Область техники, к которой относится изобретение
Настоящее изобретение относится к новым соединениям формулы I, представляющим собой модуляторы метаботропных рецепторов подтипа 5 (“mGluR5”), которые применимы для лечения расстройств центральной нервной системы, таких как, например, хроническое прогрессирующее снижение познавательной способности, как положительные, так и отрицательные симптомы при шизофрении, а также других расстройств, опосредуемых mGluR5 рецепторами.
Уровень техники
Глютамат, основной аминокислотный медиатор центральной нервной системы (ЦНС) млекопитающих, проводит возбуждающую синаптическую нейропередачу через активацию рецептор-каналов ионотропных глютаматных рецепторов (сокращенно iGluR, а именно NMDA, AMPA и каината) и метаботропных глютаматных рецепторов (mGluR). iGluR ответственны за быструю возбуждающую передачу (Nakanishi С. et al., (1998) Brain Res Brain Res Rev., 26:230-235), в то время как mGluR-рецепторы играют в большей степени модулирующую роль, которая способствует тонкому регулированию синаптической эффективности. Глютамат выполняет многочисленные физиологические функции, такие как долговременная потенциация (long-term potentiation - LTP), процесс, который, как считается, лежит в основе познавательной способности и памяти, а также сердечно-сосудистая стабилизация, чувственное восприятие и развитие синаптической пластичности. Кроме того, глютамат играет важную роль в патофизиологии различных неврологических и психиатрических заболеваний, особенно при дисбалансе глютаматергической нейропередачи.
mGluR-рецепторы представляют собой семь-трансмембранные G-белок-связанные рецепторы. Восемь представителей этого семейства подразделены на три группы (группы I, II и III) в соответствии с гомологией последовательностей и фармакологическими свойствами (Schoepp D.D. et al. (1999) Neuropharmacology, 38:1431-1476). Активация mGluR приводит к большому количеству разнообразных внутриклеточных реакций и активации различных метаболических каскадов. Среди представителей mGluR рецепторов значительный интерес представляют рецепторы подтипа mGluR5 в качестве рецепторов противодействия недостаточной или избыточной нейропередачи при нейропсихиатрических заболеваниях. mGluR5 относится к I группе, и его активация инициирует клеточные реакции через механизмы, опосредуемые G-белком. mGluR5 связан с фосфолипазой C и стимулирует фосфоинозитидный гидролиз и внутриклеточную мобилизацию кальция.
Было показано, что mGluR5 белки локализованы в постсинаптических элементах, прилегающих к постсинаптической плотности (Lujan R et al. (1996) Eur. J. Neurosci. 8:1488-500; Lujan R. et al. (1997) J. Chem. Neuroanat., 13:219-41), и редко обнаруживаются в пресинаптических элементах (Romano C. et al. (1995) J. Comp. Neurol. 355:455-69). Таким образом, mGluR5 рецепторы могут модифицировать постсинаптические реакции в нейромедиатор или регулировать высвобождение нейромедиатора.
В ЦНС mGluR5 рецепторы распространены, главным образом, в коре головного мозга, гиппокампе, в участках головного мозга Caudate-putamen и Nucleus accumbens. Поскольку было показано, что эти области мозга вовлечены в процессы возбуждения, побуждения и в большое количество аспектов познавательной функции, предполагается, что mGluR5 модуляторы представляют интерес в качестве терапевтических средств.
В качестве мишеней для разработки селективных модуляторов рецепторов подтипа mGLuR был предложен ряд возможных клинических показаний. Эти показания включают эпилепсию, невропатическую и воспалительную боль, различные психические расстройства (например, состояние тревоги и шизофрению), расстройства двигательной функции (например, болезнь Паркинсона), нейропротекция (защита от удара, повреждения головы), мигрень и аддикция/лекарственная зависимость (см. Brauner-Osborne H. et al. (2000) J. Med. Chem. 43:2609-45; Bordi F. and Ugolini A. (1999) Prog. Neurobiol. 59:55-79; Spooren W. et al. (2003) Behav. Pharmacol: 14:257-77).
Гипотеза о том, что предполагаемой причиной шизофрении является гипофункция глютаматергической системы, представляющая собой отражение гипофункции NMDA рецепторов, в последние несколько лет получает возрастающую поддержку (см. Goff D.C. and Coyle J.T. (2001) Am. J. Psychiatry, 158:1367-1377; Carlsson A. et al. (2001) Annu. Rev. Pharmacol. Toxicol., 41:237-260). Данные о причастности дисфункции глютаматергической нейротрансмиссии подтверждены выявлением того факта, что антагонисты глутаматного рецептора подтипа NMDA могут воспроизводить полный спектр симптомов, а также физиологические проявления шизофрении, такие как гипофронтальный синдром, ухудшение ингибирования пульсовых колебаний кровяного давления и повышение высвобождения субкортикального допамина. Кроме того, клинические исследования показали, что частота аллелей mGluR5 связана с шизофренией среди некоторых групп пациентов (Devon R.S. et al. (2001), Mol. Psychiatry. 6:311-4), и повышение mGluR5 передачи было обнаружено в кортикальных пирамидальных клеточных слоях мозга больного шизофренией (Ohnuma T. et al. (1998), Brain Res. Mol. Brain Res. 56:207-17).
Вовлеченность mGluR5 в неврологические и психиатрические расстройства подтверждено тем фактом, что активация mGluR рецепторов I группы индуцирует потенцирование функции NMDA в различных областях мозга, главным образом, посредством активации mGluR5 рецепторов (Mannaioni G. et al. (2001), Neurosci. 21:5925-34; Awad H. et al. (2000), J. Neurosci, 20:7871-7879; Pisani A. et al. (2001) Neuroscience 106:579-87; Benquet P. et al. (2002), J. Neurosci. 22:9679-86).
Кроме того, в последнее десятилетие была точно определена роль глютамата в процессах запоминания (Martin S.J. et al. (2000) Annu. Rev. Neurosci. 23:649-711; Baudry M. and Lynch G. (2001), Neurobiol. Learn Mem., 76:284-297). Использование mGluR5 «ноль»-мутантных мышей четко подтвердило роль mGluR5 в обучении и запоминании. Указанные мыши демонстрируют селективную утрату способности в двух задачах пространственного обучения и запоминания и сниженный CA1 LTP (Lu et al. (1997), J. Neurosci., 17:5196-5205; Schulz B. et al. (2001), Neuropharmacology, 41:1-7; Jia Z. et al. (2001) Physiol. Behav., 73:793-802; Rodrigues et al. (2002), J. Neurosci., 22:5219-5229).
Выявление того, что mGIuR5 является ответственным за потенцирование токов, проводимых NMDA рецептором, повышает вероятность возможности применения агонистов данного рецептора в качестве средств, повышающих познавательную функцию, а также в качестве новых нейролептических средств, которые воздействуют посредством селективного повышения функции NMDA рецептора.
Активация NMDAR могла бы придать силу гипофункциональным NMDAR в нейронной цепи, имеющим место при шизофрении. Последние данные исследований in vivo четко подтверждают, что активация mGluR5 может предоставить новый и эффективный подход к лечению снижения познавательной способности, а также как положительных, так и отрицательных симптомов при шизофрении (Kinney G.G. et al. (2002) 43:292).
Таким образом, MGluR5 рецептор рассматривается как потенциальная мишень лекарственного средства для лечения психиатрических и неврологических расстройств, включая заболевания, подлежащие лечению, такие как расстройства, характеризующиеся состоянием тревоги, расстройства внимания, расстройства питания, расстройства настроения, психотические расстройства, расстройства познавательной способности, личностные расстройства и расстройства, связанные с лекарственной зависимостью.
Большинство из известных в настоящее время модуляторов функции mGluR5 разрабатывались как структурные аналоги глютамата, хисквалата или фенилглицина (Schoepp D.D. et al. (1999) Neuropharmacology, 38:1431-1476), и значительную трудность представляет разработка in vivo активных и селективных модуляторов mGluR5, воздействующих на глютамат-связывающий сайт. Новым путем разработки селективных модуляторов является идентификация молекул, которые действуют посредством аллостерических механизмов, модулируя рецептор связыванием его с сайтом, отличным от труднодоступного ортостерического связывающего сайта.
Положительные аллостерические модуляторы рецепторов mGluR появились в последнее время как новые фармакологические объекты, предоставляющие такую привлекательную альтернативу. Молекулы данного типа были разработаны для mGluRl, mGluR2, mGluR4 и mGluR5 (Knoflach F. et al. (2001), Proc. Natl. Acad. Sci. USA. 98:13402-13407; O'Brien J.A. et al. (2003), Mol. Pharmacol. 64:731-40; Johnson K. et al. (2002), Neuropharmacology 43:291; Johnson M.P. et al. (2003), J. Med. Chem. 46:3189-92; Marino M.J. et al. (2003), Proc. Natl. Acad. Sci. USA. 100(23):13668-73; см. обзор Mutel V. (2002), Expert Opin. Ther. Patents 12:1-8). DFB и родственные молекулы были описаны в качестве положительного аллостерического модулятора, но с низким потенциалом in vitro ((O'Brien J.A. et al. (2003), Mol. Pharmacol. 64:731-40). Запатентованы недавно разработанные бензамидные модуляторы mGluR5 рецепторов (WO 2004/087048). Был также описан новый класс положительных аллостерических модуляторов; эти молекулы представляют собой производные аминопиразола (C. W. Lindsley et al. (2004), J. Med. Chem. Epub 10/23/2004 jm049400d).
Ни одно из конкретно описанных соединений структурно не относится к соединениям настоящего изобретения.
Настоящее изобретение относится к способу лечения или профилактики у млекопитающего, включая человека, состояния, лечение или профилактика которого опосредованы или могут быть облегчены нейромодуляторным действием модуляторов mGluR5.
Сущность изобретения
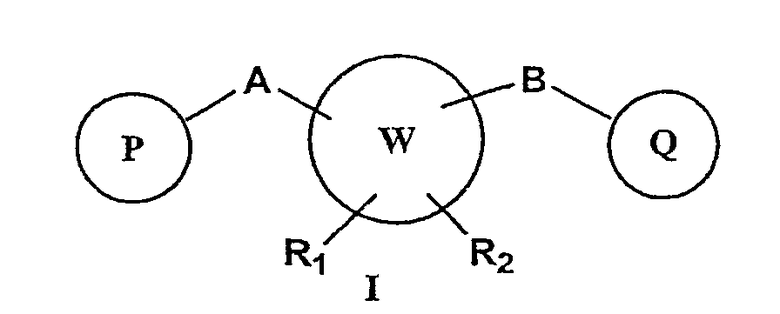
В соответствии с настоящим изобретением предоставлены новые соединения, замещенные мостиком А или В с ненасыщенным пяти- или шестичленным арильным или гетероарильным циклом, содержащим атомы, независимо выбранные из атомов углерода, азота, серы и кислорода. Кроме того, изобретение относится к фармацевтически приемлемым формам этих соединений.
Изобретение применимо также для лечения расстройств ЦНС, подверженных действию нейромодуляторного эффекта mGluR5 положительных аллостерических модуляторов, таких как расстройство познавательной способности, а также для лечения как положительных, так и отрицательных симптомов при шизофрении.
Чертежи
На фиг.1 показано повышение мобилизации Са2+, индуцированной 1 мкМ глютамата, в культивированных астроцитах крысы в присутствии 3 мкМ соединений примеров 12, 55 и 56 настоящего изобретения.
На фиг.2 показано ослабляющее действие типичного соединения настоящего изобретения на возрастание двигательной активности, индуцированной PCP (f=13,39, df=(2,45), n=16/группа), в дозе 100 мг/кг и.п.
На фиг.3 показано ослабляющее действие типичного соединения настоящего изобретения на возрастание двигательной активности, индуцированной амфетамином (f=13,04, df=(4,82), n=8-33 мышей на группу), в дозах 50 и 100 мг/кг и.п.
Подробное описание изобретения
Согласно настоящему изобретению предоставлены новые соединения общей формулы I

или фармацевтически приемлемые соли, гидраты или сольваты указанных соединений,
где
W представляет собой 5-7-членное циклоалкильное или гетероциклоалкильное кольцо;
R1 и R2 независимо представляют собой водород, C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, арилалкил, гетероарилалкил, гидрокси, амино, аминоалкил, гидроксиалкил, C1-C6-алкокси, или R1 и R2 вместе могут образовывать C3-C7-циклоалкильное кольцо, карбонильную связь C=O или двойную углерод-углеродную связь;
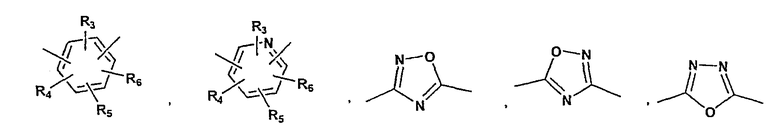
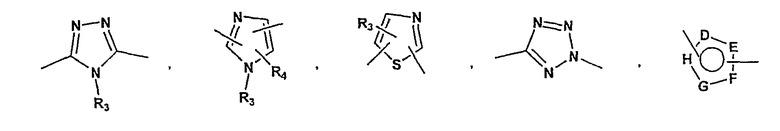
P и Q, каждый независимо, выбран из циклоалкильной, арильной или гетероарильной группы формулы
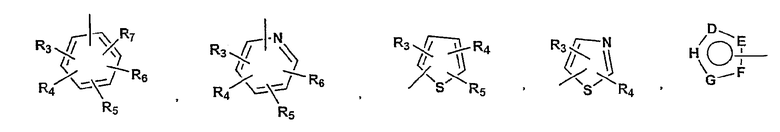
R3, R4, R5, R6 и R7 независимо представляют собой водород, галоген, -CN, нитро, C1-C6-алкил, C3-C6-циклоалкил, C3-C7-циклоалкилалкил, C2-C6-алкенил, C2-C6-алкинил, галоген-C1-C6-алкил, -гетероарил, гетероарилалкил, арилалкил, арил, -OR8, -NR8R9, -C(=NR10)NR8R9, N(=NR10)NR8R9, -NR8COR9, NR8CO2R9, NR8SO2R9,
-NR10CONR8R9, -SR8, -S(=O)R8, -S(=O)2R8, -S(=O)2NR8R9, -C(=O)R8, -C(=O)2R8, -C(=O)NR8R9,
-C(=NR8)R9 или C(=NOR8)R9 заместители; где необязательно два заместителя вместе с находящимися между ними атомами образуют бициклическое гетероциклоалкильное, арильное или гетероарильное кольцо; где каждое кольцо является необязательно дополнительно замещенным 1-5 группами, независимо выбранными из галогена, -CN, C1-C6-алкила, -O(C0-C6-алкила), -O(C3-C7-циклоалкилалкила), -O(арила), -O(гетероарила), -O(C1-C3-алкиларила), -O(C1-C3-алкилгетероарила), -N(C0-C6-алкил)(C0-C3-алкиларила) или -N(C0-C6-алкил)(C0-C3-алкилгетероарила);
R8, R9, R10, каждый независимо, представляет собой водород, C1-C6-алкил, C3-C6-циклоалкил, C3-C7-циклоалкилалкил, C2-C6-алкенил, C2-C6-алкинил,
галоген-C1-C6-алкил, гетероциклоалкил, гетероарил, гетероарилалкил, арилалкил или арил, каждый из которых является необязательно замещенным 1-5 заместителями, независимо выбранными из галогена, -CN, C1-C6-алкила, -O(C0-C6-алкила), -O(C3-C7-циклоалкилалкила), -O(арила), -O(гетероарила), -N(C0-C6-алкил)(C0-C6-алкила), -N(C0-C6-алкил)(C3-C7-циклоалкила) или -N(C0-C6-алкил)(арила);
D, E, F, G и H независимо представляют собой -C(R3)=, -C(R3)=C(R4)-, -C(=O)-,
-C(=S)-, -O-, -N=, -N(R3)- или -S-;
A представляет собой азо-N=N-, этил, этенил, этинил, -NR8C(=O)-, NR8S(=O)2-,
-C(=O)NR8-, -S-, -S(=O)-, -S(=O)2-, -S(=O)2NR8-, -C(=O)-O-, -O-C(=O)-, -C(=NR8)NR9-, C(=NOR8)NR9-, -NR8C(=NOR9)-, =N-O-, -O-N=CH- или арильную или гетероарильную группу формулы
R3, R4, R5 и R6 независимо принимают значения, определенные выше;
D, E, F, G и H независимо представляют собой атом углерода, кислорода, азота или серы или двойную связь;
B представляет собой одинарную связь, -C(=O)-C0-C2-алкил-, -C(=O)-C2-C6-алкенил-, -C(=O)-C2-C6-алкинил-, -C(=O)-O-, -C(=O)NR8-C0-C2-алкил-,
-C(=NR8)NR9-S(=O)-C0-C2-алкил-, -S(=O)2-C0-C2-алкил-, -S(=O)2NR8-C0-C2-алкил-,
C(=NR8)-C0-C2-алкил-, -C(=NOR8)-C0-C2-алкил- или -C(=NOR8)NR9-C0-C2-алкил-; где R8 и R9 независимо принимают значения, определенные выше;
любой N может представлять собой N-оксид.
Настоящее изобретение включает все возможные стереоизомеры, а также не только рацематы, но и отдельные энантиомеры соединений.
В представленном выше определении термин «C1-C6-алкил» включает группу, такую как метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил, трет-бутил, пентил, изопентил, неопентил, трет-пентил, гексил или т.п.
Термин «C2-C6-алкенил» включает такие группы, как этенил, 1-пропенил, аллил, изопропенил, 1-бутенил, 3-бутенил, 4-пентенил и т.п.
Термин «C2-C6-алкинил» включает такие группы, как этинил, пропинил, бутинил, пентинил и т.п.
Термин «галоген» включает атомы фтора, брома, хлора и йода.
Термин «циклоалкил» относится к необязательно замещенному карбоциклу, не содержащему гетероатомов, и включает моно-, би- и трициклические насыщенные карбоциклы, а также конденсированные циклические системы. Такие конденсированные циклические системы могут включать кольцо, которое является частично или полностью ненасыщенным, такое как бензольное кольцо, для образования конденсированных циклических систем, таких как бензоконденсированные карбоциклы. Циклоалкил включает такие конденсированные циклические системы, как спироконденсированные циклические системы. Примеры циклоалкила включают циклопропил, циклобутил, циклопентил, циклогексил, декагидронафталин, адамантан, инданил, флуоренил, 1,2,3,4-тетрагидронафталин и т.п.
Термин «гетероциклоалкил» относится к необязательно замещенному карбоциклу, содержащему, по меньшей мере, один гетероатом, независимо выбранный из атомов О, N и S. Он включает моно-, би- и трициклические насыщенные карбоциклы, а также конденсированные циклические системы. Такие конденсированные циклические системы могут включать цикл, который является частично или полностью ненасыщенным, такой как бензольное кольцо, для образования конденсированных циклических систем, таких как бензоконденсированные карбоциклы. Примеры гетероциклоалкила включают пиперидин, пиперазин, морфолин, тетрагидротиофен, индолин, изохинолин и т.п.
Термин «арил» относится к C6-C10 арильной группе, такой как фенил, 1-нафтил, 2-нафтил и т.п.
Термин «арилалкил» относится к C6-C10-арил-C1-C3-алкильной группе, такой как бензильная группа, 1-фенилэтильная группа, 2-фенилэтильная группа, 1-фенилпропильная группа, 2-фенилпропильная группа, 3-фенилпропильная группа, 1-нафтилметильная группа, 2-нафтилметильная группа или т.п.
Термин «гетероарил» относится к 5-10-членной гетероциклической группе, содержащей от 1 до 4 гетероатомов, выбранных из атомов кислорода, азота или серы, для образования цикла, такого как фурил (фурановое кольцо), бензофуранил (бензофуран), тиенил (тиофен), бензотиофенил (бензотиофен), пирролил (пирольное кольцо), имидазолил (имидазольное кольцо), пиразолил (пиразольное кольцо), тиазолил (тиазольное кольцо), изотиазолил (изотиазольное кольцо), триазолил (триазольное кольцо), тетразолил (тетразольное кольцо), пиридил (пиридильное кольцо), пиразинил (пиразиновое кольцо), пиримидинил (пиримидинильное кольцо), пиридазинил (пиридазинильное кольцо), индолил (индольное кольцо), изоиндолил (изоиндольное кольцо), бензимидазолил (бензимидазольное кольцо), пуринильная группа (пуринильное кольцо), хинолил (хинолиновое кольцо), фталазинил (фтализинильное кольцо), нафтиридинил (нафтиридильное кольцо), хиноксалинил (хиноксалиновое кольцо), циннолил (циннольное кольцо), птеридинил (птеридильное кольцо), оксазолил (оксазольное кольцо), изоксазолил (изоксазольное кольцо), бензоксазолил (бензоксазольное кольцо), бензотиазолил (бензотиазольное кольцо), фуразанил (фуразановое кольцо) и т.п.
Термин «гетероарилалкил» относится к гетероарил-C1-C3-алкильной группе, примеры гетероарила включают группы, представленные в приведенном выше определении, такие как 2-фурилметил, 3-фурилметил, 2-тиенилметил, 3-тиенилметил, 1-имидазолилметил, 2-имидазолилметил, 2-тиазолилметил, 2-пиридилметил, 3-пиридилметил, 1-хинолилметил или т.п.
Термин «сольват» относится к комплексу с изменяемой стехиометрией, образованному растворенным веществом (например, соединением формулы I) и растворителем. Растворитель представляет собой фармацевтически приемлемый растворитель, предпочтительно воду, который не оказывает неблагоприятного воздействия на биологическую активность растворенного вещества.
Термин «необязательно» означает, что описанное(ые) далее событие(я) может(гут) иметь или не иметь место и включает как событие(я), которое(ые) происходит(ят), так и событие(я), которое(ые) не происходит(ят).
Термин «замещенный» относится к замещению указанным заместителем или заместителями, причем за исключением особо оговоренных случаев допустимы множественные уровни замещения.
Предпочтительными соединениями настоящего изобретения являются соединения формулы I-A, представленной ниже
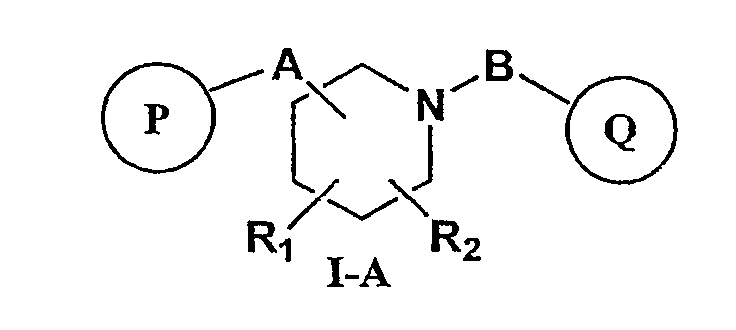
или фармацевтически приемлемые соли, гидраты или сольваты таких соединений,
где
R1 и R2 независимо представляют собой водород, C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, арилалкил, гетероарилалкил, гидрокси, амино, аминоалкил, гидроксиалкил, C1-C6-алкокси, или R1 и R2 вместе могут образовывать C3-C7-циклоалкильное кольцо, карбонильную связь C=O или двойную углерод-углеродную связь;
P и Q, каждый независимо, представляет собой циклоалкильную, арильную или гетероарильную группу формулы
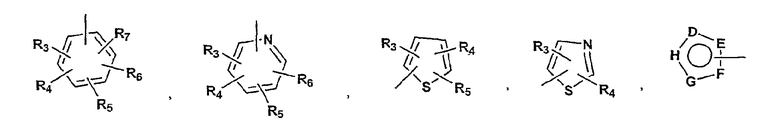
R3, R4, R5, R6 и R7 независимо представляют собой водород, галоген, -CN, нитро, C1-C6-алкил, C3-C6-циклоалкил, C3-C7-циклоалкилалкил, C2-C6-алкенил, C2-C6-алкинил, галоген-C1-C6-алкил, -гетероарил, гетероарилалкил, арилалкил, арил, -OR8, -NR8R9, -C(=NR10)NR8R9, N(=NR10)NR8R9, -NR8COR9, NR8CO2R9, NR8SO2R9,
-NR10CONR8R9, -SR8, -S(=O)R8, -S(=O)2R8, -S(=O)2NR8R9, -C(=O)R8, -C(=O)2R8, -C(=O)NR8R9,
-C(=NR8)R9 или C(=NOR8)R9 заместители; где необязательно два заместителя вместе с находящимися между ними атомами образуют бициклическое гетероциклоалкильное, арильное или гетероарильное кольцо; где каждое кольцо является необязательно дополнительно замещенным 1-5 независимыми группами, выбранными из галогена, -CN, C1-C6-алкила, -O(C0-C6-алкила), -O(C3-C7-циклоалкилалкила), -O(арила), -O(гетероарила), -O(C1-C3-алкиларила), -O(C1-C3-алкилгетероарила),
-N(C0-C6-алкил)(C0-C3-алкиларила) или -N(C0-C6-алкил)(C0-C3-алкилгетероарила);
R8, R9, R10, каждый независимо, представляет собой водород, C1-С6-алкил, C3-C6-циклоалкил, C3-C7-циклоалкилалкил, C2-C6-алкенил, C2-C6-алкинил,
галоген-C1-C6-алкил, гетероциклоалкил, гетероарил, гетероарилалкил, арилалкил или арил, каждый из которых является необязательно замещенным 1-5 заместителями, независимо выбранными из галогена, -CN, C1-C6-алкила, -O(C0-C6-алкила), -O(C3-C7-циклоалкилалкила), -O(арила), -O(гетероарила), -N(C0-C6-алкил)(C0-C6-алкила), -N(C0-C6-алкил)(C3-C7-циклоалкила) или -N(C0-C6-алкил)(арила);
D, E, F, G и H независимо представляют собой -C(R3)=, -C(R3)=C(R4)-, -C(=O)-,
-C(=S)-, -O-, -N=, -N(R3)- или -S-;
A представляет собой азо-N=N-, этил, этенил, этинил, -NR8C(=O)-, NR8S(=O)2-,
-C(=O)NR8-, -S-, -S(=O)-, -S(=O)2-, -S(=O)2NR8-, -C(=O)-O-, -O-C(=O)-, -C(=NR8)NR9-, C(=NOR8)NR9-, -NR8C(=NOR9)-, =N-O-, -O-N=CH- или арильную или гетероарильную группу формулы
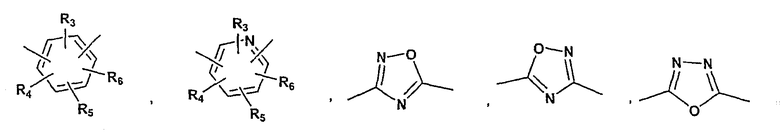
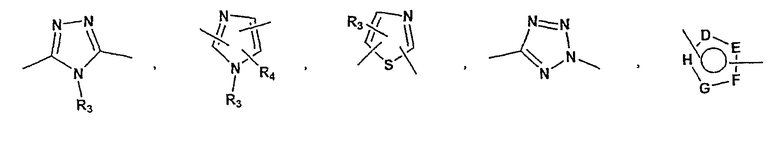
R3, R4, R5 и R6 независимо принимают значения, определенные выше;
D, E, F, G и H независимо представляют собой атом углерода, кислорода, азота или серы или двойную связь;
B представляет собой одинарную связь, -C(=O)-C0-C2-алкил-, -C(=O)-C2-C6-алкенил-, -C(=O)-C2-C6-алкинил-, -C(=O)-O-, -C(=O)NR8-C0-C2-алкил-,
-C(=NR8)NR9-S(=O)-C0-C2-алкил-, -S(=O)2-C0-C2-алкил-, -S(=O)2NR8-C0-C2-алкил-,
C(=NR8)-C0-C2-алкил-, -C(=NOR8)-C0-C2-алкил- или -C(=NOR8)NR9-C0-C2-алкил-; где R8 и R9 независимо принимают значения, определенные выше;
любой N может представлять собой N-оксид.
