Область изобретения
Настоящее изобретение относится к новым гомо-аза-стероидным сложным эфирам с алкилирующими горчицами, производными анилина, такими как бис(2-хлорэтил)аминофеноксипропановая кислота и замещенные производные.
Предпосылки к созданию изобретения
В наше время алкилирующие противораковые агенты, такие как азотные горчицы, все еще остаются эффективным классом противоопухолевых лекарственных средств в текущей клинической практике, терапевтические эффекты которой происходят из их способности присоединять алкильные группы к клеточной ДНК и приводить к значительному повреждению ДНК (Hurley LH, Nature Rev Cancer, 2002, 2:188-200; Brendel M and Ruhland A, Mutat Res, 1984; 133:51-85).
Стероидные конъюгаты ранее использовались в качестве носителей цитотоксических алкилирующих агентов, потому что они уменьшают системную токсичность и улучшают эффективность терапии рака (Wall ME et al, J Med Chem, 1969, 12:810-8; Catane R, Cancer Treat Rep, 1978; 62:1264-5). Стероидные алкилирующие агенты, такие как эстрамустин (сложный эфир эстрадиола и мехлорэтамина) и преднимустин (сложный эфир преднизолона и хлорамбуцила) в настоящее время применяются в терапии рака для лечения рака предстательной железы и лимфопролиферативных злокачественных новообразований, соответственно (Catane R, Cancer Treat Rep, 1978, 62:1264-5; Matsumoto K et al, Med Oncol, 2013, 30:717; IARC Monogr Eval Carcinog Risks Hum, 1990, 50:115-22; Hiddemann W, Eur J Cancer, 1995, 31A(13-14):2141-5).
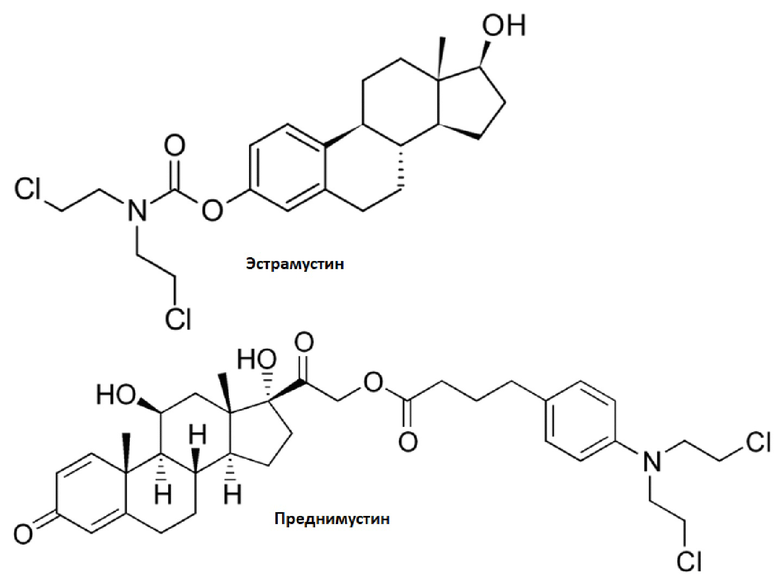
Хотя эти лекарственные средства проявляют уменьшенную острую и системную токсичность вопреки намного более высокой токсичности, которую их алкилирующие компоненты проявляют индивидуально, их противораковая активность улучшается не так сильно, также как специфичность нацеливания для раковых клеток оказывается ограниченной, несмотря на начальные оценки. Однако, даже если главные молекулярные фармакологические механизмы, через которые проявляется противораковая активность эстрамустина и преднимустина, довольно отличаются от специфического действия на стероидные рецепторы, в целом они показывают хорошую и улучшенную терапевтическую эффективность в клинической практике.
Несколько гомо-аза- или лактамовых стероидных сложных эфиров (стероиды, содержащие группу лактама -NHC=O- в стероидном кольце/кольцах, конъюгированных с алкилирующими агентами) были ранее синтезированы и протестированы на токсичность и противораковую активность в преклинических исследованиях, как in vitro, так и in vivo (Wampler GL and Catsoulacos P, Cancer Treat Rep, 1977, 61:37-41; Catsoulacos P and Catsoulacos D, Anticancer Res, 1991, 11:1773-7; Catsoulacos P and Catsoulacos D, Anticancer Res, 1993, 13(4):1203-8; Catsoulacos P et al, Oncology, 1994, 51:74-8; Catsoulacos P and Catsoulacos D, Anticancer Res, 1994, 14(6B):2525-8; Camoutsis C and Trafalis DT, Invest New Drugs, 2003, 21:47-54; Koutsourea AI et al, Bioorg Med Chem, 2008, 16:5207-15).
Было показано, что сложные алкилирующие эфиры лактама и стероида генерируют значительно уменьшенную острую токсичность in vivo, тогда как они продемонстрировали улучшенную и очень перспективную противоопухолевую активность in vitro и in vivo, в то время как соответствующие немодифицированные (нелактамные) стероидные алкилирующие агенты проявляли значительно более низкую или слабую противораковую активность против соответствующих экспериментальных опухолевых систем. Кроме повреждения клеточной ДНК, молекулярные фармакологические механизмы, значительно улучшившие противораковое действие алкилирующих сложных эфиров лактама и стероида, все еще неизведаны. Кроме того, биологическая важность положения, в котором одна или более групп лактама включены в стероидную структуру, также неизвестна. Кроме того, алкилирующий агент, конъюгированный через сложноэфирную связь с лактамовым стероидом, играет значительную роль и модулирует пропорцию острой токсичности и противоопухолевой активности, и, следовательно, степени терапевтического индекса, который генерирует лактамный стероидный алкилирующий агент. До сих пор несколько активных лактамных стероидных алкилирующих агентов были синтезированы и протестированы, но те, которые показывали более высокую противоопухолевую активность, были более токсичными, а те, которые демонстрировали более низкую токсичность, были менее активными. Эти наблюдения показывают, что существует четкая потребность в разработке и получении новых активных лактам-стероидных алкилирующих конъюгатов, генерирующих оптимальную более низкую токсичность и более высокую противораковую активность, и поэтому оптимальный терапевтический индекс.
Предыдущие исследования алкилирующих лактам-стероидных сложных эфиров производных азотной горчицы показали, что 3бета-гидрокси-13альфа-амино-13,17-секо-5альфа-андростан-17-овый-13,17-лактам-[p-[бис(2-хлорэтил)амино]фенил]ацетат (ASE, NSC 290205) показывал очень хорошо сбалансированные эффекты в преклиническом тестировании острой токсичности in vivo и противоопухолевой активности in vitro и in vivo, поддерживая существенно высокий терапевтический индекс.
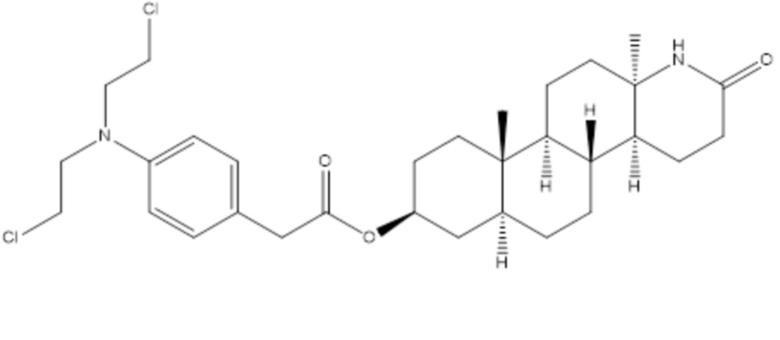
3бета-гидрокси-13альфа-амино-13,17-секо-5альфа-андростан-17-овый-13,17-лактам-[p-[бис(2-хлорэтил)амино]фенил]ацетат (ASE, NSC 290205)
Поэтому ASE представляет собой «золотой» стандарт для разработки новых молекул того же класса агентов и тестирования их в отношении терапевтической эффективности по сравнению с эффективностью ASE.
Сущность изобретения
Настоящее изобретение относится к новым сложным эфирам стероидных лактамов и алкилирующих агентов. Более конкретно, соединения согласно настоящему изобретению являются сложными эфирами стероидных лактамов с производными бис(2-хлорэтил)аминофеноксипропановой кислоты. Эти соединения показывают более высокую противоопухолевую активность и сниженную острую токсичность по сравнению со сложными алкилирующими эфирами стероидных лактамов предшествующего уровня техники и могут быть использованы в качестве противоопухолевых средств и средств для терапии рака.
Подробное описание изобретения
Настоящее изобретение относится к соединению формулы (I) или к его фармацевтически приемлемой соли
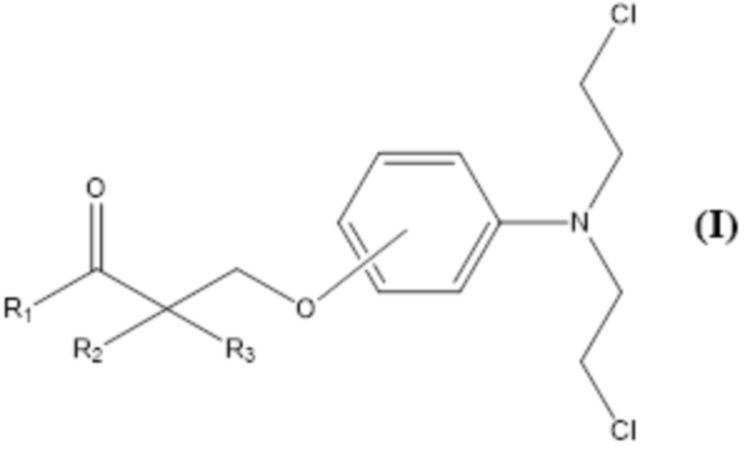
в которой
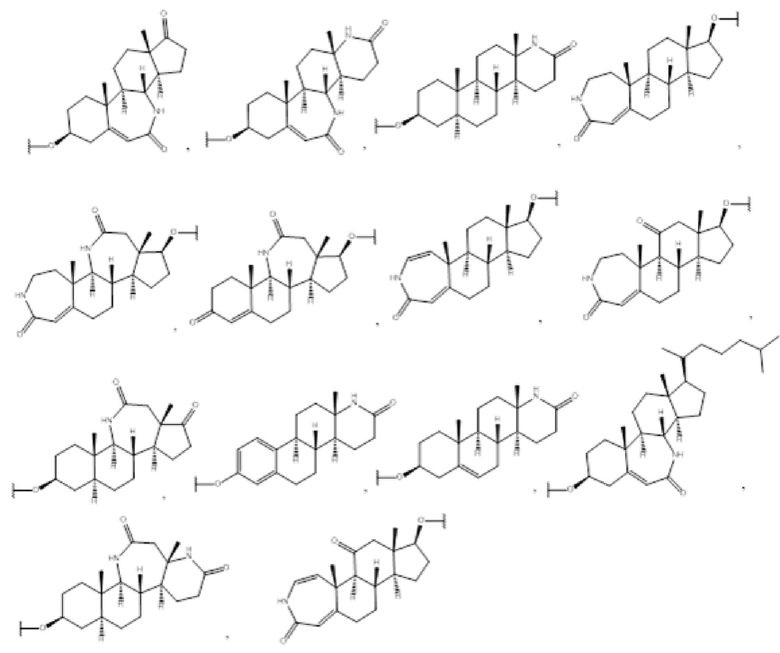
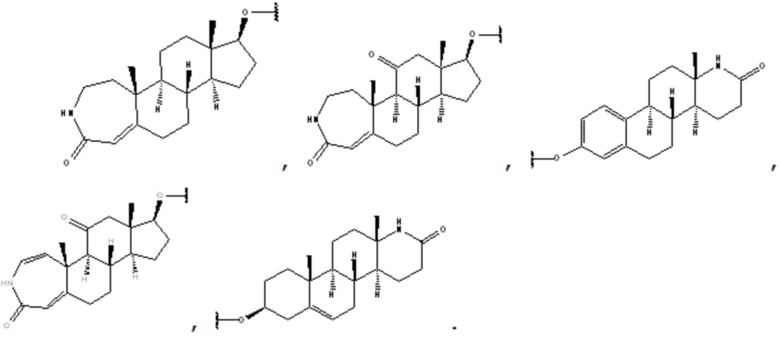
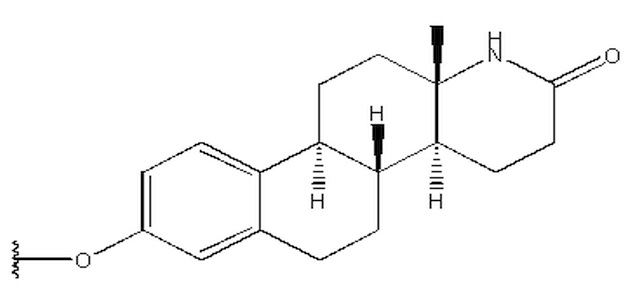
R1 выбран из группы, состоящей из
R2 выбран из группы, состоящей из H, -CH3, -CH=CH2, -CH2-CH3, -CH2CH2CH3,
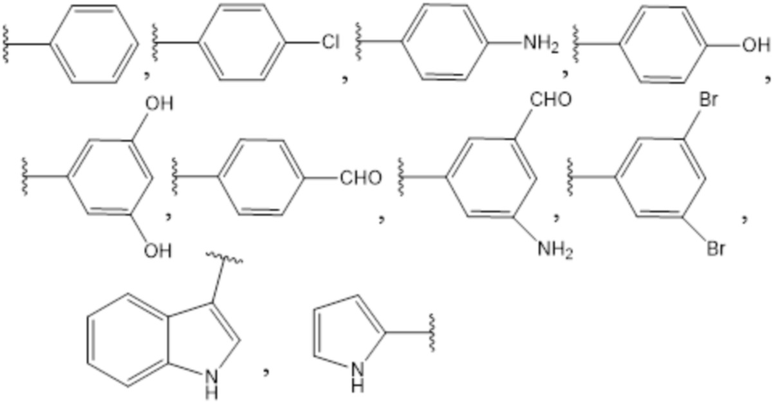
R3 выбран из группы, состоящей из H, -OH, -NH2.
