Предпосылки создания изобретения
Область изобретения
Настоящее изобретение относится к новым бифенильным соединениям, обладающим активностью в качестве антагонистов мускаринового рецептора и антихолинергической активностью. Данное изобретение также относится к фармацевтическим композициям, включающим такие бифенильные соединения, способам и промежуточным соединениям для получения таких бифенильных соединений и способам применения таких бифенильных соединений для лечения легочных заболеваний.
Уровень техники в данной области
Заболевания легких и дыхательных путей, такие как хроническое обструктивное заболевание легких (ХОЗЛ) и астма, поражают множество миллионов людей по всему миру, и такие заболевания являются основной причиной заболеваемости и смертности.
Известно, что антагонисты мускариновых рецепторов обеспечивают бронхопротекторное действие, и, следовательно, такие соединения могут использоваться для лечения заболеваний дыхательных путей, таких как ХОЗЛ и астма. При использовании для лечения таких заболеваний антагонисты мускариновых рецепторов обычно вводят путем ингаляции. Однако даже при введении путем ингаляции значительное количество антагониста мускаринового рецептора часто поглощается системой кровообращения, приводя к системным побочным действиям, таким как сухость во рту, расширение зрачка и сердечно-сосудистые побочные действия.
Кроме того, многие антагонисты мускаринового рецептора, вводимые путем ингаляции, обладают относительно короткой продолжительностью действия, что требует их введения несколько раз в день. Такой режим дозирования, включающий многократное введение в течения дня, не только неудобен, но и создает существенный риск неадекватного лечения из-за несоблюдения пациентом требуемой частоты введения дозы согласно расписанию.
Соответственно, имеется необходимость в новых антагонистах мускаринового рецептора. В частности, имеется необходимость в новых антагонистах мускаринового рецептора, которые обладали бы высокой эффективностью и пониженными побочными действиями при введении путем ингаляции. Дополнительно, имеется необходимость в новых антагонистах мускаринового рецептора, которые обладали бы продолжительным действием, давая возможность введения дозы один раз в день или даже один раз в неделю. Можно полагать, что такие соединения будут особенно эффективными для лечения легочных заболеваний, таких как ХОЗЛ и астма, при снижении или исключении побочных действий, таких как сухость во рту и запор.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к новым бифенильным соединениям, обладающим активностью в качестве антагонистов мускаринового рецептора и антихолинергической активностью. Было установлено, что наряду с другими свойствами соединения по данному изобретению обладают высокой эффективностью и пониженным и системными побочными действиями при введении путем ингаляции и имеют продолжительный период действия.
Соответственно в одном из составляющих его аспектов данное изобретение относится к соединению формулы I:
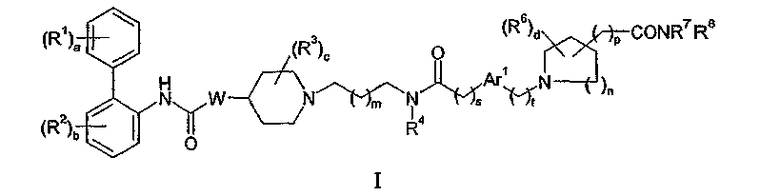
где
a равно 0 или целому числу от 1 до 5;
каждый из R1 независимо выбирают из (1-4C)алкила, (2-4C)алкенила, (2-4C)алкинила, (3-6C)циклоалкила, циано, галогена, -OR1a, -C(O)OR1b, -SR1c, -S(O)R1d, -S(O)2R1e, -NR1fR1g, -NR1hS(O)2R1i и -NR1jC(O)R1k; где каждый из R1a, R1b, R1c, R1d, R1e,
R1f, R1g, R1h, R1i, R1j и R1k независимо представляет собой водород, (1-4C)алкил или фенил(1-4C)алкил;
b равно 0 или целому числу от 1 до 4;
каждый R2 независимо выбирают из (1-4C)алкила, (2-4C)алкенила, (2-4C)алкинила, (3-6C)циклоалкила, циано, галогена, -OR2a, -C(O)OR2b, -SR2c, -S(O)R2d, -S(O)2R2e,
-NR2fR2g, -NR2hS(O)2R2i и -NR2jC(O)R2k; где каждый из R2a, R2b, R2c, R2d, R2e, R2f, R2g, R2h, R2i,
R2j и R2k независимо представляет собой водород, (1-4C)алкил или фенил(1-4C)алкил;
W представляет собой O или NWa, где Wa представляет собой водород или (1-4C)алкил;
c равно 0 или целому числу от 1 до 5;
каждый R3 независимо представляет собой (1-4C)алкил или две группы R3 объединены с образованием (1-3C)алкилена, (2-3C)алкенилена или оксиран-2,3-диила;
m равно 0 или 1;
R4 выбирают из водорода, (1-4C)алкила и (3-4C)циклоалкила;
s представляет собой 0, 1 или 2;
Ar1 представляет собой фениленовую группу или (3-5C)гетероариленовую группу, содержащую 1 или 2 гетероатома, независимо выбранных из кислорода, азота или серы; где фениленовая или гетероариленовая группа замещена (R5)q, где q равно 0 или целому числу от 1 до 4, и каждый R5 независимо выбирают из галогена, гидрокси, (1-4C)алкила или (1-4C)алкокси;
t равно 0, 1 или 2;
n равно 0 или целому числу от 1 до 3;
d равно 0 или целому числу от 1 до 4;
каждый R6 независимо представляет собой фтор или (1-4C)алкил;
p равно 0 или 1; и
R7 и R8 независимо представляют собой водород или (1-4C)алкил;
где каждая алкильная и алкоксильная группа в R1, R1a-1k, R2, R2a-2k, R3, R5, R6, R7 и
R8 необязательно замещена 1-5 фторными заместителями;
или его фармацевтически приемлемой соли или сольвату, или стереоизомеру.
В другом из составляющих его аспектов данное изобретение относится к фармацевтической композиции, включающей фармацевтически приемлемый носитель и терапевтически эффективное количество соединения формулы I или его фармацевтически приемлемой соли или сольвата, или стереоизомера. Такие фармацевтические композиции могут необязательно включать другие терапевтические агенты. Соответственно, в одном варианте осуществления, данное изобретение относится к такой фармацевтической композиции, где композиция дополнительно включает терапевтически эффективное количество стероидного противовоспалительного агента, такого как кортикостероид, агониста β2 адренергического рецептора, ингибитора фосфодиэстеразы-4 или их комбинацию.
Соединения по данному изобретению обладают антагонистической активностью в отношении мускаринового рецептора. Соответственно, предполагается, что соединения формулы I могут использоваться для лечения легочных заболеваний, таких как хроническое обструктивное заболевание легких и астма.
Соответственно в одном из аспектов данного способа данное изобретение относится к способу лечения легочного заболевания, при этом способ включает введение пациенту терапевтически эффективного количества соединения формулы I или его фармацевтически приемлемой соли или сольвата, или стереоизомера.
Дополнительно, в других аспектах данного способа данное изобретение относится к способу осуществления бронходилатации у пациента, при этом способ включает введение пациенту вызывающего бронходилатацию количества соединения формулы I или его фармацевтически приемлемой соли или сольвата, или стереоизомера.
Данное изобретение также относится к способу лечения хронического обструктивного заболевания легких или астмы, при этом способ включает введение пациенту терапевтически эффективного количества соединения формулы I или его фармацевтически приемлемой соли или сольвата, или стереоизомера.
В другом аспекте данного способа данное изобретение относится к способу антагонического воздействия на мускариновый рецептор у млекопитающего, включающего введение млекопитающему терапевтически эффективного количества соединения формулы I.
Поскольку соединения по данному изобретению обладают активностью в качестве антагонистов мускаринового рецептора, их также можно использовать в качестве инструмента при научных исследованиях. Соответственно, еще в одном из аспектов способа, данное изобретение относится к способу применения соединения формулы I или его фармацевтически приемлемой соли или сольвата, или стереоизомера в качестве инструмента при научных исследованиях для изучения биологической системы или образца, или для обнаружения новых химических соединений, обладающих антагонистической активностью по отношению к мускариновому рецептору.
Данное изобретение также относится к способам и новым промежуточным соединениям, используемым для получения соединений формулы I или их фармацевтически приемлемых солей или сольватов, или стереоизомеров.
Соответственно, в других аспектах данного способа данное изобретение относится к способу получения соединения формулы I, при этом способ включает:
(a) взаимодействие соединения формулы II с соединением формулы III; или
(b) конденсацию соединения формулы IV с соединением формулы V; или
(c) взаимодействие соединения формулы VI с соединением формулы VII; или
(d) взаимодействие соединения формулы II с соединением формулы VIII в присутствии восстановителя; или
(e) взаимодействие соединения формулы IX с соединением формулы VII в присутствии восстановителя; или
(f) взаимодействие соединения формулы XVIII с соединением формулы XIX;
и затем удаление, при необходимости, любых защитных групп для получения соединения формулы I; где соединения формулы I-IX, XVIII и XIX являются такими, как определено в данном описании.
В одном варианте осуществления вышеуказанный способ дополнительно включает стадию образования фармацевтически приемлемой соли соединения формулы I. В других вариантах осуществления данное изобретение относится к другим описанным здесь способам и к продукту, полученному любым из описанных здесь способов.
Данное изобретение также относится к соединению формулы I или его фармацевтически приемлемой соли или сольвату, или стереоизомеру, предназначенному для применения в терапии или в качестве лекарственного средства.
Дополнительно данное изобретение относится к применению соединения формулы I или его фармацевтически приемлемой соли или сольвата, или стереоизомера для получения лекарственного средства, особенно для получения лекарственного средства для лечения легочного заболевания или для антагонистического воздействия на мускариновый рецептор у млекопитающих.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В одном из составляющих его аспектов данное изобретение относится к новым бифенильным соединениям формулы I или их фармацевтически приемлемым солям, или сольватам, или стереоизомерам. Данные соединения могут содержать один или несколько хиральных центров, и, следовательно, данное изобретение относится к рацемическим смесям; чистым стереоизомерам (т.е. энантиомерам или диастереомерам), стереоизомерно обогащенным смесям и тому подобному, если не указано другого. Когда представлен или назван конкретный стереоизомер, специалистам в данной области следует понимать, что незначительные количества других стереоизомеров могут присутствовать в композициях данного изобретения, если не указано другого, при условии, что желаемая полезность композиции как целого не устраняется вследствие присутствия других изомеров.
Соединения формулы I также содержат несколько основных групп (например, аминогруппы) и, следовательно, соединения формулы I могут существовать в виде свободного основания или в виде различных солей. Все такие солевые формы включены в объем данного изобретения. Кроме того, сольваты соединений формулы I или их соли включены в объем данного изобретения.
Дополнительно, где это применимо, все цис-транс и E/Z изомеры (геометрические изомеры), таутомерные формы и топоизомерные формы соединений формулы I включены в объем данного изобретения, если не указано другого.
Соединения формулы I, а также те соединения, которые используются при их синтезе, также могут включать изотопно-меченые соединения, т.е. те, где один или несколько атомов были обогащены атомами, имеющими атомную массу, отличающуюся от атомной массы, доминирующей в природе. Примеры изотопов, которые могут быть введены в соединения формулы (I), включают, но не ограничиваются указанным, 2H, 3H, 13C, 14C, 15N, 18O и 17O.
Номенклатура, использованная в данном описании для обозначения соединений данного изобретения, проиллюстрирована в примерах. Данная номенклатура была получена с использованием коммерчески доступного программного обеспечения AutoNom (MDL, San Leandro, California). Например, соединения формулы I, где W представляет собой O, обычно называли как сложноэфирные производные бифенил-2-илкарбаминовой кислоты.
Иллюстративные варианты осуществления
Следующие заместители и величины предназначены для обеспечения иллюстративных примеров различных аспектов и вариантов осуществления данного изобретения. Данные иллюстративные величины предназначены для дополнительного определения и иллюстрации таких аспектов и вариантов осуществления, а не предназначены для исключения других вариантов осуществления или для ограничения объема данного изобретения. В данном отношении иллюстрация того, что конкретное значение или заместитель являются предпочтительными, не предназначена для исключения каким-либо образом других значений или заместителей из объема изобретения, если это специально не указано.
Значения a составляют 0, 1, 2, 3, 4 или 5; в частности, 0, 1 или 2, и еще более конкретно 0 или 1. Значения b составляют 0, 1, 2, 3, или 4; в частности, 0, 1 или 2, и еще более конкретно 0 или 1. В одном варианте осуществления, оба a и b равны 0.
Если он присутствует, каждый R1 может быть расположен в 2, 3, 4, 5 или 6-положении фенильного кольца, с которым он соединен. Каждый R1 независимо выбирают из (1-4C)алкила, (2-4C)алкенила, (2-4C)алкинила, (3-6C)циклоалкила, циано, галогена,
-OR1a, -C(O)OR1b, -SR1c, -S(O)R1d, -S(O)2R1e, -NR1fR1g, -NR1hS(O)2R1i и -NR1jC(O)R1k, примеры которых включают метил, фтор, хлор, бром, гидрокси, метокси, амино, метиламино, диметиламино и тому подобные. Конкретными представителями R1 являются фтор или хлор.
Если он присутствует, каждый R2 может быть расположен в 3, 4, 5 или 6-положении фенильного кольца, с которым он соединен (где атом углерода фенильного кольца соединен с атомом азота в положении 1). Каждый R2 независимо выбирают из (1-4C)алкила, (2-4C)алкенила, (2-4C)алкинила, (3-6C)циклоалкила, циано, галогена, -OR2a, -C(O)OR2b, -SR2c, -S(O)R2d, -S(O)2R2e, -NR2fR2g, -NR2hS(O)2R2i и -NR2jC(O)R2k, примеры которых включают метил, фтор, хлор, бром, гидрокси, метокси, амино, метиламино, диметиламино и тому подобные. Конкретными представителями R2 являются фтор или хлор.
Каждый из R1a, R1b, R1c, R1d, R1e, R1f, R1g, R1h, R1i, R1j и R1k, и R2a, R2b, R2c, R2d, R2e, R2f, R2g, R2h, R2i, R2j и R2k при использовании в R1 и R2 соответственно независимо представляет собой водород, (1-4C)алкил или фенил(1-4C)алкил, примеры которых включают водород, метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, изобутил, трет-бутил или бензил. В одном варианте осуществления данные группы независимо представляют собой водород или (1-3C)алкил. В другом варианте осуществления данные группы независимо представляют собой водород, метил или этил. Дополнительно, каждая алкильная и алкоксильная группа в R1,
R1a-1k, R2, и R2a-2k необязательно замещена 1-5 фторными заместителями.
В одном варианте осуществления данного изобретения W представляет собой O. В другом варианте осуществления W представляет собой NWa. Обычно, как было установлено, соединения, в которых W представляет собой O, проявляют особенно высокую аффинность в отношении мускариновых рецепторов. Соответственно, в конкретном варианте осуществления данного изобретения, W представляет собой O.
Когда W представляет собой NWa, Wa представляет собой водород или (1-4C)алкил, примеры которых включают водород, метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, изобутил и трет-бутил. В одном варианте осуществления Wa представляет собой водород или (1-3C)алкил. В другом варианте осуществления Wa представляет собой водород, метил или этил, в частности водород или метил. Еще в одном варианте осуществления Wa представляет собой водород, и NWa представляет собой NH.
Значения c составляют 0, 1, 2, 3, 4 или 5; в частности, 0, 1 или 2; и более конкретно 0 или 1. В одном конкретном варианте осуществления c равно 0. В другом варианте осуществления c равно 2.
Каждый R3 независимо представляет собой (1-4C)алкил или две группы R3 объединены с образованием (1-3C)алкилена, (2-3C)алкенилена или оксиран-2,3-диила. В одном варианте осуществления каждый R3 независимо представляет собой (1-4C)алкил, такой как метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, изобутил и трет-бутил. Кроме того, каждая алкильная группа в R3 необязательно замещена 1-5 фторными заместителями. В одном варианте осуществления каждый R3 независимо представляет собой (1-3C)алкил, и в другом варианте осуществления каждый R3 независимо представляет собой метил или этил.
В одном варианте осуществления каждый R3 находится в 3, 4 или 5-положении пиперидинового кольца (где атом азота пиперидинового кольца находится в положении 1). В конкретном варианте осуществления R3 находится в 4-положении пиперидинового кольца. В другом варианте осуществления R3 находится в 1-положении пиперидинового кольца, т.е. на атоме азота пиперидинового кольца, образуя таким образом четвертичную соль амина.
Еще в одном варианте осуществления две группы R3 объединены с образованием (1-3C)алкиленовой или (2-3C)алкениленовой группы. Например, две группы R3 во 2 и 6-положениях пиперидинового кольца могут быть объединены с образованием этиленового мостика (т.e. пиперидиновое кольцо и группы R3 образуют 8-азабицикло[3,2,1]октановое кольцо), или две группы R3 в 1 и 4-положениях пиперидинового кольца могут быть объединены с образованием этиленового мостика (т.e. пиперидиновое кольцо и группы R3 образуют (1-азабицикло[2,2,2]октановое кольцо)). В таком варианте осуществления другие группы R3, как они определены в данном описании, также могут присутствовать.
В другом варианте осуществления две группы R3 объединены с образованием оксиран-2,3-диильной группы. Например, две группы R3 во 2 и 6-положениях пиперидинового кольца могут быть объединены с образованием 3-оксатрицикло [3,3,1,02,4]нонанового кольца. В таком варианте осуществления другие группы R3, как они определены в данном описании, также могут присутствовать.
Значения m равны 0 или 1. В одном варианте осуществления m равно 0.
R4 представляет собой водород, (1-4C)алкил или (3-4C)циклоалкил. Примеры (1-4C)алкила включают метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, изобутил и трет-бутил. Примеры (3-4C)циклоалкильных групп включают циклопропил и циклобутил. В одном варианте осуществления R4 представляет собой водород или (1-3C)алкил, в частности водород, метил или этил. В другом варианте осуществления R4 представляет собой водород.
Значения s равны 0, 1 или 2. Конкретное значение для s равно 0 или 1. В одном варианте осуществления s равно 0. В другом варианте осуществления s равно 2.
Ar1 представляет собой фениленовую группу или (3-5C)гетероариленовую группу, содержащую 1 или 2 гетероатома, независимо выбранных из кислорода, азота или серы. Фениленовая или гетероариленовая группа может быть незамещенной (q равно 0) или замещенной 1, 2, 3 или 4 (q равно 1, 2, 3 или 4) заместителями R5, которые независимо выбраны из галогена, гидрокси, (1-4C)алкила или (1-4C)алкокси. Кроме того, каждая алкильная и алкоксильная группа в R5 необязательно замещена 1-5 фторными заместителями. Значение q составляет 0, 1, 2, 3 или 4, в частности 0, 1, 2 или 3. В одном варианте осуществления q равно 0, 1 или 2. Местом присоединения Ar1 является любой доступный кольцевой атом углерода или гетероатом. В определенных вариантах осуществления Ar1 представляет собой фениленовую группу, присоединенную в мета- или параположении.
В одном варианте осуществления Ar1 представляет собой фен-1,3-илен или фен-1,4-илен, где фениленовая группа является незамещенной или замещенной 1, 2 или 3 заместителями R5. Иллюстративные заместители R5 включают фтор, хлор, бром, метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, метокси, этокси, изопропокси, дифторметил, трифторметил, 2,2,2-трифторэтил и трифторметокси. Конкретные примеры групп Ar1 в данном варианте осуществления включают 2-фторфен-1,4-илен, 3-фторфен-1,4-илен, 2-хлорфен-1,4-илен, 3-хлорфен-1,4-илен, 2-метилфен-1,4-илен, 3-метилфен-1,4-илен, 2-метоксифен-1,4-илен, 3-метоксифен-1,4-илен, 2-трифторметоксифен-1,4-илен, 3-трифторметоксифен-1,4-илен, 2,3-дифторфен-1,4-илен, 2,5-дифторфен-1,4-илен, 2,6-дифторфен-1,4-илен, 2,3-дихлорфен-1,4-илен, 2,5-дихлорфен-1,4-илен, 2,6-дихлорфен-1,4-илен, 2-хлор-5-метоксифен-1,4-илен, 2-хлор-6-метоксифен-1,4-илен, 2-хлор-5-трифторметоксифен-1,4-илен, 2-хлор-6-трифторметоксифен-1,4-илен и 2,5-дибромфен-1,4-илен.
В другом варианте осуществления Ar1 представляет собой (3-5C)гетероариленовую группу, содержащую 1 или 2 гетероатома, независимо выбранных из кислорода, азота или серы, где гетероариленовая группа является незамещенной или замещенной 1 или 2 заместителями R5. Иллюстративная гетероариленовая группа включает двухвалентные разновидности пиррола, имидазола, тиазола, оксазола, фурана, тиофена, пиразола, изоксазола, изотиазола, пиридина, пиразина, пиридазина и пиримидина, где места присоединения расположены при любом доступном атоме углерода или азота. Более конкретные примеры таких групп Ar1 включают 2,5-фурилен, 2,4-тиенилен, 2,5-тиенилен, 2,5-пиридилен, 2,6-пиридилен и 2,5-пирролилен. Иллюстративные заместители R5 включают фтор, хлор, метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, метокси, этокси, изопропокси, дифторметил, трифторметил, 2,2,2-трифторэтил и трифторметокси. Конкретные примеры замещенных групп Ar1 включают 3-фтор-2,5-тиенилен, 3-хлор-2,5-тиенилен, 3-метил-2,5-тиенилен, 3-метокси-2,5-тиенилен и 3-метокси-6-хлор-2,5-пиридилен.
В одном конкретном варианте осуществления Ar1 представляет собой фен-1,3-илен, фен-1,4-илен, 2,4-тиенилен или 2,5-тиенилен, где фениленовая или тиениленовая группа необязательно замещена 1 или 2 заместителями R5. В другом конкретном варианте осуществления Ar1 представляет собой фен-1,4-илен или 2,4-тиенилен, необязательно замещенный 1 или 2 заместителями R5.
Значения t равны 0, 1 или 2. Конкретное значение для t равно 1.
Значения n равны 0, 1, 2, или 3. Конкретные значения для n составляют 1 или 2. В одном варианте осуществления n равно 2.
Значения d равны 0, 1, 2, 3, или 4. Конкретные значения для d составляют 0, 1 или 2. В одном варианте осуществления d равно 0.
Каждый R6 независимо представляет собой фтор или (1-4C)алкил, примеры которого включают метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, изобутил и трет-бутил. Кроме того, каждая алкильная и алкоксильная группа в R6 необязательно замещена 1-5 фторными заместителями. В одном варианте осуществления каждый R6 независимо представляет собой фтор или (1-3C)алкил, и в другом варианте осуществления каждый R6 независимо выбирают из фтора, метила, этила или трифторметила.
Значения p равны 0 или 1. В одном конкретном варианте осуществления p равно 0.
Каждый из R7 и R8 независимо представляет собой водород или (1-4C)алкил, примеры которого включают метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, изобутил и трет-бутил. В одном варианте осуществления каждый из R7 и R8 независимо представляет собой водород или (1-3C)алкил. В конкретном варианте осуществления R7 представляет собой водород, метил, этил, н-пропил или изопропил, и R8 представляет собой водород. В другом конкретном варианте осуществления оба R7 и R8 представляют собой водород или оба представляют собой этил. Кроме того, каждая алкильная и алкоксильная группа в R7 и R8 необязательно замещена 1-5 фторными заместителями.
Как отмечено в формуле I, группа -CONR7R8 может быть расположена при любом атоме углерода кольца. Например, когда n равно 2, группа -CONR7R8 может быть расположена в орто-, мета- и параположениях. В одном варианте осуществления группа -CONR7R8 расположена в мета- и параположениях; и в конкретном варианте осуществления группа -CONR7R8 расположена в параположении.
Конкретная группа представляющих интерес соединений включает соединения формулы I, где a, b, c и d равны 0; n равно 2; и R4 представляет собой водород, метил или этил.
Другая конкретная группа представляющих интерес соединений включает соединения формулы I, где a, b, c и d равны 0; R4 представляет собой водород, метил или этил; и R7 представляет собой водород.
Другая конкретная группа представляющих интерес соединений включает соединения формулы I, где a, b, c и d равны 0; R4 представляет собой водород, метил или этил; R7 представляет собой водород, метил, этил, н-пропил или изопропил, и R8 представляет собой водород.
Другая конкретная группа представляющих интерес соединений включает соединения формулы I, где a, b, c и d равны 0; R4 представляет собой водород, метил или этил; и R7 и R8 представляют собой этил.
Другая конкретная группа представляющих интерес соединений включает соединения формулы I, где a, b, c и d равны 0; R4 представляет собой водород, метил или этил; R7 и R8 представляют собой водород; и s равно 0.
Другая конкретная группа представляющих интерес соединений включает соединения формулы I, где a, b, c и d равны 0; R4 представляет собой водород, метил или этил; R7 и R8 представляют собой водород; s равно 0; и t равно 1.
Другая конкретная группа представляющих интерес соединений включает соединения формулы I, где a, b, c и d равны 0; R4 представляет собой водород, метил или этил; R7 и R8 представляют собой водород; s равно 0; t равно 1; и m равно 0.
Иллюстративные субгенерические группировки
Следующие субгенерические формулы и группировки предназначены для обеспечения иллюстративных примеров различных аспектов и вариантов осуществления данного изобретения и, как таковые, они не предназначены для исключения других вариантов осуществления или для ограничения объема данного изобретения, если не указано другого.
