Объектом изобретения являются производные 1-пиперазин- и 1-гомопиперазинкарбоксилатов, их получение и их применение в терапии.
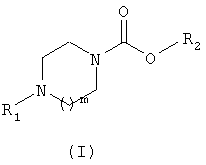
Соединения согласно изобретению соответствуют общей формуле (I)
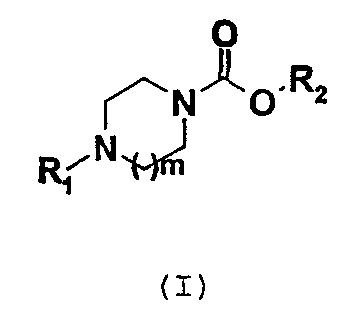
в которой m обозначает целое число, равное 1 или 2;
R1 обозначает группу, выбранную, в частности, из фенила, пиридинила, пиридазинила, пиримидинила, пиразинила, триазинила, оксазолила, тиазолила, имидазолила, оксадиазолила, тиадиазолила, триазолила, нафтила, хинолинила, тетрагидрохинолинила, изохинолинила, тетрагидроизохинолинила, фталазинила, хиназолинила, хиноксалинила, циннолинила, нафтиридинила, бензофуранила, дигидробензофуранила, бензотиенила, дигидробензотиенила, индолила, бензоксазолила, бензизоксазолила, бензотиазолила, бензизотиазолила, бензимидазолила, индазолила, пирролопиридинила, фуропиридинила, дигидрофуропиридинила, тиенопиридинила, дигидротиенопиридинила, имидазопиридинила, имидазопиримидинила, пиразолопиридинила, оксазолопиридинила, изоксазолопиридинила, тиазолопиридинила, изотиазолопиридинила,
при этом указанная группа возможно замещена одной или несколькими группами R3, одинаковыми или отличающимися друг от друга или группой R4,
R2 обозначает группу общей формулы CHR5CONHR6,
R3 обозначает атом галогена или группу: гидрокси, циано, нитро, С1-6-алкил, С1-6-алкокси, С1-6-тиоалкил, С1-6-фторалкил, С1-6-фторалкокси, -О-(С2-3-алкилен)-, -О-(С1-3-алкилен)-О-, С1-6-фтортиоалкил, С3-7-циклоалкил, С3-7-циклоалкил-С1-3-алкилен, пиперидинил, бензилокси, пиперазинил, пирролидинил, морфолинил, фенилокси, NR7R8, NHCOR7, NHSO2R7, COR7, CO2R7, CONR7R8, SO2R7 или SO2NR7R8,
R4 обозначает группу, выбранную, в частности, из фенила, бензофуранила, нафтила, пиридинила, пиримидинила, пиридазинила, пиразинила, триазинила, оксазолила, тиазолила, имидазолила, оксадиазолила, тиадиазолила, триазолила, хинолинила, тетрагидрохинолинила, изохинолинила, тетрагидроизохинолинила, фталазинила, хиназолинила, хиноксалинила, нафтиридинила, циннолинила, имидазолпиримидинила, бензотиенила, индолила, бензоксазолила, бензизоксазолила, бензотиазолила, бензизотиазолила, бензимидазолила, индазолила, пирролопиридинила, фуропиридинила, дигидрофуропиридинила, тиенопиридинила, дигидротиенопиридинила, имидазопиридинила, имидазопиримидинила, пиразопиридинила, оксазолопиридинила, изоксазолопиридинила, тиазолопиридинила, изотиазолопиридинила;
при этом одна или несколько групп R4 могут быть замещены одной или несколькими группами R3, одинаковыми или отличающимися друг от друга;
R5 обозначает атом водорода или С1-3-алкильную группу;
R6 обозначает атом водорода или С1-6-алкильную, С3-7-циклоалкильную или С3-7-циклоалкил-С1-3-алкиленовую группу;
R7 и R8 обозначают независимо друг от друга атом водорода, С1-3-алкильную группу или фенильную группу.
Из соединения общей формулы (I) первая подгруппа соединений состоит из соединений, в которых:
m обозначает целое число, равное 1 или 2; и/или
R1 обозначает группу, выбранную, в частности, из фенила, пиридинила, пиримидинила, пиразинила, нафтила, хинолинила, изохинолинила, бензизоксазолила, тиенопиридинила, причем указанная группа возможно замещена одной или несколькими группами R3, более конкретно одной или двумя группами R3, одинаковыми или отличающимися друг от друга; и/или
R2 обозначает группу общей формулы CHR5CONHR6, и/или
R3 обозначает атом галогена, более конкретно хлор, бром или фтор, или группу циано, С1-6-алкильную, более конкретно метил, этил, n-пропил, изобутил, С1-6-алкокси, более конкретно метокси, С1-6-фторалкильную, более конкретно CF3, С1-6-фторалкокси, более конкретно -OCH2CF3, -О-(С2-3-алкилен)-, более конкретно -О-(СН2)3-, фенилокси; и/или
R5 обозначает атом водорода; и/или
R6 обозначает атом водорода или С1-6-алкильную группу, более конкретно метил.
Из соединения общей формулы (I) вторая подгруппа соединений состоит из соединений, в которых:
m равно 1; и/или
R1 обозначает группу, выбранную, в частности, из пиридинила, пиримидинила, пиразинила, хинолинила, изохинолинила, причем указанная группа возможно замещена группой R3; и/или
R2 обозначает группу общей формулы CHR5CONHR6; и/или
R3 обозначает атом галогена, более конкретно хлор, или С1-6-алкильную группу, более конкретно метил, этил, н-пропил, изобутил, С1-6-алкокси, более конкретно метокси, С1-6-фторалкил, более конкретно CF3; и/или
R5 обозначает атом водорода; и/или
R6 обозначает атом водорода или С1-6-алкильную группу, более конкретно метил.
Из соединения общей формулы (I) третья подгруппа соединений состоит из соединений, в которых:
m обозначает целое число, равное 1 или 2; и/или
R1 обозначает группу, выбранную, в частности, из фенила, пиридинила, пиридазинила, пиримидинила, тиадиазолила, причем указанная группа возможно замещена группой R4; и/или
R4 обозначает группу, выбранную, в частности, из фенила, бензофуранила, нафтила; причем группа R4 возможно замещена одной или несколькими группами R3, одинаковыми или отличающимися друг от друга, более конкретно одной или двумя группами R3, одинаковыми или отличающимися друг от друга; и/или
R2 обозначает группу общей формулы CHR5CONHR6; и/или
R3 обозначает атом галогена, более конкретно хлор, бром или фтор, группу нитро, С1-6-алкильную, более конкретно метил, изопропил, С1-6-алкокси, более конкретно метокси, этокси, С1-6-фторалкильную, более конкретно CF3, С1-6-фторалкокси, более конкретно OCF3, -O-(С1-3-алкилен)-O-, более конкретно -O-CH2-O-, бензилокси; и/или
R5 обозначает атом водорода; и/или
R6 обозначает атом водорода или С1-6-алкильную группу, более конкретно метил или этил, или С3-7-циклоалкил-С1-3-алкилен, более конкретно циклопропил-СН2-.
Из соединения общей формулы (I) четвертая подгруппа соединений состоит из соединений, в которых:
m равно 1; и/или
R1 обозначает группу, выбранную, в частности, из фенила, пиридинила, пиридазинила, пиримидинила,
причем указанная группа возможно замещена группой R4; и/или
R4 обозначает группу, выбранную, в частности, из фенила, бензофуранила, нафтила; причем группа R4 возможно замещена одной или несколькими группами R3, одинаковыми или отличающимися друг от друга, более конкретно одной или двумя группами R3, одинаковыми или отличающимися друг от друга; и/или
R2 обозначает группу общей формулы CHR5CONHR6; и/или
R3 обозначает атом галогена, более конкретно хлор, бром или фтор, или группу нитро, С1-6-алкильную, более конкретно метил, изопропил, С1-6-алкокси, более конкретно метокси, этокси, С1-6-фторалкильную, более конкретно CF3, С1-6-фторалкокси, более конкретно OCF3, -O-(С1-3-алкилен)-O-, более конкретно -O-CH2-O-, бензилокси; и/или
R5 обозначает атом водорода; и/или
R6 обозначает атом водорода или С1-6-алкильную группу, более конкретно метил или этил.
Соединения общей формулы (I) могут содержать один или несколько асимметричных атомов углерода. Они могут иметь форму энантиомеров или диастереоизомеров. Эти энантиомеры или диастереоизомеры, а также их смеси, включая рацемические смеси, являются составной частью изобретения.
Соединения формулы (I) могут быть в виде оснований или солей присоединения кислот. Такие соли присоединения являются составной частью изобретения.
Эти соли преимущественно получают с использованием фармацевтически приемлемых кислот, но соли других кислот, пригодных, например, для очистки или выделения соединений формулы (I) также являются составной частью изобретения.
Соединения формулы (I) могут быть в виде гидратов или сольватов, а именно в виде ассоциатов или комбинаций с одной или несколькими молекулами воды или с сольватом. Такие гидраты и сольваты также являются составной частью изобретения.
В рамках изобретения понимают под:
-Сt-z, где t и z могут иметь значения от 1 до 7, углеродную цепь, которая может иметь от t до z атомов углерода, например, С1-3-углеродную цепь, которая может содержать от 1 до 3 атомов углерода,
-алкилом - алифатическую, насыщенную, линейную или разветвленную группу, например, С1-3-алкильная группа обозначает углеродную цепь, содержащую от 1 до 3 атомов углерода, линейную или разветвленную, более конкретно метил, этил, пропил, 1-метилэтил,
-алкиленом - двухвалентную алкильную группу, насыщенную, линейную или разветвленную, например, С1-3-алкиленовая группа обозначает двухвалентную углеродную цепь, содержащую от 1 до 3 атомов углерода, линейную или разветвленную, более конкретно метилен, этилен, 1-метилэтилен, пропилен,
-циклоалкилом - циклическую алкильную группу, например, С3-5-циклоалкильная группа обозначает циклическую углеродную группу, содержащую от 3 до 5 атомов углерода, более конкретно циклопропил, циклобутил, циклопентил,
-алкокси - группу -О-алкильную, имеющую насыщенную линейную или разветвленную алифатическую цепочку,
-тиоалкилом - группу -S-алкильную, имеющую насыщенную линейную или разветвленную алифатическую цепочку,
-фторалкилом - алкильную группу, один или несколько атомов водорода которой были замещены атомом фтора,
-фторалкокси - группу алкокси, один или несколько атомов водорода которой были замещены атомом фтора,
-фтортиоалкильную - тиоалкильную группу, один или несколько атомов водорода которые были замещены атомом фтора,
-атомом галогена - фтор, хлор, бром или иод.
Соединения согласно изобретению можно получить различными способами, которые иллюстрируют приведенные ниже схемы.
Так первый способ получения (схема 1) заключается в том, что приводят во взаимодействие амин общей формулы (II), в которой R1 и m имеют те же значения, которые определены для общей формулы (I), и карбонат общей формулы (III), в которой Z обозначает атом водорода или нитрогрупу и R2 имеет то же значение, которое определено для общей формулы (I), в растворителе, таком как толуол или дихлорэтан, при температуре от 0 до 80°С.
Схема 1
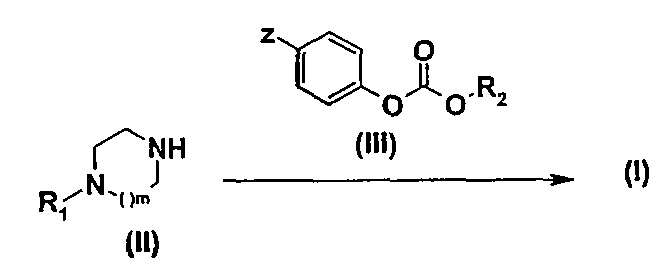
Карбонаты общей формулы (III) можно получить любыми способами, описанными в литературе, например, путем взаимодействия спирта общей формулы HOR2 и фенилхлорформиата или 4-нитрофенила в присутствии основания, такого как триэтиламин или диизопропилэтиламин при температуре от 0°С до температуры кипения растворителя.
