Соединения согласно изобретению отвечают общей формуле (I)
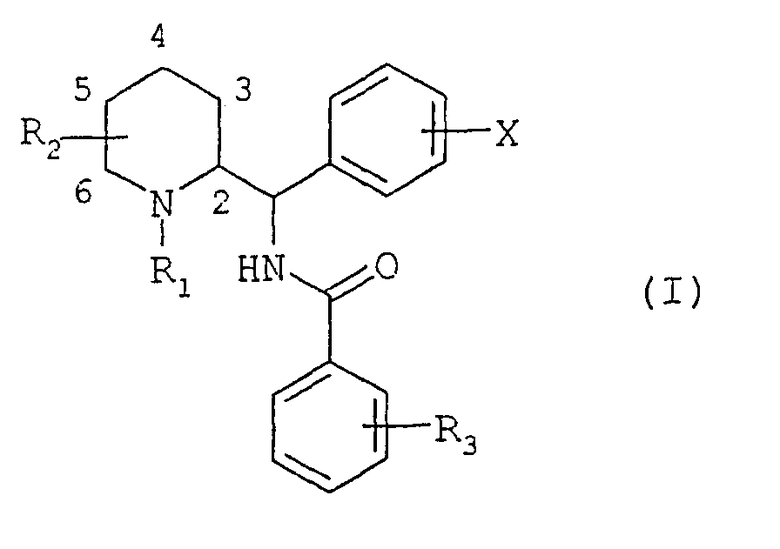
в которой R1 представляет собой либо атом водорода, либо прямой или разветвленный (С1-С7)алкил, возможно замещенный одним или несколькими атомами фтора, либо (С3-С7)циклоалкил, либо (С3-С7)циклоалкил(С1-С3)алкил, либо фенил(С1-С3)алкил, возможно замещенный одной или двумя метоксигруппами, либо (С2-С4)алкенил, либо (С2-С4)алкинил;
R2 представляет собой прямой или разветвленный (С1-С7)алкил или (С3-С7)циклоалкил, либо (С3-С7)циклоалкил(С1-С3)алкил;
Х представляет собой либо атом водорода, либо один или несколько заместителей, выбранных из атомов галогена и следующих групп: трифторметил, прямой или разветвленный (С1-С6)алкил или (С1-С6)алкоксил;
R3 представляет собой либо атом водорода, либо один или несколько заместителей, выбранных из атомов галогена и следующих групп: трифторметил, прямой или разветвленный (С1-С6)алкил, (С3-С7)циклоалкил, (С1-С6)алкоксил, фенил, циано, ацетил, бензоил, (С1-С6)тиоалкил, (С1-С6)алкилсульфонил, карбоксил или (С1-С6)алкоксикарбонил, либо группу общей формулы NR4R5 или SO2NR4R5, или CONR4R5, в которых R4 и R5 обозначают, каждый независимо друг от друга, атом водорода или прямой или разветвленный (С1-С6)алкил или (С3-С7)циклоалкил, или R4 и R5 образуют вместе с атомом азота, с которым они соединены, цикл пирролидина, пиперидина или морфолина.
Соединения формулы (I) имеют два или три асимметрических центра в зависимости от того, находится ли R2 в положении 2 или 3, 4, 5 и 6. В этой связи они могут находиться в форме энантиомеров или диастереоизомеров. Указанные энантиомеры, диастереоизомеры и их смеси, включая рацемические смеси, составляют часть изобретения.
В частности, соединения формулы (I) могут существовать в форме энантиомеров или диастереоизомеров конфигурации трео (1S, 2S) и (1R, 2R) или эритро (1S, 2R) и (1R, 2S) при стехиометрическом положении цис- и транс-заместителей пиперидина или в виде смесей таких изомеров.
Соединения формулы (I) могут находиться в форме оснований или аддитивных солей с кислотами. Такие аддитивные соли составляют часть изобретения.
Эти соли преимущественно получают при взаимодействии с фармацевтически приемлемыми кислотами, однако соли других кислот, пригодные, например, для очистки или выделения соединений формулы (I), также составляют часть изобретения.
Соединения формулы (I) могут существовать также в форме гидратов или сольватов, а именно в форме ассоциаций или комбинаций с одной или несколькими молекулами воды или с растворителем. Такие гидраты и сольваты также составляют часть изобретения.
Соединения согласно изобретению проявляют активность в качестве специфических ингибиторов переносчиков глицина glyt 1 и/или glyt 2.
Соединения формулы (I), в которой R1 отличается от атома водорода, могут быть получены способом, иллюстрируемым схемой 1, приведенной ниже:
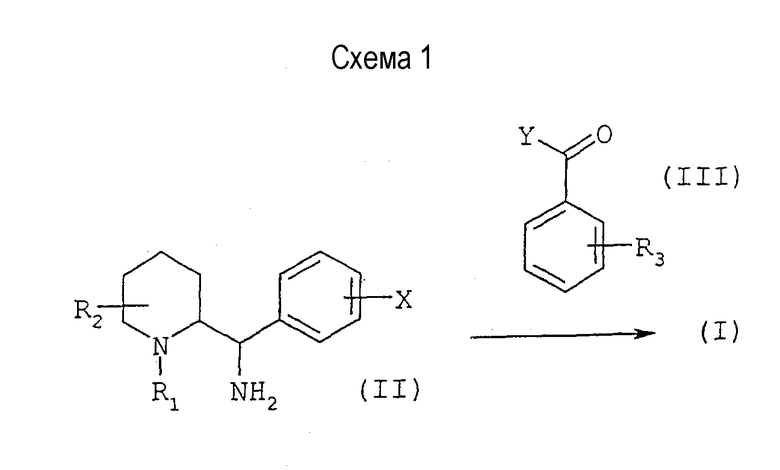
Осуществляют реакцию сочетания между диамином общей формулы (II), в которой R1, R2 и Х имеют значения, указанные выше (при условии, что R1 отличается от атома водорода), и активированной кислотой или хлорангидридом кислоты общей формулы (III), в которой Y обозначает активированную группу ОН или атом хлора, а R3 имеет значения, указанные выше, с использованием методов, известных специалисту.
Диамин общей формулы (II), в которой R2 находится в положении 3, 4, 5 или 6, может быть получен способом, иллюстрируемым схемой 2, представленной ниже.
Согласно первому варианту осуществляют реакцию образования альфа-литийпроизводного из пиперидина общей формулы (IV), в которой R2 имеет определенное выше значение и Вос обозначает группу 1,1-диметилэтоксикарбонил, при помощи втор-бутиллития в присутствии TMEDA (N,N,N',N'-тетраметилэтилендиамин) в эфирном растворителе, таком как диэтиловый эфир, при -78°С с последующим взаимодействием образовавшегося in situ литийамина с производным бензальдегида общей формулы (VI), в которой Х имеет значения, описанные выше. Получают смесь спирта общей формулы (VIII) и циклического карбамата общей формулы (IX).
