Настоящее изобретение описывает химические соединения, ингибирующие репликацию вируса иммунодефицита человека (ВИЧ), и описывает соединения бензотрополона и фармацевтические композиции на их основе, которые могут быть использованы для предотвращения или лечения инфекции, вызванной вирусом иммунодефицита человека (ВИЧ), и для предотвращения, лечения или задержки развития Синдрома Приобретенного Иммунодефицита (СПИД).
Вирус ВИЧ-1 принадлежит к числу ретровирусов и может инфицировать клетки, содержащие поверхностный рецептор CD-4. Он является этиологическим агентом, вызывающим Синдром Приобретенного Иммунодефицита и дегенеративные изменения нервной системы. Следовательно, соединения, которые способны устранять вирус из тела пациента или замедлять его размножение в теле пациента, могут быть эффективны для лечения или профилактики ВИЧ-1 инфекции и СПИД.
Как и другие ретровирусы, после прикрепления и проникновения в клетку-хозяина, вирус ВИЧ-1 высвобождает в клеточную цитоплазму вирусную РНК и три фермента, необходимые для репликации вируса: обратную транскриптазу, интегразу и вирусную протеазу. Обратная транскриптаза производит синтез вирусной ДНК на матрице вирусной РНК. Эта вирусная ДНК затем проникает в ядро и интегрируется в геном клетки-хозяина с помощью ВИЧ-1 интегразы. Вирусная ДНК, интегрированная в геном клетки, затем участвует в процессах транскрипции и трансляции, осуществляемых с помощью обычных ферментативных реакций клетки-хозяина. Синтезированные таким образом вирусные белки подвергаются окончательной обработке ВИЧ-1 протеазой, после чего вирусные белки и вирусные РНК формируют новые единицы вируса, которые покидают инфицированную клетку.
Таким образом, ферменты, специфичные для вируса ВИЧ-1 - обратная транскриптаза, протеаза и интеграза, необходимы для репликации вируса и представляют собой естественные мишени для возможной антиретровирусной терапии.
В настоящее время существует множество доступных противовирусных препаратов, способных противодействовать инфекции. Эти препараты могут быть разделены на три класса, основываясь на вирусном белке, который является их мишенью, а также на способе их действия. В частности, саквинавир, индинавир, ритонавир, нелфинавир и ампренавир являются конкурентными ингибиторами аспартильной протеазы, экспрессируемой ВИЧ. Зидовудин, диданозин, ставудин, ламивудин и абакавир являются нуклеотидными ингибиторами обратной траскриптазы, которые ведут себя как миметики субстрата и которые останавливают синтез вирусной ДНК. Ненуклеотидные ингибиторы обратной транскриптазы, такие как невирапин, делавирдин и эфавиренц, ингибируют синтез вирусной ДНК через неконкурентный механизм. Использование этих препаратов является эффективным лишь для снижения репликации вируса. Эффект является только временным, поскольку вирус легко вырабатывает устойчивость ко всем известным агентам. Однако комбинированная терапия доказала свою высокую эффективность как для ослабления репликации вируса, так и для подавления появления устойчивости у множества пациентов. В США, где комбинированная терапия широко применяется, количество связанных с ВИЧ смертельных случаев уменьшилось (Palella, F.J.; Delany, К.М.; Morman, А.С; Loveless, М. О.; Futher, J.; Satten, G. A.; Aschman, D. J.; Holmberg, S. D. N. Engl J. Med. 1998, 338, 853).
Таким образом, как было отмечено выше, в течение последних лет во врачебную практику было введено большое количество ингибиторов ВИЧ-1 специфичной обратной транскриптазы и ВИЧ-1 протеазы, которые продемонстрировали эффективность в лечении и профилактике ВИЧ-1 инфекции (в основном в форме многокомпонентной терапии). Такая многокомпонентная терапия на сегодняшний день является основным способом борьбы с Синдромом Приобретенного Иммунодефицита.
К сожалению, не все пациенты являются восприимчивыми к многокомпонентной терапии, и большое число неудач сопровождает применение этой терапии, кроме того, многие анти-ВИЧ лекарственные средства обладают побочными токсическими эффектами. В действительности, приблизительно 30-50% пациентов в конечном счете не воспринимают комбинированную терапию. Неудачи в лечении в большинстве случаев вызваны появлением вирусной устойчивости. Вирусная устойчивость, в свою очередь, вызвана быстрым оборотом ВИЧ-1 при проявлении инфекции в сочетании с высокой скоростью вирусных мутаций. При этих обстоятельствах неполная вирусная супрессия, вызванная недостаточной активностью лекарственного средства, недостаточный отклик на множество сложных лекарственных средств, так же как и внутренние фармакологические барьеры при взаимодействии, формируют основу для появления устойчивости вируса к лекарственным веществам, применяемым в составе многокомпонентной терапии ВИЧ-инфекции. Недавно полученные многочисленные данные предполагают, что низкоуровневая репликация продолжается, даже когда уровень вируса в плазме крови опускается ниже обнаруживаемых уровней (меньше 50 копий/мл) (Carpenter, С.С.J.; Cooper, D.A.; Fischi, М.A.; Garil, J.М.; Gazzard, В.G.; Hammer, S.М.; Hisch, М.S.; Jacobsen, D.M.; Katzenstein, D.A.; Montaner, J.S.; Richman, D.D.; Saag, M.S.; Schecter, M.; Schoolery, R.Т.; Thompson, M.A.; Vella, S.; Yeni, P.G.; Volberding, P.A. JAMA 2000, 283, 381). Очевидно, что появление новых мультирезистентных штаммов ВИЧ-1, устойчивых к стандартной многокомпонентной терапии, делает разработку новых анти-ВИЧ противовирусных средств крайне необходимой. Ингибиторы ВИЧ-1 интегразы могут быть эффективными лекарственными веществами для предотвращения и лечения ВИЧ-1, так как ВИЧ-1 интеграза также является ферментом, необходимым для репликации ВИЧ.
В настоящее время нет известных противовирусных лекарственных средств, структурно сходных с анти-ВИЧ средствами на основе производных бензотрополона, описываемыми в настоящем изобретении. Некоторые производные бензотрополона были описаны, как имеющие иную терапевтически полезную активность.
Так, патент WO 92/20332 описывает фармацевтические композиции, содержащие пурпурогаллин
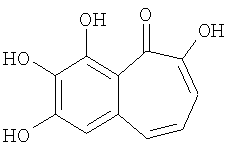
и его моно и диглюкозиды как антиоксиданты и цитопротекторы.
В патенте US 2006/0241154 описана пурпурогаллинкарбоновая кислота
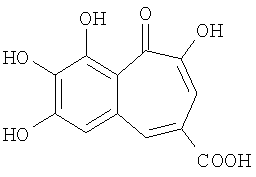
и некоторые другие природные и синтетические бензотрополоны в качестве эффективных антиоксидантов и противовоспалительных средств.
Патент US5650439 описывает природный бензотрополон состава
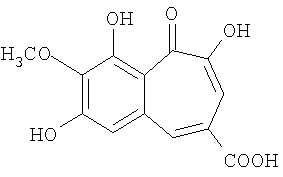
как эффективный ингибитор катехоламин-О-метилтрансферазы.
Патент JP 2004-359575 описывает бензотрополоны вида
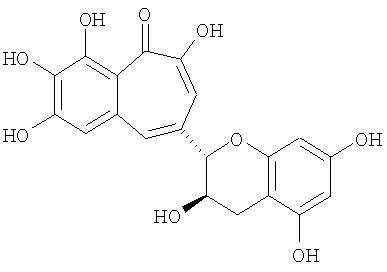
и похожие соединения как вещества, индуцирующие процесс апоптоза злокачественных клеток и пригодные для химиотерапии раковых заболеваний.
