Область изобретения
Это изобретение относится к новым фармацевтически полезным соединениям, в частности соединениям, которые сами являются, и/или соединениям, которые метаболизируются в соединения, которые являются конкурентными ингибиторами трипсиноподобных сериновых протеаз, особенно тромбина, к их применению в качестве лекарств, к фармацевтическим композициям, которые их содержат, и к синтетическим путям их получения.
Предпосылки изобретения
Коагуляция крови является ключевым процессом, участвующим как в гемостазе (то есть предотвращении потери крови из поврежденного сосуда), так и тромбозе (то есть образовании кровяного сгустка в кровеносном сосуде, иногда приводящего к закупорке сосуда).
Коагуляция является результатом сложной серии ферментативных реакций. Одной из конечных стадий этой серии реакций является превращение профермента протромбина в активный фермент тромбин.
Известно, что тромбин играет центральную роль в коагуляции. Он активирует тромбоциты, что приводит к агрегации тромбоцитов, превращает фибриноген в мономеры фибрина, которые спонтанно полимеризуются в полимеры фибрина, и активирует фактор XIII, который в свою очередь сшивает эти полимеры, образуя нерастворимый фибрин. Более того, тромбин активирует фактор V и фактор VIII, что ведет к генерации тромбина из протромбина по механизму «положительной обратной связи».
Следует ожидать, что путем ингибирования агрегации тромбоцитов и образования и сшивания фибрина эффективные ингибиторы тромбина будут проявлять антитромботическую активность. Кроме того, следует ожидать, что антитромботической активности будет способствовать эффективное ингибирование механизма положительной обратной связи.
Предшествующий уровень техники
Ранние разработки низкомолекулярных ингибиторов тромбина были описаны Claesson в Blood Coagul. Fibrinol. (1994) 5, 411.
Blombäck et al. (в J. Clin. Lab. Invest. 24, suppl. 107, 59, (1969)) сообщили об ингибиторах тромбина на основе аминокислотной последовательности, располагающейся вокруг сайта расщепления для Аα-цепи фибриногена. Эти авторы предположили, что из числа обсуждавшихся аминокислотных последовательностей самым эффективным ингибитором будет трипептидная последовательность Phe-Val-Arg (P9-P2-P1, именуемая здесь ниже как последовательность Р3-Р2-Р1).
Ингибиторы тромбина на основе дипептидильных производных с α,ω-аминоалкилгуанидином в P1-положении раскрыты в патенте США №4346078 и в международной заявке на патент WO 93/11152. Также сообщалось о сходных структурно родственных дипептидильных производных. Например, международная заявка на патент WO 94/29336 раскрывает соединения с, например, аминометилбензамидинами, циклическими аминоалкиламидинами и циклическими аминоалкилгуанидинами в P1-положении (международная заявка на патент WO 97/23499 раскрывает пролекарства этих соединений), заявка на европейский патент 0648780 раскрывает соединения с, например, циклическими аминоалкилгуанадинами в P1-положении.
Ингибиторы тромбина на основе пептидильных производных, также имеющие циклические аминоалкилгуанидины (например, 3- или 4-аминометил-1-амидинопиперидин) в P1-положении известны из заявок на европейский патент 0468231, 0559046 и 0641779.
Ингибиторы тромбина, основанные на трипептидильных производных с аргининовым альдегидом в P1-положении, впервые были раскрыты в заявке на европейский патент 0185390.
Позднее появилось сообщение о пептидильных производных на основе аргининового альдегида, модифицированных в Р3-положении. Например, в международной заявке на патент WO 93/18060 раскрыты гидроксикислоты, в заявке на европейский патент 0526877 раскрыты дезаминокислоты, а в заявке на европейский патент 0542525 раскрыты O-метилминдальные кислоты в Р3-положении.
Также известны ингибиторы сериновых протеаз (например, тромбина), основанные на электрофильных кетонах в P1-положении. Например, заявка на европейский патент 0195212 раскрывает пептидильные α-кетоэфиры и амиды, заявка на европейский патент 0362002 раскрывает фторалкиамидкетоны, заявка на европейский патент 0364344 раскрывает α,β,δ-трикетосоединения, а заявка на европейский патент 0530167 раскрывает α-алкоксикетоновые производные аргинина в P1-положении.
Другие, структурно отличающиеся, ингибиторы трипсиноподобных сериновых протеаз на основе С-терминальных бороновокислотных производных аргинина и их изотиоурониевых аналогов известны из заявки на европейский патент 0293881.
Позднее ингибиторы тромбина, основанные на пептидильных производных, были описаны в заявке на европейский патент 0669317 и в международных заявках на патент WO 95/35309, WO 95/23609, WO 96/25426, WO 97/02284, WO 97/46577, WO 96/32110, WO 96/31504, WO 96/03374, WO 98/06740, WO 97/49404, WO 98/57932, WO 99/29664, WO 00/35869 и WO 00/42059.
В частности, WO 97/02284 и WO 00/42059 раскрывают ингибиторы тромбина с замещенными миндальными кислотами в Р3-положении.
Однако сохраняется потребность в более эффективных ингибиторах трипсиноподобных сериновых протеаз, таких как тромбин. Также существует потребность в соединениях, которые имеют благоприятный фармакокинетический профиль и являются селективными в ингибировании тромбина в сравнении с другими сериновыми протеазами, в частности теми, которые участвуют в гемостазе. Следует ожидать, что соединения, которые проявляют конкурентную ингибиторную активность в отношении тромбина, будут особенно полезны в качестве антикоагулянтов и поэтому в терапевтическом лечении тромбоза и связанных с ним нарушений.
Описание изобретения
Согласно настоящему изобретению предложено соединение формулы
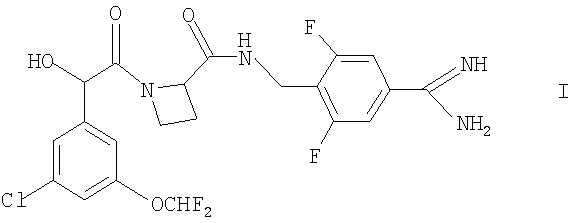
(то есть Ph(3-Cl)(5-OCHF2)-CH(OH)C(O)-Aze-Pab(2,6-diF)) или его фармацевтически приемлемое производное.
Термин "фармацевтически приемлемое производное" включает в себя фармацевтически приемлемые соли (например, соли присоединения кислот).
Использованные сокращения приведены в конце данного описания.
Соединение формулы I можно получать в соответствии с методиками, хорошо известными специалистам, например, как описано здесь ниже.
Согласно еще одному аспекту этого изобретения предложен способ получения соединения формулы I, при котором
(1) осуществляют сочетание соединения формулы II
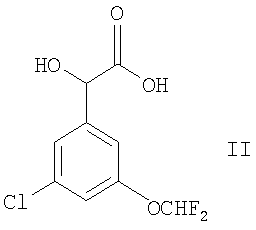
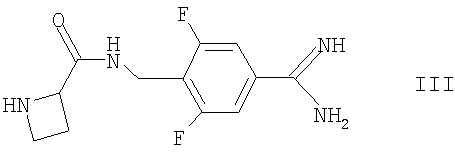
с соединением формулы III
например, в присутствии агента сочетания (например, оксалилхлорида в DMF, EDC, DCC, HBTU, HATU, РуОР или TBTU), подходящего основания (например, пиридина, DMAP, TEA, 2,4,6-коллидина или DIPEA) и подходящего органического растворителя (например, дихлорметана, ацетонитрила, EtOAc или DMF);
(2) осуществляют сочетание соединения формулы IV
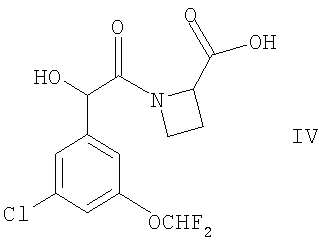
с соединением формулы V
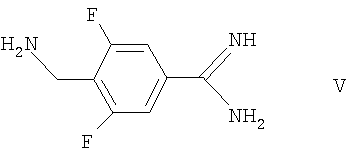
например, в условиях, описанных выше в способе (1), или
(3) осуществляют взаимодействие соответствующего соединения формулы XVI, как оно определено здесь ниже, с подходящим источником аммиака (например, ацетатом аммония или газообразным аммиаком) в условиях, известных специалистам в данной области, например, посредством взаимодействия этилимидоатного промежуточного соединения (образованного в результате взаимодействия соединения формулы XVI с HCI (газ) в этаноле) с газообразным аммиаком в этаноле или в условиях, описанных в Tetrahedron Lett. 40, 7067 (1999), описания в этом документе включены сюда ссылкой.
Соединения формулы II можно получить, используя известные и/или стандартные методики.
Например, соединения формулы II могут быть получены путем взаимодействия альдегида формулы VI
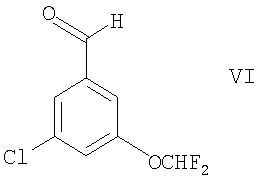
со следующими соединениями:
(а) соединением формулы VII

в которой R″ представляет собой Н или (СН3)3Si, например, при комнатной или повышенной температуре (например, ниже 100°С) в присутствии подходящего органического растворителя (например, хлороформа или метиленхлорида) и, если необходимо, в присутствии подходящего основания (например, TEA) и/или подходящей каталитической системы (например, хлорида бензиламмония или йодида цинка или путем использования хирального катализатора, например так, как описано в Chem. Rev., (1999) 99, 3649), с последующим гидролизом в условиях, которые хорошо известны специалистам в данной области (например, как описано здесь ниже);
(б) NaCN или KCN, например, в присутствии NaHSO3 и воды с последующим гидролизом;
(в) хлороформом, например, при повышенной температуре (например, выше комнатной температуры, но ниже 100°С) в присутствии подходящего органического растворителя (например, хлороформа) и, если необходимо, в присутствии подходящей каталитической системы (например, хлорида бензиламмония) с последующим гидролизом;
(г) соединением формулы VIII
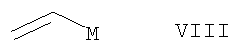
в которой М представляет собой Mg или Li, с последующим окислительным расщеплением (например, озонолизом или катализом осмием или рутением) в условиях, которые хорошо известны специалистам в данной области, или
(е) трис(метилтио)метаном в условиях, которые хорошо известны специалистам в данной области, с последующим гидролизом в присутствии, например, HgO и HBF4.
Соединения формулы II альтернативно могут быть получены окислением соединения формулы IX
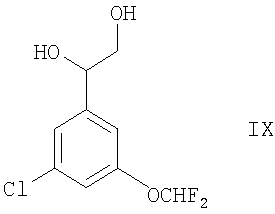
или его производного, которое возможно защищено по вторичной гидроксильной группе, в присутствии подходящего окислителя (например, комбинации подходящего оксиданта свободных радикалов (такого как TEMPO) и соответствующей гипохлоритной соли (такой как гипохлорит натрия) в условиях, известных специалистам в данной области, например при температуре между -10°С и комнатной температурой в присутствии подходящего растворителя (например, воды, ацетона или их смеси), подходящей соли (например, галогенида щелочного металла, такого как бромид калия) и подходящего основания (например, карбоната или гидрокарбоната щелочного металла, такого как гидрокарбонат натрия).
