Уровень техники изобретения
Настоящее изобретение относится к новым органическим соединениям и к фармацевтическим композициям, содержащим подобные соединения. Указанные соединения могут быть пригодны при лечении рака, в частности таких онкологических заболеваний, при которых, как известно, пригодны агенты, нацеливающиеся на сплайсосому и ее мутации.
В организмах эукариот вновь синтезированные матричные РНК, как правило, имеют несколько интронов, которые вырезают, чтобы получить зрелую мРНК. Сплайсосома представляет собой комплекс, состоящий из многих субъединиц, который решает указанную задачу. Сплайсосома состоит из пяти небольших ядерных РНК (snRNAs; U1-6) в сочетании с различными белками. Мутации в генах сплайсосомы обнаружены в различных типах онкологических заболеваний.
Например, мутации в субъединице 1 фактора сплайсинга 3B (SF3B1) сплайсосомы существует в ряде видов онкологических заболеваний и являются мишенью для противораковых агентов. Подобные онкологические заболевания включают, однако этим не ограничиваясь, миелодиспластический синдром (MDS), лейкемию, такую как хронический лимфоцитарный лейкоз (CLL), хронический миеломоноцитарный лейкоз (CMML) и острый миелоидный лейкоз (AML), а также солидные опухоли, такие как рак молочной железы и увеальная меланома.
Соединения, выделенные из бактерий Streptomyces platensis (Sakai, Takashi; Sameshima, Tomohiro; Matsufuji, Motoko; Kawamura, Naoto; Dobashi, Kazuyuki; Mizui, Yoshiharu. Pladienolides, New Substances from Culture of Streptomyces platensis Mer-11107. I. Taxonomy, Fermentation, Isolation and Screening. The Journal of Antibiotics, 2004, Vol. 57, No.3.), названные пладиенолидами и обнаруженные при скрининге промотора ингибитора фактора роста эндотелия сосудов (VEGF), ингибируют экспрессию репортерного гена, контролируемого промотором VEGF человека, а как известно, подобное ингибирование является подходящим механизмом действия для противораковых агентов.
Указанные соединения также ингибируют пролиферацию клеток глиомы U251 человека в условиях in vitro. Наиболее действенное из указанных соединений, пладиенолид B, ингибирует промотируемую VEGF экспрессию гена с величиной IC50 1,8 нМ, и ингибирует пролиферацию клеток глиомы с величиной IC50 3,5 нМ. Структура пладиенолида B известна, (Sakai, Takashi; Sameshima, Tomohiro; Matsufuji, Motoko; Kawamura, Naoto; Dobashi, Kazuyuki; Mizui, Yoshiharu. Pladienolides, New Substances from Culture of Streptomyces platensis Mer-11107. II. Physico-chemical Properties and Structure Elucidation. The Journal of Antibiotics. Vol. 57, No.3. (2004)), а также известно, что пладиенолид B нацеливается на сплайсосому SF3b, ингибируя сплайсинг и изменяя характер экспрессии генов (Kotake et al., "Splicing factor SF3b as a target of the antitumor natural product pladienolide", Nature Chemical Biology 2007, 3, 570-575).
Известны также некоторые соединения пладиенолида B, а также соединения других пладиенолидов, которые раскрыты в следующих патентных заявках WO 2002/060890; WO 2004/011459; WO 2004/011661; WO 2004/050890; WO 2005/052152; WO 2006/009276; и WO 2008/126918. Например, соединение пладиенолида, (8Е,12Е,14Е)-7-((4-циклогептилпиперазин-1-ил)карбонил)окси-3,6,16,21-тетрагидрокси-6,10,12,16,20-пентаметил-18,19-эпокситрикоза-8,12,14-триен-11-олид, также известное как E7107, представляет собой полусинтетическое производное природного пладиенолида D, и были сообщены результаты фазы I его исследования.
Тем не менее, необходимы дополнительные агенты, пригодные при лечении онкологических заболеваний, в частности, онкологического заболевания, для которого, как известно, пригодны агенты, нацеливающиеся на сплайсосому и ее мутации.
Сущность изобретения
Целью настоящего изобретения является получение соединения формулы 1 ("Соединение 1"), соединения формулы 2 ("Соединение 2"), соединения формулы 3 ("Соединение 3") и соединения формулы 4 ("Соединение 4"):
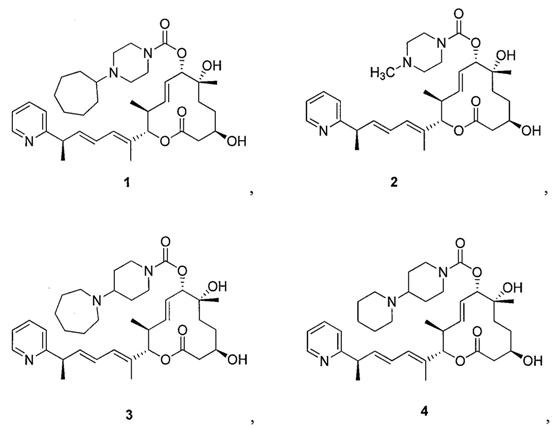
и их фармацевтически приемлемые соли.
Еще одной целью настоящего изобретения является фармацевтическая композиция, содержащая Соединение 1, Соединение 2, Соединение 3, Соединение 4 или его фармацевтически приемлемую соль. Подобные фармацевтические композиции могут быть составлены с использованием одного или нескольких фармацевтически приемлемых носителей. Подобные композиции готовят для использования путем различных обычных способов введения, в том числе внутривенного, перорального, подкожного или внутримышечного введения.
Настоящее изобретение может также относиться к способу лечения субъекта с онкологическим заболеванием, который включает введение указанному субъекту количества Соединения 1, Соединения 2, Соединения 3, Соединения 4 или его фармацевтически приемлемой соли, эффективного для получения терапевтически благоприятного ответа. Онкологическое заболевание может представлять собой миелодиспластический синдром, лейкоз, такой как хронический лимфоцитарный лейкоз, острый лимфобластный лейкоз, хронический миеломоноцитарный лейкоз или острый миелоидный лейкоз, или солидную опухоль, такую как рак толстого кишечника, рак поджелудочной железы, рак эндометрия, рак яичников, рак молочной железы, увеальная меланома, рак желудка, холангиокарцинома, рак легкого, или любую их разновидность. Онкологическое заболевание может давать положительный результат теста на одну или несколько мутаций в гене сплайсосомы или белке, таких как мутации, перечисленные в таблице 1.
Настоящее изобретение может также относиться к применению Соединения 1, Соединения 2, Соединения 3, Соединения 4 или его фармацевтически приемлемой соли в способе терапевтического воздействия, например, с целью лечения онкологического заболевания. Онкологическое заболевание может представлять собой миелодиспластический синдром, лейкоз, такой как хронический лимфоцитарный лейкоз, острый лимфобластный лейкоз, хронический миеломоноцитарный лейкоз или острый миелоидный лейкоз, или солидную опухоль, такую как рак толстого кишечника, рак поджелудочной железы, рак эндометрия, рак яичников, рак молочной железы, увеальная меланома, рак желудка, холангиокарцинома, рак легкого, или любую их разновидность. Онкологическое заболевание может давать положительный результат теста на одну или несколько мутаций в гене сплайсосомы или белке, таких как мутации, перечисленные в таблице 1.
Настоящее изобретение может также относиться к применению Соединения 1, Соединения 2, Соединения 3, Соединения 4 или его фармацевтически приемлемой соли, для изготовления лекарственного средства. В частности, лекарственное средство может быть предназначено для лечения онкологического заболевания. Онкологическое заболевание может представлять собой миелодиспластический синдром, лейкоз, такой как хронический лимфоцитарный лейкоз, острый лимфобластный лейкоз, хронический миеломоноцитарный лейкоз или острый миелоидный лейкоз, или солидную опухоль, такую как рак толстого кишечника, рак поджелудочной железы, рак эндометрия, рак яичников, рак молочной железы, увеальная меланома, рак желудка, холангиокарцинома, рак легкого, или любую их разновидность. Онкологическое заболевание может давать положительный результат теста на одну или несколько мутаций в гене сплайсосомы или белке, таких как мутации, перечисленные в таблице 1.
Настоящее изобретение может также относиться к применению Соединения 1, Соединения 2, Соединения 3, Соединения 4 или его фармацевтически приемлемой соли для нацеливания на сплайсосому, например, субъединицу 1 сплайсосомы SF3B.
Краткое описание чертежей
Фиг. 1 показывает результаты фармакокинетического (PK) исследования на CD-1 мышах, которым вводили Соединение 2 с дозой 5 мг/кг внутривенно (IV) или 10 мг/кг перорально (РО).
Фиг. 2 показывает эффективность Соединения 2 для модели ксенотрансплантата мышей Nalm-6 (линия пред-В-клеток человека) со сконструированной мутацией SF3B1K700E. Мышам вводили 2,5; 5 или 10 мг/кг Соединения 2 один раз в день (QD) в течение 14 дней, а объем опухоли измеряли в течение 40 дней.
Фиг. 3 показывает данные фармакокинетического и фармакодинамического анализа Соединения 2 для модели ксенотрансплантата мышей Nalm-6 (линия пред-В-клеток человека) со сконструированной мутацией SF3B1K700E. Мышам вводили перорально Соединения 2 с дозой 10 мг/кг и определяли концентрацию опухоли (мкг/г) и кратность изменения экспрессии Pre-EIF4A1 (пред-мРНК транскрипта EIF4A1) и SLC25A19 (зрелая мРНК транскрипта SLC25A19) относительно носителя.
Фиг. 4 показывает результаты анализа жизнеспособности клеток с Соединением 2 в линии раковых клеток PANC0504 (SF3B1MUT) (мутант PANC 05.04) по сравнению с диким типом SF3B1 панкреатических раковых клеток линий BXPC3, HPAFII, PANC0403, PANC1005, CFPAC1 и MIAPACA2.
Фиг. 5 показывает регулирование альтернативного сплайсинга для E7107 и Соединения 2 (cmp 2) на основе анализа с использованием технологии Nanostring. Значки "+" и "-" обозначают положительные или отрицательные значения, соответственно, по шкале оттенков, которая указывает на различные уровни экспрессии различных точек сплайсинга.
На фиг. 6 показаны результаты PK исследования для мышей CD-1, которым вводили Соединение 1 с дозой 5 мг/кг внутривенно (IV) или 12 мг/кг перорально (РО).
Фиг. 7 показывает эффективность Соединения 1 в модели ксенотрансплантата мышей Nalm-6 с помощью сконструированной мутации SF3B1K700E. Мышам вводили 7,5 или 10 мг/кг Соединения 1 раз в сутки (QD) в течение 14 дней, а объем опухоли измеряли в течение 30 дней.
На фиг. 8 приведены данные фармакокинетического и фармакодинамического анализа Соединения 1 для модели ксенотрансплантата мышей Nalm-6 со сконструированной мутацией SF3B1K700E. Мышам вводили перорально одну дозу 10 мг/кг Соединения 2 и определяли концентрацию опухоли (мкг/г) и кратность изменения экспрессии Pre-EIF4A1 (пред-мРНК транскрипта EIF4A1) и SLC25A19 (зрелая мРНК транскрипта SLC25A19) относительно носителя.
Фиг. 9 показывает результаты PK исследования для мышей CD-1, которым вводили Соединение 3 с дозой 5,964 мг/кг внутривенно (IV) или 13,307 мг/кг перорально (РО).
Фиг. 10 показывает результаты PK исследования для мышей CD-1, которым вводили Соединение 4 с дозой 5 мг/кг внутривенно (IV) или 10 мг/кг перорально (РО).
Подробное описание некоторых вариантов осуществления изобретения
A. Определения
Используемые в данном описании определения имеют следующие значения, если не указано иное.
Термин "изомеры" относится к соединениям, имеющим одинаковое количество и тип атомов и, следовательно, такую же молекулярную массу, но различающимся расположением или конфигурацией атомов. Термин "стереоизомеры" относится к соединениям, которые включают те же связи между атомами, но имеют различное расположение атомов в пространстве. Термин "диастереоизомеры" или "диастереомеры" относится к стереоизомерам, которые не являются энантиомерами. Термин "энантиомеры" относится к стереоизомерам, которые не являются зеркальными отражениями друг друга. Термин "геометрические изомеры" относится к цис-транс-изомерам, имеющим различные расположения групп относительно двойной связи, или цикла, или центрального атома.
Энантиомеры, рассматриваемые в данном описании, могут включать "энантиомерно чистые" изомеры, которые содержат практически один энантиомер, например, больше или равно 90%, 92%, 95%, 98% или 99%, или равно 100% одного энантиомера при конкретном асимметрическом центре или центрах. "Асимметрический центр" или "хиральный центр" относится к тетраэдрическому атому углерода, который имеет четыре различных заместителя.
"Стереомерно чистый" в данном описании означает соединение или его композицию, которая содержит один стереоизомер соединения и практически свободна от других стереоизомеров данного соединения. Например, стереоизомерно чистая композиция соединения, имеющего один хиральный центр, в значительной степени свободна от противоположного энантиомера указанного соединения. Стереомерно чистая композиция соединения, имеющего два хиральных центра, практически не содержит диастереомеры и практически свободна от противоположного энантиомера указанного соединения. Типичное стереомерно чистое соединение включает больше чем приблизительно 80% масс. одного стереоизомера указанного соединения и меньше чем приблизительно 20% масс. других стереоизомеров указанного соединения, более предпочтительно, больше чем приблизительно 90% масс. одного стереоизомера указанного соединения и меньше чем приблизительно 10% масс. других стереоизомеров указанного соединения, еще более предпочтительно, больше чем приблизительно 95% масс. одного стереоизомера указанного соединения и меньше чем приблизительно 5% масс. других стереоизомеров указанного соединения и, наиболее предпочтительно, больше чем приблизительно 97% масс. одного стереоизомера указанного соединения и меньше чем приблизительно 3% масс. других стереоизомеров указанного соединения. См., например, патент США № 7189715.
"R" и "S" в качестве терминов, описывающих изомеры, представляют собой идентификаторы стереохимической конфигурации при асимметрично замещенном атоме углерода. Обозначение асимметрично замещенного атома углерода как "R" или "S" осуществляют путем применения правил приоритета Кана-Ингольда-Прелога, которые хорошо известны специалистам в данной области техники и описаны Международным союзом теоретической и прикладной химии (IUPAC) в Правилах номенклатуры органической химии. Раздел E. Стереохимия.
Термины "терапия", "лечить" или "лечение" онкологического заболевания относится к обращению (например, преодолению блокады дифференцировки клеток), облегчению (например, облегчению одного или нескольких симптомов, таких как изнурение от анемии, низкие показатели крови и т.д.) и/или задержке развития (например, задержке развития состояния, такого как преобразование в острый миелогенный лейкоз) онкологического заболевания, рассматриваемого в данном документе.
"Субъект" в данном описании означает животное, предпочтительно, млекопитающее и, в частности, людей.
Термин "фармацевтически приемлемый носитель" в данном описании относится к нетоксичному носителю, вспомогательному веществу или наполнителю, который не нарушает фармакологическую активность соединения, вместе с которым оно образует композицию. Фармацевтически приемлемые носители, вспомогательные вещества или наполнители, которые могут быть использованы в композициях по настоящему изобретению, включают, однако этим не ограничиваясь, ионообменные смолы, оксид алюминия, стеарат алюминия, лецитин, сывороточные белки, такие как сывороточный альбумин человека, буферные вещества, такие как фосфаты, глицин, сорбиновая кислот, сорбат калия, смеси неполных глицеридов насыщенных растительных жирных кислот, вода, соли или электролиты, такие как сульфат протамина, динатрий гидрофосфат, гидрофосфат калия, хлорид натрия, соли цинка, коллоидный оксид кремния, трисиликат магния, поливинилпирролидон, вещества на основе целлюлозы, полиэтиленгликоль, циклодекстрины, натриевая соль карбоксиметилцеллюлозы, полиакрилаты, воски, блок-полимеры полиэтилена-полиоксипропилена, полиэтиленгликоль и ланолин.
