганда формулы 4. Способ может применяться как для получения R-энантиомера (формула 5), так и S-энантиомера, при этом каждый имеет ЭИ приблизительно 80%, 90%, 95% или 99% или больше. Как показано на схеме III, способ включает взаимодействие первого монофосфина (формула 9) со вторым монофосфином (формула 10) с образованием первого бисфосфинового промежуточного соединения (формула 11), при этом первый монофосфин перед проведением реакции обрабатывают основанием, Х обозначает уходящую группу (в частности, галоген, такой как хлор), а R6, как правило, обозначает ВН3, но может быть также серой или кислородом. Способ далее включает взаимодействие первого бисфосфинового промежуточного соединения (формула 11) с бораном или серой или кислородом с образованием второго бисфосфинового промежуточного соединения (формула 12), в котором R7 совпадает или отличается от R6 и обозначает ВН3, серу или кислород. Затем заместители R6 и R7 удаляют и получают хиральный бисфосфиновый лиганд формулы 4. Хотя это и не показано на схеме III, второе бисфосфиновое промежуточное соединение (формула 12) расщепляют на отдельные энантиомеры до или после удаления R6 и R7.
Схема III
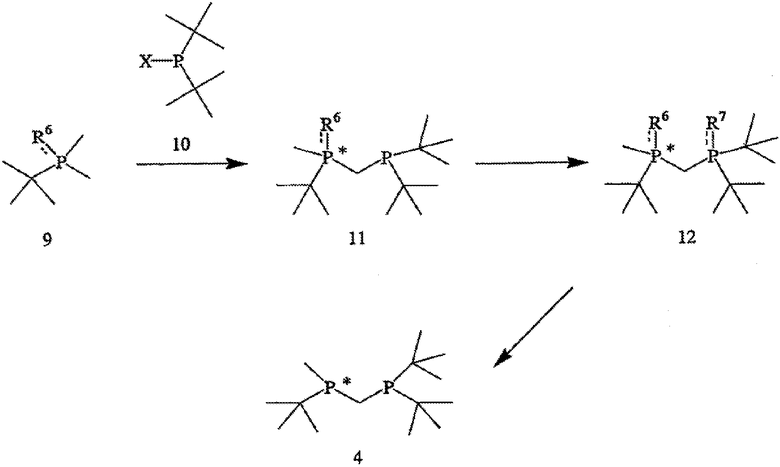
Заместители R6 и R7 могут быть удалены различными способами в зависимости от типа конкретного заместителя. Например, в том случае, когда R6 и R7, каждый, обозначают ВН3 (формула 13), они могут быть удалены взаимодействием второго бисфосфинового промежуточного соединения с амином или кислотой с образованием соединения формулы 4. Так, например, соединение формулы 13 можно ввести во взаимодействие с HBF4•Ме2О с последующим основным гидролизом и получить соединение формулы 4. Аналогично, соединение формулы 13 можно обработать с помощью DABCO, TMEDA, DBU или Et2NH или их комбинацией, с целью удаления R6 и R7. См., например, H. Bisset et al., Tetrahedron Letters 34(28): 4523-26 (1993); см. также совместную заявку на патент США № 2003/0143214 А1, опубликованную 3 октября 2002 года, и совместную заявку на патент США № 2003/0073868, опубликованную 17 апреля 2003 года, полное изложение которых для всех целей включено в данное описание в качестве ссылки.
В том случае, когда оба заместителя представляют собой атомы серы (формула 14), R6 и R7 могут быть удалены с помощью методов, приведенных на схеме IV. Один из способов включает стадии (а) взаимодействия соединения формулы 14 с R8OTf с образованием соединения формулы 15, в котором R8 обозначает С1-4 алкил (в частности, метил); (b) взаимодействия соединения формулы 15 с боргидридом (в частности, LiBH4) с получением соединения формулы 13; и (с) взаимодействия соединения формулы 13 с амином или кислотой с образованием соединения формулы 4. Другой способ включает приведенные выше стадии (а) и (b) и далее включает стадии (с) взаимодействия соединения формулы 13 с HCl, который диспергируют в полярном апротонном растворителе, с образованием соединения формулы 15, и (d) взаимодействия соединения формулы 16 с амином или кислотой с образованием соединения формулы 4.
В том случае, когда оба заместителя представляют собой атомы серы или кислорода, R6 и R7 могут быть также удалены взаимодействием соединения формулы 12 с восстановителем, включая такой перхлорполисилан, как гексахлордисилан. Для обсуждения применения перхлорполисилана для стереоспецифической дезоксигенации ациклического фосфиноксида см. K. Naumann et al., J. Am. Chem. Soc. 91(25): 7012-23 (1969), которая для всех целей полностью включена в данное описание в качестве ссылки.
Как указано выше в связи со схемой I, в способах, используемых для конверсии прохиральных субстратов формулы 3 или формулы 6 в требуемые энантиомеры формулы 1 или формулы 7, применяют хиральные катализаторы или предшественники хиральных катализаторов, которые перед использованием превращают в хиральные катализаторы. Катализаторы или прекатализаторы состоят из хирального лиганда формулы 4 или формулы 5 (или ее зеркального отображения), связанного с переходным металлом (в частности, Rh) посредством атомов фосфора.
Катализатор или прекатализатор могут быть получены с помощью способа, приведенного на схеме V. Способ включает стадии (а) удаления заместителя R9 с образованием соединения формулы 4, где R9 обозначает ВН3, серу или кислород; и (b) связывания соединения формулы 4 с переходным металлом (в частности, родием). Стадия (b) обычно включает взаимодействие соединения формулы 4 с комплексом формулы 18, в котором лигандами L1 и L2 являются соответственно диен или анионный лиганд, определение которых дано выше, А обозначает отрицательно заряженный противоион, определение которого дано выше, а m, n и p соответственно обозначают целое число от 0 до 2 включительно, целое число от 0 до 4 включительно и такое положительное нечетное число, что 4×m+2×n+p=9. Прекатализатор может обладать некоторыми преимуществами как перед свободным лигандом (формула 4), так и хиральным катализатором, такими как лучшая устойчивость при хранении, простота в обращении (в частности, он может быть твердым веществом, а не жидкостью) и т.п.
Схема IV
В общем случае химические превращения, приведенные в данном описании, могут быть осуществлены с использованием практически стехиометрических количеств реагентов, хотя для проведения некоторых реакций лучше использовать избыток одного или нескольких реагентов. Кроме того, многие реакции, приведенные в данном описании, включая асимметрическое гидрирование соединений формулы 2 и формулы 6, могут быть проведены приблизительно при комнатной температуре, но конкретные реакции, в зависимости от кинетики реакции, выходов и т.п., могут потребовать использования более высоких или более низких температур. Кроме того, любые ссылки в описании на стехиометрический диапазон, диапазон температуры, диапазон рН и т.д. включают указанные крайние значения.
Схема V
Требуемые (S)- или (R)-энантиомеры любых соединений, приведенных в данном описании, могут быть затем обогащены с помощью классических методов расщепления, хиральной хроматографии или с помощью перекристаллизации. Например, соединения формулы 1 или формулы 2 могут быть введены во взаимодействие с энантиомерно чистыми соединениями (в частности, кислотой или основанием) с получением пары диастереомеров, каждый из которых образован индивидуальным энантиомером и которые потом разделяют, например, посредством дробной перекристаллизации или хроматографии. Требуемый энантиомер затем регенерируют из соответствующего диастереоизомера. Кроме того, требуемый энантиомер можно дополнительно обогатить путем перекристаллизации в подходящем растворителе, если он имеется в достаточном количестве (в частности, ЭИ составляет немногим меньше приблизительно 85%, а в некоторых случаях ЭИ составляет немногим меньше приблизительно 90%).
Многие из приведенных в настоящем описании соединений, включая соединения, представленные формулами 1, 2, 8 и 32, способны образовывать фармацевтически приемлемые соли. Указанные соли включают, без ограничений, кислотно-аддитивные соли (включая соли дикислот) и соли оснований. Фармацевтически приемлемые кислотно-аддитивные соли включают нетоксичные соли, полученные из неорганических кислот, таких как соляная, азотная, фосфорная, серная, бромистоводородная, йодистоводородная, фтористоводородная, фосфористая и т.п., а также нетоксичные соли, полученные из органических кислот, таких как алифатические моно- и дикарбоновые кислоты, фенилзамещенные алкановые кислоты, гидроксиалкановые кислоты, алкандикислоты, ароматические кислоты, алифатические и ароматические сульфоновые кислоты и т.п. Так, подобные соли включают сульфат, пиросульфат, бисульфат, сульфит, бисульфит, нитрат, фосфат, моногидрофосфат, дигидрофосфат, метафосфат, пирофосфат, хлорид, бромид, йодид, ацетат, трифторацетат, пропионат, каприлат, изобутират, оксалат, малонат, сукцинат, суберат, себакат, фумарат, малеат, манделат, бензоат, хлорбензоат, метилбензоат, динитробензоат, фталат, бензолсульфонат, толуолсульфонат, фенилацетат, цитрат, лактат, малат, тартрат, метансульфонат и т.п.
