Область техники, к которой относится изобретение
Это изобретение относится к новому классу производных фосфоновой кислоты, которые являются ингибиторами PTP-1B и которые могут быть эффективными в лечении диабета типа 2 и других PTP-1B обусловленных заболеваний.
Уровень техники изобретения
Протеинтирозинфосфатазы представляют собой обширное семейство трансмембранных или внутриклеточных ферментов, которые дефосфорилируют субстраты, участвующие в разнообразных регуляторных процессах (Fischer et al., 1991, Science 253:401-406). Протеинтирозинфосфатаза-1B (PTP-1B) представляет собой ~50 кД внутриклеточный белок, присутствующий в достаточно больших количествах в разнообразных человеческих тканях (Charbonneau et al., 1989, Proc. Natl. Acad. Sci. USA 86:5252-5256; Goldstein, 1993, Receptor 3:1-15).
Многочисленные белки являются субстратами PTP-1B. Один такой важный субстрат представляет собой инсулиновый рецептор. Связывание инсулина с его рецептором в результате приводит к аутофосфорилированию рецептора, а именно, с наибольшей вероятностью, тирозинов 1146, 1150 и 1151 в киназном каталитическом домене (White & Kahn, 1994, J. Biol. Chem. 269:1-4). Это вызывает активацию инсулиновой рецепторной тирозинкиназы, которая фосфорилирует разнообразные инсулиновые рецепторные субстратные (IRS) белки, что распространяет сигнализирующее превращение инсулина далее на нижние уровни для опосредования различных биологических эффектов инсулина.
Kennedy et al., 1999, Science 283: 1544-1548 показал, что протеинтирозинфосфатаза PTP-1B представляет собой негативный регулятор инсулинового сигнального пути, предполагая, что ингибиторы этого фермента могут быть полезными в лечении диабета типа 2. Мыши, лишенные PTP-1B, являются устойчивыми как к диабету, так и к ожирению.
Дополнительное свидетельство в пользу применения ингибиторов PTP-1B для лечения диабета типа 2 и связанных с ним заболеваний было предоставлено посредством использования антисмысловых олигонуклиотидов, специфичных к PTP-1B, на животных моделях диабета типа 2. Ингибирование PTP-1B антисмысловыми олигонуклеотидами на животных моделях в результате привело к нормализации уровней глюкозы в крови и инсулина. Zinker et al., 2002, Proc. Natl. Acad. Sci. USA, 99: 11357.
Поэтому следует ожидать, что соединения, которые ингибируют PTP-1B, могут найти применение для лечения и/или контролирования диабета типа 2 и для улучшения толерантности к глюкозе для нуждающихся в этом пациентов. Также ожидается, что ингибиторы PTP-1B могут быть пригодными для сдерживающего действия на проявление диабета у преддиабетических пациентов и для предотвращения развития диабета у преддиабетических пациентов.
Также ингибиторы PTP-1B должны найти применение в лечении ожирения и дислипидемии. Лекарства для людей с целью лечения диабета посредством ингибирования PTP-1B до настоящего времени с достаточным успехом все еще не разработаны. Необходимы новые химические соединения, которые ингибируют PTP-1B.
Сверхэкспрессия и повышенные концентрации PTP-1B, зарегистрированные в случае некоторых линий злокачественных опухолей, включая хронический миелолейкоз (CML), рак молочной железы, рак яичников и рак предстательной железы, позволяют сделать предположение о регуляторной роли PTP-1B в контролировании киназной активности в этих и других раковых клетках. См., например, Liu, et al., J. Biol. Chem., 1996, 271: 31290-31295; Kenneth et al., Mol. Cell Biol., 1998, 18:2965-2975; Weiner et al., J. Natl. Cancer Inst., 1996, 86: 372-378. Таким образом, ингибирование PTP-1B активности может представлять собой важную мишень для лечения или предотвращения этих или других злокачественных опухолей. PTP-1B ингибиторы могут, таким образом, найти себе применение для лечения или предотвращения злокачественных опухолей и для замедления прогрессирования злокачественной опухоли, как только она обнаружена.
Исследования также дают возможность предположить, что PTP-1B ингибиторы могут найти применение для лечения или предотвращения нейродегенеративных заболеваний.
Сущность изобретения
Соединения, представленные формулой I, включая их фармацевтически приемлемые соли и их пролекарства, являются PTP-1B ингибиторами, которые могут найти применение для лечения диабета и сопутствующих медицинских показаний, а также могут найти применение для лечения других PTP-1B обусловленных заболеваний или патологических состояний.
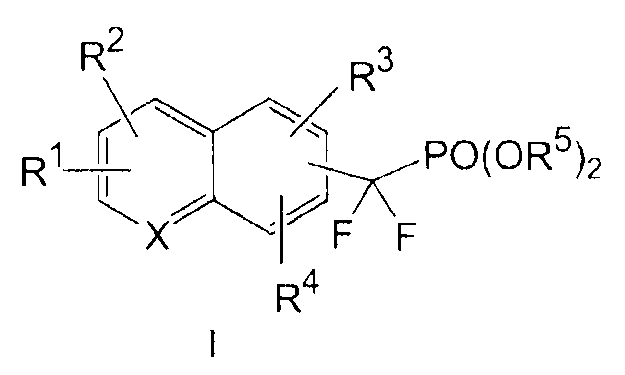
В соединениях формулы I
Х выбирают из СН и N;
R1 выбирают из группы, состоящей из (а) С1-3алкила, необязательно замещенного 1-3 галогенами и необязательно одной группой, выбранной из -ОН,
-ОС1-3алкила, необязательно замещенного 1-3 галогенами, -SOxC1-3алкила и -CN, (b) -C(=O)H, (c) -C(=O)С1-3алкила, необязательно замещенного 1-3 галогенами, (d)
-CN, (e) -HC=NOH, (f) -(CH3)C=NOH, (g) -HC=NOC1-3алкила, необязательно замещенного 1-3 галогенами, (h) -(CH3)C=NOC1-3алкила, необязательно замещенного 1-3 галогенами, (i) -C(=O)OC1-3алкила, необязательно замещенного 1-3 галогенами, (j) -C(=О)NHR6, (k) -CH=CH-фенила, в котором -CH=CH- необязательно замещен 1-2 заместителями, независимо выбираемыми из галогена и С1-2алкила, необязательно замещенного 1-3 F, (l) -CH2CH2-фенила, в котором -CH2CH2- необязательно замещен 1-4 заместителями, независимо друг от друга выбираемыми из галогена и С1-2алкила, необязательно замещенного 1-3 F, (m) фенила, (n) -HET-фенила, в котором HET представляет собой 5- или 6-членный ароматический гетероцикл, содержащий 1-3 гетероатомов, выбираемых из О, N и S, (o) -C≡С-фенила и (p) -CH2-фенила, в котором -CH2-группа в -CH2-фениле необязательно замещена 1-2 заместителями, независимо друг от друга выбираемыми из галогена и C1-2алкила, необязательно замещенного 1-3 F, где фенил и HET во всех случаях являются необязательно замещенными 1-3 заместителями, независимо друг от друга выбираемыми из (i) галогена, (ii) -C(=O)OC1-3алкила, необязательно замещенного 1-3 галогенами, (iii) -C(=O)OH, (iv) C1-3алкила, необязательно замещенного 1-3 галогенами, (v) -OC1-3алкила, необязательно замещенного 1-3 галогенами, (vi) -SOxMe и (vii) -SO2NH2;
R6 выбирают из группы, состоящей из Н, С1-3алкила, необязательно замещенного 1-3 галогенами, фенила и -CH2-фенила, в которых фенил в обоих случаях необязательно замещен 1-3 заместителями, независимо друг от друга выбираемыми из (i) галогена, (ii) -C(=O)OC1-3алкила, необязательно замещенного 1-3 галогенами, (iii) -C(=O)OH, (iv) C1-3алкила, необязательно замещенного 1-3 галогенами, и (v) -OC1-3алкила, необязательно замещенного 1-3 галогенами;
R2 и R4, независимо друг от друга, выбирают из H, галогена, -CH3, -CF3, -OCH3 и -OCF3;
R3 представляет собой галоген, в котором указанный галоген связан с конденсированным ароматическим циклом формулы I в ортоположении по отношению к -CF2PO(OR5)2 группе,
каждую R5 группу, независимо друг от друга, выбирают из группы, состоящей из H и C1-3алкила, необязательно замещенного 1-3 галогенами, и
x представляет собой 0, 1 или 2.
Способы лечения и контролирования диабета, ожирения и других заболеваний и патологических состояний, в которых применяют соединения формулы I, раскрыты в этом документе. Фармацевтические композиции и комбинированная терапия также раскрыты в данном описании.
Соединения, раскрытые в этом документе, представляют собой новый класс PTP-1B ингибиторов. Структура и название одного из соединений (пример 7B) раскрыты в двух публикациях, перечисленных ниже, в качестве PTP-1B ингибитора. Синтез соединения не был раскрыт в этих публикациях: (1) Montalibet et al., Biochemical Pharmacology, 2004, 68:1807-1814, (2) Montalibet et al., Journal of Biological Chemistry, 2006, 281, No. 8: 5258-5266.
Подробное описание изобретения
Соединения формулы I характеризуются многочисленными примерами осуществления изобретения, как описано ниже.
Изобретение включает соединения, как показано, а также (когда это возможно) индивидуальные диастереомеры, энантиомеры и эпимеры соединений и смеси диастереомеров и/или их энантиомеров, включая рацемические смеси. Несмотря на то, что конкретные стереохимические примеры, раскрытые в этом документе, являются предпочтительными, другие стереоизомеры, включая диастереомеры, энантиомеры, эпимеры, и их смеси также могут найти применение в лечении PTP-1B опосредованных заболеваний. Неактивные или менее активные диастереоизомеры и энантиомеры применимы для научных исследований, касающихся рецептора и механизма активации.
Изобретение также включает фармацевтически приемлемые соли соединений и фармацевтические композиции, включающие соединения и фармацевтически приемлемый носитель. Соединения являются главным образом пригодными для лечения резистентности к инсулину, диабета типа 2 и дислипидемии, которая вызвана диабетом типа 2 и резистентностью к инсулину. Соединения также пригодны для лечения ожирения. Они также пригодны для лечения определенных видов злокачественной опухоли и для замедления развития злокачественной опухоли, если она обнаружена у пациента. Они также пригодны для лечения, предотвращения или замедления развития нейродегенеративных заболеваний.
Соединения, раскрытые в этом документе, могут применяться в фармацевтических композициях, включающих (a) соединение(я) или их фармацевтически приемлемые соли и (b) фармацевтически приемлемый носитель. Соединения могут применяться в фармацевтических композициях, которые включают один или несколько других активных фармацевтических ингредиентов. Соединения также могут применяться в фармацевтических композициях, в которых соединение формулы I или его фармацевтически приемлемая соль представляет собой единственный активный ингредиент.
Соединение формулы I или его фармацевтически приемлемая соль может применяться в приготовлении лекарственного средства для лечения сахарного диабета типа 2 у людей или других пациентов, относящихся к млекопитающим.
Способ лечения диабета типа 2 включает введение терапевтически эффективного количества соединения формулы I или его фармацевтически приемлемой соли, или фармацевтической композиции, включающей соединение, пациенту, нуждающемуся в лечении. Другие медицинские применения соединений формулы I описываются в дальнейшем в этом документе.
Сокращения
Сокращения и термины, которые обычно используются в областях органической химии, медицинской химии, фармакологии и медицины, и которые хорошо известны практикующим специалистам в этих областях, применяются в данном документе. Типичные сокращения и определения представлены ниже.
Ac обозначает ацетил [CH3C(O)-]; Ac2O - ацетангидрид;
9-BBN - 9-борабицикло[3.3.1]нонан; Bn - бензил; BOC - трет-бутилоксикарбонил; DIAD - диизопропилазодикарбоксилат; DIBAL - диизобутилалюминийгидрид; DMF - N,N-диметилформамид; DMSO - диметилсульфоксид; EDAC (или EDC) - l-этил-3-[3-(диметиламино)пропил]-карбодиимид HCl; Et3N - триэтиламин; Et - этил; EtOAc - этилацетат; EtOH - этанол; 3-F-Ph - 3-фторфенил, HCl - хлористоводородную кислоту; HOBt - 1-гидроксибензотриазол; HPLC - высокоэффективную жидкостную хроматографию (ВЭЖХ); LCMS - ВЭЖХ с масс-спектрометрической детекцией; LG - уходящую группу; M - молярную концентрацию; ммоль - миллимоль; Me - метил; MeOH - метанол; MsCl метансульфонилхлорид; N - нормальность; NaHMDS - гексаметилдисилазид натрия; NaOAc - ацетат натрия; NaOtBu - трет-бутоксид натрия; NMO - N-метилморфолин-N-оксид; NMP - N-метилпирролидинон; Pd(dba)2 - трис(дибензилиденацетон)дипалладий; PdCl2(Ph3P)2 - дихлор-бис-(трифенилфосфин)палладий; PG обозначает неспецифическую защитную группу; Ph - фенил; PhMe - толуол;
PPh3 - трифенилфосфин; PMB - пара-метоксибензил; RT - комнатную температуру; TBAF - тетрабутиламмонийфторид; TBS - трет-бутилдиметилсилил; tBu - трет-бутил; Tf - трифлат; TFA - трифторуксусную кислоту; THF - тетрагидрофуран; TLC - тонкослойную хроматографию (ТСХ); TMS - триметилсилил; TPAP - тетрапропиламмонийперрутенат.
Определения
"Ac" обозначает ацетил, который представляет собой CH3C(=O)-.
"Алкил" обозначает насыщенные углеродные цепи, которые могут быть линейными или разветвлеными, или представлять собой их комбинацию, за исключением тех случаев, когда углеродная цепь определена иначе. Другие группы, имеющие префикс "алк", такие как алкокси и алканоил, также могут быть линейными или разветвлеными, или представлять собой их комбинацию, за исключением тех случаев, когда углеродная цепь определена иначе. Примеры алкильных групп включают метил, этил, пропил, изопропил, бутил, втор- и трет-бутил, пентил, гексил, гептил, октил, нонил и им подобные.
"Алкенил" обозначает углеродные цепи, которые содержат, по крайней мере, одну углерод-углеродную двойную связь и которые могут быть линейными или разветвленными, или представлять собой их комбинацию. Примеры алкенила включают винил, аллил, изопропенил, пентенил, гексенил, гептенил, 1-пропенил, 2-бутенил, 2-метил-2-бутенил и им подобные.
"Алкинил" обозначает углеродные цепи, которые содержат, по крайней мере, одну углерод-углеродную тройную связь и которые могут быть линейными или разветвленными, или представлять собой их комбинацию. Примеры алкинила включают этинил, пропаргил, 3-метил-l-пентинил, 2-гептинил и им подобные.
"Циклоалкил" обозначает насыщенный углеродный цикл, имеющий конкретное указанное количество углеродных атомов. Термин также может применяться для описания углеродного цикла, конденсированного с арильной группой. Примеры циклоалкила включают циклопропил, циклопентил, циклогексил, циклогептил и им подобные. Циклоалкенильные кольца включают двойную связь в цикле.
