Настоящее изобретение относится к новому способу получения иммуноглобулинов. Полученная композиция иммуноглобулинов является подходящей, например, для парентерального введения.
Иммуноглобулины представляют собой гликопротеины, которые могут быть обнаружены в растворимой форме в крови и других жидкостях организма позвоночных и которые служат в иммунной системе для распознавания и нейтрализации чужеродных объектов, таких как бактерии, вирусы или паразиты. Иммуноглобулины находят различные применения в медицине, в том числе в диагностике заболеваний, терапевтических способах лечения и пренатальной терапии. Наиболее распространенные терапевтические применения иммуноглобулинов могут быть классифицированы на три общие группы патологий: первичные иммунодефициты (гуморальный иммунодефицит), вторичные иммунодефициты или приобретенные иммунодефициты (например, в предупреждении и лечении вирусных инфекций) и аутоиммунные иммунодефициты (выработка антител).
Иммуноглобулины могут быть введены разными путями, такими как внутримышечный, внутривенный и подкожный пути в числе прочих. Из них предпочтительно применять внутривенный путь, поскольку он обеспечивает много преимуществ, в особенности более высокую терапевтическую эффективность.
Обычно иммуноглобулины выделяют из человеческой плазмы способами, основанными на методе фракционирования по Кону (Cohn EJ. et al., J Am Chem Soc, 1946, 62, 459-475), методе Кона-Онкли (Oncley JL. et al., J Am Chem Soc, 1949, 71, 541-550), или другими эквивалентными способами, основанными на фракционировании холодным этанолом, например, методе Кистлера-Ничмана (Kistler Р, Nitschmann Н, 1962, 7, 414-424). Соответственно, используя фракции, богатые иммуноглобулинами (такие как фракция II+III, или фракция II, или осадок А, или осадок гамма-глобулина GG), полученные любым из вышеизложенных способов, были введены модификации с целью более тщательной очистки иммуноглобулинов (IgG) и для придания им лучшей переносимости при введении, предпочтительно внутривенном. Указанные модификации были введены, например, для удаления агрегатов и других примесей, а также для обеспечения безопасности продукта. Однако, добавление множества стадий к методике получения иммуноглобулинов уменьшает выход при использовании этой методики и увеличивает стоимость производства. Растущая потребность в продуктах иммуноглобулинов, главным образом для внутривенного введения, сделала выход ключевым аспектом в процессе их получения в промышленном масштабе.
Из способов, описанных в уровне техники, методики для получения композиций иммуноглобулинов, которые являются приемлемыми для внутривенного пути, включают такие, в которых используются следующие стадии: осаждение полиэтиленгликолем (PEG), ионообменная хроматография, физические/химические способы, способные инактивировать вирусы, или обработка ферментами и частичная химическая модификация молекул иммуноглобулинов.
Следовательно, необходимость обеспечения безопасности продукта требует осуществления трудоемких стадий, дающих возможность устранения патогенных биологических агентов. Способ, который обычно используют, включает применение растворителя/детергента для инактивации вирусов с липидной оболочкой, поскольку это не сильно уменьшает биологическую активность белков. Однако, учитывая токсичность смесей растворителей/детергентов, этот реагент следует тщательно удалять перед получением конечного продукта, и это увеличивает время, требуемое для этого процесса, и уменьшает выход. Методики, описанные для удаления указанного растворителя/детергента, не являются простыми и обычно требуют применения хроматографических адсорбционных методов, либо прямых, основанных на гидрофобном взаимодействии, либо непрямого захвата иммуноглобулина в ионообменных смолах и отделения незахваченного растворителя/детергента. Во всех случаях эти процессы являются дорогостоящими и трудоемкими, включающими значительные потери белка.
Однако в данной области техники известны более простые и более эффективные альтернативные обработки, способные инактивировать вирусы. Например, использовали каприловую жирную кислоту (также известную как октановая кислота) или ее соли.
В патенте US 4446134 каприлат натрия использовали в комбинации с аминокислотами и тепловой обработкой в качестве методики для инактивации вирусов в способе получения фактора VIII. Хотя полагают, что вирулицидным агентом, способным разрушать липидные мембраны, является недиссоциированная каприловая кислота, методика, в которой используют указанный агент, общеизвестна как инактивация каприлатом, в соответствии с правилами в биохимии обозначать раствор кислоты и ее ионизованной формы по названию последнего, то есть каприлата.
Каприловую кислоту также использовали в качестве осаждающего агента для выделения иммуноглобулинов (Steinbuch, М. et al., Arch. Biochem. Biophys., 1969, 134(2), 279-284). Чистота и выход иммуноглобулинов зависят, главным образом, от концентрации добавленной каприловой кислоты и значения рН. Steinbuch, М. et al. также установили, что полезным является добавление эффективного количества каприлата на двух разных стадиях с удалением осадка между этими двумя стадиями. Это придает данной методике способность удалять вирусы как с оболочками, так и без них, благодаря распределению неиммуноглобулиновых белков в осадке.
Также в данной области техники обнаружены описания комбинации осаждения каприлатом с последующей ионообменной хроматографией для очистки иммуноглобулинов (Steinbuch, М. et al., см. выше).
В европейском патенте ЕР 0893450 раскрыт способ очистки IgG (иммуноглобулина G) с использованием фракции II+III (полученной путем осуществления методик, основанных на ранее упомянутом методе Кона), включающий две анионообменные колонки последовательно после стадий добавления каприлата в концентрации 15-25 мМ на стадии двойного осаждения и объединяющий оба эффекта каприлата: уменьшение содержания неиммуноглобулиновых белков путем осаждения и способность к инактивации вирусов посредством инкубирования. Последующие стадии анионообменной хроматографии, в дополнение к удалению других примесей (IgM, IgA, альбумина и других), используют для удаления каприлата, и по этой причине требуется двойная адсорбция с применением относительно больших количеств анионных смол.
В заявке на патент РСТ WO 2005/082937 также раскрыт способ осаждения композиции, которая включает иммуноглобулины, включающий стадии добавления каприлата и/или гептаноата к раствору или композиции, которая содержит иммуноглобулины, с последующим введением указанного раствора в колонку с анионообменной смолой.
Однако, авторы настоящего изобретения пришли к выводу, что применение каприлата в подходящей концентрации и при подходящем значении рН (например рН 5,0-5,2) для того, чтобы обеспечить способность указанной обработки инактивировать вирусы, как было описано в уровне техники, вызывает образование белковых агрегатов с высокой молекулярной массой, что частично необратимо при разбавлении и/или изменении рН. Более того, эти агрегаты только частично можно отделить путем фильтрования, и следовательно требуется специальная последующая стадия отделения, например, посредством хроматографии или осаждения. Отделение этих агрегатов вызывает значительные потери белка и уменьшение выхода промышленного процесса получения иммуноглобулинов.
Кроме того, авторы настоящего изобретения пришли к выводу, что присутствие агрегатов, образованных во время обработки каприлатом, даже в очень маленьких количествах, затрудняет корректное удаление каприлата путем непосредственного осуществления стадии отделения с использованием ультрафильтрационной мембраны в оптимальных условиях процесса. Эти агрегаты затрудняют или препятствуют получению раствора иммуноглобулинов в терапевтических концентрациях (например от 5% до 20%) вследствие присутствия коллоидов (мутности) или нестабильности в жидкой форме, таким образом затрудняя или препятствуя проведению последующих стадий способа получения иммуноглобулинов, таких как нанофильтрация и стерилизующая фильтрация.
Как следствие вышеизложенного, авторы настоящего изобретения разработали способ получения растворов иммуноглобулинов, который неожиданно включает обработку каприлатом, способную инактивировать вирусы при более низкой концентрации каприлата, чем та, которая описана в уровне техники, и который, при подходящим образом очищенном и разбавленном исходном растворе и в присутствии по меньшей мере одного полиэфира или полимера гликоля, ингибирует, предупреждает, позволяет избежать или не вызывает появление агрегатов.
Дополнительно авторы настоящего изобретения обнаружили, что присутствие по меньшей мере одного полиэфира или полимера гликоля в способе по настоящему изобретению не препятствует активности и эффективности каприлата в отношении его способности инактивировать оболочечные вирусы.
В дополнительном аспекте авторы настоящего изобретения впервые описали способ получения иммуноглобулинов, который, наряду с тем что включает обработку с инактивационной способностью в оптимальных условиях, предполагает возможность удаления или уменьшения содержания реагентов каприлата и полиэфира или полимера гликоля (присутствующих ранее во время указанной обработки) путем использования только лишь ультрафильтрационной методологии. Эта стадия ультрафильтрации обеспечивает возможность очистки и концентрирования продукта до уровней, которые являются приемлемыми для его введения, например внутривенным, внутримышечным или подкожным путем, без образования иммуноглобулиновых белковых агрегатов в конечном продукте. Это устраняет необходимость введения дополнительных стадий разделения после обработки каприлатом, таких как, например, хроматография. Более того, остаточные уровни полиэфира или полимера гликоля и каприлата после ультрафильтрации обеспечивают возможность достижения концентраций иммуноглобулинов, например IgG, вплоть до 20±2%, которые, будучи правильно приготовленными в форме препарата, не дестабилизируются во время их хранения в жидкой форме.
Учитывая факт упрощения способа согласно настоящему изобретению, это дает возможность существенно улучшить выход и весьма значительно снизить производственные затраты по сравнению с предыдущими способами, описанными в уровне техники, тем самым не подвергая риску уровень безопасности или чистоты продукта.
Следовательно, в первом аспекте настоящее изобретение относится к способу получения раствора иммуноглобулинов, включающему добавление каприловой кислоты или ее солей в присутствии по меньшей мере одного полиэфира или полимера гликоля к очищенному раствору иммуноглобулинов и последующее удаление или уменьшение содержания указанных реагентов посредством ультрафильтрации/диафильтрации.
В дополнительном аспекте настоящее изобретение относится к применению каприловой кислоты или ее солей в присутствии по меньшей мере одного полиэфира или полимера гликоля для инактивации вирусов в процессах получения белка и последующего удаления или уменьшения содержания указанных реагентов посредством ультрафильтрации/диафильтрации.
В еще одном аспекте настоящее изобретение относится к осуществлению одной стадии ультрафильтрации/диафильтрации для удаления или уменьшения уровней каприловой кислоты или ее солей и/или полиэфира или полимера гликоля, используемых для инактивации вирусов в процессах получения белка.
Следовательно, в настоящем изобретении раскрыт способ получения раствора иммуноглобулинов из исходного раствора иммуноглобулинов со степенью чистоты не менее 96% в присутствии полиэфира или полимера гликоля, отличающийся тем, что он включает стадии:
а) добавления каприловой кислоты или ее солей к исходному раствору;
б) подведения рН раствора, полученного на стадии а);
в) инкубирования раствора, полученного на стадии б), в течение периода времени и при температуре, необходимых для инактивации оболочечных вирусов; и
г) выполнения стадии ультрафильтрации/диафильтрации раствора, полученного на стадии в).