Имеется ряд публикаций, описывающих производные бензотрополона как потенциальные лекарственные вещества:
В статье: "Ингибирование метилирования эстрадиола бензотрополонами: кинетика и моделирование in silico" (Bioorganic and Medicinal Chemistry, Vol.13, p.2501-2507, 2005) авторы описывают природные и синтетические производные бензотрополона, которые являются ингибиторами метилирования гидроксиэстрадиола катехоламин-О-метилтрансферазой.
В статье: "Новые производные трополона, повреждающие ДНК, выделенные из Goupia glabra" (Eur. J. Med. Chem., 2003, p.4243-4247) авторы описывают выделение бензотрополона
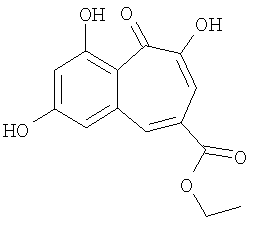
и его действие в качестве генотоксина, а также его возможное применение в качестве противоракового лекарственного средства.
В статье: "Открытие, характеризация и изучение соотношения структура-активность полифенолов, вызывающих апоптоз и воздействующих на белки В-cell Lyphocyte/Leukemia-2" (J. Med. Chem., Vol.46, p.4259-4264, 2003) было показано, что пурпурогаллин и его производные способны вызывать апоптоз злокачественных клеток.
Некоторые производные бензотрополона образуются в результате окисления катехиновых соединений при ферментации в процессе приготовления черного чая. Такие соединения получили название теафлавины, причем их доля может составлять до 2-6% от сухого веса качественного черного чая.
Недавно теафлавины привлекли значительное внимание, поскольку обнаружилось, что они обладают богатым спектром биологической активности, включая следующие воздействия на организм:
противовоспалительное и цитотоксическое (Bioorganic and Medicinal Chemistry, Vol.12, p.459-467, 2004);
антимутагенное (Mutat. Res. Vol.389, p.167, 1997);
подавление активности изоформы цитохрома Р450 1А1 в клеточной культуре (Agric. Food Chem., Vol.50, p.213,2002);
антикластогенный эффект на клетках костного мозга мышей (Life Sci., Vol.69, р.2735,2001);
подавление межклеточных взаимодействий и развития клеток (Carcinogenesis, Vol.20, р.733, 1999);
противовоспалительное и профилактическое противораковое действие (Biochem. Pharm., Vol.59, р.59, 2000).
Все перечисленные публикации не описывают производные бензотрополона как ингибиторы ВИЧ-1 интегразы и анти-ВИЧ лекарственные средства.
Технической задачей настоящего изобретения является поиск новых соединений и лекарственных средств, обладающих анти-ВИЧ активностью.
Методы получения производных бензотрополона (или их выделения из природных источников) известны и были опубликованы в соответствующей научной литературе (J.Am. Chem. Soc, Vol.52, 1930, р.3647; J. Chem. Soc, 1948, р.117; J. Chem. Soc, 1951, p.1313; J. Cem. Soc, 1951, p.1318; J. Chem. Soc, 1951, p.1325; J. Chem. Soc, 1952, p.3705; J. Chem. Soc. Japan (Pure Chem. Sect.), Vol.75, 1954, p.620; J. Chem. Soc. Japan (Pure Chem. Sect.), Vol.77, 1956, p.305; Chem., Bd.69, 1957, p.723; Naturforsch., Bd.14b, 1959, p.742; Chem. Ber., Bd.97, 1964, p.307; Chem. Ber., Bd.97, 1964, p.312; Chem., Bd.98, 1967, p.872; Tetrahedron, Vol.23, 1967, p.2829; Eur. J. Org. Chem., 2003, p.4243).
В соответствии с одним из методов бензотрополоновое ядро может быть получено совместным окислением смеси катехоловой и пирогаллоловой компонент подходящим окислителем в водной, или органической, или водно-органической среде:
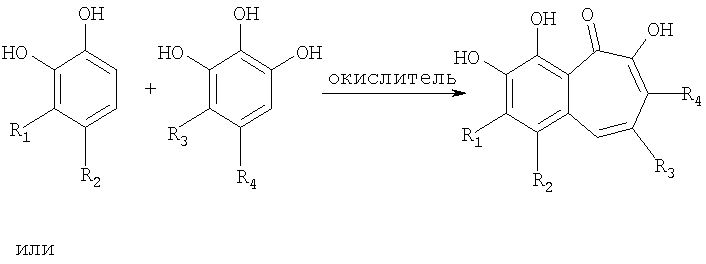
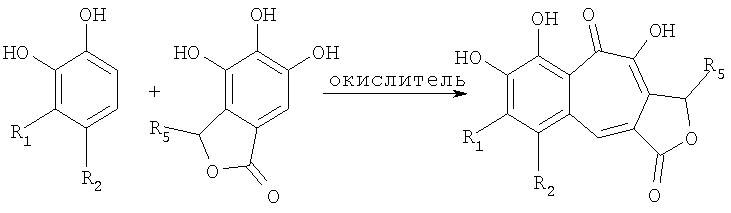
Предпочтительным окислителем является иодат калия или натрия, но многие другие органические и неорганические окислители также пригодны для проведения этой реакции: феррицианид калия, перекись водорода, перекись водорода в присутствии ферментов каталазы или пероксидазы, азотная кислота, висмутат натрия, висмутат калия, полифенолоксидаза, бихромат калия, перманганат калия, феррат бария, пара-бензохинон, тетрахлор-орто-бензохинон, тетрабром-орто-бензохинон и т.д.
Имея синтезированное бензотрополоновое ядро, другие производные бензотрополона, описанные в настоящем изобретении, могут быть приготовлены стандартными методами органической химии - электрофильным замещением в бензотрополоновом ядре, модификацией функциональных групп - окислением, восстановлением, снятием защитных групп, образованием или гидролизом сложных или простых эфиров, амидов, оксимов, гидроксамовых кислот, сульфамидов и т.д.
В настоящем изобретении, если не определено иное, используются следующие определения.
«С1-6алкил» представляет собой алкильную группу, содержащую неразветвленную или разветвленную углеводородную цепь, содержащую от 1 до 6 атомов углерода, например метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил и так далее.
«ОС1-6алкил» представляет собой алкоксильную группу, содержащую неразветвленную или разветвленную углеводородную цепь, содержащую от 1 до 6 атомов углерода, например метокси, этокси, н-пропокси, изопропокси и так далее.
«С1-6алкил, замещенный одним или более атомами галогена», представляет собой алкильную группу, содержащую неразветвленную или разветвленную углеводородную цепь, содержащую от 1 до 6 атомов углерода, замещенную одним или более атомами галогена, например фторметил, дифторметил, трифторметил, хлорметил, дихлорметил, трихлорметил и так далее.
«ОС1-6алкил, замещенный одним или более атомами галогена» представляет собой алкоксильную группу, содержащую неразветвленную или разветвленную углеводородную цепь, содержащую от 1 до 6 атомов углерода, замещенную одним или более атомами галогена, например фторметокси, дифторметокси, трифторметокси, хлорметокси и так далее.
«Галоген» означает хлор, бром, йод или фтор.
Фармацевтически приемлемой может считаться любая соль, образуемая активным соединением Формулы (А), если она не токсична и не препятствует адсорбции и фармакологическому действию активного соединения. Эта соль может быть получена действием на соединение Формулы (А) органического или неорганического основания, такого как гидроксид натрия, гидроксид калия, гидроксид аммония, метиламин, этиламин и тому подобных.
