Изобретение относится к новым гетероциклическим соединениям, обладающим биологической активностью, более конкретно, к производным бензотиофена, бензофурана, индолтиазепинона, оксазепинона и диазепинона, фармацевтической композиции, обладающей ингибирующей клеточную адгезию или ВИЧ активностью, способу торможения адгезии лейкоцитов к эндотелиальным клеткам при лечении вызванных ею болезней и способу лечения млекопитающих, зараженных ВИЧ.
Адгезия лейкоцитов к сосудистому эндотелию является неотъемлемой частью патогенеза воспаления. Процесс адгезии предшествует трансэндотелиальной миграции лейкоцитов в окружающую ткань и последующему повреждению ткани. От соединений, способных к торможению этого начального адгезионного взаимодействия, ожидают эффективность при лечении воспалительных заболеваний, таких, как, например, ревматоидный артрит, остеоартрит, астма, псориаз. Другие показания включают, например, респираторный дистресс-синдром у взрослых, травму в связи с повторной перфузией, ишемию, язвенный колит, васкулит, атеросклероз, воспалительную кишечную болезнь и метастазы опухолей.
Рецепторы адгезии можно подразделить на три главные семьи: селектины, суперсемейство иммуноглобулинов и интегрины (см. Nature, 346:426 (1990)). Члены всех трех классов содействуют адгезии лейкоцитов во время воспаления (отн. обзоров см. Thrombosis and Hemostasis, 65(3); 223 (1991); Clinical and Experimental Allergy, 20:619 (1990); Transplantation, 48:727 (1989); Biochemical Pharm., 40(8):1683 (1990)). Молекула-1 адгезии лейкоцитов к эндотелию (в дальнейшем E-селектин) является членом семьи служащих в качестве селектина гликопротеинов, способствующих межклеточной адгезии. В литературе есть сведения о максимальной экспрессии E-селектина на поверхности эндотелиальных клеток через 4 ч после стимуляции эндотелиальных клеток цитокинами, такими, как, например, интерлейкин-1, фактор α некроза опухолей или другие медиаторы воспаления как, например, липополисахариды (см. Pro. Nat. Acad. Sci., 84: 9238 (1987)).
Молекула-1 межклеточной адгезии (в дальнейшем ICAM-1) является членом суперсемейства иммуноглобулинов. Максимальная экспрессия наблюдается через 12-24 ч после стимуляции. Есть сведения о том, что через 4 ч после стимуляции эндотелиальных клеток медиатором воспаления и E-селектин, и ICAM-1 находятся на клеточной поверхности (см. J. Clin. Invest., 52:1746 (1988); J. Immun., 137:1893 (1986); Blood, 78:2721 (1991)).
Бензотиофен, бензофуран и индолтиазепиноны, оксазепиноны и диазепиноны данного изобретения тормозят адгезию нейтрофилов к эндотелиальным клеткам пупочной вены человека, стимулированным фактором α некроза опухолей в рамках испытания in vitro.
Настоящее изобретение также относится к новым тиазепинонам, оксазепинонам и диазепинонам для лечения зараженных вирусом иммунодефицита человека (ВИЧ) пациентов путем торможения активации ВИЧ, являющегося латентным в зараженных лицах.
Патогенез вируса иммунодефицита человека (ВИЧ) является сложным и пока полностью не выявлен. Жизненный цикл вируса теоретически разделяют на афферентные и эфферентные компоненты. Связывание, слияние, обратная транскрипция и в заключение интеграция вируса являются событиями афферентного компонента жизненного цикла. Именно афферентные компоненты жизненного цикла ВИЧ отвечают за первичную инфекцию ВИЧ индивидуума, после чего обычно наблюдается вспышка виремии с клиническими симптомами или же без них.
Было разработано множество терапевтических стратегий с целью вмешательства в афферентные события (см., например, Н. Mitsuya, S. Broder, Inhibition of the In Vitro Infectivity and Cytopathic Effect on Human T-lymphotropic Virus Type III/lymphadenopathy Virus-associated Virus (HTLV-III/LAV) by 2',3'-Dideoxynucleosides, Proc. Natl. Sci. (USA), 83:1911-1915 (1986)).
В то время как разные стадии афферентного компонента позволяют эффективное терапевтическое вмешательство, становилось все более очевидным, что вмешательствa лишь в этот период недостаточно. После заражения ВИЧ и прогрессирования болезни по афферентным стадиям индивидуум переживает продолжительный период клинической латентности, который может длиться несколько лет, и индивидуум является здоровым. В этот момент достигается низкая или нулевая степень виремии и вирусной репликации в периферических клетках крови. Позже, однако, болезнь в конечном счете прогрессирует до угрожающей жизни иммуносупрессии (СПИД), способ лечения которой не известен. Эти более поздние события представляют собой клинические проявления эфферентных стадий заражения ВИЧ.
Эфферентный компонент жизненного цикла ВИЧ включает события, необходимые провирусу ВИЧ для успешной транскрипции, трансляции, сборки и продукции вирионов. Начало событий, необходимых для прогрессирования ВИЧ-зараженных клеток от бессимптомной, не проявляющей ВИЧ стадии к симптоматической, проявляющей ВИЧ стадии, называют активацией. В настоящее время эфферентный компонент и клеточная основа активации полностью не выявлены. Несмотря на это, разработка новых терапевтических средств и стратегий и их применение во время клинически бессимптомного периода в целях борьбы с прогрессированием к СПИДу может вселять определенную надежду предполагаемому числу (один миллион) зараженных, но клинически латентных индивидуумов.
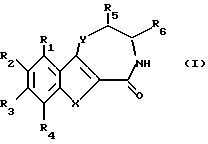
С учетом вышеизложенного первым объектом изобретения являются производные бензотиофена, бензофурана, индолтиазепинона, оксазепинона и диазепинона формулы I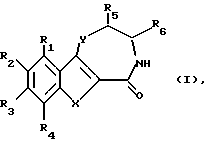
где
R1, R2, R3 и R4 независимо друг от друга означают водород, гидроксил, низший алкил, низший алкоксил,
R5 и R6 независимо друг от друга означают водород или низший алкил,
X - группа S(O)n или NH,
Y - кислород, группа S(O)n или NH,
n - 0, 1 или 2,
при условии, что
1) если X означает группу NH, Y означает группу NH, R1 означает водород и R3 - водород, то R2 не означает метил,
2) если X означает группу NH, Y означает группу NH, R1, R3 и R4 означают водород, то R2 не означает метоксил или этоксил, и
3) если X означает группу NH, Y означает серу, то по меньшей мере один из радикалов R1, R3 и R4 не означает водород, или их фармацевтически приемлемые кислотно-аддитивные соли.
Вторым объектом изобретения является фармацевтическая композиция, обладающая ингибирующей клеточную адгезию или ВИЧ активностью, содержащая активное начало и фармацевтически приемлемый носитель, которая содержит терапевтически эффективное количество соединения вышеприведенной формулы I и фармацевтически приемлемый носитель.
Третьим объектом изобретения является способ торможения адгезии лейкоцитов к эндотелиальным клеткам при лечении вызванных ею болезней, который заключается в том, что включает введение терапевтически эффективного количества предлагаемой фармацевтической композиции в виде дозировочной единицы. Предпочтительный вариант данного способа заключается в том, что лечению подвергается воспалительная болезнь.
Четвертым объектом изобретения является способ лечения млекопитающих, т. е. человека или животного, зараженных ВИЧ, который заключается в том, что включает введение терапевтически эффективного количества предлагаемой фармацевтической композиции в виде дозировочной единицы.
Соединения вышеприведенной формулы I относятся к категории малотоксичных веществ.
Фармацевтически приемлемые кислотно-аддитивные соли соединений формулы I включают соли неорганических кислот, таких, как, например, хлористоводородная кислота, азотная кислота, фосфорная кислота, серная кислота, бромистоводородная кислота, йодистоводородная кислота, фтористоводородная кислота, фосфористая кислота, а также соли нетоксических органических кислот, таких, как, например, алифатические моно- и дикарбоновые кислоты, фенил-замещенные алканкарбоновые кислоты, оксиалканкарбоновые кислоты, алкандикарбоновые кислоты, ароматические кислоты, алифатические и ароматические сульфокислоты. В качестве примеров таких солей можно назвать сульфат, пиросульфат, бисульфат, сульфит, бисульфит, нитрат, фосфат, вторичный фосфат, первичный фосфат, метафосфат, пирофосфат, хлорид, бромид, йодид, ацетат, трифторацетат, пропионат, каприлат, изобутират, оксалат, малонат, сукцинат, суберат, себакат, фумарат, малеат, манделат, бензоат, хлорбензоат, метилбензоат, динитробензоат, фталат, бензолсульфонат, толуолсульфонат, фенилацетат, цитрат, лактат, малеат, тартрат, метансульфонат. Кроме того, можно применять соли аминокислот, например аргинат и тому подобноe, а также глюконат, галактуронат, N-метилглутамин (см., например, С.М. Берге и др., Pharmaceutical Salts, Journal of Pharmaceutical Science, 66:1-19 (1977)).