Способ по настоящему изобретению может также включать стадию конечного приготовления раствора, полученного на стадии г), в форме препарата.
В способе по настоящему изобретению исходный раствор иммуноглобулинов получают из фракции I+II+III, фракции II+III или фракции II, полученных по методу Кона или Кона-Онкли, или из осадка А или I+А или GG, полученных по методу Кистлера-Ничмана, или их вариантов, которые были дополнительно очищены для получения IgG со степенью чистоты не менее 96%. Предпочтительно, исходный раствор иммуноглобулинов получают из фракции II+III, полученной по методу Кона или его модификаций, которая была затем очищена посредством осаждения полиэтиленгликолем (PEG) и анионной хроматографии, как описано в документе ЕР 1225180 В1. Согласно настоящему изобретению любая из вышеупомянутых фракций может быть подвергнута процедуре осаждения с использованием PEG с последующей фильтрацией для удаления осадка и дополнительной стадией очистки с использованием ионообменной колонки (например колонки с DEAE-сефарозой). Во всех этих случаях исходный раствор иммуноглобулинов получают из человеческой плазмы.
В наиболее предпочтительном воплощении исходный раствор иммуноглобулинов получают из фракции II+III, полученной с использованием методик, основанных на методе Кона, которую дополнительно очищают любым из способов, описанных в уровне техники, для достижения адекватного уровня очистки, требуемого при обработке каприлатом в неосаждающих условиях по настоящему изобретению, то есть показателя чистоты большего чем 96% (масс./об.) IgG или равного 96% IgG, определенного путем электрофореза в ацетате целлюлозы, с содержанием альбумина предпочтительно меньшим 1% или равным 1% (масс./об.) относительно общего содержания белков. Таким образом, указанный исходный раствор иммуноглобулинов является в достаточной степени очищенным, до и после обработки каприлатом, для пути терапевтического введения, для которого он предназначен, так что не требуется какой-либо дополнительной очистки после стадии, обеспечивающей инактивацию вирусов, по настоящему изобретению.
Иммуноглобулины исходного раствора способа по настоящему изобретению также можно получить методами генетической рекомбинации, например, путем экспрессии в культурах клеток; методами химического синтеза и методами продуцирования трансгенного белка.
В наиболее предпочтительном воплощении иммуноглобулины, упомянутые в способе по настоящему изобретению, представляют собой IgG. Предполагается, что указанные IgG могут быть моноклональными или поликлональными. В наиболее предпочтительном воплощении IgG являются поликлональными.
Предполагается, что полиэфиры или полимеры гликоля по настоящему изобретению могут представлять собой полиэфиры алкана или оксиды полиалкана, также известные как полигликоли, и относятся, например, к производным этила или этилена и пропила или пропилена, лучше известным как полиэтиленгликоль (PEG) или полипропиленгликоль (PPG), или их эквивалентам. В дополнение, указанные реагенты должны быть совместимыми с иммуноглобулинами в том смысле, что они не нарушают их стабильность или растворимость и что, благодаря их размеру, их можно легко удалить с использованием методик ультрафильтрации, или что, благодаря их пониженной токсичности, они согласуются с терапевтическим применением иммуноглобулинов.
В предпочтительном воплощении полиэфир или полимер гликоля выбран из полиэтиленгликоля (PEG), полипропиленгликоля (PPG) или их комбинаций. Предпочтительно, полиэфир или полимер гликоля представляет собой PEG, более предпочтительно PEG с номинальной молекулярной массой от 3350 Да до 4000 Да и наиболее предпочтительно PEG с номинальной молекулярной массой 4000 Да.
Содержание вышеупомянутого полиэфира или полимера гликоля в исходном растворе иммуноглобулинов составляет предпочтительно от 2% до 6% (масс./об.) и более предпочтительно от 3% до 5% (масс./об.).
Предполагается, что возможно может быть необходимо подводить концентрацию указанного полиэфира или полимера гликоля в исходном растворе иммуноглобулинов. Указанное подведение концентрации указанного полиэфира или полимера гликоля может быть выполнено путем разбавления исходного очищенного раствора иммуноглобулинов и/или путем его добавления.
В соответствии с составом исходного раствора иммуноглобулинов предполагается, что перед стадией а) способа по настоящему изобретению проводят серию стадий очистки или подведения концентраций, таких как например:
- подведение концентрации иммуноглобулинов до 1-10 мг/мл, более предпочтительно 3-7 мг/мл. Это подведение может быть выполнено с использованием любой из методик, известных в данной области техники, например путем разбавления или концентрирования белка в установленном диапазоне (определенном, например, в соответствии с общим содержанием белка по оптической плотности при 280 нм Е(1%)=13,8-14,0 UA (условных единиц) биуретовым методом, методом Бредфорда или в особенности иммунонефелометрией) в зависимости от обстоятельств. Следовательно, в предпочтительном воплощении исходный раствор иммуноглобулинов имеет концентрацию иммуноглобулинов предпочтительно 1-10 мг/мл и более предпочтительно 3-7 мг/мл; и/или
- доведение чистоты раствора иммуноглобулинов, которая предпочтительно должна достигать по меньшей мере 96% IgG относительно общего содержания белка. Эта очистка может быть выполнена с использованием методов, полностью известных специалисту в данной области техники, таких как, например, осаждение PEG, и фильтрация, и последующая анионообменная хроматография (DEAE-сефароза).
На стадии а) способа по настоящему изобретению добавляют каприловую кислоту или ее соли, предпочтительно используя их концентрированный раствор, например от 1,5 М до 2,5 М, для достижения конечной концентрации предпочтительно от 9 мМ до 15 мМ.
В предпочтительном воплощении на стадии б) значение рН полученного раствора доводят до 5,0-5,2, более предпочтительно до 5,1.
В предпочтительном воплощении на стадии в) полученный раствор инкубируют в течение по меньшей мере 10 минут, более предпочтительно 1-2 часов и еще более предпочтительно 2 часов. В дополнение, температура, при которой проводят указанную инкубацию, составляет от 2°С до 37°С, более предпочтительно от 20°С до 30°С.
В предпочтительном воплощении перед стадией г) способа по настоящему изобретению содержание полимеров или агрегатов с высокой молекулярной массой в растворе, полученном на указанной стадии в), составляет не более 0,2% и более предпочтительно менее 0,1%. Это процентное содержание полимеров или молекулярных агрегатов иммуноглобулинов относительно общего содержания белков определяют с помощью колонки с гелем для эксклюзионной HPLC (жидкостная хроматография высокого разрешения) в соответствии с величиной оптической плотности при 280 нм. Указанное процентное содержание полимеров или молекулярных агрегатов иммуноглобулинов может быть оценено, например, с использованием способа анализа, описанного в монографии Европейской Фармакопеи, относящейся к внутривенному гамма-глобулину.
Предпочтительно раствор иммуноглобулинов осветляют с помощью пористых фильтров перед осуществлением стадии г) ультрафильтрации/диафильтрации.
В отношении стадии г) предполагается, что предпочтительно ультрафильтрация/диафильтрация в способе по настоящему изобретению включает начальные стадии диафильтрации и концентрирования с уменьшением объема с последующим применением диафильтрации при постоянном объеме.
Ультрафильтрацию/диафильтрацию можно осуществлять в промышленном масштабе, предпочтительно используя способ одновременного диализа и концентрирования, уменьшающего объем продукта, и диафильтрации в свою очередь, что таким образом несколько снижает расход реагентов и делает процесс более эффективным, принимая во внимание тот факт, что концентрация белков является оптимальной и предпочтительно составляет не более 30 мг/мл. В любом случае специалист в данной области техники легко может определить наиболее подходящий и практичный путь выполнения этой стадии ультрафильтрации/диафильтрации, выбирая среди различных технологических методик, известных в данной области техники (например, разбавления/концентрирования или диафильтрации/концентрирования, диафильтрации при постоянном объеме или модификаций и комбинаций вышеизложенного).
Мембрана для ультрафильтрации/диафильтрации, используемая на стадии г) способа по настоящему изобретению, предпочтительно состоит из полисульфона, регенерированной целлюлозы или эквивалентов, таких как, например, мембраны, выпускаемые под товарными знаками Biomax® (Millipore, USA), Omega® (Pall, USA), Kvik-flow® (General Electric, USA). Однако порог отсечения молекулярной массы, выбранный для мембраны, может варьировать в зависимости от различных факторов, например выбора производителя. Специалист в данной области техники легко может определить предпочтительную мембрану, которая будет соответствовать потребностям в каждом случае в зависимости, например, от концентрации каприлата и полиэфира или полимера гликоля в обрабатываемом растворе.
Предпочтительно, стадию г) ультрафильтрации/диафильтрации осуществляют с использованием мембраны с порогом отсечения молекулярной массы не более 100 кДа, более предпочтительно составляющим 100 кДа.
В наиболее предпочтительном воплощении ультрафильтрацию/диафильтрацию на стадии г) осуществляют в два этапа:
первый этап, на котором значение рН подводят до рН 5,0-6,0 для уменьшения содержания или удаления большей части каприлата, и
второй этап, на котором значение рН подводят до рН менее 5,0, предпочтительно до рН 4,0-5,0, для уменьшения содержания или удаления большей части полиэфира или полимера гликоля.
В предпочтительном воплощении на первом этапе стадии ультрафильтрации/диафильтрации диафильтрацию осуществляют с использованием среды для диафильтрации, которая содержит щелочные соли карбоновой кислоты, например уксусной кислоты, в концентрации более 5 мМ или приблизительно равной 5 мМ. В наиболее предпочтительном воплощении вышеупомянутую диафильтрацию осуществляют с использованием раствора ацетата натрия в концентрации более 5 мМ или равной 5 мМ с рН, подведенным как указано выше, то есть до рН 5,0-6,0.
Число объемов диафильтрации, которые следует выполнить на первом этапе стадии г) ультрафильтрации/диафильтрации, может легко определить специалист в данной области техники в соответствии с количеством каприлата, взятого изначально, и приемлемым конечным количеством. Предпочтительно используют по меньшей мере три объема среды для диафильтрации, где указанная среда для диафильтрации предпочтительно представляет собой, как упоминалось ранее, 5 мМ раствор ацетата натрия с рН 5,0-6,0. Предпочтительно на этом первом этапе ультрафильтрации/диафильтрации приблизительно 90% или более исходного каприлата удаляется, так что на этом первом этапе концентрация каприлата уменьшается приблизительно до 1 мМ или ниже.