Энантиомерно чистые формы соединений формулы II (то есть соединения, имеющие разные конфигурации заместителей при атоме углерода от α- до CO2Н-группы) можно разделять стадией энантиоспецифичной дериватизации. Это можно осуществить, например, ферментативным путем. Такой ферментативный способ включает в себя, например, переэтерификацию группы α-ОН при температуре между комнатной и температурой флегмы (например, между 45 и 65°С) в присутствии подходящего фермента (например, Lipase PS Amano), соответствующего сложного эфира (например, винилацетата) и подходящего растворителя (например, метил-трет-бутилового эфира). Затем можно отделить дериватизированный изомер от непрореагировавшего изомера с помощью общепринятой методики разделения (например, хроматографически).
Группы, присоединенные к соединениям формулы II в ходе такой стадии дериватизации, можно удалять либо перед любой последующей реакцией, либо на любой более поздней стадии синтеза соединений формулы I. Дополнительные группы можно удалять с помощью общепринятых методик (например, для сложных эфиров группы α-ОН-гидролизом в условиях, известных специалистам (например, при температуре между комнатной и температурой флегмы в присутствии подходящего основания (например, NaOH) и соответствующего растворителя (например, МеОН, воды или их смесей)).
Соединения формулы III могут быть получены сочетанием азетидин-2-карбоновой кислоты с соединением формулы V, как она определена здесь ранее, например, в условиях, сходных с теми, что описаны здесь для получения соединений формулы I.
Соединения формулы IV могут быть получены сочетанием соединения формулы II, как оно определено здесь ранее, с азетидин-2-карбоновой кислотой, например, в условиях, сходных с теми, что описаны здесь для получения соединений формулы I.
Соединение формулы VI доступно с использованием известных и/или стандартных методик. Например, оно может быть получено
(1) металлированием (причем металл может быть, например, щелочным металлом, таким как Li, или предпочтительно двухвалентным металлом, таким как Mg) соединения формулы Х
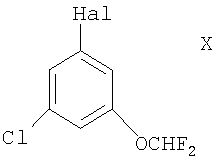
в которой Hal представляет собой атом галогена, выбранный из Cl, Br и I, с последующим взаимодействием с подходящим источником формильной группы (таким как N,N-диметилформамид), например, в условиях, описанных здесь ниже;
(2) восстановлением соединения формулы XI
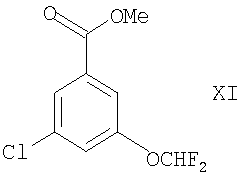
в присутствии подходящего восстановителя (например, DIBAL-H) или
(3) окислением соединения формулы XII
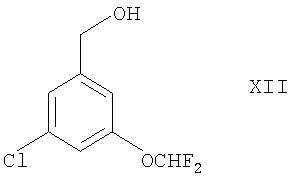
в присутствии подходящего окислителя (например, MnO2, хлорхромата пиридиния, комбинации DMSO и оксалилхлорида или комплекса SO3 и пиридина в DMSO).
Соединения формулы IX могут быть получены дигидроксилированием соответствующего соединения формулы XIII
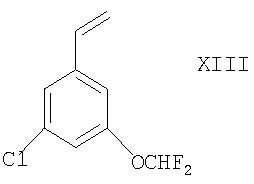
в присутствии подходящего дигидроксилирующего агента (например, реагента или смеси реагентов, дающих OsO4, таких как AD-mix-α или, в особенности, AD-mix-β), например, в условиях, которые известны специалистам в данной области, например между -10°С и комнатной температурой, в присутствии соответствующего растворителя (например, воды, трет-бутанола или их смеси). При использовании асимметричных оксидантов, таких как AD-mix-α или AD-mix-β, этот способ можно применять для получения соединений формулы IX, которые имеют специфические конфигурации групп (например, R или S) при обоих атомах углерода, к которым присоединены первичные и вторичные гидроксильные группы.
Соединение формулы XIII можно получать взаимодействием соответствующего соединения формулы X, как оно определено здесь ранее, с подходящим источником винильного аниона (например, трибутил(винил)оловом) в условиях, известных специалистам в данной области, например при температуре между комнатной и температурой флегмы (например, 50°С) в присутствии соответствующего растворителя (например, толуола), подходящего агента сочетания (например, координационного комплекса палладия(0), такого как тетракис(трифенилфосфин)палладий(0)) и, возможно, в присутствии подходящего катализатора (например, 2,6-ди-трет-бутил-4-метилфенола).
Соединения формул V, VII, VIII, X, XI, XII и азетидин-2-карбоновая кислота либо имеются в продаже, либо известны из литературы, либо их можно получать по аналогии с описанными здесь способами или общепринятыми синтетическими процедурами в соответствии со стандартными методиками из легкодоступных исходных материалов с использованием соответствующих реагентов и реакционных условий. Соединения формулы XVI можно получать способами, описанным здесь ниже.
Заместители по фенильному кольцу в соединениях формул I, II, III, IV, V, VI, IX, X, XI, XII и XIII можно вводить с использованием методик, хорошо известных специалистам в данной области, посредством стандартных взаимопревращений функциональных групп в соответствии со стандартными методиками, из легкодоступных исходных материалов с использованием соответствующих реагентов и реакционных условий.
Например, соединения формул I, II, IV, VI, X, XI и XII могут быть получены из соединений, соответствующих тем соединениям, в которых вместо группы -OCH2F присутствует группа -ОН (ниже именуемых как "релевантные фенольные соединения-предшественники"), например, посредством взаимодействия такого релевантного фенольного соединения-предшественника с соответствующим фторированным галогеноалканом (таким как ClCHF2), например, при комнатной или более высокой температуре (например, при температуре флегмы) в присутствии подходящего основания (например, трет-бутилата калия, KOH или NaOH, например, в водном растворе) и соответствующего органического растворителя (например, THF, хлороформа или изо-пропанола), например, как описано здесь ниже.
Специалисту должно быть понятно, что такие трансформации функциональных групп можно также проводить на более ранней стадии всего синтеза соединений формул II, IV, VI, X, XI или XII (то есть на соответствующих предшественниках релевантных фенольных соединений-предшественников). Релевантные фенольные соединения-предшественники либо имеются в продаже или известны из литературы, либо могут быть получены по аналогии с описанными здесь способами или посредством общепринятых синтетических процедур в соответствии со стандартными методиками, из легкодоступных исходных материалов при использовании соответствующих реагентов и реакционных условий. Например, релевантные фенольные соединения-предшественники могут быть получены снятием защиты с соответствующих защищенных фенолов (у которых защитной группой может быть, например, метил, аллил, бензил или трет-бутил) в стандартных условиях.
Соединения формулы I можно выделять из их реакционных смесей с помощью общепринятых методик.
В соответствии с настоящим изобретением фармацевтически приемлемые производные соединений формулы I также включают в себя "защищенные" производные и/или соединения, которые действуют как пролекарства соединений формулы I.
Соединения, которые могут действовать как пролекарства соединений формулы I и которые могут быть упомянуты, включают в себя соединения формулы Ia
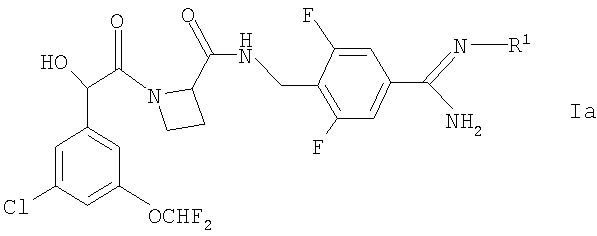
где R1 представляет собой OR2 или C(O)OR3;
R2 представляет собой Н, С1-10алкил, C1-3алкиларил или C1-3алкилоксиарил (алкильные части двух последних групп возможно прерваны одним или более чем одним атомом кислорода, а арильные части этих двух последних групп возможно замещены одним или более чем одним заместителем, выбранным из галогено, фенила, метила или метокси, причем последние три группы также возможно замещены одним или более чем одним заместителем, представляющим собой галогено) и
R3 представляет собой C1-10алкил (эта последняя группа возможно прервана одним или более чем одним атомом кислорода) или C1-3алкиларил, или C1-3алкилоксиарил (алкильные части двух последних групп возможно прерваны одним или более чем одним атомом кислорода, а арильные части двух последних групп возможно замещены одним или более чем одним заместителем, выбранным из галогено, фенила, метила или метокси, причем последние три группы также возможно замещены одним или более чем одним заместителем, представляющим собой галогено),
и их фармацевтически приемлемые производные.
Термин "фармацевтически приемлемые производные" соединений формулы Ia включает в себя фармацевтически приемлемые соли (например, соли присоединения кислот).
Алкилоксиарильные группы, которыми могут являться R2 и R3, содержат алкильную и арильную группу, связанные через атом кислорода. Алкиларильные и алкилоксиарильные группы связаны с остальной частью молекулы через алкильную часть этих групп, причем эти алкильные части могут (если есть достаточное число (то есть три) атомов углерода) иметь разветвленную цепь. Арильные части алкиларильных и алкилоксиарильных групп, которыми могут являться R2 и R3 или которыми они могут быть замещены, включают в себя карбоциклические и гетероциклические ароматические группы, такие как фенил, нафтил, пиридинил, оксазолил, изоксазолил, тиадиазолил, индолил, бензофуранил и подобные им.
Алкильные группы, которыми могут являться R2 и R3, могут быть прямоцепочечными или, когда имеется достаточное количество (то есть минимум три) атомов углерода, могут иметь разветвленную цепь и/или быть циклическими. Далее, когда имеется достаточное количество (то есть минимум четыре) атомов углерода, такие алкильные группы могут также быть частично циклическими/ациклическими. Такие алкильные группы также могут быть насыщенными или, когда имеется достаточное количество (то есть минимум два) атомов углерода, могут быть ненасыщенными.
Галогеногруппы, которыми могут быть замещены R2 и R3, включают в себя фторо, хлоро, бромо и йодо.
Когда R1 представляет собой C(O)OR3, предпочтительные группы R3 включают в себя
(а) линейный, разветвленный или циклический С3-6алкил, например С4-6циклоалкил;
(б) С1-2алкиларильные группы, такие как бензил, возможно замещенные так, как указано здесь ранее.
Предпочтительные соединения формулы Ia включают в себя те, у которых R1 представляет собой OR2.
Когда R1 представляет собой OR2, предпочтительные группы R2 включают в себя
(а) Н;
(б) незамещенный, линейный, разветвленный или циклический C1-8 (например, С1-6)алкил, такой как линейный С1-3алкил (например, этил или, особенно, метил), разветвленный С3-8алкил (например, изо-пропил, изо-бутил или 4-гептил) или циклический С4-7алкил (то есть С4-7циклоалкил, например циклобутил или циклогексил);
(в) C1-3алкилоксифенил (например, С2алкилоксифенил), причем фенильная группа возможно замещена одним или более чем одним заместителем, как указано здесь выше (например, трифторметилом);
(г) С1-2алкиларил (например, метиларил), у которого арильная группа представляет собой фенил, пиридинил, оксазолил или изоксазолил, причем последние три группы возможно замещены одним или более чем одним заместителем, как указано здесь ранее (например, метокси, метилом, бромо и/или хлоро).
Предпочтительные соединения формулы Ia включают в себя те, у которых R1 представляет собой OR2, a R2 представляет собой линейный, разветвленный (как целесообразно) или циклический (как целесообразно) С1-6 (например, С1-4)алкил, такой как метил, этил, н-пропил, изо-пропил или циклобутил.
