ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Изобретение касается феноксифенил- и феноксибензил- оксазолидин-2,4-дионов и тиазолидин-2,4-дионов, включая их фармацевтически приемлемые соли и пролекарства, которые пригодны в качестве терапевтических средств, в особенности при лечении сахарного диабета Типа 2 и состояний, которые часто сопутствуют заболеваниям, включая ожирение и нарушение метаболизма липидов.
УРОВЕНЬ ТЕХНИКИ ИЗОБРЕТЕНИЯ
Диабет является заболеванием, происходящим из-за многочисленных причинных факторов и характеризующимся повышенными уровнями глюкозы в плазме крови (гипергликемия) при состоянии голодания или при приеме внутрь глюкозы в процессе пероральной пробы на толерантность глюкозы. Обычно выделяют две формы диабета. При диабете Типа 1 или инсулинзависимом диабете (IDDM) в организме пациента вырабатывается мало инсулина или вообще не вырабатывается инсулин-гормон, который регулирует утилизацию глюкозы. При диабете Типа 2 или инсулиннезависимом диабете (NIDDM), инсулин все же продуцируется в организме. Пациенты с диабетом Типа 2 обычно имеют гиперинсулинемию (повышенные уровни инсулина в плазме крови); однако эти пациенты имеют инсулиновую резистентность, что означает, что они имеют резистентность к воздействию инсулина на усиление утилизации глюкозы и на липидный метаболизм в основных инсулин-чувствительных тканях, к которым относятся мышечная ткань, ткань печени и жировая ткань. Инсулиновая резистентность первоначально не вызывается сокращением количества инсулиновых рецепторов, но, до некоторой степени, нарушением связывания пост-инсулинового рецептора, что еще полностью не выяснено. Этот недостаток восприимчивости к инсулину приводит к недостаточной активации инсулин-опосредованного поглощения, к окислению и накоплению глюкозы в мышцах, и неадекватному инсулин-опосредованному подавлению липолиза в жировой ткани и к выработке и секретированию глюкозы в печени. Те пациенты, которые резистентны к инсулину, но не имеют диабета, компенсируют резистентность к инсулину повышенной секрецией инсулина, поэтому уровни глюкозы в плазме крови могут быть повышенными, но повышенными не настолько, чтобы достигнуть критерия диабета Типа 2, который выводится на основании содержания глюкозы в плазме крови в состоянии голодания.
Постоянная или неконтролируемая гипергликемия, которая встречается при диабете, связана с повышением уровня преждевременной заболеваемости и смертности. Часто нарушенный гомеостаз глюкозы прямо или косвенно связан с ожирением, гипертензией, изменениями в липидном, липопротеиновом и аполипопротеиновом метаболизме не только с метаболическим, но и с гемодинамическим заболеванием. Пациенты с диабетом Типа 2 имеют значительно повышенный риск макрососудистых и микрососудистых осложнений, включая атеросклероз, коронарную болезнь сердца, инсульт, болезнь периферических сосудов, гипертензию, нефропатию, нейропатию и ретинопатию. Таким образом, терапевтический контроль гомеостаза глюкозы, липидного метаболизма, ожирения и гипертензии критически важен в клиническом менеджменте и лечении диабетов.
Пациенты, имеющие инсулиновую резистентность, часто имеют несколько симптомов, которые вместе обозначают как синдром Х или метаболический синдром. Согласно одному из широко применяемых определений пациент, имеющий метаболический синдром, характеризуется как имеющий три или более симптомов, выбираемых из нижеследующей группы из пяти симптомов: (1) абдоминальное ожирение; (2) гипертриглицеридемия; (3) низкий уровень холестерина липопротеинов высокой плотности (HDL); (4) высокое кровяное давление и (5) повышенный уровень глюкозы в состоянии голодания, который может быть в интервале, характерном для диабета Типа 2, если у пациента также диабет. Каждый из этих симптомов клинически определен в недавно выпущенном издании Third Report of the National Cholesterol Education Program Expert Panel on Detection, Evaluation and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III, or ATP III), National Institutes of Health, 2001, NIH Publication No. 01-3670. Пациенты с метаболическим синдромом вне зависимости от того, есть ли у них уже клинический сахарный диабет или он только развивается, имеют повышенный риск развития макрососудистых и микрососудистых осложнений, которые выше перечислены и встречаются при диабете Типа 2, таких как атеросклероз и коронарная болезнь сердца.
Существует несколько доступных способов лечения для диабета Типа 2, каждый из которых имеет свои собственные ограничения и потенциальный риск. Физические упражнения и диетическое понижение в поступлении калорий часто наглядно улучшают патологическое состояние диабета и являются наилучшим первым направлением при лечении диабета Типа 2. Соблюдение требований такого лечения недостаточное, потому что существуют крепко укоренившийся сидячий образ жизни и избыточное потребление пищи, особенно пищевых продуктов с повышенным содержанием жира. Широко применяемое медикаментозное лечение включает в себя назначение приема меглитинида или сульфонилмочевины (например, толбутамида или глипизида), которые являются стимуляторами секреции инсулина. Эти лекарства повышают уровень инсулина в плазме крови посредством стимуляции панкреатических β-клеток к секреции большего количества инсулина. Они часто применяются индивидуально или как первое направление медикаментозного лечения диабета Типа 2, но они могут также применяться и в комбинации с другими лекарствами, которые назначаются при диабете Типа 2. Когда прием сульфонилмочевины или меглитинида становится неэффективным, количество инсулина в организме может быть дополнено путем инъекции инсулина, таким образом концентрации инсулина достаточно высоки даже для стимуляции очень инсулин-резистентных тканей. Однако опасно низкие уровни глюкозы в плазме крови могут быть результатом применения инсулина и/или стимуляторов секреции инсулина, и в конце концов возникает повышенный уровень инсулиновой резистентности даже при высоких уровнях содержания инсулина в плазме крови.
Бигуаниды являются другим классом лекарств, широко применяемых при лечении диабетов Типа 2. Два хорошо известных бигуанида фенформин и метформин вызывают некоторую коррекцию гипергликемии. Бигуаниды могут применяться как монотерапевтически, так и в комбинации с другими антидиабетическими лекарствами, такими как инсулин или стимуляторы секреции инсулина, без повышения вероятности гипогликемии. Однако фенформин и метформин могут вызывать молочный ацидоз и тошноту/диарею. Метформин имеет более низкую вероятность побочных эффектов, чем фенформин, и он широко назначается для лечения диабета Типа 2.
Глитазоны (т.е. 5-бензилтиазолидин-2,4-дионы) являются новейшим классом соединений, которые могут улучшить состояние гипергликемии и другие симптомы диабета Типа 2. Эти средства существенно повышают чувствительность к инсулину в мышечной ткани, в ткани печени и в жировой ткани в случае нескольких моделей диабета Типа 2 на животных, приводя в результате к частичной или полной коррекции повышенных уровней глюкозы в плазме крови без появления гипогликемии. Глитазоны, которые продаются в настоящее время (розиглитазон и пиоглитазон), являются агонистами рецепторов, активирующих пероксисомальную пролиферацию (PPAR) гамма-субтипа. Обычно считается, что PPAR-гамма агонисты ответственны за улучшение чувствительности к инсулину, которая наблюдается для глитазонов. Разрабатываются новые PPAR агонисты для лечения диабета Типа 2 и/или дислипидемии. Множество новейших PPAR соединений являются агонистами одного или нескольких PPAR альфа-, гамма- и -дельта субтипов. Весьма перспективны соединения, являющиеся агонистами как PPAR альфа, так и PPAR гамма субтипов (PPAR альфа/PPAR гамма двойные агонисты), потому что они снижают гипергликемию и также улучшают липидный метаболизм.
В настоящее время имеющиеся в продаже PPAR агонисты, которые относятся к глитазонам, проявили недостатки. Троглитазон был первым поступившим в продажу глитазоном, но в конце концов он был изъят из торгового оборота из-за его гепатотоксичности. Другой недостаток имеющихся в продаже в настоящее время PPAR
агонистов заключается в возможности монотерапии диабета Типа 2, обеспечивающей ограниченную эффективность - понижение уровня глюкозы в плазме крови, в среднем, на ≈20% и снижение Гемоглобина А1С от ≈9,0% до ≈8,0%. Находящиеся в обращении соединения, кроме того, незначительно улучшают липидный метаболизм и, вообще говоря, могут негативно влиять на липидные параметры. Эти недостатки обуславливают стимулирование разработки лучших инсулиновых сенсибилизаторов для диабета Типа 2, которые действуют по аналогичному механизму (механизмам).
Недавно было сообщено о соединениях, которые являются PPAR гамма-антагонистами или частичными PPAR агонистами. В патенте WO 01/30343 описано специфическое соединение, которое частично является PPAR агонистом/антагонистом и которое можно применять для лечения ожирения и диабета Типа 2. Патент WO 02/08188 раскрывает класс PPAR агонистов и частичных агонистов, являющихся производными индола и применимых в лечении диабета Типа 2 с пониженными побочными эффектами прибавки в весе для организма и для сердца. На PPAR гамма частичные агонисты часто ссылаются как на селективные PPAR модуляторы (SPPRM).
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Класс описанных здесь соединений является новым классом сильных PPAR лигандов, которые in vitro обычно являются PPARγ агонистами или частичными агонистами. Соединения могут также быть PPARγ антагонистами. В дополнение к вышесказанному, некоторые соединения могут также обладать PPARα активностью в дополнение к PPARγ активности. Соединения можно применять в лечении PPAR модулируемых заболеваний, включая диабет Типа 2, гипергликемию и резистентность к инсулину.
Соединения также могут применяться в лечении одного вида или нескольких видов нарушения липидного метаболизма, включая смешанную или диабетическую дислипидемию, локализованную гиперхолестеринемию, которые могут быть обнаружены по повышению уровня LDL-C и/или не-HDL-C, гипераповлипротеинемию, гипертриглицеридемию, повышение в содержании обогащенных триглицеридами липопротеинов и пониженные концентрации HDL холестерина. Они также могут применяться для лечения или улучшения состояния в случае ожирения. Они также могут применяться для лечения и улучшения состояния при атеросклерозе, при рестенозе сосудов, при состояниях воспаления, при псориазе и при синдроме поликистоза яичников. Они также могут быть полезны в лечении других PPAR опосредованных заболеваний, нарушениях метаболизма и состояниях.
Настоящее изобретение направлено на соединения формулы I:
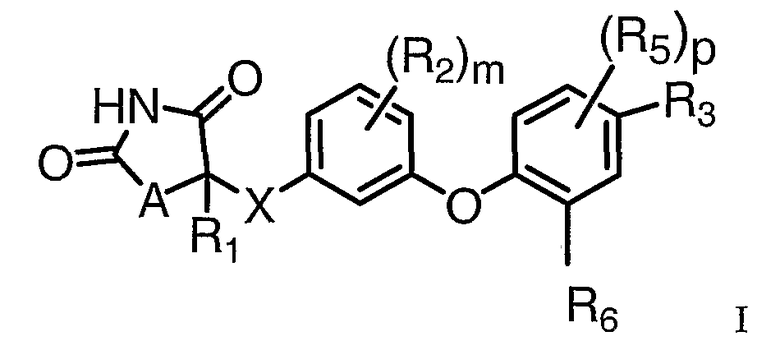
и их фармацевтически приемлемые соли и лекарственные средства.
В соединениях формулы I:
А представляет O или S;
Х представляет связь или СН2;
R1 выбирается из группы, состоящей из Н и С1-С3алкила, где С1-С3алкил необязательно замещен 1-3 F;
Каждый R2 независимо выбирается из группы, состоящей из F, Cl, CH3, CF3, -OCH3 и -OCF3;
R3 представляет
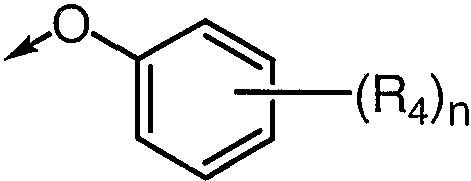
Каждый R4 независимо выбирается из группы, состоящей из галогена, С1-С3алкила, -ОС1-С3алкила, -ОС(=О)С1-С3алкила и -S(O)qС1-С3алкила, где С1-С3алкил, -ОС1-С3алкил, -ОС(=О)С1-С3алкил и -S(O)qС1-С3алкил могут быть замещены 1-3 F;
Каждый R5 независимо выбирается из группы, состоящей из F, Cl, CH3, -OCH3, CF3 и -OCF3;
R6 независимо выбирается из группы, состоящей из С2-С5алкила, -CH2циклопропила, -С(=О)С1-С3алкила, где R6 необязательно замещен 1-3 F;
m равно 0 или 1;
n равно целому числу 1-3;
p равно целому числу 0-2 и
q равно целому числу 0-2.
В вышеприведенных определениях и последующих определениях алкильные группы могут быть как линейными, так и разветвленными, если иное не предусмотрено.
Ожидается, что эти соединения будут эффективно воздействовать на понижение уровня глюкозы, на липиды и на инсулин в случае диабетических пациентов и в случае недиабетических пациентов, у которых нарушена толерантность к глюкозе и/или которые находятся в преддиабетическом состоянии. Ожидается, что соединения будут эффективны в лечении неинсулинзависимого диабета (NIDDM) у людей и других пациентов из числа млекопитающих животных, и в особенности, при лечении гипергликемии и в лечении патологических состояний, ассоциированных с NIDDM, включая гиперлипидемию, дислипидемию, ожирение, гиперхолестеринемию, гипертриглицеридемию, атеросклероз, рестеноз сосудов, состояний воспаления и других PPAR опосредованных заболеваний, нарушений метаболизма и патологических состояний.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Изобретение имеет многочисленные варианты осуществления, суммированные ниже. Эти варианты осуществления изобретения включают соединения, фармацевтически приемлемые соли этих соединений и фармацевтические композиции, содержащие эти соединения и фармацевтически приемлемые носители. Эти варианты осуществления изобретения имеют специальные полезные свойства в лечении инсулиновой резистентности, диабета Типа 2 и дислипидемии, которая ассоциирована с диабетом Типа 2 и с резистентностью к инсулину.
Один из вариантов осуществления изобретения включает соединения формулы I, в которой:
R1 представляет Н или СН3;
Каждая R4 группа независимо выбирается из группы, состоящей из F, Cl, CH3, CF3, -OCH3, -OCF3, -OCH2CH3, -ОС(=О)CH3, -ОС(=О)CF3 и -S(O)qCH3;
R5 представляет Cl или F;
R6 выбирается из н-С3Н7, -CH2циклопропила и С(=О)С2Н5;
m равно 0;
n равно 1 или 2;
p равно 0 или 1 и
q равно целому числу 0-2.
В другом варианте осуществления изобретения, соединения формулы I имеют следующие группы, в то время как другие группы определены ранее:
А представляет О;
R1 представляет СН3;
R3 определен ранее;
Каждый R4 независимо выбирается из группы, состоящей из
Cl, -OCH3, -OCF3 и -S(O)qCH3;
R5 представляет F;
R6 представляет н-С3Н7;
m равно 0;
n равно 1 или 2 и
p равно 0 или 1.
В других вариантах осуществления изобретения в соединениях формулы I R1 представляет Н или СН3 и другие группы такие же, как определены выше. В предпочтительных вариантах осуществления изобретения R1 представляет СН3.
Во многих предпочтительных вариантах А представляет О. Другие группы такие же, как определено выше.
В других предпочтительных вариантах осуществления изобретения А представляет S.
Другой вариант осуществления изобретения включает соединения формулы I, в которых R4 представляет F, Cl, CH3, CF3, -OCH3, -OCF3, -OCHF2, -ОС2H5, -ОС(=О)CH3 или -S(O)qCH3, где q равно 0, 1 или 2 и n равно 1 или 2. Другие группы такие же, как определено выше.
Во многих соединениях изобретения, как определено выше, Х представляет связь.
Во многих соединениях изобретения, как определено выше, Х представляет CH2.
Полезные подгруппы в соединениях, как определено ранее, имеют R2 группы, которые выбираются из F, Cl, CH3, CF3, -OCH3, -OCF3; где m равно 0 или 1.
В предпочтительных вариантах осуществления изобретения в соединениях, как определено ранее, R6 выбирается из н-С3Н7, -CH2Циклопропила и С(=О)С2Н5. Во многих предпочтительных соединениях и группах соединений R6 является н-С3Н7.
Предпочтительные R5 заместители выбираются из F, Cl, CH3, -OCH3, CF3 и -OCF3; где p равно 0 или 1.
Оба энантиомера (т.е. R и S) по 5 положению оксазолидиндионового и тиазолидиндионового кольца являются активными PPAR гамма-агонистами и частичными агонистами и относятся к соединениям изобретения. R энантиомеры обычно более активны и они являются предпочтительными.
Структуры специфических соединений и синтетические способы получения соединений раскрыты в примерах. Структуры специфических образцов изобретения раскрыты в нижеприведенной таблице 1, включая фармацевтически приемлемые соли соединений.