"Арил" обычно применяется как имеющий отношение к карбоциклическим ароматическим структурам. Наиболее распространенные арильные группы представляют собой фенил и нафтил. Фенил в большинстве случаев представляет собой наиболее предпочтительную арильную группу.
"Гетероцикл" обозначает насыщенное или частично ненасыщенное кольцо или циклическую систему, содержащие, по крайней мере, один гетероатом, выбираемый из N, S и O, для которых количество гетероатомов и размер цикла, и степень ненасыщенности (если таковая имеется) охарактеризованы в этом документе. Примеры гетероциклов включают тетрагидрофуран, пиперазин, пиперидин и морфолин.
"Гетероарил" обозначает гетероароматическое кольцо, содержащее, по крайней мере, один кольцевой гетероатом, выбираемый из N, O и S (включая SO и SO2), как охарактеризовано более конкретно в этом документе. Примеры гетероарила включают пирролил, изоксазолил, изотиазолил, пиразолил, пиридил, оксазолил, оксадиазолил, тиадиазолил, тиазолил, имидазолил, триазолил, тетразолил, фуранил, триазинил, тиенил, пиримидил, пиридазинил, пиразинил, бензизоксазолил, бензоксазолил, бензотиазолил, бензимидазолил, бензофуранил, бензотиофенил (включая S-оксид и диоксид), фуро-(2,3-b)-пиридил, хинолил, индолил, изохинолил, хиназолинил, дибензофуранил и им подобные.
"Галоген" включает фтор, хлор, бром и иод.
"Me" представляет собой метил.
Словосочетание "фармацевтически приемлемые" употребляется в этом документе как относящееся к таким соединениям, материалам, композициям, солям и/или дозированным лекарственным формам, которые при использовании в случае обоснованного медицинского заключения и при следовании всем надлежащим нормативным документам являются безопасными и пригодными для введения человеку или животному.
Термин "композиция", как в фармацевтической композиции, предназначается для охвата продукта, включающего активный(е) ингредиент(ы) и инертный(е) ингредиент(ы), который(ые) укомплектован(ы) носителем, а также продукта, который является прямым или косвенным результатом комбинации, комплексообразования или агрегирования каких-либо двух или нескольких ингредиентов, или образуется в результате диссоциации одного или нескольких ингредиентов, или является результатом других типов реакций или взаимодействий одного или нескольких ингредиентов. Таким образом, фармацевтические композиции настоящего изобретения охватывают любые композиции, полученные путем добавления в смесь соединения по настоящему изобретению и фармацевтически приемлемого носителя.
Заместитель "тетразол" обозначает 2H-тетразол-5-ил заместительную группу и ее таутомеры.
Оптические изомеры - диастереомеры - геометрические
изомеры - таутомеры
Соединения формулы I могут содержать один или несколько асимметрических центров и, таким образом, могут существовать в виде рацематов, рацемических смесей, одиночных энантиомеров, индивидуальных диастереомеров и смесей диастереомеров и/или энантиомеров. Изобретение предназначено охватить все подобные изомерные формы соединений формулы I. А именно соединения непосредственно этого изобретения имеют, по крайней мере, три асимметрических центра. Дополнительные асимметрические центры могут присутствовать в зависимости от природы различных заместителей в молекуле. Подразумевается, что все возможные оптические изомеры, стереоизомеры и диастереомеры в смесях и в качестве чистых или частично очищенных соединений включены в объем этого изобретения (т.e. все возможные комбинации асимметрических центров как чистых соединений, так и смесей).
Некоторые из соединений, описанных в этом документе, могут содержать олефиновые двойные связи, и, за исключением особо оговоренных случаев, подразумевается, что они включают как E, так и Z геометрические изомеры.
Некоторые из соединений, описанных в этом документе, могут существовать как соединения с различными позициями присоединения водорода, именуемые как таутомеры. Примером является кетон и его енольная форма, известные как кето-енольные таутомеры. Индивидуальные таутомеры, так же как и их смеси охватываются соединениями формулы I.
Соединения формулы I, имеющие один или несколько асимметрических центров, могут быть разделены на диастереомеры, энантиомеры и им подобные способами, которые хорошо известны из уровня техники.
Альтернативно, энантиомеры и другие соединения с хиральными центрами могут быть синтезированы путем стереоспецифического синтеза с использованием оптически чистых исходных веществ и/или реагентов известной конфигурации.
Соли
Термин "фармацевтически приемлемые соли" относится к солям, полученным из фармацевтически приемлемых нетоксичных оснований или кислот, включая неорганические или органические основания и неорганические или органические кислоты. Соли, полученные из неорганических оснований, включают соли алюминия, аммония, кальция, меди, трехвалентного железа, двухвалентного железа, лития, магния, трехвалентного марганца, двухвалентного марганца, калия, натрия, цинка и им подобные. В особенности предпочтительными являются соли аммония, кальция, магния, калия и натрия. Соли в твердой форме могут существовать в нескольких кристаллических структурах, а также могут быть в форме гидратов. Соли, полученные из фармацевтически приемлемых органических нетоксичных оснований, включают соли первичных, вторичных и третичных аминов, замещенных аминов, включая встречающиеся в природе замещенные амины, циклических аминов и основных ионообменных смол, таких как аргинин, бетаин, кофеин, холин, N,N'-дибензилэтилендиамин, диэтиламин, 2-диэтиламиноэтанол, 2-диметиламиноэтанол, этаноламин, этилендиамин, N-этилморфолин, N-этилпиперидин, глюкамин, глюкозамин, гистидин, гидрабамин, изопропиламин, лизин, метилглюкамин, морфолин, пиперазин, пиперидин, полиаминные смолы, прокаин, пурины, теобромин, триэтиламин, триметиламин, трипропиламин, трометамин и им подобные.
Если соединение настоящего изобретения является основным или если оно имеет в структуре основные заместительные группы, то соли могут быть получены из фармацевтически приемлемых нетоксичных кислот, включая неорганические и органические кислоты. Такие кислоты включают уксусную, бензолсульфоновую, бензойную, камфорсульфоновую, лимонную, этансульфоновую, фумаровую, глюконовую, глутаминовую, бромистоводородную, хлористоводородную, изэтиновую, молочную, малеиновую, яблочную, миндальную, метансульфоновую, муциновую, азотную, памовую, пантотеновую, фосфорную, янтарную, серную, виноградную, p-толуолсульфоновую кислоты и им подобные. В особенности предпочтительными являются лимонная, бромистоводородная, хлористоводородная, малеиновая, фосфорная, серная и виноградная кислоты.
Следует понимать, что при использовании в этом документе ссылки на соединения формулы I обозначают то, что они также включают фармацевтически приемлемые соли.
Метаболиты - пролекарства
Изобретение включает терапевтически активные метаболиты, в тех случаях, когда метаболиты сами по себе попадают под действие объема формулы изобретения. Изобретение также включает пролекарства, которые представляют собой соединения, которые превращены в заявленные соединения, поскольку они представляют собой соединения при их введении пациенту или после введения пациенту. Заявленные химические структуры для этих применений в некоторых случаях сами по себе могут быть пролекарствами.
Полезность
Соединения, а именно представленные в этом документе, проявляют высокую эффективность в ингибировании PTP-1B фермента, как это показано при их испытаниях in vitro. Соединения обычно имеют величину IC50 менее чем 2 мкМ при испытании с ферментом, описанным в разделе "Испытания", и, предпочтительно, имеют величину IC50 менее чем 1 мкМ.
Ингибиторы PTP-1B улучшают чувствительность к инсулину и могут найти применение в предотвращении или лечении диабета, улучшая толерантность к глюкозе и чувствительность к инсулину, если существует инсулиноустойчивость, и при лечении или предотвращении ожирения, - все это касается млекопитающих, включая людей, которые нуждаются в таких лечениях или которые получают облегчение при таких лечениях. Соединения являются наиболее широко применимыми в лечении диабета типа 2 (инсулинонезависимого диабета, или NIDDM). Соединения могут также вызывать лечебное восстановление нормальных показателей по триглицеридам и липидам.
Соединения, которые ингибируют PTP-1B, также могут быть полезными в лечении, предотвращении или контроле ряда патологических состояний, которые сопутствуют диабету типа 2, включая гиперлипидемию, гипертриглицеридемию, гиперхолестеринемию (включая существенное повышение недостаточных уровней HDL), атеросклероз, сосудистый рестеноз, панкреатит, опухоли жировых клеток, карциномы жировых клеток, такие как липосаркома, дислипидемия, воспалительные заболевания кишечника, воспаление в целом и другие заболевания, при которых резистентность к инсулину является составляющим компонентом.
Ожидается, что соединения будут эффективны в снижении уровня глюкозы и липидов у страдающих диабетом пациентов и у не болеющих диабетом пациентов, которые имеют нарушенную толерантность к глюкозе и/или находятся в преддиабетическом состоянии. Соединения могут способствовать улучшению при гиперинсулинемии, которая часто возникает у страдающих диабетом или преддиабетических пациентов, посредством регулирования колебаний уровня сывороточной глюкозы, которые часто случаются у этих пациентов. Соединения также могут быть эффективными в лечении или восстановлении нормального показателя резистентности к инсулину. Соединения могут быть эффективными в лечении или предотвращении гестационного диабета.
Соединения, композиции и лекарственные препараты, как описано в этом документе, также могут быть эффективными в снижении рисков вредных осложнений, связанных с метаболическим синдромом, и в снижении риска развития атеросклероза, замедляя проявление атеросклероза и/или снижая риск осложнений атеросклероза. Осложнения атеросклероза включают стенокардию, перемежающуюся хромоту, инфаркт миокарда, инсульт и другие.
Посредством обеспечения контроля над гипергликемией соединения также могут быть эффективными при замедлении или предотвращении сосудистого рестеноза и диабетической ретинопатии.
Соединения этого изобретения также могут найти применение для улучшения или восстановления β-клеточной функции, поэтому они могут быть пригодны для лечения диабета типа 1 или для замедления или предотвращения у пациентов диабета типа 2 при необходимости инсулинотерапии.
Сверхэкспрессия и повышенные концентрации PTP-1B, зарегистрированные в случае некоторых линий злокачественных опухолей, включая хронический миелолейкоз (CML), рак молочной железы, рак яичников и рак предстательной железы, наводят на мысль о регуляторной роли PTP-1B в контролировании киназной активности в этих и других раковых клетках. Таким образом, ингибирование PTP-1B активности может представлять собой важную мишень для лечения или предотвращения этих и других злокачественных опухолей. Соединения могут, таким образом, найти применение для лечения или предотвращения злокачественных опухолей, таких как рак предстательной железы, рак молочной железы, рак яичников, множественная миелома, лейкемия, меланома, лимфома, ренальный рак и рак мочевого пузыря.
Соединения могут также найти применение для лечения нейродегенеративных заболеваний.
Соединения обычно являются эффективными при лечении одного или нескольких следующих заболеваний: (1) диабет типа 2 (также известный как инсулинонезависимый сахарный диабет или NIDDM), (2) гипергликемия, (3) нарушенная толерантность к глюкозе, (4) резистентность к инсулину, (5) ожирение, (6) заболевания, связанные с нарушением липидного метаболизма, (7) смешанная или диабетическая дислипидемия, (8) гиперлипидемия, (9) гипертриглицеридемия, (10) гиперхолестеринемия, (11) пониженное содержание холестерина липопротеинов высокой плотности (HDL), (12) повышенное содержание холестерина липопротеинов низкой плотности (LDL), (13) гипер-апо-бета-липопротеинемия, (14) атеросклероз и его осложнения, (14) сосудистый рестеноз, (15) абдоминальное ожирение, (16) ретинопатия, (17) метаболический синдром, (18) высокое кровяное давление, (19) резистентность к инсулину, (19) злокачественная опухоль и (20) нейродегенеративные заболевания.
Один аспект изобретения предоставляет способ лечения и контроля смешанной или диабетической дислипидемии, гиперхолестеринемии, атеросклероза, низких уровней HDL, высоких уровней LDL, гиперлипидемии, и/или гипертриглицеридемии, который включает введение пациенту, нуждающемуся в таком лечении, терапевтически эффективного количества соединения, имеющего формулу I. Соединение может применяться отдельно или, предпочтительно, может быть введено с ингибитором биосинтеза холестерина, в особенности с ингибитором HMG-CoA редуктазы, таким как ловастатин, симвастатин, розувастатин, правастатин, флувастатин, аторвастатин, ривастатин или итавастатин. Соединение также может применяться, предпочтительно, в комбинации с другими понижающими уровень липидов лекарственными средствами, такими как ингибиторы абсорбции холестерина (например, сложные эфиры станола, стеролгликозиды, такие как тиквесид (tiqueside), и азетидиноны, такие как эзетимиб), ACAT ингибиторы (такие как авасимиб), CETP ингибиторы (например, торсетрапиб и такие, которые описаны в опубликованных заявках WO2005/100298, WO2006/014413 и WO2006/014357), ниацин и агонисты ниациновых рецепторов, секвестранты желчных кислот, ингибиторы микросомального триглицеридного транспорта и ингибиторы обратного захвата желчной кислоты. Эти комбинированные лечения могут быть эффективными для лечения или контроля одного или нескольких взаимосвязанных патологических состояний, включающих гиперхолестеринемию, атеросклероз, гиперлипидемию, гипертриглицеридемию, дислипидемию, повышенный уровень LDL и пониженный уровень HDL.
Соединения формулы I или их фармацевтически приемлемые соли могут применяться в способах лечения одного или нескольких вышеперечисленных заболеваний посредством введения терапевтически эффективного количества соединения пациенту, нуждающемуся в лечении. Соединения формулы I или их фармацевтически приемлемые соли также могут применяться при приготовлении лекарственных средств для лечения одного или нескольких перечисленных заболеваний.
Введение и диапазоны дозировки
Любой удобный путь введения может применяться для обеспечения млекопитающего, в частности человека, эффективной дозой соединения согласно настоящему изобретению. Например, могут применяться пероральный, ректальный, топический, парентеральный, внутриглазной, пульмональный, назальный и им подобные пути введения. Лекарственные формы включают таблетки, пастилки, дисперсии, суспензии, растворы, капсулы, кремы, мази, аэрозоли и им подобные. Предпочтительно соединения формулы I вводят перорально.
Применяемая эффективная доза активного ингредиента может изменяться в зависимости от применяемого конкретного соединения, способа введения, подлежащего лечению патологического состояния и от тяжести подлежащего лечению патологического состояния. Такая доза может быть легко установлена средним специалистом в данной области.