Предпочтительно, R1 выбран из группы, состоящей из
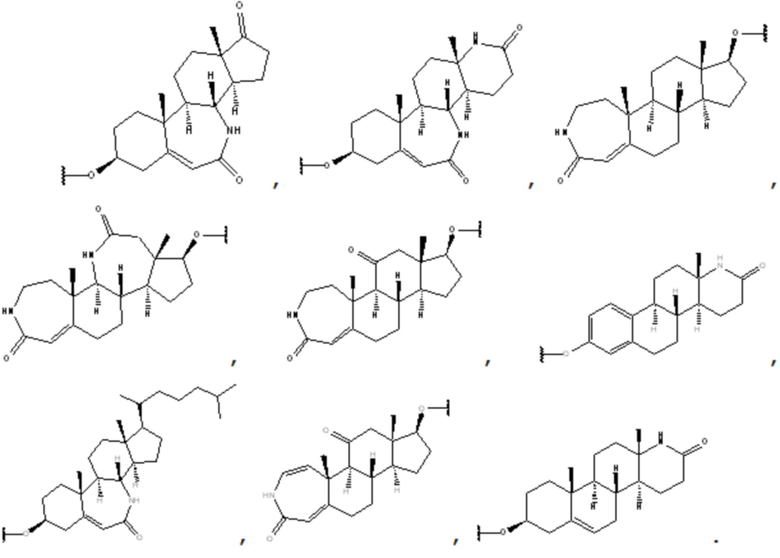
Более предпочтительно, R1 выбран из группы, состоящей из
Предпочтительно, R2 выбран из группы, состоящей из H, -CH3, -CH=CH2, -CH2-CH3, -CH2CH2CH3. Более предпочтительно, R2 обозначает H.
Предпочтительно, R3 обозначает H или NH2.
Гидроксильная группа бис(2-хлорэтил)аминофенольной группы соединений формулы (I) может быть в орто-, мета- или пара-положении относительно аминогруппы фенильного кольца.
Соединения формулы (I) содержат по меньшей мере один центр асимметрии. Если стереохимия центра асимметрии не определена, структура охватывает все отдельные стереоизомеры, а также их смеси.
Соединения формулы (I) содержат по меньшей мере одну основную функциональную группу и способны поэтому образовывать фармацевтически приемлемые соли в результате обработки подходящей кислотой. Подходящие кислоты включают фармацевтически приемлемые неорганические и фармацевтически приемлемые органические кислоты. Примеры фармацевтически приемлемых солей включают гидрохлорид, гидробромид, сульфат, фосфат, нитрат, ацетат, пропионат, бутират, малеат, фумарат, тартрат, цитрат, лактат, оксалат, сукцинат и бензоат.
Соединения формулы (I) или их фармацевтически приемлемые соли могут использоваться для лечения широкого диапазона раковых заболеваний. Предпочтительно, они используются для лечения рака яичника, молочной железы, предстательной железы или лейкоза.
Соединения формулы (I) или их фармацевтически приемлемые соли показывают более высокую противоопухолевую активность и пониженную острую токсичность по сравнению со сложными алкилирующими эфирами стероидных лактамов предшествующего уровня техники и могут быть использованы в качестве противоопухолевых средств и средств для терапии рака. Преклиническое тестирование на биологическую активность, раскрытое далее в примерах, демонстрирует превосходство новых алкилирующих стероидных сложных эфиров лактама в плане терапевтической эффективности в отношении рака над двумя положительными контролями, алкилирующим агентом (3-(4-(бис(2-хлорэтил)амино)фенокси)пропановой кислотой, pBCEAPOPA) индивидуально, и «золотым» стандартом описанного класса экспериментальных лактам-стероидных алкилирующих агентов, ASE (NSC 290205).
Настоящее изобретение относится также к фармацевтическим композициям, содержащим соединение формулы (I) или его фармацевтически приемлемую соль. Такая фармацевтическая композиция может быть составлена для введения любым подходящим путем, таким как пероральный, назальный, топический или парентеральный путь. Например, фармацевтическая композиция может быть составлена как таблетка, капсула, порошок, раствор, суспензия, крем или гель. Такая композиция обычно содержит, в дополнение к соединению формулы (I) или его фармацевтически приемлемой соли, фармацевтически приемлемый носитель. Такой носитель включает эксципиенты, известные в данной области техники, такие как разбавители, связующие, наполнители, разрыхлители, лубриканты, растворители, суспендирующие агенты, загустители, буферы, консерванты. Эти композиции могут быть получены согласно способам, известным в данной области техники.
Настоящее изобретение относится также к способам получения соединения формулы (I) или его фармацевтически приемлемой соли.
Стероидные лактамы (аза-гомо стероиды) в соединениях формулы (I) несут одну или более функциональных амидных групп в кольцах основной стероидной структуры. Известно, что такие стероидные лактамы могут быть синтезированы из кетостероида через соответствующие оксимы и перегруппировку Бекмана (Koutsourea AI et al, Steroids, 2003, 68(7-8):659-66; Mazur RH, J Org Chem, 1963, 28(1):248-250; Morzycki JW et al, Bioorg Med Chem, 1996, 4(8): 1209-15; Camoutsis C and Catsoulacos P, J Heterocycl Chem, 1983, 20(4):1093-4; Huang Y et al, Molecules, 2013, 18(7):7436-47).
Общая процедура перегруппировки Бекмана
Оксимы (1 ммоль) растворяли в 17,5 мл сухого диоксана. Смесь охлаждали до 0 °C, и тионилхлорид (1,9 мл) добавляли по каплям. Смеси давали достигнуть комнатной температуры и перемешивали в течение 24 ч. Реакцию гасили с помощью NaHCO3, и смесь экстрагировали этилацетатом (3×20 мл). Органические слои высушивали (Na2SO4) и концентрировали при пониженном давлении с получением сырого продукта, который далее очищали хроматографией на SiO2.
Производные бис(2-хлорэтил)аминофеноксипропановой кислоты и соединений согласно настоящему изобретению могут быть получены следующим образом:
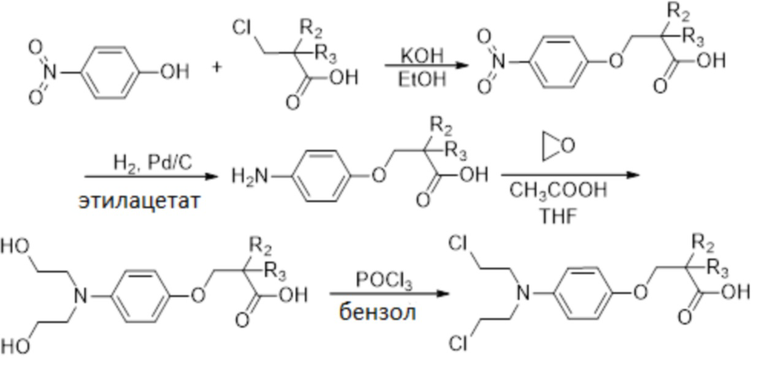
Замещенные 3-(4-(бис(2-хлорэтил)амино)фенокси)пропановые кислоты синтезировали исходя из 4-нитрофенола. Алкилирование 4-нитрофенола различными 3-хлорпропановыми кислотами дало 3-(4-нитрофенокси)пропановые кислоты, которые далее восстанавливали до аминопроизводных с использованием H2 и Pd/C в качестве катализатора. Затем аминогруппу еще раз алкилировали этиленоксидом в CH3COOH, THF согласно известной процедуре (Valu et al, J Med Chem, 1990, 33 (11): 3014-19). Наконец, спиртовые группы превращали в соответствующие хлориды с использованием POCl3 в бензоле и нагревали, получая соответствующие 3-(4-(бис(2-хлорэтил)амино)фенокси)пропановые кислоты. В некоторых случаях, когда присутствуют амино или гидроксильные группы, необходимы дополнительные стадии реакции присоединения и удаления защитных групп. Например, когда R3 обозначает группу NH2 или OH, используют Вос или ацетильные защитные группы, соответственно (Valu KK et al, J Med Chem, 1990, 33(11):3014-9). Согласно тому же способу, и исходя из 2-нитрофенола или 3-нитрофенола, могут быть синтезированы производные формулы (I), в которой гидроксильная группа находится в орто- или мета-положении относительно аминогруппы.
Для получения стероидных сложных эфиров лактама с алкилирующими агентами стероидные лактамы, содержащие группу OH, вводят в реакцию с ДНК-алкилирующим агентом. Например, стероидный лактам вводят в реакцию с 3-(4-(бис(2-хлорэтил)амино)фенокси)пропановой кислотой, с DCC, DMAP или с 3-(4-(бис(2-хлорэтил)амино)фенокси)пропаноилхлоридом или со смешанным ангидридом 3-(4-(бис(2-хлорэтил)амино)фенокси)пропановой кислоты для получения соответствующих сложных эфиров. Любой стероидный моно- или бис-лактам может быть дериватизован с использованием способа по изобретению.
Общая процедура A для этерификации стероидных лактамов
Спирт (1 ммоль) растворяли в 28 мл сухого дихлорметана. Затем добавляли кислоту (2 ммоль), DCC (2 ммоль) и каталитическое количество DMAP (3 мол.%). После перемешивания конечного раствора при комнатной температуре в течение 24 ч, растворитель выпаривали, и остаток очищали флэш-хроматографией на силикагеле.