Фармацевтически приемлемые основные соли включают нетоксичные соли, полученные из оснований, в том числе катионов металлов, таких как катионы щелочных или щелочно-земельных металлов, а также аминов. Примеры подходящих катионов металлов включают, без ограничений, катионы натрия (Na+), катионы калия (К+), катионы магния (Mg2+), катионы кальция (Са2+) и т.п. Примеры подходящих аминов включают, без ограничений, N,N'-дибензилэтилендиамин, хлорпрокаин, холин, диэтаноламин, дициклогексиламин, этилендиамин, N-метилглукамин и прокаин. Для обсуждения пригодных кислотно-аддитивных и основно-аддитивных солей см. S.M. Berge et al., "Pharmaceutical Salts," 66 J. of Pharm. Sci., 1-19 (1977); см. также Stahl и Wermuth, Handbook of Pharmaceutical Salts: Properties, Selection, and Use (2002).
Можно приготовить фармацевтически приемлемую кислотно-аддитивную соль (или соль основания) путем контактирования свободного основания (или свободной кислоты) соединения с достаточным количеством подходящей кислоты (или основания), с целью получить нетоксичную соль. Затем можно выделить соль фильтрованием, если она осаждается из раствора, или извлечь соль путем испарения. Можно также регенерировать свободное основание (или свободную кислоту) путем контактирования кислотно-аддитивной соли с основанием (или основно-аддитивной соли с кислотой). Степень ионизации полученной соли может варьировать от полностью ионизованной до практически неионизованной.
Помимо солей, заявляемые и раскрываемые соединения могут существовать как в несольватированных, так и в сольватированных формах, а также в виде других типов комплексов. Пригодные комплексы включают клатраты или комплексы включения типа лекарственный препарат-хозяин, в которых лекарственный препарат и хозяин присутствуют в стехиометрических или нестехиометрических количествах. Пригодные комплексы могут также включать два или несколько органических, неорганических или органических и неорганических компонентов в стехиометрических или нестехиометрических количествах. Полученные комплексы могут быть ионизованными, частично ионизованными или неионизованными. Для обзора подобных комплексов см. J.K. Haleblian, J. Pharm. Sci. 64(8): 1269-88 (1975).
Пригодные к применению формы заявляемых и раскрываемых соединений, в том числе соединений, представленных формулами 1, 2, 8 и 32, включают все полиморфы и характерные формы кристаллизации, а также стереоизомеры (геометрические изомеры, энантиомеры и диастереомеры), которые могут быть чистыми, практически чистыми, обогащенными или рацемическими. Пригодные формы заявляемых и раскрываемых соединений могут также включать таутомерные формы, если они возможны.
Кроме того, определенные соединения в данном описании, включая соединения, представленные формулами 1, 2, 8 и 32, могут существовать в несольватированной форме или в сольватированной форме, включая гидратированные формы. Фармацевтически приемлемые сольваты включают гидраты и сольваты, в которых растворитель кристаллизации может быть изотопно замещенным, в частности D2O, d6-ацетон, d6-ДМСО и т.п. Если специально не указано, все ссылки на свободное основание, свободную кислоту, цвиттер ион или несольватированную форму соединения также включают соответствующую кислотно-аддитивную соль, соль основания или сольватированную форму соединения.
Раскрываемые соединения включают также фармацевтически приемлемые изотопные вариации, в которых, по меньшей мере, один атом замещен на атом, имеющий тот же атомный номер, но имеющий атомную массу, отличную от атомной массы, обычно встречающейся в природе. Примеры изотопов, подходящих для введения в раскрываемые соединения, включают, без ограничений, такие изотопы водорода, как 2Н и 3Н; такие изотопы углерода, как 13С и 14С; такие изотопы азота, как 15N; такие изотопы кислорода, как 17О и 18О; такие изотопы фосфора, как 31Р и 32Р; такие изотопы серы, как 35S; такие изотопы фтора, как 18F; и такие изотопы хлора, как 36Cl. Применение изотопных вариаций (в частности, дейтерия, 2Н) может предоставить терапевтические преимущества, заключающиеся в большей метаболической устойчивости, например в увеличении полужизни препарата в условиях in vivo или уменьшении требований к дозировке. Кроме того, определенные изотопные вариации раскрываемых соединений могут включать радиоактивные изотопы (в частности, тритий, 3Н или 14С), которые могут быть полезны для проведения изучения распределения лекарственного препарата и/или субстрата в тканях.
ПРИМЕРЫ
Предполагается, что следующие примеры даны для пояснения, не ограничивают изобретения и представляют конкретные варианты осуществления настоящего изобретения.
Общие методы и вещества
Все реакции и манипуляции проводят в атмосфере азота в стандартной лабораторной посуде. Асимметрическое гидрирование осуществляют в заполненной азотом перчаточной камере. ТГФ (безводный, 99,9%), ACN (безводный, 99,8%), диэтиловый эфир (безводный, 99,8%), МеОН (безводный, 99,8%) и MeCl2 (безводный, 99,8%) получают от компании ALDRICH. Тетрафторборат бис(1,5-циклооктадиен)родия(I) синтезируют в соответствии с методикой, приведенной T.G. Schenk et al., Inorg. Chem. 24: 2334 (1985). Используют газообразный водород из демонстрационного баллона, поставляемого компанией SPECIALTY GAS. Гидрирование проводят в автоклаве Гриффина-Уордена, который поставляется компанией KIMBLE/KONTES.
Ядерный магнитный резонанс
Спектры 400 МГц 1Н-ЯМР, 100 МГц 13С-ЯМР и 162 МГц 31Р-ЯМР получают на спектрометре VARIAN INOVA 400, снабженном 4-ядерным автоматически переключаемым зондом PFG, двумя высокочастотными каналами и устройством для смены образцов SMS-100 компании ZYMARK. Спектры получают приблизительно при комнатной температуре и используют стандартный автолок и обычную автокоррекцию уровня сигнала и автоматическое выравнивание контраста. Для проведения одномерных экспериментов образцы обычно вращают с частотой 20 Гц. Спектры 1Н-ЯМР получают с помощью пульсов с углом наклона 45 градусов, время задержки повторения цикла составляет 1,0 секунды, при этом проводят 16 сканирований с разрешением 0,25 Гц/точка. Окно развертки обычно составляет 8000 Гц от +18 до -2 м.д. (стандарт - ТМС при 0 м.д.), а обработку проводят при уширении линии 0,2 Гц. Типичное время развертки составляет 80 секунд. Регулярные спектры 13С-ЯМР получают с помощью пульсов с углом наклона 45 градусов, время задержки повторения цикла составляет 2,0 секунды, при этом проводят 2048 сканирований с разрешением 1 Гц/точка. Ширина спектра обычно составляет 25 кГц от +235 до -15 м.д. (стандарт - ТМС при 0 м.д.). Постоянно применяют развязку от протонов, и при обработке используют уширение линии 2 Гц. Типичное время развертки составляет 102 минуты. Спектры 31Р-ЯМР получают с помощью пульсов с углом наклона 45 градусов, время задержки повторения цикла составляет 1,0 секунды, при этом проводят 64 сканирования с разрешением 2 Гц/точка. Ширина спектра обычно составляет 48 кГц от +200 до -100 м.д. (стандарт - 85%-ная фосфорная кислота при 0 м.д.). Постоянно применяют развязку от протонов и при обработке используют уширение линии 2 Гц. Типичное время развертки составляет 1,5 минуты.
Масс-спектрометрия
Масс-спектрометрию осуществляют с использованием платформы ЖХ системы MICROMASS с помощью находящегося в свободном доступе программного обеспечения MassLynx и OpenLynx. Жидкостный хроматограф снабжен четырехкомпонентной ЖХ системой НР 1100 и жидкостным держателем GILSON 215 в качестве автосамплера. Данные получают с помощью химической ионизации при атмосферном давлении с использованием 80:20 ACN/вода в качестве растворителя. Температурные режимы: температура зонда 450°С, температура источника 150°С. Коронный разряд составляет 3500 В для положительного иона и 3200 В для отрицательного иона.
Высокоэффективная жидкостная хроматография
Высокоэффективную жидкостную хроматографию (ВЭЖХ) проводят на приборе серии 1100 компании AGILENT TECHNOLOGIES, снабженном ручным инжектором, четырехкомпонентным насосом и УФ детектором. Контроль за процессом жидкостной хроматографии проводят с помощью персонального компьютера и программного обеспечения HP Chemstation Plus. Хиральную ВЭЖХ с нормальной фазой проводят с использованием колонки Chiracel OJ, поставляемой компанией CHIRAL TECHNOLOGIES.
Газовая хроматография
Газовую хроматографию (ГХ) проводят на 110 В установке VARIAN STAR 3400, снабженной FID детектором с электрометром, импульсным ID инжектором модели 1061/530 мкм, капиллярным инжектором модели 1077 со сбросом/без сброса потока, блоком реле, который контролирует четыре внешних события, и встроенным принтером/плоттером. Газовую хроматографию проводят, используя колонки 40 м × 0,25 мм CHIRALDEX G-TA или B-TA, поставляемые компанией ADVANCED SEPARATION TECHNOLOGIES, INC., или колонку 25 м × 0,25 мм CHIRASIL-L-VAL, поставляемую компанией CHROMPACK.