При лечении или контролировании сахарного диабета и/или гипергликемии, или гипертриглицеридемии, или других заболеваний, для которых соединения формулы I показаны, обычно удовлетворительные результаты получают, когда соединения настоящего изобретения вводят в дневной дозе, составляющей от приблизительно 0,1 миллиграмма до приблизительно 100 миллиграммов на килограмм массы тела животного, предпочтительно даваемой в виде однократной дневной дозы или в виде раздельных доз от двух до шести раз в день, или в виде лекарственной формы с замедленным высвобождением. Для большинства крупных млекопитающих суммарная дневная доза составляет от приблизительно 1,0 миллиграмма до приблизительно 1000 мг. В случае взрослого человека весом 70 кг суммарная дневная доза, как правило, будет составлять от приблизительно 1 миллиграмма до приблизительно 500 миллиграммов. Для особенно сильнодействующего соединения доза для взрослого человека может быть ниже чем 0,1 мг. В некоторых случаях дневная доза может быть выше чем 1,0 г. Дозировка по схеме лечения может быть скорректирована в рамках этого диапазона или даже может выйти за пределы этого диапазона для обеспечения оптимальной терапевтической восприимчивости.
Пероральное введение обычно осуществляют посредством применения таблеток или капсул. Примеры доз в таблетках и капсулах представляют собой 0,1 мг, 0,25 мг, 0,5 мг, 1 мг, 2 мг, 5 мг, 10 мг, 25 мг, 50 мг, 100 мг, 200 мг, 300 мг, 400 мг, 500 мг и 750 мг. Другие пероральные формы также могут содержать такие же или подобные дозы. Эти таблетки и капсулы можно принимать один раз в день, два раза в день, три раза в день или четыре раза в день. Прием один раз в день, как правило, предпочтителен.
Фармацевтические композиции
Другой аспект настоящего изобретения предоставляет фармацевтические композиции, которые включают соединение формулы I и фармацевтически приемлемый носитель. Фармацевтические композиции настоящего изобретения включают соединение формулы I или фармацевтически приемлемую соль в качестве активного ингредиента, а также фармацевтически приемлемый носитель и, необязательно, другие терапевтические ингредиенты. Термин "фармацевтически приемлемые соли" относится к солям, полученным из фармацевтически приемлемых нетоксичных оснований или кислот, включая неорганические основания или кислоты и органические основания или кислоты. Фармацевтическая композиция также может включать пролекарство или его фармацевтически приемлемую соль, если пролекарство вводится.
Композиции включают композиции, пригодные для перорального, ректального, топического, парентерального (включая подкожный, внутримышечный и внутривенный), внутриглазного (офтальмологического), пульмонального (назальная или внутриротовая ингаляция) или назального введения, хотя наиболее удобный способ применения в любом случае будет зависеть от природы и тяжести патологических состояний, подлежащих лечению, и от природы активного ингредиента. Они (композиции) могут быть удобно представлены в дозированной лекарственной форме и приготовлены любыми способами, хорошо известными из уровня техники в области фармацевтики.
При практическом использовании соединения формулы I могут быть скомбинированными в качестве активного ингредиента в однородной смеси с фармацевтическим носителем, в соответствии со стандартными фармацевтическими технологиями сочетания компонентов. Носитель может быть выбран из широкого разнообразия типов, в зависимости от лекарственной формы препарата, необходимого для введения, например, оральной или парентеральной (включая внутривенную). При приготовлении композиций в виде пероральной дозированной формы может применяться любая традиционная фармацевтическая среда, такая как, например, вода, гликоли, масла, спирты, ароматизирующее вещество, консерванты, окрашивающее вещество и им подобные в случае пероральных жидких препаратов, таких как, например, суспензии, эликсиры и растворы; или носители, такие как крахмалы, сахара, микрокристаллическая целлюлоза, разбавители, гранулирующие агенты, скользящие вещества, связующие вещества, дезинтегрирующие вещества и им подобные в случае пероральных твердых препаратов, таких как, например, порошки, твердые и мягкие капсулы и таблетки, при этом твердые пероральные препараты являются более предпочтительными по сравнению с жидкими пероральными препаратами.
Благодаря легкости введения таблетки и капсулы представляют собой наиболее предпочтительную пероральную дозированную лекарственную форму, в случае которой твердые фармацевтические носители, конечно, применяются. Если это необходимо, таблетки могут быть покрыты оболочкой посредством применения стандартных водных и неводных технологий. Такие композиции и препараты должны содержать, по крайней мере, 0,1 процент активного соединения. Процентное содержание активного соединения в этих композициях, конечно, может изменяться и обычно может быть в диапазоне от приблизительно 2 процентов до приблизительно 60 процентов от массы дозированной лекарственной формы. Количество активного соединения в таких терапевтически пригодных композициях является таким, чтобы обеспечивалось достижение эффективной дозы. Активные соединения также могут быть введены интраназально в виде, например, жидких капель или разбрызгиваемого раствора.
Таблетки, пилюли, капсулы и им подобные также могут содержать связующее вещество, такое как камедь трагакантовая, акациевая, кукурузный крахмал или желатин; эксципиенты, такие как дикальцийфосфат; дезинтегрирующий агент, такой как кукурузный крахмал, картофельный крахмал, альгиновая кислота; скользящее вещество, такое как стеарат магния; и подсластитель, такой как сахароза, лактоза или сахарин. В случае, если дозированная лекарственная форма представляет собой капсулу, она может содержать, в дополнение к веществам вышеперечисленного типа, жидкий носитель, такой как жирное масло.
В некоторых случаях, в зависимости от растворимости вводимого соединения или соли, может быть эффективным составление рецептуры соединения или соли в виде раствора в масле, таком как триглицерид, содержащий одну или несколько жирных кислот средней длины цепи, в липофильном растворителе, таком как триацетилглицерин, в гидрофильном растворителе (например, пропиленгликоле) или в смеси двух или нескольких этих растворителей, а также, необязательно, включающей один или несколько ионных или неионных поверхностно-активных веществ, таких как лаурилсульфат натрия, полисорбат 80, полиэтоксилированные триглицериды и моно- и/или диглицериды, содержащие одну или несколько жирных кислот средней длины цепи. Растворы, содержащие поверхностно-активные вещества (особенно 2 и более поверхностно-активных веществ) будут образовывать эмульсии или микроэмульсии при контакте с водой. Соединение также может быть по рецептуре в водорастворимом полимере, в котором оно диспергировано в качестве аморфной фазы такими способами, как горячее прессование расплава и сушка распылением; такие полимеры включают ацетат гидроксилприпилметилцеллюлозы (HPMCAS), гидроксилприпилметилцеллюлозу (HPMCS) и поливинилпирролидиноны, включая гомополимер и сополимеры.
Различные другие вещества могут присутствовать в качестве покрывающей оболочки или как модифицирующие физическую форму дозированной лекарственной формы. Например, таблетки могут быть покрыты оболочкой с шеллаком, сахаром или и с тем, и с другим. Сироп или эликсир могут содержать в добавление к активному ингредиенту сахарозу в качестве подсластителя, метил- и пропилпарабены как консерванты, краситель и вкусовое ароматическое вещество, такое как вишневый или апельсиновый вкусовой ароматизатор.
Соединения формулы I также могут вводиться парентерально. Растворы или суспензии этих активных соединений могут быть приготовлены в воде, соответственно смешанной с поверхностно-активным веществом или смесью поверхностно-активных веществ, таких как гидроксипропилцеллюлоза, полисорбат 80, и моно- и диглицеридами, содержащими жирные кислоты со средней и большой длиной цепи. Дисперсии также могут быть приготовлены в глицерине, в жидких полиэтиленгликолях и их смесях в маслах. При нормальных условиях хранения и применения эти препараты содержат консерванты для предотвращения развития микроорганизмов.
Фармацевтические формы, пригодные для инъекционного использования, включают стерильные водные растворы или дисперсии и стерильные порошки для приготовления непосредственно перед введением стерильных инъекционных растворов или дисперсий. Во всех случаях лекарственная форма должна быть стерильной и должна быть в виде раствора во всем объеме, что обеспечивает легкое введение с помощью шприца. Она должна быть стабильна при условиях изготовления и хранения и должна иметь предохранение против загрязнения микроорганизмами, такими как бактерии и грибы. Носитель может быть растворителем или дисперсионной средой, содержащими, например, воду, этанол, многоатомный спирт (например, глицерин, пропиленгликоль и жидкий полиэтиленгликоль), их подходящие смеси и растительные масла.
Комбинированная терапия
Соединения формулы I могут применяться в комбинации с другими лекарственными веществами, которые также могут быть пригодны для лечения или улучшения состояния при заболеваниях или патологических состояниях, для которых соединения формулы I применимы. Такие другие лекарственные вещества могут быть введены посредством способа применения и в количестве, как они обычно применяются, одновременно или последовательно с соединением формулы I. При лечении пациентов, которые имеют диабет типа 2, резистентность к инсулину, ожирение, метаболический синдром и сопутствующие патологии, которые сопровождают эти заболевания, обычно вводится более одного лекарственного средства. Соединения согласно этому изобретению, как правило, могут быть введены пациенту, который уже получает одно или несколько лекарственных средств в связи с этими патологическими состояниями. Часто соединения должны быть введены пациенту, которого уже лечили с помощью одного или нескольких антидиабетических соединений, таких как метформин, сульфонилмочевины и/или PPAR агонисты, в случае, когда у пациента гликемические уровни неадекватно среагировали на лечение.
Когда соединение формулы I применяется одновременно с одним или несколькими лекарственными средствами, фармацевтическая композиция в дозированной лекарственной форме, содержащей такие другие лекарственные средства и соединение формулы I, является предпочтительной. Тем не менее, комбинированная терапия также включает методы лечения, в которых соединение формулы I и одно или несколько других лекарственных средств вводятся при совмещении различных режимов. Также предполагается, что при использовании в комбинации с одним или несколькими другими активными ингредиентами соединение по настоящему изобретению и другие активные ингредиенты могут применяться в более низких дозах, чем тогда, когда каждое используется отдельно. Таким образом, фармацевтические композиции по настоящему изобретению включают такие, которые содержат один или несколько других активных ингредиентов в дополнение к соединению формулы I.
Примеры других активных ингредиентов, которые могут быть введены в комбинации с соединением формулы I, и либо введены раздельно, либо в той же фармацевтической композиции, включают, но не ограничиваются только ими:
(a) PPAR гамма-агонисты и частичные агонисты, включая и глитазоны, и неглитазоны (например, троглитазон, пиоглитазон, энглитазон, MCC-555, росиглитазон, балаглитазон, нетоглитазон, T-131, LY-300512, LY-818 и соединения, раскрытые в WO02/08188, WO2004/020408 и WO2004/020409;
(b) бигуаниды, такие как метформин и фенформин;
(c) GPR40 агонисты;
(d) ингибиторы дипептидилпептидазы IV (DP-IV), такие как ситаглиптин, саксаглиптин и вилдаглиптин;
(e) инсулин или инсулиномиметики;
(f) сульфонилмочевины, такие как толбутамид, глимепирид, глипизид и родственные им вещества;
(g) ингибиторы α-глюкозидазы (такие как акарбоза);
(h) агенты, которые улучшают липидный профиль пациента, такие как (i) ингибиторы HMG-CoA редуктазы (ловастатин, симвастатин, росувастатин, правастатин, флувастатин, аторвастатин, ривастатин, итавастатин, ZD-4522 и другие статины), (ii) секвестранты желчных кислот (холестирамин, колестипол и диалкиламиноалкильные производные поперечно-сшитого декстрана), (iii) агонисты ниацинового рецептора, никотиниловый спирт, никотиновая кислота или ее соль, (iv) PPARα агонисты, такие как производные фенофиброевой кислоты (гемфиброзил, клофибрат, фенофибрат и безафибрат), (v) ингибиторы абсорбции холестерина, такие как, напроимер, эзетимиб, (vi) ингибиторы ацилCoA:холестеролацилтрансферазы (ACAT), такие как авасимиб, (vii) ингибиторы CETP, такие как торсетрапиб и (viii) фенольные антиоксиданты, такие как пробукол;
(i) PPARα/γ двойственные агонисты, такие как мураглитазар, тезаглитазар, фарглитазар и JT-501;
(j) PPARδ агонисты, такие как раскрытые в WO97/28149;
(k) соединения против ожирения, такие как фенфлурамин, дексфенфлурамин, фентирамин, субитрамин, орлистат, ингибиторы нейропептида Y5, Mc4r агонисты, каннабиноидного рецептора 1 (CB-1) антагонисты/обратные агонисты, и β3 адренергического рецептора агонисты;
(l) ингибиторы подвздошно-кишечного переносчика желчной кислоты;
(m) агенты, предназначенные для использования в воспалительных состояниях, такие как аспирин, нестероидные противовоспалительные лекарственные средства, глюкокортикоиды, азулфидин и селективные ингибиторы циклооксигеназы 2;
(n) антагонисты глюкагонового рецептора;
(o) GLP-1,
(p) GIP-1,
(q) аналоги GLP-1, такие как эксендины, например эксенатид (Баета), и
(r) ингибиторы гидроксистеролдегидрогеназы-1 (HSD-1).
Вышеуказанные комбинации включают комбинации соединения согласно настоящему изобретению не только с одним другим активным соединением, но также и с двумя или несколькими другими активными соединениями. Не ограничивающие их примеры включают комбинации соединения, имеющего формулу I, с двумя или несколькими активными соединениями, выбираемыми из бигуанидов, сульфонилмочевин, ингибиторов HMG-CoA редуктазы, других PPAR агонистов, GPR40 агонистов, ингибиторов DP-IV и соединений против ожирения.
Испытания для определения биологической активности
Активность соединений этой заявки показана с применением нижеследующих испытаний активности ингибирования PTP-1B.
Протокол испытаний фосфатазы
Материалы:
EDTA - этилендиаминтетрауксусная кислота (Sigma)
DMH - N,N'-диметил-N,N'-бис(меркаптоацетил)гидразин (синтез опубликован в J. Org. Chem. 56, pp. 2332-2337, (1991), авторами R. Singh и G.M. Whitesides, и может быть заменен на DTT - дитиотреитол, Бис-Трис - 2,2-бис(гидроксиметил)-2,2',2"-нитрилтриэтанол (Sigma), Triton X-100 - октилфенолполи(этиленгликоля простой эфир) 10 (Pierce)
Антитела: фракция анти-глутатион-S-трансферазы кролика (H и L) (Molecular Probes)
Фермент: человеческий рекомбинантный PTP-1B, содержащий аминокислоты 1-320, соединенный с GST ферментом (глутатион-S-трансфераза) или с FLAG пептидом, очищенный с помощью афинной хроматографии (Huyer et al., 1997, J. Biol. Chem., 272, 843-852). Дикий тип содержит в активном центре цистеин (215), в то время как мутант содержит в активном центре серин (215).
Меченный тритием пептид: Bz-NEJJ-CONH2, молекулярная масса 808, эмпирическая формула C32H32T2O12P2F4
Базовые растворы
(10X) Буферный раствор для испытаний
500 мМ Бис-Трис (Sigma), pH 6,2,
Молекулярная масса = 209,2
20 мМ EDTA (GIBCO/BRL)
Хранить при 4°C.
Свежеприготовленные каждый день:
Буферный раствор для испытаний (IX)
(комнатная температура)
50 мМ Бис-Трис
2 мМ EDTA
5 мМ DMH (Молекулярная масса = 208)
Раствор для фермента
Буферный раствор (хранить во льду)
50 мМ Бис-Трис
2 мМ EDTA
5 мМ DMH
20% Глицерин (Sigma)
0,01 % Тритон X-100 (Pierce)
Раствор для антител
Буферный раствор (хранить во льду)
50 мМ Бис-Трис
2 мМ EDTA
Протокол испытаний связывания IC 50:
Соединения (лиганды), которые потенциально ингибируют связывание радиоактивного лиганда со специфической фосфатазой, подвергали скринингу на 96-луночной плашке следующим образом.