Общая процедура B для этерификации стероидных лактамов
В круглодонной колбе 1 ммоль кислоты разбавляли в 3,3 мл сухого бензола. Добавляли 2,4,6-трихлорбензоилхлорид (1,2 ммоль) и триэтиламин (2,4 ммоль), и смесь нагревали с обратным холодильником в атмосфере Ar в течение 1 ч. К этой смеси добавляли раствор 50 мг стероидного спирта (1 ммоль) в 3,3 мл сухого бензола и каталитическое количество 4-диметиламинопиридина. Нагревание с обратным холодильником продолжали в течение 3 ч. Бензол полностью удаляли выпариванием в вакууме, и оставшийся остаток разбавляли CH2Cl2. Полученную смесь экстрагировали 5%-м водным раствором HCl, органический слой промывали 7%-м водным раствором NaHCO3 и наконец водой, высушивали над Na2SO4, и растворитель удаляли при пониженном давлении. Остаток подвергали флэш-хроматографии на силикагеле.
Общая процедура C для этерификации стероидных лактамов
Смесь спирта (1 ммоль), Et3N (1,3 ммоль) и каталитического количества DMAP растворяли в CH2Cl2 (5 мл) с последующим добавлением бензоилхлорида (0,12 мл, 1,1 ммоль). Реакцию проверяли с помощью TLC, и реакционную смесь перемешивали при комнатной температуре в течение 24 ч, затем растворяли CH2Cl2 и гасили насыщенным водным раствором NH4Cl. Органический слой высушивали, и сырой продукт очищали флэш-хроматографией на силикагеле.
Следующие примеры служат для иллюстрации изобретения.
Пример 1
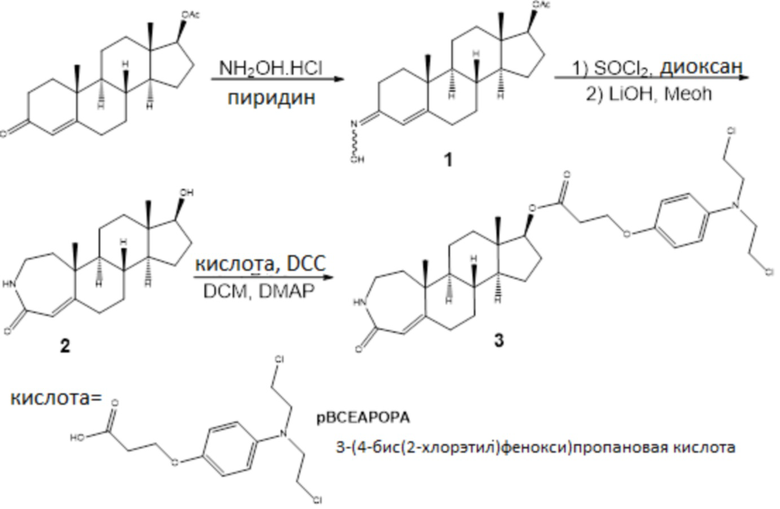
Схема 1
1: 3-Аза-17β-гидрокси-A-гомо-4α-андростен-4-он синтезировали модификацией (Camoutsis C и Catsoulacos P, J Heterocycl Chem, 1983, 20 (4):1093-4) процедуры в три стадии из тестостерон-17-β-ацетата. Тестостерон-17-β-ацетат (914 мг, 2,77 ммоль) растворяли в 10 мл сухого пиридина. Добавляли гидроксиламина гидрохлорид (461 мг, 6,64 ммоль), и раствор перемешивали при нагревании с обратным холодильником в течение 6 ч. Раствор выливали в воду, и смесь экстрагировали этилацетатом (3×30 мл). Органические слои высушивали (Na2SO4) и концентрировали при пониженном давлении с получением сырого продукта, который далее очищали хроматографией на SiO2 (элюент; гексан-этилацетат=4/1) с получением 675 мг син- и анти-оксимов (74%) в форме твердого вещества белого цвета.
2: Син- и анти-оксимы тестостерон-17-ацетата (100 мг, 0,145 ммоль) растворяли в 3,5 мл сухого диоксана. Смесь охлаждали до 0°C и добавляли по каплям тионилхлорид (0,6 мл). Смеси давали достигнуть комнатной температуры и перемешивали в течение 3 ч. Реакцию останавливали NaHCO3, и смесь экстрагировали этилацетатом (3×20 мл). Органические слои высушивали (Na2SO4) и концентрировали при пониженном давлении с получением сырого продукта, который затем очищали хроматографией на SiO2 (этилацетат) с получением 63 мг 3-аза-17β-ацетокси-A-гомо-4α-андростен-4-она (63%) в форме твердого вещества белого цвета.
3-Аза-17β-ацетокси-A-гомо-4α-андростен-4-он 1 растворяли в 4,9 мл MeOH и добавляли по каплям LiOH (1н., 2 мл). Смесь перемешивали при комнатной температуре в течение 2 ч. Реакцию останавливали NH4Cl, и смесь экстрагировали дихлорметаном (3×10 мл). Органические слои высушивали (Na2SO4) и концентрировали при пониженном давлении с получением 87 мг 3-аза-17β-гидрокси-A-гомо-4α-андростен-4-она 2 с 74%-м выходом.
3: 3-аза-17β-гидрокси-A-гомо-4α-андростен-4-он 2 растворяли в 28 мл сухого дихлорметана. Затем добавляли 3-(4-(бис(2-хлорэтил)амино)фенокси)пропановую кислоту (106 мг, 0,573 ммоль), DCC (119 мг, 0,574 ммоль) и каталитическое количество DMAP. После того, как конечный раствор перемешивали при комнатной температуре в течение 24 ч, растворитель выпаривали, и остаток очищали флэш-хроматографией на силикагеле (элюент; гексан-этилацетат=1/2) с получением конъюгата 3 (191 мг, 99%). 3: Т.пл.=53-56 ⁰C; [α]D23 +10,5 (c=0,91 CHCl3); 1H ЯМР (500 МГц, cdcl3) δ 6,92 (с, 1H), 6,83 (д, J=8,8 Гц, 2H), 6,66 (д, J=8,6 Гц, 2H), 5,72 (с, 1H), 4,66 (т, J=8,4 Гц, 1H), 4,17 (т, J=6,0 Гц, 2H), 3,63 (м, 4H), 3,59 (м, 4H), 3,32-3,04 (м, 2H), 2,75 (т, J=6,1 Гц, 2H), 2,48 (м, 1H), 2,27 (м, 1H), 2,15 (м, 2H), 1,50-1,98 (м, 10H), 1,33 (м, 2H), 1,14 (с, 3H), 1,05 (м, 1H), 0,80 (с, 3H); 13C ЯМР (126 МГц, cdcl3) δ 171,0, 170,4, 161,3, 151,3, 140,8, 118,8, 116,3, 114,4, 82,7, 64,4, 54,2, 53,2, 50,2, 44,5, 42,7, 41,9, 40,7, 36,7, 36,2, 35,3, 33,8, 33,1, 27,5, 25,6, 24,9, 23,4, 21,3, 12,1; FT-IR: 3450, 2925, 1731, 1651, 1607, 1512, 1469, 1353, 1238, 1181, 1041, 869, 813.
Пример 2
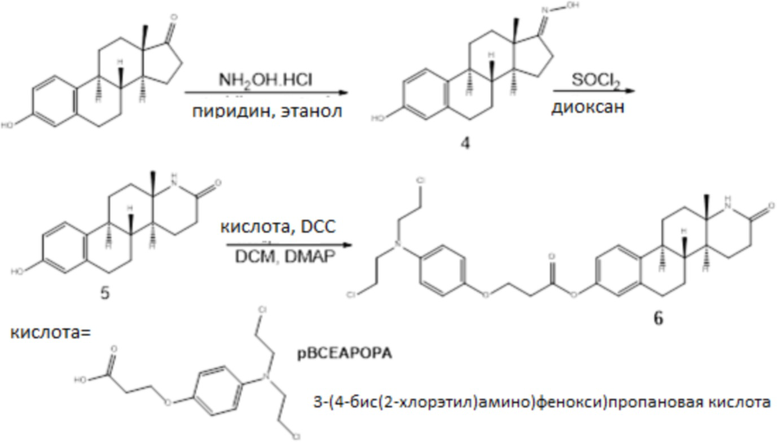
Схема 2
4: Оксим эстрона синтезировали согласно ранее описанной процедуре (Ivanenko TI et al, Pharm Chem J, 1982, 16(10):751-6). К раствору эстрона (100 мг, 0,37 ммоль) в 2,2 мл абсолютного этанола добавляли гидроксиламин гидрохлорид (62 мг, 0,88 ммоль) и пиридин (1,2 мл). Смесь нагревали с обратным холодильником в течение 6 часов. Затем добавляли воду, и смесь экстрагировали этилацетатом (3×10 мл). Органические слои высушивали (Na2SO4) и концентрировали при пониженном давлении с получением сырого продукта, который затем очищали хроматографией на SiO2 (элюент; гексан: этилацетат=3:1) с получением 105 мг оксима эстрона (100%) в форме твердого вещества белого цвета.
5: Лактам 5 синтезировали согласно ранее описанной процедуре (Regan BM and Newton Hayes F, J Am Chem Soc, 1956, 78(3): 639-43). Оксим эстрона (108 мг, 0,376 ммоль) растворяли в 6,3 мл сухого диоксана. Смесь охлаждали до 0 °C, и тионилхлорид (0,7 мл) добавляли по каплям. Смеси давали достигнуть комнатной температуры и перемешивали в течение 24 ч. Реакцию останавливали NaHCO3, и смесь экстрагировали дихлорметаном (3×20 мл). Органические слои высушивали (Na2SO4) и концентрировали при пониженном давлении с получением сырого продукта, который затем очищали хроматографией на SiO2 (элюент; гексан: этилацетат=2:1) с получением 42 мг лактама 5 (56% в расчете на рекуперированный исходный материал) вместе с рекуперированным исходным материалом [32 мг исходного материала (0,112 ммоль)].
6: Лактам 5 растворяли в 14 мл сухого DMF. Затем добавляли 3-(4-(бис(2-хлорэтил)амино)фенокси)пропановую кислоту (90 мг, 0,293 ммоль), DCC (61 мг, 0,293 ммоль) и каталитическое количество DMAP. После того, как конечный раствор перемешивали при комнатной температуре в течение 24 ч, растворитель выпаривали, и остаток очищали флэш-хроматографией на силикагеле (элюент; дихлорметан/ацетон=2/1) с получением конъюгата 6 (56 мг, 68%). Конъюгат 6: [α]D23+73,5 (c=0,90 CHCl3); 1H ЯМР (500 МГц, cdcl3) δ 7,25 (д, J=6,0 Гц, 1H), 6,89 (д, J=8,8 Гц, 2H), 6,82 (с, 1H), 6,68 (д, J=8,8 Гц, 2H), 6,31 (с, 1H), 4,30 (т, J=6,1 Гц, 2H), 3,62 (дт, J=29,2, 6,6 Гц, 8H), 2,97 (дд, J=15,1, 9,0 Гц, 2H), 2,88 (м, 2H), 2,58-2,36 (м, 4H), 2,23-2,00 (м, 2H), 1,92-1,66 (м, 3H), 1,60-1,29 (м, 4H), 1,19 (с, 3H); 13C ЯМР (126 МГц, cdcl3) δ 171,7, 169,8, 151,3, 148,5, 141,0, 137,8, 137,2, 126,1, 121,3, 118,7, 116,5, 114,5, 64,4, 54,4, 54,2, 46,6, 43,4, 40,7, 39,9, 38,9, 34,9, 30,5, 29,5, 26,5, 25,9, 22,1, 19,8; FTIR: 3329, 2927, 2850, 1757, 1626, 1577, 1512, 1437, 1311, 1244, 1157, 1088, 1045, 892.
