Родственные заявки
Данная заявка относится к предварительным заявкам США №№62/825741, поданной 28 марта 2019 г.; 62/849095, поданной 16 мая 2019 г.; 62/864306, поданной 20 июня 2019 г.; 62/938277 поданной 20 ноября 2019 г.; 62/955967, поданной 31 декабря 2019 г.; и 62/959799, поданной 10 января 2020 г.; каждая из которых полностью включена в настоящее описание посредством ссылки.
УРОВЕНЬ ТЕХНИКИ
ОБЛАСТЬ ТЕХНИКИ
Изобретение относится к модуляторам Mas-связанного G-белок-сопряженного рецептора Х4, к содержащим их продуктам, а также к способам их применения и получения.
Описание уровня техники
Mas-связанные рецепторы G-белка (MRGPR) представляют собой группу орфанных рецепторов с ограниченной экспрессией в очень специализированных тканях. О функциях большинства этих рецепторов известно очень мало. Существует восемь родственных рецепторов этого класса, экспрессируемых у людей, только четыре из которых имеют легко идентифицируемые ортологи у других видов (а именно, MRGPR D, Е, F и G). Остальные четыре рецептора (MRGPR X1, Х2, Х3 и Х4), исходя из гомологии, не имеют аналогов у других видов, кроме человека.
КРАТКОЕ ОПИСАНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Данное изобретение основано частично на определении того, что MRGPR А1 у мышей функционально соответствует, по крайней мере частично, MRGPR Х4 человека. Эти рецепторы опосредуют расстройства, включая хроническую чесотку (например, зуд), воспалительные расстройства, аутоиммунитет, кожные расстройства, сердечно-сосудистые заболевания, воспаление легких/ХОБЛ и неблагоприятные кожные реакции на лекарства. Более конкретно, как MRGPR А1, так и MRGPR Х4 экспрессируются в сенсорных нейронах, меланоцитах кожи, дендритных клетках, полиморфно-ядерных клетках, макрофагах, бронхиальных эпителиальных клетках, гладких мышцах легких и ганглиях дорсальных корешков. В настоящее время установлено, что как MRGPR А1, так и MRGPR Х4 являются рецепторами (или чувствительны к активации под действием) циркулирующего билирубина и его метаболитов и, таким образом, являются важным фактором ощущения зуда в условиях повышенного билирубина, таких как холестатический зуд. Кроме того, MRGPR Х4 дополнительно активируется множеством компонентов желчи, включая желчные кислоты и их метаболиты, а также метаболитами гема, включая билирубин и уробилин. Желчные кислоты и билирубин сильно повышены при холестатическом зуде, в то время как уробилин является мощным медиатором индукции зуда в мышиной модели и, таким образом, может иметь важное значение для ощущения зуда в условиях повышенного уробилина, таких как уремический зуд. Таким образом, модуляция MRGPR Х4 позволяет лечить аутоиммунные заболевания, такие как псориаз, рассеянный склероз, синдром Стивенса-Джонсона и другие состояния хронической чесотки, как более подробно описано ниже.
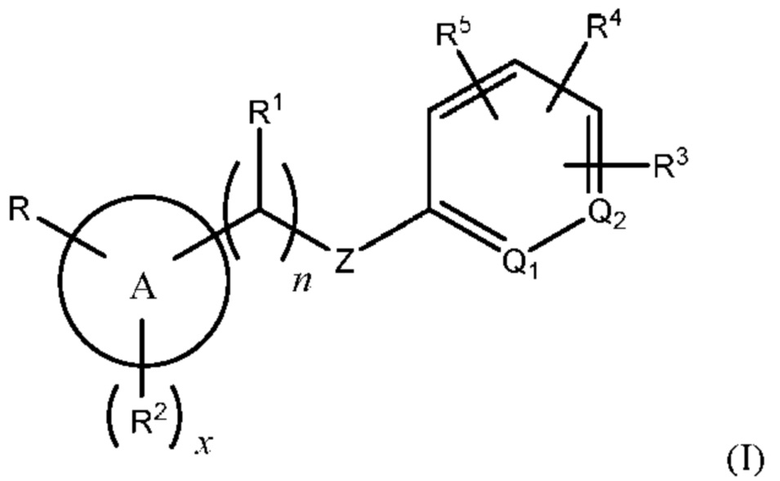
Соответственно, в варианте осуществления изобретения предусматриваются способы модуляции MRGPR Х4 посредством контакта MRGPR Х4 с эффективным количеством соединения, имеющего структуру формулы (I):
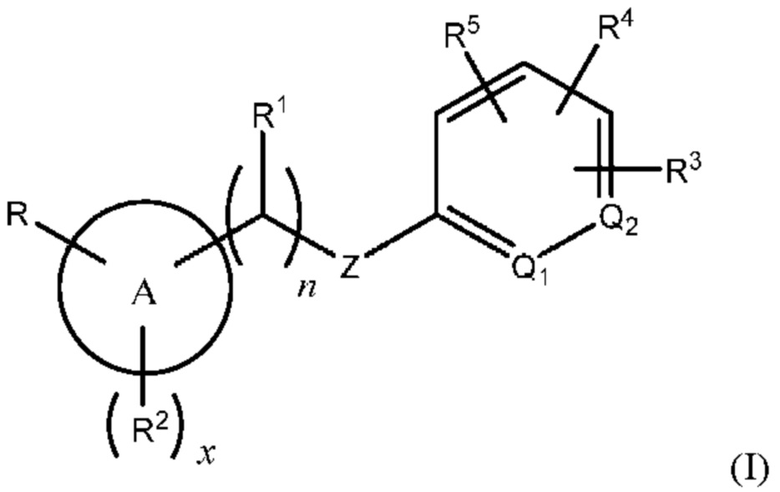
или его фармацевтически приемлемого изомера, рацемата, гидрата, сольвата, изотопа или соли, где и, х, A, Q1, Q2, Z, R, R1, R2, R3, R4 и R5 имеют значения, определенные ниже.
В другом варианте осуществления изобретения предложены способы лечения зависимого от MRGPR Х4 состояния путем введения субъекту, который нуждается в этом, эффективного количества соединения, имеющего структуру формулы (I), или его фармацевтически приемлемого изомера, рацемата, гидрата, сольвата, изотопа или соли.
В более конкретных вариантах осуществления изобретения зависимое от MRGPR Х4 состояние представляет собой одно или несколько из состояния, связанного с зудом, состояния, связанного с болью, состояния, связанного с воспалением, или аутоиммунного расстройства.
В другом варианте осуществления изобретения предусматриваются фармацевтические композиции, содержащие соединение, имеющее структуру формулы (I), или его фармацевтически приемлемый изомер, рацемат, гидрат, сольват, изотоп или соль в сочетании с фармацевтически приемлемым эксципиентом.
В другом варианте осуществления изобретения предложены соединения, имеющие одну или несколько из структур, раскрытых в данном документе, или их фармацевтически приемлемые изомеры, рацематы, гидраты, сольваты, изотопы или соли.
В дополнительных вариантах осуществления изобретения также предусматриваются пролекарства и/или метаболиты соединения, имеющего структуру формулы (I). В случае пролекарств, субъекту может быть введено соединение (т.е. пролекарство), которое затем превращают in vivo в соединение, имеющее структуру формулы (I). В случае метаболитов, после введения субъекту соединения, имеющего структуру формулы (I), такое соединение может быть преобразовано in vivo в активный метаболит.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
Фиг. 1 изображает in vitro активацию MRGPR Х4 метаболитами гема билирубином, биливердином, уробилином, уробилиногеном и стеркобилином.
Фиг. 2А-2В показывают индукцию чесотки у мышей дикого типа уробилином по сравнению с носителем (VEH) (Фиг. 2А) и уробилином, билирубином и дезоксихолевой кислотой (Фиг. 2В).
На Фиг. 3А-3В представлены стабильность билирубина (Фиг. 3А) и агонизм билирубина к MRGPR Х4 (Фиг. 3В) после 24 часов выдерживания при различных условиях температуры и освещения (нулевой момент времени (свежеприготовленный), в темноте при комнатной температуре, в темноте при -20°С, при комнатной температуре с лабораторным освещением и при комнатной температуре и освещении синим светом 400 нм).
На Фиг. 4А-4В представлены стабильность уробилина (Фиг. 4А) и агонизм уробилина к MRGPR Х4 (Фиг. 4В) после 24 часов выдерживания при различных условиях температуры и освещения (нулевой момент времени (свежеприготовленный), в темноте при комнатной температуре, в темноте при -20°С, при комнатной температуре с лабораторным освещением и при комнатной температуре и освещении синим светом).
ПОДРОБНОЕ ОПИСАНИЕ
Как упоминалось выше, данное изобретение относится к модуляторам MRGPR Х4, к содержащим их продуктам, а также к способам их применения и приготовления. Данное изобретение частично основано на определении того, что MRGPR А1 у мышей функционально соответствует MRGPR Х4 человека. Эти рецепторы опосредуют расстройства, включая хроническую и периодическую чесотку (например, зуд), воспалительные расстройства, аутоиммунитет, кожные расстройства и неблагоприятные кожные реакции на лекарства и инфекционные заболевания. Более конкретно, как MRGPR А1, так и MRGPR Х4 экспрессируются в сенсорных нейронах и ганглиях дорсальных корешков. В настоящее время установлено, что MRGPR А1 и MRGPR Х4 являются рецепторами (или чувствительны к активации под действием) циркулирующего билирубина и его метаболитов и, таким образом, важны для ощущения чесотки в условиях повышенного билирубина, таких как холестатический зуд и терминальная почечная недостаточность. Кроме того, MRGPR Х4 также активируется желчными кислотами и их метаболитами, содержание которых также повышается при холестатическом зуде. Кроме того, уробилин, окисленный продукт метаболита гема уробилиногена, который выводится исключительно почками, является мощным агонистом MRGPR Х4 и пруритогеном и, таким образом, может иметь важное значение для ощущения чесотки в условиях повышенного уробилина, таких как уремический зуд, заболевание почек и терминальная почечная недостаточность. Таким образом, модулирование MRGPR Х4 позволяет лечить аутоиммунные заболевания, такие как псориаз, рассеянный склероз, синдром Стивенса-Джонсона, атопические расстройства, такие как атопический дерматит, и другие состояния хронической чесотки, как более подробно описано ниже.
MRGPR, по-видимому, являются сенсорными рецепторами, которые распознают в своей внешней среде экзогенные или эндогенные сигналы/химические вещества. Эти рецепторы, вероятно, реагируют на несколько химических лигандов/агонистов. Например, MRGPR Х4 распознает билирубин, желчные кислоты и уробилин как агонистические сигналы. В некоторых вариантах осуществления молекулы по настоящему изобретению модулируют MRGPR Х4, функционируя как обратные агонисты, способные блокировать несколько химических соединений, и/или как конкурентные антагонисты, которые могут специфически блокировать отдельные лиганды. В одном варианте осуществления изобретения такая модуляция является селективной в отношении других MRGPR, таких как MRGPR X1, Х2 и/или Х3.
Соответственно, в одном варианте осуществления изобретения предусматриваются способы модуляции MRGPR Х4, включающие приведение MRGPR Х4 в контакт с эффективным количеством соединения, имеющего структуру формулы (I):
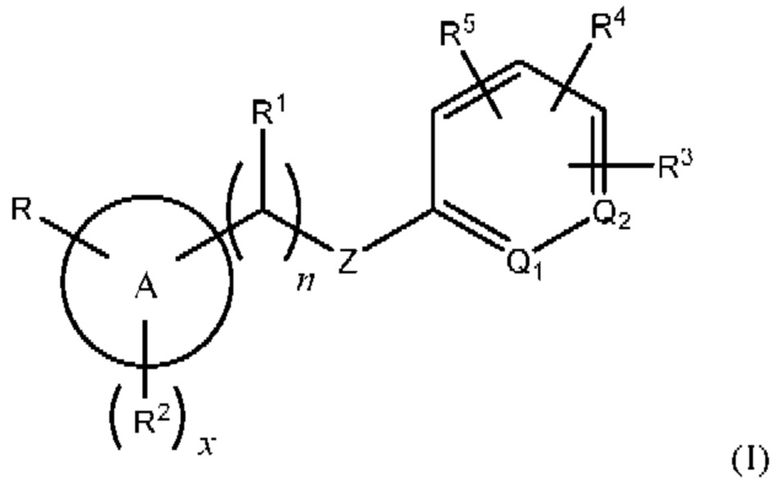
или его фармацевтически приемлемого изомера, рацемата, гидрата, сольвата, изотопа или соли, где:
n равен 0 или 1;
х равен 0, 1 или 2;
А представляет собой арил или гетероарил;
Q1 и Q2 оба представляют собой CR10, или один из Q1 или Q2 представляет собой CR10, а другой - N;
Z представляет собой -О-, -S-, -N(R11)-, -СН2- или -С≡С-;
каждый R10 представляет собой Н или алкил;
R представляет собой -(CH2)mC(=O)OR12, -(CH2)mNHR13, (C=O)NR14R15, -СН2ОН, -CN, галоидалкил, карбоцикл, гетероцикл или изостер карбоновой кислоты;
m равен 0 или 1;
R11, R12 и R13 являются одинаковыми или разными и по отдельности представляют собой Н или алкил;
R14 представляет собой Н, и R15 представляет собой Н, -SO2CH3, карбоцикл, гетероцикл или алкил, замещенный 0, 1, 2 или 3 заместителями, выбранными из ОН, CN, -NR'R'', С(=O)ОН, C(=O)NR'R'', -SO2OH, алкокси, карбоцикла или гетероцикла, где R' и R'' по отдельности представляют собой Н или алкил, или
R14 и R15, вместе с атомом азота, к которому они присоединены, образуют гетероцикл;
R1 представляет собой Н или алкил;
R2 представляет собой галоид, циано, амино, алкил, алкокси, карбоцикл или гетероцикл;
R3, R4 и R5 являются одинаковыми или разными и либо отсутствуют, либо, если они присутствуют, представляют собой циано, нитро, галоген, алкил, галоидалкил, цианоалкил, алкокси, галоидалкокси, -(С=O)алкил, -(С=O)NH-алкил, карбоцикл, гетероцикл, -О-карбоцикл или -О-гетероцикл, или
любые два R и R2, взятые вместе с атомами, к которым они присоединены, образуют гетероцикл;
любые два R3, R4, R5 и R10, взятые вместе с атомами, к которым они присоединены, образуют карбоцикл или гетероцикл;
и при этом в каждом случае карбоцикл или гетероцикл замещен 0, 1, 2 или 3 заместителями, индивидуально выбранными из галогена, гидроксила, оксо, галоида, алкила, галоидалкила, алкокси, галоидалкокси, карбоцикла или гетероцикла.
«Модуляция» MRGPR Х4 означает, что соединение взаимодействует с MRGPR Х4 таким образом, что он действует как обратный агонист рецептора и/или как конкурентный антагонист рецептора. В одном варианте осуществления изобретения такая модуляция является частично или полностью избирательной в отношении других MRGPR, таких как MRGPR X1, Х2 и/или Х3.
«MRGPR» относится к одному или нескольким Mas-связанным G-белок-сопряженным рецепторам, которые представляют собой группу орфанных рецепторов с ограниченной экспрессией в очень специализированных тканях (например, в сенсорных нейронах и ганглиях дорсальных корешков) и барьерных тканях. Существует восемь родственных рецепторов этого класса, экспрессируемых у людей, только 4 из которых имеют легко идентифицируемые ортологи у других видов (например, MRGPR D, Е, F и G). Остальные четыре рецептора (MRGPR X1, Х2, Х3 и Х4), исходя из гомологии, не имеют аналогов у других видов, не относящихся к человеку.
«Эффективное количество» относится к количеству указанного агента, достаточному для достижения желаемого эффекта у субъекта, получающего лечение этим агентом. В идеале эффективное количество агента представляет собой количество, достаточное для ингибирования или лечения заболевания, не вызывая значительной токсичности у субъекта. Эффективное количество агента будет зависеть от субъекта, получающего лечение, тяжести заболевания и способа введения фармацевтической композиции. Способы определения эффективного количества соединения по настоящему изобретению, достаточного для достижения желаемого эффекта у субъекта, будут понятны специалистам в этой области в свете данного описания.
«Алкил» означает насыщенную или ненасыщенную алкильную группу с прямой или разветвленной цепью, имеющую от 1 до 8 атомов углерода, в некоторых вариантах осуществления изобретения - от 1 до 6 атомов углерода, в некоторых вариантах осуществления изобретения - от 1 до 4 атомов углерода, и в некоторых вариантах осуществления изобретения - от 1 до 3 атомов углерода. Примеры насыщенных линейных алкильных групп включают, без ограничений, метальную, этильную, н-пропильную, н-бутильную, н-пентильную, н-гексильную, н-гептильную и н-октильную группы. Примеры разветвленных алкильных групп включают, без ограничений, изопропильную, изобутильную, втор-бутильную, трет-бутильную, неопентильную, изопентильную и 2,2-диметилпропильную группы. Ненасыщенный алкил включает алкенил и алкинил, как определено ниже.
«Алкенил» означает алкенильную группу с прямой или разветвленной цепью, имеющую от 2 до 8 атомов углерода, в некоторых вариантах осуществления изобретения - от 2 до 6 атомов углерода, в некоторых вариантах осуществления изобретения - от 2 до 4 атомов углерода, и в некоторых вариантах осуществления изобретения - от 2 до 3 атомов углерода. Алкенильные группы представляют собой ненасыщенные углеводороды, содержащие по крайней мере одну углерод-углеродную двойную связь. Примеры низших алкенильных групп включают, без ограничений, винил, пропенил, бутенил, пентенил и гексенил.
«Алкинил» означает алкинильную группу с прямой или разветвленной цепью, имеющую от 2 до 8 атомов углерода, в некоторых вариантах осуществления изобретения - от 2 до 6 атомов углерода, в некоторых вариантах осуществления изобретения - от 2 до 4 атомов углерода, и в некоторых вариантах осуществления изобретения - от 2 до 3 атомов углерода. Алкинильные группы представляют собой ненасыщенные углеводороды, содержащие по крайней мере одну углерод-углеродную тройную связь. Примеры алкинильных групп включают, без ограничений, этинил, пропинил, бутинил, пентинил и гексинил.
«Галоид» или «галоген» относится к фтору, хлору, брому и йоду.
«Гидрокси» относится к -ОН.
«Циано» относится к -CN.
Амино относится к-NH2, -NH-алкилу или -N(алкил)2, где алкил имеет указанные выше значения. Примеры амино включают, без ограничений, -NH2, -NHCH3, -N(СН3)2 и т.п.
«Галоидалкил» относится к алкилу, как определено выше, с одним или несколькими атомами водорода, замещенными галогеном. Примеры низших галоидалкильных групп включают, без ограничений, -CF3, -CHF2 и т.п.
«Алкокси» относится к алкилу, как определено выше, присоединенному через атом кислорода (т.е. -О-алкил). Примеры алкоксигрупп включают, без ограничений, метокси, этокси, н-пропокси, н-бутокси, изопропокси, втор-бутокси, трет-бутокси и т.п.
«Галоидалкокси» относится к галоидалкилу, как определено выше, присоединенному через атом кислорода (т.е. -О-галоидалкил). Примеры низших галоидалкоксигрупп включают, без ограничений, -OCF3 и т.п.
«Циклоалкил» относится к алкильным группам, образующим кольцевую структуру, которая может быть замещенной или незамещенной, причем кольцо является полностью насыщенным, частично ненасыщенным или полностью ненасыщенным, при этом в случае ненасыщенности сопряжение пи-электронов в кольце не создает ароматичности. Примеры циклоалкилов включают, без ограничений, циклопропильную, циклобутильную, циклопентильную, циклогексильную, циклогептильную и циклооктильную группы. В некоторых вариантах осуществления изобретения циклоалкильная группа имеет от 3 до 8 членов в кольце, тогда как в других вариантах осуществления количество кольцевых атомов углерода находится в диапазоне от 3 до 5, от 3 до 6, или от 3 до 7. Циклоалкильные группы дополнительно включают полициклические циклоалкильные группы, такие как, без ограничений, норборнильные, адамантильные, борнильные, камфенильные, изокамфенильные и каренильные группы, и конденсированные кольца, такие как, без ограничений, декалинил и т.п.
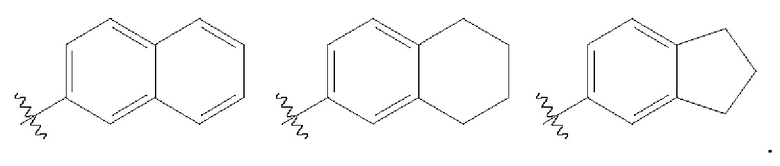
«Арильные» группы представляют собой циклические ароматические углеводороды, не содержащие гетероатомов. Типичные арильные группы включают, без ограничений, фенильные, азуленильные, гепталенильные, бифенильные, индаценильные, флуоренильные, фенантренильные, трифениленильные, пиренильные, нафтаценильные, хризенильные, бифениленильные, антраценильные и нафтильные группы. В некоторых вариантах осуществления изобретения арильные группы содержат 6-14 атомов углерода в кольцевых частях групп. Термины «арил» и «арильные группы» включают конденсированные кольца, причем по меньшей мере одно кольцо, но не обязательно все кольца, являются ароматическими, такие как конденсированные ароматически-алифатические кольцевые системы (например, инданил, тетрагидронафтил и т.п.). В одном варианте осуществления арил представляет собой фенил или нафтил, и в другом варианте осуществления арил представляет собой фенил.
«Карбоцикл» относится к алкильным группам, образующим кольцевую структуру, которая может быть замещенной или незамещенной, причем кольцо является либо полностью насыщенным, либо частично ненасыщенным, либо полностью ненасыщенным, при этом в случае ненасыщенности, сопряжение пи-электронов в кольце может создавать ароматичность. В одном варианте осуществления карбоцикл включает циклоалкил, как определено выше. В другом варианте осуществления карбоцикл включает арил, как определено выше.
«Гетероцикл» относится к ароматическим и неароматическим кольцевым фрагментам, содержащим 3 или более членов кольца, один или несколько из которых являются гетероатомами, такими как, без ограничений, N, О, S или Р. В некоторых вариантах осуществления изобретения гетероциклил включает 3-20 членов кольца, тогда как другие такие группы содержат 3-15 членов кольца. По крайней мере одно кольцо содержит гетероатом, но каждое кольцо полициклической системы не должно обязательно содержать гетероатом. Например, диоксоланильное кольцо и бенздиоксоланильная кольцевая система (метилендиоксифенильная кольцевая система) обе являются гетероциклическими группами в контексте настоящего описания.
Гетероциклические группы также включают конденсированные кольцевые группы, в том числе имеющие конденсированные ароматические и неароматические группы. Гетероциклическая группа также включает полициклические кольцевые системы, содержащие гетероатом, такие как, без ограничений, хинуклидил, а также включает гетероциклические группы, имеющие заместители, включая, без ограничений, алкильные, галоидные, амино, гидрокси, циано, карбокси, нитро, тио или алкоксигруппы, связанные с одним из членов кольца. Гетероциклическая группа, как определено в данном документе, может быть гетероарильной группой или частично или полностью насыщенной циклической группой, включающей по меньшей мере один кольцевой гетероатом. Гетероциклические группы включают, без ограничений, пирролидинильные, фуранильные, тетрагидрофуранильные, диоксоланильные, пиперидинильные, пиперазинильные, морфолинильные, пирролильные, пиразолильные, триазолильные, тетразолильные, оксазолильные, изоксазолильные, тазолильные, пиридинильные, тиофенильные, бензотиофенильные, бензофуранильные, дигидробензофуран ильные, индолильные, дигидроиндолильные, азаиндолильные, индазолильные, бензимидазолильные, азабензимидазолильные, бензоксазолильные, бензотиазолильные, бензотиадиазолильные, имидазопиридинильные, изоксазолопиридинильные, тианафталинильные, пуринильные, ксантинильные, аденинильные, гуанинильные, хинолинильные, изохинолинильные, тетрагидрохинолинильные, хиноксалинильные и хиназолинильные группы.
«Гетероарил» относится к ароматическим кольцевым фрагментам, содержащим 5 или более членов кольца, один или несколько из которых являются гетероатомами, такими как, без ограничений, N, О и S. Гетероарильные группы включают, без ограничений, такие группы, как пирролильные, пиразолильные, пиридинильные, пиридазинильные, пиримидильные, пиразильные, пиразинильные, пиримидинильные, тиенильные, триазолильные, тетразолильные, триазинильные, тиазолильные, тиофенильные, оксазолильные, изоксазолильные, бензотиофенильные, бензофуранильные, индолильные, азаиндолильные, индазолильные, бензимидазолильные, азабензимидазолильные, бензоксазолильные, бензотиазолил, бензотиазолильные, бензотиадиазолильные, имидазопиридинильные, изоксазолопиридинильные, тианафталенильные, пуринильные, ксантинильные, аденинильные, гуанинильные, хинолинильные, изохинолинильные, тетрагидрохинолинильные, тетрагидроизохинолинильные, хиноксалинильные и хиназолинильные группы. Термины «гетероарил» и «гетероарильные группы» включают соединения с конденсированными кольцами, в которых по меньшей мере одно кольцо, но не обязательно все кольца, являются ароматическими, включая тетрагидрохинолинил, тетрагидроизохинолинил, индолил и 2,3-дигидроиндолил.
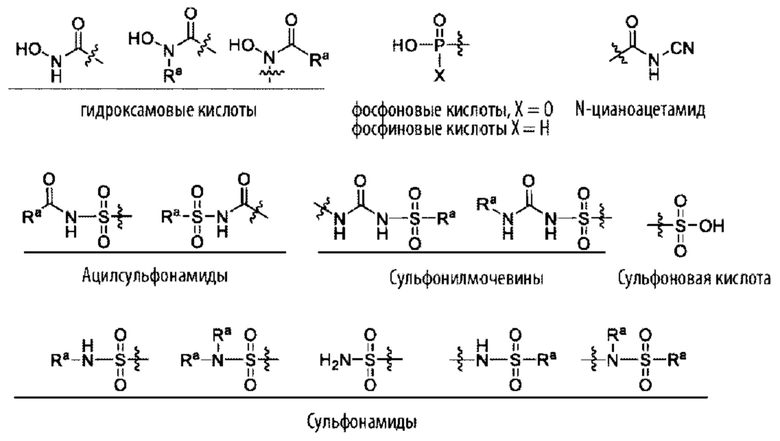
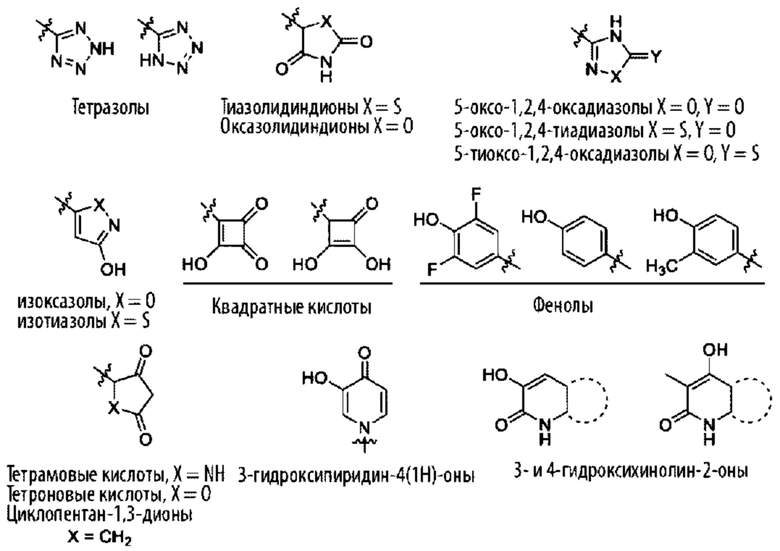
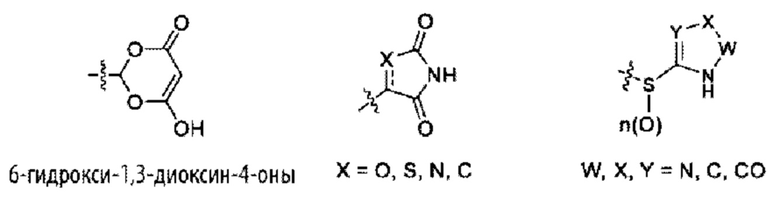
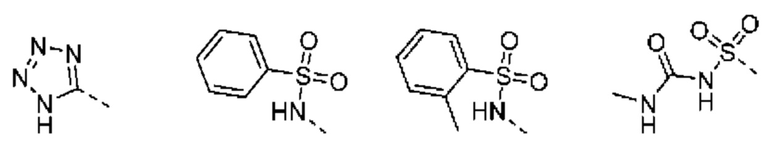
«Изостер карбоновой кислоты» относится к группе, которая служит суррогатом группы карбоновой кислоты (т.е. -СООН). Использование изостера карбоновой кислоты может быть предпочтительнее группы карбоновой кислоты по ряду причин, включая большую селективность, уменьшенные побочные эффекты, пониженную токсичность, улучшенную фармакокинетику, повышенную стабильность и/или упрощенный синтез. Изостеры карбоновых кислот включают гидроксамовые кислоты, ацилцианамиды, сульфонамиды, фосфоновые кислоты, фосфиновые кислоты, цианоацетамиды, сульфонаты, сульфонамиды, ацилсульфонамиды, арилсульфонамиды, сульфонилмочевины, тетразолы, тиазолидиндионы, оксазолидиндионы, изоксазолы, изотиазолы, квадратные кислоты, 3-гидроксихинолин-2-оны, 4-гидроксихинолин-2-оны, 5-оксо-1,2,4-оксадиазолы, 5-оксо-1,2,4-тиадиазолы, 5-тиоксо-1,2,4-оксадиазолы, гидроксиизоксазолы, фенолы, тетрамовые кислоты, тетроновые кислоты, циклопентан-1,3-дионы, 6-гидрокси-1,3-диоксин-4-оны, 3-гидроксипиридин-4(1Н)-оны и оксадиазолоны.
В одном из вариантов осуществления изостер карбоновой кислоты может быть ациклическим и иметь одну из следующих структур (где Ra представляет собой алкил, карбоцикл или гетероцикл, причем каждый из карбоцикла и гетероцикла может быть один или несколько раз замещен R2):
В другом варианте осуществления изостер карбоновой кислоты может быть циклическим и иметь одну из следующих структур:
«Изомер» используется в данном документе для охвата всех хиральных, диастереомерных или рацемических форм структуры, если специально не указана конкретная стереохимия или изомерная форма. Такие соединения могут быть обогащенными или разделенными оптическими изомерами по любому или всем асимметричным атомам, как видно на изображениях, при любой степени обогащения. Как рацемические, так и диастереомерные смеси, а также индивидуальные оптические изомеры могут быть синтезированы таким образом, чтобы они по существу не содержали их энантиомерных или диастереомерных партнеров, и все они входят в объем определенных вариантов осуществления настоящего изобретения. Изомеры, связанные с наличием хирального центра, включают пару несовмещающихся при наложении изомеров, которые называются «энантиомерами». Отдельные энантиомеры чистого соединения являются оптически активными (т.е. они способны вращать плоскость плоскополяризов энного света и обозначаются R или S).
«Выделенный оптический изомер» означает соединение, которое было по существу очищено от соответствующего оптического изомера (изомеров) той же формулы. Например, выделенный изомер может иметь чистоту по меньшей мере около 80%, по меньшей мере 80% или по меньшей мере 85% мас. В других вариантах осуществления изобретения выделенный изомер имеет чистоту по меньшей мере 90%, или по меньшей мере 98%, или по меньшей мере 99% мас.
«По существу энантиомерно или диастереомерно» чистый означает уровень энантиомерного или диастереомерного обогащения одного энантиомера по отношению к другому энантиомеру или диастереомеру, равный по меньшей мере примерно 80%, более конкретно, более 80%, 85%, 90%, 95%., 98%, 99%, 99,5% или 99,9%.
Термины «рацемат» и «рацемическая смесь» относятся к равной смеси двух энантиомеров. Рацемат обозначается «(±)», потому что он не является оптически активным (т.е. не будет вращать плоскополяризованный свет в каком-либо направлении, поскольку составляющие его энантиомеры нейтрализуют друг друга). Все соединения с отмеченными звездочкой (*) третичнымы или четвертичнымы атомами углерода являются оптически активными изомерами, которые могут быть получены методами очистки из соответствующего рацемата и/или синтезированы подходящими методами хирального синтеза.
«Гидрат» представляет собой соединение, которое существует в сочетании с молекулами воды. Такая комбинация может включать воду в стехиометрических количествах, таких как моногидрат или дигидрат, или может включать воду в произвольных количествах. Используемый в данном документе термин «гидрат» относится к твердой форме; то есть соединение в водном растворе, хотя оно может быть гидратировано, не является гидратом в значении термина, используемом в данном документе.
«Сольват» подобен гидрату, за исключением того, что присутствует растворитель, отличный от воды. Например, метанол или этанол могут образовывать «алкоголяты», которые также могут быть стехиометрическими или нестехиометрическими. Используемый в данном документе термин «сольват» относится к твердой форме; то есть раствор соединения в растворителе, хотя оно может быть сольватированным, не является сольватом в значении термина, используемом в данном документе.
«Изотоп» относится к атомам с одинаковым числом протонов, но разным числом нейтронов, и изотоп соединения формулы (I) включает любое такое соединение, в котором один или несколько атомов заменены изотопом этого атома. Например, углерод-12, наиболее распространенная форма углерода, имеет шесть протонов и шесть нейтронов, тогда как углерод-13 имеет шесть протонов и семь нейтронов, а углерод-14 имеет шесть протонов и восемь нейтронов. Водород имеет два стабильных изотопа: дейтерий (один протон и один нейтрон) и тритий (один протон и два нейтрона). Фтор имеет ряд изотопов, самым долгоживущим из которых является фтор-19. Таким образом, изотоп соединения, имеющего структуру формулы (I), включает, без ограничений, соединения формулы (I), в которых один или несколько атомов углерода-12 заменены атомами углерода-13 и/или углерода-14, в которых один или несколько атомов водорода заменены дейтерием и/или тритием, и/или в которых один или несколько атомов фтора заменены фтором-19.
«Соль» обычно относится к органическому соединению, такому как карбоновая кислота или амин, в ионной форме в сочетании с противоионом. Например, соли, образованные кислотами в их анионной форме и катионами, называются «кислотно-аддитивными солями». И наоборот, соли, образованные основаниями в катионной форме и анионами, называются «солями присоединения оснований».
Термин «фармацевтически приемлемый» относится к агенту, который одобрен для употребления человеком и, как правило, не токсичен. Например, термин «фармацевтически приемлемая соль» относится к нетоксичным солям присоединения неорганических или органических кислот и/или оснований (см., например, Lit et al., Salt Selection for Basic Drugs, Int. J. Pharm., 33, 201-217, 1986) (включена в данный документ посредством ссылки).
Фармацевтически приемлемые соли присоединения оснований соединений по изобретению включают, например, соли металлов, включая соли щелочных металлов, щелочноземельных металлов и переходных металлов, такие как, например, соли кальция, магния, калия, натрия и цинка. Фармацевтически приемлемые соли присоединения оснований также включают органические соли, полученные из основных аминов, таких как, например, N,N'-дибензилэтилендиамин, хлорпрокаин, холин, диэтаноламин, этилендиамин, меглумин (N-метилглюкамин) и прокаин.
Фармацевтически приемлемые кислотно-аддитивные соли могут быть получены из неорганической кислоты или из органической кислоты. Примеры неорганических кислот включают хлористоводородную, бромистоводородную, йодистоводородную, азотную, угольную, серную и фосфорную кислоты. Подходящие органические кислоты могут быть выбраны из классов алифатических, циклоалифатических, ароматических, ароматических алифатических, гетероциклических, карбоновых и сульфоновых органических кислот, примеры которых включают муравьиную, уксусную, пропионовую, янтарную, гликолевую, глюконовую, молочную, яблочную, винную, лимонную, аскорбиновую, глюкуроновую, малеиновую, фумаровую, пировиноградную, аспарагиновую, глутаминовую, бензойную, антраниловую, 4-оксибензойную, фенилуксусную, миндальную, гиппуровую, малоновую, щавелевую, эмбоновую (памоевую), метансульфоновую, этансульфоновую, бензолсульфоновую, пантотеновую, трифторметансульфоновую, 2-оксиэтансульфоновую, n-толуолсульфоновую, сульфаниловую, циклогексиламиносульфоновую, стеариновую, альгиновую, β-оксимасляную, салициловую, -галактаровую и галактуроновую кислоты.
Хотя фармацевтически неприемлемые соли обычно не используются в качестве лекарственных средств, такие соли могут быть полезны, например, в качестве промежуточных продуктов в синтезе соединений, имеющих структуру формулы I, например, при их очистке перекристаллизацией.
В другом варианте осуществления изобретения предусматривается способ лечения субъекта, имеющего зависимое от MRGPR Х4 состояние, который включает введение субъекту фармацевтически эффективного количества соединения, имеющего структуру формулы (I):
или его фармацевтически приемлемого изомера, рацемата, гидрата, сольвата, изотопа или соли, где:
n равен 0 или 1;
х равен 0 или 1;
А представляет собой арил или гетероарил;
Q1 и Q2 оба представляют собой CR10, или один из Q1 или Q2 представляет собой CR10, а другой - N;
Z представляет собой -О-, -S-, -N(R11)-, -СН2- или -С≡С-;
каждый R10 представляет собой Н или алкил;
R представляет собой -(CH2)mC(=O)OR12, -(CH2)mNHR13, (C=O)NR14R15, -СН2ОН, -CN, галоидалкил, карбоцикл, гетероцикл или изостер карбоновой кислоты;
m равен 0 или 1;
R11, R12 и R13 являются одинаковыми или разными и по отдельности представляют собой Н или алкил;
R14 представляет собой Н, и R15 представляет собой Н, -SO2CH3, карбоцикл, гетероцикл или алкил, замещенный 0, 1, 2 или 3 заместителями, выбранными из ОН, CN, -NR'R'', С(=O)ОН, C(=O)NR'R'', -SO2OH, алкокси, карбоцикла или гетероцикла, где R' и R'' по отдельности представляют собой Н или алкил, или
R14 и R15, вместе с атомом азота, к которому они присоединены, образуют гетероцикл;
R1 представляет собой Н или алкил;
R2 представляет собой галоид, циано, алкил, алкокси, карбоцикл или гетероцикл;
R3, R4 и R5 являются одинаковыми или разными и либо отсутствуют, либо, в случае присутствия, представляют собой циано, нитро, галоген, алкил, галоидалкил, алкокси, галоидалкокси, карбоцикл, гетероцикл, -О-карбоцикл или -О-гетероцикл, или
любые два R и R2, взятые вместе с атомами, к которым они присоединены, образуют гетероцикл;
любые два R3, R4, R5 и R10, взятые вместе с атомами, к которым они присоединены, образуют карбоцикл или гетероцикл;
и при этом в каждом случае карбоцикл или гетероцикл замещен 0, 1, 2 или 3 заместителями, индивидуально выбранными из галогена, оксо, галоида, алкила, галоидалкила, алкокси, галоидалкокси, карбоцикла или гетероцикла.
При использовании в данном документе, фраза «зависимое от MRGPR Х4 состояние» означает состояние, при котором активация, повышенная сенсибилизация или десенсибилизация MRGPR Х4 естественным или синтетическим лигандом инициирует, опосредует, поддерживает или усиливает патологическое состояние. Например, известно, что некоторые виды чесотки или болевых ощущений у пациентов, страдающих зудом, атопическими или другими аутоиммунными или воспалительными заболеваниями, вызваны повышенным билирубином и его метаболитами или желчными кислотами. Было обнаружено, что MRGPR Х4 является чувствительным (или активируется) билирубином и его метаболитами, включая уробилин, или желчными кислотами. Не ограничиваясь теорией, следует понимать, что путем модуляции MRGPR Х4 можно ослабить зуд или болевые ощущения.
В некоторых вариантах осуществления изобретения зависимое от MRGPR Х4 состояние представляет собой состояние, вызванное активацией MRGPR Х4 желчной кислотой. Используемый в данном документе термин «желчная кислота» включает первичные желчные кислоты (например, холевая кислота, хенодезоксихолевая кислота), конъюгированные желчные кислоты, также называемые солями желчных кислот (например, таурохолевая кислота, гликохолевая кислота, таурохенодезоксихолевая кислота, гликохенодезоксихолевая кислота), вторичные желчные кислоты (например, дезоксихолевая кислота, литохолевая кислота) и аналоги желчных кислот. В некоторых вариантах осуществления изобретения аналог желчной кислоты представляет собой агонист фарнезоидного Х-рецептора (ФХР). Таким образом, соединения по настоящему изобретению можно использовать для лечения MRGPR Х4-зависимого состояния, вызванного активацией MRGPR Х4 желчной кислотой, при котором модуляция MRGPR Х4 могла бы быть полезной.
В некоторых вариантах осуществления изобретения зависимое от MRGPR Х4 состояние представляет собой состояние, связанное с чесоткой, состояние, связанное с болью, аутоиммунное заболевание или аутоиммунное или воспалительное расстройство.
Используемое в данном документе выражение «состояние, связанное с чесоткой» означает зуд (включая острый и хронический зуд), связанный с любым состоянием. Ощущение чесотки может исходить, например, из периферической нервной системы (например, кожная или нейропатическая чесотка) или из центральной нервной системы (например, невропатическая, нейрогенная или психогенная чесотка). Таким образом, в одном варианте осуществления предусматривается способ по настоящему изобретению для лечения состояния, связанного с чесоткой, такого как хроническая чесотка; холестатический зуд; контактный дерматит, аллергический блефарит; анемия; атопический дерматит; буллезный пемфигоид; кандидоз; ветряная оспа; холестаз; терминальная почечная недостаточность; гемодиализ; контактный дерматит, атопический дерматит; герпетиформный дерматит, диабет, лекарственная аллергия, сухая кожа; дисгидротический дерматит; эктопическая экзема; эритразма; фолликулит; грибковая инфекция кожи; геморрой; герпес; ВИЧ-инфекция; болезнь Ходжкина; гипертиреоз; железодефицитная анемия; болезнь почек; лейкозы, порфирии; заболевание печени, включая первичный билиарный холангит, первичный склерозирующий холангит, синдром Алажилля, прогрессирующий семейный внутрипеченочный холестаз, внутрипеченочный холестаз при беременности, неалкогольный стеатогепатит (НАСГ), неалкогольную жировую болезнь печени (НАЖБП), атрезию желчных протоков, хронический гепатит В, лекарственно-хронический вирусный гепатит, лекарственно-индуцированное повреждение печени (ЛПП), фиброз печени, холестатическую болезнь печени и алкогольную болезнь печени; лимфома; злокачественные новообразования; множественная миелома; нейродермит; онхоцеркоз; болезнь Педжета; педикулез; истинная полицитемия; плоский лишай; склеротический лихен; анальный зуд; псевдобешенство; псориаз; выпадение прямой кишки; зудневая чесотка; шистосомоз; склеродермия, тяжелый стресс, стазовый дерматит; зуд купальщиков; заболевание щитовидной железы; паховый дерматомикоз; уремический зуд; розацеа; кожный амилоидоз; склеродермия; прыщи; заживление раны; глазной зуд; и крапивница.
Используемое в данном документе выражение «состояние, связанное с болью», означает любую боль, вызванную состоянием здоровья. Таким образом, в одном варианте осуществления предусматривается способ по настоящему изобретению для лечения состояния, связанного с болью, такого как острая боль, прогрессирующий рак простаты, боль, связанная со СПИДом, анкилозирующий спондилит, арахноидит, артрит, артрофиброз, атаксический церебральный паралич, аутоиммунный атрофический гастрит, аваскулярный некроз, боль в спине, болезнь (синдром) Бехчета, синдром жжения во рту, бурсит, раковая боль, туннельный запястный синдром, синдром конского хвоста, центральный болевой синдром, церебральный паралич, стеноз шейки матки, болезнь Шарко-Мари-Тута (ШМТ), синдром хронической усталости (СХУ), хроническая функциональная абдоминальная боль (ХФАБ), хроническая боль, хронический панкреатит, коллапс легкого (пневмоторакс), комплексный регионарный болевой синдром (РБС), нейропатическая боль роговицы, болезнь Крона, дегенеративная болезнь диска, болезнь Деркума, дерматомиозит, диабетическая периферическая нейропатия (ДПН), дистония, синдром Элерса-Данлоса (СЭД), эндометриоз, синдром эозинофилии-миалгии (СЭМ), эритромелалгия, фибромиалгия, подагра, головные боли, грыжа межпозвоночного диска, гидроцефалия, межреберная невралгия, интерстициальный цистит, синдром раздраженного кишечника (СРК), ювенильный дерматозит (дерматомиозит), травма колена, боль в ногах, синдром боли в пояснице-гематурии, волчанка, болезнь Лайма, медуллярная губчатая почка (МГП), парестетическая мералгия, мезотелиома, мигрень, скелетно-мышечная боль, миофасциальная боль, миозит, боль в шее, невропатическая боль, затылочная невралгия, остеоартрит, болезнь Педжета, синдром Парсонажа-Тернера, боль в тазу, периферическая нейропатия, фантомная боль в конечностях, защемление нерва, поликистозная болезнь почек, ревматическая полимиалгия, полимиозит, порфирия, болевой синдром после герниорафии, болевой синдром после мастэктомии, постинсультная боль, болевой синдром после торакотомии, постгерпетическая невралгия (опоясывающий лишай), постполиомиелитный синдром, первичный боковой склероз, псориатический артрит, пудендальная невралгия, радикулопатия, болезнь Рейно, ревматоидный артрит (РА), дисфункция крестцово-подвздошного сочленения, саркоидоз, кифоз Шейермана, ишиалгия, сколиоз, опоясывающий лишай (герпес зостер), синдром Шегрена, спастическая кривошея, дисфункция сфинктера Одди, спиноцеребеллярная атаксия (SCA-атаксия), травма спинного мозга, стеноз позвоночника, сирингомиелия, кисты Тарлова, поперечный миелит, тригеминальная невралгия, невропатическая боль, неспецифический язвенный колит, сосудистая боль и вульводиния.
Используемый в данном документе термин «аутоиммунное расстройство» или «воспалительное расстройство» означает заболевание или расстройство, возникающее из и/или направленное против собственных тканей или органов человека, или совокупность их составных элементов, или проявление, или возникающее в результате состояние. Как правило, могут существовать различные клинические и лабораторные маркеры аутоиммунных заболеваний, включая, без ограничений, гипергаммаглобулинемию, высокие уровни аутоантител, отложения комплекса антиген-антитело в тканях, клиническую пользу от лечения кортикостероидами или иммунодепрессантами и агрегаты лимфоидных клеток в пораженных тканях. Таким образом, в одном варианте осуществления предусматривается способ по настоящему изобретению для лечения аутоиммунного расстройства, такого как хроническое воспаление, рассеянный склероз, синдром Стивенса-Джонсона, аппендицит, бурсит, колит, цистит, дерматит, флебит, рефлекторная симпатическая дистрофия/комплексный регионарный болевой синдром (РСД/КРБС), ринит, тендинит, тонзиллит, вульгарные угри, нарушение реактивности дыхательных путей, астма, инфекция дыхательных путей, аутовоспалительное заболевание, глютеновая болезнь, хронический простатит, дивертикулит, гломерулонефрит, гнойный гидраденит, гиперчувствительности, кишечное расстройство, расстройство кишечного эпителия, воспалительное заболевание кишечника, синдром раздраженного кишечника, колит, интерстициальный цистит, отит, воспалительное заболевание органов малого таза, боль в эндометрии, реперфузионное повреждение, ревматическая лихорадка, ревматоидный артрит, саркоидоз, отторжение трансплантата, псориаз, воспаление легких, хроническая обструктивная болезнь легких, сердечно-сосудистые заболевания и васкулит.
Используемый в данном документе термин «введение» относится к обеспечению соединения или фармацевтической композиции, содержащей соединение, описанное в данном документе. Соединение или композиция могут быть введены субъекту другой особой или субъект может вводить их самостоятельно. Неограничивающими примерами путей введения являются пероральный, парентеральный (например, внутривенный), или местный.
Используемый в данном документе термин «лечение» относится к вмешательству, которое уменьшает признак или симптом заболевания или патологического состояния. Используемые в данном документе термины «лечение», «лечить» и «проведение лечения» применительно к заболеванию, патологическому состоянию или симптому также относятся к любому наблюдаемому положительному эффекту лечения. О положительном эффекте могут свидетельствовать, например, отсроченное начало проявления клинических симптомов заболевания у восприимчивого субъекта, снижение тяжести некоторых или всех клинических симптомов заболевания, более медленное прогрессирование заболевания, уменьшение количества рецидивов заболевания, улучшение общего состояния здоровья или самочувствия субъекта или другие параметры, хорошо известные в данной области, являющиеся специфичными для конкретного заболевания. Профилактическое лечение представляет собой лечение, назначаемое субъекту, у которого отсутствуют признаки заболевания или проявляются только ранние признаки, с целью снижения риска развития патологии. Терапевтическое лечение - это лечение, назначаемое субъекту после появления признаков и симптомов заболевания.
Используемый в данном документе термин «субъект» относится к животному (например, млекопитающему, такому как человек). Субъект, которого лечат описанными в данном документе способами, может иметь диагностированное зависимое от MRGPR Х4 состояние, такое как состояние, связанное с чесоткой, состояние, связанное с болью, или аутоиммунное расстройство. Диагностика может быть выполнена любым способом или методкой, известными в данной области. Специалисту в данной области понятно, что субъект, подлежащий лечению в соответствии с настоящим изобретением, мог быть подвергнут стандартным тестам или мог быть идентифицирован без обследования как подверженный риску из-за наличия одного или нескольких факторов риска, связанных с заболеванием или состоянием.
В другом варианте осуществления изобретения способ лечения субъекта, имеющего зависимое от MRGPR Х4 состояние (например, состояние, связанное с чесоткой, состояние, связанное с болью, аутоиммунное заболевание или аутоиммунное расстройство), описанное в данном документе, дополнительно включает введение субъекту фармацевтически эффективного количества второго терапевтического агента. В одном из вариантов осуществления состояние, связанное с чесоткой, представляет собой заболевание печени. В одном варианте осуществления изобретения, второй терапевтический агент представляет собой терапевтическое средство для лечения заболевания печени. В одном варианте осуществления терапевтическое средство для лечения заболеваний печени представляет собой урсодезоксихолевую кислоту (УДХК), норурсодезоксихолевую кислоту, холестирамин, станозолол, налтрексон, рифампицин, ализол В 23-ацетат (АВ23А), куркумин, дигидроартемизинин, фенофибрат, безафибрат, метронидазол, метотрексат, колхицин, метформин, бетаин, глюкагон, налтрексон, агонист фарнезоидных Х-рецепторов (ФХР), агонист рецептора, активируемого пролифератором пероксисом (PPAR), агонист бета-рецептора тиреоидного гормона (TRβ), или любую их комбинацию.
Примеры агонистов ФХР, которые можно использовать в описанных в данном документе способах, включают обетихолевую кислоту, турофексорат изопропил (WAY-362450), 3-(2,6-дихлорфенил)-4-(3'-карбокси-2-хлорстильбен-4-ил)оксиметил-5-изопропилизоксазол (GW4064), РХ20606 (РХ-102), РХ-101, INT-767, TNT-787, TERN-101, альтенузин, тропифексор (LJN452), нидуфексор, турофексорат изопропил, фексарамин, силимарин, силибин, гедрагоновую кислоту, кафестол, цилофексор (GS-9674 или Рх-104), EDP-305, BAR704, BAR502, EYP-001, RDX-023, AGN-242266, HPG-1860, МЕТ-409, AGN-242256, ЕР-024297, IOT-022, М-480, INV-33, RDX023-02 или любую их комбинацию. В одном варианте осуществления агонист ФХР представляет собой желчную кислоту или ее аналог (например, обетихолевую кислоту, TNT-767, INT-787, BAR502, гедрагоновую кислоту или BAR704), или агонист, не являющийся желчной кислотой (например, EDP-305, тропифексор, нидуфексор, цилофексор, GW4064, турофексорат изопропил, фексарамин, РХ20606 (РХ-102), TERN-101, альтенузин, силимарин, силибин, EYP-001, RDX023-2, AGN-242266, HPG-1860, МЕТ-409, ЕР-024297, М-480 или кафестол).
В одном из вариантов осуществления агонист PPAR представляет собой агонист PPAR-альфа, агонист PPAR-гамма, агонист PPAR-дельта, двойной агонист PPAR-альфа/гамма, двойной агонист PPAR-альфа/дельта, двойной агонист PPAR-гамма/дельта. или пан-агонист PPAR-альфа/гамма/дельта.
Примеры агонистов PPAR-альфа, которые могут быть использованы в описанных в данном документе способах, включают фенофибрат, ципрофибрат, пемафибрат, гемфиброзил, клофибрат, бинифибрат, клинофибрат, клофибриновую кислоту, никофибрат, пирифибрат, плафибрид, ронифибрат, теофибрат, токофибрати SRI 0171.
Примеры агонистов PPAR-гамма, которые можно использовать в описанных в данном документе способах, включают розиглитазон, пиоглитазон, стабилизированный дейтерием R-пиоглитазон, эфатутазон, АТх08-001, OMS-405, CHS-131, THR-0921, SER-150-DN, KDT-501, GED-0507-34-Levo, CLC-3001 и ALL-4.
Примеры агонистов PPAR-дельта, которые можно использовать в описанных в данном документе способах, включают GW501516 (эндурабол или ({4-[({4-метил-2-[4-(трифторметил)фенил]-1,3-тиазол-5-ил)метил)сульфанил]-2-метилфенокси}уксусная кислота)), МВХ8025 (селаделпар или {2-метил-4-[5-метил-2-(4-трифторметилфенил)-2Н-[1,2,3]триазол-4-илметилсульфанил]фенокси}уксусная кислота), GW0742 ([4-[[[2-[3-фтор-4-(трифторметил)фенил]-4-метил-5-тиазолил]метил]тио]-2-метилфенокси]уксусная кислота), L165041, НРР-593 и NCP-1046.
Примеры агонистов PPAR альфа/гамма, которые можно использовать в описанных в данном документе способах, включают сароглитазар, алеглитазар, мураглитазар, тесаглитазар и DSP-8658.
Примеры агонистов PPAR альфа/дельта, которые можно использовать в описанных в данном документе способах, включают элафибранор и Т913659.
Примеры агонистов PPAR гамма/дельта, которые можно использовать в описанных в данном документе способах, включают конъюгированную линолевую кислоту (КЛК) и T3D-959.
Примеры агонистов PPAR альфа/гамма/дельта, которые можно использовать в описанных в данном документе способах, включают IVA337 (ланифибранор), ТТА (тетрадецилтиоуксусная кислота), бавахинин, GW4148, GW9135, безафибрат, лобеглитазон, 2-(4-(5,6-метилендиоксибензо[d]тиазол-2-ил)-2-метилфенокси)-2-метилпропановую кислоту (MHY2013) и CS038.
Примеры агонистов бета-рецептора тиреоидного гормона, которые можно использовать в описанных в данном документе способах, включают собетиром, эпротиром, GC-24, MGL-3196, MGL-3745, VK-2809, KB141 [3,5-дихлор-4-(4-гидрокси-3-изопропилфенокси)фенилуксусная кислота] и МВ07811 (2R,4S)-4-(3-хлорфенил)-2-[(3,5-диметил-4-(4'-гидрокси-3'-изопропилбензил)фенокси)метил]-2-оксидо[1,3,2]диоксафосфонан).
Второй терапевтический агент можно вводить одновременно, отдельно или последовательно с соединениями по настоящему изобретению. При одновременном введении второй терапевтический агент и соединение по настоящему изобретению можно вводить в виде отдельных лекарственных форм или в одной лекарственной форме.
В другом варианте осуществления изобретения предусматривается способ лечения субъекта, имеющего состояние, связанное с чесоткой, при этом способ включает введение субъекту фармацевтически эффективного количества соединения, имеющего структуру формулы (I), или его фармацевтически приемлемого изомера, рацемата, гидрата, сольвата, изотопа или соли, или его фармацевтической композиции. В одном варианте осуществления изобретения связанное с чесоткой состояние представляет собой холестатический зуд, уремический зуд, атопический дерматит, сухую кожу, псориаз, контактный дерматит или экзему.
В одном из вариантов осуществления Формулы (I) n равен 1, R1 представляет собой Н, Z представляет собой О, R представляет собой -C(=O)OR12, и соединение имеет структуру формулы (II):
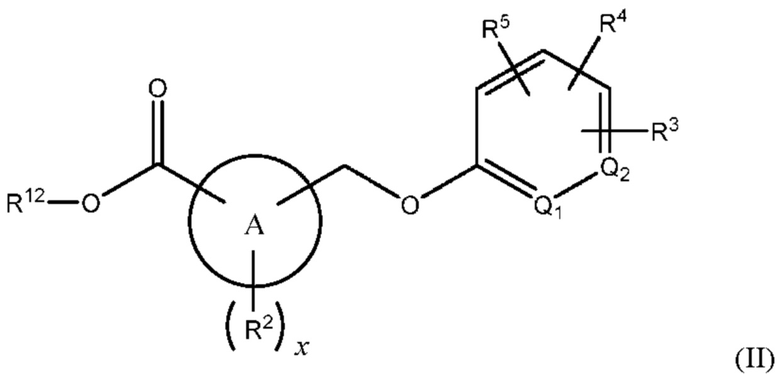
или представляет собой его фармацевтически приемлемый изомер, рацемат, гидрат, сольват, изотоп или соль, где х, A, Q1, Q2, R2, R3, R4, R5 и R12 имеют указанные выше значения.
В одном из вариантов осуществления Формулы (I) n равен О, Z представляет собой О, и соединение имеет структуру формулы (III):
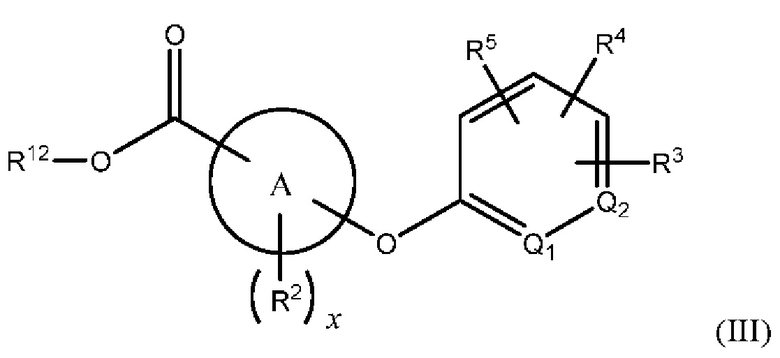
или представляет собой его фармацевтически приемлемый изомер, рацемат, гидрат, сольват, изотоп или соль, где х, A, Q1, Q2, R2, R3, R4, R5 и R12 имеют указанные выше значения.
В одном из вариантов осуществления Формулы (II) х равен 0, и соединение имеет структуру Формулы (IV):
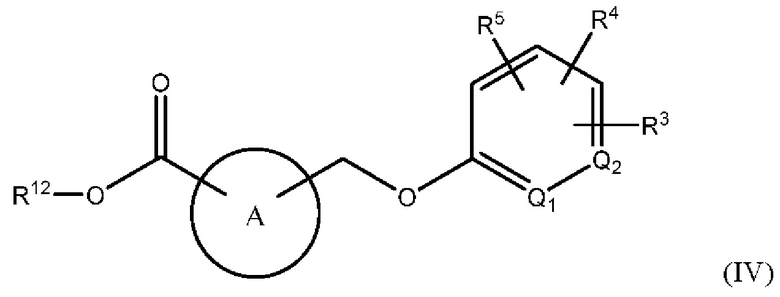
или представляет собой его фармацевтически приемлемый изомер, рацемат, гидрат, сольват, изотоп или соль, где A, Q1, Q2, R3, R4, R5 и R12 имеют указанные выше значения.
В одном варианте осуществления Формулы (II) х равен 1, и соединение имеет структуру Формулы (V):
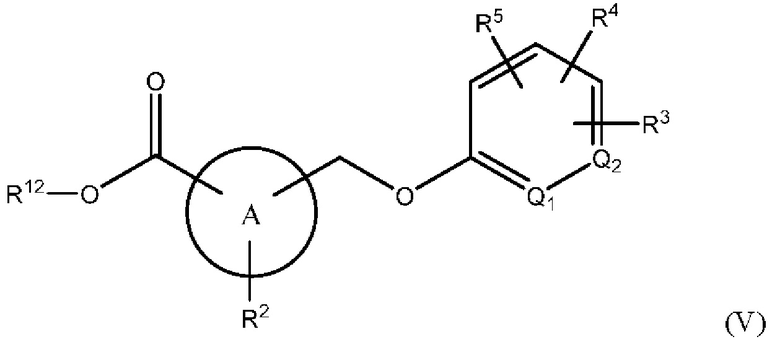
или представляет собой его фармацевтически приемлемый изомер, рацемат, гидрат, сольват, изотоп или соль, где A, Q1, Q2, R2, R3, R4, R5 и R12 имеют указанные выше значения.
В одном варианте осуществления Формулы (III) х равен 0, и соединение имеет структуру Формулы (VI):
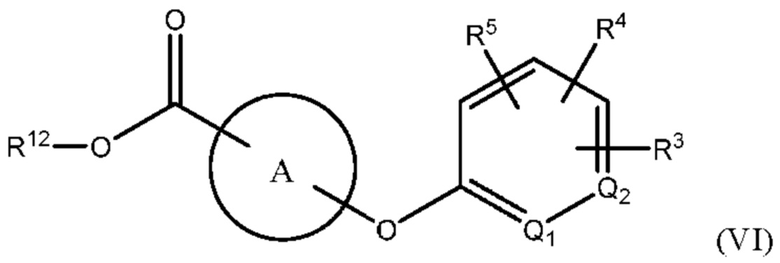
или представляет собой его фармацевтически приемлемый изомер, рацемат, гидрат, сольват, изотоп или соль, где х, A, Q1, Q2, R3, R4, R5 и R12 имеют указанные выше значения.
В одном варианте осуществления Формулы (III) х равен 1, и соединение имеет структуру Формулы (VII):
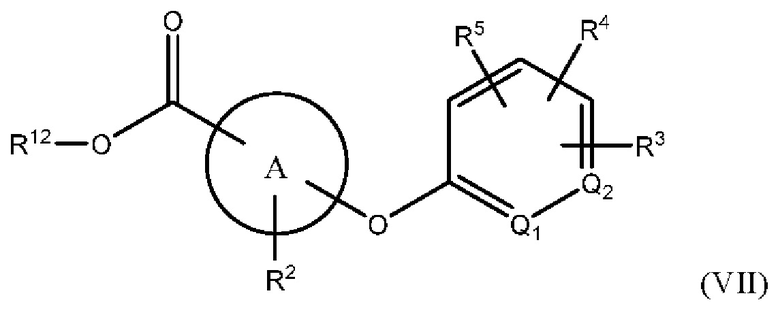
или представляет собой его фармацевтически приемлемый изомер, рацемат, гидрат, сольват, изотоп или соль, где х, A, Q1, Q2, R2, R3, R4, R5 и R12 имеют указанные выше значения.
В другом варианте осуществления, когда R12 представляет собой водород в каждой из формул (II)-(VII), образующаяся группа карбоновой кислоты (-СООН) заменяется изостером карбоновой кислоты, как определено в данном документе.
В одном из вариантов осуществления Формулы (I) n равен 1, R1 представляет собой Н, Z представляет собой О, R представляет собой -(C=O)NHR15, -СН2ОН, -CH2NH2 или -CN, и соединение имеет структуру Формулы (VIII), (IX), (X) или (XI), соответственно:
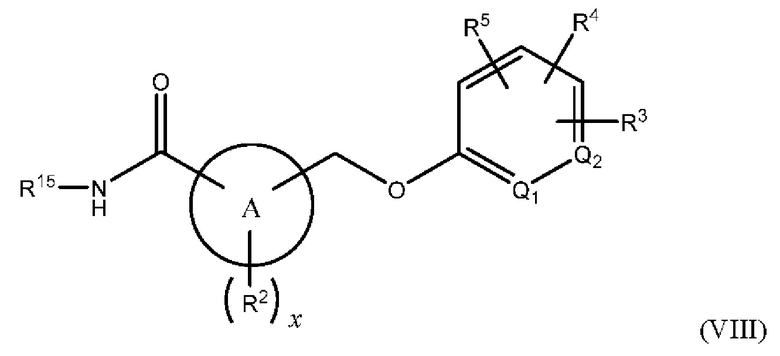
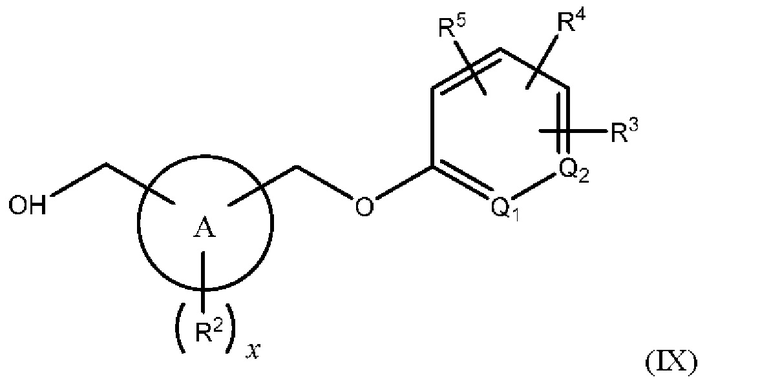
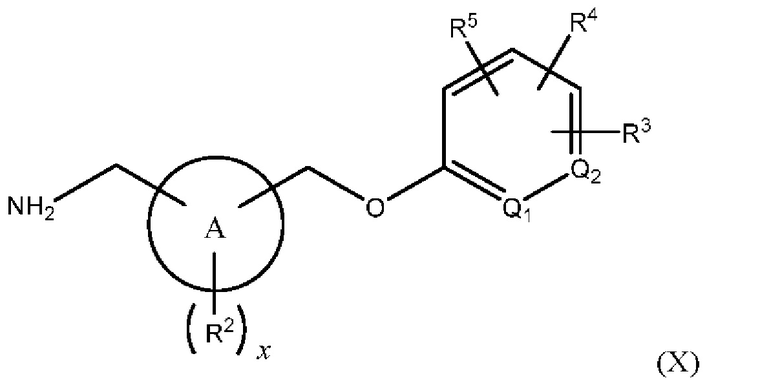
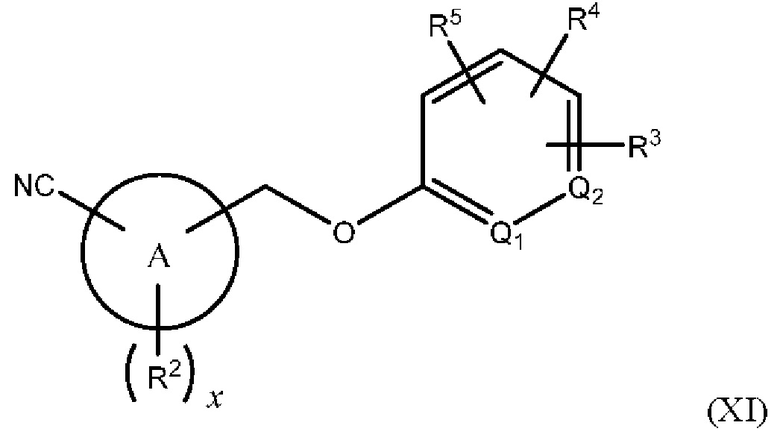
или представляет собой его фармацевтически приемлемый изомер, рацемат, гидрат, сольват, изотоп или соль, где х, A, Q1, Q2, R2, R3, R4, R5 и R15 имеют указанные выше значения.
В одном из вариантов осуществления Формулы (I) n равен О, Z представляет собой О, R представляет собой -(C=O)NHR15, -СН2ОН, -CH2NH2 или -CN, и соединение имеет структуру формулы (XII), (XIII), (XIV) или (XV), соответственно:
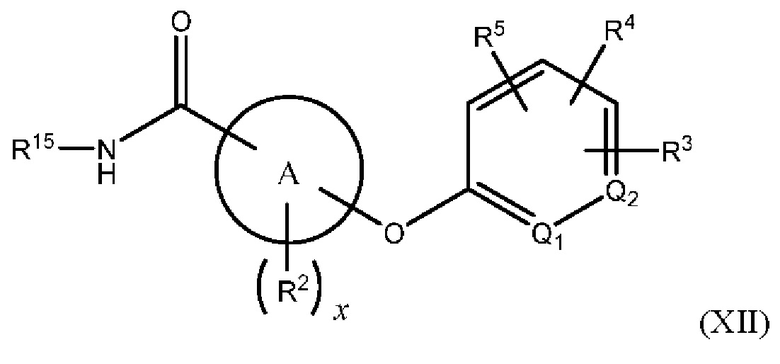
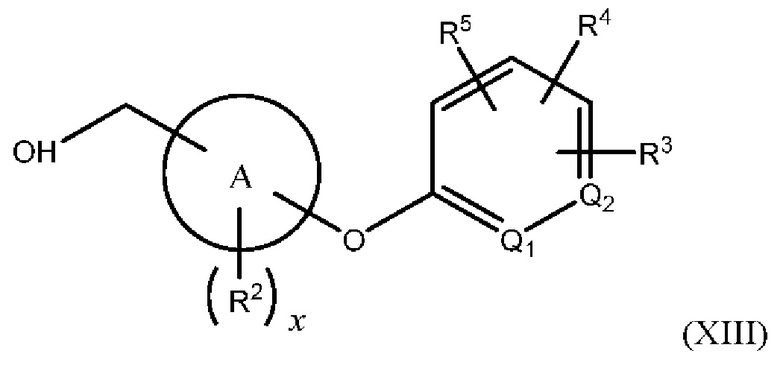
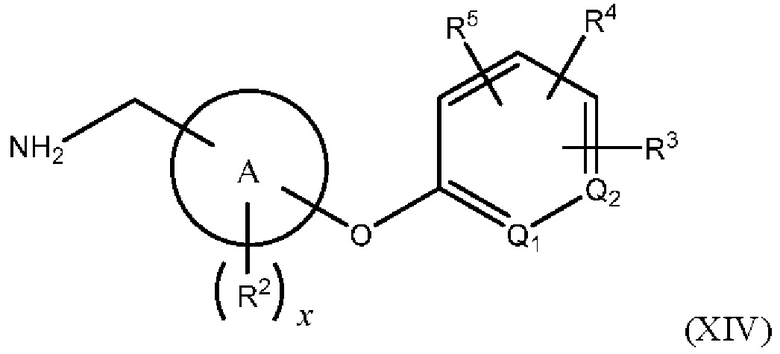
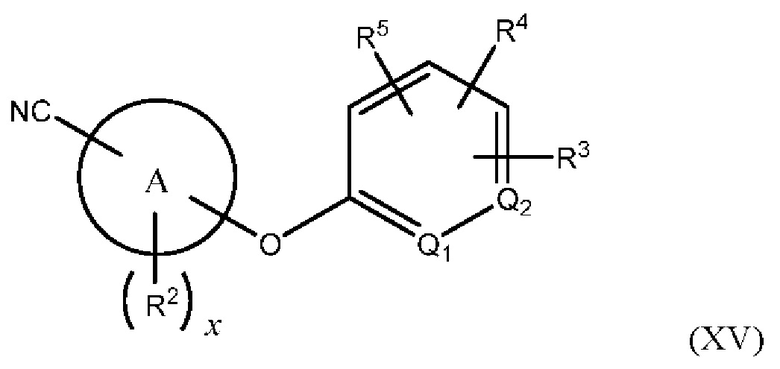
или представляет собой его фармацевтически приемлемый изомер, рацемат, гидрат, сольват, изотоп или соль, где х, A, Q1, Q2, R2, R3, R4, R5 и R15 имеют указанные выше значения.
В одном из вариантов осуществления Формулы (I) Z представляет собой -S-, N(R11)-, -СН2- или -С≡С-, и соединение имеет структуру Формулы (XVI), (XVII), (XVIII) ил (XIX), соответственно:
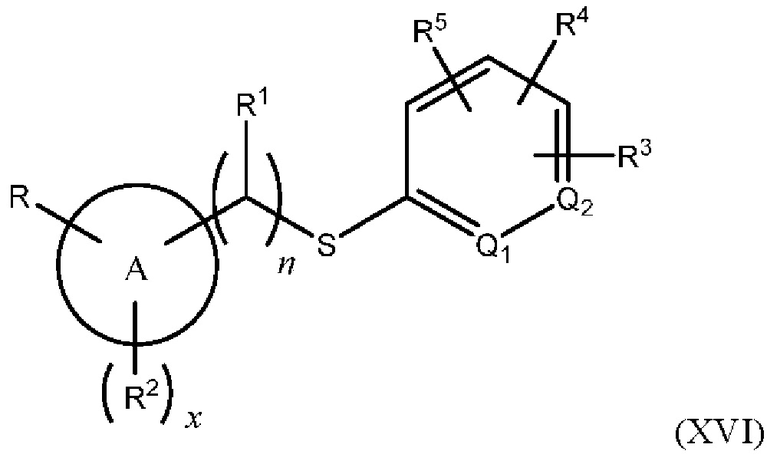
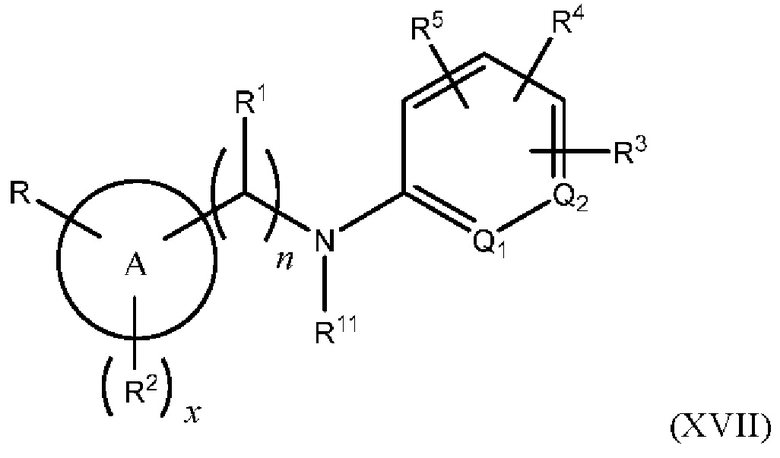
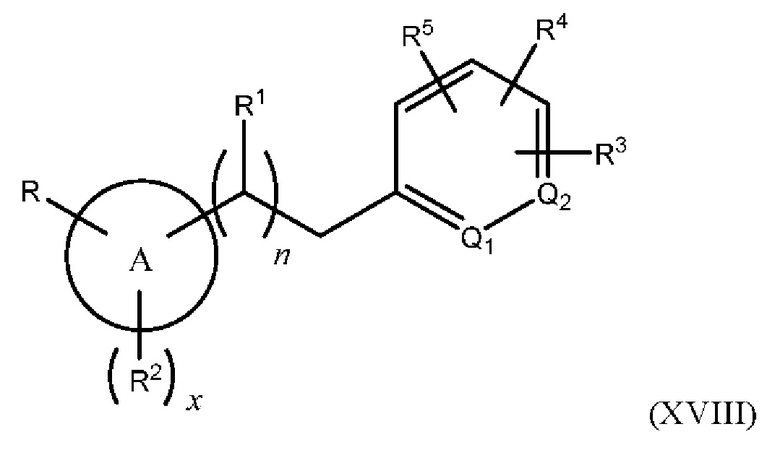
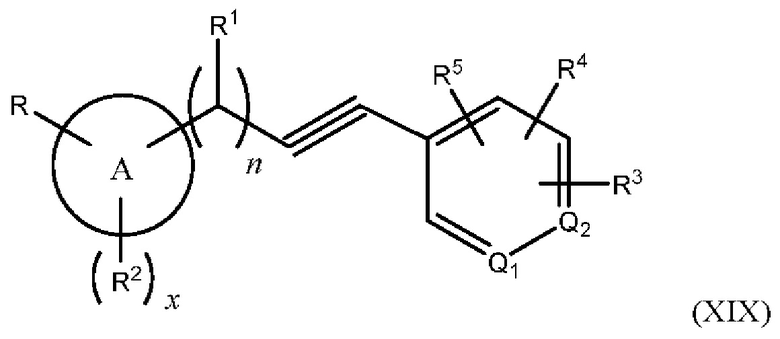
или представляет собой его фармацевтически приемлемый изомер, рацемат, гидрат, сольват, изотоп или соль, где n, х, A, Q1, Q2, R, R1, R2, R3, R4, R5 и R11 имеют указанные выше значения.
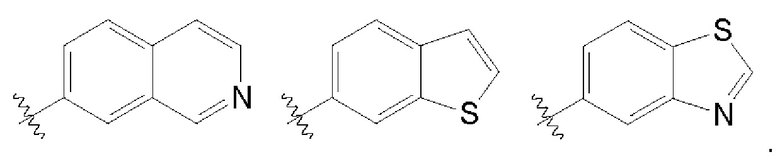
В одном варианте осуществления любой из формул (I)-(XIX) А представляет собой арил.
В одном из вариантов осуществления любой из формул (I)-(XIX) А представляет собой фенил.
В одном варианте осуществления любой из формул (I)-(XIX) А представляет собой фенил со следующими точками присоединения:
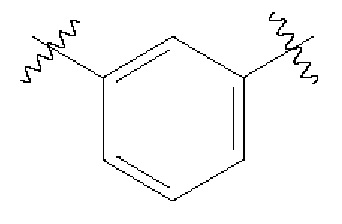
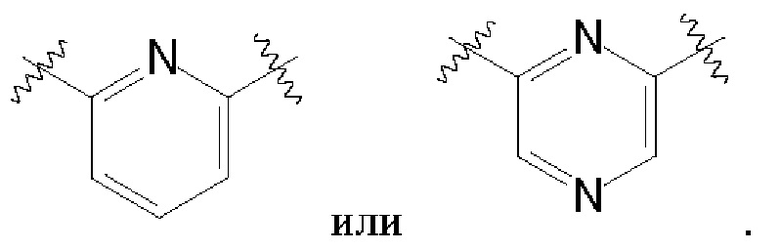
В одном варианте осуществления любой из формул (I)-(XIX) А представляет собой гетероарил.
В одном варианте осуществления любой из формул (I)-(XIX) А представляет собой пиридин или пиразин.
В одном варианте осуществления любой из формул (I)-(XIX), А представляет собой пиридин или пиразин со следующими точками присоединения, соответственно:
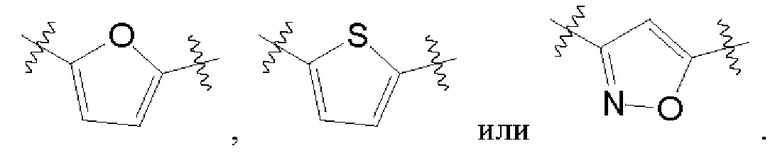
В одном варианте осуществления любой из формул (I)-(XIX) А представляет собой фуран, тиофен или изоксазол.
В одном варианте осуществления любой из формул (I)-(XIX), А представляет собой фуран, тиофен или изоксазол со следующими точками присоединения, соответственно:
В одном варианте осуществления любой из формул (I)-(XIX) Q1 и Q2 оба представляют собой СН.
В одном варианте осуществления любой из формул (I)-(XIX) Q1 представляет собой СН, и Q2 представляет собой N.
В одном варианте осуществления любой из формул (I)-(XIX) Q1 представляет собой N, и Q2 представляет собой СН.
В одном варианте осуществления любой из формул (I)-(XIX) R1 представляет собой водород.
В одном варианте осуществления любой из формул (I)-(XIX) R1 представляет собой алкил.
В одном варианте осуществления любой из формул (I)-(XIX) R1 представляет собой метил.
В одном из вариантов осуществления Формулы (I) соединение имеет структуру Формулы (XX):
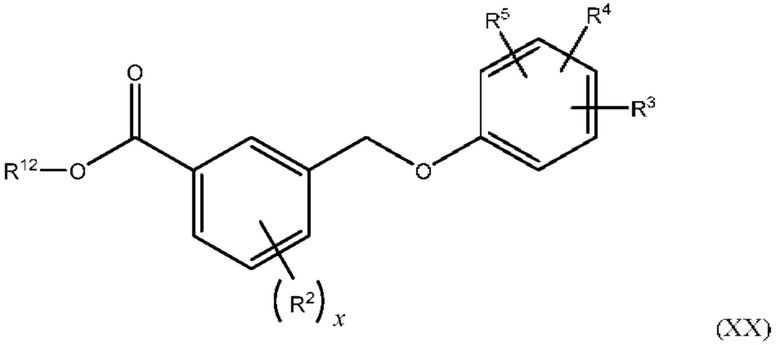
или представляет собой его фармацевтически приемлемый изомер, рацемат, гидрат, сольват, изотоп или соль, где х, R2, R3, R4, R5 и R12 имеют указанные выше значения.
В одном из вариантов осуществления Формулы (I) соединение имеет структуру Формулы (XXI):
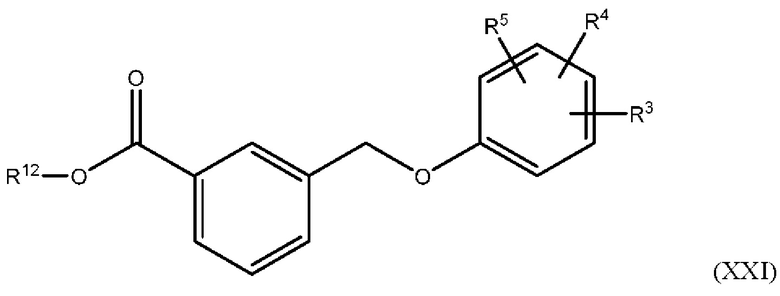
или представляет собой его фармацевтически приемлемый изомер, рацемат, гидрат, сольват, изотоп или соль, где R3, R4, R5 и R12 имеют указанные выше значения.
В одном из вариантов осуществления Формулы (I) соединение имеет структуру Формулы (XXII):
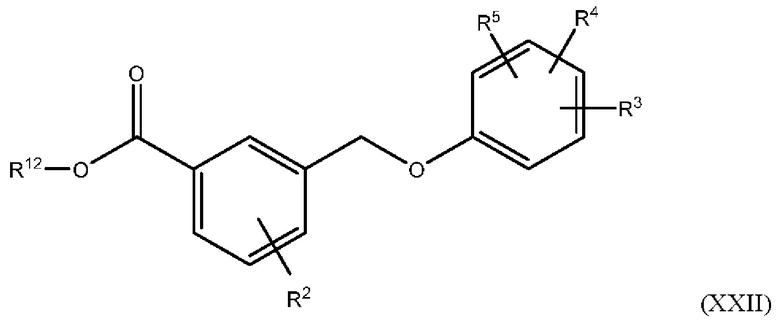
или представляет собой его фармацевтически приемлемый изомер, рацемат, гидрат, сольват, изотоп или соль, где R2, R3, R4, R5 и R12 имеют указанные выше значения.
В другом варианте осуществления, когда R12 представляет собой водород в каждой из вышеуказанных формул (ХХ)-(XXII), образующаяся группа карбоновой кислоты (-СООН) заменяется изостерой карбоновой кислоты, как определено в данном документе.
В одном из вариантов осуществления Формулы (I) соединение имеет структуру Формулы (XXIII):
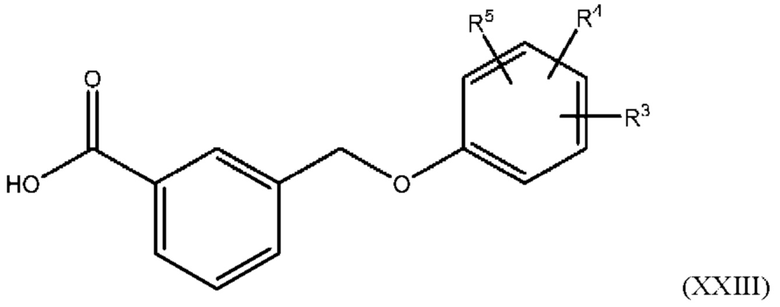
или представляет собой его фармацевтически приемлемый изомер, рацемат, гидрат, сольват, изотоп или соль, где R3, R4 и R5 имеют указанные выше значения.
В одном варианте осуществления Формулы (I) соединение имеет структуру Формулы (XXIV):
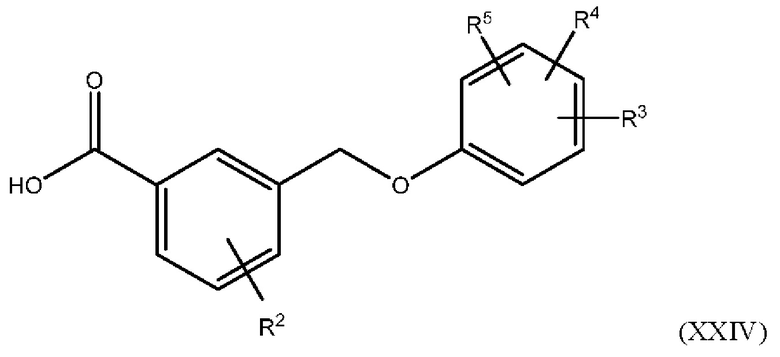
или представляет собой его фармацевтически приемлемый изомер, рацемат, гидрат, сольват, изотоп или соль, где R2, R3, R4 и R5 имеют указанные выше значения.
В другом варианте осуществления изобретения группа карбоновой кислоты (-СООН) каждой из формул (XXIII) и (XXIV) выше заменена изостером карбоновой кислоты, как определено в данном документе.
В одном варианте осуществления любой из формул (I)-(XXIV) n равен 0.
В одном варианте осуществления любой из формул (I)-(XXIV) n равен 1.
В одном варианте осуществления любой из формул (I)-(XXIV) х равен 0.
В одном варианте осуществления любой из формул (I)-(XXIV) х равен 1.
В одном варианте осуществления любой из формул (I)-(XXIV) х равен 2.
В одном варианте осуществления любой из формул (I)-(XXIV) А представляет собой арил.
В одном варианте осуществления любой из формул (I)-(XXIV) А представляет собой гетероарил.
В одном варианте осуществления любой из формул (I)-(XXIV) Z представляет собой -О-.
В одном варианте осуществления любой из формул (I)-(XXIV) Z представляет собой -S-.
В одном варианте осуществления любой из формул (I)-(XXIV) Z представляет собой -N(R11)-.
В одном варианте осуществления любой из формул (I)-(XXIV) Z представляет собой -СН2-.
В одном варианте осуществления любой из формул (I)-(XXIV) Z представляет собой или -С≡С-.
В одном варианте осуществления любой из формул (I)-(XXIV) R представляет собой -(CH2)mC(=O)OR12.
В одном варианте осуществления любой из формул (I)-(XXIV) R представляет собой -(CH2)mNHR13.
В одном варианте осуществления любой из формул (I)-(XXIV) R представляет собой -(C=O)NR14R15.
В одном варианте осуществления любой из формул (I)-(XXIV) R представляет собой -СН2ОН.
В одном варианте осуществления любой из формул (I)-(XXIV) R представляет собой -CN.
В одном варианте осуществления любой из формул (I)-(XXIV) R представляет собой галоидалкил.
В одном варианте осуществления любой из формул (I)-(XXIV) R представляет собой карбоцикл.
В одном варианте осуществления любой из формул (I)-(XXIV) R представляет собой гетероцикл.
В одном варианте осуществления любой из формул (I)-(XXIV) m равен 0.
В одном варианте осуществления любой из формул (I)-(XXIV) m равен 1.
В одном варианте осуществления любой из формул (I)-(XXIV), R14 представляет собой Н, и R15 представляет собой Н, -SO2CH3, карбоцикл, гетероцикл или алкил, замещенный 0, 1, 2 или 3 заместителями, выбранными из -ОН, -CN, -NR'R", С(=O)ОН, C(=O)NR'R", -SO2OH, алкокси, карбоцикла или гетероцикла, где R' и R" по отдельности представляют собой Н или алкил.
В одном варианте осуществления любой из формул (I)-(XXIV), R14 и R15, вместе с атомом азота, к которому они присоединены, образуют гетероцикл.
В одном варианте осуществления любой из формул (I)-(XXIV), R1 представляет собой Н.
В одном варианте осуществления любой из формул (I)-(XXIV), R1 представляет собой алкил.
В одном варианте осуществления любой из формул (I)-(XXIV), R2 представляет собой галоген.
В одном варианте осуществления любой из формул (I)-(XXIV), R2 представляет собой циано.
В одном варианте осуществления любой из формул (I)-(XXIV), R2 представляет собой амино.
В одном варианте осуществления любой из формул (I)-(XXIV), R2 представляет собой алкил.
В одном варианте осуществления любой из формул (I)-(XXIV), R2 представляет собой алкокси.
В одном варианте осуществления любой из формул (I)-(XXIV), R2 представляет собой карбоцикл.
В одном варианте осуществления любой из формул (I)-(XXIV), R2 представляет собой гетероцикл.
В одном варианте осуществления любой из формул (I)-(XXIV), R3, R4 и R5 являются одинаковыми или разными и либо отсутствуют, либо, если они присутствуют, представляют собой циано, цианоалкил, нитро, галоген, алкил, галоидалкил, алкокси, галоидалкокси, -(С=O)алкил, -(С=O)NH-алкил, карбоцикл, гетероцикл, -О-карбоцикл или -О-гетероцикл.
В одном варианте осуществления любой из Формул (I)-(XXIV), R3, R4 и R5 являются одинаковыми или разными и либо отсутствуют, либо, если они присутствуют, представляют собой циано, нитро, галоген, алкил, галоидалкил, алкокси или галоалкокси.
В одном варианте осуществления любой из формул (I)-(XXIV), R3, R4 и R5 являются одинаковыми или разными и либо отсутствуют, либо, в случае присутствия, представляют собой -CN, -NO2, -F, -Cl, -Br, -СН3, -CF3, -CHF2, -С(СН3)3, -ОСН3 или -OCF3.
В одном варианте осуществления любой из формул (I)-(XXIV) любые два из R3, R4 и R5, взятые вместе с атомами, к которым они присоединены, образуют карбоцикл или гетероцикл, который является незамещенным или замещен 1, 2 или 3 заместителями, независимо выбранными из галогена, гидроксила, оксо, галоида, алкила, галоидалкила, алкокси, галоидалкокси, карбоцикла или гетероцикла.