Соединения формулы Ia можно получать одним или более чем одним из следующих способов:
(а) взаимодействие соответствующего соединения формулы II, как оно определено здесь ранее, с соединением формулы XIV
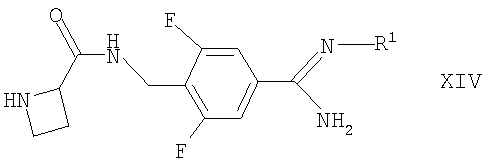
в которой R1 такой, как определено здесь ранее, например, в условиях, аналогичных раскрытым здесь ранее для синтеза соединений формулы I;
(б) взаимодействие соответствующего соединения формулы IV, как определено здесь ранее, с соединением формулы XV
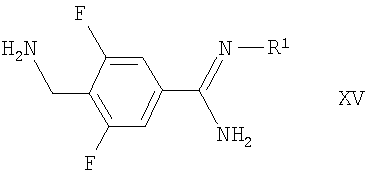
в которой R1 такой, как определено здесь ранее, например, в условиях, аналогичных раскрытым здесь ранее для синтеза соединений формулы I;
(в) для соединений формулы Ia, у которых R1 представляет собой ОН, взаимодействие соответствующего соединения формулы XVI
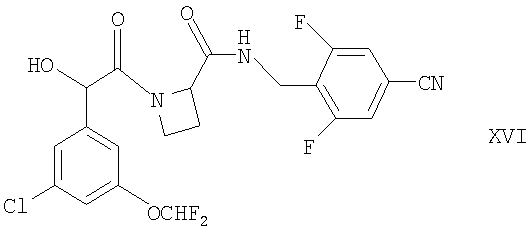
с гидроксиламином, например, в условиях, известных специалистам в данной области;
(г) для соединений формулы Ia, у которых R1 представляет собой OR2, взаимодействие защищенного производного соответствующего соединения формулы I, которое представляет собой, например, соединение формулы XVII
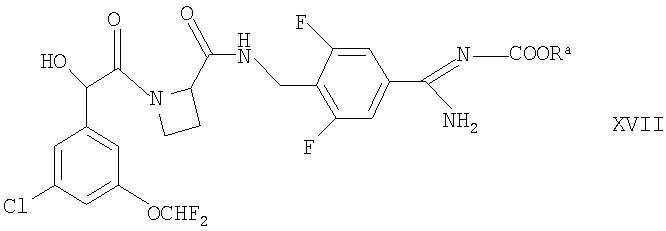
где Ra представляет собой, например, -СН2СН2-Si(CH3)3 или бензил, или его таутомер,
с соединением формулы XVIII

где R2 такой, как определено здесь ранее, или его солью присоединения кислоты, например, при температуре между комнатной и температурой флегмы, в присутствии соответствующего органического растворителя (например, THF, СН3CN, DMF или DMSO) с последующим удалением группы -С(O)ORa в условиях, известных специалистам в данной области (например, путем взаимодействия с QF или TFA (например так, как описано здесь ниже));
(д) для соединений формулы Ia, в которых R1 представляет собой ОН, взаимодействие соединения формулы XVII, как оно определено здесь выше, в которой Ra представляет собой бензил, с гидроксиламином или его солью присоединения кислоты, например, в условиях, которые должны быть хорошо известными специалистам;
(е) для соединений формулы Ia, в которой R1 представляет собой COOR3, взаимодействие соответствующего соединения формулы I, как оно определено здесь выше, с соединением формулы XIX
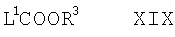
где L1 представляет собой подходящую уходящую группу, такую как галогено или нитрофенил (например, 4-нитрофенил), и R3 такой, как определено здесь ранее, например, при комнатной температуре в присутствии подходящего основания (например, NaOH, например, в водном растворе) и соответствующего органического растворителя (например, метиленхлорида) или
(ж) для соединений формулы Ia, в которых R1 представляет собой ОСН3 или OCH2СН3, взаимодействие соответствующего соединения формулы Ia, в которой R1 представляет собой ОН, с диметилсульфатом или диэтилсульфатом соответственно, например, в присутствии подходящего основания (например, гидроксида щелочного металла, такого как KOH (например, в водном растворе при, например, 50 мас.%)), и соответствующего катализатора (например, галогенида четвертичного аммония, такого как хлорид бензилтриметиламмония (например, в растворе CH2Cl2 или THF при, например, 10 мас.%)).
Соединения формулы XVI могут быть получены взаимодействием соответствующего соединения формулы II, как определено здесь ранее, с соединением формулы XX
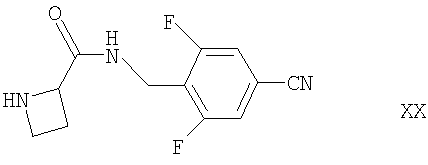
например, в условиях, аналогичных описанным здесь ранее для синтеза соединений формулы I.
Альтернативно соединения формулы XVI могут быть получены взаимодействием соответствующего соединения формулы IV, как определено здесь ранее, с соединением формулы XXI
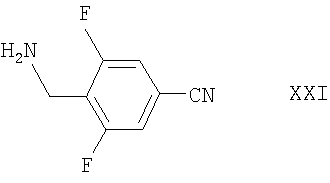
например, в условиях, аналогичных описанным здесь ранее для синтеза соединений формулы I.
Соединения формулы XVII могут быть получены взаимодействием соответствующего соединения формулы II, как определено здесь ранее, с соединением формулы XXII
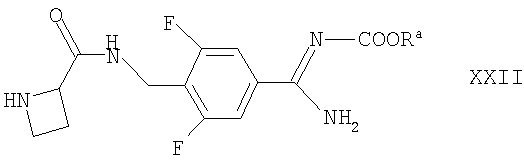
где Ra такие, как определено здесь выше, например, в условиях, аналогичных описанным здесь ранее для синтеза соединений формулы I.
Альтернативно соединения формулы XVII могут быть получены взаимодействием соответствующего соединения формулы I с соединением, соответствующим соединению формулы XIX, в которой вместо R3 присутствует группа Ra, причем Ra является такой, как определено выше, например, в условиях, описанных выше в отношении получения соединений формулы Ia.
Соединения формул XIV и XXII могут быть получены взаимодействием азетидин-2-карбоновой кислоты с соответственно соединением формулы XV, как оно определено выше, или соединением формулы XXIII
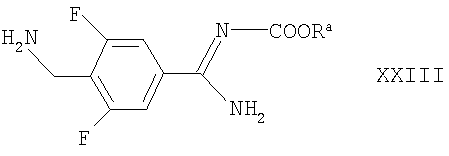
где Ra такой, как определено выше, например, в условиях, аналогичных описанным здесь ранее для синтеза соединений формулы I.
Соединения формулы XV, XVIII, XIX, XX, XXI и XXIII либо имеются в продаже или известны из литературы, либо могут быть получены по аналогии с описанными здесь способами или общепринятыми синтетическими процедурами в соответствии со стандартными методиками, из легкодоступных исходных материалов, используя соответствующие реагенты и реакционные условия. Например, соединения формулы XX могут быть получены взаимодействием соответствующего соединения формулы XXI с азетидин-2-карбоновой кислотой, например, в условиях, аналогичных описанным здесь ранее.
Соединения формул I и Ia, как они определены здесь выше, и их производные обозначены здесь ниже как "соединения по изобретению".
Соединения по изобретению могут проявлять таутомерию. Все таутомерные формы и их смеси включены в объем этого изобретения. Конкретные таутомерные формы, которые можно упомянуть, включают в себя таутомерные формы, связанные с положением двойной связи в амидиновой функциональной группе в соединении формулы Ia и с положением заместителя R1.
Соединения по этому изобретению также содержат два или более асимметричных атома углерода и поэтому могут проявлять оптическую изомерию и/или диастереоизомерию. Диастереоизомеры могут быть разделены общепринятыми методиками, например хроматографией. Разные стереоизомеры можно выделять разделением рацемической или иной смеси этих соединений общепринятыми методиками, например посредством ВЭЖХ. Альтернативно желаемые оптические изомеры можно получать взаимодействием соответствующих оптически активных исходных материалов в условиях, которые не должны вызывать рацемизацию или эпимеризацию, или дериватизацией, например, гомохиральной кислотой, с последующим разделением диастереомерных производных общепринятыми средствами (например, ВЭЖХ, хроматографией на диоксиде кремния). Все стереоизомеры включены в объем этого изобретения.
Предпочтительными являются соединения, у которых фрагмент
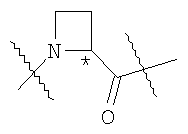
находится в S-конфигурации.
Предпочтительные соединения по изобретению также включают в себя соединения, у которых структурный фрагмент
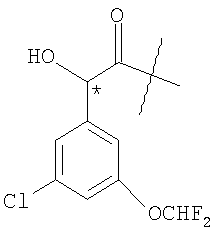
находится в R-конфигурации.
Волнистые линии на связях в двух вышеуказанных фрагментах обозначают положения связывания этих фрагментов.
Таким образом, предпочтительные соединения по изобретению включают в себя
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-Pab(2,6-diF),
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-Pab(2,6-diF)(OMe) и
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-Pab(2,6-diF)(OH).
Специалисты должны понимать, что в способах, описанных здесь выше и далее, функциональные группы промежуточных соединений могут нуждаться в защите защитными группами.
Функциональные группы, которые желательно защищать, включают в себя гидрокси, амино и карбоновую кислоту. Подходящие защитные группы для гидрокси включают в себя возможно замещенные и/или ненасыщенные алкильные группы (например, метил, аллил, бензил или трет-бутил), триалкилсилильные или диарилалкилсилильные группы (например, трет-бутилдиметилсилил, трет-бутилдифенилсилил или триметилсилил) и тетрагидропиранил. Подходящие защитные группы для карбоновой кислоты включают в себя C1-6алкиловые или бензиловые сложные эфиры. Подходящие защитные группы для амино и амидино включают в себя трет-бутилоксикарбонил, бензилоксикарбонил или 2-триметилсилилэтоксикарбонил (Теос). Атомы азота в амидино также могут быть защищены гидрокси- или алкоксигруппами, и эти атомы могут быть защищенными как однократно, так и двукратно.
Защита функциональных групп и снятие защиты с них может иметь место до или после сочетания либо до или после любой другой реакции в вышеуказанных схемах.
Защитные группы можно удалять в соответствии с методиками, которые хорошо известны специалистам, и как описано здесь ниже.
Специалисты должны понимать, что для получения соединений по изобретению альтернативным или, в некоторых случаях, более удобным образом упомянутые здесь выше индивидуальные стадии способов можно осуществлять в ином порядке, и/или индивидуальные реакции можно проводить на иной стадии всего пути (то есть можно осуществлять введение заместителей и/или химические трансформации с иными промежуточными соединениями, чем вышеуказанные в связи с конкретной реакцией). Это может исключать или создавать необходимость в потребности в защитных группах.
Тип вовлеченной химии будет диктовать потребность в защитных группах и их тип, а также последовательность выполнения синтеза.
Применение защитных групп полностью описано в "Protective Groups in Organic Chemistry", edited by J W F McOmie, Plenum Press (1973) и в "Protective Groups in Organic Synthesis", 3rd edition, T.W.Green & P.G.M. Wutz, Wiley-Interscience (1999).
Защищенные производные соединений по изобретению можно превращать химическим путем в соединения по изобретению, используя стандартные методики снятия защиты (например, гидрогенизацию). Специалист также должен понимать, что некоторые соединения формулы Ia можно также обозначать как представляющие собой "защищенные производные" соединений формулы I.
