Настоящее изобретение относится к способу получения очищенных, безопасных с вирусологической точки зрения препаратов антител из исходного раствора, содержащего антитела и загрязняющие примеси. Описывается процесс очистки гамма-глобулинов из плазмы крови человека и других первичных источников. Стадии инактивации и удаления вирусов включены в описанный здесь производственный процесс.
Преципитация и происходящее в результате удаление/инактивация вирусов
В 1940-х годах Cohn с соавторами ввели в практику фракционирование человеческой плазмы холодным этанолом. Несколько вариантов данной методики были позже разработаны для повышения чистоты и/или выхода различных интермедиатов. Было показано, что некоторые стадии в процессе фракционирования по Cohn являются эффективными в инактивации и удалении вирусов. В особенности в процессе очистки IgG отделение фракции I+III, по Cohn, чрезвычайно эффективно для данных целей. Определенные чувствительные вирусы (главным образом оболочечные вирусы) разрушаются под воздействием низких pH и EtOH, и значительная часть как оболочечных, так и безоболочечных вирусов может быть удалена путем распределения в преципитате I+III, который обычно отбрасывается.
В 1960-х годах было показано, что низкомолекулярные жирные кислоты (С6-С12) образуют нерастворимые комплексы с α- и β-глобулинами, в то время как γ-глобулины обычно столь же легко не преципитируются (Chanutin et al., 1960). Steinbruch с соавторами (1996) описали процесс очистки с каприлатом (т.е. октаноатом, солью С8-насыщенной жирной кислоты) в качестве преципитирующего агента. Неиммуноглобулиновые компоненты преципитировали из человеческой плазмы после разбавления ацетатным буфером до конечного значения pH 4,8. После добавления каприлата при интенсивном перемешивании получали раствор, обогащенный IgG. Чистота и выход зависели от количества каприловой кислоты, pH, молярности буфера и фактора разведения. Steinbruch с соавторами утверждали также, что преимущественным является добавление эффективного количества каприлата в две стадии с удалением промежуточных преципитатов. Вирусы, имеющие и не имеющие внешнюю оболочку, удаляют путем распределения в преципитате не-IgG-белков, как в случае отделения фракции I+III.
Хроматография
В нескольких патентах описана очистка растворов IgG в так называемом отрицательном режиме; IgG проходит без связывания (только в следовых количествах), в то время как бóльшая часть не-IgG-белков связывается с анионными лигандами (Bertolini et al. 1998, WO-A-98/05686; Lebing 1999, US-A-5886154; Friesen et al., 1986, CA 1201063). Комбинация преципитации каприлатом с последующей ионообменной хроматографией для очистки IgG была описана во многих публикациях. Одной из первых была работа Steinbuch с соавторами (1969). Он описал дальнейшую очистку IgG на DEAE-целлюлозе после преципитации каприлатом. В недавней работе Lebing с соавторами (2003) описано последовательное использование двух анионообменных колонок для удаления IgM, IgA, альбумина и других загрязняющих примесей. Lebing с соавторами скомбинировали оба эффекта, опосредованных действием каприлата, а именно, необходимое снижение содержания не-IgG-белков за счет преципитации, обеспечивающее отделение вирусов, и способность жирных кислот инактивировать оболочечные вирусы, реализованную на дополнительной стадии инкубации. Так называемый «pH-свинг» Lebing с соавторами (2003), заключающийся в последовательных стадиях разведения содержащей IgG пасты/преципитата при pH 4,2 и добавления каприлата одновременно с доведением значения pH до 5,2, подчеркивается как необходимый для процедуры обогащения IgG, таким образом нуждаясь в эффективном снижении содержания не-IgG-белков. Как некоторые другие загрязняющие примеси, такие как IgM и IgA, так и каприлат затем удаляют за счет выполнения вышеупомянутых последующих стадий ионообменной хроматографии.
Неожиданно авторами было обнаружено, что подобный обсуждаемый выше pH-сдвиг, описанный Lebing с соавторами, не является необходимым для достижения значительного эффекта очистки в результате добавления каприлата и удаления полученного преципитата. Вместо этого эффективное обогащение IgG достигается при поддержании постоянного pH на уровне 4,6-4,95 в течение всего процесса разведения пасты, инкубации с каприлатом и удаления преципитата. Также содержание загрязняющих примесей, в особенности альбумина, снижается более эффективно при поддержании постоянного pH на уровне 4,8-4,95. В то же время вирусы также удаляются. На последней стадии оставшиеся загрязняющие примеси и каприлат удаляют путем ионообменной хроматографии.
Классическая инактивация вирусов
Использование растворителя/детергента (S/D) и действие pH 4 являются хорошо известными способами и широко используются для (очистки) иммуноглобулинов. Обычно в подобных процессах используется обработка SD из-за превосходства этого метода в инактивации оболочечных вирусов (Biesert L. Clinical and Experimental Rheumatology 1996; 14: 47). Как оболочечные, так и безоболочечные вирусы чувствительны к действию низких значений pH, хотя оболочечные вирусы более чувствительны, нежели безоболочечные (Biesert. Clinical and Experimental Rheumatology 1996; 14: 47, Bos et al. Biologicals 1998; 26: 267, In Seop et al. J Microbiol Biotechnol 2001; 11: 619). В документе EP-A-0 525 502 описано комбинированное действие растворителя/детергента (S/D) и pH 4 в качестве стадий инактивации вирусов.