Настоящее изобретение включает все возможные стереоизомеры, а также не только рацематы, но и отдельные энантиомеры соединений.
Более предпочтительными соединениями настоящего изобретения являются соединения формулы I-B
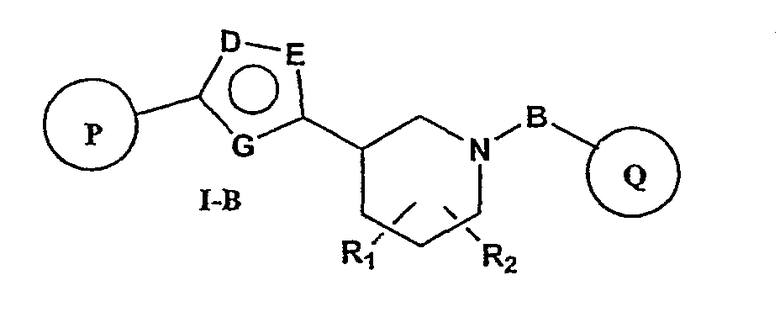
или фармацевтически приемлемые соли, гидраты или сольваты таких соединений;
где
R1 и R2 независимо представляют собой водород, C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, арилалкил, гетероарилалкил, гидрокси, амино, аминоалкил, гидроксиалкил, C1-C6-алкокси, или R1 и R2 вместе могут образовывать C3-C7-циклоалкильное кольцо, карбонильную связь C=O или двойную углерод-углеродную связь;
P и Q, каждый независимо, представляет собой циклоалкильную, арильную или гетероарильную группу формулы
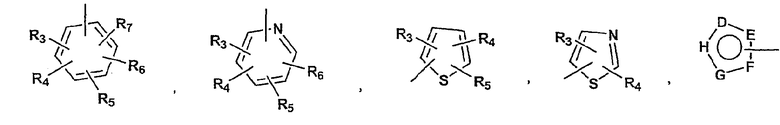
R3, R4, R5, R6 и R7 независимо представляют собой водород, галоген, -CN, нитро, C1-C6-алкил, C3-C6-циклоалкил, C3-C7-циклоалкилалкил, C2-C6-алкенил, C2-C6-алкинил, галоген-C1-C6-алкил, -гетероарил, гетероарилалкил, арилалкил, арил, -OR8, -NR8R9, -C(=NR10)NR8R9, N(=NR10)NR8R9, -NR8COR9, NR8CO2R9, NR8SO2R9, -NR10CONR8R9, -SR8, -S(=O)R8, -S(=O)2R8, -S(=O)2NR8R9, -C(=O)R8, -C(=O)2R8, -C(=O)NR8R9, -C(=NR8)R9 или C(=NOR8)R9 заместители; где необязательно два заместителя вместе с находящимися между ними атомами образуют бициклическое гетероциклоалкильное, арильное или гетероарильное кольцо; где каждое кольцо является необязательно дополнительно замещенным 1-5 группами, независимо выбранными из галогена, -CN, C1-C6-алкила, -O(C0-C6-алкила), -O(C3-C7-циклоалкилалкила), -O(арила), -O(гетероарила), -O(C1-C3-алкиларила), -O(C1-C3-алкилгетероарила), -N(C0-C6-алкил)(C0-C3-алкиларила) или -N(C0-C6-алкил)(C0-C3-алкилгетероарила);
R8, R9, R10, каждый независимо, представляет собой водород, C1-C6-алкил, C3-C6-циклоалкил, C3-C7-циклоалкилалкил, C2-C6-алкенил, C2-C6-алкинил, галоген-C1-C6-алкил, гетероциклоалкил, гетероарил, гетероарилалкил, арилалкил или арил, каждый из которых является необязательно замещенным 1-5 заместителями, независимо выбранными из галогена, -CN, C1-C6-алкила, -O(C0-C6-алкила), -O(C3-C7-циклоалкилалкила), -O(арила), -O(гетероарила), -N(C0-C6-алкил)(C0-C6-алкила), -N(C0-C6-алкил)(C3-C7-циклоалкила) или -N(C0-C6-алкил)(арила);
D, E, F, G и H в P и Q независимо представляют собой -C(R3)=, -C(R3)=C(R4)-, -C(=O)-, -C(=S)-, -O-, -N=, -N(R3)- или -S-;
D, E и G в A независимо принимают значения, определенные выше для A;
B представляет собой одинарную связь, -C(=O)-C0-C2-алкил-, -C(=O)-C2-C6-алкенил-, -C(=O)-C2-C6-алкинил-, -C(=O)-O-, -C(=O)NR8-C0-C2-алкил-, -C(=NR8)NR9-S(=O)-C0-C2-алкил-, -S(=O)2-C0-C2-алкил-, -S(=O)2NR8-C0-C2-алкил-, C(=NR8)-C0-C2-алкил-, -C(=NOR8)-C0-C2-алкил- или -C(=NOR8)NR9-C0-C2-алкил-; где R8 и R9 независимо принимают значения, определенные выше;
любой N может представлять собой N-оксид.
Настоящее изобретение включает все возможные стереоизомеры, а также не только рацематы, но и отдельные энантиомеры соединений.
Особенно предпочтительными соединениями настоящего изобретения являются соединения формулы I-C
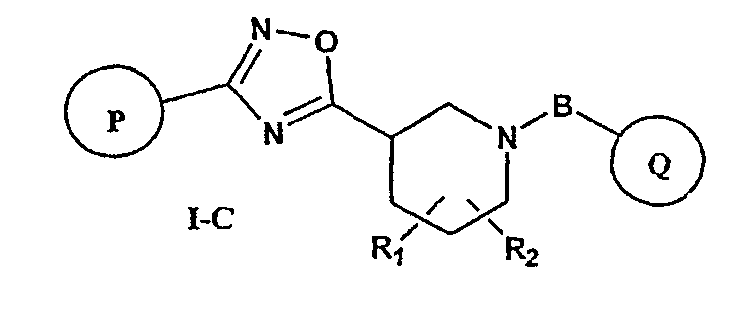
или фармацевтически приемлемые соли, гидраты или сольваты таких соединений,
где
R1 и R2 независимо представляют собой водород, C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, арилалкил, гетероарилалкил, гидрокси, гидроксиалкил, C1-C6-алкокси, или R1 и R2 вместе могут образовывать карбонильную связь C=O или двойную углерод-углеродную связь;
P и Q, каждый независимо, представляет собой циклоалкильную, арильную или гетероарильную группу формулы
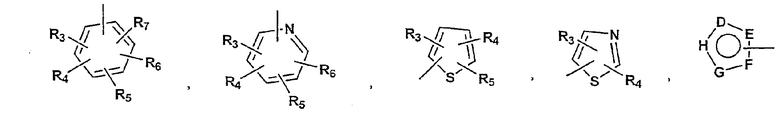
R3, R4, R5, R6 и R7 независимо представляют собой водород, галоген, -CN, нитро, C1-C6-алкил, C3-C6-циклоалкил, C3-C7-циклоалкилалкил, C2-C6-алкенил, C2-C6-алкинил, галоген-C1-C6-алкил, -гетероарил, гетероарилалкил, арилалкил, арил, -OR8, -NR8R9, -C(=NR10)NR8R9, N(=NR10)NR8R9, -NR8COR9, NR8CO2R9, NR8SO2R9, -NR10CONR8R9, -SR8, -S(=O)R8, -S(=O)2R8, -S(=O)2NR8R9, -C(=O)R8, -C(=O)2R8, -C(=O)NR8R9, -C(=NR8)R9 или C(=NOR8)R9 заместители; где необязательно два заместителя вместе с находящимися между ними атомами образуют бициклическое гетероциклоалкильное, арильное или гетероарильное кольцо; где каждое кольцо является необязательно дополнительно замещенным 1-5 группами, независимо выбранными из галогена, -CN, C1-C6-алкила, -O(C0-C6-алкила), -O(C3-C7-циклоалкилалкила), -O(арила), -O(гетероарила), -O(C1-C3-алкиларила), -O(C1-C3-алкилгетероарила), -N(C0-C6-алкил)(C0-C3-алкиларила) или -N(C0-C6-алкил)(C0-C3-алкилгетероарила);
R8, R9, R10, каждый независимо, представляет собой водород, C1-C6-алкил, C3-C6-циклоалкил, C3-C7-циклоалкилалкил, C2-C6-алкенил, C2-C6-алкинил, галоген-C1-C6-алкил, гетероциклоалкил, гетероарил, гетероарилалкил, арилалкил или арил, каждый из которых является необязательно замещенным 1-5 заместителями, независимо выбранными из галогена, -CN, C1-C6-алкила, -O(C0-C6-алкила), -O(C3-C7-циклоалкилалкила), -O(арила), -O(гетероарила), -N(C0-C6-алкил)(C0-C6-алкила), -N(C0-C6-алкил)(C3-C7-циклоалкила) или -N(C0-C6-алкил)(арила);
D, E, F, G и H независимо представляют собой -C(R3)=, -C(R3)=C(R4)-, -C(=O)-, -C(=S)-, -O-, -N=, -N(R3)- или -S-;
B представляет собой одинарную связь, -C(=O)-C0-C2-алкил-, -C(=O)-C2-C6-алкенил-, -C(=O)-C2-C6-алкинил-, -C(=O)-O-, -C(=O)NR8-C0-C2-алкил-, -C(=NR8)NR9-S(=O)-C0-C2-алкил-, -S(=O)2-C0-C2-алкил-, -S(=O)2NR8-C0-C2-алкил-, C(=NR8)-C0-C2-алкил-, -C(=NOR8)-C0-C2-алкил- или -C(=NOR8)NR9-C0-C2-алкил-; где R8 и R9 независимо принимают значения, определенные выше;
любой N может представлять собой N-оксид.
Настоящее изобретение включает все возможные стереоизомеры, а также не только рацематы, но и отдельные энантиомеры соединений.
Другими предпочтительными соединениями настоящего изобретения являются соединения формулы I-D
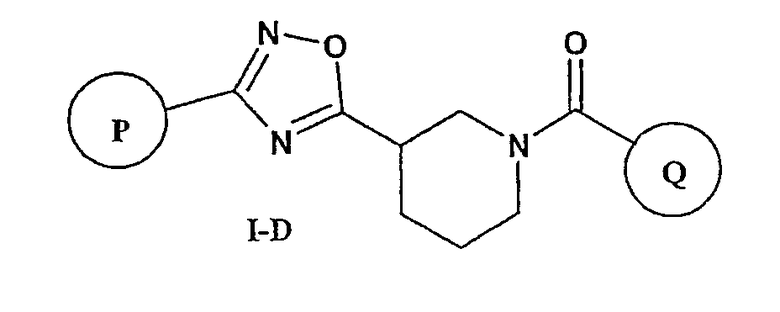
или фармацевтически приемлемые соли, гидраты или сольваты таких соединений,
где
P и Q, каждый независимо, представляет собой циклоалкильную, арильную или гетероарильную группу формулы

R3, R4, R5, R6 и R7 независимо представляют собой водород, галоген, -CN, нитро, C1-C6-алкил, C3-C6-циклоалкил, C3-C7-циклоалкилалкил, C2-C6-алкенил, C2-C6-алкинил, галоген-C1-C6-алкил, -гетероарил, гетероарилалкил, арилалкил, арил, -OR8, -NR8R9, -C(=NR10)NR8R9, N(=NR10)NR8R9, -NR8COR9, NR8CO2R9, NR8SO2R9, -NR10CONR8R9, -SR8, -S(=O)R8, -S(=O)2R8, -S(=O)2NR8R9, -C(=O)R8, -C(=O)2R8, -C(=O)NR8R9, -C(=NR8)R9 или C(=NOR8)R9 заместители; где необязательно два заместителя вместе с находящимися между ними атомами образуют бициклическое гетероциклоалкильное, арильное или гетероарильное кольцо; где каждое кольцо является необязательно дополнительно замещенным 1-5 группами, независимо выбранными из галогена, -CN, C1-C6-алкила, -O(C0-C6-алкила), -O(C3-C7-циклоалкилалкила), -O(арила), -O(гетероарила), -O(C1-C3-алкиларила), -O(C1-C3-алкилгетероарила), -N(C0-C6-алкил)(C0-C3-алкиларила) или -N(C0-C6-алкил)(C0-C3-алкилгетероарила);
R8, R9, R10, каждый независимо, представляет собой водород, C1-С6-алкил, C3-C6-циклоалкил, C3-C7-циклоалкилалкил, C2-C6-алкенил, C2-C6-алкинил, галоген-C1-C6-алкил, гетероциклоалкил, гетероарил, гетероарилалкил, арилалкил или арил, каждый из которых является необязательно замещенным 1-5 заместителями, независимо выбранными из галогена, -CN, C1-C6-алкила, -O(C0-C6-алкила), -O(C3-C7-циклоалкилалкила), -O(арила), -O(гетероарила), -N(C0-C6-алкил)(C0-C6-алкила), -N(C0-C6-алкил)(C3-C7-циклоалкила) или -N(C0-C6-алкил)(арила);
D, E, F, G и H независимо представляют собой -C(R3)=, -C(R3)=C(R4)-, -C(=O)-, -C(=S)-, -O-, -N=, -N(R3)- или -S-;
любой N может представлять собой N-оксид.
Настоящее изобретение включает все возможные стереоизомеры, а также не только рацематы, но и отдельные энантиомеры соединений.
В соответствии с другим аспектом соединение настоящего изобретения представляет собой соединение формулы (I-E) или его фармацевтически приемлемую соль
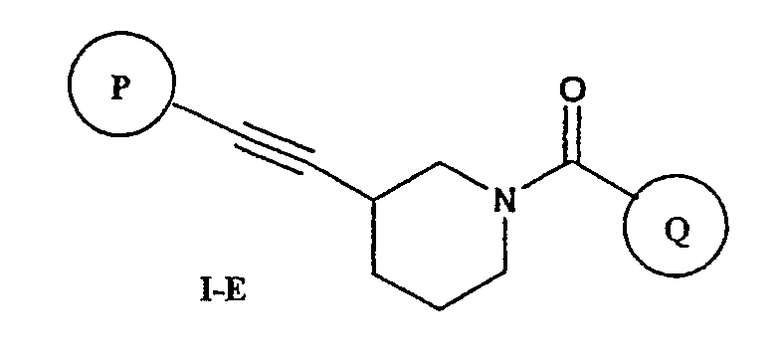
где
P и Q, каждый независимо, представляет собой циклоалкильную, арильную или гетероарильную группу формулы
R3, R4, R5, R6 и R7 независимо представляют собой водород, галоген, -CN, нитро, C1-C6-алкил, C3-C6-циклоалкил, C3-C7-циклоалкилалкил, C2-C6-алкенил, C2-C6-алкинил, галоген-C1-C6-алкил, -гетероарил, гетероарилалкил, арилалкил, арил, -OR8, -NR8R9, -C(=NR10)NR8R9, N(=NR10)NR8R9, -NR8COR9, NR8CO2R9, NR8SO2R9, -NR10CONR8R9, -SR8, -S(=O)R8, -S(=O)2R8, -S(=O)2NR8R9, -C(=O)R8, -C(=O)2R8, -C(=O)NR8R9, -C(=NR8)R9 или C(=NOR8)R9 заместители; где необязательно два заместителя вместе с находящимися между ними атомами образуют бициклическое гетероциклоалкильное, арильное или гетероарильное кольцо; где каждое кольцо является необязательно дополнительно замещенным 1-5 группами, независимо выбранными из галогена, -CN, C1-C6-алкила, -O(C0-C6-алкила), -O(C3-C7-циклоалкилалкила), -O(арила), -O(гетероарила), -O(C1-C3-алкиларила), -O(C1-C3-алкилгетероарила), -N(C0-C6-алкил)(C0-C3-алкиларила) или -N(C0-C6-алкил)(C0-C3-алкилгетероарила);
R8, R9, R10, каждый независимо, представляет собой водород, C1-C6-алкил, C3-C6-циклоалкил, C3-C7-циклоалкилалкил, C2-C6-алкенил, C2-C6-алкинил, галоген-C1-C6-алкил, гетероциклоалкил, гетероарил, гетероарилалкил, арилалкил или арил, каждый из которых является необязательно замещенным 1-5 заместителями, независимо выбранными из галогена, -CN, C1-C6-алкила, -O(C0-C6-алкила), -O(C3-C7-циклоалкилалкила), -O(арила), -O(гетероарила), -N(C0-C6-алкил)(C0-C6-алкила), -N(C0-C6-алкил)(C3-C7-циклоалкила) или -N(C0-C6-алкил)(арила);
D, E, F, G и H независимо представляют собой -C(R3)=, -C(R3)=C(R4)-, -C(=O)-, -C(=S)-, -O-, -N=, -N(R3)- или -S-;
любой N может представлять собой N-оксид.
Настоящее изобретение включает все возможные стереоизомеры, а также не только рацематы, но и отдельные энантиомеры соединений.
В соответствии с дополнительным аспектом, соединение настоящего изобретения представляет собой соединение формулы (I-F) или его фармацевтически приемлемую соль
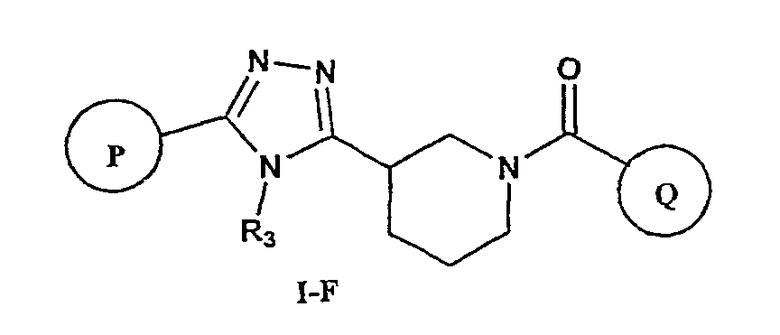
где
P и Q, каждый независимо, представляет собой циклоалкильную, арильную или гетероарильную группу формулы

R3, R4, R5, R6 и R7 независимо представляют собой водород, галоген, -CN, нитро, C1-C6-алкил, C3-C6-циклоалкил, C3-C7-циклоалкилалкил, C2-C6-алкенил, C2-C6-алкинил, галоген-C1-C6-алкил, -гетероарил, гетероарилалкил, арилалкил, арил, -OR8, -NR8R9, -C(=NR10)NR8R9, N(=NR10)NR8R9, -NR8COR9, NR8CO2R9, NR8SO2R9, -NR10CONR8R9, -SR8, -S(=O)R8, -S(=O)2R8, -S(=O)2NR8R9, -C(=O)R8, -C(=O)2R8, -C(=O)NR8R9, -C(=NR8)R9 или C(=NOR8)R9 заместители; где необязательно два заместителя вместе с находящимися между ними атомами образуют бициклическое гетероциклоалкильное, арильное или гетероарильное кольцо; где каждое кольцо является необязательно дополнительно замещенным 1-5 группами, независимо выбранными из галогена, -CN, C1-C6-алкила, -O(C0-C6-алкила), -O(C3-C7-циклоалкилалкила), -O(арила), -O(гетероарила), -O(C1-C3-алкиларила), -O(C1-C3-алкилгетероарила), -N(C0-C6-алкил)(C0-C3-алкиларила) или -N(C0-C6-алкил)(C0-C3-алкилгетероарила);
R8, R9, R10, каждый независимо, представляет собой водород, C1-C6-алкил, C3-C6-циклоалкил, C3-C7-циклоалкилалкил, C2-C6-алкенил, C2-C6-алкинил, галоген-C1-C6-алкил, гетероциклоалкил, гетероарил, гетероарилалкил, арилалкил или арил, каждый из которых является необязательно замещенным 1-5 заместителями, независимо выбранными из галогена, -CN, C1-C6-алкила, -O(C0-C6-алкила), -O(C3-C7-циклоалкилалкила), -O(арила), -O(гетероарила), -N(C0-C6-алкил)(C0-C6-алкила), -N(C0-C6-алкил)(C3-C7-циклоалкила) или -N(C0-C6-алкил)(арила);
D, E, F, G и H независимо представляют собой -C(R3)=, -C(R3)=C(R4)-, -C(=O)-, -C(=S)-, -O-, -N=, -N(R3)- или -S-;
любой N может представлять собой N-оксид.
Настоящее изобретение включает все возможные стереоизомеры, а также не только рацематы, но и отдельные энантиомеры соединений.
