Настоящее изобретение описывает новые химические соединения, применение этих соединений в качестве антикоагулянтов, фармацевтические композиции и плазмозамещающие растворы на их основе и может быть использовано для лечения тромбоэмболических осложнений при таких болезнях, как инфаркт миокарда, инсульт, тромбоз глубоких вен или легочной артерии; для профилактики возникновения гиперкоагуляционных состояний при травмах, хирургических операциях, сепсисе, различных акушерских патологиях, в медицине катастроф, в условиях реанимации и т.п.
Огромное число самых разнообразных патологических состояний организма связано с возникновением нарушений в системе гемостаза. Тромбоэмболические осложнения, возникающие при таких болезнях, как инфаркт миокарда, инсульт, тромбоз глубоких вен или легочной артерии, являются одной из основных причин смерти во всем мире. Неудивительно, что на протяжении большого количества лет ведется интенсивный поиск препаратов, которые могли бы служить в клинике эффективными и безопасными лекарствами. В первую очередь это различные антитромботические агенты, обладающие антикоагулянтными свойствами.
Тромбин является ключевым ферментом системы свертывания крови. Он не только осуществляет превращение растворимого белка плазмы фибриногена в нерастворимый фибриновый сгусток, но и запускает большинство положительных и отрицательных обратных связей в этой системе, активируя тромбоциты, факторы V, VIII и XIII, а также протеин Ц. Тромбин инициирует также разнообразные клеточные и сосудистые реакции, в том числе пролиферацию клеток эндотелия, выброс из них активаторов плазминогена и т.д. Именно из-за того, что тромбин участвует в большом числе важнейших биорегуляторных событий, ингибирование тромбина по его активному центру должно быть очень эффективным и многообещающим в контроле многих патофизиологических состояний.
Существуют три основных направления воздействия на систему гемостаза с целью предупреждения нежелательного тромбообразования: применение прямых и непрямых ингибиторов сериновых протеаз свертывающего каскада (в первую очередь тромбина и фактора Ха); применение антитромбоцитарных препаратов (антагонистов GPIIb/IIIa, аспирина, антагонистов тромбинового рецептора и т.п.), снижающих агрегационные свойства тромбоцитов и препятствующих таким образом дальнейшей активации свертывания; применение антагонистов витамина К, которые снижают синтез предшественников факторов свертывания печенью.
В настоящее время в клинике чаще всего используют три основных антитромботических препарата: нефракционированный гепарин, оральный антикоагулянт варфарин (витамин К-антагонист) и ингибитор агрегации тромбоцитов аспирин. Однако каждый из этих препаратов имеет свои ограничения применения и нежелательные побочные эффекты.
Нефракционированный гепарин (UFH) - природный анионный полисахарид, представляющий собой смесь полисахаридных цепочек различной длины, построенных из повторяющихся дисахаридных единиц, состоящих из остатков уроновой кислоты (L-идуроновой и/или D-глюкуроновой) и остатков D-глюкозамина. Его молекулярный вес распределяется по данным разных литературных источников от 3-5 до 30-40 тысяч дальтон с пиком на 12-15 тысячах дальтон.
Нефракционированный гепарин и его более легкие аналоги (гепарины низкого молекулярного веса (LMWH)) являются непрямыми антикоагулянтами. Они не ингибируют тромбин самостоятельно, а ускоряют действие природного плазменного ингибитора свертывания антитромбина III (ATIII). Таким образом, если содержание ATIII в плазме больного по каким-то причинам сильно снижено, гепарины оказывают слабое антикоагулянтное действие.
Клиническое применение нефракционированного гепарина имеет ряд недостатков:
1. Действие нефракционированного гепарина короткоживущее. Оно проходит очень быстро после отмены лекарства, поэтому использование UFH не может снизить риска повторных тромботических событий.
2. Гепарин осуществляет свой антитромботический эффект непрямо, а опосредовано, для чего ему необходимо присутствие в системе антитромбина.
3. Гепарин активен только против циркулируещего тромбина, но практически не ингибирует тромбин, сорбированный на сгустке.
4. При одинаковой дозе гепарин имеет непредсказуемый антикоагулянтный ответ у разных пациентов, что связано с целым рядом причин, в том числе уровнем ATIII в плазме, индивидуальной скоростью выведения препарата, связыванием и нейтрализацией гепарина под действием различных белков плазмы и активированных тромбоцитов (тромбоцитарный фактор 4, гепариназа и т.п.). Это определяет необходимость частого мониторинга статуса коагуляционной системы.
5. Риск геморрагических осложнений и возможность появления тромбоцитопении.
6. При длительной гепаринотерапии (более 6 месяцев) и достаточно высоких дозах гепарина (>15000 единиц) возможно проявление такого осложнения, как остеопороз.
7. Гепарин может быть введен только внутривенно в условиях стационара.
Непрямыми ингибиторами свертывания являются и антагонисты витамина К (типа варфарина). Механизм действия этих препаратов на систему свертывания крови связан с тем, что они эффективно блокируют синтез витамин К-зависимых факторов свертывания печенью. Для синтеза нормальной молекулы фактора необходимо посттрансляционное γ-карбоксилирование N-терминального конца будущего фактора. Это карбоксилирование совершенно необходимо для того, чтобы синтезированные молекулы фактора могли в ходе свертывания связываться (при посредстве ионов Са+2) с отрицательно заряженной фосфолипидной поверхностью активированных тромбоцитов и осуществлять свою функцию. Витамин К является необходимым кофактором этого карбоксилирования. В ходе реакции он попеременно переходит из своей гидроксихиноновой формы, которая и участвует в реакции карбоксилирования, в окисленную эпоксидную форму. Под действием фермента витамин К-редуктазы эта эпоксидная форма восстанавливается и может вновь участвовать в реакции карбоксилирования. Препараты группы кумаринов блокируют это восстановление.
Применение варфарина тоже имеет ряд ограничений и недостатков. Во-первых, ответ на терапию развивается медленно. Он начинает проявляться через сутки, но максимального уровня достигает только через несколько дней. Кроме того, этот препарат довольно сильно связывается с различными компонентами пищи и сильно перекрывается со многими лекарственными веществами. Существует также большая генетическая вариабельность в активности варфарин-метаболизирующего фермента. Это объясняет большую индивидуальную вариабельность в ответе на варфарин и означает, что при его приеме нужны определенные диетарные ограничения и систематический мониторинг.
Антитромбоцитарные препараты (аспирин, антагонисты GPIIb/IIIa и т.п.), как уже говорилось выше, препятствуют полноценной активации тромбоцитов и их участию в ускорении реакций свертывания, ограничивая, таким образом, дальнейшее производство тромбина. Однако они никак не влияют на работу уже образовавшегося тромбина.
Таким образом, все рассмотренные выше, стандартно применяемые антитромботические агенты имеют свои недостатки. Часть из них не является прямыми ингибиторами тромбина, поэтому либо требует для проявления своего действия присутствия в плазме антитромбина III (UFH, LMWH), либо действует медленно, ингибируя синтез необходимых факторов свертывания (варфарин и т.п.), другие вообще не действуют на уже образовавшийся тромбин (антитромбоцитарные агенты). Это способствует тому, что на протяжении многих лет ведутся интенсивные поиски "идеальных" ингибиторов, которые обладали бы не меньшей эффективностью, но были бы свободны от многих недостатков стандартных препаратов.
Очень привлекательной в этом смысле оказалась стратегия разработки небольших синтетических ингибиторов тромбина - антикоагулянтов. Подобные ингибиторы быстро действуют прямо на присутствующий в крови тромбин, что позволяет надеяться на то, что они будут эффективны для купирования остро развивающихся тромботических осложнений даже в условиях сниженного содержания ATIII в плазме.
Стратегия поиска новых прямых синтетических ингибиторов сериновых протеаз свертывающего каскада ориентируется на следующие требования, которым должен удовлетворять такой ингибитор:
- Высокое сродство к ферменту-мишени (т.е. высокая эффективность ингибирования).
- Высокая селективность по отношению к ферменту-мишени в сравнении с другими родственными сериновьми протеазами.
- Химическая и метаболическая стабильность.
- Отсутствие токсичности.
- Слабое (или не очень сильное) связывание с белками плазмы.
- Высокая биодоступность при приеме через рот.
- Достаточно большое время полужизни препарата, позволяющее при его оральном приеме поддерживать терапевтический уровень в плазме настолько долго, чтобы можно было ограничиться приемом препарата 1-2 раза в день.
- Возможность простого метода мониторинга уровня препарата. К настоящему времени в литературе опубликовано большое количество обзоров, посвященных разработке низкомолекулярных ингибиторов тромбина (Shafer J.A., Cardiovascular chemotherapy: anticoagulants, Curr. Opin. Chem. Biol., 1998, 2:458-465; Steinmetzer Т., Hauptmann J., Sturzebecher J., Advances in the development of thrombin inhibitors, Exp.Opin. Invest. Drugs, 2001, 10(5):845-864; Edmunds JJ, Rapundalo ST, Siddiqui MA, Thrombin and factor Xa inhibition, Ann. Rep. Med. Chem., 1996, 31:51-60; Wiley M.R., Fisher M.J., Small molecule direct thrombin inhibitors. Expert Opin. Ther. Patents, 1997, 7:1265-1282; Hauptmann J, Sturzebecher J., Synthetic inhibitors of thrombin and factor Xa: from bench to bedside, Thromb Res., 1999, 93(5):203-241; Vacca JP., New advances in the discovery of thrombin and factor Xa inhibitors, Curr Opin Chem Biol., 2000, 4(4):394-400).
Однако разработка лекарств на основе новых химических соединений требует не только определения их возможного фармакологического действия, но и тщательной проверки токсикологических свойств препаратов, их возможного влияния на наследственность и выявления других отдаленных последствий применения.
Задача осложняется также тем, что не каждый ингибитор, снижающий активность тромбина в водных буферных растворах, может являться реальным антикоагулянтом при свертывании крови в организме. Это может быть связано, например, с механизмом ингибирования. Так, если ингибитор является неконкурентным, то даже при 100%-ном связывании всех активных центров присутствующего в плазме тромбина с таким ингибитором активность фермента не будет подавлена полностью. Остаточная активность тромбина может быть не очень высокой, однако, в некоторых случаях, она не может быть до конца подавлена в плазме природным ингибитором тромбина - ATIII. Это происходит из-за определенного изменения конформации молекулы тромбина при связывании с таким ингибитором, которое не позволяет ATIII подойти к активному центру фермента. В результате в крови длительное время сохраняется остаточная активность тромбина. Это приводит к тому, что из-за присутствия такого соединения в организме, интегральный коагуляционный ответ не только не снизится, но может суммарно даже усилиться. Если предполагаемый ингибитор тромбина взаимодействует также с другими компонентами системы свертывания (факторами или ингибиторами свертывания), то суммарный ответ системы также невозможно предсказать заранее. Сильное связывание ингибитора с различными белками плазмы может значительно повысить дозу ингибитора, которая должна быть введена в организм для получения желаемого антикоагуляционного эффекта.
Если учесть все вышесказанное, то становится понятным, почему при существовании большого числа синтезированных соединений, которые способны ингибировать тромбин, до настоящего времени полностью прошел все необходимые испытания и реально разрешен для применения в клинике только один препарат этого типа - синтезированный в Японии ингибитор тромбина аргатробан (патент США 5214052, 1993; Schwarz R.P., The preclinical and clinical pharmacology of Novastan (Argatroban), In: "New Anticoagulants for the CardiovascularbPatient, Pifarre R., editor, Hanley and Belfus, Inc., Philadelphia, PA, US, 1997, p.231-249; Okamoto S, Hijikata A, Kikumoto R, Tonomura S, Hara Н, Ninomiya K, Maruyama A, Sugano M, Tamao Y., Potent inhibition ofthrombin by the newly synthesized arginine derivative No.805. The importance of stereo-structure of its hydrophobic carboxamide portion, Biochem Biophys Res Commun. 1981, 101(2):440-446).
Таким образом, поиск новых антикоагулянтов среди низкомолекулярных синтетических ингибиторов тромбина продолжает оставаться актуальнейшей задачей.
Эти ингибиторы - антикоагулянты могут быть прямо использованы для лечения острых тромботических состояний, возникающих в организме в результате различных патологий.
Кроме того, они могут быть использованы и для профилактики возникновения гиперкоагуляционных состояний. В настоящей заявке предлагается использовать низкомолекулярный синтетический ингибитор тромбина, обладающий антикоагулянтной активностью в плазме, для добавления в стандартный плазмозамещающий раствор.
В клинике часто возникают ситуации, когда достаточно большие объемы потерянной крови приходится быстро восполнять искусственными плазмозамещающими растворами (ПЗР). Это происходит при травмах, хирургических операциях, сепсисе, различных акушерских патологиях, в медицине катастроф, в условиях реанимации и т.п. При этом преследуются следующие основные цели:
1. Восполнение объема циркулирующей крови (ОЦК) для поддержания артериального давления, объема сердечного выброса, предупреждения коллапса сосудов, сохранения нормальных реологических характеристик крови и нормальной перфузии органов и тканей.
2. Поддержание нормального коллоидно-осмотического давления плазмы и ее кислотно-щелочного равновесия.
3. Поддержание кислородтранспортной функции крови и функций системы свертывания.
