Настоящее изобретение относится к защищенной от применения не по назначению, термоформованной путем экструзии без изменения окраски лекарственной форме, которая наряду с одним или несколькими действующими веществами (А), потенциально являющимися объектом их немедицинского употребления, а также возможными физиологически совместимыми вспомогательными веществами (Б) содержит по меньшей мере один синтетический или природный полимер (В) и необязательно по меньшей мере один воск (Г) и в которой компонент (В) и возможно присутствующий компонент (Г) обладают сопротивлением разрушению по меньшей мере по 500 Н, а также относится к способу приготовления подобной, предлагаемой в изобретении лекарственной формы.
Множество фармацевтических действующих веществ наряду со своей исключительно высокой эффективностью, которую они проявляют при соответствующих их применению показаниях, потенциально являются также объектом их немедицинского употребления, т.е. они могут использоваться с целью вызвать эффекты, не соответствующие их применению по прямому назначению. В качестве примера подобных веществ можно назвать опиаты, которые являются высокоэффективными болеутоляющими средствами, применяемыми при сильных и очень сильных болях, и которые часто используются пристрастившимися к ним или злоупотребляющими ими людьми с целью вызвать подобные опьянению, эйфорические состояния.
Для немедицинского употребления подобного рода веществ соответствующие, содержащие их лекарственные формы, например таблетки или капсулы, измельчают, например, в ступке, из полученного таким путем порошка экстрагируют действующее вещество с помощью преимущественно водной жидкости и полученный раствор, иногда после его фильтрации через вату или целлюлозу, вводят себе парентерально, обычно внутривенно. При введении действующего вещества в организм таким способом по сравнению с пероральным приемом действующего вещества в немедицинских целях дополнительно ускоряется его поступление ("приток") в организм с достижением требуемого результата, а именно так называемого быстрого "прихода" или "кайфа". Подобное быстрое наступление "кайфа" достигается также при назальном введении, т.е. при нюханье, измельченной в порошок лекарственной формы. Поскольку предназначенные для приема внутрь лекарственные ретард-формы, которые содержат действующие вещества, потенциально являющиеся объектом их немедицинского употребления, даже при пероральном приеме в преднамеренно больших количествах обычно не приводят к требуемому быстрому наступлению "кайфа", такие лекарственные формы также подвергают в целях немедицинского употребления содержащихся в них действующих веществ измельчению и экстрагируют из них действующие вещества.
Для предотвращения немедицинского употребления действующих веществ в US 4070494 было предложено добавлять к лекарственной форме набухающее вещество. Такое вещество при добавлении воды для экстракции действующего вещества набухает, образуя гель, который связывает действующее вещество, количество которого в отделенном от геля фильтрате в результате оказывается лишь на незначительном уровне.
Еще один подход по предотвращению немедицинского употребления действующих веществ путем их парентерального введения в организм предложен в публикации WO 95/20947, в которой описана многослойная таблетка, которая по отдельности содержит в разных ее слоях действующее вещество, потенциально являющееся объектом его немедицинского употребления, и по меньшей мере один гелеобразователь.
Другой подход по предотвращению немедицинского употребления действующих веществ путем их парентерального введения в организм предложен в публикации WO 03/015531 А2. В этой публикации описана лекарственная форма, содержащая аналгетический опиоид и краситель в качестве аверсивного (вызывающего отвращение) средства. Благодаря такому красителю лекарственная форма при совершении с ней недопустимых манипуляций окрашивается в цвет, при виде которого человек должен воздерживаться от дальнейшего применения такой лекарственной формы.
Еще одна известная возможность, призванная затруднить немедицинское употребление действующих веществ, заключается в добавлении к лекарственной форме антагонистов действующих веществ, например налоксона или налтексона в случае опиоидов, или средств, которые вызывают физиологические защитные реакции, например корня ипекакуаны, иначе называемого рвотным корнем.
Поскольку, однако, лекарственные формы, содержащие пригодное для немедицинского употребления действующее вещество, для возможности его введения в организм в немедицинских целях в большинстве случаев требуется, как и ранее, измельчать в порошок, в основу настоящего изобретения была положена задача осложнить предшествующее немедицинскому применению действующего вещества измельчение содержащей его лекарственной формы в порошок обычно имеющимися в распоряжении подручными средствами, соответственно воспрепятствовать подобному измельчению лекарственной формы и тем самым предложить твердую лекарственную форму для потенциально являющихся объектом немедицинского употребления действующих веществ, которая при ее применении по прямому назначению проявляла бы требуемое терапевтическое действие, но содержащиеся в которой действующие вещества невозможно было бы перевести в пригодную для их немедицинского введения в организм форму путем простого измельчения такой лекарственной формы в порошок.
Указанная задача решается с помощью предлагаемой в изобретении, защищенной от применения не по назначению, термоформованной путем экструзии без изменения окраски лекарственной формы, которая наряду с одним или несколькими действующими веществами (А), потенциально являющимися объектом их немедицинского употребления, содержит по меньшей мере один синтетический или природный полимер (В) и необязательно по меньшей мере один воск (Г) и в которой компонент (В) и возможно присутствующий компонент (Г) обладают сопротивлением разрушению по меньшей мере по 500 Н.
Благодаря применению полимеров с указанным минимальным сопротивлением разрушению (измеряемым по описанной ниже методике), предпочтительно в таких количествах, при которых и лекарственная форма имеет такое же минимальное сопротивление разрушению, составляющее по меньшей мере 500 Н, удается воспрепятствовать измельчению лекарственной формы в порошок с помощью обычных подручных средств и тем самым значительно затруднить, соответственно пресечь последующее ее применение не по назначению.
Обусловлено это тем, что без достаточного измельчения лекарственной формы исключается возможность парентерального, прежде всего внутривенного, безопасного введения содержащегося в ней действующего вещества в организм либо экстракция из нее содержащегося в ней действующего вещества продолжается слишком долго, соответственно при ее пероральном приеме в немедицинских целях не происходит быстрого наступления "кайфа" из-за отсутствия спонтанного высвобождения из нее действующего вещества.
Под измельчением лекарственной формы в порошок согласно настоящему изобретению подразумевается ее измельчение до порошкообразного состояния обычными, имеющимися в распоряжении подручными средствами, такими, например, как ступка и пестик, молоток, колотушка или иные обиходные средства, позволяющие с приложением соответствующего усилия измельчать твердые материалы в порошок.
В соответствии с этим предлагаемая в изобретении лекарственная форма пригодна для предотвращения применения действующих веществ, предпочтительно фармацевтических действующих веществ, потенциально являющихся объектом их немедицинского употребления, не по назначению путем их парентерального, назального и/или перорального введения в организм.
Фармацевтические действующие вещества, потенциально являющиеся объектом их немедицинского употребления, равно как и их применяемые количества и способы их получения известны специалистам в данной области и могут присутствовать в предлагаемой в изобретении лекарственной форме как таковые, в виде их соответствующих производных, прежде всего сложных или простых эфиров, либо в виде соответствующих физиологически совместимых соединений, прежде всего в виде их соответствующих солей или сольватов, а также в виде рацематов или стереоизомеров. Предлагаемая в изобретении лекарственная форма пригодна также для введения в организм в ее составе нескольких фармацевтических действующих веществ. В предпочтительном варианте, однако, предлагаемая в изобретении лекарственная форма содержит только одно определенное действующее вещество.
Предлагаемая в изобретении лекарственная форма пригодна прежде всего для предотвращения немедицинского употребления по меньшей мере одного фармацевтического действующего вещества, выбранного из группы, включающей опиоиды, транквилизаторы, предпочтительно бензодиазепины, барбитураты, стимуляторы и другие наркотические средства.