На втором этапе стадии г) ультрафильтрации/диафильтрации раствор иммуноглобулинов подвергают диафильтрации предпочтительно при постоянном объеме.
Предпочтительно диафильтрацию на указанном втором этапе ультрафильтрации/диафильтрации осуществляют с использованием буферного раствора, который содержит соли щелочных металлов, образованные ацетатом, фосфатом или их эквивалентами или аминокислотами и/или полиолами, например глицином и/или сорбитом, при значении рН, указанном ранее.
Как в случае с первым этапом диафильтрации, на втором этапе число диализных объемов, используемых для соответствующего уменьшения содержания полиэфира или полимера гликоля, применяемых в способе по настоящему изобретению, может легко определить специалист в данной области техники с учетом требуемого уменьшения содержания или удаления полиэфира или полимера гликоля. В предпочтительном воплощении количество буфера, подлежащего обмену при диафильтрации на втором этапе стадии г) ультрафильтрации/диафильтрации, равно не менее шести объемов. В наиболее предпочтительном воплощении на указанном втором этапе обмен происходит в соответствии с числом объемов буфера, необходимых для достижения уменьшения содержания полиэфира или полимера гликоля в 100 раз или более чем в 100 раз относительно исходного содержания указанного полиэфира или полимера гликоля перед началом стадии г) ультрафильтрации/диафильтрации.
Как только содержание каприлата и полиэфира или полимера гликоля уменьшено на стадии г) ультрафильтрации/диафильтрации, на конечной стадии приготовления в форме препарата, упомянутой ранее, раствор может быть приведен в соответствие с желаемой конечной композицией путем добавления необходимых эксципиентов и/или стабилизаторов, так чтобы сконцентрировать продукт с целью получения конечной композиции. Добавление эксципиентов и/или стабилизаторов, которое нужно выполнить в конце приготовления композиции, может быть выполнено непосредственно путем добавления указанных эксципиентов и/или стабилизаторов в твердой форме или в концентрированном растворе или, еще более предпочтительно, посредством диафильтрации с использованием необходимого числа объемов обмена раствора-композиции для обеспечения соответствующего состава конечного продукта.
В другом воплощении добавление эксципиентов и/или стабилизаторов осуществляют посредством полной или частичной замены диализного буферного раствора ацетата натрия, используемого на втором этапе стадии г), раствором, содержащим эксципиенты и/или стабилизаторы, предпочтительно с рН, подведенным до такого же значения 4,0-5,0, так что после конечного концентрирования иммуноглобулин получается уже приготовленным в форме композиции.
Специалисту в данной области техники известно, какие типы эксципиентов и/или стабилизаторов следует добавлять для достижения желаемой стабильности. Предполагают, например, что указанные эксципиенты и/или стабилизаторы могут представлять собой одну или более чем одну аминокислоту, например глицин, предпочтительно в концентрации 0,2-0,3 М, один или более чем один углевод или полиол, например сорбит, или их комбинацию.
Наконец, конечную концентрацию иммуноглобулинов, предпочтительно IgG, подводят до концентрации, подходящей для их внутривенного, внутримышечного или подкожного применения, которая будет известна специалисту в данной области техники и может составлять, например, от 5% до 22% (масс./об.). На указанную концентрацию влияет любая процедура, известная в данной области техники, например, концентрирование путем ультрафильтрации. Предполагается, что если на концентрацию иммуноглобулинов влияет ультрафильтрация, указанное концентрирование может быть осуществлено с использованием той же самой мембраны, что и в предыдущей диафильтрации. Очевидно, три упомянутые диафильтрации, также как и концентрирование, могут также быть осуществлены с использованием разных мембран.
Способ по настоящему изобретению также предполагает возможность введения стадии нанофильтрации для повышения спектра безопасности продукта. Существует множество этапов в методике, на которых продукт может быть подвергнут нанофильтрации через имеющиеся в продаже фильтры (например, Planova® и Bioex®, изготовленные Asahi-Kasei, DV® и SV4®, изготовленные Pall, Virosart®, изготовленные Sartorius, Vpro®, изготовленные Millipore или эквиваленты) с размерами пор от 20 нм или меньше и вплоть до 50 нм, предпочтительно с размерами пор 20 нм или меньше, или даже можно применять нанофильтры 15 нм. Промежуточные стадии, на которых можно выполнять стадию нанофильтрации, могут быть осуществлены, например, на исходном растворе иммуноглобулинов, или на материале, обработанном каприлатом после стадии ультрафильтрации/диафильтрации (как только содержание каприлата и полиэфира или полимера гликоля снижено), или в материале после концентрирования и приготовления раствора иммуноглобулинов, предпочтительно IgG, (конечный продукт) в форме препарата. Специалист в данной области техники выберет наилучший вариант в зависимости, среди прочего, от размера пор мембраны, площади фильтрации, требуемой в соответствии с временем процедуры, объема продукта, который подлежит нанофильтрации, и выхода белка.
Конечный продукт, полученный способом по настоящему изобретению, полностью удовлетворяет критериям Европейской Фармакопеи в отношении содержания изогемагглютининов. Однако, способ по настоящему изобретению также предполагает возможность включения стадии селективного и специфичного захвата антител крови анти-А и/или анти-В для достижения максимального уменьшения их содержания. Эту стадию предпочтительно осуществляют с использованием биоспецифичных аффинных смол, как было описано в уровне техники. Например, путем использования биоспецифичных аффинных смол с лигандами, образованными трисахаридами, может быть достигнуто значительное снижение уровня изогемагглютининов (Spalter et al., Blood, 1999, 93, 4418-4424). Этот дополнительный захват возможно может быть включен по усмотрению специалиста в данной области техники на любой стадии способа по настоящему изобретению или может быть выполнен до или после осуществления способа по настоящему изобретению.
Следовательно, в отношении способа получения раствора иммуноглобулинов по настоящему изобретению в наиболее предпочтительном воплощении используют исходный раствор иммуноглобулинов с чистотой не менее 96% IgG. Концентрацию IgG в этом растворе подводят предпочтительно до концентрации от 1 мг/мл до 10 мг/мл, и предпочтительно от 3 мг/мл до 7 мг/мл, и он содержит (вследствие добавления на предыдущих стадиях) PEG в концентрации 4±1% (масс./об.) или к нему добавляют PEG до концентрации 4±1% (масс./об.). Затем значение рН раствора подводят до 5,0-5,2 уксусной кислотой и добавляют каприлат натрия (например, используя концентрированный раствор указанного каприлата натрия). В предпочтительном воплощении концентрированный раствор каприлата медленно и при помешивании добавляют к очищенному раствору IgG. После добавления всего каприлата, рассчитанного для приведения продукта к конечной концентрации каприлата 9-15 мМ, окончательно подводят значение рН до 5,0-5,2 при необходимости и раствор инкубируют предпочтительно при температуре 2-37°С и более предпочтительно при температуре 25±5°С в течение по меньшей мере 10 минут и предпочтительно в течение 1-2 часов.
Затем выполняют очищение с использованием пористых фильтров (например Cuno 90LA, 50LA, Seitz EK, EK-1, EKS или эквивалентов).
Полученный таким образом раствор затем обрабатывают с помощью оборудования для ультрафильтрации/диафильтрации, образованного мембранами, содержащими полисульфон, например Biomax®, изготовленными Millipore, или Omega® от Pall, предпочтительно в форме составной кассеты. Раствор рециркулирует через каждый ультрафильтрационный/диафильтрационный элемент, предпочтительно в объеме приблизительно 100-500 л/ч и при температуре 5±3°С. Перепад давления между давлением на входе и на выходе (атмосферное давление) предпочтительно составляет от 1 бар (100 кПа) до 3 бар (300 кПа). Затем начинают первый этап диафильтрации стадии ультрафильтрации/диафильтрации для удаления каприлата, предпочтительно используя обмен по меньшей мере трех объемов буфера, образованного предпочтительно раствором ацетата натрия в концентрации не менее 5 мМ и рН 5,0-6,0. Предпочтительно с каждым объемом буфера, добавленного или израсходованного, объем раствора продукта уменьшается наполовину от исходного объема, за исключением последнего добавления.
После первого этапа диафильтрации (посредством разбавления и концентрирования или равнозначным путем) значение рН полученного раствора подводят до 4,0-5,0 с помощью, например, уксусной кислоты. Затем начинают диафильтрацию при постоянном объеме, предпочтительно используя шесть или более объемов буферного раствора, образованного ацетатом натрия в концентрации не менее 5 мМ и при рН 4,0-5,0.
Вышеупомянутый диализный буферный раствор, образованный ацетатом натрия, возможно может быть полностью или частично заменен раствором аминокислот, например глицина в концентрации 0,2-0,3 М, возможно в комбинации с углеводами и полиолами, например сорбитом, значение рН которого предпочтительно подведено до такого же значения 4,0-5,0, так что после конечного концентрирования иммуноглобулин получается уже приготовленным в форме препарата.
После применения предпочтительно по меньшей мере шести объемов (более предпочтительно от шести до десяти объемов) вышеупомянутых диализных растворов при рН 4,0-5,0 продукт может быть приготовлен в форме препарата, если он уже не находится в форме препарата, путем прямого добавления к полученному раствору эксципиента(ов) и/или стабилизатора(ов), таких как, например, глицин или другие аминокислоты, а также углеводы, например сорбит, или их комбинация, в твердом состоянии или в форме концентрированного раствора указанного(ых) эксципиента(ов) и/или стабилизатора(ов). Затем раствор IgG, полученный путем уменьшения объема, концентрируют для достижения концентрации IgG, подходящей для внутривенного, внутримышечного или подкожного применения.
Указанный концентрированный раствор, с подходящим образом подведенными значениями концентрации эксципиента(ов) и/или стабилизатора(ов) и рН, подвергают абсолютной фильтрации с использованием фильтров с размером пор 0,2 мкм и возможно нанофильтрации. Наконец, раствор IgG в асептических условиях дозируют в форме инъецируемых препаратов в ампулы, пробирки, флаконы или другие стеклянные контейнеры, которые затем герметически закрывают. Другая возможность представляет дозирование в совместимые твердые или гибкие пластиковые контейнеры, например пакеты или флаконы.
Дозированный продукт проходит через карантин и визуальный осмотр перед тем, как его помещают на хранение при температуре 2-30°С для сохранения вплоть до по меньшей мере 2 лет.
Более того, как упоминалось ранее, в настоящем изобретении также впервые раскрыто применение каприловой кислоты или ее солей в присутствии по меньшей мере одного полиэфира или полимера гликоля для инактивации вирусов в процессе получения белка, в котором указанный полиэфир или полимер гликоля и каприловую кислоту или ее соль впоследствии удаляют путем ультрафильтрации.