Для достижения первых двух целей обычно переливают различные плазмозамещающие растворы и растворы альбумина. Обеспечение кислородтранспортной функции достигается переливанием эритроцитов, модифицированного гемоглобина или кислородпереносящих растворов типа перфторанов, а поддержание функций системы свертывания - переливанием свежезамороженной плазмы (СЗП), концентратов тромбоцитов, концентратов факторов протромбинового комплекса или отдельных факторов свертывания.
Искусственные ПЗР делятся на два класса: кристаллоидные и коллоидные. Первые представляют собой солевые растворы (например, 0.9%-ный раствор NaCl-физиологический раствор), тогда как вторые содержат добавки высокомолекулярных полимеров (декстранов, гидроксиэтилкрахмалов, производных желатины и т.п.).
Инфузии больших объемов стандартных ПЗР приводят к разбавлению крови этими растворами (гемодилюции). Т.к. ни один стандартный ПЗР на сегодняшний день не содержит факторов и ингибиторов свертывания, то в результате гемодилюции происходит снижение концентраций компонентов системы свертывания в крови. Ранее было показано, что при массивных переливаниях стандартных ПЗР, возникающая в крови гемодилюция вызывает разбаланс работы системы свертывания и усиление коагуляции. Оно связано с тем, что при разбавлении система оказывается более чувствительной к снижению концентраций ингибиторов свертывания, в то время как прокоагулянтные предшественники факторов свертывания присутствуют в плазме в достаточно большом избытке, в результате чего при умеренных степенях разбавления снижение их концентраций практически не оказывает влияния на скорость свертывания. Для коррекции возможных гиперкоагуляционных нарушений, связанных с объемными переливаниями ПЗР, авторами был предложен новый ПЗР, в состав которого был введен природный ингибитор тромбина - антитромбин III (Патент РФ, решение о выдаче по заявке №2005140841 от 27.12.2005). Предложенный раствор является первым представителем нового поколения ПЗР, которые способны частично корректировать гиперкоагуляционные нарушения, вызываемые объемными переливаниями ПЗР. Недостатком этого раствора является то, что антитромбин III - это природный белок, который необходимо выделять из плазмы человека. Это, с одной стороны, достаточно дорого, а с другой - не позволяет полностью исключить возможность вирусного заражения препарата (ВИЧ, гепатит и т.п.). Хорошей альтернативой антитромбина III может служить синтетический низкомолекулярный ингибитор тромбина - антикоагулянт.
Отбор соединений, представляющих интерес, с точки зрения практического применения в качестве антикоагулянтов, то есть обладающих способностью замедлять и/или предотвращать свертывание крови, производился на основании следующих критериев:
1. Вещество должно являться ингибитором тромбина, т.е. предотвращать катализируемую тромбином амидолитическую реакцию расщепления молекулы фибриногена.
2. Вещество должно иметь приемлемые физико-химические свойства (липофильность, гидрофильность), чтобы присутствовать в плазме крови в свободном виде в достаточной концентрации. Иными словами, связывание с другими белками плазмы крови (альбумины, глобулины и т.д.) должно быть умеренным.
3. Вещество должно обладать достаточным временем жизни в плазме крови, чтобы мог проявиться присущий ему терапевтический эффект.
Отбор по первому критерию осуществлялся в две ступени. Сначала была построена виртуальная библиотека, сфокусированная на структурах, описываемых общей структурной формулой (I), с последующим докингом полученных структур в активный центр молекулы тромбина. В результате отбирались наиболее перспективные кандидаты («виртуальные хиты»), то есть те молекулы, которые демонстрировали значение скоринг-функции (определяемой в процессе докинга) не хуже, чем -5.0 ккал/моль. Второй ступенью отбора было экспериментальное измерение прямого ингибирующего действия отобранных соединений на активность тромбина в водном буферном растворе, где тромбин расщеплял специфичный хромогенный (или флюорогенный) субстрат. В присутствии ингибитора скорость этой реакции снижалась. Для последующих испытаний антикоагулянтного действия новых соединений в плазме крови были отобраны соединений формулы (I), которые в достаточно небольших концентрациях (<1 мМ) ингибировали активность тромбина в буферном растворе более чем на 60%.
При конструировании молекул новых ингибиторов тромбина, которые могли бы являться эффективными антикоагулянтами в организме, учитывали, что для придания молекуле ингибитора приемлемых физико-химических свойств, определяющих благоприятную фармакокинетику, желательно частично уравновесить в целом гидрофобный характер молекулы ингибитора формулы (I) путем выбора гидрофильных линкеров. С этой же целью возможна модификация гидрофобных фрагментов, размещаемых в кармане S3 молекулы тромбина, гидрофильными остатками, которые размещаются в кармане со стороны, экспонированной в растворитель.
Достаточное время жизни в плазме крови можно получить, избегая в структуре ингибитора лабильных химических групп, легко расщепляемых в химических или биохимических процессах. Примером таких нежелательных групп служит, например, сложноэфирная группа.
Таким образом, можно отобрать для химического синтеза и последующей экспериментальной проверки в качестве антикоагулянтов молекулы, оптимальным образом сочетающие в себе вышеперечисленные требования (иногда противоречивые).
Окончательно решение о синтезе принималось, исходя из оценки его возможной сложности.
В настоящем изобретении, если не определено иное, используются следующие определения:
Активный центр представляет собой участок белковой макромолекулы, играющий ключевую роль в биохимических взаимодействиях.
Белок или протеин представляет собой белковую макромолекулу.
Белок-мишень представляет собой белковую макромолекулу, участвующую в процессе связывания.
Лиганды представляют собой коллекцию низкомолекулярных химических структур.
Процесс связывания - это образование Ван-дер-Ваальсового или ковалентного комплекса лиганда с активным центром белка-мишени.
Скрининг - это выявление из коллекции химических структур набора соединений, избирательно взаимодействующих с определенным участком белковой макромолекулы.
Правильное позиционирование - это позиционирование, когда лиганд находится в положении, соответствующем минимуму свободной энергии связывания с белком.
Избирательно взаимодействующий лиганд - это лиганд, специфично связывающийся с данным белком-мишенью.
Реперный белок представляет собой белок, используемый либо для корректировки параметров скора в соответствии с экспериментальными данными, либо в процессе валидации работы системы, либо для оценки специфичности связывания данного ингибитора.
Валидация - это набор расчетов и методика сравнений, позволяющая судить о качестве работы системы, оценить насколько эффективно система отбирает из произвольного набора лигандов те из них, которые хорошо связываются с заданным белком-мишенью.
Специфично связывающийся лиганд - это лиганд, связывающийся только с данным белком и не связывающийся с другими белками.
Ингибитор - это лиганд, связывающийся с активным центром заданного белка-мишени и блокирующий нормальный ход биохимических реакций.
Докинг - это позиционирование лиганда в активном центре белка.
Скоринг - это расчет для оценки свободной энергии связывания лиганд-белок.
ΔG связывания - результат расчета выигрыша свободной энергии при связывании лиганда с белком мишенью (с помощью программы SOL).
C1-6алкил представляет собой алкильную группу, содержащую неразветвленную или разветвленную углеводородную цепь, содержащую от 1 до 6 атомов углерода, например, метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил и так далее.
С1-6алкокси представляет собой алкоксильную группу, содержащую неразветвленную или разветвленную углеводородную цепь, содержащую от 1 до 6 атомов углерода, например, метокси, этокси, н-пропокси, изопропокси и так далее.
Галоген означает хлор, бром, йод или фтор.
Фармацевтически приемлемая соль - любая соль, образуемая активным соединением Формулы (I), если она не токсична и не препятствует адсорбции и фармакологическому действию активного соединения. Эта соль может быть получена действием на соединение Формулы (I) органического или неорганического основания, такого как гидроксид натрия, гидроксид калия, гидроксид аммония, метиламин, этиламин и тому подобных.
Сольват представляет собой кристаллическую форму активного соединения Формулы (I), в состав кристаллической решетки которой входят молекулы воды или иного растворителя, из которого кристаллизовалось активное соединение Формулы (I).
Фармацевтически приемлемый носитель означает, что носитель должен являться совместимым с другими ингредиентами композиции и не наносить вреда его реципиенту, то есть быть нетоксичным для клеток или млекопитающих в тех дозах и концентрациях, в которых его применяют. Часто фармацевтически приемлемый носитель представляет собой водный рН буферный раствор. Примеры физиологически приемлемых носителей включают буферы, такие как фосфаты, цитраты и другие соли органических кислот, антиоксиданты, включающие аскорбиновую кислоту; полипептиды с низким молекулярным весом (меньше 10 остатков); протеины, такие как сывороточный альбумин, желатин или иммуноглобулины; гидрофильные полимеры, такие как поливинилпирролидон; аминокислоты, такие как глицин, глютамин, аспарагин, аргинин или лизин; моносахариды, дисахариды и другие углеводы, включая глюкозу, маннозу или декстрины; хелатообразующие агенты, такие как ЭДТА; сахарные спирты, такие как маннитол или сорбитол.
Терапевтически эффективное количество - это количество, необходимое для проявления лечебного эффекта (т.е.достижения нужной степени ингибирования тромбообразования) в организме млекопитающего.
Млекопитающее, как используется здесь, включает в себя представителей отряда приматов (например, человек, человекообразные обезьяны, нечеловекообразные обезьяны, низшие обезьяны), отряда хищных (например, кошки, собаки, медведи), отряда грызунов (например, мышь, крыса, белка), отряда насекомоядных (например, землеройка, крот) и др.
Технической задачей настоящего изобретения было получение новых низкомолекулярных соединений, обладающих высокой антикоагулянтной активностью.
В настоящей заявке описан ряд таких новых низкомолекулярных соединений, обладающих высокой антикоагулянтной активностью, а именно, соединений общей структурной формулы (I), их фармацевтически приемлемых солей или сольватов:
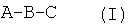
где С выбирают из группы, содержащей структуры:
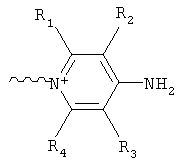
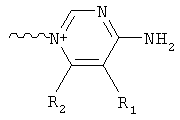
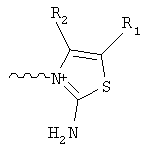
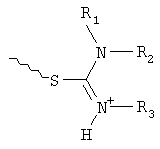
где R1, R2, R3, R4 независимо друг от друга представляют собой водород или С1-6алкил;
В представляет собой -(СН2)n-, где n принимает целые значения от 1 до 5;
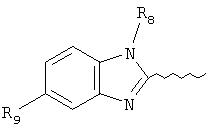
А выбирают из группы, содержащей структуры:
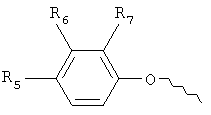
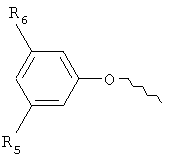
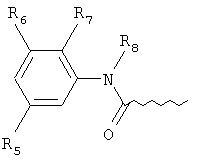
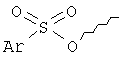
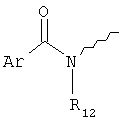
где R5 выбирают из группы, содержащей водород, С1-6-алкокси, СН2NR10R11, СН(СН3)R10R11,
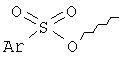 ,
,  ,
,  ,
,  ,
,
где R6 и R7 независимо представляют собой водород, С1-6алкил, С1-6алкокси, галоген;
R8 представляет собой водород или С1-6алкил;
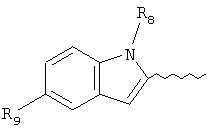
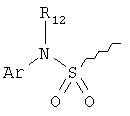
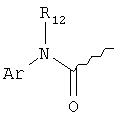
R9 выбирают из следующей группы, состоящей из:
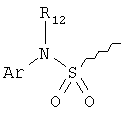 ,
, 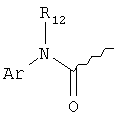 ,
,
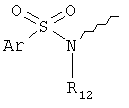
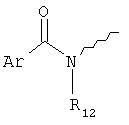
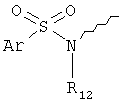
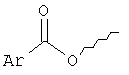
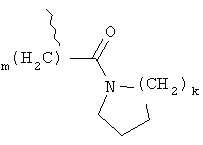
R10, R12 независимо друг от друга выбирают из группы, состоящей из водорода, С1-6алкила, (CH2)mCOOR13, (CH2)mCON(R13)2,
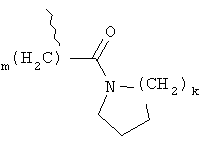 ,
, 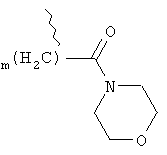 ,
, 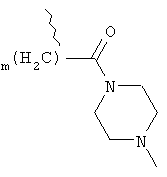
где m принимает целые значения от 1 до 4;
R13 представляет собой водород или С1-6алкил;
R11 представляет собой С1-6алкил или Ar;
Ar представляет собой фенил, пиридил, оксазолил, тиазолил, тиенил, фуранил, пиримидинил, пиридазинил, пиразинил, индолил, бензофуранил, бензотиофенил, имеющие от одного до пяти заместителей, выбранных из группы:
водород, С1-6алкил, C1-6алкокси, галоген, N(R13)2, ОН, NO2, CN, COOR13, CON(R13)2, SO2R13;
за исключением:
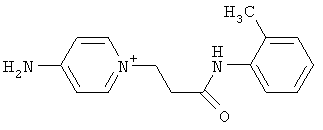
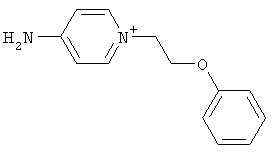
Исключенные соединения являются известными. 4-амино-1-[3-[(2-метилфенил) амино]-3-оксопропил] пиридиниум хлорид описан в Journal of Medicinal Chemistry, 17(7), 739-744, 1974 в статье Carbocyclic Derivatives related to indoramin (Карбоциклические производные, относящиеся к индорамину). 4-амино-1-(2-феноксиэтил)-пиридиниум бромид описан в Journal of Organic Chemistry, 26, 2740-7, 1961 в статье Application of sodium borohydride reduction to synthesis of substituted aminopiperidines, aminopiperazines, aminopyridines and hydrazines (Применение восстановления боргидридом натрия для синтеза замещенных аминопиперидинов, аминопиперазинов, аминопиридинов и гидразинов). Однако следует отметить, что в указанных источниках не описана возможность использования этих соединений в качестве антикоагулянтов.