Термин "фармацевтически приемлемая соль" обозначает соль, которая сохраняет требуемую биологическую активность исходного соединения и не вызывает нежелательные токсикологические эффекты. Примерами подобных солей являются: (a) кислотно-аддитивные соли, образованные с неорганическими кислотами, например, с хлористоводородной кислотой, бромистоводородной кислотой, серной кислотой, фосфорной кислотой, азотной кислотой и т.п.; и соли, образованные с органическими кислотами, например, с уксусной кислотой, щавелевой кислотой, винной кислотой, янтарной кислотой, малеиновой кислотой, фумаровой кислотой, глюконовой кислотой, лимонной кислотой, яблочной кислотой, аскорбиновой кислотой, бензойной кислотой, дубильной кислотой, пальмитиновой кислотой, альгиновой кислотой, полиглютаминовой кислотой, нафталинсульфоновой кислотой, метансульфоновой кислотой, п-толуолсульфоновой кислотой, нафталиндисульфоновой кислотой, полигалактуроновой кислотой и т.п.; и (b) соли, образованные анионами таких элементов как хлор, бром и иод. См, например, Haynes et al., "Commentary: Occurrence of Pharmaceutically Acceptable Anions and Cations in the Cambridge Structural Database," J. Pharmaceutical Sciences, vol. 94, no. 10 (2005) и Berge et al., "Pharmaceutical Salts", J. Pharmaceutical Sciences, vol. 66, no. 1 (1977), которые включены в данное описание посредством ссылки.
В. Соединения
Если не указано иное, соединения, приведенные в данном описании, могут включать смеси приведенных в данном описании соединений, а также любые энантиомерные, диастереомерные и геометрические (или конформационные) формы структур; например, R- и S-конфигурации для каждого асимметрического центра, (Z) и (Е) изомеры при двойной связи и (Z) и (Е) конформационные изомеры. Если не указано иное, соединения, приведенные в данном описании, которые сосуществуют с таутомерными формами, включены в объем настоящего изобретения. Кроме того, если не указано иное, структуры, приведенные в данном описании, также включают соединения, которые отличаются только присутствием одного или нескольких изотопно обогащенных атомов. Например, соединения, имеющие приведенные в данном описании структуры, отличающиеся заменой атома водорода на дейтерий или тритий или заменой атома углерода на 13C- или 14C-обогащенный углерод, входят в объем настоящего изобретения. Подобные соединения могут быть пригодны, например, в качестве аналитических средств или зондов в биологических анализах.
В данном описании, в соответствии с некоторыми вариантами осуществления настоящего изобретения, соединениями являются соединение формулы 1 ("Соединение 1"), соединение формулы 2 ("Соединение 2"), соединение формулы 3 ("Соединение 3") и соединение формулы 4 ("Соединение 4"):
и их фармацевтически приемлемые соли.
С. Фармацевтические препараты
Соединения по настоящему изобретению могут быть объединены с фармацевтически приемлемым носителем, с целью приготовления фармацевтических составов. Конкретный выбор носителя и композиции будет зависеть от конкретного пути введения, для которого предназначена данная композиция.
Фармацевтические композиции по настоящему изобретению, могут быть соответствующим образом приготовлены для парентерального, перорального введения, в виде спрея для ингаляции, для местного, ректального, назального, трансбуккального, вагинального введения или введения в виде имплантируемого резервуара и т.п. Термин "парентеральный" в данном описании включает методы подкожной, внутривенной, внутримышечной, внутрисуставной, внутрисиновиальной, интрастернальной, интратекальной, внутрипеченочной, внутриочаговой и внутричерепной инъекции или инфузии. В конкретных вариантах осуществления настоящего изобретения соединения вводят внутривенно, перорально, подкожно или внутримышечно. Стерильные формы композиций для инъекций по настоящему изобретению могут представлять собой водную или масляную суспензию. Указанные суспензии могут быть приготовлены в соответствии с методиками, известными в данной области техники, с использованием подходящих диспергирующих или смачивающих агентов и суспендирующих агентов. Стерильный препарат для инъекций может также представлять собой стерильный раствор или суспензию для инъекций в нетоксичном парентерально приемлемом разбавителе или растворителе, например, в виде раствора в 1,3-бутандиоле. Среди приемлемых носителей и растворителей, которые могут быть использованы в соответствии с настоящим изобретением, можно указать воду, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, в качестве растворителя или суспендирующей среды обычно используют стерильные нелетучие масла.
С этой целью может быть использовано любое нелетучее масло, в том числе синтетические моно- или диглицериды. Для получения препаратов для инъекций пригодны жирные кислоты, такие как олеиновая кислота и ее глицеридные производные в виде природных фармацевтически приемлемых масел, таких как оливковое масло или касторовое масло, в особенности в виде их полиоксиэтилированных вариантов. Указанные масляные растворы или суспензии могут также содержать разбавитель или диспергатор в виде спирта с длинной цепью, такой как карбоксиметилцеллюлоза, или подобные диспергирующие агенты, которые обычно используют в композициях фармацевтически приемлемых лекарственных форм, включая эмульсии и суспензии. Другие обычно применяемые поверхностно-активные вещества, такие как Tweens, Spans и другие эмульгирующие агенты или усилители биодоступности, которые обычно используют для приготовления фармацевтически приемлемых твердых, жидких или других лекарственных форм, также могут быть применены с целью приготовления композиций.
Для перорального введения соединение может быть приготовлено в виде перорально приемлемой лекарственной формы, в том числе, однако этим не ограничиваясь, в виде капсул, таблеток, водных суспензии или растворов. В случае таблеток для перорального применения, обычно используемые носители включают лактозу и кукурузный крахмал. Могут быть также добавлены лубриканты, такие как стеарат магния. Для перорального введения в форме капсулы пригодные разбавители включают лактозу и высушенный кукурузный крахмал. Когда для перорального применения требуются водные суспензии, активный ингредиент объединяют с эмульгаторами и/или суспендирующими агентами. При желании также могут быть добавлены некоторые подсластители, ароматизаторы или красители.
D. Субъекты и методы применения
Соединение по настоящему изобретению может быть использовано для лечения различных видов онкологических заболеваний, в том числе таких онкологических заболеваний, которые чувствительны к действию агентов, нацеливающихся на SF3B1. Как было отмечено выше, сообщалось, что противоопухолевая активность пладиенолида B связана с его нацеливанием на комплекс SF3b, с ингибированием сплайсинга и изменением характера экспрессии генов (Kotake et al., "Splicing factor SF3b as a target of the antitumor natural product pladienolide," Nature Chemical Biology 2007, 3, 570-575). Известно, что мутации в генах сплайсосомы, в таких как белок субъединицы 1 фактора сплайсинга 3B (SF3B1), принимают участвуют в ряде видов онкологических заболеваний, таких как гематологические злокачественные опухоли и солидные опухоли. Scott et al., "Acquired mutations that affect pre-mRNA splicing in hematologic malignancies and solid tumors," JNCI 105, 20, 1540-1549.
Гематологические злокачественные заболевания могут включать онкологическое заболевание крови (лейкозы) или онкологическое заболевание лимфатических узлов (лимфомы). Лейкозы могут включать острый лимфобластный лейкоз (ALL), острый миелогенный лейкоз (ALL), хронический лимфоцитарный лейкоз (CLL), хронический миелогенный лейкоз (CML), хронический миеломоноцитарный лейкоз (CMML), острый моноцитарный лейкоз (FMoL) и т.д. Лимфомы могут включать лимфому Ходжкина и неходжкинскую лимфому. Другие гематологические злокачественные опухоли могут включать миелодиспластический синдром (MDS).
Солидные опухоли могут включать карциномы, такие как аденокарцинома, в частности, рак молочной железы, рак поджелудочной железы, рак предстательной железы, рак толстого кишечника или колоректальный рак, рак легкого, рак желудка, рак матки, рак эндометрия, рак яичников, холангиокарцинома, глиома, меланома и т.д.
Соединение по настоящему изобретению также может быть использовано для лечения онкологических заболеваний, которые могут быть чувствительны к действию агентов, нацеливающихся на ген сплайсосомы или белок, отличный от SF3B1. Следующие примеры иллюстрируют некоторые из различных видов онкологических заболеваний, которые могут быть чувствительны к действию агентов, нацеливающихся на сплайсосому, и их не следует рассматривать как каким-либо образом ограничивающие объема настоящего изобретения. Так, соединения по настоящему изобретению можно вводить субъектам, с целью лечения различных подобных онкологических заболеваний или состояний, таким как пациенты или субъекты, страдающие от следующих заболеваний:
a) Миелодиспластический синдром (MDS): См., например, "SF3B1 mutations in myelodysplastic syndromes: clinical associations and prognostic implications," Damm F. et al. Leukemia, 2011, 1-4; "Frequent pathway mutations in splicing machinery in myelodysplasia," Yoshida K. et al, Nature, 2011, 478, 64-69; "Clinical significance of SF3B1 mutations in myelodysplastic syndromes and myelodysplastic/myeloproliferative neoplasms," Malcovati L. et al., Blood, 2011, 118, 24, 6239-6246; "Mutations in the spliceosome machinery, a novel and ubiquitous pathway in leukemogenesis," Makishima et al, Blood, 2012, 119, 3203-3210; "Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts," Pappaemannuil, E. et al, New England J. Med. 2011, DOI 10.1056/NEJMoa1103283.
b) Хронический лимфолейкоз (CLL): см., например, "Defects in the spliceosomal machinery: a new pathway of leukaemogenesis," Maciejewski, J.P., Padgett, R.A., Br. J. Haematology, 2012, 1-9; "Mutations in the SF3B1 splicing factor in chronic lymphocytic leukemia: associations with progression and fludarabine-refractoriness," Rossi et al, Blood, 2011, 118, 6904-6908; "Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia," Quesada et al, Nature Genetics, 2011, 44, 47-52.
c) Хронический миеломоноцитарный лейкоз (CMML): см., например, Yoshida et al, Nature 2011; "Spliceosomal gene mutations are frequent events in the diverse mutational spectrum of chronic myelomonocytic leukemia but largely absent in juvenile myelomonocytic leukemia," Kar S.A. et al, Haematologia, 2012, DOI: 10.3324/haematol.2012.064048; DeBoever et al., "Transcriptome sequencing reveals potential mechanism of cryptic 3' splice site selection in SF3B1-mutated cancers," PLOS Computational Biology, 2013, DOI: 10.1371/journal.pcbi.1004105.
d) Острый миелоидный лейкоз (AML): cм., например, Malcovati et al., Blood 2011; Yoshida et al, Nature 2011.
е) Рак молочной железы: См., например, "Whole genome analysis informs breast cancer response to aromatase inhibition," Ellis et al., Nature, 2012, 486, 353-360; DeBoever et al., "Transcriptome sequencing reveals potential mechanism of cryptic 3' splice site selection in SF3B1-mutated cancers," PLOS Computational Biology, 2013, DOI: 10.1371/journal.pcbi.1004105; Maguire et al., "SF3B1 mutations constitute a novel therapeutic target in breast cancer," J. Pathol. 2015, 235, 571-580.
f) Увеальная меланома: См. например, "SF3B1 mutations are associated with alternative splicing in uveal melanoma," Furney et al., Cancer Disc. 2013, 10, 1122-1129; DeBoever et al., "Transcriptome sequencing reveals potential mechanism of cryptic 3' splice site selection in SF3B1-mutated cancers," PLOS Computational Biology, 2013, DOI: 10.1371/journal.pcbi.1004105.
g) Рак эндометрия: См., например, Tefferi et al., "Myelodysplastic syndromes." N. Engl. J. Med. 2009; 361:1872-85.
h) Рак желудка: См., например, Int. J. Cancer. 2013 Jul; 133(1):260-5, "Mutational analysis of splicing machinery genes SF3B1, U2AF1 and SRSF2 in myelodysplasia and other common tumors." Je et al.
i) Рак яичников: См., например, Int. J. Cancer. 2013 Jul; 133(1):260-5, "Mutational analysis of splicing machinery genes SF3B1, U2AF1 and SRSF2 in myelodysplasia and other common tumors." Je et al.
j) Рак желчных протоков, такой холангиокарцинома, и рак поджелудочной железы: См., например, Biankin et al., "Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes," Nature 2012, 491, 399-405.
k) Рак легкого: См., например, "Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia," Quesada et al., Nature Genetics 44, 47-52 (2012); Scott et al., "Acquired mutations that affect pre-mRNA splicing in hematologic malignancies and solid tumors," JNCI 105, 20, 1540-1549.
Кроме того, Каталог соматических мутаций при онкологическом заболевании (COSMIC) (Wellcome Trust Sanger Institute, Genome Research Limited, Англия) сообщает, что мутации SF3B1 были обнаружены для различных типов онкологических заболеваний.
Соединение по настоящему изобретению можно вводить субъекту в эффективном для лечения или терапевтически эффективном количестве. Количество соединения по настоящему изобретению, которое можно объединять с веществом носителя для получения композиции в форме однократной дозы, меняется в зависимости от подвергаемого лечению субъекта и конкретного способа введения. Предпочтительно, композиции должны быть составлены таким образом, чтобы субъекту, получающему указанные композиции, можно было вводить дозу активного агента в диапазоне 0,01-100 мг/кг массы тела в день. В некоторых вариантах осуществления настоящего изобретения композиции по настоящему изобретению обеспечивают дозу от 0,01 мг до 50 мг. В других вариантах осуществления настоящего изобретения доза составляет от 0,1 мг до 25 мг или от 5 мг до 40 мг.
Следует также понимать, что конкретная доза и схема лечения для любого конкретного пациента будет зависеть от множества факторов, в том числе от активности конкретного используемого соединения, возраста, массы тела, общего состояния здоровья, пола, диеты, времени введения, скорости выведения из организма, сочетания лекарственных средств, решения лечащего врача и тяжести конкретного заболевания, подлежащего лечению. Количество активного агента по настоящему изобретению в композиции будет также зависеть от конкретного соединения/соли в композиции.
В некоторых вариантах осуществления настоящего изобретения онкологическое заболевание проверяют на наличие одной или нескольких мутаций и/или оно является позитивным для одной или нескольких мутаций в гене сплайсосомы или белке, причем присутствие мутации(ий) ("позитивно") может указывать на то, что онкологическое заболевание субъекта чувствительно к способу лечения, включающему введение соединения, которое нацеливается на указанный белок и/или сплайсосому. Примеры подобных генов сплайсосомы включают, однако этим не ограничиваясь, те, что представлены в таблице 1.
Таблица 1: Гены сплайсосомы и потенциально чувствительные заболевания
Условные обозначения:
MDS=миелодиспластический синдром
AML=острый миелоидный лейкоз
CMML=хронический миеломоноцитарныЙ лейкоз
LUAD=легочная аденокарцинома
UCEC=эндометриальная карцинома тела матки
PMF=массивный прогрессивный фиброз
PRAD=аденокарцинома предстательной железы
COAD=аденокарцинома толстого кишечника
OV=серозная цистаденокарцинома яичника
SKCM=меланома кожи
LUSC=карцинома плоскоклеточных клеток легкого
STAD=аденокарцинома желудка
GBM=мультиформная глиобластома
LGG=Lower Grade Glioma мозга
DLBCL=диффузная лимфома B-клеток
В некоторых вариантах осуществления настоящего изобретения онкологическое заболевание может быть чувствительно к способу лечения, включающему введение соединения, которое нацеливается на указанный белок и/или сплайсосому даже при отсутствии подобных мутаций в гене сплайсосомы или белке.
Скрининг или тестирование на предмет мутаций может быть осуществлено любым известным способом, например, путем генотипирования, фенотипирования и т.д., путем амплификации нуклеиновых кислот, методом электрофореза, с помощью микрочипов, путем блоттинга, с помощью функциональных анализов, иммунологических анализов и т.д. Методы скрининга могут включать, например, получение биологического образца от указанного субъекта, содержащего раковые клетки/ткани.
