Настоящее изобретение относится к новым кахалалидным противоопухолевым соединениям, в особенности к аналогам кахалалида F, содержащим их фармацевтическим композициям и к их применению в качестве противоопухолевых, противовирусных, противогрибковых агентов и при лечении псориаза.
Кахалалидные соединения являются пептидами, выделенными из гавайских травоядных морских видов моллюсков, Elysia rufescens, и их пищи, зеленых водорослей Bryopsis sp. Кахалалид F описан Hamann М. и др., J. Am. Chem. Soc., 1993, 115, 5825-5826.
Кахалалиды A-G описаны Hamann М. и др., J. Org. Chem., 1996, 61, 6594-6600: "Kahalalides: bioactive peptides from a marine mollusk Elysia rufescens and its algal diet Bryopsis sp.".
Кахалалиды Н и J описаны Scheuer P.J. и др., J. Nat. Prod., 1997, 60, 562-567: "Two acyclic kahalalides from the sacoglossan mollusk Elysia rufescens".
Кахалалид О описан Scheuer P.J. и др., J. Nat. Prod., 2000, 63(1), 152-4: "A new depsipeptide from the sacoglossan mollusk Elysia ornata and the green alga Bryopsis species".
В отношении кахалалида К см. Kan Y. и др., J. Nat. Prod., 1999, 62(8), 1169-72: "Kahalalide K: A new cyclic depsipeptide from the hawaiian green alga Bryopsis species".
В отношении связанных с этим сообщений также см. Goetz и др., Tetrahedron, 1999, 55, 7739-7746: "The absolute stereochemistry of Kahalalide F"; Albericio F. и др., Tetrahedron Letters, 2000, 41, 9765-9769: “Kahalalide B. Synthesis of a natural cyclodepsipeptide”; Becerro и др., J. Chem. Ecol., 2001, 27(11), 2287-99: “Chemical defenses of the sarcoglossan mollusk Elysia rufescens and its host Alga bryopsis sp.”.
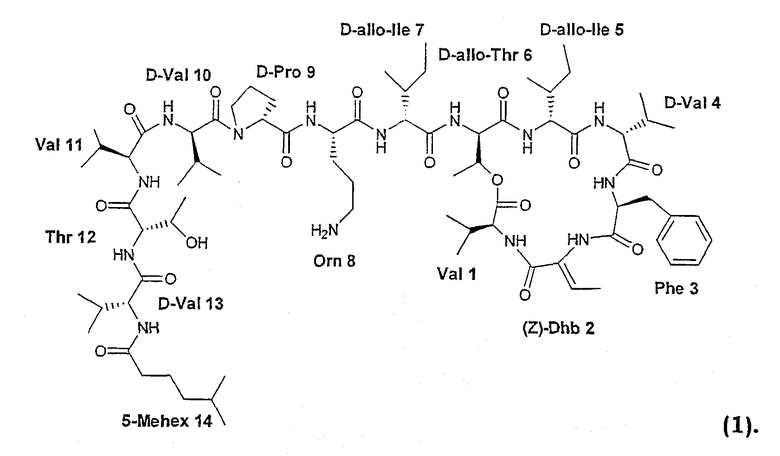
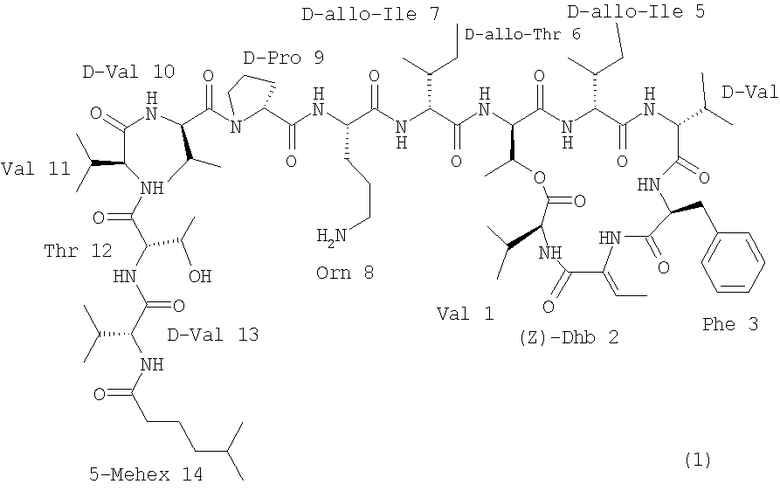
Из кахалалидных соединений кахалалид F является наиболее перспективным из-за своей противоопухолевой активности. У него сложная структура, включающая шесть аминокислот в качестве циклической части и экзоциклическую цепь из семи аминокислот с концевой группой жирной кислоты. Первоначально кахалалид F отвечает структуре (I):
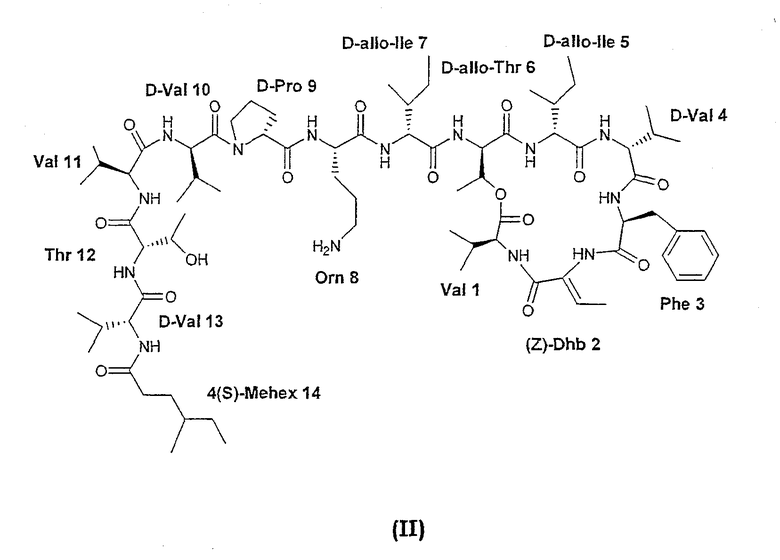
В WO 04035613 описано 4(S)-метилгексильное соединение следующей формулы (II). WO 04035613 полностью включена в данный контекст путем ссылки.
Активность кахалалида F против in vitro клеточных культур карциномы легкого человека А-549 и карциномы ободочной кишки человека НТ-29 описана в ЕР 610078. Кахалалид F также проявляет наличие противовирусных и противогрибковых свойств, а также пригоден при лечении псориаза.
В WO 0236145 описываются фармацевтические композиции, содержащие кахалалид F, новые применения этого соединения в раковой терапии, и ее содержание полностью включено в данный контекст путем ссылки.
В WO 0333012 описывается клиническое применение кахалалидных соединений в онкологии, и ее содержание полностью включено в данный контекст путем ссылки.
Синтез и цитотоксические активности природных и синтетических кахалалидных соединений описаны в WO 0158934, которая полностью включена в данный контекст в качестве ссылки. В WO 0158934 описывается синтез кахалалида F и также соединений подобной структуры, в которой концевая цепь жирной кислоты заменена другими жирными кислотами.
Тем не менее, существует потребность в получении дальнейших противоопухолевых соединений, в особенности, дальнейших кахалалидных соединений с улучшенными свойствами.
Краткое изложение сущности изобретения
Получены аналогичные кахалалиду соединения с улучшенной биологической активностью.
Настоящее изобретение относится к соединениям формулы (1):
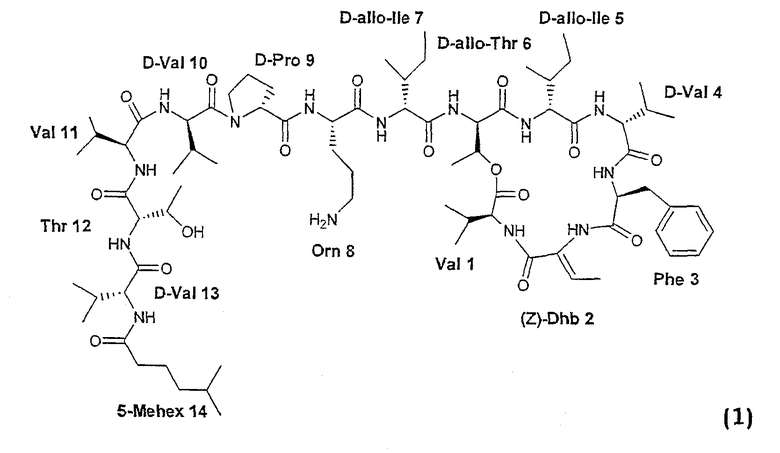
где одна или более аминокислот в циклической или экзоциклической части замещены другими природными или неприродными аминокислотами, защищенными органическими группами, или удалены. Настоящее изобретение также относится к соединениям формулы (1), где алифатическая концевая кислотная группа замещена другими ацильными группами или удалена.
Настоящее изобретение также относится к фармацевтической композиции, содержащей вышеуказанное соединение и фармацевтически приемлемый носитель, растворитель или разбавитель.
Настоящее изобретение далее относится к способу лечения любого млекопитающего, особенно человека, пораженного раком, вирусной инфекцией, грибковой инфекцией или псориазом, который включает введение пораженному индивидууму терапевтически эффективного количества соединения, как описанное выше.
Настоящее изобретение особенно можно применять для лечения пациентов с трудноизлечимыми видами рака, которые не поддаются воздействию других лечений. В частности, композиции согласно данному изобретению можно применять после того, как была испытана и не дала результатов другая химиотерапия.
Настоящее изобретение, в частности, относится к лечению пациентов, пораженных раком простаты, раком молочной железы, гепатоцеллюлярным раком, меланомой, раком ободочной и прямой кишки, раком почки, раком яичников, NSCL-раком, эпителиальным раком, раком поджелудочной железы и опухолями, которые сверхэкспрессирует онкоген Her2/neu.
В другом аспекте настоящее изобретение относится к применению соединения, как описанное выше, для получения лекарственного средства. В предпочтительном воплощении лекарственное средство является лекарственным средством для лечения рака, псориаза, вирусной инфекции или грибковой инфекции.
Данное изобретение дополнительно предусматривает фармацевтический набор, содержащий отдельные емкости, заполненные фармацевтической композицией, включающей вышеописанное соединение и восстановительный агент. Также предусматриваются способы восстановления.
Авторы идентифицировали аналоги кахалалида F, которые показывают значительное улучшение активности по отношению к кахалалиду F.
Настоящее изобретение относится к соединениям формулы (1):
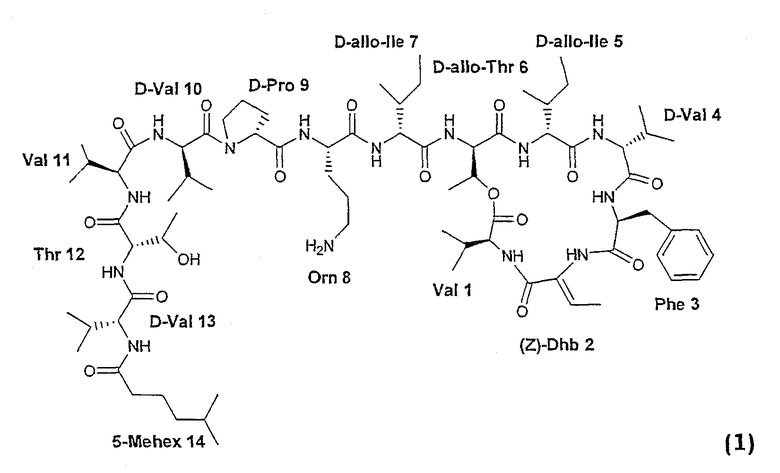
где одна или более экзоциклических или циклических аминокислот замещены другими природными или неприродными аминокислотами, защищенными органическими группами, или удалены. Настоящее изобретение также относится к соединениям формулы (1), где алифатическая метилгексанацильная группа замещена другими ацильными группами или удалена.
Экзоциклические аминокислоты
Предпочтительные соединения согласно данному изобретению отвечают формуле (1), где одна или более аминокислот экзоциклической цепи замещены другими природными или неприродными аминокислотами, защищенными органическими группами, или удалены.
В особенности, предпочтительные соединения включают таковые с заменой 1, 2 или 3 аминокислот в экзоциклической цепи и таковые с удаленными 1, 2, 3, 4, 5 или 6 аминокислотами в экзоциклической цепи.
Особенно предпочтительными являются такие соединения формулы (1), где одна экзоциклическая аминокислота замещена другой природной или неприродной аминокислотой и/или защищена одной или более замещающими органическими группами. Это особенно касается аминокислоты L-Orn в позиции 8.
Таким образом, в одном аспекте настоящего изобретения предусматриваются соединения формулы (1), где в позиции 8 L-Orn отсутствует.
L-Orn может быть заменена D-Orn или другой природной или неприродной аминокислотой. Например, L-Orn может быть заменена природной аминокислотой, такой как лизин; или защищена природной аминокислотой, такой как аргинин или лизин с одним или более алкильными, фенильными или олигометиленовыми заместителями, например N(Me)2,N'(Me)2-Arg, N(Me,Ph),N'(Me)2-Arg, N(CH2)4,N'(Me)2-Arg, N(CH2)4,N'(CH2)4-Arg, Nε(Me)3-Lys.
L-Orn может быть защищена. Например, аминогруппа L-Orn может иметь заместители, особенно алкильные заместители, которые могут быть далее замещены, особенно гетероциклическими группами, например Nδ(СНN(CH2)4,N'(CH2)4)-Orn; или более сложными заместителями, как биотинилорнитин или Orn(NδTfa).
Циклические аминокислоты
Другие предпочтительные соединения согласно данному изобретению включают соединения формулы (1), где одна или более аминокислот циклической цепи замещены другими природными или неприродными аминокислотами, защищенными органическими группами, или удалены.
В особенности, предпочтительные соединения включают соединения с заменой 1, 2 или 3 аминокислот в циклической цепи.
Особенно предпочтительными являются такие соединения формулы (1), где одна или более аминокислот замещены другими природными или неприродными аминокислотами и/или защищены органическими группами. Это, предпочтительно, особенно касается аминокислоты L-Phe в позиции 3.
Таким образом, в одном аспекте настоящего изобретения, предусматриваются соединения формулы (1), где L-Phe в позиции 3 заменена или защищена.
L-Phe может быть заменена D-Phe или другой природной или неприродной аминокислотой. Например, L-Phe может быть заменена природной аминокислотой, такой как тирозин; алкилзамещенной аминокислотой, такой как N-метилтирозин; арилзамещенной аминокислотой, такой как 2-амино-3-бифенил-4-илпропионовая кислота, 2-амино-3-нафталин-2-илпропионовая кислота или аминофенилуксусная кислота; гетероциклилзамещенной аминокислотой, такой как 2-амино-3-тиофен-2-илпропионовая кислота; или циклической аминокислотой, где аминогруппа находится в гетероциклической части, такой как 1,2,3,4-тетраизохинолин-3-карбоновая кислота или октагидроизоиндол-1-карбоновая кислота.
L-Phe может быть защищена. Например, фенильное кольцо может быть частично или полностью насыщенным и может быть замещено одним или более заместителями. Заместители фенильного кольца могут быть, как указанные, предпочтительно, галоген или нитрогруппа. Аминогруппа может иметь 1 или 2 заместителя, особенно алкил, такой как метил, особенно один метильный заместитель.
Концевая ацильная группа
Помимо модификации аминокислот экзоциклической цепи и/или циклической части также могут быть соединения с модификацией в концевой ацильной группе экзоциклической цепи.
Например, ацильная группа может включать один или более заместителей, особенно ароматические, гетероциклические или карбоциклические группы, которые, в свою очередь, могут иметь один или более заместителей, например может быть бензоильная группа, которая может иметь один или более заместителей, фенилалканоил, который может иметь один или более заместителей, или циннамоил, который может иметь один или более заместителей. Кроме того, концевая ацильная группа может быть другой жирной кислотой, обычно насыщенной или ненасыщенной жирной кислотой с 3-26 атомами углерода, более предпочтительно с 12-20 атомами углерода.
Типичные группы для введения вместо концевой ацильной группы в формуле (1) включают:
- линейную алканоильную цепь с вплоть до 26 атомами углерода и с одним или более заместителями, особенно заместителями, выбираемыми из необязательно замещенного циклоалкила, такого как 4-метилциклогексил; алкила, особенно короткоцепочечного алкила, такого как метил; необязательно замещенного арила, особенно фенила, такого как дифторфенил; галогена, такого как от фтора вплоть до перфтора; оксогруппы, аминогруппы или иминогруппы;
- необязательно замещенный бензоил, такой как сам бензоил, п-метилбензоил, п-трифторметилбензоил, пиперонилоил;
- арилалкеноильную группу, предпочтительно, с двумя атомами углерода в алкеноиле, особенно циннамоильную группу, такую как п-трифторметилциннамоил.
Кроме того, ацильная группа может быть заменена другой ацильной группой, предпочтительно, группой формулы R'CO-. R' означает вышеописанный радикал и означает, соответственно, алкил, алкенил, арил, гетероциклил или карбоциклил и сам может быть замещен.
Наконец, это могут быть соединения с единственной модификацией, находящейся в жирной кислоте экзоциклической цепи, обеспечивающей образование 5-метилгексильной концевой группы.
Комбинированные замены
Возможные замены в экзоциклических аминокислотах, циклических аминокислотах и концевой ацильной группе могут быть осуществлены в комбинации.
Таким образом, другими предпочтительными соединениями согласно данному изобретению являются такие соединения формулы (1), где одна или более аминокислот экзоциклической цепи и/или одна или более аминокислот циклической части модифицированы, как указано выше, и/или модифицирована концевая ацильная группа.
Соединения, где L-Orn в позиции 8 заменена или защищена, могут иметь другие предпочтительные модификации, включая 4(S)-метилгексильную или другую группу в концевой группе боковой цепи.
Соединения, где L-Phe в позиции 3 заменена или защищена, могут иметь другие предпочтительные модификации, включая 4(S)-метилгексильную или другую группу в концевой группе боковой цепи.
Примеры замещающих групп включают С1-С18-алкил, С2-С18-алкенил, С2-С18-алкинил, арил, гетероциклические группы, OR', SR', SOR', SO2R', NO2, NHR', N(R')2, NHCOR', N(COR')2, NHSO2R', CN, галоген, С(=О)R', CO2R', OC(=O)R', где каждая из R'-групп является независимо выбираемой из группы, состоящей из Н, ОН, NO2, NH2, SH, CN, галогена, C(=O)H, C(=O)-алкила, СO2H, замещенного или незамещенного С1-С18-алкила, замещенного или незамещенного С2-С18-алкенила, замещенного или незамещенного С2-С18-алкинила и замещенного или незамещенного арила. Подходящие галогены-заместители в соединениях согласно настоящему изобретению включают F, Cl, Br и I.
Алкильные группы представляют собой насыщенную линейную или разветвленную углеводородную группу, включая, например, метил, этил, изопропил, изобутил, трет-бутил, гептил, додецил, октадецил, амил, 2-этилгексил, 2-метилбутил, 5-метилгексил, 9-метил-3-децил и тому подобные, в особенности алкильные группы, которые имеют одну боковую метильную группу. Подходящая алкильная группа является длинной и имеет 1-20 атомов углерода, более типично 1-15 или 1-10 атомов углерода, или может быть короткой и имеет 1-6 или 1-3 атома углерода. Длинные углеродные цепи являются «претендентами» для применения в концевой группе жирной кислоты.
Предпочтительные алкенильные и алкинильные группы в соединениях согласно настоящему изобретению имеют одну или более ненасыщенных связей и 2-12 атомов углерода, более предпочтительно, 2-8 атомов углерода, еще более предпочтительно, 2-6 атомов углерода, даже более предпочтительно 2, 3 или 4 атома углерода. Термины «алкенил» и «алкинил», как используемые в данном контексте, относятся как к циклическим, так и нециклическим группам, хотя линейные или разветвленные нециклические группы обычно более предпочтительны. В общем смысле авторы включают термин «алкилиден» в термин «алкенил», поскольку они оба представляют собой заместители с двойной связью.
Подходящие арильные группы в соединениях согласно настоящему изобретению включают моноциклические или полициклические соединения, включая полициклические соединения, которые содержат отдельные и/или конденсированные арильные группы. Типичные арильные группы содержат 1-3 отдельных или конденсированных циклов и 6-18 атомов углерода в циклах. Особенно предпочтительные арильные группы включают замещенный или незамещенный фенил, нафтил, бифенил, фенантрил и антрацил.
Подходящие ацильные группы включают алканоильные группы, которые имеют 2-12 атомов углерода, более предпочтительно, 2-8 атомов углерода, еще более предпочтительно, 2-6 атомов углерода, даже более предпочтительно, 2 атома углерода. Другие ацильные группы включают алкенилацил, алкинилацил, арилацил, гетероциклилацил.
Подходящие гетероциклические группы включают гетероароматические и гетероалициклические группы. Подходящие гетероароматические группы в соединениях согласно настоящему изобретению содержат один, два или три гетероатома, выбираемые из атомов N, O или S, и включают, например, кумаринил, включая 8-кумаринил, хинолинил, включая 8-хинолинил, пиридил, пиразинил, пиримидил, фурил, пирролил, тиенил, тиазолил, оксазолил, имидазолил, индолил, бензофуранил и бензотиазол. Подходящие гетероалициклические группы в соединениях согласно настоящему изобретению содержат один, два или три гетероатома, выбираемые из атомов N, O или S, и включают, например, тетрагидрофуранил, тетрагидропиранил, пиперидинил, морфолиногруппу и пирролидинильную группу.
Вышеуказанные группы могут быть замещены в одной или более доступных позициях одним или более заместителями.
Природные аминокислоты включают аланин, глицин, валин, лейцин, изолейцин, фенилаланин, тирозин, триптофан, метионин, цистеин, аспартат, аспарагин, глутаминовую кислоту, глутамин, лизин, аргинин, пролин, серин, треонин, гистидин и гидроксипролин.
Авторы также ссылаются на WO 0158934, полностью включенную в данный контекст в качестве ссылки. Ранее приведенный текст является руководством, применимым к соединениям согласно данному изобретению, и авторы вводят определения, используемые в случае соединений согласно данному изобретению.
Аббревиатуры, используемые в последующем описании для аминокислот, и названия пептидов соответствуют правилам комиссии по биохимической номенклатуре IUPAC-IUB согласно J. Biol. Chem., 1972, 247, 977-983.
Используются следующие дополнительные аббревиатуры:
Обозначения аминокислот указаны в L-конфигурации за исключением случаев, где это указано иначе.
Примеры соединений согласно данному изобретению включают соединения формулы (1), где:
- Glu или Lys находится вместо L-Orn в позиции 8;
- Gly, Phe, Ala, Leu, D-Val, Pro, Gln, Orn, Thr или Glu находятся вместо L-Val в позиции 11;
- Val или D-Thr находится вместо L-Thr в позиции 12;
- Gly, D-Ala, D-Leu, D-Phe, D-Pro, Val, D-Glu, D-Gln или D-Thr находятся вместо D-Val в позиции 13;
- hCh находится вместо L-Val в позиции 11 и/или D-Cha находится вместо D-Val в позиции 13;
- Ala, Gly, Leu, Pro, Glu, Orn или Gln находятся вместо L-Thr в позиции 12 и отсутствует аминокислота в позиции 13;
- D-Ile или D-Val находится вместо D-allo-Ile в позиции 7;
- D-Orn находится вместо L-Orn в позиции 8 и L-Pro находится вместо D-Pro в позиции 9;
- D-Cys находится вместо D-allo-Ile в позиции 7 и L-Cys находится вместо L-Val в позиции 11; или
- D-Cys или D-homo-Cys находится вместо D-allo-Ile в позиции 7 и D-Cys или D-homo-Cys находится вместо D-Val в позиции 10.
- Icos, (c/t)-4-Me-cHexa, Und, (4R)-MeHex, (4RS)-MeHex, (4S)-MeHex, Oct, p-MeBza, Bza, p-CF3Bza, 3,5-dFPhAc, Pipe, p-CF3Cinn, p-CF3PhAc, Pfh, 6-OHep, 6,6-dFHep, 4-GuBut, AM, AO или C(=N(CH3)2) находятся вместо 5-MeHex в позиции 14 и, необязательно, Lys находится вместо L-Orn в позиции 8;
- Pro, D-Pip, D-Tic или (5R)-Ph-Pro находятся вместо D-Pro в позиции 9 и (4S)-MeHex находится вместо 5-MeHex в позиции 14;
- N(Me)2,N'(Me)2-Arg, N(Me,Ph),N'(Me)2-Arg, N(CH2)4,N'(Me)2-Arg, N(CH2)4,N'(CH2)4-Arg, Nδ(СНN(CH2)4,N'(CH2)4)-Orn, Nε(Me)3-Lys, Orn(NδTfa) или Orn(Biot) находятся вместо L-Orn в позиции 8, и, необязательно, (4S)-MeHex находится вместо 5-MeHex в позиции 14, и, необязательно, Thr(OTfa) находится вместо L-Thr в позиции 12;
- Thr(OTfa) находится вместо L-Thr в позиции 12 и (4S)-MeHex или Lit(OTfa) находится вместо 5-MeHex в позиции 14;
- N-(Hep)2-D-Val находится вместо D-Val в позиции 13 и отсутствует 5-MeHex в позиции 14; или
- отсутствуют аминокислоты в позициях 11, 12 и 13 и, необязательно, Mst находится вместо 5-MeHex в позиции 14.
- Dha или E-Dhb находится вместо Z-Dhb в позиции 2;
- D, L-Ser, Gly или Aib находятся вместо Thr в позиции 2, причем имеются гидрированные аналоги;
- D-Val находится вместо L-Val в позиции 1;
- Trp находится вместо L-Phe в позиции 3; или
- D-Thr или D-Ser находится вместо D-alo-Thr в позиции 6.
- hCh, Phe(3,4-Cl2), Phe(F5), Phe(4-I), Phe(4-NO2), Phe(4-F), Tyr(Me), Thi, Tic, Tyr, Oic, NMePhe, Phe(2-Cl), Phe(3-Cl), Phe(4-Cl), Phe(3,4-F2), NaI, Bip или Phg находятся вместо L-Phe в позиции 3 и, необязательно, (4S)-MeHex или p-CF3Cinn находится вместо 5-MeHex в позиции 14;
- D-Val или D-Cha находится вместо D-alo-Ile в позициях 5 и 7 и, необязательно, D-Cha находится вместо D-Val в позиции 4; или
- D-Val находится вместо L-Val в позиции 1, D-Phe находится вместо L-Phe в позиции 3, Val находится вместо D-Val в позиции 4, L-alo-Ile находится вместо D-alo-Ile в позиции 5, L-alo-Thr находится вместо D-alo-Thr в позиции 6, L-alo-Ile находится вместо D-alo-Ile в позиции 7, D-Orn находится вместо L-Orn в позиции 8, L-Pro находится вместо D-Pro в позиции 9, L-Val находится вместо D-Val в позиции 10, D-Val находится вместо L-Val в позиции 11, D-Thr находится вместо L-Thr в позиции 12 и L-Val находится вместо D-Val в позиции 13.
Соответственно, эти предлагаемые замены можно осуществлять в комбинации.
Настоящее изобретение также охватывает их фармацевтически приемлемые соли, пролекарства, таутомеры и сольваты.
Соединения согласно настоящему изобретению имеют асимметрические центры и, следовательно, существуют в различных энантиомерных и диастереомерных формах. Данное изобретение относится к применению всех оптических изомеров и стереоизомеров соединений согласно настоящему изобретению и их смесей и ко всем фармацевтическим композициям и способам лечения, которые можно применять или которые могут их включать.
Соединения согласно данному изобретению могут быть в кристаллической форме, или в виде свободных соединений, или в виде сольватов (например, гидраты) и подразумевают, что обе формы находятся в объеме настоящего изобретения. Способы сольватации, как правило, известны в уровне техники.
Настоящее изобретение также включает соединения согласно настоящему изобретению и их фармацевтически приемлемые соли, где один или более атомов водорода, углерода или других атомов заменены их изотопами. Такие соединения могут быть пригодны в качестве исследовательских и диагностических средств при фармакокинетическом изучении метаболизма и в анализах на связывание.
Как указано в описании, соединения согласно данному изобретению, включая соединения формулы (1), включают их фармацевтически приемлемые производные или пролекарства. Термин «фармацевтически приемлемое производное или пролекарство» означает любую фармацевтически приемлемую соль, сложный эфир, соль сложного эфира или другое производное соединения согласно данному изобретению, которое при введении реципиенту способно давать (непосредственно или косвенно) соединение согласно данному изобретению, или его метаболит, или его остаток. Особенно предпочтительными производными и пролекарствами являются такие, которые увеличивают биодоступность соединений согласно данному изобретению, когда такие соединения вводят пациенту (например, учитывая, что перорально вводимое соединение легче абсорбируется в крови), увеличивают доставку исходного соединения в данный биологический компартмент, повышают растворимость для введения путем инъекции, изменяют метаболизм или изменяют скорость экскреции.
Соли соединений согласно настоящему изобретению могут включать аддитивные соли кислот, образующиеся по атому азота в соединении формулы (1) или (2). Терапевтическая активность свойственна части, происходящей от соединения согласно данному изобретению, как указанное здесь, а идентичность другого компонента имеет меньшее значение, несмотря на то что для терапевтических и профилактических целей он является, предпочтительно, фармацевтически приемлемым для пациента. Примеры фармацевтически приемлемых аддитивных солей кислот включают таковые, происходящие от минеральных кислот, таких как соляная кислота, бромводородная кислота, фосфорная кислота, метафосфорная кислота, азотная кислота и серная кислота; и органических кислот, таких как винная кислота, уксусная кислота, трифторуксусная кислота, лимонная кислота, яблочная кислота, молочная кислота, фумаровая кислота, бензойная кислота, гликолевая кислота, глюконовая кислота, янтарная кислота, метансульфоновая кислота и арилсульфоновая кислота, например п-толуолсульфоновая кислота. Предпочтительные соли включают трифторацетат и гидрохлорид.
Соединения согласно настоящему изобретению можно получать в соответствии со способом синтеза, описанным в WO 0158934, или в соответствии с улучшенным способом, как описано в описании. Следовательно, данное изобретение также относится к способу получения соединения, соответствующего формуле (1).
Ключевыми стадиями оптимизированного способа для более экономичного и безопасного синтеза кахалалида F и его аналогов являются: (i) частичное включение Fmoc-D-Val-OH в хлортритилхлорполистирольную смолу с начальной загрузкой 0,5 ммоль/г; (ii) применение в качестве способа связывания DIPCDI-HOBt вместо HATU-DIPEA для последовательного включения защищенных аминокислот и алифатических карбоновых кислот; (iii) стадия циклизации DIPCDI/HOBt/DIPEA в CH2Cl2; в этих условиях избегают двух побочных реакций: эпимеризация остатка Val, который вовлечен в активацию, и трифторацетилирование Phe или его замена; (iv) использование диэтилдитиокарбамата натрия после удаления Alloc, чтобы избежать присутствия Pd(O) в конечном продукте.
Синтез является, предпочтительно, твердофазным синтезом.
Предпочтительное воплощение способа синтеза согласно настоящему изобретению наилучшим образом представлено на схеме 1, которая относится к образованию целевых соединений.
Схема 1