Предлагаемая в изобретении лекарственная форма наиболее пригодна для предотвращения немедицинского употребления опиоида, транквилизатора или иного наркотического средства, выбранного из группы, включающей N-{1-[2-(4-этил-5-оксо-2-тетразолин-1-ил)этил]-4-метоксиметил-4-пиперидил}пропионанилид (альфентанил), 5,5-диаллилбарбитуровую кислоту (аллобарбитал), аллилпродин, альфапродин, 8-хлор-1-метил-6-фенил-4H-[1,2,4]триазоло[4,3-а][1,4]-бензодиазепин (алпразолам), 2-диэтиламинопропиофенон (амфепрамон), (±)-α-метилфенетиламин (амфетамин), 2-(α-метилфенетиламино)-2-фенилацетонитрил (амфетаминил), 5-этил-5-изопентилбарбитуровую кислоту (амобарбитал), анилеридин, апокодеин, 5,5-диэтилбарбитуровую кислоту (барбитал), бензилморфин, безитрамид, 7-бром-5-(2-пиридил)-1H-1,4-бензодиазепин-2(3H)-он (бромазепам), 2-бром-4-(2-хлорфенил)-9-метил-6H-тиено[3,2-ƒ][1,2,4]триазоло[4,3-а][1,4]диазепин (бротизолам), 17-циклопропилметил-4,5α-эпокси-7α[(S)-1-гидрокси-1,2,2-триметилпропил]-6-метокси-6,14-эндо-этаноморфинан-3-ол(бупренорфин), 5-бутил-5-этилбарбитуровую кислоту (бутобарбитал), буторфанол, (7-хлор-1,3-дигидро-1-метил-2-оксо-5-фенил-2H-1,4-бензодиазепин-3-ил)диметилкарбамат (камазепам), (1S,2S)-2-амино-1-фенил-1-пропанол (катин/D-норпсевдоэфедрин), 4-оксид 7-хлор-N-метил-5-фенил-3H-1,4-бензодиазепин-2-иламина(хлордиазепоксид), 7-хлор-1-метил-5-фенил-1H-1,5-бензодиазепин-2,4(3H,5H)-дион (клобазам), 5-(2-хлорфенил)-7-нитро-1H-1,4-бензодиазепин-2(3H)-он (клоназепам), клонитазен, 7-хлор-2,3-дигидро-2-оксо-5-фенил-1H-1,4-бензодиазепин-3-карбоновую кислоту(хлоразепат), 5-(2-хлорфенил)-7-этил-1-метил-1H-тиено[2,3-е][1,4]диазепин-2(3Н)-он (клотиазепам), 10-хлор-11b-(2-хлорфенил)-2,3,7,11b-тетрагидрооксазоло[3,2-d][1,4]бензодиазепин-6(5H)-он (клоксазолам), (-)-метил-[3β-бензоилокси-2β(1αH,5αH)-тропанкарбоксилат] (кокаин), 4,5α-эпокси-3-метокси-17-метил-7-морфинен-6α-ол (кодеин), 5-(1-циклогексенил)-5-этилбарбитуровую кислоту (циклобарбитал), циклорфан, ципренорфин, 7-хлор-5-(2-хлорфенил)-1Н-1,4-бензодиазепин-2(3Н)-он (делоразепам), дезоморфин, декстроморамид, (+)-(1-бензил-3-диметиламино-2-метил-1-фенилпропил)пропионат (декстропропоксифен), дезоцин, диампромид, диаморфон, 7-хлор-1-метил-5-фенил-1Н-1,4-бензодиатепин-2(3Н)-он (диазепам), 4,5α-эпокси-3-метокси-17-метил-6α-морфинанол (дигидрокодеин), 4,5α-эпокси-17-метил-3,6а-морфинандиол (дигидроморфин), дименоксадол, димефептанол, диметилтиамбутен, диоксафетилбутират, дипипанон, (6аR,10аR)-6,6,9-триметил-3-пентил-6а,7,8,10а-тетрагидро-6H-бензо[с]хромен-1-ол (дронабинол), эптазоцин, 8-хлор-6-фенил-4H-[1,2,4]триазоло[4,3-a][1,4]бензодиазепин (эстазолам), этогептазин, этилметилтиамбутен, этил[7-хлор-5-(2-фторфенил)-2,3-дигидро-2-оксо-1H-1,4-бензодиазепин-3-карбоксилат] (этиллофлазепат), 4,5α-эпокси-3-этокси-17-метил-7-морфинен-6α-ол (этилморфин), этонитазен, 4,5α-эпокси-7α-(1-гидрокси-1-метилбутил)-6-метокси-17-метил-6,14-эндо-этеноморфинан-3-ол (эторфин), N-этил-3-фенил-8,9,10-тринорборнан-2-иламин (фенкамфамин), 7-[2-(α-метилфенетиламино)этил]теофиллин) (фенетиллин), 3-(α-метилфенетиламино)пропионитрил (фенпропорекс), Н-(1-фенетил-4-пиперидил)пропионанилид (фентанил), 7-хлор-5-(2-фторфенил)-1-метил-1Н-1,4-бензодиазепин-2(3H)-он (флудиазепам), 5-(2-фторфенил)-1-метил-7-нитро-1Н-1,4-бензодиазепин-2(3H)-он (флунитразепам), 7-хлор-1-(2-диэтиламиноэтил)-5-(2-фторфенил)-1Н-1,4-бензодиазепин-2(3H)-он (флуразепам), 7-хлор-5-фенил-1-(2,2,2-трифторэтил)-1H-1,4-бензодиазепин-2(3H)-он (галазепам), 10-бром-11b-(2-фторфенил)-2,3,7,11b-тетрагидро[1,3]оксазоло[3,2-d][1,4]бензодиазепин-6(5H)-он (галоксазолам), героин, 4,5а-эпокси-3-метокси-17-метил-6-морфинанон (гидрокодон), 4,5α-эпокси-3-гидрокси-17-метил-6-морфинанон (гидроморфон), гидроксипетидин, изометадон, гидроксиметилморфинан, 11-хлор-8,12b-дигидро-2,8-диметил-12b-фенил-4H-[1,3]оксазино[3,2-d][1,4]бензодиазепин-4,7(6H)-дион (кетазолам), 1-[4-(3-гидроксифенил)-1-метил-4-пиперидил]-1-пропанон (кетобемидон), (3S,6S)-6-диметиламино-4,4-дифенилгептан-3-илацетат (левацетилметадол (ЛААМ)), (-)-6-диметиламино-4,4-дифенил-3-гептанон (левометадон), (-)-17-метил-3-морфинанол (леворфанол), левофенацилморфан, лофентанил, 6-(2-хлорфенил)-2-(4-метил-1-пиперазинилметилен)-8-нитро-2H-имидазо[1,2-а][1,4]бензодиазепин-1(4H)-он (лопразолам), 7-хлор-5-(2-хлорфенил)-3-гидрокси-1H-1,4-бензодиазепин-2(3H)-он (лоразепам), 7-хлор-5-(2-хлорфенил)-3-гидрокси-1 -метил-1Н-1,4-бензодиазепин-2(3H)-он (лорметазепам), 5-(4-хлорфенил)-2,5-дигидро-3H-имидазо[2,1-а]изоиндол-5-ол (мазиндол), 7-хлор-2,3-дигидро-1-метил-5-фенил-1H-1,4-бензодиазепин (медазепам), N-(3-хлорпропил)-α-метилфенетиламин (мефенорекс), меперидин, 2-метил-2-пропилтриметилендикарбамат (мепробамат), мептазинол, метазоцин, метилморфин, N,α-диметилфенетиламин (метамфетамин), (±)-6-диметиламино-4,4-дифенил-3-гептанон (метадон), 2-метил-3-о-толил-4(3H)-хиназолинон (метаквалон), метил[2-фенил-2-(2-пиперидил)ацетат] (метилфенидат), 5-этил-1-метил-5-фенилбарбитуровую кислоту (метилфенобарбитал), 3,3-диэтил-5-метил-2,4-пиперидиндион (метиприлон), метопон, 8-хлор-6-(2-фторфенил)-1-метил-4H-имидазо[1,5-а][1,4]бензодиазепин (мидазолам), 2-(бензгидрилсульфинил)ацетамид (модафинил), 4,5α-эпокси-17-метил-7-морфинен-3,6а-диол (морфин), мирофин, (±)-транс-3-(1,1-диметилгептил)-7,8,10,10а-тетрагидро-1-гидрокси-6,6-диметил-6H-дибензо[b,d]пиран-9(6αH)-он (набилон), нальбуфен, налорфин, нарцеин, никоморфин, 1-метил-7-нитро-5-фенил-1H-1,4-бензодиазепин-2(3H)-он (ниметазепам), 7-нитро-5-фенил-1H-1,4-бензодиазепин-2(3H)-он (нитразепам), 7-хлор-5-фенил-1H-1,4-бензодиазепин-2(3H)-он (нордазепам), норлеворфанол, 6-диметиламино-4,4-дифенил-3-гексанон (норметадон), норморфин, норпипанон, свернувшийся сок растений, относящихся к виду Papaver somniferum (опий), 7-хлор-3-гидрокси-5-фенил-1H-1,4-бензодиазепин-2(3H)-он (оксазепам), (цис-транс)-10-хлор-2,3,7,11b-тетрагидро-2-метил-11b-фенилоксазоло[3,2-d][1,4]бензодиазепин-6-(5H)-он (оксазолам), 4,5а-эпокси-14-гидрокси-3-метокси-17-метил-6-морфинанон (оксикодон), оксиморфон, растения и части растений, относящихся к виду Papaver somniferum (включая подвид setigerum), папаверетум, 2-имино-5-фенил-4-оксазолидинон (пернолин), 1,2,3,4,5,6-гексагидро-6,11-диметил-3-(3-метил-2-бутенил)-2,6-метано-3-бензазоцин-8-ол (пентазоцин), 5-этил-5-(1-метилбутил)барбитуровую кислоту (пентобарбитал), этил(1-метил-4-фенил-4-пиперидинкарбоксилат) (петидин), фенадоксон, феноморфан, феназоцин, феноперидин, пиминодин, фолкодеин, 3-метил-2-фенилморфолин (фенметразин), 5-этил-5-фенилбарбитуровую кислоту (фенобарбитал), α,α-диметилфенетиламин (фентермин), 7-хлор-5-фенил-1-(2-пропинил)-1H-1,4-бензодиазепин-2(3H)-он (пиназепам), α-(2-пиперидил)бензгидриловый спирт (пипрадрол), 1'-(3-циано-3,3-дифенилпропил)[1,4'-бипиперидин]-4'-карбоксамид (пиритрамид), 7-хлор-1-(циклопропилметил)-5-фенил-1Н-1,4-бензодиазепин-2(3H)-он (празепам), профадол, прогептазин, промедол, проперидин, пропоксифен, N-(1-метил-2-пиперидиноэтил)-N-(2-пиридил)пропионамид, метил {3-[4-метоксикарбонил-4-(N-фенилпропанамидо)пиперидино]пропаноат} (ремифентанил), 5-втор-бутил-5-этилбарбитуровую кислоту (секбутабарбитал), 5-аллил-5-(1-метилбутил)барбитуровую кислоту (секобарбитал), N-{4-метоксиметил-1-[2-(2-тиенил)этил]-4-пиперидил}пропионанилид (суфентанил), 7-хлор-2-гидроксиметил-5-фенил-1H-1,4-бензодиазепин-2(3H)-он (темазепам), 7-хлор-5-(1-циклогексенил)-1-метил-1H-1,4-бензодиазепин-2(3H)-он (тетразепам), этил(2-диметиламино-1-фенил-3-циклогексен-1-карбоксилат) (тилидин (цис и транс)), трамадол, 8-хлор-6-(2-хлорфенил)-1-метил-4H-[1,2,4]триазоло[4,3-а][1,4]бензодиазепин (триазолам), 5-(1-метилбутил)-5-винилбарбитуровую кислоту (винилбитал), (1R,2R)-3-(3-диметиламино-1-этил-2-метилпропил)фенол, (1R,2R,4S)-2-[диметиламино)метил-4-(n-фторбензилокси)-1-(м-метоксифенил)циклогексанол, (1R,2R)-3-(2-диметиламинометилцикло-гексил)фенол, (1S,2S)-3(3-диметиламино-1-этил-2-метилпропил)фенол, (2R,3R)-1-диметиламино-3-(3-метоксифенил)-2-метилпентан-3-ол, (1RS,3RS,6RS)-6-диметиламинометил-1-(3-метоксифенил)циклогексан-1,3-диол, предпочтительно в виде рацемата, 3-(2-диметиламинометил-1-гидроксициклогексил)фенил-2-(4-изобутилфенил)пропионат, 3-(2-диметиламинометил-1-гидроксициклогексил)фенил-2-(6-метоксинафталин-2-ил)пропионат, 3-(2-диметиламинометилциклогекс-1-енил)фенил-2-(4-изобутилфенил)пропионат, 3-(2-диметиламинометилциклогекс-1-енил)фенил-2-(6-метоксинафталин-2-ил)пропионат, 3-(2-диметиламинометил-1-гидроксициклогексил)фениловый эфир RR-SS)-2-ацетокси-4-трифторометилбензойной кислоты, 3-(2-диметиламинометил-1-гидроксициклогексил)фениловый эфир (RR-SS)-2-гидрокси-4-трифторометилбензойной кислоты, 3-(2-диметиламинометил-1-гидроксициклогексил)фениловый эфир (RR-SS)-4-хлор-2-гидроксибензойной кислоты, 3-(2-диметиламинометил-1-гидроксициклогексил)фениловый эфир (RR-SS)-2-гидрокси-4-метилбензойной кислоты, 3-(2-диметиламинометил-1-гидроксициклогексил)фениловый эфир (RR-SS)-2-гидрокси-4-метоксибензойной кислоты, 3-(2-диметиламинометил-1-гидроксициклогексил)фениловый эфир (RR-SS)-2-гидрокси-5-нитробензойной кислоты, 3-(2-диметиламинометил-1-гидроксициклогексил)фениловый эфир (RR-SS)-2',4'-дифтор-3-гидроксидифенил-4-карбоновой кислоты, а также соответствующие стереоизомерные соединения, их соответствующие производные, прежде всего амиды, сложные или простые эфиры, и их физиологически совместимые соединения, прежде всего их соли и сольваты, наиболее предпочтительно гидрохлориды.
Предлагаемая в изобретении лекарственная форма пригодна прежде всего для предотвращения немедицинского употребления опиоидного действующего вещества, выбранного из группы, включающей оксикодон, гидроморфон, морфин, трамадол и их физиологически совместимые производные или соединения, предпочтительно их соли и сольваты, предпочтительно их гидрохлориды.
Предлагаемая в изобретении лекарственная форма наиболее пригодна также для предотвращения немедицинского употребления опиоидного действующего вещества, выбранного из группы, включающей (1R,2R)-3-(3-диметиламино-1-этил-2-метилпропил)фенол, (2R,3R)-1-диметиламино-3-(3-метоксифенил)-2-метилпентан-3-ол, (1RS,3RS,6RS)-6-диметиламинометил-1-(3-метоксифенил)циклогексан-1,3-диол, (1R,2R)-3-(2-диметиламинопэтилциклогексил)фенол, их физиологически совместимые соли, предпочтительно гидрохлориды, физиологически совместимые энантиомеры, стереоизомеры, диастереомеры и рацематы и их физиологически совместимые производные, предпочтительно простые эфиры, сложные эфиры и амиды.