В одном варианте осуществления любой из формул (I)-(XXIV), R3 и R4, взятые вместе с атомами, к которым они присоединены, образуют гетероцикл, как показано ниже, который является незамещенным или замещен 1, 2 или 3 заместителями, независимо выбранными из галогена, гидроксила, оксо, галоида, алкила, галоидалкила, алкокси, галоидалкокси, карбоцикла или гетероцикла:
В одном варианте осуществления любой из формул (I)-(XXIV), R3 и R4, взятые вместе с атомами, к которым они присоединены, образуют карбоцикл, как показано ниже, который является незамещенным или замещен 1, 2 или 3 заместителями, независимо выбранными из галогена, оксо, галоида, алкила, галоидалкила, алкокси, галоидалкокси, карбоцикла или гетероцикла:
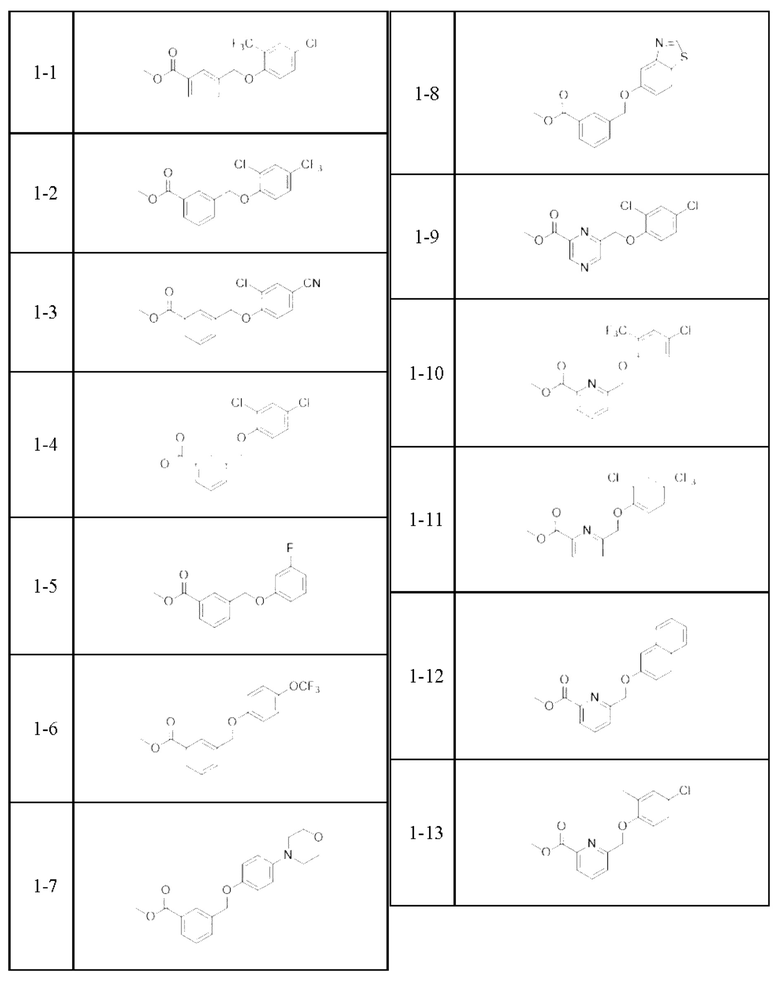
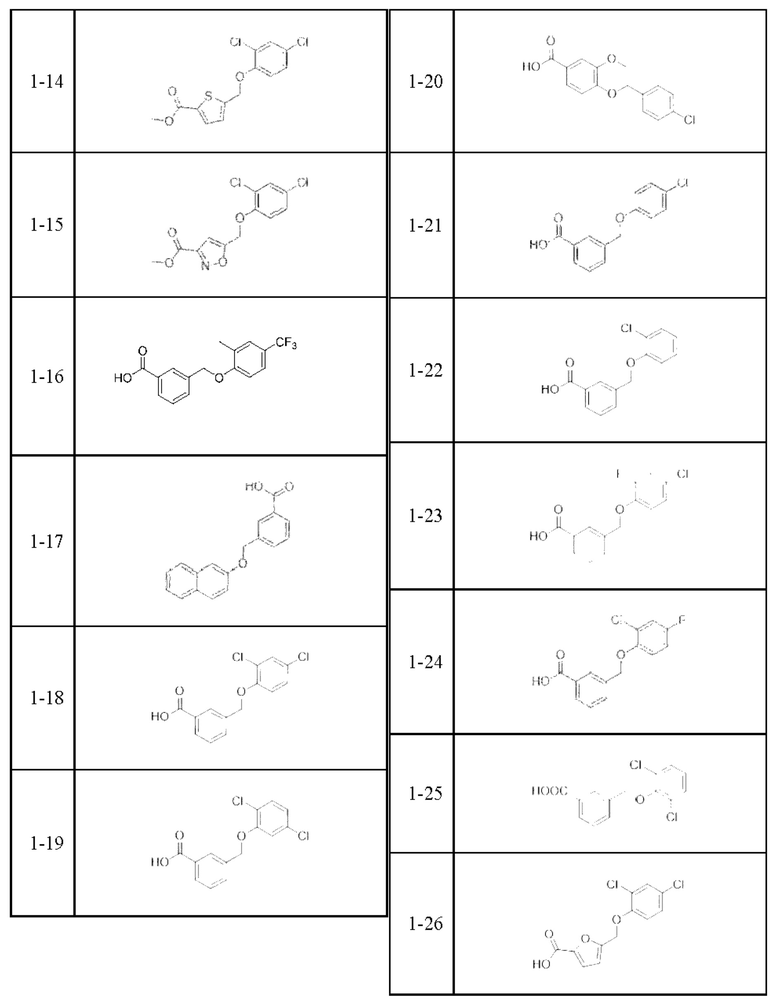
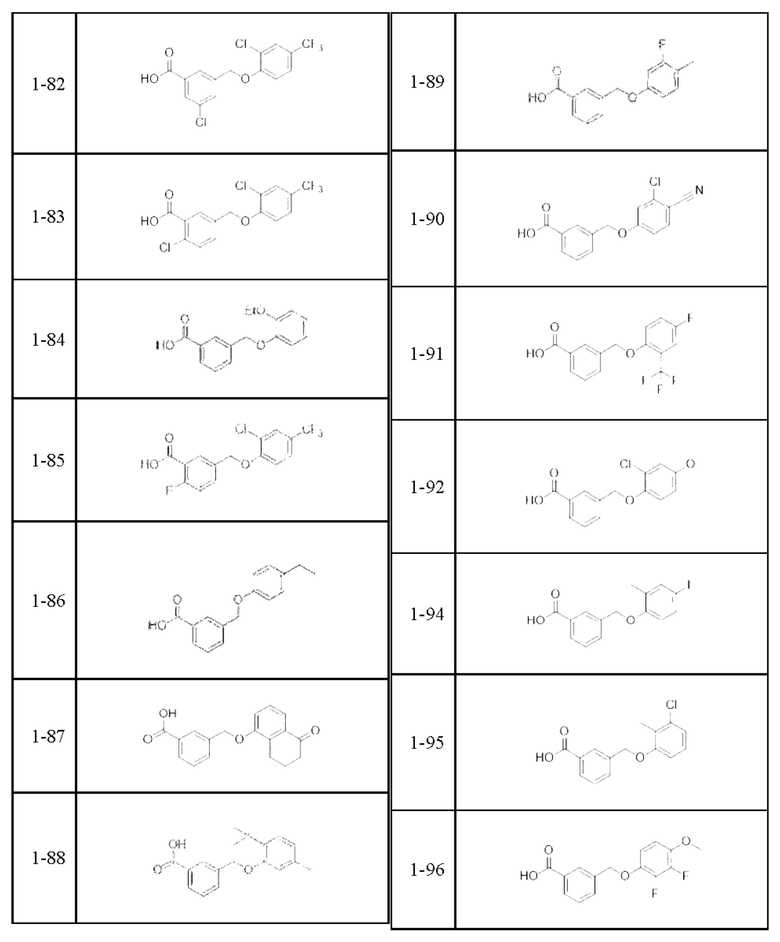
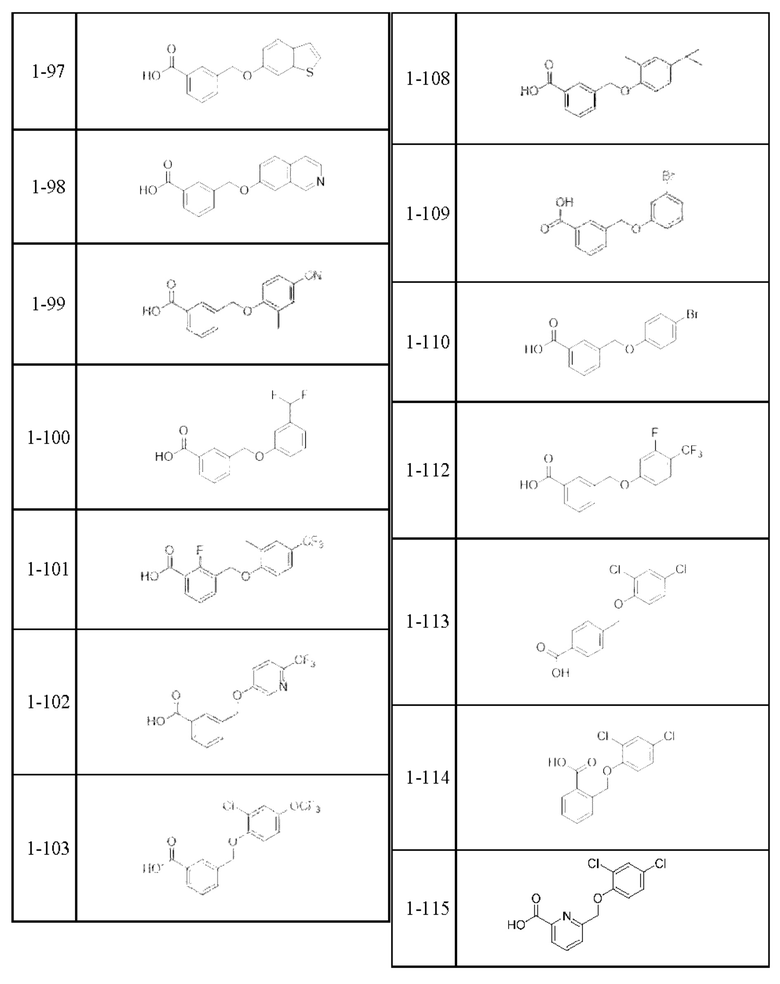
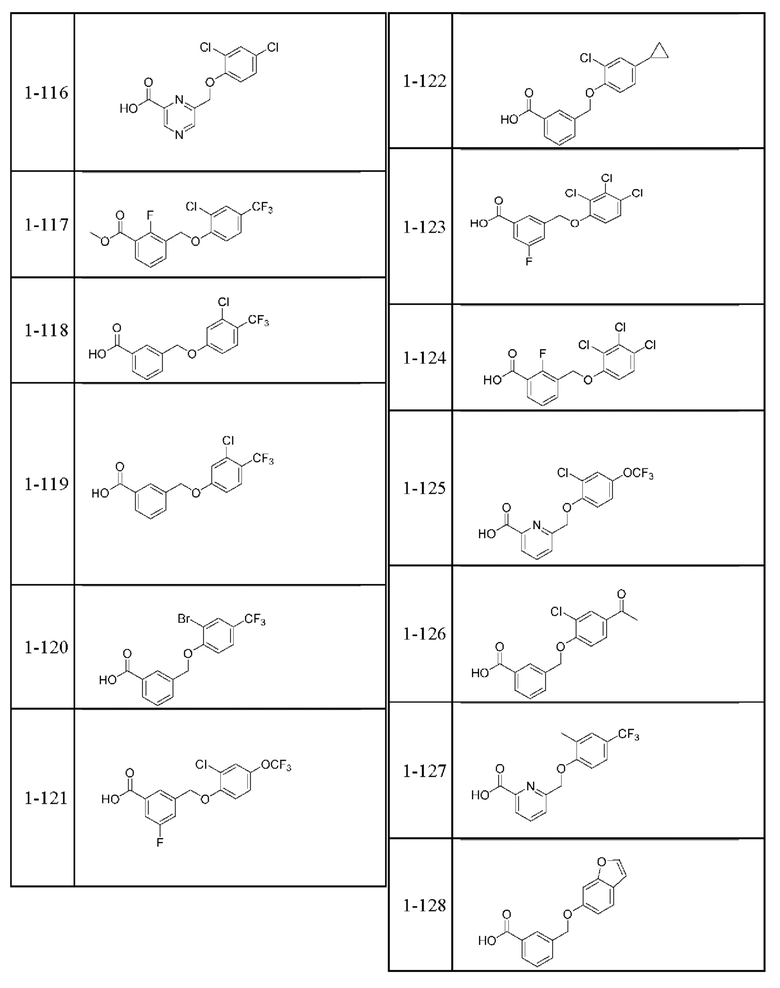
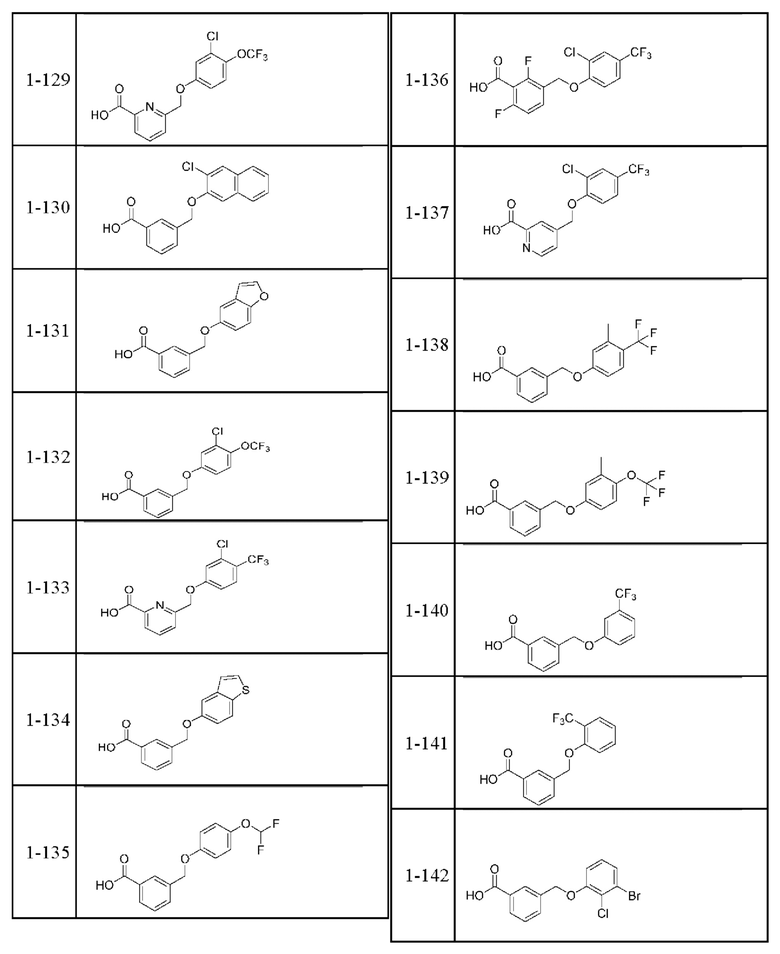
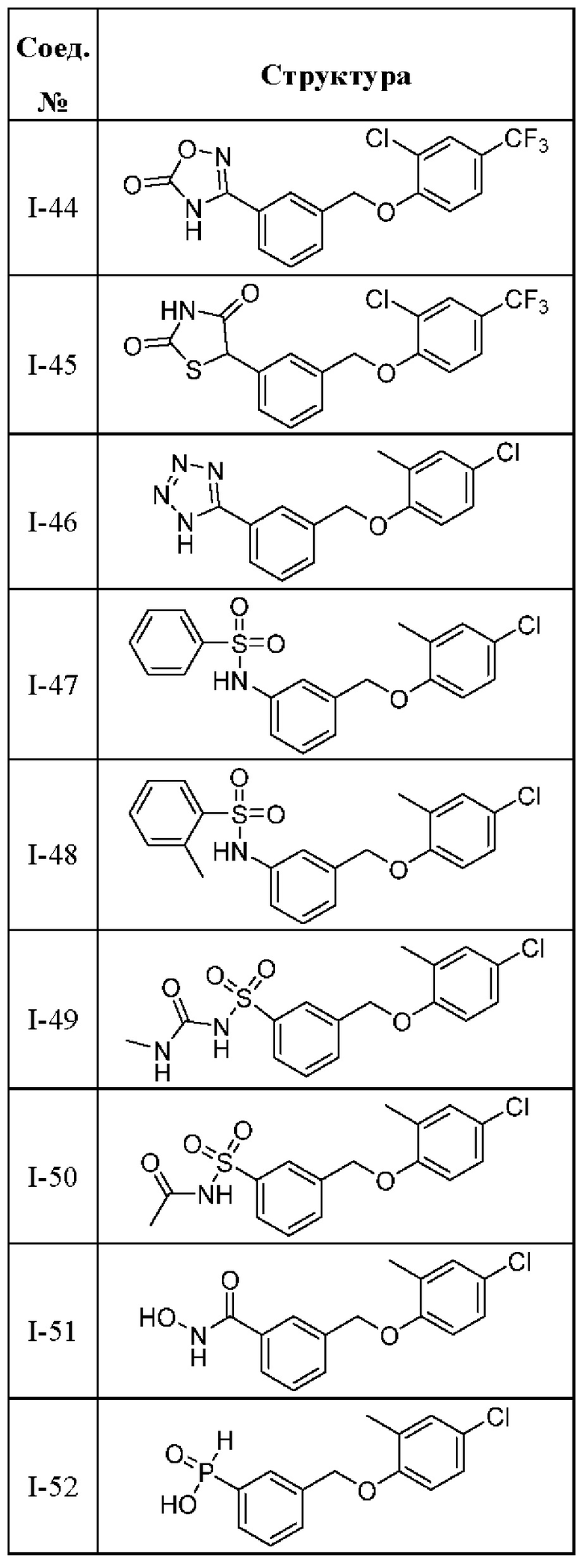
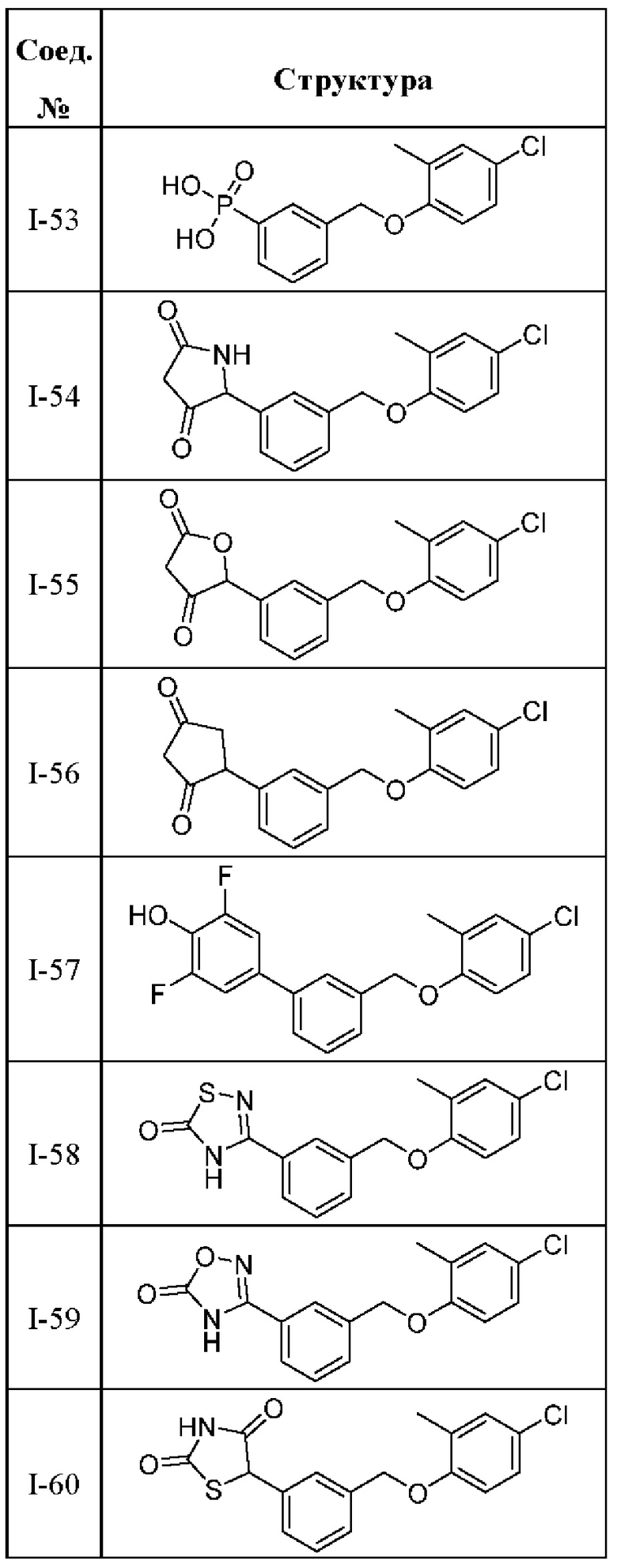
Типичные соединения формулы (I), а также формул (II)-(XXIV), когда это применимо, включают любое из соединений, перечисленных в Таблице А ниже, а также его фармацевтически приемлемый изомер, рацемат, гидрат, сольват, изотоп или соль. С этой целью репрезентативные соединения обозначаются в данном документе с помощью соответствующего «номера соединения», который иногда сокращенно указывается как «Соединение №» или «Соед. №».

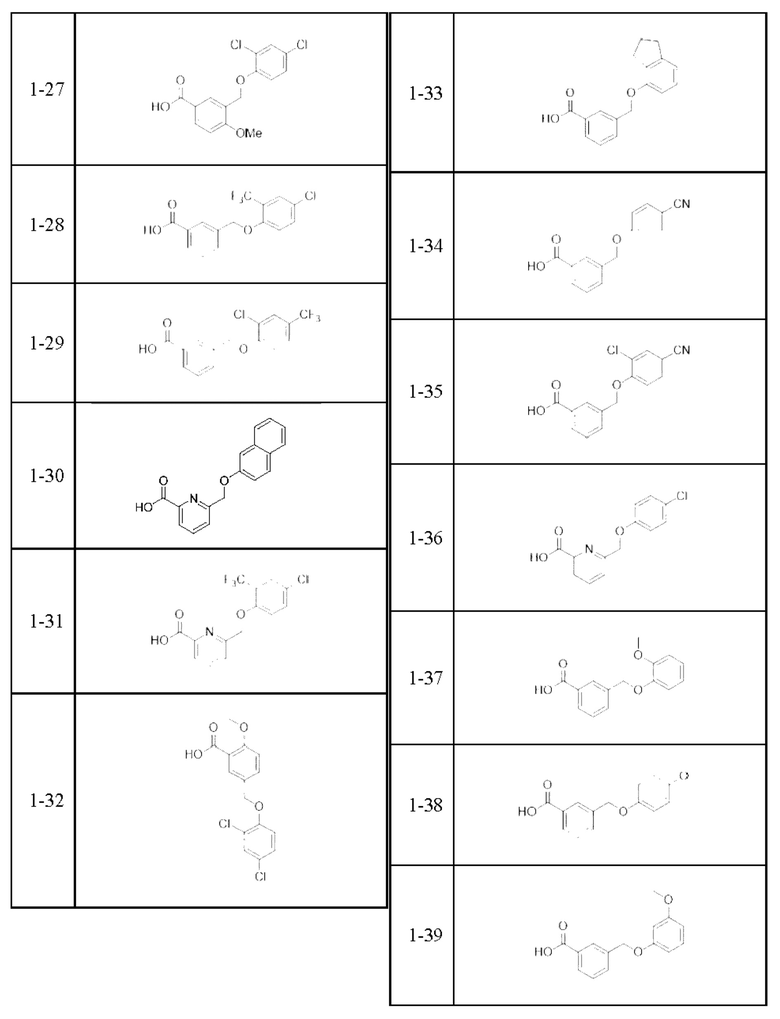
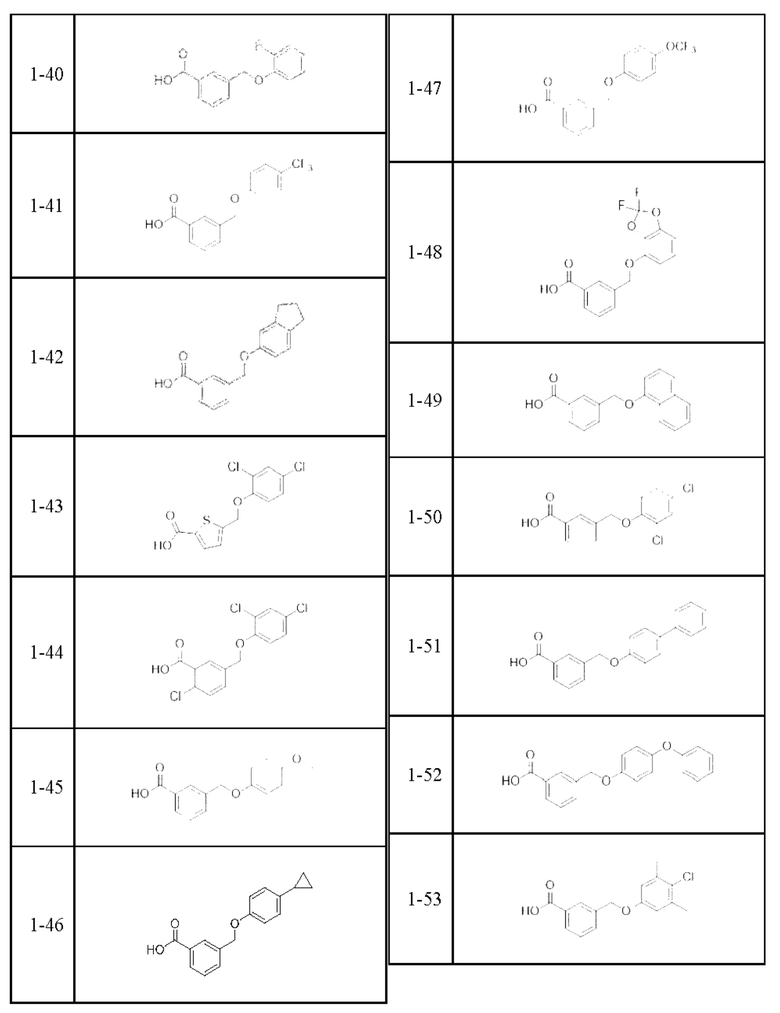
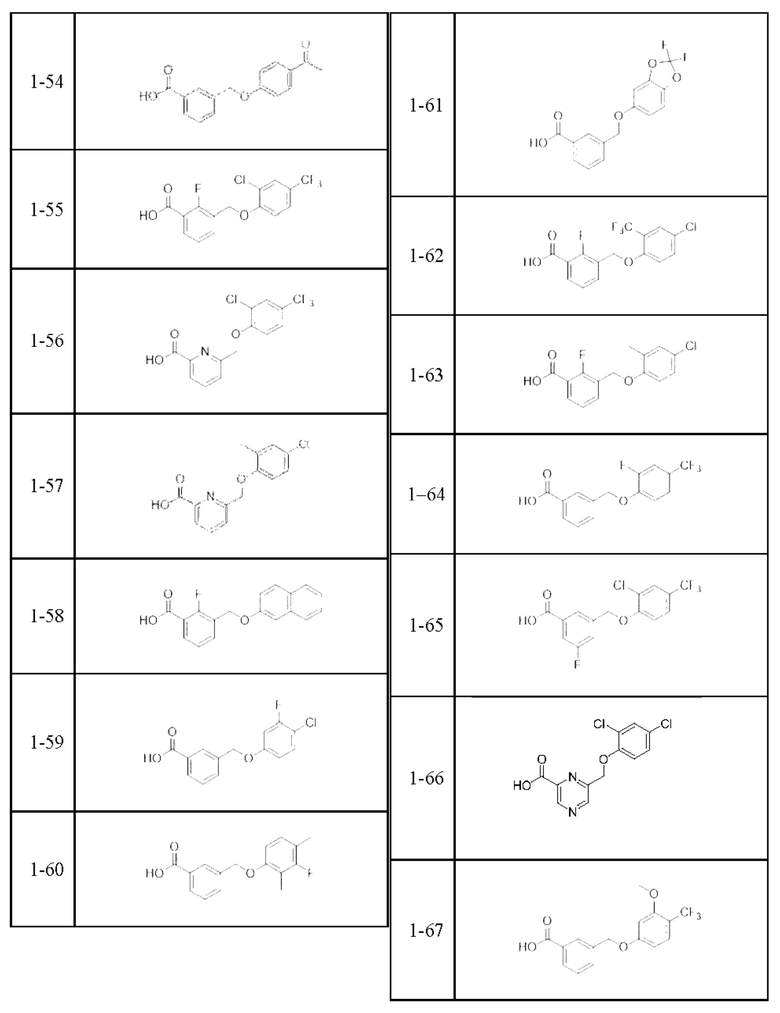
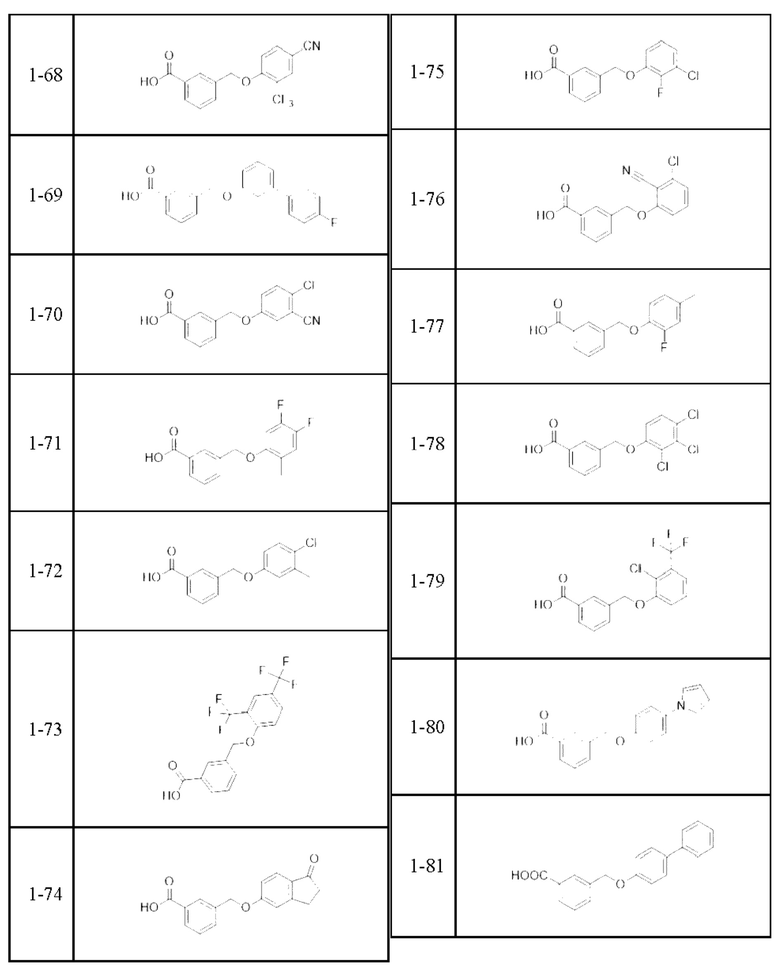
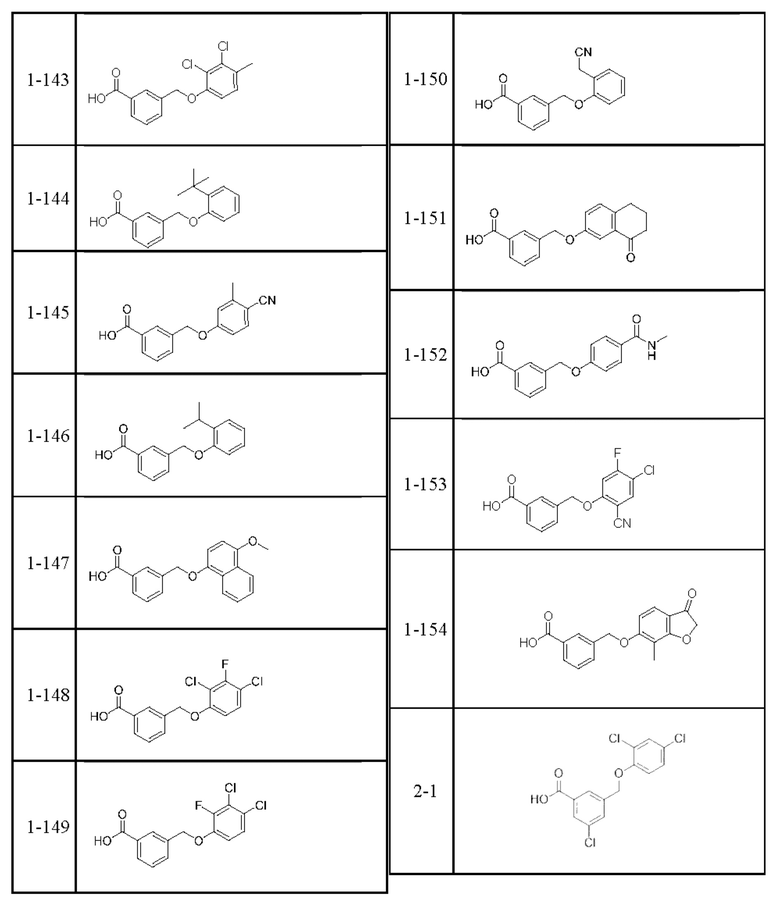
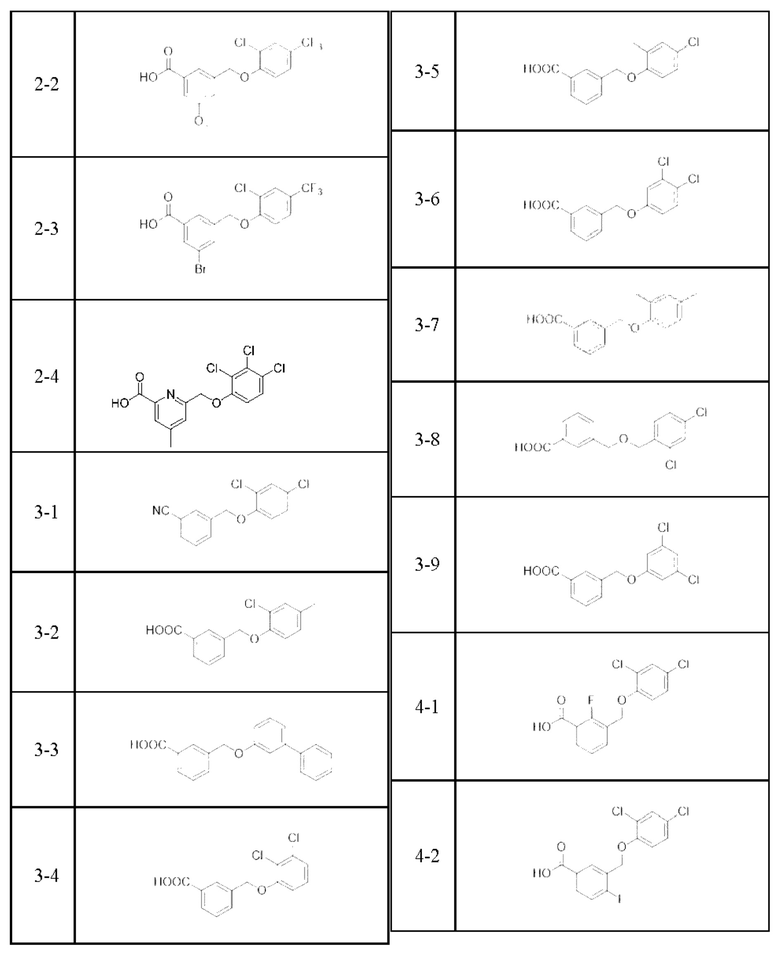
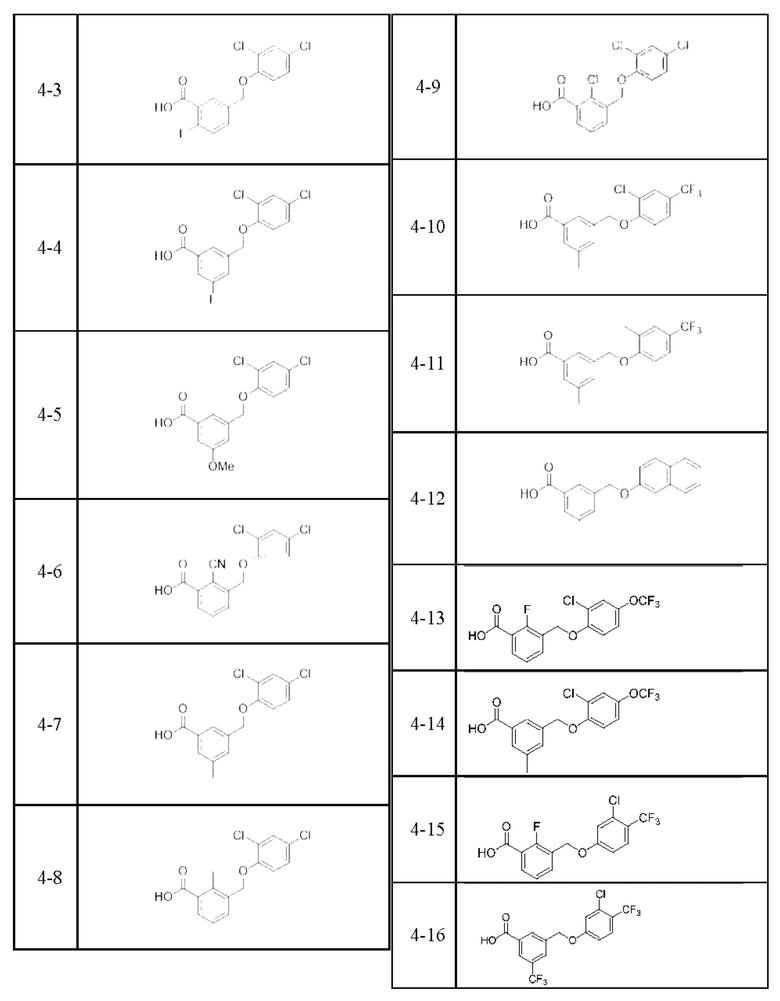
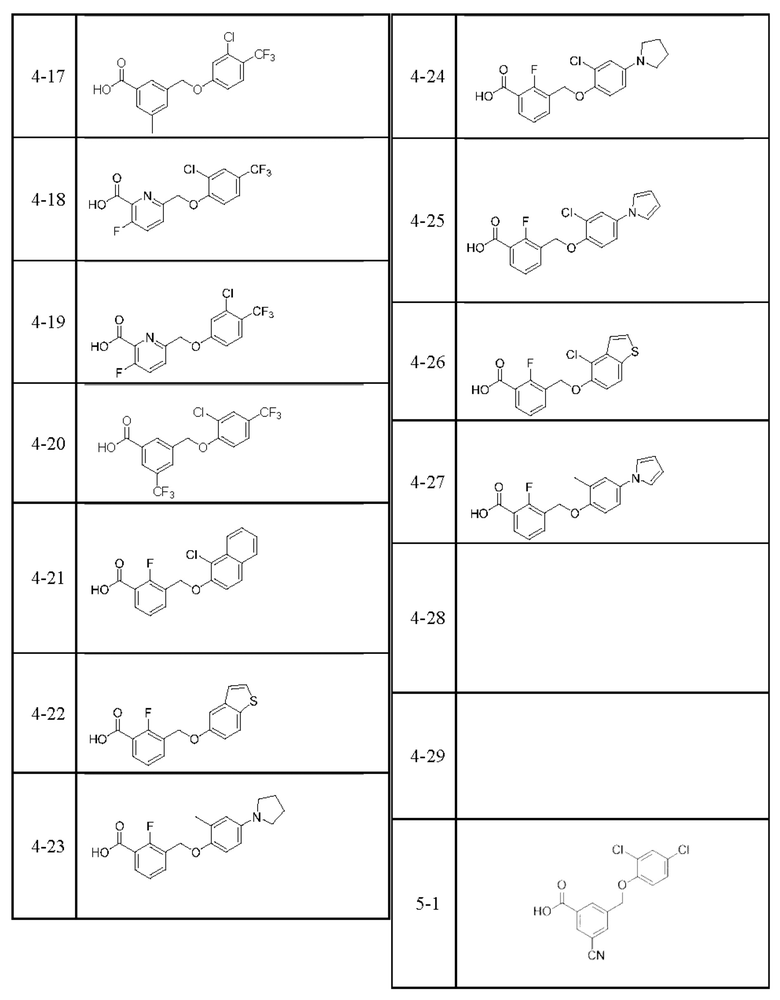
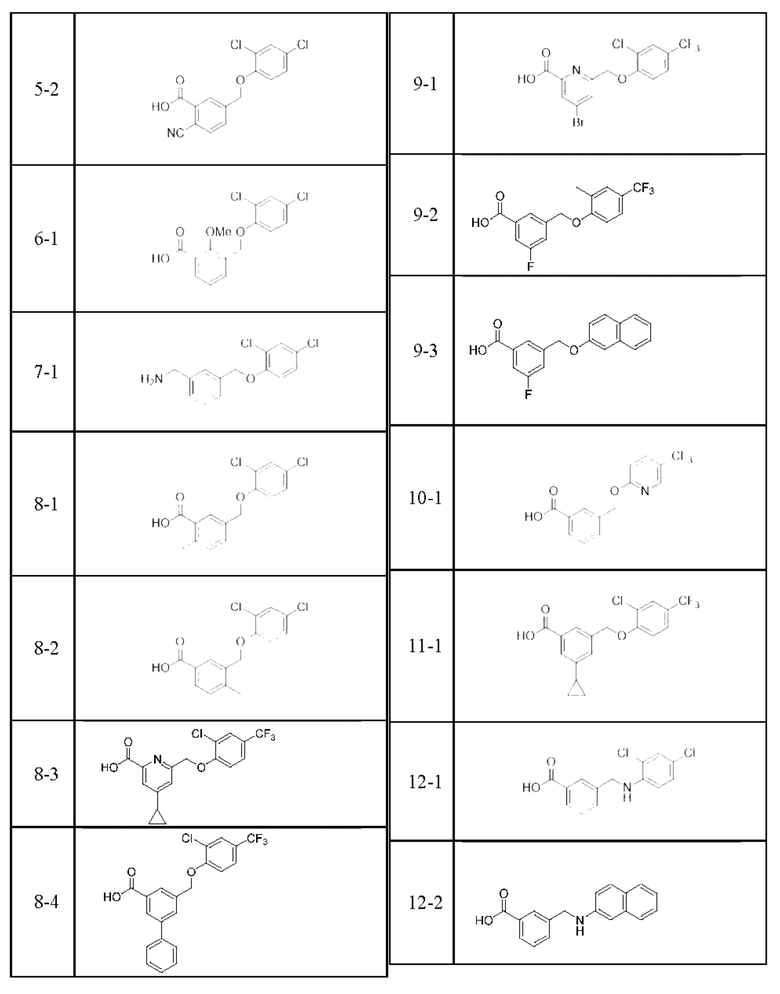
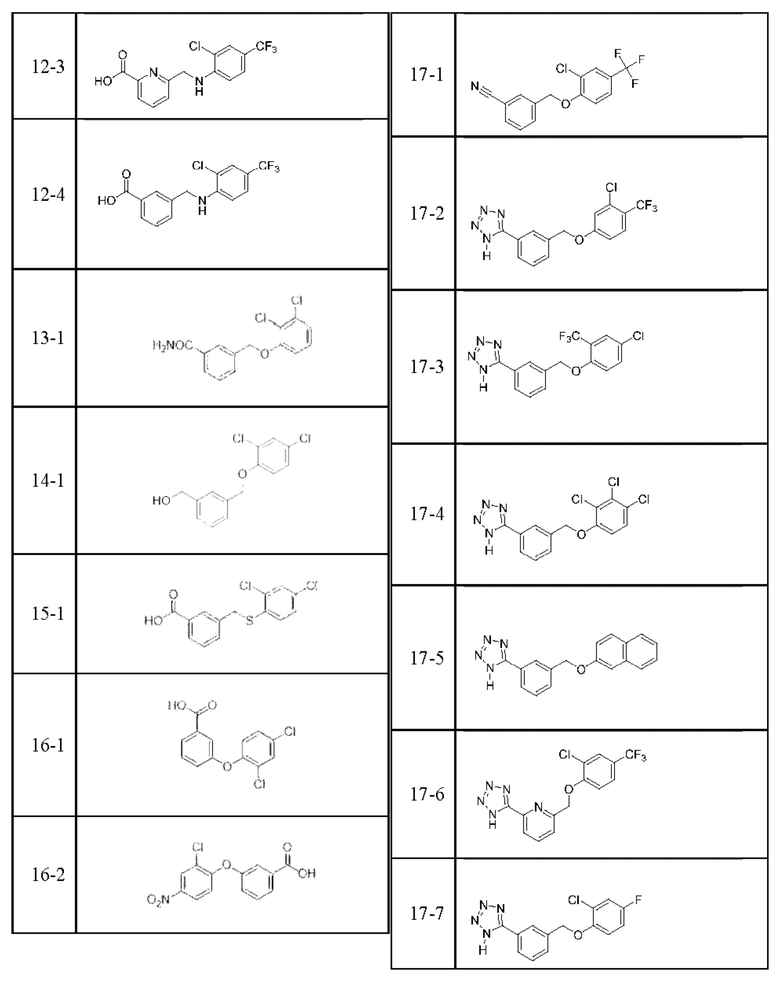
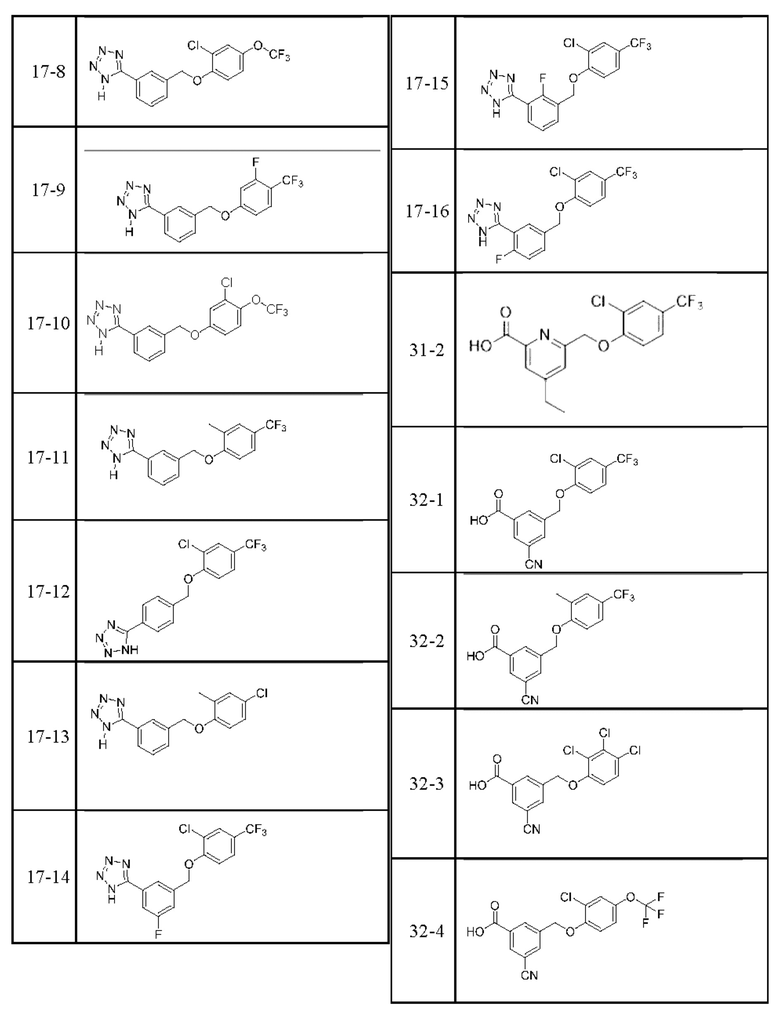
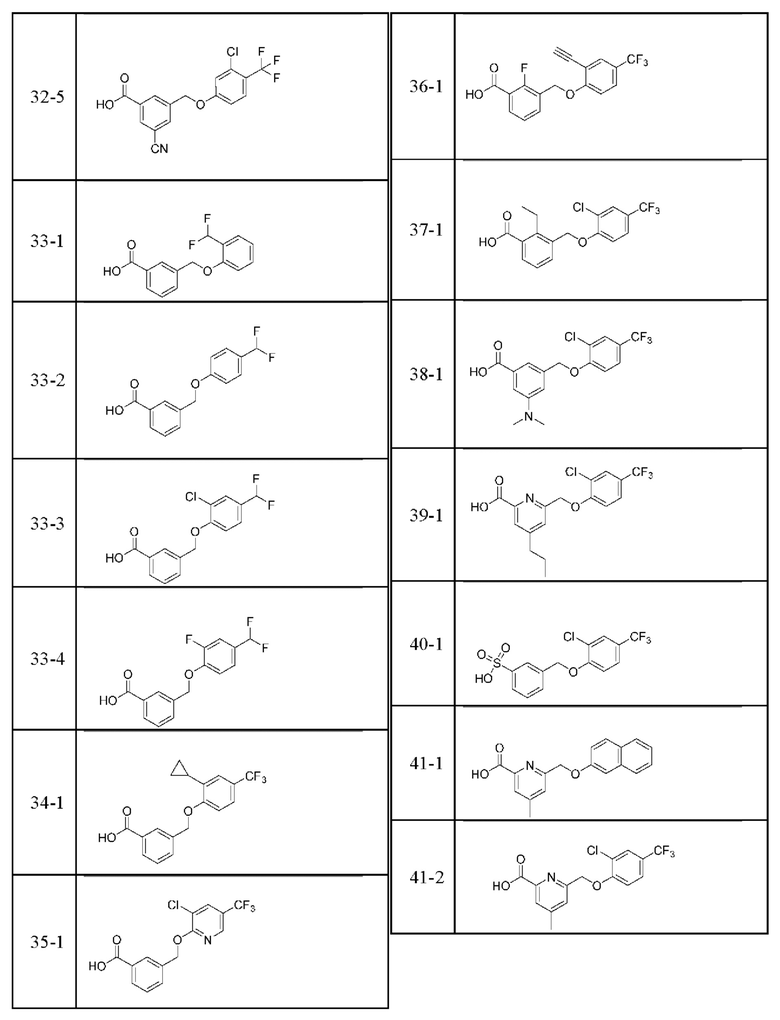
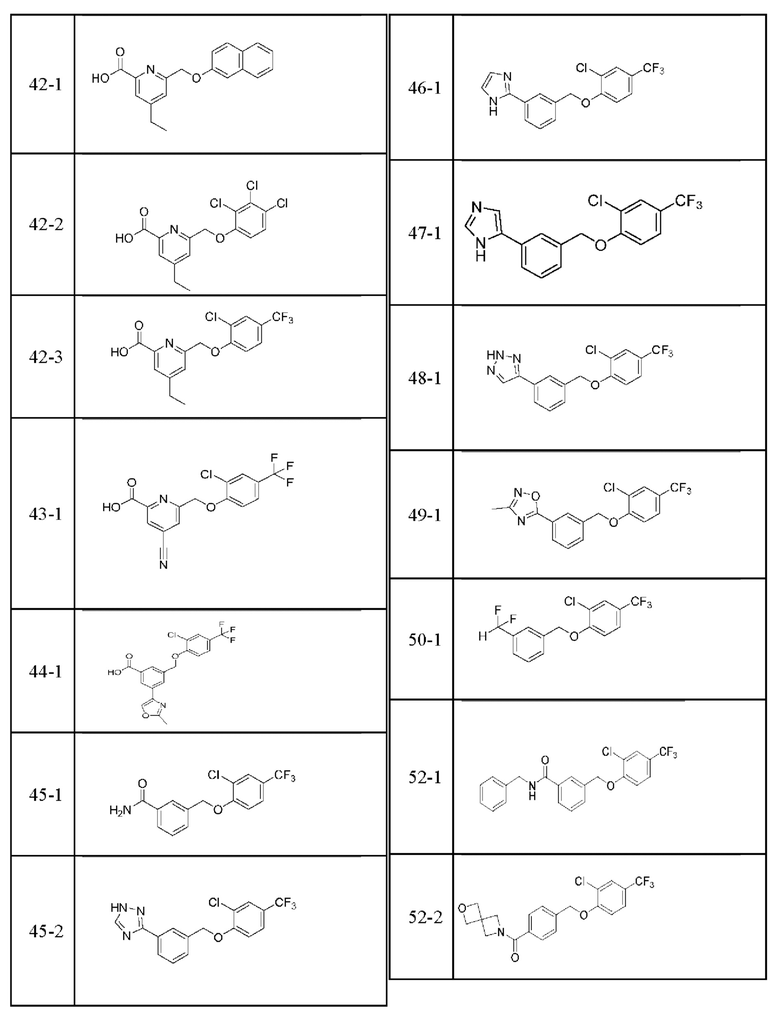
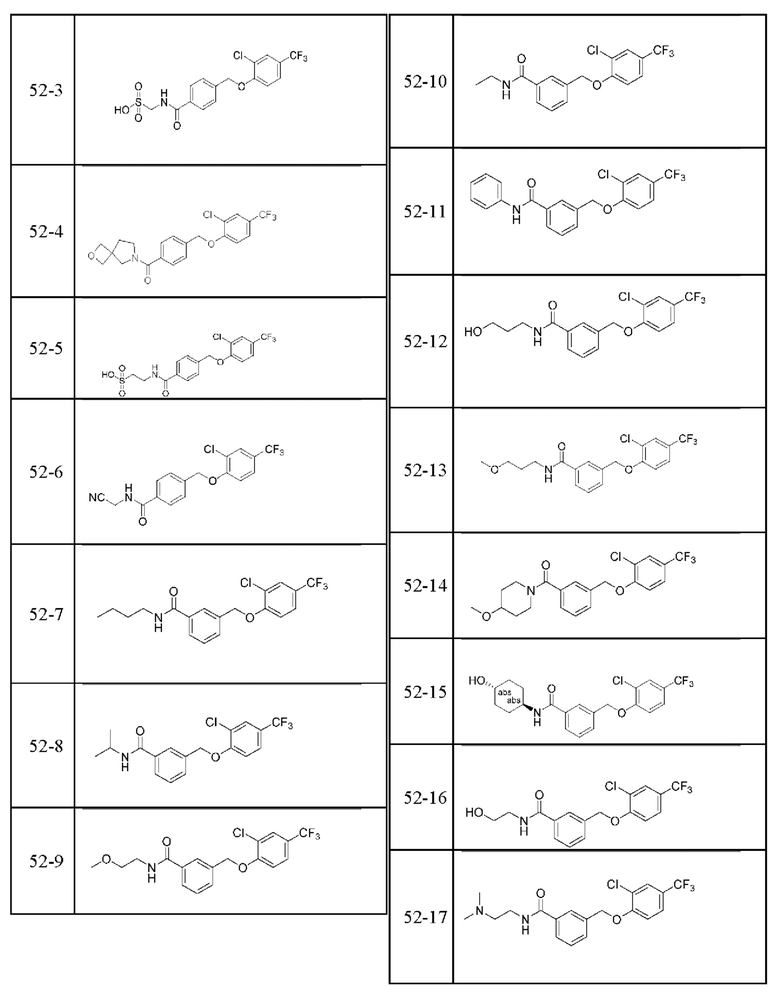
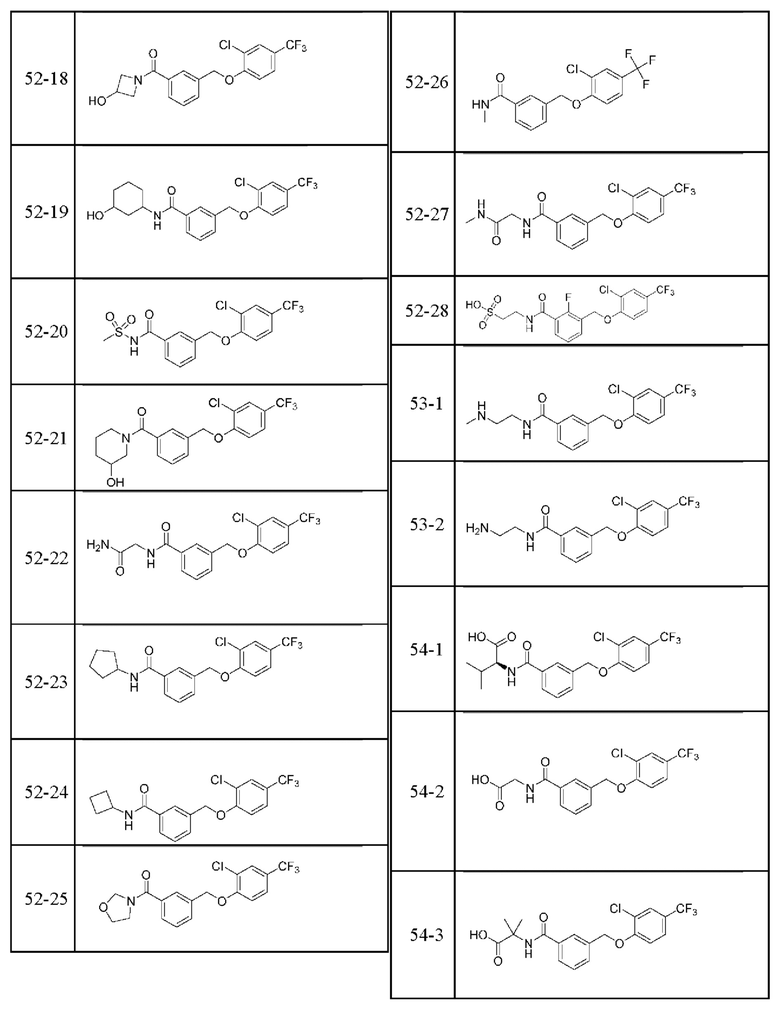
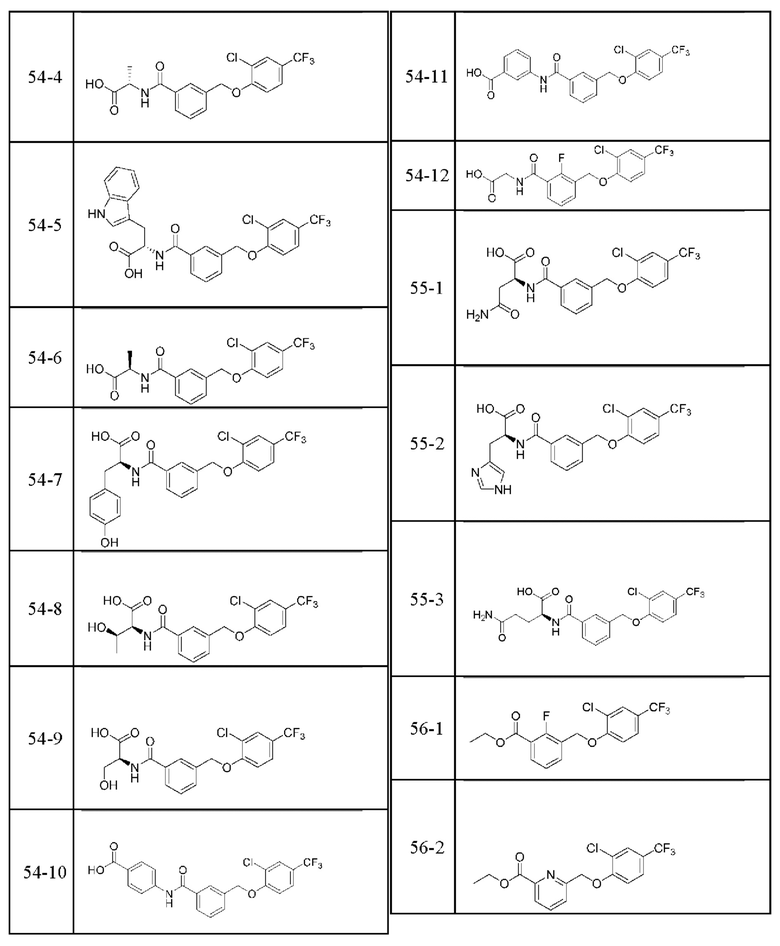
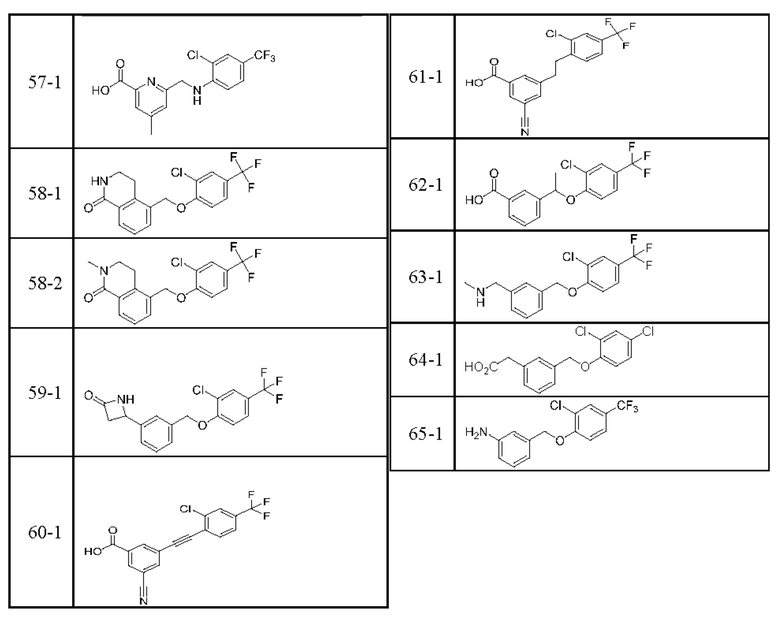
В более конкретном варианте осуществления изобретения соединение имеет следующую структуру или представляет собой его фармацевтически приемлемый изомер, рацемат, гидрат, сольват, изотоп или соль:
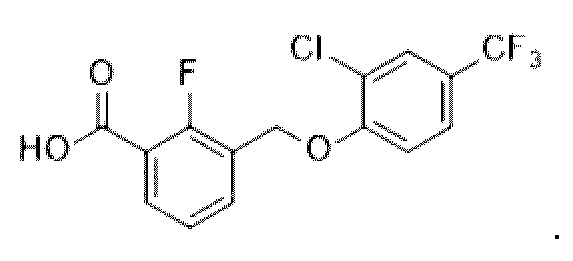
В более конкретном варианте осуществления изобретения соединение имеет следующую структуру или представляет собой его фармацевтически приемлемый изомер, рацемат, гидрат, сольват, изотоп или соль:
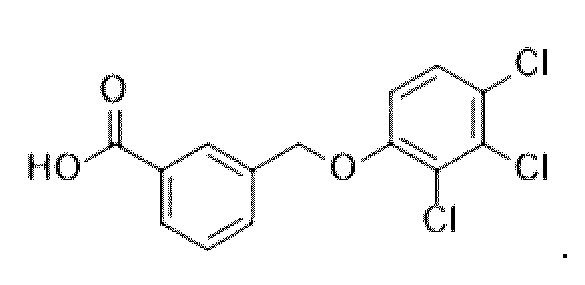
В более конкретном варианте осуществления изобретения соединение имеет следующую структуру или представляет собой его фармацевтически приемлемый изомер, рацемат, гидрат, сольват, изотоп или соль:
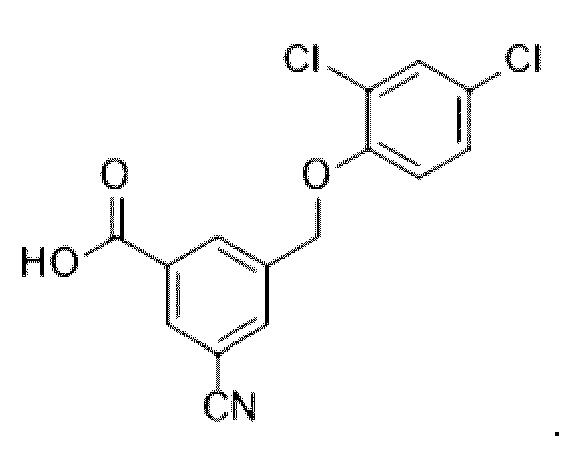
В более конкретном варианте осуществления изобретения соединение имеет следующую структуру или представляет собой его фармацевтически приемлемый изомер, рацемат, гидрат, сольват, изотоп или соль:
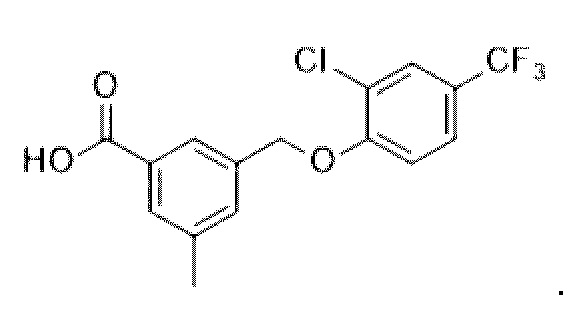
В более конкретном варианте осуществления изобретения соединение имеет следующую структуру или представляет собой его фармацевтически приемлемый изомер, рацемат, гидрат, сольват, изотоп или соль:
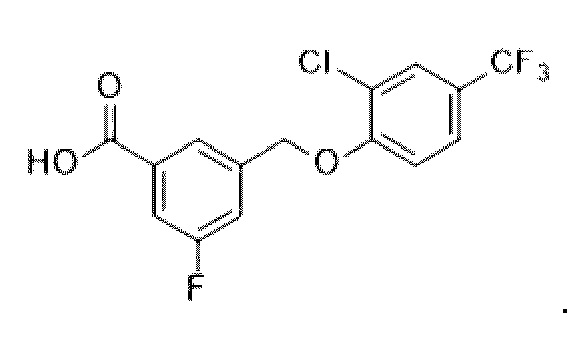
В более конкретном варианте осуществления изобретения соединение имеет следующую структуру или представляет собой его фармацевтически приемлемый изомер, рацемат, гидрат, сольват, изотоп или соль:
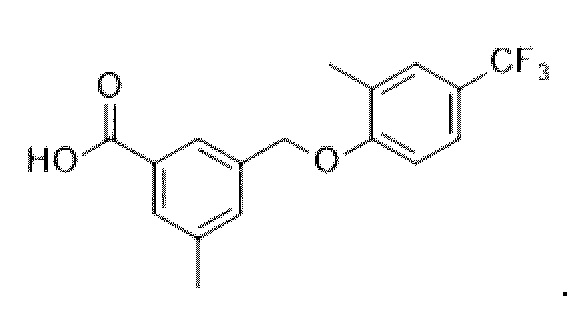
В более конкретном варианте осуществления изобретения соединение имеет следующую структуру или представляет собой его фармацевтически приемлемый изомер, рацемат, гидрат, сольват, изотоп или соль:
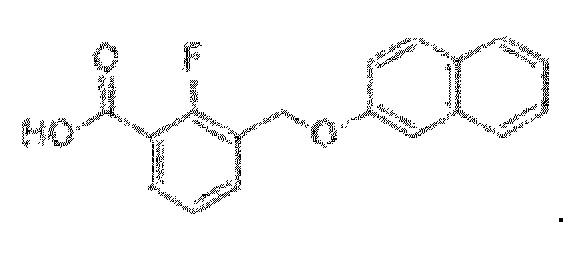
В более конкретном варианте осуществления изобретения соединение имеет следующую структуру или представляет собой его фармацевтически приемлемый изомер, рацемат, гидрат, сольват, изотоп или соль:
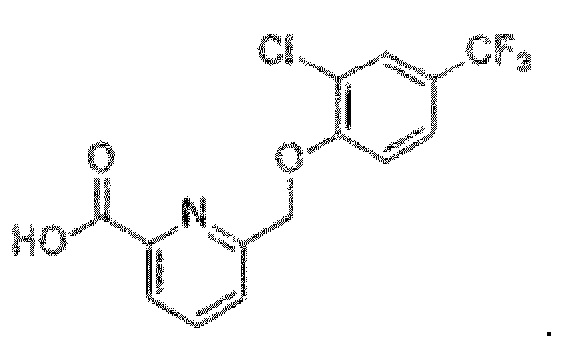
В более конкретном варианте осуществления изобретения соединение имеет следующую структуру или представляет собой его фармацевтически приемлемый изомер, рацемат, гидрат, сольват, изотоп или соль:
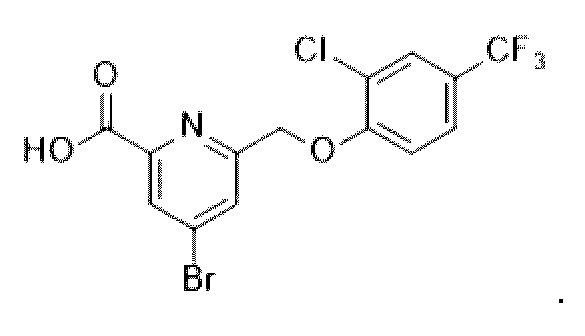
В более конкретном варианте осуществления изобретения соединение имеет следующую структуру или представляет собой его фармацевтически приемлемый изомер, рацемат, гидрат, сольват, изотоп или соль:
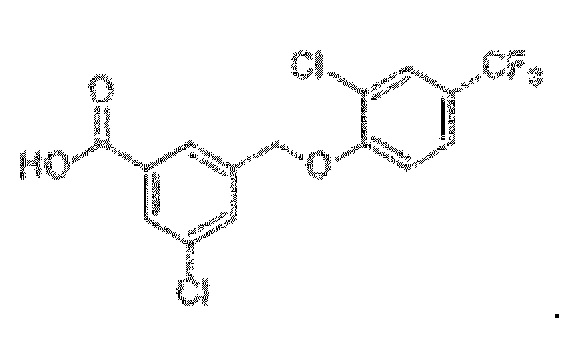
В более конкретном варианте осуществления изобретения соединение имеет следующую структуру или представляет собой его фармацевтически приемлемый изомер, рацемат, гидрат, сольват, изотоп или соль:
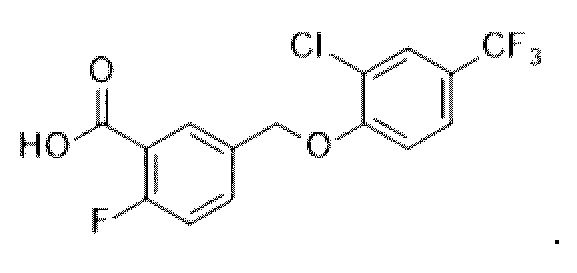
В более конкретном варианте осуществления изобретения соединение имеет следующую структуру или представляет собой его фармацевтически приемлемый изомер, рацемат, гидрат, сольват, изотоп или соль:
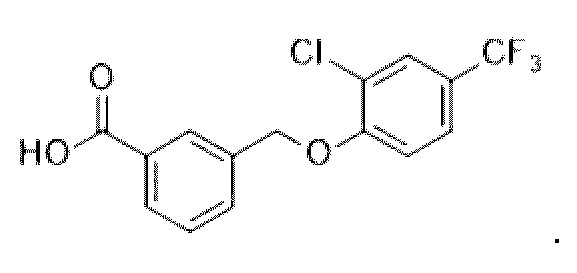
В более конкретном варианте осуществления изобретения соединение имеет следующую структуру или представляет собой его фармацевтически приемлемый изомер, рацемат, гидрат, сольват, изотоп или соль:
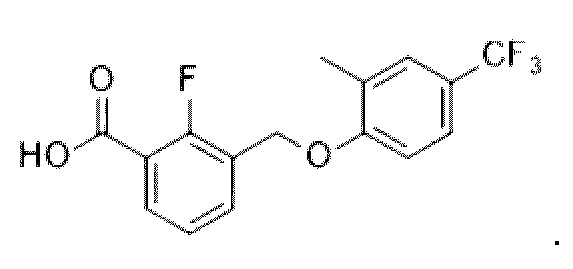
В более конкретном варианте осуществления изобретения соединение имеет следующую структуру или представляет собой его фармацевтически приемлемый изомер, рацемат, гидрат, сольват, изотоп или соль:
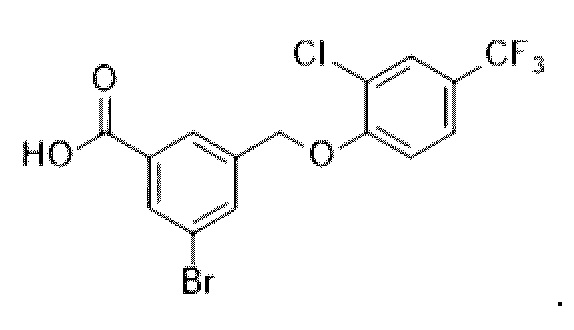
В более конкретном варианте осуществления изобретения соединение имеет следующую структуру или представляет собой его фармацевтически приемлемый изомер, рацемат, гидрат, сольват, изотоп или соль:
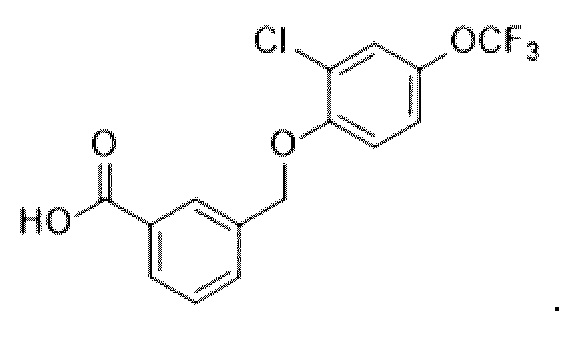
В более конкретном варианте осуществления изобретения соединение имеет следующую структуру или представляет собой его фармацевтически приемлемый изомер, рацемат, гидрат, сольват, изотоп или соль:
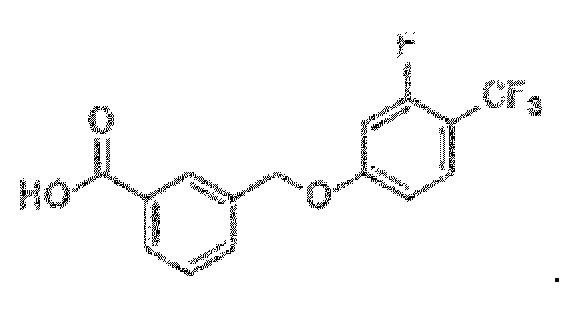
В более конкретном варианте осуществления изобретения соединение имеет следующую структуру или представляет собой его фармацевтически приемлемый изомер, рацемат, гидрат, сольват, изотоп или соль:
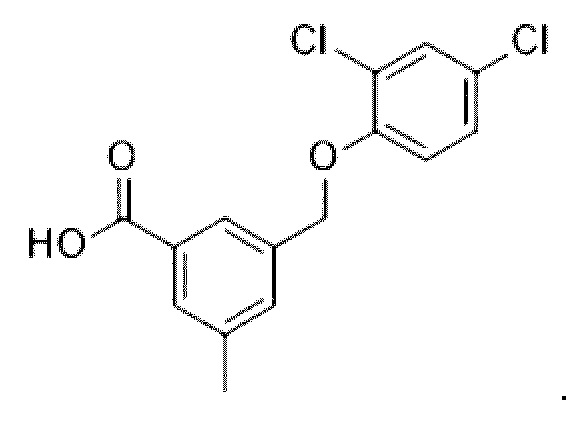
В более конкретном варианте осуществления изобретения соединение имеет следующую структуру или представляет собой его фармацевтически приемлемый изомер, рацемат, гидрат, сольват, изотоп или соль:
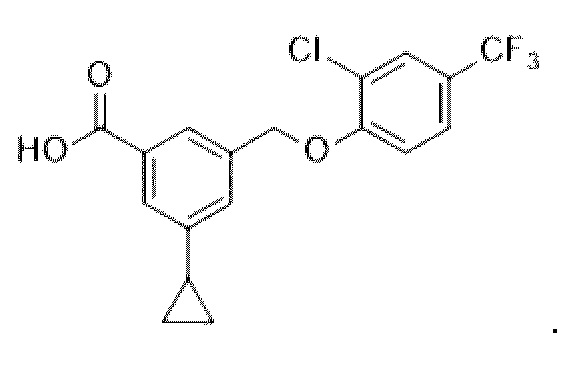
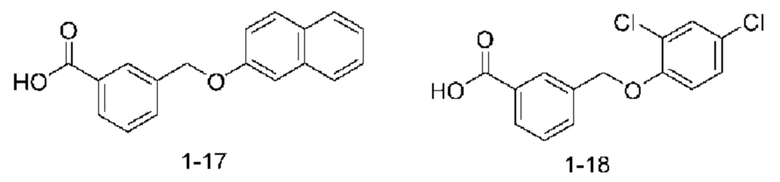
В другом варианте осуществления изобретения некоторые соединения формулы (I), а также формул (II)-(XXIV), когда это применимо, могут иметь замещение фрагмента карбоновой кислоты изостерной группой карбоновой кислоты, как описано в данном документе. Типичные соединения с изостерами карбоновых кислот, производные от типичных соединений, перечисленных ниже, представлены в Таблице В.
С этой целью в соединениях Таблицы В используются следующие изостерные группы карбоновых кислот:
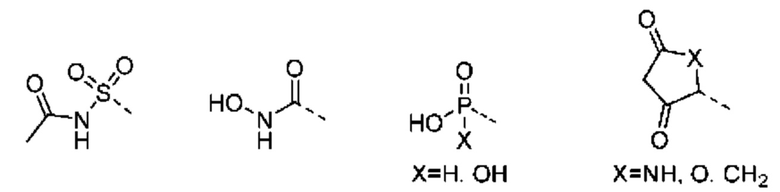
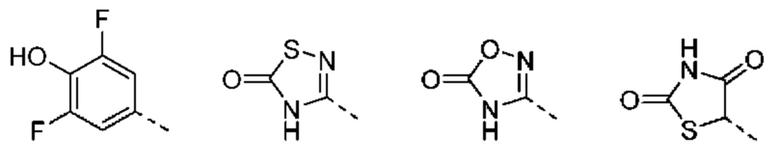
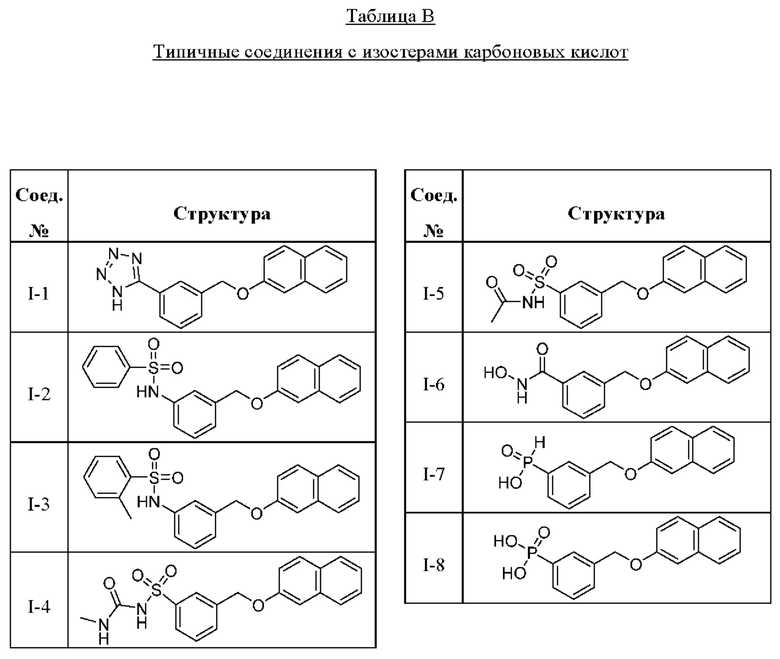
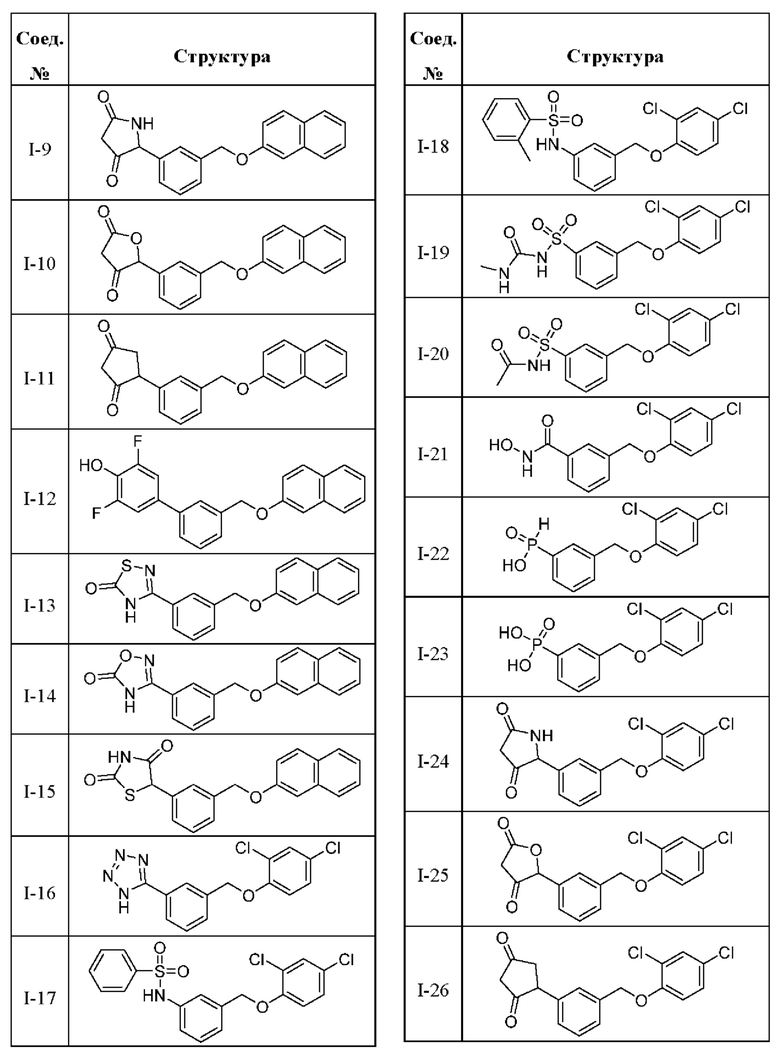
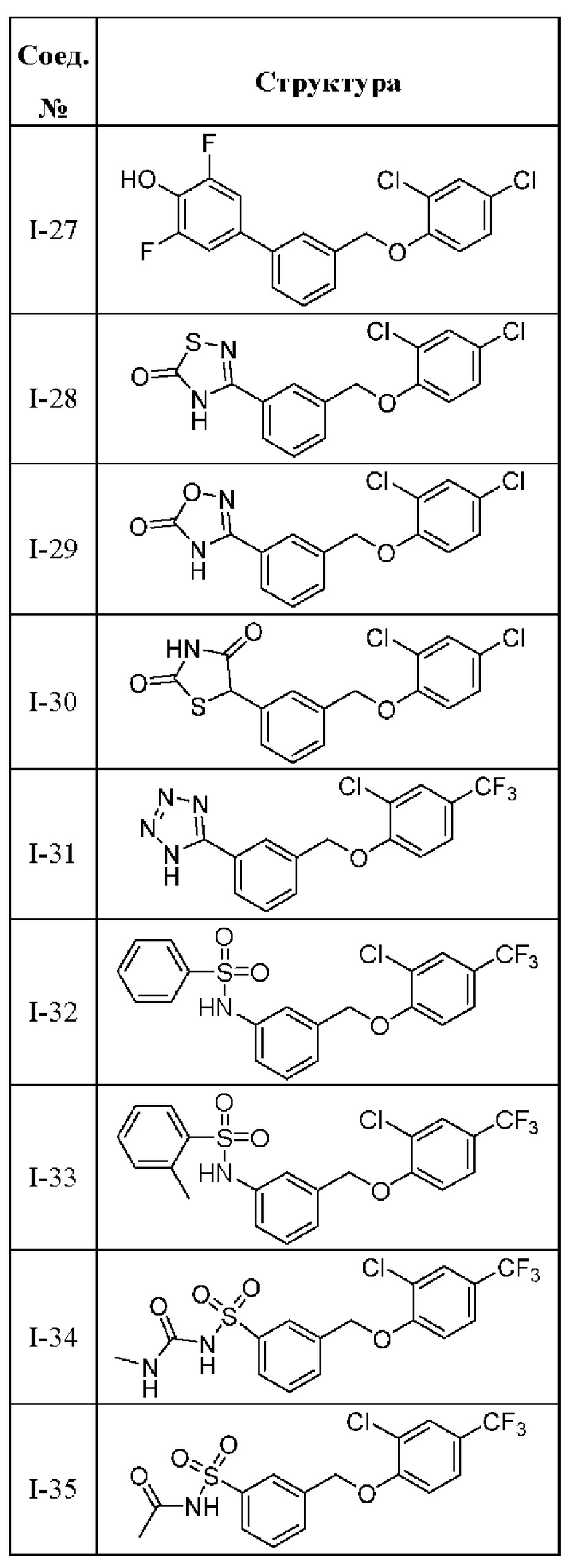
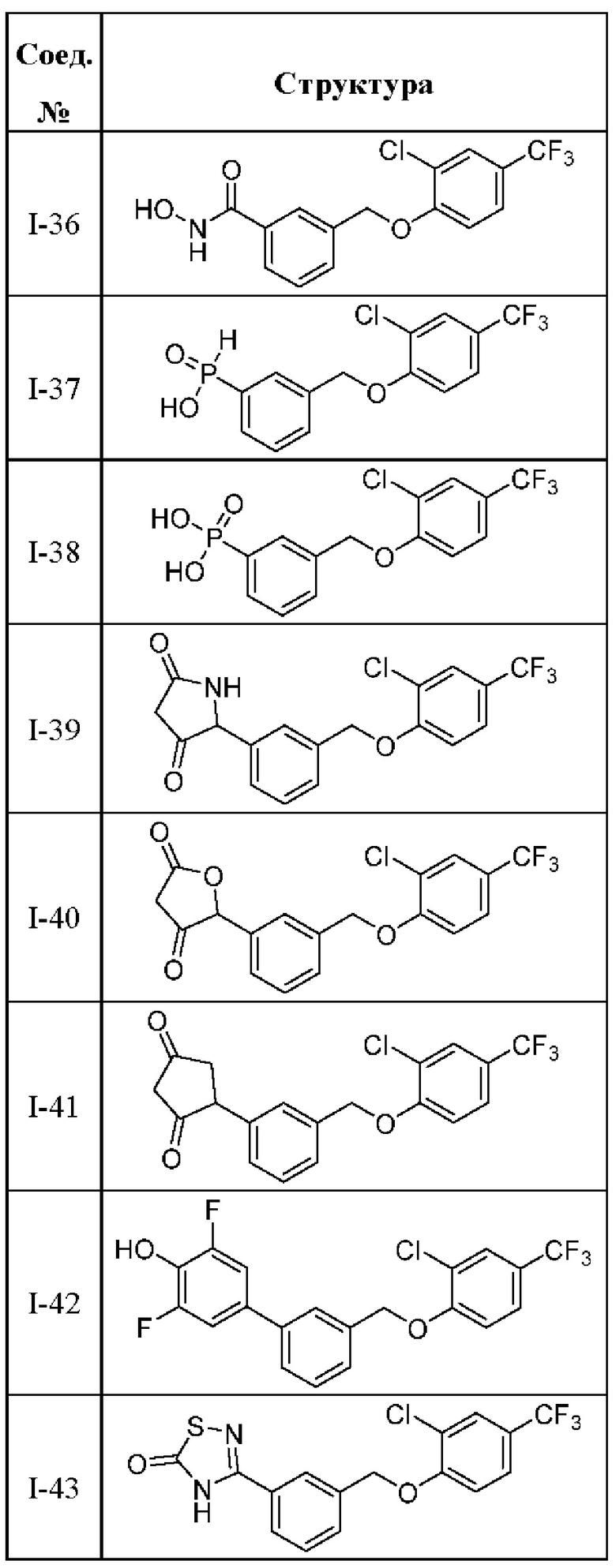
В других вариантах осуществления изобретения предусматриваются пролекарства и/или метаболиты соединений формулы (I), а также формул (II)-(XXIV).
Таким образом, в одном варианте осуществления предусматриваются пролекарства соединения по изобретению, которые при введении субъекту подвергаются химическому превращению в результате метаболических или других физиологических процессов, становясь активными фармакологическими веществами. Превращение посредством метаболических или других физиологических процессов включает, без ограничений, ферментативное (например, специфическое ферментативно катализируемое) и неферментативное (например, произвольное или специфическое кислотно- или основно-индуцируемое) химическое превращение пролекарства в активное фармакологическое вещество. В общем, такие пролекарства будут функциональными производными соединения по изобретению, которые легко превращаются in vivo в соединение по изобретению. Обычные процедуры выбора и получения подходящих производных пролекарств описаны, например, в Design of Prodrugs, ed. H. Bundgaard, Elsevier, 1985.
Соответственно, «пролекарство» представляет собой вещество, которое при введении субъекту превращают in vivo под действием биохимических веществ в организме субъекта, таких как ферменты, в активный фармацевтический ингредиент. Примеры пролекарств включают сложные эфиры групп карбоновых кислот, которые могут гидролизоваться эндогенными эстеразами, присутствующими в кровотоке людей и других млекопитающих. В одном варианте осуществления изобретения предусматриваются вещества, которые могут быть введены субъекту, и которые затем превращаются в организме субъекта с образованием соединения, имеющего структуру формулы (I) или любой из формул (II)-(XXIV).
В этом отношении пролекарства карбоновых кислот обычно представляют собой сложные эфиры и амиды, которые можно легко получить из соответствующей карбоновой кислоты известными методами. Например, в одном варианте осуществления изобретения пролекарства могут быть получены путем превращения фрагмента карбоновой кислоты соединений формул (I)-(VII) и (XVI)-(XXIV) в сложноэфирную функциональную группу, включая сложные алкиловые эфиры, такие как сложные метиловый, этиловый, изопропиловый и н-бутиловый эфиры; сложные ариловые эфиры, такие как сложные фениловый и инданиловый эфиры; двойные сложные эфиры, такие как сложные (ацилокси)алкиловые или [(алкоксикарбонил)окси]метиловые эфиры; и циклические карбонаты, такие как сложные (оксодиоксолил)метиловые эфиры. В другом варианте осуществления изобретения фрагмент карбоновой кислоты может включать карбамоилметиловый, аминоалкиловый или амидоалкиловый фрагмент с образованием карбамоилметилового, аминоалкилового и амидоалкилового сложных эфиров, соответственно. В еще одном варианте осуществления изобретения фрагмент карбоновой кислоты может включать сложные эфиры ацилглицеринов и бис(ациламино)пропан-2-олов. В другом варианте осуществления изобретения фрагмент карбоновой кислоты может включать амидные группы, включая N-гидроксиамид, N-ацилсульфонамиды и N-ацилсульфонилмочевины.
При использовании в данном документе, «метаболит» представляет собой соединение, которое после введения субъекту подвергается превращению в организме субъекта с образованием активного вещества. Такое превращение часто включает процессы гидролиза, фосфорилирования и/или окисления/восстановления и может опосредоваться любым количеством ферментов (например, эстеразами, фосфатазами, цитохромом Р450 и т.п.), а также различными средами в организме (например, изменениями рН).
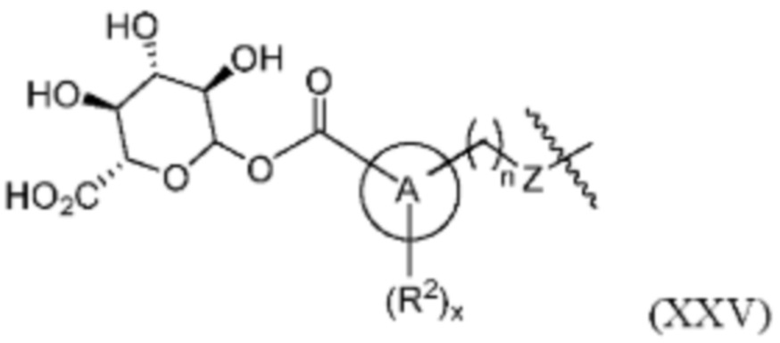
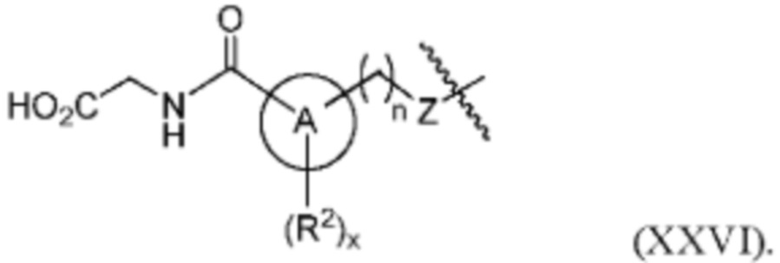
В одном варианте осуществления изобретения соединения формулы (I), а также формул (II)-(XXII), когда это применимо, модифицированы для включения метаболитов исходного соединения. В другом варианте осуществления изобретения соединений формулы (I), а также формул (II)-(XXII) модифицированы так, чтобы карбоновая кислота «А-кольца» формулы (I) была дериватизирована углеводными или аминокислотными соединениями. В дополнительном варианте осуществления изобретения фрагмент карбоновой кислоты А-кольца дериватизируют глюкуроновой кислотой или аминокислотой глицином с получением соединений формул (XXV) и (XXVI), соответственно:
В некоторых вариантах осуществления изобретение предусматривает фармацевтическую композицию, содержащую соединение любой из формул (I)-(XIV) вместе с по меньшей мере одним фармацевтически приемлемым носителем, разбавителем или вспомогательным веществом. Например, активное соединение обычно будет смешано с носителем или разбавлено носителем, или заключено в носитель, который может иметь форму ампулы, капсулы, саше, бумажного или другого контейнера. Когда активное соединение смешивают с носителем или когда носитель служит разбавителем, он может быть твердым, полутвердым или жидким материалом, который выступает в роли носителя, вспомогательного вещества или среды для активного соединения. Активное соединение может адсорбироваться на гранулированном твердом носителе, например, помещенном в саше. Некоторыми примерами подходящих носителей являются вода, солевые растворы, спирты, полиэтиленгликоли, полигидроксиэтоксилированное касторовое масло, арахисовое масло, оливковое масло, желатин, лактоза, сульфат кальция (terra alba), сахароза, декстрин, карбонат магния, сахар, циклодекстрин, амилоза, стеарат магния, тальк, желатин, агар, пектин, гуммиарабик, стеариновая кислота или низшие простые алкиловые эфиры целлюлозы, кремнекислота, жирные кислоты, амины жирных кислот, моноглицериды и диглицериды жирных кислот, сложные эфиры пентаэритрита и жирных кислот, полиоксиэтилен, гидроксиметилцеллюлоза и поливинилпирролидон. Аналогично, носитель или разбавитель может включать любой материал с замедленным высвобождением, известный в данной области техники, такой как моностеарат глицерина или дистеарат глицерина, отдельно или в смеси с воском.
Используемый в данном документе термин «фармацевтическая композиция» относится к композиции, содержащей одно или несколько соединений, описанных в данном документе, или их фармацевтически приемлемый изомер, рацемат, гидрат, сольват, изотоп или соль, составленной с фармацевтически приемлемым носителем, которая может также включать другие добавки, и производится или продается с одобрения государственного регулирующего органа как часть терапевтической схемы лечения заболевания у млекопитающего. Фармацевтические композиции могут быть составлены, например, для перорального введения в виде стандартной лекарственной формы (например, таблетки, капсулы, каплеты, желатиновой капсулы или сиропа); для местного применения (например, в виде крема, геля, лосьона или мази); для внутривенного введения (например, в виде стерильного раствора, не вызывающего эмболии дисперсными частицами, и в системе растворителей, подходящей для внутривенного применения); или в любом другом составе, описанном в данном документе. Обычные процедуры и ингредиенты для выбора и приготовления подходящих составов описаны, например, в Remington: The Science and Practice of Pharmacy, 21st Ed., Gennaro, Ed., Lippencott Williams & Wilkins (2005) и в Фармакопее США: Национальный формуляр (USP 36 NF31), опубликованном в 2013 г.
В некоторых вариантах осуществления изобретения, фармацевтическая композиция, содержащая соединение любой из формул (I)-(XIV), с по меньшей мере одним фармацевтически приемлемым носителем, разбавителем или вспомогательным веществом, дополнительно содержит второй терапевтический агент. В одном варианте осуществления изобретения, второй терапевтический агент представляет собой терапевтическое средство для лечения заболевания печени. В одном варианте осуществления терапевтическое средство для лечения заболеваний печени представляет собой урсодезоксихолевую кислоту (УДХК), норурсодезоксихолевую кислоту, холестирамин, станозолол, налтрексон, рифампицин, ализол В 23-ацетат (АВ23А), куркумин, дигидроартемизинин, фенофибрат, безафибрат, метронидазол, метотрексат, колхицин, метформин, бетаин, глюкагон, налтрексон, агонист фарнезоидных Х-рецепторов (ФХР), агонист рецептора, активируемого пролифератором пероксисом (PPAR), агонист бета-рецептора тиреоидного гормона (TRβ), или любую их комбинацию.
Примеры агонистов ФХР, которые могут использоваться в фармацевтических композициях, описанных в данном документе, включают обетихолевую кислоту, турофексорат изопропил (WAY-362450), 3-(2,6-дихлорфенил)-4-(3'-карбокси-2-хлорстильбен-4-ил)оксиметил-5-изопропилизоксазол (GW4064), РХ20606 (РХ-102), РХ-101, INT-767, INT-787, TERN-101, альтенузин, тропифексор (LJN452), нидуфексор, турофексорат изопропил, фексарамин, силимарин, силибин, гедрагоновую кислоту, кафестол, цилофексор (GS-9674 или Рх-104), EDP-305, BAR704, BAR502, EYP-001, RDX-023, AGN-242266, HPG-1860, МЕТ-409, AGN-242256, ЕР-024297, IOT-022, М-480, INV-33, RDX023-02 или любую их комбинацию. В одном из вариантов осуществления агонист ФХР представляет собой желчную кислоту или ее аналог (например, обетихолевая кислота, INT-767, INT-787, турофексорат изопропил (WAY-362450) или BAR704) или агонист, не являющийся желчной кислотой (например, EDP-305, тропифексор, нидуфексор, цилофексор, GW4064, турофексорат изопропил, фексарамин, РХ20606 (РХ-102), TERN-101, альтенузин, силимарин, силибин, гедрагоновая кислота, BAR502, EYP-001, RDX023-2, AGN-242266, HPG-1860, МЕТ-409, ЕР-024297, М-480 или кафестол).
В одном из вариантов осуществления агонист PPAR представляет собой агонист PPAR-альфа, агонист PPAR-гамма, агонист PPAR-дельта, двойной агонист PPAR-альфа/гамма, двойной агонист PPAR-альфа/дельта, двойной агонист PPAR-гамма/дельта, пан-агонист PPAR альфа/гамма/дельта или любую их комбинацию.
Примеры агонистов PPAR-альфа, которые можно использовать в фармацевтических композициях, описанных в данном документе, включают фенофибрат, ципрофибрат, пемафибрат, гемфиброзил, клофибрат, бинифибрат, клинофибрат, клофибриновую кислоту, никофибрат, пирифибрат, плафибрид, ронифибрат, теофибрат, токофибрат и SRI 0171.
Примеры агонистов PPAR-гамма, которые можно использовать в фармацевтических композициях, описанных в данном документе, включают розиглитазон, пиоглитазон, стабилизированный дейтерием R-пиоглитазон, эфатутазон, АТх08-001, OMS-405, CHS-131, THR-0921, SER-150-DN, KDT-501, GED-0507-34-Levo, CLC-3001 и ALL-4.
