ОБЛАСТЬ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Настоящее изобретение связано с новыми противоопухолевыми соединениями кахалалида, в частности с аналогами кахалалида F, где алифатическая 5-метилгексановая кислота замещена 4-метилгексановой кислотой, фармацевтическими композициями, содержащими их, и их применением в качестве противоопухолевых, антивирусных, противогрибковых агентов и при лечении псориаза.
ОСНОВА СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Соединения кахалалида представляют собой пептиды, выделенные из гавайских морских травоядных видов моллюска Elysia rufescens и его корма, зеленой водоросли Bryopsis. sp. Кахалалиды A-F описаны у Hamman et al., J. Am. Chem. Soc., 1993, 115, 5825-5826.
Кахалалиды A-G описаны у Hamann, M. et al., J. Org. Chem, 1996, 61, 6594-6600: "Kahalalides: bioactive peptides from a marine mollusk Elysia rufescens and its algal diet Bryopsis sp.".
Кахалалиды H и J описаны у Scheuer P. J. et al, J. Nat. Prod. 1997, 60, 562-567: "Two acyclic kahalalides from the sacoglossan mollusk Elysia rufescens ".
Кахалалид O описан у Scheuer P. J. et al., J. Nat. Prod. 2000, 63(1) 152-4: A new depsipeptide from the sacoglossan mollusk Elysia ornata and the green alga Bryopsis species".
В связи с кахалалидом K см. публикацию Kan, Y. et al., J. Nat. Prod. 1999 62(8) 1169-72: "Kahalalide K: A new cyclic Depsipeptide from the hawaiian green alga bryopsis species".
Что касается связанных с ним публикаций, см. также Goetz et al., Tetrahedron, 1999, 55; 7739-7746: "The absolute stereochemistry of Kahalalide F"; Albericio, F. et al. Tetrahedron Letters, 2000, 41, 9765-9769: "Kahalalide B. Synthesis of a natural cyclodepsipeptide"; Becerro et al. J. Chem. Ecol. 2001, 27(11), 2287-99: "Chemical defenses of the sarcoglossan mollusk Elysia rufescens and its host Alga bryopsis sp.".
Среди кахалалидных соединений кахалалид F является наиболее многообещающим в силу его противоопухолевой активности. Он имеет сложную структуру, включающую в себя шесть аминокислот в циклической части и экзоциклическую цепочку из семи аминокислот с концевой группой из жирной кислоты. О его активности против клеточных культур in vitro клеток A-549 легочной карциномы человека и клеток HT-29 кишечной карциномы человека сообщалось в публикации EP 610 078. Было также показано, что кахалалид F обладает противовирусным и противогрибковым свойствами.
В предклинических исследованиях in vivo было показано, что максимально переносимая доза (МПД) однократной болюсной в/в инъекции кахалалида F у самок мышей составляет 280 мкг/кг. Несмотря на то, что однократные дозы чуть выше МПД в/в были исключительно токсичны, так что у животных проявлялись признаки нейротоксичности, заканчивающиеся смертельным исходом, дозу в 280 мкг/кг кахалалида F можно было вводить повторно, по схеме пять введений по одному введению в день, без каких-либо признаков острой токсичности. См. Supko, F. et al., Proceedings of the 1999 AACR NCI EORTC International Conference, abstract 315: "Preclinical pharmacology studies with the marine natural product Kahalalide F".
В Международной заявке WO 02 36145 описаны фармацевтические композиции, содержащие кахалалид F, и новые варианты применения этого соединения в терапии рака - эта публикация во всей своей полноте включена в настоящее описание в виде ссылки.
В Международной заявке WO 03 33012, от даты подачи которой испрашивается приоритет настоящей заявки, описано клиническое применение соединений кахалалида в онкологии, и эта публикация также во всей своей полноте включена в настоящее описание в виде ссылки.
В патенте Великобритании GB 0304367, от даты подачи которого также испрашивается приоритет настоящей заявки, описано применение соединений кахалалида при лечении псориаза и родственных ему соединений, и эта публикация также во всей своей полноте включена в настоящее описание в виде ссылки.
Синтез и цитотоксические активности природных и синтетических соединений кахалалида описаны в Международной заявке WO 01 58934, которая во всей своей полноте включена в настоящее описание в виде ссылки. В документе WO 01 58934 описан синтез кахалалида F, а также соединений со сходной структурой, в которых концевая цепь из жирной кислоты заменена другими жирными кислотами.
По-прежнему ощущается необходимость в создании новых противоопухолевых соединений, в частности дополнительных соединений кахалалида с улучшенными свойствами.
КРАТКОЕ СОДЕРЖАНИЕ ИЗОБРЕТЕНИЯ
Авторы изобретения неожиданно обнаружили, что одно из соединений-аналогов кахалалида обладает весьма перспективной активностью и проявляет повышенное противоопухолевое действие на моделях in vivo.
Настоящее изобретение связано с соединением формулы 1
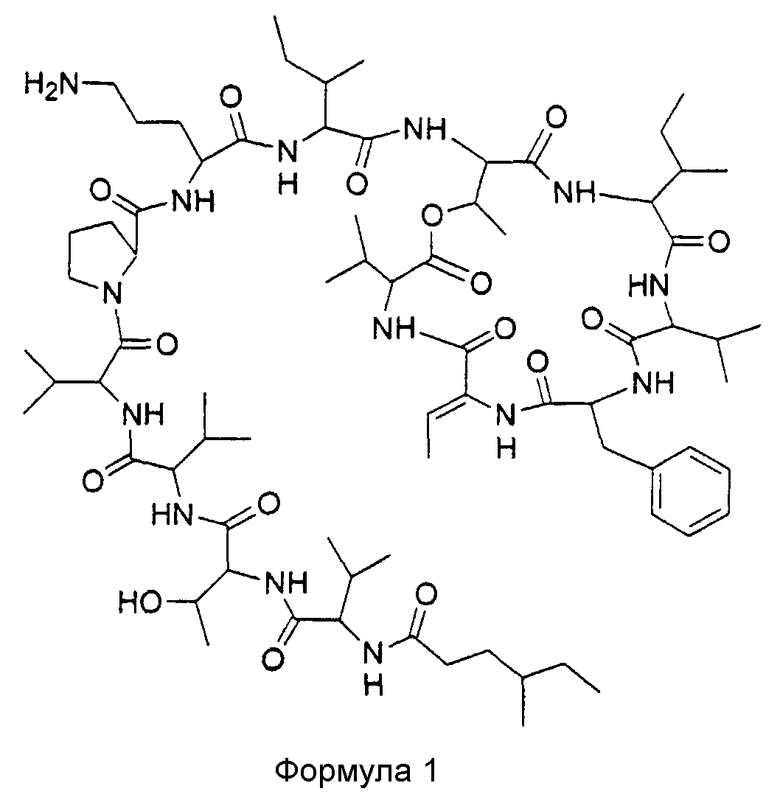
и его фармацевтически приемлемыми солями, пролекарствами, таутомерами и сольватами.