Другим аспектом настоящего изобретения являются соединения формулы I-G
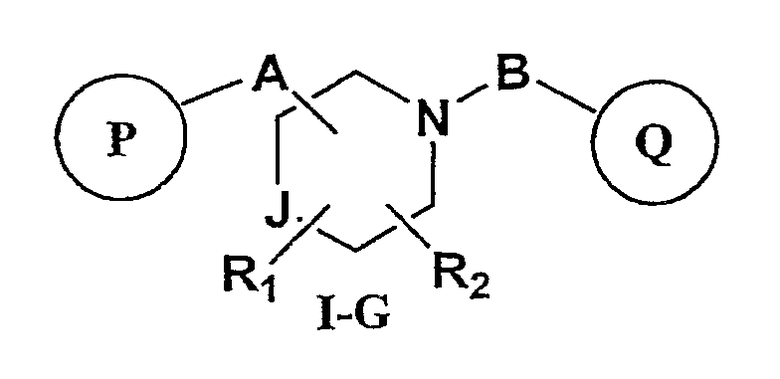
где
R1 и R2 независимо представляют собой водород, C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, арилалкил, гетероарилалкил, гидрокси, амино, аминоалкил, гидроксиалкил, C1-C6-алкокси, или R1 и R2 вместе могут образовывать C3-C7-циклоалкильное кольцо, карбонильную связь C=O или двойную углерод-углеродную связь;
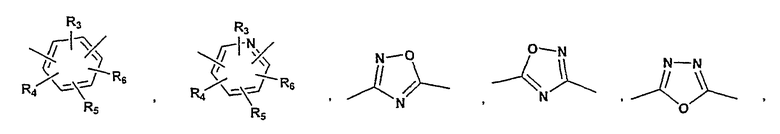
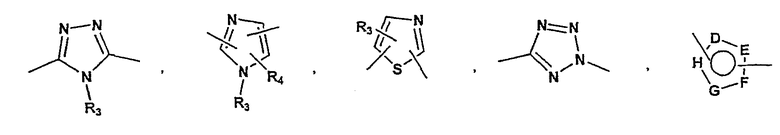
P и Q, каждый независимо, представляет собой арильную или гетероарильную группу формулы
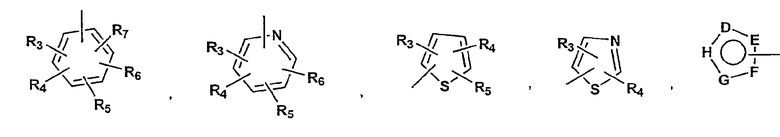
R3, R4, R5, R6 и R7 независимо представляют собой водород, галоген, -CN, нитро, C1-C6-алкил, C3-C6-циклоалкил, C3-C7-циклоалкилалкил, C1-C6-алкенил, C1-C6-алкинил, галоген-C1-C6-алкил, -гетероарил, гетероарилалкил, арилалкил, арил, -OR8, -NR8R9, -C(=NR10)NR8R9, N(=NR10)NR8R9, -NR8COR9, NR8CO2R9, NR8SO2R9, -NR10CONR8R9, -SR8, -S(=O)R8, -S(=O)2R8, -S(=O)2NR8R9, -C(=O)R8, -C(=O)2R8, -C(=O)NR8R9, -C(=NR8)R9 или C(=NOR8)R9 заместители; где необязательно два заместителя вместе с находящимися между ними атомами образуют бициклическое арильное или гетероарильное кольцо; где каждое кольцо является необязательно дополнительно замещенным 1-5 группами, независимо выбранными из галогена, -CN, C1-C6-алкила, -O(C0-C6-алкила), -O(C3-C7-циклоалкилалкила), -O(арила), -O(гетероарила), -O(C1-C3-алкиларила), -O(C1-C3-алкилгетероарила), -N(C0-C6-алкил)(C0-C3-алкиларила) или -N(C0-C6-алкил)(C0-C3-алкилгетероарила);
R8, R9, R10, каждый независимо, представляет собой водород, C1-C6-алкил, C3-C6-циклоалкил, C3-C7-циклоалкилалкил, C2-C6-алкенил, C2-C6-алкинил, галоген-C1-C6-алкил, гетероарил, гетероарилалкил, арилалкил или арил, каждый из которых является необязательно замещенным 1-5 заместителями, независимо выбранными из галогена, -CN, C1-C6-алкила, -O(C0-C6-алкила), -O(C3-C7-циклоалкилалкила), -O(арила), -O(гетероарила), -N(C0-C6-алкил)(C0-C6-алкила), -N(C0-C6-алкил)(C3-C7-циклоалкила) или -N(C0-C6-алкил)(арила);
D, E, F, G и H независимо представляют собой -C(R3)=, -C(R3)=C(R4)-, -C(=O)-, -C(=S)-, -O-, -N=, -N(R3)- или -S-;
A представляет собой азо-N=N-, этил, этенил, этинил, -NR8C(=O)-, NR8S(=O)2-, -C(=O)NR8-, -S-, -S(=O)-, -S(=O)2-, -S(=O)2NR8-, -C(=O)-O-, -O-C(=O)-, -C(=NR8)NR9-, C(=NOR8)NR9-, -NR8C(=NOR9)-, =N-O-, -O-N=CH- или арильную или гетероарильную группу формулы
R3, R4, R5 и R6 независимо принимают значения, определенные выше;
D, E, F, G и H независимо принимают значения, определенные выше в A;
B представляет собой одинарную связь, -C(=O)-C0-C2-алкил-, -C(=O)-C2-C6-алкенил-, -C(=O)-C2-C6-алкинил-, -C(=O)-O-, -C(=O)NR8-C0-C2-алкил-, -C(=NR8)NR9-S(=O)-C0-C2-алкил-, -S(=O)2-C0-C2-алкил-, -S(=O)2NR8-C0-C2-алкил-, C(=NR8)-C0-C2-алкил-, -C(=NOR8)-C0-C2-алкил- или -C(=NOR8)NR9-C0-C2-алкил-; где R8 и R9, независимо, принимают значения, определенные выше;
J представляет собой -C(R11, R12), -O-, -N(R11)- или -S-;
R11, R12 независимо представляют собой водород, C1-C6-алкил, C3-C6-циклоалкил, C3-C7-циклоалкилалкил, C2-C6-алкенил, C2-C6-алкинил, галоген-C1-C6-алкил, гетероарил, гетероарилалкил, арилалкил или арил, каждый из которых является необязательно замещенным 1-5 заместителями, независимо выбранными из галогена, -CN, C1-C6-алкила, -O(C0-C6-алкила), -O(C3-C7-циклоалкилалкила), -O(арила), -O(гетероарила), -N(C0-C6-алкил)(C0-C6-алкила), -N(C0-C6-алкил)(C3-C7-циклоалкила) или -N(C0-C6-алкил)(арила);
любой N может представлять собой N-оксид.
Настоящее изобретения включает все возможные стереоизомеры, а также не только рацематы, но и отдельные энантиомеры соединений.
Один вариант осуществления настоящего изобретения включает соединения формулы I-H
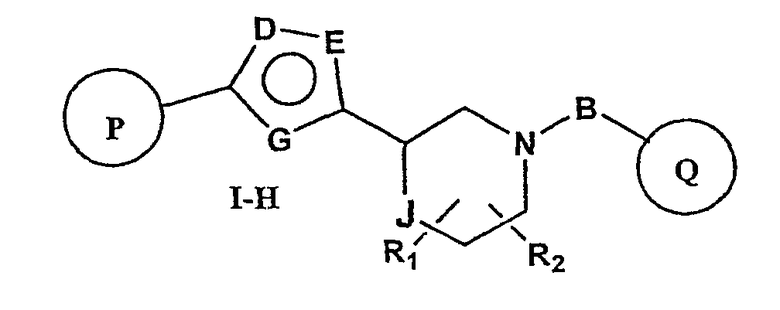
где
R1 и R2 независимо представляют собой водород, C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, арилалкил, гетероарилалкил, гидрокси, амино, аминоалкил, гидроксиалкил, C1-C6-алкокси, или R1 и R2 вместе могут образовывать C3-C7-циклоалкильное кольцо, карбонильную связь C=O или двойную углерод-углеродную связь;
Р и Q, каждый независимо, представляет собой арильную или гетероарильную группу формулы
R3, R4, R5, R6 и R7 независимо представляют собой водород, галоген, -CN, нитро, C1-C6-алкил, C3-C6-циклоалкил, C3-C7-циклоалкилалкил, C2-C6-алкенил, C2-C6-алкинил, галоген-C1-C6-алкил, гетероарил, гетероарилалкил, арилалкил, арил, -OR8, -NR8R9, -C(=NR10)NR8R9, N(=NR10)NR8R9, -NR8COR9, NR8CO2R9, NR8SO2R9, -NR10CONR8R9, -SR8, -S(=O)R8, -S(=O)2R8, -S(=O)2NR8R9, -C(=O)R8, -C(=O)2R8, -C(=O)NR8R9, -C(=NR8)R9 или C(=NOR8)R9 заместители; где необязательно два заместителя вместе с находящимися между ними атомами образуют бициклическое арильное или гетероарильное кольцо; где каждое кольцо является необязательно дополнительно замещенным 1-5 группами, независимо выбранными из галогена, -CN, C1-C6-алкила, -O(C0-C6-алкила), -O(C3-C7-циклоалкилалкила), -O(арила), -O(гетероарила), -O(C1-C3-алкиларила), -O(C1-C3-алкилгетероарила), -N(C0-C6-алкил)(C0-C3-алкиларила) или -N(C0-C6-алкил)(C0-C3-алкилгетероарила);
R8, R9, R10, каждый независимо, представляет собой водород, C1-С6-алкил, C3-C6-циклоалкил, C3-C7-циклоалкилалкил, C2-C6-алкенил, C2-C6-алкинил, галоген-C1-C6-алкил, гетероарил, гетероарилалкил, арилалкил или арил, каждый из которых является необязательно замещенным 1-5 заместителями, независимо выбранными из галогена, -CN, C1-C6-алкила, -O(C0-C6-алкила), -O(C3-C7-циклоалкилалкила), -O(арила), -O(гетероарила), -N(C0-C6-алкил)(C0-C6-алкила), -N(C0-C6-алкил)(C3-C7-циклоалкила) или -N(C0-C6-алкил)(арила);
D, E, F, G и H в P и Q независимо представляют собой -C(R3)=, -C(R3)=C(R4)-, -C(=O)-, -C(=S)-, -O-, -N=, -N(R3)- или -S-;
D, E и G в A независимо принимают значения, определенные выше для А;
B представляет собой одинарную связь, -C(=О)-C0-C2-алкил-, -C(=О)-C2-C6-алкенил-, -C(=O)-C2-C6-алкинил-, -C(=O)-O-, -C(=O)NR8-C0-C2-алкил-, -C(=NR8)NR9-S(=O)-C0-C2-алкил-, -S(=O)2-C0-C2-алкил-, -S(=O)2NR8-C0-C2-алкил-, C(=NR8)-C0-C2-алкил-, -C(=NOR8)-C0-C2-алкил- или -C(=NOR8)NR9-C0-C2-алкил-; где R8 и R9, независимо, принимают значения, определенные выше;
J представляет собой -C(R11, R12), -O-, -N(R11)- или -S-;
R11, R12 независимо представляют собой водород, C1-C6-алкил, C3-C6-циклоалкил, C3-C7-циклоалкилалкил, C2-C6-алкенил, C2-C6-алкинил, галоген-C1-C6-алкил, гетероарил, гетероарилалкил, арилалкил или арил, каждый из которых является необязательно замещенным 1-5 заместителями, независимо выбранными из галогена, -CN, C1-C6-алкила, -O(C0-C6-алкила), -O(C3-C7-циклоалкилалкила), -O(арила), -O(гетероарила), -N(C0-C6-алкил)(C0-C6-алкила), -N(C0-C6-алкил)(C3-C7-циклоалкила) или -N(C0-C6-алкил)(арила);
любой N может представлять собой N-оксид.
Настоящее изобретение включает все возможные стереоизомеры, а также не только рацематы, но и отдельные энантиомеры соединений.
Другой вариант осуществления настоящего изобретения включает соединения формулы I-I
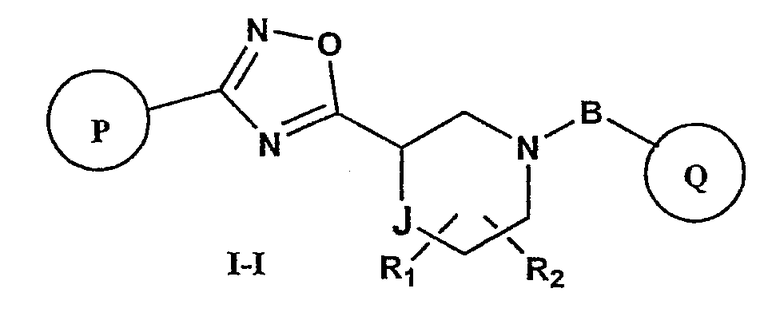
где
R1 и R2 независимо представляют собой водород, C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, арилалкил, гетероарилалкил, гидрокси, гидроксиалкил, C1-C6-алкокси, или R1 и R2 вместе могут образовывать карбонильную связь C=O или двойную углерод-углеродную связь;
Р и Q, каждый независимо, представляет собой арильную или гетероарильную группу формулы
R3, R4, R5, R6 и R7 независимо представляют собой водород, галоген, -CN, нитро, C1-C6-алкил, C3-C6-циклоалкил, C3-C7-циклоалкилалкил, C2-C6-алкенил, C2-C6-алкинил, галоген-C1-C6-алкил, гетероарил, гетероарилалкил, арилалкил, арил, -OR8, -NR8R9, -C(=NR10)NR8R9, N(=NR10)NR8R9, -NR8COR9, -NR8CO2R9, NR8SO2R9, -NR10CONR8R9, -SR8, -S(=O)R8, -S(=O)2R8, -S(=O)2NR8R9, -C(=O)R8, -C(=O)2R8, -C(=O)NR8R9, -C(=NR8)R9 или C(=NOR8)R9 заместители; где необязательно два заместителя вместе с находящимися между ними атомами образуют бициклическое арильное или гетероарильное кольцо; где каждое кольцо является необязательно дополнительно замещенным 1-5 группами, независимо выбранными из галогена, -CN, C1-C6-алкила, -O(C0-C6-алкила), -O(C3-C7-циклоалкилалкила), -O(арила), -O(гетероарила), -O(C1-C3-алкиларила), -O(C1-C3-алкилгетероарила), -N(C0-C6-алкил)(C0-C3-алкиларила) или -N(C0-C6-алкил)(C0-C3-алкилгетероарила);
R8, R9, R10, каждый независимо, представляет собой водород, C1-C6-алкил, C3-C6-циклоалкил, C3-C7-циклоалкилалкил, C2-C6-алкенил, C2-C6-алкинил, галоген-C1-C6-алкил, гетероарил, гетероарилалкил, арилалкил или арил, каждый из которых является необязательно замещенным 1-5 заместителями, независимо выбранными из галогена, -CN, C1-C6-алкила, -O(C0-C6-алкила), -O(C3-C7-циклоалкилалкила), -O(арила), -O(гетероарила), -N(C0-C6-алкил)(C0-C6-алкила), -N(C0-C6-алкил)(C3-C7-циклоалкила) или -N(C0-C6-алкил)(арила);
D, E, F, G и H независимо представляют собой -C(R3)=, -C(R3)=C(R4)-, -C(=O)-, -C(=S)-, -O-, -N=, -N(R3)- или -S-;
B представляет собой одинарную связь, -C(=О)-C0-C2-алкил-, -C(=О)-C2-C6-алкенил-, -C(=O)-C2-C6-алкинил-, -C(=O)-O-, -C(=O)NR8-C0-C2-алкил-, -C(=NR8)NR9-S(=O)-C0-C2-алкил-, -S(=O)2-C0-C2-алкил-, -S(=O)2NR8-C0-C2-алкил-, C(=NR8)-C0-C2-алкил-, -C(=NOR8)-C0-C2-алкил- или -C(=NOR8)NR9-C0-C2-алкил-; где R8 и R9 независимо принимают значения, определенные выше;
J представляет собой -C(R11, R12), -O-, -N(R11)- или -S-;
R11, R12 независимо представляют собой водород, C1-C6-алкил, C3-C6-циклоалкил, C3-C7-циклоалкилалкил, C2-C6-алкенил, C2-C6-алкинил, галоген-C1-C6-алкил, гетероарил, гетероарилалкил, арилалкил или арил, каждый из которых является необязательно замещенным 1-5 заместителями, независимо выбранными из галогена, -CN, C1-C6-алкила, -O(C0-C6-алкила), -O(C3-C7-циклоалкилалкила), -O(арила), -O(гетероарила), -N(C0-C6-алкил)(C0-C6-алкила), -N(C0-C6-алкил)(C3-C7-циклоалкила) или -N(C0-C6-алкил)(арила);
любой N может представлять собой N-оксид.
Настоящее изобретение включает все возможные стереоизомеры, а также не только рацематы, но и отдельные энантиомеры соединений.
Вариант осуществления настоящего изобретения включает соединения формулы I-J
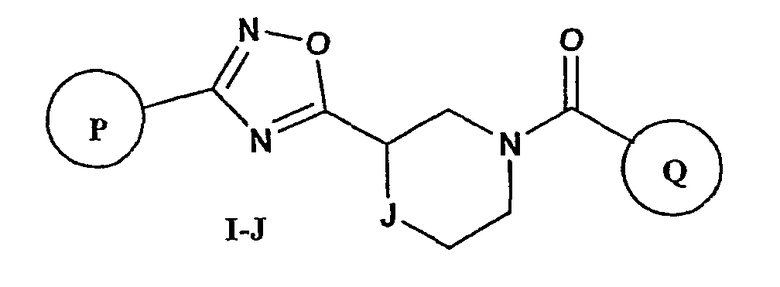
где
Р и Q, каждый независимо, представляет собой арильную или гетероарильную группу формулы
R3, R4, R5, R6 и R7 независимо представляют собой водород, галоген, -CN, нитро, C1-C6-алкил, C3-C6-циклоалкил, C3-C7-циклоалкилалкил, C2-C6-алкенил, C2-C6-алкинил, галоген-C1-C6-алкил, гетероарил, гетероарилалкил, арилалкил, арил, -OR8, -NR8R9, -C(=NR10)NR8R9, N(=NR10)NR8R9, -NR8COR9, NR8CO2R9, NR8SO2R9, -NR10CONR8R9, -SR8, -S(=O)R8, -S(=O)2R8, -S(=O)2NR8R9, -C(=O)R8, -C(=O)2R8, -C(=O)NR8R9, -C(=NR8)R9 или C(=NOR8)R9 заместители; где необязательно два заместителя вместе с находящимися между ними атомами образуют бициклическое арильное или гетероарильное кольцо; где каждое кольцо является необязательно дополнительно замещенным 1-5 группами, независимо выбранными из галогена, -CN, C1-C6-алкила, -O(C0-C6-алкила), -O(C3-C7-циклоалкилалкила), -O(арила), -O(гетероарила), -O(C1-C3-алкиларила), -O(C1-C3-алкилгетероарила), -N(C0-C6-алкил)(C0-C3-алкиларила) или -N(C0-C6-алкил)(C0-C3-алкилгетероарила);
R8, R9, R10, каждый независимо, представляет собой водород, C1-C6-алкил, C3-C6-циклоалкил, C3-C7-циклоалкилалкил, C2-C6-алкенил, C2-C6-алкинил, галоген-C1-C6-алкил, гетероарил, гетероарилалкил, арилалкил или арил, каждый из которых является необязательно замещенным 1-5 заместителями, независимо выбранными из галогена, -CN, C1-C6-алкила, -O(C0-C6-алкила), -O(C3-C7-циклоалкилалкила), -O(арила), -O(гетероарила), -N(C0-C6-алкил)(C0-C6-алкила), -N(C0-C6-алкил)(C3-C7-циклоалкила) или -N(C0-C6-алкил)(арила);
D, E, F, G и H независимо представляют собой -C(R3)=, -C(R3)=C(R4)-, -C(=O)-, -C(=S)-, -O-, -N=, -N(R3)- или -S-;
J представляет собой -C(R11, R12), -O-, -N(R11)- или -S-;
R11, R12 независимо представляют собой водород, C1-C6-алкил, C3-C6-циклоалкил, C3-C7-циклоалкилалкил, C2-C6-алкенил, C2-C6-алкинил, галоген-C1-C6-алкил, гетероарил, гетероарилалкил, арилалкил или арил, каждый из которых является необязательно замещенным 1-5 заместителями, независимо выбранными из галогена, -CN, C1-C6-алкила, -O(C0-C6-алкила), -O(C3-C7-циклоалкилалкила), -O(арила), -O(гетероарила), -N(C0-C6-алкил)(C0-C6-алкила), -N(C0-C6-алкил)(C3-C7-циклоалкила) или -N(C0-C6-алкил)(арила);
любой N может представлять собой N-оксид.
Настоящее изобретение включает все возможные стереоизомеры, а также не только рацематы, но и отдельные энантиомеры соединений.
Еще один вариант осуществления настоящего изобретения включает соединения формулы I-K
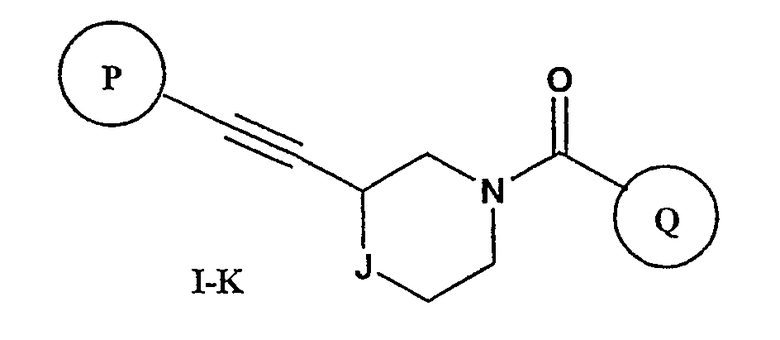
где
Р и Q, каждый независимо, представляют собой арильную или гетероарильную группу формулы
где
R3, R4, R5, R6 и R7 независимо представляют собой водород, галоген, -CN, нитро, C1-C6-алкил, C3-C6-циклоалкил, C3-C7-циклоалкилалкил, C2-C6-алкенил, C2-C6-алкинил, галоген-C1-C6-алкил, гетероарил, гетероарилалкил, арилалкил, арил, -OR8, -NR8R9, -C(=NR10)NR8R9, N(=NR10)NR8R9, -NR8COR9, NR8CO2R9, NR8SO2R9, -NRl0CONR8R9, -SR8, -S(=O)R8, -S(=O)2R8, -S(=O)2NR8R9, -C(=O)R8, -C(=O)2R8, -C(=O)NR8R9, -C(=NR8)R9 или C(=NOR8)R9 заместители; где необязательно два заместителя вместе с находящимися между ними атомами образуют бициклическое арильное или гетероарильное кольцо; где каждое кольцо является необязательно дополнительно замещенным 1-5 группами, независимо выбранными из галогена, -CN, C1-C6-алкила, -O(C0-C6-алкила), -O(C3-C7-циклоалкилалкила), -O(арила), -O(гетероарила), -O(C1-C3-алкиларила), -O(C1-C3-алкилгетероарила), -N(C0-C6-алкил)(C0-C3-алкиларила) или -N(C0-C6-алкил)(C0-C3-алкилгетероарила);
R8, R9, R10, каждый независимо, представляет собой водород, C1-C6-алкил, C3-C6-циклоалкил, C3-C7-циклоалкилалкил, C2-C6-алкенил, C2-C6-алкинил, галоген-C1-C6-алкил, гетероарил, гетероарилалкил, арилалкил или арил, каждый из которых является необязательно замещенным 1-5 заместителями, независимо выбранными из галогена, -CN, C1-C6-алкила, -O(C0-C6-алкила), -O(C3-C7-циклоалкилалкила), -O(арила), -O(гетероарила), -N(C0-C6-алкил)(C0-C6-алкила), -N(C0-C6-алкил)(C3-C7-циклоалкила) или -N(C0-C6-алкил)(арила);
D, E, F, G и H независимо представляют собой -C(R3)=, -C(R3)=C(R4)-, -C(=O)-, -C(=S)-, -O-, -N=, -N(R3)- или -S-;
J представляет собой -C(R11, R12), -O-, -N(R11)- или -S-;
R11, R12 независимо представляют собой водород, C1-C6-алкил, C3-C6-циклоалкил, C3-C7-циклоалкилалкил, C2-C6-алкенил, C2-C6-алкинил, галоген-C1-C6-алкил, гетероарил, гетероарилалкил, арилалкил или арил, каждый из которых является необязательно замещенным 1-5 заместителями, независимо выбранными из галогена, -CN, C1-C6-алкила, -O(C0-C6-алкила), -O(C3-C7-циклоалкилалкила), -O(арила), -O(гетероарила), -N(C0-C6-алкил)(C0-C6-алкила), -N(C0-C6-алкил)(C3-C7-циклоалкила) или -N(C0-C6-алкил)(арила);
любой N может представлять собой N-оксид.
Настоящее изобретение включает все возможные стереоизомеры, а также не только рацематы, но и отдельные энантиомеры соединений.
Еще один вариант осуществления настоящего изобретения включает соединения формулы I-L
где
Р и Q, каждый независимо, представляет собой арильную или гетероарильную группу формулы
R3, R4, R5, R6 и R7 независимо представляют собой водород, галоген, -CN, нитро, C1-C6-алкил, C3-C6-циклоалкил, C3-C7-циклоалкилалкил, C2-C6-алкенил, C2-C6-алкинил, галоген-C1-C6-алкил, гетероарил, гетероарилалкил, арилалкил, арил, -OR8, -NR8R9, -C(=NR10)NR8R9, N(=NR10)NR8R9, -NR8COR9, NR8CO2R9, NR8SO2R9, -NRl0CONR8R9, -SR8, -S(=O)R8, -S(=O)2R8, -S(=O)2NR8R9, -C(=O)R8, -C(=O)2R8, -C(=O)NR8R9, -C(=NR8)R9 или C(=NOR8)R9 заместители; где необязательно два заместителя вместе с находящимися между ними атомами образуют бициклическое арильное или гетероарильное кольцо; где каждое кольцо является необязательно дополнительно замещенным 1-5 группами, независимо выбранными из галогена, -CN, C1-C6-алкила, -O(C0-C6-алкила), -O(C3-C7-циклоалкилалкила), -O(арила), -O(гетероарила), -O(C1-C3-алкиларила), -O(C1-C3-алкилгетероарила), -N(C0-C6-алкил)(C0-C3-алкиларила) или -N(C0-C6-алкил)(C0-C3-алкилгетероарила);
R8, R9, R10, каждый независимо, представляет собой водород, C1-С6-алкил, C3-C6-циклоалкил, C3-C7-циклоалкилалкил, C2-C6-алкенил, C2-C6-алкинил, галоген-C1-C6-алкил, гетероарил, гетероарилалкил, арилалкил или арил, каждый из которых является необязательно замещенным 1-5 заместителями, независимо выбранными из галогена, -CN, C1-C6-алкила, -O(C0-C6-алкила), -O(C3-C7-циклоалкилалкила), -O(арила), -O(гетероарила), -N(C0-C6-алкил)(C0-C6-алкила), -N(C0-C6-алкил)(C3-C7-циклоалкила) или -N(C0-C6-алкил)(арила);
D, E, F, G и H независимо представляют собой -C(R3)=, -C(R3)=C(R4)-, -C(=O)-, -C(=S)-, -O-, -N=, -N(R3)- или -S-;
J представляет собой -C(R11, R12), -O-, -N(R11)- или -S-;
R11, R12 независимо представляют собой водород, C1-C6-алкил, C3-C6-циклоалкил, C3-C7-циклоалкилалкил, C2-C6-алкенил, C2-C6-алкинил, галоген-C1-C6-алкил, гетероарил, гетероарилалкил, арилалкил или арил, каждый из которых является необязательно замещенным 1-5 заместителями, независимо выбранными из галогена, -CN, C1-C6-алкила, -O(C0-C6-алкила), -O(C3-C7-циклоалкилалкила), -O(арила), -O(гетероарила), -N(C0-C6-алкил)(C0-C6-алкила), -N(C0-C6-алкил)(C3-C7-циклоалкила) или -N(C0-C6-алкил)(арила);
любой N может представлять собой N-оксид.