Эти соединения, соответственно способы их получения описаны в заявках ЕР-А-693475, соответственно ЕР-А-780369. В соответствии с этим указанные публикации включены в настоящее описание в качестве ссылки и составляют его часть.
Для придания предлагаемой в изобретении лекарственной форме необходимого сопротивления разрушению используют по меньшей мере один синтетический или природный полимер (В) с сопротивлением разрушению, измеренным в соответствии с представленной ниже в настоящем описании методикой, по меньшей мере 500 Н. В предпочтительном варианте для этого используют по меньшей мере один полимер, выбранный из группы, включающей полиалкиленоксиды, предпочтительно полиметиленоксид, полиэтиленоксид и полипропиленоксид, полиэтилен, полипропилен, поливинилхлорид, поликарбонат, полистирол, полиакрилат, их сополимеры и смеси по меньшей мере двух указанных полимеров. Предпочтительны при этом высокомолекулярные, термопластичные полиалкиленоксиды. Наиболее предпочтительны высокомолекулярные полиэтиленоксиды с молекулярной массой, определенной путем реологических измерений, по меньшей мере 0,5 млн, предпочтительно от по меньшей мере 1 до 15 млн. Вязкость таких полимеров при 25°С при измерении на 5%-ном по массе водном растворе с помощью вискозиметра Брукфилда, модель RVF (шпиндель №2, скорость вращения 2 об/мин), составляет от 4500 до 17600 сП, при измерении на 2%-ном по массе водном растворе с помощью указанного вискозиметра (шпиндель №1, соответственно 3, скорость вращения 10 об/мин) составляет от 400 до 4000 сП, соответственно при измерении на 1%-ном по массе водном растворе с помощью указанного вискозиметра (шпиндель №2, скорость вращения 2 об/мин), составляет от 1650 до 10000 сП.
Подобные полимеры предпочтительно использовать в виде порошков. Они могут быть растворимы в воде.
Для придания предлагаемой в изобретении лекарственной форме необходимого сопротивления разрушению дополнительно можно использовать, кроме того, по меньшей мере один природный или синтетический воск (Г) с сопротивлением разрушению, измеренным в соответствии с представленной ниже в настоящем описании методикой, по меньшей мере 500 Н. Предпочтительны при этом воски с температурой размягчения, составляющей по меньшей мере 60°С. Более предпочтительны карнаубский воск и пчелиный воск. Наиболее предпочтителен карнаубский воск. Карнаубский воск представляет собой природный воск, который получают из листьев карнаубы (восконосной пальмы) и температура размягчения которого составляет по меньшей мере 80°С. При дополнительном использовании воскового компонента его совместно с по меньшей мере одним полимеров (В) применяют в таких количествах, чтобы сопротивление лекарственной формы разрушению составляло по меньшей мере 500 Н.
Компонент (В) предпочтительно использовать в количестве от 20 до 99,9 мас.%, более предпочтительно в количестве по меньшей мере 30 мас.%, наиболее предпочтительно в количестве по меньшей мере 40 мас.%, в пересчете на всю массу лекарственной формы.
В качестве вспомогательных веществ (Б) можно использовать обычно применяемые при приготовлении твердых лекарственных форм вспомогательные вещества. В предпочтительном варианте к ним относятся пластификаторы, такие как полиэтиленгликоль, вспомогательные вещества, влияющие на высвобождение действующего вещества, предпочтительно гидрофобные или гидрофильные, более предпочтительно гидрофильные, полимеры, наиболее предпочтительно гидроксипропилцеллюлоза, и/или антиокислители. Для применения в качестве антиокислителей пригодны аскорбиновая кислота, бутилгидроксианизол, бутилгидрокситолуол, соли аскорбиновой кислоты, монотиоглицерин, фосфористая кислота, витамин С, витамин Е и его производные, бисульфит натрия, наиболее предпочтительно бутилгидрокситолуол (БГТ) или бутилгидроксианизол (БГА) и α-токоферол.
Антиокислитель предпочтительно использовать в количестве от 0,01 до 10 мас.%, более предпочтительно от 0,03 до 5 мас.%, в пересчете на всю массу лекарственной формы.
Предлагаемые в изобретении лекарственные формы отличаются тем, что их благодаря их твердости невозможно измельчить в порошок обычными, имеющимися в распоряжении подручными средствами, такими как ступка и пестик. Тем самым практически полностью исключается возможность немедицинского употребления содержащихся в них действующих веществ путем их перорального, парентерального, прежде всего внутривенного, или назального введения в организм. Вместе с тем предлагаемые в изобретении лекарственные формы с целью предотвратить любое их возможное применение не по назначению в одном из предпочтительных вариантов могут содержать в качестве вспомогательных веществ (Б) дополнительные затрудняющие, соответственно предотвращающие немедицинское употребление входящих в их состав действующих веществ средства.
Так, в частности, предлагаемая в изобретении, защищенная от применения не по назначению лекарственная форма наряду с одним или несколькими действующими веществами, потенциально являющимися объектом их немедицинского употребления, по меньшей мере одним повышающим ее твердость полимером (В) и возможно по меньшей мере одним воском (Г) может также содержать в качестве вспомогательных веществ (Б) по меньшей мере один из следующих компонентов (а)-(е):
(а) по меньшей мере одно раздражающее полость носа и/или глотки вещество,
(б) по меньшей мере одно повышающее вязкость средство (загуститель), которое в извлеченном из лекарственной формы с помощью минимально необходимого количества водной жидкости экстракте образует гель, который в предпочтительном варианте при его добавлении в дополнительное количество водной жидкости остается визуально различим,
(в) по меньшей мере один антагонист для каждого из действующих веществ, потенциально являющихся объектом их немедицинского употребления,
(г) по меньшей мере одно рвотное средство.
(д) по меньшей мере один краситель в качестве аверсивного средства,
(е) по меньшей мере одно горькое вещество.
Каждый из вышеуказанных компонентов (а)-(е) индивидуально пригоден для дополнительной защиты предлагаемой в изобретении лекарственной формы от применения не по назначению. Так, в частности, компонент (а) предпочтительно использовать для защиты лекарственной формы от немедицинского употребления содержащегося в ней действующего вещества путем его назального, перорального и/или парентерального, предпочтительно внутривенного, введения в организм, компонент (б) предпочтительно использовать в целях воспрепятствовать немедицинскому употреблению действующего вещества путем его парентерального, преимущественно внутривенного, и/или назального введения в организм, компонент (в) предпочтительно использовать для защиты от немедицинского употребления действующего вещества путем его назального и/или парентерального, преимущественно внутривенного, введения в организм, компонент (г) предпочтительно использовать для защиты от немедицинского употребления действующего вещества путем его парентерального, преимущественно внутривенного, и/или перорального и/или назального введения в организм, компонент (д) предпочтительно использовать в качестве визуального отталкивающего средства для защиты от немедицинского употребления действующего вещества путем его перорального или парентерального введения в организм, а компонент (е) предпочтительно использовать для защиты от немедицинского употребления действующего вещества путем его перорального или назального введения в организм. Предлагаемое в изобретении использование по меньшей мере одного из указанных выше компонентов в дополнение к остальным ингредиентам предлагаемых в изобретении лекарственных форм позволяет дополнительно осложнить их применение не по назначению.
В одном из вариантов предлагаемая в изобретении лекарственная форма может также содержать комбинацию из двух или более компонентов (а)-(е), предпочтительно комбинацию из компонентов (а), (б) и необязательно (в) и/или (е) и/или (д), соответственно из компонентов (а), (б) и необязательно (г) и/или (е) и/или (д).
В другом варианте предлагаемая в изобретении лекарственная форма может содержать все компоненты (а)-(е).
Если предлагаемая в изобретении лекарственная форма содержит защищающий ее от применения не по назначению компонент (а), то в качестве раздражающих полость носа и/или глотки веществ согласно изобретению могут использоваться все те вещества, которые при соответствующем попадании в организм через полость носа и/или глотки провоцируют ответную реакцию организма, которая либо проявляется в возникновении столь неприятных ощущений, например жжения, что пропадает дальнейшее желание или становится невозможным продолжить введение соответствующего действующего вещества в организм, либо на физиологическом уровне препятствует приему соответствующего действующего вещества, например проявляется в повышенном выделении носового секрета или чиханье. Подобные обычно раздражающие полость носа и/или глотки вещества и при их парентеральном, прежде всего внутривенном, попадании в организм вызывают очень неприятные ощущения вплоть до появления невыносимых болей, отбивая всякое желание или делая невозможным дальнейшее введение соответствующего действующего вещества в организм.
К наиболее пригодным для применения в предусмотренных изобретением целях раздражающим полость носа и/или глотки веществам относятся вещества, которые вызывают жжение, зуд, позыв к чиханию, повышенное выделение секрета или одновременно оказывают по меньшей мере два таких раздражающих действия. Соответствующие вещества и их обычно применяемые количества известны специалистам в данной области либо могут быть определены путем проведения простых предварительных опытов.
Основу раздражающего полость носа и/или глотки вещества, используемого в качестве компонента (а), предпочтительно составляет одно или несколько раздражающих ("жгучих") веществ из по меньшей мере одного содержащего его(их) лекарственного или употребляемого в пищу растения либо одна или несколько содержащих его(их) частей такого растения.
Соответствующие лекарственные или употребляемые в пищу растения, содержащие раздражающие вещества, известны специалистам в данной области и описаны, например, в книге "Pharmazeutische Biologic - Drogen und ihre Inhaltsstoffe", Prof. Dr. Hildebert Wagner, 2-е, переработанное, изд., изд-во Gustav Fischer Verlag, Stuttgart-New York, 1982, cc.82 и далее. Соответствующие разделы этой книги тем самым включены в настоящее описание в качестве ссылки и являются его частью.
Под лекарственной формой подразумевается отдельная, соответственно отделяемая разовая дозированная форма (дозированная единица), как, например, таблетка или капсула.
В предпочтительном варианте к предлагаемой в изобретении лекарственной форме можно добавлять в качестве компонента (а) одно или несколько раздражающих веществ из по меньшей мере одного соответствующего содержащего его(их) лекарственного или употребляемого в пищу растения, выбранного из группы, включающей Allii sativi Bulbus, Asari Rhizoma с. Herba, Calami Rhizoma, Capsici Fructus (стручковый перец), Capsici Fructus acer (кайенский перец), Curcumae longae Rhizoma, Curcumae xanthorrhizae Rhizoma, Galangae Rhizoma, Myristicae Semen, Piperis nigri Fructus (перец), Sinapis albae (Erucae) Semen, Sinapis nigri Semen, Zedoariae Rhizoma и Zingiberis Rhizoma, наиболее предпочтительно из группы, включающей Capsici Fructus (стручковый перец), Capsici Fructus acer (кайенский перец) и Piperis nigri Fructus (перец).
Под раздражающими веществами из содержащих их лекарственных или употребляемых в пищу растений предпочтительно подразумеваются о-метокси(метил)фенольные соединения, амиды кислот, горчичные масла или сульфидные соединения либо их производные.
По меньшей мере одно раздражающее вещество из содержащих его лекарственных или употребляемых в пищу растений наиболее предпочтительно выбирать из группы, включающей миристицин, элемицин, изоэвгенол, α-асарон, сафрол, джинджиролы, ксанторизол, капсаициноиды, предпочтительно капсаицин, производные капсаицина, такие как N-ваниллил-9Е-октадеценамид, дигидрокапсаицин, нордигидрокапсаицин, гомокапсаицин, норкапсаицин и номоркапсаицин, пиперин, предпочтительно транс-пиперин, глюкозинолатов, предпочтительно на основе нелетучих горчичных масел, наиболее предпочтительно на основе n-гидроксибензилированного горчичного масла, содержащего метилмеркаптогруппы горчичного масла или метилсульфонилированного горчичного масла, и производные указанных веществ.