Примеры агонистов PPAR-дельта, которые можно использовать в описанных в данном документе фармацевтических композициях, включают GW501516 (эндурабол или ({4-[({4-метил-2-[4-(трифторметил)фенил]-1,3-тиазол-5-ил}метил)сульфанил]-2-метилфенокси}уксусная кислота)), МВХ8025 (селаделпар или {2-метил-4-[5-метил-2-(4-трифторметилфенил)-2Н-[1,2,3]триазол-4-илметилсульфанил]фенокси}уксусная кислота), GW0742 ([4-[[[2-[3-фтор-4-(трифторметил)фенил]-4-метил-5-тиазолил]метил]тио]-2-метилфенокси]уксусная кислота), L165041, НРР-593 и NCP-1046.
Примеры агонистов PPAR альфа/гамма, которые можно использовать в фармацевтических композициях, описанных в данном документе, включают сароглитазар, алеглитазар, мураглитазар, тесаглитазар и DSP-8658.
Примеры агонистов PPAR альфа/дельта, которые можно использовать в фармацевтических композициях, описанных в данном документе, включают элафибранор и Т913659.
Примеры агонистов PPAR гамма/дельта, которые можно использовать в фармацевтических композициях, описанных в данном документе, включают конъюгированную линолевую кислоту (КЛК) и T3D-959.
Примеры агонистов PPAR альфа/гамма/дельта, которые могут использоваться в фармацевтических композициях, описанных в данном документе, включают IVA337 (ланифибранор), ТТА (тетрадецилтиоуксусная кислота), бавахинин, GW4148, GW9135, безафибрат, лобеглитазон, 2-(4-(5,6-метилендиоксибензо[d]тиазол-2-ил)-2-метилфенокси)-2-метилпропановую кислоту (MHY2013) и CS038.
Примеры агонистов бета-рецептора тиреоидного гормона, которые можно использовать в фармацевтических композициях, описанных в данном документе, включают собетиром, эпротиром, GC-24, MGL-3196, MGL-3745, VK-2809, KB141 [3,5-дихлор-4-(4-гидрокси-3-изопропилфенокси)фенилуксусная кислота] и МВ07811 (2R,4S)-4-(3-хлорфенил)-2-[(3,5-диметил-4-(4'-гидрокси-3''-изопропилбензил)фенокси)метил]-2-оксидо-[1,3,2]-диоксафосфонан).
Используемый в данном документе термин «фармацевтически приемлемый носитель» относится к любому ингредиенту, отличному от раскрытых соединений или их фармацевтически приемлемых изомеров, рацематов, гидратов, сольватов, изотопов или солей (например, к носителю, способному суспендировать или растворять активное соединение), и являющемуся нетоксичным и невоспалительным для пациента. Вспомогательные вещества могут включать, например: антиадгезивы, антиоксиданты, связующие, покрытия, добавки для прессования, разрыхлители, красители (красящие вещества), умягчители, эмульгаторы, наполнители (разбавители), пленкообразователи или покрытия, вкусовые вещества, ароматизаторы, глиданты (вещества, способствующие скольжению), смазки, консерванты, печатные краски, сорбенты, суспендирующие или диспергирующие агенты, подсластители или воду гидратации. Примеры вспомогательных веществ включают, без ограничений: бутилированный гидрокситолуол (ВНТ), карбонат кальция, фосфат кальция (двухосновный), стеарат кальция, кроскармеллозу, сшитый поливинилпирролидон, лимонную кислоту, кросповидон, цистеин, этилцеллюлозу, желатин, гидроксипропилцеллюлозу, гидроксипропилметилцеллюлозу, лактозу, стеарат магния, мальтит, маннит, метионин, метилцеллюлозу, метилпарабен, микрокристаллическую целлюлозу, полиэтиленгликоль, поливинилпирролидон, повидон, прежелатинизированный крахмал, пропилпарабен, ретинилпальмитат, шеллак, диоксид кремния, карбоксиметилцеллюлозу натрия, цитрат натрия, натрия крахмалгликолят, сорбит, крахмал (кукурузный), стеариновую кислоту, стеариновую кислоту, сахарозу, тальк, диоксид титана, витамин А, витамин Е, витамин С и ксилит.
Композиции можно смешивать со вспомогательными агентами, которые не вступают в нежелательные реакции с активными соединениями. Такие добавки могут включать смачивающие агенты, эмульгирующие и суспендирующие агенты, соли для воздействия на осмотическое давление, буферы и/или красящие вещества, консерванты, подсластители или вкусовые вещества. При желании композиции также можно стерилизовать.
Путь введения может представлять собой любой путь, который эффективно доставляет активное соединение по изобретению к подходящему или желаемому месту действия, например пероральный, назальный, легочный, буккальный, субдермальный, интрадермальный, трансдермальный или парентеральный, включая внутривенный, подкожный и/или внутримышечный. В одном из вариантов осуществления путь введения -пероральный. В другом варианте осуществления способ введения является местным.
Лекарственные формы можно вводить один раз в день или более одного раза в день, например, два или три раза в день. Альтернативно, лекарственные формы можно вводить реже, чем ежедневно, например, через день или еженедельно, если это будет рекомендовано лечащим врачом или указано в информации о назначении лекарственного средства. Схемы дозирования включают, например, титрование дозы до степени, необходимой или полезной при показании, подлежащем лечению, что позволяет организму пациента адаптироваться к лечению, минимизировать или избегать нежелательных побочных эффектов, связанных с лечением, и/или увеличить до максимума терапевтический эффект соединений по настоящему изобретению. Другие лекарственные формы включают формы с отсроченным или контролируемым высвобождением. Подходящие схемы дозирования и/или лекарственные формы включают указанные, например, в последнем издании Physicians' Desk Reference (Настольный справочник врача), включенного в данный документ посредством ссылки.
В одном варианте осуществления изобретение обеспечивает пероральную фармацевтическую композицию, содержащую соединение любой из формул (I)-(XXIV) вместе с по меньшей мере одним фармацевтически приемлемым пероральным носителем, разбавителем или вспомогательным веществом. В другом варианте осуществления изобретение предусматривает фармацевтическую композицию для местного применения, содержащую соединение любой из формул (I)-(XXIV) вместе с по меньшей мере одним фармацевтически приемлемым носителем, разбавителем или вспомогательным веществом для местного применения. Например, предусматривается пероральная фармацевтическая композиция для лечения холестатического зуда, при этом схемой дозирования является введение, например, один раз в день. В одном варианте осуществления изобретения предусматривается фармацевтическая композиция для местного применения для лечения атопического дерматита.
В другом варианте осуществления изобретения предусматриваются способы приготовления композиции соединения, описанного в данном документе, включая составление композиции соединения по изобретению с фармацевтически приемлемым носителем или разбавителем. В некоторых вариантах осуществления изобретения фармацевтически приемлемый носитель или разбавитель является пригодным для перорального введения. В некоторых таких вариантах осуществления изобретения способы могут дополнительно включают стадию составления композиции в виде таблетки или капсулы. В других вариантах осуществления изобретения фармацевтически приемлемый носитель или разбавитель является пригодным для парентерального введения. В некоторых таких вариантах осуществления изобретения способы дополнительно включают стадию лиофилизации композиции с образованием лиофилизированного препарата.
В некоторых вариантах осуществления изобретение относится к соединению, имеющему структуру любой из формул (I)-(XXIV). Такие соединения можно синтезировать с использованием стандартных методов синтеза, известных специалистам в данной области. Например, соединения по настоящему изобретению можно синтезировать с использованием соответствующим образом модифицированных методик синтеза, изложенных в приведенных далее примерах и схемах реакций. При этом, изостеры карбоновых кислот и замещение ими карбоновых кислот, как раскрыто в данном документе, также могут быть выполнены с использованием стандартных методов синтеза, известных специалистам в данной области.
При этом, описанные в данном документе реакции, процессы и методы синтеза не ограничиваются конкретными условиями, указанными в приведенном далее экспериментальном разделе, а должны использоваться скорее в качестве руководства для специалистов в этой области. Например, реакции можно проводить в любом подходящем растворителе или других реагентах для выполнения необходимого превращения (превращений). Обычно подходящими растворителями являются протонные или апротонные растворители, которые по существу не вступают в реакцию с реагентами, промежуточными соединениями или продуктами при температурах проведения реакций (т.е. температурах, которые могут варьироваться от температур замерзания до температур кипения). Какая-либо данная реакция может быть проведена в одном растворителе или в смеси нескольких растворителей. В зависимости от конкретной реакции могут использоваться подходящие растворители для конкретной доработки после проведения реакции.
Все реагенты, синтез которых не описан в экспериментальной части, либо коммерчески доступны, либо являются известными соединениями или могут быть получены из известных соединений известными методами специалистом в данной области техники. Соединения и промежуточные продукты, полученные в соответствии со способами по изобретению, могут потребовать очистки. Очистка органических соединений хорошо известна специалистам в данной области, и может существовать несколько способов очистки одного и того же соединения. В некоторых случаях очистка может не потребоваться. В некоторых случаях, соединения можно очищать кристаллизацией. В некоторых случаях, примеси можно удалить перемешиванием с использованием подходящего растворителя. В некоторых случаях соединения могут быть очищены с помощью хроматографии, особенно флэш-хроматографии на колонке, с использованием специально изготовленных или предварительно набитых картриджей с силикагелем и элюентов, таких как градиенты растворителей, таких как гептан, эфир, этилацетат, ацетонитрил, этанол и т.п. В некоторых случаях соединения могут быть очищены препаративной ВЭЖХ с использованием описанных способов.
Способы очистки, как описано в данном документе, могут обеспечить соединения по настоящему изобретению, обладающие достаточно основной или кислотной функциональностью, в форме соли, например, в случае соединения по настоящему изобретению, являющегося достаточно основным - в виде трифторацетатной или формиатной соли, или, в случае соединения по настоящему изобретению, являющегося достаточно кислым - соли аммония. Соль такого типа может быть преобразована либо в форму свободного основания, либо в форму свободной кислоты, соответственно, различными способами, известными специалисту в данной области, или может использоваться в солевой форме в последующих биологических анализах. Следует понимать, что конкретная форма соединения по настоящему изобретению, выделенная и описанная в данном документе, не обязательно является единственной формой, в которой указанное соединение может применяться в биологическом анализе для количественной оценки конкретной биологической активности.
Химические названия были созданы с использованием программы для генерирования названий ChemDraw (версия 17.0.0.206) фирмы PerkinElmer Informatics, Inc. В некоторых случаях вместо названий, созданных программой генерирования названий, использовались общепринятые названия коммерчески доступных реагентов.
ПРИМЕРЫ
Общие способы
Спектры 1Н ЯМР (400 МГц) были получены в растворе дейтерохлороформа (CDCl3), дейтерометанола (CD3OD) или диметилсульфоксида-D6 (ДМСО). Хроматографические времена удерживания, чистоты и масс-спектры (ЖХ-МС) были получены с использованием одного из следующих методов:
Метод 1: Система Agilent 1260 Infinity II, оснащенная колонкой Agilent Poroshell 120 ЕС-18, 2,7 мкм, 4,6×100 мм, с использованием H2O с 0,1% муравьиной кислоты в качестве подвижной фазы А и MeCN с 0,1% муравьиной кислоты в качестве подвижной фазы В. Использовали детектор с ионизацией электрораспылением (ESI) в положительном режиме. Градиент составлял 20-95% подвижной фазы В в течение 5 минут, затем поддерживался на уровне 95% в течение 3,8 минут, затем возвращался к 20% мобильной фазы В в течение 0,2 минуты. Скорость потока составляла 1 мл/мин.
Метод 2: Система Agilent 1260 Infinity II, оснащенная колонкой Agilent Poroshell 120 ЕС-18, 2,7 мкм, 4,6×100 мм, с использованием Н2О с 0,1% муравьиной кислоты в качестве подвижной фазы А и MeCN с 0,1% муравьиной кислоты в качестве подвижной фазы В. Использовали детектор ESI в отрицательном режиме. Градиент составлял 20-95% подвижной фазы В в течение 5 минут, затем поддерживался на уровне 95% в течение 3,8 минут, затем возвращался к 20% мобильной фазы В в течение 0,2 минуты. Скорость потока составляла 1 мл/мин.
Метод 3: Система Agilent 1260 Infinity II, оснащенная колонкой Agilent Poroshell 120 ЕС-18, 2,7 мкм, 4,6×100 мм, с использованием H2O с 0,1% муравьиной кислоты в качестве подвижной фазы А и MeCN с 0,1% муравьиной кислоты в качестве подвижной фазы В. Использовали детектор ESI в положительном режиме. Градиент составлял 20-95% подвижной фазы В в течение 5 минут, затем поддерживался на уровне 95% в течение 3,8 минут, затем возвращался к 20% мобильной фазы В в течение 0,2 минуты. Скорость потока составляла 1 мл/мин.
Метод 4: Система Agilent 1260 Infinity II, оснащенная колонкой Agilent Poroshell 120 ЕС-18, 2,7 мкм, 4,6×100 мм, с использованием H2O с 0,1% муравьиной кислоты в качестве подвижной фазы А и MeCN с 0,1% муравьиной кислоты в качестве подвижной фазы В. Использовали детектор ESI в отрицательном режиме. Градиент составлял 10-95% подвижной фазы В в течение 12 минут, затем поддерживался на уровне 95% в течение 2 минут, возвращался к 10% мобильной фазы В в течение 1 минуты. Скорость потока составляла 1 мл/мин.
Метод 5: Система Shimadzu LCMS-2020, оснащенная колонкой KinetiX EVO С18, 2,1×30 мм, 5 мкм, с использованием Н2О с 0,025% NH3-H2O в качестве подвижной фазы А и MeCN в качестве подвижной фазы В. Скорость потока составляла 1,5 мл/мин. Использовался масс-детектор ESI в отрицательном режиме. Градиент составлял 0-60% В в течение 0,8 мин, затем поддерживался при 60% В в течение 0,4 мин, затем возвращался к 0% В в течение 0,01 мин и поддерживался при 0% В в течение 0,34 мин.
Метод 6: Система Agilent 1200/G6110A, оснащенная колонкой Chromolith Flash RP-18e 25×2,0 мм, с использованием Н2О с 0,0375% ТФУК в качестве подвижной фазы А и MeCN с 0,01875% ТФУК в качестве подвижной фазы В. Использовался масс-детектор ESI в положительном режиме. Градиент составлял 5-95% В в течение 0,8 мин, выдерживание при 95% В в течение 0,4 мин, затем возврат к 5% В в течение 0,01 мин и выдерживание на уровне 5% В в течение 0,29 мин.
Метод 7: Система Shimadzu LCMS-2020, оснащенная колонкой KinetiX EVO C18 2,1×30 мм, 5 мкм, с использованием H2O с 0,025% NH3 в качестве подвижной фазы А и MeCN в качестве подвижной фазы В. Скорость потока составляла 1,5 мл/мин. Использовался масс детектор ESI в отрицательном режиме. Градиент составлял 5-95% В в течение 0,8 мин, выдерживание при 95% В в течение 0,4 мин, затем возврат к 5% В в течение 0,01 мин и выдерживание на уровне 5% В в течение 0,34 мин.
Метод 8: Система Agilent 1200/G6110A, оснащенная колонкой АСЕ Excel С18, 2,1×30 мм, 5 мкм, с использованием H2O с 0,025% NH3 в качестве подвижной фазы А и MeCN в качестве подвижной фазы В. Использовали масс-детектор ESI, установленный в отрицательном режиме. Градиент составлял 10-80% В в течение 1,2 мин, выдерживание на уровне 80% В в течение 0,4 мин, затем возврат к 5% В в течение 0,01 мин и выдерживание при 5% В в течение 0,39 мин.
Метод 9: Система Agilent 1100, оснащенная колонкой Agilent Eclipse XDB-C18, 3,5 мкм, 4,6×150 мм, с использованием H2O с 0,1% трифторуксусной кислоты в качестве подвижной фазы А и метанола с 0,1% трифторуксусной кислоты в качестве подвижной фазы В. Градиент составлял 5-95% подвижной фазы В в течение 12 минут, затем выдерживание при 95% подвижной фазы В в течение 3 мин, затем возврат к 5% подвижной фазы В в течение 1 мин. Скорость потока составляла 1 мл/мин.
Метод 10: Система Shimadzu SCL-10A, оснащенная колонкой Agilent Eclipse XDB-C18, 3,5 мкм, 4,6×150 мм и РЕ Sciex API 150 EX, с использованием H2O с 0,1% трифторуксусной кислоты в качестве подвижной фазы А и метанола с 0,1% трифторуксусной кислоты в качестве подвижной фазы В. Градиент составлял 5-95% подвижной фазы В в течение 12 мин, затем выдерживание при 95% подвижной фазы В в течение 3 мин, затем возврат к 5% подвижной фазы В в течение 1 мин. Скорость потока составляла 1 мл/мин.
Метод 11: Система Shimadzu SCL-10A, оснащенная колонкой Agilent Eclipse XDB-C18, 3,5 мкм, 4,6×150 мм и РЕ Sciex API 150 EX, с использованием H2O с 0,1% трифторуксусной кислоты в качестве подвижной фазы А и метанол с 0,1% трифторуксусной кислоты в качестве подвижной фазы В. Градиент составлял 50-95% подвижной фазы В в течение 4 мин, затем выдерживание при 95% подвижной фазы В в течение 4 мин, затем возврат к 50% подвижной фазы В в течение 0,1 мин. Скорость потока составляла 1 мл/мин.
Метод 12: Система Waters Acquity, оснащенная колонкой Acquity UPLC ВЕН С18 1,7 мкм, 2,1×50 мм, с использованием H2O с 0,1% формиатом аммония, доведенной до рН 3,8 муравьиной кислотой, в качестве подвижной фазы А и ацетонитрилом в качестве подвижной фазы В. Градиент составлял 5-100% в течение 9 минут, затем выдерживание при 100% подвижной фазы В в течение 1 минуты. Скорость потока 0,7 мл/мин.
Метод 13: Система Waters Acquity, оснащенная EVO С18 (5 мкм, 3,0×50 мм) с использованием градиента буфера с низким рН от 5% до 100% MeCN в H2O (0,1% НСООН) в течение 2,5 мин при 2,2 мл/мин и выдерживания при 100% в течение в общей сложности 3,5 мин.
Пиридин, дихлорметан (ДХМ), тетрагидрофуран (ТГФ) и толуол, использованные в процедурах, брали из бутылей Aldrich Sure-Seal, хранимых под азотом (N2). Все реакции перемешивали с помощью магнитной мешалки, и температуры являются внешними температурами реакции. Хроматографии обычно выполняли с использованием системы флэш-очистки Combiflash Rf (Teledyne Isco), оснащенной колонками Redisep (Teledyne Isco) Rf Gold с нормальной фазой силикагеля (SiO2), или с использованием аналогичной системы.
Очистку методом препаративной ВЭЖХ обычно проводили с использованием одной из следующих систем: 1) система Waters, оснащенная детектором УФ/видимой области Waters 2489, детектором Aquity QDA, Waters xBridge Prep C18 5 мкм OBD, колонка 30×1560 мм, при элюировании с различными градиентами H2O/MeCN (0,1% муравьиной кислоты) при скорости потока 30 мл/мин, или 2) колонка: Phenomenex Synergi C18 150×30 мм-4 мкм; подвижная фаза: [H2O (0,225% муравьиной кислоты)-MeCN]; В%: 55%-85%, 12 мин) и обычно концентрировали с использованием Genevac EZ-2.
Используются следующие дополнительные сокращения: этилацетат (ЭА), триэтиламин (ТЭА), диметилсульфоксид (ДМСО), силикагель (SiO2), азобисизобутиронитрил (AIBN), гидрид диизобутилалюминия (DIBAL), трифторуксусная кислота (ТФУК), 4-диметиламинопиридин (DMAP), дифенилфосфорилазид (DPPA), пероксид бензоила (ВРО), 1,1'-бис(дифенилфосфино)ферроцен (dppf или DPPF), тетрагидрофуран (ТГФ), аддукт 1,4-диазабицикло[2.2.2]октан-бис(диоксид серы) (DABSO), гексафторфосфат азабензотриазолтетраметилурония (HATU), гидроксибензотриазол (HOBt), N-метилморфолин (NMM), N-бромсукцинимид (NBS), диизопропилэтиламин (DIPEA), диэтилазодикарбоксилат (DEAD), 2-[2-(дициклогексилфосфино)фенил]-N-метилиндол (CM-Phos), 1-этил-3-(3-диметиламинопропил)карбодиимида гидрохлорид (EDC), изопропанол (IPA), диметилформамид (ДМФА), диметилацетамид (ДМА), ацетонитрил (MeCN или ACN), 1,1'-тиокарбонилдиимидазол (TCDI), петролейный эфир (ПЭ), не определено (н.о.), время удержания (RT), молекулярный вес (м.в.), комнатная температура (к.т.), час (ч) и нет данных (н.д.).
ПРИМЕР
1 Синтез Соединения 1-0, Соединения 1-16 и других типичных соединений
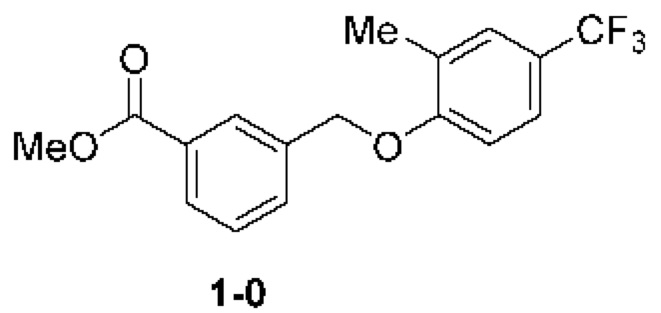
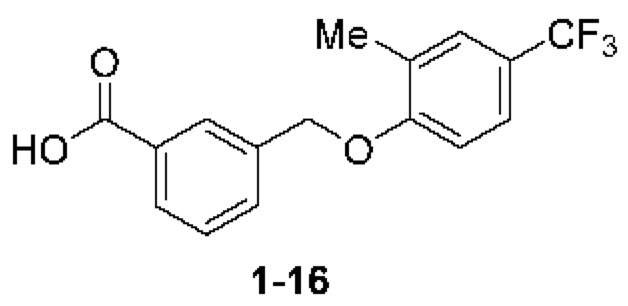
Схема 1
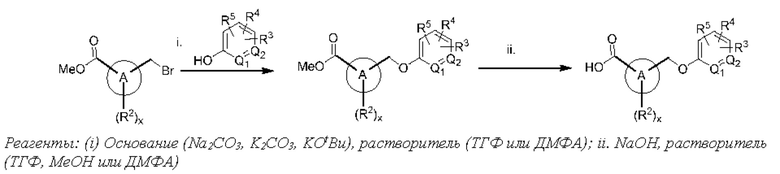
Стадия 1-1. Синтез метил-3-((4-хлор-2-(трифторметил)фенокси)метил)бензоата (Соединение 1-0)
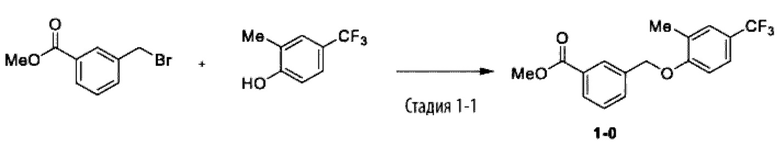
К перемешиваемому раствору метил-3-(бромметил)бензоата (150 мг, 655 мкмоль) в MeCN (3 мл) добавляли 2-метил-4-(трифторметил)фенол (115 мг, 655 мкмоль) и K2CO3 (118 мг, 0,85 ммоль). Реакционную смесь нагревали при 60°С в течение 3 часов, затем охлаждали до комнатной температуры и разбавляли H2O (3 мл). Водный слой экстрагировали Et2O (2×6 мл) и ЭА (1×6 мл), и объединенные органические слои осушали (Na2SO4), фильтровали, концентрировали и очищали хроматографией на SiO2 (ЭА/гексаны), получая 203 мг (77,4%) метил-3-((2-метил-4-(трифторметил)фенокси)метил)бензоата (Соединение 1-0) в виде белого твердого вещества. ЖХ-MC-ESI (m/z) вычислено для C17H15F3O3: 324,3; найдено 346,1 [M+Na]+, tR=6,68 мин (Метод 1).
Соединения, перечисленные в Таблице 1А, были получены с использованием процедур Схемы 1.
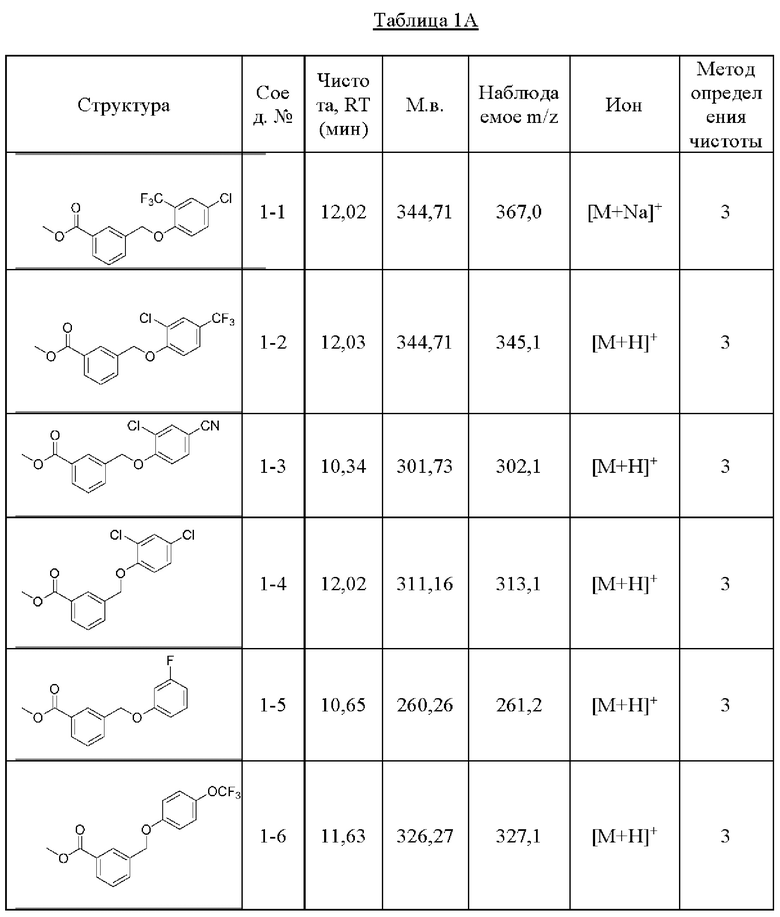
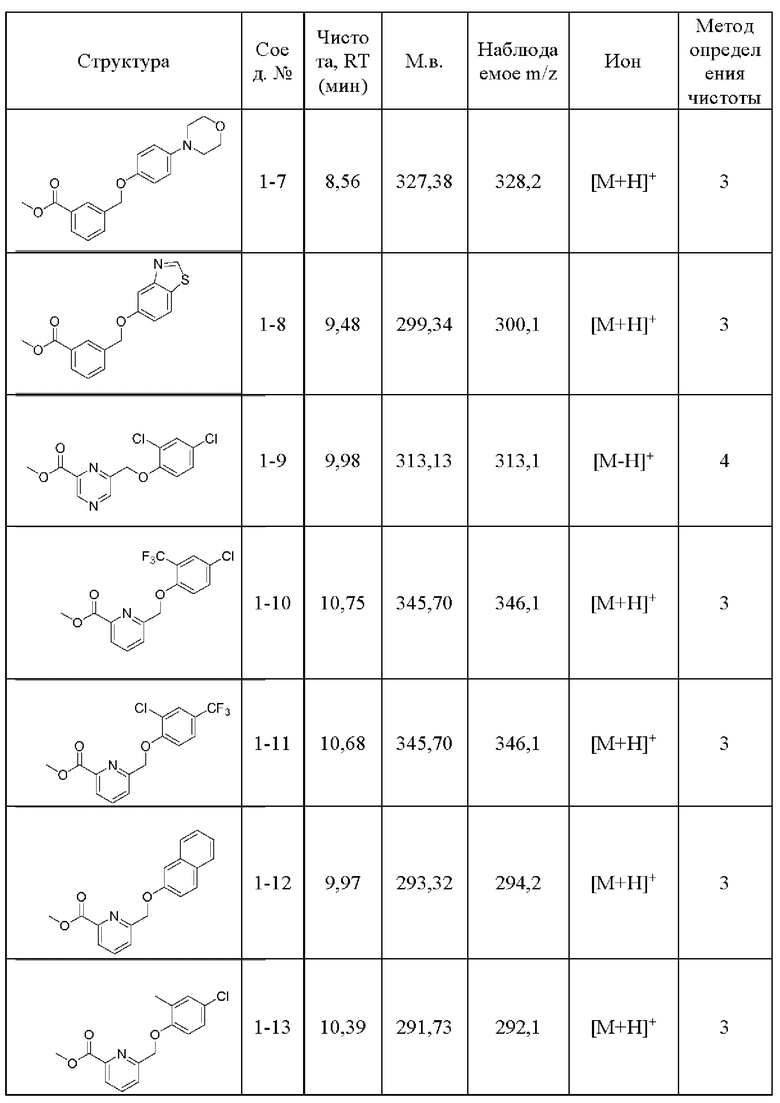
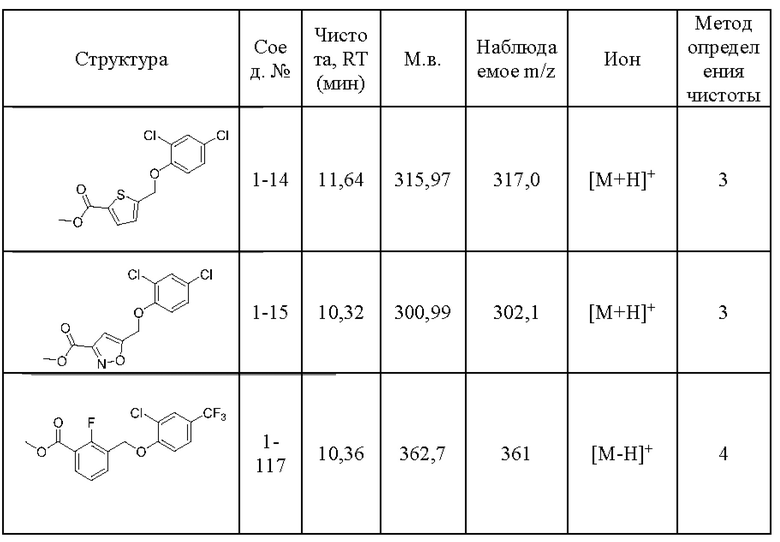
Стадия 1-2. Синтез 3-((2-метил-4-(трифторметилфенокси)метил)бензойной кислоты (Соединение 1-16)
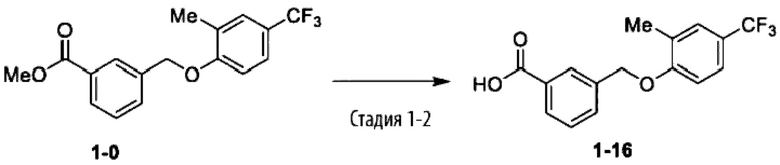
К перемешиваемому раствору метил-3-((2-метил-4-(трифторметил)фенокси)метил)бензоата (Соединение 1-0) (206 мг, 0,635 ммоль) в ТГФ (3 мл) добавляли 1М NaOH (3 мл, 3,18 ммоль). Реакционную смесь нагревали при 60°С в течение ночи, летучие вещества удаляли в вакууме и полученный водный слой подкисляли 3М HCl. Полученный раствор экстрагировали этилацетатом и Et2O, осушали (Na2SO4), фильтровали и концентрировали с получением неочищенного твердого вещества, которое очищали обращенно-фазовой хроматографией на SiO2, получая 155 мг (79%) 3-((2-метил-4-(трифторметил)фенокси)метил)бензойной кислоты (Соединение 1-16) в виде белого твердого вещества. ЖХ-MC-ESI (m/z) вычислено для C16H13F3O3: 310,2; найдено 333,1 [M+Na]+, tR=10,4 мин. (Метод 3).
Соединения, перечисленные в Таблице 1В, были получены с использованием процедур Схемы 1.
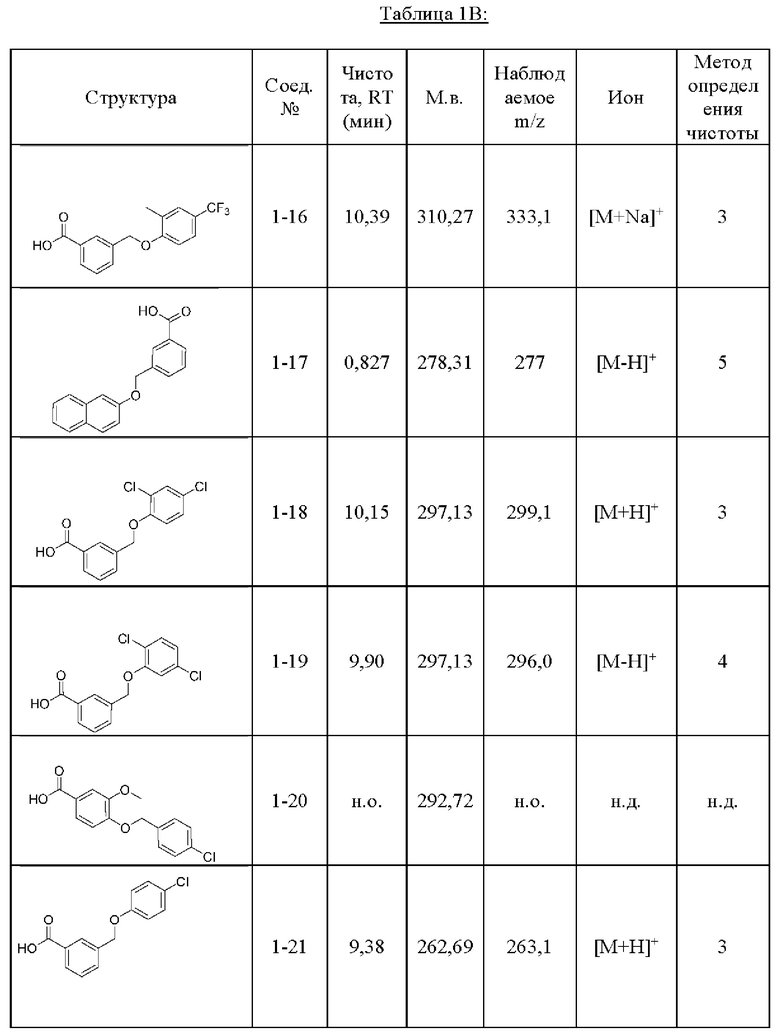
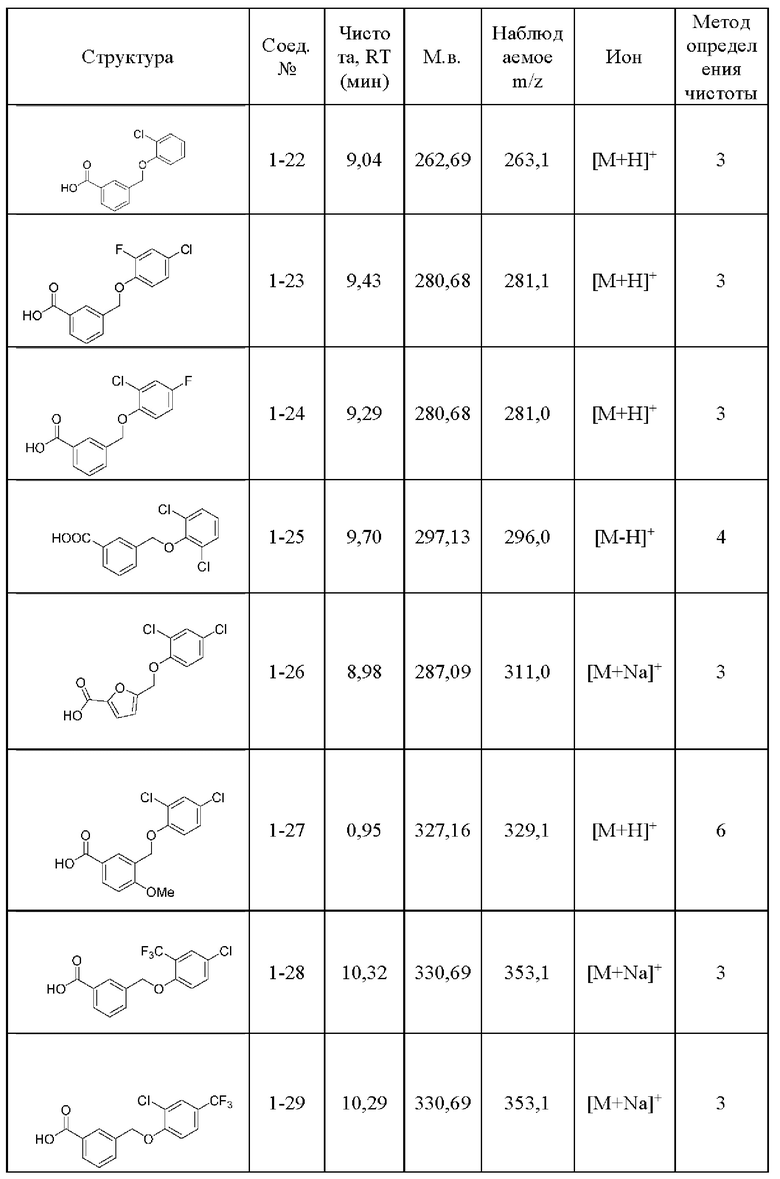
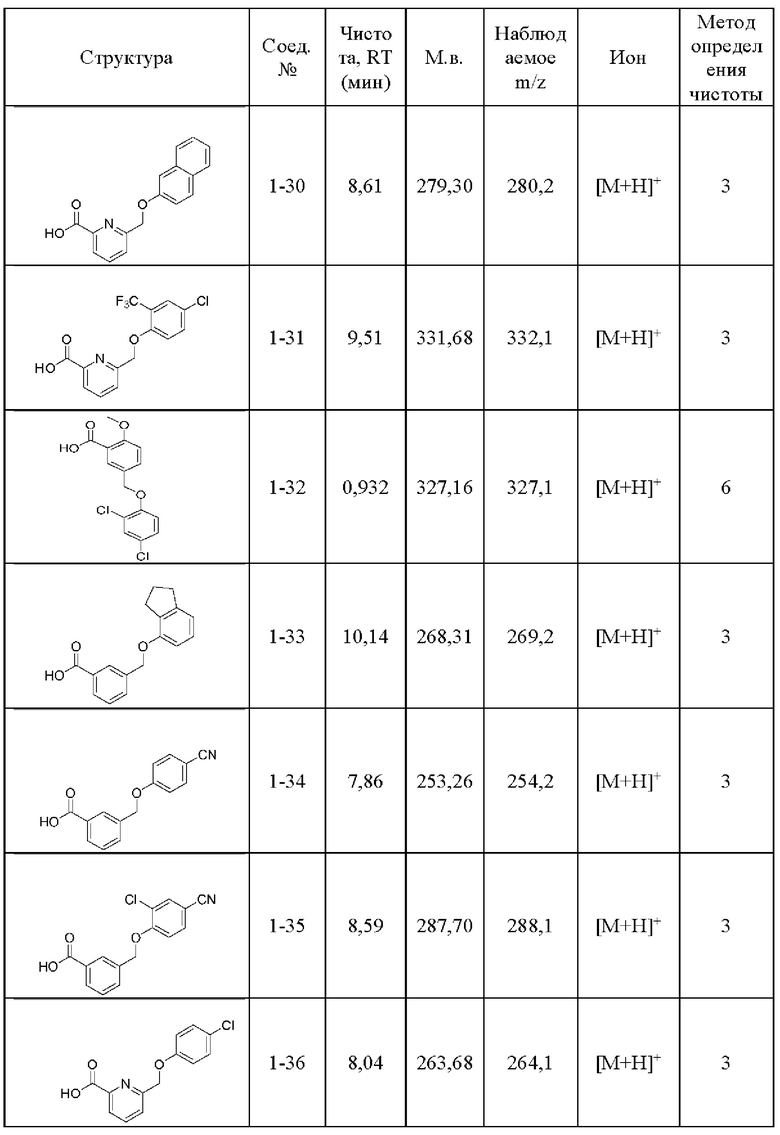
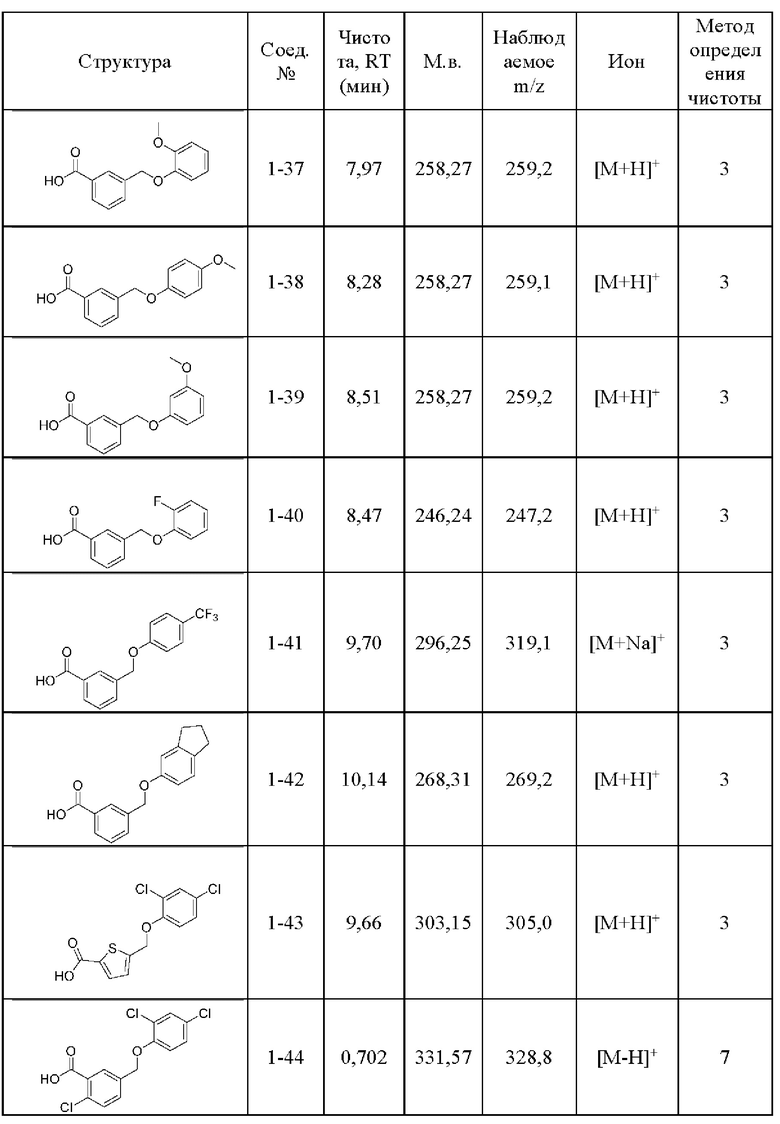
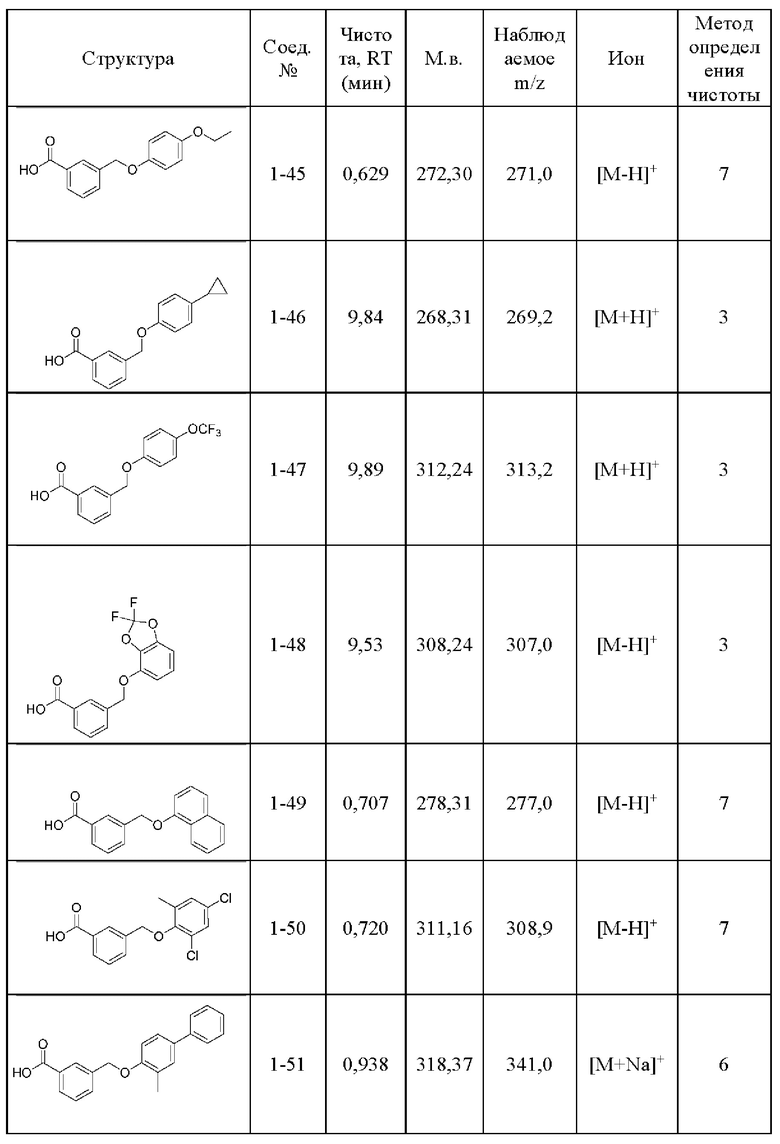
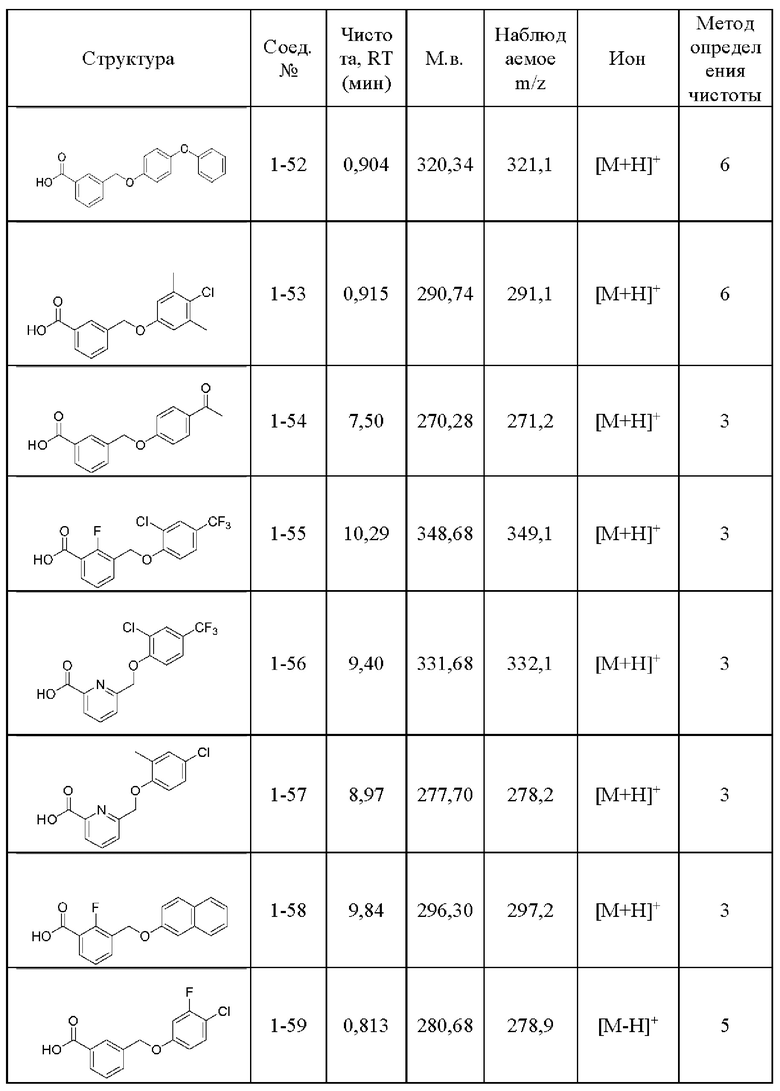
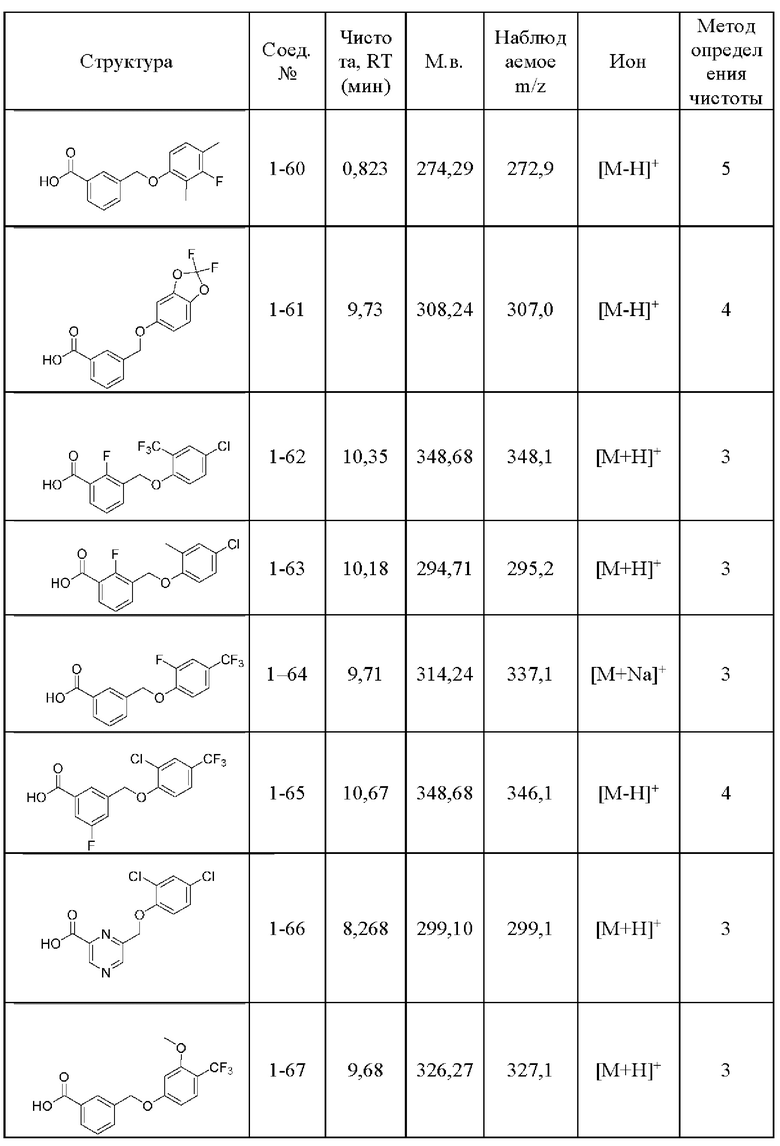
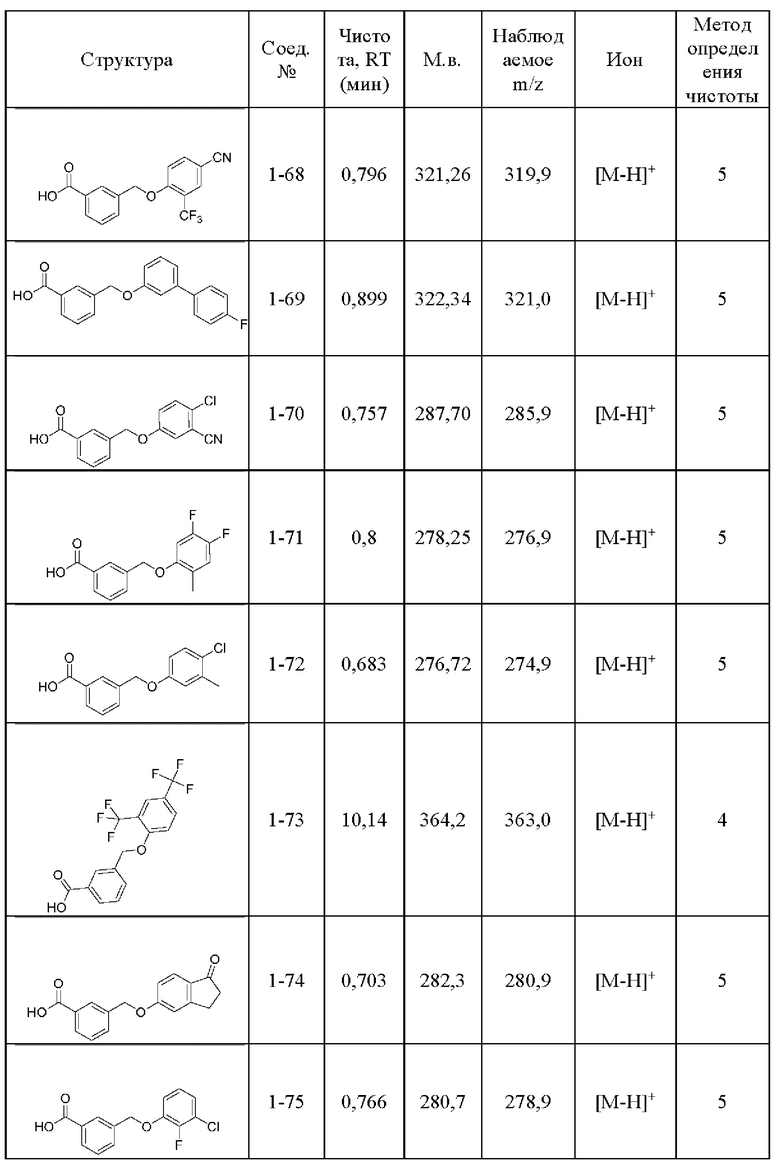
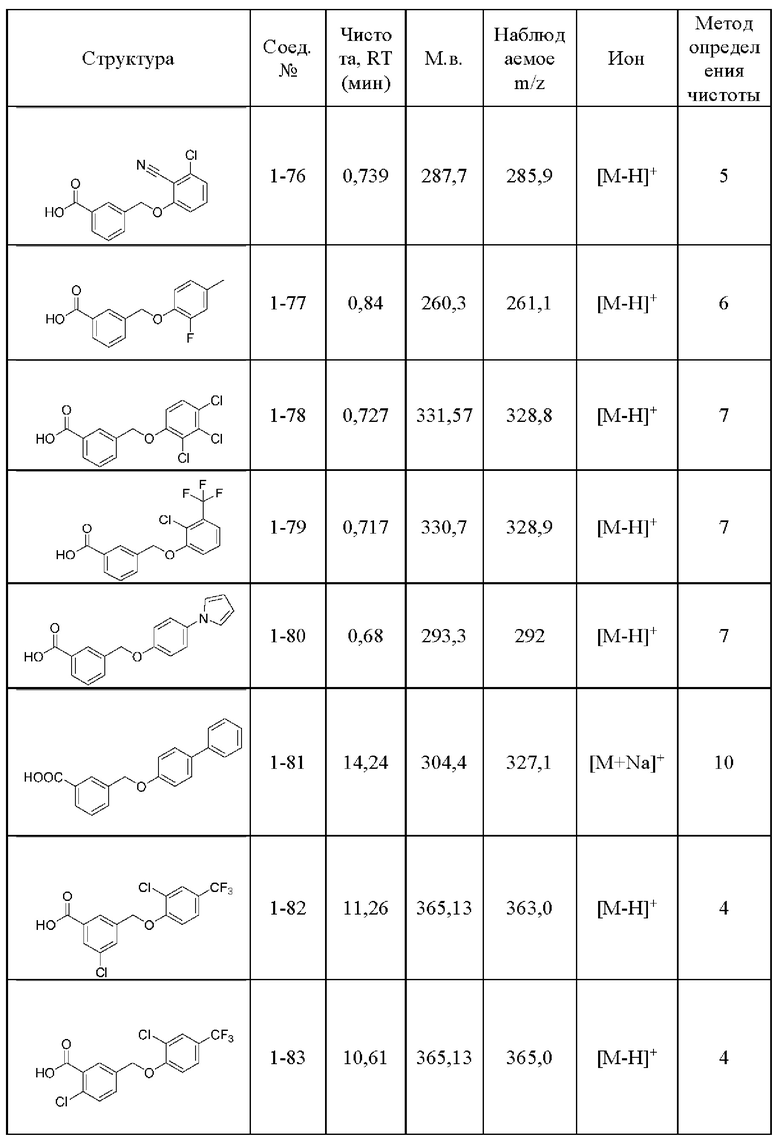
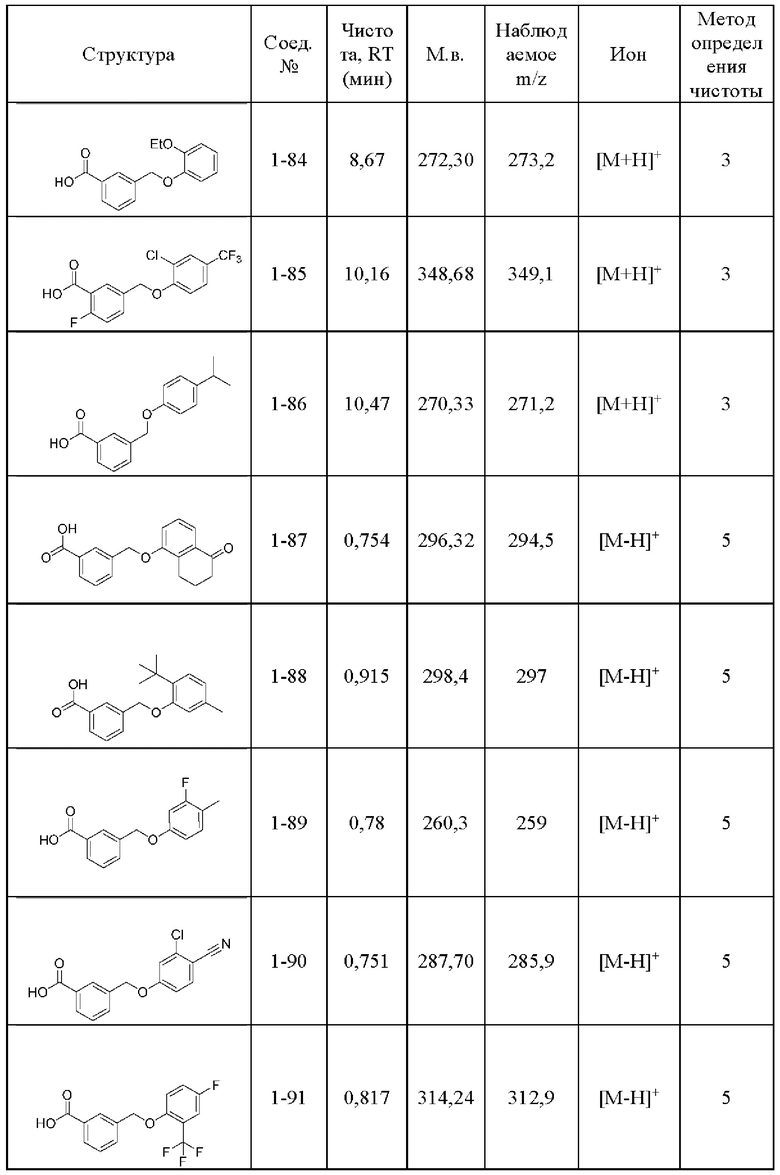
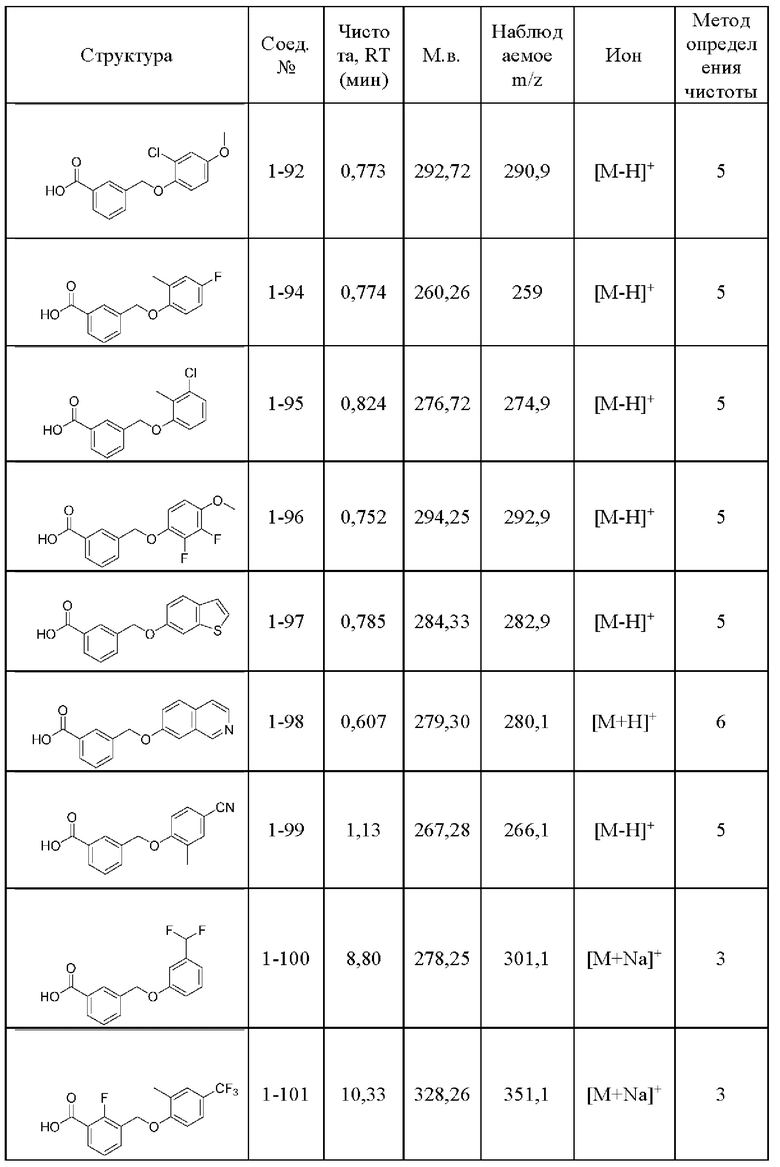
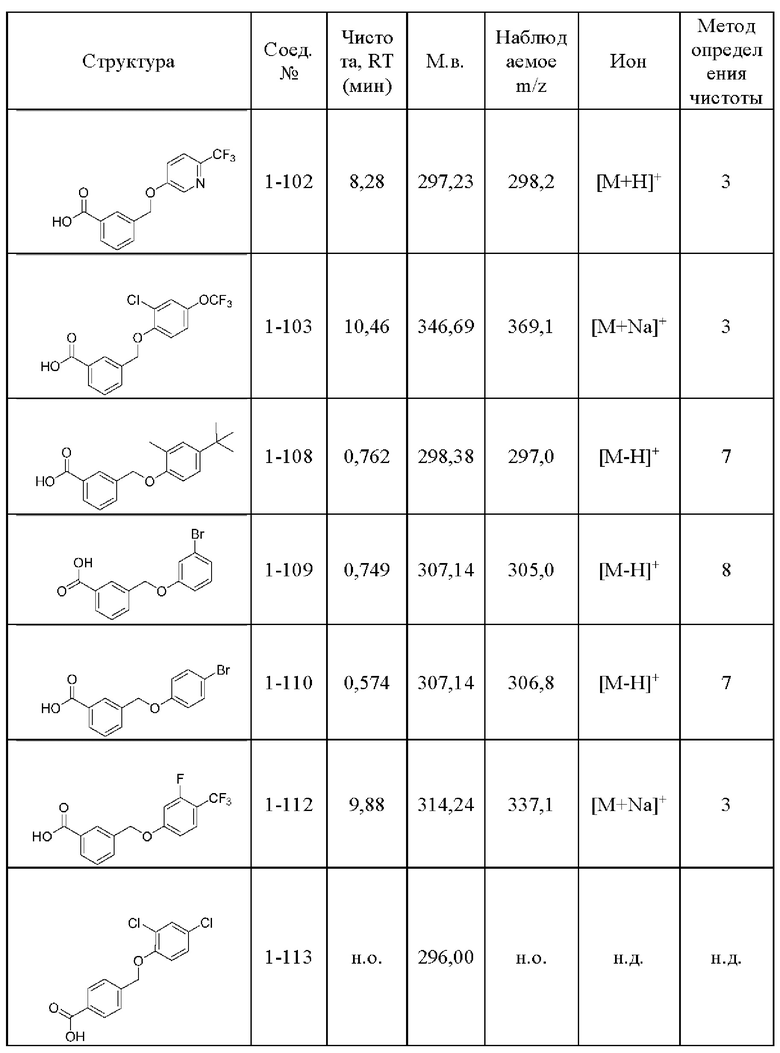
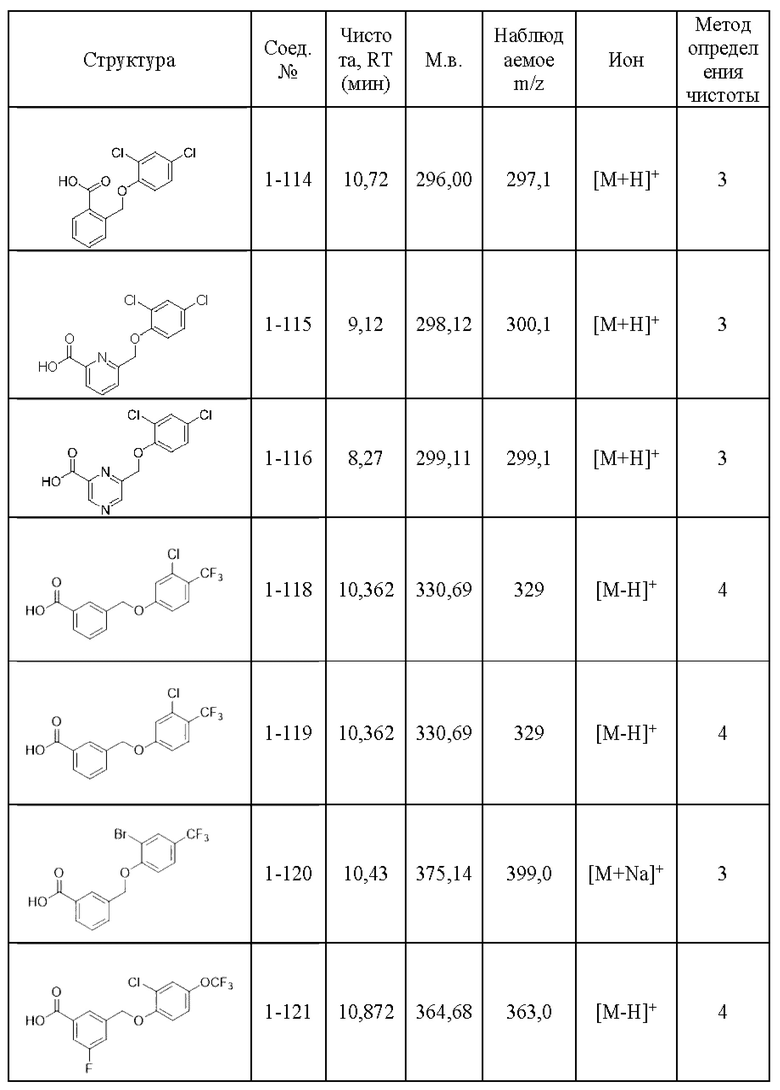
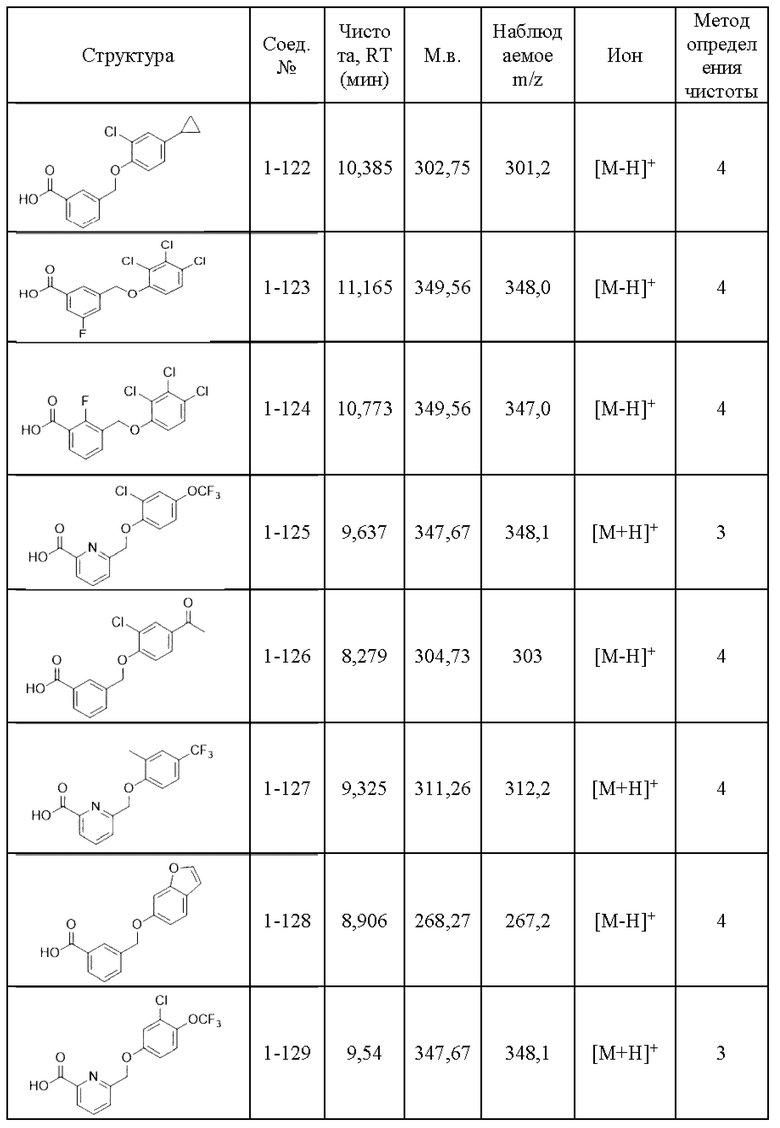
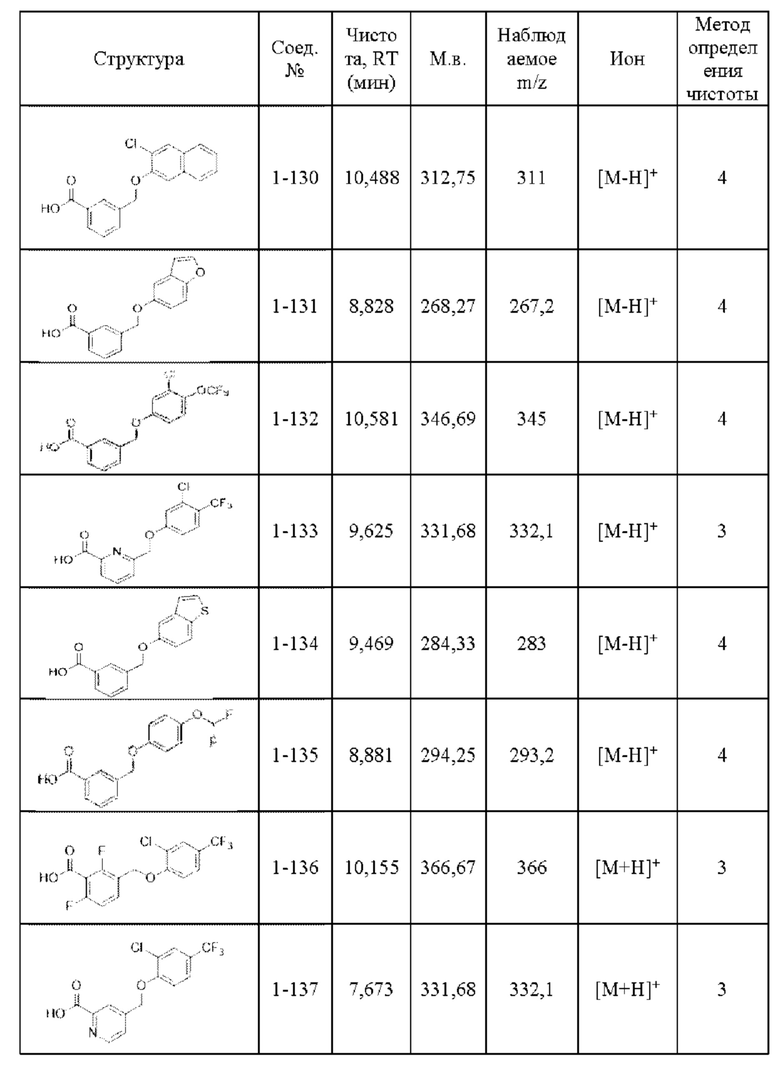
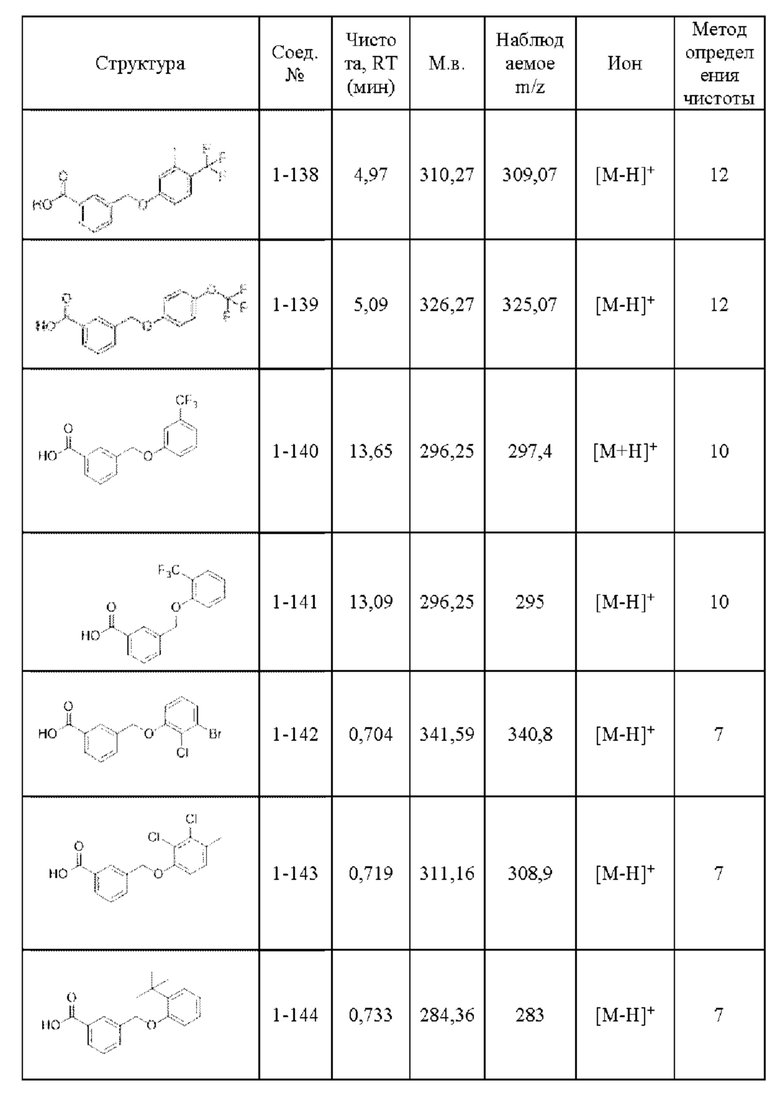
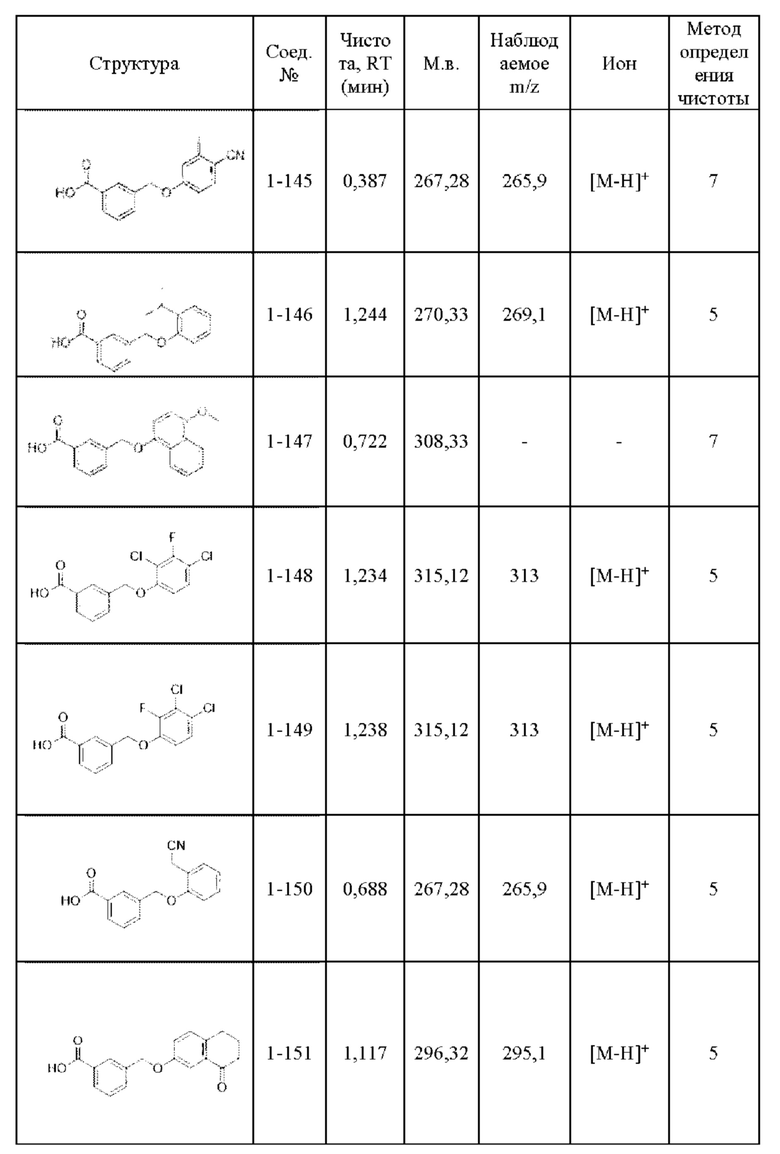
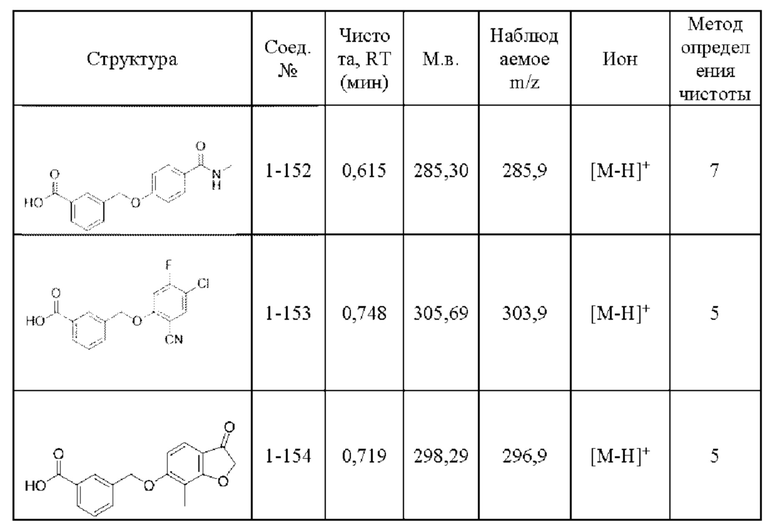
ПРИМЕР 2
Синтез Соединения 2-1 и других типичных соединений
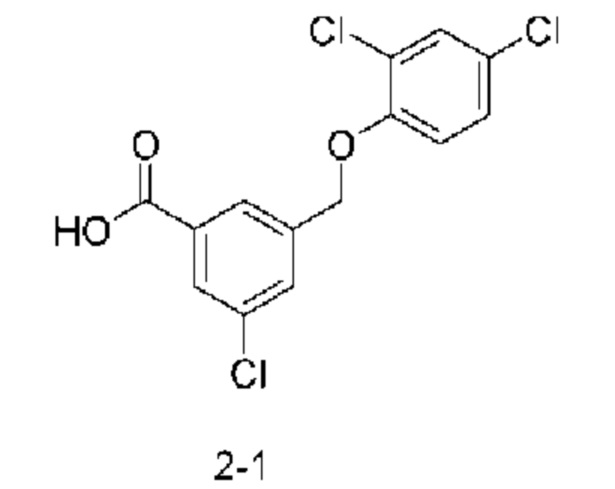
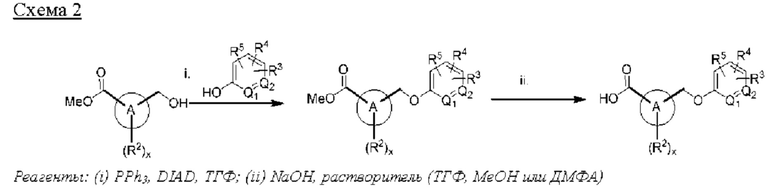
Стадия 2-1. Синтез метил-3-хлор-5-((2,4-дихлорфенокси)метил)бензоата (ΊΝΤ 2-A)
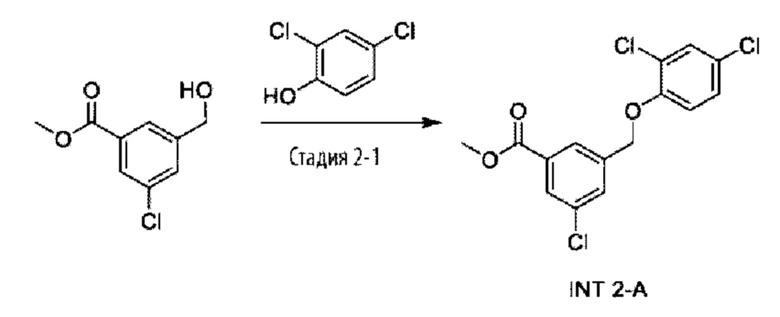
К перемешиваемому раствору метил-3-хлор-5-гидроксибензоата (100 мг, 0,50 ммоль) в ДХМ (5 мл) добавляли трифенилфосфин (131 мг, 0,50 ммоль) и DEAD (108,7 мкл, 0,60 ммоль). Реакционную смесь продували N2 (3 раза) и перемешивали при 10°С в течение 16 часов (в атмосфере N2), затем концентрировали и очищали флэш-хроматографией на SiO2 (ЭА/петролейный эфир) с получением 150 мг (87,0%) метил-3-хлор-5-((2,4-дихлорфенокси)метил)бензоата (INT 2-А) в виде твердого вещества розового цвета. ТСХ (10% ЭА/петролейный эфир): Rf = 0,50. 1Н ЯМР (400 МГц, CDCl3-d) δ 7,99 (д, J = 1,8 Гц, 2Н), 7,70-7,66 (м, 1H), 7,42 (д, J = 2,6 Гц, 1H), 7,34 (д, J = 2,4 Гц, 1H), 6,87 (д, J = 8,8 Гц, 1Н),5,14 (с, 2Н), 3,95 (с, 3Н).
Стадия 2-2. Синтез 3-хлор-5-((2,4-дихлорфенокси)метил)бензойной кислоты (Соединение 2-1)
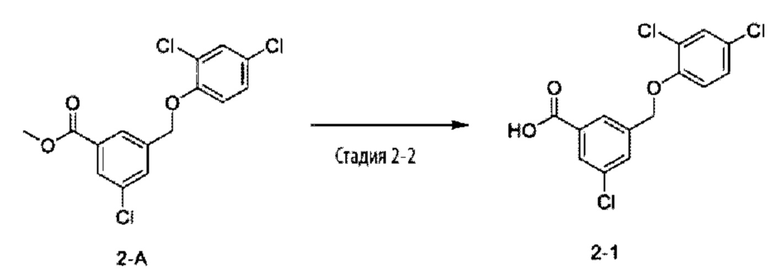
К перемешиваемому раствору метил-3-хлор-5-((2,4-дихлорфенокси)метил)бензоата (INT 2-А) (100 мг, 0,29 ммоль) в МеОН (1 мл) и ТГФ (1 мл) добавляли 2М NaOH (0,43 мл, 0,87 ммоль). Реакционную смесь нагревали при 30°С в течение 1 часа, а затем концентрировали в вакууме. Полученный остаток очищали с помощью ВЭЖХ с обращенной фазой, получая 12,6 мг (13%) 3-хлор-5-((2,4-дихлорфенокси)метил)бензойной кислоты (Соединение 2-1) в виде белого твердого вещества. ЖХ-MC-ESI (m/z) вычислено для C14H9Cl3O3: 331,5; найдено 328,8 [М-Н]+, tR = 0,72 мин. 1Н ЯМР (400 МГц, ДМСО-d6) δ 7,99 (с, 1Н), 7,86 (с, 1H), 7,77 (с, 1H), 7,62 (д, J = 2,6 Гц, 1Н), 7,40 (дд, J = 2,6, 8,8 Гц, 1H), 7,25 (д, J = 8,9 Гц, 1H), 5,31 (с, 2Н).
Соединения, перечисленные в Таблице 2, были получены с использованием процедур Схемы 2.
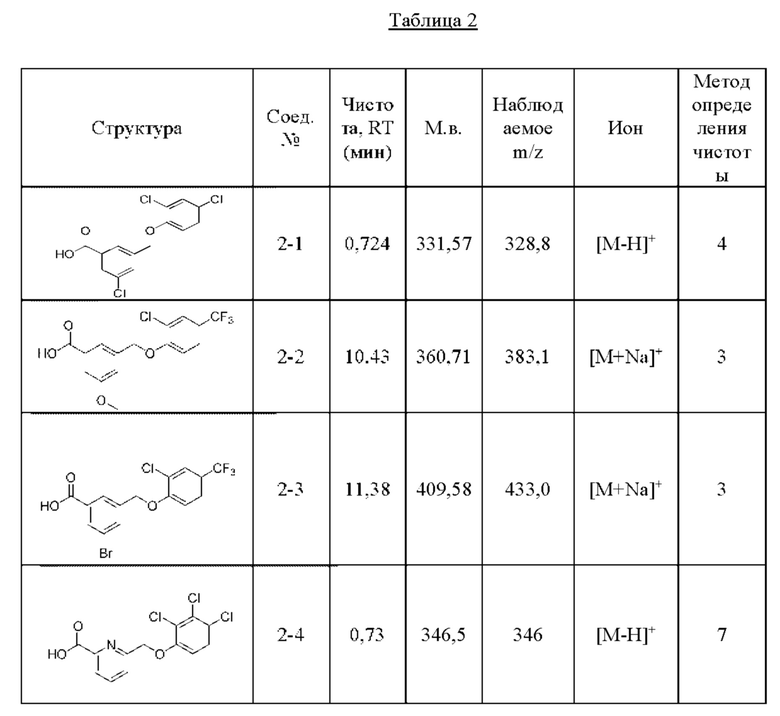
ПРИМЕР 3
Синтез Соединения 3-1. Соединения 3-2 и других типичных соединений 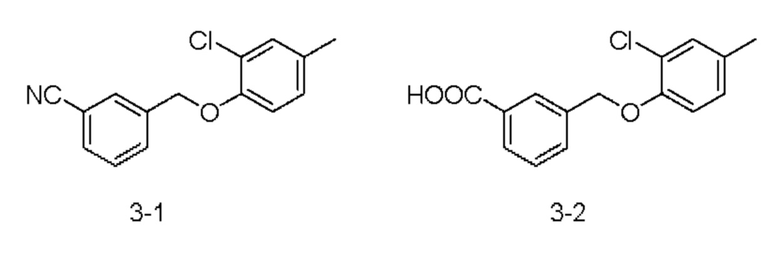
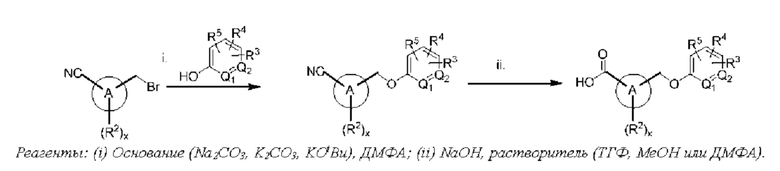
Стадия 3-1. Синтез 3-((2-хлор-4-метилфенокси)метил)бензонитрила (ΊΝΤ 3-А)
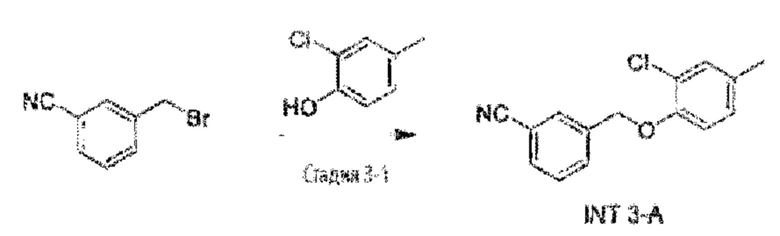
К перемешиваемому раствору 3-(бромметил)бензонитрила (500 мг, 2,55 ммоль) в ДМФА (8 мл) добавляли 2-хлор-4-метилфенол (360 мг, 2,5 ммоль) а также Na2CO3 (0,81 г, 7,65 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи, затем гасили 150 мл Н2О. Образовавшийся осадок собирали, промывали Н2О (2 × 20 мл) и осушали с получением 600 мг (91,3%) 3-((2-хлор-4-метилфенокси)метил)бензонитрила (ΙΝΤ 3-А). ЖХ-MC-ESI (m/z) вычислено для C15H12ClNO: 257,7; найдено 258,0 [М+Н]+, tR = 5,43 мин (Метод 11).
Соединение, указанное в Таблице 3А, было получено с использованием процедур Схемы 3, стадия 3-1, с использованием 3-(бромметил)бензонитрила и 2,4-дихлорфенола.
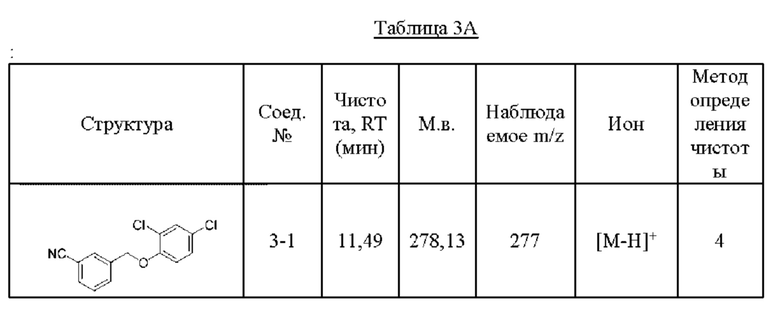
Стадия 3-2. Синтез 3-((2-хлор-4-метилфенокси)метил)бензойной кислоты (Соединение 3-2)

К перемешиваемому раствору 3-((2-хлор-4-метилфенокси)метил)бензонитрила (INT 3-А) (300 мг, 0,12 ммоль) в МеОН (5 мл) добавляли раствор NaOH (375 мг, 9,4 ммоль) в H2O (8 мл). Реакционный сосуд герметично закрывали и перемешивали при 90°С в течение ночи, затем охлаждали до комнатной температуры и концентрировали для удаления МеОН. Водный слой промывали ЭА и подкисляли 4 н. HCl. Полученный осадок собирали, получая 210 мг (65%) 3-((2-хлор-4-метилфенокси)метил)бензойной кислоты (Соединение 3-2). ЖХ-MC-ESI (m/z) вычислено для C15H13Cl1O3: 276,7; найдено 277,3 [М+Н]+, tR = 14,01 мин. 1Н ЯМР (400 МГц, CDCl3): 8,19 (с, 1Н), 8,06 (д, J = 8 Гц, 1Н), 7,77 (д, J = 8 Гц, 1H), 7,53 (т, J = 8 Гц, 1H), 7,22 (с, 1H), 6,99 (д, J = 8 Гц, 1H), 6,86 (д, J = 8 Гц, 1H), 5,18 (с, 2Н), 2,27 (с, 3Н).
Соединения, перечисленные в Таблице 3 В, были получены с использованием процедур Схемы 3, стадия 3-2.