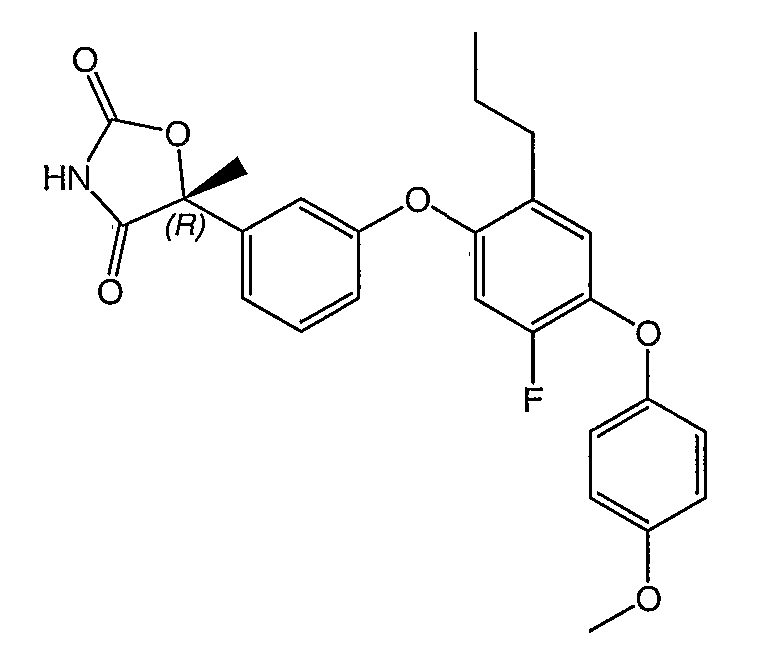
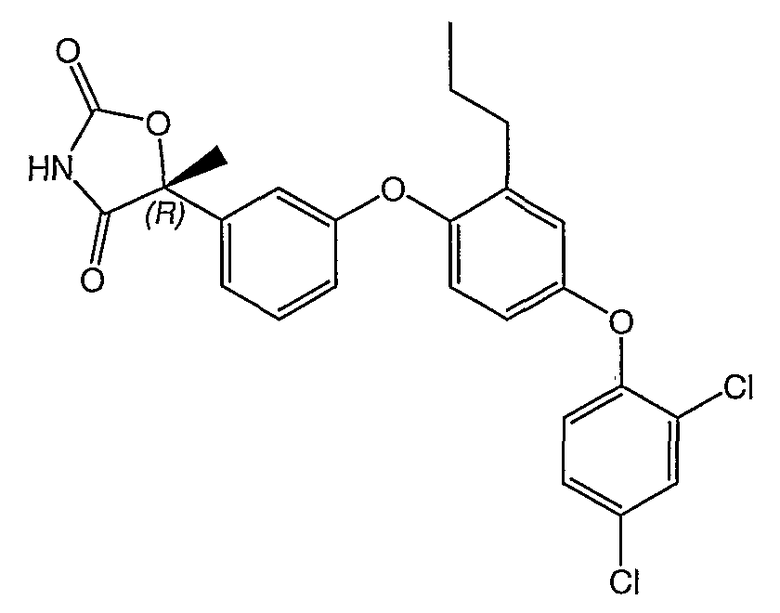
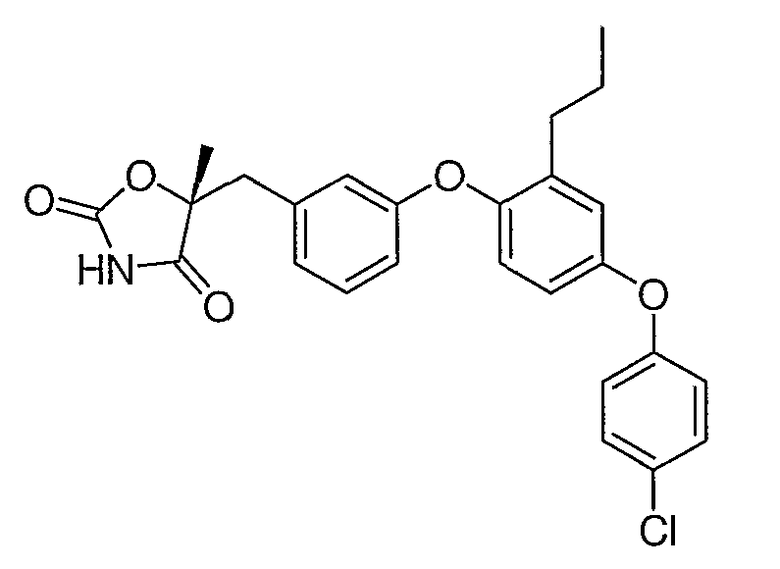
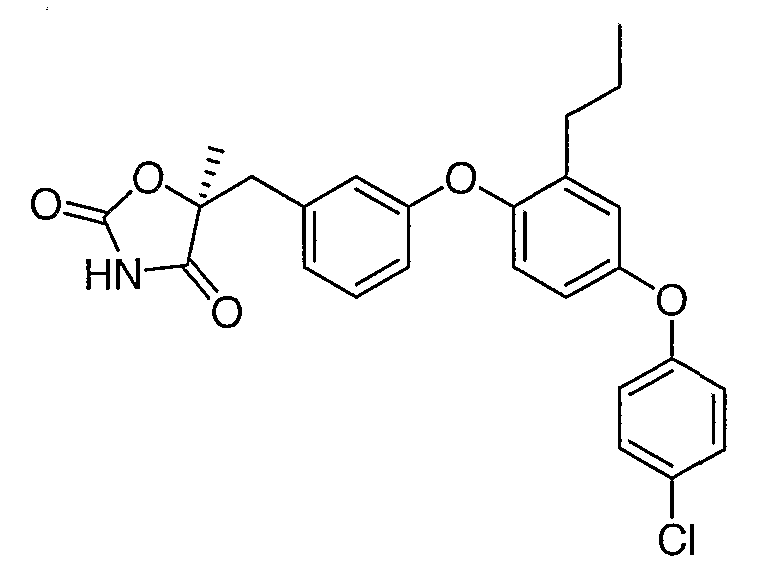
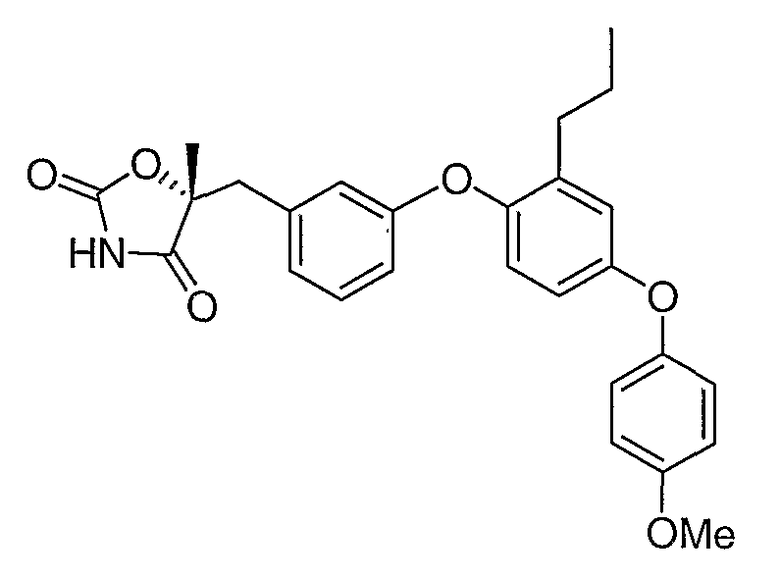
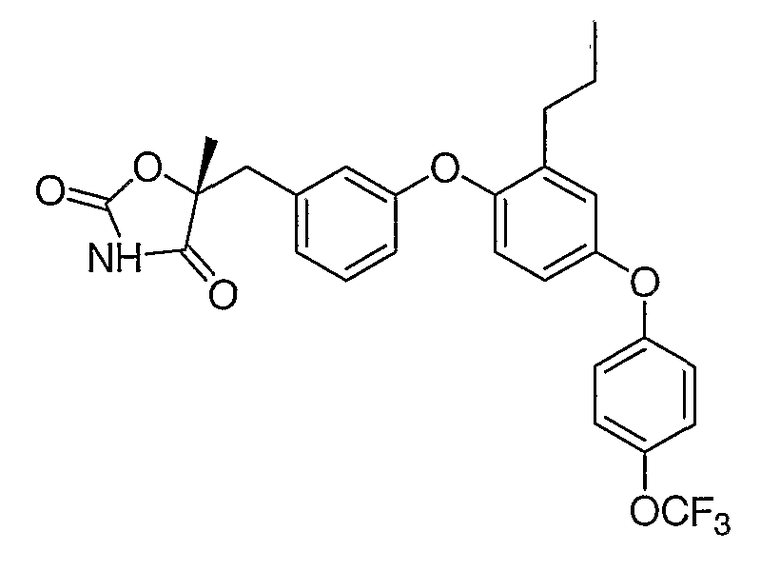
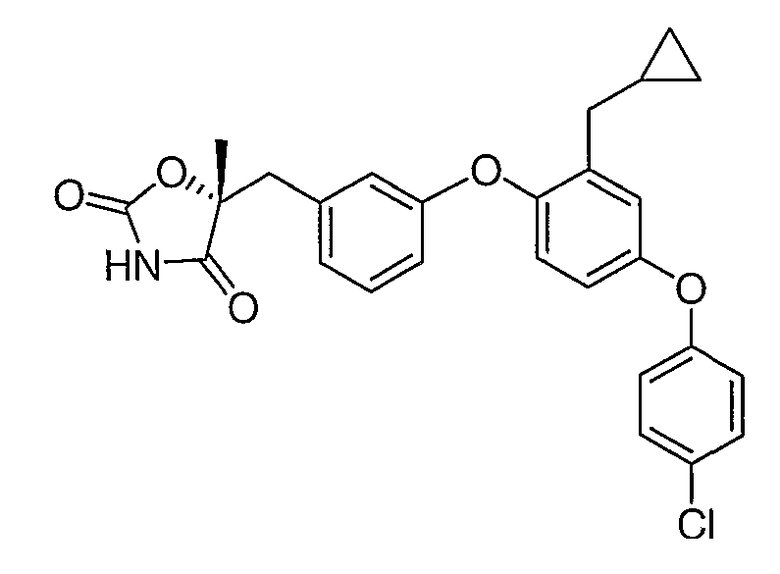
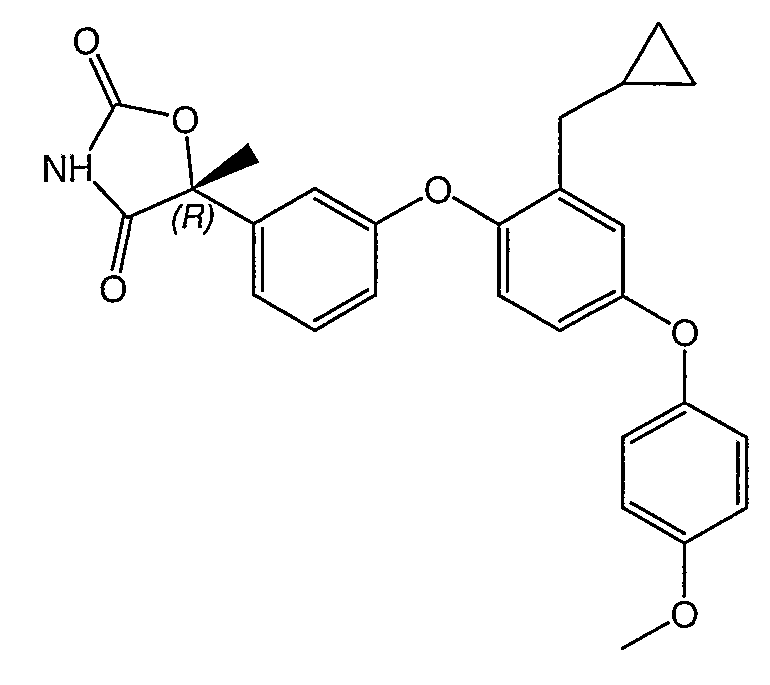
Соединения этого изобретения могут применяться в фармацевтических композициях, включающих соединение или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель. Соединения настоящего изобретения могут использоваться в фармацевтических композициях, которые включают один или несколько других активных фармацевтических ингредиентов. Соединение этого изобретения может также использоваться в фармацевтических композициях, в которых соединение формулы I или его фармацевтически приемлемая соль является единственным активным ингредиентом.
Соединения изобретения и их фармацевтически приемлемые соли могут применяться в производстве лекарственных препаратов для лечения диабета Типа 2 у человека или других пациентов из числа млекопитающих.
Соединения, как они определены здесь, могут применяться для лечения заболеваний согласно нижеследующим способам, а также и других заболеваний, ниже не перечисленных:
(1) Способ лечения инсулиннезависимого диабета (диабет Типа 2) у человека или других млекопитающих пациентов, нуждающихся в таком лечении, который включает назначение пациенту терапевтически эффективного количества соединения формулы I.
(2) Способ лечения или уменьшения гипергликемии у человека или других млекопитающих пациентов, нуждающихся в таком лечении, который включает назначение пациенту терапевтически эффективного количества соединения формулы I.
(3) Способ лечения или уменьшения метаболического синдрома у человека или других млекопитающих пациентов, нуждающихся в таком лечении, который включает назначение пациенту терапевтически эффективного количества соединения формулы I.
(4) Способ лечения или уменьшения ожирения у человека или других млекопитающих пациентов, нуждающихся в таком лечении, который включает назначение пациенту терапевтически эффективного количества соединения формулы I.
(5) Способ лечения или уменьшения гиперхолестеринемии у человека или других млекопитающих пациентов, нуждающихся в таком лечении, который включает назначение пациенту терапевтически эффективного количества соединения формулы I.
(6) Способ лечения или уменьшения гипертриглицеридемии у человека или других млекопитающих пациентов, нуждающихся в таком лечении, который включает назначение пациенту терапевтически эффективного количества соединения формулы I.
(7) Способ лечения или уменьшения одного или большего количества нарушений липидного метаболизма, включая смешанную или диабетическую дислипидемию, низкий уровень HDL холестерина, высокий уровень LDL холестерина, гиперлипидемию, гиперхолестеринемию и гипертриглицеридемию у человека или других млекопитающих пациентов, нуждающихся в таком лечении, который включает назначение пациенту терапевтически эффективного количества соединения формулы I.
(8) Способ снижения вероятности возникновения неблагоприятных осложнений, ассоциированных с метаболическим синдромом, у человека или других млекопитающих пациентов, нуждающихся в таком лечении, который включает назначение пациенту терапевтически эффективного количества соединения формулы I.
(9) Способ лечения атеросклероза для снижения вероятности развития атеросклероза, для задержки начала развития склероза и/или снижения вероятности возникновения осложнений атеросклероза у человека или других млекопитающих пациентов, нуждающихся в таком лечении или находящихся в состоянии риска развития атеросклероза или осложнений атеросклероза, который включает назначение пациенту терапевтически эффективного количества соединения формулы I. Осложнения атеросклероза включают, например, ангину, перемежающуюся хромоту, инфаркт миокарда, инсульт и др.
Соединения особенно полезны в лечении нижеследующих заболеваний путем назначения терапевтически эффективных количеств пациенту, нуждающемуся в лечении:
(1) диабета Типа 2 и, в частности, гипергликемии;
(2) метаболического синдрома;
(3) ожирения и
(4) гиперхолестеринемии.
Определения
“Ac” означает ацетил, который является СН3С(=О)-.
“Алкил” означает насыщенную углеродную цепь, которая может быть линейной или разветвленной, или являться их комбинацией, если только углеродная цепь не определена иным образом. Другие группы, имеющие в названии приставку “Алк”, такие как алкокси, алканоил, также могут быть линейными или разветвленными или являться их комбинацией, если только углеродная цепь не определена иным образом. Примеры алкильных групп включают метил, этил, пропил, изопропил, бутил, втор- и трет-бутил, пентил, гексил, гептил, октил, нонил и им подобные.
“Алкенил” означает углеродные цепи, которые содержат, как минимум, одну углерод-углеродную двойную связь и которые могут быть линейными или разветвленными, или являться их комбинацией. Примеры алкенилов включают винил, аллил, изопропенил, пентенил, гексенил, гептенил, 1-пропенил, 2-бутенил, 2-метил-2-бутенил и им подобные.
“Алкинил” означает углеродные цепи, которые содержат, как минимум, одну углерод-углеродную тройную связь и которые могут быть линейными или разветвленными, или являться их комбинацией. Примеры алкинилов включают этинил, пропаргил, 3-метил-1-пентинил, 2-гептинил и им подобные.
“Циклоалкил” обозначает моно- или бициклические насыщенные карбоциклические кольца, которые содержат от 3 до 10 атомов углерода, если только не установлено иным образом. Термин также включает моноциклические кольца, объединенные с арильной группой. Примеры циклоалкилов включают циклопропил, циклопентил, циклогексил, циклогептил и им подобные.
“Арил” (и “арилен”), когда применяется для описания заместителя или группы в структуре, означает моноциклическое, бициклическое или трициклическое соединение, в котором все кольца ароматические и которое содержит только углеродные атомы. Термин “арил” также относится к арильной группе, которая объединена с циклоалкилом или с гетероциклом. “Гетероциклил”, “гетероцикл” и “гетероциклическая” означает полностью или частично насыщенную моноциклическую, бициклическую или трициклическую кольцевую систему, содержащую, как минимум, один гетероатом, выбираемый из N, S и O, каждое из упомянутых колец имеет от 3 до 10 атомов. Примеры арильных заместителей включают фенил и нафтил. Арильные кольца, объединенные с циклоалкилами, встречаются в инданиле, индениле и тетрагидронафтиле. Примеры арила, объединенного с гетероциклической группой встречаются в 2,3-дигидробензофураниле, бензопираниле, 1,4-бензодиоксаниле и им подобных. Примеры гетероциклов включают тетрагидрофуран, пиперазин, пиперидин и морфолин. Предпочтительными арильными группами являются фенил или нафтил. Обычно фенил является наиболее предпочтительной арильной группой.