Как показано выше на схеме 1, предпочтительный синтетический способ образования аналогов кахалалида F основан на твердофазном методе, см., например, Lloyd-Williams P. и др., Chemical Approaches to the Synthesis of Peptides and Proteins. CRC Press, Boca Raton (FL), 1997, и следуя модификациям способа, уже описанным для получения кахалалида F и некоторых его аналогов (WO 0158934).
Способ на схеме 1 включает следующие стадии:
(а) введение Fmoc-D-Val-OH в хлортритилхлорсмолу с образованием сложноэфирной связи;
(b) удлинение пептидной цепи за счет трех аминокислот [DaIle, DaThr(свободное OH), DaIle] с применением стратегии Fmoc/tBu;
(с) введение [Val(1)] с применением стратегии Alloc/tBu;
(d) удлинение пептидной цепи за счет остаточных аминокислот и алифатических карбоновых кислот с применением стратегии Fmoc/tBu;
(е1) введение дипептида Alloc-Phe-ZDhb-OH, который комбинируют и дегидратируют в растворе;
(е2а) удлинение пептидной цепи за счет двух аминокислот, предпочтительно, Thr и Phe. Группа ОН аминогруппы Thr является незащищенной, а аминогруппа Phe, или ее замена, защищена с помощью Fmoc или, предпочтительно, с помощью Alloc; в некоторых случаях, если она защищена с помощью группы Fmoc, ее удаляют и Alloc вводят в твердой фазе;
(е2b) дегидратация в твердой фазе для получения дидегидропептида;
(f) удаление Alloc/Fmoc-группы Phe, или ее замены, в то время как пептид по-прежнему присоединен к твердой основе;
(g) отщепление защищенного в боковой цепи пептида от твердой основы;
(h) циклизация пептида в растворе;
(i) удаление лабильных TFA защитных групп боковой цепи;
(j) дальнейшие модификации функциональных(ой) групп(ы) в фазе раствора.
Следовательно, способ можно проводить следующим образом:
Fmoc-D-Val-OH вводят, предпочтительно, в хлортритилполистирольную смолу, см. Barlos K.; Gatos D.; Schäfer W., Angew. Chem. Int. Ed. Engl., 1991, 30, 590-593, в присутствии DIPEA, сохраняя степень замещения приблизительно 0,5 ммоль/г. Применение более высоких загрузок вызывает присутствие концевых пептидов в конечном продукте, см. Chiva C.; Vilaseca M.; Giralt E.; Albericio F.; J. Pept. Sci., 1999, 5, 131-140.
Все относительные расходы растворителей приводятся в об./об. за исключением, если указано иначе.
Удаление Fmoc-группы осуществляют с помощью пиперидин-ДМФА (2:8, об./об.)(1×2 мин, 2×10 мин). Связывания Fmoc-аа-ОН (4-5 экв.) и кислот в позиции 14 можно осуществлять с помощью DIPCDI-HOBt (эквимолярные количества каждого из них относятся к карбоксильному компоненту) или PyBOP-DIPEA (эквимолярное количество PyBOP и двойное количество DIPEA) в ДМФА или ДМФА-толуоле (1:1) в течение 90 мин. После связывания осуществляют нингидриновый или хлораниловый тесты и, если тест положительный, связывание повторяют в тех же условиях, иначе, процесс продолжают. Промывки между стадиями удаления защиты, связывания и снова удаления защиты осуществляют с помощью ДМФА (5×0,5 мин) и CH2Cl2 (5×0,5 мин), используя, например, каждый раз 10 мл растворителя на грамм смолы.
Введение Alloc-Val-OH (5 экв.) можно осуществлять с эквимолярным количеством DIPCDI и 10% DMAP. Это связывание повторяют, по меньшей мере, дважды.
Удаление Alloc-группы можно осуществлять с помощью Pd(PPh3)4 (0,1 экв.) в присутствии PhSiH3 (10 экв.), см. Gómez-Martínez P.; Thieriet N.; Albericio F.; Guibé F.; J. Chem. Soc. Perkin I, 1999, 2871-2874, и промывки смолы 0,02 М раствором диэтилдитиокарбамата натрия в ДМФА (3×15 мин).
Дипептид Alloc-Phe-ZDhb-OH (4 экв.), который получают в растворе из Alloc-Phe-ОН и Н-Thr-OtBu с EDC·HCl, с последующей дегидратацией и обработкой TFA, можно связывать с DIPCDI-HOАt (4 экв. каждого) в течение от 5 часов до в течение ночи. Применение других реагентов связывания, основанных на HOВt, таких как HBTU или DIPCDI-HOВt, приводит к неполным включениям дипептида.
Дегидратацию можно осуществлять в твердой фазе с помощью EDC·HCl (водорастворимый карбодиимид, 20 экв.) в присутствии CuCl (12 экв.) в CH2Cl2-ДМФА (9:1) в течение 7 дней. EDC·HCl/CuCl используют при дегидратации в растворе остатка Thr во фрагменте низина (Fukase K.; Kitazawa M.; Sano A.; Shimbo K.; Horimoto S.; Fujita H.; Kubo A.; Wakamiya T.; Shibe A. Bull. Chem. Soc. Jpn., 1992, 65, 2227-2240) и в твердой фазе для получения дидегидропептидов из Thr, Ser и фенилсерина (Royo M.; Jiménez J.C.; López-Maciá A.; Giralt E.; Albericio F.; Eur. J. Org. Chem., 2001, 45-48).
Отщепление защищенного пептида от смолы можно осуществлять с помощью TFA-CH2Cl2 (1:99)(5×30 сек).
Стадию циклизации можно осуществить с помощью DIPCDI/HOBt/DIPEA в CH2Cl2. В этих условиях избегают двух побочных реакций: эпимеризация остатка Val, который вовлечен в активацию, и трифторацетилирование Phe или его замена.
Окончательное удаление защиты можно осуществлять с помощью TFA-Н2О (95:5) в течение 1 часа.
Следует принимать во внимание, что конкретный выбор защитных групп не является критическим и широко доступны другие альтернативы. Например, группы типа Bzl можно заменять tBu/Boc; Boc вместо Fmoc; Fmoc вместо Alloc; смолу Wang вместо хлортритила.
Дальнейшие детали синтеза приведены в примерах.
Способ согласно данному изобретению можно осуществлять из исходных материалов энантио-, стереоконтролируемым и быстро проводимым образом, используя преимущества в методологии твердофазного синтеза, где молекула в конструкции связана с нерастворимой основой в течение всех операций синтеза.
Фармацевтические готовые лекарственные формы соединений согласно данному изобретению могут быть приспособлены для введения любым соответствующим путем, например пероральным (включая буккальный или подъязычный), ректальным, назальным, местным (включая буккальный, подъязычный или трансдермальный), вагинальным или парентеральным (включая подкожный, внутримышечный, внутривенный или интрадермальный) путем. Такие готовые лекарственные формы можно получать любым способом, известным в области фармации, например, путем ассоциации активного ингредиента с носителем(ями) или эксципиентом(ами).
Предпочтительно, фармацевтические композиции, содержащие соединения согласно данному изобретению, включают жидкие композиции (растворы, суспензии или эмульсии) с подходящим составом для внутривенного введения, и они могут содержать чистое соединение или находиться в комбинации с любым носителем или другими фармакологически активными соединениями. Дальнейшее руководство относительно фармацевтических композиций можно найти в WO 0236145, которая полностью включена в данный контекст путем ссылки.
Таким образом, комбинация неионогенного поверхностно-активного вещества и органической кислоты пригодна для применения с наполнителем для получения лиофилизированной формы соединения согласно данному изобретению, подходящей для восстановления. Восстановление предпочтительно осуществляют в смеси эмульгирующего солюбилизатора, алканола и воды.
Лиофилизированная композиция предпочтительно включает, главным образом, наполнитель, например, по меньшей мере, 90% или, по меньшей мере, 95% наполнителя. Примеры наполнителей хорошо известны и включают сахарозу и маннит. Можно применять другие наполнители.
Неионогенным поверхностно-активным веществом в лиофилизированной композиции является, предпочтительно, сложный сорбитановый эфир, более предпочтительно, полиэтиленсорбитановый сложный эфир, такой как полиоксиэтиленсорбитаналканоат, особенно, полиоксиэтиленсорбитанмоноолеат, например полисорбат 80. Неионогенное поверхностно-активное вещество обычно составляет несколько процентов композиции, как, например, 0-5% композиции, например 2, 3 или 4% композиции.
Органическая кислота в лиофилизированной композиции обычно представляет собой алифатическую кислоту, предпочтительно, гидроксикарбоновую кислоту и, более предпочтительно, гидроксиполикарбоновую кислоту, особенно, лимонную кислоту. Органическая кислота обычно составляет несколько процентов композиции, как, например, 0-5% композиции, например 2, 3 или 4% композиции.
Количество соединения согласно данному изобретению в лиофилизированной композиции обычно составляет менее чем 1% или, чаще, менее чем 0,1%, в расчете на смесь. Подходящее количество находится в диапазоне 50-200 мкг, приблизительно около 100 мкг, на 100 мг композиции.
Эмульгирующий солюбилизатор для восстановительного агента, соответственно, включает сложный эфир полиэтиленгликоля, особенно, эфир жирной кислоты, более предпочтительно, олеат PEG, такой как олеат PEG-35. Эмульгирующий солюбилизатор составляет, соответственно, 0-10% восстановительного агента, обычно примерно 3-7%, приблизительно около 5%. Алканол обычно является этанолом и составляет, соответственно, 0-10% восстановительного агента, обычно примерно 3-7%, приблизительно около 5%. Остальной частью восстановительного агента является вода, с помощью которой получают восстановительный раствор, пригодный для внутривенной инъекции.
Дальнейшее разбавление восстановительного раствора 0,9%-ным солевым раствором может быть подходящим для инфузии кахалалидного соединения. Пригодное оборудование для инфузии предпочтительно включает стеклянную емкость, которая предпочтительнее, чем таковая из полиэтилена. Трубка предпочтительно является силиконовой.
Предпочтительный восстановительный агент затем содержит 2-7%, приблизительно около 5%, эмульгирующего солюбилизатора; 2-7%, приблизительно около 5%, спирта; и остальное составляет вода.
Готовые лекарственные формы могут быть представлены в виде упаковок со стандартной дозой или множественными дозами, например герметичные ампулы и пузырьки, и они могут храниться в подвергнутом сушке вымораживанием (лиофилизированном) состоянии, непосредственно перед использованием, требующем только добавления стерильного жидкого носителя, например воды для инъекций.
Данное изобретение дополнительно предусматривает фармацевтический набор, содержащий отдельные емкости, заполненные лиофилизированной композицией и восстановительным агентом. Также предусматриваются способы восстановления.
Введение соединений или композиций согласно настоящему изобретению осуществляют путем внутривенной инфузии. Время инфузии может составлять вплоть до 72 часов, более предпочтительно до 1-24 часов или наиболее предпочтительно до примерно 1 или до примерно 3 часов. Особенно желательны кратковременные инфузии, которые дают возможность осуществлять лечение всю ночь вне клиники. Однако инфузию можно осуществлять около 24 часов или даже дольше, если необходимо.
Введение выполняют циклами, предпочтительным применяемым способом; внутривенной инфузии соединения согласно данному изобретению пациентов подвергают в первую неделю каждого цикла и пациентам предоставляют возможность выздоравливать в течение остатка цикла. Предпочтительная продолжительность каждого цикла составляет или 1, 3 или 4 недели; множественные циклы можно проводить по необходимости. В альтернативном протоколе дозирования соединение согласно данному изобретению вводится в продолжение приблизительно 1 часа в течение 5 последовательных дней все 3 недели. Другие протоколы можно составлять в качестве вариаций.
Замедление дозирования и/или уменьшение дозирования и регулировки схем применения выполняют как необходимые в зависимости от индивидуальной переносимости пациентом лечения, в особенности, уменьшение дозирования рекомендуют пациентам с более высокими, чем нормальные, уровнями в сыворотке печеночных трансаминаз или щелочной фосфатазы.
В одном аспекте настоящее изобретение относится к способу лечения человека, пораженного раком, включая введение вышеуказанному пациенту соединения согласно данному изобретению в дозе менее чем 1200 мкг два раза в день, предпочтительно менее чем 930 мкг два раза в день и, более предпочтительно, менее чем 800 мкг два раза в день. Подходящим образом, доза составляет, по меньшей мере, 320 мкг два раза в день. Предпочтительно, доза находится в диапазоне 400-900 мкг два раза в день, предпочтительно, 500-800 мкг два раза в день, более предпочтительно, 600-750 мкг два раза в день. Особенно предпочтительными являются дозы приблизительно 650-700 мкг два раза в день.
В дальнейшем аспекте данное изобретение относится к способу лечения человека, пораженного раком, включающему введение вышеуказанному больному соединения согласно данному изобретению, ежедневно в течение 5 дней в дозе менее чем 930 мкг два раза в день, с последующим перерывом от 1 до 4 недель, в которые кахалалидное соединение не вводят. Доза составляет, предпочтительно, 650-750 мкг два раза в день, более предпочтительно, примерно 700 мкг два раза в день. Время инфузии составляет, предпочтительно, 1-24 часа, более предпочтительно, 1-3 часа. Особенно предпочтительно, время инфузии составляет примерно 1 или примерно 3 часа. Перерыв составляет, предпочтительно, 2-3 недели, более предпочтительно, примерно 2 недели.
Настоящее изобретение также относится к способу лечения человека, пораженного раком, включающему введение вышеуказанному больному соединения согласно данному изобретению один раз в неделю в дозе менее чем 800 мкг два раза в день. Доза составляет, предпочтительно, 600-700 мкг два раза в день, более предпочтительно, 650 мкг два раза в день. Время инфузии составляет, предпочтительно, 1-24 часа, более предпочтительно, 1-3 часа.