Эти же соединения могут быть также получены согласно второму варианту следующим образом: альдегид общей формулы (V) получают либо методами, описанными в литературе, либо исходя из пиперидина общей формулы (IV) с последующим образованием литий-производного и реакции конденсации с диметилформальдегидом в условиях, описанных выше. Затем его подвергают взаимодействию с производным общей формулы (VII), в которой Х имеет значения, описанные выше, а М обозначает металл, такой как литий, в эфирном растворителе, таком как диэтиловый эфир, при температуре в интервале от -30°С до комнатной температуры с образованием соединений общих формул (VIII) и (IX). Спирт общей формулы (VIII) конфигурации трео/эритро превращают в спирт общей формулы (XI) конфигурации трео в две стадии следующим образом: окисляют спирт до кетона общей формулы (Х) при помощи окисляющего агента, такого как хлорхромат пиридиния, в хлорированном растворителе, таком как дихлорметан, при комнатной температуре, затем осуществляют диастереоселективное восстановление кетона до спирта трео-конфигурации общей формулы (XI) c помощью восстановителя, такого как K-Selectride® или L-Selectride® (три-втор-бутилборогидрид калия или лития) в эфирном растворителе, таком как тетрагидрофуран, при температуре от -78°С до комнатной температуры.
Карбамат общей формулы (XI) трео-конфигурации может быть далее восстановлен до трео-N-метиламиноспирта общей формулы (XII) действием смешанного гидрида, такого как двойной гидрид алюминия и лития, в эфирном растворителе, таком как тетрагидрофуран, при температуре от комнатной до температуры кипения с обратным холодильником. При таких же условиях восстановления циклический трео-карбамат общей формулы (IX) также приводит к получению трео-N-метиламиноспиртовому производному общей формулы (XII).
Смесь трео/эритро циклического карбамата общей формулы (IX) приводит к смеси производных трео/эритро общей формулы (XII), которую можно очистить и разделить с помощью хроматографии на силикагеле с получением чистого трео-соединения и чистого эритро-соединения.
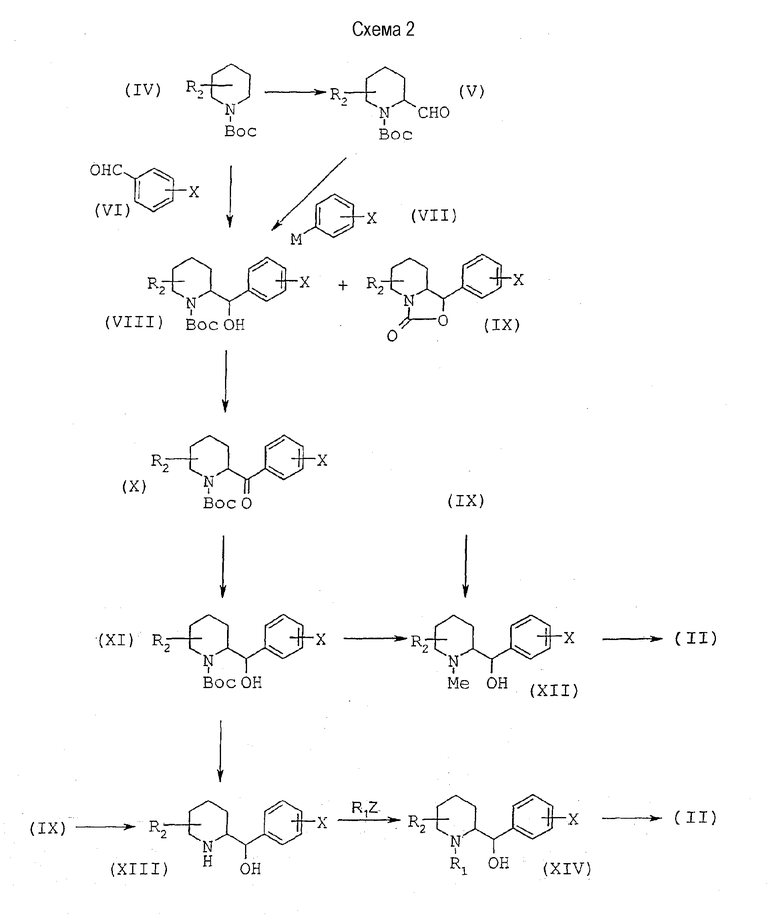
Затем превращают в две стадии трео-спирт общей формулы (XII) в промежуточное трео-соединение общей формулы (II), в которой R1 обозначает группу метил, следующим образом: сначала превращают спиртовую группу в нуклеофобную группу, например в группу метансульфонат, при действии метилсульфонилхлорида в хлорированном растворителе, таком как дихлорметан, в присутствии основания, такого как триэтиламин, при температуре от 0°С до комнатной температуры, затем нуклеофобную группу подвергают взаимодействию с жидким аммиаком при -50°С в спирте, таком как этанол, в закрытой среде, такой как автоклав, при температуре от -50°С до комнатной температуры.
Можно также снять защитную группу с карбамата общей формулы (XI) трео-конфигурации с помощью сильного основания, такого как водный раствор щелочи калия, в спирте, таком как метанол, для получения трео-аминоспирта общей формулы (XIII). В таких же условиях гидролиза циклический карбамат общей формулы (IX) приводит к получению аминоспирта общей формулы (XIII).
Затем осуществляют N-алкилирование с помощью галогенпроизводного формулы R1Z, в которой R1 имеет значения, определенные выше, но при условии, что он не является водородом, и Z является атомом галогена, в присутствии основания, такого как карбонат калия, в полярном растворителе, таком как N,N-диметилформамид, при температуре от комнатной до 100°С с получением алкилпроизводного общей формулы (XIV). Затем превращают алкилпроизводное общей формулы (XIV) в промежуточное соединение общей формулы (II), как описано выше в отношении спирта общей формулы (XII).
Работая таким же образом, как описано выше, но используя производные общей формулы (IX) эритро-конфигурации, можно получить соединения общей формулы (I) эритро-конфигурации.
Диамин общей формулы (II), где R2 находится в положении 2, может быть получен способом, иллюстрированным схемой 3, представленной ниже.
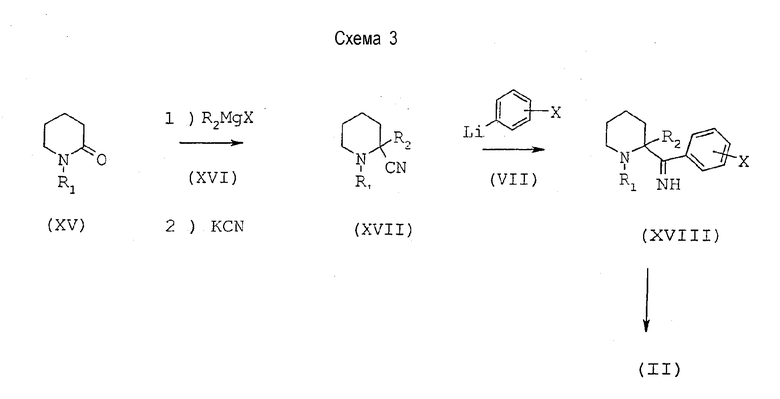
Пиперидинон общей формулы (XV), в которой R1 имеет значения, определенные выше, подвергают взаимодействию с металлорганическим соединением общей формулы (XVI) при нагревании с обратным холодильником в тетрагидрофуране и обрабатывают реакционную среду раствором цианида калия для получения аминонитрила общей формулы (XVII). Это полученное соединение вводят затем во взаимодействие с литийпроизводным общей формулы (VII), в которой Х имеет значения, описанные выше, в эфирном растворителе, таком как диэтиловый эфир или тетрагидрофуран, при температуре от -90 до -30°С; получают промежуточный имин общей формулы (XVIII), который восстанавливают до первичного трео-амина общей формулы (II) с помощью восстановителя, такого как борогидрид натрия, в протонном растворителе, таком как метанол, при температуре от 0°С до комнатной температуры.