Термин "пролекарство" означает производное соединения, описанного в настоящем изобретении, которое имеет группы, легко отщепляемые в результате химических или метаболических процессов и которое после применения пациентом в его организме отщепляет эти группы, давая исходное соединение по настоящему изобретению, проявляющее присущую ему анти-ВИЧ активность in vivo. "Пролекарство" соединения структурной формулы (А) может быть получено обычным способом с помощью модификации функциональных групп соединений, таких как гидрокси или карбоксигруппа.
"Пролекарства" применяются для улучшения таких характеристик лекарственных средств, как растворимость, тканевая совместимость, контролируемое высвобождение в организме млекопитающего, всасывание из желудочно-кишечного тракта и транспортировка лекарственного вещества в организме к месту его действия. Пролекарства включают производные кислот, хорошо известные средним специалистам в данной области, такие как, например, сложные эфиры, полученные путем взаимодействия исходного кислотного соединения с соответствующим спиртом, или амиды, полученные взаимодействием исходного кислотного соединения с соответствующим амином. Сложные алифатические или ароматические эфиры, производные от кислотных боковых групп соединений по настоящему изобретению, являются предпочтительными пролекарствами. В некоторых случаях желательно получать эфирные пролекарства по другим группам, такие как алкилсложные эфиры или ((алкоксикарбонил)окси)алкилсложные эфиры.
Термин "фармацевтически приемлемый носитель" означает, что носитель должен являться совместимым с другими ингредиентами композиции и не наносить вреда его реципиенту, то есть быть нетоксичным для клеток или млекопитающих в тех дозах и концентрациях, в которых его применяют. Часто фармацевтически приемлемый носитель представляет собой водный рН буферный раствор. Примеры физиологически приемлемых носителей включают буферы, такие как фосфаты, цитраты и другие соли органических кислот, антиоксиданты, включающие аскорбиновую кислоту; полипептиды с низким молекулярным весом (меньше 10 остатков); протеины, такие как сывороточный альбумин, желатин или иммуноглобулины; гидрофильные полимеры, такие как поливинилпирролидон; аминокислоты, такие как глицин, глютамин, аспарагин, аргинин или лизин; моносахариды, дисахариды и другие углеводы, включая глюкозу, маннозу или декстрины; хелатообразующие агенты, такие как ЭДТА; сахарные спирты, такие как маннитол или сорбитол.
Терапевтически эффективное количество - это количество, необходимое для достижения эффекта подавления репликации ВИЧ в организме млекопитающего.
«Млекопитающее», как используется здесь, включает в себя представителей отряда приматов (например, человек, человекообразные обезьяны, нечеловекообразные обезьяны, низшие обезьяны), отряда хищных (например, кошки, собаки, медведи), отряда грызунов (например, мышь, крыса, белка), отряда насекомоядных (например землеройка, крот) и др.
По термином "профилактика СПИД" понимается введение анти-ВИЧ лекарственных средств ВИЧ-позитивными субъектами, у которых, однако, еще не развился Синдром Приобретенного Иммунодефицита, а также применение анти-ВИЧ лекарственных средств субъектами с симптомами СПИД, подвергавшимися антиретровирусной терапии, чье состояние улучшилось, во избежание повторного ухудшения, а также употребление анти-ВИЧ лекарственных средств при угрозе возможного заражения ВИЧ-инфекцией.
В результате решения указанной выше технической задачи заявитель предлагает к защите ряд производных бензотрополона, обладающих анти-ВИЧ активностью, общей структурной формулы (А)
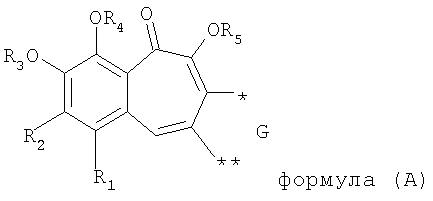
где R1 выбирают из группы, состоящей из водорода, галогена;
R2 выбирают из группы, состоящей из водорода, ОС1-6алкила;
R3, R4, R5 каждый независимо представляет собой водород;
G выбирают из группы, состоящей из (структуры I-II):
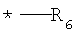
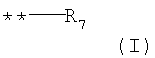
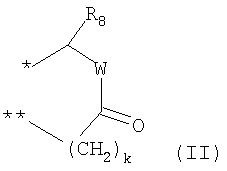
где k=0,1
R6 представляет собой водород;
R7 представляет собой группу COOR9;
R8 выбирают из С1-6алкила, замещенного одним или более атомами галогена;
W представляет собой О;
R9 выбирают из водорода, C1-6алкила;
или их фармацевтически приемлемые соли.
Наиболее предпочтительными являются производные бензотрополона формулы (А) или их фармацевтически приемлемые соли, где
R1 представляет собой водород, Br;
R2 выбирают из водорода, ОСН3;
R3, R4, R5 представляют собой водород;
G представляет собой структуру (I);
R6 представляет собой водород;
R7 представляет собой Y;
Y выбирают из группы, состоящей из СООН, СООС1-6алкила.
Настоящее изобретение также включает фармацевтические композиции, предназначенной для лечения ВИЧ инфекций, содержащих терапевтически эффективное количество производного бензотрополона по пункту 1 или его фармацевтически приемлемой соли и фармацевтически приемлемый носитель.
Активный ингредиент в таких составах включает от 0.1 процента до 99.9 процента от массы состава, предпочтительно от 5 до 90 процента.
Фармацевтические композиции могут быть получены в соответствии с известными методиками, использующими известные и легко получаемые ингредиенты. Такие композиции по настоящему изобретению могут быть разработаны, чтобы обеспечить быстрое, устойчивое или замедленное введение активного ингредиента после введения пациенту в соответствии с применяемыми процедурами, известными среднему специалисту. При получении фармацевтической композиции активный ингредиент обычно смешивают с носителем или разбавляют носителем или заключают в носитель, который может быть в форме капсулы, драже или в таблетированной форме. Когда носитель является разбавителем, то он может быть твердым веществом, полутвердым или жидким материалом, который действует как связующее средство или инертный наполнитель для активного ингредиента.
Таким образом, лекарственные средства на основе фармацевтических композиций могут быть в форме таблеток, пилюль, порошков, шариков, суспензий, эмульсий, растворов, сиропов, аэрозолей мягких и твердых капсул желатина, свечей, растворов для инъекций и тому подобные. Лекарственные средства и фармацевтические композиции изготавливаются обычными технологическими методами. Лекарственные средства могут применяться в качестве внутренних или наружных медицинских средств. Лекарственные средства могут применяться перорально, парэнтерально или интраназально.
При применении соединений, описанных в настоящем изобретении в составе фармацевтических композиций, они могут быть смешаны с подходящими добавками, инертными наполнителями, разбавителями, диспергирующими веществами, стабилизаторами, консервантами, буферными составами, эмульгаторами, отдушками, красителями, подсластителями и другими известными фармацевтическими добавками, такими как вода, растительные масла, спирты (например, этанол, бензиловый спирт и т.д.), полиэтиленгликоль, триацетат глицерина, желатин, полиэтоксилированные растительные масла, углеводы (например, лактоза, крахмал) стеарат магния, тальк, ланолин, петролатум и тому подобными.
Настоящее изобретение также включает способ ингибирования ВИЧ-1 интегразы, включающий введение млекопитающим терапевтически эффективного количества соединения по формуле (А) или его фармакологически приемлемой соли.