Такое соединение соответствует кахалалиду F с 4-метилгексановой цепью в виде концевой жирной кислоты, и в дальнейшем в настоящем описании будет обозначаться как 4-метилгексановый KF.
В предпочтительном воплощении данное изобретение связано с соединением, содержащим (4S)-метилгексановую кислоту, имеющую формулу 2
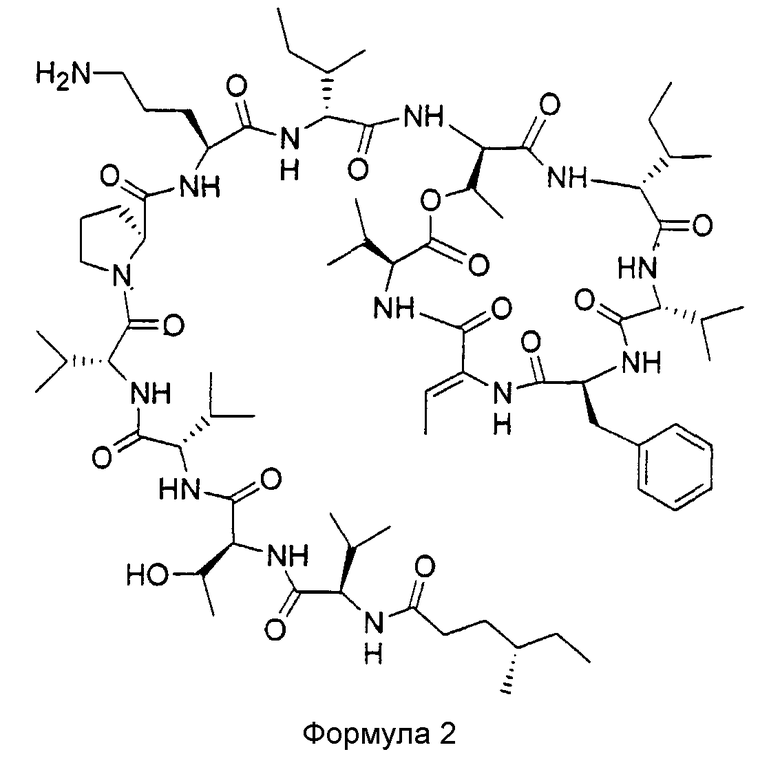
и его фармацевтически приемлемыми солями, пролекарствами, таутомерами и сольватами. Такое соединение будет в дальнейшем в настоящем описании обозначаться как (4S)-метилгексановый KF.
Настоящее изобретение связано с фармацевтической композицией, содержащей определяемое выше соединение и фармацевтически приемлемый носитель, наполнитель или разбавитель.
Настоящее изобретение дополнительно связано со способом лечения любого животного, особенно человека, пораженного злокачественной опухолью или псориазом, причем этот способ включает в себя введение пораженному заболеванием индивиду терапевтически эффективного количества соединения, определяемого выше.
Настоящее изобретение может быть использовано, в частности, для лечения пациентов с рефрактерными злокачественными опухолями, которые отличаются неудовлетворительной реакцией на другие терапевтические воздействия. В частности, композиции согласно изобретению могут быть использованы после того как были использованы другие виды химиотерапии, которые не оказали должного действия.
Настоящее изобретение направлено, в частности, на лечение пациентов, пораженных злокачественной опухолью предстательной железы, злокачественной опухолью молочной железы, гепатоклеточной карциномой, меланомой, злокачественной опухолью ободочной и прямой кишки, злокачественной опухолью почки, злокачественной опухолью яичника, злокачественной опухолью NSCL (карцинома легких), злокачественной опухолью эпителия, злокачественной опухолью поджелудочной железы и опухолями, для которых характерна повышенная экспрессия онкогена Her2/neu.
В другом аспекте настоящее изобретение направлено на применение определяемого выше соединения для производства лекарственного средства. В предпочтительном воплощении указанное лекарственное средство предназначено для лечения злокачественной опухоли, псориаза, вирусной инфекции или грибковой инфекции.
Настоящее изобретение дополнительно связано с наборами, включающими в себя раздельные контейнеры, содержащие фармацевтическую композицию, содержащую определяемое выше соединение и восстанавливающий агент. Способы восстановления также обеспечиваются настоящим изобретением.
Настоящее изобретение направлено также на способ получения определяемого выше соединения. В способе получения предпочтительно используется в качестве исходного соединения 4-метилгексановая кислота. В предпочтительном воплощении 4-метилгексановая кислота представляет собой (4S)-метилгексановую кислоту. В наиболее предпочтительном воплощении этот способ представляет собой твердофазный синтез.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Авторы изобретения идентифицировали аналоги кахалалида F, которые проявляют значительно более улучшенную активность по сравнению с кахалалидом F. Как показано в сравнительных примерах, 4-метилгексановый кахалалид F необъяснимым образом проявляет улучшенную эффективность на in vivo моделях злокачественных опухолей. Это представляется еще более неожиданным с точки зрения малых структурных различий между 4-метилгексановым кахалалидом F и 5-метилгексановым кахалалидом F.