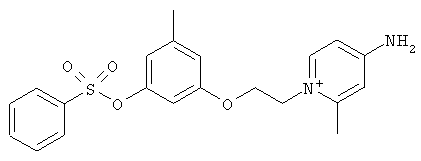
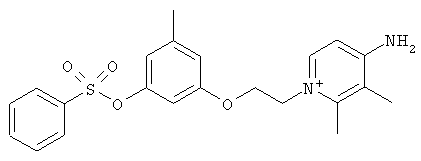
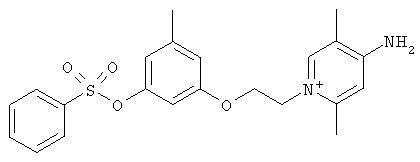
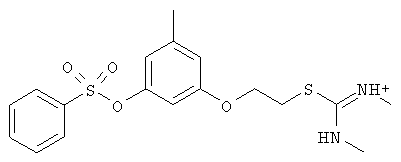
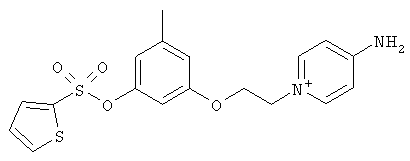
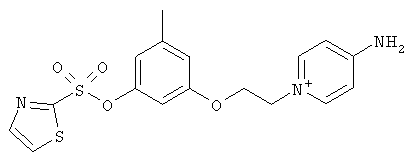
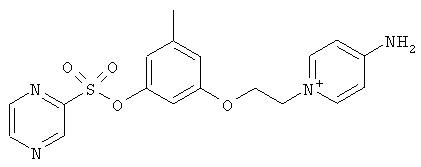
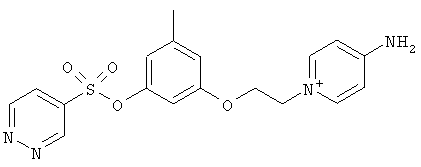
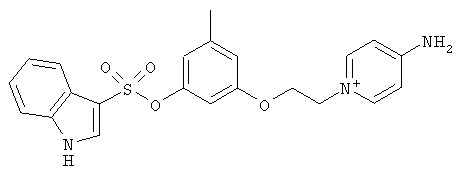
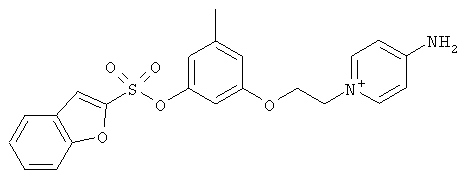
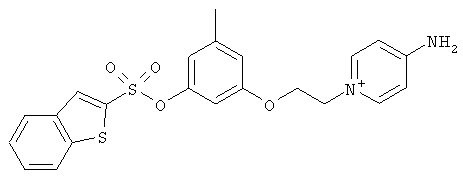
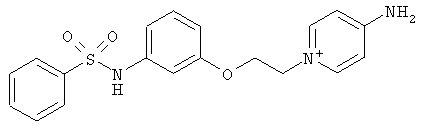
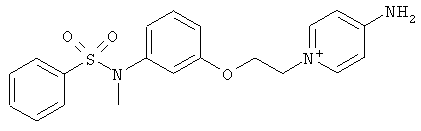
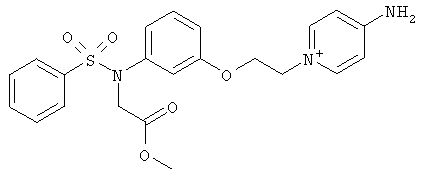
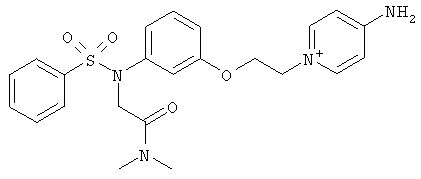
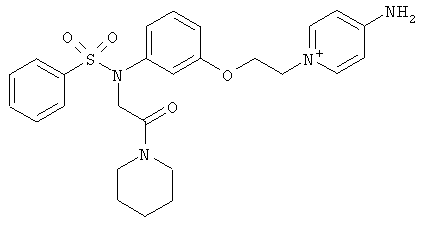
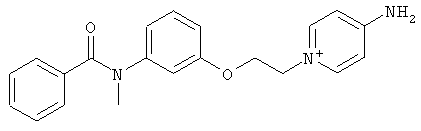
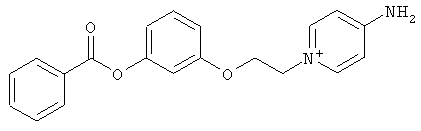
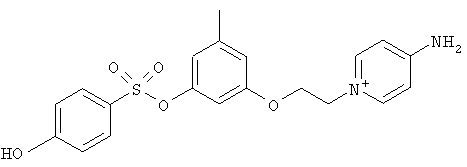
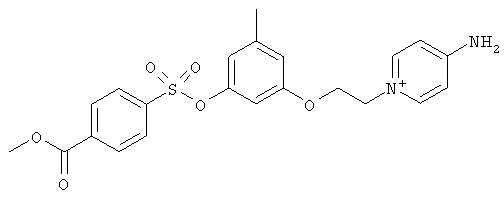
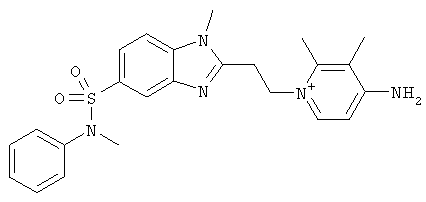
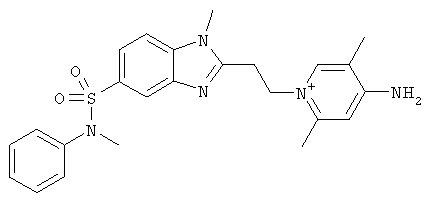
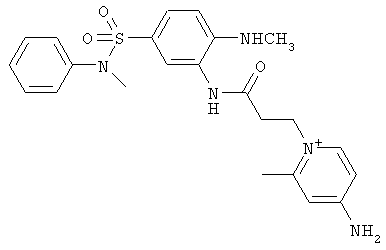
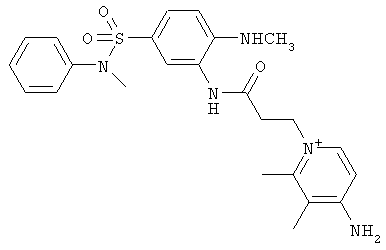
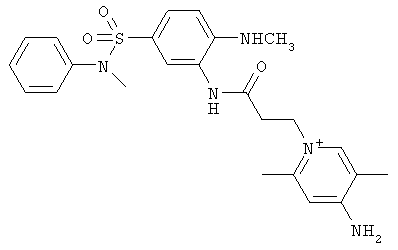
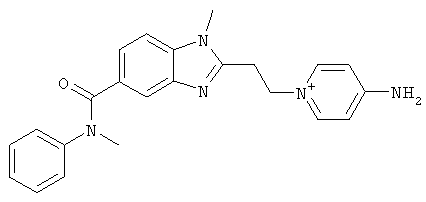
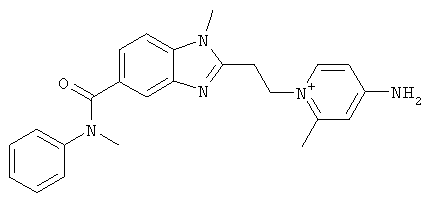
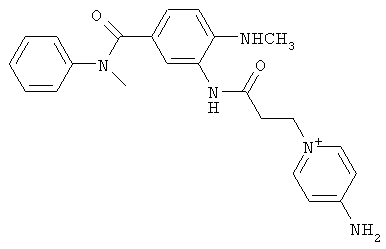
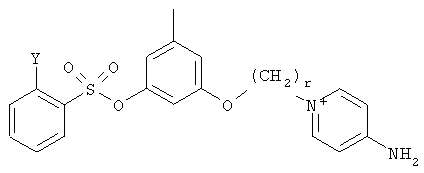
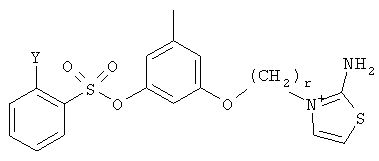
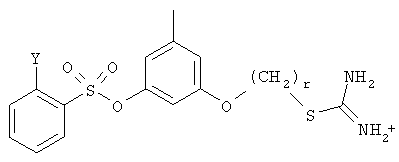
В предпочтительном варианте осуществления изобретения описаны следующие соединения по пункту 1, их фармацевтически приемлемые соли или сольваты:
а)
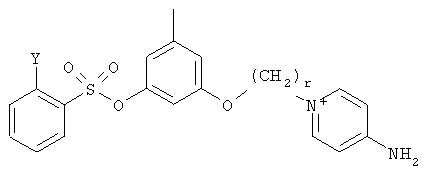 ,
,
б)
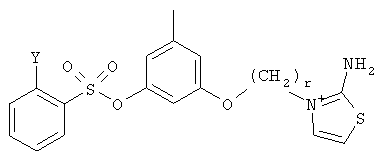 ,
,
в)
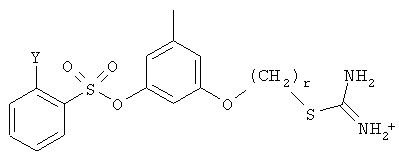
где Y выбирают из группы, состоящей из водорода, галогена, COOR13, CON(R13)2, SO2R13;
r принимает целые значения от 2 до 5.
Настоящее изобретение описывает также применение соединения формулы (I), его фармакологически приемлемых солей и сольватов в качестве антикоагулянтов для лечения и профилактики различных тромботических состояний, а также фармацевтическую композицию для лечения тромботических состояний, содержащую терапевтически эффективное количество соединения по пункту 1, и/или его фармацевтически приемлемых солей и/или сольватов, и фармацевтически приемлемый носитель.
Изобретение описывает также новый плазмозамещающий раствор, содержащий в качестве антикоагулянта соединение формулы (I), его приемлемые фармакологические соли или сольваты. Для создания такого раствора в стандартные плазмозамещающие растворы добавляют соединение формулы (I), его фармакологически приемлемые соли или сольваты. Концентрация добавляемого антикоагулянта зависит от его ингибирующей способности, и может меняться для разных соединений в широких пределах (от 0.01 нМ до 1 мМ). Раствор, содержащий антикоагулянт, позволяет частично корректировать те гиперкоагуляционные нарушения, которые возникают в организме при объемных переливаниях стандартных ПЗР, не содержащих факторов, а главное, ингибиторов свертывания. Предлагаемый новый ПЗР, содержащий искусственный синтетический низкомолекулярный антикоагулянт, выгодно отличается от аналогичного раствора, содержащего природный ингибитор тромбина ATIII, т.к. содержит вместо дорогого природного белка (ATIII) стандартный по составу и более дешевый ингибитор, который, кроме того, не создает опасности вирусного заражения пациента при инфузии ПЗР.
Соединения по изобретению могут быть введены любым подходящим образом, который обеспечил бы их бионакопление в крови. Это может быть достигнуто парентеральными способами введения, включая внутривенные, внутримышечные, внутрикожные, подкожные и внутрибрюшинные инъекции. Могут быть использованы и другие способы введения. Например, абсорбцией через желудочно-кишечный тракт, осуществляемой путем перорального введения соответствующих составов. Пероральное введение имеет преимущество за счет более легкого использования. Альтернативно, могут быть использованы способы введение через мышечную ткань - вагинальный и ректальный. Кроме того, соединения по изобретению могут быть введены через кожу (например, трансдермально) или ингаляцией. Понятно, что предпочтительный способ введения зависит от состояния, возраста и восприимчивости пациента.