Предпочтительно, указанные белки выбраны из группы белков, которая включает иммуноглобулины, альбумин, факторы коагуляции, такие как фактор VII, фактор VIII и фактор IX, и фактор фон Виллебранда. Еще более предпочтительно указанные белки представляют собой иммуноглобулины. В наиболее предпочтительном воплощении указанные белки представляют собой IgG.
Далее настоящее изобретение будет описано более подробно со ссылкой на различные примеры воплощений. Однако эти примеры не предназначены ограничивать объем настоящего изобретения, а предназначены только иллюстрировать его описание.
ПРИМЕРЫ
Пример 1. Способ по настоящему изобретению для получения из плазмы раствора иммуноглобулинов, который является вирусологически безопасным, не содержит агрегатов и имеет достаточный выход для промышленного применения
Исходный материал представлял собой 16 литров раствора иммуноглобулинов, который содержал IgG в качестве основного белкового компонента, полученный способом, описанным в Европейском патенте ЕР 1225180 В1. Вкратце, указанный раствор был получен путем экстрагирования гаммаглобулина из фракции II+III с использованием метода Кона. Для осуществления этой экстракции гаммаглобулина из фракции II+III указанную фракцию предварительно выделяли путем фракционирования человеческой плазмы с использованием этанола. Затем ее суспендировали в присутствии углевода и содержание большей части сопровождающих белков уменьшали путем осаждения с использованием PEG-4000. Наконец, выполняли конечную очистку фракции путем адсорбции на колонке с ионообменной смолой (DEAE-сефарозой). Полученный таким образом колоночный эффлюент (фракция, не адсорбированная на смоле, то есть DEAE) имел электрофоретическую чистоту в ацетате целлюлозы (АСЕ) 98±2% иммуноглобулинов, рН 6,0, мутность 2,6 нефелометрических единиц мутности (NTU) и концентрацию IgG приблизительно 5 мг/мл.
Значение рН полученного раствора подводили до рН 5,1 путем добавления уксусной кислоты, а его температуру до 2-8°С. Затем концентрацию этого раствора иммуноглобулина подводили до конечной концентрации 13 мМ путем добавления концентрированного раствора каприлата натрия.
Раствор иммуноглобулинов с каприлатом нагревали до 25°С и инкубировали при этой температуре в течение 2 часов при медленном перемешивании. Во время процедуры инкубирования поддерживали значение рН при 5,10±0,05. Мутность полученного раствора составляла 17,3 NTU.
Раствор, обработанный каприлатом, охлаждали приблизительно до температуры 8°С для последующего осветления с использованием пористого фильтра (CUNO®, Ultrafilter, Дания). Примерно 20 литров профильтрованной жидкости было получено после указанного осветления (включая промывку) с концентрацией IgG приблизительно 4 мг/мл и мутностью менее 3 NTU.
Вышеупомянутый осветленный раствор подвергали диализу путем ультрафильтрации с использованием мембран с номинальным порогом отсечения молекулярной массы 100 кДа (Biomax®, Millipore, USA). Ультрафильтрацию выполняли в два дифференцированных этапа: на первом этапе материал, который имел значение рН 5,1, подвергали трем стадиям последовательного диализа и концентрирования посредством диафильтрации с использованием 5 мМ раствора ацетата, рН которого подводили до 5,1, и посредством концентрирования приблизительно до 30 UA. На втором этапе рН раствора, который имел достаточную концентрацию белков, каприлата и PEG, доводили до 4,5±0,1 и затем начинали диализ с использованием восьми объемов 5 мМ раствора ацетата с рН 4,5. Далее продукт приготавливали в форме препарата посредством диализа с использованием приблизительно 20 литров 200 мМ раствора глицина с рН 4,2 и концентрировали в том же самом ультрафильтрационном элементе до величины 140,5 UA с целью получения раствора IgG с концентрацией 10% (масс./об.).
Наконец, указанный раствор фильтровали с использованием пористого фильтра (CUNO®, Ultrafilter, Дания) и абсолютных фильтров или мембран с размером пор 0,22 мкм (CVGL®, Millipore, США или DFL®, PALL, США).
В таблице 1 представлена характеристика исходного материала, нескольких промежуточных продуктов и конечного продукта в соответствии со способом, описанным выше. Относительно результатов, включенных в указанную таблицу, следует отметить, что мутность измеряли нефелометрией; процентное содержание полимера или молекулярных агрегатов иммуноглобулинов относительно общего содержания обнаруженных белков определяли на колонке с гелем для эксклюзионной ВЭЖХ в соответствии с величиной оптической плотности при 280 нм; концентрацию каприлата определяли ферментативным методом путем количественного определения колориметрического субстрата; концентрацию PEG определяли с помощью ВЭЖХ-колонки для гель-фильтрации с использованием рефрактометрического детектора; и процент выхода этого процесса рассчитывали в соответствии с концентрацией IgG, количественно определенной посредством нефелометрии.
Результаты, полученные в этом примере, показывают, что обработка вышеупомянутого очищенного раствора каприлатом не вызывает какого-либо образования иммуноглобулиновых агрегатов или других осадков, поддерживая неизменным молекулярное распределение в продукте. Следовательно, после обработки каприлатом не требовались никакие стадии очистки для удаления агрегатов и/или осадков. Этот факт значительно облегчил процесс получения и сделал возможным прямое нанесение материала на ультрафильтрационную мембрану.
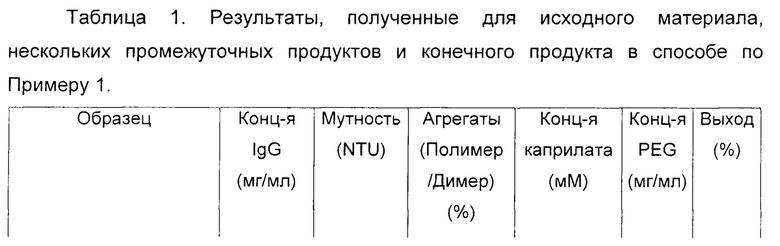
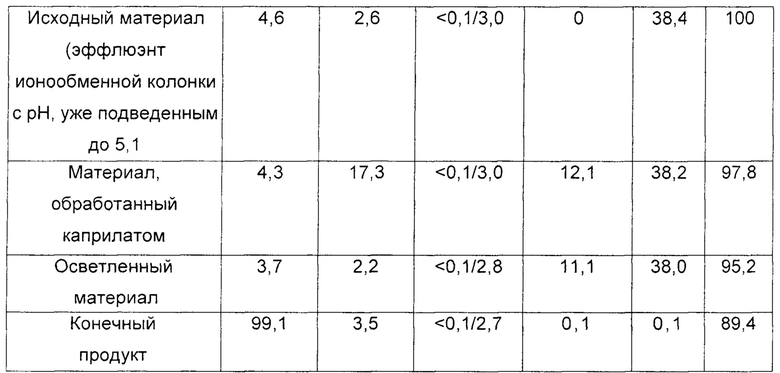
Таким образом, последующий процесс ультрафильтрации удовлетворительно достиг цели эффективного уменьшения содержания химических реагентов производственного процесса (то есть PEG и каприлата), а также обеспечил возможность последующего приготовления в форме препарата и концентрирования очищенного раствора иммуноглобулина с получением подходящей композиции для его терапевтического применения.
Как видно из таблицы 1, выход белка, полученного в этом случае, от исходного эффлюента до 10% концентрированного продукта составил 89,4%, демонстрируя эффективность этого процесса в промышленном масштабе. Этот выход был выше, чем величина, полученная традиционными способами в соответствии с уровнем техники и как описано в заявке на патент РСТ WO 2005/073252 (выход 70% из расчета выхода 4,8 г/л по сравнению с исходными 6,8 г/л).
Пример 2. Влияние чистоты исходного раствора иммуноглобулинов при обработке каприлатом
В этом примере оценивали влияние чистоты исходного раствора иммуноглобулинов и присутствия сопутствующих белков в исходном материале, подвергаемому способу по настоящему изобретению.
Были образованы две независимые экспериментальные тестируемые группы:
- в группе А исходный материал представлял собой эффлюент колонки DEAE-сефарозы с электрофоретической чистотой (АСЕ) 98±2% IgG, то есть исходный материал, описанный в Примере 1;
- в группе Б исходный материал, обозначенный как 4% PEG-фильтрат, был получен путем такого же процесса, описанного в Примере 1, вплоть до стадии перед хроматографией на DEAE-сефарозе. Таким образом, материал Б был получен после осаждения с помощью PEG экстракционной суспензии фракции II+III и имел электрофоретическую чистоту (АСЕ) приблизительно 90% IgG.
Оба исходных материала (группа А и группа Б) с эквивалентным содержанием PEG приблизительно 4% подвергали обработке каприлатом в концентрации 13 мМ и с рН 5,0-5,2 и очищали как показано в Примере 1.
В таблице 2 подробно описаны основные характеристики исходного материала, используемого в обеих тестируемых группах (А и Б, соответственно), а также характеристики материала, полученного на стадиях, следующих после обработки каприлатом.
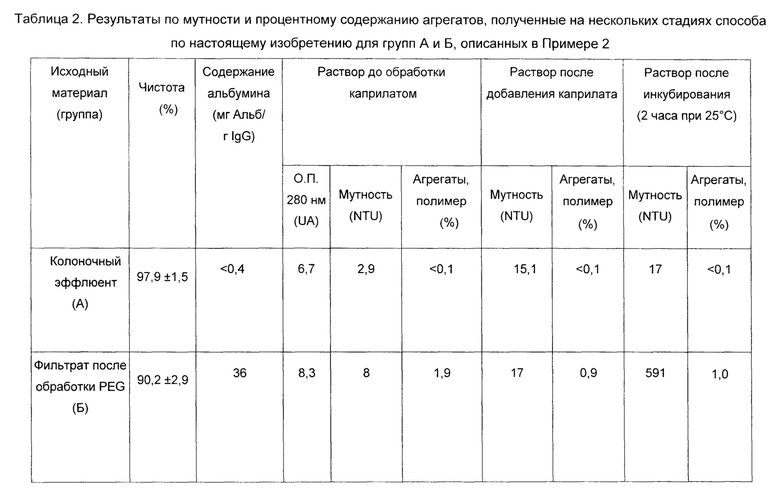
Результаты, полученные и сведенные в Таблицу 2, показывают, что добавление каприлата в эффективной концентрации для инактивации (13 мМ) к материалу низкой степени чистоты (приблизительно 90% IgG, см. группа Б) вызывает осаждение компонентов раствора, приводя к резкому повышению мутности (выше 500 NTU). Таким образом, результаты по молекулярному распределению для этого раствора показали осаждение части сопутствующих белков с высокой молекулярной массой.