Пример 3
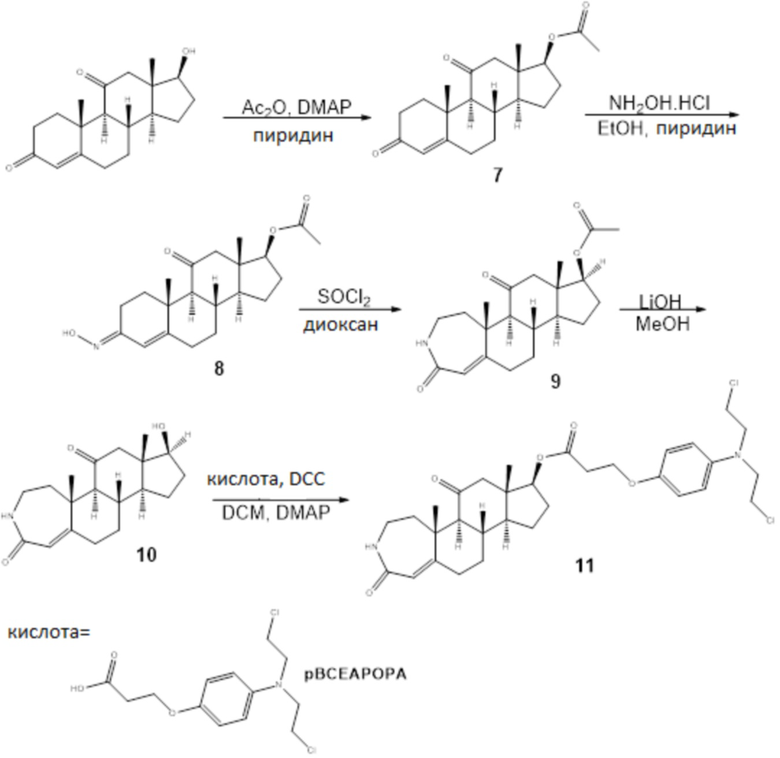
Схема 3
7: 17-Гидроксиандрост-4-ен-3,11-дион (484 мг, 1,59 ммоль) растворяли в 2,2 мл уксусного ангидрида. Затем добавляли 4 мг (0,037 ммоль) DMAP и 0,25 мл сухого пиридина. Смесь перемешивали при комнатной температуре в течение 24 ч. Реакцию останавливали водой, и смесь экстрагировали этилацетатом (3×30 мл). Органические слои высушивали (Na2SO4) и концентрировали при пониженном давлении с получением сырого продукта, который затем очищали хроматографией на SiO2 (элюент; гексан: этилацетат=6:1) с получением 472 мг 17-ацетоксиандрост-4-ен-3,11-диона с 86%-м выходом. 7: Т.пл.=162-164°C [α]D23+148,0 (c 1,68 CHCl3); 1H ЯМР (500 МГц, cdcl3) δ 5,69 (с, 1H), 4,76 (т, J=8,6 Гц, 1H), 2,83-2,69 (м, 1H), 2,54-2,20 (м, 6H), 2,01 (д, J=1,2 Гц, 3H), 1,92 (м, 3H), 1,85-1,55 (м, 4H), 1,51-1,41 (м, 1H), 1,44-1,34 (м, 3H), 1,32-1,10 (м, 2H), 0,85-0,69 (м, 3H); 13C ЯМР (126 МГц, cdcl3) δ 208,3, 199,5, 170,8, 168,3, 124,6, 80,2, 62,6, 54,8, 49,4, 46,2, 38,2, 37,0, 34,7, 33,7, 32,1, 31,7, 27,6, 22,9, 20,9, 17,2, 12,8; FT-IR: 3443, 2958, 2935, 2850, 1732,1702, 1677, 1618, 1426, 1373, 1360, 1343, 1271, 1238, 1224, 1045, 1027
8: К раствору 17-ацетоксиандрост-4-ен-3,11-диона (465 мг, 1,35 ммоль) в 7 мл абсолютного этанола добавляли гидроксиламин гидрохлорид (100 мг, 1,44 ммоль) и сухой пиридин (4,2 мл). Смесь перемешивали при комнатной температуре в течение 24 часов. Затем добавляли воду, и смесь экстрагировали этилацетатом (3×40 мл). Органические слои высушивали (Na2SO4) и концентрировали при пониженном давлении с получением сырого продукта, который затем очищали хроматографией на SiO2 (элюент; дихлорметан: этилацетат=20:1) с получением 461 мг оксимов 8 (95%).
9: Оксим 8 (264 мг, 0,74 ммоль) растворяли в 13 мл сухого диоксана. Смесь охлаждали до 0°C и добавляли по каплям тионилхлорид (1,4 мл). Смеси давали достигнуть комнатной температуры и перемешивали в течение 24 ч. Реакцию останавливали NaHCO3, и смесь экстрагировали этилацетатом (3×20 мл). Органические слои высушивали (Na2SO4) и концентрировали при пониженном давлении с получением сырого продукта, который затем очищали хроматографией на SiO2 (элюент; этилацетат: метанол=1: 0,03) с получением 163 мг лактама 9 с 62%-м выходом. 9: 1H ЯМР (500 МГц, cdcl3) δ 6,39 (с, 1H), 5,75 (с, 1H), 4,78 (т, J=8,6 Гц, 1H), 3,35-3,18 (м, 1H), 3,09 (дт, J=14,7, 7,2 Гц, 1H), 2,67 (дд, J=14,9, 8,2 Гц, 1H), 2,48 (тд, J=13,6, 3,9 Гц, 1H), 2,34-2,20 (м, 3H), 2,14 (дд, J=9,3, 6,5 Гц, 1H), 2,03 (с, 3H), 2,01-1,86 (м, 2H), 1,83-1,53 (м, 5H), 1,48-1,37 (м, 1H), 1,38 (с, 3H), 1,28-1,08 (м, 1H), 0,76 (с, 3H); 13C ЯМР (126 МГц, cdcl3) δ 209,0, 170,8, 169,5, 158,4, 120,1, 80,1, 62,4, 55,1, 49,9, 46,7, 43,6, 40,4, 36,8, 36,8, 35,5, 33,2, 27,6, 22,8, 21,1, 20,9, 12,8; FT-IR: 3428, 2971, 2920, 2878, 2364, 2341, 1736, 1701, 1664, 1639, 1599, 1444, 1375, 1339, 1245, 1127, 1089, 1046.
10: Лактам 9 76 мг (0,21 ммоль) растворяли в 3 мл MeOH и добавляли по каплям LiOH (1н., 1,2 мл). Смесь перемешивали при комнатной температуре в течение 1 ч. Реакцию останавливали NH4Cl, и смесь экстрагировали дихлорметаном (3×5 мл). Органические слои высушивали (Na2SO4) и концентрировали при пониженном давлении с получением 67 мг лактама 10. 10: 1H ЯМР (500 МГц, ДМСО) δ 7,72 (с, 1H), 5,51 (с, 1H), 4,66 (д, J=4,7 Гц, 1H), 3,66 (дд, J=13,4, 8,4 Гц, 1H), 3,08-2,90 (м, 2H), 2,47-2,32 (м, 2H), 2,29 (д, J=11,5 Гц, 1H), 2,21 (д, J=11,2 Гц, 1H), 2,14-2,01 (м, 2H), 2,01-1,78 (м, 3H), 1,74-1,50 (м, 3H), 1,40 (м, 1H), 1,28 (с, 3H), 1,23 (с, 1H), 1,15-1,02 (м, 1H), 0,55 (с, 3H); 13C ЯМР (126 МГц, ДМСО) δ 210,2, 167,8, 157,3, 120,3, 78,1, 61,0, 54,5, 48,8, 47,1, 43,1, 40,4, 36,8, 35,5, 34,9, 33,1, 29,9, 22,3, 20,9, 11,8; FT-IR: 3423, 3262, 2952, 2923, 2853, 1693, 1647, 1609, 1458, 1407, 1375, 1353, 1261, 1062.
11: Лактам 10 растворяли в 8,2 мл сухого DCM. Затем добавляли 3-(4-(бис(2-хлорэтил)амино)фенокси)пропановую кислоту (51 мг, 0,17 ммоль), DCC (51 мг, 0,25 ммоль) и каталитическое количество DMAP. После того, как конечный раствор перемешивали при комнатной температуре в течение 24 ч, растворитель выпаривали, и остаток очищали флэш-хроматографией на силикагеле (элюент; этилацетат) с получением конъюгата 11 (48,5 мг, 96%). Конъюгат 11: 1H ЯМР (500 МГц, cdcl3) δ 6,83 (д, J=9,0 Гц, 2H), 6,67 (д, J=9,0 Гц, 2H), 6,11 (с, 1H), 5,76 (с, 1H), 4,86 (т, J=8,6 Гц, 1H), 4,17 (т, J=6,2 Гц, 2H), 3,61 (м, 8H), 3,25 (м, 1H), 3,18-3,01 (м, 1H), 2,84-2,59 (м, 2H), 2,59-2,38 (м, 1H), 2,37-2,25 (м, 3H), 2,16 (м, 1H), 2,10-1,89 (м, 3H), 1,87-1,55 (м, 4H), 1,39 (с, 3H), 1,26 (м, 2H), 1,12 (м, 1H), 0,76 (с, 3H); 13C ЯМР (126 МГц, cdcl3) δ 208,9, 170,9, 169,5, 158,7, 151,3, 140,9, 119,9, 116,2, 114,5, 80,4, 64,2, 62,4, 55,1, 54,3, 49,9, 46,8, 43,6, 40,7, 36,8, 35,5, 34,9, 33,9, 27,6, 25,6, 24,9, 22,8, 21,2, 12,8; FT-IR: 3432, 3328, 2927, 2850, 1733, 1701, 1664, 1626, 1599, 1513, 1444, 1389, 1369, 1310, 1273, 1243, 1179, 1087, 1041, 999.
Пример 4
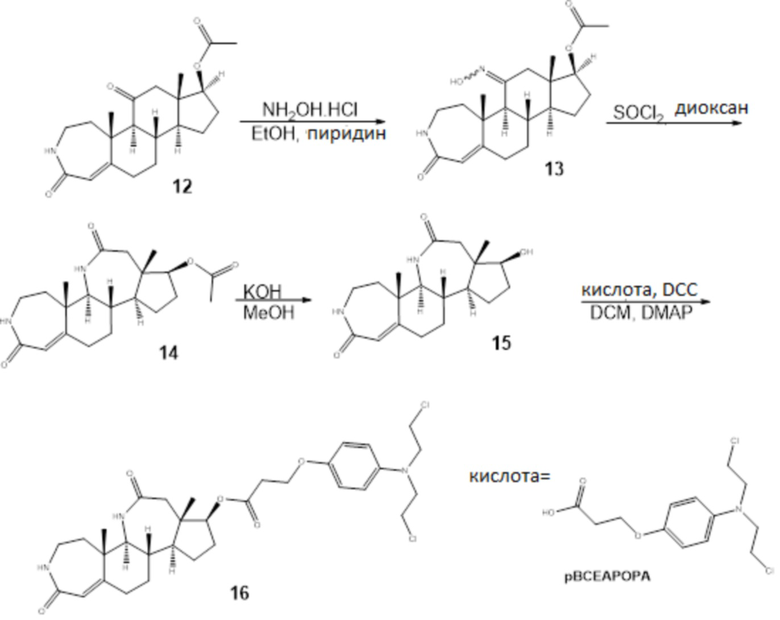
Схема 4
13: К раствору 12 (100 мг, 0,28 ммоль) в 1,5 мл абсолютного этанола в закупоренную пробирку добавляли гидроксиламина гидрохлорид (21 мг, 0,31 ммоль) и сухой пиридин (0,9 мл). Смесь нагревали при 140°C в течение 7 дней. Затем добавляли воду, и смесь экстрагировали этилацетатом (3×5 мл). Органические слои высушивали (Na2SO4) и концентрировали при пониженном давлении с получением сырого продукта, который использовали на следующей стадии без дополнительной очистки.