Соединением согласно изобретению является кахалалид F с цепью 4-метилгексановой жирной кислоты вместо 5-метилгексановой, имеющий структуру в соответствии с формулой 1
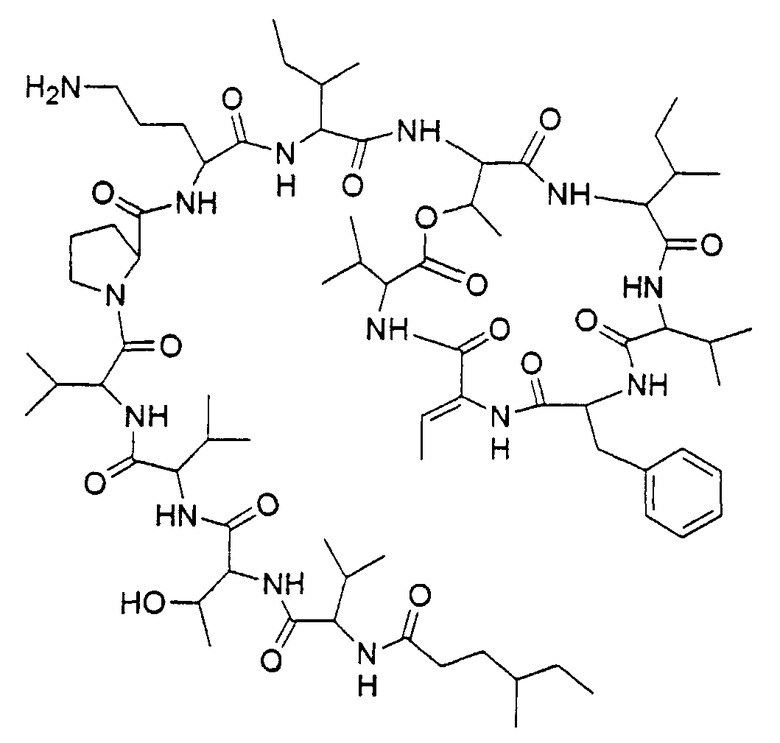
В частности, является предпочтительным, чтобы это соединение имело стереохимию, определяемую формулой 2
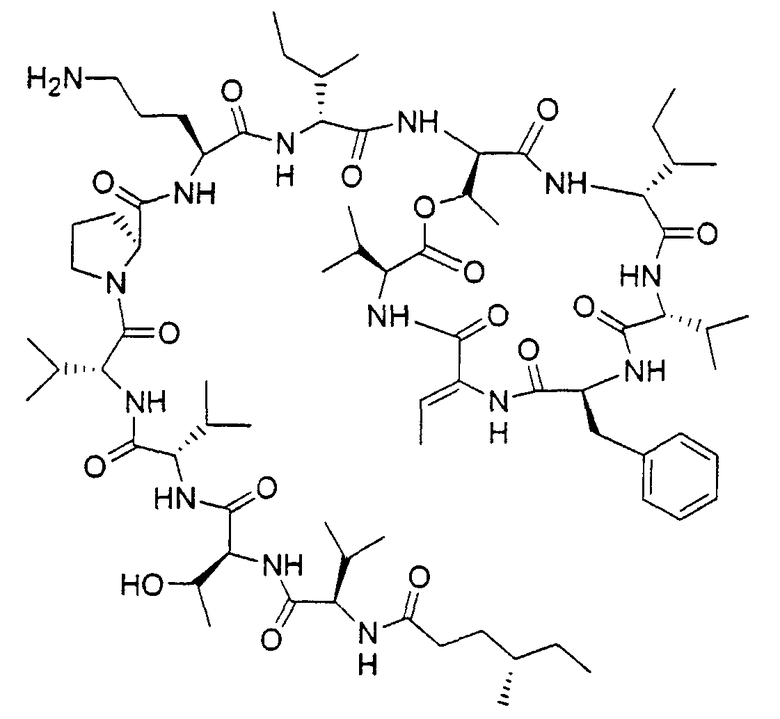
Тем не менее соединения согласно изобретению имеют асимметричные центры и, следовательно, существуют в различных энантиомерных и диастереомерных формах. Данное изобретение связано с применением всех оптических изомеров и стереоизомеров соединений согласно изобретению и их смесей, а также с фармацевтическими композициями, содержащими их, и со способами лечения, в которых они используются.
Для удобства авторы изобретения ссылаются на соединения согласно изобретению, а именно на соединения 1 и 2, как на соединения 4-метилгексилкахалалида F, или соединения 4-мегексKF. Предпочтительно соединения 4-мегексKF согласно изобретению являются соединениями, которые в большой степени свободны, по существу свободны или полностью свободны от других соединений кахалалида. Например, соединение 4-мегексKF согласно изобретению предпочтительно свободно от кахалалида F, имеющего 5-метилгексильную боковую цепь. В частности, 4-мегексKF согласно изобретению предпочтительно содержит самое большее 25%, 10%, 5%, 2%, 1% или 0,5%, или менее чем 0,5% любого другого кахалалида, особенно кахалалида F. В близком аспекте, 4-мегексKF согласно изобретению обеспечивается по существу в чистом виде. Такое соединение 4-мегексKF, в некоторой степени свободное от других кахалалидов, особенно подходит для фармацевтических композиций и способов лечения согласно изобретению.
Настоящее изобретение включает в себя соединения согласно изобретению и их фармацевтически приемлемые соли, в которых один или более атомов водорода, углерода или других атомов замещены их изотопами. Такие соединения могут быть использованы в качестве средств для научных исследований и диагностики при изучении метаболизма и фармакокинетики, а также в анализах связывания.
В данном описании соединения согласно изобретению, включая соединения формулы 1 и 2, определяются как соединения, которые включают в себя также и фармацевтически приемлемые производные или пролекарства. "Фармацевтически приемлемое производное или пролекарство" означает любую фармацевтически приемлемую соль, сложный эфир, соль сложного эфира или иное производное соединения согласно изобретению, которое при введении реципиенту способно обеспечить (прямо или косвенно) соединение согласно изобретению или его метаболит или остаток. Особенно предпочтительными производными и пролекарствами являются такие, которые увеличивают биодоступность соединений согласно изобретению, когда такие соединения вводятся пациенту (например, придавая перорально вводимому соединению свойство повышенной абсорбции в крови), улучшают доставку родительского соединения к конкретному биологическому компартменту, повышают растворимость, создавая возможность введения путем инъекции, изменяют метаболизм или изменяют скорость экскреции.
Соли соединений согласно изобретению включают в себя аддитивные соли, полученные из азота в соединении формулы 1 или 2. Терапевтическая активность, как определено в настоящем описании, связана с фрагментом, полученным из соединения согласно изобретению, а идентичность оставшегося компонента не столь важна, хотя для терапевтических и профилактических целей предпочтительно, чтобы он являлся фармацевтически приемлемым для пациента. Примеры фармацевтически приемлемых кислотно-аддитивных солей включают в себя такие кислотно-аддитивные соли, которые получены из минеральных кислот, таких как хлористоводородная, бромистоводородная, фосфорная, метафосфорная, азотная и серная кислоты, органических кислот, таких как винная, уксусная, трифторуксусная, лимонная, яблочная, молочная, фумаровая, бензойная, гликолевая, глюконовая, янтарная и метансульфоновая и арилсульфоновая, например, пара-толуолсульфоновая, кислоты. Предпочтительной солью является соль трифторуксусной кислоты.
Соединения согласно изобретению могут быть получены в соответствии с синтетическими процессами, описанными в международной заявке WO 01 58934, например, при добавлении соответствующей (S) или (R) 4-метилгексановой кислоты вместо 5-метилгексановой в примере 3 международной заявки WO 01 58934. Следовательно, данное изобретение распространяется также на способ получения соединения формулы 1 или 2. В этом способе в качестве исходного материала используется 4-метилгексановая кислота. Наиболее предпочтительным исходным материалом является (4S)-метилгексановая кислота. Способ синтеза предпочтительно представляет собой способ твердофазного синтеза. Дополнительные подробности синтеза приведены в примерах.