Добавление каприлата в количествах и в условиях, описанных ранее (13 мМ каприлата, рН 5,0-5,2) к материалу низкой степени чистоты приводило к осажденной суспензии, что делало необходимым включение дополнительных стадий разделения и очистки для отделения белков с высокой молекулярной массой и осажденных агрегатов. Следовательно, молекулярный состав продукта группы А, обработанного каприлатом, то есть с содержанием агрегата, превышающим 1%, демонстрировал неэффективность переработки этого продукта в очищенный конечный продукт без включения дополнительных стадий очистки или разделения, таких как стадии осаждения PEG, хроматографии или эквивалентных способов. Наконец, этот факт демонстрирует эффективность использования каприлата в качестве агента, обладающего способностью инактивировать вирусы в неосаждающих условиях, только при добавлении его к материалу достаточной степени чистоты.
Пример 3. Влияние состава исходного материала на образование агрегатов
Задача данного эксперимента заключалась в оценке влияния состава исходного раствора иммуноглобулинов, для которого применяют обработку каприлатом.
Были образованы две независимые экспериментальные тестируемые группы А и Б, исходя из материалов с эквивалентной степенью чистоты (97,9±1,5%), но различного состава.
В группе А исходный материал представлял собой колоночный эффлюент (полученный в соответствии с исходным способом, описанным в Примере 1), с концентрацией белка 5±2 мг/мл и концентрацией PEG-4000 4±1%.
В группе Б исходный материал, обозначенный как концентрированный и диализированный эффлюент, представлял собой тот же самый колоночный эффлюент, упомянутый для группы А, но после концентрирования и диализа. Следовательно, эффлюент с колонки DEAE (упомянутый выше в Примере 1 и соответствующий группе А настоящего примера) подвергали дополнительной стадии диализа и концентрирования путем ультрафильтрации, так что содержание PEG было уменьшено приблизительно в 6 раз, и белок был сконцентрирован приблизительно до величины 4%, то есть 40 мг/мл.
Материал, полученный в обеих экспериментальных группах А и Б, подвергали обработке каприлатом в концентрации 13 мМ и рН 5,0-5,2 и ультрафильтрации в условиях, описанных в Примере 1, для получения продукта с концентрацией IgG 10%.
В Таблице 3 подробно описаны основные характеристики материала, обработанного в вышеупомянутых экспериментальных группах А и Б, а также характеристики материала, полученного на стадии, следующей за обработкой каприлатом, и в подвергнутом диафильтрации и концентрированном конечном продукте для каждой экспериментальной группы.
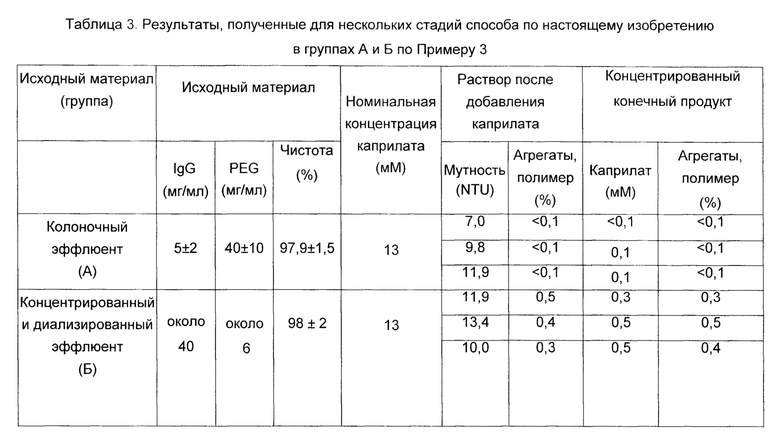
Как видно из Таблицы 3, результаты подтверждают, что обработка каприлатом очищенного раствора иммуноглобулина в концентрации 5±2 мг/мл и в присутствии PEG в концентрации 40±10 мг/мл (4±1%) (группа А, колоночный эффлюент) в определенных условиях не вызывает какого-либо изменения или агрегации раствора иммуноглобулинов, поддерживая неизменным молекулярное распределение продукта во время и после добавления каприлата с недетектируемой долей агрегатов, составляющей менее 0,1%.
Однако, когда те же самые условия обработки каприлатом применяли к материалу с низким содержанием PEG (менее 1%) (группа Б), наблюдали значительное увеличение количества иммуноглобулиновых агрегатов после добавления каприлата. Более того, было невозможно удалить это агрегированное содержимое путем ультрафильтрации в применяемых условиях, и соизмеримые уровни полимера были обнаружены в конечном продукте.
Учитывая, что главные отличительные характеристики исходных материалов, применяемых в экспериментальных группах А и Б, представляли собой концентрацию белка и концентрацию PEG, был проведен дополнительный тест с целью убедиться в влиянии каждого из этих параметров на последующую обработку каприлатом.
В этом эксперименте исходная точка представляла собой одну партию концентрированного и диализированного эффлюента (исходный материал предыдущей группы Б), которую разделили на четыре отдельные экспериментальные группы: группы Б1, Б2, Б3 и Б4.
Материал группы Б1 обрабатывали при концентрации белка приблизительно 4% и концентрации PEG приблизительно 0,6%.
Материал группы Б2 обрабатывали при такой же концентрации белка приблизительно 4%, но содержание PEG было изменено до величины 4±1% (масс./масс.).
В группах Б3 и Б4 материал разбавляли до 0,5±0,2% белка. В отношении содержания PEG в группе Б3, его доводили до концентрации приблизительно 0,6% (масс./масс.), в то время как в группе Б4 содержание PEG было изменено до 4±1% (масс./масс.).
Значение рН полученного в результате материала в этих четырех экспериментальных группах подводили до 5,10±0,05 и концентрацию каприлата в нем до 15 мМ, а затем инкубировали при 25°С в течение 2 часов. Полученные результаты показаны в Таблице 4.
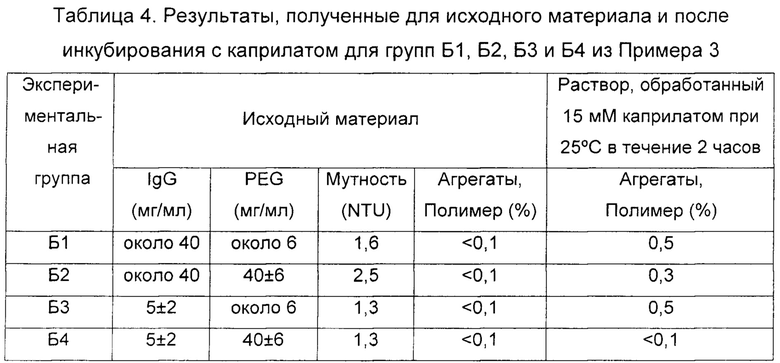
Результаты, представленные в Таблице 4, показывают, что во время обработки каприлатом в установленных условиях защитное действие PEG наблюдалось в комбинации с достаточным разбавлением белка. Заслуживает внимания тот факт, что когда исходный материал имел приблизительную концентрацию белка 5±2 мг/мл и концентрацию PEG 4%, после обработки каприлатом уровни полученных агрегатов были недектируемыми (менее 0,1%).
Пример 4. Влияние рН на растворимость раствора иммуноглобулинов, обработанных каприлатом
Известно, что удаление PEG из растворов иммуноглобулинов, также как и концентрирование указанных иммуноглобулинов до подходящих концентраций для их внутривенного применения должно происходить предпочтительно при значениях рН примерно 4,5.
Более того, учитывая нерастворимость каприловой кислоты при значениях рН ниже ее рКа (4,89), в настоящем эксперименте оценивали влияние рН на растворимость раствора иммуноглобулинов, обработанного каприлатом, с целью определения подходящего значения рН для начала его ультрафильтрации.
С этой целью партию колоночного эффлюента, полученного в соответствии с изначальным способом, подробно описанным в Примере 1, подвергали обработке для получения раствора иммуноглобулинов, обработанного 13 мМ каприлатом, и осветляли.
Этот промежуточный продукт, который составляет материал до стадии ультрафильтрации, подкисляли добавлением уксусной кислоты, чтобы довести его значение рН при обработке каприлатом (5,1) до значений рН около 4,5. Затем оценивали внешний вид и растворимость раствора для каждого из оцениваемых значений рН, и образование коллоидных частиц количественно измеряли путем нефелометрического измерения мутности.
В Таблице 5 показаны результаты оценки внешнего вида и мутности, полученные для каждого из оцениваемых значений рН.
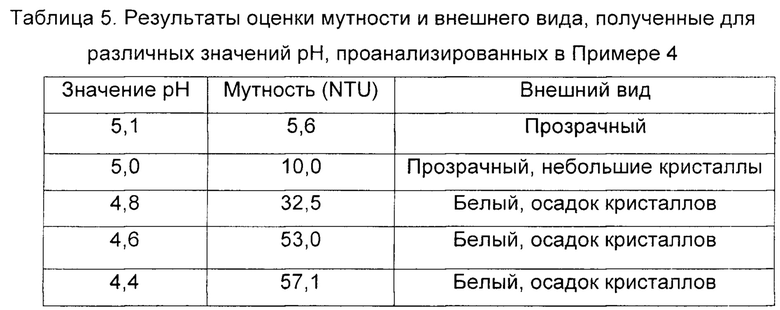
Полученные результаты, как видно из Таблицы 5, демонстрируют, что когда раствор иммуноглобулинов, обработанный 13 мМ каприлатом, подкисляли до рН 5,0 или ниже, наблюдалось появление беловатого осадка наряду с явным увеличением мутности. Наиболее вероятно, этот эффект возникал благодаря образованию нерастворимой каприловой кислоты в коллоидной форме, которая делала невозможным начало процесса ультрафильтрации при значениях рН ниже 5,0.
Полученные результаты доказывают, что когда очищенный раствор подвергали обработке каприлатом в диапазоне концентраций, эффективных для инактивации вирусов (9-15 мМ каприлата), и в условиях, описанных ранее, предпочтительно начинать последующую стадию ультрафильтрации при значении рН не менее чем рН обработки для инактивации вирусов, то есть 5,1, с целью увеличения концентрации ионизированной и растворимой формы каприлата и, следовательно, облегчения его проникновения через ультрафильтрационную мембрану.