Соединения общей формулы (I), в которой R1 обозначает атом водорода, могут быть получены исходя из соединения общей формулы (I), в которой R1 представляет собой
- либо фенилметил, возможно замещенный, путем снятия защитной группы с атома азота пиперидинового цикла, например, с помощью окислительного агента или кислоты Льюиса, такой как трибромид бора, или путем гидрогенолиза,
- либо алкенил, предпочтительно аллил, путем снятия защитной группы с атома азота пиперидинового цикла, например, комплексом палладия Pd°,
и получают соединение общей формулы (I), в которой R1 обозначает атом водорода.
Пиперидинон общей формулы (XV) является коммерчески доступным продуктом.
Кроме того, хиральные соединения общей формулы (I) могут быть получены путем разделения рацемических соединений методом высокоэффективной жидкостной хроматографии (ВЭЖХ) на хиральной колонке или путем расщепления рацемического амина общей формулы (II) с использованием хиральной кислоты, такой как винная кислота, камфорсульфоновая кислота, дибензоилвинная кислота, N-ацетиллейцин, путем фракционной и предпочтительной перекристаллизации диастереоизомерной соли в растворителе спиртового типа.
Пиперидины общей формулы (IV) получают путем защиты, например, с помощью защитной группы Вос атома азота соответствующих пиперидинов, имеющихся в продаже или описанных в литературе, согласно методам, известным специалисту. Способ образования литийпиперидина исходя из пиперидина общей формулы (IV) и его взаимодействие с бензальдегидом общей формулы (VI) аналогичен способу, описанному в J.O.C., 55 (1990), 2578-2580. Фениллитиевое соединение общей формулы (VII), в которой Х обозначает атом водорода, является коммерчески доступным продуктом. Замещенные производные этого соединения могут быть получены аналогично методам, описанным в Tetrahedron Lett., 57, 33 (1996), 5905-5908. Альдегиды общей формулы (V), в которой R2 обозначает метил в положении 2, 4, 5 и 6, описаны в J.O.C. 58 (1993), 1109-117, и J.Chem.Soc., Perkin Trans.1 (2002), 1438-1443. Галогенпроизводные формулы R1Z являются коммерчески доступными продуктами. Некоторые кислоты и хлорангидриды кислот общей формулы (III) являются коммерчески доступными продуктами или, если они являются новыми соединениями, могут быть получены аналогично методам, описанным в патентах ЕР-0556672, US-3801636 и в J.Chem.Soc., (1927), 25, Chem. Pharm. Bull. (1992), 1789-1792, Aust. J.Chem. (1984), 1938-1950 и J.O.C. (1980), 527.
Примеры, представленные ниже, иллюстрируют получение некоторых соединений согласно изобретению. Элементные микроанализы, ИК-спектры и ЯМР-анализ, а также ВЭЖХ на хиральной колонке подтверждают структуру и чистоту энантиомеров полученных соединений.
Номера, указанные в скобках в названиях примеров, соответствуют номерам 1-ой колонки приведенной ниже таблицы.
Пример 1 (соединение № 1)
Гидрохлорид цис-трео-2-хлор-N-[(1,6-диметилпиперидин-2-ил)(фенил)метил]-3-(трифторметил)бензамида, 1:1.
1.1. 1,1-Диметилэтил-цис-[2-гидрокси(фенил)метил-6-метил]пиперидин-1-карбоксилат.
В колбу емкостью 50 мл вводят в атмосфере азота 1 г (4,4 ммоль) 1,1-диметилэтил-цис-2-формил-6-метилпиперидин-1-карбоксилата в 15 мл безводного тетрагидрофурана, охлаждают среду до -78°С, прикапывают 4,4 мл (4,4 ммоль) 1 М раствора фенилмагнийбромида в тетрагидрофуране и оставляют смесь при перемешивании в течение 2 часов для достижения температуры -50°С.
После гидролиза насыщенным водным раствором хлорида аммония отделяют водную фазу и экстрагируют ее этилацетатом. Объединенные органические фазы сушат над сульфатом натрия, фильтруют, концентрируют при пониженном давлении и очищают остаток хроматографией на колонке с силикагелем, элюируя смесью этилацетата с циклогексаном. Получают 1,15 г спирта в виде смеси трео/эритро-диастереоизомеров.
1.2. 1,1-Диметилэтил-цис-2-метил-6-(фенилкарбонил)пиперидин-1-карбоксилат.
В колбу емкостью 100 мл последовательно вводят 0,12 г (1,5 ммоль) ацетата натрия, суспензированного в 20 мл дихлорметана, и 1,2 г (5,5 ммоль) хлорхромата пиридиния, затем добавляют раствор 1,15 г (3,76 ммол) 1,1-диметилэтил-цис-[2-гидрокси(фенил)метил-6-метил]пиперидин-1-карбоксилата в 20 мл дихлорметана. Смесь быстро становится черной и ее оставляют на 4 часа с перемешиванием при комнатной температуре.
Добавляют 30 мл этилового эфира, фильтруют, промывают, концентрируют при пониженном давлении и очищают остаток хроматографией на колонке с силикагелем, элюируя смесью этилацетата с циклогексаном. Получают 0,46 г кетона в виде твердого продукта белого цвета. Т.пл. 92-93°С.
1.3. 1,1-Диметилэтил-цис-трео-[2-гидрокси(фенил)метил-6-метил]пиперидин-1-карбоксилат.
В колбу емкостью 100 мл вводят в атмосфере аргона 0,46 г (1,5 ммоль) 1,1-диметилэтил-цис-2-метил-6-(фенилкарбонил)пиперидин-1-карбоксилата в 40 мл безводного тетрагидрофурана, охлаждают раствор до -78°С, прикапывают 4,6 мл (4,6 ммоль) 1 М раствора L-Selectride® (три-втор - бутилборогидрид лития) в тетрагидрофуране и перемешивают смесь при температуре -78°С в течение 5 часов.
Медленно гидролизуют на холоде с помощью 3,2 мл воды и 3,2 мл 35%-ного водного раствора перекиси водорода и оставляют смесь при перемешивании на 2 часа для возвращения ее к комнатной температуре.
Разбавляют смесь водой и этилацетатом, разделяют фазы и водную фазу экстрагируют этилацетатом. После промывки объединенных органических фаз, сушки над сульфатом натрия, фильтрации и выпаривания остаток очищают хроматографией на колонке с силикагелем, элюируя смесью этилацетата с циклогексаном. Получают 0,38 г трео-цис-изомера в виде бесцветного масла.