Фильтрация вирусов
Для повышения вирусологической безопасности растворы IgG фильтруют через мембраны с чрезвычайно малым размером пор (обычно от 15 до 50 нм) в условиях, удерживающих вирусы по механизму, в основном основанному на гель-фильтрации (Burnouf and Radosevich. Haemophilia 2003; 9: 24). Также для фильтрации иммуноглобулинов используются глубинные фильтры, удерживающие вирусы за счет ионообменной адсорбции.
Краткое описание изобретения
Настоящее изобретение относится к процессу очистки IgG с высоким выходом и сниженным временем процесса по сравнению с классическим процессом фракционирования по Кону-Онкли. IgG разводят в буфере в условиях кислого pH в диапазоне от 4,6 до 4,95, предпочтительно 4,9. Не-IgG-белки отделяют путем двухстадийной инкубации с каприлатом в диапазоне концентраций от 10 до 30 мМ, предпочтительно 20 мМ.
Для эффективной инактивации оболочечных вирусов и повышения инактивирующей активности всего процесса в целом может быть добавлена стадия инкубации, известная как обработка растворителем/детергентом, т.е. одновременная обработка растворителем (таким как TnBP) и детергентом, таким как Triton X-100, Tween 80 и т.д. К процедурам удаления вирусов также может быть добавлено удаление вирусов, к примеру, путем так называемой нанофильтрации либо использования заряженных глубинных фильтров. Далее в комбинации с вышеупомянутыми способами может быть использована обработка УФ-С. Подобные способы описаны, к примеру, в документах EP-A-0 840 624 и EP-A-0 422 007.
Далее каприлат/каприловая кислота могут быть заменены или скомбинированы с гептаноатом/гептановой кислотой для выполнения вышеупомянутых стадий преципитации и инкубации.
В продукте, получаемом описанным в заявке способом, вирусы инактивированы (либо удалены). Прионы также инактивируют/удаляют в процессе обработки каприлатом.
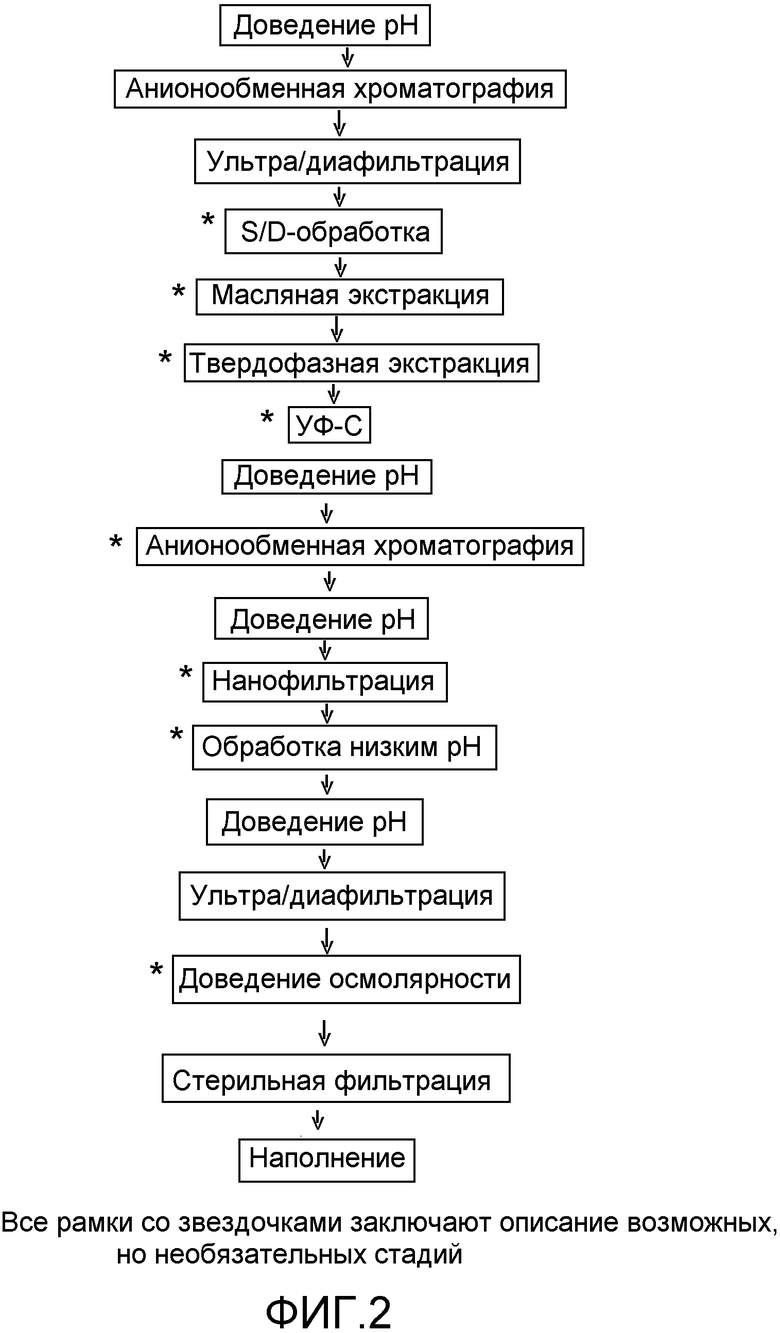
Фиг.1 и 2 представляют из себя блок-схему, иллюстрирующую специфические воплощения процесса согласно настоящему изобретению.