Для перорального введения фармацевтические композиции могут представлять собой, например, таблетки или капсулы с фармацевтически приемлимыми добавками, такими как связующие агенты (например, пептизированный маисовый крахмал, поливинилпирролидинон или гидроксипропил метилцеллюлоза); наполнители (например, лактоза, микрокристаллическая целлюлоза, гидрофосфат кальция; стеарат магния, тальк или оксид кремния; картофельный крахмал или крахмальный гликолят натрия); или увлажнители (например, лаурилсульфат натрия). Таблетки могут быть покрыты оболочкой. Жидкие составы для орального введения могут быть выполнены в форме, например, растворов, сиропов или суспензий. Такие жидкие составы могут быть получены обычными средствами с фармацевтически приемлемыми добавками, такими как суспендирующие агенты (например, производные целлюлозы); эмульгаторы (например, лецитин), разбавители (очищенные растительные масла); и консерванты (например, метил или пропил-п-гидроксибензоаты или сорбиновая кислота). Составы могут также содержать соответствующие буферные соли, ароматизаторы, красители и подсластители.
Токсичность представленных антикоагулянтов была определена, используя стандартные фармацевтические процедуры на экспериментальных животных для определения LD50 (доза, летальная для 50% популяции). Для предпочтительных соединений по изобретению LD50 оказалось больше 367 мк/кг, что соизмеримо с летальной дозой прошедшего клинические испытания аргатробана, имеющего LD50=475 мк/кг.
Для того чтобы предмет настоящего изобретения был более понятен, ниже приведены некоторые примеры получения новых соединений, описаны методы исследования антикоагулянтной активности этих соединений и результаты данных исследований. Примеры носят иллюстративный характер, причем сущность изобретения ни в коей мере не ограничивается представленными примерами.
Пример 1
Синтез промежуточного продукта 3-(3-хлорпропокси)-5-метилфенола
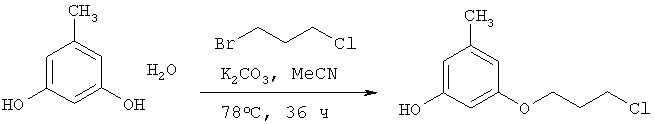
Смесь 3.8 г (27 ммоль) орцин гидрата, 4.8 г (30 ммоль) 1-бром-3-хлорпропана и 4.0 г (29 ммоль) карбоната калия кипятили в 30 мл ацетонитрила при перемешивании в течение 36 часов. Затем реакционную смесь упарили, растворили в 20 мл эфира, промыли два раза по 15 мл насыщенным раствором карбоната калия, водный слой отбросили, эфирный слой экстрагировали 3 раза по 15 мл 10% раствора гидроксида натрия. Эфирный слой отбросили, водный осторожно подкислили конц. HCl и затем экстрагировали 3×15 мл эфира. Эфирные экстракты объединили, промыли небольшим количеством насыщенного раствора гидрокарбоната натрия и сушили безводным сульфатом натрия, разбавили примерно 1/3 частью (по объему) гексана и профильтровали через слой силикагеля. После упаривания получили 1.7 г желтого масла, представляющего из себя смесь, состоящую из ~70% орцина (Rf 0.10), ~30% 3-(2-хлорпропокси)-5-метилфенола (Rf 0.26, выход ~1.2 г (22% на чистое вещество)).
Аналогичным образом из орцин гидрата и 1-бром-2-хлорэтана получен 3-(2-хлорэтокси)-5-метилфенол (Rf 0.26, выход ~1.1 г (20% на чистое вещество)), а из орцин гидрата и 1-бром-4-хлорбутана получен 3-(4-хлорбутокси)-5-метилфенол.
Пример 2
Синтез промежуточного продукта 3-(3-хлорпропокси)-5-метилфенилового эфира 2-фторбензолсульфокислоты.
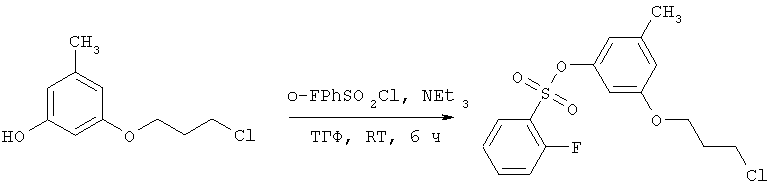
К раствору 1.3 г смеси из опыта 1А в 30 мл сухого ТГФ добавили 2.05 г (10 ммоль) 2-фторбензолсульфохлорида и 1.1 г (11 ммоль) триэтиламина. Смесь перемешивали в течение 6 часов, отфильтровали осадок гидрохлорида триэтиламмония и упарили. Полученное масло растворили в 20 мл эфира и несколько раз промыли 10 мл ~10-12% водным раствором аммиака для отделения избытка непрореагировавшего бензолсульфохлорида (контроль по ТСХ) и затем 10 мл ~20% соляной кислоты. После сушки безводным сульфатом натрия и упаривания получено 2.4 г светло-желтого масла, представляющего смесь целевого продукта и дибензолсульфонилированного орцина в соотношении ~2:1, выход на чистый целевой продукт 1.6 г (97% по реакции) (ТСХ пластинки Мерк 60, гексан-этилацетат 2:1. Rf 0.35 - продукт, Rf 0.25 - примесь дибензилового эфира.
Аналогичным образом из 3-(2-хлорэтокси)-5-метилфенола, 3-(3-хлорпропокси)-5-метилфенола, 3-(4-хлорбутокси)-5-метилфенола и соответствующих арилсульфохлоридов получены:
3-(3-хлорпропокси)-5-метилфениловый эфир 2-хлор бензолсульфокислоты (77% на чистое вещество).
3-(3-хлорпропокси)-5-метилфениловый эфир бензолсульфокислоты (60%).
3-(3-хлорпропокси)-5-метилфениловый эфир 2-карбметокси бензолсульфокислоты (56%).
3-(2-хлорэтокси)-5-метилфениловый эфир бензолсульфокислоты (72%).
3-(2-хлорэтокси)-5-метилфениловый эфир 2-хлор бензолсульфокислоты (35%).
3-(2-хлорэтокси)-5-метилфениловый эфир 2-фтор бензолсульфокислоты (34%).
3-(2-хлорэтокси)-5-метилфениловый эфир 2-карбметоксибензолсульфокислоты (37%).
3-(4-хлорбутокси)-5-метилфениловый эфир бензолсульфокислоты (45%).
3-(4-хлорбутокси)-5-метилфениловый эфир 2-хлор бензолсульфокислоты (27%).
3-(4-хлорбутокси)-5-метилфениловый эфир 2-фтор бензолсульфокислоты (32%).
3-(4-хлорбутокси)-5-метилфениловый эфир 2-карбметокси бензолсульфокислоты (21%).
Пример 3
Синтез промежуточного продукта 3-(3-иодпропокси)-5-метилфенилового эфира 2-фторбензолсульфокислоты.
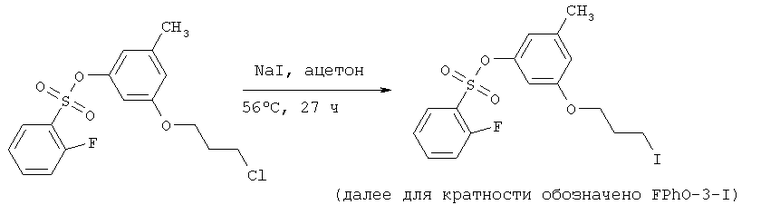
К 2.4 г смеси из предыдущего опыта (2А) в 30 мл сухого ацетона добавили 2 г (13 ммоль) прокаленного иодида натрия и кипятили в течение 27 часов. Затем разбавили реакционную смесь 10 мл гексана и профильтровали и упарили. Полученное темно-желтое масло растворили в 20 мл смеси эфир-гексан (2:3), профильтровали через слой силикагеля (2 см, Lancaster) и упарили. Получили 2.45 г желтого масла, содержащего 3-(2-иодэтокси)-5-метилфениловый эфир 2-фторбензолсульфокислоты (Rf 0.35), и дибензоилсульфоновый эфир орцина (Rf 0.25).
Аналогичным образом из соответствующих хлоридов получены:
3-(3-иодпропокси)-5-метилфениловый эфир бензолсульфокислоты
3-(3-иодпропокси)-5-метилфениловыйэфир 2-хлорбензолсульфокислоты
3-(3-иодпропокси)-5-метилфениловый эфир 2-карбметокси бензолсульфокислоты
3-(2-иодэтокси)-5-метилфениловый эфир бензолсульфокислоты
3-(2-иодэтокси)-5-метилфениловый эфир 2-хлор бензолсульфокислоты
3-(2-иодэтокси)-5-метилфениловый эфир 2-фтор бензолсульфокислоты
3-(2-иодэтокси)-5-метилфениловыйэфир 2-карбметоксибензолсульфокислоты
3-(4-иодбутокси)-5-метилфениловый эфир бензолсульфокислоты
3-(4-иодбутокси)-5-метилфениловый эфир 2-хлор бензолсульфокислоты
3-(4-иодбутокси)-5-метилфениловый эфир 2-фтор бензолсульфокислоты
3-(4-иодбутокси)-5-метилфениловый эфир 2-карбметокси бензолсульфокислоты
Пример 4
Синтез 4-амино-1-(3-(3-метил-5-(2-фторбензолсульфонилокси)фенокси)пропил)-пиридиний иодида (НС_029S_IOC).
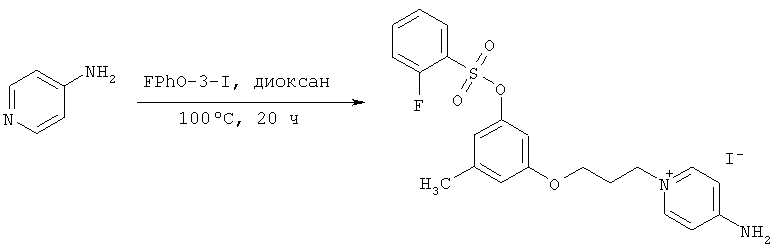
Смесь 0.65 г "сырого иодида" (из опыта 3А) (в рассчете на 70% содержание активного вещества) и 0.09 г (0.95 ммоль) 4-аминопиридина в 10 мл сухого диоксана кипятили в течение 20 часов. После того как смесь остыла, раствор упарили, полученное масло затирали с несколькими порциями эфира до затвердевания. Твердый остаток отфильтровали и перекристаллизовывали из смеси диоксан-ацетонитрил (5:1), осадок соли отфильтровали, промыли эфиром. После сушки в вакууме получено 0.4 г (72%) светло-бежевой соли, содержавшей ок. 4-5% неидентифицированных примесей. Для их удаления вещество было еще раз перекристализовано из той же системы. Получено 0.25 г светлого порошка.
Аналогичным образом из соответствующих иодидов и гетероциклических соединений, тиомочевины и производных тиомочвины были получены:
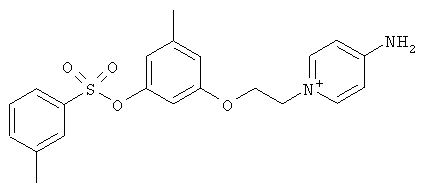
4-амино-1-(3-(3-метил-5-(бензолсульфонилокси)фенокси)пропил)-пиридиний иодид (НС_016s_IOC).
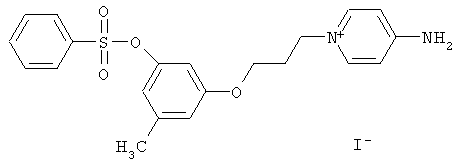
Выход 78%.
ЯМР 1Н (Bruker DRX500, 500 МГц, ДМСО-d6, м.д.,. J Гц); 2.20 с, 3H; 3.88 т, 2Н, J=5.50; 2.16 м, 2Н, J=6.11; 4.25 т, 2Н, J=6.71; 6.31 с, 1Н, 6.44 с, 1Н, 6.66 с, 1Н; 7.68 т, 2Н, J=7.94, 7.82 т, 1Н, J=7.94, 7.87 д, 2Н, J=7.32; 6.81 д, 2Н, J=6.72, 8.17 д, 2Н, J=6.72; 8.09 с, 2Н.
2-амино-1-(3-(3-метил-5-(бензолсульфонилокси)фенокси)пропил)-тиазолий иодид (НС_017s_IOC).
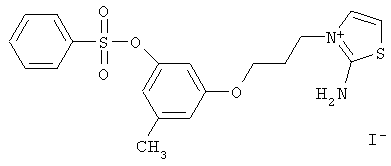
Выход 65%.
ЯМР 1Н (Bruker DRX500, 500 МГц, ДМСО-d6, м.д., J Гц): 2.21 с, 3Н; 3.93 т, 2Н, J=6.11; 2.11 м, 2Н, J=6.10; 4.15 т, 2Н, J=6.71; 6.35 с, 1Н, 6.44 с, 1Н, 6.68 с, 1Н; 7.69 т, 2Н, J=7.33, 7.84 т, 1Н, J=7.32, 7.88 д, 2Н, J=7.93; 7.02 д, 1Н, J=4.27, 7.42 д, 1Н, J=4.27; 9.42 с, 2Н.
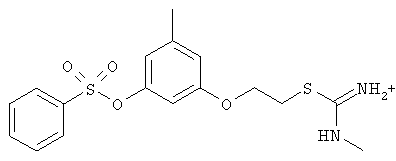
3-(3-метил-5-(бензолсульфонилокси)фенокси)пропил-изотиоуроний иодид (НС_018s_IOC).
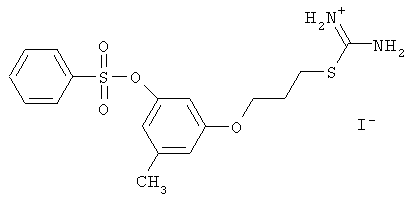
Выход 80%.
ЯМР 1Н (Bruker DRX500, 500 МГц, ДМСО-d6, м.д., J Гц): 2.21 с, 3Н; 3.95 т, 2Н, J=6.10; 2.00 м, 2Н, J=6.71; 3.25 т, 2Н, J=7.32; 6.40 с, 1Н, 6.25 с, 1Н, 6.74 с, 1Н; 7.69 т, 2Н, J=7.94, 7.84 т, 1Н, J=7.93, 7.89 д, 2Н, J=7.33; 9.03 с, 4Н.
4-амино-1-(2-(3-метил-5-(бензолсульфонилокси)фенокси)этил)-пиридиний иодид (НС_019s_IOC).
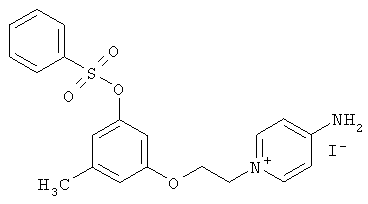
Выход 60%.
ЯМР 1Н (Bruker DRX500, 500 МГц, ДМСО-d6, м.д., J Гц): 2.20 с, 3Н; 4.24 т, 2Н, J=4.88; 4.48 т, 2Н, J=4.89; 6.39 с, 1Н, 6.45 с, 1Н, 6.73 с, 1Н; 7.68 т, 2Н, J=7.93, 7.82 т, 1Н, J=7.93, 7.87 д, 2Н,1=7.32; 6.82 д, 2Н, J=7.32, 8.18 д, 2Н, J=7.33; 8.14 с, 2Н.
2-(3-метил-5-(бензолсульфонилокси)фенокси)этил-изотиоуроний иодид (НС_020s_IOC).
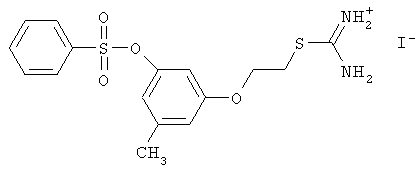
Выход 45%.
ЯМР 1Н (Bruker DRX500, 500 МГц, ДМСО-d6, м.д., J Гц): 2.22 с, 3Н; 4.11 т, 2Н, J=5.49; 3.54 т, 2Н, J=5.49; 6.41 с, 1Н, 6.48 с, 1Н, 6.76 с, 1Н; 7.69 т, 2Н, J=7.93, 7.84 т, 1Н, J=7.93, 7.89 д, 2Н, J=7.32; 9.10 с, 4Н.
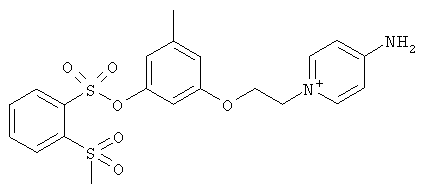
2-(3-метил-5-(2-хлорбензолсульфонилокси)фенокси)этил-изотиоуроний иодид (НС_024s_IOC)
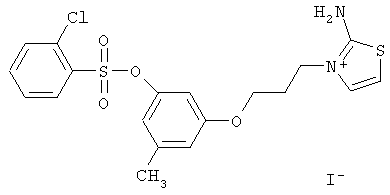
Выход 53%.
ЯМР 1Н (Bruker DRX500, 500 МГц, ДМСО-d6, м.д., J Гц): 2.21 с, 3Н; 3.95 т, 2Н, J=5.50; 2.12 м, 2Н, J=5.50; 4.15 т, 2Н, J=6.10; 6.42 с, 1Н, 6.51 с, 1Н, 6.70 с, 1Н; 7.59 т, 1Н, J=7.32, 7.83 т, 1Н, J=7.94, 7.88 д, 1Н, J=7.94, 7.95 д, 1Н, J=7.94; 7.01 д, 1Н, J=4.27, 7.42 д, 1Н, J=4.27; 9.39 с, 2Н.
3-(3-метил-5-(2-хлорбензолсульфонилокси)фенокси)пропил-изотиоуроний иодид (НС_026s_IOC).
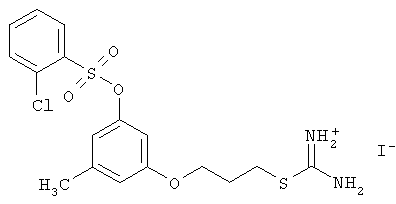
Выход 55%.
ЯМР 1Н (Bruker DRX500, 500 МГц, ДМСО-d6, м.д., J Гц): 2.22 с, 3Н; 3.97 т, 2Н, J=6.10; 2.01 м, 2Н, J=7.33, J=6.10; 4.26 т, 2Н, J=7.33; 6.47 с, 1Н, 6.51 с, 1Н, 6.75 с, 1Н; 7.60 т, 1Н, J=7.93, 7.84 т, 1Н, J=7.94, 7.88 д, 1Н, J=7.93, 7.96 д, 1Н, J=7.94; 8.95 с, 2Н, 9.07 с, 2Н.
4-амино-1-(2-(3-метил-5-(2-хлорбензолсульфонилокси)фенокси)этил)-пиридиний иодид (НС_025s_IOC).
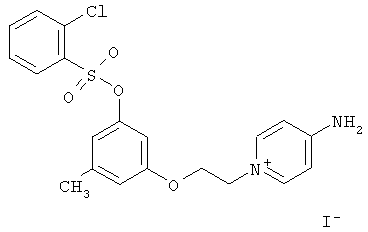
Выход 58%.
ЯМР 1Н (Bruker DRX500, 500 МГц, ДМСО-d6, м.д., J Гц): 2.20 с, 3Н; 4.26 т, 2Н, J=4.88; 4.49 т, 2Н, J=4.88; 6.45 с, 1Н, 6.51 с, 1Н, 6.74 с, 1Н; 7.58 т, 1Н, J=7.93, 7.84 т, 1Н, J=7.94, 7.88 д, 1Н, J=7.93, 7.94 д, 1Н, J=7.94; 6.82 д, 2Н, J=7.32, 8.18 д, 2Н, J=7.33; 8.14 с, 2Н.
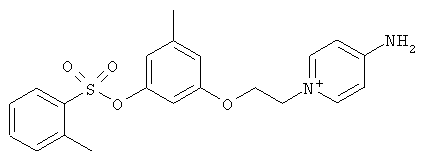
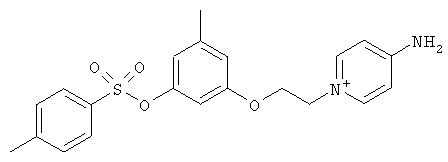
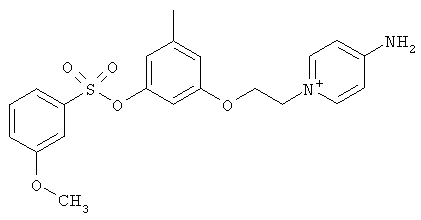
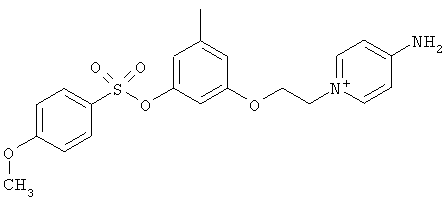
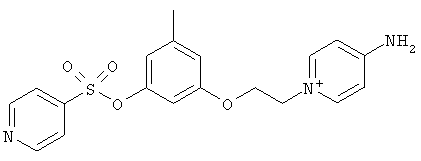
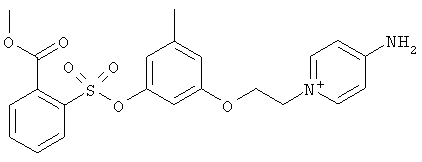
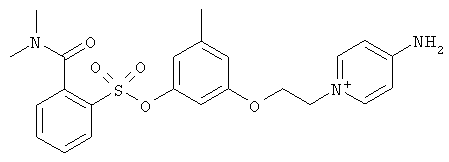
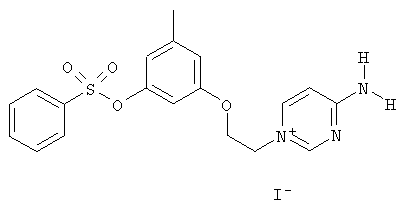
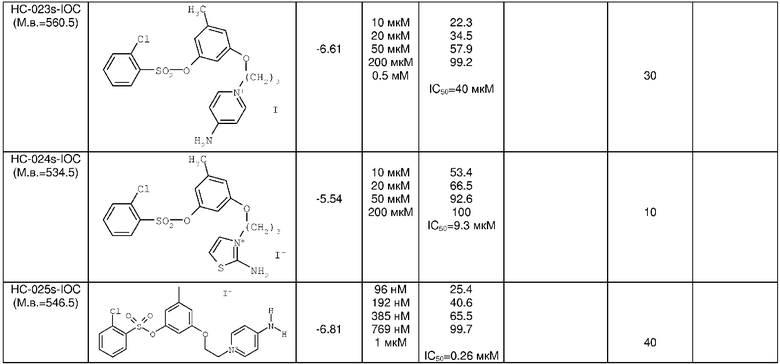
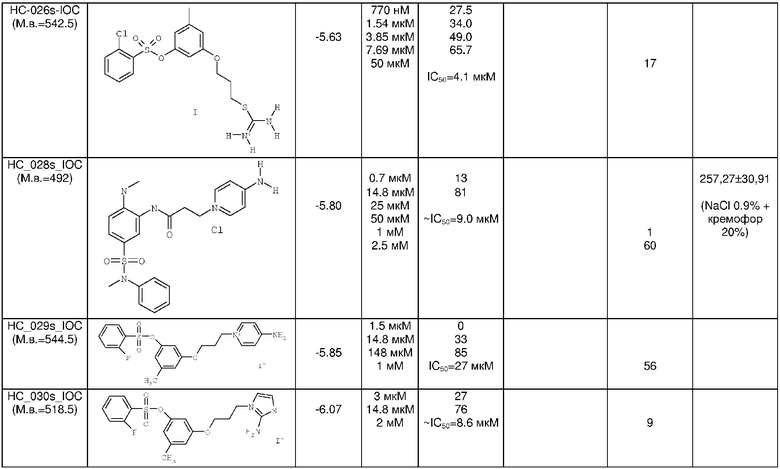
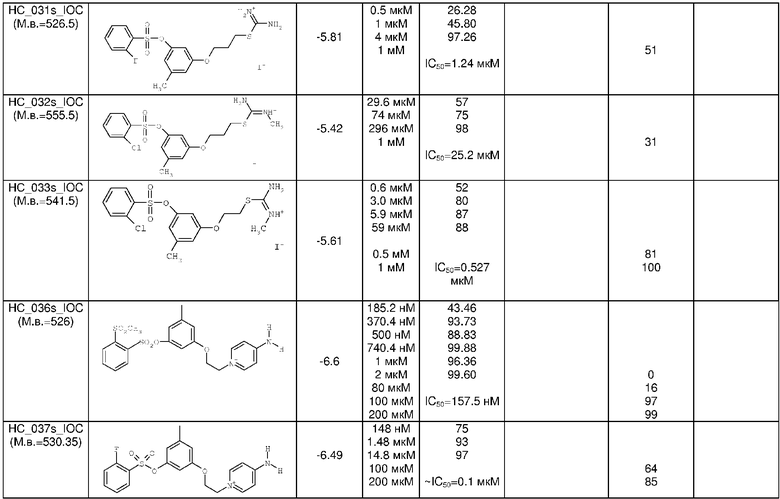
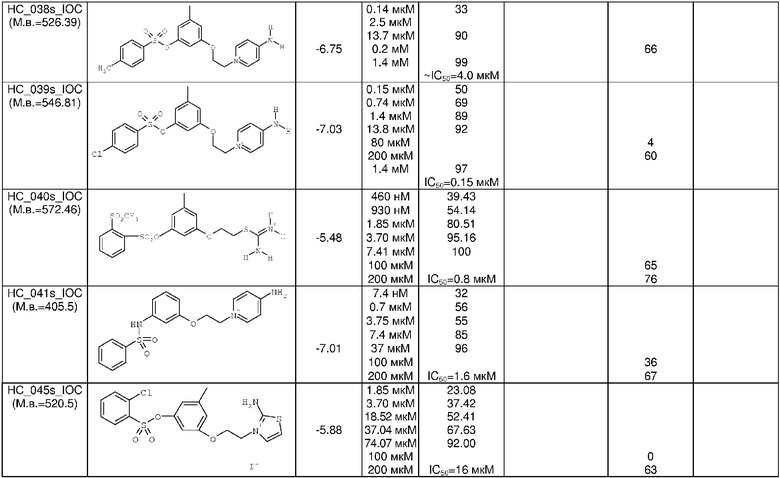
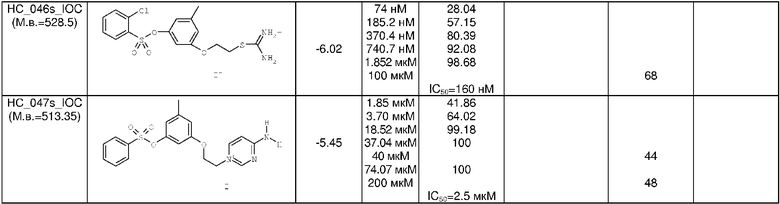
Аналогичным образом, по методикам, изложенным в примерах 1-4 с использованием различных арилсульфохлоридов и гетероциклилсульфохлоридов, был получен еще ряд веществ, химические формулы которых, а также параметры масс-спектра и рассчитанная величина скоринг-функции приведены в таблице 1. Вещества могут быть получены в виде соответствующих иодидов, бромидов, хлоридов или иных солей.
