Данная заявка испрашивает приоритет согласно предварительной заявке США №60/612457, поданной 22 сентября 2004, содержание которой полностью включено сюда путем ссылки.
Область изобретения
Настоящее изобретение главным образом относится к способам получения соединений, которые ингибируют поли(АДФ-рибоза)полимеразы и таким образом замедляют восстановление повреждений цепей ДНК. Способ по изобретению особенно полезен для получения соединений, которые полезны для потенцирования противораковой терапии.
Предпосылки изобретения
Соединение 8-фтор-2-{4-[(метиламино)метил]фенил}-1,3,4,5-тетрагидро-6Н-азепино[5,4,3-cd]индол-6-он, изображенное формулой
 ,
,
представляет собой ингибитор поли(АДФ-рибоза)полимеразы (PARP) с малым размером молекулы. 8-Фтор-2-{4-[(метиламино)метил]фенил}-1,3,4,5-тетрагидро-6Н-азепино[5,4,3-cd]индол-6-он и его соли раскрыты в патенте США №6495541 и заявке РСТ №PCT/IB2004/000915, международная публикация WO 2004/087713, описания которых включены здесь посредством ссылки во всей их полноте. Предварительные заявки на патент США №№60/612459 и 60/679296 под названием "Полиморфные формы фосфатной соли 8-фтор-2-{4-[(метиламино)метил]фенил}-1,3,4,5-тетрагидро-6Н-азепино[5,4,3-cd]индол-6-она," описания которых включены здесь посредством ссылки во всей их полноте, описывают новые полиморфные формы фосфатной соли 8-фтор-2-{4-[(метиламино)метил]фенил}-1,3,4,5-тетрагидро-6Н-азепино[5,4,3-cd]индол-6-она и способы их получения. Предварительные заявки на патент США №№60/612458 и 60/683006 под названием "Терапевтические комбинации, включающие ингибитор поли(АДФ-рибоза)полимераз," описания которых включены здесь посредством ссылки во всей их полноте, описывают фармацевтические комбинации 8-фтор-2-{4-[(метиламино)метил]фенил}-1,3,4,5-тетрагидро-6Н-азепино[5,4,3-cd]индол-6-она.
К настоящему времени идентифицировано восемнадцать ферментов по гомологии последовательности ДНК в семействе PARP, и были исследованы биохимические и ферментативные свойства семи из них: PARP-1 и PARP-2 стимулируются разрывами цепей ДНК, PARP-3 взаимодействует с PARP-1 и центросомой, PARP-4, также известная как PARP «хранилищам (vault PARP, VPARP), является самой большой PARP и связана с цитоплазматическими «хранилищами», танкиразы 1 и 2 (PARP-5a и 5b) ассоциированы с телеметрическими белками, а функция PARP-7 (TiPARP) в настоящее время не ясна, но она может быть вовлечена в функционирование Т-клеток и может являться гистоном поли(АДФ-рибозилата) (Ame JC, Splenlehauer С и de Murcia G. The PARP Superfamily. Boiessays 26 882-893 (2004)). Фармакологические исследование показали, что соединение формулы 1 представляет собой ингибитор PARP-1 (Кi=1,4 нМ) и PARP-2 (Кi=0,17 нМ). Исходя из структурного сходства в последовательностях аминокислот между ферментами PARP, соединение формулы 1, вероятно, связывается с высоким аффинитетом также и с другими членами данного семейства.
Фермент-опосредованное восстановаление разрывов одно- и двуцепочечных ДНК представляет собой потенциальный механизм устойчивости к радиотерапии или цитотоксическим лекарствам, чей механизм зависит от повреждения ДНК. Таким образом, ингибирование ферментов репарации ДНК является стратегией для потенцирования этих агентов. PARP-1, наилучшим образом охарактеризованный член семейства PARP, является ядерным ферментом, который при активации посредством повреждения ДНК опосредует перенос фрагментов АДФ-рибозы от НАД+ к ряду акцепторных белков. В зависимости от величины нанесенных ДНК повреждений активация PARP-1 и последующее поли(АДФ-рибозил)ирование опосредуют репарацию поврежденной ДНК или индуцируют клеточную гибель. Когда повреждения ДНК являются незначительными, PARP-1 играет значительную роль в процесе репарации ДНК. И наоборот, в случае значительного повреждения ДНК избыточная активация PARP-1 истощает пулы АТФ (в попытке пополнить НАД+), что в конечном счете ведет к гибели клетки путем некроза (Tentori L, Portarena I, Graziani G. Potential applications of poly(ADP-ribose)polymerase (PARP) inhibitors. Pharmacol Res 2002; 45:73-85).
Как результат двойственной роли PARP-1, ингибиторы данного фермента, такие как 8-фтор-2-{4-[(метиламино)метил]фенил}-1,3,4,5-тетрагидро-6Н-азепино[5,4,3-cd]индол-6-он, могут играть роль химиосесибилизирующих агентов (путем предотвращения репарации ДНК, например, после противораковой терапии), или в качестве лекарственных средств при различных заболеваниях и токсических состояниях, в которые вовлечены стресс, вызванный окисленем или оксидом азота, и последующая гиперактивация PARP. Такие состояния включают неврологические и нейродегенеративные нарушения (например, болезнь Паркинсона, болезнь Альцгеймера) (Love S, Barber R, Wilcock GK. Increased poly(ADP-ribosyl)ation of nuclear proteins in Alzheimer's disease. Brain 1999; 122:247-53; Mandir AS, Przedborski S, Jackson-Lewis V, et al. Poly(ADP-ribose)polymerase activation mediates 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced parkinsonism, Proc Natl Acad Sci USA 1999; 96:5774-9), сердечно-сосудистые расстройства (например, инфаркт миокарда, ишемически-реперфузионное повреждение) (Pieper AA, Walles Т, Wei G, et al. Myocardial postischemic injury is reduced by poly(ADP-ribose)полимераза-1 gene disruption. J Mol Med 2000; 6:271-82; Szabó G, Bährle S, Stumpf N, et al. Poly(ADP-ribose)polymerase inhibition reduces injury after heart transplantation, Circ Res 2002; 90:100-6; патент США 6423705), воспалительные заболевания (Szabó С, Dawson V. Role of poly(ADP-ribose)synthetase in inflammation and ischaemia-reperfusion. TIPS 1998; 19:287-98), диабетическую сосудистую дисфункцию (Soriano FG, Virág L, Szabó C. Diabetic endothelial dysfunction: role of reactive oxygen and nitrogen species production and poly(ADP-ribose)polymerase activation. J Mol Med 2001; 79:437-48), артрит (Szabó С, Virág L, Cuzzocrea S, et al. Protection against peroxynitrite-induced fibroblast injury and arthritis development by inhibition of poly(ADP-ribose)synthase. Proc Natl Acad Sci USA 1998; 95:3867-72) и нефротоксичность, вызванную цисплатином (Racz I, Tory К, Gallyas F, et al. BGP-15 - a novel poly(ADP-ribose)polymerase inhibitor - protects against nephrotoxicity of cisplatin without compromising its antitumor activity. Biochem Pharmacol 2002; 63:1099-111). Более того, было показано, что клетки опухоли с дефицитом BRCA2 особенно чувствительны к ингибиторам PARP-1 (Bryant et al. "Specific kitting of BRCA2 deficient tumors with inhibitors of poly(ADP-ribose)polymerase," представлено для публикации). Ингибиторы PARP также вовлечены в усиление индукции экспрессии гена Reg в β клетках и HGF гена и соответственно способствуют пролиферации панкреатических β-клеток островков Лангерганса и подавляют апоптоз клеток (публикация заявки на патент США 2004/0091453; РСТ публикация WO 02/00665). Кроме того, ингибиторы PARP также используются в косметических препаратах, особенно в лосьонах после загара (РСТ публикация WO 01/82877). В настоящее время ингибиторы PARP не представлены на рынке.