1.4. цис-трео-(1,6-Диметилпиперидин-2-ил)фенилметанол.
В двухгорлую колбу емкостью 25 мл вводят в атмосфере азота 0,24 г (6,3 ммоль) двойного гидрида алюминия и лития в 7 мл безводного тетрагидрофурана, нагревают смесь с обратным холодильником, добавляют 0,38 г (1,2 ммоль) раствора 1,1-диметилэтил-цис-трео-[2-гидрокси(фенил)метил-6-метил]пиперидин-1-карбоксилата в 3 мл тетрагидрофурана и выдерживают смесь при нагревании с обратным холодильником в течение 3,5 часов.
Смесь охлаждают, медленно гидролизуют 0,1 М раствором двойного тартрата калия и натрия и оставляют смесь при перемешивании в течение ночи. Фильтруют, промывают осадок тетрагидрофураном, затем концентрируют фильтрат при пониженном давлении и очищают остаток хроматографией на колонке с силикагелем, элюируя смесью дихлорметана с метанолом. Получают 0,11 г маслянистого бесцветного продукта.
1.5. цис-трео-(1,6-Диметилпиперидинил-2-ил)фенилметанамин.
В колбу емкостью 25 мл вводят в атмосфере азота 0,11 г (0,52 ммоль) цис-трео-(1,6-диметилпиперидин-2-ил)фенилметанола и 0,11 мл (0,78 ммоль) триэтиламина в 7 мл безводного дихлорметана, охлаждают среду до 0°С, добавляют 0,06 мл (0,78 ммоль) метансульфонилхлорида, оставляют смесь на 2 часа для постепенного доведения ее до комнатной температуры и концентрируют смесь при пониженном давлении.
В автоклав, снабженный магнитной мешалкой и охлажденный до -50°С, вводят жидкий аммиак, добавляют раствор сырого, полученного выше метансульфоната в 30 мл абсолютного этанола, закрывают автоклав и поддерживают перемешивание в течение 48 часов. Выливают смесь в колбу, концентрируют досуха, разбавляют остаток водой и дихлорметаном, разделяют фазы и экстрагируют водную фазу дихлорметаном. После промывки объединенных органических фаз, сушки над сульфатом натрия, фильтрации и выпаривания выделяют 0,1 г амина в виде маслянистого соединения, которое используют как таковое на следующей стадии.
1.6. Гидрохлорид цис-трео-2-хлор-N-[(1,6-диметилпиперидин-2-ил)(фенил)метил]-3-(трифторметил)бензамида, 1:1.
В колбу емкостью 25 мл последовательно вводят 0,13 г (0,58 ммоль) 2-хлор-3-трифторметанбензойной кислоты, 0,11 г (0,59 ммоль) гидрохлорида 1-[3-(диметиламино)пропил]-3-этилкарбодиимида, 0,03 г (0,24 ммоль) диметиламинопирина, растворенного в 4 мл дихлорметана, добавляют раствор 0,10 г (0,48 ммоль) цис-трео-(1,6-диметилпиперидинил-2-ил)фенилметанамина в 1 мл дихлорметана и оставляют смесь при перемешивании на 5 часов.
Смесь обрабатывают водой, экстрагируют несколько раз дихлорметаном. После промывки органических фаз водой, затем 1 н. водным раствором щелочи натрия, сушки над сульфатом магния, фильтрации и испарения растворителя при пониженном давлении остаток очищают с помощью хроматографии на колонке с силикагелем, элюируя смесью дихлорметана с метанолом. Получают 0,18 г маслянистого продукта, который выделяют в форме гидрохлорида из 0,1 н. раствора хлористоводородной кислоты в пропан-2-оле. В заключение выделяют 0,12 г гидрохлорида в виде твердого вещества белого цвета. Т.пл. 208-209°С.
Пример 2 (соединение № 9)
Гидрохлорид цис-трео-2-хлор-N-[(1,4-диметилпиперидинил-2-ил)(фенил)метил]-3-(трифторметил)бензамида, 1:1.
2.1. цис-7-Метил-1-фенилгексагидро[1,3]оксазоло[3,4-а]пиридин-3-он.
В колбу емкостью 1 л вводят в атмосфере аргона 14,2 г (71,2 ммоль) 1,1-диметилэтил-4-метилпиперидин-1-карбоксилата, растворенного в 130 мл безводного диэтилового эфира, и охлаждают среду до -70°С. Добавляют 14 мл (92,5 ммоль) TMEDA (N,N,N',N'-тетраметилэтилендиамин), затем 70 мл (92,5 ммоль) 1,3 М раствора втор-бутиллития в циклогексане и оставляют на 0,5 часа при перемешивании до возвращения к температуре -30°С.
Затем добавляют раствор 11,33 мл (106,8 ммоль) бензальдегида (предварительно отогнанного) в 40 мл безводного диэтилового эфира и оставляют смесь при перемешивании в течение 12 часов до возвращения ее к комнатной температуре.
После гидролиза водой отделяют водную фазу и экстрагируют этилацетатом. Сушат объединенные органические фазы над сульфатом натрия, фильтруют, концентрируют при пониженном давлении и очищают остаток путем хроматографии на колонке с силикагелем, элюируя смесью дихлорметана с метанолом.
Получают 3,3 г бесцветного масла, состоящего из смеси цис-трео/эритро-изомеров.
2.2. цис-трео-(1,4-Диметилпиперидин-2-ил)фенилметанол.
В двухгорлую колбу емкостью 500 мл вводят в атмосфере азота 2,71 г (71,5 ммоль) двойного гидрида алюминия и лития в 120 мл безводного тетрагидрофурана, нагревают смесь с обратным холодильником, добавляют 3,33 г (14,3 ммоль) раствора цис-7-метил-1-фенилгексагидро[1,3]оксазоло[3,4-а]пиридин-3-она в 40 мл тетрагидрофурана и выдерживают смесь с обратным холодильником в течение 5,5 часов.
Смесь охлаждают, медленно гидролизуют 0,1 М раствором двойного тартрата калия и натрия и оставляют смесь при перемешивании в течение ночи. Фильтруют и осадок промывают тетрагидрофураном, затем фильтрат концентрируют при пониженном давлении и очищают остаток хроматографией на колонке с силикагелем, элюируя смесью дихлорметана с метанолом. Получают 0,95 г бесцветного маслянистого продукта.
2.3. цис-трео-(1,4-Диметилпиперидинил-2-ил)фенилметанамин.
В колбу емкостью 10 мл вводят в атмосфере азота 0,95 г (4,3 ммоль) цис-трео-(1,4-диметилпиперидин-2-ил)фенилметанола и 0,9 мл (6,5 ммоль) триэтиламина в 40 мл безводного дихлорметана, охлаждают среду до 0°С, добавляют 0,5 мл (6,5 ммоль) метансульфонилхлорида, оставляют смесь в течение 2 часов для медленного возвращения ее к комнатной температуре и затем концентрируют при пониженном давлении.
В автоклав, снабженный магнитной мешалкой и охлажденный до -50°С, вводят жидкий аммиак, добавляют раствор полученного выше сырого метансульфоната в 30 мл абсолютного этанола, закрывают автоклав и перемешивают содержимое в течение 48 часов.
