Согласно 35 USC §119 (е) данная патентная заявка имеет преимущество до приоритета условной патентной заявки 60/502, 562, зарегистрированной 12 сентября 2003 г., на которую, таким образом, здесь дана ссылка.
Область изобретения
В целом, изобретение относится к молекулярной медицине, а более подробно к α-2 адренергическим агонистам, которые высоко селективны по отношению к α-2А адренергическому рецептору по сравнению с α-1А адренергическим рецептором.
Введение
Разнообразие состояний может быть, по крайней мере частично, обусловлено симпатической нервной системой, включая многообразие состояний, связанных со стрессом. Симпатически усиленные состояния включают, без ограничения, сенсорную гиперчувствительность, такую как сенсорная гиперчувствительность, обусловленная фибромиалгией или головной болью, например мигренью; болезни желудочно-кишечного тракта, такие как синдром раздраженной толстой кишки и диспепсия; дерматологические болезни, например псориаз; сердечно-сосудистые нарушения; тахикардии; болезни периферического сужения кровеносных сосудов, включая синдром Рейно и склеродерму; приступ паники; метаболические расстройства, такие как диабет II типа, резистентность к инсулину и ожирение; нарушения сокращения мышц, включая расстройства сокращения скелетных мышц, нарушения сокращения гладких мышц, мышечную спастичность и нарушения сокращения мышц, обусловленные головной болью напряжения; расстройства поведения, например переедание и лекарственная зависимость, но не ограничиваясь ими, и половую дисфункцию.
Несмотря на то, что α-2 адренергические агонисты являются перспективными в лечении симптомов симпатически усиленных состояний, использование этих α-2 адренергических агонистов может быть непригодным вследствие сопутствующих седативных эффектов. По этой причине ограничено эффективное лечение α-2 адренергическим агонистом других состояний, включая неврологические состояния, глазные состояния и хроническую боль. Таким образом, существует необходимость в новых эффективных, не обладающих седативным эффектом α-2 адренергических агонистов для использования в терапии. Настоящее изобретение удовлетворяет этой необходимости, а также имеет ряд взаимосвязанных преимуществ.
Краткое изложение сущности изобретения
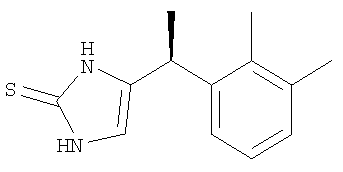
В настоящем изобретении предложен α-2А/α-1А селективный агонист, который представляет собой следующее соединение:

или его фармацевтически приемлемую соль, эфир, амид, стереоизомер или рацемическую смесь. Кроме того, в настоящем изобретении предложена фармацевтическая композиция, которая содержит фармацевтический носитель и терапевтически эффективное количество α-2А/α-1А селективного агониста, который представляет собой следующее соединение:
(структура 1) или его фармацевтически приемлемую соль, эфир, амид, стереоизомер или рацемическую смесь.
Краткое описание чертежей
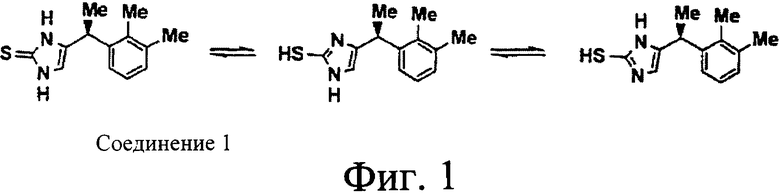
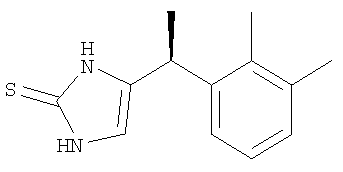
На фиг.1 изображено соединение 1 ((+)-(S)-4-[1-(2,3-диметилфенил)этил]-1,3-дигидроимидазол-2-тион) из (+)-(S)-4-[1-(2,3-диметилфенил)этил]-1Н-имидазола.
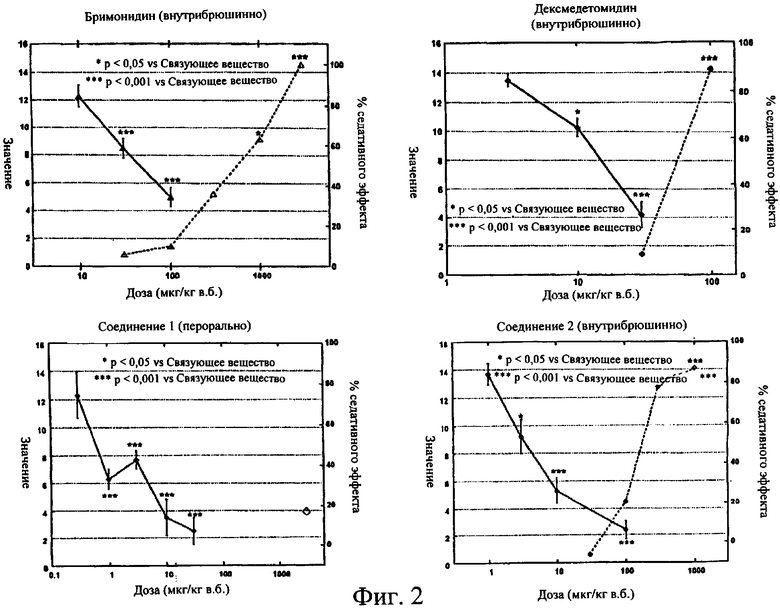
Фиг.2 показывает, что соединение 1 превосходит бримонидин в его способности ослаблять сульпростон-индуцированную тактильную гиперчувствительность в отсутствие седативного эффекта. Антигиперчувствительность к дозе и седативные эффекты четырех α-2 агонистов сравнивали на моделях сульпростон-индуцированной тактильной гиперчувствительности и двигательной активности. Верхняя левая область: внутрибрюшинное введение бримонидина. Верхняя правая область: внутрибрюшинное введение дексмедетомидина. Нижняя левая область: пероральное введение соединения 1. Нижняя правая область: внутрибрюшинное введение соединения 2. Вычислены среднее значение общего показателя чувствительности и среднеквадратичная погрешность этого значения (см. сплошную линию и заштрихованные символы, левая ось). Двигательная активность животных с введенным препаратом выражена в процентах, и процент седативного эффекта рассчитан как 100% минус процент двигательной активности (см. пунктирную линию и незаштрихованные символы, правая ось).
Подробное описание изобретения
Адренергические рецепторы опосредуют физиологические отклики на катехоламины, норадреналин и адреналин, и являются членами надсемейства связывающих G-белок рецепторов, имеющих семь трансмембранных доменов. Эти рецепторы, которые в фармацевтике подразделяют на типы α-1, α-2 и β-адренергических рецепторов, вовлечены в различные физиологические функции, включая функции сердечно-сосудистой и центральной нервной систем. α-Адренергические рецепторы опосредуют возбуждающую и подавляющую функции: α-1 адренергические рецепторы представляют собой типичные возбуждающие постсинаптические рецепторы, которые, в основном, опосредуют отклики в эффекторном нервном отростке, тогда как α-2 адренергические рецепторы расположены как постсинаптически, так и пресинаптически, где они подавляют высвобождение нейротрансмиттеров. В настоящее время агонисты α-2 адренергических рецепторов используют клинически при лечении повышенного кровяного давления, глаукомы, мышечной спастичности и при синдроме нарушенного внимания, при прекращении приема наркотических препаратов, а также дополнительно к общей анестезии и при лечении боли, вызванной раковым заболеванием.
На данный момент α-2 адренергические рецепторы классифицированы на три подтипа на основе их фармакологической и молекулярной характеристики: α-2A/D (α-2А в человеке и α-2D в крысе); α-2 В и α-2С (Bylund et al., Pharmacol. Rev. 46: 121-136 (1994) и Hein and Kobilka, Neuropharmacol. 34: 357-366 (1995)). α-2А и α-2 В подтипы могут управлять артериальным сокращением в некоторых сосудистых ложах, и α-2А и α-2С подтипы передают реакцию торможения высвобождения норадреналина из симпатических нервных окончаний по принципу обратной связи. α-2А подтип также служит медиатором многих из основных эффектов α-2 адренергических агонистов (Calzada and Artinano, Pharmacol. Res. 44: 195-208 (2001); Hein et al., Ann. NY Acad. Science 881: 265-271 (1999); Ruffolo (Ed.), α-Adrenoreceptors: Molecular Biology, Biochemistry and Pharmacology S. Karger Publisher′s Inc. Farmington, CT (1991)).
Как здесь раскрыто, некоторые α-2 агонисты анализировали на α-2А/α-1А функциональную селективность, используя клеточные анализы in vitro. Пример 1 показывает получение α-2 адренергического агониста ((+)-(S)-4-[1-(2,3-диметилфенил)этил]-1,3-дигидроимидазол-2-тион) из (+)-(S)-4-[1-(2,3-диметилфенил)этил]-1Н-имидазола (также см. фиг.1). Как показано в таблице 1, этот α-2 адренергический агонист, обозначенный как соединение 1, проявлял высокую селективность по отношению к α-2А/α-1А, что подтверждается недиагностированным уровнем α-1А активности, наблюдаемой у этого соединения в функциональном клеточном анализе (также см. пример II). Напротив, дексмедетомидин был менее α-2А/α-1А селективен, чем бримонидин (см. таблицу 1). Эти результаты показывают, что соединение 1 является высоко селективным для активации α-2А рецептора по сравнению с α-1А рецептором.