Настоящее изобретение включает все возможные стереоизомеры, а также не только рацематы, но и отдельные энантиомеры соединений.
Еще один вариант осуществления настоящего изобретения включает соединения формулы I-M
где
Р и Q, каждый независимо, представляет собой арильную или гетероарильную группу формулы
R3, R4, R5, R6 и R7 независимо представляют собой водород, галоген, -CN, нитро, C1-C6-алкил, C3-C6-циклоалкил, C3-C7-циклоалкилалкил, C2-C6-алкенил, C2-C6-алкинил, галоген-C1-C6-алкил, гетероарил, гетероарилалкил, арилалкил, арил, -OR8, -NR8R9, -C(=NR10)NR8R9, N(=NR10)NR8R9, -NR8COR9, NR8CO2R9, NR8SO2R9, -NRl0CONR8R9, -SR8, -S(=O)R8, -S(=O)2R8, -S(=O)2NR8R9, -C(=O)R8, -C(=O)2R8, -C(=O)NR8R9, -C(=NR8)R9 или C(=NOR8)R9 заместители; где необязательно два заместителя вместе с находящимися между ними атомами образуют бициклическое арильное или гетероарильное кольцо; где каждое кольцо является необязательно дополнительно замещенным 1-5 группами, независимо выбранными из галогена, -CN, C1-C6-алкила, -O(C0-C6-алкила), -O(C3-C7-циклоалкилалкила), -O(арила), -O(гетероарила), -O(C1-C3-алкиларила), -O(C1-C3-алкилгетероарила), -N(C0-C6-алкил)(C0-C3-алкиларила) или -N(C0-C6-алкил)(C0-C3-алкилгетероарила);
R8, R9, R10, каждый независимо, представляет собой водород, C1-C6-алкил, C3-C6-циклоалкил, C3-C7-циклоалкилалкил, C2-C6-алкенил, C2-C6-алкинил, галоген-C1-C6-алкил, гетероарил, гетероарилалкил, арилалкил или арил, каждый из которых является необязательно замещенным 1-5 заместителями, независимо выбранными из галогена, -CN, C1-C6-алкила, -O(C0-C6-алкила), -O(C3-C7-циклоалкилалкила), -O(арила), -O(гетероарила), -N(C0-C6-алкил)(C0-C6-алкила), -N(C0-C6-алкил)(C3-C7-циклоалкила) или -N(C0-C6-алкил)(арила);
D, E, F, G и H независимо представляют собой -C(R3)=, -C(R3)=C(R4)-, -C(=O)-, -C(=S)-, -O-, -N=, -N(R3)- или -S-;
J представляет собой -C(R11, R12), -O-, -N(R11)- или -S-;
R11, R12 независимо представляют собой водород, C1-C6-алкил, C3-C6-циклоалкил, C3-C7-циклоалкилалкил, C2-C6-алкенил, C2-C6-алкинил, галоген-C1-C6-алкил, гетероарил, гетероарилалкил, арилалкил или арил, каждый из которых является необязательно замещенным 1-5 заместителями, независимо выбранными из галогена, -CN, C1-C6-алкила, -O(C0-C6-алкила), -O(C3-C7-циклоалкилалкила), -O(арила), -O(гетероарила), -N(C0-C6-алкил)(C0-C6-алкила), -N(C0-C6-алкил)(C3-C7-циклоалкила) или -N(C0-C6-алкил)(арила);
любой N может представлять собой N-оксид.
Настоящее изобретение включает все возможные стереоизомеры, а также не только рацематы, но и отдельные энантиомеры соединений.
Еще один вариант осуществления настоящего изобретения включает соединения формулы I-N
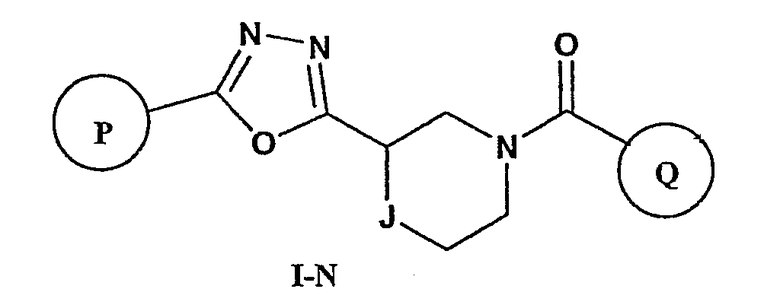
где
Р и Q, каждый независимо, представляет собой арильную или гетероарильную группу формулы
R3, R4, R5, R6 и R7 независимо представляют собой водород, галоген, -CN, нитро, C1-C6-алкил, C3-C6-циклоалкил, C3-C7-циклоалкилалкил, C2-C6-алкенил, C2-C6-алкинил, галоген-C1-C6-алкил, гетероарил, гетероарилалкил, арилалкил, арил, -OR8, -NR8R9, -C(=NR10)NR8R9, N(=NR10)NR8R9, -NR8COR9, NR8CO2R9, NR8SO2R9, -NRl0CONR8R9, -SR8, -S(=O)R8, -S(=O)2R8, -S(=O)2NR8R9, -C(=O)R8, -C(=O)2R8, -C(=O)NR8R9, -C(=NR8)R9 или C(=NOR8)R9 заместители, где необязательно два заместителя вместе с находящимися между ними атомами образуют бициклическое арильное или гетероарильное кольцо; где каждое кольцо является необязательно дополнительно замещенным 1-5 группами, независимо выбранными из галогена, -CN, C1-C6-алкила, -O(C0-C6-алкила), -O(C3-C7-циклоалкилалкила), -O(арила), -O(гетероарила), -O(C1-C3-алкиларила), -O(C1-C3-алкилгетероарила), -N(C0-C6-алкил)(C0-C3-алкиларила) или -N(C0-C6-алкил)(C0-C3-алкилгетероарила);
R8, R9, R10, каждый независимо, представляет собой водород, C1-C6-алкил, C3-C6-циклоалкил, C3-C7-циклоалкилалкил, C2-C6-алкенил, C2-C6-алкинил, галоген-C1-C6-алкил, гетероарил, гетероарилалкил, арилалкил или арил, каждый из которых является необязательно замещенным 1-5 заместителями, независимо выбранными из галогена, -CN, C1-C6-алкила, -O(C0-C6-алкила), -O(C3-C7-циклоалкилалкила), -O(арила), -O(гетероарила), -N(C0-C6-алкил)(C0-C6-алкила), -N(C0-C6-алкил)(C3-C7-циклоалкила) или -N(C0-C6-алкил)(арила);
D, E, F, G и H независимо представляют собой -C(R3)=, -C(R3)=C(R4)-, -C(=O)-, -C(=S)-, -O-, -N=, -N(R3)- или -S-;
J представляет собой -C(R11, R12), -O-, -N(R11)- или -S-;
R11, R12 независимо представляют собой водород, C1-C6-алкил, C3-C6-циклоалкил, C3-C7-циклоалкилалкил, C2-C6-алкенил, C2-C6-алкинил, галоген-C1-C6-алкил, гетероарил, гетероарилалкил, арилалкил или арил, каждый из которых является необязательно замещенным 1-5 заместителями, независимо выбранными из галогена, -CN, C1-C6-алкила, -O(C0-C6-алкила), -O(C3-C7-циклоалкилалкила), -O(арила), -O(гетероарила), -N(C0-C6-алкил)(C0-C6-алкила), -N(C0-C6-алкил)(C3-C7-циклоалкила) или -N(C0-C6-алкил)(арила);
любой N может представлять собой N-оксид.
Настоящее изобретение включает все возможные стереоизомеры, а также не только рацематы, но и отдельные энантиомеры соединений.
Конкретными предпочтительными соединениями являются:
(4-фторфенил)-[3-(4-фторфенилэтинил)пиперидин-1-ил]метанон;
(4-фторфенил)-{3-[5-(4-фторфенил)-4H-[1,2,4]триазол-3-ил]пиперидин-1-ил}метанон;
(S)-(4-фторфенил)-{3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон;
(S)-(тиофен-2-ил)-{3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон;
{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}-(4-метил-2-пиразин-2-илтиазол-5-ил)метанон;
(2,4-дифторфенил)-{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон;
{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}-(3,4,5-трифторфенил)метанон;
{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}-(5-пиридин-2-илтиофен-2-ил)метанон;
циклопентил-{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон;
(3,4-дифторфенил)-{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон;
бензотиазол-6-ил-{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон;
(3,5-диметилизоксазол-4-ил)-{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон;
(4-фторфенил)-{(S)-3-[3-(2,4,6-трифторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон;
(4-фторфенил)-[(S)-3-(3-пиридин-3-ил-[1,2,4]оксадиазол-5-ил)пиперидин-1-ил]метанон;
(4-фторфенил)-[(S)-3-(3-пиридин-4-ил-[1,2,4]оксадиазол-5-ил)пиперидин-1-ил]метанон;
{(S)-3-[3-(2,4-дифторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}-(4-фторфенил)метанон;
(4-фторфенил)-[(S)-3-(3-п-толил-[1,2,4]оксадиазол-5-ил)пиперидин-1-ил]метанон;
(4-фторфенил)-{(S)-3-[3-(2-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон;
(4-фторфенил)-[(S)-3-(3-пиридин-2-ил-[1,2,4]оксадиазол-5-ил)пиперидин-1-ил]метанон;
(4-фторфенил)-{3-[5-(4-фторфенил)-[1,3,4]оксадиазол-2-ил]пиперидин-1-ил}метанон;
(2-фторфенил)-{(S)-3-[2-(3,4-дифторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон;
(4-фторфенил)-{2-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]морфолин-4-ил}метанон;
{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}тиофен-3-илметанон;
(4-фторфенил)-[3-(5-фенилтетразол-2-ил)пиперидин-1-ил]метанон;
(4-фторфенил)-[(S)-3-(3-фенил-[1,2,4]оксадиазол-5-ил)пиперидин-1-ил]метанон;
(3,4-дифторфенил)-[(S)-3-(3-фенил-[1,2,4]оксадиазол-5-ил)пиперидин-1-ил]метанон;
{3-[3-(4-метоксифенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}фенилметанон;
{3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}фенилметанон;
(4-фторфенил)-[3-(3-фенил-[1,2,4]оксадиазол-5-ил)пиперидин-1-ил]метанон;
(3-фторфенил)-[3-(3-фенил-[1,2,4]оксадиазол-5-ил)пиперидин-1-ил]метанон;
(4-фторфенил)-{3-[3-(3-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон;
(3-фторфенил)-{3-[3-(3-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон;
(4-фторфенил)-{3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон;
(3-фторфенил)-{3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон;
(R)-(4-фторфенил)-{3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон;
(4-фторфенил)-{3-[5-(4-фторфенил)-[1,2,4]оксадиазол-3-ил]пиперидин-1-ил}метанон;
(4-фторфенил)-{3-[5-(4-фторфенил)-4-метил-4H-[1,2,4]триазол-3-ил]пиперидин-1-ил)метанон;
{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}-(2-фенилтиазол-4-ил)метанон;
{{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}-(2-метил-6-трифторметилпиридин-3-ил)метанон;
{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}-[1,2,3]тиадиазол-4-илметанон;
бензотиазол-2-ил-{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон;
{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}-(5-метилизоксазол-3-ил)метанон;
(1,5-диметил-1H-пиразол-3-ил)-{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон;
{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}-(4-трифторметилфенил)метанон;
4-{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-карбонил}бензонитрил;
{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}изоксазол-5-илметанон;
(3-хлор-4-фторфенил)-{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон;
{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}-(2-фенил-2H-пиразол-3-ил)метанон;
{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}-(5-метил-2-фенил-2H-[1,2,3]триазол-4-ил)метанон;
(4-фтор-3-метилфенил)-{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон;
(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}-(3-метилтиофен-2-ил)метанон;
{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}-(1-метил-1H-пиррол-2-ил)метанон;
{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}тиазол-2-илметанон;
{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}-(4-метилтиазол-5-ил)метанон;
{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}-(6-морфолин-4-илпиридин-3-ил)метанон;
{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}-(1Н-индол-5-ил)метанон;
2-(4-фторфенил)-1-{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}этанон;
3-(4-фторфенил)-1-{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}пропан-1-он;
{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}изохинолин-3-илметанон;
{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}хиноксалин-6-илметанон;
{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}бензимидазол-6-илметанон;
(4-фторфенил)-[(S)-3-(3-нафталин-1-ил-[1,2,4]оксадиазол-5-ил)пиперидин-1-ил]метанон;
{(S)-3-[3-(2,6-дифторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}-(4-фторфенил)метанон;
(4-фторфенил)-{(S)-3-[3-(2-метоксифенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон;
(4-фторфенил)-[(S)-3-(3-нафталин-2-ил-[1,2,4]оксадиазол-5-ил)пиперидин-1-ил]метанон;
(4-фторфенил)-{3-[5-(4-фторфенил)-[1,2,4]оксадиазол-3-ил]пиперидин-1-ил}метанон;
(4-фторфенил)-{3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]-4-метилпиперазин-1-ил}метанон;
(4-фторфенил)амид (S)-1-(4-фторбензоил)пиперидин-3-карбоновой кислоты;
(4-фторфенил)метиламид (S)-1-(4-фторбензоил)пиперидин-3-карбоновой кислоты;
(E)-3-(4-фторфенил)-1-{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}пропенон;
1-(4-{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил}пиперидин-1-карбонил}пиперидин-1-ил)этанон;
{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}-(4-имидазол-1-илфенил)метанон;
(4-фторфенил)-{(S)-3-[3-(4-нитрофенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон;
(3,4-дифторфенил)-{(S)-3-[3-(4-нитрофенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон.
Другими иллюстративными примерами соединений формулы I являются следующие соединения:
(4-фторфенил)(3-(5-(4-фторфенил)изоксазол-3-ил)пиперидин-1-ил)метанон;
(4-фторфенил)(3-(5-(4-фторфенил)-1H-имидазол-2-ил)пиперидин-1-ил)метанон;
(4-фторфенил)(3-(4-(4-фторфенил)-1H-имидазол-1-ил)пиперидин-1-ил)метанон;
(4-фторфенил)(3-(4-(4-фторфенил)-1H-пиразол-1-ил)пиперидин-1-ил)метанон;
N-(1-(4-фторбензоил)пиперидин-3-ил)-4-фторбензамид;
(2-фторфенил)-{3-[2-(4-фторфенил)оксазол-5-ил]пиперидин-1-ил}метанон;
(2-фторфенил)-{3-[5-(4-фторфенил)оксазол-2-ил]пиперидин-1-ил}метанон;
(2-фторфенил)-{3-[5-(4-фторфенил)тиазол-2-ил]пиперидин-1-ил}метанон;
(2-фторфенил)-{3-[2-(4-фторфенил)тиазол-5-ил]пиперидин-1-ил}метанон;
(2-фторфенил)-{3-[5-(4-фторфенил)-[1,3,4]тиадиазол-2-ил]пиперидин-1-ил}метанон;
(2-фторфенил)-{3-[5-(4-фторфенил)-[1,2,4]оксадиазол-3-ил]пиперидин-1-ил}метанон;
(2-фторфенил)(3-(5-(4-фторфенил)изоксазол-3-ил)пиперидин-1-ил)метанон;
(2-фторфенил)(3-(5-(4-фторфенил)-1H-имидазол-2-ил)пиперидин-1-ил)метанон;
(2-фторфенил)(3-(4-(4-фторфенил)-1H-имидазол-1-ил)пиперидин-1-ил)метанон;
(2-фторфенил)(3-(4-(4-фторфенил)-1H-пиразол-1-ил)пиперидин-1-ил)метанон;
N-(1-(4-фторбензоил)пиперидин-3-ил)-2-фторбензамид;
(2-фторфенил)-{3-[2-(3,4-фторфенил)оксазол-5-ил]пиперидин-1-ил}метанон;
(2-фторфенил)-{3-[5-(3,4-фторфенил)оксазол-2-ил]пиперидин-1-ил}метанон;
(2-фторфенил)-{3-[5-(3,4-фторфенил)тиазол-2-ил]пиперидин-1-ил}метанон;
(2-фторфенил)-{3-[2-(3,4-фторфенил)тиазол-5-ил]пиперидин-1-ил}метанон;
(2-фторфенил)-{3-[5-(3,4-фторфенил)-[1,3,4]тиадиазол-2-ил]пиперидин-1-ил}метанон;
(2-фторфенил)-{3-[5-(3,4-фторфенил)-[1,2,4]оксадиазол-3-ил]пиперидин-1-ил}метанон;
(2-фторфенил)(3-(5-(3,4-фторфенил)изоксазол-3-ил)пиперидин-1-ил)метанон;
(2-фторфенил)(3-(5-(3,4-фторфенил)-1H-имидазол-2-ил)пиперидин-1-ил)метанон;
(2-фторфенил)(3-(4-(3,4-фторфенил)-1H-имидазол-1-ил)пиперидин-1-ил)метанон;
(2-фторфенил)(3-(4-(3,4-фторфенил)-1H-пиразол-1-ил)пиперидин-1-ил)метанон;
N-(1-(3,4-фторбензоил)пиперидин-3-ил)-2-фторбензамид.
Настоящее изобретение относится к фармацевтически приемлемым кислотно-аддитивным солям соединений формулы (I) или фармацевтически приемлемым носителям или наполнителям.
Настоящее изобретение относится к способу лечения или профилактики у млекопитающего, включая человека, состояния, лечение или профилактика которого опосредованы или могут быть облегчены нейромодуляторным действием аллостерических модуляторов mGluR5 и особенно положительных аллостерических модуляторов.
Настоящее изобретение относится к способу лечения или профилактики расстройств периферийной и центральной нервной системы, выбранных из группы, включающей толерантность или зависимость, состояние беспокойства, депрессию, психиатрическое заболевание, такое как психоз, воспалительная или невропатическая боль, ухудшение памяти, болезнь Альцгеймера, ишемия, злоупотребление лекарственными средствами и лекарственная зависимость (наркомания).
Настоящее изобретение относится к фармацевтическим композициям, включающим примерно от 0,01 до 1000 мг активного ингредиента на единичную дозу. Композиции могут вводиться любым подходящим способом, например перорально в форме капсул и т.д., парентерально в форме растворов для инъекции, местно в форме мазей (onguents) или лосьонов, в глаза в форме глазного лосьона, ректально в форме свечей.
Фармацевтические препараты согласно настоящему изобретению могут быть получены стандартными способами данной области техники; форма применяемой фармацевтической композиции будет зависеть от желаемого способа введения. Общая суточная доза обычно находится в интервале примерно от 0,05 до 2000 мг.
Способы синтеза
Соединения общей формулы I могут быть получены способами, известными в области органического синтеза, которые кратко описаны ниже с помощью схем. Совершенно ясно, что на всех схемах, описанных ниже, для чувствительных или реакционноспособных групп, когда это необходимо, применяются защитные группы в соответствии с общими принципами химии. Введение защитных групп осуществляется в соответствии со стандартными способами органического синтеза (T.W. Green and P.G.M. Wuts (1991), Protective Groups in Organic Synthesis, John Wiley et Sons). Такие группы удаляются на подходящей стадии синтеза соединения с использованием способов, которые известны квалифицированному специалисту данной области техники. Выбор способа, а также условий реакции и порядка выполнения работ должны быть совместимы с получением соединений формулы I.
Соединение формулы I может быть представлено в виде смеси энантиомеров, которая может разделяться на отдельные чистые R- или S-энантиомеры. Если, например, желателен конкретный энантиомер соединения формулы I, он может быть получен асимметрическим синтезом или деривацией с хиральным вспомогательным веществом, где образующаяся диастереомерная смесь разделяется и вспомогательная группа расщепляется для получения желательных чистых энантиомеров. Альтернативно, когда молекула содержит основную функциональную группу, такую как аминогруппа или кислотная функциональная группа, например карбоксильная группа, разделение может удобно осуществляться фракционной кристаллизацией соединений формулы I с оптически активной кислотой из различных растворителей солей или другими способами, описанными в литературе, например хиральной колоночной хроматографией. Выделение конечного продукта, промежуточного продукта или исходного вещества может осуществляться любым подходящим способом, известным в данной области техники, которые описаны в публикации E.L. Eliel, S.H. Wilen and L.N. Mander (1984), Stereochemistry of Organic Compounds, Wiley-Interscience.
Большое количество гетероциклических соединений формулы I, где А представляет собой гетероароматическую группу, могут быть получены с использованием способов синтеза, хорошо известных в данной области техники (A.R. Katrizky A.R. and C.W. Rees (1984) Comprehensive Heterocyclic Chemistry, Pergamon Press).
Продукт реакции может быть выделен и очищен с использованием стандартных методик, таких как экстракция, хроматография, кристаллизация, отгонка и т.п.
Соединение формулы I-A в случае, когда А представляет собой триазольную группу формулы 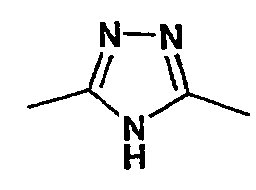 и W представляет собой 3-замещенный пиперидиновый цикл, могут быть получены в соответствии с последовательностями синтеза, представленными на схемах 1-3,
и W представляет собой 3-замещенный пиперидиновый цикл, могут быть получены в соответствии с последовательностями синтеза, представленными на схемах 1-3,
где
Р и Q, каждый независимо, представляет собой арил или гетероарил, которые описаны выше,
В представляет собой -С(=О)-С0-С2-алкил-; -S(=O)2-С0-С2-алкил-.

В соответствии со схемой 1 предшественник нипекотиновой кислоты (например, этилнипекотат) подвергается взаимодействию с арил- или гетероарил-производными, например, 4-фторбензоилхлоридом, с использованием способа, известного квалифицированным специалистам в данной области техники. На схеме 1 В принимает значения, определенные выше, Х представляет собой галоген, PG1 представляет собой защитную группу, такую как бензил, трет-бутил, этил, аллил и т.п. Реакция может быть ускорена основанием, таким как триэтиламин, диизопропиламин, пиридин, в подходящем растворителе (например, тетрагидрофуране, дихлорметане). Реакция обычно проводится при условии медленного нагрева реакционной смеси от 0°С до комнатной температуры в течение периода времени в интервале от 4 до 12 часов. Защитные группы PG1 удаляются стандартными способами.
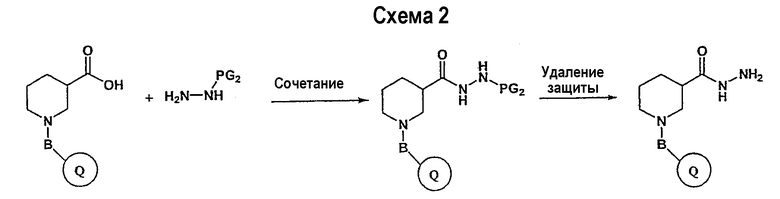
В свою очередь, замещенное производное кислоты (представленное на схеме 1) может подвергаться превращению в гидразид-производное с использованием подхода, представленного на схеме 2. PG2 на схеме 2 представляет аминозащитную группу, такую как трет-бутилоксикарбонил, бензилоксикарбонил, этоксикарбонил, бензил и т.п. Реакция может быть ускорена с помощью агента реакции сочетания, известного в области органического синтеза, такого как EDCI (1-(3-диметиламинопропил)-3-этилкарбодиимид), DCC (N,N'-дициклогексилкарбодиимид), в подходящем растворителе (например, таком как тетрагидрофуран, дихлорметан, N,N-диметилформамид, диоксан). Обычно в реакционной смеси будет также присутствовать второй катализатор, такой как HOBT (гидроксибензотриазол). Реакция обычно проводится при комнатной температуре в течение периода времени в интервале примерно от 4 до 12 часов. Защитные группы PG2 удаляются стандартными способами.
Схема 3 иллюстрируют заключительную стадию синтеза.