“Гетероарил” (и гетероарилен) означает моно-, би- или трициклическое ароматическое кольцо, содержащее, как минимум, один гетероатом в цикле, выбираемый из N, S и O (включая SO и SO2), вместе с которым кольцо содержит от 5 до 6 атомов. Примеры гетероарила включают пирролил, изоксазолил, изотиазолил, пиразолил, пиридил, оксазолил, оксадиазолил, тиадиазолил, тиазолил, имидазолил, триазолил, тетразолил, фуранил, триазинил, тиенил, пиримидил, пиридазинил, пиразинил, бензизоксазолил, бензоксазолил, бензотиазолил, бензимидазолил, бензофуранил, бензотиофенил (включая S-оксид и диоксид), фуро(2,3-b)пиридил, хинолил, индолил, изохинолил, хиназолинил, дибензофуранил и им подобные.
“Галоген” включает фтор, хлор, бром и иод.
“Ме” представляет собой метил.
Термин “композиция”, как фармацевтическая композиция, предназначается для определения продукта, включающего активный ингредиент(ы), инертный ингредиент(ы), который осуществляет перенос, а также какой-либо продукт, действия которого в результате осуществляются направленно или ненаправленно, посредством соединения, комплексообразования или агрегации каких-либо двух или более ингредиентов, или из-за диссоциации одного или нескольких ингредиентов, или из-за других типов реакций или взаимодействий одного или нескольких ингредиентов. В соответствии с этим фармацевтическая композиция настоящего изобретения охватывает любую композицию, полученную добавлением в смесь соединения настоящего изобретения и фармацевтически приемлемого носителя.
Заместитель “тетразол” означает 2Н-тетразол-5-ил заместительную группу и ее таутомеры.
Оптические изомеры - Диастериомеры - Геометрические изомеры - Таутомеры
Соединения формулы I могут иметь один или несколько асимметрических центров и могут поэтому существовать в виде рацематов, рацемических смесей, индивидуальных энантиомеров, диастереомерных смесей и индивидуальных диастереомеров. Настоящее изобретение подразумевает охват всех таких изомерных форм соединений формулы I.
Некоторые из описанных здесь соединений могут содержать олефиновые двойные связи, если это не указано специально иным образом, что означает, что они включают как E, так и Z геометрические изомеры.
Некоторые из описанных здесь соединений могут существовать с различными положениями присоединения водорода, так как они относятся к таутомерам. Примером являются кетон и его енольная форма, известные как кето-енольные таутомеры. Индивидуальные таутомеры, так же как и их смеси, также охватываются соединениями формулы I.
Соединения формулы I, имеющие один или несколько асимметрических центров, могут быть разделены на диастереомеры, энантиомеры и им подобные с помощью способов, хорошо известных из уровня техники.
В качестве альтернативы, энантиомеры и другие соединения с хиральными центрами могут быть синтезированы стереоспецифическим синтезом при использовании оптически чистых исходных веществ и/или реагентов с известной конфигурацией.
Соли
Термин “фармацевтически приемлемые соли” относится к солям, полученным из фармацевтически приемлемых нетоксичных оснований или кислот, включая неорганические или органические основания и неорганические или органические кислоты. Соли, полученные из неорганических оснований, включают в свой состав алюминий, аммоний, кальций, медь, трехвалентное железо, двухвалентное железо, литий, магний, соли трехвалентного марганца, двухвалентный марганец, калий, натрий, цинк и им подобные. Особенно предпочтительными являются аммониевые, кальциевые, магниевые, калиевые и натриевые соли. Соли в твердом состоянии могут существовать в более чем одной кристаллической форме и могут также быть в форме гидратов. Соли, полученные из фармацевтически приемлемых органических нетоксичных оснований, включают соли первичных, вторичных и третичных аминов, замещенных аминов, включая встречающиеся в природе замещенные амины, циклических аминов и основных ионообменных смол, таких как аргинин, бетаин, кофеин, холин, N,N'-дибензилэтилендиамин, диэтиламин, 2-диэтиламиноэтанол, 2-диметиламиноэтанол, этаноламин, этилендиамин, N-этилморфолин, N-этилпиперидин, глюкамин, глюкозамин, гистидин, гидрабамин, изопропиламин, лизин, метилглюкамин, морфолин, пиперазин, пиперидин, полиаминные смолы, прокаин, пурины, теобромин, триэтиламин, триметиламин, трипропиламин, трометамин и им подобные.
Если соединения настоящего изобретения являются основными, соли могут быть получены из фармацевтически приемлемых нетоксичных кислот, включая неорганические и органические кислоты. Такие кислоты включают следующие: уксусная, бензолсульфоновая, бензойная, камфорсульфоновая, лимонная, этансульфоновая, фумаровая, глюконовая, глутаминовая, бромистоводородная, хлористоводородная, изэтионовая, молочная, малеиновая, яблочная, миндальная, метансульфоновая, муциновая, азотная, памовая, пантотеновая, фосфорная, янтарная, серная, виноградная, п-толуолсульфоновая и им подобные. Особенно предпочтительными являются лимонная, бромистоводородная, хлористоводородная, малеиновая, фосфорная, серная и виноградная кислоты.
Следует понимать, что, как это используется здесь, ссылки на соединения формулы I подразумевают также включение фармацевтически приемлемых солей.
Метаболиты - Пролекарства
Терапевтически активные метаболиты в тех случаях, когда сами метаболиты попадают в поле зрения объема, охватываемого заявленным изобретением, также являются соединениями настоящего изобретения. Пролекарства, которые являются соединениями, превращающимися в заявленные соединения либо в процессе приема пациентом, или после их приема пациентом, также являются соединениями этого изобретения.
Применимость
Соединения настоящего изобретения являются сильными лигандами, имеющими активность агонистов, частичных агонистов или антагонистов по отношению к одному или к нескольким субтипам рецепторов активации пролиферации пероксисом, в особенности, к PPARγ. Некоторые соединения могут также быть агонистами, частичными агонистами или антагонистами PPARα субтипов так же успешно, как и для PPARγ субтипов, в результате проявляется смешанный PPARα/γ агонизм. Некоторые соединения (обычно менее предпочтительные) могут также быть PPARδ лигандами и иметь PPARδ активность в дополнение к их PPARγ активности. Соединения этого изобретения применимы для лечения или контролирования заболеваний, нарушений метаболизма или состояний, которые опосредованы одним или большим количеством лигандов индивидуальных PPAR субтипов (например, γ) или комбинацией PPAR субтипов (например, α/γ).
Одним из аспектов настоящего изобретения является предоставление способа лечения и контролирования заболеваний, что может быть осуществлено посредством PPAR агониста или частичного агониста так же, как при диабете Типа 2. Одним из аспектов настоящего изобретения является предоставление способа лечения и контролирования таких заболеваний, нарушений метаболизма или состояний у млекопитающих пациентов или у человека, нуждающихся в лечении, который включает назначение такому млекопитающему пациенту терапевтически эффективного количества соединения формулы I. Соединения настоящего изобретения могут применяться в лечении и контролировании многих PPAR опосредованных заболеваний и состояний, включая следующие, но не ограничиваясь ими: (1) диабет Типа 2 (также известный как инсулиннезависимый диабет, или NIDDM), (2) гипергликемия, (3) низкая толерантность к глюкозе, (4) резистентность к инсулину, (5) ожирение, (6) нарушения липидного метаболизма, (7) дислипидемия, (8) гиперлипидемия, (9) гипертриглицеридемия, (10) гиперхолестеринемия, (11) низкие уровни HDL, (12) высокие уровни LDL, (13) атеросклероз и его осложнения, (14) рестеноз сосудов, (15) синдром раздраженной толстой кишки, (16) воспалительные заболевания кишечника, включая болезнь Крона и язвенный колит, (17) другие воспалительные состояния, (18) панкреатит, (19) абдоминальное ожирение, (20) нейродегенеративные заболевания, (21) ретинопатия, (22) псориаз, (23) метаболический синдром, (24) овариальный гиперандрогенизм (синдром поликистоза яичников) и другие заболевания, при которых резистентность к инсулину является составляющим компонентом. Они также могут найти применение в лечении высокого кровяного давления, неопластических состояний, опухолей жировых клеток, карцином жировых клеток, таких как липосаркома, рак простаты и другие виды рака, включая рак пищевода и желудка, рак молочной железы, рак мочевого пузыря и рак толстой кишки, ангиогенеза, остеопороза и болезни Альцгеймера.
Соединения могут найти применение в лечении остеопороза. Соединения этого изобретения могут лечить остеопороз или снижать вероятность развития остеопороза посредством замедления или остановки потери плотности кости у пациентов, имеющих остеопороз или вероятность развития остеопороза. Соединения этого изобретения могут также реверсировать потерю костной массы для пациентов, у которых уже началась потеря костной массы.
Одним из аспектов настоящего изобретения является предоставление способа лечения и контролирования смешанной или диабетической дислипидемии, гиперхолестеринемии, атеросклероза, низких уровней HDL, высоких уровней LDL, гиперлипидемии и/или гипертриглицеридемии, который предусматривает назначение пациенту, нуждающемуся в таком лечении, терапевтически эффективных количеств соединения, имеющего формулу I. Соединение может применяться только одно или успешно может приниматься с ингибитором биосинтеза холестерина, в особенности с ингибитором HMG-CoA редуктазы, таким как ловастатин, симвастатин, росувастатин, правастатин, флувастатин, аторвастатин, ривастатин, итавастатин или ZD-4522. Соединение может также успешно применяться в комбинации с другими липидпонижающими лекарствами, такими как ингибиторы абсорбции холестерина (например, эфиры станола, стеролгликозиды, такие как тикуесид, и азетидиноны, такие как эзетимиб), ингибиторы ACAT (такие как авасимиб), ингибиторы CETP, ниацин, вещества, усиливающие экскрецию желчной кислоты, ингибиторы микросомального транспорта триглицеридов, ингибиторы обратного захвата желчной кислоты. Эти комбинированные лечения могут также быть эффективными при лечении или контролировании одного или нескольких сопутствующих состояний, выбираемых из группы, включающей гиперхолестеринемию, атеросклероз, гиперлипидемию, гипертриглицеридемию, дислипидемию, высокий уровень LDL и низкий уровень HDL.
Другой аспект изобретения заключается в предоставлении способа лечения воспалительных состояний, включая воспалительные заболевания кишечника, болезнь Крона и язвенный колит, путем назначения эффективных количеств соединения этого изобретения пациенту, нуждающемуся в лечении. Дополнительные воспалительные заболевания, которые можно лечить с помощью рассматриваемого в данный момент изобретения, включают подагру, ревматоидный артрит, остеоартрит, множественный склероз, астму, ARDS, псориаз, васкулит, ишемию/реперфузное поражение, обморожение и сопутствующие заболевания.
Введение и диапазон доз
Любой удобный способ введения может быть использован для предоставления млекопитающим, в особенности людям, эффективных доз соединения настоящего изобретения. Например, пероральный, ректальный, местный, парентеральный, окулярный, пульмональный, назальный и им подобные могут применяться. Дозированные формы включают таблетки, пастилки, дисперсии, суспензии, растворы, капсулы, кремы, мази, аэрозоли и им подобные. Предпочтительно соединения формулы I назначаются перорально.
Эффективные дозы применяемых активных ингредиентов могут изменяться в зависимости от особенностей применяемого соединения, способа введения, патологического состояния, подвергаемого лечению, и тяжести патологического состояния, подвергаемого лечению. Такие дозы могут быть легко определены специалистом в данной области техники.
При лечении или контролировании сахарного диабета и/или гипергликемии или гипертриглицеридемии или других заболеваний, для которых соединения формулы I предписаны, обычно удовлетворительные результаты получаются в том случае, когда соединения настоящего изобретения назначаются для введения в суточных дозах, составляющих от, приблизительно, 0,1 мг до, приблизительно, 100 мг на 1 кг веса животного, предпочтительно даваемых в виде одноразовой суточной дозы или дробными дозами от двух до шести раз в день, или в форме с замедленным высвобождением. Для большинства больших млекопитающих общая суточная доза составляет от, приблизительно, 1 мг до, приблизительно, 1000 мг, предпочтительно от, приблизительно, 1 мг до, приблизительно, 50 мг. В случае взрослого человека весом 70 кг общая суточная доза обычно будет составлять от, приблизительно, 1 мг до, приблизительно, 350 мг. Для особенно сильного соединения доза в случае взрослого человека может быть минимально возможной 0,1 мг. Дозированный режим может быть отрегулирован в пределах этого диапазона или даже за пределами этого диапазона для обеспечения оптимальной терапевтической восприимчивости.
Пероральный прием обычно осуществляется с использованием таблеток. Примеры доз в таблетках следующие: 0,5 мг, 1 мг, 2 мг, 5 мг, 10 мг, 25 мг, 50 мг, 100 мг и 250 мг. Другие пероральные формы также могут содержать те же дозы (например, капсулы).
Фармацевтические композиции
Другой аспект настоящего изобретения заключается в предоставлении фармацевтических композиций, которые включают соединение формулы I и фармацевтически приемлемый носитель. Фармацевтические композиции настоящего изобретения включают соединение формулы I или его фармацевтически приемлемую соль в качестве активного ингредиента, а также и фармацевтически приемлемый носитель, и добавочно могут содержать другие терапевтические ингредиенты. Термин “фармацевтически приемлемые соли” относится к солям, приготовленным из фармацевтически приемлемых нетоксичных оснований или кислот, включая неорганические основания или кислоты и органические основания или кислоты. Фармацевтическая композиция может также включать пролекарство или его фармацевтически приемлемую соль, если пролекарство назначено для приема.
Композиции включают композиции, пригодные для перорального, ректального, местного, парентерального (включая подкожный, внутримышечный и внутривенный), окулярного (офтальмологического), пульмонального (носовая или ротовая ингаляция) или назального приема, хотя наиболее удобный способ применения в каком-либо случае будет зависеть от природы и тяжести патологических состояний, подвергаемых лечению, и от природы и активности ингредиента. Они могут быть удобно представлены в единичной дозированной форме и приготовлены одним из методов, хорошо известных в фармацевтической технологии.
При практическом применении соединения формулы I могут комбинироваться как активный ингредиент в однородной смеси с фармацевтическими носителями согласно промышленным фармацевтическим технологическим инструкциям по приготовлению многокомпонентной смеси. Носитель может быть выбран из обширного множества форм в зависимости от формы лекарственного препарата, требуемой для приема, например пероральной или парентеральной (включая внутривенную). При приготовлении композиции для пероральной дозированной формы могут применяться любые обычные фармацевтические среды, такие как, например, вода, гликоли, масла, спирты, ароматизаторы, консервирующие добавки, окрашивающие средства и им подобные, пригодные в случае пероральных жидких лекарственных препаратов, таких как, например, суспензии, эликсиры и растворы; или носители, такие как крахмалы, сахара, микрокристаллическая целлюлоза, разбавители, грануляторы, смазывающие вещества, связующие вещества, дезинтегрирующие вещества и им подобные, пригодные в случае пероральных твердых лекарственных препаратов, таких как, например, порошки, мягкие и твердые капсулы и таблетки, с учетом того, что твердые пероральные лекарственные препараты предпочтительнее жидких лекарственных препаратов.
Из-за облегченного приема таблетки и капсулы представляют собой предпочтительную пероральную дозированную единичную форму, в случае которой безусловно применяются твердые фармацевтические носители. Если необходимо, таблетки могут быть покрыты оболочкой с использованием стандартной водной или неводной методики. Такие композиции и лекарственные препараты должны содержать, как минимум, 0,1% активного соединения. Процентное содержание активного соединения в этих композициях конечно может варьироваться и удобно, чтобы оно составляло от, приблизительно, 2% до, приблизительно, 60% от веса единицы. Количество активного вещества в такой терапевтически приемлемой композиции является таким, что будет обеспечена эффективная доза его. Активные соединения могут также приниматься интраназально, как, например, в виде жидких капель или спрея.
Таблетки, пилюли, капсулы и им подобные могут также содержать связующее вещество, такое, как трагакант, аравийская камедь, кукурузный крахмал или желатин; инертные наполнители, такие как фосфат кальция, дезинтегрирующие вещества, такие как кукурузный крахмал, картофельный крахмал, альгиновая кислота, смазывающие вещества, такие как стеарат магния; и подсластители, такие как сахароза, лактоза или сахарин. Если дозированная единичная форма является капсулой, она может, дополнительно к вышеперечисленным веществам, содержать жидкий носитель, такой как жирное масло.
Различные другие вещества могут быть предложены как для покрытия оболочкой, так и для модификации физической формы дозированной единицы. Например, таблетки могут быть покрыты шеллаком, сахаром или и тем, и другим. Сироп или эликсир может содержать, в дополнение к активному ингредиенту, сахарозу как подсластитель, метил или пропилпарабен как консервирующее вещество, окрашивающее вещество и ароматизатор, такой как вишневый или апельсиновый ароматизатор.
Соединения формулы I могут также вводиться парентерально. Растворы или суспензии этих активных соединений могут быть приготовлены в водных смесях с поверхностно-активными веществами, такими как гидроксипропилцеллюлоза. Дисперсии также могут быть приготовлены в глицерине, жидких полиэтиленгликолях и их смесях с маслами. При обычных условиях хранения и использования эти лекарственные препараты содержат консервирующее вещество для предотвращения роста микроорганизмов.