Подробное описание изобретения
Данное изобретение относится к способу получения очищенных, вирусологически безопасных и инактивированных с вирусологической точки зрения препаратов антител из исходного раствора, содержащего антитела и загрязняющие примеси, способ включает в себя следующие стадии:
(а) доведение pH исходного раствора приблизительно до 4,6-4,95, в частности приблизительно до 4,8-4,95, для получения промежуточного раствора;
(b) добавление каприлат- и/или гептаноат-ионов в промежуточный раствор при поддерживании pH на уровне приблизительно 4,6-4,95, в частности на уровне приблизительно 4,8-4,95, что приводит к образованию преципитата, при этом антитела присутствуют в основном в супернатанте;
(с) инкубирование раствора супернатанта в условиях, включающих в себя определенные концентрации каприлат- и/или гептаноат-ионов, время, pH и температуру; необязательно концентрирование и диафильтрацию раствора до доведения pH;
(d) нанесение фильтрованного раствора по меньшей мере на одну анионообменную смолу и необязательно - на две различные анионообменные смолы в условиях, обеспечивающих связывание загрязняющих примесей со смолой в отсутствие значительного связывания антител со смолой, с получением очищенных, вирусологически безопасных и инактивированных с вирусологической точки зрения препаратов антител.
В одном из воплощений настоящего изобретения инактивированный с вирусологической точки зрения раствор на стадии (d) приводят в контакт по меньшей мере с одной анионообменной смолой в диапазоне pH приблизительно 5,0-5,2. В случае применения двух хроматографических стадий с двумя анионообменниками вторая стадия должна выполняться в диапазоне pH от 6,7 до 6,9. Стадии (b) и (с) могут быть (но необязательно) повторены по меньшей мере один раз. Обработка каприлатом при pH 4,9 приводит к значительному удалению нежелательных белков.
В типичном случае исходный раствор содержит антитела, полученные из плазмы.
На стадии (d) может быть полезно привести инактивированный раствор в контакт с двумя различными анионообменными смолами в условиях, обеспечивающих связывание загрязняющих примесей со смолой в отсутствие значительного связывания антител со смолой.
Предпочтительно, антитела представляют собой иммуноглобулины G-типа.
Между двумя стадиями анионообменной хроматографии (AEX) pH может быть изменен, в частности, до 6,8±0,1. Сошедший с хроматографической колонки раствор может быть сконцентрирован до 60-90 мг/мл и подвергнут диафильтрации против, к примеру, фосфатного буфера. В другом воплощении настоящего изобретения сошедший после первой AEX с хроматографической колонки раствор одновременно обрабатывают растворителем и детергентом, предпочтительно TnBP и Triton X-100 соответственно, в предпочтительных концентрациях 1% Triton X-100 и 0,3% TnBP на протяжении от 4,5 до 8 часов, для инактивации вирусов с липидной оболочкой. Данный способ известен как обработка растворителем и раскрыт в документе EP-A-0 131 740 (включен посредством ссылки). Согласно изобретению детергенты инкубируемой смеси, в частности, удаляют путем экстракции из твердой и жидкой фаз. После экстракции твердой фазы pH раствора доводят до 6,7-6,9. Комбинация S/D-обработки и инактивации вирусов каприлатом приводит к получению более (вирусологически) безопасного продукта.
Раствор далее может быть подвергнут AEX на второй колонке, и pH сошедшего с колонки в результате AEX раствора может быть доведен, к примеру, до 3,5-4,5, в частности - до 4,0±0,1. Согласно изобретению раствор IgG с доведенным pH далее приводят в контакт с фильтром, удерживающим вирусы. Эта необязательная стадия в комбинации с обработкой каприлатом приводит к получению более вирусологически безопасного продукта.
Раствор IgG также может быть проинкубирован при 37°С±1 на протяжении по меньшей мере 24 часов. Для повышения уровня вирусологической безопасности препарата антител обработка УФ-C-излучением может быть использована в комбинации с обработкой фракции, содержащей антитела, каприлатом. Инактивационные методы по отдельности либо в комбинации с такими методами, как обработка TnBP, УФ-C, фильтрация вирусов либо нагревание, могут быть скомбинированы с процессом согласно настоящему изобретению.
Способ согласно настоящему изобретению может быть скомбинирован с методами удаления или инактивации прионов, к примеру, с фильтрационными, адсорбционными или хроматографическими методами согласно ранее раскрытому в данной области техники, к примеру, в документе EP-A-0 954 528, Trejo, S.R. et al., Vox Sanguinis, (2003) 84, 176-187. Раствор IgG, получаемый согласно настоящему изобретению, концентрируют для дальнейшего терапевтического использования, обычно до концентраций 5 или 10%, и осмолярность концентрата доводят до 200-400 мОсмоль/кг подходящей добавкой. Тем не менее, любые другие значения также допустимы в случае сохранения фармакологической приемлемости. Подобные добавки хорошо известны экспертам и включают в себя (без ограничения общности) сахара, сахарные спирты и аминокислоты. pH раствора IgG может быть доведен до 3,5-6,0, в частности до 4,0-5,5. Далее раствор IgG стерильно фильтруют и разливают в стеклянные бутыли или пластиковые контейнеры. Альтернативно раствор, сошедший с колонки после первого либо второго этапа AEX, подвергают фильтрованию на нанофильтрах для достижения еще большей безопасности продукта.