В соответствии со вторым способом (схема 2) соединения общей формулы (I) можно получить путем взаимодействия амина общей формулы (II), такой как описана выше, и карбоната общей формулы (IIIa), в которой Z обозначает атом водорода или нитрогруппу, R5 имеет то же значение, которое определено для общей формулы (1), и R обозначает метильную или этильную группу.
Полученный таким образом сложный карбаматный эфир общей формулы (Ia) затем превращают в соединение общей формулы (I) путем аминолиза с помощью амина общей формулы R6NH2, в которой R6 имеет тоже значение, которое определено для общей формулы (I). Реакцию аминолиза можно провести в растворителе, таком как метанол или в смеси растворителей, таких как метанол и терагидрофуран или метанол и диоксан.
Схема 2
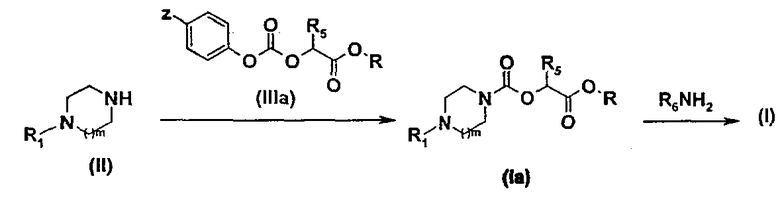
Карбонаты общей формулы (IIIa) можно получить любым способом, описанным в литературе, например, путем взаимодействия спирта общей формулы HOCHR5COOR, где R обозначает метильную или этильную группу, и фенилхлорформиата или 4-нитрофенила в присутствии основания, такого как триэтиламин или диизопропилэтиламин.
Соединения общей формулы (I), в которой R1 обозначает группу, замещенную группой R3 типа С1-6-алкила, С3-7-циклоалкила или С3-7-циклоалкил-С1-3-алкилена, или группой R4, такой как определена в общей формуле (I), можно также получить путем реакции Судзуки с использованием соответствующих соединений общей формулы (I), в которых R1 замещен атомом хлора, брома, йода или трифталатной группой в положении, в котором должна быть введена группа R3 или R4 , например, с помощью бороновой кислоты алкила, циклоалкила, арила или гетероарила.
Для соединений общей формулы (I), в которой R1 обозначает группу, замещенную группой R3 типа С1-6-алкила, С3-7-циклоалкила или С3-7-циклоалкил-С1-3-алкилена, или группой R4, такой как определена в общей формуле (I), и R2 более конкретно обозначает группу общей формулы CHR5CONHR6, реакцию Судзуки, описанную выше, можно провести с карбаматным эфиром общей формулы (Ia), такой как описана выше. Действием амина общей формулы R6NH2, такой как описана выше, на полученный таким образом карбонат - эфир можно получить соединения общей формулы (I).
Соединения общей формулы (II), если способ их получения не описан, являются коммерчески доступными или описаны в литературе, или их можно получить методами, которые там описаны, или которые известны специалисту в данной области.
Амины общей формулы R6NH2 являются коммерчески доступными в соответствии с другим аспектом изобретения его объектом являются также соединения общей формулы (Ia). Эти соединения пригодны в качестве промежуточных продуктов синтеза соединений общей формулы (I).
Приведенные ниже примеры иллюстрируют получение нескольких соединений согласно изобретению. Эти примеры только иллюстрируют изобретение, не ограничивая его. Микроанализы, инфракрасные спектры и спектры ЯМР и/или ЖХ-МС (жидкостная хроматография в сочетании с масс-спектроскопией) подтверждают структуры и степень чистоты полученных соединений.
ТП(°С) обозначает точку плавления в градусах Цельсия.
Номера, указанные в скобках в названии примеров, соответствуют номерам первой колонки приведенной ниже таблицы.
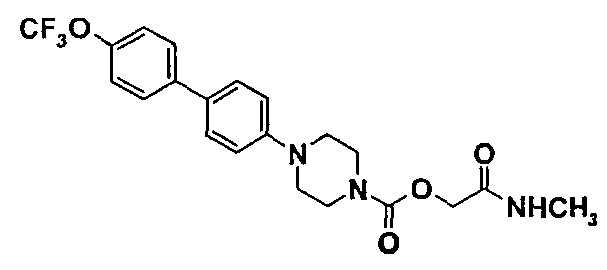
Пример 1 (соединение №44)
2-(метиламино)-2-оксоэтил-4-{4'-[(трифторметил)окси]-4-бифенилил}-1-пиперазинкарбоксилат
1.1. Этил[(фенилоксикарбонил)окси]ацетат
В раствор 25 г (240 ммоль) этилгликолята и 55 мл (315 ммоль) диизопропилэтиламина в 500 мл толуола медленно вводят при комнатной температуре 32 мл (256 ммоль) фенилхлорформиата. Перемешивают в течение 2 часов при комнатной температуре.
Отделяют образовавшуюся соль и концентрируют фильтрат при пониженном давлении.
Получают 53,7 г маслянистого продукта, который используют как таковой на следующей стадии.
1.2. 2-(этилокси)-2-оксоэтил-4-(4-бромфенил)-1-пиперазинкарбоксилат
В течение 12 часов нагревают до 80°С раствор 5,81 г (24,08 ммоль) 1-(4-бромфенил)пиперазина и 6г (26,76 ммоль) этил[(фенилоксикарбонил)окси]ацетата, полученного на стадии 1.1, в 50 мл толуола.
Охлаждают до комнатной температуры, концентрируют при пониженном давлении, затем очищают полученный таким образом остаток хроматографией на колонке с силикагелем, элюируя смесью 20/80, затем 30/70 этилацетата и циклогекана.
Таким образом, получают 7,75 г чистого продукта в виде масла, которое кристаллизуется при комнатной температуре.
ТП(°С): 80-82°С.
1.3. 2-(этилокси)-2-оксоэтил-4-{4'-[(трифторметил)окси]-4-бифенилил}-1-пиперазинкарбоксилат
В инертной атмосфере вводят 2 г (5,39 ммоль) 2-(этилокси)-2-оксоэтил-4-(4-бромфенил)-1-пиперазинкарбоксилата, полученного на стадии 1.2., 3,33 г (16,16 ммоль) 4-(трифторметокси)фенил бороновой кислоты и 4,57 г (21,55 ммоль) гидратированного фосфатата калия, суспендированного в 18 мл 1,2-диметоксиэтана. Затем вводят 0,62 г (0,54 ммоль) тетракис(трифенилфосфин) палладия. Реакционную смесь нагревают до 80°С в течение 12 часов.
Концентрируют при пониженном давлении. Обрабатывают остаток дихлорметаном и водой, отделяют водную фазу, дважды экстрагируют дихлорметаном, сушат объединенные органические фазы на сульфате натрия и концентрируют фильтрат при пониженном давлении. Полученный таким образом остаток очищают хроматографией на силикагеле, элюируя смесью 30/70 этилацетата и циклогексана.
Получают 1,65 г продукта в виде белого твердого вещества.
ТП(°С): 112-116°С.
1.4. 2-(метиламино)-2-оксоэтил-4-{4'-[(трифторметил)окси]-4-бифенилил}-1-пиперазинкарбоксилат
В раствор 1,60 г (3,54 ммоль) 2-(этилокси)-2-оксоэтил-4-{4'-[(трифторметил)окси]-4-бифенилил}-1-пиперазинкарбоксилата, полученного на стадии 1.3., в 14 мл метанола вводят 7,10 мл (14,15 ммоль) раствора метиламина (2М) в тетрагидрофуране. Перемешивают при комнатной температуре в течение 12 часов.
После концентрирования при пониженном давлении очищают полученный остаток хроматографией на силикагеле, элюируя смесью 97/3 дихлорметана и метанола. Получают твердое вещество, которое перекристаллизовывают в смеси этилацетата и простого диизопропилового эфира.
Получают таким образом 0,86 г чистого продукта в виде белого твердого вещества.
ЖХ-МС: М+Н=438.
ТП(°С): 187-198°С.
ЯМР1Н (CDCL3)δ (ч./млн): 2,90 (д, 3Н); 3,25 (м, 4Н); 3,70 (м, 4Н); 4,60 (с, 2Н); 6,10 (уш.с., 1Н); 7,0 (д, 2Н); 7,30 (д, 2Н); 7,50 (д,2Н); 7,60 (д, 2Н).
Пример 2 (Соединение № 37)
2-(метиламино)-2-оксоэтил-4-[3'-(трифторметил)-4-бифенилил]-1-пиперазинкарбоксилат
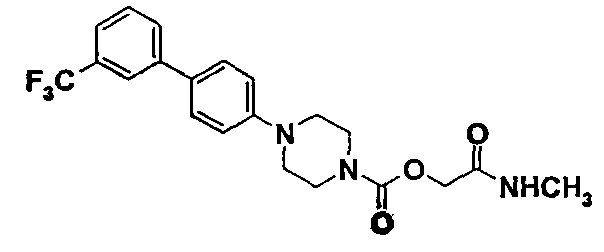
2.1. 2-(метиламино)-2-оксоэтил-4-нитрофенилкарбонат
В суспензию 2,62 г (29,4 ммоль) 2-гидрокси-N-метилацетамида и 16,5 г (58,7 ммоль) диизопропилэтиламина на носителе (Ps-DIEA Argonaut, загрузка = 3,56 ммоль/г) в 250 мл дихлорметана вводят маленькими порциями и при комнатной температуре 5,93 г (29,4 ммоль) 4-нитрофенилхлорформиата. Продолжают орбитальное перемешивание при комнатной температуре в течение 16 часов.
Фильтруют смолу, ополаскивают 150 мл дихлорметана и концентрируют фильтрат при пониженном давлении.
Получают 6 г продукта в виде светло-желтого твердого вещества (приблизительная степень очистки 70%), которое используют как таковое на следующей стадии.
2.2. 2-(метиламино)-2-оксоэтил-4-(4-бромфенил]-1-пиперазинкарбоксилат
В раствор 1,47 г (4 ммоль) 2-(метиламино)-2-оксоэтил-4-нитрофенилкарбоната, полученного на стадии 2.1., в 18 мл 1,2-дихлорэтана вводят 1,17 г (4,85 ммоль) 1-(4-бромфенил)пиперазина. Эту реакционную смесь нагревают до 65°С в течение 2,25 часов.
Охлаждают до комнатной температуры, затем концентрируют при пониженном давлении. Желтый маслянистый остаток обрабатывают дихлорметаном и последовательно промывают гидроксидом натрия (1н), водой, 5%-ным водным раствором лимонной кислоты, водой, затем рассолом. Эту органическую фазу сушат на сульфате натрия и концентрируют при пониженном давлении. После промывания простым диизопропиловым эфиром получают 1,3 г продукта в виде белого твердого вещества.
2.3. Синтез катализатора, содержащего палладий, привитый на смоле Merrifield
В инертной атмосфере в суспензию 5г (3,5 ммоль) смолы Merrifield (Fluka, 200-400 Mesh, сшитой с 2% дивинилбензола (ДВБ), загрузка = 0,7 ммоль/г) в 50 мл безводного тетрагидрофурана (ТГФ) вводят 54,6 мл (27,3 ммоль) раствора коммерческого дифенилфосфина лития в концентрации 0,5М в ТГФ. Орбитальное перемешивание продолжают в течение 24 часов при комнатной температуре, затем вводят 60 мл ацетона и 20 мл воды. Фильтруют смолу и последовательно промывают водой, ацетоном, ТГФ, смесью ТГФ/Н2О (2/1), ТГФ, толуолом, дихлорметаном и простым этиловым эфиром, затем сушат в вакууме в течение 2 часов.
Полученную таким образом суспензию смолы нагревают до 70°С в течение 24 часов в 47 мл этанола и 23 мл толуола. После фильтрования смолу последовательно промывают ацетоном, ТГФ и простым этиловым эфиром. В целом эту обработку повторяют четыре раза для удаления растворимых фракций полимера. Полученную таким образом смолу сушат в вакууме в течение 2 часов.
В суспензию этой смолы в 60 мл толуола вводят 0,18 г (0,16 ммоль) тетракис(трифенилфосфина) палладия и нагревают эту реакционную смесь до 95°С в течение 24 часов.
Охлаждают до комнатной температуры, фильтруют смолу и последовательно промывают ацетоном, ТГФ, затем простым этиловым эфиром. Получают 5,135 г смолы, которую используют как таковую на следующей стадии.
