Изобретение относится к фармацевтической лекарственной форме, проявляющей прочность на разрыв по меньшей мере 500 Н, указанная лекарственная форма содержит фармакологически активный компонент (А); неорганическую соль (В); и полиалкилен оксид (С), имеющий средневесовую молекулярную массу по меньшей мере 200,000 г/моль, где содержание полиалкилен оксида (С) составляет по меньшей мере 20 мас.%, исходя из общей массы лекарственной формы; где фармакологически активный компонент (А) присутствует в матриксе с контролируемым высвобождением, содержащем неорганическую соль (В) и полиалкилен оксид (С) и где, в условиях in vitro, профиль высвобождения фармакологически активного компонента (А) из указанного матрикса включает по меньшей мере временной интервал, в течение которого высвобождение соответствует кинетике нулевого порядка.
Многие фармакологически активные компоненты потенциально могут подвергаться злоупотреблению и, следовательно, благоприятным является обеспечение в форме устойчивых к разрушению фармацевтических лекарственных форм. Известными примерами таких фармакологически активных компонентов являются опиоиды.
Известно, что лица, злоупотребляющие наркотиками, дробят общепринятые таблетки, которые содержат опиоиды, для разрушения замедленного высвобождения "микро-инкапсулирования" и после этого принимают полученный порошок орально, интраназально, ректально, или путем инъекции.
Были разработаны различные концепции для избежания злоупотребления фармакологически активного компонента. Одна концепция основывается на механических свойствах фармацевтических лекарственных форм, в особенности повышенная прочность на разрыв (сопротивление раздавливанию). Основным преимуществом таких фармацевтических лекарственных форм является то, что измельчение, в особенности пульверизация, с помощью общепринятых средств, таких как перетирание в ступке или раздробление с помощью молотка, невозможно или по меньшей мере существенно затруднено.
Такие фармацевтические лекарственные формы пригодны для избегания злоупотребления фармакологически активного компонента, который в них содержится, так как они не могут быть превращены в порошок с помощью общепринятых средств и таким образом, не могут быть введены в порошкообразной форме, например, назально. Механические свойства, в особенности высокая прочность на разрыв этих фармацевтических лекарственных форм придает им устойчивость к разрушению. В контексте таких устойчивых к разрушению фармацевтических лекарственных форм можно привести ссылки, например, на WO 2005/016313, WO 2005/016314, WO 2005/063214, WO 2005/102286, WO 2006/002883, WO 2006/002884, WO 2006/002886, WO 2006/082097, WO 2006/082099, WO 2008/107149, и WO 2009/092601.
Кинетики высвобождения фармакологически активных компонентов из таких устойчивых к разрушению лекарственных форм являются важным фактором. Хорошо известно, что в зависимости от того, каким образом фармацевтически активный компонент приготовлен в лекарственном препарате в таблетке, характер его высвобождения может быть модифицирован.
С одной стороны, препараты, обеспечивающие быстрое высвобождение при пероральном введении, имеют преимущество, что они приводят к быстрому высвобождению фармакологически активного компонента в желудочно-кишечном тракте. Вследствие этого, сравнительно высокая доза фармакологически активного компонента быстро абсорбируется, что приводит к высоким уровням в плазме крови в течение короткого периода времени и приводит к быстрому началу действия лекарственного средства, то есть действие лекарственного средства начинается вскоре после введения. Тем не менее, в то же самое время наблюдается быстрое уменьшение действия лекарственного средства, поскольку метаболизация и/или экскреция фармакологически активного компонента вызывают снижение уровней в плазме. В связи с этим, препараты, обеспечивающие быстрое высвобождение фармакологически активных компонентов, обычно необходимо вводить часто, например, шесть раз в сутки. Это может вызывать сравнительно высокие пики концентраций фармакологически активного компонента и значительные колебания между пиком и минимумов концентрации в плазме фармакологически активного компонента, что, в свою очередь, может ухудшать переносимость.
Контролируемое высвобождение (например, замедленное высвобождение, пролонгированное высвобождение, длительное высвобождение, и другие) могут основываться на различных концепциях, таких как покрытие оболочкой фармацевтической лекарственной формы с мембраной с контролируемым высвобождением, заделывание фармакологически активного компонента в матрикс, связывание фармакологически активный компонент с ионообменной смолой, формирование комплекса фармакологически активного компонента, и другие. В этом контексте можно привести ссылки, например, на W.A. Ritschel, Die Tablette, 2. Auflage, Editio Cantor Verlag Aulendorf, 2002.
По сравнению с препаратами, обеспечивающими быстрое высвобождение, препараты, обеспечивающие пролонгированное высвобождение при пероральном введении имеют преимущество - что их необходимо вводить менее часто, типично один раз в сутки или два раза в сутки. Это может уменьшать пик концентрации фармакологически активного компонента в плазме и колебания между пиком и минимумом концентрации фармакологически активного компонента в плазме, что, в свою очередь, может улучшать переносимость.
Идеальной задачей создания системы с пролонгированным высвобождением является доставка фармакологически активного компонента в желательный сайт со скоростью в соответствии с потребностями организма. При отсутствии управления с обратной связью, может наблюдаться простой пролонгированный эффект, где основным вопросом является при какой скорости фармакологически активный компонент будет доставляться для поддержания постоянного уровня фармакологически активного компонента в крови. Эта постоянная скорость должна быть такой же, что и достигается при непрерывной внутривенной инфузии, где фармакологически активный компонент доставляет пациенту при постоянной скорости, равной его скорости элиминации. Это подразумевает, что скорость доставки должна быть независимой от количества фармакологически активного компонента, оставшегося в лекарственной форме, и постоянной в зависимости от времени.
Полностью инвариантные уровни фармакологически активного компонента в крови или ткани относительно временного профиля является идеальной исходной задачей системы с пролонгированным высвобождением. Возможным путем достижения этого, в наиболее простом случае, является применение поддерживающей дозы, которая высвобождает ее фармакологически активный компонент путем кинетики нулевого порядка.
В US 5,082,668 описана управляемая осмосом лекарственная форма, а именно устройство, содержащее стенку, которая окружает компартмент. Компартмент содержит композицию с лечебным средством и стимулирующую композицию.
Проход в стенке связывает компартмент с наружной стороной устройства для доставки лечебного средства со скоростью, регулируемой, в комбинации, стенкой, композицией с лечебным средством и стимулирующей композицией через проход устройства в зависимости от времени.
US 7,300,668 относится к лекарственной форме, содержащей: пространственно напечатанный самый внутренний участок, содержащий первую региональную концентрацию по меньшей мере одного активного фармацевтического компонента; и множество пространственных напечатанных не-внутренних участков в гнездовом расположении и содержащий: а) одним или несколько гнездовых внутренних участков, где внутренний участок полностью окружен и контактирует с самыми внутренними участками, и любой другой присутствующий внутренний участок полностью окружен другим внутренним участком, расположенным у него внутри; и Ь) наружный участок полностью окружает внутренний участок, где внутренний и наружный участки находятся в гнездовом расположении, где по меньшей мере один активный фармацевтический компонент высвобождается приблизительно в высвобождением нулевого порядка.
В WO 2008/086804 описаны устойчивые к злоупотреблению фармацевтические композиции на основе полигликоля. Композиция содержит один или несколько полигликолей и одно или несколько активных веществ и она устойчива к разрушению, расплавлению и/или экстракции. Кроме того, такие композиции имеют аналогичную или более низкую растворимость в этанольно-водной среде, то есть они не подвергаются индуцированному этанолом эффекту сбрасывания дозы.
В WO 2008/148798 описана многослойная фармацевтическая композиция, пригодная для перорального применения, для лечения заболеваний, где абсорбция происходит в большой части желудочно-кишечного тракта.
В WO 03/024426 описана фармацевтическая композиция с контролируемым высвобождением для перорального применения, которая содержит твердую дисперсию: i) по меньшей мере одного терапевтически, профилактически и/или диагностически активного вещества, которое по меньшей мере частично представлено в аморфной форме, ii) фармацевтически приемлемого полимера, который имеет пластифицирующие свойства, и iii) необязательно, стабилизирующего средства, по меньшей мере одно активное вещество имеет ограниченную растворимость в воде, и композиция создана для высвобождения активного вещества с по существу высвобождением нулевого порядка. Высвобождение нулевого порядка обеспечивается оболочкой, которая остается интактной в течение фазы высвобождения и охватывает матричную композицию таким образом, что только специфическая площадь поверхности подвергается эрозии. Следовательно, площадь поверхности, с которой высвобождается активное вещество, поддерживается существенно постоянной в течение периода времени.
В WO 2010/057036 описана твердая композиция и представлены способы ее получения и применения твердой композиции. Твердая композиция содержит: (а) по меньшей мере один активный компонент с растворимостью меньше, чем приблизительно 0,3 мг/мл в водном растворе с pH самое большее приблизительно 6,8 при температуре приблизительно 37°C; и (b) гидрофильный полимерный матриксный состав, содержащий: i) гидрофильный полимер, выбранный из группы, включающей METHOCEL®, POLYOX® WSR 1105 и их комбинации; и необязательно ii) гидрофобный полимер, выбранный из группы, включающей Ethocel 20 premium; и (c) подщелачивающий агент, выбранный из группы, включающей карбонат кальция, тяжелый оксид магния и бикарбонат натрия; где композиция обеспечивает по меньшей мере приблизительно 70% высвобождение активного вещества между от приблизительно 7 до приблизительно 12 часов после перорального введения.
V. Pillay и др., Journal of Controlled Release, 67 (2000) 67-78 описан подход для доставки с постоянной скоростью высоко растворимых биологически активных веществ из простой монолитной системы, приготовленной путем прямого прессования в условиях окружающей среды.
М.Е. McNeill и др., J Biomater Sci Polym 1996, 7 (11), 953-63 относится к свойствам, контролирующим диффузию и высвобождение водорастворимых растворенных веществ из поли(этилен оксид) гидрогелей. В части 4 рассматривается продленное высвобождение с постоянной скоростью из частично покрытых оболочкой сфер.
D. Henrist и др. относится к оценке in vitro и in vivo двойных матричных систем на основе крахмала, экструдированных при высоких температурах. Задачей разработки двойной матричной системы, содержащей экструдированную при высоких температурах крахмальную трубку, окруженную экструдированным при высоких температурах и содержащим лекарственное средство крахмальным ядром, было получение монолитной матриксной системы, применимой в области длительного высвобождения лекарственного средства. Поведение систем оценивали путем тестирования растворения и путем рандомизированного перекрестного исследования биодоступности на девяти добровольцах мужского пола. Все двойные матриксные системы показали in vitro практически постоянный профиль высвобождения лекарственного средства после начальной более медленной фазы высвобождения в течение 4 часов. Этой начальной более медленной фазы высвобождения избегали путем загрузки крахмальной трубки небольшим количеством лекарственного средства.
L. Yang и др., J. Pharm. Sciences, 85 (2), 1996, 170-173 относится к кинетике высвобождения нулевого порядка из самокорректирующейся поддающейся флотации системы доставки лекарственного средства с ассиметричной конфигурацией.
Задачей изобретения является обеспечение фармацевтических лекарственных форм, которые имеют преимущества по сравнению с фармацевтическими лекарственными формами из уровня техники.
Эта задача решается с помощью объектов пунктов формулы изобретения.
Неожиданно было обнаружено, что относительно низкие количества неорганических солей, содержащихся в полимерном матриксе, обеспечивают дополнительную задержку высвобождения фармакологически активных компонентов из устойчивых к разрушению лекарственных форм, не приводя к существенному повышению общего веса. Кроме того, неожиданно было обнаружено, что инкорпорация неорганической соли в полимерный матрикс существенно не изменяет механические свойства устойчивой к разрушению лекарственной формы, которая основана на полимерном матриксе, в особенности прочности на разрыв. Также, неожиданно было обнаружено, что профиль высвобождения соответствует кинетике нулевого порядка и не зависит от значения pH среды высвобождения.
На фигуре 1 представлен профиль высвобождения in vitro фармацевтической лекарственной формы в соответствии с изобретением, содержащей 30 мас.% (Вариант Е) и 40 мас.% (Вариант F), соответственно, карбоната натрия по сравнению с эталонными таблетками.
На фигуре 2 представлены профили высвобождения in vitro фармацевтической лекарственной формы в соответствии с изобретением, содержащей 15 мас.% (Вариант F) и 20 мас.% (Вариант G), соответственно, каждая карбонат натрия и пятизамещенный трифосфат натрия по сравнению с эталонными таблетками.
На фигуре 3 представлены профили высвобождения in vitro фармацевтической лекарственной формы в соответствии с изобретением в кислой среде, содержащей 30 мас.% карбонат натрия (Вариант Е) и 15 мас.% карбонат натрия с 15 мас.% пятизамещенным трифосфатом натрия (Вариант F) по сравнению с эталонными таблетками.
На фигуре 4 представлены профили высвобождения in vitro фармацевтической лекарственной формы в соответствии с изобретением в кислой среде (pH 1,2) и с фосфатным буфером (рН 4,5), содержащей оксиморфон и 30 мас.% карбонат натрия (Пример II) по сравнению с эталонными таблетками.
На фигуре 5 представлены профили высвобождения in vitro фармацевтической лекарственной формы в соответствии с изобретением в кислой среде (pH 1,2) и с фосфатным буфером (pH 4,5), содержащей оксиморфон и 15 мас.% каждая карбонат натрия и пятизамещенный трифосфат натрия. (Пример III) по сравнению с эталонными таблетками.
Первый аспект изобретения относится к фармацевтической лекарственной форме, проявляющей прочность на разрыв по меньшей мере 500 Н, указанная лекарственная форма содержит
- фармакологически активный компонент (А);
- неорганическую соль (В); и
- полиалкилен оксид (С), имеющий средневесовую молекулярную массу по меньшей мере 200,000 г/моль, где содержание полиалкилен оксида (С) составляет по меньшей мере 20 мас.%, исходя из общей массы лекарственной формы;
где фармакологически активный компонент (А) присутствует в матриксе с контролируемым высвобождением, содержащем неорганическую соль (В) и полиалкилен оксид (С), и где, в условиях in vitro, профиль высвобождения фармакологически активного компонента (А) из указанного матрикса включает по меньшей мере временной интервал, в течение которого высвобождение соответствует кинетике нулевого порядка.
Лекарственная форма в соответствии с изобретением содержит один или несколько фармакологически активных компонентов (А).
В целом отсутствуют ограничения относительно использования в качестве фармакологически активного компонента (А) (фармакологически активного соединения), которое может быть включено в таблетку согласно изобретению.
В предпочтительном варианте осуществления, фармацевтическая лекарственная форма содержит только один фармакологически активный компонент (А). В другом предпочтительном варианте осуществления, фармацевтическая лекарственная форма содержит комбинацию двух или более фармакологически активных компонентов (А).
Предпочтительно, фармакологически активный компонент (А) потенциально может подвергаться злоупотреблению. Активные компоненты, которыми потенциально можно злоупотребить, известны специалисту в данной области техники и включают, например, транквилизаторы, стимуляторы, барбитураты, наркотические средства, опиоиды или производные опиоидов.
Предпочтительно, фармакологически активный компонент (А) проявляет психотропное действие.
Предпочтительно, фармакологически активный компонент (А) выбирают из группы, включающей опиаты, опиоиды, стимуляторы, транквилизаторы, и другие наркотические средства.
Особенно предпочтительно, фармакологически активный компонент (А) представляет собой опиоид. В соответствии с индексом АТС, опиоиды подразделяются на натуральные алкалоиды опия, производные фенилпиперидина, производные дифенилпропиламина, производные бензоморфана, производные орипавина, производные морфинана и другие.
Следующие опиаты, опиоиды, транквилизаторы или другие наркотические средства являются веществами с психотропным действием, то есть потенциально могут разрушаться, и, следовательно, предпочтительно содержатся в фармацевтической лекарственной форме в соответствии с изобретением: альфентанил, аллобарбитал, аллилпродин, альфапродин, алпразолам, амфепрамон, амфетамин, амфетаминил, амобарбитал, анилеридин, апокодеин, аксомадол, барбитал, бемидон, бензилморфин, безитрамид, бромазепам, бротизолам, бупренорфин, бутобарбитал, буторфанол, камазепам, карфентанил, катин/D-норпсевдоэфедрин, хлордиазепоксид, клобазам клофеданол, клоназепам, клонитазен, клоразепат, клотиазепам, клоксазолам, кокаин, кодеин, циклобарбитал, циклорфан, ципренорфин, делоразепам, дезоморфин, декстроморамид, декстропропоксифен, дезоцин, диампромид, диаморфон, диазепам, дигидрокодеин, дигидроморфин, дигидроксиморфон, дименоксадол, димефетамол, диметилтиамбутен, диоксафетилбутират, дипипанон, дронабинол, эптазоцин, эстазолам, этогептазин, этилметилтиамбутен, этил лофлазепат, этилморфин, этонитазен, эторфин, факселадол, фенкамфамин, фенетилин, фенпипрамид, фенпропорекс, фентанил, флудиазепам, флунитразепам, флуразепам, галазепам, галоксазолам, героин, гидрокодон, гидроксиморфон, гидроксипетидин, изометадон, гидроксиметилморфинан, кетазолам, кетобемидон, левацетилметадол (LAAM), левометадон, леворфанол, левофенацилморфан, левоксемацин, лиздексамфетамин димезилат, лофентанил, лопразолам, лоразепам, лорметазепам, мазиндол, медазепам, мефенорекс, мепиридин, мепробамат, метапон, мептазинол, метазоцин, метилморфин, метамфетамин, метадон, метаквалон, 3-метилфентанил, 4-метилфентанил, метилргенидат, метилфенобарбитал, метиприлон, метопон, мидазолам, модафинил, морфин, мирофин, набилон, налбуфен, налорфин, нарцеин, никоморфин, ниметазепам, нитразепам, нордамепам, норлеворфанол, норметадон, норморфин, норпипанон, опий, оксазепам, оксазолам, оксикодон, оксиморфон, Мак опийный, пантопон, пернолин, пентазоцин, пентобарбитал, петидин, фенадоксон, феноморфан, феназоцин, феноперидин, пиминодин, фолькодеин, фенметразин, фенобарбитал, фентермин, пиназепам, пипрадрол, пиритрамид, празепам, профадол, прогептазин, промедол, проперидин, пропоксифен, ремифентанил, секбутабарбитал, секобарбитал, суфентанил, тапентадол, темазепам, тетразепам, тилидин (цис и транс), трамадол, триазолам, винилбитал, N-(1-метил-2-пиперидиноэтил)-N-(2-пиридил)пропионамид, (1R,2R)-3-(3-диметиламино-1-этил-2-метил-пропил)фенол, (1R,2R,4S)-2-(диметиламино)метил-4-(п-фторбензилокси)-1-(м-метоксифенил)циклогексанол, (1R,2R)-3-(2-диметиламинометил-циклогексил)фенол, (1S,2S)-3-(3-диметиламино-1-этил-2-метил-пропил)фенол, (2R,3R)-1-диметиламино-3(3-метоксифенил)-2-метил-пентан-3-ол, (1RS,3RS,6RS)-6-диметиламинометил-1-(3-метоксифенил)-циклогексан-1,3-диол, предпочтительно в виде рацемата, 3-(2-диметиламинометил-1-гидрокси-циклогексил)фенил 2-(4-изобутил-фенил)пропионат, 3-(2-диметиламинометил-1-гидрокси-циклогексил)фенил 2-(6-метокси-нафталин-2-ил)пропионат, 3-(2-диметиламинометил-циклогекс-1-енил)-фенил 2-(4-изобутил-фенил)пропионат, 3-(2-диметиламинометил-циклогекс-1-енил)-фенил 2-(6-метокси-нафталин-2-ил)пропионат, 3-(2-диметиламинометил-1-гидрокси-циклогексил)-фениловый эфир (RR-SS)-2- ацетокси-4-трифторметил-бензойной кислоты, 3-(2-диметиламинометил-1 -гидрокси-циклогексил)-фениловый эфир (RR-SS)-2-гидрокси-4-трифторметил-бензойной кислоты, 3-(2-диметиламинометил-1-гидрокси-циклогексил)-фениловый эфир (RR-SS)-4-хлор-2-гидрокси-бензойной кислоты, 3-(2-диметиламинометил-1-гидрокси-циклогексил)-фениловый эфир (RR-SS)-2-гидрокси-4-метил-бензойной кислоты, 3-(2-диметиламинометил-1-гидрокси-циклогексил)-фениловый эфир (RR-SS)-2-гидрокси-4-метокси-бензойной кислоты, (RR-SS)-2-гидрокси-5-нитро-бензойной кислоты 3-(2-диметиламинометил-1-гидрокси-циклогексил)-фениловый эфир, 3-(2-диметиламинометил-1-гидрокси-циклогексил)-фениловый эфир (RR-SS)-1′,4′-дифтор-3-гидрокси-бифенил-4-карбоновой кислоты, и соответствующие стереоизомерные соединения, в каждом случае их соответствующие производные, физиологически приемлемые энантиомеры, стереоизомеры, диастереомеры и рацематы и их физиологически приемлемые производные, например, простые эфиры, сложные эфиры или амиды, и в каждом случае их физиологически приемлемые соединения, в частности их соли присоединения кислот и оснований и сольваты, например, дигидрохлориды.
В предпочтительном варианте осуществления фармацевтическая лекарственная форма в соответствии с изобретением содержит опиоид, выбранный из группы, включающей DPI-125, M6G (СЕ-04-410), ADL-5859, CR-665, NRP290 и себакоил диналбуфиновый эфир.
В предпочтительном варианте осуществления, фармацевтическая лекарственная форма в соответствии с изобретением содержит один фармакологически активный компонент (А) или несколько фармакологически активных компонентов (А), выбранных из группы, включающей оксиморфон, гидроксиморфон, морфин и их физиологически приемлемые соли.
В другом предпочтительном варианте осуществления, фармакологически активный компонент (А) выбирают из группы, включающей тапентадол, факселадол, аксомадол и их физиологически приемлемые соли.
В еще другом предпочтительном варианте осуществления,, фармакологически активный компонент (А) выбирают из группы, включающей 1,1-(3-диметиламино-3-фенилпентаметилен)-6-фтор-1,3,4,9-тетрагидропирано[3,4-b]индол, в особенности его гемицитрат; 1,1-[3-диметиламино-3-(2-тиенил)пентаметилен]-1,3,4,9-тетрагидропирано[3,4-b]индол, в особенности его цитрат; и 1,1-[3-диметиламино-3-(2-тиенил)пентаметилен]-1,3,4,9-тетрагидропирано[3,4-b]-6-фториндол, в особенности его гемицитрат. Эти соединения известны, например, из WO 2004/043967, WO 2005/066183.
Фармакологически активный компонент (А) может присутствовать в форме физиологически приемлемой соли, например, физиологически приемлемой соли присоединения кислоты.
Физиологически приемлемые соли присоединения кислот включают солевые формы присоединения кислот, которые легко получают путем обработки формы основания активного компонента с подходящими органическими и неорганическими кислотами. Активные компоненты, содержащие кислотообразующий протон, могут быть превращены в их нетоксичные солевые формы присоединения металла или амина путем обработки с подходящими органическими и неорганическими основаниями. Термин соль присоединения также включает гидраты и формы присоединения растворителей, которые способны образовывать активные компоненты. Примерами таких форм являются, например, гидраты, алкоголяты и другие.
Фармакологически активный компонент (А) присутствует в лекарственной форме в терапевтически эффективном количестве. Количество, которое составляет терапевтически эффективное количество, изменяется в соответствии с используемыми активными компонентами, условием, подвергаемым лечению, тяжестью указанного условия, пациентом, подвергаемым лечению, и создана ли лекарственная форма для быстрого или замедленного высвобождения. Количество активного (ых) компонента (ов), используемых в настоящем изобретении, предпочтительно находится в диапазоне от приблизительно 0,01 мас.% до приблизительно 95 мас.%, более предпочтительно от приблизительно 0,1 мас.% до приблизительно 80 мас.%, еще более предпочтительно от приблизительно 1,0 мас.% до приблизительно 50 мас.%, также более предпочтительно от приблизительно 1,5 мас.% до приблизительно 30 мас.%, и наиболее предпочтительно от приблизительно 2,0 мас.% до 20 мас.%, исходя из общей массы фармацевтической лекарственной формы.
Содержание фармакологически активного компонента (А) в фармацевтической лекарственной форме не ограничено. Доза фармакологически активного компонента (А) которая приспособлена для введения, предпочтительно находится в диапазоне от 0,1 мг до 500 мг, более предпочтительно в диапазоне от 1,0 мг до 400 мг, еще более предпочтительно в диапазоне от 5,0 мг до 300 мг, и наиболее предпочтительно в диапазоне от 10 мг до 250 мг. В предпочтительном варианте осуществления, общее количество фармакологически активного компонента (А), которое содержится в фармацевтической лекарственной форме, находится в диапазоне от 0,01 до 200 мг, более предпочтительно 0,1 до 190 мг, еще более предпочтительно 1,0 до 180 мг, также более предпочтительно 1,5 до 160 мг, наиболее предпочтительно 2,0 до 100 мг и в частности 2,5 до 80 мг.
Предпочтительно, содержание фармакологически активного компонента (А) находится в диапазоне от 0,01 до 80 мас.%, более предпочтительно 0,1 до 50 мас.%, еще более предпочтительно 1 до 25 мас.%, исходя из общей массы фармацевтической лекарственной формы. В предпочтительном варианте осуществления, содержание фармакологически активного компонента (А) находится в диапазоне 7±6 мас.%, более предпочтительно 7±5 мас.%, еще более предпочтительно 5±4 мас.%, 7±4 мас.% или 9±4 мас.%, наиболее предпочтительно 5±3 мас.%; 7±3 мас.% или 9±3 мас.%, и в частности 5±2 мас.%, 7±2 мас.% или 9±2 мас.%, исходя из общей массы фармацевтической лекарственной формы. В другом предпочтительном варианте осуществления, содержание фармакологически активного компонента (А) находится в диапазоне 11±10 мас.%, более предпочтительно 11±9 мас.%, еще более предпочтительно 9±6 мас.%, 11±6 мас.%, 13±6 мас.% или 15±6 мас.%, наиболее предпочтительно 11±4 мас.%, 13±4 мас.% или 15±4 мас.%, и в частности 11±2 мас.%, 13±2 мас.% или 15±2 мас.%, исходя из общей массы фармацевтической лекарственной формы. В дальнейшем предпочтительном варианте осуществления, содержание фармакологически активного компонента (А) находится в диапазоне 20±6 мас.%, более предпочтительно 20±5 мас.%, еще более предпочтительно 20±4 мас.%, наиболее предпочтительно 20±3 мас.%, и в частности 20±2 мас.%, исходя из общей массы фармацевтической лекарственной формы.
В предпочтительном варианте осуществления, фармакологически активный компонент (А) содержится в фармацевтической лекарственной форме в количестве 7,5±5 мг, 10±5 мг, 20±5 мг, 30±5 мг, 40±5 мг, 50±5 мг, 60±5 мг, 70±5 мг, 80±5 мг, 90±5 мг, 100±5 мг, 110±5 мг, 120±5 мг, 130±5, 140±5 мг, 150±5 мг, 160±5 мг, 170±5 мг, 180±5 мг, 190±5 мг, 200±5 мг, 210±5 мг, 220±5 мг, 230±5 мг, 240±5 мг, или 250±5 мг. В другом предпочтительном варианте осуществления, фармакологически активный компонент (А) содержится в фармацевтической лекарственной форме в количестве 5+2,5 мг, 7,5±2,5 мг, 10±2,5 мг, 15±2,5 мг, 20±2,5 мг, 25±2,5 мг, 30±2,5 мг, 35±2,5 мг, 40±2,5 мг, 45±2,5 мг, 50±2,5 мг, 55±2,5 мг, 60±2,5 мг, 65±2,5 мг, 70±2,5 мг, 75±2,5 мг, 80±2,5 мг, 85±2,5 мг, 90±2,5 мг, 95±2,5 мг, 100±2,5 мг, 105±2,5 мг, 110±2,5 мг, 115±2,5 мг, 120±2,5 мг, 125±2,5 мг, 130±2,5 мг, 135±2,5 мг, 140±2,5 мг, 145±2,5 мг, 150±2,5 мг, 155±2,5 мг, 160±2,5 мг, 165±2,5 мг, 170±2,5 мг, 175±2,5 мг, 180±2,5 мг, 185±2,5 мг, 190±2,5 мг, 195±2,5 мг, 200±2,5 мг, 205±2,5 мг, 210±2,5 мг, 215±2,5 мг, 220±2,5 мг, 225±2,5 мг, 230±2,5 мг, 235±2,5 мг, 240±2,5 мг, 245±2,5 мг, или 250±2,5 мг.
Предпочтительно, фармацевтическая лекарственная форма обеспечивает высвобождение фармакологически активного компонента (А) через 1 час предпочтительно самое большее 60%, более предпочтительно самое большее 40%, также более предпочтительно самое большее 30%, еще более предпочтительно самое большее 20% и наиболее предпочтительно самое большее 17%; через 2 часа предпочтительно самое большее 80%, более предпочтительно самое большее 60%, также более предпочтительно самое большее 50%, еще более предпочтительно самое большее.40% и наиболее предпочтительно самое большее 32%; через 3 часа предпочтительно самое большее 85%, более предпочтительно самое большее 65%, также более предпочтительно самое большее 55%, еще более предпочтительно самое большее 48% и наиболее предпочтительно самое большее 42%; через 4 часа предпочтительно самое большее 90%, более предпочтительно самое большее 75%, также более предпочтительно самое большее 65%, еще более предпочтительно самое большее 55% и наиболее предпочтительно самое большее 49%; через 7 часов предпочтительно самое большее 95%, более предпочтительно самое большее 85%, также более предпочтительно самое большее 80%, еще более предпочтительно самое большее 70% и наиболее предпочтительно самое большее 68%; через 10 часов предпочтительно самое большее 99%, более предпочтительно самое большее 90%, также более предпочтительно самое большее 88%, еще более предпочтительно самое большее 83% и наиболее предпочтительно самое большее 80%; и через 13 часов предпочтительно самое большее 99%, более предпочтительно самое большее 95%, также более предпочтительно самое большее 93%, еще более предпочтительно самое большее 91% и наиболее предпочтительно самое большее 89%.
В особенно предпочтительном варианте осуществления, фармакологически активный компонент (А) представляет собой тапентадол, предпочтительно его HCl соль, и фармацевтическая лекарственная форма адаптирована для введения один раз в сутки или два раза в сутки. В этом варианте осуществления, фармакологически активный компонент (А) предпочтительно содержится в фармацевтической лекарственной форме в количестве от 25 до 250 мг.
В другом особенно предпочтительном варианте осуществления, фармакологически активный компонент (А) представляет собой оксиморфон, предпочтительно его HCI соль, и фармацевтическая лекарственная форма адаптирована для введения два раза в сутки. В этом варианте осуществления, фармакологически активный компонент (А) предпочтительно содержится в фармацевтической лекарственной форме в количестве от 5 до 40 мг. В другом особенно предпочтительном варианте осуществления, фармакологически активный компонент (А) представляет собой оксиморфон, предпочтительно его HCl, и фармацевтическая лекарственная форма адаптирована для введения один раз в сутки. В этом варианте осуществления, фармакологически активный компонент (А) предпочтительно содержится в фармацевтической лекарственной форме в количестве от 10 до 80 мг.
В другом особенно предпочтительном варианте осуществления, фармакологически активный компонент (А) представляет собой оксикодон, предпочтительно его HCl соль, и фармацевтическая лекарственная форма адаптирована для введения два раза в сутки. В этом варианте осуществления, фармакологически активный компонент (А) предпочтительно содержится в фармацевтической лекарственной форме в количестве от 5 до 80 мг. В другом особенно предпочтительном варианте осуществления, фармакологически активный компонент (А) представляет собой оксикодон, предпочтительно его HCl, и фармацевтическая лекарственная форма адаптирована для введения один раз в сутки. В этом варианте осуществления, фармакологически активный компонент (А) предпочтительно содержится в фармацевтической лекарственной форме в количестве от 10 до 320 мг.
В еще другом особенно предпочтительном варианте осуществления, фармакологически активный компонент (А) представляет собой гидроксиморфон, предпочтительно его HCl, и фармацевтическая лекарственная форма адаптирована для введения два раза в сутки. В этом варианте осуществления, фармакологически активный компонент (А) предпочтительно содержится в фармацевтической лекарственной форме в количестве от 2 до 52 мг. В другом особенно предпочтительном варианте осуществления, фармакологически активный компонент (А) представляет собой гидроксиморфон, предпочтительно его HCl, и фармацевтическая лекарственная форма адаптирована для введения один раз в сутки. В этом варианте осуществления, фармакологически активный компонент (А) предпочтительно содержится в фармацевтической лекарственной форме в количестве от 4 до 104 мг.
Фармацевтическая лекарственная форма в соответствии с изобретением характеризуется чрезвычайной устойчивостью фармакологически активного компонента (А). Предпочтительно, после хранения в течение 4 недель при 40°C и 75% отн. влажн., содержание фармакологически активного компонента (А) составляет по меньшей мере 98,0%, более предпочтительно по меньшей мере 98,5%, еще более предпочтительно по меньшей мере 99,0%, также более предпочтительно по меньшей мере 99,2%, наиболее предпочтительно по меньшей мере 99,4% и в частности по меньшей мере 99,6%, его исходного содержания перед хранением. Подходящие методы измерения содержания фармакологически активного компонента (А) в фармацевтической лекарственной форме известны специалистам в данной области техники. В этом отношении можно привести ссылку на Европейскую фармакопею или фармакопею США, в особенности на ВЭЖХ анализ с обращенной фазой. Предпочтительно, фармацевтическую лекарственную форму хранят в закрытых, предпочтительно запечатанных контейнерах, предпочтительно как описано в экспериментальном разделе, наиболее предпочтительно оборудованных поглотителем кислорода, в частности поглотителем кислорода, который эффективен при низкой относительной влажности.
Лекарственная форма в соответствии с изобретением содержит фармакологически активный компонент (А) в матриксе с контролируемым высвобождением, содержащем неорганическую соль (В), где, в условиях in vitro, профиль высвобождения фармакологически активного компонента (А) из указанного матрикса включает по меньшей мере временной интервал, в течение которого высвобождение соответствует кинетике нулевого порядка.
Специалист в данной области техники известно, каким требованиям необходимо соответствовать для того, чтобы квалифицировать профиль высвобождения in vitro фармацевтической лекарственной формы как нулевого порядка. Растворение фармакологически активного компонента из твердых лекарственных форм описано с помощью кинетических моделей, в которых растворенной количество фармакологически активного компонента (Q) зависит от времени тестирования, t или Q=f(t). Обычно используются некоторые аналитические определения Q(t) функции, такие как нулевой порядок, первый порядок, модели Hixson-Crowell, Weibull, Higuchi, Baker-Lonsdale, Korsmeyer-Peppas и Hopfenberg. Другие параметры высвобождения, такие как время растворения (tx%), время исследования (tx мин), эффективность растворения (ED), фактор отличия (f1), фактор подобия (f2) и индекс Rescigno (xi1 и xi2) могут использоваться для характеристики профилей растворения/высвобождения фармакологически активного компонента.
Для целей настоящей заявки термин "кинетика нулевого порядка" предпочтительно определяется с помощью уравнения W0-Wt=Kt, где W0 представляет собой исходное количество фармакологически активного компонента (А) в фармацевтической лекарственной форме, Wt представляет собой количество фармакологически активного компонента (А) в фармацевтической лекарственной форме во время f и K представляет собой константу пропорциональности. Разделяя это уравнение на W0 и упрощая ft=K0t, где ft=1-(Wt/W0) и ft представляют собой фракцию фармакологически активного компонента (А), растворенную во время t и К0 видимую константу скорости растворения или константу высвобождения нулевого порядка. Таким образом, графически растворенная фракция фармакологически активного компонента относительно времени может быть линейной. Эта зависимость может использоваться на описания растворения нескольких типов модифицированного высвобождения фармацевтических лекарственных форм, как в случае матриксных таблеток с фармакологически активными компонентами с низкой растворимостью, покрытых оболочкой форм, осмотических систем и др. Фармацевтические лекарственные формы, следующие этому профилю, высвобождают аналогичное количество фармакологически активного компонента за единицу времени и он является идеальным методом высвобождения фармакологически активного компонента для достижения фармакологического пролонгированного действия. Последующая взаимосвязь может, простым образом, выражать эту модель:Q1=Q0+K0t, где Qt представляет собой количество фармакологически активного компонента, растворенное во время t, Q0 представляет собой исходное количество фармакологически активного компонента в растворе (многократно, Q0=0) и К0 представляет собой константу высвобождения нулевого порядка (ср. например, P. Costa и др., Eur J Pharm Sci. 2001, 13 (2), 123-33).
Для специалиста в данной области техники является очевидным, что на практике фармацевтические лекарственные формы обычно не обеспечивают точного высвобождения нулевого порядка, в частности не в течение полного периода высвобождения, то есть от самого начала до высвобождения 100% фармакологически активного компонента (А), который изначально присутствует в фармацевтической лекарственной форме. В значительной степени, на практике профили высвобождения in vitro могут быть описаны со значительной степенью точности с помощью этих математических моделей, в частности, когда не принимать во внимание начальную фазу, а также конечную фазу высвобождения.
Предпочтительно, профиль высвобождения in vitro фармакологически активного компонента (А) из фармацевтической лекарственной формы в соответствии с изобретением содержит временной интервал, в течение которого высвобождение соответствует существенно кинетике нулевого порядка, где временной интервал предпочтительно представляет собой время, необходимое для высвобождения 50±5%, более предпочтительно 50±10%, еще более предпочтительно 50±15%, также более предпочтительно 50±20%, еще более предпочтительно 50±25%, наиболее предпочтительно 50±30%, и в частности 50±35%, фармакологически активного компонента (А). Например, время, необходимое для высвобождения 50±30% фармакологически активного компонента (А), начинается с высвобождения 20% (например, через 2,5 часа) и оканчивается с высвобождением 80% (например, через 10,5 часов) фармакологически активного компонента (А). В течение такого временного интервала, профиль высвобождения in vitro фармакологически активного компонента (А) из фармацевтической лекарственной формы существенно следует кинетике нулевого порядка, то есть существенно является линейным.
В предпочтительном варианте осуществления, кинетики высвобождения in vitro фармакологически активного компонента (А) из фармацевтической лекарственной формы приблизительно определяются уравнением Mt/М0=ktn где t представляет собой время, Mt представляет собой количество фармакологически активного компонента (А), которое высвобождается во время t, M0 представляет собой общее количество фармакологически активного компонента (А), которое изначально содержится в лекарственной форме, то есть перед экспозицией фармацевтической лекарственной формы со средой высвобождения, к представляет собой константу, и n представляет собой экспоненту кинетики высвобождения. Предпочтительно, профиль высвобождения in vitro фармацевтической лекарственной формы в соответствии с изобретением обеспечивает кривую, которая определяет ретардированное высвобождение в процентах относительно времени. Для определенного периода времени, предпочтительно от начала или от точки времени после начала, например, от времени, где высвободилось 20%, до времени, где высвободилось 95% фармакологически активного компонента (А) из лекарственной формы в соответствии с изобретением, профиль высвобождения является существенно линейным.
Предпочтительно, временной интервал, в течение которого высвобождение соответствует кинетике нулевого порядка, например, где вторая производная графика является существенно линейной, составляет по меньшей мере 20%, более предпочтительно по меньшей мере 30%, еще более предпочтительно по меньшей мере 40%, также более предпочтительно по меньшей мере 50%, еще более предпочтительно по меньшей мере 60%, наиболее предпочтительно по меньшей мере 70% и в частности по меньшей мере 80% общего времени высвобождения, необходимого для высвобождения 95 мас.% фармакологически активного компонента (А), который изначально присутствует в фармацевтической лекарственной форме.
Предпочтительно, границы (пределы) "существенно линейного" могут быть оценены на основе второй производной кривой, подогнанные к точкам измерений. Тем не менее, предпочтительно, определенная степень отклонения также присутствует в значении "существенно линейный" в соответствии с изобретением. Предпочтительно, указанные отклонения от идеального линейного поведения могут быть количественно определенны с помощью критерия хиквадрат, который известен специалисту в данной области техники. Предпочтительно, значение, определенной в соответствии с критерием хиквадрат, составляет самое большее 2,5, более предпочтительно самое большее 1,75, еще более предпочтительно самое большее 1,0, также более предпочтительно самое большее 0,75, еще более предпочтительно самое большее 0,5, наиболее предпочтительно самое большее 0,25, и в частности самое большее 0,1.
Предпочтительно, нулевой порядок in vitro кинетики высвобождения в достаточной мере может быть описан с помощью Mt/M∞=k0tn, где Mt и М∞ - представляют собой количества высвобожденного лекарственного средства во время t и общее высвобожденное количество, соответственно, n представляет собой экспоненту высвобождения, указывающую на форму профиля, и k0 представляет собой is константа скорости высвобождения нулевого порядка.
В предпочтительном варианте осуществления, где подгоняют релевантную часть суммарного профиля высвобождения in vitro, который показывает нулевой порядок кинетики высвобождения к уравнению Mt/M∞=k0tn (то есть где n=1), коэффициент корреляции подгонки предпочтительно составляет по меньшей мере 0,75, более предпочтительно по меньшей мере 0,80, еще более предпочтительно по меньшей мере 0,85, также более предпочтительно по меньшей мере 0,90, еще более предпочтительно по меньшей мере 0,925, наиболее предпочтительно по меньшей мере 0,95 и в частности по меньшей мере 0,975.
В предпочтительном варианте осуществления, константа скорости высвобождения нулевого порядка ко находится в диапазоне 0,030±0,028 ч-1, более предпочтительно 0,030±0,026 ч-1, еще более предпочтительно 0,030±0,024 ч-1 также более предпочтительно 0,030±0,020 ч-1, еще более предпочтительно 0,030±0,015 ч-1, наиболее предпочтительно 0,030±0,010 ч-1 и в частности 0,030±0,005 ч-1. В другом предпочтительном варианте осуществления, константа скорости высвобождения нулевого порядка ко находится в диапазоне 0,040±0,035 ч-1 более предпочтительно 0,040±0,030 ч-1, еще более предпочтительно 0,040±0,025 ч-1 также более предпочтительно 0,040±0,020 ч-1, еще более предпочтительно 0,040±0,015 ч-1, наиболее предпочтительно 0,040±0,010 ч-1, и в частности 0,040±0,005 ч-1. В еще другом предпочтительном варианте осуществления, константа скорости высвобождения нулевого порядка ко находится в диапазоне 0,050±0,035 ч-1 более предпочтительно 0,050±0,030 ч-1, еще более предпочтительно 0,050±0,025 ч-1, также более предпочтительно 0,050±0,020 ч-1, еще более предпочтительно 0,050±0,015 ч-1, наиболее предпочтительно 0,050±0,010 ч-1, и в частности 0,050±0,005 ч-1. В еще другом предпочтительном варианте осуществления, константа скорости высвобождения нулевого порядка ко находится в диапазоне 0,060±0,035 ч-1, более предпочтительно 0,060±0,030 ч-1, еще более предпочтительно 0,060±0,025 ч-1, также более предпочтительно 0,060±0,020 ч-1, еще более предпочтительно 0,060±0,015 ч-1, наиболее предпочтительно 0,060±0,010 ч-1, и в частности 0,060±0,005 ч-1. В дальнейшем предпочтительном варианте осуществления, константа скорости высвобождения нулевого порядка ко находится в диапазоне 0,070±0,035 ч-1, более предпочтительно 0,070±0,030 ч-1, еще более предпочтительно 0,070±0,025 ч-1, также более предпочтительно 0,070±0,020 ч-1, еще более предпочтительно 0,070±0,015 ч-1, наиболее предпочтительно 0,070±0,010 ч-1, и в частности 0,070±0,005 ч-1. В еще более предпочтительном варианте осуществления, константа скорости высвобождения нулевого порядка k0 находится в диапазоне 0,080±0,035 ч-1, более предпочтительно 0,080±0,030 ч-1, еще более предпочтительно 0,080±0,025 ч-1, также более предпочтительно 0,080±0,020 ч-1, еще более предпочтительно 0,080±0,015 ч-1, наиболее предпочтительно 0,080±0,010 ч-1, и в частности 0,080±0,005 ч-1. В другом также предпочтительном варианте осуществления, константа скорости высвобождения нулевого порядка ко находится в диапазоне 0,090±0,035 ч-1, более предпочтительно 0,090±0,030 ч-1, еще более предпочтительно 0,090±0,025 ч-1, также более предпочтительно 0,090±0,020 ч-1, еще более предпочтительно 0,090±0,015 ч-1, наиболее предпочтительно 0,090±0,010 ч-1, и в частности 0,090±0,005 ч-1. В другом предпочтительном варианте осуществления, константа скорости высвобождения нулевого порядка ко находится в диапазоне 0,100±0,035 ч-1, более предпочтительно 0,100±0,030 ч-1, еще более предпочтительно 0,100±0,025 ч-1, также более предпочтительно 0,100±0,020 ч-1, еще более предпочтительно 0,100±0,015 ч-1, наиболее предпочтительно 0,100±0,010 ч-1, и в частности 0,100±0,005 ч-1.
В предпочтительном варианте осуществления, экспонента высвобождения n составляет по меньшей мере 0,65, более предпочтительно по меньшей мере 0,70, еще более предпочтительно по меньшей мере 0,75, также более предпочтительно по меньшей мере 0,80, еще более предпочтительно по меньшей мере 0,85, наиболее предпочтительно по меньшей мере 0,90 и в частности по меньшей мере 0,95.
Кинетика высвобождения нулевого порядка фармацевтической лекарственной формы в соответствии с изобретением предпочтительно не зависит от оболочки, которая остается интактной в процессе фазы высвобождения и охватывает матриксную композицию таким образом, что только специфическая площадь поверхности подвергается эрозии. Таким образом, площадь поверхности фармацевтической лекарственной формы в соответствии с изобретением, из которой высвобождается активное вещество, предпочтительно не поддерживается существенно постоянной с помощью такой оболочки. Наоборот, кинетика высвобождения нулевого порядка фармацевтической лекарственной формы в соответствии с изобретением предпочтительно основана на свойствах матрикса, в который фармакологически активный компонент (А) заделан таким образом, что инертные оболочки могут полностью отсутствовать. Таким образом, в то время как фармацевтическая лекарственная форма в соответствии с изобретением может быть покрыта с помощью общепринятых материалов для оболочки, таких как поливиниловый спирт, она предпочтительно не покрыта с помощью инертных материалов для оболочки, которые служат для постоянного покрытия существенной части наружной поверхности лекарственной формы для предоставления возможности высвобождения лекарственного средства только через заранее определенную, непокрытую часть. Таким образом, в предпочтительном варианте осуществления, фармацевтическая лекарственная форма в соответствии с изобретением не покрыта оболочкой, или она покрыта материалом оболочки, который охватывает существенно полную наружную поверхность лекарственной формы, но не оставляет покрытой определенную часть.
Фармацевтическая лекарственная форма в соответствии с изобретением содержит неорганическую соль (В).
В предпочтительном варианте осуществления, фармацевтическая лекарственная форма содержит единственную неорганическую соль (В).
В другом предпочтительном варианте осуществления, фармацевтическая лекарственная форма содержит смесь двух или более неорганических солей (В). Если фармацевтическая лекарственная форма в соответствии с изобретением содержит две различные неорганические соли (В), например, пятизамещенный трифосфат натрия и карбонат натрия, то их относительное весовое соотношение предпочтительно находится в диапазоне от 8:1 до 1:8, более предпочтительно 7:1 до 1:7, еще более предпочтительно 6:1 до 1:6, также более предпочтительно 5:1 до 1:5, еще более предпочтительно 4:1 до 1:4, наиболее предпочтительно 3:1 до 1:3, и в частности 2:1 до 1:2.
В другом предпочтительном варианте осуществления, фармацевтическая лекарственная форма содержит смесь двух неорганических солей (В). Если фармацевтическая лекарственная форма в соответствии с изобретением содержит две различные неорганические соли (В), например, пятизамещенный трифосфат натрия и карбонат натрия, то стабильность при хранении при 5°C и 25°C существенно повышается. Относительно этого предмета, снижение содержания Витамина Е, содержащегося в фармацевтической лекарственной форме, является более медленным в отличие от этого для фармацевтической лекарственной формы, содержащей только одну неорганическую соль, например, карбонат натрия, и профиль высвобождения фармакологически активного компонента (А) не изменяется по сравнению с профилем высвобождения, который восстанавливается перед тестированием стабильности при хранении.
Предпочтительно, неорганическая соль (В) представляет собой соль, предпочтительно соль щелочного металла или щелочноземельного металла, сильной неорганической кислоты, имеющей pKA значение самое большее 3, предпочтительно самое большее 2, более предпочтительно самое большее 1, еще более предпочтительно самое большее 0 и в частности самое большее - 1. Если указанная неорганическая кислоту является мультипротонной кислотой, то предпочтительно по меньшей мере первый протон соответствует вышеуказанным требованиям.
Предпочтительно, неорганическая соль (В) представляет собой соль угольной кислоты (H2CO3), фосфорной кислоты (H3PO4), фосфористой кислоты (H3PO3), пирофосфорной кислоты (H4P2O7), или трифосфорной кислоты (H5P3O10), предпочтительно их соль щелочного и/или щелочно-земельного металла и/или их гидрированную соль.
Предпочтительно, неорганическую соль (В) выбирают из группы, включающей карбонаты щелочных металлов (например, Na2CO3, K2CO3, NaKCO3), карбонаты щелочно-земельных металлов (например, MgCO3, CaCO3), гидрокарбонаты щелочных металлов (например, NaHCO3, KHCO3), гидрокарбонаты щелочноземельных металлов (например, Mg(HCO3)2, Ca(HCO3)2), фосфаты щелочных металлов (например, Na3PO4, Na2KPO4, NaK2PO4, K3PO4), фосфаты щелочноземельных металлов (например, Mg3(PO4)2, Ca3(PO4)2,), пирофосфаты щелочных металлов (например, Na4P2O7, Na3KP2O7, Na2K2P2O7, NaK3P2O7, K4P2O7), пирофосфаты щелочно-земельных металлов (например, Mg2P2O7, CaMgP2O7, Ca2P2O7), пента щелочные три(поли)фосфаты (трехосновный трифосфат щелочного металла) (например, Na5P3O10, Na4KP3O10, Na3K2P3O10, Na2K3P3O10, NaK4P3O10, K5P3O10, Na4KP3O10), гидрофосфаты щелочных металлов (например, Na2HPO4, NaKHPO4, K2HPO4), гидрофосфаты щелочноземельных металлов (например, MgHPO4, CaHPO4), дигидрофосфаты щелочных металлов (например, NaH2PO4, KH2PO4), дигидрофосфаты щелочноземельных металлов (например, Mg(H2PO4)2, Ca(H2PO4)2.
Предпочтительно, неорганическую соль (В) представляет собой карбонат натрия или пятизамещенный трифосфат натрия или их смеси.
Неожиданно было обнаружено, что неорганическая соль (В) может дополнительно удлинять профиль высвобождения фармацевтической лекарственной формы по сравнению с эталонной лекарственной формы, не содержащей неорганическую соль (В).
В предпочтительном варианте осуществления, содержание неорганической соли (В) составляет от 1 до 80 мас.%, более предпочтительно от 5 до 70 мас.%, еще более предпочтительно от 12 до 60 мас.%, также более предпочтительно от 17 до 50 мас.% и наиболее предпочтительно от 25 до 45 мас.% и в частности от 29 до 41 мас.%, исходя из общей массы фармацевтической лекарственной формы.
В предпочтительном варианте осуществления, содержание неорганической соли (В) находится в диапазоне 30±9 мас.%, более предпочтительно 30±8 мас.%, еще более предпочтительно 30±7 мас.%, также более предпочтительно 30±6 мас.%, наиболее предпочтительно 30±5 мас.%, и в частности 30±2,5 мас.%, исходя из общей массы фармацевтической лекарственной формы.