В каждую лунку добавляли следующие растворы при 25°C в следующем порядке:
1. 110 мкл буферного раствора для испытаний.
2. 10 мкл 50 нМ меченного тритием BzN-EJJ-CONH2 в буферном растворе для испытаний (IX) при 25°C.
3. 10 мкл тестируемого соединения в DMSO при 10 различных концентрациях при серийном разбавлении (конечный DMSO приблизительно 5% об./об.) с дублирующей пробой при 25°C.
4. 10 мкл 3,75 мкг/мл очищенного человеческого рекомбинантного GST-PTP-1B в ферментном буферном растворе.
5. Плашку встряхивали в течение 2 минут.
6. 10 мкл 0,3 мкг/мкл антиглутатион-S-трансферазы (анти-GST) кролика IgG (Molecular Probes) разбавляли в буферном растворе для разбавления антител при 25°C.
7. Плашку встряхивали в течение 2 минут.
8. 50 мкл протеин A-PVT SPA гранулы (Amersham) при 25°C.
9. Плашку встряхивали в течение 5 минут. Показатель связывания количественно определяли датчиком Microbeta для 96-луночной плашки.
10. Показатель неспецифического связывания определяли как связывание фермент-лиганд в отсутствие анти-GST антител.
11. 100% связывающую активность определяли как связывание фермент-лиганд в присутствии анти-GST антител, но в отсутствие тестируемых лигандов, за вычетом неспецифического связывания.
12. Процент ингибирования рассчитывали в соответствии с этим.
13. IC50 величину находили аппроксимацией нелинейной регрессионной функциональной зависимости с 4-параметровым/многомерным уравнением (описано в: "Robust Statistics", New York, Wiley, by P.J. Huber (1981)), и выражали в единицах нМ.
14. Тестируемые лиганды (соединения) с более чем 90% ингибированием при 10 мкМ охарактеризовывали как активные.
Ферментативное испытание PTP-1B
Буферный раствор для испытания
50 мМ Бис-Трис (pH=6,3)
2 мМ EDTA
5 мМ N,N'-диметил-N,N'-бис(меркаптоацетил)гидразин (DMH)
Субстрат
10 мМ флуоресцеин дифосфат (FDP) хранится при -20°C (также можно использовать 10 мМ DiFMUP)
Буферный раствор для фермента
50 мМ Бис-Трис (pH=6,3)
2 мМ EDTA
5 мМ DMH
20 %(об./об.) глицерин
0,01% Тритон X-100
Испытания проводили при комнатной температуре в 96-луночных плашках. Реакционная смесь в 170 мкл содержала 50 мМ Бис-Трис (pH=6,3), 2 мМ EDTA, 5 мМ N,N'-диметил-N,N'-бис(меркаптоацетил)гидразина (DMH) и 10 мкМ флуоресцеина дифосфата (FDP) или 6,8-дифтор-4-метилумбеллиферил фосфата (DiFMUP). По 10 мкл с 10 концентрациями (серийное разведение) тестового соединения (ингибитор), разбавленные DMSO или только один DMSO для контроля добавляли в каждую лунку, и плашку подвергали помешиванию в течение 2 мин. Реакцию инициировали посредством добавления 20 мкл растворенного PTP-1B (50 нМ для FDP, 0,5 нМ для DiFMUP в 50 мМ Бис-Трис (pH=6,3), 2 мМ EDTA, 5 мМ DMH, 20% глицерина и 0,01% Тритона X-100. Фосфатазная активность сопровождалась непрерывной регистрацией появления флуоресцирующего продукта флуоресцеин монофосфата (FMP) или 6,8-дифтор-7-гидрокси-4-кумарина (DiFMU), продолжающейся в течение 15-30 мин, с использованием Spectromax Gemini считывающего устройства флуоресцирующей плашки (Molecular probes) с возбуждением при 440 нм и испусканием при 530 нм (отсекающий фильтр при 525 нм) для FDP и с возбуждением при 360 нм и испусканием при 450 нм (отсекающий фильтр при 435 нм) для DiFMUP. Все испытания осуществляли, по крайней мере, в двукратной повторности. Начальная скорость образования FMP или DiFMU наносилась на график напротив концентрации ингибитора, и данные согласовывались с 4-параметровым уравнением, и точка перегиба кривой графика соответствия представляет собой IC50.
Испытания на способность к обратимости процесса
Такие же реагенты, как в ферментативных испытаниях для PTP1B. Значения IC50 определяли для соединений, используя 10 мкМ FDP и 5 нМ PTP1B (конечная концентрация) в 96-луночной плашке, как это описано выше. Фосфатазная активность проявлялась в течение 10 минут. 40-кратное разбавление реакционной смеси получали путем отбора 5 мкл FDP реакционной смеси и добавлением их к 195 мкл буферного раствора для испытаний, содержащего 10 мкМ DiFMUP, в другой 96-луночной плашке. Образование DiFMU проявлялось в течение 30 мин. Данные как для FDP реакции, так и для DiFMUP реакции согласовывались с 4-параметровым уравнением, и значения IC50 определяли в точке перегиба кривой графика соответствия как для FDP, так и для DiFMUP реакций. Соединения допускали обратимость процесса, если значения IC50 изменялись >20 кратно от FDP до разбавления в буферном растворе DiFMUP.
Фармакокинетика у крыс
Фармакокинетика у крыс при пероральном введении
Методика проведения анализа
Животных содержали, кормили и ухаживали за ними в соответствии с рекомендациями Канадского Совета по уходу за животными (Guidelines of the Canadian Council on Animal Care).
Самцы крыс линии Спраг Доули (Sprague Dawley) (325-375 г) не получали пищу в течение ночи перед каждым исследованием крови в случае перорального введения.
Крыс помещали в клетку для иммобилизации поодиночке, и бокс прочно запирали. Нулевой образец крови получали отрезанием кончика хвоста в виде небольшого кусочка (1 мм или менее). Хвост затем плотно зажимали, но мягко приводили в движение от верхнего конца до нижней части для выдаивания крови. Приблизительно 1 мл крови собирали в гепаринизированную пробирку с вакуумом. Соединения готовили, как требуется, в стандартном дозированном объеме 10 мл/кг, и вводили перорально шунтированием зонда для принудительного кормления с иглой 16 диаметра, 3" в желудок.
Последующие отборы крови осуществляли таким же способом, как и нулевой отбор крови, за исключением того, что уже не было необходимости снова надрезать хвост. Хвост очищали кусочком марли и выдаивали/зажимали, как описано выше, в соответствующим образом помеченные пробирки.
Незамедлительно после отбора образца кровь центрифугировали, разделяли, помещали в четко обозначенные флаконы и хранили в морозильной камере до проведения анализа.
Обычные моменты времени для определения показателей крови крыс после пероральной дозировки следующие:
0,15 мин, 30 мин, 1 ч, 2 ч, 4 ч, 6 ч.
После отбора крови в 4 ч момент времени пищу предоставляли крысам неограниченно. Воду предоставляли в любой момент в течение исследования.
Наполнители (среды для лекарственных средств)
Следующие наполнители (среды для лекарственных средств) могут применяться при их пероральном введении для определения показателей крови крыс:
PEG 200/300/400: с ограничением до 2 мл/кг
Метилцеллюлоза 0,5-1,0%: 10 мл/кг
Твин 80:10 мл/кг
Соединения при пероральном введении лекарства для определения показателей крови могут быть в суспендированной форме. Для лучшего растворения раствор может быть помещен в ультразвуковой аппарат приблизительно на 5 минут. Для проведения анализа аликвоты разбавляли равным объемом ацетонитрила и центрифугировали для удаления белкового осадка. Супернатант непосредственно наносили на C-18 колонку ВЭЖХ с УФ-детектором. Количественное определение осуществляли относительно чистого образца крови с добавлением известного количества лекарственного вещества. Биодоступность (F) оценивали посредством сравнения площади под кривой (AUC) внутривенного введения в сравнении с пероральным.
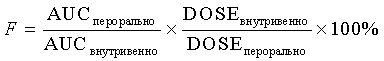
Скорости выведения из организма рассчитывали из нижеприведенного соотношения:
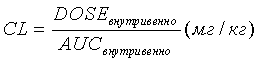
Единицами измерения CL являются мл/ч·кг (миллилитры в час на килограмм)
Фармакокинетика у крыс при внутривенном введении
Методика проведения анализа
Животных содержали, кормили и ухаживали за ними в соответствии с рекомендациями Канадского Совета по уходу за животными (Guidelines of the Canadian Council on Animal Care).
Самцов крыс линии Спраг Доули (Sprague Dawley) (325-375 г) помещали в пластиковые клетки с навесными дном, верхней частью клетки, водяным баллоном и пищей.
Соединение готовили, как требуется, в стандартном дозированном объеме 1 мл/кг. У крыс отбирали кровь для нулевого образца крови и давали дозами лекарство при CO2 седативном воздействии. Крыс поодиночке помещали в камеру, подвергшуюся предварительной обработке CO2, и вынимали как только они утрачивали свой установочный рефлекс. Крысу затем помещали на доску для фиксации, носовой конус с подачей CO2 надевали как намордник, и крысу закрепляли на доске эластичными резиновыми тягами. При применении хирургических щипцов и ножниц яремную вену вскрывали и брали нулевой образец крови, вслед за этим отмеряли дозу соединения, которое вводили в яремную вену. Легким давлением пальцев прижимали место инъекции и убирали носовой конус. Момент времени отмечали. Он устанавливался как точка нулевого момента времени.
Отбор крови в момент времени 5 мин осуществляли отрезанием кончика хвоста в виде небольшого кусочка (1-2 мм). Хвост затем плотно зажимали, но мягко приводили в движение от верхнего конца до нижней части для выдаивания крови из хвоста. Приблизительно 1 мл крови собирали в гепаринизированную пробирку. Последующие отборы крови осуществлялись таким же способом, как и нулевой отбор крови, за исключением того, что уже не было необходимости снова надрезать хвост. Хвост очищали кусочком марли и отбирали кровь, как описано выше, в соответствующим образом помеченные пробирки.
Обычные моменты времени для определения показателей крови крыс после внутривенной дозировки следующие:
0,5 мин, 15 мин, 30 мин, 1 ч, 2 ч, 6 ч
или 0,5 мин, 30 мин, 1 ч, 2 ч, 4 ч, 6 ч.
Наполнители:
Следующие наполнители (среды для лекарственных средств) могут применяться при их внутривенном введении для определения показателей крови крыс:
Декстроза: 1 мл/кг
2-гидроксипропил-b-циклодекстрин 1 мл/кг
DMSO (диметилсульфоксид, ДМСО): ограничение для объема дозы составляет 0,1 мл на животное
PEG 200: не более чем 60%, смешанные с 40% стерильной воды - 1 мл/кг
Вместе с декстрозой можно добавлять либо бикарбонат натрия, либо карбонат натрия, если раствор замутненный.
Для проведения анализа аликвоты разбавляли равным объемом ацетонитрила и центрифугировали для удаления белкового осадка. Супернатант непосредственно наносили на C-18 колонку ВЭЖХ с УФ-детектором. Количественное определение осуществляли относительно чистого образца крови с добавлением известного количества лекарственного вещества. Биодоступность (F) оценивали посредством сравнения площади под кривой (AUC) внутривенного введения в сравнении с пероральным.
Скорости выведения из организма рассчитывали из нижеприведенного соотношения:
Единицами измерения CL являются мл/ч·кг (миллилитры в час на килограмм)
Исследование PTP-1B интактной клетки
Конструирование рекомбинантных бакуловирусных трансфекционных векторов и клетки насекомых
Кратко, используя Bac-to-Bac бакуловирусную экспрессионную систему (Gibco-BRL, Mississauga, Ontario, Canada), PTP 1B кДНК (cDNA) (полученную от Dr. R. L. Erikson, Harvard University, USA), клонировали в составе pF ASTB AC донорной генноинженерной плазмиды, чтобы включить FLAG последовательность по 5' концу кДНК (PTP1B-FL). Рекомбинантную плазмиду трансформировали в компетентные клетки DH10BAC E. Coli. Далее следовала транспозиция и селекция в присутствии антибиотика; рекомбинантную бактериальную ДНК выделяли из селекционированных колоний E. Coli и использовали для трансфекции sf9 клеток насекомых (Invitrogen, San Diego, CA, USA). Клетки sf9 культивировали во вращающихся колбах при 28°C на Graces среде с добавками (Gibco-BRL, Mississauga, Ontario, Canada), с 10% термоинактивированной эмбриональной бычьей сыворотки (Gibco-BRL), следуя рекомендациям протокола Summers и Smith (A manual for Methods for Baculovirus Vectors and Insect Culture Procedures (Bulletin No. 1555). Texas A & M University, Texas Agricultural Experiment Station, College Station, TX, 1987).
Испытание на интактных клетках
Инфицированные sf9 клетки, экспрессирующие PTP1B-FL, и проверочные суррогатно-инфицированные клетки получали при 29 hpi (часы после инфицирования - hours post infection) путем центрифугирования в щадящем режиме (Beckman GS-6R) при 460 об/мин, (48 g) в течение 5 мин. Клетки однократно промывали в буферном растворе для испытаний (раствор Хенкса, забуференный 15 мМ Hepes (N-2-гидроксиэтилпиперазин-N-2-этансульфоновая кислота), pH 7,4, полученный от фирмы Sigma, St. Louis, MO, USA) и повторно центрифугировали при 300 об/мин (21 g) в течение 10 мин. Клетки затем ресуспендировали в щадящем режиме в буферном растворе для испытаний и подвергали проверке, используя гемоцитометр, определяя для клеток плотность и жизнеспособность по эксклюзии трипанового синего. Испытания осуществляли, используя прибор с автоматической пипеткой Tomtec Quadra 96 pipeting robot, запрограммированный для смешивания клеток в щадящем режиме после каждого добавления. В 200 мкл буферного раствора для испытаний, 2×105 PTP экспрессирующих клеток или проверочных суррогатно-инфицированных клеток распределяли в каждую лунку 96-луночных полипропиленовых плашек и проводили преинкубирование либо с тестируемым соединением, либо в среде DMSO (3 мкл) в течение 15 мин при 37°C. Преинкубированные клетки оценивали по конечной концентрации с 10 мМ pNPP (p-нитрофенилфосфатом, полученным от фирмы Sigma-Aldrich Canada Ltd., Oakville, Ontario) в течение 15 мин, центрифугировали при 4°C, и количество гидролизованного субстрата определяли спектрофотометрически по оптической плотности OD405.