ПРИМЕР 1. Получение (2-{[(ди-трет-бутилфосфанил)метил]метил-фосфанил}-2-метилпропан)диборана (формула 13)
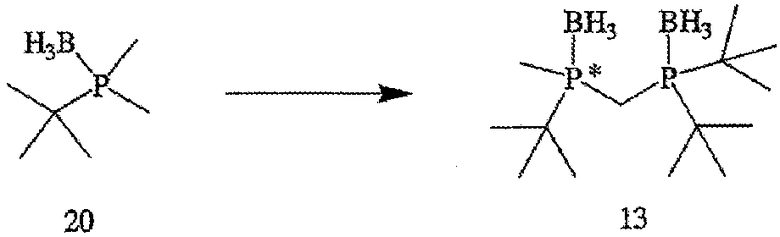
Раствор трет-бутилдиметилфосфинборана (формула 20) (20,1 г, 152 ммоль) в ТГФ (50 мл) перемешивают при 0°С. К раствору добавляют s-BuLi (104 мл, 145 ммоль) в течение 20 минут, поддерживая температуру реакции ниже 20°С. По окончании добавления раствор становится слегка мутноватым и меняет цвет на оранжевый. Реакционную смесь перемешивают в течение 1 часа при 0°С. Полученный раствор затем через канюлю переносят в течение 20 минут в предварительно охлажденный раствор ди-трет-бутилхлорфосфина (25 г, 138 ммоль) в ТГФ (50 мл) при 0°С, который сразу же по окончании добавления становится красным. При добавлении температуру поддерживают ниже 20°С. По окончании добавления смесь перемешивают при 0°С в течение 2 часов. К полученному раствору добавляют BH3•Me2S (14,4 мл, 152 ммоль) в течение 10 минут, поддерживая температуру реакции ниже 20°С. Реакционную смесь перемешивают в течение 1 часа, после чего ее выливают в 100 г льда в 1 н. растворе HCl (100 мл) и перемешивают 30 минут. Водную фазу экстрагируют с помощью EtOAc (2×100 мл), органические слои объединяют, сушат над MgSO4 и отфильтровывают. Летучие компоненты затем удаляют с помощью роторного испарителя. Остаток перекристаллизовывают из горячего гептана и получают указанное в названии соединение (рацемат) в виде кристаллического вещества белого цвета. Масса твердого вещества составляет 25 г (63%); т.пл.=150-152°С; 1Н-ЯМР (400 МГц, CDCl3) δ 1,88 (т, J=12 Гц, 2H), 1,56 (д, J=10 Гц, 3H), 1,33 (д, J=13 Гц, 9H), 1,27 (д, J=13 Гц, 9H), 1,19 (д, J=13 Гц, 9H), 0,61 (шир. кв, 6H); 13С-ЯМР (100 МГц, CDCl3) δ 34,29 (д, J=25 Гц), 33,41 (д, J=25 Гц), 30,00 (д, J=25 Гц), 28,30 (c), 27,89 (c), 25,21 (c), 9,12 (дд, J=21 и 15 Гц), 6,52 (д, J=32 Гц); 31Р-ЯМР (162 МГц, CDCl3) δ 49,70-48,15 (м), 33,03-31,56 (м). Элементный анализ: вычислено для С14Н38В2Р2: С, 57,98; Н, 13,21. Найдено: С, 57,64; Н, 13,01.
ПРИМЕР 2. Получение (R)-(-)- и (S)-(+)-(2-{[(ди-трет-бутилфосфанил)метил]метилфосфанил}-2-метилпропан)диборана (формулы 21 и 22)
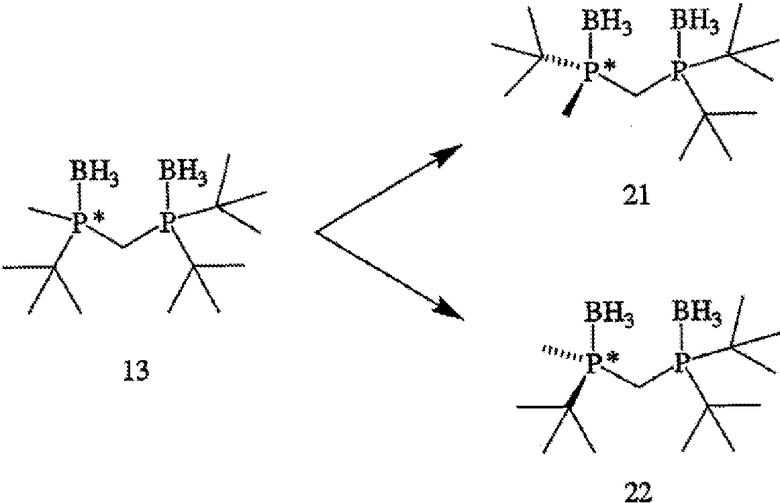
(R)-(-)- и (S)-(+)-энантиомеры (формулы 21 и 22 соответственно) (2-{[(ди-трет-бутилфосфанил)метил]метил-фосфанил}-2-метилпропан)диборана (формула 13) разделяют методом ВЭЖХ с помощью препаративной хиральной колонки и условий, приведенных ниже в таблице 2. Поскольку отсутствовал детектор по показателю преломления для осуществления приготовлений в препаративном масштабе, то детектирование по показателю преломления нельзя было использовать для контролирования времени удерживания энантиомеров. Вместо этого растворитель фракционируют, используя коллектор фракций, и индивидуальные фракции анализируют методом ВЭЖХ с помощью хиральной аналитической колонки в условиях, приведенных в таблице 2. Время удерживания для (R)- и (S)-энантиомеров составляет 6,8 минуты, [α]24 D=-5,5° (с 0,5, МеОН) и 8,2 минуты соответственно.
Условия ВЭЖХ для разделения и анализа энантиомеров (2-{[(ди-трет-бутилфосфанил)метил]метилфосфанил}-2-метилпропан)диборана
10 мкм)
ПРИМЕР 3. Получение (R)-2-{[(ди-трет-бутилфосфанил)метил]-метилфосфанил}-2-метилпропана (формула 5)
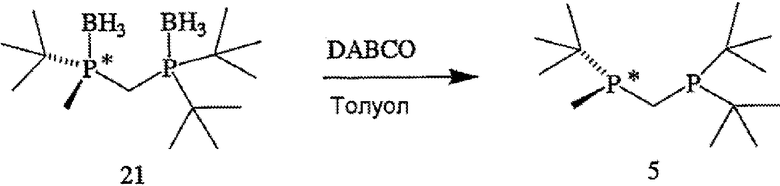
(R)-(-)-(2-{[(ди-трет-бутилфосфанил)метил]метилфосфанил}-2-метилпропан)диборан (формула 21, 290 мг, 1,0 ммоль) и DABCO (135 мг, 1,2 ммоль) растворяют в дегазированном толуоле (10 мл) при 20°С. Раствор перемешивают при 80°С в течение 4 часов. Растворитель удаляют в вакууме, и полученный остаток экстрагируют гексаном (3×20 мл). Органические вытяжки объединяют и сушат, получая (R)-(2-{[(ди-трет-бутилфосфанил)метил]метилфосфанил}-2-метилпропан (формула 5, 228 мг, 87%) в виде бесцветной жидкости. 1Н-ЯМР (400 МГц, CDCl3) δ 1,47-1,41 (м, 2Н), 1,09 (д, J=11 Гц, 9H), 1,03 (д, J=11 Гц, 9H), 0,94 (д, J=11 Гц, 9H), 0,93 (д, J=3 Гц, 3H); 13С-ЯМР (100 МГц, CDCl3) δ 7,44 (дд, J=19 и 6 Гц), 16,09 (дд, J=32 и 25 Гц), 26,63 (д, J=14 Гц), 27,95 (дд, J=23 и 3 Гц), 29,73 (д, J=14 Гц), 30,16 (дд, J=13 и 4 Гц), 31,70 (дд, J=23 и 9 Гц), 32,16 (дд, J=23 и 3 Гц); 31Р-ЯМР (162 МГц, CDCl3) δ -13,66 (шир. м), 18,35 (шир. м).
ПРИМЕР 4. Получение тетрафторбората (S)-(+)-(2-{[(ди-трет-бутилфосфанил)метил]метилфосфанил}-2-метилпропан)-(1,5-цикло-октадиен)родия(I) (формула 23)
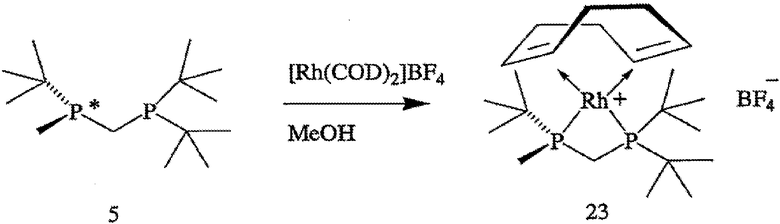
Раствор (R)-2-{[(ди-трет-бутилфосфанил)метил]метилфосфанил}-2-метилпропана (формула 5, 66 мг, 0,25 ммоль) в ТГФ (5 мл) при перемешивании по каплям добавляют к раствору [Rh(COD)2]BF4 (102 мг, 0,25 ммоль) в МеОН (10 мл) при 20°С. После добавления реакционную смесь перемешивают в течение 1 часа и растворитель удаляют в вакууме, получая твердое вещество красного цвета. После перекристаллизации продукта из теплого ТГФ получают тетрафторборат (S)-(+)-(2-{[(ди-трет-бутилфосфанил)метил]метил-фосфанил}-2-метилпропан)-(1,5-циклооктадиен)родия(I) (формула 23, 89 мг, 64%) в виде кристаллического вещества красного цвета. [α]24 D=+52,4° (с 0,9, МеОН); 1Н-ЯМР (400 МГц, CDCl3) δ 5,63-5,51 (м, 2Н), 5,11 (шир. с, 2H), 3,48-3,328 (м, 1H), 3,14 (дт, J=17 и 10 Гц, 1H), 2,49-2,25 (м, 4H), 2,21-2,09 (м, 4H), 1,69 (д, J=9 Гц, 3Н), 1,39 (д, J=14 Гц, 9H), 1,33 (д, J=14 Гц, 9H), 1,13 (д, J=16 Гц, 9H); 13С-ЯМР (100 МГц, CDCl3) (100 МГц, CDCl3) δ 100,20 (дд, J=9 и 6 Гц), 97,70 (дд, J=9 и 6 Гц), 92,95 (т, J=8 Гц), 92,27 (д, J=8 Гц), 37,68 (м), 36,04 (д, J=9 Гц), 32,54 (м), 31,48 (с), 30,94 (с), 30,09 (д, J=5 Гц), 29,81 (д, J=5 Гц), 29,32 (с), 29,16 (с), 26,57 (д, J=5 Гц), 9,58 (д, J=21 Гц); 31Р-ЯМР (162 МГц, CDCl3) δ -3,97 (дд, J=126 и 56 Гц), -29,36 (дд, J=126 и 56 Гц). Элементный анализ: вычислено для С21Н42В1F4Р2Rh1: С, 46,18; Н, 7,75. Найдено: С, 45,66; Н, 7,19.