Для того чтобы описанное здесь изобретение было более понятным, приведены следующие примеры. Следует понимать, что указанные примеры приведены только в иллюстративных целях и не должны быть истолкованы как каким-либо образом ограничивающие настоящее изобретение.
ПРИМЕРЫ
Получение соединений 1, 2, 3 и 4
Общие замечания:
ВЧ нагрев осуществляли с использованием микроволновой печи Biotage Emrys Liberator или Initiator. Колоночную хроматографию проводили с использованием Isco Rf200d. Растворитель удаляли, используя либо роторный испаритель Büchi, либо центробежный испаритель Genevac. Препаративную LC/MS проводили с использованием автоочистителя Waters и 19×100 мм колонки XTerra 5 мкм MS C18 с кислой подвижной фазой. Спектры ЯМР регистрировали на спектрометре Varian 400 МГц.
Когда термин "забалластированный инертным газом" используется для описания реактора (например, реакционного сосуда, колбы, стеклянного реактора и т.п.), это означает, что воздух в реакторе был замещен практически не содержащим влагу или сухим инертным газом (например, азотом, аргоном и т.п.).
Ниже приведены общие методы и экспериментальные разработки для получения соединений по настоящему изобретению.
В данном описании используются следующие сокращения:
МеОН: метанол
ДМФА: диметилформамид
KHMDS: бис(триметилсилил)амид калия
LC/MS: жидкостная хроматография-масс-спектрометрия
TBSCl: трет-бутилдиметилсилилхлорид
ТГФ: тетрагидрофуран
ТСХ: тонкослойная хроматография
Вещества: Следующие соединения коммерчески доступны и/или могут быть получены несколькими способами, хорошо известными специалистам в области органического синтеза. В частности, раскрытые соединения могут быть получены с использованием реакций и методик, приведенных в данном описании. При описании способов синтеза, приведенных ниже, следует понимать, что все предлагаемые условия реакции, в том числе выбор растворителя, реакционной атмосферы, температуры реакции, продолжительности эксперимента, а также процедуры выделения продуктов, могут быть выбраны из условий, которые являются стандартными для указанной реакции, если не указано иное. Специалисту в области органического синтеза должно быть понятно, что функциональные группы, присутствующие в различных участках молекулы, должны быть совместимы с реагентами и предложенными реакциями. Заместители, которые не совместимы с условиями реакции, очевидны для специалиста в данной в данной области техники, а поэтому возможны альтернативные методы. Исходные вещества для примеров либо коммерчески доступны, либо могут быть легко получены с помощью стандартных способов из известных веществ.
Данные LC/MS
Подвижные фазы: А (0,1%-ная муравьиная кислота в H2O) и В (0,1%-ная муравьиная кислота в ацетонитриле).
Градиент: В 5% → 95% в течение 1,8 мин.
Колонка: Acquity BEH C18 (1,7 мкм, 2,1×50 мм).
В патентах США № 7884128 и 7816401, которые оба имеют название: Способ полного синтеза пладиенолида В и пладиенолида D, описываются способы, известные в данной области техники, для синтеза пладиенолида В и D. Синтез пладиенолидов В и D также может быть осуществлен с использованием методов, известных в данной области техники и описанных в Kanada et al., "Total Synthesis of the Potent Antitumor Macrolides Pladienolide B and D," Angew. Chem. Int. Ed. 46:4350-4355 (2007). Kanada et al. и патентной заявке PCT WO 2003/099813 с названием: Новые физиологически активные вещества, описывают способы, известные из области техники для синтеза E7107 (Соединение 45 из WO '813) из пладиенолида D (11107D из WO '813). Соответствующий патент США № 7550503 выдан на имя Kotake et al.
Примеры синтеза соединений
Синтез Соединения 1
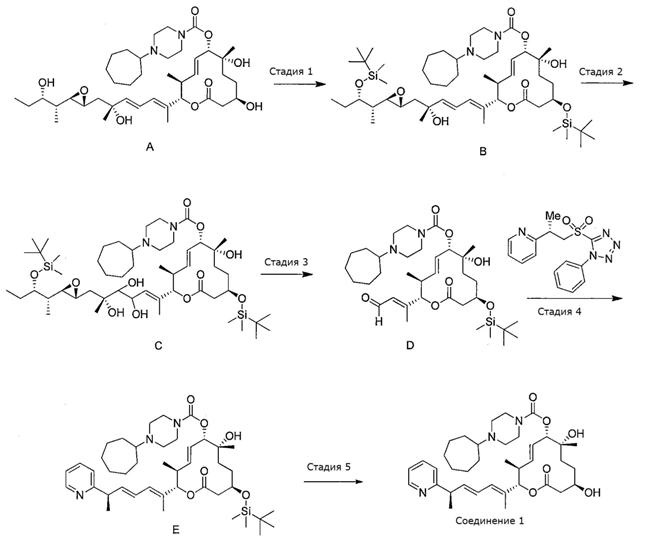
Схема I
Стадия 1: Синтез (2S,3S,6S,7R,10R,Е)-10-((трет-бутилдиметилсилил)окси)-((R,2Е,4Е)-7-((2R,3R)-3-((2S,3S)-3-((трет-бутилдиметилсилил)окси)пентан-2-ил)-6-гидрокси-6-метилгепта-2,4-диен-2-ил)-7-гидрокси-3,7-диметил-12-оксооксациклодец-4-ен-6-ил 4-циклогептилпиперазин-1-карбоксилата.
К раствору E7107 (A, 3,7 г, 5,1 ммоль, 1,0 экв.) в атмосфере азота в ДМФА (100 мл, 0,05 М) при 0°С добавили имидазол (2,5 г, 36,1 ммоль, 7,0 экв.) и TBSCl (3,9 г, 25,7 ммоль, 5,0 экв.). Реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали в течение 20 час или до тех пор, пока реакция не закончится, что определяли с помощью LC/MS или ТСХ. Реакционную смесь разбавляли этилацетатом, и органический слой промывали насыщенным раствором соли, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Полученное масло очищали колоночной хроматографией на силикагеле (гексан/этилацетат в качестве элюента), получая требуемый продукт (B, 4,7 г, 5,0 ммоль, 96%).
Стадия 2: Синтез (2S,3S,6S,7R,10R,Е)-10-((трет-бутилдиметилсилил)окси)-2-((6R,Е)-7-((2R,3R)-3-((2S,3S)-3-((трет-бутилдиметилсилил)окси)пентан-2-ил)оксиран-2-ил)-4,5,6-тригидрокси-6-метилгепт-2-ен-2-ил)-7-гидрокси-3,7-диметил-12-оксооксациклодец-4-ен-6-ил 4-циклогептилпиперазин-1-карбоксилата.
К раствору олефина В (4,7 г, 5,0 ммоль, 1,0 экв.) в смеси ТГФ:Н2О (10:1, 133 мл:13 мл, 0,03 М) в атмосфере азота при 0°С добавляли тетраоксид осмия (12,4 мл, 1,0 ммоль, 0,2 экв., 2,5%-ный раствор) с последующим добавлением N-оксида N-метилморфолина (1,16 г, 9,9 ммоль, 2,0 экв.). Реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали в течение 13 час или до тех пор, пока реакция не завершится по данным LC/MS или ТСХ. Реакцию прерывали, добавив сульфит натрия, смесь разбавляли этилацетатом и органический слой промывали водой, сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Полученное масло очищали с помощью колоночной хроматографии на силикагеле (смесь дихлорметан/метанол в качестве элюента) и получали требуемый продукт (C, 4,8 г, 4,9 ммоль, 99%).
Стадия 3: Синтез (2S,3S,6S,7R,10R,Е)-10-((трет-бутилдиметилсилил)окси-7-гидрокси-3,7-диметил-12-оксо-2-((Е)-4-оксобут-2-ен-2-ил)оксациклододец-4-ен-6-ил 4-циклогептилпиперазин-1-карбоксилата.
К раствору диола C (4,4 г, 4,5 ммоль, 1,0 экв.) в бензоле (100 мл, 0,05М) в атмосфере азота при комнатной температуре добавляли тетраацетат свинца (4,0 г, 9,0 ммоль, 2,0 экв.). Реакционную смесь перемешивали в течение 30 мин или до тех пор, пока реакция не завершится, что определяли с помощью LC/MS или ТСХ. Реакцию прерывали, добавив сульфит натрия, и смесь разбавляли дихлорметаном. Органический слой промывали водой, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Требуемый продукт (D, 1,5 г, 2,3 ммоль, 52%) использовали в сыром виде.
Стадия 4: Синтез (2S,3S,6S,7R,10R,Е)-10-((трет-бутилдиметилсилил)окси-7-гидрокси-3,7-диметил-12-оксо-2-((R,2Е,4E)-6-(пиридин-2-ил)гепта-2,4-диен-2-ил)оксациклододец-4-ен-6-ил 4-циклогептилпиперазин-1-карбоксилата.
Примечание: Синтез (S)-2-(1-((1-фенил-1Н-тетразол-5-ил)сульфонил)пропан-2-ил)пиридина описан ниже и приведен на схеме V.
К раствору (S)-2-(1-((1-фенил-1Н-тетразол-5-ил)сульфонил)пропан-2-ил)пиридина (1,67 г, 5,08 ммоль, 2,5 экв.) в сухом ТГФ (30,0 мл, 0,05М) в атмосфере азота при температуре -78°С по каплям добавляли KHMDS (8,53 мл, 4,265 ммоль, 2,1 экв.) и реакционную смесь перемешивали в течение 10 мин. Затем по каплям добавляли альдегид D (2S,3S,6S,7R,10R,Е)-10-((трет-бутилдиметилсилил)окси)-7-гидрокси-3,7-диметил-12-оксо-2-((Е)-4-оксобут-2-ен-2-ил)оксациклододец-4-ен-6-ил 4-циклогептилпиперазин-1-карбоксилат (1,318 г, 2,031 ммоль, 1,0 экв.) в ТГФ (10 мл). Реакционную смесь перемешивали при температуре -78°С в течение одного часа и затем оставляли на ночь нагреваться до комнатной температуры. Реакционную прерывали, добавив воду, и разбавляли этилацетатом. Органический слой промывали водой и насыщенным раствором соли, сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Полученное масло очищали с помощью колоночной хроматографии на силикагеле (смесь гексан/этилацетат в качестве элюента) получали желаемый продукт (Е, 1,20 г, 2,03 ммоль, 79%).
Стадия 5: Синтез (2S,3S,6S,7R,10R,Е)-7,10-дигидрокси-3,7-диметил-12-оксо-2-((R,2Е,4Е)-6-(пиридин-2-ил)гепта-2,4-диен-2-ил)оксациклододец-4-ен-6-ил 4-циклогептилпиперазин-1-карбоксилата (Соединение 1).
К раствору силилового эфира Е (1,80 г, 2,39 ммоль, 1,0 экв.) в МеОН (10,0 мл, 0,24М) в атмосфере азота при комнатной температуре добавили pTsOH (1,14 г, 5,98 ммоль, 2,5 экв.). Реакционную смесь перемешивали в течение 2 час или до тех пор, пока реакция не завершится, что определяли по данным LC/MS или ТСХ. Затем реакционную смесь разбавляли этилацетатом и промывали насыщенным раствором соли, сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Полученное масло очищали с помощью препаративной ТСХ (смесь дихлорметан/метанол в качестве элюента) и получали желаемый продукт (Соединение 1, 1,19 г, 1,83 ммоль, 76%). Спектр 1H-ЯМР (400 МГц, хлороформ-d) δ: 0,88 (д, J=6,65 Гц, 6Н) 1,23 (с, 3H), 1,34-1,78 (м, 12 Н) 1,44 (д, J=7,03 Гц, 3Н) 1,73 (с, 3H), 2,28-2,39 (м, 1Н), 2,45-2,66 (м, 8Н) 3,48 (шир. с, 5Н) 3,72 (м, 2H), 5,01 (д, J=9,54 Гц, 1Н), 5,14 (д, J=10,67 Гц, 1Н), 5,55-5,72 (м, 2H), 6,00 (дд, J=15,00, 7,47 Гц, 1Н), 6,11 (д, J=11,29 Гц, 1Н) 6,28-6,35 (м, 1Н), 7,12 (ддд, J=7,47, 4,89, 1,07 Гц, 1Н), 7,16 (д, J=7,78 Гц, 1Н), 7,61 (т, J=7,65 Гц, 1Н) 8,55 (д, J=4,91 Гц, 1Н). Масс-спектр (ES+)=638,4 [М+Н]+.
Синтез Соединения 2
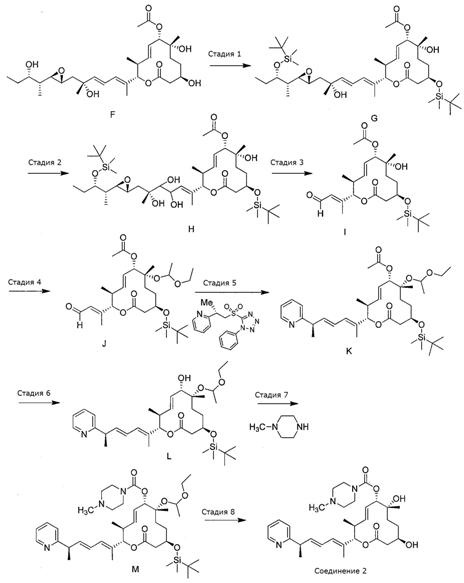
Схема II
Стадия 1: Синтез (2S,3S,6S,7R,10R,Е)-10-((трет-бутилдиметилсилил)окси)-2-((R,2Е,4Е)-7-((2R,3R)-3-((2S,3S)-3-((трет-бутилдиметилсилил)окси)пентан-2-ил)оксиран-2-ил)-6-гидрокси-6-метилгепта-2,4-диен-2-ил)-7-гидрокси-3,7-диметил-12-оксооксациклодец-4-ен-6-ил ацетата.
К раствору пладиенолида D (F, 5,3 г, 9,7 ммоль, 1,0 экв.) в атмосфере азота в ДМФА (80 мл, 0,1 М) при 0°С добавили имидазол (4,6 г, 67,8 ммоль, 7,0 экв.) и TBSCl (7,3 г, 48,4 ммоль, 5,0 экв.). Реакционную смесь оставили нагреваться до комнатной температуры и перемешивали в течение 20 час или до тех пор, пока реакция не завершится по данным LC/MS или ТСХ. Реакционную смесь экстрагировали этилацетатом, и органический слой промывали насыщенным раствором соли, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Полученное масло очищали колоночной хроматографией на силикагеле (смесь гексан/этилацетат в качестве элюента) и получали желаемый продукт (G, 7,5 г, 9,6 ммоль, 99%).
Стадия 2: Синтез (2S,3S,6S,7R,10R,Е)-10-((трет-бутилдиметилсилил)окси)-2-((6R,Е)-7-((2R,3S)-3-((трет-бутилдиметилсилил)окси)пентан-2-ил)оксиран-2-ил)-4,5,6-тригидрокс-6-метилгепт-2-ен-2-ил)-7-гидрокси-3,7-диметил-12-оксооксациклодец-4-ен-6-ил ацетата.
К раствору олефина G (7,6 г, 9,7 ммоль, 1,0 экв.) в дегазированной смеси ТГФ:H2O (210 мл:21 мл, 0,01М) в атмосфере азота при 0°С добавляли тетраоксид осмия (24,4 мл, 1,9 ммоль, 0,2 экв., 2,5%-ный раствор в трет-бутаноле), а затем N-оксид N-метилморфолина (2,3 г, 19,5 ммоль, 2,0 экв.). Реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали в течение 13 час или до тех пор, пока реакция не завершится, что определяли методом LC/MS или ТСХ. Реакцию прерывали сульфитом натрия, смесь разбавляли этилацетатом и органический слой промывали водой, сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Полученное масло очищали с помощью колоночной хроматографии на силикагеле (смесь дихлорметан/метанол в качестве элюента), получая желаемый продукт (H, 6,8 г, 8,3 ммоль, 86%).