В другом предпочтительном варианте осуществления, содержание неорганической соли (В) находится в диапазоне 40±9 мас.%, более предпочтительно 40±8 мас.%, еще более предпочтительно 40±7 мас.%, также более предпочтительно 40±6 мас.%, наиболее предпочтительно 40±5 мас.%, и в частности 40±2,5 мас.%, исходя из общей массы фармацевтической лекарственной формы.
Неожиданно было обнаружено, что механические свойства (устойчивой к разрушению) фармацевтической лекарственной формы в соответствии с изобретением, в особенности ее повышенная прочность на разрыв, не уменьшаются при добавлении существенных количеств неорганической соли (В). Это является особенно неожиданным, поскольку специалист в данной области техники будет ожидать, что высокая прочность на разрыв может быть достигнута только с помощью подходящих полимеров в подходящих количествах и обработки при определенных условиях (типично давление и нагревание). Неорганическая соль (В), тем не менее, не является полимером.
Кроме того, неожиданно было обнаружено, что неорганическая соль (В) может оказывать влияние на характеристики высвобождения матрикса с контролируемым высвобождением, включающего полиалкилен оксид (С), хотя во всех фармацевтических лекарственных форм в соответствии с изобретением указанный полиалкилен оксид (С) обеспечивает прочность на разрыв по меньшей мере 500 Н для всей фармацевтической лекарственной формы. Это указывает на то, что в общепринятых гидрофильных монолитных полимерных матриксах ни проявляющая прочность на разрыв по меньшей мере 500 Н, ни набухание матрикса, ни повышение жесткости матрикса, ни поддержка матрикса путем взаимодействия электролитов и постоянного изменения периферического уплотнения не играют центральную роль в индуцированной электролитами гетерогенности композиции. Неожиданно, такой процесс также происходит в лекарственных формах в соответствии с изобретением, хотя специалист в данной области техники будет ожидать совершенно другого поведения вследствие специфических механических свойств.
Кроме того указанная фармацевтическая лекарственная форма может быть получена с помощью существенно уменьшенного количества стадий способа без потери способностей устойчивости к разрушению.
Кроме того, неожиданно было обнаружено, что профиль высвобождения in vitro фармацевтической лекарственной формы существенно может быть существенно независимым от значения pH. Предпочтительно, профиль высвобождения in vitro фармацевтической лекарственной формы следует кинетике нулевого порядка в диапазоне от pH 1 до pH 7.
В предпочтительном варианте осуществления, неорганическая соль (В) гомогенно распределена в фармацевтической лекарственной форме в соответствии с изобретением. Предпочтительно, фармакологически активный компонент (А) и неорганическая соль (В) чрезвычайно гомогенно распределены в фармацевтической лекарственной форме таким образом, что фармацевтическая лекарственная форма не содержит каких-либо сегментов, где либо фармакологически активный компонент (А) присутствует при отсутствии неорганической соли (В) или где неорганическая соль (В) присутствует при отсутствии фармакологически активного компонента (А).
Если фармацевтическая лекарственная форма покрыта оболочкой, то неорганическая соль (В) предпочтительно гомогенно распределена в ядре фармацевтической лекарственной формы, то есть пленочная оболочка предпочтительно не содержит неорганической соли (В).
Фармацевтическая лекарственная форма в соответствии с изобретением содержит полиалкилен оксид (С). Активный компонент (А) присутствует, предпочтительно заделан в матриксе с контролируемым высвобождением, содержащим указанный полиалкилен оксид, а также неорганическую соль (В).
Предпочтительно, полиалкилен оксид (С) выбирают из полиметилен оксида, полиэтилен оксида и полипропилен оксида, или их сополимеров или смесей.
Полиалкилен оксид (С) имеет средневесовую молекулярную массу (MW), предпочтительно также средневязкостную молекулярную массу (Мη) по меньшей мере 200,000 г/моль или по меньшей мере 500,000 г/моль, предпочтительно по меньшей мере 1,000,000 г/моль или по меньшей мере 2,500,000 г/моль, более предпочтительно в диапазоне от приблизительно 1,000,000 г/моль до приблизительно 15,000,000 г/моль, и наиболее предпочтительно в диапазоне от приблизительно 5,000,000 г/моль до приблизительно 10,000,000 г/моль. Подходящие методы определения MW и Мη известны специалисту в данной области техники. Мη предпочтительно определяют с помощью реологических измерений, тогда как MW можно определить с помощью гель-проникающей хроматографии (GPC).
Предпочтительно, содержание полиалкилен оксида (С) находится в диапазоне от 20 до 99 мас.%, более предпочтительно 25 до 95 мас.%, еще более предпочтительно 30 до 90 мас.%, также более предпочтительно 30 до 85 мас.%, наиболее предпочтительно 30 до 80 мас.% и в частности 30 до 75 мас.% или 45 до 70 мас.%, исходя из общей массы фармацевтической лекарственной формы. Содержание полиалкилен оксида составляет по меньшей мере 20 мас.%, предпочтительно по меньшей мере 25 мас.%, более предпочтительно по меньшей мере 30 мас.%, еще более предпочтительно по меньшей мере 35 мас.% и в частности по меньшей мере 40 мас.%, исходя из общей массы фармацевтической лекарственной формы.
В предпочтительном варианте осуществления, общее содержание полиалкилен оксида (С) находится в диапазоне 25±5 мас.%. В другом предпочтительном варианте осуществления, общее содержание полиалкилен оксида (С) находится в диапазоне 35±15 мас.%, более предпочтительно 35±10 мас.%, и в частности 35±5 мас.%. В еще другом предпочтительном варианте осуществления, общее содержание полиалкилен оксида (С) находится в диапазоне от 45±20 мас.%, более предпочтительно 45±15 мас.%, наиболее предпочтительно 45±10 мас.%, и в частности 45±5 мас.%. В еще другом предпочтительном варианте осуществления, общее содержание полиалкилен оксида (С) находится в диапазоне 55±20 мас.%, более предпочтительно 55±15 мас.%, наиболее предпочтительно 55±10 мас.%, и в частности 55±5 мас.%. В дальнейшем предпочтительном варианте осуществления, общее содержание полиалкилен оксида (С) находится в диапазоне 65±20 мас.%, более предпочтительно 65±15 мас.%, наиболее предпочтительно 65±10 мас.%, и в частности 65±5 мас.%. В дальнейшем предпочтительном варианте осуществления, общее содержание полиалкилен оксида (С) находится в диапазоне 75±20 мас.%, более предпочтительно 75±15 мас.%, наиболее предпочтительно 75±10 мас.%, и в частности 75±5 мас.%. В другом предпочтительном варианте осуществления, общее содержание полиалкилен оксида (С) находится в диапазоне 80±15 мас.%, более предпочтительно 80±10 мас.%, и наиболее предпочтительно 80±5 мас.%.
Полиалкилен оксид (С) может включать простой полиалкилен оксид, имеющий частную среднюю молекулярную массу, или смесь (композицию) различных полимеров, таких как два, три, четрые или пять полимеров, например, полимеров с одинаковой химической природой, но с разной средней молекулярной массой, полимеров с разной химической природой, но с аналогичной средней молекулярной массой, или полимеров с разной химической природой, а также с разной молекулярной массой.
Для целей настоящей заявки, полиалкилен гликоль имеет молекулярную массу вплоть до 20,000 г/моль, тогда как полиалкилен оксид имеет молекулярную массу больше, чем 20,000 г/моль. В предпочтительном варианте осуществления, средневесовые суммарные молекулярные массы всех полиалкилен оксидов, которые содержатся в фармацевтической лекарственной форме, составляют по меньшей мере 200,000 г/моль. Таким образом, полиалкилен гликоли, если таковые имеются, предпочтительно не принимают во внимание при определении средневесовой молекулярной массы полиалкилен оксида (С).
В предпочтительном варианте осуществления, полиалкилен оксид (С) гомогенно распределен в фармацевтической лекарственной форме в соответствии с изобретением. Предпочтительно, фармакологически активный компонент (А) и полиалкилен оксид (С) чрезвычайно гомогенно распределены в фармацевтической лекарственной форме таким образом, что фармацевтическая лекарственная форма не содержит каких-либо сегментов, где либо фармакологически активный компонент (А) присутствует при отсутствии полиалкилен оксида (С) или где полиалкилен оксид (С) присутствует при отсутствии фармакологически активного компонента (А).
Если фармацевтическая лекарственная форма покрыта оболочкой, то полиалкилен оксид (С) предпочтительно гомогенно распределен в ядре фармацевтической лекарственной формы, то есть пленочная оболочка предпочтительно не содержит полиалкилен оксида (С). Тем не менее, пленочная оболочка сама по себе может содержать, конечно, один или несколько полимеров, которые, однако, предпочтительно отличаются от полиалкилен оксида (С), содержащегося в ядре.
Полиалкилен оксид (С) можно комбинировать с одним или несколькими различными полимерами, выбранными из группы, включающей полиалкилен оксид, предпочтительно полиметилен оксид, полиэтилен оксид, полипропилен оксид; полиэтилен, полипропилен, поливинил хлорид, поликарбонат, полистирол, поливинилпирролидон, поли(алк)акрилат, поли(гидрокси жирные кислоты), такие как, например, поли(3-гидроксибутират-со-3-гидроксивалерат) (Биопол®), поли(гидроксивалериановая кислота); поликапролактон, поливиниловый спирт, полиэстерамид, полиэтилен сукцинат, полилактон, полигликолид, полиуретан, полиамид, полилактид, полиацеталь (например, полисахариды необязательно с модифицированными боковыми цепями), полилактид/гликолид, полилактон, полигликолид, полиортоэфир, полиангидрид, блок полимеры полиэтилен гликоля и полибутилен терефталата (Полиактив®), полиангидрид (Полифепросан), их сополимеры, их блок-сополимеры, и смеси по меньшей мере двух указанных полимеров, или другие полимеры с вышеприведенными характеристиками.
Предпочтительно, дисперсность молекулярной массы Mw/Mn полиалкилен оксида (С) находится в диапазоне 2,5±2,0, более предпочтительно 2,5±1,5, еще более предпочтительно 2,5±1,0, также более предпочтительно 2,5±0,8, наиболее предпочтительно 2,5±0,6, и в частности 2,5±0,4.
Полиалкилен оксид (С) предпочтительно имеет вязкость при 25°C от 30 до 17,600 сП, более предпочтительно от 55 до 17,600 сП, еще более предпочтительно от 600 до 17,600 сП и наиболее предпочтительно от 4,500 до 17,600 сП, измеренную в 5 мас.% водной растворе, используя модель вискозиметра RVF Brookfield (№ веретена 2/частота вращения 2 об./мин.); от 400 до 4,000 сП, более предпочтительно 400 до 800 сП или 2,000 до 4,000 сП, измеренную в 2 мас.% водном растворе, используя указанный вискозиметр (№ веретена 1 или 3/частота вращения 10 об./мин.); или от 1,650 до 10,000 сП, более предпочтительно 1,650 до 5,500 сП, 5,500 до 7,500 сП или 7,500 до 10,000 сП, измеренную в 1 мас.% водном растворе, используя указанный вискозиметр (№ веретена 2/частота вращения 2 об./мин.).
В предпочтительном варианте осуществления, относительное весовое соотношение полиалкилен оксида (С) к неорганической соли (В) находится в диапазоне от 20:1 до 0,1:1, более предпочтительно от 15:1 до 0,25:1, еще более предпочтительно от 10:1 до 0,4:1, также более предпочтительно от 5:1 до 0,5:1, наиболее предпочтительно от 3:1 до 0,75:1 и в частности 1,6:1 до 0,85:1. В предпочтительном варианте осуществления, содержание полиалкилен оксида (С) в фармацевтической лекарственной форме превышает содержание неорганической соли (В). В другом предпочтительном варианте осуществления, содержание неорганической соли (В) в фармацевтической лекарственной форме превышает содержание полиалкилен оксида (С).
Предпочтительно, относительное весовое соотношение полиалкилен оксида (С) к фармакологически активному компоненту (А) составляет по меньшей мере 0,5:1, более предпочтительно по меньшей мере 1:1, по меньшей мере 2:1, по меньшей мере 3:1, по меньшей мере 4:1, по меньшей мере 5:1, по меньшей мере 6:1, по меньшей мере 7:1, по меньшей мере 8:1 или по меньшей мере 9:1; еще более предпочтительно по меньшей мере 10:1 или по меньшей мере 15:1, также более предпочтительно по меньшей мере 20:1, наиболее предпочтительно по меньшей мере 30:1 и в частности по меньшей мере 40:1. В предпочтительном варианте осуществления, относительное весовое соотношение полиалкилен оксида (С) к фармакологически активному компоненту (А) находится в диапазоне от 3:1 до 50:1, более предпочтительно 3:1 до 40:1 и, в частности, 3:1 до 30:1.
Кроме фармакологически активного компонента (А), неорганической соли (В) и полиалкилен оксида (С), фармацевтическая лекарственная форма в соответствии с изобретением может содержать дополнительные компоненты, например, один или несколько общепринятых фармакологических наполнителей, например, заполнители, вещества, способствующие скольжению, связующие вещества, гранулирующие вещества, агенты, предотвращающие слипание, смазывающие вещества, ароматизаторы, красители, и/или консерванты.
Предпочтительно, фармацевтическая лекарственная форма дополнительно содержит пластификатор. Пластификатор улучшает способность к обработке полиалкилен оксида (С) и необязательно, также неорганической соли (В). Предпочтительный пластификатор представляет собой полиалкилен гликоль, также полиэтилен гликоль, триацетин, жирные кислоты, сложные эфиры жирных кислот, воски и/или микрокристаллические воски. Особенно предпочтительные пластификаторы представляют собой полиэтилен гликоли, такие как PEG 6000.
Предпочтительно, содержание пластификатора находится в диапазоне от 0,1 до 25 мас.%, более предпочтительно от 0,5 до 22,5 мас.%, еще более предпочтительно от 1,0 до 20 мас.%, также более предпочтительно от 2,5 до 17,5 мас.%, наиболее предпочтительно от 5,0 до 15 мас.% и в частности от 7,5 до 12,5 мас.%, исходя из общей массы фармацевтической лекарственной формы.
В предпочтительном варианте осуществления, пластификатор представляет собой полиалкилен гликоль, имеющий содержание в диапазоне 10±8 мас.%, более предпочтительно 10±6 мас.%, еще более предпочтительно 10±5 мас.%, также более предпочтительно 10±4 мас.%, наиболее предпочтительно 10±3 мас.%, и в частности 10±2 мас.%, исходя из общей массы фармацевтической лекарственной формы.
В другом предпочтительном варианте осуществления, пластификатор представляет собой полиалкилен гликоль, имеющий содержание в диапазоне 15±8 мас.%, более предпочтительно 15±6 мас.%, еще более предпочтительно 15±5 мас.%, также более предпочтительно 15±4 мас.%, наиболее предпочтительно 15±3 мас.%, и в частности 15±2 мас.%, исходя из общей массы фармацевтической лекарственной формы.
В предпочтительном варианте осуществления, относительное весовое соотношение полиалкилен оксида (С) к полиалкилен гликолю находится в диапазоне 4,2±2:1, более предпочтительно 4,2±1,5:1, еще более предпочтительно 4,2±1:1, также более предпочтительно 4,2±0,5:1, наиболее предпочтительно 4,2±0,2:1, и в частности 4,2±0,1:1. Это соотношение соответствует требованиям относительно высокого содержания полиалкилен оксида (С) и хорошей экструдируемости.
При приготовлении лекарственных форм со срезов, которые получают путем нарезания экструдируемой ленты, вес срезов определяет вес полученной лекарственной формы. Явно выраженное колебание веса этих срезов приводит к соответствующим отклонениям веса лекарственных форм от целевого веса. Колебание веса срезов существенным образом зависит поверхностных свойств экструдируемой ленты. Лента с совершенно гладкой поверхностью предоставляет возможность получить срезы, характеризующиеся незначительным колебанием веса. В отличие от этого, волнистая лента или лента типа «акульей кожи» приводит к срезам, проявляющим более значительные колебания веса, таким образом повышая количество брака. Неожиданно было обнаружено, что поверхностные свойства экструдируемой ленты можно управлять с помощью весового соотношения полиалкилен оксид: полиалкилен гликоль.
Предпочтительно, фармацевтическая лекарственная форма дополнительно содержит антиоксидант. Подходящие антиоксиданты включают аскорбиновую кислоту, бутилированный гидроксианизол (ВНА), бутилированный гидрокситолуол (ВНТ), соли аскорбиновой кислоты, монотиоглицерин, фосфористую кислоту, витамин С, витамин Е и их производные, кониферил бензоат, нордигидрогваяретовую кислоту, сложные эфиры галлюсовой кислоты, бисульфит натрия, особенно предпочтительно бутилгидрокситолуол или бутилгидроксианизол и α-токоферол. Антиоксидант предпочтительно используют в количествах от 0,01 до 10 мас.%, предпочтительно от 0,03 до 5 мас.%, относительно общего веса фармацевтической лекарственной формы.
В предпочтительном варианте осуществления, фармацевтическая лекарственная форма дополнительно содержит кислоту, предпочтительно лимонную кислоту. Количество кислоты предпочтительно находится в диапазоне от 0,01 до приблизительно 20 мас.%, более предпочтительно в диапазоне от 0,02 до приблизительно 10 мас.%, и наиболее предпочтительно в диапазоне от 0,05 до приблизительно 5 мас.%.
В предпочтительном варианте осуществления, фармацевтическая лекарственная форма содержит природный, полусинтетический или синтетический воск. Воски с температурой размягчения по меньшей мере 50°C, более предпочтительно 60°C являются предпочтительными. Карнаубский воск и пчелиный воск являются особенно предпочтительными, в особенности карнаубский воск.
В предпочтительном варианте осуществления, фармацевтическая лекарственная форма дополнительно содержит другой полимер, который предпочтительно выбирают из сложных эфиров целлюлозы и простых эфиров целлюлозы, в частности гидроксипропил метилцеллюлозы (НРМС). Количество дополнительного полимера, предпочтительно гидроксипропил метилцеллюлозы, предпочтительно находится в диапазоне от 0,1 мас.% до приблизительно 30 мас.%, более предпочтительно в диапазоне от 1,0 мас.% до приблизительно 20 мас.%, и наиболее предпочтительно в диапазоне от 2,0 мас.% до приблизительно 15 мас.%.
В другом предпочтительном варианте осуществления, фармацевтическая лекарственная форма в соответствии с изобретением не содержит какого-либо дополнительного полимера, кроме полиалкилен оксида (С) и необязательно, полиэтилен гликоля.
Фармацевтическая лекарственная форма в соответствии с изобретением предпочтительно представляет собой пероральную лекарственную форму, в особенности таблетку. Тем не менее, также представляется возможным, вводить фармацевтическую лекарственную форму различными путями и таким образом, фармацевтическая лекарственная форма альтернативно может быть приспособлена для буккального, лингвального, ректального или вагинального введения. Также возможны импланты.
Предпочтительно, фармацевтическая лекарственная форма является монолитной. Предпочтительно, фармацевтическая лекарственная форма не представлена в пленочной форме, а также в виде множества частиц.
В предпочтительном варианте осуществления, фармацевтическая лекарственная форма в соответствии с изобретением представляет собой круглую таблетку. Таблетки согласно этому варианту осуществления предпочтительно имеют диаметр в диапазоне от приблизительно 1 мм до приблизительно 30 мм, в частности в диапазоне от приблизительно 2 мм до приблизительно 25 мм, более предпочтительно от приблизительно 5 мм до приблизительно 23 мм, также в частности от приблизительно 7 мм до приблизительно 13 мм; и толщину в диапазоне от приблизительно 1,0 мм до приблизительно 12 мм, в частности в диапазоне от приблизительно 2,0 мм до приблизительно 10 мм, еще более предпочтительно от 3,0 мм до приблизительно 9,0 мм, также предпочтительно от приблизительно 4,0 мм до приблизительно 8,0 мм.
В другом предпочтительном варианте осуществления, фармацевтическая лекарственная форма в соответствии с изобретением представляет собой продолговатую таблетку. Таблетки согласно этому варианту осуществления предпочтительно имеют удлинение в длину (продольное удлинение) от приблизительно 1 мм до приблизительно 30 мм, в частности в диапазоне от приблизительно 2 мм до приблизительно 25 мм, более предпочтительно от приблизительно 5 мм до приблизительно 23 мм, также в частности от приблизительно 7 мм до приблизительно 20 мм; и толщину в диапазоне от приблизительно 1,0 мм до приблизительно 12 мм, в частности в диапазоне от приблизительно 2,0 мм до приблизительно 10 мм, еще более предпочтительно от 3,0 мм до приблизительно 9,0 мм, также предпочтительно от приблизительно 4,0 мм до приблизительно 8,0 мм.
Фармацевтическая лекарственная форма в соответствии с изобретением предпочтительно имеют вес в диапазоне от 0,01 до 1,5 г, более предпочтительно в диапазоне от 0,05 до 1,2 г, еще более предпочтительно в диапазоне от 0,1 г до 1,0 г, также более предпочтительно в диапазоне от 0,2 г до 0,9 г, и наиболее предпочтительно в диапазоне от 0,25 г до 0,8 г.
Фармацевтическую лекарственную форму в соответствии с изобретением предпочтительно приготавливают путем высокотемпературного формообразования, предпочтительно путем экструзии горячего расплава, хотя также можно использовать другие методы высокотемпературного формообразования для приготовления фармацевтической лекарственной формы в соответствии с изобретением, такие как прессование при повышенной температуре или нагревание таблеток, которые были приготовлены путем общепринятого сжатия, на первой стадии и затем нагревания выше температуры размягчения полимера в таблетке на второй стадии с образованием твердых таблеток. В этом отношении, высокотемпературное формообразование обозначает формирование или отливку в форме массы после применения тепла. В предпочтительном варианте осуществления, фармацевтическая лекарственная форма термоформируема путем экструзии горячего расплава.
В предпочтительном варианте осуществления, фармацевтическая лекарственная форма приготовлена путем экструзии горячего расплава, предпочтительно с помощью двухшнекового экструдера. Экструзия расплава предпочтительно обеспечивает экструдированную расплавом ленту, которую предпочтительно разрезают на монолиты, которые после этого спрессовывают и формирируют в таблетки. В этом отношении, термин "таблетки" предпочтительно не понимается как лекарственные формы, полученные путем прессования порошка или гранул (compressi), а как формированные экструдаты. Предпочтительно, сжатие осуществляют с помощью штампа и пуансона, предпочтительно монолитной массы, полученной путем экструзии расплава. Если получение осуществляют путем экструзии расплава, стадию сжатия предпочтительно осуществляют с монолитной массой, имеющей повышенную температуру, то есть температуру в диапазоне от 20 до 25°C. Ленты, полученные путем экструзии, могут быть либо подвергнуты стадии сжатия сами по себе или могут быть разрезаны перед стадией сжатия. Это разрезание может быть осуществлено с помощью общепринятых технологий, например, используя вращающиеся ножи или сжатый воздух. Альтернативно, можно формировать, как описано в EP-A 240906, путем пропускания экструдата между двумя вращающимися в противоположных направлениях плющильных валиках и формируя непосредственно таблетки. Также, разумеется, можно подвергать эктрудированные ленты стадии сжатия или стадии нарезания, пока они еще теплые, то есть сразу или практически сразу после стадии экструзии. Экструзию предпочтительно осуществляют с помощью двухшнекового экструдера.
Фармацевтическая лекарственная форма в соответствии с изобретением необязательно может быть обеспечена, частично или полностью, общепринятой оболочкой. Лекарственные формы согласно настоящему изобретению предпочтительно покрыты оболочкой с помощью общепринятых композиций для пленочных оболочек. Особенно предпочтительно, лекарственные формы в соответствии с изобретением являются либо не покрытыми оболочкой вообще или полностью покрыты оболочкой, но предпочтительно не частично покрыты.
Подходящие материалы оболочек являются коммерчески доступными, например, под торговыми марками Opadry® и Eudragit®.
Примеры подходящих материалов включают сложные эфиры целлюлозы и простые эфиры целлюлозы, такие как метилцеллюлоза (МС), гидроксипропилметилцеллюлоза (НРМС), гидроксипропилцеллюлоза (НРС), гидроксиэтилцеллюлоза (НЕС), натрий карбоксиметилцеллюлоза (Na-CMC), этилцеллюлоза (ЕС),. целлюлоза ацетат фталат (САР), гидроксипропилметилцеллюлоза фталат (НРМСР); поли(мет)акрилаты, такие как аминоалкилметакрилатные сополимеры, этилакрилат метилметакрилатные сополимеры, метилметакрилатные сополимеры метакриловой кислоты, метилметакрилатные сополимеры метакриловой кислоты; виниловые полимеры, такие как поливинилпирролидон, поливинилацетатфталат, поливиниловый спирт, поливинилацетат; и природные пленкообразователи, такие как шеллак.
В особенно предпочтительном варианте осуществления, оболочка является водорастворимой. В предпочтительном варианте осуществления, оболочка основана на поливиниловом спирте, таком как поливиниловый спирт-част, гидролизованный, и может дополнительно содержать полиэтилен гликоль, такой как макрогол 3350, и/или пигменты. В другом предпочтительном варианте осуществления, оболочка основана на гидроксипропилметилцеллюлозе, предпочтительно гипромеллозе типа 2910, имеющей вязкость от 3 до 15 мПаскаль.
Оболочка может быть устойчива к желудочному соку и растворяться в зависимости от значения pH среды высвобождения. С помощью этой оболочки, представляется возможным обеспечить, чтобы фармацевтическая лекарственная форма в соответствии с изобретением проходила через желудок нерастворенной и активный компонент высвобождается только в кишечнике. Оболочка, которая устойчива к желудочному соку, предпочтительно растворяется при значении pH в диапазоне от 5 до 7,5. Соответствующие материалы и методы для замедленного высвобождения активных компонентов и для применения оболочек, которые устойчивы к желудочному соку, известны специалисту в данной области техники, например, из «Coated Pharmaceutical dosage forms - Fundamentals, Manufacturing Techniques, Biopharmaceutical Aspects, Test Methods and Raw Materials" под ред. Kurt H. Bauer, K. Lehmann, Hermann P. Osterwald, Rothgang, Gerhart, 1-ое изд., 1998, Medpharm Scientific Publishers.
Оболочка также может применяться, например, для улучшения эстетического впечатления и/или вкуса лекарственных форм и легкости, с которой они могут проглатываться. Оболочка лекарственных форм согласно настоящему изобретению также может служить для других целей, например, улучшить стабильность и срок годности. Подходящие составы для нанесения оболочек включают пленкообразующий полимер, такой как, например, поливиниловый спирт или гидроксипропил метилцеллюлозу, например, гипромеллозу, пластификатор, такой как, например, гликоль, например, пропилен гликоль или полиэтилен гликоль, замутнитель, такой как, например, диоксид титана, и разглаживатель пленки, такой как, например, тальк. Подходящие растворители оболочек представляют собой воду, а также органические растворители. Примерами органических растворителей являются спирты, например, этанол или изопропанол, кетоны, например, ацетон, или галогенированные углеводороды, например, метилен хлорид. Необязательно, оболочка может содержать терапевтически эффективное количество одного или нескольких активных компонентов для обеспечения быстрого высвобождения указанного активного компонента (А) и, следовательно, для быстрого облегчения симптомов, леченных с помощью указанного активного компонента (А). Покрытие оболочкой лекарственные формы согласно настоящему изобретению предпочтительно приготавливают сначала путем получения ядер и после этого указанные ядра покрывают оболочкой, используя общепринятые техники, такие как нанесение оболочки в установке для нанесения оболочки.
В соответствии с изобретением, активный компонент (А) присутствует, предпочтительно заделан в матриксе с контролируемым высвобождением, содержащем неорганическую соль (В) и полиалкилен оксид (С).
Контролируемое высвобождение активного компонента из пероральной лекарственной формы известно специалисту в данной области техники. Для целей настоящей заявки, контролируемое высвобождение охватывает замедленное высвобождение, ретардированное высвобождение, длительное высвобождение, продленное высвобождение, пролонгированное высвобождение, и другие.
Контролированное или пролонгированное высвобождение в соответствии с изобретением предпочтительно обозначает профиль высвобождения, с которым фармакологически активный компонент (А) освобождается в течение относительно долгого периода с уменьшенной частотой введения для удлиненного терапевтического действия. Предпочтительно, значение термина "пролонгированное высвобождение" соответствует Европейским требованиям относительно номенклатуры профиля высвобождения фармацевтических лекарственных форм (СНМР). Это достигается, в частности, при пероральном введении. Выражение "по меньшей мере частично замедленное или пролонгированное высвобождение" охватывает в соответствии с изобретением любые фармацевтические лекарственные формы, которые обеспечивают модифицированное высвобождение опиоидов (А), которые в них содержатся. Фармацевтические лекарственные формы предпочтительно включают покрытые оболочкой или непокрытые оболочкой фармацевтические лекарственные формы, которые изготавливаются со специфическими вспомогательными веществами, с помощью специальных процессов или путем комбинации двух возможных опций для преднамеренного изменения скорости высвобождения или локализации высвобождения.
В случае фармацевтических лекарственных форм в соответствии с изобретением, временная зависимость высвобождения формы с контролируемым высвобождением может быть модифицирована, например, следующим образом: продленное высвобождение, поэтапное высвобождение, пролонгированное высвобождение и длительное высвобождение.
Для целей настоящей заявки "контролируемое высвобождение" предпочтительно обозначает продукт, в котором высвобождение активного компонента в зависимости от времени контролируется типом и составом препарата. Для целей настоящей заявки "продленное высвобождение" предпочтительно обозначает продукт, в котором высвобождение активного компонента замедлено на время с конечным лагом, после чего высвобождение осуществляется беспрепятственно. Для целей настоящей заявки "поэтапное высвобождение" предпочтительно обозначает продукт, в котором первая порция активного компонента высвобождается сначала, с последующим высвобождением по меньшей мере одной дальнейшей порции активного компонента. Для целей настоящей заявки "пролонгированное высвобождение" предпочтительно обозначает продукт, в котором скорость высвобождения активного компонента из препарата после введения уменьшена в зависимости от времени, для поддержания терапевтической активности, для уменьшения токсических эффектов, или для других терапевтических целей. Для целей настоящей заявки "длительное высвобождение" предпочтительно обозначает путь приготовления лекарственного средства таким образом, что его высвобождение в организме стабильно, в течение продолжительного периода времени, таким образом уменьшая частоту дозирования. Для более подробного описания, можно привести ссылку, например, на K.Н. Bauer, Lehrbuch der Pharmazeutischen Technologie, бое издание, WVG Stuttgart, 1999; и Европейскую фармакопею
Предпочтительно, при физиологических условиях фармацевтическая лекарственная форма в соответствии с изобретением высвобождает через 30 минут от 0,1 до 75%, через 240 минут от 0,5 до 95%, через 480 минут от 1,0 до 100% и через 720 минут от 2,5 до 100% фармакологически активного компонента (А). Другие предпочтительные профили высвобождения R1-R6 обобщены в таблице, представленной ниже [все данные в мас.% высвобожденного фармакологически активного компонента (А)]:
Другие предпочтительные профили высвобождения R1 до R6 обобщены в таблице, представленной ниже [все данные в мас.% высвобожденного фармакологически активного компонента (А)]:
Предпочтительно, профиль высвобождения фармацевтической лекарственной формы в соответствии с настоящим изобретением стабильный при хранении, предпочтительно при хранении при повышенной температуре, например, 40°C, в течение 3 месяцев в запечатанных контейнерах. В этом отношении "стабильный" обозначает, что при сравнении профиля исходного высвобождения с профилем высвобождения после хранения, в любую заданную временную точку профили высвобождения отличаются друг от друга не более, чем на 20%, более предпочтительно не более, чем на 15%, еще более предпочтительно не более, чем на 10%, также более предпочтительно не более, чем на 7,5%, наиболее предпочтительно не более, чем на 5,0% и в частности не более, чем на 2,5%.
Предпочтительно, в условиях in vitro фармацевтическая лекарственная форма высвобождает через 0,5 ч. от 1,0 до 35 мас.%, через 1 ч. от 5,0 до 45 мас.%, через 2 ч. от 10 до 60 мас.%, через 4 ч. по меньшей мере 15 мас.%, через 6 ч. по меньшей мере 20 мас.%, через 8 ч. по меньшей мере 25 мас.% и через 12 ч. по меньшей мере 30 мас.% фармакологически активного компонента (А), который изначально присутствует в фармацевтической лекарственной форме.
Подходящие условия in vitro известны специалистам в данной области техники. В этом отношении можно привести ссылки, например, на Европейскую фармакопею. Предпочтительно, профиль высвобождения определяют в следующих условиях: Прибор с лопастью, оборудованный грузом, 75 об./мин., 37±5°C, 600 мл искусственного кишечного сока pH 6,8 (фосфатный буфер) или pH 4,5. В предпочтительном варианте осуществления, частота вращения лопасти повышена до 100 об./мин.
В предпочтительном варианте осуществления, фармацевтическая лекарственная форма в соответствии с изобретением адаптирована для введения один раз в сутки. В другом предпочтительном варианте осуществления, фармацевтическая лекарственная форма в соответствии с изобретением адаптирована для введения два раза в сутки. В еще другом предпочтительном варианте осуществления, фармацевтическая лекарственная форма в соответствии с изобретением адаптирована для введения три раза в сутки.
Для целей настоящей заявки, "два раза в сутки" обозначает равные или практически равные временные интервалы, то есть, приблизительно каждые 12 часов, или различные временные интервалы, например, 8 и 16 часов или 10 и 14 часов, между индивидуальными введениями.
Для целей настоящей заявки, "три раза в сутки" обозначает равные или практически равные временные интервалы, то есть, приблизительно каждые 8 часов, или различные временные интервалы, например, 6, 6 и 12 часов; или 7, 7 и 10 часов, между индивидуальными введениями.
Предпочтительно, фармацевтическая лекарственная форма в соответствии с изобретением высвобождает через 5 ч. самое большее 99%, более предпочтительно самое большее 90%, еще более предпочтительно самое большее 75%, и наиболее предпочтительно самое большее 60% активного компонента (А).
Неорганическая соль (В) предпочтительно является гидрофильной, обозначая, что матрикс, содержащий неорганическую соль (В) и полиалкилен оксид (С) имеет склонность к набуханию при контактировании с водными жидкостями после введения, и предпочтительно приводит к образованию вязкого, гелевого слоя, регулирующего высвобождение фармакологически активного компонента.
В предпочтительном варианте осуществления, матрикс, содержащий неорганическую соль (В) и полиалкилен оксид (С) содержит неорганическую соль (В) в таком количестве, что в условиях in vitro высвобождение активного компонента (А) дополнительно замедлена, и профиль высвобождения фармакологически активного компонента (А) из указанного матрикса включает по меньшей мере временной интервал, в течение которого высвобождение соответствует кинетике нулевого порядка, сравнивают с идентичной, сравнительной фармацевтической лекарственной формой, где неорганическая соль (В) заменена соответствующим количеством гидроксилпропил метил целлюлозы (НРМС) или лактозы.
В особенно предпочтительном варианте осуществления,
- фармацевтическая лекарственная форма термоформирована, предпочтительно путем экструзии горячего расплава; и/или
- фармацевтическая лекарственная форма проявляет прочность на разрыв по меньшей мере 1500 Н; и/или
- фармацевтическая лекарственная форма адаптирована для введения один раз в сутки, два раза в сутки или три раза в сутки; и/или
- фармакологически активные компоненты (А) выбирают из группы опиоидов и опиатов; и или
- содержание неорганической соли (В) находится в диапазоне от 2,0 мас.% до 50 мас.%; и/или
- полиалкилен оксид (С) выбирают из полиметилен оксида, полиэтилен оксида и полипропилен оксида, или их сополимеров или смесей; который имеет средневесовую молекулярную массу (Mw) по меньшей мере 500,000 г/моль, более предпочтительно в диапазоне от 1,000,000 г/моль до 10,000,000 г/моль; и/или - содержание полиалкилен оксида (С) составляет по меньшей мере 30 мас.%, исходя из общей массы лекарственной формы.
В предпочтительном варианте осуществления, фармацевтическая лекарственная форма в соответствии с изобретением не содержит веществ, которые раздражают носовые ходы и/или глотку, то есть веществ, которые, при введении через носовые ходы и/или глотку, обуславливают физическую реакцию, которая либо настолько неприятна для пациента, что он/она не желает или не может продолжить введение, например, жжение, или физиологически препятствует приему соответствующего активного компонента, например, следствие повышенной носовой секреции или чиханья. Другими примерами веществ, которые раздражают носовые ходы и/или глотку, являются те, которые вызывают жжение, зуд, позыв к чиханию, повышенное образовании секрета или комбинацию по меньшей мере двух из этих воздействий. Соответствующие вещества и их количества, которые обычно используются, известны специалисту в данной области техники. Некоторые вещества, которые раздражают носовые ходы и/или глотку, соответственно, основаны на одном или нескольких составляющих или одной или нескольких частях растений горячего вещества фармакологически активного компонента. Соответствующие горячие вещества фармакологически активного компонента известны per se специалисту в данной области техники и описаны, например, в "Pharmazeutische Biologie - Drogen und ihre Inhaltsstoffe" ред. Prof. Dr. Hildebert Wagner, 2oe, пересмотренное издание, Gustav Fischer Verlag, Stuttgart-New York, 1982, стр.82 и далее. Соответствующее описание таким образом включено в качестве ссылки и является частью описания.
Фармацевтическая лекарственная форма в соответствии с изобретением, кроме того, предпочтительно не содержит антагонистов для фармакологически активного компонента (А), предпочтительно не содержит антагонистов к психотропным веществам, в частности не содержит антагонистов к опиоидам (А). Антагонисты, пригодные для данного фармакологически активного компонента (А), известны специалисту в данной области техники и могут быть представлены как таковые или в форме соответствующих производных, в частности сложные эфиры или простые эфиры, или в каждом случае в форме соответствующих физиологически приемлемых соединений, в частности в форме их солей или сольватов. Фармацевтическая лекарственная форма в соответствии с изобретением предпочтительно не содержит антагонистов, выбранных, в частности, из группы, включающей налоксон, налтрексон, намелфен, налид, налмексон, налорфин или налуфин, в каждом случае необязательно в форме соответствующего физиологически приемлемого соединения, в частности в форме основания, соли или сольвата; и не содержит нейролептиков, например, соединения, выбранного, в частности, из группы, включающей галоперидол, прометацин, флуфеназин, перфеназин, левомепромазин, тиоридазин, перазин, хлорпромазин, хлорпротиксин, зуклопентиксол, флупентиксол, протипендил, зотепин, бенперидол, пипамперон, мелперон и бромперидол.
Фармацевтическая лекарственная форма в соответствии с изобретением, кроме того, предпочтительно не содержит средства, вызывающего рвоту. Средство, вызывающие рвоту, известны специалисту в данной области техники и могут быть представлены как таковые или в форме соответствующих производных, в частности сложные эфиры или простые эфиры, или в каждом случае в форме соответствующих физиологически приемлемых соединений, в частности в форме их солей или сольватов. Фармацевтическая лекарственная форма в соответствии с изобретением предпочтительно не содержит средства, вызывающего рвоту, на основе на одного или нескольких составляющих рвотного корня (ипекуана), например, на основе составляющего эметин, как, например, описано в "Pharmazeutische Biologie - Drogen und ihre Inhaltsstoffe" ред. Prof. Dr. Hildebert Wagner, 2oe пересмотренное издание, Gustav Fischer Verlag, Stuttgart, New York, 1982. Соответствующее описание в литературе таким образом включено в качестве ссылки и является частью описания. Фармацевтическая лекарственная форма в соответствии с изобретением предпочтительно также не содержит апоморфина в качестве средства, вызывающего рвоту.
В завершение, фармацевтическая лекарственная форма в соответствии с изобретением предпочтительно также не содержит горького вещества. Горькие вещества и количества, эффективные для применения, могут быть обнаружены в US-2003/0064099 A1, соответствующее раскрытие которого следует рассматривать как описание настоящей заявки и таким образом включено в качестве ссылки. Примерами горьких веществ являются ароматические масла, такие как мятное масло, эвкалиптовое масло, масло горького миндаля, ментол, ароматические фруктовые вещества, ароматические вещества из лимонов, апельсинов, лаймов, грейпфрутов или их смеси, и/или денатониум бензоат.
Фармацевтическая лекарственная форма в соответствии с изобретением таким образом предпочтительно не содержит ни веществ, которые раздражают носовые ходы и/или глотку, ни антагонистов для фармакологически активного компонента (А), ни средства, вызывающего рвоту, ни горьких веществ.
Фармацевтическая лекарственная форма в соответствии с изобретением имеет прочность на разрыв по меньшей мере 500 Н.
Фармацевтическая лекарственная форма в соответствии с изобретением предпочтительно устойчива к разрушению. Предпочтительно, устойчивость к разрушению достигается на основе механические свойства фармацевтической лекарственной формы таким образом, что раздробления избегается или по меньшей мере существенно затруднено. В соответствии с изобретением, термин раздробление обозначает пульверизацию фармацевтической лекарственной формы, используя общепринятые средства, обычно доступные для злоумышленника, например, пестик и ступка, молоток, деревянный молоток или другие общепринятые средства для пульверизации под воздействием силы. Таким образом, устойчивая к разрушению предпочтительно обозначает, что пульверизация фармацевтической лекарственной формы с использованием общепринятых средств исключена или по меньшей мере существенно затруднена.
Предпочтительно, механические свойства фармацевтической лекарственной формы в соответствии с изобретением, в особенности ее прочность на разрыв, существенным образом основано на присутствии и пространственном распределении неорганической соли (В) и полиалкилен оксид (С), хотя их простого присутствия типично недостаточно для достижения указанных свойств. Благоприятные механические свойства фармацевтической лекарственной формы в соответствии с изобретением автоматически не могут быть достигнуты путем простой обработки фармакологически активный компонент (А), неорганическую соль (В), полиалкилен оксид (С), и необязательно дополнительных наполнителей с помощью общепринятых методов для приготовления фармацевтических лекарственных форм. В действительности, обычно пригодные инструменты приборы быть отобраны для приготовления и основные параметры обработки должны быть подогнаны, в особенности давление/сила, температура и время. Таким образом, даже если используются общепринятые приборы, то протоколы процесса обычно должны быть адаптированы для соответствия необходимым критериям.
В целом, лекарственные формы, проявляющие желательные свойства, могут быть получены, только если, при приготовлении лекарственной формы,
- подходящие компоненты
- в подходящих количествах
подвергаются воздействию
- достаточного давления
- при достаточной температуре
- в течение достаточного периода времени.
Таким образом, независимо от используемого прибора, протоколы процесса должны быть адаптированы для соответствия необходимым критериям. Следовательно, прочность на разрыв отделима от композиции.
Фармацевтическая лекарственная форма в соответствии с изобретением имеет прочность на разрыв по меньшей мере 500 Н, предпочтительно по меньшей мере 600 Н, более предпочтительно по меньшей мере 700 Н, еще более предпочтительно по меньшей мере 800 Н, также более предпочтительно по меньшей мере 1000 Н, наиболее предпочтительно по меньшей мере 1250 N и в частности по меньшей мере 1500 Н.
«Прочность на разрыв" (сопротивление раздавливанию) фармацевтической лекарственной формы известно специалисту в данной области. В этом отношении можно привести ссылки, например, на W.A. Ritschel, Die Tablette, 2. Auflage, Editio Cantor Verlag Aulendorf, 2002; H Liebermann и др., Pharmaceutical dosage forms: Tablets, Том 2, Informa Healthcare; 2 издание, 1990; и Encyclopedia of Pharmaceutical Technology, Informa Healthcare; 1 издание.
Для целей настоящей заявки, прочность на разрыв предпочтительно определяется как количество силы, которое необходимо для разрушения фармацевтической лекарственной формы (= разрывное усилие). Следовательно, для целей настоящей заявки фармацевтическая лекарственная форма предпочтительно не проявляет желательной прочности на разрыв, когда она распадается, то есть, разламывается на по меньшей мере две независимые части, которые отделены друг от друга. В другом предпочтительном варианте осуществления, тем не менее, фармацевтическая лекарственная форма считается разрушенной, если сила снижается на 25% (пороговое значение) наибольшей силы, измеренной в процессе измерения (см. ниже).
Фармацевтические лекарственные формы в соответствии с изобретением отличаются от общепринятых фармацевтических лекарственных форм тем, что благодаря их прочности на разрыв, они не могут пульверизоваться путем применения силы с помощью общепринятых средств, таких как, например, пестик и ступка, молоток, деревянный молоток или других обычных средств для пульверизации, в частности устройств, разработанных для этой цели (дробилки таблеток). В этом отношении "пульверизация» означает дробление на мелкие частицы, которые сразу будут высвобождать фармакологически активный компонент (А) в подходящую среду. Устранение пульверизации фактически исключает оральное или парентеральное, в частности внутривенное или назальное злоупотребление.
Общепринятые таблетки обычно имеют прочность на разрыв значительно ниже 200 Н в любом направлении растяжения. Прочность на разрыв общепринятых круглых таблеток может быть оценена в соответствии со следующей эмпирической формулой: Прочность на разрыв [в Н]=10 × Диаметр таблетки [в мм]. Таким образом, в соответствии с указанной эмпирической формулой, круглая таблетка, имеющая прочность на разрыв по меньшей мере 300 Н должна иметь диаметр по меньшей мере 30 мм). Тем не менее такая таблетка не может быть проглочена. Вышеописанная эмпирическая формула предпочтительно не применяется к фармацевтическим лекарственным формам в соответствии с изобретением, которые не являются общепринятыми, а скорее специальными.
Кроме того, действительная средняя сила разжевывания составляет приблизительно 220 Н (ср., например, Р.А. Proeschel и др., J Dent Res, 2002, 81 (7), 464-468). Это обозначает, что общепринятые таблетки, имеющие прочность на разрыв значительно ниже 200 Н, могут быть разрушены при самопроизвольном жевании, тогда как фармацевтические лекарственные формы в соответствии с изобретением не могут быть разрушены.
Кроме того, при применении гравитационного ускорения приблизительно 9,81 м/с2, 500 Н соответствует силы тяжести больше, чем 50 кг, то есть фармацевтические лекарственные формы в соответствии с изобретением предпочтительно могут выдержать вес больше, чем 50 кг без пульверизации.
Способы измерения прочности на разрыв фармацевтической лекарственной формы известны специалистам в данной области техники. Подходящие устройства являются коммерчески доступными.
Например, прочность на разрыв (сопротивление раздавливанию) может быть определена в соответствии с Европейской фармакопеей 5.0, 2.9.8 или 6.0, 2.09.08 "Сопротивление раздавливанию таблеток". Тест предназначен для определения, в указанных условиях, Сопротивление раздавливанию таблеток измеряется с помощью силы, необходимой для деструкции их путем разрушения. Аппарат состоит из 2 зажимов лицом друг к другу, которые движутся навстречу друг другу. Плоские поверхности зажимов перпендикулярны направлению движения. Разрушающие поверхности зажимов являются плоскими и больше, чем зона контакта с таблеткой. Аппарат откалиброван с использованием системы с погрешностью 1 Ньютон. Таблетку помещали между зажимами, принимая во внимание, в соответствующих случаях, форму, метку для разрыва и надпись; для каждого измерения таблетку ориентировали аналогично по отношению к направлению применения силы (и направление растяжения, в котором измеряют прочность на разрыв). Измерения осуществляли на 10 таблетках, обращая внимания на то, чтобы все фрагменты таблеток были удалены перед каждым определением. Результат представляли в виде среднего, минимального и максимального значений измеренной силы, все выражали в Ньютонах.
Аналогичное описание прочности на разрыв (разрывное усилие) можно найти в Фармакопее США. Прочность на разрыв альтернативно может быть измерена в соответствии с методом, описанном там, где указано, что прочность на разрыв представляет собой силу, необходимую для вызывания повреждения таблетки (то есть, треснуть) в специфической плоскости. Таблетки обычно помещают между двумя плитами, одна из которых движется для приложения достаточной силы на таблетку для вызывания трещины. Для обычных, круглых (круглое поперечное сечение) таблеток, нагрузка оказывается вдоль их диаметра (в некоторых случаях обозначается как диаметральная нагрузка), и излом наступает в плоскости. Разрывное усилие таблеток обычно называется твердость в фармацевтической литературе; тем не менее, использование этого термина вводит в заблуждение. В научных материалах, термин твердость относится к устойчивости поверхности к проникновению или вдавливанию небольшим зондом. Термин прочность при раздавливании также часто используется для описания устойчивости таблеток к применению нагрузки при сжатии. Хотя этот термин описывает истинную природу теста более точно, чем твердость, он подразумевает, что таблетки в действительности разрушаются при проведении теста, хотя часто этого не происходит.
Альтернативно, прочность на разрыв (сопротивление раздавливанию) может быть определена в соответствии с WO 2005/016313, WO 2005/016314, и WO 2006/082099, который может быть обозначен как модификация метода, описанного в Европейской фармакопее. Аппарат, используемый для измерения, предпочтительно представляет собой "Zwick Z 2.5" установку для испытания материалов, Fmax=2,5 кН с максимальным вытяжением 1150 мм, который должен быть установлен с одной колонной и одним веретеном, клиренсом за 100 мм и тестовой скоростью, регулируемой в диапазоне от 0,1 до 800 мм/мин совместно с программным обеспечением testControl. Измерение осуществляли, используя нагнетательный поршень с ввинченными вкладками и цилиндр (диаметр 10 мм), датчик силы, Fmax. 1 кН, диаметр=8 мм, класс 0,5 от 10 Н, класс 1 от 2 Н до ISO 7500-1, со свидетельством об испытании производителя М в соответствии с DIN 55350-18 (Zwick главная сила Fmax=1,45 кН) (все аппараты от Zwick GmbH & Co. KG, Ulm, Germany) с Номером заказа BTC-FR 2,5 TH. D09 для тестера, Номером заказа BTC-LC 0050N. Р01 для датчика силы, Номером заказа ВО 70000 S06 для центрирующего устройства.
В предпочтительном варианте осуществления изобретения, прочность на разрыв оценивают с помощью тестера прочности, например, Sotax®, тип НТ100 или тип НТ1 (Allschwil, Switzerland). Оба Sotax® НТ100 и Sotax® НТ1 могут измерять прочность на разрыв в соответствии с двумя различными принципами измерения: постоянная скорость (где контрольный зажим движется с постоянной скоростью, регулируемой в пределах 5-200 мм/мин) или постоянная сила (где контрольный зажим повышает силу линейно, регулируемой в пределах 5-100 Н/с). В принципе, оба принципа измерения пригодны для измерения прочности на разрыв фармацевтической лекарственной формы в соответствии с изобретением. Предпочтительно, прочность на разрыв измеряют при постоянной скорости, предпочтительно при постоянной скорости 120 мм/мин.
В предпочтительном варианте осуществления, фармацевтическая лекарственная форма считается разрушенной, если она треснула по меньшей мере на два отдельных куска.
Фармацевтическая лекарственная форма в соответствии с изобретением предпочтительно проявляет механическую прочность в широком температурном диапазоне, дополнительно к прочности на разрыв (сопротивление раздавливанию) необязательно также достаточную твердость, сопротивление удару, ударную упругость, прочность при растяжении и/или модуль упругости, необязательно также при низких температурах (например, ниже -24°C, ниже -40°C или в жидком азоте), для ее фактически невозможной пульверизации путем спонтанного жевания, измельчения в ступке, дробления и т.д.. Таким образом, предпочтительно, в направлении растяжения E1 сравнительно высокая прочность на разрыв фармацевтической лекарственной формы в соответствии с изобретением поддерживается даже при низких или очень низких температурах, например, если фармацевтическую лекарственную форму изначально вымораживают для повышения ее ломкости, например, до температур ниже -25°C, ниже -40°C или даже в жидком азоте.
Фармацевтическая лекарственная форма в соответствии с изобретением характеризуется определенной степенью прочности на разрыв. Это не обозначает, что фармацевтическая лекарственная форма также должна проявлять определенную степень твердости. Твердость и прочность на разрыв являются различными физическими свойствами. Следовательно, устойчивость к разрушению фармацевтической лекарственной формы не обязательно зависит от твердости фармацевтической лекарственной формы. Например, благодаря ее прочности на разрыв, прочности при ударной нагрузке, модулю упругости и прочности при растяжении, соответственно, фармацевтическая лекарственная форма предпочтительно может быть деформирована, например, пластично, при приложении внешней силы, например, используя молоток, но не может быть пульвелизирована, то есть раскрошена на большое число фрагментов. Другими словами, фармацевтическая лекарственная форма в соответствии с изобретением характеризуется определенной степенью прочности на разрыв, но необязательно также определенной степенью стабильности формы.
Следовательно, в контексте настоящей заявки, фармацевтическая лекарственная форма, которая деформирована при воздействии силы в конкретном направлении растяжения, но которая не разламывается (пластическая деформация или пластическая текучесть) предпочтительно обозначается как имеющая желательную прочность на разрыв в указанном направлении растяжения.
В предпочтительном варианте осуществления изобретение относится к устойчивой к разрушению фармацевтической лекарственной форме, имеющей профиль замедленного высвобождения, в особенности устойчивой к разрушению пероральной лекарственной форме, имеющей профиль замедленного высвобождения, в особенности устойчивой к разрушению таблетке, имеющей профиль замедленного высвобождения, содержащей по меньшей мере один фармацевтически активный компонент (А) (фармакологически активное соединение) с возможностью злоупотребления.
Фармацевтическая лекарственная форма в соответствии с изобретением может быть получена с помощью различных процессов, особенно предпочтительные из которых более подробно представлены ниже. Некоторые подходящие процессы уже были описаны в уровне техники. В этом отношении можно привести ссылки, например, на WO 2005/ 016313, WO 2005/016314, WO 2005/063214, WO 2005/102286, WO 2006/002883, WO 2006/002884, WO 2006/002886, WO 2006/082097, и WO 2006/082099.
Настоящее изобретение также относится к фармацевтическим лекарственным формам, которые получены с помощью любого из процессов, описанных ниже.
В целом, процесс получения фармацевтической лекарственной формы в соответствии с изобретением предпочтительно включает следующие стадии:
(a) смешивание всех компонентов;
(b) необязательно предварительное формирование смеси, полученной на стадии (а), предпочтительно путем применения тепла и/или силы к смеси, полученной на стадии (а), количества подаваемого тепла предпочтительно не достаточно для нагревания полиалкилен оксида (С) вплоть до его температуры размягчения;
(c) закаливание смеси путем применения тепла и силы, возможно подводить тепло в процессе и/или перед применением силы и количества подаваемого тепла достаточно для нагревания полиалкилен оксида (С) по меньшей мере вплоть до его температуры размягчения;
(d) необязательно разделение отвердевшей смеси;
(e) необязательно придание формы фармацевтической лекарственной формы; и
(f) необязательно обеспечение пленочной оболочкой.
Нагревание может быть осуществлено непосредственно, например, путем контакта или с помощью горячего газа, такого как горячий воздух, или с помощью ультразвука. Можно применять силу и/или фармацевтической лекарственной форме можно придавать форму, например, путем прямого таблетирования или с помощью подходящего экструдера, в особенности с помощью шнекового экструдера, оборудованного двумя шнеками (двухшнековый экструдер) или с помощью планетарного зубчатого экструдера.
Предпочтительно, экструзию горячего расплава осуществляют при отсутствии воды.
Конечное формирование фармацевтической лекарственной формы можно либо осуществлять в процессе закаливания смеси путем применения тепла и силы (стадия (с)) или на последующей стадии (стадия (е)). В обоих случаях, смесь всех компонентов предпочтительно находится в пластифицированном состоянии, то есть предпочтительно, формировку осуществляют при температуре по меньшей мере выше температуры размягчения полиалкилен оксида (С). Тем не менее, также возможна экструзия при более низких температурах, например, температуре окружающей среды и может быть предпочтительной.
Формировку можно осуществлять, например, с помощью таблеточного пресса, содержащего штамп и пуансоны подходящей формы.
Особенно предпочтительный процесс приготовления фармацевтической лекарственной формы изобретения включает экструзию горячего расплава. В этом процессе, фармацевтическую лекарственную форму в соответствии с изобретением получают путем высокотемпературного формообразования с помощью экструдера, предпочтительно без какого-либо наблюдаемого последующего обесцвечивания экструдата.
Этот процесс характеризуется тем, что
a) все компоненты смешивают,
b) полученную смесь нагревают в экструдере по меньшей мере вплоть до температуры размягчения полиалкилен оксида (С) и экструдируют через выпускное отверстие экструдера путем применения силы,
c) еще пластический экструдат разделывают и формируют в фармацевтическую лекарственную форму или
d) охлажденный и необязательно повторно нагретый разделанный экструдат формируют в фармацевтическую лекарственную форму.
Смешивание компонентов в соответствии с процессом на стадии а) также может осуществляться в экструдере.
Компоненты также могут быть смешаны в миксере, известном специалисту в данной области техники. Миксер может представлять собой, например, ротационный миксер, встряхивающий миксер, миксер, обеспечивающий высокие напряжения сдвига, или смеситель принудительного действия.
Предпочтительно расплавленную смесь, которую нагрели в экструдере по меньшей мере вплоть до температуры размягчения полиалкилен оксида (С) экструдировали из экструдера через штамп с по меньшей мере одним отверстием.
Для процесса экструзии в соответствии с изобретением необходимо применение подходящих экструдеров, предпочтительно шнековых экструдеров. Шнековые экструдеры, которые оборудованы двумя шнеками (двухшнековые экструдеры) являются особенно предпочтительными.
Экструзию предпочтительно осуществляют таким образом, что расширение ленты вследствие экструзии составляет не более, чем на 30%, то есть, если используют штамп с отверстием, имеющим диаметр, например, 6 мм, то экструдируемая лента должна иметь диаметр не более, чем 8 мм. Более предпочтительно, расширение ленты происходит не более, чем на 25%, еще более предпочтительно не более, чем на 20%, наиболее предпочтительно не более, чем на 15% и в частности не более, чем на 10%.
Предпочтительно, экструзию осуществляют при отсутствии воды, то есть воду не добавляют. Тем не менее, следы воды (например, вызванные атмосферной влажностью) могут присутствовать.
Экструдер предпочтительно содержит по меньшей мере две температурные зоны, с нагреванием смеси по меньшей мере вплоть до температуры размягчения полиалкилен оксида (С), осуществляемом в первой зоне, которая расположена ниже от питающей зоны и необязательно зону смешивания. Производительность смеси составляет предпочтительно от 1,0 кг до 15 кг/ч. В предпочтительном варианте осуществления, производительность составляет от 1 до 3,5 кг/ч. В другом предпочтительном варианте осуществления, производительность составляет от 4 до 15 кг/ч.
В предпочтительном варианте осуществления, давление экструзионной головки находится в диапазоне от 25 до 100 бар. Давление экструзионной головки можно регулировать, в частности, с помощью геометрии штампа, температурного режима и скорости экструзии.
Геометрия штампа или геометрия отверстий выбирается произвольно. Штамп или отверстия может, следовательно, проявлять круглое, удлиненное или овальное поперечное сечение, где круглое поперечное сечение предпочтительно имеет диаметр от 0,1. мм до 15 мм и удлиненное поперечное сечение предпочтительно имеет максимальную протяженность в продольном направлении 21 мм и протяженность в поперечном направлении 10 мм. Предпочтительно, штамп или отверстия имеют круглое поперечное сечение. Корпус используемого экструдера в соответствии с изобретением может быть нагретым или охлажденным. Соответствующий температурный контроль, то есть нагревание или охлаждение, регулируют таким образом, что экструдируемая смесь проявляет по меньшей мере среднюю температуру (температуру продукта), которая соответствует температуре размягчения полиалкилен оксида (С) и не повышается выше температуры, при которой фармакологически активный компонент (А), подлежащей обработке, может быть поврежден. Предпочтительно, температуру экструдируемой смеси регулируют ниже 180°C, предпочтительно ниже 150°C, но по меньшей мере до температуры размягчения полиалкилен оксида (С). Типичные температуры экструзии составляют 120°C и 130°C.
В предпочтительном варианте осуществления, крутящийся момент экструдера находится в диапазоне от 30 до 95%. Крутящийся момент экструдера можно регулировать, в частности, с помощью геометрии штампа, температурного режима и скорости экструзии.
После экструзии расплавленной смеси и необязательного охлаждения экструдированной ленты или экструдированных лент, экструдаты предпочтительно разделяют. Это разделение предпочтительно можно осуществить путем разрезания экструдатов с помощью поворотных или вращающихся ножей, станков гидроабразивной резки, проволоки, лезвий или с помощью лазерных резаков.
Предпочтительно, промежуточное или конечное хранение необязательно разделенного экструдата или конечное формирование фармацевтической лекарственной формы в соответствии с изобретением осуществляют в бескислородной атмосфере, которую можно получить, например, с помощью поглотителей кислорода.
Разделенный экструдат может быть пресс-формирирован в таблетки для придания конечной формы фармацевтической лекарственной форме.
Применение силы в экструдере на по меньшей мере пластифицированную смесь регулируют путем контролирования частоты вращения транспортного приспособления в экструдере и его геометрии и задания размеров выпускного отверстия таким образом, что давление, необходимое для экструдирования пластифицированной смеси, создается в экструдере, предпочтительно непосредственно перед экструзией. Параметры экструзии, которые, для каждой конкретной композиции, необходимы для получения фармацевтической лекарственной формы с желательными механическими свойствами, могут быть установлены путем простого предварительного тестирования.
Например, но не ограничиваясь, экструзию можно осуществить с помощью двухшнекового экструдера типа ZSE 18 или ZSE27 (Leistritz, Nürnberg, Germany), диаметры шнеков 18 или 27 мм. Можно использовать шнеки, имеющие эксцентричные концы. Можно использовать нагреваемый штамп с круглым отверстием, имеющим диаметр 7, 8 или 9 мм. Параметры экструзию можно доводить, например, до следующих значений: частота вращения шнеков: 120 Upm; скорость доставки 2 кг/ч для ZSE 18 или 8 кг/ч для ZSE27; температуру продукта: спереди штампа 125°C и на задней части штампа 135°C; и температура оболочки: 110°C.
Предпочтительно, экструзию осуществляют с помощью двухшнековых экструдеров или планетарных зубчатых экструдеров, особенно предпочтительными являются двухшнековые экструдеры (вращающиеся в одном или в противоположных направлениях).
Фармацевтическая лекарственная форма в соответствии с изобретением предпочтительно получена путем высокотемпературного формообразования с помощью экструдера без какого-либо наблюдаемого последующего обесцвечивания экструдатов.
Способ приготовления фармацевтической лекарственной формы в соответствии с изобретением предпочтительно осуществляют непрерывно. Предпочтительно, способ включает экструзию гомогенной смеси всех компонентов. Это является особенно благоприятным, если таким образом полученная промежуточная, например, лента, полученная путем экструзии, проявляет однородные свойства. Особенно предпочтительными являются однородная плотность, однородное распределение активного компонента, однородные механические свойства, однородная пористость, однородный внешний вид поверхности, и др. Только при таких обстоятельствах можно обеспечить однородность фармацевтических свойств, таких как стабильность профиля высвобождения, и количество брака можно поддерживать на низком уровне.
Дальнейший аспект изобретения относится к применению фармакологически активного компонента (А) для приготовления фармацевтической лекарственной формы, как описано выше, для лечения боли.
Дальнейший аспект изобретения относится к применению фармацевтической лекарственной формы, как описано выше, для избегания или затруднений злоупотребления фармакологически активного компонента (А), который в ней содержатся.
Дальнейший аспект изобретения относится к применению фармацевтической лекарственной формы, как описано выше, для избегания или препятствования непреднамеренной передозировке фармакологически активного компонента (А), который в ней содержатся.
В этом отношении, изобретение также относится к применению фармакологически активного компонента (А), как описано выше, и/или полиалкилен оксида (С), как описано выше, для приготовления фармацевтической лекарственной формы в соответствии с изобретением для профилактики и/или лечения нарушения, таким образом предотвращая передозировку фармакологически активного компонента (А), в частности благодаря раздроблению фармацевтической лекарственной формы путем механического воздействия.
Кроме того, изобретение относится к способу профилактики и/или лечения нарушения, который включает введение фармацевтической лекарственной формы в соответствии с изобретением, таким образом предотвращая передозировку фармакологически активного компонента (А), в частности благодаря раздроблению фармацевтической лекарственной формы путем механического воздействия. Предпочтительно, механическое воздействие выбирают из группы, включающей жевание, измельчение в ступке, дробление, и используя приборы для пульверизации общепринятых фармацевтических лекарственных форм.
Последующие примеры дополнительно иллюстрируют изобретение, но не должны рассматриваться как ограничивающие его объем:
Во всех примерах лекарственные формы представляли собой таблетки с предположительно круглой формой с диаметром 12 мм.
Общая процедура
Полиэтилен оксид, α-токоферол, гидрохлорид трамадола и все другие наполнители взвешивали и просеивали друг с другом. Порошок смешивали и дозировали гравиметрически на экструдер. Экструзию горячего расплава осуществляли с помощью двушнекового экструдера типа Micro 27 GL 40 D (Leistritz, Nürnberg, Germany), который оборудован способным к нагреванию круглым штампом, имеющим диаметр 10 мм.
Горячий экструдат охлаждали на ленте транспортера и охлажденную экструзионную ленту измельчали, нарезая на куски. Нарезанным кускам придавали форму с помощью эксцентричного пресса.
Прочность на разрыв фармацевтических лекарственных форм измеряли с помощью Sotax® НТ100 при постоянной скорости 120 мм/мин и/или Zwick Z 2.5 при постоянной скорости 10 мм/мин. Таблетку обозначали как такую, которая не прошла тест прочности на разрыв, если при измерении сила падала ниже пороговое значение 25% максимальной силы, которую наблюдали при осуществлении измерения, независимо от того, распадалась ли лекарственная форма на отдельные куски или нет. Все значения представлены в виде среднего 3 измерений (Zwick; n=3) или в виде среднего 10 измерений (Sotax, n=10).
Пример I:
Формировали таблетки следующего состава, содержащие трамадол:
Для каждой композиции, измеряли профиль высвобождения in vitro фармакологически активного компонента в 600 мл искусственного желудочного сока (pH 6,8, фосфатный буфер) при температуре 37°C с грузом (тип 4). Скорость вращения лопасти доводили до 75/мин. Фармакологически активный компонент определяли путем спектрометрического измерения при длине волны 271 нм.
В соответствии с вышеприведенной таблицей, вариант D и Е тестировали с карбонатом натрия. Кривые растворения таблеток с 30 мас.% и 40 мас.% карбонатом натрия по сравнению с эталонными таблетками проиллюстрированы на Фигуре 1. Замедленный эффект более выражен для препарата с 30 мас.% карбонатом натрия.
Вариант F и G тестировали с карбонатом натрия и пятизамещенном трифосфатом натрия. Кривые растворения таблеток с 20 мас.% и 15 мас.% каждого карбоната натрия и пятизамещенного трифосфата натрия по сравнению с эталоном проиллюстрированы на Фигуре 2. И в этом случае, профиль высвобождения проявляет существенное замедление и линейное высвобождение фармакологически активного компонента. Профиль высвобождения с более низким содержанием общих солей снова проявляет наилучшие результаты.
Сравнение растворения в кислой среде с кривыми растворения таблеток с 30 мас.% карбоната натрия и 15 мас.% каждого карбоната натрия и пятизамещенного трифосфата натрия по сравнению с эталонными таблетками проиллюстрировано на Фигуре 3. Профиль высвобождения 4 кривых сопоставим и, следовательно, не зависит от значения pH.
Пример II:
Формировали таблетки следующего состава, содержащие оксиморфон:
Профиль высвобождения in vitro фармакологически активного компонента измеряли в 900 мл кислой среды (pH 1,2) и в 900 мл среды, забуференной ацетатом (pH 4,5), оба при температуре 37°C с грузом (тип 4). Скорость вращения лопасти доводили до 50/мин. Фармакологически активный компонент определяли путем спектрометрического измерения при длине волны 271 нм.
В соответствии с вышеприведенной таблицей, препарат готовили с оксиморфоном вместо трамадола. Как показано на Фигуре 4, профиль высвобождения проявляет замедленное и линейное высвобождение фармакологически активного компонента. В кислой среде высвобождение ускоряется по сравнению с высвобождение в среде, забуференной ацетатом (pH 4,5).
Пример III:
Готовили таблетки следующего состава:
В соответствии с Примером II, измеряли профиль высвобождения in vitro фармакологически активного компонента в 900 мл кислой среды (pH 1,2) и в 900 мл среды, забуференной ацетатом (pH 4,5).
В соответствии с вышеприведенной таблицей препарат готовили с оксиморфоном вместо трамадола. Кроме того, препарат содержит карбонат натрия и пятизамещенный трифосфат натрия каждого 15 мас.%. Как показано на Фигуре 5, профиль высвобождения проявляет замедленное и линейное высвобождение фармакологически активного компонента. В кислой среде высвобождение ускоряется по сравнению с высвобождением в среде, забуференной ацетатом (pH 4,5).
| название | год | авторы | номер документа |
|---|---|---|---|
| УСТОЙЧИВАЯ К РАЗРУШЕНИЮ ЛЕКАРСТВЕННАЯ ФОРМА, КОТОРАЯ СОДЕРЖИТ АНИОННЫЙ ПОЛИМЕР | 2011 |
|
RU2607499C2 |
| ЛЕКАРСТВЕННАЯ ФОРМА | 2009 |
|
RU2493830C2 |
| ЗАЩИЩЕННАЯ ОТ ПРИМЕНЕНИЯ НЕ ПО НАЗНАЧЕНИЮ ЛЕКАРСТВЕННАЯ ФОРМА ДЛЯ ЧУВСТВИТЕЛЬНЫХ К ОКИСЛЕНИЮ ОПИОИДОВ | 2010 |
|
RU2555531C2 |
| СТАБИЛЬНАЯ ПРИ ОКИСЛЕНИИ, ПРОЧНАЯ НА ИЗЛОМ ЛЕКАРСТВЕННАЯ ФОРМА | 2010 |
|
RU2567723C2 |
| ЭКСТРУДИРОВАННАЯ ИЗ ГОРЯЧЕГО РАСПЛАВА ФАРМАЦЕВТИЧЕСКАЯ ЛЕКАРСТВЕННАЯ ФОРМА | 2010 |
|
RU2547555C2 |
| Пероральная твердая лекарственная форма с микофенольной кислотой или ее солью для использования в качестве иммунодепрессанта для лечения или предупреждения отторжения трансплантата органа или ткани и способ ее получения | 2016 |
|
RU2670447C2 |
| ФАРМАЦЕВТИЧЕСКИЙ СОСТАВ | 2018 |
|
RU2809144C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИМАТИНИБ И ЗАМЕДЛИТЕЛЬ ВЫСВОБОЖДЕНИЯ | 2006 |
|
RU2404775C2 |
| Экструдат с микофенолятом натрия для получения пероральной твердой лекарственной формы | 2018 |
|
RU2723255C2 |
| ЛЕКАРСТВЕННАЯ ДОЗИРОВАННАЯ ФОРМА, КОТОРАЯ СОДЕРЖИТ 6'-ФТОР-(N-МЕТИЛ- ИЛИ N, N-ДИМЕТИЛ-)-4-ФЕНИЛ-4', 9'-ДИГИДРО-3'Н-СПИРО[ЦИКЛОГЕКСАН-1, 1'-ПИРАНО[3, 4, b]ИНДОЛ]-4-АМИН | 2011 |
|
RU2589831C2 |

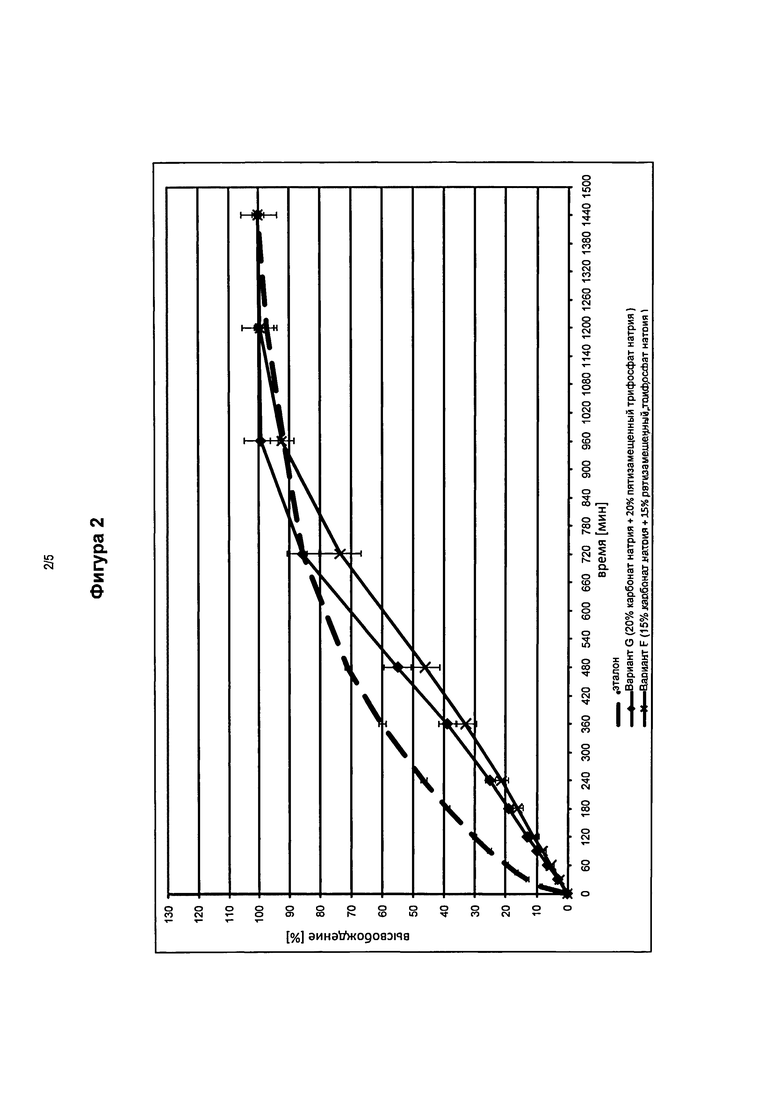
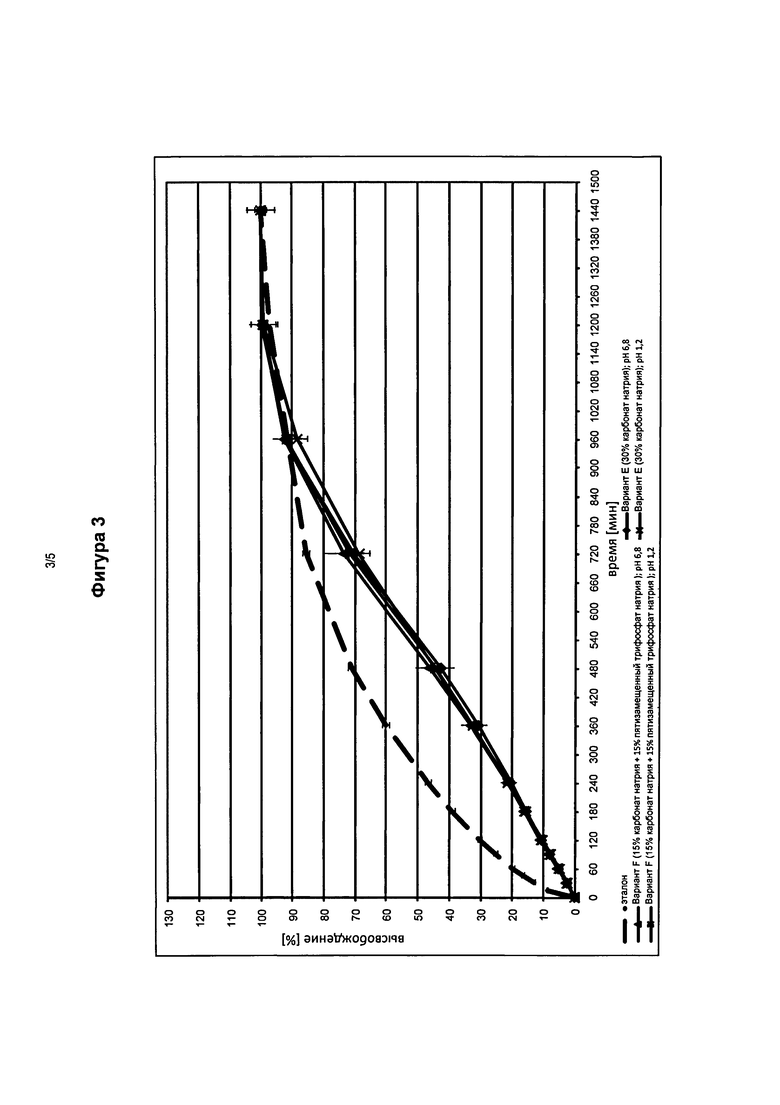
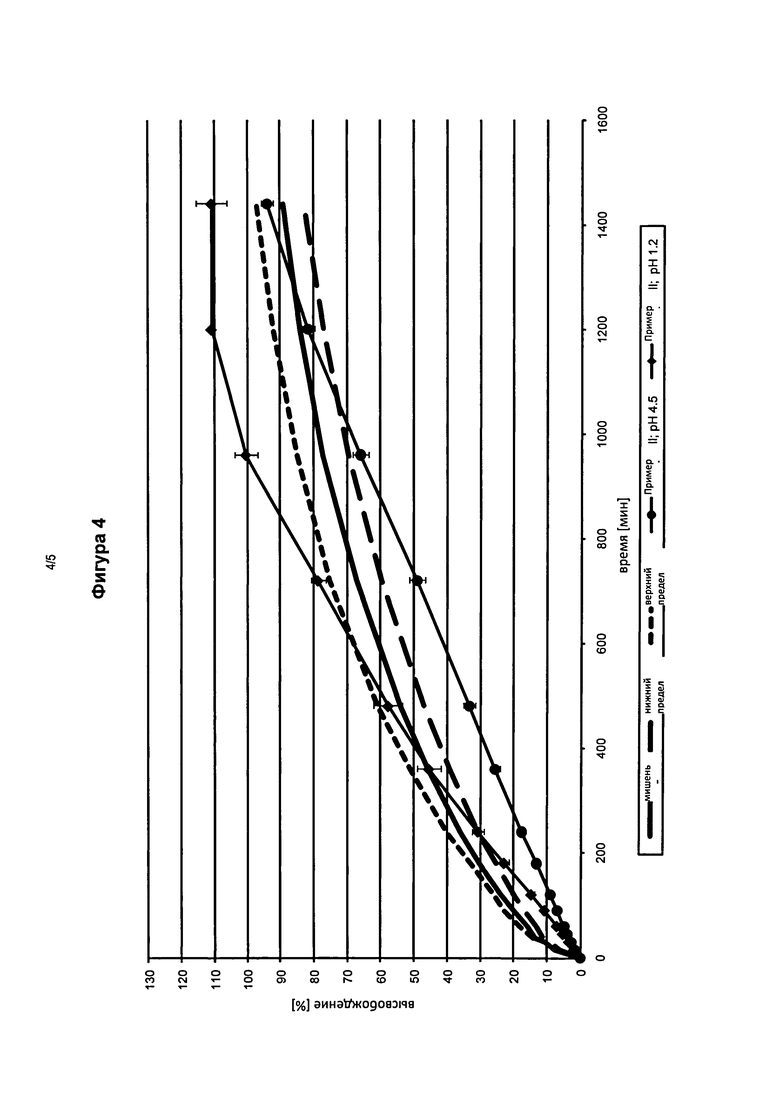
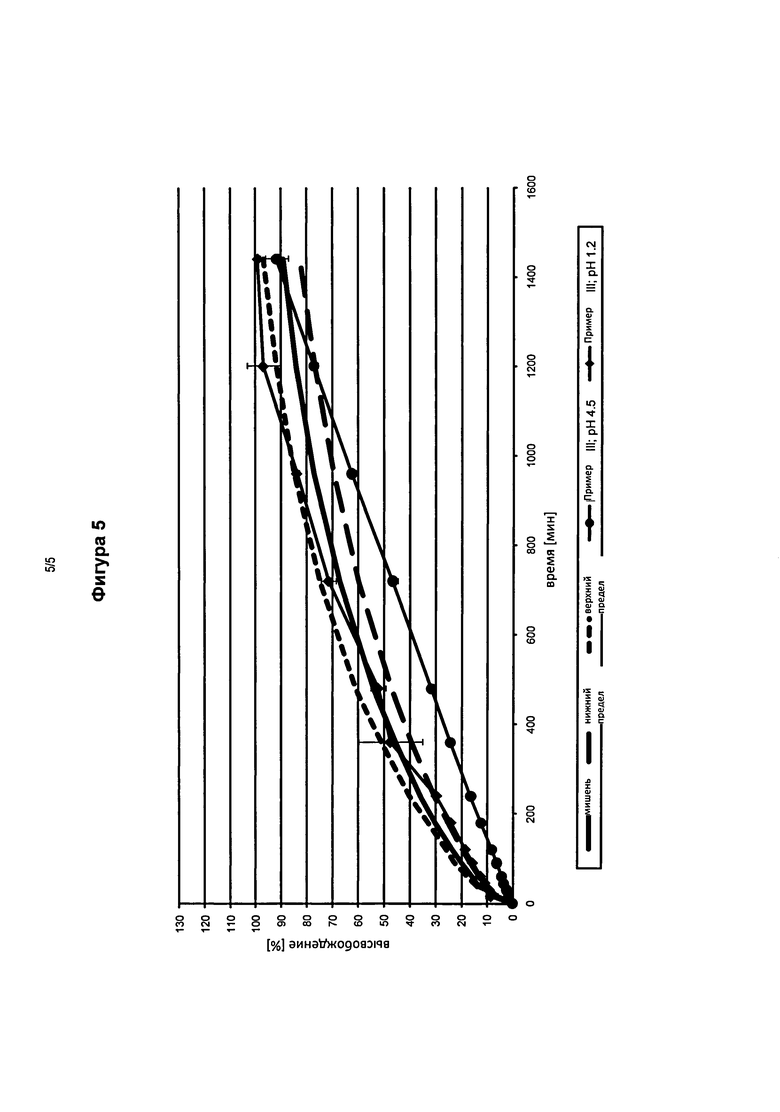
Изобретение относится к фармацевтике, а именно к фармацевтической лекарственной форме, проявляющей прочность на разрыв по меньшей мере 500 Н, указанная лекарственная форма содержит фармакологически активный компонент (А); неорганическую соль (В), где неорганическая соль (В) представляет собой карбонат натрия или пятизамещенный трифосфат натрия или их смесь, и содержание неорганической соли (В) составляет от 15 до 40 мас. %, исходя из общей массы лекарственной формы; и полиалкилен оксид (С), имеющий средневесовую молекулярную массу по меньшей мере 200,000 г/моль, где содержание полиалкиленоксида (С) составляет по меньшей мере 30 мас. %, исходя из общей массы лекарственной формы; где фармакологически активный компонент (А) присутствует в матриксе с контролируемым высвобождением, содержащим неорганическую соль (В) и полиалкилен оксид (С) и где, в условиях in vitro, профиль высвобождения фармакологически активного компонента (А) из указанного матрикса включает по меньшей мере временной интервал, в течение которого высвобождение соответствует кинетике нулевого порядка. Заявленная фармацевтическая лекарственная форма пригодна для избегания злоупотребления фармакологически активного компонента, который в ней содержится. 9 з.п. ф-лы, 3 пр., 5 ил.
1. Дозированная лекарственная форма, которая представляет собой таблетку, проявляющая прочность на разрыв по меньшей мере 500 Н и содержащая
- фармакологически активный компонент (А);
- неорганическую соль (В), где неорганическая соль (В) представляет собой карбонат натрия или пятизамещенный трифосфат натрия или их смесь, и содержание неорганической соли (В) составляет от 15 до 40 мас. %, исходя из общей массы лекарственной формы;
- полиалкиленоксид (С), имеющий средневесовую молекулярную массу по меньшей мере 200,000 г/моль, где содержание полиалкиленоксида (С) составляет по меньшей мере 30 мас. %, исходя из общей массы лекарственной формы;
где фармакологически активный компонент (А) заделан в матрикс с контролируемым высвобождением, содержащий неорганическую соль (В) и полиалкиленоксид (С), и
где, в условиях in vitro, профиль высвобождения фармакологически активного компонента (А) из указанного матрикса включает по меньшей мере временной интервал, в течение которого высвобождение соответствует кинетике нулевого порядка.
2. Дозированная лекарственная форма в соответствии с п. 1, где временной интервал, в течение которого высвобождение соответствует кинетике нулевого порядка, составляет по меньшей мере 20% общего времени высвобождения, необходимого для высвобождения 95 мас. % фармакологически активного компонента (А), который изначально присутствует.
3. Дозированная лекарственная форма в соответствии с п. 1 или 2, где профиль высвобождения соответствует кинетике нулевого порядка в диапазоне от рН 1 до рН 7.
4. Дозированная лекарственная форма в соответствии с пунктом 1 или 2, которую получают методом экструзии горячего расплава.
5. Дозированная лекарственная форма в соответствии с п. 1 или 2, где фармакологически активный компонент (А) представляет собой опиоид, выбранный из группы, включающей тапентадол, оксиморфон, гидроксиморфон, оксикодон, морфин и их физиологически приемлемые соли.
6. Дозированная лекарственная форма в соответствии с п. 1 или 2, где полиалкиленоксид (С) имеет молекулярную массу по меньшей мере 0,5 миллион г/моль.
7. Дозированная лекарственная форма в соответствии с п. 6, где полиалкиленоксид (С) имеет молекулярную массу по меньшей мере 1 миллион г/моль.
8. Дозированная лекарственная форма в соответствии с пунктом 7, где полиалкиленоксид (С) имеет молекулярную массу в диапазоне от 1 до 15 миллион г/моль.
9. Дозированная лекарственная форма в соответствии с п. 1 или 2, которая содержит полиалкиленгликоль.
10. Дозированная лекарственная форма в соответствии с п. 9, где полиалкиленгликоль имеет молекулярную массу по меньшей мере 1000 г/моль.
| WO2010057036, 20.05.2010;US 2006/0002860 A1, 05.01.2006;WO03024426, 27.03.2003 | |||
| Алексеева И.В | |||
| и др | |||
| / Биофармацевтические исследования биорастворимых лекарственных пленок с анилокаином / Химико-фармацевтический журнал | |||
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| Механический грохот | 1922 |
|
SU41A1 |
| Разборный с внутренней печью кипятильник | 1922 |
|
SU9A1 |
| Способ смешанной растительной и животной проклейки бумаги | 1922 |
|
SU49A1 |
| Saleem M.A | |||
| et al | |||
| / Formulation and Evaluation of Tramadol hydrochloride Rectal | |||