ПРИМЕРЫ 5-9. Получение хиральных соединений (формула 2) посредством асимметрического гидрирования прохиральных субстратов (формула 3) с использованием тетрафторбората (S)-(+)-(2-{[(ди-трет-бутилфосфанил)метил]метилфосфанил}-2-метилпропан)-(1,5-циклооктадиен)родия(I) (формула 23)
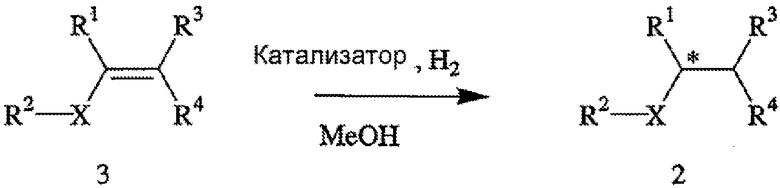
В таблице 3 приводится список субстратов (формула 3), ЭИ и абсолютная стереохимическая конфигурация хиральных продуктов (формула 2), полученных посредством асимметрического гидрирования с использованием предшественника хирального катализатора, тетрафторбората (S)-(+)-(2-{[(ди-трет-бутилфосфанил)метил]метилфосфанил}-2-метилпропан)-(1,5-циклооктадиен)родия(I) (формула 23). Для каждого внесенного в таблицу 3 соединения предшественник катализатора (0,01 ммоль) растворяют в дегазированном МеОН (1 мл) в автоклаве Гриффина-Уордена, снабженном приспособлениями, необходимыми для подсоединения демонстрационного баллона. Субстрат (1 ммоль) вначале растворяют в МеОН (4 мл), а потом с помощью шприца добавляют к раствору катализатора в МеОН. Автоклав герметизируют и повышают давление Н2 до 50 psig. Время, необходимое для завершения реакции, определяют по прекращению поглощения газообразного Н2.
Энантиоселективность хиральных соединений (формула 2), полученных асимметрическим гидрированием прохиральных субстратов (формула 3)
Для каждой из реакций, приведенных в таблице 3, энантиомерный избыток определяют с помощью хиральной ГХ или хиральной ВЭЖХ. В таблице 4 приведены детали ЭИ методологии. С целью определения энантиомерных избытков для N-ацетилаланина (пример 5) и N-ацетилфенилаланина (Пример 6) каждое соединение обрабатывают триметилсилилдиазометаном для превращения в соответствующий сложный метиловый эфир, который анализируют, как описано в примере 7 или примере 8, соответственно. Абсолютную стереохимическую конфигурацию определяют путем сравнения знаков вращения плоскости поляризации со значениями, приведенными в литературных источниках: сложный (S)-N-ацетилаланин метиловый эфир [α]20 D=-91,7° (с 2, Н2О), J.P. Wolf III & C. Neimann, Biochemistry 2: 493 (1963); сложный (S)-N-ацетилфенилаланин метиловый эфир [α]20 D=+16,4° (с 2, МеОН), B.D. Vineyard et al., J. Am. Chem. Soc. 99: 5946 (1997); сложный (S)-N-ацетилциклогексилглицин метиловый эфир [α]20 D=-4,6° (с 0,13, EtOH), M.J. Burk et al., J. Am. Chem. Soc. 117: 9375 (1995).
Условия определения энантиомерного избытка
ПРИМЕРЫ 10-13. Получение хирального предшественника прегабалина (формула 25) посредством асимметрического гидрирования прохирального субстрата (формула 24) с использованием тетрафторбората (S)-(+)-(2-{[(ди-трет-бутилфосфанил)метил]метилфосфанил}-2-метилпропан)-(1,5-циклооктадиен)родия(I) (формула 23)
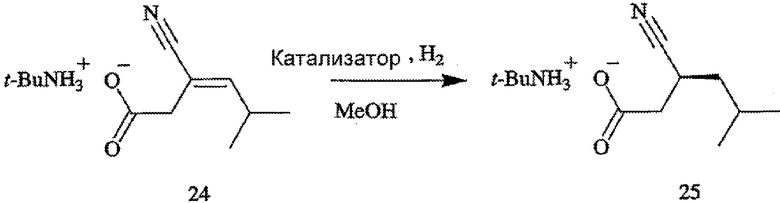
В таблице 5 приведены катализатор (или предшественник катализатора), концентрация субстрата (в МеОН, % мас.), s/c, время удерживания, давление Н2, время, необходимое для окончания процесса, и ЭИ при получении трет-бутиламмониевой соли (S)-3-циано-5-метилгексановой кислоты (формула 25) асимметрическим гидрированием трет-бутиламмониевой соли 3-циано-5-метилгекс-3-еновой кислоты (формула 24). Для каждого внесенного в таблицу 5 соединения субстрат (формула 24, 100 г, 442 ммоль) отвешивают на воздухе в сосуд для гидрирования. Затем сосуд для гидрирования переносят в перчаточную камеру ([O2] < 5 м.д.). К субстрату при перемешивании добавляют дегазированный МеОН (500 мл), чтобы растворить субстрат. К раствору субстрата добавляют необходимое количество предшественника катализатора - либо тетрафторбората (S)-(+)-(2-{[(ди-трет-бутилфосфанил)метил]метилфосфанил}-2-метилпропан)-(1,5-циклооктадиен)родия(I) (формула 23), либо (R,R)-Rh-Me-DuPhos. Сосуд для гидрирования герметизируют, повышают давление Н2 до 50 psig и энергично перемешивают с помощью магнитной мешалки, покрытой защитным чехлом из TEFLON®. Давление реакции поддерживают на уровне 50 psig Н2. Время, необходимое для завершения реакции, определяют по прекращению поглощения газообразного Н2.
С целью определения энантиомерного избытка хиральный предшественник прегабалина (формула 25 или ее зеркальный образ) подкисляют in situ с помощью 1 н. раствора HCl. Органические компоненты экстрагируют в MeCl2. После сушки над MgSO4 летучие вещества удаляют в вакууме. Карбоновые кислоты обрабатывают триметилсилилдиазометаном, чтобы превратить их в соответствующие сложные метиловые эфиры, которые затем анализируют методом капиллярной ГХ (Astec GTA (30 м), 140°С, изотермический процесс, R t1=8,8 минуты, S t2=9,5 минуты). Абсолютные конфигурации хиральных предшественников прегабалина определяют сравнением порядка элюирования с аутентичными образцами, имеющими S-конфигурацию.
ПРИМЕР 14. Получение 2-(диметилфосфинотиоил)-2-метилпропана (формула 27)
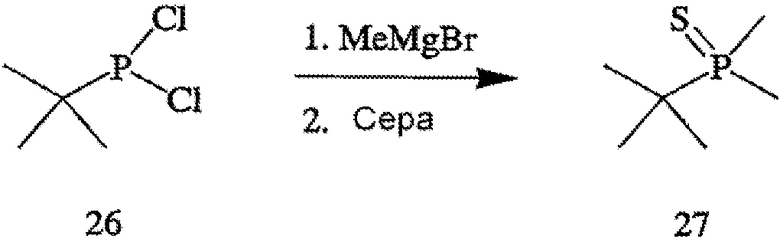
Дихлор-трет-бутилфосфин (формула 26, 10,0 г, 62,9 ммоль) растворяют в ТГФ (100 мл) в атмосфере N2 и полученный раствор охлаждают до 0°С. С помощью шприца в течение 10 минут добавляют MeMgBr (16,5 г, 138 ммоль). Процесс протекает с выделением тепла. Реакционную смесь нагревают до комнатной температуры и затем в один прием добавляют серу (2,22 г, 69,2 ммоль), при этом выделяется тепло. После перемешивания в течение 1 часа проводят обычную обработку реакционной смеси. Проводят перекристаллизацию продукта из гептана и получают 2-(диметилфосфинотиоил)-2-метилпропан (формула 27, 8,0 г, выход 85%).