Фармацевтические формы, пригодные для инъекционного применения, содержат в своем составе стерильные водные растворы или дисперсии и стерильные порошки для приготовления, с целью немедленного приема, стерильных инъекционных растворов или дисперсий. Во всех случаях форма должна быть стерильной и должна представлять собой до такой степени жидкую текучую среду, какая просто может существовать. Она должна быть стабильной в условиях производства и хранения и должна быть предохранена от порчи под действием микроорганизмов, таких как бактерии и грибы. Носитель может быть растворителем или дисперсионной средой, содержащей, например, воду, этанол, полиол (например, глицерин, пропиленгликоль и жидкий полиэтиленгликоль), подходящие смеси их и растительные масла.
Комбинированная терапия
Соединения формулы I могут применяться в комбинации с другими лекарствами, которые также могут быть полезны для лечения или улучшения состояния при заболеваниях или патологических состояниях, для которых полезны соединения формулы I. Такие дополнительные лекарства могут назначаться для приема в соответствии с обычно используемыми для них способом приема и количеством, одновременно или последовательно с веществом формулы I. В том случае, если вещество формулы I применяется одновременно с одним или с несколькими другими дополнительными лекарствами, предпочтительной является фармацевтическая композиция с единичной дозированной формой, содержащей такие дополнительные лекарства и соединение формулы I. Однако комбинированная терапия также включает в себя методы лечения, в которых соединение формулы I и одно или большее количество дополнительных лекарств принимаются с различной суперпозицией схем применения. Также предполагается, что при использовании в комбинации с одним или с несколькими другими дополнительными активными ингредиентами соединение настоящего изобретения и дополнительные активные ингредиенты могут применяться в более низких дозах, чем в тех случаях, когда каждое из них применяется отдельно. Следовательно, фармацевтические композиции настоящего изобретения включают такие, которые содержат один или несколько дополнительных активных ингредиентов в дополнение к соединению формулы I.
Примеры дополнительных активных ингредиентов, которые могут назначаться для введения либо в комбинации с соединением формулы I, либо назначаться отдельно или в той же фармацевтической композиции, включают следующие ингредиенты, но не ограничиваются ими:
(а) дополнительные PPAR гамма-агонисты и частичные агонисты, включая как глитазоны, так и не глитазоны (например, троглитазон, пиоглитазон, энглитазон, МСС-555, розиглитазон, балаглитазон, нетоглитазон, Т-131, LY-300512 и LY-818;
(b) бигуаниды, такие как метформин и фенформин;
(с) ингибиторы белковой тирозинфосфатазы-1В (РТР-1В);
(d) ингибиторы дипептидилпептидазы IV (DP-IV);
(е) инсулин или миметики инсулина;
(f) сульфонилмочевины, такие как толбутамид и глипизид, или родственные им вещества;
(g) ингибиторы α-глюкозидазы (такие как акарбоза);
(h) средства, которые улучшают липидные характеристики пациента, такие как (i) ингибиторы HMG-CoA редуктазы (ловастатин, симвастатин, розувастатин, правастатин, флувастатин, аторвастатин, ривастатин, итавастатин, ZD-4522 и другие статины), (ii) вещества, усиливающие экскрецию желчных кислот (холестирамин, холестипол и диалкиламиноалкильные производные поперечно сшитого декстрана), (iii) никотиниловый спирт, никотиновая кислота или ее соли, (iv) PPARα агонисты, такие как производные фенофибриновой кислоты (гемфиброзил, клофибрат, фенофибрат и безафибрат), (v) ингибиторы абсорбции холестерина, такие как, например, эзетимиб, (vi) ингибиторы ацилCoA:холестерин ацилтрансферазы (АСАТ), такие как авасимиб, (vii) ингибиторы СЕТР и (viii) фенольные антиоксиданты, такие как пробукол;
(i) PPARα/γ двойные агонисты, такие как KRP-297, мураглитазар, тезаглитазар, фарглитазар и JT-501;
(j) PPARδ агонисты, такие как раскрытые в WO 97/28149;
(k) соединения против ожирения, такие как фенфлюрамин, дексфенфлюрамин, фентриамин, субитрамин, орлистат, ингибиторы нейропептида Y5, Mc4r агонисты, антагонисты/инверсные агонисты каннабиноидного рецептора 1 (СВ-1) и агонисты β3 адренергического рецептора;
(l) ингибиторы физиологического носителя желчных кислот;
(m) средства, предназначенные к применению при воспалительных патологических состояниях, такие как аспирин, нестероидные противовоспалительные лекарства, глюкокортикоиды, азульфидин и селективные ингибиторы циклооксигеназы 2;
(n) антагонисты глюкагонового рецептора;
(o) GLP-1;
(p) GIP-1; и
(q) аналоги GLP-1, такие как эксендины, например эксенитид.
Вышеприведенные комбинации включают комбинации соединения настоящего изобретения не только с одним дополнительным активным соединением, но также с двумя и с большим количеством других активных соединений. Нелимитирующие примеры включают комбинации соединений, имеющих формулу I, с двумя или с большим количеством активных соединений, выбранных из числа бигуанидов, сульфонилмочевины, ингибиторов HMG-CoA редуктазы, других PPAR агонистов, ингибиторов РТР-1В, ингибиторов DP-IV и соединений против ожирения.
БИОЛОГИЧЕСКИЕ ИСПЫТАНИЯ
А) Испытания связывания PPAR
Для получения рекомбинантных человеческих PPARγ, PPARδ и PPARα: человеческий PPARγ2, человеческий PPARδ и человеческий PPARα были экспрессированы как gst-слитые белки в E.coli. Полноразмерная человеческая кДНК для PPARγ2 была субклонирована в экспрессионный вектор pGEX-2T (Pharmacia). Полноразмерные человеческие кДНК для PPARδ и PPARα были субклонированы в экспрессионный вектор pGEX-KT (Pharmacia). E.coli, содержащая соответствующие плазмиды, была репродуцирована, индуцирована и собрана центрифугированием. Бляшка ресуспендированного остатка была разрушена с использованием Французского пресса и дебрис был удален центрифугированием при 12000×g. Рекомбинантные человеческие PPAR рецепторы были очищены аффинной хроматографией на глутатион-сефарозе. После нанесения на колонку и однократной промывки рецептор элюировался с глутатионом. Глицерин (10%) добавляли для стабилизации рецептора и аликвоты хранились при -80°С.
Для связывания с PPARγ аликвоту рецептора инкубировали в TEGM (10 мМ Tris, pH 7,2, 1 мМ EDTA, 10% глицерина, 7 мкл/100 мл β-меркаптоэтанола, 10 мМ молибдата Na, 1 мМ дитиотреитола, 5 мкг/мл апротинина, 2 мкг/мл леупептина, 2 мкг/мл бензамидина и 0,5 мМ PMSF), содержащего 0,1% обезжиренного сухого молока и 10 нМ [3H2]AD5075, (21 Ci/ммоль), ± тестируемое соединение, как описано у Berger и др. (Novel peroxisome proliferator-activated receptor (PPARγ) and PPARδ ligands produce distinct biological effects. J. Biol. Chem. (1999), 274: 6718-6725). Образцы инкубировали ~16 часов при 4°С в конечном объеме 150 мкл. Несвязанный лиганд удаляли инкубацией со 100 мкл имеющего декстран/желатиновое покрытие активированного угля при охлаждении льдом в течение ~10 мин. После центрифугирования при 3000 об/мин в течение 10 мин при 4°С 50 мкл фракции супернатанта было зафиксировано в верхней части.
Для связывания PPARδ аликвоту рецептора инкубировали в TEGM (10 мМ Tris, pH 7,2, 1 мМ EDTA, 10% глицерина, 7 мкл/100 мл β-меркаптоэтанола, 10 мМ молибдата Na, 1 мМ дитиотреитола, 5 мкг/мл апротинина, 2 мкг/мл леупептина, 2 мкг/мл бензамида и 0,5 мМ PMSF), содержащего 0,1% обезжиренного сухого молока и 2,5 нМ [3H2]L-783483, (17 Ci/ммоль), ± тестируемое соединение, как описано у Berger и др. (Novel peroxisome proliferators-activated receptor γ (PPARγ) and PPARδ ligands produce distinct biological effects. 1999 J. Biol. Chem. 274: 6718-6725). (L-783483 представляет собой 3-хлор-4-(3-(7-пропил-3-трифторметил-6-бенз-[4,5]-изоксазолокси)пропилтио)фенилуксусную кислоту, пример 20 в патенте WO 97/28137). Образцы инкубировали в течение ~16 часов при 4°С в конечном объеме 150 мкл. Несвязанный лиганд удаляли инкубацией со 100 мкл имеющего декстран/желатиновое покрытие активированного угля при охлаждении льдом в течение ~10 мин. После центрифугирования при 3000 об/мин в течение 10 мин при 4°С 50 мкл фракции супернатанта было зафиксировано в верхней части.
Для связывания PPARα аликвоту рецептора инкубировали в TEGM (10 мМ Tris, pH 7,2, 1 мМ EDTA, 10% глицерина, 7 мкл/100 мл β-меркаптоэтанола, 10 мМ молибдата Na, 1 мМ дитиотреитола, 5 мкг/мл апротинина, 2 мкг/мл леупептина, 2 мкг/мл бензамида и 0,5 мМ PMSF), содержащего 0,1% обезжиренного сухого молока и 5,0 нМ [3H2]L-797773, (34 Ci/ммоль), ± тестируемое соединение. (L-797773 представляет собой (3-(4-(3-фенил-7-пропил-6-бенз-[4,5]-изоксазолокси)бутилокси)фенилуксусную кислоту, пример 62 в патенте WO 97/28137). Образцы инкубировали в течение ~16 часов при 4°С в конечном объеме 150 мкл. Несвязанный лиганд удаляли инкубацией со 100 мкл имеющего декстран/желатиновое покрытие активированного угля при охлаждении льдом в течение ~10 мин. После центрифугирования при 3000 об/мин в течение 10 мин при 4°С 50 мкл фракции супернатанта было зафиксировано в верхней части.
В) Испытания Gal-4 hPPAR трансактивации
Химерные рецепторные экспрессирующиеся конструкции pcDNA3-hPPARγ/GAL4, pcDNA3-hPPARδ/GAL4, pcDNA3-hPPARα/GAL4 были получены путем инсерции дрожжам GAL4 транскрипционных факторов DBD, примыкающих к лигандсвязывающим доменам (LBD) hPPARγ, hPPARδ, hPPARα соответственно. Репортерная конструкция pUAS(5X)-tk-luc была создана путем инсерции 5 копий GAL4 зависимого элемента в обратном направлении вируса герпеса минимального тримидинкиназного промотора и люциферазного репортерного гена. pCMV-lacZ содержит галактозидазный Z ген, регулируемый цитомегаловирусным промотором. COS-1 клетки были посеяны по 12×103 клеток/на ячейку в 96-ячеечный планшет для клеточной культуры в богатую глюкозой среду, предложенную Dulbecco и модифицированную Eagle (DMEM), содержащую 10% активированный уголь, десорбированный эмбриональной телячьей сывороткой (Gemini Bio-Products, Calabasas, CA), неэссенциальные аминокислоты, 100 единиц/мл Пенициллина G и 100 мг/мл Стрептомицина сульфата, при 37°С во влажной атмосфере, содержащей 10% СО2. После 24 часов осуществляли трансфекцию с Липофектамином (GIBCO BRL, Gaithersburg, MD) согласно инструкциям производителя. В нескольких словах, трансфекционные смеси для каждой ячейки, содержащие 0,48 мкл Липофектамина, 0,00075 мкг pcDNA3-hPPAR/GAL4 экспрессируемого вектора, 0,045 мкг pUAS(5X)-tk-luc репортерного вектора и 0,0002 мкг pCMV-lacZ для промежуточного контроля эффективности трансактивации. Клетки инкубировали в трансфекционной смеси в течение 5 часов при 37°С в атмосфере, содержащей 10% СО2. Затем клетки инкубировали в течение ~48 часов в свежей богатой глюкозой среде (DMEM), содержащей 5% активированного угля, десорбированного эмбриональной телячьей сывороткой, неэссенциальные аминокислоты, 100 единиц/мл Пенициллина G и 100 мг/мл Стрептомицина сульфата ± увеличивающиеся концентрации тестируемого соединения. Поскольку тестируемые соединения были растворены в ДМСО, контрольные клетки инкубировали с эквивалентными концентрациями ДМСО; конечные концентрации ДМСО составляли ≤0,1%, что соответствует концентрации, которая не проявила воздействия на активность трансактивации. Клеточные лизаты получали с использованием Репортерного Лизисного Буфера (Promega, Madison, WI), согласно инструкциям производителя. Люциферазную активность в клеточных экстрактах определяли с использованием Образца Люциферазного Буфера (Promega, Madison, WI) в ML3000 люминометре (Dynatech Laboratories, Chantilly, VA). β-Галактозидазную активность определяли с использованием β-D-галактопиранозида (Calbiochem, San Diego, CA).
Агонизм определяли посредством сравнения максимальной трансактивационной активности с полным PPAR агонистом, таким как розиглитазон. Обычно, в том случае, если максимальная стимуляция трансактивации составляет менее чем 50% от эффекта, наблюдаемого с полным агонистом, то соединение определяется как частичный агонист. Если максимальная стимуляция трансактивации составляет более чем 50% от эффекта, наблюдаемого с полным агонистом, то соединение определяется как полный агонист. Соединения этого изобретения имели величины EC50 в пределах от 1 нМ до 3000 нМ.
C). Исследования in vivo
Самцов db/db мышей (возраста 10-11 недель, C57B1/KFJ, Jackson Labs, Bar Harbor, ME)
размещали по 5 в клетке и обеспечивали свободным доступом к корму для грызунов Purina и воде. Животных и их пищу взвешивали каждые 2 дня и им вводили дозированное зондовое питание с наполнителем (0,5% карбоксиметилцеллюлоза) ± тестовое соединение в указанной дозе. Суспензии лекарств готовили ежедневно. Концентрации глюкозы в плазме и триглицеридов определяли для крови, полученной при обрезке хвоста с 3-5 дневными интервалами в течение периода исследований. Определения глюкозы и триглицеридов осуществляли на автоматическом анализаторе Boehringer Mannheim Hitachi 911 (Boehringer Mannheim, Indianapolis, IN) с использованием гепаринизированной плазмы, разбавленной 1:6 (об./об.) физиологическим раствором. Худые животные были по возрастным группам гетерозиготными мышами, содержащимися таким же образом.
ПРИМЕРЫ
Нижеследующие примеры предусмотрены для иллюстрации изобретения и не должны быть истолкованы, как каким-либо образом ограничивающие изобретение. Объем изобретения определяется в прилагаемой формуле изобретения.
Процесс получения соединений, рассматриваемого в данный момент изобретения, в общем представлен на нижеприведенной схеме 1.
Схема 1
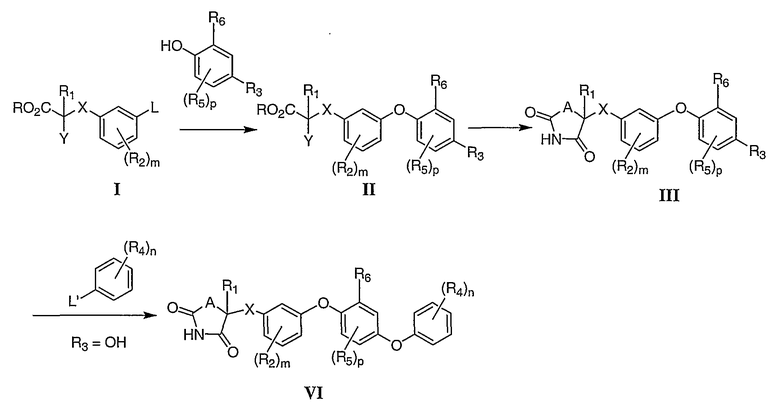
Соответствующие α-замещенные фенилацетаты или их гомологи (например, в которых Х является СН2 или связью) I сочетают с 2-замещенными фенолами с образованием диарилэфирных производных II. α-Замещенная эфирная часть соединения II затем превращается в 1,3-оксазолидин-2,4-дионовое (OZD) или в 1,3-тиазолидин-2,4-дионовое (TZD) кольцо, давая соединение III (А является O или S). Соединение III может быть либо конечным продуктом, либо ключевым промежуточным соединением во множестве синтетических трансформаций. Так, в том случае, если R3 является гидроксильной группой, соединение III может быть связано через гидроксильную группу с предназначенными для сочетания партнерами, например арилбороновыми кислотами, галогенарилами, давая соединение IV. L и L' на схеме 1 являются уходящими группами.
Промежуточное соединение 1
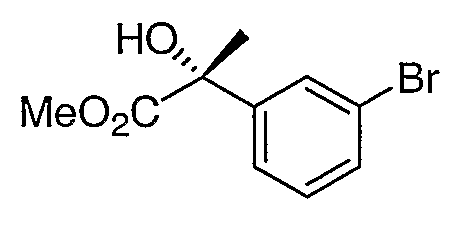
Стадия 1. Получение 1-бром-3-(2-пропенил)бензола
Раствор NaHMDS в ТГФ (1,0 М, 18,0 мл, 18,0 ммоль) добавляли к суспензии бромида метилтрифенилфосфония (6,4 г, 18,0 ммоль) в ТГФ (60 мл), которую охлаждали на ледяной бане. Получившуюся в результате суспензию оранжевого цвета перемешивали 30 мин и затем охлаждали до -78°С. 3-Бромацетофенон (3,0 г, 15,0 ммоль) добавляли по каплям. После выдерживания 30 мин при -78°С реакционную смесь нагревали до 25°С, обрабатывали уксусной кислотой (1,0 мл). После удаления растворителя остаток растирали со смесью этилацетат/гексан (3:7, 100 мл) и фильтровали через короткую колонку с силикагелем. Концентрирование фильтрата приводило к получению соединения, указанного в заголовке.