Один способ синтеза 8-фтор-2-{4-[(метиламино)метил]фенил}-1,3,4,5-тетрагидро-6Н-азепино[5,4,3-cd]индол-6-она раскрыт в упомянутом выше патенте США №6495541. Данный способ представляет собой линейный синтез из 10 стадий, который включает в себя ключевую стадию образования индола Леймгрубера-Батчо (Leimgruber-Batcho) и реакцию сочетания Сузуки. Хотя данный путь является эффективным синтетическим путем, используемым в синтезе токсикологичесих и клинических партий 8-фтор-2-{4-[(метиламино)метил]фенил}-1,3,4,5-тетрагидро-6Н-азепино[5,4,3-cd]индол-6-она, было бы желательным иметь альтернативный конвергентный путь для возможного коммерческого производства. Настоящее изобретение предлагает новый и конвергентный путь к получению 8-фтор-2-{4-[(метиламино)метил]фенил}-1,3,4,5-тетрагидро-6Н-азепино[5,4,3-cd]индол-6-она, который разработан через ключевую реакцию сочетания Соногаширы и CuI-промотированное образование индола.
Сущность изобретения
В одном воплощении настоящего изобретения предложен способ получения соединения формулы I
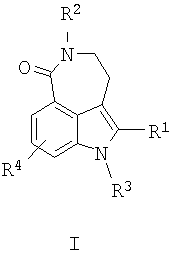
где R1 представляет собой:
H;
циано;
возможно незамещенную или замещенную алкильную, алкенильную, алкинильную, циклоалкильную, гетероциклоалкильную, арильную или гетероарильную группу; или
-C(O)-R5, где R5 представляет собой: Н; возможно незамещенную или замещенную алкильную, алкенильную, алкинильную, циклоалкильную, гетероциклоалкильную, арильную или гетероарильную группу; или OR6 или NR6R7, где каждый из R6 и R7 независимо представляет собой Н или возможно незамещенную или замещенную алкильную, алкенильную, алкинильную, циклоалкильную, гетероциклоалкильную, арильную или гетероарильную группу;
R2 представляет собой Н или алкил;
R3 представляет собой Н или алкил;
R4 представляет собой Н, галоген или алкил,
включающий:
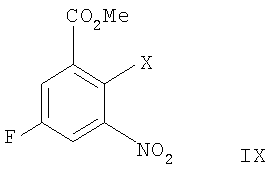
а) сочетание Соногаширы соединения формулы II
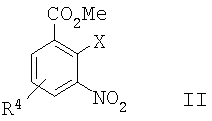
где Х представляет собой галоген или CF3SO2-O-,
с соединением формулы III

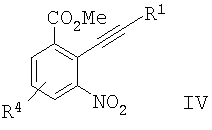
с образованием соединения формулы IV
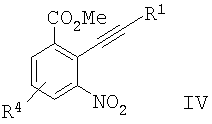
б) восстановление соединения формулы IV с образованием соединения формулы V
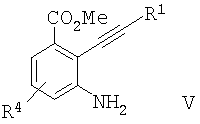
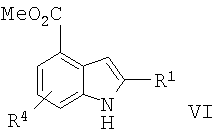
в) превращение соединения формулы V в соединение формулы VI
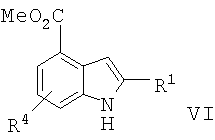
г) превращение соединения формулы VI в соединение формулы I.
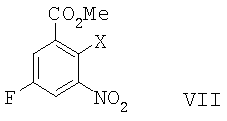
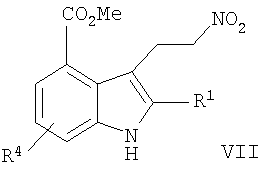
В другом воплощении данного изобретения предложен способ получения 8-фтор-2-{4-[(метиламино)метил]фенил}-1,3,4,5-тетрагидро-6Н-азепино[5,4,3-cd]индол-6-она, включающий сочетание Соногаширы соединения формулы VII
 ,
,
где Х представляет собой галоген или CF3SO2-O-,
с метиловым эфиром (4-этинил-бензил)-метил-карбаминовой кислоты с образованием метилового эфира 5-фтор-2-{4-[(метоксикарбонил-метил-амино)-метил]-фенилэтинил}-3-нитро-бензойной кислоты.
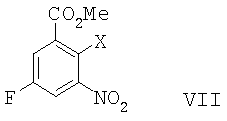
В другом воплощении данного изобретения предложен способ получения 8-фтор-2-{4-[(метиламино)метил]фенил}-1,3,4,5-тетрагидро-6Н-азепино[5,4,3-cd]индол-6-она, включающий сочетание Соногаширы соединения формулы VII
,
где Х представляет собой галоген или CF3SO2-O-,
с метиловым эфиром (4-этинил-бензил)-метил-карбаминовой кислоты с образованием метилового эфира 5-фтор-2-{4-[(метоксикарбонил-метил-амино)-метил]-фенилэтинил}-3-нитро-бензойной кислоты; восстановление метилового эфира 5-фтор-2-{4-[(метоксикарбонил-метил-амино)-метил]-фенилэтинил}-3-нитро-бензойной кислоты до метилового эфира 3-амино-5-фтор-2-{4-[(метоксикарбонил-метил-амино)-метил]-фенилэтинил}-бензойной кислоты; превращение метилового эфира 3-амино-5-фтор-2-{4-[(метоксикарбонил-метил-амино)-метил]-фенилэтинил}-бензойной кислоты в метиловый эфир 6-фтор-2-{4-[(метоксикарбонил-метил-амино)-метил]-фенил}-1H-индол-4-карбоновой кислоты в результате Cul-промотированного образования индола;
обработку метилового эфира 6-фтор-2-{4-[(метоксикарбонил-метил-амино)-метил]-фенил}-1H-индол-4-карбоновой кислоты N,N-диметил-2-нитроэтиленамином с образованием метилового эфира 6-фтор-2-{4-[(метоксикарбонил-метил-амино)-метил]-фенил}-3-((Е)-2-нитро-винил)-1H-индол-4-карбоновой кислоты;
восстановление метилового эфира 6-фтор-2-{4-[(метоксикарбонил-метил-амино)-метил]-фенил}-3-((Е)-2-нитро-винил)-1H-индол-4-карбоновой кислоты до метилового эфира 6-фтор-2-{4-[(метоксикарбонил-метил-амино)-метил]-фенил}-3-(2-нитро-этил)-1Н-индол-4-карбоновой кислоты;
гидрирование метилового эфира 6-фтор-2-{4-[(метоксикарбонил-метил-амино)-метил]-фенил}-3-(2-нитро-этил))-1H-индол-4-карбоновой кислоты с получением метилового эфира [4-(8-фтор-6-охо-3,4,5,6-тетрагидро-1H-азепино[5,4,3-cd]индол-2-ил)-бензил]-метил-карбаминовой кислоты; и
удаление защиты с метилового эфира [4-(8-фтор-6-охо-3,4,5,6-тетрагидро-1H-азепино[5,4,3-cd]индол-2-ил)-бензил]-метил-карбаминовой кислоты с получением 8-фтор-2-{4-[(метиламино)метил]фенил}-1,3,4,5-тетрагидро-6Н-азепино[5,4,3-cd]индол-6-она.
Определения и сокращения
При использовании здесь термин "алкил" означает разветвленную или прямоцепочечную (линейную) алкановую углеводородную группу (насыщенную алифатическую группу), имеющую от 1 до 10 атомов углерода в своей цепи, которая в общем может быть представлена формулой CkH2k+1, где k означает целое число от 1 до 10. Примеры алкильных групп включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, пентил, н-пентил, изопентил неопентил и гексил, и их простые алифатические изомеры. "Низший алкил" означает алкильную группу, имеющую от 1 до 4 атомов углерода в своей цепи.
Термин "алкенил" означает разветвленную или прямоцепочечную алкеновую углеводородную группу (ненасыщенную алифатическую группу, имеющую одну или более чем одну двойную связь), содержащую от 2 до 10 атомов углерода в своей цепи. Примеры алкенилов включают этенил, 1-пропенил, 2-пропенил, 1-бутенил, 2-бутенил, изобутенил и различные изомерные пентенилы и гексенилы (включая как цис-, так и транс-изомеры).
Термин "алкинил" означает разветвленную или прямоцепочечную углеводородную группу, имеющую одну или более чем одну углерод-углеродную тройную связь и содержащую от 2 до 10 атомов углерода в своей цепи. Примеры алкинилов включают этинил, пропинил, 1-бутинил, 2-бутинил и 1-метил-2-бутинил.