Кроме того, как раскрыто в примере II, α-2А/α-1А функциональная селективность, показанная в in vitro клеточном анализе, связана обратно пропорционально с in vivo седативной активностью при терапевтической дозе. Как показано на фиг.2, α-2 агонист, который был наиболее селективен к α-2А/α-1А функции, in vitro также показал наибольший интервал между терапевтической дозой, облегчающей сульпростон-индуцированную тактильную гиперчувствительность, и дозой, приводящей к значительному седативному эффекту. В частности, соединение 1, вводимое перорально при дозе 1 мкг/кг, вызвало 50% уменьшение в сенсибилизации (сплошная линия, левая ось) с менее, чем 30% седативным эффектом (незаштрихованные ромбы, правая ось) при дозах, стократно или даже тысячекратно превышающих терапевтически эффективную дозу 1 мкг/кг (см. фиг.2, нижняя левая область). Это разделение между терапевтически эффективной дозой и седативной дозой было больше, чем наблюдаемое для любого другого исследуемого α-2 агониста. Эти результаты показывают, что α-2А/α-1А селективность адренергического рецептора α-2 агонистов, определяемая с использованием функциональных клеточных анализов in vitro, обратно пропорционально связана с седативной активностью при терапевтических дозах системного или периферического введения in vivo. Кроме того, эти результаты показывают, что особенно полезные α-2 агонисты, с широким интервалом между терапевтически эффективной дозой и седативной дозой, проявляют α-2А/α-1А функциональную селективность адренергического рецептора.
Основываясь на этих открытиях, настоящее изобретение предлагает α-2А/α-1А селективный агонист, который включает следующее соединение:
(структура 1) или его фармацевтически приемлемую соль, эфир, амид, стереоизомер или рацемическую смесь. Селективный агонист по изобретению может, например, проявлять α-1А эффективность, меньшую, чем бримонидин, или иметь соотношение α-1А/α-2А эффективностей, большее по сравнению с бримонидином. В одном воплощении α-2А/α-1А селективный агонист этого изобретения включает соединение, представленное формулой 1.
"α-2А/α-1А селективный агонист" по изобретению можно отчасти охарактеризовать, как, во-первых, обладающий более чем 25% эффективностью по сравнению с бримонидином для одного или нескольких α-2 адренергических рецепторов, включая α-2А адренергический рецептор, и, во-вторых, обладающий α-1А эффективностью меньшей, чем бримонидин, иными словами, соотношение α-1А/α-2А эффективностей больше по сравнению с бримонидином. В отдельных воплощениях этого изобретения α-2А/α-1А селективный агонист имеет α-1А/α-2А ЕС50 соотношение, по крайней мере, в два раза превышающее α-1А/α-2А ЕС50 соотношение для бримонидина, или α-1А/α-2А ЕС50 соотношение, которое, по крайней мере, в пять, десять, двадцать, тридцать, сорок, пятьдесят, шестьдесят, семьдесят, восемьдесят, девяносто или сто раз больше, чем α-1А/α-2А ЕС50 соотношение для бримонидина. Понятно, что в дополнение к активности α-2А агониста, α-2А/α-1А селективный агонист по изобретению может, по выбору, обладать активностью агониста или антагониста для одного или нескольких дополнительных адренергических или других рецепторов, при условии, что селективный агонист удовлетворяет вышеизложенному критерию в отношении α-2А/α-1А селективности.
Эффективность, также известная как внутренняя активность, является мерой максимальной активации рецептора, достигнутой агентом. С целью определения α-2А/α-1А селективности эффективность предпочтительно определять, используя любой функциональный анализ, который не увеличивает значительно отклик рецептора. Эффективность можно представить как отношение или процент максимального эффекта агента к максимальному эффекту стандартного агониста для каждого подтипа рецептора. Обычно в качестве стандартного агониста для α-2А, α-2В и α-2С рецепторов используют бримонидин (UK 14304), при определении относительной эффективности α-2 рецептора в данном изобретении в качестве стандарта также использовали бримонидин. Фенилэфрин является общепринятым стандартным агонистом для α-1А, α-1В и α-1D рецепторов, и использовался здесь в качестве стандарта при определении относительной эффективности α-1 рецептора.
При функциональном определении α-2А/α-1А селективного агониста по изобретению α-1А эффективность, или соотношение α-1А/α-2А эффективностей, или обе величины сравнивали с таковыми значениями для бримонидина. Термин "бримонидин" используется здесь для обозначения соединения, имеющего следующую формулу:
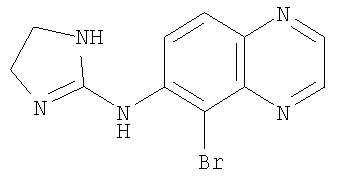
или его фармацевтически приемлемого производного. Бримонидин представляет собой, без ограничения, 5-бром-6-(2-имидазолин-2-иламино)хиноксалин D-тартрат (1:1), Alphagan™ и UK 14304. Бримонидин и его фармацевтически приемлемые производные можно приобрести из промышленных источников или получить рутинными методами, например, как описано в патенте США №6323204.
Любой из множества анализов является пригодным для определения α-2А/α-1А функциональной селективности. Не ограничиваясь примерами, эффективность, активность или ЕС50 у α-2А рецептора можно определить, анализируя подавление активности аденилатциклазы. Кроме того, подавление активности аденилатциклазы можно анализировать, без ограничения, в клетках PC 12, стабильно экспрессирующих α-2А рецептор, например α-2А рецептор человека. Более того, не ограничиваясь примерами, эффективность, активность или ЕС50 для α-1А рецептора можно определить, анализируя внутриклеточный кальций. Внутриклеточный кальций, без ограничения, можно анализировать в клетках НЕК293, стабильно экспрессирующих α-1А рецептор, например бычий α-1А рецептор.
Таким образом, понятно, что α-2А/α-1А функциональную селективность можно охарактеризовать, используя любой рутинный функциональный анализ, например клеточные анализы in vitro, которые измеряют отклик проксимального агента к возбуждению рецептора. Пригодные анализы, без ограничения, включают анализы in vitro, например анализы циклического АМФ (аденозинмонофосфата), или анализы включения ГТФγS (γ-тиогуанозинтрифосфата) для определения проксимальной к активации α-2 рецептора функции (Shimizu et а1., J.Neurochem. 16: 1609-1619 (1969); Jasper et al., Biochem. Pharmacol. 55: 1035-1043 (1998); анализы внутриклеточного кальция, например FLIPR анализы и определение изменений кальция при помощи fluo-3 для определения проксимальной к активации α-1 рецептора функции (Sullivan et al., Methods Mol. Biol. 114: 125-133 (1999); Kao et al., J. Biol. Chem. 264: 8179-8184 (1989)). Далее в примере II раскрыты анализы α-2А селективности, основанные на подавлении форсколин-индуцированной аккумуляции цАМФ в клетках PC12, стабильно экспрессирующих α-2А рецептор, и увеличении во внутриклеточном кальции в клетках НЕК293, стабильно экспрессирующих α-1А рецептор. Дополнительные полезные анализы включают, без ограничения, анализы инозитолфосфата, например анализы совпадения сцинтилляции (Brandish et al., Anal. Biochem. 313: 311-318 (2003); определения для β-аррестин секвестрации GPCR (рецепторов, сопряженных с G-белками), например анализы резонансного переноса энергии биолюминесценции (Bertrand et al., J. Receptor Signal Transduc. Res. 22: 533-541 (2002)) и анализы циточувствительной микрофизиометрии (Neve et al., J. Biol. Chem. 267: 25748-25753 (1992)). Эти и дополнительные анализы для проксимальной функции α-2 и α-1 рецептора являются стандартными и хорошо известны из предшествующего уровня техники.
Для другого не ограничивающего изобретение примера определение ГТФγS представляет собой анализ, полезный для определения α-2А/α-1А функциональной селективности, α-2 адренергические рецепторы передают включение гуанозин 5′-O-(гамма-тио)трифосфата ([35S]ГТФγS) в G-белки в выделенные мембраны посредством катализируемого рецептором обмена [35S]ГТФγS для ГДФ (гуанозиндифосфата). По существу, анализ, основанный на включении [35S]ГТФγS, можно провести, как ранее описано у Jasper и сотр. в 1998. Коротко, сливающиеся клетки, обработанные агентом, который следует протестировать, собирают с чашек культуры клеток тканей в фосфатный буферный физиологический раствор перед центрифугированием при 300 об/мин в течение 5 мнут при 4°С. Осадок клеток после центрифугирования ресуспендируют в холодном лизирующем буфере (5 мМ Трис/HCl, 5 мМ ЭДТК, 5 мМ ЭГТК (этиленгликольтетрауксусная кислота), 0,1 мМ ФМСФ (фенилметилсульфонилфторида, рН 7,5), используя Дезинтегратор Политрон (установка #6, пять секунд) и центрифугируют при 34000×g в течение 15 минут при 4°С перед ресуспендированием в холодном лизирующем буфере и повторным центрифугированием, как описано выше. После второй стадии промывки аликвотные пробы мембранного препарата помещают в мембранный буфер (50 мМ Трис/HCl, 1 мМ ЭДТК, 5 мМ MgCl2, 0,1 мМ ФМСФ, рН 7,4) и замораживают при -70°С до использования в анализе связывания.