Способ согласно изобретению может быть осуществлен на основе исходных материалов в энантио-, стереоконтролируемом и быстром режиме, с использованием преимуществ методологии твердофазного синтеза, где на протяжении всех операций синтеза молекула в конструкции связана с нерастворимой подложкой.
Фармацевтические композиции соединений согласно изобретению могут быть адаптированы для любого желаемого пути введения, например перорального (включая буккальное или подъязычное), ректального, назального, местного (включая буккальное, подъязычное или трансдермальное), вагинального или парентерального (включая подкожное, внутримышечное, внутривенное или внутридермальное) путей введения. Такие композиции могут быть получены любым способом, известным в области фармации, например путем ассоциации активного ингредиента с носителем (носителями) или наполнителем (наполнителями).
Предпочтительные фармацевтические композиции соединений согласно изобретению включают в себя жидкость (растворы, суспензии или эмульсии) с соответствующим составом для внутривенного введения, и они могут содержать соединение в чистом виде или в комбинации с любым носителем или другими фармацевтически активными соединениями. Дальнейшим путеводителем в связи с фармацевтическими композициями может служить международная заявка WO 02 36145, которая во всей ее полноте включена в настоящее описание в виде ссылки.
Таким образом, комбинация неионного поверхностно-активного вещества и органической кислоты является подходящей для использования с агентом-наполнителем для получения лиофилизированной формы соединения согласно изобретению, подходящей для восстановления. Восстановление предпочтительно производят путем смешивания эмульсифицирующего солюбилизатора, алканола и воды.
Лиофилизированные композиции предпочтительно включают в себя в основном наполнитель, например, по меньшей мере 90% или по меньшей мере 95% агента-наполнителя. Примеры агентов-наполнителей хорошо известны и включают в себя сахарозу и маннит. Могут быть использованы также и другие агенты-наполнители.
Неионным поверхностно-активным веществом в лиофилизированной композиции предпочтительно является сложный эфир сорбитана, более предпочтительно - сложный эфир полиэтиленсорбитана, такой как полиоксиэтиленсорбитаналканоат, в частности полиоксиэтиленсорбитанмоноолеат, например полисорбат-80. Неионное поверхностно-активное вещество обычно составляет небольшой процент композиции, например от 0 до 5% композиции, например от 2 до 3 или 4% композиции.
Органической кислотой в составе лиофилизированной композиции обычно является алифатическая кислота, предпочтительно гидроксикарбоновая кислота, а более предпочтительно - гидроксиполикарбоновая кислота, в частности лимонная кислота. Органическая кислота обычно составляет небольшой процент композиции, например, от 0 до 5% композиции, например, от 2 до 3 или 4% композиции.
Количество соединения согласно изобретению в составе лиофилизированной композиции обычно составляет менее 1% или чаще менее чем 0,1% смеси. Подходящее количество заключено в пределах от 50 до 200 мкг, например приблизительно 100 мкг на 100 мг композиции.
Эмульсифицирующий солюбилизатор для восстанавливающего агента в приемлемом случае содержит сложный эфир полиэтиленгликоля, в особенности сложный эфир жирной кислоты, более предпочтительно - ПЭГ-олеат, такой как ПЭГ-олеат-35. Эмульсифицирующий солюбилизатор в приемлемом случае составляет от 0 до 10% восстанавливающего агента, обычно приблизительно от 3 до 7%, например приблизительно 5%. Алаканол обычно является этанолом, который в приемлемом случае составляет от 0 до 10% восстанавливающего агента, обычно приблизительно от 3 до 7%, например приблизительно 5%. Остальную часть восстанавливающего агента составляет вода, и в результате получают восстановленный раствор, подходящий для внутривенной инъекции.
Дальнейшее разбавление восстановленного раствора 0,9% физиологическим раствором может быть предпринято для инфузии кахалалидного соединения. Соответствующее для инфузии оборудование предпочтительно включает в себя стеклянный контейнер, который является предпочтительнее полиэтиленового. Трубки предпочтительно должны быть сделаны из силикона.
Далее, предпочтительный восстанавливающий агент составляет от 2 до 7%, например приблизительно 5%, эмульсифицирующего солюбилизатора; от 2 до 7%, например приблизительно 5%, спирта; а остальное - вода.
Композиции могут быть представлены в контейнерах с единичной дозой или в контейнерах с многократной дозой, например запечатанных ампулах и флаконах, и могут храниться в замороженно-высушенном (лиофилизированном) состоянии, требуя лишь дополнительного стерильного жидкого носителя, например воды для инъекций, используемого непосредственно перед употреблением.
Таким образом, настоящее изобретение дополнительно связано с наборами, включающими в себя отдельные контейнеры, содержащие лиофилизированную композицию и восстанавливающий агент. Описание способов восстановления также включено в набор.
Введение соединений или композиций согласно изобретению производится путем внутривенной инфузии. Время инфузирования может достигать 72 часов, более предпочтительно от 1 до 24 часов, наиболее предпочтительно либо приблизительно 1 час, либо приблизительно 3 часа. Особенно предпочтительными являются короткие времена инфузирования, которые позволяют производить терапевтическое воздействие без необходимости пребывания больного в госпитале в течение ночного времени. Однако в случае необходимости инфузия может приблизительно продолжаться в течение 24 часов и даже дольше.
Предпринимается цикличное введение, в предпочтительном способе введения пациентам производят внутривенную инфузию соединения согласно изобретению в первую неделю каждого цикла, затем в остальное время в рамках цикла пациентам дают время для восстановления. Предпочтительная длительность каждого цикла составляет либо 1, либо 3, либо 4 недели; в случае необходимости могут применяться множественные циклы. В альтернативном протоколе дозирования соединения согласно изобретению вводят, например, приблизительно в течение 1 часа в течение 5 дней подряд в течение 3 недель. В качестве вариантов могут быть прописаны и другие схемы.
В случае необходимости прибегают к отсрочиванию дозы или снижению доз и изменению схемы введения, в зависимости от толерантности индивидуального пациента к лечению, в частности снижение доз рекомендуется больным, у которых уровни трансаминазы или щелочной фосфатазы в сыворотке выше нормы.
В одном из аспектов настоящего изобретения обеспечивается способ лечения человека, пораженного злокачественной опухолью, включающий в себя введение указанному больному соединения согласно изобретению в дозе ниже 1200 мкг/м2/день, предпочтительно ниже 930 мкг/м2/день, а более предпочтительно ниже 800 мкг/м2/день. Подходящей дозой является по меньшей мере 320 мкг/м2/день. Предпочтительной является доза в интервале от 400 до 900 мкг/м2/день, предпочтительно от 500 до 800 мкг/м2/день, более предпочтительно от 600 до 750 мкг/м2/день. Особенно предпочтительными являются дозы приблизительно от 650 до 700 мкг/м2/день.