Несмотря на то что инструкция по дозировке приведена выше, точная дозировка соединения будет меняться в соответствии с конкретной готовой лекарственной формой, способом применения и конкретным местоположением, реципиентом и обрабатываемой опухолью. Другими факторами, которые нужно учитывать, являются возраст, масса тела, пол, режим питания, время введения, скорость экскреции, состояние реципиента, комбинации лекарственных средств, чувствительности к воздействию и тяжесть заболевания. Введение осуществляют непрерывно или периодически в пределах максимально допустимой дозы.
Настоящее изобретение, в частности, относится к лечению пациентов, пораженных раком простаты, раком молочной железы, гепатоцеллюлярным раком, меланомой, раком ободочной и прямой кишки, раком почки, раком яичников, NSCL-раком, эпителиальным раком, раком поджелудочной железы и опухолями, которые сверхэкспрессируют онкоген Her2/neu. Более предпочтительно, оно относится к лечению гепатоцеллюлярного рака, меланомы, рака молочной железы, рака поджелудочной железы и рака простаты.
Настоящее изобретение также относится к способу лечения кожного заболевания, вызываемого гиперпролиферацией клеток кожи у млекопитающего, который включает введение млекопитающему эффективного нетоксичного количества соединения согласно данному изобретению. Кожным заболеванием, предпочтительно, является псориаз. Настоящее изобретение относится, предпочтительно, к лечению людей, пораженных псориазом, в частности тяжелой формой псориаза.
Соединения и композиции согласно данному изобретению можно применять с другими лекарственными средствами для осуществления комбинированной терапии. Другие лекарственные средства могут составлять часть одной и той же композиции или предусматриваться в виде отдельной композиции для введения в то же самое время или в другое время. Идентичность другого лекарственного средства особенно не ограничена, несмотря на то что предусматривается комбинация с другими химиотерапевтическими агентами, гормональными агентами или антителами. Количества соединения согласно данному изобретению и другого фармацевтически активного агента или агентов и относительное регулирование введения выбирают для того, чтобы достичь желательного комбинированного терапевтического эффекта.
ПРИМЕРЫ
Общие методики
Cl-TrtCl-смола, защищенные Fmoc-аминокислотные производные, HOBt, HOAt выпускаются фирмами ABI (Framingham, MA), Bachem (Bubendorf, Швейцария), NovaBiochem (Läufelfingen, Швейцария), 4-МеНех-производные - фирмой Narchem; HATU и другие гуанидилированные производные выпускаются фирмой ABI (Framingham, MA), или их получают согласно del Fresno M., El-Faham A.; Carpino L.A.; Royo M.; Albericio F. Organic Lett., 2000, 2, 3539-3542.
Alloc-аминокислоты, по существу, получают, как описано Dangles и др., см. Dangles O.; Guibé F.; Balavoine G.; Lavielle S.; Marquet. A. J. Org. Chem., 1987, 52, 4984-4993; и Alloc-Z-Dhb-Phe-OH и кахалалид F получают, как описано в WO 0158934; DIPEA, DIPCDI, EDC·HCl, пиперидин, TFA выпускаются фирмой Aldrich (Milwaukee, WI). ДМФА и CH2Cl2 выпускаются фирмой SDS (Peypin, Франция). Ацетонитрил (степень чистоты для ВЭЖХ) выпускается фирмой Scharlau (Барселона, Испания). Все торговые реагенты и растворители используют как полученные, за исключением CH2Cl2, который пропускают через колонку с оксидом алюминия для удаления загрязняющих примесей кислотного характера.
Твердофазный синтез осуществляют в полипропиленовых шприцах (10-50 мл), снабженных пористым полиэтиленовым диском. Растворители и растворимые реагенты удаляют путем отсасывания. Удаление Fmoc-группы осуществляют с помощью пиперидин-ДМФА (2:8, об./об.) (1×2 мин, 2×10 мин). Промывки между стадиями удаления защиты, связывания и снова удаления защиты осуществляют с помощью ДМФА (5×0,5 мин) и CH2Cl2 (5×0,5 мин), используя каждый раз 10 мл растворителя на грамм смолы. Превращения пептидного синтеза и промывки выполняют при 25оС. Синтезы, осуществляемые на твердой фазе, контролируют путем ВЭЖХ промежуточных продуктов, получаемых после отщепления с помощью TFA-H2O (1:99) за 1 мин в аликвоте (приблизительно 2 мг) пептидильной смолы. ВЭЖХ-колонки с обращенной фазой [Nucleosil-C18 4,6×250 мм, 10 мкм (колонка А); Nucleosil-C4 4,6×250 мм, 10 мкм (колонка В) выпускаются фирмой Sharlau (Испания); Symmetry™ C18 4,6×150 мм, 5 мкм (колонка С); Symmetry300™ C18 4,6×50 мм, 5 мкм (колонка D) выпускаются фирмой Waters (Ирландия), а Zorbax SB C18 4,6×150 мм, 3,5 мкм (колонка Е) выпускается фирмой Agilent (США)]. Аналитическую ВЭЖХ осуществляют на приборе Waters, содержащем два нагнетательных насоса для растворителя (Waters 1525), автоматический инжектор (автоматический пробоотборник Waters 717), детектор для двух длин волн (Waters 2487) и системный контроллер (Breeze V3.20); и на приборе Agilent 1100, содержащем два нагнетательных насоса для растворителя (G1311А), автоматический инжектор (G1329А), DAD (G1315В). УФ-детектирование проводят при 215 или 220 нм, и линейные градиенты CH3CN (+0,036% TFA) в Н2О (+0,045% TFA) действуют в следующих условиях:
(% CH3CN)
Анализы путем времяпролетной масс-спектрометрии с лазерной десорбцией и ионизацией из матрицы (MALDI-TOF) и масс-спектрометрию с ионизацией электронным распылением (ES-MС) пептидных образцов выполняют на PerSeptive Biosystems Voyager DE RP, используя DHB-матрицу, и на спектрометре Waters Micromass ZQ, и на Agilent Ion Trap 1100 серии LC/MSDTrap. Образцы содержащей пептид смолы гидролизуют в смеси 12 н. водного раствора HCl-пропионовой кислоты (1:1) при 155оС в течение 1-3 часов и не содержащие пептида образцы гидролизуют в 6 н. водном растворе HCl при 155оС в течение 1 часа. Последующие аминокислотные анализы выполняют на автоанализаторе Beckman System 6300. 1Н-ЯМР-спектроскопию [1H, NOESY, TOCSY при (278К)] выполняют на Varian Unity Plus (500 МГц). Химические сдвиги (δ), отсчитываемые от ТМS в сторону менее сильного поля, выражают в миллионных долях (м.д.). Константы взаимодействия выражают в герцах.
Обозначения аналогов делают относительно 5-метилгексильного изомера кахалалида F формулы (I), указывая в квадратных скобках модифицированный остаток; суффикс «no» относится к исключению природного остатка из последовательности.
Пример 1
(4S)-MeHex-D-Val-ThrVal-D-Val-D-Pro-Orn-D-allo-Ile-цикло[D-allo-Thr-D-allo-Ile-D-Val-Phe-(Z)Dhb-Val], [(4S)-MeHex 14 ]-кахалалид F (соединение 1)
Стадия 1
H-D-Val-O-TrtCl-смола
Cl-TrtCl-смолу (1 г, 1,64 ммоль/г) помещают в 20-мл полипропиленовый шприц, снабженный полиэтиленовым фильтровальным диском. Смолу затем промывают CH2Cl2 (5×0,5 мин) и добавляют раствор Fmoc-D-Val-OH (238 мг, 0,7 ммоль, 0,7 экв.) и DIPEA (0,41 мл) в CH2Cl2 (2,5 мл) и смесь перемешивают в течение 15 мин, тогда вводят дополнительный DIPEA (0,81 мл, всего 7 экв., 7 ммоль) и смесь перемешивают в течение 45 мин. Взаимодействие завершают добавлением МеОН (800 мкл) после перемешивания в течение 10 мин. Fmoc-D-Val-O-TrtCl-смолу подвергают последующим промывкам/обработкам CH2Cl2 (3×0,5 мин), ДМФА (3×0,5 мин), пиперидином, как указано в общих методиках, и ДМФА (5×0,5 мин). Загрузка, рассчитываемая путем определения Fmoc, составляет 0,50 ммоль/г.
Стадия 2
Fmoc-D-allo-Ile-D-allo-Thr(Val-Alloc)-D-allo-Ile-D-Val-O-TrtCl-смола
Fmoc-D-allo-Ile-ОН (707 мг, 2 ммоль, 4 экв.), Fmoc-D-allo- Thr-ОН (свободная гидроксильная группа) (683 мг, 2 ммоль, 4 экв.) и Fmoc-D-allo-Ile-ОН (707 мг, 2 ммоль, 4 экв.) последовательно добавляют к вышеполученной H-D-Val-O-TrtCl-смоле, используя DIPCDI (310 мкл, 2 ммоль, 4 экв.) и HOBt (307 мг, 2 ммоль, 4 экв.) в ДМФА (2,5 мл). Во всех случаях после 90 мин реакции связывания нингидриновый тест является отрицательным. Удаление Fmoc-группы и промывки осуществляют, как описано в общих методиках. Alloc-Val-OH (502 мг, 2,5 ммоль, 5 экв.) связывают с DIPCDI (387 мг, 2,5 ммоль, 5 экв.) в присутствии DMAP (30,6 мг, 0,25 ммоль, 0,5 экв.) и DIPEA (88 мкл, 0,5 ммоль, 1 экв.) в течение 45 мин. Это связывание повторяют в тех же условиях дважды. Аликвоту пептидильной смолы обрабатывают TFA и ВЭЖХ (tR 7,8 мин, условия S, колонка D) сырого продукта, получаемого после выпаривания, показывает чистоту >98%.
ES-MС, рассчитано для С45Н63N5O11: 849,45.
Найдено: m/z 850,1 [М+Н]+.
Стадия 3
Fmoc-D-Val-D-Pro-Orn(Boc)-D-allo-Ile-D-allo-Thr(Val-Alloc)-D-allo-Ile-D-Val-O-TrtCl-смола
Fmoc-группу удаляют и Fmoc-Orn(Boc)-ОН (912 мг, 2 ммоль, 4 экв.), Fmoc-D-Pro-ОН (843 мг, 2,5 ммоль, 5 экв.) и Fmoc-D-Val-ОН (255 мг, 2,5 ммоль, 5 экв.) последовательно добавляют к вышеуказанной пептидильной смоле (стадия 2), используя DIPCDI (310 мкл, для 2,0 ммоль и 4 экв.; и 388 мкл, для 2,5 ммоль, 5 экв.) и HOBt (307 мг, для 2,0 ммоль и 4 экв.; и 395 мг, 2,5 ммоль, 5 экв.) в течение 90 мин. Нингидриновый тест после введения Orn и D-Pro является отрицательным. Хлораниловый тест после введения D-Val является слегка положительным, и, следовательно, осуществляют повторное связывание этого остатка с Fmoc-D-Val-ОН (678 мг, 2,0 ммоль, 4 экв.), DIPCDI (310 мкл, 2,0 ммоль, 4 экв.) и HOBt (307 мг, 2,0 ммоль, 4 экв.) в течение 90 мин. Аликвоту пептидильной смолы обрабатывают TFA и ВЭЖХ (tR=10,1 мин, условия S, колонка D) сырого продукта, получаемого после выпаривания, показывает чистоту >98%.
MALDI-TOF-MS, рассчитано для С65Н97N9O16: 1259,71.
Найдено: m/z 1282,16 [М+Na]+.
Стадия 4
(4S)-MeHex-D-Val-Thr(tBu)-Val-D-Val-D-Pro-Orn(Boc)-D-allo-Ile-D-allo-Thr(Val-Alloc)-D-allo-Ile-D-Val-O-TrtCl-смола
Fmoc-группу удаляют и Fmoc-Val-ОН (678 мг, 2 ммоль, 4 экв.), Fmoc-Thr(tBu)-ОН (992 мг, 2,5 ммоль, 5 экв.), Fmoc-D-Val-ОН (678 мг, 2 ммоль, 4 экв.) и (4S)-MeHex-ОН (195 мг, 1,5 ммоль, 3 экв.) последовательно добавляют к вышеуказанной пептидильной смоле (стадия 3), используя DIPCDI (233 мкл, для 1,5 ммоль и 3 экв.; и 310 мкл, для 2 ммоль и 4 экв.; и 388 мкл, для 2,5 ммоль, 5 экв.) и HOBt (230 мг, для 1,5 ммоль и 3 экв.; 307 мг, 2 ммоль и 4 экв.; и 395 мг, 2,5 ммоль, 5 экв.) в течение 90 мин. Во всех случаях после 90 мин связывания нингидриновый тест является отрицательным. Удаление Fmoc-группы и промывки осуществляют, как описано в общих методиках.
Стадия 5
(4S)-MeHex-D-Val-Thr(tBu)-Val-D-Val-D-Pro-Orn(Boc)-D-allo-Ile-D-allo-Thr(Val-Z-Dhb-Phe-Alloc)-D-allo-Ile-D-Val-O-TrtCl-смола
Alloc-группу удаляют с помощью Pd(PPh3)4 (58 мг, 0,05 ммоль, 0,1 экв.) в присутствии PhSiH3 (617 мкл, 5 ммоль, 10 экв.) в атмосфере Ar и Alloc-Phe-Z-Dhb-ОН (666 мг, 2 ммоль, 4 экв.) и HOAt (273 мг, 2 ммоль, 4 экв.) растворяют в ДМФА (1,25 мл) и добавляют к пептидильной смоле, затем добавляют DIPCDI (310 мкл, 2 ммоль, 4 экв.) и смесь перемешивают в течение 5 часов, после чего нингидриновый тест является отрицательным. После промывок с помощью ДМФА и CH2Cl2 аликвоту пептидильной смолы обрабатывают TFA-H2O (1:99) в течение 1 мин и продукт характеризуют с помощью MALDI-TOF-MS.
Рассчитано для С88Н146N14O21: 1735,08.
Найдено: m/z 1758,67 [М+Na]+, 1774,62 [М+К]+.
Стадия 6
(4S)-MeHex-D-Val-Thr(tBu)-Val-D-Val-D-Pro-Orn(Boc)-D-allo-Ile-D-allo-Thr(Val-Z-Dhb-Phe-Н)-D-allo-Ile-D-Val-OН
После промывок с помощью ДМФА и CH2Cl2 Alloc-группу удаляют с помощью Pd(PPh3)4 (58 мг, 0,05 ммоль, 0,1 экв.) в присутствии PhSiH3 (617 мкл, 5 ммоль, 10 экв.) в атмосфере Ar. Защищенный пептид отщепляют от смолы с помощью TFA-CH2Cl2 (1:99) (5×30 сек). Фильтрат собирают в Н2О (4 мл) и Н2О частично удаляют в роторном испарителе. Затем добавляют AСN для растворения твердого вещества, которое появляется во время удаления Н2О, и раствор лиофилизируют с получением 639 мг (387 мкмоль, выход 77%) указанного в заголовке соединения со степенью чистоты >95%, как проверено с помощью ВЭЖХ (условие R, колонка С, tR=10,5 мин).
Стадия 7
(4S)-MeHex-D-Val-Thr-Val-D-Val-D-Pro-Orn-D-allo-Ile-цикло(D-allo-Thr-D-allo-Ile-D-Val-Phe-Z-Dhb-Val)
Защищенный пептид (стадия 6) (639 мг, 387 мкмоль) растворяют в CH2Cl2 (390 мл, 1 мМ) и добавляют HOBt (237 мг, 1,55 ммоль), растворенный в минимальном объеме ДМФА для растворения HOBt, добавляют DIPEA (203 мкл, 1,16 ммоль, 3 экв.) и DIPCDI (240 мкл, 1,55 ммоль, 4 экв.). Смесь оставляют перемешиваться в течение 1 часа, затем протекание стадии циклизации контролируют с помощью ВЭЖХ. Растворитель удаляют выпариванием при пониженном давлении. Защищенный циклический пептид растворяют в TFA-H2O (19:1, 85 мл) и смесь оставляют перемешиваться в течение 1 часа. Растворитель удаляют выпариванием при пониженном давлении, добавляют диоксан (30 мл) и растворитель удаляют выпариванием при пониженном давлении (процесс повторяют три раза), затем добавляют H2O (40 мл) и лиофилизируют. Сырой продукт очищают с помощью ВЭЖХ (Kromasil C8, 5 мкм, 205×50 мм), изократическим 44%-ным ацетонитрилом (+0,05% TFA) в воде (+0,05% TFA), 55 мл/ч, детектирование проводят при 220 нм, с получением указанного в заголовке продукта (192 мг, 0,13 ммоль, выход 26%, 92,3%).
MALDI-TOF-MS, рассчитано для С75Н124N14O16: 1476,93.
Найдено: m/z 1500,12 [М+Na]+, 1515,97 [М+К]+.
1Н-ЯМР-спектр соединения (2,5 мМ, 500 МГц, H2O-D2O (9:1)) представлен в таблице II.
Пример 2
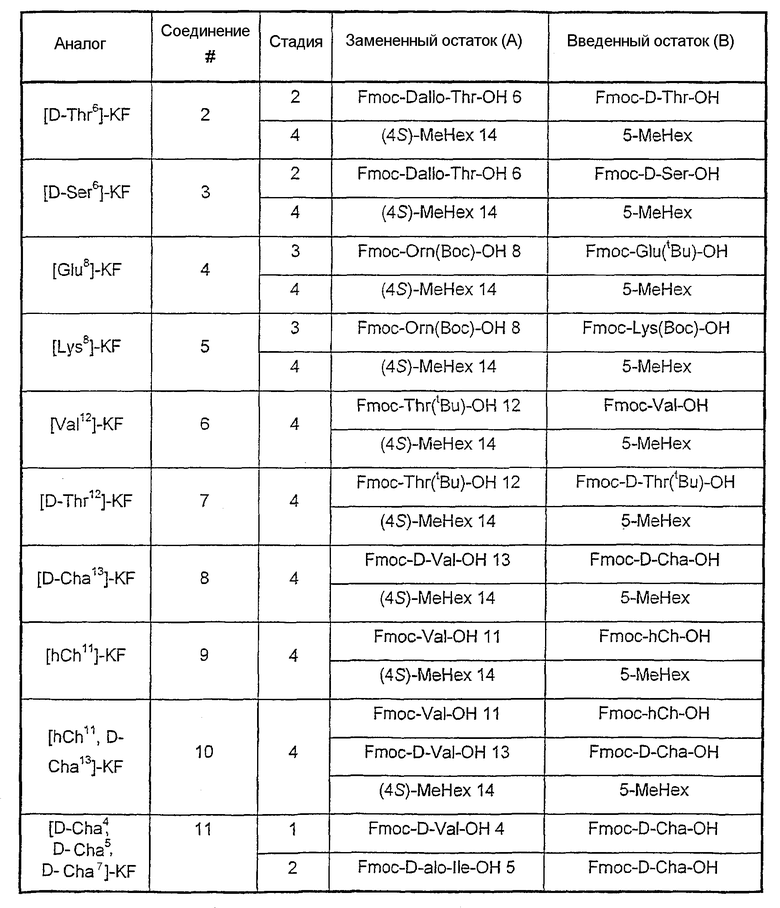
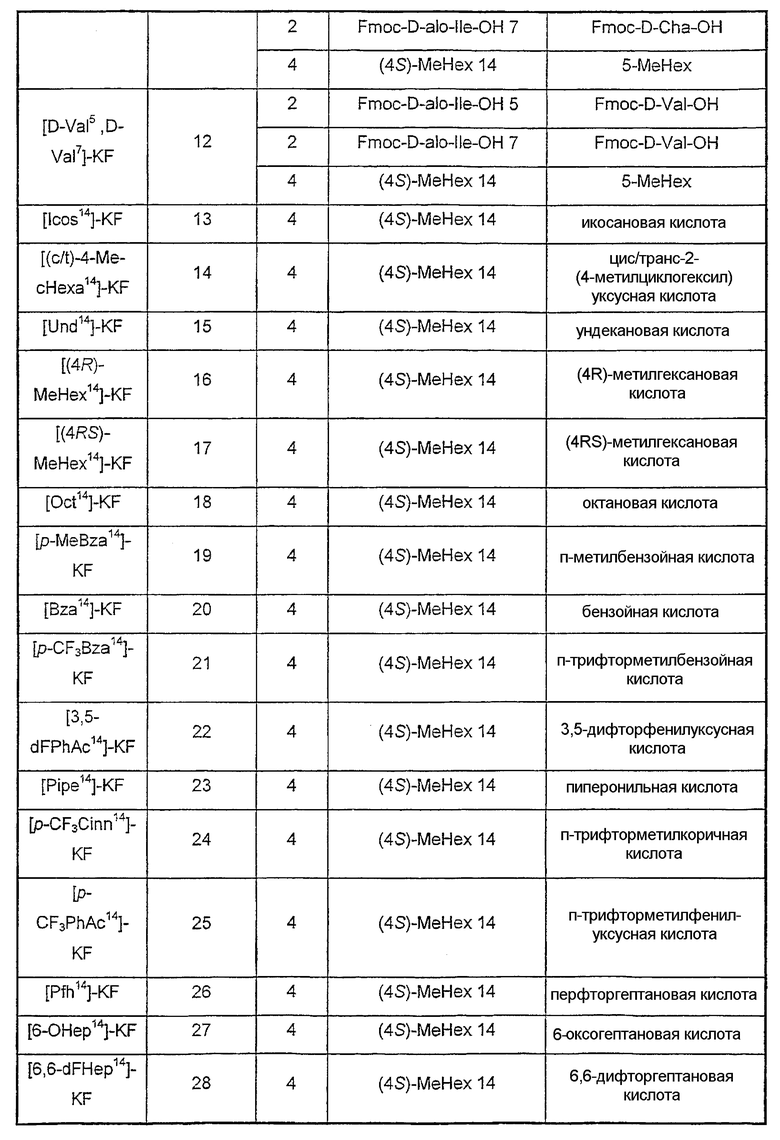
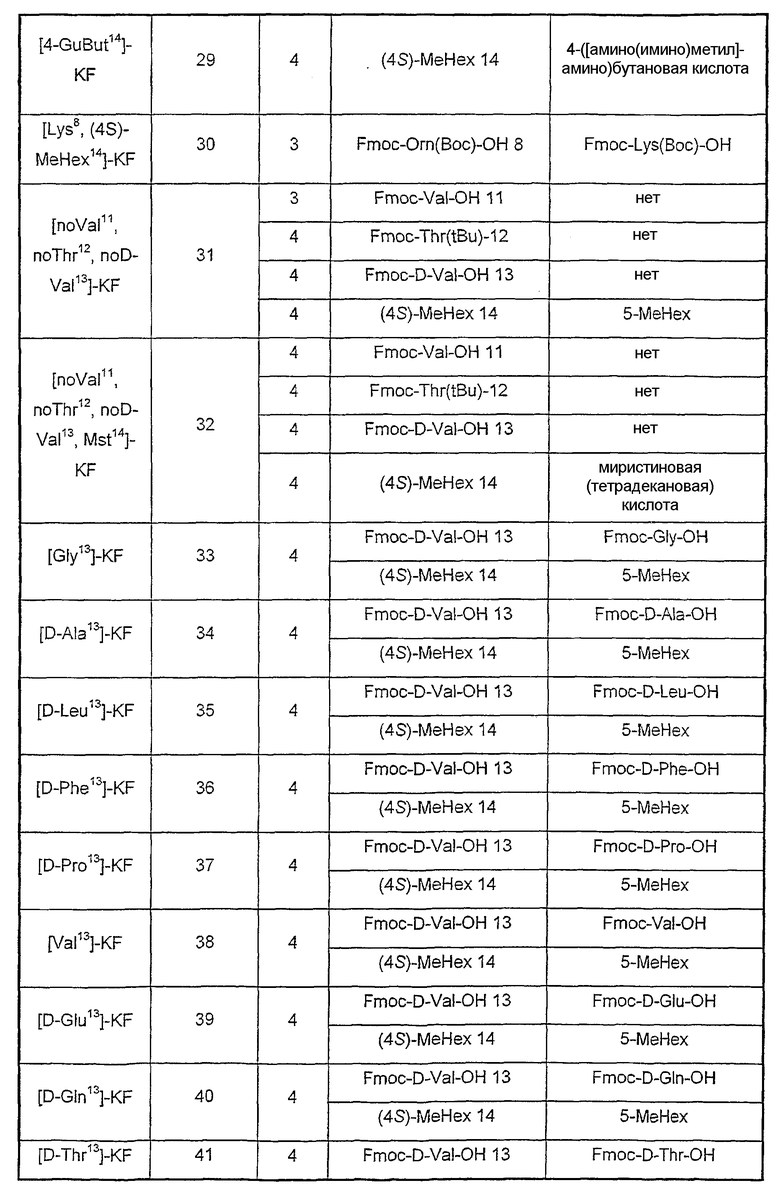
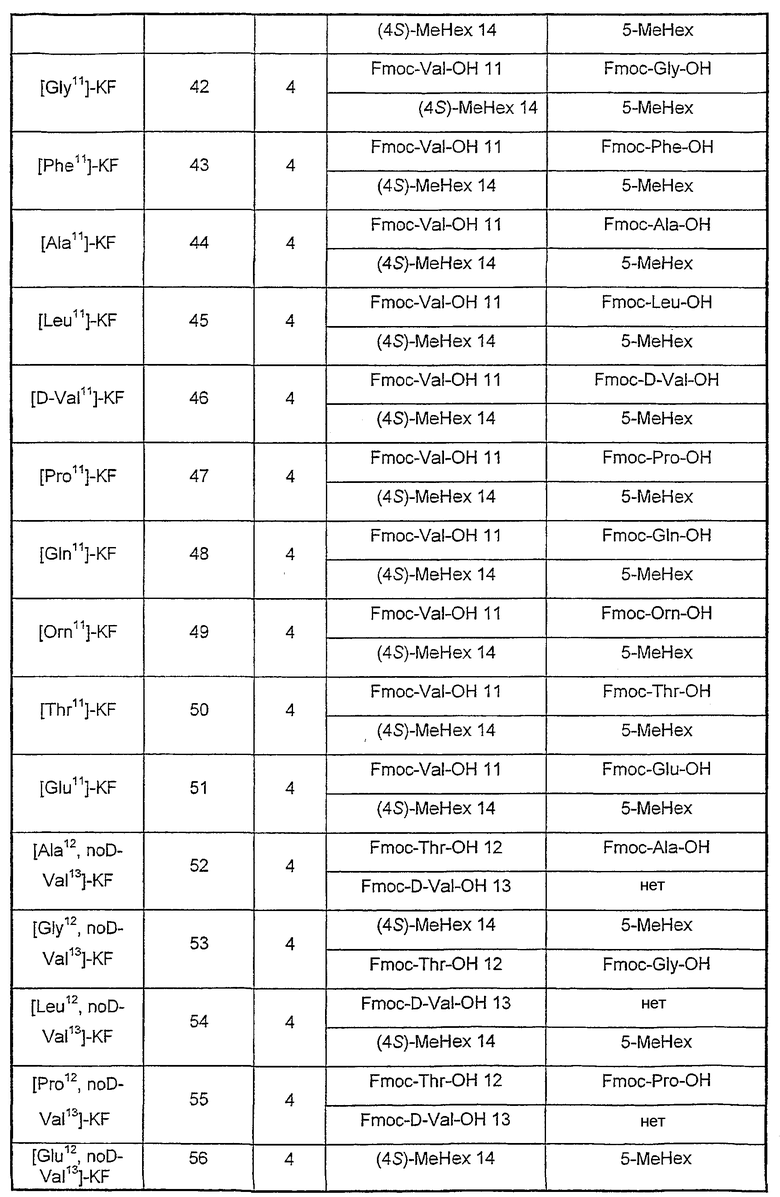
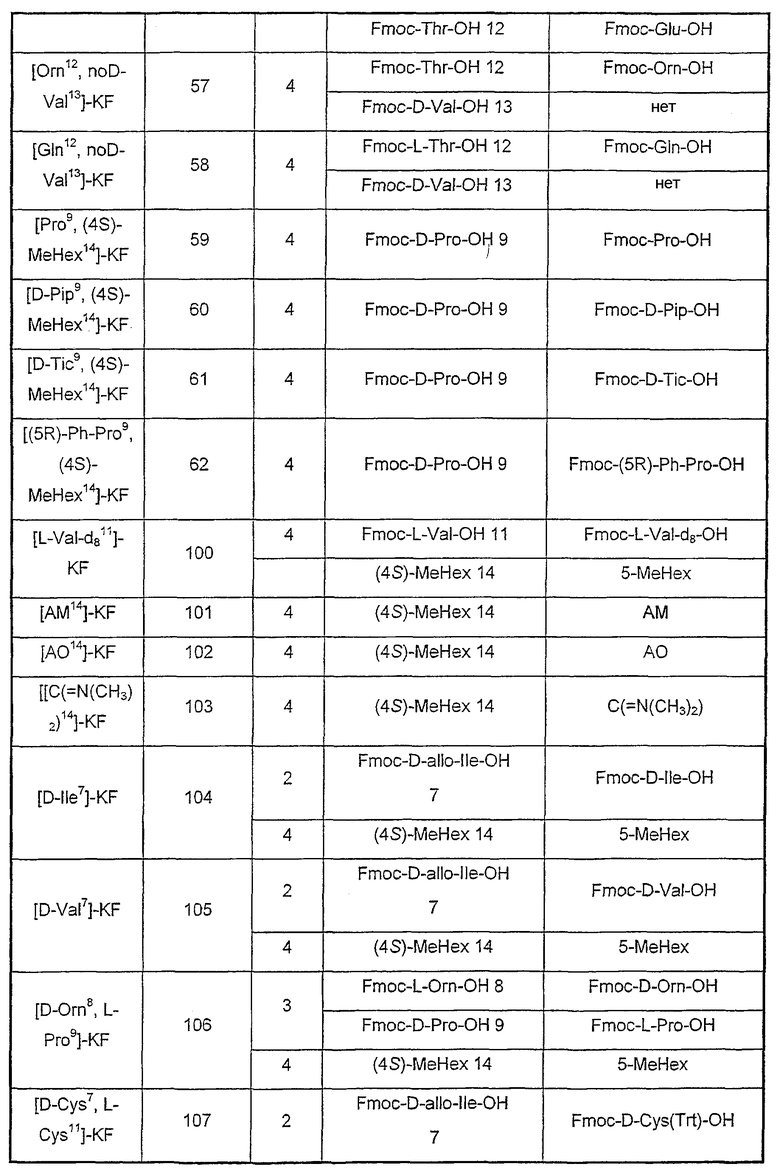
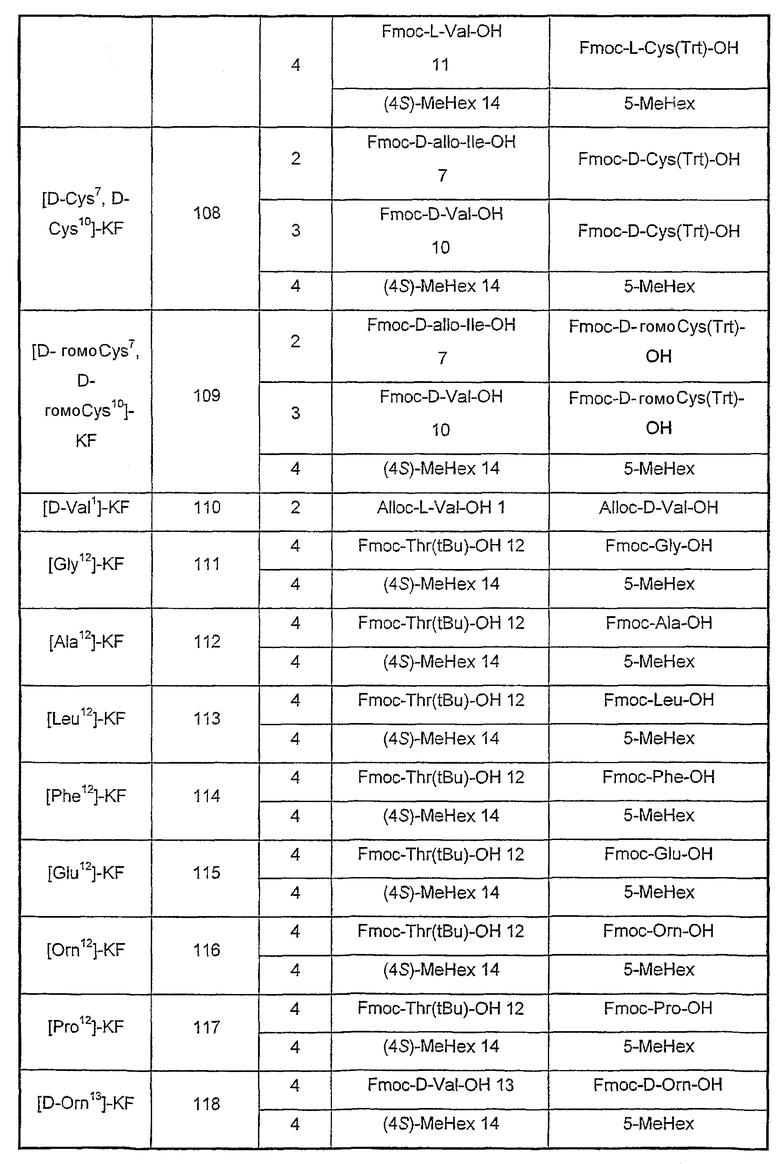
Аналоги, описанные в таблице III, синтезированы с помощью экспериментальных методик, как описанные в примере 1, за исключением стадии, указанной в колонке (стадия), где остаток(ки) (А) заменен(ы) другим(и) остатком(ами) (В) или удалены (нет).
Таблица III