ПРИМЕР 15. Получение 2-[(ди-трет-бутилфосфинотиоилметил)-метилфосфинотиоил]-2-метилпропана (формула 14)
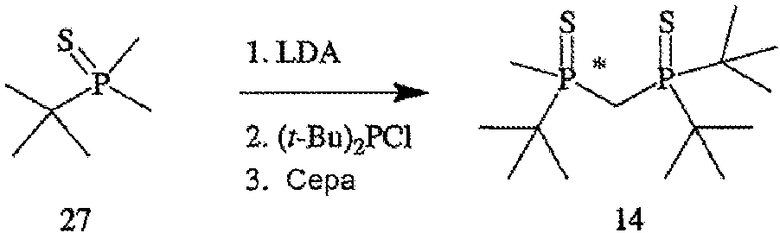
В колбу помещают диизопропиламин (74,2 г, 102,8 мл, ммоль) и ТГФ (100 мл) и охлаждают до -10°С в атмосфере аргона. К раствору добавляют n-BuLi (44,8 г, 280 мл, 700 ммоль) с помощью капельной воронки, поддерживая при этом температуру ниже 0°С. К полученному раствору в LDA в атмосфере аргона через капельную воронку добавляют раствор 2-(диметилфосфинотиоил)-2-метилпропана (формула 27, 50,07 г, 333,3 ммоль) в ТГФ (300 мл). В процессе добавления температуру поддерживают ниже -5°С. К полученному раствору в атмосфере аргона с помощью капельной воронки добавляют раствор хлор-ди-трет-бутилфосфина (60,2 г, 333 ммоль) в ТГФ (80 мл), при этом температура остается ниже -3°С. Реакционную смесь перемешивают в течение 1 часа при -10°С, а затем реакцию прерывают в атмосфере аргона добавлением 6 н. раствора HCl (290 мл), при этом температуру поддерживают ниже -5°С. После добавления рН раствора составляет около 2. В один прием добавляют серу (11,8 г, 367 ммоль) и реакционную смесь без охлаждения оставляют перемешиваться в течение ночи. Органический слой отделяют и промывают 6 н. раствором HCl, а затем дистиллированной Н2О. Водную фазу экстрагируют с помощью EtOAc. Органические вытяжки объединяют и промывают насыщенным раствором соли, сушат над MgSO4, отфильтровывают и упаривают в вакууме. Остаток суспендируют при 40°С в ИПС (60 мл) и охлаждают до 5°С. Твердое вещество отделяют и трижды промывают с помощью ИПС и сушат в вакууме при комнатной температуре в течение ночи. Указанным способом получают 2-[(ди-трет-бутилфосфинотиоилметил)метилфосфинотиоил]-2-метилпропан (формула 14) в виде твердого вещества белого цвета (64,6 г, выход 59%).
ПРИМЕР 16. Получение (R)- и (S)-2-[(ди-трет-бутилфосфино-тиоилметил)метилфосфинотиоил]-2-метилпропана (формулы 28 и 29)
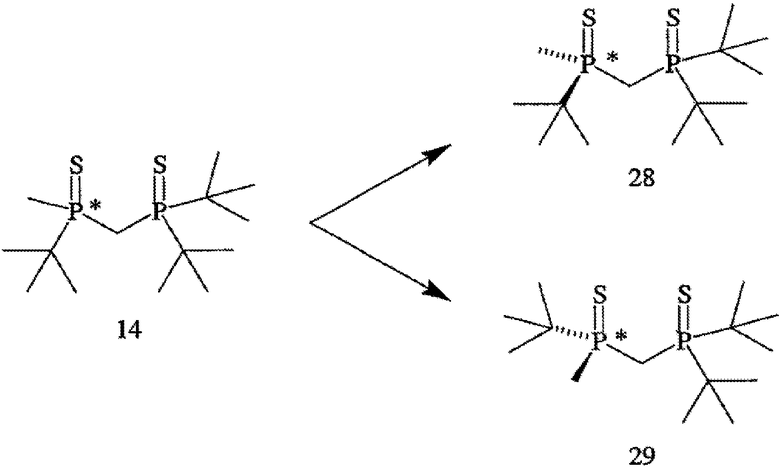
R- и S-энантиомеры (формулы 28 и 29, соответственно) 2-[(ди-трет-бутилфосфинотиоилметил)метилфосфинотиоил]-2-метилпропана (формула 14) разделяют с помощью ВЭЖХ, используя хиральную препаративную колонку и условия, приведенные ниже в таблице 5. Как показано в таблице 5, ВЭЖХ используют также для проверки хиральной чистоты и химической чистоты.
Разделение и анализ энантиомеров 2-[(ди-трет-бутилфосфинотиоилметил)метилфосфинотиоил]-2-метилпропана методом ВЭЖХ
Одна инжекция через каждые 10 минут
ПРИМЕР 17. Получение (S)-(ди-трет-бутилметилтиофосфониумил-метил)-трет-бутилметилметилтиофосфониум дитрифлата (формула 30)
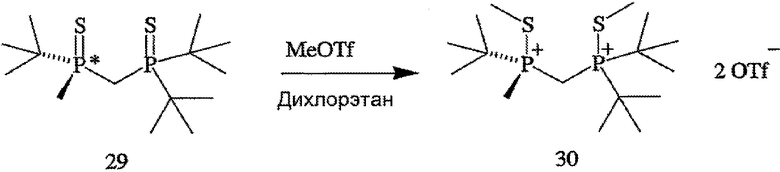
(S)-2-[(ди-трет-бутилфосфинотиоилметил)метилфосфинотиоил]-2-метилпропан (формула 29, 5,10 г, 15,6 ммоль) растворяют в 1,2-дихлорэтане (50 мл). К раствору добавляют метилтрифлат (7,69 г, 46,9 ммоль). Реакционную смесь в атмосфере аргона перемешивают при комнатной температуре. Через 10 минут масс-спектр показывает лишь монометилированный продукт. Реакционную смесь оставляют перемешиваться в течение ночи, при этом образуется осадок (продукт диметилирования). Твердое вещество собирают, трижды промывают 1,2-дихлорэтаном и сушат в вакуумном шкафу при комнатной температуре, получая после сушки (S)-(ди-трет-бутилметилтиофосфониумилметил)-трет-бутилметилметилтиофосфониум дитрифлат (формула 30) в виде твердого вещества белого цвета (6,90 г, выход 67%).
ПРИМЕР 18. Получение (R)-(2-{[(ди-трет-бутилфосфанил)метил]-метилфосфанил}-2-метилпропан)диборана (формула 21)
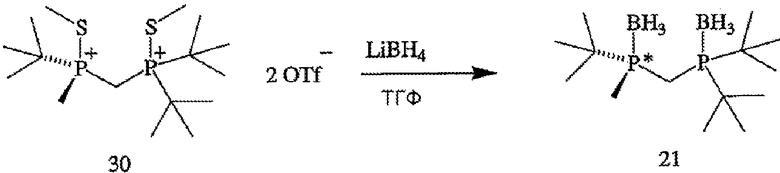
(S)-(ди-трет-бутилметилтиофосфониумилметил)-трет-бутилметил-метилтиофосфониум дитрифлат (2,005 г, 3,063 ммоль) суспендируют в ТГФ (25 мл). С помощью бани со льдом реакционную смесь в атмосфере аргона охлаждают до 0°С. Через капельную воронку добавляют LiBH4 (0,400 г, 18,4 ммоль), поддерживая температуру ниже 5°С. В процессе добавления наблюдается выделение газа. После добавления баню со льдом убирают и реакционную смесь оставляют перемешиваться в течение ночи при комнатной температуре. Спектр 1Н-ЯМР показывает, что сохраняется некоторое количество исходных веществ. Добавляют дополнительное количество LiBH4 (3 мл). Не наблюдается выделение газа или протекание экзотермической реакции. Реакционную смесь оставляют перемешиваться в течение ночи, после чего в соответствии с данными 1Н-ЯМР реакцию считают закончившейся. Реакционную смесь охлаждают на бане со льдом и прерывают реакцию добавлением 1 н. раствора HCl (15 мл). Наблюдается энергичное выделение газа. При перемешивании добавляют EtOAc. Органический слой отделяют и промывают 1 н. раствором HCl и Н2О. Водный слой экстрагируют с помощью EtOAc. Органические вытяжки объединяют и промывают насыщенным раствором соли, сушат над MgSO4, отфильтровывают и упаривают в вакууме, получая (R)-(2-{[(ди-трет-бутилфосфанил)метил]метилфосфанил}-2-метилпропан)диборан (формула 21, 0,492 г, выход 55%). Энантиомерный избыток определяют, используя аналитическую методику, описанную выше в таблице 2: ЭИ 98,7%; т.пл.=150-152°С; элементный анализ: вычислено для С14Н38В2Р2: С, 57,98; Н, 13,21. Найдено: С, 57,64; Н, 13,01.