Медицинское и фармацевтическое применение
Соединения по изобретению могут обладать фармакологической активностью как таковые. Соединения по изобретению, которые могут обладать такой активностью, включают в себя соединения формулы I, но не ограничены ими.
Однако другие соединения по этому изобретению (в том числе соединения формулы Ia) могут не обладать такой активностью, но их можно вводить парентерально или перорально, после чего они в организме могут быть метаболизированы с образованием соединений, которые фармакологически активны (включая соответствующие соединения формулы I, но не ограничиваясь ими). Такие соединения (которые также включают в себя соединения, которые могут обладать некоторой фармакологической активностью, но эта активность значительно ниже, чем таковая у "активных" соединений, в которые они метаболизируются) можно поэтому описать как "пролекарства" таких активных соединений.
Таким образом, соединения по этому изобретению полезны, потому что они обладают фармакологической активностью и/или после перорального или парентерального введения метаболизируются в организме с образованием соединений, которые обладают фармакологической активностью. Поэтому соединения по изобретению показаны в качестве фармацевтических средств.
Согласно еще одному аспекту этого изобретения предложены соединения по изобретению для применения в качестве фармацевтических средств.
В частности, соединения по изобретению являются мощными ингибиторами тромбина как таковые и/или (например, в случае пролекарств) после введения метаболизируются с образованием мощных ингибиторов тромбина, например, как продемонстрировано в нижеописанных тестах.
В термин "пролекарство ингибитора тромбина" включены соединения, которые образуют ингибитор тромбина в экспериментально обнаруживаемом количестве и в течение предопределенного промежутка времени (например, около 1 часа) после перорального или пар ентерального введения (смотри, например, Тест Д далее) или, альтернативно, после инкубации в присутствии микросом печени (смотри, например, Тест Ж далее).
Таким образом, ожидается, что соединения по изобретению полезны при тех состояниях, когда требуется ингибирование тромбина, и/или в состояниях, когда показана антикоагулянтная терапия, включая следующее.
Лечение и/или профилактика тромбоза и гиперкоагуляции в крови и/или тканях животных, включая человека. Известно, что гиперкоагуляция может приводить к тромбоэмболическим заболеваниям. Состояния, ассоциированные с гиперкоагуляцией и тромбоэмболическими заболеваниями, которые могут быть упомянуты, включают в себя наследственную или приобретенную резистентность к активированному протеину С, такую как мутация фактора V (фактор V Лейдена), и наследственные или приобретенные дефициты антитромбина III, протеина С, протеина S, гепаринового кофактора II. Другие состояния, о которых известно, что они ассоциированы с гиперкоагуляцией и тромбоэмболическим заболеванием, включают в себя циркулирующие антифосфолипидные антитела (антикоагулянт волчанки), гомоцистеинемию, индуцированную гепарином тромбоцитопению и дефекты фибринолиза, а также коагуляционные синдромы (например, диссеминированное внутрисосудистое свертывание (ДВС)) и повреждение сосудов в общем (например, из-за хирургического вмешательства).
Лечение состояний, когда имеет место нежелательный избыток тромбина без признаков гиперкоагуляции, например при нейродегенеративных заболеваниях, таких как болезнь Альцгеймера.
Конкретные болезненные состояния, которые можно упомянуть, включают в себя терапевтическое и/или профилактическое лечение венозного тромбоза (например, тромбоза глубоких вен (ТГВ)) и эмболии легких, артериального тромбоза (например, при инфаркте миокарда, нестабильной стенокардии, ударе в результате тромбоза и периферическом артериальном тромбозе) и системной эмболии, обычно из предсердия во время фибрилляции предсердий (например, невальвулярной фибрилляции предсердий) или из левого желудочка после трансмурального инфаркта миокарда, или вызванной застойной сердечной недостаточностью, профилактику реокклюзии (то есть тромбоза) после тромболиза, чрезкожной транслюминальной ангиопластики (ЧТА) и коронарного шунтирования; предупреждение повторного тромбоза после микрохирургии и сосудистой хирургии в целом.
Дополнительные показания включают в себя терапевтическое и/или профилактическое лечение диссеминированной внутрисосудистой коагуляции, вызванной бактериями, множественной травмой, интоксикацией или любым другим механизмом; антикоагулянтное лечение, когда кровь находится в контакте с чужеродными поверхностями в организме, такими как сосудистые трансплантаты, сосудистые стенты, сосудистые катетеры, механические и биологические протезы клапанов или любое другое медицинское приспособление; антикоагулянтное лечение, когда кровь находится в контакте с медицинскими приспособлениями вне организма как при сердечно-сосудистой хирургической операции с использованием аппарата "сердце-легкие" или при гемодиализе; терапевтическое и/или профилактическое лечение идиопатического или имеющего место у взрослых респираторного дистресс-синдрома, фиброза легких после лечения облучением или химиотерапии, септического шока, септицемии, воспалительных реакций, которые включают в себя, без ограничений, отек, острый или хронический атеросклероз, такой какой как коронарное артериальное заболевание и образование атеросклеротических бляшек, церебральное артериальное заболевание, церебральный инфаркт, церебральный тромбоз, церебральную эмболию, периферическое артериальное заболевание, ишемию, стенокардию (включая нестабильную стенокардию), реперфузионное повреждение, рестеноз после чрескожной транслюминальной ангиопластики (ЧТА) и шунтирование коронарных артерий.
Соединения по изобретению, которые ингибируют трипсин и/или тромбин, также могут быть полезными в лечении панкреатита.
Таким образом, соединения по изобретению показаны как для терапевтического, так и профилактического лечения этих состояний.
В соответствии с еще одним аспектом настоящего изобретения предложен способ лечения состояния, при котором требуется ингибирование тромбина, при котором лицу, страдающему таким состоянием или подверженному такому состоянию, вводят терапевтически эффективное количество соединения по изобретению.
Соединения по изобретению обычно вводят перорально, внутривенно, подкожно, трансбуккально, ректально, дермально, назально, трахеально, бронхиально и любым другим парентеральным путем или посредством ингаляции в форме фармацевтических препаратов, содержащих соединение по изобретению в фармацевтически приемлемой лекарственной форме.
В зависимости от расстройства или пациента, подлежащего лечению, и пути введения эти композиции можно вводить в варьирующих дозах.
Соединения по этому изобретению можно также комбинировать и/или вводить совместно с любым(и) антитромботическим(и) агентом(ами) с другим механизмом действия, такими как один или более чем один из числа следующих: антитромбоцитарные агенты - ацетилсалициловая кислота, тиклопидин и клопидогрел; ингибиторы рецепторов или синтетазы тромбоксана; антагонисты рецепторов фибриногена; миметики простациклина; ингибиторы фосфодиэстеразы; антагонисты ADP-рецепторов (P2Т) и ингибиторы карбоксипептидазы U (CPU).
При лечении тромботических заболеваний, в частности инфаркта миокарда, соединения по изобретению можно дополнительно комбинировать и/или вводить совместно с тромболитиками, такими как один или более чем один из числа следующего: активатор тканевого плазминогена (природный, рекомбинантный или модифицированный), стрептокиназа, урокиназа, проурокиназа, анизоилированный плазминоген-стрептокиназный активаторный комплекс (АПСАК), активаторы плазминогена слюнных желез животных и тому подобное.
Согласно еще одному аспекту изобретения предложен фармацевтический препарат, включающий в себя соединение по изобретению в смеси с фармацевтически приемлемым адъювантом, разбавителем или носителем.
Подходящие суточные дозы соединений по изобретению в терапевтическом лечении людей составляют, исключая массу какого-либо противоиона кислоты, около 0,001-100 мг/кг массы тела при пероральном введении и 0,001-50 мг/кг массы тела при парентеральном введении.
Во избежание сомнения термин "лечение" при использовании здесь включает в себя терапевтическое и/или профилактическое лечение.
При сравнении с соединениями, известными из уровня техники, соединения по изобретению имеют то преимущество, что они могут быть более эффективными, менее токсичными, обладать большей продолжительностью действия, иметь более широкий диапазон активности, быть более сильнодействующими, продуцировать меньше побочных эффектов, легче абсорбироваться и/или иметь лучший фармакокинетический профиль (например, более высокую пероральную биодоступность и/или более низкий клиренс), и/или иметь другие полезные фармакологические, физические или химические свойства.
Соединения по изобретению могут иметь еще и то преимущество, что их можно вводить менее часто, чем соединения, известные из уровня техники.
Биологические тесты
Можно применять следующие процедуры тестов.
Тест А
Определение тромбинового времени свертывания (ТВ)
Раствор ингибитора (25 мкл) инкубируют с плазмой (25 мкл) в течение трех минут. Затем добавляют человеческий тромбин (Т 6729; Sigma Chem. Co. или Hematologic Technologies) в буферном растворе с рН 7,4 (25 мкл, 4,0 ед. NIH/мл) и измеряют время свертывания в автоматическом устройстве (КС 10; Amelung).
Тромбиновое время свертывания (ТВ) выражают в абсолютных величинах (секундах), так же как и отношение ТВ без ингибитора (ТВ0) к ТВ с ингибитором (ТВи). Эти последние отношения (диапазон 1-0) наносят на график против концентрации ингибитора (преобразованной в логарифм) и подгоняют к сигмоидальным кривым доза-ответ в соответствии с уравнением
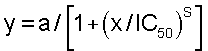
где а = максимальный диапазон, то есть 1; s = наклон кривой доза-ответ; IC50 = концентрация ингибитора, которая удваивает время свертывания. Вычисления осуществляют на ПК, используя программное обеспечение GraFit Version 3, принимая установки уравнения: начало при (Start at) 0, конец определения (define end) = 1 (Erithacus Software, Robin Leatherbarrow, Imperial College of Science, London, UK).
Тест Б
Определение ингибирования тромбина посредством хромогенного автоматизированного анализа
Активность ингибитора тромбина измеряют способом хромогенного субстрата в автоматизированном микропланшетном процессоре Plato 3300 (Rosys AG, CH-8634, Hombrechtikon, Switzerland), используя 96-луночные титрационные микропланшеты половинного объема (Costar, Cambridge, MA, USA; Cat No 3690). Маточные растворы тестируемого вещества в DMSO (72 мкл), 0,1-1 ммоль/л, серийно разводят DMSO в соотношении 1:3 (24+48 мкл) с получением десяти разных концентраций, которые анализируют в качестве проб в анализе. Разбавляют 2 мкл тестируемой пробы буфером для анализа (124 мкл), добавляют 12 мкл раствора хромогенного субстрата (S-2366, Chromogenix, Mölndal, Sweden) в буфере для анализа и, наконец, 12 мкл раствора α-тромбина (человеческий α-тромбин, Sigma Chemical Co. или Hematologic Technologies) в буфере для анализа и перемешивают пробы. Конечными концентрациями в анализе являются тестируемое вещество 0,00068-13,3 мкмоль/л; S-2366 0,30 ммоль/л; α-тромбин 0,020 ед. NIH/мл. Для вычисления процентов ингибирования в тестируемых пробах при сравнении с пустым контролем без ингибитора используют линейный абсорбционный инкремент в течение 40 минут инкубации при 37°С. Полученное автоматизированным способом значение IC50, соответствующее концентрации ингибитора, которая вызывает 50%-ное ингибирование активности тромбина, вычисляют по кривой зависимости между логарифмом концентрации и % ингибирования.