Способ профилактики или лечения ВИЧ инфекции у млекопитающего включает введение такому пациенту терапевтически эффективного количества производного бензотрополона по формуле (А) или его фармацевтически приемлемой соли. Применяемые дозы зависят от возраста млекопитающего, его веса, симптомов, эффекта от лечения, метода введения и т.п. Обычно дозы составляют от 0.1 мг до 5 г, предпочтительно от 10 мг до 2 г, на одну особь млекопитающего за одно введение. Таких введений может быть от одного до 10 в течение суток, либо перорально, либо путем внутривенных, внутримышечных или подкожных инъекций, либо внутривенных инфузий.
Дополнительно вместе с активным соединением могут вводить другие анти-ВИЧ агенты, в частности конкурентные ингибиторами ВИЧ-протеазы (например, саквинавир, индинавир, ритонавир, нелфинавир и ампренавир), нуклеотидные ингибиторы обратной траскриптазы (например, зидовудин, диданозин, ставудин, ламивудин, абакавир), ненуклеотидные ингибиторы обратной транскриптазы (например, невирапин, делавирдин и эфавиренц).
Описание изобретения сопровождается чертежами.
На фиг.1 схематически изображен 3'-процессинг U5-дуплекса, осуществляемый ВИЧ-1 интегразой.
На фиг.2 показан радиоавтограф денатурирующего полиакриламидного геля, демонстрирующий влияние ингибиторов на 3'-процессинг активность интегразы в соответствии с методикой, описанной далее в Примере 10.
На фиг.3 показана графическая зависимость ингибирования 3'-процессинга от концентрации ингибитора, получаемая в процессе обработки экспериментальных результатов, в ходе которого определяются параметры экспоненциальной кривой ингибирования первого порядка, наилучшим образом проходящей через экспериментальные точки. Параметры экспоненциальной кривой используются для определения величины IC50 ингибирования 3'-процессинг активности ВИЧ-1 интегразы данным ингибитором.
На фиг.4 схематически изображен процесс переноса полинуклеотидной цепи, осуществляемый ВИЧ-1 интегразой.
На фиг.5 показан радиоавтограф денатурирующего полиакриламидного геля, демонстрирующий влияние ингибитора на активность интегразы относительно стадии переноса цепи в соответствии с методикой, описанной далее в Примере 10.
На фиг.6 показана графическая зависимость ингибирования реакции переноса цепи от концентрации ингибитора, получаемая в процессе обработки экспериментальных результатов, в ходе которого определяются параметры экспоненциальной кривой ингибирования первого порядка, наилучшим образом проходящей через экспериментальные точки. Параметры экспоненциальной кривой используются для определения величины IC50, характеризующей ингибирующую активность данного вещества по отношению к процессу переноса цепи, катализируемому ВИЧ-1 интегразой.
Для того чтобы предмет настоящего изобретения был более понятен, ниже приведены некоторые примеры получения производных бензотрополона (Примеры 1-10) и их использования в качестве ингибиторов интегразы и анти-ВИЧ лекарственных соединений. Примеры носят иллюстративный характер, причем содержание данного изобретения ни в коей мере не ограничивается представленными примерами.
Пример 1
Синтез метилового эфира 7-метокси-8,9-дигидроксибензотрополон-4-карбоновой кислоты
Раствор 1.176 грамм иодата калия (5.495 ммоль) в воде (40 мл) быстро добавляют к раствору, содержащему 1.011 грамм (5.495 ммоль) метилгаллата и 0.769 грамм (5.495 ммоль) 3-метоксипирокатехина в смеси воды (40 мл) и ацетона (10 мл) при непрерывном интенсивном перемешивании при комнатной температуре. Реакционная смесь становится коричневой, и вскоре начинается выделение углекислого газа и осаждение продукта реакции. Перемешивание продолжают 1.5 часа и затем оставляют реакционную смесь стоять еще 30 минут. Отфильтровывают полученный осадок, промывают 3 раза водой, сушат на воздухе, перекристаллизовывают из 95% уксусной кислоты (3 мл), промывают уксусной кислотой (0.5 мл) и наконец сушат на воздухе. Получают 1.323 грамм (82%) метилового эфира 7-метокси-8,9-дигидроксибензотрополон-4-карбоновой кислоты. Температура плавления 229-231°С (из уксусной кислоты). 1Н-ЯМР (500 МГц; ДМСО-д6): δ 3.88 (с, 3Н, 4-СООСН3), 4.00 (с, 3Н, 7-ОСН3), 7.40 (с, 1H, 6-Н), 7.54 (с, 1H, 3-Н), 8.33 (с, 1H, 5-Н), 9.68 (с, 1Н, 8(9)-ОН), 9.88 (с, 1Н, 9(8)-ОН), 14.93 (с, 1Н, 2-ОН).
Пример 2
Синтез метилового эфира 8,9-дигидроксибензотрополон-4-карбоновой кислоты
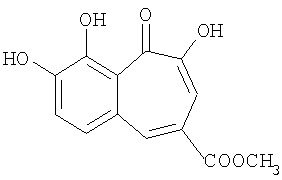
Вещество приготовлено по методике, описанной в Примере 1 из метилгаллата и пирокатехина. Выход 51%. Температура плавления 214-215°С (из уксусной кислоты). 1Н-ЯМР (500 МГц; ДМСО-д6): δ 3.88 (с, 3Н, 4-СООСН3), 7.45 (дд, J=0.87 Гц, 1H, 7-Н), 7.60 (с, 1H, 3-Н), 7.65 (дд, J=0.87 Гц, 1Н, 6-Н), 8.30 (с, 1H, 5-Н), 9.65 (уш. с, 1Н, 8(9)-ОН), 10.42 (уш. с, 1H, 9(8)-ОН), 14.77 (уш.с, 1H, 2-ОН).
Пример 3
Синтез метилового эфира 6-бромо-7-метокси-8,9-дигидроксибензотрополон-4-карбоновой кислоты
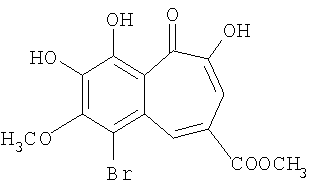
0.162 грамм (0.556 ммоль) метилового эфира 7-метокси-8,9-дигидроксибензотрополон-4-карбоновой кислоты добавляют к перемешиваемому раствору 0.089 грамм (0.556 ммоль) брома в 3.345 грамм ледяной уксусной кислоты. Перемешивают реакционную смесь еще 20 часов при комнатной температуре. Продукт реакции осаждают добавлением воды (40 мл), отфильтровывают, трижды промывают водой (3×5 мл) и сушат на воздухе. Получают 0.165 грамм (80%) метилового эфира 6-бромо-7-метокси-8,9-дигидроксибензотрополон-4-карбоновой кислоты. Температура плавления 233-235°С (из уксусной кислоты). 1Н-ЯМР (500 МГц; ДМСО-д6): δ 3.88 (с, 3Н, 4-СООСН3), 3.96 (с, 3Н, 7-ОСН3), 7.52 (с, 1H, 3-Н), 8.95 (с, 1H, 5-Н), 10.03 (с, 1H, 8(9)-ОН), 10.59 (с, 1Н, 9(8)-ОН), 15.18 (с, 1H, 2-ОН).