2.4. 2-(метиламино)-2-оксоэтил-4-[3'-(трифторметил)-4-бифенилил]-1-пиперазинкарбоксилат
Вводят 0,18 г (0,5 ммоль) 2-(метиламино)-2-оксоэтил-4-(4-бромфенил]-1-пиперазинкарбоксилата, полученного на стадии 2.2., 0,21 г (1,1 ммоль) 3-(трифторметил)фенилбороновой кислоты и 0,16 г (1,5 ммоль) карбоната натрия, суспендированного в 3 мл толуола и 0,3 мл этанола. Затем вводят 0,14 г (приблизительно 10 мол. %) катализатора, содержащего палладий на носителе, полученного на стадии 2.3, и продолжают орбитальное перемешивание при 80°С в течение 48 часов.
Охлаждают до комнатной температуры, фильтруют смолу, ополаскивают дихлорметаном и концентрируют фильтрат при пониженном давлении.
Остаток обрабатывают 5 мл дихлорметана и промывают водой, затем насыщенным водным раствором бикарбоната натрия. Органическую фазу фильтруют через гидрофобный элемент, затем концентрируют фильтрат при пониженном давлении.
Получают маслянистый остаток, который кристаллизуется в простом диизопропиловом эфире.
Получают 0,15 г белых кристаллов.
ЖХ-МС: М+Н=422.
ТП(°С): 129-130°С.
ЯМР1Н (CDCL3)δ (ч./млн): 2,95 (д, 3Н); 3,20-3,25 (м, 4Н); 3,65-3,80 (м, 4Н); 4,65 (с, 2Н); 6,05 (уш.с. 1Н); 7,05 (д, 2Н); 7,50-7,60 (м, 4Н); 7,65-7,80 (м, 2Н).
Пример 3 (Соединение № 76)
2-(метиламино)-2-оксоэтил-4-{5-[3-(трифторметил)фенил]-2-пиридинил}-1-пиперазинкарбоксилат
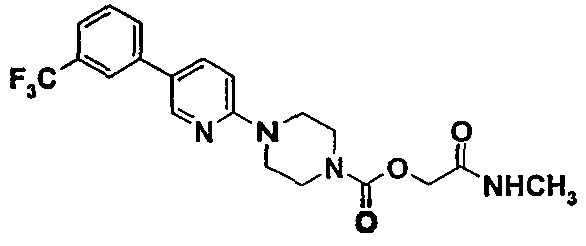
3.1. 1,1-диметилэтил-4-(5-бром-2-пиридинил)-1-пиперазинкарбоксилат
В автоклав вводят 29,2 г (157 ммоль) 1,1-диметилэтил-1-пиперазинкарбоксилата, 37 г (157 ммоль) 2,5-дибромпиридина и 21,7 г (157 ммоль) карбоната калия, суспендированного в 27 мл диметилсульфоксида (ДМСО). Затем нагревают до 150°С в течение 21 часа.
Охлаждают до комнатной температуры, обрабатывают реакционную смесь этилацетатом и водой, затем отделяют не растворившееся вещество фильтрованием. Отделяют водную фазу, экстрагируют два раза этилацетатом, промывают объединенные органические фазы насыщенным водным раствором хлорида натрия, сушат их на сульфате натрия и концентрируют фильтрат при пониженном давлении. Остаток очищают хроматографией на силикагеле, элюируя смесью 99/1 дихлорметана и метанола.
Таким образом получают 44 г продукта в виде белого твердого вещества.
ТП(°С): 83-85°С.
3.2. 1-(5-бром-2-пиридинил)пиперазин
В раствор 18,60 г (54,40 ммоль) 1,1-диметилэтил-4-(5-бром-2-пиридинил)-1-пиперазинкарбоксилата, полученного на стадии 3.1., в 100 мл 1,4-диоксана вводят при комнатной температуре 49 мл (272 ммоль) раствора соляной кислоты (6н) в изопропаноле. Затем реакционную смесь нагревают до 60°С в течение 3 часов. Концентрируют досуха при пониженном давлении. Полученный дигидрохлорид обрабатывают 200 мл дихлорметана и 200 мл воды, затем добавляют маленькими порциями при перемешивании 10 г гидрокарбоната натрия. Декантируют, водную фазу дважды экстрагируют дихлорметаном, промывают объединенные органические фазы насыщенным водным раствором хлорида натрия, сушат их на сульфате натрия и концентрируют при пониженном давлении.
Получают 12 г продукта в виде белого твердого вещества.
ТП(°С): 72°С.
3.3. 2-(этилокси)-2-оксоэтил-4-(5-бром-2-пиридинил)-1-пиперазинкарбоксилат
Порядок выполнения работы такой же, как описан в примере 1 (стадия 1.2). Из 6 г (24,80 ммоль) 1-(5-бром-2-пиридинил)пиперазина, полученного на стадии 3.2, и 10,88 г (48,52 ммоль) этил[(фенилоксикарбонил)окси]ацетата, полученного на стадии 1.1. примера 1, и после проведения хроматографии на силикагеле, элюируя смесью 15/85, затем 30/70 этилацетата и циклогексана, получают 6,70 г продукта в виде масла, которое кристаллизуется в белое твердое вещество.
3.4. 2-(этилокси)-2-оксоэтил-4-{5-[3-(трифторметил)фенил] -2-пиридинил}-1-пиперазинкарбоксилат
Порядок выполнения работы такой же, как описан в примере 1 (стадия 1.3.). Из 3 г (8,06 ммоль) 2-(этилокси)-2-оксоэтил-4-(5-бром-2-пиридинил)-1-пиперазинкарбоксилата, полученного на стадии 3.3., 4,59 г (24,17 ммоль) 3-(трифторметил)фенилбороновой кислоты, 6,84 г (32,23 ммоль) гидратированного фосфата калия и 0,93 г (0,806 ммоль) тетракис(трифенилфосфин) палладия и после проведения хроматографии на силикагеле, элюируя смесью 30/70 этилацетата и циклогексана, получают 2,22 г продукта в виде белого твердого вещества.
3.5. 2-(метиламино)-2-оксоэтил-4-{5-[3-(трифторметил)фенил]-2-пиридинил}-1-пиперазинкарбоксилат
Порядок выполнения работы такой же, как описан в примере 1 (стадия 1.4.). Из 1,50 г (3,43 ммоль) 2-(этилокси)-2-оксоэтил-4-{5-[3-(трифторметил)фенил]-2-пиридинил}-1-пиперазинкарбоксилата, полученного на стадии 3.4, и 8,6 мл (17,15 ммоль) раствора метиламина (2М) в тетрагидрофуране и после проведения хроматографии на силикагеле, элюируя смесью 97/3 дихлорметана и метанола, с последующим промыванием простым диизопрпиловым эфиром, получают 1,18 г продукта в виде белого твердого вещества.
ЖХ-МС: М+Н=423.
ТП(°С): 158-160°С.
ЯМР1Н (CDCL3)δ (ч./млн): 2,90 (д, 3Н); 3,75 (уш.с., 8Н); 4,65 (с, 2Н); 6,05 (уш.с. 1Н); 6,75 (д, 1Н); 7,50-7,80 (массив, 5Н); 8,50 (д, 1Н).
Пример 4 (соединение №79)
2-(метиламино)-2-оксоэтил-4-{5-[4-(трифторметил)фенил]-2-пиридинил}-1-пиперазинкарбоксилат
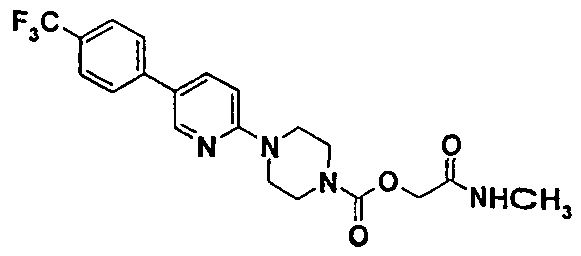
4.1. 2-(этилокси)-2-оксоэтил-4-{5-[4-(трифторметил)фенил]-2-пиридинил}-1-пиперазинкарбоксилат
Порядок выполнения работы такой же, как описан в примере 1 (стадия 1.3.). Из 4 г (10,75 ммоль) 2-(этилокси)-2-оксоэтил-4-(5-бром-2-пиридинил)-1-пиперазинкарбоксилата, полученного на стадии 3.3. примера 3, 5,50 г (28,96 ммоль) 4-(трифторметил)фенилбороновой кислоты, 9,12 г (42,99 ммоль) гидриратированного фосфата калия и 1,24 г (1,07 ммоль) тетракис(трифенилфосфин) палладия и после проведения хроматографии на силикагеле, элюируя смесью 30/70 этилацетата и циклогексана, получают 2,78 г продукта в виде белого твердого вещества.
4.2. 2-(метиламино)-2-оксоэтил-4-{5-[4-(трифторметил)фенил]-2-пиридинил}-1-пиперазинкарбоксилат
Порядок выполнения работы такой же, как описан в примере 1 (стадия 1.4.). Из 2,77 г (6,33 ммоль) 2-(этилокси)-2-оксоэтил-4-{5-[4-(трифторметил)фенил]-2-пиридинил}-1-пиперазинкарбоксилата, полученного на стадии 4.1, и 15,80 мл (31,67 ммоль) раствора метиламина (2М) в тетрагидрофуране и после проведения хроматографии на силикагеле, элюируя смесью 97/3 дихлорметана и метанола, с последующей перекристаллизацией в этилацетате, получают 1,69 г продукта в виде белого твердого вещества.
ЖХ-МС: М+Н=423.
ТП(°С): 206-209°С.
ЯМР1Н (CDCL3)δ (ч./млн): 2,90 (д, 3Н); 3,70 (уш.с., 8Н); 4,65 (с, 2Н); 6,05 (уш.с. 1Н); 6,75 (д, 1Н); 7,60-7,75 (м, 4Н); 7,80 (дд, 1Н); 8,50 (д, 1Н).
Пример 5 (соединение №83)
2-(метиламино)-2-оксоэтил-4-{5-[4-(трифторметил)окси]фенил}-2-пиридинил}-1-пиперазинкарбоксилат
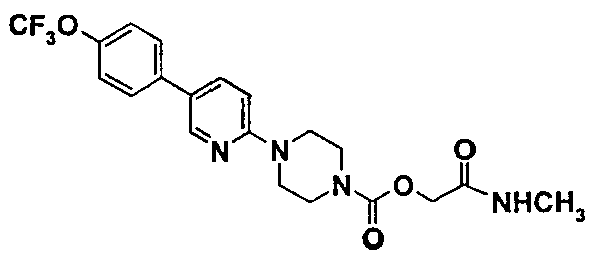
5.1. 1,1-диметилэтил-2-(метиламино)-2-оксоэтил-1,4-пиперазиндикарбоксилат
В охлажденный до 0°С раствор 1,1 г (3 ммоль) 2-(метиламино)-2-оксоэтил-4-нитрофенилкарбоната, полученного на стадии 2.2. примера 2, в 10 мл 1,2-дихлорэтана вводят по каплям и примерно при 0°С раствор 0,53 г (2,85 ммоль) 1,1-диметилэтил-1-пиперазинкарбоксилата в 5 мл 1,2-дихлорэтана. Перемешивают при 0°С в течение 1 часа, затем при комнатной температуре в течение 3 часов.
Концентрируют при пониженном давлении и очищают полученный таким образом остаток хроматографией на силикагеле, элюируя смесью 20/80 этилацетата и циклогексана. Постепенно увеличивают градиент с тем, чтобы завершить элюирование этилацетатом.
Получают маслянистый остаток, который кристаллизуется в простом диизопропиловом эфире.
Получают 0,61 г продукта в виде белого твердого вещества, которое используют как таковое на следующей стадии.
5.2. Гидрохлорид 2-(метиламино)-2-оксоэтил-1-пиперазинкарбоксилата
В раствор 2,68 г (8,9 ммоль) 1,1-диметилэтил-2-(метиламино)-2-оксоэтил-1,4-пиперазиндикарбоксилата, полученного на стадии 5.1., в 25 мл дихлорметана вводят 25 мл раствора 6н соляной кислоты в изопропаноле. Перемешивают при комнатной температуре в течение 1 часа.