Тест В
Определение константы ингибирования Кi для человеческого тромбина
Осуществляют определения Ki, используя метод хромогенного субстрата, который реализуют при 37°С на центрифужном анализаторе Cobas Bio (Roche, Basel, Switzerland). Остаточную активность фермента после инкубации человеческого α-тромбина определяют с разными концентрациями тестируемого соединения при трех разных концентрациях субстрата и измеряют как изменение оптического поглощения при 405 нм.
Смешивают растворы тестируемого соединения (100 мкл, обычно в буфере или физиологическом растворе, содержащем 10 г/л БСА) с 200 мкл человеческого α-тромбина (Sigma Chemical Co.) в буфере для анализа (0,05 моль/л Трис-HCl, рН 7,4; ионная сила 0,15 установлена с помощью NaCl), содержащем БСА (10 г/л), и анализируют в качестве проб в Cobas Bio. Добавляют пробу объемом 60 мкл вместе с 20 мкл воды в 320 мкл субстрата S-2238 (Chromogenix AB, Mölndal, Sweden) в буфере для анализа и регистрируют изменение поглощения (ΔА/мин). Конечные концентрации для S-2238 составляют 16, 24 и 50 мкмоль/л, а для тромбина 0,125 ед. NIH/мл.
Для построения графиков Диксона (Dixon), то есть диаграмм концентрации ингибитора против 1/(ΔА/мин) используют скорость реакции в стационарном состоянии. Для обратимых конкурентных ингибиторов точки данных для разных концентраций субстрата обычно образуют прямые линии, которые пересекаются при х=-Ki.
Тест Г
Определение активированного парциального тромбопластинового времени (АПТВ)
АПТВ определяют в пулированной нормальной стабилизированной цитратом человеческой плазме с реагентом РТТ Automated 5, изготавливаемым Stago. Добавляют к плазме ингибиторы (10 мкл раствора ингибитора к 90 мкл плазмы) и инкубируют с реагентом АРТТ в течение 3 минут с последующим добавлением 100 мкл раствора хлорида кальция (0,025 М) и определяют АПТВ, используя анализатор коагуляции КС10 (Amelung) в соответствии с инструкциями производителя реагента.
Время свертывания выражают в абсолютных величинах (секунды), так же как и соотношение АПТВ без ингибитора (АПТВ0) и АПТВ с ингибитором (АПТВи). Эти последние соотношения (диапазон 1-0) наносят на график против концентрации ингибитора (преобразованной в логарифм) и подгоняют к сигмоидальным кривым доза-ответ в соответствии с уравнением
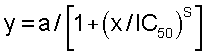
где а = максимальный диапазон, то есть 1; s = наклон кривой доз-ответ; IC50 = концентрация ингибитора, которая удваивает время свертывания. Вычисления осуществляют на ПК, используя программное обеспечение GraFit Version 3, принимая установки уравнения: начало при (Start at) 0, конец определения (define end) = 1 (Erithacus Software, Robin Leatherbarrow, Imperial College of Science, London, UK). IC50АПТВ определяют как концентрацию ингибитора в человеческой плазме, которая удваивает активированное парциальное тромбопластиновое время.
Тест Д
Определение тромбинового времени ex vivo
Ингибирование тромбина после перорального или парентерального введения соединений по изобретению, растворенных в смеси этанол:SolutolK:вода (5:5:90), оценивают у находящихся в сознании крыс, которых за день или два дня до эксперимента снабжают катетером для взятия крови из сонной артерии. В день эксперимента в фиксированные моменты времени после введения соединения берут пробы крови в пластиковые пробирки, содержащие 1 часть раствора цитрата натрия (0,13 моль/л) и 9 частей крови. Для получения обедненной тромбоцитами плазмы центрифугируют пробирки.
Пробы плазмы, имеющие объем 50 мкл, подвергают осаждению холодным ацетонитрилом (100 мкл). Центрифугируют пробы в течение 10 минут при 4000 об/мин. Разбавляют 75 мкл супернатанта 0,2%-ной муравьиной кислотой (75 мкл). Анализируют объемы, равные 10 мкл, полученных растворов методом ЖХ-МС/МС и определяют концентрации ингибитора тромбина по стандартным кривым.
Тест Е
Определение клиренса из плазмы у крыс
Клиренс из плазмы оценивают у самцов крыс Sprague Dawley. Соединение растворяют в воде и вводят в виде болюсной подкожной инъекции в дозе 4 мкмоль/кг. Собирают пробы крови через частые интервалы в течение интервала времени вплоть до 5 часов после введения лекарства. Центрифугируют пробы крови и отделяют плазму от клеток крови и переносят во флаконы, содержащие цитрат (конечная концентрация 10%). Пробы плазмы, имеющие объем 50 мкл, подвергают осаждению холодным ацетонитрилом (100 мкл). Центрифугируют пробы в течение 10 минут при 4000 об/мин. Разбавляют 75 мкл супернатанта 0,2%-ной муравьиной кислотой (75 мкл). Анализируют объемы полученных растворов, равные 10 мкл, методом ЖХ-МС/МС и определяют концентрации ингибитора тромбина по стандартным кривым. Оценивают площадь под профилем концентрация-время (ППК) с использованием логарифмического/линейного правила трапеций и экстраполируют в бесконечное время. Затем определяют клиренс из плазмы (КП) для соединения как
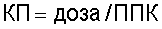
Выражают величины в мл/мин/кг.
Тест Ж
Определение стабильности in vitro
Микросомы печени получают из образцов печени крыс Sprague Dawley и печени человека в соответствии с внутренними стандартными рабочими протоколами (SOP). Инкубируют соединения при 37°С при концентрации общего микросомального белка, равной 3 мг/мл, в 0,05 моль/л буфера ТРИС при рН 7,4 в присутствии кофакторов НАДН (2,5 ммоль/л) и НАДФН (0,8 ммоль/л). Исходная концентрация соединений составляет 5 или 10 мкмоль/л. Затем в период до 60 минут после начала инкубации берут пробы для анализа. Ферментативную активность собранных проб немедленно останавливают добавлением 20% миристиновой кислоты в объеме, соответствующем 3,3% общего объема пробы. Концентрацию соединения, остающуюся в 60-минутной пробе (FINAL CONC), определяют методом ЖХМС с использованием проб, собранных в нулевое время, в качестве контроля (START CONC). Процент разложившегося ингибитора тромбина вычисляют как

Тест З
Модель артериального тромбоза
Повреждение сосудов индуцируют местным нанесением хлорида трехвалентного железа (FeCl3) на сонную артерию. Крыс анестезируют внутрибрюшинной инъекцией пентобарбитала натрия (80 мг/кг; Apoteksbolaget; Umee, Sweden) с последующей непрерывной инфузией (12 мг/кг/ч) в течение всего эксперимента. Температуру тела крысы в течение всего эксперимента поддерживают внешним подогревом на уровне 38°С. Эксперимент начинают с 5-минутного контрольного периода. Спустя пять минут дают внутривенно человеческий 125I-фибриноген (80 кБк, IМ53, Amersham International, Buckinghamshire, UK) и используют как маркер для последующего включения фибрина (фибриногена) в тромб. Проксимальный конец сегмента сонной артерии помещают в продольно открытую пластиковую пробирку (6 мм, Silastic®, Dow Corning, MI, USA), содержащую пропитанную FeCl3 (2 мкл, 5% мас., Merck, Darmstadt, Germany) фильтровальную бумагу (диаметр 3 мм, 1F, Munktell, Grycksbo, Sweden). Левую сонную артерию подвергают действию FeCl3 в течение 10 минут, затем извлекают из пластиковой пробирки и замачивают в физиологическом растворе. Спустя пятьдесят минут удаляют сонную артерию и промывают в физиологическом растворе. Также через 10 минут после инъекции 125I-фибриногена и в конце эксперимента берут контрольные пробы крови для определения 125I-активности крови, 125I-активность в контрольных пробах крови и в сегменте сосуда измеряют в гамма-счетчике (1282 Compugamma; LKB Wallac Oy, Turku, Finland) в тот же день, когда выполняют эксперимент. Размер тромба определяют как количество 125I-активности, включенной в сегмент сосуда, относительно 125I-активности в крови (импульсы в минуту на мг).
Общие экспериментальные детали
Тонкослойную хроматографию (ТСХ) выполняли на силикагеле. Анализ методом хиральной ВЭЖХ выполняли с использованием колонки Chiralcel OD 46 мм × 250 мм с пятисантиметровой предохранительной колонкой. Температуру колонки поддерживали на уровне 35°С. Использовали скорость потока 1,0 мл/мин. Использовали детектор Gilson 115 UV при 228 нм. Подвижная фаза состояла из гексанов, этанола и трифторуксусной кислоты, причем соответствующие соотношения указаны для каждого соединения. Обычно продукт растворяли в минимальном количестве этанола и разбавляли подвижной фазой.
ЖХ-МС/МС выполняли с использованием прибора HP-1100, снабженного инжектором CTC-PAL и колонкой ThermoQuest, Hypersil BDS-C18, 5 мкм, 4×100 мм. Использовали масс-спектрометрический детектор API-3000 (Sciex). Скорость потока составляла 1,2 мл/мин, подвижная фаза (градиент) состояла из 10-90% ацетонитрила с 90-10% 4 мМ водного ацетата аммония, причем оба содержали 0,2% муравьиной кислоты.
Спектры 1H-ЯМР записывали с использованием тетраметилсилана в качестве внутреннего стандарта. Спектры 13С-ЯМР записывали с использованием перечисленных дейтерированных растворителей в качестве внутренних стандартов.
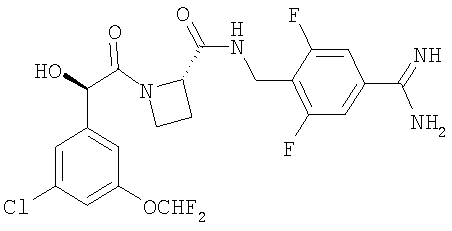
Пример 1
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-Pab(2.6-diF)
(1) 3-Хлор-5-метоксибензальдегид
К металлу, представляющему собой магний (14,2 г; 585 ммоль, предварительно промыт 0,5 н. HCl), в THF (100 мл) при 25°С добавляли 3,5-дихлоранизол (74,0 г; 419 мл) в THF (200 мл). После этого добавления добавляли каплями 1,2-дибромэтан (3,9 г; 20,8 ммоль). Полученную темно-коричневую смесь кипятили с обратным холодильником в течение 3 ч. Охлаждали эту смесь до 0°С и добавляли одной порцией N,N-диметилформамид (60 мл). Эту смесь фракционировали диэтиловым эфиром (3×400 мл) и 6 н. HCl (500 мл). Промывали объединенные органические экстракты рассолом (300 мл), сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением масла. Флэш-хроматография (2×) на силикагеле с элюцией смесью Нех:EtOAc (4:1) давала указанное в подзаголовке соединение (38,9 г; 54%) в виде желтого масла.
1H-ЯМР (300 МГц, CDCl3) δ 9.90 (s, 1H), 7.53 (s, 1H), 7.38 (s, 1H), 7.15 (s, 1H), 3.87 (s, 3Н).