14: Сырые оксимы 13, описанные выше (0,28 ммоль), растворяли в 4,9 мл сухого диоксана. Смесь охлаждали до 0°C и добавляли по каплям тионилхлорид (0,54 мл). Смеси давали достигнуть комнатной температуры и перемешивали в течение 24 ч. Реакцию останавливали NaHCO3, и смесь экстрагировали этилацетатом (3×20 мл). Органические слои высушивали (Na2SO4) и концентрировали при пониженном давлении с получением сырого продукта, который затем очищали хроматографией на SiO2 (элюент; этилацетат: метанол=1: 0,1) с получением 52 мг лактама с 50%-м выходом. 14: 1H ЯМР (500 МГц, cdcl3) δ 7,00 (с, 1H), 5,77 (с, 1H), 5. 59 (с, 1H), 4,61 (т, J=8,3 Гц, 1H), 3,20 (м, 2H), 3,02 (дд, J=9,6, 5,0 Гц, 1H), 2,48-2,40 (м, 2H), 2,31 (д, J=13,7 Гц, 1H), 2,16 (м, 2H), 2,09-2,0-1,97 (м, 5H), 1,74-1,84 (м, 2H), 1,51-1,40 (м, 2H), 1,35-1,30 (м, 1H), 1,24 (с, 3H), 1,23 (м, 1H), 1,08 (м, 1H), 0,95 (с, 3H); 13C ЯМР (126 МГц, cdcl3) δ 175,1, 170,9, 169,4, 156,3, 120,4, 80,1, 64,2, 55,5, 45,2, 44,6, 41,0, 40,8, 38,0, 36,3, 34,4, 31,0, 25,4, 25,2, 21,9, 21,0, 11,7.
15: Лактам 14 28 мг (0,084 ммоль) растворяли в 1,2 мл MeOH и добавляли по каплям LiOH (1 Н, 0,5 мл). Смесь перемешивали при комнатной температуре в течение 1 ч. Реакцию останавливали NH4Cl, и смесь экстрагировали этилацетат (3×5 мл). Органические слои высушивали (Na2SO4) и концентрировали при пониженном давлении. Сырой продукт далее очищали хроматографией на SiO2 (элюент; этилацетат: метанол=1: 0,1) с получением 28 мг лактама 15 со 100%-м выходом. 15: 1H ЯМР (500 МГц, ДМСО) δ 7,75 (с, 1H), 6,13 (д, J=3,9 Гц, 1H), 5,53 (с, 1H), 4,66 (д, J=5,3 Гц, 1H), 3,40 (м, 2H), 3,11-2,93 (м, 2H), 2,44-2,34 (м, 1H), 2,29 (с, 2H), 2,04-1,72 (м, 5H), 1,65 (м, 2H), 1,24 (м, 3H), 1,20 (с, 3H), 0,89 (м, 1H), 0,66 (с, 3H); 13C ЯМР (126 МГц, ДМСО) δ 174,6, 167,7, 156,1, 120,1, 78,1, 69,8, 63,4, 44,9, 44,2, 41,2, 40,4, 37,7, 35,3, 33,7, 31,3, 27,7, 24,4, 20,9, 10,7.
16: Лактам 15 (30 мг, 0,09 ммоль) растворяли в 9 мл сухого DCM. Затем добавляли 3-(4-(бис(2-хлорэтил)амино)фенокси)пропановую кислоту (67 мг, 0,22 ммоль), DCC (60 мг, 0,29 ммоль) и каталитическое количество DMAP. После того, как конечный раствор перемешивали при комнатной температуре в течение 24 ч, растворитель выпаривали, и остаток очищали флэш-хроматографией на силикагеле (элюент; этилацетат/MeOH=10/1) с получением конъюгата 16 (39 мг, 70%). 1H ЯМР (500 МГц, cdcl3) δ 6,85 (д, J=9,0 Гц, 2H), 6,76 (с, 1H), 6,65 (д, J=9,0 Гц, 2H), 5,79 (с, 1H), 5,46 (с, 1H), 4,68 (т, J=8,3 Гц, 1H), 4,17 (т, J=6,2 Гц, 2H), 3,60 (м, 4H), 3,51-3,42 (м, 1H), 3,18 (м, 2H), 3,02 (дд, J=9,5, 5,0 Гц, 1H), 2,85-2,67 (м, 2H), 2,51 (д, J=13,8 Гц, 1H), 2,44 (дд, J=13,6, 10,1 Гц, 1H), 2,33 (д, J=13,8 Гц, 1H), 2,25-2,06 (м, 2H), 2,07-1,73 (м, 5H), 1,73-1,28 (м, 5H), 1,25 (с, 3H), 1,17-1,03 (м, 2H), 0,96 (с, 3H); 13C ЯМР (126 МГц, cdcl3) δ 174,7, 170,9, 169,1, 155,6, 151,3, 140,9, 120,7, 116,3, 114,4, 80,5, 64,2, 64,1, 55,5, 54,2, 45,3, 44,5, 41,1, 40,7, 38,1, 36,3, 34,8, 34,4, 33,9, 31,0, 25,6, 25,5, 25,2, 24,9, 21,9, 11,8. FT-IR: 3410, 3330, 2926, 2850, 1734, 1654, 1627, 1577, 1513, 1445, 1349, 1273, 1243, 1180, 1133, 1110, 1087, 1044, 890.
Пример 5
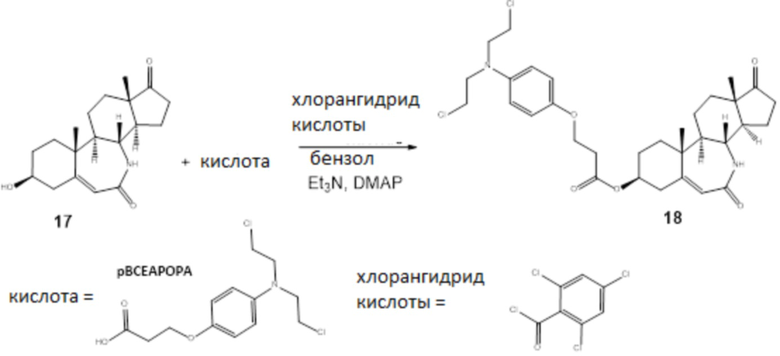
Схема 5
Лактам 17 синтезировали согласно Koutsourea et al (Steroids, 2003, 68(7-8):659-66).
18: В круглодонной колбе 48 мг (0,157 ммоль) кислоты разбавляли в 0,5 мл сухого бензола. Добавляли 2,4,6-трихлорбензоилхлорид (30 мкл, 0,189 ммоль) и триэтиламин (53 мкл, 0,378 ммоль), и смесь нагревали с обратным холодильником под Ar в течение 1 ч. К этой смеси раствор добавляли стероидного спирта 50 мг (0,157 ммоль) в 0,5 мл сухого бензола и каталитическое количество 4-диметиламинопиридина. Нагревание продолжали в течение 3 ч. Бензол полностью удаляли выпариванием в вакууме, и остаток разбавляли CH2Cl2. Полученную смесь экстрагировали 5%-м водным раствором HCl, органический слой промывали 7%-м водным раствором NaHCO3 и наконец водой, высушивали над Na2SO4, и растворитель удаляли при пониженном давлении. Остаток хроматографировали на колонке с силикагелем (элюент; этилацетат/MeOH=100/1) с получением 46 мг конъюгата 18 с 48%-м выходом. Конъюгат 18: 1H ЯМР (500 МГц, cdcl3) δ 6,84 (д, J=8,5 Гц, 2H), 6,67 (д, J=8,5 Гц, 2H), 5,90 (с, 1H), 5,86 (с, 1H), 4,78 (м, 1H), 4,19 (м, 2H), 3,58-3,59 (м, 8H), 3,48 (м, 1H), 2,61-1,25 (18-й), 1,29 (с, 3H), 0,88 (с, 3H); [M+H]+ =605.
Пример 6
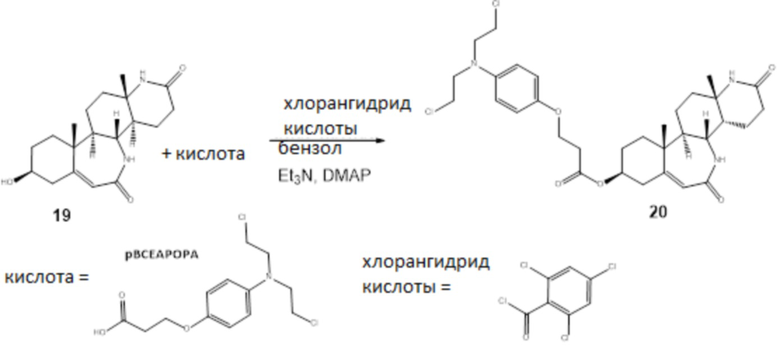
Схема 6
Лактам 19 синтезировали согласно Koutsourea et al (Steroids, 2003, 68(7-8):659-66).
20: В круглодонной колбе 46 мг (0,15 ммоль) кислоты разбавляли в 0,5 мл сухого бензола. Добавляли 2,4,6-трихлорбензоилхлорид (28 мкл, 0,18 ммоль) и триэтиламин (50 мкл, 0,36 ммоль), и смесь нагревали с обратным холодильником под Ar в течение 1 ч. К этой смеси добавляли раствор 50 мг (0,150 ммоль) стероидного спирта в 0,5 мл сухого бензола и каталитическое количество 4-диметиламинопиридина. Нагревание продолжали в течение 3 ч. Бензол полностью удаляли выпариванием в вакууме, и остаток разбавляли CH2Cl2. Полученную смесь экстрагировали 5%-м водным раствором HCl, органический слой промывали 7%-м водным раствором NaHCO3 и наконец водой, высушивали над Na2SO4, и растворитель удаляли при пониженном давлении. Остаток хроматографировали на колонке с силикагелем (элюент; этилацетат/MeOH=100/2) с получением 19 мг конъюгата 20 с 20%-м выходом. 20: 1H ЯМР (500 МГц, cdcl3) δ 7,18 (с, 1H), 6,84 (д, J=8,5 Гц, 2H), 6,65 (д, J=8,5 Гц, 2H), 6,60 (с, 1H), 5,82 (с, 1H), 4,80 (1H, m), 4,21 (2H, m), 3,50 (м, 8H), 3,20 (1H, m), 2,80-1,30 (19Н), 1,20 (с, 3H), 0,9 (с, 3H); [M+H]+=621.
Пример 7
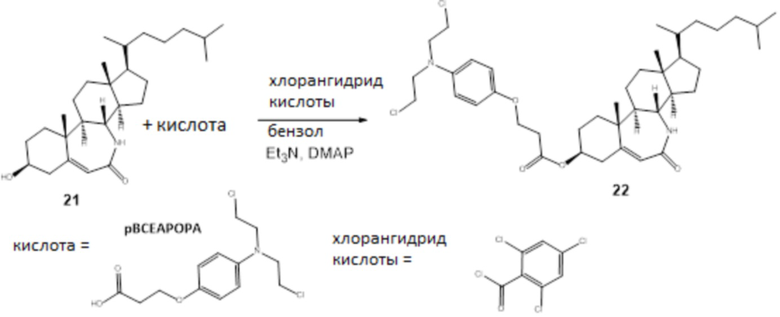
Схема 7
Лактам 21 синтезировали согласно Koutsourea et al (Steroids, 2003, 68(7-8):659-66).
22: В круглодонной колбе 37 мг (0,12 ммоль) кислоты разбавляли в 0,4 мл сухого бензола. Добавляли 2,4,6-трихлорбензоилхлорид (22 мкл, 0,144 ммоль) и триэтиламин (40 мкл, 0,288 ммоль), и смесь нагревали с обратным холодильником под Ar в течение 1 ч. К этой смеси добавляли раствор 50 мг (0,120 ммоль) стероидного спирта в 0,4 мл сухого бензола и каталитическое количество 4-диметиламинопиридина. Нагревание продолжали в течение 3 ч. Бензол полностью удаляли выпариванием в вакууме, и остаток разбавляли CH2Cl2. Полученную смесь экстрагировали 5%-м водным раствором HCl, органический слой промывали 7%-м водным раствором NaHCO3 и наконец водой, высушивали над Na2SO4, и растворитель удаляли при пониженном давлении. Остаток хроматографировали на колонке с силикагелем (элюент; этилацетат) с получением 34 мг конъюгата 22 с 40%-м выходом 22: 1H ЯМР (500 МГц, cdcl3) δ 6,84 (д, J=8,5 Гц, 2H), 6,60 (д, J=8,5 Гц, 2H), 5,90 (с, 1H), 5,79 (с, 1H), 4,80 (м, 1H), 4,15 (м, 2H), 3,5 (м, 8H), 3,25 (м, 1H), 2,8-0,8 (22Н); [M+H]+=605.
Пример 8
In vitro и in vivo биологическое тестирование на противораковая активность
A) In vitro противораковая активность
Девять устойчивых человеческих линий раковых клеток (таблица 1) обрабатывали для тестирования цитостатической и цитотоксической активности недавно синтезированных соединений. Клеточные линии получали из American Type Culture Collection (ATCC) и выращивали в другой культуральной среде согласно инструкциям. Тест с использованием MTT ((3-(4,5-диметилтиазол-2-ил)-2,5-дифенилтетразолий бромид) является известным и стандартным методом для оценки цитостатической и цитотоксической активности лекарственных средств и химикатов (Trafalis DT et al, J BUON, 2003, 8:333-9; Trafalis DT et al, J BUON, 2004, 9(3):275-82; Trafalis DT et al, J BUON, 2005; 10:227-34; Trafalis DT et al, Breast Cancer Res Treat, 2006, 97:17-31). Кратко, клетки высевали в планшет с 96 лунками при плотности -3×104 клеток/мл на лунку и сохраняли в течение 72 ч при 37°C в инкубаторе с 5% CO2, и выращивали в виде монослоев или суспензий. Через 24 часа клетки обрабатывали 0,1-100 мкмоль/л соединений в течение 48 ч. Жизнеспособность культивируемых клеток оценивали с помощью метаболического теста MTT (Sigma, Сент-Луис, Миссури, США), как описано ранее. Поглощение конвертированного красителя измеряли при длине волны 540 нм на считывателе ELISA (Versamax, Орлеан, США). Средние концентрации каждого лекарственного средства, генерирующие 50% или общее (100%) ингибирование роста (GI50 и TGI, соответственно), а также концентрации лекарственного средства, приводящие к цитотоксичности против 50% культивируемых клеток [(половина максимальной цитотоксической концентрации (IC50)], вычисляли с помощью способа линейной регрессии. Используя семь измерений поглощения [время 24 ч (Ct24), контрольный рост 72 ч (Ct72) и тестовый рост в присутствии лекарственного средства на пяти уровнях концентрации (Tt72x)], процент роста вычисляли на каждом уровне концентраций лекарственного средства. Процент ингибирования роста вычисляли согласно National Cancer Institute (NCI) как: [(Tt72x)-(Ct24)/(Ct72)-(Ct24)] x 100 для концентраций, для которых Tt72x>Ct24 и [(Tt72x)-(Ct24)/Ct24] x 100 для концентраций, для который Tt72x<Ct24; GI50 вычисляли от [(Tt72x)-(Ct24)/(Ct72)-(Ct24)] x 100=50, TGI от [(Tt72x)-(Ct24)/(Ct72)-(Ct24)] x 100=0, и IC50 от [(Tt72x)-(Ct24)/Ct24] x 100=50. Все эксперименты проводили в трех экземплярах.
Таблица 1
Адриамицин, Мелфалан и Цисплатин резистентная
BRCA1 - (видоизмененный)
BRCA1 +
Белки, связывающие инсулиноподобный фактор роста (IGFBP) BP 2; BP 4; BP 5
Результаты in vitro цитостатического (GI50, TGI) и цитотоксического эффектов (IC50) протестированных соединений в отношении человеческих линий раковых клеток представлены в Таблицах 2, 3, 4.
Таблица 2
Таблица 3
Таблица 4
B) In vivo острая токсичность
Для внутрибрюшинного (i.p.) лечения сток-растворы тестируемых соединений готовили непосредственно перед использованием. Их суспендировали в кукурузном масле в желаемой концентрации после начального растворения в 10%-м диметилсульфоксиде (DMSO). Эта концентрация отдельно не оказывала заметного токсического эффекта.
Самки мыши C57Bl/6 использовались для исследований токсичности. Мышей получали из экспериментальной секции Греческого Института Пастера.
Кратко, острую токсичность, вызванную тестируемыми соединениями, определяли, как ранее было очень хорошо описано (Catsoulacos P et al, Cancer Chemother Pharmacol, 1979, 3(1):67-70; Catsoulacos P et al, J Pharm Sci, 1978, 67(9):1342-3; Catsoulacos P et al, Anticancer Res, 1995; 15:827-30) после единственной внутрибрюшинной (i.p.) инъекции в группах из десяти (10) мышей C57Bl/6 в четырех различных дозировках; мышей наблюдали в течение 30 дней, и терапевтическую дозу соединений, обычно определяемую как LD10 (летальная доза для 10% животных), а также LD50 (летальная доза для 50% животных) определяли после графической оценки (30-дневные кривые). Токсичность протестированных соединений оценивали по летальности у мышей C57Bl/6. Значения LD50 и LD10 были оценены графически, где процент смертей вследствие токсичности каждой дозы был показан на оси ординат, в то время как введенные дозы были показаны на оси абсцисс (таблица 5).
C) In vivo противоопухолевая активность
Эксперимент инициировали в день 0 путем внутрибрюшинной (i.p.) имплантации 106 клеток асцита лимфолейкоза P388 согласно протоколу National Cancer Institute (NCI), США. Для i.p. лечения сток-растворы тестируемых соединений готовили непосредственно перед использованием. Противоопухолевую активность оценивали по онкостатическому параметру T/C%, который означает среднее время выживания (MST) обработанных лекарственным средством животных (T) по отношению к обработаному солевым раствором контролю (C). Согласно NCI (США), минимальный критерий активности составляет T/C выше, чем 125%. Кроме того, противоопухолевую активность оценивали по числу долговременно выживших (излечение: определенное как мыши, живые в течение 90 дней после инокуляции опухоли) (Golidim A et al, Nat Cancer Inst Monogr, 1980, 55: 25-26; NCI Monograph, NIH publication 1986, 55:80-193).
Самки мышей BALB/c scid использовались для оценки противоопухолевой активности. Эти животные несут тяжелую комбинированную мутацию иммунодефицита (scid) на фоне BALB/c, и они были получены от NCSR ʺDemokritosʺ, Институт Биологии. Мышей сохраняли в условиях постоянной температуры и влажности, в стерильных клетках, с водой и едой. Шесть мышей были включены в каждую группу лечения и восемь в контрольную группу.
Тестируемую терапевтическую дозу соединений определяли при соответствующей LD10 (мг/кг).
Таблица 5. Острая токсичность соединений у C57Bl/6. LD50 и LD10=летальные дозы для 50% и 10% популяции леченных мышей.
Таблица 6. Антилейкозная активность тестируемых соединений против мышиного лимфолейкоза P388 in vivo.
(мг/кг)
(дни)
*p <0,001
Самки мышей BALB/c scid использовались для in vivo оценки противоопухолевой активности тестируемых соединений против рака яичника человека SCOV-3. Суспензии 3×106 раковых клеток SCOV-3/0,2 мл/мышь имплантировали подкожно в правый или левый бок каждого животного. Мышей содержали в условиях постоянной температуры и влажности в стерильных клетках с водой и едой. Десять мышей были включены в каждую группу лечения и контроля. Тестирование осуществляли согласно известным лабораторным протоколам. Эффективность лекарственных средств определяли по среднему изменению объема опухоли у подвергнутых лечению животных (T) по сравнению с контролем (C) (T/C%= TI, Ингибирование опухоли) и увеличению среднего времени выживания, согласно кинетике опухолевых клеток и биологическим свойствам. Объемы или массы опухоли вычисляли как 0,52×a2×b, где a и b являются меньшей и большей осями опухоли, и данные выстраивали на полулогарифмическом графике как средние объемы опухоли ± стандартное отклонение от среднего (±SEM) по сравнению с временем после лечения. Когда опухоли достигали в объеме 0,085-0,1 мм3, мышей разделяли на контрольную группу и группу медикаментозного лечения (10 мышей/группа) со схожими средними объемами опухоли в каждой группе. Тестируемые соединения вводили внутрибрюшинно в дозах LD10/4 соответственно в дни 1, 5 и 9.
Для оценки противоопухолевого эффекта (a) определяли среднее изменение массы опухоли или объема опухоли за неделю, и ингибирование опухоли (TI) вычисляли формулой: TI(%)= [1)(TWT)TWZ)/(TWC)TWZ)]x100, где TWT определяется как масса опухоли (мг) или объем опухоли (мм3) у подвергнутых лечению животных во время оценки, TWZ определяется как масса опухоли (мг) или объем опухоли (мм3) во время инициирования лечения (нулевое время или день 1), TWC определяется как масса опухоли (мг) или объем опухоли (мм3) у не подвергнутых лечению животных (контроль) во время оценки, (b) процент выживших (OS %) в День 70, (c) процент выживших без развития опухоли (PFS %) в день 70. Результаты продемонстрированы в Таблице 7.
Таблица 7. Противоопухолевая активность тестируемых соединений против человеческого рака яичника SCOV-3 in vivo.