Выливают смесь в колбу, концентрируют досуха, разбавляют остаток водой и дихлорметаном, разделяют фазы и экстрагируют водную фазу дихлорметаном. После промывки объединенных органических фаз, сушки над сульфатом натрия, фильтрации и выпаривания выделяют 0,8 г амина в виде маслянистого соединения, которое используют как таковое на следующей стадии.
2.4. Гидрохлорид цис-трео-2-хлор-N-[(1,4-диметилпиперидин-2-ил)(фенил)метил]-3-(трифторметил)бензамида, 1:1.
В колбу емкостью 50 мл последовательно вводят 0,98 г (4,38 ммоль) 2-хлор-3-трифторметанбензойной кислоты, 0,85 г (4,46 ммоль) гидрохлорида 1-[3-(диметиламино)пропил]-3-этилкарбодиимида, 0,22 г (1,83 ммоль) диметиламинопирина, растворенного в 20 мл дихлорметана, добавляют 0,8 мл (3,66 ммоль) цис-трео-(1,4-диметилпиперидин-2-ил)фенилметанамина, растворенного в 4 мл дихлорметана, и перемешивают смесь в течение 12 часов.
Обрабатывают смесь водой и экстрагируют несколько раз дихлорметаном. После промывки органических фаз водой, затем 1 н. водным раствором щелочи натрия, сушки над сульфатом магния, фильтрации и испарения растворителя при пониженном давлении остаток очищают хроматографией на колонке из силикагеля, элюируя смесью дихлорметана с метанолом.
Получают 0,32 г маслянистого продукта, который выделяют в форме гидрохлорида из 0,1 н. раствора хлористоводородной кислоты в пропан-2-оле.
В заключение выделяют 0,28 г гидрохлорида в виде твердого белого вещества. Т.пл. 209-211°С.
Пример 3 (соединение № 5).
Гидрохлорид транс-трео-2-хлор-N-[(1,5-диметилпиперидинил-2-ил)(4-фторфенил)метил]-3-(трифторметил)бензамида, 1:1.
Работают, как описано в примере 2, но заменяя 1,1-диметилэтил-4-метилпиперидин-1-карбоксилат на 1,1-диметилэтил-5-метилпиперидин-1-карбоксилат, а бензальдегид на 4-фторбензальдегид, получают смесь соответствующих спирта и изоксазолидона. Восстановление полученного изоксазолидона двойным гидридом алюминия и лития приводит к получению транс-трео-аминоспиртовому соединению, которое используют в способах, описанных на стадиях 2.3 и 2.4 примера 2, при использовании 2-хлор-3-трифторметанбензойной кислоты. Т.пл. 220-222°С.
Пример 4 (соединение № 10).
Гидрохлорид трео-2-хлор-N-[(1,2-диметилпиперидин-2-ил)(фенил)метил]-3-(трифторметил)бензамида, 1:1.
4.1. 1,2-Диметилпиперидин-2-карбонитрил.
В трехгорлую колбу емкостью 500 мл, снабженную холодильником и магнитной мешалкой, вводят в атмосфере азота 22 мл (31 ммоль) 1,4 М раствора метилмагнийбромида в тетрагидрофуране, затем раствор 5 г (44,2 ммоль) 1-метилпиперидин-2-она в 20 мл тетрагидрофурана и нагревают смесь при перемешивании с обратным холодильником в течение 2 часов.
Смесь оставляют охлаждаться, добавляют 50 мл 2 н. раствора хлористоводородной кислоты и экстрагируют этилацетатом. Водную фазу доводят до рН 6 с помощью бикарбоната натрия и добавляют 2,9 г (2,8 ммоль) цианида калия. Оставляют смесь при перемешивании при температуре 25°С в течение 12 часов.
Добавляют 10%-ный раствор бикарбоната натрия и экстрагируют этилацетатом. После промывки объединенных органических фаз, сушки над сульфатом натрия, фильтрации и выпаривания получают 3 г продукта в виде маслянистого соединения, которое используют как таковое на следующей стадии.
4.2. 1-(1,2-Диметилпиперидин-2-ил)-1-фенилметанамин.
В колбу емкостью 250 мл, снабженную магнитной мешалкой, в атмосфере аргона получают при -78°С раствор фениллития из 6,8 г (43,4 ммоль) бромбензола в 50 мл тетрагидрофурана и 17,4 мл бутиллития (2,5 М в гексане). Вводят при -78°С раствор 3 г (21,71 ммоль) 1,2-диметилпиперидин-2-карбонитрила в 50 мл тетрагидрофурана и оставляют смесь на 1 час при перемешивании (раствор желтого цвета) до возвращения ее к комнатной температуре. Добавляют воду и экстрагируют этилацетатом. Сушат объединенные органические фазы над сульфатом натрия, фильтруют и концентрируют имин при пониженном давлении. Обрабатывают остаток в колбе емкостью 250 мл при помощи 50 мл метанола. Охлаждают смесь до 0°С и медленно прибавляют 4 г (108 ммоль) борогидрида натрия. Продолжают перемешивание в течение 1 часа для возвращения смеси к комнатной температуре. Концентрируют смесь при пониженном давлении и обрабатывают ее водой и этилацетатом. Разделяют фазы и экстрагируют водную фазу этилацетатом. После промывки объединенных органических фаз, сушки над сульфатом натрия, фильтрации и выпаривания получают 3,2 г продукта в виде маслянистого соединения, которое используют как таковое на следующей стадии.
4.3. Гидрохлорид трео-2-хлор-N-[(1,2-диметилпиперидин-2-ил)(фенил)метил]-3-(трифторметил)бензамида, 1:1.
В колбу емкостью 100 мл последовательно вводят в 20 мл дихлорметана 0,41 г (1,8 ммоль) 1-(1,2-диметилпиперидин-2-ил)-1-фенилметанамина, 0,3 мл (2,25 ммоль) триэтиламина, 0,54 г (2,25 ммоль) хлорангидрида 2-хлор-3-(трифторметил)бензойной кислоты и перемешивают смесь при комнатной температуре в течение 1 часа.
Обрабатывают смесь водой и экстрагируют несколько раз дихлорметаном. После промывки органических фаз водой, затем 1 н. водным раствором щелочи натрия, сушки над сульфатом магния, фильтрации и испарения растворителя при пониженном давлении остаток очищают хроматографией на колонке с силикагелем, элюируя смесью дихлорметана с метанолом. Получают 0,36 г маслянистого продукта.
Продукт превращают в гидрохлорид из 0,1 н. раствора хлористоводородной кислоты в пропан-2-оле. В заключение выделяют 0,14 г гидрохлорида в виде твердого вещества белого цвета. Т.пл. 239-241°С.
Таблица, представленная ниже, иллюстрирует химическую структуру и физические свойства нескольких соединений согласно изобретению.
В колонке “Соль” символ HCl обозначает гидрохлорид.
Соединения согласно изобретению были подвергнуты серии фармакологических испытаний и проявили себя интересными как вещества с терапевтической активностью.