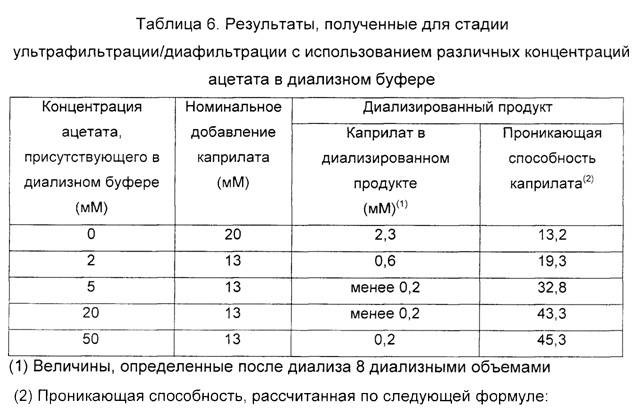
Пример 5. Влияние содержания ацетата в диализном растворе на уменьшение содержания каприлата путем ультрафильтрации/диафильтрации Серию независимых процессов ультрафильтрации/диафильтрации проводили в присутствии различных концентраций ацетата в буферном растворе, используемом для диализа продукта.
Используемый исходный материал, обозначенный как концентрированный и диализированный эффлюент, был таким же как для группы Б Примера 3. Указанный исходный материал с чистотой IgG 98±2%, приблизительной концентрацией белка 40 мг/мл и приблизительным содержанием PEG 0,6%, подвергали обработке каприлатом и затем ультрафильтрации/диафильтрации с использованием мембран с номинальным порогом отсечения молекулярной массы приблизительно 100 кДа.
Применяемая стадия ультрафильтрации/диафильтрации включала первый этап концентрирования приблизительно до 4% (масс./об.) IgG, второй этап диализа с использованием восьми объемов диализного раствора и, наконец, концентрирование до приблизительно величины 9-10% (масс./об.) IgG.
Первый из тестов ультрафильтрации/диафильтрации был выполнен с использованием воды для инъекций, в то время как последующие тесты проводили с использованием буферных растворов с повышающимися концентрациями ацетата, более конкретно 2, 5, 20 или 50 мМ ацетата, соответственно, и с рН, подведенным до 5,0-5,5 во всех случаях.
Число диализных объемов = In(Cf/Co)/(R-1); где Cf представляет собой концентрацию после диализа с рассматриваемым числом диализных объемов, Со представляет собой концентрацию до диализа, и R представляет собой коэффициент удерживания.
Результаты в Таблице 6 показывают, что методика ультрафильтрации/диафильтрации с использованием мембран с порогом отсечения молекулярной массы приблизительно 100 кДа с использованием 8 диализных объемов буферного раствора с ацетатом при рН 5,0-5,5 и с минимальной концентрацией ацетата около 5 мМ и по меньшей мере 50 мМ позволяет удовлетворительно достичь цели эффективного уменьшения содержания каприлата до подходящих уровней в конечном концентрированном продукте.
Наоборот, когда раствор, используемый для диализа, представлял собой воду для инъекций или буферный раствор с уровнями ацетата 2 мМ, не происходило эффективного удаления каприлата из фильтрата.
Это доказывает, что способ ультрафильтрации/диафильтрации с использованием мембраны с порогом отсечения молекулярной массы приблизительно 100 кДА в описанных ранее условиях эффективен в уменьшении содержания каприлата, происходящего из предыдущей обработки, с учетом того, что в конечном концентрированном продукте были определены корректные уровни указанного реагента.
Пример 6. Одновременное удаление химических реагентов (PEG и каприлата) посредством одной стадии ультрафильтрации
Партию IgG обрабатывали в соответствии со способом, описанным в Примере 1, для получения раствора, инактивированного каприлатом и осветленного. Указанный раствор с приблизительной концентрацией белка 0,5% и рН 5,1 обрабатывали с использованием оборудования для ультрафильтрации/диафильтрации, образованного полисульфоновыми мембранами типа Biomax® (Millipore, USA) с порогом отсечения молекулярной массы 100 кДа. Ультрафильтрацию/диафильтрацию проводили в два отдельных этапа как описано в Примере 5:
- на первом этапе, проводимом при рН 5,1, 5,6 или 5,8, материал подвергали стадиям последовательного диализа и концентрирования посредством диафильтрации не менее чем тремя объемами буферного раствора 5 мМ ацетата с рН, подведенным до 5,1, 5,6 или 5,8, и концентрирования белка до приблизительной величины 2%;
- на втором этапе, как только содержание каприлата было снижено приблизительно до одной десятой, значение рН раствора подводили до 4,5±0,1 или 5,1. Затем продукт доводили до адекватных концентраций белка, чтобы начать диализ, и начинали диализ с восемью объемами буферного раствора 5 мМ ацетата с рН 4,5 или 5,1.
Наконец, продукт приготавливали в форме препарата путем диализа с шестью объемами раствора глицина с концентрацией 200 мМ и рН 4,2 и концентрировали для получения 10% раствора IgG.
В Таблице 7 показано процент прохождения PEG и каприлата, полученных в начале каждого этапа ультрафильтрации/диафильтрации и при различных значениях рН.
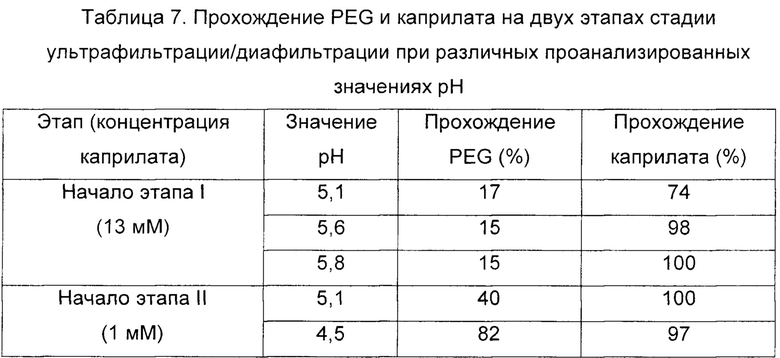
Результаты в Таблице 7 показывают, что в начале стадии ультрафильтрации/диафильтрации на этапе I каприлат демонстрировал очень высокие величины прохождения при рН 5,1-5,8. Эти величины привели к очень сильному уменьшению содержания каприлата во время указанного этапа I стадии ультрафильтрации/диафильтрации (было получено уменьшение содержания каприлата более чем в 10 раз относительно исходного содержания). Наоборот, прохождение PEG было очень низким (менее 20%) на указанном этапе I, и его полное удаление было практически невозможным при рН более 5 в присутствии каприлата.
С другой стороны, на этапе II, как видно из Таблицы 7, прохождение PEG было очень высоким при рН 4,5, с величиной 82%. Дополнительно, было обнаружено, что во время этого этапа II содержание каприлата также уменьшалось, с учетом того, что в начале этого этапа он присутствовал на остаточном уровне менее или равном 1 мМ, что обеспечивает практически 100%-ное прохождение.
В Таблице 8 подробно описано изменение концентрации белка, PEG и каприлата на каждом из этапов стадии ультрафильтрации/диафильтрации и на конечном этапе приготовления в форме препарата.
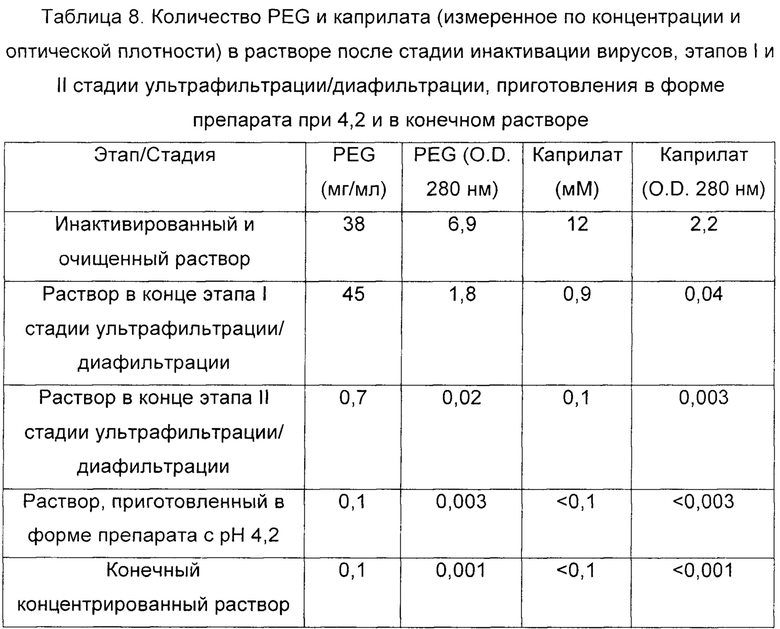
В соответствии с величинами PEG и каприлата, зарегистрированными на каждой стадии и каждом этапе, и принимая во внимание концентрацию белка на каждом этапе, коэффициент уменьшения PEG составил 4 на этапе I (при рН 5,1) стадии ультрафильтрации/диафильтрации и 90 на этапе II (при рН 4,5 стадии ультрафильтрации/диафильтрации, давая общий коэффициент уменьшения (этап I и этап II) приблизительно в 350 раз (начальное поглощение 6,9 по сравнению с поглощением 0,02, полученным в конце стадии ультрафильтрации/диафильтрации).
В случае каприлата, коэффициент уменьшения составил 55 на этапе I (при рН 5,1) стадии ультрафильтрации/диафильтрации и 13 на этапе II (при рН 4,5) стадии ультрафильтрации/диафильтрации, давая общий коэффициент уменьшения (этап I и этап II) приблизительно в 700 раз (начальное поглощение 2,2 по сравнению с поглощением 0,003, полученным в конце стадии ультрафильтрации/диафильтрации).
Результаты показали, что содержание реагента, обладающего способностью инактивировать вирусы (каприловая кислота или каприлат), а также осадителя (PEG) можно эффективно уменьшить посредством одной стадии ультрафильтрации, используя мембрану с порогом отсечения молекулярной массы приблизительно 100 кДа, выбирая физические и химические условия, которые будут применять на каждом этапе стадии ультрафильтрации/диафильтрации (среди которых рН, концентрация белка, число диализных объемов, диализный буфер) и приводящие к получению конечного продукта IgG, сконцентрированного до 10%, с небольшими остаточными концентрациями обоих реагентов, подходящими для внутривенного применения.
Пример 7. Оценка инактивирующей вирусы способности каприлата в присутствии PEG
Были выполнены различные независимые эксперименты, в которых в качестве исходного материала брали колоночный эффлюент или Анализированный и концентрированный эффлюент (полученный в соответствии с Примерами 1 и 3, соответственно) для оценки способности каприловой кислоты или каприлата в присутствии PEG элиминировать или инактивировать вирусы с липидной оболочкой.
Оба материала имели иммунологическую чистоту 98±2% и концентрацию белка 5-10 мг/мл, в то время как содержание в них PEG отличалось, составляя 40 мг/мл и 1,5 мг/мл, соответственно.
Тесты инактивации вирусов выполняли с использованием вируса диареи крупного рогатого скота (BVDV) семейства флавивирусов (Flaviviridae), 40-60 нм, с липидной оболочкой и средней устойчивостью к физическим и химическим агентам.