В предпочтительном варианте содержание частей соответствующих лекарственных или употребляемых в пищу растений, содержащих раздражающие вещества, в предлагаемой в изобретении лекарственной форме может составлять от 0,01 до 30 мас.%, наиболее предпочтительно от 0,1 до 0,5 мас.%, в пересчете на всю массу дозированной лекарственной формы. При использовании же одного или нескольких раздражающих веществ из соответствующих лекарственных или употребляемых в пищу растений содержание такого(их) вещества(веществ) в предлагаемой в изобретении дозированной лекарственной форме предпочтительно должно составлять от 0,001 до 0,005 мас.% в пересчете на всю ее массу.
Другая возможность воспрепятствовать применению предлагаемой в изобретении лекарственной формы не по назначению состоит в добавлении к ней по меньшей мере одного повышающего вязкость средства в качестве еще одного компонента (б), который предотвращает немедицинское употребление входящего в состав лекарственной формы действующего вещества и которое в извлеченном из лекарственной формы с помощью минимально необходимого количества водной жидкости экстракте, предпочтительно в виде водного экстракта, образует гель, безопасное введение которого в организм маловероятно и который в предпочтительном варианте при его добавлении в дополнительное количество водной жидкости остается визуально различим.
Выражение "визуально различимый" согласно настоящему изобретению означает, что образовавшийся при добавлении минимально необходимого количества водной жидкости содержащий действующее вещество гель при его добавлении, предпочтительно с помощью инъекционной иглы, в дополнительное количество водной жидкости с температурой 37°С практически не растворяется в ней и сохраняет свою целостность и не поддается простым путем диспергированию, после которого стало бы возможным его парентеральное, прежде всего внутривенное, безопасное введение в организм. В предпочтительном варианте подобный гель должен оставаться визуально различим по меньшей мере в течение одной минуты, более предпочтительно в течение по меньшей мере десяти минут.
Повышение вязкости экстракта приводит к ухудшению его проходимости через иглу, соответственно его инъецируемости или даже полностью делает невозможным его инъекцию. Остающийся визуально различимым образовавшийся гель при его добавлении в дополнительное количество водной жидкости, например, при впрыскивании в кровь, сначала сохраняет форму практически сплошной нити, которая хотя и дробится при механическом воздействии на сравнительно мелкие фрагменты, однако не поддается диспергированию или даже растворению, после которого стало бы возможным парентеральное, прежде всего внутривенное, безопасное введение такого геля в организм. Тем самым в сочетании с по меньшей мере одним другим возможным компонентом (а), (в)-(е) применение гелеобразующего компонента (б) благодаря подобному его свойству дополнительно приводит к появлению неприятного жжения, возникновению рвоты, приданию лекарственной форме или извлеченному из нее экстракту неприятного вкуса и/или приданию лекарственной форме или извлеченному из нее экстракту отталкивающего внешнего вида.
Поэтому внутривенное введение соответствующего геля с высокой вероятностью привело бы к закупорке сосудов и тем самым к нанесению серьезного вреда здоровью.
Для проверки того или иного повышающего вязкость средства на его пригодность для применения в предлагаемой в изобретении лекарственной форме в качестве компонента (б) действующее вещество смешивают с повышающим вязкость средством и суспендируют в 10 мл воды при температуре 25°С. Если при этом образуется гель, который отвечает указанным выше условиям, то соответствующее повышающее вязкость средство пригодно для применения в предлагаемых в изобретении лекарственных формах в целях дополнительного предотвращения, соответственно предупреждения немедицинского употребления содержащихся в них действующих веществ.
При добавлении к предлагаемой в изобретении лекарственной форме компонента (б) в качестве него предпочтительно использовать одно или несколько повышающих вязкость средств, выбранных из группы, включающей микрокристаллическую целлюлозу с 11 мас.% натриевой соли карбоксиметилцеллюлозы (Avicel® RC 591), натриевую соль карбоксиметилцеллюлозы (Blanose®, CMC-Na С300Р, Frimulsion BLC-5®, Tyiose С300Р®), полиакриловую кислоту (Carbopol® 980 NF, Carbopol® 981), камедь плодов рожкового дерева (Cesagum® LA-200, Cesagum® LID/150, Cesagum® LN-1), пектины, предпочтительно из цитрусовых плодов или яблок (Cesapectin® HM Medium Rapid Set), крахмал восковидной кукурузы (C*Gel 04201®), альгинат натрия (Frimulsion ALG (E401) ®), гуаровую камедь (Frimulsion ВМ®, Polygum 26/1-75®), ι-каррагинан (Frimulsion D021®), камедь карайи, геллановую камедь (Kelcogel F®, Kelcogel LT100®), галактоманнан (Meyprogat 150®), камедь тары (Polygum 43/1®), пропиленгликольальгинат (Protanal-Ester SD-LB®), гиалуронат натрия, трагант, камедь тары (Vidogum SP 200®), велановую камедь (К1А96), представляющую собой получаемый ферментативным путем полисахарид, и ксантановую камедь (Xantural 180®).
Наиболее предпочтительны при этом ксантаны. Приведенные в скобках обозначения представляют собой торговые наименования, под которыми соответствующие материалы поставляются на рынок. Для соблюдения указанных выше условий повышающее(ие) вязкость средство(а) в целом достаточно использовать в количестве от 0,1 до 20 мас.%, наиболее предпочтительно от 0,1 до 15 мас.%, в пересчете на всю массу лекарственной формы.
Содержание повышающих вязкость средств при их использовании в качестве компонента (б) в предлагаемой в изобретении лекарственной форме предпочтительно должно составлять не менее 5 мг в пересчете на каждую разовую дозированную лекарственную форму, т.е. на каждую дозированную единицу.
В наиболее предпочтительном варианте осуществления настоящего изобретения в качестве компонента (б) используют те повышающие вязкость средства, которые при экстракции из лекарственной формы минимально необходимым количеством водной жидкости образуют гель с пузырьками воздуха в его объеме. Подобные гели внешне выглядят мутными, служа дополнительным визуальным предостережением, удерживающим от их парентерального введения в организм.
Повышающим вязкость средством, которое при добавлении минимально необходимого количества водной жидкости образует гель, в некоторых случаях дополнительно может служить также компонент (В).
Повышающие вязкость средства и остальные ингредиенты предлагаемой в изобретении лекарственной формы можно также располагать в ней пространственно отдельно друг от друга.
Предлагаемая в изобретении лекарственная форма для предупреждения ее применения не по назначению и для защиты от такого применения не по назначению может далее содержать компонент (в), а именно: один или несколько антагонистов действующего вещества, соответственно действующих веществ, потенциально являющегося(имися) объектом его(их) немедицинского употребления, при этом в предпочтительном варианте антагонист(ы) присутствует(ют) в предлагаемой в изобретении лекарственной форме пространственно отдельно от остальных ее ингредиентов и при ее применении по прямому назначению не проявляет(ют) никакого действия.
Соответствующие антагонисты для предупреждения немедицинского употребления действующих веществ известны специалистам в данной области и могут содержаться в предлагаемой в изобретении лекарственной форме как таковые либо в виде соответствующих производных, прежде всего сложных или простых эфиров, или же в виде соответствующих физиологически совместимых соединений, прежде всего в виде их солей или сольватов.
В состав лекарственной формы, содержащей в качестве действующего вещества опиоид, предпочтительно включать антагонист, выбранный из группы, включающей налоксон, налтрексон, налмефен, налид, налмексон, налорфин или налуфин, в каждом случае необязательно в виде соответствующего физиологически совместимого соединения, прежде всего в виде основания, соли или сольвата. Соответствующие антагонисты, если в состав предлагаемой в изобретении лекарственной формы предусмотрено включение компонента (в), используют в количестве не менее 1 мг, более предпочтительно в количестве от 3 до 100 мг, наиболее предпочтительно в количестве от 5 до 50 мг, из расчета на одну лекарственную форму, т.е. на одну дозированную единицу.
В состав предлагаемой в изобретении лекарственной формы, содержащей в качестве действующего вещества стимулятор, в качестве его антагониста предпочтительно включать нейролептик, предпочтительно по меньшей мере одно соединение, выбранное из группы, включающей галоперидол, прометацин, флуфеназин, перфеназин, левомепромазин, тиоридазин, перазин, хлорпромазин, хлорпротиксин, зуклопентиксол, флупентиксол, протипендил, зотепин, бенперидол, пипамперон, мелперон и бромперидол.
Указанные антагонисты предпочтительно включать в состав предлагаемой в изобретении лекарственной формы в обычной, известной специалистам в данной области терапевтической дозировке, а наиболее предпочтительно - в дозировке, которая из расчета на одну дозированную единицу в 2-3 раза превышает обычную дозировку.
Предлагаемая в изобретении лекарственная форма для предупреждения ее применения не по назначению и для защиты от такого применения не по назначению может также содержать компонент (г), которым является по меньшей мере одно рвотное средство, которое в предпочтительном варианте должно присутствовать в предлагаемой в изобретении лекарственной форме пространственно отдельно от остальных ее ингредиентов и при ее применении по прямому назначению не должно проявлять в организме никакого действия.
Соответствующие рвотные средства для предупреждения немедицинского употребления действующего вещества известны специалистам в данной области и могут содержаться в предлагаемой в изобретении лекарственной форме как таковые либо в виде соответствующих производных, прежде всего сложных или простых эфиров, или же в виде соответствующих физиологически совместимых соединений, прежде всего в виде их солей или сольватов.
В составе предлагаемой в изобретении лекарственной формы в предпочтительном варианте можно использовать рвотное средство на основе одного или нескольких веществ, содержащихся в корне ипекакуаны (рвотном корне), предпочтительно на основе содержащегося в нем эметина, и описанных, например, в книге "Pharmazeutische Biologic - Drogen и ihre Inhaltsstoffe", Prof. Dr. Hildebert Wagner, 2-е, переработанное, изд., изд-во Gustav Fischer Verlag, Stuttgart, New York, 1982. Соответствующие разделы этой книги тем самым включены в настоящее описание в качестве ссылки и являются его частью.
В предпочтительном варианте предлагаемая в изобретении лекарственная форма может содержать в качестве компонента (г) рвотное средство эметин, предпочтительно в количестве не менее 3 мг, более предпочтительно не менее 10 мг, наиболее предпочтительно в количестве не менее 20 мг, из расчета на одну лекарственную форму, т.е. на одну дозированную единицу.
Равным образом в другом предпочтительном варианте в качестве рвотного средства в предлагаемой в изобретении лекарственной форме для ее защиты от применения не по назначению можно использовать апоморфин, предпочтительно в количестве не менее 3 мг, более предпочтительно не менее 5 мг, наиболее предпочтительно не менее 7 мг, из расчета на одну дозированную единицу.
Если предлагаемая в изобретении лекарственная форма содержит компонент (д) в качестве еще одного препятствующего ее применению не по назначению вспомогательного вещества, то подобный краситель прежде всего при попытке экстрагировать из лекарственной формы действующее вещество для его парентерального, преимущественно внутривенного, введения в организм придает соответствующему водному раствору интенсивную окраску, которая может вызвать зрительный отталкивающий эффект. Подобное изменение окраски позволяет воспрепятствовать немедицинскому употреблению действующего вещества и путем его приема внутрь, которому обычно предшествует водная экстракция действующего вещества из содержащей его лекарственной формы. Соответствующие красители, а также количества, в которых их требуется использовать для достижения необходимого зрительного отталкивающего эффекта, рассмотрены в публикации WO 03/015531, соответствующие разделы которой тем самым включены в настоящее описание в качестве ссылки и являются его частью.