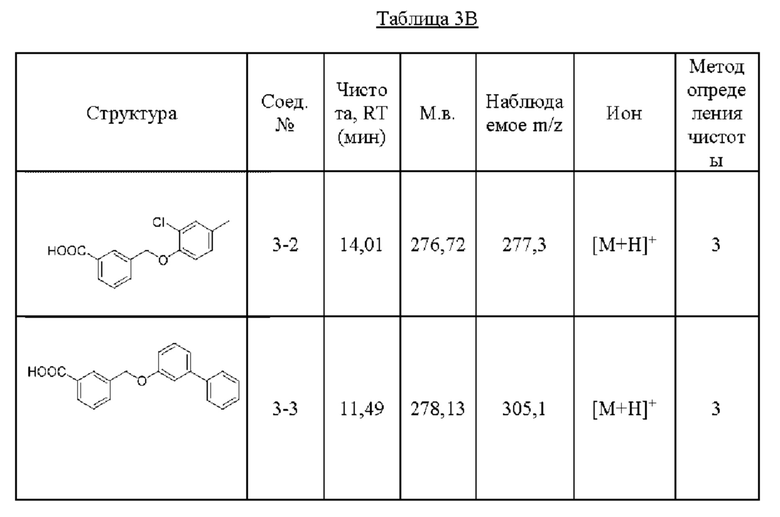
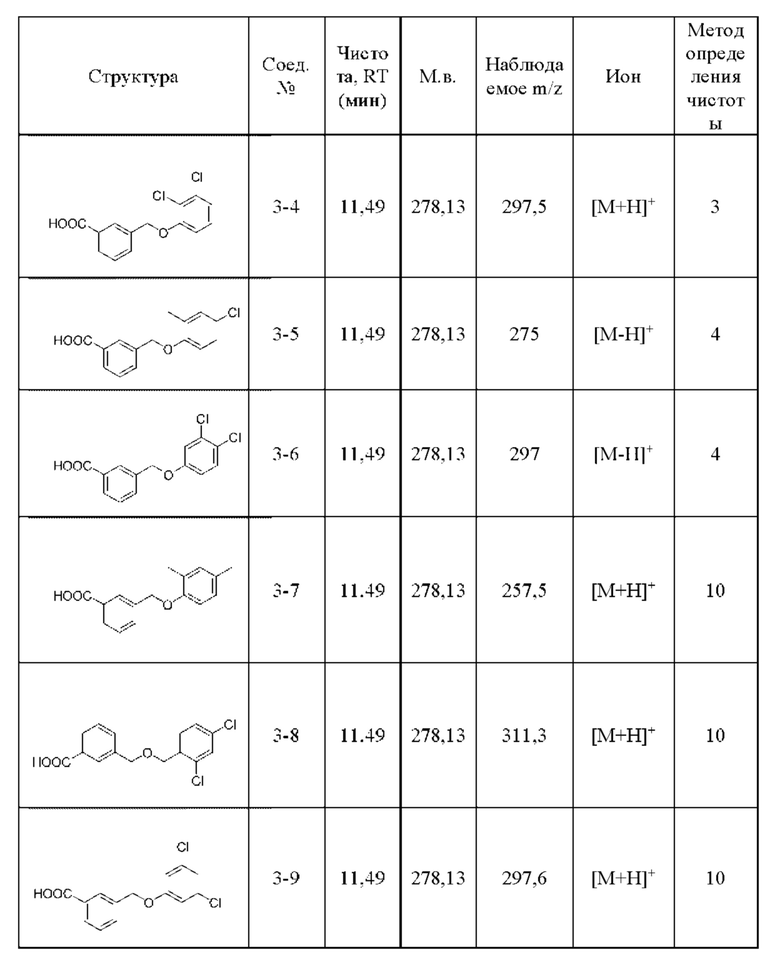
ПРИМЕР 4
Синтез Соединения 4-1 и других типичных соединений
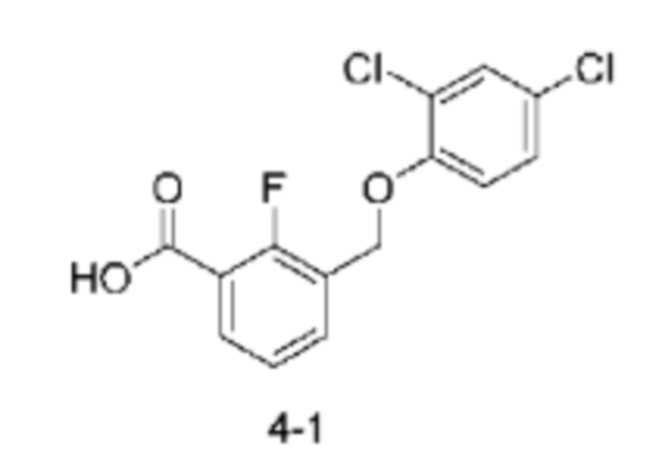
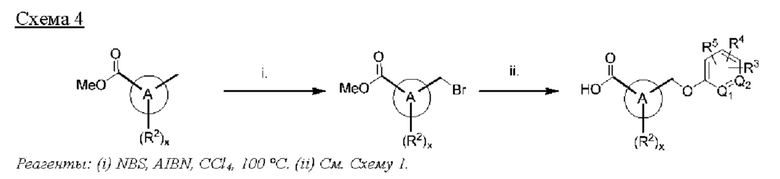
Стадия 4-1. Синтез метил-3-(бромметил)-2-фторбензоата (ΊΝΤ-4Α)
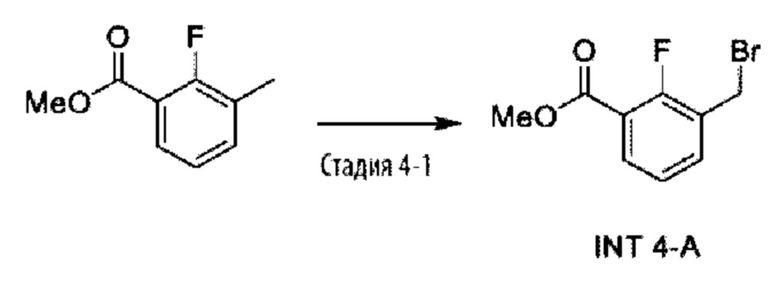
К перемешиваемому раствору метил-2-фтор-3-метилбензоата (1,0 г, 5,95 ммоль) в CCl4 (5 мл) добавляли NBS (1,06 г, 5,95 ммоль) и AIBN (19,6 мг, 119 ммоль). После перемешивания при 100°С в течение 2 часов реакционную смесь концентрировали, и полученный остаток очищали хроматографией на SiO2, получая 858 мг (58%) метил-3-(бромметил)-2-фторбензоата (INT-4A) в виде белого твердого вещества. ТСХ (10% ЭА/петролейный эфир): Rf = 0,50. ЖХ-MC-ESI (m/z) вычислено для C9H8BrFO2: 245,97; найдено 247,0 [М+Н]+, tR = 0,86 мин (Метод 6).
Стадия 4-2. Синтез метил-3-((2,4-дихлорфенокси)метил)-2-фторбензоата (IΝΤ 4-В)
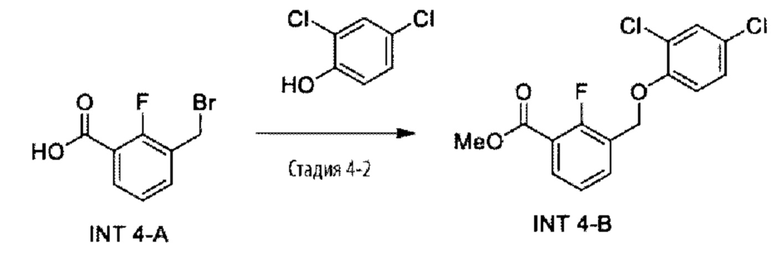
К перемешиваемому раствору метил-3-(бромметил)-2-фгорбензоата (INT-4A) (500 мг, 2,02 ммоль) в MeCN (2 мл) добавляли K2CO3 (559,4 мг, 4,05 ммоль) и 2,4-дихлорфенол (329,9 мг, 2,02 ммоль). После перемешивания при 50°С в течение 16 ч реакционную смесь концентрировали и полученный остаток очищали флэш-хроматографией на SiO2, получая 537 мг (81%) метил-3-((2,4-дихлорфенокси)метал)-2-фторбензоата (INT 4-В) в виде белого твердого вещества. ТСХ (10% ЭА/петролейный эфир): Rf = 0,45. ЖХ-MC-ESI (m/z) вычислено для C15H11Cl2BrFO3: 328,01; найдено 329,1 [М+Н]+, tR = 1,03 мин (Метод 6).
Стадия 4-3. Синтез 3-((2,4-дихлорфенокси)метил)-2-фторбензойной кислоты (Соединение 4-1)
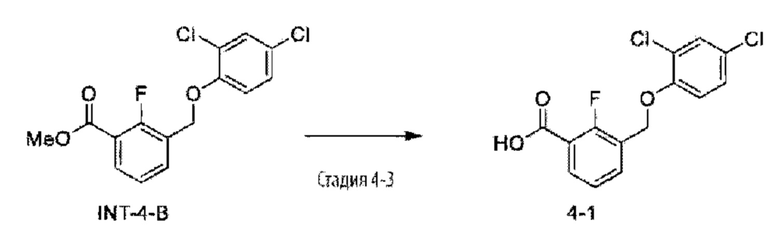
К перемешиваемому раствору метил-3-((2,4-дихлор фенокси)метил)-2-фтор бензоата (INT 4-В) (100 мг, 0,303 ммоль) в МеОН (1 мл) и ТГФ (1 мл) добавляли раствор 2М NaOH (455,7 мкл, 0,9 ммоль). После перемешивания при 10°С в течение 16 ч реакционную смесь концентрировали и полученный остаток очищали препаративной ВЭЖХ, получая 6,4 мг (7%) 3-((2,4-дихлорфенокси)метил)-2-фторбензойной кислоты (Соединение 4-1) в виде белого твердого вещества. ЖХ-MC-ESI (m/z), вычислено для C14H9Cl2FO3: 313,99; найдено 312,9 [М-Н]+, tR = 0,663 мин (Метод 7). 1Н ЯМР (400 МГц, ДМСО-d6) δ 7,84 (т, J = 6,7 Гц, 1Н), 7,75 (шир.т, J = 7,0 Гц, 1H), 7,60 (д, J = 2,4 Гц, 1H), 7,42-7,30 (м, 3Н), 5,28 (с, 2Н).
Синтез Соединения 1-55
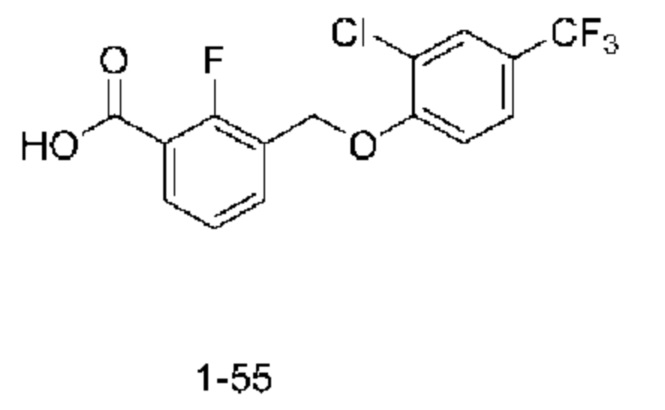
Стадия 4-4. Синтез метил-3-(бромметил)-2-фторбензоата (INT 4-С)
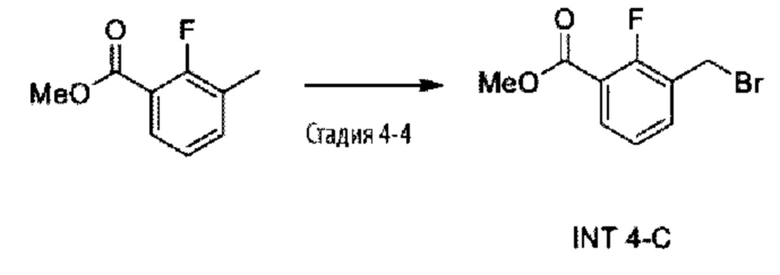
К перемешиваемому раствору метил-2-фтор-3-метилбензоата (1,0 г, 5,9 ммоль) в CCl4 (20 мл) добавляли NBS (1,2 г, 6,5 ммоль) и AIBN (98 мг, 0,59 ммоль). Реакционную смесь кипятили с обратным холодильником в течение 3 часов, затем охлаждали до комнатной температуры и концентрировали в вакууме с получением сырого продукта. Неочищенный продукт очищали хроматографией на SiO2 (ЭА/гексаны) с получением 399 мг (27%) метил-3-(бромметил)-2-фторбензоата (INT 4-С) в виде белого твердого вещества. ЖХ-MC-ESI (m/z) не наблюдается, tR = 5,05 мин. (Метод 7, минутный).
Стадия 4-5. Синтез метил-3-((2-хлор-4-(трифторметил)фенокси)метил)-2-фгорбензоата(INT 4-D)
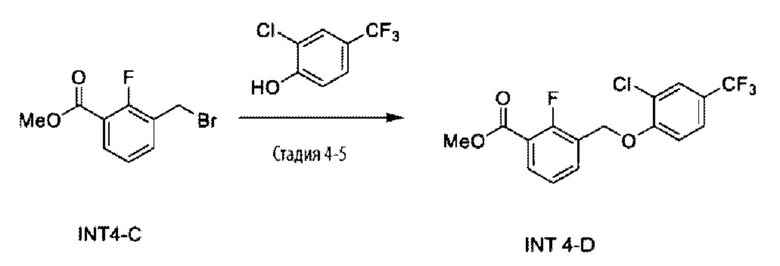
К перемешиваемому раствору INT 4-С (449 мг, 1,82 ммоль) в MeCN (4 мл) добавляли 2-хлор-4-(трифторметил)фенол (357 мг, 1,82 ммоль) и K2CO3 (327 мг, 2,36 ммоль). После нагревания в течение 18 ч при 60°С реакционную смесь охлаждали до комнатной температуры и разбавляли H2O (3 мл). Водный слой экстрагировали Et2O (2 × 6 мл) и ЭА (6 мл). Объединенные органические слои осушали (Na2SO4), фильтровали и концентрировали в вакууме, получая неочищенное белое твердое вещество, которое очищали хроматографией на SiO2 (ЭА/гексаны) с получением 551,6 мг (83,7%) метил-3-((2-хлор-4-(трифторметил)фенокси)метил)-2-фторбензоата (INT 4-D) в виде белого твердого вещества. ЖХ-MC-ESI (m/z) вычислено для C16H11ClF4O3: 362,7; найдено 363,1 [М+Н]+ (Метод 7 минут).
Стадия 4-6. Синтез 3-((2-хлор-4-(трифторметил)фенокси)метил)-2-фторбензойной кислоты (1-55)
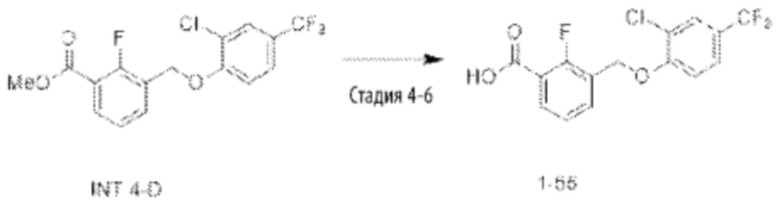
К перемешиваемому раствору INT 4-D (551 мг, 1,52 ммоль) в ТГФ (8 мл) добавляли 1М NaOH (7,6 мл, 7,60 ммоль). Реакционную смесь нагревали при 60°С в течение ночи, затем концентрировали в вакууме, разбавляли 3М HCl, экстрагировали ЭА и Et2O, осушали (Na2SO4), фильтровали и концентрировали в вакууме. Полученное белое твердое вещество растворяли в MeCN (5 мл) и H2O (5 мл) и лиофилизировали с получением 460,5 мг (86,9%) 3-((2-хлор-4-(трифторметил)фенокси)метил)-2-фторбензойной кислоты (Соединение 1-55) в виде белого твердого вещества. ЖХ-MC-ESI (m/z) вычислено для C15H9ClF4O3: 348,68; найдено 349,1 [М+Н]+, tR = 10,28 мин (чистота 15 мин). 1Н ЯМР (500 МГц, ДМСО-d6) δ 13,36 (шир.с, 1Н), 7,92-7,87 (м, 2Н), 7,83-7,80 (м, 1Н), 7,73 (дд, J = 8,5, 2,0 Гц, 1Н), 7,51 (д, J = 8,5 Гц, 1H), 7,36 (кажущ.т, J = 7,5 Гц, 1Н), 5,40 (с, 2Н).
Синтез Соединения 1-65
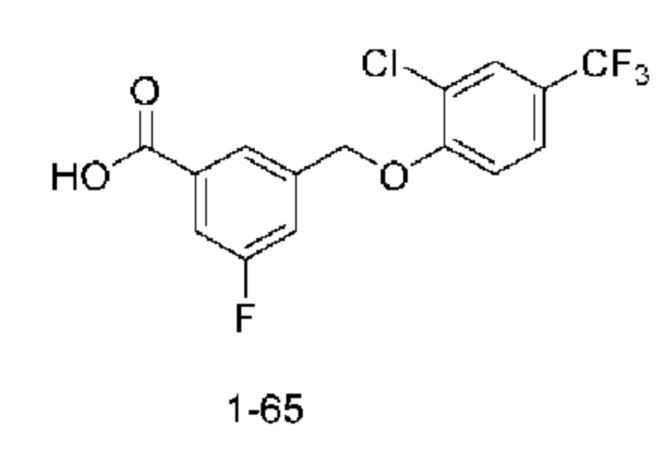
Стадия 4-7. Синтез метил-3-фтор-5-метилбензоата (ΊΝΤ 4-Е)
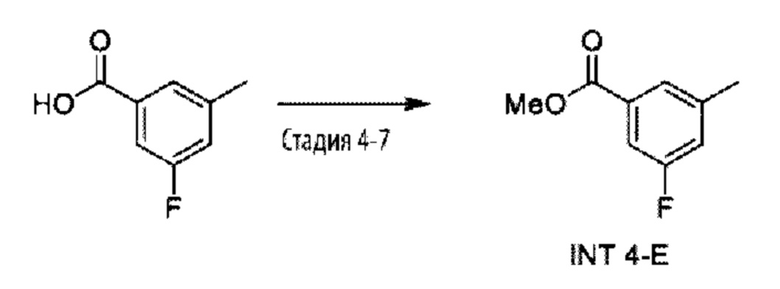
Раствор 3-фтор-5-метилбензойной кислоты (5 г, 32,4 ммоль) и H2SO4 (15,91 г, 162,2 ммоль, 8,65 мл) в МеОН (30 мл) перемешивали при 70°С в течение 12 часов. Реакционную смесь выливали в Н2О (100 мл) и экстрагировали ЭА. Объединенные органические фазы осушали и концентрировали с получением остатка, который очищали хроматографией на SiO2 (ПЭ/ЭА) с получением 4,5 г (82,5%) метил-3-фтор-5-метилбензоата (INT 4-Е) в виде желтой маслянистой жидкости. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 2,40 (с, 3Н), 3,92 (с, 3Н), 7,08 (шир.д, J = 9,26 Гц, 1H), 7,51 (шир.д, J = 9,13 Гц, 1H), 7,65 (с, 1H).
Стадия 4-8. Синтез метил-3-(бромметил)-5-фторбензоата (TNT 4-F)
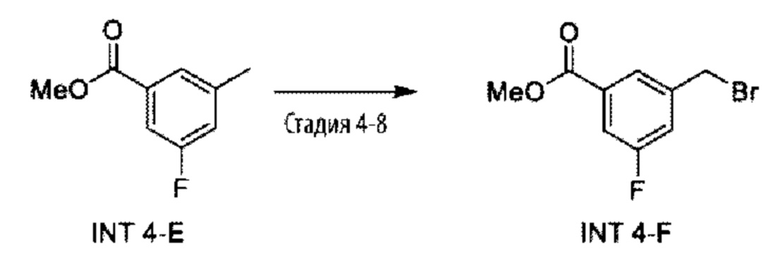
Раствор INT 4-Е (4,5 г, 26,76 ммоль), NBS (5,24 г, 29,44 ммоль) и AIBN (219,71 мг, 1,34 ммоль) в CCl4 (50 мл) перемешивали при 70°С в течение 12 часов. Реакционную смесь концентрировали и очищали хроматографией на SiO2 (ПЭ/ЭА) с получением 4,9 г (74%) сырого метил-3-(бромметил)-5-фторбензоата (INT 4-F) в виде желтой маслянистой жидкости. ТСХ (10:1 петролейный эфир:ЭА): Rf = 0,70. 1Η ЯМР (400 МГц, CDCl3) δ м.д. 7,86 (т, J = 1,41 Гц, 1H), 7,64-7,67 (м, 1Н), 7,31 (дт, J = 8,71, 2,06 Гц, 1H), 4,48 (с, 2Н), 3,94 (с, 3Н).
Стадия 4-9. Синтез метил-3-((2-хлор-4-(трифторметил)фенокси)метил)-5-фторбензоата(INT 4-G)
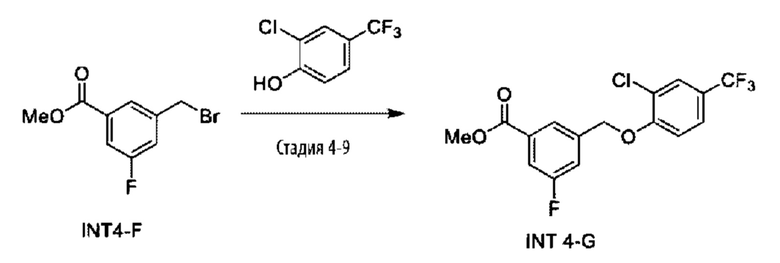
Смесь INT 4-F (3 г, 12,1 ммоль), 2-хлор-4-(трифторметил)фенола (3,58 г, 18,2 ммоль) и K2CO3 (5,03 г, 36,4 ммоль) в MeCN (50 мл) перемешивали при 30°С в течение 12 часов. Реакционную смесь фильтровали и концентрировали с получением остатка, который очищали хроматографией на SiO2 (ПЭ/ЭА) с получением 2,4 г (55%) метил-3-((2-хлор-4-(трифторметил)фенокси)метил)-5-фторбензоата (INT 4-G) в виде белого твердого вещества. ТСХ (5:1 петролейный эфир:ЭА): Rf = 0,60.
Стадия 4-10. Синтез 3-((2-хлор-4-(трифторметил)фенокси)метил)-5-фторбензойной кислоты (1-65)
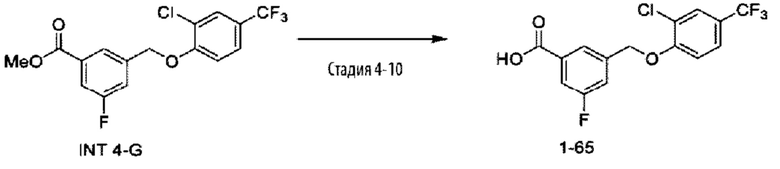
Смесь INT 4-G (2,4 г, 6,6 ммоль) и NaOH (794 мг, 19,9 ммоль) в ТГФ (1 мл) и H2O (0,5 мл) перемешивали при 30°С в течение 2 часов. Реакционную смесь подкисляли 1 н. HCl и экстрагировали ЭА. Объединенные органические слои осушали (Na2SO4) и концентрировали, чтобы получить остаток, который растворяли в ПЭ/ЭА и фильтровали. Осадок на фильтре разбавляли MeCN/Н2О и лиофилизировали с получением 1,91 г (82%) 3-((2-хлор-4-(трифторметил)фенокси)метил)-5-фторбензойной кислоты (Соединение 1-56) в виде белого твердого вещества. ЖХ-MC-ESI (m/z) вычислено для C15H9ClF4O3: 348,6; найдено 347,0 [М-Н]+, tR = 0,958 мин (Метод 8). 1Н ЯМР (400 МГц, CDCl3-d) δ м.д. 7,99 (с, 1H), 7,77 (шир.д, J = 8,19 Гц, 1H), 7,70 (д, J = 1,96 Гц, 1H), 7,51 (шир.д, J = 8,68 Гц, 2Н), 7,03 (д, J = 8,56 Гц, 1H), 5,26 (с, 2Н).
Синтез Соединения 1-85
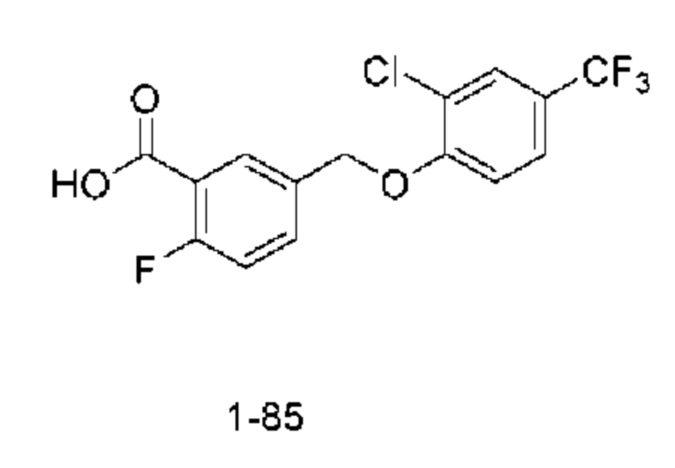
Стадия 4-11. Синтез метил-2-фтор-5-метилбензоата (INT 4-H)
К раствору 2-фтор-5-метилбензойной кислоты (10 г, 64,9 ммоль) в МеОН (200 мл) по каплям при 25°С добавляли тионилхлорид (23,53 мл, 324,4 ммоль). Через 0,5 ч при 25°С смесь концентрировали и очищали хроматографией на SiO2 (ПЭ/ЭА) с получением 10,6 г (97%) метил-2-фтор-5-метилбензоата (INT 4-Н) в виде бесцветной маслянистой жидкости. ТСХ(1:1 петролейный эфир:ЭА): Rf = 0,90.
Стадия 4-12. Синтез метил-5-(бромметил)-2-фторбензоата (TNT 4-I)
К раствору INT 4-Н (8 г, 47,6 ммоль) в CHCl3 (200 мл) добавляли NBS (10,16 г, 57,1 ммоль) и AIBN (781,2 мг, 4,76 ммоль). Через 12 ч при 70°С реакционную смесь разбавляли Н2О (200 мл) и экстрагировали ЭА (3 × 100 мл). Объединенные органические слои осушали (Na2SO4), концентрировали и очищали хроматографией на SiO2 (ПЭ/ЭА) с получением 10,6 г (97%) метил-5-(бромметил)-2-фгорбензоата (INT 4-I) в виде белого твердого вещества, загрязненного вторым неидентифицированным продуктом. ТСХ (10:1 петролейный эфир:ЭА): Rf=0,4, 0,35. ЖХ-MC-ESI (m/z) вычислено для C9H8BrFO2: 247,06; найдено 248,8 [М-Н]+, tR = 0,702 мин. 1Н ЯМР (400 МГц, CDCl3-d) δ 8,01-7,94 (м, 1Н), 7,62-7,52 (м, 1H), 7,17-7,10 (м, 1H), 4,49 (с, 2Н), 3,95 (с, 3Н).
Стадия 4-13. Синтез метил-5-((2-хлор-4-(трифторметил)фенокси)метил)-2-фторбензоата (IΝΤ4-J)
К раствору INT 4-1 (4 г, 16,19 ммоль) и 2-хлор-4-(трифторметил)фенола (3,18 г, 16,19 ммоль) в MeCN (30 мл) добавляли K2CO3 (6,71 г, 48,57 ммоль). Через 2 ч при 50°С реакционную смесь фильтровали, концентрировали и очищали хроматографией на SiO2 (ПЭ), получая 1,7 г (29%) метил-5-((2-хлор-4-(трифторметил)фенокси)метил)-2-фторбензоата (INT 4-J) в виде белого твердого вещества. ТСХ (10:1 петролейный эфир:ЭА): Rf = 0,40. ЖХ-MC-ESI (m/z) вычислено для C16H11ClF4O3: 362,7; найдено 363,0 [М-Н]+, tR = 1,07 мин (Метод 6). 1Н ЯМР (400 МГц, CDCl3-d) δ 8,05 (дд, J = 2,3, 6,7 Гц, 1H), 7,72-7,63 (м, 2Н), 7,55-7,45 (м, 1H), 7,26-7,15 (м, 1H), 7,04 (д, J = 8,6 Гц, 1H), 5,20 (с, 2Н), 3,97 (с, 3Н).
Стадия 4-14. Синтез 5-((2-хлор-4-(трифторметил)фенокси)метил)-2-фторбензойной кислоты (1-85)
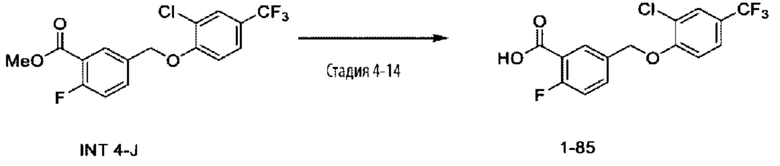
К раствору INT 4-J (1,7 г, 4,63 ммоль) в H2O (10 мл), ТГФ (10 мл) и МеОН (5 мл) добавляли LiOH⋅H2O (582,33 мг, 13,88 ммоль). Через 2 ч при 25°С добавляли в реакционную смесь Н2О (30 мкл) и органический растворитель удаляли при пониженном давлении с получением 1,57 г (97%) 5-((2-хлор-4-(трифторметил)фенокси)метил)-2-фторбензойной кислоты (Соединение 1-85) в виде белого твердого вещества. ЖХ-MC-ESI (m/z) вычислено для C15H9ClF4O3: 348,68; найдено 349,0 [М-Н]+, tR = 0,925 мин (Метод 6). 1Н ЯМР (400 МГц, ДМСО-d6) δ 7,96-7,90 (м, 1H), 7,87 (д, J = 2,0 Гц, 1H), 7,72 (дд, J = 1,7, 8,7 Гц, 1H), 7,66 (дт, J = 2,3, 5,3 Гц, 1H), 7,44 (д, J = 8,6 Гц, 1H), 7,36-7,28 (м, 1H), 5,34 (с, 2Н).
Синтез Соединения 1-101
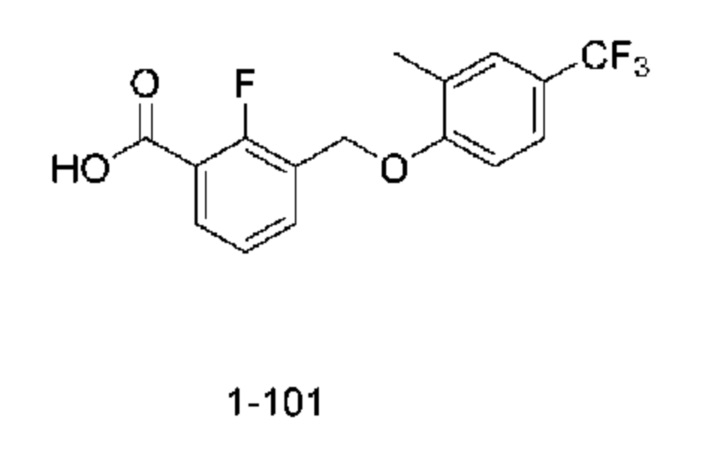
Стадия 4-15. Синтез 4,4,5,5 -тетраметил-2-(2-метил-4-(трифторметил)фенил)-1,3,2-диоксаборолана (INT 4-K)
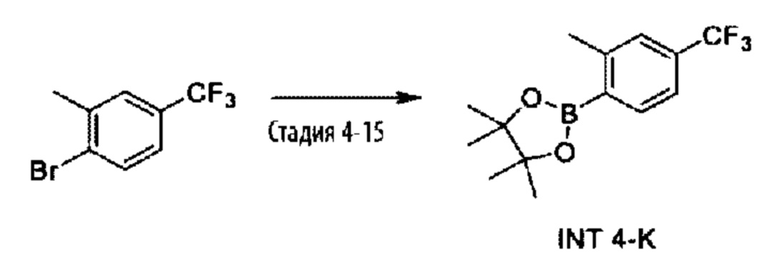
В раствор 1-бром-2-метил-4-(трифторметил)бензола (7,3 г, 30,5 ммоль) в диоксане (100 мл) добавляли AcOK (11,99 г, 122,16 ммоль, 4 экв.), 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолан) (15,51 г, 61,1 ммоль) и Pd(dppf)Cl2⋅CH2Cl2 (2,49 г, 3,05 ммоль). После перемешивания в течение 12 ч при 100°С в атмосфере N2 смесь фильтровали и фильтрат концентрировали с получением сырого продукта, который очищали хроматографией на SiO2 (ПЭ) с получением 6,3 г (72%) 4,4,5,5-тетраметил-2-(2-метил-4-(трифторметил)фенил)-1,3,2-диоксаборолана(INT 4-K) в виде желтой маслянистой жидкости. ТСХ (ПЭ): Rf = 0,90. 1Н ЯМР (400 МГц, CDCl3-d) δ 7,93-7,84 (м, 1Н), 7,46-7,39 (м, 2Н), 2,61 (с, 3Н), 1,38 (с, 13Н).
Стадия 4-16. Синтез 2-метил-4-(трифторметил)фенола (INT 4-L)
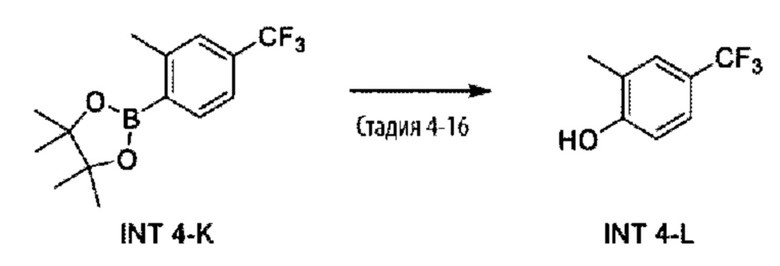
К раствору INT 4-K (5,8 г, 20,27 ммоль) в EtOH (40 мл) и Η2O (20 мл) добавляли m-СРВА (6,17 г, 30,41 ммоль, чистота 85%). После перемешивания в течение 12 часов при 25°С смесь выливали в насыщенный Na2SO3 (100 мл) и концентрировали для удаления летучих веществ. Полученный раствор разбавляли Н2О (50 мл) и экстрагировали ЭА (3 × 80 мл). Объединенные органические слои промывали насыщенным раствором NaHCO3 (2 × 50 мл) и рассолом (100 мл × 2), затем осушали (Na2SO4), концентрировали и очищали хроматографией на SiO2 с получением 2,5 г (70%) 2-метил-4-(трифторметил)фенола (INT 4-L) в виде бесцветной маслянистой жидкости. ТСХ (5:1 ПЭ:ЭА): Rf = 0,50. 1Н ЯМР (400 МГц, CDCl3-d) δ 7,45-7,39 (м, 1Н), 7,38-7,32 (м, 1H), 6,90-6,79 (м, 1H), 5,87-5,77 (м, 1H), 2,31 (с, 3Н).
Стадия 4-17. Синтез метил-2-фтор-3-((2-метил-4-(трифторметил)фенокси)метил)бензоата (INT 4-М)
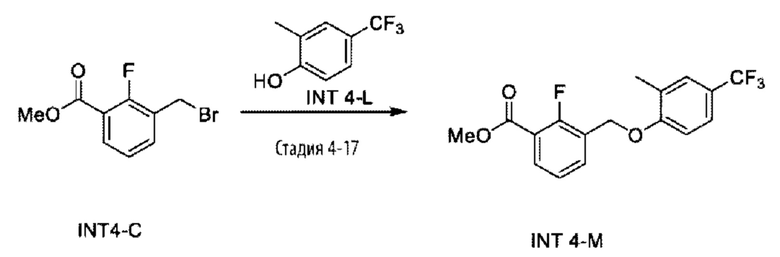
К раствору INT 4-С (2,8 г, 11,33 ммоль) и INT 4-L (2,4 г, 13,6 ммоль) в MeCN (30 мл) добавляли K2CO3 (2,04 г, 14,7 ммоль). Через 12 ч при 60°С реакционную смесь фильтровали, концентрировали и очищали хроматографией на SiO2 (ЭА/ПЭ) с получением 3,0 г (77%) метил-2-фтор-3-((2-метил-4-(трифторметил)фенокси)метил)бензоата (INT 4-М) в виде бесцветной маслянистой жидкости. ЖХ-MC-ESI (m/z) вычислено для C17H14F4O3: 342,29; найдено 343,0 [М+Н]+, tR = 1,04 мин (Метод 6). 1Н ЯМР (400 МГц, CDCl3-d) δ 7,98-7,91 (м, 1H), 7,77-7,69 (м, 1H), 7,49-7,41 (м, 2Н), 7,31-7,22 (м, 1H), 7,03-6,92 (м, 1H), 5,40-5,15 (м, 2Н), 3,98 (с, 3Н), 2,34 (с, 3Н).
Стадия 4-18. Синтез 5-((2-метил-4-(трифторметил)фенокси)метил)-2-фторбензойной кислоты (1-101)
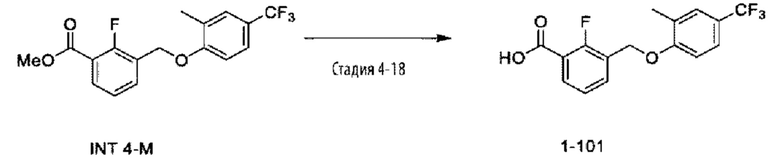
К раствору ΙΝΤ 4-М (3,0 г, 8,76 ммоль) в ТГФ (30 мл) и МеОН (30 мл) добавляли 2М NaOH (30 мл, 60 ммоль). Через 12 ч при 40°С рН доводили до рН 5 с помощью HCl (1М) для получения твердого осадка, который фильтровали и собирали. Полученный продукт растворяли в ЭА (500 мл), осушали (Na2SO4), фильтровали и концентрировали с получением 2,35 г (81%) 5-((2-метил-4-(трифторметил)фенокси)метил)-2-фторбензойной кислоты (1-101) в виде светло-желтого твердого вещества. ЖХ-MC-ESI (m/z) вычислено для C16H12F4O3: 328,2; найдено 326,9 [М-Н]+, tR = 0,73 мин (Метод 7). 1Н ЯМР (400 МГц, CD4OD) δ 7,90-7,82 (м, 1H), 7,74-7,66 (м, 1H), 7,52-7,42 (м, 2Н), 7,32-7,24 (м, 1H), 7,20-7,13 (м, 1H), 5,44-5,15 (м, 2Н), 2,30 (с, 3Н).
Синтез Соединения 4-10
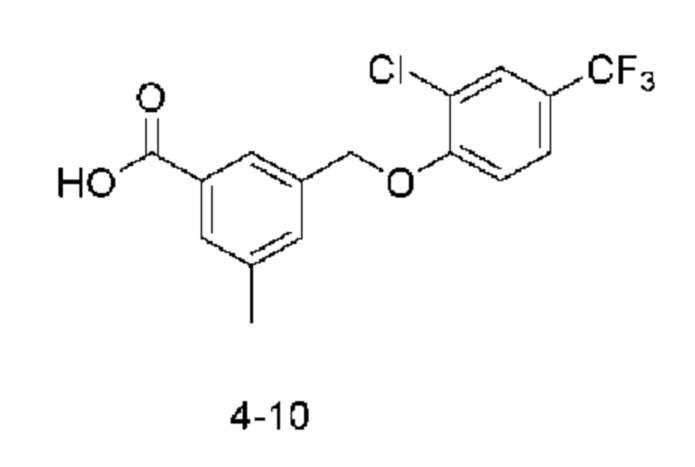
Стадия 4-19. Синтез метил-3-(бромметил)-5-метилбензоата (ΊΝΤ 4-Ν)
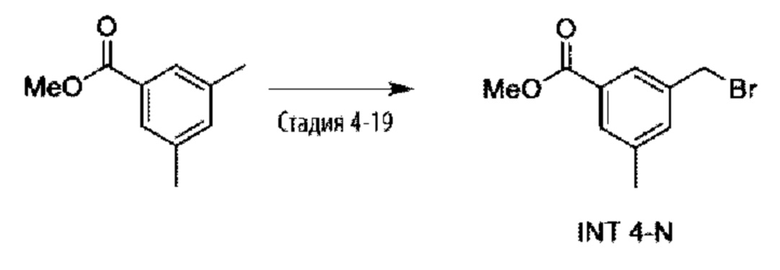
К раствору метил-3,5-диметилбензоата (5 г, 30,5 ммоль) в CCl4 (200 мл) добавляли NBS (5,96 г, 33,5 ммоль) и ΑΙΒΝ (1,00 г, 6,1 ммоль). После перемешивания в течение 12 часов при 80°С реакционную смесь концентрировали и очищали хроматографией на SiO2 (ПЭ/ЭА) с получением 8,4 г (79%) сырого метил-3-(бромметил)-5-метилбензоата (ΙΝΤ 4-Ν) в виде бесцветной маслянистой жидкости с чистотой 70%. PX-MC-ESI (m/z), вычислено для С10Н11 BrO2: 243,1; найдено 245 [М+Н]+, tR = 0,873 мин. 1Н ЯМР (400 МГц, CDCl3-d) δ 7,87 (с, 1H), 7,80 (с, 1H), 7,48-7,37 (м, 1H), 4,49 (с, 2Н), 3,96-3,89 (м, 4Н), 2,41 (с, 5Н).
Стадия 4-20. Синтез метил-3-((2-хлор-4-(трифторметил)фенокси)метил)-5-метилбензоата (IΝΤ 4-O)
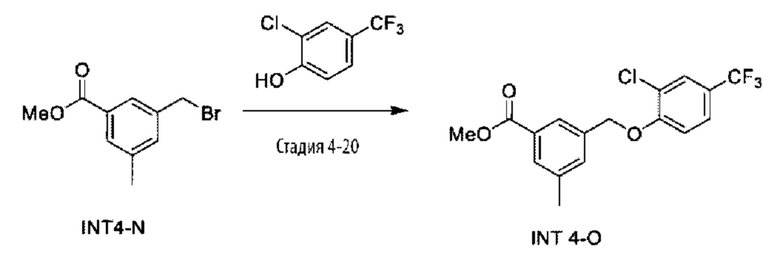
Смесь INT 4-N (3 г, 8,64 ммоль), 2-хлор-4-(трифторметил)фенола (1,7 г, 8,64 ммоль) и K2CO3 (5,03 г, 36,4 ммоль) в MeCN (30 мл) перемешивали при 60°С в течение 12 часов. Реакционную смесь фильтровали и концентрировали с получением остатка, который очищали хроматографией на SiO2 (ПЭ/ЭА) с получением 2,8 г (90%) метил-3-((2-хлор-4-(трифторметил)фенокси)метил)-5-метилбензоата (INT 4-O) в виде белого твердого вещества. ТСХ (5:1 петролейный эфир:ЭА): Rf = 0,60. ЖХ-MC-ESI (m/z) вычислено для C17H14ClF3O3: 358,7; найдено 359 [М+Н]+, tR = 1,06 мин.
Стадия 4-21. Синтез 3-((2-хлор-4-(трифторметил)фенокси)метил)-5-метилбензойной кислоты (4-10)
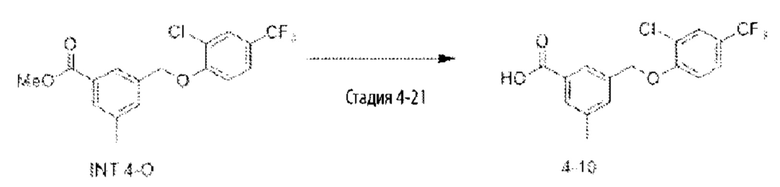
Смесь INT 4-0 (2,8 г, 7,81 ммоль) и 2М NaOH (30 мл, 30 ммоль) в ТГФ (30 мл) и МеОН (30 мл) перемешивали при 30°С в течение 12 часов. Летучие растворители удаляли в вакууме и полученный раствор подкисляли 1 н. HCl до рН 5. Полученный осадок собирали фильтрованием, и сырой продукт растирали с 10:1 ПЭ:ЭА с получением 2,1 г (72%) 3-((2-хлор-4-(трифторметил)фенокси)метил)-5-метилбензойной кислоты (Соединение 4-10) в виде белого твердого вещества. ЖХ-MC-ESI (m/z) вычислено для C16H12ClF3O3: 344,71; найдено 342,9 [М+Н]+, tR = 0,761 мин (Метод 7). 1Н ЯМР (400 МГц, CD4OD) δ 8,02-7,95 (м, 1H), 7,86-7,81 (м, 1H), 7,75-7,68 (м, 1H), 7,62-7,52 (м, 2Н), 7,36-7,27 (м, 1Н), 5,29 (с, 2Н), 2,44 (с, 3Н).
Соединения, перечисленные в Таблице 4, были получены с использованием процедур Схемы 4:
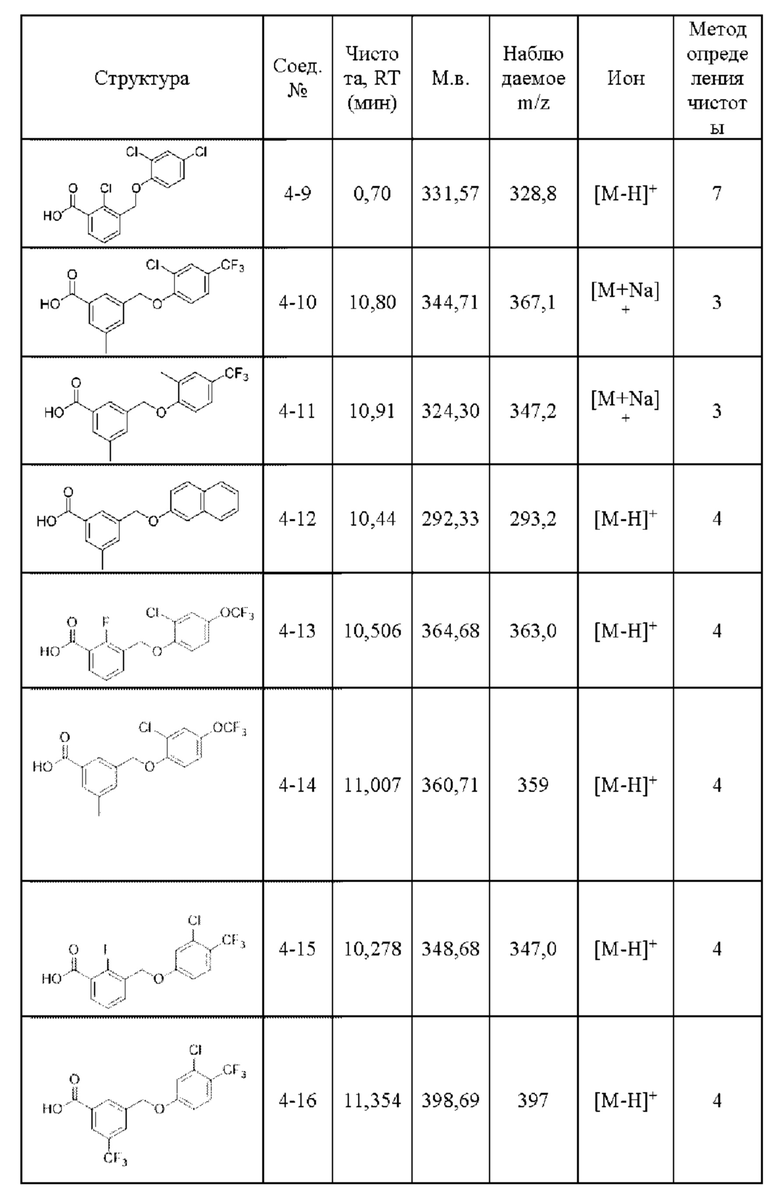
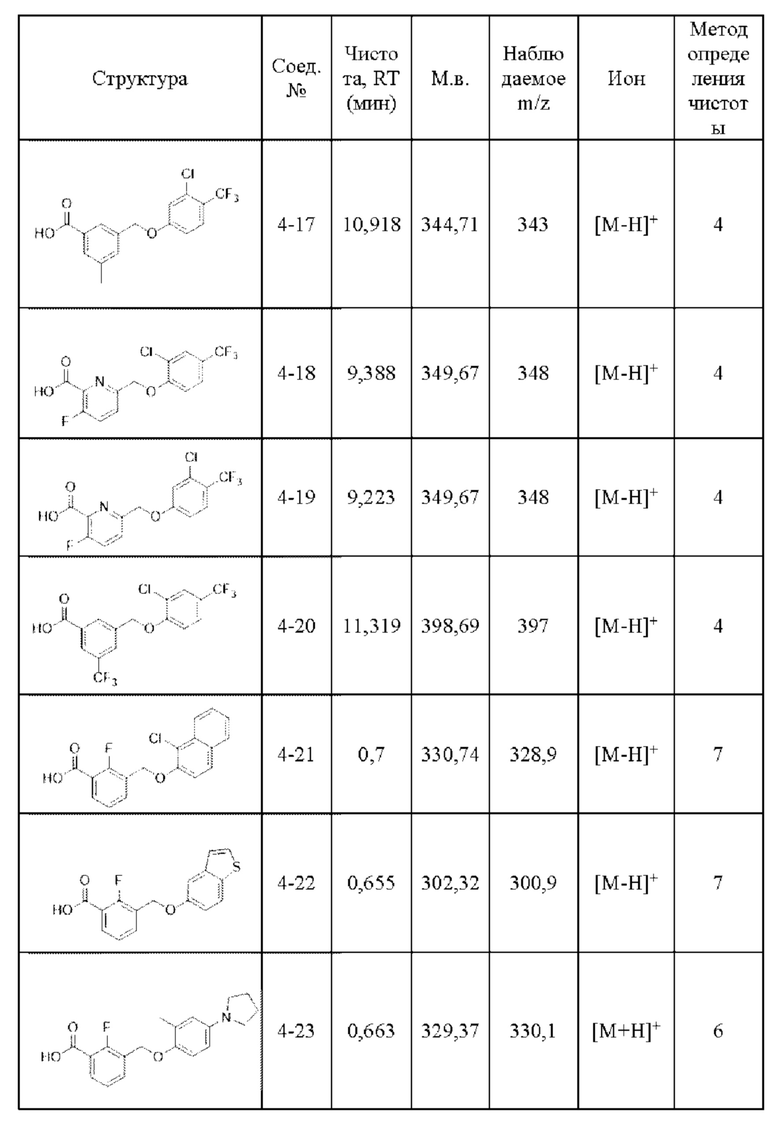
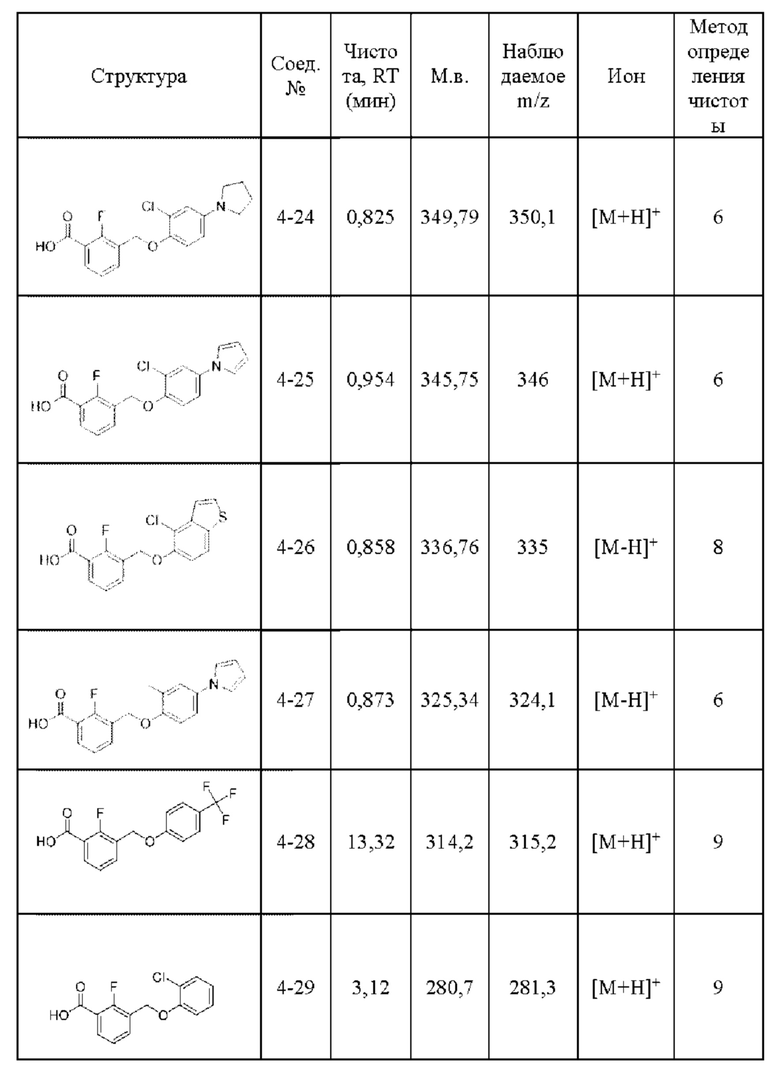
ПРИМЕР 5
Синтез Соединения 5-1 и других типичных соединений
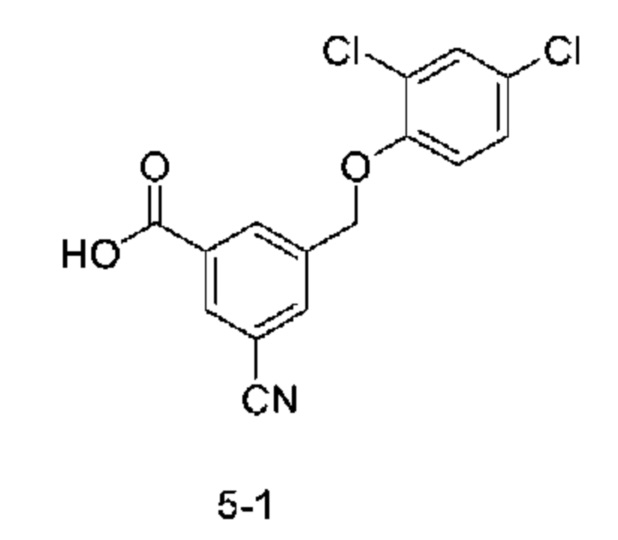
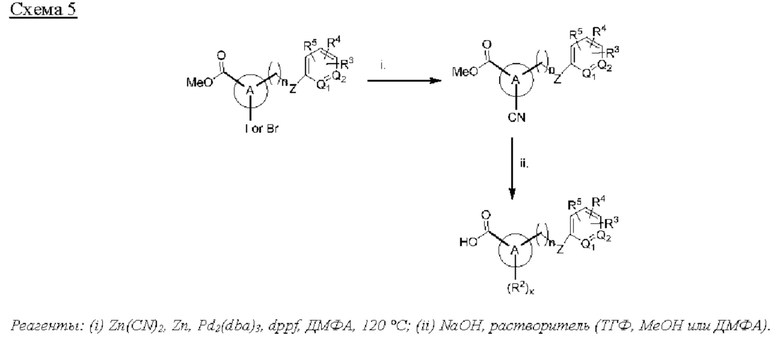
Стадия 5-1. Синтез метил-3-1щано-5-((2,4-дихлорфенокси)метил)бензоата (INT 5-В).
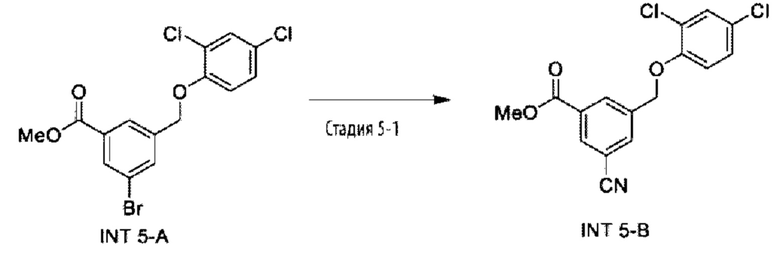
К перемешиваемому раствору метил-3-бром-5-((2,4-
дихлорфенокси)метил)бензоата IΝΤ 5-А (100 мг, 256,37 мкмоль, получен из метил-3-бром-5-(бромметил)бензоата и 2,4-дихлорфенола по Схеме 1) в ДМФА (2 мл), добавляли Zn (33,53 мг, 512,75 мкмоль), Pd2(dba)3 (23,48 мг, 25,64 мкмоль), DPPF (28,43 мг, 51,27 мкмоль) и Zn(CN)2 (60,21 мг, 512,75 мкмоль, 32,55 мкл). Смесь перемешивали при 120°С в течение 2 часов, фильтровали, концентрировали и очищали с помощью препаративной тонкослойной хроматографии с получением 60 мг (69,2%) метил-3-циано-5-((2,4-дихлорфенокси)метил)бензоата (INT 5-В) в виде белого твердого вещества. ЖХ-MC-ESI (m/z) вычислено для C16H11Cl2NO3: 336,2; m/z не наблюдается, tR = 1,1 мин (Метод 6). 1Н ЯМР (400 МГц, CDCl3) δ 8,34 (c, 1H), 8,31 (с, 1H), 7,99 (с, 1H), 7,44 (д, J = 2,4 Гц, 1H), 7,24-7,17 (м, 1H), 6,89 (д, J = 8,8 Гц, 1H), 5,19 (с, 2Н), 3,99 (с, 3Н).
Стадия 5-2. Синтез 3-циано-5-((2,4-дихлорфенокси)метил)бензойной кислоты (Соединение 5-1)
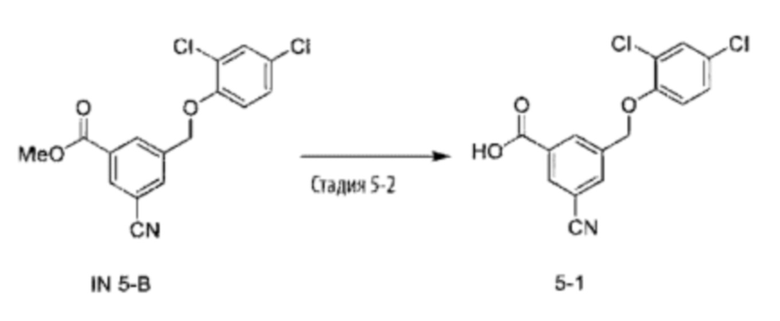
К перемешиваемому раствору метил-3-циано-5-[(2,4-дихлорфенокси)метил] бензоата (INT 5-В) (60 мг, 178,48 мкмоль) в МеОН (1 мл) и ТГФ (1 мл) добавляли NaOH (2М, 267,72 мкл). Смесь перемешивали при 10°С в течение 16 ч, затем концентрировали. Полученный остаток растворяли в H2O (20 мл) и подкисляли (1Μ HCl) до рН 5, и полученный осадок собирали и очищали препаративной ВЭЖХ с получением 3,2 мг (5,6%) 3-циано-5-((2,4-дихлорфенокси)метил)бензойной кислоты (Соединение 5-1) в виде белого твердого вещества. ЖХ-MC-ESI (m/z) вычислено для C15H9Cl2NO3: 322,14; найдено 319,9 [М-Н]+, tR = 0,727 мин (Метод 6). 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,32 (с, 1H), 8,25 (с, 1H), 8,11 (с, 1H), 7,62 (с, 1H), 7,41 (шир.д, J = 8,8 Гц, 1H), 7,27 (д, J = 8,9 Гц, 1H), 5,34-5,32 (м, 1H), 5,35 (с, 1H).
Соединения, перечисленные в Таблице 5, были получены с использованием процедур Схемы 5.
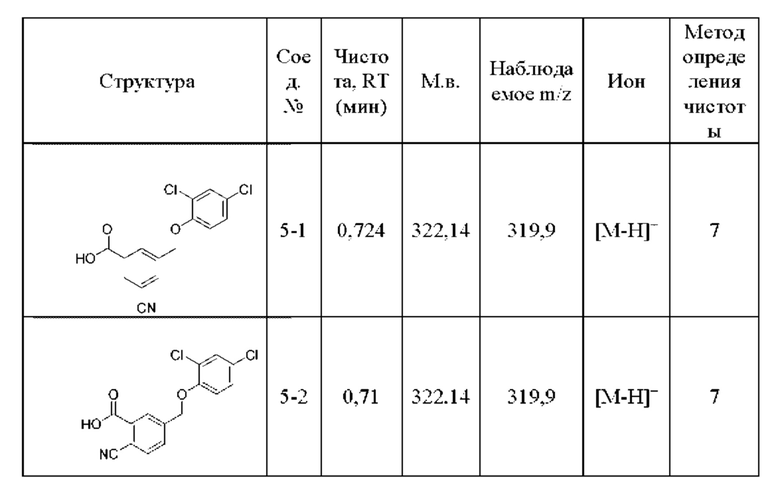
ПРИМЕР 6
Синтез Соединения 6-1
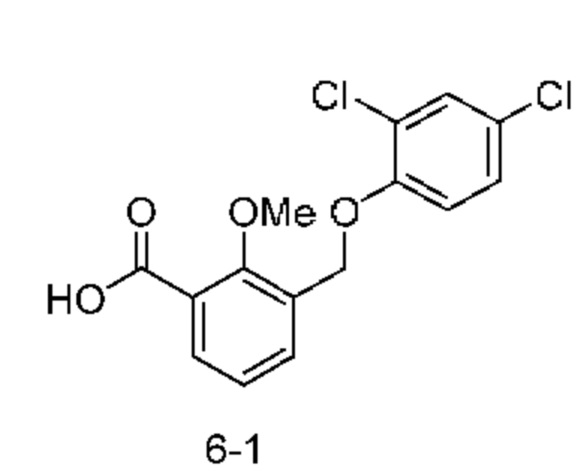
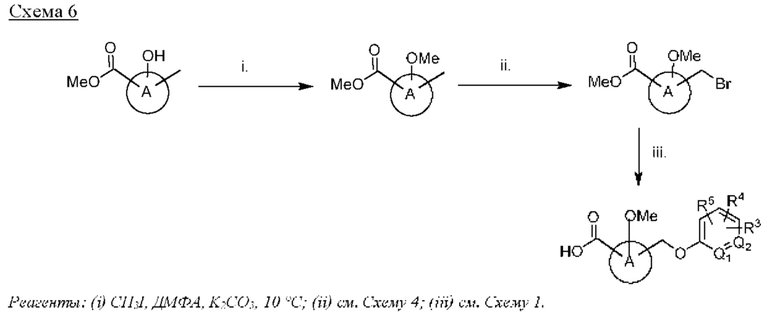
Стадия 6-1. Синтез метил-2-метокси-3-метилбензоата (INT 6-А)
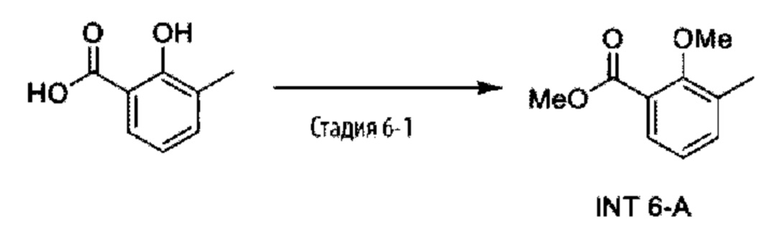
К раствору 2-гидрокси-3-метилбензойной кислоты (1 г, 6,6 ммоль) в ДМФА (15 мл) добавляли K2CO3 (2,73 г, 19,7 ммоль) и CH3I (4,66 г, 32,86 ммоль, 2,1 мл). Смесь перемешивали при 10°С в течение 2 часов. Добавляли дополнительный CH3I (2,33 г, 16,43 ммоль, 1,0 мл) и смесь перемешивали в течение еще 16 часов. Реакционную смесь гасили добавлением Н2О (50 мл), а затем экстрагировали ЭА (100 мл × 3). Объединенные органические слои осушали (Na2SO4), фильтровали и концентрировали в вакууме с получением остатка, который очищали хроматографией на SiO2 с получением 1,0 г (85%) метил-2-метокси-3-метилбензоата (INT 6-А) в виде бесцветной маслянистой жидкости. ТСХ (33% ЭА/петролейный эфир), Rf = 0,45. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 2,33 (с, 3Н), 3,84 (с, 3Н), 3,92 (с, 3Н), 7,06 (т, J = 7,64 Гц, 1H), 7,35 (д, J = 7,46 Гц, 1H), 7,64 (д, J = 7,70 Гц, 1H).
Стадия 6-2. Синтез 3-((2,4-дихлорфенокси)метил)-2-метоксибензойной кислоты (Соединение 6-1)
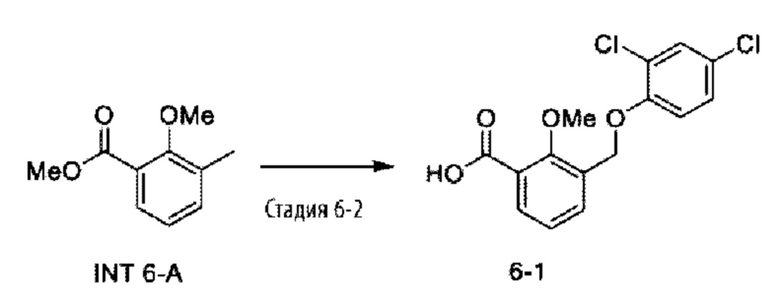
Соединение 6-1 получали из INT 6-А в соответствии с процедурами Схемы 4, а затем Схемы 1, с получением 1,0 г (85%) 3-((2,4-дихлорфенокси)метил)-2-метоксибензойной кислоты (6-1) в виде бесцветной маслянистой жидкости. ЖХ-MC-ESI (m/z) вычислено для С10Н12О3: 180,2; m/z не наблюдается, tR = 0,70 мин (Метод 7). 1Н ЯМР (400 МГц, ДМСО-d6) δ м.д. 3,82 (с, 3Н), 5,22 (с, 2Н), 7,25 (т, J = 7,64 Гц, 1H), 7,30-7,35 (м, 1H), 7,37-7,43 (м, 1Н), 7,60 (д, J = 2,57 Гц, 1H), 7,68 (дд, J = 7,52, 1,65 Гц, 1H), 7,73 (дд, J = 7,76, 1,77 Гц, 1H), 13,03 (шир.с, 1Н).
Соединение, указанное в Таблице 6, было получено с использованием процедур
Схемы 6.
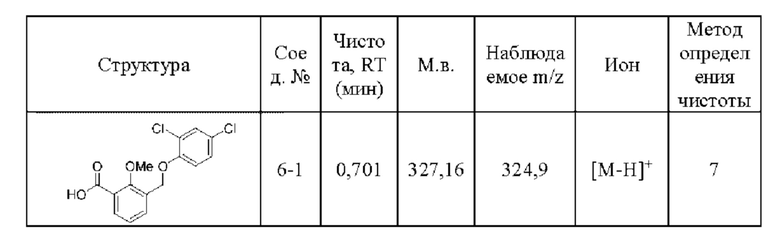
ПРИМЕР 7
Синтез Соединения 7-1
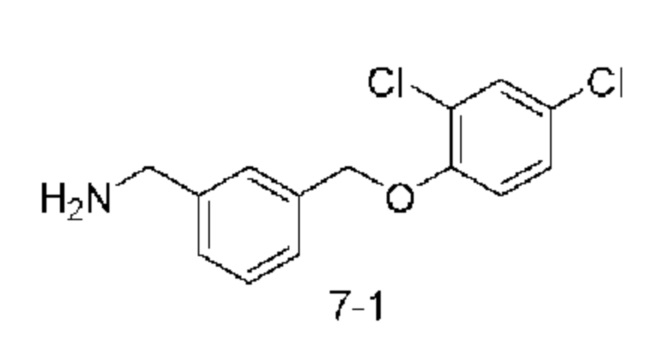
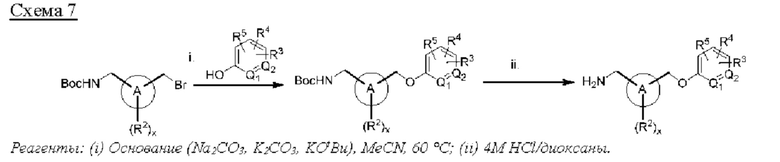
Стадия 7-1. Синтез трет-бутил-(3-((2,4-дихлорфенокси)метил)бензил)карбамата (INT 7-A)
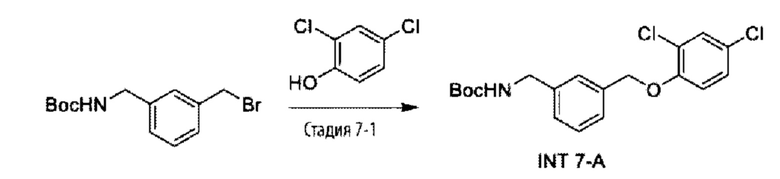
К перемешиваемому раствору 2,4-дихлорфенола (271 мг, 1,67 ммоль) в MeCN (7 мл) добавляли трет-бутил-(3-(бромметил)бензил)карбамат (500 мг, 1,67 ммоль) и K2CO3 (299 мг, 2,17 ммоль). Колбу закрывали, и полученную белую суспензию нагревали при 60°С в течение 3 часов. Реакционную смесь охлаждали до комнатной температуры, разбавляли Н2О (10 мл), экстрагировали Et2O (2 × 10 мл), осушали (Na2SO4), фильтровали через целит и концентрировали в вакууме с получением 627 мг (96%) трет-бутил-(3-((2,4-дихлорфенокси)метил)бензил)карбамата (INT 7-А). ЖХ-MC-ESI (m/z) вычислено для C19H21Cl2NO3: 381; найдено 404,1 [M+Na]+, tR = 12,2 мин (Метод 3). 1H ЯМР (500 Гц, CDCl3) 7,37 (д, J = 2,5, 1H), 7,34-7,33 (м, 3H), 7,25-7,22 (м, 1H), 7,134 (дд, J = 9,0, 2,5, 1H), 6,858 (д, J = 9,0, 1Η), 5,10 (с, 2Н), 4,33 (д, J = 5,5, 2Н), 1,45 (с, 9Н).
Стадия 7-2. Синтез (3-((2,4-дихлорфенокси)метил)фенил)метанамина (Соединение 7-1)
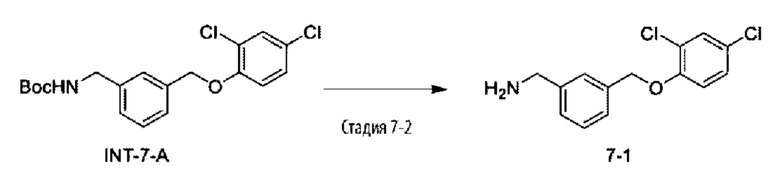
К перемешиваемому раствору трет-бутил-(3-((2,4-
дихлорфенокси)метил)бензил)карбамата (INT 7-А) (200 мг, 523 мкмоль) в диоксане (5 мл) добавляли 4М хлористый водород в диоксанах (5 мл, 20,9 ммоль). Через 3 часа реакционная смесь превращалась в суспензию и ее фильтровали. Фильтрат концентрировали с получением 101 мг неочищенного белого твердого вещества, которое перекристаллизовывали из EtOH (0,7 мл) с получением 15,5 мг (10,5%) (3-((2,4-дихлорфенокси)метил)фенил)метанамина (Соединение 7-1) в виде белого твердого вещества. ЖХ-MC-ESI (m/z) вычислено для C14H13Cl2NO: 281; найдено 282,1 [М+Н]+, tR = 6,345 мин (Метод 3). 1Н ЯМР (500 Гц, ДМСО-d6) 8,26 (шир.с, 3Н), 7,61 (д, J = 3,0, 1H), 7,55 (с, 1H), 7,49-7,47 (м, 3Н), 7,39 (дд, J = 9,0, 2,5, 1H), 7,28 (д, J = 9,0, 1Н), 5,22 (с, 2Н), 4,05 (с, 2Н).
Соединение, указанное в Таблице 7, было получено с использованием процедур Схемы 7.

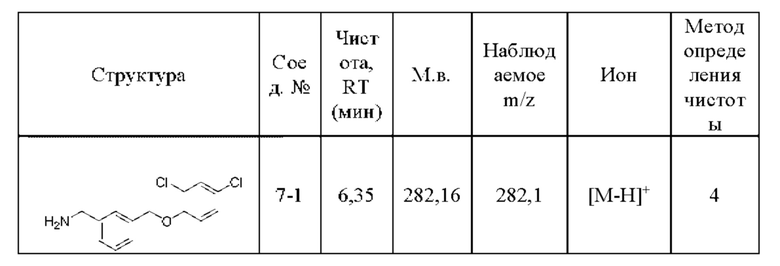
ПРИМЕР 8
Синтез Соединения 8-1 и других типичных соединений
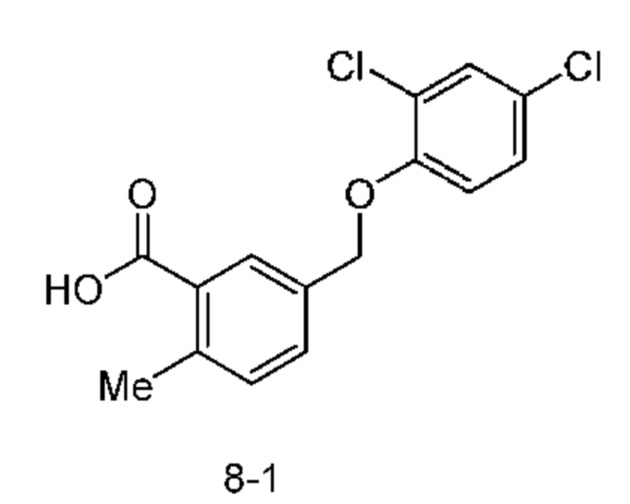
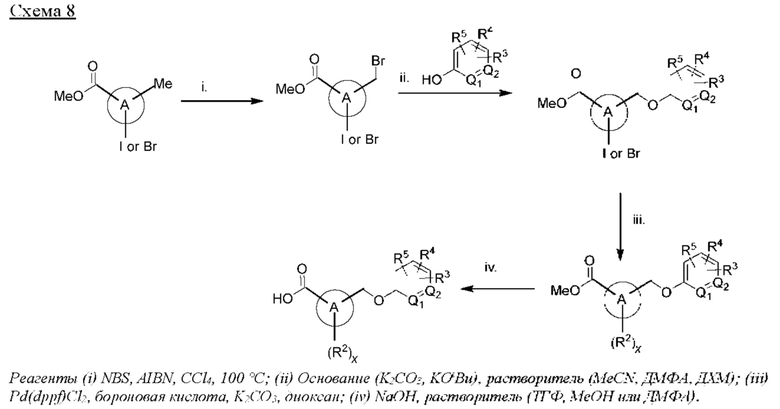
Стадия 8-1. Синтез метил-5-(бромметил)-2-йодбензоата (INT 8-А)
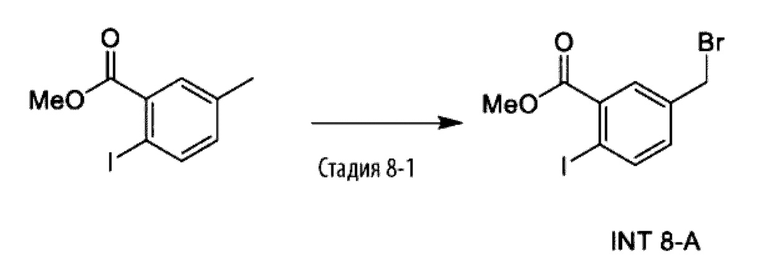
К раствору метил-2-йод-5-метилбензоата (1 г, 3,62 ммоль) в CCl4 (10 мл) добавляли NBS (644,7 мг, 3,62 ммоль) и AIBN (11,9 мг, 72,5 мкмоль). Смесь перемешивали при 100°С в течение 2 часов и концентрировали при пониженном давлении с получением остатка, который очищали флэш-хроматографией на SiO2 (ЭА/петролейный эфир) с получением 733 мг (57,0%) метил-5-(бромметил)-2-йодбензоата (INT 8-А) в виде твердого вещества коричневого цвета. ТСХ: (10% ЭА/петролейный эфир) Rf: 0,5. 1H ЯМР (400 МГц, CDCl3) δ 7,98 (д, J=8,1 Гц, 1Н), 7,84 (д, J=2,3 Гц, 1Н), 7,20 (дд, J=2,3, 8,1 Гц, 1Н), 4,46-4,43 (м, 2Н), 3,95 (с, 3Н).
Стадия 8-2. Синтез метил-5-((2,4-дихлорфенокси)метил)-2-йодбензоата (INT 8-B)
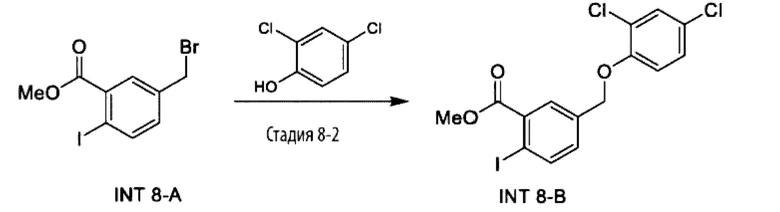
К раствору метил-5-(бромметил)-2-йодбензоата (INT 8-А) (733 мг, 2,06 ммоль) в MeCN (5 мл) добавляли K2CO3 (571 мг, 4,13 ммоль) и 2,4-дихлорфенол (337 мг, 2,06 ммоль). После перемешивания при 50°С в течение 1 6 ч реакционную смесь концентрировали в вакууме и очищали флэш-хроматографией на SiO2, получая 780 мг (выход 86,4%) метил-5-((2,4-дихлорфенокси)метил)-2-йодбензоата (INT 8-B) в виде белого твердого вещества. ТСХ: (10% ЭА/петролейный эфир) R: 0,3. 1H ЯМР (400 МГц, CDCl3) δ 8,02 (д, J=8,2 Гц, 1Н), 7,88 (д, J=2,1 Гц, 1Н), 7,41 (д, J=2,4 Гц, 1Н), 7,30-7,28 (м, 1Н), 7,17 (дд, J=2,4, 8,8 Гц, 1Н), 6,86 (д, J=8,8 Гц, 1Н), 5,10 (с, 2Н), 3,96 (с, 3Н).
Стадия 8-3. Синтез метил-5-((2,4-дихлорфенокси)метил)-2-метилбензоата (INT 8-С)
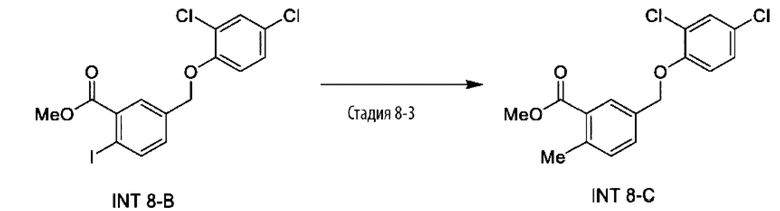
К раствору метил-5-((2,4-дихлорфенокси)метил)-2-йодбензоата (INT 8-В) (200 мг, 457,6 мкмоль) в диоксане (1 мл) и H2O (1 мл) добавляли Pd(dppf)Cl2 (16,7 мг, 22,9 мкмоль), K2CO3 (189,7 мг, 1,4 ммоль) и МеВ(ОН)2 (54,8 мг, 915 мкмоль). Смесь перемешивали при 100°С в течение 2 часов, концентрировали и очищали флэш-хроматографией на SiO2 (ЭА/петролейный эфир) с получением 100 мг (67,2%) метил-5-((2,4-дихлорфенокси)метил)-2-метилбензоата (INT 8-С) в виде белого твердого вещества. ТСХ: (10% ЭА/петролейный эфир) Rf=0,4. 1H ЯМР (400 МГц, CDCl3) δ 7,98 (д, J=1,6 Гц, 1Н), 7,51 (дд, J=1,7, 7,8 Гц, 1Н), 7,40 (д, J=2,6 Гц, 1Н), 7,31-7,28 (м, 1Н), 7,16 (дд, J=2,4, 8,8 Гц, 1Н), 6,88 (д, J=8,8 Гц, 1Н), 5,12 (с, 2Н), 3,96-3,90 (м, 3Н), 2,61 (с, 3Н).
Стадия 8-4. Синтез 5-((2,4-дихлорфенокси)метил)-2-метилбензойной кислоты (Соединение 8-1)
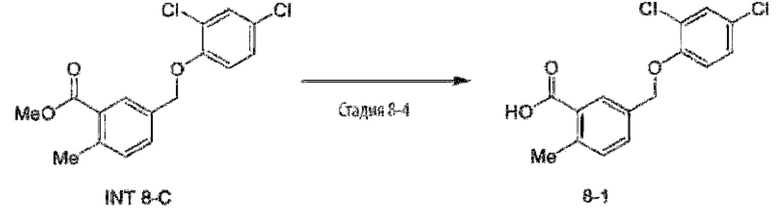
К раствору метил-5-[(2,4-дихлорфенокси)метил]-2-метилбензоата (INT 8-C) (100 мг, 307,52 мкмоль) в МеОН (1 мл) и ТГФ (1 мл) добавляли NaOH (2M, 461,27 мкл). После перемешивания при 10°С в течение 16 ч смесь концентрировали в вакууме и очищали препаративной ВЭЖХ с получением 29 мг (30,3%) 5-((2,4-дихлорфенокси)метил)-2-метилбензойной кислоты (Соединение 8-1) в виде белого твердого вещества. ЖХ-MC-ESI (m/z) вычислено для C15H12Cl2O3: 310,02; найдено 308,9 [M-H]+, tr=0,718 мин (Метод 7). 1H ЯМР (400 МГц, CDCl3) δ 8,12 (с, 1Н), 7,57 (шир. д, J=7,9 Гц, 1Н), 7,40 (д, J=2,4 Гц, 1Н), 7,32 (д, J=7,5 Гц, 1Н), 7,17 (дд, J=2,4, 8,8 Гц, 1Н), 6,89 (д, J=8,8 Гц, 1Н), 5,14 (с, 2Н), 2,67 (с, 3Н).
Соединения, перечисленные в Таблице 8, были получены с использованием процедур Схемы 8.
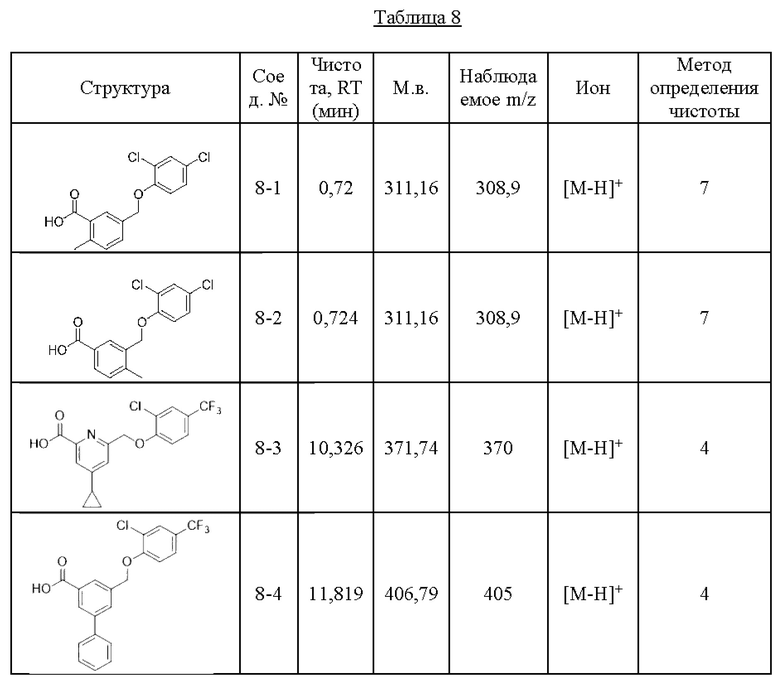
ПРИМЕР 9
Синтез Соединения 9-1
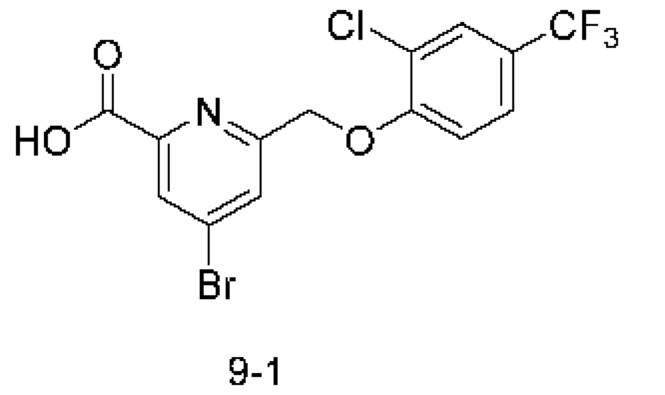
Схема 9
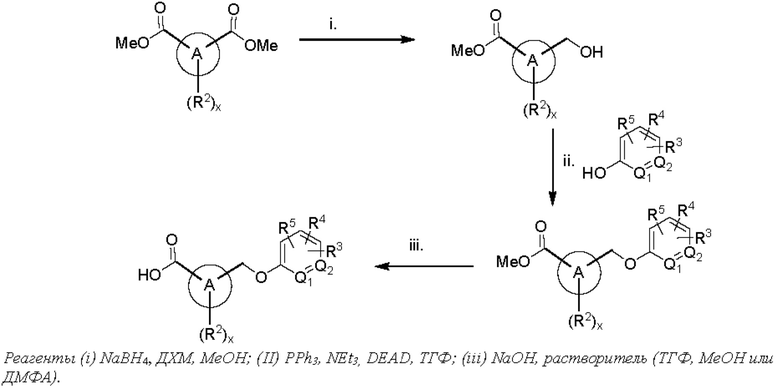
Стадия 9-1. Синтез метил-4-бром-6-(гидроксиметил)пиколината (INT 9-А)
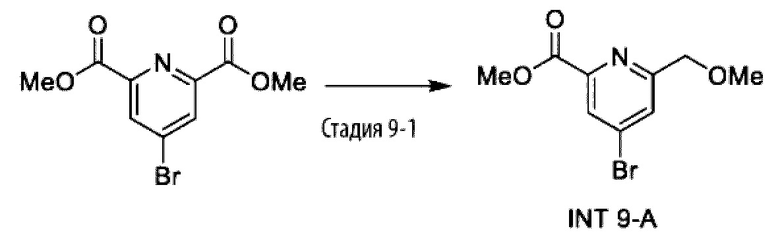
К перемешиваемому раствору диметил-4-бромпиридин-2,6-дикарбоксилата (1,0 г, 3,6 ммоль) в МеОН (12 мл) и ДХМ (6 мл) при 0°С добавляли боргидрид натрия (0,17 г, 4,4 ммоль) 3-мя порциями. Реакционную смесь нагревали до комнатной температуры и перемешивали в течение ночи. Добавляли дополнительно боргидрид натрия (0,17 г, 4,4 ммоль). Через 2 часа реакционную смесь разбавляли NH4Cl (водн.) (10 мл) и ДХМ (10 мл). Водный слой экстрагировали ДХМ (2×10 мл) и ЭА (10 мл), осушали (Na2SO4), фильтровали через целит и концентрировали в вакууме, получая неочищенное белое твердое вещество, которое очищали хроматографией на SiO2 (10% МеОН в ЭА/гексанах) с получением 514 мг (57%) метил-4-бром-6-(гидроксиметил)пиколината (INT 9-А) в виде белого твердого вещества. ЖХ-MC-ESI (m/z) вычислено для C9H10BrNO3: 258,98; m/z не наблюдается, tR=3,21 мин (Метод 1).
Стадия 9-2. Синтез метил-4-бром-6-((2-хлор-4-(трифторметил)фенокси)метил)пиколината (INT 9-В)
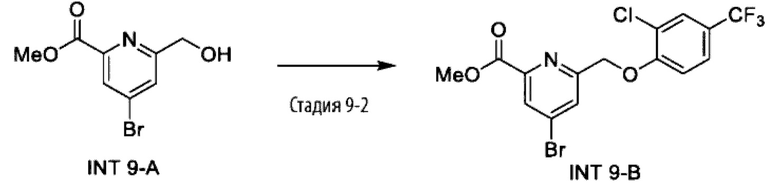
К перемешиваемому раствору 2-хлор-4-(трифторметил)фенола (87,9 мг, 0,447 ммоль) в ТГФ (10 мл) добавляли метил-4-бром-6-(гидроксиметил)пиколинат (INT 9-A) (100 мг., 0,406 ммоль), трифенилфосфин (107 мг, 0,406 ммоль) и TEA (56,7 мкл, 406 мкмоль). Реакционную смесь охлаждали до 0°С и добавляли по каплям диизопропилазодикарбоксилат (82,2 мг, 80,0 мкл, 0,406 ммоль). Реакционную смесь перемешивали при 0°С в течение 30 минут, нагревали до комнатной температуры и перемешивали в течение ночи. Реакционную смесь концентрировали в вакууме с получением сырого продукта, который очищали хроматографией на SiO2 (ЭА/гексаны) с получением 94 мг (55%) метил-4-бром-6-((2-хлор-4-(трифторметил)) фенокси)метил)пиколинат (INT 9-B) в виде грязно-белого твердого вещества. ЖХ-MC-ESI (m/z) вычислено для C15H10BrClF3NO3: 422,95; m/z не наблюдается, tR=6,73 мин (Метод 1).
Стадия 9-3. Синтез 4-бром-6-((2-хлор-4-(трифторметил)фенокси)метил)пиколиновой кислоты (Соединение 9-1)
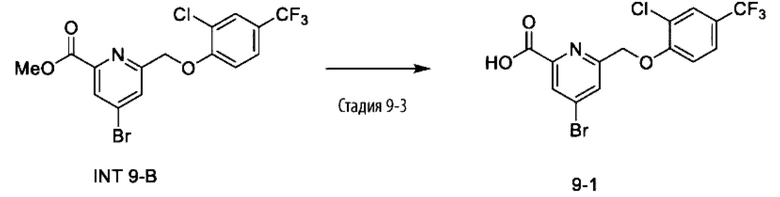
К перемешиваемому раствору метил-4-бром-6-((2-хлор-4-(трифторметил)фенокси)метил)пиколината (INT 9-В) (94,5 мг, 223 мкмоль) в ТГФ (2 мл) добавляли 1М NaOH (1 мл, 1,11 ммоль). Реакционную смесь нагревали при 60°С в течение ночи, охлаждали и подкисляли 3М HCl. Смесь экстрагировали этилацетатом и Et2O, и объединенные органические вещества осушали (Na2SO4), фильтровали и концентрировали в вакууме с получением 77,6 мг (85%) 4-бром-6-((2-хлор-4-(трифторметил)фенокси)метил)пиколиновой кислоты (Соединение 9-1) в виде белого твердого вещества. ЖХ-MC-ESI (m/z) вычислено для C14H18BrClF3NO3: 408,93; найдено 410,0 [M+H]+, tR=10,7 мин (Метод 3).
Соединения, перечисленные в Таблице 9, были получены с использованием процедур Схемы 9:
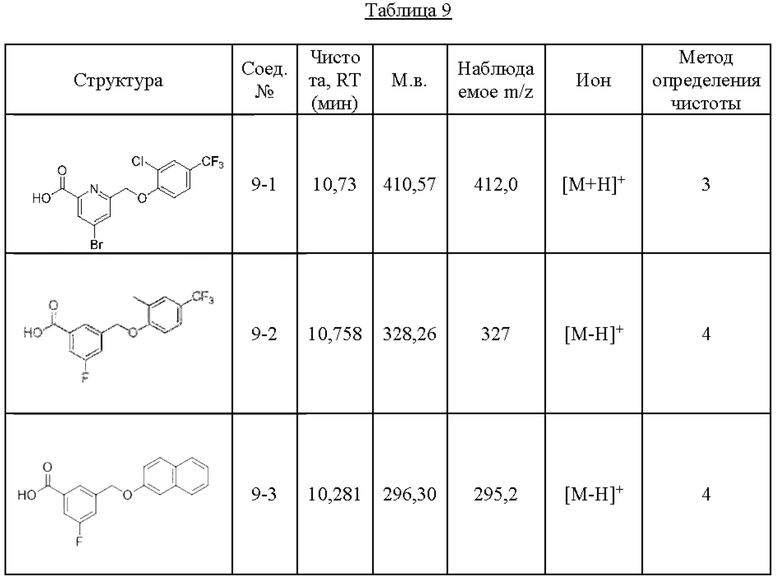
ПРИМЕР 10
Синтез Соединения 10-1
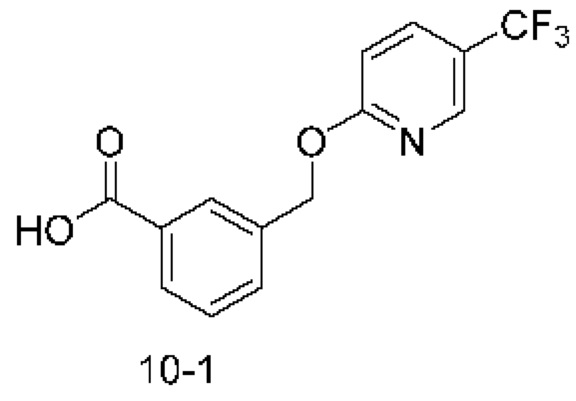
Схема 10
Стадия 10-1. Синтез метил-3-(((5-(трифторметил)пиридин-2-ил)окси)метил)бензоата (INT 10-А)
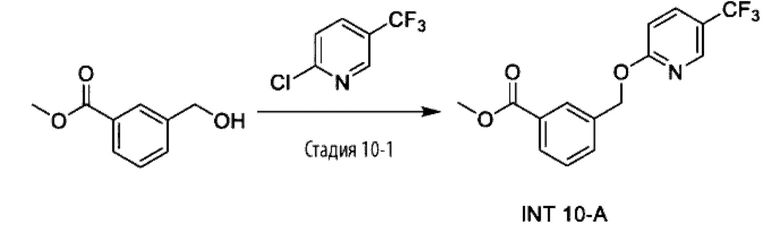
В сосуд высокого давления, содержащий раствор метил-3-(гидроксиметил)бензоата (499 мг, 3,00 ммоль) в 1,4-диоксане (9 мл), добавляли 2-хлор-5-(трифторметил)пиридин (363 мг, 2,00 ммоль) и трет-бутоксид калия (337 мг, 3,00 ммоль). Сосуд герметично закрывали, и реакционную смесь нагревали и перемешивали в течение ночи при 90°С, затем охлаждали до комнатной температуры. Реакционную смесь распределяли между Et2O и H2O. Фазы разделяли, и водный слой дополнительно экстрагировали диэтиловым эфиром (2Х). Органические фазы объединяли, промывали рассолом, осушали (Na2SO4), фильтровали и концентрировали при пониженном давлении. Полученную бесцветную маслянистую жидкость очищали флэш-хроматографией на SiO2 (ЭА/гексаны) с получением 177 мг (28,4%) метил-3-(((5-(трифторметил)пиридин-2-ил)окси)метил)бензоата (INT 10-А) в виде бесцветной маслянистой жидкости. ЖХ-MC-ESI (m/z) вычислено для C15H12F3NO3: 311,1; найдено 312,1 [М+Н]+, tR=6,25 мин (Метод 1). 1Н ЯМР (500 МГц, ДМСО-d6) δ 8,608 (с, 1Н), 8,114 (дд, J=8,5, 2,5 Гц, 1Н), 8,050 (с, 1Н), 7,934 (д, J=7,5 Гц, 1Н), 7,753 (д, J=8,0 Гц, 1Н), 7,551 (т, J=7,5 Гц, 1Н), 7,129 (д, J=9,0 Гц, 1Н), 5,512 (с, 2Н), 3,858 (с, 3Н). 19F ЯМР (470 МГц, ДМСО-d6) δ 60,140 (с).
Стадия 10-2. Синтез 3-(((5-(трифторметил)пиридин-2-ил)окси)метил)бензойной кислоты (Соединение 10-1)
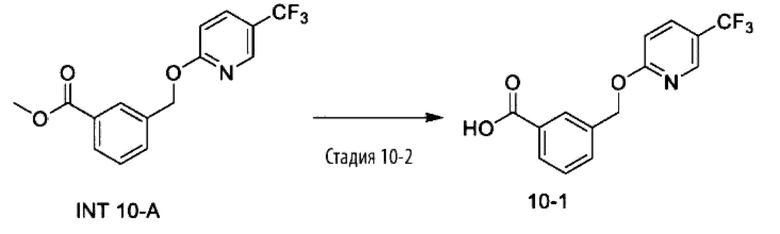
Во флакон на 20 мл, содержащий перемешиваемый раствор метил-3-(((5-(трифторметил)пиридин-2-ил)окси)метил)бензоата (INT 10-А) (177 мг, 0,569 ммоль) в ТГФ (6 мл) загружали 1М NaOH (2,27 мл, 2,27 ммоль). После перемешивания в течение 12 ч при 50°С реакционную смесь концентрировали в вакууме, а остаток растворяли в H2O и подкисляли до рН 4-5 с помощью 3М HCl. Полученный белый осадок экстрагировали Et2O (3 раза). Объединенные органические слои промывали рассолом, осушали (Na2SO4) и концентрировали в вакууме, получая 151 мг (89,3%) 3-(((5-(трифторметил)пиридин-2-ил)окси)метил)бензойной кислоты (Соединение 10-1) в виде белого твердого вещества. ЖХ-MC-ESI (m/z) вычислено для C14H11F3NO3: 297,2; найдено 298,1 [M+H]+, tR=9,33 мин (Метод 3). 1H ЯМР (500 МГц, ДМСО-d6) δ 13,006 (шир.с, 1Н), 8,595 (с, 1Н), 8,100 (дд, J=9,0, 2,5 Гц, 1Н), 8,015 (с, 1Н), 7,900 (д, J=8,0 Гц, 1Н), 7,702 (д, 8,0 Гц, 1Н), 7,522 (т, J=7,5 Гц, 1Н), 7,115 (д, J=8,5 Гц, 1Н), 5,493 (с, 2Н). 19F ЯМР (470 МГц, ДМСО-d6) 5 60,126 (с).
Соединение, указанное в Таблице 10, было получено с использованием процедур Схемы 10:
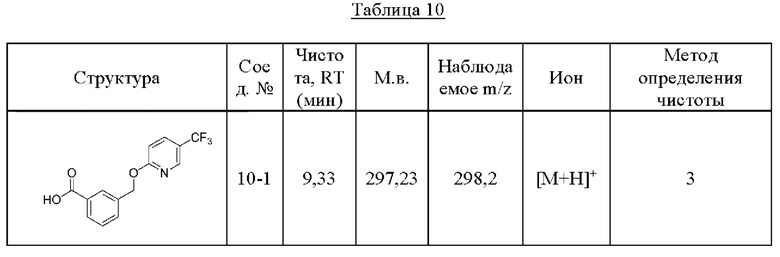
ПРИМЕР 11
Синтез Соединения 11-1
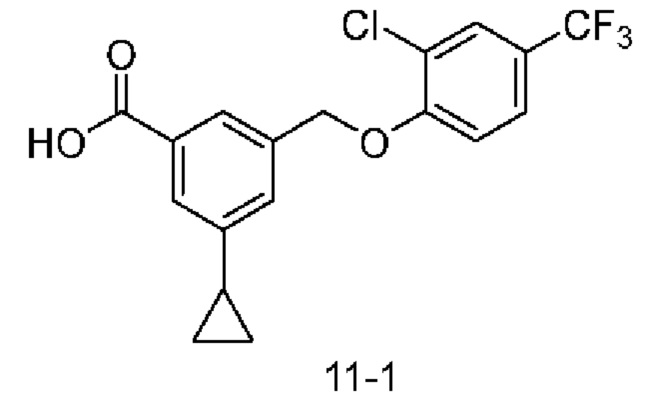
Схема 11
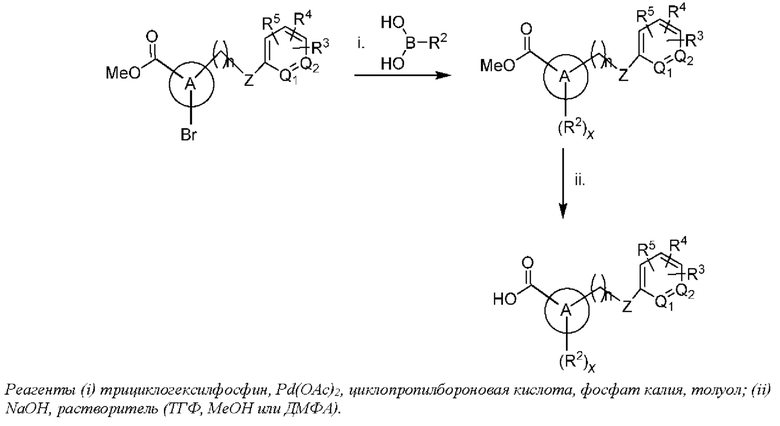
Стадия 11-1. Синтез метил-3-((2-хлор-4-(трифторметил)фенокси)метил)-5-циклопропилбензоата (INT 11-В)
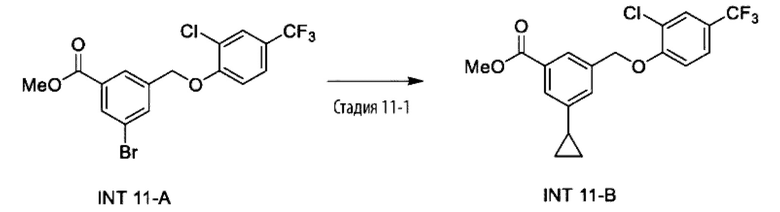
К дегазированному раствору метил-3-бром-5-((2-хлор-4-(трифторметил)фенокси)метил)бензоата (INT 11-А) (200 мг, 472 мкмоль, получен по Схеме 2 из метил-3-бром-5-(гидроксиметил)бензоата и 2-хлор-4-(трифторметил)фенола)), трициклогексилфосфина (6,62 мг, 23,6 мкмоль), фосфата калия (230 мг, 1,09 ммоль) и циклопропилбороновой кислоты (52,7 мг, 614 мкмоль) в толуоле (4 мл) добавляли диацетат палладия (5,30 мг, 23,6 мкмоль). Реакционный сосуд закрывали крышкой и нагревали при 100°С в течение ночи. Реакционную смесь дополнительно дегазировали и добавляли дополнительно трициклогексилфосфин (6,62 мг, 23,6 мкмоль), циклопропилбороновую кислоту(52,7 мг, 614 мкмоль) и диацетат палладия (5,30 мг, 23,6 мкмоль). После нагревания при 100°С в течение 4 часов реакционную смесь фильтровали через целит, промывали ЭА и концентрировали в вакууме. Остаток разводили в ЭА, промывали насыщенным бикарбонатом натрия и рассолом, осушали (Na2SO4), фильтровали и концентрировали в вакууме с получением сырого материала, который очищали хроматографией на SiO2 (ЭА/гексаны), получая 132 мг (72%) метил-3-((2-хлор-4-(трифторметил)фенокси)метил)-5-циклопропилбензоата (INT 11-В). ЖХ-MC-ESI (m/z) вычислено для C19H16ClF3O3: 384,1; m/z не наблюдается, tr=7,02 мин (Метод 1). 1H ЯМР (500 Гц, ДМСО-d6) δ 7,86 (д, J=2,5, 1Н), 7,85 (с, 1Н),7,71 (дд, J=8,8, 2,5, 1Н), 7,64 (с, 1Н), 7,46, (с, 1Н), 7,41 (д, J=9,0, 1Н), 5,35 (с, 2Н), 3,85 (с, 3Н), 2,07-2,02 (м, 1Н), 1,03-1,00 (м, 2Н) 0,73-0,70 (м, 2Н).
Стадия 11-2. Синтез 3-((2-хлор-4-(трифторметил)фенокси)метил)-5-циклопропилбензойной кислоты (Соединение 11-1).
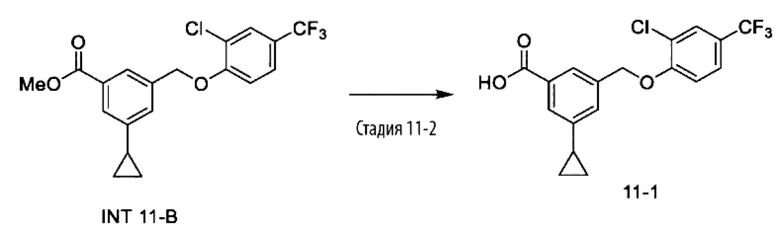
К перемешиваемому раствору метил-3-((2-хлор-4-(трифторметил)фенокси)метил)-5-циклопропилбензоата (INT 11-В, 132 мг, 0,343 ммоль) в ТГФ (2 мл) добавляли 1 М NaOH (2 мл, 1,72 ммоль). После нагревания при 60°С в течение ночи реакционную смесь охлаждали и подкисляли 3М HCl. Смесь экстрагировали ЭА и Et2O, осушали (Na2SO4), фильтровали, концентрировали в вакууме. Неочищенное твердое вещество очищали с помощью ВЭЖХ с обращенной фазой с получением 71,2 мг (56%) 3-((2-хлор-4-(трифторметил)фенокси)метил)-5-циклопропилбензойной кислоты (Соединение 11-1) в виде белого твердого вещества. ЖХ-MC-ESI (m/z) вычислено для C18H14ClF3O3: 370,7; найдено 393,1 [M+Na]+, tR=11,25 мин (Метод 3). 1H ЯМР (500 Гц, ДМСО-d6) δ 13,00 (с, 1Н), 7,87 (с, 1Н), 7,82 (с, 1Н), 7,71 (д, J=8,0, 1Н), 7,62 (с, 1Н)), 7,43-7,41 (м, 2Н), 5,34 (с, 2Н), 2,04 (м, 1Н), 1,01 (д, J=7,0, 2Н), 0,72-0,71 (м, 2Н).
Соединение, указанное в Таблице 11, было получено с использованием процедур Схемы 11.
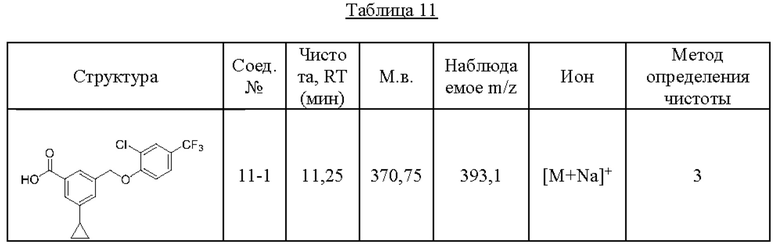
ПРИМЕР 12
Синтез Соединения 12-1
Схема 12
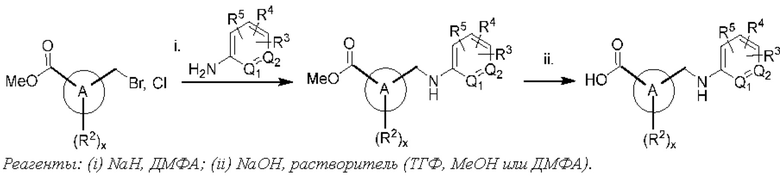
Стадия 12-1. Синтез метил-3-(((2,4-дихлорфенил)амино)метил)бензоата (INT 12-А)
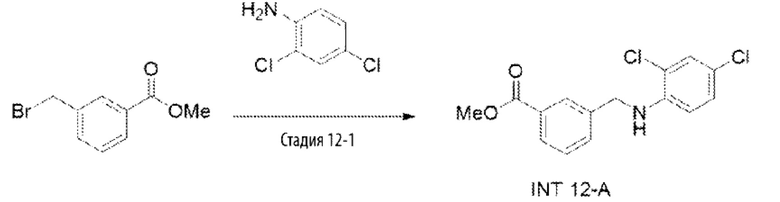
К перемешиваемому раствору метил-3-(бромметил)бензоата (300 мг, 1,31 ммоль) и 2,4-дихлоранилина (0,23 г, 1,44 ммоль) в ДМ ФА (2 мл) при 0°С добавляли NaH (60% в минеральном масле, 38 мг, 0,95 ммоль). Через 1 час при 0°С смесь разбавляли H2O (20 мл) и экстрагировали ЭА (2×50 мл). Органические слои объединяли, промывали рассолом, осушали (Na2SO4), фильтровали и концентрировали с получением сырого продукта, который очищали хроматографией на SiO2 с получением 230 мг (56%) метил-3-(((2,4-дихлорфенил)амино)метил)бензоата (INT 12-А), который имел чистоту 30% и использовался без дополнительной очистки. ЖХ-MC-ESI (m/z) вычислено для C15H13Cl2NO2: 310,2; найдено 311,2 [М+Н]+, tR=5,7 мин (Метод 11).
Стадия 12-2. Синтез 3-(((2,4-дихлорфенил)амино)метил)бензойной кислоты (Соединение 12-1)
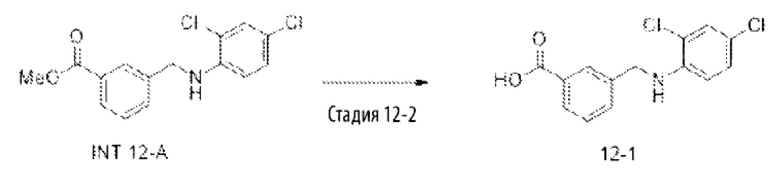
К перемешиваемому раствору сырого метил-3-(((2,4-дихлорфенил)амино)метил)бензоата(INT 12-А) (230 мг, 0,74 ммоль)в МеОН (3 мл) добавляли раствор NaOH (290 мг, 7,4 ммоль) в H2O (1 мл). Реакционную смесь кипятили с обратным холодильником в течение 2 часов, охлаждали до комнатной температуры и концентрировали в вакууме для удаления МеОН. Водный слой подкисляли до рН 2 с помощью 4 н. HCl (водн.) и экстрагировали ЭА (2×50 мл). Объединенные органические слои промывали H2O, рассолом, осушали (MgSO4), фильтровали и концентрировали в вакууме, получая неочищенный материал, который очищали хроматографией на SiO2 с получением 5 мг (2,3%) 3-(((2,4-дихлорфенил)амино)метил)бензойной кислоты (Соединение 12-1). ЖХ-MC-ESI (m/z) вычислено для C14H11Cl1NO2: 296,2; найдено 296,5 [M+H]+, tR=13,88 мин (Метод 9). 1H ЯМР (400 МГц, ДМСО-d6) δ 7,88 (с, 1Н), 7,76 (д, J=8 Гц, 1Н), 7,42 (д, J=8 Гц, 1Н), 7,35 (м, 2Н), 7,06 (д, J=8 Гц, 1Н), 6,51 (д, J=8 Гц, 1Н), 6,39 (т, J=8 Гц, 1Н), 4,43 (д, J=4 Гц, 2Н).
Соединения, перечисленные в Таблице 12, были получены с использованием процедур Схемы 12.
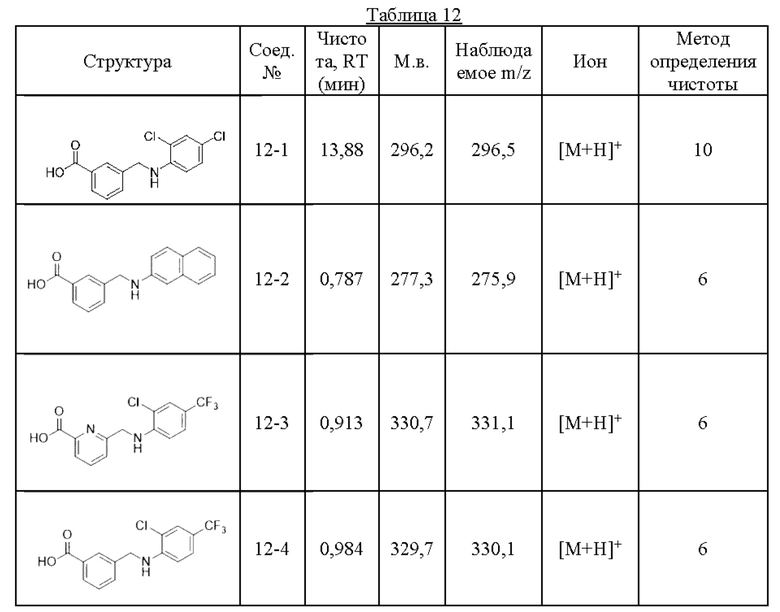
ПРИМЕР 13
Синтез Соединения 13-1
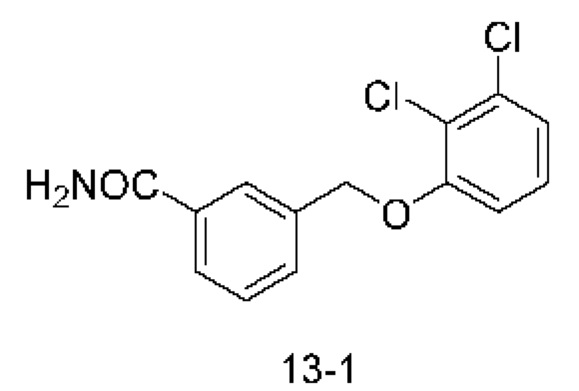
Схема 13
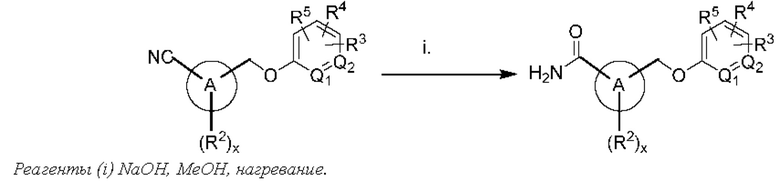
Стадия 13-1. Синтез 3-((2,3-дихлорфенокси)метил)бензамида (Соединение 13-1)
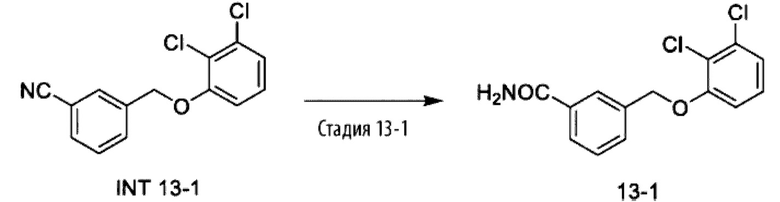
К перемешиваемому раствору INT 13-1 (0,3 г, 1,1 ммоль, получено по Схеме 3 из 3-(бромметил)бензонитрила и 2,3-дихлорфенола) в МеОН (5 мл) добавляли раствор NaOH (0,34 г, 8,6 ммоль) в H2O (5 мл). После нагревания в течение 4 ч при 90°С реакционную смесь охлаждали до комнатной температуры и полученное твердое вещество собирали и промывали H2O (10 мл). Материал сушили в высоком вакууме, получая 68,8 мг (21%) 3-((2,3-дихлорфенокси)метил)бензамида (Соединение 13-1). ЖХ-MC-ESI (m/z) вычислено для C14H11Cl2NO2: 296,2; найдено 297,2 [М+Н]+, tr=11,8 мин (Метод 10). 1H ЯМР (400 МГц, CDCl3) δ 7,92 (с, 1Н), 7,78 (д, J=8 Гц, 1Н), 7,65 (д, J=8 Гц, 1Н), 7,50 (т, J=8 Гц, 1Н), 7,10 (м, 2Н), 6,86 (д, J=8 Гц, 1Н), 6,22 (шир.с, 1Н), 5,59 (шир.с, 1Н), 5,21 (с, 2Н).
Соединение, указанное в Таблице 13, было получено с использованием процедур Схемы 13.
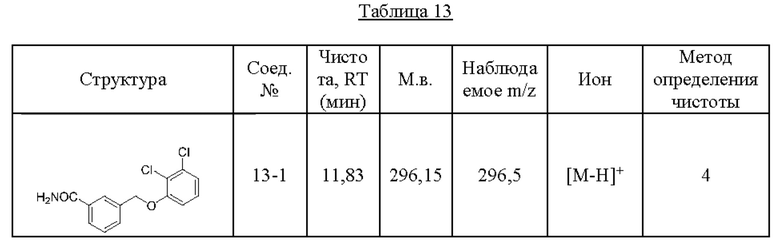
ПРИМЕР 14
Синтез Соединения 14-1
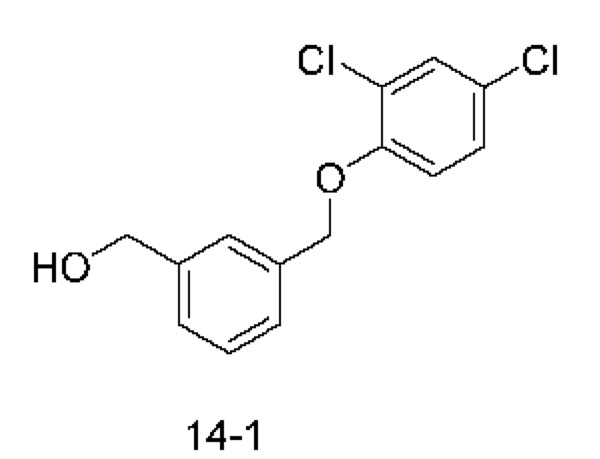
Схема 14
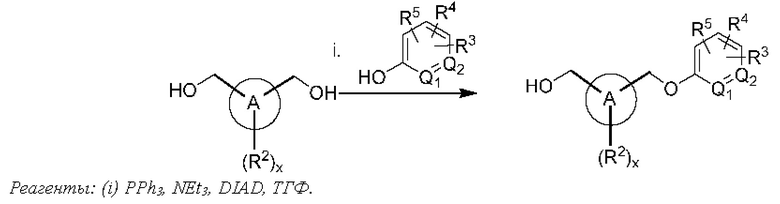
Стадия 14-1. Синтез (3-((2,4-дихлорфенокси)метил)фенил)метанола (Соединение 14-1)
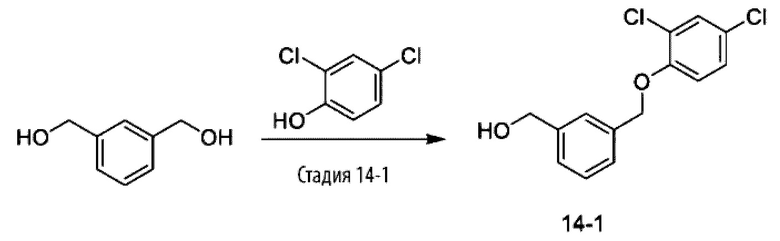
К перемешиваемому раствору 2,4-дихлорфенола (324 мг, 1,99 ммоль) в ТГФ (15 мл) добавляли 1,3-фенилендиметанол (250 мг, 1,81 ммоль), трифенилфосфин (475 мг, 1,81 ммоль) и TEA (183 мг, 252 мкл, 1,81 ммоль). Реакционную смесь охлаждали до 0°С и добавляли по каплям диизопропилазодикарбоксилат (356 мкл, 1,81 ммоль). Реакционную смесь перемешивали при 0°С в течение 30 минут, нагревали до комнатной температуры и перемешивали в течение ночи. Реакционную смесь концентрировали в вакууме с получением сырого продукта, который очищали хроматографией на SiO2 (ЭА/гексаны) с получением 90,9 мг (17,7%) (3-((2,4-дихлорфенокси)метил)фенил)метанола (Соединение 14-1) в виде грязно-белого твердого вещества. ЖХ-MC-ESI (m/z), вычислено для C14H12Cl2O2: 282,0; найдено 282,21 [М+Н]+, tR=10,08 мин (Метод 3).
Соединение, указанное в Таблице 14, было получено с использованием процедур Схемы 14:
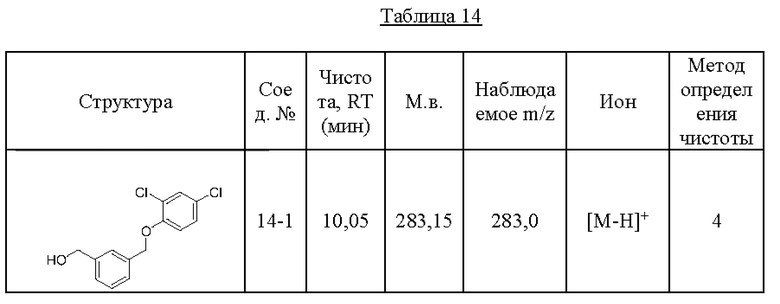
ПРИМЕР 15
Синтез Соединения 15-1
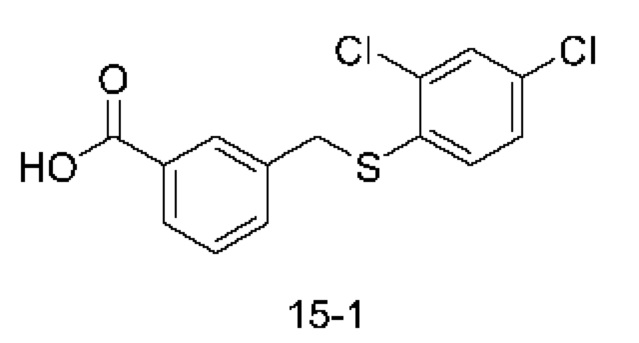
Схема 15
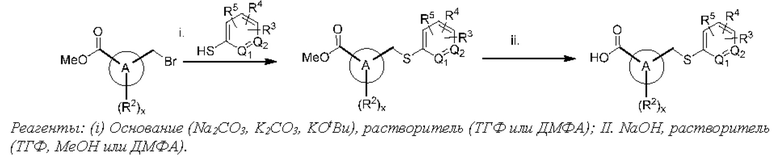
Стадия 15-1. Синтез метил-3-(((2,4-дихлорфенил)тио)метил)бензоата (INT 15-А)
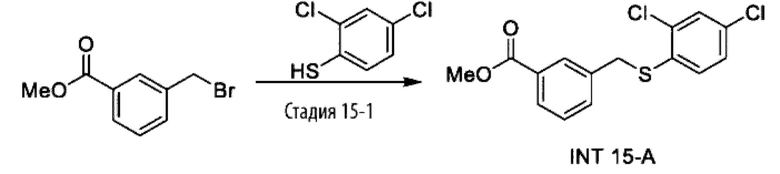
К перемешиваемому раствору 2,4-дихлорбензолтиола (750 мг, 4,19 ммоль) в MeCN (20 мл) добавляли метил-3-(бромметил)бензоат (959 мг, 4,19 ммоль) и карбонат калия (753 мг, 5,45 ммоль). Реакционную смесь нагревали при 60°С в течение 3 часов, охлаждали до комнатной температуры, разбавляли H2O (20 мл) и экстрагировали Et2O (2×20 мл). Объединенные органические слои осушали (Na2SO4), фильтровали и концентрировали с получением 1,3 г (94%) метил-3-(((2,4-дихлорфенил)тио)метил)бензоата (INT 15-А) в виде желтой маслянистой жидкости, которая затвердевает при стоянии. ЖХ-MC-ESI (m/z) вычислено для C15H12Cl2O2S: 325,9; найдено 327,1 [М+Н]+, tR=12,5 мин (Метод 3).
Стадия 15-2. Синтез 3-(((2,4-дихлорфенил)тио)метил)бензойной кислоты (15-1)
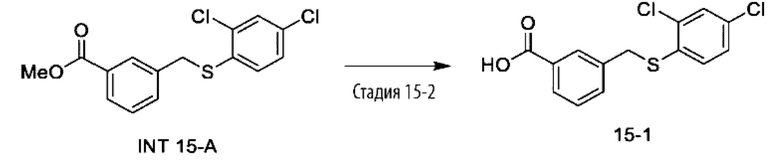
К перемешиваемому раствору метил-3-(((2,4-дихлорфенил)тио)метил)бензоата (INT 15-А) (250 мг, 0,764 ммоль) в ТГФ (3 мл) добавляли 1М NaOH (4 мл, 3,82 ммоль). Реакционную смесь нагревали при 60°С в течение 3 часов, водный слой экстрагировали ЭА (2×5 мл), осушали (Na2SO4), фильтровали и концентрировали, получая неочищенное твердое вещество, которое очищали обращенно-фазовой ВЭЖХ с получением 240,3 мг (99%) 3-(((2,4-дихлорфенил)тио)метил)бензойной кислоты (Соединение 15-1) в виде грязно-белого твердого вещества. ЖХ-MC-ESI (m/z), вычислено для C14H10Cl2O2S: 311,97; найдено 313,1 [M+Na]+, tR=10,59 мин (Метод 3).
Соединение, указанное в Таблице 15, было получено с использованием процедур Схемы 15.
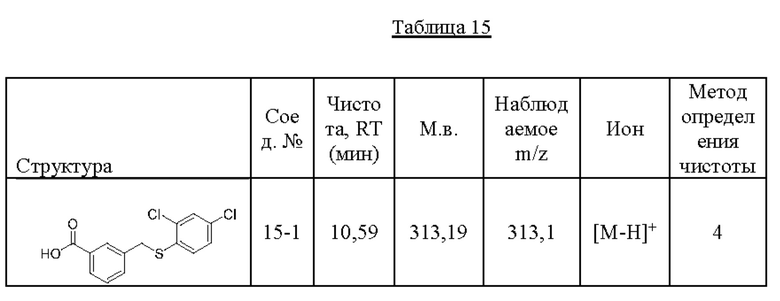
ПРИМЕР 16
Синтез Соединения 16-1 и других типичных соединений
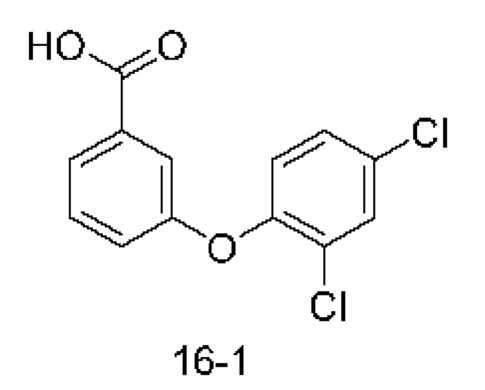
Схема 16
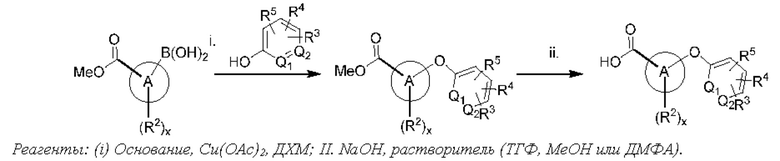
Стадия 16-1. Синтез метил-3-(2,4-дихлорфенокси)бензоата (INT 16-А).
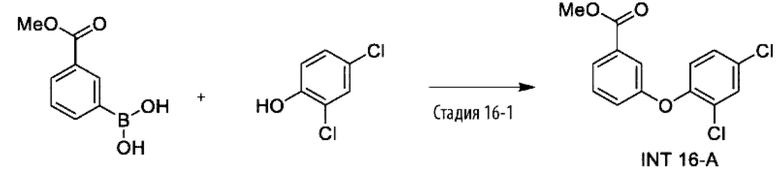
К перемешиваемому раствору (3-(метоксикарбонил)фенил)бороновой кислоты (221 мг, 1,2 ммоль) и 2,4-дихлорфенола (100 мг, 0,61 ммоль) в безводном ДХМ (5 мл) добавляли Cu(OAc)2 (111 мг, 0,61 ммоль) и TEA (0,86 мл, 0,61 ммоль). После перемешивания в течение ночи реакционную смесь фильтровали, концентрировали и очищали хроматографией на SiO2 (ЭА/гексан) с получением 182 мг (27%) метил-3-(2,4-дихлорфенокси)бензоата (INT 16-А). ЖХ-MC-ESI (m/z) вычислено для C14H10Cl2O3: 296; найдено 297,5 [M+H]+, tR=5,96 мин (Метод 11).
Стадия 16-2. Синтез 3-(2,4-дихлорфенокси)бензойной кислоты (16-1)
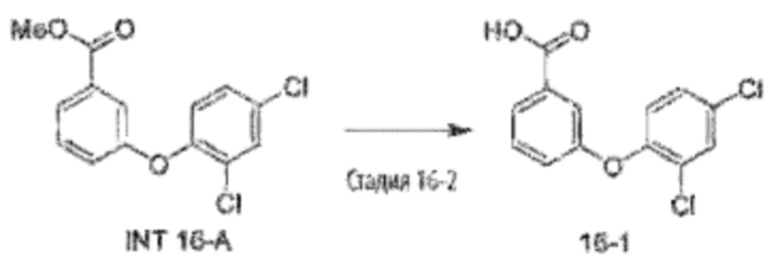
К перемешиваемому раствору метил-3-(2,4-дихлорфенокси)бензоата (INT 16-А) (50 мг, 0,17 ммоль) в МеОН (3 мл) добавляли 1М NaOH (67 мг, 1,68 ммоль). Реакционную смесь кипятили с обратным холодильником в течение 2 часов. Доводили рН до 2 добавлением 4 н. HCl (водн.) и затем экстрагировали ЭА. Органический слой промывали рассолом, осушали (MgSO4), фильтровали и концентрировали в вакууме, получая неочищенное твердое вещество, которое очищали хроматографией на SiO2 (ЭА/гексаны), получая 10 мг (21%) 3-(2,4-дихлорфенокси)бензойной кислоты (Соединение 16-1) в виде белого твердого вещества. ЖХ-MC-ESI (m/z), вычислено для C13H8Cl2O3: 281,99; найдено 283,6 [М+Н]+, tR=14,15 мин (Метод 10).
Соединения, перечисленные в Таблице 16, были получены с использованием процедур Схемы 16.
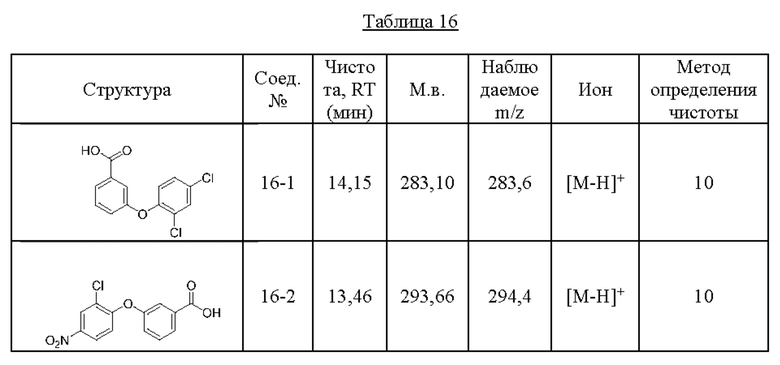
ПРИМЕР 17
Синтез Соединения 17-1, Соединения 1-31 и других типичных изостерных соединений тетразола
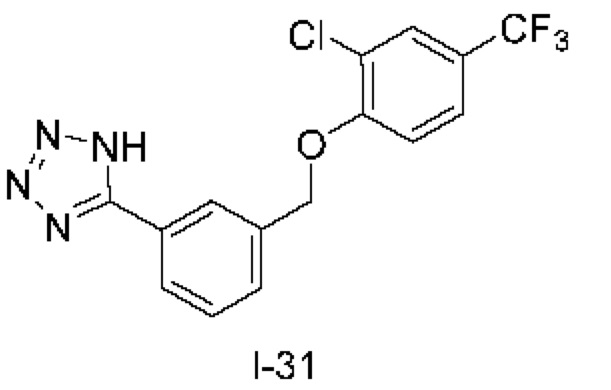
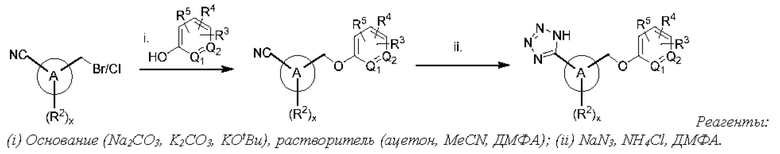
Стадия 17-1: 3-((2-Хлор-4-(трифторметил)фенокси)метил)бензонитрил (INT-17-1)
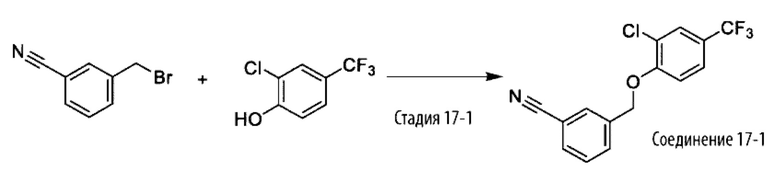
Смесь 3-(бромметил)бензонитрила (500 мг, 2,55 ммоль), 2-хлор-4-(трифторметил)фенола (0,551 г, 2,81 ммоль) и K2CO3 (1,06 г, 7,65 ммоль) в ацетоне (10,0 мл) нагревали при 80°С в течение 1 часа. Смесь фильтровали, и фильтрат концентрировали при пониженном давлении. Остаток очищали хроматографией на силикагеле (картридж 40 г), элюируя смесью гексанов и ЭА, с получением 750 мг (94%) 3-((2-хлор-4-(трифторметил)фенокси)метил)бензонитрила (Соединение 17-1) в виде твердого вещества. 1H ЯМР (400 МГц, ДМСО) δ 7,96-7,92 (м, 1Н), 7,90-7,87 (м, 1Н), 7,87-7,80 (м, 2Н), 7,72 (ддд, J=8,7, 2,3, 0,7 Гц, 1Н), 7,66 (т, J=7,7 Гц, 1Н), 7,43 (д, J=8,5 Гц, 1Н), 5,38 (с, 2Н); ЖХ-МС: m/z (ES-), [М-Н]-: 310,15; ВЭЖХ tR=5,78 мин (Метод 12).
Стадия 17-2: 5-(3-((2-хлор-4-(трифторметил)фенокси)метил)фенил)-1Н-1,2,3,4-тетразол (INT-31)
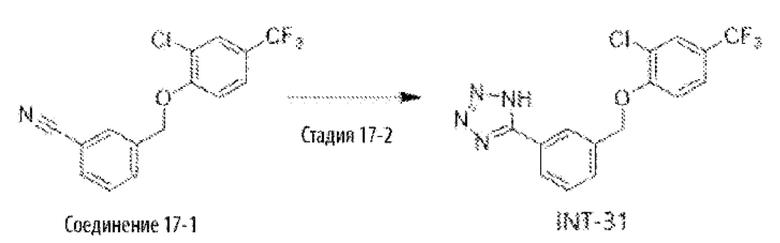
Смесь соединения 17-1 (100 мг, 0,321 ммоль), NaN3 (31,3 мг, 0,481 ммоль) и NH4Cl (27,5 мг, 0,513 ммоль) в ДМФА (1,00 мл) нагревали при 130°С в течение 12 часов. Смесь охлаждали до комнатной температуры и выливали в 2М HCl при 0°С. Смесь фильтровали, и твердое вещество сушили, получая 5-(3-((2-хлор-4-(трифторметил)фенокси)метил)фенил)-1Н-1,2,3,4-тетразол (INT-31) в виде твердого вещества (102 мг, 90%). 1H ЯМР (400 МГц, CD3OD) δ 8,17 (с, 1Н), 8,00 (д, J=7,8 Гц, 1Н), 7,72 (дд, J=8,1, 5,0 Гц, 2Н), 7,67-7,53 (м, 2Н), 7,33 (д, J=8,7 Гц, 1Н), 5,37 (с, 2Н); ЖХ-МС: вычислено для C15H10ClF3N4O: 354; найдено 355,06, [M-H]+, tR=4,35 мин (Метод 12).
Соединения, перечисленные в Таблице 17, были получены с использованием процедур Схемы 17.
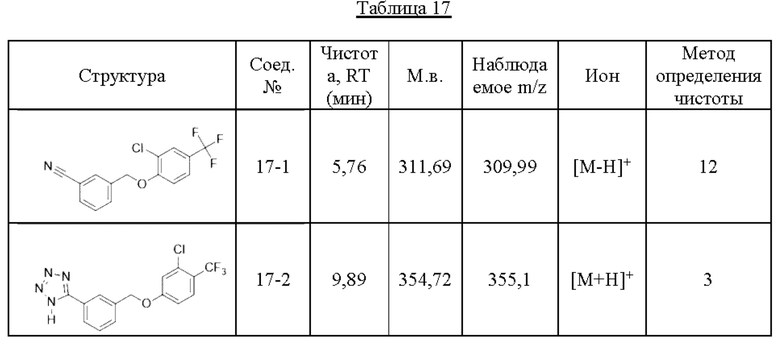
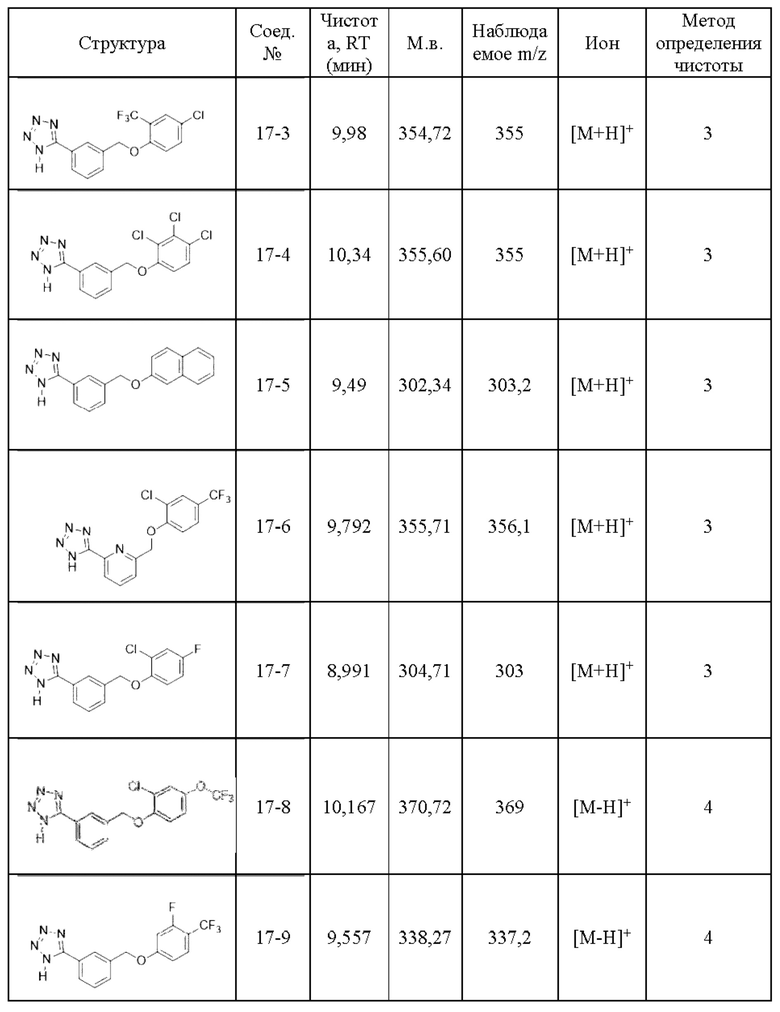
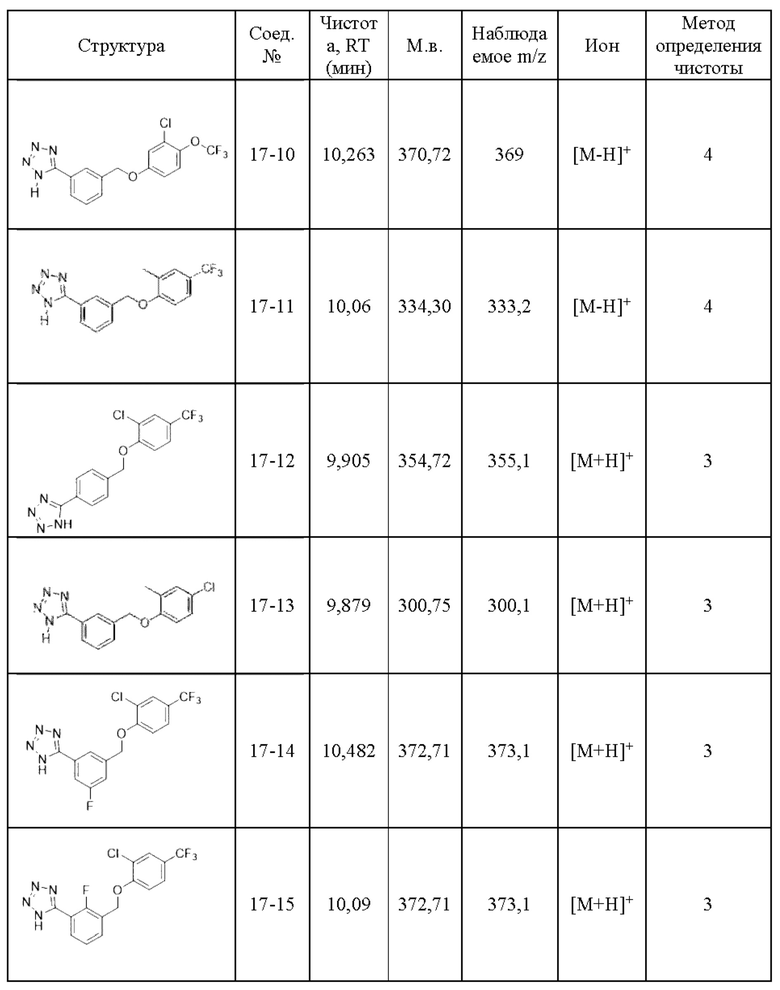
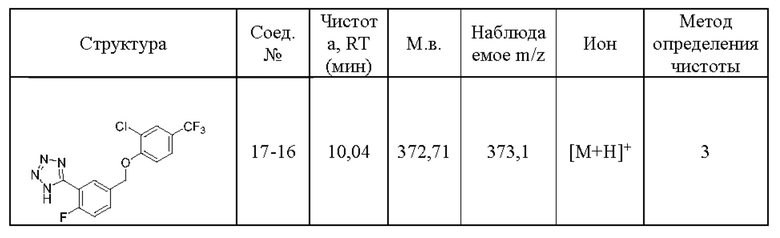
ПРИМЕР 18
Общий синтез типичных изостерных соединений арилсульфонамида
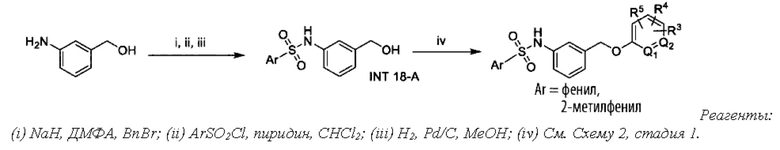
Раствор 3-амино бензинового спирта в ДМФА обрабатывают NaH и бензилбромидом с образованием 3-((бензилокси)метил)анилина. После выделения 3-((бензилокси)метил)анилин растворяют в CH2Cl2, обрабатывают пиридином и арилсульфонилхлоридом (ArSO2Cl) с получением арилсульфонамида, защищенного O-бензилом, и защитную группу удаляют каталитическим гидрированием с получением Промежуточного соединения 18-А в виде спирта. Получение конечного соединения осуществляют в соответствии с условиями реакции Мицунобу, описанными на Схеме 2, стадия 1.
ПРИМЕР 19
Общий синтез типичных соединений-изостеров сульфонилмочевины
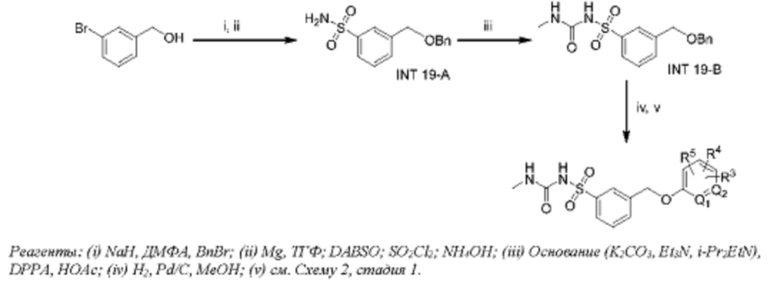
Раствор 3-бромбензилового спирта в ДМФА обрабатывают NaH и бензилбромидом с образованием 1-((бензилокси)метил)-3-бромбензола, который превращают в реактив Гриньяра на отдельной стадии путем растворения в сухом ТГФ и обработки Mg. Проводят реакцию реагента Гриньяра с DABSO, сульфурилхлоридом и гидроксидом аммония согласно Woolven, Н. etal. (Org. Lett. 13:4876, 2011) для получения защищенного 0-бензильной группой промежуточного соединения 19-А в форме сульфонамида. INT 19-А обрабатывают основанием, DPPA и уксусной кислотой в соответствии с методом Lockhwst, CA et al. (Tet. Lett. 48:8878, 2007) с получением защищенной O-бензилом сульфонилмочевины, промежуточного соединения 19-В. С INT 19-B снимают защиту каталитическим гидрированием с получением промежуточного свободного спирта, который превращают в конечное соединение в соответствии с условиями реакции Мицунобу, описанными на Схеме 2, стадия 1.
ПРИМЕР 20
Общий синтез типичных изостерных соединений N-ацилсульфонамида
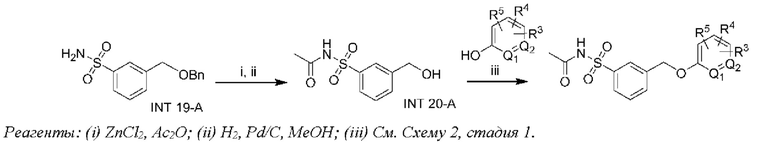
Промежуточное соединение 19-А (см. Пример 19) взаимодействует с ZnCl2 и уксусным ангидридом (Ac2O) согласно Pham, M.V. et al. (Angew. Chem I.E. 51:10610, 2012) с получением O-бензил-защищенного промежуточного N-ацилсульфонамида, с которого снимают защиту каталитическим гидрированием с образованием свободного спирта, промежуточного соединения 20-А. Спирт превращают в конечное соединение в соответствии с условиями реакции Мицунобу, описанными на Схеме 2, стадия 1.
ПРИМЕР 21
Общий синтез типичных изостерных соединений N-гидроксиамида
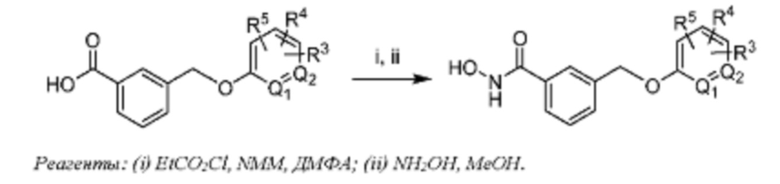
Соединение формулы (I), где А представляет собой фенил и R представляет собой карбоновую кислоту, растворяют в ДМФА и охлаждают до 0°С. Последовательно добавляют этилхлорформиат (1,2 экв.) и N-метилморфолин (1,3 экв.) и смесь перемешивают 10 минут. Добавляют гидроксиламин (2 экв.) в метаноле, и реакционной смеси дают нагреться до комнатной температуры и перемешивают в течение ночи. Обычная доработка и очистка дают желаемый продукт в виде N-гидроксиамида.
ПРИМЕР 22
Общий синтез типичных изостерных соединений фосфиновой кислоты
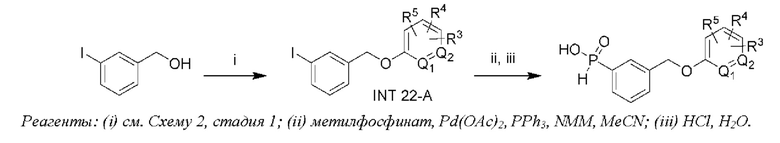
3-Иодбензиловый спирт превращают в промежуточное соединение 22-А в соответствии с условиями реакции Мицунобу, описанными на Схеме 2, стадия 1. Арилйодид превращают в алкилфосфинат с использованием катализируемой палладием реакции перекрестного сочетания (Pd(OAc)2 и PPh3 в качестве лиганда), описанной Grady, H.L. ("Preparation of arylphosphinic acid derivatives as building blocks for binding sites". Retrospective Theses and Dissertations, 10373, 1992), в которой в качестве основания используется метилфосфинат в ацетонитриле в присутствии NMM. Продукт в виде арилфосфиновой кислоты получают гидролизом алкилфосфината в водной HCl.
ПРИМЕР 23
Общий синтез типичных изостерных соединений фосфоновой кислоты
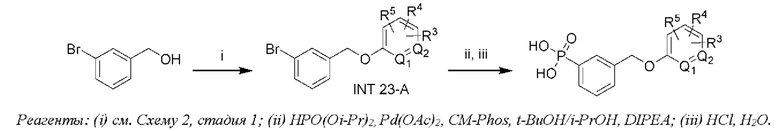
3-Бромбензиловый спирт превращают в промежуточное соединение 23-А в соответствии с условиями реакции Мицунобу, описанными на Схеме 2, стадия 1. Арилбромид превращают в диалкилфосфонат с использованием реакции перекрестного сочетания, катализируемой палладием (Pd(OAc)2 и CM-Phos в качестве лиганда), описанной Fu, C.W. et al. (Org. Lett. 17:5906, 2015), в которой используется диизопропилфосфит в спиртовом растворителе с DIPEA в качестве основания. Продукт в виде арилфосфоновой кислоты получают гидролизом диалкилфосфоната в водной HCl.
ПРИМЕР 24
Общий синтез типичных изостерных соединений пирролидин-2,4-диона
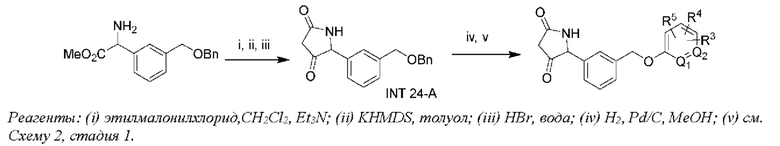
Превращение исходного производного аминокислоты фенилглицина в пирролидин-2,4-дион осуществляется посредством трехстадийной последовательности реакций присоединения/циклизации/декарбоксилирования, описанной в WO 2007/063010, с получением промежуточного соединения 24-А. С этого промежуточного продукта снимают защиту каталитическим гидрированием с получением свободного спирта, который превращают в конечное соединение в соответствии с условиями реакции Мицунобу, описанными на Схеме 2, стадия 1. Исходный материал, представляющий собой метиловый эфир аминокислоты, может быть получен различными способами, известными специалистам в данной области; например, из 3-((бензилокси)метил)бензальдегида (описан ниже) путем синтеза аминокислот по Штреккеру и эстерификации.
ПРИМЕР 25
Общий синтез типичных изостерных соединений фуран-2,4-диона
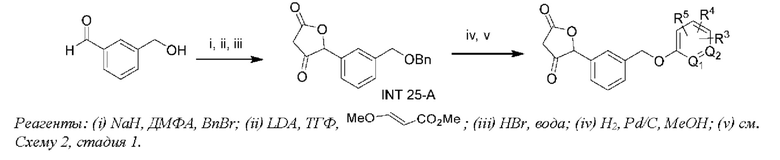
Проводят реакцию коммерчески доступного 3-гидроксиметилбензальдегида с NaH и бензилбромидом с образованием 3-((бензилокси)метил)бензальдегида. Это промежуточное соединение превращают в циклопентан-1,3-дион путем двухступенчатой последовательности, описанной в WO 2007/063010, с получением промежуточного соединения 25-А. С этого промежуточного продукта снимают защиту каталитическим гидрированием с получением свободного спирта, который превращают в конечное соединение в соответствии с условиями реакции Мицунобу, описанными на Схеме 1, стадия 1.
ПРИМЕР 26
Общий синтез типичных изостерных соединений циклопентан-1,3-диона
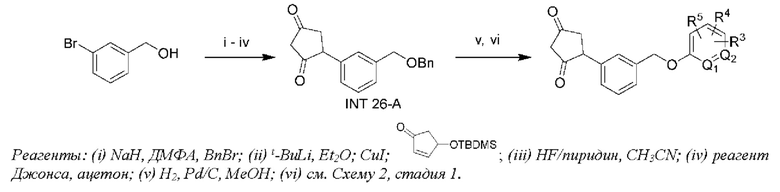
Раствор 3-бромбензилового спирта в ДМФА обрабатывают NaH и бензилбромидом с образованием 1-((бензилокси)метил)-3-бромбензола. Это промежуточное соединение превращают в циклопентан-1,3-дион путем трехстадийной последовательности сопряженного присоединения/снятия защиты/окисления, описанной Lassalas, P. et al. (ACS Med. Chem. Lett. 8:864, 2017), с получением промежуточного соединения 26-А. С этого промежуточного продукта снимают защиту каталитическим гидрированием с получением свободного спирта, который превращают в конечное соединение в соответствии с условиями реакции Мицунобу, описанными на Схеме 2, стадия 1.
ПРИМЕР 27
Общий синтез типичных изостерных соединений дифторфенола
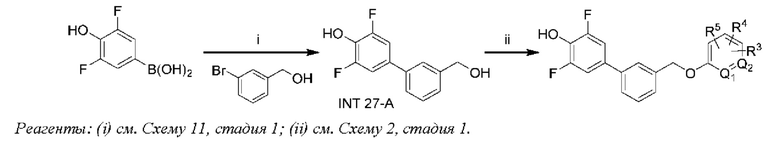
Проводят реакцию сочетания 3-бромбензилового спирта и коммерчески доступной 4-окси-3,5-дифторфенилборной кислоты в соответствии с методом сочетания Сузуки, описанным в Примере 11, с получением промежуточного соединения 27-А, которое превращают в конечное соединение в соответствии с условиями реакции Мицунобу, описанными на Схеме 2, стадия 1.
ПРИМЕР 28
Общий синтез типичных изостерных соединений 3-замещенного 5-оксотиадиазола
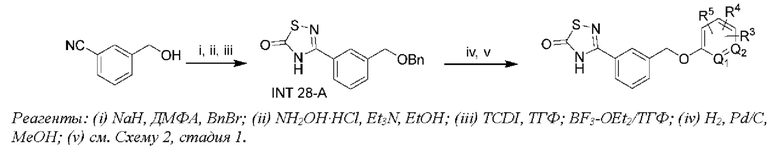
Раствор 3-цианобензилового спирта в ДМФА обрабатывают NaH и бензилбромидом с образованием 3-((бензилокси)метил)бензонитрила. Затем арилцианид превращают в 3-замещенный 5-оксотиадиазол в две стадии согласно Kohara, Y. et al. (J. Hetercyclic Chem. 37:1419, 2000) с получением промежуточного соединения 28-А. С этого промежуточного продукта снимают защиту каталитическим гидрированием с получением свободного спирта, который превращают в конечное соединение в соответствии с условиями реакции Мицунобу, описанными на Схеме 2, стадия 1.
ПРИМЕР 29
Общий синтез типичных изостерных соединений 3-замещенного оксадиазолона
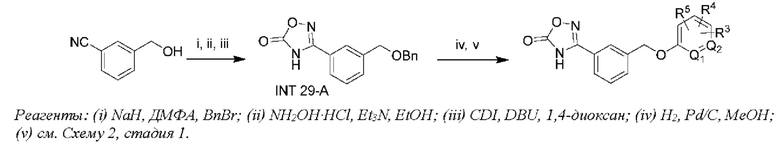
Раствор 3-цианобензилового спирта в ДМФА обрабатывают NaH и бензилбромидом с образованием 3-((бензилокси)метил)бензонитрила. Затем арилцианид превращают в 3-замещенный оксадиазолон в две стадии согласно Yu, X. et al. (Org. Lett. 18:5412, 2-016) с получением промежуточного соединения 29-А. С этого промежуточного продукта снимают защиту каталитическим гидрированием с получением свободного спирта, который превращают в конечное соединение в соответствии с условиями реакции Мицунобу, описанными на Схеме 2, стадия 1.
ПРИМЕР 30
Общий синтез типичных изостерных соединений тиазолидин-2,4-диона
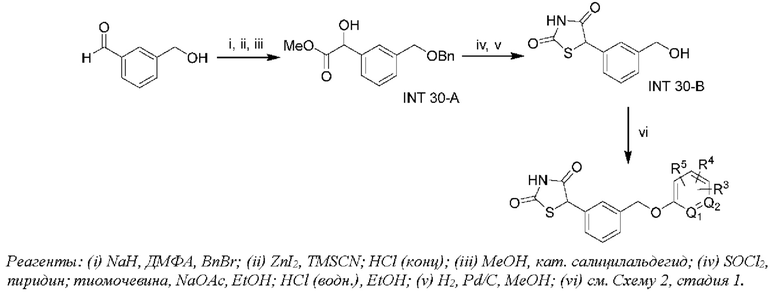
Проводят реакцию коммерчески доступного 3-гидроксиметилбензальдегида с NaH и бензилбромидом с образованием 3-((бензилокси)метил)бензальдегида. Полученный эфиральдегид превращают в манделат по Сириманну и Паттерсону (Sirimanne and Patterson) (J. Label. Cmpd. Radiopharm. 33:725, 1993) с получением сначала производного миндальной кислоты, которое эстерифицируют с образованием промежуточного соединения 30-А. Промежуточное соединение 30-А превращают в тиазолидин-2,4-дион согласно Koyama et al. (Biorg. Med. Chem. Lett. 13:1801, 2003), а каталитическое гидрирование позволяет снять защиту со спирта с получением промежуточного соединения 30-В. Спирт 30-В превращают в конечное соединение в соответствии с условиями реакции Мицунобу, описанными на Схеме 2, стадия 1.
ПРИМЕР 31
Альтернативный синтез Соединения 1-56 и синтез 31-2
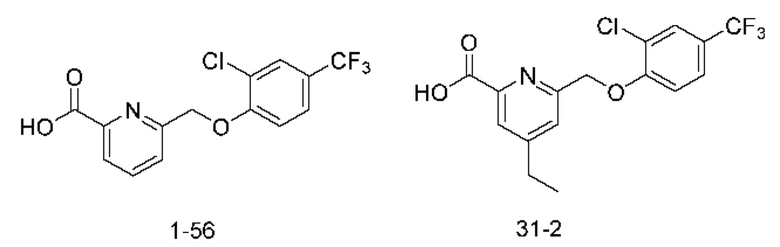
Схема 31
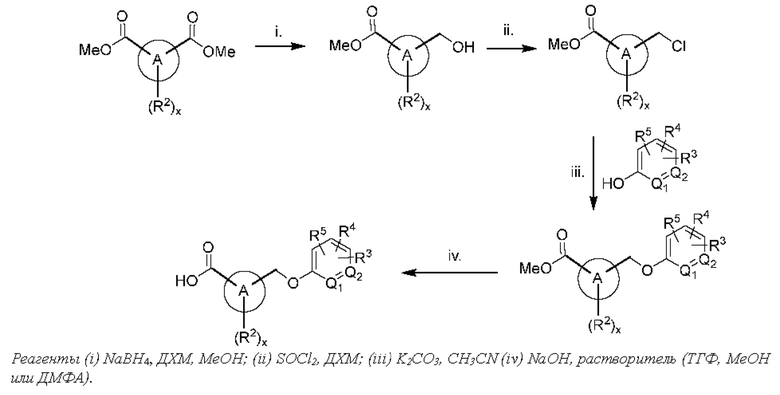
Стадия 31-1. Синтез метил-6-(гидроксиметил)пиколината(INT 31-А)
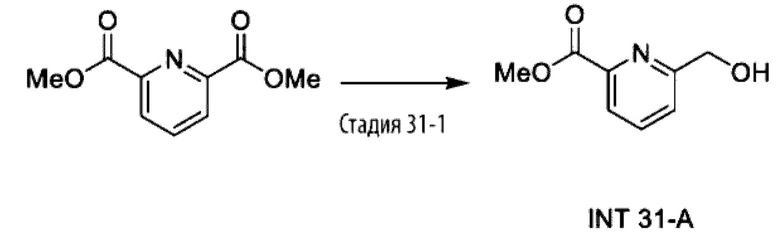
К перемешиваемому раствору диметилпиридин-2,6-дикарбоксилата (20 г, 102,5 ммоль) в МеОН (20 мл) при 0°С добавляли боргидрид натрия (5,81 г, 153,7 ммоль) 3 порциями. Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 1 часа. Реакционную смесь разбавляли NH4Cl (водн.) (10 мл) и экстрагировали ЭА (3×500 мл) и ЭА (10 мл). Объединенные органические слои осушали (Na2SO4), концентрировали в вакууме и очищали хроматографией на SiO2 (10% МеОН в ЭА/гексанах) с получением 12 г (70%) метил-6-(гидроксиметил)пиридин-2-карбоксилата (INT 31-A) в виде белого твердого вещества. ТСХ (ЭА): Rf=0,60.
Стадия 31-2. Синтез метил-6-(хлорметил)пиколината (INT 31-В)
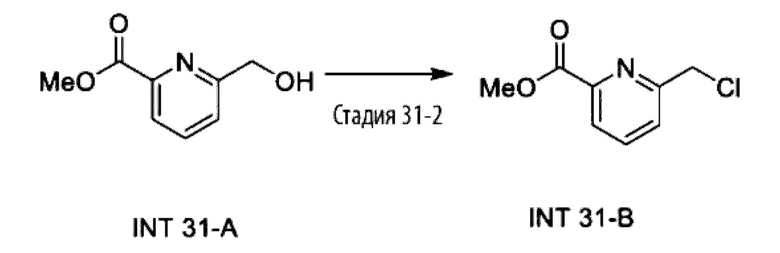
В колбу, содержащую перемешиваемый раствор INT 31-A (5,00 г, 29,9 ммоль) в ДХМ (62,5 мл), загружали тионилхлорид (4,36 мл, 59,8 ммоль) при комнатной температуре. После перемешивания в течение 14 часов в реакционную смесь по каплям добавляли насыщенный водный K2CO3, чтобы довести рН до 10-11. Органический слой собирали, а водный слой дважды экстрагировали ДХМ. Органические слои объединяли, промывали рассолом, осушали (Na2SO4) и очищали хроматографией на SiO2 (ЭА/гексаны) с получением 3,9 г (69,7%) метил-6-(хлорметил)пиколината (INT 31-В) в виде бесцветной маслянистой жидкости, которая затвердевает при стоянии с образованием белого кристаллического порошка. ЖХ-MC-ESI (m/z) вычислено для C8H8ClNO2: 185,61; m/z 186,1 (М+Н)+, tR=3,64 мин (Метод 1). 1H ЯМР (500 МГц, ДМСО-d6) δ 8,09-7,99 (м, 2Н), 7,81 (дд, J=7,5, 1,3 Гц, 1Н), 4,86 (с, 2Н), 3,89 (с, 3Н).
Стадия 31-3. Синтез метил-6-((2-хлор-4-(трифторметил)фенокси)метил)пиколината (Соединение 31-С)
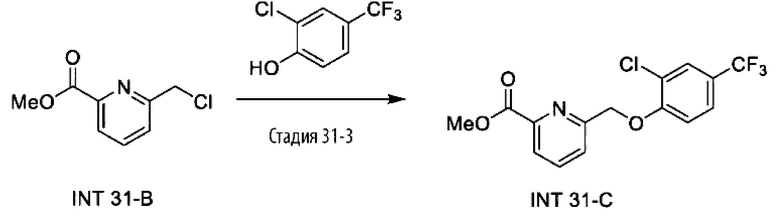
В колбу, содержащую INT 31-В (3,861 г, 20,8 ммоль), загружали раствор 2-хлор-4-(трифторметил)фенола (4,497 г, 22,9 ммоль) в MeCN (70 мл), а затем K2CO3 (4,312 г, 31,2 ммоль) и йодид калия (345,3 мг, 2,08 ммоль). Полученную суспензию нагревали до 60°С. После перемешивания в течение 16 часов реакционную смесь охлаждали до комнатной температуры, разбавляли H2O и экстрагировали 3 раза Et2O. Органические слои объединяли, промывали рассолом, осушали (Na2SO4) и очищали хроматографией на SiO2 (ЭА/гексаны) с получением 6,64 г (92,4%) метил-6-((2-хлор-4-(трифторметил)фенокси)метил)пиколината (INT 31-С) в виде белого твердого вещества. ЖХ-MC-ESI (m/z) вычислено для C15H11ClF3NO3: 345,70; найдено 346,1 [М+Н]+, tR=5,99 мин (Метод 1).
Стадия 31-4. Синтез 6-((2-хлор-4-(трифторметил)фенокси)метил)пиколиновой кислоты (Соединение 1-56)

В колбу, содержащую перемешиваемый раствор INT 31-С (500 мг, 1,45 ммоль) в ТГФ (7,23 мл), загружали 1М NaOH (7,23 мл, 7,23 ммоль). После перемешивания в течение 17 часов при 50°C смесь разбавляли ТГФ и H2O, однако четких слоев не наблюдалось. Добавляли Et2O для разделения органического и водного слоев. Водный слой собирали и подкисляли до рН 3-4, используя 3М HCl. Полученный белый осадок экстрагировали 3 раза Et2O, и объединенные органические экстракты промывали рассолом, осушали (Na2SO4) и концентрировали при пониженном давлении, получая 310 мг (64,6%) 6-((2-хлор-4-(трифторметил)фенокси)метил)пиколиновой кислоты (Соединение 1-56) в виде белого твердого вещества. ЖХ-MC-ESI (m/z) вычислено для C14H9ClF3NO3: 331,68; найдено 332,1 [М+Н]+, tR=5,34 мин (Метод 1).
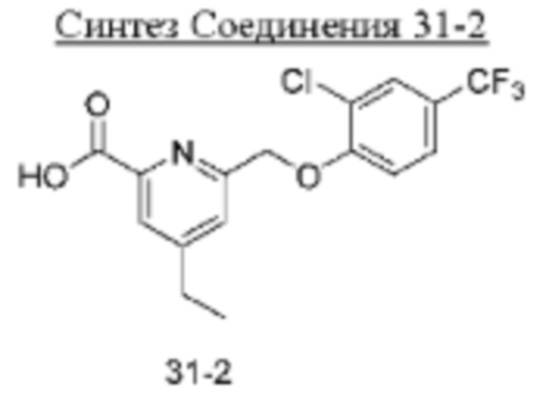
Стадия 31-5. Синтез диметил-4-этилпиридин-2,6-дикарбоксилата (INT 31-D)
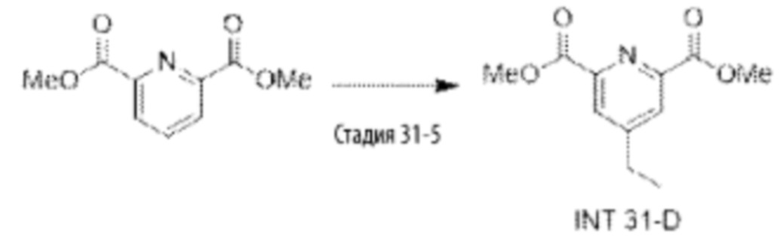
К раствору диметилгафидин-2,6-дикарбоксилата (10 г, 51,2 ммоль) и пропаналя (18,7 мл, 256,2 ммоль) в H2SO4 (100 мл) добавляли FeSO4 (5,70 г, 20,49 ммоль) и 30% Н2О2 (9,9 мл, 102,5 ммоль) по каплям на протяжении 15 мин. После перемешивания при 0°C в течение 15 минут смесь разбавляли насыщенным K2CO3 (водн.) и экстрагировали ЭА. Органический слой осушали (Na2SO4), концентрировали в вакууме и очищали хроматографией на SiO2 (петролейный эфир/ЭА) с получением 4,5 г (39%) диметил-4-этилпиридин-2,6-дикарбоксилата (INT 31-D) в виде желтого твердого вещества. ТСХ (3:1 петролейный эфир : ЭА): Rf=0,6. 1Н ЯМР (400 МГц, CDCl3) δ 8,13-8,20 (м, 2Н), 4,00-4,03 (м, 6Н), 2,78-2,87 (м, 2Н), 1,29-1,37 (м, 3Н).
Стадия 31-6. Синтез метил-4-этил-6-(гидроксиметил)пиколината (1NT 31-Е)
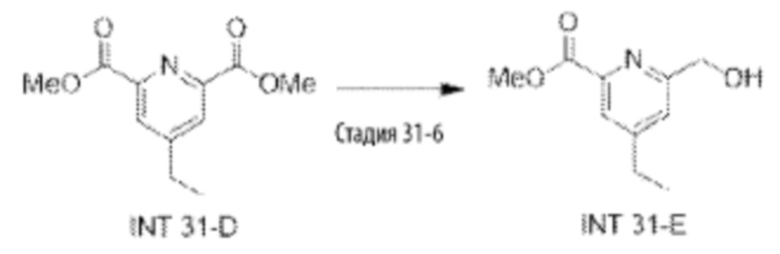
К раствору INT-31-D (4,5 г, 20,2 ммоль) в МеОН (80 мл) и ДХМ (20 мл) добавляли NaBH4 (1,14 г, 30,24 ммоль) при 0°C. После перемешивания в течение 12 ч при 20°C смесь разбавляли насыщенным водн. NH4Cl и экстрагировали ЭА. Органический слой осушали (Na2SO4), концентрировали в вакууме и очищали хроматографией на SiO2 (петролейный эфир/ЭА) с получением 2,8 г (71%) метил-4-этил-6-(гидроксиметил)пиколината (INT 31-Е) в виде желтого твердого вещества. ТСХ (1:1 петролейный эфир : ЭА): Rf=0,4. 1Н ЯМР (400 МГц, CDCl3) δ 7,84-7,93 (м, 1H), 7,34-7,43 (м, 1H), 4,79-4,86 (м, 2Н), 3,93-4,00 (м, 3Н), 2,68-2,77 (м, 2Н), 1,27 (т, J=7,64 Гц, 3Н).
Стадия 31-7. Синтез метил-6-(хлорметил)-4-этилпиколината (INT 31-D
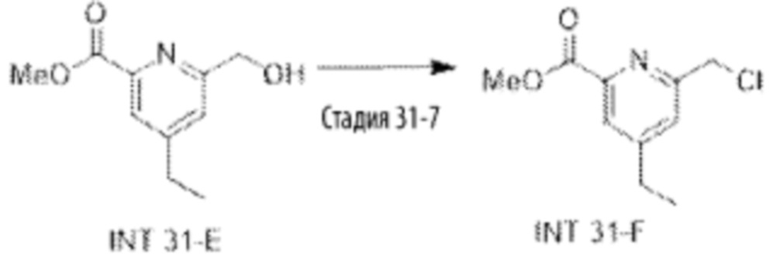
К раствору INT-31-E (2,8 г, 14,34 ммоль) в ДХМ (100 мл) при 0°C добавляли SOCl2 (14,01 мл, 193 ммоль). Через 1,5 ч реакционную смесь концентрировали с получением 2,5 г (82%) метил-6-(хлорметил)-4-этилпиколината (INT-31F) в виде желтой маслянистой жидкости, которую использовали на следующей стадии без какой-либо дополнительной очистки. ЖХ-MC-ESI (m/z) вычислено для C10H12ClNO2: 213,66; найдено 214,0 [М+Н]+, tR=0,842 мин (Метод 6).
Стадия 31-8. Синтез метил-6-((2-хлор-4-(трифторметил)фенокси)метил)-4-этилпиколината (INT 31-G)
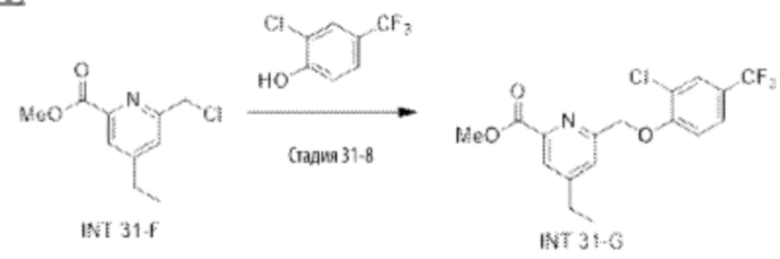
К раствору INT-31-F (2,5 г, 11,70 ммоль) и 2-хлор-4-(трифторметил)фенола (2,0 г, 10,18 ммоль) в MeCN (160 мл) добавляли K2CO3 (4,85 г, 35,10 ммоль). Суспензию перемешивали при 80°C в течение 12 часов, охлаждали и фильтровали для сбора остатка, который очищали хроматографией на SiO2 (ПЭ:ЭА), получая 4 г (82%) метил-6-((2-хлор-4-(трифторметил)фенокси)метил)-4-этилпиколината (INT-31-G) в виде светло-желтого твердого вещества. ТСХ (3:1 петролейный эфир : ЭА): Rf=0,55. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 1,32 (т, J=7,58 Гц, 3Н), 2,79 (кв, J=7,62 Гц, 2Н), 4,03-4,05 (м, 3Н), 5,40 (с, 2Н), 7,06 (д, J=8,56 Гц, 1H), 7,50 (дд, J=8,68, 1,59 Гц, 1H), 7,67-7,74 (м, 2Н), 7,97 (с, 1H).
Стадия 31-9. Синтез 6-((2-хлор-4-(трифторметил)фенокси)метил)-4-этилпиколиновой кислоты (Соединение 31-2)
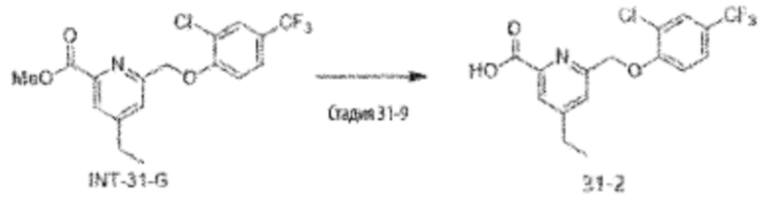
Раствор INT-31-G (3,4 г, 9,1 ммоль) и LiOH-H2O (1,15 г, 27,3 ммоль) в ТГФ (5 мл) и H2O (1 мл) перемешивали при 30°C в течение 12 час. Реакционную смесь подкисляли 1 н. HCl до рН 6, затем экстрагировали ЭА. Объединенные органические экстракты осушали (Na2SO4), концентрировали и растворяли в MeCN. Добавляли H2O, чтобы образовался белый осадок, который собирали фильтрацией и промывали H2O. Полученный осадок на фильтре лиофилизировали с получением 1,93 г (58%) 6-((2-хлор-4-(трифторметил)фенокси)метил)-4-этилпиколиновой кислоты (Соединение 31-2) в виде белого твердого вещества. ЖХ-MC-ESI (m/z) вычислено для C16H13ClF3NO3: 359,7; найдено 360,0 [М+Н]+, tR=0,95 мин (Метод 5-95AB_R_220 & 254.1cm). 1Н ЯМР (400 МГц, CDCl3) δ м.д. 8,07 (с, 1H), 7,76 (с, 1H), 7,72 (д, J=1,88 Гц, 1H), 7,53 (дд, J=8,63, 1,63 Гц, 1H), 7,06 (д, J=8,63 Гц, 1H), 5,34 (с, 2Н), 2,84 (кв, J=7,63 Гц, 2Н), 1,33 (т, J=7,57 Гц, 3Н).
ПРИМЕР 32
Синтез Соединения 32-1
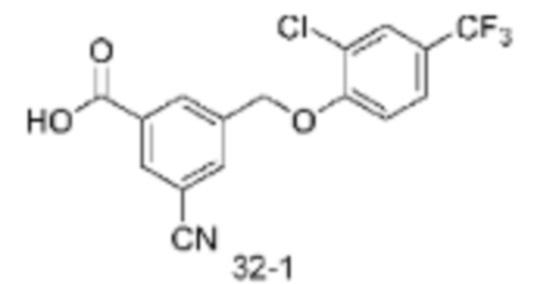
Схема 32
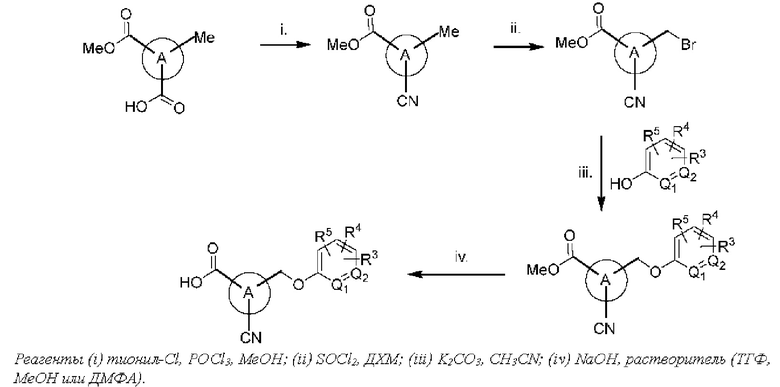
Стадия 32-1. Синтез метил-3-циано-5-метилбензоата (INT 32-А)
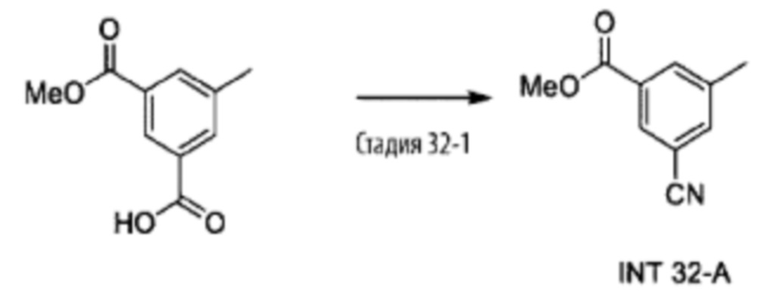
Тионилхлорид (7 мл) добавляли к 3-(метоксикарбонил)-5-метил бензойной кислоте (1,5 г, 7,7 ммоль). После перемешивания при кипячении с обратным холодильником в течение 1 часа реакционную смесь растворяли и концентрировали 3 раза с толуолом. Остаток растворяли в ДХМ (5 мл) и добавляли к NH4OH (5 мл) при 0°C, получая белый осадок. Реакционную смесь перемешивали 5 мин при 0°C. Добавляли H2O и ЭА, и смесь фильтровали, получая 1,40 г белого твердого вещества. К отфильтрованному твердому веществу добавляли POCl3 (4,7 мл) и реакционную смесь нагревали при 100°C в течение 1 часа. Реакционную смесь охлаждали, концентрировали в вакууме, растворяли в ДХМ и обрабатывали насыщ. NaHCO3. Смесь экстрагировали ЭА (2×20 мл), осушали (Na2SO4), фильтровали и концентрировали в вакууме, получая 1,15 г сырого материала. Неочищенный материал очищали хроматографией на SiO2 (ЭА/гексаны) с получением 952 мг (70%) метил-3-циано-5-метилбензоата (INT 32-А) в виде белого твердого вещества. ЖХ-MC-ESI (m/z) вычислено для C10H9NO2: 175,19; найдено 176,2 [М+Н]+, tR=4,87 мин (Метод 1). 1Н ЯМР (500 МГц, CDCl3) 5 8,12 (шир.с, 1H), 8,07 (шир.с, 1H), 7,63 (шир.с, 1H), 3,94 (с, 3Н), 2,45 (с, 3Н).
Стадия 32-2. Синтез метил-3-(бромметил)-5-цианобензоата (TNT 32-В)
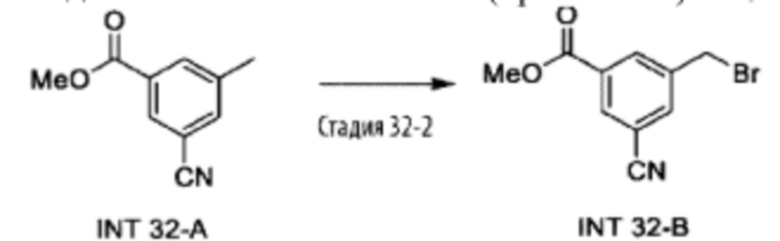
К перемешиваемому раствору INT 32-А (0,50 г, 2,9 ммоль) в CCl4 (10 мл) добавляли NBS (0,56 г, 3,1 ммоль) и AIBN (94 мг, 0,57 ммоль). Реакционную смесь нагревали до 77°C (с обратным холодильником) в течение 4 часов, затем концентрировали в вакууме и очищали хроматографией на SiO2 (ЭА/гексаны) с получением 247 мг (34%) метил-3-(бромметил)-5-цианобензоата (INT 32-В) в виде белого твердого вещества. ЖХ-MC-ESI (m/z) вычислено для C10H8BrNO2: 254,08; m/z 255,2 (М+Н)+, tR=5,05 мин (Метод 1).
Стадия 32-3. Синтез метил-3-((2-хлор-4-(трифторметил)фенокси)метил)-5-цианобензоата (INT 32-С)
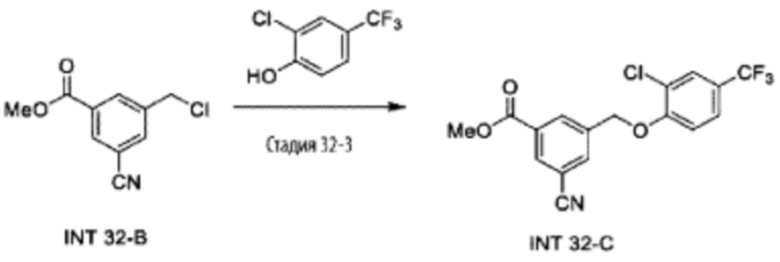
К перемешиваемому раствору INT 32-В (124 мг, 488 мкмоль) в MeCN (3 мл) добавляли 2-хлор-4-(трифторметил)фенол (95,9 мг, 488 мкмоль) и K2CO3 (87,7 мг, 634 мкмоль). После нагревания при 60°C в течение 12 часов реакционную смесь охлаждали до комнатной температуры и разбавляли H2O (6 мл). Водный слой экстрагировали Et2O (2×6 мл) и ЭА (6 мл), осушали (Na2SO4), фильтровали через целит и концентрировали в вакууме с получением 146,1 мг (81%) метил-3-((2-хлор-4-(трифторметил)фенокси)метил)-5-цианобензоата (INT 32-С) в виде твердого вещества бежевого цвета. ЖХ-MC-ESI (m/z) вычислено для C17H11ClF3NO3: 369,72; найдено 370,0 [М+Н]+, tR=6,39 мин (Метод 1).
Стадия 32-4. Синтез 3-((2-хлор-4-(трифторметил)фенокси)метил)-5-цианобензойной кислоты (Соединение 32-1)
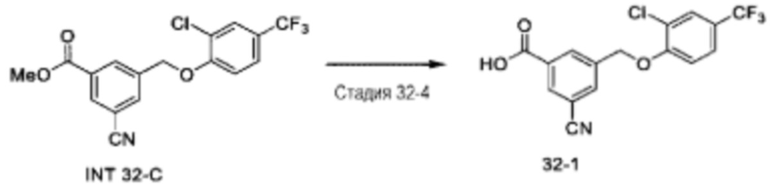
Во флакон, содержащий перемешиваемый раствор INT 32-С (146,1 мг, 395,2 мкмоль) в МеОН (2 мл) и ТГФ (2 мл), загружали твердый NaOH (79 мг, 1,98 ммоль). После перемешивания при 50°C в течение 12 часов реакционную смесь разбавляли H2O и подкисляли до рН 4-5, используя 3М HCl. Полученный белый осадок экстрагировали Et2O (3×10 мл) и ЭА (2×10 мл). Органические слои объединяли, промывали рассолом, осушали (Na2SO4) и концентрировали при пониженном давлении с получением неочищенного твердого вещества, которое очищали с помощью ВЭЖХ с обращенной фазой (H2O/CH3CN). Лиофилизация объединенных чистых фракций дала 82,6 мг (59%) 3-((2-хлор-4-(трифторметил)фенокси)метил)-5-цианобензойной кислоты (Соединение 32-1) в виде белого твердого вещества. ЖХ-MC-ESI (m/z) вычислено для C16H9ClF3NO3: 355,0; найдено 354,0 [М-H]+, tR=10,07 мин (Метод 4).]Н ЯМР (500 МГц, ДМСО-d6) δ 13,60 (шир.с, 1Н), 8,36 (с, 1H), 8,29 (с, 1H), 8,18 (с, 1H), 7,89 (с, 1H), 7,74 (дд, J=8,5, 2,0 Гц, 1H), 7,43 (д, J=9,0 Гц, 1Н), 5,46 (с, 2Н).
Соединения, перечисленные в Таблице 32, были получены с использованием процедур Схемы 32.
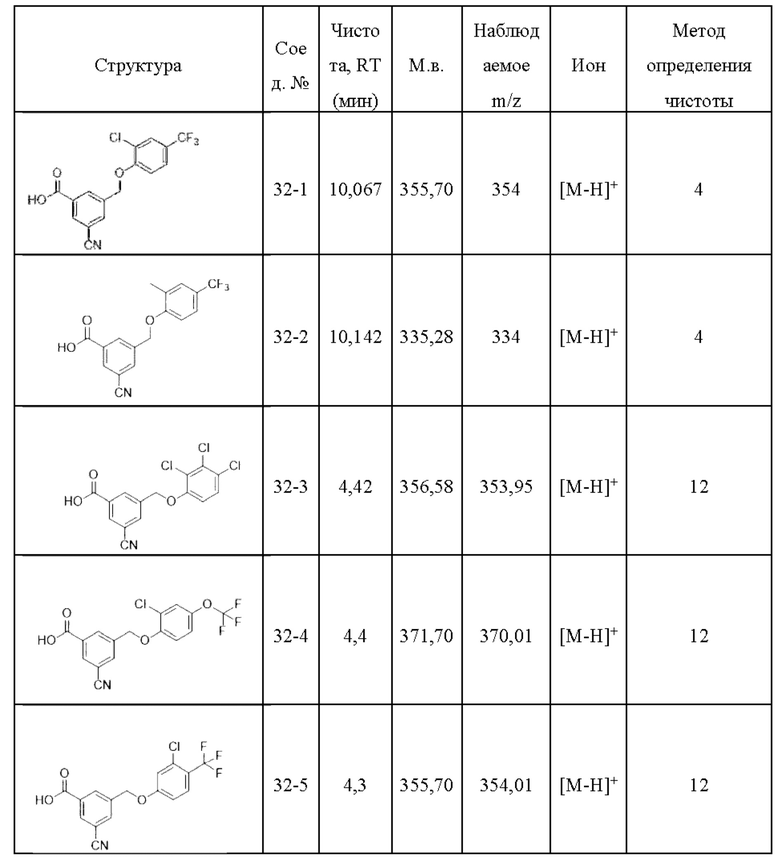
ПРИМЕР 33
Синтез Соединения 33-1
Схема 33
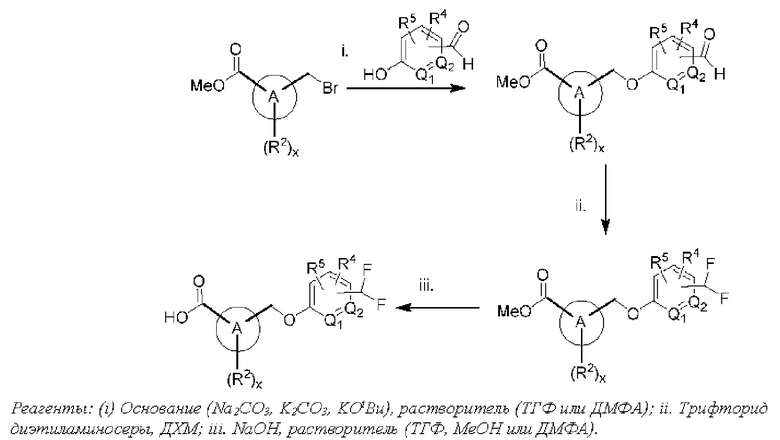
Стадия 33-1. Синтез метил-3-((2-формилфенокси)метил)бензоата (INT 33-А)
К перемешиваемому раствору метил-3-(бромметил)бензоата (300 мг, 1,31 ммоль) в MeCN (6 мл) добавляли 2-гидроксибензальдегид (160 мг, 1,31 ммоль) и K2CO3 (235 мг, 1,70 ммоль). После нагревания при 60°C в течение 18 часов смесь охлаждали до комнатной температуры, разбавляли H2O (6 мл) и водный слой экстрагировали Et2O (2×6 мл) и ЭА (6 мл). Объединенные органические слои осушали (Na2SO4), фильтровали через целит и очищали хроматографией на SiO2 (ЭА/гексаны) с получением 315 мг (89%) метил-3-((2-формилфенокси)метил)бензоата (INT 33-А) в виде белого твердого вещества. ЖХ-MC-ESI (m/z) вычислено для С16Н14О4: 270,1; найдено 271,5 (М+Н)+, tR=5,4 мин (Метод 1).
Стадия 33-2. Синтез метил-3-((2-(дифторметил)фенокси)метил)бензоата (INT 33-В)
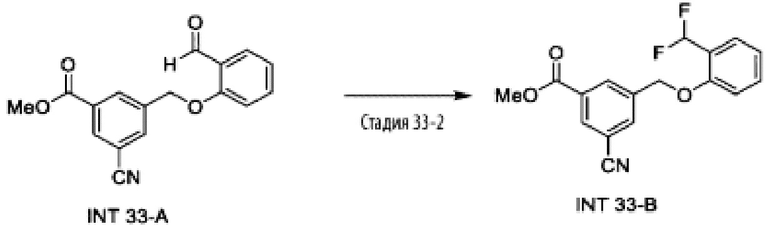
В перемешиваемый раствор INT 33-А (50 мг, 0,18 ммоль) в ДХМ (2 мл) добавляли трифторид диэтиламиносеры (0,12 мл, 0,92 ммоль). После нагревания при 40°C в течение ночи добавляли дополнительное количество трифторида диэтиламиносеры (0,12 мл, 0,92 ммоль) и реакционную смесь перемешивали при 40°C в течение ночи. Реакционную смесь охлаждали до комнатной температуры, разбавляли H2O и экстрагировали ДХМ (3×5 мл). Объединенные органические экстракты осушали (Na2SO4), фильтровали, концентрировали в вакууме и очищали с помощью хроматографии на SiO2 (ЭА/гексаны) с получением 28,7 мг (53%) метил-3-((2-(дифторметил)фенокси)метил)бензоата (INT 33-В). ЖХ-MC-ESI (m/z) вычислено для C16H14F2O3: 292,28; найдено 273,2 (М+Н)+, tR=5,94 мин (Метод 1). 1Н ЯМР (500 МГц, CDCl3) δ 8,09 (с, 1H), 8,03-8,01 (м, 1Н), 7,65-7,63 (м, 1H), 7,60 (д, J=10 Гц, 1Н),7,48 (д, J=10,0 Гц, 1H), 7,41-7,39 (м, 1H), 7,06 (т, J=10,0 Гц, 1Н),7,02 (т, J=55 Гц, 1H), 6,99-6,97 (м, 1Н), 5,17 (с, 2Н), 3,94 (с, 3Н).
Стадия 33-3. Синтез 3-((2-(дифторметил)фенокси)метил)бензойной кислоты (Соединение 33-1)
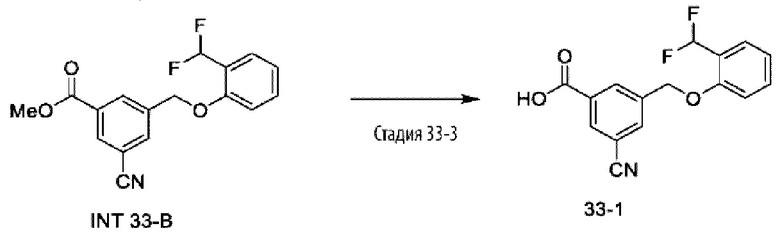
К перемешиваемому раствору INT 33-В (28,7 мг, 98,2 мкмоль) в ТГФ (2 мл) добавляли 1М NaOH (0,5 мл, 491 мкмоль). Реакционную смесь нагревали при 60°C в течение ночи, концентрировали в вакууме, разбавляли 3М HCl и экстрагировали (ЕА и Et2O). Объединенные органические слои осушали (Na2SO4), фильтровали и концентрировали в вакууме, получая 20,0 мг (73%) 3-((2-(дифторметил)фенокси)метил)бензойной кислоты (Соединение 33-1) в виде белого твердого вещества. ЖХ-MC-ESI (m/z) вычислено для C15H12F2O3: 278,3; найдено 277,2 [М-Н]+, tR=8,02 мин (Метод 4). 1Н ЯМР (500 МГц, ДМСО-d6) δ 12,99 (шир.с, 1Н), 8,05 (с, 1H), 7,91 (д, J=7,5 Гц, 1H), 7,73 (д, J=7,0 Гц, 1H), 7,54-7,45 (м, 3Н), 7,26-7,04 (м, 3Н), 5,30 (с, 2Н).
Соединения, перечисленные в Таблице 33, были получены с использованием процедур Схемы 33.