Пример 3
5-MeHex-D-Val-Thr-Val-D-Val-D-Pro-Orn-D-allo-Ile-цикло[D-allo-Thr-D-alo-Ile-D-Val-hCh-(Z)-Dhb-Val] (соединение 63)
Экспериментальные методики, как описано в примере 1, за исключением того, что на стадии 4 (4S)-MeHex заменен 5-MeHex и стадию 5 осуществляют в соответствии со следующей экспериментальной методикой:
Alloc-группу удаляют из 5-MeHex-D-Val-Thr(tBu)-Val-D-Val-D-Pro-Orn(Boc)-D-allo-Ile-D-alo-Thr(Val-Alloc)-D-allo-Ile-D-Val-O-2-хлортритил-Ps (250 мг, начальная загрузка = 0,5 ммоль/г смолы) с помощью, как указано выше, Pd(PPh3)4 в присутствии PhSiH3 в атмосфере Ar. Fmoc-Thr-OH (свободная гидроксильная группа) (213,2 мг, 0,63 ммоль, 5 экв.) и Fmoc-hСhа-OH (254,0 мг, 0,63 ммоль, 5 экв.) последовательно добавляют к вышеуказанной пептидильной смоле, используя DIPCDI (96,8 мг, 0,63 ммоль, 5 экв.) и HOBt (85 мг, 0,63 ммоль, 5 экв.) в ДМФА. После экстенсивных промывок с помощью ДМФА (5×30 сек) пептидильную смолу обрабатывают EDC·HCl (479 мг, 2,5 ммоль, 20 экв.), CuCl (184 мг, 1,5 ммоль, 12 экв.) в CH2Cl2-ДМФА (1:9) в течение 6 дней. После экстенсивных промывок с помощью ДМФА, CH2Cl2 и ДМФА следуют экспериментальным протоколам, описанным в примере 1, для получения KF-аналога.
Пример 4
Аналоги, описанные в таблице IV, синтезированы с помощью экспериментальных методик, как описано в примере 1, за исключением того, что на стадии 4 (4S)-MeHex заменен 5-MeHex; стадию 5 проводят, как описано в примере 3, но включая Fmoc-Рhе-OH вместо Fmoc-hСh-OH, и на стадии, указанной в колонке (Стадия), остаток(ки) (А) заменен(ы) другим(и) остатком(ами) (В) или удален(ы) (нет). В случае аналогов 64-66 не осуществляют реакцию дегидратации, потому что аналоги гидрированы.
Таблица IV