Если предлагаемая в изобретении лекарственная форма содержит в качестве еще одного препятствующего ее применению не по назначению вспомогательного вещества компонент (е), то подобное добавление к лекарственной форме по меньшей мере одного горького вещества позволяет благодаря вызываемому им ухудшению вкуса лекарственной формы дополнительно воспрепятствовать немедицинскому употреблению содержащегося в ней действующего вещества путем его перорального и/или назального введения в организм.
Соответствующие горькие вещества, а также их необходимые для достижения требуемого эффекта количества описаны в заявке US 2003/0064099 А1, соответствующие разделы которой тем самым включены в настоящее описание в качестве ссылки и являются его частью. В качестве горьких веществ предпочтительно применять ароматические масла, предпочтительно масло перечной мяты, эвкалиптовое масло, масло горького миндаля, ментол, ароматические вещества плодов, предпочтительно ароматические вещества цитронов, апельсинов, лимонов, грейпфрутов или смеси ароматических веществ указанных плодов, и/или бензоат денатония (Bitrex®). Особенно предпочтителен при этом бензоат денатония.
Предлагаемая в изобретении твердая лекарственная форма пригодна для перорального, вагинального или ректального, предпочтительно перорального, применения. В предпочтительном варианте предлагаемая в изобретении лекарственная форма не является пленочной. Предлагаемая в изобретении лекарственная форма может быть представлена в дисперсном (сыпучем) виде, предпочтительно в виде микротаблеток, микрокапсул, микропеллетов, гранулятов, сфероидов, крупинок или пеллетов, необязательно в расфасованном в капсулы или в спрессованном в таблетки виде, и предпочтительно предназначена для перорального применения. В предпочтительном варианте размер, соответственно полидисперсность указанных микротаблеток, микрокапсул, микропеллетов, гранулятов, сфероидов, крупинок или пеллетов лежит в пределах от 0,1 до 3 мм, наиболее предпочтительно от 0,5 до 2 мм. В зависимости от типа и требуемых свойств лекарственной формы для ее приготовления в некоторых случаях можно также использовать обычные вспомогательные вещества (Б).
Предлагаемую в изобретении защищенную от применения не по назначению лекарственная форму изготавливают путем термоформования с помощью экструдера без изменения при этом окраски экструдата.
Для определения степени обусловленного подобным термоформованием изменения окраски сначала определяют цвет смеси исходных компонентов, входящих в состав лекарственной формы, но без добавления придающего окраску компонента, например цветного пигмента или обладающего собственной окраской компонента (такого как α-токоферол). Этот состав затем подвергают согласно изобретению термоформованию, проводя все стадии процесса, включая охлаждение экструдата, в атмосфере инертного газа. Для сравнения такой же состав подвергают термоформованию тем же способом, но не в атмосфере инертного газа. Далее определяют окраску обеих приготовленных из одинаковых исходных составов лекарственных форм, одну из которых получали в соответствии с изобретением, а другую - в сравнительных целях. Для определения окраски используют атлас цветов "Munsell Book of Colour", изданный в 1966 г. компанией Munsell Colour Company Baltimore, шт.Мэриленд, США. Если термоформованная в соответствии с изобретением лекарственная форма имеет цвет с идентификационным номером N 9,5/, а максимум имеет цвет с идентификационным номером 5Y 9/1, то процесс термоформования классифицируют как не сопровождающийся изменением окраски. Если же лекарственная форма после термоформования имеет определенный по атласу цветов "Munsell Book of Colour" цвет с идентификационным номером 5Y 9/2 или более, то процесс термоформования классифицируют как сопровождающийся изменением окраски.
При создании изобретения неожиданно было установлено, что защищенные от применения не по назначению предлагаемым в изобретении способом твердые лекарственные формы не проявляют никакого классифицируемого по описанной выше методике изменения цвета, если весь процесс их приготовления проводят в атмосфере инертного газа, предпочтительно азота, с использованием экструдера для их термоформования.
В соответствии с этим еще одним объектом настоящего изобретения является способ приготовления предлагаемых в изобретении защищенных от применения не по назначению лекарственных форм, отличающийся тем, что
1) компоненты (А), (Б), (В) и возможно используемый компонент (Г) смешивают между собой, а также смешивают с ними либо при необходимости смешивают отдельно с добавлением компонента (В) и возможно используемого компонента (Г) возможно используемые дополнительные компоненты (а)-(е),
2) полученную смесь или полученные смеси нагревают в экструдере по меньшей мере до температуры размягчения компонента (В) и путем приложения усилия экструдируют через выходное отверстие экструдера,
3) еще пластичный экструдат разделяют на отдельные части, из которых формуют лекарственные формы, или
4) из охлажденного и при необходимости вновь нагретого разделенного на отдельные части экструдата формуют лекарственные формы, проводя при этом стадии 2) и 3) и необязательно стадии 1) и 4) в атмосфере инертного газа, предпочтительно азота.
Смешение компонентов на стадии 1) может также происходить уже в экструдере.
Для смешения компонентов (А), (Б), (В) и возможно (Г), а также смешения с ними, соответственно отдельного смешения возможно используемых дополнительных компонентов (а)-(е) и при необходимости компонентов (В) и возможно используемого компонента (Г) используют, соответственно в каждом случае используют известный смеситель. Такой смеситель может представлять собой, например, валковый смеситель, вибросмеситель, работающий с созданием сдвигового усилия смеситель или смеситель принудительного перемешивания.
Компонент (В) и возможно используемый компонент (Г) перед их смешением с остальными компонентами согласно изобретению предпочтительно объединять с антиокислителем. С этой целью оба компонента (В) и (Г) можно смешивать с антиокислителем, для чего в предпочтительном варианте антиокислитель растворяют или суспендируют в легко летучем растворителе, полученный раствор, соответственно полученную суспензию затем до гомогенности смешивают с компонентом (В) и возможно используемым компонентом (Г) и в завершение путем сушки удаляют растворитель, предпочтительно в атмосфере инертного газа.
Для приготовления предлагаемых в изобретении лекарственных форм, которые содержат дозированные субъединицы с дополнительными предотвращающими, соответственно затрудняющими немедицинское употребление входящих в их состав действующих веществ компонентами, полученные на стадии 1) смеси можно экструдировать совместно либо раздельно.
В любом случае нагретую(ые) в экструдере по меньшей мере до температуры размягчения компонента (В), а предпочтительно расплавленную(ые) смесь(и) выдавливают из экструдера через его головку с по меньшей мере одним каналом.
Для осуществления предлагаемого в изобретении способа предпочтительно использовать обычные экструдеры, наиболее предпочтительно одно- или двухчервячные экструдеры.
В предпочтительном варианте в экструдере должно иметься по меньшей мере две температурные зоны, в первой из которых, примыкающей к загрузочной и возможно предусмотренной смесительной зоне, происходит нагрев смеси по меньшей мере до температуры размягчения компонента (В). Расход формуемой смеси в предпочтительном варианте составляет от 2,0 до 8,0 кг/ч.
После нагрева до по меньшей мере температуры размягчения компонента (В) расплавленная смесь перемещается в экструдере его червяком, продолжает гомогенизироваться, компримируется, соответственно уплотняется, достигая непосредственно перед выходом из экструзионной головки минимального давления в 5 бар, предпочтительно по меньшей мере 10 бар, и в зависимости от количества каналов (отверстий) в экструзионной головке выдавливается через нее в виде экструдата или экструдатов. Экструдер можно оснащать головкой с любым профилем канала, соответственно каналов в ней. Так, в частности, канал, соответственно каналы в экструзионной головке может(гут) иметь круглое сечение, предпочтительно диаметром от 0,1 до 15 мм, продолговатое сечение, предпочтительно с максимальной продольной протяженностью 21 мм и поперечной протяженностью 10 мм, или овальное сечение. Предпочтительно использовать экструзионную головку с каналом, соответственно каналами круглого сечения. Используемый экструдер может иметь нагреваемый или охлаждаемый корпус. Соответствующее термостатирование корпуса экструдера, т.е. нагрев или охлаждение, зависит от средней температуры экструдируемой смеси (температуры продукта), которая (температура) должна быть не меньше температуры размягчения компонента (В), но не должна превышать температуру, выше которой может произойти недопустимое изменение свойств перерабатываемого действующего вещества, потенциально являющегося объектом его немедицинского употребления. С учетом этого температуру экструдируемой смеси предпочтительно поддерживать ниже 180°С, более предпочтительно ниже 150°С, но не ниже по меньшей мере температуры размягчения компонента (В).
После экструзии расплавленной смеси и при необходимости охлаждения полученного экструдата, соответственно экструдатов его(их) предпочтительно подвергать измельчению. Подобное измельчение в предпочтительном варианте может заключаться в разрезании экструдатов летучими или вращающимися ножами, водоструйными резаками, проволоками, лезвиями или лазерными резаками.
Для промежуточного, соответственно окончательного хранения экструдата или разделенного на отдельные части экструдата, соответственно готовых предлагаемых в изобретении лекарственных форм не требуется создание атмосферы инертного газа.
Разделенный на отдельные части экструдат можно обычными методами подвергать гранулированию или таблетированию для придания лекарственной форме окончательной формы. В другом варианте имеющий форму прутка экструдат можно не разделять на отдельные части, а подвергать каландрованию пропусканием через вращающиеся во взаимно противоположных направлениях валки, которые на своих боковых поверхностях имеют расположенные друг против друга профильные углубления, с приданием экструдату таким путем профиля, который соответствует окончательному контуру лекарственной формы, предпочтительно таблетки, отделяемой затем от остального экструдата обычными методами.
В том случае, когда экструдат или разделенный на отдельные части экструдат не подвергают сразу же формованию с приданием ему профиля, который соответствует окончательной форме лекарственной формы, а сначала охлаждают для промежуточного хранения, дальнейшую переработку экструдата после такого его промежуточного хранения необходимо проводить в атмосфере инертного газа, предпочтительно азота, которую требуется сохранять от момента нагрева промежуточно хранившегося экструдата вплоть до завершения его пластикации и окончательного формования из него лекарственной формы.
Усилие, прикладываемое в экструдере к по меньшей мере пластицированной смеси, устанавливают регулированием скорости вращения и подбором геометрии транспортирующего устройства (червяка) в экструдере, а также подбором размеров выходного отверстия экструдера на значение, при котором давление в экструдере предпочтительно перед непосредственным экструдированием пластицированной смеси достигает необходимого для этого уровня. Параметры экструзии, необходимые для получения лекарственной формы с сопротивлением разрушению по меньшей мере 500 Н, можно определять для каждого конкретного состава проведением простых предварительных экспериментов.
В следующем предпочтительном варианте предлагаемая в изобретении лекарственная форма представлена в виде таблетки, капсулы или в виде пероральной осмотической терапевтической системы (ПОТС), предпочтительно при использовании по меньшей мере одного дополнительного, препятствующего применению лекарственной формы не по назначению компонента (а)-(е).
При включении в состав предлагаемой в изобретении лекарственной формы компонента (в) и/или компонента (г) и/или компонента (е) каждый из них необходимо использовать в такой форме или в столь низкой дозировке, чтобы при применении лекарственной формы по прямому назначению практически полностью исключить возможность проявления им причиняющего вред пациенту или снижающего эффективность действующего вещества действия.
При включении в состав предлагаемой в изобретении лекарственной формы компонента (г) и/или компонента (е) дозировку каждого из них необходимо подбирать с таким расчетом, чтобы он при применении лекарственной формы по прямому назначению путем ее приема внутрь не вызывал никакой отрицательной реакции организма. При превышении же предусмотренной дозировки указанных компонентов в случае применения лекарственной формы не по назначению они вызывают тошноту, соответственно позывы на рвоту, соответственно неприятные вкусовые ощущения. Конкретную дозу компонента (г) и/или компонента (е), при которой пациент при применении лекарственной формы по прямому назначению путем ее приема внутрь все еще остается толерантен к каждому из этих компонентов, можно определить путем проведения простых предварительных опытов.
Если же для защиты предлагаемой в изобретении лекарственной формы от применения не по назначению вне зависимости от практически полной невозможности ее измельчения в порошок предусмотрено применение компонента (в) и/или компонента (г) и/или компонента (е), то каждый из таких компонентов предпочтительно использовать в столь высокой дозировке, чтобы при применении лекарственной формы не по прямому назначению он проявлял ярко выраженное отрицательное действие. В предпочтительном варианте обеспечить соблюдение этого условия удается за счет пространственного отделения по меньшей мере действующего вещества, соответственно действующих веществ от компонента (в) и/или компонента (г) и/или компонента (е) предпочтительно путем включения действующего вещества, соответственно действующих веществ в состав по меньшей мере одной дозированной субъединицы (X), а компонента (в) и/или компонента (г) и/или компонента (е) - в состав по меньшей мере одной другой дозированной субъединицы (Y), в которой компоненты (в), (г) и (е) при применении лекарственной формы по прямому назначению не проявляют при приеме внутрь и/или при попадании в организм своего действия, при этом в обеих дозированных субъединицах применяются идентичные остальные компоненты лекарственной формы, прежде всего компонент (В) и возможно используемый компонент (Г).
При включении в состав предлагаемой в изобретении лекарственной формы по меньшей мере двух компонентов (в) и (г), соответственно (е) они могут содержаться в одной и той же дозированной субъединице (Y) или в разных дозированных субъединицах (Y). В предпочтительном варианте все компоненты (в), (г) и (е) при их использовании включают в состав одной и той же дозированной субъединицы (Y).
Под дозированными субъединицами согласно настоящему изобретению подразумеваются твердые формы, каждая из которых наряду с обычными, известными вспомогательными веществами содержит действующее(ие) вещество(а), по меньшей мере один полимер (В) и возможно используемый компонент (Г) и при необходимости по меньшей мере один из возможно используемых компонентов (а) и/или (б) и/или (д), соответственно каждая из которых содержит по меньшей мере один полимер (В) и возможно используемый компонент (Г) и антагонист(ы) и/или рвотное(ые) средство(а) и/или компонент (д) и/или компонент (е) и при необходимости по меньшей мере один из возможно используемых компонентов (а) и/или (б). При этом следует отметить, что каждую из указанных выше дозированных субъединиц приготавливают описанным выше способом.
Существенное преимущество, связанное с отделением в предлагаемой в изобретении лекарственной форме действующих веществ от компонентов (в), соответственно (г), соответственно (е) путем включения тех и других в состав разных дозированных субъединиц (X) и (Y), состоит в том, что при применении лекарственной формы по прямому назначению компонент (в) и/или компонент (г) и/или компонент (е) при приеме внутрь и/или при попадании в организм практически не высвобождаются из содержащих их дозированных субъединиц или высвобождаются из них лишь в столь малых количествах, при которых они не проявляют никакого причиняющего вред пациенту или снижающего эффективность лечения действия, либо при прохождении через организм пациента высвобождаются из содержащих их дозированных субъединиц только в тех местах, где они не резорбируются в достаточной для проявления ими своего действия степени. В предпочтительном варианте компонент (в) и/или компонент (г) и/или компонент (е) при применении лекарственной формы по прямому назначению практически не высвобождаются в организме пациента или не ощущаются им.
Для специалиста в данной области очевидно, что эти указанные выше условия могут варьироваться в зависимости от конкретно применяемых компонентов (в), (г) и/или (е), а также от состава и типа дозированных субъединиц, соответственно лекарственной формы. Оптимальные для каждой конкретной лекарственной формы ее состав и тип можно определить путем проведения простых предварительных опытов. Решающее же значение имеет лишь включение в состав каждой конкретной дозированной субъединицы полимера (В) и при необходимости компонента (Г) и ее приготовление описанным выше способом.
Если вопреки ожиданиям подобную предлагаемую в изобретении лекарственную форму, которая содержит компонент (в) и/или компонент (д) и/или компонент (г) и/или компонент (е) в дозированных субъединицах (Y), и удастся в целях немедицинского употребления действующего вещества измельчить в порошок и экстрагировать из него действующее вещество с помощью пригодного для этого экстрагента, то наряду с действующим веществом будет получен и соответствующий компонент (в) и/или (д) и/или (е) и/или (г) в форме, в которой его невозможно простым путем отделить от действующего вещества, и поэтому он при применении полученного из лекарственной формы экстракта, прежде всего при его пероральном и/или парентеральном введении в организм, будет при приеме внутрь и/или при попадании в организм проявлять свое действие, причем один из компонентов (в) и/или (г) и/или (е) дополнительно вызовет соответствующую отрицательную реакцию организма, а компонент (д) при попытке экстрагировать действующее вещество вызовет визуально отталкивающее изменение окраски и таким путем воспрепятствует применению лекарственной формы не по назначению.
Предлагаемая в изобретении лекарственная форма с пространственным отделением в ней действующего вещества, соответственно действующих веществ от компонентов (в), (г) и/или (д), предпочтительно за счет включения того, соответственно тех и других в состав разных дозированных субъединиц, может иметь самую разнообразную структуру, при этом соответствующие дозированные субъединицы могут иметь в предлагаемой в изобретении лекарственной форме любое пространственное расположение друг относительно друга при условии соблюдения указанных выше условий касательно высвобождения компонента (в) и/или компонента (г).
Для специалиста в данной области очевидно, что компонент (а) и/или компонент (б) при его, соответственно их использовании в предлагаемой в изобретении лекарственной форме в предпочтительном варианте можно включать в состав не только каждой из соответствующих дозированных субъединиц (X) и (Y), но и самостоятельных дозированных субъединиц, соответствующих дозированным субъединицам (X) и (Y), при условии, что форма приготовления таких самостоятельных дозированных субъединиц не сказывается отрицательно на защите лекарственной формы от применения не по назначению и на высвобождении действующего вещества при применении лекарственной формы по прямому назначению и содержит в своем составе полимер (В) и возможно используемый компонент (Г), а сами эти дозированные субъединицы для достижения необходимой твердости получают описанным выше способом.
В одном из предпочтительных вариантов дозированные субъединицы (X) и (Y), составляющие предлагаемую в изобретении лекарственную форму, представлены в дисперсной форме, предпочтительно в виде микротаблеток, микрокапсул, микропеллетов, гранулятов, сфероидов, крупинок или пеллетов, при этом и дозированная субъединица (X), и дозированная субъединица (Y) должны иметь одну и ту же форму, т.е. внешний вид, чтобы дозированную субъединицу (X) невозможно было отделить от дозированной субъединицы (Y), например, путем механической сортировки. В предпочтительном варианте размер указанных микротаблеток, микрокапсул, микропеллетов, гранулятов, сфероидов, крупинок или пеллетов лежит в пределах от 0,1 до 3 мм, наиболее предпочтительно от 0,5 до 2 мм.
Дозированные субъединицы (X) и (Y), представленные в дисперсной форме, в предпочтительном варианте можно также расфасовывать в капсулы или спрессовывать в таблетки, при этом завершающая стадия приготовления лекарственной формы должна обеспечивать сохранение дозированных субъединиц (X) и (Y) и в готовой лекарственной форме.
Соответствующие дисперсные дозированные субъединицы (X) и (Y) не только должны иметь идентичную внешнюю форму, но и должны быть визуально неотличимы одна от другой, чтобы их невозможно было отделить друг от друга путем простой сортировки. Для этого на дозированные субъединицы обоих типов можно, например, наносить идентичные покрытия, которые наряду с подобной функцией, состоящей в придании дозированным субъединицам обоих типов одинакового внешнего вида, могут выполнять и другие функции, например, ретардацию высвобождения одного или нескольких действующих веществ или придавать дозированным субъединицам каждого конкретного типа устойчивость к действию желудочного сока.
Из дисперсных дозированных субъединиц путем их суспендирования в фармацевтически приемлемых суспендирующих агентах можно также приготавливать взвеси или суспензии, представляющие собой лекарственные формы для приема внутрь.
В другом предпочтительном варианте осуществления настоящего изобретения дозированные субъединицы (X) и (Y) предлагается располагать слоями друг относительно друга.
В этом варианте слои из дозированных субъединиц (X) и (Y) предпочтительно располагать друг относительно друга в предлагаемой в изобретении лекарственной форме вертикально или горизонтально, при этом в лекарственной форме может также присутствовать один или несколько слоев из дозированных субъединиц (X) и один или несколько слоев из дозированных субъединиц (Y), и поэтому наряду с предпочтительными последовательностями расположения слоев типа (X)-(Y), соответственно (X)-(Y)-(X) возможны и любые иные последовательности расположения слоев, в том числе и в сочетании со слоями, содержащими компонент (а) и/или компонент (б).
Равным образом предпочтительна предлагаемая в изобретении лекарственная форма, в которой дозированная субъединица (Y) образует сердцевину, которая полностью заключена в оболочку из дозированной субъединицы (X) и которая может быть отделена от нее разделительным слоем (Z). Такую же структуру предпочтительно использовать и в указанных выше дисперсных формах, причем в этом случае обе дозированные субъединицы (X) и (Y), а также возможно предусмотренный между ними разделительный слой (Z), который по своей твердости должен удовлетворять соответствующим изобретению требованиям, приготавливают в одной и той же дисперсной форме.
В следующем предпочтительном варианте дозированная субъединица (X) образует в предлагаемой в изобретении лекарственной форме сердцевину, заключенную в оболочку из дозированной субъединицы (Y), которая имеет по меньшей мере один канал, ведущий от сердцевины лекарственной формы к ее поверхности.
Между слоем из дозированной субъединицы (X) и слоем из дозированной субъединицы (Y) предлагаемая в изобретении лекарственная форма для пространственного отделения дозированной субъединицы (X) от дозированной субъединицы (Y) может иметь по одному или нескольким разделительным слоям (Z), предпочтительно по одному разделительному слою (Z), который в некоторых случаях может быть образован набухающим материалом.
Предлагаемая в изобретении лекарственная форма с по меньшей мере частичным вертикальным или горизонтальным расположением слоев из дозированных субъединиц (X) и (Y), а также возможно предусмотренного между ними разделительного слоя (Z) предпочтительно должна быть представлена в виде таблетки или ламината.
При этом в наиболее предпочтительном варианте свободная поверхность дозированной субъединицы (Y) полностью и в некоторых случаях по меньшей мере часть свободной поверхности дозированной(ых) субъединицы (субъединиц) (X), а также в некоторых случаях по меньшей мере часть свободной поверхности возможно предусмотренного(ых) разделительного(ых) слоя(ев) (Z) могут быть покрыты по меньшей мере одним препятствующим высвобождению компонента (в) и/или компонента (д) и/или компонента (г) и/или (е) компонента барьерным слоем (Z'). Такой барьерный слой (Z') по своей твердости также должен удовлетворять соответствующим изобретению требованиям.
Равным образом особенно предпочтительна предлагаемая в изобретении лекарственная форма с вертикальным или горизонтальным расположением слоев из дозированных субъединиц (X) и (Y) и по меньшей мере одного расположенного между ними "выталкивающего" слоя (р) ("push"-слоя) (р), а также возможно предусмотренного разделительного слоя (Z), у которой все свободные поверхности состоящей из дозированных субъединиц (X) и (Y), "выталкивающего" слоя ("push"-слоя) и возможно предусмотренного разделительного слоя (Z) слоистой структуры покрыты полупроницаемым покрытием (Е), которое проницаемо для среды, в которую высвобождается действующее вещество, т.е. обычно для физиологической жидкости, но в основном не проницаемо для действующего вещества и для компонента (в) и/или компонента (г) и/или компонента (е) и которое в зоне дозированной субъединицы (X) имеет по меньшей мере одно отверстие для высвобождения действующего вещества.
Соответствующая лекарственная форма, которая известна под названием пероральной осмотической терапевтической системы (ПОТС), равно как и пригодные для ее изготовления материалы и способы ее изготовления описаны помимо прочего в патентах US 4612008, US 4765989 и US 4783337. В соответствии с этим указанные патенты включены в настоящее описание в качестве ссылки и составляют его часть.
В следующем предпочтительном варианте дозированная субъединица (X) в предлагаемой в изобретении лекарственной форме имеет вид таблетки, у которой ее боковая грань и необязательно одна из обоих основных поверхностей покрыта барьерным слоем (Z'), содержащим компонент (в) и/или компонент (г) и/или компонент (е).
Для специалиста в данной области очевидно, что используемые в каждом случае для приготовления предлагаемой в изобретении лекарственной формы вспомогательные вещества, включаемые в состав дозированной(ых) субъединицы (субъединиц) (X), соответственно (Y), а также возможно предусмотренного(ых) разделительного(ых) слоя(ев) (Z) и/или барьерного(ых) слоя(ев) (Z'), выбираются в зависимости от расположения указанных субъединиц и слоев в предлагаемой в изобретении лекарственной форме, от предусмотренного пути ее введения в организм, а также в зависимости от конкретного вещества, используемого в качестве возможно предусмотренного компонента (а) и/или компонента (б) и/или компонента (д) и в качестве компонента (в) и/или компонента (г) и/или компонента (е). Материалы, обладающие необходимыми в каждом конкретном случае свойствами, известны специалистам в данной области.
При заключении в предлагаемой в изобретении лекарственной форме дозированной субъединицы (Y) в препятствующую высвобождению из нее компонента (в) и/или компонента (г) и/или компонента (е) оболочку, предпочтительно в виде барьерного слоя, для приготовления такой дозированной субъединицы можно использовать обычные, известные специалистам в данной области материалы при условии включения в ее состав по меньшей мере одного полимера (В) и возможно компонента (Г) для соблюдения требований, предъявляемых к твердости предлагаемой в изобретении лекарственной формы.
Если же применение соответствующего барьерного слоя (Z') для предотвращения высвобождения компонента (в) и/или компонента (г) и/или компонента (е) не предусмотрено, то материалы для приготовления дозированных субъединиц (Y) должны выбираться таким образом, чтобы практически полностью исключить высвобождение соответствующего компонента (в) и/или компонента (г) из дозированной субъединицы (Y). В предпочтительном варианте для соблюдения этого требования можно использовать перечисленные ниже материалы, которые могут использоваться и для образования барьерного слоя.
К предпочтительным в этом отношении материалам относятся материалы, выбранные из группы, включающей алкилцеллюлозы, гидроксиалкилцеллюлозы, глюканы, склероглюканы, маннаны, ксантаны, сополимеры поли[бис-(n-карбоксифенокси)пропана и себациновой кислоты, предпочтительно в молярном соотношении 20:80 (выпускаемые под названием Polifeprosan 20®), карбоксиметилцеллюлозы, простые эфиры целлюлозы, сложные эфиры целлюлозы, нитроцеллюлозы, полимеры на основе (мет)акриловой кислоты, а также ее эфиров, полиамиды, поликарбонаты, полиалкилены, полиалкиленгликоли, полиалкиленоксиды, полиалкилентерефталаты, поливиниловые спирты, простые поливиниловые эфиры, сложные поливиниловые эфиры, галогенированные поливинилы, полигликолиды, полисилоксаны, а также полиуретаны и их сополимеры.
К наиболее пригодным для приготовления дозированных субъединиц (Y) материалам относятся материалы, выбранные из группы, включающей метилцеллюлозу, этилцеллюлозу, гидроксипропилцеллюлозу, гидроксипропилметилцеллюлозу, гидроксибутилметилцеллюлозу, ацетат целлюлозы, пропионат целлюлозы (с низкой, средней или высокой молекулярной массой), ацетопропионат целлюлозы, ацетобутират целлюлозы, ацетофталат целлюлозы, карбоксиметилцеллюлозу, триацетат целлюлозы, натриевый сульфат целлюлозы, полиметилметакрилат, полиэтилметакрилат, полибутилметакрилат, полиизобутилметакрилат, полигексилметакрилат, полиизодецилметакрилат, полилаурилметакрилат, полифенилметакрилат, полиметилакрилат, полиизопропилакрилат, полиизобутилакрилат, полиоктатдецилакрилат, полиэтилен, полиэтилен низкой плотности, полиэтилен высокой плотности, полипропилен, полиэтиленгликоль, полиэтиленоксид, полиэтилентерефталат, поливиниловый спирт, поливинилизобутиловый эфир, поливинилацетат и поливинилхлорид.
К наиболее пригодным для применения в указанных выше целях сополимерам относятся сополимеры, выбранные из группы, включающей сополимеры бутилметакрилата и изобутилметакрилата, сополимеры метилвинилового эфира и малеиновой кислоты с высокой молекулярной массой, сополимеры метилвинилового эфира и моноэтилового эфира малеиновой кислоты, сополимеры метилвинилового эфира и малеинового ангидрида, а также сополимеры винилового спирта и винилацетата.
К числу других, наиболее пригодных для выполнения барьерного слоя материалов относятся наполненный крахмалом поликапролактон (WO 98/20073), алифатические полиэфирамиды (DE 19753534 A1, DE 19800698 A1, ЕР 0820698 A1), алифатические и ароматические прлиэфируретаны (DE 19822979), полигидроксиалканоаты, прежде всего полигидроксибутираты и полигидроксивалериаты, казеин (DE 4309528), полилактиды и сополилактиды (ЕР 0980894 A1). Указанные публикации в соответствии с этим включены в настоящее описание в качестве ссылки и составляют его часть.
Рассмотренные выше материалы можно при необходимости использовать в смеси с другими обычными известными специалистам в данной области вспомогательными веществами, предпочтительно вспомогательными веществами, выбранными из группы, включающей пластификаторы, скользящие вещества, антиокислители, например моностеарат глицерина, полусинтетические производные триглицеридов, полусинтетические глицериды, гидрированное касторовое масло, пальмитостеарат глицерина, бегенат глицерина, поливинилпирролидон, желатин, стеарат магния, стеариновую кислоту, стеарат натрия, тальк, бензоат натрия, борную кислоту и коллоидный кремнезем, жирные кислоты, замещенные триглицериды, глицериды, полиоксиалкиленгликоли, полиалкиленгликоли и их производные.
При наличии в предлагаемой в изобретении лекарственной форме разделительного слоя (Z) его, равно как и не заключенную в оболочку дозированную субъединицу (Y), предпочтительно выполнять из указанных выше для барьерного слоя материалов. Для специалиста в данной области очевидно, что регулировать высвобождение действующего вещества, соответственно компонента (в) и/или компонента (г) из соответствующей дозированной субъединицы можно также варьированием толщины разделительного слоя.
Предлагаемая в изобретении лекарственная форма представляет собой лекарственную форму с контролируемым высвобождением из нее действующего вещества. При этом в предпочтительном варианте она пригодна для двукратного в сутки введения в организм пациента.
Помимо этого предлагаемая в изобретении лекарственная форма может представлять собой ретард-форму с по меньшей мере частично замедленным (ретардированным) высвобождением из нее одного или нескольких действующих веществ, которые потенциально являются объектом их немедицинского употребления и ретардация высвобождения которых может обеспечиваться с помощью обычных, известных специалистам в данной области материалов и способов, например, за счет внедрения действующего вещества в задерживающую его высвобождение матрицу или за счет нанесения одного или нескольких задерживающих его высвобождение покрытий. Вместе с тем при регулируемом высвобождении действующего вещества всегда должно обеспечиваться соблюдение указанных выше условий, т.е., например, при применении лекарственной формы по прямому назначению практически полное высвобождение действующего вещества, соответственно действующих веществ из нее должно завершаться еще до того момента, как возможно содержащиеся в ней компонент (в) и/или компонент (г) начнут проявлять свое отрицательное действие на организм. Помимо этого добавление задерживающих высвобождение действующих веществ материалов не должно снижать необходимую твердость лекарственной формы.
Контролируемое высвобождение действующего вещества из предлагаемой в изобретении лекарственной формы предпочтительно обеспечивать за счет внедрения действующего вещества в матрицу. Служащие матрицей вспомогательные вещества контролируют высвобождение действующих веществ. Образующие матрицу материалы могут представлять собой, например, гидрофильные гелеобразующие материалы, высвобождение из которых действующих веществ происходит преимущественно в результате диффузии, либо гидрофобные материалы, высвобождение из которых действующих веществ происходит преимущественно в результате диффузии из пор в матрице.
В качестве образующих матрицу материалов могут использоваться физиологически совместимые гидрофильные материалы, известные специалистам в данной области. В качестве гидрофильных образующих матрицу материалов предпочтительно использовать полимеры, наиболее предпочтительно простые эфиры целлюлозы, сложные эфиры целлюлозы и/или акриловые смолы. К особенно предпочтительным образующим матрицу материалам относятся этилцеллюлоза, гидроксипропилметилцеллюлоза, гидроксипропилцеллюлоза, гидроксиметилцеллюлоза, поли(мет)акриловая кислота и/или ее производные, такие как ее соли, амиды или эфиры.
Равным образом в качестве образующих матрицу материалов предпочтительно использовать гидрофобные материалы, такие как гидрофобные полимеры, воски, жиры, жирные кислоты с длинной цепью, жирные спирты или соответствующие сложные или простые эфиры либо смеси указанных материалов. К особенно предпочтительным для применения в качестве матрицы гидрофобным материалам относятся моно- или диглицериды жирных кислот с С12-С30 и/или жирные спирты с C12-C30 и/или воски либо смеси указанных материалов.
В качестве образующих матрицу материалов возможно также использовать смеси указанных выше гидрофильных и гидрофобных материалов.
Помимо этого и компонент (В), а также возможно предусмотренный компонент (Г), которые согласно изобретению используются для придания лекарственной форме необходимого сопротивления разрушения, составляющего по меньшей мере 500 Н, уже могут служить дополнительными образующими матрицу материалами.
Предлагаемая в изобретении лекарственная форма, когда она предназначена для приема внутрь, в предпочтительном варианте может иметь также устойчивое к действию желудочного сока покрытие, которое растворяется в зависимости от значения рН среды, в которой происходит высвобождение действующего вещества из лекарственной формы. При наличии такого покрытия предлагаемая в изобретении лекарственная форма проходит через желудок без растворения в нем и начинает растворяться и высвобождать действующее вещество лишь в кишечнике. В предпочтительном варианте устойчивое к действию желудочного сока покрытие должно растворяться при значении рН в пределах от 5 до 7,5.
Соответствующие материалы и способы, используемые для ретардации высвобождения действующих веществ, а также для нанесения устойчивых к действию желудочного сока покрытий, известны, например, из книги "Coated Pharmaceutical Dosage Forms - Fundamentals, Manufacturing Techniques, Biopharmaceutical Aspects, Test Methods and Raw Materials", Kurt Н. Bauer, K. Lehmann, Hermann P. Osterwald, Rothgang, Gerhart, 1-е изд., изд-во Medpharm Scientific Publishers, 1998. В соответствии с этим указанная публикация включена в настоящее описание в качестве ссылки и составляют его часть.