Органическую фазу удаляют фильтрованием через гидрофобный элемент и кислую водную фазу концентрируют при пониженном давлении.
После кристаллизации в изопропаноле получают 2,05 г продукта в виде белого твердого вещества, которое используют как таковое на следующей стадии.
ТП(°С): 167-169°С.
5.3. 2-(метиламино)-2-оксоэтил-4-(5-нитро-2-пиридинил)-1-пиперазинкарбоксилат
В раствор 2,05 г (8,62 ммоль) гидрохлорида 2-(метиламино)-2-оксоэтил-1-пиперазинкарбоксилата, полученного на стадии 5.2., и 3,85 мл (22,4 ммоль) N,N-диизопропилэтиламина в 55 мл 1,2-дихлорэтана вводят 1,84 г (11,6 ммоль) 2-хлор-5-нитропиридина. Реакционную смесь нагревают до 70°С в течение 5 часов.
Охлаждают до комнатной температуры, концентрируют при пониженном давлении и полученный таким образом остаток очищают хроматографией на силикагеле, элюируя смесью 98/2 дихлорметана и метанола.
Получают 2,48 г продукта в виде светло-желтого твердого вещества, которое используют как таковое на следующей стадии.
5.4. 2-(метиламино)-2-оксоэтил-4-(5-амино-2-пиридинил)-1-пиперазинкарбоксилат
В суспензию 0,64 г (1,98 ммоль) 2-(метиламино)-2-оксоэтил-4-(5-нитро-2-пиридинил)-1-пиперазинкарбоксилата, полученного на стадии 5.3., в 90 мл этилацетата вводят 0,24 г 10%-ного палладия на угле. Перемешивают при комнатной температуре в атмосфере водорода 60 psi в течение 14 часов. После фильтрования катализатора фильтрат концентрируют при пониженном давлении и полученный таким образом остаток очищают хроматографией на силикагеле, элюируя смесью 98/2 дихлорметана и метанола.
Получают 0,47 г продукта в виде фиолетового маслянистого вещества, которое используют как таковое на следующей стадии.
5.5. 2-(метиламино)-2-оксоэтил-4-(5-йод-2-пиридинил)-1-пиперазинкарбоксилат
В охлажденный до 0°С раствор 0,47 г (1,5 ммоль) 2-(метиламино)-2-оксоэтил-4-(5-амино-2-пиридинил)-1-пиперазинкарбоксилата, полученного на стадии 5.4., в 15 мл водного раствора серной кислоты (0,33н) медленно вводят раствор 0,16 г (2,2 ммоль) нитрата натрия, растворенного в 3,5 мл воды. Перемешивают при 0°С в течение 0,5 часа и медленно вводят 0,83 г (5 ммоль) йодида калия. Продолжают перемешивать при указанной температуре в течение 0,5 часа, затем реакционную смесь нагревают до 85°С в течение 2 часов.
После охлаждения до комнатной температуры реакционную смесь подщелачивают до рН 14 путем добавления насыщенного водного раствора бикарбоната натрия. Водную фазу трижды экстрагируют дихлорметаном, промывают объединенные органические фазы 35%-ным водным раствором тиосульфита, воды, рассола и сушат их на сульфате натрия. Фильтрат концентрируют досуха и полученный таким образом осадок очищают хроматографией на силикагеле, элюируя смесью 98/2 дихлорметана и метанола.
Промывают простым диизопропиловым эфиром и получают 0,35 г продукта в виде бежевого твердого вещества, которое используют как таковое на следующей стадии.
5.6. 2-(метиламино)-2-оксоэтил-4-{5-[4-(трифторметил)окси]фенил}-2-пиридинил}-1-пиперазинкарбоксилат
Порядок выполнения работы такой же, как описан в примере 2 (стадия 2.4.). Из 0,250 г (0,61 ммоль) 2-(метиламино)-2-оксоэтил-4-(5-йодо-2-пиридинил)-1-пиперазинкарбоксилата, полученного на стадии 5.5., 0,51 г (2,44 ммоль) 4-(трифторметокси)фенилбороновой кислоты, 0,61 г (примерно 8% мол.) катализатора, содержащего палладий на твердом носителе, полученного на стадии 2.1 примера 2, и 2,9 мл (7,32 ммоль) водного раствора карбоната натрия (2,5М), суспендированного в 12 мл толуола и 3 мл этанола, и после проведения хроматографии на силикагеле, элюируя смесью 98/2 дихлорметана и метанола, с последующим промыванием простым диизопропиловым эфиром, получают 0,092 г продукта в виде белого твердого вещества.
ЖХ-МС: М+Н=439.
ТП(°С): 188-190°С.
ЯМР1Н (CDCL3)δ (ч./млн): 2,90 (д, 3Н); 4,70 (уш.с., 8Н); 4,65 (с, 2Н); 6,05 (уш.с. 1Н); 6,75 (дд, 1Н); 7,30 (д, 2Н); 7,55 (дд, 2Н); 7,75 (дд, 1Н);8,45 (дд, 1Н).
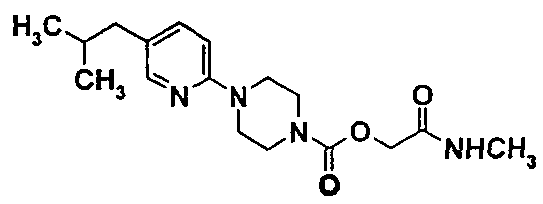
Пример 6 (соединение №63)
2-(метиламино)-2-оксоэтил-4-[5-(2-метилпропил)-2-пиридинил]-1-пиперазинкарбоксилат
6.1. 2-(метиламино)-2-оксоэтил-4-(5-бром-2-пиридинил)-1-пиперазинкарбоксилат
Порядок выполнения работы тот же, что описан в примере 1 (стадия 1.4). Из 2,20 г (5,91 ммоль) 2-(этилокси)-2-оксоэтил-4-(5-бром-2-пиридинил)-1-пиперазинкарбоксилата, полученного на стадии 3.3. примера 3, и 14,80 мл (29,55 ммоль) раствора метиламина (2М) в тетрагидрофуране и после кристаллизации в простом диизопропиловом эфире получают 1,974 г чистого продукта в виде твердого белого вещества.
6.2. 2-(метиламино)-2-оксоэтил-4-[5-(2-метилпропил)-2-пиридинил]-1-пиперазинкарбоксилат
В инертной атмосфере помещают 0,88 г (2,47 ммоль) 2-(метиламино)-2-оксоэтил-4-(5-бром-2-пиридинил)-1-пиперазинкарбоксилата, полученного на стадии 6.1., 0,33 г (3,22 ммоль) изобутилбороновой кислоты, 1,16 г (5,44 ммоль) гидратированного фосфата калия и 0,07 г (0,25 ммоль) трициклогексилфосфина, суспендированного в 11 мл толуола. Затем вводят 0,028 г (0,12 ммоль) диацетата палладия. Затем реакционную смесь нагревают с обратным холодильником в течение 3 часов.
Охлаждают до комнатной температуры, затем вводят 15 мл воды и 15 мл этилацетата. Отделяют соли фильтрованием через фритту, декантируют, экстрагируют водную фазу два раза этилацетатом, промывают объединенные органические фазы насыщенным водным раствором хлорида натрия и сушат их на сульфате натрия. После выпаривания растворителя полученный остаток очищают хроматографией на силикагеле, элюируя смесью 97/3 дихлорметана и метанола. После кристаллизации в простом диизопропиловом эфире получают 0,17 г продукта в виде твердого белого вещества.
ЖХ-МС: М+Н=335.
ТП(°С): 127-129°С.
ЯМР1Н (CDCL3)δ (ч./млн): 0,90 (д, 6Н); 1,80 (м, 1Н); 2,35 (д, 2Н); 2,90 (д, 3Н); 3,60 (м,8Н); 4,65 (с, 2Н); 6,10 (уш.с. 1Н); 6,60 (д, 1Н); 7,35 (дд, 1Н); 8,0 (д, 1Н).
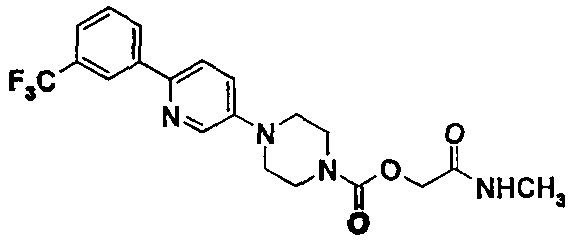
Пример 7 (соединение №85)
2-(метиламино)-2-оксоэтил-4-{6-[3-(трифторметил)фенил]-3-пиридинил}-1-пиперазинкарбоксилат
7.1. 1,1-диметилэтил-4-(3-пиридинил}-1-пиперазинкарбоксилат
В инертной атмосфере вводят 7,07 г (44,74 ммоль) 3-бромпиридина, 10 г (53,69 ммоль) 1,1-диметилэтил-1-пиперазинкарбоксилата, 6,02 г (62,64 ммоль) третбутилата натрия и 0,836 г (1,34 ммоль) (2,2'-бис(дифенилфосфино)-1,1'-бинафтил)(BINAP), суспендированного в 100 мл толуола. Затем вводят 0,41 г (0,45 ммоль) [дипалладия три(дибензилиденацетона)](Pd2(dba)3). Реакционную смесь затем нагревают с обратным холодильником в течение 22 часов.
Охлаждают до комнатной температуры, отделяют соли фильтрованием через стекловолокно, затем концентрируют фильтрат при пониженном давлении. Остаток обрабатывают 100 мл этилацетата и 100 мл воды, отделяют водную фазу, несколько раз экстрагируют ее этилацетатом, промывают объединенные органические фазы насыщенным водным раствором хлорида натрия, сушат их на сульфате натрия и концентрируют фильтрат при пониженном давлении. Очищают полученный таким образом остаток хроматографией на силикагеле, элюируя смесью 98/2, затем 95/5 дихлорметана и метанола.
Получают 9,80 г продукта в виде масла, которое кристаллизуется при комнатной температуре.
7.2. 1,1-диметилэтил-4-(6-бром-3-пиридинил)-1-пиперазинкарбоксилат
В раствор 4 г (15,19 ммоль) 1,1-диметилэтил-4-(3-пиридинил)-1-пиперазинкарбоксилата, полученного на стадии 7.1, в 50 мл ацетонитрила, охлажденный примерно до 0°С, вводят маленькими порциями 2,70 г (15,19 ммоль) N-бромсукцинимида (NBS). Перемешивают при 0°С в течение 15 минут, затем при комнатной температуре в течение 2 часов.
В реакционную среду вводят 100 мл водного раствора гидроксида натрия (1М) и 100 мл этилацетата. Отделяют водную фазу, экстрагируют ее два раза этилацетатом, промывают объединенные органические фазы насыщенным водным раствором хлорида натрия, сушат их на сульфате натрия и концентрируют фильтрат при пониженном давлении.
Получают таким образом 5,16 г продукта в виде твердого вещества желто-оранжевого цвета, который используют как таковой на следующей стадии.
7.3. 1-(6-бром-3-пиридинил)пиперазин
В суспензию 5,16 г (15,08 ммоль) 1,1-диметилэтил-4-(6-бром-3-пиридинил)-1-пиперазинкарбоксилата, полученного на стадии 7.2., в 70 мл дихлорметана медленно вводят 11,20 мл (150,77 ммоль) трифторуксусной кислоты. Перемешивают при комнатной температуре в течение 16 часов. Концентрируют при пониженном давлении, остаток обрабатывают 40 мл хлороформа, затем медленно вводят 4 мл водного раствора гидроксида натрия (10М). Отделяют водную фазу, затем экстрагируют ее два раза хлороформом. Объединяют органические фазы и промывают их насыщенным водным раствором хлорида натрия. Сушат органическую фазу на сульфате натрия и концентрируют фильтрат при пониженном давлении.
Получают таким образом 5,16 г продукта в виде оранжевого масла, которое кристаллизуется при комнатной температуре. Этот продукт используют как таковой на следующей стадии.