Пример 5
Аналоги, описанные в таблице V, синтезированы с помощью экспериментальных методик, как описано в примере 1, за исключением того, что стадию 5 проводят, как описано в примере 3, и на стадии, указанной в колонке (Стадия), остаток(ки) (А) заменен(ы) другим(и) остатком(ами) (В). Кроме того, перед твердофазной дегидратацией Fmoc-группу удаляют, как описано в общих методиках, и затем аминогруппу защищают в форме Alloc путем взаимодействия с Alloc-OSu (5 экв.) в присутствии DIPEA (5 экв.), используя ДМФА в качестве растворителя (2 часа).
Таблица V


Пример 6
[N(Me) 2 ,N'(Me) 2 -Arg 8 ]-кахалалид F (соединение 89)
DIPEA (1,73 мкл, 10,17 мкмоль) и HATU (3,86 мг, 10,15 мкмоль) добавляют к кахалалиду F (10,0 мг, 6,76 мкмоль), растворенному в ДМФА (5 мл). За взаимодействием следят с помощью ВЭЖХ и спустя 4 часа ДМФА удаляют при пониженном давлении и остаток растворяют в CH3CN-H2O-AcOH (4,5:4,5:1, 20 мл), лиофилизируют и очищают путем полупрепаративной ВЭЖХ с получением указанного в заголовке аналога (4,5 мг, выход 40%).
Пример 7
[N(Me,Ph),N'(Me) 2 -Arg 8 ]-кахалалид F (соединение 90)
Экспериментальные методики, как описано в примере 6, но HAPyU (4,87 мг, 10,15 мкмоль) используют вместо HATU, получая 1,63 мг, 12%.
Пример 8
[N(СН 2 ) 4 ,N'(Me) 2 -Arg 8 ]-кахалалид F (соединение 91)
Экспериментальные методики, как описано в примере 6, но М2А (4,12 мг, 10,14 мкмоль) используют вместо HATU, получая 5,2 мг, 46%.
Пример 9
[N(СН 2 ) 4 ,N'(СН 2 ) 4 -Arg 8 ]-кахалалид F (соединение 92а) и [N δ (СНN(СН 2 ) 4 ,N'(СН 2 ) 4 )-Оrn 8 ]-кахалалид F (соединение 92b)
Экспериментальные методики, как описано в примере 6, но BTFFH (содержащий приблизительно 50% примеси 1,1'-(фторметилен)дипирролидина) (3,16 мг, 10,16 мкмоль) используют вместо HATU. Получают два продукта, 92а и 92b, и их невозможно разделить (4,0 мг смеси в пропорции 1:1, 35,6%).
ВЭЖХ (условие А, колонка А), tR: 20,8 мин (92b) с выходом 45% и tR: 20,9 мин (92а) с выходом 46%.
EM (MALDI-TOF, m/z): (92а) рассчитано 1627,05; найдено 1630,8 [M+H]+; 1652,6 [M+Na]+. (92b) рассчитано 1629,06; найдено 1632,7 [M+H]+; 1670,6 [M+К]+.
Пример 10
[N ε (Me) 3 -Lys 8 ,(4S)-MeHex 14 ]-КF (соединение 93)
DIPEA (10 мкл, 58,8 мкмоль) и MeI (6 мкл, 0,100 ммоль) добавляют к [Lys8,(4S)-MeHex14]-кахалалиду F (соединение 30) (5,0 мг, 3,35 мкмоль), растворенному в ДМФА/DCM (1:1, 5 мл). За взаимодействием следят с помощью ВЭЖХ и спустя 12 часов растворители удаляют при пониженном давлении и остаток растворяют в CH3CN-H2O-AcOH (4,5:4,5:1, 20 мл), лиофилизируют и очищают путем полупрепаративной ВЭЖХ с получением указанного в заголовке аналога (2,2 мг, выход 44%).
Пример 11
[Thr(OTfa) 12 ,4(S)-MeHex 14 ]-кахалалид F (соединение 94)
[4(S)-MeHex14]-кахалалид F (соединение 1; 10,0 мг, 6,7 мкмоль) растворяют в TFA/DCM (1:1, 20 мл) и оставляют перемешиваться в течение 3 дней при комнатной температуре. Затем растворитель удаляют при пониженном давлении и остаток растворяют в H2O-CH3CN (1:9) и немедленно очищают при минимальном времени, в течение которого образец растворяется в H2O. Соответствующие фракции указанного в заголовке аналога собирают в круглодонную колбу, погруженную в жидкий азот, и лиофилизируют (2,5 мг, выход 25%).
Пример 12
[Orn(N δ Tfa) 8 ,Thr(OTfa) 12 ,4(S)-MeHex 14 ]-кахалалид F (соединение 95)
[(4S)-MeHex14]-KF (соединение 1; 10 мг, 6,7 мкмоль) растворяют в DCM (8 мл), добавляют TFAA (18,9 мкл, 134 мкмоль) и DIPEA (22,8 мкл, 134 мкмоль) и оставляют для взаимодействия в течение 12 часов. Затем растворитель удаляют при пониженном давлении, снова растворяют в H2O-CH3CN (1:1) и немедленно очищают (2,0 мг, выход 20%).
Пример 13
[Orn(N δ Tfa) 8 ,4(S)-MeHex 14 ]-кахалалид F (соединение 96)
Экспериментальные методики, как описано в примере 12, но перед очисткой аналог растворяют в H2O-CH3CN (1:1), оставляют раствор на 2 часа и очищают (4,3 мг, выход 43%).
Пример 14
[Thr(OTfa) 12 ,Lit(OTfa) 14 ]-кахалалид F (соединение 97)
Экспериментальные методики, как описано в примере 1, за исключением того, что на стадии 4 (4S)-MeHex заменен Lit-OH и на стадии 7 циклический пептид растворяют в TFA/DCM (1:1, 20 мл) и оставляют перемешиваться в течение 3 дней при комнатной температуре. После этого времени растворитель удаляют при пониженном давлении из аликвоты (10%) и твердый остаток растворяют в H2O-CH3CN (1:9) и немедленно очищают при минимальном времени, в течение которого образец растворяется в Н2О. Фракции, соответствующие указанному в заголовке аналогу, собирают в круглодонную колбу, погруженную в жидкий азот, и лиофилизируют (5,6 мг, выход 6% от исходной смолы).
Пример 15
[no5-MeHex 14 -N-(Hep) 2 -D-Val 13 ]-кахалалид F (соединение 98)
Экспериментальные методики, как описано в примере 1, за исключением того, что на стадии 4 гептанальдегид (60,65 мкл, 0,75 ммоль, 5 экв.), растворенный в ДМФА-АсОН (99:1, 2 мл), добавляют к Н-D-Val-Thr(tBu)-Val-D-Val-D-Pro-Orn(Boc)-D-allo-Ile-D-alo-Thr(Val-Alloc)-D-allo-Ile-D-Val-O-2-хлортритил-Ps (300 мг, начальная загрузка = 0,5 ммоль/г смолы) и спустя 5 мин добавляют NaBH3CN (28,28 мл, 0,45 ммоль, 3 экв.), растворенный в ДМФА-АсОН (99:1, 1 мл), и смесь оставляют для взаимодействия в течение 2 часов. Эту обработку повторяют еще два раза при мониторинге взаимодействия с помощью ВЭЖХ.
Пример 16
[Orn(Biot) 8 ]-кахалалид F (соединение 99)
Кахалалид F (150,0 мг, 94,3 мкмоль), d-биотин (37,0 мг, 151,5 мкмоль) и HATU (114,0 мг, 299,8 мкмоль) растворяют в безводном DCM (6,0 мл) в атмосфере Ar и добавляют NMM (58 мкл, 524,0 мкмоль). Смесь оставляют перемешиваться в течение 20 часов. Затем растворитель удаляют при пониженном давлении и остаток растворяют в МеОН и очищают. Фракции, соответствующие указанному в заголовке аналогу, лиофилизируют (74,0 мг, выход 43%).
Характеристика аналогов кахалалида F
Характеристика показана в таблице VI и таблице VII. ЕМ в таблице VI соответствует MALDI-TOF, и точную массу рассчитывают с помощью Chemwind 6,0, а ЕМ в таблице VII соответствует химической ионизации при атмосферном давлении (АРСI) с образованием положительных ионов.
Таблица VI