Стадия 3: Синтез (2S,3S,6S,7R,10R,Е)-10-((трет-бутилдиметилсилил)окси)-7-гидрокси-3,7-диметил-12-оксо-2-((Е)-4-оксобут-2-ен-2-ил)оксациклододец-4-ен-6-ил ацетата.
К раствору диола Н (7,9 г, 9,7 ммоль, 1,0 экв.) в бензоле (350 мл, 0,03 М) в атмосфере азота при комнатной температуре добавляли тетраацетат свинца (8,6 г, 19,4 ммоль, 2,0 экв.). Реакционную смесь перемешивали в течение 30 мин или до тех пор, пока реакция не закончится, что определяли методом LC/MS или ТСХ. Реакционную смесь концентрировали и очищали с помощью колоночной хроматографии на силикагеле (смесь гексан/этилацетат в качестве элюента) и получали требуемый продукт (I, 2,5 г, 5,26 ммоль, 54%).
Стадия 4: Синтез (2S,3S,6S,7R,10R,Е)-10-((трет-бутилдиметилсилил)окси)-7-(1-этоксиэтокси)-3,7-диметил-12-оксо-2-((Е)-4-оксобут-2-ен-2-ил)оксациклододец-4-ен-6-ил ацетата.
К раствору альдегида I (1,4 г, 2,9 ммоль, 1,0 экв.) в ТГФ (9,5 мл, 0,5 М) при комнатной температуре добавляли этоксиэтилен (11,1 мл, 40,0 экв.) и п-толуолсульфонат пиридиния (0,07 г, 0,3 ммоль, 0,1 экв.). Реакционную смесь перемешивали в течение 24 час или до тех пор, пока реакция не закончится, что определяли методом LC/MS или ТСХ. Реакцию прерывали с помощью бикарбоната натрия и разбавляли этилацетатом. Этилацетатный раствор промывали водой, насыщенным раствором соли, сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Полученное масло очищали с помощью колоночной хроматографии на силикагеле (смесь гексан/этилацетат в качестве элюента) и получали требуемый продукт (J, 1,2 г, 2,2 ммоль, 75%).
Стадия 5: Синтез (2S,3S,6S,7R,10R,Е)-10-((трет-бутилдиметилсилил)окси)-7-(1-этоксиэтокси)-3,7-диметил-12-оксо-2-((R,2Е,4Е)-6-(пиридин-2-ил)гепта-2,4-диен-2-ил)оксациклододец-4-ен-6-ил) ацетата.
К раствору (S)-2-(1-((1-фенил-1Н-тетразол-5-ил)сульфонил)пропан-2-ил)пиридина (695,0 мг, 2,1 ммоль, 1,5 экв.) в ТГФ (20 мл, 0,06 М) в атмосфере азота при температуре -78°С по каплям добавляли KHMDS (4,2 мл, 2,1 ммоль, 1,5 экв.) и реакционную смесь перемешивали в течение 20 мин. Затем по каплям добавляли альдегид J (780,0 мг, 1,4 ммоль, 1,0 экв.) в ТГФ (1,0 мл). Реакционную смесь перемешивали при -78°С в течение 90 мин, а затем в течение 1 час дали нагреться до температуры -20°С. Реакцию прерывали раствором хлорида аммония, разбавляли смесь этилацетатом и нагревали до комнатной температуры. Органический слой промывали водой, насыщенным раствором соли, сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Полученное масло очищали с помощью колоночной хроматографии на силикагеле (смесь гексан/этилацетат в качестве элюента) и получали требуемый продукт (K, 490 мг, 0,7 ммоль, 53%).
Стадия 6: Синтез (4R,7R,8S,11S,Е)-4-((трет-бутилдиметилсилил)окси)-7-(1-этоксиэтокси)-8-гидрокси-7,11-диметил-12-((R,2Е,4Е)-6-(пиридин-2-ил)гепта-2,4-диен-2-ил)оксациклододец-9-ен-2-она.
К раствору ацетата К (490 мг, 0,7 ммоль, 1,0 экв.) в метаноле (15 мл, 0,05 моль) при комнатной температуре добавляли карбонат калия (155 мг, 0,4 ммоль, 1,5 экв.). Реакцию проводили в течение 24 час или до тех пор, пока реакция не закончится, что определяли методом LC/MS или ТСХ. Реакцию прерывали, добавив воду, разбавляли смесь этилацетатом, промывали насыщенным раствором соли, сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Полученное в результате пенообразное твердое вещество (L, 459 мг, 0,7 ммоль, 100%) использовали на следующей стадии без дополнительной очистки.
Стадия 7: Синтез (2S,3S,6S,7R,10R,Е)-10-((трет-бутилдиметилсилил)окси)-7-(1-этоксиэтокси)-3,7-диметил-12-оксо-2-((R,2Е,4Е)-6-(пиридин-2-ил)гепта-2,4-диен-2-ил)оксациклододец-4-ен-6-ил 4-метилпиперазин-1-карбоксилата.
К раствору спирта L (459 мг, 0,7 ммоль, 1,0 экв.) в дихлорметане (0,5 мл, 0,1 М) при комнатной температуре добавляли N,N-диметиламинопиридин (27,3 мг, 0,2 ммоль, 0,3 экв.) и триэтиламин (1,0 мл, 7,4 ммоль, 10,0 экв.), а затем 4-нитрофенил хлорформиат (451 мг, 02,2 ммоль, 3,0 экв.). Реакционную смесь перемешивали при комнатной температуре в течение трех часов. Затем добавляли N-метилпиперазин (299 мг, 2,98 ммоль, 4,0 экв.) при комнатной температуре. После перемешивания в течение одного часа реакционную смесь прерывали водой и разбавляли дихлорметаном. Органический слой промывали 1N раствором гидроксида натрия, и органический слой концентрировали. Полученное масло очищали с помощью колоночной хроматографии на силикагеле (смесь гексан/этилацетат в качестве элюента) и получали требуемый продукт (М, 553 мг, 0,75 ммоль, 100%).
Стадия 8: Синтез (2S,3S,6S,7R,10R,Е)-7,10-дигидрокси-3,7-диметил-12-оксо-2-((R,2Е,4Е)-6-(пиридин-2-ил)гепта-2,4-диен-2-ил)оксациклододец-4-ен-6-ил 4-метилпиперазин-1-карбоксилата (Соединение 2).
К раствору силилового эфира (М, 553 мг, 0,74 ммоль, 1,0 экв.) в метаноле (20 мл, 0,04 М) при комнатной температуре добавляли п-метокситолуолсульфоновую кислоту (425 мг, 2,2 ммоль, 3,0 экв.). Реакционную смесь перемешивали в течение 3 час или до тех пор, пока реакция не закончится, что определяли методом LC/MS или ТСХ. Реакцию прерывали с помощью бикарбоната натрия и разбавляли этилацетатом. Органический слой промывали водой, насыщенным раствором соли, сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Полученное масло очищали с помощью колоночной хроматографии на силикагеле (смесь гексан/этилацетат в качестве элюента) и получали требуемый продукт (Соединение 2, 184 мг, 0,33 ммоль, 44%). Спектр 1H ЯМР (400 МГц, хлороформ-d) δ: 0,82-1,00 (м, 3H), 1,22-1,48 (м, 8H) 1,50-1,63 (м, 1H), 1,66-1,83 (м, 4H), 1,97 (с, 1H) 2,07 (с, 1H), 2,33 (с, 3H) 2,40 (шир. с, 3H) 2,45-2,68 (м, 3H) 3,44-3,61 (м, 5H) 3,74 (дд, J=14,2, 7,2 Гц, 2H) 5,04 (д, J=9,3 Гц, 1H), 5,17 (д, J=10,5 Гц, 1H), 5,57-5,76 (м, 2H), 6,02 (дд, J=15,1, 7,5 Гц, 1H), 6,13 (д, J=10,8 Гц, 1H), 6,34 (ддд, J=15,1, 10,7, 1,0 Гц, 1H), 7,14 (т, J=6,2 Гц, 1H), 7,18 (д, J=7,4 Гц, 1H), 7,63 (т, J=7,3 Гц, 1H), 8,57 (д, J=5,1 Гц, 1H). Масс-спектр (ES+)=556,4 [М+Н].
Синтез Соединения 3
Стадии 1-6 проводили так же, как указано выше при синтезе Соединения 2, при этом получали спирт L.
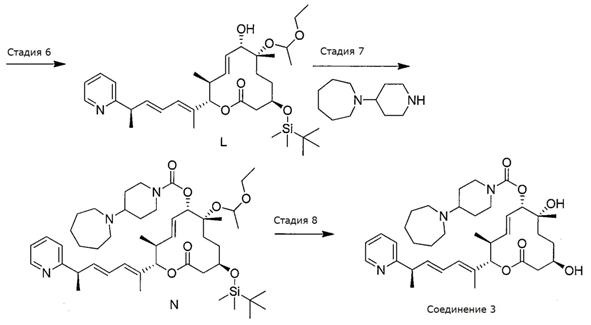
Схема III
Стадия 7: Синтез (2S,3S,6S,7R,10R,Е)-10-((трет-бутилдиметилсилил)окси)-7-(1-этоксиэтокси)-3,7-диметил-12-оксо-2-((R,2Е, 4Е) -6- (пиридин-2-ил) гепта-2,4-диен-2-ил) оксациклододец-4-ен-6-ил 4-(азепан-1-ил)пиперидин-1-карбоксилата.
К раствору спирта L (300 мг, 0,49 ммоль, 1,0 экв.) в дихлорметане (3,0 мл, 0,15 М) при комнатной температуре добавили N,N-диметиламинопиридин (71,4 мг, 0,58 ммоль, 1,2 экв.) и триэтиламин (0,27 мл, 1,95 ммоль, 4,0 экв.), а затем 4-нитрофенил хлорформиат (196 мг, 0,97 ммоль, 2,0 экв.). Реакционную смесь перемешивали при комнатной температуре в течение трех часов. Затем при комнатной температуре добавили 1-(пиперидин-4-ил)азепан (265 мг, 1,46 ммоль, 3,0 экв.). После перемешивания в течение одного часа реакцию прерывали водой и разбавили смесь дихлорметаном. Органический слой промыли 1N раствором гидроксида натрия и органический слой концентрировали. Полученное масло очищали с помощью колоночной хроматографии на силикагеле (смесь гексан/этилацетат в качестве элюента) и получали требуемый продукт (N, 400 мг, 0,48 ммоль, 100%).
Стадия 8: Синтез (2S,3S,6S,7R,10R,Е)-7,10-дигидрокси-3,7-диметил-12-оксо-2-((R,2Е,4Е)-6-(пиридин-2-ил)гепта-2,4-диен-2-ил)оксациклододец-4-ен-6-ил 4-(азепан-1-ил)пиперидин-1-карбоксилата (Соединение 3).
К раствору силилового эфира (N, 400 мг, 0,48 ммоль, 1,0 экв.) в метаноле (4,0 мл, 0,1 М) при комнатной температуре добавили п-метокситолуолсульфоновую кислоту (231 мг, 1,2 ммоль, 2,5 экв.). Реакционную смесь перемешивали в течение 3 час или до тех пор, пока реакция не закончится, что определяли методом LC/MS или ТСХ. Реакцию прерывали с помощью бикарбоната натрия и разбавляли этилацетатом. Органический слой промывали водой, насыщенным раствором соли, сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Полученное масло очищали с помощью колоночной хроматографии на силикагеле (смесь гексан/этилацетат в качестве элюента) и получали требуемый продукт (Соединение 3, 226 мг, 0,35 ммоль, 73%). Спектр 1H ЯМР (400 МГц, хлороформ-d) δ: 0,88 (д, J=6,53 Гц, 3H), 1,20-1,28 (м, 4H), 1,35 (с, 3H), 1,45 (д, J=7,03 Гц, 4 Н) 1,59 (шир. с, 10 Н) 1,74 (д, J=0,75 Гц, 3H), 1,75-1,83 (м, 2Н), 1,99 (с, 1Н), 2,46-2,62 (м, 3H), 2,62-2,71 (м, 4Н), 2,79 (шир. с, 2Н), 3,51 (д, J=9,79 Гц, 1Н), 3,63-3,82 (м, 2H), 4,03-4,26 (м, 2H), 5,01 (д, J=9,54 Гц, 1Н), 5,16 (д, J=10,79 Гц, 1Н), 5,54-5,64 (м, 1Н), 5,65-5,75 (м, 1Н), 6,01 (дд, J=15,06, 7,53 Гц, 1Н), 6,12 (д, J=11,04 Гц, 1Н), 6,25-6,39 (м, 1Н), 7,12 (ддд, J=7,47, 4,83, 1,25 Гц, 1Н), 7,17 (дт, J=8,03, 1,00 Гц, 1Н), 7,62 (тд, J=7,65, 1,76 Гц, 1Н), 8,56 (ддд, J=4,96, 1,82, 1,00 Гц, 1Н). Масс-спектр (ES+)=638,6 [М+Н].
Синтез Соединения 4
Стадии 1-6 проводили так же, как указано выше при синтезе Соединения 2, при этом получали спирт L.
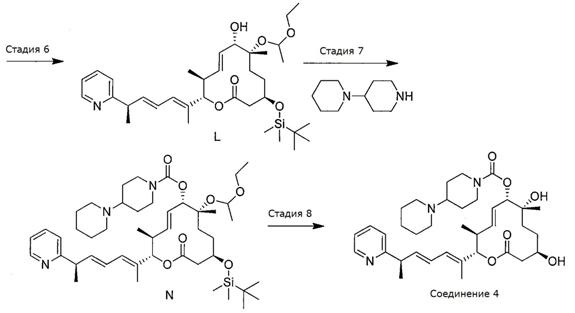
Схема IV
Стадия 7: Синтез (2S,3S,6S,7R,10R,Е)-10-((трет-бутилдиметилсилил)окси)-7-(1-этоксиэтокси)-3,7-диметил-12-оксо-2-((R,2Е,4Е)-6-(пиридин-2-ил)гепта-2,4-диен-2-ил)оксациклододец-4-ен-6-ил [1,4'-бипиперидин]-1'-карбоксилата.
К раствору спирта L (20 мг, 0,032 ммоль, 1,0 экв.) в дихлорметане (0,3 мл, 0,1 М) при комнатной температуре добавляли N,N-диметиламинопиридин (4,8 мг, 0,04 ммоль, 1,2 экв.) и триэтиламин (0,02 мл, 0,13 ммоль, 4,0 экв.), а затем 4-нитрофенил хлорформиат (13,1 мг, 0,065 ммоль, 2,0 экв.). Реакционную смесь перемешивали при комнатной температуре в течение трех часов. Затем при комнатной температуре добавили 1,4'-бипиперидин (16,4 мг, 0,97 ммоль, 3,0 экв.). После перемешивания в течение одного часа реакцию прерывали, добавив воду, и реакционную смесь разбавляли дихлорметаном. Органический слой промывали 1N раствором гидроксидом натрия, и органический слой концентрировали. Полученное масло очищали с помощью колоночной хроматографии на силикагеле (смесь гексан/этилацетат в качестве элюента) и получали требуемый продукт (N, 18 мг, 0,22 ммоль, 68,4%).
Стадия 8: Синтез (2S,3S,6S,7R,10R,Е)-7,10-дигидрокси-3,7-диметил-12-оксо-2-((R,2Е,4Е)-6-(пиридин-2-ил)гепта-2,4-диен-2-ил)оксациклододец-4-ен-6-ил [1,4'-бипиперидин]-1'-карбоксилата (Соединение 4).