Пример 4
Синтез метилового эфира 6-бромо-8,9-дигидроксибензотрополон-4-карбоновой кислоты
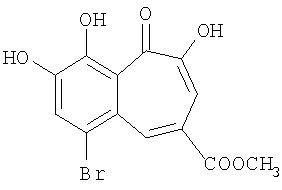
Вещество получено по методике, описанной в примере 3, путем бромирования метилового эфира 8,9-дигидроксибензотрополон-4-карбоновой кислоты в ледяной уксусной кислоте. Выход 20%. Температура плавления 214-215°С (из уксусной кислоты). 1Н-ЯМР (500 МГц; ДМСО-д6): δ 3.90 (с, 3Н, 4-СООСН3), 7.53 (с, 1H, 3-Н), 7.83 (с, 1H, 7-Н), 8.80 (с, 1H, 5-Н), 10.04 (уш.с, 1H, 8(9)-ОН), 10.82 (уш.с, 1H, 9(8)-ОН), 14.74 (с, 1H, 2-ОН).
Пример 5
Синтез 8,9-дигидроксибензотрополон-4-карбоновой кислоты
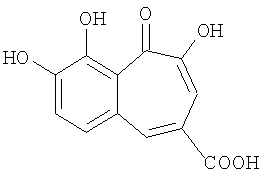
Кипятят с обратным холодильником 0.131 грамм (0.500 ммоль) метилового эфира 8,9-дигидроксибензотрополон-4-карбоновой кислоты в смеси диоксана (3 мл) и конц. HCl (2 мл) в течение 4.5 часа. Реакционную смесь разбавляют водой (10 мл). Отфильтровывают образовавшийся осадок, трижды промывают водой (3×5 мл), сушат на воздухе, перекристаллизовывают из 95% уксусной кислоты (1.5 мл), промывают уксусной кислотой (0.5 мл), затем снова трижды промывают водой (3×5 мл) и наконец сушат на воздухе. Получают 0.092 грамм (74%) 8,9-дигидроксибензотрополон-4-карбоновой кислоты. Температура разложения >300°С. 1Н-ЯМР (500 МГц; ДМСО-д6): δ 7.48 (дд, J=0.87 Гц, 1Н, 7-Н), 7.64 (дд, J=0.87 Гц, 1H, 6-Н), 7.66 (с, 1H, 3-Н), 8.33 (с, 1H, 5-Н), 9.69 (с, 1H, 8(9)-ОН), 10.38 (с, 1Н, 9(8)-ОН), 13.34 (уш.с, 1H, 4-СООН), 14.82 (с, 1Н, 2-ОН).
Пример 6
Синтез 7-метокси-8,9-дигидроксибензотрополон-4-карбоновой кислоты
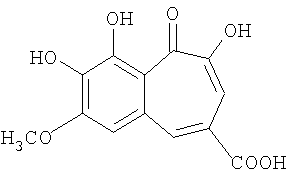
Вещество приготовлено гидролизом метилового эфира 7-метокси-8,9-дигидроксибензотрополон-4-карбоновой кислоты в условиях, описанных в Примере 5. Выход 95%. Температура разложения >300°С. 1Н-ЯМР (500 МГц; ДМСО-д6): δ 4.00 (с, 3Н, 7-ОСН3), 7.42 (с, 1Н, 6-Н), 7.62 (с, 1H, 3-Н), 8.40 (с, 1Н, 5-Н), 9.60 (уш.с, 1H, 8(9)-ОН), 14.93 (с, 1H, 2-ОН). Сигналы от 9(8)-ОН и 4-СООН протонов не видны.
Пример 7
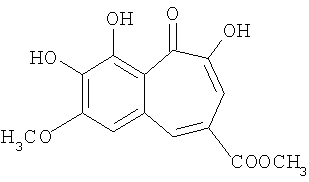
Синтез метилового эфира 7-метокси-8,9-дигидроксибензотрополон-4-метиленкарбоновой кислоты
(i) метиловый эфир 3,4,5-тригидроксифенилуксусной кислоты
Кипятят 1.130 грамм (5 ммоль) 3,4,5-триметоксифенилуксусной кислоты в 25 мл конц. HBr, в атмосфере аргона на протяжении 12.5 часов. Затем растворитель отгоняют в вакууме (10 торр, 80-90°С). Остаток сушат в вакууме (2 торр, 20°С) над NaOH в течение 3 дней. Технический продукт, содержащий 3,4,5-тригидроксифенилуксусную кислоту, растворяют в метаноле (10 мл) и охлаждают до 0÷+5°С. К охлажденному метанольному раствору добавляют 0.51 мл (0.833 грамм, 7 ммоль) тионилхлорида. Кипятят реакционную смесь с обратным холодильником 2.5 часа, затем растворитель отгоняют, остаток сушат в вакууме (2 торр, комнатная температура) над NaOH в течение 2 часов. Полученный метиловый эфир 3,4,5-тригидроксифенилуксусной кислоты может быть использован в последующем синтезе без дополнительной очистки.
(ii) метиловый эфир 7-метокси-8,9-дигидроксибензотрополон-4-метиленкарбоновой кислоты:
Растворяют метиловый эфир 3,4,5-тригидроксифенилуксусной кислоты, полученный на предыдущей стадии, и 0.686 грамм (4.900 ммоль) 3-метоксипирокатехина в смеси воды (25 мл) и ацетона (5 мл). К полученному раствору при комнатной температуре и интенсивном перемешивании добавляют раствор 1.049 грамм (4.900 ммоль) иодата калия в 35 мл воды. Реакционная смесь становится коричневой, и вскоре начинается выделение углекислого газа и осаждение продукта реакции. Перемешивание продолжают 1.5 часа и затем оставляют реакционную смесь стоять еще 30 минут. Отфильтровывают осадок, промывают его трижды небольшим количеством воды (3×5 мл) и сушат на воздухе. Технический бензотрополон растворяют в ацетоне и упаривают с 2 мл перлита, высушивают смесь в вакууме 2 мм рт.ст. Загружают смесь в аппарат для непрерывной экстрактивной хроматографии (нейтральный силикагель 100/160, 3 мл) и экстрагируют гексаном 2 суток. Получают 0.300 г (20%) чистого метилового эфира 7-метокси-8,9-дигидроксибензотрополон-4-метиленкарбоновой кислоты. Температура плавления 172-173°С. 1Н-ЯМР (500 МГц; ДМСО-д6): δ 3.65 (с, 3Н, 4-СООСН3), 3.60 (с, 2Н, 4-СН2), 3.97 (с, 3Н, 7-ОСН3), 7.09 (с, 1H, 3-Н), 7.10 (с, 1Н, 5-Н), 7.50 (с, 1H, 6-Н), 9.38 (с, 1H, 8(9)-ОН), 9.53 (с, 1H, 9(8)-ОН), 15.00 (с, 1Н, 2-ОН).
Пример 8
Синтез 7-метокси-8,9-дигидроксибензотрополон-4-метиленкарбоновой кислоты
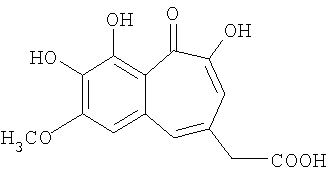
Кипятят 0.240 г (0.667 ммоль) метилового эфира 7-метокси-8,9-дигидроксибензотрополон-4-метиленкарбоновой кислоты в смеси 3 мл уксусной кислоты и 0.1 мл конц. соляной кислоты 4 часа, затем упаривают в вакууме (10 торр) при 90-95°С и сушат на воздухе. Получают 0.129 г (66%) 7-метокси-8,9-дигидроксибензотрополон-4-метиленкарбоновой кислоты. Температура плавления 201-203°С.