Конкретная группа соединений формулы I представляет собой соединения, описанные в предварительной заявке на патент США No. 60/552,443, поданной 11 марта 2004. Данная группа включает соединения формулы Ia:
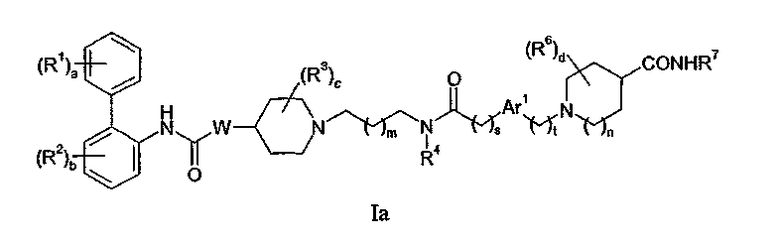
где
a равно 0 или целому числу от 1 до 3; каждый R1 независимо выбирают из (1-4C)алкила, (2-4C)алкенила, (2-4C)алкинила, (3-6C)циклоалкила, циано, галогена,
-OR1a, -C(O)OR1b, -SR1c, -S(O)R1d, -S(O)2R1e и -NR1fR1g; каждый из R1a, R1b, R1c, R1d, R1e, R1f и R1g независимо представляет собой водород, (1-4C)алкил или фенил(1-4C)алкил;
b равно 0 или целому числу от 1 до 3; каждый R2 независимо выбирают из (1-4C)алкила, (2-4C)алкенила, (2-4C)алкинила, (3-6C)циклоалкила, циано, галогена,
-OR2a, -C(O)OR2b, -SR2c, -S(O)R2d, -S(O)2R2e и -NR2fR2g; каждый of R2a, R2b, R2c, R2d, R2e, R2f и R2g независимо представляет собой водород, (1-4C)алкил или фенил(1-4C)алкил;
W представляет собой O или NWa, где Wa представляет собой водород или (1-4C)алкил;
c равно 0 или целому числу от 1 до 4; каждый R3 независимо представляет собой (1-4C)алкил;
m равно 0 или 1;
R4 представляет собой водород или (1-4C)алкил;
s равно 0 или 1;
Ar1 представляет собой фениленовую группу или (3-5C)гетероариленовую группу, содержащую 1 или 2 гетероатома, независимо выбранных из кислорода, азота или серы, где фениленовая или гетероариленовая группа замещена (R5)q, где q равно 0 или целому числу от 1 до 4, и каждый R5 независимо выбирают из галогена, гидрокси,
(1-4C)алкила или (1-4C)алкокси;
t равно 0 или 1;
n равно 0, 1 или 2;
d равно 0 или целому числу от 1 до 4; каждый R6 независимо представляет собой фтор или (1-4C)алкил; и
R7 представляет собой водород или (1-4C)алкил;
где каждая алкильная и алкоксильная группа в R1, R1a-1g, R2, R2a-2g, R3, R5, R6 или R7 необязательно замещена 1-5 фторными заместителями; или их фармацевтически приемлемые соли или сольваты, или стереоизомеры.
Данная группа также включает соединения формулы Ib:
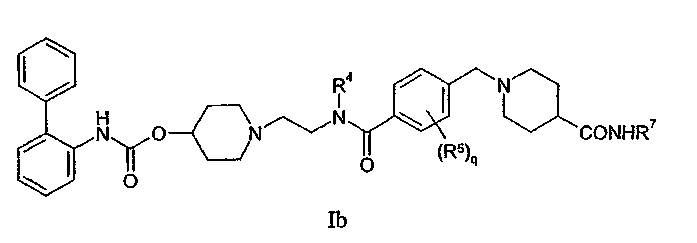
где R4, q, R5 и R7 являются такими, как определено для формулы Ia; или их фармацевтически приемлемые соли или сольваты, или стереоизомеры. Конкретный вариант осуществления включает соединения формулы Ib, где q равно 0, 1 или 2, и R5 независимо выбирают из галогена, (1-4C)алкила или (1-4C)алкокси, где каждая алкильная и алкоксильная группа необязательно замещена 1-3 фторными заместителями.
Дополнительно, конкретные соединения формулы I, представляющие интерес, включают:
1-(2-{[4-(4-карбамоилпиперидин-1-илметил)бензоил]метиламино}этил)пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты;
1-(2-{[4-(4-карбамоилпиперидин-1-илметил)бензоил]этиламино}этил)пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты;
1-(2-{метил-[4-(4-метилкарбамоилпиперидин-1-илметил)бензоил]амино}этил)пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты;
1-(2-{[4-(4-этилкарбамоилпиперидин-1-илметил)бензоил]метиламино}этил)пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты;
1-(2-{метил-[4-(4-пропилкарбамоилпиперидин-1-илметил)бензоил]амино}этил)пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты;
1-(2-{[4-(4-изопропилкарбамоилпиперидин-1-илметил)бензоил]метиламино}этил)пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты;
1-{2-[4-(4-карбамоилпиперидин-1-илметил)-2-фторбензоиламино]этил}пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты;
1-(2-{[2,5-дибром-4-(4-карбамоилпиперидин-1-илметил)бензоил]метиламино}этил)пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты;
1-(2-{[4-(4-карбамоилпиперидин-1-илметил)-2-фторбензоил]метиламино}этил)пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты;
1-{2-[4-(4-диэтилкарбамоилпиперидин-1-илметил)-2-фторбензоиламино]этил}пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты;
1-(2-{[4-(4-диэтилкарбамоилпиперидин-1-илметил)-2-фторбензоил]метиламино}этил)пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты;
1-(2-{[4-(4-диэтилкарбамоилпиперидин-1-илметил)бензоил]метиламино}этил)пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты;
1-(2-{[4-(3-(S)-диэтилкарбамоилпиперидин-1-илметил)бензоил]метиламино}этил)пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты;
1-(2-{[4-(2-карбамоилпиперидин-1-илметил)бензоил]метиламино}этил)пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты;
1-(2-{[4-(4-карбамоилпиперидин-1-илметил)-2-метоксибензоил]метиламино}этил)пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты;
1-(2-{[5-(4-карбамоилпиперидин-1-илметил)тиофен-2-карбонил]метиламино}этил)пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты;
1-(2-{[5-((R)-3-диэтилкарбамоилпиперидин-1-илметил)тиофен-2-карбонил]метиламино}этил)пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты;
1-(2-{[5-((R)-3-диэтилкарбамоилпиперидин-1-илметил)тиофен-2-карбонил]амино}этил)пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты;
1-(2-{[5-(4-карбамоилпиперидин-1-илметил)тиофен-2-карбонил]амино}этил)пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты;
1-(2-{[5-((R)-3-диэтилкарбамоилпиперидин-1-илметил)-1H-пиррол-2-карбонил]метиламино}этил)пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты;
1-(2-{[5-(4-карбамоилпиперидин-1-илметил)-1H-пиррол-2-карбонил]метиламино}этил)пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты;
1-(2-{[5-((R)-3-диэтилкарбамоилпиперидин-1-илметил)фуран-2-карбонил]метиламино}этил)пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты;
1-(2-{[5-(4-диэтилкарбамоилпиперидин-1-илметил)фуран-2-карбонил]метиламино}этил)пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты;
1-(2-{[5-(4-карбамоилпиперидин-1-илметил)фуран-2-карбонил]-амино}этил)пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты;
1-(2-{[5-((R)-3-диэтилкарбамоилпиперидин-1-илметил)фуран-2-карбонил]амино}этил)пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты;
1-[2-({3-[4-(3-карбамоилпиперидин-1-илметил)фенил]пропионил}метиламино)этил]пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты;
1-[2-({3-[4-(4-карбамоилпиперидин-1-илметил)фенил]пропионил}метиламино)этил]пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты;
1-(2-{3-[4-(4-карбамоилпиперидин-1-илметил)фенил]пропиониламино}этил)пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты;
1-(2-{3-[4-(4-диэтилкарбамоилпиперидин-1-илметил)фенил]пропиониламино}этил)пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты;
1-(2-{3-[4-(3-диэтилкарбамоилпиперидин-1-илметил)фенил]пропиониламино}этил)пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты;
1-{2-[4-(4-карбамоилпиперидин-1-илметил)бензоиламино]этил}пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты;
1-(2-{[4-(4-карбамоилпиперидин-1-илметил)-2-хлор-бензоил]метиламино}этил)пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты;
1-(2-{[4-(4-карбамоилпиперидин-1-илметил)-2-хлор-5-метоксибензоил]метиламино}этил)пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты; и
1-[2-({2-[4-(4-карбамоилпиперидин-1-илметил)фенил]ацетил}метиламино)этил]пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты;
или их фармацевтически приемлемую соль или сольват.
Определения
При описании соединений, композиций, методов и способов данного изобретения следующие термины имеют следующие значения, если не указано другого.
Термин «алкил» означает одновалентную насыщенную углеводородную группу, которая может быть линейной или разветвленной. Если не указано другого, такие алкильные группы обычно содержат от 1 до 10 атомов углерода. Иллюстративные алкильные группы включают, в качестве примера, метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, изобутил, трет-бутил, н-пентил, н-гексил, н-гептил, н-октил, н-нонил, н-децил и тому подобные.
Термин «алкилен» означает двухвалентную насыщенную углеводородную группу, которая может быть линейной или разветвленной. Если не указано другого, такие алкиленовые группы обычно содержат от 1 до 10 атомов углерода. Иллюстративные алкиленовые группы включают, в качестве примера, метилен, этан-1,2-диил (“этилен”), пропан-1,2-диил, пропан-1,3-диил, бутан-1,4-диил, пентан-1,5-диил и тому подобные.
Термин «алкокси» означает одновалентную группу формулы (алкил)-O-, где алкил является таким, как определено в данном описании. Иллюстративные алкоксильные группы включают, в качестве примера, метокси, этокси, н-пропокси, изопропокси, н-бутокси, втор-бутокси, изобутокси, трет-бутокси и тому подобные.
Термин «алкенил» означает одновалентную ненасыщенную углеводородную группу, которая может быть линейной или разветвленной и которая содержит по меньшей мере одну, и обычно 1, 2 или 3, углерод-углеродную двойную связь. Если не указано другого, такие алкенильные группы обычно содержат от 2 до 10 атомов углерода. Иллюстративные алкенильные группы включают, в качестве примера, этенил, н-пропенил, изопропенил, н-бут-2-енил, н-гекс-3-енил и тому подобные.
Термин «алкенилен» означает двухвалентную алкенильную группу.
Термин «алкинил» означает одновалентную ненасыщенную углеводородную группу, которая может быть линейной или разветвленной и которая содержит по меньшей мере одну, и обычно 1, 2 или 3, углерод-углеродную тройную связь. Если не указано другого, такие алкинильные группы обычно содержат от 2 до 10 атомов углерода. Иллюстративные алкинильные группы включают, в качестве примера, этинил, н-пропинил, н-бут-2-инил, н-гекс-3-инил и тому подобные.
Термин «алкинилен» означает двухвалентную алкинильную группу.
Термин «арил» означает одновалентный ароматический углеводород, имеющий единственное кольцо (т.е. фенил) или конденсированные кольца (т.е. нафталин). Если не определено другого, такие арильные группы обычно содержат от 6 до 10 кольцевых атомов углерода. Иллюстративные арильные группы включают, в качестве примера, фенил и нафталин-1-ил, нафталин-2-ил и тому подобные.
Термин «арилен» означает двухвалентную арильную группу.
Термин «азациклоалкил» означает одновалентное гетероциклическое кольцо, содержащее один атом азота, т.е. циклоалкильную группу, в которой один атом углерода был заменен на атом азота. Если не определено другого, такие азациклоалкильные группы обычно содержат от 2 до 9 атомов углерода. Иллюстративными примерами азациклоалкильной группы являются пирролидинильная и пиперидинильная группы.
Термин «азациклоалкилен» означает двухвалентную азациклоалкильную группу. Иллюстративными примерами азациклоалкиленовой группы являются пирролидиниленовая и пиперидиниленовая группы.
Термин «циклоалкил» означает одновалентную насыщенную карбоциклическую углеводородную группу. Если не определено другого, такие циклоалкильные группы обычно содержат от 3 до 10 атомов углерода. Иллюстративные циклоалкильные группы включают, в качестве примера, циклопропил, циклобутил, циклопентил, циклогексил и тому подобные.
Термин «циклоалкилен» означает двухвалентную циклоалкильную группу.
Термин «галоген» означает фтор, хлор, бром и иод.
Термин «гетероарил» означает одновалентную ароматическую группу, имеющую одно кольцо или два конденсированных кольца и содержащую в кольце по крайней мере один гетероатом (обычно от 1 до 3 гетероатомов), выбранный из азота, кислорода или серы. Если не определено иначе, такие гетероарильные группы обычно содержат всего от 5 до 10 кольцевых атомов. Иллюстративные гетероарильные группы включают, в качестве примера, одновалентные разновидности пиррола, имидазола, тиазола, оксазола, фурана, тиофена, триазола, пиразола, изоксазола, изотиазола, пиридина, пиразина, пиридазина, пиримидина, триазина, индола, бензофурана, бензoтиофена, бензимидазола, бензтиазола, хинолина, изохинолина, хиназолина, хиноксалина и тому подобные, где местом присоединения является любой доступный атом углерода или азота кольца.
Термин «гетероарилен» означает двухвалентную гетероарильную группу.
Термин «гетероциклил» или «гетероциклический» означает одновалентную насыщенную или ненасыщенную (неароматическую) группу, имеющую одно кольцо или множество конденсированных колец и содержащую в кольце по крайней мере один гетероатом (обычно от 1 до 3 гетероатомов), выбранным из азота, кислорода или серы. Если не определено другого, такая гетероциклическая группа обычно содержит всего от 2 до 9 кольцевых атомов углерода. Иллюстративные гетероциклические группы включают, в качестве примера, одновалентные разновидности пирролидина, имидазолидина, пиразолидин, пиперидина, 1,4-диоксана, морфолина, тиоморфолина, пиперазина, 3-пирролина и тому подобные, местом присоединения является любой доступный атом углерода или азота кольца.
Термин «гетероциклен» означает двухвалентную гетероциклильную или гетероциклическую группу.
Когда определенное число атомов углерода предназначено для конкретного термина, используемого в данном описании, число атомов углерода показано в скобках, предшествующих термину. Например, термин «(1-4C)алкил» означает алкильную группу, имеющую от 1 до 4 атомов углерода.
Термин «фармацевтически приемлемая соль» означает соль, которая является приемлемой для введения пациенту, такому как млекопитающее (например, соли, обладающие приемлемой безопасностью для млекопитающего при данном режиме дозировки). Такие соли могут быть получены из фармацевтически приемлемых неорганических и органических оснований и из фармацевтически приемлемых неорганических и органических кислот. Соли, полученные из фармацевтически приемлемых неорганических оснований, включают соли аммония, кальция, меди, двухвалентного железа, трехвалентного железа, лития, магния, трехвалентного марганца, марганца, калия, натрия, цинка и тому подобные. Особенно предпочтительными являются соли аммония, кальция, магния, калия и натрия. Соли, полученные из фармацевтически приемлемых органических оснований, включают соли первичных, вторичных и третичных аминов, таких как аргинин, бетаин, каффеин, холин, N,N'-дибензилэтилендиамин, диэтиламин, 2-диэтиламиноэтанол, 2-диметиламиноэтанол, этаноламин, этилендиамин, N-этилморфолин, N-этилпиперидин, глюкамин, глюкозамин, гистидин, гидрабамин, изопропиламин, лизин, метилглюкамин, морфолин, пиперазин, пиперадин, полиаминовые смолы, прокаин, пурины, теобромин, триэтиламин, триметиламин, трипропиламин, трометамин и тому подобные.
Соли, полученные из фармацевтически приемлемых кислот, включают соли уксусной, аскорбиновой, бензольсульфоновой, бензойной, камфорсульфоновой, лимонной, этансульфоновой, эдисиловой, фумаровой, гентизиновой, глюуоновой, глюкуроновой, глутамовой, гиппуровой, бромистоводородной, хлористоводородной, изетионовой, молочной, лактобионовой, малеиновой, яблочной, миндальной, метансульфоновой, муциновой, нафталинсульфоновой, нафталин-1,5-дисульфоновой, нафталин-2,5-дисульфоновой, никотиновой, азотной, оротовой, памовой, пантотеновой, фосфорной, янтарной, серной, винной, п-толуолсульфоновой, ксинафоевой кислот и тому подобных. Особенно предпочтительными являются лимонная, бромистоводородная, хлористоводородная, изетионовая, малеиновая, нафталин-1,5-дисульфоновая, фосфорная, серная и винная кислоты.
Термин «соль соединения» означает соединение, образованное при замене атома водорода кислоты на катион, такой как катион металла или органический катион и тому подобное. Предпочтительно соль представляет собой фармацевтически приемлемую соль, хотя это не требуется для солей промежуточных соединений, которые не предназначены для введения пациенту.
Термин «сольват» означает комплекс или агрегат, образованный одной или несколькими молекулами растворенного вещества, т.е. соединения формулы I или его фармацевтически приемлемой соли, и одной или несколькими молекулами растворителя. Такие сольваты обычно представляют собой кристаллические твердые вещества, имеющие по существу фиксированное молярное соотношение растворенного вещества и растворителя. Иллюстративные растворители включают, в качестве примера, воду, метанол, этанол, изопропанол, уксусную кислоту и тому подобное. Когда растворитель представляет собой воду, образованный сольват представляет собой гидрат.
Следует понимать, что термин «или его фармацевтически приемлемая соль, или сольват, или стереоизомер» предназначен для включения всех измененных форм солей, сольватов и стреоизомеров, таких как сольват фармацевтически приемлемой соли стереоизомера соединения формулы I.
Термин «терапевтически эффективное количество» означает количество, достаточное для эффективного лечения при введении пациенту, нуждающемуся в лечении.
Термин «лечить» или «лечение», как он использован в данном описании, означает лечение заболевания или медицинского состояния (такого как COPD) у пациента, такого как млекопитающее (в частности, человек), которое включает:
(a) предотвращение возникновения заболевания или медицинского состояния, т.е. профилактическое лечение пациента;
(b) облегчение заболевания или медицинского состояния, т.е. ликвидацию или вызов регрессии заболевания или медицинского состояния у пациента;
(c) подавление заболевания или медицинского состояния, т.е. замедление или прекращение развития заболевания или медицинского состояния у пациента; или
(d) облегчение симптомов заболевания или медицинского состояния у пациента.
Термин «уходящая группа» означает функциональную группу или атом, которые могут быть замещены на другую функциональную группу или атом в реакции замещения, такой как реакция нуклеофильного замещения. В качестве примера, иллюстративные уходящие группы включают группы хлора, брома и иода; сульфоновые сложноэфирные группы, такие как мезилат, тозилат, брозилат, нозилат и тому подобные; и ацилокси группы, такие как ацетокси, трифторацетокси и тому подобные.
Термин «их защищенные производные» относится к производному конкретного соединения, в котором одна или несколько функциональных групп соединения защищены от нежелательных реакций с помощью защитных или блокирующих групп. Функциональные группы, которые могут быть защищены, включают, например, группы карбоновых кислот, аминогруппы, гидроксильные группы, тиольные группы, карбонильные группы и тому подобные. Иллюстративные защитные группы для карбоновых кислот включают сложные эфиры (такие как п-метоксибензиловый эфир), амиды и гидразиды; для аминогрупп - карбаматы (такие как трет-бутоксикарбонил) и амиды; для гидроксильных групп - простые эфиры и сложные эфиры; для тиоловых группы простые тиоэфиры и сложные тиоэфиры; для карбонильных групп - ацетали и кетали; и тому подобные. Такие защитные группы хорошо известны специалистам в данной области и описаны, например, в T. W. Greene and G. M. Wuts, Protecting Groups in Organic Synthesis, Third Edition, Wiley, New York, 1999 и процитированных там ссылках.
Термин «защитная группа аминогруппы» означает защитную группу, подходящую для предотвращения нежелательных реакций по аминогруппе. Иллюстративные защитные группы аминогруппы включают, но не ограничиваются указанным, трет-бутоксикарбонил (BOC), тритил (Tr), бензилоксикарбонил (Cbz), 9-флуоренилметоксикарбонил (Fmoc), формил, триметилсилил (TMS), трет-бутилдиметилсилил (TBS), и тому подобные.
Термин «защитная группа карбоксигруппы» означает защитную группу, подходящую для предотвращения нежелательных реакций по карбоксильной группе. Иллюстративные защитные группы карбоксигруппы включают, но не ограничиваются указанным, сложные эфиры, такие как метиловый, этиловый, трет-бутиловый, бензиловый (Bn), p-метоксибензиловый (PMB), 9-флуоренилметиловый (Fm), триметилсилиловый (TMS), трет-бутилдиметилсилилиловый (TBS), дифенилметиловый (бензгидриловый, DPM) и тому подобные.
Термин «защитная группа гидроксила» означает защитную группу, подходящую для предотвращения нежелательных реакций по гидроксильной группе. Иллюстративные защитные группы карбоксигруппы включают, но не ограничиваются указанным, силильные группы, включая три(1-6C)алкилсилильные группы, такие как триметилсилил (TMS), триэтилсилил (TES), трет-бутилдиметилсилил (TBS) и тому подобные; сложные эфиры (ацильные группы), включая (1-6C)алканоильные группы, такие как формил, ацетил и тому подобные; арилметильные группы, такие как бензил (Bn), п-метоксибензил (PMB), 9-флуоренилметил (Fm), дифенилметил (бензгидрил, DPM) и тому подобные. Дополнительно, две гидроксильные группы также могут быть защищены алкилиденовой группой, такой как проп-2-илидин, образованной, например, при реакции с кетоном, таким как ацетон.
Общие методы синтеза
Бифенильные соединения данного изобретения могут быть получены из легко доступных исходных веществ с использованием следующих общих методов и способов и с использованием другой информации, легко доступной специалистам в данной области. Хотя конкретные варианты осуществления настоящего изобретения могут быть продемонстрированы и описаны в данном описании, специалистам в данной области будет понятно, что все варианты осуществления или аспекты настоящего изобретения могут быть получены с использованием описанных здесь способов или с использованием других способов, реагентов и исходных веществ, известных специалистам в данной области. Также будет очевидно, что здесь приведены типичные или предпочтительные условия проведения способов (т.е. реакционные температуры, время, мольные соотношения реагентов, растворители, давления и т.д.), если не указано другого, можно использовать другие реакционные условия. Хотя оптимальные условиях реакций могут изменяться в зависимости от конкретных использованных реагентов и растворителей, такие условия легко могут быть определены специалистом в данной области с помощью методов оптимизации.
Кроме того, как будет очевидно специалистам в данной области, для предотвращения протекания нежелательных реакций некоторых функциональных групп могут оказаться необходимыми или желательными обычные защитные группы. Выбор подходящей защитной группы для конкретной функциональной группы, а также подходящие условия для введения и снятия защиты для таких функциональных групп хорошо известны. При желании, можно использовать защитные группы, отличающиеся от тех, которые проиллюстрированы в приведенных в данном описании способах. Например, многочисленные защитные группы и их введение и удаление описаны в T. W. Greene and G. M. Wuts, Protecting Groups in Organic Synthesis, Third Edition, Wiley, New York, 1999 и процитированных там ссылках.
В качестве иллюстрации соединения формулы I могут быть получены способом, включающим:
(a) взаимодействие соединения формулы II:
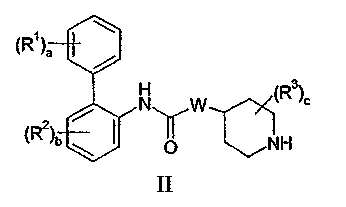
или его соли с соединением формулы III:
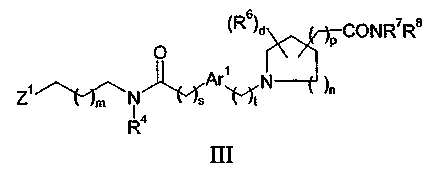
где Z1 представляет собой уходящую группу; или
(b) конденсацию соединения формулы IV:
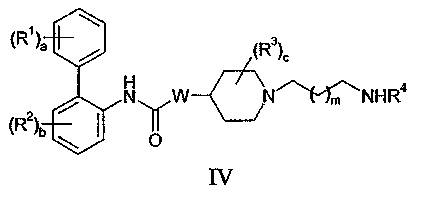
с соединением формулы V:
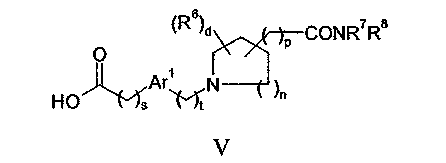
или его реакционноспособным производным; или
(c) взаимодействие соединения формулы VI:
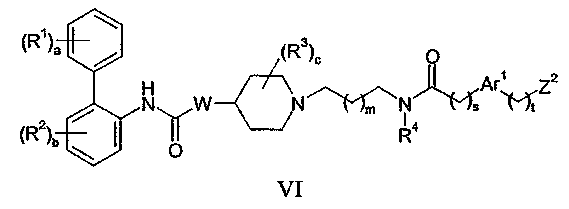
где Z2 представляет собой уходящую группу; с соединением формулы VII:
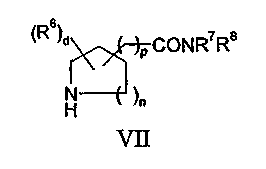
или
(d) взаимодействие соединения формулы II с соединением формулы VIII:
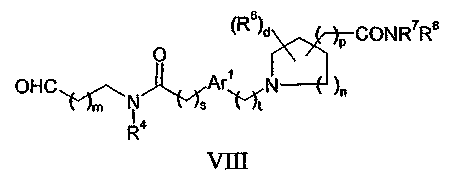
в присутствии восстановителя; или
(e) взаимодействие соединения формулы IX:
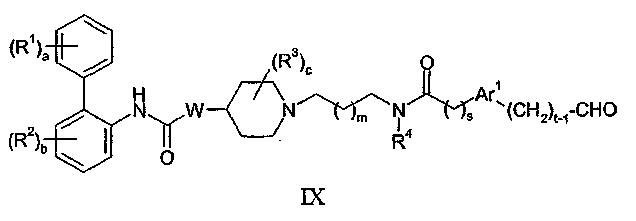
с соединением формулы VII в присутствии восстановителя; или
(f) взаимодействие соединения формулы XVIII:
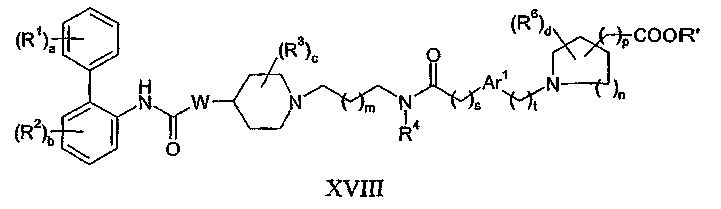
где R' представляет собой H, -CH3 или -CH2CH3, с соединением формулы XIX:
NHR 7 R 8
XIX
и затем
(g) удаление любых защитных групп, которые могут присутствовать, с получением соединения формулы I; и необязательно образование его фармацевтически приемлемой соли.