В каждом тесте соответствующий исходный материал инокулировали вирусом до величины не более 0,5% и подвергали обработке, инактивирующей вирусы, в течение двух часов при температуре 15°С или 25°С, используя концентрации каприлата 9 мМ или 13 мМ.
Количественную оценку вирусной нагрузки BVDV в различных полученных примерах выполняли посредством теста TCID50 (50% доза заражения культуры ткани) с использованием клеточной линии MBDK. Фактор ослабления вирусов (RF) стадии инактивации вирусов определяли как показатель вирусной нагрузки, определенный в инокулированном исходном материале, деленный на количество вируса, обнаруженного в полученном в результате образце в конце обработки, выраженный в log10.
В Таблице 9 подробно описаны характеристики исходного материала каждого теста, а также полученное значение RF.
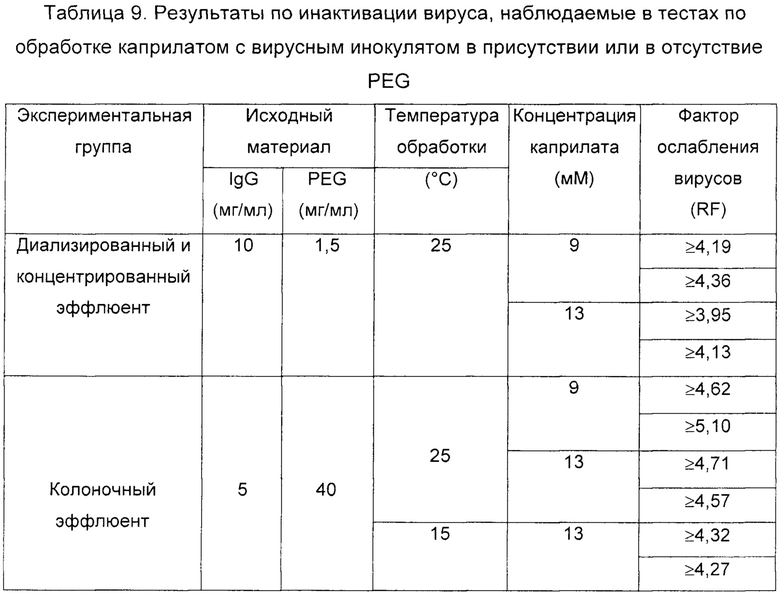
Результаты по ослаблению вируса, полученные во всех тестах (см. Таблицу 9), показали высокую способность к инактивации BVDV в обоих исходных материалах даже для минимальной 9 мМ концентрации каприлата после обработки при различных температурах (15 и 25°С). Более того, эти тесты показали, что для каждой из проанализированных концентраций PEG никакого вмешательства в отношении способности каприлата инактивировать вирус со стороны PEG не было обнаружено, учитывая что эквивалентные результаты были получены с обоими анализируемыми материалами.
Пример 8. Характеризация внутривенного раствора иммуноглобулинов, полученного в соответствии со способом получения по настоящему изобретению
Данный пример предназначен для установления биохимических и функциональных свойств раствора иммуноглобулинов с 10% (масс./об.) белков, полученного способом по настоящему изобретению.
Две партии эффлюента с DEAE-колонки обрабатывали в соответствии со способом, подробно описанным в Примере 1, для получения инактивированного вирусного раствора с каприлатом приблизительно в объеме 200 литров плазмы.
Указанный раствор с каприлатом после осветления подвергали диализу и концентрированию путем ультрафильтрации на отдельных этапах, как описано в Примере 6, с целью достижения удаления основных производственных остатков (PEG и каприлата). Затем указанный очищенный раствор с приблизительной концентрацией белка 2,5% приготавливали в форме препарата путем диализа при постоянном объеме, составляющем приблизительно 6 объемов буферного раствора, состоящего из 1% сорбита и 240 мМ глицина, с рН, подведенным до 4,5±0,1. Наконец, указанный раствор концентрировали путем ультрафильтрации и значение оптической плотности доводили до 140±5 UA (280 нм), эквивалентной 10% (масс./об.) белков, а его рН доводили до конечного значения рН 5,25±0,25.
Полученный продукт (IGIV 10% (масс./об.)), стабилизированный сорбитом и глицином и однократно осветленный и профильтрованный с использованием стерилизующих мембран (0,22 мкм), дозировали в стеклянные флаконы с хлорбутиловыми пробками для определения наиболее релевантных аналитических параметров качества, неизменности и стабильности раствора иммуноглобулина для внутривенного введения. Средние аналитические величины, полученные для двух партий, а также стандартные показатели по Европейской Фармакопее показаны в Таблице 10.
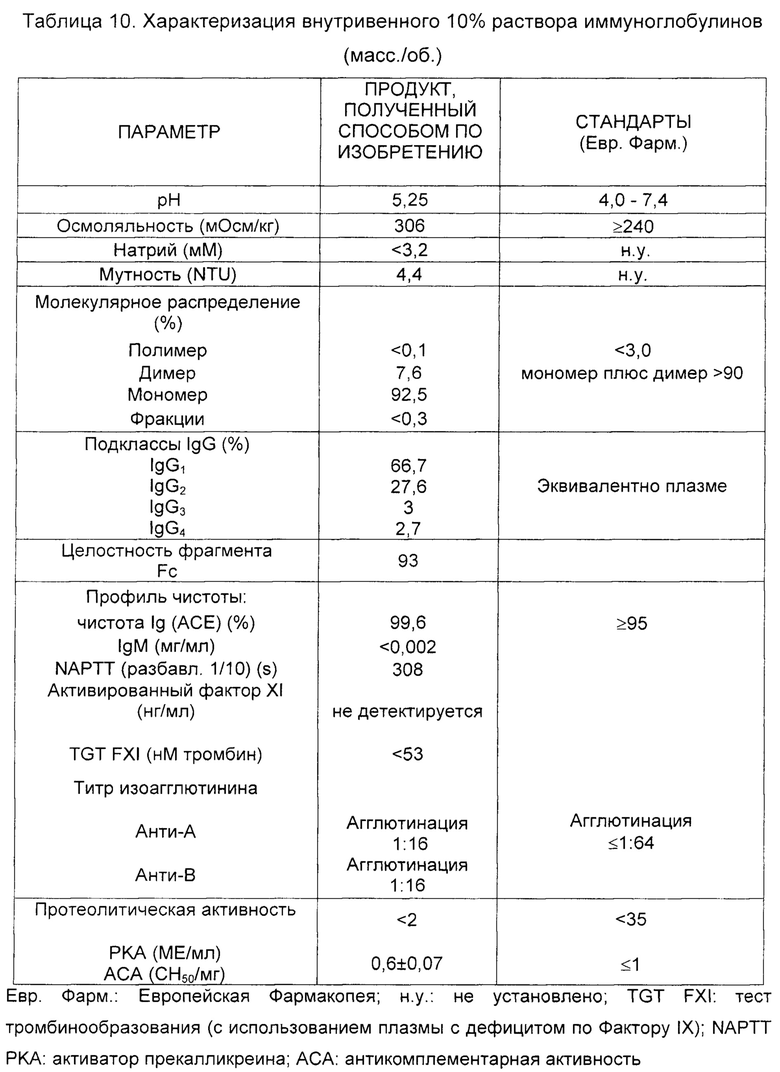
Вышеизложенные результаты подтверждают, что полученный продукт остается по существу неизмененным в результате процесса очистки по настоящему изобретению в отношении таких параметров как отсутствие полимера, нежелательная биологическая активность, такая как активность РКА или АСА, среди других, сохраняя интактными относительно плазмы несколько функциональных свойств, таких как соотношение подклассов IgG и целостность фрагмента Fc, и одновременно демонстрируя превосходный профиль чистоты (низкие титр анти-А/анти-В изогемагглютининов, концентрацию IgM, прокоагулянтную активность и так далее).
Был сделан вывод, что в целом способ по настоящему изобретению для получения 10% (масс./об.) IGIV, включающий стадию инактивации вирусов с использованием каприлата в присутствии PEG и его последующее отделение, а также конечное приготовление в форме препарата, является полностью выполнимым и масштабируемым в отношении конечного продукта, приготовленного в форме препарата и сконцентрированного в форме раствора белка 10% IGIV (масс./об.), дающего конечный продукт, который полностью соответствует показателям, установленным в Европейской Фармакопее.
Проведенные исследования стабильности, которые существенны для коммерческой жизнеспособности продукта, продемонстрировали пригодность композиций с сорбитом (до 5%), глицином (до изотоничности) или их комбинации в диапазоне рН 4,2-6,0 для стабилизации 10% (масс./об.) внутривенных растворов иммуноглобулина при температуре окружающей среды (25°С-30°С) в течение двух лет.
Пример 9. Применимость обработки каприлатом к обогащенной IgG фракции, полученной альтернативными способами
Оценивали обоснованность применения обработки каприлатом в условиях, описанных в настоящем изобретении, с использованием других промежуточных продуктов процесса, полученных с применением альтернативных способов очистки.
Было выполнено два независимых эксперимента с использованием в качестве исходного материала промежуточного продукта на основе плазмы, обогащенного IgG, обозначенного как суспензия фракции II от фракционирования этанолом по Кону-Онкли.
Этот промежуточный продукт был получен таким же способом фракционирования плазмы, описанного в настоящем изобретении вплоть до фракции II+III. Затем эту процедуру продолжали с использованием спиртового переосаждения экстракционной суспензии фракции II+III с последующим отделением фракции III, наконец получая фракцию II со степенью чистоты более 96%. Суспензия указанной фракции II, будучи очищенной бентонитом и подвергнутой диализу с водой для удаления спиртового содержимого, служила в качестве исходного материала для этих экспериментов.
В двух проведенных экспериментах материал, полученный из двух партий плазмы, разделяли на две разные группы А и Б, в соответствии с содержанием в них PEG. В группе Б концентрацию PEG в исходном материале доводили до номинальной концентрации 40 мг/мл путем добавления концентрированного раствора PEG-4000.
Затем оба материала, полученные из обеих групп (А и Б), разбавляли до концентрации белка приблизительно 5 мг/мл, значение рН доводили до 5,1 и подвергали обработке каприлатом до достижения номинальной концентрации 13 мМ и рН 5,0-5,2, как описано в способе по данному изобретению.
В Таблице 11 подробно описаны основные характеристики исходного материала, используемого в обеих экспериментальных группах (А и Б, соответственно), а также характеристики материала, полученного после обработки каприлатом.