Исследование переноса глицина в клетках SK-N-MC, экспрессирующих нативный переносчик glyt1 человека.
Захват [14С]глицина изучался на клетках SK-N-MC (нейроэпителиальные клетки человека), экспрессирующих нативный переносчик glyt1 человека, путем измерения включенной радиоактивности в присутствии или в отсутствие исследуемого соединения. Клетки культивировали в монослое в течение 48 часов в планшетах, предварительно обработанных 0,02%-ным фибронектином. В день эксперимента культуральную среду удаляли и клетки промывали буфером Krebs-HEPES (4-(2-гидроксиэтил)пиперазин-1-этансульфоновая кислота) с рН 7,4. После 10 минут предварительной инкубации при 37°С в присутствии либо буфера (контрольная партия), либо исследуемого соединения при различных концентрациях или 10 мМ глицина (определение неспецифического захвата) вводили 10 мкМ [14С]глицина (специфическая активность =112 mCi/ммоль). Инкубирование продолжали 10 минут при 37°С и реакцию останавливали путем 2 промывок буфером Krebs-HEPES с рН 7,4. Включенную в клетки радиоактивность измеряли после введения 100 мкл сцинтилляционной жидкости и перемешивания в течение 1 часа. Подсчет осуществляли на счетчике Microbeta Tri-luxTM. Эффективность соединения определяли по значению CI50, представляющему собой концентрацию соединения, при которой снижается на 50% специфический захват глицина, выражаемый разностью между уровнем радиоактивности, включенной в контрольную партию и в партию, в которой находился глицин в концентрации 10 мМ.
Наиболее активные соединения согласно изобретению имели в данном тесте значение CI50 порядка от 0,001 до 10 мкМ.
Конкретные результаты нескольких соединений следующие (CI50 в мкМ):
Изучение ex vivo ингибирующей активности соединения на захват [ 14 C]глицина в гомогенате клеток коры головного мозга.
Самцам мышей породы OF1 Iffa Credo, весом 20-25 г в день эксперимента, вводили перорально возрастающие дозы исследуемого соединения (препарат получен растиранием в ступке исследуемой молекулы в 0,5%-ном растворе Tween/MethocelTM в дистиллированной воде) или интраперитонально (растворение исследуемой молекулы в физиологической сыворотке или растирание в ступке в 0,5%-ном растворе Tween/MethocelTM в воде в зависимости от растворимости молекулы). Контрольной группе вводили носитель. Дозы введения в мг/кг, метод введения и продолжительность обработки устанавливались с учетом исследуемой молекулы. После умерщвления животных путем обезглавливания, осуществляемого в заданное время после введения, быстро извлекали у каждого животного кору головного мозга и помещали на лед, взвешивали и хранили при 4°С или замораживали при -80°С (в обоих случаях образцы хранили не более 1 суток). Каждый образец гомогенизировали в буфере Krebs-HEPES с рН 7,4 из расчета 10 мл/г ткани. Каждый гомогенат в количестве 20 мкл инкубировали в течение 10 минут при комнатной температуре в присутствии 10 мМ L-аланина и буфера. Неспецифический захват определяли введением 10 мМ глицина в контрольную группу. Реакцию останавливали путем фильтрации в вакууме и измеряли удержанную радиоактивность путем сцинтилляции твердой пробы и подсчета на счетчике Microbeta Tri-luxTM. Ингибитор захвата [14С]глицина снижает количество радиолиганда, включенного в каждый гомогенат. Активность соединения оценивалась по параметру DE50, представляющему собой дозу, которая ингибирует на 50% захват [14С]глицина по сравнению с контрольной группой.
Наиболее эффективные соединения согласно изобретению имели показатель DE50 в интервале от 0,1 до 5 мг/кг при интраперитональном или пероральном введении.
Изучение переноса глицина в гомогенате клеток спинного мозга мыши.
Захват [14С]глицина переносчиком glyt2 изучался на гомогенате клеток спинного мозга мыши путем измерения включенной радиоактивности в присутствии или в отсутствие исследуемого соединения.
После умерщвления животных (самцы мышей породы OF1 Iffa Credo весом 20-25 г в день эксперимента) быстро извлекали у каждого животного спинной мозг, взвешивали и хранили на льду. Образцы гомогенизировали в буфере Krebs-HEPES (4-(2-гидроксиэтил)пиперазин-1-этансульфоновая кислота) с рН 7,4 из расчета 25 мл/г ткани.
Предварительно инкубировали 50 мкл гомогената в течение 10 минут при 25°С в присутствии буфера Krebs-HEPES с рН 7,4 и исследуемого соединения при различных концентрациях, или 10 мМ глицина для определения неспецифического захвата. Затем добавляли [14C]глицин (специфическая активность=112mCi/ммоль) в течение 10 минут при 25°С до конечной концентрации 10 мкМ. Реакцию останавливали фильтрацией в вакууме и радиоактивность определяли путем сцинтилляции твердой пробы и подсчета на счетчике Microbeta Tri-luxTM. Эффективность соединения определяли по концентрации CI50, при которой снижается на 50% специфический захват глицина, выражаемый разностью между уровнем радиоактивности, включенной в контрольную партию и в партию, в которой находился глицин в концентрации 10 мМ.
Наиболее активные соединения согласно изобретению в данном тесте имели значение CI50 порядка от 0,001 до 10 мкМ.
Конкретные результаты некоторых соединений следующие (CI50 в мкМ):
Исследование ex vivo ингибирующей активности соединения на захват [ 14 С]глицина в гомогенате спинальных клеток мыши.
Самцам мышей породы OF1 Iffa Credo весом 20-25 г в день эксперимента вводили перорально возрастающие дозы исследуемого соединения (препарат получен растиранием в ступке исследуемого соединения в 0,5%-ном растворе Tween/MethocelTM в дистиллированной воде) или интраперитонально (растворяли исследуемое соединение в физиологической сыворотке или растирали в ступке в 0,5%-ном растворе Tween/MethocelTM в дистиллированной воде). Контрольной группе животных вводили носитель. Дозы введения в мг/кг, метод введения, продолжительность обработки, а также время умерщвления устанавливались с учетом исследуемого соединения. После умерщвления животных путем обезглавливания в определенное время после введения у каждого животного быстро извлекали спинной мозг, взвешивали и помешали в склянки для сцинтилляции, хранили на колотом льду или замораживали при -80°С (в обоих случаях образцы хранили не более 1 суток). Каждый образец гомогенизировали в буфере Krebs-HEPES с рН 7,4 из расчета 25 мл/г ткани. Каждый гомогенат в количестве 50 мкл инкубировали в течение 10 минут при комнатной температуре в присутствии буфера. Неспецифический захват определяли введением 10 мМ глицина в контрольную группу. Реакцию останавливали путем фильтрации в вакууме и измеряли радиоактивность путем сцинтилляции твердой пробы и подсчета на счетчике Microbeta Tri-luxTM. Ингибитор захвата [14С]глицина снижает количество радиолиганда, включенного в каждый гомогенат. Активность соединения оценивалась по параметру DE50, представляющему собой дозу, которая ингибирует на 50% захват [14С]глицина по сравнению с контрольной группой.
