ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
1. Область техники
Настоящее изобретение относится к новому нанесенному на носитель катализатору, к новому фосфазеновому соединению и к применению катализатора. Говоря более конкретно, изобретение относится к фосфазеновому нанесенному на носитель катализатору, в котором носитель образует связь с группой, описывающейся общей формулой (1); новому фосфазеновому соединению и новой фосфазениевой соли, которые являются подходящими для использования при получении нанесенного на носитель катализатора; и способу полимеризации циклического мономера, способу замещения заместителя и способу проведения реакции с получением связи углерод-углерод при использовании нанесенного на носитель катализатора.
2. Уровень техники
Фосфазениевая соль, описывающаяся следующей далее общей формулой (9):
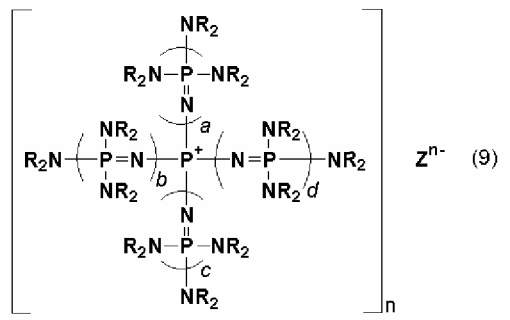
(где n является целым числом в диапазоне от 1 до 8 и представляет собой количество фосфазениевых катионов, а Zn- представляет собой анион n-валентного соединения, содержащего атомы активного водорода, в форме, полученной в результате высвобождения n протонов из соединения, содержащего атомы активного водорода, в котором имеется, самое большее, 8 атомов активного водорода на атоме кислорода или атоме азота, a, b, c и d представляют собой положительное целое число, равное 3 или менее или 0 соответственно при том условии, что они все не равны 0 одновременно, R представляют собой одинаковые или различные углеводородные группы, содержащие от 1 до 10 атомов углерода, и два R, расположенные на каждом общем атоме азота, могут быть связаны друг с другом с образованием кольцевой структуры), известна как соединение, которое способствует протеканию различных каталитических реакций благодаря образованию стабильных катионов и подбору противоионов (смотрите документ JP-A № 10-77289). Такое соединение является эффективным при проведении различных каталитических реакций, но его относительно трудно получить, оно дорого стоит, и, таким образом, желательным является повторное использование катализатора. В то же самое время известно то, что фосфазеновое соединение, имеющее центр связывания (так называемое фосфазеновое основание), образует связь с функциональной группой носителя и получается в форме, нанесенной на носитель, а также известно то, что такой нанесенный на носитель катализатор используют для полимеризации алкиленоксида (смотрите публикации международной патентной заявки № WO01/90220).
Описанный выше нанесенный на носитель катализатор является подходящим для использования, но в нем сшитый органический полимерный носитель образует связь непосредственно или опосредованно с атомом азота, связанным с атомом фосфора в центре. Это приводит к возникновению проблемы, заключающейся в том, что стабильность катиона невысока с точки зрения его химической структуры, и поэтому, вероятно, он будет разлагаться. Катион фосфазениевой соли, имеющей скелет, описывающийся общей формулой (9), характеризуется высокой стабильностью, проявляющейся в его химическом строении. Однако по обычно используемому способу было невозможно фосфазениевую соль, имеющую скелет, описывающийся общей формулой (9), нанести на носитель, чтобы, тем самым, получить такой нанесенный на носитель катализатор. Кроме того, совершенно было неизвестно введение центра связывания в фосфазениевую соль, описывающуюся общей формулой (9), а также было неизвестно эффективное нанесение фосфазениевой соли, описывающейся общей формулой (9), на носитель в результате получения и использования фосфазениевой соли, содержащей центр связывания, при сохранении ее эксплуатационных характеристик как таковой.
Поэтому возникает необходимость получить нанесенный на носитель катализатор, обладающий функцией фосфазениевой соли, описывающейся общей формулой (9), как таковой, и, таким образом, желательно разработать такой нанесенный на носитель катализатор.
КРАТКОЕ ИЗЛОЖЕНИЕ ИЗОБРЕТЕНИЯ
Изобретатели настоящего изобретения провели интенсивные исследования катализатора с целью решения вышеупомянутой задачи и в результате обнаружили то, что для получения нанесенного на носитель катализатора, имеющего скелет, описывающийся общей формулой (9), и, таким образом, для решения вышеупомянутой задачи может быть использован специфический продукт частичной трансформации структуры, полученный из фосфазениевой соли, описывающейся общей формулой (9), и, таким образом, создали данное изобретение во всей его полноте.
Говоря конкретно, изобретение относится к фосфазеновому нанесенному на носитель катализатору, в котором носитель образует связь с группой, описывающейся общей формулой (1):
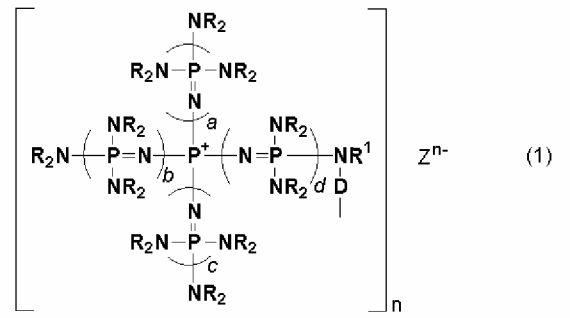
(где n является целым числом в диапазоне от 1 до 8 и представляет собой количество фосфазениевых катионов, а Zn- представляет собой анион соединения, содержащего атомы активного водорода, в форме, полученной в результате высвобождения n протонов из соединения, содержащего атомы активного водорода, в котором имеется, самое большее, 8 атомов активного водорода. Каждый из a, b, c и d представляет собой положительное целое число, равное 3 или менее. R представляют собой одинаковые или различные углеводородные группы, содержащие от 1 до 10 атомов углерода, и два R, расположенные на каждом общем атоме азота, могут быть связаны друг с другом с образованием кольцевой структуры. R1 представляет собой атом водорода или углеводородную группу, содержащую от 1 до 10 атомов углерода. D представляет собой непосредственную связь или двухвалентную группу, способную связывать N с носителем).
Кроме того, изобретение относится к новому фосфазеновому соединению, описывающемуся общей формулой (2):
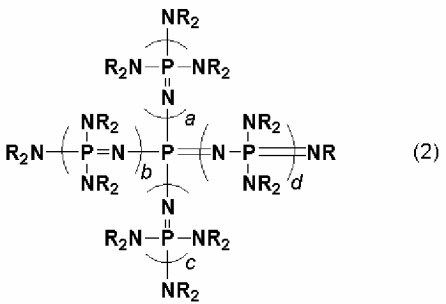
(где a, b, c и d представляют собой положительное целое число, равное 3 или менее соответственно. R представляют собой одинаковые или различные углеводородные группы, содержащие от 1 до 10 атомов углерода, и два R, расположенные на каждом общем атоме азота, могут быть связаны друг с другом с образованием кольцевой структуры);
новому фосфазеновому соединению, описывающемуся общей формулой (3):
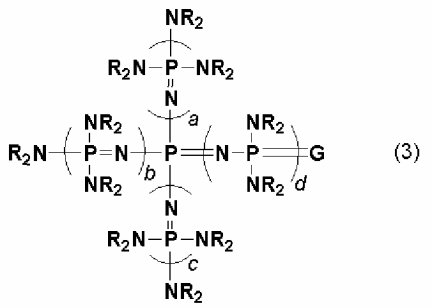
(где каждый из a, b, c и d представляет собой положительное целое число, равное 3 или менее. G представляет собой атом кислорода или атом серы. R представляют собой одинаковые или различные углеводородные группы, содержащие от 1 до 10 атомов углерода, и два R, расположенные на каждом общем атоме азота, могут быть связаны друг с другом с образованием кольцевой структуры);
новой фосфазениевой соли, описывающейся общей формулой (4):
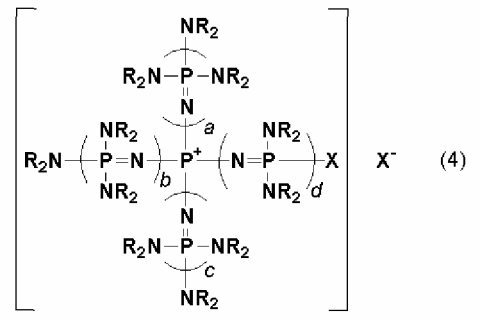
(где каждый из a, b, c и d представляет собой положительное целое число, равное 3 или менее соответственно. R представляют собой одинаковые или различные углеводородные группы, содержащие от 1 до 10 атомов углерода, и два R, расположенные на каждом общем атоме азота, могут быть связаны друг с другом с образованием кольцевой структуры. Х представляет собой атом галогена, а Х- представляет собой анион атома галогена, который может быть идентичным или отличным в сопоставлении с Х); и
новой фосфазениевой соли, описывающейся общей формулой (5):
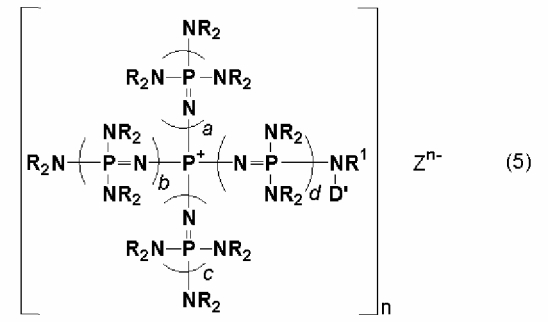
(где n является целым числом в диапазоне от 1 до 8 и представляет собой количество фосфазениевых катионов, а Zn- представляет собой анион соединения, содержащего атомы активного водорода, в форме, полученной в результате высвобождения n протонов из соединения, содержащего атомы активного водорода, в котором имеется, самое большее, 8 атомов активного водорода. Каждый из a, b, c и d представляет собой положительное целое число, равное 3 или менее. R представляют собой одинаковые или различные углеводородные группы, содержащие от 1 до 10 атомов углерода, и два R, расположенные на каждом общем атоме азота, могут быть связаны друг с другом с образованием кольцевой структуры. R1 представляет собой атом водорода или углеводородную группу, содержащую от 1 до 10 атомов углерода. D' представляет собой одновалентную группу, которая образует связь с N, при том условии, что она отличается от атома водорода и насыщенной углеводородной группы), которые являются подходящими для использования при получении катализатора, нанесенного на носитель.
Изобретение также относится к новой фосфазениевой соли, описывающейся общей формулой (6):
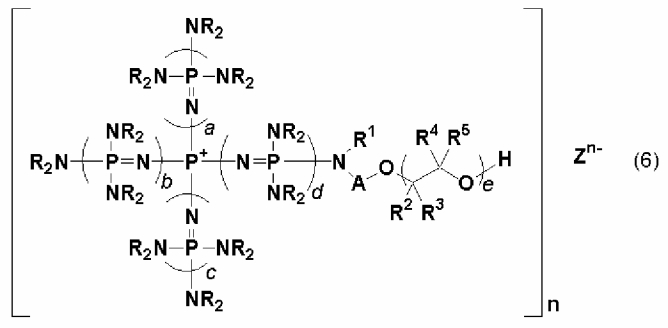
(где n является целым числом в диапазоне от 1 до 8 и представляет собой количество фосфазениевых катионов, а Zn- представляет собой анион соединения, содержащего атомы активного водорода, в форме, полученной в результате высвобождения n протонов из соединения, содержащего атомы активного водорода, в котором имеется, самое большее, 8 атомов активного водорода. Каждый из a, b, c и d представляет собой положительное целое число, равное 3 или менее. R представляют собой одинаковые или различные углеводородные группы, содержащие от 1 до 10 атомов углерода, и два R, расположенные на каждом общем атоме азота, могут быть связаны друг с другом с образованием кольцевой структуры. А представляет собой углеводородную группу, содержащую от 1 до 20 атомов углерода. Кроме того, R1 представляет собой атом водорода или углеводородную группу, содержащую от 1 до 10 атомов углерода. Каждый из R2, R3, R4 и R5 представляет собой атом водорода или углеводородную группу содержащую от 1 до 8 атомов углерода, е находится в диапазоне от 0 до 200), в качестве одного предпочтительного варианта реализации фосфазениевой соли, описывающейся общей формулой (5);
новой фосфазениевой соли, описывающейся общей формулой (7):
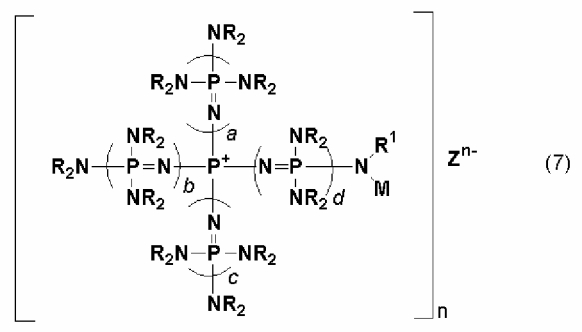
(где n является целым числом в диапазоне от 1 до 8 и представляет собой количество фосфазениевых катионов, а Zn- представляет собой анион соединения, содержащего атомы активного водорода, в форме, полученной в результате высвобождения n протонов из соединения, содержащего атомы активного водорода, в котором имеется, самое большее, 8 атомов активного водорода. Каждый из a, b, c и d представляет собой положительное целое число, равное 3 или менее. R представляют собой одинаковые или различные углеводородные группы, содержащие от 1 до 10 атомов углерода, и два R, расположенные на каждом общем атоме азота, могут быть связаны друг с другом с образованием кольцевой структуры. R1 представляет собой атом водорода или углеводородную группу, содержащую от 1 до 10 атомов углерода. М представляет собой группу, содержащую ненасыщенную связь углерод-углерод), в качестве еще одного предпочтительного варианта реализации фосфазениевой соли, описывающейся общей формулой (5); и
новой фосфазениевой соли, описывающейся общей формулой (8):
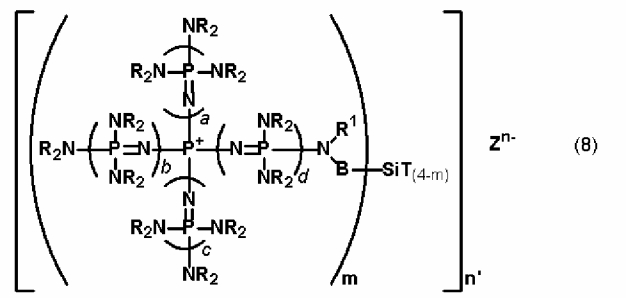
(где m является целым числом в диапазоне от 1 до 3 и представляет собой количество фосфазениевых катионов, связанных с атомом кремния, n' является целым числом в диапазоне от 1 до 8 и представляет собой количество соединений кремния, с которыми фосфазениевые катионы образуют связь, n представляет собой множитель для m и n', а Zn- представляет собой анион соединения, содержащего атомы активного водорода, в форме, полученной в результате высвобождения n протонов из соединения, содержащего атомы активного водорода, в котором имеется, самое большее, 24 атома активного водорода. Каждый из a, b, c и d представляет собой положительное целое число, равное 3 или менее. R представляют собой одинаковые или различные углеводородные группы, содержащие от 1 до 10 атомов углерода, и два R, расположенные на каждом общем атоме азота, могут быть связаны друг с другом с образованием кольцевой структуры. В представляет собой углеводородную группу, содержащую от 1 до 20 атомов углерода. Кроме того, R1 представляет собой атом водорода или углеводородную группу, содержащую от 1 до 10 атомов углерода. Т представляет собой функциональную группу, в которой связь Si-T может быть разрушена в результате гидролиза), в качестве предпочтительного варианта реализации фосфазениевой соли, описывающейся общей формулой (5).
Изобретение также относится к способу полимеризации циклического мономера, в котором используют описанный выше нанесенный на носитель катализатор, способу замещения заместителя, в котором используют описанный выше нанесенный на носитель катализатор, и способу проведения реакции, в котором описанный выше нанесенный на носитель катализатор используют в реакциях с образованием связи углерод-углерод.
Фосфазеновый нанесенный на носитель катализатор, соответствующий настоящему изобретению, является высокоэффективным при проведении различных органических реакций и, кроме того, не демонстрирует уменьшения активности даже после извлечения и повторного использования катализатора, и, таким образом, он может быть эффективно использован повторно, и он также является экономически выгодным. Кроме того, фосфазеновое соединение и фосфазениевая соль изобретения не только являются промежуточными соединениями, которые легко могут обеспечить получение описанного выше нанесенного на носитель катализатора изобретения, но также являются и катализаторами, которые сами по себе являются подходящими для использования при проведении различных органических реакций. В дополнение к этому, в соответствии со способом изобретения полимеризацию циклических мономеров, замещение заместителей, реакции с образованием связи углерод-углерод и тому подобное можно проводить с чрезвычайно высокой эффективностью.
ОПИСАНИЕ ВАРИАНТОВ РЕАЛИЗАЦИИ
Далее в настоящем документе изобретение будет описываться подробно.
В общих формулах от (1) до (8) R представляют собой одинаковые или различные углеводородные группы, содержащие от 1 до 10 атомов углерода. На углеводородную группу, представляемую символом R, особенных ограничений не накладывается, и она может представлять собой алифатическую углеводородную группу или ароматическую углеводородную группу. Алифатическая углеводородная группа включает, например, алкильную группу, содержащую от 1 до 10 атомов углерода, такую как метильная, этильная и пропильная; алкенильную группу, содержащую от 2 до 10 атомов углерода, такую как винильная и аллильная; алкинильную группу, содержащую от 2 до 10 атомов углерода, такую как этинильная и пропинильная, а ароматическая углеводородная группа включает, например, арильную группу, содержащую от 6 до 10 атомов углерода, такую как фенильная и нафтильная; и аралкильную группу, содержащую от 7 до 10 атомов углерода, такую как бензильная и фенэтильная. R предпочтительно представляет собой алифатическую углеводородную группу, а более предпочтительно метильную группу и этильную группу.
В общих формулах от (1) до (8) два R, расположенные на каждом общем атоме азота, могут быть связаны друг с другом с образованием кольцевой структуры. Группа, полученная в результате объединения друг с другом двух R, расположенных на каждом общем атоме азота, включает, например, алкиленовую группу, содержащую от 2 до 10 атомов углерода, такую как этиленовая, тетраметиленовая и пентаметиленовая; циклоалкиленовую группу, содержащую от 3 до 10 атомов углерода, такую как циклогексиленовая; алкениленовую группу, содержащую от 2 до 10 атомов углерода, такую как виниленовая; циклоалкениленовую группу, содержащую от 3 до 10 атомов углерода, такую как циклогексениленовая; ариленовую группу, содержащую от 6 до 20 атомов углерода, такую как фениленовая и нафтиленовая; и аралкиленовую группу, содержащую от 8 до 20 атомов углерода, такую как фенилэтиленовая. В их числе предпочтительными являются тетраметиленовая и пентаметиленовая. Такую кольцевую структуру может образовывать часть или все количество каждых общих атомов азота, с которыми два атома R образуют связь.
В общих формулах от (1) до (8) каждый из а, b, c и d представляет собой положительное целое число, равное 3 или менее. Предпочтительно они представляют собой положительное целое число, равное 2 или менее, а предпочтительная комбинация a, b, c и d включает (2, 1, 1, 1) и (1, 1, 1, 1) вне зависимости от порядка а, b, c и d, а в особенности предпочтительная комбинация представляет собой (1, 1, 1, 1).
В общих формулах (1), (5), (6), (7) и (8) R1 представляет собой атом водорода или углеводородную группу, содержащую от 1 до 10 атомов углерода. На углеводородную группу, представляемую символом R1, особенных ограничений не накладывается, и она может являться алифатической углеводородной группой или ароматической углеводородной группой. Алифатическая углеводородная группа и ароматическая углеводородная группа включают те же самые специфические примеры, что и перечисленные выше для R из общих формул от (1) до (8). R1 предпочтительно представляет собой алифатическую углеводородную группу, а более предпочтительно метильную группу или этильную группу.
В общей формуле (1) D представляет собой непосредственную связь или двухвалентную группу, способную образовывать связь между N и носителем. Двухвалентная группа, представляемая символом D, может образовывать связь между атомом азота в фосфазениевом катионе и носителем, и на нее особенных ограничений не накладывается до тех пор, пока она не начнет препятствовать достижению цели изобретения.
Как описывалось выше, D может быть любым D до тех пор, пока он не начнет препятствовать достижению цели изобретения, и он может образовывать связь с атомом азота фосфазениевых катионов через атом углерода или образовывать связь с атомом азота фосфазениевых катионов через гетероатом. Однако с точки зрения прочности связи предпочитается, чтобы он образовывал бы связь с атомом азота фосфазениевых катионов через атом углерода.
На расстояние между атомом азота, присутствующим в фосфазениевых катионах, и носителем особенных ограничений не накладывается, что понятно, исходя из духа изобретения. Однако количество атомов, составляющих основную цепь D, зависит от размера носителя, но в общем случае оно находится в диапазоне приблизительно от 1 до 600, а с точки зрения увеличения концентрации катализатора в нанесенном на носитель катализаторе оно предпочтительно находится в диапазоне от 1 до 300, а более предпочтительно от 1 до 100.
В дополнение к этому, с точки зрения получения предпочитается, чтобы фосфазеновые соединения (2) и (3), фосфазениевые соли (4), (5), (6), (7) и (8), которые являются подходящими для использования при получении нанесенного на носитель катализатора изобретения, или соединение, которое, кроме того, имеет реакционно-способную группу, соединенную с ними, вступали бы в реакцию с носителем, в который предварительно ввели бы функциональную группу, способную вступать в реакцию с вышеупомянутыми соединениями в мягких условиях с получением D. В случае получения с использованием такого способа получения их центры связывания представляют собой связь, содержащую гетероатом, обычно атом кислорода, атом азота, атом серы и тому подобное, такую как в случае простого эфира, сложного эфира, простого тиоэфира, сложного тиоэфира, амина, амида и тому подобного.
Кроме того, например, в качестве одного примера может оказаться предпочтительным, чтобы фосфазениевая соль (7), подходящая для использования при получении нанесенного на носитель катализатора изобретения, и соединение, имеющее полимеризуемую функциональную группу, были бы заполимеризованы, или чтобы фосфазениевая соль (8) и соединение кремния, имеющее гидролизуемую группу, такую как алкоксисилановая и тому подобное, были бы заполимеризованы, что обеспечило бы синтез носителя и одновременно получение D.