Кислотно-аддитивные соли приведенных основных соединений получают путем контактирования свободного основания с достаточным количеством желаемой кислоты с получением соли стандартным образом. Свободное основание можно высвобождать путем контактирования соли с основанием и выделением свободного основания стандартным образом. Свободные основания немного отличаются от их соответствующих солей по некоторым физическим свойствам, например по растворимости в полярных растворителях, но в остальном биологические свойства солей являются эквивалентными соответствующему свободному основанию.
Некоторые из соединений данного изобретения могут иметься и в несольватированных формах, и в сольватированных формах, включая гидрированные формы. В общем сольватированные формы, включая гидрированные формы, являются эквивалентными несольватированным формам, и все указанные формы охватываются настоящим изобретением.
Предпочтительными соединениями формулы I являются те, у которых R1, R3 и R4 означают водород.
Кроме того, предпочтительными соединениями формулы I являются еще те, у которых R1, R3 и R4 означают водород, R2 означает водород или низший алкоксил, X означает группу S(O)n или NH, Y означает кислород или группу S(O)n, а n означает 0, 1 или 2.
В частности, предпочитаются соединения из группы, включающей 2,3-дигидро-9-метокси-[1] -бензотиено[2,3-f] -1,4-тиазепин-5(4H)-он, 2,3-дигидро-[1] бензотиено[2,3-f] -1,4-оксазепин-5(4H)-он, 2,3-дигидро-9-метокси-[1]бензотиено[2,3-f] -1,4-тиазепин-5(4H)-он-1-оксид, 3,4-дигидро-9-метокси-6-метил-2H-1,4-оксазепино[6,7-b] -индол-5(6H)-он, 2,3-дигидро-1H-бензотиено-[3,2-e] -1,4-диазепин-5-он, 2,3-дигидро-9-метокси-1H-бензотиено-[2,3-f] -1,4-оксазепин-5-он, 2,3-дигидро-9-метокси-6-оксид-1H-бензотиено-[2,3-f]оксазепин-5-он, 2,3-дигидро-9-метокси-2-метил-1H-бензотиено-[2,3-f]-1,4-оказепин-5-он.
При определении показания к ингибитору клеточной адгезии или ингибитору активации ВИЧ лечащий врач будет учитывать, конечно, среди прочего соответствующее состояние пациента, серьезность состояния, а также возраст, пол, вес и т.п.
Для достижения терапевтического эффекта необходимое количество соединения формулы I или его фармакологически приемлемой кислотно-аддитивной соли, конечно, варьирует в зависимости от соответствующего соединения, пути введения средства, проходящего курс лечения пациента, определенного расстройства или соответствующей болезни. Предпочтительный вариант изобретения охватывает метод лечения лиц, страдающих от воспалительной болезни, такой, как, например артрит или припухлость, включающий дачу соединения в эффективном противовоспалительном количестве. Подходящей дозой соединения формулы I или его фармацевтически приемлемой кислотно-аддитивной соли для пациента, страдающего или предрасположенного к страданию от любого из вышеописанных состояний, является 0,1 мкг - 500 мг соединения на 1 кг веса тела. В случае системной дачи доза может составлять 0,5 - 500 мг соединения на 1 кг веса тела, предпочтительно 0,5 - 50 мг/кг веса тела пациента два-три раза в день. В случае местного применения, например на коже или на глазах, подходящая доза может составлять 0,1 нг - 100 мкг соединения на 1 кг, обычно приблизительно 0,1 мкг/кг.
В случае оральной дачи соединения для лечения или профилактики артрита или воспаления, вызванных любой причиной, подходящая доза соединения формулы I или его физиологически приемлемой кислотно-аддитивной соли может быть той же самой, что и в предыдущем абзаце, но предпочтительно применяют 1 - 10 мг соединения на 1 кг, а более предпочтительно 1 - 5 мг/кг веса тела пациента, например 1 - 2 мг/кг.
Само собой разумеется, что специалист в данной области (врач или ветеринар) будет определять и прописывать эффективное количество соединения для предотвращения или прекращения прогрессирования соответствующего состояния. При этом врач или ветеринар может поступать так, что в начале курса лечения дает относительно низкие дозы, после чего повышает дозу до достижения максимальной реакции.
Активное начало можно, правда, давать отдельно, но предпочтительно дают его в виде фармацевтической композиции, включающей соединение формулы I или его фармацевтически приемлемую кислотно-аддитивную соль и фармацевтически приемлемый носитель. Такие композиции являются дополнительным объектом настоящего изобретения.
Для применения в области ветеринарии, а также в области медицины композиции данного изобретения содержат активное начало вместе с фармацевтически приемлемым носителем и факультативно другой (другие) терапевтический(ие) ингредиент(ы). Носитель(и) должен(ны) быть "приемлемым(и)" в том смысле, что он(и) должен(ны) быть совместимым(и) с другими ингредиентами композиций и невредным(и) для пациента.
Композиции включают препараты, пригодные для орального, легочного, глазного, ректального, парентерального (включая подкожноe, внутримышечноe и внутривенноe), внутрисуставного, местного, носового или трансбуккального введения. Такие композиции включают известные препараты длительного или продленного действия.
Композиции можно выпускать в виде препарата, включающего дозировочные единицы. Их можно получать любым известным в области фармацевтики способом. Эти способы могут охватывать стадию контактирования активного начала с носителем, представляющим собой по меньшей мере одно вспомогательное вещество. Обычно композиции получают путем равномерного и гомогенного контактирования активного начала с жидким носителем и/или тонкоизмельченным твердым носителем и, в случае необходимости, переведения продукта в желаемый препарат.
Пригодные для оральной аппликации препараты согласно изобретению могут иметься в виде дискретных единиц, таких, как, например, капсулы, крахмальные капсулы, таблетки, лепешки, содержащие предопределенное количество активного начала; в виде порошка или гранул; в виде раствора или суспензии в водной или неводной жидкости; или же в виде эмульсии типа масла в воде или эмульсии типа воды в масле. Активное начало может также иметься в виде болюса, электуария или пасты.
Пригодность соединений данного изобретения в качестве ингибиторов адгезии лейкоцитов к васкулярному эндотелию и, следовательно, пригодность для лечения связанных с воспалением болезней или состояний можно определять на основе их эффективности в рамках различных стандартных испытаний.
Hиже приведены описание хода анализов и результаты испытаний.
Испытание экспрессии молекулы-1 межклеточной адгезии и E-селектина в эндотелиальных клетках пупочной вены человека.
Клеточная культура.
Эндотелиальные клетки пупочной вены человека (поставляемые фирмой "Клонетикс") в склянках T-25 культивируют в течение 1 - 3 дней при температуре 37oC в атмосфере, содержащей 5% двуокиси углерода. Потом клетки расщепляют путем обработки 10 мл раствора, содержащего 0,025% трипсина и 0,01% этилендиаминотетрауксусной кислоты (далее ЭДТУК), в течение 5 - 10 с. После декантирования промывочного раствора добавляют еще 10 мл раствора трипсина и ЭДТУК и клетки перемешивают в течение 2 - 4 мин, слегка ударяя резинкой по стенке склянки. Затем содержимое склянки подают в пробирку для центрифугирования емкостью 50 мл, содержащую 40 мл среды. Среда представляет собой эндотелиальную базальную среду (поставляемую фирмой "Клонетикс"), содержащую 2 мг/л гидрокортизона, 0,05 мкг/л эпидермального фактора роста, 12 мг/л экстракта бычьего мозга и 6%-ную термически инактивированную фетальную телячью сыворотку (поставляемую фирмой "Хайклон"). Клетки центрифугируют при 15oC в течение 10 - 15 мин, надосадочную жидкость удаляют и клетки ресуспендируют в свежей среде. Клетки вышеописанным образом еще раз обрабатывают, после чего их высеивают в пластинку с 96 чашками.
Стимулирование цитокином.
Через 5 дней после достижения слияния клетки стимулируют фактором α некроза опухолей (поставляемым фирмой "Гензайм") для получения окончательной концентрации среды, равной 140 ед/мл, и инкубируют при 37oC в течение 4 ч. Потом среду удаляют из инкубатора и хранят для анализа производства хемокина. Клетки три раза промывают фосфатсодержащим буфером солевого раствора, не содержащего кальция или магния. Затем монокультуры фиксируют путем добавления к чашкам буферсодержащего 10%-ного формалина в течение 15 мин. Потом клетки три раза промывают модифицированной по способу Дульбекко средой Игла (далее среда ДМЕМ) (поставляемой фирмой "Гибко"), содержащей 2% альбумина бычьей сыворотки, и хранят в холодильнике в течение ночи.
Твердофазный иммуноферментный анализ.
Мышиные моноклональные античеловеческие молекулы-1 межклеточной адгезии (поставляемые фирмой R & D Systems, кат. N BBA-4) или мышиный моноклональный античеловеческий E-селектин (фирмы R & D Systems, кат. N BBA-2) растворяют в среде ДМЕМ, содержащей 2% альбумина бычьей сыворотки. Указанные молекулы-1 или E-селектин в концентрации 0,5 мкг/мл добавляют к каждой чашке и инкубируют при 37oC в течение 2 ч. Монокультуры 4 раза промывают средой ДМЕМ, содержащей 2% альбумина бычьей сыворотки. Добавляют разведенный в соотношении 1:3000 конъюгированный пероксидазой, выделившийся из овцы противомышиный иммуноглобулин Г (поставляемый фирмой "Каппел") и инкубируют при 37oC в течение часа. Клетки четыре раза промывают средой ДМЕМ. Затем к зафиксированным клеткам добавляют окрашивающий реактив и инкубируют 15 мин при комнатной температуре. Реакцию прекращают добавлением 2%-ного раствора щавелевой кислоты. Абсорбция составляет 414 нм на аппарате для чтения титрационных пластинок.
Испытание соединений.
Соединения растворяют в диметилсульфоксиде при концентрации 30 ммоль и разбавляют средой для достижения конечной концентрации. К клеткам добавляют растворенное в среде соединение за 30 мин до стимуляции фактором α некроза опухолей. Абсорбцию нестимулированных клеток вычитают из значений абсорбции стимулированных фактором α некроза опухолей клеток перед определением процента торможения. Процент торможения определяют путем сравнения абсорбции обработанных только носителем соединений клеток (контрольный опыт) с клетками, обработанными исследуемым соединением. Значения KT50 определяют с помощью линейно-регрессионного анализа.
Метод определения торможения адгезии нейтрофилов человека к стимулированным фактором α некроза опухолей эндотелиальным клеткам пупочной вены человека.
Клеточная культура.
Эндотелиальные клетки пупочной вены человека второго пассирования (поставляемые фирмой "Клонетикс Корпорейшн", Сан-Диeго, Калифорния, СС-2617) высеивают в пластинки с 96 чашками (поставляемые фирмой "Корнинг гласс веркс", Корнинг, Нью-Йорк) плотностью приблизительно 5•103 клеток на чашку и выращивают до слияния в полной эндотелиальной базальной среде (EBM, MCDB, поставляемой фирмой Клонетикс), содержащей 10 нг/мл эпидермального фактора роста, 1 мкг/мл гидрокортизона, 0,4% экстракта бычьего мозга и 6%-ную фетальную телячью сыворотку. За один день до проведения испытания, обычно через 3 дня после высеивания клеток, к культурам добавляют еще 0,2 мл полной эндотелиальной базальной среды на чашку.
Изготовление испытуемых соединений.
Испытуемые соединения переводят в маточный раствор объемом 10 мл, концентрация которого составляет 1,0 ммоль. Сначала соединения солюбилизируют в 0,1 мл диметилсульфона, после чего добавляют 9,9 мл полной эндотелиальной базальной среды. Затем испытуемые соединения в одну стадию разбавляют до достижения концентрации 66,6 мкм. Операции солюбилизации и разбавления осуществляют в полистирольных сосудах.
Стимулирование эндотелиальных клеток пупочной вены человека.
Рекомбинантный человеческий фактор α некроза опухолей (поставляемый фирмой "Гензайм", Бостон, Массачусетс, код TNF-H) получают в концентрации, равной 400 ед/мл в полной эндотелиальной базальной среде. Маточный раствор фактора α некроза опухолей состоит из 20000 ед/мл фосфатсодержащего буфера солевого раствора (PBS, поставляемого фирмой "Гибко", Грэнд Айлэнд, Нью-Йорк) и 0,1% альбумина бычьей сыворотки. Его хранят при температуре -70oC. Эндотелиальные клетки пупочной вены человека один раз промывают 0,2 мл теплой неполной эндотелиальной базальной среды, после чего их стимулируют фактором α некроза опухолей, взятым в концентрации 200 ед/мл, в присутствии 33,3 мкм испытуемого соединения при температуре 37oC в течение 4 ч. Эту операцию осуществляют путем добавления 0,1 мл фактора α некроза опухолей, взятым в концентрации 400 ед/мл, и 0,1 мл (66,6 мкм) испытуемого соединения. Вещества добавляют медленно для предотвращения разрыва монослоя эндотелиальных клеток. Каждое соединение испытывают в шести чашках. Кроме того, в каждой пластинке проводят контрольный опыт с нестимулированными клетками и опыт с клетками, стимулированными фактором α некроза опухолей без обработки испытуемым соединением.
Мечение нейтрофилов.
За час до добавления нейтрофилов к эндотелиальным клеткам нейтрофилы в концентрации 5•106/мл подвергают мечению 5 мкм кальцеина-AM (поставляемого фирмой "Молекулар Пробе", Юджин, Орегон) в сбалансированном солевом растворе Хэнкса, содержащем 0,45% альбумина бычьей сыворотки, при температуре 37oC в течение 30 мин. Маточный кальцеин получают в концентрации 5 ммоль в безводном диметилсульфоксиде, высушивают и хранят при -20oC. После инкубации клетки два раза промывают холодным сбалансированным солевым раствором Хэнкса и ресуспендируют до конечной концентрации, равной 1•106 клеток/мл в полной эндотелиальной базальной среде.
Добавление нейтрофилов к эндотелиальным клеткам.
После четырехчасового стимулирования и непосредственно до добавления нейтрофилов к монослою эндотелиальных клеток пластинки обрабатывают 0,2 мл теплой неполной эндотелиальной базальной среды для удаления фактора α некроза опухолей и испытуемого соединения. К каждой обрабатываемой чашке медленно добавляют нейтрофилы (1•105 клеток) и инкубируют при 37oC в течение 30 мин. Потом пластинки два раза промывают 0,2 мл теплой неполной эндотелиальной базальной среды, после чего добавляют еще 0,1 мл среды для сканирования пластинки.
Определение относительной флюоресценции.
Относительную флюоресценцию определяют с помощью системы Millipore Cytofluor 300 (возбуждение = 480, эмиссия = 530, чувствительность = 4).
Расчеты.
Испытание считается действительным, если стимулирование клеток фактором α некроза опухолей приводит к 300%-ному повышению адгезии нейтрофилов по сравнению с нестимулированными клетками. Результаты приведены в процентах торможения стимулированной фактором α некроза опухолей адгезии.

Некоторые соединения согласно изобретению подвергали испытанию в концентрациях 33,3, 10,0, 3,3 и 1,0 мкмоль для определения средних значений KT50, которые определялись с помощью линейно-регрессионного анализа.
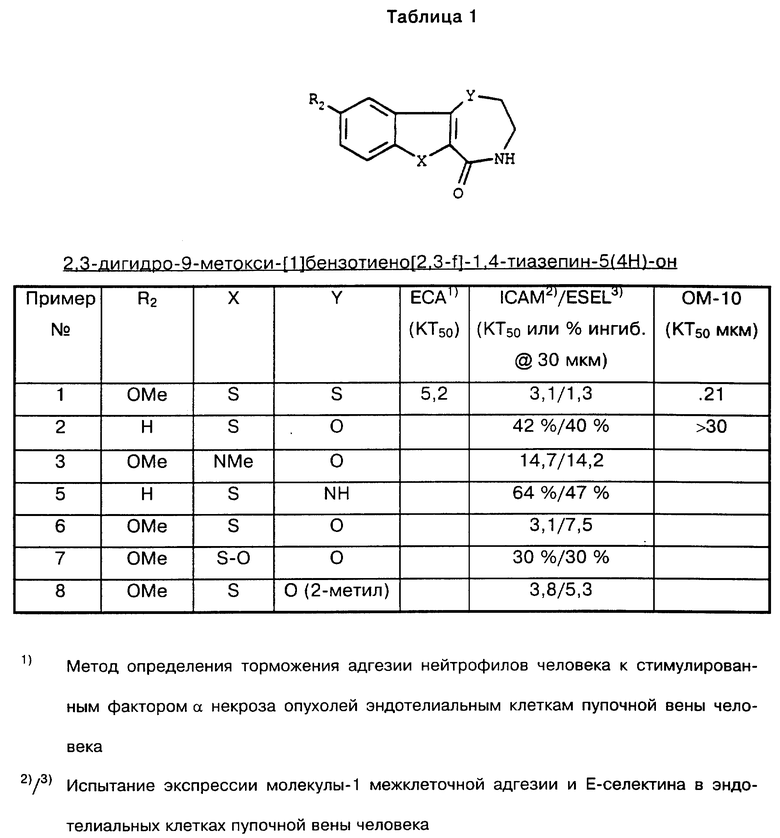
Результаты испытаний соединений согласно изобретению приведены в табл. 1.
Было найдено, что соединения согласно изобретению, в частности соединения формулы III, тормозят активацию вируса иммунодефицита человека (ВИЧ), являющегося латентным в зараженных людях и животных, и, следовательно, являются пригодными для лечения СПИДа.
Попытки понять вирусные и клеточные основы клинического бессимптомного периода показывают, что ВИЧ представляет собой покоящийся или неэкспримирующийся провирус в популяции хронически зараженных клеток. Специфический вид вируса ВИЧ, ВИЧ-1, подвергали ряду различных испытаний, в результате которых стало ясно, что вирус является покоящимся или неэкспримирующимся провирусом в популяции хронически зараженных T-лимфоцитов. Однако в рамках данных испытаний не излагаются подробности ядерных и биохимических механизмов, которые являются основой латентного вирусного состояния. Подробную информацию см. в статье Беднарика и др. Mechanisms of HIV-1 Latency в журнале Aids, N 6, с. 3-16, 1992 г.
Вплоть до недавнего времени предполагалось, что во время клинического бессимптомного периода ВИЧ является покоящимся или неэкспримирующимся во всех популяциях хронически зараженных клеток. Вследствие наблюдений низких или отсутствующих степеней вирусемии и репликации вируса в периферических гемоцитах предполагалось, что ВИЧ не является активным во время клинического бессимптомного периода. Однако было найдено, что настоящего скрытого состояния во время заражения вирусом ВИЧ не существует (см. статью Фоки и др. HIV Infection is Active and Progressive in Lymphoid Tissue During the Clinically Latent Stage of disease, в журнале Nature, N 362, с. 355-358, 1993 г.).
Во время клинического скрытого состояния наблюдается дихотомия между степенями вирусного груза и репликации вируса в периферических гемоцитах по отношению к лимфатическим органам. На основе данных наблюдений было установлено, что периферические гемоциты неточно отражают настоящую стадию заболевания вирусом ВИЧ, в частности в начале клинического хода ВИЧ-инфекции. На самом деле, заболевание вирусом ВИЧ является активным и развивается, даже в том случае, если активность, измеряемая в периферических гемоцитах вирусными показателями, низкая и пациент находится в клиническом скрытом состоянии.
Неизбежно стадия заболевания развивается начиная с клинически латентного бессимптомного периода до экспрессионного и активного симптомного периода. Согласно Бутера и др. (см. Aids, N 6, с. 994, 1992 г.) были разработаны различные клеточные модели, которые при обработки цитокинами могут экспримировать ВИЧ-1. Значит, на стадии микробиологического скрытого состояния ВИЧ-1 начинает репликацию только после получения внеклеточного сигнала. Этот сигнал может быть вызван не только взаимодействием растворимого цитокина с рецептором, но и межрецепторным взаимодействием, происходящим во время межклеточной коммуникации или воздействия на клетки ультрафиолетовым излучением или температурным шоком. Кроме того, внеклеточный сигнал может быть вызван аутокринным или паракринным способом, так что активированная вирусом ВИЧ-1 клетка может продолжать экспримироваться и одновременно активировать смежную латентную клетку.
Предполагается, что и дополнительные факторы участвуют в активации вируса ВИЧ. Было установлено, что применение 12-0-тетрадеканоилфорбол-13-ацетата приводит к снижению количества рецепторов CD4 и к вирусной экспрессии в зараженных вирусом ВИЧ клетках (см. Хамамото и др. в журнале Biochem. Biophys. Res. Commun., N 164, с. 339-344, 1989 г.). Интересно, что Хамамото исследовал также эффект высокоактивных ингибиторов протеинкиназы C, таких, как, например, стауроспорин, H-7, UCN-01, на вызываемое 12-0-тетрадеканоилфорбо-13-ацетатом снижение количества рецепторов CD4 и повышение экспрессии ВИЧ. Найдено, что стауроспорин представляет собой эффективный ингибитор как снижения количества рецептора CD4, так и экспрессии вируса ВИЧ.
Клеточные пути, передающие сигнал активации от плазматической мембраны к вирусу, что приводит к экспрессии ВИЧ-1, менее известны. Недавно, на симпозиуме National Cooperative Discovery Grant (NCDDG)/AIDS, состоявшемся 3 - 7 ноября 1991 г., П. Феорино, С.Т. Бутера, Т.М. Фолкс и Р.Ф. Шинаци сообщили о разработке надежной и простой системы определения соединений, способных предотвращать активацию латентного вируса ВИЧ. Испытательная система включает клеточную линию ОМ-10.1, т.е. уникальный хронически зараженный промиелоциточный клон, который сохраняет уровень рецептора CD4+ до активации ВИЧ-1 фактором α некроза опухолей. Экспрессия рецептора CD4+ на поверхности клеток, а также активность обратной транскриптазы используют для определения экспрессии вируса.
Альтернативно для определения экспрессии вируса ВИЧ можно применять и активность протеазы. Клетки ОМ-10.1 сохраняют уровень рецептора CD4+ до активации вируса и реагируют на индукцию фактора α некроза опухолей. Поэтому эти культуры используют для удобного и быстрого исследования фармакологических свойств соединений в отношении их способности предотвращать снижение количества рецептора CD4+ на поверхности клеток и экспрессию вируса ВИЧ.
Исследовали ряд соединений, проявляющих антивирусную активность в отношении остро или хронически зараженных клеток, проверяя их способность тормозить экспрессию вируса ВИЧ в клетках ОМ-10.1. Кроме того, исследовали ряд соединений, которые взаимодействуют с биохимическими путями, которые могут отрицательно влиять на реактивацию. Результаты исследования были показаны на плакате, представленном на симпозиуме NCDDG/AIDS, состоявшемся 3 - 7 ноября 1991 г. в Сан-Диего в шт. Калифорния, США. Из числа приблизительно 48 исследованных соединений низкодействующими ингибиторами активации ВИЧ-1 считают 3'-фтор-3'-деокситимидин, интерферон Y и десферриоксамин.
Охватываемое формулой I соединение 2,3-дигидро-9-метокси-[1]бензотиено[2,3-f] -1,4-тиазепин-5(4H)-он в концентрации 0,21 мкм проявляет 50%-ное торможение в клетках ОМ-10.1 (см. табл. 1).
Предлагаемые соединения проявляют активность и в обычных опытах in vivo, в которых измеряют способность веществ предотвращать втекание нейтрофилов, и соответственно их полезность для лечения воспалений. В одном опыте крыс-самцов типа Вистар весом 220 - 245 г не кормят в течение 16 - 18 ч. Им внутривенно дают буфер (смесь этанола и солевого раствора в соотношении, равном 1:1) или предлагаемое соединение в буфере. Крыс слабо анестезируют диэтиловым эфиром, им дают внутривенную инъекцию раствора 2,5 мг альбумина бычьей сыворотки в солевом растворе. Непосредственно после внутривенной инъекции делают маленький разрез между ребрами и с помощью подходящей иглы в плевральную полость инъецируют раствор 0,2 мл выделенного из кроличьего иммуноглобулина G антиальбумина бычьей сыворотки в фосфатсодержащем солевом растворе (концентрация: 10 мг/мл солевого раствора).
Затем разрез закрывают зажимом из нержавеющей стали размером 9 мм. Через 4 ч крыс умерщвляют с помощью двуокиси углерода и плевральную полость промывают 2 мл 0,325%-ного раствора красного фенола в фосфатсодержащем солевом растворе. Выходящий из плевральной полости поток буфера подвергают анализу. Лейкоциты (> 90% нейтрофилов) подсчитают счетчиком фирмы "Каультер". Объем выходящего потока буфера измеряют способом разведения с применением красителя (см. статью Г.В. Картера и др. в журнале J. Pharm. Pharmacol., N 34, с. 66-67, 1982 г.). Животных, которым дали предлагаемое соединение, сравнивают с животными, которым дали только буфер. При этом статистическую значимость активности испытуемых соединений определяют с помощью t-опыта по Стьюденту.
Результаты данного анализа, которому подвергают соединение примера 1, приведены в табл. 2.
В другом опыте in vivo мышей-самок типа Balb/c помещают в клетках группами по семь. Во время проведения опыта мыши имеют свободный доступ к пище и воде. Им орально дают носитель (0,5%-ный раствор гидроксипропилметилцеллюлозы, содержащий 0,2% Твина 80) или предлагаемое соединение, растворенное или суспендированное в указанном носителе. Через час после оральной подачи мышей анестезируют путем ингаляции диэтилового эфира, и им внутрибрюшинно инъецируют 1,0 мл 3%-ного тиогликоллата в солевом растворе. Через 2 ч после инъекции тиогликоллата мышей умерщвляют с помощью двуокиси углерода, и им инъецируют 6 мл фосфатсодержащего солевого раствора, содержащего 10 ед/мл гепарина натрия и 0,1% альбумина бычьей сыворотки. Брюшинную полость массируют и разрезают, и вытекающую жидкость собирают в центрифужных пробирках емкостью 15 мл. От каждой мыши берут аликвотную пробу и с помощью счетчика фирмы "Каультер" (модель ZBi, поставляемая фирмой "Каультер Инструменте", г. Hialeah, шт. Флорида, США) подсчитают общее количество клеток в каждой аликвотной пробе. Берут другую аликвотную пробу с целью ее исследования под микроскопом с последующим окрашиванием модифицированным красителем фирмы "Райт". С помощью гематологического анализа определяют процентное количество нейтрофилов, вытекших в брюшинную полость.
В данном опыте соединение примера 1 проявляет следующее торможение втекания нейтрофилов: 26,1% при концентрации 10 мг/кг, 31,9% при концентрации 30 мг/кг и соответственно 34,3% при концентрации 100 мг/кг.
Предлагаемые соединения могут получаться следующими способами.
