Настоящее изобретение относится к молекулярной биологии, биохимии, медицине, в частности к антивирусной терапии, и описывает применение соединений ненуклеозидной природы: производных 1,2,5-оксадиазолов (фуразанов), N-оксидов 1,2,5-оксадиазолов (фуроксанов), 1,2,5-бензоксадиазолов (бензофуразанов), N-оксидов 1,2,5-бензоксадиазолов (бензофуроксанов) следующей общей структурной формулы
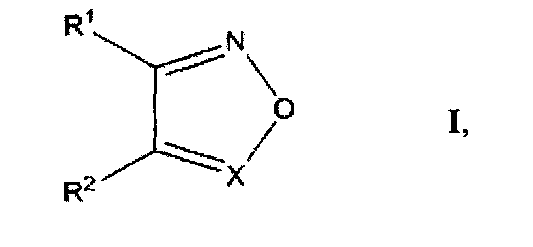
а также фармацевтические композиции на их основе для ингибирования репликации вируса иммунодефицита человека (ВИЧ), или предотвращения или лечения инфекции, вызванной ВИЧ, или для предотвращения, лечения или задержки развития Синдрома Приобретенного Иммунодефицита (СПИД).
Вирус ВИЧ-1 принадлежит к числу ретровирусов и может инфицировать клетки, содержащие поверхностный рецептор CD-4. Он является этиологическим агентом, вызывающим Синдром Приобретенного мунодефицита и дегенеративные изменения нервной системы. Следовательно, соединения, которые способны устранять вирус из тела пациента или замедлять его размножение в теле пациента, могут быть эффективны для лечения или профилактики ВИЧ-1 инфекции и СПИД.
Как и другие ретровирусы, после прикрепления и проникновения в клетку-хозяина, вирус ВИЧ-1 высвобождает в клеточную цитоплазму вирусную РНК и три фермента, необходимые для репликации вируса: обратную транскриптазу, интегразу и вирусную протеазу. Обратная транскриптаза производит синтез вирусной ДНК на матрице вирусной РНК. Эта вирусная ДНК затем проникает в ядро и интегрируется в геном клетки-хозяина с помощью ВИЧ-1 интегразы. Вирусная ДНК, интегрированная в геном клетки, затем участвует в процессах транскрипции и трансляции, осуществляемых с помощью обычных ферментативных реакций клетки-хозяина. Синтезированные таким образом вирусные белки подвергаются окончательной обработке ВИЧ-1 протеазой, после чего вирусные белки и вирусные РНК формируют новые единицы вируса, которые покидают инфицированную клетку.
Таким образом, ферменты, специфичные для вируса ВИЧ-1 - обратная транскриптаза, протеаза и интеграза, необходимы для репликации вируса и представляют собой естественные мишени для возможной антиретровирусной терапии.
В настоящее время существует множество доступных противовирусных препаратов, способных противодействовать инфекции. Эти препараты могут быть разделены на три класса, основываясь на вирусном белке, который является их мишенью, а также на способе их действия. В частности, саквинавир, индинавир, ритонавир, нелфинавир и ампренавир являются конкурентными ингибиторами аспартильной протеазы, экспрессируемой ВИЧ. Зидовудин, диданозин, ставудин, ламивудин и абакавир являются нуклеотидными ингибиторами обратной транскриптазы, которые ведут себя как миметики субстрата и которые останавливают синтез вирусной ДНК. Ненуклеотидные ингибиторы обратной транскриптазы, такие как невирапин, делавирдин и эфавиренц, ингибируют синтез вирусной ДНК через неконкурентный механизм. Использование этих препаратов является эффективным лишь для снижения репликации вируса. Эффект является только временным, поскольку вирус легко вырабатывает устойчивость ко всем известным агентам. Однако комбинированная терапия доказала свою высокую эффективность как для ослабления репликации вируса, так и для подавления появления устойчивости у множества пациентов. В США, где широко применяется комбинированная терапия, количество смертельных случаев, связанных с ВИЧ, уменьшилось (Palella, F.J.; Delany, К.М.; Morman, А.С.; Loveless, M.O.; Futher, J.; Satten, G.A.; Aschman, D.J.; Holmberg, S.D. N. Engl. J. Med. 1998, 338; 853).
Стадия интеграции вирусной ДНК в клеточную является одной из ключевых в репликативном цикле ВИЧ, поэтому катализирующий ее вирусный фермент интеграза (ИН) представляет собой одну из самых привлекательных мишеней для разработки ингибиторов ВИЧ-1. Показано, что вирус, содержащий дефектную ИН, не способную осуществлять интеграцию вирусной ДНК, не размножается в культуре клеток. Кроме того, ИН не имеет клеточного эквивалента, и, следовательно, ингибиторы, специфично подавляющие ее каталитическую активность, не должны влиять на клеточные процессы и, как следствие, должны быть менее токсичны для клетки и всего организма в целом, чем ингибиторы других стадий репликативного цикла ВИЧ. Интеграция протекает в два этапа: сначала интеграза катализирует реакцию 3'-концевого процессинга, которая представляет собой отщепление динуклеотида GT с 3'-конца каждой цепи вирусной кДНК; затем ИН встраивает концы вирусной ДНК в клеточную ДНК. По механизму действия ингибиторы интеграции можно разделить на две группы: ингибиторы 3'-концевого процессинга и ингибиторы встраивания вирусной ДНК в геном клетки [Приказчикова Т.А., Сычева A.M., Агапкина Ю.Ю., Александров Д.А., Готтих М.Б. Успехи химии, 2008, 77, 445-459]. Разрешенный к применению в качестве компонента анти-ретровирусной терапии препарат Исентресс или ралтегравир относится к ингибиторам встраивания. Этот препарат показал меньшую токсичность, чем ингибиторы обратной транскриптазы и протеазы ВИЧ-1, он лучше переносится пациентами.
Таким образом, во врачебную практику введено большое количество ингибиторов обратной транскриптазы и протеазы ВИЧ-1 и только один ингибитор ИН ВИЧ-1, хотя ИН, несомненно, является крайне привлекательной мишенью для терапии ВИЧ-1 инфекции. Все перечисленные препараты применяются в форме многокомпонентной терапии. Такая многокомпонентная терапия на сегодняшний день является основным способом борьбы с Синдромом Приобретенного Иммунодефицита.
К сожалению, не все пациенты являются восприимчивыми к многокомпонентной терапии, и ее применение часто может оказаться неудачным, кроме того, многие анти-ВИЧ лекарственные средства обладают побочными токсическими эффектами. Известно, что приблизительно 30-50% пациентов, в конечном счете, не воспринимают комбинированную терапию, причем, основные проблемы обычно связаны с появлением вирусной устойчивости. Очевидно, что появление новых мультирезистентных штаммов ВИЧ-1, устойчивых к стандартной многокомпонентной терапии, делает разработку новых анти-ВИЧ противовирусных средств крайне необходимой. Следует отметить также их высокую стоимость и отсутствие (за редким исключением) отечественных лекарств. Таким образом, поиск новых соединений, обладающих анти-ВИЧ активностью как в отношении дикого штамма ВИЧ-1, так и в отношении резистентных изолятов вируса, с целью создания лекарственных анти-ВИЧ препаратов представляет крайне важную и перспективную проблему современной вирусологии и медицинской химии.