Пероральный тест на толерантность к глюкозе
Пероральные тесты на толерантность к глюкозе проводили на бодрствующих страдающих ожирением fa/fa крысах Цукера (Zucker) или страдающих ожирением ob/ob мышах (возраст 12 недель или старше). Животные не получали пищу в течение 16-18 часов перед их использованием для экспериментов. Тестируемое соединение или наполнитель давали либо интраперитониально, либо перорально за 60 минут до перорального введения раствора глюкозы в дозе 2 г/кг массы тела. Уровень глюкозы в крови измеряли, используя Medisense глюкометр для образцов крови из хвоста, взятых в различные моменты времени до и после введения глюкозы. Составляли кривые графической зависимости уровней глюкозы в крови от времени, и рассчитывали площадь под кривой (AUC)) за 120 минут (момент времени введения глюкозы представлял собой нулевой момент времени). Процент ингибирования определяли, используя AUC в случае наполнителя, - контрольная группа с нулевым процентом ингибирования.
В отдельных исследованиях C57BL/6J мышей откармливали пищей с высоким содержанием жира (35%) и высоким содержанием углеводов (36%), полученной от фирмы Bioserv (Frenchtown, NJ), в течение от 3 до 4 недель, за это время мыши прибавляли 50-100% от исходной массы тела. Пероральные тесты на толерантность к глюкозе выполняли таким же образом, как описано выше.
ПРИМЕРЫ
Нижеприведенные примеры предоставлены для иллюстрирования изобретения и не должны истолковываться как ограничивающие изобретение каким-либо образом. Объем изобретения определяется прилагаемой формулой изобретения.
Способы получения соединений, раскрытые в этом документе, иллюстрируются нижеприведенными схемами и примерами. Исходные вещества либо являются коммерчески доступными, либо их получают по известным из литературы методикам или как это проиллюстрировано. Настоящее изобретение также предоставляет процессы для получения соединений формулы I, как определено выше. В некоторых случаях порядок осуществления нижеследующих реакционных схем может быть изменен для облегчения проведения реакции или чтобы избежать нежелательных реакционных продуктов. Нижеследующие примеры предоставлены только с иллюстративной целью, и их не следует истолковывать как ограничения раскрытого изобретения.
Пример 1
3-((E)-2-{5-бром-6-[дифтор(фосфоно)метил]-2-нафтил}этенил)бензойная кислота
Схема 1
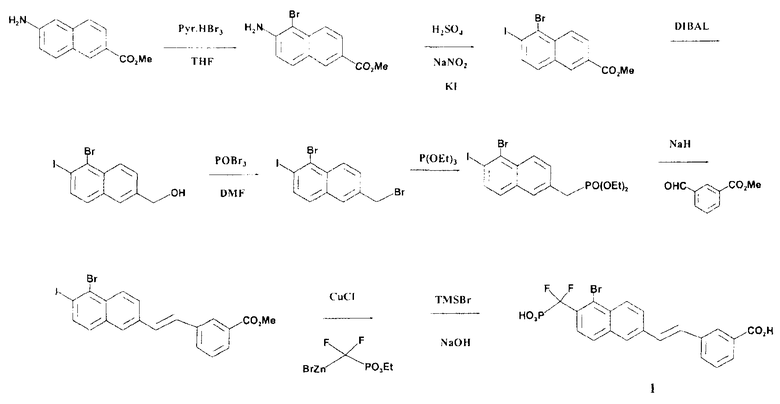
Стадия 1: Метил-6-амино-5-бром-2-нафтоат
К раствору метил-6-амино-2-нафтоата (0,5 г) в ТГФ (THF) (10 мл) добавляли пиридинийтрибромид (0,87 г). Реакционную смесь перемешивали при 0°C в течение 1 ч, после этого ее фильтровали через слой SiO2 и промывали гексаном. Органические промывные фазы упаривали досуха и остаток очищали флэш-хроматографией, элюируя гексаном, что позволило получить указанное в заголовке соединение.
Стадия 2: Метил-5-бром-6-иод-2-нафтоат
К раствору метил-6-амино-5-бром-2-нафтоата (700 мг) в воде (5 мл) при 0°C добавляли H2SO4. Реакционную смесь перемешивали в течение 30 мин. Затем по каплям добавляли раствор NaNO2 (0,3 г) в 5 мл воды и смесь перемешивали в течение 90 мин. К раствору при 0°C добавляли раствор KI (1,1 г в 5 мл воды). Реакционную смесь перемешивали в течение ночи при комнатной температуре, после чего насыщенный раствор NH4Cl добавляли к смеси. Смесь экстрагировали EtOAc и экстракт сушили над Na2SO4. Органические экстракты упаривали досуха и остаток очищали флэш-хроматографией, элюируя гексаном, что позволило получить указанное в заголовке соединение.
Стадия 3: (5-бром-6-иод-2-нафтил)метанол
К раствору метил-5-бром-6-иод-2-нафтоата (0,37 г, 0,95 ммоль) в толуоле (10 мл) при -78°C по каплям добавляли DIBAL (3 мл 1M раствора в PhMe, 3 ммоль). Температуру поднимали до 0°C в течение 1 ч. Реакцию останавливали добавлением 10 мл 1M HCl, экстрагировали EtOAc и сушили над Na2SO4. Органические экстракты упаривали досуха, что позволило получить указанное в заголовке соединение.
Стадия 4: 1-бром-6-(бромметил)-2-иоднафталин
К раствору POBr3 (662 мг, 2,3 ммоль) в 4,5 мл CH2Cl2 при 0°C по каплям добавляли DMF (2,25 мл). Реакционную смесь перемешивали в течение 10 минут и затем добавляли раствор (5-бром-6-иод-2-нафтил)метанола (280 мг, 0,77 ммоль) в 5 мл CH2Cl2. Реакционную смесь перемешивали в течение 30 минут, останавливали реакцию добавлением насыщенного раствора NH4Cl, экстрагировали EtOAc и сушили над Na2SO4. Органические экстракты упаривали досуха, что позволило получить указанный в заголовке продукт, который в таком виде использовали для следующей стадии.
Стадия 5: Диэтил-(5-бром-6-иод-2-нафтил)метилфосфонат
К 1-бром-6-(бромметил)-2-иоднафталину (270 мг) со стадии 4 добавляли триэтилфосфит (4 мл). Реакционную смесь нагревали с обратным холодильником в течение 1 часа, после чего удаляли избыток триэтилфосфита отгонкой в высоком вакууме, получая указанный в заголовке продукт.
Стадия 6: Метил-3-[(E)-2-(5-бром-6-иод-2-нафтил)этенил]бензоат
К раствору диэтил-(5-бром-6-иод-2-нафтил)метилфосфоната (250 мг) со стадии 5 в THF (5 мл) при 0°C добавляли NaH (60% в вазелиновом масле, 17 мг). Реакционную смесь перемешивали в течение 1 часа, после чего добавляли метил-3-формилбензоат (85 мг) и перемешивание продолжали в течение 1 часа при комнатной температуре. Смесь обрабатывали насыщенным раствором NH4Cl, экстрагировали EtOAc, сушили над Na2SO4 и упаривали. Остаток очищали флэш-хроматографией, элюируя смесью 5% EtOAc/гексан, что позволило получить указанный в заголовке продукт.
Стадия 7: Метил-3-((E)-2-{5-бром-6-[(диэтоксифосфорил)(дифтор)метил]-2-нафтил}этенил)бензоат
Этот продукт получали из метил-3-[(E)-2-(5-бром-6-иод-2-нафтил)этенил]бензоата в результате его реакции с бромидом ((диэтоксифосфинил)дифторметил)цинка, в соответствии с методикой, предложенной S. Shibuya в Tetrahedron 1997, 53.3, 815.
Стадия 8: 3-((E)-2-{5-бром-6-[дифтор(фосфоно)метил]-2-нафтил}этенил)бензойная кислота
Гидролиз метил-3-((E)-2-{5-бром-6-[(диэтоксифосфорил)(дифтор)метил]-2-нафтил}этенил)бензоата (35 мг) со стадии 7 осуществляли, используя TMSBr (2 мл) в 1 мл CH2Cl2 при комнатной температуре в течение ночи. Смесь упаривали досуха и остаток растворяли в этаноле. Этот раствор опять упаривали досуха и процесс повторяли 3 раза. Реакционный остаток растворяли в воде и обрабатывали NaOH 1N, что позволило получить указанный в заголовке продукт в виде натриевой соли.
1H ЯМР (500 МГц, CD3OD): δ 8,52 (д, 1H), 8,30 (с, 1H), 8,15 (м, 2H), 7,95-8,05 (м, 3H), 7,72 (д, 1H), 7,60 (м, 3H).
Пример 2 3-((E)-2-{6-бром-7-{дифтор(фосфоно)метил]-2-нафтил}этенил)бензойная кислота
Схема 2
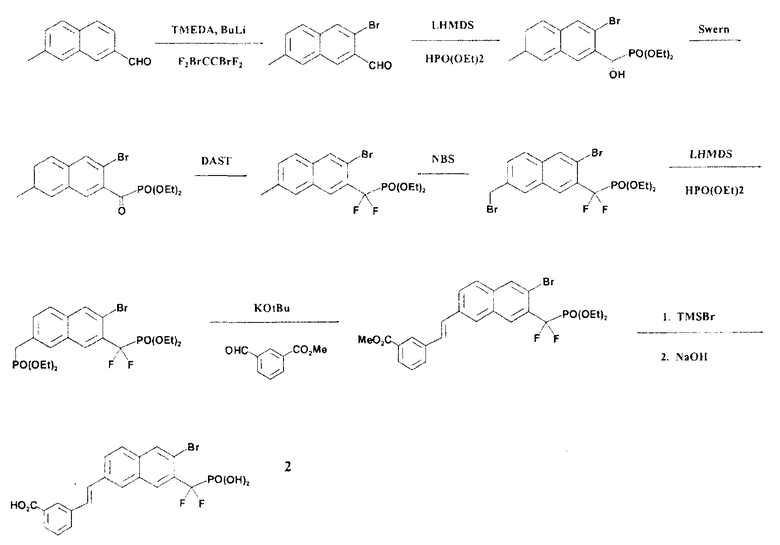
Стадия 1: 3-бром-7-метил-2-нафтальдегид
Из 7-метил-2-нафтальдегида (430 мг), N,N,N'-триметилэтилендиамина (500 мг), BuLi (1,6 M в гексане, 4,95 мл) и тетрафтордибромэтана (2,5 мл) получали указанный в заголовке продукт, как это описано в литературе (Sun, Q., Lavoie E. J.; Heterocycles; 1996, 43, (4), 131-743).
Стадия 2: Диэтил-(3-бром-7-метил-2-нафтил)(гидрокси)метилфосфонат
К раствору диэтилфосфита (0,22 мл) в THF (5 мл) при -78°C добавляли LiHMDS (1 эквивалент 1M раствора в THF). Реакционную смесь перемешивали в течение 1 ч при -78°C. Раствор 3-бром-7-метил-2-нафтальдегида добавляли по каплям и реакционную смесь перемешивали в течение ночи при 0°C. Реакцию останавливали обработкой насыщенным раствором NH4Cl, экстрагировали EtOAc и сушили над Na2SO4. Органические экстракты упаривали досуха и остаток очищали флэш-хроматографией, элюируя смесью 50-100% EtOAc/гексан, что позволило получить указанный в заголовке продукт.
Стадия 3: Диэтил-3-бром-7-метил-2-нафтоилфосфонат
К раствору оксалилхлорида (0,15 мл) в 2,5 мл CH2Cl2 при -78°C добавляли DMSO (0,23 мл). Реакционную смесь перемешивали в течение 10 минут, после чего раствор диэтил-(3-бром-7-метил-2-нафтил)(гидрокси)метилфосфоната (160 мг) в 2,5 мл CH2Cl2 добавляли по каплям. Реакционную смесь перемешивали в течение 1 ч при -78°C, после чего к смеси добавляли триэтиламин (0,66 мл) и температуру поднимали до комнатной. Добавляли воду (5 мл) и смесь экстрагировали CH2Cl2. Органические экстракты объединяли, сушили над Na2SO4 и упаривали досуха, что позволило получить указанный в заголовке продукт, который в таком виде использовался на следующей стадии.
Стадия 4: Диэтил-(3-бром-7-метил-2-нафтил)(дифтор)метилфосфонат
К раствору диэтил-3-бром-7-метил-2-нафтоилфосфоната (160 мг) в CHCl3 (3 мл) при -78°C добавляли (диэтиламино)сератрифторид (0,44 мл). Реакционную смесь перемешивали при комнатной температуре в течение 5 ч и затем выливали в смесь лед/вода/CH2Cl2. Органические экстракты промывали 50% водным раствором NH4OH и солевым раствором. Затем экстракты сушили над Na2SO4 и упаривали досуха. Остаток очищали флэш-хроматографией, элюируя смесью 40% гексан/EtOAc, что позволило получить указанный в заголовке продукт.
Стадия 5: Диэтил-[3-бром-7-(бромметил)-2-нафтил](дифтор)метилфосфонат
К раствору диэтил-(3-бром-7-метил-2-нафтил)(дифтор)метилфосфоната (200 мг) в CCl4 (12 мл) добавляли NBS (90 мг) и каталитическое количество бензоилпероксида. Смесь кипятили с обратным холодильником в течение 2 часов и затем разбавляли гексаном. Раствор фильтровали через слой целита и промывали гексаном. Гексановые смывы упаривали досуха, что позволило получить указанный в заголовке продукт.
Стадия 6: Диэтил-{6-бром-7-[(диэтоксифосфорил)(дифтор)метил]-2-нафтил}метилфосфонат
К раствору диэтилфосфита (0,22 мл) в толуоле (5 мл) при 0°C добавляли NaH (60% в вазелиновом масле, 20 мг). Реакционную смесь перемешивали в течение 1 часа и затем по каплям добавляли раствор диэтил-[3-бром-7-(бромметил)-2-нафтил](дифтор)метилфосфоната (220 мг) в толуоле (2 мл). Реакционную смесь перемешивали в течение 1 ч при 0°C, реакцию останавливали обработкой насыщенным раствором NH4Cl, экстрагировали EtOAc и сушили над Na2SO4. Органические экстракты упаривали досуха и остаток очищали флэш-хроматографией, элюируя смесью 50% EtOAc/гексан, что позволило получить указанное в заголовке соединение.
Стадия 7: Метил-3-((Е)-2-{6-бром-7-[(диэтоксифосфорил)(дифтор)метил]-2-нафтил}этенил)бензоат
К раствору диэтил-{6-бром-7-[(диэтоксифосфорил)(дифтор)метил]-2-нафтил}метилфосфоната (190 мг) и метил-3-формилбензоата (60 мг) в дегазированном THF (5 мл) при -78°C добавляли трет-бутоксид калия (0,35 мл 1M раствор в THF) и реакционную смесь перемешивали в течение 1 ч при 0°C. Смесь обрабатывали насыщенным раствором NH4Cl, экстрагировали EtOAc, сушили над Na2SO4 и упаривали досуха. Остаток очищали флэш-хроматографией, элюируя смесью 25% EtOAc/гексан, что позволило получить указанный в заголовке продукт.