1H ЯМР (500 МГц, CDCl3) δ 7,62 (т, J=2,5 Гц, 1H), 7,50 (м, 1H), 7,41 (м, 1H), 7,22 (т, J=8,5 Гц, 1H), 5,40 (c, 1H), 5,15 (c, 1H), 2,16 (c, 3H).
Стадия 2. Получение (2R)-2-(3-бромфенил)-1,2-пропандиола
Смесь продукта, полученного на стадии 1, (2,9 г, 15 ммоль) и AD-mix-β (Aldrich, 21,0 г) в t-BuOH-H2O (1:1, 150 мл) интенсивно перемешивали при 4°С в течение 16 часов. Реакционную смесь обрабатывали твердым Na2SO3 (5,0 г) и разбавляли этилацетатом (150 мл). Водную фазу отделяли и экстрагировали этилацетатом. Объединенную органическую фазу промывали солевым раствором, сушили и фильтровали через невысокий слой силикагеля. Удаление растворителя приводило к получению фактически чистого соединения, указанного в заголовке.
1H ЯМР (500 МГц, CDCl3) 8 7,63 (т, J=2,5 Гц, 1H), 7,40 (м, 1H), 7,36 (м, 1H), 7,23 (т, 8,4 Гц, 1H), 3,75 (д, J=11,8 Гц, 1H), 3,62 (д, J=11,8 Гц, 1H), 1,50 (c, 3H).
Стадия 3. Получение (2R)-2-(3-бромфенил)-2-гидроксипропионата
Диол со стадии 2 (3,3 г, 15 ммоль) и 10% Pt на угле (1,5 г) перемешивали в 0,1М буферном растворе K2HPO4 (300 мл). Реакционную смесь нагревали при 80°С и барботировали потоком воздуха в течение 6 часов. Горячую реакционную смесь фильтровали через слой целита и слой осадка на фильтре промывали этилацетатом, содержащим 5% уксусной кислоты (100 мл). Водный фильтрат подкисляли концентрированной хлористоводородной кислотой до рН 2 и экстрагировали этилацетатом (3×80 мл). Объединенную органическую фазу промывали солевым раствором, сушили и концентрировали. Остаток растворяли в смеси 7:1 (об./об.) бензол-метанол (75 мл) и обрабатывали триметилсилилдиазометаном (1,0 М в гептане) до тех пор, пока прекратится выделение газа. Летучие компоненты удаляли и остаток очищали с помощью хроматографии на силикагеле, элюируя смесью 7:3 гексан:этилацетат, с получением соединения, указанного в заголовке.
1H ЯМР (500 МГц, CD3OD) 5 7,71 (т, J=2,0 Гц, 1H), 7,49 (дт, J=8,0 Гц, 1,0 Гц, 1H), 7,44 (ддд, J=8,0 Гц, 2,0 Гц, 1,0 Гц, 1H), 7,26 (т, J=8,0 Гц, 1H), 3,72 (c, 3H), 1,72 (c, 3H).
MC (ESI, m/z): 281,0 (М+Na+).
Промежуточное соединение 2
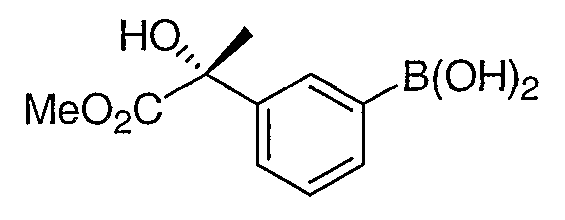
Стадия 1. Получение метил (2R)-2-гидрокси-2-[3-(4,4,5,5,-тетраметил-1,3,2-диоксаборолан-2-ил)фенил]пропионата
Смесь промежуточного соединения 1 (2,6 г, 10 ммоль), бис(пинаколато)дибора (2,8 г, 11 ммоль), ацетата калия (2,9 г, 30 ммоль) и Pd(dppf)Cl2 (0,49 г 0,6 ммоль) в ДМСО (50 мл) дегазировали и нагревали в атмосфере азота при 80°С в течение 2 часов. Реакционную смесь разбавляли диэтилацетатом (100 мл) и фильтровали через невысокий слой силикагеля. Фильтрат промывали водой (2×100 мл) и концентрировали. Остаток очищали с помощью хроматографии на силикагеле, элюируя смесью 2:8 этилацетат:гексан, с получением соединения, указанного в заголовке.
Стадия 2. Получение (2R)-2-гидрокси-2-[3-(бороно)фенил]пропионата
Смесь продукта со стадии 1 (0,61 г, 2,0 ммоль), периодата натрия (1,3 г, 6,0 ммоль) и ацетата аммония (0,31 г, 4,0 ммоль) в смеси ацетон:вода (1:1, 20 мл) перемешивали при 25°С в течение 16 часов. Осадок отфильтровывали и фильтрат упаривали. Водную фазу подкисляли 2н. HCl до рН 3 и экстрагировали этилацетатом (3Ч20 мл). Органическую фазу промывали солевым раствором и концентрировали, что приводило к получению фактически чистого промежуточного соединения 2.
1H ЯМР (500 МГц, CDCl3) δ 8,45 (c, 1H), 8,22 (д, J=7,5 Гц, 1H), 7,84 (д, J=7,5 Гц, 1H), 7,56 (т, J=7,5 Гц, 1H), 3,88 (c, 3H).
MC (ESI, m/z): 247,1 (М+Na+).
Промежуточное соединение 3
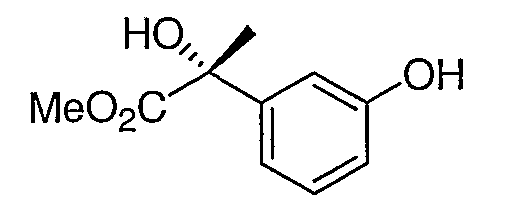
К раствору промежуточного соединения 2 (2,47 г, 10 ммоль) в дихлорметане (50 мл) добавляли по каплям 30% раствор перекиси водорода в воде (1,7 мл, 15 ммоль). Спустя 30 мин реакционную смесь обрабатывали водным сульфитом натрия и экстрагировали дихлорметаном. После удаления растворителя неочищенный продукт очищали с помощью хроматографии на силикагеле, элюируя смесью 7:3 гексана и этилацетата, что обеспечивало получение промежуточного соединения 3.
1H ЯМР (500 МГц, CD3OD) δ 7,25 (т, J=8,0 Гц, 1H), 7,00 (дт, J=8,0, 2,0 Гц, 1H), 6,98 (т, J=2 Гц, 1H), 6,81 (дт, J=8,0, 2,0 Гц, 1H), 1,87 (c, 3H).
MC (ESI, m/z): 208,2 (М+1).
Промежуточное соединение 4
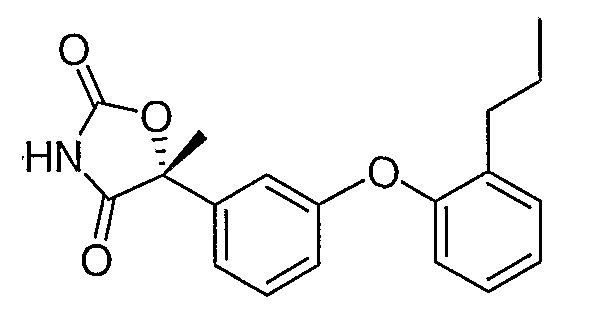
Стадия 1. Получение (2R)-2-гидрокси-2-[3-(2-пропилфенокси)фенил]пропионата
Смесь промежуточного соединения 1 (2,6 г, 10 ммоль), 2-пропилфенола (2,0 г, 15 ммоль), ацетата палладия (90 мг, 0,04 ммоль), ди(трет-бутил)(2-бифенил)фосфина (179 мг, 0,06 ммоль) и фосфата калия (4,2 г, 20 ммоль) в толуоле (30 мл) дегазировали и нагревали в атмосфере N2 при 100°С в течение 16 часов. Реакционную смесь разбавляли эфиром (50 мл) и фильтровали через невысокий слой силикагеля, что приводило к получению неочищенного указанного в заголовке продукта, который немедленно использовался на следующей стадии.
Стадия 2. Получение (2R)-2-гидрокси-2-[3-(2-пропилфенокси)фенил]пропионамида
Раствор неочищенного продукта со стадии 1 в метаноле (35 мл) охлаждали до 0°С и насыщали газообразным аммиаком. Раствор выдерживали при 25°С в течение 2 дней и затем концентрировали, остаток хроматографировали на силикагеле, элюируя сначала смесью 3:7 этилацетат:гексан и затем 100% этилацетатом. Этилацетатную фракцию концентрировали, что приводило к получению соединения, указанного в заголовке.
Стадия 3. Получение (5R)-5-[3-(2-пропилфенокси)фенил]-5-метил-1,3-оксазолидин-2,4-диона
Амид со стадии 2 (2,1 г, 7,0 ммоль) растворяли в диэтилкарбонате (35 мл). 1,1'-Карбонилдиимидазол (3,4 г, 21 ммоль) и гидрид натрия (60% дисперсия в минеральном масле, 0,84 г, 21 ммоль) были последовательно добавлены. Полученную в результате реакционную смесь перемешивали при 50°С в течение 2 часов и выливали в ледяную воду. Водную смесь подкисляли концентрированной хлористоводородной кислотой до рН 2 и экстрагировали этилацетатом. Объединенную органическую фазу промывали солевым раствором, сушили и концентрировали. Остаток очищали с помощью хроматографии на силикагеле, элюируя смесью 3:7 этилацетат:гексан, содержащей 1% уксусной кислоты, что обеспечивало получение промежуточного соединения 4 в виде белого твердого вещества.
1H ЯМР (500 МГц, CD3OD) δ 7,37 (т, J=8,0 Гц, 1H), 7,30 (дд, J=7,5 Гц, 2,0 Гц, 1H), 7,23 (м, 1H), 7,20 (дд, J=8,0 Гц, 2,0 Гц, 1H), 7,13 (тд, J=7,5 Гц, 1,5 Гц, 1H), 7,07 (т, J=2,5 Гц, 1H), 6,89 (дд, J=8,0 Гц, 1 Гц, 1H), 6,87 (м, 1H), 2,56 (т, J=7,5 Гц, 2H), 1,84 (c, 3H), 1,60 (м, 2H), 0,90 (т, J=7,5 Гц, 3H).
MC (ESI, m/z): 326,1 (М++1).
Промежуточное соединение 5
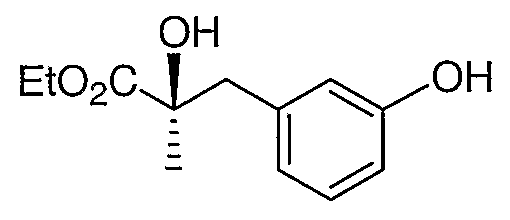
Стадия 1. Получение этил (E)-2-метил-3-(3-бензилоксифенил)пропеноата
Раствор 3-бензилоксибензальдегида (10 г, 50 ммоль) и (карбэтоксиэтилиден)трифенилфосфорана (20 г, 55 ммоль) в ТГФ (200 мл) нагревали с обратным холодильником в течение 2 часов. Реакционную смесь концентрировали и остаток растирали со смесью 7:3 этилацетат:гексан и фильтровали через невысокий слой силикагеля. Удаление растворителя из фильтрата приводило к получению продукта, указанного в заголовке.
Стадия 2. Получение этил (2R,3R)-3-(3-бензилоксифенил)-2,3-дигидрокси-2-метилпропионата
Смесь продукта со стадии 1 (5,9 г, 20 ммоль) и AD-mix-α (Aldrich, 28,0 г) перемешивали в смеси 1:1 t-BuOH:Н2О (200 мл). Полученную в результате смесь перемешивали при 4°С в течение 2 дней и обрабатывали добавлением водного раствора Na2SO3 (2н, 20 мл). Смесь разбавляли этилацетатом (200 мл), промывали солевым раствором (2×100 мл) и сушили. Удаление растворителя приводит к получению соединения, указанного в заголовке.
1H ЯМР (500 МГц, CDCl3) δ 7,46 (м, 2H), 7,41 (м, 2H), 7,36 (м, 1H), 7,29 (т, J=8,4 Гц, 1H), 7,09 (т, J=2,4 Гц, 1H), 7,01 (д, J=8,4 Гц, 1H), 6,96 (дд, J=8,4, 2,4 Гц, 1H), 5,1 (c, 2H), 4,8 (д, J=7,1 Гц, 1H), 4,3 (м, 2H), 3,50 (c, 1H), 2,70 (д, J=7,1 Гц, 1H), 1,35 (т, J=7,5 Гц, 3H), 1,22 (c, 3H).
Стадия 3. Получение этил (2R,3R)-3-(3-бензилоксифенил)-2,3-дигидрокси-2-метилпропионат 2,3-карбоната
Раствор продукта со стадии 2 (6,6 г, 20 ммоль) и карбонилдиимидазола (6,5 г, 40 ммоль) в толуоле (100 мл) нагревали при 60°С в течение 1 часа. После охлаждения до комнатной температуры реакционную смесь фильтровали через небольшую колонку с силикагелем. Оставшийся на фильтре осадок промывали смесью 3:7 этилацетат:гексан, что приводило к получению указанного в заголовке циклического карбоната с количественным выходом.
Стадия 4. Получение этил (2R)-2-гидрокси-3-(3-гидроксифенил)-2-метилпропионата
Раствор продукта со стадии 3 (7,1 г, 20 ммоль) в этаноле (100 мл) перемешивали с 10% Pd/C (1,4 г) в атмосфере водорода (1 атм.) в течение 16 часов. После удаления катализатора раствор концентрировали и остаток хроматографировали на силикагеле, элюируя смесью 3:7 этилацетат:гексан, что обеспечивало получение промежуточного соединения 5.
1H ЯМР (500 МГц, CDCl3) δ 7,16 (т, J=8,4 Гц, 1H), 6,76 (м, 3H), 4,80 (шир.с, 1H), 4,20 (м, 2H), 3,08 (д, J=15,0 Гц, 1H), 2,91 (д, J=15,0, 1H), 1,54 (c, 3H), 1,31 (т, J=7,2 Гц, 3H).
MC (ESI, m/z): 247,1 (М+Na+).
Промежуточное соединение 6
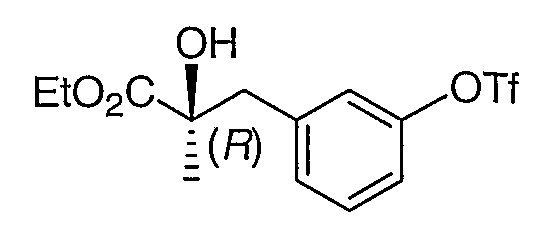
К раствору промежуточного соединения 5 (2,2 г, 10 ммоль) и этилдиизопропиламина (3,5 мл, 20 ммоль) в дихлорметане (50 мл), охлажденному до -75°С, добавляли трифторметансульфоновый ангидрид (1,77 мл, 10,5 ммоль). После перемешивания в течение 30 мин при -75°С реакционную смесь выливали в воду (50 мл) и экстрагировали дихлорметаном (1×20 мл). Органическую фазу промывали солевым раствором и концентрировали. Остаток помещали в эфир и фильтровали через невысокий слой силикагеля, что приводило к получению промежуточного соединения 6.
1H ЯМР (500 МГц, CD3OD) δ 7,05 (т, J=8,5 Гц, 1H), 6,62-6,69 (м, 3H), 4,13 (м, 2H), 2,95 (д, J=13,5, 1H), 2,86 (д, J=13,5 Гц, 1H), 1,38 (c, 3H), 1,23 (т, J=7,5 Гц, 3H)
MC (ESI, m/z): 379,0 (М+Na+).
Промежуточное соединение 7
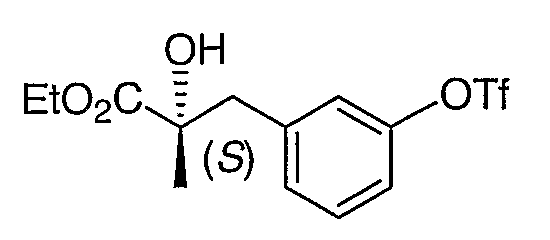
Стадия 1. Получение этил (2S)-2-гидрокси-3-(3-гидроксифенил)-2-метилпропионата
Указанное в заголовке соединение было получено согласно методике, описанной для промежуточного соединения 5, заменяя AD-mix-β на AD-mix-α на стадии 1.
Стадия 2. Получение этил (2S)-2-гидрокси-3-(3-(трифторметансульфонилокси)фенил)-2-метилпропионата
Продукт стадии 1 превращали в указанное в заголовке соединение согласно методике, описанной для его энантиомера, промежуточного соединения 6.
1H ЯМР (500 МГц, CD3OD) 7,05 (т, J=8,5 Гц, 1H), 6,62-6,69 (м, 3H), 4,13 (м, 2H), 2,95 (д, J=13,5, 1H), 2,86 (д, J=13,5 Гц, 1H), 1,38 (c, 3H), 1,23 (т, J=7,5 Гц, 3H).
MC (ESI, m/z): 379,0 (М+Na+).