Область применения разработки - терапия ВИЧ-инфекции и СПИД. Ингибиторы ВИЧ-1 ИН могут быть эффективными лекарственными веществами для предотвращения и лечения ВИЧ-1 инфекции, так как ВИЧ-1 ИН является ключевым ферментом, необходимым для репликации ВИЧ.
В настоящее время имеется большое число патентных публикаций, касающихся различных аспектов синтеза новых веществ, которые могут быть использованы против вируса иммунодефицита человека. Однако применение всех этих веществ имеет ограничения, главным из которых является образование в процессе терапии устойчивых форм вируса, что делает необходимой постоянную смену препаратов. Лекарственное вещество должно быть эффективно в отношении огромного количества различных видов ВИЧ-1, в то время как большинство известных соединений проявляют недостаточно высокую эффективность по отношению к мутантным формам ВИЧ. Таким образом, существует большая потребность в разработке новых структур, которые могли бы использоваться при устойчивых мутациях вируса.
Разработка ингибиторов интеграции ВИЧ-1 ведется более 10 лет, но к настоящему времени только один ингибитор интеграции - ралтегравир - допущен к применению в качестве лекарственного средства. Этот ингибитор хелатирует ионы металла, находящиеся в активном центре фермента, и блокирует одну из реакций, катализируемых интегразой, - перенос цепи. Ралтегравир успешно прошел все стадии клинических испытаний и в октябре 2007 г. был одобрен FDA в качестве лекарственного средства для больных, приобретших устойчивость к компонентам ВААРТ [FDA approves raltegravir tablets. AIDS Patient Care STDS 2007, 21, 889], а в июле 2009 г. FDA разрешило использовать ралтегравир и при первичной терапии ВИЧ-инфицированных [FDA notifications. Raltegravir indication extended for treatment-naive patients. AIDS Alert 2009. 24, 93; WO 2003/035077; WO 2006/060730]. Одним из недостатков терапии с использованием этого лекарственного средства является то, что у некоторых больных вирус достаточно быстро приобретает устойчивость к ралтегравиру. На стадии клинических испытаний находятся еще несколько ингибиторов интеграции ВИЧ-1. Однако все они являются аналогами ралтегравира как по структуре, так и по механизму действия. Возникновение устойчивости вируса к некоторым из них вызвано мутациями в интегразе, сходными с мутациями, обуславливающими устойчивость к ралтегравиру.
В 2005 году компании «Japan Тоbассо» и «Gilead Sciences)) начали клинические испытания ингибитора интегразы, названного элвитегравир, [Al-Mawsawi, L.Q.; Al-Safi, R.I.; Neamati, N. Expert Opin. Emerg. Drugs 2008, 13, 213; WO2007/089030; US 2010/0285122 A1], который является наиболее активным представителем ингибиторов ИН структурного класса 4-оксохинолина. По результатам исследований элвитегравир показал высокую эффективность, сравнимую с эффективностью широко применяемого противовирусного средства эфавиренца.
Все известные аналоги ралтегравира действуют на интегразу ВИЧ-1 по одному и тому же механизму, содержат сходный структурный мотив и проявляют сравнимую активность in vitro и в клеточных испытаниях. Идентичность подобного рода ставит под сомнение успех будущего применения данных ингибиторов в качестве лекарственных средств. Эти опасения вызваны перекрестной резистентностью вируса к этим ингибиторам. Возникновение перекрестной устойчивости можно объяснить, в первую очередь, схожим механизмом связывания ингибиторов переноса цепи с комплексом ИН и вирусной ДНК [Mouscadet, J.F.; Delelis, О.; Marcelin, A.G.; Tchertanoy, L. Drug Resist Updat. 2010, 13, 139]. Эти соединения связываются таким образом, что «выталкивают» 3'-концевой гидроксил процессированной цепи ДНК из активного центра фермента и тем самым блокируют интеграцию.
Еще одна проблема, способная осложнить успешное применение ингибиторов интеграции, - недостаток наших знаний о полиморфизме ИН у ВИЧ-1 различных подтипов. В опытах in vitro показано, что ИН вируса подтипа С, содержащая мутацию E92Q/N155H, в 10 раз более чувствительна к ралтегравиру и элвитегравиру, чем фермент ВИЧ-1 подтипа В [Bar-Magen, Т.; Donahue, D.A.; McDonough, E.I. AIDS 2010, 24, 2171]. Показано также, что у вируса подтипа CR F02 AG реже возникают мутации остатка G140, чем у подтипа В [Maiga, A.I.; Malet, I.; Soulie, С; Derache, A.; Koita, V.; Amellal, В.; Tchertanov, L.; Delelis, O.; Morand-Joubert, L.; Mouscadet, J.-F., et al. Antivir. Ther. 2009, 14. 123]. Есть данные и о том, что ралтегравир чаще оказывается неэффективным у лиц, инфицированных вирусом не-В-подтипа [Sichtig, N.; Sierra, S.; Kaiser, R.; Daumer, M.; Reuter, S.; Schulter, E.; Altmarm, A.; Fatkenheuer, G.; Dittmer, U.;, Pfister, H., et al. J. Antimicrob. Chemother. 2009, 64, 25]. Этот результат особенно важен, учитывая, что в России наиболее распространенным является вирус подтипа А.
Таким образом, задачей настоящего изобретения является применение химических соединений ненуклеозидной природы общей структурной формулы I, включающих производные 1,2,5-оксадиазолов (фуразанов), N-оксидов 1,2,5-оксадиазолов (фуроксанов), 1,2,5-бензоксадиазолов (бензофуразанов), JV-оксидов 1,2,5-бензоксадиазолов (бензофуроксанов), а также фармацевтических композиций на их основе, для ингибирования репликации вируса иммунодефицита человека (ВИЧ), для предотвращения или лечения инфекции, вызванной вирусом иммунодефицита человека (ВИЧ), а также для предотвращения, лечения или задержки развития Синдрома Приобретенного Иммунодефицита (СПИД).