Пример 5
Синтез соединений
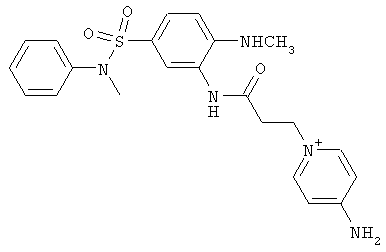
1. 4-хлор-3-нитробензол-1-сульфонилхлорид
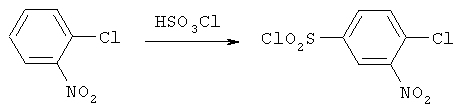
15 г о-нитрохлоранилина вносят в 30 мл хлорсульфоновой кислоты при перемешивании и нагревают 2 часа при 100°С, 2 часа при 110°С и 5 часов при 127°С. Реакционную массу охлаждают до комнатной температуры и выливают на 140 г колотого льда. Осадок отфильтровывают, промывают ледяной водой и сушат на воздухе. Получают 15 г 4-хлор-3-нитробензол-1-сульфонилхлорида.
2. 4-хлор-N-метил-3-нитро-N-фенилбензолсульфонамид
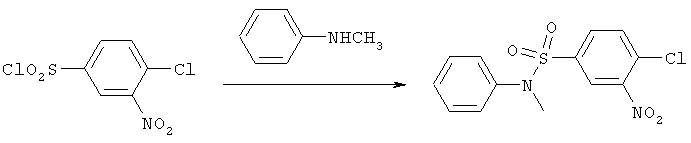
10.6 г (0.041 моль) 4-хлор-3-нитробензол-1-сульфонилхлорида растворяют в 50 мл толуола, добавляют 4,14 г (0,041 моль) триэтиламина и при перемешивании вносят 4,4 г (0,041 моль) N-метиланилина. Реакционную смесь нагревают при 70-80°С в течение 1 часа, после охлаждения толуольный раствор дважды промывают 30 мл воды. Реакционную массу концентрируют в вакууме и осадок перекристаллизовывают из этанола. Выход 4-хлор-N-метил-3-нитро-N-фенилбензолсульфонамида - 9,4 г (61%).
3. N-метил-4-(метиламино)-3-нитро-N-фенилбензолсульфонамид
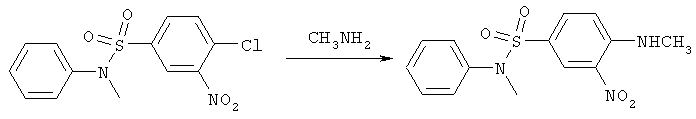
9,4 г (0,029 моль) 4-хлор-N-метил-3-нитро-N-фенилбензолсульфонамида растворяют в 50 мл этанола и добавляют 25 мл 40% водного раствора метиламина. Реакционную смесь нагревают до 70°С и перемешивают при этой температуре в течение часа. После охлаждения реакционной массы осадок отфильтровывают, промывают этанолом и сушат при 60°С. Выход N-метил-4-(метиламино)-3-нитро-N-фенилбензолсульфонамида - 9.0 г (97%).
4. 3-амино-N-метил-4-(метиламино)-N-фенилбензолсульфонамид
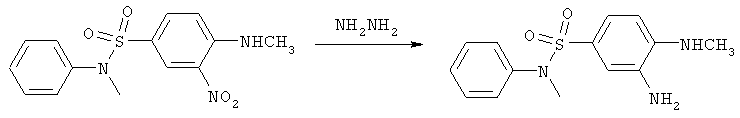
9 г (0,028 моль) N-метил-4-(метиламино)-3-нитро-N-фенилбензолсульфонамида растворяют в 90 мл изопропилового спирта. Добавляют 11 мл гидразин-гидрата, 2 г активированного угля и раствор 0,5 г FeCl3·6Н2О в 10 мл спирта. Реакционную массу кипятят в течение 8 часов. Уголь отфильтровывают, фильтрат упаривают досуха. Получают 8,1 г (99%) 3-амино-N-метил-4-(метиламино)-N-фенилбензолсульфонамида.
5. 3-хлор-N-(5-N-метил-N-фенилсульфамоил)-2-(метиламино)фенил)пропанамид
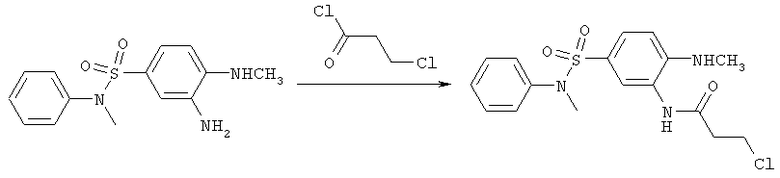
К раствору 5,4 г (0,018 моль) 3-амино-N-метил-4-(метиламино)-N-фенилбензолсульфонамида и 1,81 г (0,018 моль) триэтиламина в 16 мл диметилформамида добавляют 2,32 г (0,018 моль) хлорпропионилхлорида при охлаждении на ледяной бане (~5°С). Реакционную массу перемешивают в течение 5-ти часов при комнатной температуре. Добавляют 14 мл воды и 5 мл ацетонитрила. Осадок отфильтровывают через 4-5 часов.
Выход 3-хлор-N-(5-(N-метил-N-фенилсульфамоил)-2-(метиламино)фенил)пропанамида-3,1 г (45%).
6. 4-амино-1-(3-(5-(N-метил-N-фенилсульфамоил)-2-(метиламино)фениламино)-3-оксопропил)пиридинийхлорид.
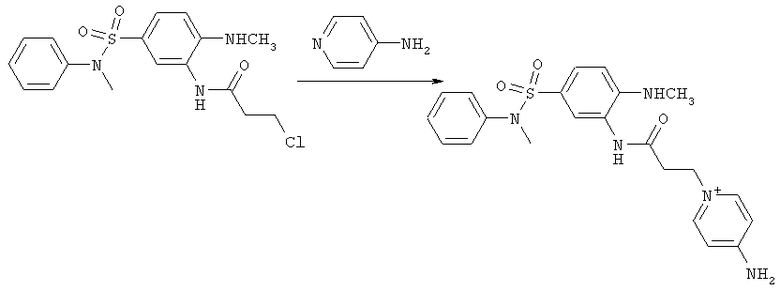
1 г (0,0026 моль) 3-хлор-N-(5-(N-метил-N-фенилсульфамоил)-2-(метиламино)фенил)пропанамида и 0,73 г (0,0078 моль) 4-аминопиридина кипятят в 50 мл безводного ацетона в течение 50 часов. Выделившийся осадок отфильтровывают и перекристаллизовывают из смеси ацетонитрил/этанол - 10/1.
Выход 4-амино-1-(3-(5-(N-метил-N-фенилсульфамоил)-2-(метиламино)фениламино)-3-оксопропил)пиридинийхлорида - 0,54 г (43%).
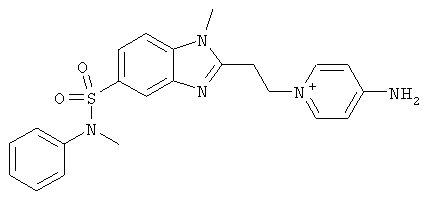
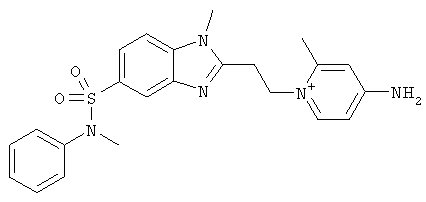
7. 4-амино-1-(2-(1-метил-5-(N-метил-N-фенилсульфамоил)-1Н-бензо[d]имидазол-2-ил)этил)пиридинийхлорид.
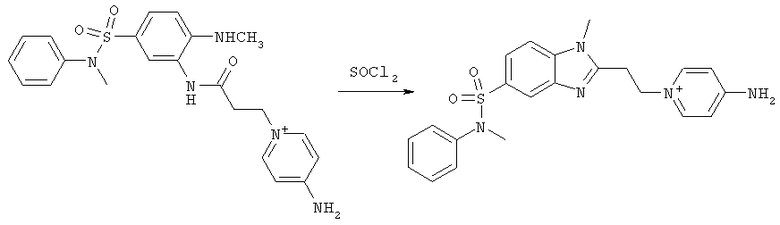
К суспензии 0, 2 г (0,00042 моль) 4-амино-1-(3-(5-(N-метил-N-фенилсульфамоил)-2-(метиламино)фениламино)-3-оксопропил)пиридинийхлорида в 8 мл ацетонитрила добавляют 0,2 мл тионилхлорида и реакционную массу кипятят в течение 10 мин. Оставляют при комнатной температуре на 24 часа. После выдержки реакционную массу разбавляют 8 мл диэтилового эфира, выделившийся осадок отфильтровывают и перекристаллизовывают из смеси ацетонитрил/этанол (абс) в соотношении 10/1. Выход 4-амино-1-(2-(1-метил-5-(N-метил-N-фенилсульфамоил)-1Н-бензо[d]имидазол-2-ил)этил)пиридинийхлорида - 0,055 г (26%).
Аналогичным образом, по методикам, изложенным в примере 5, был получен ряд веществ, химические формулы которых, а также параметры масс-спектра и рассчитанная величина скоринг-функции приведены в таблице 2. Вещества могут быть получены в виде соответствующих иодидов, бромидов, хлоридов или иных солей.
Пример 6
Оценка влияния синтезированных соединений на состояние гемостаза.
Для оценки статуса системы свертывания в присутствии вновь синтезированных соединений в настоящей работе был использован стандартный коагулогический тест - тромбиновое время свертывания (ТВ), а также два современных in vitro теста: тест генерации тромбина и измерение скорости роста сгустка в пространстве. В отличие от стандартных тестов измерения времен свертывания (ПВ и АЧТВ), которые проводятся в условиях максимальной активации свертывания по внешнему или внутреннему пути, соответственно, и, следовательно, не способны обнаружить состояние гиперкоагуляции в исследуемой системе, данные тесты проводятся при гораздо более слабой исходной активации, близкой к существующей в организме. Это делает их чувствительными как к состоянию гипо-, так и к состоянию гиперкоагуляции в исследуемых образцах плазмы.
Измерение тромбинового времени
Тест тромбинового времени свертывания характеризует последний этап в каскаде реакций свертывания - превращение фибриногена плазмы под действием добавляемого в систему тромбина в нерастворимый фибриновый сгусток. К бедной тромбоцитами плазме (РРР), которую получают путем центрифугирования (15 минут при 1300 g) крови, заготовленной на 3.8%-ном цитрате натрия (рН 5.5) (соотношение кровь:цитрат=9:1), добавляют фиксированное количество тромбина со стандартной активностью. Измеряемое время образования сгустка в этом тесте зависит от количества ингибиторов тромбина в плазме. Оно удлиняется, когда в системе присутствует сильный тромбиновый ингибитор, который уменьшает активность добавляемого фермента и, таким образом, задерживает свертывание.
Тромбиновое время измеряли по стандартной методике (З.С.Баркаган, А.П.Момот.Диагностика и контролируемая терапия нарушений гемостаза. М.: "Ньюдиамед", 2001, стр.87-89). В кювету агрегометра фирмы Biola Ltd. (Россия) вносили 90 мкл РРР, прогретой на водяной бане при 37°С в течение 3 минут, добавляли 10 мкл смеси тестируемого вещества с буфером (до достижения конечной концентрации в кювете от 0.005 мМ до 5 мМ для разных соединений) и 100 мкл раствора тромбина (стандартизированного по активности на контрольной нормальной плазме). Измеряли время образования сгустка. Результаты усредняли по трем независимым экспериментам.
Измерение эндогенного тромбинового потенциала (тест генерации тромбина)
Метод был описан подробно в целом ряде работ (Hemker НС, Giesen PL, Ramjee М, Wagenvoord R, Beguin S. The thrombogram: monitoring thrombin generation in platelet-rich plasma. Thromb. Haemost, 2000; 83(4): 589-591; Hemker НС, AlDieri R, Beguin S. Thrombin generation assays: accruing clinical relevance. Curr. Opin. Hematol., 2004, 11(3): 170-175; Hemker НС, Giesen P, AlDieri R, Regnault V, de Smed E, Wagenvoord R, Lecompte T, Beguin S. The calibrated automated thrombogram (CAT): a universal routine test for hyper- and hypocoagulability. Pathophysiol. Haemost. Thromb., 2002, 32(5-6):249-253). При проведении этого теста измеряют кинетику и суммарное количество активного тромбина, образовавшегося в образце плазмы за определенное время, при стандартной величине активации свертывания в пробе. В нашем варианте метода измерение концентрации тромбина осуществляется с помощью медленного флюорогенного субстрата (ВОС-Ile-Gly-Arg-AMC), при расщеплении которого тромбином образуется сильно флюоресцирующий продукт 7-амино-4-метилкумарин (АМС). Использование именно флюорогенного, а не хромогенного субстрата вызвано тем, что на величину флюоресценции в образце практически не влияет образование в измерительной ячейке твердого фибринового сгустка, в то время, как для хромогенного субстрата это является условием, ограничивающим его применение (Hemker НС, Giesen P, AlDieri R, Regnault V, de Smed E, Wagenvoord R, Lecompte T, Beguin S. The calibrated automated thrombogram (CAT): a universal routine test for hyper- and hypocoagulability. Pathophysiol. Haemost. Thromb., 2002, 32(5-6):249-253). С другой стороны, известно, что основная часть тромбина в образце (около 95%) нарабатывается после того, как первый сгусток уже образовался (Hemker НС, AlDieri R, Beguin S. Thrombin generation assays: accruing clinical relevance. Curr. Opin. Hematol., 2004, 11(3):170-175).