ПРИМЕР 19. Получение (R)-(2-{[(ди-трет-бутилфосфанил)метил]-метилфосфанил}-2-метилпропан)-ди-(хлорборана) (формула 31)
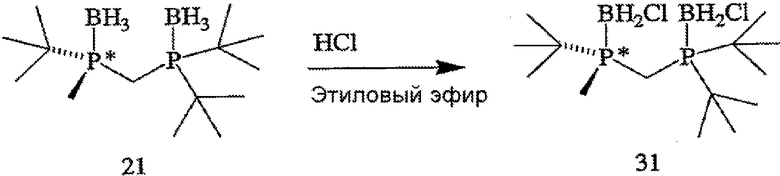
(R)-(2-{[(ди-трет-бутилфосфанил)метил]метилфосфанил}-2-метилпропан)диборан (формула 21, 0,200 г, 0,690 ммоль) помещают в толстостенную трубку, армированную нитью из #15 ACE. В трубку помещают 2 М раствор HCl (0,438 г, 12 ммоль), диспергированный в этиловом эфире (6 мл). Пространство над реакционной смесью продувают аргоном и трубку герметизируют пробкой из #15 ACE с прокладкой из TEFLON®. Содержимое трубки нагревают до 85°С в течение 12 часов, а затем охлаждают до комнатной температуры, получая (R)-(2-{[(ди-трет-бутилфосфанил)метил]метилфосфанил}-2-метилпропан)-ди-(хлорборан) (формула 31) в виде твердого вещества белого цвета. Поскольку в процессе протекания реакции выделяется газообразный Н2, то принимают меры против избыточного давления в трубке в процессе реакции и после проведения реакции. Растворитель удаляют пипеткой, а твердые вещества трижды растирают в этиловом эфире. Твердое вещество сушат в вакууме и получают продукт в виде твердого вещества белого цвета (0,222 г, выход 90%). Поскольку указанное в названии соединение является гигроскопичным, избегают контакта с воздухом и продукт хранят в вакууме или в перчаточной камере до использования.
ПРИМЕР 20. Получение тетрафторбората (S)-(+)-(2-{[(ди-трет-бутилфосфанил)метил]метилфосфанил}-2-метилпропан)-(1,5-циклооктадиен)родия(I) (формула 23)
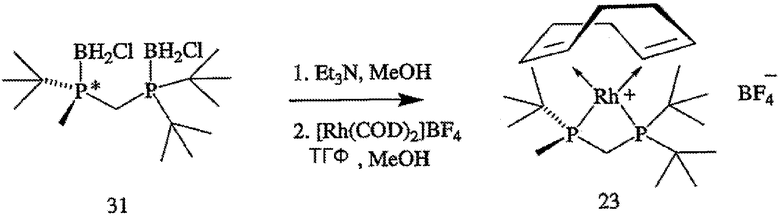
(R)-(2-{[(ди-трет-бутилфосфанил)метил]метилфосфанил}-2-метилпропан)-ди-(хлорборан) (формула 31, 179 мг, 0,5 ммоль) растворяют в МеОН (5 мл) и охлаждают до 0°С. К полученному раствору по каплям добавляют Et3N (505 мг, 5,0 ммоль). По окончании добавления смесь нагревают до 20°С и перемешивают в течение 30 минут. МеОН удаляют в вакууме, а остаток экстрагируют гексаном (3×20 мл). Органические слои объединяют, отфильтровывают и концентрируют, получая (R)-(2-{[(ди-трет-бутилфосфанил)метил]метилфосфанил}-2-метилпропан (формула 5, 66 мг). 31Р и 1Н-ЯМР показывают небольшие пики примесей. Хиральный лиганд (формула 5) растворяют в ТГФ (5 мл) и при перемешивании при комнатной температуре по каплям добавляют к раствору [Rh(COD)2]BF4 (102 мг, 0,25 ммоль) в МеОН (10 мл). По окончании добавления реакционную смесь перемешивают в течение 1 часа. Растворитель удаляют в вакууме и получают твердый продукт красного цвета. Твердое вещество перекристаллизовывают из теплого ТГФ, получая кристаллический продукт красного цвета. Кристаллы промывают смесью 5:1 гексан/диэтиловый эфир и сушат в вакууме, получая тетрафторборат (S)-(+)-(2-{[(ди-трет-бутилфосфанил)метил]метилфосфанил}-2-метилпропан)-(1,5-циклооктадиен)родия(I) (формула 23, 89 мг, выход 48% из 31). [α]D 24=+52,4° (с 0,9, МеОН); элементный анализ: вычислено для С21Н42В1F4Р2Rh1: С, 46,18; Н, 7,75; найдено: С, 45,66; Н, 7,19.
ПРИМЕР 21. Получение (R)-2-{[(ди-трет-бутилфосфанил)метил]-метилфосфанил}-2-метилпропана (формула 5)
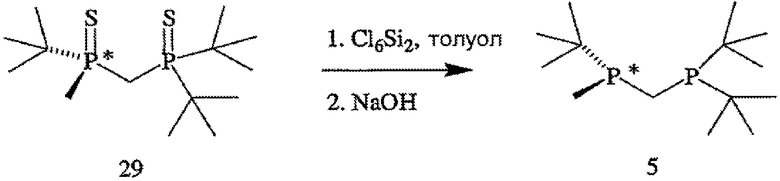
Гексахлордисилан (2,0 г, 7,5 ммоль) добавляют с помощью шприца к раствору (S)-2-[(ди-трет-бутилфосфинотиолметил)метил-фосфинотиоил]-2-метилпропана (формула 29, 0,5 г, 1,5 ммоль) в дегазированном толуоле (5 мл). Раствор нагревают при перемешивании при 80°С в течение 3 часов, после чего образуется осадок желтого цвета. Затем смесь охлаждают до 0°С и течение реакции прерывают, медленно добавляя при перемешивании 6,5 н. раствор NaOH в воде (8 мл), при этом температуру реакционной смеси поддерживают ниже 70°С. После добавления NaOH смесь перемешивают 1 час при 50°С, пока реакционная смесь не станет прозрачной. Органическую фазу отделяют в разделительной воронке, а водную фазу промывают диэтиловым эфиром (2×15 мл). Органические слои объединяют и сушат над MgSO4, отфильтровывают и концентрируют в вакууме, получая (R)-2-{[(ди-трет-бутилфосфанил)метил]метилфосфанил}-2-метилпропан (формула 5) в виде бесцветного масла (0,25 г, выход 64%). Свободный фосфин сразу же без дальнейшей очистки используют на стадии получения родиевого катализатора (Пример 22). Масштаб получения свободного фосфина (формула 5) увеличивают до 2,2 г исходного вещества (формула 29), 5,0 г исходного вещества и 10,0 г исходного вещества, получая выходы 82%, 80% и 98% соответственно.
ПРИМЕР 22. Получение тетрафторбората (S)-(+)-(2-{[(ди-трет-бутилфосфанил)метил]метилфосфанил}-2-метилпропан)-(1,5-цикло-октадиен)родия(I) (формула 23)
Раствор (R)-2-{[(ди-трет-бутилфосфанил)метил]метилфосфанил}-2-метилпропана (формула 5, 0,32 г, 1,2 ммоль) в дегазированном ТГФ (5 мл) при комнатной температуре и при перемешивании по каплям со скоростью 1 мл/мин добавляют к раствору [Rh(COD)2]BF4 (0,49 г, 1,2 ммоль) в дегазированном МеОН (10 мл). Цвет раствора меняется с коричневого на красный. После добавления смесь перемешивают в течение 1 часа и концентрируют в вакууме. Остаток перемешивают с дегазированным ТГФ (5 мл) и затем отфильтровывают. Фильтрат промывают смесью 1:1 диэтиловый эфир/ТГФ (2×5 мл) и сушат в вакууме, получая мелкий, как пыль, порошок твердого вещества, тетрафторбората (S)-(+)-(2-{[(ди-трет-бутилфосфанил)метил]метилфосфанил}-2-метилпропан)-(1,5-циклооктадиен)родия(I) (формула 23, 0,5 г, выход 75%). Масштаб получения родиевого комплекса (формула 23) увеличивают до 1,51 г исходного вещества (формула 5), 3,27 г исходного вещества и 8,15 г исходного вещества, получая выходы 87%, 92% и 91% соответственно.
ПРИМЕРЫ 23-46. Получение хиральных соединений (формула 32) посредством асимметрического гидрирования прохиральных олефинов (формула 33) с использованием тетрафторбората (S)-(+)-(2-{[(ди-трет-бутилфосфанил)метил]метилфосфанил}-2-метилпропан)-(1,5-циклооктадиен)родия(I) (формула 23)
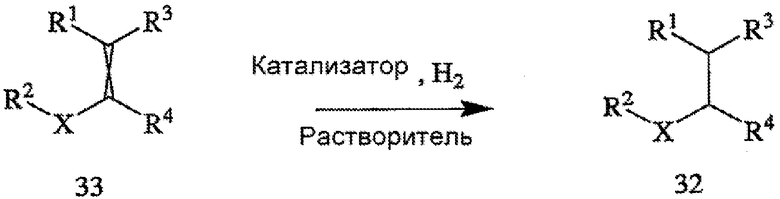
В таблице 6 приведен перечень субстратов (формула 33) и стереохимическая конфигурация их двойных связей, давление водорода, растворитель, ЭИ и абсолютная стереохимическая конфигурация хиральных продуктов (формула 32), полученных асимметрическим гидрированием с использованием предшественника хирального катализатора, тетрафторбората (S)-(+)-(2-{[(ди-трет-бутилфосфанил)метил]метилфосфанил}-2-метилпропан)-(1,5-циклооктадиен)родия(I) (формула 23). Для каждого внесенного в таблицу 6 соединения предшественник катализатора (примеры 23-45, 0,005 ммоль; пример 46, 0,025 ммоль) и субстрат (0,50 ммоль, 0,2 М) растворяют в растворителе (2,5 мл) в автоклаве Гриффина-Уордена, который герметизируют, и поднимают давление Н2 до требуемого значения. Смесь энергично перемешивают при 25°С на магнитной мешалке с покрытием из политетрафторэтилена до тех пор, пока не прекратится поглощение Н2 (менее 15 минут для примеров 23-45; 6 часов для примера 46, как следует из данных капиллярной ГХ). Затем давление Н2 в сосуде снижают и реакционную смесь анализируют методом хиральной ГХ для определения процента конверсии в продукт и энантиомерного избытка.
Энантиоселективность хиральных соединений (формула 32, R1=AcNH, X=связь), полученных асимметрическим гидрированием прохиральных олефинов (формула 33, R1=AcNH, X=связь)
Конфигурация
Каждый из Z- и Е-β-ацетамидо-β-замещенных акрилатов (формула 33) получают из соответствующих сложных β-кетоэфиров. Раствор требуемого сложного β-кетоэфира (24 ммоль) и NH4ОАс (9,2 г, 120 ммоль) в МеОН (30 мл) перемешивают при 20°С в течение 3 дней. После упаривания растворителя к остатку добавляют хлороформ (50 мл), при этом образуется твердое вещество белого цвета, которое отфильтровывают и промывают хлороформом (2×50 мл). Объединенный фильтрат промывают водой и насыщенным раствором соли и сушат над сульфатом натрия. После упаривания растворителя получают β-амино-β-замещенный акрилат. К раствору β-амино-β-замещенного акрилата в ТГФ (24 мл) добавляют пиридин (12 мл) и безводный уксусный ангидрид (36 мл). Смесь кипятят с обратным холодильником в течение 18 часов. Затем смесь охлаждают до комнатной температуры и летучие компоненты отгоняют. Полученный остаток растворяют в EtOAc (40 мл) и получают раствор, который промывают водой (20 мл), 1 н. раствором HCl (20 мл), 1 М раствором КН2РО4 (20 мл), насыщенным раствором NaHCO3 (20 мл) и насыщенным раствором соли (30 мл). Раствор сушат над сульфатом натрия и остатки растворителя упаривают при пониженном давлении. Быстрая хроматография на силикагеле с мобильной фазой 5:1 и 3:1 гексан/этилацетат соответственно позволяет получить Z- и Е-изомеры β-ацетамидо-β-замещенного акрилата, которые подтверждают сравнением с данными 1Н-ЯМР.
В таблице 7 приведены детали методики, использованной для определения стереохимической конфигурации продуктов реакций, приведенных в таблице 6. Энантиомерный избыток (ЭИ) определяют с помощью хиральной ГХ с использованием гелия в качестве газа-носителя под давлением 20 psig. В таблице 7 "Столбец А" относится к CP Chirasil-Dex CB (30 м × 0,25 мм), а "Столбец В" относится к ChiralDex-gamma-TA (25 м × 0,25 мм). Рацемические продукты получают гидрированием соответствующих енаминов, катализируемым 10% Pd/C, в МеОН под давлением Н2 50 psig при комнатной температуре в течение 2 часов.
Абсолютные стереохимические конфигурации определяют, сравнивая знаки вращения плоскости поляризации с данными, приведенными в литературе G. Zhu et al., J. Org. Chem. 64: 6907-10 (1999): метил 3-ацетамидобутаноат, [α]D 20=+8,09 (с 1,24, МеОН), литературные данные +21,4 (с 1,4, CHCl3); этил 3-ацетамидогексаноат, [α]D 20=+18,26 (с 1,02, МеОН), литературные данные, этиловый эфир, +42,8 (с 1,86, CHCl3); этил 3-ацетамидо-4-метилпентаноат, [α]D 20=+9,05 (с 1,15, МеОН), литературные данные, этиловый эфир, +52,8 (с 1,2, CHCl3); этил 3-ацетамидо-5-метилгексаноат, [α]D 20=+24,44 (с 0,95, МеОН), литературные данные, +44,6 (с 1,56, CHCl3); метил 3-ацетамидо-4,4-диметилпентаноат, [α]D 20=+4,86 (с 0,93, МеОН), литературные данные отсутствуют; этил 3-ацетамидо-3-фенилпропаноат, [α]D 20=-47,66 (с 0,91, МеОН), литературные данные, -40,5 (с 2,15, МеОН).
Условия определения энантиомерного избытка
с помощью хиральной ГХ
Следует отметить, что применяемые в данном описании и прилагаемой формуле изобретения указания на какой-либо объект могут относиться, если из контекста однозначно не следует другое, как к одному объекту, так и к множеству объектов. Так, например, ссылка на состав, содержащий "соединение", может включать единственное соединение или же два или несколько соединений.
Следует понимать, что приведенное выше описание дано в качестве пояснения и не ограничивает настоящее изобретение. После прочтения приведенного выше описания для специалистов в данной области техники будут очевидны многочисленные варианты осуществления настоящего изобретения. Таким образом, объем настоящего изобретения должен определяться не ссылкой на приведенное выше описание, а, напротив, должен определяться ссылкой на прилагаемую далее формулу изобретения вместе с полным набором эквивалентов, на которые указанная формула изобретения дает право. Содержание всех статей и ссылок, включая патенты, заявки на патенты и публикации, для всех целей целиком включено в настоящее описание в качестве ссылок.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЗАМЕЩЕННЫЕ ФЕРРОЦЕНИЛДИФОСФИНЫ В КАЧЕСТВЕ ЛИГАНДОВ ДЛЯ ГОМОГЕННЫХ КАТАЛИЗАТОРОВ ГИДРИРОВАНИЯ | 2004 |
|
RU2352577C2 |
| ДИФОСФИНЫ И МЕТАЛЛОКОМПЛЕКСЫ | 2006 |
|
RU2408600C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЭНАНТИОМЕРНО И ДИАСТЕРЕОМЕРНО ОБОГАЩЕННЫХ ЦИКЛОБУТАНАМИНОВ И -АМИДОВ | 2018 |
|
RU2793738C2 |
| СПОСОБ ПОЛУЧЕНИЯ (S)-3-АМИНОМЕТИЛ-5-МЕТИЛГЕКСАНОВОЙ КИСЛОТЫ (ПРЕГАБАЛИНА) | 2013 |
|
RU2544859C1 |
| СПОСОБ ПОЛУЧЕНИЯ БИАРИЛЗАМЕЩЕННОЙ 4-АМИНОМАСЛЯНОЙ КИСЛОТЫ ИЛИ ЕЕ ПРОИЗВОДНЫХ И ИХ ПРИМЕНЕНИЕ В ИЗГОТОВЛЕНИИ ИНГИБИТОРОВ НЭП | 2007 |
|
RU2469019C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПИРИМИДИНИЛЦИКЛОПЕНТАНОВЫХ СОЕДИНЕНИЙ | 2014 |
|
RU2702355C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПИРИМИДИНИЛЦИКЛОПЕНТАНОВЫХ СОЕДИНЕНИЙ | 2014 |
|
RU2826179C2 |
| СПОСОБ СИНТЕЗА ПРОИЗВОДНЫХ АМИНО-МЕТИЛТЕТРАЛИНА | 2009 |
|
RU2512285C2 |
| Способ и промежуточные соединения для получения прегабалина | 2014 |
|
RU2628298C2 |
| СПОСОБ ЭПОКСИДИРОВАНИЯ ПРОХИРАЛЬНОГО ОЛЕФИНА | 1995 |
|
RU2204562C2 |
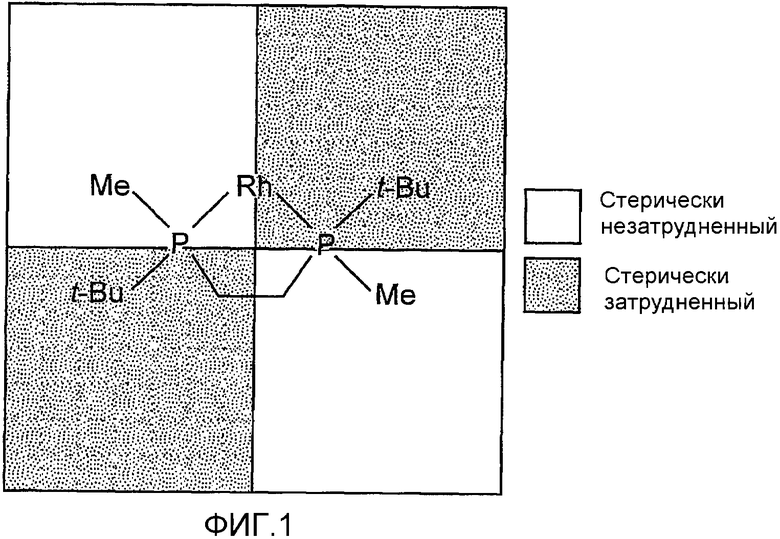
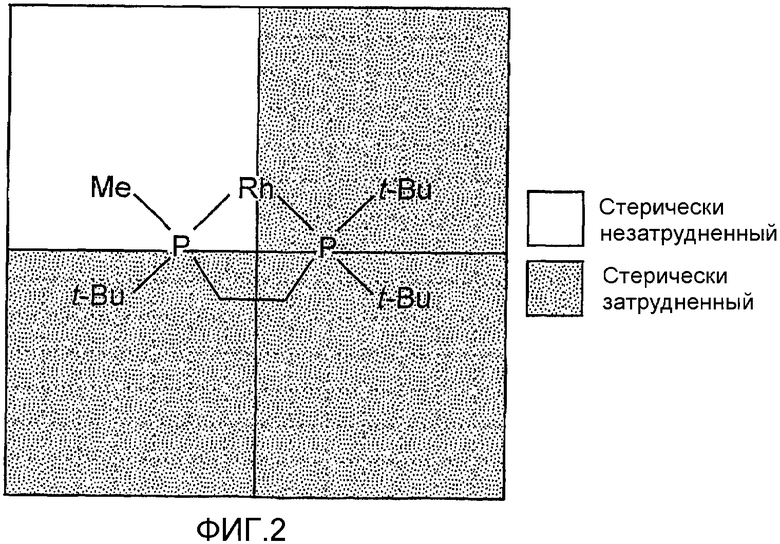
Настоящее изобретение относится к С1-симметричным бисфосфиновым лигандам и соответствующим катализаторам и к их применению в асимметрических синтезах, включая энантиоселективное гидрирование прохиральных олефинов, с целью получения фармацевтически полезных соединений, в том числе (S)-(+)-3-(аминометил)-5-метилгексановой кислоты, которая известна как прегабалин, и структурно родственных ей соединений. В способах применяют новые хиральные катализаторы или предшественники катализаторов, для их получения, которые включают хиральный лиганд, связанные с родием посредством атомов фосфора, при этом хиральный лиганд имеет структуру, представленную формулой 4. 6 н. и 13 з.п. ф-лы, 7 табл. 2 фиг.