В следующем аспекте настоящего изобретения предлагается способ лечения человека, пораженного злокачественной опухолью, включающий в себя введение указанному больному соединения согласно изобретению ежедневно в течение 5 дней в дозе ниже 930 мкг/м2/день, после чего следует период восстановления, составляющий от 1 до 4 недель, в течение которого соединение кахалалида больному не вводят. Доза предпочтительно составляет 650-750 мкг/м2/день, более предпочтительно - приблизительно 700 мкг/м2/день. Время инфузии предпочтительно составляет между 1 и 24 часами, более предпочтительно - между 1 и 3 часами. Особенно предпочтительным является время инфузии, составляющее приблизительно 1 час или примерно 3 часа. Оставшийся период предпочтительно составляет 2-3 недели, более предпочтительно - приблизительно 2 недели.
Настоящее изобретение также обеспечивает способ лечения человека, пораженного злокачественной опухолью, включающий в себя введение указанному больному соединения согласно изобретению еженедельно в дозе ниже 800 мкг/м2/день. Доза предпочтительно составляет 600-700 мкг/м2/день, более предпочтительно - 650 мкг/м2/день. Время инфузии предпочтительно составляет от 1 до 24 часов, более предпочтительно - между 1 и 3 часами. Особенно предпочтительным является время инфузии, составляющее приблизительно 1 час.
Несмотря на приведенные выше ориентировочные дозировки, точная подборка дозировки соединения будет варьировать, в зависимости от конкретной композиции, способа введения и с учетом конкретной ситуации, связанной с состоянием самого больного и опухоли, которые подвергаются лечению. Должны быть приняты во внимание и другие факторы, такие как возраст, вес тела, пол, диета, время введения, скорость выведения, состояние больного, комбинация лекарственных средств, реакция чувствительности и тяжесть заболевания. Введение может быть произведено в непрерывном режиме или же периодически, в рамках максимально переносимой дозы.
Настоящее изобретение в особенности направлено на лечение пациентов, пораженных злокачественной опухолью предстательной железы, злокачественной опухолью молочной железы, гепатоклеточной карциномой, меланомой, злокачественной опухолью прямой кишки, злокачественной опухолью почки, злокачественной опухолью яичника, карциномой легких NSCL, злокачественной опухолью эпителия, злокачественной опухолью поджелудочной железы опухолями, которые в избытке экспрессируют онкоген Her2/neu. В наиболее предпочтительном варианте оно направлено на лечение гепатоклеточной злокачественной опухоли, меланомы, злокачественной опухоли молочной железы и злокачественной опухоли предстательной железы.
Настоящее изобретение также относится к способу лечения кожного заболевания, включающего в себя гиперпролиферацию дермальных клеток у млекопитающего, где указанный способ включает в себя введение указанному млекопитающему эффективного, нетоксичного количества соединения согласно изобретению. Предпочтительно, если кожным заболеванием является псориаз. Настоящее изобретение предпочтительно направлено на лечение больного человека, пораженного псориазом, в особенности тяжелой формой псориаза.
Соединения и композиции согласно изобретению могут быть использованы и в сочетании с другими лекарственными средствами для обеспечения комбинированной терапии. Указанные другие лекарственные средства могут составлять часть одной и той же композиции или же могут быть представлены в виде отдельной композиции для одновременного введения или же раздельного введения в разное время. Тождественность другого лекарственного средства не является особенно лимитированной, хотя комбинации с другими химиотерапевтическими, гормональными средствами и антителами должны учитываться. Количества соединения согласно изобретению и другого фармацевтически активного агента или агентов и относительные времена введения должны быть отработаны с целью достижения желаемого эффекта комбинированной терапии.
ПРИМЕРЫ
Пример 1: получение (4S)-метилгексанового KF
Основные процедуры и начальные стадии процесса соответствуют таковым, описанным в международной заявке WO 01 58934.
(4S)-MeHex-D-Val-Thr(tBu)-Val-D-Val-D-Pro-Orn(Boc)-D-allo-Ile-D-allo-Thr(Val-Alloc)-D-allo-Ile-D-Val-O-TrtCl-полимерная смола:
Fmoc-группу удаляли, и к указанному выше пептидил-полимеру последовательно добавляли Fmoc-Val-OH (678 мг, 2 ммоль, 4 эквив.), Fmoc-Thr(tBu)-OH (992 мг, 2,5 ммоль, 5 эквив.), Fmoc-D-Val-OH (678 мг, 2 ммоль, 4 эквив.) и (4S)-MeHex-OH (195 мг, 1,5 ммоль, 3 эквив.) (пример 3) с использованием DIPCDI (233 мкл на 1,5 ммоль и 3 эквив.; 310 мкл на 2 ммоль и 4 эквив.; и 388 мкл на 2,5 ммоль, 5 эквив.) и HOBt (230 мг на 1,5 ммоль и 3 эквив.; 307 мг на 2 ммоль и 4 эквив.; и 395 мг, 2,5 ммоль, 5 эквив.) в течение 90 мин. Во всех случаях через 90 мин связывания нингидриновый тест был отрицательным. Удаление Fmoc-группы и промывание проводили, как описано в разделе General Procedures.
(4S)-MeHex-D-Val-Thr(tBU)-Val-D-Val-D-Pro-Orn(Boc)-D-allo-Ile-D-allo-Thr(Val-Z-Dhb-Phe-Alloc)-D-allo-Ile-D-Val-O-TrtCl-полимерная смола:
Alloc-группу удаляли с помощью Pd(PPh3)4 (58 мг, 0,05 ммоль, 0,1 эквив.) в присутствии PhSiH3 (617 мкл, 5 ммоль, 10 эквив.) в атмосфере Ar, а Alloc-Phe-Z-Dhb-OH (666 мг, 2 ммоль, 4 эквив.) и HOAt (273 мг, 2 ммоль, 4 эквив.) растворяли в DMF (1,25 мл) и добавляли к пептидил-полимерной смоле, затем добавляли DIPCDI (310 мкл, 2 ммоль, 4 эквив.), и смесь перемешивали в течение 5 часов, когда нингидриновый тест был отрицательным.
После промывания с использованием DMF и CH2Cl2 аликвоту пептидил-полимерной смолы обрабатывали смесью ТФУ-H2О (1:99) в течение 1 мин, и полученный продукт охарактеризовали с использованием метода MALDI-TOF-MS, рассчитывая для C88H146N14О21, 1736,18. Получено: m/z 1758,67 [M+NA]+, 1774,62 1618,2 [M+K]+.
(4S)-MeHex-D-Val-Thr(tBU)-Val-D-Val-D-Pro-Orn(Boc)-D-allo-Ile-D-allo-Thr(Val-Z-Dhb-Phe-H)-D-allo-Ile-D-Val-OH:
После промывания с использованием DMF и CH2Cl2 Alloc-группу удаляли с помощью Pd(PPh3)4 (58 мг, 0,05 ммоль, 0,1 эквив.) в присутствии PhSiH3 (617 мкл, 5 ммоль, 10 эквив.) в атмосфере Ar. Защищенный пептид отщепляли от полимерной смолы с помощью смеси ТФУ-CH2Cl2 (1:99) (5Ч30 сек). Фильтрат собирали на H2О (4 мл), и H2О частично удаляли на роторном испарителе. Затем добавляли ACN для растворения твердого вещества, образовавшегося в процессе удаления H2О, и раствор лиофилизировали, получая 639 мг (387 мкмоль, выход 77%) указанного в заголовке соединения со степенью чистоты >95%, что подтверждено методом HPLC (Кондиция A, tR 10,5 мин).
(4S)-MeHex-D-Val-Thr-Val-D-Val-D-Pro-Orn-D-allo-Ile-цикло(D-allo-Thr-D-allo-Ile-D-Val-Phe-Z-Dhb-Val)= (4S)метилгексановый KF
Защищенный пептид (пример 6) (639 мг, 387 мкмоль) растворяли в CH2Cl2 (390 мл, 1 мМ), и HOBt (237 мг, 1,55 ммоль) растворяли в минимальном объеме DMF для растворения HOBt, добавляли DIEA (203 мкл, 1,16 ммоль, 3 эквив.) и DIPCDI (240 мкл, 1,55 ммоль, эквив.). Смесь оставляли на мешалке в течение 1 часа, затем результат выполнения стадии циклизации контролировали методом HPLC. Растворитель удаляли путем выпаривания в условиях пониженного давления. Защищенный циклический пептид растворяли в смеси ТФУ-H2O (19:1, 85 мл), и образовавшуюся смесь оставляли на мешалке в течение 1 часа. Растворитель удаляли путем выпаривания в условиях пониженного давления, и добавляли диоксан (30 мл), и растворитель удаляли путем выпаривания в условиях пониженного давления (этот процесс повторяли трижды), затем добавляли H2О (40 мл), и осуществляли лиофилизацию. Грубый продукт очищали методом HPLC (Kromasil C8 5 мкм, 205×50 мм), изократически 44% ацетонитрила (+0,05% ТФУ) в воде (+0,05% ТФУ), 55 мл/час, детектировали при 220 нм, получая указанный в заголовке продукт (192 мг, 0,13 ммоль, выход 26%, 92,3%). MALDI-TOF-MS, рассчитывая для C75H124N14О16, 1477,9. Получено: m/z 1500,12 [M+Na]+, 1515,97 [M+K]+.
Спектр 1H-ЯМР (2,5 мМ; 500 МГц, H2О-D2О (9:1) этого соединения указан в таблице I).
7,25 (3Н Ar, д)
(2Н)
1H-ЯМР-спектроскопию [1H, NOESY, TOCSY при (278K)] проводили на установке Varian Unity Plus (500 МГц). Химические сдвиги (d) выражены в частях на миллион в сторону уменьшения относительно тетраметилсилана (TMS). Константы связывания выражены в Герцах.
Пример 2: получение (4R)-метилгексанового KF
Экспериментальные процедуры проводились, как в примере 1, начиная с 1 г полимерной смолы, с одним только исключением, что на соответствующей стадии (4S)-MexHex был заменен на (4R)-MexHex. Полученный продукт (220 мг, 0,15 ммоль, 30%, 92,3% чистоты) был охарактеризован с помощью ES-MS, C75H124N14О16, 1477,9. Получено: m/z 1499,07 [M+Na]+, 1514,94 [M+K]+.
Пример 3: Цитотоксическая активность in vitro
Целью данных исследований является прекращение роста культуры опухолевых клеток "in vitro" путем непрерывного контакта этих клеток с тестируемым образцом.
КЛЕТОЧНЫЕ ЛИНИИ
андрогенных рецепторов
Разновидность колориметрического анализа с использованием реакции сульфородамина B (SRB) была адаптирована для количественного измерения роста клеток и их жизнеспособности [в соответствии с технологией, описанной Philip Skehan, et al. (1990), New colorimetric cytotoxicity assay for anticancer drug screening, J. Natl. Cancer Inst., 82: 1107-1112].
В этом способе анализа используют 96-луночные культуральные микропланшеты диаметром 9 мм (Faircloth, 1988; Mosmann, 1983). Большинство клеточных линий получено из Американской Коллекции Типовых Культур (ATCC), образованной из различных типов раковых клеток человека.
Клетки поддерживали в культуральной среде RPMI 1640, 10% PBS, обогащенной 0,1 г/л пенициллина и 0,1 г/л стрептомицинсульфата, а затем инкубировали при 37°C в атмосфере 5% CO2 и 98% влажности. Для проведения экспериментов клетки собирали из субконфлуэнтных культур с помощью трипсина и перед посевом ресуспендировали их в свежей среде.
Клетки высевали в 96-луночные культуральные микропланшеты, по 5×103 клеток на лунку, в аликвоты из 195 мкл среды, и оставляли их, давая им прикрепиться к поверхности планшета в условиях роста в культуральной среде, не содержащей лекарственного средства, в течение 18 часов. Затем добавляли образцы [испытуемого соединения] в аликвотах по 5 мкл в количествах, варьирующих от 10 до 10-8 мкг/мл, растворенных в DMSO/EtOH/PBS (0,5:0,5:99). После 48-часовой экспозиции противоопухолевый эффект измеряли, используя метод SRB: клетки фиксировали путем добавления 50 мкл холодной 50% (вес/объем) трихлоруксусной кислоты (ТХУ) и инкубировали в течение 60 минут при 4°C. Планшеты промывали деионизированной водой и сушили. Сто микролитров раствора SRB (0,4% вес/объем в 1% уксусной кислоте) добавляли в каждую лунку микропланшета и инкубировали в течение 10 минут при комнатной температуре. Несвязанный SRB удаляли путем промывания 1% уксусной кислотой. Планшеты сушили на воздухе, и связанный краситель солюбилизировали Трис-буфером. Оптические плотности считывали на автоматическом спектрофотометрическом устройстве для считывания планшетов при одной длине волны в 490 нм.
На основании данных, полученных в трех параллельных лунках, вычисляли величины средних значений +/- SD. Были вычислены некоторые параметры, характеризующие ответы клеток: GI = ингибирование роста, TGI = общее ингибирование роста (цитостатический эффект) и LC = клеточная гибель (цитотоксический эффект).
Результаты приведены в таблице II, существенных различий между приведенными соединениями не отмечается.
Пример 4: токсичность in vitro
С целью установления цитотоксичности указанных лекарственных средств в отношении нормальных клеток использовали 96-луночные планшеты, засеянные с плотностью по 5000 клеток на лунку клетками нормальных клеточных линий, установленных в соответствии с указаниями ATCC: AML-12, нормальные клетки печени мыши, и NRK-52E, нормальные клетки почки крысы. После высевания клеток в каждом планшете их выдерживали в течение ночи перед добавлением тестируемого лекарственного средства. К каждой лунке (100 мкл среды) добавляли 10 мкл лекарственного средства в среде в различных концентрациях (в конечной концентрации 1×10-10-0,01 мг/мл), а затем инкубировали в течение ночи при 37°C в присутствии 5% CO2. Все эксперименты повторяли по меньшей мере 3 раза, причем в каждом эксперименте использовали по две параллельные пробы. Через 24 часа осуществляли анализ MTS (водный CellTiter 96), в соответствии с инструкциями производителя (PROMEGA) (для всех типов клеток). Жизнеспособность клеток (митохондриальную активность) определяли путем ферментативного превращения формазанового субстрата.
Как видно из результатов, приведенных в таблице III, существенных различий между соединениями 5-метилгексанового KF и (4S)-метилгексанового KF не наблюдается.
IC50 (мкМ)
IC50 (мкМ)
Пример 5: МПД in vivo у мыши CD-1 и у бестимусных животных
Максимально переносимую дозу определяли у мышей CD-1 и у бестимусных мышей (обоих полов) для каждого лекарственного средства после однократного введения болюса и после 5 ежедневных введений дозы. Результаты приведены в таблице IV, где существенных различий между соединениями 5-метилгексанового KF и (4S)-метилгексанового KF не наблюдается.
Болюс МПД
Болюс МПД
5DD МПД
5DD МПД
Пример 6: эффективность in vivo при ксенотрансплантации молочной железы (5DD)
Эффективность 5-метилгексанового KF и (4S)-метилгексанового KF, каждый из которых вводили в количестве трех четвертей от максимально переносимой дозы (МПД), изучали у бестимусных самок мышей, которым после ксенотрансплантации молочной железы внутривенно (в/в) вводили в течение пяти дней подряд (т.е. в режиме QDx5) болюсные инъекции (дни 0-4). Соединения применяли в виде свежеприготовленных запаянных в ампулы растворов в составе с носителем [кремофор-EL/этанол/вода (5:5:90)]. Каждую суточную дозу (в режиме QDx5) вводили внутривенно в объеме 0,2 мл раствора для инъекций на 20 граммов веса животного.
Оценка собственно размера опухоли в группах, обработанных соответствующим образом, относительно контрольной группы, которой вводили носитель (т.е. % ΔT/ΔC), показала, что самое низкое (оптимальное) значение достигалось на день 3 после начала введения лекарственного средства во всех группах. Кроме того, с помощью попарного статистического анализа (с использованием непараметрического метода Mann-Whitney) удалось выяснить, что существует значительная разница в эффективности между двумя указанными соединениями, что является весьма неожиданным с той точки зрения, что эти соединения имеют очень схожую структуру, а также в свете результатов, приведенных в предшествующих примерах.
На основе описанных здесь исследований эффективности и проведенных ранее экспериментов на токсичность (пример 4) терапевтический показатель по меньшей мере 1,33 (1 × МПД/0,75 × МПД, доза, при которой лекарственное средство является токсичным/дозу, при которой лекарственное средство является эффективным) может быть утвержден для соединения (4S)-метилгексановый KF. Кроме того, еще одним связанным с этим биологическим результатом явилось то, что противоопухолевый эффект (4S)-метилгексанового KF в отношении молочной железы был более продолжительным, чем в отношении трансплантатов предстательной железы (пример 7), при одной и той же относительной дозе МПД. Резюмируя, можно утверждать, что (4S)-метилгексановый KF определенно является более мощным изомером в отношении трансплантатов молочной железы, а продолжительность его биологического действия дает основания полагать, что он имеет более длительное влияние на опухоли такого типа.
Пример 7: эффективность in vivo при ксенотрансплантации предстательной железы (5DD)
Эффективность одинаковой дозы каждого из 5-метилгексанового KF и (4S)-метилгексанового KF изучали у бестимусных самцов мышей, которым после ксенотрансплантации предстательной железы внутривенно (в/в) вводили в течение пяти дней подряд (т.е. в режиме QDx5) болюсные инъекции (дни 0-4). Соединения применяли в виде свежеприготовленных запаянных в ампулы растворов в составе с носителем [кремофор-EL/этанол/вода (5:5:90)]. Каждую суточную дозу (в режиме QDx5) вводили внутривенно в объеме 0,2 мл раствора для инъекций на 20 граммов веса животного.
Оценка собственно размера опухоли в группах, обработанных соответствующим образом, относительно контрольной группы, которой вводили носитель (т.е. % ΔT/ΔC), показала, что самое низкое (оптимальное) значение достигалось на день 3 после начала введения лекарственного средства во всех группах. Кроме того, с помощью попарного статистического анализа (с использованием непараметрического метода Mann-Whitney) удалось выяснить, что существенно большая эффективность достигалась с помощью (4S)-метилгексанового KF в дозе 262 мкг/кг/день. На основе описанных здесь исследований эффективности и проведенных ранее экспериментов на токсичность (пример 4) терапевтический показатель по меньшей мере 1,33 (1 × МПД/0,75 × МПД, доза, при которой лекарственное средство является токсичным/дозу, при которой лекарственное средство является эффективным) может быть утвержден для соединения (4S)-метилгексановый KF. Результаты приведены ниже в таблице VI.
Пример 8: противоопухолевая активность аналогов кахалалида F, исследуемая на полых волокнах с использованием панели линий опухолевых клеток человека
Противоопухолевая активность аналогов кахалалида F, обсуждаемая выше, была проверена на системе полых волокон (HF) с помощью панели линий опухолевых клеток человека, а именно клеток SK-Hep-1 (гепатомы), HepG2 (гепатоклеточной карциномы), Panc-1 (поджелудочной железы) и Mel-28 (меланомы). Опухолевые клетки человека были инкапсулированы в полые волокна in vitro, а затем имплантированы бестимусным мышам-самкам in vivo.
Дозы 5-метилгексанового KF и (4S)-метилгексанового KF подбирали на основе предварительных МПД-экспериментов, проведенных на бестимусных мышах, в результате которых была выбрана дозировка 325 мкг/кг/день (см. пример 5). Пять дней подряд дозу вводили внутрибрюшинно (в/б) в объеме 0,2 мл раствора для инъекций на 20 граммов веса животного.
В целом соединение KF-4(S)-Met проявляет статистически значимую противоопухолевую активность против клеток гепатомы (подкожно, п/к), гепатоклеточной карциномы (как подкожно, так и внутрибрюшинно), поджелудочной железы (внутрибрюшинно) и проявляет тенденцию в сторону значимости эффекта (т.е. P=0,059) при подкожном введении при опухоли поджелудочной железы и меланоме. В отличие от этого соединения, KF-5-Met активен в отношении меньшего количества типов опухолей, а именно только в отношении опухолей поджелудочной железы (как подкожно, так и внутрибрюшинно) и меланомы (подкожно), но не в отношении всех типов рака печени, которые были протестированы. Ниже в таблице приведены суммарные результаты, которые ясно показывают различия между этими соединениями.
количество клеток/HF)
* Статистически значимо, P<0,05.
§ Тенденция к значимости, т.е. P=0,059.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ КАХАЛАЛИДА F | 2004 |
|
RU2395520C2 |
| КАХАЛАЛИДНЫЕ СОЕДИНЕНИЯ | 2001 |
|
RU2280039C2 |
| КАХАЛАЛИД F | 2001 |
|
RU2292216C2 |
| ПРОТИВООПУХОЛЕВЫЕ СОЕДИНЕНИЯ ДИГИДРОПИРАН-2-ОНА | 2007 |
|
RU2444519C2 |
| ПРОИЗВОДНЫЕ ЭКТЕИНАСЦИДИНА, ИХ ПРИМЕНЕНИЕ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2002 |
|
RU2283842C2 |
| ПРОТИВОРАКОВЫЕ СТЕРОИДНЫЕЛАКТОНЫ, НЕНАСЫЩЕННЫЕ В ПОЛОЖЕНИИ 7(8) | 2011 |
|
RU2572595C2 |
| СЛОЖНЫЕ ЭФИРЫ СТЕРОИДНОГО ЛАКТАМА И ПРОИЗВОДНЫХ БИС(2-ХЛОРЭТИЛ)АМИНОФЕНОКСИПРОПАНОВОЙ КИСЛОТЫ | 2016 |
|
RU2719457C2 |
| ПИРИДИНОВЫЕ СОЕДИНЕНИЯ ПЛАДИЕНОЛИДА И СПОСОБЫ ПРИМЕНЕНИЯ | 2015 |
|
RU2807278C2 |
| ПРОТИВООПУХОЛЕВЫЕ СОЕДИНЕНИЯ | 2008 |
|
RU2493147C2 |
| ПИРИДИНОВЫЕ СОЕДИНЕНИЯ ПЛАДИЕНОЛИДА И СПОСОБЫ ПРИМЕНЕНИЯ | 2015 |
|
RU2707730C2 |
Настоящее изобретение связано с новыми противоопухолевыми соединениями кахалалида, в частности с аналогами кахалалида F, используемыми в качестве противоопухолевых агентов. 6 н. и 7 з.п. ф-лы, 6 табл.
1. Соединение формулы 1

и его фармацевтически приемлемые соли, сложные эфиры, соли сложных эфиров или таутомеры.
2. Соединение по п.1, представляющее собой (4S)-MeHex-D-Val-L-Thr-L-Val-D-Val-D-Pro-L-Orn-D-allo-Ile-цикло(D-allo-Thr-D-allo-Ile-D-Val-L-Phe-Z-Dhb-L-Val), или его фармацевтически приемлемые соли, сложные эфиры, соли сложных эфиров или таутомеры.
3. Соединение по п.1, представляющее собой (4S)-MeHex-D-Val-L-Thr-L-Val-D-Val-D-Pro-L-Orn-D-allo-Ile-цикло(D-allo-Thr-D-allo-Ile-D-Val-L-Phe-Z-Dhb-L-Val), или его фармацевтически приемлемые соли.
4. Фармацевтическая композиция, обладающая противоопухолевой активностью, содержащая терапевтически эффективное количество соединения, определяемого в любом из пп.1-3, и фармацевтически приемлемый носитель, наполнитель или разбавитель.
5. Способ лечения млекопитающего, предпочтительно человека, пораженного злокачественной опухолью, включающий в себя введение указанному индивиду терапевтически эффективного количества соединения по любому из пп.1-3.
6. Способ по п.5, где млекопитающим является человек.
7. Способ по п.6, где пациент поражен стойкой формой злокачественной опухоли, которая не дает благоприятного ответа на другие виды терапии.
8. Способ по п.6, где злокачественная опухоль выбрана из злокачественной опухоли предстательной железы, злокачественной опухоли молочной железы, гепатоклеточной карциномы, меланомы, злокачественной опухоли прямой кишки, злокачественной опухоли почки, злокачественной опухоли яичника, карциномы легких NSCL, злокачественной опухоли эпителия, злокачественной опухоли поджелудочной железы и опухолей, которые экспрессируют в повышенном количестве онкоген Her2/neu.
9. Соединение, определяемое в любом из пп.1-3, для применения в качестве лекарственного средства для лечения злокачественной опухоли.
10. Применение соединения, определяемого в любом из пп.1-3, в производстве лекарственного средства для лечения злокачественной опухоли.
11. Набор, содержащий отдельные контейнеры, которые содержат фармацевтическую композицию, обладающую противоопухолевой активностью, которая содержит соединение, определяемое в любом из пп.1-3, и восстанавливающий агент.
12. Способ получения соединения, определяемого в любом из пп.1-3, путем твердофазного синтеза, где в качестве исходного соединения используют 4-метилгексановую кислоту.
13. Способ по п.12, отличающийся тем, что используют (4S)-метилгексановую кислоту.
Приоритет по пунктам:
18.10.2002, 24.06.2003 по п.1;
24.06.2003 по пп.2 и 3;
18.10.2002, 24.06.2003, 20.10.2003 по пп.4-11;
24.06.2003, 20.10.2003 по пп.12 и 13.
| АНАЛОГ-АГОНИСТ АМИЛИНА, ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, КОМПОЗИЦИЯ, ПРОЯВЛЯЮЩАЯ СВОЙСТВА АГОНИСТА АМИЛИНА | 1992 |
|
RU2130463C1 |
| ШАРНИРНАЯ МУФТА | 0 |
|
SU236145A1 |
| US 6011010 А, 04.01.2000 | |||
| Устройство для регулирования температуры | 1976 |
|
SU610078A1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |