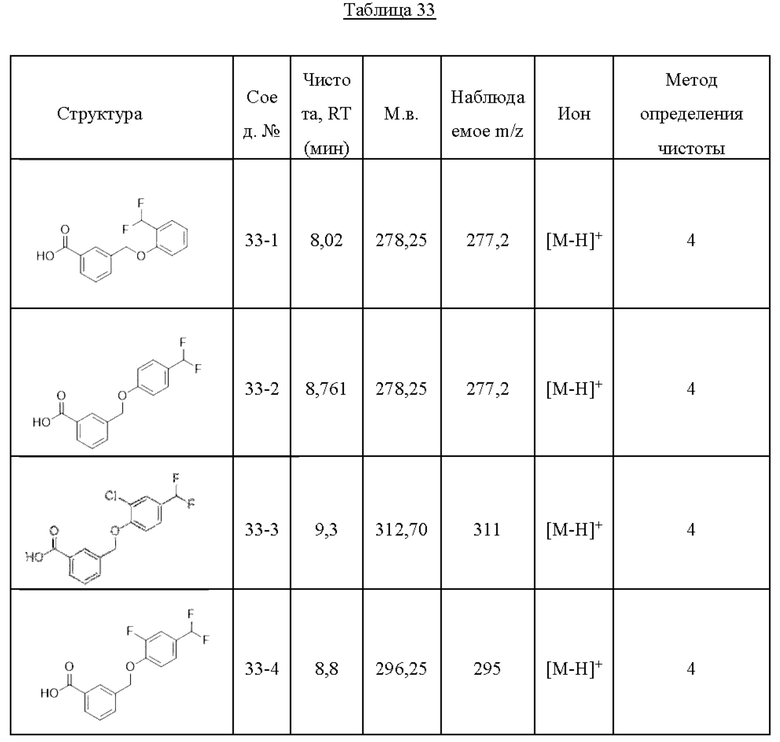
ПРИМЕР 34
Синтез соединения 34-1
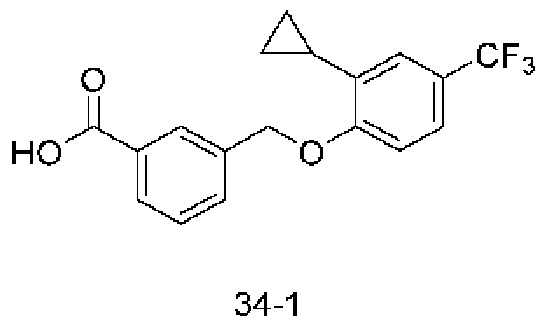
Схема 34
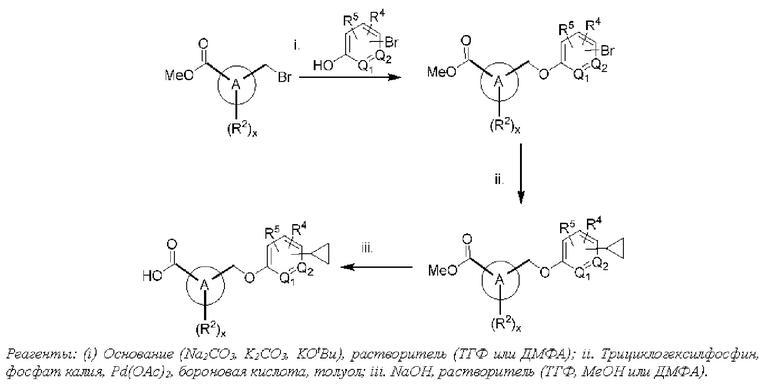
Стадия 34-1. Синтез метил-3-((2-бром-4-(трифторметил)фенокси)метил)бензоата (INT 34-А)
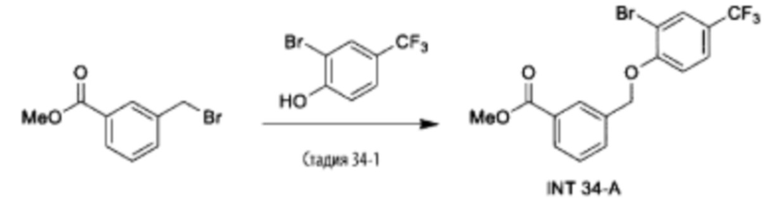
Во флакон, содержащий перемешиваемый раствор 2-бром-4-(трифторметил)фенола (316 мг, 1,31 ммоль) в MeCN (5 мл), загружали метил-3-(бромметил)бензоат (300 мг, 1,31 ммоль) и K2CO3 (235 мг, 1,70 ммоль). Полученную желтую суспензию перемешивали при 60°C в течение 16 часов, охлаждали до комнатной температуры, разбавляли H2O и экстрагировали 3 раза Et2O. Органические слои объединяли, промывали рассолом, концентрировали при пониженном давлении и очищали хроматографией на SiO2 (ЭА/гексаны) с получением 451 мг (88,5%) метил-3-((2-бром-4-(трифторметил)фенокси)метил)бензоат (INT 34-А) в виде белого твердого вещества. ЖХ-МС-ESI (m/z) вычислено для C16H12BrF3O3: 389,2; найдено 391,0 (М+Н)+, tR=6,7 мин (Метод 1).
Стадия 34-2. Синтез метил-3-((2-циклопропил-4-(трифторметил)фенокси)метил)бензоата (TNT 34-В)
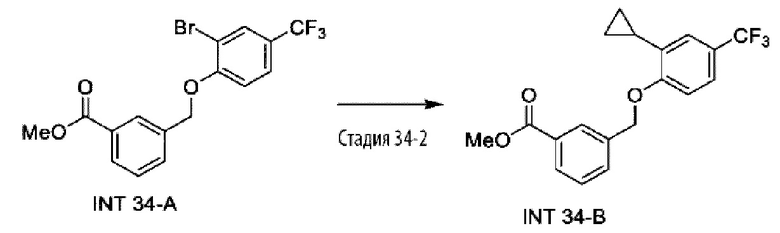
В пробирку высокого давления на 15 мл, содержащую смесь INT 34-А (300 мг, 771 мкмоль) в толуоле (4 мл), загружали фосфат калия (491 мг, 2,31 ммоль), трициклогексилфосфин (32,4 мг, 116 мкмоль), циклопропилбороновую кислоту (132 мг, 1,54 ммоль) и ацетат палладия(II) (17,3 мг, 77,1 мкмоль). Пробирку закрывали, и полученную оранжевую суспензию перемешивали при 100°C в течение 13,5 часов, затем охлаждали до комнатной температуры и распределяли между Et2O и H2O. Водный слой подвергали обратной экстракции Et2O (2Х). Органические слои объединяли, промывали рассолом, осушали (Na2SO4), концентрировали при пониженном давлении и очищали хроматографией на SiO2 (ЭА/гексаны) с получением 201 мг (74%) метил-3-((2-циклопропил-4-(трифторметил)фенокси)метил)бензоата (INT 34-В). ЖХ-MC-ESI (m/z) вычислено для C19H17F3O3: 350,34; найдено 373,2 (M+Na)+, tR=6,8 мин (Метод 1). 1Н ЯМР (500 МГц, ДМСО-d6) δ 8,11 (с, 1Н), 7,93 (д, J=7,7 Гц, 1H), 7,78 (д, J=7,4 Гц, 1Н), 7,58 (т, J=7,7 Гц, 1H), 7,49 (дд, J=8,2, 2,0 Гц, 1Н), 7,21 (д, J=8,6 Гц, 1H), 7,15 (д, J=2,3 Гц, 1H), 5,34 (с, 2Н), 3,86 (с, 3Н), 2,18 (тт, J=8,5, 5,3 Гц, 1Н), 1,01-0,91 (м, 2Н), 0,76-0,69 (м, 2Н).
Стадия 34-3. Синтез 3-((2-циклопропил-4-(трифторметил)фенокси)метил)бензойной кислоты (Соединение 34-1)
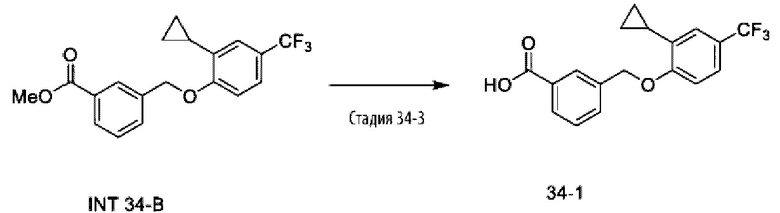
В перемешиваемый раствор INT 34-В (195 мг, 557 мкмоль) в ТГФ (5 мл) добавляли раствор 1М NaOH (2,23 мл, 2,23 ммоль). Раствор перемешивали в течение ночи при 50°C в течение 12,5 часов, концентрировали при пониженном давлении, растворяли в H2O и подкисляли до рН 4-5 с использованием 3М HCl. Полученный белый осадок экстрагировали 3 раза Et2O. Органические слои объединяли, промывали рассолом, осушали (Na2SO4) и концентрировали при пониженном давлении с получением сырого продукта, который очищали методом препаративной ВЭЖХ (CH3CN/H2O, содержащая 0,1% муравьиной кислоты) с получением 66 мг (35%) 3-((2-циклопропил-4-(трифторметил)фенокси)метил)бензойной кислоты (Соединение 34-1) в виде белого твердого вещества. ЖХ-MC-ESI (m/z) вычислено для C18H15F3O3: 336,3; найдено 335,2 [М-Н]+, tR=10,83 мин (Метод 4). 1Н ЯМР (499 МГц, ДМСО-d6) δ 13,03 (с, 1H), 8,09 (с, 1H), 7,91 (д, J=7,8 Гц, 1H), 7,74 (д, J=7,7 Гц, 1H), 7,55 (т, J=7,7 Гц, 1H), 7,50 (дд, J=8,7, 2,3 Гц, 1H), 7,21 (д, J=8,6 Гц, 1H), 7,15 (д, J=2,3 Гц, 1H), 5,33 (с, 2Н), 2,18 (тт, J=8,5, 5,3 Гц, 1Н), 0,99-0,91 (м, 2Н), 0,76-0,69 (м, 2Н). 19F ЯМР (376 МГц, ДМСО-d6) δ -59,98.
ПРИМЕР 35
Синтез соединения 35-1
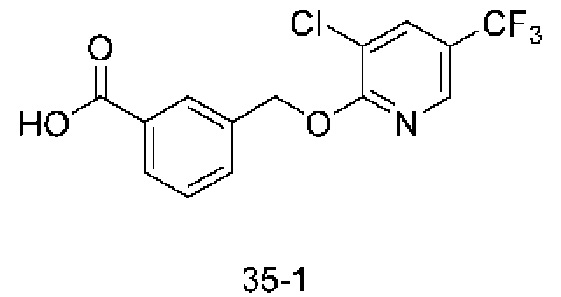
Схема 35
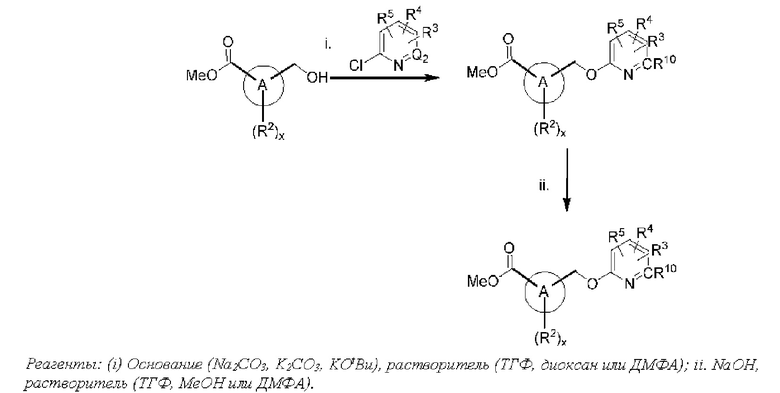
Стадия 35-1. Синтез метил-3-(((3-хлор-5-(трифторметил)пиридин-2-ил)окси)метил)бензоата (TNT35-A)
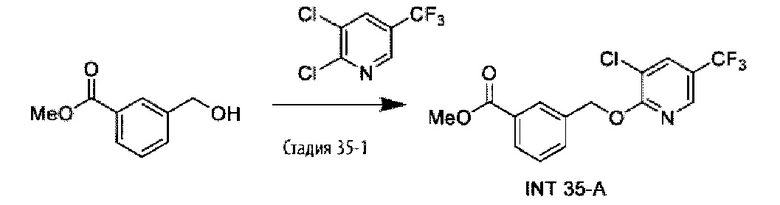
В сосуд высокого давления на 48 мл, содержащий раствор 2,3-дихлор-5-(трифторметил)пиридина (433 мг, 2,01 ммоль) в 1,4-диоксане (9 мл), добавляли метил-3-(гидроксиметил)бензоат (500 мг, 3,01 ммоль) и трет-бутоксид калия (338 мг, 3,01 ммоль). Сосуд герметично закрывали, реакционную смесь нагревали при 90°C в течение 15,5 часов, а затем охлаждали до комнатной температуры. Реакционную смесь распределяли между Et2O и H2O. Фазы разделяли, и водный слой экстрагировали Et2O (2Х). Органические фазы объединяли, промывали рассолом, осушали (Na2SO4), концентрировали и очищали хроматографией на SiO2 (ЭА/гексаны) с получением 198 мг (28,6%) метил-3-(((3-хлор-5-(трифторметил)пиридин-2-ил)окси)метил)бензоата (INT 35-А) в виде белого твердого вещества. ЖХ-MC-ESI (m/z) вычислено для C15H11ClF3NO3: 345,7; найдено 346,1 (М+Н)+, tR=6,6 мин (Метод 1). 1Н ЯМР (500 МГц, ДМСО-d6) δ 8,59 (дд, J=2,2, 1,1 Гц, 1H), 8,44 (д, J=1,9 Гц, 1H), 8,09 (т, J=1,8 Гц, 1H), 7,94 (дт, J=1,1, 1,5 Гц, 1Н), 7,76 (дт, J=1,6, 1,5 Гц, 1Н), 7,57 (т, J=1,1 Гц, 1H), 5,60 (с, 2Н), 3,86 (с, 3Н).
Стадия 35-2. Синтез 3-(((3-хлор-5-(трифторметил)пиридин-2-ил)окси)метил) бензойной кислоты (Соединение 35-1)
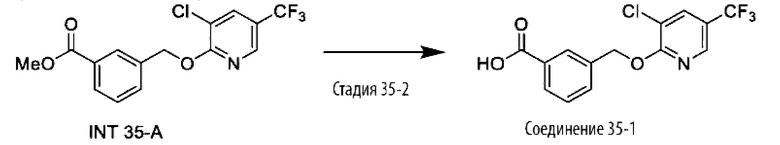
В сосуд на 20 мл, содержащий перемешиваемый раствор INT 35-А (190 мг, 550 мкмоль) в ТГФ (5 мл), загружали 1М NaOH (2,20 мл, 2,20 ммоль). После перемешивания в течение 22,5 часов при 50°C реакционную смесь концентрировали при пониженном давлении, полученный остаток растворяли в H2O и подкисляли до рН 4-5 с использованием 3М HCl. Полученный белый осадок экстрагировали Et2O (3 раза). Органические слои объединяли, промывали рассолом, осушали (Na2SO4) и концентрировали при пониженном давлении с получением 150 мг (82,3%) 3-(((3-хлор-5-(трифторметил)пиридин-2-ил)окси)метил)бензойной кислоты (Соединение 35-1) в виде белого порошка. ЖХ-MC-ESI (m/z) вычислено для C14H9ClF3NO3: 331,7; найдено 333,2 (M+Na)+, tR=10,1 мин (Метод 3). 1Н ЯМР (500 МГц, ДМСО-d6) δ 13,05 (с, 1Н), 8,60 (д, J=1,1 Гц, 1Н), 8,44 (д, J=2,2 Гц, 1Н), 8,06 (с, 1Н), 7,92 (д, J=7,8 Гц, 1H), 7,73 (д, J=7,7 Гц, 1Н), 7,54 (т, J=1,1 Гц, 1H), 5,59 (с, 2Н). 19F ЯМР (376 МГц, ДМСО-d6) δ -60,02.
ПРИМЕР 36
Синтез Соединения 36-1
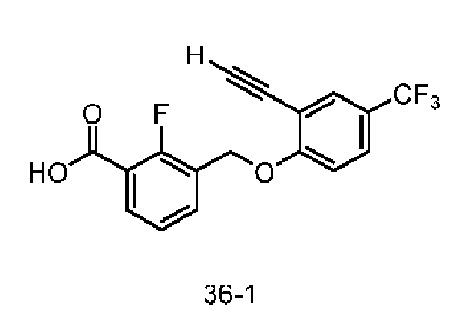
Схема 36
Стадия 36-1. Синтез метил-2-фтор-3-((2-йод-4-(трифторметил)фенокси)метил)бензоата (INT 36-А)
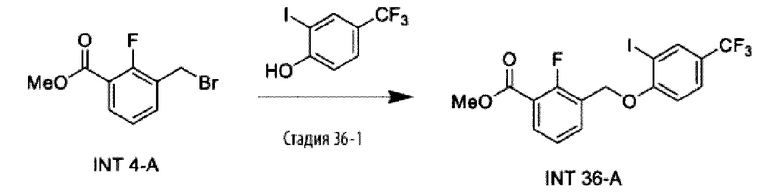
В раствор INT 4-А (300 мг, 1,21 ммоль) и 2-йод-4-(трифторметил)фенола (349,72 мг, 1,21 ммоль) в CH3CN (10 мл) добавляли K2CO3 (218,17 мг, 1,58 ммоль). После перемешивания при 60°C в течение 12 ч реакционную смесь фильтровали и фильтрат концентрировали с получением 500 мг (91%) метил-2-фтор-3-((2-йод-4-(трифторметил)фенокси)метил)бензоата (INT 36-А), который использовали без дополнительной очистки. ЖХ-MC-ESI (m/z) вычислено для C16H11F4IO3: 454,16; найдено 454,9 (М+Н)+, tR=1,04 мин (Метод 6).
Стадия 36-2. Синтез метил-2-фтор-3-((4-(трифторметил)-2-((триметилсилил)зтинил)фенокси)метил)бензоата (TNT 36-В)
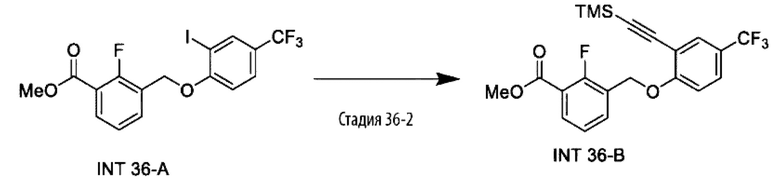
В раствор INT 36-А (500 мг, 1,10 ммоль) в ТГФ (10 мл) добавляли этинил(триметил)силан (167,7 мкл, 1,21 ммоль), дихлорпалладия трифенилфосфин (77,28 мг, 110,09 мкмоль), CuI (20,97 мг, 110,09 мкмоль) и TEA (459,72 мкл, 3,30 ммоль). После перемешивания при 40°C в течение 12 часов смесь выпивали в H2O (20 мл) и экстрагировали ЭА (3×20 мл). Органический слой осушали (Na2SO4), концентрировали и очищали хроматографией на SiO2 (ПЭ, ЭА) с получением 200 мг (42%) метил-2-фтор-3-((4-(трифторметил)-2-((триметилсилил)этинил)фенокси)метил)бензоата (INT 36-В) в виде белого твердого вещества. ТСХ (5:1 ПЭ:ЭА, Rf=0,7) 1Н ЯМР (400 МГц, CDCl3) δ 7,95-7,90 (м, 2Н), 7,74 (с, 1H), 7,57-7,53 (м, 1H), 7,33-7,27 (м, 1H), 7,05-7,01 (м, 1H), 5,29 (с, 2Н), 3,96 (с, 3Н), 0,31-0,27 (м, 9Н).
Стадия 36-3. Синтез 3-((2-зтинил-4-(трифторметил)фенокси)метил)-2-фторбензойнои кислоты (Соединение 36-1).
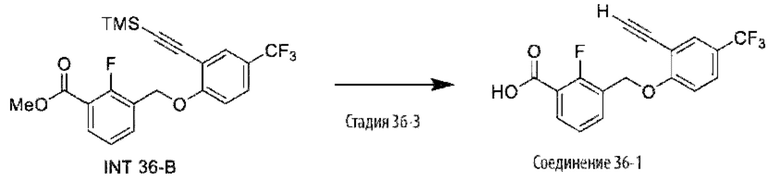
К суспензии INT 36-В (185 мг, 0,44 ммоль) в МеОН (5 мл) и H2O (5 мл) и ТГФ (5 мл) добавляли NaOH (52,3 мг, 1,4 ммоль). После перемешивания при 30°C в течение 1,5 ч реакционную смесь концентрировали, растворяли в МеОН (5 мл), фильтровали и очищали препаративной ВЭЖХ (H2O/CH3CN с муравьиной кислотой) с получением 59 мг (48%) 3-((2-этинил-4-(трифторметил)фенокси)метил)-2-фторбензойной кислоты (Соединение 36-1) в виде белого твердого вещества. ЖХ-MC-ESI (m/z) вычислено для C17H10F4O3: 338,3; найдено 339,1 (M+Na)+, tR=0,786 мин (Метод 6). 1H ЯМР (400 МГц, ДМСО-d6) δ м.д. 4,44 (с, 1Н), 5,38 (с, 2Н), 7,36 (т, J=7,69 Гц, 1H), 7,43 (д, J=8,50 Гц, 1H), 7,75-7,84 (м, 3Н), 7,88 (тд, J=7,38, 1,75 Гц, 1H).
ПРИМЕР 37
Синтез Соединения 37-1
Схема 37
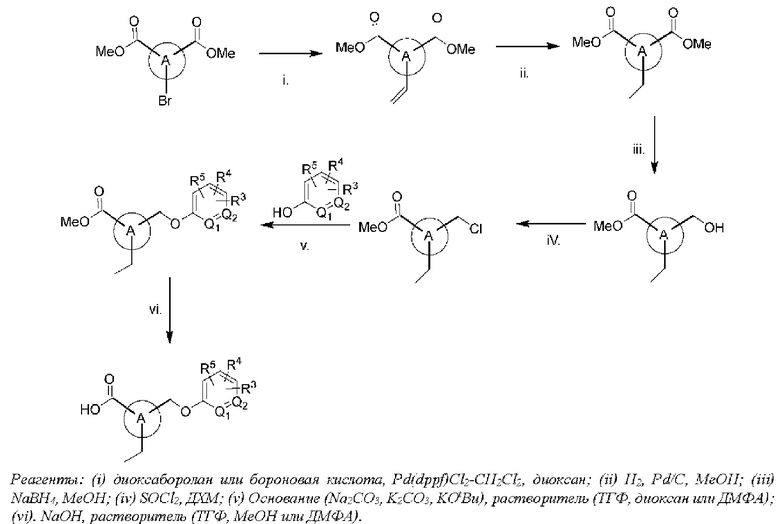
Стадия 37-1. Синтез метил-3-(гидроксиметил)-2-винилбензоата (INT 37-А)

К суспензии диметил-2-бромбензол-1,3-дикарбоксилата (1 г, 3,66 ммоль) и Na2CO3 (776,25 мг, 7,32 ммоль) в 1,4-диоксане (20 мл) и H2O (4 мл) добавляли 4,4,5,5-тетраметил-2-винил-1,3,2-диоксаборолан (683,25 мкл, 4,03 ммоль). Реакционную смесь обрабатывали Pd(dppf)Cl2-CH2Cl2 (149,5 мг, 183,1 мкмоль) и перемешивали при 100°C в течение 12 часов. Смесь фильтровали. Фильтрат распределяли между ЭА (30 мл) и H2O (30 мл). Водный слой подвергали обратной экстракции ЭА (30 мл). Объединенные органические экстракты осушали (Na2SO4), фильтровали и концентрировали в вакууме с получением остатка, который очищали хроматографией на SiO2, получая 680 мг (84,3%) метил-3-(гидроксиметил)-2-винилбензоата (INT 37-А) в виде бесцветной маслянистой жидкости. ТСХ (5:1 ПЭ:ЭА): Rf=0,7.
Стадия 37-2. Синтез метил-2-этил-3-(гидроксиметил)бензоата (TNT 37-В)
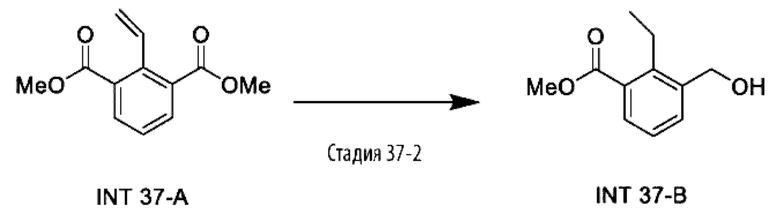
Барботировали H2 (103 кПа (15 psi)) в растворе INT 37-А (680 мг, 3,09 ммоль), Pd/C (70 мг, 308,8 мкмоль, чистота 10%) в МеОН (10 мл) при 30°C в течение 12 час. Реакционную смесь фильтровали и фильтрат концентрировали с получением сырого продукта, который очищали хроматографией на SiO2 (ЭА/ПЭ), получая 560 мг (81,6%) метил-2-этил-3-(гидроксиметил)бензоата (INT 37-В) в виде бесцветной маслянистой жидкости. ТСХ (5:1 ПЭ:ЭА): Rf=0,4. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 1,25 (т, J=7,40 Гц, 3Н), 3,15 (кв, J=7,46 Гц, 2Н), 3,92 (с, 6Н), 7,30 (т, J=7,76 Гц, 1H), 7,85 (д, J=7,70 Гц, 2Н).
Стадия 37-3. Синтез метил-2-этил-3-(гидроксиметил)бензоата (TNT 37-С)
В раствор INT 37-В (0,4 г, 1,80 ммоль) в ТГФ (10 мл) при 0°C добавляли NaBH4 (102,13 мг, 2,70 ммоль) и МеОН (2 мл). После перемешивания в течение 12 ч при 70°C смесь выливали в насыщенный NH4Cl (водн., 20 мл) и экстрагировали ЭА (3×20 мл). Объединенные органические слои осушали (Na2SO4), концентрировали и очищали препаративной ТСХ с получением 170 мг (48,6%) метил-2-этил-3-(гидроксиметил)бензоата (INT 37-С) в виде маслянистой жидкости желтого цвета. ТСХ(5:1 ПЭ:ЭА): Rf=0,5.
Стадия 37-4. Синтез метил-3-(хлорметил)-2-этилбензоата (TNT 37-D)

В раствор INT 37-С (70 мг, 360,4 мкмоль) в ДХМ (2 мл) добавляли SOCl2 (130,7 мкл, 1,80 ммоль) при 0°C. После перемешивания при 30°C в течение 1 ч реакционную смесь концентрировали в вакууме, получая 72 мг (94%) метил-3-(хлорметил)-2-этилбензоата (INT 37-D) в виде коричневой смолы, которую использовали на следующей стадии без дополнительной очистки. ТСХ (5:1 ПЭ:ЭА): Rf=0,7.
Стадия 37-5. Синтез метил-3-((2-хлор-4-(трифторметил)фенокси)метил)-2-этилбензоата (TNT 37-Е)
В суспензию INT 37-D (70 мг, 329,15 мкмоль) и K2CO3 (136,47 мг, 987,44 мкмоль) в CH3CN (2 мл) добавляли 2-хлор-4-(трифторметил)фенол (71,16 мг, 362,06 мкмоль, 1,1 экв.). После перемешивания при 80°C в течение 12 ч реакционную смесь фильтровали и фильтрат концентрировали в вакууме с получением остатка, который очищали хроматографией на SiO2 (ЭА/ПЭ) с получением 59 мг (48%) метил-3-((2-хлор-4-(трифторметил)фенокси)метил)-2-этилбензоата (INT 37-Е) в виде светло-желтой смолы. ТСХ (10:1 ПЭ:ЭА): Rf=0,75. ЖХ-MC-ESI (m/z) вычислено для C18H16ClF3O3: 372,7; найдено 373,4 (М+Н)+, tR=1,14 мин (Метод 6). 1Н ЯМР (400 МГц, CDCl3) δ 7,78 (дд, J=1,3, 7,8 Гц, 1H), 7,66-7,63 (м, 1H), 7,62 (с, 1Н), 7,46 (дд, J=1,6, 8,7 Гц, 1Н), 7,30-7,22 (м, 1H), 7,03 (д, J=8,6 Гц, 1H), 5,22 (с, 2Н), 3,89 (с, 3Н), 2,97 (кв, J=7,5 Гц, 2Н), 1,23 (т, J=7,5 Гц, 3Н).
Стадия 37-6. Синтез 3-((2-хлор-4-(трифторметил)фенокси)метил)-2-этилбензойной кислоты (Соединение 37-1)
В раствор INT 37-Е (156 мг, 418,5 мкмоль) в ТГФ (3 мл), МеОН (1 мл) и Н2О (1 мл) добавляли NaOH (42,92 мг, 1,07 ммоль). После перемешивания при 50°C в течение 12 часов смесь подкисляли 3М соляной кислотой, затем распределяли между ЭА (10 мл) и H2O (10 мл). Органический слой осушали (Na2SO4), фильтровали и концентрировали в вакууме. Полученный остаток очищали препаративной ВЭЖХ (H2O (0,225% муравьиной кислоты (FA))-CH3CN) с получением 144 мг (77%) 3-((2-хлор-4-(трифторметил)фенокси)метил)-2-этилбензойной кислоты (Соединение 37-1). ЖХ-MC-ESI (m/z) вычислено для C17H14ClF3O3: 358,7; найдено 357,0 (М-Н)+, tR=0,95 мин (Метод 8). 1Н ЯМР (400 МГц, CDCl3) δ 8,02 (дд, J=1,1, 7,8 Гц, 1H), 7,74 (д, J=7,0 Гц, 1H), 7,69 (д, J=2,1 Гц, 1H), 7,52 (дд, J=1,7, 8,7 Гц, 1H), 7,36 (т, J=7,8 Гц, 1H), 7,09 (д, J=8,6 Гц, 1Н), 5,28 (с, 2Н), 3,12 (кв, J=7,5 Гц, 2Н), 1,31 (т, J=7,5 Гц, 3Н
ПРИМЕР 38
Синтез Соединения 38-1
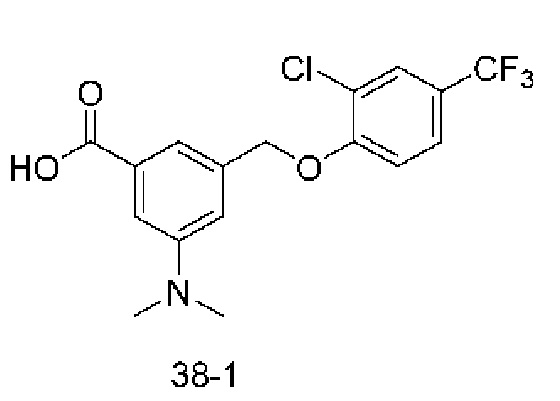
Схема 38
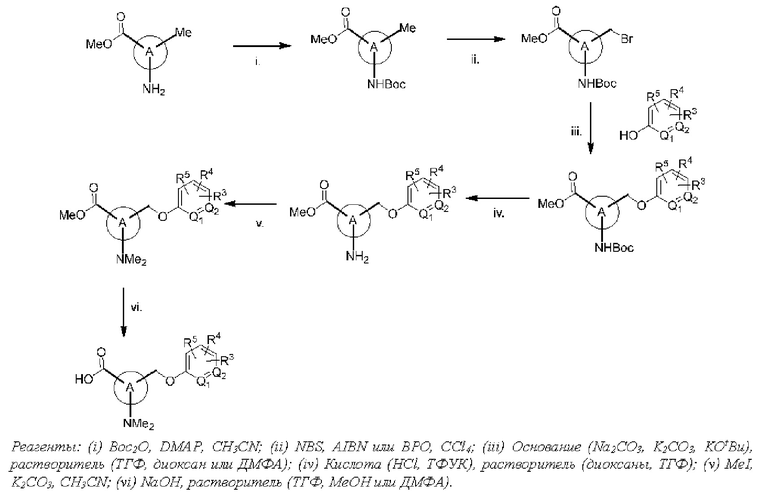
Стадия 38-1. Синтез метил-3-((трет-бутоксикарбонил)амино)-5-метилбензоата (TNT 38-А)
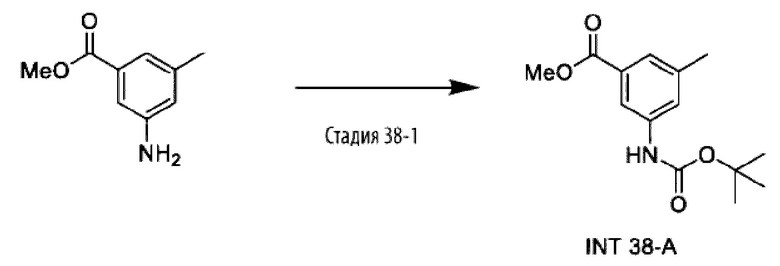
В раствор метил-3-амино-5-метилбензоата (1 г, 6,05 ммоль, 1), ди-трет-бутилдикарбоната (2,64 г, 12,11 ммоль) и TEA (1,69 мл, 12,11 ммоль) в CH3CN (15 мл) добавляли 4-диметиламинопиридин (73,96 мг, 605,37 мкмоль). Реакционную смесь перемешивали при 50°C в течение 12 ч, затем фильтровали. Фильтрат концентрировали и остаток очищали хроматографией на SiO2 (ЭА/ПЭ) с получением 850 мг (53%) метил-3-((трет-бутоксикарбонил)амино)-5-метилбензоата (INT 38-А) в виде желтой смолы. ТСХ (5:1 ПЭ:ЭА): Rf=0,7. 1Н ЯМР (400 МГц, CDCl3) δ м.д. 1,46 (с, 9Н), 2,40 (с, 3Н), 3,91 (с, 3Н), 7,28 (с, 1H), 7,75 (с, 1H), 7,82 (с, 1 Н).
Стадия 38-2. Синтез метил-3-(бромметил)-5-((трет-бутоксикарбонил)амино)бензоата (TNT 38-В)
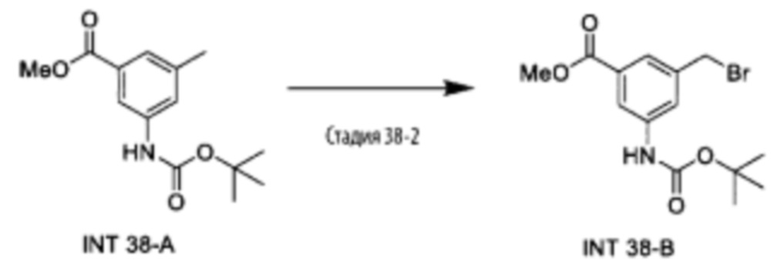
В раствор INT 38-А (750 мг, 2,83 ммоль) и NBS (604 мг, 3,39 ммоль) в CCl4 (10 мл) добавляли AIBN (46 мг, 282 мкмоль). После перемешивания при 80°C в течение 12 ч реакционную смесь фильтровали и очищали хроматографией на SiO2 (ЭА/ПЭ), получая 800 мг (82%) метил-3-(бромметил)-5-((трет-бутоксикарбонил)амино)бензоата (INT 38-В) в виде коричневой смолы. ТСХ (10:1 ПЭ:ЭА): Rf=0,45.
Стадия 38-3. Синтез метил-3-((трет-бутоксикарбонил)амино)-5-((2-хлор-4-(трифторметил)фенокси)метил)бензоата (TNT 38-С)
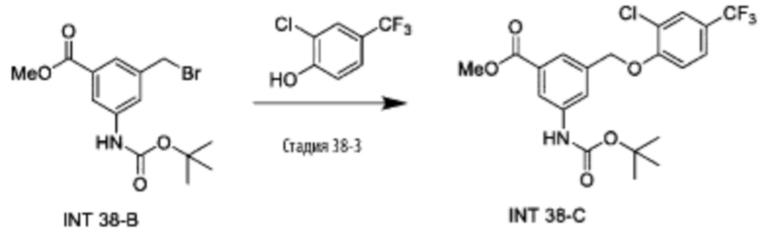
В суспензию INT 38-В (560 мг, 1,63 ммоль) и K2CO3 (674,57 мг, 4,88 ммоль) в CH3CN (10 мл) добавляли 2-хлор-4-(трифторметил)фенол (351,76 мг, 1,79 ммоль). После перемешивания при 80°C в течение 12 ч реакционную смесь фильтровали, концентрировали и очищали хроматографией на SiO2, получая 140 мг (18%) метил-3-((трет-бутоксикарбонил)амино)-5-((2-хлор-4-(трифторметил)фенокси)метил)бензоата (INT 38-С) в виде светло-желтой смолы. ТСХ (10:1 ПЭ:ЭА): Rf=0,60. ЖХ-MC-ESI (m/z) вычислено для C21H21ClF3NO5: 459,85; найдено 458 (М-Н)+, tR=1,12 мин (Метод 6). 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,72 (с, 1H), 8,15 (с, 1Н), 7,87 (д, J=1,9 Гц, 1H), 7,78 (с, 1H), 7,72-7,68 (м, 2Н), 7,39 (д, J=8,9 Гц, 1H), 5,35 (с, 2Н), 3,85 (с, 3Н), 1,48 (с, 9Н).
Стадия 38-4. Синтез метил-3-амино-5-((2-хлор-4-(трифторметил)фенокси)метил)бензоата (INT 38-Р)
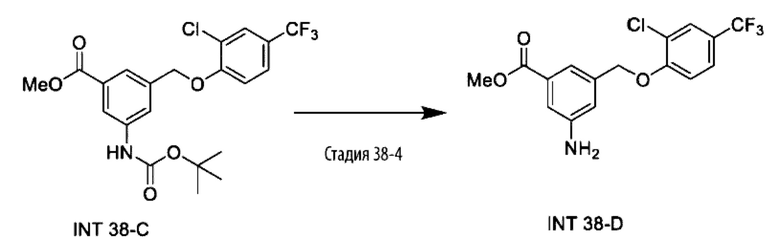
Раствор INT 38-С (60 мг, 130,48 мкмоль) в HCl/диоксане (4 М, 1 мл) перемешивали при 30°C в течение 1 часа. Реакционную смесь концентрировали в вакууме, получая 55 мг сырого метил-3-амино-5-((2-хлор-4-(трифторметил)фенокси)метил)бензоата (INT 38-D) в виде серого твердого вещества, которое использовали на следующей стадии без дополнительной очистки. ТСХ (10:1 ПЭ:ЭА): Rf=0,65.
Стадия 38-5. Синтез метил-3-((2-хлор-4-(трифторметил)фенокси)метил)-5-(диметиламино)бензоата (TNT 38-Е)
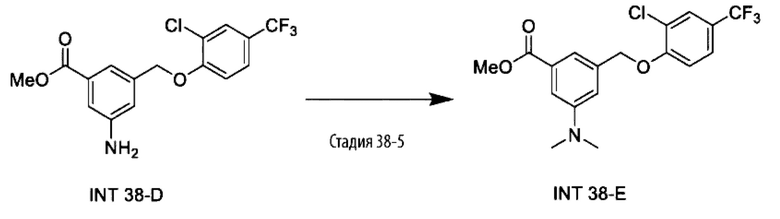
В суспензию INT 38-D (50 мг, 138,99 мкмоль) и K2CO3 (38,42 мг, 278 мкмоль) в MeCN (3 мл) добавляли Mel (17,31 мкл, 277,99 мкмоль). Реакционную смесь перемешивали при 30°C в течение 12 ч, фильтровали и очищали хроматографией на SiO2, получая 12 мг (22%) метил-3-((2-хлор-4-(трифторметил)фенокси)метил)-5-.(диметиламино)бензоата (INT 38-Е) в виде светло-желтой смолы. ТСХ (5:1 ПЭ:ЭА): Rf=0,8. ЖХ-MC-ESI (m/z) вычислено для C18H17ClF3NO3: 387,8; найдено 388 (М-Н)+, tR=0,99 мин (Метод 6). 1Н ЯМР (400 МГц, CDCl3) 5 м.д. 7,66 (д, J=1,88 Гц, 1H), 7,46 (дд, J=8,63, 1,63 Гц, 1H), 7,42 (с, 1H), 7,36 (с, 1H)) 7,00-7,06 (м, 2Н), 5,21 (с, 2Н), 3,92 (с, 3Н), 3,02 (с, 6Н).
Стадия 38-6. Синтез 3-((2-хлор-4-(трифторметил)фенокси)метил)-5-(диметиламино)бензойной кислоты (Соединение 38-1)
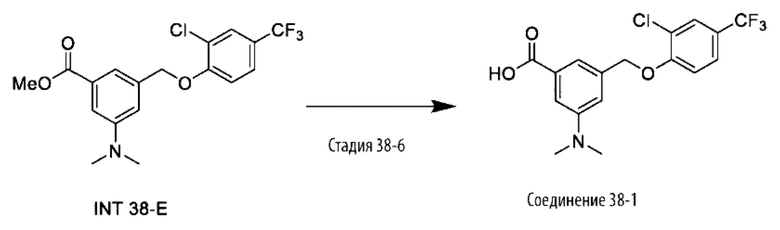
К раствору INT 38-Е (12 мг, 30,95 мкмоль) в ТГФ (1 мл), МеОН (0,5 мл) и H2O (0,5 мл), добавляли NaOH (4,95 мг, 123,78 мкмоль). После перемешивания при 50°C в течение 4 ч смесь подкисляли 3М соляной кислотой. Смесь распределяли между ЭА (10 мл) и H2O (10 мл), и полученный органический слой осушали (Na2SO4), фильтровали и очищали препаративной ВЭЖХ (H2O (0,225% муравьиной кислоты (FA))-CH3CN) с получением 3,6 мг (31%) 3-((2-хлор-4-(трифторметил)фенокси)метил)-5-(диметиламино)бензойной кислоты (Соединение 38-1) в виде белого твердого вещества. ЖХ-MC-ESI (m/z) вычислено для C17H15ClF3NO3: 373,76; найдено 374,1 (М+Н)+, tR=0,93 мин (Метод 6). 1Н ЯМР (400 МГц, CDCl3) δ 7,67 (д, J=1,8 Гц, 1H), 7,50-7,44 (м, 2Н), 7,41 (с, 1H), 7,09-7,02 (м, 2Н), 5,23 (с, 2Н), 3,03 (с, 6Н).
ПРИМЕР 39
Синтез Соединения 39-1
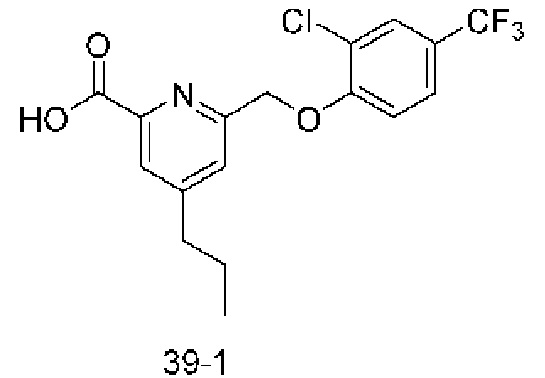
Схема 39
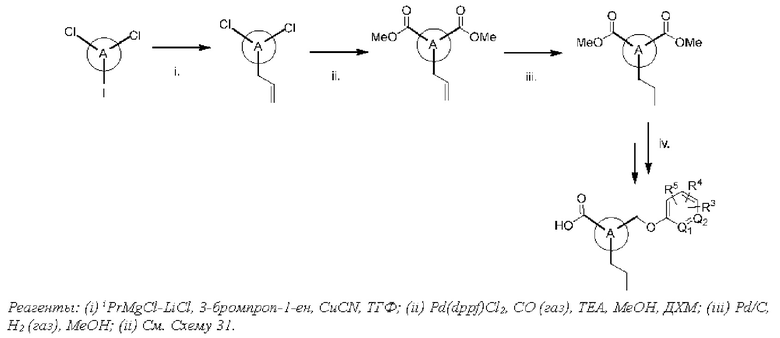
Стадия 39-1. Синтез 4-аллил-2,6-дихлорпиридина (TNT 39-А)
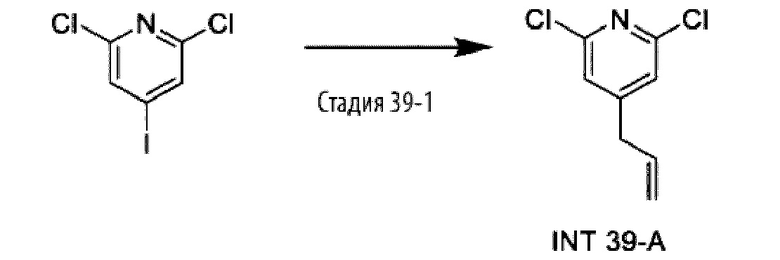
В смесь iPrMgCl-LiCl (1 М, 12,78 мл) в ТГФ (100 мл) при -60°C добавляли 2,6-дихлор-4-йодпиридин (2,8 г, 10,22 ммоль). Смесь перемешивали при -60°C в течение 0,5 часа, затем добавляли 3-бромпроп-1-ен (1,55 г, 12,78 ммоль) и CuCN (1,14 г, 12,78 ммоль) и смесь перемешивали в течение 16 часов при 25°C. Реакционную смесь гасили добавлением H2O (100 мл) и экстрагировали ЭА (3×50 мл). Объединенные органические слои осушали и концентрировали с получением остатка, который очищали с помощью препаративной ТСХ (ПЭ), получая 1,4 г (73%) 4-аллил-2,6-дихлорпиридина (INT 39-А) в виде желтой маслянистой жидкости. ТСХ (ПЭ): Rf=0,50.
Стадия 39-2. Синтез диметил-4-аллилпиридин-2,6-дикарбоксилата (TNT 39-В)
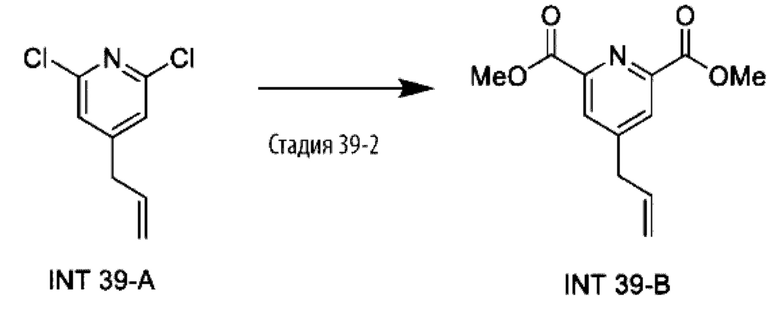
В растворе INT 39-А (1,4 г, 7,44 ммоль) Pd(dppf)Cl2-CH2Cl2 (3,04 г, 3,72 ммоль) и TEA (6,22 мл, 44,67 ммоль) в МеОН (10 мл) барботировали газообразный СО (20,85 г, 744,47 ммоль). Смесь перемешивали при 70°C в течение 12 ч, затем фильтровали. Полученный остаток очищали хроматографией на SiO2 (ЭА/ПЭ), получая 1,75 г (60%) диметил-4-аллилпиридин-2,6-дикарбоксилата (INT 39-В) в виде черного твердого вещества. ТСХ (1:1 ЭА:ПЭ): Rf=0,50.
Стадия 39-3. Синтез диметил-4-пропилпиридин-2,6-дикарбоксилата (INT 39-С)
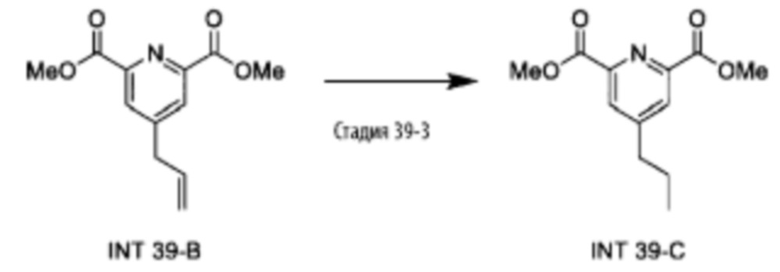
Раствор INT 39-В (2,9 г, 12,33 ммоль) и Pd/C (0,3 г, 1,23 ммоль, чистота 10%) в МеОН (80 мл) перемешивали при 25°C в течение 12 часов в атмосфере H2 (345 кПа (50 psi)). Реакционную смесь фильтровали и концентрировали, получая остаток, который очищали хроматографией на SiO2 (ЭА), получая 2,5 г (85%) диметил-4-пропилпиридин-2,6-дикарбоксилата (INT 39-С) в виде желтого твердого вещества. ТСХ (1:1 ЭА:ПЭ): Rf=0,80. 1Н ЯМР (400 МГц, CDCl3) 5 м.д. 8,14 (с, 2Н), 4,02 (с, 6Н), 2,76 (т, J=7,64 Гц, 2Н), 1,75 (секстет, J=7,46 Гц, 2Н), 0,98 (т, J=7,34 Гц, 3Н).
Стадия 39-4. Синтез 6-((2-хлор-4-(трифторметил)фенокси)метил)-4-пропилпиколиновой кислоты (Соединение 39-1)
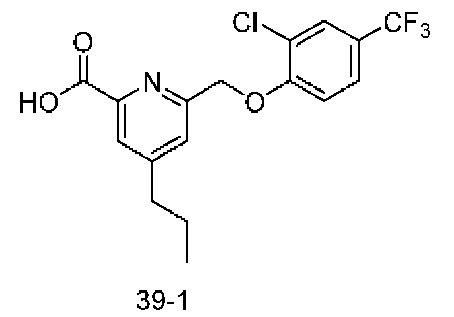
Синтез Соединения 39-1 был завершен из INT 39-С, как показано на Схеме 31. ЖХ-MC-ESI (m/z) вычислено для C17H15ClF3NO3: 373,76; найдено 373,8 (М+Н)+, tR=0,786 мин (Метод 6). 1Н ЯМР (400 МГц, ДМСО-d6) δ 7,85-7,91 (м, 2Н), 7,71 (дд, J=8,76, 1,63 Гц, 1H), 7,61 (с, 1Н), 7,45 (д, J=8,63 Гц, 1Н), 5,42 (с, 2Н), 2,70 (т, J=7,50 Гц, 2Н), 1,62 (м, J=7,40 Гц, 2Н), 0,87 (т, J=7,32 Гц, 3Н).
ПРИМЕР 40
Синтез Соединения 40-1
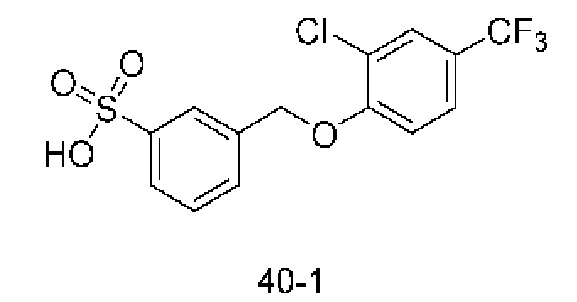
Схема 40
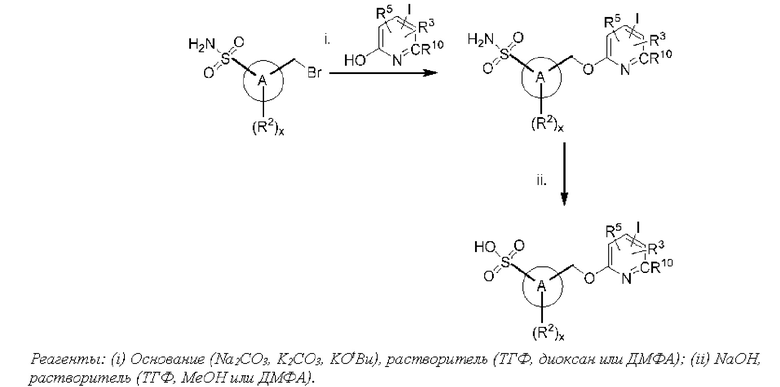
Стадия 40-1. Синтез 3-((2-хлор-4-(трифторметил)фенокси)метил)бензолсульфонамида (INT 40-А)
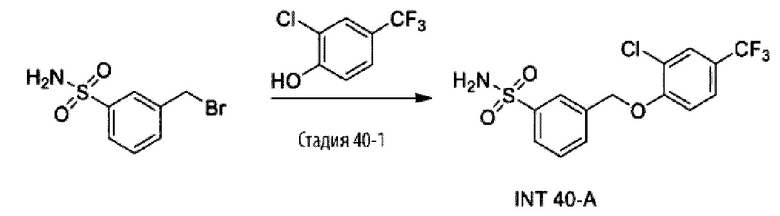
К раствору 3-(бромметил)бензолсульфонамида (100 мг, 400 мкмоль) в CH3CN (3 мл) добавляли 2-хлор-4-(трифторметил)фенол (78,6 мг, 400 мкмоль) и K2CO3 (111 мг, 800 мкмоль). После перемешивания при 30°C в течение 12 ч смесь концентрировали с получением сырого продукта, который очищали с помощью препаративной ВЭЖХ (H2O (0,225% муравьиной кислоты (FA))/CH3CN), получая 70 мг (48%) 3-((2-хлор-4-(трифторметил)фенокси)метил)бензолсульфонамида (INT 40-А) в виде белого твердого вещества. ЖХ-MC-ESI (m/z), вычислено для C14H11ClF3NO3S: 365,8; найдено 364,0 (М-Н)+, tR=0,967 мин (Метод 7).
Стадия 40-2. Синтез 3-((2-хлор-4-(трифторметил)фенокси)метил)бензоилсульфоновой кислоты (Соединение 40-1)

К раствору INT 40-А (30 мг, 82 мкмоль) в ТГФ (3 мл) добавляли HCl (2 М, 41,01 мкл) и NaNO2 (9,6 мг, 139 мкмоль). После перемешивания при 40°C в течение 12 часов смесь концентрировали с получением сырого продукта, который очищали препаративной ВЭЖХ (H2O (0,1% ТФУК)/CH3CN) с получением 2,6 мг (8,5%) 3-((2-хлор-4-(трифторметил)фенокси)метил)бензолсульфоновой кислоты (Соединение 40-1) в виде коричневой смолы. ЖХ-MC-ESI (m/z) вычислено для C14H10ClF3O4S: 366,7; найдено 365,0 (М-Н)+, tR=0,708 мин (Метод 7).
ПРИМЕР 41
Синтез Соединения 41-1
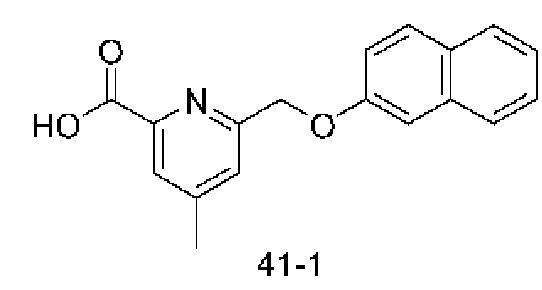
Схема 41
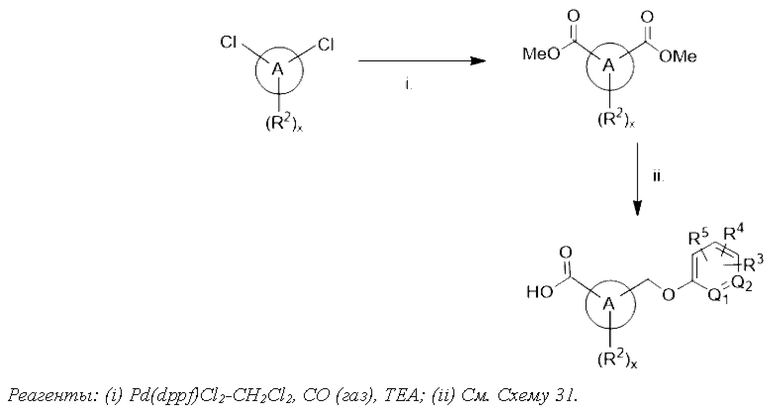
Стадия 41-1. Синтез диметил-4-метилпиридин-2,6-дикарбоксилата (TNT 41-А)
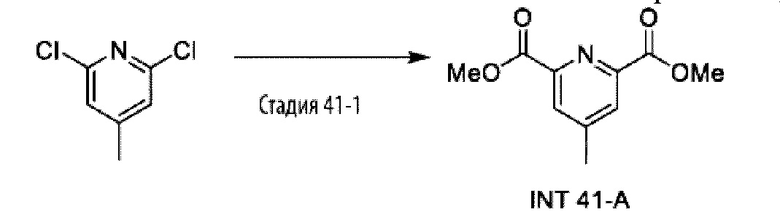
К раствору 2,6-дихлор-4-метилпиридина (1 г, 6,17 ммоль) в ДМФА (20 мл) и МеОН (10 мл) добавляли Pd(dppf)Cl2-CH2Cl2 (504,05 мг, 617,22 мкмоль) и TEA (3,44 мл, 24,69 ммоль). После перемешивания при 80°C в атмосфере СО (345 кПа (50 PSI)) в течение 16 часов реакционную смесь фильтровали, концентрировали, разбавляли H2O и экстрагировали ЭА (2×50 мл). Объединенные органические слои осушали (Na2SO4), фильтровали, концентрировали и очищали хроматографией на SiO2 (ЭА/ПЭ) с получением 900 мг (68%) диметил-4-метилпиридин-2,6-дикарбоксилата (INT 41-А) в виде желтого твердого вещества. ТСХ (2:1 ЭА:ПЭ): Rf=0,20. ЖХ-MC-ESI (m/z) вычислено для C10H11NO4: 209,2; найдено 210,6 (M+H)+, tR=0,756 мин (Метод 7). 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,12 (д, J=0,7 Гц, 2Н), 3,91 (с, 6Н), 2,49-2,48 (с, 3Н).
Стадия 41-2. Синтез 4-метил-6-((нафталин-2-илокси)метил)пиколиновой кислоты (Соединение 41-1)
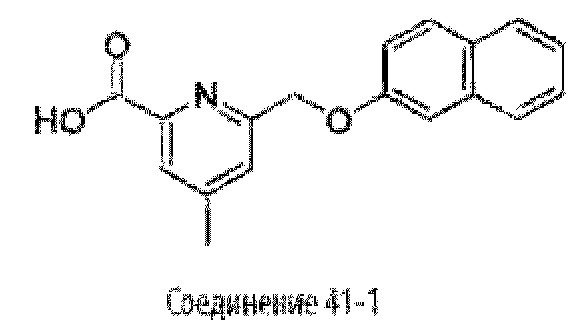
4-Метил-6-((нафталин-2-илокси)метил)пиколиновую кислоту (Соединение 41-1) синтезировали согласно Схеме 31 (стадии i-iv) из INT 41-А и нафталин-2-ола, получая ее в виде белого твердого вещества. ЖХ-MC-ESI (m/z) вычислено для C18H15NO3: 293,3; найдено 294,2 (М+Н)+, tR=0,789 мин (Метод 7). 1Н ЯМР (400 МГц, ДМСО-d6) δ 12,93-13,50 (м, 1Н), 7,77-7,90 (м, 4Н), 7,64 (с, 1Н), 7,42-7,50 (м, 2Н), 7,33-7,40 (м, 1H), 7,30 (дд, J=8,94, 2,44 Гц, 1H), 5,22-5,41 (м, 2Н), 2,38-2,45 (м, 3Н).
Соединения, перечисленные в Таблице 41, были получены с использованием процедур Схемы 41.
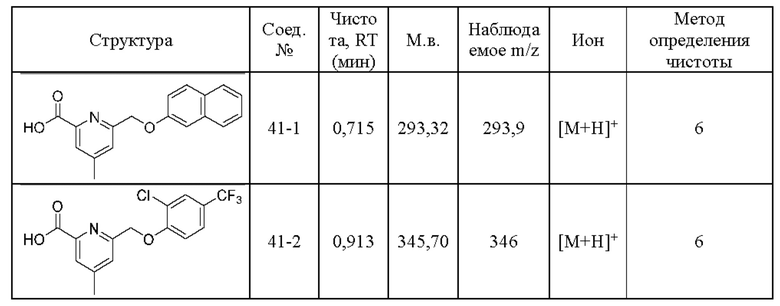
ПРИМЕР 42
Синтез Соединения 42-1
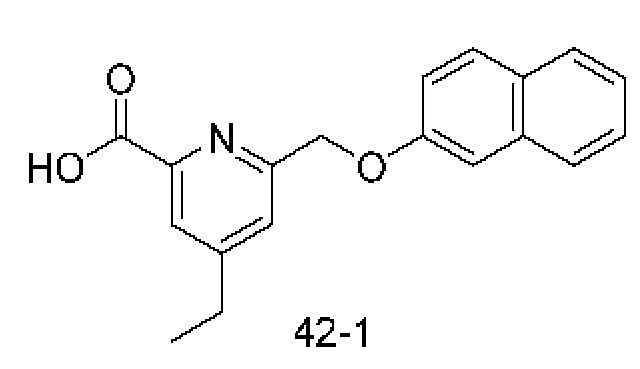
Схема 42
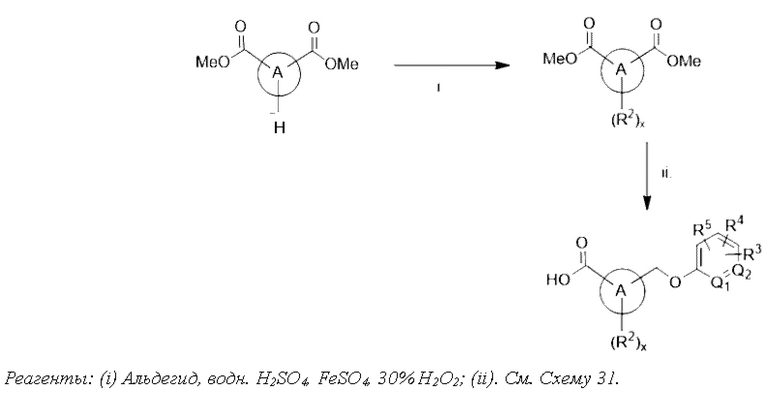
Стадия 42-1. Синтез диметил-4-этилпиридин-2,6-дикарбоксилата (INT 42-А)

К раствору диметилпиридин-2,6-дикарбоксилата (10 г, 51,2 ммоль) и пропаналя (18,65 мл, 256,2 ммоль) в H2SO4 (100 мл) добавляли FeSO4 (5,70 г, 20,49 ммоль) и 30% H2O2 (9,85 мл, 102,47 ммоль) по каплям в течение 15 мин. После перемешивания при 0°С в течение 15 минут смесь разбавляли насыщенным К2СО3 (водн.) и экстрагировали ЭА (3 × 200 мл). Объединенные органические слои осушали (Na2SO4), концентрировали, очищали хроматографией на SiO2 (ЭА/ПЭ) с получением 4,5 г (39%) диметил-4-этилпиридин-2,6-дикарбоксилата (INT 42-А) в виде желтого твердого вещества. ТСХ (3:1 ЭА:ПЭ): Rf=0,60. 1Н ЯМР (400 МГц, CDCl3) δ 8,13-8,20 (м, 2Н), 4,00-4,03 (м, 6Н), 2,78-2,87 (м, 2Н), 1,29-1,37 (м, 3Н).
Стадия 42-2. Синтез 4-этил-6-((нафталин-2-илокси)метил)пиколиновой кислоты (Соединение 42-1)
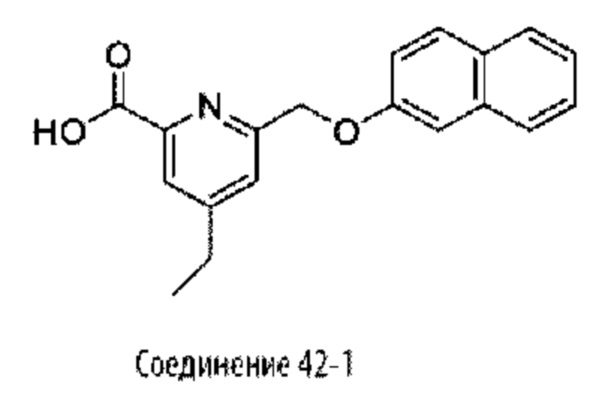
4-Этил-6-((нафталин-2-илокси)метил)пиколиновая кислота (соединение 42-1) была синтезирована согласно Схеме 31 (стадии i-iv) из INT 42-А и нафталин-2-ола, и была получена в виде светло-желтого твердого вещества. ЖХ-MC-ESI (m/z) вычислено для C19H17NO3: 307,4; найдено 307,9 (М+Н)+, tR=0,743 мин (Метод 6). 1Н ЯМР (400 МГц, ДМСО-d6) δ 12,82-13,61 (м, 1H), 7,78-7,91 (м, 4Н), 7,68 (с, 1H), 7,43-7,50 (м, 2Н), 7,33-7,39 (м, 1H), 7,31 (дд, J=8,88, 2,50 Гц, 1Н), 5,26-5,41 (м, 2Н), 2,74 (кв, J=7,63 Гц, 2Н), 1,13-1,30 (м, 3Н).
Соединения, перечисленные в Таблице 42, были получены с использованием процедур Схемы 42.
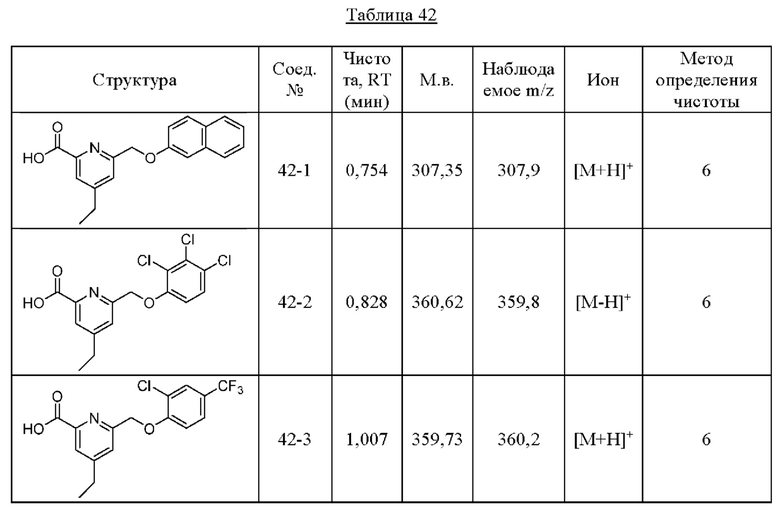
ПРИМЕР 43.
Синтез Соединения 43-1
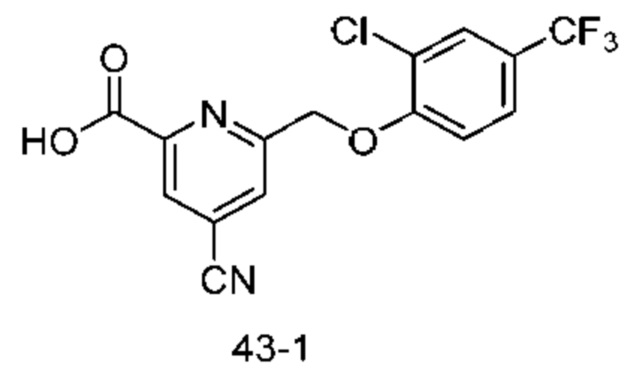
Схема 43
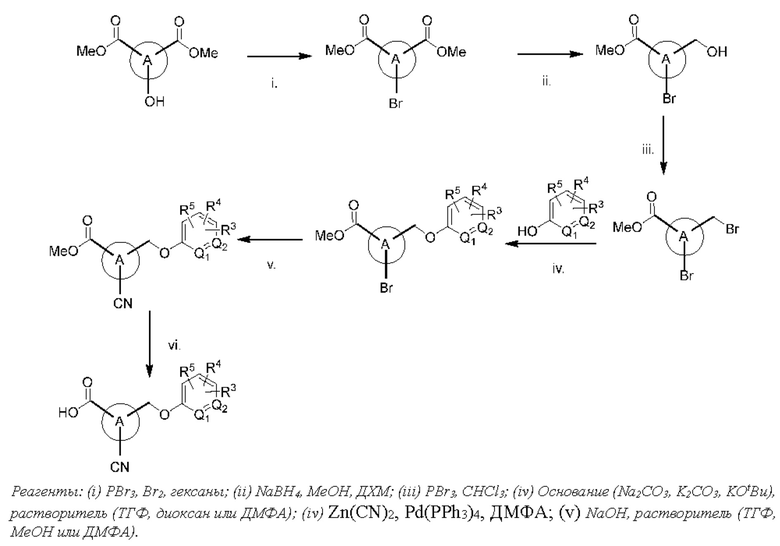
Стадия 43-1. Синтез диметил-4-бромпиридин-2,6-дикарбоксилата (TNT 43-А)
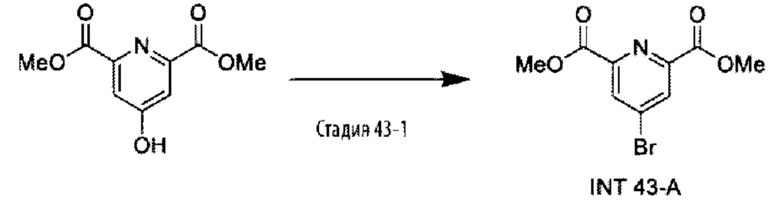
Добавляли PBr3 (1,61 мл, 16,9 ммоль) к раствору Br2 (700 мкл, 13,7 ммоль) в гексанах (10,0 мл) при 0°С. Смесь перемешивали при 22°С в течение 1 часа. Добавляли 4-гидроксипиридин-2,6-дикарбоновую кислоту (1,00 г, 5,46 ммоль) и смесь перемешивали при 90°С в течение 6 часов. Смесь охлаждали и разбавляли CHCl3 (50 мл). Безводный МеОН (50 мл) добавляли по каплям при 0°С и смесь перемешивали при 22°С в течение 1 часа. Смесь концентрировали и остаток растворяли в ДХМ (50 мл) и разбавляли насыщ. водн. NaHCO3 (50 мл). Водную фазу экстрагировали ДХМ (4 × 50 мл), и объединенные органические слои осушали (Na2SO4), фильтровали и концентрировали при пониженном давлении. Остаток очищали хроматографией на SiO2 (ЭА/гексаны), получая 924 мг (62%) диметил-4-бромпиридин-2,6-дикарбоксилата (INT 43-А) в виде твердого вещества. ЖХ-MC-ESI (m/z) вычислено для C9H8BrNO4: 274,07; найдено 276,1 (М+Н)+, tR=2,O4 мин (Метод 13).
Стадия 43-2. Синтез метил-4-бром-6-(гадроксиметил)пиридин-2-карбоксилата (TNT 43-В)
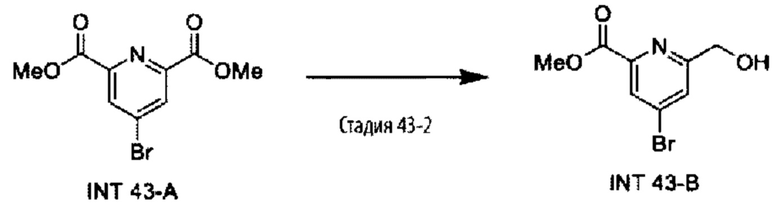
Добавляли NaBH4 (191 мг, 5,06 ммоль) к раствору INT 43-А (924 мг, 3,37 ммоль) в МеОН и ДХМ (4:1 об./об., 50 мл) при 0°С. Смесь перемешивали при 0°С в течение 30 мин. Добавляли насыщенный водн. NaHCO3 (50 мл) и водную фазу экстрагировали ДХМ (4 × 30 мл). Объединенные органические слои осушали (Na2SO4), фильтровали и концентрировали с получением 690 мг (83%) метил-4-бром-6-(гидроксиметил)пиридин-2-карбоксилата (INT 43-В) в виде твердого вещества. ЖХ-MC-ESI (m/z) вычислено для C8H8BrNO3: 246,06; найдено 246,1 (М+Н)+, tR=1,78 мин (Метод 13).
Стадия 43-3. Синтез метил-4-бром-6-(бромметил)пиридин-2-карбоксилата (TNT 43-С)
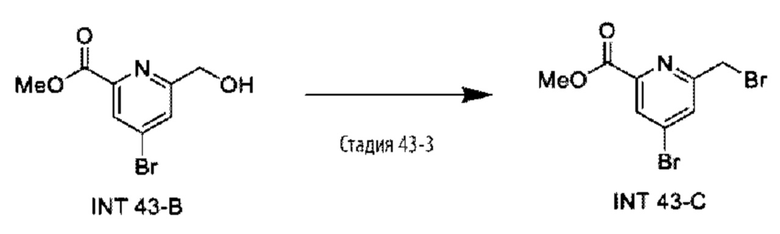
Добавляли PBr3 (450 мкл, 4,74 ммоль) к раствору INT 43-В (690 мг, 2,80 ммоль) в CHCl3 (35,0 мл) при 0°С. Смесь перемешивали при 22°С в течение 5 ч, охлаждали до 0°С и разбавляли насыщ. водн. К2СО3 (25,0 мл). Водную фазу экстрагировали ЭА (3 × 30 мл), и объединенные органические слои промывали рассолом (20 мл), осушали (Na2SO4), фильтровали и концентрировали. Остаток очищали хроматографией на SiO2 (ЭА/гексаны) с получением 700 мг (81%) диметил-4-бромпиридин-2,6-дикарбоксилата (INT 43-С) в виде твердого вещества. ЖХ-MC-ESI (m/z) вычислено для C8H7Br2NO2: 308,96; найдено 246,1 (М+Н)+, tR=2,22 мин (Метод 13).
Стадия 43-4. Синтез метил-4-бром-6-((2-хлор-4-(трифторметил)фенокси)метил)пиридин-2-карбоксилата (INT 43-D)
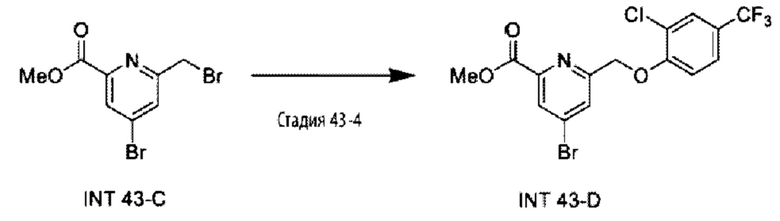
2-Хлор-4-(трифторметил)фенол (490 мг, 2,49 ммоль) и Cs2CO3 (1,48 г, 4,53 ммоль) добавляли к раствору INT 43-С (700 мг, 2,27 ммоль) в безводном ДМФА (5 мл) при 22°С. Смесь перемешивали при 50°С в течение 18 ч, охлаждали до комнатной температуры и разбавляли Н2О (25 мл). Водную фазу экстрагировали ЭА (3 × 30 мл), и объединенные органические слои промывали рассолом (20 мл), осушали (Na2SO4), фильтровали и концентрировали. Остаток очищали хроматографией на SiO2 (ЭА/гексаны), получая 750 мг (78%) метил-4-бром-6-((2-хлор-4-(трифторметил)фенокси)метил)пиридин-2-карбоксилата (INT 43-D) в виде твердого вещества. ЖХ-MC-ESI (m/z) вычислено для C15H10BrClF3NO3: 422,95; найдено 424,4 (М+Н)+, tR=2,78 мин (Метод 13).
Стадия 43-5. Синтез метил-4-бром-6-((2-хлор-4-(трифторметил)фенокси)метил)пиридин-2-карбоксилата (INT 43-Е)
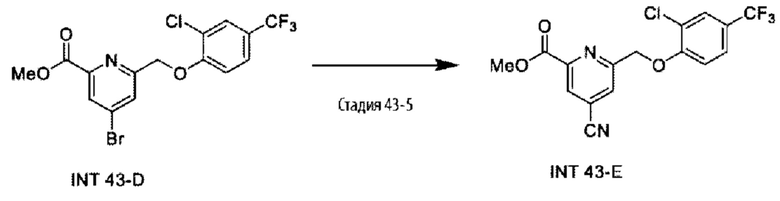
Добавляли Zn(CN)2 (59,7 мг, 0,509 ммоль) и Pd(PPh3)4 (44,1 мг, 0,038 ммоль) к раствору INT 43-D (108 мг, 0,254 ммоль) в дегазированном ДМФА (2,00 мл) при 22°С. Смесь продували N2 в течение 5 минут и перемешивали при 150°С в течение 6 часов. Смесь концентрировали, и остаток очищали хроматографией на SiO2 (ЭА/гексаны), получая 74 мг (79%) метил-4-бром-6-((2-хлор-4-(трифторметил)фенокси)метил)пиридин-2-карбоксилата (INT 43-Е) в виде твердого вещества. ЖХ-MC-ESI (m/z) вычислено для C16H10ClF3N2O3: 370,03; m/z не наблюдается, tR=2,78 мин (Метод 13).
Стадия 43-6. Синтез 6-((2-хлор-4-(трифторметил)фенокси)метил)-4-цианопиридин-2-карбоновой кислоты (Соединение 43-1)
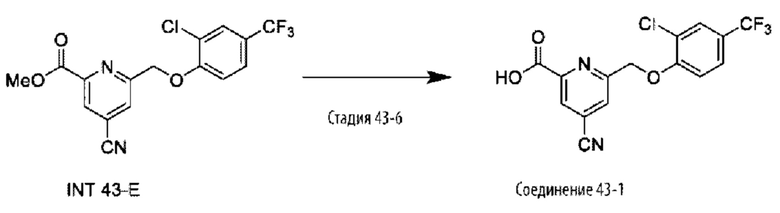
Водный 2М NaOH (299 мкл, 0,150 ммоль) добавляли к раствору INT 43-Е (74,0 мг, 0,20 ммоль) в МеОН (1 мл) и ТГФ (1 мл) при 22°С. Смесь перемешивали при 22°С в течение 2 ч и концентрировали. Остаток подкисляли водн. 2 М HCl (рН ~2) и разбавляли H2O (10 мл). Водную фазу экстрагировали ЭА (3 × 10 мл), и объединенные органические слои промывали рассолом (10 мл), осушали (Na2SO4), фильтровали и концентрировали. Остаток очищали обращенно-фазовой хроматографией (H2O (+0,1% муравьиной кислоты)/CH3CN) с получением 63 мг (89%) 6-((2-хлор-4-(трифгорметил)фенокси)метил)-4-цианопиридин-2-карбоновой кислоты (Соединение 43-1) в виде твердого вещества. ЖХ-MC-ESI (m/z) вычислено для C15H8ClF3N2O3: 356,7; найдено 357,1 (М+Н)+, tR=3,91 мин (Метод 12). 1Н ЯМР (500 МГц, ДМСО-d6) δ 13,84 (шир.с, 1Н), 8,39 (д, J=1,4 Гц, 1Н), 8,17 (д, J=1,4 Гц, 1Н), 7,92 (д, J=2,2 Гц, 1H), 7,73 (дд, J=2,3, 8,7 Гц, 1H), 7,48 (д, J=8,7 Гц, 1H), 5,51 (с, 2Н).
ПРИМЕР 44
Синтез Соединения 44-1
Схема 44
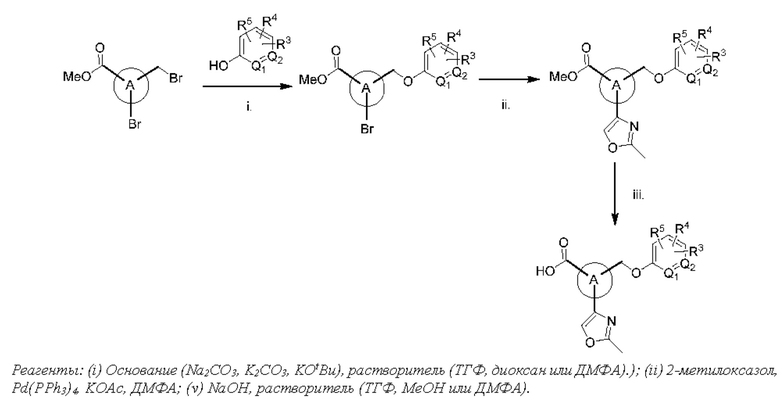
Стадия 44-1. Синтез метил-3-бром-5-((2-хлор-4-(трифторметил)фенокси)метил)бензоата (TNT 44-А)
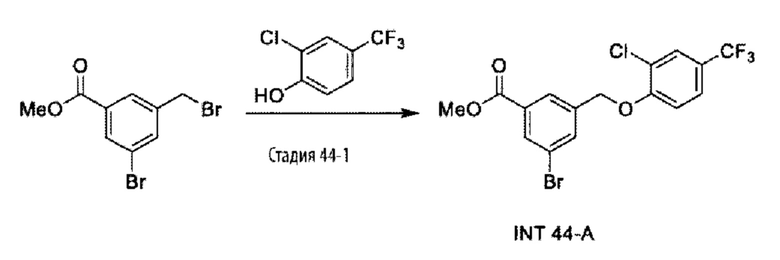
Смесь метил-3-бром-5-(бромметил)бензоата (3,60 г, 11,7 ммоль), 2-хлор-4-(трифторметил)фенола (1,48 мл, 11,1 ммоль) и К2СО3 (4,85 г, 35,1 ммоль) в ацетоне (30 мл) перемешивали при 90°С в течение 1 часа. Смесь фильтровали, и фильтрат концентрировали. Остаток очищали хроматографией на SiO2 (ЭА/гексаны) с получением 2,36 г (48%) метил-3-бром-5-((2-хлор-4-(трифторметил)фенокси)метил)бензоата (INT 44-А) в виде твердого вещества. ЖХ-MC-ESI (m/z) вычислено для C16H11BrClF3O3: 423,61; найдено 442,2 (М+H2O)+, tR=3,29 мин (Метод 13). 1Н ЯМР (400 МГц, CDCl3) δ 8,18-8,13 (м, 1Н), 8,05 (тт, J=1,5, 0,7 Гц, 1H), 7,84 (тд, J=1,7, 0,8 Гц, 1H), 7,68 (дт, J=2,3, 0,7 Гц, 1H), 7,52-7,45 (м, 1Н), 7,06-6,94 (м, 1H), 5,20 (с, 2Н), 3,94 (с, 3Н).
Стадия 44-2. Синтез метил-3-((2-хлор-4-(трифторметил)фенокси)метил)-5-(2-метилоксазол-5-ил)бензоата (INT 44-В)
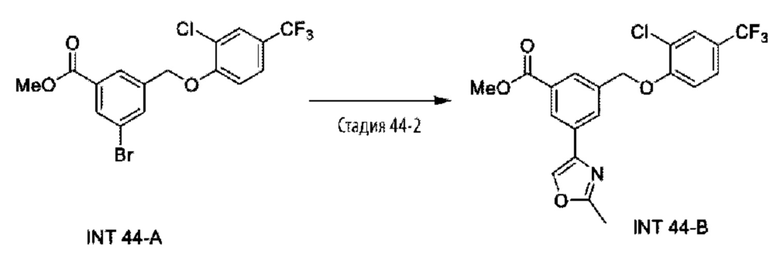
Смесь INT 44-А (150 мг, 0,354 ммоль), 2-метилоксазола (58,0 мкл, 0,708 ммоль), Pd(PPh3)4 (41,0 мг, 0,035 ммоль) и КОАс (70,0 мг, 0,708 ммоль) в ДМФА (4,00 мл) перемешивали при 110°С в течение 16 часов. Смесь охлаждали и разбавляли Н2О (20 мл). Водную фазу экстрагировали ЭА (3 × 20,0 мл), и объединенные органические слои промывали рассолом (20 мл), осушали (MgSO4), фильтровали и концентрировали. Остаток очищали хроматографией на SiO2 (ЭА/гексаны), получая 98 мг (65%) метил-3-((2-хлор-4-(трифторметил)фенокси)метил)-5-(2-метилоксазол-5-ил)бензоата (INT 44-В) в виде твердого вещества. ЖХ-MC-ESI (m/z) вычислено для C20H15ClF3NO4: 425,06; найдено 426,25 (М+Н)+, tR=2,79 мин (Метод 13). 1Н ЯМР (500 МГц, CDCl3) δ 8,25 (т, J=1,6 Гц, 1Н), 8,07-8,03 (м, 1H), 7,95-7,89 (м, 1H), 7,71-7,65 (м, 1H), 7,49 (ддд, J=8,7, 2,3, 0,8 Гц, 1H), 7,33 (с, 1H), 7,08-7,00 (м, 1H), 5,26 (с, 2Н), 3,97 (с, 3H), 2,56 (с, 3H).
Стадия 44-3. Синтез 3-((2-хлор-4-(трифторметил)фенокси)метил)-5-(2-метилоксазол-5-ил)бензойной кислоты (Соединение 44-1)
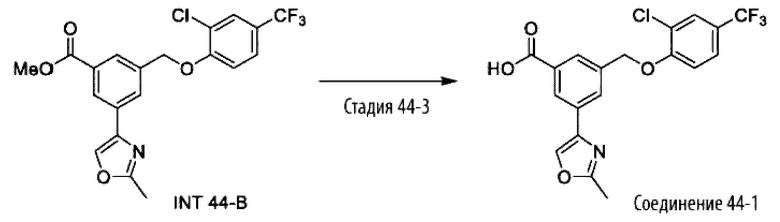
Раствор NaOH (1 М в H2O, 676 мкл, 0,676 ммоль) добавляли к смеси INT 44-В (96,0 мг, 0,225 ммоль) в ТГФ и H2O (3:1 об./об., 4,00 мл). Смесь перемешивали при 22°С в течение 4 часов. Смесь подкисляли водн. 1 М HCl (рН 2) и разбавили ЭА (20 мл). Водную фазу экстрагировали ЭА (2 × 30 мл), и объединенные органические слои промывали рассолом (20 мл), осушали (MgSO4), фильтровали и концентрировали. Остаток очищали хроматографией на SiO2 (МеОН/ДХМ), получая 77,5 мг (83%) 3-((2-хлор-4-(трифторметил)фенокси)метил)-5-(2-метилоксазол-5-ил)бензойной кислоты (соединение 44-1) в виде твердого вещества. ЖХ-MC-ESI (m/z) вычислено для C19H13ClF3NO4: 411,05; найдено 412,2 (М+Н)+, tR=4,59 мин (Метод 12). 1Н ЯМР (500 МГц, ДМСО-d6) δ 13,35 (с, 1H), 8,17 (дд, J=1,7 Гц, 1H), 8,04-7,98 (м, 2Н), 7,88 (д, J=2,3 Гц, 1H), 7,73 (дд, J=8,9, 2,3 Гц, 1H), 7,66 (с, 1H), 7,45 (д, J=8,7 Гц, 1H), 5,43 (с, 2Н), 2,50 (с, 3H).
ПРИМЕР 45
Синтез Соединений 45-1 и 45-2
Схема 45
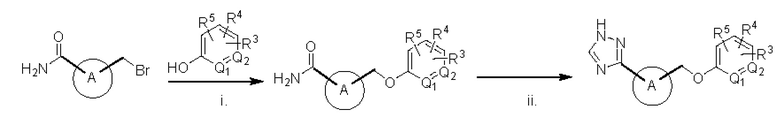
Стадия 45-1. Синтез 3-((2-хлор-4-(трифторметил)фенокси)метил)бензамида (Соединение 45-1)
В раствор 3-(бромметил)бензамида (254 мг, 1,29 ммоль) в ДМФА (5 мл) при 0°С добавляли NaH. После перемешивания реакционной смеси и ее нагревания в течение 30 минут до комнатной температуры добавляли 2-хлор-4-(трифторметил)фенол (250 мг, 1,17 ммоль). Через 4 часа реакционную смесь разбавляли ЭА и промывали H2O, 1 М HCl, 1М NaOH, Н2О, рассолом, осушали (Na2SO4), фильтровали и концентрировали. Полученный сырой остаток очищали обращенно-фазовой хроматографией на SiO2 (МеОН/H2O), получая материал, который растирали с МеОН/Н2О, фильтровали и сушили в вакууме с получением 260 мг (67%) 3-((2-хлор-4-(трифторметил)фенокси)метил)бензамид (соединение 45-1). ЖХ-MC-ESI (m/z) вычислено для C15H11ClF3NO2: 329,O4; найдено 330,1 (М+Н)+, tR=12,14 мин (Метод 10). 1Н ЯМР (300 МГц, CDCl3) δ 7,96 (с, 1H), 7,80 (д, J=7,7 Гц, 1H), 7,69 (с, 1H), 7,68 (д, J=8,3 Гц, 1H), 7,54 (д, J=7,7 Гц, 1H), 7,51 (д, J=6,6 Гц, 1Н), 7,05 (д, J=8,7 Гц, 1H), 6,11 (шир.с, 1H), 5,69 (шир.с, 1H), 5,28 (с, 2Н).
Стадия 45-2. Синтез 3-(3-((2-хлор-4-(трифторметил)фенокси)метил)фенил)-1Н-1,2,4-триазола (Соединение 45-2)
Раствор Соединения 45-1 (260 мг, 0,079 ммоль) в диметилформамида диметилацетале (4 мл) нагревали при 120°С в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры и концентрировали в вакууме. Полученный остаток растворяли в АсОН (4 мл) и по каплям добавляли N2H4-H2O (47 мг, 0,946 ммоль). После перемешивания при 90°С в течение 2 часов смесь концентрировали, разбавляли Et2O и охлаждали до 0°С. Полученный осадок собирали фильтрованием и очищали хроматографией на SiO2 (ЭА/гексан) с получением 125 мг (45%) 3-((2-хлор-4-(трифторметил)фенокси)метил)бензамида (Соединение 45-2). ЖХ-MC-ESI (m/z) вычислено для C16H11ClF3N3O: 353,05; найдено 354,5 (М+Н)+, tR=13,56 мин (Метод 10). 1Н ЯМР (300 МГц, CDCl3) δ 8,30 (с, 1Н), 8,16 (с, 1Н), 8,04 (д, J=7,5 Гц, 1H), 7,76 (д, J=2,9 Гц, 1H), 7,56-7,44 (м, 3H), 7,03 (д, J=8,6 Гц, 1Н), 5,24 (с, 2Н).
Соединения, перечисленные в Таблице 45, были получены с использованием процедур Схемы 45.
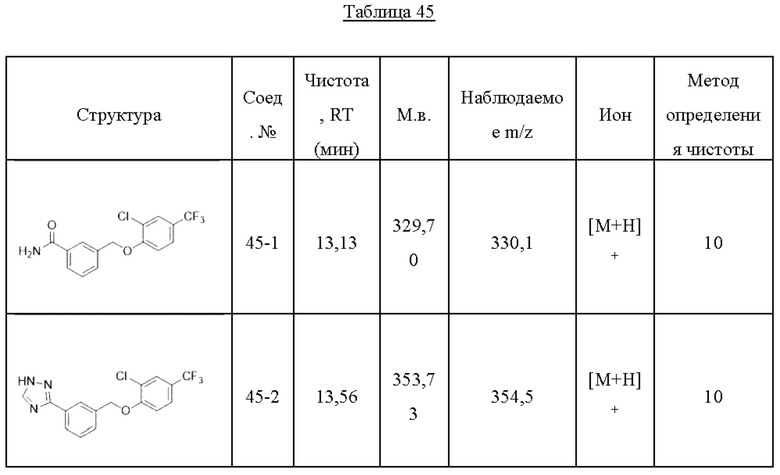
ПРИМЕР 46
Синтез Соединения 46-1
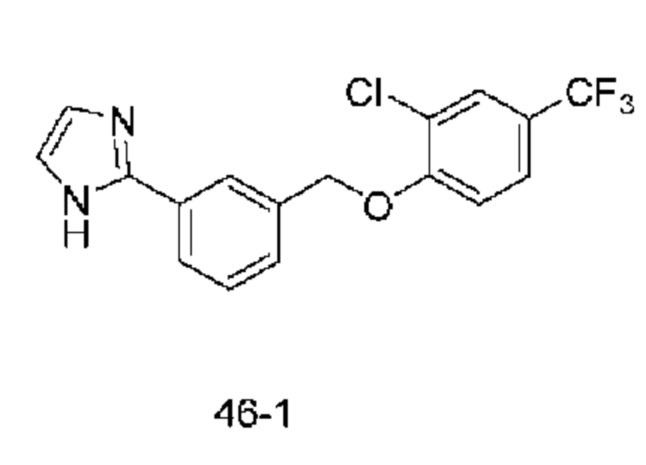
Схема 46
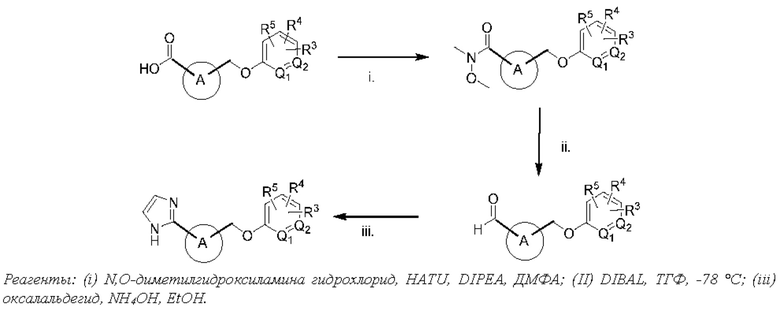
Стадия 46-1. Синтез 3-((2-хлор-4-(трифторметил)фенокси)метил)-N-метокси-N-метилбензамида (TNT 46-А)
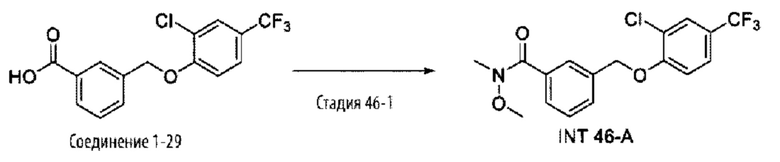
В раствор Соединения 1-29 (500 мг, 1,51 ммоль) в ДМФА (10 мл) добавляли гидрохлорид N,O-диметилгидроксиламина (161 мг, 1,66 ммоль), HATU (632 мг, 1,66 ммоль) и DIPEA (585 мг, 4,5 ммоль). После перемешивания реакционной смеси в течение 18 ч реакционную смесь подкисляли ТФУК и очищали хроматографией на SiO2 с обращенной фазой (МеОН/H2O, 0/1% ТФУК) с получением 530 мг (94%) 3-((2-хлор-4-(трифторметил)фенокси)метил)-N-метокси-N-метилбензамида (INT 46-А). ЖХ-MC-ESI (m/z) вычислено для C17H15ClF3NO3: 373,07; m/z не наблюдается; tR=6,2 мин (Метод 11).
Стадия 46-2. Синтез 3-((2-хлор-4-(трифторметил)фенокси)метил)бензальдегида (TNT 46-В)
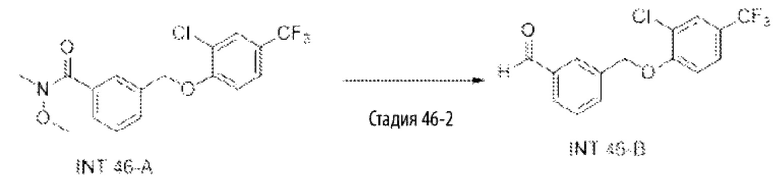
В раствор INT 46-А (1000 мг, 2,68 ммоль) в ТГФ (10 мл) при -78°С добавляли DIBAL (3,21 мл 1М раствора в ТГФ, 3,21 ммоль). После перемешивания реакционной смеси в течение 30 мин добавляли H2O и раствор экстрагировали ЭА, осушали (Na2SO4) и очищали хроматографией на SiO2 (ЭА/гексаны) с получением 600 мг (71,1%) 3-((2-хлор-4-(трифторметил)фенокси)метил)бензальдегида (INT 46-В). ЖХ-MC-ESI (m/z) вычислено для C15H10ClF3O2: 314,03; m/z не наблюдается, tR=6,3 мин (Метод 11).
Стадия 46-3. Синтез 2-(3-((2-хлор-4-(трифторметил)фенокси)метил)фенил)-1H-имидазола (Соединение 46-1)
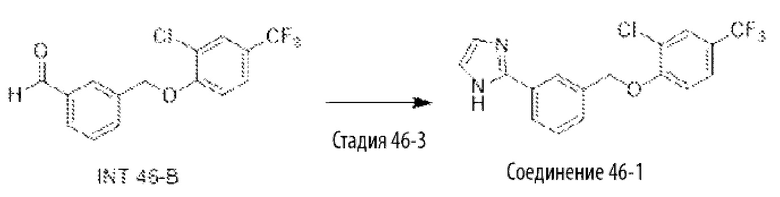
В раствор INT 46-В (100 мг, 0,32 ммоль) в EtOH (5 мл) при 0°С добавляли оксалальдегид (0,04 мл раствора 8,8 М в H2O, 0,35 ммоль) и NH4OH (0,053 мл 29% раствора в EtOH, 0,44 ммоль). После перемешивания в течение 48 часов смесь концентрировали и очищали хроматографией на SiO2 (ЭА/гексаны), получая 40 мг (35%) 2-(3-((2-хлор-4-(трифторметил)фенокси)метил)фенил)-1Н-имидазола (Соединение 46-1) в виде белого твердого вещества. ЖХ-MC-ESI (m/z) вычислено для C17H12ClF3N2O: 352,74; найдено 353,2 (М+Н)+, tR=11,97 мин (Метод 10). 1Н ЯМР (400 МГц, ДМСО-d6) δ 12,6 (с, 1H), 8,08 (с, 1H), 8,00-7,75 (м, 2Н), 7,72 (д, J=6 Гц, 1H), 7,60-7,35 (м, 3H), 7,26 (шир.с, 1Н), 7,04 (шир.с, 1Н),5,38 (с, 2Н).