К раствору силилового эфира (N, 18 мг, 0,022 ммоль, 1,0 экв.) в метаноле (0,5 мл, 0,04 М) при комнатной температуре добавляли п-метокситолуолсульфоновую кислоту (10,6 мг, 0,56 ммоль, 2,5 экв.). Реакционную смесь перемешивали в течение 3 час или до тех пор, пока реакция не закончится, что определяли методом LC/MS или ТСХ. Реакцию прерывали с помощью бикарбоната натрия и разбавляли смесь этилацетатом. Органический слой промывали водой, насыщенным раствором соли, сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Полученное масло очищали с помощью колоночной хроматографии на силикагеле (смесь гексан/этилацетат в качестве элюента) и получали требуемый продукт (Соединение 4, 4,0 мг, 0,006 ммоль, 29%). Спектр 1H ЯМР (400 МГц, хлороформ-d) δ: 0,90 (д, J=6,8 Гц, 3H), 1,17-1,42 (м, 5H), 1,46 (д, J=7,0 Гц, 6H) 1,51-1,65 (м, 6H) 1,65-1,78 (м, 5H), 1,85 (д, J=11,5 Гц, 2H), 2,44 (д, J=11,3 Гц, 2H), 2,49-2,66 (м, 6H), 2,80 (шир. с, 2H), 3,42-3,62 (м, 1H), 3,63-3,82 (м, 2H), 4,18 (шир. с, 2H), 5,02 (д, J=9,5 Гц, 1H), 5,17 (д, J=10,8 Гц, 1H), 5,57-5,75 (м, 2H), 6,02 (дд, J=15,2, 7,4 Гц, 1H), 6,14 (д, J=11,0 Гц, 1H), 6,34 (ддд, J=15,1, 10,8, 1,0 Гц, 1H), 7,14 (т, J=6,1 Гц, 1H), 7,18 (д, J=7,5 Гц, 1H), 7,29 (с, 2H), 7,63 (тд, J=7,7, 1,9 Гц, 1H), 8,57 (д, J=5,1 Гц, 1H). Масс-спектр (ES+)=624,6 [М+Н].
Синтез (S)-2-(1-((1-фенил-1Н-тетразол-5-ил)сульфонил)пропан-2-ил)пиридина
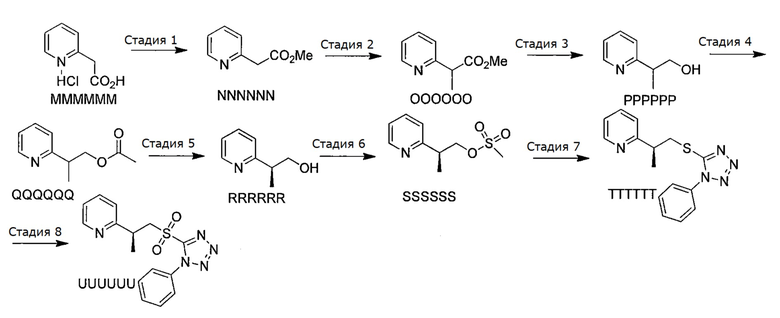
Схема V
Стадия 1: К раствору гидрохлоридной соли 2-(пиридин-2-ил)уксусной кислоты MMMMMM (50,0 г, 288,0 ммоль, 1,0 экв.) в метаноле (500 мл, 0,5 М) при температуре 0°С по каплям добавили тионилхлорид (31,5 мл, 432,0 ммоль, 1,5 экв.). Реакционную смесь перемешивали при 0°С в течение 60 мин или до тех пор, пока реакция не завершится, что определяли с помощью LC/MS или ТСХ. Реакцию осторожно прерывали с помощью карбоната натрия, и водный слой экстрагировали этилацетатом. Органические слои объединяли, промывали водой, насыщенным раствором соли, сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Полученный в результате продукт (NNNNNN, 41,5 г, 275,0 ммоль, 95%) использовали на следующей стадии без дополнительной очистки.
Стадия 2: К раствору сложного эфира NNNNNN (41,5 г, 275,0 ммоль, 1,0 экв.) в ТГФ (1500 мл, 0,2 М) при температуре 0°С добавили 2-метилпропан-2-олат натрия (28,6 г, 288,3 ммоль, 1,05 экв.) и реакционную смесь перемешивали в течение 30 мин при 0°С, а затем добавили иодметан (34,3 мл, 549,1 ммоль, 2,0 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 1 час или до тех пор, пока реакция не завершится, что определяли с помощью LC/MS или ТСХ. Реакцию прерывали раствором хлорида аммония, и избыток растворителя удаляли в вакууме. Сырой продукт затем экстрагировали этилацетатом. Органические слои объединяли, промывали насыщенным раствором соли и сушили над сульфатом магния. После фильтрации смесь концентрировали в вакууме. Полученный сложный метиловый эфир (OOOOOO, 41,3 г, 250 ммоль, 91%) использовали без дополнительной очистки.
Стадия 3: К раствору метилового эфира OOOOOO (43,0 г, 260,3 ммоль, 1,0 экв.) в ТГФ (1500 мл, 0,1 М) при 0°С по каплям добавили алюмогидрид лития (312 мл, 312,4 ммоль, 1,2 экв., раствор в ТГФ). Реакционную смесь оставляли постепенно нагреваться до 0°C в течение 30 мин, а затем до комнатной температуры в течение 1 час или до тех пор, пока реакция не завершится, что определяли с помощью LC/MS или ТСХ. Реакционную смесь осторожно гасили водой, гидроксидом натрия и водой. После перемешивания смеси в течение 30 мин образовавшийся белый осадок отфильтровывали и растворитель удаляли в вакууме. Затем реакционную смесь экстрагировали диэтиловым эфиром, органические вытяжки объединяли и промывали водой, насыщенным раствором соли, сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Полученный спирт (PPPPPP, 30,0 г, 219,0 ммоль, 84%) использовали без дополнительной очистки.
Стадия 4: К раствору спирта PPPPPP (30,0 г, 219,0 ммоль, 1,0 экв.) в дихлорметане (700 мл, 0,3 М) при температуре 0°С добавили триэтиламин (61,5 мл, 437,4 ммоль, 2,0 экв.) и DMAP (2,7 г, 21,9 ммоль, 0,1 экв.). Добавили уксусный ангидрид (24,8 мл, 262,4 ммоль, 1,2 экв.) и реакционную смесь перемешивали в течение 30 мин или до тех пор, пока реакция не завершится, что определяли с помощью LC/MS или ТСХ. Реакцию прерывали раствором хлорида аммония, органический слой промывали насыщенным раствором соли, сушили над сульфатом магния и фильтровали. Полученный раствор затем выпаривали и неочищенный ацетат (QQQQQQ, 37,0 г, 206,0 ммоль, 94%) использовали на следующей стадии без дополнительной очистки.
Стадия 5: К раствору ацетата QQQQQQ (39,4 г, 219,8 ммоль, 1,0 экв.) в диэтиловом эфире (100 мл) добавили 118 г силикагеля. Избыток эфира удаляли в вакууме, и неочищенный твердый продукт затем разбавляли водным буферным раствором с рН 7 (1970 мл, 0,1 М) (гидроксид натрия/одноосновной фосфат натрия/вода). Добавили свиную панкреатическую липазу типа II (3,3 г, (15 мг/ммоль)) и реакционную смесь перемешивали при температуре 37°С в течение четырех часов или до тех пор, пока реакция не завершится, что определяли с помощью ТСХ или LC/MS. (Через четыре часа степень превращения достигла 40%, согласно ELSD, а энантиомерный избыток определяли с помощью хиральной SFC, которая дала соотношение энантиомеров 13:1 S:R). (Условия проведения SFC: SFC Investigator (Waters/Thar), программное обеспечение: Chromscope v1.2, метод: изократический 15% сорастворитель 95:5 гептан:изопропанол+0,1% DEA в течение 10 мин, колонка: Lux-Amylose-2, 4,6×250 мм, 5 мкм, суммарный расход: 4 мл/мин (3,80 мл из насоса СО2, 0,20 мл из модифицирующего насоса). Температуру в термостате устанавливали равной 35°С, а давление в системе - 100 бар; время удерживания: требуемый и основной (S)-энантиомер 6,9 мин, минорный (R)-энантиомер 8,4 мин). Силикагель отфильтровывали, и водный слой экстрагировали три раза этилацетатом. Органические слои объединяли, промывали насыщенным раствором соли, сушили над сульфатом магния и концентрировали. Продукт очищали с помощью колоночной хроматографии на силикагеле (смесь гексан:этилацетат в качестве элюента) и получали требуемый спирт (RRRRRR, 12,5 г, 91 ммоль, 41%).
Стадия 6: К раствору спирта RRRRRR (12,5 г, 91,0 ммоль, 1,00 экв.) в дихлорметане (570 мл, 0,16M) при комнатной температуре, добавили триэтиламин (13,9 мл, 100,1 ммоль, 1,1 экв.). Реакционную смесь охладили до температуры 0°С и затем добавили метансульфонилхлорид (7,44 мл, 95,5 ммоль, 1,05 экв.). Реакционную смесь перемешивали при температуре 0°С в течение 30 мин или до тех пор, пока реакция не завершится, что определяли с помощью ТСХ или LC/MS. Реакцию прерывали с помощью бикарбоната натрия и слои разделяли. Водный слой затем экстрагировали дихлорметаном. Органические слои объединяли, промывали насыщенным раствором соли, сушили над сульфатом магния и концентрировали в вакууме. Полученный сульфонат SSSSSS (19,2 г, 89 ммоль, 98%) использовали далее без дополнительной очистки.
Стадия 7: К раствору сульфоната SSSSSS (19,2 г, 89 ммоль, 1,0 экв.) в ДМФА (120 мл, 0,1 М) при комнатной температуре добавили карбонат цезия (40,7 г, 125,0 ммоль, 1,4 экв.) и 1-фенил-1Н-тетразол-5-тиол (19,1 г, 107,1 ммоль, 1,2 экв.). Полученную смесь перемешивали при температуре 50°С в течение 48 час или до тех пор, пока реакция не завершится, что определяли с помощью ТСХ или LC/MS. После охлаждения смеси до комнатной температуры, добавили насыщенный раствор соли и водный слой трижды экстрагировали диэтиловым эфиром. Органические слои объединяли, промывали водой, насыщенным раствором соли и сушили над сульфатом магния. После фильтрации растворитель удаляли в вакууме, а остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: гексан/этилацетат) и получали требуемый продукт (TTTTTT, 28,9 г, 88 ммоль, 99%).
Стадия 8: К раствору сульфида TTTTTT (31,5 г, 105,9 ммоль, 1,0 экв.) в EtOH (700 мл, 0,1 М) при температуре -10°С добавили тетрагидрат молибдата аммония (6,5 г, 5,3 ммоль, 0,05 экв.) и пероксид водорода (108 мл, 1060 ммоль, 5,0 экв., 33%-ный водный раствор). Реакционную смесь перемешивали при температуре -10°С в течение четырех часов или до тех пор, пока реакция не завершится, что определяли с помощью ТСХ или LC/MS. Реакционную смесь гасили водой и раствором метабисульфита натрия. Сырой продукт выделяли фильтрованием и очищали с помощью колоночной хроматографии на силикагеле (смесь гексан:этилацетат в качестве элюента) и получали требуемый продукт (UUUUUU, 23,2 г, 70,4 ммоль, 66%). Спектр 1H ЯМР (400 МГц, хлороформ-d) δ: 1,50 (д, J=7,03 Гц, 3H), 1,66 3,75 (м, 1H), 3,94 (дд, J=14,81, 5,02 Гц, 1Н), 4,55 (дд, J=14,68, 7,91 Гц, 1Н), 7,14-7,22 (м, 2H), 7,29 (с, 1Н), 7,57-7,70 (м, 6H), 8,44-8,49 (м, 1 Н).
Бесцветное масло затем перекристаллизовывали, используя смесь толуол/гептан (1/1) (1 мл толуола и 1 мл гептана на 100 мг соединения). Осторожно нагревали смесь, чтобы смешать два растворителя. Дали смеси остыть до комнатной температуры в течение 12 час. (Если не наблюдалась кристаллизация, добавляли один кристаллик в раствор. Кристаллик помогает образованию кристаллического осадка, действуя как затравка.) Кристаллы образуются медленно с течением времени. Их можно выделить с помощью фильтрации или путем удаления слоя жидкости пипеткой. Полученные кристаллы промывали гептаном, а затем быстро толуолом. Значение er сульфона анализировали до и после перекристаллизации (условия SFC:
SFC Investigator (Waters/Thar), программное обеспечение: Chromscope v1.2, метод: изократический 10% сорастворитель MeOH в течение 10 мин, колонка: ChiralPak IC 4,6×250 мм, 5 мкм, суммарный расход: 4 мл/мин (3,80 мл из насоса СО2, 0,20 мл из модифицирующего насоса). Температуру в термостате устанавливали равной 35°С, а давление в системе - 100 бар; время удерживания: требуемый и основной (S)-энантиомер 3,5 мин, минорный (R)-энантиомер 3,8 мин).
Определение рН стабильности
Соединения помещали на 96-луночные планшеты и проводили исследования в виде трех серий. Четыре микролитра 10 мМ исходного раствора соединения в ДМСО помещали в каждую из трех лунок. Планшет хранили при температуре, равной или ниже -20°С до дня проведения анализа. Для разведений использовали метанол (степени чистоты для ВЭЖХ) и 0,1 N раствор HCl (HX0603A-6 по каталогу EMD). Для приготовления подвижной фазы для двух анализов использовали ацетонитрил (степени чистоты для ВЭЖХ), воду (профильтрованную через элемент Milli-Q), трифторуксусную кислоту (спектральной чистоты) и 0,2 М фосфатный буфер (№163-14471 по каталогу Wako).
Данные о стабильности получали, используя ВЭЖХ-хроматограф Waters Acquity, снабженный УФ-детектором (Waters TUV) и одним квадрупольным масс-спектрометрическим детектором (Waters SQD). 96-луночный планшет, содержащий представляющее(ие) интерес соединение(ия), извлекали из холодильника и давали ему(им) нагреться до комнатной температуры в течение одного часа. ВЭЖХ-хроматограф готовили, уравновешивали и характеристики системы проверяли путем введения стандарта. Через 1 час каждую из трех лунок разбавляли, используя 266 мкл 0,1 N раствора HCl до рН=1. Планшет закрывали и помещали на качалку (Eppindorf Thermomixer R) на 45 минут при скорости 600 об./мин. Планшет удаляли с качалки и содержимое каждой лунки фильтровали через фильтрующую пластину (№ MSSLBPC50 по каталогу Millipore), используя вакуум, и инжектировали в ВЭЖХ-хроматограф. Приблизительно через 24 час содержимое лунок вновь инжектировали в ВЭЖХ-хроматограф.
Параметры ВЭЖХ-хроматографа (Определение растворимости и стабильности)
В=0,05% TFA в СН3СN
Стабильность в различных буферах определяли путем сравнения площади пика - % анализируемого вещества в метаноле по сравнению с площадью пика - % анализируемого вещества при том же времени удерживания в буфере 0,1 N HCl с контрольный моментом времени инжекции 24 час. Данные о стабильности, приведенные в таблице 2, показали, что соединения 1-4 имеют большую устойчивость при рН 1, чем соединение E7107 в течение периода 24 час.
Таблица 2: Результаты анализов на стабильность
(рН=1, 24 часа)
Биологические анализы
Методика анализа жизнеспособности клеток
Клетки (WiDr и Panc 05.04, полученные из АТСС) высевали в 96-луночные планшеты по 2000 клеток/100 мкл/лунка и инкубировали в течение ночи. Отработанную среду удаляли и добавляли свежую среду, содержащую 9 различных концентраций соединения (100 мкл/лунка), при этом концентрацию в ДМСО исходного раствора соединения доводили до 0,1%. Каждую обработку соединением проводили в виде двух или трех серий для каждой концентрации.
Еще один планшет с высеянными клетками обозначали как планшет нулевого момента времени (Tz), к которому добавили 0,1% ДМСО в среде (100 мкл/лунка), а затем реагент CellTiter-Glo® (Promega Corporation, Мэдисон, Висконсин) (50 мкл/лунка) для измерения АТФ в качестве суррогата жизнеспособности клеток. Среднее значение измерений нескольких лунок данного планшета использовали в качестве Tz.