Процесс согласно настоящему изобретению более подробно описан и предпочтительно выполнен в соответствии с нижеследующей схемой.
В качестве исходного материала использовали фракции плазмы I+II+III либо II+III. Данные фракции были получены согласно описанному Cohn с соавторами (1946). Доведение pH в процессе проводили при помощи 1M уксусной кислоты, 0,1M NaOH или 0,3M HCl. Каприлат добавляли в виде 1M сток-раствора каприлата натрия. Данный раствор готовили путем растворения 166 г каприлата натрия в 1 л воды для инъекций (WFI) и перемешивания до полного растворения каприлата натрия.
В качестве контроля для SD-обработки использовали Triton X-100 и TnBP. Для удаления S/D-реагентов использовали растительное масло, такое как масло бобов сои либо касторовое масло.
Все используемые реагенты имели степень чистоты «USP» и выше.
Количественную хроматографию с исключением по размеру и ELISA использовали для определения концентрации IgG. Аналитическую ВЭЖХ проводили при помощи Agilent HPLC System на колонках TosoHaas G3000SW.
Схематическая иллюстрация процесса приведена на фиг.1. Процесс начинается с растворения преципитата IgG, называемого пастой, в очищенной воде. Обычно чем больший объем воды используется для разведения пасты, тем выше выход IgG. pH раствора доводят до 4,60-4,95, предпочтительно до 4,90 1М уксусной кислотой. Раствор перемешивают несколько часов для растворения максимально возможного количества IgG. Далее добавляют каприлат в виде сток-раствора до концентрации от 10 до 30 мМ, предпочтительно до 20 мМ каприлата. pH в процессе добавления каприлата поддерживают постоянным на уровне 4,80-4,95, предпочтительно на уровне 4,90. В процессе инкубации раствора IgG с каприлатом не-IgG-белки и липиды преципитируют. Полученный преципитат удаляют из раствора IgG фильтрованием. После первой стадии преципитации некоторые загрязняющие примеси могут остаться в растворе IgG. Подобно первой стадии, далее добавляют каприлат приблизительно до 20 мМ при постоянном pH на уровне 4,80-4,95, предпочтительно на уровне 4,9. После инкубации преципитат удаляют путем фильтрования или центрифугирования. Для более эффективного фильтрования используют сорбент для нанесения на фильтр. pH фильтрованного раствора доводят до 5,0-5,2, предпочтительно до 5,1, и помещают на анионообменную колонку. В качестве анионообменных колонок были выбраны такие сильные анионообменники, как Q-Sepharose-FF, Q-Sepharose HP, Q-Sepharose-XL, Source Q 15 или 30 (Amersham Biosience), Q- Thruput, Q-Thruput plus (Sterogene), Macro Prep Q и Macro Prep High Q (Bio- Rad), Q Hyper D (BioSepra) и Poros HQ (PerSeptive Biosystem).
При выбранных условиях IgG проходит через колонку, в то время как определенные добавки/загрязняющие примеси, такие как каприлат и IgA, связываются со смолой. Белковый раствор помещают на колонку из расчета от 40 до 120 г, в частности от 40 до 90 мг на мл смолы. Прошедший через колонку раствор концентрируют до содержания белка от 60 до 90 мг/мл, предпочтительно до 70 мг/мл, и подвергают диафильтрации против 5 объемов фосфатного буфера с концентрацией от 5 до 20 мМ, предпочтительно 10 мМ фосфата натрия. В качестве необязательной дополнительной стадии инактивации вирусов после диафильтрации может быть выполнена S/D-обработка. Подвергнутый диафильтрации раствор подвергают инактивации вирусов путем S/D-обработки согласно описанному Horowitz, к примеру, в документе EP-A-0 131 740. В качестве S/D-реагентов использовали TnBP и Triton X-100. После перемешивания раствор инкубировали вплоть до 8 часов при температуре от 4 до 10°С. Далее добавляли растительное масло, такое как масло бобов сои или касторовое масло, предпочтительно касторовое масло добавляли в раствор до концентрации от 3 до 5% (мас./мас.). После разделения водной и масляной фаз водную фазу фильтруют. Для этого используют подходящий глубинный фильтр. Примерами подобных фильтров могут служить Polysep II (Millipore), Sartofine PP и Sartobran P (Sartorius). Дальнейшую экстракцию водной фазы выполняют предпочтительно с использованием гидрофобного «support»-буфера, используемого также в хроматографии с обращенной фазой с гелевой матрицей, сделанной из диоксида кремния, сополимеров стирола и дивинилбензола (SDVB), глицидилметакрилата и этилендиметакрилата, либо полиароматики.
Примерами подобных буферов могут служить μBondapak (Waters), Amberchrom CG-161 M и S, Amberchrom CG-070 (Tosoh Biosep), PLRP-S (Polymer Laboratories), RPC-1 и Toyopearl Hexyl 650C (Tosoh Biosep), Source 15 RPC (Amersham Biosiences), LiChroprep Si60 (Merck), Chromabond Sorbent HR-P and EASY (Machery-Nagel), ProntoSORB SPE (Bischoff Chrom). Раствор белка наносят на колонку в расчете от 0,5 до 1,5 мг/мл сухой смолы. Прошедший раствор, полученный в результате экстракции твердой фазы (или стадии хроматографии соответственно), подвергают обработке УФ-C, после чего его pH доводят до 6,7-6,9, предпочтительно до 6,8 путем добавления 0,1M NaOH при температуре от 4 до 10°С. Далее раствор IgG помещают на вторую анионообменную колонку. IgG проходит через колонку, не связываясь, в то время как загрязняющие примеси и полимеры связываются с колонкой. В качестве анионообменных колонок были выбраны такие сильные анионообменники, как Q-Sepharose-FF, Q-Sepharose HP, Q-Sepharose-XL, Source Q 15 или 30 (Amersham Biosience), Q-Thruput, Q-Thruput plus (Sterogene), Macro Prep Q и Macro Prep High Q (Bio- Rad), Q Hyper D (BioSepra) и Poros HQ (PerSeptive Biosystem). Колонку уравновешивают 10 мМ буфера фосфата натрия. После нанесения раствора IgG колонку промывают уравновешивающим буфером для того, чтобы весь не связавшийся IgG сошел с колонки. Раствор белка помещают на колонку из расчета от 120 до 300 мг белка/мл смолы. pH собранного раствора IgG доводят до значений 3,9-4,1, предпочтительно до 4,0 0,3M HCl при температуре от 4 до 10°С. Далее раствор стерильно фильтруют и инкубируют при 37°С по меньшей мере 24 часа. После обработки низкими pH pH раствора доводят до 4,7 0,1M NaOH при температуре от 4 до 10°С. В качестве дополнительного этапа снижения количества вирусов может быть использован подходящий фильтр для очистки от вирусов. Для фильтрования от вирусов раствор IgG фильтровали через 0,1 мкм фильтр с последующей фильтрацией через фильтры для очистки от вирусов с размером пор от 200 до 15 нм. Примерами подобных фильтров могут служить DVD, DV 50, DV 20 (Pall), Viresolve NFP, Viresolve NFR (Millipore), Planova 75, 35, 20, 15N (Asahi Kasei Pharma). Также может быть использован заряженный глубинный фильтр, такой как Zeta Plus VR (Cuno). Стадия фильтрации также может быть проведена после инкубации при pH 4. Предпочтительно эта стадия будет включена в процесс до обработки низкими pH. Высокоочищенный раствор IgG подвергают диафильтрации и концентрируют до требуемого в данном препарате содержания IgG. В качестве конечных концентраций белка в жидком препарате были выбраны концентрации 5 или 10% (мас./об.). После доведения концентрации осмолярность раствора также доводят до значений, совместимых с применением препарата путем внутривенных инъекций, за счет подходящей добавки. Могут быть использованы сахара, сахарные спирты и аминокислоты. pH снова проверяют и доводят до значений 4,5-5,0, предпочтительно 4,7. Далее выполняется стерильное фильтрование, и раствор разливают в инфузионные бутыли.
Нижеследующие примеры более подробно поясняют процесс согласно настоящему изобретению:
Пример 1
Фракцию I+II+III либо II+III, по Cohn, растворяли в 12 объемах воды, pH доводили до 4,9 1М уксусной кислотой и раствор перемешивали вплоть до 5 часов, пока большая часть IgG не перешла в раствор при температуре от 2 до 8°С. Далее в раствор IgG добавляли каприлат в виде 1M сток-раствора каприлата натрия до концентрации 20 мМ каприлата при поддержании постоянного pH на уровне 4,9 путем добавления 1М уксусной кислоты. Раствор перемешивали на протяжении одного часа. Липиды и загрязняющие примеси в этих условиях преципитировали, их удаляли фильтрованием. Далее в раствор снова добавляли каприлат до концентрации 20 мМ каприлата при поддержании постоянного pH на уровне 4,9. Образовавшийся преципитат снова отделяли фильтрованием. После этого был получен прозрачный раствор. pH раствора доводили до 5,1 0,1M NaOH при температуре 7±3°С и раствор помещали на колонку Source Q 30 column. Раствор IgG проходил через колонку, в то время как загрязняющие примеси и каприлат связывались с колонкой. Собранный раствор IgG концентрировали до содержания 70 мг/мл и подвергали диафильтрации против 5 объемов фосфатного буфера, pH 5,1. Далее в раствор добавляли 0,3% (мас./мас.) TnBP и 1% (мас./мас.) Triton X-100, после чего интенсивно перемешивали. После по меньшей мере 4,5 часов перемешивания при температуре 7±3°С, в раствор добавляли 5% (мас./мас.) касторового масла. Масляную экстракцию проводили при комнатной температуре. Масляную и водную фазы разделяли и водную фазу фильтровали через фильтр Millipore Opticap Polysep. Фильтрованный раствор помещали на колонку с матрицей обращенной фазы μBondapak (Waters). pH раствора доводили до 6,8 0,1M NaOH при температуре 7±3°С и помещали на колонку с сильным анионообменником, а именно на Q-Sepharose-XL. IgG проходил через колонку, в то время как загрязняющие примеси связывались с колонкой. pH собранного раствора доводили до 4,7 0,1M NaOH при температуре 7±3°С. После этого вновь проводили ультрафильтрацию для достижения конечной концентрации белка 50 или 100 мг/мл, после чего добавляли мальтозу до концентрации от 2 до 10% по массе, предпочтительно 8%, либо глицин до концентрации 0,1-0,5М, предпочтительно 0,3М, в особенности 0,2. После стерильной фильтрации раствор разливали в стерилизованные и силиконизированные инфузионные бутыли различных объемов (50, 100, 200 мл). Бутыли закупоривали пробками.