Кинетика возникновения и последующего исчезновения тромбина в плазме после активации свертывания представлена на Фиг.1. Здесь показана зависимость концентрации активного тромбина в образце плазмы от времени. Эндогенным тромбиновым потенциалом (ЭТП) называют площадь под кривой генерации тромбина (в нашем случае от 0 до 50 минут инкубации). Характеристиками кривой являются также такие параметры, как tmax - время достижения максимальной концентрации тромбина в пробе, Amax - максимальная концентрация тромбина в системе и tлаг - время до начала свертывания, за которое условно принимают время достижения тромбином концентрации 5 нМ.
Очевидно, что важную информацию о свертывании несет не только интегральная величина ЭТП, но и сам вид кривых зависимости концентрации тромбина от времени. Так в условиях сниженной концентрации факторов и ингибиторов свертывания максимумы концентрации тромбина могут стать более низкими, но широкими, хотя суммарный ЭТП может не измениться. Присутствие в плазме дополнительных ингибиторов тромбина должно снижать величину ЭТП и максимальной достигаемой концентрации тромбина, а также увеличивать лаг-период свертывания и время достижения максимальной концентрации тромбина.
Измерения проводили следующим образом. В лунки 96-луночного планшета помещали по 90 мкл нормальной донорской плазмы (РРР), от 0 до 20 мкл раствора испытуемого вещества и от 20 до 0 мкл раствора буфера (20 мМ HEPES, 140 мМ NaCl, pH 7.5), таким образом, чтобы суммарный объем добавленного вещества и буфера составлял всегда 20 мкл. После этого в каждую лунку планшета добавляли 20 мкл раствора флюорогенного субстрата (исходная концентрация 5 мМ) и в течение 3-5 минут прогревали плазму при 37°С. После этого во все лунки одновременно (многоканальной пипеткой) вносили по 25 мкл раствора активатора свертывания. Активатором служил раствор тромбопластина, приготовленный из стандартного реагента тромбопластина для измерения протромбинового времени (ПВ) (РЕНАМ, Россия), разбавленный в 250 раз тем же буфером, содержащим дополнительно 80 мМ CaCl2. Момент внесения активатора являлся началом отсчета. Запись кинетики накопления флюоресцирующего продукта реакции (АМС) проводили в течение 60 минут. Скорость накопления продукта в каждый момент времени пропорциональна концентрации тромбина, присутствующего в плазме в данный момент. Таким образом, продифференцировав кривую накопления АМС, можно получить зависимость концентрации тромбина в системе от времени и затем определить параметры, характеризующие эту кривую. Условные единицы флюоресценции для каждого образца были переведены в абсолютные концентрации АМС с помощью калибровки сигнала по величинам сигналов известных концентраций АМС в данных образцах плазмы. Линейность метода в широком диапазоне концентраций АМС была проверена предварительно.
Измерение скорости роста сгустка в пространстве.
Скорость роста сгустка в пространстве характеризует динамику свертывания, которое является процессом, развивающимся не только во времени, но и в пространстве. В данном методе с помощью измерения светорассеяния определяется размер сгустка в разные моменты времени после начала свертывания. Активация свертывания проводится в системе без перемешивания строго локализованным в пространстве активатором, в качестве которого использовали либо просто стеклянную пластинку со шлифованным краем для контактной активации свертывания по внутреннему пути, либо полиэтилентерфталатные пленки, покрытые фибробластами, несущими на поверхности тканевой фактор, активирующий свертывание по внешнему пути. В случае активации свертывания по внешнему пути в исходную плазму добавляли ингибитор трипсина из кукурузы (CTI) в концентрации 200 мкг/мл, который предотвращал контактную активацию свертывания в системе. Измерения проводили в плазме, свободной от тромбоцитов (PFP), которую получали путем дополнительного центрифугирования РРР в течение 10 минут при 10000 g.
Микрокамеру собирали в 35 мм полистироловой чашке Петри. При активации свертывания по внешнему пути торец стеклянной пластинки толщиной 1 мм оборачивали полиэтилентетфталатной пленкой, на которой были выращены фибробласты. После этого пластинку с пленкой фиксировали на дне чашки 2-сторонним скотчем. Торец стекла представлял собой боковой край микрокамеры, покрытый активатором. На верхнюю плоскость стекла таким же образом прикрепляли выступающую за край стекла, закрашенную с внешней стороны черным полистироловую пластинку, которая формировала верхнюю поверхность микрокамеры. Рекальцифицированную плазму свободную от тромбоцитов (финальная концентрация добавленного CaCl2 20 мМ) с добавкой или без предполагаемого ингибитора тромбина аккуратно заливали между верхней пластинкой и дном камеры таким образом, чтобы она пришла в соприкосновение с боковой активирующей свертывание стенкой. Момент соприкосновения плазмы с активатором принимали за t=0. Затем чашку герметично закрывали и помещали в термостатируемую при 37°С кювету с прозрачным дном, через которое плазма в чашке освещалась светом красных диодов (λ=660 нм). Изображение постоянной области микрокамеры (7.2×5.4 мм) каждые 30 сек фиксировалось видеокамерой OS-75D (Mintron Enterprise, Тайвань), сопряженной с платой захвата изображений EZ98 (Lifeview Inc., USA), позволяющей оцифровку и ввод в память компьютера изображений с видеокамеры.
При обработке серии записанных изображений были получены профили светорассеяния, соответствующие различным временам от начала эксперимента. В процессе роста сгусток все дальше распространялся от поверхности активатора вглубь плазмы. Размер сгустка в каждом кадре был определен как расстояние от активатора до края сгустка, за который принимали точку, где величина светорассеяния составляла половину максимальной. Скорость роста сгустка была затем определена как тангенс угла наклона прямой зависимости размеры сгустка от времени. Подробнее метод описан в работах: Ovanesov MV, Krasotkina JV, Ul'yanova LI, Abushinova KV, Plyushch OP, Domogatskii SP, Vorob'ev AI, Ataullakhanov FI. Hemophilia A and В are associated with abnormal spatial dynamics of clot growth. Biochim. Biophys. Acta, 2002, 1572(1):45-57; Ovanesov MV, Lopatina EG, Saenko EL, Ananyeva NM, Ul'yanova LI, Plyushch OP, Butilin AA, Ataullakhanov FI. Effect of factor VIII on tissue factor-initiated spatial clot growth. Thromb. Haemost, 2003, 89(2):235-242.
На Фиг.2 представлен ряд последовательных исходных фотографий сгустка, растущего в нормальной плазме (полоса слева - стеклянный активатор, фибрин - светлые области). На первом кадре выбрана полоса, перпендикулярная активатору, вдоль которой вычислялись профили светорассеяния.
Профили светорассеяния растущего сгустка в различные моменты времени после начала свертывания можно видеть на Фиг.3.
Последняя стадия обработки результатов представляет собой расчет скорости роста сгустка на основании полученных профилей светорассеяния. Размер сгустка в каждый момент времени оценивали как расстояние соответствующего профиля светорассеяния от активатора (на половине максимальной высоты профиля). На Фиг.4 представлена полученная зависимость размера сгустка от времени. Скорость квазистационарного роста сгустка вычислялась по наклону линейного участка этой кривой.
Проведенные испытания показали, что заявляемые в данной работе вновь синтезированные соединения обладают антикоагулянтными свойствами, т.е. замедляют свертывание плазмы.
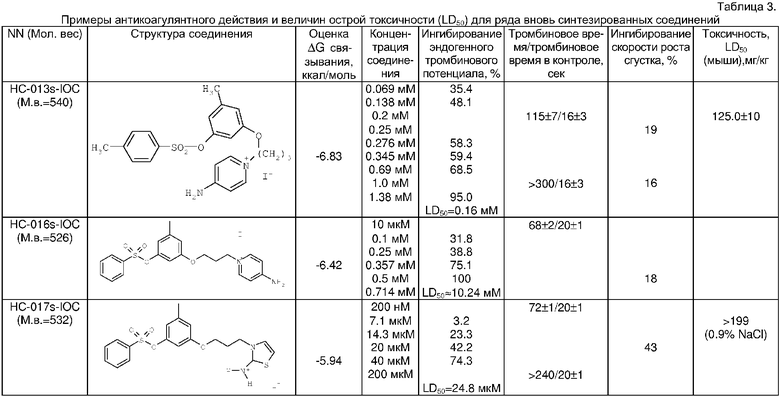
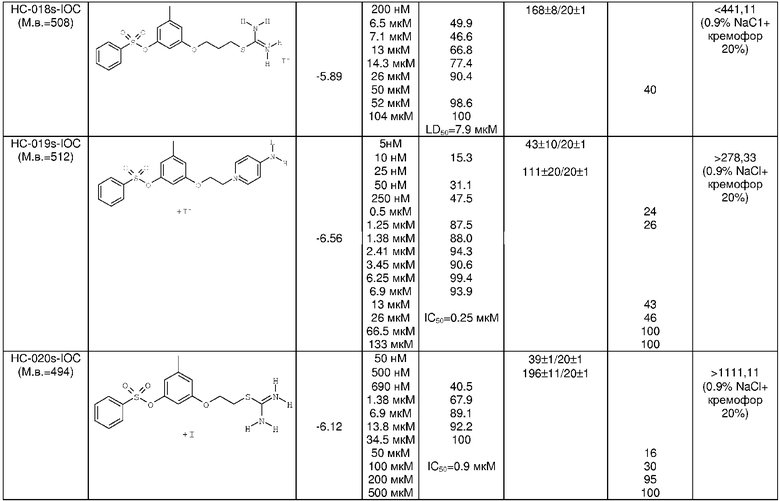
Рассмотрим в качестве примеров изменения ряда параметров, измеряемых в тесте генерации тромбина, в присутствии различных концентраций соединения HC-020s-IOC в плазме (см. Таблицу 3). Фиг.5 представляет кривые накопления флюоресцирующего продукта реакции расщепления флюорогенного субстрата тромбином, образующимся в ходе свертывания плазмы, непосредственно записанные прибором (условные единицы флюоресценции пересчитаны в абсолютные концентрации АМС с помощью специально измеренных калибровок). На Фиг.6 приведены кинетические профили изменения концентрации тромбина в ходе эксперимента, полученные дифференцированием кривых, представленных на Фиг.5. Уменьшение тромбинового потенциала и удлинение времени достижения максимума на кривой генерации тромбина при возрастании концентрации ингибитора в системе приведены на Фиг.7 и Фиг.8, соответственно. Представленные результаты показывают, что при повышении концентрации исследованного соединения (HC-020s-IOC) свертывание плазмы сильно замедляется. Концентрация этого соединения, вызывающая 50%-ное уменьшение эндогенного тромбинового потенциала (IC50) составляла 0.9 мкМ. Таким образом, данное соединение является сильным антикоагулянтом.
Следующие три фигуры демонстрируют изменение пространственной динамики роста сгустка в присутствии соединения HC-025s-IOC. На Фиг.9 и Фиг.10 можно видеть профили светорассеяния, регистрируемые в ходе свертывания в исходной нормальной плазме, а также в той же плазме в присутствии 1 мкМ данного соединения, соответственно. Фиг.11 представляет зависимости увеличения размера сгустка (выраженного как расстояние от активатора, на которое распространился сгусток) от времени для контрольной плазмы и плазмы в присутствии 1 мкМ соединения HC-025s-IOC. Сравнение полученных кривых также показывает, что присутствие соединения HC-025s-IOC в плазме ингибирует свертывание.
Примеры антикоагулянтного действия ряда вновь синтезированных соединений представлены в Таблице 3.
Пример 7
Усиление свертывания при разбавлении нормальной донорской плазмы кристаллоидным плазмозамещающим раствором (NaCl 0.9%) и его коррекция при введении в замещающий раствор различных концентраций соединения HC-025s-IOC, ингибирующего активность тромбина
Кровь донора, взятая на 3.8%-ном цитрате натрия в соотношении кровь:цитрат=9:1, (20 мл), была центрифугирована 15 мин при 1300g. Часть полученной таким образом бедной тромбоцитами плазмы (РРР) была далее центрифугирована 10 мин при 10000 g для получения свободной от тромбоцитов плазмы (PFP). Полученная PFP была использована для определения эндогенного тромбинового потенциала.
PFP была разбавлена в 1.5, 2, 3 и 4 раза либо физиологическим раствором для переливаний (NaCl 0.9%), либо тем же раствором, но содержащим добавленный ингибитор тромбина HC-025s-IOC (в концентрациях 0.25, 0.5 или 1 мкМ). Для того, чтобы поддерживать постоянную концентрацию ионов Са+2 в тестах после рекальцификации, концентрация цитрата натрия во всех разведениях плазм была выдержана постоянной, соответствующей этой концентрации в исходной неразбавленной плазме, для чего исходные плазмозамещающие растворы, которыми разбавляли плазму, были сначала смешаны с 3.8%-ным цитратом натрия аналогично исходной крови в соотношении раствор:цитрат=9:1.
В описанном выше методе измерения ЭТП образец плазмы в ходе измерения всегда разбавляется в 1.5 раза. Однако в опытах по разведению плазм ПЗР необходимо иметь возможность измерить ЭТП в неразбавленной плазме. Поэтому для измерения ЭТП в данных экспериментах была разработана специальная методика, позволяющая практически не разбавлять образец плазмы в ходе измерения. Для проведения измерения в ячейки 96-луночного планшета помещали по 200 мкл образцов плазмы (неразбавленной или разбавленной в желаемое количество раз ПЗР). Затем в каждую ячейку было добавлено по 2 мкл раствора флюорогенного субстрата в ДМСО (исходной концентрации 30.75 мМ). Свертывание запускали, добавляя в каждую ячейку по 3 мкл активатора. Активатором служил раствор, приготовленный из стандартного раствора тромбопластина для измерения протромбинового времени (фирма Ренам, Россия), разбавленный в 20 раз буфером, содержащим 20 мМ HEPES, 140 мМ NaCl и 1.235 М CaCl2, pH 7.5. Далее измерение проводили описанным ранее способом. Эндогенный тромбиновый потенциал был измерен для каждого из разведений плазм в присутствии различных концентраций соединения HC-025s-IOC.
На Фиг.12 представлены результаты по измерению ЭТП в сериях разбавлений плазмы раствором NaCl (0.9%), содержащим различные концентрации ингибитора. Видно, что в отсутствии добавленного ингибитора при увеличении степени разведения плазмы замещающим раствором наблюдается существенное увеличение эндогенного тромбинового потенциала, что говорит об усилении свертывания в данных условиях. Необходимо отметить, что уже изменено ЭТП на 20% считается фактором риска для возникновения тромбоза (Hemker НС, Al Dieri R, Beguin S. Thrombin generation assays: accruing clinical relevance. Curr. Opin. Hematol. 2004, 11(3):170-175). В тоже время, в наших экспериментах при разведении исходной плазмы в 1.5-4 раза максимальное увеличение ЭТП составляет в различных плазмах от 1.5 до более 4-х раз. Добавление ингибитора не отменяет полностью эффекта усиления свертывания из-за разведения плазмы, но значительно корректирует его, снижая величину ЭТП для различных разведении плазмы. При этом концентрации HC-025s-IOC в растворе, равные 0.25-0.5 мкМ, дают кривые, для которых значения ЭТП при различных разведениях плазмы наиболее близки к нормальным.
| название | год | авторы | номер документа |
|---|---|---|---|
| Диметил 7-метил-2-n-толил-4-фенилпирроло[2,1-f] [2,1,4]триазин-5,6-дикарбоксилат, обладающий антикоагулянтным действием | 2022 |
|
RU2790375C1 |
| ПРЯМОЙ ИНГИБИТОР ТРОМБИНА, ОБЛАДАЮЩИЙ АНТИПРОЛИФЕРАТИВНЫМ ДЕЙСТВИЕМ | 2007 |
|
RU2369615C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ МИНДАЛЬНОЙ КИСЛОТЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ТРОМБИНА | 2002 |
|
RU2341516C2 |
| РАСТВОР ДЛЯ КОРРЕКЦИИ ПЕРВИЧНЫХ НАРУШЕНИЙ ГЕМОСТАЗА ПЛАЗМОЗАМЕЩАЮЩИМИ РАСТВОРАМИ НОВОГО СОСТАВА | 2005 |
|
RU2300385C1 |
| ПРОИЗВОДНЫЕ ТРИПЕПТИДОВ В ВИДЕ R- ИЛИ RS-ФОРМЫ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ НЕТОКСИЧНЫЕ СОЛИ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1991 |
|
RU2077538C1 |
| АПТАМЕРНЫЙ ОЛИГОНУКЛЕОТИД - ПРЯМОЙ ИНГИБИТОР ТРОМБИНА | 2008 |
|
RU2401306C2 |
| МОДИФИЦИРОВАННЫЕ ДНК АПТАМЕРЫ, ИНГИБИРУЮЩИЕ АКТИВНОСТЬ ТРОМБИНА | 2009 |
|
RU2410432C1 |
| СТАНДАРТНЫЕ ЛЕКАРСТВЕННЫЕ ПРЕПАРАТЫ И СПОСОБЫ ЛЕЧЕНИЯ ТРОМБОЗА ПЕРОРАЛЬНЫМ ВВЕДЕНИЕМ ИНГИБИТОРА ФАКТОРА ХА | 2007 |
|
RU2452484C2 |
| Применение пинанилсульфида с метилмеркаптоацетатным фрагментом - метил 2-({ [(1S, 2R, 5S)-6,6-диметилбицикло[3.1.1]гепт-2-ил]метил} тио)ацетата, обладающий ингибирующим действием на активацию тромбоцитов | 2015 |
|
RU2671567C2 |
| СИНТЕТИЧЕСКОЕ ПРОИЗВОДНОЕ ПЕПТИДА, ПРОЯВЛЯЮЩЕЕ АНТИКОАГУЛЯНТНУЮ АКТИВНОСТЬ | 2007 |
|
RU2377246C2 |
Настоящее изобретение описывает новые химические соединения, обладающие свойством замедлять свертывание плазмы крови, общей структурной формулы (I), его фармацевтически приемлемые соли или сольваты: 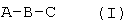 , где значение радикалов А, С, R9, такие как указано в формуле изобретения, R1, R2, R3, R4 независимо друг от друга представляют собой водород или C1-6 алкил; В представляет собой -(СН2)n-, где n принимает целые значения от 1 до 5; где R5 выбирают из группы, содержащей водород, C1-6алкокси, CH2NR10R11, СН(СН3)HR10R11 и другие значения, указанные в формуле изобретения, R6 и R7 независимо представляют собой водород, C1-6алкил; C1-6алкокси; галоген; R8 представляет собой водород или C1-6алкил; R10, R12 независимо друг от друга выбирают из группы, состоящей из водорода, C1-6алкила; (СН2)mCOOR13, (CH2)mCON(R13)2 и др. значения, указанные в формуле изобретения, где m принимает целые значения от 1 до 4, R13 представляет собой водород или C1-6алкил, R11 представляет собой C1-6алкил; Ar; Ar представляет собой фенил, пиридил, оксазолил, тиазолил, тиенил, фуранил, пиримидинил, пиридазинил, пиразинил, индолил, бензофуранил, бензотиофенил, замещенные от одного до пяти заместителей, выбранных из группы: водород, C1-6алкил, C1-6алкокси, галоген, N(R13)2, ОН, NO2, CN, COOR13, CON(R13)2, SO2R13; применение этих соединений в качестве антикоагулянтов для лечения тромботических состояний, фармацевтическая композиция на их основе и плазмозаменяющий раствор для коррекции гиперкоагуляционных нарушений. Технический результат: получены новые соединения, которые могут быть полезны для лечения тромбоэмболических осложнений и профилактики возникновения гиперкоагуляционных состояний. 4 н.и 1 з.п. ф-лы, 3 табл., 12 ил.
, где значение радикалов А, С, R9, такие как указано в формуле изобретения, R1, R2, R3, R4 независимо друг от друга представляют собой водород или C1-6 алкил; В представляет собой -(СН2)n-, где n принимает целые значения от 1 до 5; где R5 выбирают из группы, содержащей водород, C1-6алкокси, CH2NR10R11, СН(СН3)HR10R11 и другие значения, указанные в формуле изобретения, R6 и R7 независимо представляют собой водород, C1-6алкил; C1-6алкокси; галоген; R8 представляет собой водород или C1-6алкил; R10, R12 независимо друг от друга выбирают из группы, состоящей из водорода, C1-6алкила; (СН2)mCOOR13, (CH2)mCON(R13)2 и др. значения, указанные в формуле изобретения, где m принимает целые значения от 1 до 4, R13 представляет собой водород или C1-6алкил, R11 представляет собой C1-6алкил; Ar; Ar представляет собой фенил, пиридил, оксазолил, тиазолил, тиенил, фуранил, пиримидинил, пиридазинил, пиразинил, индолил, бензофуранил, бензотиофенил, замещенные от одного до пяти заместителей, выбранных из группы: водород, C1-6алкил, C1-6алкокси, галоген, N(R13)2, ОН, NO2, CN, COOR13, CON(R13)2, SO2R13; применение этих соединений в качестве антикоагулянтов для лечения тромботических состояний, фармацевтическая композиция на их основе и плазмозаменяющий раствор для коррекции гиперкоагуляционных нарушений. Технический результат: получены новые соединения, которые могут быть полезны для лечения тромбоэмболических осложнений и профилактики возникновения гиперкоагуляционных состояний. 4 н.и 1 з.п. ф-лы, 3 табл., 12 ил.
1. Соединение общей структурной формулы (I), его фармацевтически приемлемые соли или сольваты:
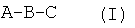
структуры, где С выбирают из группы, содержащей
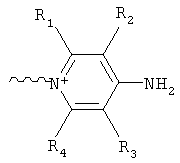
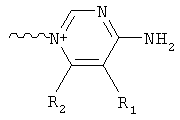
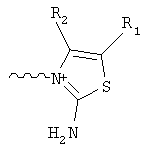
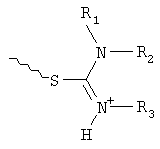
где R1, R2, R3, R4 независимо друг от друга представляют собой водород или C1-6алкил;
В представляет собой -(СН2)n-, где n принимает целые значения от 1 до 5;
А выбирают из группы, содержащей структуры:
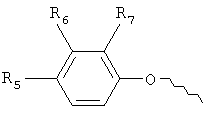
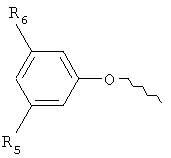
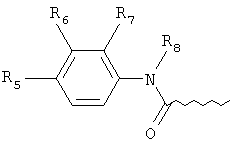
где R5 выбирают из группы, содержащей водород, C1-6алкокси, CH2NR10R11, CH(CH3)NR10R11,
 ,
,  ,
,  ,
,  ,
,
R6 и R7 независимо представляют собой водород, C1-6алкил; C1-6алкокси; галоген;
R8 представляет собой водород или C1-6алкил;
R9 выбирают из следующей группы, состоящей из:
 ,
,  ,
,
R10, R12 независимо друг от друга выбирают из группы, состоящей из водорода, C1-6алкила; (СН2)mCOOR13, (CH2)mCON(R13)2,
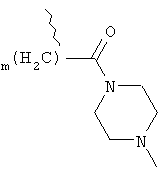
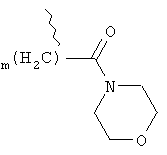 ,
, 
где m принимает целые значения от 1 до 4,
R13 представляет собой водород или C1-6алкил,
R13 представляет собой C1-6алкил; Ar;
Ar представляет собой фенил, пиридил, оксазолил, тиазолил, тиенил, фуранил, пиримидинил, пиридазинил, пиразинил, индолил, бензофуранил, бензотиофенил, имеющие от одного до пяти заместителей, выбранных из группы:
водород, C1-6алкил, C1-6алкокси, галоген, N(R13)2, ОН, NO2, CN, COOR13, CON(R13)2, SO2R13;
за исключением
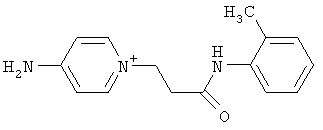
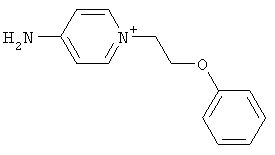
обладающие свойством замедлять свертывание плазмы крови.
2. Соединение по п.1, его фармацевтически приемлемые соли или сольваты, а именно
 ,
,
 ,
,
где Y выбирают из группы, состоящей из водорода, галогена, COOR13, CON(R13)2, SO2R13,
r принимает целые значения от 2 до 5.
3. Применение соединения по п.1, его фармацевтически приемлемых солей или сольватов в качестве антикоагулянтов для лечения тромботических состояний млекопитающего.
4. Фармацевтическая композиция для лечения тромботических состояний, содержащая терапевтически эффективное количество соединения по п.1, и/или его фармацевтически приемлемых солей и/или сольватов, а также фармацевтически приемлемый носитель.
5. Плазмозаменяющий раствор для коррекции гиперкоагуляционных нарушений при гемодилюции, характеризующийся тем, что содержит соединение по п.1, и/или его фармацевтически приемлимые соли и/или сольваты.
| US 5792799, 11.08.1998 | |||
| 4-АМИНОПИРИДИНЫ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1994 |
|
RU2126388C1 |
| US 5510369, 23.04.1996 | |||
| РАСТВОР ДЛЯ КОРРЕКЦИИ ПЕРВИЧНЫХ НАРУШЕНИЙ ГЕМОСТАЗА ПЛАЗМОЗАМЕЩАЮЩИМИ РАСТВОРАМИ НОВОГО СОСТАВА | 2005 |
|
RU2300385C1 |