Обработанные соединениями планшеты инкубировали в течение 72 час при 37°С. Затем добавили реагент CellTiter-Glo® (50 мкл/лунка) и измеряли АТФ. Среднее значение измерения двух или трех серий обработанных соединениями лунок использовали в качестве Ti, а высеянные планшеты с носителем, содержащим 0,1% ДМСО без соединения, использовали в качестве контроля роста (C).
Процент ингибирования роста/Процент жизнеспособности рассчитывали как:
[(Ti-Tz)/(C-Tz)] × 100 для концентраций, где Ti >/= Тz
[(Ti-Tz)/Tz] × 100 для концентраций, где Ti < Tz.
* нулевой момент времени (Tz), контроль роста (С) и контроль роста в присутствии соединения (Ti).
Значения отношения Процент ингибирования роста/Процент жизнеспособности отложили на графике в зависимости от концентрации соединения, чтобы определить Emax.
Ингибирование роста на 50% (GI50) рассчитывали из значения [(Ti-Тz)/(C-Tz)] х 100=50, которое представляет собой концентрацию лекарственного средства, приводящую к 50%-ному уменьшению общего прироста АТФ при контроле роста (C) в процессе обработки соединением.
Методика in vitro анализа сплайсинга (биохимического)
Путем in vitro транскрипции получали содержащую метку биотина пред-мРНК генетической конструкции аденовируса типа 2 с делецией интрона (Ad2) (Berg, M.G., et al. 2012 Mol. Cell Bio., 32(7):1271-83). Ad2 конструкцию, содержащую экзон 1 (41 нуклеотидов), интрон (231 нуклеотидов) и экзон 2 (72 нуклеотидов), конструировали путем синтеза генов и клонировали на участки EcoRI и XbaI вектора pGEM®-3Z (Promega) с помощью Genewiz® (South Plainfield, Нью-Джерси). Затем плазмиду преобразовывали к линейному виду путем XbaI ферментации и очищали. In vitro транскрипцию и очистку транскрибированной пред-мРНК осуществляли с использованием набора для транскрипции MEGAscript® Т7 (InvitrogenTM, Life TechnologiesTM, Гранд-Айленд, штат Нью-Йорк) и набора для очистки транскрипта MEGAclearTM (InvitrogenTM, Life TechnologiesTM, Гранд-Айленд, штат Нью-Йорк), соответственно, следуя инструкциям изготовителя. Отношение biotin-16-UTP (Roche Diagnostics Corporation, Индианаполис, Индиана) к холодному UTP составляло 1:13, что позволяет включать приблизительно две молекулы биотина на одну подвергнутую сплайсингу Ad2 мРНК.
In vitro анализ сплайсинга проводили при 30°C, используя реакционные смеси объемом 25 мкл, содержащие 95 мкг ядерного экстракта HeLa (Promega Corporation, Мэдисон, штат Висконсин), 47 нМ пред-мРНК, 25 единиц ингибитора РНКазы Rnasin (Promega Corporation, Мэдисон, штат Висконсин), буфер 1X SP (0,5 мМ АТФ, 20 мМ фосфата креатина, 1,6 мМ MgCl2) и соединения в ДМСО (с конечной концентрацией 1% в ДМСО). После 90-минутной инкубации реакцию прекращали добавлением 18 мкл 5 М раствора NaCl и смеси инкубировали вместе с 10 мкл магнитных шариков М-280, покрытых стрептавидином (InvitrogenTM, Life TechnologiesTM, Гранд-Айленд, штат Нью-Йорк), в течение 30 мин при комнатной температуре, чтобы связать Ad2, пред-мРНК и сплайсированные мРНК. Шарики дважды промывали, используя 100 мкл буфера, содержащего 10 мМ Tris с рН=7,5, 1 мМ ЭДТК и 2М NaCl, а затем инкубировали в геле загрузочного буфера РНК, содержащем 95%-ный формамид, при 70°C в течение 10 мин, чтобы элюировать РНК. Ad2 РНК расщепляли с помощью 6%-ного геля ТВЕ-UREA, переносили на УФ-сшитую нейлоновую мембрану и испытывали содержащим метку IRDye® стрептавидином (LI-COR, Линкольн, штат Небраска). Количество сплайсированных РНК оценивали количественно путем измерения диапазона интенсивности флуоресценции, используя программное обеспечение LI-COR Image Studio.
Результаты
Данные представлены в ниже таблице 3. Emax относится к максимально достижимой ответной реакции на соединение в тестируемом интервале доз, при этом отрицательное значение указывает на клеточную летальность. Большее отрицательное значение Emax указывает на большую клеточную летальность для конкретного соединения. Например, в клетках Panc 05.04 мутантной клеточной линии SF3B1 большее отрицательное значение Emax указывает на то, что Соединение 1 обладает большей клеточной летальностью, чем Соединение 2.
Клетки WiDr-R представляют собой клетки рака толстого кишечника, которые содержат химически индуцированную мутацию R1074H и, как было показано, резистентны к пладиенолиду B с точки зрения ингибирования роста (Yokoi, A., et al., 2011 FEBS Journal, 278: 4870-4880). Контр-скрининг соединений в данном исследовании жизнеспособности с "резистентной" клеточной линией WiDr-R может показать, обладают ли указанные соединения побочным(и) эффектом(ами). Указанные соединения, у которых отсутствует ингибирующая рост активность (GI50) в резистентной клеточной линии WiDr-R, но сохраняется активность в родительской клеточной линии WiDr, заставляют предположить, что действенный механизм модуляции сплайсинга отвечает за ингибирование роста, которое наблюдается в родительской клеточной линии WiDr.
Описанный выше in vitro анализ сплайсинга (IVS) представляет собой биохимический анализ, который контролирует ингибирование сплайсинга в рассмотренных в примере пред-мРНК в мРНК. Данный биохимический анализ позволяет исследователям оценить, при какой концентрации соединения сплайсинг конкретного транскрипта ингибируется во внеклеточном окружении, и используется для демонстрации механизмов ингибирующей сплайсинг активности.
Таблица 3: Биологическая активность Соединений 1, 2, 3 и 4
Условные обозначения:
Клетки Panc 05.04: клетки рака поджелудочной железы, мутантная клеточная линия SF3B1 (мутации Q699H и K700E в SF3B1)
Клетки WiDr: клетки рака толстого кишечника (дикий тип SF3B1)
Клетки WiDr-R: клетки рака толстого кишечника (химически индуцированный мутант SF3B1, который устойчив к E7107 (мутация R1074H))
Дополнительные испытания соединений
Фармакокинетическое (PK) исследование на мышах
Соединение 2 вводили мышам CD-1 внутривенно (IV) с дозой 5 мг/кг или с дозой 10 мг/кг перорально (PO). После введения отбирали образцы крови в заранее определенные моменты времени от пяти мышей путем периодических кровопусканий из хвостовой вены. Кровь собирали через 0,083 (0,167 только перорально), 0,5, 1, 2, 4, 6, 8 и 24 час после введения. Образцы крови центрифугировали со скоростью 5000 об./мин в течение 5 мин, чтобы собрать плазму, в пределах 30 мин после отбора крови. После экстракции образцы анализировали методом LC/MS. Параметры PK рассчитывали путем некомпартментного анализа с применением WinNonlin v6.3.
Полученные данные показывают, что Соединение 2 обладает биодоступность при пероральном введении и благоприятными фармакокинетическими свойствами в мышиной модели (Фиг. 1, таблица 4).
Таблица 4
Мышиная модель ксенотрансплантата
Эффективность Соединения 2 исследовали в мышиной модели ксенотрансплантата. Изогенные клетки Nalm-6 SF3B1K700E (линия пред-В-клеток человека, 10*106 клеток) имплантировали подкожно в бок самкам мышей СВ17-SCID. Мышам назначали Соединение 2 (10% этанола, 5% TWEEN-80, 85% физиологического раствора) или контрольный носитель. Животным вводили ежедневно перорально в течение 14 дней (QDx14PO) дозу, указанную на фиг. 2, и осуществляли контроль до тех пор, пока они не достигли какой-либо из следующих конечных точек: 1) чрезмерный объем опухоли, который измеряли три раза в неделю (объем опухоли рассчитывается с использованием формулы эллипсоида: (длина × ширина2)/2); или 2) возникновение любых проблем со здоровьем, таких как паралич или чрезмерная потеря массы тела. Все исследования на животных были проведены в соответствии с руководством H3 Biomedicine Guide for the Care and Use of Laboratory Animals.
Результаты показывают, что Соединение 2 эффективно при введении пероральным путем и демонстрирует сокращение роста опухоли в мышиной модели ксенотрансплантата (Фиг. 2).
PK/PD исследование в мышиной модели ксенотрансплантата
Фармакокинетику (PK)/фармакодинамику (PD) Соединения 2 также анализировали в модели ксенотрансплантата мышей Nalm-6. Изогенные клетки Nalm-6 SF3B1K700E (линия пред-В-клеток человека, 10*106 клеток) имплантировали подкожно в бок самкам мышей СВ17-SCID. Мышам вводили разовую оральную дозу Соединения 1 (10% этанола, 5% TWEEN-80, 85% физиологического раствора), и опухоли отбирали в указанные промежутки времени после введения для проведения анализа.
РНК выделяли, используя набор RiboPure™ для очистки РНК (Ambion®) и использовали для анализа КПЦР. РНК ретротранскрибировали в соответствии с инструкциями, которые прилагаются к набору SuperScript® VILO™ (Invitrogen™) для синтеза кДНК, и 0,04 мкл кДНК использовали для количественной ПЦР (КПЦР). КПЦР для пред-мРНК EIF4A1 и зрелой мРНК SLC24A19 и оценку PK проводили, как сообщалось ранее (Eskens, F. A. et al. Phase I pharmacokinetic and pharmacodynamic study of the first-in-class spliceosome inhibitor E7107 in patients with advanced solid tumors. Clin. Cancer Res. 19, 6296-6304, doi:10.1158/1078-0432.CCR-13-0485 (2013)). Все исследования на животных были проведены в соответствии с руководством H3 Biomedicine Guide for the Care and Use of Laboratory Animals.
Результаты, приведенные на Фиг. 3, показывают, что Соединение 2 демонстрирует PD ответные реакции для переносимой дозы при пероральном пути введения.
Анализ жизнеспособности клеток
Для оценки жизнеспособности Panc 05.04 раковых клеток (SF3B1MUT) (мутации Q699H и K700E в SF3B1) в присутствии Соединения 2 клетки высевали в количестве 750 клеток на лунку в 384-луночный планшет и обрабатывали Соединением 2 с концентрациями, указанными на Фиг. 4, в течение 72 час при температуре 37°С. Относительное количество жизнеспособных или апоптотических клеток измеряли, используя Celltiter-GLO® для проведения люминесцентного анализа жизнеспособности клеток (Promega).
Результаты указывают на дифференцированную клеточную летальность в мутантной клеточной линии SF3B1 рака поджелудочной железы, по сравнению с клеточной линией SF3B1 рака поджелудочной железы дикого типа (Фиг. 4).
Сравнение альтернативного сплайсинга для E7107 и Соединения 2
Регулирование альтернативного сплайсинга для E7107 и Соединения 2 определяли с использованием системы анализа nCounter® (NanoString Techologies, Inc., Сиэтл, штат Вашингтон). Изогенные клетки Nalm-6 обрабатывали Соединением 2 или E7107 (получали от Eisai, Inc.) при 10 х GI50 в течение 6 час. РНК выделяли с помощью набора RiboPure™ для очистки РНК (Ambion®) и использовали для анализа. РНК ретротранскрибировали в соответствии с инструкциями, прилагаемыми к набору SuperScript® VILO™ для синтеза кДНК (Invitrogen™), и 0,04 мкл кДНК использовали для проведения кПЦР.
Результаты, приведенные на Фиг. 5, показывают, что профиль регулирования сплайсинга для Соединения 2 отличается от профиля E7107.
Фармакокинетические (PK) исследования Соединения 1 на мышах
Соединение 1 вводили мышам CD-1 с дозой 5 мг/кг внутривенно или 12 мг/кг перорально. После введения отбирали образцы крови в заранее определенные моменты времени от пяти мышей путем периодических кровопусканий из хвостовой вены. Кровь собирали через 0,083 (0,167 только перорально), 0,5, 1, 2, 4, 6, 8 и 24 час после введения. Образцы крови центрифугировали со скоростью 5000 об./мин в течение 5 мин, чтобы собрать плазму, в пределах 30 мин после отбора крови. После экстракции образцы анализировали методом LC/MS. Параметры PK рассчитывали путем некомпартментного анализа с применением WinNonlin v6.3.
Полученные данные показывают, что Соединение 1 обладает биодоступностью при пероральном введении и благоприятными фармакокинетическими свойствами в мышиной модели (Фиг. 6, таблица 5).
Таблица 5
Эффективность соединения 1 в мышиной модели ксенотрансплантата
Эффективность Соединения 1 исследовали в мышиной модели ксенотрансплантата. Изогенные клетки Nalm-6 SF3B1K700E (линия пред-В-клеток человека, 10*106 клеток) имплантировали подкожно в бок самкам мышей СВ17-SCID. Мышам назначали Соединение 1 (10% этанола, 5% TWEEN-80, 85% физиологического раствора) или контрольный носитель. Животным вводили ежедневно перорально в течение 14 дней (QDx14PO) дозу 7,5 мг/кг или 10 мг/кг Соединения 1 и осуществляли контроль до тех, пока они не достигли какой-либо из следующих конечных точек: 1) чрезмерный объем опухоли, который измеряли три раза в неделю (объем опухоли рассчитывали с использованием формулы эллипсоида: (длина × ширина2)/2); или 2) возникновение любых проблем со здоровьем, таких как паралич или чрезмерная потеря массы тела. Все исследования на животных были проведены в соответствии с руководством H3 Biomedicine Guide for the Care and Use of Laboratory Animals.
Результаты показывают, что Соединение 1 эффективно при введении пероральным путем и демонстрирует сокращение роста опухоли в мышиной модели ксенотрансплантата (Фиг. 7).
PK/PD исследование Соединения 1 в мышиной модели ксенотрансплантата
Фармакокинетику (PK)/фармакодинамику (PD) Соединения 1 также анализировали в модели ксенотрансплантата мышей Nalm-6. Изогенные клетки Nalm-6 SF3B1K700E (линия пред-В-клеток человека, 10*106 клеток) имплантировали подкожно в бок самкам мышей СВ17-SCID. Мышам вводили разовую оральную дозу 10 мг/кг Соединения 2 (10% этанола, 5% TWEEN-80, 85% физиологического раствора), и опухоли отбирали в указанные промежутки времени после введения для проведения анализа.
РНК выделяли, используя набор RiboPure™ для очистки РНК (Ambion®) и использовали для анализа методом КПЦР. РНК ретротранскрибировали в соответствии с инструкциями, которые прилагаются к набору SuperScript® VILO™ (Invitrogen™), для синтеза кДНК, и 0,04 мкл кДНК использовали для количественной ПЦР (КПЦР). КПЦР для пред-мРНК EIF4A1 и зрелой мРНК SLC24A19 и оценку PK проводили, как сообщалось ранее (Eskens, F. A. et al. Phase I pharmacokinetic and pharmacodynamic study of the first-in-class spliceosome inhibitor E7107 in patients with advanced solid tumors. Clin. Cancer Res. 19, 6296-6304, doi:10.1158/1078-0432.CCR-13-0485 (2013)). Все исследования на животных были проведены в соответствии с руководством H3 Biomedicine Guide for the Care and Use of Laboratory Animals.
Результаты, приведенные на Фиг. 8, показывают, что Соединение 1 демонстрирует PD ответные реакции для переносимой дозы при пероральном пути введения.
Фармакокинетические (PK) исследования Соединения 3 на мышах
Соединение 3 вводили мышам CD-1 с дозой 5,964 мг/кг внутривенно или 13,307 мг/кг перорально. После введения отбирали образцы крови в заранее определенные моменты времени от пяти мышей путем периодических кровопусканий из хвостовой вены. Кровь собирали через 0,083 (0,167 только перорально), 0,5, 1, 2, 4, 6, 8 и 24 час после введения. Образцы крови центрифугировали со скоростью 5000 об./мин в течение 5 мин, чтобы собрать плазму, в пределах 30 мин после отбора крови. После экстракции образцы анализировали методом LC/MS. Параметры PK рассчитывали путем некомпартментного анализа с применением WinNonlin v6.3.