Стадия 8: 3-((E)-2-{6-бром-7-{дифтор(фосфино)метил]-2-нафтил}этенил)бензойная кислота
Гидролиз метил-3-((E)-2-{6-бром-7-[(диэтоксифосфорил)(дифтор)метил]-2-нафтил}этенил)бензоата (120 мг) со стадии 7 осуществляли, используя TMSBr (2 мл) в 1 мл CH2Cl2 при комнатной температуре в течение ночи. Смесь упаривали досуха и остаток растворяли в этаноле. Этот раствор снова упаривали досуха и процесс повторяли 3 раза. Реакционный остаток растворяли в воде и обрабатывали NaOH 1N, что позволило получить указанный в заголовке продукт в виде натриевой соли.
1H ЯМР (500 МГц, CD3OD): δ 8,84 (с, 1H), 8,22 (с, 1H), 8,12 (с, 1H), 8,05 (с, 1H), 7,88 (м, 2H), 7,78 (д, 1H), 7,70 (д, 1H), 7,40 (м, 3H).
Примеры 3-6
Схема 3
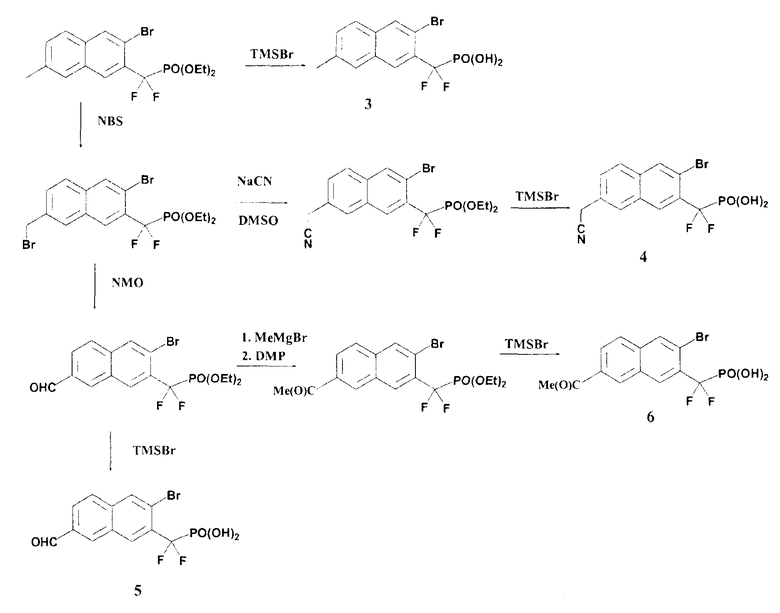
Пример 3 (3-бром-7-метил-2-нафтил)(дифтор)метилфосфоновая кислота
Диэтил-(3-бром-7-метил-2-нафтил)(дифтор)метилфосфонат (0,1 г со стадии 4, пример 2) гидролизовали в присутствии 2 мл TMSBr в 1 мл CH2Cl2 при комнатной температуре в течение ночи. Смесь упаривали досуха и остаток растворяли в этаноле. Этот раствор снова упаривали досуха и процесс повторяли 3 раза. Реакционный остаток растворяли в воде и обрабатывали NaOH 1N, что позволило получить указанный в заголовке продукт в виде натриевой соли.
1H ЯМР (500 МГц, CD3OD): δ 8,15 (д, 2H), 7,70 (м, 2H), 7,45 (д, 1H), 2,50 (с, 3H).
Пример 4 [3-бром-7-(цианометил)-2-нафтил](дифтор)метилфосфоновая кислота
К раствору диэтил-[3-бром-7-(бромметил)-2-нафтил](дифтор)метилфосфоната (0,06 г со стадии 5, пример 2) в 3 мл DMSO добавляли NaCN (18 мг). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Реакцию останавливали добавлением воды и дважды экстрагировали диэтиловым эфиром. Органические экстракты сушили над Na2SO4 и упаривали. Остаток очищали флэш-хроматографией, элюируя смесью 20% EtOAc/гексан, что позволило получить сложный эфир фосфоновой кислоты (20 мг). Диэтил-[3-бром-7-(цианометил)-2-нафтил](дифтор)метилфосфонат гидролизовали в 2 мл TMSBr при комнатной температуре в течение ночи. Смесь упаривали досуха и остаток растворяли в этаноле. Этот раствор снова упаривали досуха и процесс повторяли 3 раза. Реакционный остаток растворяли в воде и обрабатывали NaOH 1N, что позволило получить указанный в заголовке продукт в виде натриевой соли.
1H ЯМР (500 МГц, CD3OD): δ 8,40 (д, 1H), 8,34 (с, 1H), 8,13 (с, 1H), 8,05 (д, 1H), 7,72 (д, 1H), 4,20 (с, 2H).
Пример 5 (3-бром-7-формил-2-нафтил)(дифтор)метилфосфоновая кислота
К раствору диэтил-[3-бром-7-(бромметил)-2-нафтил](дифтор)метилфосфонат (0,2 г со стадии 5, пример 2) в 5 мл диоксана добавляли N-метилморфолин-N-оксид (0,17 г). Реакционную смесь кипятили с обратным холодильником в течение 1 ч. Смесь обрабатывали для остановки реакции насыщенным раствором NH4Cl и экстрагировали EtOAc, экстракт сушили над Na2SO4 и упаривали. Остаток очищали флэш-хроматографией, элюируя смесью 10-20% EtOAc/гексан, что позволило получить диэтил-(3-бром-7-формил-2-нафтил)(дифтор)метилфосфонат (0,15 грамма), который гидролизовали в присутствии TMSBr (2 мл) в CH2Cl2 (1 мл) при комнатной температуре в течение ночи. Смесь упаривали досуха и остаток растворяли в этаноле. Этот раствор снова упаривали досуха и процесс повторяли 3 раза. Реакционный остаток растворяли в воде и обрабатывали NaOH 1N, что позволило получить указанный в заголовке продукт в виде натриевой соли.
1H ЯМР (500 МГц, CD3OD): δ 10,22 (с, 1H), 8,70 (с, 1H), 8,51 (с, 1H), 8,42 (с, 1H), 8,09 (м, 2H).
Пример 6 (7-ацетил-3-бром-2-нафтил)(дифтор)метилфосфоновая кислота
Стадия 1: Диэтил-[3-бром-7-(1-гидроксиэтил)-2-нафтил](дифтор)метилфосфонат
К раствору диэтил-(3-бром-7-формил-2-нафтил)(дифтор)метилфосфоната (0,1 г из примера 5) в THF (1 мл) при -78°C добавляли MeMgBr (79 мкл 3N раствора в THF). Температуру увеличивали до 0°C и перемешивали в течение 1 ч. Смесь обрабатывали для остановки реакции насыщенным раствором NH4Cl, экстрагировали EtOAc, органические экстракты сушили над Na2SO4 и упаривали досуха. Остаток очищали флэш-хроматографией, используя смесь 10-30% гексан/EtOAc, что позволило получить указанный в заголовке продукт.
Стадия 2: Диэтил-(7-ацетил-3-бром-2-нафтил)(дифтор)метилфосфонат
К раствору диэтил-[3-бром-7-(1-гидроксиэтил)-2-нафтил](дифтор)метилфосфоната (20 мг) со стадии 1 в CH2Cl2 (2 мл) при 0°C добавляли Dess-Martin реагент (24 мг). Температуру увеличивали до комнатной и реакционную смесь перемешивали в течение 1 ч. Реакционную смесь фильтровали через слой SiO2, элюируя смесью 30% EtOAc/гексан, органическую фазу упаривали досуха. Остаток растворяли и гидролизовали в присутствии чистого TMSBr (3 мл) и перемешивали при комнатной температуре в течение ночи. Смесь упаривали досуха и остаток растворяли в этаноле. Этот раствор снова упаривали досуха и процесс повторяли 3 раза. Реакционный остаток растворяли в воде, перегоняли совместно с толуолом и откачивали высоким вакуумом, что позволило получить указанный в заголовке продукт.
1H ЯМР (500 МГц, CD3OD) δ 8,79 (д, 1H), 8,50 (с, 1H), 8,39 (с, 1H), 8,15 (д, 1H), 8,02 (д, 1H), 2,75(с, 3H).
Пример 7 [(3-бром-6-циано-2-нафтил)(дифтор)метил]фосфоновая кислота
Схема 4
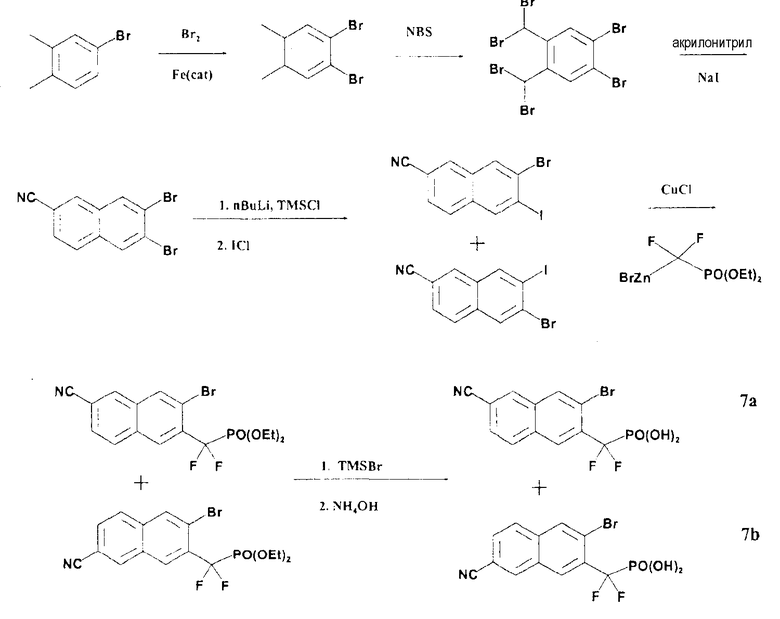
Стадии 1-3: 6,7-дибром-2-нафтонитрил
В соответствии методикой, описанной в литературе (Hanack, M., Grobhans, R., Chem. Ber. 1992, 125, 1243-1247), 6,7-дибром-2-нафтонитрил может быть получен в 2 стадии из коммерчески доступного 4,5-дибром-o-ксилена или в 3 стадии из коммерчески доступного 4-бром-o-ксилена.
Стадия 4: 7-бром-6-иод-2-нафтонитрил и 6-бром-7-иод-2-нафтонитрил
К раствору 6,7-дибром-2-нафтонитрила (15 г) и TMSCl (6,73 мл) в THF (250 мл) при -78°C быстро, при интенсивном перемешивании, добавляли n-BuLi (53 мл, 1,6 M в гексане, предварительно охлажденный до -20°C) и смесь перемешивали дополнительно в течение 5 мин и обрабатывали для остановки реакции насыщенным раствором NH4Cl. Затем смесь экстрагировали этилацетатом и органический слой промывали водой и солевым раствором, сушили над MgSO4 и фильтровали. Фильтрат концентрировали и неочищенный продукт очищали колоночной хроматографией, получая требуемый продукт в виде желтого твердого вещества.
1H ЯМР (400 МГц, ацетон-d6) (смесь двух региоизомеров): δ 8,53 (с, 1H), 8,42 (с, 1H), 8,33 (с, 1H), 8,30 (с, 1Н), 8,23 (с, 1Н), 8,20 (с, 1Н), 8,18 (д, 1H), 8,07 (д, 1H), 7,82-7,77 (м, 2H), 0,50 (с, 18H).
К раствору вышеуказанного продукта в дихлорметане (250 мл) добавляли избыток ICl и смесь перемешивали при комнатной температуре в течение 1 ч. Затем раствор промывали 10% Na2S2O3 до тех пор, пока весь ICl не был израсходован. Затем раствор промывали водой, солевым раствором, сушили над MgSO4 и фильтровали. Фильтрат концентрировали и остаток перекристаллизовывали из смеси диэтиловый эфир/гексан, получая желаемый продукт.
1H ЯМР (400 МГц, ацетон-d6) (смесь двух региоизомеров): δ 8,74 (с, 1H), 8,73 (с, 1H), 8,46-8,44 (м, 4H), 8,10-8,07 (м, 2Н), 7,84-7,81 (м, 2H).
Стадия 5: Диэтил-[(3-бром-6-циано-2-нафтил)(дифтор)метил]фосфонат и диэтил-[(3-бром-7-циано-2-нафтил)(дифтор)метил]фосфонат
В высушенную пламенем круглодонную колбу загружали CuBr (99,999%) и THF (10 мл), затем добавляли бромид ((диэтоксифосфинил)дифторметил)цинка (29 мл, 1,72 M в THF), согласно методике S. Shibuya, опубликованной в Tetrahedron 1997, 53.3, 815). Суспензию перемешивали в атмосфере N2 в течение 15 минут. 7-Бром-6-иод-2-нафтонитрил (7,1 г) затем добавляли в виде твердого вещества и смесь нагревали до 45°C в течение ночи и охлаждали до комнатной температуры. Суспензию затем обрабатывали для остановки реакции раствором NH4Cl с концентрацией, вдвое меньшей насыщенного, и экстрагировали смесью 1:1 диэтиловый эфир/этилацетат (3×). Экстракты подвергали обычной обработке, получая неочищенный продукт, который сначала очищали флэш-хроматографией (40% этилацетат в гексане). Два региоизомера затем разделяли с помощью ВЭЖХ. Элюируя смесью 50% этилацетат/гексан, сначала получали менее полярный изомер диэтил-[(3-бром-7-циано-2-нафтил)(дифтор)метил]фосфонат.
1H ЯМР (400 МГц, ацетон-d6): δ 8,72 (с, 1H), 8,54 (с, 1H), 8,46 (с, 1H), 8,19 (д, 1H), 7,95 (д, 1H), 4,26 (м, 4H), 1,33 (т, 6H).
Продолжая элюирование, получали более полярный изомер, диэтил-[(3-бром-6-циано-2-нафтил)(дифтор)метил]-фосфонат.
1H ЯМР (400 МГц, ацетон-d6): δ 8,56 (с, 2H), 8,43 (с, 1H), 8,35 (д, 1H), 7,93 (д, 1H), 4,26 (м, 4H), 1,33 (т, 6H).
Стадия 6: [(3-бром-7-циано-2-нафтил)(дифтор)метил]фосфоновая кислота (7a)
Раствор диэтил-[(3-бром-7-циано-2-нафтил)(дифтор)метил]фосфоната (2,2 г) в дихлорметане (5 мл) и TMSBr (7 мл) перемешивали в течение ночи и концентрировали. Остаток совместно упаривали с дихлорметаном (2×), со смесью этанол/вода (2×) и затем растворяли в 20 мл метанола. Затем добавляли аммиак (30%) при интенсивном перемешивании и смесь концентрировали и совместно упаривали с метанолом (3×). Твердый остаток промывали диэтиловым эфиром, получая требуемый продукт в виде белого порошка. Масс-спектр (-ионизация электрораспылением): m/z 360,0 и 361,9 (M-1)-.
Примечание: [(3-бром-6-циано-2-нафтил)(дифтор)метил]фосфоновую кислоту (7b) получали таким же образом, как описано на стадии 6. Масс-спектр (-ионизация электрораспылением): m/z 360,0, 361,9.