(2) 3-Хлор-5-гидроксибензальдегид
Раствор 3-хлор-5-метоксибензальдегида (22,8 г; 134 ммоль; см. стадию (1) выше) в СН2Cl2 (250 мл) охлаждали до 0°С. В течение 15 мин добавляли каплями трибромид бора (15,8 мл; 167 ммоль). После 2 часов перемешивания к этой реакционной смеси медленно добавляли Н2O (50 мл). Затем экстрагировали этот раствор Et2O (2×100 мл). Объединяли органические слои, сушили их (Na2SO4) фильтровали и концентрировали в вакууме. Флэш-хроматография на силикагеле с элюцией смесью Нех:EtOAc (4:1) давала указанное в подзаголовке соединение (5,2 г; 25%).
1H-ЯМР (300 МГц, CDCl3) δ 9.85 (s, 1H), 7.35 (s, 1H), 7.20 (s, 1H), 7.10 (s, 1H), 3.68 (s, 1H).
(3) 3-Хлор-5-дифторметоксибензальдегид
Раствор 3-хлор-5-гидроксибензальдегида (7,5 г; 48 ммоль; см. стадию (2) выше) в 2-пропаноле (250 мл) и 30%-ном KOH (100 мл) нагревали до начала флегмообразования. В реакционную смесь при перемешивании в течение 2 часов барботировали CHClF2. Реакционную смесь охлаждали, подкисляли 1 н. HCl и экстрагировали EtOAc (2×100 мл). Органику промывали рассолом (100 мл), сушили (Na2SO4) фильтровали и концентрировали в вакууме. Флэш-хроматография на силикагеле с элюцией смесью Hex:EtOAc (4:1) давала указанное в подзаголовке соединение (4,6 г; 46%).
1H-ЯМР (300 МГц, CDCl3) δ 9.95 (s, 1H), 7.72 (s, 1H), 7.52 (s, 1H), 7.40 (s, 1Н), 6,60 (t, JH-F=71.1 Гц, 1Н).
(4) Ph(3-Cl)(5-OCHF2)-(R,S)CH(OTMS)CN
Раствор 3-хлор-5-дифторметоксибензальдегида (4,6 г; 22,3 ммоль; см. стадию (3) выше) в CH2Cl2 (200 мл) охлаждали до 0°С. Добавляли Znl2 (1,8 г; 5,6 ммоль) и триметилсилилцианид (2,8 г; 27,9 ммоль), давали реакционной смеси нагреться до комнатной температуры и перемешивали ее в течение 15 ч. Смесь частично концентрировали в вакууме с получением указанного в подзаголовке соединения в виде жидкости, которую прямо использовали в стадии (5) ниже без дальнейшей очистки или характеризации.
(5) Ph(3-Cl)(5-OCHF2)-(R,S)CH(OH)C(NH)OEt
Ph(3-Cl)(5-OCHF2)-(R,S)CH(OTMS)CN (6,82 г; принято за 22,3 ммоль; см. стадию (4) выше) добавляли каплями к смеси HCl/EtOH (500 мл). Эту реакционную смесь перемешивали в течение 15 ч, затем частично концентрировали в вакууме с получением указанного в подзаголовке соединения в виде жидкости, которую использовали в стадии (6) без дальнейшей очистки или характеризации.
(6) Ph(3-Cl)(5-OCHF2)-(R,S)CH(OH)C(O)OEt
Растворяли Ph(3-Cl)(5-OCHF2)-(R,S)CH(OH)C(NH)OEt (6,24 г; 22,3 ммоль; см. стадию (5) выше) в THF (250 мл), добавляли 0,5 М H2SO4 (400 мл) и перемешивали реакционную смесь при 40°С в течение 65 ч, охлаждали ее и затем частично концентрировали в вакууме для удаления большей части THF. Затем экстрагировали реакционную смесь Et2О (3×100 мл), сушили (Na2SO4), фильтровали и концентрировали в вакууме, что давало указанное в подзаголовке соединение в виде твердого вещества, которое затем использовали в стадии (7) без дальнейшей очистки или характеризации.
(7) Ph(3-Cl)(5-OCHF2)-(R,S)CH(OH)C(O)OH
Раствор Ph(3-Cl)(5-OCHF2)-(R,S)CH(OH)C(O)OEt (6,25 г; принято за 22,3 ммоль; см. стадию (6) выше) в 2-пропаноле (175 мл) и 20%-ном KOH (350 мл) перемешивали при комнатной температуре в течение 15 часов. Затем частично концентрировали реакционную смесь в вакууме для удаления большей части 2-пропанола. Оставшуюся часть подкисляли 1 М Н2SO4, экстрагировали Et2O (3×100 мл), сушили (Na2SO4) и концентрировали в вакууме с получением твердого вещества. Флэш-хроматография на силикагеле с элюцией смесью CHCl3:МеОН:концентрированный NH4OH (6:3:1) давала аммониевую соль указанного в подзаголовке соединения. Эту аммониевую соль затем растворяли в смеси EtOAc (75 мл) и Н2O (75 мл) и подкисляли 2 н. HCl. Органический слой отделяли и промывали рассолом (50 мл), сушили (Na2SO4) и концентрировали в вакууме, что давало указанное в подзаголовке соединение (3,2 г; 57% от стадии (4) до стадии (7)).
1H-ЯМР (300 МГц, CD3OD) δ 7.38 (s, 1Н), 7.22 (s, 1H), 7.15 (s, 1H), 6.89 (t, JH-F=71,1 Гц, 1H), 5.16(s, 1H).
(8) Ph(3-Cl)(5-OCHF2)-(R)СН(OH)C(O)OH (а) и
Ph(3-Cl)(5-OCHF2)-(S)CH(OAc)C(O)OH (б)
Смесь Ph(3-Cl)(5-OCHF2)-(R,S)CH(OH)C(O)OH (3,2 г; 12,7 ммоль; см. стадию (7) выше) и Lipase PS "Amano" (около 2 г) в винилацетате (125 мл) и МТВЕ (125 мл) кипятили с обратным холодильником в течение 48 ч. Реакционную смесь охлаждали, фильтровали через Celite® и осадок на фильтре промывали EtOAc. Фильтрат концентрировали в вакууме и подвергали флэш-хроматографии на силикагеле с элюцией смесью CHCl3:МеОН:концентрированный NH4OH (6:3:1) с получением аммониевых солей указанных в подзаголовке соединений (а) и (б). Соединение (а) в виде соли растворяли в H2O, подкисляли 2 н. HCl и экстрагировали EtOAc. Органический слой промывали рассолом, сушили (Na2SO4) и концентрировали в вакууме, что давало указанное в подзаголовке соединение (а) (1,2 г; 37%).
Для указанного в подзаголовке соединения (а)
1H-ЯМР (300 МГц, CD3OD) δ 7.38 (s, 1H), 7.22 (s, 1H), 7.15 (s, 1H), 6.89 (t, JH-F=71,1 Гц, 1Н), 5.17 (S, 1Н).
(9) 2,6-Дифтор-4-[(метилсульфинил)(метилтио)метил]бензонитрил
(Метилсульфинил)(метилтио)метан (7,26 г; 0,0584 моль) растворяли в 100 мл сухого THF под аргоном и охлаждали до -78°С. Добавляли каплями при перемешивании бутиллитий в гексане (16 мл; 1,6 М; 0,0256 моль). Эту смесь перемешивали в течение 15 минут. Тем временем охлаждали раствор 3,4,5-трифторбензонитрила (4,0 г; 0,025 ммоль) в 100 мл сухого THF под аргоном до -78°С и добавляли первый раствор ко второму раствору через канюлю на протяжении периода 35 минут. Спустя 30 минут убирали охлаждающую баню и, когда реакционная смесь достигала комнатной температуры, ее вливали в 400 мл воды. Выпаривали THF и трижды экстрагировали оставшийся водный слой диэтиловым эфиром. Объединенную эфирную фазу промывали водой, сушили (Na2SO4) и упаривали. Выход 2,0 г (30%).
1H-ЯМР (500 МГц, CDCl3) δ 7.4-7.25 (m, 2H), 5.01 (s, 1H, диастереомер), 4.91 (s, 1H, диастереомер), 2.88 (s, 3Н, диастереомер), 2.52 (s, 3Н, диастереомер), 2.49 (s, 3Н, диастереомер), 2.34 (s, 3Н, диастереомер), 1.72 (широкий, 1H).
(10) 2,6-Дифтор-4-формилбензонитрил
Растворяли 2,6-дифтор-4-[(метилсульфинил)(метилтио)метил]бензонитрил (2,17 г; 8,32 ммоль; см. стадию (9) выше) в 90 мл THF и добавляли 3,5 мл концентрированной серной кислоты. Эту смесь оставляли при комнатной температуре на 3 дня и затем вливали в 450 мл воды. Затем трижды экстрагировали EtOAc и объединенную эфирную фазу дважды промывали водным бикарбонатом натрия и затем рассолом, сушили (Na2SO4) и упаривали. Выход 1,36 г (98%). Положение формильной группы устанавливали методом 13С ЯМР. Сигнал от фторированных атомов углерода при 162,7 м.д. проявлял ожидаемый паттерн взаимодействия с двумя константами взаимодействия порядка 260 Гц и 6,3 Гц соответственно, что соответствует ипсо и мета взаимодействию от атомов фтора.
1H-ЯМР (400 МГц, CDCl3) δ 10.35 (s, 1H), 7.33 (m, 2H).
(11) 2,6-Дифтор-4-гидроксиметилбензонитрил
2,6-Дифтор-4-формилбензонитрил (1,36 г; 8,13 ммоль; см. стадию (10) выше) растворяли в 25 мл метанола и охлаждали на ледяной бане. Добавляли порциями при перемешивании боргидрид натрия (0,307 г; 8,12 ммоль) и оставляли реакционную смесь на 65 мин. Выпаривали растворитель и распределяли остаток между диэтиловым эфиром и водным бикарбонатом натрия. Эфирный слой промывали дополнительным количеством водного бикарбоната натрия и рассолом, сушили (Na2SO4) и упаривали. Неочищенный продукт вскоре кристаллизовался, и его можно было использовать без дальнейшей очистки. Выход 1,24 г (90%).
1Н ЯМР (400 МГц, CDCl3) δ 7.24 (m, 2H), 4.81 (s, 2H), 2.10 (широкий, 1Н).
(12) 4-Циано-2,6-дифторбензилметансульфонат
К охлажденному на льду раствору 2,6-дифтор-4-гидроксиметилбензонитрила (1,24 г; 7,32 ммоль; см. стадию (11) выше) и метансульфонилхлорида (0,93 г; 8,1 ммоль) в 60 мл метиленхлорида добавляли при перемешивании триэтиламин (0,81 г; 8,1 ммоль). Спустя 3 часа при 0°С эту смесь промывали дважды 1 M HCl и одни раз водой, сушили (Na2SO4) и упаривали. Продукт можно было использовать без дальнейшей очистки. Выход 1,61 г (89%).
1H ЯМР (300 МГц, CDCl3) δ 7.29 (m, 2H), 5.33 (s, 2H), 3.07 (s, 3Н).
(13) 4-Азидометил-2,6-дифторбензонитрил
Смесь 4-циано-2,6-дифторбензилметансульфоната (1,61 г; 6,51 ммоль; см. стадию (12) выше) и азида натрия (0,72 г; 0,0111 моль) в 10 мл воды и 20 мл DMF перемешивали при комнатной температуре в течение ночи. Затем вливали полученную смесь в 200 мл воды и экстрагировали три раза диэтиловым эфиром. Объединенную эфирную фазу промывали пять раз водой, сушили (Na2SO4) и упаривали. Небольшой образец упаривали для целей ЯМР и продукт кристаллизовали. Остальную часть упаривали осторожно, но не до полной сухости. Выход (теоретически 1,26) считали почти количественным на основании ЯМР и аналитической ВЭЖХ.