Наиболее эффективные соединения согласно изобретению имели показатель DE50 в интервале от 1 до 20 мг/кг при интраперитональном или пероральном введении.
Результаты испытаний, проведенных с соединениями согласно изобретению, показывают, что они являются ингибиторами переносчиков глицина glyt1, находящихся в головном мозге, и переносчиков глицина glyt2, находящихся в головном мозге или в спинном мозге.
Следовательно, соединения согласно изобретению могут быть использованы для получения лекарственных средств, в частности лекарственных ингибиторов переносчиков глицина glyt1 и/или glyt2.
В этой связи, в соответствии с другим аспектом изобретения объектом изобретения являются лекарственные средства, которые содержат соединение формулы (I), или аддитивную соль этого соединения с фармацевтически приемлемой кислотой, или гидрат или сольват соединения формулы (I).
Соединения согласно изобретению могут быть использованы, в частности, для лечения поведенческих нарушений, ассоциированных с деменцией, психозов, в частности шизофрении (дефицитная форма и продуктивная форма), и острых или хронических экстрапирамидальных симптомов, индуцируемых нейролептиками, для лечения различных форм тревожных состояний, приступов паники, фобий, обсессивно-компульсивных расстройств, для лечения различных форм депрессии, включая психотическую депрессию, для лечения расстройств, связанных со злоупотреблением алкоголем или с прекращением принятия алкоголя, нарушений полового поведения, нарушений приема пищи и для лечения мигрени.
Кроме того, соединения согласно изобретению могут быть использованы для лечения болезненных мышечных контрактур в ревматологии и в случае острых спинальных патологий, для лечения спастических контрактур мозгового и спинно-мозгового происхождения, для симптоматического лечения острых и подострых болей от легкой до умеренной интенсивности, для лечения интенсивных и/или хронических болей, нейрогенных и непрекращающихся болей, для лечения болезни Паркинсона и симптомов Паркинсона нейродегенеративного происхождения или индуцируемых нейролептиками, для лечения генерализованных первичных и вторичных эпилепсий, очаговых эпилепсий с простой или сложной симптоматикой, смешанных и других эпилептических синдромов в сочетании с другим антиэпилептическим лечением, или в монотерапии, для лечения апное во сне и в качестве нейропротекторов.
В этой связи настоящее изобретение относится также к фармацевтическим композициям, содержащим эффективную дозу по меньшей мере одного соединения согласно изобретению, в форме фармацевтически приемлемого основания или соли или сольвата, при необходимости в смеси с одним или несколькими подходящими эсципиентами. Указанные эксципиенты выбирают в зависимости от фармацевтической формы и желаемого способа введения лекарства.
Таким образом, фармацевтические композиции согласно изобретению могут быть предназначены для перорального подъязычного, подкожного, внутримышечного, внутривенного введения, для топического нанесения, для интратрахеального, назального, трансдермального, ректального, внутриглазного введения.
Единичные вводимые формы могут быть представлены, например, в виде таблеток, желатиновых капсул, гранул, порошков, растворов или суспензий для перорального приема или для инъекций, трансдермальных наклеек (“пластырей“), суппозиториев. Для топического нанесения можно использовать мази, лосьоны и глазные капли. Указанная единичная форма, выпускаемая, например, в виде таблетки, может содержать следующие компоненты:
Указанные единичные формы выпускаются в дозах, позволяющих ежедневно принимать от 0,01 до 20 мг активного начала на 1 кг веса тела пациента в зависимости от галеновой формы.
Возможны особые ситуации, когда требуется более высокая или более низкая дозы; такие дозировки не выходят за рамки настоящего изобретения. В соответствии с имеющейся практикой соответствующая доза для каждого больного устанавливается врачом в зависимости от способа введения, массы тела и индивидуальной чувствительности указанного пациента.
Согласно другому аспекту изобретения настоящее изобретение относится также к способу лечения перечисленных выше патологий, который заключается во введении пациенту эффективного количества соединения согласно изобретению, или одной из его фармацевтически приемлемых солей, или одного из его гидратов или сольватов.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ N-[ГЕТЕРОАРИЛ(ПИПЕРИДИН-2-ИЛ)МЕТИЛ]БЕНЗАМИДА И ИХ ПРИМЕНЕНИЕ В ТЕРАПИИ | 2004 |
|
RU2351596C2 |
| ПРОИЗВОДНЫЕ N-ФЕНИЛ(ПИПЕРИДИН-2-ИЛ)МЕТИЛ-БЕНЗАМИДА И ИХ ПРИМЕНЕНИЕ В ТЕРАПИИ | 2004 |
|
RU2351588C2 |
| ПРОИЗВОДНЫЕ N-[ФЕНИЛ(ПИРРОЛИДИН-2-ИЛ)МЕТИЛ]БЕНЗАМИДА И N-[(АЗЕПАН-2-ИЛ)ФЕНИЛМЕТИЛ]БЕНЗАМИДА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ В ТЕРАПИИ | 2004 |
|
RU2386614C2 |
| ПРОИЗВОДНЫЕ N-ГЕТЕРОЦИКЛИЛМЕТИЛБЕНЗАМИДОВ, ИХ ПОЛУЧЕНИЕ И ИХ ПРИМЕНЕНИЕ В ТЕРАПИИ | 2004 |
|
RU2346945C2 |
| ПРОИЗВОДНЫЕ ТЕТРАГИДРОПИРАНА ПРОТИВ НЕВРОЛОГИЧЕСКИХ ЗАБОЛЕВАНИЙ | 2011 |
|
RU2554344C2 |
| АРОИЛАМИНО- И ГЕТЕРОАРОИЛАМИНО-ЗАМЕЩЕННЫЕ ПИПЕРИДИНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ GLYT-1 | 2010 |
|
RU2517701C2 |
| ПРОИЗВОДНЫЕ ПИПЕРИДИНБЕНЗОЛСУЛЬФАМИДА | 2004 |
|
RU2330842C2 |
| ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ ФЕНИЛМЕТАНОНА | 2007 |
|
RU2437872C2 |
| ПРОИЗВОДНЫЕ ДИАЗАСПИРОПИПЕРИДИНА | 2004 |
|
RU2355690C2 |
| ЗАМЕЩЕННЫЕ БЕНЗАМИДНЫЕ ПРОИЗВОДНЫЕ | 2010 |
|
RU2595902C2 |
Изобретение относится к новым соединениям общей формулы (I)
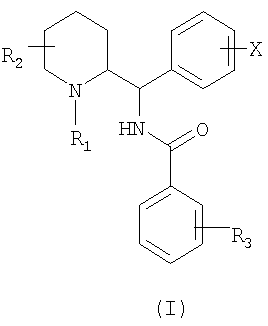
в которой R1 представляет собой либо атом водорода, либо прямой или разветвленный (С1-С7)алкил; R2 представляет собой прямой или разветвленный (С1-С7)алкил, или (С3-С7)циклоалкил, либо (С3-С7)циклоалкил(С1-С3)алкил; X представляет собой атом водорода или галоген; R3 представляет собой либо атом водорода, либо один или несколько заместителей, выбранных из атомов галогена и следующих групп: трифторметил, прямой или разветвленный (С1-С6)алкил, (С1-С6)алкоксил, либо группу общей формулы NR4R5, в которой R4 и R5 обозначают атом водорода; в форме свободного основания или аддитивной соли с кислотой.