Пример 2
Данный пример отличается от примера 1, в частности, использованием стадии инкубации при pH 4. Фракцию I+II+III либо II+III, по Cohn, растворяли в 12 объемах воды, pH доводили до 4,9 1М уксусной кислотой и раствор перемешивали вплоть до 5 часов, пока большая часть IgG не перешла в раствор при температуре от 2 до 8°С. Далее в раствор IgG добавляли каприлат в виде 1M сток-раствора каприлата натрия до концентрации 20 мМ каприлата при поддержании постоянного pH на уровне 4,9 путем добавления 1М уксусной кислоты. Раствор перемешивали на протяжении одного часа. Липиды и загрязняющие примеси в этих условиях преципитировали, их удаляли фильтрованием. Далее в раствор снова добавляли каприлат до концентрации 20 мМ каприлата при поддержании постоянного pH на уровне 4,9. Образовавшийся преципитат снова отделяли фильтрованием. После этого был получен прозрачный раствор. Собранный раствор IgG концентрировали до 70 мг/мл и подвергали диафильтрации против 5 объемов фосфатного буфера pH 5,1. pH раствора доводили до 5,1 0,1M NaOH при температуре 7±3°С и раствор помещали на колонку c сильным анионообменником Q-Sepharose-XL. Раствор IgG проходил через колонку, в то время как загрязняющие примеси и каприлат связывались с колонкой. Далее в раствор добавляли 0,3% (мас./мас.) TnBP и 1% (мас./мас.) Triton X-100, после чего интенсивно перемешивали. После по меньшей мере 4,5 часов перемешивания при температуре от 4 до 10°С в раствор добавляли 5% (мас./мас.) касторового масла. Масляную экстракцию проводили при комнатной температуре. Масляную и водную фазы разделяли и водную фазу фильтровали через фильтр Millipore Opticap Polysep. Фильтрованный раствор помещали на колонку, наполненную Amberchrom CG-161M. pH раствора доводили до 6,8 0,1M NaOH при температуре 7±3°С и помещали на колонку с сильным анионообменником Q-Hyper D. IgG проходил через колонку, в то время как загрязняющие примеси связывались с колонкой. pH собранного раствора доводили до 4,0 0,3M HCl при температуре 7±3°С. Раствор стерильно фильтровали и хранили при температуре 37±3°С по меньшей мере 24 часа. Далее pH раствора доводили до 4,7 0,1M NaOH при температуре 7±3°С. После этого вновь проводили ультрафильтрацию для достижения концентрации белка 50 или 100 мг/мл после смешивания с раствором мальтозы или глицина. После стерильной фильтрации раствор разливали в стерилизованные и силиконизированные инфузионные бутыли различных объемов (50, 100, 200 мл). Бутыли закупоривали пробками.
Пример 3
Данный пример отличается от предыдущих концентрированием раствора IgG до 70 мг/мл белка перед первой AEX, а также использованием стадии нанофильтрации.
Фракцию I+II+III либо II+III, по Cohn, растворяли в 12 объемах воды, pH доводили до 4,9 1М уксусной кислотой и раствор перемешивали вплоть до 5 часов, пока большая часть IgG не перешла в раствор при температуре от 2 до 8°С. Далее в раствор IgG добавляли каприлат в виде 1M сток-раствора каприлата натрия до концентрации 20 мМ каприлата при поддержании постоянного pH на уровне 4,9 путем добавления 1М уксусной кислоты. Раствор перемешивали на протяжении одного часа. Липиды и загрязняющие примеси в этих условиях преципитировали, и их удаляли фильтрованием. Далее в раствор снова добавляли каприлат до концентрации 20 мМ каприлата при поддержании постоянного pH на уровне 4,9. Образовавшийся преципитат снова отделяли фильтрованием. После этого был получен прозрачный раствор. Собранный раствор IgG концентрировали до 70 мг/мл и подвергали диафильтрации против 5 объемов фосфатного буфера pH 5,1. pH раствора доводили до 5,1 0,1M NaOH при температуре 7±3°С и раствор помещали на колонку c сильным анионообменником. Раствор IgG проходил через колонку, в то время как загрязняющие примеси и каприлат связывались с колонкой. Далее pH раствора доводили до 6,8 0,1M NaOH при температуре 7±3°С и раствор помещали на колонку со вторым сильным анионообменником. Раствор IgG проходил через колонку, в то время как загрязняющие примеси связывались с колонкой. pH собранного раствора доводили до 4,0 0,3M HCl при температуре 7±3°С. Раствор фильтровали через 0,1 мкм фильтр, после чего для нанофильтрации применяли каскад фильтров PALL, а именно - PALL DVD, DV 50 и DV 20 с размером пор, последовательно уменьшающимся до 20 нм. Нанофильтрованный раствор инкубировали при температуре 37±3°С по меньшей мере 24 часа. Далее pH раствора доводили до 4,7 0,1M NaOH при температуре 7±3°С. После этого вновь проводили ультрафильтрацию для достижения концентрации белка 50 или 100 мг/мл после смешивания с раствором мальтозы или глицина. После стерильного фильтрования раствор разливали в стерилизованные и силиконизированные инфузионные бутыли различных объемов (50, 100, 200 мл). Бутыли закупоривали пробками.