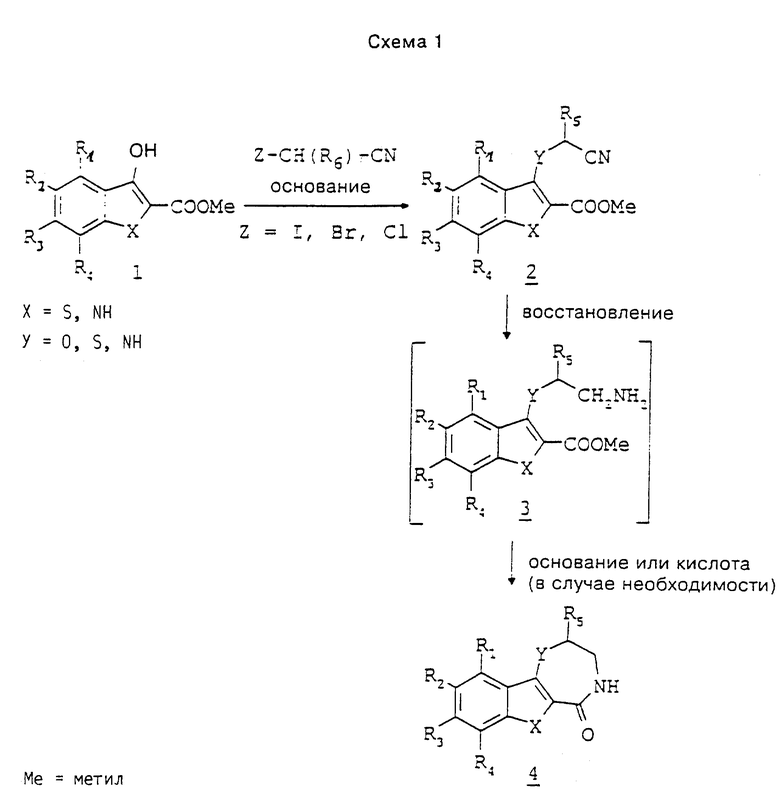
Согласно первому способу в качестве исходных соединений применяют 3-гидрокси-, 3-тиол-, 3-аминобензо[b] тиофен-, бензофуран- или индол-2-карбоксилат 1 (схема 1). Сложные эфиры 3-гидрокси-бензо[b]тиофена-2 получают по способу, описанному Коннором и др. в журнале J. Med. Chem., N 35, с. 958, 1992 г. 3-тиобензо[b]тиофен-2-карбоксилат получают взаимодействием соответствующего 3-хлор-производного (см. Коннор и др. в журнале J. Med. Chem., N 35, с. 958, 1992 г.) с тиоацетамидом в присутствии основания, такого, как 1,8-диазабицикло[5.4.0] -ундек-7-ен, в среде растворителя, такого, как N, N'-диметилформамид или тетрагидрофуран. 3-аминобензо[b]тиофен-2-карбоксилат получают по общеизвестному методу, описанному Бекком в журнале J. Org. Chem. , N 37, с. 3224, 1972 г. 3-гидроксииндол-2-карбоксилат получают известными способами, описанными, например, Унангстом и др. в журнале J. Heterocyclic Chem., N 24, с. 811, 1987 г., и Мойером и др. в журнале J. Org. Chem., N 51, с. 5106, 1986 г. 3-тиоиндол-2-карбоксилат получают по известным методам, описанным, например, Унангстом и др. в журнале J. Heterocyclic Chem., N 24, с. 811, 1987 г.; Аткинсоном и др. в журнале Synthesis, стр. 480, 1988 г.; Нагараяном и др. в журнале Indian J. Chem., N 20B, с. 672, 1981 г. 3-аминоиндол-2-карбоксилат получают известными способами, описанными С.В. Симаковым и др. в Химико-фармацевтическом журнале, N 17, с. 1183, 1983 г.
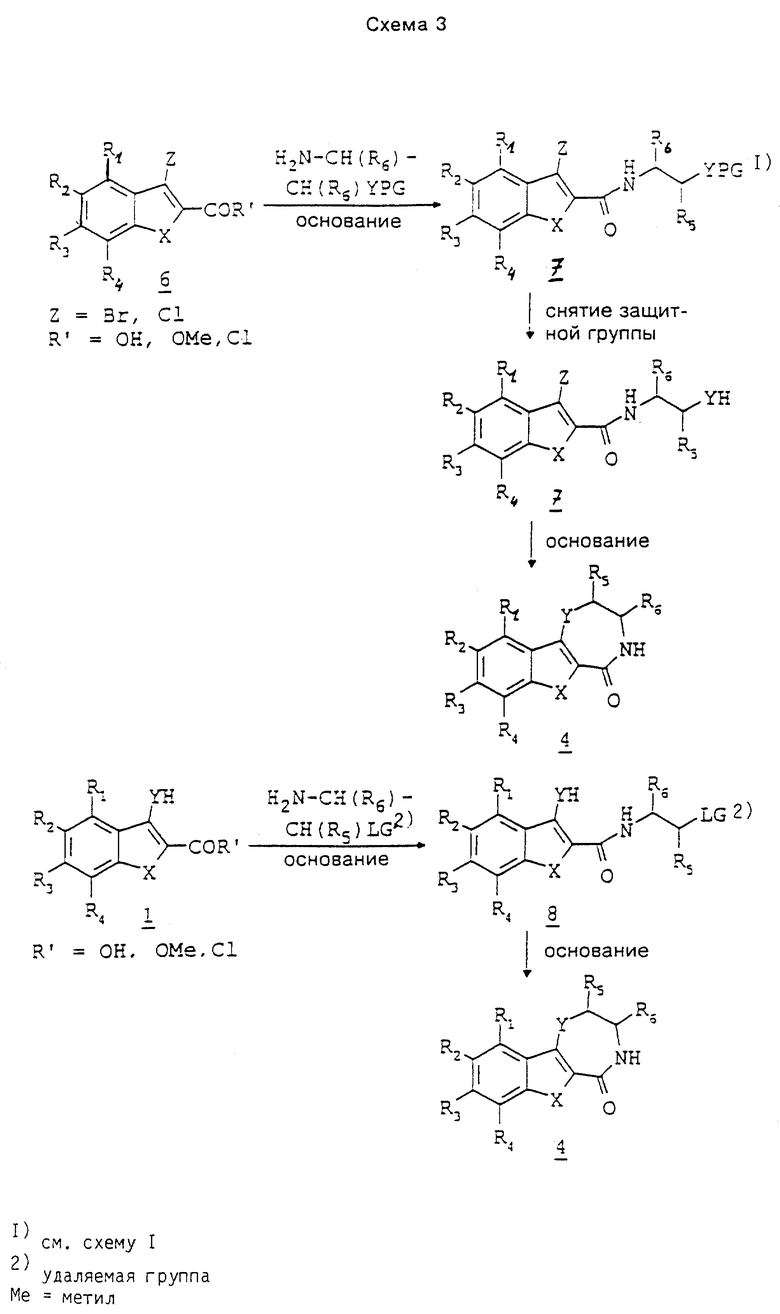
Схема 1 поясняет превращение соединений 1 до предлагаемых соединений. Сложные эфиры подвергают взаимодействию с производным замещенного α-галогеном ацетонитрила, таким как бромацетонитрил, в присутствии основания, такого как трет-бутилат калия, в тетрагидрофуране, ацетонитриле или диметилсульфоксиде при температурах 0 - 80oC. Получают сложные эфиры 2. Нитрил восстанавливают до соответственного первичного амина и получаемый при этом промежуточный продукт 3 циклизуют с получением лактама 4. Предпочтительной реакцией является гидрирование сложных эфиров 2 на кобальтовом катализаторе типа Ренея в среде растворителя, такого как тетрагидрофуран, в присутствии основания, такого как триэтиламин, при повышенных температурах и под давлением. В таких условиях сложные эфиры 2 превращаются непосредственно до лактама 4. В случае предварительного выделения промежуточный продукт 3 циклизуют до лактама 4 в основных условиях, предпочтительно в присутствии метилата натрия в метаноле, или в кислотных условиях, предпочтительно в присутствии полифосфорной кислоты, при повышенных температурах.
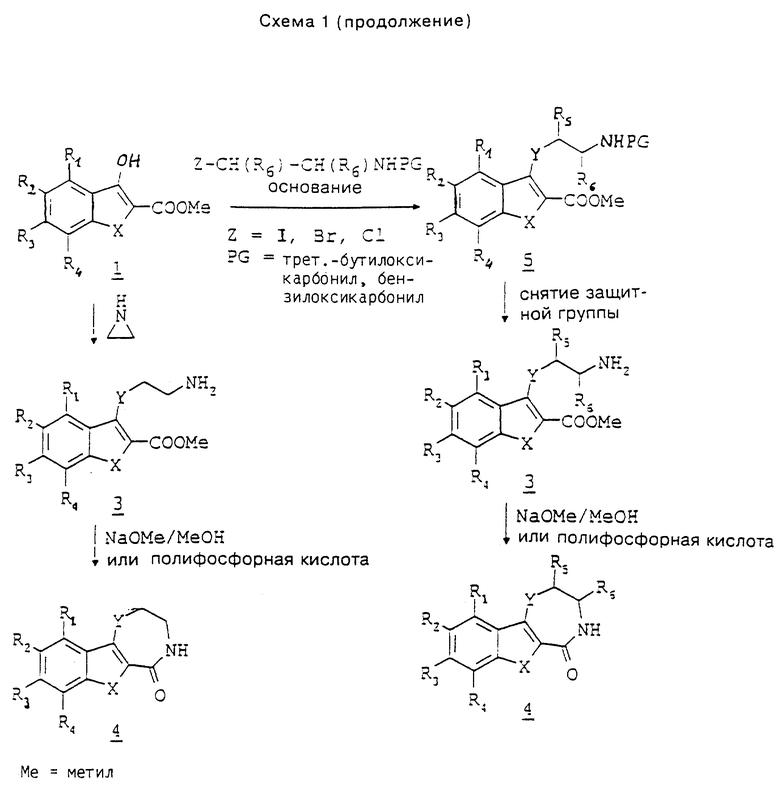
При синтезе некоторых предлагаемых соединений необходимо или желательно переводить реакционноспособные группы, как гидроксил, амино или карбоксил, в производные, защищающие их от нежелательных побочных реакций. Защитные группы снимают с гидроксила, амино-группы или карбоксила стандартными методами. Общепринятые химические группы, способные защищать реакционноспособные группы, как гидроксил, амино и карбоксил, а также методы их введения в молекулу и последующего снятия описаны Грином и Вутсом в источнике Protective Groups in Organic Synthesis, изд-во Джон Уайлей и Санс, Инк., Нью-Йорк, 1991 г.