Пример 9
Получение натриевой соли 7-метокси-8,9-дигидроксибензотрополон-4-метиленкарбоновой кислоты
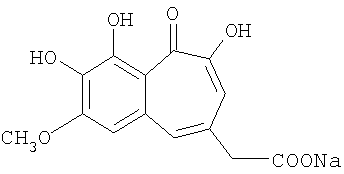
Растворяют 0.278 г (1 ммоль) 7-метокси-8,9-дигидроксибензотрополон-4-метиленкарбоновой кислоты в растворе метилата натрия, приготовленном заранее из 0.023 г натрия и 30 мл метанола. Метанол отгоняют на роторном испарителе, остаток сушат в вакууме (10 торр). Получают 0.3 г натриевой соли 7-метокси-8,9-дигидроксибензотрополон-4-метиленкарбоновой кислоты.
Пример 10
Синтез 7,8,10-тригидрокси-6-метокси-1-трихлорметил-2-оксобензо[f]азулен-3,9-диона
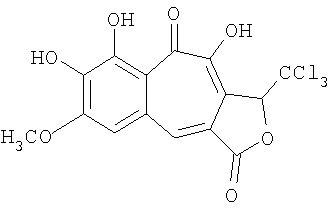
К раствору 0.7 г (5.000 ммоль) о-метоксипирокатехина и 1.498 г (5.000 ммоль) 4,5,6-тригидрокси-3-трихлорметил-3H-изобензофуран-1-она (синтезирован согласно J. Chem. Soc. 119, 208 (1921)) в 6 мл ацетона при перемешивании быстро прибавляют раствор 1.070 г (5 ммоль) иодата калия в 60 мл воды; смесь становится темно-фиолетового цвета, затем начинает выпадать продукт и выделяется диоксид углерода. Перемешивают смесь 1.5 ч, дают постоять без перемешивания 30 мин, отфильтровывают осадок, промывают три раза по 5 мл воды и сушат на воздухе. Технический бензотрополон растворяют в ацетоне и упаривают с 2 мл перлита, высушивают смесь в вакууме (2 торр.) 1 час при 90°С. Загружают смесь в аппарат для непрерывной экстрактивной хроматографии (нейтральный силикагель 100/160, 5 мл) и экстрагируют МТБЭ 7 ч, получают 1.427 г 7,8,10-тригидрокси-6-метокси-1-трихлорметил-2-оксобензо[f]азулен-3,9-диона красного цвета. Его промывают на фильтре два раза по 0.2 мл ацетона, затем три раза по 0.2 мл гексана и сушат на воздухе. Получают 0.588 г (57%) чистого вещества. Температура разложения >253°С. 1Н-ЯМР (500 МГц; ДМСО-д6): δ 4.00 (с, 3Н, 6-ОСН3), 6.57 (с, 1H, 1-Н), 7.57 (с, 1Н, 5-Н), 8.19 (с, 1H, 4-Н), 10.15 (уш.с, 1Н, 7(8)-ОН), 11.03 (с, 1H, 8(7)-ОН), 14.50 (с, 1Н, 10-ОН).
Приведенные ниже примеры 11-13 иллюстрируют измерения ингибирующей способности описываемых в настоящем изобретении производных бензотрополона по отношению к биохимическим реакциям, катализируемым ВИЧ-1 интегразой, а также измерения ингибирующей способности этих соединений по отношению к репликации ВИЧ в культуре человеческих лимфоидных клеток. Также приводится пример испытания описываемых в настоящем изобретении соединений на острую летальность на мышах при внутрибрюшинном введении.
Пример 11
Способность соединений ингибировать работу ВИЧ-1 интегразы определялась по следующей методике.
Влияние на 3'-процессинг активность интегразы.
Свободная от детергента рекомбинантная ВИЧ-1 интеграза нарабатывалась в культуре Escherichia coli, последующие выделение и очистка производились в соответствии с методикой: (Leh, Н., Brodin, P., Bischerour, J., Deprez, Е., Tauc, P., Brochon, J.C., LeCam, E., Coulaud, D., Auclair, C. & Mouscadet, J.F., 2000, Determinants of
Mg2+ - dependent activities of recombinant human immunodeficiency virus type 1 integrase. Biochemistry 39, 9285-9294). В олигонуклеотид U5B (10 pmol) была введена радиоактивная метка при помощи Т4 полинуклеотидкиназы (Fermentas) и 50 µCi [γ-32Р]АТР (3000 Ci/ммоль). После инкубации при 37°С в течение 1 часа Т4 полинуклеотидкиназа инактивировалась добавлением EDTA и нагреванием до 65°С в течение 5 минут. Затем производился отжиг олигонуклеотида U5B с эквимолярным количеством комплементарного олигонуклеотида U5A. Образовавшийся дуплекс U5B/U5A окончательно отмывался от непрореагировавшего [γ-32Р]АТР на колонке MicroSpin G-25 (Amersham Biosciences).
Влияние на 3'-процессинг активность интегразы в условиях конкурентного ингибирования.
В 10 мкл буфера (20 мМ HEPES 7.5 рН, 1 мМ ДТТ, 7.5 мМ хлорид магния) готовили 3 нМ раствор 32Р-меченного субстрата U5B/U5A, содержащего радиоактивно меченную процессируемую цепь U5B (смесь I) и тестируемый ингибитор в диапазоне концентраций 0.1-100 µМ. 10 мкл 200 нМ раствора интегразы в этом же буфере добавляли к смеси I и выдерживали в термостате 2 часа при температуре 37°С. По завершении реакции к смеси добавляли 80 мкл стоп-смеси (9 мМ Tris-HCl, 6 мМ ЭДТА, 0.4 М СН3COONa, 0.125 мг/мл гликогена), фермент экстрагировали 100 мкл смеси фенол/хлороформ/изоамиловый спирт (25:24:1). Нуклеотидный материал осаждали 5-кратным избытком этилового спирта при температуре 0°С и анализировали с помощью электрофореза в 20%-ном денатурирующем ПААГ с последующей визуализацией геля на приборе STORM 840ТМ Phosphorlmager (Molecular Dynamics, США) и обсчетом средствами программы ImageQuant 4.1. О степени протекания реакции 3'-концевого процессинга судили по появлению на радиоавтографе полосы, соответствующей укороченной на два нуклеотида цепи U5B (19-звенный продукт). По соотношению интенсивностей излучения полос, соответствующих 21- и 19-звенным олигонуклеотидам, определяли эффективность процессинга. По полученным значениям эффективности рассчитывали концентрацию продукта реакции. Данные усредняли по трем независимым экспериментам. Значения IC50 ингибиторов были рассчитаны по экспериментальным зависимостям ингибирования каталитического превращения U5-субстрата интегразы от концентрации взятого ингибитора. При оценке степени превращения субстрата учитывалась суммарная концентрация ДНК, подвергшейся 3'-концевому процессингу, а также следующей за ним стадии переноса цепи - /Р/Σ. Добавление в реакционную смесь ингибитора приводило к снижению суммарной концентрации продуктов вследствие подавления каталитического превращения субстрата. Остаточная концентрация продуктов при добавлении ингибитора в наибольшей взятой концентрации обозначена как [P]fin. Экспериментальные зависимости образования продуктов каталитического превращения U5-субстрата от концентрации ингибитора аппроксимировали экспоненциальной функцией вида
[P]Σ=[P]fin+A × exp(-[I]/B),
где [I] и [P] - общие концентрации ингибитора и суммы продуктов реакции соответственно, а А, В - вычисляемые параметры.
Эмпирические параметры А и В использовались для нахождения значения IC50, были рассчитаны значения IC50 ингибиторов как
IC50=В×ln(2A/(A-[P]fin)).