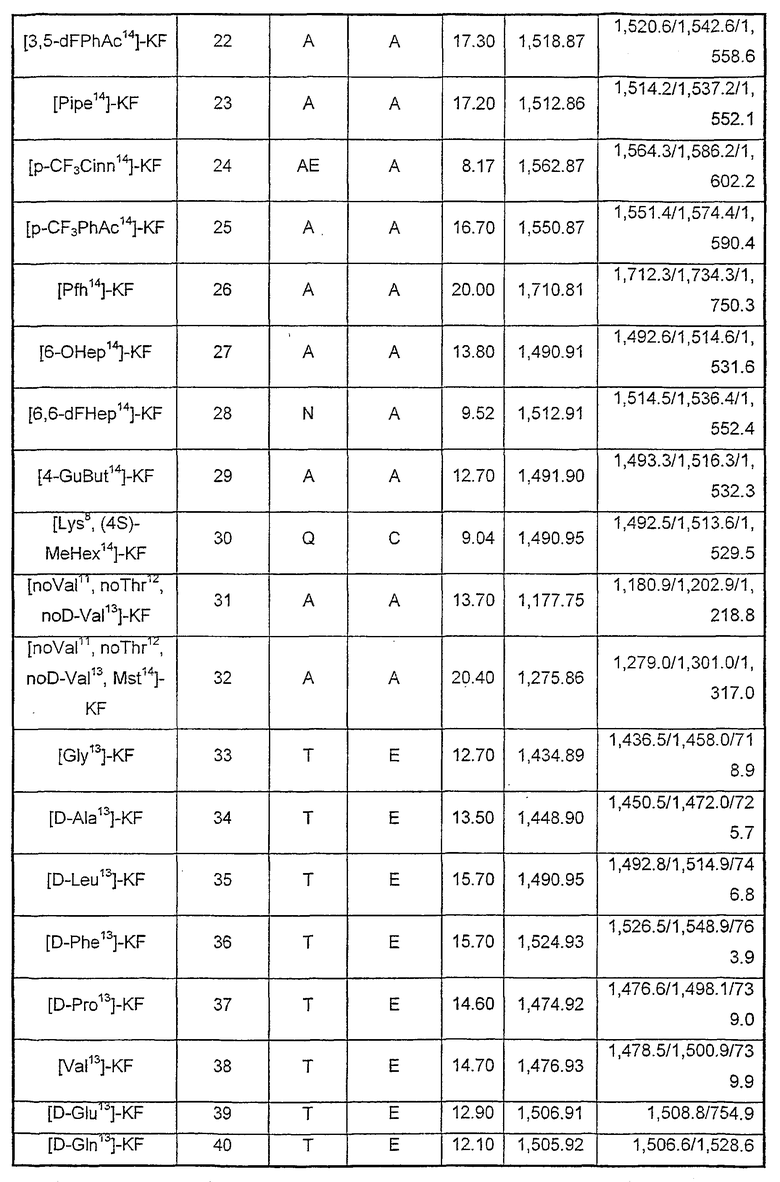



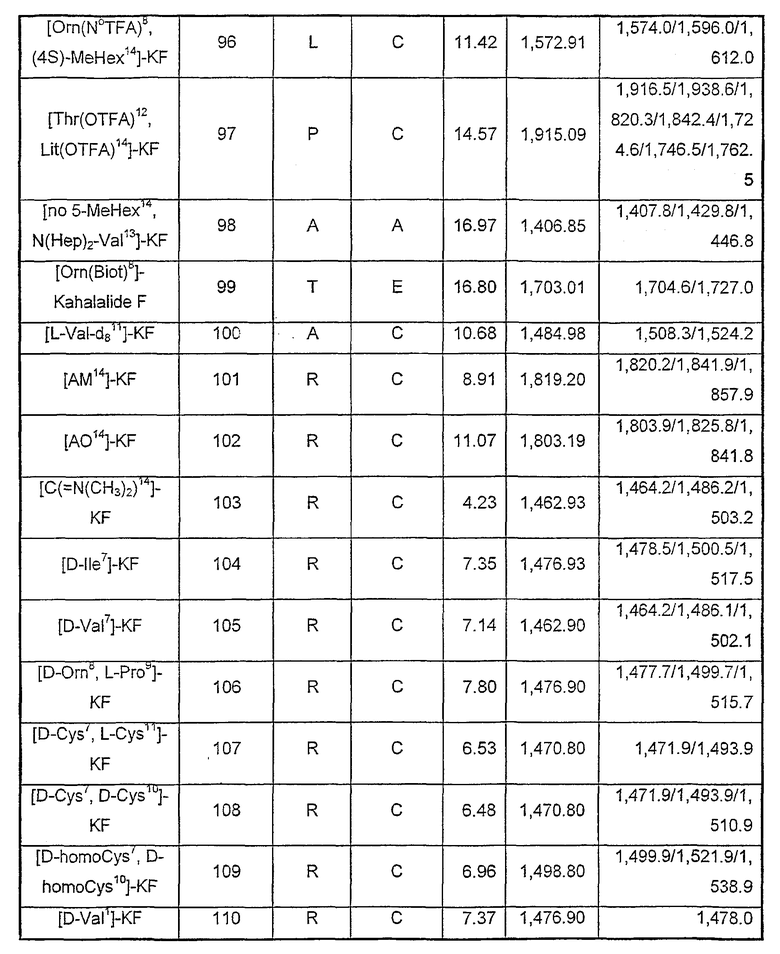
Таблица VII


Пример 17
Биологическая активность
Биоактивность соединений согласно данному изобретению продемонстрирована с помощью результатов в следующих таблицах, полученных в соответствии с методикой, описанной следующим образом.
Завершением этого анализа считается прекращение роста «in vitro» опухолевой клеточной культуры посредством непрерывной обработки клеток исследуемым образцом.
Линии клеток
(плевральный выпот)
Колориметрический тип анализа с использованием реакции сульфородамина В (SRB) приспособлен для количественного определения роста и жизнеспособности клеток [следуя методике, описанной Philip Skehan и др. (1990), New colorimetric cytotoxicity assay for anticancer drug screening, J. Natl. Cancer Inst., 82:1107-1112].
В этом виде анализа используют 96-луночные микропланшеты для клеточных культур, имеющие диаметр 9 мм (Faircloth, 1988; Mosmann, 1983). Большинство линий клеток получали из американской коллекции типовых культур (АТСС), происходящих от различных типов рака человека.
Клетки содержали в среде RPMI 1640 с 10% фетальной телячьей сывороткой, дополненной 0,1 г/л пенициллина и 0,1 г/л сульфата стрептомицина, и затем инкубировали при 37оС, 5% СО2 и 98% влажности. Для экспериментов клетки собирали из субконфлюэнтных культур, используя трипсин, и повторно суспендировали в свежей среде перед культивированием.
Клетки высевали в 96-луночные титрационные микропланшеты, по 5×103 клеток на лунку, в аликвотах среды по 195 мкл, и оставляли их для прикрепления к поверхности планшета путем культивирования в свободной от лекарственного средства среде в течение 18 часов. Потом образцы добавляли в аликвотах по 5 мкл в диапазоне от 10 до 10-8 мкг/мл, растворенные в ДМСО:EtOH:PBS (0,5:0,5:99). После 48 часов воздействия противоопухолевый эффект измеряли с помощью методологии SRB: клетки фиксировали добавлением 50 мкл холодной 50%-ной (мас./об.) трихлоруксусной кислоты (ТСА) и инкубировали в течение 60 мин при 4оС. Планшеты промывали деионизированной водой и сушили. Добавляли 100 мкл раствора SRB (0,4 мас./об.% в 1% уксусной кислоте) в каждую лунку на микротитрационном планшете и инкубировали в течение 10 мин при комнатной температуре. Несвязанный SRB удаляли путем промывки 1% уксусной кислотой. Планшеты высушивали на воздухе и связанный краситель солюбилизировали с помощью Трис-буфера. Оптические плотности считывали на автоматическом спектрофотометрическом планшет-ридере при одной длине волны 490 нм.
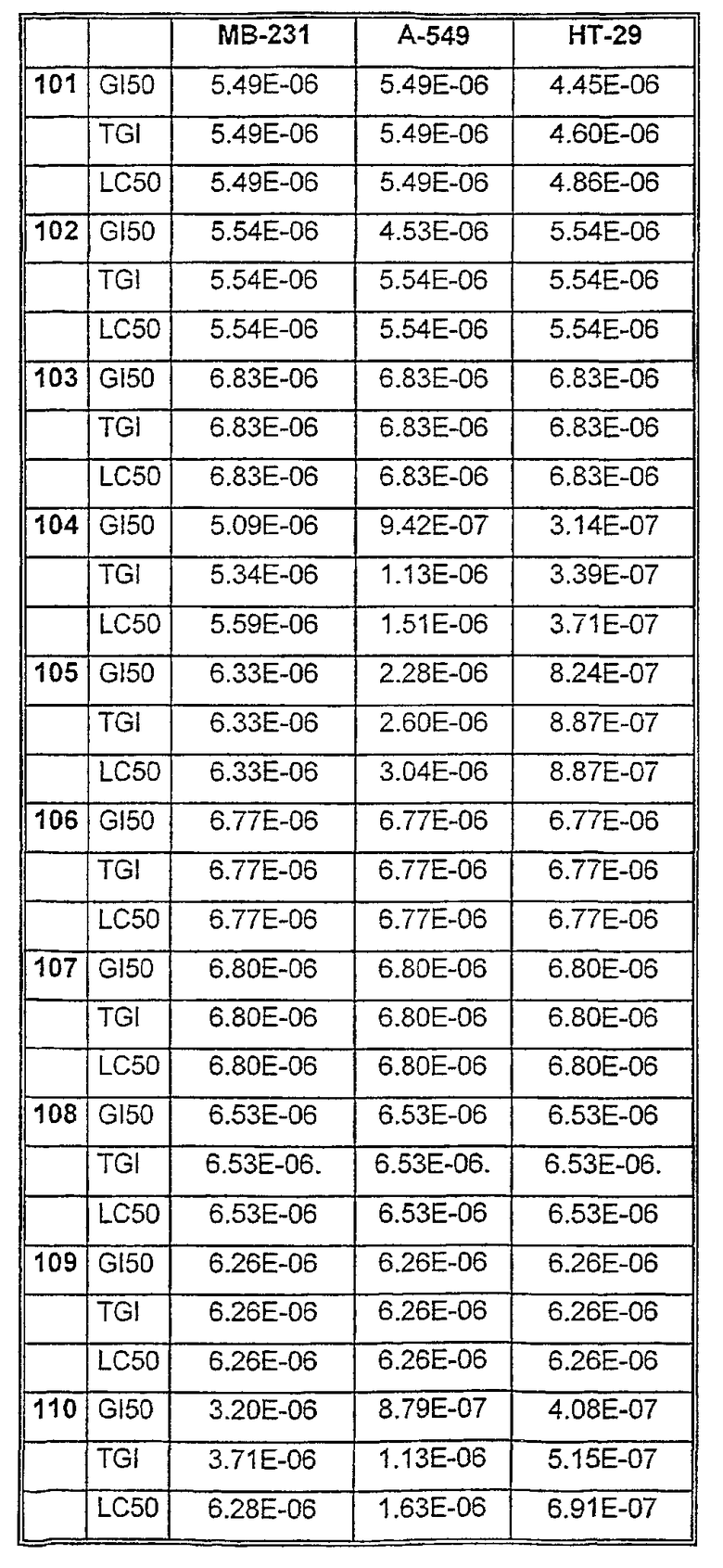
Средние значения ± стандартное отклонение данных рассчитывали из трех лунок. Некоторые параметры клеточных ответов могут быть рассчитаны: GI = ингибирование роста, TGI = полное ингибирование роста (цитостатический эффект) и LC = лизис клеток (цитотоксический эффект).
Таблицы VIII-IX иллюстрируют данные о биологической активности соединений согласно настоящему изобретению.
Таблица VIII
Данные об активности (мол.)
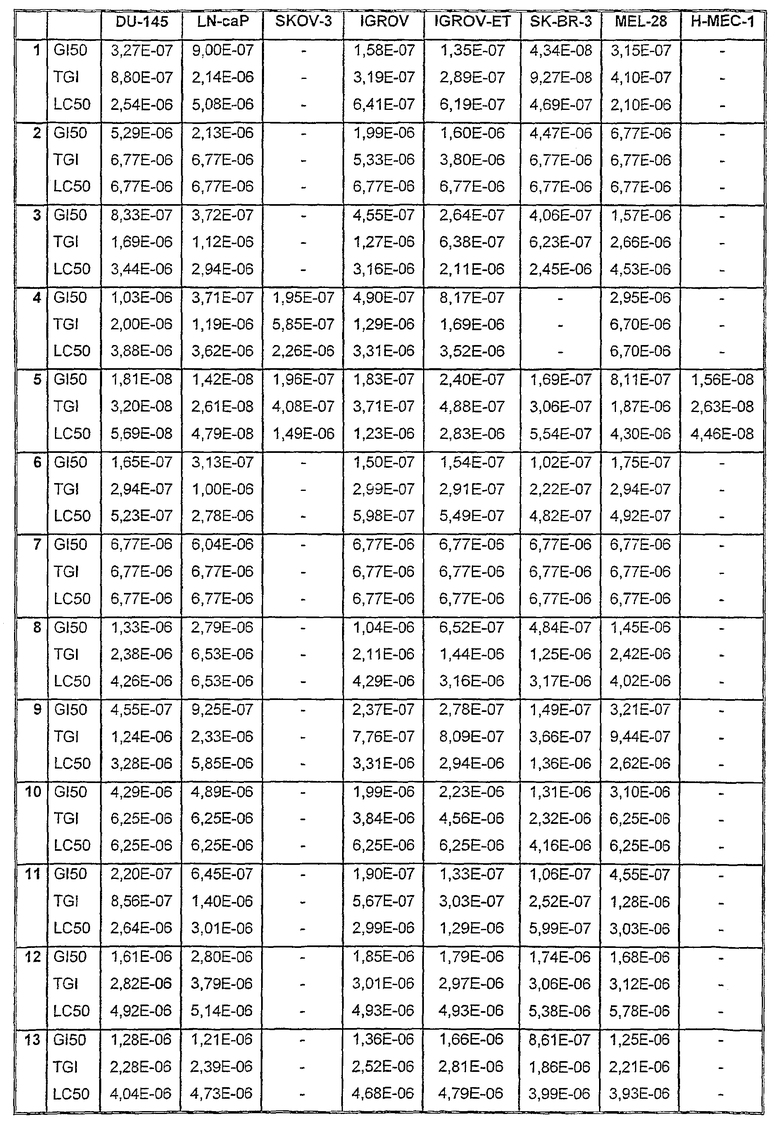

Таблица VIII (продолжение)
Данные об активности (мол.)
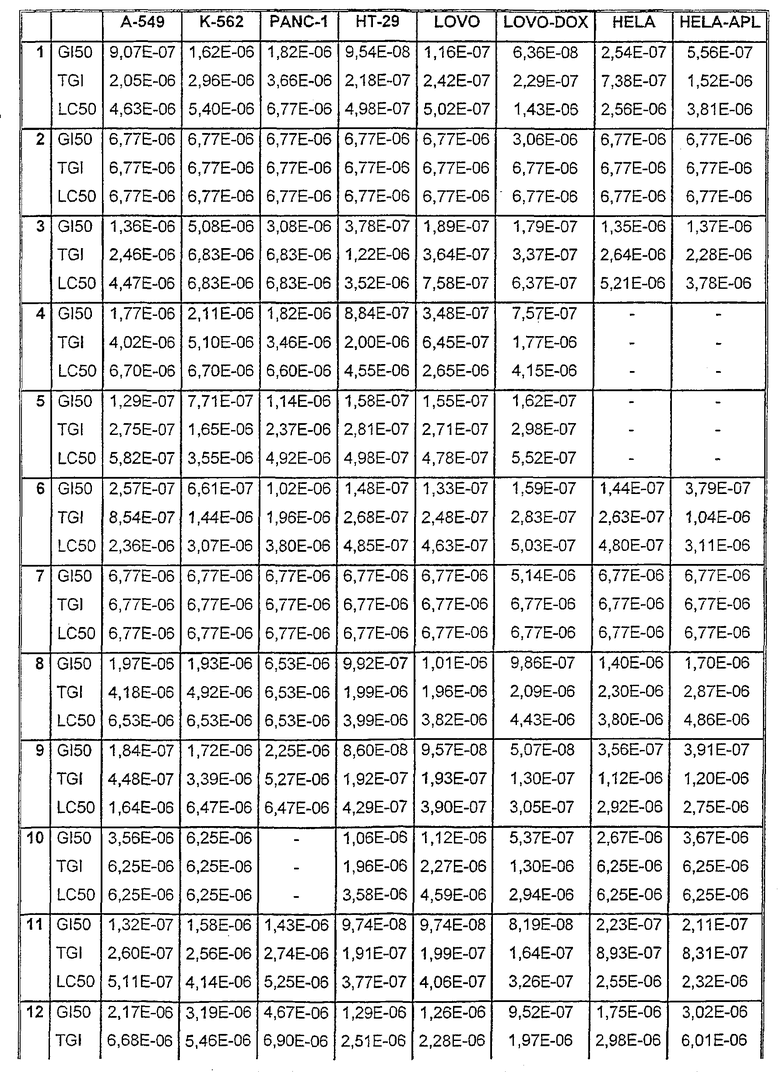

Таблица VIII (продолжение)
Данные об активности (мол.)