Полученные данные показывают, что Соединение 3 обладает биодоступностью при пероральном введении и благоприятными фармакокинетическими свойствами в мышиной модели (Фиг. 9, таблица 6).
Таблица 6
Фармакокинетические (PK) исследования Соединения 4 на мышах
Соединение 4 вводили мышам CD-1 с дозой 5 мг/кг внутривенно или 10 мг/кг перорально. После введения отбирали образцы крови в заранее определенные моменты времени от пяти мышей путем периодических кровопусканий из хвостовой вены. Кровь собирали через 0,083 (0,167 только перорально), 0,5, 1, 2, 4, 6, 8 и 24 час после введения. Образцы крови центрифугировали со скоростью 5000 об./мин в течение 5 мин, чтобы собрать плазму, в пределах 30 мин после отбора крови. После экстракции образцы анализировали методом LC/MS. Параметры PK рассчитывали путем некомпартментного анализа с применением WinNonlin v6.3.
Полученные данные показывают, что Соединение 4 обладает биодоступностью при пероральном введении и благоприятными фармакокинетическими свойствами в мышиной модели (Фиг. 10, таблица 7).
Таблица 7
Приведенные выше результаты показывают, что каждое из Соединений 1, 2, 3 и 4 обладает биодоступностью при пероральном введении и благоприятными фармакокинетическими свойствами. Указанные свойства улучшены, по сравнению с E7107, который вводят пациентам в виде внутривенного вливания вследствие его недостаточной биодоступности при пероральном введении (Hong et al. (2014), Invest. New Drugs 32, 436-444).
| название | год | авторы | номер документа |
|---|---|---|---|
| ПИРИДИНОВЫЕ СОЕДИНЕНИЯ ПЛАДИЕНОЛИДА И СПОСОБЫ ПРИМЕНЕНИЯ | 2015 |
|
RU2807278C2 |
| КОМБИНАЦИЯ, ВКЛЮЧАЮЩАЯ ПО МЕНЬШЕЙ МЕРЕ ОДИН МОДУЛЯТОР СПЛАЙСОСОМЫ И ПО МЕНЬШЕЙ МЕРЕ ОДИН ИНГИБИТОР, ВЫБРАННЫЙ ИЗ ИНГИБИТОРОВ BCL2, ИНГИБИТОРОВ BCL2/BCLXL И ИНГИБИТОРОВ BCLXL, А ТАКЖЕ СПОСОБЫ ПРИМЕНЕНИЯ | 2018 |
|
RU2783239C2 |
| ТВЕРДЫЕ ФОРМЫ ПЛАДИЕНОЛИДПИРИДИНОВЫХ СОЕДИНЕНИЙ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2743349C2 |
| ОПРЕДЕЛЕННЫЕ СОЕДИНЕНИЯ ПЛАДИЕНОЛИДА И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2019 |
|
RU2813597C2 |
| ПРОИЗВОДНЫЕ ПЛАДИЕНОЛИДА В КАЧЕСТВЕ СРЕДСТВ, ОКАЗЫВАЮЩИХ ЦЕЛЕНАПРАВЛЕННОЕ ВОЗДЕЙСТВИЕ НА СПЛАЙСОСОМУ, ДЛЯ ЛЕЧЕНИЯ РАКА | 2019 |
|
RU2815064C2 |
| КОНЪЮГАТЫ АНТИТЕЛА И ЛЕКАРСТВЕННОГО СРЕДСТВА В КАЧЕСТВЕ МОДУЛЯТОРА СПЛАЙСИНГА И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2019 |
|
RU2799438C2 |
| СПОСОБ ЛЕЧЕНИЯ СОЛИДНЫХ ЗЛОКАЧЕСТВЕННЫХ ОПУХОЛЕЙ МЛЕКОПИТАЮЩИХ | 2024 |
|
RU2841088C2 |
| Производные 2H-[1,4]оксазино- и 2H-[1,4]тиазино[3,2-c]хинолин-3(4H)-онов в качестве ингибиторов ATM и DNA-PK киназ для лечения и/или профилактики онкологических заболеваний | 2024 |
|
RU2835676C1 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ МОДУЛЯТОРЫ СИНТЕЗА ЛИПИДОВ ДЛЯ ПРИМЕНЕНИЯ ПРОТИВ РАКА И ВИРУСНЫХ ИНФЕКЦИЙ | 2015 |
|
RU2766087C2 |
| ДИАРИЛТИОГИДАНТОИНОВЫЕ СОЕДИНЕНИЯ | 2007 |
|
RU2449993C2 |
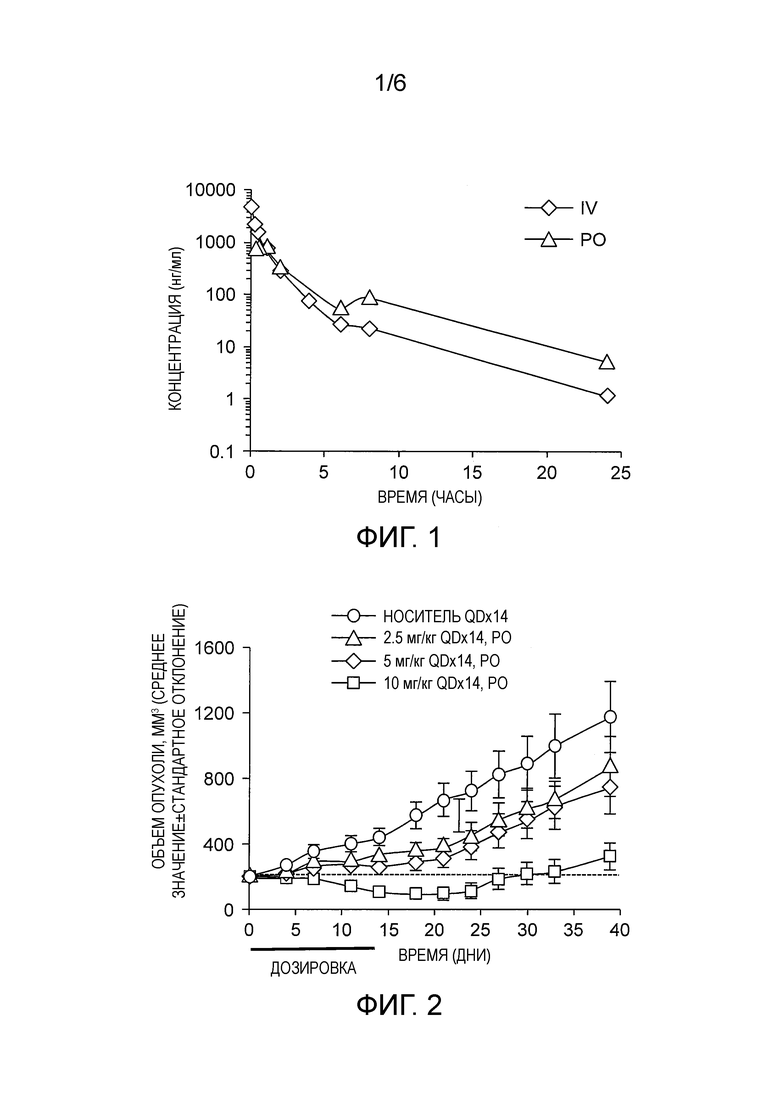

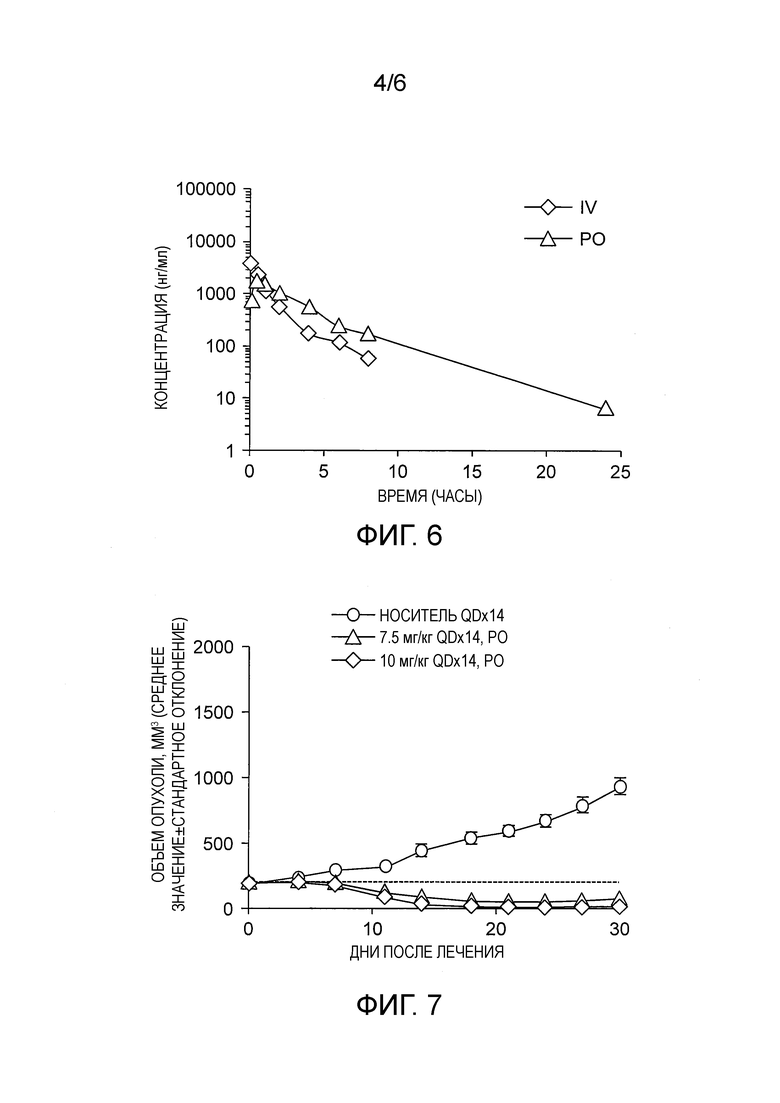
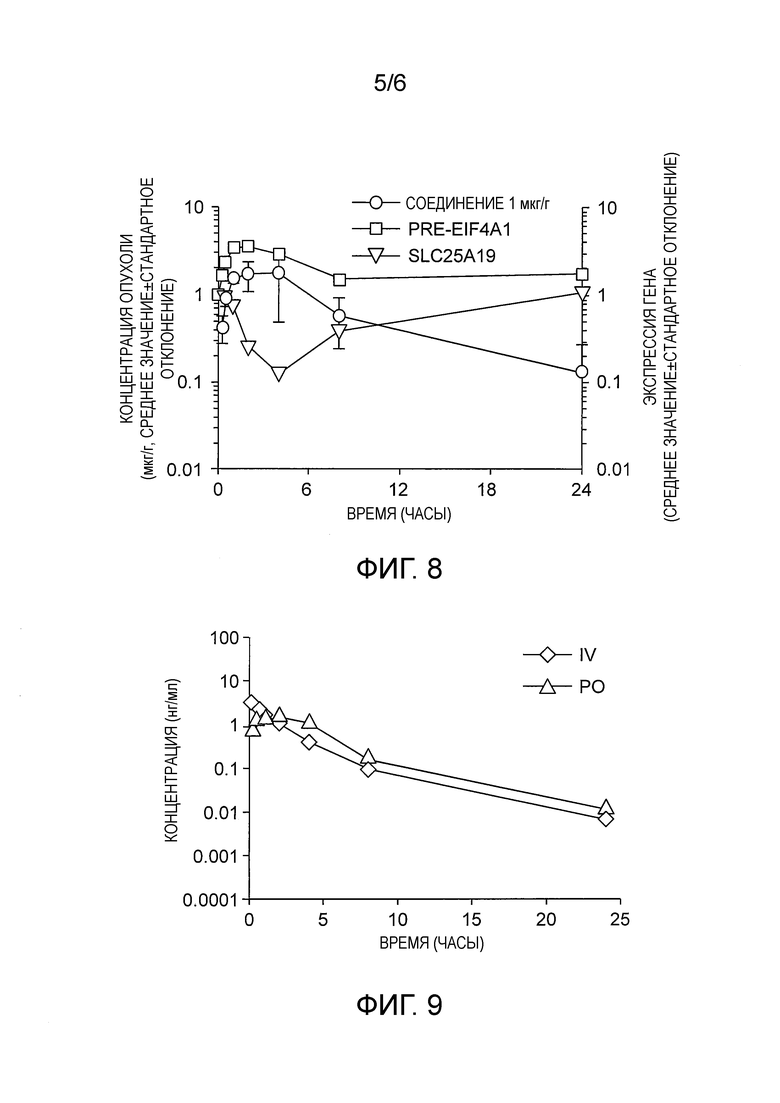
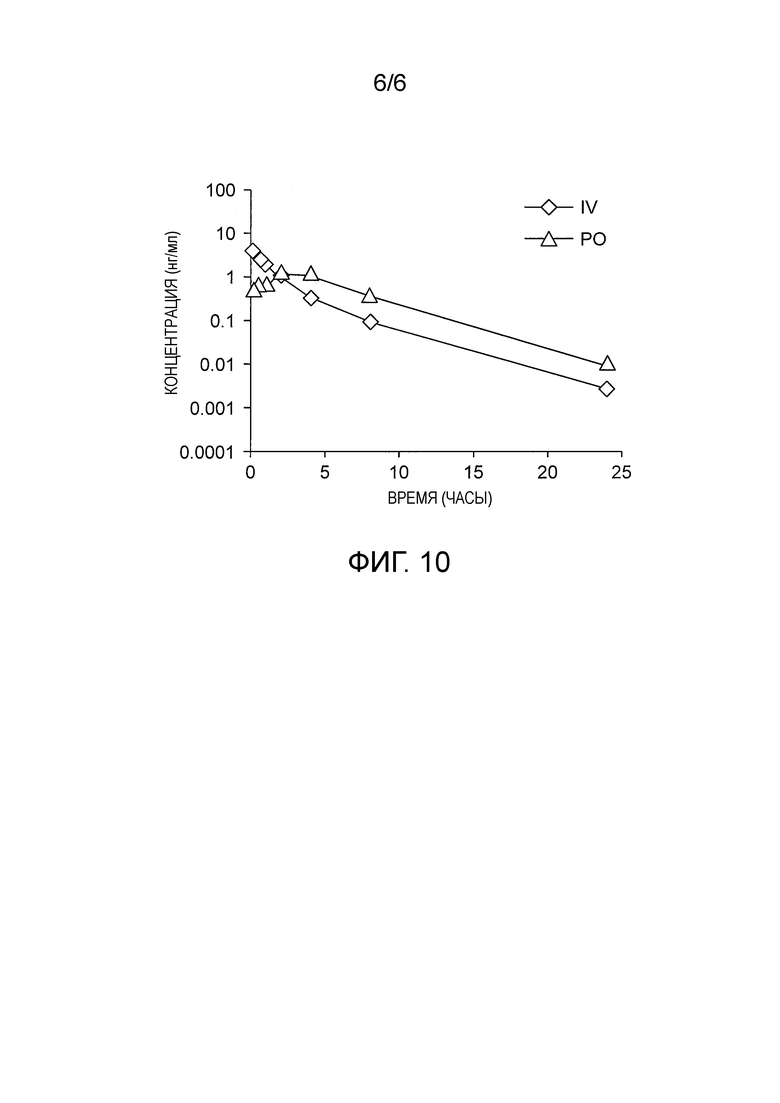
Изобретение относится к пиридиновым соединениям пладиенолида формул 1-4, к фармацевтическим композициям, содержащим подобные соединения, и к способам применения указанных соединений в качестве терапевтических агентов. Технический результат – получены новые соединения и фармацевтические композиции на их основе, которые могут найти применение в медицине для лечения онкологических заболеваний, в частности, при которых пригодны агенты, нацеливающиеся на сплайсосому и ее мутации. 9 н. и 44 з.п. ф-лы, 7 табл., 10 ил., 1 пр.