7.4. 2-(этилокси)-2-оксоэтил-4-(6-бром-3-пиридинил)-1-пиперазинкарбоксилат
Порядок выполнения работы тот же, что описан в примере 1 (стадия 1.2.). Из 3,57 г (14,76 ммоль) 1-(6-бром-3-пиридинил)пиперазина, полученного на стадии 7.3., и 3,97 г (17,71 ммоль) этил[(фенилоксикарбонил)окси]ацетата, полученного на стадии 1.1. примера 1, и после проведения хроматографии на силикагеле, элюируя смесью 99/1, затем 98/2 дихлорметана и метанола, получают 3,75 г продукта в виде желтого масла, которое кристаллизуют при комнатной температуре.
7.5. 2-(этилокси)-2-оксоэтил-4-{6-[3-(трифторметил)фенил]-3-пиридинил)-1-пиперазинкарбоксилат
Порядок выполнения работы тот же, что описан в примере 1 (стадия 1.3.). Из 1,28 г (3,43 ммоль) 2-(этилокси)-2-оксоэтил-4-(6-бром-3-пиридинил)-1-пиперазинкарбоксилата, полученного на стадии 7.4., 1,96 г (10,29 ммоль) 3-(трифторметил)фенилбороновой кислоты, 2,91 г (13,72 ммоль) гидратированного фосфата калия и 0,40 г (0,34 ммоль) тетракис(трифенилфосфин) палладия и после проведения хроматографии на силикагеле, элюируя смесью 35/65 этилацетата и циклогексана, получают 0,98 г чистого продукта в виде желтого масла, которое кристаллизуется при комнатной температуре.
7.6. 2-(метиламино)-2-оксоэтил-4-{6-[3-(трифторметил)фенил]-3-пиридинил}-1-пиперазинкарбоксилат
Порядок выполнения работы тот же, что описан в примере 1 (стадия 1.4). Из 0,60 г (1,37 ммоль) 2-(этилокси)-2-оксоэтил-4-{6-[3-(трифторметил)фенил]-3-пиридинил}-1-пиперазинкарбоксилата, полученного на стадии 7.5, и 3,40 мл (6,86 ммоль) раствора метиламина (2М) в тетрагидрофуране и после проведения хроматографии на силикагеле, элюируя смесью 98/2, затем 97/3 дихлорметана и метанола, с последующим промыванием простым диизопропиловым эфиром, получают 0,36 г продукта в виде белого твердого вещества.
ЖХ-МС: М+Н=423.
ТП(°С): 146-150°С.
ЯМР1Н (CDCL3)δ (ч./млн): 2,90 (д, 3Н); 3,35 (м, 4Н); 3,80 (м, 4Н); 4,65 (с, 2Н); 6,05 (уш.с. 1Н); 7,30 (м, 1Н); 7,65 (м, 2Н); 7,70 (д, 1Н); 8,10 (д, 1Н); 8,25 (с, 1Н); 8,45 (д, 1Н).
Пример 8 (соединение №86)
2-амино-2-оксоэтил-4-{6-[3-(трифторметил)фенил]-3-пиридинил}-1-пиперазинкарбоксилат
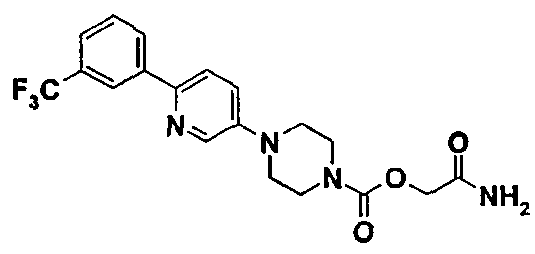
В раствор 0,30 г (0,69 ммоль) 2-(этилокси)-2-оксоэтил-4-{6-[3-(трифторметил)фенил]-3-пиридинил}-1-пиперазинкарбоксилата, полученного на стадии 7.5. примера 7, в 6 мл смеси 1/1 метанола и тетрагидрофурана водят 5,90 мл (41,40 ммоль) раствора аммиака (7н) в метаноле. Перемешивают при комнатной температуре в течение 22 часов.
Концентрируют при пониженном давлении, затем полученный остаток очищают хроматографией на силикагеле, элюируя смесью 96/4 дихлорметана и метанола, с последующим промыванием простым диизопропиловым эфиром. Получают 0,19 г продукта в виде желтого твердого вещества.
ЖХ-МС: М+Н=409.
ТП(°С): 155-157°С.
ЯМР1Н (CDCL3)δ (ч./млн): 3,35 (м, 4Н); 3,75 (м, 4Н); 4,70 (с, 2Н); 5,50 (уш.с. 1Н); 6,0 (уш.с. 1Н); 7,30 (м. 1Н); 7,55 (м, 2Н); 7,70 (д, 1Н); 8,10 (д, 1Н); 8,25 (с, 1Н); 8,40 (д, 1Н).
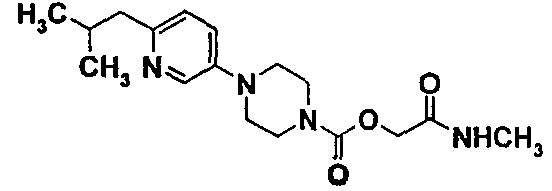
Пример 9 (соединение №66)
2-(метиламино)-2-оксоэтил-4-[6-(метилпропил)-3-пиридинил]-1-пиперазинкарбоксилат
9.1. 2-(метиламино)-2-оксоэтил-4-(6-бром-3-пиридинил)-1-пиперазинкарбоксилат
Порядок выполнения работы тот же, что описан в примере 1 (стадия 1.4.). Из 2,35 г (6,32 ммоль) 2-(этилокси)-2-оксоэтил-4-(6-бром-3-пиридинил)-1-пиперазинкарбоксилата, полученного на стадии 7.4 примера 7, и 15,80 мл (31,61 ммоль) раствора метиламина (2М) в терагидрофуране и после проведения хроматографии на силикагеле, элюируя смесью 98/2, затем 97/3 дихлорметана и метанола, получают 1,779 г продукта в виде белого твердого вещества.
ТП(°С): 164°С.
9.2. 2-(метиламино)-2-оксоэтил-4-[6-(метилпропил)-3-пиридинил]-1-пиперазинкарбоксилат
в инертной атмосфере вводят 1,25 г (3,50 ммоль) 2-(метиламино)-2-оксоэтил-4-(6-бром-3-пиридинил)-1-пиперазинкарбоксилата, полученного на стадии 9.1., и 0,12 г (0,17 ммоль) дихлорбис(трифенилфосфин) палладия (Pd(PPh3)2Cl2), суспендированного в 7 мл тетрагидрофурана. Затем вводят 17,50 мл (8,74 ммоль) раствора бромизобутил цинка (0,5М) в терагидрофуране. Перемешивают при комнатной температуре в течение 19 часов.
Реакционную смесь выливают в 25 мл воды и 25 мл этилацетата. Нерастворившееся твердое вещество фильтруют через стекловолокно. Декантируют, дважды экстрагируют водную фазу этилацетатом, сушат объединенные органические фазы на сульфате натрия и концентрируют фильтрат при пониженном давлении. Остаток, полученный таким образом, очищают хроматографией на силикагеле, элюируя смесью 95/5 дихлорметана и метанола, с последующей перекристаллизацией в простом диизопропиловом эфире.
Получают 0,36 г чистого продукта в виде коричневого твердого вещества.
ЖХ-МС: М+Н=335.
ТП(°С): 87-89°С.
ЯМР1Н (CDCL3)δ (ч./млн): 0,90 (д, 6Н); 2,05 (м, 1Н); 2,60 (д, 2Н); 2,90 (д, 3Н); 3,20 (м, 4Н); 3,70 (м, 4Н); 4,65 (с, 2Н); 6,05 (уш.с. 1Н); 7,0-7,20 (м, 2Н); 8,25 (д, 1Н).
Пример 10 (соединение №87)
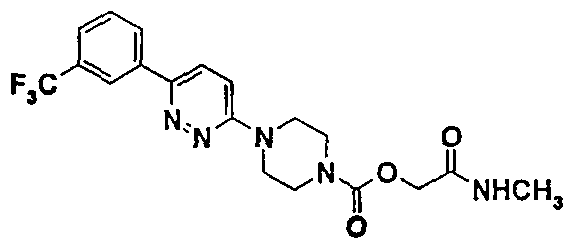
2-(метиламино)-2-оксоэтил-4-{6-[3-(трифторметил)фенил]-3-пиридазинил}-1-пиперазинкарбоксилат
10.1. 2-(этилокси)-2-оксоэтил-4-(6-хлор-3-пиридазинил)-1-пиперазинкарбоксилат
Порядок выполнения работы тот же, что описан в примере 1 (стадия 1.2.). Из 1,60 г (8,05 ммоль) 3-хлор-6-(1-пиперазинил)пиридазина (J. Med. Chem., 18, 2002, 4011-4017) и 1,99 г (8,86 ммоль) этил[(фенилоксикарбонил)окси]ацетата, полученного на стадии 1.1. примера 1, и после проведения хроматнографии на силикагеле, элюируя смесью 98/2 дихлорметана и метанола, получают 1,70 г продукта в виде белого твердого вещества.
ТП(°С): 149-151°С.
10.2. 2-(этилокси)-2-оксоэтил-4-{6-[3-(трифторметил)фенил]-3-пиридазинил}-1-пиперазинкарбоксилат
Порядок выполнения работы тот же, что описан в примере 1 (стадия 1.3.). Из 1,15 г (3,50 ммоль) 2-(этилокси)-2-оксоэтил-4-(6-хлор-3-пиридазинил)-1-пиперазинкарбоксилата, полученного на стадии 10.1., 1,99 г (10,49 ммоль) 3-(трифторметил)фенилбороновой кислоты, 2,97 г (13,99 ммоль) гидратированного фосфата калия и 0,40 г (0,35 ммоль) тетракис(трифенилфосфин) палладия и после проведения хроматографии на силикагеле, элюируя смесью 35/65, затем 45/55 этилацетата и циклогексана, получают 0,67 г чистого продукта в виде твердого вещества.
ТП(°С): 126-128°С.
10.3. 2-(метиламино)-2-оксоэтил-4-{6-[3-(трифторметил)фенил]-3-пиридазинил}-1-пиперазинкарбоксилат
Порядок выполнения работы тот же, что описан в примере 1 (стадия 1.4.). Из 0,66 г (1,51 ммоль) 2-(этилокси)-2-оксоэтил-4-{6-[3-(трифторметил)фенил]-3-пиридазинил}-1-пиперазинкарбоксилата, полученного на стадии 10.2., и 3 мл (6,02 ммоль) раствора метиламина (2М) в тетрагидрофуране и после проведения хроматнографии на силикагеле, элюируя смесью 96/4 дихлорметана и метанола, с последующим промыванием простым диизопропиловым эфиром, получают 0,50 г продукта в виде белого твердого вещества.
ЖХ-МС: М+Н=424.
ТП(°С): 151-153°С.
ЯМР1Н (DMSO)δ (ч./млн): 2,60 (д, 3Н); 3,55 (м, 4Н); 3,75 (м, 4Н); 4,45 (с, 2Н); 7,40 (д, 1Н); 7,80 (м, 3Н); 8,10 (д, 1Н); 8,35 (м, 2Н).
Пример 11 (соединение №103)
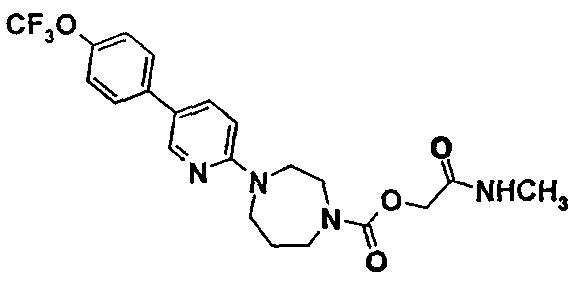
2-(метиламино)-2-оксоэтил-4-(5-{4-[(трифторметил)окси]фенил}-2-пиридинил)-1,4-диазепин-1-карбоксилат
11.1. 1,1-диметилэтил-4-(5-бром-2-пиридинил)-1,4-диазепин-1-карбоксилат
В автоклав вводят 1,03 г (5 ммоль) 1,1-диметилэтил-1,4-диазепин-1-карбоксилат, 1,19 г (5 ммоль) 2,5-дибромпиридина и 0,7 г (5 моль) карбоната калия, суспендированного в 0,90 мл диметилсульфоксида (ДМСО). Нагревают до 150°С в течение 22 часов.