Включение ГТФγS анализируют, используя [35S]ГТФγS при удельной активности 1250 кюри/ммоль. Замороженные мембранные аликвотные пробы размораживают и растворяют в инкубационном буфере (50 мМ Трис/HCl, 5 мМ MgCl2, 100 мМ NaCl, 1 мМ ЭДТК, 1 мМ ДТТ (дитиотреитол), 1 мМ пропранолол, 2 мМ ГДФ, рН 7,4) и инкубируют с радиолигандом при конечной концентрации 0,3 нМ при 25°С в течение 60 минут. После инкубирования образцы фильтруют через стеклянные волоконные фильтры (Whatman GF/B, обработанные с 0,5% бычьим сывороточным альбумином) в 96-луночном коллекторе клеток и быстро промывают четыре раза с четырьмя миллилитрами ледяного промывочного буфера (50 мМ Трис/HCl, 5 мМ MgCl2, 100 мМ NaCl, pH 7,5). После сушки в сушильном аппарате фильтры переносят в пробирки сцинтилляции, содержащие пять миллилитров сцинтилляционной смеси Beckman′s Ready Proteins® для подсчета. Затем для α-2А рецептора определяют ЕС50 и максимальный эффект (эффективность).
Понятно, что пригодные анализы, как правило, осуществляют, используя клетки, которые естественным образом экспрессируют значительные уровни только единственного подтипа α-адренергического рецептора, или используя трансфектированные клетки, которые экспрессируют значительные уровни только единственного подтипа рекомбинантного α-адренергического рецептора. В качестве не ограничивающего изобретение примера, адренергический рецептор может быть рецептором человека или его гомологом, обладающим сходной фармакологией. Как здесь раскрыто, α-2А/α-1А селективность предпочтительно определяют рецептор-проксимальными анализами, т.е. такими, при которых реакция рецептора не увеличивается или увеличивается лишь минимально, или такими, при которых обнаруживается быстрый сигнал. С точки зрения вышеизложенного, квалифицированный специалист в данной области будет предпочитать использовать анализы, отличные от анализов Технологии Рецепторной Селекции и Амплификации (RSAT) и подобных анализов, в которых четко не дифференцированы частичный или полный агонизм.
Фармацевтически приемлемую соль, эфир, амид, стереоизомер или рацемическую смесь соединения 1 можно получить традиционными способами. α-2А/α-1А селективный агонист, имеющий структуру 1, является просто характерным из ряда солей, эфиров, амидов и др. этого соединения, которое квалифицированным специалистом в данной области может быть легко получено раскрытым здесь способом при использовании хорошо известных способов химического синтеза, включая способы, подобные тем, что здесь показаны (см. пример I).
Квалифицированный специалист в данной области понимает, что в дополнение к схеме синтеза, показанной в примере I, можно использовать ряд способов для получения, например, системы с имидазольным циклом соединения 1. Такие синтезы хорошо известны из предшествующего уровня техники, как описано, например, в Grimmett, "Imidazole and Benzimidazole Synthesis", Ross Academic Press (1997). Кроме того, также могут быть пригодными при получении селективного агониста соединения 1 альтернативные способы получения имидазол-2-тионов из имидазолов. В качестве не ограничивающего изобретение примера, циклическую систему имидазол-2-тиона можно получить из имидазольного кольца, избирательно защищая атом азота N1 тритильной группой, после чего депротонируя сильным основанием, например н-BuLi или ЛДА (диизопропиламидом лития), с образованием аниона в позиции С2. Затем анион может вступать в реакцию с серой с образованием требуемого имидазол-2-тиона. В качестве дополнительного не ограничивающего изобретение примера, имидазольный цикл может реагировать с фенилхлорформиатом с образованием 2-имидазолона, который можно превратить в тион, используя, например, реагент Лавессона. Эти и подобные способы получения соединения 1 и других α-2А/α-1А селективных агонистов по изобретению хорошо известны из предшествующего уровня техники.
Предлагаемый здесь α-2А/α-1А селективный агонист может быть полезным, например, для предотвращения или облегчения симпатически усиленного состояния без сопутствующего седативного эффекта при периферическом введении. Любое из ряда симпатически усиленных состояний можно предотвратить или облегчить без сопутствующего седативного эффекта под действием α-2А/α-1А селективного агониста этого изобретения, включая, без ограничения, сенсорную гиперчувствительность, такую как обусловленную фибромиальгией или головной болью, например мигренями; болезни желудочно-кишечного тракта, такие как синдром раздраженной толстой кишки и диспепсия; дерматологические болезни, например псориаз; сердечно-сосудистые нарушения; тахикардии; болезни периферического сужения кровеносных сосудов, включая синдром Рейно и склеродерму; приступы паники; метаболические расстройства, такие как диабет типа II, резистентность к инсулину и ожирение; нарушения сокращения мышц, включая расстройства сокращения скелетных мышц, нарушения сокращения гладких мышц, мышечную спастичность и нарушения сокращения мышц, обусловленные головной болью напряжения; расстройства поведения, например, но, не ограничиваясь ими, переедание и лекарственная зависимость, а также половую дисфункцию.
Предлагаемый здесь α-2А/α-1А селективный агонист может также быть полезным, например, для предотвращения или облегчения хронической боли без сопутствующего седативного эффекта при периферическом введении. Хроническая боль представляет собой термин, который означает боль, отличную от острой боли, и включает, без ограничения, невропатическую боль, внутреннюю боль, воспалительную боль, головную боль, мышечную боль и реперкуссионную боль. Понятно, что хроническая боль продолжается относительно долго, например несколько лет, и может быть постоянной или периодической. Хроническая боль отличается от острой боли, которая происходит в данный момент, как правило, высокой пороговой величиной, боль обусловлена обычно повреждениями, такими как порез, удар, ожог или химическое раздражение, например, которое испытываешь при воздействии капсаицина, активного компонента красного стручкового перца.
Любую из многообразия типов хронической боли можно предотвратить или облегчить без сопутствующего седативного эффекта под действием α-2А/α-1А селективного агониста этого изобретения, включая, но, не ограничиваясь ими, невропатическую боль, например невропатическую боль, обусловленную диабетической невропатией или постгерпетической невралгией; хроническую боль, обусловленную раковым заболеванием; послеоперационную боль; аллодинию, например, боль, вызываемую фибромиалгией; хроническую боль, обусловленную комплексным региональным болевым синдромом (КРБС); хроническую внутреннюю боль, например, обусловленную синдромом раздраженной толстой кишки или дисменореей; хроническую головную боль, такую как мигрень, аваскулярная головная боль, "гистаминовая" головная боль или ежедневная головная боль напряжения; хроническую мышечную боль, например, однако не ограничиваясь ей, боль, обусловленную спазмом спины.
Предлагаемый здесь α-2А/α-1А селективный агонист может быть полезным, например, для предотвращения или облегчения неврологического состояния без сопутствующего седативного эффекта при периферическом введении. Таким неврологическим состоянием, без ограничения, может быть острое или хроническое неврологическое состояние. В качестве неогранчивающих изобретение примеров, можно привести острые неврологические состояния, которые можно предотвратить или облегчить без сопутствующего седативного эффекта под действием α-2А/α-1А селективного агониста этого изобретения, включают инсульт; травму головного и спинного мозга и эпилептический припадок. Более того, хронические неврологические состояния, которые можно предотвратить или облегчить без сопутствующего седативного эффекта под действием α-2А/α-1А селективного агониста этого изобретения, включают, но не ограничены приведенными примерами, нейродегенеративные заболевания, например болезнь Альцгеймера; болезнь Паркинсона; болезнь Хантингтона; боковой амиотрофический склероз и рассеянный склероз; деменцию и невропатию, обусловленные ВИЧ; глазные заболевания, например глаукому, диабетическую невропатию и возрастную дегенерацию желтого пятна; шизофрению, наркоманию, синдром отмены и лекарственную зависимость, депрессию и тревогу.
Термин неврологическое состояние включает все острые и хронические расстройства, которые поражают, по крайней мере частично, нейроны. Таким образом, термин неврологическое состояние включает, без ограничения, гипоксию-ишемию (инсульт); повреждение головного и спинного мозга; эпилепсию; нейродегенеративные заболевания, такие как болезнь Альцгеймера, болезнь Паркинсона, паркинсонизм; болезнь Хантингтона; боковой амиотрофический склероз и рассеянный склероз; глазные невропатии, например глаукому, светоиндуцированную дегенерацию сетчатки, такую как дегенерация фоторецептора, и дегенерацию желтого пятна; расстройства дегенерации фоторецептора, например пигментную дегенерацию сетчатки; деменцию, обусловленную ВИЧ, (комплекс слабоумия и синдрома приобретенного иммунодефицита) и невропатию, обусловленную ВИЧ; метаболические, митохондриальные и инфекционные умственные отклонения, например, но не ограничиваясь этим, энцефалит; невропатические болевые синдромы, такие как каузалгия или болезненные периферические невропатии; оливомостомозжечковую атрофию; митохондриальные отклонения и другие биохимические нарушения, например синдром MELAS (митохондриальная энцефаломиопатия, лактатацидоз, инсультоподобные эпизоды), синдром MERRF (миоклонус-эпилепсия с разорванными красными волокнами), болезнь Лебера, болезнь Вернике, синдром Ретта, гомоцистинурия, гипергомоцистеинемия, гиперпролинемия, некетотическая гиперглицинемия, гидроксимасляная аминоацидурия, недостаточность сульфитоксидазы, болезнь комбинированных систем, свинцовая энцефалопатия; печеночная энцефалопатия, синдром Туретта; наркоманию и лекарственную зависимость; синдром отмены лекарственного средства, например прекращение приема алкоголя или успокоительных; синдромы депрессии или беспокойства (см., например, Lipton and Rosenberg, New Enql. J. Med. 330:613 (1994)).