Пример 8a: [{2-[(фениламино)карбонил]-6-бромхинолин-7-ил} (дифтор)метил]фосфоновая кислота
Схема 5
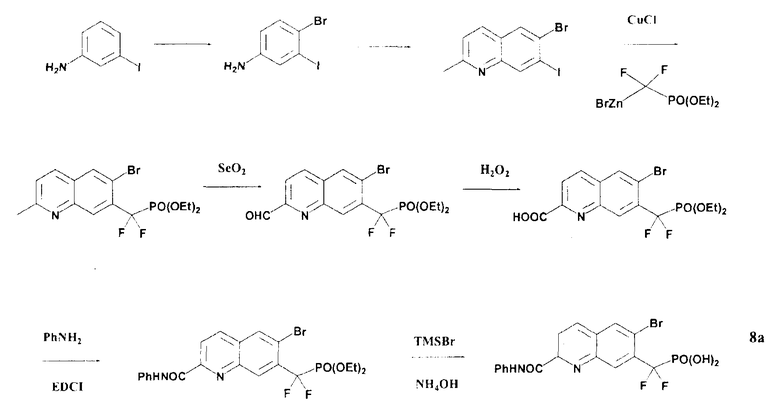
Стадия 1: (4-бром-3-иодфенил)амин
К раствору 3-иоданилина (12 мл, 100 ммоль) в 400 мл CH2Cl2 при -10°C добавляли по частям 2,4,4,6-тетрабром-2,5-циклогексадиенон (45,1 г, 110 ммоль), при этом поддерживая внешнюю температуру -10°C. После перемешивания в течение 4 часов добавляли 150 мл 1N NaOH и продукт экстрагировали CH2Cl2. Объединенные экстракты промывали водой и затем солевым раствором и сушили над Na2SO4. После концентрирования в вакууме неочищенный продукт перекристаллизовывали из смеси 2:1 гексан:толуол, получая требуемый продукт.
Стадия 2: 6-бром-7-иод-2-метилхинолин
К (4-бром-3-иодфенил)амину (11,93 г, 40 ммоль) добавляли концентрированную HCl (6 мл), пара-хлоранил (9,83 г, 1 эк.) и изопропанол (20 мл) и смесь нагревали до кипения с обратным холодильником. Раствор кротональдегида (3,98 мл) в изопропаноле (3,8 мл) затем добавляли со скоростью 0,1 мл/мин, используя поршневой насос, и смесь перемешивали при кипении с обратным холодильником еще в течение 40 мин после завершения добавления. Смесь охлаждали до комнатной температуры, разбавляли EtOAc и 5% водн. NH4OH. Продукты экстрагировали в EtOAc и органический слой промывали несколько раз водой, солевым раствором и сушили над Na2SO4. Неочищенное вещество растворяли по мере возможности в 300 мл кипящего толуола и очищали флэш-хроматографией на силикагеле в градиенте EtOAc/толуол от 0 до 5%. Первым продуктом был 6-бром-7-иод-2-метилхинолин, затем следовал его изомер 6-бром-5-иод-2-метилхинолин.
6-бром-7-иод-2-метилхинолин: 1H ЯМР (400 МГц, ацетон-d6): δ 8,58 (с, 1H), 8,32 (с, 1H), 8,20 (д, 1H), 7,48 (д, 1H), 2,68 (с, 3H).
6-бром-5-иод-2-метилхинолин: 1H ЯМР (400 МГц, ацетон-d6): δ 8,45 (д, 1H), 8,01 (д, 1H), 7,92 (д, 1H), 7,53 (д, 1H), 2,72 (с, 3H).
Стадия 3: Диэтил-[(6-бром-2-метилхинолин-7-ил)(дифтор)метил]фосфонат
Этот продукт получали из 6-бром-7-иод-2-метилхинолин при его реакции с бромидом ((диэтоксифосфинил)дифторметил)цинка, согласно методике S. Shibuya, опубликованной в Tetrahedron 1997, 53.3, 815.
Стадия 4: Диэтил-[(6-бром-2-формилхинолин-7-ил)(дифтор)метил]фосфонат
К продукту со стадии 3 (1,24 г, 3,04 ммоль) в 15 мл диоксана добавляли диоксид селена (388 мг, 1,15 эквив., высушенный в вакууме на пламени горелки) и смесь нагревали до 100°C в течение 1,3 ч. Растворитель упаривали и остаток очищали флэш-хроматографией на силикагеле смесью 30% EtOAc/толуол, получая указанный в заголовке продукт в виде желтого твердого вещества.
1H ЯМР (500 МГц, ацетон-d6) δ 10,16 (с, 1H), 8,63, (d, 1H), 8,48 (с, 1H), 8,53 (с, 1H), 8,13 (д, 1H), 4,30 (м, 4H), 1,33 (т, 6H).
Стадия 5: 6-бром-7-[(диэтоксифосфорил)(дифтор)метил]хинолин-2-карбоновая кислота
Как описано в литературе (Synthesis, 1993, 295), к продукту со стадии 4 (105 мг, 0,25 ммоль) в 1 мл муравьиной кислоты по каплям добавляли 30% перекись водорода (0,13 мл, 1 ммоль) и смесь перемешивали в течение ночи при комнатной температуре. Растворитель упаривали и добавляли абсолютный этанол. Этот процесс повторяли 3 раза и после упаривания оставшегося этанола получали указанный в заголовке продукт в виде масла.
1H ЯМР (500 МГц, ацетон-d6) δ 8,63, (с, 1H), 8,49 (с, 2H), 8,45 (с, 1H), 4,30 (м, 4H), 1,33 (т, 6H).
Стадия 6: Диэтил-[{2-[(фениламино)карбонил]-6-бромхинолин-7-ил}(дифтор)метил]фосфонат
К продукту со стадии 5 (109 мг) в 5 мл CH2Cl2 добавляли EDCI (96 мг), анилин (0,1 мл) и основание Хунига (0,1 мл). Раствор перемешивали при комнатной температуре в течение 3 часов и после концентрирования и флэш-хроматографии на силикагеле в градиенте 10-25% EtOAc/толуол получали амид.
ЯМР (500 МГц, CDCl3) δ 10,2, (с, 1H), 8,6 (с, 1H), 8,5 (д, 1H), 8,45 (д, 1H), 8,4 (с, 1H), 7,4-7,0 (м, 5H), 4,30 (м, 4H), 1,33 (т, 6H).
Стадия 7: [{2-[(фениламино)карбонил]-6-бромхинолин-7-ил} (дифтор)метил]фосфоновая кислота (8)
Раствор диэтил-[(3-бром-7-циано-2-нафтил)(дифтор)метил]фосфоната (2,2 г) в дихлорметане (5 мл) и TMSBr (7 мл) перемешивали в течение ночи и концентрировали. Остаток совместно упаривали с дихлорметаном (2×), со смесью этанол/вода (2×) и затем растворяли в 20 мл метанола. Затем добавляли аммиак (30%) при интенсивном перемешивании и смесь концентрировали и совместно упаривали с метанолом (3×). Твердый остаток промывали диэтиловым эфиром, получая требуемый продукт в виде белого порошка. Масс-спектр (-ионизация электрораспылением): m/z 457,2 и 456,3 (M-1)-.
В приведенной ниже таблице показаны производные, аналогичные примеру 8a, которые были получены с использованием способа, аналогичного приведенному на схеме:
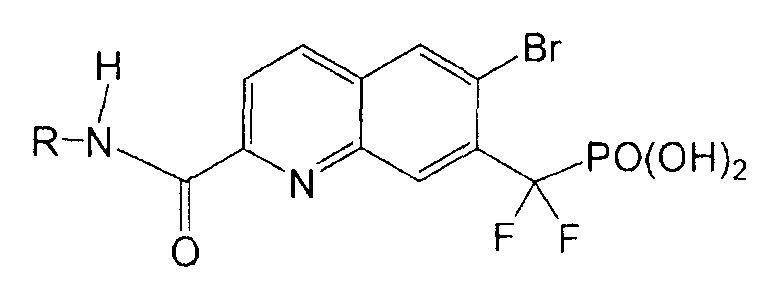
Примеры 9a-9i: (6-бром-2-замещенный хинолин-7-ил)(дифтор)метил]фосфоновые кислоты
Соединения из нижеприведенной таблицы получали, как показано в примечаниях к промежуточным соединениям, описанным в схеме 5 (пример 8) и схеме 6 (для примера 9-1, который представлен в последней графе таблицы 2).
Пример 9i (таблица 2): Диаммоний-[(6-бром-2-цианохинолин-7-ил)(дифтор)метил]фосфат
Схема 6
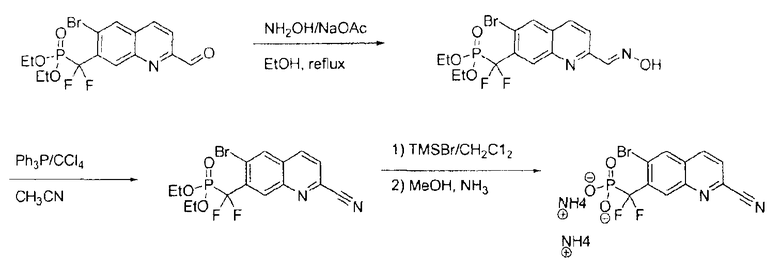
Стадия 1: Диэтил-[(6-бром-2-((гидроксиимино)метил)хинолин-7-ил)(дифтор)метил]фосфонат
К перемешиваемому раствору диэтил-[(6-бром-2-формилхинолин-7-ил)(дифтор)метил]фосфоната (525 мг, 1,244 ммоль) [пример 8, стадия 4] в этаноле (12 мл) при комнатной температуре добавляли гидроксиламина гидрохлорид (130 мг, 1,865 ммоль) и ацетат натрия (508 мг, 3,73 ммоль). Реакционную смесь перемешивали при 70°C в течение 1 часа. Затем ее выливали в водный раствор гидрокарбоната натрия и экстрагировали этилацетатом (100 мл), промывали водным раствором гидрокарбоната натрия (2×), солевым раствором, сушили с MgSO4 и концентрировали в вакууме, получая неочищенный диэтил-[(6-бром-2-[(Е)-(гидроксиимино)метил]хинолин-7-ил)(дифтор)метил]фосфонат.
1H ЯМР δ (м.д.)(ацетон-d6) 11,19 (1H, с), 8,44 (1H, с), 8,39 (1H, д, J=8,72 Гц), 8,36 (1H, с), 8,29 (1H, с), 8,13 (1H, д, J=8,72 Гц), 4,34-4,26 (4H, м), 1,36-1,29 (6H, м)
Стадия 2: Диэтил-[(6-бром-2-цианохинолин-7-ил)(дифтор)метил]фосфонат
К перемешиваемому раствору диэтил-[(6-бром-2-[(E)-(гидроксиимино)метил]хинолин-7-ил)(дифтор)метил]фосфоната (520 мг, 1,189 ммоль) в ацетонитриле (30 мл) при комнатной температуре добавляли трифенилфосфин (1,248 г, 4,76 ммоль) и четыреххлористый углерод (230 мкл, 2,383 ммоль). Реакционную смесь перемешивали при 100°C в течение 1 часа. Ее концентрировали досуха в вакууме, предварительно адсорбировали на силикагеле для проведения флэш-хроматографии с элюированием этилацетатом в толуоле (от 20% до 30%), что позволило получить диэтил-[(6-бром-2-цианохинолин-7-ил)(дифтор)метил]фосфонат.
1H ЯМР δ (м.д.)(ацетон-d6): 8,72 (1H, д, J=8,52 Гц), 8,63 (1H, с), 8,43 (1H, с), 8,13 (1H, д, J=8,52 Гц), 4,36-4,26 (4H, м), 1,41-1,29 (6H, м). Масс-спектр (+ионизация электрораспылением) = 419,0 и 421,0.
Стадия 3: Диаммоний-[(6-бром-2-цианохинолин-7-ил)(дифтор)метил]фосфат
К перемешиваемому раствору диэтил-[(6-бром-2-цианохинолин-7-ил)(дифтор)метил]фосфоната (370 мг, 0,883 ммоль) в дихлорметане (9) при 0°C добавляли по каплям бромтриметилсилан (1,15 мл, 8,83 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Ее концентрировали досуха и совместно упаривали с дихлорметаном (2×). Этанол (5 мл) добавляли к остатку, перемешивали в течение 20 минут. Раствор концентрировали досуха и совместно упаривали с этанолом (2×). Остаток растворяли в метаноле (8 мл) и по каплям добавляли 2,0 M аммиак в метаноле (4,4 мл, 8,80 ммоль). Смесь перемешивали в течение 20 минут, концентрировали досуха и суспендировали в диэтиловом эфире. Осадок фильтровали, что позволило получить диаммоний [(6-бром-2-цианохинолин-7-ил)(дифтор)метил]фосфонат.
1H ЯМР δ (м.д.) (CD3OD): 8,69 (1H, с), 8,48 (1H, д, J=8,50 Гц), 8,39 (1H, с), 7,89 (1H, д, J=8,50 Гц), Масс-спектр
(-ионизация электрораспылением) = 361,0 и 363,0.
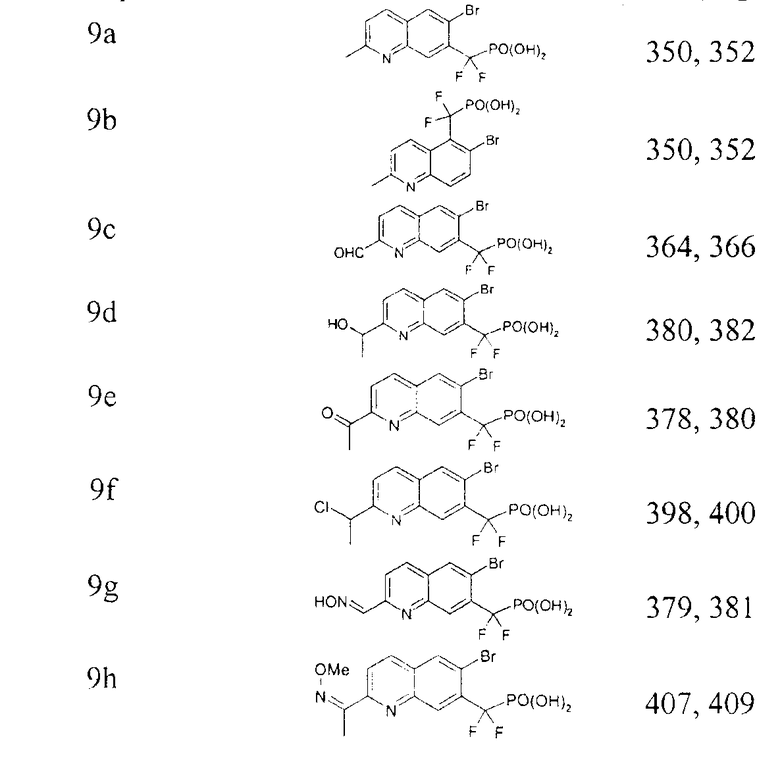
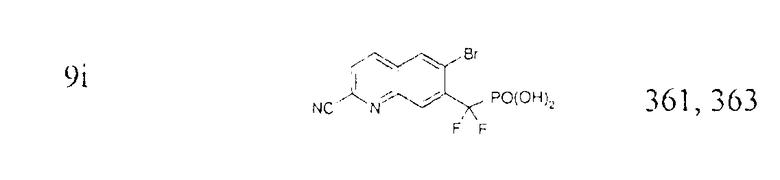
9a) Получали в присутствии TMSBr гидролизом диэтил-[(6-бром-2-метилхинолин-7-ил)(дифтор)метил]фосфоната (пример 8, стадия 3), как в примере 1, стадия 8.