Как правило, если в описанном здесь способе используется соль одного из исходных веществ, такая как кислотно-аддитивная соль, соль обычно нейтрализуют перед или во время процесса реакции. Такую реакцию нейтрализации обычно проводят путем контактирования соли с одним мольным эквивалентом основания на каждый мольный эквивалент кислотно-аддитивной соли.
В способе (а), при взаимодействии между соединениями формул II и III, уходящая группа, обозначенная Z1, может представлять собой, например, галоген, такой как хлор, бром или йод, или сульфоновую сложноэфирную группу эфира, такую как мезилат или тозилат. Реакцию обычно проводят в присутствии основания, например, третичного амина, такого как диизопропилэтиламин. Обычные растворители включают нитрилы, такие как ацетонитрил. Реакцию обычно проводят при температуре в диапазоне от 0 до 100°C.
Соединения формулы II обычно известны в данной области или могут быть получены снятием защиты в соединении формулы X:
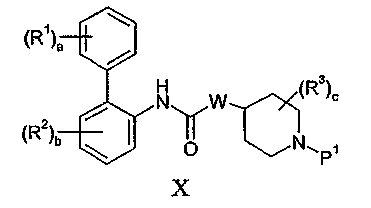
где P1 представляет собой защитную группу аминогруппы, такую как бензильная группа. Бензильные группы обычно удаляют восстановлением с использованием водорода или формиата аммония и катализатора на основе металла VIII Группы, такого как палладий. Когда W представляет собой NWa, гидрирование обычно проводят с использованием катализатора Перлмана (Pd(OH)2).
Соединения формулы X могут быть получены взаимодействием изоцианата формулы XI:
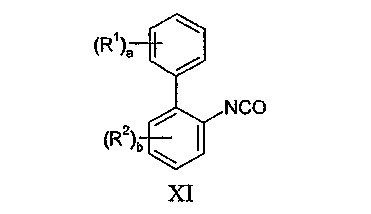
с соединением формулы XII:
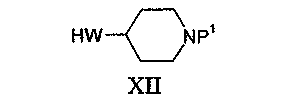
Соединения формулы III могут быть получены, исходя из соответствующего соединения, в котором Z1 представляет собой гидроксильную группу, например, при реакции с галогенирующим агентом, таким как хлористый тионил, давая соединение формулы III, в котором Z1 представляет собой галоген, такой как хлор. Соединения, в которых Z1 представляет собой гидроксильную группу, могут быть получены, например, взаимодействием соединения формулы V с подходящим аминозамещенным спиртом, таким как 2-аминоэтанол или 3-аминопропан-1-ол.
В способе (b) соединение формулы IV подвергают взаимодействию с соединением формулы V или его реакционноспособным производным. Под выражением «реакционноспособное производное» соединения V подразумевается, что карбоновая кислота является активированной, например, путем образования ангидрида или галогенангидрида карбоновой кислоты, такого как хлорангидрид карбоновой кислоты. Альтернативно карбоновая кислота может быть активирована с использованием обычных конденсирующих агентов карбоновая кислота/амин, таких как карбодиимиды, гексафторфосфат O-(7-азабензoтриaзол-1-ил-N,N,N',N'-тетраметилурония (HATU) и тому подобные. Реакцию обычно проводят в обычных условиях образования амидной связи. Способ обычно осуществляют при температуре в диапазоне от -10 до 100°C.
Соединения формулы IV могут быть получены взаимодействием соединения формулы II с соединением формулы XIII:
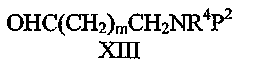
где P2 представляет собой водород или защитную группу аминогруппы, такую как бензил, в присутствии восстановителя, такого как триацетоксиборгидрид натрия с последующим удалением, при необходимости, защитной группы аминогруппы P2, например, гидрированием в присутствии палладия.
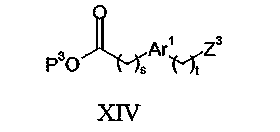
Соединения формулы V могут быть получены взаимодействием соединения формулы VII с соединением формулы XIV:
где P3 представляет собой водород или защитную группу карбоксила, такую как метил или этил, и Z3 представляет собой уходящую группу с последующим удалением, при необходимости, защитной группы карбоксила P3. Альтернативно такие соединения могут быть получены восстановительным аминированием соединения формулы XV:
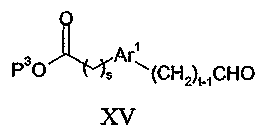
в присутствии соединения формулы VII в обычных условиях реакции, таких как описанные для способов (d) и (e).
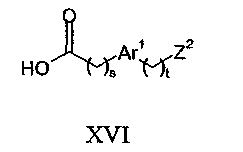
Что касается способа (с), уходящая группа, представленная Z2, может представлять собой, например, галоген, такой как хлор, бром или йод, или сульфоновую сложноэфирную группу, такую как мезилат или тозилат. Данную реакцию обычно проводят в присутствии основания, например, третичного амина, такого как диизопропилэтиламин. Обычные растворители включают нитрилы, такие как ацетонитрил. Реакцию обычно проводят при температуре в диапазоне от 0 до 100°C. Соединения формулы VI могут быть получены взаимодействием соединения формулы IV с соединением формулы XVI:
или его реакционноспособным производным, таким как хлорангидрид или ангидрид кислоты. Взаимодействие обычно проводят в соответствии с методом осуществления способа (b), приведенным в данном описании. Соединения формулы VII обычно представляют собой известные соединения или могут быть получены из легко доступных исходных веществ с использованием хорошо известных методов синтеза.
В способе (d) восстановитель может представлять собой, например, водород в присутствии катализатора на основе металла VIII Группы, такого как палладий, или восстановитель типа гидрида металла, такой как боргидрид, включая триацетоксиборгидрид натрия. Обычные растворители включают спирты, такие как метанол. Реакцию обычно проводят при температуре в диапазоне от 0 до 100°C.
Соединения формулы VIII могут быть получены окислением соединения, соответствующего формуле III, в котором Z1 представляет собой гидроксильную группу. Такие реакции окисления могут быть проведены с использованием, например, комплекса диоксида серы с пиридином в диметилсульфоксиде в присутствии третичного амина, такого как диизопропилэтиламин.
В способе (e) восстановитель может представлять собой, например, водород в присутствии катализатора на основе металла VIII Группы, такого как палладий, или восстановитель типа гидрида металла, включая боргидриды, такие как триацетоксиборгидрид натрия, необязательно используемый в сочетании с тетраалкоксидом титана, таким как тетраизопропоксид титана. Обычные растворители включают спирты, такие как метанол и галогенированные углеводороды, такие как дихлорметан. Реакцию обычно проводят при температуре в диапазоне от 0 до 100°C. Соединения формулы IX могут быть получены взаимодействием соединения формулы IV с соединением формулы XVII:
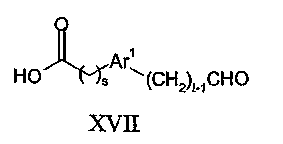
в присутствии конденсирующего агента карбоновая кислота/амин, такого как 1-(3-диметиламинопропил)-3-этилкарбодиимид (EDC) и гидрат 1-гидроксибензoтриазола (HOBT) и тому подобные.
Относительно способа (f), соединения формулы XVIII могут быть получены взаимодействием соединения формулы IX с соединением формулы VII в присутствии восстановителя, такого как триацетоксиборгидрид натрия, аналогично тому, как это проводят в способе (e).
Как будет очевидно специалистам в данной области, соединения формулы I, полученные в соответствии с указанными здесь стадиями (a) - (f), могут быть дополнительно превращены в другие производные с образованием других соединений формулы I с использованием способов и реагентов, хорошо известных в данной области. В качестве иллюстрации, соединение формулы I можно подвергнуть взаимодействию с бромом с получением соответствующего соединения формулы I, в котором R2, например, представляет собой группу брома. Дополнительно, соединения формулы I, в которых R4 представляет собой атом водорода, могут быть проалкилированы с получением соответствующего соединения формулы I, в котором
R4 представляет собой (1-4C) алкильную группу.
Предполагается, что некоторые из описанных здесь промежуточных соединений являются новыми, и, соответственно, такие соединения представляют собой дополнительные аспекты изобретения, включая, например, соединения формулы III, V и VIII и их соли.
Дополнительные подробности, касающиеся конкретных реакционных условий и других способов получения иллюстративных соединений данного изобретения или промежуточных продуктов их синтеза, описаны в приведенных далее примерах.
Фармацевтические композиции и препараты
Бифенильные соединения данного изобретения обычно вводят пациенту в виде фармацевтической композиции или препарата. Такие фармацевтические композиции могут вводиться пациенту с использованием любого подходящего пути введения, включая, но не ограничиваясь указанным, ингаляции, оральный, назальный, наружный (включая чрескожный) и парентеральный способы введения.
Следует понимать, что в обсуждающихся в данном описании фармацевтических композициях можно использовать любую форму соединения данного изобретения (т.e. свободное основание, фармацевтически приемлемую соль, сольват и т.д.), которая является подходящей для конкретного способа введения.
Соответственно, в одном из составляющих его аспектов, данное изобретение относится к фармацевтической композиции, включающей фармацевтически приемлемый носитель или эксципиент и терапевтически эффективное количество соединения формулы I, или его фармацевтически приемлемой соли, или сольвата, или стереоизомера. Такие фармацевтические композиции, при желании, могут содержать другие терапевтические агенты и/или агенты для получения препаратов.
Фармацевтические композиции данного изобретения обычно содержат терапевтически эффективное количество соединения настоящего изобретения или его фармацевтически приемлемой соли, или сольвата, или стереоизомера. Обычно такие фармацевтические композиции будут содержать примерно от 0,01 до примерно 95% по весу активного агента; включая примерно от 0,01 до примерно 30% по весу, как, например, примерно от 0,01 до примерно 10% по весу активного агента.
В фармацевтических композициях данного изобретения можно использовать любой обычный носитель или эксципиент. Выбор конкретного носителя или эксципиента, или комбинации носителей или эксципиентов, будет зависеть от способа введения, используемого для лечения конкретного пациента, или типа медицинского состояния, или состояния заболевания. В данном контексте, получение подходящей фармацевтической композиции для конкретного способа введения входит в объем компетенции специалистов в области фармацевтики. Кроме того, ингредиенты для таких композиций являются коммерчески доступными, например, от Sigma, P.O. Box 14508, St. Louis, MO 63178. В качестве дополнительной иллюстрации, обычные методы получения препаратов описаны в Remington: The Science и Practice of Pharmacy, 20th Edition, Lippincott Williams & White, Baltimore, Maryland (2000); и H.C. Ansel et al., Pharmaceutical Dosage Forms и Drug Delivery Systems, 7th Edition, Lippincott Williams & White, Baltimore, Maryland (1999).
Иллюстративные примеры веществ, которые могут служить в качестве фармацевтически приемлемых носителей, включают, но не ограничиваются указанным, следующие: (1) сахара, такие как лактоза, глюкоза и сахароза; (2) крахмалы, такие как кукурузный крахмал и картофельный крахмал; (3) целлюлоза и ее производные, такие как карбоксиметилцеллюлоза натрия, этилцеллюлоза и ацетат целлюлозы; (4) порошкообразная трагакантовая камедь; (5) солод; (6) желатин; (7) тальк; (8) эксципиенты, такие как масло какао и воски для суппозиториев; (9) масла, такие как арахисовое масло, хлопковое масло, сафлоровое масло, кунжутное масло, оливковое масло, кукурузное масло и соевое масло; (10) гликоли, такие как пропиленгликоль; (11) полиатомные спирты, такие как глицерин, сорбит, маннит и полиэтиленгликоль; (12) сложные эфиры, такие как этилолеат и этиллаурат; (13) агар; (14) буферные агенты, такие как гидроксид магния и гидроксид алюминия; (15) альгиновую кислоту; (16) апирогенную воду; (17) изотонический физиологический раствор; (18) раствор Рингера; (19) этиловый спирт; (20) фосфатные буферные растворы; (21) сжатые газообразные пропелленты, такие как хлорфторуглероды и гидрофторуглероды; и (22) другие нетоксичные совместимые вещества, используемые в фармацевтических композициях.
Фармацевтические композиции данного изобретения обычно получают путем тщательного и равномерного перемешивания или смешивания соединения по изобретению с фармацевтически приемлемым носителем и одним или несколькими необязательными ингредиентами. При необходимости или при желании, полученная равномерно перемешанная смесь может быть сформована или загружена в таблетки, капсулы, пилюли, емкости, картриджи, дозаторы и тому подобное с использованием обычных способов и оборудования.
В одном варианте осуществления фармацевтические композиции по данному изобретению являются подходящими для введения путем ингаляции. Подходящие фармацевтические композиции для введения путем ингаляции обычно будут находиться в виде аэрозоля или порошка. Такие композиции обычно вводят с использованием хорошо известных устройств доставки, таких как распыляющий ингалятор (небулайзер), ингалятор с нормируемым дозированием (MDI), ингалятор сухих порошков или аналогичных устройств доставки.
В конкретном варианте осуществления данного изобретения фармацевтическую композицию, содержащую активный агент, вводят путем ингаляции с использованием распыляющего ингалятора (небулайзера). Такой небулайзер обычно дает поток воздуха высокой скорости, который вызывает распыление фармацевтической композиции, включающей активный агент, в виде тумана, который переносится в дыхательные пути пациента. Соответственно, когда агент вводят в состав композиции для применения в небулайзере, активный агент обычно растворяют в подходящем носителе для образования раствора. Альтернативно активный агент может быть мелко измельчен и объединен с подходящим носителем с образованием суспензии тонкодисперсных частиц пригодного для вдыхания размера, при этом термин «мелко измельчен» определяют как получение примерно 90% или более частиц с диаметром менее примерно 10 мкм. Подходящие устройства для распыления (небулайзеры) доступны коммерчески, например, от PARI GmbH (Starnberg, German). Другие небулайзеры включают Respimat (Boehringer Ingelheim) и устройства, описанные, например, в патенте США No. 6123068, выданном Lloyd et al., и WO 97/12687 (Eicher et al.).
Иллюстративная фармацевтическая композиция для применения в небулайзере включает изотонический водный раствор, включающий примерно от 0,05 мкг/мл до примерно 10 мг/мл соединения формулы I или его фармацевтически приемлемой соли, или сольвата, или стереоизомера.
В другом конкретном варианте осуществления данного изобретения фармацевтическую композицию, содержащую активный агент, вводят путем ингаляции с использованием ингалятора для сухого порошка. Такие ингаляторы для сухих порошков обычно вводят активный агент в виде свободно текущего порошка, который распределяется в потоке воздуха, вдыхаемом пациентом. Для получения свободно текущего порошка активный агент обычно вводят в состав вместе с подходящим эксципиентом, таким как лактоза или крахмал.
Иллюстративная фармацевтическая композиция для применения в ингаляторе для сухого порошка включает сухую лактозу, имеющую размер частиц между примерно 1 мкм и примерно 100 мкм и тонко измельченные частицы соединения формулы I, или его фармацевтически приемлемой соли, или сольвата, или стереоизомера.
Такие препараты сухих порошков могут быть получены, например, путем объединения лактозы с активным ингредиентом с последующим сухим смешиванием компонентов. Альтернативно, при желании, активный агент можно использовать в препарате без эксципиента. Фармацевтическую композицию затем обычно загружают в дозатор сухого порошка, или в картриджи для ингаляции, или в капсулы для использования в устройствах для доставки сухих порошков.
Примеры устройств ингаляторов для сухих порошков включают Diskhaler (GlaxoSmithKline, Research Triangle Park, NC) (см., например, патент США No. 5035237, выданный Newell et al.); Diskus (GlaxoSmithKline) (см., например, патент США No. 6378519, выданный Davies et al.); Turbuhaler (AstraZeneca, Wilmington, DE) (см., например, патент США No. 4524769, выданный Wetterlin); Rotahaler (GlaxoSmithKline) (см., например, патент США No. 4353365, выданный Hallworth et al.) и Handihaler (Boehringer Ingelheim). Дополнительные примеры подходящих устройств DPI описаны в патентах США № 5415162, выданном Casper et al., № 5239993, выданном Evans, и № 5715810, выданном Armstrong et al., и процитированных там ссылках.
Еще в одном конкретном варианте осуществления данного изобретения фармацевтическую композицию, содержащую активный агент, вводят путем ингаляции с использованием ингалятора с нормируемым дозированием. Такие ингаляторы с нормируемым дозированием обычно выпускают измеренное количество активного агента или его фармацевтически приемлемой соли, или сольвата, или стереоизомера с использованием сжатого газа-пропеллента. Соответственно, фармацевтические композиции, вводимые с использованием ингалятора с нормируемым дозированием, обычно включают раствор или суспензию активного агента в сжиженном пропелленте. Можно использовать любой подходящий сжиженный пропеллент, включая хлорфторуглероды, такие как CCl3F, и гидрофторалканы (HFAs), такие как 1,1,1,2-тетрафтоэтан (HFA 134a) и 1,1,1,2,3,3,3-гептафтор-н-пропан, (HFA 227). В связи с озабоченностью тем фактом, что хлорфторуглероды поражают озоновый слой, обычно предпочтительными являются препараты, содержащие HFAs. Дополнительные необязательные компоненты препаратов HFA включают сорастворители, такие как этанол или пентан и поверхностно-активные вещества, такие как триолеат сорбита, олеиновая кислота, лецитин и глицерин. См., например, патент США 5225183, выданный Purewal et al., EP 0717987 A2 (Minnesota Mining and Manufacturing Company), и WO 92/22286 (Minnesota Mining and Manufacturing Company).
Иллюстративная фармацевтическая композиция для применения в ингаляторе с нормируемым дозированием включает примерно от 0,01 % до примерно 5 % по весу соединения формулы I, или его фармацевтически приемлемой соли, или сольвата, или стереоизомера; примерно от 0 % до примерно 20 % по весу этанола; и примерно от 0 % до примерно 5 % по весу поверхностно-активного вещества, при этом оставшуюся часть составляет HFA пропеллент.
Такие композиции обычно получают добавлением охлажденного или сжатого гидрофторалкана в подходящий контейнер, содержащий активный агент, этанол (если присутствует) и поверхностно-активное вещество (если присутствует). Для получения суспензии активный агент мелко измельчают и затем объединяют с пропеллентом. Препарат затем загружают в аэрозольный баллончик, который составляет часть устройства ингалятора с нормируемым дозированием. Пример устройств ингаляторов с нормируемым дозированием, разработанных специально для использования HFA пропеллентов, приведены в патентах США 6006745, выданном Marecki, и 6143277, выданном Ashurst et al. Альтернативно препарат суспензии может быть получен путем сушки распылением нанесенного в виде покрытия на мелкоизмельченные частицы активного агента поверхностно-активного вещества. См., например, WO 99/53901 (Glaxo Group Ltd.) и WO 00/61108 (Glaxo Group Ltd.).
Дополнительные примеры способов получения приспособленных для вдыхания частиц, и препаратов, и устройств, подходящих для дозирования при ингаляции, приведены в патентах США № 6268533, выданном Gao et al., № 5983956, выданном Trofast, № 5874063, выданном Briggner et al., и № 6221398, выданном Jakupovic et al.; и WO 99/55319 (Glaxo Group Ltd.) и WO 00/30614 (AstraZeneca AB).
В другом варианте осуществления фармацевтические композиции данного изобретения являются подходящими для перорального введения. Подходящие фармацевтические композиции для перорального введения могут быть в виде капсул, таблеток, пилюль, пастилок, саше, драже, порошков, гранул, или в виде раствора, или суспензии в водной или неводной жидкости; или в виде жидкой эмульсии масло-в-воде или вода-в-масле; или в виде эликсира или сиропа и тому подобного; где каждая из форм содержит определенное количество соединения по настоящему изобретению в качестве активного ингредиента.
В том случае если они предназначены для перорального введения в виде твердой препаративной лекарственной формы (т.е. капсул, таблеток, пилюль и тому подобного), фармацевтические композиции данного изобретения обычно будут включать соединение по настоящему изобретению в качестве активного ингредиента и один или несколько фармацевтически приемлемых носителей, таких как цитрат натрия или дикальцийфосфат. Необязательно или альтернативно такие твердые препаративные лекарственные формы также могут включать: (1) наполнители или наполняющие агенты, такие как крахмалы, лактоза, сахароза, глюкоза, манит и/или кремневая кислота; (2) связующие вещества, такие как карбоксиметилцеллюлоза, альгинаты, желатин поливинилпирролидон, сахароза и/или аравийская камедь; (3) увлажнители, такие как глицерин; (4) дезинтегрирующие агенты, такие как агар-агар, карбонат кальция, картофельный или тапиоковый крахмал, альгиновая кислота, некоторые силикаты и/или карбонат натрия; (5) замедляющие растворение агенты, такие как парафин; (6) ускорители поглощения, такие как четвертичные аммониевые соединения; (7) смачивающие агенты, такие как цетиловый спирт и/или моностеарат глицерина; (8) абсорбенты, такие как каолин и/или бентонитовая глина; (9) смазочные вещества, такие как тальк, стеарат кальция, стеарат магния, твердые полиэтиленгликоли, лаурилсульфат натрия и/или их смеси; (10) красители и (11) буферные агенты.
В фармацевтических композициях данного изобретения также могут присутствовать агенты высвобождения, смачивающие агенты, агенты оболочки, подсластители, вкусовые добавки и ароматизаторы, консерванты и антиоксиданты. Примеры фармацевтически приемлемых антиоксидантов включают: (1) растворимые в воде антиоксиданты, такие как аскорбиновая кислота, гидрохлорид цистеина, бисульфат натрия, метабисульфат натрия, сульфит натрия и тому подобные; (2) растворимые в масле антиоксиданты, такие как аскорбилпальмитат, бутилированный гидроксианизол (BHA), бутилированный гидрокситолуол (BHT), лецитин, пропилгаллат, альфа-токоферол и тому подобные; и (3) агенты, образующие комплексы с металлами, такие как лимонная кислота, этилендиаминтетрауксусная кислота (EDTA), сорбит, винная кислота, фосфорная кислота и тому подобные. Образующие оболочку агенты для таблеток, капсул, пилюль и тому подобного включают те, которые используются для получения оболочек, растворимых в кишечнике, такие как ацетатфталат целлюлозы, поливинилацетатфталат (PVAP), фталат гидроксипропилметилцеллюлозы, сополимеры метакриловой кислоты со сложными эфирами метакриловой кислоты, ацетаттримеллитат целлюлозы (CAT), карбоксиметилэтилцеллюлоза (CMEC), ацетатсукцинат гидроксипропилметилцеллюлозы (HPMCAS), и тому подобные.
При желании фармацевтические композиции по настоящему изобретению также могут быть получены в виде препаратов для обеспечения замедленного или контролируемого высвобождения активного ингредиента с использованием, в качестве примера, гидроксипропил метилцелюлозы в различных пропорциях или других полимерных матриц, липосомов и/или микросфер.
Дополнительно, фармацевтические композиции по настоящему изобретению могут необязательно содержать контрастные агенты и могут быть составлены таким образом, что они будут высвобождать только активный ингредиент только или главным образом в определенной части желудочно-кишечного тракта, необязательно замедленным образом. Примеры встроенных композиций, которые можно использовать, включают полимерные вещества и воски. Активный ингредиент также может находиться в микроинкапсулированном виде, если это является подходящим, вместе с одним или несколькими вышеуказанными эксципиентами.
Подходящие жидкие препаративные лекарственные формы для перорального введения включают, в качестве иллюстрации, фармацевтически приемлемые эмульсии, микроэмульсии, растворы, суспензии, сиропы и эликсиры. Такие жидкие препаративные лекарственные формы обычно содержат активный ингредиент и инертный разбавитель, такой как, например, вода или другие растворители, солюбилизирующие агенты и эмульгаторы, такие как этиловый спирт, изопропиловый спирт, этилкарбонат, этилацетат бензиловый спирт бензилбензoат, пропиленгликоль, 1,3-бутиленгликоль, масла (например, хлопковое, арахисовое, кукурузное, зародышевое, оливковое, касторовое и кунжутное масла), глицерин, тетрагидрофуриловый спирт, полиэтиленгликоли и сложные эфиры жирных кислот и сорбитана и их смеси. Суспензии в дополнение к активному ингредиенту могут содержать суспендирующие агенты, такие как например, этоксилированные изостеариловые спирты, полиоксиэтиленовые сложные эфиры сорбита и сорбитана, микрокристаллическую целлюлозу, метагидроксид алюминия, бентонит, агар-агар и трагакантовую камедь и их смеси.
В том случае если они предназначены для перорального введения, фармацевтические композиции данного изобретения предпочтительно упаковывают в виде единичной дозированной формы. Термин «единичная дозированная форма» означает физически дискретную единицу, подходящую для дозирования пациенту, т.е. каждая единица содержит предварительно определенное количество активного ингредиента, рассчитанное для оказания желательного терапевтического действия или по отдельности или в сочетании с одной или несколькими дополнительными единицами. Например, такая единичная дозированная форма может представлять собой капсулы, таблетки, пилюли и тому подобное.
Соединения данного изобретения также можно вводить чрескожно с использованием известных систем чрескожной доставки. Например, соединение данного изобретения может быть смешано с усилителями проницаемости, такими как пропиленгликоль, монолаурат полиэтиленгликоля, азациклоалкaн-2-оны и тому подобные, и введено в пластырь или аналогичную систему доставки. При желании, в таких чрескожных композициях можно использовать дополнительные эксципиенты, включая гелеобразующие агенты, эмульгаторы и буферы.
Фармацевтические композиции данного изобретения также могут содержать другие терапевтические агенты, которые вводят совместно с соединением формулы I, или его фармацевтически приемлемой солью, или сольватом, или стереоизомером. Например, фармацевтические композиции данного изобретения могут дополнительно включать один или несколько терапевтических агентов, выбранных из других бронходилататоров (например, ингибиторов PDE3, модуляторов аденозина 2b и агонистов β2 адренергического рецепторов), противовоспалительных агентов (например, стероидных противовоспалительных агентов, таких как кортикостероиды; нестероидных противовоспалительных средств (НСПВС) и ингибиторов PDE4); других антагонистов мускариновых рецепторов (например, антихолинергических агентов); противоинфекционных агентов (например, грамположительных и грамотрицательных антибиотиков и противовирусных агентов); антигистаминов; ингибиторов протеазы и иммунорегуляторных блокаторов (например, агонистов D2 и модуляторов нейрокинина). В одном конкретном аспекте изобретения соединение по изобретению вводят совместно с агонистом β2 адренергического рецептора и стероидным противовоспалительным агентом. Другие терапевтические агенты можно использовать в виде фармацевтически приемлемых солей или сольватов. Кроме того, если это является подходящим, другие терапевтические агенты можно использовать в виде оптически чистых стереоизомеров.
Иллюстративные агонисты β2 адренергического рецептора, которые можно использовать в сочетании с соединениями данного изобретения, включают, но не ограничиваются указанным, салметерол, салбутамол, формотерол, салмефамол, фенотерол, тербуталин, албутерол, изоэтарин, метапротеренол, битолтерол, пирбутерол, лавалбутерол и тому подобные или их фармацевтически приемлемые соли. Другие агонисты β2 адренергического рецептора, которые можно использовать в сочетании с соединениями данного изобретения, включают, но не ограничиваются указанным, 3-(4-{[6-({(2R)-2-гидрокси-2-[4-гидрокси-3-(гидроксиметил)-фенил]этил}амино)гексил]окси}бутил)бензолсульфонамид и 3-(-3-{[7-({(2R)-2-гидрокси-2-[4-гидрокси-3-(гидроксиметил)фенил]-этил}амино)гептил]окси}пропил)бензолсульфонамид и родственные соединения, описанные в WO 02/066422 (Glaxo Group Ltd.); 3-[3-(4-{[6-([(2R)-2-гидрокси-2-[4-гидрокси-3-(гидроксиметил)фенил]-этил}амино)гексил]окси}бутил)фенил]имидазолидин-2,4-дион и родственные соединения, описанные в WO 02/070490 (Glaxo Group Ltd.); 3-(4-{[6-({(2R)-2-[3-(формиламино)-4-гидроксифенил]-2-гидроксиэтил}амино)гексил]окси}бутил)бензолсульфонамид, 3-(4-{[6-({(2S)-2-[3-(формиламино)-4-гидроксифенил]-2-гидроксиэтил}амино)гексил]окси}бутил)бензолсульфонамид, 3-(4-{[6-({(2R/S)-2-[3-(формиламино)-4-гидроксифенил]-2-гидроксиэтил}амино)гексил]окси}бутил)бензолсульфонамид, N-(трет-бутил)-3-(4-{[6-({(2R)-2-[3-(формиламино)-4-гидроксифенил]-2-гидроксиэтил}амино)гексил]окси}бутил)бензолсульфонамид, N-(трет-бутил)-3-(4-{[6-({(2S)-2-[3-(формиламино)-4-гидроксифенил]-2-гидроксиэтил}амино)гексил]окси}бутил)бензолсульфонамид, N-(трет-бутил)-3-(4-{[6-({(2R/S)-2-[3-(формиламино)-4-гидроксифенил]-2-гидроксиэтил}амино)гексил]окси}бутил)бензолсульфонамид и родственные соединения, описанные в WO 02/076933 (Glaxo Group Ltd.); 4-{(1R)-2-[(6-{2-[(2,6-дихлорбензил)окси]этокси}гексил)амино]-1-гидроксиэтил}-2-(гидроксиметил)фенол и родственные соединения, описанные в WO 03/024439 (Glaxo Group Ltd.); N-{2-[4-((R)-2-гидрокси-2-фенилэтиламино)фенил]этил}-(R)-2-гидрокси-2-(3-формамидо-4-гидроксифенил)этиламин и родственные соединения, описанные в патенте США No. 6576793, выданном Moran et al.; N-{2-[4-(3-фенил-4-метоксифенил)аминофенил]этил}-(R)-2-гидрокси-2-(8-гидрокси-2(1H)-[хинолинoн-5-ил)этиламин и родственные соединения, описанные в патенте США No. 6653323, выданном Moran et al.; и их фармацевтически приемлемые соли. В конкретном варианте осуществления агонист β2-адренорецептора представляет собой кристаллическую моногидрохлоридную соль N-{2-[4-((R)-2-гидрокси-2-фенилэтиламино)фенил]этил}-(R)-2-гидрокси-2-(3-формамидо-4-гидроксифенил)этиламина. Если он используется, агонист β2-адренорецептора будет присутствовать в фармацевтической композиции в терапевтически эффективном количестве. Обычно агонист β2-адренорецептора будет присутствовать в количестве, достаточном для обеспечения примерно от 0,05 мкг до примерно 500 мкг на дозу.
Иллюстративные стероидные противовоспалительные агенты, которые можно использовать в сочетании с соединениями данного изобретения, включают, но не ограничиваются указанным, метилпреднизолон, преднизолон, дексаметазон, флутиказон пропионат, S-фторметиловый эфир 6α,9α-дифтор-17α-[(2-фуранилкарбонил)окси]-11β-гидрокси-16α-метил-3-оксоандроста-1,4-диен-17β-карботиовой кислоты, S-(2-оксо-тетрагидрофуран-3S-иловый) эфир 6α,9α-дифтор-11β-гидрокси-16α-метил-3-oxo-17α-пропионилокси-андроста-1,4-диен-17β-карботиовой кислоты, сложные эфиры беклометазона (например, 17-пропионатный сложный эфир или 17,21-дипропионатный сложный эфир), будезонид, флунизолид, сложные эфиры мометазона (например, фуроатный сложный эфир), триамцинолон ацетонид, рофлепонид, циклезонид, бутиксокорт пропионат, RPR-106541, ST-126 и тому подобные или их фармацевтически приемлемые соли. При использовании стероидный противовоспалительный агент будет присутствовать в фармацевтической композиции в терапевтически эффективном количестве. Обычно, стероидный противовоспалительный агент будет присутствовать в количестве, достаточном для обеспечения примерно от 0,05 мкг до примерно 500 мкг на дозу.
Иллюстративная комбинация представляет собой соединение формулы I или его фармацевтически приемлемую соль, или сольват, или стереоизомер, вводимое совместно с салметеролом в качестве агониста β2 адренергического рецептора и пропионатом флутиказона в качестве стероидного противовоспалительного агента. Другая иллюстративная комбинация представляет собой соединение формулы I или его фармацевтически приемлемую соль, или сольват, или стереоизомер, вводимое совместно с кристаллической моногидрохлоридной солью N-{2-[4-((R)-2-гидрокси-2-фенилэтиламино)фенил]этил}-(R)-2-гидрокси-2-(3-формамидо-4-гидроксифенил)этиламина в качестве агониста β2 адренергического рецептора и S-фторметиловым эфиром 6α,9α-дифтор-17α-[(2-фуранилкарбонил)окси]-11β-гидрокси-16α-метил-3-оксоандроста-1,4-диен-17β-карботиовой кислоты в качестве стероидного противовоспалительного агента.
Другие подходящие комбинации включают, например, другие противовоспалительные агенты, например, нестероидные противовоспалительные лекарственные средства (НСПВС) (такие как хромогликат натрия, недокромил натрия; ингибиторы фосфордиэстеразы (PDE) (например, теофилин, ингибиторы PDE4 или смешанные ингибиторы PDE3/PDE4); антагонисты лейкотриена (например, монтелейкаст); ингибиторы синтеза лейкотриена; ингибиторы iNOS; ингибиторы протеазы, такие как ингибиторы триптазы и эластазы; антагонисты бета-2-интегрина и агонисты или антагонисты аденозинового рецептора (например, агонисты аденозин 2а); антагонисты цитокина (например, антагонисты хемокина, такие как антитело против интерлейкина (αIL антитело), в частности, αIL-4 терапия, αIL-13 терапия или их комбинация); или ингибиторы синтеза цитокина.
Например, иллюстративные ингибиторы фосфодиэстеразы-4 (PDE4) или смешанные ингибиторы PDE3/PDE4, которые можно использовать в сочетании с соединениями данного изобретения, включают, но не ограничиваются указанным, цис 4-циано-4-(3-циклопентилокси-4-метоксифенил)циклогексан-1-карбоновую кислоту, 2-карбометокси-4-циано-4-(3-циклопропилметокси-4-дифторметоксифенил)циклогексан-1-он; цис-[4-циано-4-(3-циклопропилметокси-4-дифторметоксифенил)циклогексан-1-ол]; цис-4-циано-4-[3-(циклопентилокси)-4-метоксифенил]циклогексан-1-карбоновую кислоту и тому подобные или их фармацевтически приемлемые соли. Другие иллюстративные ингибиторы PDE4 или смешанные ингибиторы PDE4/PDE3 включают AWD-12-281 (элбион); NCS-613 (INSERM); D-4418 (Chiroscience и Schering-Plough); CI-1018 или PD-168787 (Pfizer); соединения бензодиоксола, описанные в WO99/16766 (Kyowa Hakko); K-34 (Kyowa Hakko); V-11294A (Napp); рофлумиласт (Byk-Gulden); соединения фталазинона, описанные в WO99/47505 (Byk-Gulden); Pumafentrine (Byk-Gulden, в настоящее время Altana); арофиллин (Almirall-Prodesfarma); VM554/UM565 (Vernalis); T-440 (Tanabe Seiyaku); и T2585 (Tanabe Seiyaku).
Иллюстративные антагонисты мускаринового рецептора (т.е. антихолинергические агенты), которые можно использовать в сочетании и в дополнение к соединениям данного изобретения, включают, но не ограничиваются указанным, атропин, сульфат атропина, оксид атропина, нитрат метилатропина, гидробромид гоматропина, гидробромид гиосциамина (d,l), гидробромид скополамина, бромид ипратропия, бромид окситропия, бромид тиотропия, метантелин, бромид пропантелина, метилбромид анизотропина, бромид клидиния, копирролят (Робинул (Robinul)), йодид изопропамида, бромид мепензолята, хлорид тридигексетила (Патилон (Pathilone)), метилсульфат гексоциклия, гидрохлорид циклопентолят, тропикамид, гидрохлорид тригексифенидила, пирензепин, телензепин, AF-DX 116 и метоктрамин и тому подобные или их фармацевтически приемлемые соли; или, для тех соединений, которые перечислены в виде соли, их альтернативные фармацевтически приемлемые соли.
Иллюстративные антигистамины (т.е. антагонисты H1-рецептора), которые можно использовать в сочетании с соединениями данного изобретения, включают, но не ограничиваются указанным, этаноламины, такие как малеат карбиноксамина, фумарат клемастина, гидрохлорид дифенилнидрамина и дименгидринат; этилендиамины, такие как малеат пириламина, гидрохлорид трипеленнамина и цитрат трипеленнамина; алкиламины, такие как хлорфенирамин и акривастин; пиперазины, такие как гидрохлорид гидроксизина, памоат гидроксизина, гидрохлорид циклизина, лактат циклизина, гидрохлорид меклизина и гидрохлорид цетиризина; пиперидины, такие как астемизол, гидрохлорид левокабастина, лоратидин и его дезкарбэтоксильный аналог, терфенадин и гидрохлорид фексофенадина; гидрохлорид азеластина и тому подобные, или их фармацевтически приемлемые соли; или, для тех соединений, которые перечислены в виде соли, их альтернативные фармацевтически приемлемые соли.
Подходящие дозировки для других терапевтических агентов, вводимых в сочетании с соединением по изобретению, находятся в диапазоне от примерно 0,05 мкг/день до примерно 100 мг/день.
Следующие составы показывают иллюстративные фармацевтические композиции по настоящему изобретению.
Пример состава A
Сухой порошок для введения путем ингаляции получают следующим образом.
Иллюстративный способ: Соединение по изобретению мелко измельчают и затем смешивают с лактозой. Данную перемешанную смесь затем загружают в желатиновый картридж для ингаляции. Содержимое картриджа вводят с использованием порошкового ингалятора.
Пример состава B
Препарат в виде сухого порошка для применения в ингаляторе для сухих порошков получают следующим образом.
Иллюстративный способ: Получают фармацевтическую композицию, имеющую объемное соотношение по составу тонко измельченного соединения по изобретению к лактозе, равное 1:200. Композицию упаковывают в ингалятор для сухих порошков, способный доставлять примерно от 10 мкг до примерно 100 мкг соединения по изобретению на дозу.
Пример состава C
Сухой порошок для введения путем ингаляции с помощью ингалятора с нормируемым дозированием получают следующим образом.
Иллюстративный способ: Суспензию, содержащую 5 вес.% соединения по изобретению и 0,1 вес.% лецитина, получают путем распределения 10 г соединения по изобретению в виде тонко измельченных частиц со средним размером менее примерно 10 мкм в растворе, образованном при растворении 0,2 г лецитина в 200 мл деминерализованной воды. Суспензию сушили распылением, и полученное вещество измельчали, получая частицы, имеющие средний диаметр менее чем 1,5 мкм. Частицы загружали в картриджи со сжатым 1,1,1,2-тетрафторэтаном.
Пример состава D
Фармацевтическую композицию для применения в ингаляторе с нормируемым дозированием получают следующим образом.
Иллюстративный способ: Суспензию, содержащую 5 вес.% соединения по изобретению, 0,5 вес.% лецитина и 0,5 вес.% трегалозы, получают путем распределения 5 г активного ингредиента в виде тонко измельченных частиц со средним размером менее примерно 10 мкм в коллоидном растворе, образованном при растворении 0,5 г трегалозы и 0,5 г лецитина в 100 мл деминерализованной воды. Суспензию сушили распылением, и полученное вещество измельчали, получая частицы, имеющие средний диаметр менее чем 1,5 мкм. Частицы загружали в картриджи со сжатым 1,1,1,2-тетрафторэтаном.
Пример состава E
Фармацевтическую композицию для применения в небулайзере (ультразвуковой ингалятор) получают следующим образом:
Иллюстративный способ: Водный аэрозольный препарат для применения в небулайзере получают путем растворения 0,1 мг соединения по изобретению в 1 мл
0,9 %-ного раствора хлорида натрия, подкисленного лимонной кислотой. Смесь перемешивают и обрабатывают ультразвуком до растворения активного ингредиента. рН раствора доводят до значения в диапазоне примерно 3-8 путем медленного добавления NaOH.
Пример состава F
Твердые желатиновые капсулы для перорального введения получают следующим образом.
Иллюстративный способ: Ингредиенты тщательно перемешивают и затем загружают в твердые желатиновые капсулы (460 мг композиции на капсулу).
Пример состава G
Суспензию для перорального введения получают следующим образом.
Иллюстративный способ: Ингредиенты смешивают с образованием суспензии, содержащей 100 мг активного ингредиента на 10 мл суспензии.
Пример состава H
Препарат для инъекций получают следующим образом.
Иллюстративный способ: Вышеуказанные ингредиенты смешивают, и pH доводят до 4 ± 0,5 с использованием 0,5 н. HCl или 0,5 н. NaOH.
Применение
Ожидается, что бифенильные соединения по данному изобретению могут использоваться в качестве антагонистов мускариновых рецепторов и, следовательно, предполагается, что такие соединения будут полезными для лечения медицинских состояний, опосредованных мускариновыми рецепторами, т.е. медицинских состояний, которые облегчаются при лечении антагонистом мускаринового рецептора. Такие медицинские состояния включают, в качестве примера, легочные нарушения и заболевания, включая те, которые связаны с обратимой непроходимостью дыхательных путей, такие как хроническое обструктивное заболевание легких (например, хронический и сопровождаемый шумами бронхит и эмфизема), астма, фиброз легких, аллергический ринит, ринорея и тому подобные. Другие медицинские состояния, которые можно лечить с помощью антагонистов мускариновых рецепторов, представляют собой нарушения мочеполовых путей, такие как повышенная активность мочевого пузыря и гиперактивность мышцы-сжимателя и их симптомы; нарушения желудочно-кишечного тракта, такие как синдром воспаленного кишечника, заболевание дивертикула, ахалазия (нарушение способности расслабления гладкомышечных сфинктеров), нарушения, связанные с гиперподвижностью желудочно-кишечного тракта, и диарея; сердечная аритмия, такая как синусная брадикардия; болезнь Паркинсона; нарушения сознания, такие как болезнь Альцгеймера, дисменорея и тому подобные.
В одном варианте осуществления соединения данного изобретения используются для лечения нарушений гладкой мускулатуры у млекопитающих, включая человека и его животных-спутников (например, собаки, кошки и т.д.). Такие нарушения гладкой мускулатуры включают, в качестве иллюстрации, сверхактивный мочевой пузырь, хроническое обструктивное заболевание легких и синдром воспаленного кишечника.
При использовании для лечения нарушений гладкой мускулатуры и других состояний, опосредованных мускариновыми рецепторами, соединения данного изобретения обычно будут вводить перорально, ректально, парентерально или путем ингаляции в виде единственной дневной дозы и множества доз в сутки. Количество активного ингредиента, вводимого с одной дозой, и общее количество, вводимое в сутки, обычно будет определяться лечащим врачом пациента и будет зависеть от таких факторов, как природа и серьезность состояния пациента, состояние, подвергаемое лечению, возраст и общее состояние здоровья пациента, переносимость пациентом активного агента, путь введения и тому подобные.
Обычно, подходящие дозы для лечения нарушений гладкой мускулатуры или других нарушений, опосредованных мускариновыми рецепторами, будут колебаться примерно от 0,14 мкг/кг/день до примерно 7 мг/кг/день активного агента; включая примерно от 0,15 мкг/кг/день до примерно 5 мг/кг/день. Для среднего человека весом 70 кг это будет количество от примерно 10 мкг в день до примерно 500 мг в день активного агента.
В конкретном варианте осуществления соединения по данному изобретению используются для лечения заболеваний легких и дыхательных путей, таких как ХОЗЛ или астма, у млекопитающих, включая человека. При использовании для лечения таких заболеваний соединения по данному изобретению обычно будут вводить путем ингаляции в виде множества доз в день, в виде единичной дневной дозы или в виде единичной недельной дозы. Обычно доза для лечения легочного заболевания будет колебаться примерно от 10 мкг/день до примерно 200 мкг/день. Как использовано в данном описании, ХОЗЛ включает хронический обструктивный бронхит и эмфизему (см., например, Barnes, Chronic Obstructive Pulmonary Disease, N Engl J Med 343:269-78 (2000)).
При использовании для лечения заболевания легких соединения по данному изобретению необязательно вводят в сочетании с другими терапевтическими агентами, такими как агонист β2-адренорецептора, кортикостероид, нестероидное противовоспалительное средство или их комбинация.
При введении путем ингаляции соединения по данному изобретению обычно оказывают действие, вызывающее бронходилатацию. Соответственно, в другом аспекте составляющих его способов, данное изобретение относится к способу продуцирования бронходилатации у пациента, где способ включает введение пациенту вызывающее бронходилатацию количество соединения по изобретению. Обычно терапевтически эффективная доза, вызывающая бронходилатацию, будет колебаться примерно от 10 мкг/день до примерно 200 мкг/день.
В другом варианте осуществления соединения по данному изобретению используют для лечения сверхактивного мочевого пузыря. При использовании для лечения сверхактивного мочевого пузыря соединения по данному изобретению обычно будут вводить перорально в виде единичной дневной дозы или в виде множества доз в день, предпочтительно в виде единичной дневной дозы. Предпочтительно доза для лечения сверхактивного мочевого пузыря будет колебаться примерно от 1,0 до примерно 500 мг/день.
Еще в одном варианте осуществления соединения по данному изобретению используют для лечения синдрома воспаленного кишечника. При использовании для лечения синдрома воспаленного кишечника соединения по данному изобретению будут вводиться орально или ректально либо раз в день, либо несколько раз в день. Предпочтительно доза для лечения синдрома воспаленного кишечника будет колебаться примерно от 1,0 до примерно 500 мг/день.
Поскольку соединения по данному изобретению представляют собой антагонисты мускаринового рецептора, такие соединения также можно использовать в качестве научно-исследовательского инструмента для исследования и изучения биологических систем и образцов, обладающих мускариновыми рецепторами. Такие биологические системы или образцы могут включать мускариновые рецепторы M1, M2, M3, M4 и/или M5. В таких исследованиях, которые можно проводить или in vitro, или in vivo можно использовать любую подходящую биологическую систему или образец, имеющие мускариновые рецепторы. Иллюстративные биологические системы или образцы, подходящие для таких исследований включают, но не ограничиваются указанным, клетки, клеточные экстракты, плазменные мембраны, образцы тканей млекопитающих (таких как мыши, крысы, морские свинки, кролики, собаки, свиньи и т.д.) и тому подобное.
В данном варианте осуществления биологическую систему или образец, содержащий мускариновый рецептор, приводят в контакт с количеством соединения по данному изобретению, вызывающим антагонистическое воздействие на мускариновый рецептор. Эффект антагонистического воздействия на мускариновый рецептор затем определяют с использованием обычных процедур и оборудования, таких как анализ связывания с радиоактивным лигандом и функциональный анализ. Такие функциональные анализы включают лиганд-опосредованные изменения внутриклеточного циклического аденозинмонофосфата (цАМФ), лиганд-опосредованные изменения активности фермента аденилциклазы (который синтезирует цАМФ), лиганд-опосредованные изменения во введении гуанозин 5'-O-(γ-тио)трифосфата ([35S]GTPγS) в выделенные мембраны через катализируемый рецептором обмен [35S]GTPγS на GDP, лиганд-опосредованные изменения в свободных внутриклеточных ионах кальция (измеренных, например, с использованием связанного с флуоресценцией визуализирующего планшетного ридера или FLIPR® от Molecular Devices, Inc.). Соединения данного изобретения будут оказывать антагонистическое действие или уменьшать активацию мускариновых рецепторов в любом из перечисленных выше функциональных анализов или в анализах аналогичной природы. Количество соединения данного изобретения, достаточное для антагонистического воздействия на мускариновый рецептор, обычно будет колебаться примерно от 0,1 наномолярного до примерно 100 наномолярного.
Дополнительно соединения по данному изобретению можно использовать в качестве научно-исследовательских инструментов для открытия новых соединений, которые обладают антагонистической активностью в отношении мускаринового рецептора. В данном варианте осуществления данные по связыванию мускаринового рецептора (например, определенные с помощью анализов вытеснения радиоактивного лиганда in vitro) для тестируемого соединения или группы тестируемых соединений сравнивают с данными по связыванию для соединения данного изобретения, чтобы выявить те тестируемые соединения, которые проявляют примерно равное или превосходящее связывание мускаринового рецептора, если вообще его связывают. Данный аспект изобретения включает, в качестве отдельных вариантов осуществления, как получение данных сравнения (с использованием подходящих анализов), так и анализ данных тестирования для идентификации представляющих интерес тестируемых соединений.
В другом варианте осуществления соединения по данному изобретению используют для антагонистического воздействия на мускариновый рецептор в биологической системе и, в частности, у млекопитающего, такого как мыши, крысы, морские свинки, кролики, собаки, свиньи, люди и так далее. В данном варианте осуществления терапевтически эффективное количество соединения формулы I вводят млекопитающему. Действие, связанное с антагонистическим воздействием на мускариновый рецептор, затем определяют с использованием обычных процедур и оборудования, примеры которого описаны выше.
Было установлено, что наряду с другими свойствами соединения по данному изобретению являются сильными ингибиторами активности M3 мускаринового рецептора. Соответственно, в конкретном варианте осуществления данное изобретение относится к соединениям формулы I, обладающим ингибирующей константой диссоциации (Ki) в отношении подтипа M3 рецептора, меньшей чем или равной 10 нМ; предпочтительно, меньшей чем или равной 5 нМ (как определено, например, с помощью анализа вытеснения радиоактивного лиганда in vitro).
Дополнительно было выявлено, что соединения по данному изобретению обладают удивительной и неожиданной продолжительностью действия. Соответственно в другом конкретном варианте осуществления данное изобретение относится к соединениям формулы I, обладающим продолжительностью действия, превышающей или равной примерно 24 часам.
Более того, было установлено, что при введении путем ингаляции соединения по данному изобретению в эффективных дозах обладают сниженными побочными действиями, такими как сухость во рту, по сравнению с другими известными антагонистами мускариновых рецепторов, вводимыми путем ингаляции (такими как тиотропий).
Данные свойства, также как применение соединений по данному изобретению, могут быть продемонстрированы с использованием различных анализов in vitro и in vivo, хорошо известных специалистам в данной области. Например, иллюстративные анализы дополнительно описаны подробно в следующих примерах.
ПРИМЕРЫ
Следующие получения и примеры иллюстрируют конкретные варианты осуществления данного изобретения. В данных примерах следующие сокращения имеют следующие значения:
AC аденилциклаза
ACh ацетилхолин
ACN ацетонитрил
BSA бычий сывороточный альбумин
цAMФ 3'-5' циклический аденозинмонофосфат
CHO яичник китайского хомячка
cM5 клонированный рецептор M5 шимпанзе
ДХМ дихлорметан (т.е. хлористый метилен)
DIBAL диизобутилалюминийгидрид
DIPEA N,N-диизопропилэтиламин
dPBS забуференный фосфатом физиологический раствор Дульбекко
ДМФ диметилформамид
ДМСО диметилсульфоксид
EDC 1-этил-3-(3-диметиламинопропил)карбодиимид
EDCI гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида
EDTA этилендиаминотетрауксусная кислота
EtOAc этилацетат
EtOH этанол
FBS фетальная бычья сыворотка
FLIPR планшетный ридер флуориметрического отображения
HATU гексафторфосфат O-(7-азабензотриазол-1-ил-N,N,N',N'-тетраметилурония
HBSS сбалансированный солевой раствор Хенка
HEPES 4-(2-гидроксиэтил)-1-пиперазинэтансульфоновая кислота
HOAt 1-гидрокси-7-азабензотриазол
hM1 клонированный рецептор M1 человека
hM2 клонированный рецептор M2 человека
hM3 клонированный рецептор M3 человека
hM4 клонированный рецептор M4 человека
hM5 клонированный рецептор M5 человека
HOBT гидрат 1-гидроксибензотриазола
ВЭЖХ высокоэффективная жидкостная хроматография
IPA изопропанол
MCh метилхолин
MTBE метил-трет-бутиловый эфир
ТФУК трифторуксусная кислота
ТГФ тетрагидрофуран
Любые другие использованные, но не определенные в данном описании сокращения, имеют стандартные общепринятые значения. Если не указано другого, все вещества, такие как реагенты, исходные вещества и растворители, были получены из коммерческих источников (такие как Sigma-Aldrich, Fluka, и тому подобные) и использованы без дополнительной очистки.
Если не указано другого, ВЭЖХ анализ проводили с использованием прибора Agilent (Palo Alto, CA) серия 1100, оборудованного колонкой Zorbax Bonus RP 2,1 × 50 мм (Agilent) с размером частиц 3,5 микрона. Обнаружение проводили с помощью УФ-поглощения при 214 нм. Использовали следующие подвижные фазы (по объему): A представляет собой ACN (2%), воду (98%) и ТФУК (0,1%); и B представляет собой ACN (90%), воду (10%) и ТФУК (0,1%). Данные ВЭЖХ 10-70 получали с использованием скорости потока 0,5 мл/минуту при градиенте от 10 до 70% B в течение 6 минут (остальная часть представляла собой A). Аналогично данные ВЭЖХ 5-35 и данные ВЭЖХ 10-90 получали с использованием градиента от 5 до 35% B; или от 10 до 90% B в течение 5 минут.
Данные жидкостной хроматографии в комплексе с масс-спектрометрическим анализом (ЖХМС) получали с использованием прибора Applied Biosystems (Foster City, CA) модель API-150EX. Данные ЖХМС 10-90 получены с использованием от 10 до 90% подвижной фазы В при 5 минутном градиенте.
Очистку в небольшом масштабе проводили с использованием системы для препаративной жидкостной хроматографии API 150EX Prep Workstation от Applied Biosystems. Используемые подвижные фазы были следующими (по объему): A представляет собой воду и 0,05% ТФУК; и B представляет собой ACN и 0,05% ТФУК. Для большинства случаев (обычно для выделяемого образца массой примерно от 3 до 50 мг) были использованы следующие условия: скорость потока 20 мл/мин; градиент в течение 15 минут и колонка 20 мм × 50 мм Prism RP с частицами размером 5 микрон (Thermo Hypersil-Keystone, Bellefonte, PA). Для очистки в большем масштабе (обычно более чем 100 мг неочищенного образца) были использованы следующие условия: скорость потока 60 мл/мин; градиент в течение 30 минут и колонка 41,4 мм × 250 мм Microsorb BDS с частицами размером 5 микрон (Varian, Palo Alto, CA).
Получение 1
Пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты
Бифенил-2-изоцианат (97,5 г, 521 ммоль) и 4-гидрокси-N-бензилпиперидин (105 г, 549 ммоль) нагревали совместно при 70°C в течение 12 часов. Реакционную смесь затем охлаждали до 50°C и добавляли EtOH (1 л) и затем медленно добавляли 6M HCl (191 мл). Полученную смесь затем охлаждали до температуры окружающей среды и добавляли формиат аммония (98,5 г, 1,56 моль) и затем интенсивно барботировали через раствор газообразный азот в течение 20 минут. Затем добавляли палладий на активированном угле (20 г, 10 вес.% из расчета на сухой вес) и реакционную смесь нагревали при 40°C в течение 12 часов, а затем фильтровали через слой целита. Растворитель затем удаляли при пониженном давлении и добавляли к неочищенному остатку 1M HCl (40 мл). Затем pH смеси доводили с использованием 10н NaOH до pH 12. Водный слой экстрагировали этилацетатом(2 × 150 мл), органический слой сушили (сульфат магния), фильтровали и растворитель удаляли при пониженном давлении, получая 155 г указанного в заголовке промежуточного соединения (100% выход). ВЭЖХ (10-70) Rt = 2,52; m/z: [M+H+] вычислено для C18H20N2O2, 297,15; найдено 297,3.
Получение 2
N-Бензил-N-метиламиноацетальдегид
В трехгорлую колбу емкостью 2 л добавляли N-бензил-N-метилэтаноламин (30,5 г, 0,182 моль), ДХМ (0,5 л), DIPEA (95 мл, 0,546 моль) и ДМСО (41 мл, 0,728 моль). С использованием ледяной бани смесь охлаждали примерно до -10°C и добавляли комплекс триоксида серы с пиридином (87 г, 0,546 моль) в виде 4 порций с 5-минутными интервалами. Реакционную смесь перемешивали при -10°C в течение 2 часов. Перед удалением ледяной бани реакцию гасили путем добавления воды (0,5 л). Водный слой отделяли, органический слой промывали водой (0,5 л) и насыщенным раствором соли (0,5 л) и затем сушили над сульфатом магния и фильтровали, получая указанное в заголовке соединение, которое использовали без дополнительной очистки.
Получение 3
1-[2-(Бензилметиламино)этил]пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты
В 2-литровую колбу, содержащую продукт получения 2 в ДХМ (0,5 л), добавляли продукт получения 1 (30 г, 0,101 моль) с последующим добавлением триацетоксиборгидрида натрия (45 г, 0,202 моль). Реакционную смесь перемешивали в течение ночи и затем гасили путем добавления 1 н. соляной кислоты (0,5 л) при интенсивном перемешивании. Наблюдали три слоя и водный слой удаляли. После промывания 1 н. NaOH (0,5 л) получали гомогенный органический слой, который затем промывали насыщенным водным раствором NaCl (0,5 л), сушили над сульфатом магния, фильтровали и растворитель удаляли при пониженном давлении. Остаток очищали, растворяя его в минимальном количестве IPA и охлаждая данный раствор до 0°C для образования твердого вещества, которое отфильтровывали и промывали холодным IPA, получая 42,6 г указанного в заголовке соединения (95% выход). Масс-спектр m/z: [M+H+] вычислено для C28H33N3O2, 444,3; найдено 444,6. R f = 3,51 мин (10-70 ACN:H2O, ВЭЖХ с обращенной фазой).
Получение 4
1-(2-Метиламиноэтил)пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты
В колбу Парра для гидрирования добавляли продукт получения 3 (40 г, 0,09 моль) и EtOH (0,5 л). Колбу продували газообразным азотом и добавляли палладий-на-активированном угле (15 г, 10 вес. % (из расчета на сухую массу), 37% вес./вес.) вместе с уксусной кислотой (20 мл). Смесь выдерживали в аппарате Парра для гидрирования в атмосфере водорода (~50 фт/кв.дюйм) в течение 3 часов. Смесь затем фильтровали и промывали EtOH. Фильтрат конденсировали и остаток растворяли в минимальном количестве ДХМ. Медленно добавляли изопропилацетат (10 объемов) для образования твердого вещества, которое собирали, получая 22,0 г указанного в заголовке соединения (70% выход). Масс-спектр m/z: [M+H+] вычислено для
C21H27N3O2, 354,2; найдено 354,3. R f = 2,96 мин (10-70 ACN:H2O, ВЭЖХ с обращенной фазой).
Получение 5
1-{2-[(4-Формилбензоил)метиламино]этил}пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты
В трехгорлую колбу емкостью 1 л добавляли 4-карбоксибензальдегид (4,77 г, 31,8 ммоль), EDC (6,64 г, 34,7 ммоль), HOBT (1,91 г, 31,8 ммоль) и ДХМ (200 мл). Когда смесь становилась гомогенной, медленно добавляли продукт получения 4 (10 г, 31,8 ммоль) в ДХМ (100 мл). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов и затем промывали водой (1 × 100 мл), 1 н. HCl (5 × 60 мл), 1 н. NaOH (1 × 100 мл) насыщенным раствором соли (1 × 50 мл), сушили над сульфатом натрия, фильтровали и концентрировали, получая 12,6 г указанного в заголовке соединения (92% выход; 85% чистота по данным ВЭЖХ). Масс-спектр m/z: [M + H+] вычислено для C29H31N3O4, 486,2; найдено 486,4. R f 3,12 мин (10-70 ACN:H2O, ВЭЖХ с обращенной фазой).
Пример 1
1-(2-{[4-(4-Карбамоилпиперидин-1-илметил)бензоил]метиламино}этил)пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты
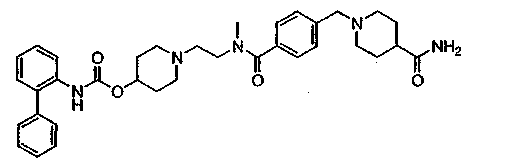
В трехгорлую колбу емкостью 2 л добавляли изонипекотамид (5,99 г, 40,0 ммоль), уксусную кислоту (2,57 мл), сульфат натрия (6,44 г) и IPA (400 мл). Реакционную смесь охлаждали до 0-10°C с использованием ледяной бани и медленно добавляли к ней раствор продукта получения 5 (11 г, 22,7 ммоль) в IPA (300 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов и затем охлаждали до 0-10°C. Добавляли порциями триацетоксиборгидрид натрия (15,16 г, 68,5 ммоль) и перемешивали данную смесь при комнатной температуре в течение 16 часов. Реакционную смесь затем концентрировали при пониженном давлении до объема примерно 50 мл и данную смесь подкисляли 1 н. HCl (200 мл) до pH 3. Полученную смесь перемешивали при комнатной температуре в течение 1 часа и затем экстрагировали ДХМ (3 × 250 мл). Водную фазу затем охлаждали до 0-5°C на ледяной бане и добавляли 50% водный раствор NaOH, доводя pH смеси до 10. Данную смесь затем экстрагировали изопропилацетатом (3 × 300 мл) и объединенные органические слои промывали водой (100 мл), насыщенным раствором соли (2 × 50 мл), сушили над сульфатом натрия, фильтровали и концентрировали, получая 10,8 г указанного в заголовке соединения (80% выход). Масс-спектр m/z: [M+H+] вычислено для C35H43N5O4, 598,3; найдено 598,6. R f = 2,32 мин (10-70 ACN:H2O, ВЭЖХ с обращенной фазой).
Пример 1A
1-(2-{[4-(4-Карбамоилпиперидин-1-илметил)бензоил]метиламино}этил)пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты также получали в виде дифосфатной соли с использованием следующего способа.
5,0 г продукта примера 1 объединяли с 80 мл смеси IPA:ACN (1:1). Добавляли 4,0 мл воды и смесь нагревали до 50°C при перемешивании, получая прозрачный раствор. К нему добавляли по каплям при 50°C 16 мл 1M фосфорной кислоты. Полученный мутный раствор перемешивали при 50°C в течение 5 часов, затем оставляли охлаждаться до температуры окружающей среды, при медленном перемешивании, в течение ночи. Полученные кристаллы собирали фильтрованием и сушили на воздухе в течение 1 часа, затем в вакууме в течение 18 часов, получая дифосфатную соль указанного в заголовке соединения (5,8 г, 75% выход) в виде белого кристаллического твердого вещества (98,3% чистота по ВЭЖХ).
Пример 1B
1-(2-{[4-(4-карбамоилпиперидин-1-илметил)бензоил]метиламино}этил)пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты также получали в виде моносульфатной соли с использованием следующего способа.
442 мг продукта примера 1 (0,739 ммоль вещества 96% чистоты) помещали в 5 мл смеси H2O:ACN (1:1) и медленно добавляли 1,45 мл 1 н. серной кислоты, контролируя pH. pH доводили приблизительно до pH 3,3. Прозрачный раствор фильтровали через 0,2 микронный фильтр, замораживали и лиофилизовали досуха.
161 г лиофилизованного вещества растворяли в 8,77 мл смеси IPA:ACN (10:1). Суспензию нагревали в течение 1,5 часов, помещая пузырек в предварительно нагретую до 70°C водяную баню. В течение 5 минут образовывались масляные капельки. Нагрев снижали до 60°C и смесь нагревали дополнительно в течение 1,5 часов с последующим нагреванием при 50°C в течение 40 минут, при 40°C в течение 40 минут, затем при 30°C в течение 45 минут. Нагрев выключали, и смесь оставляли медленно охлаждаться до комнатной температуры. На следующий день при исследовании вещества под микроскопом обнаружены иголки и пластины. Вещество затем нагревали при 40°C в течение 2 часов, при 35°C в течение 30 минут и затем при 30°C в течение 30 минут. Нагрев выключали, и смесь оставляли медленно охлаждаться до комнатной температуры. Твердое вещество затем отфильтровывали и сушили с использованием вакуумного насоса в течение 1 часа, получая моносульфатную соль указанного в заголовке соединения (117 мг, 73% выход).
Пример 1C
1-(2-{[4-(4-Карбамоилпиперидин-1-илметил)бензоил]метиламино}этил)пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты также получали в виде диоксалатной соли с использованием следующего способа.
510 мг продукта примера 1 (0,853 ммоль вещества 96% чистоты) помещали в 5 мл смеси H2O:ACN (1:1) и медленно добавляли 1,7 мл 1M водной щавелевой кислоты, контролируя pH. pH доводили приблизительно до pH 3,0. Прозрачный раствор фильтровали через 0,2 микронный фильтр, замораживали и лиофилизовали досуха. 150 мг лиофилизованного вещества растворяли в 13,1 мл смеси 94% IPA/6% H2О. Смесь перемешивали на предварительно нагретой до 60°C водяной бане в течение 2,5 часов. Нагрев выключали, и смесь оставляли охлаждаться до комнатной температуры. Пузырек замораживали при 4°C. Через 6 дней наблюдали образование маслянистого вещества, которое на стенке пузырька оказалось кристаллическим. Пузырек оставляли нагреваться до комнатной температуры и добавляли точечную затравку для кристаллизации (синтез описан ниже) и оставляли на 16 дней. За это время из раствора выпадало дополнительное количество кристаллов. Твердое вещество затем отфильтровывали и сушили с использованием вакуумного насоса в течение 14 часов, получая диоксалатную соль указанного в заголовке соединения (105 мг, 70% выход).
Синтез затравки для кристаллизации
510 мг продукта примера 1 (0,853 ммоль вещества 96% чистоты) помещали в 5 мл смеси H2O:ACN (1:1) и медленно добавляли 1,7 мл 1M водной щавелевой кислоты, контролируя pH. pH доводили приблизительно до pH 3,0. Прозрачный раствор фильтровали через 0,2 микронный фильтр, замораживали и лиофилизовали досуха, получая диоксалатную соль. 31,5 мг данной диоксалатной соли растворяли в 2,76 мл смеси 94% IPA/6% H2О. Смесь перемешивали на предварительно нагретой до 60°C водяной бане в течение 2,5 часов. Нагрев выключали, и смесь оставляли охлаждаться до комнатной температуры. Через 25 минут весь образец переходил в раствор. Нагрев выключали, и смесь оставляли охлаждаться до комнатной температуры. На следующий день наблюдалось небольшое количество вязкого вещества. Пузырек замораживали при 4°C. Через 4 дня все еще присутствовало вязкое вещество. Затем пузырек помещали храниться при комнатной температуре и осматривали через месяц. Вещество оказывалось твердым и кристаллическим при исследовании под микроскопом. Твердое вещество затем отфильтровывали и сушили с использованием вакуумного насоса в течение 1 часа, получая диоксалатную соль (20 мг, 63,5% выход).
Пример 1D
1-(2-{[4-(4-Карбамоилпиперидин-1-илметил)бензоил]метиламино}этил)пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты также получали в виде кристаллов свободного основания с использованием следующего способа.
230 мг продукта примера 1 растворяли в 0,2 мл смеси H2O:ACN (1:1) с использованием легкого нагрева. Затем смесь нагревали на водяной бане с температурой 70°C в течение 2 часов. Нагрев выключали и смесь оставляли охлаждаться до комнатной температуры, затем помещали в холодильник при 4°C на 1 час. Затем добавляли 50 мкл воды (выпадение в осадок масла) с последующим добавлением 40 мкл ACN, чтобы перевести образец обратно в раствор. Добавляли при медленном перемешивании при комнатной температуре затравку для кристаллизации (синтез описан ниже). Начинали образовываться кристаллы, и смесь оставляли на ночь при медленном перемешивании. На следующий день проводили цикл нагрев-охлаждение (30°C в течение 10 минут, 40°C в течение 10 минут, затем 50°C в течение 20 минут). Нагрев выключали, и смесь оставляли охлаждаться на ночь при медленном перемешивании. На следующий день проводили второй цикл нагрев/охлаждение (60°C в течение 1 часа при растворении, наблюдающемся при 70°C). Нагрев выключали, и смесь оставляли охлаждаться на ночь при медленном перемешивании. На следующий день присутствовали кристаллы, и проводили третий цикл нагрев/охлаждение (60°C в течение 3 часов). Нагрев выключали, и смесь оставляли охлаждаться на ночь при медленном перемешивании. На следующий день проводили цикл нагрев/охлаждение (60°C в течение 3 часов, медленное охлаждение, затем 60°C в течение 3 часов). Нагрев выключали, и смесь оставляли охлаждаться на ночь при медленном перемешивании. Через 3 дня твердое вещество отфильтровывали и помещали в высоковакуумную линию для удаления всех растворителей и получения кристаллов свободного основания указанного в заголовке соединения.
Синтез затравки для кристаллизации
109 мг продукта примера 1 растворяли в 0,56 мл смеси H2O:ACN (1:1). Суспензию оставляли в пробирке (пробка свободно размещена в верней части), чтобы дать возможность медленно выпариваться со временем. Пробирку помещали в ток азота, хотя азот использовали не для выпаривания, а только в качестве окружающей среды. Осадок становился видимым в течение 1 дня, который представлял собой кристаллическое вещество при исследовании под микроскопом. Твердое вещество затем помещали в высоковакуумную линию для удаления всех растворителей и получения кристаллов свободного основания. Количественное выделение, чистота 97,8% по данным ВЭЖХ.
Пример 1E
1-(2-{[4-(4-Карбамоилпиперидин-1-илметил)бензоил]метиламино}этил)пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты также получали в виде кристаллов свободного основания с использованием следующего альтернативного способа.
70 мг продукта примера 1 растворяли в 0,1 мл ACN. После добавления 0,3 мл MTBE раствор медленно становился мутным. Добавляли дополнительно 50 мкл ACN, чтобы раствор стал прозрачным (155 мг/мл ACN:MTBE = 1:2). Смесь оставляли в пробирке и закрывали. На следующий день появлялось твердое вещество. Затем твердое вещество отфильтровывали и помещали в высоковакуумную линию для удаления всех растворителей и получения кристаллов свободного основания указанного в заголовке соединения.
Пример 2
1-(2-{[4-(4-Карбамоилпиперидин-1-илметил)бензоил]этиламино}этил)пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты
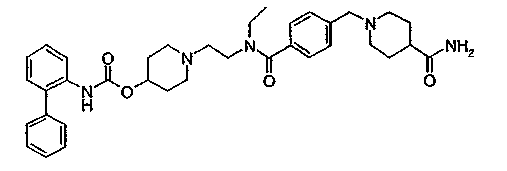
С использованием способа примера 1 и используя в получении 2 N-бензил-N-этилэтаноламин вместо N-бензил-N-метилэтаноламина, получали указанное в заголовке соединение. Масс-спектр m/z: [M+H+] вычислено для C36H45N5O4, 612,3; найдено 612,6.
Получение 6
1-(2-{[4-(4-(Метиловый сложный эфир)пиперидин-1-илметил)бензоил]метиламино}этил)пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты
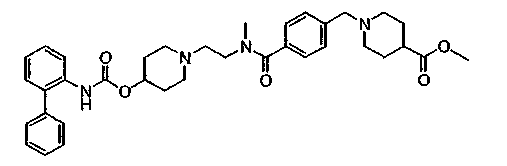
В трехгорлую колбу емкостью 100 мл добавляли метилизонипекотат (344 мг, 2,4 ммоль), уксусную кислоту (136 мкл), сульфат натрия (341 мг) и IPA (20 мл). Реакционную смесь охлаждали до 0-10°C на ледяной бане и медленно добавляли раствор продукта получения 5 (600 мг, 1,24 ммоль) в IPA (10 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа и затем охлаждали до 0-10°C. Добавляли порциями триацетоксиборгидрид натрия (763 мг, 3,6 ммоль). После перемешивания при комнатной температуре в течение 16 часов реакционную смесь затем концентрировали при пониженном давлении до объема примерно 5 мл и разбавляли ДХМ (50 мл). Органические слои промывали 0,5 н. HCl (2 × 30 мл), водой
(2 × 30 мл), насыщенным раствором соли (2 × 30 мл), сушили над сульфатом натрия, фильтровали и концентрировали, получая 700 мг указанного в заголовке соединения (92% выход). Масс-спектр m/z: [M+H+] вычислено для C36H44N4O5, 612,8; найдено, 613,5).
Пример 3
1-(2-{Метил-[4-(4-метилкарбамоилпиперидин-1-илметил)бензоил]амино}этил)пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты
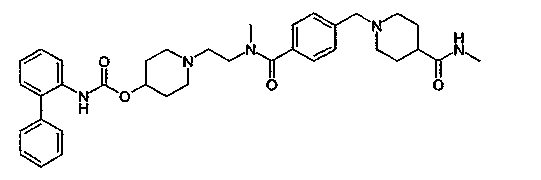
В пробирку емкостью 4 мл добавляли продукт получения 6 (61,2 мг, 0,1 ммоль) и метиламин (1 мл, 2M в MeOH). Реакционную смесь нагревали при 60°C в течение 72 часов и очищали препаративной ВЭЖХ, получая 46,9 мг указанного в заголовке соединения. (Масс-спектр m/z: [M+H+] вычислено для C36H45N5O4, 611,8; найдено 612,4.)
Пример 4
1-(2-{[4-(4-Этилкарбамоилпиперидин-1-илметил)бензоил]метиламино}этил)пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты
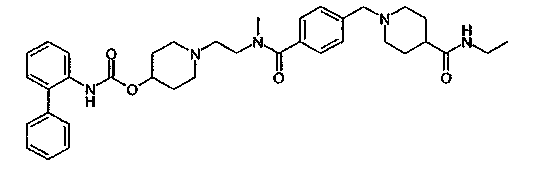
С использованием способа примера 3 и используя этиламин (1 мл, 2M в EtOH) вместо метиламина (1 мл, 2M в MeOH), получали 17 мг указанного в заголовке соединения. (Масс-спектр m/z: [M+H+] вычислено для C37H47N5O4, 625,8; найдено 626,4.)
Пример 5
1-(2-{Метил-[4-(4-пропилкарбамоилпиперидин-1-илметил)бензоил]амино}этил)пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты
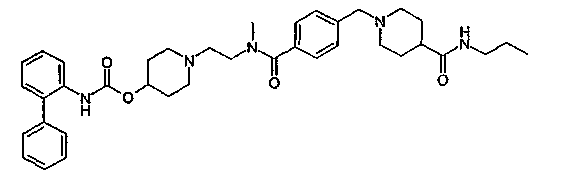
В пробирку емкостью 4 мл добавляли продукт получения 6 (61,2 мг, 0,1 ммоль) и пропиламин (1 мл). Реакционную смесь нагревали при 60°C в течение 24 часов и очищали препаративной ВЭЖХ, получая 39,5 мг указанного в заголовке соединения. (Масс-спектр m/z: [M+H+] вычислено для C38H49N5O4, 639,8; найдено 640,4.)
Пример 6
1-(2-{[4-(4-Изопропилкарбамоилпиперидин-1-илметил)бензоил]метиламино}этил)пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты
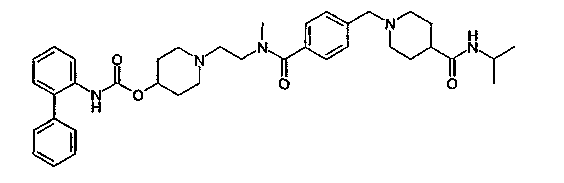
С использованием способа примера 5 и используя изопропиламин (1 мл) вместо пропиламина (1 мл), получали 27,8 мг указанного в заголовке соединения. (Масс-спектр m/z: [M+H+] вычислено для C38H49N5O4, 639,8; найдено 640,4.)
Получение 7
1-(2-Boc-аминоэтил)пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты
В колбу емкостью 1 л, содержащую продукт получения 1 (25,4 г, 85,6 ммоль) в ДХМ (0,43 л), добавляли DIPEA (29,9 мл, 171,1 ммоль) и 2-(Boc-Амино)этилбромид (21,8 г, 94,4 ммоль). Реакционную смесь затем нагревали при 50°C в течение ночи (~18 часов). По окончании данного промежутка времени реакционную смесь затем охлаждали до 0°C для того, чтобы вызвать осаждение продукта. Осадок отфильтровывали и собирали, получая указанное в заголовке соединение с 42% выходом (15,8 г). Масс-спектр m/z: [M+H+] вычислено для
C25H33N3O4, 439,3; найдено 440,4.
Получение 8
1-(2-Аминоэтил)пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты
Продукт получения 7 (3,5 г, 8,1 ммоль) добавляли к смеси ДХМ:ТФУК в соотношении 1:1 (50 мл) и реакционную смесь оставляли перемешиваться при комнатной температуре в течение 30 минут. По завершении реакции смесь разбавляли ДХМ (125 мл) и промывали 1 н. NaOH (200 мл). Органический слой затем промывали водой (200 мл), NaCl (насыщ.) (200 мл), сушили над Na2SO4 и затем фильтровали. Растворитель удаляли при пониженном давлении. Неочищенное вещество было достаточно чистым для использования без дополнительной очистки. Указанное в заголовке соединение получали с 94% выходом (2,6 г, 7,6 ммоль). Масс-спектр m/z: [M+H+] вычислено для C20H25N3O2, 339,2; найдено 339,6.
Получение 9
2-Фтор-4-формилбензойная кислота
Перемешиваемый раствор 4-циано-2-фторбензoйной кислоты (2,5 г, 15,2 ммоль) в ДХМ (100 мл) охлаждали до -78°C и осторожно, из-за выделения H2, добавляли к нему по каплям DIBAL (30 мл, 45,4 ммоль, 25% в толуоле). Смесь оставляли перемешиваться при -78°C в течение 4 часов. Реакцию осторожно, из-за выделения H2, гасили добавлением MeOH (10 мл). Органический слой затем промывали 1 н. HCl (100 мл), водой (100 мл), NaCl (насыщ.) (100 мл), сушили над MgSO4 и затем фильтровали. Растворитель удаляли при пониженном давлении. Неочищенное вещество было достаточно чистым для использования без дополнительной очистки. Указанное в заголовке соединение получали с 78% выходом (2,0 г, 11,9 ммоль).
Пример 7
1-{2-[4-(4-Карбамоилпиперидин-1-илметил)-2-фторбензоиламино]этил}пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты
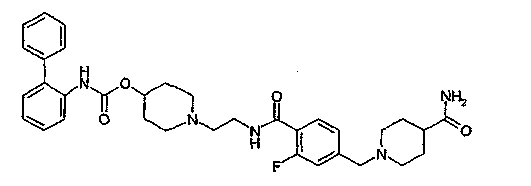
С использованием способа примера 1 и используя в получении 5 вместо продукта получения 4 продукт получения 8 и продукт получения 9 вместо 4-карбоксибензальдегида, получают указанное в заголовке соединение. Масс-спектр m/z: [M+H+] вычислено для C34H40FN5O4, 601,7; найдено 602,2.
Пример 8
1-(2-{[4-(4-Карбамоилпиперидин-1-илметил)-2-фторбензоил]метиламино}этил)пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты
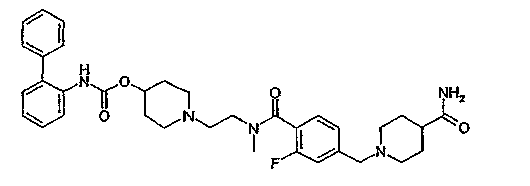
С использованием способа примера 1 и используя в получении 5 вместо 4-карбоксибензальдегида продукт получения 9, получают указанное в заголовке соединение. Масс-спектр m/z: [M+H+] вычислено для C35H42FN5O4, 615,8; найдено 616,2.
Получение 10
Диэтиламид пиперидин-4-карбоновой кислоты
К перемешиваемому раствору 1-трет-бутоксикарбонилпиперидинe-4-карбоновой кислоты (5 г, 22,0 ммоль) в ДМФ (100 мл) добавляли диэтиламин (4,6 мл, 44 ммоль), триэтиламин (9,1 мл, 66,0 ммоль), HOAt (22,0 мл, 0,5 M в ДМФ, 22,0 ммоль) и, наконец, EDCI (8,4 г, 44 ммоль). Эту смесь оставляли перемешиваться в течение 14 часов при комнатной температуре. Растворитель затем удаляли при пониженном давлении. Смесь помещали в ДХМ (100 мл). Органический слой затем промывали водой (100 мл), 1 н. HCl (100 мл), NaCl (насыщ.) (100 мл), сушили над MgSO4 и затем фильтровали. К органическому слою добавляли ТФУК (33 мл). Реакционную смесь оставляли перемешиваться при комнатной температуре в течение 2 часов. Растворитель затем удаляли при пониженном давлении. Смесь помещали в ДХМ (100 мл). Органический слой затем промывали 1 н. NaOH (100 мл), водой (100 мл), NaCl (насыщ.) (100 мл), сушили над MgSO4 и затем фильтровали. Растворитель удаляли при пониженном давлении. Неочищенное вещество было достаточно чистым для применения без дополнительной очистки. Указанное в заголовке соединение получали с 86%-ным выходом (3,5 г, 19,0 ммоль).
Пример 9
1-{2-[4-(4-Диэтилкарбамоилпиперидин-1-илметил)-2-фторбензоиламино]этил}пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты
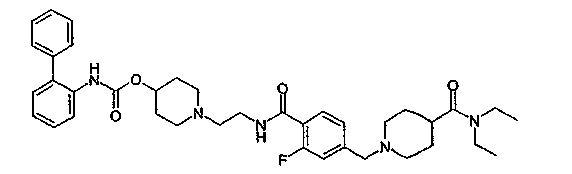
С использованием способа примера 1, используя в получении 5 вместо 4-карбоксибензальдегида продукт получения 9 и вместо продукта получения 4 продукт получения 8, и в способе примера 1 продукт получения 10 вместо изонипекотамида, получают указанное в заголовке соединение. Масс-спектр m/z:
[M+H+] вычислено для C38H48FN5O4, 657,8; найдено 658,4.
Пример 10
1-(2-{[4-(4-Диэтилкарбамоилпиперидин-1-илметил)-2-фторбензоил]метиламино}этил)пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты
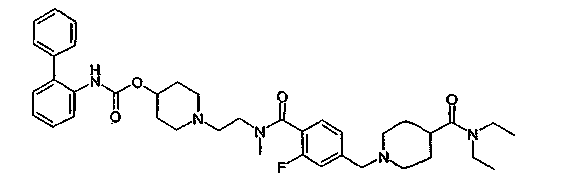
С использованием способа примера 1, заменяя в получении 5 4-карбоксибензальдегид на продукт получения 9 и используя продукт получения 10 вместо изонипекотамида в способе примера 1, получают указанное в заголовке соединение. Масс-спектр m/z: [M+H+] вычислено для C39H50FN5O4, 671,9; найдено 672,4.
Пример 11
1-(2-{[4-(4-Диэтилкарбамоилпиперидин-1-илметил)бензоил]метиламино}этил)пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты
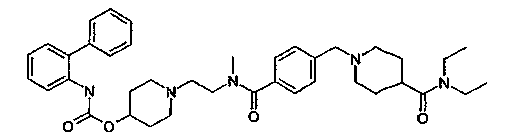
С использованием способа примера 1, но заменяя изонипекотамид на продукт получения 10, получают указанное в заголовке соединение. Масс-спектр m/z: [M+H+] вычислено для C39H51N5O4, 653,9; найдено 654,4.
Пример 12
1-(2-{[4-(3-(S)-Диэтилкарбамоилпиперидин-1-илметил)бензоил]метиламино}этил)пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты
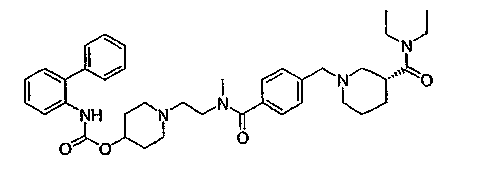
С использованием способа примера 1, но заменяя изонипекотамид на диэтиламид пиперидин-3-(S)-карбоновой кислоты, получают указанное в заголовке соединение. Масс-спектр m/z: [M + H+] вычислено для C39H51N5O4, 653,9; найдено 654,4.
Диэтиламид пиперидин-3-(S)-карбоновой кислоты получали в соответствии с публикацией Chirality 7(2): 90-95 (1995).
Получение 11
N-{2-[4-(Бифенил-2-илкарбамоилокси)пиперидин-1-ил]этил}-2,5-дибром-N-метилтерефталаминовая кислота
В колбу емкостью 100 мл, содержащую продукт получения 1 (2,5 г, 7,1 ммоль) в ДМФ (20 мл), добавляли 2,5-дибромтерефталевую кислоту (6,88 г, 21,2 ммоль) с последующим добавлением DIPEA (1,6 мл, 9,2 ммоль) и HATU (3,23 г, 8,5 моль). Желтую суспензию перемешивали при комнатной температуре в течение 3 часов (все вещество находится в растворе после завершения реакции). Реакционную смесь разбавляли ДХМ (200 мл). К раствору добавляли 1 н. NaOH (150 мл) и MeOH (добавляли минимальное количество для растворения мелкого белого осадка, который наблюдался после добавления основания). Раствор переносили в делительную воронку и водный слой отбрасывали. Органический слой промывали 1 н. HCl (1 × 150 мл), сушили над сульфатом натрия, фильтровали и растворитель удаляли при пониженном давлении, получая 7 г указанного в заголовке соединения (>100% выход за счет присутствия остатка ДМФ). Данное вещество использовали без дополнительной очистки. Масс-спектр m/z: [M + H+] вычислено для C29H29Br2N3O5, 659,4; найдено 660,3. R f = 3,39 мин (2-90 ACN: H2O, ВЭЖХ с обращенной фазой).
Получение 12
Метиловый эфир N-{2-[4-(бифенил-2-илкарбамоилокси)пиперидин-1-ил]этил}-2,5-дибром-N-метилтерефталаминовой кислоты
В колбу емкостью 100 мл, содержащую продукт получения 11 (7,0 г, 10,6 ммоль), добавляли раствор смеси толуол/MeOH (9:1, 70 мл). Поскольку все твердое вещество не растворялось, добавляли дополнительно 3 мл MeOH. Раствор охлаждали до 0°C на ледяной бане и добавляли с помощью шприца триметилсилилдиазометан (2,0 M раствор в гексанах, 6,3 мл, 12,7 ммоль). Реакционную смесь оставляли нагреваться до комнатной температуры. После перемешивания в течение 2 часов по данным ВЭЖХ и масс-спектрального анализа установлено, что реакция не завершилась. Дополнительно добавляли триметилсилилдиазометан (10,0 мл) и реакционную смесь перемешивали при комнатной температуре в течение 70 часов. Хотя ВЭЖХ-анализ показал, что реакция не завершилась, к реакционной смеси добавляли уксусную кислоту (15 мл) и полученный раствор концентрировали при пониженном давлении. Неочищенный продукт очищали хроматографией на силикагеле с использованием градиента от 2% до 5% MeOH в ДХМ в качестве элюента, получая 2,32 г указанного в заголовке соединения (49% выход). Масс-спектр m/z: [M+H+] вычислено для C30H31Br2N3O5, 673,4; найдено 674,3. R f =4,26 мин (2-90 ACN:H2O, ВЭЖХ с обращенной фазой).
Получение 13
1-{2-[(2,5-Дибром-4-гидроксиметилбензоил)метиламино]этил}пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты
В колбу емкостью 100 мл, содержащую продукт получения 12 (2,2 г, 3,3 ммоль), добавляли ТГФ (35 мл). С использованием ледяной бани смесь охлаждали примерно до 0°C и добавляли шприцом литийалюминийгидрид (1,0M раствор в ТГФ, 6,6 мл, 6,6 ммоль). Полученную суспензию перемешивали при комнатной температуре в течение 4 часов. Реакцию гасили добавлением 1 н. NaOH (100 мл). Водный слой отделяли, и органический слой промывали насыщенным раствором соли (50 мл), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении (80,2 % чистота по данным ВЭЖХ). Часть неочищенного продукта очищали препаративной ВЭЖХ (20-40 ACN:H2O, ВЭЖХ с обращенной фазой), получая 317 мг ТФУК соли. ТФУК соль целевого продукта распределяли между EtOAc (10 мл) и насыщенным раствором бикарбоната натрия (10 мл). Органический слой промывали насыщенным раствором соли (5 мл), сушили над сульфатом натрия, фильтровали и концентрировали, получая 223,6 мг указанного в заголовке соединения. Масс-спектр m/z: [M + H+] вычислено для C29H31Br2N3O4, 645,4; найдено 646,3. R f = 3,56 мин (10-70 ACN:H2O, ВЭЖХ с обращенной фазой).
Получение 14
4-({2-[4-(Бифенил-2-илкарбамоилокси)пиперидин-1-ил]этил}метилкарбамоил)-2,5-дибромбензиловый эфир метансульфоновой кислоты
В колбу емкостью 25 мл, содержащую продукт получения 13 (223,6 мг, 0,346 ммоль), добавляли ДХМ (10 мл) с последующим добавлением DIPEA (135,5 мкл, 0,778 ммоль) и метансульфонилхлорида (41 мкл, 0,528 ммоль). Реакционную смесь перемешивали в течение 30 минут при комнатной температуре. К реакционной смеси затем добавляли насыщенный раствор бикарбоната натрия (10 мл). Водный слой отбрасывали, и органический слой промывали насыщенным раствором соли (5 мл), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении, получая 229 мг указанного в заголовке соединения (91 % выход). Масс-спектр m/z: [M+H+] вычислено для C30H33Br2N3O6S, 723,5; найдено 724,3. R f =3,77 мин (10-70 ACN:H2O, ВЭЖХ с обращенной фазой).
Пример 13
1-(2-{[2,5-Дибром-4-(4-карбамоилпиперидин-1-илметил)бензоил]метиламино}этил)пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты
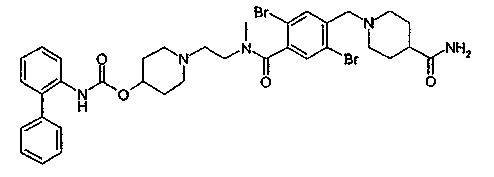
В колбу емкостью 25 мл, содержащую продукт получения 14 (229 мг, 0,316 ммоль), добавляли изонипекотамид (48,7 мг, 0,380 ммоль), DIPEA (110,2 мкл, 0,633 ммоль) и ACN (4 мл). Реакционную смесь перемешивали при комнатной температуре в течение 63 часов в атмосфере азота. Реакционную смесь затем разбавляли ДХМ (15 мл) и промывали насыщенным водным раствором бикарбоната натрия. Продукт экстрагировали в водный слой с использованием 1,0 н. HCl (2 × 10 мл). Водный слой промывали ДХМ (2 × 15 мл) и pH доводили до 10-11 с использованием 1,0 н. NaOH. Данную смесь затем экстрагировали ДХМ (3 × 20 мл) и объединенные органические слои промывали насыщенным раствором соли (10 мл), сушили над сульфатом натрия, фильтровали и концентрировали, получая указанное в заголовке соединение. Масс-спектр m/z: [M+H+] вычислено для C35H41Br2N5O4, 756,5; найдено 756,3. R f =2,65 мин (10-70 ACN:H2O, ВЭЖХ с обращенной фазой).
Пример 14
1-(2-{[4-(2-Карбамоилпиперидин-1-илметил)бензоил]метиламино}этил)пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты
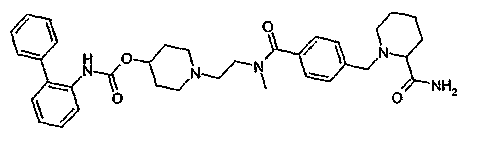
Указанное в заголовке соединение получали с использованием способов, описанных в примере 1, и применяя подходящие исходные вещества. Масс-спектр m/z: [M+H+] вычислено для C35H43N5O4, 598,3; найдено 597,8.
Пример 15
1-(2-{[4-(4-Карбамоилпиперидин-1-илметил)-2-метоксибензоил]метиламино}этил)пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты
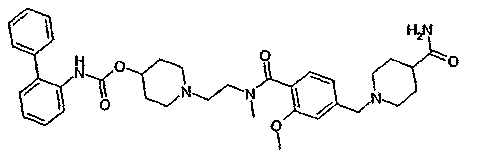
К перемешиваемому раствору 4-бром-3-метоксибензойной кислоты (15,0 г, 58 ммоль) в ДМСО (150 мл) добавляли NaHCO3 (20,0 г, 230 ммоль). Эту смесь нагревали при 80°C в течение 18 часов. Реакционную смесь затем охлаждали до комнатной температуры, и растворитель удаляли при пониженном давлении. Неочищенную реакционную смесь затем растворяли в ДХМ (200 мл) и промывали 1 н. HCl (100 мл), водой (100 мл), NaCl (насыщ.) (100 мл), сушили над MgSO4 и затем фильтровали. Растворитель удаляли при пониженном давлении. Неочищенное вещество было достаточно чистым для использования без дополнительной очистки. Продукт, метиловый эфир 4-формил-3-метоксибензойной кислоты, получали с выходом 79%
(8,9 г, 45,8 ммоль).
К перемешиваемому раствору метилового эфира 4-формил-3-метоксибензойной кислоты (5,0 г, 26 ммоль) в трет-бутиловом спирте (200 мл) добавляли NaH2PO4-2H2O (3,6 г, 26 ммоль), воду (50 мл), 2-метил-2-бутен (11 мл, 104 ммоль), и, наконец, NaClO2 (7,02 г, 78 ммоль). Реакционную смесь оставляли перемешиваться при комнатной температуре в течение 4 часов. Растворитель затем удаляли при пониженном давлении. Неочищенную реакционную смесь затем растворяли в ДХМ (200 мл) и продукт экстрагировали с использованием 1 н. NaOH (200 мл). Водный слой промывали ДХМ (200 мл) и затем нейтрализовали 6н HCl (~40 мл) и продукт экстрагировали ДХМ (200 мл). Органический слой затем промывали водой (100 мл), NaCl (насыщ.) (100 мл), сушили над MgSO4 и затем фильтровали. Растворитель удаляли при пониженном давлении. Неочищенное вещество было достаточно чистым для использования без дополнительной очистки. Продукт, 4-метиловый эфир 2-метокситерефталевой кислоты, получали с 47% выходом (2,4 г, 12,3 ммоль).
К перемешиваемому раствору 4-метилового эфира 2-метокситерефталевой кислоты (450 мг, 2,1 ммоль) в ДМФ (10 мл) добавляли EDC (630 мг, 3,3 ммоль), HOAt (2,4 мл, 1,18 ммоль, 0,5M в ДМФ) и DIPEA (1,3 мл, 7,05 ммоль). Когда смесь становилась гомогенной, медленно добавляли раствор продукта получения 4 (830 мг, 2,4 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов и затем промывали водой (100 мл), 1 н. HCl (100 мл), 1 н. NaOH (100 мл), насыщенным раствором соли (100 мл), сушили над MgSO4, фильтровали и концентрировали, получая продукт сложного эфира с 89% выходом (1,04 г, 1,9 ммоль). Масс-спектр m/z: [M+H+] вычислено для C31H35N3O6, 545,6; найдено 546,6.
К перемешиваемому раствору данного продукта сложного эфира (1,0 г, 1,8 ммоль) в ТГФ (100 мл) при 0°C добавляли метанол (57 мкл, 1,8 ммоль), с последующим добавлением LiAlH4 (1,8 мл, 1,8 ммоль, 1,0M в ТГФ). Ледяную баню удаляли, и реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Реакцию гасили 1 н. HCl (водн.) при 0°C до прекращения выделения газа, затем перемешивание продолжали в течение 10 минут. Растворитель удаляли при пониженном давлении. Неочищенную реакционную смесь помещали в ДХМ (100 мл) и промывали водой (100 мл), NaCl (насыщ.) (100 мл), сушили над MgSO4 и затем фильтровали. Растворитель удаляли при пониженном давлении. Неочищенное вещество было достаточно чистым для использования без дополнительной очистки. Продукт спирта получали с 89% выходом (831 мг, 1,6 ммоль). Масс-спектр m/z: [M+H+] вычислено для C30H35N3O5, 517,6; найдено 518,6.
К перемешиваемому раствору данного продукта спирта (78 мг, 1,5 ммоль) в ДХМ (2,5 мл) при -15°C добавляли ДМСО (130 мкл, 22,5 ммоль), DIPEA (130 мкл, 7,5 ммоль). К раствору добавляли комплекс триоксида серы с пиридином (240 мг, 15 ммоль). Через 30 минут реакционную смесь гасили H2O (~3 мл). Два слоя разделяли, органический слой сушили над MgSO4, фильтровали и продукт альдегида непосредственно использовали в следующей реакции.
С использованием способа примера 1, но используя вместо продукта получения 5 продукт альдегида получали указанное в заголовке соединение. Масс-спектр m/z:[M+H+] вычислено для C36H45N5O5, 627,3; найдено 628,2.
С использованием описанных здесь способов и применяя подходящие исходные вещества, получали следующие соединения:
Пример 16 - 1-(2-{[5-(4-карбамоилпиперидин-1-илметил)тиофен-2-карбонил]метиламино}этил)пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты. Масс-спектр m/z: [M+H+] вычислено для C33H41N5O4S, 604,3; найдено 604,2.
Пример 17 - 1-(2-{[5-((R)-3-диэтилкарбамоилпиперидин-1-илметил)тиофен-2-карбонил]метиламино}этил)
пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты. Масс-спектр m/z: [M+H+] вычислено для C37H49N5O4S, 660,4; найдено 660,4.
Пример 18 - 1-(2-{[5-((R)-3-диэтилкарбамоилпиперидин-1-илметил)тиофен-2-карбонил]амино}этил)пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты. Масс-спектр m/z: [M+H+] вычислено для C36H47N5O4S, 646,3; найдено 646,4.
Пример 19 - 1-(2-{[5-(4-карбамоилпиперидин-1-илметил)тиофен-2-карбонил]амино}этил)пиперидин-4-
иловый эфир бифенил-2-илкарбаминовой кислоты. Масс-спектр m/z: [M+H+] вычислено для
C32H39N5O4S, 590,3; найдено 590,2.
Пример 20 - 1-(2-{[5-((R)-3-диэтилкарбамоилпиперидин-1-илметил)-1H-пиррол-2-карбонил]метиламино}этил)пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты. Масс-спектр m/z: [M+H+] вычислено для
C37H50N6O4, 643,4; найдено 643,2.
Пример 21 - 1-(2-{[5-(4-карбамоилпиперидин-1-илметил)-1H-пиррол-2-карбонил]метиламино}этил)пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты. Масс-спектр m/z: [M+H+] вычислено для
C33H42N6O4, 587,3; найдено 587,2.
Пример 22 - 1-(2-{[5-((R)-3-диэтилкарбамоилпиперидин-1-илметил)фуран-2-карбонил]метиламино}этил)
пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты. Масс-спектр m/z: [M+H+] вычислено для C37H49N5O5, 644,4; найдено 644,4.
Пример 23 - 1-(2-{[5-(4-диэтилкарбамоилпиперидин-1-илметил)фуран-2-карбонил]метиламино}этил)
пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты. Масс-спектр m/z: [M+H+] вычислено для C37H49N5O5, 644,4; найдено 644,4.
Пример 24 - 1-(2-{[5-(4-карбамоилпиперидин-1-илметил)фуран-2-карбонил]амино}этил)пиперидин-4-
иловый эфир бифенил-2-илкарбаминовой кислоты. Масс-спектр m/z: [M+H+] вычислено для
C32H39N5O5, 574,3; найдено 574,2.
Пример 25 - 1-(2-{[5-((R)-3-диэтилкарбамоилпиперидин-1-илметил)фуран-2-карбонил]амино}этил)пиперидин-4-
иловый эфир бифенил-2-илкарбаминовой кислоты. Масс-спектр m/z: [M+H+] вычислено для C36H47N5O5, 630,4.
Пример 26 - 1-[2-({3-[4-(3-карбамоилпиперидин-1-илметил)фенил]пропионил}метиламино)этил]пиперидин-4-
иловый эфир бифенил-2-илкарбаминовой кислоты. Масс-спектр m/z: [M+H+] вычислено для C37H47N5O4, 626,4; найдено 625,8.
Пример 27 - 1-[2-({3-[4-(4-карбамоилпиперидин-1-илметил)фенил]пропионил}метиламино)этил]пиперидин-4-
иловый эфир бифенил-2-илкарбаминовой кислоты. Масс-спектр m/z: [M+H+] вычислено для C37H47N5O4, 626,4; найдено 625,8.
Пример 28 - 1-(2-{3-[4-(4-карбамоилпиперидин-1-илметил)фенил]пропиониламино}этил)пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты. Масс-спектр m/z: [M+H+] вычислено для C36H45N5O4, 612,4; найдено 611,8.
Пример 29 - 1-(2-{3-[4-(4-диэтилкарбамоилпиперидин-1-илметил)фенил]пропиониламино}этил)пиперидин-4-
иловый эфир бифенил-2-илкарбаминовой кислоты. Масс-спектр m/z: [M+H+] вычислено для C40H53N5O4, 668,4; найдено 667,9.
Пример 30 - 1-(2-{3-[4-(3-диэтилкарбамоилпиперидин-1-илметил)фенил]пропиониламино}этил)пиперидин-4-
иловый эфир бифенил-2-илкарбаминовой кислоты. Масс-спектр m/z: [M+H+] вычислено для C40H53N5O4, 668,4; найдено 667,9.
С использованием описанных здесь способов и применяя подходящие исходные вещества, могут быть получены следующие соединения:
Пример 31 - 1-{2-[4-(4-карбамоилпиперидин-1-илметил)бензоиламино]этил}пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты;
Пример 32 - 1-(2-{[4-(4-карбамоилпиперидин-1-илметил)-2-хлор-бензоил]метиламино}этил)пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты;
Пример 33 - 1-(2-{[4-(4-карбамоилпиперидин-1-илметил)-2-хлор-5-метоксибензоил]метиламино}этил)пиперидин-4-иловый эфир бифенил-2-илкарбаминовой кислоты; и
Пример 34 - 1-[2-({2-[4-(4-карбамоилпиперидин-1-илметил)фенил]ацетил}метиламино)этил]пиперидин-4-
иловый эфир бифенил-2-илкарбаминовой кислоты.
Анализ 1
Анализ радиолигандного связывания
A. Получение мембран клеток, экспрессирующих подтипы hM 1 , hM 2 , hM 3 и hM 4 мускариновых рецепторов
CHO клеточные линии, стабильно экспрессирующие клонированные подтипы мускариновых рецепторов hM1, hM2, hM3 и hM4 человека, соответственно, выращивали почти до слияния в среде, состоящей из HAM's F-12, дополненного 10% FBS и 250 мкг/мл генетицина. Клетки выращивали в инкубаторе в атмосфере 5% CO2, при 37°C и способствовали росту с использованием 2 мM EDTA в dPBS. Клетки собирали путем центрифугирования в течение 5 минут при 650×g, и осадок клеток либо хранили замороженным при -80°C, или же сразу получали мембраны. Для получения мембран осадок клеток повторно суспендировали в лизисном буфере и гомогенизировали с использованием разрушителя тканей Polytron PT-2100 (Kinematica AG; 20 секунд x 2 разрыва). Неочищенные мембраны центрифугировали при 40000×g в течение 15 минут при 4°C. Осадок мембран затем повторно суспендировали в буфере для повторного суспендирования и опять гомогенизовали с использованием разрушителя тканей Polytron. Концентрацию белка в суспензии мембран определяли способом, описанным Lowry, O. et al., Journal of Biochemistry 193:265 (1951). Все мембраны замораживали в аликвотах при -80°C или же сразу же использовали. Аликвоты полученных мембран рецептора hM5 закупали непосредственно от Perkin Elmer и хранили при -80°C до использования.
B. Анализ радиолигандного связывания на подтипах hM 1 , hM 2 , hM 3 , hM 4 и hM 5 мускаринового рецептора
Анализы радиолигандного связывания проводили в 96-луночных планшетах для микротитрования при общем объеме для анализа 100 мкл. Мембраны клеток СНО, стабильно экспрессирующие либо hM1, hM2, hM3, hM4, либо hM5 подтип мускаринового рецептора, разводили в буфере для анализа до следующих определенных концентраций целевого белка (мкг/лунку): 10 мкг для hM1, 10-15 мкг для hM2, 10-20 мкг для hM3, 10-20 мкг для hM4 и 10-12 мкг для hM5. Мембраны гомогенизировали в течение короткого промежутка времени с использованием разрушителя тканей Polytron (10 секунд) перед добавлением в планшеты для анализа. Исследования насыщения связывания для определения величин KD радиолиганда проводили с использованием L-[N-метил-3H]скополаминметилхлорида ([3H]-NMS) (TRK666, 84,0 Ки/ммоль, Amersham Pharmacia Biotech, Buckinghamshire, Англия) в концентрациях, колеблющихся от 0,001 нM до 20 нM. Анализ замещения для определения величин Ki тестируемых соединений проводили с использованием
[3H]-NMS в концентрации 1 нM и при одиннадцати различных концентрациях тестируемого соединения. Тестируемые соединения первоначально растворяли в концентрации 400 мкМ в буфере для разведения и затем серийно разводили 5× с использованием буфера для разведения до конечных концентраций, колеблющихся от 10 пкМ до 100 мкМ. Порядок добавления в планшеты для анализа и добавляемые объемы были следующими: 25 мкл радиолиганда, 25 мкл разведенного тестируемого соединения и 50 мкл мембран. Планшеты для анализа инкубировали в течение 60 минут при 37°C. Реакции связывания заканчивали с помощью быстрого фильтрования через фильтровальные планшеты из стекловолокна (PerkinElmer Inc., Wellesley, MA), предварительно обработанные в 1% BSA. Фильтровальные планшеты ополаскивали три раза промывочным буфером (10 мМ HEPES) для удаления несвязанной радиоактивности. Планшеты затем сушили на воздухе и в каждую лунку добавляли 50 мкл сцинтиляционной жидкости Microscint-20 (PerkinElmer Inc., Wellesley, MA). Планшеты затем считывали с помощью сцинтилляционного счетчика PerkinElmer Topcount (PerkinElmer Inc., Wellesley, MA). Данные связывания анализировали с использованием нелинейного регрессионного анализа с помощью пакета программного обеспечения GraphPad Prism (GraphPad Software, Inc., San Diego, CA) с использованием односайтовой модели конкурирования. Величины Ki для тестируемых соединений рассчитывали из наблюдаемых значений IC50 и значение KD радиолиганда с использованием уравнения Ченга-Прусова (Cheng-Prusoff) (Cheng Y; Prusoff W. H. Biochemical Pharmacology 22(23):3099-108 (1973)). Значения Ki преобразовывали в величины pKi для определения среднего геометрического значения и 95% доверительных интервалов. Данные суммарные статистические данные затем преобразовывали обратно в величины Ki для описания данных.
В данном анализе более низкие величины Ki указывают на то, что тестируемое соединение обладает более высокой аффинностью связывания в отношении исследуемого рецептора. Например, в данном анализе было найдено, что соединения примеров 1 и 2 обладают величиной Ki менее примерно 5 нМ в отношении подтипа M3 мускаринового рецептора.
Анализ 2
Анализы функциональной эффективности мускариновых рецепторов
А. Блокирование опосредованного агонистом ингибирования накопления цАМФ
В данном анализе функциональную эффективность тестируемого соединения определяли путем измерения способности тестируемого соединения блокировать ингибирование оксотреморином форсколин-опосредованного накопления цАМФ в клетках CHO-K1, экспрессирующих рецептор hM2.
Анализы цАМФ проводили в формате радиоиммуноанализа с использованием Flashplate системы для анализа активирования аденилциклазы с использованием 125I-цАМФ (NEN SMP004B, PerkinElmer Life Sciences Inc., Boston, MA) в соответствии с инструкциями производителя.
Клетки ополаскивали один раз dPBS и поднимали раствором Trypsin-EDTA (0,05% трипсина/0,53 мM EDTA), как описано выше в разделе «Получение клеточных культур и мембран». Изолированные клетки дважды промывали путем центрифугирования при 650×g в течение пяти минут в 50 мл dPBS. Осадок клеток затем повторно суспендировали в 10 мл dPBS, и клетки подсчитывали с использованием счетчика Coulter Z1 Dual Particle (Beckman Coulter, Fullerton, CA). Клетки затем еще раз центрифугировали при 650×g в течение пяти минут и повторно суспендировали в стимулирующем буфере для получения концентрации для анализа 1,6×106 - 2,8×106 клеток/мл.
Тестируемое соединение первоначально разводили в концентрации 400 мкМ в буфере для разведения (dPBS, дополненный 1 мг/мл BSA (0,1%)), и затем серийно разводили буфером для разведения до конечных концентраций в диапазоне от 100 мкМ до 0,1 нМ. Оксотреморин разводили аналогичным образом.
Для измерения ингибирования оксотреморином активности AC 25 мкл форсколина (конечная концентрация 25 мкM получена разведением в dPBS), 25 мкл разведенного оксотреморина и 50 мкл клеток добавляли в лунки для анализа агониста. Для измерения способности тестируемого соединения блокировать ингибируемую оксотреморином активность AC 25 мкл форсколина и оксотреморина (конечные концентрации 25 мкМ и 5 мкМ, соответственно, разведенные в dPBS), 25 мкл разведенного тестируемого соединения и 50 мкл клеток добавляли в оставшиеся лунки для анализа.
Реакции инкубировали в течение 10 минут при 37°C и останавливали добавлением 100 мкл охлажденного на льду буфера для обнаружения. Планшеты герметично закрывали, инкубировали в течение ночи при комнатной температуре и считывали на следующее утро с использованием жидкостного сцинтилляционного счетчика PerkinElmer TopCount (PerkinElmer Inc., Wellesley, MA). Количество продуцированного цАМФ (пмоль/лунку) подсчитывали на основе числа импульсов для образцов и стандартов цАМФ, как описано в руководстве производителя для пользователя. Данные анализировали методом нелинейного регрессионного анализа с помощью пакета программного обеспечения GraphPad Prism (GraphPad Software, Inc., San Diego, CA) с использованием нелинейной регрессии и односайтового уравнения конкурирования. Для расчета Ki использовали уравнение Ченга-Прусова с использованием кривой концентрации оксотреморина-ответ и концентрации оксотреморина в анализе в качестве KD и [L] соответственно. Значения Ki преобразовывали в величины pKi для определения среднего геометрического значения и 95% доверительных интервалов. Данные суммарные статистические данные затем преобразовывали обратно в величины Ki для описания данных.
В данном анализе более низкие величины Ki указывали, что тестируемое соединение обладало более высокой функциональной активностью в отношении тестируемого рецептора. Как установлено, иллюстративные соединения по изобретению, которые были протестированы в данном анализе, имели значения Ki меньше чем 10 нМ для блокады ингибирования оксотреморином форсколин-опосредованного накопления цАМФ в клетках СНО-К1, стабильно экспрессирующих рецептор hM2. Например, было установлено, что соединение примера 1 имеет величину Ki меньше чем 5 нМ.
B. Блокирование агонист-опосредованного [ 35 S]GTPγS-связывания
Во втором функциональном анализе функциональную эффективность тестируемых соединений можно определить путем измерения способности соединений блокировать стимулированное оксотреморином [35S]GTPγS-связывание в клетках CHO-K1, экспрессирующих рецептор hM2.
К моменту использования замороженные мембраны размораживали и затем разбавляли в буфере для анализа при конечной концентрации целевой ткани 5-10 мкг белка на лунку. Мембраны гомогенизировали в течение короткого промежутка времени с использованием разрушителя для тканей Polytron PT-2100 и затем добавляли в планшеты для анализа.
В каждом эксперименте определяли величину EC90 (эффективная концентрация для 90%-ной максимальной ответной реакции) для стимуляции связывания [35S]GTPγS с помощью агониста оксотреморина.
Для определения способности тестируемого соединения ингибировать стимулированное оксотреморином связывание [35S]GTPγS в каждую лунку 96-луночного планшета добавляли следующее: 25 мкл буфера для анализа с [35S]GTPγS (0,4 нM), 25 мкл оксотреморина (EC90) и GDP (3 мкM), 25 мкл разведенного тестируемого соединения и 25 мкл мембран клеток CHO, экспрессирующих рецептор hM2. Планшеты для анализа затем инкубировали при 37°C в течение 60 минут. Планшеты для анализа фильтровали через предварительно обработанные BSA фильтры GF/B с использованием 96-луночного харвестера PerkinElmer. Планшеты ополаскивали охлажденным на льду промывочным буфером в течение 3×3 секунд и затем сушили на воздухе и в вакууме. В каждую лунку добавляли сцинтилляционную жидкость Microscint-20 (50 мкл), и каждый планшет герметически закрывали, и радиоактивность регистрировали с использованием верхнего счетчика (PerkinElmer). Данные анализировали методом нелинейной регрессии с помощью пакета программ GraphPad Prism (GraphPad Software, Inc., San Diego, CA) с использованием нелинейной регрессии, односайтового уравнения конкурирования. Уравнение Ченга-Прусова (Cheng-Prusoff) применяли для расчета Ki с использованием величин IC50 для кривой концентрация-ответ для тестируемого соединения и концентрации оксотреморина в анализе в качестве KD и [L], концентрации лиганда соответственно.
В данном анализе более низкие величины Ki указывали, что тестируемое соединение обладало более высокой функциональной активностью в отношении тестируемого рецептора. Как установлено, иллюстративные соединения по изобретению, которые были протестированы в данном анализе, имели значения Ki меньше чем 10 нМ для блокады стимулированного оксотреморином [35S]GTPγS-связывания в клетках CHO-K1, экспресирующих рецептор hM2. Например, было установлено, что соединение примера 1 имеет величину Ki меньше чем 5 нМ.
C. Определение блокирования агонист-опосредованного высвобождения кальция с помощью анализа FLIPR
Подтипы мускариновых рецепторов (рецепторы M1, M3 и M5), которые связываются с белками Gq, активируют путь фосфолипазы С (PLC) при связывании агониста с рецептором. В результате, активированный PLC гидролизует фосфатилинозитолдифосфат (PIP2) до диацилглицерина (DAG) и фосфатидил-1,4,5-трифосфата (IP3), который в свою очередь генерирует высвобождение кальция из внутриклеточных источников, т.е. эндоплазматической и саркоплазматической сети. В анализе FLIPR (Molecular Devices, Sunnyvale, CA) используется данное увеличение внутриклеточного кальция за счет кальцийчувствительного красителя (Fluo-4AM, Molecular Probes, Eugene, OR), который является флуоресцентным при связывании со свободным кальцием. Такая возникающая флуоресценция измеряется в режиме реального времени с помощью FLIPR, который обнаруживает изменение во флуоресценции от монослоя клонированных клеток, содержащих рецепторы M1 и M3 человека и M5 шимпанзе. Антагонистическая эффективность может быть определена по способности антагонистов ингибировать агонист-опосредованное увеличение внутриклеточного кальция.
Для FLIPR анализа стимуляции кальция СНО клетки стабильно экспрессирующие рецепторы hM1, hM3 и cM5 высевали в 96-луночные планшеты FLIPR за ночь до проведения анализа. Размещенные клетки дважды промывали с помощью устройства Cellwash (MTX Labsystems, Inc.) с использованием буфера FLIPR (10 мM HEPES, pH 7,4, 2 мM хлорида кальция, 2,5 мM пробенецида в HBSS без кальция и магния) для удаления среды для роста клеток и оставляли 50 мкл/лунку буфера FLIPR. Клетки затем инкубировали в присутствии 50 мкл/лунку 4 мкM FLUO-4AM (готовили 2X раствор) в течение 40 минут при 37°C, 5% диоксида углерода. После периода инкубации с красителем клетки промывали дважды буфером FLIPR, оставляя в конце объем 50 мкл/лунку.
Для определения эффективности антагониста первоначально определяют дозазависимую стимуляцию внутриклеточного высвобождения Ca2+ для оксотреморина таким образом, что эффективность антагониста позднее может быть измерена относительно стимуляции оксотреморином при концентрации EC90. Клетки первоначально инкубировали с буфером для разведения соединения в течение 20 минут с последующим добавлением агониста, которое проводили с помощью FLIPR. Значения EC90 для оксотреморина получали в соответствии со способом, подробно изложенным далее в разделе, посвященном измерению и предварительной обработке данных FLIPR, в сочетании с формулой ECF = ((F/100-F)^1/H)*EC50. Концентрацию оксотреморина 3×ECF получали в планшетах для стимуляции таким образом, что EC90 концентрацию оксотреморина добавляли в каждую лунку в планшеты для анализа ингибирования антагониста.
Параметры, использованные для FLIPR, являются следующими: длина экспозиции 0,4 секунды, мощность лазера 0,5 ватт, длина волны возбуждения 488 нм и длина волны испускания 550 нм. Базовую линию определяют путем измерения изменения флуоресценции в течение 10 секунд перед добавлением агониста. После стимуляции с помощью агониста FLIPR непрерывно измерял изменение флуоресценции каждые 0,5-1 секунду в течение 1,5 минут для фиксирования максимального изменения флуоресценции.
Изменение флуоресценции выражали как максимальную флуоресценцию за вычетом базовой флуоресценции для каждой лунки. Полученные необработанные данные анализировали относительно логарифма концентрации лекарственного средства методом нелинейной регрессии с помощью программы GraphPad Prism (GraphPad Software, Inc., San Diego, CA) с использованием встроенной модели сигмоидальная доза-ответная реакция.
Ki значения антагониста определяли с помощью программы Prism с использованием значения EC50 для оксотреморина в качестве KD и значения EC90 для концентрации лиганда в соответствии с уравнением Ченга-Прусова (Cheng & Prusoff, 1973).
В данном анализе более низкие величины Ki указывали, что тестируемое соединение обладало более высокой функциональной активностью в отношении тестируемого рецептора. Как установлено, иллюстративные соединения по изобретению, которые были протестированы в данном анализе, имели значения Ki меньше чем 10 нМ для блокады агонист-опосредованного высвобождения кальция в клетках СНО, стабильно экспрессирующих рецептор hM3. Например, было установлено, что соединение примера 1 имеет величину Ki меньше чем 10 нМ для рецептора hM3.
Анализ 3
Определение продолжительности защиты бронхов на модели индуцированного ацетилхолином сужения бронхов у морской свинки
Данный анализ in vivo использовали для оценки бронхопротекторного действия тестируемых соединений, проявляющих антагонистическую активность в отношении мускариновых рецепторов.
Группы из шести самцов морских свинок Duncan-Hartley (HsdPoc:DH) Harlan, Madison, WI) весом от 250 до 350 г идентифицировали индивидуально с помощью карточек на клетках. Во время исследования животные имели свободный доступ к пище и воде. Тестируемые соединения вводили путем ингаляции в течение 10 минут в камере дозирования для воздействия на все тело (R&S Molds, San Carlos, CA). Камеры дозирования были расположены таким образом, что аэрозоль одновременно доставлялся в 6 отдельных камер от центрального трубопровода. Морских свинок подвергали действию аэрозоля тестируемого соединения или носителя (WFI). Данные аэрозоли генерировали из водных растворов с использованием набора небулайзеров LC Star Nebulizer Set (модель 22F51, PARI Respiratory Equipment, Inc. Midlothian, VA), управляемых смесью газов (CO2 = 5%, O2 = 21% и N2 = 74%) при давлении 22 фт/кв.дюйм. Поток газа через небулайзер при таком управляющем давлении был приблизительно 3 л/минуту. Генерируемые аэрозоли поступали в камеры под положительным давленим. Во время доставки аэрозольных растворов воздух для разбавления не использовался. В течение 10 минут распыления расходовалось приблизительно 1,8 мл раствора. Это измеряли гравиметрически путем сравнения веса наполненных небулайзеров перед распылением и после распыления.
Бронхопротекторное действие тестируемых соединений, вводимых путем ингаляции, оценивали с использованием плетизмографии всего тела через 1, 5, 24, 48 и 72 часа после введения дозы.
За сорок пять минут перед началом оценки легких каждую морскую свинку анестезировали путем внутримышечной инъекции кетамина (43,75 мг/кг), ксилазина (3,50 мг/кг) и ацепромазина (1,05 мг/кг). После того, как место для хирургического вмешательства выбривали и очищали 70%-ным спиртом, делали 2-3 см надрез средней линии вентрального аспекта шеи. Затем выделяли шейную вену и канюлировали с использованием наполненного физиологическим раствором катетера (PE-50, Becton Dickinson, Sparks, MD), чтобы иметь возможность внутривенной инфузии ACh (Sigma-Aldrich, St. Louis, MO) в физиологическом растворе. Трахею затем рассекали для освобождения и канюлировали с использованием тефлоновой трубочки 14G (#NE- 014, Small Parts, Miami Lakes, FL). Если требовалось, анестезию поддерживали дополнительными внутримышечными инъекциями вышеуказанной анестезирующей смеси. Глубину анестезии контролировали и регулировали в том случае, если животное проявляло ответную реакцию на щипание его лапы, или если частота дыхания была более 100 вдохов/минуту.
После завершения канюлирования животных помещали в плетизмограф (#PLY3114, Buxco Electronics, Inc., Sharon, CT) и вставляли пищеводную канюлю давления (PE-160, Becton Dickinson, Sparks, MD) для измерения управляющего легочного давления (давление). Тефлоновую трахейную трубку присоединяли к входному отверстию плетизмографа, чтобы морская свинка имела возможность дышать комнатным воздухом снаружи камеры. Камеру затем плотно закрывали. Для поддержания температуры тела использовали нагревающую лампу, и легкие морской свинки 3 раза продували 4 мл воздуха с использованием 10 мл калиброванного шприца (#5520 Series, Hans Rudolph, Kansas City, MO), чтобы иметь гарантию того, что нижняя часть дыхательных путей не разрушена и что животное не страдает от гипервентиляции.
После того, как было определено, что значения для базовой линии находятся в диапазоне 0,3-0,9 мл/cм H2O для поддатливости и в диапазоне 0,1-0,199 cм H2O/мл в секунду для сопротивляемости, начинали проводить оценку легких. Компьютерная программа Buxco для проведения легочных измерений делала возможным сбор и обработку легочных параметров.
При включении данная программа начинала экспериментальный протокол и сбор данных. Изменения объема во времени, которые происходили в плетизмографе при каждом вдохе, измеряли с помощью датчика давления Buxco. При интегрировании данного сигнала во времени для каждого вдоха рассчитывали размеры потока. Данный сигнал вместе с изменениями управляющего легочного давления, данные для которого собирали с использованием датчика давления Sensym (#TRD4100), был связан через предусилитель Buxco (MAX 2270) с интерфейсом сбора данных (#'s SFT3400 и SFT3813). Все другие легочные параметры были производными от этих двух видов информации на входе.
Данные для базовой линии получали в течение 5 минут, после чего морским свинкам вводили ACh. ACh (0,1 мг/мл) вводили в виде инфузии внутривенно в течение 1 минуты с использованием насоса для шприцов (sp210iw, World Precision Instruments, Inc., Sarasota, FL) в следующих дозах и в течение определенного времени от начала эксперимента: 1,9 мкг/минуту через 5 минут, 3,8 мкг/минуту через 10 минут, 7,5 мкг/минуту через 15 минут, 15,0 мкг/минуту через 20 минут, 30 мкг/минуту через 25 минут и 60 мкг/минуту через 30 минут. Если сопротивляемость и поддатливость не возращались к значениям базовой линии в течение 3 минут после каждой дозы ACh, легкие морской свинки надували 3 раза 4 мл воздуха из 10 мл калиброванного шприца. Регистрируемые параметры легких включали частоту дыхания (вдохов в минуту), поддатливость (мл/cм H2O) и легочную сопротивляемость (cм H2O/ мл в секунду). После измерения легочной функции на 35-й минуте данного протокола морскую свинку удаляли из плетизмографа и проводили эвтаназию, вызывая асфиксию диоксидом углерода.
Данные оценивали одним или обоими следующими способами:
(а) Легочную сопротивляемость (RL, см H2O/мл в секунду) рассчитывали из соотношения «изменение давления» к «изменению потока». RL ответ на ACh (60 мкг/мин, IH) обсчитывали для групп носителя и тестируемого соединения. Вычисляли средний ACh-ответ в группах животных, обработанных носителем и тестируемым соединением. Рассчитывали средний ответ на Ach для животных, обработанных носителем, в каждый момент времени обработки и использовали для расчета % ингибирования ответа на Ach в соответствующий момент обработки для каждой дозы тестируемого соединения. Кривые ингибирования доза-ответ для 'RL' хорошо соответствовали четырем параметрам логистического уравнения при использовании программы GraphPad Prism, версия 3.00 для Windows (GraphPad Software, San Diego, California) для оценки бронхопротекторной ID50 (дозы, требуемой для ингибирования на 50% ответной реакции сужения бронхов на введение ACh (60 мкг/мин)). Использованное уравнение было следующим:
Y = Мin + (Max-Мin)/(1 + 10 ((log ID50-X)* Hillslope))
где X представляет собой логарифм дозы, Y представляет собой ответную реакцию (% ингибирования индуцированного ACh повышения RL). Y начинается при минимуме (Мin) и приближается асимптотически к максимуму (Мax) по кривой сигмоидальной формы.