Термин "карбоцикл" относится к насыщенной, частично насыщенной, ненасыщенной или ароматической, моноциклической, конденисированной или неконденсированной полициклической кольцевой структуре, имеющей в кольце только атомы углерода (нет гетероатомов, т.е. неуглеродных кольцевых атомов). Примеры карбоциклов включают циклоалкильные, арильные и циклоалкил-арильные группы.
Термин "гетероцикл" относится к насыщенной, частично насыщенной, ненасыщенной или ароматической, моноциклической, конденсированной или неконденсированной полициклической кольцевой структуре, имеющей один или более чем один гетероатом, выбранный из N, О и S. Примеры гетероциклов включают гетероциклоалкильные, гетероарильные и гетероциклоалкил-гетероарильные группы.
"Циклоалкильная группа" означает неароматическую моновалентную, моноциклическую или конденсированную полициклическую кольцевую структуру, имеющую в целом от 3 до 18 кольцевых атомов углерода (но не гетероатомов). Примеры циклоалкилов включают циклопропил, циклобутил, циклопентил, циклопентенил, циклогексил, циклогептил, адамантил, фенантренил и подобные группы.
"Гетероциклоалкильная группа" означает неароматическую моновалентную, моноциклическую или конденсированную полициклическую кольцевую структуру, имеющую в целом от 3 до 18 кольцевых атомов, включая от 1 до 5 гетероатомов, выбранных из азота, кислорода и серы. Иллюстративные примеры гетероциклоалкильных групп включают пирролидинил, тетрагидрофурил, пиперидинил, пиперазинил, морфолинил, тиоморфолинил, азиридинил и подобные группы.
Термин "арил" означает ароматическую моноциклическую или конденсированную полициклическую кольцевую структуру, имеющую в целом от 4 до 18, предпочтительно от 6 до 18 кольцевых атомов углерода (гетероатомы отсутствуют). Примеры арильных групп включают фенил, нафтил, антраценил и подобные группы.
«Гетероарильная группа» означает ароматическую моновалентную, моноциклическую или конденсированную полициклическую кольцевую структуру, имеющую от 4 до 18, предпочтительно от 5 до 18 кольцевых атомов, включая от 1 до 5 гетероатомов, выбранных из азота, кислорода и серы. Иллюстративные примеры гетероарильных групп включают пирролил, тиенил, оксазолил, пиразолил, тиазолил, фурил, пиридинил, пиразинил, триазолил, тетразолил, индолил, хинолинил, хиноксалинил и подобные группы.
Термин "возможно замещенный" указывает, что конкретная группа является незамещенной или замещена одним или более чем одним подходящим заместителем, если возможные заместители не оговорены точно, в таком случае данный термин указывает, что эта группа является незамещенной или замещена конкретными заместителями. Если не указано иначе (например, путем указания, что конкретная группа является незамещенной), различные группы, указанные выше, могут быть обычно незамещенными или замещены (т.е. они являются возможно замещенными) одним или более чем одним подходящим заместителем.
Термин "заместитель" или "подходящий заместитель" означает любой заместитель для группы, который может быть распознан или легко выбран специалистом в данной области техники, например, путем обычного тестирования, как являющийся фармацевтически приемлемым. Иллюстративные примеры подходящих заместителей включают гидрокси, галоген (F, Cl, I или Br), оксо, алкил, ацил, сульфонил, меркапто, нитро, алкилтио, алкокси, циклоалкил, гетероциклоалкил, арил, гетероарил, карбокси, амино (первичный, вторичный или третичный), карбамоил, арилокси, гетероарилокси, арилтио, гетероарилтио и тому подобные (например, как продемонстрировано соединениями из примеров, описанными здесь). Подходящие заместители очевидны из примеров соединений, приведенных ниже.
Предпочтительные возможные заместители для алкильных и арильных групп в соединениях по изобретению включают галогены и арильные группы. Особенно предпочтительными для замещенных алкильных групп являются перфторзамещенные алкилы. Особенно предпочтительные возможные заместители для арильных группировок включают галоген, низший алкил, -ОН, -NO2, -CN, -CO2H, O-низший алкил, арил, -O-арил, арил-низший алкил, -CO2СН3, -CONH2, -OCH2CONH2, -NH2, -SO2NH2, -OCHF2, -CF3, -OCF3 и тому подобные. Арильные группировки могут также быть возможно замещены двумя заместителями, образующими мостик, например -O-(CH2)z-O-, где z пердставляет собой целое число 1, 2 или 3.
При использовании в данной заявке "Et" означает этил, "Ас" означает ацетил, "Me" означает метил, "Ms" означает метансульфонил (СН3SO2), "iPr" означает изопропил, "HATU" означает 2-(7-аза-1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилуроний гексафторфосфат, "PD" означает фенил, "Boc" означает трет-бутоксикарбонил, "EtOAc" означает этилацетат, "НОАс" означает уксусную кислоту, "NEt3" или "Et3N" означает триэтиламин, «Tf» означает трифторметансульфонил, "THF" означает тетрагидрофуран, "DIC" означает диизопропилкарбодиимид, "HOBt" означает гидроксибензотриазол, "МеОН" означает метанол, "i-PrOAc" означает изопропилацетат, "КОАс" означает ацетат калия, "DMSO" означает диметилсульфоксид, "AcCI" означает ацетилхлорид, "CDCl3" означает дейтерированный хлороформ, "МТВЕ" означает метил-трет-бутиловый эфир, "DMF" означает диметилформамид, "Ас2O" означает уксусный ангидрид, "Ме3SOI" означает триметилсульфоксония иодид, "DMAP" означает 4-диметиламинопиридин, "dppf" означает дифенилфосфиноферроцен, "DME" означает диметиловый эфир этиленгликоля (1,2-диметоксиэтан), HOBT означает 1-гидроксибензотриазол, EDC означает 1-этил-3-(3-диметиламинопропил)-карбодиимид.
Подробное описание изобретения
Настоящее изобретение предлагает новый и конвергентный путь синтеза трициклических ингибиторов поли(АДФ-рибоза)полимеразы (PARP), которые раскрыты в упомянутом выше патенте США №6495541. Только в качестве иллюстрации способ по настоящему изобретению демонстрируется примером способа получения 8-фтор-2-{4-[(метиламино)метил]фенил}-1,3,4,5-тетрагидро-6Н-азепино[5,4,3-cd]индол-6-она. Реагенты и условия реакций, описанные здесь, иллюстрируют большое разнообразие исходных материалов, их количества и условий, которые могут быть использованы подходящим образом в настоящем изобретении, что будет понятно специалисту в данной области техники, и ни в коем случае не являются ограничивающими.
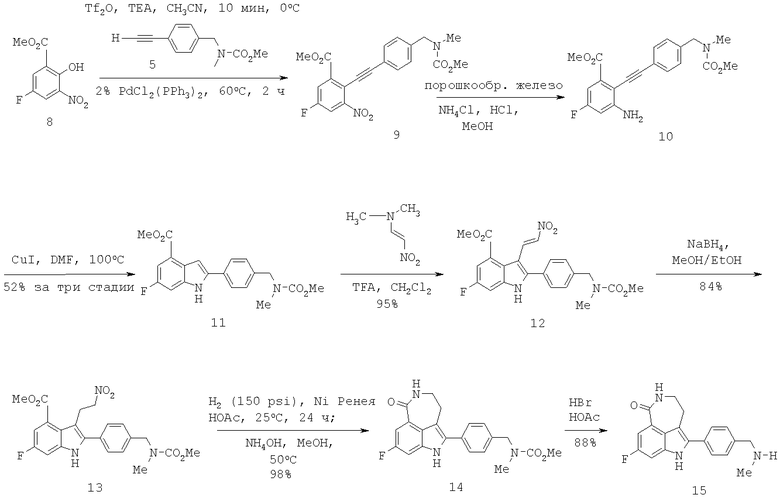
Способ по настоящему изобретению включает в себя получение 8-фтор-2-{4-[(метиламино)метил]фенил}-1,3,4,5-тетрагидро-6Н-азепино[5,4,3-cd]индол-6-она (соединение 15) через ключевую реакцию сочетания Соногаширы и Cul-индуцируемое образование индола согласно Схеме 1.
Схема 1

I. Получение алкина 5 и трифлатного предшественника 8 для сочетания Соногаширы
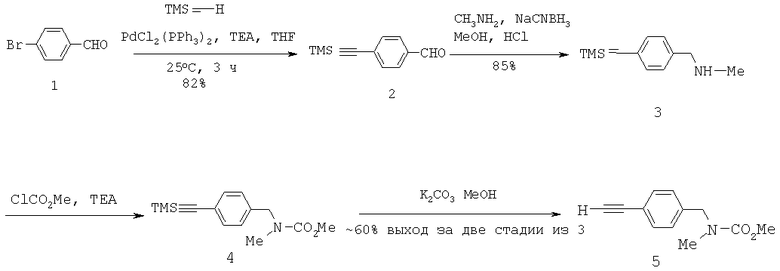
Синтез алкина 5 показан на Схеме 2. Альдегид 2, хотя он имеется в продаже от Aldrich, является дорогим, поэтому будет более экономичным получить его из относительно недорогого 4-бромбензальдегида 1 согласно процедуре, описанной в литературе (Thorand, S. and Krause, N. J. Org. Chem. 1998, 63, 8551). Далее восстановительное аминирование альдегида 2 метиламином позволяет получить амин 3, который далее может быть защищен с полученим метилкарбамата 4. Удаление триметилсилильной группы (TMS) в основных условиях позволяет получить 5 с выходом 90%.
Схема 2
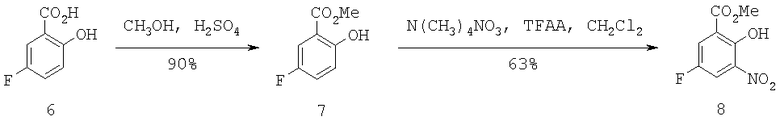
Трифлатный предшественник 8 можно получить через нитрование 5-фторсалициловой кислоты 6 с использованием нитрата тетраметиламмония и ангидрида трифторуксусной кислоты в стандартных условиях (Схема 3). Желаемый продукт 8 может быть получен региоспецифично в виде желтого твердого вещества после кристаллизации из CH3CN/H2O с выходом 63%.
Схема 3
II. Синтез индольного промежуточного соединения 11
Как показано на Схеме 4, хотя трифлат 16 и может быть выделен, более выгодно получать его in situ.
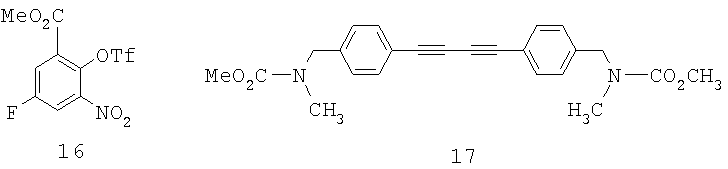
Образование трифлата 16 и его сочетание с алкином 5 может быть проведено в одной емкости с получением промежуточного соединения 9. Предпочтительными для этого сочетания являются следующие условия: CH3CN-раствор, содержащий алкин 5, и заранее полученный трифлат 16 добавляют к раствору, содержащему 2% PdCl2(PPh3)2 в CH3CN, при 60°С. Как определено, в этих условиях главным побочным продуктом является димер 17. Далее добавление сокатализатора CuI, как обнаружено, является нежелательным, так как способствует образованию большего количества димера. После завершения сырой продукт 9 может быть восстановлен порошкообразным железом. Добавление нескольких капель концентрированной HCl является очень благоприятным для активации порошкообразного железа. Последующая циклизация неочищенного соединения 10 в индол 11 может быть осуществлена с каталитичеким количеством Cul в DMF. Индол 11 может быть получен с выходом 51% за три стадии после растирания со смесью метиленхлорид/гексаны с тем, чтобы удалить побочный продукт 17.
Схема 4
III. Превращение индольного промежуточного соединения 11 в целевое соединение 15
Обработка индола 11 N,N-диметил-2-нитроэтиленамином (Mahboobi, S.; Elbler, E.; Roller, M.; Kumar, S.; Popp, A. J. Org. Chem. 1999, 64; 4697) в смеси TFA/CH2Cl2 дает нитроалкен 12 (Схема 4). Нитроалкен 12 затем может быть восстановлен боргидридом натрия в смеси 9:1 EtOH/MeOH до нитроалкана 13 с выходом 84%. Из-за низкой растворимости 12 и 13 в EtOH необходим большой объем растворителя для обеспечения полного превращения. Гидрирование 13 до лактама 14 с использованием Pd/C или Pt/C в нейтральных условиях позволяет получить N-гидроксилактам 18 в качестве основного побочного продукта. Однако когда гидрирование проводят в кислых условиях с Pd/C или Pt/C, образование 18 подавляется, но это вызывает расщепление связи C-N в 13 с получением 19 в качестве другого основного побочного продукта. Так как Ni Ренея не катализирует расщепление связей C-N, следовало протестировать реакции гидрирования в условиях, опосредованных Ni Ренея. Неожиданно было обнаружено, что гидрирование 13 с использованием Ni Ренея хорошо протекает как в нейтральных, так и кислых условиях. Так, нитроалкан 13 при восстановлении Ni Ренея в уксусной кислоте дает соответствующий аминацетат, который при обработке основанием циклизуется в 14 с выходом 96% в масштабе 20 г без каких-либо побочных продуктов.
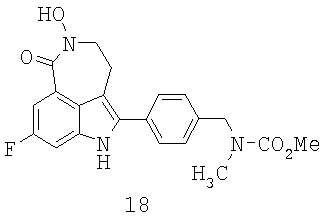
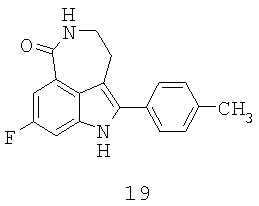
Наконец, с карбаматной группы в 14 легко снимается защита с помощью HBr/уксусной кислоты при температуре окружающей среды с получением 15. Первоначальные попытки удалить карбаматную группу щелочным гидролизом, например 8 н. КОН в EtOH при 80°С, приводят к нежелательному расщеплению лактамного кольца в 15. Хотя метилкарбаматы также могут быть расщеплены триметилсилилиодидом, образование побочного продукта метилиодида поднимает проблему безопасности и делает этот способ менее привлекательным. С другой стороны, токсичный метилбромид, образующийся во время расщепления смесью HBr/уксусная кислота, может эффективно улавливаться с помощью этаноламинной скрубберной системы, как описано в Hettenbach, К.; Am Ende, D.J.; Leeman, К.; Dias, E.; Kasthurikhshnan, N.; Brenek, S.J.; Ahlijanian, P. Органический Process Research & Development, 2002, 6, 407. Таким образом получают целевое соединение 15 в количестве 20 г, которое идентично аутентичному образцу, синтезированному предыдущим способом.
Описания всех цитированных ссылок включены сюда путем ссылки во всей полноте.
Примеры
Примеры и получения, приведенные ниже, иллюстрируют и служат примерам сопсобов по настоящему изобретению. Следует понимать, что объем настоящего изобретения никоим образом не ограничивается следующими примерами.
Пример 1. Синтез 4-триметилсиланилэтинил-бензальдегида (2)
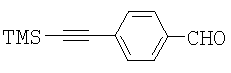
4-Бромбензальдегид (1) (185 г, 1,0 моль) растворяли в THF (1 л) с последующим добавлением иодида меди(I) (7,6 г, 0,04 моль), дихлорбис(трифенилфосфин)палладия(II) (14,02 г, 0,02 моль) и триэтиламина (151,5 г, 1,5 моль). Этинилтриметилсилан (109,1 г, 1,11 моль) добавляли из капельной воронки в виде раствора в THF (0,2 л). Реакционную смесь перемешивали при 30°С в течение 30 минут, а затем при 25°С в течение 20 часов. Анализ посредством HPLC (высокоэффективная жидкостная хроматография) показал завершение реакции. THF удаляли и остаток обрабатывали гексаном (1,8 л). Твердое вещество удаляли фильтрацией, осадок на фильтре промывали гексаном (0,3 л). Объединенный гексановый раствор промывали водой (2×0,5 л). Гексан удаляли в аппарате rotovap. Остаток растворяли в EtOH (0,5 л) при 50°С. Раствор затем медленно охлаждали до 16°С и перемешивали в течение 30 минут. Продукт начинал кристаллизоваться. Смесь далее охлаждали до 5°С. Медленно добавляли смесь 1:1 EtOH/Н2O (0,24 л). Смесь перемешивали при 5°С в течение 30 минут. Твердое вещество собирали фильтрацией, промывали смесью 4:1 EtOH/H2O (0,2 л) и сушили с получением 137,0 г продукта. Маточный раствор концентрировали досуха. Остаток разделяли между гексаном (0,5 л) и рассолом (0,25 л). Гексановый слой отделяли и концентрировали досуха. Остаток кристаллизовали из гексана (40 мл) и затем перекристаллизовывали из смеси 4:1 EtOH/H2O (0,1 л) с получением второй порции альдегида 2 (27,0 г). Общий выход альдегида 2 составил 81%. 1Н ЯМР (300 МГц, CDCl3) δ 0.081 (s, 9H), 7.41 (d, 2H, J=8,1 Гц), 7.63 (d, 2H, J=8,4 Гц), 9.81 (s, 1H).
Пример 2. Синтез метил-(4-триметилсиланилэтинил-бензил)-амина (3)
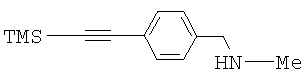
Метиламин (8 М в МеОН, 135 мл) и гидрохлорид метиламина (44,0 г, 0,65 моль) растворяли в метаноле (900 мл). Добавляли альдегид 2 (44,0 г, 0,22 моль) и перемешивали при комнатной температуре в течение 30 минут. Затем добавляли циноборгидрид натрия (17,42 г, 0,28 моль). После завершения добавления добавляли метанольный раствор гидрохлорида для доведения значения рН до 5, при этом температуру поддерживали равной приблизительно 30°С. Реакционную смесь перемешивали в течение 2 часов. Значение рН реакционной смеси поддерживали между 4 и 6 добавлением раствора гидрохлорида в МеОН. Реакционный растворитель удаляли. Остаток переносили в воду (400 мл) и рассол (50 мл). Смесь экстрагировали метиленхлоридом (2×300 мл). Объединенный органический раствор концентрировали досуха с получением сырого продукта (40,6 г, выход приблизительно 85%), который использовали непосредственно на следующей стадии без дальнейшей очистки. 1Н ЯМР (300 МГц, CDCl3) δ 0.254 (s, 9H), 2.434 (s, 3Н), 3.736 (s, 2H), 7.25 (d, 2H, J=9 Гц), 7.43 (d, 2H, J=9 Гц). Точная масса, подсчитанная для C13H20NSi: 218,1365. Обнаружено: 218,1357.
Пример 3. Синтез метилового эфира метил-(4-триметилсиланилэтинил-бензил)-карбаминовой кислоты (4)
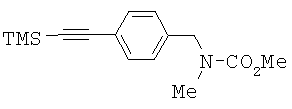
Амин 3 (90,0 г, приблизит.0,41 моль) растворяли в метиленхлориде (810 мл). Добавляли триэтиламин (66,6 г, 0,66 моль) и раствор охлаждали до 5°С. Затем медленно добавляли метилхлорформиат (47,0 г, 0,50 моль) в метиленхлориде (100 мл) и температуру реакции поддерживали между 10°С и 14°С. После завершения добавления реакционный раствор перемешивали при комнатной температуре в течение 12 часов. Добавляли воду (540 мл). Водную фазу отделяли. Органическую фазу концентрировали досуха с получением сырого соединения 4. Сырой продукт использовали на следующей стадии без дополнительной очистки. 1Н ЯМР (300 МГц, CDCl3) δ 0.279 (s, 9H), 2.86 (d, ушир., 3Н), 3.774 (s, 3Н), 4.484 (s, ушир., 2H), 7.190 (s, ушир., 2H), 7.46 (d, 2H, J=8.10 Гц).
Пример 4. Синтез метилового эфира (4-этинил-бензил)-метил-карбаминовой кислоты (5)
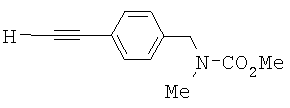
Карбамат 4 со стадии 3 растворяли в метаноле (630 мл). Добавляли карбонат калия (10,5 г, 0,08 моль). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Анализ посредством TLC (тонкослойная хроматография) показал завершение реакции. Отфильтровали белое твердое вещество. Метанол удаляли перегонкой при пониженном давлении с получением желтого масла. Это масло далее очищали колоночной хроматографией (силикагель, гексан/EtOAc) с выходом 50,2 г соединения 5 (выход приблизительно 60% из двух стадий из амина 3). 1Н ЯМР (300 МГц, CDCl3) δ 2.85 (d, ушир., 3Н), 3.068 (s, 1H), 3.744 (s, 3H), 4.468 (s, ушир., 2Н), 7.195 (s, ушир., 2Н), 7.46 (d, 2Н, J=8.1 Гц). Точная масса, подсчитанная для C12H14NO2: 204,1025. Обнаружено: 204,1022.
Пример 5. Синтез метилового эфира 5-Фторсалициловой кислоты (7)

5-Фторсалициловую кислоту (272,6 г, 1,74 моль) растворяли в метаноле (1,3 л) с образованием прозрачного раствора. Концентрированную серную кислоту (50 мл) медленно добавляли к метанольному раствору при активном перемешивании. Раствор нагревали до температуры дефлегмации в течение 4 часов. Триметилортоформиат (200 мл) медленно добавляли к реакционному раствору. Отогнали 300 мл растворителей (метилформиат и метанол). Остаток реакционного раствора нагревали при 66°С (температура дефлегмации) в течение 16 часов. Анализ посредством HPLC показал, что реакция завершена. Реакционный раствор охлаждали до температуры окружающей среды. Растворитель удаляли при пониженном давлении. Остаток разделяли между водой (140 мл) и метиленхлоридом (220 мл). Органический слой отделяли. Водный слой экстрагировали метиленхлоридом (3×220 мл). Объединенный органический слой промывали водой (270 мл), рассолом (270 мл) и сушили MgSO4. Органический раствор концентрировали досуха с получением сырого продукта (293,8 г). Этот сырой продукт использовали на следующей стадии без дополнительной очистки. 1Н ЯМР (300 МГц, CDCl3) δ 3.96 (s, 3H), 6.94 (dd, 1H, J=4,5 Гц и J=9,0 Гц), 7.19 (m, 1H), 7.50 (dd, 1H, J=3,3 Гц и J=8,7 Гц), 10.508 (s, 1H).
Пример 6. Синтез метилового эфира 5-фтор-2-гидрокси-3-нитро-бензойной кислоты (8)
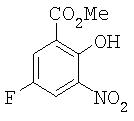
Тетраметиламмония нитрат (98,4 г, 0,72 моль) суспендировали в метиленхлориде (800 мл). К смеси при перемешивании добавляли трифторуксусный ангидрид (180,3 г, 0,857 моль). Смесь охлаждали до 8°С. Затем в реакционную колбу медленно добавляли раствор эфира 7 (106 г, 0,624 моль) в СН2Cl2 (150 мл) в течение 25 минут, температуру в это время поддерживали между 5°С и 10°С. Реакционную смесь перемешивали при 8°С в течение дополнительных 40 минут после завершения добавления. Насыщенный водный бикарбонат натрия (500 мл) медленно добавляли для гашения реакции. Органический слой отделяли, промывали водой (2×500 мл) и концентрировали досуха с получением сырого твердого вещества. Это твердое вещество растворяли в ацетонитриле (300 мл) при 55°С. Медленно добавяли воду (100 мл) к ацетонитрильному раствору при перемешивании и твердое вещество выпадало в осадок. Суспензию охлаждали до 18°С и перемешивали при этой температуре в течение 30 минут. Затем ее охлаждали до 3°С и перемешивали при этой температуре в течение 30 минут. Твердое вещество собирали фильтрованием. Осадок на фильтре промывали холодным растворителем CH3CN/H2O (2/1, 100 мл) и сушили с получением 78,49 г светло-желтого продукта (57%-ный выход за две стадии). 1Н ЯМР (300 МГц, CDCl3) δ 4.035 (s, 3Н), 7.88 (dd, 1H, J=3,6 Гц и J=9,0 Гц), 7.94 (dd, 1H, J=3,3 Гц и J=7,5 Гц), 11.71 (s, 1H). Точная масса, подсчитанная для С8Н5FNO5: 214,0152. Обнаружено: 214,0141.
Пример 7. Синтез метилового эфира 5-фтор-2-{4-[(метоксикарбонил-метил-амино)-метил1-фенилэтинил}-3-нитро-бензойной кислоты (9)

Нитросоединение 8 (47,87 г, 0,223 моль) растворяли в ацетонитриле (240 мл) и охлаждали до -8°С. Добавляли триэтиламин (33,8 г, 0,334 моль). Образовывался прозрачный темный раствор. Медленно добавляли ангидрид трифторметансульфоновой кислоты (69,17 г, 0,245 моль) и температуру реакции поддерживали равной -10°С. Через 10 минут поле завершения добавления анализ посредством TLC показал завершение образования трифлата. Охлаждающую баню удаляли. Добавляли алкин 5 (47,46 г, 0,234 моль). Раствор дегазировали трижды путем откачивания газа из колбы и повторного заполнения ее азотом. Затем раствор перенесли в капельную воронку. В отдельную реакционную колбу добавили ацетонитрил (240 мл) и триэтиламин (22,5 г, 0,223 моль). Раствор дегазировали трижды, подвергая реакционную колбу воздействию вакуума и азота альтернативно. К раствору добавили дихлорбис(трифенилфосфин)палладий (II) (3,13 г, 0,0045 моль) и раствор снова дегазировали трижды. Этот раствор нагревали до 65°С в атмосфере азота. Когда температура достигла 65°С, одну четверть раствора алкина 5 и трифлата из капельной воронки быстро добавляли в реакционную колбу. Оставшийся раствор в капельной воронке добавляли в течение 30 минут при 65°С. После завершения добавления реакционный раствор перемешивали при 65°С в течение 2 часов. HPLC анализ показал завершение реакции. Реакционную смесь охлаждали до комнатной температуры. Растворитель удаляли при пониженном давлении. Остаток растворяли в этилацетате (500 мл) и промывали водой (300 мл). Водную фазу отделяли и снова экстрагировали этилацетатом (200 мл). Объединенный органический раствор промывали водой (200 мл). Затем его концентрировали досуха с получением темного масляного остатка (140 г). Этот сырой продукт использовали на следующей стадии без дополнительной очистки. 1Н ЯМР (300 МГц, CDCl3) δ 2.87 (d, ушир., 3Н), 3.755 (s, 3Н), 4.011 (s, 3Н), 4.50 (s, ушир., 2Н), 7.21 (s, ушир., 2Н), 7.55 (d, 2H, J=9 Гц), 7.80 (dd, 1H, J=2,7 Гц и J=7.2 Гц), 7.86 (dd, 1 H, J=2,7 Гц и J=8,1 Гц).
Пример 8. Синтез метилового эфира 3-амино-5-фтор-2-{4-[(метоксикарбонил-метил-амино)-метил]-фенилэтинил}-бензойной кислоты (10)

Сырое соединение 9 (140 г) растворяли в метаноле (1,05 л). Добавляли порошкообразное железо (325 меш, 76,2 г, 1,36 моль) с последующим добавлением насыщенного водного хлорида аммония (210 мл). Раствор нагревали до 65°С. Добавляли водную соляную кислоту (16% масс., 32 мл). Реакционную смесь нагревали при 65°С в течение 2 часов. HPLC анализ показал завершение реакции. Реакционную смесь охлаждали до комнатной температуры. Твердое вещество удаляли фильтрованием. Осадок на фильтре промывали метанолом (2 л). Объединенный метанольный раствор концентрировали досуха. Остаток разделяли между этилацетатом (800 мл) и разбавленным водным хлористоводородным раствором (0,5 М, 300 мл). Органический слой отделяли. Водный слой экстрагировали этилацетатом (3×200 мл). Объединенный органический раствор промывали рассолом (200 мл). Раствор затем концентрировали досуха с получением 122,8 г сырого продукта в виде темного масла. Этот сырой продукт использовали на следующей стадии без дополнительной очистки. 1Н ЯМР (300 МГц, CDCl3) δ 2.87 (d, ушир., 3Н), 3.491 (s, 3Н), 3.932 (s, 3Н), 4.486 (s, ушир., 2Н), 6.60 (dd, 1H, J=2,7 Гц и J=9,9 Гц), 7.04 (dd, 1H, J=2,4 Гц и J=9,0 Гц), 7.237 (s, ушир., 2Н), 7.52 (d, 2Н, J=8,1 Гц).
Пример 9. Синтез метилового эфира 6-фтор-2-{4-[(метоксикарбонил-метил-амино)-метил]-фенил}-1H-индол-4-карбоновой кислоты (11)

Сырое соединение 10 (114,37 г, приблизит.0,309 моль) растворяли в DMF (1,5 л). Добавляли иодид меди (I) (99,9999%-ная чистота, 5,8 г, 0,0309 моль). Реакционную смесь дегазировали 4 раза, подвергая реакционную колбу воздействию вакуума и азота альтернативно. Реакционную смесь затем нагревали при 100°С в течение 44 часов в атмосфере азота. HPLC анализ показал исчезновение исходного материала. Реакционную смесь охлаждали до температуры окружающей среды. Растворитель удаляли при пониженном давлении (9 мбар, (9 кПа)) при 35°С. Черный остаток растворяли в CH2Cl2 (0,15 л). Смесь пропускали сквозь подушку из целита. Затем органический раствор пропускали через подушку из силикагеля (300 г силикагеля). Для элюции соединение из силикагелевой подушки использовали CH2Cl2 (2,3 л). Органический раствор концентрировали досуха. Остаток растворяли в СН2Cl2 (0,15 л). К раствору при перемешивании медленно добавляли гексан (1 л). Выпавший в осадок продукт собирали фильтрованием, промывали EtOAc (0,2 л) и сушили с получением 31,34 г продукта. Промывочный раствор EtOAc соединяли с маточным раствором. Этот раствор концентрировали досуха. Остаток растворяли в СН2Cl2 (0,15 л). Раствор затем пропускали через подушку из силикагеля (300 г). Силикагелевую подушку промывали CH2Cl2 для элюирования соединения. Органический раствор концентрировали досуха. Остаток растворяли в СН2Cl2 (0,07 л). Добавляли гептан (0,22 л) для осаждения продукта. Твердое вещество собирали, промывали раствором 1:3 СН2Cl2/гептан (0,06 л) и сушили с получением дополнительных 27,51 г продукта. Объединенный продукт имел массу 58,85 г, что составляет 51%-ный выход с трех стадий из соединения 8. 1Н ЯМР (300 МГц, CDCl3) δ 2.896 (s, 3Н), 3.779 (s, 3Н), 4.006 (s, 3Н), 4.516 (s, 2H), 7.31 (dd, 2H, J=1,5 Гц и J=8,7 Гц). 7.429 (s, 1H), 7.65 (m, 3Н), 9.17 (d, ушир., 1H). Точная масса, подсчитанная для C20H20FN2O4: 371,1407. Обнаружено: 371,1418.
Пример 10. Синтез метилового эфира 6-фтор-2-{4-[(метоксикарбонил-метил-амино)-метил]-фенил}-3-((Е)-2-нитро-винил)-1H-индол-4-карбоновой кислоты (12)
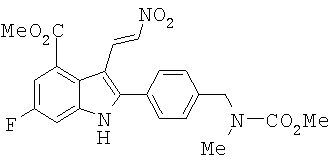
Трифторуксусную кислоту (0,14 л) охлаждали до 5°С. К TFA порциями добавляли 1-N,N-диметиламино-2-нитроэтилен (15,0 г, 0,129 моль) при 5-8°С. Образовался прозрачный раствор. Индол 11 добавляли несколькими порциями. Затем добавляли СН2Cl2 (0,02 л). Реакционный раствор перемешивали при температуре окружающей среды в течение 44 часов. TLC анализ показал завершение реакции. Растворитель удаляли при пониженном давлении при 23°С (47 мбар (47кПа)). Остаток медленно вливали в колбу, содержащую СН2Cl2 (0,4 л) и насыщенный водный NaHCO3 (0,5 л). Конечное значение рН водной фазы составляло 5,0. Органическую фазу отделяли. Водную фазу экстрагировали СН2Cl2 (2×100 мл). Органический раствор объединяли и концентрировали в аппарате rotovap, пока не образовалось твердое вещество. К суспензии в СН2Cl2 добавляли гептан (0,2 л) и смесь перемешивали в течение 30 минут. Собирали твердое вещество, промывали смесью 1:4 СН2Cl2/гептан (100 мл) и сушили на воздухе с получением 49,18 г продукта (95%-ный выход). 1Н ЯМР (300 МГц, DMSO-d6) δ 2.88 (s, 3Н), 3.661 (s, 3Н), 3.920 (s, 3Н), 4.554 (s, 2H), 6.86 (d, 1H, J=13,5 Гц), 7.46 (d, 2H, J=7,5 Гц), 7.52 (d, 2H, J=9,3 Гц), 7.65 (d, 2H, J=8,1 Гц), 8.69 (d, 1H, J=13,5 Гц), 12.895 (s, ушир., 1H). Точная масса, подсчитанная для С22Н21FN3О6: 442,1414. Обнаружено: 442,1420.