(мг/кг)
Все различия в TI%, OS%, PFS% были значимыми на уровнях p<0,05-0,001
D) Фармакологические эффекты
Новые лактам-стероидные алкилирующие агенты оказывают значительное ингибирующее действие на активность поли(АДФ-рибоза)полимеразы (PARP1/2), демонстрируя полумаксимальные ингибирующие концентрации (IC50) менее 1,7 мкM, лучше, чем известный ингибитор PARP1/2 3-аминобензамид (3-AB). Кроме того, новые лактам-стероидные алкилирующие агенты вызывают существенные изменения в транскрипции и экспрессии мРНК PARP1 и PARP2 зависимым от дозы и времени образом, in vitro и in vivo. Сначала или в более низких дозах они могут вызвать увеличение экспрессии мРНК PARP1 и PARP2, которое вплоть до 5-400 раз выше, чем контрольные значения, генерируя изменения внутриклеточной концентрации NAD+ и клеточное истощение ATФ, и позднее или в более высоких дозах они вызывают уменьшение экспрессии мРНК PARP1 и PARP2, которое может достигнуть значений, близких к 100%. Новые лактам-стероидные алкилирующие агенты вызывают значительное повреждение ДНК, сопоставимое с вызываемым одним только их алкилирующим компонентом, при оценке in vitro в тесте Сестринского хроматидного обмена (SCE) и in vivo путем продукции аддуктов 8-гидрокси-2'-дезоксигуанозина (8-OHdG) в сыворотке или моче, в то время как они генерируют значительно более высокую противоопухолевую активность. Кроме того, новые лактам-стероидные алкилирующие агенты значительно ингибируют (> 60%) фосфорилирование ERK1/2 и AKT1/2, и, следовательно, активацию молекулярных сигнальных путей PI3K и MAPK. Впервые молекулярные фармакологические эффекты лактам-стероидных алкилирующих агентов были исследованы подробно.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ ПРОИЗВОДНЫЕ 1,2,4-ТРИАЗОЛО-[3,4-B]-1,3,4-ТИАДИАЗОЛА | 2017 |
|
RU2765180C2 |
| ПРОТИВООПУХОЛЕВЫЕ СОЕДИНЕНИЯ ДИГИДРОПИРАН-2-ОНА | 2007 |
|
RU2444519C2 |
| ПРОТИВООПУХОЛЕВЫЕ СОЕДИНЕНИЯ | 2008 |
|
RU2493147C2 |
| ПРОТИВООПУХОЛЕВЫЕ СОЕДИНЕНИЯ | 2008 |
|
RU2528393C9 |
| ПРОТИВООПУХОЛЕВЫЕ СОЕДИНЕНИЯ | 2008 |
|
RU2489429C2 |
| ПРОИЗВОДНЫЕ ТАКСАНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ | 1994 |
|
RU2137764C1 |
| НОВЫЕ СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, СВЯЗАННЫХ С АМИЛОИДНЫМИ ИЛИ АМИЛОИДОПОДОБНЫМИ БЕЛКАМИ | 2011 |
|
RU2603008C2 |
| НОВОЕ ТРИЦИКЛИЧЕСКОЕ ПРОИЗВОДНОЕ ИЛИ ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, СПОСОБ ИХ ПОЛУЧЕНИЯ И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2009 |
|
RU2470934C1 |
| 7-ДЕЗОКСИПРОИЗВОДНЫЕ ТАКСОЛА, ПРОИЗВОДНЫЕ БАККАТИНА, 10-ПЕНТАФТОРФЕНИЛБАККАТИН, 7-[(МЕТИЛТИО)КАРБОНОТИОИЛОКСИ]-13-ТРИЭТИЛСИЛИЛОКСИБАККАТИН, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1993 |
|
RU2122545C1 |
| ПРОИЗВОДНЫЕ ХИНОКСАЛИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЙ СОСТАВ | 1995 |