(b) Количество PD2, которое определяли как количество ACh или гистамина, требуемое для того, чтобы вызвать удвоение базовой линии легочной сопротивляемости, рассчитывали с использованием величин легочной сопротивляемости, полученных из величин потока и давления для диапазона инъекций ACh или гистамина, в соответствии со следующим уравнением (которое получено из уравнения, использованного для расчета величин PC20, описанного в American Thoracic Society. Guidelines for methacholine и exercise challenge testing - 1999, Am J Respir Crit Care Med. 161: 309-329 (2000)):
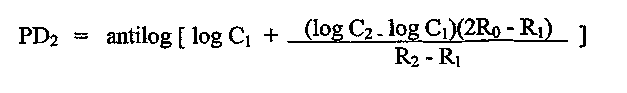
где
C1 = концентрация ACh или гистамина, предшествующая C2
C2 = концентрация ACh или гистамина, приводящая по крайней мере к 2-кратному увеличению сопротивляемости легких (RL)
R0 = базовое значение RL
R1 = значение RL после C1
R2 = значение RL после C2
Эффективную дозу определяли как дозу, которая ограничивала ответную реакцию сужения бронхов на дозу ACh в 50 мкг/мл до удвоения базовой линии сопротивляемости легких (PD2(50)).
Статистический анализ данных проводили с использованием двухстороннего t-теста Стьюдента. Величина P <0,05 рассматривалась как значимая.
Обычно предпочтительными в данном анализе являлись тестируемые соединения, имевшие PD2(50) менее чем примерно 200 мкг/мл для ACh-индуцированного сужения бронхов через 1,5 часа после введения дозы. Например, было установлено, что соединение примера 1 имеет PD2(50) менее чем примерно 200 мкг/мл для ACh-индуцированного сужения бронхов через 1,5 часа после введения дозы.
Анализ 4
Анализ слюноотделения у морских свинок при ингаляции
Морским свинкам (Charles River, Wilmington, MA) весом 200-350 г предоставляли возможность акклиматизации в колонии морских свинок собственного производства в течение по крайней мере 3 дней после прибытия. Тестируемое соединение или носитель дозировали путем ингаляции (IH) в течение 10-минутного промежутка времени в секторной камере для дозирования (R&S Мolds, San Carlos, CA). Тестируемые растворы растворяли в стерильной воде и доставляли с использованием небулайзера, наполненного 5,0 мл дозируемого раствора. Морских свинок удерживали в камере для ингаляции в течение 30 минут. В это время морские свинки были ограничены площадью примерно в 110 кв. см. Такое пространство было нормальным для того, чтобы животные могли свободно поворачиваться и чистить себя. Через 20 минут аклиматизации морских свинок подвергали действию аэрозоля, генерируемого из небулайзера LS Star Nebulizer Set (модель 22F51, PARI Respiratory Equipment, Inc. Midlothian, VA), управляемого обычным воздухом при давлении 22 фт/кв.дюйм. После завершения распыления моских свинок обследовали через 1,5, 6, 12, 24, 48 или 72 часа после обработки.
Морских свинок анестезировали за один час перед тестированием с помощью внутримышечной инъекции смеси кетамина 43,75 мг/кг, ксилазина 3,5 мг/кг и ацепромазина 1,05 мг/кг в объеме 0,88 мл/кг. Животных помещали брюшком вверх на нагретую (37°C) подстилку при 20-градусном наклоне головы сверху вниз. Cложенный в 4 раза марлевый тампон 2×2 дюйма (Марлевые тампоны для общего применения (Nu-Gauze General-use sponges), Johnson и Johnson, Arlington, TX) вставляли в рот морской свинки. Через пять минут вводили мускариновый агонист пилокарпин (3,0 мг/кг, подкожно) и марлевый тампон выбрасывали и заменяли на новый, который был предварительно взвешен. Слюну собирали в течение 10 минут, и в этот момент времени марлевый тампон взвешивали и разницу в весе регистрировали для определения количества накопившейся слюны (в мг). Рассчитывали среднее количество собранной слюны для животных, получавших носитель и каждую дозу тестируемого соединения. Среднее значение для группы носителя рассматривали как 100% слюноотделение. Результаты рассчитывали с использованием средних значений (n = 3 или больше). Доверительные интервалы (95%) рассчитывали для каждой дозы в каждый момент времени с использованием двухстороннего ANOVA. Данная модель представляет собой модифицированную версию способа, описанного в публикации Rechter, «Estimation of anticholinergic drug effects in mice by antagonism against pilocarpine-induced salivation», Ata Pharmacol Toxicol 24:243-254 (1996).
Рассчитывали средний вес слюны у обработанных носителем животных в каждый момент времени при обработке, и использовали этот параметр для расчета % ингибирования слюноотделения в соответствующий момент обработки для каждой дозы. Данные ингибирования доза-ответ хорошо соответствовали четырем параметрам логистического уравнения при использовании программы GraphPad Prism, версия 3.00 для Windows (GraphPad Software, San Diego, California) для оценки ID50 действия, антистимулирующего слюноотделение (доза, требуемая для 50%-ного ингибирования, вызванного пилокарпином слюноотделения). Используемое уравнение было следующим:
Y = Min + (Max-Min)/(1 + 10 ((log ID50-X)* Hillslope))
где X представляет собой логарифм дозы, Y представляет собой ответную реакцию (% ингибирования слюноотделения). Y начинается в минимуме (Min) и приближается асимптотически к максимуму (Max) по кривой сигмоидальной формы.
Соотношение ID50, антистимулирующей слюноотделение, к бронхопротекторной
ID50 использовали для расчета индекса кажущейся селективности легких для тестируемого соединения. Обычно, предпочтительными являлись соединения, имеющие индекс кажущейся селективности легких больше примерно 5. Например, в данном анализе, соединение примера 1 имело индекс кажущейся селективности легких больше примерно 5.
Анализ 5
Метахолин-индуцированные депрессорные ответные реакции у находящихся в сознании морских свинок
В данных исследованиях использовали здоровых взрослых самцов морских свинок Sprague-Dawley (Harlan, Indianapolis, IN), весом между 200 и 300 г. При анестезии изофлураном (для эффективности) животным вводили обычные катетеры в сонную артерию и шейную вену (трубки PE-50). Катетеры выводили наружу с использованием подкожного прохода в подлопаточную область. Все хирургические надрезы зашивали шелком 4-0 Ethicon и катетеры закрывали гепарином (1000 единиц/мл). Каждому животному вводили физиологический раствор (3 мл, подкожно) в конце хирургического вмешательства, а также бупренорфин (0,05 мг/кг, внутримышечно). Животных оставляли приходить в сознание на нагреваемой подстилке перед возвращением в места их содержания.
Приблизительно через 18-20 часов после хирургии животных взвешивали, и катетер сонной артерии на каждом животном соединяли с датчиком для регистрации артериального давления. Артериальное давление и частоту сердечных сокращений регистрировали с использованием системы учета Biopac MP-100. Животным давали возможность акклиматизироваться и стабилизироваться в течение 20 минут.
Каждому животному делали инъекцию MCh (0,3 мг/кг, внутривенно) через линию шейной вены, и контролировали сердечно-сосудистую ответную реакцию в течение 10 минут. Затем животных помещали в камеру дозирования, где помещалось все тело, которая была соединена с небулайзером, содержащим тестируемое соединение или раствор-носитель. Раствор затем распыляли в течение 10 минут с использованием газовой смеси, состоящей из пригодного для дыхания воздуха и 5% диоксида углерода со скоростью потока 3 литра/минуту. Животных затем удаляли из камеры для размещения всего тела и возвращали в соответствующие клетки. Через 1,5 и 24 часа после дозирования животным повторно делали инъекцию MCh (0,3 мг/кг, внутривенно) и определяли гемодинамический ответ. После этого проводили эвтаназию животных с помощью пентобарбитала натрия (150 мг/кг, внутривенно).
MCh вызывает снижение среднего артериального давления (MAP) и уменьшение частоты сердечных сокращений (брадикардия). Пик снижения MAP от базовой линии (ответные реакции на депрессор) измеряли для каждой инъекции MCh (перед и после дозирования путе мингаляции (IH)). Брадикардическое действие для анализа не использовали, поскольку данные ответные реакции не были сильными и воспроизводимыми. Влияние обработки на ответные реакции на MCh выражали как % ингибирования (среднее значение +/- SEM) контрольных ответных реакций на депрессор. Для исследования влияния обработки и времени обработки использовали двухсторонний ANOVA с подходящим последующим тестом. Депрессорные ответные реакции на MCh были относительно неизменными через 1,5 и 24 часа после ингаляционного дозирования носителя.
Соотношение ID50 антидепрессора к ID50 бронхопротектора использовали для расчета кажущейся селективности легких для тестируемого соединения. Обычно, предпочтительными являются соединения, имеющие кажущийся индекс селективности легких больше 5. Например, в данном анализе соединение примера 1 имело кажущийся индекс селективности легких больше 5.
Хотя настоящее изобретение было описано со ссылкой на его конкретные аспекты или варианты осуществления, специалистам в данной области следует понимать, что могут быть сделаны различные изменения, или разные эквиваленты могут быть заменены, не отступая от истинной сути и объема изобретения. Дополнительно, в степени, допускаемой применяемыми патентными состояниями и правилами, все процитированные здесь публикации, патенты и патентные заявки включены в данное описание путем ссылки во всей своей полноте в той степени, как если бы каждый документ был по отдельности включен в данное описание путем ссылки.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ БИФЕНИЛА | 2004 |
|
RU2330841C2 |
| ДИАМИДНЫЕ СОЕДИНЕНИЯ, ОБЛАДАЮЩИЕ АКТИВНОСТЬЮ АНТАГОНИСТА МУСКАРИНОВОГО РЕЦЕПТОРА И АКТИВНОСТЬЮ АГОНИСТА β АДРЕНЕРГИЧЕСКОГО РЕЦЕПТОРА | 2010 |
|
RU2676686C2 |
| ДИАМИДНЫЕ СОЕДИНЕНИЯ, ОБЛАДАЮЩИЕ АКТИВНОСТЬЮ АНТАГОНИСТА МУСКАРИНОВОГО РЕЦЕПТОРА И АКТИВНОСТЬЮ АГОНИСТА В2 АДРЕНЕРГИЧЕСКОГО РЕЦЕПТОРА | 2010 |
|
RU2543716C2 |
| ПРОИЗВОДНЫЕ БИЦИКЛО[2,2,1]ГЕПТ-7-ИЛАМИНА И ИХ ПРИМЕНЕНИЯ | 2006 |
|
RU2442771C2 |
| ГУАНИДИНСОДЕРЖАЩИЕ СОЕДИНЕНИЯ, ПРИМЕНИМЫЕ В КАЧЕСТВЕ АНТАГОНИСТОВ МУСКАРИНОВЫХ РЕЦЕПТОРОВ | 2008 |
|
RU2480458C2 |
| КРИСТАЛЛИЧЕСКАЯ ФОРМА БИФЕНИЛЬНОГО СОЕДИНЕНИЯ | 2005 |
|
RU2381223C2 |
| ПРОИЗВОДНЫЕ 4-ПИПЕРИДИНИЛАЛКИЛАМИНА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ АНТАГОНИСТИЧЕСКИМ ДЕЙСТВИЕМ В ОТНОШЕНИИ МУСКАРИНОВЫХ РЕЦЕПТОРОВ | 2002 |
|
RU2300524C2 |
| ПРОИЗВОДНЫЕ 1-(АЛКИЛАМИНОАЛКИЛ-ПИРРОЛИДИН/ПИПЕРИДИНИЛ)-2,2-ДИФЕНИЛАЦЕТАМИДА В КАЧЕСТВЕ АНТАГОНИСТОВ МУСКАРИНОВЫХ РЕЦЕПТОРОВ | 2004 |
|
RU2337095C2 |
| ФЕНОКСИЗАМЕЩЕННЫЕ ПИРИМИДИНЫ ДЛЯ ИСПОЛЬЗОВАНИЯ В КАЧЕСТВЕ МОДУЛЯТОРОВ ОПИОИДНЫХ РЕЦЕПТОРОВ | 2010 |
|
RU2554870C2 |
| АЗОЛЬНЫЕ И ТИАЗОЛЬНЫЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЯ | 2006 |
|
RU2436779C2 |
Изобретение относится к новым соединениям формулы I:
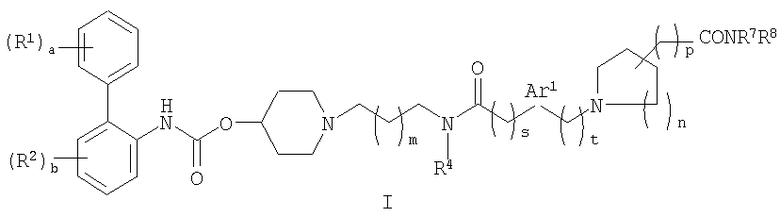
где а равно 0 или 1; R1 означает галоген; b равно 0 или 1; R2 означает галоген; m равно 0 или 1; R4 выбран из водорода, (1-4С)алкила и (3-4С)циклоалкила; s равно 0, 1 или 2; Ar1 представляет собой фениленовую группу или (3-5С)гетероариленовую группу, содержащую 1 или 2 гетероатома, независимо выбранных из кислорода, азота или серы; где фениленовая или гетероариленовая группа замещена (R5)q, где q равно 0 или целому числу от 1 до 4, и каждый R5 независимо выбирают из галогена или (1-4С)алкокси; t равно 0, 1 или 2; n равно 0 или целому числу от 1 до 3; p равно 0 или 1; и R7 и R8 независимо представляют собой водород или (1-4С)алкил; или к его фармацевтически приемлемым солям или сольватам, или к его стереоизомерам. Изобретение также относится к фармацевтической композиции, способу получения соединений по любому одному из п.п.1-13, к лекарственному средству, а также к применению соединений. Технический результат - получение новых биологически активных соединений, обладающих активностью в качестве антагонистов мускаринового рецептора. 8 н. и 19 з.п. ф-лы.
1. Соединение формулы I:
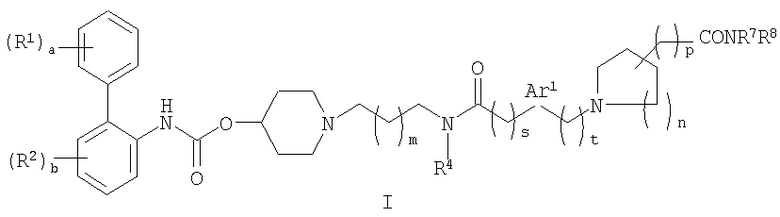
где а равно 0 или 1;
R1 означает галоген;
b равно 0 или 1;
R2 означает галоген;
m равно 0 или 1;
R4 выбран из водорода, (1-4С)алкила и (3-4С)циклоалкила;
s равно 0, 1 или 2;
Ar1 представляет собой фениленовую группу или (3-5С)гетероариленовую группу, содержащую 1 или 2 гетероатома, независимо выбранные из кислорода, азота или серы; где фениленовая или гетероариленовая группа замещена (R5)q, где q равно 0 или целому числу от 1 до 4, и каждый R5 независимо выбирают из галогена или (1-4С)алкокси;
t равно 0, 1 или 2;
n равно 0 или целому числу от 1 до 3;
p равно 0 или 1; и
R7 и R8 независимо представляют собой водород или (1-4С)алкил;
или его фармацевтически приемлемая соль или сольват, или стереоизомер.
2. Соединение по п.1, где каждый из а и b равен 0.
3. Соединение по п.1, где m равно 0, s равно 0, и t равно 1.
4. Соединение по п.1, где группа -CONR7R8 находится в пара-положении, и n равно 2.
5. Соединение по п.1, где Ar1 представляет собой фен-1,3-илен, фен-1,4-илен, 2,4-тиенилен или 2,5-тиенилен; где фениленовая или тиениленовая группа необязательно замещена одним или двумя заместителями R5.
6. Соединение по п.5, где Ar1 представляет собой фен-1,4-илен или 2,4-тиенилен, необязательно замещенный одним или двумя заместителями R5.
7. Соединение по любому одному из пп.1-5, где R4 выбран из водорода, метила и этила.
8. Соединение по любому одному из пп.1-5, где R7 выбран из водорода, метила, этила, н-пропила и изопропила; и R8 представляет собой водород.
9. Соединение по любому одному из пп.1-5, где R7 и R8 представляют собой этил.
10. Соединение по п.1, где каждый a и b равен 0; m равно 0; s равно 0; t равно 1; Ar1 представляет собой фен-1,4-илен, необязательно замещенный одним или двумя заместителями R5; n равно 2, группа -CONR7R8 находится в пара-положении; и R8 представляет собой водород.
11. Соединение по п.10, где R5 независимо выбирают из галогена или (1-4С)алкокси, где каждая алкоксильная группа необязательно замещена 1-3 фторными заместителями.
12. Соединение по п.1, выбранное из:
1-(2-{[4-(4-карбамоилпиперидин-1-илметил)бензоил]метиламино}этил)пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты;
1-(2-{[4-(4-карбамоилпиперидин-1-илметил)бензоил]этиламино}этил)пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты;
1-(2-{метил-[4-(4-метилкарбамоилпиперидин-1-илметил)бензоил]амино}этил)пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты;
1-(2-{[4-(4-этилкарбамоилпиперидин-1-илметил)бензоил]метиламино}этил)пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты;
1-(2-{метил-[4-(4-пропилкарбамоилпиперидин-1-илметил)бензоил]амино}этил)пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты;
1-(2-{[4-(4-изопропилкарбамоилпиперидин-1-илметил)бензоил]метиламино}этил)пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты;
1-{2-[4-(4-карбамоилпиперидин-1-илметил)-2-фторбензоиламино]этил}пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты;
1-(2-{[2,5-дибром-4-(4-карбамоилпиперидин-1-илметил)бензоил]метиламино}этил)пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты;
1-(2-{[4-(4-карбамоилпиперидин-1-илметил)-2-фторбензоил]метиламино}этил)пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты;
1-{2-[4-(4-диэтилкарбамоилпиперидин-1-илметил)-2-фторбензоиламино]этил}пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты;
1-(2-{[4-(4-диэтилкарбамоилпиперидин-1-илметил)-2-фторбензоил]метиламино}этил)пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты;
1-(2-{[4-(4-диэтилкарбамоилпиперидин-1-илметил)бензоил]метиламино}этил)пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты;
1-(2-{[4-(3-(S)-диэтилкарбамоилпиперидин-1-илметил)бензоил]метиламино}этил)пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты;
1-(2-{[4-(2-карбамоилпиперидин-1-илметил)бензоил]метиламино}этил)пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты;
1-(2-{[4-(4-карбамоилпиперидин-1-илметил)-2-метоксибензоил]метиламино}этил)пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты;
1-(2-{[5-(4-карбамоилпиперидин-1-илметил)тиофен-2-карбонил]метиламино}этил)пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты;
1-(2-{[5-((R)-3-диэтилкарбамоилпиперидин-1-илметил)тиофен-2-карбонил]метиламино}этил)пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты;
1-(2-{[5-((R)-3-диэтилкарбамоилпиперидин-1-илметил)тиофен-2-карбонил]амино}этил)пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты;
1-(2-{[5-(4-карбамоилпиперидин-1-илметил)тиофен-2-карбонил]амино}этил)пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты;
1-(2-{[5-((R)-3-диэтилкарбамоилпиперидин-1-илметил)-1Н-пиррол-2-карбонил]метиламино}этил)пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты;
1-(2-{[5-(4-карбамоилпиперидин-1-илметил)-1Н-пиррол-2-карбонил]метиламино}этил)пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты;
1-(2-{[5-((R)-3-диэтилкарбамоилпиперидин-1-илметил)фуран-2-карбонил]метиламино}этил)пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты;
1-(2-{[5-(4-диэтилкарбамоилпиперидин-1-илметил)фуран-2-карбонил]метиламино}этил)пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты;
1-(2-{[5-(4-карбамоилпиперидин-1-илметил)фуран-2-карбонил]амино}этил)пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты;
1-(2-{[5-((R)-3-диэтилкарбамоилпиперидин-1-илметил)фуран-2-карбонил]амино}этил)пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты;
1-[2-({3-[4-(3-карбамоилпиперидин-1-илметил)фенил]пропионил}метиламино)этил]пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты;
1-[2-({3-[4-(4-карбамоилпиперидин-1-илметил)фенил]пропионил}метиламино)этил]пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты;
1-(2-{3-[4-(4-карбамоилпиперидин-1-илметил)фенил]пропиониламино}этил)пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты;
1-(2-{3-[4-(4-диэтилкарбамоилпиперидин-1-илметил)фенил]пропиониламино}этил)пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты;
1-(2-{3-[4-(3-диэтилкарбамоилпиперидин-1-илметил)фенил]пропиониламино}этил)пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты;
1-{2-[4-(4-карбамоилпиперидин-1-илметил)бензоиламино]этил}пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты;
1-(2-{[4-(4-карбамоилпиперидин-1-илметил)-2-хлор-бензоил]метиламино}этил)пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты;
1-(2-{[4-(4-карбамоилпиперидин-1-илметил)-2-хлор-5-метоксибензоил]метиламино}этил)пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты; и
1-[2-({2-[4-(4-карбамоилпиперидин-1-илметил)фенил]ацетил}метиламино)этил]пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты;
или его фармацевтически приемлемая соль или сольват.
13. Соединение по п.12, выбранное из: 1-(2-{[4-(4-карбамоилпиперидин-1-илметил)бензоил]метиламино}этил)пиперидин-4-илового эфира бифенил-2-илкарбаминовой кислоты или его фармацевтически приемлемой соли или сольвата.
14. Фармацевтическая композиция, обладающая активностью в качестве антагонистов мускаринового рецептора, включающая фармацевтически приемлемый носитель и терапевтически эффективное количество соединения по любому из пп.1-13.
15. Способ получения соединения по любому одному из пп.1-13, включающий взаимодействие соединения формулы VI:
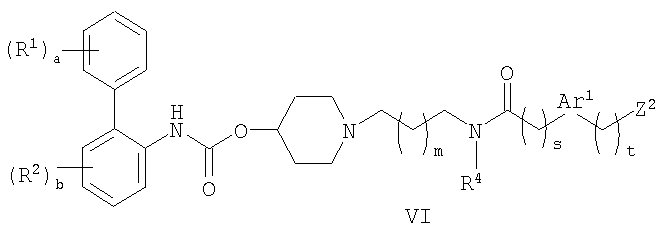
где Z2 представляет собой уходящую группу, с соединением формулы VII:
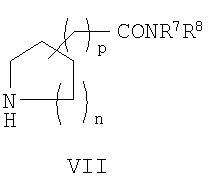
16. Способ получения соединения по любому одному из пп.1-13, включающий взаимодействие соединения формулы IX:
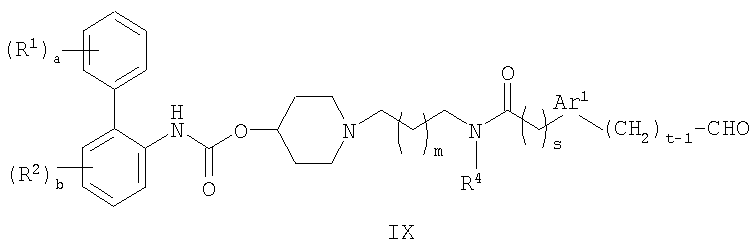
с соединением формулы VII в присутствии восстановителя.
17. Способ получения соединения по любому одному из пп.1-13,
включающий взаимодействие соединения формулы XVIII:
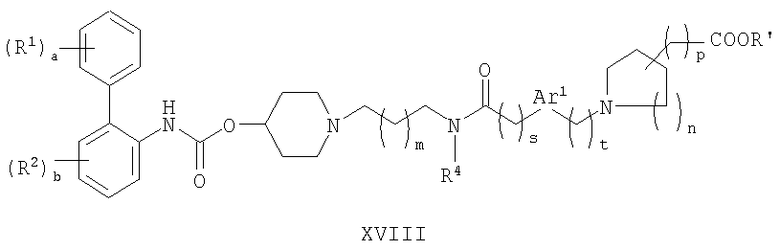
где R' представляет собой Н, -СН3 или -СН2СН3, с соединением формулы XIX:
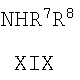
с получением соединения формулы I.
18. Способ по любому из пп.15-17, дополнительно включающий образование фармацевтически приемлемой соли соединения формулы I.
19. Соединение формулы I по п.1, полученное способом по любому из пп.15-18.
20. Соединение по любому из пп.1-13, предназначенное для применения в терапии или в качестве лекарственного средства, обладающего активностью в качестве антагонистов мускаринового рецептора.
21. Соединение по любому из пп.1-13, предназначенное для лечения легочного заболевания.
22. Соединение по любому из пп.1-13, предназначенное для антагонистического воздействия на мускариновый рецептор у млекопитающего.
23. Лекарственное средство, обладающее активностью в качестве антагонистов мускаринового рецептора и содержащее соединение по любому из пп.1-13.
24. Применение соединения по любому из пп.1-13 для получения лекарственного средства, обладающего активностью в качестве антагонистов мускаринового рецептора.
25. Применение по п.24, где лекарственное средство предназначено для лечения легочного заболевания.
26. Применение по п.24, где лекарственное средство предназначено для антагонистического воздействия на мускариновый рецептор у млекопитающего.
27. Лекарственное средство, предназначенное для лечения легочного заболевания и содержащее соединение по любому из пп.1-13.
| US 6693202 B1, 17.02.2004 | |||
| Топчак-трактор для канатной вспашки | 1923 |
|
SU2002A1 |
| ПРОИЗВОДНЫЕ ПИПЕРАЗИНА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1996 |
|
RU2126001C1 |