Промежуточное соединение 8
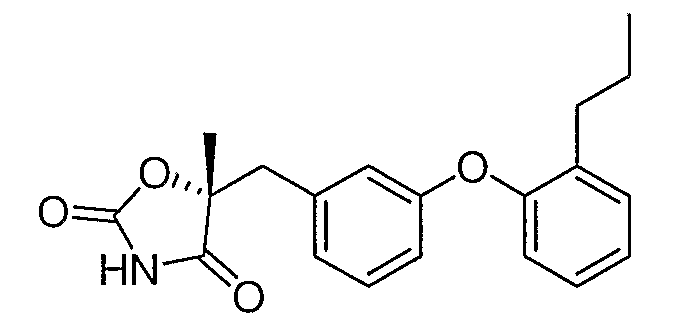
Указанное в заголовке соединение было получено согласно методике, описанной для промежуточного соединения 4, стадии с 1 по 3, используя промежуточное соединение 6 вместо промежуточного соединения 1 на стадии 1.
1H ЯМР (500 МГц, CDCl3) δ 8,14 (шир.с, 1H), 7,28 (м, 1H), 7,25 (т, J=7,9 Гц, 1H), 7,20 (м, 1H), 7,17 (м, 1H), 6,92 (д, J=7,6 Гц, 1H), 6,86 (м, 2H), 6,80 (д, J=1,6 Гц, 1H), 3,15 (д, J=14,3 Гц, 1H), 3,07 (д, J=14,3 Гц, 1H), 2,60 (т, J=7,6 Гц, 2H), 1,64 (м, 5H), 0,94 (т, J=7,6 Гц, 3H).
MC (ESI, m/z): 340,1 (М++1).
Промежуточное соединение 9
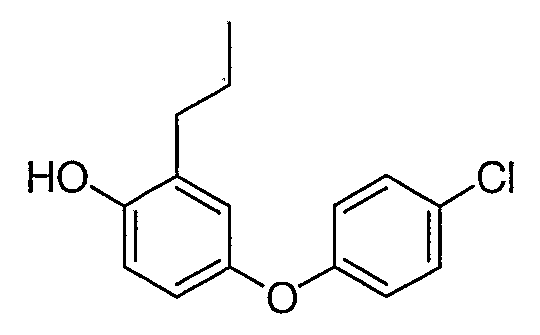
Стадия 1. Получение 4-хлорфеноксибензальдегида
Гетерогенную смесь 4-хлорфенола (14,1 г, 0,11 ммоль), 4-фторбензальдегида (12,4 г, 0,1 ммоль) и Cs2CO3 (65,0 г, 0,20 ммоль) в DMF (400 мл) перемешивали при 90°С в течение 6 часов. Реакционную смесь выливали в воду (1,2 л) и экстрагировали этилацетатом (2×200 мл). Органическую фазу промывали водой (2×100 мл), сушили и концентрировали, что приводило к получению фактически чистого 4-хлорфеноксибензальдегида, который немедленно использовали на следующей стадии.
Стадия 2. Получение 4-(4-хлорфенокси)фенола
Неочищенный альдегид со стадии 1 (23,3 г, 0,10 ммоль) растворяли в дихлорметане (500 мл) и добавляли м-хлорпербензойную кислоту (70%, 50,0 г, 0,20 ммоль) и бикарбонат натрия (25,2 г, 0,30 ммоль). Полученную в результате гетерогенную смесь перемешивали и нагревали с обратным холодильником 2 часа и затем обрабатывали водным раствором сульфита натрия (0,5М, 500 мл). После перемешивания при 25°С в течение 30 мин органическую фазу отделяли и водную фау экстрагировали дихлорметаном (2×200 мл). Объединенную органическую фазу промывали насыщенным раствором бикарбоната натрия (2×200 мл), сушили и концентрировали. Остаток хроматографировали на силикагеле, элюируя смесью 8:2 гексана и этилацетата, что приводило к получению указанного в заголовке фенола.
Стадия 3. Получение 3-[4-(4-хлорфенокси)фенокси]-1-пропена
Смесь фенола со стадии 2 (16,5 г, 75 ммоль), аллилбромида (10,8 г, 90 ммоль) и карбоната цезия (48,7 г, 150 ммоль) в DMF (300 мл) перемешивали при 25°С в течение 6 часов. Смесь выливали в воду (1,0 л) и экстрагировали этилацетатом (2×200 мл). Объединенную органическую фазу промывали водой (3×100 мл), сушили и концентрировали. Неочищенный продукт немедленно использовали на следующей стадии.
Стадия 4. Получение 4-(4-хлорфенокси)-2-(2-пропенил)фенола
Неочищенный аллильный эфир со стадии 3 (20,0 г) растворяли в 2,4,6-трихлорбензоле (60 мл) и раствор нагревали с обратным холодильником в течение 4 часов. После охлаждения до комнатной температуры раствор немедленно наносили на колонку с силикагелем и элюировали последовательно гексаном и смесью 8:2 гексана и этилацетата, что приводило к получению 4-(4-хлорфенокси)-2-(2-пропенил)фенола.
Стадия 5. Получение 4-(4-хлорфенокси)-2-пропилфенола
Смесь продукта со стадии 4 (15,7 г, 60 ммоль) и 10% Pd/C (3,1 г) в этилацетате (300 мл) перемешивали в атмосфере водорода (1 атм). После завершения реакции (приблизительно через 30 мин) смесь фильтровали через целит и фильтрат концентрировали, что приводило к получению промежуточного соединения 9 в виде масла, которое затвердевает при хранении.
1H ЯМР (500 МГц, CDCl3) δ 7,26 (д, J=8,5 Гц, 2H), 6,88 (д, J=8,5 Гц, 2H), 6,84 (м, 1H), 6,77 (м, 2H), 2,58 (т, J=7,5 Гц, 2H), 1,65 (м, 2H), 0,99 (т, J=7,5 Гц, 3H).
MC (ESI, m/z): 263,0 (М++1).
Промежуточное соединение 10
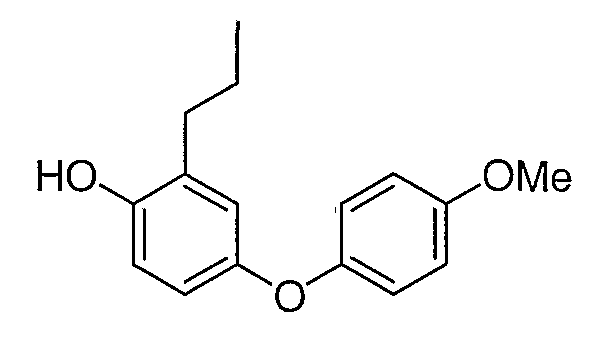
Промежуточное соединение 10 было получено согласно методике, аналогичной описанной для промежуточного соединения 9, стадии с 1 по 5, используя 4-метоксифенол вместо 4-хлорфенола на стадии 1.
Промежуточное соединение 11
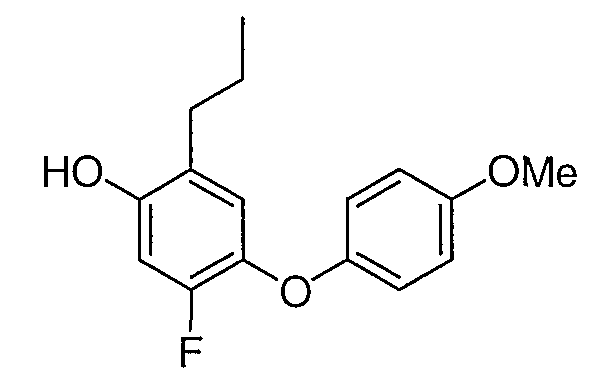
Промежуточное соединение 11 было получено согласно методике, аналогичной описанной для промежуточного соединения 9, стадии с 1 по 5, заменяя 4-метоксифенол на 4-хлорфенол и 3,4-дифторбензальдегид на 4-фторбензальдегид на стадии 1.
Пример 1
(5R)-5-{3-[4-(4-Хлорфенокси)-2-пропилфенокси]фенил}-5-метил-1,3-оксазолидин-2,4-дион
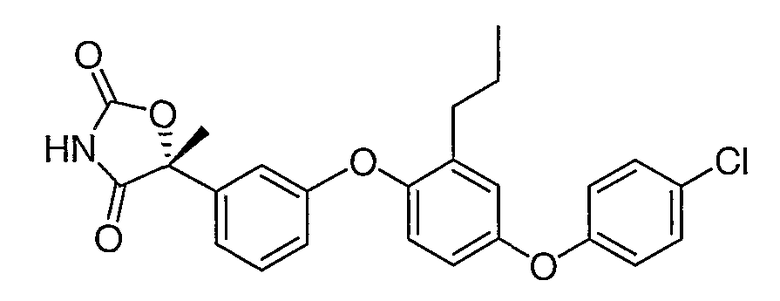
Стадия 1. Получение метил (2R)-2-{3-[4-(4-хлорфенокси)-2-пропилфенокси]фенил}-2-гидроксипропионат
Смесь промежуточного соединения 1 (2,6 г, 10 ммоль), промежуточного соединения 9 (3,9 г, 15 ммоль), ацетата палладия (90 мг, 0,04 ммоль), ди(трет-бутил)(2-бифенил)фосфина (179 мг, 0,06 ммоль) и фосфата калия (4,2 г, 20 ммоль) в толуоле (30 мл) дегазировали и нагревали в атмосфере N2 до 100°С в течение 16 часов. Реакционную смесь разбавляли эфиром (50 мл) и фильтровали через невысокий слой силикагеля, что приводило к получению неочищенного указанного в заголовке продукта, который немедленно использовался на следующей стадии.
Стадия 2. Получение (2R)-2-{3-[4-(4-хлорфенокси)-2-пропилфенокси]фенил}-2-гидроксипропионамида
Раствор неочищенного продукта со стадии 1 в метаноле (35 мл) охлаждали до 0°С и насыщали газообразным аммиаком. Раствор выдерживали при 25°С в течение 2 дней и затем концентрировали. Остаток хроматографировали на силикагеле, элюируя сначала смесью 3:7 этилацетат:гексан, и затем 100% этилацетатом. Этилацетатную фракцию концентрировали с получением продукта, указанного в заголовке.
Стадия 3. Получение(5R)-5-{3-[4-(4-хлорфенокси)-2-пропилфенокси]фенил}-5-метил-1,3-оксазолидин-2,4-диона
Амид со стадии 2 (3,0 г, 7,0 ммоль) растворяли в диэтилкарбонате (35 мл). 1,1'-Карбонилдиимидазол (3,4 г, 21 ммоль) и гидрид натрия (60% дисперсия в минеральном масле, 0,84 г, 21 ммоль) были последовательно добавлены. Полученную в результате реакционную смесь перемешивали при 50°С в течение 2 часов и выливали в ледяную воду. Водную смесь подкисляли концентрированной хлористоводородной кислотой до рН 2 и экстрагировали этилацетатом. Объединенную органическую фазу промывали солевым раствором, сушили и концентрировали. Остаток очищали с помощью хроматографии на силикагеле, элюируя смесью 3:7 этилацетат:гексан, содержащей 1% уксусной кислоты, что обеспечивало получение соединения, указанного в заголовке.
1H ЯМР (500 МГц, CDCl3) δ 8,40 (шир.с, 1H), 7,35 (т, J=8,4 Гц, 1H), 7,31 (д, J=8,5 Гц, 2H), 7,26 (м, 1H), 7,20 (т, J=2,5 Гц, 1H), 6,94-6,99 (м, 3H), 6,87-6,90 (м, 2H), 6,84 (дд, J=8,4, 2,5, 1H), 2,55 (т, J=7,2 Гц, 2H), 1,95 (c, 3H), 1,62 (м, 2H), 0,93 (т, J=7,2 Гц, 3H).
MC (ESI, m/z): 494,9 (М++1).
Пример 2
(5R)-5-{3-[4-(4-Метоксифенокси)-2-пропилфенокси]фенил}-5-метил-1,3-оксазолидин-2,4-дион
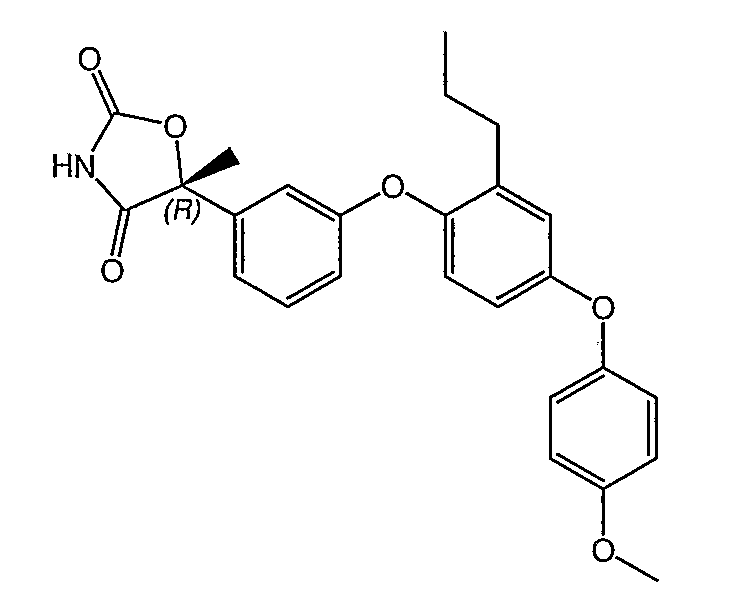
Указанное в заголовке соединение было получено согласно такой же методике, которая описана для примера 1, стадии с 1 по 3, заменяя промежуточное соединение 10 на промежуточное соединение 9 на стадии 1.
1H ЯМР (600 МГц, CD3OD) δ 7,26 (т, J=7,8 Гц, 1H), 7,19 (д, J=7,8 Гц, 1H), 7,07 (т, J=1,8 Гц, 1H), 6,94 (дд, J=8,4 Гц, 2,4 Гц, 1H), 6,93 (д, J=12,0 Гц, 2H), 6,91 (дд, J=7,2 Гц, 2,4 Гц, 1H), 6,83 (д, J=3,6 Гц, 1H), 6,82 (д, J=1,8 Гц, 1H), 6,74 (дд, J=8,4 Гц, 2,4 Гц, 1H), 6,72 (дд, J=8,0 Гц, 3,0 Гц, 1H), 3,77 (c, 3H), 2,47 (т, J=7,8 Гц, 2H), 1,69 (c, 3H), 1,54 (м, 2H), 0,87 (т, J=7,8 Гц, 3H).
MC (ESI, m/z): 447,9 (М+1).
Пример 3
(5R)-5-{3-[5-Фтор-4-(4-метоксифенокси)-2-пропилфенокси]фенил}-5-метил-1,3-оксазолидин-2,4-дион
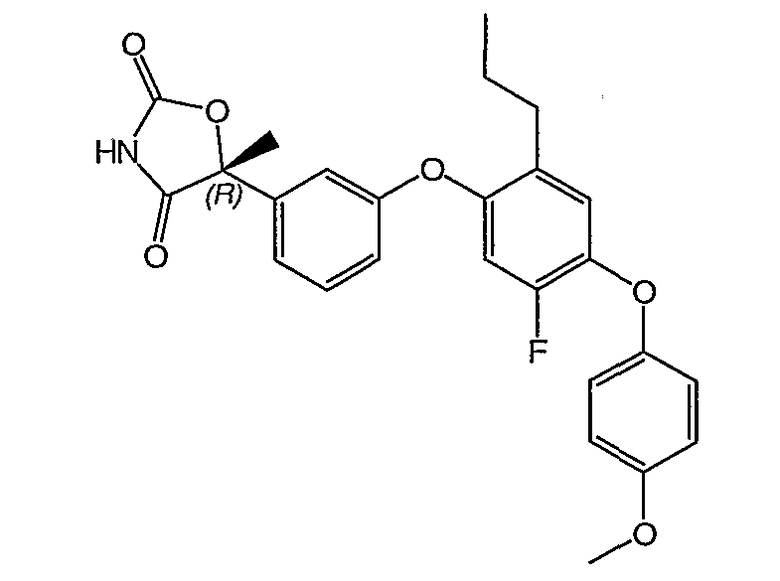
Указанное в заголовке соединение было получено согласно такой же методике, которая описана для примера 1, стадии с 1 по 3, заменяя промежуточное соединение 11 на промежуточное соединение 9 на стадии 1.
1H ЯМР (500 МГц, CD3OD) δ 7,44 (т, J=8,0 Гц, 1H), 7,27 (д, J=7,5 Гц, 1H), 7,11 (т, J=2,0 Гц, 1H), 6,97 (д, J=9,0 Гц, 2H), 6,96 (д, J=8,0 Гц, 1H), 6,941 (c, 1H), 6,940 (c, 1H), 6,93 (м, 1H), 6,83 (д, J=12 Гц, 1H), 3,80 (c, 3H), 2,50 (т, J=7,5 Гц, 2H), 1,88 (c, 3H), 1,55 (м, 2H), 0,88 (т, J=7,5 Гц, 3H).
MC (ESI, m/z): 465,0 (М+1).