Методика определения сопротивления разрушению
Для проверки пригодности того или иного материала для применения в качестве компонента (В) или (Г) из проверяемого материала при температуре, равной по меньшей мере температуре его размягчения, определяемой по ДСК-диаграмме материала, прессуют при усилии прессования, равном 150 Н, таблетку диаметром 10 мм и толщиной (высотой) 5 мм. Затем в соответствии с методом определения сопротивления таблеток разрушению, опубликованным в европейской фармакопее 1997-го года издания, сс. 143, 144, метод №2.9.8, с использованием описанного ниже оборудования определяют сопротивление изготовленных таким путем таблеток разрушению. В качестве измерительного оборудования используют машину для испытания материалов типа "Zwick Z 2.5" фирмы Zwick с максимальным развиваемым усилием Fmax, равным 2,5 кН, с ходом поперечины, равным максимум 1150 мм и выставляемым с помощью регулировочного механизма, состоящего из колонны и ходового винта, со свободным рабочим пространством сзади, равным 100 мм, со скоростью испытания, регулируемой в пределах от 0,1 до 800 мм/мин, и программным обеспечением "testControl". Для измерений используют пуансон с ввинчиваемыми вставками и цилиндром (диаметром 10 мм) и месдозу с максимальным измеряемым усилием Fmax 1 кН, с диаметром 8 мм, с точностью измерения согласно стандарту ISO 7500-1, соответствующей классу 0.5, начиная с 10 Н, и классу, 1 начиная с 2 Н, и с выдаваемым производителем проверочным сертификатом М согласно стандарту DIN 55350-18 (рассчитываемое по методике фирмы Zwick максимальное суммарное усилие Fmax 1,45 кН) (все оборудование фирмы Zwick GmbH & Со. KG, Ульм, Германия, со следующими номерами для заказа: BTC-FR 2.5 ТН. D09 для испытательной машины, BTC-LC 0050N. Р01 для месдозы и ВО 70000 S06 для центрирующего приспособления).
На прилагаемом к описанию чертеже в предельно упрощенном виде схематично показано устройство для измерения сопротивления таблеток разрушению и, в частности, показано используемое для этого юстировочное приспособление (6) для выверки положения таблетки (4) до и в процессе измерения. Для проведения измерений таблетку (4) помещают между верхней нажимной пластиной (1) и нижней нажимной пластиной (3) не показанного на чертеже устройства для приложения нагрузки и фиксируют с помощью двух состоящих из двух частей зажимов (2), каждый из которых после выставления необходимого для закрепления и центрирования анализируемой таблетки расстояния (5) прочно соединяют с верхней, соответственно нижней нажимной пластиной (не показано). Для регулирования расстояния (5) обе части каждого из зажимов можно сдвигать или раздвигать в горизонтальном направлении перемещением по соответствующей нажимной пластине, на которой они установлены.
К стойким к разрушению под воздействием определенной механической нагрузки относят также таблетки, которые при приложении заданного усилия не разрушаются, но в некоторых случаях пластически деформируются.
Описанным выше методом измерений, которым можно испытывать не только таблетки, но и отличные от них лекарственные формы, определяли сопротивление предлагаемых в изобретении лекарственных форм разрушению.
Ниже изобретение поясняется на примере. Все рассмотренные в этом примере пояснения носят исключительно иллюстративный характер и не ограничивают объема изобретения (см. таблицу).
Указанное количество БГТ растворяли в этаноле (96%-ном) с получением 7,7%-ного (по массе) этанольного раствора. Этот раствор сначала в течение 30 мин перемешивали в скоростном смесителе с 150 г полиэтиленоксида, после чего добавляли остальное количество полиэтиленоксида и перемешивали еще в течение 30 мин. Затем полученную смесь сушили при 40°С в течение 12 ч. К этой высушенной смеси добавляли все остальные компоненты и перемешивали в течение 15 мин в гравитационном смесителе. Полученную порошковую смесь подавали в дозированных количествах в экструдер. Для экструзии использовали двухчервячный экструдер типа Micro 27 GL 40 D фирмы Leistritz (Нюрнберг, Германия) с диаметром шпинделя 18 мм. В экструдере использовали червяки с тупыми концами, имеющиеся на которых шестигранные углубления под соответствующий ключ закрывали заглушками. В качестве экструзионной головки использовали обогреваемую головку с каналом круглого сечения и диаметром 8 мм. Весь процесс проводили в атмосфере азота.
Для экструзии использовали следующие параметры:
Еще горячий экструдат охлаждали в атмосфере азота. От охлажденного экструдата отделяли двоякоплоские таблетки.
Сопротивление таблеток разрушению определяли описанным выше методом. При воздействии на таблетки усилием в 500 Н не происходило никакого их разрушения. Помимо этого таблетки невозможно было измельчить ни молотком, ни с помощью ступки и пестика.
Охлажденный экструдат, соответственно 10 отделенных от него таблеток имели определенный с помощью атласа цветов "Munsell Book of Colour" цвет с идентификационным номером N 9,5/, что указывало на отсутствие изменения окраски приготовленной предлагаемым в изобретении способом лекарственной формы при ее термоформовании с помощью экструдера.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЗАЩИЩЕННАЯ ОТ ПРИМЕНЕНИЯ НЕ ПО НАЗНАЧЕНИЮ ЛЕКАРСТВЕННАЯ ФОРМА | 2004 |
|
RU2354357C2 |
| ЗАЩИЩЕННАЯ ОТ ПРИМЕНЕНИЯ НЕ ПО НАЗНАЧЕНИЮ ПЕРОРАЛЬНАЯ ЛЕКАРСТВЕННАЯ ФОРМА | 2005 |
|
RU2396944C2 |
| ЗАЩИЩЕННАЯ ОТ ПРИМЕНЕНИЯ НЕ ПО НАЗНАЧЕНИЮ ПЕРОРАЛЬНАЯ ЛЕКАРСТВЕННАЯ ФОРМА, СОДЕРЖАЩАЯ (1R, 2R)-3-(3-ДИМЕТИЛАМИНО-1-ЭТИЛ-2-МЕТИЛПРОПИЛ)ФЕНОЛ | 2005 |
|
RU2393847C2 |
| СТОЙКАЯ К РАЗРУШЕНИЮ ЛЕКАРСТВЕННАЯ ФОРМА С ЗАМЕДЛЕННЫМ ВЫСВОБОЖДЕНИЕМ ОКСИКОДОНА ГИДРОХЛОРИДА | 2010 |
|
RU2461381C2 |
| СТОЙКИЕ К РАЗРУШЕНИЮ ЛЕКАРСТВЕННЫЕ ФОРМЫ С ЗАМЕДЛЕННЫМ ВЫСВОБОЖДЕНИЕМ ФИЗИОЛОГИЧЕСКИ АКТИВНОГО ВЕЩЕСТВА | 2006 |
|
RU2399371C2 |
| Фармацевтическая композиция, содержащая вещество с анальгетической активностью | 2023 |
|
RU2831791C2 |
| САМОРЕГУЛИРУЕМОЕ ВЫСВОБОЖДЕНИЕ ФАРМАЦЕВТИЧЕСКОГО ИНГРЕДИЕНТА | 2013 |
|
RU2673818C2 |
| ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМАЯ СОЛЮБИЛИЗИРУЮЩАЯ КОМПОЗИЦИЯ И СОДЕРЖАЩАЯ ЕЕ ФАРМАЦЕВТИЧЕСКАЯ ДОЗИРОВАННАЯ ФОРМА | 2007 |
|
RU2469708C2 |
| УСТОЙЧИВАЯ К РАЗРУШЕНИЮ ЛЕКАРСТВЕННАЯ ФОРМА, СОДЕРЖАЩАЯ НЕОРГАНИЧЕСКУЮ СОЛЬ | 2011 |
|
RU2604676C2 |
| ФАРМАЦЕВТИЧЕСКАЯ ДОЗИРОВАННАЯ ФОРМА ДЛЯ ПЕРОРАЛЬНОГО ВВЕДЕНИЯ ИНГИБИТОРА СЕМЕЙСТВА BCL-2 | 2010 |
|
RU2568599C2 |
Настоящее изобретение относится к области лекарственных средств, в частности к защищенной от применения не по назначению, термоформованной путем экструзии без изменения окраски лекарственной форме, которая включает действующее вещество, потенциально являющееся объектом их немедицинского употребления и вспомогательные вещества, также содержит синтетический или природный полимер с сопротивлением разрушению по меньшей мере 500 Н. Кроме того, описан способ приготовления такой лекарственной формы. Технический результат заключается в обеспечении защиты от введения в организм в немедицинских целях. 2 н. и 33 з.п. ф-лы, 1 ил., 1 табл.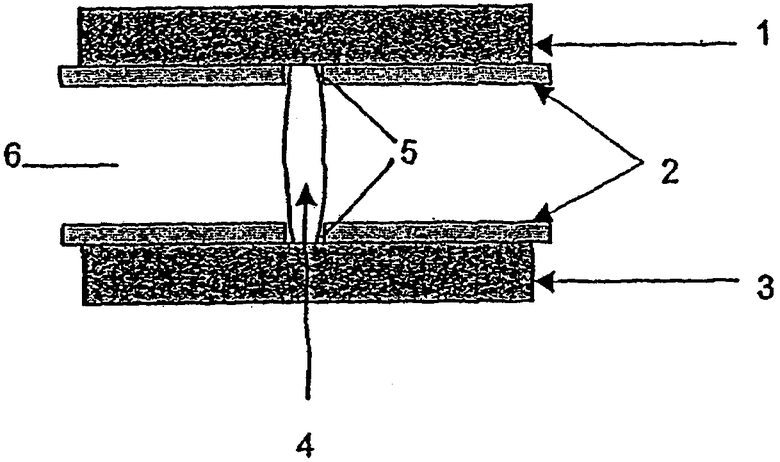
(а) по меньшей мере одно раздражающее полость носа и/или глотки вещество,
(б) по меньшей мере одно повышающее вязкость средство, которое в извлеченном из лекарственной формы с помощью минимально необходимого количества водной жидкости экстракте образует гель, который предпочтительно при его добавлении в дополнительное количество водной жидкости остается визуально различим,
(в) по меньшей мере один антагонист для действующего вещества, соответственно для действующих веществ, потенциально являющихся объектом их немедицинского употребления,
(г) по меньшей мере одно рвотное средство,
(д) по меньшей мере один краситель в качестве аверсивного средства,
(е) по меньшей мере одно горькое вещество.
1) компоненты (А), (Б), (В) и возможно используемый компонент (Г) смешивают между собой, а также смешивают с ними либо при необходимости смешивают отдельно с добавлением компонента (В) и возможно используемого компонента (Г) возможно используемые дополнительные компоненты (а)-(е),
2) полученную смесь или полученные смеси нагревают в экструдере по меньшей мере до температуры размягчения компонента (В) и путем приложения усилия экструдируют через выходное отверстие экструдера,
3) еще пластичный экструдат разделяют на отдельные части, из которых формуют лекарственные формы, или
4) из охлажденного и при необходимости вновь нагретого разделенного на отдельные части экструдата формуют лекарственные формы, проводя при этом стадии 2) и 3) и необязательно стадии 1) и 4) в атмосфере инертного газа.
| US 2003068392 A1, 10.04.2003 | |||
| US 2003124185 А1, 03.07.2003 | |||
| WO 03015531 А2, 27.02.2003. |