Предлагаемый здесь α-2А/α-1А селективный агонист, кроме того, может быть полезным, например, для предотвращения или облегчения глазного состояния без сопутствующего седативного эффекта при периферическом введении. Глазные состояния, которые можно предотвратить или облегчить без сопутствующего седативного эффекта под действием α-2А/α-1А селективного агониста по изобретению, включают, без ограничения, глаукому; дегенерацию желтого пятна и дегенерации сетчатки, например диабетическую ретинопатию.
Любое из многообразия глазных состояний можно предотвратить или облегчить без сопутствующего седативного эффекта после периферического введения α-2А/α-1А селективного агониста этого изобретения. Такие состояния включают, не ограничиваясь лишь ими, диабетическую ретинопатию; отек макулы, например, обусловленный диабетом; состояния дегенерации сетчатки, например глаукому, дегенерацию желтого пятна, такую как возрастная дегенерация желтого пятна (ARMD) и пигментная дегенерация сетчатки; дистрофии сетчатки; воспалительные расстройства сетчатки; состояния окклюзии сосудов сетчатки, например окклюзии вены сетчатки, или ветви сосуда, или окклюзии центральной артерии сетчатки; ретинопатию раннего развития; ретинопатию, обусловленную болезнями крови, например серповидноклеточной анемией; повышенное внутриглазное давление; глазной зуд; нарушение после отслоения сетчатки; повреждение или кровоизлияние вследствие витрэктомии, хирургии сетчатки и другой хирургии; другие повреждения сетчатки, включающие терапевтическое повреждение, такое как полученное вследствие лазерной обработки сетчатки, например панретинальной фотокоагуляции для диабетической ретинопатии или фототерапии сетчатки. Кроме того, глазные состояния, которые можно предотвратить или облегчить без сопутствующего седативного эффекта при периферическом введении α-2А/α-1А селективного агониста этого изобретения, включают, без ограничения, генетические и приобретенные невропатии зрительного нерва, такие как невропатии зрительного нерва, характеризуемые, главным образом, потерей центрального зрения, например наследственной атрофией зрительного нерва (LHON), аутосомной доминантной атрофией зрительного нерва (болезнь Кьера) и другими невропатиями зрительного нерва, например, включающими митохондриальные дефекты, аберрантные динамин-родственные белки или несвойственный апоптоз; неврит зрительного нерва, например, обусловленный рассеянным склерозом, окклюзией вены сетчатки, или фототерапией или лазерной терапией. См., например, Carelli et al., Neurochem. Intl. 40: 573-584 (2002), и Olichon et al., J. Biol. Chem. 278: 7743-7746 (2003). Понятно, что эти и другие зрительные отклонения, особенно отклонения нейросенсорной сетчатки, можно предотвратить или облегчить без сопутствующего седативного эффекта, используя селективные агонисты по изобретению.
В дополнение к предотвращению или облегчению симпатически усиленных состояний, неврологических состояний, глазных состояний и хронической боли, α-2А/α-1А селективный агонист может быть полезным для предотвращения или облегчения других расстройств без сопутствующего седативного эффекта. Таким расстройством может быть, например, синдром дефицита внимания (СДВ/СДВГ, синдром дефицита внимания с гиперактивностью), который представляет собой расстройство, характеризуемое, главным образом, невнимательностью, патологически повышенной отвлекаемостью и импульсивностью, начинающимися до семилетнего возраста. Симптомы могут включать, без ограничения, проявление нетерпения и беспокойное движение, трудность пребывания в сидячем положении, легкую отвлекаемость, трудность ожидания изменения положения, трудность в удерживании ответов, неспособность следовать инструкциям, чрезмерную болтливость и другое разрушительное поведение (Anderson, ранее, 1994). Более того, СДВГ/СДВ сохраняются в период полового созревания у многих индивидуумов, несмотря на то что первоначально были установлены у детей (см., например. Block, Pediatr. Clin. North Am. 45:1053-1083 (1998), и Pary et al., Ann. Clin. Psychiatry 14:105-111 (2002)). Квалифицированному специалисту в данной области понятно, что способ по изобретению может быть полезным для предотвращения и ослабления СДВГ/СДВ у детей и взрослых, имеющих как легкие, так и тяжелые формы расстройства.
α-2А/α-1А селективный агонист также может быть полезным для предотвращения или ослабления заложенности носа; диареи; расстройств мочеиспускания, таких как сверхактивное мочеиспускание и сверхактивный мочевой пузырь; застойной сердечной недостаточности или психоза, например маниакального расстройства. Более того, α-2А/α-1А селективный агонист может быть полезным для предотвращения или ослабления одного или более симптомов, связанных с анестезией, например тошноты, рвоты, дрожи или тревоги; для улучшения памяти и процессов развития, без сопутствующего седативного эффекта.
Как здесь раскрыто, α-2А/α-1А селективный агонист этого изобретения отчасти характеризуется способностью предотвращать или облегчать любое многообразие симпатически усиленных состояний, неврологических состояний, глазных состояний и типов хронической боли без сопутствующего седативного эффекта. Используемый здесь термин "облегчать" означает ослабление по меньшей мере около 50% по крайней мере одного симптома отдельного состояния или типа хронической боли при лечении.
Как хорошо известно из предшествующего уровня техники, термин седативный эффект означает уменьшение двигательной активности. Используемая здесь относительно селективного агониста фраза "без сопутствующего седативного эффекта" означает, что при периферическом введении селективный агонист вызывает менее чем 30% появления седативного эффекта при дозе в десять раз выше, чем доза селективного агониста, требуемая для 50% ослабления одного или более симптомов отдельного состояния или типов хронической боли при лечении. Например, как показано на фиг.2 (нижняя левая область), соединение 1 вводили перорально при дозе 1 мкг/кг, чтобы вызвать 50% уменьшение в показателе сенсибилизации (сплошная линия, левая ось) с менее чем 30% седативным эффектом (незаштрихованный ромб, правая ось) при дозах в 100 и даже 1000 раз больше, чем терапевтически эффективная доза 1 мкг/кг. Таким образом, α-2А/α-1А селективный агонист, представленный формулой соединения 1, имеет эффективную терапевтическую активность "без сопутствующего седативного эффекта". Наоборот, многие α-2 агонисты, такие как дексмедетомидин, полностью проявляют седативный эффект при дозах в 10 раз больше, чем доза, требуемая для 50% уменьшения показателя сенсибилизации.
В качестве не ограничивающих изобретение примеров, доза α-2А/α-1А селективного агониста, требуемая для создания приблизительно 30% седативного эффекта (уменьшение в двигательной активности), может быть, по меньшей мере, в 25, 50, 100, 250, 500, 1000, 2500, 5000 или 10000 раз больше, чем доза, требуемая для 50% ослабления одного или более симптомов отдельного состояния или типов хронической боли при лечении. Способы для определения как степени ослабления симптома, так и степени седативного эффекта описаны здесь и, кроме того, хорошо известны из предшествующего уровня техники.
Кроме того, согласно настоящему изобретению создана фармацевтическая композиция, которая содержит фармацевтический носитель и терапевтически эффективное количество α-2А/α-1А селективного агониста, которое включает следующее соединение:
(структура 1) или его фармацевтически приемлемую соль, эфир, амид, стереоизомер или рацемическую смесь. В фармацевтической композиции этого изобретения селективный агонист может, например, иметь α-1А эффективность, меньшую по сравнению с бримонидином, или соотношение α-1А/α-2А эффективностей, большее по сравнению с бримонидином. В одном воплощении α-2А/α-1А селективный агонист этого изобретения, входящий в фармацевтическую композицию этого изобретения, содержит соединение, представленное структурой 1.
Таким образом, согласно изобретению предлагается фармацевтическая композиция, содержащая эффективное количество фармацевтического носителя и терапевтически эффективное количество α-2А/α-1А селективного агониста этого изобретения. Такая фармацевтическая композиция может быть полезна для предотвращения или облегчения, например, любого из рассмотренных выше симпатически усиленных, неврологических или глазных состояний или типов хронической боли без сопутствующего седативного эффекта. Фармацевтическая композиция изобретения включает α-2А/α-1А селективный агонист и, кроме того, включает фармацевтически приемлемый носитель, который представляет собой любой носитель, эксципиент или разбавитель, в основном не обладающий длительным периодом или вредным остаточным воздействием при введении пациентуу. Эксципент, как правило, смешивают с активным 2А/α-1А селективным агонистом, или дают ему возможность разбавить или окружить селективный агонист. Носитель может быть твердым, полутвердым или жидким агентом, который действует в качестве эксципиента или связующего вещества для активного селективного агониста. Примеры твердых носителей, пригодных в фармацевтических композициях этого изобретения, включают, без ограничения, фармацевтические сорта маннитола, лактозы, крахмала; стеарат магния, сахарин натрия, полиалкиленгликоли, тальк, целлюлозу, глюкозу, сахарозу и карбонат магния. В состав суппозиториев могут, например, входить пропиленгликоль в качестве носителя. Кроме того, примеры фармацевтически приемлемых носителей включают, без ограничения, воду, например дистиллированную или деионизированную воду; физиологический раствор; водную декстрозу, глицерин, этанол и т.п. Понятно, что активные компоненты в фармацевтической композиции могут находиться в растворенном состоянии или в виде суспензии в требуемом носителе или растворителе.