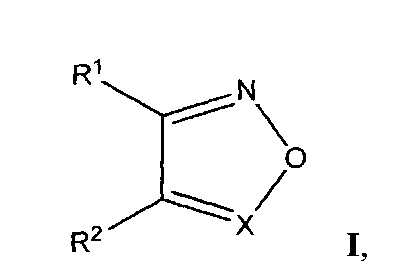
где R1 и R2 выбирают из фенилсульфонила, замещенного одним или более атомами галогена, нитрогруппами, карбоксигруппами, алкилгалогенидами, СН3, ОСН3, OCF3;
X выбирают из N или N→O;
либо R1 и R2 образуют группу
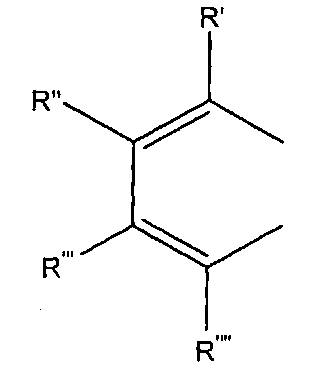
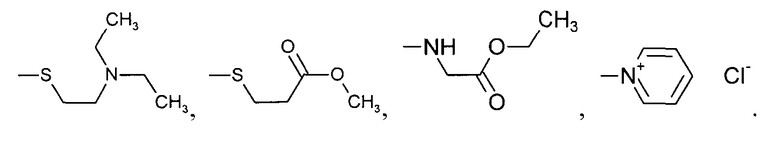
где R', R'', R''' и R'''' независимо выбирают из водорода; галогенов; нитрогруппы, гидроксигруппы, карбоксигруппы, СН3; CH2Br; ОСН3; фенилсульфонила; фенилтиогруппы; или следующих групп: 
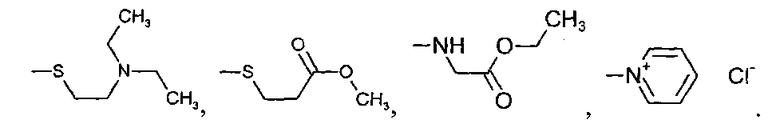
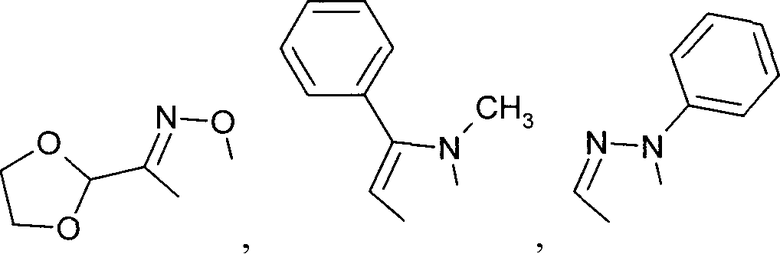
R' и R'' также могут быть объединены в один из следующих общих циклов:
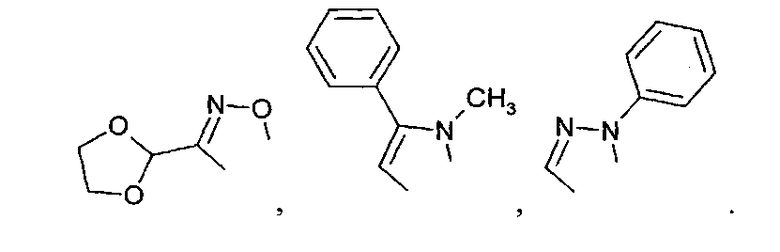
В настоящем изобретении, если не определено иное, используются следующие определения:
«Галоген» означает хлор, бром, йод или фтор.
Термин «пролекарство» означает производное соединения, описанного в настоящем изобретении, которое имеет группы, легко отщепляемые в результате химических или метаболических процессов, и которое после применения пациентом в его организме отщепляет эти группы, давая исходное соединение по настоящему изобретению, проявляющее присущую ему анти-ВИЧ активность in vivo. «Пролекарство» соединений структурной формулы I может быть получено обычным способом с помощью модификации функциональных групп соединений, таких как гидрокси- или карбоксигруппа.
«Пролекарства» применяются для улучшения таких характеристик лекарственных средств, как растворимость, тканевая совместимость, контролируемое высвобождение в организме млекопитающего, всасывание из желудочно-кишечного тракта и транспортировка лекарственного вещества в организме к месту его действия.
Пролекарства включают производные кислот, хорошо известные средним специалистам в данной области, такие как, например, сложные эфиры, полученные путем взаимодействия исходного кислотного соединения с соответствующим спиртом или амиды, полученные взаимодействием исходного кислотного соединения с соответствующим амином. Сложные алифатические или ароматические эфиры, производные от кислотных боковых групп соединений по настоящему изобретению, являются предпочтительными пролекарствами. В некоторых случаях желательно получать другие эфирные производные, такие как алкил-замещенные сложные эфиры или ((алкоксикарбонил)окси)алкил-замещенные сложные эфиры.
Термин «фармацевтически приемлемый» носитель означает, что носитель должен являться совместимым с другими ингредиентами композиции и не наносить вреда его реципиенту, то есть быть нетоксичным для клеток или млекопитающих в тех дозах и концентрациях, в которых его применяют. Часто фармацевтически приемлемый носитель представляет собой водный буферный раствор. Примеры физиологически приемлемых носителей включают буферы, например растворы фосфатов, цитратов и других солей органических и неорганических кислот, антиоксиданты, включающие аскорбиновую кислоту; полипептиды с низким молекулярным весом (меньше 10 остатков); белки, такие как сывороточный альбумин, желатин или иммуноглобулины; гидрофильные полимеры, такие как поливинилпирролидон; аминокислоты, такие как глицин, глютамин, аспарагин, аргинин или лизин; моносахариды, дисахариды и другие углеводы, включая глюкозу, маннозу или декстрины; хелатообразующие агенты, такие как ЭДТА; сахарные спирты, такие как маннитол или сорбитол.
Терапевтически эффективное количество - это количество, необходимое для достижения эффекта подавления репликации ВИЧ в организме млекопитающего.
«Млекопитающее», включает в себя представителей отряда приматов (например, человек, человекообразные обезьяны, нечеловекообразные обезьяны, низшие обезьяны), отряда хищных (например, кошки, собаки, медведи), отряда грызунов (например, мышь, крыса, белка), отряда насекомоядных (например, землеройка, крот) и др.
Под термином «профилактика СПИД» понимается введение анти-ВИЧ лекарственных средств ВИЧ-позитивными субъектами, у которых, однако, еще не развился Синдром Приобретенного Иммунодефицита, а также применение анти-ВИЧ лекарственных средств субъектами с симптомами СПИД, подвергавшимися антиретровирусной терапии, чье состояние улучшилось, во избежание повторного ухудшения, а также употребление анти-ВИЧ лекарственных средств при угрозе возможного ВИЧ-заражения.