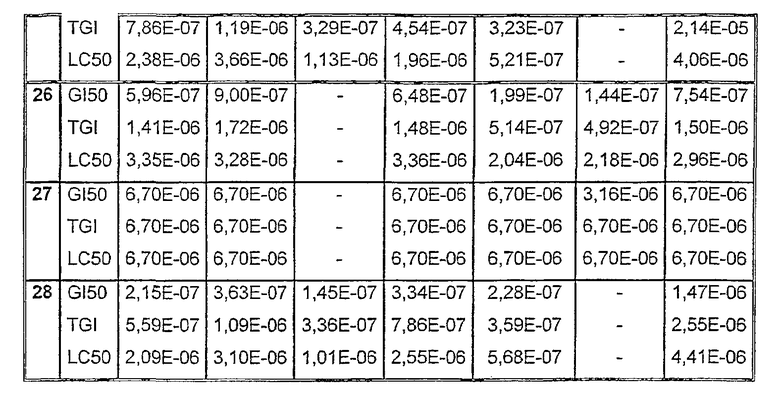
Таблица VIII (продолжение)
Данные об активности (мол.)
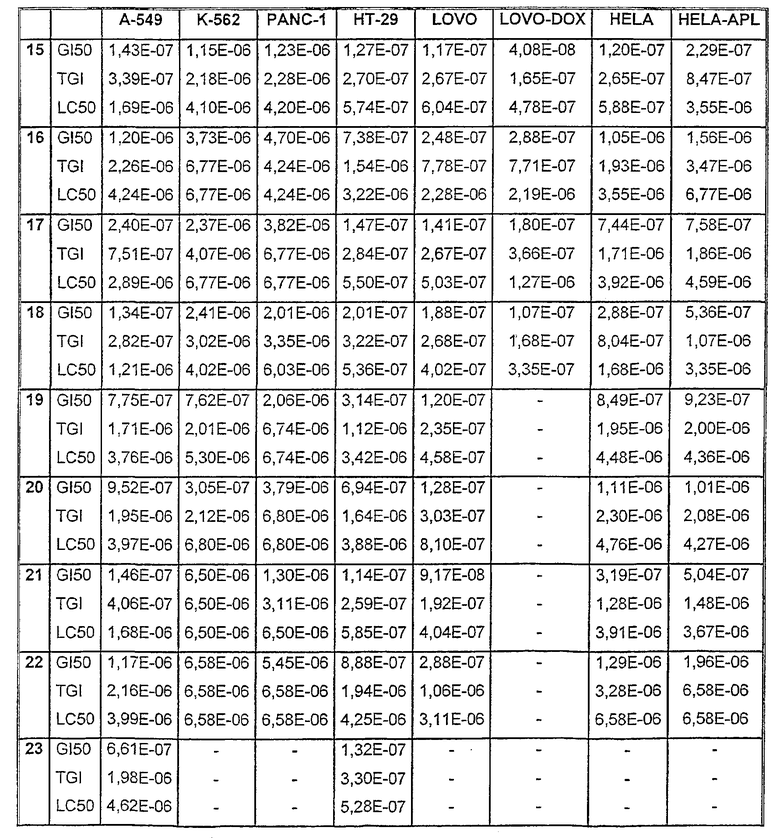

Таблица VIII (продолжение)
Данные об активности (мол.)

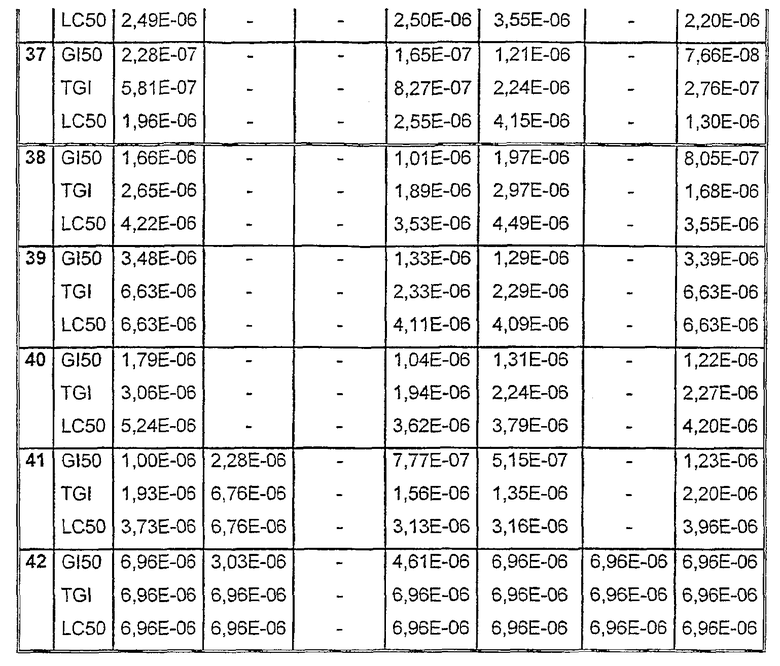
Таблица VIII (продолжение)
Данные об активности (мол.)


Таблица VIII (продолжение)
Данные об активности (мол.)

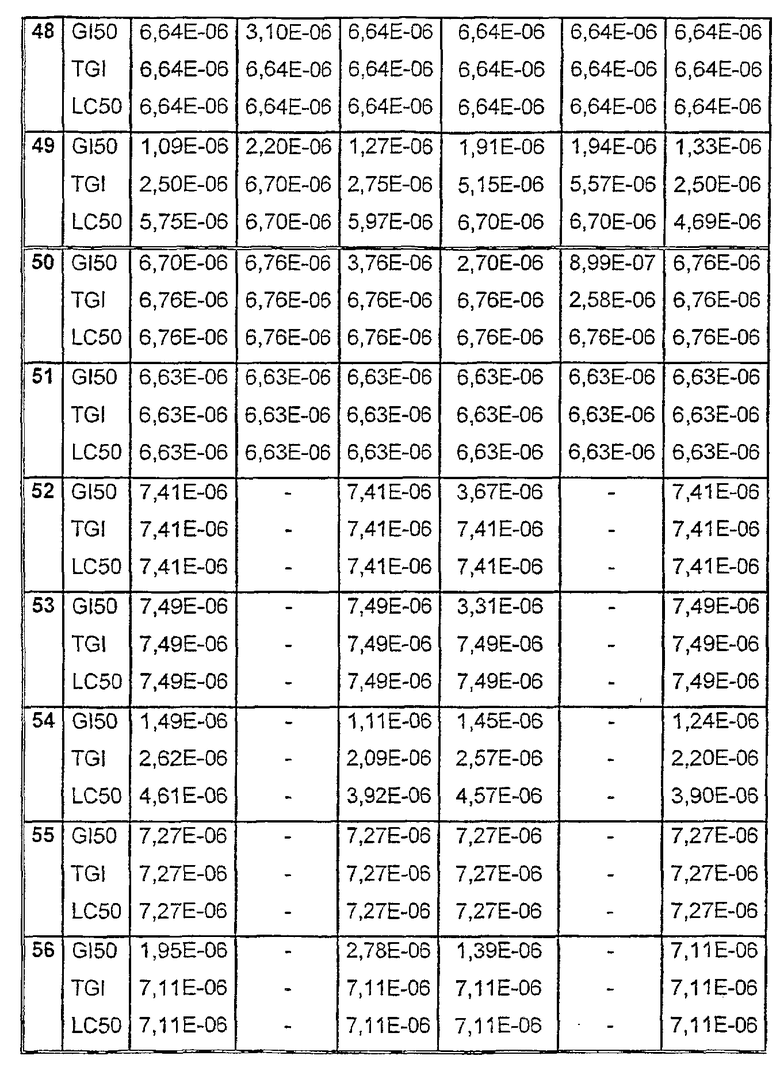
Таблица VIII (продолжение)
Данные об активности (мол.)
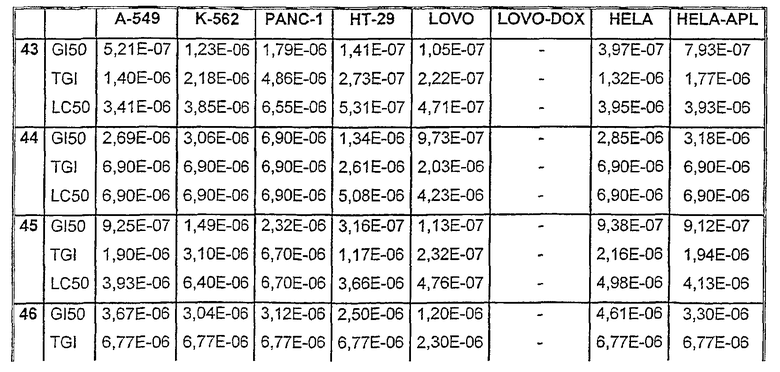
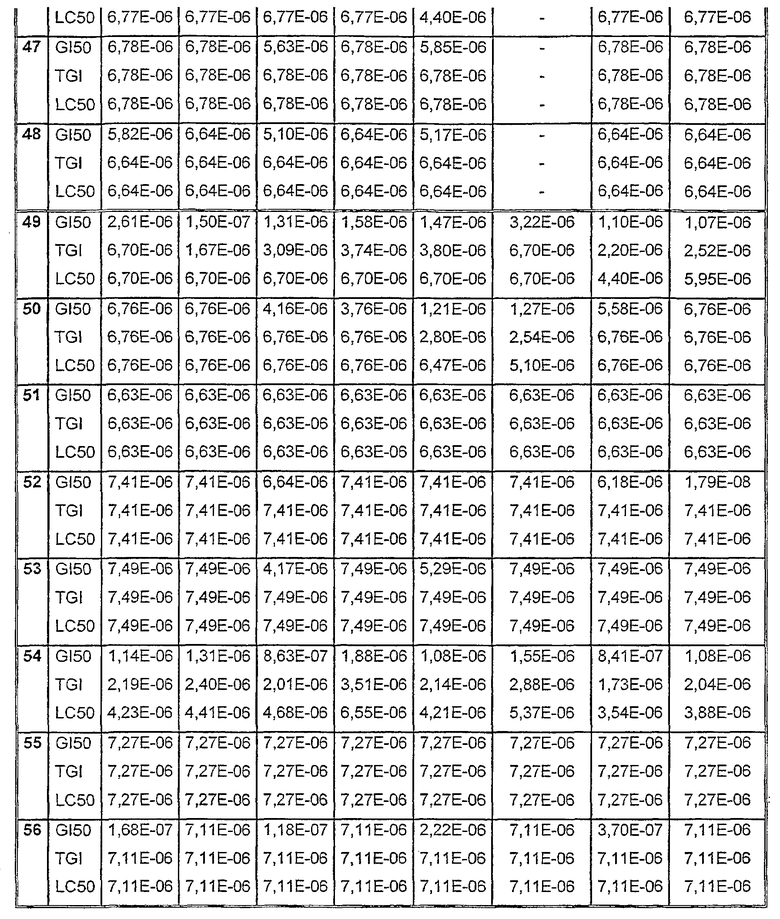
Таблица VIII (продолжение)
Данные об активности (мол.)

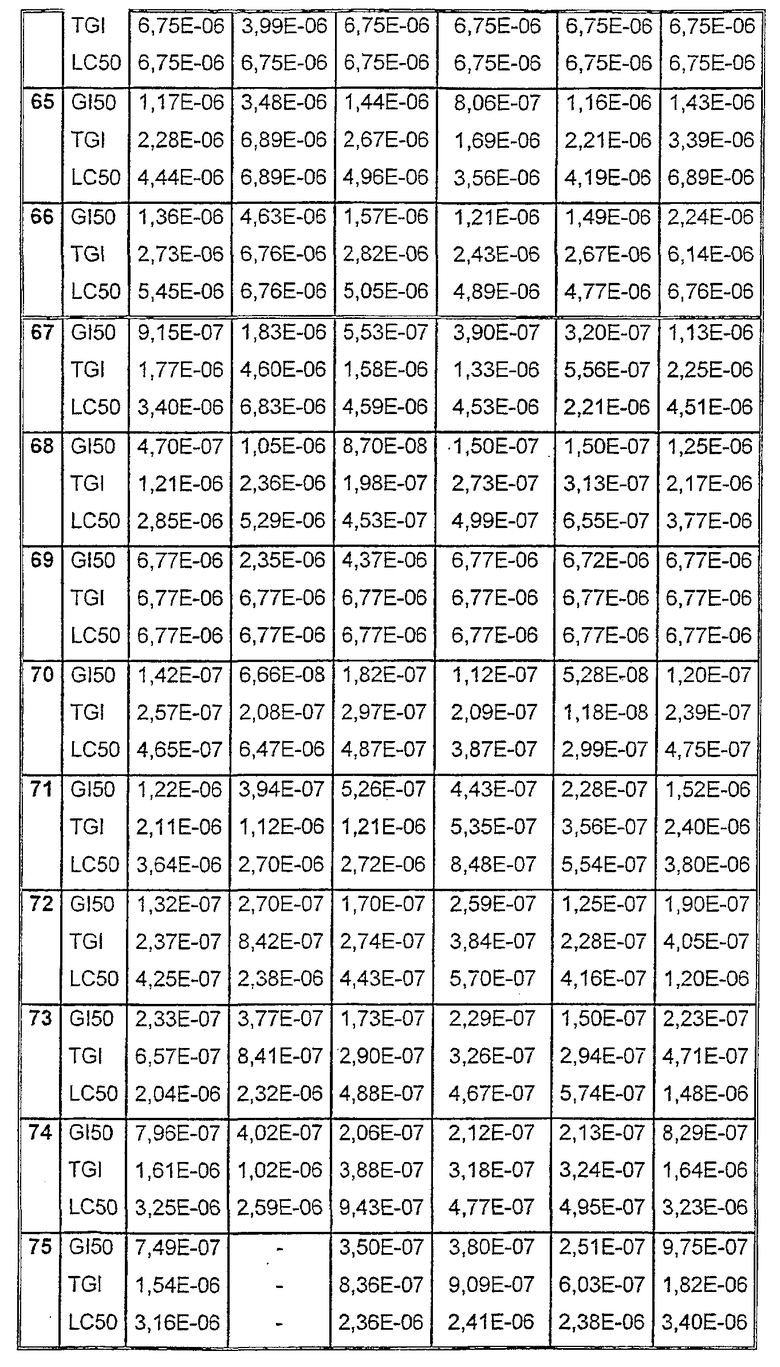
Таблица VIII (продолжение)
Данные об активности (мол.)

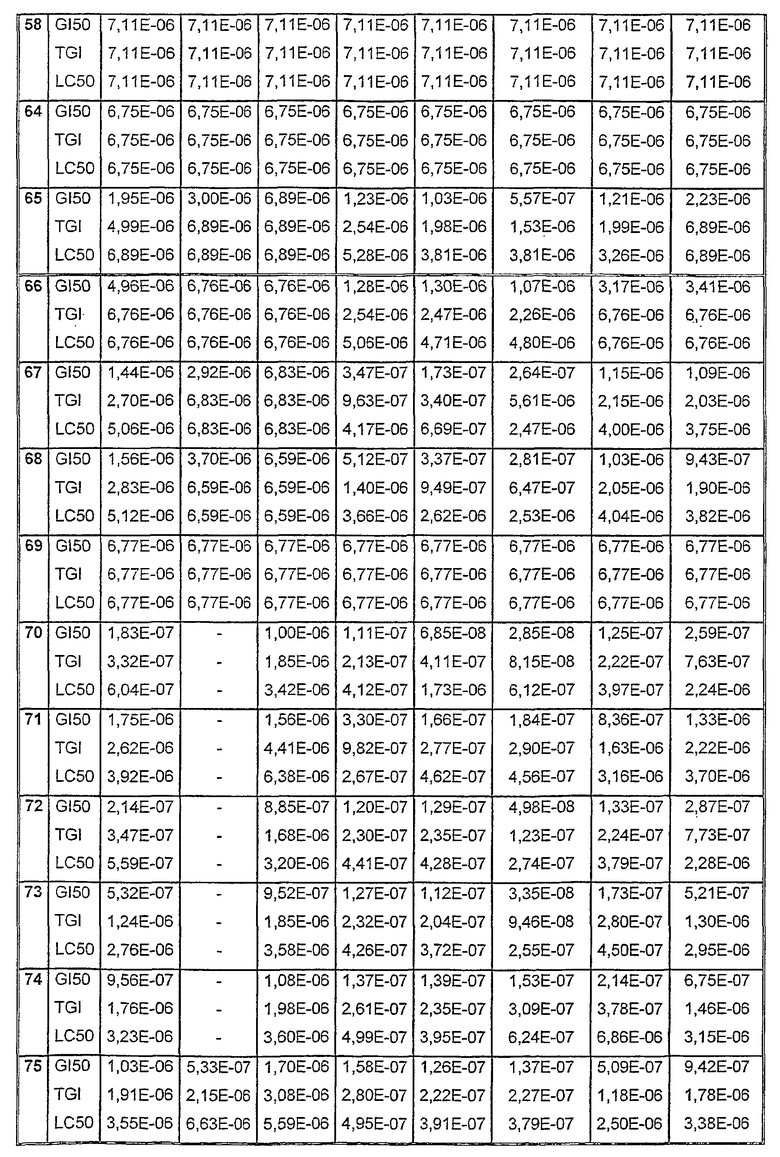
Таблица VIII (продолжение)
Данные об активности (мол.)
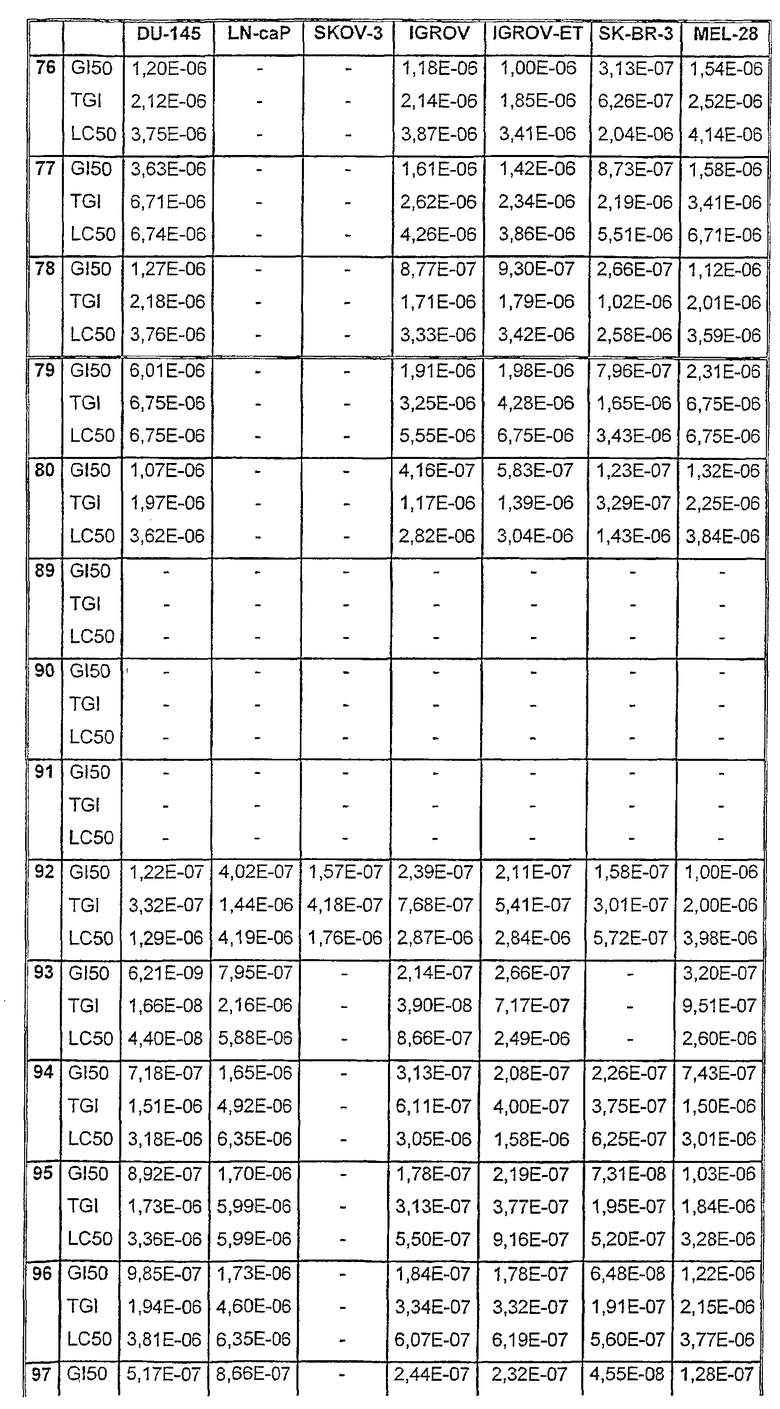

Таблица VIII (продолжение)
Данные об активности (мол.)


Таблица VIII (продолжение)
Данные об активности (мол.)
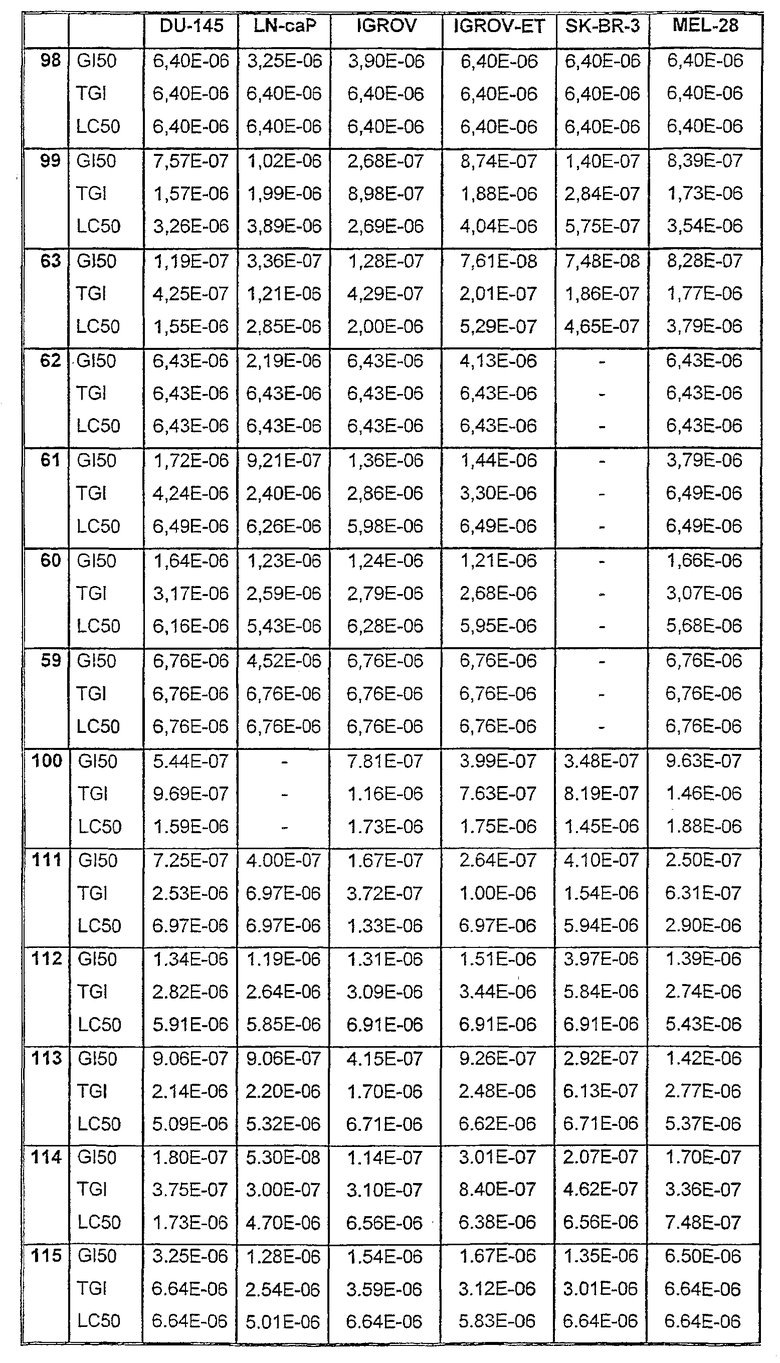
Таблица VIII (продолжение)
Данные об активности (мол.)
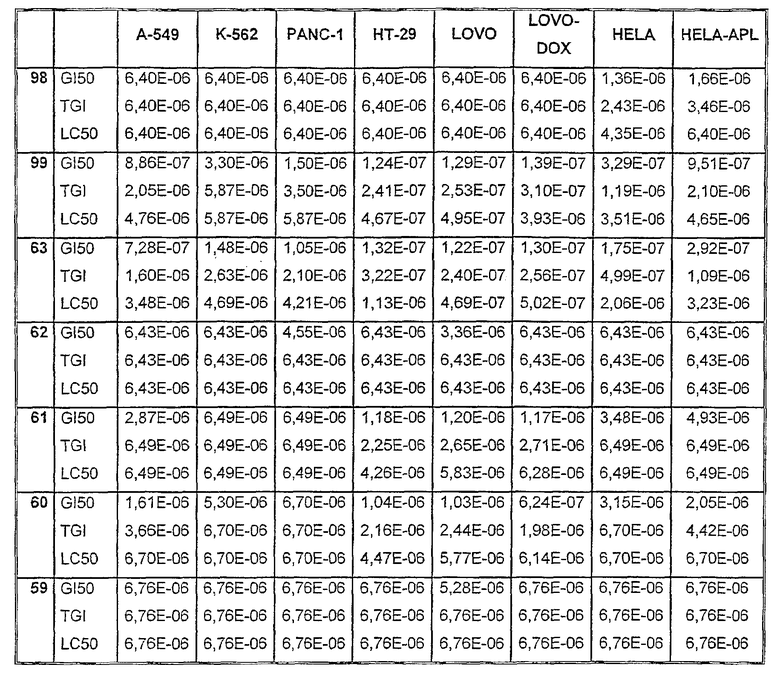
Таблица VIII (продолжение)
Данные об активности (мол.)
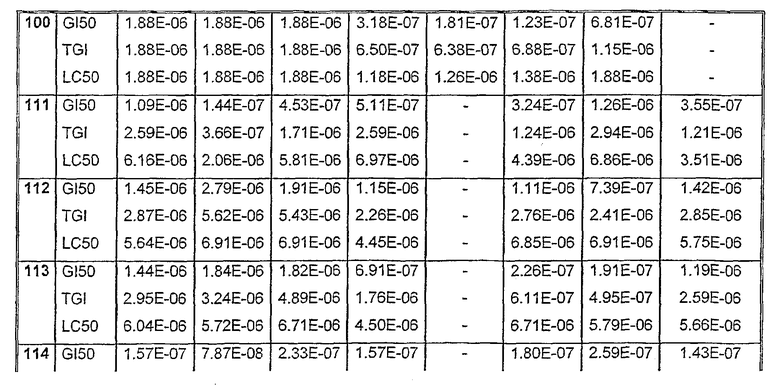

Таблица VIII (продолжение)
Данные об активности (мол.)
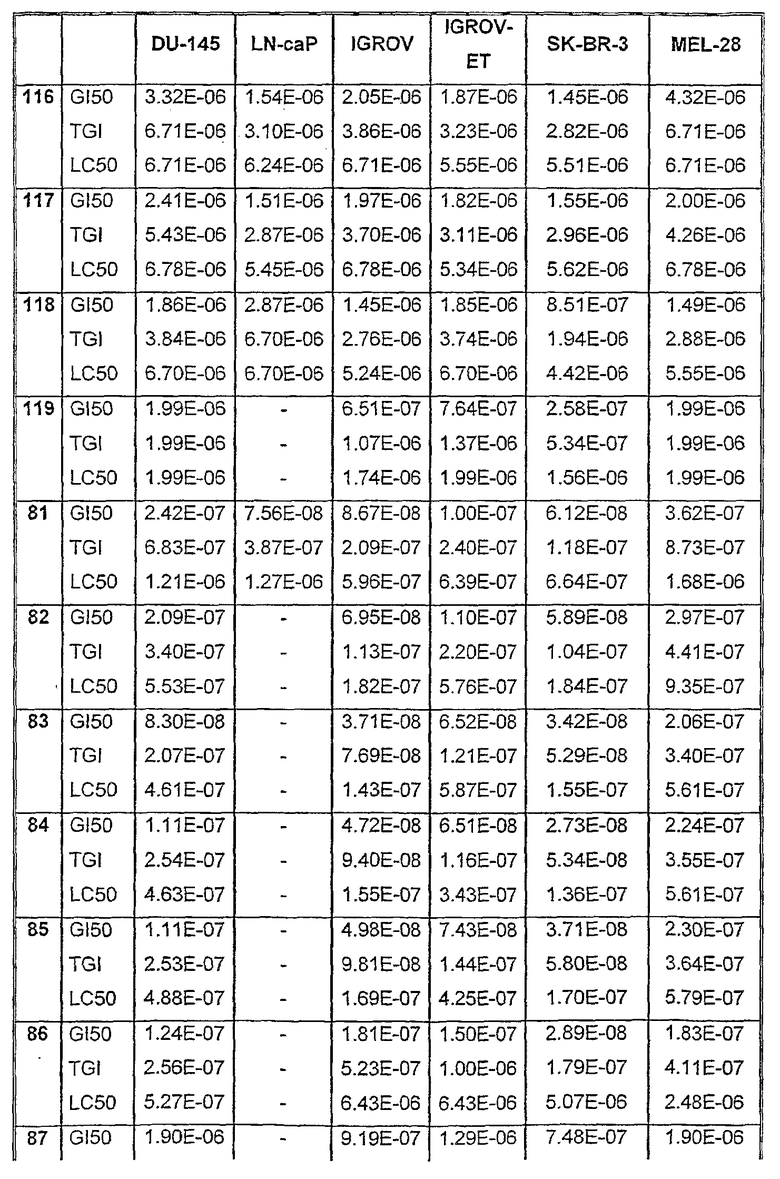

Таблица VIII (продолжение)
Данные об активности (мол.)

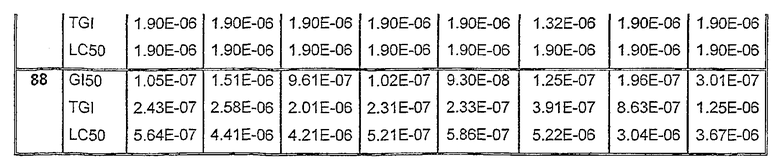
Таблица IХ
Данные об активности (мол.)
| название | год | авторы | номер документа |
|---|---|---|---|
| КАХАЛАЛИДНЫЕ СОЕДИНЕНИЯ | 2001 |
|
RU2280039C2 |
| НОВЫЕ ПРОТИВООПУХОЛЕВЫЕ СОЕДИНЕНИЯ | 2003 |
|
RU2356908C2 |
| Ингибиторы фактора VIIa | 1999 |
|
RU2223967C2 |
| ПЕПТИДНЫЕ АНАЛОГИ GH-RH С АНТАГОНИСТИЧЕСКИМ ДЕЙСТВИЕМ, СПОСОБ СНИЖЕНИЯ УРОВНЯ GH, СПОСОБ СНИЖЕНИЯ УРОВНЯ IGF-I И IGF-II, ПРИМЕНЕНИЕ ДЛЯ ИНГИБИРОВАНИЯ РОСТА РАКОВЫХ КЛЕТОК, ФАРМАКОЛОГИЧЕСКИ ПРИЕМЛЕМАЯ КОМПОЗИЦИЯ (ВАРИАНТЫ) | 2004 |
|
RU2335506C2 |
| СОЕДИНЕНИЯ, ИНГИБИРУЮЩИЕ MASP, И ИХ ПРИМЕНЕНИЯ | 2020 |
|
RU2840441C2 |
| АНАЛОГ ЭКСЕНАТИДА | 2020 |
|
RU2823623C1 |
| ПОЛИПЕПТИД, ИМЕЮЩИЙ ММР-2 ИНГИБИРУЮЩЕЕ ДЕЙСТВИЕ | 2020 |
|
RU2828218C1 |
| АНАЛОГИ ИНСУЛИНОПОДОБНОГО ФАКТОРА РОСТА-1 (IGF-1), СОДЕРЖАЩИЕ АМИНОКИСЛОТНУЮ ЗАМЕНУ В ПОЛОЖЕНИИ 59 | 2010 |
|
RU2511577C2 |
| АНТАГОНИСТИЧЕСКИЕ АНАЛОГИ РИЛИЗИНГ-ГОРМОНА ГОРМОНА РОСТА (GH-RH), ИНГИБИРУЮЩИЕ ИНСУЛИНОПОДОБНЫЕ ФАКТОРЫ РОСТА (IGF-I И-II) | 1999 |
|
RU2235099C2 |
| RGD-СОДЕРЖАЩИЕ ПЕПТИДОМИМЕТИКИ И ИХ ПРИМЕНЕНИЕ | 2009 |
|
RU2519736C2 |
Изобретение относится к новым противоопухолевым соединениям, представляющим собой производные кахалалида F, содержащим их фармацевтическим композициям и их применению для приготовления противоопухолевого лекарственного средства. 4 н.п. ф-лы, 11 табл.
1. Соединение на основе структуры кахалалида F формулы (1)

обозначаемого KF, и выбираемое из группы, состоящей из [D-Ser6]-KF, [Glu8]-KF, [Lys8]-KF, [Val12]-KF,
[hCh11]-KF, [D-Cha4], D-Cha5, D-Cha7]-KF, [Icos14]-KF, [(c/t)-4-Me-cHexa14]-KF, [Und14]-KF, [Oct14]-KF, [p-MeBza14]-KF, [Bza14]-KF, [р-CF3Bza14]-KF, [Pipe14]-KF, [р-CF3Cinn14]-КF, [p-CF3PhAcl4]-KF, [Pfh14]-KF, [6,6-dFHep14]-KF, [Lys8], (4S)-MeHexl4]-KF, [Gly13]-KF, [D-Ala13]-KF, [D-Leu13]-KF, [D-Phe13]-KF, [D-Pro13]-KF, [Val13]-KF, [D-Glu13]-KF, [D-Gln13]-KF, [D-Thr13]-KF, [Phe11]-KF, [Leu11]-KF, [Orn11]-KF, [Ala12, noD-Val13]-KF, [Gly12, noD-Val13]-KF, [Leu12, noD-Val13]-KF, [Glu12, noD-Val13]-KF, [D-Tic9, (4S)-MeHex14]-KF, [hCh3]-KF, [Dha2]-KF, [Trp3]-KF, [Phe(3,4-Cl2)3, (4S)-MeHex14]-KF, [Phe(F5)3, (4S)-MeHexl4]-KF, [Phe(4-I)3, (4S)-MeHexl4]-KF, [Phe(4-NO2)3, (4S)-MeHexl4]-KF, [Phe(4-F)3, (4S)-MeHex14]-KF, [Tyr(Me)3, (4S)-MeHex14]-KF, [Thi3, (4S)-MeHex14]-KF, [Tyr3, (4S)-MeHex14]-KF, [NMePhe3, (4S)-MeHex14]-KF, [Phe-(2-Cl)3]-KF, [Phe(3-Cl)3]-KF, [Phe(4-Cl)3]-KF, [Phe(3,4-F2)3]-KF, [Nal3]-KF, [Bip3]-KF, [Phe(3,4-Cl2)3, р-CF3Cinn14]-KF, [N(Me)2,N'(Me)2-Arg8]-KF, [N(Me,Ph),N'(Me)2-Arg8]-KF, [N(CH2)4,N'(Me)2-Arg8]-KF, [N(CH2)4,N'(CH2)4-Arg8]-KF, [Nδ(CHN(CH2)4-N'(CH2)4)-Orn8]-KF, [Nε(Me)3-Lys8, (4S)-MeHex14]-KF, [Thr(OTFA)12, (4S)-MeHex14]-KF, [Orn(NδTFA)8, Thr(OTFA)12, (4S)-MeHex14]-KF, [Orn(NδTFA)8, (4S)-MeHex14]-KF, [Thr(OTFA)12, Lit(OTFA)14]-KF, и [Orn(Biot)8]-KF,
где аминокислота или группа, указанная в квадратных скобках, является модификацией, введенной в структуру кахалалида
F, или его фармацевтически приемлемая соль.
2. Соединение на основе структуры кахалалида F формулы (I):
обозначаемого KF, и выбираемое из группы, состоящей из [Gly12]-KF, [Ala12]-KF, [Leu12]-KF, [Phe12]-KF, [Orn12]-KF, [Pro12]-KF, [Gln12]-KF и [D-Val1]-KF, где аминокислота или группа, указанная в квадратных скобках, является модификацией, введенной в структуру кахалалида F, или его фармацевтически приемлемая соль.
3. Фармацевтическая композиция, обладающая противоопухолевой активностью, содержащая эффективное количество соединения по любому из предшествующих пунктов и фармацевтически приемлемый носитель, растворитель или разбавитель.
4. Применение соединения по п.1 или 2 в получении лекарственного средства для лечения рака.
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| M | |||
| Hamann et al "Kahalalides: Bioactive peptides from a marine mollusk Elysia rufescens and its algal diet Briopsis sp.", Journal of Organic Chemistry, 1996, 61, 6594-6600. | |||