1H ЯМР (400 МГц, CDCl3) δ 7.29 (m, 2H), 4.46 (s, 2H).
(14) 4-Аминометил-2,6-дифторбензонитрил
Реакцию проводили в соответствии с процедурой, описанной в J. Chem. Res. (M) (1992) 3128. К суспензии 520 мг 10%-ного Pd/C (50%-ная влажность) в 20 мл воды добавляли раствор боргидрида натрия (0,834 г; 0,0221 моль) в 20 мл воды. Результатом было выделение некоторого количества газа. 4-Азидометил-2,6-дифторбензонитрил (1,26 г; 6,49 ммоль; см. стадию (13) выше) растворяли в 50 мл THF и добавляли к этой водной смеси на ледяной бане в течение 15 мин. Смесь перемешивали в течение 4 ч, после чего добавляли 20 мл 2 М HCl и фильтровали смесь через целит. Целит промывали дополнительным количеством воды и объединенную водную фазу промывали EtOAc и затем подщелачивали 2 М NaOH. Затем проводили трехкратную экстракцию метиленхлоридом и объединенную органическую фазу промывали водой, сушили (Na2SO4) и упаривали. Выход 0,87 г (80%).
1H ЯМР (400 МГц, CDCl3) δ 7.20 (m, 2H), 3.96 (s, 2H), 1.51 (широкая, 2H).
(15) 2,6-Дифтор-4-трет-бутоксикарбониламинометилбензонитрил
Растворяли раствор 4-аминометил-2,6-дифторбензонитрила (0,876 г; 5,21 ммоль; см. стадию (14) выше) в 50 мл THF и добавляли ди-трет-бутилдикарбонат (1,14 г; 5,22 ммоль) в 10 мл THF. Смесь перемешивали в течение 3,5 ч. Выпаривали THF и распределяли остаток между водой и EtOAc. Органический слой трижды промывали 0,5 М HCl и водой, сушили (Na2SO4) и упаривали. Продукт мог быть использован без дополнительной очистки. Выход 1,38 г (99%).
1H ЯМР (300 МГц, CDCl3) δ 7.21 (m, 2H), 4.95 (широкий, 1Н), 4.43 (широкий, 2H), 1.52 (s, 9H).
(16) Boc-Pab(2,6-diF)(OH)
Смесь 2,6-дифтор-4-трет-бутоксикарбониламинометилбензонитрила (1,38 г; 5,16 ммоль; см. стадию (15) выше), гидрохлорида гидроксиламина (1,08 г; 0,0155 моль) и триэтиламина (1,57 г; 0,0155 моль) в 20 мл этанола перемешивали при комнатной температуре в течение 36 часов. Выпаривали растворитель и распределяли остаток между водой и метиленхлоридом. Органический слой промывали водой, сушили (Na2SO4) и упаривали. Продукт можно было использовать без дальнейшей очистки. Выход 1,43 г (92%).
1H ЯМР (500 МГц, CD3OD) δ 7.14 (m, 2Н), 4.97 (широкая, 1Н), 4.84 (широкая, 2Н), 4.40 (широкий, 2Н), 1.43 (s, 9H).
(17) Вос-Pab(2,6-diF)×НОАс
Эту реакцию проводили в соответствии с процедурой, описанной Judkins et al. в Synth. Comm. (1998) 4351. Boc-Pab(2,6-diF)(OH) (1,32 г; 4,37 ммоль; см. стадию (16) выше), уксусный ангидрид (0,477 г; 4,68 ммоль) и 442 мг 10% Pd/C (50%-ная влажность) в 100 мл уксусной кислоты гидрировали при давлении 5 атм (0,51·106 Па) в течение 3,5 часов. Смесь фильтровали через целит, промывали этанолом и упаривали. Остаток лиофилизировали из ацетонитрила и воды и нескольких капель этанола. Указанный в подзаголовке продукт можно было использовать без дальнейшей очистки. Выход 1,49 г (99%).
1H ЯМР (400 МГц, CD3OD) δ 7.45 (m, 2Н), 4.34 (s, 2Н), 1.90 (s, ЗН), 1.40 (s, 9H).
(18) Boc-Pab(2,6-diF)(Teoc)
К раствору Boc-Pab(2,6-diF)×НОАс (1,56 г; 5,49 ммоль; см. стадию (17) выше) в 100 мл THF и 1 мл воды добавляли 2-(триметилсилил)-этил-п-нитрофенилкарбонат (1,67 г; 5,89 ммоль). Раствор карбоната калия (1,57 г; 0,0114 моль) в 20 мл воды добавляли каплями в течение 5 минут. Смесь перемешивали в течение ночи. Выпаривали THF и распределяли остаток между водой и метиленхлоридом. Водный слой экстрагировали метиленхлоридом и объединенную органическую фазу дважды промывали водным бикарбонатом натрия, сушили (Na2SO4) и упаривали. Флэш-хроматография на силикагеле со смесью гептан/этилацетат = 2/1 давала 1,71 (73%) чистого соединения.
1H ЯМР (400 МГц, CDCl3) δ 7.43 (m, 2H), 4.97 (широкий, 1Н), 4.41 (широкий, 2H), 4.24 (m, 2H), 1.41 (s, 9H), 1.11 (m, 2H), 0.06 (s, 9H).
(19) Boc-(S)Aze-Pab(2,6-diF)(Teoc)
Boc-Pab(2,6-diF)(Teoc) (1,009r; 2,35 ммоль; см. стадию (18) выше) растворяли в 50 мл EtOAc, насыщенного газообразным HCl. Эту смесь оставляли на 10 мин, упаривали и растворяли в 18 мл DMF, затем охлаждали на ледяной бане. Добавляли Boc-(S)-Aze-OH (0,450 г; 2,24 ммоль), РуВОР (1,24 г; 2,35 ммоль) и, наконец, диизопропилэтиламин (1,158 г; 8,96 ммоль). Реакционную смесь перемешивали в течение двух часов, затем вливали в 350 мл воды и экстрагировали три раза EtOAc. Объединенную органическую фазу промывали рассолом, сушили (Na2SO4) и упаривали. Флэш-хроматография на силикагеле со смесью гептан:этилацетат (1:3) давала 1,097 г (96%) желаемого соединения.
1H ЯМР (500 МГц, CDCl3) δ 7.46 (m, 2H), 4.65-4.5 (m, 3Н), 4.23 (m, 2H), 3.87 (m, 1Н), 3.74 (m, 1H), 2.45-2.3 (m, 2H), 1.40 (s, 9Н), 1.10 (m, 2H), 0.05 (s, 9H).
(20) Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-Pab(2,6-diF)(Teoc)
Boc-(S)Aze-Pab(2,6-diF)(Teoc) (0,256 г; 0,500 ммоль; см. стадию (19) выше) растворяли в 20 мл EtOAc, насыщенного газообразным HCl. Эту смесь оставляли на 10 мл, упаривали и растворяли в 5 мл DMF. Добавляли Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)OH (0,120 г; 0,475 ммоль; см. стадию (8) выше), РуВОР (0,263 г; 0,498 ммоль) и, наконец, диизопропилэтиламин (0,245 г; 1,89 ммоль). Реакционную смесь перемешивали в течение двух часов и затем вливали в 350 мл воды и экстрагировали три раза EtOAc. Объединенную органическую фазу промывали рассолом, сушили (Na2SO4) и упаривали. Флэш-хроматография на силикагеле с EtOAc давала 0,184 г (60%) желаемого соединения, указанного в подзаголовке.
1H ЯМР (400 МГц, CD3OD, смесь ротамеров) δ 7.55-7.45 (m, 2H), 7.32 (m, 1H, основной ротамер), 7.27 (m, 1H, минорный ротамер), 7.2-7.1 (m, 2H), 6.90 (t, 1H, основной ротамер), 6.86 (t, 1H), минорный ротамер), 5.15 (s, 1H, основной ротамер), 5.12 (m, 1H, минорный ротамер), 5.06 (s, 1H, минорный ротамер), 4.72 (m, 1H, основной ротамер), 4.6-4.45 (m, 2H), 4.30 (m, 1H, основной ротамер), 4.24 (m, 2H), 4.13 (m, 1H, основной ротамер), 4.04 (m, 1H, минорный ротамер), 3.95 (m, 1H, минорный ротамер), 2.62 (m, 1H, минорный ротамер), 2.48 (m, 1H, основной ротамер), 2.22 (m, 1H, основной ротамер), 2.10 (m, 1H, минорный ротамер), 1.07 (m, 2H), 0.07 (m, 9H).
(21) Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-Pab(2,6-diF)
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-Pab(2,6-diF)(Teoc) (81 мг; 0,127 ммоль; см. стадию (20) выше) растворяли в 0,5 мл метиленхлорида и охлаждали на ледяной бане. Добавляли TFA (3 мл) и оставляли реакционную смесь на 75 мин. Выпаривали TFA и лиофилизировали остаток из воды и ацетонитрила. Неочищенный продукт очищали препаративной ЖХОФ со смесью СН3CN : 0,1 М NH4OAc (35:65) с получением 39 мг (55%) соединения, указанного в заголовке, в виде его НОАс соли, чистота 99%.
1H ЯМР (400 МГц, CD3OD, смесь ротамеров) δ 7.5-7.4 (m, 2H), 7.32 (m, 1H, основной ротамер), 7.28 (m, 1H, минорный ротамер), 7.2-7.1 (m, 3Н) 6.90 (t, 1H, основной ротамер), 6.86 (t, минорный ротамер), 5.15 (s, 1H, основной ротамер), 5.14 (m, 1H, минорный ротамер), 5.07 (s, 1H, минорный ротамер), 4.72 (m, 1H, основной ротамер), 4.65-4.45 (m, 2H), 4.30 (m, 1H, основной ротамер), 4.16 (m, 1H, основной ротамер), 4.03 (m, 1H, минорный ротамер), 3.95 (m, 1H, минорный ротамер), 2.63 (m, 1H, минорный ротамер), 2.48 (m, 1H, основной ротамер), 2.21 (m, 1H, основной ротамер), 2.07 (m, 1H, минорный ротамер), 1.89 (s, 3Н).
13С-ЯМР (75 МГц, CD3OD) (карбонильные и/или амидиновые атомы углерода, смесь ротамеров) δ 171.9, 171.2, 165.0, 162.8, 160.4.
ХИАД-МС:(М+1)=503/505 m/z.
Пример 2
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-Pab(2,6-diF)(OMe)
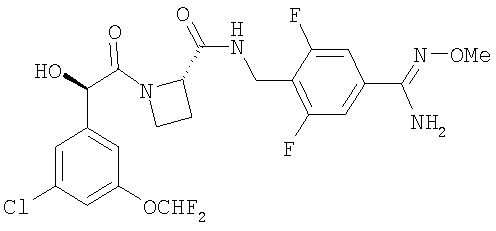
(1) Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-Pab(2,6-diF)(OMe,Teoc)
Смесь Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-Pab(2,6-diF)(Teoc) (64 мг; 0,099 ммоль; см. пример 1 (20) выше) и гидрохлорида O-метилгидроксиламина (50 мг; 0,60 ммоль) в 4 мл ацетонитрила нагревали при 70°С в течение 3 часов. Растворитель выпаривали и остаток распределяли между водой и EtOAc. Водный слой дважды экстрагировали EtOAc и объединенную органическую фазу промывали водой, сушили (Na2SO4) и упаривали. Продукт можно было использовать без дальнейшей очистки. Выход 58 мг (87%).