Сравнительные примеры
Способ согласно изобретению
Около 310 г фракции I+II+III (включая пре-сорбент для фильтрования) разводят в 12 объемах WFI, рассчитанных на основе теоретической массы фракции I+II+III (без пре-сорбента для фильтрования). Раствор перемешивают 1 час при 5°С. Далее pH доводят до 4,9±0,1 (прибл. 950 г). Разведение продолжают на протяжении 2 часов при 5°С. Содержание альбумина и IgA в пробе «разведенная фракция I+II+III» приведено в таблицах в конце сравнительных примеров. Далее в раствор добавляют одномолярный раствор каприлата до достижения концентрации в растворе 20 мМ. pH поддерживают постоянным на уровне 4,9. Раствор инкубируют 1 час при 5°С. Раствор фильтруют через глубинный фильтр и листок бумаги при 5°С (прибл. 1050 г). Фильтр промывают 10 мМ раствором хлорида натрия. Фильтрат доводят до температуры 25°С и добавляют одномолярный раствор каприлата для достижения дополнительной 10 мМ концентрации. Раствор инкубируют 1 час при 25°С. Далее раствор центрифугируют, и супернатант фильтруют через фильтр (1110 г) c размером пор 1 мкм и 0,5 мкм. Содержание альбумина и IgA в пробе «после каприлата» приведено в таблицах в конце сравнительных примеров.
Согласно EP 0 893 450
Около 310 г фракции I+II+III (включая пре-сорбент для фильтрования) разводят в 7 объемах WFI, рассчитанных на основе теоретической массы фракции I+II+III (без пре-сорбента для фильтрования). Раствор перемешивают 1 час при 5°С. Далее pH доводят до 4,1±0,1. Разведение продолжают на протяжении 2 часов при 5°С. Содержание альбумина и IgA в пробе «разведенная фракция I+II+III» приведено в таблицах в конце сравнительных примеров. Далее в раствор добавляют одномолярный раствор каприлата до достижения концентрации в растворе 20 мМ. pH изменяется до 4,9 в ходе добавления каприлата, и его доводят до 5,1. Раствор инкубируют 1 час при 5°С. Раствор фильтруют через глубинный фильтр и листок бумаги при 5°С. Фильтр промывают 10 мМ раствором хлорида натрия (прибл. 1050 г). Фильтрат доводят до температуры 25°С и добавляют одномолярный раствор каприлата для достижения дополнительной 10 мМ концентрации. Раствор инкубируют 1 час при 25°С. Далее раствор центрифугируют и супернатант фильтруют через фильтр (прибл. 1110 г) c размером пор 0,45 мкм. Содержание альбумина и IgA в пробе «после каприлата» приведено в таблицах в конце сравнительных примеров.
Удаление альбумина и IgA
Изобретение
EP 0893450
Изобретение
EP 0893450
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ РАСТВОРА ГАММА-ГЛОБУЛИНА, ПРЕДНАЗНАЧЕННОГО ДЛЯ ВНУТРИВЕННОГО ВВЕДЕНИЯ, И ПРОДУКТ, ПОЛУЧАЕМЫЙ ЭТИМ СПОСОБОМ | 1998 |
|
RU2198668C2 |