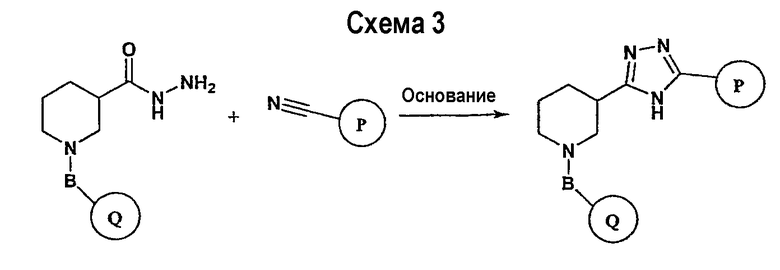
Гидразид-производное подвергается взаимодействию с нитрилпроизводным (например, 4-фторбензонитрилом) в основных условиях, таких как метилат натрия или этилат натрия и т.п., в подходящем растворителе (например, метиловом спирте, этиловом спирте). Реакция обычно проводится при условии медленного нагрева реакционной смеси от комнатной температуры до 65°С в течение периода времени в интервале примерно от 24 часов до 48 часов (см., например, Alcalde, Ermitas; Gisbert, Maria; Perez-Garcina. Lluisa; Tetrahedron; 51; 48; 1955; 13365-13378).
В соответствии с другим вариантом осуществления настоящего изобретения соединения формулы I-A, где А представляет собой  и W представляет собой 3-замещенный пиперидиновый цикл, могут быть получены в соответствии с последовательностями синтеза, представленными на схемах 4-6,
и W представляет собой 3-замещенный пиперидиновый цикл, могут быть получены в соответствии с последовательностями синтеза, представленными на схемах 4-6,
где
Р и Q, каждый независимо, представляет собой арил или гетероарил, которые описаны выше,
В представляет собой -С(=О)-С0-С2-алкил; -S(=O)2-С0-С2-алкил-.
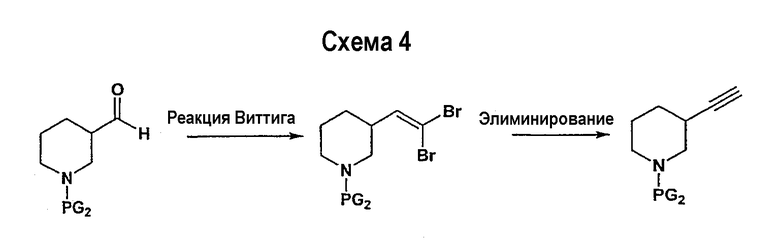
В соответствии с настоящим изобретением ацетиленовые производные могут быть получены способами, известными в данной области техники, например, описанным выше способом. Свободный азот пиперидинового цикла защищается защитной группой аминогруппы PG2.
Подходящее альдегидное производное, например, трет-бутиловый эфир 3-формилпиперидин-1-карбоновой кислоты, подвергается превращению в соответствующий ненасыщенный гем-дибромид-производное посредством реакции Виттига в соответствии со способом, описанным в патенте WO 02/088114. Реакция Виттига может быть ускорена с помощью смеси метиленовых предшественников (например, тетрабромида углерода) и фосфина, такого как трифенилфосфин, в подходящем растворителе (например, дихлорметане, тетрагидрофуране, диэтилэфире). Если необходимо, в реакционной смеси будет присутствовать катализатор, такой как цинковый порошок. Реакцию обычно проводят, поддерживая комнатную температуру в течение периода времени в интервале примерно от 12 часов до 24 часов. Ненасыщенное гем-дибромидное соединение затем подвергается взаимодействию с металлоорганическими соединениями, такими как н-бутиллитий, трет-бутиллитий и т.п., которые могут подвергаться реакции обмена металла с последующим дегидрогалогенированием. Реакция может быть ускорена в подходящем растворителе (например, тетрагидрофуране, эфире и т.п.) при температуре около -78°С в течение 1 часа.
Схема 5 иллюстрирует получение дизамещенных ацетиленовых производных взаимодействием алкиленового производного (получение которого описано на схеме 4), например трет-бутилового эфира 3-этинилпиперидин-1-карбоновой кислоты, с замещенным Р, например 1-фтор-4-йодбензолом. Таким образом, на схеме 5 Х означает галогениды, такие как Cl, Br, I, или трифторметансульфонил и паратолуолсульфонил. Такой общий путь синтеза был описан в публикации J. Med. Chem. 2000, 43, 4288-4312.
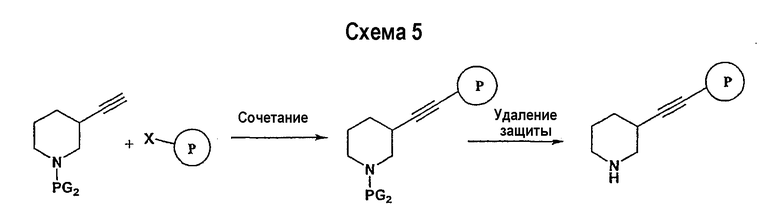
Для осуществления данной реакции С-С-связывания, катализируемой палладием, необходим катализатор, такой как PdCl2(PPh3)2, Pd(PPh3)4, Pd(OAc)2 или Pd на углероде, в подходящем растворителе, таком как ДМФА, ацетонитрил или бензол. Обычно в реакционной смеси будет также присутствовать второй катализатор, такой как йодид меди (I), и основание (например, триэтиламин, диизопропиламин, ацетат калия и т.д.). Реакция сочетания обычно проводится при обеспечении медленного нагрева от примерно 0°С до комнатной температуры или при нагреве до температуры в интервале от 30°С до 150°С. После этого реакционная смесь выдерживается при подходящей температуре в течение периода времени в интервале примерно от 1 часа до 24 часов, причем обычно достаточно примерно 12 часов. Защитные группы PG2 удаляются стандартными способами.
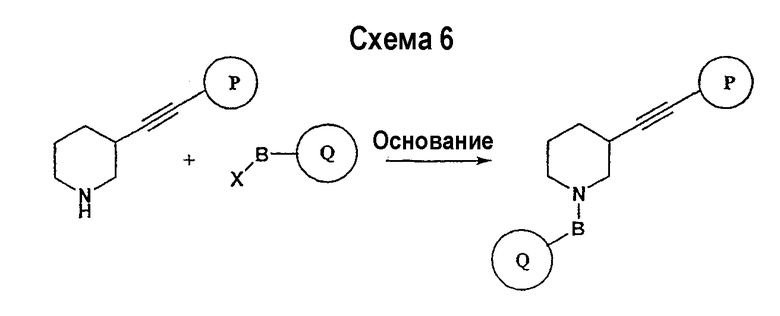
Схема 6 иллюстрирует последнюю стадию способа, аналогичного способу, представленному на схеме 1.
Соединения формулы I-A, где A представляет собой 
и W представляет собой 3-замещенный пиперидиновый цикл, могут быть получены в соответствии с последовательностями синтеза, представленными на схемах 7-10,
где
Р и Q, каждый независимо, представляет собой арил или гетероарил, которые описаны выше,
В представляет собой -С(=О)-С0-С2-алкил-; -S(=O)2-С0-С2-алкил-.
Исходный амидоксим может быть получен способами, известными в области органического синтеза, как показано на представленной далее схеме синтеза 7.
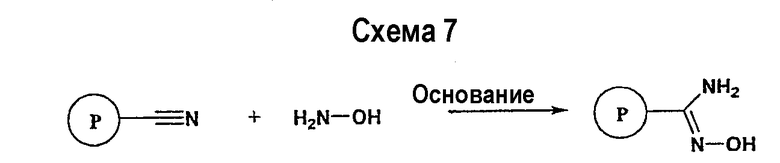
Нитрильное производное (например, 4-фторбензонитрил) подвергается взаимодействию с гидроксиламином в нейтральных или основных условиях, таких как триэтиламин, диизопропилэтиламин, карбонат натрия, гидроксид натрия и т.п., в подходящем растворителе (например, метиловом спирте, этиловом спирте). Реакция обычно проводится при условии медленного нагрева от температуры окружающей среды до температуры в интервале от 70°С до 80°С включительно в течение периода времени в интервале примерно от 1 часа до 48 часов включительно (см., например, Lucca, George V. De; Kim, Ui T.; Liang, Jing; Cordova, Beverly; Klabe, Ronald M.; et al.; J. Med. Chem.; EN; 41; 13; 1998; 2411-2423; Lila, Christine; Gloanec, Philippe; Cadet, Laurence; Herve, Yolande; Fournier, Jean; et al.; Synth. Commun.; EN; 28; 23; 1998; 4419-4430; см. также: Sendzik, Martin; Hui, Hon C.; Tetrahedron Lett.; EN; 44; 2003; 8697-8700 и ссылки, приведенные в данной публикации для реакции в нейтральных условиях).
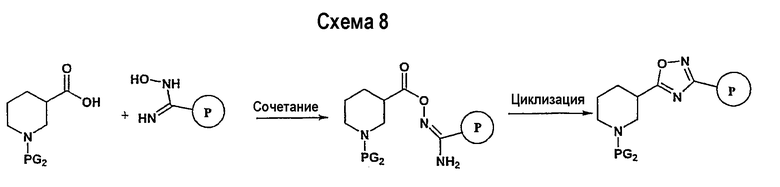
Замещенное амидоксим-производное (описанное на схеме 7) может подвергаться превращению в ациламидоксим-производное с использованием подхода, представленного на схеме 8. PG2 на схеме 8 представляет собой защитную группу, как описано выше. Реакция сочетания может быть ускорена с помощью агента реакции сочетания, известного в области органического синтеза, такого как EDCI (1-(3-диметиламинопропил)-3-этилкарбодиимид), DCC (N,N'-дициклогексилкарбодиимид), в присутствии подходящего основания, такого как триэтиламин, диизопропилэтиламин, в подходящем растворителе (например, тетрагидрофуране, дихлорметане, N,N-диметилформамиде, диоксане). Обычно в реакционной смеси может также присутствовать второй катализатор, такой как НОВТ (гидроксибензотриазол), НОАТ (1-гидрокси-7-азабензотриазол). Реакция обычно проводится при температуре в интервале от температуры окружающей среды до 60°С, включительно в течение периода времени в интервале примерно от 2 часов до 12 часов для получения промежуточного ациламидоксима. Реакция циклизации может осуществляться нагревом реакционной смеси до температуры в интервале от примерно 80°С до примерно 150°С в течение периода времени в интервале примерно от 2 часов до 18 часов (см., например, Suzuki, Takeshi; Iwaoka, Kiyoshi; Imanishi, Naoki; Nagakura, Yukinori; Miyata, Keiji; et al.; Chem. Pharm. Bull.; EN; 47; 1; 1999; 120-122). Продукт, полученный в результате реакции, может быть выделен и очищен с применением стандартных методик, таких как экстракция, хроматография, кристаллизация, отгонка и т.п.
Заключительная стадия может осуществляться способом, описанным на схеме 9, или способом, описанным на схеме 10.
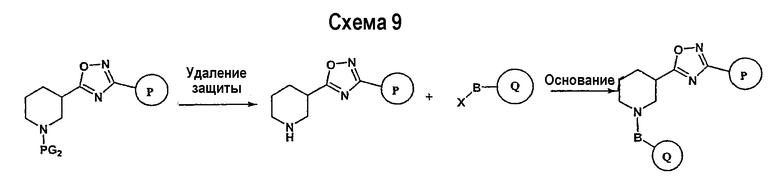
Как показано на схеме 9, защитные группы PG2 удаляются с использованием стандартных способов. Сочетание, показанное на схеме 9, аналогично сочетанию, представленному на схеме 1.
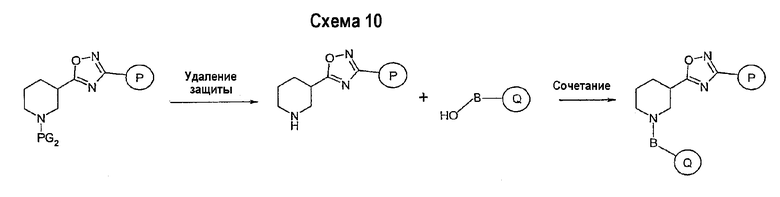
Как показано на схеме 10, защитные группы PG2 удаляются стандартными способами. Реакция сочетания может быть ускорена с помощью агентов реакции сочетания, известных в области органического синтеза, таких как EDCI (1-(2-диметиламинопропил)-3-этилкарбодиимид), DCC (N,N'-дициклогексилкарбодиимид), или агентов реакции сочетания, нанесенных на полимерный носитель, таких как карбодиимид, нанесенный на полимерный носитель (PS-DCC, например, Argonaut Technologies), в присутствии подходящего основания, такого как триэтиламин, диизопропилэтиламин, в подходящем растворителе (например, тетрагидрофуране, дихлорметане, N,N-диметилформамиде, диоксане). Обычно в реакционной смеси может также присутствовать второй катализатор, такой как НОВТ (1-гидроксибензотриазол), НОАТ (1-гидрокси-7-азабензотриазол) и т.п. Реакция обычно проводится при температуре окружающей среды в течение периода времени в интервале примерно от 2 часов до 12 часов.
Соединения формулы I, где А представляет собой
и W представляет собой 2-замещенный морфолиновый цикл, могут быть получены в соответствии с последовательностями синтеза, представленными на схемах 11-12,
где
Р и Q, каждый независимо, представляет собой арил или гетероарил, которые описаны выше,
В представляет собой -С(=О)-С0-С2-алкил-; -S(=O)2-С0-С2-алкил-.

В соответствии со схемой 11 замещенное амидооксим-производное (описанное на схеме 7) может подвергаться превращению в ациламидоксим-производное взаимодействием с морфолин-производным посредством способа, аналогичного представленному на схеме 8. Аналогично, ациламидоксим-производное может подвергаться циклизации до [1,2,4]оксадиазол-производного в соответствии со способом, представленным на схеме 8.
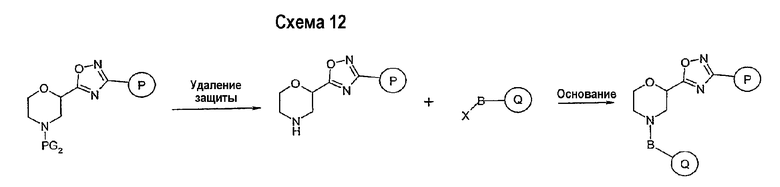
На схеме 12 группы PG2 удаляются стандартными способами. Сочетание, представленное на схеме 12, аналогично реакциям, представленным на схеме 9 и схеме 10.
Соединения формулы I, где А представляет собой
и W представляет собой 2-замещенный пиперазиновый цикл, могут быть получены в соответствии с последовательностями синтеза, представленными на схемах 13-15,
где
Р и Q, каждый независимо, представляет собой арил или гетероарил, которые описаны выше,
В представляет собой -С(=О)-С0-С2-алкил-; -S(=O)2-С0-С2-алкил-.
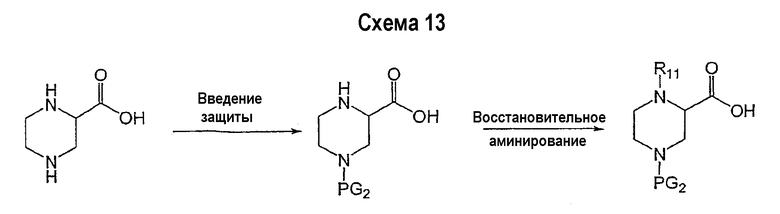
На схеме 13 пиперазин-2-карбоновая кислота выборочно защищена по атому азота в положении 4. PG2 представляет собой защитную группу аминогруппы, такую как трет-бутилоксикарбонил и т.п. Данная реакция может проводиться с использованием таких соединений, как 2-(Вос-оксимино)-2-фенилацетонитрил, ди-третбутилдикарбонат и т.п., в подходящем органическом растворителе (например, диоксане, тетрагидрофуране) в смеси с водой. Обычно рН реакционной смеси будет доводиться до значения в интервале от 8 до 12 добавлением подходящего основания, такого как гидроксид натрия, гидроксид калия, триэтиламин и т.п. Реакция обычно протекает при комнатной температуре в течение периода времени в интервале примерно от 1 часа до 4 часов (см., например: Bigge, Christopher F.; Hays, Sheryl J.; Novak, Perry M.; Drummond, James T. et al.; Tetrahedron Letters; 30, 39; 1989; 5193-5196 и WO 2004/022061). N4-защищенное пиперазин-производное может подергаться превращению в пиперазин-производное, замещенное в положение 1, с использованием стандартных условий восстановительного аминирования. R11 может представлять собой, например, С1-С6-алкил, С3-С6-циклоалкил, С3-С7-циклоалкилалкил, арилалкил, гетероарилалкил. Реакция может проводиться взаимодействием N4-защищенного пиразин-производного с альдегидом или кетоном (например, формальдегидом) в присутствии подходящего восстановителя, такого как триацетоксиборгидрид натрия, цианоборгидрид натрия, боргидрид натрия и т.п., в подходящем растворителе, таком как ацетонитрил, тетрагидрофуран, метанол, этанол, 1,2-дихлорэтан и т.п. Обычно для проведения реакции может быть необходимо добавление кислоты для снижения значения рН реакционной смеси до менее примерно 7, и такая кислота представляет собой уксусную кислоту, соляную кислоту и т.п. Реакция обычно протекает при комнатной температуре в течение периода времени в интервале примерно от 2 часов до 4 часов.
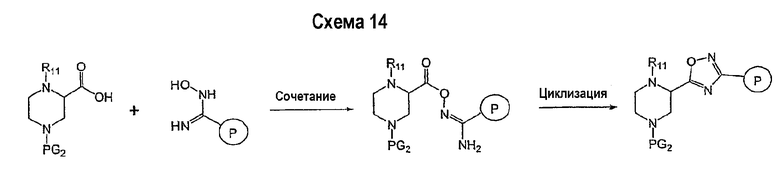
На схеме 14 замещенное амидоксим-производное (полученное, как показано на схеме 7) может подвергаться превращению до ациламидоксим-производного взаимодействием с пиперазин-производным (описанным на схеме 13) способом, аналогичным описанному на схеме 8. Ациламидоксим-производное может подвергаться циклизации до [1,2,4]оксадиазол-производного в соответствии со способом, описанным на схеме 8.
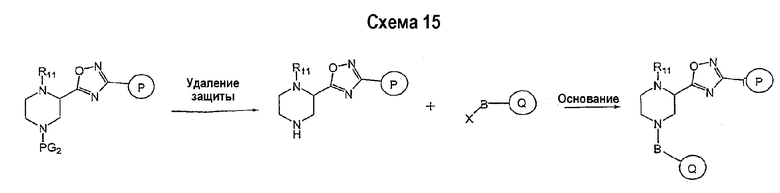
На схеме 15 PG2 группы удаляются с использованием стандартных способов. Сочетание, представленное на схеме 15, аналогично реакциям, представленным на схемах 9-10.
Соединения формулы I-A, где А представляет собой группу формулы  и W представляет собой 3-замещенный пиперидиновый цикл, могут быть получены в соответствии с последовательностями синтеза, представленными на схеме 16,
и W представляет собой 3-замещенный пиперидиновый цикл, могут быть получены в соответствии с последовательностями синтеза, представленными на схеме 16,
где
Р и Q, каждый независимо, представляет собой арил или гетероарил, которые описаны выше,
В представляет собой -С(=О)-С0-С2-алкил-; -S(=O)2-С0-С2-алкил-.
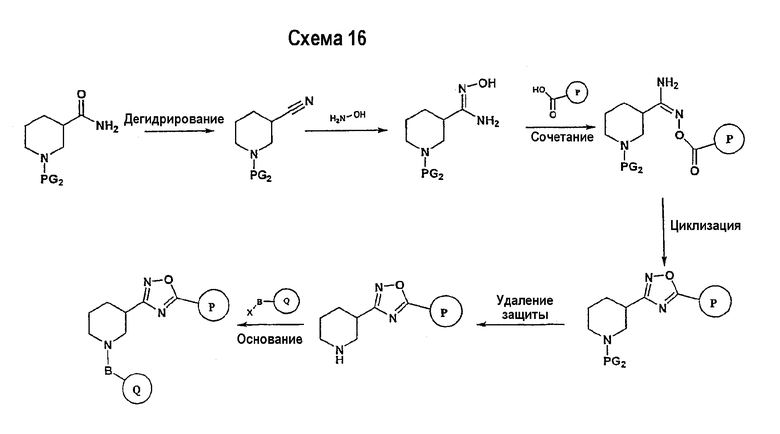
Оксадиазольный цикл, описанный выше, получают в соответствии с последовательностями синтеза, хорошо известными в данной области техники (A.R. Katrizky and C.W. Rees (1984) Comprehensive Heterocyclic Chemistry, Pergamon Press).
Соединения формулы I-A, где А представляет собой группу формулы  и W представляет собой 3-замещенный пиперидиновый цикл, могут быть получены в соответствии с последовательностями синтеза, представленными на схемах 17-19,
и W представляет собой 3-замещенный пиперидиновый цикл, могут быть получены в соответствии с последовательностями синтеза, представленными на схемах 17-19,
где
Р и Q, каждый независимо, представляет собой арил или гетероарил, которые описаны выше,
В представляет собой -С(=О)-С0-С2-алкил-; -S(=O)2-С0-С2-алкил-.
Алкилирование триазол-производных (полученных, как показано на схемах 1-3) алкилирующим агентом, таким как метилйодид и т.п., в основных условиях (например, NaH, MeONa и т.п.) приводит к получению N-алкилтриазол-производного (см., например, Tarrago, Georges; Marzin, Claude; Najiumi, Ouafa; Pellegrin, Valdo; J. Org. Chem.; 55; 2; 1990; 420-425). Альтернативно, для получения этих производных может применяться другая последовательность реакций, как показано на схемах 18-19.
Вторичный амид (например, N-метил-4-фторбензамид) подвергается превращению в имидоилхлорид с использованием подхода, представленного на схеме 18. R3 может представлять собой, например, С1-С6-алкил, С3-С6-циклоалкил, С3-С7-циклоалкилалкил, арилалкил, гетероарилалкил, арил, гетероарил. Обычно вторичный амид подвергается взаимодействию с хлорирующим агентом, таким как тионилхлорид, пентахлорид фосфора, оксихлорид фосфора и т.п., в подходящем растворителе (например, дихлорметане, хлороформе, тетрагидрофуране) или с использованием хлорирующего агента в большом избытке в качестве растворителя. Реакция обычно проводится при обеспечении медленного нагрева от температуры окружающей среды до температуры в интервале от 40°С до 110°С включительно в течение периода времени в интервале примерно от 1 часа до 4 часов.
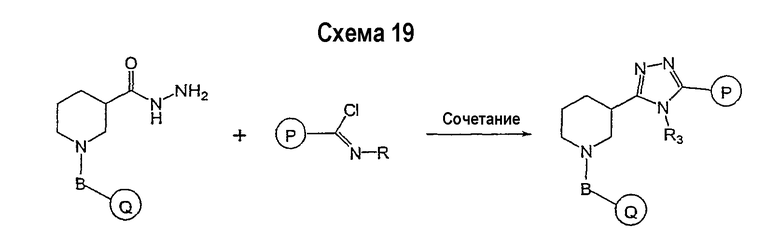
На схеме 19 производное гидразида (описанное на схеме 2) может подвергаться взаимодействию с имидоилхлоридом (описанным на схеме 18) в соответствии со способом, описанным на схеме 19. Реакция может быть ускорена с помощью подходящего основания, такого как триэтиламин, диизопропилэтиламин, карбонат кальция, в подходящем растворителе (например, толуоле, диоксане, тетрагидрофуране). Реакция обычно проводится при условии медленного нагрева реакционной смеси от температуры окружающей среды до температуры в интервале от 60°С до 110°С, включительно, в течение периода времени в интервале примерно от 1 часа до 8 часов (см., например; Clemence, Francois; Joliveau-Maushart, Claudine; Meier, Jean; Cerede, Jean et al.; Eur. J. Med. Chem.; 20; 2; 1985; 257-266, а также ссылки, приведенные в данных публикациях).
Соединения формулы I-A, где А представляет собой группу формулы  и W представляет собой 3-замещенный пиперидиновый цикл, могут быть получены в соответствии с последовательностями синтеза, представленными на схемах 20-21.
и W представляет собой 3-замещенный пиперидиновый цикл, могут быть получены в соответствии с последовательностями синтеза, представленными на схемах 20-21.