1H ЯМР (400 МГц, CDCl3) δ 7.90 (bt, 1H), 7.46 (m, 1H), 7.25-6.95 (m, 5H), 6.51, t, 1H), 4.88 (s, 1H), 4.83 (m, 1H), 4.6-4.5 (m, 2H), 4.4-3.9 (m, 4H), 3.95 (s, 3Н), 3.63 (m, 1H), 2.67 (m, 1Н), 2.38 (m, 1H), 1.87 (широкий, 1Н), 0.98 (m, 2H), 0.01, s, 9Н).
(2) Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-Pab(2,6-diF)(OMe)
Растворяли Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-Pab(2,6-diF)(OMe, Теос) (58 мг; 0,086 ммоль; см. стадию (1) выше) в 3 мл TFA, охлаждали на ледяной бане и оставляли взаимодействовать в течение 2 часов. Выпаривали TFA и растворяли остаток в EtOAc. Органический слой дважды промывали водным карбонатом натрия и водой, сушили (Na2SO4) и упаривали. Остаток лиофилизировали из воды и ацетонитрила с получением 42 мг (92%) соединения, указанного в заголовке. Чистота 94%.
1H ЯМР (300 мГц, CDCl3) δ 7.95 (bt, 1H), 7.2-71 (m, 4H), 6.99 (m, 1H), 6.52 (t, 1H), 4.88 (s, 1H), 4.85-4.75 (m, 3Н), 4.6-4.45 (m, 2H), 4.29 (широкий, 1H), 4.09 (m, 1H), 3.89 (s, 3Н), 3.69 (m, 1H), 2.64 (m, 1H), 2.38 (m, 1H), 1.85 (широкий, 1H).
13С ЯМР (100 мГц, CDCl3) (карбонильные и/или амидиновые атомы углерода) δ 172.1, 169.8, 151.9.
ХИАД-МС: (М+1)=533/535 m/z.
Пример 3
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-Pab(2,6-diF)(OH)
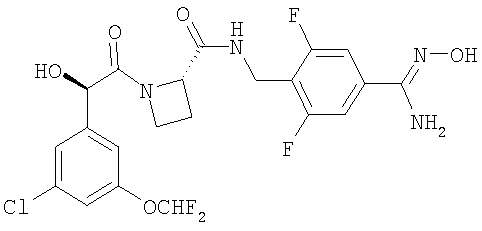
(1) Boc-(S)Aze-NHCH2-Ph(2,6-diF,4-CN)
Boc-(S)Aze-OH (1,14 г; 5,6 ммоль) растворяли в 45 мл DMF. Добавляли 4-аминометил-2,6-дифторбензонитрил (1,00 г; 5,95 моль, см. пример 1 (14) выше), РуВОР (3,10 г; 5,95 ммоль) и DIPEA (3,95 мл; 22,7 ммоль) и перемешивали этот раствор при комнатной температуре в течение 2 часов. Растворитель выпаривали и остаток распределяли между Н2O и EtOAc (по 75 мл каждого). Водную фазу экстрагировали EtOAc (2×50 мл) и объединенную органическую фазу промывали рассолом и сушили над Na2SO4. Флэш-хроматография (SiO2, EtOAc/гептан (3/1)) давала указанное в подзаголовке соединение (1,52 г; 77%) в виде масла, которое кристаллизовалось в холодильнике.
1H-ЯМР (400 мГц, CD3OD): δ 7.19 (m, 2H), 4.65-4.5 (m, 3Н), 3.86 (m, 1H), 3.73 (m, 1Н), 2.45-2.3 (m, 2H), 1.39 (s, 9H).
(2) H-(S)Aze-NHCH2-Ph(2,6-diF,4-CN)×HCl
Boc-(S)Aze-NHCH2-Ph(2,6-diF,4-CN) (0,707 г; 2,01 ммоль, см. стадию (1) выше) растворяли в 60 мл EtOAc, насыщенного газообразным HCl. После перемешивания при комнатной температуре в течение 15 минут выпаривали растворитель. Остаток растворяли в смеси СН3СМ/Н2О (1/1) и лиофилизировали, что давало указанное в подзаголовке соединение (0,567 г; 98%) в виде беловатого аморфного порошка.
1H ЯМР (400 мГц, CD3OD): δ 7.49 (m, 2H). 4.99 (m, 1H), 4.58 (m, 2H), 4.12 (m, 1H), 3.94 (m, 1H), 2.80 (m, 1H), 2.47 (m, 1H).
MC (m/z) 252,0 (M+1)+.
(3) Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-NHCH2-Ph(2,6-diF,4-CN)
Растворяли Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)OH (0,40 г; 1,42 ммоль, см. пример 1 (8) выше) в 10 мл DMF и добавляли H-(S)Aze-NHCH2-Ph(2,6-diF,4-CN)×HCl (0,43 г; 1,50 ммоль, см. стадию (2) выше) и РуВОР (0,779 г; 1,50 ммоль) и затем DIPEA (1,0 мл; 5,7 ммоль). После перемешивания при комнатной температуре в течение 2 часов растворитель выпаривали. Остаток распределяли между Н2O (200 мл) и EtOAc (75 мл). Водную фазу экстрагировали EtOAc (2×75 мл) и объединенную органическую фазу промывали рассолом и сушили над Na2SO4. Флэш-хроматография (SiO2, EtOAc/гептан (4/1)) давала указанное в подзаголовке соединение (0,56 г; 81%) в виде масла.
1H ЯМР (400 мГц, CD3OD) ротамеры: δ 7.43 (m, 2H), 7.31 (m, 1H, основной ротамер), 7.26 (m, 1H, минорный ротамер), 7.2-7 A (m, 2H), 6.90 (t, 1H, основной ротамер), 6.86 (t, 1H, минорный ротамер), 5.14 (s, 1H, основной ротамер), 5.11 (m, 1H, минорный ротамер), 5.04 (s, 1H, минорный ротамер), 4.71 (m, 1H, основной ротамер), 4.6-4.45 (m, 2H), 4.30 (m, 1H, основной ротамер), 4.2-3.9 (m, 1H; и 1H, минорный ротамер), 2.62 (m, 1H, минорный ротамер), 2.48 (m, 1H, основной ротамер), 2.21 (m, 1H, основной ротамер), 2.09 (m, 1H, минорный ротамер).
13С ЯМР (100 мГц, CD3OD) (карбонильные атомы углерода) δ 171.9, 171.8.
МС (m/z) 484.0, 485.9 (М-1)-, 486.0, 487.9 (M+1)+.
(4) Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-Pab(2,6-diF)(OH)
Ph(3-Cl)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-NHCH2-Ph(2,6-diF,4-CN) (0,555 г; 1,14 ммоль, со стадии (3), указанной выше) растворяли в 10 мл EtOH (95%). К этому раствору добавляли гидрохлорид гидроксиламина (0,238 г; 3,42 ммоль) и Et3N (0,48 мл; 3,44 ммоль). После перемешивания при комнатной температуре в течение 14 часов растворитель удаляли и остаток растворяли в EtOAc. Органическую фазу промывали рассолом и H2O и сушили над Na2SO4. Неочищенный продукт очищали препаративной ЖХОФ со смесью СН3СМ:0,1 М NH4OAc в качестве элюента с получением соединения, указанного в заголовке, в виде аморфного порошка (0,429 г; 72%) после лиофилизации.
1H ЯМР (400 мГц, CD3OD) ротамеры: δ 7.35-7.1 (m, 5Н), 6.90 (t, 1H, основной ротамер), 6.85 (t, 1H, минорный ротамер), 5.15 (s, 1H, основной ротамер), 5.12 (m, 1H, минорный ротамер), 5.08 (s, 1H, минорный ротамер), 4.72 (m, 1H, основной ротамер), 4.6-4.4 (m, 2H), 4.30 (m, 1H, основной ротамер), 4.12 (m, 1H, основной ротамер), 4.04 (m, 1H, минорный ротамер), 3.94 (m, 1H, минорный ротамер), 2.62 (m, 1H, минорный ротамер), 2.48 (m, 1H, основной ротамер), 2.22 (m, 1H, основной ротамер), 2.10 (m, 1H, минорный ротамер)
13С ЯМР (100 мГц, CD3OD) (карбонильные и амидиновые атомы углерода, ротамеры) δ 172.4, 171.9, 171.0, 152.3, 151.5
МС (m/z) 517.1, 519.0 (M-1)-, 519.1, 521.0 (M+1)+.
Пример 4
Соединение заголовка примера 1 тестировали в вышеуказанном Тесте А и обнаружили, что оно имеет величину IC50TB, составляющую менее 0,02 мкМ.
Пример 5
Соединение заголовка примера 1 тестировали в вышеуказанном Тесте Г и обнаружили, что оно имеет величину IC50АПТВ, составляющую менее 1 мкМ.
Пример 6
Соединение заголовка примера 2 тестировали в вышеуказанном Тесте Д и обнаружили, что оно имеет пероральную и/или парентеральную биодоступность у крысы такую же, как у соответствующего активного ингибитора (свободный амидин).
Пример 7
Соединение, указанное в заголовке примера 2, тестировали в вышеуказанном Тесте Ж и обнаружили, что оно превращается в соответствующий активный ингибитор (свободный амидин) в микросомах печени людей и крыс. Такое же превращение в свободный амидин примера 1 имеет место в Тесте Ж для соединения, указанного в заголовке примера 3.
Сокращения
Префиксы н, втор, изо и трет имеют их обычные значения: нормальный, вторичный, изо и третичный. Префикс ц означает цикло.
Изобретение относится к новому соединению Ph(3-CI)(5-OCHF2)-(R)CH(OH)C(O)-(S)Aze-Pab(2,6-diF)(OH) формулы
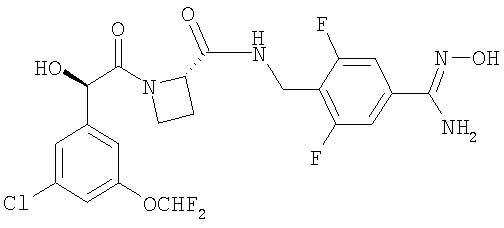
или его фармацевтически приемлемой соли, которое может быть использовано в качестве активного ингредиента для изготовления лекарства для лечения состояния, при котором требуется ингибирование тромбина. 5 н. и 7 з.п.ф-лы.
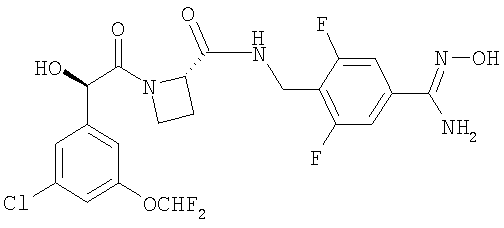
или его фармацевтически приемлемая соль.
| WO 00/42059 A1, 20.07.2000 | |||
| Бесколесный шариковый ход для железнодорожных вагонов | 1917 |
|
SU97A1 |
| RU 92004425 A, 19.06.1995. | |||