Охлаждают до комнатной температуры, обрабатывают реакционную смесь этилацетатом, промывают водой, затем рассолом и сушат на сульфате натрия. Концентрируют фильтрат при пониженном давлении и полученный таким образом остаток очищают хроматографией на силикагеле, элюируя смесью 99,5/0,5 дихлорметана и метанола. Получают 1,63 г продукта в виде масла, которое используют как таковое на следующей стадии.
11.2. 1-(5-бром-2-пиридинил)-1,4-диазепин
В раствор 1,63 г (4,4 ммоль) 1,1-диметилэтил-4-(5-бром-2-пиридинил)-1,4-диазепин-1-карбоксилата, полученного на стадии 11.1., в 12 мл диоксана и 4 мл этанола вводят 6 мл раствора соляной кислоты (6н) в изопропаноле. Эту реакционную смесь нагревают до 70°С в течение 3 часов.
Охлаждают до комнатной температуры, затем концентрируют при пониженном давлении. После кристаллизации в ацетоне получают 1,32 г твердого белого вещества. Эти кристаллы обрабатывают в 10 мл дихлорметана и подщелачивают реакционную среду до получения рН 14 путем добавления 28%-ного раствора аммиака. Органическую фазу отделяют фильтрованием через гидрофобный элемент и концентрируют фильтрат при пониженном давлении.
Получают 0,96 г продукта в виде масла, которое используют как таковое на следующей стадии.
11.3. 2-(метиламино)-2-оксоэтил-4-(5-бром-2-пиридинил)-1,4-диазепин-1-карбоксилат
Порядок выполнения работы тот же, что описан в примере 1 (стадия 1.2.). Из 0,95 г (3,70 ммоль) 1-(5-бром-2-пиридинил)-1,4-диазепина, полученного на стадии 11.2., и 0,94 г (3,70 ммоль) 2-(метиламино)-2-оксоэтил-4-нитрофенилкарбоната, полученного на стадии 2.2. примера 2, и после проведения хроматнографии на силикагеле, элюируя смесью 30/70 этилацетата и циклогексана, затем смесью 95/5 дихлорметана и метанола, с последующей кристаллизацией в простом диизопропиловыом эфире, получают 0,97 г продукта в виде белого твердого вещества.
11.4. 2-(метиламино)-2-оксоэтил-4-(5-{4-[(трифторметил)окси]фенил}-2-пиридинил)-1,4-диазепин-1-карбоксилат
В реактор из Pyrex вводят 0,12 г (0,3 ммоль) 2-(метиламино)-2-оксоэтил-4-(5-бром-2-пиридинил)-1,4-диазепин-1-карбоксилата, полученного на стадии 11.3., 0,25 г (1,2 ммоль) 4-(трифторметокси)фенилбороновой кислоты и 0,9 мл (1,8 ммоль) водного раствора карбоната натрия (2М), суспендированного в 3,5 мл толуола и 0,8 мл этанола. Затем добавляют 0,07 г (0,06 ммоль) тетракис(трифенилфосфин) палладия и после герметизации реактора температуру поднимают до 150°С в течение 15 минут в условиях микроволнового излучения.
Органическую фазу отделяют декантацией, и фильтрат концентрируют при пониженном давлении. Полученный таким образом остаток очищают хроматографией на силикагеле, элюируя смесью 30/70/5 этилацетата, циклогексана и метанола.
После кристаллизации в простом диизопропиловом эфире получают 0,078 г продукта в виде белого твердого вещества.
ЖХ-МС: М+Н=452.
ТП(°С): 191-193°С.
ЯМР1Н (DMSO)δ (ч./млн): 1,70-2,00 (м, 2Н); 2,55 (д, 3Н); 3,25-3,40 (м, 2Н); 3,40-3,90 (м, 6Н); 4,35 (д, 2Н); 6,75 (д, 1Н); 7,35 (д, 2Н); 7,70 (уш.д, 2Н+NH); 7,80 (дд, 1Н); 8,45 (д, 1Н).
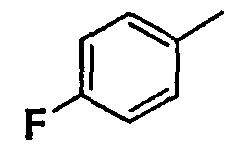
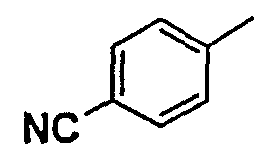
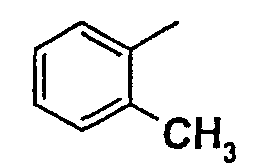
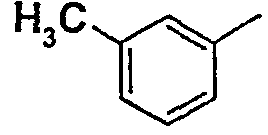
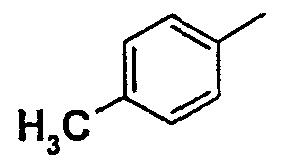
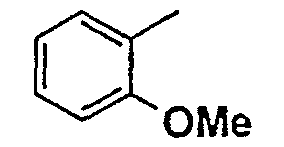
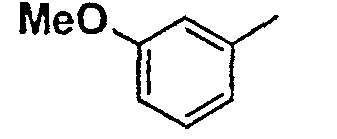
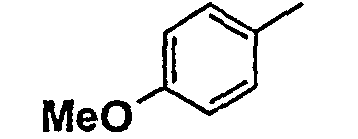
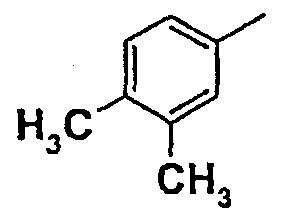
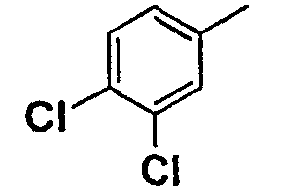
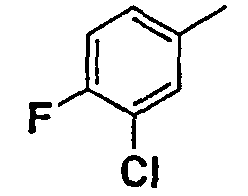
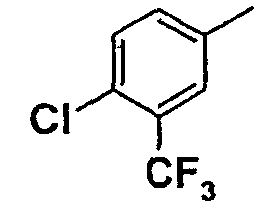
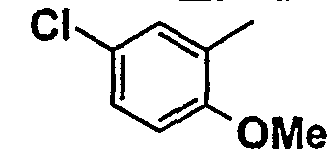
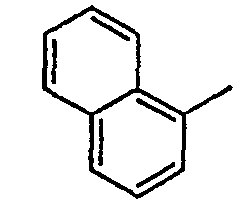
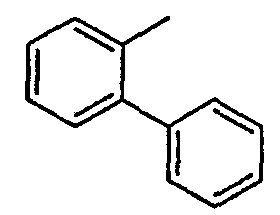
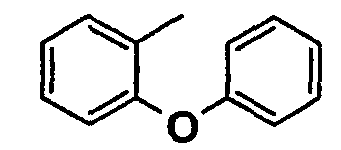
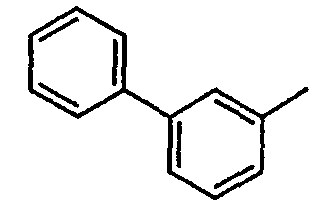
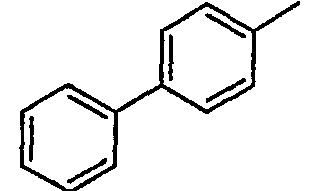
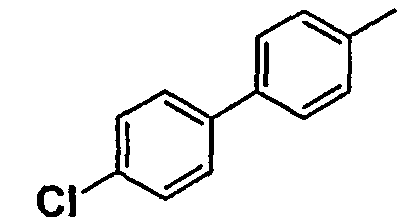
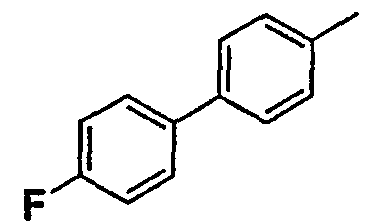
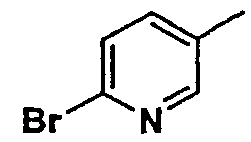
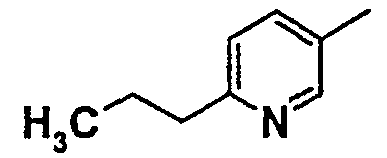
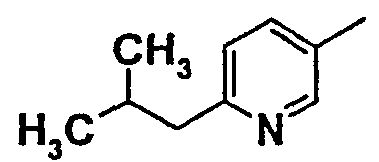
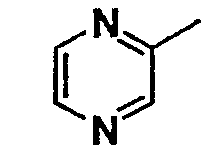
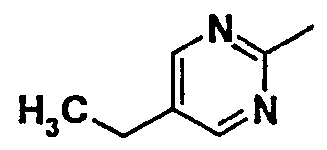
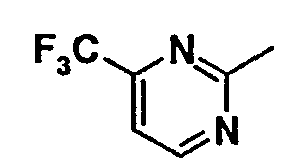
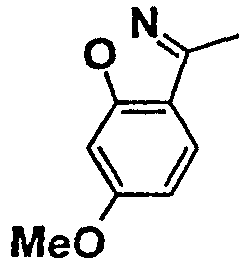
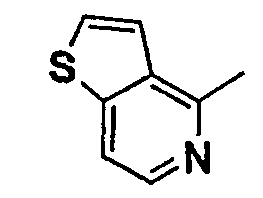
Нижеследующая таблица 1 иллюстрирует химические структуры и физические свойства нескольких соединений согласно изобретению.
В колонке «основание или соль» «основание» обозначает соединение в виде свободного основания, тогда как “HCl” обозначает соединение в виде гидрохлорида. В таблице ОМе обозначает группу метокси.
В колонке “ТП(°С) или М+Н” ТП(°С) обозначает температуру плавления соединения в градусах Цельсия и М+Н обозначает значение массы соединения, протонированного атомом водорода (масса соединения + 1), определяемого методом ЖХ-МС (Жидкостная хроматография-масс-спектроскопия).