Пример 4
(5R)-5-{3-[4-(2,4-Дихлорфенокси)-2-пропилфенокси]фенил}-5-метил-1,3-оксазолидин-2,4-дион
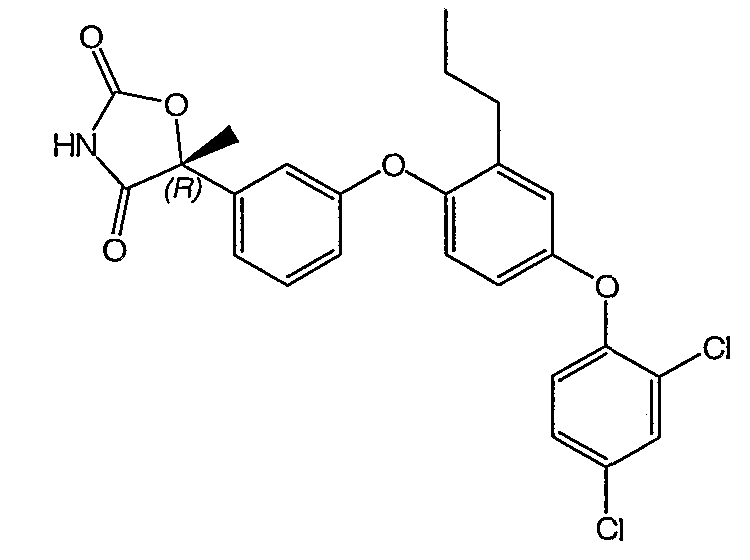
Стадия 1. Получение (2R)-5-[3-(4-ацетил-2-пропилфенокси)фенил]-5-метил-1,3-оксазолин-2,4-диона
Промежуточное соединение 4 (3,3 г, 10 ммоль) и ацетат натрия (2,5 г, 30 ммоль) растворяли в трифторметансульфоновой кислоте (30 мл). Получившийся в результате темно-оранжевый раствор перемешивали при 55°С в течение 1 часа. Реакционную смесь затем разбавляли этилацетатом и выливали медленно в лед. Органический слой отделяли и промывали последовательно солевым раствором и водным NaHCO3. Удаление растворителя приводило к получению неочищенного соединения, указанного в заголовке.
Стадия 2. Получение (2R)-5-[3-(4-гидрокси-2-пропилфенокси)фенил]-5-метил-1,3-оксазолин-2,4-диона
Смесь неочищенного продукта со стадии 1 (3,7 г, 10 моль), м-хлорпербензойной кислоты (70%, 4,9 г, 20 ммоль) и бикарбоната натрия (2,5 г, 30 ммоль) в дихлорметане (100 мл) нагревали с обратным холодильником 2 часа. Реакционную смесь выливали в водный сульфит натрия (2н, 100 мл) и экстрагировали дихлорметаном. Органическую фазу промывали насыщенным водным бикарбонатом натрия и концентрировали. Остаток растворяли в метаноле (50 мл) и обрабатывали гидроксидом калия (5н, 10 мл). Через 30 мин метанольный раствор подкисляли уксусной кислотой до рН 4 и концентрировали. Остаток очищали с помощью хроматографии на силикагеле, элюируя смесью 4:6 этилацетат:гексан, содержащей 1% уксусной кислоты, что обеспечивало получение соединения, указанного в заголовке.
Стадия 3. Получение (5R)-5-[3-(4-(2,4-дихлорфенокси)-2-пропилфенокси)фенил]-5-метил-1,3-оксазолидин-2,4-диона
Смесь продукта стадии 2 (0,34 г, 1,0 ммоль), 2,4-дихлорфенилбороновой кислоты (0,58 г, 3,0 ммоль), ацетата меди (0,27 г, 1,5 ммоль), триэтиламина (0,68 мл, 5,0 ммоль) и молекулярных сит 4Е (0,7 г) в дихлорметане (8 мл) перемешивали при 25°С на воздухе. Спустя 16 часов реакционную смесь разбавляли диэтиловым эфиром (24 мл) и фильтровали через невысокий слой силикагеля. Фильтрат концентрировали и остаток очищали препаративной ВЭЖХ с обращенной фазой, что и давало возможность получить соединение, указанное в заголовке.
1H ЯМР (500 МГц, CD3OD) δ 7,56 (д, J=2,5 Гц, 1H), 7,39 (т, J=8,0 Гц, 1H), 7,32 (дд, J=8,5 Гц, 2,5 Гц, 1H), 7,24 (м, 1H), 7,04 (т, J=2,0 Гц, 1H), 7,00 (д, J=9 Гц, 1H), 6,95 (д, J=4,0 Гц, 1H), 6,94 (д, J=1,5 Гц, 1H), 6,92 (м, 1H), 6,83 (дд, J=9 Гц, 3 Гц, 1H), 2,53 (т, J=7,5 Гц, 2H), 1,85 (c, 3H), 1,58 (м, 2H), 0,88 (т, J=7,5 Гц, 3H).
MC (ESI, m/z): 485,9 (М+1).
Пример 5
(5R)-5-{3-[4-(4-Хлорфенокси)-2-пропилфенокси]бензил}-5-метил-1,3-оксазолидин-2,4-дион
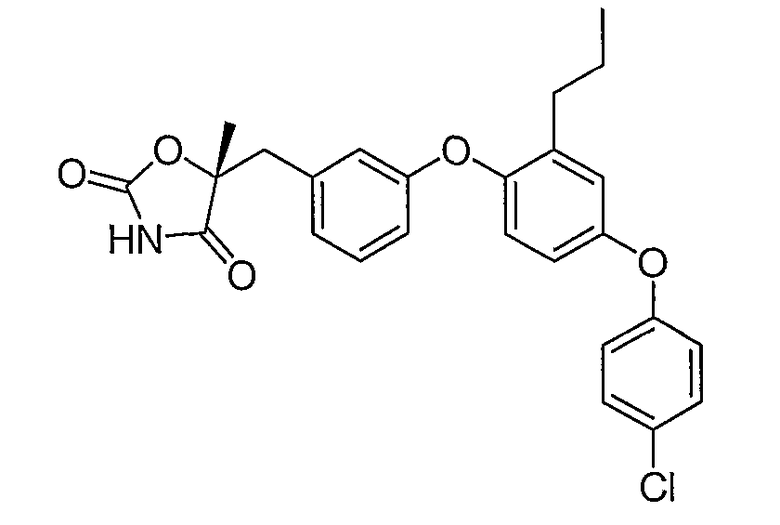
Стадия 1. Получение этил (2R)-3-[4-(4-хлорфенокси)-2-пропилфенил]-2-гидрокси-2-метилпропионата
Смесь промежуточного соединения 6 (3,6 г, 10 ммоль), промежуточного соединения 9 (3,9 г, 15 ммоль), ацетата палладия (90 мг, 0,04 ммоль), ди(трет-бутил)(2-бифенил)фосфина (179 мг, 0,06 ммоль) и фосфата калия (4,2 г, 20 ммоль) в толуоле (30 мл) дегазировали и нагревали в атмосфере N2 при 100°С в течение 16 часов. Реакционную смесь разбавляли эфиром (50 мл) и фильтровали через невысокий слой силикагеля с получением неочищенного указанного в заголовке продукта, который немедленно использовался на следующей стадии.
Стадия 2. Получение (2R)-3-[4-(4-хлорфенокси)-2-пропилфенил]-2-гидрокси-2-метилпропамида
Раствор неочищенного продукта со стадии 1 в метаноле (35 мл) охлаждали до 0°С и насыщали газообразным аммиаком. Раствор выдерживали в герметичном сосуде при 55°С 2 дня и затем концентрировали. Остаток хроматографировали на силикагеле, элюируя сначала смесью 3:7 этилацетат:гексан и затем 100% этилацетатом. Этилацетатную фракцию концентрировали, что приводило к получению соединения, указанного в заголовке.
Стадия 3. Получение (5R)-5-{3-[4-(4-хлорфенокси)-2-пропилфенокси]бензил}-5-метил-1,3-оксазолидин-2,4-диона
Амид со стадии 2 (2,7 г, 6,0 ммоль) растворяли в диэтилкарбонате (30 мл). 1,1'-Карбонилдиимидазол (2,9 г, 18 ммоль) и гидрид натрия (60% дисперсия в минеральном масле, 0,72 г, 18 ммоль) были последовательно добавлены. Полученную в результате реакционную смесь перемешивали при 50°С в течение 2 часов и выливали в ледяную воду. Водную смесь подкисляли концентрированной хлористоводородной кислотой до рН 2 и экстрагировали этилацетатом. Объединенную органическую фазу промывали солевым раствором, сушили и концентрировали. Остаток очищали с помощью хроматографии на силикагеле, элюируя смесью 3:7 этилацетат:гексан, содержащей 1% уксусной кислоты, что обеспечивало получение соединения, указанного в заголовке.
1H ЯМР (500 МГц, CDCl3) δ 7,69 (шир.с, 1H), 7,31 (м, 2H), 7,26 (м, 1H), 6,97 (дд, J=8,5, 2,5 Гц, 1H), 6,94 (д, J=2,5 Гц, 1H), 6,92 (д, J=7,5 Гц, 1H), 6,86 (м, 3H), 6,80 (м, 1H), 3,17 (д, J=14,3 Гц, 1H), 3,09 (д, J=14,3 Гц, 1H), 2,54 (т, J=7,6 Гц, 2H), 1,66 (c, 3H), 1,61 (м, 2H), 0,92 (т, J=7,6 Гц, 3H).
MC (ESI, m/z): 466,2 (М++1).
Пример 6
(5S)-5-{3-[4-(4-Хлорфенокси)-2-пропилфенокси]бензил}-5-метил-1,3-оксазолидин-2,4-дион
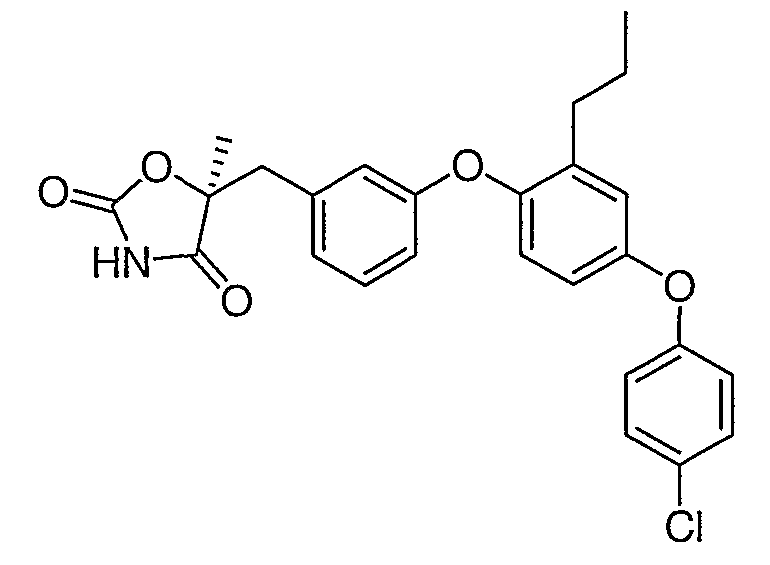
Указанное в заголовке соединение было получено согласно такой же методике, которая описана для примера 5, стадии с 1 по 3, заменяя промежуточное соединение 7 на промежуточное соединение 6 на стадии 1.
1H ЯМР (500 МГц, CDCl3) δ 7,69 (шир.c, 1H), 7,31 (м, 2H), 7,26 (м, 1H), 6,97 (дд, J=6,9, 2,3 Гц, 1H), 6,94 (д, J=2,5 Гц, 1H), 6,92 (д, J=7,5 Гц, 1H), 6,86 (м, 3H), 6,80 (м, 1H), 3,17 (д, J=14,3 Гц, 1H), 3,09 (д, J=14,3 Гц, 1H), 2,54 (т, J=7,6 Гц, 2H), 1,66 (c, 3H), 1,61 (м, 2H), 0,92 (т, J=7,3 Гц, 3H).
MC (ESI, m/z): 466,2 (М++1).
Пример 7
(5R)-5-{3-[4-(4-Метоксифенокси)-2-пропилфенокси]бензил}-5-метил-1,3-оксазолидин-2,4-дион
Указанное в заголовке соединение было получено согласно такой же методике, которая описана для примера 5, стадии с 1 по 3, заменяя промежуточное соединение 10 на промежуточное соединение 9 на стадии 1.
1H ЯМР (500 МГц, CD3OH) δ 7,17 (т, J=7,5, 1H), 6,95 (м, 5H), 6,80-6,85 (м, 3H), 6,74 (дд, J=7,5, 3,0 Гц, 1H), 6,71 (дд, J=8,0, 2,5 Гц, 1H), 3,80 (c, 3H), 3,01 (д, J=14,0 Гц, 1H), 2,95 (д, J=14,0 Гц, 1H), 2,50 (т, J=7,2 Гц, 2H), 1,57 (м, 2H), 1,44 (c, 3H), 0,88 (т, J=7,2 Гц, 3H).
MC (ESI, m/z): 462,0 (М++1).
Пример 8
(5R)-5-{3-{4-[4-(Трифторметокси)фенокси]-2-пропилфенокси}бензил}-5-метил-1,3-оксазолидин-2,4-дион
Стадия 1. Получение 5(R)-5-[3-(4-гидрокси-2-пропилфенокси)бензил]-5-метил-1,3-оксазолидин-2,4-диона
Указанное в заголовке соединение было получено согласно такой же методике, которая описана для примера 4, стадии с 1 по 2, заменяя промежуточное соединение 8 на промежуточное соединение 4 на стадии 1.
Стадия 2. Смесь продукта со стадии 1 (0,35 г, 1,0 ммоль), 4-(трифторметил)фенилбороновой кислоты (0,57 г, 3,0 ммоль), ацетата меди (0,27 г, 1,5 ммоль), триэтиламина (0,68 мл, 5,0 ммоль) и молекулярных сит 4Е (0,7 г) в дихлорметане (8 мл) перемешивали при 25°С на воздухе. Спустя 16 часов реакционную смесь разбавляли диэтиловым эфиром (24 мл) и фильтровали через невысокий слой силикагеля. Фильтрат концентрировали и остаток очищали препаративной ВЭЖХ с обращенной фазой, что и обеспечивало получение соединения, указанного в заголовке.
1H ЯМР (500 МГц, CDCl3) δ 7,12 (д, J=8,7 Гц, 2H), 7,06 (м, 1H), 6,93 (д, J=8,7 Гц, 2H), 6,84 (м, 3H), 6,73 (м, 2H), 6,62 (д, J=7,7 Гц, 1H), 2,87 (м, 2H), 2,44 (т, J=7,4 Гц, 2H), 1,50 (м, 2H), 1,23 (c, 3H),0,79 (т, J=7,2 Гц, 3H).
MCS (ESI, m/z): 516,1 (М++1).
Пример 9
(5R)-5-{3-[4-(4-Хлорфенокси)-2-(циклопропилметил)фенокси]бензил}-5-метил-1,3-оксазолидин-2,4-дион
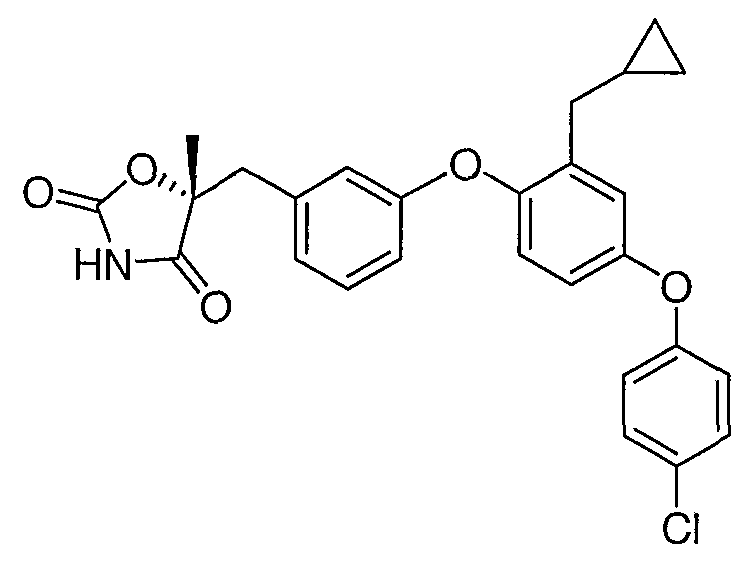
Стадия 1. Получение 5-(4-хлорфенокси)-2-фторбензальдегида
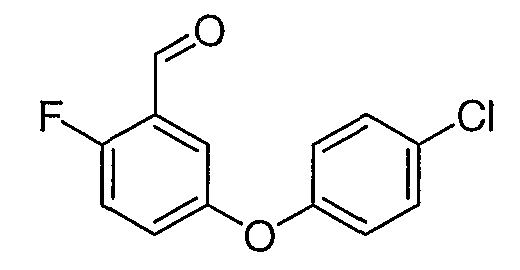
К раствору 2,2,6,6-тетраметилпиперидина (5,1 г, 40 ммоль) в ТГФ (150 мл), охлажденному до 0°С, добавляли н.-бутиллитий (1,6М в гексане, 25 мл, 40 ммоль). Полученный раствор охлаждали до -75°С и добавляли 4-хлор-4'-фтордифениловый эфир (4,5 г, 20 ммоль) в ТГФ (40 мл). После выдерживания при -75°С в течение 1,5 часов реакционную смесь обрабатывали диметилформамидом (4,4 г, 60 ммоль) и нагревали до 0°С. Смесь выливали в разбавленную хлористоводородную кислоту (0,5н, 200 мл) и экстрагировали этилацетатом (2×100 мл). Удаление растворителя и хроматорафирование на силикагеле, элюируя смесью 1:9 этилацетат:гексан, обеспечивало получение соединения, указанного в заголовке.
1H ЯМР (500 МГц, CDCl3) δ 10,38 (c, 1H), 7,45 (дд, J=8,5, 2,5 Гц, 1H), 7,35 (д, J=8,5 Гц, 2H), 7,28 (м, 1H), 7,21 (т, J=8,5 Гц, 1H), 6,95 (д, J= 2H).