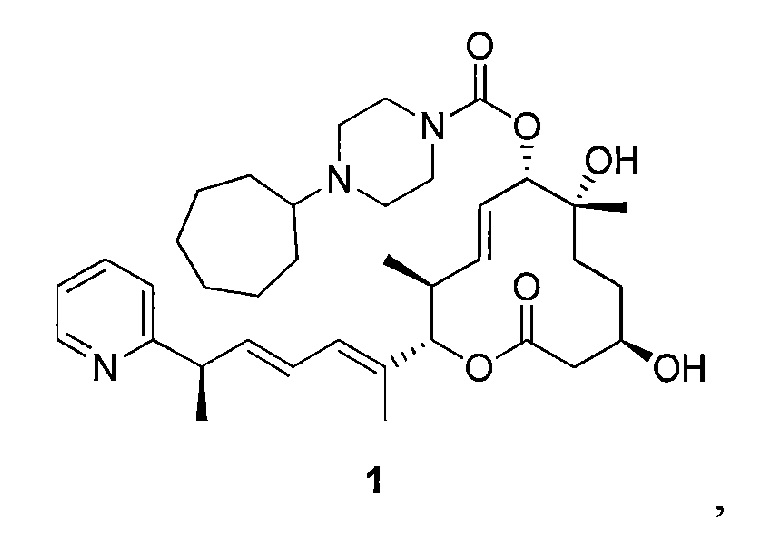
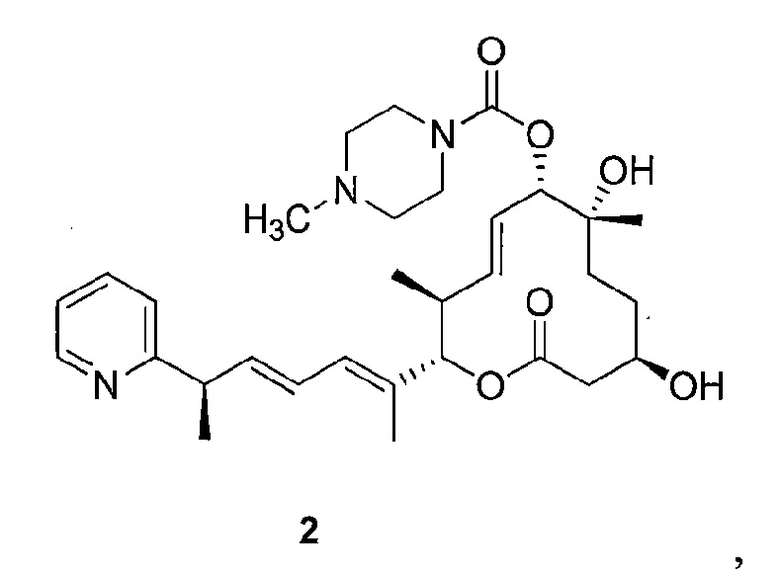
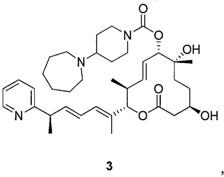
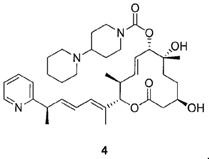
1. Соединение, которое выбрано из:
соединения формулы 1

соединения формулы 2
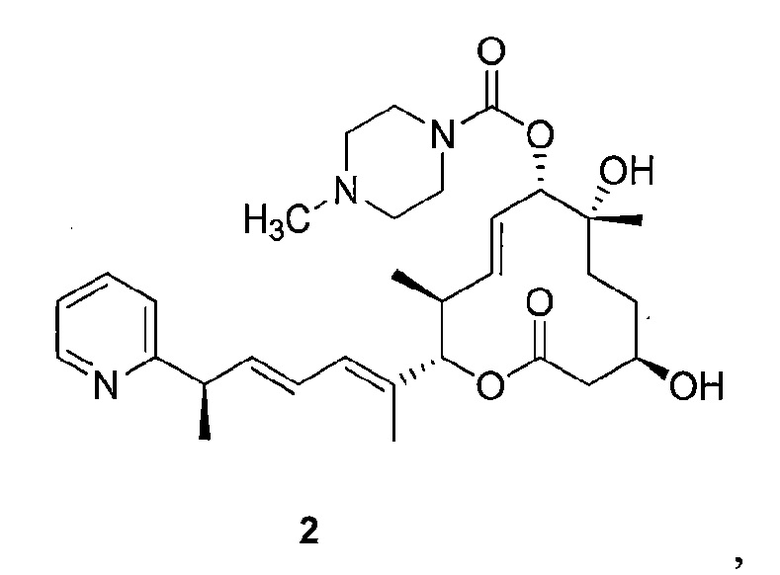
соединения формулы 3
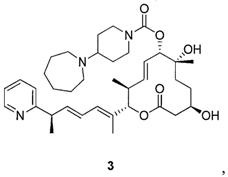
соединения формулы 4
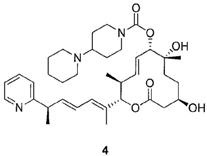
и фармацевтически приемлемых солей этих соединений.
2. Соединение, которое выбрано из соединения формулы 1
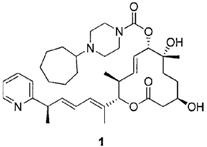
и его фармацевтически приемлемых солей.
3. Соединение, которое выбрано из соединения формулы 2

и его фармацевтически приемлемых солей.
4. Соединение, которое выбрано из соединения формулы 3
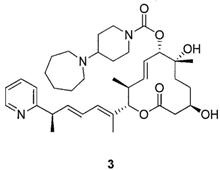
и его фармацевтически приемлемых солей.
5. Соединение, которое выбрано из соединения формулы 4
и его фармацевтически приемлемых солей.
6. Соединение по п.1, которое содержит больше чем приблизительно 80 мас.% одного стереоизомера указанного соединения.
7. Соединение по п.1, которое содержит больше чем приблизительно 90 мас.% одного стереоизомера указанного соединения.
8. Соединение по п.1, которое содержит больше чем приблизительно 95 мас.% одного стереоизомера указанного соединения.
9. Соединение по п.1, которое содержит больше чем приблизительно 97 мас.% одного стереоизомера указанного соединения.
10. Фармацевтическая композиция, содержащая эффективное количество соединения и/или его фармацевтически приемлемой соли по любому из пп. 1-9, предназначенная для лечения рака, выбранного из миелодиспластического синдрома, хронического лимфоцитарного лейкоза, острого лимфобластного лейкоза, хронического миеломоноцитарного лейкоза, острого миелоидного лейкоза, рака толстого кишечника, рака поджелудочной железы, рака эндометрия, рака яичников, рака молочной железы, увеальной меланомы, рака желудка, холангиокарциномы и рака легкого, где указанная композиция дополнительно содержит один или несколько фармацевтически приемлемых носителей.
11. Фармацевтическая композиция по п. 10, где указанная композиция составлена для внутривенного, перорального, подкожного или внутримышечного введения.
12. Фармацевтическая композиция по п. 11, где указанная композиция составлена для перорального введения.
13. Применение соединения или его фармацевтически приемлемой соли по любому из пп. 1-9 или фармацевтической композиции по любому из пп. 10-12 для изготовления лекарственного средства, которое предназначено для лечения рака, выбранного из миелодиспластического синдрома, хронического лимфоцитарного лейкоза, острого лимфобластного лейкоза, хронического миеломоноцитарного лейкоза, острого миелоидного лейкоза, рака толстого кишечника, рака поджелудочной железы, рака эндометрия, рака яичников, рака молочной железы, увеальной меланомы, рака желудка, холангиокарциномы и рака легкого.
14. Применение по п. 13, где указанное лекарственное средство предназначено для лечения рака толстого кишечника.
15. Применение по п. 13, где указанное лекарственное средство предназначено для лечения рака поджелудочной железы.
16. Применение по п. 13, где указанный рак выбран из миелодиспластического синдрома, хронического лимфоцитарного лейкоза, острого лимфобластного лейкоза, хронического миеломоноцитарного лейкоза и острого миелоидного лейкоза.
17. Применение по п.13, где рак представляет собой острый миелоидный лейкоз.
18. Применение по п.13, где рак представляет собой миелодиспластический синдром.
19. Применение по п.13, где рак представляет собой хронический лимфоцитарный лейкоз.
20. Применение по п.13, где рак представляет собой острый лимфобластный лейкоз.
21. Применение по п.13, где рак представляет собой миеломоноцитарный лейкоз.
22. Применение по п.13, где рак представляет собой рак эндометрия.
23. Применение по п.13, где рак представляет собой рак яичников.
24. Применение по п.13, где рак представляет собой рак молочной железы.
25. Применение по п.13, где рак представляет собой увеальную меланому.
26. Применение по п.13, где рак представляет собой рак желудка.
27. Применение по п.13, где рак представляет собой холангиокарциному.
28. Применение по п.13, где рак представляет собой рак легкого.
29. Применение по любому из пп. 13-28, где указанный рак позитивен для одной или нескольких мутаций в гене сплайсосомы или белке.
30. Применение по п.29, где указанный ген сплайсосомы или белок выбраны из субъединицы 1 фактора сплайсинга 3B (SF3B1), вспомогательного фактора 1 U2 малой РНК (U2AF1), обогащенного серином/аргинином фактора сплайсинга 2 (SRSF2), цинксодержащей кольцеобразной области в ДНК (тип CCCH), РНК-связывающего фрагмента и обогащенного серином/аргинином 2 (ZRSR2), фактора процессинга-сплайсинга 8 пред-мРНК (PRPF8), вспомогательного фактора 2 U2 малой РНК (U2AF2), фактора сплайсинга 1 (SF1), субъединицы 1 фактора сплайсинга 3a (SF3A1), PRP40 гомолога B фактора процессинга 40 пред-мРНК (PRPF40B), белка 10 связывающего фрагмента РНК (RBM10), связывающего белка 1 поли(rC) (PCBP1), фактора сплайсинга 1 искривленной пред-мРНК (CRNKL1), DEAH (Asp-Glu-Ala-His) бокс-геликаза 9 (DHX9), цис-транс изомеразоподобного 2 пептидил-пролила (PPIL2), белка 22 связывающего фрагмента РНК (RBM22), малого ядерного рибонуклеопротеида Sm D3 (SNRPD3), вероятной АТФ-зависимой РНК-геликазы DDX5 (DDX5), фактора сплайсинга АТФ-зависимой РНК-геликазы DHX15пред-мРНК (DHX15) и полиаденилат-связывающего белка 1 (PABPC1).
31. Применение по п. 30, где указанным геном сплайсосомы или белком является субъединица 1 фактора сплайсинга 3B(SF3B1).
32. Фармацевтическая композиция, включающая эффективное количество соединения формулы 2
и его фармацевтически приемлемых солей, предназначенная для лечения рака, выбранного из миелодиспластического синдрома, хронического лимфоцитарного лейкоза, острого лимфобластного лейкоза, хронического миеломоноцитарного лейкоза, острого миелоидного лейкоза, рака толстого кишечника, рака поджелудочной железы, рака эндометрия, рака яичников, рака молочной железы, увеальной меланомы, рака желудка, холангиокарциномы и рака легкого, где указанная композиция дополнительно содержит один или несколько фармацевтически приемлемых носителей.
33. Фармацевтическая композиция по п. 32, где указанная композиция составлена для внутривенного, перорального, подкожного или внутримышечного введения.
34. Фармацевтическая композиция по п. 33, где указанная композиция составлена для перорального введения.
35. Применение фармацевтической композиции по любому из пп. 32-34 для приготовления лекарственного средства, предназначеного для лечения рака, выбранного из миелодиспластического синдрома, хронического лимфоцитарного лейкоза, острого лимфобластного лейкоза, хронического миеломоноцитарного лейкоза, острого миелоидного лейкоза, рака толстого кишечника, рака поджелудочной железы, рака эндометрия, рака яичников, рака молочной железы, увеальной меланомы, рака желудка, холангиокарциномы и рака легкого.
36. Применение по п.35, где рак представляет собой рак толстого кишечника.
37. Применение по п. 35, где рак представляет собой рак поджелудочной железы.
38. Применение по п. 35, где рак выбран из миелодиспластического синдрома, хронического лимфоцитарного лейкоза, острого лимфобластного лейкоза, хронического миеломоноцитарного лейкоза и острого миелоидного лейкоза.
39. Применение по п.35, где рак представляет собой острый миелоидный лейкоз.
40. Применение по п.35, где рак представляет собой миелодиспластический синдром.
41. Применение по п.35, где рак представляет собой хронический лимфоцитарный лейкоз.
42. Применение по п.35, где рак представляет собой острый лимфобластный лейкоз.
43. Применение по п.35, где рак представляет собой миеломоноцитарный лейкоз.
44. Применение по п.35, где рак представляет собой рак эндометрия.
45. Применение по п.35, где рак представляет собой рак яичников.
46. Применение по п.35, где рак представляет собой рак молочной железы.
47. Применение по п.35, где рак представляет собой увеальную меланому.
48. Применение по п.35, где рак представляет собой рак желудка.
49. Применение по п.35, где рак представляет собой холангиокарциному.
50. Применение по п.35, где рак представляет собой рак легкого.
51. Применение по любому из пп. 35-50, где указанный рак позитивен для одной или нескольких мутаций в гене сплайсосомы или белке.
52. Применение по п.51, где указанный ген сплайсосомы или белок выбраны из субъединицы 1 фактора сплайсинга 3B (SF3B1), вспомогательного фактора 1 U2 малой РНК (U2AF1), обогащенного серином/аргинином фактора сплайсинга 2 (SRSF2), цинксодержащей кольцеобразной области в ДНК (тип CCCH), РНК-связывающего фрагмента и обогащенного серином/аргинином 2 (ZRSR2), фактора процессинга-сплайсинга 8 пред-мРНК (PRPF8), вспомогательного фактора 2 U2 малой РНК (U2AF2), фактора сплайсинга 1 (SF1), субъединицы 1 фактора сплайсинга 3a (SF3A1), PRP40 гомолога B фактора процессинга 40 пред-мРНК (PRPF40B), белка 10 связывающего фрагмента РНК (RBM10), связывающего белка 1 поли(rC) (PCBP1), фактора сплайсинга 1 искривленной пред-мРНК (CRNKL1), DEAH (Asp-Glu-Ala-His) бокс-геликаза 9 (DHX9), цис-транс изомеразоподобного 2 пептидил-пролила (PPIL2), белка 22 связывающего фрагмента РНК (RBM22), малого ядерного рибонуклеопротеида Sm D3 (SNRPD3), вероятной АТФ-зависимой РНК-геликазы DDX5 (DDX5), фактора сплайсинга АТФ-зависимой РНК-геликазы DHX15пред-мРНК (DHX15) и полиаденилат-связывающего белка 1 (PABPC1).
53. Применение по п. 52, где указанный ген сплайсосомы или белок представляет собой субъединицу 1 фактора сплайсинга 3B (SF3B1).
| EP 1380579 A4, 14.09.2005 | |||
| СПОСОБ ВЫЯВЛЕНИЯ ПОСТУРАЛЬНОГО ДИСБАЛАНСА | 1997 |
|
RU2136209C1 |
| RU 97105695 A, 27.04.1999 | |||
| ГЕНЕТИЧЕСКИ МОДИФИЦИРОВАННЫЙ МИКРООРГАНИЗМ И СПОСОБ ПОЛУЧЕНИЯ МАКРОЛИДНОГО СОЕДИНЕНИЯ С ГИДРОКСИЛЬНОЙ ГРУППОЙ В 16-ПОЛОЖЕНИИ С ИСПОЛЬЗОВАНИЕМ ТАКИХ МИКРООРГАНИЗМОВ | 2006 |
|
RU2394906C2 |