Пример 11 Синтез метилового эфира 6-фтор-2-{4-[(метоксикарбонил-метил-амино)-метил]-фенил}-3-(2-нитро-этил)-1H-индол-4-карбоновой кислоты (13)
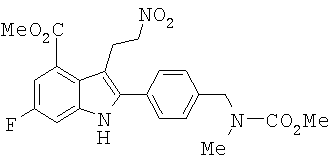
EtOH (900 мл) и МеОН (90 мл) добавляли в колбу емкостью 2 л. Затем добавляли порошкообразый боргидрид натрия (20,5 г, 0,55 моль). Суспензию охлаждали до 15°С. Порциями добавляли нитроалкен 12 (49,0 г, 0,11 моль) в течение 45 минут, при этом температуру поддерживали равной приблизит. 15°С. После завершения добавления добавляли дополнительное количество EtOH (800 мл). Реакционную смесь перемешивали в течение 15 минут. TLC анализ показал завершение реакции. Медленно добавляли смесь уксусной кислоты (40 мл) и воды (40 мл) для гашения избытка боргидрида натрия. Получали красноватую суспензию. Растворитель удаляли в аппарате rotovap. Остаток разделяли между EtOAc (600 мл) и водой (300 мл). Водную фазу отделяли. Твердое вещество в органическом слое собирали фильтрацией и сушили с получением 21,94 г продукта 13 (45%-ный выход). Органический фильтрат концентрировали досуха. Остаток затем ресуспендировали в смеси EtOAC (70 мл)/гептан (200 мл). Твердое вещество собрали фильтрацией и сушили с получением второй порции продукта (21,74 г, 44%-ный выход). Общий выход для этой стадии составил 89%. 1Н ЯМР (300 МГц, DMSO-d6) δ 2.87 (s, 3Н), 3.584 (t, 2H, J=7.8 Гц), 3.661 (s, 3Н), 3.903 (s, 3Н), 4.519 (s, 2H), 4.607 (t, 2H, J=7,8 Гц), 7.41 (m, 4H), 7.58 (d, 2H, J=8,10 Гц), 12.0 (s, ушир., 1Н). Точная масса, подсчитанная для C22H23FN3O6: 444,1571. Обнаружено: 444,1559.
Пример 12. Синтез метилового эфира [4-(8-фтор-6-оксо-3,4,5,6-тетрагидро-1H-азепино[5,4,3-cd]индол-2-ил)-бензил]-метил-карбаминовой кислоты (14)

Никель Ренея (30 мл), А-5000 от Active Metals) промывали водой (2×50 мл), МеОН (2×50 мл) и НОАс (50 мл). Затем его смешали с НОАс (400 мл) и перенесли в гидрогенизатор емкостью 2 л. Добавляли нитросоединение 13 (19,33 г, 0,044 моль). Суспензию гидрировали при 150 фунт-сила/кв. дюйм (1035 кПа) при температуре окружающей среды в течнение 20 часов. HPLC анализ показал исчезновение исходного материала 13. Катализатор отфильтровали и осадок на фильтре промывали EtOH (200 мл). (Осторожно: никель Ренея может воспламеняться на воздухе. Никогда не позволяйте осадку на фильтре высыхать). Промывной раствор в EtOH объединяли с фильтратом. Раствор концентрировали досуха в аппарате rotovap с получением зеленого остатка. Этот зеленый остаток растворяли в МеОН (200 мл). Добавляли гидроксид аммония (40 мл) для доведения значения рН раствора до приблизит. 10. Раствор затем нагревали при 45°С в течение 8 часов для циклизации промежуточного амина в лактам 14. Белое твердое вещество осаждалось в процессе циклизации. HPLC анализ показал завершение реакции. Суспензию охлаждали до комнатной температуры. Белое твердое вещество собирали фильтрованием, промывали МеОН (20 мл) и сушили на воздухе с получением 8,9 г продукта (98%-ный выход). 1Н ЯМР (300 МГц, DMSO-d6) δ 2.85 (s, 3H), 3.05 (m, 2Н), 3.39 (m, 2H), 3.66 (s, 3H), 4.486 (s, 2H), 7.39 (m, 4H), 7.76 (d, 2H, J=8,10 Гц), 8.24 (t, 1H, J=5,7 Гц), 11.66 (s, 1H). Точная масса, подсчитанная для C21H21FN3O3: 382,1567. Обнаружено: 382,1552.
Пример 13. Синтез 8-фтор-2-(4-метиламинометил-фенил)-1,3,4,5-тетрагидро-азепино[5,4,3-cd]индол-6-она (15)
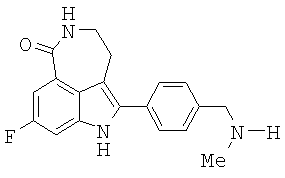
Лактам 14 (14,42 г, 0,038 моль) растворяли в бромистоводородной кислоте в уксусной кислоте (30%-32%, 140 мл). Реакционный раствор перемешивали в течение 46 часов при комнатной температуре в колбе емкостью 500 мл, соединенной с этаноламиновой скрубберной системой. HPLC анализ показал завершение реакции. Добавляли лед (30 г) к реакционному раствору с последующим добавлением водного NaOH (327 мл, 10 M, 3,7 моль), при этом температуру поддерживали между 25°С и 35°С. Когда добавление NaOH было завершено, значение рН составляло 10. Полученное в результате твердое вещество собрали фильтрованием, промывали водой (2×50 мл). Осадок на фильтре затем суспендировали в воде (125 мл) и перемешивали в течение 2 часов. Твердое вещество собирали фильтрованием, промывали водой (2×25 мл) и сушили с получением 10,76 г продукта (88%-ный выход). 1Н ЯМР (300 МГц, DMSO-d6) δ 2.577 (s, 3Н), 3.053 (m, 2H), 3.406 (m, 2H), 4.159 (s, 2H), 7.36 (dd, 1H, J=2,4Гц и J=9,3 Гц), 7.44 (dd, 1H, J=2,4 Гц и J=11,1 Гц), 7.63 (d, 2H, J=8,1 Гц), 7.70 (d, 2H, J=8,1 Гц), 8.265 (t, 1H, J=5,7 Гц), 11.77 (s, 1H). Точная масса, подсчитанная для C19H19FN3O: 324,1512. Обнаружено: 324,1497.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПОЛИМОРФНЫЕ И АМОРФНАЯ ФОРМЫ ФОСФАТНОЙ СОЛИ 8-ФТОР-2-{4-[(МЕТИЛАМИНО)МЕТИЛ]ФЕНИЛ}-1,3,4,5-ТЕТРАГИДРО-6Н-АЗЕПИНО[5.4.3-CD]ИНДОЛ-6-ОНА | 2005 |
|
RU2355691C2 |
| ПРОИЗВОДНЫЕ АЗОТСОДЕРЖАЩИХ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБЫ ЛЕЧЕНИЯ ВОСПАЛИТЕЛЬНЫХ ЗАБОЛЕВАНИЙ И ЗАБОЛЕВАНИЙ ДЫХАТЕЛЬНЫХ ПУТЕЙ | 2001 |
|
RU2265011C2 |
| 6-ОКСО-АЗЕПИНОИНДОЛОВЫЕ СОЕДИНЕНИЯ И ИХ ФИЗИОЛОГИЧЕСКИ ПЕРЕНОСИМЫЕ КИСЛОТНЫЕ СОЛИ ПРИСОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 1992 |
|
RU2078765C1 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ АЗОТИСТЫЕ ПРОИЗВОДНЫЕ ПИРРОЛА, ИХ ПОЛУЧЕНИЕ И ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2008 |
|
RU2473543C2 |
| Производные тетрагидротриазолопиримидина в качестве ингибиторов нейтрофильной эластазы человека | 2012 |
|
RU2622643C2 |
| МОДУЛЯТОРЫ ТРАНСПОРТЕРОВ АТФ-СВЯЗЫВАЮЩЕЙ КАССЕТЫ | 2005 |
|
RU2528046C2 |
| СОЕДИНЕНИЯ ИНДОЛА В КАЧЕСТВЕ ИНГИБИТОРОВ КЛЕТОЧНОГО НЕКРОЗА | 2008 |
|
RU2477282C2 |
| МОДУЛЯТОРЫ ТРАНСПОРТЕРОВ АТФ-СВЯЗЫВАЮЩЕЙ КАССЕТЫ | 2005 |
|
RU2556984C2 |
| МОДУЛЯТОРЫ ТРАНСПОРТЕРОВ АТФ-СВЯЗЫВАЮЩЕЙ КАССЕТЫ | 2005 |
|
RU2382779C2 |
| СПОСОБ МОДУЛЯЦИИ ТРАНСПОРТЕРОВ АТФ-СВЯЗЫВАЮЩЕЙ КАССЕТЫ | 2005 |
|
RU2525115C2 |
Настоящее изобретение относится к новому способ получения соединения формулы I, где R1 представляет собой: Н; циано; возможно незамещенную или замещенную алкильную, алкенильную, алкинильную, циклоалкильную, гетероциклоалкильную, арильную или гетероарильную группу; или -C(O)-R5, где R5 представляет собой Н; возможно незамещенную или замещенную алкильную, алкенильную, алкинильную, циклоалкильную, гетероциклоалкильную, арильную или гетероарильную группу; или OR6 или NR6R7, где каждый из R6 и R7 независимо представляет собой Н или возможно незамещенную или замещенную алкильную, алкенильную, алкинильную, циклоалкильную, гетероциклоалкильную, арильную или гетероарильную группу; R2 представляет собой Н; R3 представляет собой Н или алкил; R4 представляет собой Н, галоген или алкил, включающий: а) сочетание Соногаширы соединения формулы II, где Х представляет собой галоген или CF3SO2-O-, с соединением формулы III с образованием соединения формулы IV; б) восстановление соединения формулы IV с образованием соединения формулы V; в) превращение соединения формулы V в соединение формулы VI; г) обработку соединения формулы VI N,N-диметил-2-нитроэтиленамином с образованием соединения формулы VII; д) восстановление соединения формулы VII с образованием соединения формулы VIII; е) превращение соединения формулы VIII путем восстановления никелем Ренея и последующей циклизацией основанием в соединение формулы I. Также описывается способ получения 8-фтор-2-{4-[(метиламино)метил]фенил}-1,3,4,5-тетрагидро-6Н-азепино[5,4,3-cd1]индол-6-она через ключевую реакцию сочетания Соногаширы и CuI-промотированное образование индола. Технический результат: разработан новый способ получения соединений формулы I, полезных в качестве ингибиторов поли(АДФ-рибоза)полимеразы. 2 н. и 6 з.п. формулы.


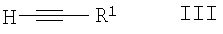

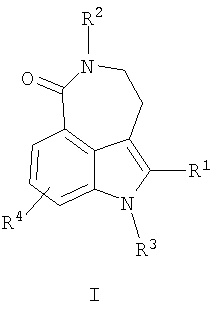
где R1 представляет собой Н; циано; возможно незамещенную или замещенную алкильную, алкенильную, алкинильную, циклоалкильную, гетероциклоалкильную, арильную или гетероарильную группу; или
-C(O)-R5, где R5 представляет собой Н; возможно незамещенную или замещенную алкильную, алкенильную, алкинильную, циклоалкильную, гетероциклоалкильную, арильную или гетероарильную группу; или OR6 или NR6R7, где каждый из R6 и R7 независимо представляет собой Н или возможно незамещенную или замещенную алкильную, алкенильную, алкинильную, циклоалкильную, гетероциклоалкильную, арильную или гетероарильную группу;
R2 представляет собой Н;
R3 представляет собой Н или алкил;
R4 представляет собой Н, галоген или алкил,
включающий
а) сочетание Соногаширы соединения формулы II
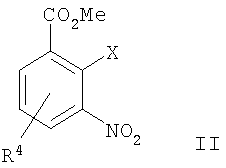 ,
,
где Х представляет собой галоген или CF3SO2-О-,
с соединением формулы III

с образованием соединения формулы IV
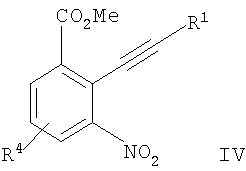 ;
;
б) восстановление соединения формулы IV с образованием соединения формулы V
 ;
;
в) превращение соединения формулы V в соединение формулы VI
 ;
;
г) обработку соединения формулы VI N,N-диметил-2-нитроэтиленамином с образованием соединения формулы VII
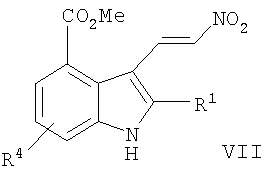 ;
;
д) восстановление соединения формулы VII с образованием соединения формулы VIII
 ;
;
е) превращение соединения формулы VIII путем восстановления никелем Ренея и последующей циклизацией основанием в соединение формулы I.
где Х представляет собой CF3SO2-О- или галоген,
с метиловым эфиром (4-этинил-бензил)-метил-карбаминовой кислоты с образованием метилового эфира 5-фтор-2-{4-[(метоксикарбонил-метил-амино)-метил]-фенилэтинил}-3-нитро-бензойной кислоты.
а) восстановление метилового эфира 5-фтор-2-{4-[(метоксикарбонил-метил-амино)-метил]-фенилэтинил}-3-нитро-бензойной кислоты до метилового эфира 3-амино-5-фтор-2-{4-[(метоксикарбонил-метил-амино)-метил]-фенилэтинил}-бензойной кислоты;
б) превращение метилового эфира 3-амино-5-фтор-2-{4-[(метоксикарбонил-метил-амино)-метил]-фенилэтинил}-бензойной кислоты в метиловый эфир 6-фтор-2-{4-[(метоксикарбонил-метил-амино)-метил]-фенил}-1H-индол-4-карбоновой кислоты;
в) обработку метилового эфира 6-фтор-2-{4-[(метоксикарбонил-метил-амино)-метил]-фенил}-1H-индол-4-карбоновой кислоты N,N-диметил-2-нитроэтиленамином до образования метилового эфира 6-фтор-2-{4-[(метоксикарбонил-метил-амино)-метил]-фенил}-3-((Е)-2-нитро-винил)-1H-индол-4-карбоновой кислоты;
г) восстановление метилового эфира 6-фтор-2-{4-[(метоксикарбонил-метил-амино)-метил]-фенил}-3-((Е)-2-нитро-винил)-1H-индол-4-карбоновой кислоты до метилового эфира 6-фтор-2-{4-[(метоксикарбонил-метил-амино)-метил]-фенил}-3-(2-нитро-этил)-1H-индол-4-карбоновой кислоты;
д) превращение метилового эфира 6-фтор-2-{4-[(метоксикарбонил-метил-амино)-метил]-фенил}-3-(2-нитро-этил)-1Н-индол-4-карбоновой кислоты в метиловый эфир [4-(8-фтор-6-оксо-3,4,5,6-тетрагидро-1Н-азепино[5,4,3-cd]индол-2-ил)-бензил]-метил-карбаминовой кислоты; и
е) удаление защиты с метилового эфира [4-(8-фтор-6-оксо-3,4,5,6-тетрагидро-1H-азепино[5,4,3-cd]индол-2-ил)-бензил]-метил-карбаминовой кислоты с получением 8-фтор-2-{4-[(метиламино)метил]фенил}-1,3,4,5-тетрагидро-6H-азепино[5,4,3-cd]индол-6-она.
| 6-ОКСО-АЗЕПИНОИНДОЛОВЫЕ СОЕДИНЕНИЯ И ИХ ФИЗИОЛОГИЧЕСКИ ПЕРЕНОСИМЫЕ КИСЛОТНЫЕ СОЛИ ПРИСОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 1992 |
|
RU2078765C1 |
| US 6495541 B1, 17.12.2002. | |||