Фармацевтическая композиция по выбору может включать один или несколько агентов, таких как, без ограничения, эмульгаторы, поверхностно-активные вещества, подсластители или ароматизаторы, тонизирующие вещества, консерванты, буферы или антиоксиданты. Тонизирующие вещества, пригодные в фармацевтической композиции этого изобретения, включают, но не ограничиваясь только ими, соли, например, ацетат натрия, хлорид натрия, хлорид калия, маннитол или глицерин и другие фармацевтически приемлемые тонизирующие вещества. Консерванты, пригодные в фармацевтических композициях этого изобретения, включают, без ограничения, бензалкония хлорид, хлорбутанол, тимеросал, фенилртути ацетат и фенилртути нитрат. Можно использовать различные буферы и способы регулирования рН для получения фармацевтической композиции, включающей, но не ограничивающейся ими, ацетатные буферы, цитратные буферы, фосфатные буферы и боратные буферы. Подобным образом антиоксиданты, пригодные в фармацевтических композициях, хорошо известны из предшествующего уровня техники и включают, например, метабисульфит натрия, тиосульфат натрия, ацетилцистеин, бутилированный гидроксианизол и бутилированный гидрокситолуол. Понятно, что эти и другие вещества, известные из предшествующего уровня техники фармакологии, можно включить в фармацевтическую композицию этого изобретения. См., например, Remington′s Pharmaceutical Sciences Mack Publishing Company, Easton, PA 16th Edition 1980. Кроме того, понятно, что фармацевтическую композицию, содержащую α-2А/α-1А селективный агонист, можно, по выбору, вводить в сочетании с одним или более другими терапевтическими веществами, в той же самой или другой фармацевтической композиции и теми же самыми или другими способами введения.
α-2А/α-1А селективный агонист вводят пациентуу периферически в терапевтически эффективном количестве. Такое терапевтически эффективное количество обычно является минимальной дозой, необходимой для достижения требуемого предотвращения или облегчения одного или более симптомов, например, симпатически усиленного состояния, неврологического состояния, глазного состояния или хронической боли, например, такое количество, которое приблизительно необходимо, чтобы ослабить до приемлемых уровней физическое недомогание, вызванное симпатически усиленным состоянием, неврологическим состоянием, глазным состоянием или хронической болью. Подобной дозой может быть количество, которое ослабляет по крайней мере один симптом состояния или тип боли по меньшей мере на приблизительно 50% и, как правило, составляет 0,1-1000 мг/день и может составлять, например, 0,1-500 мг/день, 0,5-500 мг/день, 0,5-100 мг/день, 0,5-50 мг/день, 0,5-20 мг/день, 0,5-10 мг/день или 0,5-5 мг/день, с фактическим назначенным врачом количеством, которое следует ввести, принимая во внимание существенные условия, включающие тяжесть и тип симпатически усиленного состояния, неврологического состояния, глазного состояния или хронической боли; возраст и вес пациента; общее физическое состояние пациента; фармацевтический состав и способ введения. Как будет обсуждаться далее, фармацевтическая композиция также может быть пригодной в форме суппозиториев или состава пролонгированного высвобождения, например, без ограничения, кожного пластыря, состава для нанесения на или под кожу, или состава для внутримышечной инъекции.
В одном воплощении фармацевтическая композиция этого изобретения представляет собой глазную композицию. Офтальмологическая композиция содержит приемлемый офтальмологический носитель, который является любым носителем, в основном не обладающим длительным периодом или вредным остаточным воздействием на глаза при его введении. Примеры приемлемых офтальмологических носителей включают, без ограничения, воду, например, дистиллированную или деионизированную воду; физиологический раствор и другую водную среду. Глазные композиции могут включать, например, растворимый α-2А/α-1А селективный агонист или α-2А/α-1А селективный агонист в виде суспензии в соответствующем носителе.
Также полезны местные офтальмологические композиции. Подобные композиции представляют собой, без ограничения, глазные капли, глазные мази, глазные гели и глазные кремы. Такие фармацевтические композиции удобно использовать, и они эффективно высвобождают селективный агонист. Ниже в таблице 2 показаны компоненты не ограничивающей изобретение, типичной местной офтальмологической композиции.
Если требуется, то в офтальмологическую композицию этого изобретения можно включить консервант. Таким консервантом может быть, без ограничения, бензалкония хлорид, хлорбутанол, тимеросал, фенилртути ацетат и фенилртути нитрат. Связующие вещества, пригодные для местной офтальмологической композиции, включают, не ограничиваясь этим, поливиниловый спирт, повидон, гидроксипропилметилцеллюлозу, полоксамеры, карбоксиметилцеллюлозу, гидроксиэтилцеллюлозу и очищенную воду.
Также, если требуется, можно включить в офтальмологическую композицию этого изобретения тонизирующее вещество. Таким тонизирующим средством может быть, без ограничения, соль, например хлорид натрия, хлорид калия, маннитол или глицерин, или другое фармацевтически приемлемое или приемлемое офтальмологическое тонизирующее вещество.
Для получения анофтальмической композиции по этому изобретению можно использовать различные буферы и способы регулирования рН при условии, что полученный в результате препарат будет приемлемым для офтальмологии. Такие буферы включают, но не ограничиваясь этим, ацетатные буферы, цитратные буферы, фосфатные буферы и боратные буферы. Понятно, что для достижения необходимого рН композиции можно использовать кислоты и основания. Приемлемые офтальмологические антиоксиданты, пригодные при получении глазной композиции, включают, но не ограничиваются ими, метабисульфит натрия, тиосульфат натрия, ацетилцистеин, бутилированный гидроксианизол и бутилированный гидрокситолуол.
α-2А/α-1А селективный агонист по изобретению или фармацевтическую композицию, содержащую такой селективный агонист, вводят пациентуу периферически. Используемый здесь термин "вводимый периферически" или "периферическое введение" относительно α-2А/α-1А селективного агониста означает введение пациенту α-2А/α-1А селективного агониста в обход центральной нервной системы. Таким образом, периферическое введение осуществляется любым способом, отличным от прямого введения в позвоночник или мозг.
Терапевтически эффективное количество α-2А/α-1А селективного агониста можно ввести пациенту периферически любым способом, в зависимости, например, от типа состояния или хронической боли, которые следует предотвратить или облегчить, от фармацевтического состава, и от анамнеза, факторов риска и симптомов пациента. Подходящие способы периферического введения включают как системное, так и местное введение. В качестве не ограничивающих изобретение примеров, терапевтически эффективное количество α-2А/α-1А селективного агониста можно вводить перорально; парентерально; насосом под кожу; через кожный пластырь; внутривенной, внутрисуставной, подкожной или внутримышечной инъекцией; местными каплями, кремами, гелями или мазями; имплантированным или впрыснутым составом пролонгированного высвобождения; или мининасосом под кожу или другим имплантированным аппаратом.
Квалифицированному специалисту в данной области понятно, что периферическое введение может быть локальным или системным. Локальное введение приводит к значительно большему высвобождению α-2А/α-1А селективного агониста в и около места локального введения, чем в областях, расположенных далеко от места введения. По существу, системное введение приводит к высвобождению α-2А/α-1А селективного агониста, по меньшей мере по всей целой периферийной системе пациента.
Способы периферического введения, пригодные для доставки α-2А/α-1А селективного агониста или фармацевтической композиции этого изобретения, заключаются, без ограничения, в пероральном введении, локальном введении, внутривенной или другой инъекции, и в имплантированных насосах или других аппаратах или составах пролонгированного высвобождения. α-2А/α-1А селективный агонист или фармацевтическую композицию этого изобретения можно вводить периферически, без ограничения, перорально в любой приемлемой форме, например, в виде таблетки, пилюли, капсулы, порошка, раствора, суспензии, эмульсии и т.п.; в виде аэрозоля; в виде суппозитория; внутривенной, внутрибрюшинной, внутримышечной, подкожной или парентеральной инъекцией; трансдермальной диффузией или электрофорезом; локально в любой приемлемой форме, например в виде капель, кремов, гелей или мазей; и мининасосом или другим имплантированным аппаратом или составом пролонгированного высвобождения. α-2А/α-1А селективный агонист по выбору можно формировать в виде лекарственной формы, подходящей для однократного введения, или в лекарственную форму пролонгированного высвобождения для непрерывного контролируемого введения.
Хроническое введение α-2А/α-1А селективного агониста или фармацевтической композиции по изобретению может, например, использоваться для предотвращения или облегчения хронической боли или другого хронического состояния, например, без ограничения, хронического неврологического состояния. Способы для повторного или непрерывного периферического введения включают, без ограничения, повторное пероральное или локальное введение и введение посредством подкожного мининасоса. В качестве не ограничивающих изобретение примеров, α-2А/α-1А селективный агонист или фармацевтическую композицию этого изобретения можно вводить периферически и постоянно непрерывным внутривенным введением посредством имплантированного инфузионного мининасоса или используя состав с пролонгированным высвобождением.
Понятно, что составы медленного высвобождения могут быть полезны для предупреждения или облегчения хронической боли или другого хронического состояния, например, без ограничения, хронического неврологического состояния. Более того, понятно, что частота и продолжительность дозирования такого состава медленного высвобождения должны отчасти зависеть от предотвращения или степени требуемого облегчения и полураспада α-2А/α-1А селективного агониста и что, как упоминалось выше, разнообразные способы введения пригодны для составов медленного высвобождения.
α-2А/α-1А селективный агонист или офтальмологическую композицию по изобретению можно вводить пациенту периферически для предотвращения или облегчения глазного состояния любым способом, зависимым, отчасти, от характеристик селективного агониста, которого следует ввести, и от анамнеза, факторов риска и симптомов пациента. Периферические способы введения, пригодные для предотвращения или облегчения глазного состояния, включают как системное, так и локальное введение. В отдельных воплощениях офтальмологическая композиция, содержащая α-2А/α-1А селективный агонист, вводится локально, например, в виде глазных капель, или локальной инъекцией, или высвобождается из внутриглазного или окологлазного имплантата.
Системные и локальные способы введения, полезные при предотвращении или облегчении глазного состояния при введении α-2А/α-1А селективного агониста или глазной композиции этого изобретения, заключаются, без ограничения, в пероральном зондовом питании; внутривенной инъекции; внутрибрюшинной инъекции; внутримышечной инъекции; подкожной инъекции; трансдермальной диффузии и электрофорезе; локальных глазных каплях и мазях; окологлазной и внутриглазной инъекции, включающей субконъюнктивальную инъекцию; аппаратах пролонгированного высвобождения, например локально имплантированных аппаратах пролонгированного высвобождения, и внутриглазных и окологлазных имплантатах, включающих биоразлагаемый и резервуар-размещенный имплантаты.
В одном воплощении офтальмологическая композиция, содержащая α-2А/α-1А селективный агонист, вводится локально в глаз. α-2А/α-1А селективный агонист можно вводить, например, как часть офтальмологического раствора, такого как глазные капли. В другом воплощении офтальмологическая композиция, содержащая α-2А/α-1А селективный агонист этого изобретения, непосредственно вводится в глаз. В еще другом воплощении офтальмологическая композиция, содержащая α-2А/α-1А селективный агонист этого изобретения, высвобождается из внутриглазного или окологлазного имплантата, например, биоразлагаемого или резервуар-размещенного имплантата.
Как указывалось выше, офтальмологическую композицию, содержащую α-2А/α-1А селективный агонист, можно вводить локально посредством внутриглазного или окологлазного имплантата, который, без ограничения, может быть биоразлагаемым или резервуар-размещенным. Используемый здесь термин "имплантат" относится к любому веществу, которое распространяется незначительно от места введения после имплантации. Имплантат может быть биоразлагаемым, не биоразлагаемым или состоять как из биоразлагаемых, так и не биоразлагаемых материалов; не биоразлагаемый имплантат при необходимости может включать сменный резервуар. Имплантаты, пригодные для предотвращения или облегчения глазного состояния, включают, например, кусочки, частицы, слои, бляшки, микрокапсулы и т.п. и могут иметь любые форму и размер, совместимые с выбранным местом введения, которое, без ограничения, может быть задней камерой глаза, передней камерой глаза, надсосудистой пластинкой и субконъюнктивой глаза. Понятно, что пригодный имплантат обычно высвобождает имплантированную офтальмологическую композицию при терапевтически эффективной дозе в глазу пациента в течение длительного периода времени. Различные глазные имплантаты и составы пролонгированного высвобождения, пригодные для глазного высвобождения, хорошо известны из предшествующего уровня техники, как описано, например, в патентах США №5869079 и 5443505.
Следующие примеры предназначены дли иллюстрации настоящего изобретения, не ограничивая его.
Пример I
Получение соединения 1
Данный пример описывает получение 2А/α-1А селективного агониста, соединение 1.
А. Получение соединения 1. ((+)-(S)-4-[1-(2,3-диметилфенил)этил]-1,3-дигидроимидазол-2-тиона)

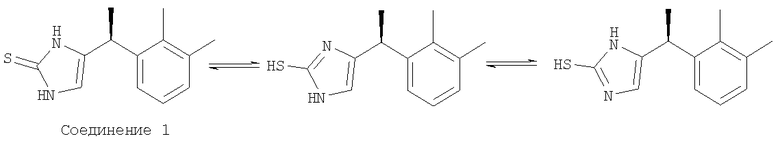
Смесь (+)-(S)-4-[1-(2,3-диметилфенил)этил]-1Н-имидазола (дексмедетомидина; 2,00 г, 10,0 ммоль), полученного, как описано у Cordi и сотр., Synth. Comm. 26: 1585 (1996), в ТГФ (45 мл) и воде (40 мл) обрабатывали NaHCO3 (8,4 г, 100 ммоль) и фенилхлортионоформиатом (3,7 мл, 27,4 ммоль). После перемешивания в течение четырех часов при комнатной температуре смесь разбавляли водой (30 мл) и эфиром (75 мл). Органический слой отделяли и водный слой дважды экстрагировали 50 мл эфира. Органические слои сушили над MgSO4 и фильтровали. Остаток концентрировали под вакуумом, разбавляли МеОН (54 мл), и остаток реагировал с NEt3 (6,5 мл) при комнатной температуре в течение 16 часов. Растворитель удаляли под вакуумом и разбавляли 30% CH2Cl2 : гексан. Снова удаляли растворитель, и образовывался сухой остаток. После еще одного суспендирования в 30% CH2Cl2 : гексан, остаток собирали на фильтре, промывали смесью CH2Cl2 : гексан и сушили под вакуумом с получением соединения 1 ((+)-(S)-4-[1-(2,3-диметилфенил)этил]-1,3-дигидроимидазол-2-тиона) массой 1,23 г (53%). Выше схематически показано получение соединения 1.
Характеристики продукта следующие. Оптическое вращение: [α]D 20=+14° (с 1,25 в МеОН). 1H-ЯМР: (300 МГц, ДМСО): d 11,8 (s, 1Н), 11,6 (s, 1H), 7,03-7,01 (m, 2H), 6,95-6,91 (m, 1H), 6,50 (s, 1H), 4,15 (q, J=6,9 Гц, 1H), 2,25 (s, 3Н), 2,20 (s, 3Н), 1,38 (d, J=6,9 Гц, 3Н).
Б. Способ получения соединения 2 (5-(1Н-имидазол-4-илметил)-циклогекс-1-енилметанола)
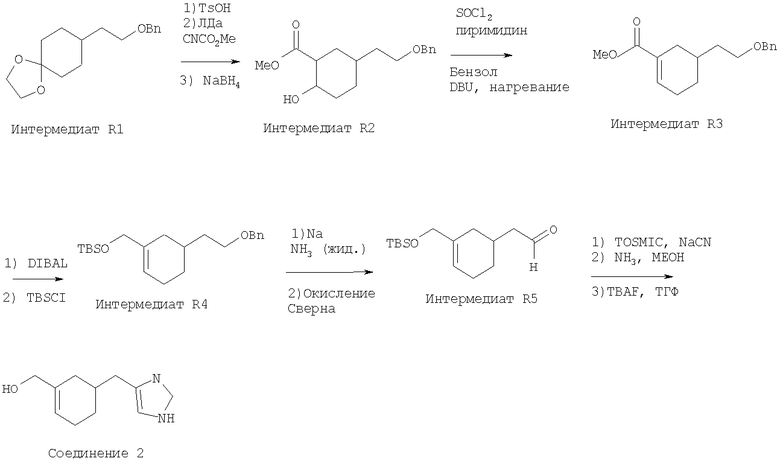
8-(2-Бензилоксиэтил)-1,4-диоксаспиро[4.5]декан (интермедиат R1; 1,02 г, 3,70 ммоль) получали, как описано у Ciufolini и сотр., J. Amer. Chem. Soc. 113:8016 (1991). Это соединение растворяли в смеси ацетона (100 мл) и воды (5 мл), проводили реакцию с TsOH (140 мг, 0,74 ммоль) при 45°С в течение 5 часов. После стандартной водной обработки вещество очищали хроматографически на SiO2 с получением 4-(2-бензилоксиэтил)циклогексанона в виде бесцветного масла (97%).
Раствор ЛДА (33 мл, 1,5 М в Et2O) в ТГФ (50 мл) при -78°С обрабатывали с 4-(2-бензилоксиэтил)циклогексаноном (9,5 г, 40,2 ммоль). Смесь нагревали до 0°С в течение 30 минут перед повторным охлаждением до -78°С и добавлением ГМФА (гексаметилфосфотриамида, 7 мл). Добавляли метилцианоформиат (4,1 мл, 85 ммоль), и смесь перемешивали в течение 15 минут перед водным охлаждением и обработкой. Продукт очищали хроматографически на SiO2 с 10% EtOAc : гексан. Выделяли метиловый эфир 5-(2-бензилоксиэтил)-2-оксоциклогексанкарбоновой кислоты, 5,8 г (49%), который восстанавливали с эквивалентным количеством NaBH4 в МеОН при -10°С. Спирт (вышеуказанный интермедиат R2) очищали хроматографически на SiO2 с 30-50% EtOAc : гексан (выход приблизительно 90%).
Раствор метилового эфира 5-(2-бензилоксиэтил)-2-гидроксициклогексанкарбоновой кислоты (интермедиат R2; 0,72 г, 2,48 ммоль) в пиридине (10 мл) обрабатывали SOCl2 (0,73 мл, 12,4 ммоль) при -20°С. Давали смеси прореагировать в течение 15 минут и затем нагревали до 55°С в течение 16 часов. Растворители удаляли под вакуумом и остаток растворяли в эфире при 0°С. Раствор охлаждали водой и промывали с 1 М HCl, 5% NaOH и солевым раствором. Органический слой сушили над MgSO4, фильтровали и отделяли растворитель. Смесь растворяли в бензоле и удаляли воду азеотропной дистилляцией под вакуумом. Остаток растворяли в бензоле (15 мл) и добавляли DBU (1,8-диазабицикло[5.4.0]ундец-7-ен, 0,76 мл, 5 ммоль). Смесь реагировала в течение 30 минут при комнатной температуре. После обработки хроматографически на SiO2 с 20% EtOAc : гексан выделяли 0,56 г (82%) метилового эфира 5-(2-бензилоксиэтил)циклогекс-1-енкарбоновой кислоты (интермедиат R3).
Интермедиат R3 растворяли в ТГФ (100 мл) и добавляли к раствору DIBAL (гидрида диизобутилалюминия, 70 мл, 1 М в гексане) в ТГФ (160 мл) при -35°С в течение 35 минут. Смесь охлаждали с раствором сегнетовой соли и экстрагировали эфиром. Сухой остаток очищали хроматографически на SiO2 с 30% EtOAc : гексан с получением [5-(2-бензилоксиэтил)циклогекс-1-енил]метанола массой 4,6 г (80%). Раствор спирта (4,0 г, 18,7 ммоль) в ДМФ (60 мл) обрабатывали триэтиламином (3 мл), а затем TBSC1 (трет-бутилдиметилсилилхлоридом, 3,0 г, 22,4 моль) в течение 20 минут при комнатной температуре. Остаток выделяли водной обработкой и очищали хроматографически с получением 3,6 г (63%) [5-(2-бензилоксиэтил)циклогекс-1-енилметокси]-трет-бутилдиметилсилана (интермедиат R4).
Спирт с бензильной защитой (интермедиат R4) (2,0 г, 5,55 ммоль) в ТГФ (20 мл) охлаждали до -70°С и в этой колбе конденсировали NH3 (около 20 мл). Добавляли кусочки натрия и смесь перемешивали при -70°С в течение 15 минут. Смесь нагревали до -30°С в течение 20 минут, охлаждали NH4Cl и выделяли экстракцией. Остаток очищали хроматографически на SiO2 с 25% EtOAc : гексан (99%). Спирт окисляли по стандартной реакции Сверна. Спирт 2-[3-(трет-бутилдиметилсиланилоксиметил)циклогекс-3-енил]этанол (1,3 г, 4,8 ммоль) добавляли к раствору оксалилхлорида (3,55 мл, 7,1 ммоль) в СН2Cl2 (30 мл) с ДМСО (0,63 мл, 8,9 ммоль) при -78°С. Через 40 минут добавляли NEt3 (2,51 мл) и смесь нагревали до комнатной температуры. После стандартной водной обработки и очистки выделяли [3-(трет-бутилдиметилсиланилоксиметил)циклогекс-3-енил]ацетальдегид (интермедиат R5) (около 95%).
Следующее получение проводили по методике Horne и сотр., Heterocycles 39: 139 (1994). Раствор альдегида (интермедиат R5, 0,34 г, 1,3 ммоль) в EtOH (5 мл) обрабатывали тозилметилизоцианидом (TosMIC; Aldrich; 0,25 г; 1,3 ммоль) и NaCN (около 15 мг, катализатор) и перемешивали при комнатной температуре в течение 20 минут. Растворитель удаляли в вакууме; остаток растворяли в ˜7 М NH3 в МеОН и переносили в закрывающуюся пробирку, затем нагревали до 100°С в течение 15 часов. Смесь концентрировали и очищали хроматографически на SiO2 с 5% МеОН (насыщ. масса / NH3) : СН2Cl2. Раствор продукта в ТГФ перемешивали с TBAF (фторидом тетрабутиламмония, 1,5 экв.) при комнатной температуре после водной обработки. Грязный продукт хроматографировали (5-7% NH3/МеОН в CH2Cl2) и обозначали как соединение 2.
Характеристики соединения 2 следующие. 1H-ЯМР: (300 МГц, ДМСО-d6): d 7,52 (s, 1Н), 6,72 (s, 1H), 5,54 (brs, 1H), 3,73 (s, 2H), 2,46 (d, J=6 Гц, 2H), 1,5-2,1 (m, 6H), 1,0-1,55 (m, 1H).
Пример II
Характеристика α-2 агониста с большей α-2А/α-1А функциональной селективностью, чем у бримонидина
Этот пример показывает, что α-2А/α-1А селективность в рецепторных проксимальных функциональных анализах коррелирует с активностью, не обладающей седативным эффектом in vivo.
A. In vitro функциональные анализы
Проксимальную функциональную активность в α-1А и α-2А адренергических рецепторах сравнивали с таковыми для бримонидина, дексмедетомидина, соединения 1 и соединения 2. Бримонидин получали из Sigma; дексмедетомидин получали, как описано ранее у Cordi и сотр. в 1996, и соединения 1 и 2 синтезировали, как описано выше в примере I. Фармакологические профили α-адренергического рецептора анализировали в пробах, используя описанные ниже клеточные линии, стабильно экспрессирующие α-2А и α-1А рецепторы.
Для определения α-1А активности соединения функционально тестировали на способность усиливать увеличение внутриклеточного кальция в НЕК293 клетках, стабильно экспрессирующих бычий α-1А рецептор. α-1А относительную эффективность определяли относительно полного агониста, фенилэфрина, как описано ниже. Как показано в представленной выше таблице 1, α-1А относительные эффективности дексмедетомидина и соединения 2 больше, чем у бримонидина, тогда как α-1А относительная эффективность соединения 1 была так мала, что была недиагностируемой в этой пробе.
Также эти соединения были функционально проверены на проксимальную α-2А функцию, анализируя подавление форсколин-индуцированной аккумуляции цАМФ в клетках PC12, стабильно экспрессирующих α-2А рецептор человека. Внутриклеточные уровни цАМФ определяли, используя систему иммуноферментного анализа цАМФ Биотрак, описанную далее. ЕС50 для α-2А подавления цАМФ выражали как отношение с α-1А ЕС50 для получения соотношения α-1А/α-2А эффективностей. Как показано выше в таблице 1, α-2 адренергический агонист, обозначенный как соединение 1, был высоко α-2А/α-1А селективен, что подтверждается недиагностированным уровнем α-1А активности, наблюдаемой у этого соединения. Наоборот, дексмедетомидин, например, был менее α-2А/α-1А селективен, чем бримонидин. Данные результаты показывают, что соединение 1 является высоко селективным для активации α-2А рецептора по сравнению с α-1А рецептором.
Стабильные клеточные линии, экспрессирующие адренергический рецептор, установили следующим образом. кДНК (комплементарные ДНК) бычьего α-1А рецептора, α-1 В рецептора хомяка, α-2А и α-2С рецепторов человека субклонировали тупым концом в NheI-EcoRI местах в ретровирусном векторе pCL BABE Puro. Конструкции на основе ретровирусного вектора проверяли по установлению последовательности двухцепочечной ДНК. Псевдотипичные ретровирусные частицы с высоким титром генерировали котрансфекцией HEK293GP, линии клеток НЕК293, стабильно экспрессирующих Gag-Pol Maloney leukemia virus, с соответствующим ретровирусным вектором и pMD.G, экспрессирующим вектором для G-белка вируса везикулярного стоматита, VSV-G. Через шестнадцать часов после трансфекции изменяли среду (DMEM (модифицированная по способу Дульбекко среда Игла), 10% FCS (сыворотка плода коровы)); еще через 48 часов получали среду с высоким титром (˜1×106 pfu/мл). Супернатант фильтровали через 0,4 мкм фильтр.
Добавляли различные количества супернатанта α-2А рецептора человека к простым PC12 клеткам, которые затем культивировали в течение 48 часов. Преобразованную клеточную популяцию повторно сеяли при низкой плотности и культивировали в среде, содержащей 100 мкг/мл пуромицина. Непреобразованные клетки убивали через три дня, и отдельный центр рос в течение двух месяцев. Центр собирали, расширяли и анализировали на рецепторную плотность радиолигандным связыванием с бримонидином. Функциональную активность α-2 рецептора усиливали подавлением форсколин-индуцированной аккумуляции цАМФ.
Добавляли различные количества супернатанта бычьего α-1А рецептора к простым PC12 клеткам, которые затем культивировали в течение 48 часов. Преобразованную клеточную популяцию повторно сеяли при низкой плотности и культивировали в среде, содержащей 0,25 мкг/мл пуромицина. Существенный некроз клеток явно происходил в течение трех дней, отдельный центр появился через две недели. Затем центр собирали и расширяли, субклоны функционально анализировали на экспрессию α-1А рецептора, измеряя фенилэфрин-индуцированную аккумуляцию внутриклеточного Ca+2, как описано ниже. Рецепторную плотность измеряли при анализе радиолигандного связывания празозина.