|
RU2135484C1 |
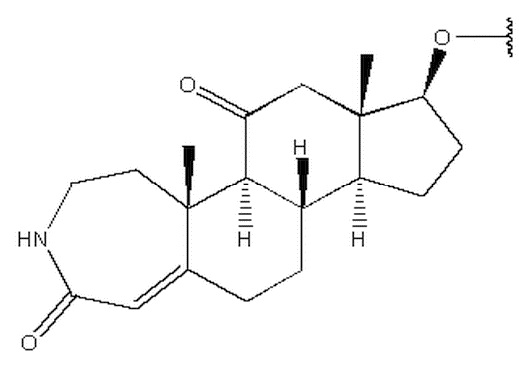
Изобретение относится к соединению формулы (I) или его фармацевтически приемлемой соли. В формуле (I) R1 выбран из группы, состоящей из
 ,
, 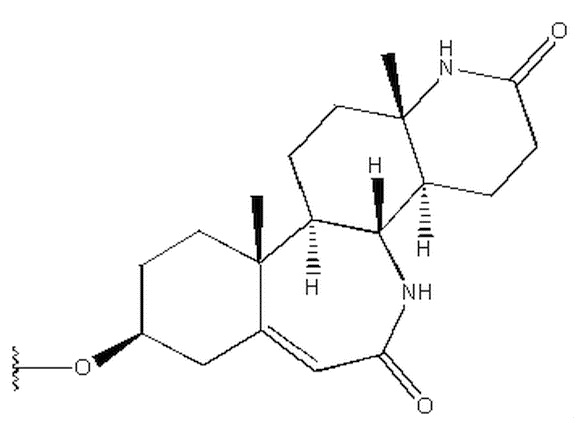 ,
, 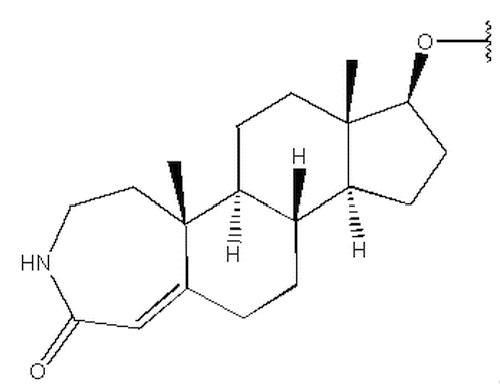 ,
, 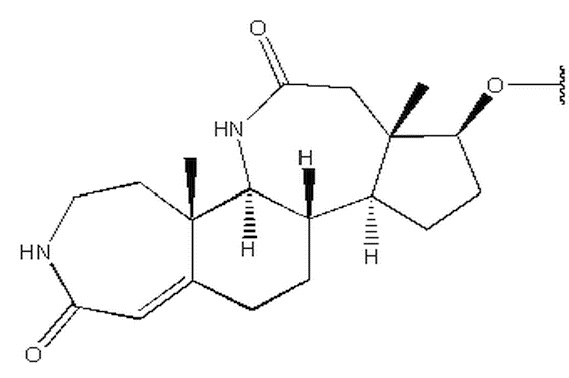 ,
,  ,
,  и
и 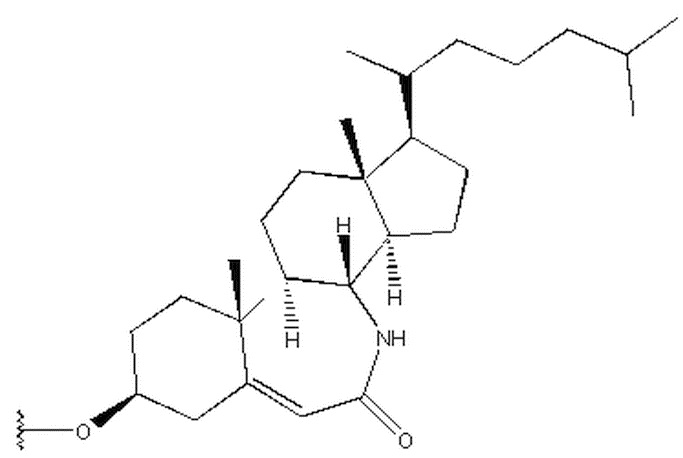 ,
,
R2 представляет собой H, R3 представляет собой H. Изобретение также относится к фармацевтической композиции, обладающей противораковой активностью. Технический результат: получены новые соединения формулы (I), которые могут применяться для лечения рака яичника, рака молочной железы, рака предстательной железы или лейкоза. 2 н. и 4 з.п. ф-лы, 7 табл., 8 пр.
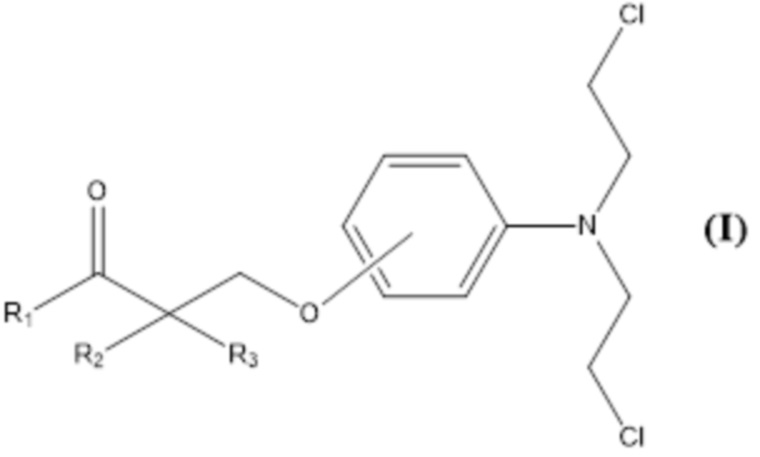
1. Соединение формулы (I) или его фармацевтически приемлемая соль
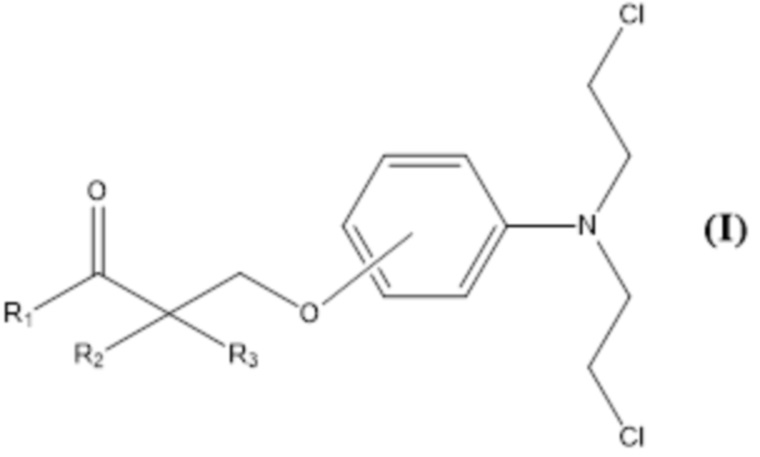 ,
,
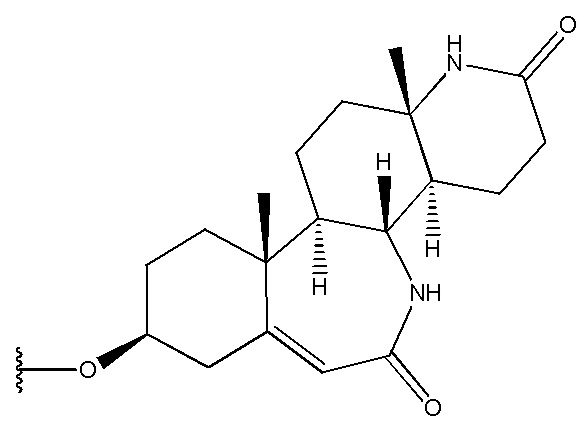
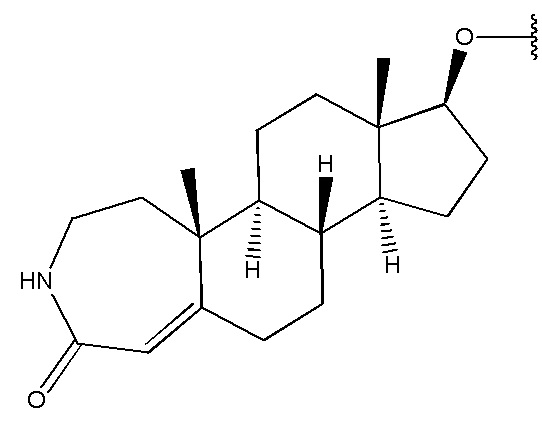
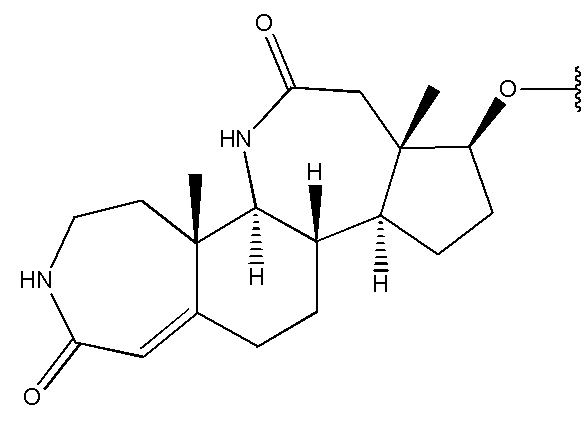
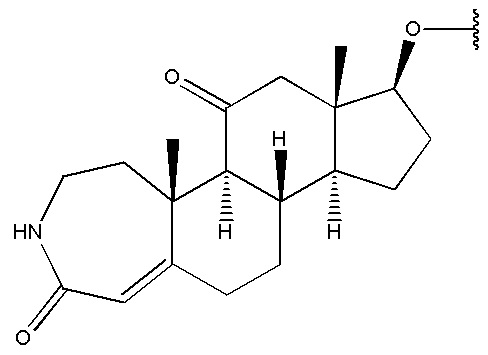
в которой R1 выбран из группы, состоящей из
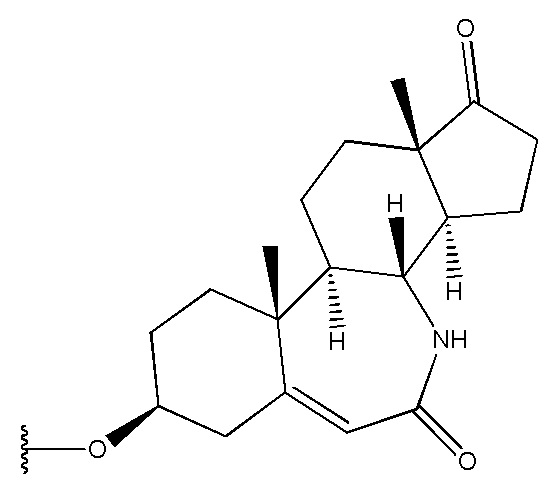 ,
, 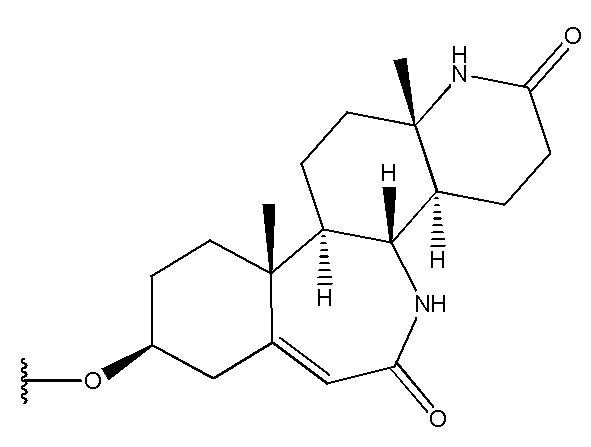 ,
, 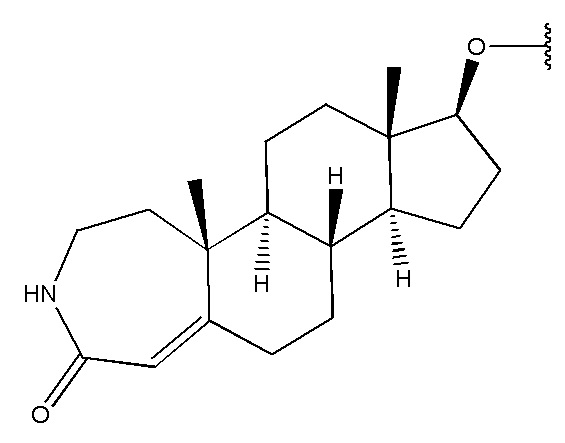 ,
, 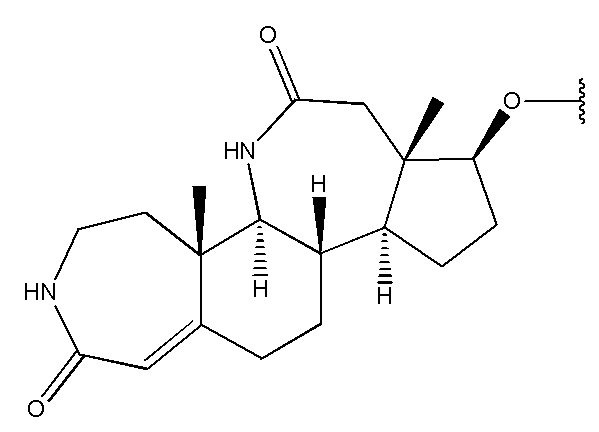 ,
, 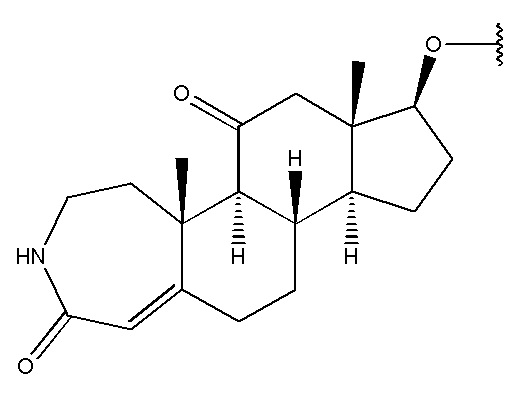 ,
,  и
и 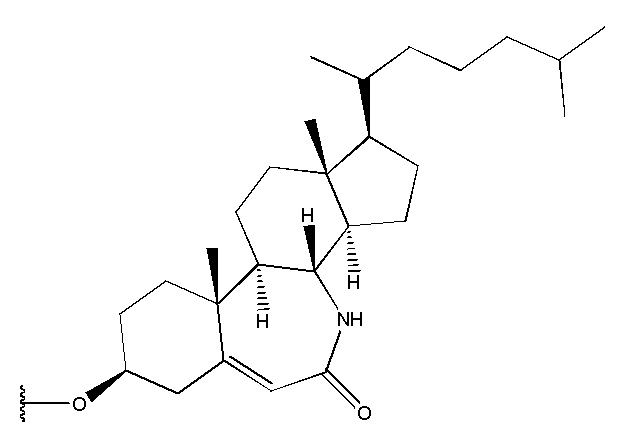 ,
,
R2 представляет собой H,
R3 представляет собой H.
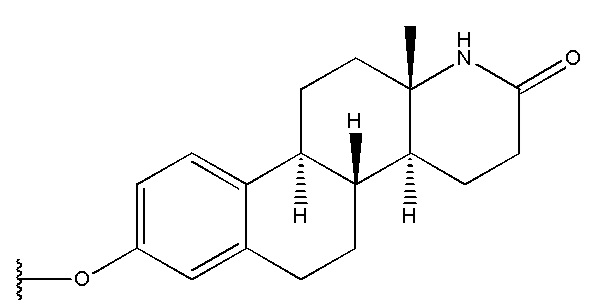
2. Соединение по п. 1 или его фармацевтически приемлемая соль, в котором R1 выбран из группы, состоящей из
 ,
,  ,
,  ,
,  ,
,  и
и 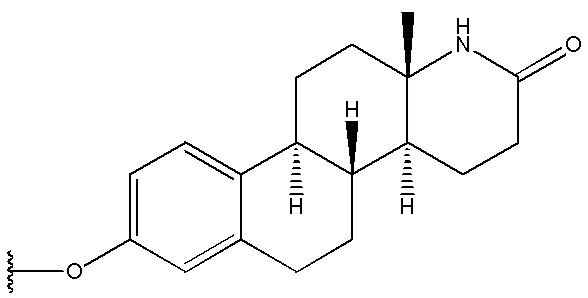 .
.
3. Соединение по п. 2 или его фармацевтически приемлемая соль, в котором R1 выбран из группы, состоящей из
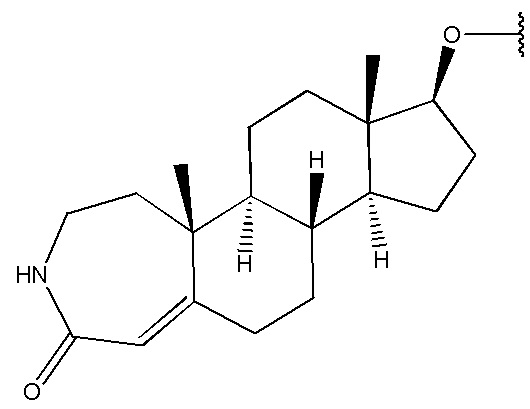 ,
, 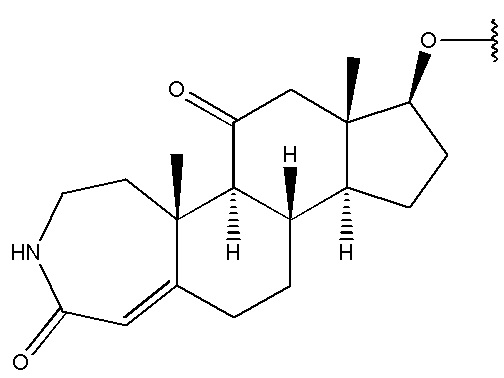 и
и 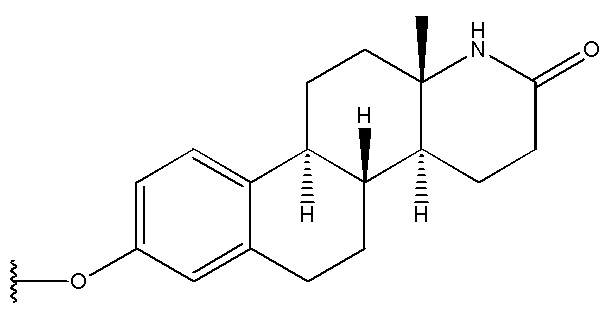 .
.
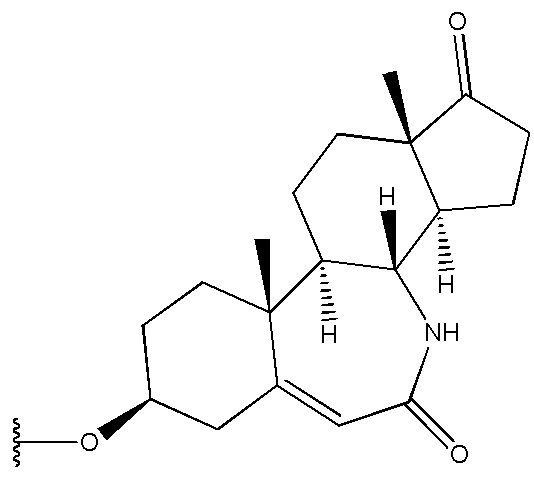
4. Соединение по п. 2 или его фармацевтически приемлемая соль, в котором R1 выбран из группы, состоящей из
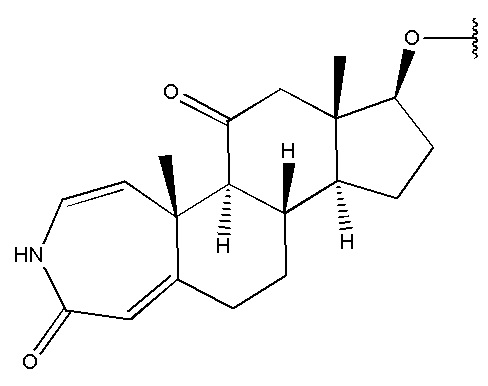 и
и 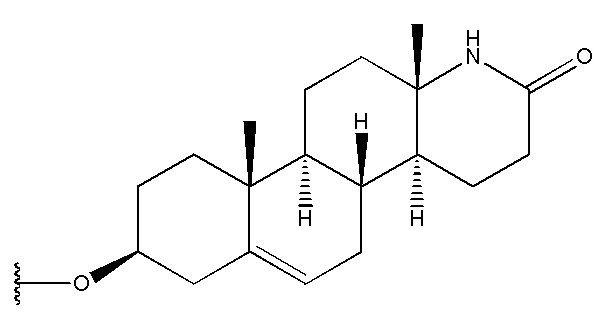 .
.
5. Соединение по любому из пп. 1-4 или его фармацевтически приемлемая соль для применения для лечения рака яичника, рака молочной железы, рака предстательной железы или лейкоза.
6. Фармацевтическая композиция, обладающая противораковой активностью, содержащая в качестве активного ингредиента соединение по любому из пп. 1-4 или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
| P | |||
| Catsoulacos et al | |||
| "Comparison of Current Alkylating Agents with a Homo-aza-Steroidal Ester for Antineoplastic Activity" Oncology, vol.51, N1, 1994, 74-78 | |||
| Kapou et al | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |