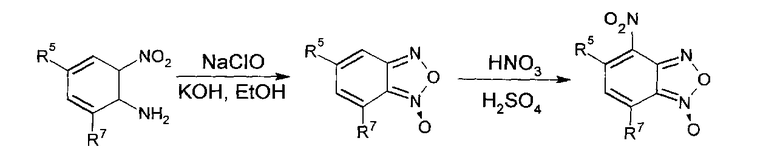
Из анализа данных литературы известен способ получения 4-нитробензофуроксанов (4-НБФС) [Ghosh Р.В., Whitehouse M.W. // J. Med. Chem. 1968, 11(2), 305-311]. Для синтеза было выбрано окисление ароматических орто-нитроаминов гипохлоритами, как способ, дающий наиболее высокие выходы, с последующим окислением полученных бензофуроксанов азотной кислотой. Таким образом, синтез 4-НБФС был проведен по следующей схеме:
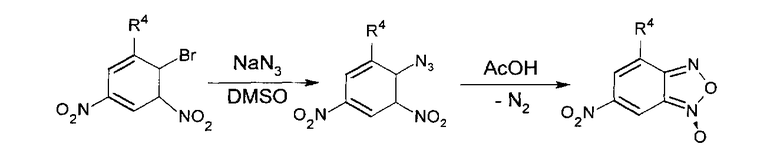
Получение 6-нитробензофуроксанов (6-НБФС) нитрованием соответствующих бензофуроксанов, как и окисление производных 2,4-динитроанилина гипохлоритами приводит к получению большого числа побочных продуктов [Terrier F., Lakhdar S., Boubaker Т., Goumont R. // J. Org. Chem., 2005, 70, 6242-6253]. Поэтому для синтеза ингибиторов ИН, относящихся к классу 6-НБФС, было решено использовать пиролиз орто-нитрофенил азидов, получаемых реакцией нуклеофильного замещения атома галогена в соответствующих динитрофенил-галагенидах азидом натрия в диметилсульфоксиде [Ghosh Р.В., Whitehouse M.W. // J. Med. Chem. 1968, 11(2), 305-311; Mallory F.B., Varinibi S.P. // J. Org. Chem. 1963, 28 (6), 1656-1659]. Общая схема получения 6-НБФС, использованная для их синтеза, представлена ниже:
Для получения 4,6-динитробензофуроксанов (4, 6-НБФС), как и 6-нитробензофуроксанов, целесообразно использовать пиролиз орто-нитрофенилазидов, а затем проводить окисление азотной кислотой [Ghosh Р.В., Whitehouse M.W. // J. Med. Chem. 1968, 11(2), 305-311; Mallory F.B., Varinibi S.P. //J. Org. Chem. 1963, 28 (6), 1656-1659]. Незамещенный 4,6-динитробензофуроксан и его производные были синтезированы по следующей схеме:
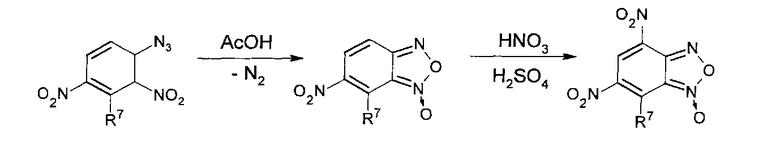
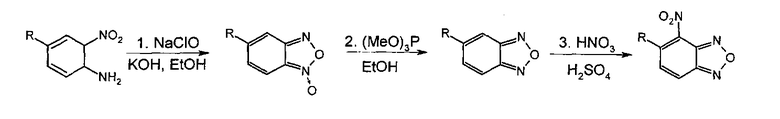
Из анализа литературы [Ghosh Р.В., Whitehouse M.W. II J. Med. Chem. 1968, 11(2), 305-311] известно, что 4-нитробензофуразаны (4-НБФЗ) наиболее удобно получать из соответствующих бензофуроксанов с последующим их восстановлением действием триметилфосфитом и нитрованием концентрированной азотной кислотой. Для проведения синтеза блокаторов интегразы использовалось окисление ароматических орто-нитроаминов гипохлоритами. Выбранная схема получения веществ представлена ниже:
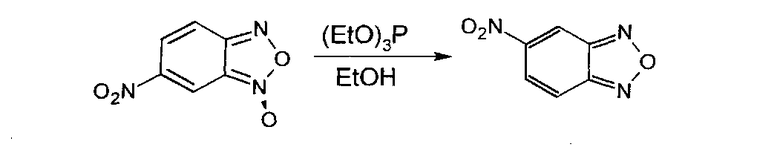
Для получения незамещенного 5-нитробензофуразана (5-НБФЗ) мы также решили использовать восстановление соответствующего 6-нитробензофуроксана согласно методике [Ghosh Р.В., Whitehouse M.W. // J. Med. Chem. 1968, 11(2), 305-311]. Схема получения потенциального блокатора ИН 5-нитробензофуразана представлена ниже:
Для подтверждения объявленных структур химических соединений были изучены спектры ЯМР 1Н, а также был проведен анализ степени чистоты новых соединений хроматографированием методом UPLC. Структуры всех синтезированных соединений были успешно подтверждены. Большая часть отобранных соединений по результатам анализа демонстрировала 80-98%-ную чистоту.
Настоящее изобретение также включает фармацевтические композиции, предназначенные для лечения ВИЧ-инфекций, содержащие терапевтически эффективное количество ненуклеозидных производных по пункту 1 и фармацевтически приемлемый носитель.
Активный ингредиент в таких составах включает от 0.1 до 99.9% от массы состава, предпочтительно от 5 до 90%.
Фармацевтические композиции могут быть получены в соответствии с известными методиками, использующими известные и легко получаемые ингредиенты. Такие композиции по настоящему изобретению обеспечивают быстрое, устойчивое или замедленное введение активного ингредиента после введения пациенту в соответствии с применяемыми процедурами, известными среднему специалисту. При получении фармацевтической композиции активный ингредиент обычно смешивают с носителем или разбавляют носителем или заключают в носитель, который может быть в форме капсулы, драже или в таблетированной форме. Когда носитель является разбавителем, то он может быть твердым веществом, полутвердым или жидким материалом, который действует как связующее средство или инертный наполнитель для активного ингредиента. Таким образом, лекарственные средства на основе фармацевтических композиций могут быть в форме таблеток, пилюль, порошков, шариков, суспензий, эмульсий, растворов, сиропов, аэрозолей мягких и твердых капсул желатина, свечей, растворов для инъекций и тому подобные. Лекарственные средства и фармацевтические композиции изготавливаются обычными технологическими методами. Лекарственные средства могут применяться в качестве внутренних или наружных медицинских средств. Лекарственные средства могут применяться перорально, парентерально или интраназально.
При применении соединений, описанных в настоящем изобретении, в составе фармацевтических композиций, они могут быть смешаны с подходящими добавками, инертными наполнителями, разбавителями, диспергирующими веществами, стабилизаторами, консервантами, буферными составами, эмульгаторами, отдушками, красителями, подсластителями и другими известными фармацевтическими добавками, такими как вода, растительные масла, спирты (например, этанол, бензиловый спирт и т.д.), полиэтиленгликоль, триацетат глицерина, желатин, полиэтоксилированные растительные масла, углеводы (например, лактоза, крахмал) стеарат магния, тальк, ланолин, петролатум и тому подобными.
Настоящее изобретение также включает способ ингибирования ВИЧ-1 интегразы, включающий введение млекопитающим терапевтически эффективного количества соединения формул I.
Способ профилактики или лечения ВИЧ-инфекции у млекопитающего включает введение такому пациенту терапевтически эффективного количества ненуклеозидных производных формул I. Применяемые дозы зависят от возраста млекопитающего, его веса, симптомов, эффекта от лечения, метода введения и т.п.Обычно дозы составляют от 0.1 мг до 5 г, предпочтительно от 10 мг до 2 г, на одну особь млекопитающего за одно введение. Таких введений может быть от одного до 10 в течение суток, либо перорально, либо путем внутривенных, внутримышечных или подкожных инъекций, либо внутривенных инфузий.
Дополнительно вместе с активным соединением могут вводить другие анти-ВИЧ агенты, в частности конкурентные ингибиторами ВИЧ-протеазы (например, саквинавир, индинавир, ритонавир, нелфинавир и ампренавир), нуклеотидные ингибиторы обратной траскриптазы (например, зидовудин, диданозин, ставудин, ламивудин, абакавир), ненуклеотидные ингибиторы обратной транскриптазы (например, невирапин, делавирдин и эфавиренц).
Доказанный биологический эффект (Ингибирования интегразы ВИЧ-1)
Для того чтобы предмет настоящего изобретения был более понятен, ниже приведены некоторые примеры использования ненуклеозидных производных в качестве ингибиторов интегразы и анти-ВИЧ лекарственных соединений. Примеры носят иллюстративный характер, причем содержание данного изобретения ни в коей мере не ограничивается представленными примерами.
Приведенные ниже примеры иллюстрируют измерения ингибирующей способности описываемых в настоящем изобретении производных ненуклеозидной природы по отношению к биохимическим реакциям, катализируемым ВИЧ-1 интегразой.
Примеры исследования биологического эффекта
Пример 1.
Влияние на активность интегразы из вируса подтипа В в реакции 3'-процессинга В 10 мкл буфера (20 мМ HEPES 7.5 рН, 1 мМ ДТТ, 7.5 мМ хлорид магния) готовили 6 нМ раствор 32Р-меченного субстрата U5B/U5A, содержащего радиоактивно меченную процессируемую цепь U5B (смесь I) и тестируемый ингибитор в диапазоне концентраций 0.02-200 мкМ. Добавляли к полученной смеси 10 мкл 250 нМ раствора интегразы в этом же буфере, перемешивали и выдерживали в термостате 2 часа при температуре 37°C. По завершении реакции к смеси добавляли 80 мкл стоп-смеси (9 мМ Tris-HCl, 6 мМ ЭДТА, 0.4 М CH3COONa, 0.125 мг/мл гликогена), фермент экстрагировали 100 мкл смеси фенол/хлороформ/изоамиловый спирт (25:24:1). Нуклеотидный материал осаждали 5-кратным избытком этилового спирта при температуре 0°C и анализировали с помощью электрофореза в 20%-ном денатурирующем ПААГ с последующей визуализацией геля на приборе STORM 840ТМ Phosphorlmager (Molecular Dynamics. США) и обсчетом средствами программы ImageQuant 4.1. О степени протекания реакции 3'-концевого процессинга судили по появлению на радиоавтографе полосы, соответствующей укороченной на два нуклеотида цепи U5B (19-звенный продукт). По соотношению интенсивностей излучения полос, соответствующих 21- и 19-звенным олигонуклеотидам, определяли эффективность процессинга. По полученным значениям эффективности рассчитывали концентрацию продукта реакции. Данные усредняли по трем независимым экспериментам. Значения IC50 ингибиторов были рассчитаны по экспериментальным зависимостям ингибирования каталитического превращения U5-субстрата интегразы от концентрации взятого ингибитора. При оценке степени превращения субстрата учитывалась суммарная концентрация ДНК, подвергшейся 3'-концевому процессингу, а также следующей за ним стадии переноса цепи -[Р]Σ. Добавление в реакционную смесь ингибитора приводило к снижению суммарной концентрации продуктов вследствие подавления каталитического превращения субстрата. Остаточная концентрация продуктов при добавлении ингибитора в наибольшей взятой концентрации обозначена как [P]fin. Экспериментальные зависимости образования продуктов каталитического превращения U5-субстрата от концентрации ингибитора аппроксимировали экспоненциальной функцией вида
[P]Σ=[P]fin+А × exp(-[I]/B),
где [I] и [Р] - общие концентрации ингибитора и суммы продуктов реакции соответственно, а А, В - вычисляемые параметры.
Эмпирические параметры А и В использовались для нахождения значения IC50. Значения IC50 ингибиторов рассчитывали как
IC50=В×ln(2A/(A-[P]fin)).
Результаты эффективности ингибиторов представлены в таблицах 1-4.
Пример 2
Влияние на активность интегразы из вируса подтипа А в реакции 3'-процессинга В 10 мкл буфера (20 мМ HEPES 7.5 рН, 1 мМ ДТТ, 7.5 мМ хлорид магния) готовили 6 нМ раствор 32Р-меченного субстрата U5B/U5A, содержащего радиоактивно меченную процессируемую цепь U5B и тестируемый ингибитор в диапазоне концентраций 0.02-200 мкМ. Добавляли к полученной смеси 10 мкл 200 нМ раствора интегразы в этом же буфере, перемешивали и выдерживали в термостате 2 часа при температуре 37°C. По завершении реакции к смеси добавляли 80 мкл стоп-смеси (9 мМ Tris-HCl, 6 мМ ЭДТА, 0.4 М CH3COONA, 0.125 мг/мл гликогена), фермент экстрагировали 100 мкл смеси фенол/хлороформ/изоамиловый спирт (25:24:1). Нуклеотидный материал осаждали 5-кратным избытком этилового спирта при температуре 0°C и анализировали с помощью электрофореза в 20%-ном денатурирующем ПААГ с последующей визуализацией геля на приборе STORM 840ТМ Phosphorlmager (Molecular Dynamics. США) и обсчетом средствами программы ImageQuant 4.1. О степени протекания реакции 3'-концевого процессинга судили как описано в примере 1.
Результаты эффективности ингибиторов представлены в таблицах 1-4.
Пример 3
Влияние на активность интегразы из вируса подтипа В в процессе переноса цепи
В 10 мкл буфера (20 мМ HEPES 7.5 рН, 1 мМ ДТТ, 7.5 мМ хлорид магния) готовили 20 мкМ раствор 32Р-меченного субстрата U5B-2/U5A, содержащего радиоактивно меченную процессированную цепь U5B-2, укороченную на два нуклеотида, и тестируемый ингибитор в диапазоне концентраций 0.02-200 мкМ. Добавляли к полученной смеси 10 мкл 250 нМ раствора интегразы в этом же буфере, перемешивали и выдерживали в термостате 2 часа при температуре 37°C. По завершении реакции к смеси добавляли 80 мкл стоп-смеси (9 мМ Tris-HCl, 6 мМ ЭДТА, 0.4 М CH3COONa, 0.125 мг/мл гликогена), фермент экстрагировали 100 мкл смеси фенол/хлороформ/изоамиловый спирт (25:24:1). Нуклеотидный материал осаждали 5-кратным избытком этилового спирта при температуре 0°C и анализировали с помощью электрофореза в 20%-ном денатурирующем ПААГ с последующей визуализацией геля на приборе STORM 840ТМ Phosphorlmager (Molecular Dynamics. США) и обсчетом средствами программы imageQuant 4.1. О степени протекания реакции переноса цепи судили по появлению радиоактивных полос, соответствующих олигонуклеотидам, обладающим в процессе электрофореза меньшей подвижностью, чем исходная 19-звенная олигонуклеотидная цепь. По соотношению интенсивностей излучения полос, соответствующих продуктам реакции с меньшей электрофоретической подвижностью, и 19-звенному исходному олигонуклеотиду, определяли эффективность реакции переноса цепи. Величина IC50 определялась в соответствии с процедурой, описанной в примере 1. Для реакции переноса цепи использовалось уравнение аналогичного вида, за исключением того, что в качестве переменной [Р] рассматривалась концентрация только продуктов данной реакции. Результаты эффективности ингибиторов представлены в таблицах 1-4.
Пример 4
Влияние на активность интегразы, содержащей аминокислотные замены, придающие ВИЧ-1 устойчивость к ралтегравиру, в процессе переноса цепи
Использовалась методика, аналогичная описанной в примере 3. При этом использовались препараты интегразы из вируса подтипа В, содержащие аминокислотные замены N155H или G140S/Q148K. Определялась активность соединений, показавших наибольшую ингибирующую активность по отношению к интегразе, не содержащей аминокислотных замен (Пример 3).
Результаты эффективности ингибиторов представлены в таблице 5.
Соединения, описанные в настоящем изобретении, демонстрируют ингибирующую активность в отношении препаратов интегразы из ВИЧ-1 подтипов А и В в обеих реакциях: 3'-процессинга и переноса цепи. Некоторые из соединений, описанные ниже, характеризуются величинами IC50 менее 5 мкМ. Наиболее активные ингибиторы интеграз сохраняют свою активность по отношению к препаратам интегразы из вируса подтипа В, содержащим аминокислотные замены N155H или G140S/Q148K, которые придают вирусу устойчивость к ралтегравиру.
Таблица 1
4-Нитрозамещенные производные бензофуразанов и их N-оксидов
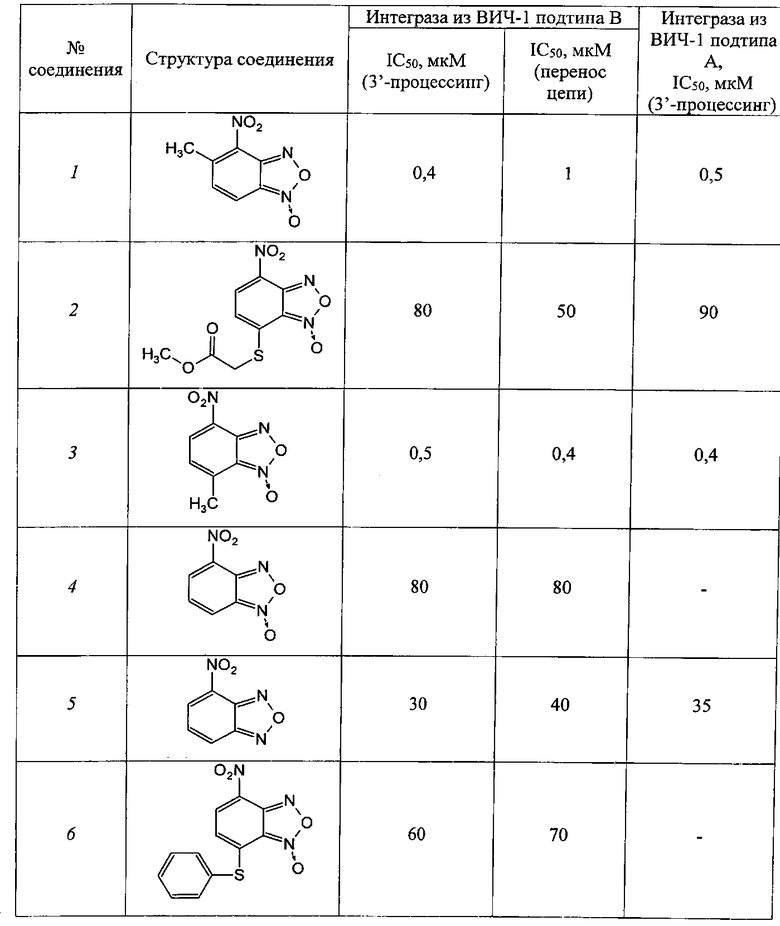
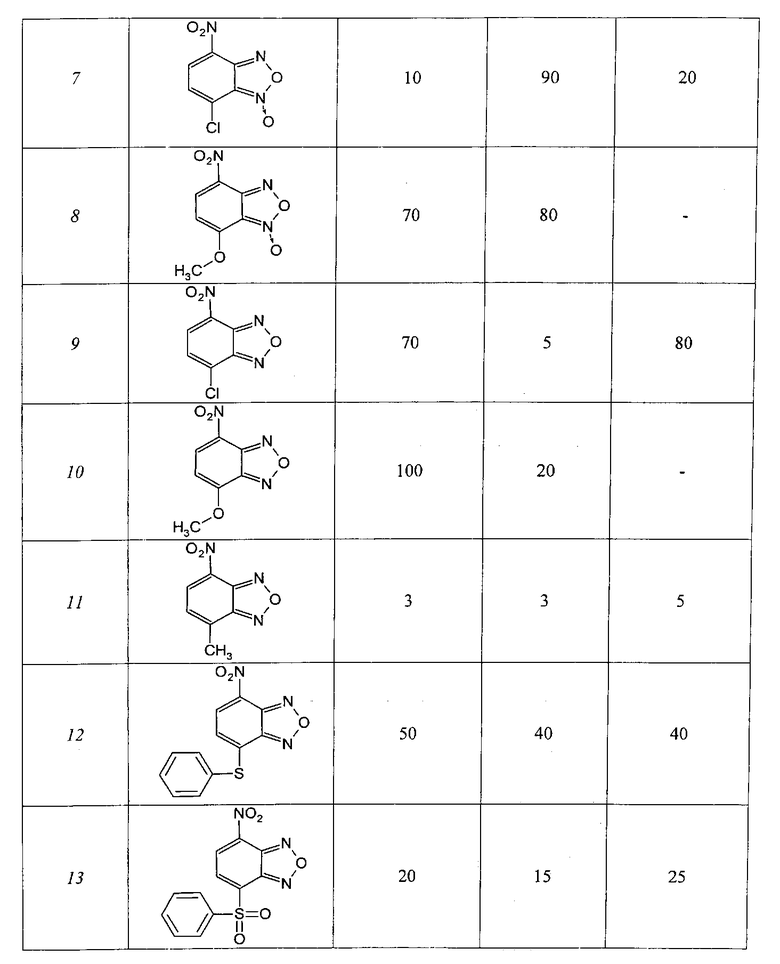

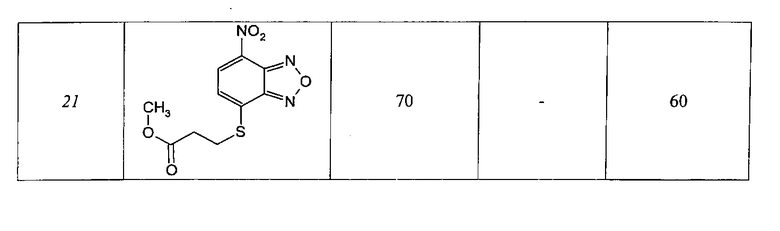
Таблица 2
5-Нитро, 6-нитрозамещенные производные бензофуразанов и их N-оксидов

Таблица 3
Динитрозамещенные производные бензофуразанов и их N-оксидов
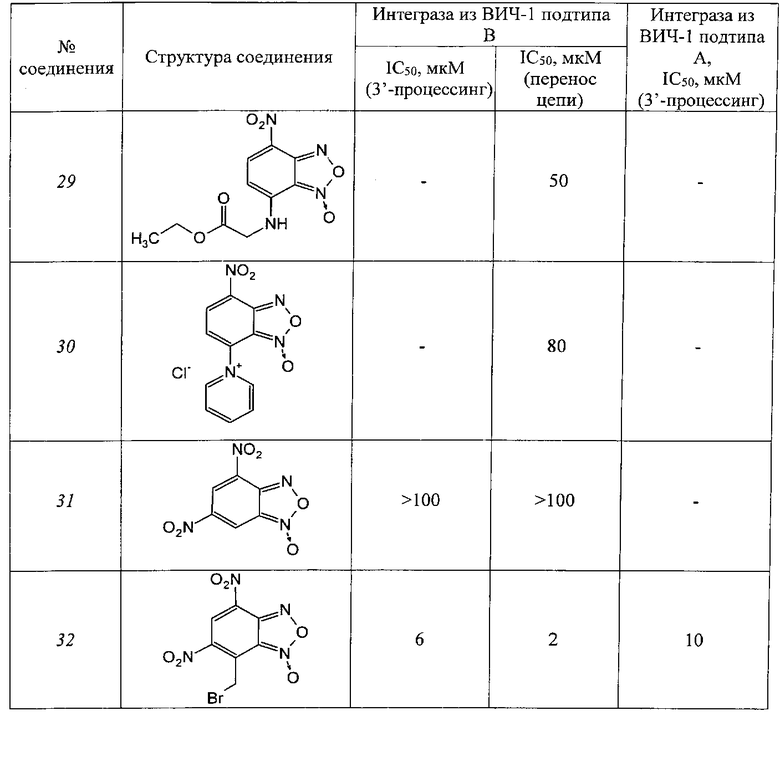
Таблица 4
Производные фуроксанов
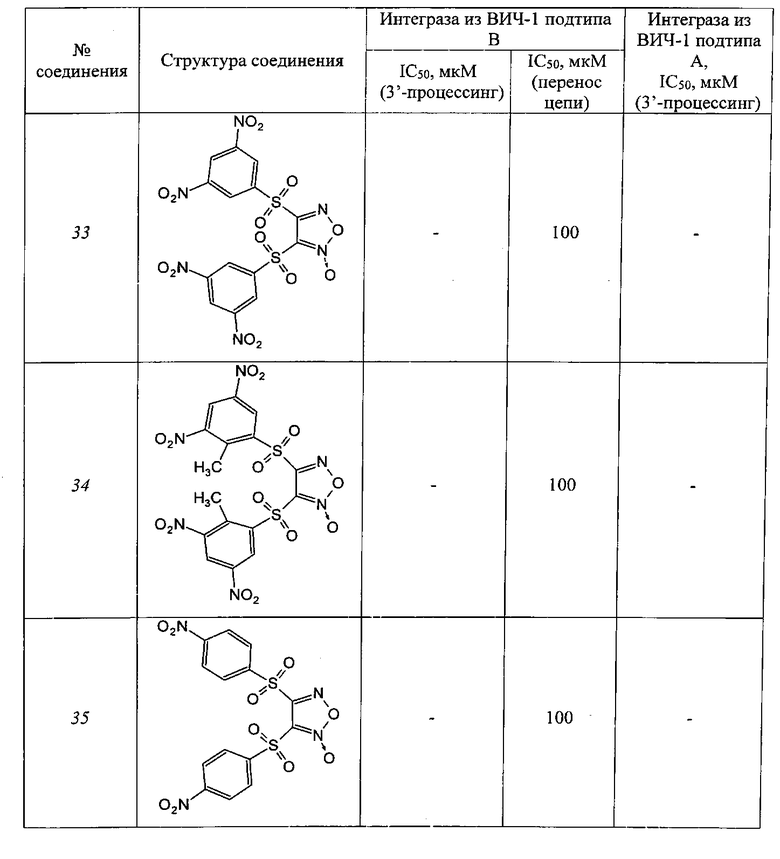
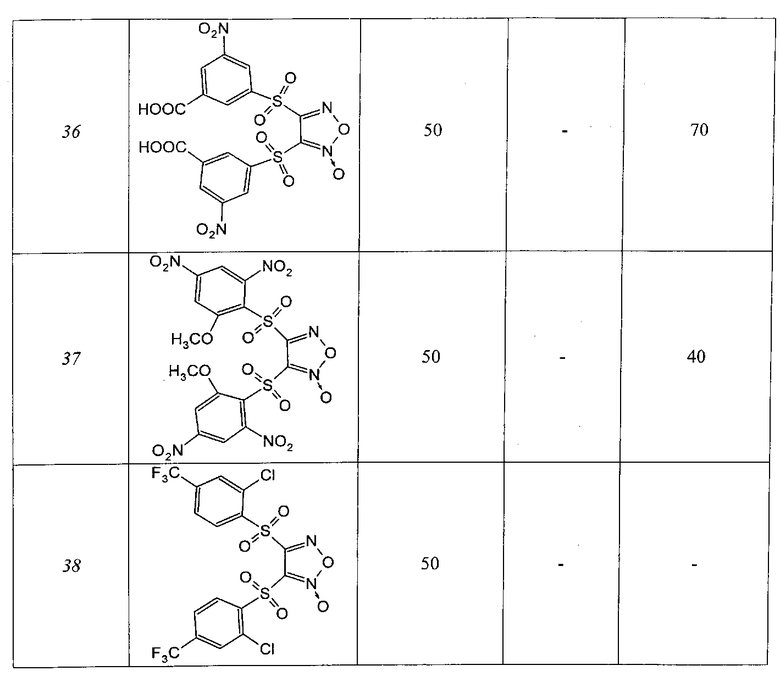
Таблица 5
Ингибирование препаратов интегразы, содержащих аминокислотные замены, придающие ВИЧ-1 устойчивость к ралтегравиру

| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ БЕНЗОТРОПОЛОНА, ОБЛАДАЮЩИЕ АНТИ-ВИЧ АКТИВНОСТЬЮ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ИНГИБИРОВАНИЯ ВИЧ-1 ИНТЕГРАЗЫ | 2007 |
|
RU2359954C2 |
| ПРОИЗВОДНЫЕ ИНГЕНОЛА ДЛЯ РЕАКТИВАЦИИ ЛАТЕНТНОГО ВИРУСА ВИЧ | 2013 |
|
RU2609512C2 |
| ПРОИЗВОДНЫЕ БЕНЗОТРОПОЛОНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ИНГИБИРОВАНИЯ РЕПЛИКАЦИИ ВИРУСОВ | 2007 |
|
RU2359955C2 |
| ФАРМАЦЕВТИЧЕСКОЕ СРЕДСТВО ДЛЯ ЛЕЧЕНИЯ ВИЧ-ИНФЕКЦИИ, СОДЕРЖАЩАЯ ЕГО КОМПОЗИЦИЯ И СПОСОБЫ ЕГО ПРИМЕНЕНИЯ | 2002 |
|
RU2290197C2 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ОПТИМАЛЬНОЙ ХИМИОТЕРАПИИ ПАЦИЕНТОВ, СЕРОПОЗИТИВНЫХ ПО ВИЧ, ОСНОВАННЫЙ НА ФЕНОТИПИЧЕСКОЙ ЛЕКАРСТВЕННОЙ ЧУВСТВИТЕЛЬНОСТИ ЧЕЛОВЕЧЕСКИХ ШТАММОВ ВИЧ | 1997 |
|
RU2174014C2 |
| ИНГИБИТОР РЕПРОДУКЦИИ ВИРУСА ИММУНОДЕФИЦИТА ЧЕЛОВЕКА | 2009 |
|
RU2396278C1 |
| ПРОИЗВОДНЫЕ ХИНОЛИНА ДЛЯ ПРИМЕНЕНИЯ В ЛЕЧЕНИИ ИЛИ ПРОФИЛАКТИКЕ ВИРУСНОЙ ИНФЕКЦИИ | 2016 |
|
RU2723016C2 |
| ИНГИБИРУЮЩИЕ ВИЧ 2-(4-ЦИАНОФЕНИЛ)-6-ГИДРОКСИЛАМИНОПИРИМИДИНЫ | 2006 |
|
RU2401261C2 |
| Схемы лечения ВИЧ-инфекций и СПИД | 2018 |
|
RU2784810C2 |
| ФАРМАЦЕВТИЧЕСКАЯ АНТИРЕТРОВИРУСНАЯ КОМПОЗИЦИЯ | 2013 |
|
RU2648457C2 |
Изобретение относится к применению производных ненуклеозидной природы - 1,2,5-оксадиазолов общей структурной формулы I
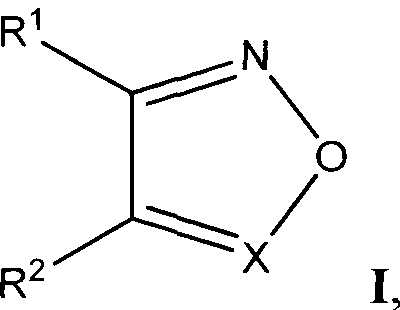
где R1 и R2 выбирают из фенилсульфонила, замещенного одним или более атомами галогена, нитрогруппами, карбоксигруппами, алкилгалогенидами, СН3, ОСН3, OCF3; Х выбирают из N или N→O; либо R1 и R2 образуют группу
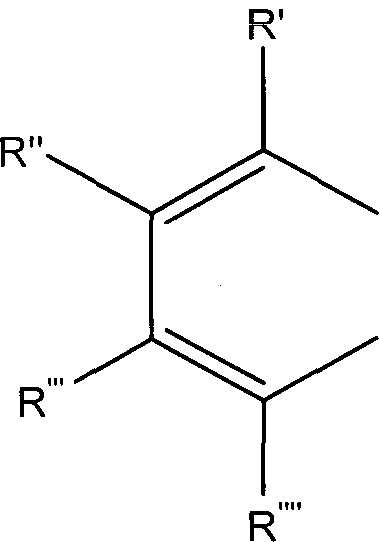
где R', R", R'" и R'''' независимо выбирают из водорода; галогенов; нитрогруппы, гидроксигруппы, карбоксигруппы, СН3; СН2Вr; ОСН3; фенилсульфонила; фенилтиогруппы; или следующих групп: 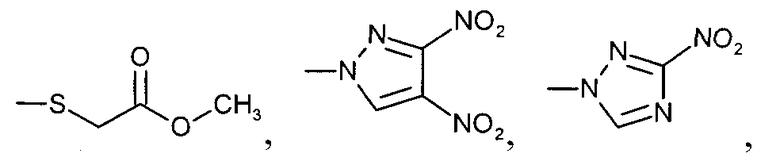
R' и R" также могут быть объединены в один из следующих общих циклов:
для ингибирования репликации вируса иммунодефицита человека (ВИЧ).
Изобретение также относится к фармацевтической композиции на основе соединений формулы I и к способу ингибирования интегразы ВИЧ-1 подтипов А и В, в том числе форм, устойчивых к ралтегравиру. Технический результат: выявлена новая активность у соединений формулы I, которые могут найти применение в медицине в качестве ингибиторов репликации ВИЧ. 3 н.п. ф-лы, 5 табл., 4 пр.
1. Применение соединений ненуклеотидной природы - производных 1,2,5-оксадиазолов, структурная формула которых представлена ниже:
где R1 и R2 выбирают из фенилсульфонила, замещенного одним или более атомами галогена, нитрогруппами, карбоксигруппами, алкилгалогенидами, СН3, ОСН3, OCF3;
Х выбирают из N или N→O;
либо R1 и R2 образуют группу
где R', R", R'" и R'''' независимо выбирают из водорода; галогенов; нитрогруппы, гидроксигруппы, карбоксигруппы, СН3; СН2Вr; ОСН3; фенилсульфонила; фенилтиогруппы; или следующих групп:
R' и R" также могут быть объединены в один из следующих общих циклов:
для ингибирования репликации вируса иммунодефицита человека (ВИЧ).
2. Фармацевтическая композиция, обладающая ингибирующей активностью в отношении репликации вируса ВИЧ-1, содержащая терапевтически эффективное количество производного ненуклеотидной природы по пункту 1 и фармацевтически приемлемый носитель.
3. Способ ингибирования ВИЧ-1 интегразы, включающий введение млекопитающему терапевтически эффективного количества фармацевтической композиции по пункту 2.
| WO 2003053439 A1, 03.07.2003; | |||
| Колосоуборка | 1923 |
|
SU2009A1 |
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| US 3279988 A, 18.10.1966 | |||
| US 3594388 A, 20.07.1971 | |||
| ПРИМЕНЕНИЕ ПРОИЗВОДНЫХ БЕНЗОФУРОКСАНА ПРИ ЛЕЧЕНИИ СТЕНОКАРДИИ | 1999 |
|
RU2209065C2 |
| Реагент для проявления пятен в тонкослойной хроматографии ароматических аминов | 1989 |
|
SU1642372A1 |