Двухвалентная группа, представляемая символом D, включает, например, углеводородную группу, которая может содержать гетероатом, такой как атом кислорода, атом серы, атом азота и атом кремния, говоря конкретно, алкиленовую группу, содержащую от 1 до 50 атомов углерода, такую как метиленовая, этиленовая, 1,2-диметилэтиленовая и пентаметиленовая; циклоалкиленовую группу, содержащую от 3 до 50 атомов углерода, такую как циклогексиленовая; алкениленовую группу, содержащую от 2 до 50 атомов углерода, такую как виниленовая и пропениленовая; циклоалкениленовую группу, содержащую от 3 до 50 атомов углерода, такую как циклогексениленовая; ариленовую группу, содержащую от 6 до 100 атомов углерода, такую как фениленовая и нафтиленовая; аралкиленовую группу, содержащую от 7 до 100 атомов углерода, такую как фенилметиленовая; углеводородную группу, включающую комбинацию углеводородных групп, такую как фениленметиленовая; тех из них, в которых часть атомов водорода в вышеупомянутой углеводородной группе замещают гетероатомом, таким как атом кислорода, атом азота, атом серы и атом кремния, или углеводородной группой, содержащей вышеупомянутый гетероатом; тех из них, в которых часть атомов углерода в вышеупомянутой углеводородной группе замещают гетероатомом, таким как атом кислорода, атом азота, атом серы и атом кремния, например, группу алкилендиокси, содержащую от 1 до 50 атомов углерода, такую как тетраметилендиокси; группу циклоалкилендиокси, содержащую от 3 до 50 атомов углерода, такую как циклогексилендиокси; группу алкилендитио, содержащую от 1 до 50 атомов углерода, такую как тетраметилендитио; группу алкилендиамино, содержащую от 1 до 50 атомов углерода, такую как N,N-диметилтетраметилендиамино; группу арилендиокси, содержащую от 6 до 100 атомов углерода, такую как фенилендиокси; и двухвалентную группу, описывающуюся следующей далее общей формулой (10):
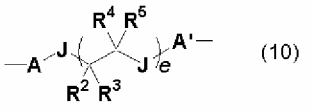
(где А представляет собой углеводородную группу, содержащую от 1 до 20 атомов углерода. R2, R3, R4 и R5 представляют собой атом водорода или углеводородную группу, содержащую от 1 до 8 атомов углерода. J представляют собой атом кислорода, атом серы или NR6, которые могут быть идентичными или различными, а R6 представляет собой атом водорода или углеводородную группу, содержащую от 1 до 8 атомов углерода. А' представляет собой непосредственную связь или углеводородную группу, содержащую от 1 до 20 атомов углерода, е находится в диапазоне от 0 до 200). В дополнение к этому, конкретные описания А, R2, R3, R4, R5 и е представляют собой то же самое, что и в общей формуле (6), как это описывается далее. R6 представляет собой то же самое, что и в случае символов от R2 до R5. На углеводородную группу, представляемую символом А', особенных ограничений не накладывается, и она может представлять собой алифатическую углеводородную группу или ароматическую углеводородную группу. Конкретные примеры алифатической углеводородной группы и ароматической углеводородной группы представляют собой то же самое, что и в случае А в общей формуле (6), как это описывается далее.
Кроме того, в качестве двухвалентной группы, представляемой описанным выше символом D, углеводородная группа, которая может содержать гетероатом, такой как атом кислорода, атом серы, атом азота и атом кремния, может иметь структуру фосфазениевой соли, описывающейся следующей далее общей формулой (11):
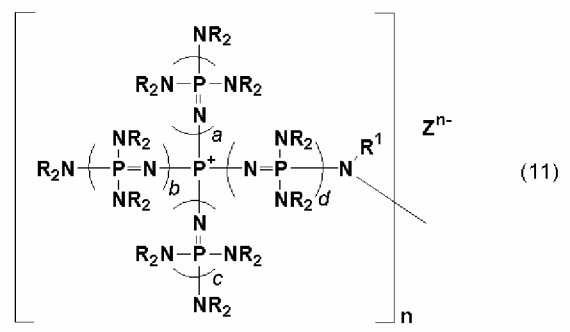
(где n, Zn-, a, b, c, d, R и R1 имеют те же самые значения, что и в общей формуле (1)).
D предпочтительно представляет собой двухвалентную группу, описывающуюся общей формулой (10), а более предпочтительно группу, в которой J представляет собой кислород и е находится в диапазоне от 0 до 30.
В общих формулах (1), (5), (6), (7) и (8) Zn- представляет собой анион соединения, содержащего атомы активного водорода, в форме, полученной в результате высвобождения n протонов из соединения, содержащего атомы активного водорода, в котором имеется, самое большее, 8 атомов активного водорода (общие формулы (1), (5), (6) и (7)) или, самое большее, 24 атома активного водорода (общая формула (8)). На анион соединения, содержащего атомы активного водорода, представляемый символом Zn-, особенных ограничений не накладывается, и он может представлять собой любой анион, который может образовывать ионную пару с фосфазениевым катионом. Соединение, содержащее атомы активного водорода и образующее Zn-, включает соединение, имеющее атом активного водорода на атоме кислорода, атоме азота или атоме серы, неорганическую кислоту и тому подобное.
В числе соединений, из которых получают Zn-, соединение, которое имеет атом активного водорода на атоме кислорода, включает, например, воду; карбоновые кислоты, такие как монокарбоновые кислоты, содержащие от 1 до 20 атомов углерода, и многовалентные карбоновые кислоты, содержащие от 2 до 20 атомов углерода, которые имеют от 2 до 6 карбоксильных групп; карбаминовые кислоты, содержащие от 1 до 20 атомов углерода; сульфоновые кислоты, содержащие от 1 до 20 атомов углерода; спирты, такие как одноатомные спирты, содержащие от 1 до 20 атомов углерода, и многоатомные спирты, содержащие от 2 до 20 атомов углерода, которые имеют от 2 до 8 гидроксильных групп; фенолы, содержащие от 6 до 20 атомов углерода; которые имеют от 1 до 3 гидроксильных групп; сахариды или их производные; и полиалкиленоксиды, имеющие атомы активного водорода в своих концевых положениях.
Монокарбоновые кислоты, содержащие от 1 до 20 атомов углерода, включают, например, алифатические монокарбоновые кислоты, такие как муравьиная кислота, уксусная кислота, трифторуксусная кислота, стеариновая кислота и олеиновая кислота; алифатические монокарбоновые кислоты, содержащие ароматическое кольцо, такие как фенилуксусная кислота; алициклические монокарбоновые кислоты, такие как циклогексанкарбоновая кислота; и ароматические монокарбоновые кислоты, такие как бензойная кислота и 2-карбоксинафталин.
Многовалентные карбоновые кислоты, содержащие от 2 до 20 атомов углерода, которые имеют от 2 до 6 карбоксильных групп, включают, например, алифатические многовалентные карбоновые кислоты, такие как щавелевая кислота и малоновая кислота; и ароматические многовалентные карбоновые кислоты, такие как фталевая кислота и тримеллитовая кислота.
Карбаминовые кислоты, содержащие от 1 до 20 атомов углерода, включают, например, N,N-диэтилкарбаминовую кислоту, N-карбоксианилин и N,N'-дикарбокси-2,4-толуилендиамин. Сульфоновая кислота, содержащая от 1 до 20 атомов углерода, включает, например, алифатические сульфоновые кислоты, такие как метансульфоновая кислота и трифторметансульфоновая кислота; алифатические сульфоновые кислоты, содержащие гетероциклы, такие как 2-морфолиноэтансульфоновая кислота и 3-(N-морфолино)пропансульфоновая кислота; ароматические сульфоновые кислоты, такие как бензолсульфоновая кислота, п-толуолсульфоновая кислота, 4-нитробензолсульфоновая кислота, 4,4'-бифенилдисульфоновая кислота, 2-нафталинсульфоновая кислота и пикрилсульфоновая кислота; и гетероциклические сульфоновые кислоты, такие как 3-пиридинсульфоновая кислота.
Одноатомные спирты, содержащие от 1 до 20 атомов углерода, включают, например, алифатические одноатомные спирты, такие как метанол, аллиловый спирт и кротиловый спирт; алициклические одноатомные спирты, такие как циклопентанол; и алифатические одноатомные спирты, содержащие ароматическое кольцо, такие как бензиловый спирт. Многоатомные спирты, содержащие от 2 до 20 атомов углерода, которые имеют от 2 до 8 гидроксильных групп, включают, например, алифатические многоатомные спирты, такие как этиленгликоль, пропиленгликоль, диэтиленгликоль, бутандиол, триметилолпропан, глицерин, диглицериновый спирт и пентаэритрит; и алициклические многоатомные спирты, такие как 1,4-циклогександиол.
Фенолы, содержащие от 6 до 20 атомов углерода, которые имеют от 1 до 3 гидроксильных групп, включают, например, одновалентные фенолы, такие как фенол, крезол, нитрофенол, хлорфенол, нафтол, антрарубин, 9-фенантрол и 1-гидроксипирен; и двухвалентные фенолы, такие как катехин, дигидроксинафталин и бисфенол А. Сахариды или их производные включают, например, сахариды, такие как глюкоза, сорбит, декстроза, фруктоза и сахароза или их производные; и тому подобное. Полиалкиленоксиды, имеющие атомы активного водорода в своих концевых положениях, включают, например, полиэтиленоксид, полипропиленоксид и полиалкиленоксиды, которые представляют собой сополимеры таких оксидов, характеризующиеся среднечисленной молекулярной массой в диапазоне от 100 до 50000 и имеющие от 2 до 8 концевых положений и от 1 до 8 гидроксильных групп в концевых положениях.
В числе соединений, из которых получают Zn-, соединение, содержащее атомы активного водорода, которое имеет атом активного водорода на атоме азота, включает, например, аммиак; амины, такие как первичные амины, содержащие от 1 до 20 атомов углерода, вторичные амины, содержащие от 2 до 20 атомов углерода, многовалентные амины, содержащие от 2 до 20 атомов углерода, которые имеют 2 или 3 первичные или вторичные аминогруппы, насыщенные циклические вторичные амины, содержащие от 4 до 20 атомов углерода, ненасыщенные циклические вторичные амины, содержащие от 4 до 20 атомов углерода, и циклические многовалентные амины, содержащие от 4 до 20 атомов углерода, которые имеют 2 или 3 вторичные аминогруппы; и амиды, такие как незамещенные или N-монозамещенные амиды кислот, содержащие от 2 до 20 атомов углерода, циклические амиды, содержащие 5-7-членные кольца, и имиды дикарбоновой кислоты, содержащей от 4 до 10 атомов углерода.
Первичные амины, содержащие от 1 до 20 атомов углерода, включают, например, алифатические первичные амины, такие как метиламин, этиламин и пропиламин; алициклические первичные амины, такие как циклогексиламин; алифатические первичные амины, содержащие ароматическое кольцо, такие как бензиламин и β-фенилэтиламин; и ароматические первичные амины, такие как анилин и толуидин.
Вторичные амины, содержащие от 2 до 20 атомов углерода, включают, например, алифатические вторичные амины, такие как диметиламин, метилэтиламин и дипропиламин; алициклические вторичные амины, такие как дициклогексиламин; и ароматические вторичные амины, такие как N-метиланилин и дифениламин. Многовалентные амины, содержащие от 2 до 20 атомов углерода, которые имеют 2 или 3 первичные или вторичные аминогруппы, включают, например, этилендиамин, ди(2-аминоэтил)амин, гексаметилендиамин, три(2-аминоэтил)амин и N,N'-диметилэтилендиамин. Насыщенные циклические вторичные амины, содержащие от 4 до 20 атомов углерода, включают, например, пирролидин, пиперидин, морфолин и 1,2,3,4-тетрагидрохинолин. Ненасыщенные циклические вторичные амины, содержащие от 4 до 20 атомов углерода, включают, например, 3-пирролин, пиррол, индол, карбазол, имидазол, пиразол и пурин.
Циклические многовалентные амины, содержащие от 4 до 20 атомов углерода, которые имеют 2 или 3 вторичные аминогруппы, включают, например, пиперазин, пиразин и 1,4,7-триазациклононан. Незамещенные или N-монозамещенные амиды кислот, содержащие от 2 до 20 атомов углерода, включают, например, ацетамид, N-метилпропионамид, амид N-метилбензойной кислоты и амид N-этилстеариновой кислоты. Циклические амиды, содержащие 5-7-членные кольца, включают, например, 2-пирролидон и ε-капролактам. Имиды дикарбоновых кислот, содержащих от 4 до 10 атомов углерода, включают, например, имид янтарной кислоты, имид малеиновой кислоты и фталимид.
В числе соединений, из которых получают Zn-, соединение, содержащее атомы активного водорода, которое имеет атом активного водорода на атоме серы, включает, например, сульфид водорода; тиоспирты, такие как одноатомные тиоспирты, содержащие от 1 до 20 атомов углерода, и многоатомные тиоспирты, содержащие от 2 до 20 атомов углерода; и тиофенолы, содержащие от 6 до 20 атомов углерода. Одноатомные тиоспирты, содержащие от 1 до 20 атомов углерода, включают, например, алифатические одноатомные тиоспирты, такие как метантиол, этантиол и аллилмеркаптан; алифатические одноатомные тиоспирты, содержащие ароматическое кольцо, такие как бензилмеркаптан; и алициклические одноатомные тиоспирты, такие как циклопентилмеркаптан и циклогексилмеркаптан. Многоатомные тиоспирты, содержащие от 2 до 20 атомов углерода, включают, например, 1,2-этандитиол, 1,3-пропандитиол, 1,2,3-пропантритиол и 2,3-ди(меркаптометил)-1,4-бутандитиол.
Тиофенолы, содержащие от 6 до 20 атомов углерода, включают, например, одноатомные тиофенолы, такие как тиофенол, тиокрезол и тионафтол; и двухатомные тиофенолы, такие как 1,2-бензолдитиол.
В числе соединений, из которых получают Zn-, неорганические кислоты включают галогениды водорода, такие как фторид водорода, хлорид водорода, бромид водорода, иодид водорода; борную кислоту, тетрафторборную кислоту, фосфорную кислоту, фосфористую кислоту, гексафторфосфорную кислоту, цианид водорода, тиоциановую кислоту, азотную кислоту, серную кислоту, угольную кислоту и перхлорную кислоту.
В числе соединений, содержащих атомы активного водорода, предпочтительными являются описанные выше неорганические кислоты и описанные выше соединения, содержащие атомы активного водорода, имеющие атом активного водорода на атоме кислорода, а более предпочтительными являются описанные выше галогениды водорода, алифатические одноатомные спирты, алициклические одноатомные спирты, алифатические одноатомные спирты, содержащие ароматическое кольцо, алифатические многоатомные спирты, алициклические многоатомные спирты, сахариды или их производные, полиэтиленоксид, полипропиленоксид или полиалкиленоксиды, которые представляют собой сополимеры таких оксидов, характеризующиеся среднечисленной молекулярной массой в диапазоне от 100 до 50000 и имеющие от 2 до 8 концевых положений и от 1 до 8 гидроксильных групп в концевых положениях.
Что касается Zn-, то в подходящем случае анионы можно выбирать в соответствии с реакциями, поскольку предпочтительность анионов варьируется в зависимости от типов реакций, в которых используют катализатор изобретения. Например, для полимеризации циклических мономеров, таких как алкиленоксид и тому подобное, предпочтительными являются анионы, полученные в результате высвобождения атома активного водорода из соединения, имеющего атом активного водорода на атоме кислорода, а для алкилирования фенольной гидроксильной группы предпочтительными являются анионы атомов галогенов.
В общих формулах (1), (5), (6) и (7) n представляет собой количество фосфазениевых катионов, а также количество протонов, высвобожденных из соединения, содержащего атомы активного водорода, в котором имеется, самое большее, 8 атомов активного водорода, n представляет собой целое число в диапазоне от 1 до 8, а предпочтительно целое число в диапазоне от 1 до 3. В дополнение к этому, в качестве конкретных примеров фосфазенового скелета, описывающегося общей формулой (1), различные варианты описываются в документах JP-A № 10-77289, JP-A № 2000-355606, JP-A № 2004-107266 и тому подобных, в которых изобретение может быть применено к такому известному фосфазеновому скелету.
В общей формуле (8) m представляет собой количество фосфазениевых катионов, связанных с атомом кремния, m представляет собой целое число в диапазоне от 1 до 3. Кроме того, n' представляет собой количество соединений кремния, с которым группа, включающая скелет фосфазениевого катиона, образует связь, n' представляет целое число в диапазоне от 1 до 8, а предпочтительно целое число в диапазоне от 1 до 3. Кроме того, n является множителем для m и n' и представляет собой совокупное количество фосфазениевых катионов и количества протонов, высвобожденных из соединения, содержащего атомы активного водорода, в котором имеется, самое большее, 24 атома активного водорода, n представляет собой целое число в диапазоне от 1 до 24, а предпочтительно целое число в диапазоне от 1 до 9.
В общей формуле (4) Х представляет собой атом галогена, а Х- представляет собой анион атома галогена. Атом галогена, представляемый символом Х, включает, например, атом фтора, атом хлора и атом брома, но в их числе предпочтительными являются атом хлора и атом брома. Анион атома галогена, представляемый символом Х-, включает, например, анионы атома фтора, атома хлора и атома брома, но в их числе предпочтительными являются анионы атома хлора и атома брома. Х- может представлять собой анион атома галогена, который является тем же самым, что и Х, или может представлять собой анион атома галогена, который отличается от Х.
В общей формуле (5) D' представляет собой одновалентную группу, способную образовывать связь с N (при том условии, что атом водорода и насыщенная углеводородная группа исключаются). На одновалентную группу, представляемую символом D', особенных ограничений не накладывается до тех пор, пока она будет представлять собой группу, отличную от атома водорода и насыщенной углеводородной группы, которая способна образовывать связь с атомом азота, присутствующим в фосфазениевом катионе. D' включает, например, углеводородную группу, содержащую гетероатом, такой как атом кислорода, атом серы, атом азота и атом кремния, и группу, включающую реакционно-способную функциональную группу, содержащую ненасыщенную связь углерод-углерод и тому подобное, и в числе тех из них, которые приводятся в качестве примеров описанного выше D, D' может соответствовать вариантам, в которых один центр связывания углеводородной группы, содержащей гетероатом, блокируется водородом, атомом галогена, кремнием и тому подобным.
Предпочтительные варианты реализации, описывающиеся общей формулой (5), приводятся далее.
(i) Фосфазениевая соль, в которой D' представляет собой одновалентную группу, описывающуюся следующей далее общей формулой (12):
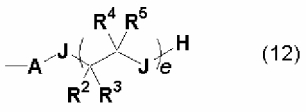
(где А, R2, R3, R4, R5, J и е имеют то же самое значение, что и в представленной выше общей формуле (10)) в углеводородной группе, содержащей гетероатом, такой как атом кислорода, атом серы, атом азота и атом кремния. В дополнение к этому, конкретные описания А, R2, R3, R4, R5 и е представляют собой то же самое, что и в общей формуле (6), представленной далее. Если J будет представлять собой NR6, то тогда R6 будет представлять собой то же самое, что и радикал в диапазоне от R2 до R5.
(ii) Фосфазениевая соль, описывающаяся общей формулой (6).
В общей формуле (6) А представляет собой углеводородную группу, содержащую от 1 до 20 атомов углерода. Углеводородная группа, содержащая от 1 до 20 атомов углерода, представляемая символом А, может являться алифатической углеводородной группой или ароматической углеводородной группой до тех пор, пока она будет представлять собой двухвалентную углеводородную группу. Двухвалентная углеводородная группа включает, например, алкиленовую группу, содержащую от 1 до 20 атомов углерода, такую как метиленовая, этиленовая, триметиленовая и метилэтиленовая; циклоалкиленовую группу, содержащую от 3 до 20 атомов углерода, такую как циклогексиленовая; алкениленовую группу, содержащую от 2 до 20 атомов углерода, такую как виниленовая и пропениленовая; циклоалкениленовую группу, содержащую от 3 до 20 атомов углерода, такую как циклогексениленовая; ариленовую группу, содержащую от 6 до 20 атомов углерода, такую как фениленовая и нафтиленовая; аралкиленовую группу, содержащую от 7 до 20 атомов углерода, такую как фенилметиленовая; и группу, включающую комбинацию данных групп, такую как фениленметиленовая и ксилиленовая. В их числе предпочтительными являются алкиленовая группа, ариленовая группа, аралкиленовая группа и группа, включающая комбинацию данных групп, а более предпочтительными являются метиленовая группа, этиленовая группа, фениленовая группа и ксилиленовая группа.
В общей формуле (6) R2, R3, R4 и R5 представляют собой атом водорода или углеводородную группу, содержащую от 1 до 8 атомов углерода. Углеводородная группа, представляемая символами R2, R3, R4 и R5, может являться алифатической углеводородной группой или ароматической углеводородной группой. В числе конкретных примеров R и R1 в общих формулах от (1) до (6) алифатическая углеводородная группа и ароматическая углеводородная группа включают, например, те из них, которые содержат от 1 до 8 атомов углерода. R2, R3, R4 и R5 предпочтительно представляют собой водород или алифатическую углеводородную группу, а более предпочтительно - водород или метильную группу. В общей формуле (6) е находится в диапазоне от 0 до 200. Предпочтительно е находится в диапазоне от 0 до 100, а более предпочтительно от 0 до 30.
(iii) Фосфазениевая соль, описывающаяся общей формулой (7).
В общей формуле (7) М представляет собой группу, содержащую ненасыщенную связь углерод-углерод. На такую группу особенных ограничений не накладывается до тех пор, пока она будет содержать ненасыщенную связь углерод-углерод, и она включает, например, алифатическую углеводородную группу, содержащую ненасыщенную связь углерод-углерод, в том числе алкенильную группу, такую как винильная, кротильная и аллильная, и алкинильную группу, такую как этинильная и пропинильная; ароматическую углеводородную группу, содержащую ненасыщенную связь углерод-углерод, в том числе стирильную, винилбензильную и стирилэтильную; группу, содержащую ненасыщенную связь углерод-углерод и карбонильную группу, такую как акрильная группа, метакрильная группа, циннамильная группа и ацетиленкарбонильная группа. В дополнение к этому, М включает группу, с которой в дополнение к вышеупомянутым группам образует связь углеводородная группа и тому подобное. В их числе предпочтительной является группа, содержащая двойную связь углерод-углерод, такая как винильная, кротильная, аллильная, стирильная, винилбензильная, стирилэтильная, акрильная, метакрильная, или группа, с которой в дополнение к вышеупомянутым группам образует связь углеводородная группа и тому подобное, а более предпочтительной является группа, содержащая двойную связь углерод-углерод в своем концевом положении, такая как винильная, аллильная, стирильная, винилбензильная, стирилэтильная, акрильная, метакрильная, или группа, с которой в дополнение к вышеупомянутым группам образует связь углеводородная группа и тому подобное.
(iv) Фосфазениевая соль, описывающаяся общей формулой (8).
В общей формуле (8) В представляет собой углеводородную группу, содержащую от 1 до 20 атомов углерода. Углеводородная группа, содержащая от 1 до 20 атомов углерода, представляемая символом В, является двухвалентной углеводородной группой, и она может представлять собой алифатическую углеводородную группу или ароматическую углеводородную группу. Двухвалентная углеводородная группа включает, например, алкиленовую группу, содержащую от 1 до 20 атомов углерода, такую как метиленовая, этиленовая, триметиленовая и метилэтиленовая; циклоалкиленовую группу, содержащую от 3 до 20 атомов углерода, такую как циклогексиленовая; алкениленовую группу, содержащую от 2 до 20 атомов углерода, такую как виниленовая и пропениленовая; циклоалкениленовую группу, содержащую от 3 до 20 атомов углерода, такую как циклогексениленовая; ариленовую группу, содержащую от 6 до 20 атомов углерода, такую как фениленовая и нафтиленовая; аралкиленовую группу, содержащую от 7 до 20 атомов углерода, такую как фенилметиленовая; и группу, включающую комбинацию данных групп, такую как фениленметиленовая и ксилиленовая. В их числе предпочтительными являются алкиленовая группа, ариленовая группа, аралкиленовая группа и группа, включающая комбинацию данных групп, а более предпочтительными являются метиленовая группа, этиленовая группа, фениленовая группа и ксилиленовая группа. В дополнение к этому, Т представляет собой функциональную группу, в которой связь Si-T может быть разрушена в результате гидролиза, и Т включает, например, атом галогена, такой как F, Cl, Br и I, или группу алкокси, такую как группа метокси, группа этокси, группа пропокси и группа бутокси.
Далее в настоящем документе фосфазеновый нанесенный на носитель катализатор изобретения, который имеет группу, описывающуюся общей формулой (1), связанную с носителем, описывается более подробно с приведением разъяснения способа его получения.
Что касается фосфазенового нанесенного на носитель катализатора изобретения, то на носитель, с которым группа, описывающаяся общей формулой (1), образует связь, особенных ограничений не накладывается до тех пор, пока он будет нерастворим в использованном растворителе реакции, и может быть использован любой из них, который имеет группу, способную образовывать связь с группой, описывающейся общей формулой (1). В качестве таких носителей известны различные варианты, например различные типы таких носителей описываются на страницах от 133 до 163 в работе "Catalyst Lecture Vol. 10 (Industrial Catalyst Reaction 4) Detailed Exposition on Catalyst", Catalyst Institute Ed. First release, Kodansha (1986). Говоря конкретно, неорганический носитель, типичными представителями которого являются оксиды металлов, такие как SiO2, Al2O3, MgO, TiO2, SnO2, ZnO и ZrO2; сложные оксиды металлов, такие как SiO2-Al2O3, SiO2-MgO, SiO2-ZrO2 и цеолит; металлическая соль твердой кислоты, такая как металлическая соль гетерополикислоты и металлическая соль твердой фосфорной кислоты, слоистое соединение, такое как слюда и монтмориллонит; и глинистый минерал, такой как диатомовая земля; органический полимерный носитель, типичными представителями которого являются органический полимер, в котором основную цепь, такую как у полистирола, поливинилпиридина, полибутадиена и поливинилхлорида, составляет связь углерод-углерод; органический полимер, содержащий в своей основной цепи атом кислорода, такой как полиакриловая кислота и поли(мет)акрилат; органический полимер, содержащий в своей основной цепи атом азота, такой как полиамид, полиуретан и полиимид; органический полимер, содержащий в своей основной цепи атом кремния, такой как полисилоксан и полисилан; и органический полимер, содержащей в своей основной цепи атом серы, такой как полисульфид и полисульфон; и сшитый органический полимерный носитель, типичными представителями которого являются полимеры, в которых описанный выше органический полимерный носитель имеет подходящую сшитую структуру. В числе таких носителей предпочтительным является носитель, такой как оксид металла, органический полимер, в котором его основную цепь составляет связь углерод-углерод, сшитый органический полимер, в котором его основную цепь составляет связь углерод-углерод, а более предпочтительными являются SiO2, сшитый и несшитый полистирол и сшитый и несшитый полиэтилен. Используют носитель, в котором в такой носитель вводят группу, способную образовывать связь с группой, описывающейся общей формулой (1) в изобретении. Что касается способа введения данных групп в носитель, то на страницах от 136 до 137 и страницах от 149 до 150 литературной ссылки продемонстрированы различные примеры, представителями которых являются способ, в котором на поверхности SiO2 гидроксильную группу вводят в реакцию с SOCl2 и замещают хлором, а после этого вводят во взаимодействие, способ, в котором гидроксильную группу на поверхности SiO2 подвергают хлорированию, вводят в реакцию с фениллитием, а после этого замещают фенильной группой, которую подвергают хлорметилированию и вводят во взаимодействие, способ, в котором полистирол подвергают хлорметилированию и вводят во взаимодействие, и тому подобное. Кроме того, также можно упомянуть и способ, в котором алкоксисилан, имеющий хлорметильную группу, используют для проведения гидролиза-поликонденсации с участием другого алкоксисилана и тому подобного или вводят в силанольную группу в силикагеле в результате проведения силилирования. В качестве альтернативного способа образования связи с носителем используют способ, в котором при использовании алкоксисилана, имеющего хлорметильную группу, алкоксисилильной группе дают возможность образовать связь для того, чтобы провести синтез фосфазениевой соли, а после этого полученный в результате продукт подвергают гидролизу-поликонденсации с участием другого алкоксисилана и тому подобного, или вводят на носитель в результате силилирования при использовании силанольной группы в силикагеле.
В конкретном способе получения нанесенного на носитель катализатора сначала получают фосфазеновое соединение, описывающееся общей формулой (2) (далее в настоящем документе называемое "фосфазеновым соединением (2)"), фосфазеновое соединение, описывающееся общей формулой (3) (далее в настоящем документе называемое "фосфазеновым соединением (3)"), фосфазениевую соль, описывающуюся общей формулой (4) (далее в настоящем документе называемую "фосфазениевой солью (4)"), фосфазениевую соль, описывающуюся общей формулой (5) (далее в настоящем документе называемую "фосфазениевой солью (5)"), фосфазениевую соль, описывающуюся общей формулой (6) (далее в настоящем документе называемую "фосфазениевой солью (6)"), фосфазениевую соль, описывающуюся общей формулой (7) (далее в настоящем документе называемую "фосфазениевой солью (7)") и фосфазениевую соль, описывающуюся общей формулой (8) (далее в настоящем документе называемую "фосфазениевой солью (8)"), которые представляют собой промежуточные соединения, подходящие для использования при получении фосфазенового нанесенного на носитель катализатора изобретения. В соответствии с этим далее сначала будет описываться способ получения фосфазеновых соединений (2) и (3) и фосфазениевых солей (4), (5), (6), (7) и (8).
Фосфазеновое соединение (2) можно получать в результате проведения реакции между фосфазениевой солью, описывающейся следующей далее общей формулой (13), например, и соединением, полученным в результате замещения водорода в соединении, содержащем атомы активного водорода, щелочным металлом или щелочноземельным металлом, при относительно повышенной температуре.
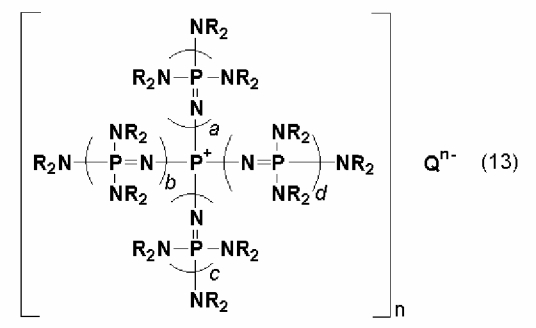
(где n является целым числом в диапазоне от 1 до 8 и представляет собой количество фосфазениевых катионов, Qn - представляет собой анион, способный образовывать ионную пару с фосфазениевым катионом. Каждый из a, b, c и d представляет собой положительное целое число, равное 3 или менее. R представляют собой одинаковые или различные углеводородные группы, содержащие от 1 до 10 атомов углерода, а два R, расположенные на каждом общем атоме азота, могут быть связаны друг с другом с образованием кольцевой структуры).
На такой анион Qn- особенных ограничений не накладывается, и им может быть любой Qn-, который образует фосфазеновое соединение, описывающееся общей формулой (2). В качестве фосфазениевых солей известны фосфазениевые соли, описанные в документах JP-A № 10-77289 и JP-A № 2000-355606, те из них, у которых анионами являются анионы атомов галогенов, таких как хлор, описанные в документе "Furka Comprehensive Catalog 1995/96" Furka Fine Chemical, и тому подобное. Qn- может быть любым Qn-, который не препятствует прохождению описанной далее реакции, и им также могут являться неорганические анионы.
В соединении, полученном в результате замещения водорода в описанном выше соединении, содержащем атомы активного водорода, щелочным металлом или щелочноземельным металлом, щелочной металл или щелочноземельный металл включает металлический литий, металлический натрий, металлический калий, металлический цезий, металлический магний, металлический кальций, металлический стронций и металлический барий.
Соединение, содержащее атомы активного водорода, включает соединение, содержащее атомы активного водорода, из которого получают Zn-, а в особенности предпочтительными являются спирты, фенолы, тиоспирты, тиофенолы и амины.
Реакцию между фосфазениевой солью, описывающейся общей формулой (13), и соединением, полученным в результате замещения водорода в описанном выше соединении, содержащем атомы активного водорода, щелочным металлом или щелочноземельным металлом, можно проводить по тому же самому способу, что и реакцию получения фосфазениевой соли, описывающейся представленной выше общей формулой (9), описанную в документе JP-A № 10-77289, за исключением того, что температуру реакции устанавливают равной относительно повышенной температуре, как это описывается выше. Говоря конкретно, например, ее можно провести в соответствии со следующими далее условиями.
Используемое количество соединения, полученного в результате замещения водорода в соединении, содержащем атомы активного водорода, щелочным металлом или щелочноземельным металлом, находится в диапазоне от обычно от 1 до 10 эквивалентов, предпочтительно от 1 до 5 эквивалентов, а более предпочтительно от 1 до 2 эквивалентов, при расчете на один эквивалент фосфазениевой соли, описывающейся общей формулой (13).
На растворитель реакции особенных ограничений не накладывается до тех пор, пока он не будет препятствовать протеканию реакции, и может быть использован любой известный растворитель. Их конкретные примеры включают алифатические или ароматические углеводороды, такие как н-гексан, бензол, толуол и тетралин; алифатические или ароматические галогенированные углеводороды, такие как метиленхлорид, хлороформ и о-дихлорбензол; простые эфиры, такие как диэтиловый эфир и тетрагидрофуран; нитрилы, такие как ацетонитрил и пропионитрил; полярные апротонные растворители, такие как N,N-диметилформамид, диметилсульфоксид, сульфолан, триамид гексаметилфосфорной кислоты и 1,3-диметил-2-имидазолидинон, и их можно использовать индивидуально или в комбинации из двух или более типов.
Температуру реакции можно надлежащим образом регулировать в зависимости от типа, концентрации и тому подобного, что связано с реагентом, но она является относительно повышенной температурой, другими словами, находится в диапазоне в общем случае от 80 до 300°С, предпочтительно от 100 до 250°С, а более предпочтительно от 120 до 200°С. Давлением во время проведения реакции может являться любое давление, выбираемое из пониженного давления, нормального давления и повышенного давления, но оно находится в диапазоне предпочтительно от 10 до 500 кПа (абсолютное давление; это равным образом будет использоваться и далее в настоящем документе), а более предпочтительно от 100 до 300 кПа. Время реакции можно надлежащим образом регулировать в зависимости от температуры реакции, типа реакционной системы и тому подобного, но оно находится в диапазоне в общем случае от 0,1 до 100 часов, предпочтительно от 1 до 50 часов, а более предпочтительно от 2 до 20 часов.
Выделение фосфазенового соединения (2) из реакционного раствора по завершении реакции можно проводить в соответствии с обычно используемым способом. Например, раствор, содержащий фосфазеновое соединение (2), может быть получен в результате выделения твердого содержимого, присутствующего в реакционном растворе, в результате проведения фильтрования, центрифугирования и тому подобного. Раствор концентрируют до сухости для того, чтобы получить фосфазеновое соединение (2) в виде твердой фазы. Кроме того, при желании его можно дополнительно очистить в результате проведения перекристаллизации и тому подобного.
Фосфазеновое соединение (2), полученное так, как это описывается выше, можно дополнительно вводить в реакцию с соединением, описывающимся следующей формулой: Х-D' (где Х представляет собой атом галогена, D' представляет собой то же самое, что и D' в общей формуле (5)), с получением фосфазениевой соли (5).
Например, фосфазеновое соединение (2) можно дополнительно вводить в реакцию с соединением, описывающимся представленной выше формулой: X-D', которое является соединением (а), описывающимся следующей формулой: X-E-Y (где Х представляет собой атом галогена, Е представляет собой углеводородную группу, которая может содержать атом кислорода, атом серы или атом азота, Y представляет собой гидроксильную группу, группу меркапто или аминогруппу, которая защищена защитной группой), например соединение, где защитная группа Y представляет собой алкилсилильную группу, с получением фосфазениевой соли (5), а после этого снятием защиты и, таким образом, получением структуры, имеющей гидроксильную группу, группу меркапто или аминогруппу, то есть фосфазениевой соли (5) и фосфазениевой соли (6) в качестве ее предпочтительного варианта реализации. В альтернативном варианте в соответствии с известным способом Y после снятия защиты получает возможность вступать в реакцию или полимеризоваться с заместителем, таким как алкиленоксид или замещенный алкиленоксид, кислород которого замещен на серу, азот и тому подобное, с получением фосфазениевой соли (5) и фосфазениевой соли (6) в качестве ее предпочтительного варианта реализации, которые в своих концевых положениях имеют гидроксильную группу, группу меркапто или аминогруппу.
Реакцию между фосфазеновым соединением (2) и соединением (а) можно проводить при следующих далее условиях. Растворитель реакции представляет собой то же самое, что и в случае получения описанного выше фосфазенового соединения (2). Температуру реакции можно надлежащим образом регулировать в зависимости от типа, концентрации и тому подобного, что связано с реагентом, но она находится в диапазоне в общем случае от - 78 до 100°С, предпочтительно от - 50 до 80°С, а более предпочтительно от 0 до 50°С. Давлением во время проведения реакции может являться любое давление, выбираемое из пониженного давления, нормального давления или повышенного давления, но оно находится в диапазоне предпочтительно от 10 до 500 кПа, а более предпочтительно от 100 до 300 кПа. Время реакции можно надлежащим образом регулировать в зависимости от температуры реакции, типа реакционной системы и тому подобного, но оно находится в диапазоне в общем случае от 0,1 до 100 часов, предпочтительно от 1 до 80 часов, а более предпочтительно от 2 до 50 часов. Фосфазениевые соли (5) и (6) можно выделять из реакционного раствора по обычно используемому способу. Например, фосфазениевые соли (5) и (6) можно получать в твердой форме или в виде вязкой жидкости в результате выделения твердого содержимого, присутствующего в реакционном растворе, в результате проведения фильтрования, центрифугирования и тому подобного, а после этого концентрирования фильтрата до сухости. При необходимости их можно дополнительно очистить при использовании перекристаллизации, колоночной хроматографии и тому подобного.
Кроме того, фосфазеновое соединение (2), например, можно вводить в реакцию с соединением, описывающимся представленной выше формулой: X-D', которое представляет собой соединение (а'), описывающееся следующей формулой: X-L (где Х представляет собой атом галогена, а L представляет собой группу, содержащую ненасыщенную связь углерод-углерод), с получением фосфазениевой соли (7).
Реакцию между фосфазеновым соединением (2) и соединением (а') можно проводить, например, при следующих далее условиях. Растворитель реакции представляет собой то же самое, что и в случае описанного выше получения фосфазенового соединения (2). Температуру нагревания можно надлежащим образом регулировать в зависимости от типа, концентрации и тому подобного, что связано с реагентом, но она находится в диапазоне в общем случае от - 78 до 100°С, предпочтительно от - 50 до 80°С, а более предпочтительно от 0 до 50°С. Давлением во время проведения нагревания может являться любое давление, выбираемое из пониженного давления, нормального давления или повышенного давления, но оно находится в диапазоне предпочтительно от 10 до 500 кПа, а более предпочтительно от 100 до 300 кПа. Время реакции можно надлежащим образом регулировать в зависимости от температуры реакции, типа реакционной системы и тому подобного, но оно находится в диапазоне в общем случае от 0,1 до 100 часов, предпочтительно от 0,5 до 80 часов, а более предпочтительно от 2 до 50 часов. Фосфазениевую соль (7) можно выделять из реакционного раствора по обычно используемому способу. Например, фосфазениевую соль (7) можно получать в твердой форме в результате выделения твердого содержимого, присутствующего в реакционном растворе, в результате проведения фильтрования, центрифугирования и тому подобного, а после этого концентрирования фильтрата до сухости. При необходимости их можно дополнительно очистить при использовании перекристаллизации, колоночной хроматографии и тому подобного.
Кроме того, фосфазеновое соединение (2) можно вводить в реакцию с соединением, описывающимся представленной выше формулой: X-D', которое является кремнийорганическим соединением (а''), описывающимся следующей формулой: X-E'-Y' (где Х представляет собой атом галогена, Е' представляет собой углеводородную группу, Y' представляет собой силильную группу, включающую гидролизуемую группу, такую как, по меньшей мере, один атом галогена или группа алкокси), с получением фосфазениевой соли (8), имеющей силильную группу, включающую гидролизуемую группу, связанную с ней.
Реакцию между фосфазеновым соединением (2) и соединением (а'') можно проводить, например, при следующих далее условиях. Растворитель реакции представляет собой то же самое, что и в случае описанного выше получения фосфазенового соединения (2). Температуру реакции можно надлежащим образом регулировать в зависимости от типа, концентрации и тому подобного, что связано с реагентом, но она находится в диапазоне в общем случае от - 78 до 100°С, предпочтительно от - 50 до 80°С, а более предпочтительно от 0 до 50°С. Давлением во время проведения нагревания может являться любое давление, выбираемое из пониженного давления, нормального давления или повышенного давления, но оно находится в диапазоне предпочтительно от 10 до 500 кПа, а более предпочтительно от 100 до 300 кПа. Время реакции можно надлежащим образом регулировать в зависимости от температуры реакции, типа реакционной системы и тому подобного, но оно находится в диапазоне в общем случае от 0,1 до 100 часов, предпочтительно от 1 до 80 часов, а более предпочтительно от 2 до 50 часов. В дополнение к этому, для получения высокополярного продукта реакции (8) и после этого немедленного удаления продукта из системы в качестве растворителя реакции используют неполярный растворитель, таким образом, данный вариант является предпочтительным с точки зрения режима проведения реакции, упрощения способа очистки, улучшения селективности и тому подобного, и предпочтительно он является режимом проведения реакции.
С другой стороны, фосфазеновое соединение (3) можно получать в результате нагревания фосфазениевой соли, описывающейся общей формулой (13), где анионом являются гидроксильный анион или меркапто-анион, при температуре, превышающей комнатную температуру, в присутствии или в отсутствие растворителя. Нагревание, например, можно проводить в соответствии со следующими далее условиями. Растворитель реакции используют так же, как и в случае получения фосфазенового соединения (2). Температуру нагревания можно надлежащим образом регулировать в зависимости от типа, концентрации и тому подобного, что связано с реагентом, но можно упомянуть температуру, превышающую комнатную температуру, то есть находящуюся в диапазоне в общем случае от 50 до 300°С, предпочтительно от 80 до 250°С, а более предпочтительно от 100 до 200°С. Давлением во время проведения нагревания может являться любое давление, выбираемое из пониженного, нормального или повышенного давления, но оно находится в диапазоне предпочтительно от 10 до 500 кПа, а более предпочтительно от 100 до 300 кПа. Время нагревания можно надлежащим образом регулировать в зависимости от температуры нагревания, типа реакционной системы и тому подобного, но оно находится в диапазоне в общем случае от 1 до 240 часов, предпочтительно от 2 до 200 часов, а более предпочтительно от 5 до 150 часов.
Выделение фосфазенового соединения (3) из реакционного раствора по завершении нагревания можно проводить в соответствии с обычно используемыми способами. Например, содержащий раствор можно получать в результате добавления к реагенту алифатического или ароматического углеводородов, таких как н-пентан, н-гексан, циклогексан, бензол, толуол, ксилол и тетралин, и выделения нерастворимых фракций в результате проведения фильтрования, центрифугирования и тому подобного. Раствор можно концентрировать до сухости с получением твердого фосфазенового соединения (3) в виде твердой фазы. Кроме того, при необходимости также можно осуществить и очистку в результате проведения перекристаллизации и тому подобного.
Фосфазениевую соль (4) можно получать в результате проведения реакции между полученным ранее фосфазеновым соединением (3) и, например, галогенирующим агентом, таким как фосген, тионилхлорид, тионилбромид, пентахлорид фосфора, трихлорид фосфора, 2,2-дихлор-1,3-диметилимидазолидин, 2,2-дифтор-1,3-диметилимидазолидин. Реакцию проводят в соответствии со следующими далее условиями.
Уровень содержания галогенирующего агента находится в диапазоне в общем случае от 1 до 10 эквивалентов, предпочтительно от 1 до 5 эквивалентов, а более предпочтительно от 1 до 2 эквивалентов, при расчете на 1 эквивалент фосфазенового соединения (3). Растворитель реакции представляет собой то же самое, что и в случае получения описанного выше фосфазенового соединения (2). Температуру реакции можно надлежащим образом регулировать в зависимости от типа, концентрации и тому подобного, что связано с реагентом, но она находится в диапазоне в общем случае от - 78 до 200°С, предпочтительно от - 50 до 150°С, а более предпочтительно от 0 до 100°С. Давлением во время проведения реакции может являться любое давление, выбираемое из пониженного, нормального или повышенного давления, но оно находится в диапазоне предпочтительно от 10 до 500 кПа, а более предпочтительно от 100 до 300 кПа.
Время реакции можно надлежащим образом регулировать в зависимости от температуры реакции, типа реакционной системы и тому подобного, но оно находится в диапазоне в общем случае от 0,1 до 300 часов, предпочтительно от 0,5 до 200 часов, а более предпочтительно от 2 до 150 часов. Выделение фосфазениевой соли (4) из реакционного раствора по завершении реакции можно проводить в соответствии с обычно используемыми способами. Например, реакционный раствор концентрируют до сухости с получением фосфазениевой соли (4) в виде твердой фазы. Кроме того, при необходимости также можно осуществить и очистку в результате проведения перекристаллизации и тому подобного.
Между тем, фосфазениевую соль (4) вводят в реакцию с соединением (b), примером которого является соединение, имеющее защитную группу, такую как алкилсилильная группа, описывающаяся следующей формулой: R1-NH-E-Y (где R1 представляет собой то же самое, что и в общих формулах (1), (5), (6), (7) и (8), Е и Y представляют собой то же самое, что и в соединении (а)), с получением фосфазениевой соли (5); после этого с Y снимают защиту и получают структуру соединения, имеющую гидроксильную группу, группу меркапто или аминогруппу, то есть фосфазениневую соль (5) и фосфазениевую соль (6) в качестве ее предпочтительного варианта реализации. Кроме того, Y после снятия защиты делает возможным протекание реакции алкиленоксида или его кислорода с заместителем, таким как сера, азот и тому подобное, при использовании широко известного способа; или в результате создания условий для полимеризации можно получить фосфазениевую соль (5) и фосфазениевую соль (6) в качестве ее предпочтительного варианта реализации, концевые положения которых имеют вид гидроксильной группы, группы меркапто, аминогруппы и тому подобного.
Реакцию между фосфазеновой солью (4) и соединением (b), например, можно проводить в соответствии со следующими далее условиями. Растворитель реакции представляет собой то же самое, что и в случае получения описанного выше фосфазенового соединения (2). Температуру реакции можно надлежащим образом регулировать в зависимости от типа, концентрации и тому подобного, что связано с реагентом, но она находится в диапазоне в общем случае от - 78 до 200°С, предпочтительно от - 50 до 150°С, а более предпочтительно от 0 до 100°С. Давлением во время проведения реакции может являться любое давление, выбираемое из пониженного, нормального или повышенного давления, но оно находится в диапазоне предпочтительно от 10 до 500 кПа, а более предпочтительно от 100 до 300 кПа. Время реакции можно надлежащим образом регулировать в зависимости от температуры реакции, типа реакционной системы и тому подобного, но оно находится в диапазоне в общем случае от 0,1 до 200 часов, предпочтительно от 0,5 до 150 часов, а более предпочтительно от 2 до 100 часов. Выделение фосфазениевой соли (5) или (6) из реакционного раствора можно проводить в соответствии с обычно используемыми способами. Например, твердое содержимое из реакционного раствора выделяют в результате проведения фильтрования, центрифугирования и тому подобного. Раствор концентрируют до сухости с получением фосфазениевой соли (5) или (6) в виде твердой фазы или вязкой жидкости. Кроме того, при необходимости также можно осуществить и очистку при использовании перекристаллизации, колоночной хроматографии и тому подобного.
В фосфазениевых солях (5), (6), (7) и (8), полученных так, как описывается выше, Zn- при необходимости может быть замещен на желательный анион в соответствии с широко известным способом. Например, замещение можно проводить по способу введения в контакт с соединением, имеющим желательный анион, в частности по способу ионного обмена при использовании ионообменной смолы, содержащей желательный анион и тому подобное, по способу обработки солью щелочного металла или солью щелочноземельного металла, содержащей желательный анион, и тому подобному.
Фосфазеновый нанесенный на носитель катализатор изобретения можно получать в результате проведения реакции с носителем, который был модифицирован для проведения реакции с фосфазеновым соединением (2) или (3) или фосфазениевой солью (4), (5), (6), (7) или (8) с фосфазеновым соединением (2) или (3) или фосфазениевой солью (4), (5), (6), (7) или (8). В частности, например, фосфазеновое соединение (2) или фосфазениевую соль (5) или (6) вводят в реакцию с галогенированным углеводородным остатком и тому подобным в носителе, благодаря чему происходит нанесение на носитель, что, таким образом, делает возможным получение. Кроме того, в случае использования в качестве носителя аминометилированного носителя нанесенный на носитель катализатор можно получить в результате проведения реакции между фосфазеновой солью (4) и аминированным углеводородным остатком. Кроме того, в случае использования в качестве носителя силикагеля фосфазениевую соль (8) можно подвергнуть силилированию при использовании силанольной группы, которая присутствует на поверхности силикагеля, в соответствии с известным способом получения нанесенного на носитель катализатора.
Реакцию между фосфазеновым соединением (2) или фосфазениевыми солями (4), (5) или (6) и носителем проводят, например, при следующих далее условиях.
Растворитель реакции представляет собой то же самое, что и в случае получения описанного выше фосфазенового соединения (2). Температуру реакции можно надлежащим образом регулировать в зависимости от типа, концентрации и тому подобного, что связано с реагентом, но она находится в диапазоне в общем случае от 78 до 200°С, предпочтительно от -50 до 150°С, а более предпочтительно от 0 до 100°С. Давлением во время проведения реакции может являться любое давление, выбираемое из пониженного, нормального или повышенного давления, но оно находится в диапазоне предпочтительно от 10 до 500 кПа, а более предпочтительно от 100 до 300 кПа. Время реакции можно надлежащим образом регулировать в зависимости от температуры реакции, типа реакционной системы и тому подобного, но оно находится в диапазоне в общем случае от 0,1 до 500 часов, предпочтительно от 0,5 до 300 часов, а более предпочтительно от 2 до 200 часов. Выделение фосфазенового нанесенного на носитель катализатора из реакционного раствора по завершении реакции можно проводить в соответствии с обычно используемыми способами. Например, фосфазеновый нанесенный на носитель катализатор (1), в качестве твердой фазы содержащийся в реакционном растворе, выделяют в результате проведения фильтрования, центрифугирования и тому подобного. Кроме того, при необходимости также можно осуществить и очистку в результате проведения промывания водой, подходящим растворителем и тому подобным.
В дополнение к этому, реакцию между фосфазениевой солью (8) и носителем можно проводить, например, при следующих далее условиях.
Фосфазениевую соль (8) наносят на носитель в результате введения ее в контакт с коммерчески доступным силикагелем и нагревания. В данный момент можно использовать растворитель реакции. На растворитель реакции особенных ограничений не накладывается до тех пор, пока он будет инертным по отношению к силикагелю и фосфазениевой соли (8), но предпочтительным является неполярный растворитель, такой как бензол, толуол, гексан и тому подобное. Температура реакции находится в диапазоне в общем случае от 0 до 200°С, предпочтительно от 20 до 150°С, а более предпочтительно от 40 до 120°С. Давлением во время проведения реакции может являться любое давление, выбираемое из пониженного давления, нормального давления или повышенного давления, но оно находится в диапазоне предпочтительно от 10 до 500 кПа, а более предпочтительно от 100 до 300 кПа. Время реакции можно надлежащим образом регулировать в зависимости от температуры реакции, типа реакционной системы и тому подобного, но оно находится в диапазоне в общем случае от 0,1 до 100 часов, предпочтительно от 1 до 50 часов, а более предпочтительно от 2 до 20 часов. После выделения силикагеля в результате проведения фильтрования получающийся в результате продукт промывают растворителем и тому подобным, а после этого высушивают до получения фосфазенового нанесенного на носитель катализатора.
После проведения реакции между фосфазеновым соединением (2) или фосфазениевой солью (4), (5), (6) или (8) и носителем, который модифицируют галогенированными углеводородными остатками, аминированными углеводородными остатками и тому подобным, не вступившие в реакцию галогенированные углеводородные остатки или аминированные углеводородные остатки можно дезактивировать при использовании обычного способа. Например, галогенированный углеводородный остаток можно дезактивировать в результате обработки алкоголятом щелочного металла или алкоголятом щелочноземельного металла и тому подобным с замещением, таким образом, атома галогена на группу алкокси и получением простого эфира. Кроме того, аминированный углеводородный остаток можно дезактивировать в результате замещения с использованием соли щелочного металла или щелочноземельного металла и алкилирования аминогруппы алкилгалогенидом и тому подобным с получением третичной аминоструктуры.
С другой стороны, например, для синтеза носителя и в то же самое время получения фосфазенового нанесенного на носитель катализатора фосфазениевую соль (7) и соединение, содержащее полимеризуемую функциональную группу, такое как так называемый винильный мономер, такой как стирол, сложный эфир (мет)акриловой кислоты и тому подобное, можно заполимеризовать в соответствии с обычно используемым способом (например, "4th Edition Experimental Chemistry Lecture Vol. 28 (Polymer Synthesis)", The Chemical Society of Japan, Maruzen, 1992, pp. 31 to 38, pp. 120 to 152).
Кроме того, в соответствии с известным способом фосфазениевую соль (8) и другой алкоксисилан можно подвергнуть гидролизу-поликонденсации и синтезировать носитель и в то же самое время получить фосфазеновый нанесенный на носитель катализатор. Настоящий способ можно реализовать, например, в соответствии со следующими далее условиями.
В способе нанесения на носитель при использовании гидролиза-поликонденсации из алкоксисилана, который может формировать матрицу силикагеля, и фосфазениевой соли (8) получают гомогенный раствор при использовании полярного растворителя, такого как метанол, этанол и тому подобное, который является инертным по отношению к алкоксисилану и воде и является водорастворимым. Сюда же добавляют хлористоводородную кислоту и получают кислую среду, после этого сюда добавляют приблизительно 1 эквивалент воды при расчете на гидролизуемые группы, а смесь нагревают при перемешивании. Температуру реакции можно надлежащим образом регулировать в зависимости от типа, концентрации и тому подобного, что связано с реагентом, но она находится в диапазоне в общем случае от 0 до 200°С, предпочтительно от 20 до 150°С, а более предпочтительно от 40 до 100°С. Давлением во время проведения реакции может являться любое давление, выбираемое из пониженного давления, нормального давления или повышенного давления, но оно находится в диапазоне предпочтительно от 10 до 500 кПа, а более предпочтительно от 100 до 300 кПа. Время реакции можно надлежащим образом регулировать в зависимости от температуры реакции, типа реакционной системы и тому подобного, но оно находится в диапазоне в общем случае от 0,1 до 100 часов, предпочтительно от 1 до 50 часов, а более предпочтительно от 2 до 20 часов.
После этого в реакционную смесь добавляют избыток воды, а реакционную смесь немедленно превращают в гель в результате ее помещения в щелочную среду, такую как в случае аммиака. В данный момент с точки зрения улучшения прочности каталитической структуры эффективными являются продолжение нагревания в щелочной среде и выдерживание в течение продолжительного периода времени. Фосфазеновый нанесенный на носитель катализатор по завершении реакции можно выделять при использовании обычного способа. Например, фосфазеновый нанесенный на носитель катализатор можно получать в результате выделения при помощи фильтрования, центрифугирования и тому подобного в отношении твердого содержимого, присутствующего в реакционном растворе, а кроме того, промывания водой и высушивания.
В фосфазеновом нанесенном на носитель катализаторе, полученном так, как это описывается выше, Zn- в соответствии с широко известным способом может быть замещен на желательный анион, который окажется подходящим для того типа реакции, в котором используют катализатор. Например, замещение можно проводить по способу введения в контакт с соединением, имеющим желательный анион, в частности по способу ионного обмена при использовании ионообменной смолы, содержащей желательный анион и тому подобное, по способу обработки солью щелочного металла или солью щелочноземельного металла, содержащей желательный анион, и тому подобному.
Фосфазеновый нанесенный на носитель катализатор, полученный так, как это описывается выше, является подходящим для использования в качестве катализатора различных органических реакций, и, в частности, он является подходящим для использования в качестве катализатора полимеризации циклического мономера и замещения заместителя. В дополнение к этому, фосфазеновые соединения (2) и (3) и фосфазениевые соли (4), (5), (6), (7) и (8) изобретения, которые являются подходящими для использования при получении фосфазенового нанесенного на носитель катализатора изобретения, сами по себе могут оказаться подходящими для использования в качестве катализатора при проведении различных реакций.
Кроме того, фосфазениевые катионы в фосфазеновом нанесенном на носитель катализаторе, описывающемся общей формулой (1), и в фосфазениевых солях, описывающихся общей формулой (4), общей формулой (5), общей формулой (6), общей формулой (7), общей формулой (8), общей формулой (9), общей формулой (11) и общей формулой (13), описывались ограничивающими структурными формулами, в каждой из которых положительный заряд локализован на центральном атоме фосфора. Помимо этого, может быть предложено несколько ограничивающих структурных формул. Фактический положительный заряд делокализован, соответственно, по всем структурам всех скелетов.
В способе полимеризации циклического мономера изобретения примеры циклического мономера включают алкиленоксиды, лактоны, лактамы, лактиды, циклические карбонаты, ангидриды (α-аминокислота)-N-карбоновых кислот, циклические фосфаты, сложные эфиры циклических фосфоновых кислот и циклические силоксаны. В качестве способа полимеризации циклического мономера с использованием фосфазенового нанесенного на носитель катализатора изобретения в качестве примера далее будет описываться полимеризация алкиленоксида.
На алкиленоксид особенных ограничений не накладывается, но примеры могут включать эпоксидные соединения, такие как этиленоксид, пропиленоксид, 1,2-бутиленоксид, 2,3-бутиленоксид, оксид стирола, циклогексеноксид, эпихлоргидрин, эпибромгидрин, метилглицидиловый эфир, аллилглицидиловый эфир, фенилглицидиловый эфир, и их можно использовать индивидуально или в смеси из двух и более компонентов. На уровень содержания фосфазенового нанесенного на носитель катализатора, предусматриваемый для проведения полимеризации алкиленоксида, особенных ограничений не накладывается, но он находится в диапазоне в общем случае от 1 х 10-15 до 5 х 10-1 моль, а предпочтительно от 1 х 10-7 до 1 х 10-2 моль.
На способ полимеризации алкиленоксида особенных ограничений не накладывается, но предпочтительным является использование обычных способов, продемонстрированных, например, в документах JP-A №№ 10-77289 и 2000-327769. В общем случае фосфазеновый нанесенный на носитель катализатор добавляют в реактор при необходимости совместно с активными гидридами, растворителем и тому подобным, а побочные продукты при необходимости удаляют. После этого используют способ подачи необходимого количества алкиленоксида в один заход или способ периодической или непрерывной подачи.
Замещение заместителя с использованием фосфазенового нанесенного на носитель катализатора изобретения может включать, например, алкилирование или тиоалкилирование фенольной гидроксильной группы, замещение в случае галогена алифатических или ароматических галогенидов и группы алкокси, тиоалкильной группы, аминогруппы, группы фторида, цианогруппы, карбоксильной группы и группы арилокси, замещение в случае алифатического или ароматического сульфонатного соединения и группы алкокси, тиоалкильной группы, аминогруппы, группы фторида, цианогруппы, карбоксильной группы и группы арилокси и переэтерификацию в случае алифатического или ароматического карбоксилатного соединения и группы алкокси. В способе замещения заместителя с использованием фосфазенового нанесенного на носитель катализатора изобретения в качестве примера далее будет описываться алкилирование фенольной гидроксильной группы.
В результате проведения реакции между ароматическим соединением, имеющим, по меньшей мере, одну гидроксильную группу, связанную с его ароматическим кольцом, и сложным диэфиром угольной кислоты в присутствии фосфазенового нанесенного на носитель катализатора изобретения можно получить ароматические простые эфиры, в которых гидроксильная группа в ароматическом соединении будет подвергнута этерификации с получением простого эфира. Например, в результате проведения реакции между соединением, имеющим одну гидроксильную группу в ароматическом кольце, таким как фенол, и сложным диалкиловым эфиром угольной кислоты, таким как диметилкарбонат, можно получить ароматический простой эфир, в случае которого гидроксильная группа ароматического соединения будет замещена группой алкокси. В том случае, если ароматическое соединение, имеющее, по меньшей мере, одну гидроксильную группу, связанную с его ароматическим кольцом, будет представлять собой соединение, имеющее несколько гидроксильных групп, то тогда в соответствии с условиями реакции для реакции с ароматическим кольцом может быть получено соединение, имеющее одну или несколько гидроксильных групп, подвергнутых этерификации с получением простого эфира.
Описанное выше ароматическое соединение и сложный диэфир угольной кислоты, который можно использовать в качестве материалов исходного сырья, описываются в документе JP-A № 2004-107266. На уровень содержания фосфазенового нанесенного на носитель катализатора, предусматриваемый для проведения алкилирования фенольной гидроксильной группы, особенных ограничений не накладывается, но он находится в диапазоне вобщем случае от 1 х 10-7 до 10 моль при расчете на 1 моль фенольной гидроксильной группы, и особенных ограничений не накладывается и на способ проведения реакции. Например, предпочитается использовать обычный способ, продемонстрированный в документе JP-A № 2004-107266. В общем случае способ реализуют в результате проведения реакции между ароматическим соединением, имеющим, по меньшей мере, одну гидроксильную группу в своем ароматическом кольце, и сложным диэфиром угольной кислоты в растворителе сложного диэфира угольной кислоты. При необходимости могут быть использованы и другие растворители.
На уровень содержания растворителя ограничений не накладывается, но его можно будет определить в зависимости от условий проведения реакции, таких как тип и количество ароматического соединения, имеющего, по меньшей мере, одну гидроксильную группу в своем ароматическом кольце, тип и количество сложного диэфира угольной кислоты, температура реакции и давление. На условия, такие как температура реакции, давление и тому подобное, особенных ограничений не накладывается до тех пор, пока при использовании данных условий будут получать ароматический простой эфир. Температура реакции и давление в общем случае находятся в диапазонах от 0°С до 250°С и от 1 атм. до 100 атм. Время реакции не является постоянным в соответствии с типом и количеством соединения, имеющего, по меньшей мере, одну гидроксильную группу, непосредственно связанную с его ароматическим кольцом, типом и количеством сложного диэфира угольной кислоты, типом и количеством катализатора, температурой реакции, давлением реакции и типом и количеством использованного растворителя, но в общем случае оно находится в диапазоне от 15 минут до 100 часов. В результате использования фосфазенового нанесенного на носитель катализатора изобретения в качестве катализатора в описанной выше реакции можно очень эффективно получать целевой продукт.
Реакция образования связи углерод-углерод при использовании фосфазенового нанесенного на носитель катализатора изобретения включает, например, альдольную реакцию, реакцию Михаэля, реакцию Кновенагеля, реакцию Петерсона, реакцию Перкина, реакцию Дарценса, реакцию Толленса и реакцию Торпа, но в том, что касается реакции образования связи углерод-углерод при использовании фосфазенового нанесенного на носитель катализатора изобретения, в качестве примера далее приводятся разъяснения для альдольной реакции.
В случае нахождения карбонильного соединения, такого как альдегид и кетон, в щелочной среде происходит так называемая альдольная реакция (конденсация) с образованием β-гидроксикарбонильного соединения или α,β-ненасыщенного карбонильного соединения.
На количество фосфазенового нанесенного на носитель катализатора, используемого в реакции, особенных ограничений не накладывается, но оно находится в диапазоне в общем случае от 1 х 10-7 до 10 моль, а предпочтительно от 1 х 10-3 до 1 моль, при расчете на один моль карбонильного соединения. На способ проведения реакции особенных ограничений не накладывается, и может быть использован известный способ. Обычно карбонильное соединение в качестве реагента в присутствии катализатора вводят в реакцию при использовании инертного растворителя или самого карбонильного соединения в качестве растворителя.
На количество использованного растворителя ограничений не накладывается, и его можно будет надлежащим образом определить в зависимости от условий проведения реакции, таких как тип и количество карбонильного соединения, температура реакции, давление реакции и тому подлобное. Температура и давление для реакции обычно находятся в диапазонах от -78°С до 250°С и от 1 атм до 100 атм соответственно. Время реакции варьируется в соответствии с типом и количеством карбонильного соединения, типом и количеством катализатора, температурой реакции, давлением реакции, типом и количеством растворителя и тому подобным, но в общем случае оно находится в диапазоне от 15 минут до 100 часов. В результате использования фосфазенового нанесенного на носитель катализатора изобретения в качестве катализатора в описанной выше реакции можно эффективно получать целевой продукт.
Примеры
Изобретение будет подробно описываться в связи со следующими примерами, но изобретение ими не ограничивается.
Пример 1
Синтез 1,1,1-трис{[трис(диметиламино)фосфоранилиден]амино}-3,3-бис(диметиламино)-3-метиламино-1λ5,3λ5-дифосфазена (далее в настоящем документе сокращенно обозначаемого как PZNB)
В стеклянную колбу объемом 1 л, оснащенную перемешивающим устройством, которую выдерживали в атмосфере азота, вводили 54 г хлорида тетракис[трис(диметиламино)фосфоранилиденамино]фосфония и 13 г калий-1-октилтиолята. Сюда же добавляли 450 мл тетралина и получали суспензию. При перемешивании суспензию нагревали до 185°С в течение 5 часов, а после этого охлаждали до комнатной температуры. Полученную суспензию отфильтровывали в атмосфере азота и промывали с использованием 100 мл тетралина с получением 531 г желтого раствора. Часть раствора добавляли к бензолу-d6, а в качестве внутреннего стандарта при проведении измерений по методу 31Р ЯМР использовали триамид гексаметилфосфорной кислоты. В данном случае наблюдали квинтуплет, соответствующий 1 атому фосфора, в области - 30,3 м.д., дублет, соответствующий 3 атомам фосфора, в области 6,79 м.д., дублет, соответствующий 1 атому фосфора, в области 16,04 м.д., а концентрация PZNB в твердой фазе составляла 0,163 ммоль/г. Кроме того, согласно результатам анализа по методу FD-MS (масс-спектроскопия с полевой десорбцией) наблюдали пик 725, соответствующий недиссоциированным молекулам PZNB. При проведении для желтого раствора отгонки при пониженном давлении получали 62,0 г светло-желтой твердой фазы. Результаты анализа твердой фазы по методам 31Р ЯМР и FD-MS представляли собой то же самое, что и результаты для желтого раствора.
Пример 2
Синтез нанесенного на полимерный носитель хлорида фосфазения
В стеклянную колбу объемом 1 л, оснащенную перемешивающим устройством, которую выдерживали в атмосфере азота, добавляли и перемешивали при комнатной температуре в течение 1 часа 51 г смолы на основе полистирола, подвергнутого хлорметилированию, (54 ммоль при расчете на атомы хлора) (произведенной в компании Argonaut Technologies, Inc., ArgoPore-Cl, 1,05 ммоль Cl/г) и 430 г тетралина. После этого дополнительно добавляли 343 г раствора PZNB, полученного в примере 1 (56 ммоль при расчете на PZNB), в тетралине и смесь непрерывно перемешивали в течение 4 дней. Таким образом полученную суспензию отфильтровывали в атмосфере азота и промывали с использованием 500 мл тетралина и 2 л смешанного растворителя, характеризующегося соотношением 1:1 для молекулярных масс в смеси 1,4-диоксан:метанол. Оставшуюся твердую фазу высушивали при 70°С при пониженном давлении, равном 1 мм ртутного столба, и получали 72 г нанесенного на полимерный носитель хлорида фосфазения. Для части твердой фазы проводили измерения по методу 31Р ЯМР при использовании тетрафторбората тетракис(диметиламино)фосфония в качестве внутреннего стандарта. В данном случае наблюдали пик, соответствующий 1 атому фосфора, в области - 35,4 м.д., пик, соответствующий 4 атомам фосфора, в области 5,8 м.д., а концентрация фосфазениевых катионов в твердой фазе согласно измерению по методу 31Р ЯМР составляла 0,427 ммоль/г.
Пример 3
Синтез нанесенного на полимерный носитель гидроксида фосфазения
В стеклянную колбу объемом 1 л, оснащенную перемешивающим устройством и холодильником, которую выдерживали в атмосфере азота, вводили и перемешивали при комнатной температуре в течение 1 часа 49 г нанесенного на полимерный носитель хлорида фосфазения (21 ммоль при расчете на фосфазениевые катионы), полученного в примере 2, и 400 мл метанола. После этого добавляли раствор, полученный в результате растворения 9,7 г метоксида натрия в 100 мл метанола, и в течение 8 часов проводили нагревание при кипячении в условиях флегмообразования, а после этого охлаждение до комнатной температуры. Полученную суспензию отфильтровывали, промывали водой, подвергали обработке в результате введения в контакт с 880 г 4%-ного водного раствора гидроксида натрия и промывали водой дополнительно. Оставшуюся твердую фазу высушивали при нагревании при 70°С при пониженном давлении, равном 1 мм ртутного столба, и получали 49 г нанесенного на полимерный носитель гидроксида фосфазения. Концентрация фосфазениевых катионов в твердой фазе согласно измерению по методу 31Р ЯМР составляла 0,431 ммоль/г. В дополнение к этому, согласно результатам элементного анализа атомов хлора не наблюдали, а анионом количественно являлся гидроксид.
Пример 4
Синтез нанесенного на полимерный носитель иодида фосфазения
8 г нанесенного на полимерный носитель гидроксида фосфазения (3,4 ммоль при расчете на фосфазениевые катионы), полученного в примере 3, в качестве набивки помещали в колонку. Колонку подвергали обработке в результате введения в контакт с 210 г 4%-ного водного раствора хлорида натрия и дополнительно промывали водой. После этого колонку подвергали обработке в результате введения в контакт с 61 г 4%-ного водного раствора иодида натрия и еще раз промывали водой. По завершении обработки полученную твердую фазу высушивали при нагревании при 70°С при пониженном давлении, равном 1 мм ртутного столба, и получали нанесенный на полимерный носитель иодид фосфазения. Концентрация фосфазениевых катионов в твердой фазе согласно измерению по методу 31Р ЯМР составляла 0,352 ммоль/г. В дополнение к этому, концентрация атомов иода в твердой фазе согласно результатам элементного анализа составляла 0,358 ммоль/г.
Пример 5
Синтез иодида фосфазения, имеющего гидроксильную группу
В стеклянную колбу объемом 300 мл, оснащенную перемешивающим устройством, которую выдерживали в атмосфере азота, вводили 4,3г (16 ммоль) простого трет-бутилдиметилсилил(4-хлорметилбензил)эфира и 79 г раствора PZNB, полученного в примере 1 (13 ммоль при расчете на PZNB), в тетралине и смесь перемешивали в течение ночи при комнатной температуре. По завершении реакции тетралин отгоняли при пониженном давлении и при перемешивании добавляли 100 мл гексана. Гексан удаляли в результате декантации. Такое промывание гексаном в совокупности проводили пять раз, а получающуюся в результате смесь высушивали при нагревании при 70°С при пониженном давлении, равном 1 мм ртутного столба. После этого при проведении охлаждения колбы на льду добавляли раствор (1,0 М, 14 ммоль) фторида тетрабутиламмония в тетрагидрофуране (далее в настоящем документе обозначаемом как ТГФ) и смесь перемешивали при 40°С в течение 3 часов, а после этого добавляли 14 мл (14 ммоль) 1 н. водного раствора хлористоводородной кислоты и, кроме того, непрерывно перемешивали. После этого при пониженном давлении ТГФ и воду отгоняли. К остатку добавляли 250 мл метиленхлорида с получением раствора и раствор промывали водой при использовании делительной воронки. Слой метиленхлорида конденсировали и получали 10,8 г вязкой жидкости. После этого к полученной вязкой жидкости добавляли 70%-ный водный раствор этиламина, добиваясь его растворения. Полученный раствор помещали в стеклянную колбу объемом 300 мл, оснащенную перемешивающим устройством. Сюда же при перемешивании добавляли 4,6 г иодида натрия в 70%-ном растворе этиламина и 80 мл воды и реакции в смеси давали возможность протекать в течение 5 дней. Осажденные кристаллы отфильтровывали, промывали водой и высушивали при нагревании при 70°С при пониженном давлении, равном 1 мм ртутного столба, с получением 7,0 г иодида фосфазения, имеющего гидроксильную группу, в виде белых кристаллов. Часть раствора добавляли к бензолу-d6, а в качестве внутреннего стандарта при проведении измерений по методу 31Р ЯМР использовали триамид гексаметилфосфорной кислоты. В данном случае наблюдали квинтуплет, соответствующий 1 атому фосфора, в области - 33,2 м.д., дублет, соответствующий 3 атомам фосфора, в области 7,45 м.д., дублет, соответствующий 1 атому фосфора, в области 8,68 м.д., а степень чистоты составляла 96,5%. Кроме того, согласно результатам анализа по методу FD-MS наблюдали пик 846, соответствующий недиссоциированным фосфазениевым катионам.
Пример 6
Синтез иодида фосфазения, содержащего полипропиленоксид в качестве боковой цепи
В автоклав объемом 70 мл, который выдерживали в атмосфере азота, вводили 5,1 г (5,2 ммоль) иодида фосфазения, полученного в примере 5, 0,01 г (0,26 ммоль) гидрида калия и 30 мл ТГФ и смесь нагревали до 80°С при перемешивании в течение 3 часов, а после этого охлаждали до комнатной температуры. Затем добавляли 3,0 г (52 ммоль) пропиленоксида и проводили нагревание до 80°С при перемешивании в течение 20 часов. По завершении реакции реакционный раствор охлаждали до комнатной температуры и добавляли 0,3 мл (0,3 ммоль) 1 н. водного раствора хлористоводородной кислоты. По завершении промывания водой реакционный раствор высушивали при нагревании при 70°С при пониженном давлении, равном 1 мм ртутного столба, и получали 8,0 г вязкой жидкости. Часть раствора добавляли к диметилсульфоксиду-d6 (далее в настоящем документе обозначаемому как ДМСО-d6), а в качестве внутреннего стандарта при проведении измерений по методу 31Р ЯМР использовали триамид гексаметилфосфорной кислоты. В данном случае наблюдали квинтуплет, соответствующий 1 атому фосфора, в области - 33,1 м.д., дублет, соответствующий 3 атомам фосфора, в области 7,56 м.д., дублет, соответствующий 1 атому фосфора, в области 8,79 м.д., а степень чистоты составляла 100,6%. Кроме того, согласно результатам анализа по методу FD-MS наблюдали пик, соответствующий среднечисленной молекулярной массе (Mn) 1412 и молекулярно-массовому распределению (Mw/Mn) 1,02. Поэтому было обнаружено, что в результате проведения добавления в рамках "живой" полимеризации происходило введение боковой цепи с получением иодида фосфазения, содержащего декамер пропиленоксида.
Примеры от 7 до 11
Синтез иодида фосфазения, содержащего полиалкиленоксид в качестве боковой цепи
Синтез проводили по тому же самому способу, что и в примере 6, за исключением того, что изменяли количество и типы полиалкиленоксида, благодаря чему в качестве боковой цепи производили введение широкого ассортимента полиалкиленоксидов, таким образом, синтезируя иодид фосфазения. Результаты продемонстрированы в таблице 1.
Пример 12
Синтез нанесенного на полимерный носитель гидроксида фосфазения
В стеклянную колбу объемом 50 мл, которую выдерживали в атмосфере азота, вводили 0,2 г (5,2 ммоль) гидрида калия и 9 мл N,N-диметилформамида (далее в настоящем документе обозначаемого как ДМФА), кроме того, 7,3 г раствора иодида фосфазения, полученного в примере 6, (5,2 ммоль) в ДМФА, а смесь перемешивали при комнатной температуре в течение 3 часов. В еще одну стеклянную колбу объемом 100 мл, оснащенную перемешивающим устройством, которую выдерживали в атмосфере азота, вводили 5,0 г смолы на основе полистирола, подвергнутого хлорметилированию (5,2 ммоль при расчете на атомы хлора) (произведенной в компании Argonaut Technologies, Inc., ArgoPore-Cl, 1,05 ммоль Cl/г), и 50 мл ДМФА, а смесь перемешивали при комнатной температуре в течение 1 часа. После этого добавляли все количество раствора предварительно полученной калиевой соли иодида фосфазения в ДМФА и, кроме того, проводили непрерывное перемешивание в течение 20 часов. По завершении реакции проводили отфильтровывание. Смолу, полученную в виде оставшейся твердой фазы, промывали в аппарате Сокслета 1,4-диоксаном, используемым в качестве растворителя, и высушивали при нагревании при 70°С при пониженном давлении, равном 1 мм ртутного столба. По завершении высушивания смолу и 35 мл метанола помещали в стеклянную колбу объемом 100 мл, оснащенную перемешивающим устройством и холодильником, которую выдерживали в атмосфере азота, а смесь перемешивали при комнатной температуре в течение 1 часа, сюда же добавляли раствор (20 мл) 2,5 г метоксида натрия в метаноле и в течение 8 часов проводили нагревание при кипячении в условиях флегмообразования, а после этого охлаждение до комнатной температуры. Полученную суспензию отфильтровывали, промывали водой, подвергали обработке в результате введения в контакт с 34 г 4%-ного водного раствора гидроксида натрия и промывали водой дополнительно. После этого оставшуюся твердую фазу высушивали при 70°С при пониженном давлении, равном 1 мм ртутного столба, и получали 5,3 г нанесенного на полимерный носитель гидроксида фосфазения. Для части твердой фазы проводили измерения по методу 31Р ЯМР при использовании в качестве внутреннего стандарта тетрафторбората тетракис(диметиламино)фосфония. В данном случае наблюдали пик, соответствующий 1 атому фосфора, в области - 35,7 м.д., пик, соответствующий 4 атомам фосфора, в области 5,7 м.д., а концентрация фосфазениевых катионов в твердой фазе составляла 0,122 ммоль/г согласно измерениям по методу 31Р ЯМР.
Примеры от 13 до 17
Синтез нанесенного на полимерный носитель гидроксида фосфазения
Реакцию проводили по тому же самому способу, что и в примере 12, за исключением того, что вместо иодида фосфазения, полученного в примере 6 и использованного в примере 12, использовали иодид фосфазения, полученный в примерах от 7 до 11, поэтому синтезировали широкий ассортимент нанесенных на полимерный носитель гидроксидов фосфазения. Результаты продемонстрированы в таблице 2.
Пример 18
Синтез бис(диметиламино){[трис({[трис(диметиламино)фосфоранилиден]амино})фосфоранилиден]амино}фосфиноксида (далее в настоящем документе сокращенно обозначаемого как PZND)
При использовании способа, описанного в документе JP-A № 11-240893, в водном растворе получали 7607 г гидроксида тетракис[трис(диметиламино)фосфоранилиденамино]фосфония. Отгонку для водного раствора проводили при пониженном давлении в диапазоне от 50 до 100 мм ртутного столба при 60°С с получением в качестве твердой фазы 180 г гидроксида тетракис[трис(диметиламино)фосфоранилиденамино]фосфония. Твердую фазу переводили в стеклянную колбу объемом 5 л и нагревали при 130°С в течение 5 дней при подаче потока азота через трубку для ввода азота с расходом 10 мл/мин. После охлаждения приблизительно до комнатной температуры добавляли 3 л гексана и при помощи перемешивающего устройства смесь перемешивали в течение 30 минут. По завершении перемешивания содержимое оставляли в состоянии покоя для осаждения нерастворимых фракций. Нерастворимые фракции отделяли в результате декантации и получали прозрачный и бесцветный раствор в гексане. н-Гексан из раствора отгоняли при нормальном давлении. В тот момент, когда собирали приблизительно 2,8 л н-Гексана, отгонку прекращали. н-Гексан дополнительно удаляли при пониженном давлении в диапазоне от 1 до 20 мм ртутного столба и получали 110 г белой твердой фазы. К части твердой фазы добавляли ДМСО-d6, а в качестве внутреннего стандарта при проведении измерений по методу 31Р ЯМР использовали триамид гексаметилфосфорной кислоты. В данном случае наблюдали квинтуплет, соответствующий 1 атому фосфора, в области - 25,46 м.д., дублет, соответствующий 1 атому фосфора, в области 7,08 м.д., дублет, соответствующий 3 атомам фосфора, в области 8,64 м.д., а степень чистоты составляла 97,5%. Кроме того, согласно результатам анализа по методу FD-MS наблюдали пик 711, соответствующий недиссоциированным молекулам PZND.
Пример 19
Синтез хлорида 1,1,1-трис{[трис(диметиламино)фосфоранилиден]амино}-3,3-бис(диметиламино)-3-хлор-3λ5-дифосфазе-1-ния (далее в настоящем документе сокращенно обозначаемого как PZND-Cl)
В стеклянной колбе объемом 500 мл, оснащенной перемешивающим устройством, которую выдерживали в атмосфере азота, в 50 мл ТГФ растворяли 30 г (42 ммоль) PZND, полученного в примере 18. При комнатной температуре добавляли 7,3 г (43 ммоль) 2,2-дихлор-1,3-диметилимидазолидина в виде твердой фазы, а после этого в течение приблизительно 3 часов проводили кипячение в условиях флегмообразования. После охлаждения до комнатной температуры ТГФ отгоняли при пониженном давлении, а после этого последовательно добавляли 50 мл ТГФ и 200 мл диэтилового эфира. После проведения кипячения в условиях флегмообразования в течение 30 минут получающуюся в результате смесь тщательно перемешивали и охлаждали до комнатной температуры для осаждения белой твердой фазы. Твердую фазу отфильтровывали в атмосфере азота и высушивали при пониженном давлении, равном 1 мм ртутного столба, с получением 26 г белой твердой фазы. Часть твердой фазы добавляли к ДМСО-d6, а в качестве внутреннего стандарта при проведении измерений по методу 31Р ЯМР использовали триамид гексаметилфосфорной кислоты. В данном случае наблюдали октеты, соответствующие 1 атому фосфора, в области - 29,81 м.д., дублет, соответствующий 1 атому фосфора, в области 6,22 м.д. и дублет, соответствующий 3 атомам фосфора, в области 12,04 м.д., а степень чистоты составляла 99,5%. Кроме того, согласно результатам анализа по методу FD-MS наблюдали пик 730, соответствующий недиссоциированным молекулам PZND-Cl.
Пример 20
Синтез хлорида фосфазения, имеющего группу силокси
В стеклянную колбу объемом 300 мл, оснащенную перемешивающим устройством, которую выдерживали в атмосфере азота, вводили 21 г (27 ммоль) PZND-Cl, полученного в примере 19, 150 мл о-дихлорбензола, 9,1 г (52 ммоль) простого трет-бутилдиметилсилил(2-аминоэтил)эфира и 4,7 г (27 ммоль) трис(диметиламино)фосфолана и реакции в смеси при 120°С давали возможность протекать в течение 24 часов, после этого проводили охлаждение до комнатной температуры. Реакционную смесь промывали водой, для слоя о-дихлорбензола проводили отгонку при пониженном давлении и после этого промывание с использованием 200 мл диэтилового эфира. Промытую твердую фазу высушивали при нормальном давлении и получали 82 г белой твердой фазы. Часть твердой фазы добавляли к ТГФ-d8, а в качестве внутреннего стандарта при проведении измерений по методу 31Р ЯМР использовали сложный три-н-бутиловый эфир фосфорной кислоты. В данном случае наблюдали октеты, соответствующие 1 атому фосфора, в области - 27,52 м.д., дублет, соответствующий 1 атому фосфора, в области 9,77 м.д. и дублет, соответствующий 3 атомам фосфора, в области 10,45 м.д.
Пример 21
Синтез иодида фосфазения, имеющего гидроксильную группу
В стеклянную колбу объемом 500 мл, оснащенную перемешивающим устройством, которую выдерживали в атмосфере азота, вводили 200 мл ТГФ, 10,9 г (455 ммоль) гидрида натрия и 67 г (470 ммоль) метилиодида, а смесь перемешивали при комнатной температуре в течение 2 часов. После этого добавляли 41 г раствора хлорида фосфазения, полученного в примере 20 (45 ммоль), в ТГФ и смесь дополнительно перемешивали при комнатной температуре в течение 16 часов. По завершении реакции суспензию отфильтровывали и промывали с использованием ТГФ, а ТГФ отгоняли из фильтрата при пониженном давлении. Полученную твердую фазу еще раз растворяли в 300 мл метиленхлорида и нерастворимые фракции отфильтровывали, а метиленхлорид из фильтрата отгоняли при пониженном давлении с получением 41 г белой твердой фазы. В стеклянную колбу объемом 500 мл, оснащенную перемешивающим устройством, которую выдерживали в атмосфере азота, вводили вышеупомянутую белую твердую фазу и 200 мл ТГФ, а смесь охлаждали на льду при перемешивании. Сюда же добавляли 41 мл раствора фторида тетрабутиламмония (1,0 М, 41 ммоль) в ТГФ и смесь перемешивали при комнатной температуре в течение 30 минут. Для разделения жидкости к реакционному раствору добавляли 300 мл воды и 300 мл метиленхлорида и из слоя органического растворителя растворитель отгоняли при пониженном давлении с получением твердой фазы, которую подвергали перекристаллизации из 70%-ного водного раствора этиламина, таким образом получая 29 г белой твердой фазы. Часть твердой фазы добавляли к ДМСО-d6, а в качестве внутреннего стандарта при проведении измерений по методу 31Р ЯМР использовали триамид гексаметилфосфорной кислоты. В данном случае наблюдали квинтуплет, соответствующий 1 атому фосфора, в области - 33,34 м.д., дублет, соответствующий 3 атомам фосфора, в области 7,60 м.д. и дублет, соответствующий 1 атому фосфора, в области 7,79 м.д., а степень чистоты составляла 100%.
Пример 22
Синтез нанесенного на полимерный носитель гидроксида фосфазения
В стеклянную колбу объемом 100 мл, которую выдерживали в атмосфере азота, вводили 0,7 г (29 ммоль) гидрида натрия и 50 мл ДМФА и сюда же дополнительно добавляли раствор 25,2 г иодида фосфазения, полученного в примере 21 (28 ммоль), в ДМФА, а смесь перемешивали при комнатной температуре в течение 3 часов. В еще одну стеклянную колбу объемом 500 мл, оснащенную перемешивающим устройством, которую выдерживали в атмосфере азота, вводили 26,6 г смолы на основе полистирола, подвергнутого хлорметилированию (28 ммоль при расчете на атомы хлора) (произведенной в компании Argonaut Technologies, Inc., ArgoPore Cl, 1,05 ммоль Cl/г), и 300 мл ДМФА, а смесь перемешивали при комнатной температуре в течение 2 часов. После этого сюда же добавляли все количество раствора предварительно полученной натриевой соли иодида фосфазения в ДМФА и смесь дополнительно перемешивали в течение 20 часов. По завершении реакции получающуюся в результате смесь отфильтровывали, а смолу, полученную в виде оставшейся твердой фазы, пять раз промывали с использованием 50 мл метанола и высушивали при нагревании при 70°С при пониженном давлении, равном 1 мм ртутного столба. В стеклянную колбу объемом 500 мл, оснащенную перемешивающим устройством и холодильником, которую выдерживали в атмосфере азота, вводили высушенную смолу и 200 мл метанола, а смесь перемешивали при комнатной температуре в течение 1 часа. Сюда же добавляли раствор (100 мл) 7,9 г метоксида натрия в метаноле и в течение 8 часов смесь нагревали при кипячении в условиях флегмообразования, а затем охлаждали до комнатной температуры. Полученную суспензию отфильтровывали и промывали водой, подвергали обработке в результате введения в контакт с 527 г 4%-ного водного раствора гидроксида натрия и промывали водой дополнительно. Таким образом, оставшуюся твердую фазу высушивали при 70°С при пониженном давлении, равном 1 мм ртутного столба, с получением 35 г нанесенного на полимерный носитель гидроксида фосфазения. Для части твердой фазы проводили измерения по методу 31Р ЯМР при использовании тетрафторбората тетракис(диметиламино)фосфония в качестве внутреннего стандарта. В данном случае наблюдали пик, соответствующий 1 атому фосфора, в области - 35,5 м.д. и пик, соответствующий 4 молям фосфора, в области 5,9 м.д., а концентрация фосфазениевых катионов в твердой фазе согласно измерению по методу 31Р ЯМР составляла 0,371 ммоль/г.
Пример 23
Синтез нанесенного на полимерный носитель хлорида фосфазения
17,4 г нанесенного на полимерный носитель гидроксида фосфазения (6,6 ммоль при расчете на фосфазениевые катионы), полученного в примере 22, в качестве набивки помещали в колонку и подвергали обработке в результате введения в контакт с 398 г 4%-ного водного раствора хлорида натрия и дополнительно промывали водой. Затем по завершении обработки твердую фазу высушивали при 70°С при пониженном давлении, равном 1 мм ртутного столба, и получали нанесенный на полимерный носитель хлорид фосфазения. Концентрация фосфазениевых катионов в твердой фазе согласно измерению по методу 31Р ЯМР составляла 0,368 ммоль/г.
Пример 24
Синтез хлорида фосфазения, имеющего группу силокси
Реакцию проводили по тому же самому способу, что и в примере 20, за исключением того, что вместо простого трет-бутилдиметилсилил(2-аминоэтил)эфира, использованного в примере 20, использовали простой диэтиленгликоль(трет-бутилдиметилсилил)(2-аминоэтил)эфир. Согласно результатам анализа по методу FD-MS наблюдали пик 958, соответствующий недиссоциированным молекулам хлорида фосфазения, имеющего группу силокси.
Пример 25
Синтез гексафторфосфата фосфазения, имеющего гидроксильную группу
Реакцию проводили по тому же способу, что и в примере 21, за исключением того, что вместо хлорида фосфазения, использованного в примере 21, использовали хлорид фосфазения, полученный в примере 24, и получали белую твердую фазу. В стеклянную колбу объемом 50 мл вводили 3,0 г вышеупомянутой твердой фазы, 20 мл 70%-ного раствора этиламина и 0,5 г гексафторфосфата натрия и смесь в течение некоторого времени перемешивали. Суспензию получающейся в результате смеси отфильтровывали, а фильтрату давали возможность отстояться для прохождения перекристаллизации с получением 1,3 г белых твердых кристаллов. Часть твердой фазы добавляли к ДМСО-d6, а в качестве внутреннего стандарта при проведении измерений по методу 31Р ЯМР использовали триамид гексаметилфосфорной кислоты. В данном случае наблюдали квинтуплет, соответствующий 1 атому фосфора, в области - 33,31 м.д., дублет, соответствующий 3 атомам фосфора, в области 7,58 м.д., дублет, соответствующий 1 атому фосфора, в области 7,72 м.д. и гептет, соответствующий 1 атому фосфора, в области 57,0 м.д., а степень чистоты составляла 91,4%. Кроме того, согласно результатам анализа по методу FD-MS наблюдали пик 858, соответствующий недиссоциированной части фосфазениевых катионов.
Пример 26
Синтез нанесенного на полимерный носитель гексафторфосфата фосфазения
В стеклянную колбу объемом 50 мл, которую выдерживали в атмосфере азота, вводили 0,07 г (1,7 ммоль) гидрида калия и 10 мл ДМФА, сюда же дополнительно добавляли 1,3 г (1,3 ммоль) гексафторфосфата фосфазения, полученного в примере 25, а смесь перемешивали при комнатной температуре в течение 3 часов. В еще одну стеклянную колбу объемом 100 мл, оснащенную перемешивающим устройством, которую выдерживали в атмосфере азота, вводили 1,1 г смолы на основе полистирола, подвергнутого хлорметилированию (1,3 ммоль при расчете на атомы хлора) (произведенной в компании Argonaut Technologies, Inc., ArgoPore Cl, 1,20 ммоль Cl/г), и 15 мл ДМФА, а смесь перемешивали при комнатной температуре в течение 1 часа. После этого сюда же добавляли все количество раствора предварительно полученной калиевой соли гексафторфосфата фосфазения в ДМФА и смесь дополнительно перемешивали в течение 24 часов. По завершении реакции получающуюся в результате смесь отфильтровывали, а смолу, полученную в виде оставшейся твердой фазы, промывали в аппарате Сокслета 1,4-диоксаном, используемым в качестве растворителя, а после этого высушивали при нагревании при 70°С при пониженном давлении, равном 1 мм ртутного столба, с получением 1,1 г нанесенного на полимерный носитель гексафторфосфата фосфазения. Для части твердой фазы проводили измерения по методу 31Р ЯМР при использовании тетрафторбората тетракис(диметиламино)фосфония в качестве внутреннего стандарта. В данном случае наблюдали пик, соответствующий 1 атому фосфора, в области - 35,7 м.д. и пик, соответствующий 4 молям фосфора, в области 5,9 м.д., а концентрация фосфазениевых катионов в твердой фазе согласно измерению по методу 31Р ЯМР составляла 0,266 ммоль/г.
Пример 27
Синтез нанесенного на полимерный носитель гидроксида фосфазения
В стеклянную колбу объемом 50 мл, оснащенную перемешивающим устройством и холодильником, которую выдерживали в атмосфере азота, вводили все количество нанесенного на полимерный носитель гексафторфосфата фосфазения, полученного в примере 26, и 10 мл метанола, а смесь перемешивали при комнатной температуре в течение 1 часа. Сюда же добавляли 10 мл раствора 1,0 г метоксида натрия в метаноле и в течение 8 часов смесь нагревали при кипячении в условиях флегмообразования, а после этого охлаждали до комнатной температуры. Полученную суспензию отфильтровывали и промывали водой, подвергали обработке в результате введения в контакт с 34 г 4%-ного водного раствора гидроксида натрия и промывали водой. После этого оставшуюся твердую фазу высушивали при 70°С при пониженном давлении, равном 1 мм ртутного столба, и получали 1,1 г нанесенного на полимерный носитель гидроксида фосфазения. Для части твердой фазы проводили измерения по методу 31Р ЯМР при использовании в качестве внутреннего стандарта тетрафторбората тетракис(диметиламино)фосфония. В данном случае наблюдали пик, соответствующий 1 атому фосфора, в области - 35,7 м.д. и пик, соответствующий 4 молям фосфора, в области 5,9 м.д., а концентрация фосфазениевых катионов в твердой фазе составляла 0,297 ммоль/г согласно измерениям по методу 31Р ЯМР.
Пример 28
Синтез нанесенного на полимерный носитель гидроксида фосфазения
В стеклянную колбу объемом 100 мл, оснащенную перемешивающим устройством, которую выдерживали в атмосфере азота, вводили 4,7 г смолы на основе полистирола, подвергнутого хлорметилированию (2,0 ммоль при расчете на атомы хлора) (произведенной в компании Argonaut Technologies, Inc., Argo-Gel-Wang Cl, 0,43 ммоль Cl/г), и 20 г тетралина, а смесь перемешивали при комнатной температуре в течение 1 часа. После этого сюда же добавляли 17,8 г раствора PZNB, полученного в примере 1 (2,7 ммоль при расчете на PZNB), в тетралине и смесь, кроме того, непрерывно перемешивали в течение 4 дней. Таким образом полученную суспензию отфильтровывали в атмосфере азота и промывали с использованием 50 мл тетралина и 200 мл смешанного растворителя, характеризующегося массовым соотношением 1:1 в смеси 1,4-диоксан: метанол. Полученную оставшуюся твердую фазу высушивали при 70°С при пониженном давлении, равном 1 мм ртутного столба, и получали 5,0 г нанесенного на полимерный носитель хлорида фосфазения. Для части твердой фазы проводили измерения по методу 31Р ЯМР при использовании тетрафторбората тетракис(диметиламино)фосфония в качестве внутреннего стандарта. В данном случае наблюдали пик, соответствующий 1 атому фосфора, в области - 35,4 м.д. и пик, соответствующий 4 молям фосфора, в области 5,8 м.д., а концентрация фосфазениевых катионов в твердой фазе согласно измерению по методу 31Р ЯМР составляла 0,316 ммоль/г. После этого в стеклянную колбу объемом 50 мл, оснащенную перемешивающим устройством и холодильником, которую выдерживали в атмосфере азота, вводили 2,3 г полученного предварительно нанесенного на полимерный носитель хлорида фосфазения (6,1 ммоль при расчете на фосфазениевые катионы) и 10 мл метанола, а смесь перемешивали при комнатной температуре в течение 1 часа. После этого 0,37 г метоксида натрия растворяли в 10 мл метанола и раствор добавляли, в течение 8 часов нагревали при кипячении в условиях флегмообразования, а после этого охлаждали до комнатной температуры. Полученную суспензию отфильтровывали и промывали водой, подвергали обработке в результате введения в контакт с 80 г 4%-ного водного раствора гидроксида натрия и промывали водой. После этого оставшуюся твердую фазу высушивали при нагревании при 70°С при пониженном давлении, равном 1 мм ртутного столба, и получали 2,3 г нанесенного на полимерный носитель гидроксида фосфазения. Концентрация фосфазениевых катионов в твердой фазе согласно измерению по методу 31Р ЯМР составляла 0,312 ммоль/г. В дополнение к этому, согласно результатам элементного анализа атомов хлора не наблюдали, а анионом количественно являлся гидроксид.
Примеры от 29 до 30
Синтез нанесенного на полимерный носитель гидроксида фосфазения
Реакцию проводили по тому же самому способу, что и в примере 28, за исключением того, что изменяли типы смолы на основе полистирола, подвергнутого хлорметилированию, использованной в примере 28, таким образом, синтезируя различные нанесенные на полимерный носитель гидроксиды фосфазения. Результаты продемонстрированы в таблице 3.
Advanced ChemTech
Пример 31
Синтез нанесенного на полимерный носитель гидроксида фосфазения
В стеклянную колбу объемом 50 мл, которую выдерживали в атмосфере азота, вводили 0,18 г (4,6 ммоль) гидрида калия и 20 мл ДМФА, сюда же добавляли раствор 5,0 г иодида фосфазения, полученного в примере 9, (3,7 ммоль) в ДМФА, а смесь перемешивали при комнатной температуре в течение 5 часов. В еще одну стеклянную колбу объемом 200 мл, оснащенную перемешивающим устройством, которую выдерживали в атмосфере азота, вводили 3,6 г смолы на основе полистирола, подвергнутого хлорметилированию (3,6 ммоль при расчете на атомы хлора) (произведенной в компании Advanced ChemTech, смола 2-pyridine-co-Merrifield, 1,0 ммоль Cl/г), и 50 мл ДМФА, а смесь перемешивали при комнатной температуре в течение 1 часа. После этого сюда же добавляли все количество раствора предварительно полученной калиевой соли иодида фосфазения в ДМФА и смесь дополнительно перемешивали в течение 62 часов. По завершении реакции получающуюся в результате смесь отфильтровывали, а смолу, полученную в виде оставшейся твердой фазы, промывали в аппарате Сокслета 1,4-диоксаном, используемым в качестве растворителя, а после этого высушивали при нагревании при 70°С при пониженном давлении, равном 1 мм ртутного столба. Высушенную смолу и 50 мл метанола вводили в стеклянную колбу объемом 200 мл, оснащенную перемешивающим устройством и холодильником, которую выдерживали в атмосфере азота, и смесь перемешивали при комнатной температуре в течение 1 часа. Сюда же добавляли раствор (10 мл) 2,6 г метоксида натрия в метаноле и в течение 8 часов смесь нагревали при кипячении в условиях флегмообразования, а после этого охлаждали до комнатной температуры. Полученную суспензию отфильтровывали и промывали водой, подвергали обработке в результате введения в контакт с 161 г 4%-ного водного раствора гидроксида натрия и промывали водой. После этого оставшуюся твердую фазу высушивали при 70°С при пониженном давлении, равном 1 мм ртутного столба, и получали 3,6 г нанесенного на полимерный носитель гидроксида фосфазения. Для части твердой фазы проводили измерения по методу 31Р ЯМР при использовании в качестве внутреннего стандарта тетрафторбората тетракис(диметиламино)фосфония. В данном случае наблюдали пик, соответствующий 1 атому фосфора, в области - 34,7 м.д. и пик, соответствующий 4 молям фосфора, в области 6,0 м.д., а концентрация фосфазениевых катионов в твердой фазе составляла 0,138 ммоль/г согласно измерениям по методу 31Р ЯМР.
Пример 32
Синтез нанесенного на полимерный носитель гидроксида фосфазения
В стеклянную колбу объемом 50 мл, которую выдерживали в атмосфере азота, вводили 0,1 г (2,4 ммоль) гидрида калия и 10 мл ДМФА, сюда же добавляли раствор 3,3 г иодида фосфазения, полученного в примере 9 (2,6 ммоль), в ДМФА, а смесь перемешивали при комнатной температуре в течение 5 часов. В еще одну стеклянную колбу объемом 100 мл, оснащенную перемешивающим устройством, которую выдерживали в атмосфере азота, вводили 3,1 г смолы на основе полистирола, подвергнутого хлорметилированию (2,2 ммоль при расчете на атомы хлора) (произведенной в компании Aldrich, JandaJels C1, 0,70 ммоль Cl/г), и 30 мл ДМФА, а смесь перемешивали при комнатной температуре в течение 1 часа. После этого сюда же добавляли все количество раствора предварительно полученной калиевой соли иодида фосфазения в ДМФА и смесь дополнительно перемешивали в течение 45 часов. По завершении реакции получающуюся в результате смесь отфильтровывали, а смолу, полученную в виде оставшейся твердой фазы, промывали в аппарате Сокслета 1,4-диоксаном, используемым в качестве растворителя, а после этого высушивали при нагревании при 70°С при пониженном давлении, равном 1 мм ртутного столба. Высушенную смолу и 40 мл метанола вводили в стеклянную колбу объемом 100 мл, оснащенную перемешивающим устройством и холодильником, которую выдерживали в атмосфере азота, и смесь перемешивали при комнатной температуре в течение 1 часа. Сюда же добавляли раствор (10 мл) 2,4 г метоксида натрия в метаноле и в течение 8 часов смесь нагревали при кипячении в условиях флегмообразования, а после этого охлаждали до комнатной температуры. Полученную суспензию отфильтровывали и промывали водой, подвергали обработке в результате введения в контакт с 89 г 4%-ного водного раствора гидроксида натрия и промывали водой. После этого оставшуюся твердую фазу высушивали при 70°С при пониженном давлении, равном 1 мм ртутного столба, и получали 2,2 г нанесенного на полимерный носитель гидроксида фосфазения. Для части твердой фазы проводили измерения по методу 31Р ЯМР при использовании в качестве внутреннего стандарта тетрафторбората тетракис(диметиламино)фосфония. В данном случае наблюдали пик, соответствующий 1 атому фосфора, в области - 34,8 м.д. и пик, соответствующий 4 молям фосфора, в области 6,2 м.д., а концентрация в твердой фазе составляла 0,078 ммоль/г согласно измерениям по методу 31Р ЯМР.
Пример 33
Синтез иодида фосфазения, имеющего стирильную группу
В стеклянную колбу объемом 300 мл, оснащенную перемешивающим устройством, которую выдерживали в атмосфере азота, вводили 96 г раствора PZNB (15 ммоль при расчете на PZNB) в тетралине и 73 мл о-дихлорбензола, а смесь охлаждали на льду при перемешивании. Сюда же по каплям добавляли 2,7 г (18 ммоль) 4-винилбензилхлорида и получающуюся в результате смесь перемешивали при комнатной температуре в течение ночи. По завершении реакции тетралин и о-дихлорбензол отгоняли при пониженном давлении и получали вязкую жидкость с оранжевой окраской. Сюда же добавляли 60 мл гексана и после перемешивания гексан удаляли в результате декантации. Промывание гексаном проводили 3 раза, а осажденную твердую фазу растворяли в результате добавления 230 мл 70%-ного водного раствора этиламина. Сюда же добавляли 2,4 г (16 ммоль) иодида натрия и 15 мл воды, смесь перемешивали при комнатной температуры в течение 1 часа, а после этого получающейся в результате смеси давали возможность отстояться в течение 4 дней. Осажденные кристаллы отфильтровывали, а после этого промывали водой и гексаном с получением светло-желтых кристаллов. Таким образом полученные кристаллы подвергали перекристаллизации при использовании смешанного растворителя, полученного из этилацетата и гексана, а осажденные кристаллы отфильтровывали, промывали гексаном и после этого высушивали при нагревании при 70°С при пониженном давлении, равном 1 мм ртутного столба, с получением в качестве белых кристаллов 8,7 г иодида фосфазения, имеющего стирильную группу. Часть твердой фазы добавляли к бензолу-d6, а в качестве внутреннего стандарта при проведении измерений по методу 31Р ЯМР использовали триамид гексаметилфосфорной кислоты. В данном случае наблюдали квинтуплет, соответствующий 1 атому фосфора, в области - 33,7 м.д., дублет, соответствующий 3 атомам фосфора, в области 7,50 м.д. и дублет, соответствующий 1 атому фосфора, в области 8,77 м.д., а степень чистоты составляла 96,1%. Кроме того, согласно результатам анализа по методу FD-MS наблюдали пик 841, соответствующий недиссоциированному фосфазениевому катиону.
Пример 34
Синтез иодида фосфазения, имеющего аллильную группу
Реакцию проводили по тому же самому способу, что и в примере 33, за исключением того, что вместо 4-винилбензилхлорида, использованного в примере 33, использовали эквимолярное количество аллилхлорида. Часть полученного соединения добавляли к бензолу-d6, а в качестве внутреннего стандарта при проведении измерений по методу 31Р ЯМР использовали триамид гексаметилфосфорной кислоты. В данном случае наблюдали квинтуплет, соответствующий 1 атому фосфора, в области - 33,6 м.д., дублет, соответствующий 3 атомам фосфора, в области 7,58 м.д. и дублет, соответствующий 1 атому фосфора, в области 8,83 м.д., а степень чистоты составляла 97,1%.
Пример 35
Синтез нанесенных на полимерный носитель иодида и гидроксида фосфазения
В стеклянную колбу объемом 50 мл, оснащенную перемешивающим устройством, которую выдерживали в атмосфере азота, вводили 0,082 г (0,50 ммоль) 2,2'-азобисизобутиронитрила, 0,97 г (0,96 ммоль) иодида фосфазения, полученного в примере 33, и добавляли 20 мл толуола, а смесь перемешивали. Сюда же помещали 10,52 г (101,0 ммоль) мономерного стирола и после перемешивания получающейся в результате смеси при 100°С в течение 7 часов смесь охлаждали до -77°С для гашения реакции. Реакционную смесь выливали в 1,5 л метанола и получали белый осадок. Осадок отфильтровывали и высушивали при нагревании при 70°С при пониженном давлении, равном 1 мм ртутного столба, с получением 4,4 г нанесенного на полимерный носитель иодида фосфазения в виде белой твердой фазы.
Полученные 3,6 г нанесенного на полимерный носитель иодида фосфазения в качестве набивки помещали в колонку и подвергали обработке в результате введения в контакт с 4%-ным водным раствором смеси хлорид натрия/метанол и дополнительно промывали метанолом и водой. По завершении обработки твердую фазу высушивали при нагревании при 70°С при пониженном давлении, равном 1 мм ртутного столба, с получением нанесенного на полимерный носитель гидроксида фосфазения. Для части твердой фазы проводили измерения по методу 31Р ЯМР при использовании тетрафторбората тетракис(диметиламино)фосфония в качестве внутреннего стандарта. В данном случае наблюдали пик, соответствующий 1 атому фосфора, в области - 35,1 м.д., пик, соответствующий 4 атомам фосфора, в области 5,6 м.д., а концентрация фосфазениевых катионов в твердой фазе согласно измерению по методу 31Р ЯМР составляла 0,1039 ммоль/г. Твердая фаза представляла собой сополимер, который получали в результате проведения сополимеризации при молярном соотношении 1:84 в системе фосфазениевая соль, имеющая стирильную группу:мономерный стирол. Кроме того, согласно результатам анализа по методу ГПХ (при расчете на стандартный полистирол) наблюдали полимер, характеризующийся среднечисленной молекулярной массой (Mn) 8536 и молекулярно-массовым распределением (Mw/Mn) 1,76.
Пример 36
Синтез хлорида фосфазения, имеющего триметоксисилильную группу - 1
В стеклянной колбе объемом 100 мл, которую выдерживали в атмосфере азота, в 100 мл сухого гексана растворяли 3,0 мл (12,2 ммоль) 4-(хлорметил)фенэтилтриметоксисилана. К раствору при комнатной температуре по каплям добавляли 50 г раствора PZNB (от 0,220 ммоль/г до 11,0 ммоль/г PZNB) в тетралине. Одновременно с покапельным добавлением производили отделение светло-коричневого маслянистого вещества. По завершении покапельного добавления получающуюся в результате смесь дополнительно перемешивали в течение 30 минут. Светло-коричневое маслянистое вещество отделяли от бесцветного жидкого супернатанта, а после этого сюда добавляли 5 мл сухого метанола. Смесь четыре раза промывали с использованием 15 мл сухого гексана. Раствор отгоняли при пониженном давлении и получали 10,5 г маслянистого вещества с оранжевой окраской. На основании результатов, полученных в соответствии с методами 1Н, 13С и 31Р ЯМР, основным компонентом полученного маслянистого вещества являлся желательный хлорид фосфазения, имеющий триметоксисилильную группу. Идентификация результатов, полученных в соответствии с методами 1Н, 13С и 31Р ЯМР, продемонстрирована далее.
1Н ЯМР (CDCl3, 270 МГц): 7,15 (м, 4H), 4,13 (д, 2H), 3,57 (с, 9H), 2,8-2,5 (м, 71H), 1,00 (м, 2H).
13C ЯМР (CDCl3, 270 МГц): 143,2, 135,8, 127,9, 127,8, 53,2, 50,5, 37,4-36,8, 34,0, 28,3, 11,3.
31P ЯМР (CDCl3, 109,3 МГц): 7,25 (д, 1P), 5,92 (д, 3P), -34,9 (кв, 1P).
Исходя из результатов, полученных в соответствии с методом 31Р ЯМР, маслянистое вещество, содержащее в качестве основного компонента вышеупомянутый хлорид фосфазения, имеющий триметоксисилильную группу, в качестве побочного продукта содержит соединение, описывающееся общей формулой (5), где a = b = c = d = 1, R = R1 = Me, D' = H, n = 1 и Z = Cl. Кроме того, наблюдаемая степень чистоты хлорида фосфазения, имеющего триметоксисилильную группу, в маслянистом веществе, определяемая в методе 1Н ЯМР на основании интегрального соотношения между пиками с использованием группы NMe2, составляла приблизительно 80%.
Пример 37
Синтез хлорида фосфазения, имеющего триметоксисилильную группу - 2
В стеклянной колбе объемом 100 мл, которую выдерживали в атмосфере азота, в 30 мл сухого гексана растворяли 0,87 мл (4,0 ммоль) 4-(хлорметил)фенилтриметоксисилана. К раствору при комнатной температуре по каплям добавляли 15 г раствора PZNB (от 0,220 ммоль/г до 3,3 ммоль/г PZNB) в тетралине. Одновременно с покапельным добавлением производили отделение светло-коричневого маслянистого вещества. По завершении покапельного добавления получающуюся в результате смесь дополнительно перемешивали в течение 30 минут. Светло-коричневое маслянистое вещество отделяли от бесцветного жидкого супернатанта, а после этого сюда добавляли 2 мл сухого метанола. Смесь четыре раза промывали с использованием 5 мл сухого гексана. Раствор отгоняли при пониженном давлении и получали 2,90 г маслянистого вещества с оранжевой окраской. На основании результатов, полученных в соответствии с методами 1Н, 13С и 31Р ЯМР, основным компонентом полученного маслянистого вещества являлся желательный хлорид фосфазения, имеющий триметоксисилильную группу. Идентификация результатов, полученных в соответствии с методами 1Н, 13С и 31Р ЯМР, продемонстрирована далее.
1Н ЯМР (CDCl3, 270 МГц): 7,61 (м, 2H), 7,26 (д, 2Н), 4,21 (д, 2H), 3,64 (с, 9H), 2,8-2,5 (м, 69H).
13C ЯМР (CDCl3, 270 МГц): 141,4, 134,9, 134,7, 127,3, 53,6, 50,1, 37,5-36,8, 34,3.
31P ЯМР (CDCl3, 109,3 МГц): 7,33 (д, 1P), 5,94 (д, 3P), -34,9 (кв, 1P).
Исходя из результатов, полученных в соответствии с методом 31Р ЯМР, маслянистое вещество, содержащее в качестве основного компонента вышеупомянутый хлорид фосфазения, имеющий триметоксисилильную группу, в качестве побочного продукта содержит соединение, описывающееся общей формулой (5), где a = b = c = d = 1, R = R1 = Me, D' = H, n = 1 и Z = Cl. Кроме того, наблюдаемая степень чистоты хлорида фосфазения, имеющего триметоксисилильную группу, в маслянистом веществе, определяемая в методе 1Н ЯМР на основании интегрального соотношения между пиками с использованием группы NMe2, составляла приблизительно 50%.
Пример 38
Синтез бромида фосфазения, имеющего триметоксисилильную группу
В стеклянной колбе объемом 100 мл, которую выдерживали в атмосфере азота, в 30 мл сухого гексана растворяли 0,75 мл (4,0 ммоль) 3-бромпропилтриметоксисилана. К раствору при комнатной температуре по каплям добавляли 15 г раствора PZNB (от 0,220 ммоль/г до 3,3 ммоль/г PZNB) в тетралине. Одновременно с покапельным добавлением производили отделение светло-коричневого маслянистого вещества. По завершении покапельного добавления получающуюся в результате смесь дополнительно перемешивали в течение 30 минут. Светло-коричневое маслянистое вещество отделяли от бесцветного жидкого супернатанта, а после этого сюда добавляли 2 мл сухого метанола. Смесь четыре раза промывали с использованием 5 мл сухого гексана. Раствор отгоняли при пониженном давлении и получали 3,10 г маслянистого вещества с оранжевой окраской. На основании результатов, полученных в соответствии с методами 1Н, 13С и 31Р ЯМР, основным компонентом полученного маслянистого вещества являлся желательный бромид фосфазения, имеющий триметоксисилильную группу. Идентификация результатов, полученных в соответствии с методами 1Н, 13С и 31Р ЯМР, продемонстрирована далее.
lН ЯМР (CDCl3, 270 МГц): 3,58 (с, 9H), 2,9-2,5 (м, 71H), 1,56 (м, 2H), 0,53 (м, 2Н).
13C ЯМР (CDCl3, 270 МГц): 52,2, 50,6, 37,3-36,9, 34,0, 21,5, 6,5.
31P ЯМР (CDCl3, 109,3 МГц): 6,20 (д, 1P), 6,00 (д, 3P), -35,1 (кв, 1P).
Исходя из результатов, полученных в соответствии с методом 31Р ЯМР, маслянистое вещество, содержащее в качестве основного компонента бромид фосфазения, имеющий триметоксисилильную группу, в качестве побочного продукта содержит соединение, описывающееся общей формулой (5), где a = b = c = d = 1, R = R1 = Me, D' = H, n = 1 и Z = Br. Кроме того, наблюдаемая степень чистоты бромида фосфазения, имеющего триметоксисилильную группу, в маслянистом веществе, определяемая в методе 1Н ЯМР на основании интегрального соотношения между пиками с использованием группы NMe2, составляла приблизительно 70%.
Пример 39
Получение силикагеля, являющегося носителем для хлорида фосфазения, по способу гидролиза-поликонденсации - 1
В двугорлую круглодонную стеклянную колбу объемом 100 мл, оснащенную перемешивающим устройством, термометром, холодильником и тому подобным, вводили 7,40 г (6,2 ммоль) сырого неочищенного хлорида фосфазения, имеющего триметоксисилильную группу, полученного в примере 36, 50,0 г (0,24 моль) тетраэтоксисилана, 50 мл этанола и 0,80 мл (9,2 ммоль) 36%-ной хлористоводородной кислоты. Сюда же в течение 10 минут по каплям добавляли 10,0 г воды, а после этого смесь в течение 3 часов перемешивали при 60°С. После того как получающейся в результате смеси давали возможности охладиться, добавляли 50,0 г воды и по каплям добавляли 2,0 мл 28%-ного водного раствора аммиака, что обеспечивало быстрое затвердевание реакционной смеси. Получающейся в результате смеси давали возможность отстояться при комнатной температуре в течение 3 дней и ее два раза промывали с использованием 100 мл воды, подвергнутой ионному обмену. Полученную твердую фазу в течение 4 часов высушивали при 100°С при пониженном давлении, равном 1 мм ртутного столба, и получали 20,9 г силикагеля, являющегося носителем для хлорида фосфазения (количество нанесенного на носитель хлорида фосфазения: 0,30 ммоль/г). Площадь удельной поверхности, измеренная по способу адсорбирования газообразного азота, составляла 432 м2/г, а объем пор для пор, имеющих диаметр в диапазоне от 9 до 500 Å, составлял 0,30 см3/г. Согласно методу 31Р ЯМР в твердом состоянии наблюдали пики в областях 35,2 (1Р) м.д., 13,4-5,8 (3Р) м.д. и - 36,4 (1Р) м.д.
Пример 40
Получение силикагеля, являющегося носителем для хлорида фосфазения, по способу гидролиза-поликонденсации - 2
В двугорлую круглодонную стеклянную колбу объемом 100 мл, оснащенную перемешивающим устройством, термометром, холодильником и тому подобным, вводили 5,95 г (5,0 ммоль) сырого неочищенного хлорида фосфазения, имеющего триметоксисилильную группу, полученного в примере 36, 20,8 г (0,10 моль) тетраэтоксисилана, 20 мл этанола и 0,60 мл (6,9 ммоль) 36%-ной хлористоводородной кислоты. Сюда же в течение 10 минут по каплям добавляли 5,0 г воды, а после этого смесь в течение 3 часов перемешивали при 60°С. После того как получающейся в результате смеси давали возможности охладиться, добавляли 20,0 г воды и по каплям добавляли 1,0 мл 28%-ного водного раствора аммиака, что обеспечивало быстрое затвердевание реакционной смеси. Получающейся в результате смеси давали возможность отстояться при комнатной температуре в течение 3 дней и ее два раза промывали с использованием 50 мл воды, подвергнутой ионному обмену. Полученную твердую фазу в течение 4 часов высушивали при 80°С при пониженном давлении, равном 1 мм ртутного столба, и получали 11,1 г силикагеля, являющегося носителем для хлорида фосфазения (количество нанесенного на носитель хлорида фосфазения: 0,45 ммоль/г). Площадь удельной поверхности, измеренная по способу адсорбирования газообразного азота, составляла 277 м2/г, а объем пор для пор, имеющих диаметр в диапазоне от 9 до 500 Å, составлял 0,59 см3/г.
Пример 41
Получение силикагеля, являющегося носителем для хлорида фосфазения, по способу силилирования
В двугорлую круглодонную стеклянную колбу объемом 50 мл, оснащенную перемешивающим устройством, термометром, холодильником и тому подобным, в атмосфере азота вводили 2,26 г (1,9 ммоль) сырого неочищенного хлорида фосфазения, имеющего триметоксисилильную группу, полученного в примере 36, 5,00 г силикагеля (произведенного в компании Kanto Kagaku Chemical Co. Ltd., 60N) и 20 мл сухого толуола, а смесь нагревали и перемешивали при кипячении в условиях флегмообразования в течение 12 часов. Получающуюся в результате смесь отфильтровывали и после двукратного промывания с использованием 20 мл метанола полученную твердую фазу в течение 4 часов высушивали при 80°С при пониженном давлении, равном 1 мм ртутного столба, с получением 6,50 г силикагеля, являющегося носителем для хлорида фосфазения (количество нанесенного на носитель хлорида фосфазения: 0,24 ммоль/г). Площадь удельной поверхности, измеренная по способу адсорбирования газообразного азота, составляла 447 м2/г, а объем пор для пор, имеющих диаметр в диапазоне от 9 до 500 Å, составлял 0,47 см3/г. Согласно методу 31Р ЯМР в твердом состоянии наблюдали пики в областях 5,71 (4Р) м.д. и -36,1 (1Р) м.д.
Пример 42
Получение силикагеля, являющегося носителем для гидроксида фосфазения - 2
11,1 г силикагеля, являющегося носителем для хлорида фосфазения (3,33 ммоль при расчете на фосфазениевые катионы), полученного в примере 39, в качестве набивки помещали в колонку. После пропускания через колонку 30,0 мл (30 ммоль) водного раствора аммиака с концентрацией 1 моль/л (SV = 3) колонку промывали водой, подвергнутой ионному обмену, и метанолом. По завершении обработки твердую фазу в течение 6 часов высушивали при 80°С при пониженном давлении, равном 1 мм ртутного столба, и получали 11,0 г силикагеля, являющегося носителем для гидроксида фосфазения, (количество нанесенного на носитель гидроксида фосфазения: 0,30 ммоль/г). Согласно результатам элементного анализа атомов хлора не наблюдали. Кроме того, площадь удельной поверхности, измеренная по способу адсорбирования газообразного азота, составляла 454 м2/г, а объем пор для пор, имеющих диаметр в диапазоне от 9 до 500 Å, составлял 0,36 см3/г. Согласно методу 31Р ЯМР в твердом состоянии наблюдали пики в областях 5,75 (4Р) м.д. и -36,1 (1Р) м.д.
Пример 43
Получение силикагеля, являющегося носителем для гидроксида фосфазения - 3
3,24 г силикагеля, являющегося носителем для хлорида фосфазения (1,46 ммоль при расчете на фосфазениевые катионы), полученного в примере 40, в качестве набивки помещали в колонку. После пропускания через колонку 29,2 мл (29,2 ммоль) водного раствора аммиака с концентрацией 1 моль/л (SV = 4) колонку промывали водой, подвергнутой ионному обмену, и метанолом. По завершении обработки твердую фазу в течение 6 часов высушивали при 80°С при пониженном давлении, равном 1 мм ртутного столба, и получали 3,15 г силикагеля, являющегося носителем для гидроксида фосфазения, (количество нанесенного на носитель гидроксида фосфазения: 0,45 ммоль/г).
Пример 44
Получение силикагеля, являющегося носителем для гидроксида фосфазения - 1
2,72 г силикагеля, являющегося носителем для хлорида фосфазения (0,65 ммоль при расчете на фосфазениевые катионы), полученного в примере 41, в качестве набивки помещали в колонку. После пропускания через колонку 6,3 мл (6,3 ммоль) водного раствора аммиака с концентрацией 1 моль/л (SV = 4) колонку промывали водой, подвергнутой ионному обмену, и метанолом. По завершении обработки твердую фазу в течение 6 часов высушивали при 80°С при пониженном давлении, равном 1 мм ртутного столба, и получали 2,65 г силикагеля, являющегося носителем для гидроксида фосфазения (количество нанесенного на носитель гидроксида фосфазения: 0,24 ммоль/г). Согласно результатам элементного анализа атомов хлора не наблюдали. Кроме того, площадь удельной поверхности, измеренная по способу адсорбирования газообразного азота, составляла 428 м2/г, а объем пор для пор, имеющих диаметр в диапазоне от 9 до 500 Å, составлял 0,50 см3/г. Согласно методу 31Р ЯМР в твердом состоянии наблюдали пики в областях 5,95 (4Р) м.д. и -36,1 (1Р) м.д.
Пример 45
Алкилирование фенольной гидроксильной группы при использовании в качестве катализатора нанесенного на полимерный носитель иодида фосфазения
В стеклянную колбу объемом 100 мл, оснащенную перемешивающим устройством и холодильником, которую выдерживали в атмосфере азота, вводили 2,7 г нанесенного на полимерный носитель иодида фосфазения (1,0 ммоль при расчете на фосфазениевые катионы), полученного в примере 4, 0,49 г (5,2 ммоль) фенола и 30 мл диметилкарбоната, а смесь в течение 18 часов нагревали при кипячении в условиях флегмообразования при перемешивании и после этого охлаждали до комнатной температуры. По завершении реакции жидкий супернатант из суспензии частично отбирали и подвергали анализу по методу газовой хроматографии. Таким образом, было обнаружено то, что степень превращения фенола составляла 99,4%, а выход целевого анизола был равен 91,0%. В дополнение к этому, по завершении реакции суспензию отфильтровывали, промывали в аппарате Сокслета диметилкарбонатом, используемым в качестве растворителя, и высушивали при нагревании при 70°С при пониженном давлении, равном 1 мм ртутного столба, с извлечением 2,7 г нанесенного на полимерный носитель иодида фосфазения.
Сравнительный пример 1
Реакцию проводили по тому же самому способу, что и в примере 45, за исключением того, что 2,7 г нанесенного на полимерный носитель иодида фосфазения (1,0 ммоль при расчете на фосфазениевые катионы), использованного в примере 45, не использовали. Проводили анализ по методу газовой хроматографии, и, таким образом, было обнаружено то, что превращение фенола не происходило полностью, и полностью же нельзя было добиться получения целевого анизола.
Сравнительный пример 2
Реакцию проводили по тому же самому способу, что и в примере 45, за исключением того, что вместо 2,7 г нанесенного на полимерный носитель иодида фосфазения (1,0 ммоль при расчете на фосфазениевые катионы), использованного в примере 45, использовали 1,0 ммоль иодида тетракис[трис(диметиламино)фосфоранилиденамино]фосфония. Проводили анализ по методу газовой хроматографии, и, таким образом, было обнаружено то, что превращение фенола происходило полностью, а выход целевого анизола составлял 93,4%. Было установлено то, что нанесенный на полимерный носитель иодид фосфазения содержал активный катализатор, эквивалентный тому, что имеет место в случае не нанесенного на полимерный носитель иодида фосфазения.
Примеры от 46 до 54
Отправление извлеченного катализатора на рецикл
Реакцию проводили по тому же самому способу, что и в примере 45, за исключением того, что вместо нанесенного на полимерный носитель иодида фосфазения, использованного в примере 45, использовали извлеченный нанесенный на полимерный носитель иодид фосфазения, полученный в примере 45, и по завершении реакции нанесенный на полимерный носитель иодид фосфазения извлекали. Кроме того, извлеченный нанесенный на полимерный носитель иодид фосфазения в реакции использовали неоднократно. Результаты продемонстрированы в таблице 4.
Кроме того, согласно измерениям, проведенным по методу 31Р ЯМР, концентрация фосфазениевых катионов в нанесенном на полимерный носитель иодиде фосфазения, извлеченном в примере 54, составляла 0,345 ммоль/г, и никакого отщепления иодида фосфазения, обусловленного неоднократным использованием, не наблюдалось.
Пример 55
Полимеризация полиалкиленоксида при использовании в качестве катализатора нанесенного на полимерный носитель гидроксида фосфазения
В автоклав объемом 70 мл, оснащенный термометром, датчиком давления, перемешивающим устройством и впускным патрубком для алкиленоксида, который выдерживали в атмосфере азота, вводили 1,0 г (11 ммоль) глицерина, 0,3 г нанесенного на полимерный носитель гидроксида фосфазения (0,13 ммоль при расчете на фосфазениевые катионы), полученного в примере 3, и 34 г пропиленоксида, автоклав герметично закрывали и смесь нагревали до 80°С при перемешивании. В данный момент давление в автоклаве увеличивалось до 0,3 МПа (избыточное давление). После этого давление уменьшалось в результате расходования пропиленоксида, но реакция продолжалась до тех пор, пока уменьшение давления не прекращалось. По завершении реакции получающуюся в результате смесь охлаждали до комнатной температуры, а остаточный пропиленоксид отгоняли при пониженном давлении. Суспензию из автоклава отбирали, разбавляли с использованием ТГФ, а после этого отфильтровывали. Кроме того, оставшуюся твердую фазу в достаточной мере промывали с использованием ТГФ, а ТГФ из фильтрата отгоняли и получали 32 г полипропиленоксида без цвета и запаха. Активность при полимеризации (количество полученного полипропиленоксида на один моль катализатора и в единицу времени) составляла 10,9 г/ммоль·час.
Сравнительный пример 3
Реакцию проводили по тому же самому способу, что и в примере 55, за исключением того, что вместо нанесенного на полимерный носитель гидроксида фосфазения, использованного в примере 55, использовали 0,06 г (1,6 ммоль) гидроксида калия. Можно было получить 30 г полипропиленоксида, но активность при полимеризации составляла только лишь 0,29 г/ммоль·час.
Примеры от 56 до 58
Реакцию проводили по тому же самому способу, что и в примере 55, за исключением того, что использовали автоклав объемом 300 мл, и вместо глицерина, использованного в примере 55, использовали типы и количество спирта и количество пропиленоксида, продемонстрированные в таблице 5. Результаты продемонстрированы в таблице 5.
Примеры от 59 до 69
Реакцию проводили по тому же самому способу, что и в примере 55, за исключением того, что использовали автоклав объемом 70 мл или 300 мл, а вместо нанесенного на полимерный носитель гидроксида фосфазения, использованного в примере 55, использовали количество глицерина и количество пропиленоксида, продемонстрированные в таблице 6. Результаты продемонстрированы в таблице 6.
ммоль·
час)
Примеры от 70 до 72
Извлечение и отправление на рецикл нанесенного на полимерный носитель гидроксида фосфазения
В автоклав объемом 300 мл, оснащенный термометром, датчиком давления, перемешивающим устройством и впускным патрубком для алкиленоксида, который выдерживали в атмосфере азота, вводили 3,2 г глицерина, 2,5 г нанесенного на полимерный носитель гидроксида фосфазения, полученного в примере 17, и 104 г пропиленоксида, автоклав герметично закрывали и смесь нагревали до 80°С при перемешивании. В данный момент давление в автоклаве увеличивалось до 0,32 МПа (избыточное давление). После этого давление уменьшалось в результате расходования пропиленоксида, но реакция продолжалась до тех пор, пока уменьшение давления не прекращалось. По завершении реакции получающуюся в результате смесь охлаждали до комнатной температуры, а остаточный пропиленоксид отгоняли при пониженном давлении. Суспензию из автоклава отбирали, разбавляли с использованием ТГФ, а после этого отфильтровывали. Кроме того, оставшуюся твердую фазу в достаточной мере промывали с использованием ТГФ, а ТГФ из фильтрата отгоняли при пониженном давлении и получали 103 г полипропиленоксида без цвета и запаха. Активность при полимеризации (количество полученного полипропиленоксида на один моль катализатора и в единицу времени) составляла 2,0 г/ммоль·час. Кроме того, по завершении фильтрования оставшуюся твердую фазу высушивали при нагревании при 70°C при пониженном давлении, равном 1 мм ртутного столба, для извлечения катализатора. После этого все количество извлеченного катализатора, 2,7 г глицерина и 85 г пропиленоксида вводили в автоклав объемом 300 мл, а смесь подвергали полимеризации при 80°С и переработке по завершении реакции с получением 87 г полипропиленоксида и извлеченного катализатора. Кроме того, извлеченный катализатор подавали на полимеризацию совместно с 2,7 г глицерина и 86 г пропиленоксида с получением 85 г полипропиленоксида. Было обнаружено то, что вторая и третья каталитические активности составляли 1,9 г/ммоль·час и 2,0 г/ммоль·час соответственно, и для нанесенного на полимерный носитель гидроксида фосфазения в изобретении никакого уменьшения активности не наблюдалось даже после извлечения и отправления на рецикл в качестве катализатора.
Примеры от 73 до 81
Извлечение и отправление на рецикл для нанесенного на полимерный носитель гидроксида фосфазения
Реакцию проводили по тому же самому способу, что и в примере 69, за исключением того, что после извлечения использовали все количество нанесенного на носитель катализатора, использованного при полимеризации из примера 69. Кроме того, по завершении полимеризации катализатор извлекали и неоднократно использовали в последующих реакциях по тому же самому способу. Результаты продемонстрированы в таблице 7.
Даже по завершении десяти циклов повторявшихся процессов полимеризации, как было обнаружено, катализатор обладал достаточной каталитической активностью.
Пример 82
Полимеризация полиалкиленоксида при использовании в качестве катализатора нанесенного на полимерный носитель гидроксида фосфазения
В автоклав объемом 70 мл, оснащенный термометром, датчиком давления, перемешивающим устройством и впускным патрубком для алкиленоксида, который выдерживали в атмосфере азота, вводили 0,6 г (7,0 ммоль) глицерина, 0,9 г нанесенного на полимерный носитель гидроксида фосфазения (0,09 ммоль при расчете на фосфазениевые катионы), полученного в примере 35, и 33 г пропиленоксида, автоклав герметично закрывали и смесь нагревали до 80°С при перемешивании. В данный момент давление в автоклаве увеличивалось до 0,3 МПа (избыточное давление). После этого давление уменьшалось в результате расходования пропиленоксида, но реакция продолжалась до тех пор, пока уменьшение давления не прекращалось. По завершении реакции получающуюся в результате смесь охлаждали до комнатной температуры, а остаточный пропиленоксид отгоняли при пониженном давлении. Суспензию из автоклава отбирали, разбавляли нормальным гексаном, а после этого отфильтровывали. Кроме того, фильтрат в достаточной мере промывали нормальным гексаном, а нормальный гексан из фильтрата отгоняли при пониженном давлении и получали 33 г полипропиленоксида без цвета и запаха. Активность при полимеризации (количество полученного полипропиленоксида на один моль катализатора и в единицу времени) составляла 12,4 г/ммоль·час.
Примеры от 83 до 84
Извлечение и отправление на рецикл нанесенного на полимерный носитель гидроксида фосфазения
Оставшуюся твердую фазу, полученную в примере 82, по завершении фильтрования высушивали при нагревании при 70°C при пониженном давлении, равном 1 мм ртутного столба, для извлечения катализатора. После этого все количество извлеченного катализатора, 0,6 г глицерина и 30 г пропиленоксида вводили в автоклав объемом 70 мл и по тому же самому способу, что и в примере 82, смесь подвергали полимеризации при 80°С и переработке по завершении реакции с получением 30 г полипропиленоксида и извлеченного катализатора. Кроме того, извлеченный катализатор подавали на полимеризацию совместно с 0,5 г глицерина и 29 г пропиленоксида с получением 29 г полипропиленоксида. Было обнаружено то, что вторая и третья каталитические активности составляли 10,6 г/ммоль·час и 14,7 г/ммоль·час соответственно, и для нанесенного на полимерный носитель гидроксида фосфазения в изобретении никакого уменьшения активности не наблюдалось даже после извлечения и отправления на рецикл в качестве катализатора.
Пример 85
Альдольная конденсация ацетона при использовании в качестве катализатора силикагеля, являющегося носителем для гидроксида фосфазения
В круглодонную стеклянную колбу объемом 50 мл вводили 1,0 г силикагеля, являющегося носителем для гидроксида фосфазения, (0,45 ммоль при расчете на фосфазениевые катионы), полученного в примере 43, и 26,5 г (0,45 моль) ацетона, а смесь перемешивали при комнатной температуре в атмосфере азота. По истечении 8 часов часть жидкого супернатанта из суспензии отбирали и подвергали анализу по методу газовой хроматографии. Таким образом, было обнаружено то, что степень превращения ацетона составляла 3,5%, а селективности по получению диацетонового спирта и метилоксида были равны 97,2% и 2,8% соответственно.
Пример 86
Альдольная конденсация ацетона при использовании в качестве катализатора силикагеля, являющегося носителем для гидроксида фосфазения
В круглодонную стеклянную колбу объемом 50 мл вводили 1,0 г силикагеля, являющегося носителем для гидроксида фосфазения (0,24 ммоль при расчете на фосфазениевые катионы), полученного в примере 44, и 14,0 г (0,24 моль) ацетона, а смесь перемешивали при комнатной температуре в атмосфере азота. По истечении 8 часов часть жидкого супернатанта из суспензии отбирали и подвергали анализу по методу газовой хроматографии. Таким образом, было обнаружено то, что степень превращения ацетона составляла 1,3%, а селективность по получению диацетонового спирта была равна 100%.
Сравнительный пример 4
Реакцию проводили по тому же самому способу, что и в примере 85, за исключением того, что вместо силикагеля, являющегося носителем для гидроксида фосфазения, использованного в примере 85, использовали 0,34 г (0,45 ммоль) гидроксида тетракис[трис(диметиламино)фосфоранилиденамино]фосфония. По истечении 8 часов часть жидкого супернатанта из суспензии отбирали и подвергали анализу по методу газовой хроматографии. Таким образом, было обнаружено то, что степень превращения ацетона составляла 8,0%, а селективности по получению диацетонового спирта и метилоксида были равны 78,2% и 21,3% соответственно.
Схема химической реакции для каждого примера
Пример 1. PZNB
Пример 2. Нанесенный на полимерный носитель хлорид фосфазения
Пример 3. Нанесенный на полимерный носитель гидроксид фосфазения
Пример 4. Нанесенный на полимерный носитель иодид фосфазения
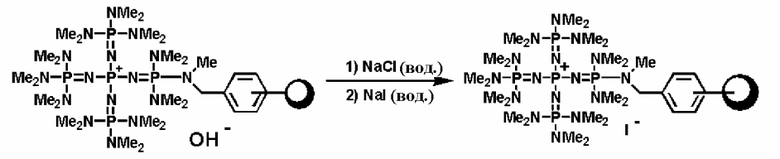
Пример 5. Иодид фосфазения, имеющий гидроксильную группу
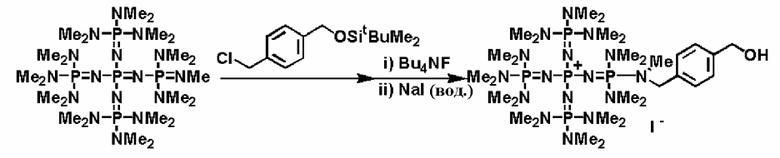
Примеры от 6 до 11. Иодид фосфазения, содержащий полиалкиленоксид в своей боковой цепи
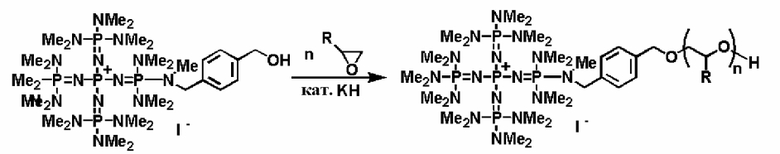
Примеры от 12 до 17. Нанесенный на полимерный носитель гидроксид фосфазения
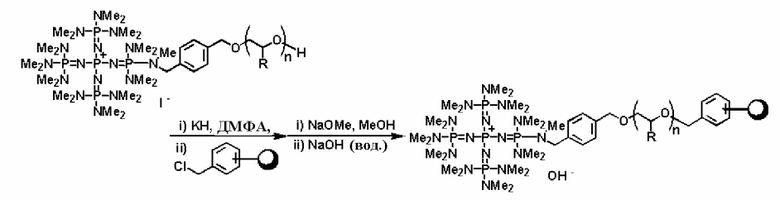
Пример 18. PZND
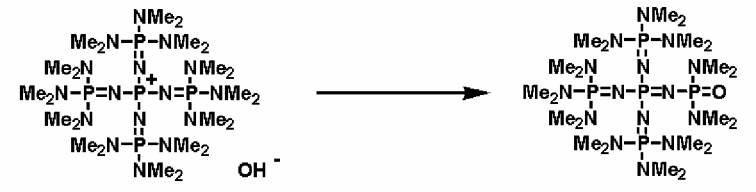
Пример 19. PZND-Cl
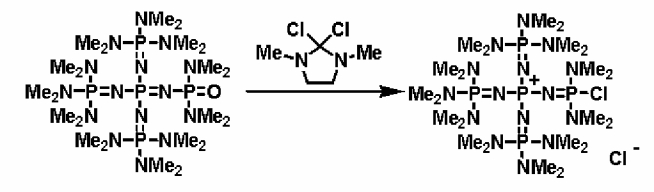
Пример 20. Хлорид фосфазения, имеющий группу силокси
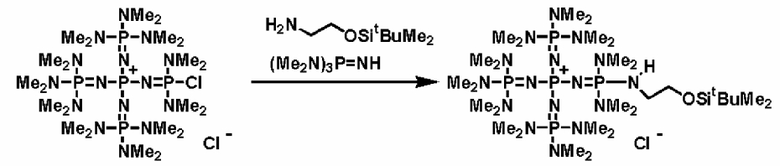
Пример 21. Иодид фосфазения, имеющий гидроксильную группу
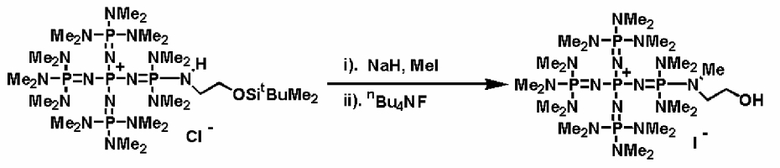
Пример 22. Нанесенный на полимерный носитель гидроксид фосфазения
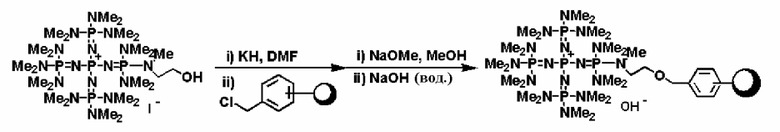
Пример 23. Нанесенный на полимерный носитель хлорид фосфазения
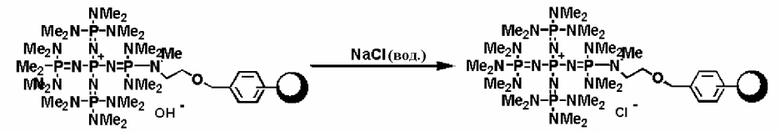
Пример 24. Хлорид фосфазения, имеющий группу силокси
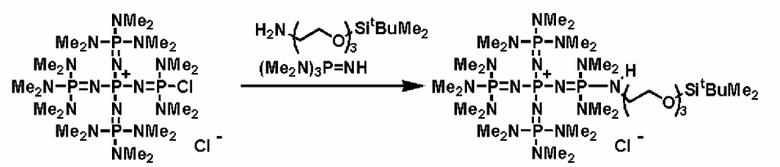
Пример 25. Гексафторфосфат фосфазения, имеющий гидроксильную группу
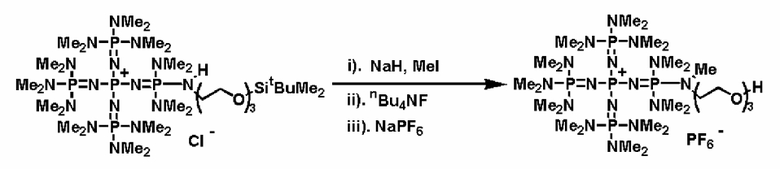
Пример 26. Нанесенный на полимерный носитель гексафторфосфат фосфазения
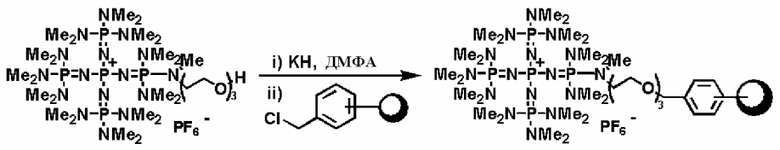
Пример 27. Нанесенный на полимерный носитель гидроксид фосфазения

Примеры от 28 до 30. Нанесенный на полимерный носитель гидроксид фосфазения
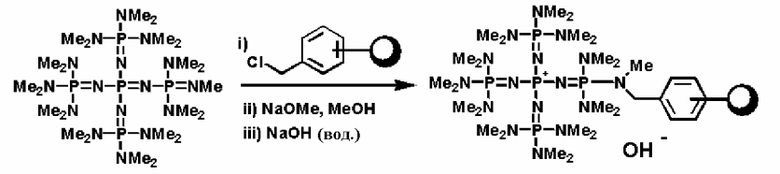
Примеры от 31 до 32. Нанесенный на полимерный носитель гидроксид фосфазения
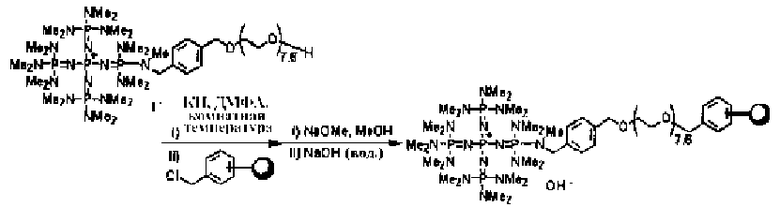
Пример 33. Иодид фосфазения, имеющий стирильную группу
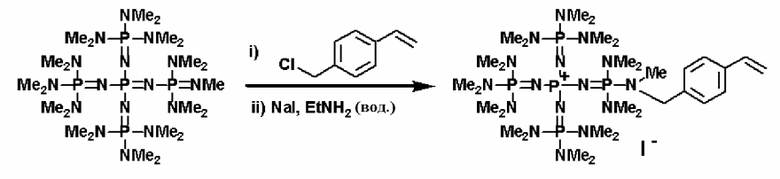
Пример 34. Иодид фосфазения, имеющий аллильную группу
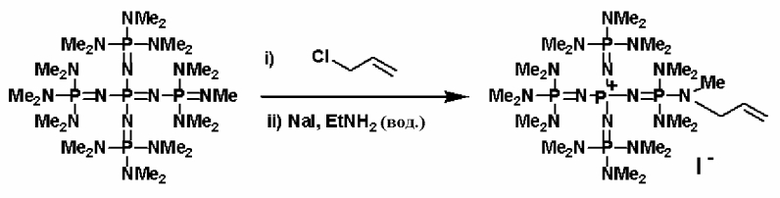
Пример 35. Нанесенные на полимерный носитель иодид и гидроксид фосфазения
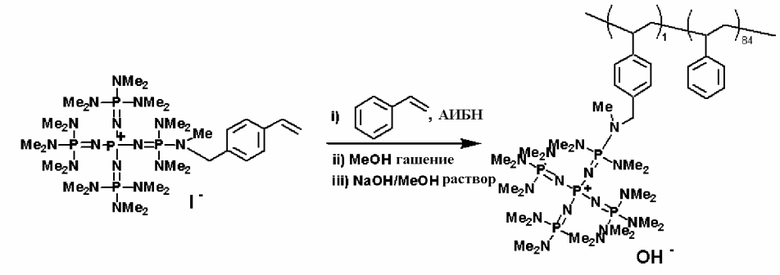
Пример 36. Хлорид фосфазения, имеющий триметоксисилильную группу, 1
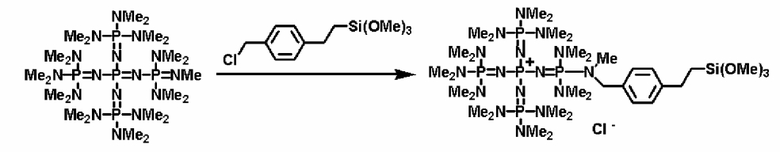
Пример 37. Хлорид фосфазения, имеющий триметоксисилильную группу, 2
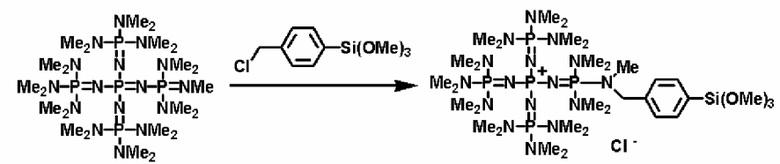
Пример 38. Бромид фосфазения, имеющий триметоксисилильную группу
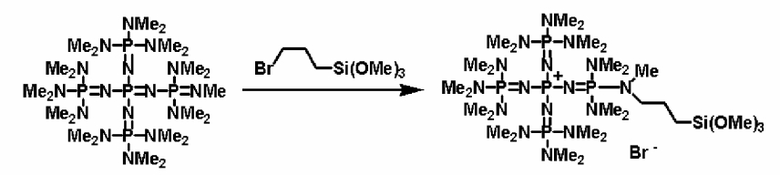
Примеры от 39 до 40. Силикагель, являющийся носителем для хлорида фосфазения (способ гидролиза-поликонденсации)
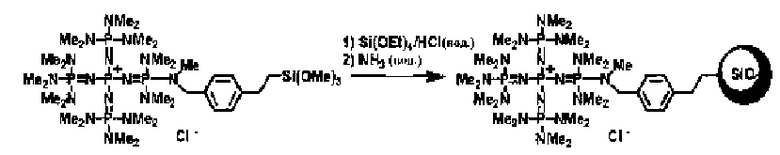
Пример 41. Силикагель, являющийся носителем для хлорида фосфазения (способ силилирования)
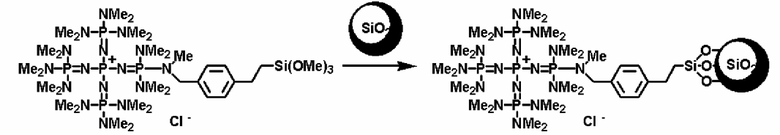
Примеры от 42 до 44. Силикагель, являющийся носителем для гидроксида фосфазения

Фосфазеновый нанесенный на носитель катализатор изобретения является подходящим для использования в качестве катализатора различных органических реакций и, в частности, катализатора полимеризации циклических мономеров и замещения заместителей. Кроме того, каждое соединение, выбираемое из фосфазенового соединения и фосфазениевой соли изобретения, является промежуточным соединением, которое является подходящим для использования при получении фосфазенового нанесенного на носитель катализатора изобретения, а также катализатором, который сам по себе является подходящим для использования при проведении различных органических реакций.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ГЛИФОСАТА И КАТАЛИЗАТОР ОКИСЛЕНИЯ | 1998 |
|
RU2184118C2 |
| НОВЫЕ ФОСФОНАМИДЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2003 |
|
RU2314314C2 |
| СПОСОБ СИНТЕЗА АМИНОАЛКИЛЕНФОСФОНОВОЙ КИСЛОТЫ | 2013 |
|
RU2694047C2 |
| НОВЫЕ ПОЛИМЕРЫ И ИХ ИСПОЛЬЗОВАНИЕ ПРИ ПОЛУЧЕНИИ ВЫСОКОУДАРНЫХ ПОЛИМЕРНЫХ КОМПОЗИЦИЙ | 2009 |
|
RU2533206C2 |
| КАТАЛИЗАТОР ПОЛИМЕРИЗАЦИИ ОЛЕФИНА И СПОСОБ ПОЛИМЕРИЗАЦИИ ОЛЕФИНА С ЕГО ИСПОЛЬЗОВАНИЕМ | 2009 |
|
RU2469046C2 |
| НОВЫЕ ПУТИ К ПОЛИАКРИЛАТАМ | 2011 |
|
RU2588569C2 |
| СПОСОБ ОЧИСТКИ α-ОЛЕФИНОВ ДЛЯ ПОЛИМЕРИЗАЦИИ И СПОСОБ ПОЛУЧЕНИЯ ПОЛИ-α-ОЛЕФИНОВ | 1995 |
|
RU2152421C1 |
| НОВЫЕ СОЕДИНЕНИЯ С АНТИБАКТЕРИАЛЬНЫМ ДЕЙСТВИЕМ | 2004 |
|
RU2397982C2 |
| КАТАЛИТИЧЕСКАЯ КОМПОЗИЦИЯ, МЕТАЛЛОЦЕНОВЫЙ КОМПЛЕКС И СПОСОБ ПОЛИМЕРИЗАЦИИ ОЛЕФИНОВ | 1994 |
|
RU2156253C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ГЛУТАМИНОВОЙ КИСЛОТЫ, СПОСОБЫ ПОЛУЧЕНИЯ ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ И НОВОЕ ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ ДЛЯ ДАННЫХ СПОСОБОВ | 2002 |
|
RU2305677C2 |
Изобретение относится к фосфазеновому нанесенному на носитель катализатору, к фосфазеновому соединению и фосфазениевой соли, которые используются при получении катализатора, и к применению катализатора. Предлагается фосфазеновый нанесенный на носитель катализатор для полимеризации циклического мономера, или для замещения заместителя в соединении, или для проведения реакции с образованием связи углерод-углерод, в котором носитель является нерастворимым в использованном растворителе и имеет группу, способную образовывать связь с группой, описывающейся общей формулой (1):
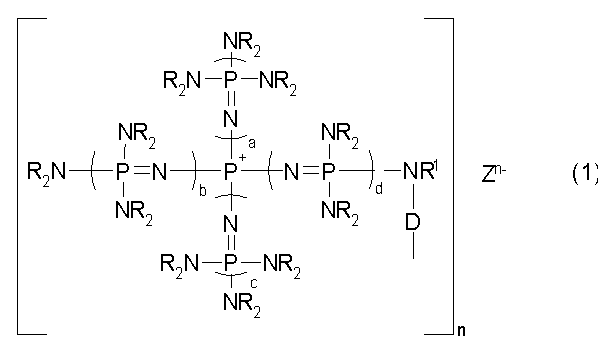
где n является целым числом в диапазоне от 1 до 8 и представляет собой количество фосфазениевых катионов, а Zn- представляет собой анион соединения, содержащего атомы активного водорода, в форме, полученной в результате высвобождения n протонов из соединения, содержащего атомы активного водорода, в котором имеется, самое большее, 8 атомов активного водорода; каждый из а, b, с и d представляет собой положительное целое число, равное 3 или менее; R представляют собой одинаковые или различные углеводородные группы, содержащие от 1 до 10 атомов углерода, и два R, расположенные на каждом общем атоме азота, могут быть связаны друг с другом с образованием кольцевой структуры; R1 представляет собой атом водорода или углеводородную группу, содержащую от 1 до 10 атомов углерода; D представляет собой непосредственную связь или двухвалентную группу, способную связывать N с носителем. Описаны фосфазеновое соединение и фосфазениевые соли и способы полимеризации циклического мономера, замещения заместителя в соединении и проведения реакции с образованием связи углерод-углерод при использовании нанесенного на носитель катализатора. Технический эффект - повышение эффективности при проведении различных органических реакций и отсутствие уменьшения активности даже после извлечения и повторного использования катализатора, что является экономически выгодным. Кроме того, в соответствии со способом изобретения полимеризацию циклических мономеров, замещение заместителей, реакции с образованием связи углерод-углерод и тому подобное можно проводить с чрезвычайно высокой эффективностью. 10 н.п. ф-лы, 7 табл.
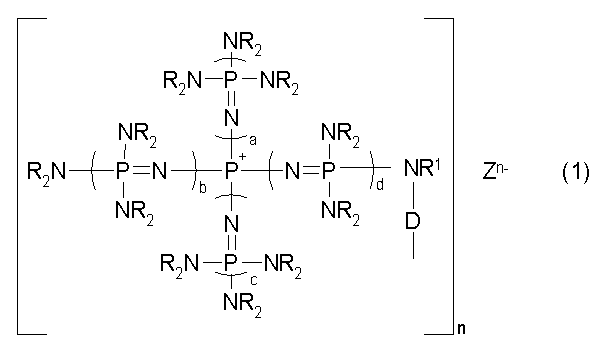
где n является целым числом в диапазоне от 1 до 8 и представляет собой количество фосфазениевых катионов, a Zn- представляет собой анион соединения, содержащего атомы активного водорода, в форме, полученной в результате высвобождения n протонов из соединения, содержащего атомы активного водорода, в котором имеется, самое большее, 8 атомов активного водорода; каждый из а, b, с и d представляет собой положительное целое число, равное 3 или менее; R представляют собой одинаковые или различные углеводородные группы, содержащие от 1 до 10 атомов углерода, и два R, расположенные на каждом общем атоме азота, могут быть связаны друг с другом с образованием кольцевой структуры; R1 представляет собой атом водорода или углеводородную группу, содержащую от 1 до 10 атомов углерода; D представляет собой непосредственную связь или двухвалентную группу, способную связывать N с носителем.
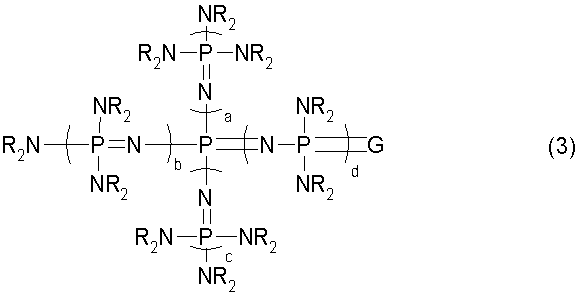
где каждый из a, b, с и d представляет собой положительное целое число, равное 3 или менее; G представляет собой группу =NR, атом кислорода или атом серы; R представляют собой одинаковые или различные углеводородные группы, содержащие от 1 до 10 атомов углерода, и два R, расположенные на каждом общем атоме азота, могут быть связаны друг с другом с образованием кольцевой структуры.
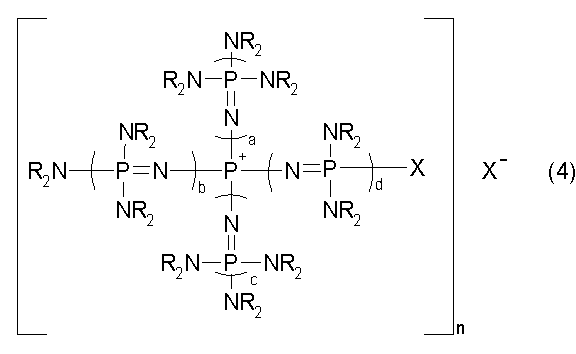
где каждый из а, b, с и d представляет собой положительное целое число, равное 3 или менее, соответственно; R представляют собой одинаковые или различные углеводородные группы, содержащие от 1 до 10 атомов углерода, и два R, расположенные на каждом общем атоме азота, могут быть связаны друг с другом с образованием кольцевой структуры; Х представляет собой атом галогена, а X- представляет собой анион атома галогена, который может быть идентичным или отличным в сопоставлении с Х.
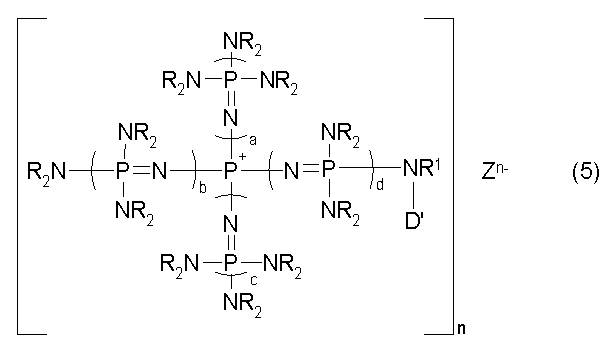
где n является целым числом в диапазоне от 1 до 8 и представляет собой количество фосфазениевых катионов, a Zn- представляет собой анион соединения, содержащего атомы активного водорода, в форме, полученной в результате высвобождения n протонов из соединения, содержащего атомы активного водорода, в котором имеется, самое большее, 8 атомов активного водорода; каждый из а, b, с и d представляет собой положительное целое число, равное 3 или менее; R представляют собой одинаковые или различные углеводородные группы, содержащие от 1 до 10 атомов углерода, и два R, расположенные на каждом общем атоме азота, могут быть связаны друг с другом с образованием кольцевой структуры; R1 представляет собой атом водорода или углеводородную группу, содержащую от 1 до 10 атомов углерода; D' представляет собой одновалентную группу, которая образует связь с N, при том условии, что она отличается от атома водорода и насыщенной углеводородной группы.
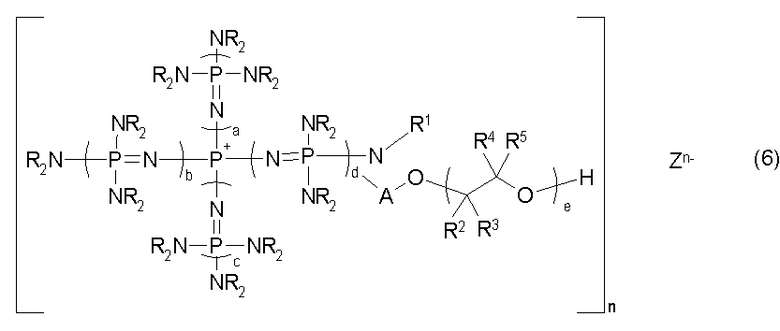
где n является целым числом в диапазоне от 1 до 8 и представляет собой количество фосфазениевых катионов, a Zn- представляет собой анион соединения, содержащего атомы активного водорода, в форме, полученной в результате высвобождения n протонов из соединения, содержащего атомы активного водорода, в котором имеется, самое большее, 8 атомов активного водорода; каждый из а, b, с и d представляет собой положительное целое число, равное 3 или менее; R представляют собой одинаковые или различные углеводородные группы, содержащие от 1 до 10 атомов углерода, и два R, расположенные на каждом общем атоме азота, могут быть связаны друг с другом с образованием кольцевой структуры; А представляет собой углеводородную группу, содержащую от 1 до 20 атомов углерода; кроме того, R1 представляет собой атом водорода или углеводородную группу, содержащую от 1 до 10 атомов углерода; каждый из R2, R3, R4 и R5 представляет собой атом водорода или углеводородную группу, содержащую от 1 до 8 атомов углерода, е находится в диапазоне от 0 до 200.
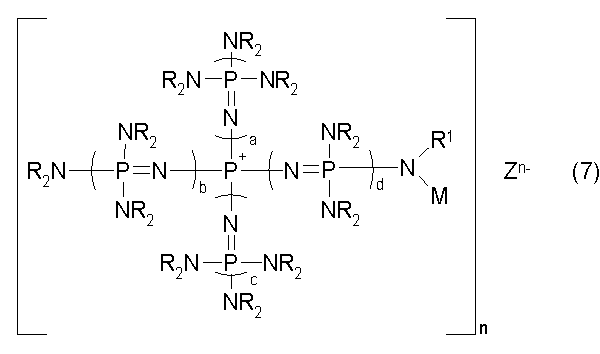
где n является целым числом в диапазоне от 1 до 8 и представляет собой количество фосфазениевых катионов, a Zn- представляет собой анион соединения, содержащего атомы активного водорода, в форме, полученной в результате высвобождения n протонов из соединения, содержащего атомы активного водорода, в котором имеется, самое большее, 8 атомов активного водорода; каждый из а, b, с и d представляет собой положительное целое число, равное 3 или менее; R представляют собой одинаковые или различные углеводородные группы, содержащие от 1 до 10 атомов углерода, и два R, расположенные на каждом общем атоме азота, могут быть связаны друг с другом с образованием кольцевой структуры; R1 представляет собой атом водорода или углеводородную группу, содержащую от 1 до 10 атомов углерода; М представляет собой группу, содержащую ненасыщенную связь углерод-углерод.
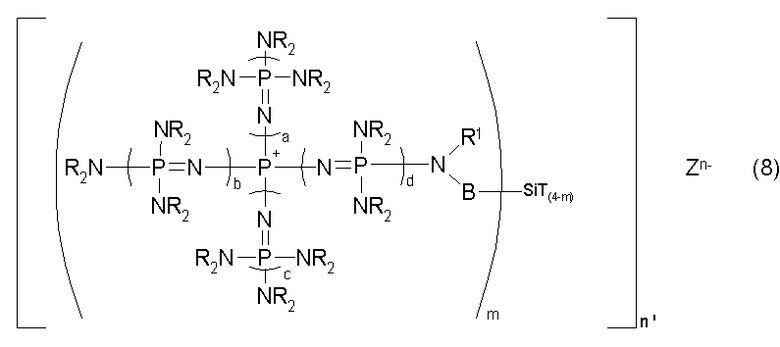
где m является целым числом в диапазоне от 1 до 3 и представляет собой количество фосфазениевых катионов, связанных с атомом кремния, n' является целым числом в диапазоне от 1 до 8 и представляет собой количество соединений кремния, с которыми фосфазениевые катионы образуют связь, n представляет собой множитель для m и n', a Zn-представляет собой анион соединения, содержащего атомы активного водорода, в форме, полученной в результате высвобождения n протонов из соединения, содержащего атомы активного водорода, в котором имеется, самое большее, 24 атома активного водорода; каждый из а, b, с и d представляет собой положительное целое число, равное 3 или менее, R представляют собой одинаковые или различные углеводородные группы, содержащие от 1 до 10 атомов углерода, и два R, расположенные на каждом общем атоме азота, могут быть связаны друг с другом с образованием кольцевой структуры; В представляет собой углеводородную группу, содержащую от 1 до 20 атомов углерода; кроме того, R1 представляет собой атом водорода или углеводородную группу, содержащую от 1 до 10 атомов углерода; Т представляет собой функциональную группу, в которой связь Si-T может быть разрушена в результате гидролиза.
| Способ изготовления фильтрующей пенокерамики | 1985 |
|
SU1294794A1 |
| Способ получения двуокиси титана для наполнения полимеров | 1975 |
|
SU791600A1 |
| US 20030144521 A1, 31.07.2003 | |||
| Teng, Conan J.; Cai, Guoping; Weber, William P | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |