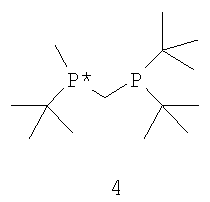
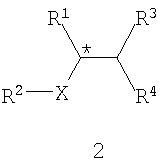 ,
,
или его фармацевтически приемлемый клатрат, соль, сольват или гидрат,
где R1 обозначает C1-6алкил, С1-7алканоиламино, C1-6алкоксикарбонил,
С1-6алкоксикарбониламино, амино, амино-С1-6алкил, С1-6алкиламино, циано, циано-С1-6алкил, карбокси или -CO2-Y;
R2 обозначает С1-7алканоил, C1-6алкоксикарбонил, карбокси или -CO2-Y;
R3 и R4 независимо обозначают атом водорода, C1-6алкил, С3-7циклоалкил, фенил, фенил-С1-6алкил, или же R3 и R4 вместе обозначают С2-6алкандиил;
X обозначает -NH-, -О-, -СН2- или связь; и
Y обозначает катион; при этом способ включает: взаимодействие соединения формулы 3,
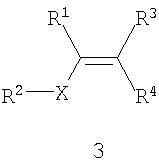 ,
,
с водородом в присутствии хирального катализатора с образованием соединения формулы 2; и
необязательно превращение соединения формулы 2 в фармацевтически приемлемую соль, клатрат, сольват или гидрат;
где хиральный катализатор включает хиральный лиганд, связанный с родием посредством атомов фосфора, при этом хиральный лиганд имеет структуру, представленную формулой 4,
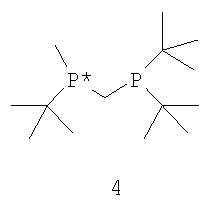
и где заместители R1, R2, R3, R4 и X в формуле 3 те же, что и определенные в формуле 2.
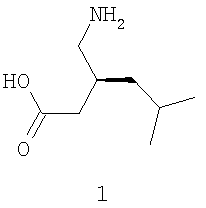
или его фармацевтически приемлемого клатрата, соли, сольвата или гидрата, при этом способ включает: взаимодействие соединения формулы 6,
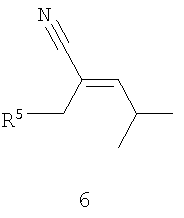 ,
,
соответствующего Z-изомера соединения формулы 6 или их смеси, с водородом в присутствии хирального катализатора с образованием соединения формулы 7,
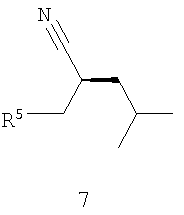 ,
,
где R5 обозначает карбоксильную группу или -CO2-Y, Y обозначает катион, а хиральный катализатор включает хиральный лиганд, связанный с родием посредством атомов фосфора, при этом хиральный лиганд имеет структуру, представленную формулой 4,
;
восстановление цианового фрагмента соединения формулы 7 с получением соединения формулы 8,
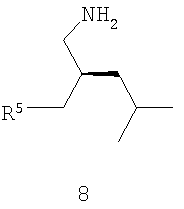 ;
;
необязательно обработку соединения формулы 8, когда R5 обозначает группу -СО2-Y, кислотой с получением соединения формулы 1; и необязательно превращение соединения формулы 8 или формулы 1 в фармацевтически приемлемый клатрат, соль, сольват или гидрат.
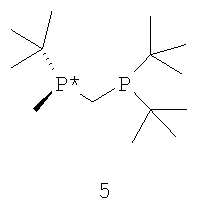
,
при этом способ включает:
взаимодействие соединения формулы 9,
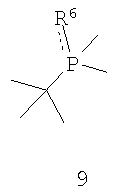 ,
,
с соединением формулы 10,
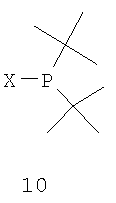 ,
,
с образованием соединения формулы 11,
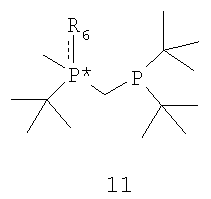
где соединение формулы 9 обрабатывают основанием перед взаимодействием с соединением формулы 10, X обозначает уходящую группу, a R6 обозначает ВН3, серу; и
взаимодействие соединения формулы 11 с бораном или серой с образованием соединения формулы 12,
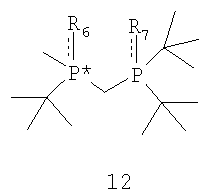 ,
,
где R7 такой же, как R6, или отличается от R6 и обозначает ВН3 или серу; и удаление R6 и R7 из соединения формулы 12 с образованием соединения формулы 4, при этом соединение формулы 12 расщепляют на отдельные энантиомеры перед удалением или после удаления R6 и R7.
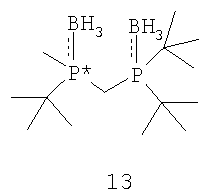 ,
,
с амином или кислотой с получением соединения формулы 4; или
обработку соединения формулы 12 восстановителем, когда R6 и R7, каждый обозначают серу; или
взаимодействие соединения формулы 14,
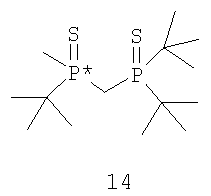 ,
,
с R8OTf с получением соединения формулы 15,
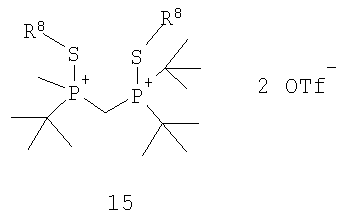 ,
,
в котором R8 обозначает С1-4алкил;
взаимодействие соединения формулы 15 с боргидридом с образованием соединения формулы 13;
; и либо
взаимодействие соединения формулы 13 с амином или кислотой с получением соединения формулы 4; либо
взаимодействие соединения формулы 13 с HCl с образованием соединения формулы 16,
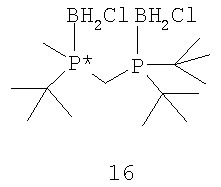 ; и
; и
взаимодействие соединения формулы 16 с амином или кислотой с получением соединения формулы 4.
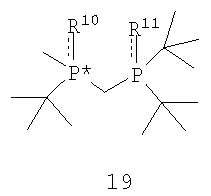 ,
,
где R10 и R11 независимо обозначают ВН3, ВН2Cl, серу, С1-4алкилтио или же отсутствуют, при условии, что R10 и R11, оба не являются ВН3.
2-{[(ди-трет-бутилфосфанил)метил]метилфосфанил}-2-метилпропан;
(R)-2-{[(ди-трет-бутилфосфанил)метил]метилфосфанил}-2-метилпропан;
(S)-2-{[(ди-трет-бутилфосфанил)метил]метилфосфанил}-2-метилпропан;
2-[(ди-трет-бутилфосфинотиоилметил)метилфосфинотиоил]-2-метилпропан;
(R)-2-[(ди-трет-бутилфосфинотиоилметил)метилфосфинотиоил]-2-метилпропан;
(S)-2-[(ди-трет-бутилфосфинотиоилметил)метилфосфинотиоил]-2-метилпропан;
(ди-трет-бутилметилтиофосфониумилметил)-трет-бутилметилметилтиофосфониум;
(R)-(ди-трет-бутилметилтиофосфониумилметил)-трет-бутилметилметилтиофосфониум;
(S)-(ди-трет-бутилметилтиофосфониумилметил)-трет-бутилметилметилтиофосфониум.
хиральный лиганд представляет собой энантиомер, который имеет структуру, представленную формулой 5,
| Оптически активный катализатор для асимметрического гидрирования замещенных акриловых кислот | 1975 |
|
SU663273A3 |
| СИСТЕМА ИЗ ИЗМЕРИТЕЛЬНОГО УСТРОЙСТВА УРОВНЯ АНАЛИЗИРУЕМЫХ ВЕЩЕСТВ В БИОЛОГИЧЕСКИХ ЖИДКОСТЯХ И КАССЕТЫ ДЛЯ ВЫПОЛНЕНИЯ КОМБИНИРОВАННЫХ ОБЩИХ ХИМИЧЕСКИХ И СПЕЦИФИЧЕСКИХ АНАЛИЗОВ СВЯЗЫВАНИЯ | 2005 |
|
RU2377069C2 |
| Топчак-трактор для канатной вспашки | 1923 |
|
SU2002A1 |