Стадия 2. Получение метил (2R)-2-гидрокси-3-{3-[4-(4-хлорфенокси)-2-формилфенокси]фенил}-2-метилпропионат
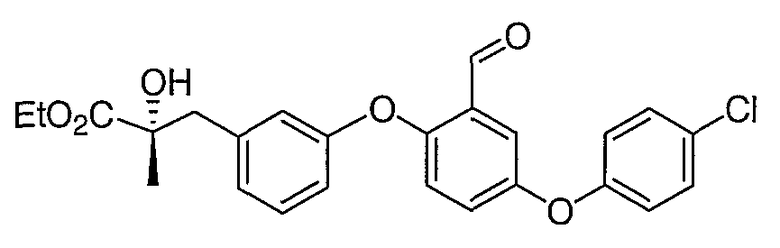
Смесь продукта со стадии 1 (2,2 г, 10 ммоль), промежуточного соединения 4 (2,5 г, 10 ммоль) и Cs2CO3 (5,9 г, 18 ммоль) в DMF (80 мл) нагревали при 85°С в течение 6 часов. Реакционную смесь выливали в воду (200 мл) и экстрагировали этилацетатом (2×100 мл). Удаление растворителя и хроматографирование остатка на силикагеле, элюируя смесью 2:8 этилацетат:гексан, приводило к получению продукта, указанного в заголовке.
Стадия 3. Получение (5R)-5-{3-[4-(4-хлорфенокси)-2-формилфенокси]бензил}-5-метил-1,3-оксазолидин-2,4-диона
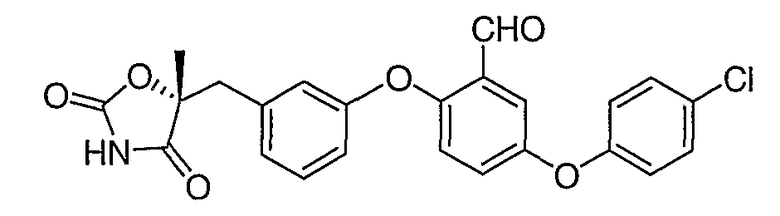
Продукт стадии 2 превращали в указанное в заголовке соединение согласно методике примера 5, стадии 2 и 3.
1H ЯМР (500 МГц, CDCl3) δ 10,38 (c, 1H), 8,20 (шир.с, 1H), 7,52 (д, J=2,5 Гц, 1H), 7,25-7,36 (м, 4H), 6,89-7,05 (м, 6H), 3,18 (д, J=14,0 Гц, 1H), 3,12 (д, J=14,0 Гц, 1H), 1,68 (c, 3H).
MC (ESI, m/z): 452,0 (М++1).
Стадия 4. Получение (5R)-5-{3-[4-(4-хлорфенокси)-2-(циклопропилметил)фенокси]бензил}-5-метил-1,3-оксазолидин-2,4-диона
К раствору продукта со стадии 3 (0,45 г, 1,0 ммоль) в ТГФ (10 мл) добавляли циклопропилметилмагнийбромид (1,0 М в Et2O, 2,5 мл, 2,5 ммоль). Реакционную смесь нагревали до 0°С и выливали в насыщенный раствор хлорида аммония (20 мл). Органическую фазу отделяли и водную фазу экстрагировали этилацетатом (2×10 мл). Объединенную органическую фазу промывали солевым раствором, сушили и концентрировали. Остаток растворяли в дихлорметане (5,0 мл), охлаждали до 0°С и обрабатывали триэтилсиланом (1,6 г, 10 ммоль) и трифторуксусной кислотой (0,23 мл, 3,0 ммоль). Через 30 мин при 0°С смесь медленно выливали в насыщенный водный раствор бикарбоната натрия (10 мл) и обрабатывали, как обычно. Неочищенный продукт очищали препаративной ВЭЖХ, что обеспечивало получение соединения, указанного в заголовке.
1H ЯМР (500 МГц, CD3OD) δ 7,23 (д, J=8,5 Гц, 2H), 7,06 (м, 2H), 6,89 (д, J=8,5, 2H), 6,82 (м, 2H), 6,73 (т, J=8,5 Гц, 1H), 6,71 (дд, J=8,5, 2,5 Гц, 1H), 6,61 (м, 1H), 2,86 (д, J=12,0 Гц, 1H), 2,82 (д, J=12,0 Гц, 1H), 2,39 (д, J=7,0 Гц, 2H), 1,22 (c, 3H), 0,85 (м, 1H), 0,38 (м, 2H), 0,04 (м, 2H).
MS (ESI, m/z): 478,0 (М++1).
Пример 10
(5R)-5-{3-[2-(Циклопропилметил)-4-(4-метоксифенокси)фенокси]фенил}-5-метил-1,3-оксазолидин-2,4-дион
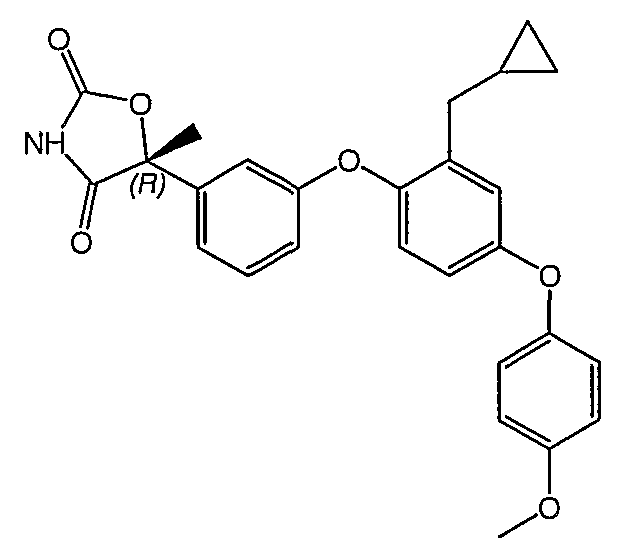
Указанное в заголовке соединение было получено согласно методике примера 9, заменяя 4-фтор-4'-метоксидифениловый эфир на 4-хлор-4'-фтордифениловый эфир на стадии 1 и промежуточное соединение 3 на промежуточное соединение 5 на стадии 2.
1H ЯМР (500 МГц, CD3OD) δ 7,28 (т, J=8,0 Гц, 1H), 7,27 (м, 1H), 7,07 (т, J=2,0 Гц, 1H), 7,00 (д, J=2,5 Гц, 1H), 6,97 (м, 2H), 6,94 (м, 2H), 6,85 (д, J=8,5 Гц, 1H), 6,77 (м, 1H), 6,75 (дд, J=8,5 Гц, 3,0 Гц, 1H), 3,79 (c, 3H), 2,43 (д, J=7,0 Гц, 2H), 1,70 (c, 3H), 0,92 (м, 1H), 0,42 (м, 2H), 0,10 (м, 2H).
MC (ESI, m/z): 460,1 (М+1).
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНДОЛЫ, ОБЛАДАЮЩИЕ ПРОТИВОДИАБЕТИЧЕСКОЙ АКТИВНОСТЬЮ | 2003 |
|
RU2328483C2 |
| НОВОЕ ПРОИЗВОДНОЕ ГЛЮЦИТОЛА, ЕГО ПРОЛЕКАРСТВО И ЕГО СОЛЬ И СОДЕРЖАЩИЙ ИХ ТЕРАПЕВТИЧЕСКИЙ АГЕНТ ДЛЯ ЛЕЧЕНИЯ ДИАБЕТА | 2005 |
|
RU2386631C2 |
| 1,3,4-ОКСАДИАЗОЛ-2-ОНЫ В КАЧЕСТВЕ МОДУЛЯТОРОВ PPAR-ДЕЛЬТА И ИХ ПРИМЕНЕНИЕ | 2005 |
|
RU2375358C2 |
| МОДУЛЯТОРЫ PPAR | 2007 |
|
RU2449999C2 |
| МОДУЛЯТОРЫ ROR-ГАММА | 2012 |
|
RU2658013C2 |
| КОНЪЮГИРОВАННЫЕ ЛИПИДНЫЕ ПРОИЗВОДНЫЕ | 2007 |
|
RU2480447C2 |
| ПРОИЗВОДНЫЕ СЕРИНА В КАЧЕСТВЕ АГОНИСТОВ ГРЕЛИНОВЫХ РЕЦЕПТОРОВ | 2015 |
|
RU2695649C2 |
| КОНДЕНСИРОВАННЫЕ АРОМАТИЧЕСКИЕ ДИФТОРМЕТАНФОСФОНАТЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНТИРОЗИНФОСФАТАЗЫ IB (PTP-1B) | 2008 |
|
RU2462469C2 |
| ПРОИЗВОДНЫЕ СПИРОКЕТАЛЕЙ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ЛЕКАРСТВЕННОГО СРЕДСТВА ПРОТИВ ДИАБЕТА | 2006 |
|
RU2416617C2 |
| АМИНОТЕТРАГИДРОПИРАНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ДИПЕПТИДИЛПЕПТИДАЗЫ-IV ДЛЯ ЛЕЧЕНИЯ ИЛИ ПРЕДУПРЕЖДЕНИЯ ДИАБЕТА | 2010 |
|
RU2550508C2 |
Изобретение относится к новым соединениям формулы I и их фармацевтически приемлемым солям. Соединения настоящего изобретения обладают свойствами PPAR-агониста. В общей формуле I

А представляет О; Х представляет связь или СН2; R1 выбирается из C1-С3алкила; каждый R4 независимо выбирается из группы, состоящей из галогена или -OC1-С3алкила, где -OC1-С3алкил может быть необязательно замещен 1-3 F; каждый R5 независимо выбирается из группы, состоящей из F, Cl; R6 выбирается из группы, состоящей из С2-С5алкила и -СН2циклопропила; m равно 0; n равно целому числу 1-2; р равно целому числу 0-1. Изобретение также относится к фармацевтической композиции, включающей в качестве активного ингредиента соединение изобретения. 2 н. и 9 з.п. ф-лы, 1 табл.
1. Соединение формулы I
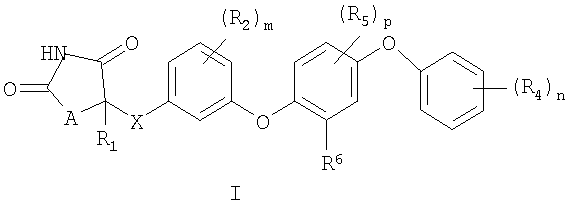
или его фармацевтически приемлемая соль, где
А представляет О;
Х представляет связь или СН2;
R1 выбирается из C1-С3алкила;
каждый R4 независимо выбирается из группы, состоящей из галогена или -OC1-С3алкила, где -OC1-С3алкил может быть необязательно замещен 1-3 F;
каждый R5 независимо выбирается из группы, состоящей из F, Cl;
R6 выбирается из группы, состоящей из С2-С5алкила и -СН2циклопропила;
m равно 0;
n равно целому числу 1-2;
р равно целому числу 0-1.
2. Соединение по п.1, в котором R1 представляет СН3.
3. Соединение по п.1, в котором каждый R4 независимо выбирается из группы, состоящей из F, Cl, -ОСН3, -OCF3, -OCHF2, -ОС2Н5, и n равно 1 или 2.
4. Соединение по п.1, в котором Х представляет связь.
5. Соединение по п.1, в котором Х представляет СН2.
6. Соединение по п.1, в котором R6 выбирается из группы, состоящей из н-С3Н7 и -СН2циклопропила.
7. Соединение по п.1, в котором R6 представляет н-С3Н7.
8. Соединение по п.1, в котором
R1 представляет СН3;
каждый R4 независимо выбирается из группы, состоящей из F, Cl, -ОСН3, -OCF3, -ОСН2СН3 и -OCHF2;
R5 представляет Cl или F;
R6 выбирается из группы, состоящей из н-С3H7, -СН2циклопропила;
m равно 0;
n равно 1 или 2; и
р равно 0 или 1.
9. Соединение по п.1, в котором
А представляет О;
R1 представляет СН3;
каждый R4 независимо выбирается из группы, состоящей из Cl, -ОСН3 и -OCF3;
R5 представляет F;
R6 представляет н-С3Н7;
m равно 0;
n равно 1 или 2; и
р равно 0 или 1.
10. Фармацевтическая композиция, обладающая свойствами PPAR-агониста, включающая в качестве активного ингредиента соединение по п.1, или его фармацевтически приемлемую соль, и фармацевтически приемлемый носитель.
11. Соединение по п.1, выбранное из перечисленных ниже соединений, или его фармацевтически приемлемая соль
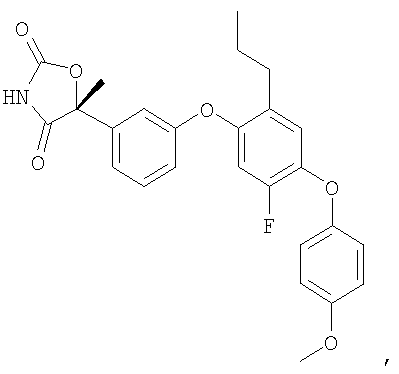
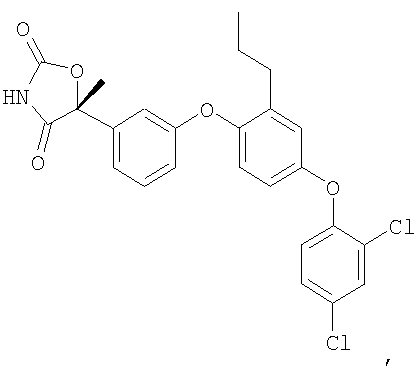
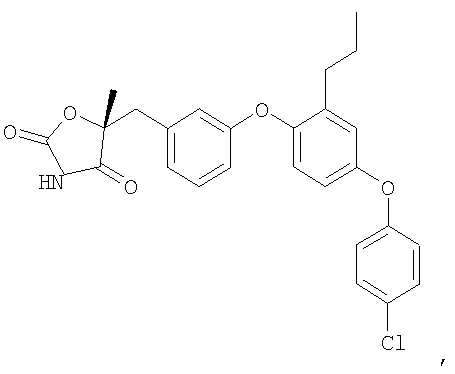
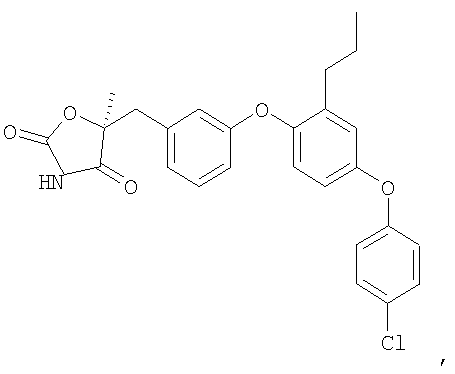
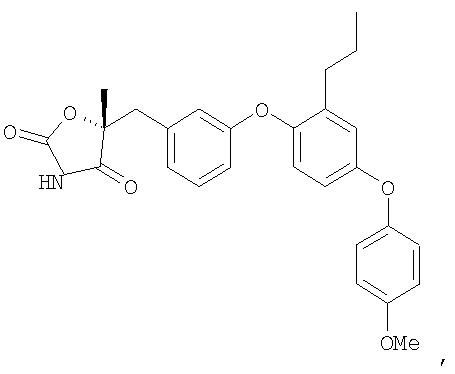
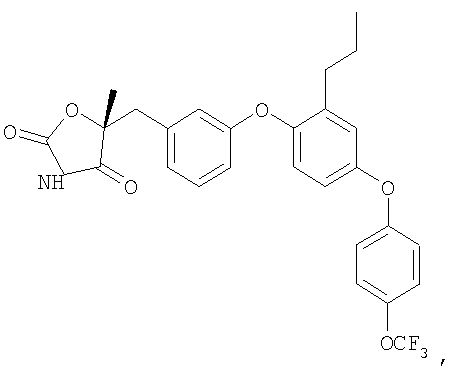

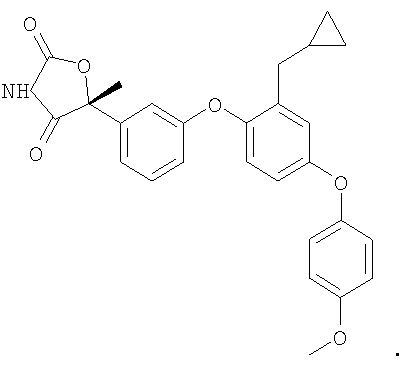
| Rami et al | |||
| Прибор для охлаждения жидкостей в зимнее время | 1921 |
|
SU1994A1 |
| ПРОИЗВОДНЫЕ 2,4-ОКСАЗОЛИДИНДИОНА ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМАЯ СОЛЬ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ЛЕКАРСТВЕННАЯ КОМПОЗИЦИЯ, ОКАЗЫВАЮЩАЯ ГИПОГЛИКЕМИЧЕСКОЕ И ГИПОЛИПИДЕМИЧЕСКОЕ ДЕЙСТВИЕ, СПОСОБ СНИЖЕНИЯ СОДЕРЖАНИЯ САХАРА И ЛИПИДОВ В КРОВИ ПРИ ЛЕЧЕНИИ МЛЕКОПИТАЮЩИХ, СТРАДАЮЩИХ ДИАБЕТОМ ИЛИ ГИПЕРЛИПИДИМИЕЙ | 1994 |
|
RU2126797C1 |
| WO 9932465 A1, 01.07.1999. | |||