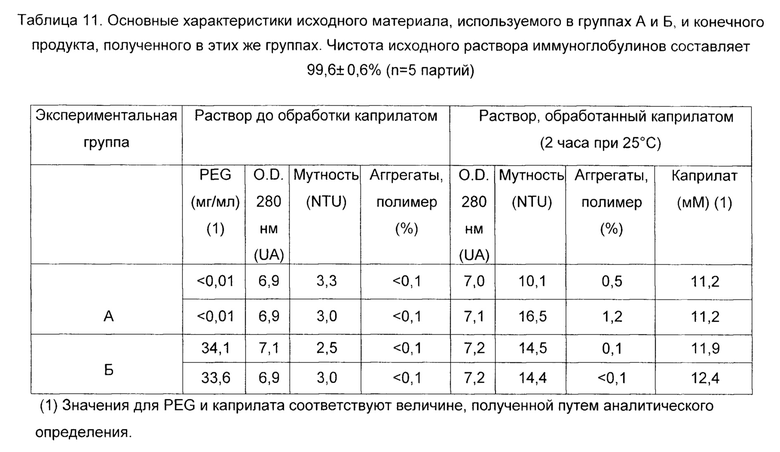
Результаты показывают эффективность инактивирующей обработки каприлатом в определенных условиях с использованием раствора иммуноглобулинов, в достаточной степени очищенного различными способами, где не происходит индуцирования образования агрегатов иммуноглобулина или других необратимых осадков, что значительно облегчает последующий процесс очистки.
Результаты демонстрируют, что в комбинации с достаточным разбавлением белка и достаточной степенью чистоты, защитный эффект PEG в отношении образования иммуноглобулиновых полимеров явно виден.
Этот экспериментальный пример демонстрирует эффективность применения каприлата только в качестве реагента со способностью инактивировать вирусы в неосаждающих условиях и/или условиях, стимулирующих агрегацию, когда его добавляют к материалу с достаточной степенью чистоты и соответствующему определенным условиям, относящимся к белку и концентрации PEG.
Хотя изобретение было представлено и описано со ссылкой на его воплощения, следует понимать, что эти воплощения не ограничивают изобретение, поскольку может быть множество переменных с точки зрения производства или других деталей, которые будут очевидными для специалиста в данной области техники после освещения предмета изобретения, раскрытого в настоящем описании и формуле изобретения. Следовательно, все варианты или эквиваленты будут включены в объем настоящего изобретения, если их можно считать попадающими в самый широкий объем следующей формулы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНАКТИВАЦИЯ ВИРУСОВ С ПРИМЕНЕНИЕМ КАПРИЛАТА | 2013 |
|
RU2633059C2 |
| ПРЕПАРАТ ИММУНОГЛОБУЛИНА IgG И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2005 |
|
RU2337109C2 |
| СПОСОБ ПОЛУЧЕНИЯ КОМПОЗИЦИИ IgG ПОСРЕДСТВОМ ТЕПЛОВОЙ ОБРАБОТКИ | 2013 |
|
RU2636049C2 |
| СПОСОБ ПОЛУЧЕНИЯ ИММУНОГЛОБУЛИНОВ ДЛЯ ВНУТРИВЕННОГО ВВЕДЕНИЯ И ДРУГИЕ ИММУНОГЛОБУЛИНОВЫЕ ПРОДУКТЫ | 1999 |
|
RU2197500C2 |
| СПОСОБ ПОЛУЧЕНИЯ ИММУНОГЛОБУЛИНА ЧЕЛОВЕКА | 2013 |
|
RU2614119C9 |
| СПОСОБЫ СНИЖЕНИЯ И/ИЛИ УДАЛЕНИЯ FXI и FXIa ИЗ РАСТВОРОВ, СОДЕРЖАЩИХ УКАЗАННЫЕ ФАКТОРЫ СВЕРТЫВАНИЯ | 2011 |
|
RU2649363C2 |
| СПОСОБ ОЧИСТКИ РАСТВОРА АЛЬФА-1-АНТИТРИПСИНА | 2004 |
|
RU2370500C2 |
| СПОСОБ ПОЛУЧЕНИЯ РАСТВОРА ГАММА-ГЛОБУЛИНА, ПРЕДНАЗНАЧЕННОГО ДЛЯ ВНУТРИВЕННОГО ВВЕДЕНИЯ, И ПРОДУКТ, ПОЛУЧАЕМЫЙ ЭТИМ СПОСОБОМ | 1998 |
|
RU2198668C2 |
| СПОСОБ ПРИГОТОВЛЕНИЯ ВИРУСИНАКТИВИРОВАННЫХ РАСТВОРОВ ИММУНОГЛОБУЛИНОВ С НИЗКИМ ОСТАТОЧНЫМ СОДЕРЖАНИЕМ КАПРИЛОВОЙ КИСЛОТЫ | 2013 |
|
RU2561596C2 |
| ПРЕПАРАТ ИММУНОГЛОБУЛИНА ЧЕЛОВЕКА ПРОТИВ ЦИТОМЕГАЛОВИРУСА ДЛЯ ВНУТРИВЕННОГО ВВЕДЕНИЯ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2009 |
|
RU2404250C1 |
Группа изобретений относится к способу получения раствора иммуноглобулинов. Способ получения раствора иммуноглобулинов из исходного раствора иммуноглобулинов со степенью чистоты не менее 96%, концентрацией от 1 до 10 мг/мл, содержащего от 2 до 6% (мас./об.) полиэтиленгликоля (PEG) или полипропиленгликоля (PPG), включает стадии а) добавления каприловой кислоты или ее солей к исходному раствору до достижения концентрации от 9 до 15 мМ; б) подведения pH раствора, полученного на стадии а), до рН 5,0-5,2; в) инкубирования раствора, полученного на стадии б), в течение периода времени и при температуре, необходимых для инактивации оболочечных вирусов; и г) выполнения стадии ультрафильтрации/диафильтрации раствора, полученного на стадии в). Также раскрыто применение каприловой кислоты для инактивации вирусов. Группа изобретений обеспечивает использование более низкой концентрации каприлата для инактивации вирусов при получении раствора иммуноглобулинов, которая позволяет избежать или не вызывает появление агрегатов. 2 н. и 17 з.п. ф-лы, 11 табл., 9 пр.
1. Способ получения раствора иммуноглобулинов из исходного раствора иммуноглобулинов со степенью чистоты не менее 96%, концентрацией от 1 до 10 мг/мл и содержащего от 2 до 6% (мас./об.) полиэтиленгликоля (PEG) или полипропиленгликоля (PPG), отличающийся тем, что он включает стадии:
а) добавления каприловой кислоты или ее солей к исходному раствору до достижения концентрации от 9 до 15 мМ;
б) подведения pH раствора, полученного на стадии а), до рН 5,0-5,2;
в) инкубирования раствора, полученного на стадии б), в течение периода времени и при температуре, необходимых для инактивации оболочечных вирусов; и
г) выполнения стадии ультрафильтрации/диафильтрации раствора, полученного на стадии в).
2. Способ по п. 1, отличающийся тем, что указанный способ также включает стадию конечного приготовления раствора иммуноглобулинов, полученного на стадии г), в форме препарата.
3. Способ по п. 1 или 2, отличающийся тем, что исходный раствор иммуноглобулинов получают из фракции I+II+III, фракции II+III или фракции II, полученных по методу Кона или Кона-Онкли, или из осадка A, или I+A, или GG, полученных по методу Кистлера-Ничмана, или их вариантов, которые были дополнительно очищены для достижения степени чистоты не менее 96% IgG.
4. Способ по п. 3, отличающийся тем, что исходный раствор иммуноглобулинов получают из фракции II+III, полученной по методу Кона или его модификаций, которая была затем очищена посредством осаждения полиэтиленгликолем (PEG) и анионной хроматографии.
5. Способ по п. 4, отличающийся тем, что концентрация иммуноглобулинов в исходном растворе иммуноглобулинов составляет 3-7 мг/мл.
6. Способ по п. 1, отличающийся тем, что концентрация PEG в исходном растворе составляет от 3 до 5% (мас./об.).
7. Способ по п. 1 или 6, отличающийся тем, что PEG представляет собой PEG с номинальной молекулярной массой 4000 Да.
8. Способ по п. 1, отличающийся тем, что на стадии б) значение pH полученного раствора подводят до рН 5,1.
9. Способ по любому из пп. 1-8, отличающийся тем, что на стадии в) раствор инкубируют в течение по меньшей мере 10 минут при температуре от 2 до 37ºC.
10. Способ по п. 8, отличающийся тем, что на стадии в) раствор инкубируют в течение 2 часов при температуре от 20 до 30ºC.
11. Способ по любому из пп. 1-10, отличающийся тем, что содержание альбумина в исходном растворе иммуноглобулинов составляет не более 1% (мас./об.) относительно общего содержания белков.
12. Способ по любому из пп. 1-11, отличающийся тем, что исходный раствор иммуноглобулинов получают из человеческой плазмы.
13. Способ по пп. 1-12, отличающийся тем, что иммуноглобулины исходного раствора иммуноглобулинов получены методами генетической рекомбинации, методами химического синтеза или методами продуцирования трансгенных белков, или в культурах клеток.
14. Способ по любому из пп. 1-13, отличающийся тем, что стадию г) ультрафильтрации/диафильтрации осуществляют с использованием мембраны 100 кДа.
15. Способ по любому из пп. 1-14, отличающийся тем, что стадию г) ультрафильтрации/диафильтрации осуществляют в два этапа:
- первый этап, на котором значение pH подводят до рН 5,0-6,0 для уменьшения содержания или удаления большей части каприлата;
- и второй этап, на котором значение pH подводят до значения не выше 5,0 для уменьшения содержания или удаления большей части указанного гликоля.
16. Способ по п. 15, отличающийся тем, что на втором этапе стадии г) ультрафильтрации/диафильтрации значение pH подводят до рН 4,0-5,0.
17. Способ по п. 2, отличающийся тем, что на стадии конечного приготовления в форме препарата добавляют эксципиенты и/или стабилизаторы, выбранные из одной или более чем одной аминокислоты, одного или более чем одного углевода или полиола или их комбинаций.
18. Способ по любому из пп. 1-17, отличающийся тем, что конечную концентрацию иммуноглобулинов доводят до концентрации, подходящей для их внутривенного, внутримышечного или подкожного применения.
19. Применение каприловой кислоты или ее солей с концентрацией от 9 до 15 мМ в присутствии от 2 до 6% (мас./об.) полиэтиленгликоля (PEG) или полипропиленгликоля (PPG) для инактивации вирусов в способе получения раствора иммуноглобулинов по п. 1, где указанные PEG или PPG и каприловую кислоту или ее соли впоследствии удаляют посредством ультрафильтрации.
| US 4164495 A, 14.08.1979 | |||
| US 4880913 A, 14.11.1989 | |||
| US 2014113355 A1, 24.04.2014 | |||
| ПРЕПАРАТ ИММУНОГЛОБУЛИНА IgG И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2005 |
|
RU2337109C2 |