Изобретение также относится к лекарственному средству, к фармацевтической композиции, а также к применению.
Технический результат - получение новых биологически активных соединений, обладающих активностью в отношении специфических ингибиторов - переносчиков глицина glyt 1 и/или glyt 2. 5 н. и 5 з.п. ф-лы, 1 табл.
1. Соединение общей формулы (I):
в которой R1 представляет собой либо атом водорода, либо прямой или разветвленный (С1-С7)алкил;
R2 представляет собой прямой или разветвленный (С1-С7)алкил, или (С3-С7)циклоалкил, либо (С3-С7)циклоалкил(С1-С3)алкил;
X представляет собой атом водорода или галоген;
R3 представляет собой либо атом водорода, либо один или несколько заместителей, выбранных из атомов галогена и следующих групп: трифторметил, прямой или разветвленный (С1-С6)алкил, (С1-С6)алкоксил, либо группу общей формулы NR4R5, в которой R4 и R5 обозначают атом водорода; в форме свободного основания или аддитивной соли с кислотой.
2. Соединение по п.1, отличающееся тем, что оно выбрано из следующих соединений:
3-Хлор-N-[(1,2-диметил-пиперидин-2-ил)-фенил-метил]-2-метил-бензамид;
N-[(1,2-Диметил-пиперидин-2-ил)-фенил-метил]-2-метил-3-трифторметил-бензамид;
2,6-Дихлор-N-[(1,2-диметил-пиперидин-2-ил)-фенил-метил]-3-трифторметил-бензамид;
2-Хлор-N-[(1,2-диметил-пиперидин-2-ил)-фенил-метил]-6-фтор-3-метил-бензамид;
2-Хлор-N-[(1,2-диметил-пиперидин-2-ил)-фенил-метил]-3-трифторметил-бензамид;
N-[(1-Аллил-2-метил-пиперидин-2-ил)-фенил-метил]-3-хлор-2-метил-бензамид;
N-[(1,2-Диметил-пиперидин-2-ил)-фенил-метил]-3-метокси-2-метил-бензамид;
2-Метил-N-[(2-метил-пиперидин-2-ил)-фенил-метил]-3-трифторметил-бензамид;
N-[(1-Аллил-2-метил-пиперидин-2-ил)-фенил-метил]-2-хлор-3-трифторметил-бензамид;
2-Метил-N-[(2-метил-пиперидин-2-ил)-фенил-метил]-3-трифторметил-бензамид;
3-Хлор-2-метил-N[(2-метил-пиперидин-2-ил)-фенил-метил]-бензамид;
3-Хлор-2-метил-N-[(2-метил-пиперидин-2-ил)-фенил-метил]-бензамид;
2-Хлор-N-[(1,2-диметил-пиперидин-2-ил)-фенил-метил]-5-трифторметил-бензамид;
2-Хлор-N-[(1,4-диметил-пиперидин-2-ил)-фенил-метил]-3-трифторметил-бензамид;
2-Хлор-N-[(1,5-диметил-пиперидин-2-ил)-(4-фтор-фенил)-метил]-3-трифторметил-бензамид;
N-[(1,5-Диметил-пиперидин-2-ил)-фенил-метил]-2-метил-3-трифторметил-бензамид;
2-Хлор-N-[(2-метил-пиперидин-2-ил)-фенил-метил]-3-трифторметил-бензамид;
2,6-Дихлор-N-[(2-метил-пиперидин-2-ил)-фенил-метил]-3-трифторметил-бензамид;
2-Метил-N-[(2-метил-пиперидин-2-ил)-фенил-метил]-3-трифторметил-бензамид;
N-[(1,5-Диметил-пиперидин-2-ил)-фенил-метил]-2-метил-3-трифторметил-бензамид;
2-Хлор-N-[(1,6-диметил-пиперидин-2-ил)-фенил-метил]-3-трифторметил-бензамид;
2-Хлор-N-[(1,6-диметил-пиперидин-2-ил)-фенил-метил]-3-трифторметил-бензамид;
2-Хлор-N-[(6-циклогексилметил-1-метил-пиперидин-2-ил)-фенил-метил]-3-трифторметил-бензамид;
2-Метил-N-[(2-метил-пиперидин-2-ил)-фенил-метил]-3-трифторметил-бензамид;
3-Хлор-2-метил-N-[(2-метил-пиперидин-2-ил)-фенил-метил]-бензамид;
4-Амино-3,5-дихлор-N-[(1,6-диметил-пиперидин-2-ил)-фенил-метил]-бензамид;
4-Амино-3,5-дихлор-N-[(1,6-диметил-пиперидин-2-ил)-(4-фтор-фенил)-метил]-бензамид.
3. Соединение по п.1, отличающееся тем, что оно имеет конфигурацию трео в форме свободного основания или аддитивной соли с кислотой.
4. Соединение по п.3, отличающееся тем, что оно имеет конфигурацию (1S, 2S) или (1R, 2R) в форме свободного основания или аддитивной соли с кислотой.
5. Соединение по п.1, отличающееся тем, что оно имеет конфигурацию эритро в форме свободного основания или аддитивной соли с кислотой.
6. Соединение по п.5, отличающееся тем, что оно имеет конфигурацию (1S, 2R) или (1R, 2S) в форме свободного основания или аддитивной соли с кислотой.
7. Лекарственное средство, которое обладает активностью в отношении специфических ингибиторов переносчиков глицина glyt 1 и/или glyt 2, отличающееся тем, что оно включает соединение по одному из пп.1-6 или аддитивную соль этого соединения с фармацевтически приемлемой кислотой.
8. Фармацевтическая композиция, которая обладает активностью в отношении специфических ингибиторов переносчиков глицина glyt 1 и/или glyt 2, отличающаяся тем, что она включает соединение по одному из пп.1-6 или фармацевтически приемлемую соль этого соединения, а также по меньшей мере один фармацевтически приемлемый эксципиент.
9. Применение соединения формулы (I) по одному из пп.1-6 для получения лекарственного средства, предназначенного для лечения поведенческих нарушений, ассоциированных с деменцией, психозов, различных форм тревожных состояний, приступов паники, фобий, обсессивно-компульсивных расстройств, различных форм депрессии, расстройств, связанных со злоупотреблением алкоголя или с прекращением принятия алкоголя, нарушений полового поведения, нарушений приема пищи и мигрени.
10. Применение соединения формулы (I) по одному из пп.1-6 для получения лекарственного средства, предназначенного для лечения контрактур, боли, болезни Паркинсона и симптомов Паркинсона, эпилепсий, смешанных и других эпилептических синдромов в сочетании с другим антиэпилептическим лечением, или в монотерапии, для лечения апное во сне и в качестве нейропротекторов.
| Прибор, замыкающий сигнальную цепь при повышении температуры | 1918 |
|
SU99A1 |
| RU 97117370 А, 10.07.1999. | |||