9b) Получали из 6-бром-5-иод-2-метилхинолина (пример 8, стадия 2) при реакции с бромидом ((диэтоксифосфинил)дифторметил)цинка (S. Shibuya в Tetrahedron 1997, vol 53, no 3, 815), затем проводили в присутствии TMSBr гидролиз, как в примере 1, стадия 8.
9c) Получали в присутствии TMSBr гидролизом диэтил-[(6-бром-2-формилхинолин-7-ил)(дифтор)метил]фосфоната (пример 8, стадия 4), как в примере 1, стадия 8.
9d) Получали при добавлении метилмагния к диэтил-[(6-бром-2-формилхинолин-7-ил)(дифтор)метил]фосфонату (пример 8, стадия 4) при температуре от -78 до -10°C в THF, с последующим гидролизом в присутствии TMSBr, как в примере 1, стадия 8.
9e) Получали окислением диэтил-[(6-бром-2-(1-гидроксиэтил)хинолин-7-ил)(дифтор)метил]фосфоната (пример 9d) с помощью MnO2 в EtOAc при комнатной температуре в течение 1,5 ч, с последующим гидролизом в присутствии TMSBr, как в примере 1, стадия 8.
9f) Получали при реакции диэтил-[(6-бром-2-(1-гидроксиэтил)хинолин-7-ил)(дифтор)метил]фосфоната (пример 9d) с метансульфонилхлоридом и в присутствии значительного избытка DBU в CH2Cl2 при комнатной температуре в течение нескольких часов, с последующим гидролизом в присутствии TMSBr, как в примере 1, стадия 8.
9g) Получали при реакции диэтил-[(6-бром-2-формилхинолин-7-ил)(дифтор)метил]фосфоната (пример 8, стадия 4) с гидрохлоридом гидроксиламина (2 экв.) и тригидратом ацетата натрия (4 экв.) в кипящем этаноле в течение 5 ч, с последующим гидролизом в присутствии TMSBr, как в примере 1, стадия 8.
9h) Получали при реакции диэтил-[(6-бром-2-ацетилхинолин-7-ил)(дифтор)метил]фосфоната (пример 9e) с гидрохлоридом метоксиамина (3 экв.) в пиридине при 80°C в течение 2 ч, с последующим гидролизом в присутствии TMSBr, как в примере 1, стадия 8.
9i) Получали дегидрированием диэтил-[(6-бром-2-((гидроксиимино)метил)хинолин-7-ил)(дифтор)метил]фосфоната (пример 9g) в присутствии трифенилфосфина и CCl4 в CH3CN (Synth. Commun. 1990, 2785), с последующим гидролизом в присутствии TMSBr (пример 9i).
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ТРИАЗОЛА КАК ИНГИБИТОРЫ 11-БЕТА-ГИДРОКСИСТЕРОИДДЕГИДРОГЕНАЗЫ-1 | 2003 |
|
RU2360910C2 |
| ПРОЛЕКАРСТВА СОПРЯЖЕННО-БИЦИКЛИЧЕСКИХ АНТАГОНИСТОВ C5aR | 2019 |
|
RU2794327C2 |
| ИНГИБИТОРЫ ВИРУСА ГЕПАТИТА С, ИМЕЮЩИЕ ЖЕСТКУЮ ВЫТЯНУТУЮ ЦЕПЬ И СОДЕРЖАЩИЕ ФРАГМЕНТ { 2-[4-(БИФЕНИЛ-4-ИЛ)-1Н-ИМИДАЗО-2-ИЛ]ПИРРОЛИДИН-1-КАРБОНИЛМЕТИЛ} АМИНА | 2012 |
|
RU2625787C2 |
| ТИОФЕНИЛЬНЫЕ И ПИРРОЛИЛЬНЫЕ АЗЕПИНЫ В КАЧЕСТВЕ ЛИГАНДОВ СЕРОТОНИНОВОГО 5-HT РЕЦЕПТОРА И ИХ ПРИМЕНЕНИЕ | 2007 |
|
RU2434872C2 |
| СОЕДИНЕНИЕ НА ОСНОВЕ ДИГИДРОНАФТИРИДИНОНА, И СПОСОБ ЕГО ПОЛУЧЕНИЯ, И ЕГО ПРИМЕНЕНИЕ В МЕДИЦИНЕ | 2021 |
|
RU2809869C1 |
| ИНДОЛЫ, ОБЛАДАЮЩИЕ ПРОТИВОДИАБЕТИЧЕСКОЙ АКТИВНОСТЬЮ | 2003 |
|
RU2328483C2 |
| ПРОИЗВОДНЫЕ ТЕТРАГИДРОПИРИДО[3,4-b]ИНДОЛА, ПОЛЕЗНЫЕ ПРИ ЛЕЧЕНИИ РАКА | 2014 |
|
RU2664109C2 |
| ИНГИБИТОРЫ ПРОТЕИНКИНАЗЫ MKK4 ДЛЯ СТИМУЛЯЦИИ РЕГЕНЕРАЦИИ ПЕЧЕНИ ИЛИ УМЕНЬШЕНИЯ ИЛИ ПРЕДОТВРАЩЕНИЯ ГИБЕЛИ ГЕПАТОЦИТОВ | 2019 |
|
RU2788000C2 |
| НОВЫЕ СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ ИНФЕКЦИЙ МЛЕКОПИТАЮЩИХ | 2019 |
|
RU2798336C2 |
| ПРОИЗВОДНЫЕ 3-КАРБОКСИПРОПИЛ-АМИНОТЕТРАЛИНА И РОДСТВЕННЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ АНТАГОНИСТОВ MU-ОПИОИДНОГО РЕЦЕПТОРА | 2008 |
|
RU2482107C2 |
Изобретение относится к ингибиторам протеинтирозинфосфатазы 1В формулы, пригодным для лечения диабета 2 типа и рака
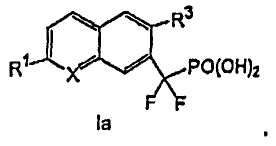
или его фармацевтически приемлемым солям, в которых X выбирают из СН и N; R1 выбирают из С1-3алкила, необязательно замещенного 1-3 галогенами или одной группой -ОН, -CN, -С(=О)Н, -С(=О)С1-3алкилом, -HC=NOH, -(CH3)C=NOH,
-НС=NОС1-3алкилом, -(СН3)С=NOC1-3алкила,
-С(=О)ОС1-3алкила, -C(=O)NHR6, -СН=СН-фенила, в котором фенил замещен -С(=О)ОН; R3 - галоген; R6 выбран из Н, С1-3алкила, фенила, и СН2-фенила, где фенил в обоих случаях необязательно замещен галогеном. Получены новые биологически активные соединения для лечения диабета 2 типа и рака, а также фармацевтические композиции на их основе. 5 н. и 5 з.п. ф-лы, 2 табл., 9 пр.
1. Соединение формулы Ia
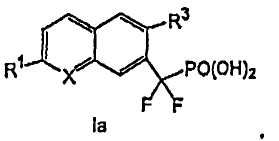
или его фармацевтически приемлемая соль,
в которых X выбирают из СН и N;
R1 выбирают из группы, состоящей из (а) С1-3алкила, необязательно замещенного 1-3 галогенами и необязательно одной группой, выбранной из -ОН и -CN, (b) -С(=О)Н, (с) -С(=О)С1-3алкила, (d) -HC=NOH, (e) -(CH3)C=NOH, (f) -НС=НОС1-3алкила, (g) -(СН3)С=NOC1-3алкила, (h) -С(=О)ОС1-3алкила, (i) -C(=O)NHR6, (j) -СН=СН-фенила, в котором фенил замещен -С(=О)ОН; и
R3 представляет собой галоген,
R6 выбран из группы, состоящей из Н, С1-3алкила, фенила и СН2-фенила,
где фенил в обоих случаях необязательно замещен галогеном.
2. Соединение формулы Ia, как определено в п.1, или его фармацевтически приемлемая соль, выбранные из следующих соединений:
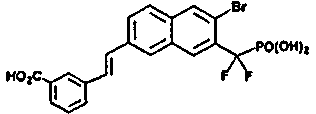
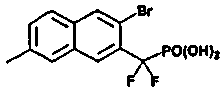
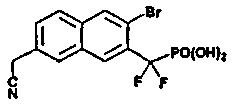
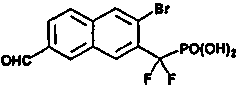
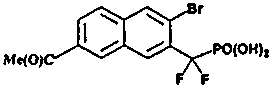
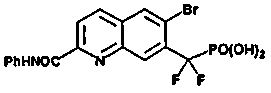
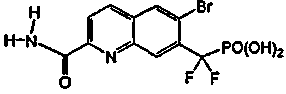
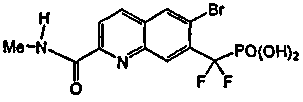
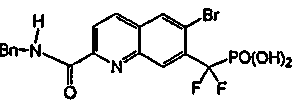
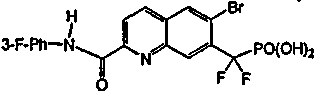
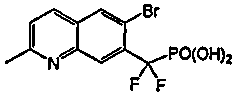
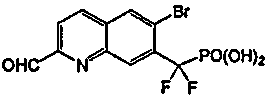
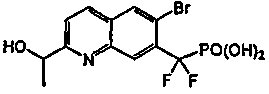
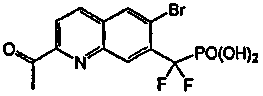
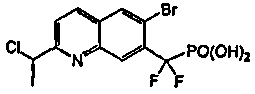
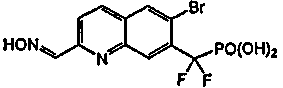
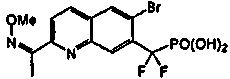
3. Фармацевтическая композиция для лечения или предупреждения заболевания, обусловленного РТР-1В, включающая соединение по п.1 или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
4. Применение соединения по п.1 или его фармацевтически приемлемой соли для изготовление лекарственного средства для лечения сахарного диабета типа 2.
5. Способ лечения сахарного диабета типа 2 у пациента, нуждающегося в лечении, включающий введение пациенту терапевтически эффективного количества соединения по п. 1 или его фармацевтически приемлемой соли.
6. Способ лечения рака у пациента, нуждающегося в лечении, включающий введение пациенту терапевтически эффективного количества соединения по п.1 или его фармацевтически приемлемой соли.
7. Соединение по п.1, выбранное из группы, состоящей из
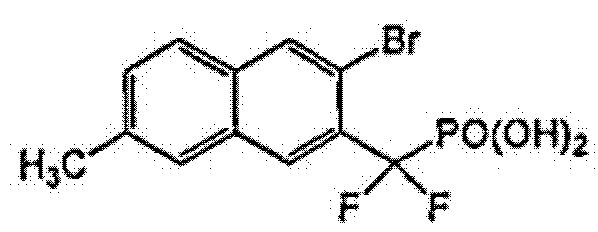
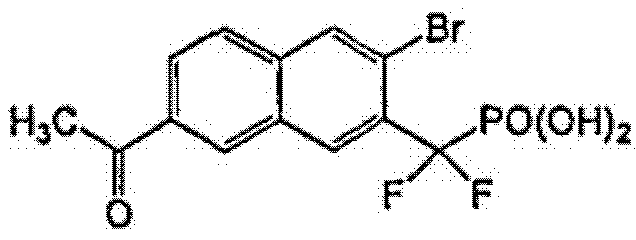
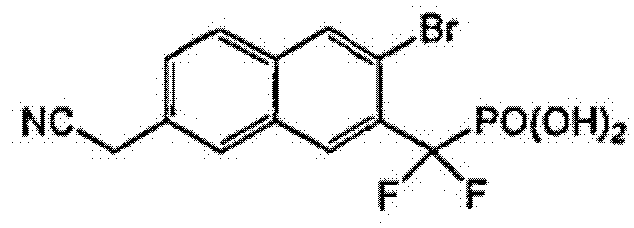
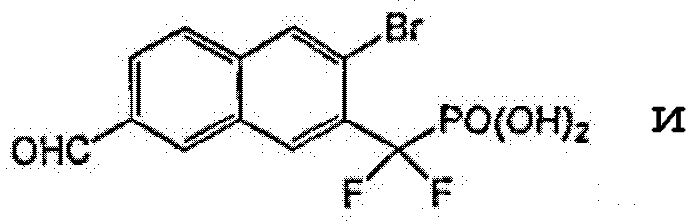
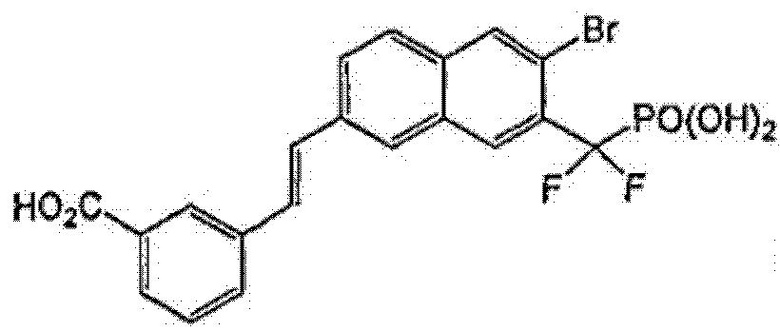
или его фармацевтически приемлемой соли.
8. Соединение по п.1, которое представляет собой
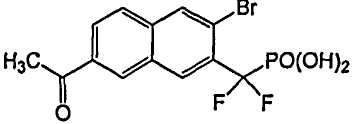
или его фармацевтически приемлемую соль.
9. Соединение по п.7, которое представляет собой
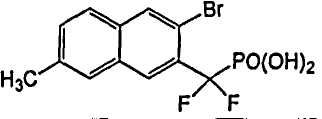
или его фармацевтически приемлемую соль.
10. Соединение по п.1, которое представляет собой
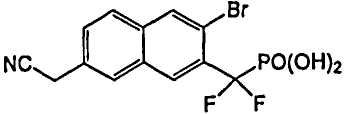
или его фармацевтически приемлемую соль.
| Jacqueline Montalibet et al | |||
| Journal of biological chemistry | |||
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| Jacqueline Montalibet et al | |||
| Biochemical pharmacology | |||
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Quingping Wang et al | |||
| Bioorganic & Medicinal Chemistry Letters | |||
| Способ и аппарат для получения гидразобензола или его гомологов | 1922 |
|
SU1998A1 |
| Tsutomu Yokomatu et al | |||
| Bioorganic & Medicinal Chemistry Letters | |||
| Металлический водоудерживающий щит висячей системы | 1922 |
|
SU1999A1 |
| WO | |||