| СПОСОБ ОЧИСТКИ РАСТВОРА АЛЬФА-1-АНТИТРИПСИНА | 2004 |
|
RU2370500C2 |
| ОЧИСТКА И ПРИМЕНЕНИЕ ФАКТОРА, СПОСОБСТВУЮЩЕГО ЗАЖИВЛЕНИЮ РАН | 2007 |
|
RU2520817C2 |
| СПОСОБ ПОЛУЧЕНИЯ ФИБРИНОГЕНА С ИСПОЛЬЗОВАНИЕМ СИЛЬНОЙ АНИОНООБМЕННОЙ СМОЛЫ И СОДЕРЖАЩИЙ ФИБРИНОГЕН ПРОДУКТ | 2011 |
|
RU2603103C2 |
| СПОСОБ ПОЛУЧЕНИЯ ИММУНОГЛОБУЛИНОВ ДЛЯ ВНУТРИВЕННОГО ВВЕДЕНИЯ И ДРУГИЕ ИММУНОГЛОБУЛИНОВЫЕ ПРОДУКТЫ | 1999 |
|
RU2197500C2 |
| Способ получения иммуноглобулинов | 2016 |
|
RU2708394C2 |
| СПОСОБ ПОЛУЧЕНИЯ ИММУНОГЛОБУЛИНА ЧЕЛОВЕКА | 2013 |
|
RU2614119C9 |
| СПОСОБ ПОЛУЧЕНИЯ КОМПОЗИЦИИ IgG ПОСРЕДСТВОМ ТЕПЛОВОЙ ОБРАБОТКИ | 2013 |
|
RU2636049C2 |
| СПОСОБ ОЧИСТКИ ЛЕЧЕБНЫХ БЕЛКОВ | 2013 |
|
RU2685956C2 |
| ПОЛУЧЕНИЕ ИММУНОГЛОБУЛИНОВ С ПОНИЖЕННЫМ СОДЕРЖАНИЕМ ТРОМБОГЕННЫХ АГЕНТОВ И ИММУНОГЛОБУЛИНОВАЯ КОМПОЗИЦИЯ | 2012 |
|
RU2627162C2 |
Изобретение относится к биотехнологии. Способ осуществляют следующим образом: доводят рН исходного раствора до 4,6-4,95 и добавляют к нему каприлат- и/или гептаноат-ионы, поддерживая рН на уровне 4,8-4,95. Инкубируют раствор супернатанта в условиях, при которых концентрация каприлат- и/или гептаноат-ионов составляет 10-30 мМ. Наносят фильтрованный раствор на анионообменную смолу в условиях, обеспечивающих связывание загрязняющих примесей со смолой в отсутствие значительного связывания антител со смолой. Способ позволяет получить очищенный, вирусологически безопасный и инактивированный с вирусологической точки зрения препарат антител. 2 н. и 20 з.п. ф-лы, 2 ил., 2 табл.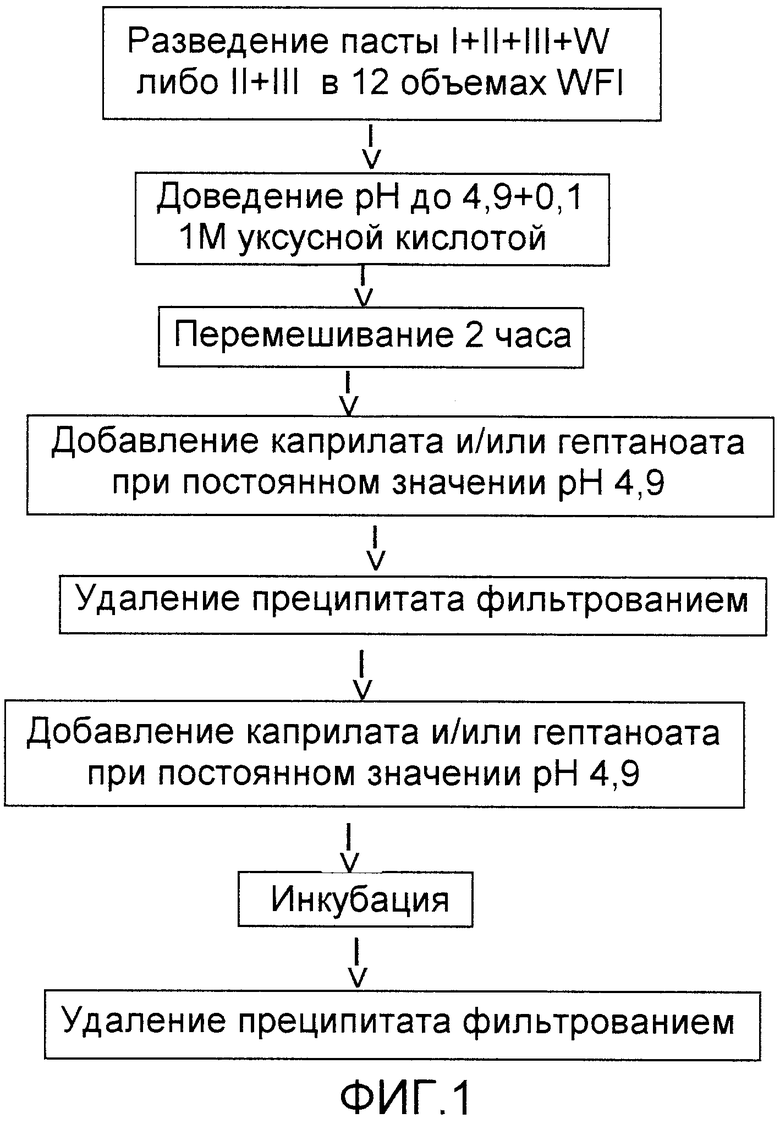
(a) доведение рН исходного раствора до 4,6-4,95 для получения промежуточного раствора;
(b) добавление каприлат- и/или гептаноат-ионов к промежуточному раствору и поддержание рН на уровне 4,8-4,95, что приводит к образованию преципитата, при этом антитела главным образом присутствуют в супернатанте;
(c) инкубирование раствора супернатанта в условиях, когда концентрация каприлат- и/или гептаноат-ионов составляет 10-30 мМ;
(d) проведение по меньшей мере одной хроматографии инактивированного фильтрованного раствора с использованием анионообменной смолы при рН от 5,0 до 5,2 в условиях, обеспечивающих связывание преципитатов со смолой в отсутствие значительного связывания IgG, с получением препарата IgG.
| STEINBUCH М | |||
| ЕТ AL | |||
| The isolation of IgG from mammalian sera with the aid of caprylic acid | |||
| Arch Biochem biophys, 1969 Nov., 134(2), p.279-84 | |||
| HABEEV A.F.S.A | |||
| ET AL | |||
| Preparation of human immunoglobulin by caprylic acid preparation, Preparative Biochemistry, 1984, 14(1), p.1-17 | |||
| Способ получения иммуноглобулинового препарата | 1978 |
|
SU836831A1 |