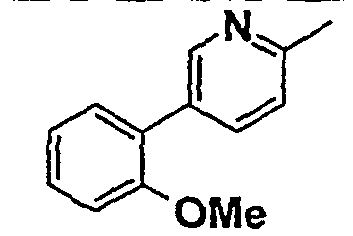
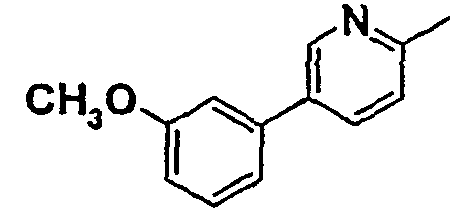
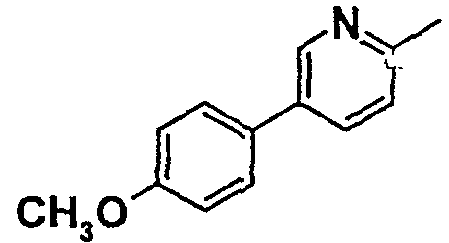
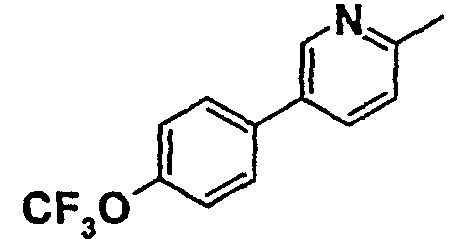
Таблица 1
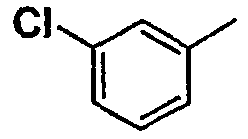
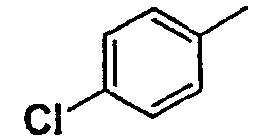
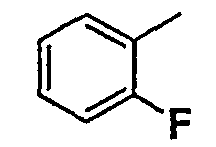
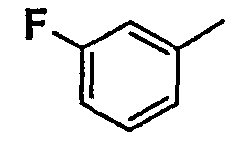

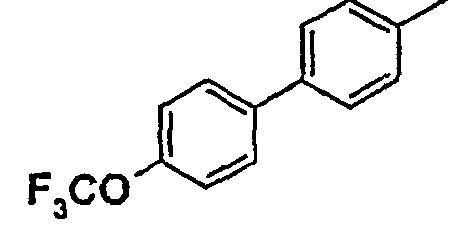
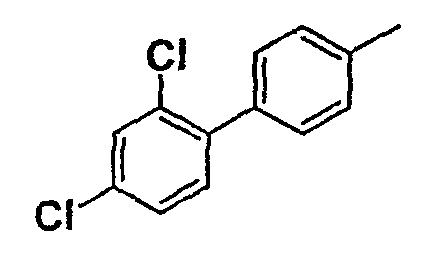
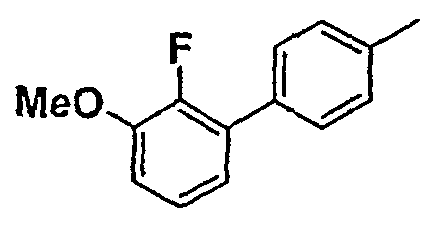
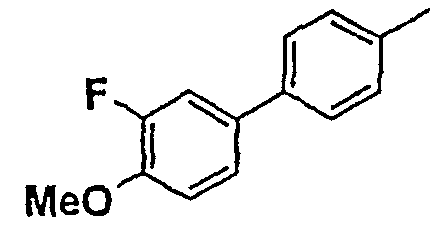
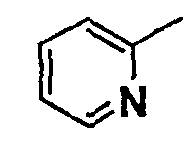
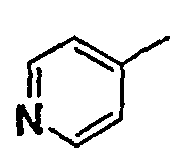
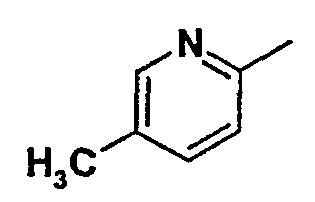
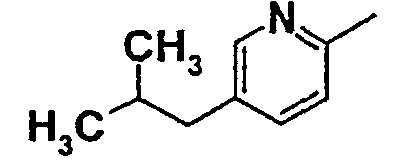
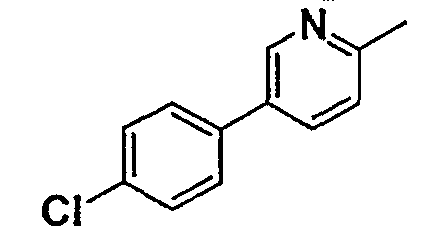
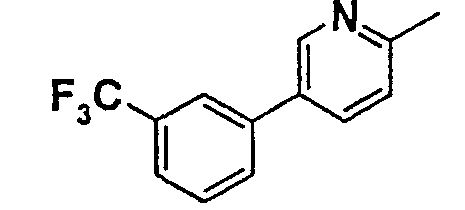
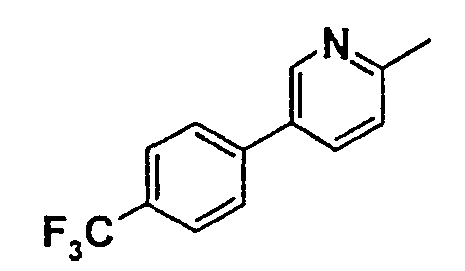
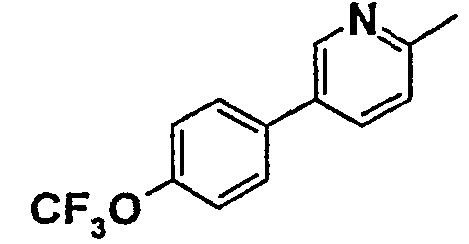
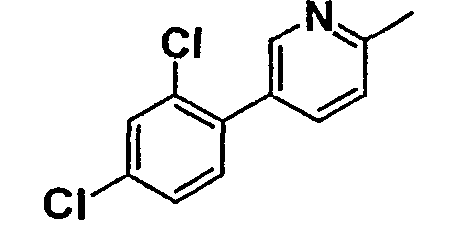
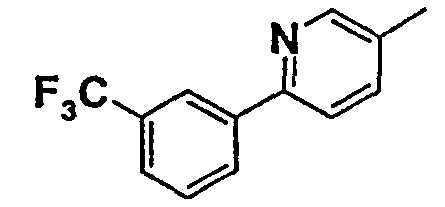
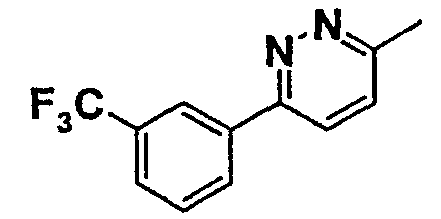
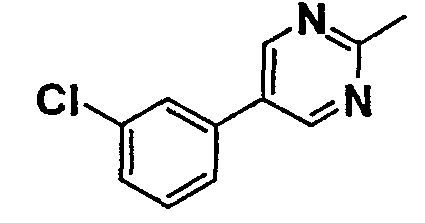
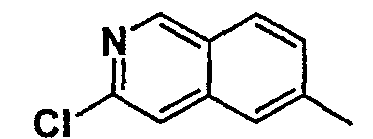
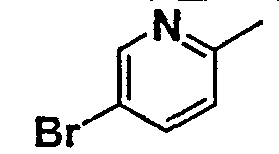
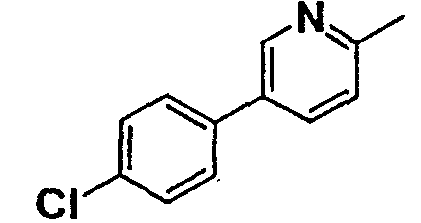
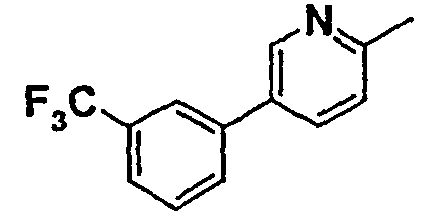

Соединения согласно изобретению подвергались фармакологическим опытным испытания, позволяющим определить их ингибирующее действие в отношении фермента FAAH (Fatty Acid Amido Hydrolase).
Ингибирующая активность была выявлена радиоферментативным тестом, основанным на измерении продукта гидролиза (этаноламин [1-3Н]) анандамида [этаноламин 1-3Н] ферментом FAAH (Life Sciences (1995), 56, 1999-2005 Journal of Pharmacology and Experimental Therapeutics (1997), 283, 729-734). Так, брали мозг мышей (кроме мозжечка) и хранили при -80°С. Мембранные гомогенаты готовили непосредственно перед использованием путем гомогенизации тканей с помощью Polytron в буфере три-HCl 10 мМ (рН 8), содержащем 150 мМ NaCl и 1 мМ EDTA. Затем проводили ферментативную реакцию в 70 мкл буфера, содержащего альбумин бычьей сыворотки без жирных кислот (1 мг/мл). Последовательно вводили тестируемые соединения в разных концентрациях, анандамид [этаноламин 1-3Н] (специфическая активность 15-20 Ci/ммоль), разведенный холодным анандамидом до концентрации 10 мкМ и мембранный препарат (400 мкг замороженной ткани для каждого опытного испытания). Через 15 минут при 25°С ферментативную реакцию останавливали путем добавления 140 мкл хлороформа/метанола (2:1). Смесь перемешивали в течение 10 минут, затем центрифугировали в течение 15 минут, 3500 г. Аликвоту (30мкл) водной фазы, содержащей этаноламин [1-3Н], вычисляли путем жидкостной сцинтилляции.
В указанных условиях наиболее активные соединения согласно изобретению имели CI50 (концентрация, ингибирующая на 50% контрольную ферментативную активность FAAH) от 0,001 до 1 мкМ.
Ниже в таблице 2 приведены CI50 нескольких соединений согласно изобретению.
Таким образом очевидно, что соединения согласно изобретению обладают ингибирующей активностью в отношении фермента FAAH.
Активность in vivo соединений согласно изобретению оценивали тестом на анальгезию.
Так интраперитонеальное введение (i.p.) PBQ (фенилбензохинона, 2 мг/кг в 0,9%-ном растворе натрия хлорида, содержащем 5% этанола) самцам мышей OF1, весом от 25 до 30 г, вызывает абдоминальные спазмы, в среднем 30 скручиваний или сокращений в течение 5-15 минут после инъекции. Тестируемые соединения вводили перорально в виде суспензии в Tween 80 в концентрации 0,5% за 60 или 120 минут перед введением PBQ. В этих условиях наиболее сильные соединения согласно изобретению уменьшали на 35-70% число спазмов, вызванных PBQ, в интервале доз от 1 до 30 мг/кг.
Ниже в таблице 3 приведены результаты теста на анальгезию в отношении нескольких соединений согласно изобретению.
(а) 1 мг/кг p.o. за 2 часа
Фермент FAAH (Chemistry and Physics of Lipids, (2000), 108, 107-121) катализирует гидролиз эндогенных производных амидов и сложных эфиров различных жирных кислот, таких как N-арахидоноилэтаноламин (анандамид), N-пальмитоилэтаноламин, N-олеоилэтаноламин, олеамид или 2-арахидоноилглицерин. Эти производные обладают разной фармакологической активностью, взаимодействуя кроме прочих с каннабиоидными и ванилоидными рецепторами.
Соединения согласно изобретению блокируют этот путь деградации и повышают тканевый показатель этих эндогенных веществ. Их можно использовать в этом качестве для профилактики и лечения патологий, в которых участвуют эндогенные каннабиоиды и/или любые другие субстраты, метаболизированные ферментом FAAH. Можно, например, назвать следующие заболевания и нарушения:
боль, в частности, острые или хронические боли нейрогенного типа: мигрень, нейропатические боли, включая формы, связанные с вирусом герпеса и диабетом; острые или хронические боли, связанные с воспалительными заболеваниями: артрит, ревматоидный артрит, остеоартрит, спондилит, подагра, васкулит, болезнь Крона, синдром раздражения ободочной кишки, периферические острые или хронические боли; головокружения; тошнота; рвота, в частности, являющаяся последствием химиотерапии, нарушения в области приема пищи, в частности, анорексия и кахексия различного происхождения; нейрологические и психиатрические патологии: дрожание, дискинезии, дистонии, спастичность, компульсивные и навязчивые побуждения, синдром Туретта, все формы депрессии и тревожности любого происхождения, перепады настроения, психозы; острые и хронические нейро-дегенеративные заболевания: болезнь Паркинсона, болезнь Альцгеймера, старческое слабоумие, хорея Гентингтона, патологические изменения, связанные с ишемической болезнью мозга и черепно-мозговыми травмами; эпилепсия; нарушения сна, включая приступы апноэ во сне; сердечно-сосудистые заболевания, в частности, гипертензия, нарушения сердечного ритма, атеросклероз, сердечный приступ, ишемическая болезнь сердца; ишемия почки; онкологические заболевания; доброкачественные опухоли кожи, папилломы и опухоли мозга, опухоли простаты, опухоли мозга (глиобластомы, медуллоэпителиомы, медуллобластомы, нейробластомы, опухоли эмбрионального происхождения, астроцитомы, астробластомы, эпендиомы, олигодендроглиомы, опухоль сплетения, нейроэпителиомы, опухоль эпифиза, эпендибластомы, злокачественные менингиомы, саркоматозы, злокачественные меланомы, невриномы); нарушения иммунной системы, в частности, аутоиммунные заболевания: псориаз, красная волчанка, заболевания соединительных тканей, синдром Шергена, анкилозирующий спондилоартрит, недифференцированный спондилоартрит, болезнь Behcet's, аутоиммунные гемолитические анемии, рассеяный склероз, боковой амиотрофический склероз, амилозы, отторжение трансплантатов, заболевания, поражающие плазмоцитные линии; аллергические заболевания: быстрая или медленная гиперчувствительность, алергические риниты или конъюктивиты, контактные дерматиты; инфекционные болезни, вызванные паразитами, вирусами или бактериями: СПИД, менингиты; воспалительные заболевания, в частности, заболевания суставов: артрит, ревматоидный артрит, остеоартрит, спондилит, подагра, васкулит, болезнь Крона, синдром раздражения ободочной кишки; остеопороз; глазные болезни: гипертензия глаза, глаукома; легочные заболевания: заболевания дыхательных путей, бронхоспазм, кашель, астма, хронический бронхит, хроническая обструкция дыхательных путей, энфизема; заболевания желудочно-кишечного тракта: синдром раздражения ободочной кишки, воспалительные заболевания кишечника, язвы, диарея; недержание мочи и воспаление мочевого пузыря.
Применение соединений согласно изобретению в виде основания, соли присоединения кислоты, фармацевтически приемлемого гидрата или сольвата, для получения лекарственного средства, предназначенного для лечения вышеуказанных патологий, является неотъемлемой частью изобретения.
Объектом изобретения также являются лекарственные средства, которые содержат соединение формулы (I) или соль присоединения кислоты или фармацевтически приемлемый гидрат или сольват соединения формулы (I). Эти лекарственные средства применяются в терапии, в частности, для лечения вышеуказанных патологий.
В соответствии с одним из своих аспектов настоящее изобретение относится к фармацевтическим композициям, содержащим в качестве активного вещества по меньшей мере одно соединение согласно изобретению. Эти фармацевтические композиции содержат эффективную дозу соединения согласно изобретению, или соль присоединения кислоты, или фармацевтически приемлемый гидрат, или сольват указанного соединения и возможно один или несколько фармацевтически приемлемых эксципиентов.
Указанные эксципиенты выбирают из обычных эксципиентов, известных специалисту, в соответствии с фармацевтической формой и требуемым способом введения.
В фармацевтических соединениях согласно настоящему изобретению для перорального, сублингвального, подкожного, внутримышечного, внутривенного, местного, локального, интратрахеального, интраназального, чрескожного, легочного, глазного или ректального введения указанное активное вещество формулы (I) или его соль присоединения кислоты, возможный гидрат или сольват, можно вводить в виде разовой дозы приема в смеси с традиционными фармацевтическими эксципиентами животным и человеку для профилактики или лечения указанных выше нарушений или заболеваний.
Соответствующие разовые формы для введения содержат пероральные формы, такие как таблетки, мягкие или твердые желатиновые капсулы, порошки, гранулы, жевательные резинки и растворы или суспензии для перорального введения, формы для сублингвального, орального, интратрахеального, внутриглазного, интраназального введения, путем ингаляции, формы для подкожного, внутримышечного или внутривенного введения и формы для ректального или вагинального введения. Для местного применения можно использовать соединения согласно изобретению в составе кремов, мазей или лосьонов.
В качестве примера разовая форма для введения соединения компоненты: согласно изобретению в виде таблетки может содержать следующие компоненты:
Указанные разовые формы имеют такую дозировку, чтобы обеспечить дневное введение от 0,01 до 20 мг активного вещества на кг веса тела в соответствии с галеновой формой.
В возможных частных случаях, когда требуются большие или меньшие дозировки, такие дозировки также относятся к изобретению. В соответствии с обычной практикой соответствующая дозировка для каждого пациента подбирается врачом согласно способу введения, весу и ответу данного пациента.
В соответствии с другим аспектом настоящее изобретение также относится к методу лечения указанных выше патологий, который содержит введение эффективной дозы соединения согласно изобретению, одной из его солей присоединения кислоты, фармацевтически приемлемой, сольвата или гидрата указанного соединения.
Настоящее изобретение относится к соединению формулы (I), в которой m
обозначает целое число, равное 1 или 2; R1 обозначает группу, выбранную, в частности, из фенила, пиридинила, пиридазинила, пиримидинила, пиразинила, тиадиазолила, нафтила, хинолинила, изохинолинила, бензизоксазолила, тиенопиридинила, при этом указанная группа возможно замещена одной или несколькими группами R3, одинаковыми или отличающимися друг от друга или группой R4, R2 обозначает группу общей формулы CHR5CONHR6, R3 обозначает атом галогена или одну из следующих групп: циано, нитро, С1-6-алкил, С1-6-алкокси, С1-6-трифторалкил, С1-6-трифторалкокси, бензилокси, фенилокси, R4 обозначает группу, выбранную, в частности, из фенила, бензофуранила, нафтила; при этом одна или несколько групп R4 могут быть замещены одной или несколькими группами R3, одинаковыми или отличающимися друг от друга; R5 обозначает атом водорода или С1-3алкильную группу; R6 обозначает атом водорода или С1-6-алкильную, С3-7-циклоалкильную или С3-7-циклоалкил-С1-3-алкиленовую группу; в виде основания, соли присоединения кислоты, гидрата или сольвата. Также изобретение относится к способу получения соединения формулы I, его применению в качестве лекарственного средства и к фармацевтической композиции на его основе. Технический результат - получены новые производные 1-пиперазин- и 1-гомопиперазинкарбоксилатов, полезных для профилактики или лечения патологии, в которой участвуют эндогенные каннабиноиды и/или любые другие субстраты, метаболизированные ферментом FAAH. 8 н. и 4 з.п. ф-лы, 3 табл.
1. Соединение формулы (I)
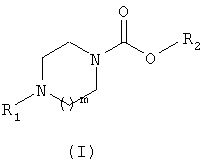
в которой m обозначает целое число, равное 1 или 2;
R1 обозначает группу, выбранную, в частности, из фенила, пиридинила, пиридазинила, пиримидинила, пиразинила, тиадиазолила, нафтила, хинолинила, изохинолинила, бензизоксазолила, тиенопиридинила,
при этом указанная группа возможно замещена одной или несколькими группами
R3, одинаковыми или отличающимися друг от друга или группой R4,
R2 обозначает группу общей формулы CHR5CONHR6,
R3 обозначает атом галогена или одну из следующих групп: циано, нитро, С1-6-алкил, C1-6-алкокси, C1-6-трифторалкил, С1-6-трифторалкокси, бензилокси, фенилокси,
R4 обозначает группу, выбранную, в частности, из фенила, бензофуранила, нафтила;
при этом одна или несколько групп R4 могут быть замещены одной или несколькими группами R3, одинаковыми или отличающимися друг от друга;
R5 обозначает атом водорода или С1-3-алкильную группу;
R6 обозначает атом водорода или С1-6-алкильную, С3-7-циклоалкильную или С3-7-циклоалкил-С1-3-алкиленовую группу;
в виде основания, соли присоединения кислоты, гидрата или сольвата.
2. Соединение формулы (I) по п.1, отличающееся тем, что m обозначает целое число, равное 1 или 2;
R1 обозначает группу, выбранную, в частности, из фенила, пиридинила, пиримидинила, пиразинила, нафтила, хинолинила, изохинолинила, бензизоксазолила, тиенопиридинила, причем указанная группа возможно замещена одной или несколькими группами R3, одинаковыми или отличающимися друг от друга;
R2 обозначает группу общей формулы CHR5CONHR6,
R3 обозначает атом галогена или циано, C1-6-алкильную, C1-6-алкокси, С1-6-трифторалкильную, С1-6-трифторалкокси, фенилокси;
R5 обозначает атом водорода;
R6 обозначает атом водорода или С1-6-алкильную группу.
3. Соединение формулы (I) по п.1, отличающееся тем, что m равно 1;
R1 обозначает группу, выбранную, в частности, из пиридинила, пиримидинила, пиразинила, хинолинила, изохинолинила, причем указанная группа возможно замещена группой R3,
R2 обозначает группу общей формулы CHR5CONHR6,
R3 обозначает атом галогена или С1-6-алкильную, С1-6-алкокси, C1-6-фторалкильную группу,
R5 обозначает атом водорода;
R6 обозначает атом водорода или С1-6-алкильную группу.
4. Соединение формулы (I) по п.1, отличающееся тем, что
m обозначает целое число, равное 1 или 2;
Р1 обозначает группу, выбранную, в частности, из фенила, пиридинила, пиридазинила, пиримидинила, тиадиазолила, причем указанная группа возможно замещена группой R4,
R4 обозначает группу, выбранную, в частности, из фенила, бензофуранила, нафтила; причем группа R4 возможно замещена одной или несколькими группами R3, одинаковыми или отличающимися друг от друга,
R2 обозначает группу общей формулы CHR5CONHR6,
R3 обозначает атом галогена или нитро, С1-6-алкильную, С1-6-алкокси, С1-6-трифторалкильную, С1-6-трифторалкокси, бензилоксигруппу;
R5 обозначает атом водорода;
R6 обозначает атом водорода или группу С1-6-алкильную или С3-7-циклоалкил-С1-3-алкилен.
5. Соединение формулы (1) по п.1 или 4, отличающееся тем, что m равно 1;
R1 обозначает группу, выбранную, в частности, из фенила, пиридинила, пиридазинила, пиримидинила,
причем указанная группа возможно замещена группой R4,
R4 обозначает группу, выбранную, в частности, из фенила, бензофуранила, нафтила; причем группа R4 возможно замещена одной или несколькими группами R3, одинаковыми или отличающимися друг от друга,
R2 обозначает группу общей формулы CHR5CONHR6,
R3 обозначает атом галогена или нитро, С1-6-алкильную, С1-6-алкокси, С1-6-трифторалкильную, С1-6-трифторалкокси, бензилокси-группу; R5 обозначает атом водорода;
R6 обозначает атом водорода или С1-6-алкильную группу.
6. Способ получения соединения формулы (I) по любому из пп.1-5, содержащий стадию, в ходе которой вводят во взаимодействие амин общей формулы (II)
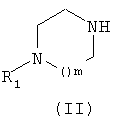
в которой R1 и m такие, как определены в общей формуле (I) по п.1, с карбонатом общей формулы (III)
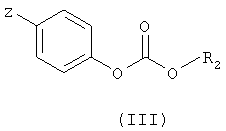
в которой Z обозначает атом водорода или нитрогруппу и R2 такой, как определен в общей формуле (I) по п.1.
7. Способ получения соединения формулы (I) по любому из пп.1-5, содержащий стадию, в ходе которой превращают сложный карбаматный эфир общей формулы (Ia)
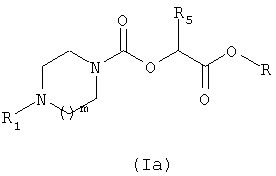
в которой m, R1 и R2 такие, как определены в общей формуле (I) по п.1, и R обозначает метильную или этильную группу,
путем аминолиза с помощью амина общей формулы R6NH2, в которой R6 такой, как определен в общей формуле (I) по п.1.
8. Соединение общей формулы (Ia)
в которой m, R1 и R5 такие, как определены в общей формуле (I) по п.1, и R обозначает метильную или этильную группу.
9. Применение соединения формулы (I) по любому из пп.1-5 в виде фармацевтически приемлемого основания, соли присоединения кислоты, гидрата или сольвата в качестве лекарственного средства, предназначенного для профилактики или лечения патологии, в которой участвуют эндогенные каннабиноиды и/или любые другие субстраты, метаболизированные ферментом FAAH.
10. Фармацевтическая композиция для профилактики или лечения патологии, в которой участвуют эндогенные каннабиноиды и/или любые другие субстраты, метаболизированные ферментом FAAH, содержащая по меньшей мере одно соединение формулы (I) по любому из пп.1-5 в виде фармацевтически приемлемого основания, соли присоединения кислоты, гидрата или сольвата и возможно один или несколько фармацевтически приемлемых эксципиентов.
11. Применение соединения формулы (1) по любому из пп.1-5 в виде фармацевтически приемлемого основания, соли присоединения кислоты, гидрата или сольвата для получения лекарственного средства, предназначенного для профилактики или лечения патологии, в которой участвуют эндогенные каннабиноиды и/или любые другие субстраты, метаболизированные ферментом FAAH.
12. Применение соединения формулы (I) по любому из пп.1-5 в виде фармацевтически приемлемого основания, соли присоединения кислоты, гидрата или сольвата для получения лекарственного средства, предназначенного для профилактики или лечения острых или хронических болей, головокружения, тошноты, рвоты, нарушений в области приема пищи, нейрологических и психиатрических патологий, острых или хронических нейро-дегенеративных заболеваний, эпилепсии, нарушения сна, сердечно-сосудистых заболеваний, ишемии почки, онкологических заболеваний, нарушений иммунной системы, аллергических заболеваний, инфекционных заболеваний, вызванных паразитами, вирусами или бактериями, воспалительных заболеваний, остеопороза, глазных болезней, легочных заболеваний, заболеваний желудочно-кишечного тракта или недержания мочи.
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Емкостный первичный преобразователь | 1975 |
|
SU548798A1 |
| Способ получения изохинолинов или их солей | 1975 |
|
SU557756A3 |