ПРИМЕР 47
Синтез Соединения 47-1
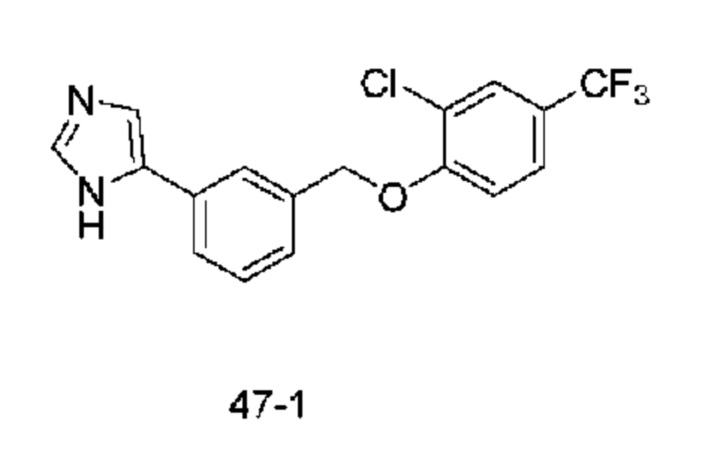
Схема 47
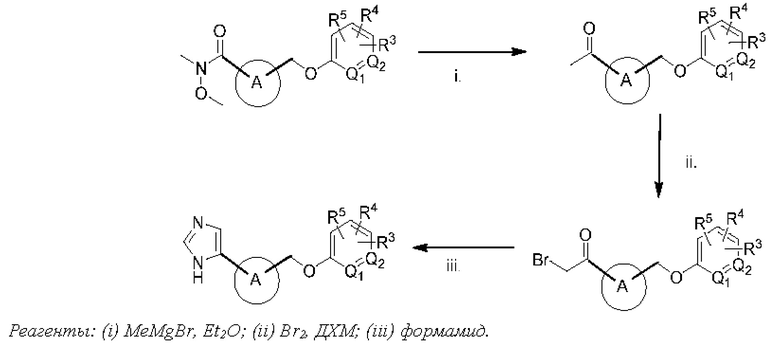
Стадия 47-1. Синтез 1-(3-((2-хлор-4-(трифторметил)фенокси)метил)фенил)этан-1-она (TNT 47-А)
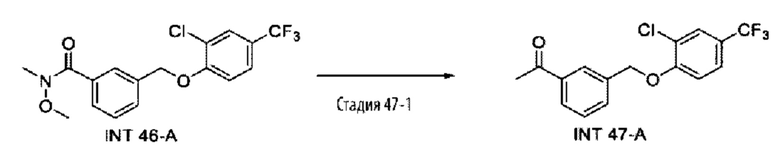
В раствор INT 46-А (480 мг, 1,28 ммоль) в Et2O (20 мл) добавляли MeMgBr (0,557 мл 3М раствора в Et2O, 1,67 ммоль). После перемешивания реакционной смеси в течение 6 часов реакционную смесь гасили 0,1 м HCl (50 мл) и экстрагировали ЭА. Полученный органический слой промывали рассолом, осушали (Na2SO4), фильтровали, концентрировали и очищали хроматографией на SiO2 (ЭА/гексаны) с получением 224 мг (53%) 1-(3-((2-хлор-4-(трифторметил)фенокси)метил)фенил)этан-1-она (INT 47-А). ЖХ-MC-ESI (m/z) вычислено для C17H15ClF3NO3: 373,07; m/z не наблюдается; tR=6,2 мин (Метод 11).
Стадия 47-2. Синтез 2-бром-1-(3-((2-хлор-4-(трифторметил)фенокси)метил)фенил)этан-1-она (INT 47-В)
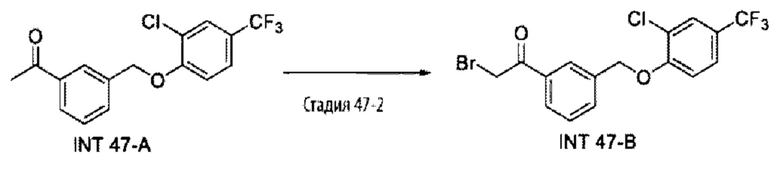
В раствор INT 47-А (224 мг, 0,68 ммоль) в ДХМ (15 мл) добавляли Br2 (0,035 мл, 0,68 ммоль). После перемешивания реакционной смеси в течение 30 минут реакционную смесь гасили NH4Cl (водн.) и экстрагировали ДХМ. Полученный органический слой промывали NaHCO3 (насыщ., водный), водой и рассолом, осушали (Na2SO4), концентрировали и очищали хроматографией на SiO2 (ЭА/гексаны) с получением 140 мг (50%) 2-бром-1-(3-((2-хлор-4-(трифторметил)фенокси)метил)фенил)этан-1-она (INT 47-В). ЖХ-MC-ESI (m/z) вычислено для C16H11BrClF3O2: 405,96; найдено 407,2 (М+Н)+; tR=5,529 мин (Метод 11).
Стадия 47-3. Синтез 5-(3-((2-хлор-4-(трифторметил)фенокси)метил)фенил)-1Н-имидазола (Соединение 47-1)
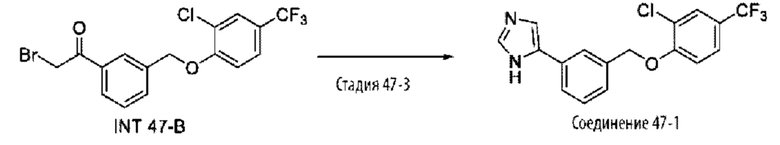
Раствор INT 47-В (100 мг, 0,3 ммоль) в формамиде (5 мл) перемешивали в течение 4 часов при 170°С, реакционную смесь концентрировали и очищали обращенно-фазовой хроматографией с С18 (H2O/МеОН), получая 6 мг (5%) 5-(3-((2-хлор-4-(трифторметил)фенокси)метил)фенил)-1Н-имидазола (Соединение 47-1). ЖХ-MC-ESI (m/z) вычислено для C17H12ClF3N2O: 352,06; найдено 353,4 (М+Н)+; tR=12,14 мин (Метод 10).
ПРИМЕР 48
Синтез Соединения 48-1
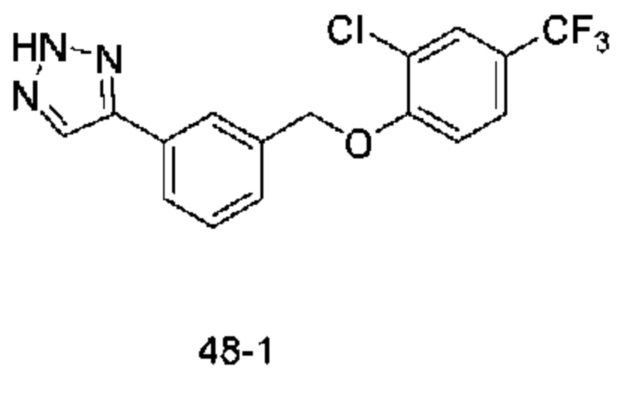
Схема 48
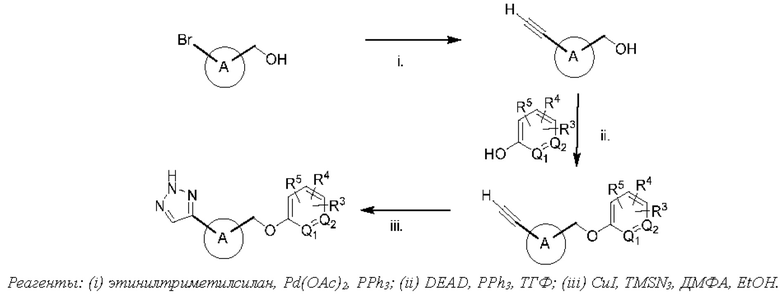
Стадия 48-1. Синтез (3-зтинилфенил)метанола (TNT 48-А)
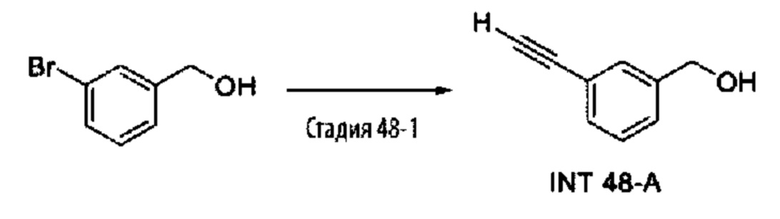
В раствор (2-бромфенил)метанола (1,5 г, 8,02 ммоль) в TEA (8 мл) добавляли этинилтриметилсилан (1,58 г, 16 ммоль), ацетат палладия (180 мг, 0,8 ммоль) и PPh3 (422 мг, 1,6 ммоль). После перемешивания в течение 1 ч при 95°С реакционную смесь фильтровали через целит и промывали ЭА. Полученный органический слой промывали H2O (3Х) и рассолом, осушали (Na2SO4), фильтровали, концентрировали и очищали хроматографией на SiO2 (ЭА/гексаны) с получением 800 мг (76%) (3-этинилфенил)метанола (INT 48-А). ЖХ-МС-ESI (m/z), вычислено для C9H8O: 132,06; найдено 133,3 (М+Н)+, tR=3,1 мин (Метод 11).
Стадия 48-2. Синтез 2-хлор-1-((3-зтинилбешил)окси)-4-(трифторметил)бензола (INT 48-В)
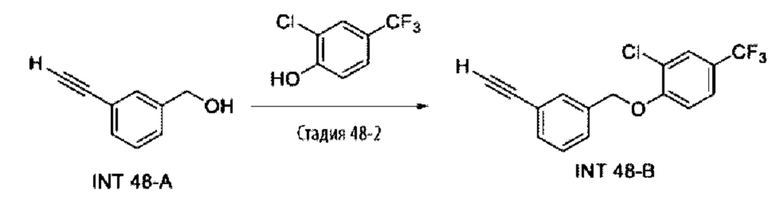
В раствор DEAD (55 мг 40% раствора, 2,7 ммоль) в ТГФ (3 мл) при 0°С добавляли PPh3 (71,2 мг, 2,7 ммоль) и INT 48-А (30 мг, 8,02 ммоль). После перемешивания реакционной смеси в течение 1 ч реакционную смесь концентрировали и очищали хроматографией на SiO2 (ЭА/гексаны) с получением 65 мг (93%) сырого 2-хлор-1-((3-этинилбензил)окси)-4-(трифторметил)бензола (INT 48-В), который переносили на следующую стадию без дополнительной очистки.
Стадия 48-3. Синтез 4-(3-((2-хлор-4-(трифторметил)фенокси)метил)фенил)-2Н-1,2,3-триазола (Соединение 48-1)
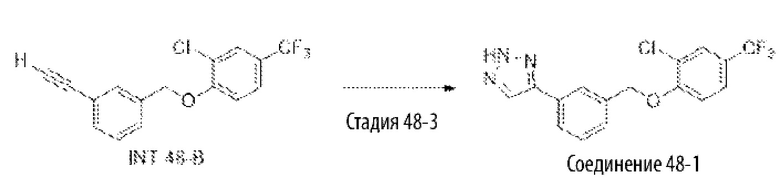
В раствор INT 48-В (65 мг, 2 ммоль) в ДМФА (8 мл) и EtOH (1 мл) добавляли CuI (122 мг, 0,64 ммоль). Смесь продували N2 и добавляли TMSN3 (741 мг, 6,4 ммоль). После перемешивания в течение 18 ч при 120°С реакционную смесь фильтровали через целит, концентрировали и очищали хроматографией на SiO2 (ЭА/гексаны) и обращенно-фазовой хроматографией (МеОН/Н2О с 0,1% ТФУК) с получением 65 мг (93%) 4-(3-((2-хлор-4-(трифторметил)фенокси)метил)фенил)-2Н-1,2,3-триазола (Соединение 48-1). ЖХ-MC-ESI (m/z) вычислено для C16H11ClF3N3O: 353,05; найдено 354,4 (М+Н)+, tR=5,39 мин (Метод 11).
ПРИМЕР 49
Синтез Соединения 49-1
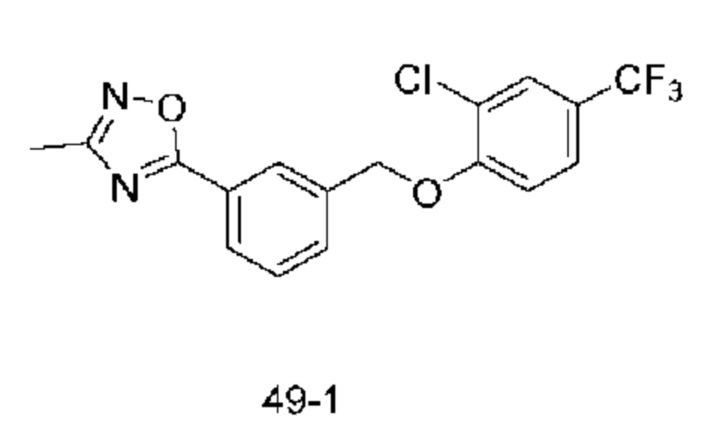
Схема 49
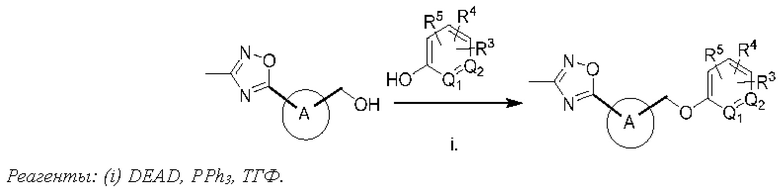
Стадия 49-1. Синтез 5-(3-((2-хлор-4-(трифторметил)фенокси)метил)фенил)-3-метил-1,2,4-оксадиазола (Соединение 49-1)
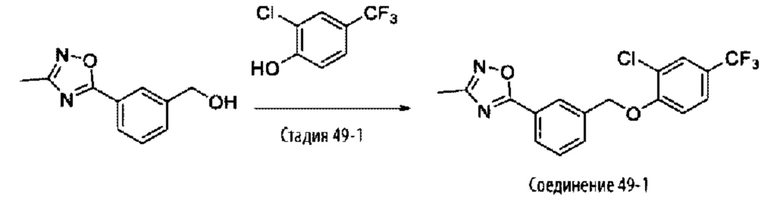
К перемешиваемому раствору (3-(3-метил-1,2,4-оксадиазол-5-ил)фенил)метанола (500 мг, 2,6 ммоль) в ТГФ (5 мл) добавляли трифенилфосфин (830 мг, 3,1 ммоль) и DEAD (911 мкл 70% раствора, 3,2 ммоль). После перемешивания в течение 2 часов реакционную смесь разбавляли ЭА и промывали насыщ. NaHCO3, H2O и рассолом, затем осушали (Na2SO4), концентрировали и очищали хроматографией на SiO2 (ЭА/гексан) до остатка, который растирали с МеОН/H2O, получая 670 мг (69,1%) 5-(3-((2-хлор-4-(трифторметил)фенокси)метил)фенил)-3-метил-1,2,4-оксадиазола (Соединение 49-1). ЖХ-МС-ESI (m/z) вычислено для C17H12ClF3N2O2: 368,05; найдено 369,1 (M+H)+, tR=14,59 мин (50-95-4 мин). 1Н ЯМР (300 МГц, ДМСО-d6) δ 8,23 (с, 1H), 8,08 (д, J=7,8 Гц, 1Н), 7,89 (шир.с, 1H), 7,80 (д, J=6,5 Гц, 1H), 7,50-7,30 (м, 2Н), 7,46 (д, J=8,5 Гц, 1H), 5,46 (с, 2Н), 2,43 (с, 3H).
ПРИМЕР 50
Синтез Соединения 50-1
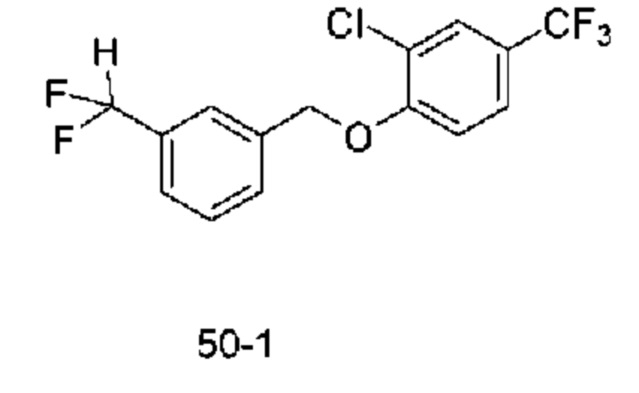
Схема 50
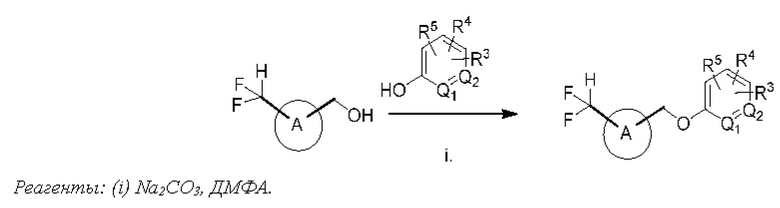
Стадия 50-1. Синтез 2-хлор-1-((3-(дифторметил)бензил)окси)-4-(трифторметил)бензола (Соединение 50-1)
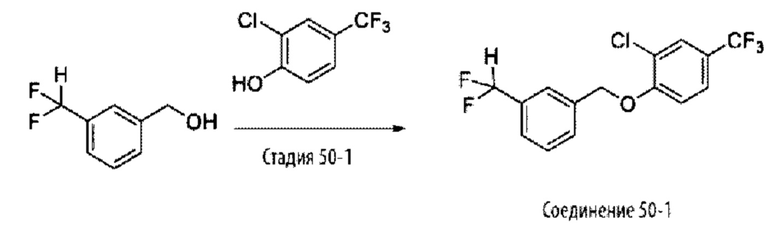
К перемешиваемому раствору (3-(дифгорметил)фенил)метанола (500 мг, 2,26 ммоль) в ДМФА (8 мл) добавляли 2-хлор-4-(трифторметил)фенол (450 мг, 2,26 ммоль) и Na2CO3 (720 мг, 6,79 ммоль). После перемешивания в течение 18 часов при 50°С реакционную смесь разбавляли H2O, экстрагировали ЭА, промывали Н2О и рассолом. Органические слои концентрировали хроматографией на SiO2 (ЭА/гексан) с получением 165 мг (22%) 2-хлор-1-((3-(дифторметил)бензил)окси)-4-(трифторметил)бензола (Соединение 50-1). ЖХ-MC-ESI (m/z) вычислено для C15H10ClF5O: 336,03; найдено 359,2 (M+Na)+, tR=5,68 мин (Метод 11). 1Н ЯМР (300 МГц, CDCl3) 7,70 (с, 1H), 7,63 (с, 1H), 7,625-7,45 (м, 4Н), 7,04 (д, J=8,6 Гц, 1H), 6,70 (т, JH-F=56 Гц, 1H), 5,27 (с, 2Н).
ПРИМЕР 52
Синтез Соединения 52-1
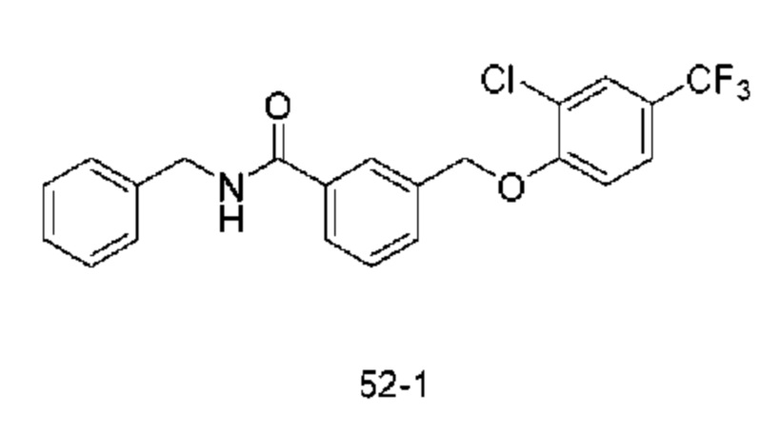
Схема 52
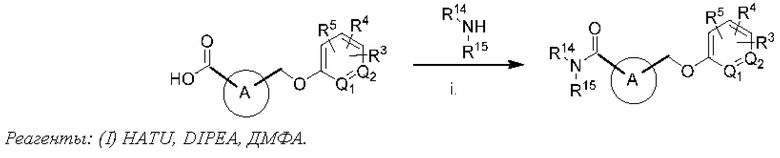
Стадия 52-1. Синтез N-бензил-3-((2-хлор-4-(трифторметил)фенокси)метил)бензамида (Соединение 52-1)
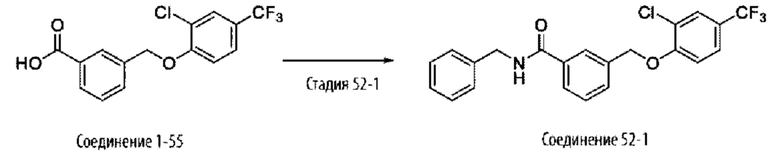
К перемешиваемому раствору Соединения 1-55 (113 мг, 0,34 ммоль) в ДМФА (5 мл) добавляли HATU (137 мг, 0,36 ммоль), DIPEA (126 мг, 0,98 ммоль) и фенилметанамин (35 мг, 0,33 ммоль). После перемешивания в течение 16 часов при комнатной температуре реакционную смесь разбавляли ЭА, промывали 30 мл каждого из H2O, 1М HCl, 1М NaOH, NaHCO3 и рассола. Органические слои осушали (Na2SO4), концентрировали и очищали хроматографией на SiO2 (ЭА/гексан) с получением 62 мг (54%) N-бензил-3-((2-хлор-4-(трифторметил)фенокси)метил)бензамида (Соединение 52-1). ЖХ-MC-ESI (m/z) вычислено для C22H17ClF3NO2: 419,1; найдено 420,3 (М+Н)+, tR=12,98 мин (Метод 10). 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,11 (с, 1H), 8,02 (с, 1H), 7,90-7,88 (м, 2Н), 7,72 (д, J=8, 1H), 7,65 (д, J=8, 1Н), 7,54 (т, J=8, 1H), 7,45 (д, J=8, 1H), 7,33 (т, J=4, 4Н), 7,26 (д, J=4, 1H), 5,38 (с, 2Н), 4,49 (д, J=4,2Н).
Соединения, перечисленные в Таблице 52, были получены с использованием процедур Схемы 52.
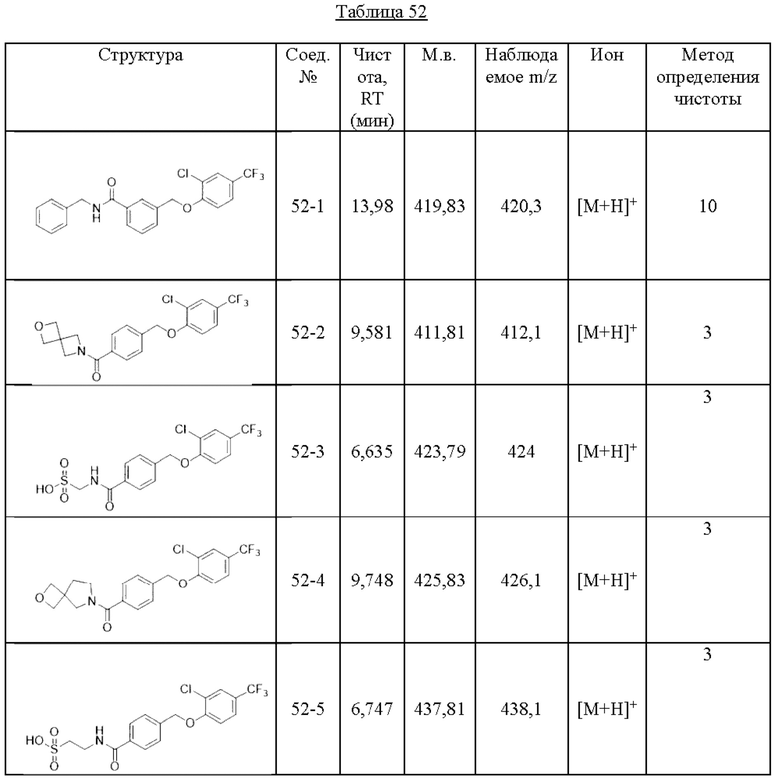
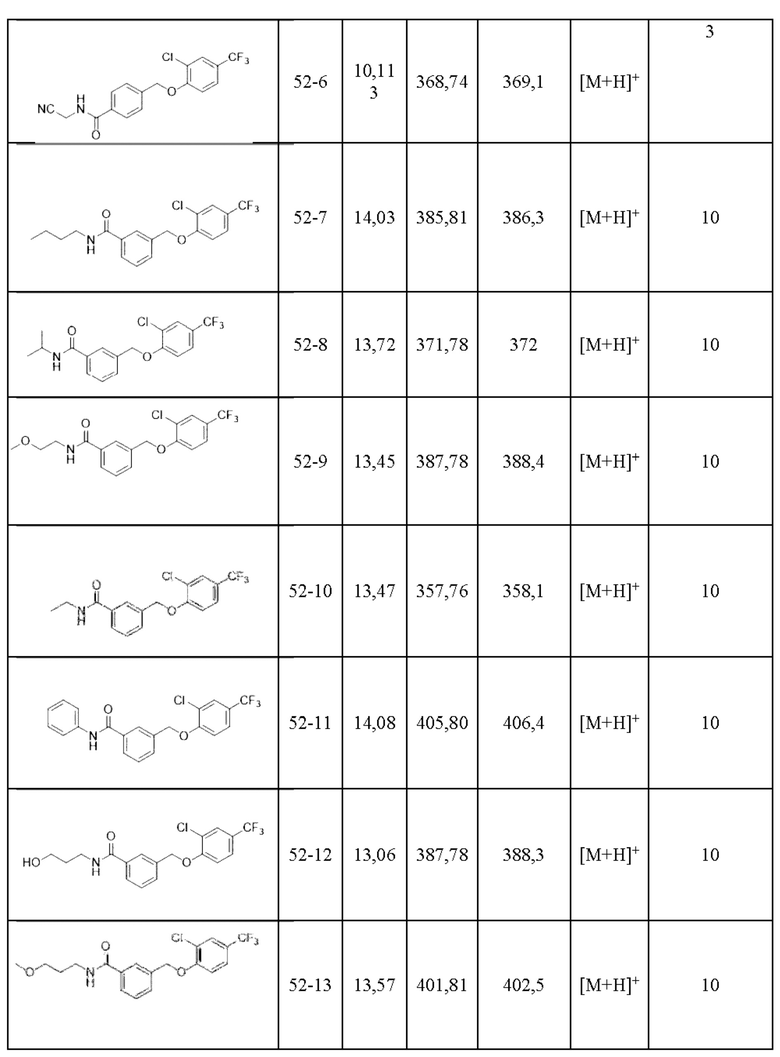
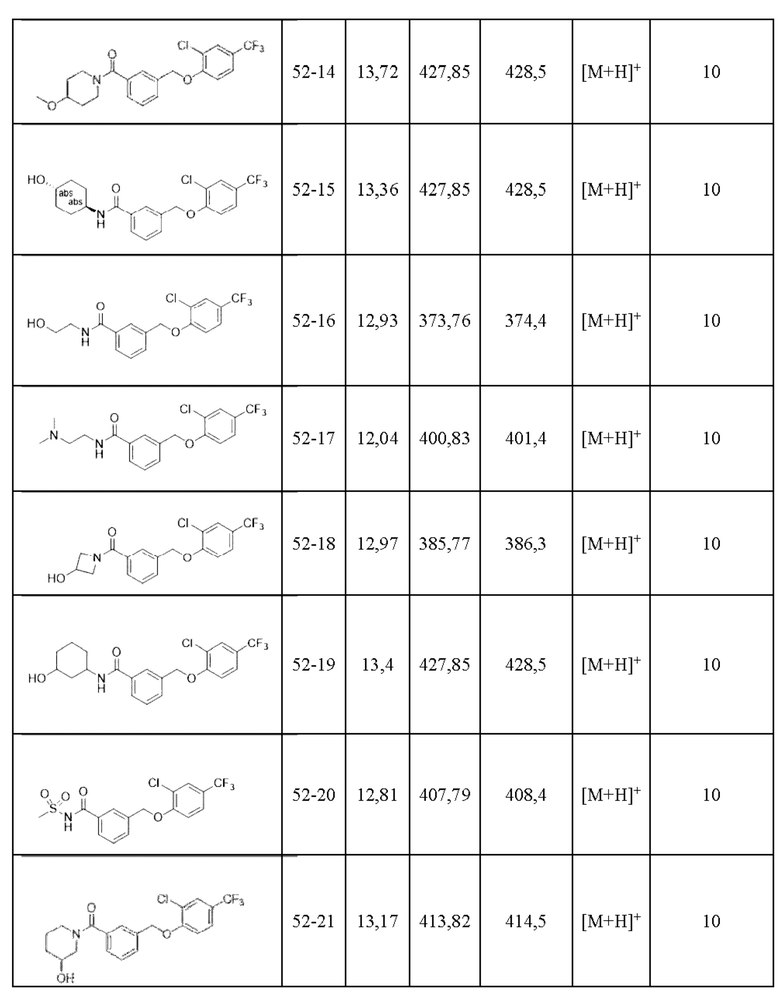
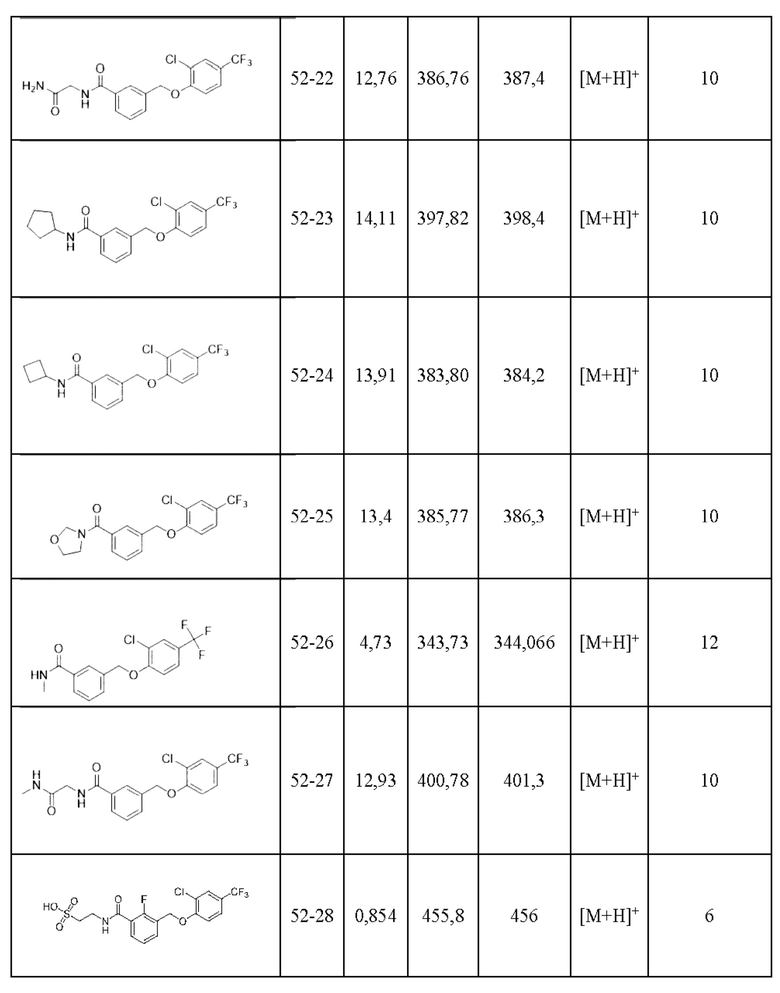
ПРИМЕР 53
Синтез Соединения 53-1
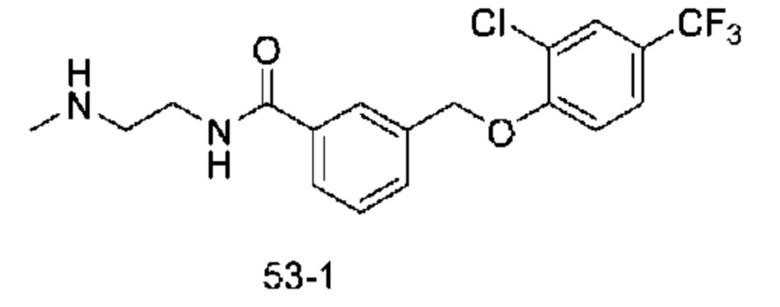
Схема 53
Стадия 53-1. Синтез 3-((2-хлор-4-(трифторметил)фенокси)метил)-N-(2-(метиламино)этил)бензамида (Соединение 53-1)
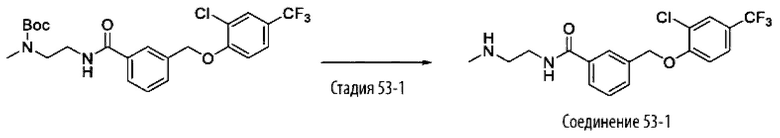
Раствор трет-бутил-(2-(3-((2-хлор-4-(трифторметил)фенокси) m) метил)бензамидо)этил)(метил)карбамата (50 мг, 0,099 ммоль) (получен из Соединения 1-55 и трет-бутил-(2-аминоэтил)(метил)карбамата по Схеме 52) в 1:1 ДХМ:ТФУК (5 мл) перемешивали в течение 16 часов при комнатной температуре. Реакционную смесь концентрировали, растворяли в H2O/CH3CN и лиофилизировали с получением 30 мг (60%) 3-((2-хлор-4-(трифторметил[)фенокси)метил)-N-(2-(метиламино)этил)бензамида (Соединение 53-1). ЖХ-MC-ESI (m/z) вычислено для C18H18ClF3N2O2: 386,1; найдено 387,4 (М+Н)+, tR=12,98 мин (Метод 10).
Соединения, перечисленные в Таблице 53, были получены с использованием процедур Схемы 53.
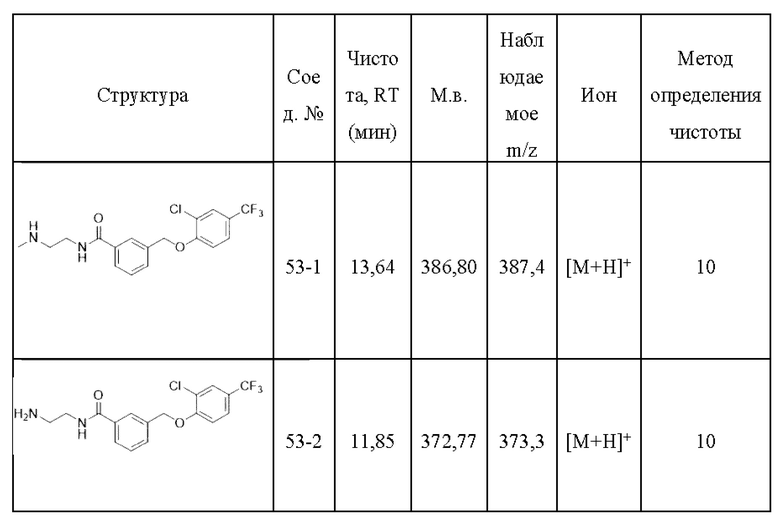
ПРИМЕР 54
Синтез Соединения 54-1
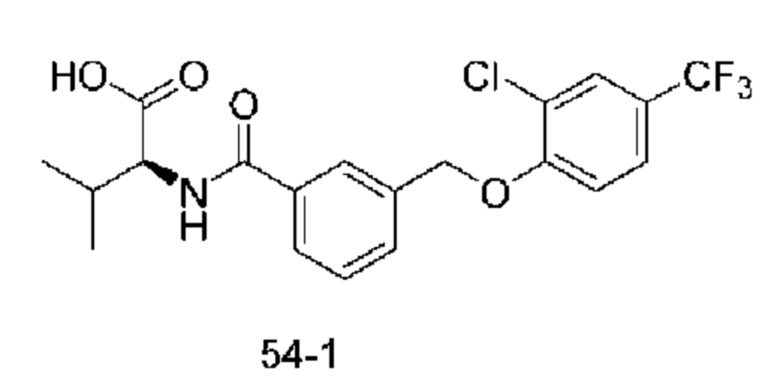
Схема 54
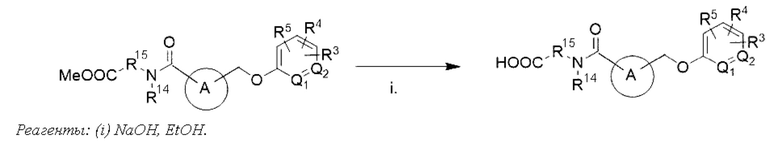
Стадия 54-1. Синтез (3-((2-хлор-4-(трифторметил)фенокси)метил)бензоил)-L-валина (Соединение 54-1)
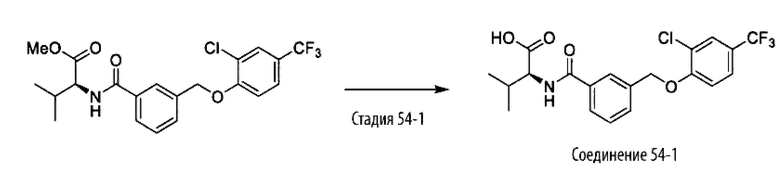
В раствор метил-(3-((2-хлор-4-(трифторметил)фенокси)метил)бензоил)-L-валината (250 мг, 0,56 ммоль) (получен из Соединения 1-55 и метил-L-валината по Схеме 52) в EtOH (5 мл) добавляли 2М NaOH (1,12 г, 1,1 ммоль). После перемешивания в течение 16 часов при комнатной температуре реакционную смесь разбавляли ЭА и подкисляли 1М HCl. Органический слой собирали и промывали рассолом, осушали (Na2SO4), концентрировали и очищали обращенно-фазовой хроматографией на SiO2 (МеОН/Н2О, с 0,1% муравьиной кислоты) с получением 36 мг (15%) (3-((2-хлор-4-(трифторметил)фенокси)метил)бензоил)-L-валина (Соединение 54-1). ЖХ-MC-ESI (m/z) вычислено для C20H19ClF3NO4: 429,1; найдено 430,6 (М+Н)+, tR=5,31 мин (Метод 11).
Соединения, перечисленные в Таблице 54, были получены с использованием процедур Схемы 54.
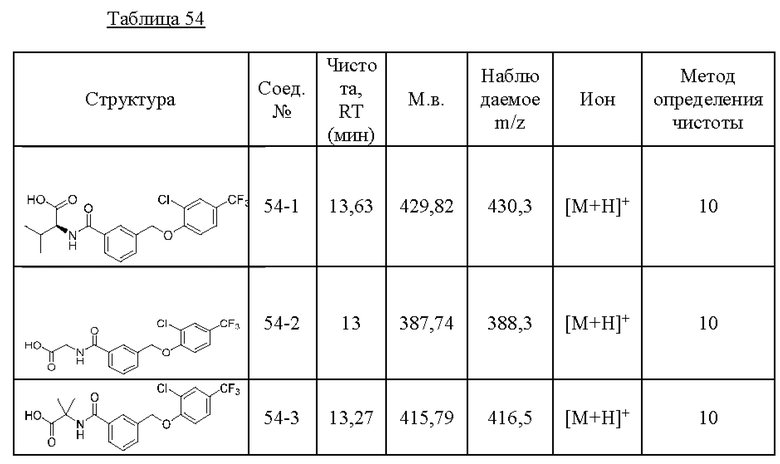
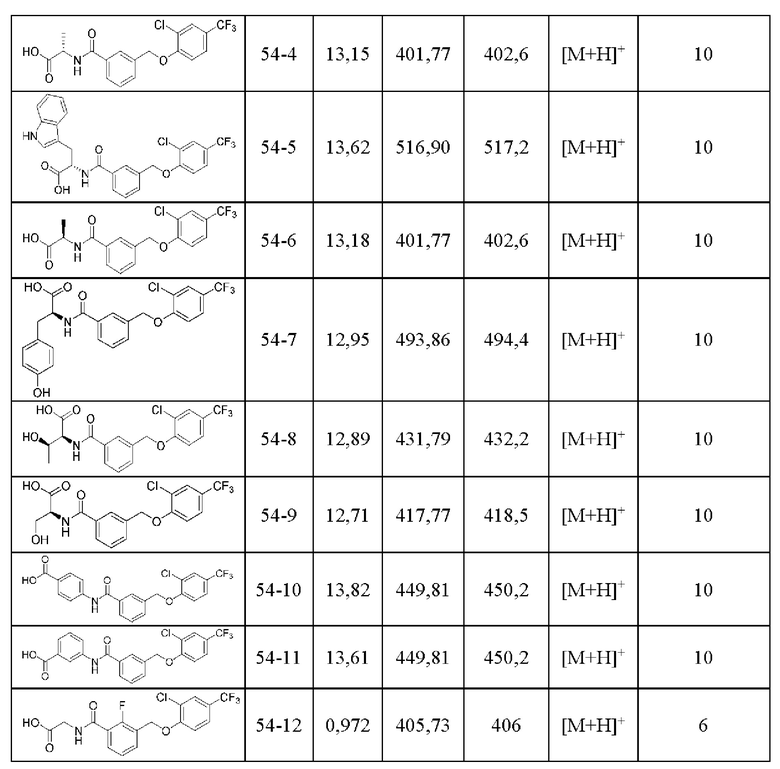
ПРИМЕР 55
Синтез Соединения 55-1
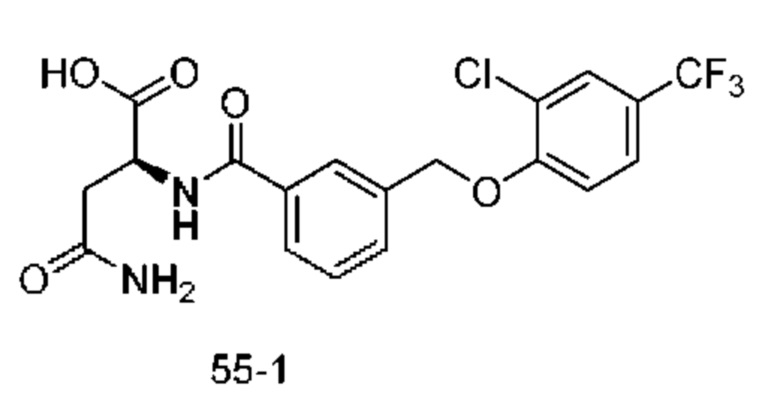
Схема 55
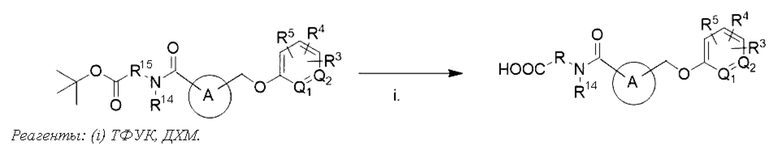
Стадия 55-1. Синтез (3-((2-хлор-4-(трифторметил)фенокси)метил)бензоил)-L-аспарагина (Соединение 55-1)
Раствор трет-бутил-(3-((2-хлор-4-(трифторметил)фенокси)метил)бензоил)-L-аспарагината (200 мг, 0,4 ммоль) (получен из Соединения 1-55 и трет-бутил-L-аспарагината по Схеме 52) в 1:1 ТФУК:ДХМ (5 мл) перемешивали в течение 16 часов при комнатной температуре. Смесь концентрировали и очищали с помощью о бращенно-фазовой хроматографии на SiO2 (МеОН/H2O, с 0,1% муравьиной кислоты) с получением 107 мг (60%) (3-((2-хлор-4-(трифторметил)фенокси)метил)бензоил)-L-аспарагина (Соединение 55-1). ЖХ-MC-ESI (m/z) вычислено для C19H16ClF3N2O5: 444,1; найдено 445,4 (М+Н)+, tR=4,70 мин (Метод 11).
Соединения, перечисленные в Таблице 55, были получены с использованием процедур Схемы 55.
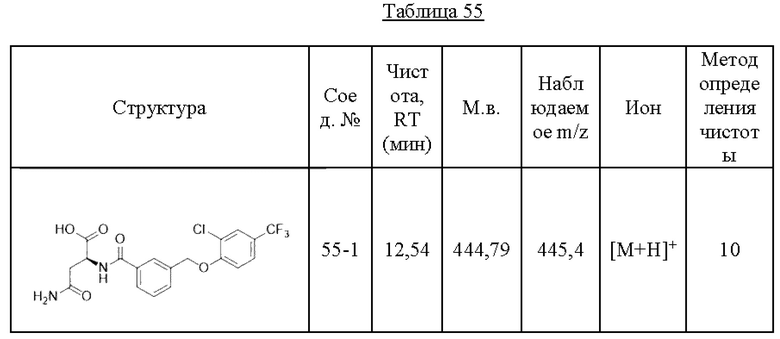
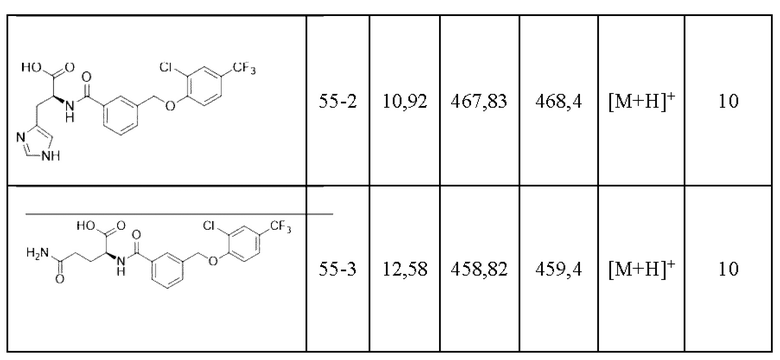
ПРИМЕР 56
Синтез Соединения 56-1
Схема 56
Стадия 56-1. Синтез этил-3-((2-хлор-4-(трифторметил)фенокси)метил)-2-фторбензоата (Соединение 56-1)
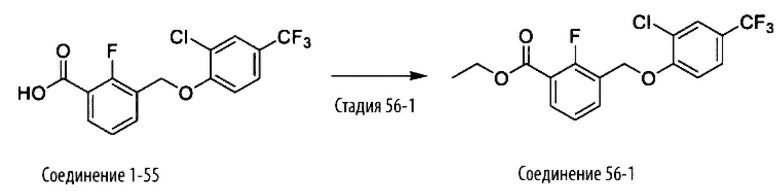
В раствор Соединения 1-55 (30 мг, 0,086 ммоль) в ДХМ (3 мл) добавляли тионилхлорид (19 мкл, 0,26 ммоль). После перемешивания в течение 2 часов реакционную смесь концентрировали и растворяли в EtOH (1 мл). Через 1 час смесь концентрировали и очищали хроматографией методом ОФ-ВЭЖХ с получением 32 мг (31%) этил-3-((2-хлор-4-(трифторметил)фенокси)метил)-2-фторбензоата (Соединение 56-1). ЖХ-MC-ESI (m/z) вычислено для C17H13ClF4O3: 376,7; найдено 378,1 (М+Н)+, tR=12,5 мин (чистота EsClent).
Соединения, перечисленные в Таблице 56, были получены с использованием процедур Схемы 56.
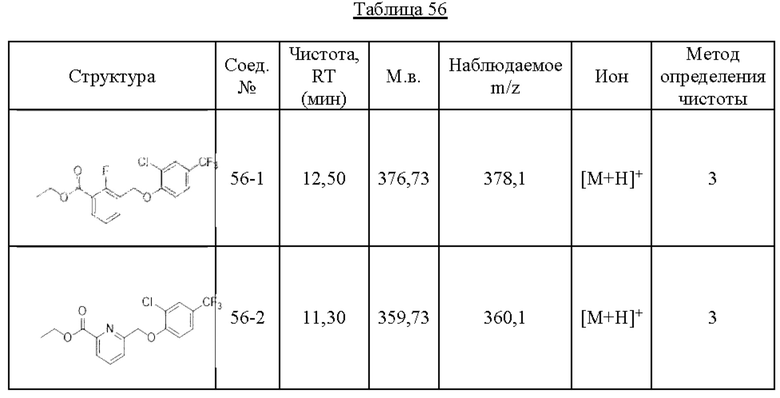
ПРИМЕР 57
Синтез Соединения 57-1
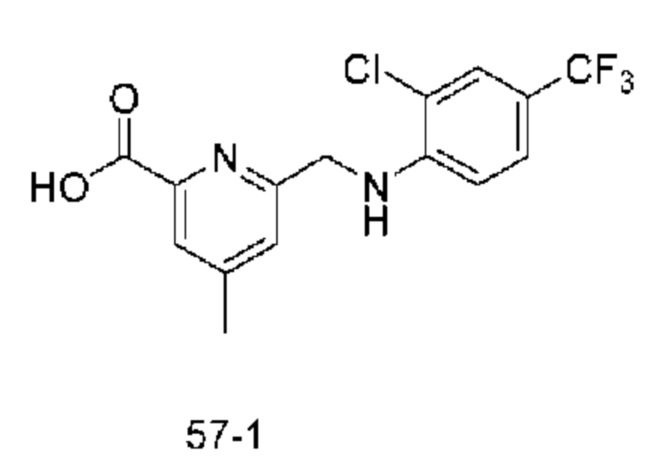
Схема 57
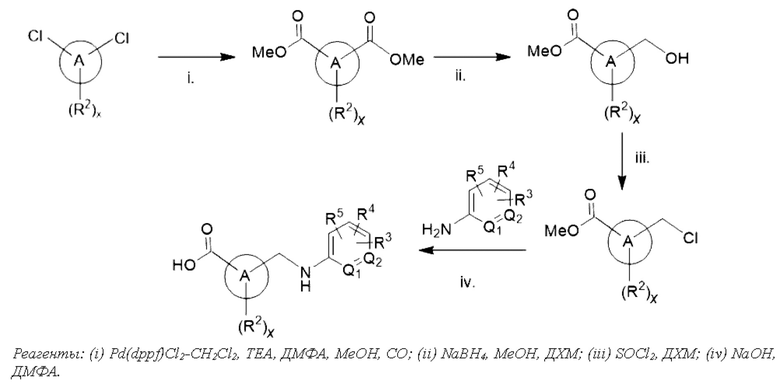
Стадия 57-1. Синтез диметил-4-метилтфидин-2,6-дикарбоксилата (INT 57-1)
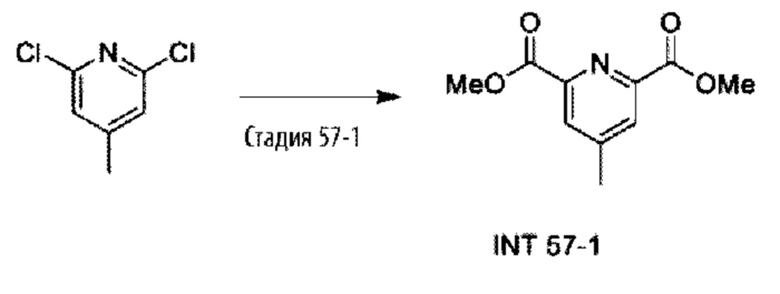
В раствор 2,6-дихлор-4-метилпиридина (6,8 г, 41,97 ммоль) в ДМФА (100 мл) и МеОН (50 мл) добавляли Pd(dppf)Cl2-CH2Cl2 (3,43 г, 4,20 ммоль), TEA (23,37 мл, 167,9 ммоль). Реакционную смесь перемешивали при 80°С в атмосфере СО (1,18 г, 41,97 ммоль, 345 кПа (50 Psi)) в течение 16 часов. Реакционную смесь фильтровали, концентрировали, разбавляли H2O (100 мл) и экстрагировали ЭА (2 × 100 мл). Органический слой собирали, сушили, фильтровали, концентрировали и очищали хроматографией на SiO2 (ЭА/петролейный эфир) с получением 6,7 г (76%) диметил-4-метилпиридин-2,6-дикарбоксилата (INT 57-1) в виде желтого твердого вещества. ЖХ-MC-ESI (m/z) вычислено для C10H11NO4: 209,07; найдено 210,1 (М+Н)+, tR=0,742 мин (Метод 6).
Стадия 57-2. Синтез метил-6-(гидроксиметил)-4-метилпиколината (TNT 57-2)
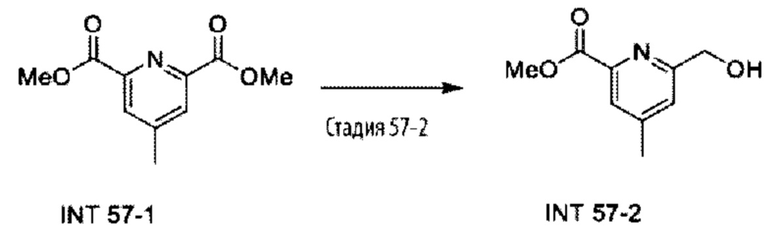
В раствор INT 57-1 (6,7 г, 31,39 ммоль) в МеОН (400 мл) и ДХМ (100 мл) при 0°С добавляли NaBH4 (1,78 г, 47,08 ммоль) небольшими порциями. После перемешивания в течение 12 ч при 0°С, реакцию гасили добавлением водного NH4Cl (200 мл) и экстрагировали ЭА (3×200 мл). Объединенные органические слои осушали (Na2SO4), фильтровали, концентрировали и очищали хроматографией на SiO2 с получением 3,7 г (64%) метил-6-(гидроксиметил)-4-метилпиколината (INT 57-2). ЖХ-MC-ESI (m/z) вычислено для C9H11NO3: 181,2; найдено 182,7 (М+Н)+, tR = 0,323 мин (Метод 6). 1Н ЯМР (400 МГц, CDCl3) δ 7,85 (с, 1Н), 7,35 (с, 1Н), 4,81 (с, 2Н), 3,97 (с, 3Н), 3,81-3,26 (м, 1Н), 2,49-2,38 (с, 3Н).
Стадия 57-3. Синтез метил-6-(хлорметил)-4-метилпиколината (TNT 57-3)
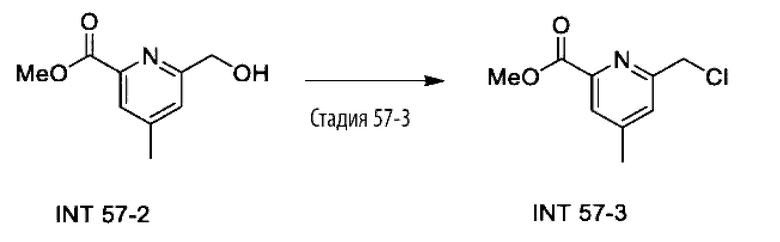
В раствор INT 57-2 (200 мг, 1,1 ммоль) в ДХМ (7 мл) при 0°С добавляли SOCl2 (1 мл, 13,8 ммоль). После перемешивания в течение 1,5 ч при комнатной температуре реакционную смесь концентрировали с получением 32 мг (31%) метил-6-(хлорметил)-4-метилпиколината (INT 57-3) в виде белого твердого вещества, которое использовали без дополнительной очистки. ЖХ-MC-ESI (m/z) вычислено для C9H10ClNO2: 199,04; найдено 200,0 (М+Н)+, tR = 0,755 мин (Метод 6).
Стадия 57-4. Синтез 6-(((2-хлор-4-(трифторметил)фенил)амино)метилУ4-метилпиколиновой кислоты (Соединение 57-1)
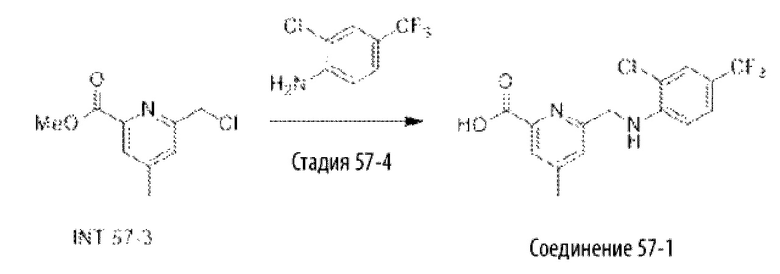
В раствор INT 57-3 (200 мг, 1,0 ммоль) и 2-хлор-4-(трифторметил)анилина (195,9 мг, 1,00 ммоль) в ДМФА (3 мл) добавляли NaOH (400,7 мг, 10,02 ммоль). Реакционную смесь перемешивали при 25°С в течение 0,5 часа. Смесь разбавляли Н2О (10 мл), доводили до рН = 7 с помощью HCl (36%), затем фильтровали и концентрировали с получением остатка, который очищали с помощью препаративной ВЭЖХ с обращенной фазой (Н2О /CH3CN с 0,225% муравьиной кислоты (FA)) получая 2,2 мг (0,67%) 6-(((2-хлор-4-(трифторметил)фенил)амино)метил)-4-метилпиколиновой кислоты (Соединение 57-1) в виде белого твердого вещества. ЖХ-MC-ESI (m/z) вычислено для C15H12ClF3N2O2: 344,05; найдено 345,0 (М+Н)+, tR = 0,873 мин (Метод 6). 1Н ЯМР (400 МГц, MeOD4) δ м.д. 2,35-2,51 (с, 3Н 4,58-4,70 (с, 2Н), 6,60-6,81 (м, 1Н), 7,26-7,35 (м, 1Н), 7,39-7,46 (м, 1Н), 7,50-7,60 (с, 1Н), 7,80-8,05 (с, 1Н).
ПРИМЕР 58
Синтез Соединений 58-1 и 58-2
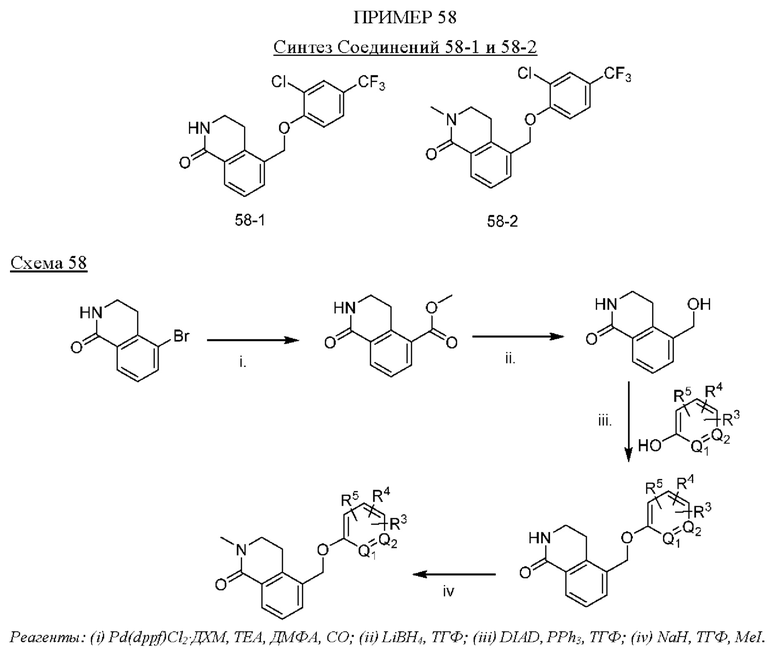
Стадия 58-1. Синтез метил-1-оксо-1,2,3,4-тетрагидроизохинолин-5-карбоксилата (INT 58-1)
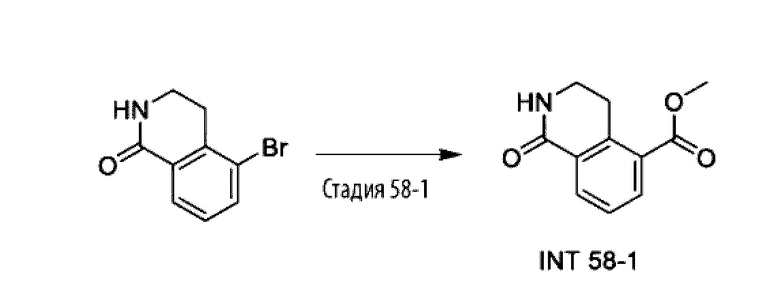
Добавляли Pd(dppf)Cl2⋅ДХМ (285 мг, 0,35 ммоль) к раствору 5-бром-3,4-дигидро-2Н-изохинолин-1-она (395 мг, 1,75 ммоль) и TEA (1,22 мл, 8,74 ммоль) в ДМФА (6,00 мл) при 22°С. Смесь откачивали и снова заполняли СО в течение 3 циклов. Добавляли МеОН (3,08 мл) и смесь нагревали до 85°С в атмосфере СО (1 атм) в течение 16 часов. Смесь разбавляли ЭА (25 мл) и фильтровали через слой целита. Фильтрат концентрировали при пониженном давлении. Остаток разбавляли ЭА (100 мл) и H2O (100 мл). Водную фазу экстрагировали ЭА (3×25,0 мл). Объединенные органические слои промывали рассолом (50 мл), осушали (Na2SO4), фильтровали и концентрировали. Остаток очищали хроматографией на SiO2 (гексаны и ЭА) с получением 285 мг (80%) метил-1-оксо-1,2,3,4-тетрагидроизохинолин-5-карбоксилата (INT 58-1). ЖХ-MC-ESI (m/z), вычислено для C11H11NO3: 205,07; найдено 205,74 (М+Н)+, tR = 1,82 мин (Метод 13).1Н ЯМР (400 МГц, CDCl3) δ 8,30 (дд, J=7,7, 1,5 Гц, 1Н), 8,09 (дд, J=7,8,1,5 Гц, 1Н), 7,42 (дд, J=7,8 Гц, 1Н), 5,95 (с, 1Н), 3,92 (с, 3Н), 3,58-3,52 (м, 2Н), 3,49-3,40 (м, 2Н).
Стадия 58-2. Синтез 5-(гадроксиметил)-3,4-дигидроизохинолин-1(2Н)-она (INT 58-2)
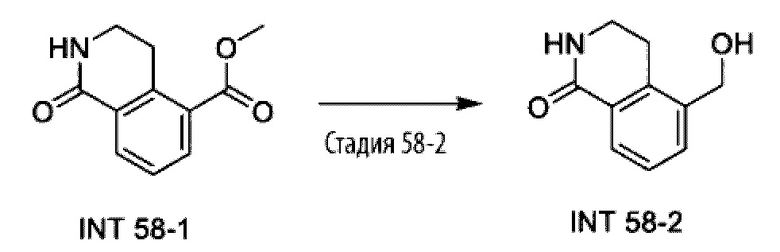
Добавляли LiBH4 (2М в ТГФ, 2,66 мл, 5,32 ммоль) к раствору INT 58-1 (182 мг, 0,887 ммоль) в ТГФ (5,00 мл) при 22°С в атмосфере N2. Смесь перемешивали при 22°С в течение 20 часов. Смесь разбавляли насыщ. водн. NH4Cl (10 мл). Водную фазу экстрагировали ЭА (3×20 мл), и объединенные органические фазы промывали рассолом (50 мл), осушали (Na2SO4), фильтровали и концентрировали с получением 88 мг (56%) 5-(гидроксиметил)-3,4-дигидроизохинолин-1(2Н)-она (INT 58-2) в виде маслянистой жидкости. ЖХ-MC-ESI (m/z), вычислено для C10H11NO2: 177,08; найдено 178,13 (М+Н)+, tR = 1,39 мин (Метод 13). 1Н ЯМР (500 МГц, CDCl3) δ 8,07 (дд, J=7,8, 1,4 Гц, 1Н), 7,52 (дд, J=7,6, 1,4 Гц, 1Н), 7,36 (т, J=7,7 Гц, 1Н), 5,95 (с, 1Н), 4,75 (с, 2Н), 3,57 (тд, J=6,7, 2,9 Гц, 2Н), 3,07 (т, J=6,6 Гц, 2Н), 1,72 (с, 1Н).
Стадия 58-3. Синтез 5-((2-хлор-4-(трифтормегил)фенокси)метил)-3,4-дигидроизохинолин-1(2Н)-она (Соединение 58-1)
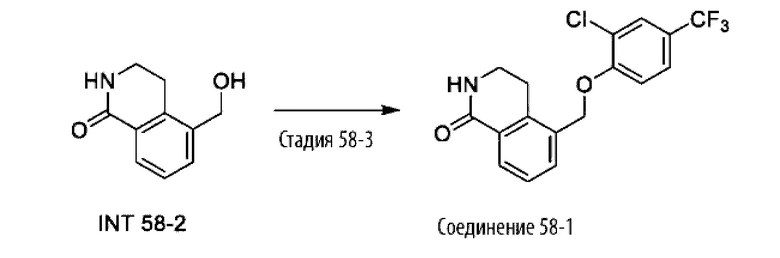
Добавляли DIAD (108 мкл, 0,55 ммоль) к смеси INT 58-2 (88,0 мг, 0,497 ммоль), 2-хлор-4-(трифторметил)фенола (69,7 мкл, 0,521 ммоль) и PPh3 (143 мг, 0,546 ммоль) в ТГФ (5,00 мл) при 0°С в атмосфере N2 Смесь перемешивали при 22°С в течение 18 часов. Смесь концентрировали и остаток очищали обращенно-фазовой хроматографией (Н2О (+ 0,1% муравьиной кислоты) и MeCN) с получением 32,7 мг (19%) 5-((2-хлор-4-(трифторметил)фенокси)метил)-3,4-дигидроизохинолин-1(2Н)-она (Соединение 58-1) в виде твердого вещества. ЖХ-MC-ESI (m/z) вычислено для C17H13ClF3NO2: 355,06; найдено 356,07 (М+Н)+, tR = 4,69 мин (Метод 12). 1Н ЯМР (500 МГц, CDCl3) δ 8,00 (с, 1Н), 7,90 (дд, J=7,8, 1,4 Гц, 1Н), 7,88-7,85 (м, 1Н), 7,73 (ддд, J=8,7, 2,3, 0,9 Гц, 1Н), 7,68 (дд, J=7,6, 1,4 Гц, 1Н), 7,52 (д, J=8,6 Гц, 1Н), 7,40 (дд, J=7,6 Гц, 1Н), 5,38 (с, 2Н), 3,38 (тд, J=6,6, 2,8 Гц, 2Н), 2,96 (т, J=6,6 Гц, 2Н).
Стадия 58-4. Синтез 5-((2-хлор-4-(трифторметил)фенокси)метил)-2-метил-3,4-дигидроизохинолин-1(2Н)-она (Соединение 58-2).
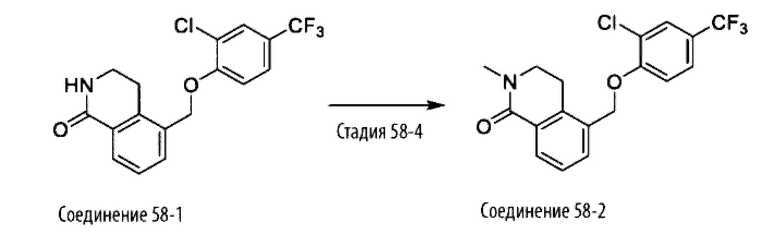
Добавляли NaH (24,9 мг, 1,08 ммоль) к раствору Соединения 58-1 (154 мг, чистота 50%, 0,216 ммоль) в ТГФ (5,00 мл) при 0°С в атмосфере N2. Смесь перемешивали при 22°С в течение 30 минут. Добавляли йодметан (67,4 мкл, 1,08 ммоль) и смесь перемешивали при 70°С в течение 1 часа. Смесь разбавляли МеОН (10,0 мл) и концентрировали. Продукт очищали обращенно-фазовой хроматографией (Н2О (+ 0,1% муравьиной кислоты) и MeCN) с получением 60 мг (76%) 5-((2-хлор-4-(трифторметил)фенокси)метил)-2-метил-3,4-дигидроизохинолин-1(2Н)-она (Соединение 58-2) в виде твердого вещества. ЖХ-MC-ESI (m/z) вычислено для C18H15ClF3NO2: 369,77; найдено 370,08 (М+Н)+, tR = 5,08 мин (Метод 12). 1Н ЯМР (500 МГц, CDCl3) δ 8,00 (с, 1Н), 7,92 (дд, J=7,8, 1,4 Гц, 1Н), 7,86 (дд, J=2,3, 0,7 Гц, 1Н), 7,73 (дд, J=8,7, 2,3, 0,8 Гц, 1Н), 7,66 (дд, J=7,6, 1,4 Гц, 1Н), 7,52 (д, J=8,7 Гц, 1Н), 7,39 (дд, J=7,7 Гц, 1Н), 5,38 (с, 2Н), 3,56 (т, J=6,7 Гц, 2Н), 3,07-2,99 (м, 4Н).
ПРИМЕР 59
Синтез Соединения 59-1
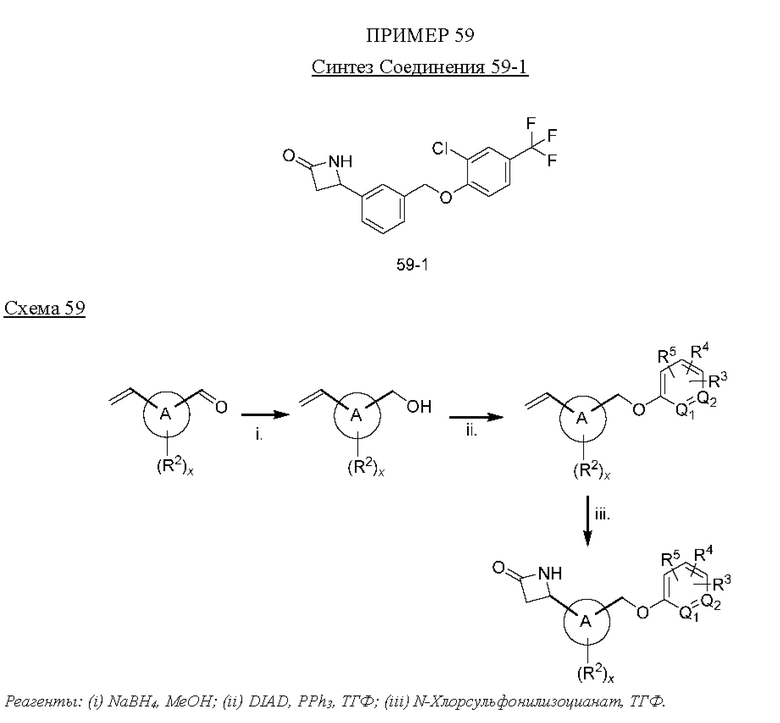
Стадия 59-1. Синтез (3-винилфенил)метанола (INT 59-1)
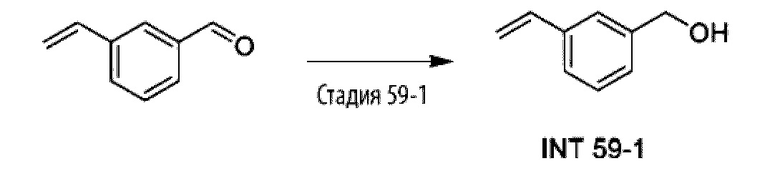
Медленно добавляли NaBH4 (327 мг, 8,66 ммоль) к раствору 3-винилбензальдегида (1,00 мл, 7,87 ммоль) в МеОН (20 мл) при 22°С в атмосфере N2. Смесь перемешивали при 22°С в течение 1 ч и концентрировали при пониженном давлении. Остаток очищали хроматографией на SiO2 (ЭА/гексаны), получая 1,05 г (99%) (3-винилфенил)метанола (INT 59-1). PX-MC-ESI (m/z) масса не наблюдается, tR = 2,00 мин (Метод 13). 1Н ЯМР (400 МГц, CDCl3) δ 7,42 (с, 1Н), 7,37-7,30 (м, 2Н), 7,26 (д, J=3,1 Гц, 1Н), 6,73 (дд, J=17,6, 10,9 Гц, 1Н), 5,78 (дд, J=17,6, 0,9 Гц, 1Н), 5,27 (дд, J=10,9, 0,9 Гц, 1Н), 4,70 (с, 2Н), 1,67 (с, 1Н).
Стадия 59-2. Синтез 2-хлор-4-(трифторметил)-1-((3-винилбензил)окси)бензола (INT 59-2)
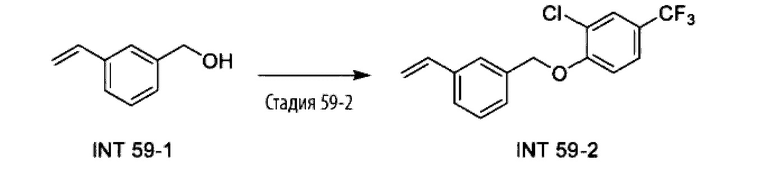
Добавляли DIAD (1,29 мл, 6,56 ммоль) по каплям к смеси INT 59-1 (800 мг, 5,96 ммоль), 2-хлор-4-(трифторметил)фенола (793 мл, 5,93 ммоль) и PPI13 (2,35 г, 8,94 ммоль) в ТГФ (15,0 мл) при 22°С в атмосфере N2 Смесь перемешивали при 22°С в течение 6 ч и концентрировали. Остаток очищали хроматографией на SiO2 (ЭА/гексаны), получая 1,73 г (93%) 2-хлор-4-(трифторметил)-1-((3-винилбензил)окси)бензола (INT 59-2) в виде маслянистой жидкости. PX-MC-ESI (m/z) масса не наблюдается, tR = 2,95 мин (Метод 13). 1Н ЯМР (500 МГц, CDCl3) δ 7,66 (дд, J=2,3, 0,7 Гц, 1Н), 7,49 (с, 1Н), 7,48-7,43 (м, 1Н), 7,41-7,35 (м, 3Н), 7,06-6,99 (м, 1Н), 6,74 (дд, J=17,6, 10,9 Гц, 1Н), 5,78 (дд, J=17,6, 0,9 Гц, 1Н), 5,29 (дд, J=10,9, 0,8 Гц, 1Н), 5,21 (с, 2Н).
Стадия 59-3. Синтез 4-(3-((2-хлор-4-(трифторметил)фенокси)метил)фенил)азетидин-2-она (Соединение 59-1)
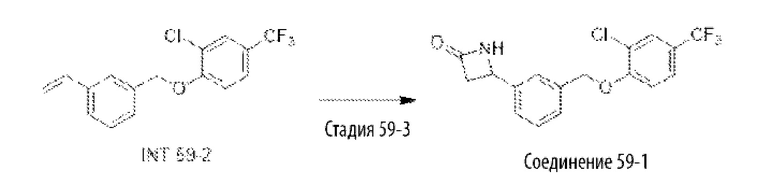
Добавляли N-хлорсульфонилизоцианат (578 мкл, 6,64 ммоль) в течение 10 минут к раствору INT 59-2 (1,73 г, 5,53 ммоль) в ТГФ (5,00 мл) при 22°С в атмосфере N2. Смесь перемешивали при 22°С в течение 16 часов. Смесь добавляли в течение 20 минут к интенсивно перемешиваемой смеси H2O (10,0 мл), карбоната натрия (1,93 г, 18,3 ммоль) и сульфита натрия (1,05 г, 8,30 ммоль) при 0°С. Смесь перемешивали при 22°С в течение 2 часов. Смесь подкисляли водн. 1М HCl (рН ~5) и разбавляли ЭА (100 мл). Водную фазу экстрагировали ЭА (3×50,0 мл) и объединенные органические слои осушали (MgSO4), фильтровали и концентрировали. Остаток очищали обращенно-фазовой хроматографией (Н2О (+ 0,1% муравьиной кислоты) и MeCN (50-100%)), получая 189 мг (10%) 4-(3-((2-хлор-4-(трифторметил)фенокси)метил)фенил)азетидин-2-она (Соединение 59-1) в виде твердого вещества. ЖХ-MC-ESI (m/z) вычислено для C17H13ClF3NO2: 355,06; найдено 356,07 (М+Н)+, tR = 4,93 мин (Метод 12). 1Н ЯМР (500 МГц, CDCl3) δ 8,41 (с, 1Н), 7,86 (дд, J=2,3, 0,7 Гц, 1Н), 7,75-7,67 (м, 1Н), 7,50 (д, J=1,9 Гц, 1Н), 7,48-7,38 (м, 3Н), 7,36 (дт, J=6,8, 2,1 Гц, 1Н), 5,33 (с, 2Н), 4,67 (дд, J=5,3, 2,5 Гц, 1Н), 3,36 (ддд, J=14,6, 5,3, 2,2 Гц, 1Н), 2,67 (ддд, J=14,6, 2,5, 1,0 Гц, 1Н).
ПРИМЕР 60
Синтез Соединения 60-1
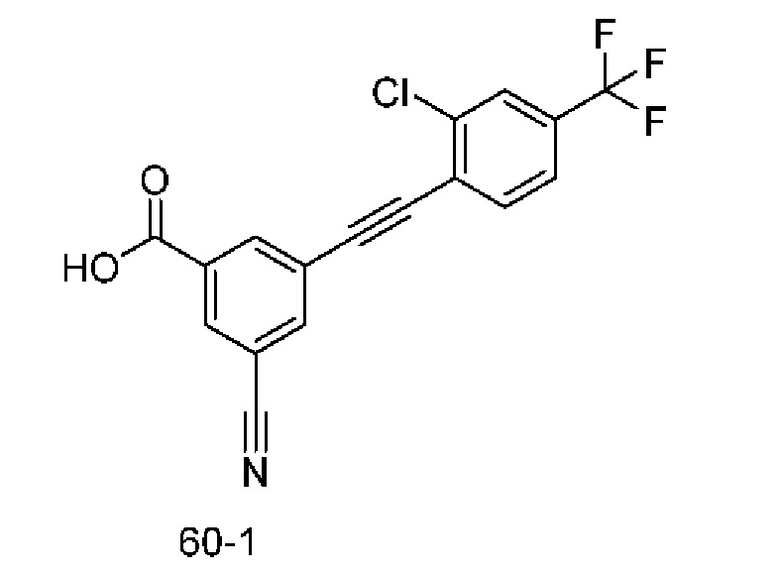
Схема 60
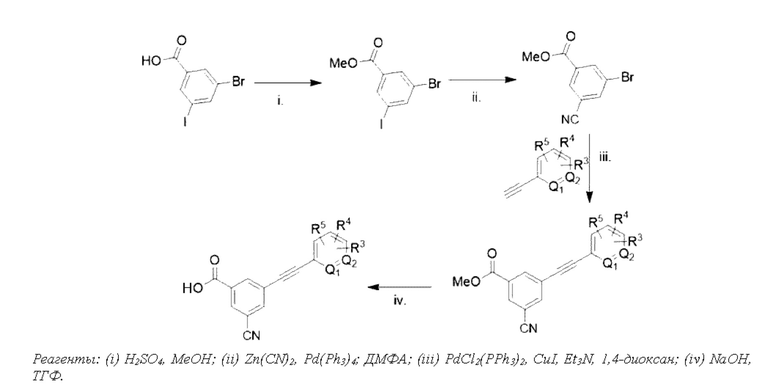
Стадия 60-1. Синтез метил-3-бром-5-йодбензоата (INT 60-1)
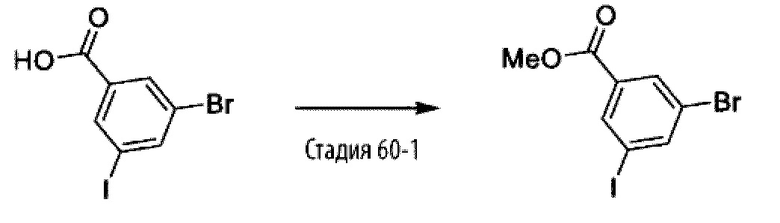
H2SO4 (600 мкл, 11,3 ммоль) добавляли к раствору 3-бром-5-йод бензойной кислоты (10,0 г, 30,6 ммоль) в МеОН (65 мл). Смесь перемешивали при 75°С в течение 18 часов. Смесь охлаждали до 22°С и концентрировали. Остаток разбавляли ЭА (100 мл), промывали насыщ. водн. NaHCO3 (100 мл), осушали (Na2SO4), фильтровали и концентрировали, получая 9,90 г (95%) метил-3-бром-5-йодбензоата(INT 60-1) в виде твердого вещества. ЖХ-MC-ESI (m/z) вычислено для C8H6BrIO2: 339,86; найдено 339,6 (М-Н)+, tR = 2,72 мин (Метод 13).
Стадия 60-2. Синтез метил-3-бром-5-цианобензоата (INT 60-2)
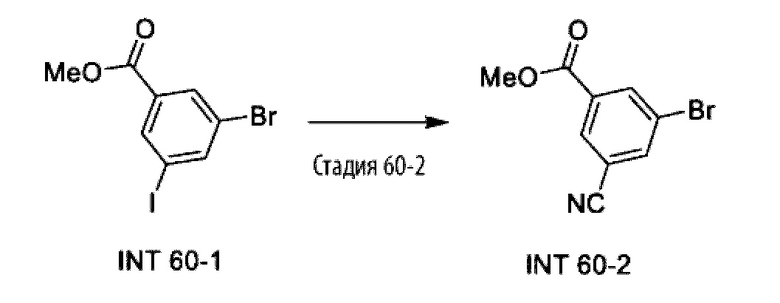
Цианид цинка (1,76 г, 15,0 ммоль) и Pd(PPh3)4 (2,88 г, 2,49 ммоль) добавляли к раствору INT 60-1 (8,50 г, 2,49 ммоль) в ДМФА (60 мл). Смесь перемешивали при 80°С в течение 2 часов. Смесь охлаждали до 22°С и концентрировали. Остаток разбавляли ЭА (100 мл). Органический слой промывали H2O (3×50,0 мл) и рассолом (150 мл), осушали (Na2SO4), фильтровали и концентрировали. Остаток очищали хроматографией на SiO2 (ЭА/гексаны) с получением 3,00 г (50%) метил-3-бром-5-цианобензоата (INT 60-2) в виде твердого вещества. ЖХ-MC-ESI (m/z): масса не наблюдается, tR = 2,38 мин (Метод 13).
Стадия 60-3. Синтез метил-3-((2-хлор-4-(трифторметил)фенил)этинил)-5-цианобензоата (TNT 60-3)
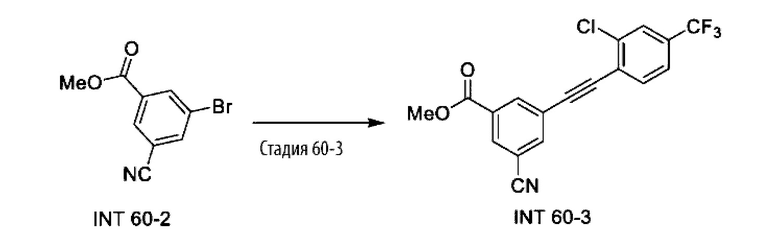
2-Хлор-1-этинил-4-(трифторметил)бензол (551 мкл, 1,87 ммоль), PdCl2(PPh3)2 (132 мг, 0,19 ммоль) и CuI (17,9 мг, 0,094 ммоль) добавляли в раствор INT 60-2 (225 мг, 0,94 ммоль) в 1,4-диоксане (2 мл) и Et3N (2,0 мл). Смесь перемешивали при 80°С в течение 24 часов. Смесь охлаждали до 22°С и разбавляли водн. насыщ. NH4Cl (20 мл). Водную фазу экстрагировали ЭА (3×50 мл), и объединенные органические слои концентрировали. Остаток очищали хроматографией на SiO2 (ЭА/гексаны) с получением 250 мг (73%) метил-3-((2-хлор-4-(трифторметил)фенил)этинил)-5-цианобензоата (INT 60-3) в виде твердого вещества. ЖХ-MC-ESI (m/z) вычислено для C18H9ClF3NO2: 363,03; найдено 365,5 (М+Н)+, tR = 2,92 мин (Метод 13). 1Н ЯМР (400 МГц, CDCl3) δ 8,42 (т, J=1,5 Гц, 1Н), 8,30 (т, J=1,4 Гц, 1Н), 8,00 (т, J=1,4 Гц, 1Н), 7,72 (с, 1Н), 7,68 (д, J=8,1 Гц, 1Н), 7,54 (д, J=7,6 Гц, 1Н), 3,99 (с, 3Н).
Стадия 60-4. Синтез 3-((2-хлор-4-(трифторметил)фенил)этинил)-5-цианобензойной кислоты (Соединение 60-01)
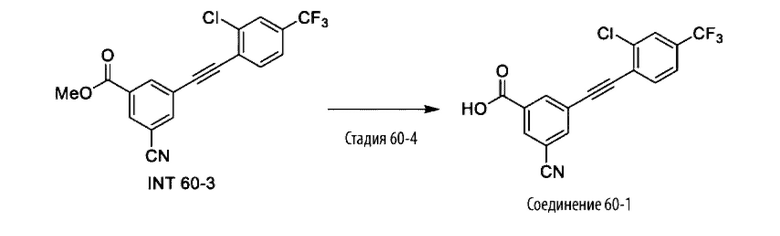
Водный 2М NaOH (165 мкл, 0,330 ммоль) добавляли к раствору INT 60-3 (60,0 мг, 0,165 ммоль) в ТГФ (2 мл) при 22°С. Смесь перемешивали при 22°С в течение 12 ч и концентрировали. Остаток разбавляли Н2О (10,0 мл) и подкисляли водн. 2М HCl (рН ~2). Водную фазу экстрагировали ЭА (3×20,0 мл), и объединенные органические слои осушали (Na2SO4), фильтровали и концентрировали. Остаток очищали хроматографией на SiO2 (МеОН / ДХМ), получая 55,0 мг (95%) 3-((2-хлор-4-(трифторметил)фенил)этинил)-5-цианобензойной кислоты (Соединение 60-1) в виде твердого вещества. ЖХ-MC-ESI (m/z) вычислено для C17H7ClF3NO2: 349,01; найдено 348,49 (М-Н)+, tR = 4,55 мин (Метод 12). 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,37-8,32 (м, 2Н), 8,30 (с, 1Н), 8,08 (с, 1Н), 7,97 (д, J=8,0 Гц, 1Н), 7,82 (дд, J=8,1, 1,1 Гц, 1Н).
ПРИМЕР 61
Синтез Соединения 61-1
Стадия 61-1. Синтез метил-3-(2-(2-хлор-4-(трифторметил)фенил)этил)-5-цианобензоата (INT 61-1)
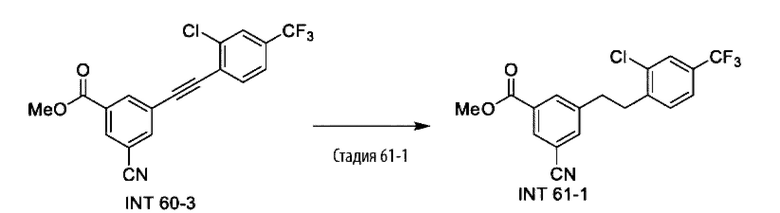
INT 60-3 (100 мг, 0,275 ммоль) и Pd/C (100 мг, 0,0940 ммоль) в ЭА (10,0 мл) перемешивали в атмосфере водорода (1 атм) при 22°С в течение 4 часов. Смесь фильтровали через целит, промывали ЭА (100 мл), и фильтрат концентрировали. Остаток очищали хроматографией на SiO2 (ЭА/гексаны) с получением 101 мг (100%) метил-3-[2-[2-хлор-4-(трифторметил)фенил]этил]-5-цианобензоата (INT 61-1) в виде твердого вещества. ЖХ-МС-ESI (m/z) вычислено для C18H13ClF3NO2: 367,06; найдено 367,3 (МН+, tR = 2,85 мин (Метод 13).
Стадия 61-2. Синтез 3-(2-хлор-4-(трифторметил)фенетил)-5-цианобензойной кислоты (Соединение 61-2)
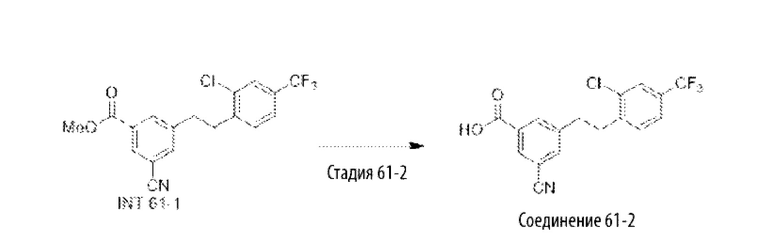
Раствор 2М NaOH (197 мкл, 0,156 ммоль) добавляли к раствору INT 61-1 (60,0 мг, 0,156 ммоль) в ТГФ (2 мл). Смесь перемешивали при 22°С в течение 12 ч и концентрировали. Остаток подкисляли 2М HCl (рН ~2) и разбавляли Н2О (10 мл). Водную фазу экстрагировали ЭА (3×20 мл), и объединенные органические слои осушали (Na2SO4), фильтровали и концентрировали. Остаток очищали обращенно-фазовой хроматографией (H2O (+ 0,1% муравьиной кислоты) и ACN) с получением 47,0 мг (85%) 3-(2-хлор-4-(трифторметил)фенетил)-5-цианобензойной кислоты (Соединение 61-2) в виде твердого вещества. ЖХ-MC-ESI (m/z) вычислено для C17H11ClF3NO2: 353,04; найдено 352,1 (М-Н)+, tR = 4,73 мин (Метод 12). 1Н ЯМР (500 МГц, ДМСО-d6) δ 13,54 (с, 1Н), 8,13 (т, J=1,5 Гц, 1Н), 8,05 (т, J=1,6 Гц, 1Н), 7,98 (т, J=1,6 Гц, 1Н), 7,83 (д, J=1,2 Гц, 1Н), 7,66 (дд, J=8,0, 1,3 Гц, 1Н), 7,58 (д, J=8,0 Гц, 1Н), 3,07 (дкв, J=9,8, 6,3 Гц, 4Н).
ПРИМЕР 62
Синтез Соединения 62-1
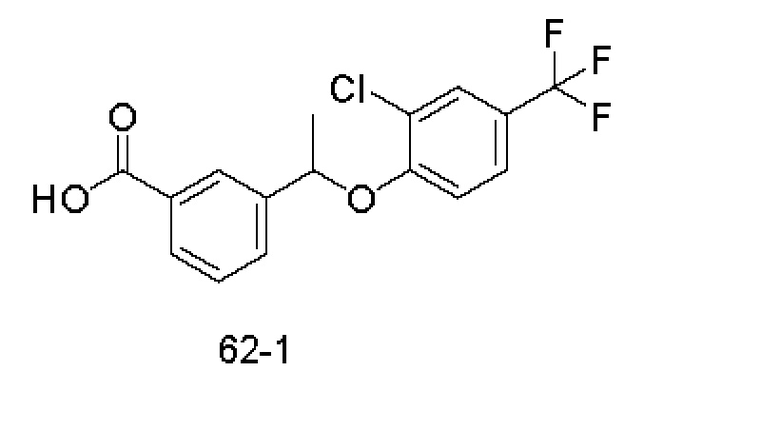
Стадия 62-1. Синтез метил-3-(1-тдроксизтил)бензоата (INT 62-1)
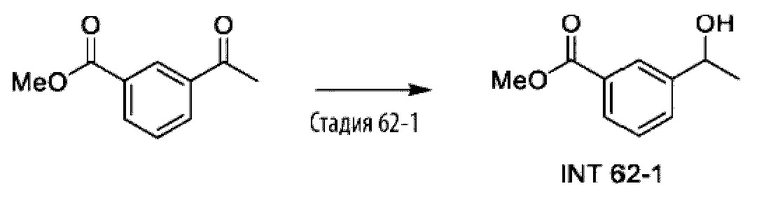
Добавляли NaBH4 (752 мг, 19,9 ммоль) к смеси метил-3-ацетилбензоата (1,18 г, 6,62 ммоль) в EtOH (15,0 мл) при 22°С. Смесь перемешивали при 0°С в течение 30 минут и при 22°С в течение 1 часа. Смесь разбавляли насыщ. водн. NH4Cl (30,0 мл). Водную фазу экстрагировали ЭА (3×30,0 мл), и объединенные органические слои осушали над Na2SO4, фильтровали и концентрировали. Остаток очищали хроматографией на SiO2 (ЭА/гексаны) с получением 830 мг (70%) метил-3-(1-гидроксиэтил)бензоата (INT 62-1) в виде твердого вещества. PX-MC-ESI (m/z) масса не наблюдается, tR = 1,93 мин (Метод 12). 1Н ЯМР (400 МГц, CDCl3) δ 8,05 (тт, J=1,8, 0,6 Гц, 1Н), 7,98-7,92 (м, 1Н), 7,59 (дддд, J=7,7, 1,8, 1,2, 0,6 Гц, 1Н), 7,43 (тт, J=7,7, 0,4 Гц, 1Н), 4,97 (кв, J=6,5 Гц, 1Н),3,92 (с, 3Н), 1,84 (с, 1Н), 1,52 (д, J=6,5 Гц, 3Н).
Стадия 62-2. Синтез метил-3-(1-(2-хлор-4-(трифторметил)фенокси)этил)бензоата (TNT 62-2)
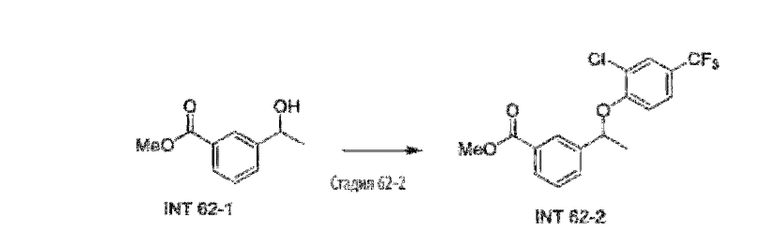
Добавляли DIAD (981 мкл, 4,98 ммоль) к смеси INT 62-1 (816 мг, 4,53 ммоль), 2-хлор-4-(трифторметил)фенола (886 мг, 4,51 ммоль) и PPh3 (1,78 г, 6,79 ммоль) в ТГФ (25,0 мл) при 22°С. Смесь перемешивали при 22°С в течение 3 часов. Смесь концентрировали и остаток очищали хроматографией на SiO2 (ЭА/гексан), получая 1,40 г (86%) метил-3-(1-(2-хлор-4-(трифторметил)фенокси)этил)бензоат (INT 62-2) в виде твердого вещества. ЖХ-MC-ESI (m/z) вычислено для C17H14ClF3O3: 358,06; найдено 357,06 (МН+, tR = 2,87 мин (Метод 13).