Так, например, 3-амино-, 3-гидрокси- или 3-тиоиндол, бензотиофен или бензофуран (соединение 1 в схеме 1) можно подвергать взаимодействию β-галогенэтиленамином, причем амино-группа защищена пригодным остатком, таким как трет-бутоксикарбонил или бензилоксикарбонил. При осуществлении реакции в вышеуказанных условиях получают соединения 5. Снятием защитных групп в соединении 5 общеизвестными способами, такими как применение трифторуксусной кислоты или водной кислоты в случае трет-бутоксикарбонила, или гидрогенолиз в случае бензилоксикарбонила, получают соединения 3, которые циклизуют вышеуказанным способом. Другим способом получения соединений 3 является взаимодействие соединений 1 с этиленимином в среде спиртового растворителя (см. статью Нагараяна и др. в журнале Indian J. Chem., N 20B, с. 672, 1981 г.).
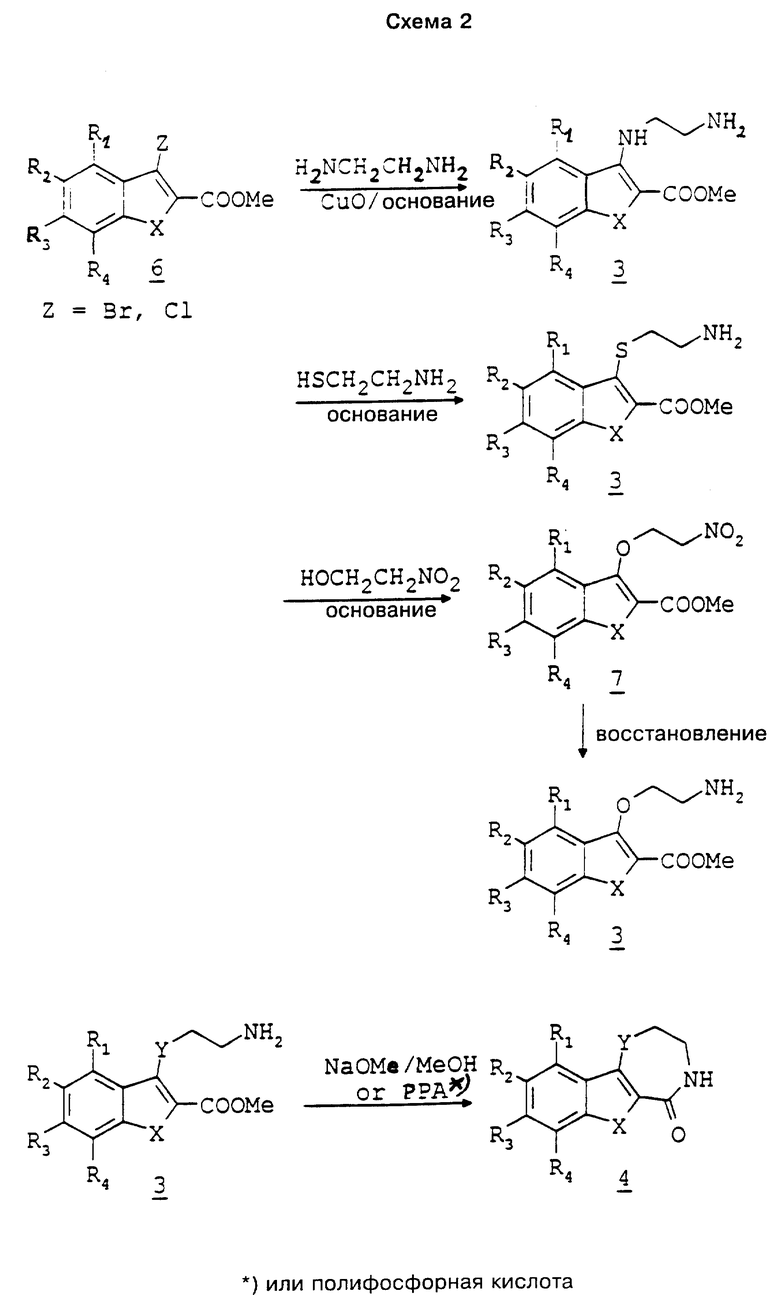
Вторым общим способом получения соединений 4 (см. схему 2) является взаимодействие соответственных 3-галогенпроизводных 6 с этилендиамином и окисью меди в среде растворителя, такого как пиридин, в присутствии основания, такого как карбонат калия, с получением соединений 3, где Y означает группу NH (см. С.М. Хайермат и др. в журнале Proc. Nat. Acad. Sci., India, N 60, с. 367, 1990 г.). Взаимодействием 3-галогенпроизводных с 2-аминоэтанолом в среде растворителя, такого как диметилформамид, в присутствии основания, такого как диазабициклоундекан, получают соединения 3, где Y означает серу. Взаимодействием 3-галогенпроизводных с нитроэтанолом в среде растворителя, такого как тетрагидрофуран, в присутствии основания, такого как трет-бутилат калия или гидрид калия, получают соединения 7. Последующим восстановлением нитро-группы до амина получают соединения 3, где Y - кислород. В некоторых случаях соединения 3 не выделяют, а непосредственно превращают до соединений 4.
В третьем общем способе получения соединения 4 тоже используют 3-галогенпроизводные 6 (см. схему 3), которые подвергают взаимодействию с первичным амином, содержащим в β-положении пригодную защищенную амино-группу, гидроксильную или тиол-группу. Получают промежуточный продукт 7. Снятием защитной группы с последующей циклизацией получают соединения 4. В аналогичном способе осуществляется взаимодействие 3-гидроксил-, 3-тиол- или 3-амино-производного с амином, имеющим в β-положении пригодную удаляемую группу. Получаемые при этом промежуточные продукты 8 циклизуют с получением соединения 4.
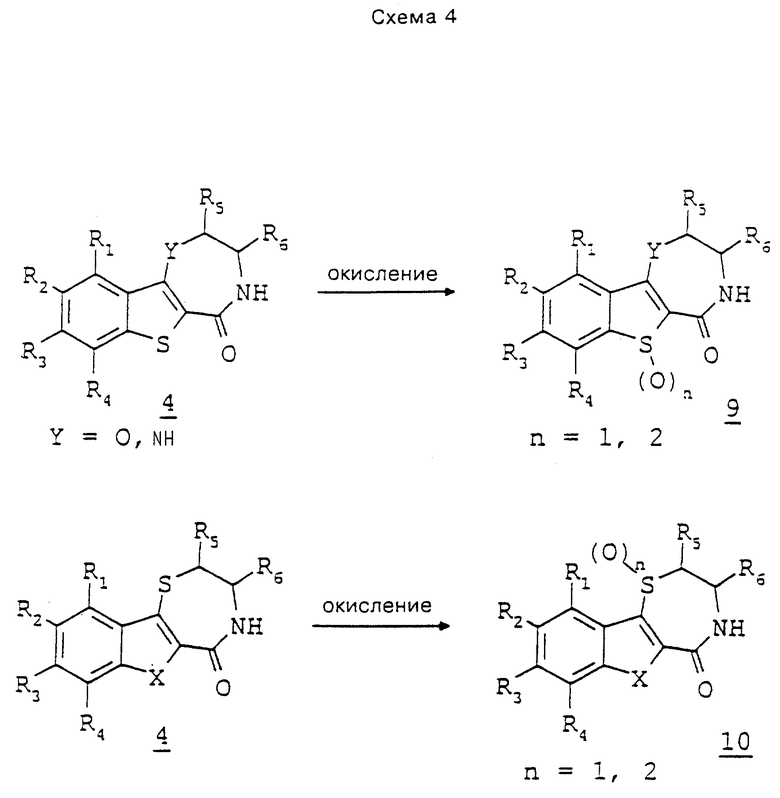
Соединения 4, где X означает серу, а Y - кислород или группу NR, могут быть превращены до соответствующих сульфоксидов и/или сульфонов 9 с помощью агента окисления, такого как хлорнадбензойная кислота, или оксазиридина, причем степень окисления зависит от условий реакции (схема 4). Аналогичным окислением соединений 4, где Y означает серу, получают либо сульфоксид, либо сульфон 10.
Условия реакций по схемам 1-4 широко известны или могут определяться простым образом.
Нижеследующие примеры поясняют получение соединений настоящего изобретения.
Пример 1
2,3-дигидро-9-метокси[1]бензотиено[2,3-f]-1,4-тиазепин-5(4H)-он
К 500 мг (1,95 ммоль) имеющего комнатную температуру раствора метил-3-хлор-5-метокси-бензо[b] тиофен-2-карбоксилата, полученного в результате взаимодействия известного 3-хлор-5-метокси-бензо[b]тиофен-2-карбонилхлорида с метанолом (см. J. Med. Chem., 35:958 (1992)) в 20 мл диметилформамида добавляют сначала 885 мг (7,79 ммоль) 2-аминоэтантиола в виде гидрохлорида, а потом 2,33 мл (15,58 ммоль) диазабициклоундекана. Реакционную смесь перемешивают при комнатной температуре в течение 1,5 ч, после чего нагревают до 70oC. Смесь разбавляют этилацетатом и промывают водной соляной кислотой, водой и рассолом. Органический слой сушат над сульфатом магния, фильтруют и сгущают в вакууме. Сырой продукт перекристаллизуют из смеси гексана и этилацетата с получением 2,3-дигидро-9-метокси[1] бензотиено[2,3-f]-1,4-тиазепин-5(4H)-она с выходом 74%. Т.п. 209 - 209,5oC.
Пример 2
2,3-дигидро-[1]бензотиено[2,3-f]-1,4-оксазепин-5(4H)-он
Смесь 405 мг (1,64 ммоль) сложного метилового эфира 3-(цианометокси)бензо[b]тиофен-2-карбоновой кислоты (см. J. Hetero. Chem., 12: 1037 (1975)), 0,5 мл триэтиламина и 0,50 г кобальта Ренея в 50 мл тетрагидрофурана нагревают при 100oC в атмосфере 84,372 кг/см2 водорода. Реакционную смесь сгущают в вакууме. В результате колоночной градиентной хроматографии с применением в качестве элюента сначала смеси гексана и этилацетата в соотношении 1: 1 и затем этилацетата получают 2,3-дигидро-[1]бензотиено[2,3-f]-1,4-оксазепин-5(4H)-она с выходом 55%. Т.п. 244 - 245oC.
Пример 3
2,3-дигидро-9-метокси-[1]бензотиено[2,3-f]-1,4-тиазепин-5(4H)-он-1-оксид
Смесь 200 мг (0,75 ммоль) 2,3-дигидро-9-метокси-[1]бензотиено[2,3-f]-1,4-тиазепин-5(4H)-она и 116 мг (0,75 ммоль) NaBO3-4H2O в 18 мл уксусной кислоты перемешивают при комнатной температуре в течение ночи. Реакционную смесь фильтруют, после чего к фильтрату добавляют 60 мл воды. В результате фильтрации получают 2,3-дигидро-9-метокси-[1] бензотиено[2,3-f]-1,4-тиазепин-5(4H)-он-1-оксид с выходом 69%. Т.п. 215 - 216oC (разл.).
Пример 4
3,4-дигидро-9-метокси-6-метил-2H-1,4-оксазепино[6,7-b]-индол-5(6H)-он
А. Метил-3-(цианометокси)-5-метокси-1-метил-1H-индол-2-карбоксилат
Суспензию 3,2 г (29 ммоль) трет-бутилата калия в 60 мл диметилсульфоксида обрабатывают порциями 5,6 г (24 ммоль) метил-3-гидрокси-5-метокси-1-метил-1H-индол-2-карбоксилата (см. Унангст и др., J. Heterocyclic Chem., 24: 811 (1987)). Смесь перемешивают в течение 15 мин, после чего прикапывают 4,8 мл (5,7 г, 76 ммоль) хлорацетонитрила. Смесь нагревают при 80oC в течение 90 мин, охлаждают и подают в 800 г льда и воды. Осажденное твердое вещество фильтруют, промывают 10%-ным раствором метанола в воде и перекристаллизуют из водного ацетонитрила с получением 3,9 г (60%) продукта. Т.п. 136 - 137oC.
Б.
Смесь 0,60 г (2,2 ммоль) метил-3-(цианометокси)-5-метокси-1-метил-1Н-индол-2-карбоксилата и 0,40 мл (0,29 г, 2,9 ммоль) триэтиламина в 35 мл тетрагидрофурана в автоклаве обрабатывают 0,40 г кобальта Ренея в качестве катализатора. В автоклаве создают повышенное давление с применением водорода (41,4829 кг/см2) и нагревают при 80oC в течение 10 ч. Реакционную смесь охлаждают, фильтруют и фильтрат упаривают. Масляный остаток растворяют в 50 мл метанола, после чего к раствору добавляют 0,80 г (15 ммоль) метилата натрия. Смесь нагревают с обратным холодильником в течение 3 ч, охлаждают и упаривают. Остаток распределяют между 75 мл этилацетата и 150 мл рассола. Водный слой экстрагируют несколько раз свежим этилацетатом. Объединенные органические слои промывают рассолом, сушат над безводным сульфатом натрия и упаривают. Остаточный сырой продукт очищают путем флеш-хроматографии на силикагеле. Элюацию осуществляют 5%-ным раствором метанола в дихлорметане с получением 0,18 г (33%) продукта. Образец, перекристаллизованный из смеси этилацетата и гексана, имеет т.п. 184 - 186oC.
Пример 5
2,3-дигидро-1H-бензотиено-[3,2-e]-1,4-диазепин-5-он
Гидрохлорид сложного метилового эфира 3-(2-аминоэтиламино)бензо[b]тиофен-2-карбоновой кислоты
Раствор 1,00 г (5,62 ммоль) 2-(4,5-дигидро-1H-имидазол-2-ил)бензолтиола (см. H. Hegen, H. Fleig, Justus Liebigs Ann. Chem., 11:1994 (1975)) и 610 мг (5,62 ммоль) хлорметилацетата в 15 мл метанола нагревают с обратным холодильником в течение 90 мин. Реакционную смесь охлаждают до комнатной температуры и фильтруют. Фильтрат сгущают досуха, и остаток растворяют в горячем хлороформе. По истечении нескольких часов полученный осадок собирают и сушат. Маточный раствор позволяет получать второй выход кристаллов в качестве гидрохлорида сложного метилового эфира 3-(2-аминоэтиламино)бензо[b]тиофен-2-карбоновой кислоты. Общий выход: 61%. Т.п. 219 - 220oC.
2,3-дигидро-1H-бензотиено-[3,2-e]-1,4-диазепин-5-он
Раствор 339 мг (1,18 ммоль) гидрохлорида сложного метилового эфира 3-(2-аминоэтиламино)бензо[b] тиофен-2-карбоновой кислоты и свежеизготовленного метилата натрия (из 134 мг (2,48 ммоль) натрия) в 5 мл метанола нагревают с обратным холодильником в течение 18 ч. Смесь охлаждают, нейтрализуют 25 мл 1 н соляной кислоты и охлаждают до 0oC в течение часа. Получаемый желтый кристаллический продукт фильтруют и сушат в вакууме при 60oC в течение нескольких часов с получением 2,3-дигидро-1H-бензотиено-[3,2-e]-1,4-диазепин-5-она. Выход: 64%. В результате колоночной градиентной хроматографии с применением в качестве элюента сначала 2%-ного раствора метанола в этилацетате и затем 5%-ного раствора метанола в этилацетате получают аналитически чистый 2,3-дигидро-1H-бензотиено-[3,2-e] -1,4-диазепин-5-он. Т. п. 210 - 212oC.
Пример 6
2,3-дигидро-9-метокси-1H-бензотиено-[3,2-f]-1,4-оксазепин-5-он
Сложный метиловый эфир 3-цианометокси-5-метокси-бензо[b]тиофен-2-карбоновой кислоты
К имеющему комнатную температуру раствору 1,00 г (4,2 ммоль) метил-3-окси-5-метоксибензо[b]тиофен-2-карбоксилата (см. Коннор и др., J. Med. Chem., 25:959 (1992)) в 20 мл диметилсульфоксида добавляют 494 мг (4,41 ммоль) трет-бутилата калия, а потом 878 мкл (12,58 ммоль) бромацетонитрила. Смесь перемешивают при комнатной температуре в течение 1,5 ч, после чего ее подают в этилацетат и 1 н соляную кислоту. Органический слой сначала промывают 1 н соляной кислотой, потом несколькими порциями рассола, после чего сушат над сульфатом магния. В результате фильтрации, удаления растворителя в вакууме и перекристаллизации остатка из смеси этилацетата и гексана получают 413 мг продукта. Дополнительный выход в количестве 112 мг можно получать из маточного раствора. Т.п. 159,5 - 160oC.
2,3-дигидро-9-метокси-1H-бензотиено-[2,3-f]-1,4-оксазепин-5-он
Раствор 2,5 г (9,0 ммоль) сложного метилового эфира 3-цианометокси-5-метокси-бензо[b]-тиофен-2-карбоновой кислоты в 50 мл тетрагидрофурана нагревают до интенсивного кипения. Быстро добавляют 9,0 мл (90,2 ммоль) борандиметилсульфида, продолжают нагревание в течение 25 мин и добавляют тетрагидрофуран в момент испарения. Добавляют еще 4,0 мл борандиметилсульфида и нагревание продолжают в течение 10 мин. Реакционную смесь охлаждают до 0oC и осторожно добавляют 50 мл 6 н соляной кислоты. Водородный газ выделяется и температура реакционной смеси повышается. Полученный осадок собирают путем фильтрации, промывают водой и сушат в вакууме в течение ночи.
2,3 г (8,2 ммоль) твердого вещества подают в свежеизготовленный раствор метилата натрия (из 1,9 г (82,0 ммоль) натрия) в 40 мл метанола. Реакционную смесь нагревают до 50oC в течение 2 ч, после чего нагревают с обратным холодильником в течение 2 ч. После охлаждения до комнатной температуры осадок собирают и промывают сначала холодным метанолом, а потом холодным диэтиловым эфиром. Твердое вещество сушат в вакууме в течение ночи с выходом 1,18 г (52%) продукта. Аналитический образец 2,3-дигидро-9-метокси-1H-бензотиено-[2,3-f] -1,4-оксазепин-5-она получают в результате перекристаллизации из смеси этилацетата и гексана. Т.п. 264 - 265oC.
Пример 7
2,3-дигидро-9-метокси-6-оксид-1H-бензотиено-[2,3-f]-1,4-оксазепин-5-он
К суспензии 1,00 г (4,0 ммоль) 2,3-дигидро-9-метокси-1H-бензотиено-[2,3-f] -1,4-оксазепин-5-она в 100 мл теплого метанола добавляют сначала 8,0 мл (80 ммоль) 30%-ной перекиси водорода, а потом 445 мг (4,01 ммоль) двуокиси селена. Реакционную смесь перемешивают при комнатной температуре в течение 3 ч, после чего нагревают при 30oC в течение 1,5 ч, а потом при 40oC в течение 2 ч. Реакционную смесь охлаждают до -40oC и полученный осадок собирают путем фильтрации. Остаток подвергают колоночной градиентной хроматографии с применением в качестве элюента 5%-ного метанола в этилацетате при постепенном увеличении концентрации метанола до получения его смеси с этилацетатом в соотношении 1:1. Получают 338 мг продукта. Аналитический образец 2,3-дигидро-9-метокси-6-оксид-1H-бензотиено-[2,3-f]-1,4-оксазепин-5-она получают путем перекристаллизации из смеси метанола и этилацетата. Т.п. 273 - 274oC.
Пример 8
2,3-дигидро-9-метокси-2-метил-1H-бензотиено-[2,3-f]-1,4-оксазепин-5-он
Сложный метиловый эфир 3-(1-цианоэтокси)-5-метокси-бензо(b]-тиофен-2-карбоновой кислоты
К имеющему комнатную температуру раствору 1,00 г (4,2 ммоль) метил-3-окси-5-метоксибензо[b]тиофен-2-карбоксилата (см. Коннор и др., J. Med. Chem. , 35: 958 (1992)) в 20 мл диметилсульфоксида добавляют сначала 494 мг (4,41 ммоль) трет-бутилата натрия, а потом 1,1 мл (12,6 ммоль) 2-хлорпропионитрила. Смесь перемешивают при комнатной температуре в течение 1,5 ч, после чего нагревают до 82oC в течение 3 ч. Реакционную смесь подают в этилацетат и 1 н соляную кислоту. Органический слой сначала промывают 1 н соляной кислотой, потом несколькими порциями рассола, после чего сушат над сульфатом магния. В результате фильтрации, удаления растворителя в вакууме и перекристаллизации остатка из смеси этилацетата и гексана получают 853 мг продукта. Т.п. 127 - 129oC.
2,3-дигидро-9-метокси-2-метил-1H-бензотиено-[2,3-f]-1,4-оксазепин-5-он
Раствор 400 мг (1,37 ммоль) сложного метилового эфира 3-(1-цианоэтокси)-5-метоксибензо[b] -тиофен-2-карбоновой кислоты в 10 мл тетрагидрофурана нагревают до интенсивного кипения. Прикапывают 1,4 мл (13,7 ммоль) борандиметилсульфида и нагревание продолжают в течение 20 мин с добавлением тетрагидрофурана в момент испарения. Реакционную смесь охлаждают до комнатной температуры и осторожно добавляют 7,5 мл 6 н соляной кислоты. Через 5 мин реакционную смесь охлаждают до 0oC и добавляют 68,5 мл 1 н гидроокиси натрия, а затем этилацетат. Слои разделяют, и органическую фазу промывают сначала смесью рассола и воды в соотношении 1:1, потом дополнительным рассолом. Органическую фазу сушат над сульфатом магния, фильтруют и сгущают в вакууме. Остаток подвергают колоночной градиентной хроматографии с применением в качестве элюента смеси метанола, гексана и хлороформа в соотношении 5:25:70, потом смеси метанола и хлороформа в соотношении 10:90, а затем смеси метанола и хлороформа в соотношении 30:70, в результате чего получают 135 мг продукта. Аналитический образец 2,3-дигидро-9-метокси-2-метил-1H- бензотиено-[2,3-f] -1,4-оксазепин-5-она получают путем перекристаллизации из смеси этилацетата и гексана. Т.п. 185 - 186oC.
Соединения согласно изобретению легко поддаются переработке со стандартными разбавителями и носителями для удобной оральной или парентеральной дачи людям и животным для лечения болезней, таких, как, например, воспаление, в частности артрит и тому подобное. Нижеследующие примеры поясняют получение типичных фармацевтических композиций.
Пример 9
Получение капсулы емкостью 250 мг
250 мг 2,3-дигидро-9-изопропокси-7-хлор-1H-бензотиено-[2,3-f] -1,4-оксазепина-5-она смешивают со 150 мг лактозы и 150 мг кукурузного крахмала до получения гомогенной смеси. Смесь подают в желатиновые капсулы, которые дают орально в количестве 1 - 3 в сутки для лечения артрита.
Пример 10
Композиция для оральной суспензии
Ингредиент - Количество
2,3-дигидро-8-этил-10-трифторметил-6-оксид-1H-бензотиено-[2,3-f]-1,4-оксазепин-5-он - 500 мг
Раствор сорбита (70%) - 40 мл
Бензоат натрия - 150 мг
Сахарин - 10 мг
Вишневый аромат - 50 мг
Дистиллированная вода - 100 мл
Раствор сорбита подают в 40 мл дистиллированной воды, и оксазепинон суспендируют. К смеси добавляют сахарин, бензоат натрия и аромат и растворяют. Объем доводят до 100 мл добавлением дистиллированной воды. Каждый миллилитр сиропа содержит 5 мг оксазепинона. Данная композиция для оральной дачи идеально годится для лечения воспаления в области педиатрии.
Пример 11
Получение растворов для парентеральной дачи
В растворе 700 мл пропиленгликоля и 200 мл дистиллированной воды для инъекции растворяют 20,0 г 2,3-дигидро-7-диметиламино-1H-бензотиено-[3,2-e] -1,4-диазепин-5-она. Значение pH раствора хлористоводородной кислотой доводят до 5,5, и объем доводят до 1000 мл добавлением дистиллированной воды. Композицию стерилизуют, разливают в ампулы емкостью 5,0 мл, каждая из которых содержит 2,0 мл композиции (что представляет собой 40 мг активного диазепинона) и герметизируют в атмосфере азота. Композицию внутривенно дают пациентам, страдающим от воспаления или СПИДа.
Пример 12
Получение крема для местного применения
500 мг 2,3-дигидро-7-этокси-бензофурано-[2,3-f]-1,4-оксазепин-5-она смешивают с 15 г цетилового спирта, 1 г лаурилсульфата натрия, 40 г жидкого силикона марки D.C. 200 (торговый продукт фирмы "Дау Корнинг Ко.", Мидленд, Мичиган), 43 г стерильной воды, 0,25 г метилпарабена и 0,15 г пропилпарабена. Смесь нагревают приблизительно до 75oC при постоянном перемешивании, после чего охлаждают до комнатной температуры, при которой смесь затвердевает. Препарат наносят на поверхность кожи пациента, страдающего от воспаления.

Описываются новые производные бензотиофена, бензофурана, индолтиазепинона, оксазепинона и диазепинона формулы I, где R1-R6, Х и Y имеют указанные в п. 1 формулы изобретения значения, и их фармацевтически приемлемые кислотно-аддитивные соли. Данные гетероциклические соединения могут представлять собой активное начало фармацевтической композиции, обладающей ингибирующей клеточную адгезию или ВИЧ активностью. Кроме того, данные соединения можно применять для торможения адгезии лейкоцитов к эндотелиальным клеткам при лечении вызванных ею болезней, например воспалительных заболеваний, а также для лечения человека и животных, зараженных ВИЧ. 4 с. и 11 з.п. ф-лы, 2 табл.
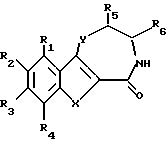
где R1, R2, R3 и R4 независимо друг от друга означают водород, гидроксил, галоген, низший алкил, низший алкоксил;
R5 и R6 независимо друг от друга означают водород или низший алкил,
X - группа S(O)n или NH,
Y - кислород, группа S(O)n или NH,
n - 0, 1 или 2,
при условии, что
1) если X означает NH, Y - NH, R1 - водород и R3 - водород, то R2 не означает метил,
2) если X означает NH, Y - NH, R1, R3 и R4 - водород, то R2 не означает метокси или этокси, и
3) если X означает NH, Y - серу, то по меньшей мере один из радикалов R1, R3 и R4 не означает водород,
или их фармацевтически приемлемые кислотно-аддитивные соли.