Процесс определения IC50 для ингибирования активности 3'-процессинга ВИЧ-1 интегразы проиллюстрирован на фиг.1, 2 и 3, которые демонстрируют полученный гель и графическую зависимость ингибирования 3'-процессинга одним из соединений. На фиг.2 показан радиоавтограф геля, причем дорожка А соответствует исходной 32Р-меченной олигонуклеотидной цепи, дорожка В соответствует продуктам реакции в присутствии интегразы без добавления ингибитора и ДМСО, дорожка С-продуктам реакции в присутствии интегразы без добавления ингибитора, но в присутствии 10% ДМСО. Остальные дорожки соответствуют продуктам реакции в присутствии интегразы и ингибитора в последовательно увеличивающихся концентрациях (показано треугольником). Так как измерения с участием ингибиторов проводятся в присутствии 10% (объемных) ДМСО, то эксперимент, представленный дорожкой С, необходим для контроля активности интегразы в присутствии 10% ДМСО.
Влияние на 3'-процессинг активность интегразы в условиях предварительно сформированного комплекса ДНК-интеграза.
Для формирования комплекса интегразы с субстратной ДНК 21-звенный меченый субстрат 32P-U5B/U5A (3 нМ) инкубировали вместе со 100 нМ интегразой в течение 30 минут при комнатной температуре в 18 мкл буфера (20 мМ HEPES 7.5 рН, 1 мМ ДТТ, 7.5 мМ хлорид магния). Затем к предварительно сформированному таким образом комплексу интегразы с субстратом добавляли 2 µL раствора ингибитора в возрастающих концентрациях, чтобы после прибавления раствора концентрация интегразы составила 100 nМ, концентрация субстрата 3 nМ, а концентрация ингибитора 0.1-100 µM. После добавления ингибитора реакционная смесь инкубировалась при 37°С в течение 2 часов. Последующие операции аналогичны описанным выше в разделе: Влияние на 3'-процессинг активность интегразы в условиях конкурентного ингибирования.
Влияние на активность интегразы в процессе переноса цепи (strand transfer). Исследование влияния ингибиторов на процесс переноса полинуклеотидной цепи, катализируемый ВИЧ-1 интегразой, проводили в условиях, аналогичных ингибированию 3'-процессинга в конкурентных условиях. Субстрат для изучения процесса переноса цепи представлял собой процессированый аналог U5 дуплекса, меченный 32Р. Концентрация субстрата составляла 10 nМ. Все другие компоненты присутствовали в реакционной смеси в приведенных выше концентрациях (см. Раздел Влияние на 3'-процессинг активность интегразы в условиях конкурентного ингибирования). Степень конверсии субстрата оценивали по появлению радиоактивных полос, соответствующих полинуклеотидам, обладающим в процессе электрофореза меньшей подвижностью, чем исходная 19-мерная полинуклеотидная цепь. Отношение между радиоактивностью, связанной с этими медленными полосами и радиоактивностью, содержащейся в полосе, обусловленной наличием исходной 19-мерной полинуклеотидной цепи, использовалось для оценки эффективности реакции переноса цепи. Величина IC50 определялась в соответствии с вышеописанной процедурой (см. Раздел: Влияние на 3'-процессинг активность интегразы в условиях конкурентного ингибирования). Для реакции переноса цепи использовалось уравнение аналогичного вида, за исключением того, что в качестве переменной [Р] рассматривалась концентрация только продуктов данной реакции. Процесс определения величины IC50 для ингибирования стадии переноса цепи проиллюстрирован на фиг.4, 5 и 6. На фиг.5 представлен радиоавтограф геля, а фиг.6 представляет графическую зависимость ингибирования стадии переноса цепи от концентрации одного из ингибиторов. На фиг.5 дорожка А соответствует исходной 32Р-меченной олигонуклеотидной цепи, дорожка В соответствует продуктам реакции в присутствии интегразы без добавления ингибитора и ДМСО, дорожка С - продуктам реакции в присутствии интегразы без добавления ингибитора, но в присутствии 10% ДМСО.
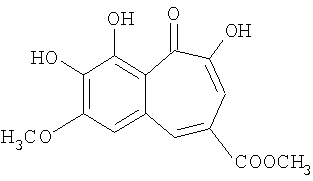
Соединения, описанные в настоящем изобретении, демонстрируют ингибирующую активность в отношении ВИЧ-1 интегразы во всех трех тестах. Некоторые из соединений, описанных в примерах 1-10, характеризуются величинами IC50 менее 5 microM во всех трех тестах. Так, для 8,9-дигидроксибензотрополон-4-карбоновой кислоты (получение см. Пример 5) получено IC50=2 мкМ. Для 7-метокси-8,9-дигидроксибензотрополон-4-метиленкарбоновой кислоты (получение см. Пример 9) получено IC50=4.8 мкМ.
Пример 12.
Ингибирующая активность соединений, описанных в настоящем изобретении, по отношению к репродукции вируса ВИЧ-1 определялась в соответствии с описанной ниже процедурой. Человеческие лимфобластоидные клетки МТ-4 (3.0-5.0··105 клеток на мл) инфицировали вирусом ВИЧ-1 (штамм BRU) в дозе 100 ТЦИД50 и инкубировали затем 5 дней в культуральной среде RPMI 1640, содержащей 10% (объемных) сыворотки новорожденных телят и 100 мкг/мл гентамицина при 37°С в атмосфере, содержащей 5% углекислоты при 98% влажности. Одновременно с внесением ВИЧ-1 инфекции в культуральную среду добавляли тестируемое соединение в виде растворов, полученных в результате последовательного разбавления. По истечении 5 дней количество живых клеток определяли по методу окрашивания трипановым синим. Для тестируемого соединения величина EC50 определялась как концентрация этого соединения в культуральной среде, при которой происходило подавление на 50% цитопатического эффекта, вызванного размножением вируса в культуре клеток МТ-4.
Соединения, описанные в настоящем изобретении, демонстрируют ингибирующую активность в отношении репликации ВИЧ-1 в культуре лимфобластоидных клеток МТ-4.
Некоторые из соединений, описанных в примерах 1-10, характеризуются величинами ЕС50 менее 10 мкМ в описанном выше тесте на ингибирование репродукции ВИЧ-1. Так, для 8,9-дигидроксибензотрополон-4-карбоновой кислоты (получение см. Пример 5) получено EC50=9 мкМ. Для 7-метокси-8,9-дигидроксибензотрополон-4-метиленкарбоновой кислоты (получение см. Пример 9) получено EC50=7 мкМ.
Пример 13
Соединения, описанные в настоящем изобретении, испытывались на острую летальность на мышах (самки линии С57В16) - то есть смертность в течение первых двух часов после внутрибрюшинного введения препарата. Соединения переводились в натриевые соли путем взаимодействия с эквимолярным количеством метилата натрия в метиловом спирте с последующей отгонкой растворителя, как это описано в Примере 9.
Затем натриевая соль растворялась в изотоническим растворе хлорида натрия до получения необходимой концентрации. Каждая мышь получала инъекцию 0.5 см3 полученного раствора. Величины испытанных доз выбирались в соответствии с «Табличным экспресс-методом» по В.Б.Прозоровскому «Практическое пособие по ускоренному определению средних эффективных доз и концентраций биологически активных веществ». Байкальск, СпбГУ, 1994, 46 с. Использовалось по две мыши на дозу.
Расчет величины LD50, характеризующей степень токсичности, проводился также в соответствии с «Табличным экспресс-методом» по В.Б. Прозоровскому.
В результате было показано, что соединения, описанные в настоящем изобретении, обладают низкой токсичностью. Так, для 8,9-дигидроксибензотрополон-4-карбоновой кислоты (получение см. Пример 5) получено LD50=0.55 г/кг. Для 7-метокси-8,9-дигидроксибензотрополон-4-метиленкарбоновой кислоты (получение см. Пример 9) получено LD50 >0.75 г/кг.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ БЕНЗОТРОПОЛОНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ИНГИБИРОВАНИЯ РЕПЛИКАЦИИ ВИРУСОВ | 2007 |
|
RU2359955C2 |
| ПРОИЗВОДНЫЕ 1,2,5-ОКСАДИАЗОЛОВ, ОБЛАДАЮЩИЕ АНТИ-ВИЧ АКТИВНОСТЬЮ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ИНГИБИРОВАНИЯ ИНТЕГРАЗЫ ВИЧ-1 | 2012 |
|
RU2515413C2 |
| ПРОИЗВОДНЫЕ, РОДСТВЕННЫЕ ЛИЗИНУ, КАК ИНГИБИТОРЫ АСПАРТИЛПРОТЕАЗЫ ВИЧ | 2007 |
|
RU2458916C2 |
| ПРОТИВОВИРУСНЫЕ СРЕДСТВА ДЛЯ ЛЕЧЕНИЯ И ПРОФИЛАКТИКИ ВИЧ ИНФЕКЦИИ | 2021 |
|
RU2780103C1 |
| ПРОИЗВОДНЫЕ БЕНЗОТИОФЕНА, БЕНЗОФУРАНА, ИНДОЛТИАЗЕПИНОНА, ОКСАЗЕПИНОНА И ДИАЗЕПИНОНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ ИНГИБИРУЮЩЕЙ КЛЕТОЧНУЮ АДГЕЗИЮ ИЛИ ВИЧ-АКТИВНОСТЬЮ, СПОСОБ ТОРМОЖЕНИЯ АДГЕЗИИ ЛЕЙКОЦИТОВ К ЭНДОТЕЛИАЛЬНЫМ КЛЕТКАМ ПРИ ЛЕЧЕНИИ ВЫЗВАННЫХ ЕЮ БОЛЕЗНЕЙ, СПОСОБ ЛЕЧЕНИЯ МЛЕКОПИТАЮЩИХ, ЗАРАЖЕННЫХ ВИЧ | 1995 |
|
RU2144033C1 |
| ИНГИБИТОРЫ ВИЧ-ИНТЕГРАЗЫ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2002 |
|
RU2284315C2 |
| СОЕДИНЕНИЯ НА ОСНОВЕ ЛИЗИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ЭТИ СОЕДИНЕНИЯ, ПРИМЕНЕНИЕ УКАЗАННЫХ СОЕДИНЕНИЙ ДЛЯ ЛЕЧЕНИЯ ИЛИ ПРОФИЛАКТИКИ ВИЧ ИНФЕКЦИИ | 2004 |
|
RU2379312C2 |
| ТЕТРАГИДРО-4Н-ПИРИДО[1,2-а]ПИРИМИДИНЫ И РОДСТВЕННЫЕ СОЕДИНЕНИЯ, ПОЛЕЗНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ВИЧ-ИНТЕГРАЗЫ | 2003 |
|
RU2329265C2 |
| 1,5,6-ЗАМЕЩЕННЫЕ 2-ОКСО-3-ЦИАНО-1,6А-ДИАЗАТЕТРАГИДРОФЛУОРАНТЕНЫ | 2006 |
|
RU2389730C2 |
| 5-АМИДО-ЗАМЕЩЕННЫЕ ПИРИМИДИНЫ, ИНГИБИРУЮЩИЕ ВИЧ | 2007 |
|
RU2480464C2 |
Настоящее изобретение относится к новым производным бензотрополона общей структурной формулы (А), а также к их фармацевтически приемлемым солям, обладающим анти-ВИЧ активностью, к фармацевтической композиции на их основе и к способу ингибирования ВИЧ-1 интегразы
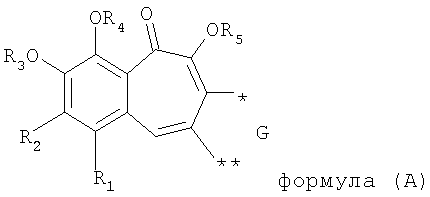
где R1 выбирают из группы, состоящей из водорода, галогена; R2 выбирают из группы, состоящей из водорода, ОС1-6алкила; R3, R4, R5 каждый независимо представляет собой водород; G выбирают из группы, состоящей из структур I, II; R6 представляет собой водород; R7 представляет собой группу COOR9; R8 выбирают из С1-6алкила, замещенного одним или более атомами галогена; W выбирают из О; R9 выбирают из водорода, С1-6алкила. Структуры I, II представлены в формуле изобретения. 3 н. и 2 з.п. ф-лы, 6 ил.
1. Производные бензотрополона, обладающие анти-ВИЧ активностью, общей структурной формулы (А):
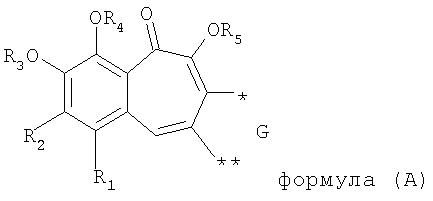
где R1 выбирают из группы, состоящей из водорода, галогена;
R2 выбирают из группы, состоящей из водорода, ОС1-6алкила;
R3, R4, R5 каждый независимо представляет собой водород;
G выбирают из группы, состоящей из (структуры I-II):
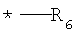
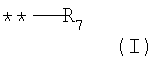
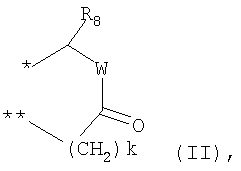
где k=0,1;
R6 представляет собой водород;
R7 представляет собой группу COOR9;
R8 выбирают С1-6алкила, замещенного одним или более атомами галогена;
W выбирают из О;
R9 выбирают из водорода, С1-6алкила;
или их фармацевтически приемлемые соли.
2. Производные бензотрополона по п.1, их фармацевтически приемлемые соли,
где R1 представляет собой водород, Br;
R2 выбирают из водорода, ОСН3;
R3, R4, R5 представляют собой водород;
G представляет собой структуру (I);
R6 представляет собой водород;
R7 представляет собой Y;
Y выбирают из группы, состоящей из СООН, СООС1-6алкил.
3. Производные бензотрополона по п.1, их фармацевтически приемлемые соли,
где R1 представляет собой водород, Br;
R2 представляет собой водород, ОСН3;
R3, R4, R5 представляют собой водород;
G представляет собой структуру (II);
R8 представляет собой группу CCl3, CF3;
W представляет собой О.
4. Фармацевтическая композиция, обладающая ингибирующей активностью в отношении репликации вируса ВИЧ-1, содержащая терапевтически эффективное количество производного бензотрополона по п.1 или его фармацевтически приемлемой соли, и фармацевтически приемлемый носитель.
5. Способ ингибирования ВИЧ-1 интегразы, включающий введение млекопитающему терапевтически эффективного количества соединения по п.1 или его фармацевтически приемлемой соли.
| HORNER L | |||
| ET AL | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Chemische Berichte, 1961, том 94, №5, с.1267-1276 | |||
| HORNER L | |||
| ET AL | |||
| Приспособление для точного наложения листов бумаги при снятии оттисков | 1922 |
|
SU6A1 |
| Chemische Berichte, 1961, том 94, №5, с.1276-1291 | |||
| Устройство для переключения подачи рабочего стола в металлорежущих станках | 1981 |
|
SU1091114A1 |
| АРОМАТИЧЕСКИЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ОБЛАДАЮЩИЕ АКТИВНОСТЬЮ В ОТНОШЕНИИ ВИЧ-ИНТЕГРАЗЫ | 1999 |
|
RU2225860C2 |