Отклики внутриклеточного Са+2 измеряли в клетках НЕК293, стабильно экспрессирующих бычий α-1А адренергический рецептор. За один день до использования от 40000 до 50000 клеток размещали в одну лунку 96-лунковых планшетов, покрытых поли-D-лизином, в 0,2 мл DMEM, содержащей 10% термоинактивированной сыворотки плода коровы, 1% антибиотического антигрибкового средства и 0,25 г/мл пуромицина. Клетки дважды промывали с HBSS (сбалансированным солевым раствором Хенкса), содержащим 10 мМ HEPES, 2,0 мМ CaCl2 и 2,5 мМ пробенецида, и затем культивировали при 37°С в течение 60 минут с 4 М Fluo-4 (молекулярные зонды; Eugene, Oregone). Внеклеточный краситель дважды смывали с планшетов до размещения планшетов во флуориметре для считывания микропланшетов (FLIPR; Molecular Devices; Sunnyvale, California). Соединения, которые требовалось проанализировать, растворяли в HBSS и помещали на 96-лунковый микропланшет; соединения тестировали в интервале концентраций от 0,64 до 10000 нМ. Данные откликов Са+2 получали в случайных единицах флуоресценции.
Процент α-1А эффективности (% Е) определяли, сравнивая максимальный эффект каждого агониста с максимальным эффектом стандартного полного агониста фенилэфрина. Значения представляли среднюю величину и SEM из 3-15 независимых экспериментов. Разовую селективность агонистов для α-2 рецепторов относительно α-1 рецепторов вычисляли из соотношения их значений ЕС50 для активации α-1А и α-2А рецепторов.
Измерение внутриклеточного цАМФ проводили следующим образом. PC12 клетки, стабильно экспрессирующие α-2А адренергический рецептор человека, размещали на 96-луночном планшете, покрытом поли-D-лизином, при плотности 30000 клеток на лунку в 100 мкл DMEM, содержащей 10% лошадиной сыворотки, 5% термоинактивированной сыворотки плода коровы, 1% антибиотического антигрибкового средства и 100 мкг/мл пуромицина. Клетки культивировали в течение всей ночи при 37°С и 5% CO2. Клетки дозировали, добавляя равные объемы среды, содержащей IBMX (до конечной концентрации 1 мМ), форсколин (до конечной концентрации 10 М) и разбавитель соответствующего лекарственного препарата (до конечной концентрации от 10-5 М до 10-12 М). После 10 минут культивирования среду отсасывали и клетки растворяли в 200 мкл лизирующего буфера (Amersham Biosciences; Piscataway, New Jersey). Планшеты хранили при -20°С до анализа вплоть до 24 часов. Внутриклеточный цАМФ определяли, используя систему иммуноферментного анализа цАМФ Биотрак (Amersham Biosciences), согласно заводским инструкциям. С планшетов снимали показания на аппарате для считывания планшетов при 450 нм.
Кривые зависимости "доза-эффект" для анализов in vitro получали, используя KaleidaGraph (Synergy Software; Reading, PA) при минимальных квадратных отклонениях от уравнения, эффект = максимальный эффект + ((минимальный эффект - максимальный эффект) / (1 + (концентрация лиганда / EC50)). Процент α-1А эффективности определяли, сравнивая максимальный эффект соединения с эффектом стандартного полного агониста фенилэфрина.
Б. In vivo эффективность и седативные эффекты
Дополнительно к вышеописанным клеточным анализам, анализировали различные α-2 агонисты на способность облегчать сульпростон-индуцированную тактильную гиперчувствительность и на седативную активность при разных дозах. Тактильную гиперчувствительность 5-6 мышей на группу оценивали каждые пять минут между 15 и 50 минутами после внутрибрюшинного введения. Животные с введенным растворителем обычно имели показатель около 4. Кроме того, двигательную активность 5-6 мышей на группу измеряли в течение пяти минут через 30 минут после внутрибрюшинного введения. Двигательную активность относительно животных с введенным препаратом выражали в процентах; процент седативного эффекта вычисляли как 100% минус процент двигательной активности.
Как показано на фиг.2 (верхняя левая область), бримонидин проявляет приблизительно 60% седативного эффекта при дозе в 10 раз больше, чем доза 100 мкг/кг, которая дает 50% уменьшение в сенсибилизации сульпростона. Более того, дексмедетомидин, показанный на верхней правой области фиг.2, был полностью седативным при дозе в 10 раз больше, чем доза, необходимая для получения 50% уменьшения в показателе сенсибилизации. Наоборот, соединение 1, вводимое перорально, при дозе 1 мкг/кг вызывало 50% уменьшение в показателе сенсибилизации (сплошная линия, левая ось) с менее чем 30% седативным эффектом (незаштрихованный ромб, правая ось) при дозах в 100 и даже 1000 раз больше, чем доза 1 мкг/кг (см. фиг.2, нижняя левая область). Подобные результаты наблюдали и после внутрибрюшинного введения соединения 1. Внутрибрюшинное введение соединения 2 также вызывало более чем 50% уменьшение в показателе сенсибилизации при 10 мкг/кг (сплошная линия, левая ось) с менее чем 30% седативным эффектом при дозах в 10 раз больше. Таким образом, соединение 1, которое имеет чрезвычайно низкую (недиагностированную) α-1А относительную эффективность, уменьшает тактильную гиперчувствительность без сопутствующего седативного эффекта при периферическом введении. Подобным образом, соединение 2, которое имеет соотношение α-1А/α-2А эффективностей больше, чем бримонидин, также облегчает тактильную гиперчувствительность без сопутствующего седативного эффекта при периферическом введении.
Эксперименты in vivo проводили следующим образом. Сульпростон (Cayman Chemical; Ann Arbor; Michigan) растворяли в диметилсульфоксиде (ДМСО), бримонидин, фенилэфрин и клонидин получали из Sigma (St. Louis, МО) и растворяли в физиологическом растворе. Спинальные инъекции лекарственного препарата проводили следующим образом. Вводили спинально мышам (20-30 мкг), как описано у Hylden и Wilcox, Eur. J. Pharmacol. 67: 313-316 (1980). Вкратце, стерильную 1/2-дюймовую 30-калибр иглу, присоединенную к микрошприцу, вставляли между L5 и L6 позвонками. Мышь крепко держали за тазовый пояс одной рукой, пока шприц держали в другой руке под углом приблизительно в 20° над позвоночником. Иглу вставляли в ткань с одной стороны L6 шиповидного отростка, в бороздку между шиповидным и поперечным отростками. Угол иглы уменьшали до приблизительно 10° и иглу медленно двигали вперед в межпозвоночное пространство до тех пор, пока не почувствовали толчок и явное змеевидное движение хвоста. Соединения медленно вводили в субарахноидальное пространство в объеме 5 мкл. Каждое соединение тестировали при многократных дозах. Для всех последующих экспериментов использовали минимальную эффективную дозу.
Чувствительность к легкому прикосновению измеряли, отмечая реакцию мышей на легкое поглаживание их боков маленькой кисточкой, которая обычно не причиняет боль. Мышей оценивали по следующей шкале один раз каждые пять минут между 15 и 50 минутами после инъекции: показатель "2" присваивали животным, показывающим энергичные реакции к бегству вместе с писком и укусами кисточки; показатель "1" присваивали животным, показывающим легкий писк с попытками к бегству и показатель "0" присваивали, если животное не проявляло реакции на легкое поглаживание кисточкой. Показатели суммировали с получением совокупного балльного показателя от 0 до 16, как описано у Minami и сотр., Pain 57: 217-223 (1994). Статистические расчеты значения для исследований in vivo проводили, используя критерий Стьюдента на основе двойной выборки.
В итоге, эти результаты показывают, что α-2А/α-1А функциональная селективность адренергического рецептора α-2 агонистов в функциональных клеточных анализах in vitro связана с отсутствием седативной активности при терапевтической дозе после системного или периферического введения. Кроме того, эти результаты показывают, что соединение 1, которое проявляет лучшую, чем селективность бримонидина, α-2А/α-1А in vitro функциональную селективность адренергического рецептора, является особенно полезным α-2 агонистом вследствие отсутствия седативных эффектов in vivo при терапевтических дозах.
Все журнальные статьи, ссылки и патентные цитаты, приведенные выше в скобках или иным образом, как прежде изложенные, так и нет, объединены здесь относительно их цельности.
Несмотря на то что изобретение описано относительно вышеприведенных примеров, должно быть понятно, что можно осуществить различные изменения без отклонения от сущности изобретения. Таким образом, изобретение ограничено только пунктами формулы изобретения.
Изобретение относится к α-2А/α-1А селективному агонисту, который
представляет собой соединение формулы
а также к фармацевтической композиции, которая содержит фармацевтический носитель и терапевтически эффективное количество α-2A/α-1А селективного агониста, который включает соединение, представленное структурой 1. Технический результат: получен α-2А/α-1А селективный агонист, который может быть полезным для предотвращения или облегчения неврологического состояния без сопутствующего седативного эффекта при периферическом введении. 2 н. и 4 з.п. ф-лы, 2 табл., 2 ил.
или его фармацевтически приемлемую соль или изомер.
или его фармацевтически приемлемую соль или изомер.
| АВТОНОМНЫЙ ПРИБОР ДЛЯ КАРОТАЖА | 0 |
|
SU284925A1 |
| УСТРОЙСТВО для ИЗМЕРЕНИЯ МАГНИТНОГО КУРСА | 0 |
|
SU302603A1 |
| US 4868197 А, 19.09.1989 | |||
| RU 2000114169 А, 10.07.2002 | |||
| RU 94021649 А, 20.04.1996. | |||