Стадия 62-3. Синтез 3-(1-(2-хлор-4-(трифторметил)фенокси)этил)бензойной кислоты (Соединение 62-1)
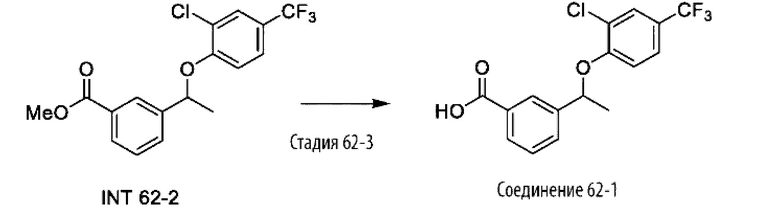
Добавляли 2М NaOH (4,68 ммоль, 2,34 мл) к раствору INT 62-2 (1,40 г, 3,90 ммоль) в МеОН (12 мл) и ТГФ (12 мл) при 22°С. Через 12 ч смесь концентрировали и остаток разбавляли Н2О (10,0 мл) и 2М HCl (рН ~4). Водную фазу экстрагировали ЭА (3×25,0 мл). Объединенные органические слои осушали (Na2SO4), фильтровали и концентрировали, а остаток очищали хроматографией на SiO2 (МеОН/ДХМ), получая 1,34 г (99%) 3-(1-(2-хлоро)-4-(трифторметил)фенокси)этил)бензойной кислоты (Соединение 62-1) в виде твердого вещества. ЖХ-MC-ESI (m/z) вычислено для C16H12ClF3O3: 344,04; найдено 343,04 (МН+, tR = 5,17 мин (Метод 12). 1Н ЯМР (400 МГц, ДМСО-d6) δ 13,04 (с, 1Н), 8,03 (т, J=1,6 Гц, 1Н),7,90-7,84 (м, 1Н), 7,82 (д, J=2,0 Гц, 1Н), 7,71-7,64 (м, 1Н), 7,60-7,54 (м, 1Н), 7,50 (т, J=7,7 Гц, 1Н), 7,24 (д, J=8,7 Гц, 1Н), 5,89 (кв, J=6,3 Гц, 1Н), 1,62 (д, J=6,3 Гц, 3Н).
ПРИМЕР 63
Синтез Соединения 63-1
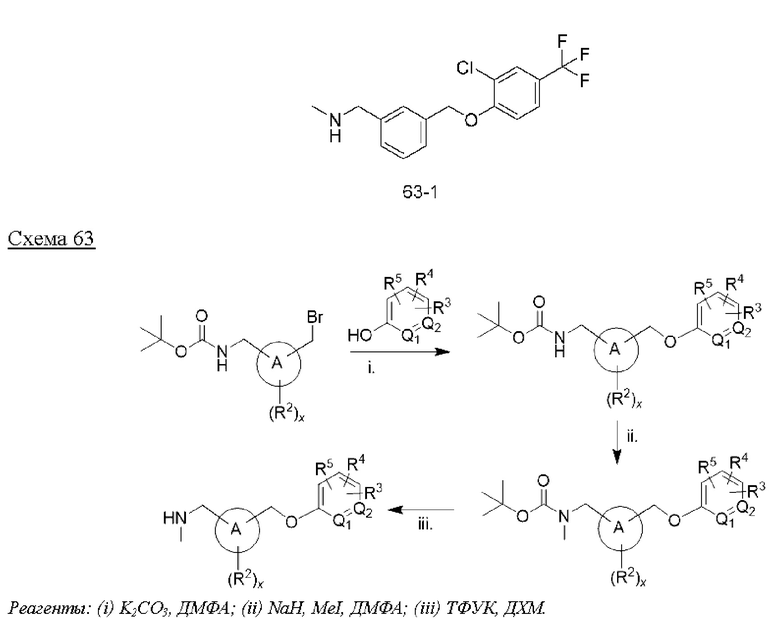
Стадия 63-1. Синтез трет-бутил-N-(3-((2-хлор-4-(трифторметил)фенокси)метил)фенил)метил)карбамата TINT 63-1)
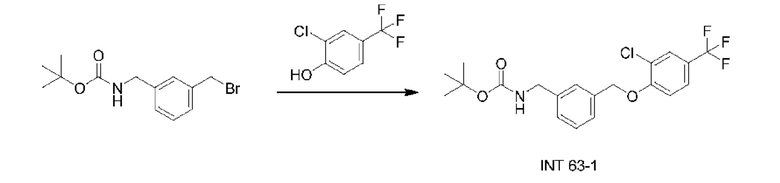
Добавляли трет-бутил-N-((3-(бромметил)фенил)метил)карбамат (100 мг, 0,333 ммоль) к смеси 2-хлор-4-(трифторметил)фенола (50,0 мкл, 0,366 ммоль) и K2CO3 (51,0 мг, 0,366 ммоль) в ДМФА (1 мл) при 22°С в атмосфере N2. Смесь перемешивали при 40°С в течение 16 часов. Смесь разбавляли Н2О (20 мл) и водный слой экстрагировали ДХМ (3×20 мл). Объединенные органические слои осушали (MgSO4), фильтровали и концентрировали. Остаток очищали хроматографией на SiO2 (ЭА/гексаны), получая 122 мг (88%) трет-бутил-N-((3-((2-хлор-4-(трифторметил)фенокси)метил)фенил)метил)карбамата (INT 63-1). ЖХ-МС-ESI (m/z) вычислено для C20H21ClF3NO3: 415,12; найдено 414,17 (М+Н)+, tR = 2,86 мин (Метод 13). 1Н ЯМР (400 МГц, CDCl3) δ 7,66 (д, J=2,3 Гц, 1Н), 7,45 (ддд, J=8,6, 2,2, 0,9 Гц, 1Н), 7,40-7,33 (м, 3Н), 7,28-7,27 (м, 1Н), 7,01 (д, J=8,6 Гц, 1Н), 5,20 (с, 2Н), 4,86 (с, 1Н), 4,34 (д, J=6,0 Гц, 2Н), 1,46 (с, 9Н).
Стадия 63-2. Синтез трет-бутил-N-((3-((2-хлор-4-(трифторметил)фенокси)метил)фенил)метил)-N-метилкарбамата (INT 63-2)
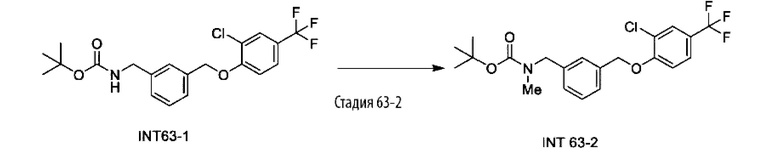
Добавляли NaH (60% мас., 12,2 мг, 0,317 ммоль) к раствору INT 63-1 (120 мг, 0,289 ммоль) в ТГФ (2 мл) при 22°С в атмосфере N2 Смесь перемешивали при 22°С в течение 15 минут. Добавляли йодметан (21,6 мкл, 0,346 ммоль), и смесь перемешивали при 50°С в течение 24 часов. Смесь концентрировали, и остаток очищали хроматографией на SiO2 (ЭА/гексаны), получая 71,2 мг (57%) трет-бутил-N-((3-((2-хлор-4-(трифторметил)фенокси)метил)фенил)метил)-N-метилкарбамата (INT 63-2). ЖХ-MC-ESI (m/z) вычислено для C21H23ClF3NO3: 429,13; найдено 428,17 (М-Н)+, tR = 3,29 мин (Метод 13). 1Н ЯМР (500 МГц, CDCl3) δ 7,66 (д, J=2,2 Гц, 1Н), 7,48-7,43 (м, 1Н), 7,38-7,33 (м, 2Н), 7,30 (с, 1Н), 7,21 (с, 1Н), 7,01 (д, J=8,6 Гц, 1Н), 5,20 (с, 2Н), 4,45 (с, 2Н), 2,82 (д, J=26,0 Гц, 3Н), 1,47 (д, J=15,5 Гц, 9Н).
Стадия 63-3. Синтез 1-(3-((2-хлор-4-(трифторметил)фенокси)метил)фенил)-N-метилметанамина (Соединение 63-1)
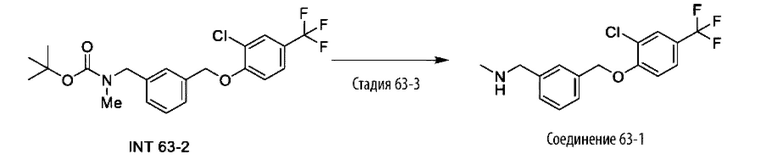
Раствор ТФУК (500 мкл) добавляли по каплям к раствору INT 63-2 (70,0 мг, 0,745 ммоль) в ДХМ (1,5 мл). Смесь перемешивали при 22°С в течение 5 часов. Смесь подщелачивали водн. 1М NaOH (10 мл) и перемешивали при 22°С в течение 15 минут. Водную фазу экстрагировали ДХМ (3×20 мл), и объединенные органические слои промывали рассолом (20 мл), осушали (MgSO4), фильтровали и концентрировали. Остаток очищали обращенно-фазовой хроматографией (H2O (+ 0,03% карбоната аммония) / MeCN) с получением свободной формы в виде маслянистой жидкости. Добавляли HCl (2М в Et2O, 121 мкл, 0,121 ммоль) к раствору свободной формы (40,0 мг, 0,121 ммоль) в Et2O (2 мл) при 22°С. Через 10 мин смесь концентрировали, получая 42,2 мг (70%) 1-(3-((2-хлор-4-(трифторметил)фенокси)метил)фенил)-N-метилметанамина (Соединение 63-1). ЖХ-MC-ESI (m/z) вычислено для C16H15ClF3NO: 329,08; найдено 330,09 (М+Н)+, tR = 3,87 мин (Метод 12). 1Н ЯМР (500 МГц, CDCl3) δ 8,98 (с, 2Н), 7,88 (д, J=2,5 Гц, 1Н), 7,71 (дд, J=8,7, 2,3 Гц, 1Н), 7,59 (д, J=1,5 Гц, 1Н), 7,54 (квд, J=4,0, 3,5, 1,6 Гц, 1Н), 7,53-7,50 (м, 2Н), 7,45 (д, J=8,7 Гц, 1Н), 5,34 (с, 2Н), 4,15 (с, 2Н), 2,56 (с, 3Н).
ПРИМЕР 64
Синтез Соединения 64-1
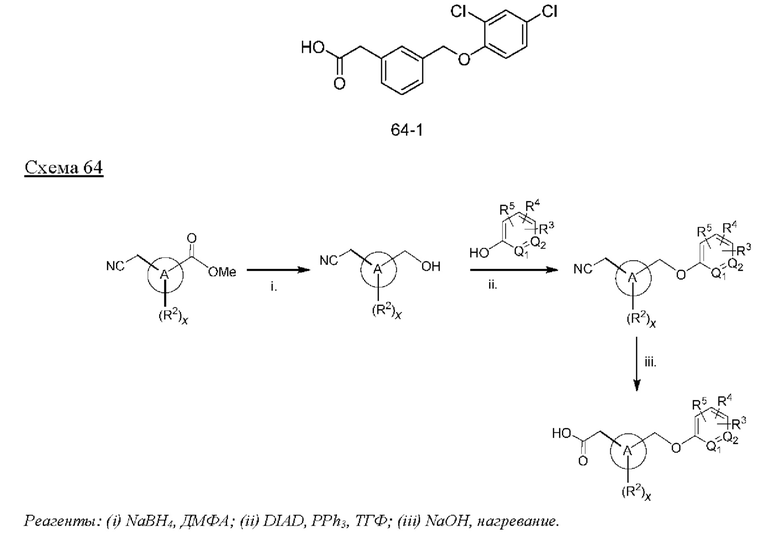
Стадия 64-1. Синтез 2-(3-(гидроксиметил)фенил)ацетонитрила (INT 64-1)
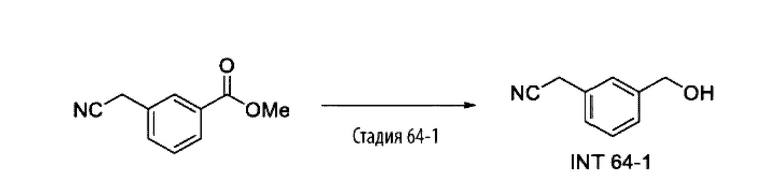
К раствору метил-3-(цианометил)бензоата (3 г, 17,1 ммоль) в ТГФ (150 мл) добавляли NaBH4 (1,3 г, 34 ммоль) δ порциями. Смесь нагревали до 80°С и перемешивали 30 мин. После охлаждения до комнатной температуры добавляли по каплям МеОН и смесь перемешивали при 80°С в течение 30 минут и при комнатной температуре в течение 16 часов. Раствор гасили Н2О (30 мл) и концентрировали. Полученный остаток разбавляли Н2О и экстрагировали ЭА. Органический слой промывали Н2О, затем рассолом, осушали (Na2SO4), фильтровали и концентрировали. Полученный сырой остаток очищали хроматографией на SiO2 (ЭА/гексан), получая 1,0 г (40%) 2-(3-(гидроксиметил)фенил)ацетонитрила (INT 64-1). ЖХ-MC-ESI (m/z), вычислено для C9H9NO: 147,07; найдено 170,3 (M+H20)+, tR = 2 мин (Метод 11).
Стадия 64-2. Синтез 2-(3-((2,4-дихлорфенокси)метил)фенил)ацетонитрила (INT 64-2)
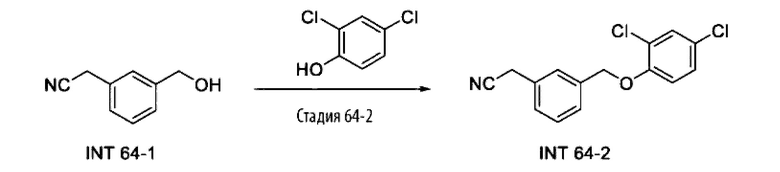
К раствору DIAD (495 мг, 2,4 ммоль) в ТГФ (10 мл) добавляли PPh3 (641 мг, 2,4 ммоль) и перемешивали в течение 10 минут. Затем добавляли раствор INT 64-1 (300 мг, 0,2 ммоль) в ТГФ (5 мл), а затем раствор 2,4-дихлорфенола (332 мг, 2 ммоль) в ТГФ (5 мл). Реакционную смесь перемешивали при комнатной температуре в течение 16 ч, разбавляли ЭА и последовательно промывали насыщенным NaHCO3 (водн.) и рассолом, затем осушали над (Na2SO4), фильтровали и концентрировали. Полученный сырой остаток дважды очищали хроматографией на SiO2 (ЭА/гекс), получая 0,21 г (35%) 2-(3-((2,4-дихлорфенокси)метил)фенил)ацетонитрила (INT 64-2). 1Н ЯМР (400 МГц, CDCl3) δ 7,27-7,5 (м, 5Н), 7,16 (д, J=8 Гц, 1Н), 6,87 (д, J=8 Гц, 1Н), 5,13 (с, 2Н), 3,77 (с, 2Н).
Стадия 64-3. Синтез 2-(3-((2,4-дихлорфенокси)метил)фенил)уксусной кислоты (Соединение 64-1)
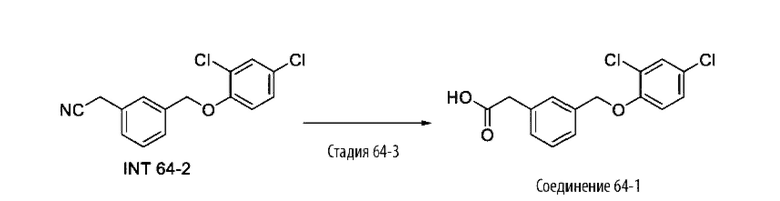
INT 64-2 (100 мг, 0,34 ммоль) растворяли в растворе NaOH (водный, 2М, 5 мл) и нагревали до 130°С в запаянной пробирке в течение 24 часов. Реакционную смесь подкисляли 1М HCl и экстрагировали ЭА. Органический слой сушили, промывали рассолом, осушали (Na2SO4), фильтровали и концентрировали. Полученный остаток дополнительно сушили в высоком вакууме, получая 20 мг (19%) 2-(3-((2,4-дихлорфенокси)метил)фенил)уксусной кислоты (Соединение 64-1). PX-MC-ESI (m/z) масса не наблюдается, tR = 13,8 мин (Метод 10). 1Н ЯМР (400 МГц, ДМСО-d6) δ 7,60 (с, 1Н), 7,40-7,30 (м, 4Н), 7,30-7,20 (м, 2Н), 5,20 (с, 2Н), 3,59 (с, 2Н).
ПРИМЕР 65
Синтез Соединения 65-1
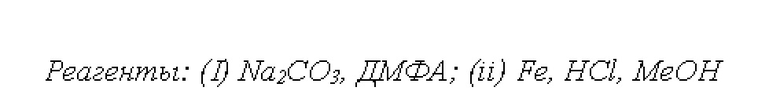
Стадия 65-1. Синтез 2-хлор-1-((3-нитробешил)окси)-4-(трифторметил)бензола (INT 65-1)
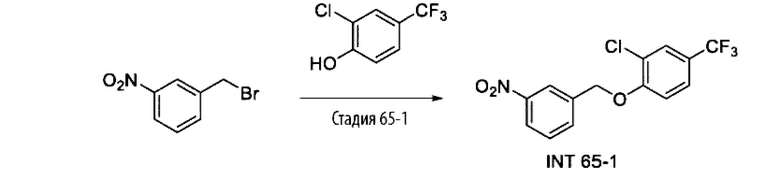
В колбу на 250 мл добавляли 1-(бромметил)-3-нитробензол (1,0 г, 4,37 ммоль), 2-хлор-4-(трифторметил)фенол (858 мг, 4,37 ммоль), Na2CO3 (1,39 г, 13 ммоль) и ДМФА (50 мл). После перемешивания при 50°С в течение 18 часов реакцию гасили H2O и экстрагировали ЭА. Органические слои осушали (Na2SO4), концентрировали и очищали хроматографией на SiO2 (ЭА/гексан) с получением 1,0 г (69%) 2-хлор-1-((3-нитробензил)окси)-4-(трифторметил)бензола (INT 65-1). ЖХ-MC-ESI: масса не наблюдается, tR = 5,66 мин (Метод 11).
Стадия 65-2. Синтез 3-((2-хлор-4-(трифторметил)фенокси)метил)анилина (Соединение 65-1)
Растворяли INT 65-1 (1,0 г, 3,02 ммоль) в МеОН (10 мл) и добавляли избыток 2н. HCl и порошок Fe (210 мг, 3,77 ммоль). Реакционную смесь нагревали до 80°С в течение 18 часов, фильтровали через целит, концентрировали и очищали над SiO2 (ЭА/гексан). Полученный материал дополнительно очищали обращенно-фазовой хроматографией (МеОН/H2O с 0,1% муравьиной кислоты) с получением 800 мг (60%) 3-((2-хлор-4-(трифторметил)фенокси)метил)анилина (Соединение 65-1). ЖХ-MC-ESI (m/z) вычислено для C14H11ClF3NO: 301,05; найдено 302,1 (М+Н)+, fR = 2 мин (Метод 11).
ПРИМЕР 66 Активность MRGPRX4
Клетки НЕК, стабильно трансфицированные для экспрессии MRGPR Х4 человека, поддерживали в инкубаторе при 37°С с 5% СО2 и выращивали в среде DMEM с 10% фетальной бычьей сыворотки (FBS) и по 1% каждого из пирувата натрия, глутамакса, пенициллина/стрептомицина и генетицина. Клетки HEK, стабильно трансфицированные для экспрессии мышиного MRGPR А1, поддерживали в том же инкубаторе и выращивали в среде DMEM с 10% FBS, по 1% пирувата натрия, глутамакса, пенициллина/стрептомицина, генетицина и 2,2 мг/мл гигромицина.
Клетки высевали в 384-луночный аналитический планшет из расчета 20000 клеток на лунку в 12 мкл Opti-MEM и выдерживали в инкубаторе в течение ночи. В день проведения анализа соединения, солюбилизированные в 10 мМ в ДМСО, добавляли с помощью цифрового дозатора Tecan D300E для получения кривой из 10 точек (конечная максимальная концентрация 10 мкМ при серийных разведениях 1:3). Агонисты разводили в буфере для анализа (конечные концентрации 5,7 мМ Трис-HCl, 43 мМ NaCl, 50 мМ LiCl, рН = 8) и добавляли в каждую лунку 2 мкл соответствующего агониста. Конечные концентрации агонистов составляли 10 мкМ билирубина, 20 мкМ дезоксихолевой кислоты или 100 мкМ конъюгированного билирубина (получен от фирмы Lee Biosolutions, № кат. 910-12). Конечные концентрации ДМСО поддерживали постоянными по всему планшету. Планшеты инкубировали в темноте в течение 1 ч при 37°С, а затем в течение 30 минут при комнатной температуре. Стандарты ИФ1 (инозитол-1-фосфат) и реагенты для обнаружения методом HTRF (гомогенная флуоресценция с временным разрешением) добавляли в соответствии с набором IP-One-Gq, приобретенным у фирмы Cisbio (№ кат. 62IPAPEJ), и инкубировали в темноте в течение 1 часа при комнатной температуре. Планшет считывали на планшет-ридере Molecular Devices SpectraMax iD5. Отношение HTRF рассчитывали на основе необработанных данных и отображали графически с использованием GraphPad Prism для расчета значения IC50 для каждого соединения.
Данные об активности выбранных антагонистов MRGPR Х4 (по сравнению с 10 мкМ агониста билирубина) представлены в Таблице 66А. Диапазоны активности обозначены следующим образом: «+++++» обозначает антагонистическую активность <100 нМ; «++++» обозначает антагонистическую активность от 100 до 500 нМ; «+++» обозначает активность от 500 до 1000 нМ; «++» обозначает активность от 1000 до 2500 нМ; и «+» означает активность >2500 нМ.
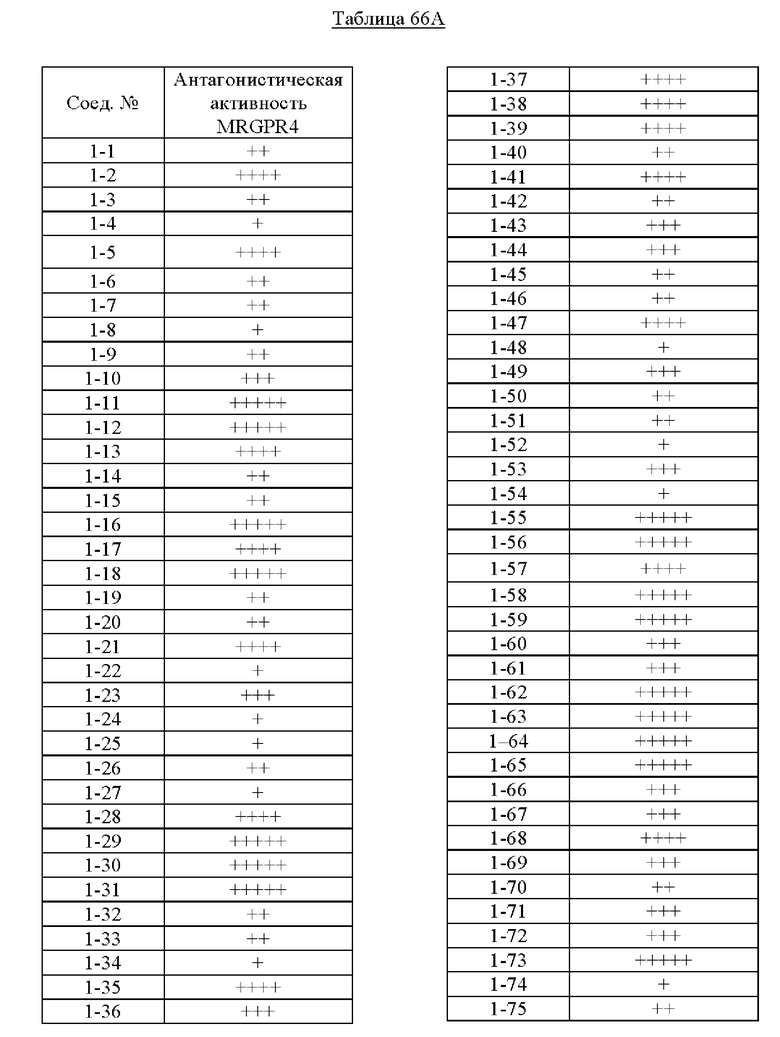
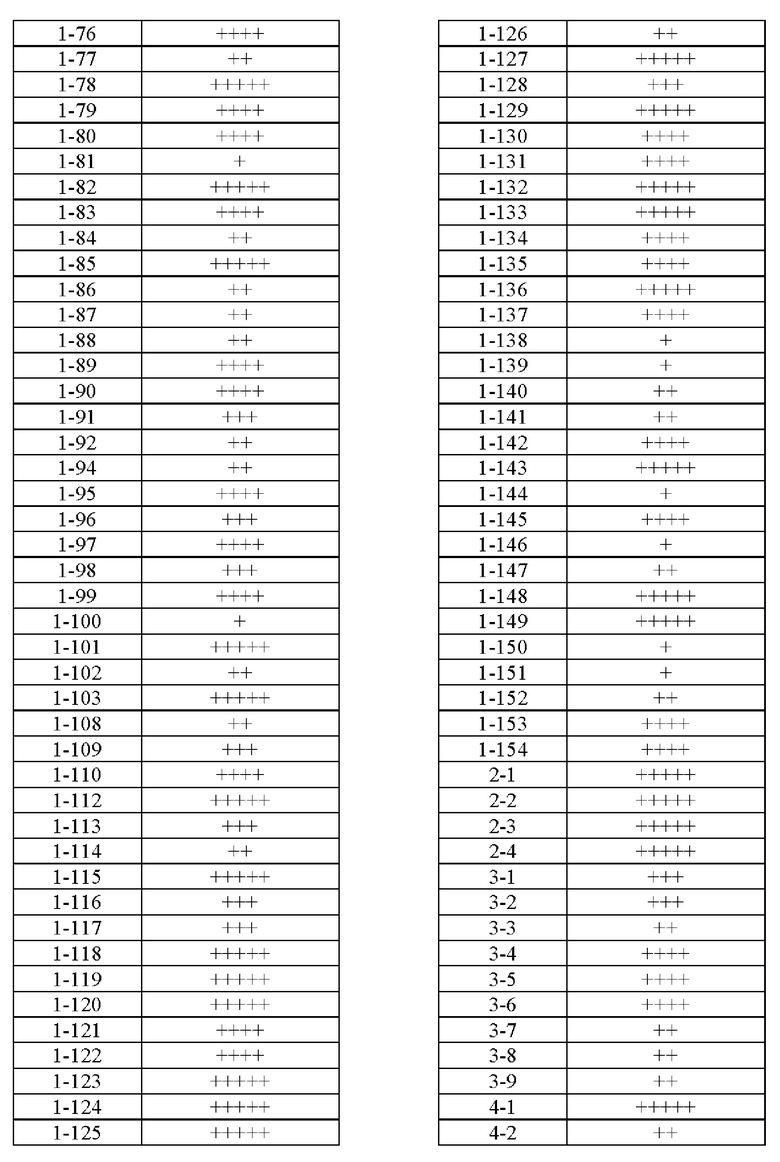
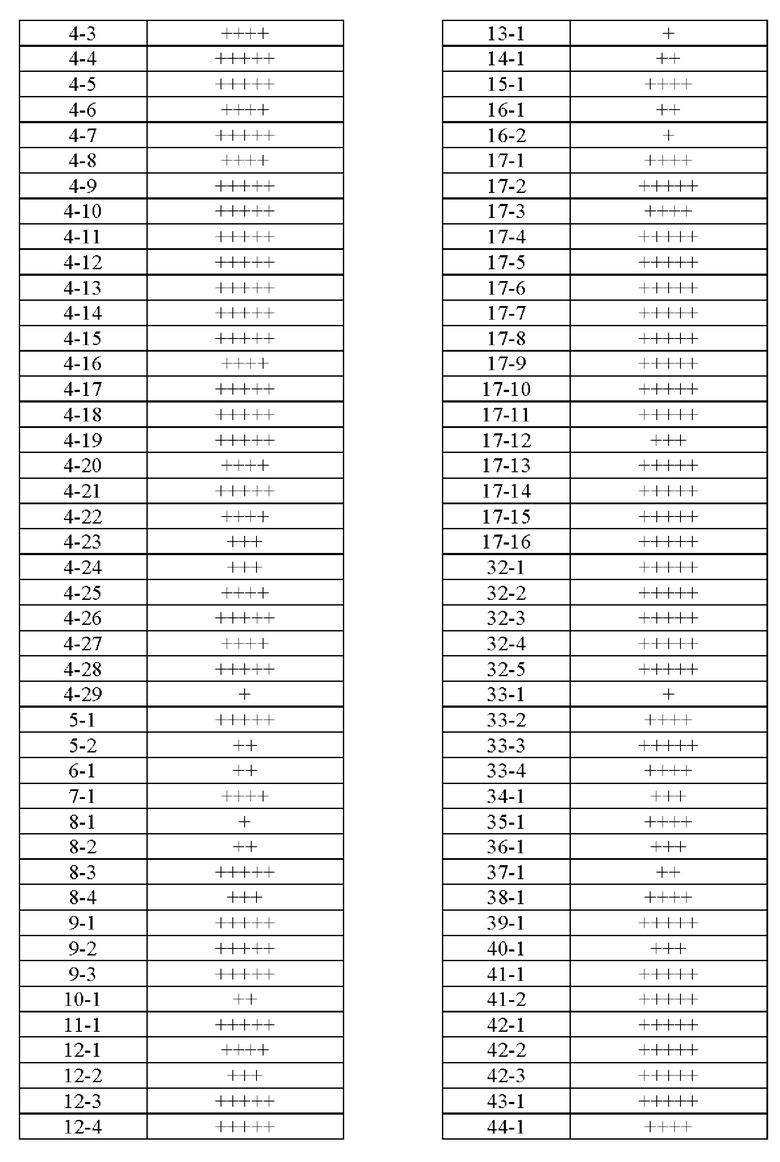
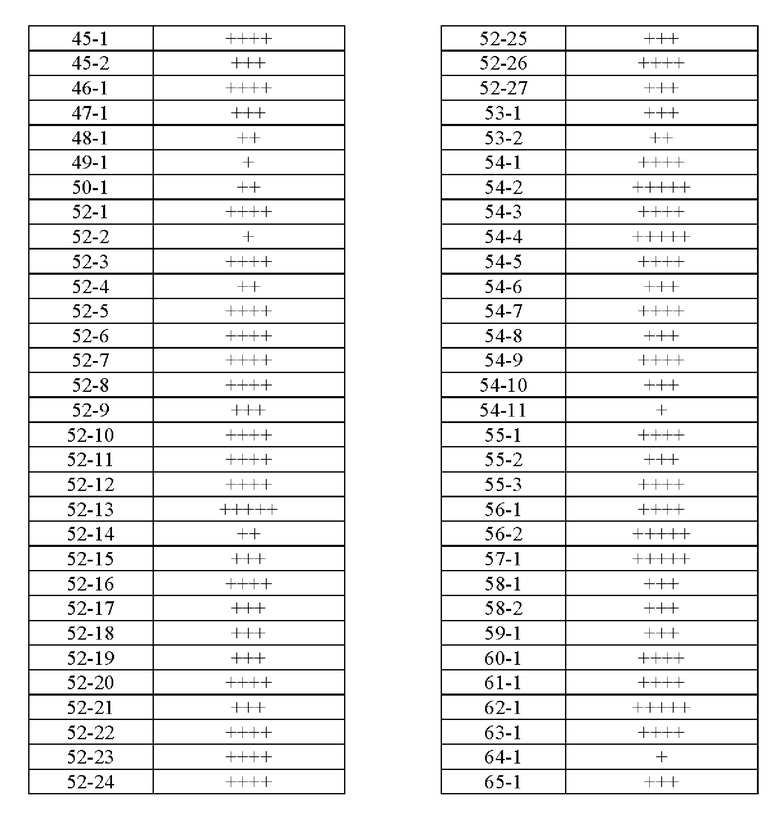
Данные об активности выбранных антагонистов MRGPR Х4 (по сравнению с 10 мкМ агониста билирубина, 20 мкМ дезоксихолевой кислоты, 100 мкМ конъюгированного билирубина, 50 мкМ уробилина, или 20 мкМ обетихолевой кислоты) представлены в Таблице 66 В.
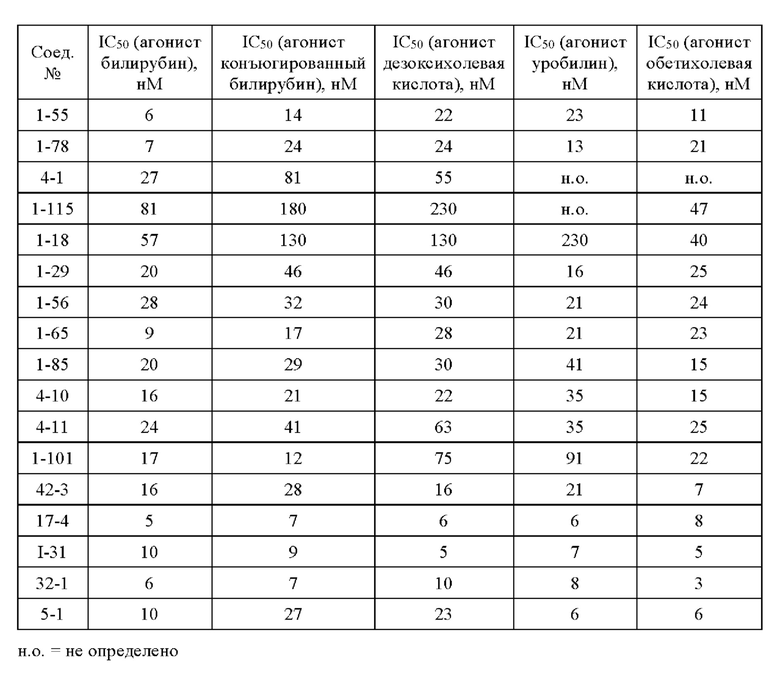
ПРИМЕР 67
Фармакокинетические исследования на мышах
Соединения были приготовлены в 5% ДМСО, 5% солютола (Solutol) и 90% физиологического раствора с фосфатным буфером в концентрации 5 мг/мл, и обычно представляли собой высокодисперсную однородную суспензию. Самцам мышей C57BL/6 (n = 3/соединение) вводили дозу 50 мг/кг каждого соединения через желудочный зонд без голодания. Образцы крови брали из подкожной вены в K2-ЭДТА через 0,25, 0,5, 1, 2, 4, 8 и 24 часа после введения дозы, получали плазму и хранили при ≤60°С до анализа. Подготовка образцов плазмы для анализа проводилась путем осаждения белка с использованием ацетонитрила (включая целекоксиб в качестве внутреннего стандарта) с последующим центрифугированием. Определяли концентрации соединения в извлеченной плазме методом ЖХ-МС/МС с использованием 8-точечной калибровочной кривой, охватывающей диапазон от 1 до 3000 нг/мл. Некомпартментный анализ с использованием Phoenix WinNonlin использовали для оценки фармакокинетических параметров, включая площадь под кривой (ППК), клиренс и период полувыведения. Введенную дозу подтверждали анализом остаточного дозируемого материала с помощью СВЭЖХ-УФ относительно образца для одноточечной калибровки. Результаты этих исследований представлены в Таблице 67.
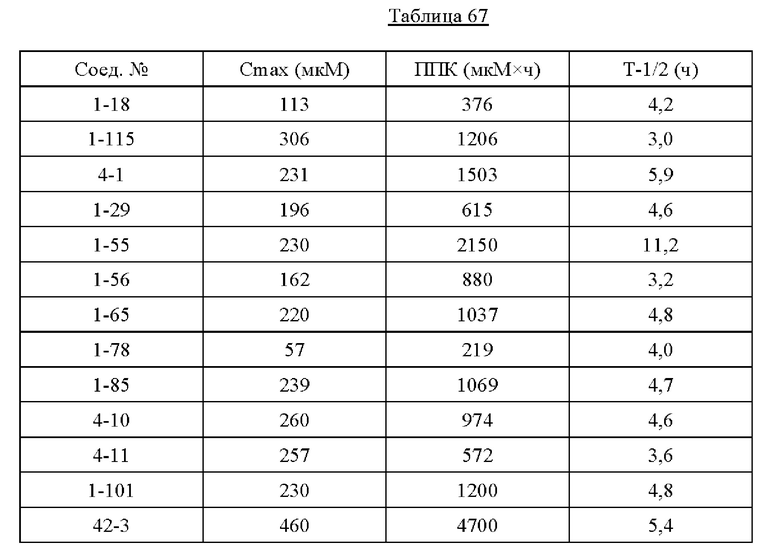
ПРИМЕР 68
Уробилин - сильный агонист MRGPR Х4 и пруритоген
Плазменный уробилин является окислительным продуктом метаболита гема уробилиногена. Уробилиноген является побочным продуктом восстановления билирубина в кишечнике. Часть уробилиногена остается в толстом кишечнике, где он превращается в стеркобилин. Некоторое количество уробилиногена реабсорбируется в кровоток и доставляется в почки, где окисляется до уробилина при контакте с воздухом.
Метаболиты гема (билирубин, биливердин, уробилин, уробилиноген и стеркобилин) анализировали на активацию MRGPR Х4 in vitro. Клетки высевали в 384-луночный аналитический планшет из расчета 20000 клеток на лунку в 12 мкл Opti-MEM и выдерживали в инкубаторе в течение ночи. В день проведения анализа добавляли различные агонисты, солюбилизированные в концентрации 10 мМ в 0,1% NaOH, для получения 10-точечной кривой (конечная максимальная концентрация 10 мМ при серийных разведениях 1:3) с использованием цифрового дозатора Tecan D300E. Агонисты разводили в буфере для анализа (конечные концентрации 5,7 мМ Трис-HCl, 43 мМ NaCl, 50 мМ LiCl, рН = 8) и добавляли в каждую лунку 2 мкл соответствующего агониста. Планшеты инкубировали в темноте в течение 1 ч при 37°С, а затем в течение 30 минут при комнатной температуре. Стандарты ИФ1 (инозитол-1-фосфат) и реагенты для обнаружения методом HTRF (гомогенная флуоресценция с временным разрешением) добавляли в соответствии с набором IP-One-Gq, приобретенным у фирмы Cisbio (№ кат. 62IPAPEJ), и инкубировали в темноте в течение 1 часа при комнатной температуре. Планшет считывали на планшет-ридере Molecular Devices SpectraMax iD5. Отношение HTRF рассчитывали на основе исходных данных и наносили на график с использованием GraphPad Prism для расчета значения IC50 для каждого соединения.
Результаты этого исследования представлены на Фиг. 1. Было показано, что уробилин как минимум в 10 раз более эффективен при активации MRGPR Х4, чем билирубин.
Также была проверена способность уробилина вызывать зуд у мышей дикого типа. Типичные исследования зуда на мышах проводили следующим образом: самцов мышей C57B6J содержали в клетках с несколькими отделениями с нормальным световым циклом (включение света в 6 часов утра, выключение в 6 часов вечера) в условиях контролируемых температуры и влажности. За мышами ухаживали и приучали к испытательным камерам перед тестированием, а затем помещали в отдельные испытательные камеры SCLABA. Через 20 минут мышам перорально вводили носитель (физиологический раствор, рН 7-8). Через 30 минут исследуемый пруритоген (100 мл в физиологическом растворе) или физиологический раствор вводили подкожно (SC) в шею по средней линии за ушами. Записывали видео с момента первой инъекции пруритогена в течение 30 минут с использованием системы SCLABA. Приступы чесания оценивали по кадровым изображениям SCLABA с использованием критериев формы волны 12/45/85/100. Размер группы обычно составлял 9-10 животных на группу.
Как показано на Фиг. 2А, уробилин индуцировал у мышей ответную реакцию чесания дозозависимым образом. Индукцию зуда уробилином также сравнивали с агонистом дезоксихолевой кислотой и агонистом билирубином. Как показано на Фиг. 2В, уробилин является мощным индуктором реакции чесания у мышей.
ПРИМЕР 69
Билирубин и уробилин могут разрушаться под действием света, что снижает их агонистическую активность по отношению к MRGPR Х4
Билирубин и уробилин представляют собой агонисты MRGPR Х4, которые, как было продемонстрировано, являются активными пруритогенами. Было показано, что световая терапия уменьшает зуд у пациентов с холестатическим пруритом, что связывают с индуцированным светом разложением или химической модификацией билирубина. Для дальнейшего изучения вклада световой деградации в снижение агонизма MRGPR Х4 билирубин и уробилин предварительно обрабатывали при разном освещении и измеряли их активность.
Были приготовлены маточные растворы билирубина и уробилина с концентрацией 210 мкМ в 0,1 н. NaOH (водн.). Образцы хранили при комнатной температуре в темноте, в морозильнике при -20°С в темноте, при комнатной температуры на столешнице в условиях нормального лабораторного освещения, или при комнатной температуре под лампой синего света с длиной волны 400 нМ (по аналогии с медицинскими лампами, применяемыми для лечения желтухи). Образцы оценивали через 24 часа и процент оставшегося уробилина и билирубина относительно стандарта в начальный момент времени определяли путем измерения деградации аналита, содержащегося в образцах, определяемого методом тандемной масс-спектроскопии (ЖХ-МС/МС). Образцы (через 24 ч) также оценивали на их способность агонизировать MRGPR Х4.
Наибольшее количество остаточного билирубина (44% от начального момента времени) было обнаружено через 24 часа в маточном растворе, хранившемся в морозильной камере, тогда как при всех других условиях (в темноте при комнатной температуре, с лабораторным освещением при комнатной температуре и с синим светом при комнатной температуре) остаточного билирубина обнаружено не было (Фиг. 3А). Все образцы, которые хранились при комнатной температуре (в темноте, при комнатном освещении и при синем свете), продемонстрировали значительно сниженную агонистическую активность по сравнению с замороженным образцом (Фиг. 3В).
Уробилин показал большую стабильность, чем билирубин, но деградация все равно наблюдалась во всех условиях. Через 24 часа в образце, хранившемся в темноте, количество остаточного уробилина было наибольшим по сравнению с измерением в начальный момент времени (40%), в то время как в образце, хранившемся в темноте в морозильной камере, остаточный уровень составлял только 21%. В образце, хранившемся при комнатной температуре и окружающем освещении, оставалось 34%, но в образце, хранившемся при комнатной температуре с синим светом, остаточный уробилин обнаружен не был, что указывает на более высокую чувствительность к свету этой длины волны (Фиг. 4А). Образец, хранившийся с синим светом, продемонстрировал очень низкую агонистическую активность по сравнению с другими тремя исследованными группами (в темноте при комнатной температуре, в темноте в морозильнике, и при комнатных освещении и температуре), что соответствует результатам измерений остаточного уробилина в этих образцах (Фиг. 4В).
ПРИМЕР 70
Проявление агонизма MRGPR Х4 агонистом ФХР может быть заблокировано типичными антагонистами MRGPR Х4
BAR502, двойной агонист ФХР и GPBAR1, также обладает агонистической активностью (5700 нМ) по отношению к MRGPR Х4. Данные об активности выбранных антагонистов MRGPR Х4 по сравнению с 10 мкМ BAR502 в Таблице 70 демонстрируют диапазон антагонизма от 11 до 48 нМ.
Все патенты США, публикации патентных заявок США, заявки на патенты США, зарубежные патенты, зарубежные патентные заявки и непатентные публикации, упомянутые в данном описании и/или перечисленные в листе данных заявки, включены в данный документ посредством ссылки в полном объеме. Кроме того, термины, используемые в приведенной ниже формуле изобретения, не должны толковаться как ограниченные конкретными вариантами осуществления, раскрытыми в данном описании, и должны включать все возможные варианты осуществления вместе с полным объемом эквивалентов, на которые распространяется такая формула изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОРЫ MAGL | 2017 |
|
RU2754536C1 |
| СОЕДИНЕНИЕ ТРИАЗИНОНА И ИНГИБИТОР КАЛЬЦИЕВЫХ КАНАЛОВ Т-ТИПА | 2013 |
|
RU2645158C2 |
| ТРИЦИКЛИЧЕСКИЕ АЗОТСОДЕРЖАЩИЕ ПРОИЗВОДНЫЕ ИМИДАЗО[4,5-с]ПИРИДИНА, ОБЛАДАЮЩИЕ ИНГИБИРУЮЩЕЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ РЕЦЕПТОРА ГИСТАМИНА 4 (hH4R) | 2012 |
|
RU2628074C2 |
| БЕНЗОКСЕПИНОВЫЕ ИНГИБИТОРЫ PI3 И СПОСОБЫ ПРИМЕНЕНИЯ | 2010 |
|
RU2557658C2 |
| НОВОЕ ПРОИЗВОДНОЕ КУМАРИНА, ОБЛАДАЮЩЕЕ ПРОТИВООПУХОЛЕВОЙ АКТИВНОСТЬЮ | 2007 |
|
RU2428420C2 |
| ПРОИЗВОДНЫЕ АРИЛАМИДОВ В КАЧЕСТВЕ БЛОКАТОРОВ TTX-S | 2011 |
|
RU2535671C1 |
| СПОСОБ И ПРОЦЕСС ПРИГОТОВЛЕНИЯ И ПРОИЗВОДСТВА ДЕЙТЕРИРОВАННОЙ ОМЕГА-ДИФЕНИЛМОЧЕВИНЫ | 2011 |
|
RU2527037C2 |
| НОВЫЙ ИНГИБИТОР AURORA КИНАЗЫ И ЕГО ПРИМЕНЕНИЕ | 2020 |
|
RU2835080C1 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ | 2015 |
|
RU2711502C2 |
| АНТИБИОТИЧЕСКИЕ ПРОИЗВОДНЫЕ 2-ОКСО-ОКСАЗОЛИДИН-3, 5-ДИИЛА | 2012 |
|
RU2616609C2 |
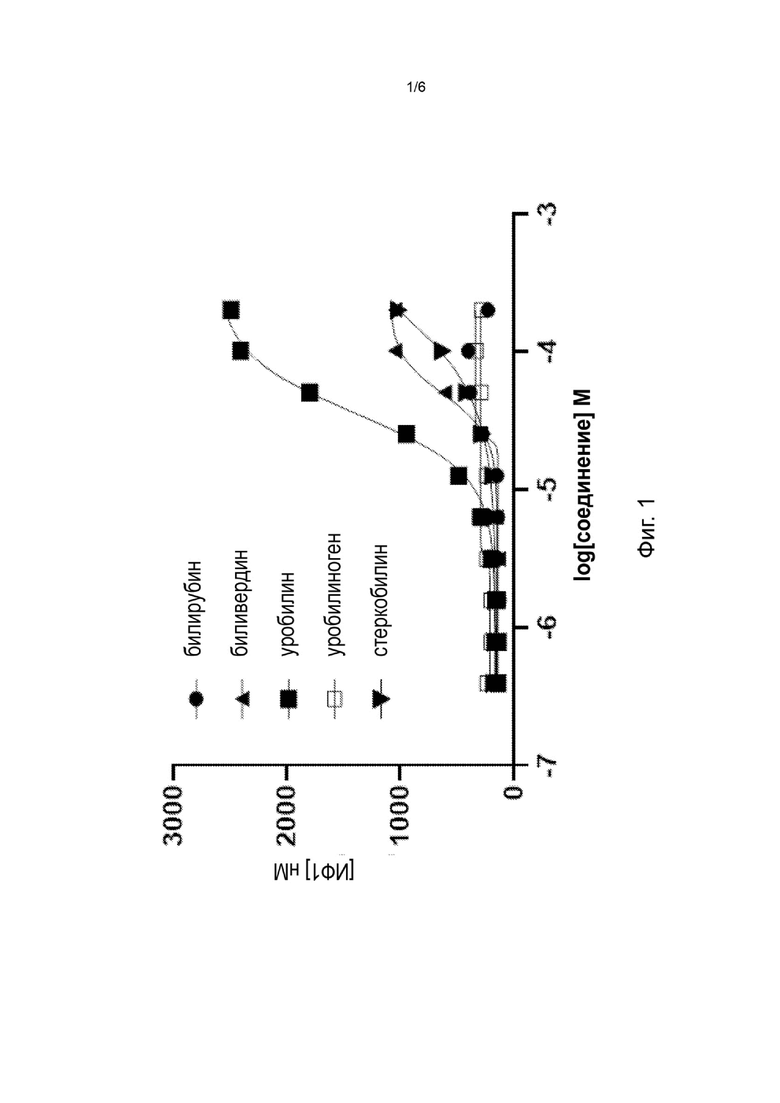
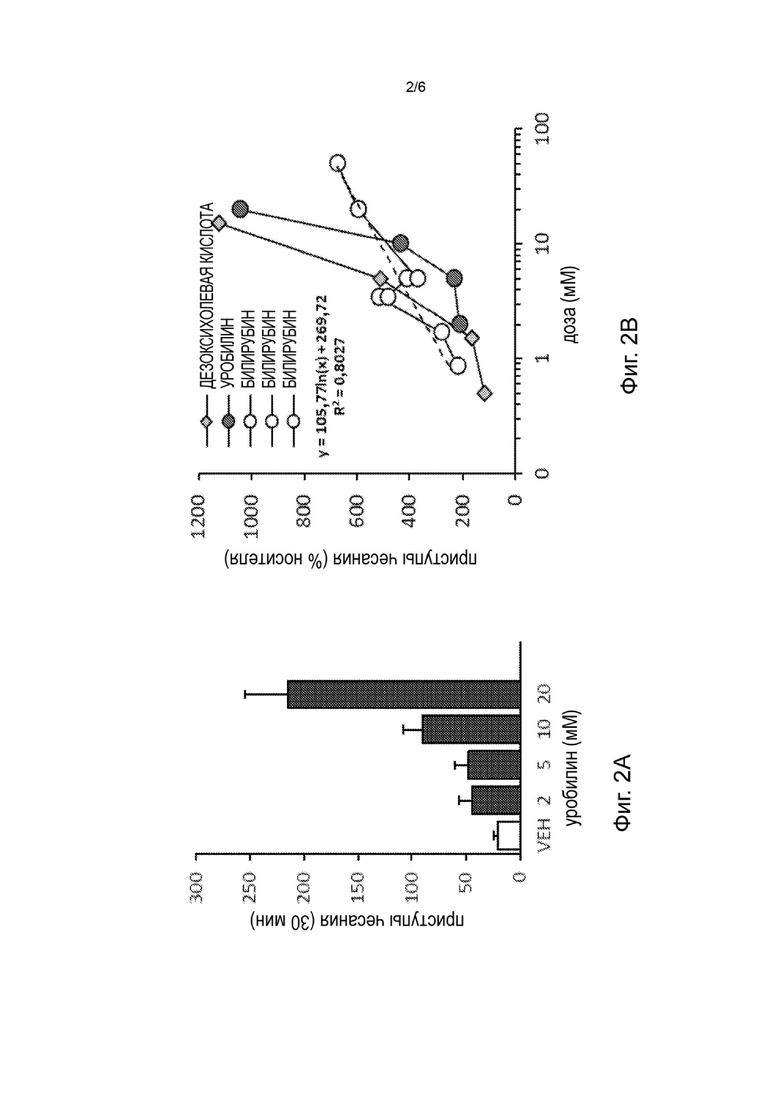
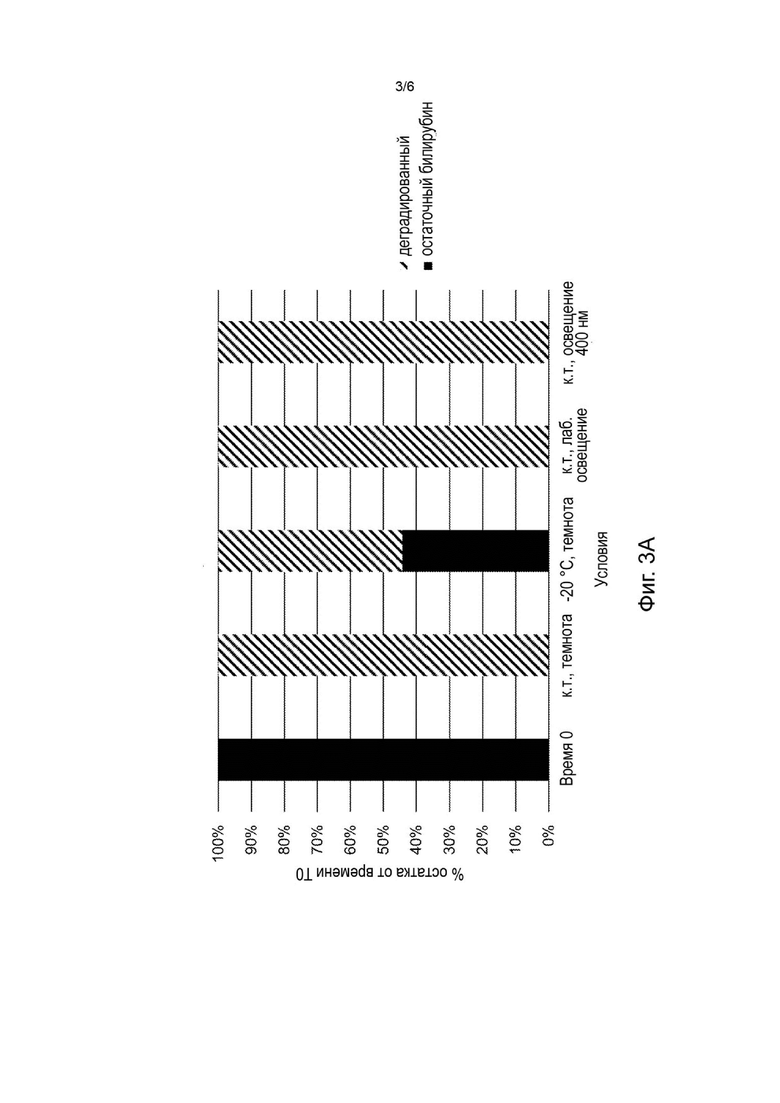
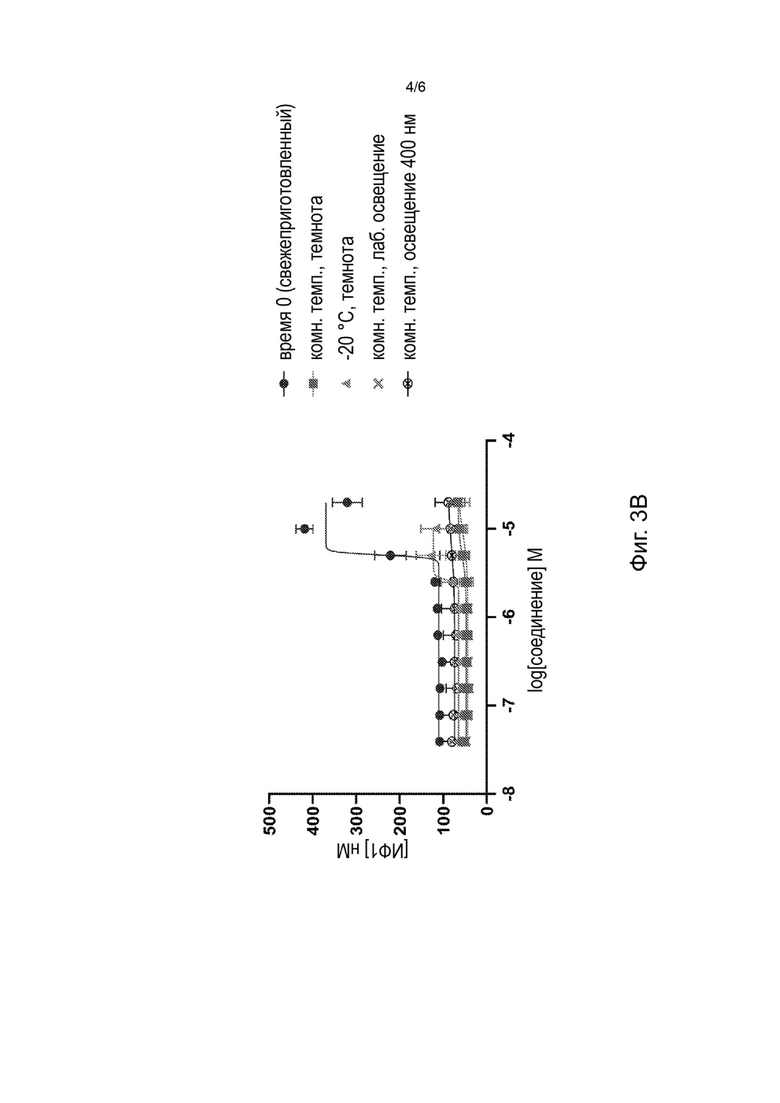
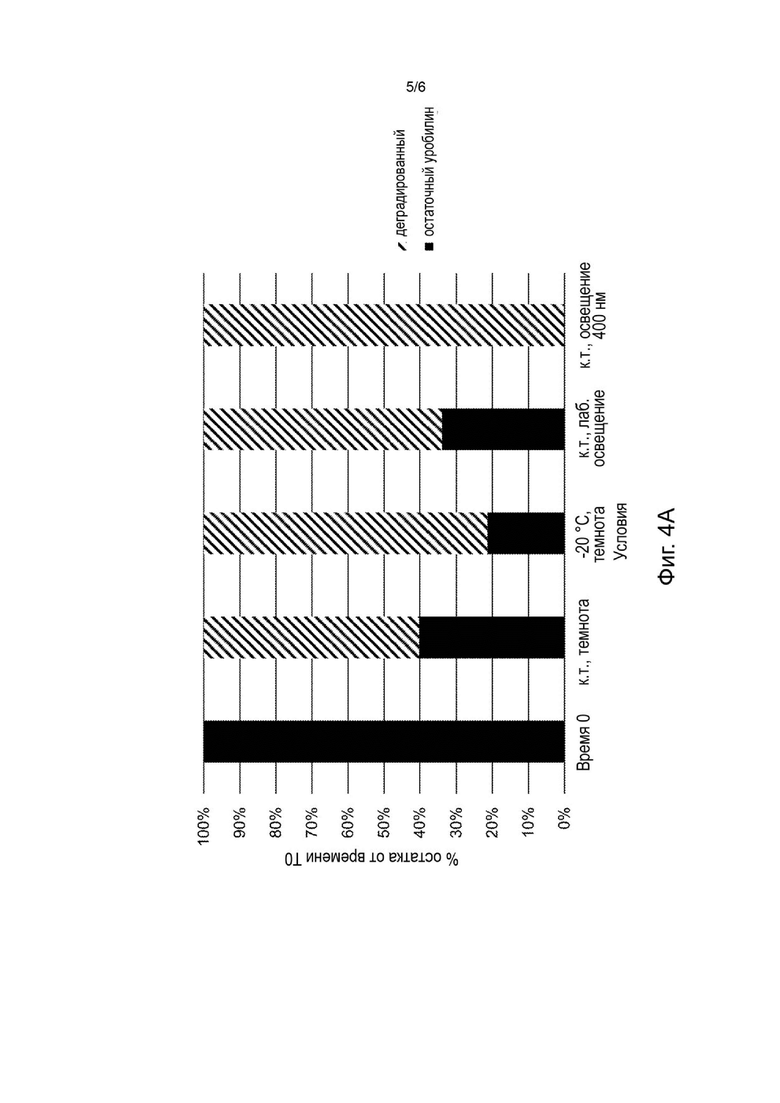
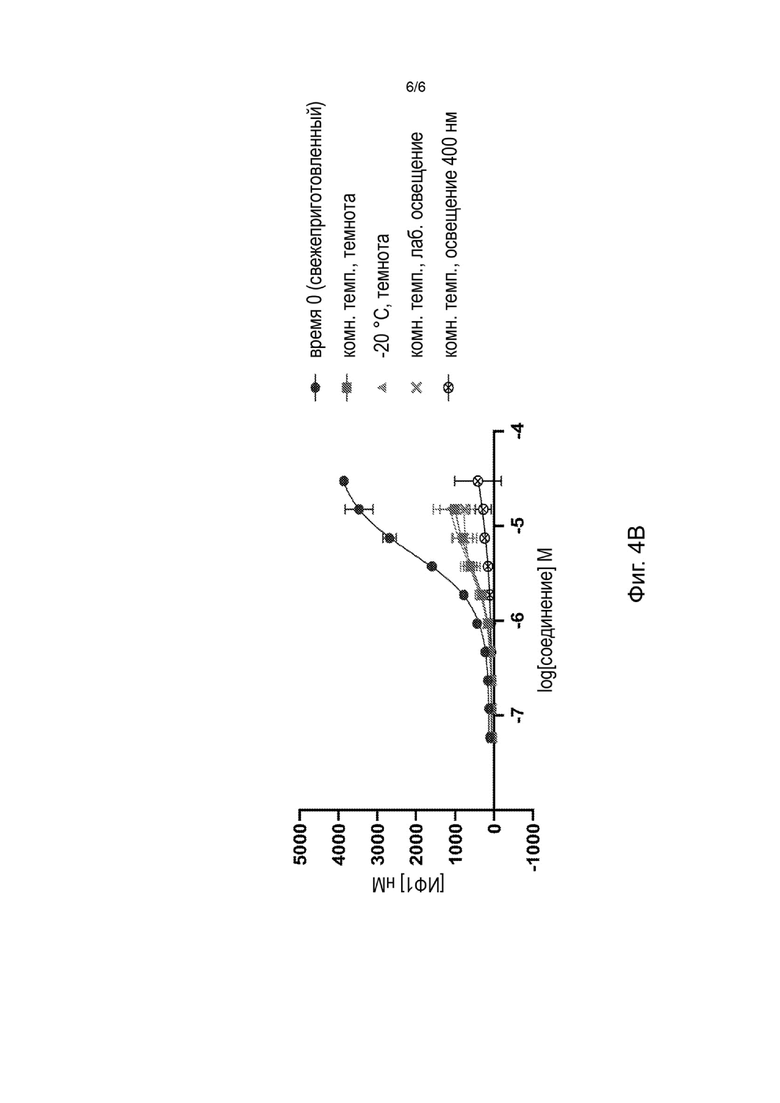
Изобретение относится к соединению, имеющему одну из следующих структур, или его фармацевтически приемлемой соли:
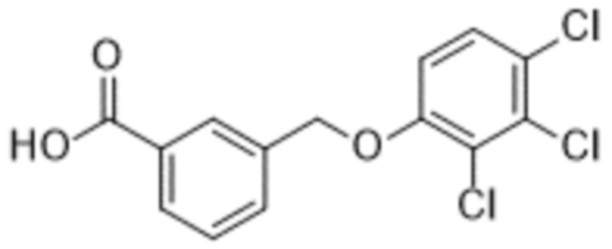
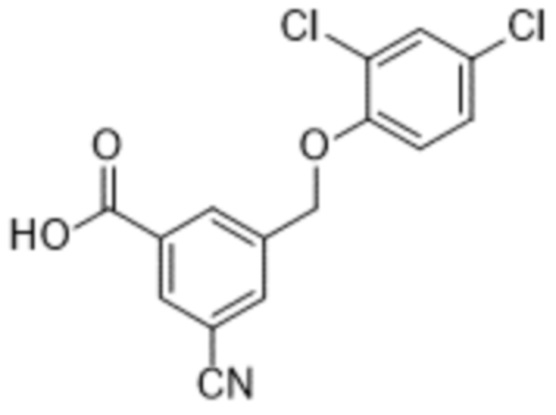
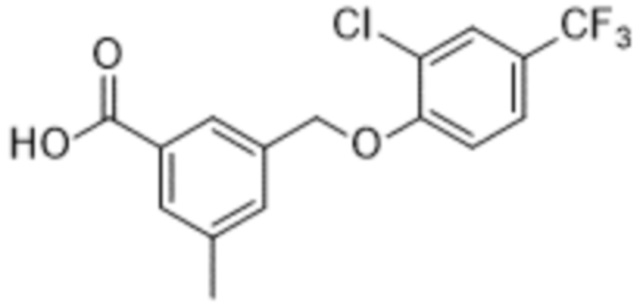
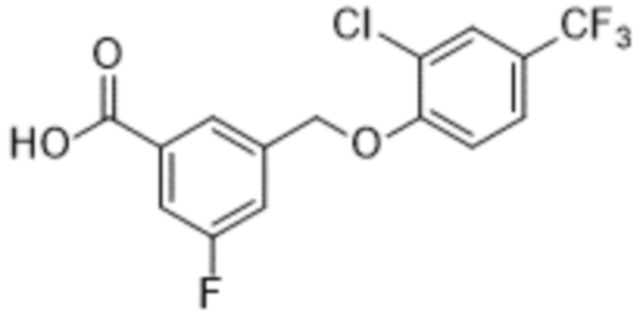
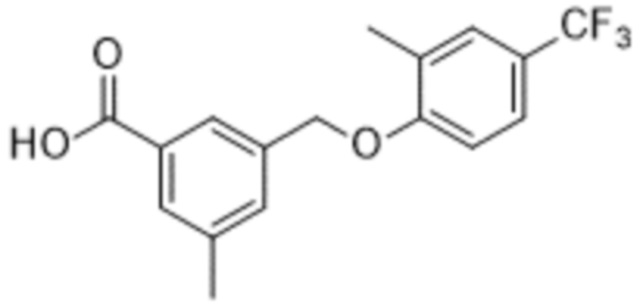
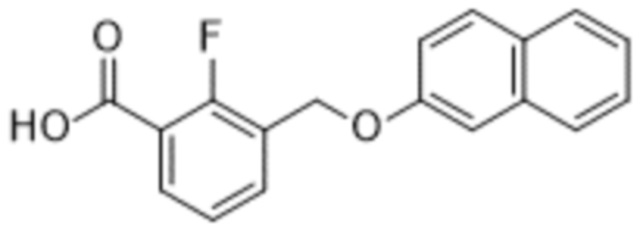
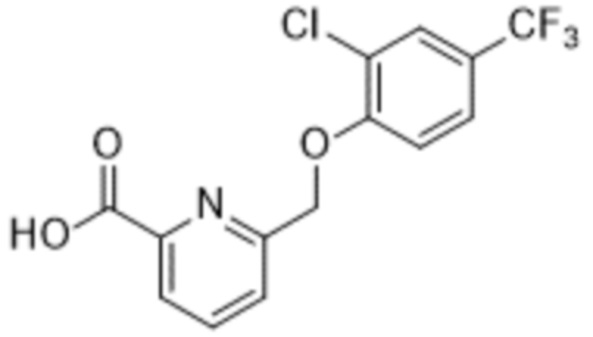
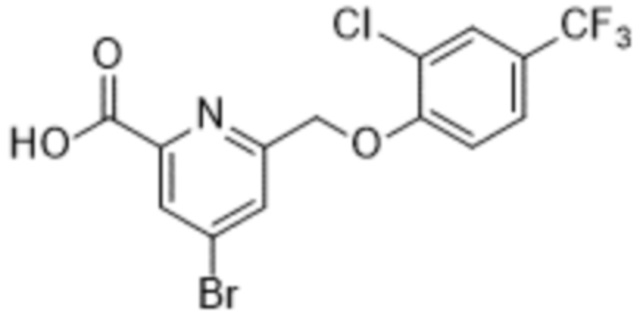
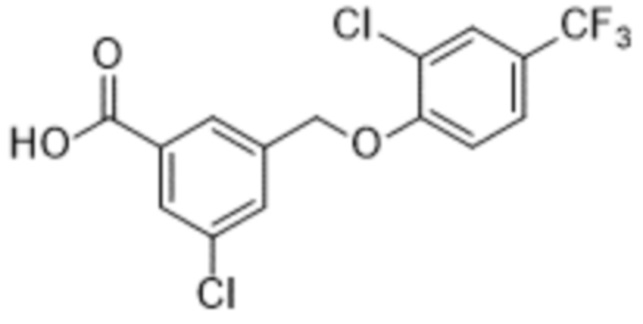
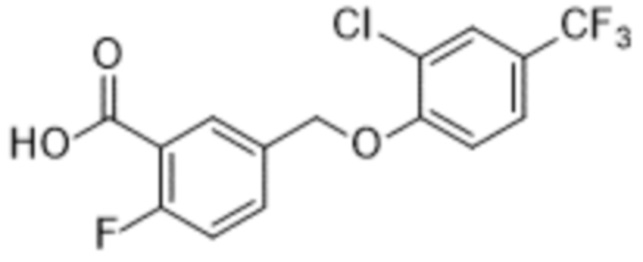
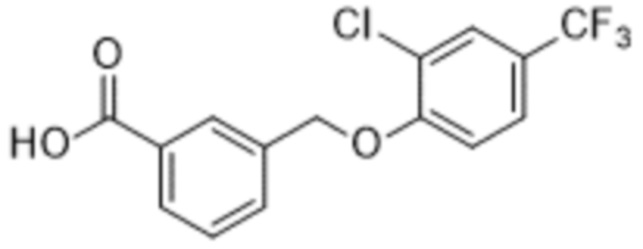
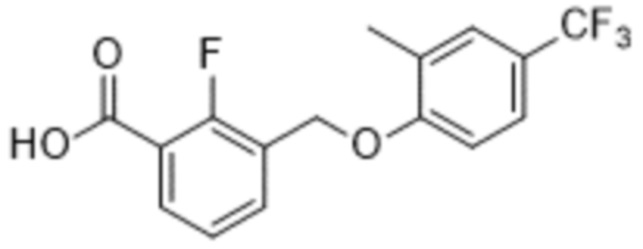
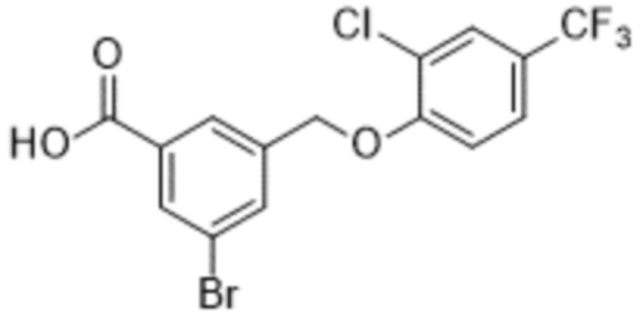
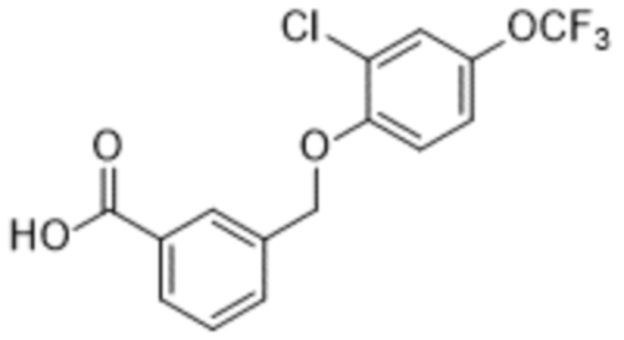
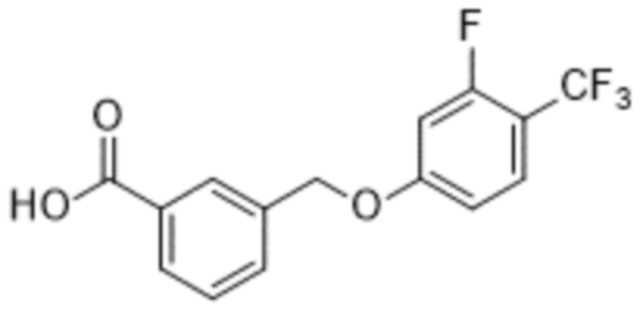
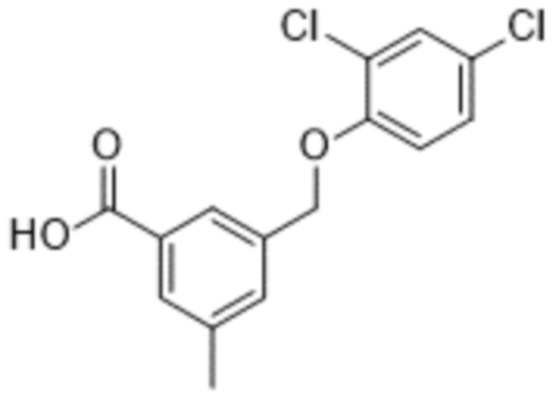
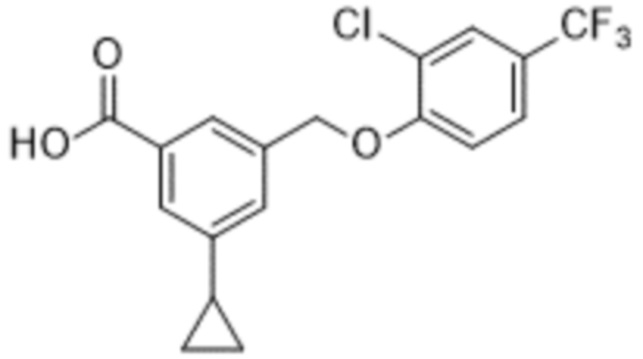
Изобретение также относится к фармацевтической композиции и способу лечения MRGPR X4-зависимых состояний на основе указанного соединения. Технический результат: получено новое соединение и фармацевтическая композиция на его основе, которые могут найти применение в медицине для лечения MRGPR X4-опосредованных заболеваний. 4 н. и 35 з.п. ф-лы, 7 ил., 70 табл., 70 пр.
1. Соединение, имеющее одну из следующих структур, или его фармацевтически приемлемая соль:
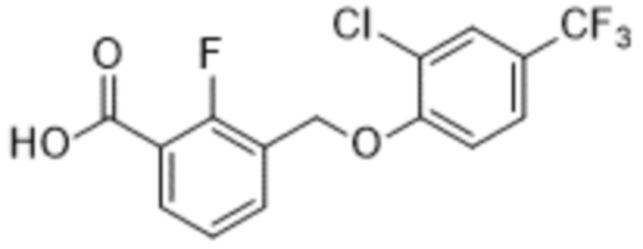
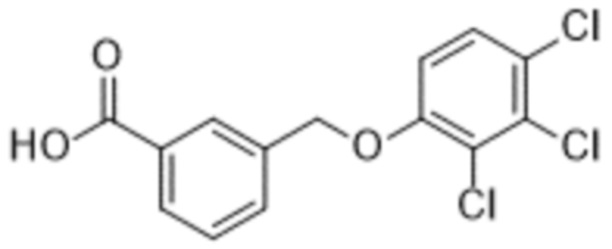
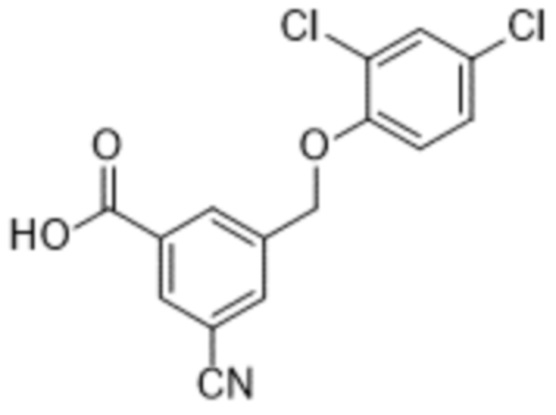
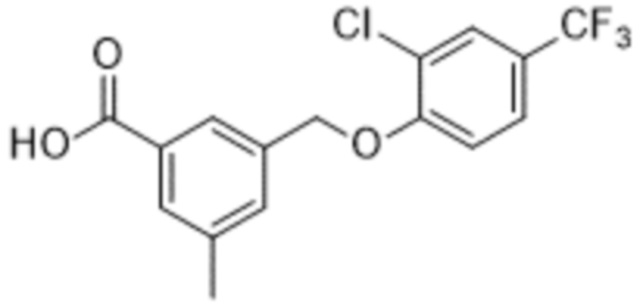
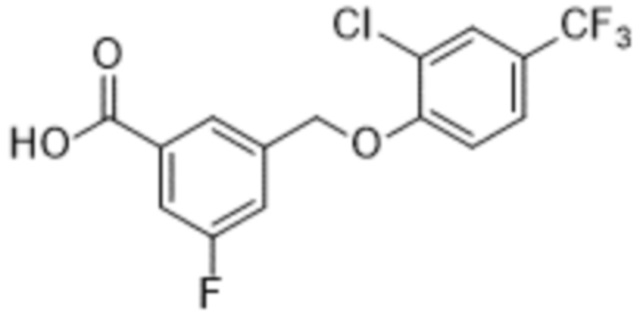
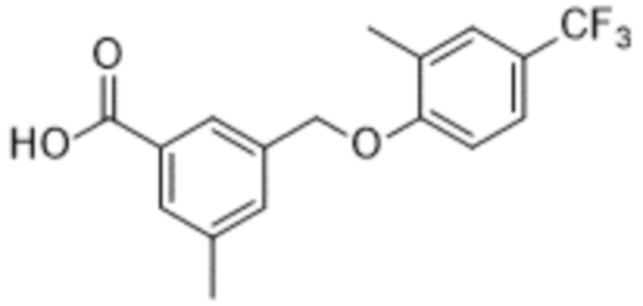
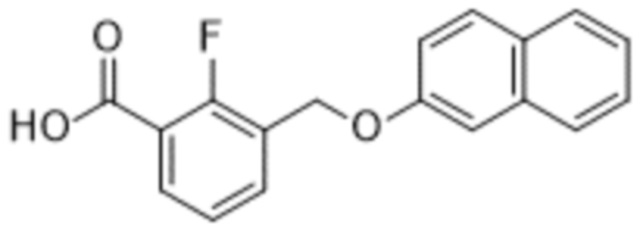
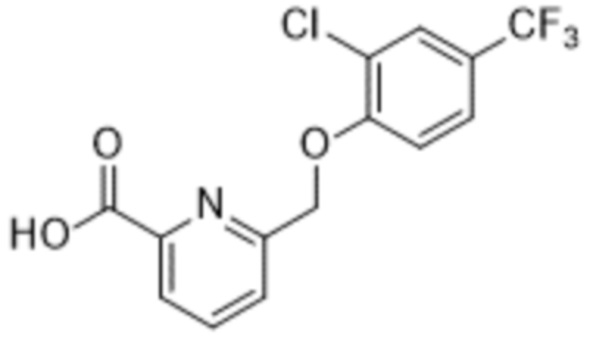
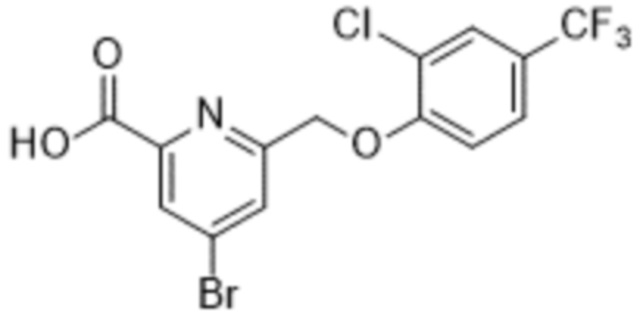
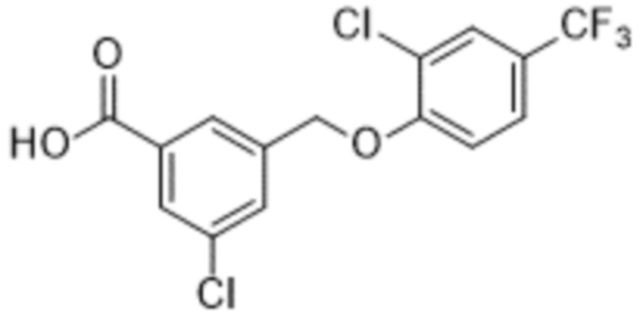
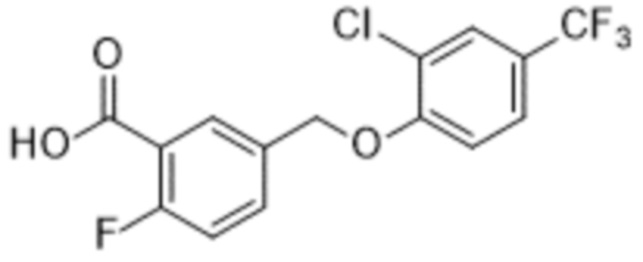
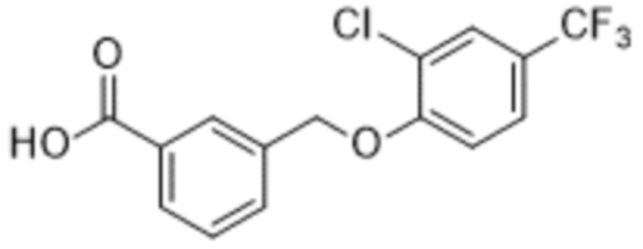
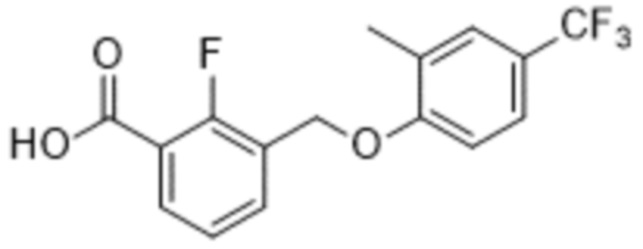
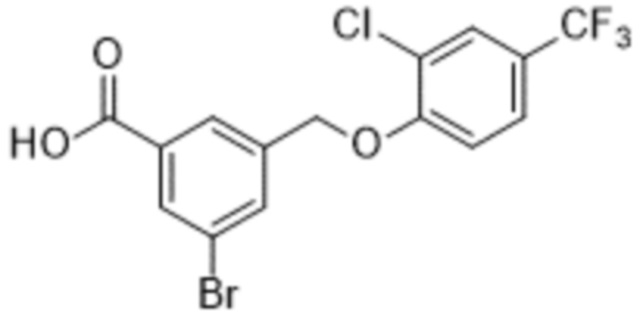
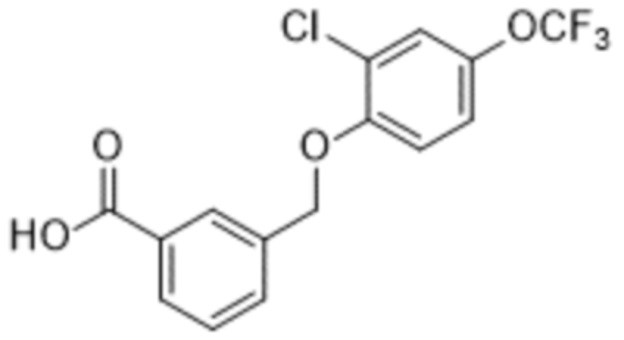
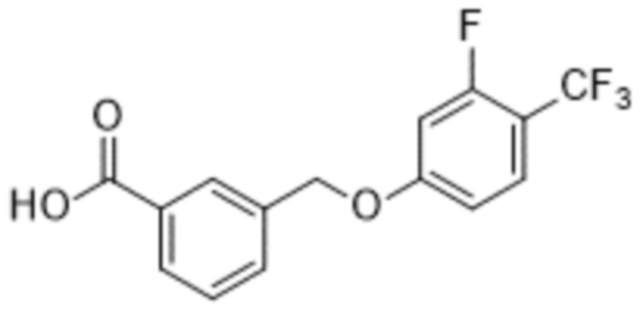
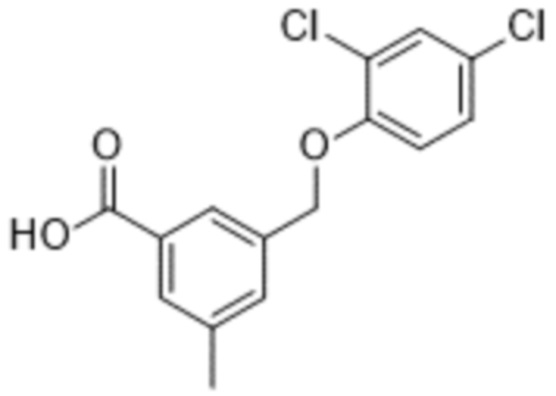
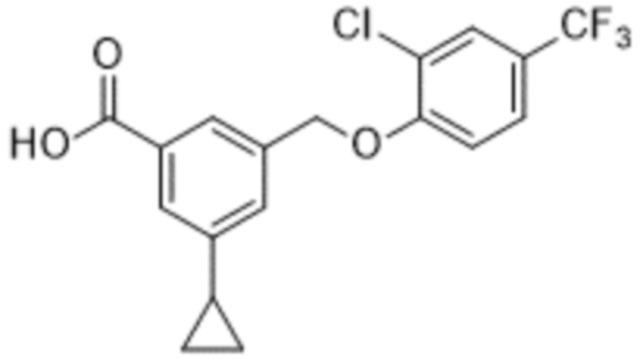
2. Соединение по п. 1, где указанное соединение представляет собой соединение 1-55 и имеет следующую структуру, или его фармацевтически приемлемая соль:
.
3. Соединение по п. 1, где указанное соединение представляет собой соединение 1-78 и имеет следующую структуру, или его фармацевтически приемлемая соль:
.
4. Соединение по п. 1, где указанное соединение представляет собой соединение 5-1 и имеет следующую структуру, или его фармацевтически приемлемая соль:
.
5. Соединение по п. 1, где указанное соединение представляет собой соединение 4-10 и имеет следующую структуру, или его фармацевтически приемлемая соль:
.
6. Соединение по п. 1, где указанное соединение представляет собой соединение 1-65 и имеет следующую структуру, или его фармацевтически приемлемая соль:
.
7. Соединение по п. 1, где указанное соединение представляет собой соединение 4-11 и имеет следующую структуру, или его фармацевтически приемлемая соль:
.
8. Соединение по п. 1, где указанное соединение представляет собой соединение 1-58 и имеет следующую структуру, или его фармацевтически приемлемая соль:
.
9. Соединение по п. 1, где указанное соединение представляет собой соединение 1-56 и имеет следующую структуру, или его фармацевтически приемлемая соль:
.
10. Соединение по п. 1, где указанное соединение представляет собой соединение 9-1 и имеет следующую структуру, или его фармацевтически приемлемая соль:
.
11. Соединение по п. 1, где указанное соединение представляет собой соединение 1-82 и имеет следующую структуру, или его фармацевтически приемлемая соль:
.
12. Соединение по п. 1, где указанное соединение представляет собой соединение 1-85 и имеет следующую структуру, или его фармацевтически приемлемая соль:
.
13. Соединение по п. 1, где указанное соединение представляет собой соединение 1-29 и имеет следующую структуру, или его фармацевтически приемлемая соль:
.
14. Соединение по п. 1, где указанное соединение представляет собой соединение 1-101 и имеет следующую структуру, или его фармацевтически приемлемая соль:
.
15. Соединение по п. 1, где указанное соединение представляет собой соединение 2-3 и имеет следующую структуру, или его фармацевтически приемлемая соль:
.
16. Соединение по п. 1, где указанное соединение представляет собой соединение 1-103 и имеет следующую структуру, или его фармацевтически приемлемая соль:
.
17. Соединение по п. 1, где указанное соединение представляет собой соединение 1-112 и имеет следующую структуру, или его фармацевтически приемлемая соль:
.
18. Соединение по п. 1, где указанное соединение представляет собой соединение 4-7 и имеет следующую структуру, или его фармацевтически приемлемая соль:
.
19. Соединение по п. 1, где указанное соединение представляет собой соединение 11-1 и имеет следующую структуру, или его фармацевтически приемлемая соль:
.
20. Фармацевтическая композиция для модуляции Mas-связанного рецептора G-белка (MRGPR) X4, содержащая эффективное количество соединения, имеющего одну из следующих структур, или его фармацевтически приемлемой соли, и фармацевтически приемлемое вспомогательное вещество:
при этом указанное соединение или его фармацевтически приемлемая соль представляет собой антагонист MRGPR X4.
21. Способ модуляции Mas-связанного рецептора G-белка (MRGPR) X4 посредством контакта MRGPR X4 с эффективным количеством соединения, имеющего одну из следующих структур, или его фармацевтически приемлемой соли:
при этом указанное соединение или его фармацевтически приемлемая соль представляет собой антагонист MRGPR X4.
22. Способ лечения MRGPR X4-зависимого состояния путем введения субъекту, который нуждается в этом, эффективного количества соединения, имеющего одну из следующих структур, или его фармацевтически приемлемой соли:
23. Способ по п. 22, отличающийся тем, что зависимое от MRGPR X4 состояние представляет собой состояние, которое вызвано активацией MRGPR X4 желчной кислотой или ее аналогом.
24. Способ по п. 22, отличающийся тем, что зависимое от MRGPR X4 состояние представляет собой состояние, связанное с чесоткой, состояние, связанное с болью, или аутоиммунное расстройство.
25. Способ по п. 24, отличающийся тем, что состояние, связанное с чесоткой, представляет собой хроническую чесотку, холестатический зуд, контактный дерматит, аллергический блефарит, анемию, атопический дерматит, буллезный пемфигоид, кандидоз, ветряную оспу, холестаз, терминальную почечную недостаточность, гемодиализ, контактный дерматит, герпетиформный дерматит, диабет, лекарственную аллергию, сухую кожу, дисгидротический дерматит, эктопическую экзему, экзему, эритразму, фолликулит, грибковую инфекцию кожи, геморрой, герпес, ВИЧ-инфекцию, болезнь Ходжкина, гипертиреоз, железодефицитную анемию, болезнь почек, лейкемию, болезнь печени, лимфому, злокачественные новообразования, множественную миелому, нейродермит, онхоцеркоз, болезнь Педжета, педикулез, истинную полицитемию, анальный зуд, псевдобешенство, псориаз, выпадение прямой кишки, зудневую чесотку, шистосомоз, склеродермию, тяжелый стресс, стазовый дерматит, зуд купальщиков, заболевание щитовидной железы, паховый дерматомикоз, уремический зуд или крапивницу.
26. Способ по п. 25, отличающийся тем, что связанное с чесоткой состояние представляет собой холестатический зуд, уремический зуд, атопический дерматит, сухую кожу, псориаз, контактный дерматит или экзему.
27. Способ по п. 25, отличающийся тем, что состояние, связанное с чесоткой, представляет собой заболевание печени, при этом заболевание печени представляет собой первичный билиарный холангит, первичный склерозирующий холангит, синдром Алагилля, прогрессирующий семейный внутрипеченочный холестаз, внутрипеченочный холестаз при беременности, неалкогольный стеатогепатит (НАСГ), неалкогольную жировую болезнь печени (НАЖБП), атрезию желчевыводящих путей, хронический гепатит B, лекарственный хронический вирусный гепатит, индуцированное повреждение печени (ЛПП), фиброз печени, холестатическую болезнь печени или алкогольную болезнь печени.
28. Способ по п. 24, отличающийся тем, что состояние, связанное с болью, представляет собой острую боль, прогрессирующий рак простаты, боль, связанную со СПИДом, анкилозирующий спондилит, арахноидит, артрит, артрофиброз, атаксический церебральный паралич, аутоиммунный атрофический гастрит, аваскулярный некроз, боль в спине, болезнь (синдром) Бехчета, синдром жжения во рту, бурсит, раковую боль, туннельный запястный синдром, синдром конского хвоста, центральный болевой синдром, церебральный паралич, стеноз шейки матки, болезнь Шарко-Мари-Тута (ШМТ), синдром хронической усталости (СХУ), хроническую функциональную абдоминальную боль (ХФАБ), хроническую боль, хронический панкреатит, коллапс легкого (пневмоторакс), комплексный регионарный болевой синдром (РБС), нейропатическую боль роговицы, болезнь Крона, дегенеративную болезнь диска, болезнь Деркума, дерматомиозит, диабетическую периферическую нейропатию (ДПН), дистонию, синдром Элерса-Данлоса (СЭД), эндометриоз, синдром эозинофилии-миалгии (СЭМ), эритромелалгию, фибромиалгию, подагру, головные боли, грыжу межпозвоночного диска, гидроцефалию, межреберную невралгию, интерстициальный цистит, синдром раздраженного кишечника (СРК), ювенильный дерматозит, травму колена, боль в ногах, синдром боли в пояснице-гематурии, волчанку, болезнь Лайма, медуллярную губчатую почку (МГП), парестетическую мералгию, мезотелиому, мигрень, скелетно-мышечную боль, миофасциальную боль, миозит, боль в шее, невропатическую боль, затылочную невралгию, остеоартрит, болезнь Педжета, синдром Парсонажа-Тернера, боль в тазу, периферическую нейропатию, фантомную боль в конечностях, защемление нерва, поликистозную болезнь почек, ревматическую полимиалгию, полимиозит, порфирию, болевой синдром после герниорафии, болевой синдром после мастэктомии, постинсультную боль, болевой синдром после торакотомии, постгерпетическую невралгию (опоясывающий лишай), постполиомиелитный синдром, первичный боковой склероз, псориатический артрит, пудендальную невралгию, радикулопатию, болезнь Рейно, ревматоидный артрит (РА), дисфункцию крестцово-подвздошного сочленения, саркоидоз, кифоз Шейермана, ишиалгию, сколиоз, опоясывающий лишай (герпес зостер), синдром Шегрена, спастическую кривошею, дисфункцию сфинктера Одди, спиноцеребеллярную атаксию (SCA-атаксию), травму спинного мозга, стеноз позвоночника, сирингомиелию, кисты Тарлова, поперечный миелит, тригеминальную невралгию, невропатическую боль, неспецифический язвенный колит, сосудистую боль и вульводинию.
29. Способ по п. 24, отличающийся тем, что аутоиммунное заболевание представляет собой хроническое воспаление, рассеянный склероз, синдром Стивенса-Джонсона, аппендицит, бурсит, колит, цистит, дерматит, флебит, рефлекторную симпатическую дистрофию/комплексный регионарный болевой синдром (РСД/КРБС), ринит, тендинит, тонзиллит, вульгарные угри, нарушение реактивности дыхательных путей, астму, инфекцию дыхательных путей, аутовоспалительное заболевание, глютеновую болезнь, хронический простатит, дивертикулит, гломерулонефрит, гнойный гидраденит, гиперчувствительности, кишечное расстройство, расстройство кишечного эпителия, воспалительное заболевание кишечника, синдром раздраженного кишечника, колит, интерстициальный цистит, отит, воспалительное заболевание органов малого таза, боль в эндометрии, реперфузионное повреждение, ревматическую лихорадку, ревматоидный артрит, саркоидоз, отторжение трансплантата или васкулит.
30. Способ по п. 24, отличающийся тем, что указанное состояние, связанное с чесоткой, представляет собой терминальную почечную недостаточность.
31. Способ по п. 24, отличающийся тем, что указанное состояние, связанное с чесоткой, представляет собой заболевание почек.
32. Способ по п. 24, отличающийся тем, что указанное состояние, связанное с болью, представляет собой медуллярную губчатую почку (МГП).
33. Способ по п. 24, отличающийся тем, что указанное состояние, связанное с болью, представляет собой поликистозную болезнь почек.
34. Способ по п. 24, отличающийся тем, что указанное состояние, связанное с чесоткой, связано с метаболитом гема.
35. Способ по п. 34, отличающийся тем, что указанный метаболит гема представляет собой билирубин.
36. Способ по п. 34, отличающийся тем, что указанный метаболит гема представляет собой уробилин.
37. Способ по п. 34, отличающийся тем, что указанный метаболит гема представляет собой биливердин.
38. Способ по п. 34, отличающийся тем, что указанный метаболит гема представляет собой что уробилиноген.
39. Способ по п. 34, отличающийся тем, что указанный метаболит гема представляет собой стеркобилин.
| WO 2011127388 A3, 13.10.2011 | |||
| WO 2013088315 A1, 20.06.2013 | |||
| WO 2018232316 A1, 20.12.2018 | |||
| WO2019045035 A1, 07.03.2019 | |||
| WO2013025425 A1, 21.03.2013 | |||
| WO 2014008458 A2, 09.01.2014 | |||
| Нивелир | 1928 |
|
SU16887A1 |