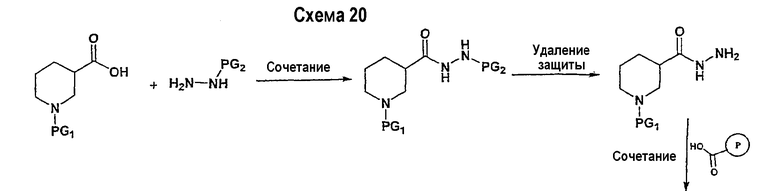
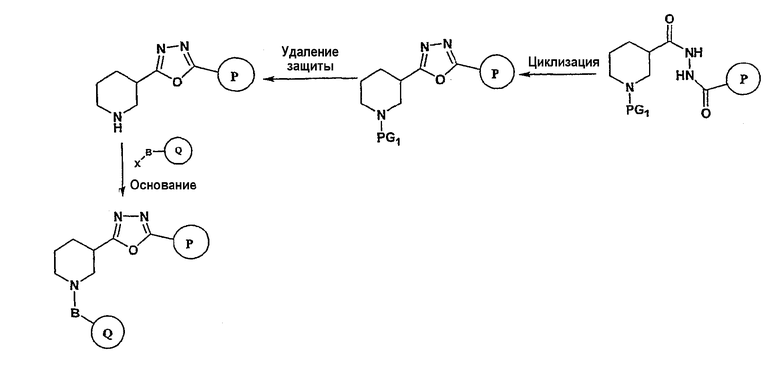
Оксадиазольный цикл, описанный выше, получают в соответствии со способами синтеза, хорошо известными в данной области техники (A.R. Katrizky and C.W. Rees (1984) Comprehensive Heterocyclic Chemistry, Pergamon Press).
В качестве альтернативы может использоваться другая методика синтеза, представленная на схеме 21.
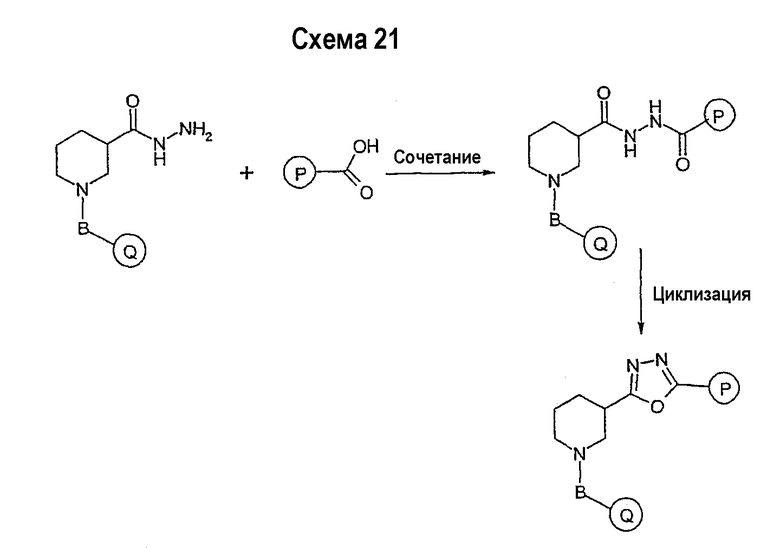
Производное гидразида (полученное, как описано на схеме 2) может подвергаться взаимодействию с кислотой в подходящих условиях сочетания. Сочетание может быть ускорено с помощью агентов реакции сочетания, известных в области органического синтеза, таких как EDCI (1-(3-диметиламинопропил)-3-этилкарбодиимид), DCC (N,N'-дициклогексилкарбодиимид), или связующих агентов, нанесенных на полимерный носитель, таких как карбодиимид, нанесенный на полимерный носитель (PS-DCC, ex Argonaur Technologies), в присутствии подходящего основания, такого как триэтиламин, диизопропилэтиламин, в подходящем растворителе (например, тетрагидрофуране, дихлорметане, N,N-диметилформамиде, диоксане). Обычно в реакционной смеси может присутствовать также второй катализатор, такой как НОВТ (1-гидроксибензотриазол), НОАТ (1-гидрокси-7-азабензотриазол) и т.п. Реакция обычно протекает при температуре окружающей среды в течение периода времени в интервале примерно от 2 часов до 12 часов.
Стадия циклизации может проводиться в присутствии дегидратирующего агента, такого как оксихлорид фосфора, тионилхлорид и т.п., в подходящем растворителе (например, ацетонитриле, пиридине) или с применением дегидратирующего агента, взятого в избытке, в качестве растворителя. Обычно реакция протекает при температуре в интервале от 70°С до 110°С в течение периода времени в интервале от 2 часов до 4 часов. Альтернативно и более предпочтительно, стадия циклизации может проводиться в присутствии 4-толуолсульфонилхлорида и ВЕМР, нанесенного на твердый носитель, в подходящем растворителе, таком как ТГФ, диоксан и т.п., с использованием традиционного или микроволнового нагрева в соответствии с методикой, описанной в литературе (см.: Brain, Christopher T., Brunton, Shirley A.; Synlett, 3, 2001, 382-384).
Соединения формулы I-A, где А представляет собой группу формулы  и W представляет собой 3-замещенный пиперидиновый цикл, могут быть получены в соответствии с последовательностями синтеза, представленными на схемах 28-29,
и W представляет собой 3-замещенный пиперидиновый цикл, могут быть получены в соответствии с последовательностями синтеза, представленными на схемах 28-29,
где
Р и Q, каждый независимо, представляет собой арил или гетероарил, которые описаны выше,
В представляет собой -С(=О)-С0-С2-алкил-; -S(=O)2-С0-С2-алкил-.
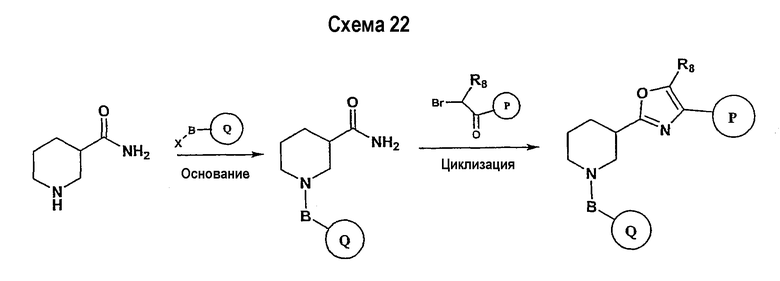
Оксазольный цикл, описанный выше, получают в соответствии со способами синтеза, хорошо известными в данной области техники (A.R. Katrizky and C.W. Rees (1984) Comprehensive Heterocyclic Chemistry, Pergamon Press).
Соединения формулы I-A, где А представляет собой группу формулы  и W представляет собой 3-замещенный пиперидиновый цикл, могут быть получены в соответствии с последовательностями синтеза, представленными на схеме 23,
и W представляет собой 3-замещенный пиперидиновый цикл, могут быть получены в соответствии с последовательностями синтеза, представленными на схеме 23,
где
Р и Q, каждый независимо, представляет собой арил или гетероарил, которые описаны выше,
В представляет собой -С(=О)-С0-С2-алкил-; -S(=O)2-С0-С2-алкил-.
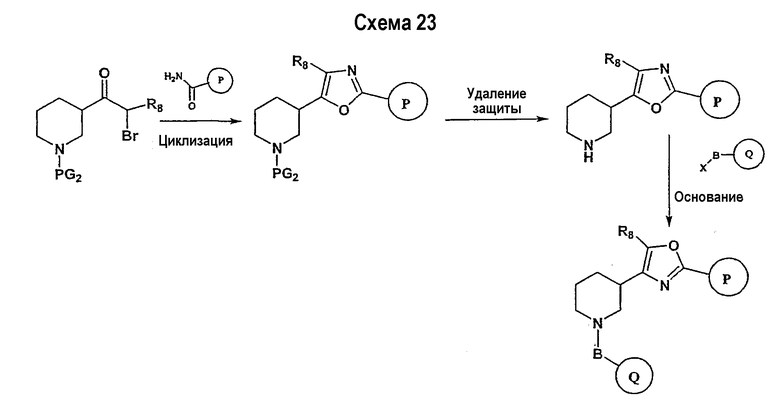
Исходные α-бромкетон-производные, показанные выше, получают в соответствии со способами синтеза, хорошо известными в данной области техники.
Соединения формулы I-A, где А представляет собой группу формулы  и W представляет собой 3-замещенный пиперидиновый цикл, могут быть получены в соответствии с последовательностями синтеза, представленными на схеме 24,
и W представляет собой 3-замещенный пиперидиновый цикл, могут быть получены в соответствии с последовательностями синтеза, представленными на схеме 24,
где
Р и Q, каждый независимо, представляет собой арил или гетероарил, которые описаны выше,
В представляет собой -С(=О)-С0-С2-алкил-; -S(=O)2-С0-С2-алкил-.
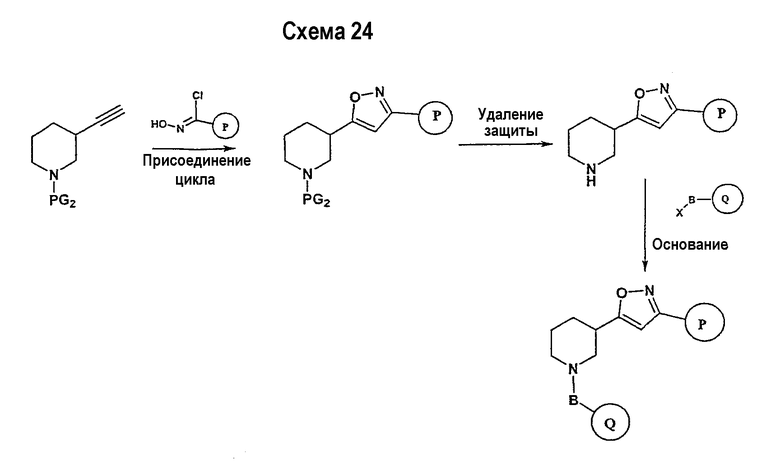
В соответствии с другим вариантом осуществления настоящего изобретения замещенное ацетиленовое производное (описанное на схеме 4) может подвергаться превращению в оксазол-производное взаимодействием с иминохлоридом арилоксима в соответствии со способами синтеза, хорошо известными в данной области техники (см, например, Diana, Guy D.; Volkots, Deborah L., Nitz, Theodore J.; Bailey, Thomas R.; Long, Melody A.; et al.; J. Med. Chem. 37; 15; 1994; 2421-2436).
Соединения формулы I-A, где А представляет собой группу формулы  и W представляет собой 3-замещенный пиперидиновый цикл, могут быть получены в соответствии с последовательностями синтеза, представленными на схеме 25,
и W представляет собой 3-замещенный пиперидиновый цикл, могут быть получены в соответствии с последовательностями синтеза, представленными на схеме 25,
Р и Q, каждый независимо, представляет собой арил или гетероарил, которые описаны выше,
В представляет собой -С(=О)-С0-С2-алкил-; -S(=O)2-С0-С2-алкил-.
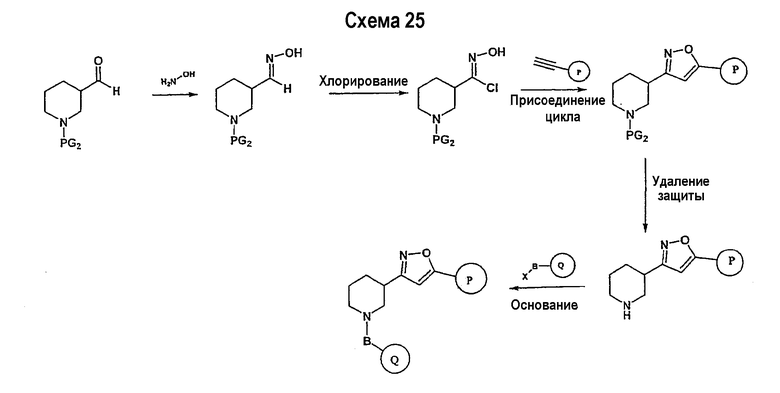
В соответствии с настоящим изобретением замещенное ацетилен-производное может подвергаться превращению в оксазол-производное взаимодействием с иминохлоридом арилоксима в соответствии со способами, хорошо известными в данной области техники (см., например., Diana, Guy D.; Volkots, Deborah L., Nitz, Theodore J.; Bailey, Thomas R.; Long, Melody A.; et al.; J. Med. Chem.; 37; 15; 1994; 2421-2436).
Соединения формулы I-A, где А представляет собой группу формулы  и W представляет собой 3-замещенный пиперидиновый цикл, могут быть получены в соответствии с последовательностями синтеза, представленными на схемах 26,
и W представляет собой 3-замещенный пиперидиновый цикл, могут быть получены в соответствии с последовательностями синтеза, представленными на схемах 26,
где
Р и Q, каждый независимо, представляет собой арил или гетероарил, которые описаны выше,
В представляет собой -С(=О)-С0-С2-алкил-; -S(=O)2-С0-С2-алкил-.
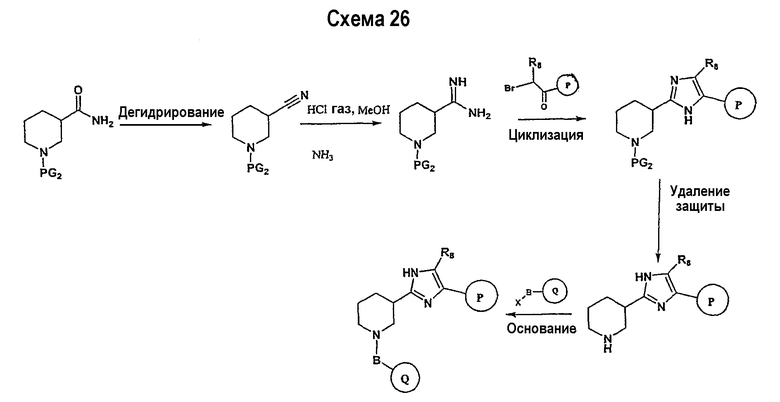
Согласно настоящему изобретению, замещенное амидин-производное может подвергаться превращению в имидазол-производное взаимодействием с α-бромкетоном в соответствии со способами синтеза, хорошо известными в данной области техники (A.R. Katrizky and C.W. Rees (1984) Comprehensive Heterocyclic Chemistry, Pergamon Press).
Соединения формулы I-A, где А представляет собой группу формулы  и W представляет собой 3-замещенный пиперидиновый цикл, могут быть получены в соответствии с последовательностями синтеза, представленными на схеме 27,
и W представляет собой 3-замещенный пиперидиновый цикл, могут быть получены в соответствии с последовательностями синтеза, представленными на схеме 27,
где
Р и Q, каждый независимо, представляет собой арил или гетероарил, которые описаны выше,
В представляет собой -С(=О)-С0-С2-алкил-; -S(=O)2-С0-С2-алкил-.
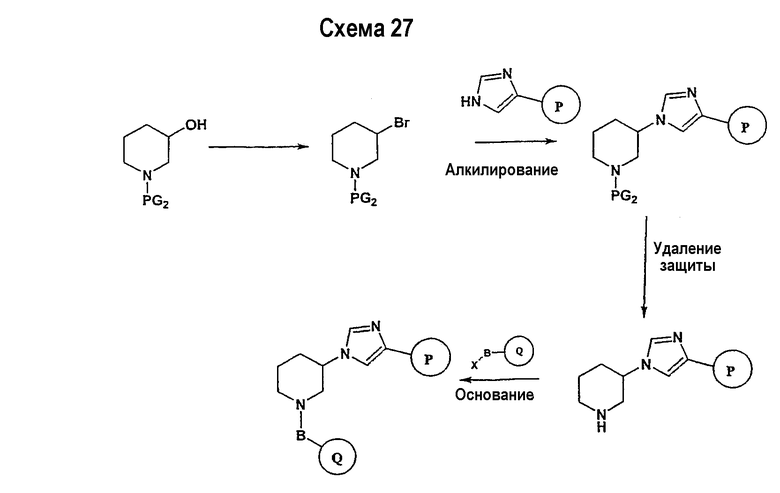
Исходные N-арилимидазол-производные получают в соответствии со способами синтеза, хорошо известными в данной области техники (A.R. Katrizky and C.W. Rees (1984) Comprehensive Heterocyclic Chemistry, Pergamon Press).
Соединения формулы I-A, где А представляет собой группу формулы и W представляет собой 3-замещенный пиперидиновый цикл, могут быть получены в соответствии с последовательностями синтеза, представленными на схемах 28-29,
где
Р и Q, каждый независимо, представляет собой арил или гетероарил, которые описаны выше,
В представляет собой -С(=О)-С0-С2-алкил-; -S(=O)2-С0-С2-алкил-.
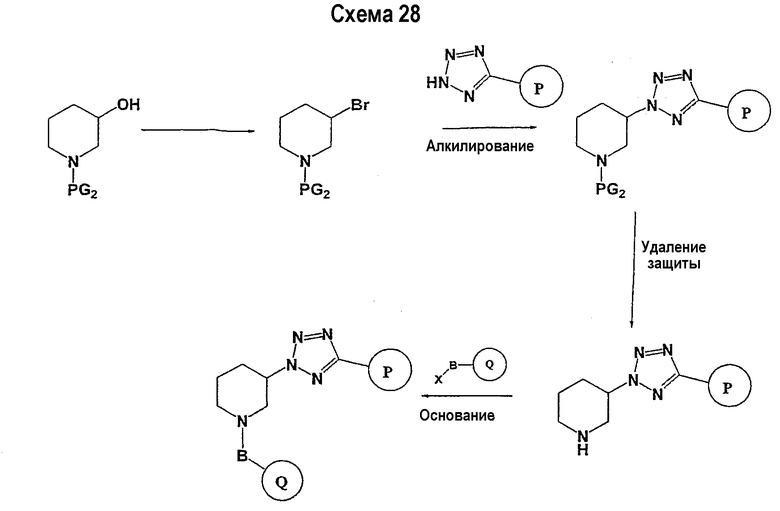
Исходные арилтетразол-производные получают в соответствии со способами синтеза, хорошо известными в данной области техники (A.R. Katrizky and C.W. Rees (1984) Comprehensive Heterocyclic Chemistry, Pergamon Press).
Альтернативно, данные производные могут быть синтезированы в соответствии с последовательностью синтеза, представленной на схеме 29.
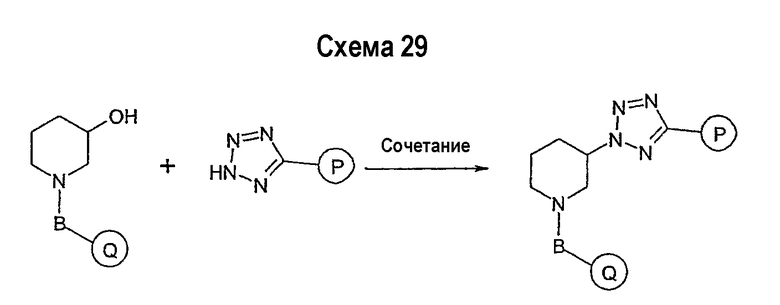
Арилтетразол, полученный в соответствии с методиками синтеза, хорошо известными в данной области техники, может подвергаться алкилированию 3-гидроксипеперидин-производным в условиях реакции сочетания Митсунобу, как описано в литературе (см., например: Synthetic Commun.; 26; 14; 1996; 2687-2694).
Соединения формулы I-A, где А представляет собой группу формулы  и W представляет собой 3-замещенный пиперидиновый цикл, могут быть получены в соответствии с методиками синтеза, представленными на схеме 30,
и W представляет собой 3-замещенный пиперидиновый цикл, могут быть получены в соответствии с методиками синтеза, представленными на схеме 30,
где
Р и Q, каждый независимо, представляет собой арил или гетероарил, которые описаны выше,
В представляет собой -С(=О)-С0-С2-алкил-; -S(=O)2-С0-С2-алкил-.
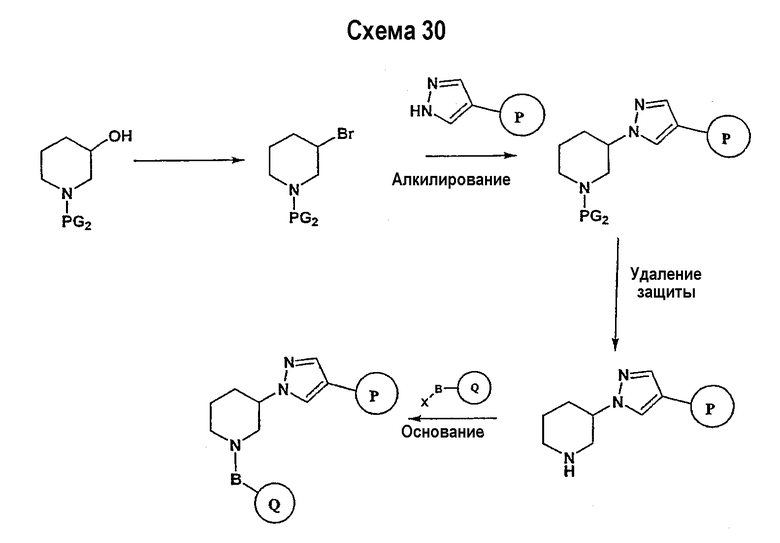
Исходные арилпиразол-производные получают в соответствии со способами синтеза, хорошо известными в данной области техники (A.R. Katrizky and C.W. Rees (1984) Comprehensive Heterocyclic Chemistry, Pergamon Press).
Соединения формулы I-A, где А представляет собой группу формулы 
и W представляет собой 3-замещенный пиперидиновый цикл, могут быть получены в соответствии с последовательностью синтеза, представленной на схеме 31,
где
Р и Q, каждый независимо, представляет собой арил или гетероарил, которые описаны выше,
В представляет собой -С(=О)-С0-С2-алкил-; -S(=O)2-С0-С2-алкил-.
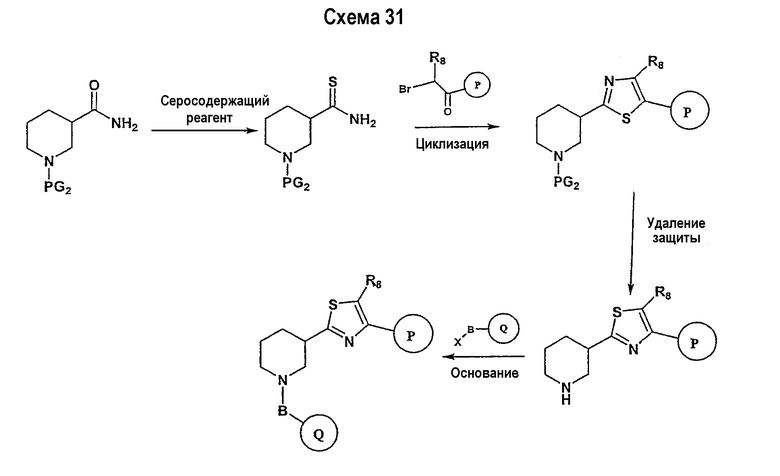
Соединения формулы I-A, где А представляет собой группу формулы  и W представляет собой 3-замещенный пиперидиновый цикл, могут быть получены в соответствии с последовательностью синтеза, представленной на схеме 32,
и W представляет собой 3-замещенный пиперидиновый цикл, могут быть получены в соответствии с последовательностью синтеза, представленной на схеме 32,
где
Р и Q, каждый независимо, представляет собой арил или гетероарил, которые описаны выше,
В представляет собой -С(=О)-С0-С2-алкил-; -S(=O)2-С0-С2-алкил-.
Схема 32 иллюстрирует получение дизамещенных этилен-производных взаимодействием защищенного винилпиперидина с замещенным Р, например 1-фтор-4-йодбензолом. Таким образом, на схеме 5 Х означает галогениды, такие как Cl, Br, I, или трифторметансульфонил и п-толуолсульфонил. Такой общий путь синтеза описан в публикации Arthzhur D: Brosius and al.; JACS. 1999, 121, 700-709.
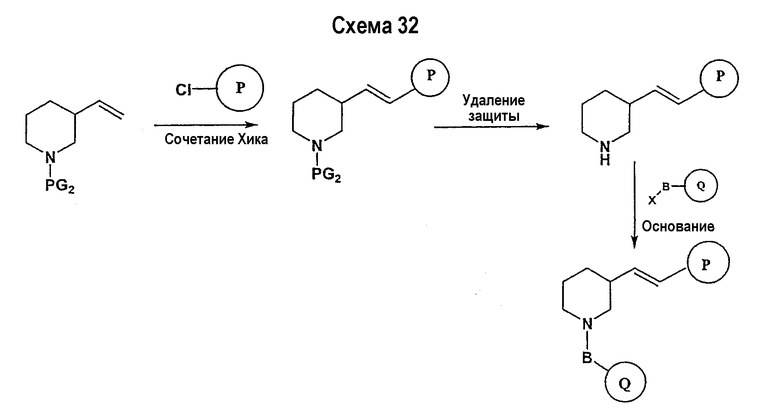
Для осуществления данной реакции С-С-связывания, катализируемой палладием, необходим катализатор, такой как PdCl2(PPH3)2, Pd(PPH3)4, Pd(OAc)2 или Pd на углероде, в подходящем растворителе, таком как ДМФА, ацетонитрил или бензол. Обычно в реакционной смеси присутствуют также второй катализатор, такой как йодид меди (I), и основание (например, триэтиламин, диизопропиламин, ацетат калия). Реакция сочетания обычно проводится при условии медленного нагрева от температуры примерно 0°С до температуры окружающей среды или нагрева до температуры в интервале от 30°С до 150°С. Затем реакционная смесь выдерживается при подходящей температуре в течение периода времени в интервале примерно от 1 до 24 часов, причем обычно достаточно бывает примерно 12 часов. Защитные группы PG2 удаляются стандартными способами.
Исходное защищенное винилпиперидин-производное получают в соответствии со способами синтеза, хорошо известными в данной области техники (см., например, Arthzhur D: Brosius and al.; JACS. 1999, 121, 700-709).
Соединения формулы I-A, где А представляет собой группу формулы  и W представляет собой 3-замещенный пиперидиновый цикл, могут быть получены в соответствии с последовательностью синтеза, представленной на схеме 33,
и W представляет собой 3-замещенный пиперидиновый цикл, могут быть получены в соответствии с последовательностью синтеза, представленной на схеме 33,
где
Р и Q, каждый независимо, представляет собой арил или гетероарил, которые описаны выше,
В представляет собой -С(=О)-С0-С2-алкил-; -S(=O)2-С0-С2-алкил-.
На схеме 33 PG2 представляет собой защитную группу аминогруппы, такую как трет-бутоксикарбонил, бензилоксикарбонил, этоксикарбонил, бензил и т.п., В принимает значения, определенные выше, Х представляет собой галоген. Образование амида может быть ускорено с помощью агентов реакции связывания, известных в области органического синтеза, таких как EDCI (1-(3-диметиламинопропил)-3-этилкарбодиимид), DCC (N,N'-дициклогексилкарбодиимид), в подходящем растворителе (например, тетрагидрофуране, дихлорметане, N,N-диметилформамиде, диоксане). Обычно в реакционной смеси может присутствовать также второй катализатор, такой как НОВТ (гидроксибензотриазол) и т.п. Реакция обычно протекает либо при комнатной температуре, либо при температуре в интервале от 40°С до 80°С включительно, в течение периода времени в интервале от примерно 4 часов до 48 часов. Защитные группы PG2 удаляются стандартными способами. Заключительная стадия, представленная на схеме 33, может быть ускорена с помощью основания, такого как триэтиламин, диизопропиламин, пиридин, в подходящем растворителе (например, тетрагидрофуране, дихлорметане). Реакция обычно проводится при условии медленного нагрева реакционной смеси от температуры 0°С до температуры окружающей среды в течение периода времени в интервале примерно от 2 часов до 12 часов.
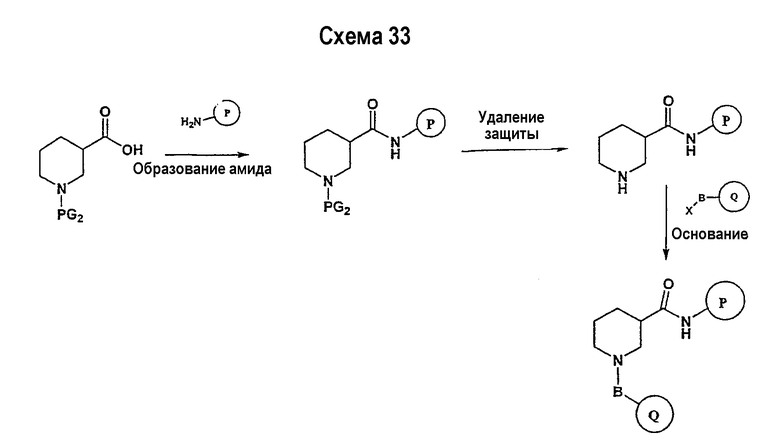
Соединения формулы I-A, где А представляет собой группу формулы  и W представляет собой 3-замещенный пиперидиновый цикл, могут быть получены в соответствии с последовательностью синтеза, представленной на схеме 34,
и W представляет собой 3-замещенный пиперидиновый цикл, могут быть получены в соответствии с последовательностью синтеза, представленной на схеме 34,
где
Р и Q, каждый независимо, представляет собой арил или гетероарил, которые описаны выше,
В представляет собой -С(=О)-С0-С2-алкил-; -S(=O)2-С0-С2-алкил-.
Соединения формулы I, которые по природе являются основными, могут образовывать широкий спектр различных фармацевтически приемлемых солей с различными неорганическими и органическими кислотами. Такие соли легко получают обработкой основных соединений по существу эквивалентным количеством выбранной минеральной или органической кислоты в подходящем органическом растворителе, таком как метанол, этанол или изопропанол (см. P. Heinrich Stahl, Camille G: Wermuth, Handbook of Pharmaceuticals Salts, Properties, Selection and Use, Wiley, 2002).
Приведенные далее примеры, не являющиеся ограничением области данного изобретения, предназначены только для пояснения настоящего изобретения. Данные физических свойств соединений, представленные в примерах, согласуются со структурой этих соединений.
ПРИМЕРЫ
За исключением особо оговоренных случаев, все исходные вещества были получены от коммерческих поставщиков и использовались без дополнительной очистки.
В примерах и описании могут применяться следующие аббревиатуры:
Все ссылки на водный раствор NaCl относятся к насыщенному водному раствору NaCl. За исключением особо оговоренных случаев все температуры выражены в °С (в градусах Цельсия). Все реакции проводятся в атмосфере, которая не является инертной, при комнатной температуре, за исключением особо оговоренных случаев.
lH-ЯМР спектры записаны на спектрометре Brucker 500 MHz или Brucker 300 MHz. Химические сдвиги выражены в миллионных долях (м.д., δ). Константы связывания представлены в герцах (Гц). Характеристики расщепления описаны мультиплетностью и обозначены как с (синглет), д (дуплет), т (триплет), кв. (квартет), квинт. (квинтет), м (мультиплет).
ЖХМС записаны в следующих условиях:
Метод A): система Waters 1525u Micromass ZQ. Колонка из нержавеющей стали 2,1×50 мм, заполненная 2,5 мкм XTerra RP C-18; скорость истечения 0,25 мл/мин; подвижная фаза: фаза A = вода/ацетонитрил 95/5 + 0,1% ТФУК, фаза B = вода/ацетонитрил 5/95 + 0,1% ТФУК. 0-1,5 мин (A: 98%, B: 2%), 1,5-8,0 мин (A: 0%, B: 100%), 8,0-11,0 мин (A: 0%, B: 100%), 11,0-11,1 мин (A: 98%, B: 2%); диодный детектор УФ-обнаружения: 200-400 нм; объем впрыска: 5 мкл.
Метод B): Waters 2795 Alliance HT Micromass ZQ. Колонка 4,6×75 мм из нержавеющей стали, заполненная 3,5 мкм Symmetry RP C-18; скорость истечения 1,0 мл/мин; подвижная фаза: фаза A = вода/ацетонитрил 95/5 + 0,05 ТФУК, фаза B = вода/ацетонитрил 5/95 + 0,05 ТФУК. 0-1,0 мин (A: 95%, B: 5%), 1,0-11,0 мин (A: 0%, B: 100%), 11,0-12,0 мин (A: 0%, B: 100%), 12,0-12,1 мин (A: 95%, B: 5%); диодный детектор УФ-обнаружения: 200-400 нм; объем впрыска: 5 мкл.
Метод C): система Waters Micromass ZQ 2996. Колонка 3,0×50 мм из нержавеющей стали, заполненная 5 мкм XTerra RP C-18; скорость истечения 1 мл/мин; подвижная фаза: фаза A=0,1% муравьиная кислота в воде, фаза B=0,07% муравьиная кислота в ацетонитриле. 0-0,5 мин (A: 95%, B: 5%), 0,5-6,0 мин (A: 0%, B: 100%), 6,0-6,5 мин (A: 95%, B: 5%), 6,5-7 мин (A: 95%, B: 5%); диодный детектор УФ-обнаружения: 200-400 нм; объем впрыска: 3 мкл.
Все масс-спектры записаны методом электрораспыления ионизацией в виде положительно заряженного иона (ESI-метод).
Большинство реакций контролировались с помощью тонкослойной хроматографии на силикагельных пластинах 0,25 мм Macherey-Nagel (60F-2254) с визуализацией в УФ-свете. Флэш-хроматография выполнялась на силикагеле (220-440 меш, Fluka). Температуры плавления измерялись на аппарате Buchi B-540.
Пример 1
(4-Фторфенил)-{3-[5-(4-фторфенил)-4H-[1,2,4]триазол-3-ил]пиперидин-1-ил}метанон
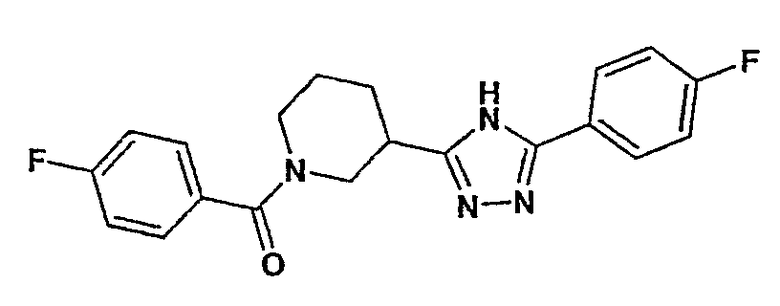
1(A). Этиловый эфир 1-(4-фторбензоил)пиперидин-3-карбоновой кислоты
К смеси этилнипекотата (2 г, 12,72 ммоль) в ТГФ (25 мл, 0,5M) добавляют DIEA (4,79 мл, 27,99 ммоль). Реакционную смесь охлаждают до 0°C и к смеси медленно добавляют 4-фторбензоилхлорид (2,5 г - 15,76 ммоль). Реакционной смеси дают нагреться до комнатной температуры и смесь перемешивают в течение 24 часов. Раствор концентрируют и последовательно добавляют ДХМ и 1 н. HCl. Водную фазу отделяют и органическую фазу экстрагируют дважды 1 н. HCl и дважды водой, сушат над Na2SO4, фильтруют и концентрируют, получая 1,15 г (33%) этилового эфира 1-(4-фторбензоил)пиперидин-3-карбоновой кислоты в виде бесцветного масла, которое используют далее без дополнительной очистки.
1(B). 1-(4-Фторбензоил)пиперидин-3-карбоновая кислота
Этиловый эфир 1-(4-фторбензоил)пиперидин-3-карбоновой кислоты (1,15 г, 4,42 ммоль) добавляют к смеси EtOH/NaOH 3 н. :1/1 (8 мл) и полученный гетерогенный раствор перемешивают при комнатной температуре в течение 1 часа. К смеси добавляют дымящую HCl до достижения pH=1. Раствор выливают в ДХМ. Органический слой отделяют и водную фазу дважды экстрагируют ДХМ. Объединенную органическую фазу дважды промывают водой. Раствор сушат над Na2SO4, фильтруют и концентрируют, получая 1,13 г (100%) 1-(4-фторбензоил)пиперидин-3-карбоновой кислоты в виде масла оранжевого цвета, которое используют далее без дополнительной очистки.
1(C). трет-Бутиловый эфир N'-[1-(4-Фторбензоил)пиперидин-3-карбонил]гидразинкарбоновой кислоты
К раствору 1-(4-Фторбензоил)пиперидин-3-карбоновой кислоты (1,13 г, 4,50 ммоль) в ДХМ (6,5 мл) добавляют избыточное количество трет-бутилового эфира гидразинкарбоновой кислоты (0,59 г, 4,50 ммоль), HOBT (0,69 г, 4,50 г) и EDCI·HCl (1,29 г, 6,758 ммоль). Смесь перемешивают при комнатной температуре в течение 72 часов. Растворитель удаляют при пониженном давлении и остаток разбавляют ДХМ. Органический слой промывают дважды водой, дважды 1 н. HCl и снова дважды водой. Органический слой сушат над Na2SO4, фильтруют и упаривают, получая 1,19 г (73%) трет-бутилового эфира N'-[1-(4-фторбензоил)пиперидин-3-карбонил]гидразинкарбоновой кислоты в виде бесцветного полутвердого вещества.
1(D). Гидразид 1-(4-фторбензоил)пиперидин-3-карбоновой кислоты
трет-Бутиловый эфир N'-[1-(4-фторбензоил)пиперидин-3-карбонил]гидразинкарбоновой кислоты растворяют в 8 мл 4 н. HCl (раствор в диоксане). Полученную реакционную смесь перемешивают при комнатной температуре в течение 1 часа и концентрируют, получая 0,88 г (100%) гидрохлорида гидразида 1-(4-фторбензоил)пиперидин-3-карбоновой кислоты в виде твердого белого вещества.
1(E). (4-Фторфенил)-{3-[5-(4-фторфенил)-4H-[1,2,4]триазол-3-ил]пиперидин-1-ил}метанон.
Раствор 4-фторбензонитрила (0,48 г, 4,02 ммоль) в метаноле (3 мл) обрабатывают металлическим натрием (77 мг, 0,35 ммоль) и перемешивают при температуре окружающей среды в течение 1 часа. После этого смесь добавляют к раствору гидразида 1-(4-фторбензоил)пиперидин-3-карбоновой кислоты (0,89 г, 3,35 ммоль) в метаноле (2 мл) и полученный раствор кипятят с обратным холодильником в течение 72 часов. Смесь концентрируют, растворяют в воде и нейтрализуют 1 н. HCl. Водную фазу экстрагируют ДХМ и органический слой сушат над Na2SO4, фильтруют и концентрируют. Очистка SPE с обращенной фазой (вода/ACN 45/55) приводит к получению 87 мг (7%) (4-фторфенил)-{3-[5-(4-фторфенил)-4H-[1,2,4]триазол-3-ил]пиперидин-1-ил}метанона в виде твердого белого вещества.
Rf=0,16 (ДХМ/MeOH:98/2); ЖХМС (Tr): 3,66 мин (метод C); МС (ES+) m/z: 369,2; т.пл.=95°C;
1H-ЯМР (CDCl3), δ (м.д.): 8,50 (с, NH), 8,05 (м, 2H), 7,52 (м, 2H), 7,15-7,03 (м, 4H), 3,65-3,30 (м, 4H), 2,45 (м, H), 1,85-1,52 (м, 4H).
Пример 2
(4-Фторфенил)-[3-(4-фторфенилэтинил)пиперидин-1-ил]метанон
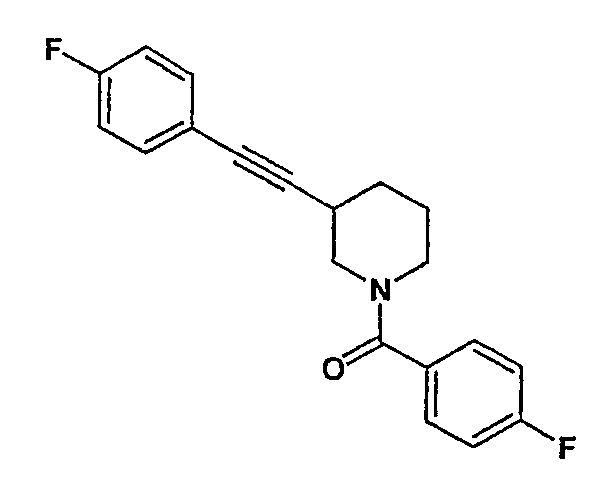
2(A). трет-Бутиловый эфир 3-(2,2-дибромвинил)пиперидин-1-карбоновой кислоты
К смеси CBr4 (1,63 г, 4,92 ммоль) и PPh3 (1,29 г, 4,92 ммоль) в ДХМ (25 мл) при комнатной температуре добавляют 1 г (4,69 ммоль) трет-бутилового эфира 3-формилпиперидин-1-карбоновой кислоты (коммерчески доступен). Реакционную смесь перемешивают при комнатной температуре в течение 24 часов и растворитель удаляют. Сырой продукт очищают флэш-хроматографией (циклогексан/AcOEt 90/10), получая 0,15 г (9%) трет-бутилового эфира 3-(2,2-дибромвинил)пиперидин-1-карбоновой кислоты в виде бесцветного масла.
2(B). трет-Бутиловый эфир 3-этинилпиперидин-1-карбоновой кислоты
К раствору трет-бутилового эфира 3-(2,2-дибромвинил)пиперидин-1-карбоновой кислоты (0,15 г, 0,42 ммоль) в ТГФ (1 мл) при -78°C добавляют 0,5 мл 2,5 M н-BuLi в гексане (1,23 ммоль). Смесь выдерживают в течение 1 часа при -78°C, затем гасят 1 мл воды и водную фазу экстрагируют AcOEt. Объединенную органическую фазу сушат над K2CO3, фильтруют и упаривают, получая 80 мг (93%) трет-бутилового эфира 3-этинилпиперидин-1-карбоновой кислоты в виде твердого белого вещества.
2(C). трет-Бутиловый эфир 3-(4-фторфенилэтинил)пиперидин-1-карбоновой кислоты
К суспензии CuI (4 мг, 0,02 ммоль) в Et3N (1 мл) добавляют трет-бутиловый эфир 3-этинилпиперидин-1-карбоновой кислоты (80 мг, 0,38 ммоль) с последующим добавлением PdCl2(PPh3)2 (13 мг, 0,02 ммоль) и 1-йод-4-фторбензола (85 мг, 0,38 ммоль). Смесь перемешивают в течение 1 часа при комнатной температуре, затем нагревают до 60°C и выдерживают при этой температуре в течение 12 часов. Et3N удаляют выпариванием. Продукт очищают флэш-хроматографией (ДХМ 100%), получая 0,1 г (89%) трет-бутилового эфира 3-(4-фторфенилэтинил)пиперидин-1-карбоновой кислоты в виде масла желтого цвета.
2(D). 3-(4-Фторфенилэтинил)пиперидин
трет-Бутиловый эфир N'-3-(4-фторфенилэтинил)пиперидин-1-карбоновой кислоты (80,1 мг, 0,34 ммоль) растворяют в 0,2 мл 4 н. HCl (раствор в диоксане). Полученную реакционную смесь перемешивают при комнатной температуре в течение 1 часа и концентрируют, получая 0,14 г (100%) гидрохлорида 3-(4-фторфенилэтинил)пиперидина в виде твердого вещества коричневого цвета.
2(E). (4-Фторфенил)-[3-(4-фторфенилэтинил)пиперидин-1-ил]метанон
К смеси гидрохлорида 3-(4-фторфенилэтинил)пиперидина (0,14 г, 0,58 ммоль) в ТГФ (2,3 мл, 0,5M) добавляют DIEA (0,5 мл, 2,92 ммоль). Реакционную смесь охлаждают до 0°C и к смеси медленно добавляют 4-фторбензоилхлорид (0,139 г - 0,87 ммоль). Реакционной смеси дают нагреться до комнатной температуры и смесь перемешивают в течение 24 часов. Раствор концентрируют и последовательно добавляют ДХМ и 1 н. HCl. Водную фазу отделяют и органическую фазу экстрагируют дважды 1 н. HCl и дважды водой, сушат над Na2SO4, фильтруют и концентрируют. Сырой продукт очищают флэш-хроматографией (ДХМ/MeOH:98/2), получая 0,86 мг (45%) (4-фторфенил)-[3-(4-фторфенилэтинил)пиперидин-1-ил]метанона в виде масла коричневого цвета. Rf=0,39 (ДХМ/MeOH:98/2); ЖХМС (Tr): 4,14 мин (метод C); МС (ES+) m/z: 326,2;
1H-ЯМР (CDCl3), δ (м.д.): 8,10 (д, 2H), 7,60 (д, 2H), 7,15 (д, 2H), 6,94 (д, 2H), 3,47-3,30 (м, 4H), 2,54 (м, H), 1,63-1,50 (м, 4H).
Пример 3
{3-[3-(4-Метоксифенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}фенилметанон
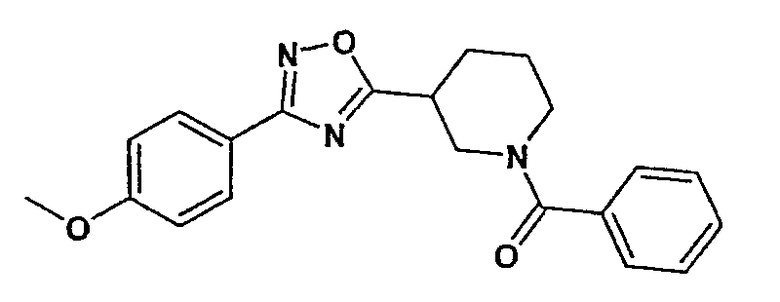
3(A). N-Гидрокси-4-метоксибензамидин
К смеси 4-метоксибензонитрила (1,07 г, 8 ммоль) и DIEA (4,11 мл, 24 ммоль) в EtOH (12,5 мл) добавляют 1,7 г гидрохлорида гидроксиламина (24 ммоль), реакционную смесь нагревают до 70°C и выдерживают при этой температуре в течение 48 часов. Половину объема растворителя удаляют при пониженном давлении. Смесь выливают в ДХМ (100 мл) и воду (30 мл). К смеси добавляют 2,5 мл 1 н. NaOH до достижения pH=9-10. Органическую фазу отделяют и водную фазу экстрагируют ДХМ. Органические слои объединяют, промывают водой, сушат над MgSO4, фильтруют и упаривают при пониженном давлении, получая 1,3 г (98%) N-гидрокси-4-метоксибензамидина в виде бесцветного масла, которое используют далее без дополнительной очистки.
3(B). трет-Бутиловый эфир 3-[3-(4-метоксифенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-карбоновой кислоты
Смесь N-гидрокси-4-метоксибензамидина (0,20 г, 1,50 ммоль), 1-Boc-пиперидин-3-карбоновой кислоты (0,34 г, 1,50 ммоль), HOBT (0,23 г, 1,50 ммоль) и EDCI·HCl (0,43 г, 2,25 ммоль) в диоксане (2,5 мл) перемешивают при комнатной температуре в течение 7 часов. После этого смесь нагревают до 80°C и выдерживают при этой температуре в течение ночи с использованием карусели Радлея (the carousel Radley's). Смесь концентрируют. Органический слой промывают водой, 1 н. NaOH и снова водой. Органический слой сушат над Na2SO4, фильтруют и упаривают. Сырой продукт очищают флэш-хроматографией (ДХМ/MeOH:99/1), получая 0,39 мг (72%) трет-бутилового эфира 3-[3-(4-метоксифенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-карбоновой кислоты в виде твердого белого вещества.
3(C). 3-[3-(4-Метоксифенил)-[1,2,4]оксадиазол-5-ил]пиперидин
трет-Бутиловый эфир 3-[3-(4-метоксифенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-карбоновой кислоты (0,39 г, 1,08 ммоль) растворяют в 2 мл 4 н. HCl (раствор в диоксане). Реакционную смесь перемешивают при комнатной температуре в течение 1 часа и концентрируют, получая 0,320 г (100%) гидрохлорида 3-[3-(4-метоксифенил)-[1,2,4]оксадиазол-5-ил]пиперидина в виде твердого вещества коричневого цвета.
3(D). {3-[3-(4-Метоксифенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}фенилметанон
К смеси гидрохлорида 3-[3-(4-метоксифенил)-[1,2,4]оксадиазол-5-ил]пиперидина (0,320 г, 1,08 ммоль) и ТГФ (2,3 мл, 0,5M) добавляют пиридин (0,3 мл, 3,78 ммоль). Реакционную смесь охлаждают до 0°C и к смеси медленно добавляют 4-фторбензоилхлорид (0,172 г, 0,87 ммоль). Реакционной смеси дают нагреться до комнатной температуры и смесь перемешивают в течение 24 часов. Раствор концентрируют и к раствору последовательно добавляют ДХМ и 1 н. HCl. Водную фазу отделяют и органическую фазу экстрагируют дважды 1 н. HCl и дважды водой, сушат над Na2SO4, фильтруют и концентрируют. Сырой продукт очищают флэш-хроматографией (ДХМ/MeOH:99/1), получая 0,26 мг (66%) {3-[3-(4-метоксифенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}фенилметанона в виде белого порошка.
Rf=0,40 (ДХМ/MeOH:99/2); ЖХМС (Tr): 4,35 мин (метод C); т.пл.=121°C; МС (ES+) m/z: 364,5;
1H-ЯМР (CDCl3), δ (м.д.): 7,95 (д, 2H), 7,51 (м, H), 7,44-7,37 (м, 4H), 6,83 (д, 2H), 3,75 (с, 3H), 3,63-3,34 (м, 4H), 2,48 (м, H), 1,90-1,50 (м, 4H).
Пример 4
(4-Фторфенил)-{3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон
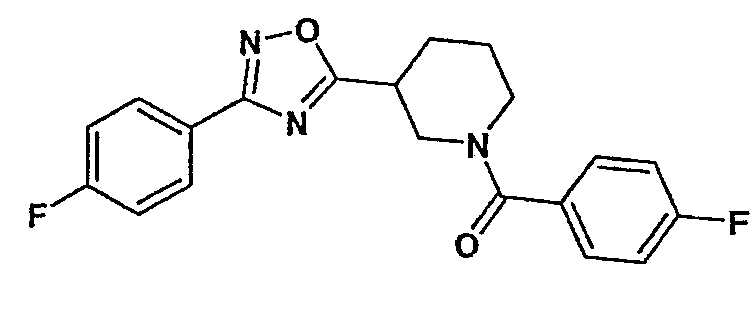
4(A). трет-Бутиловый эфир 3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-карбоновой кислоты
Соединение получают в соответствии с методикой, описанной в примере 3, используя 4-фтор-N-гидроксибензамидин (коммерчески доступен) и 1-Boc-пиперидин-3-карбоновую кислоту (выход: 60%).
4(B). 3-[3-(4-Фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин
Соединение получают в соответствии с методикой, описанной в примере 3(C) (выход: 100%).
4(C). (4-Фторфенил)-{3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон
Соединение получают в соответствии с методикой, описанной в примере 3(D), используя 4-фторбензоилхлорид в качестве выбранного ацилхлорида. Выход: 22%; т.пл.=118,5-121,5°C (белый порошок); Rf=0,30 (ДХМ/MeOH:98/2); ЖХМС (Tr): 4,87 мин (метод C); МС (ES+) m/z: 370,1;
1H-ЯМР (CDCl3), δ (м.д.): 8,10 (д, 2H), 7,51 (м, 2H), 7,15-7,03 (м, 4H), 3,63-3,34 (м, 4H), 2,48 (м, H), 1,90-1,50 (м, 4H).
Элементный анализ. Вычислено для C20H17F2N3O2: C, 65,03; H, 4,64; N, 11,38; F, 10,29. Найдено: C, 64,89; H, 4,75; N, 11,26; F, 10,36.
Пример 5
{3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}фенилметанон
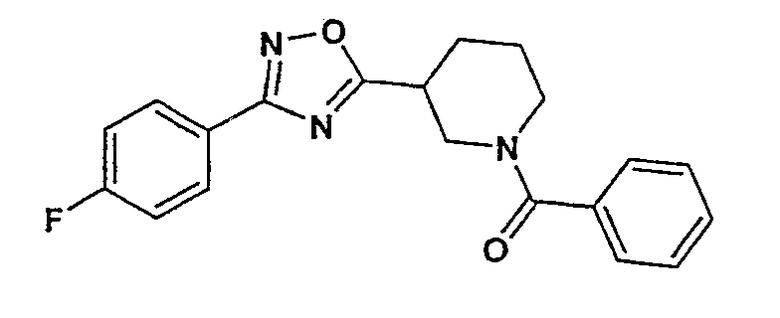
Соединение получают в соответствии с методикой, описанной в примере 3(D), используя 4-фторбензоилхлорид в качестве ацилхлорида и гидрохлорид 3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидина (полученный ранее в примере 4(B)). Выход: 30% (белый порошок); т.пл.=82-83°C; Rf=0,25 (ДХМ/MeOH:98/2);
ЖХМС (Tr): 4,70 мин (метод C); МС (ES+) m/z: 352,3.
1H-ЯМР (CDCl3), δ (м.д.): 8,10 (д, 2H), 7,51 (м, 3H), 7,15-7,03 (м, 4H), 3,63-3,34 (м, 4H), 2,48 (м, H), 1,90-1,50 (м, 4H).
Пример 6
(3-Фторфенил)-{3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон
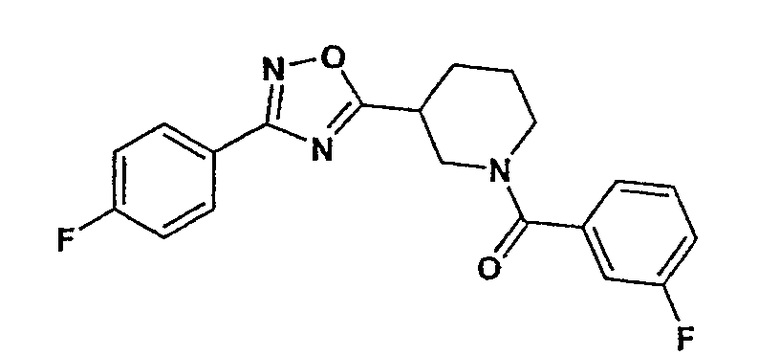
Соединение получают в соответствии с методикой, описанной в примере 3(D), используя 3-фторбензоилхлорид в качестве выбранного ацилхлорида и гидрохлорид 3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидина (полученный в соответствии с методикой примера 4(B).
Выход: 42%; т.пл.=139-140°C (порошок бежевого цвета); Rf=0,32 (ДХМ/MeOH:98/2); ЖХМС (Tr): 4,87 мин (метод C); МС (ES+) m/z: 370,1;
1H-ЯМР (CDCl3), δ (м.д.): 8,05 (д, 2H), 7,40-7,50 (м, 3H), 7,22-7,00 (м, 3H), 3,65-3,32 (м, 4H), 2,53 (м, H), 1,86-1,45 (м, 4H).
Пример 7
(4-Фторфенил)-[3-(3-фенил-[1,2,4]оксадиазол-5-ил)пиперидин-1-ил]метанон
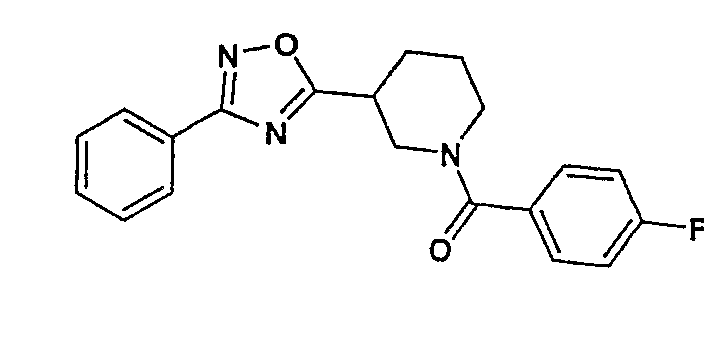
7(A). трет-Бутиловый эфир 3-(3-фенил-[1,2,4]оксадиазол-5-ил)пиперидин-1-карбоновой кислоты
Соединение получают в соответствии с методикой, описанной в примере 3(B), используя N-гидроксибензамидина (коммерчески доступен) и 1-Boc-пиперидин-3-карбоновую кислоту (выход: 58%).
7(B). 3-(3-Фенил-[1,2,4]оксадиазол-5-ил)пиперидин
Соединение получают в соответствии с методикой, описанной в примере 3(C) (выход: 94%).
7(C). (4-Фторфенил)-[3-(3-фенил-[1,2,4]оксадиазол-5-ил)пиперидин-1-ил]метанон
Соединение получают в соответствии с методикой, описанной в примере 3(D), используя 4-фторбензоилхлорид в качестве выбранного ацилхлорида. Выход: 35%; т.пл.=78-79°C (белый порошок); Rf=0,24 (ДХМ/MeOH:98/2); ЖХМС (Tr): 4,75 мин (метод C); МС (ES+) m/z: 352,3.
1H-ЯМР (CDCl3), δ (м.д.): 8,10 (д, 2H), 7,48-7,32 (м, 4H), 7,22-7,15 (м, 3H), 3,65-3,32 (м, 4H), 2,53 (м, H), 1,86-1,45 (м, 4H).
Пример 8
(3-Фторфенил)-[3-(3-фенил-[1,2,4]оксадиазол-5-ил)пиперидин-1-ил]метанон
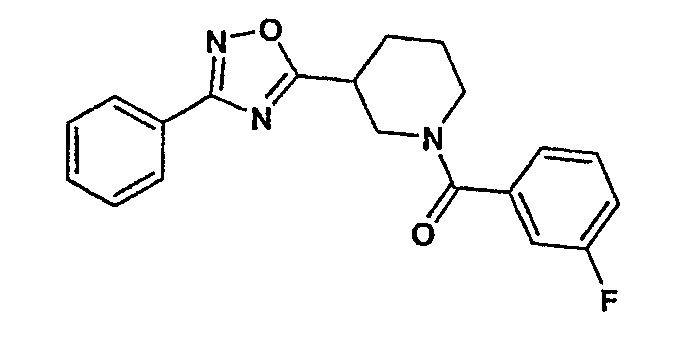
Соединение получают в соответствии с методикой, описанной в примере 3(D), используя 3-фторбензоилхлорид в качестве выбранного ацилхлорида и гидрохлорид 3-(3-фенил-[1,2,4]оксадиазол-5-ил)пиперидина (получение которого описано в примере 7). Выход: 53% (масло желтого цвета); Rf=0,25 (ДХМ/MeOH:98/2); ЖХМС (Tr): 4,77 мин (метод C); МС (ES+) m/z: 352,3.
1H-ЯМР (CDCl3), δ (м.д.): 8,00-7,72 (м, 2H), 7,48-7,40 (м, 3H), 7,32-7,22 (м, 4H), 3,65-3,32 (м, 4H), 2,53 (м, H), 1,86-1,45 (м, 4H).
Пример 9
(3-Фторфенил)-{3-[3-(3-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон
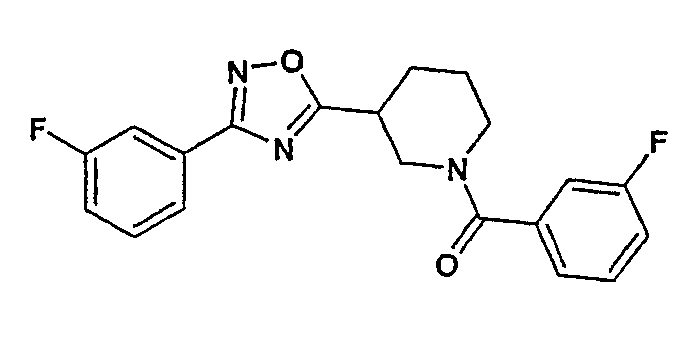
9(A). N-Гидрокси-3-фторбензамидин
К смеси 3-фтор-бензонитрила (1,21 г, 10 ммоль) и DIEA (5,20 мл, 30 ммоль) в EtOH (20 мл) добавляют 2,08 г гидрохлорида гидроксиламина (30 ммоль), реакционную смесь нагревают до 70°C и выдерживают при этой температуре в течение 48 часов. Половину объема растворителя удаляют при пониженном давлении. Смесь выливают в ДХМ (100 мл) и воду (30 мл). К смеси добавляют 2,5 мл 1 н. NaOH до достижения pH=9-10. Органическую фазу отделяют и водную фазу экстрагируют ДХМ. Органические слои объединяют, промывают водой, сушат над MgSO4, фильтруют и упаривают при пониженном давлении, получая 1,48 г (96%) N-гидрокси-3-фторбензамидина в виде твердого белого вещества, которое используют далее без дополнительной очистки.
9(B). трет-Бутиловый эфир 3-[3-(3-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-карбоновой кислоты
Соединение получают в соответствии с методикой, описанной в примере 3(B), используя N-гидрокси-3-фторбензамидин и 1-Boc-пиперидин-3-карбоновую кислоту (выход: 78%).
9(C). 3-[3-(3-Фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин
Соединение получают в соответствии с методикой, описанной в примере 3(C) (выход: 96%).
9(D). (3-Фторфенил)-{3-[3-(3-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон
Соединение получают в соответствии с методикой, описанной в примере 3(D), используя 3-фторбензоилхлорид в качестве выбранного ацилхлорида (выход: 54%).
Выход: 53% (масло желтого цвета); Rf=0,31 (ДХМ/MeOH:98/2); ЖХМС (Tr): 4,88 мин (метод C); МС (ES+) m/z: 370,3;
1H-ЯМР (CDCl3), δ (м.д.): 7,96-7,72 (м, 2H), 7,66 (м, H), 7,42-7,30 (м, 2H), 7,30-7,25 (м, 2H), 7,00 (м, 1H), 3,65-3,32 (м, 4H), 2,53 (м, H), 1,86-1,45 (м, 4H).
Пример 10
(4-Фторфенил)-{3-[3-(3-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон
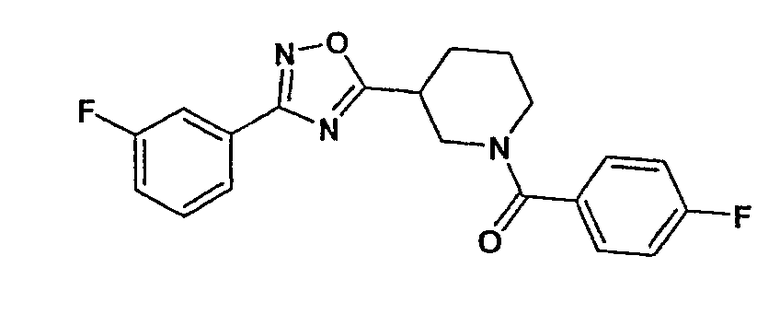
Соединение получают в соответствии с методикой, описанной в примере 3(D), используя 4-фторбензоилхлорид в качестве выбранного ацилхлорида и гидрохлорид 3-[3-(3-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидина (получение которого описано в примере 9). Выход: 50% (масло желтого цвета); т.пл.=86-89°C (порошок бежевого цвета); Rf=0,28 (ДХМ/MeOH:98/2); ЖХМС (Tr): 4,88 мин (метод C); МС (ES+) m/z: 370,3;
1H-ЯМР (CDCl3), δ (м.д.): 8,05-7,90 (м, 2H), 7,30-7,23 (м, 2H), 7,25-7,15 (м, 3H), 7,00 (м, 1H), 3,65-3,32 (м, 4H), 2,53 (м, H), 1,86-1,45 (м, 4H).
Пример 11
R -(4-Фторфенил)-{3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон
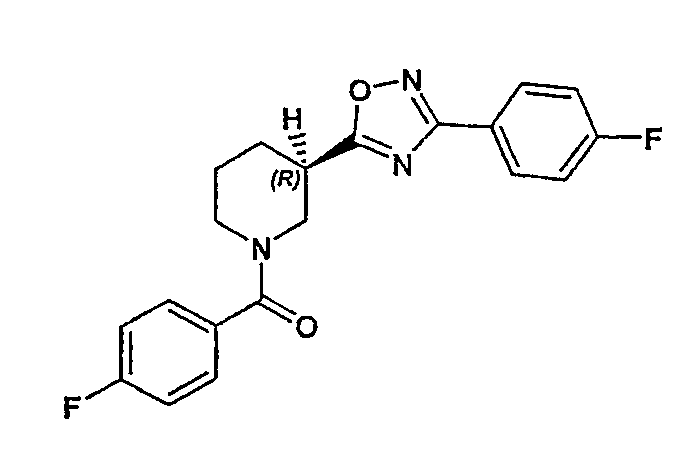
11(A). трет-Бутиловый эфир R-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-карбоновой кислоты
Соединение получают в соответствии с методикой, описанной в примере 3(B), используя 4-фтор-N-гидроксибензамидин (коммерчески доступен) и R-1-Boc-пиперидин-3-карбоновую кислоту (выход: 79%).
11(B). R-3-[3-(4-Фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин
Соединение получают в соответствии с методикой, описанной в примере 3C) (выход: 68%).
11(C). R-(4-Фторфенил)-{3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон
Соединение получают в соответствии с методикой, описанной в примере 3(D), используя 4-фторбензоилхлорид в качестве выбранного ацилхлорида. Выход: 28%; т.пл.=98°C (белый порошок); Rf=0,30 (ДХМ/MeOH:98/2); ЖХМС (Tr): 4,87 мин (метод C); МС (ES+) m/z: 370,1;
1H-ЯМР CDCl3, δ (м.д.): 8,05 (д, 2H), 7,48 (м, 2H), 7,15-7,03 (м, 4H), 3,63-3,34 (м, 4H), 2,48 (м, H), 1,90-1,50 (м, 4H).
Пример 12
S -(4-Фторфенил)-{3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон
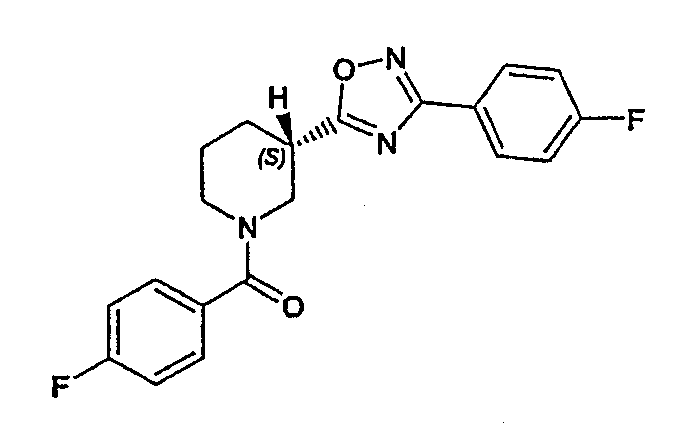
12(A). трет-Бутиловый эфир S-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-карбоновой кислоты
Соединение получают в соответствии с методикой, описанной в примере 3(B), используя 4-фтор-N-гидроксибензамидин (коммерчески доступен) и S-1-Boc-пиперидин-3-карбоновую кислоту (выход: 84%)
12(B). S-3-[3-(4-Фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин
Соединение получают в соответствии с методикой, описанной в примере 3(C) (выход: 63%).
12(B). S-(4-Фторфенил)-{3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон
Соединение получают в соответствии с методикой, описанной в примере 3(D), используя 4-фторбензоилхлорид в качестве выбранного ацилхлорида. Выход: 51%; т.пл.=99°C (белый порошок); Rf=0,30 (ДХМ/MeOH:98/2); [α]D 20=+103° (c=1, CHCl3); ЖХМС (Tr): 4,87 мин (метод C); МС (ES+) m/z: 370,1.
1H-ЯМР (CDCl3), δ (м.д.): 8,05 (д, 2H), 7,48 (м, 2H), 7,15-7,03 (м, 4H), 3,63-3,34 (м, 4H), 2,48 (м, H), 1,90-1,50 (м, 4H).
Пример 13
S -(тиофен-2-ил)-{3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон
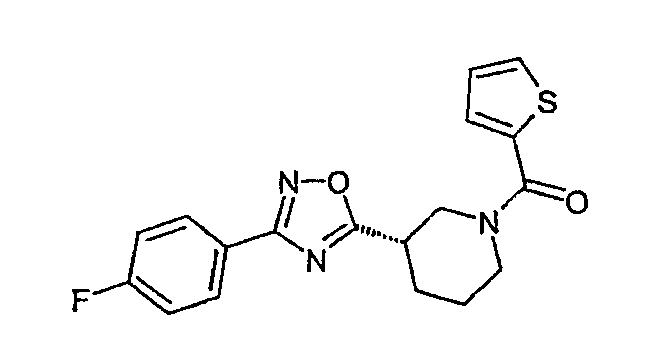
К суспензии гидрохлорида S-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидина (100 мг, 0,52 ммоль, получение которого описано в примере 12) в сухом дихлорметане (3 мл) по каплям добавляют триэтиламин (123 мкл, 0,881 ммоль) и тиофен-2-карбонилхлорид (79 мкл, 0,352 ммоль) при 0°C. Реакционной смеси дают возможность нагреться до комнатной температуры и смесь перемешивают в течение ночи в атмосфере азота. После этого раствор обрабатывают 1 н. HCl (5 мл) и фазы разделяют. Органический слой промывают последовательно 1 н. HCl (5 мл), насыщенным водным раствором NaHCO3 (5 мл) и водой (5 мл), затем сушат над Na2SO4 и упаривают при пониженном давлении. Сырой продукт пропускают через силикагелевый картридж (картридж: VARIAN HF, Mega Bond Elut SI, 5 г; элюирование с градиентом: от ДХМ 100% до ДХМ/MeOH 95/5), растворитель выпаривают при пониженном давлении, получая 97 мг S-(тиофен-2-ил)-{3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанона в виде твердого белого вещества.
Выход: 78%; т.пл.=82-83°C; [α]D 20=+11° (c=1,08, CHCl3); ЖХМС (Tr): 9,86 мин (метод A); МС (ES+) m/z: 358,1.
1H-ЯМР (CDCl3, 338 K, 300 МГц), δ (м.д.): 8,06 (дд, 2H); 7,43 (дд, 1H); 7,34 (дд, 1H); 7,14 (дд, 2H); 7,04 (дд, 1H); 4,62 (м, 1H); 4,24 (м, 1H); 3,56 (дд, 1H); 3,36-3,22 (м, 2H); 2,41-2,29 (м, 1H); 2,10-1,89 (м, 2H); 1,81-1,65 (м, 1H).
Пример 14
{(S)-3-[3-(4-Фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-4-ил}-(4-метил-2-пиразин-2-илтиазол-5-ил)метанон

Соединение получают в соответствии с методикой, описанной в примере 13, используя 4-метил-2-(2-пиразинил)-1,3-тиазол-5-карбонилхлорид в качестве выбранного ацилхлорида и гидрохлорид S-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидина (полученный в соответствии с методикой примера 12).
Выход: 47% (не совсем белое твердое вещество); т.пл.=147-148°C; [α]D 20=+120° (c=0,34, CHCl3); ЖХМС (Tr): 9,41 мин (метод A); МС (ES+) m/z: 45H,0.
1H-ЯМР (CDCl3, 338 K, 300 МГц), δ (м.д.): 9,40 (д, 1H); 8,59 (д, 1H); 8,52 (дд, 1H); 8,07 (дд, 2H); 7,14 (дд, 2H); 4,45 (дд уш., 1H); 4,04 (уш.дт, 1H); 3,62 (дд, 1H); 3,39-3,23 (м, 2H); 2,55 (с, 3H); 2,42-2,30 (м, 1Н); 2,12-1,91 (м, 2H); 1,80-1,64 (м, 1H).
Пример 15
{(S)-3-[3-(4-Фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}-(2-фенилтиазол-4-ил)метанон
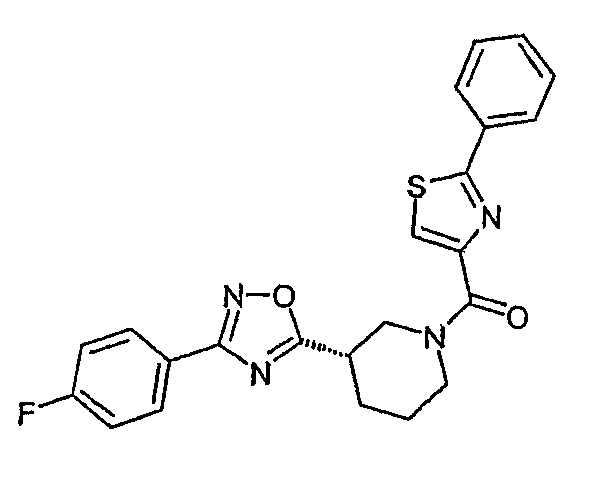
Соединение получают в соответствии с методикой, описанной в примере 13, используя 2-фенил-1,3-тиазол-4-карбонилхлорид в качестве выбранного ацилхлорида и гидрохлорид S-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидина (полученный в соответствии с методикой примера 12).
Выход: 26% (масло оранжевого цвета); [α]D 20=+176° (c=1,13, CHCl3); ЖХМС (Tr): 10,83 мин (метод A); МС (ES+) m/z: 4H5,1.
1H-ЯМР (CDCl3, 338 K, 300 МГц), δ (м.д.): 8,06 (дд, 2H); 7,96 (м, 2H); 7,95 (с, 1H); 7,43 (м, 3H); 7,14 (дд, 2H); 5,04 (уш.д, 1H); 4,59 (уш.д, 1H); 3,70-3,38 (м, 2H); 3,27 (дд, 1H); 2,46-2,34 (м, 1H); 2,15-1,92 (м, 2H); 1,91-1,74 (м, 1H).
Пример 16
{(S)-3-[3-(4-Фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}-(2-метил-6-трифторметилпиридин-3-ил)метанон
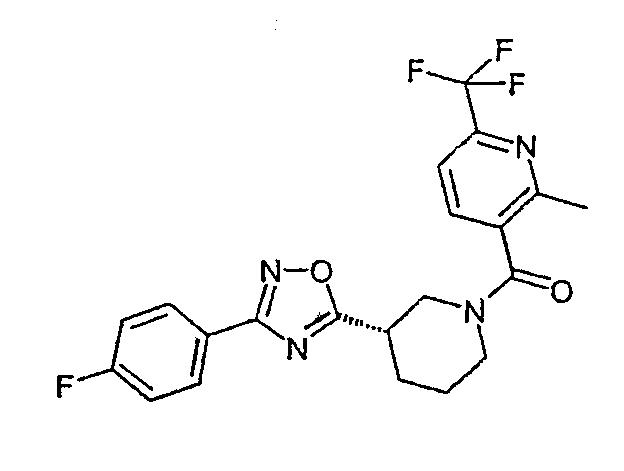
Соединение получают в соответствии с методикой, описанной в примере 13, используя 2-метил-6-(трифторметил)пиридин-3-карбонилхлорид в качестве выбранного ацилхлорида и гидрохлорид S-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидина (полученный в соответствии с методикой примера 12).
Виход: 54% (твердое воскообразное вещество желтого цвета; [α]D 20=+80° (c=1,15, CHCl3); ЖХМС (Tr): 10,06 мин (метод A); МС (ES+) m/z: 4H5,1.
1H-ЯМР (CDCl3, 338 K, 300 МГц), δ (м.д.): 8,05 (м, 2H); 7,63 (д, 1H); 7,52 (д, 1H); 7,15 (дд, 2H); 4,70 (уш.м, 1H); 4,22 (уш.м, 1H); 3,64 (м, 1Н); 3,54-3,04 (уш.м, 2H); 2,60 (с, 3H); 2,40-2,26 (м, 1H); 2,17-2,00 (м, 1H); 2,00-1,81 (уш.м, 1Н); 1,81-1,52 (уш.м, 1H).
Пример 17
(3,5-Диметилизоксазол-4-ил)-{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон
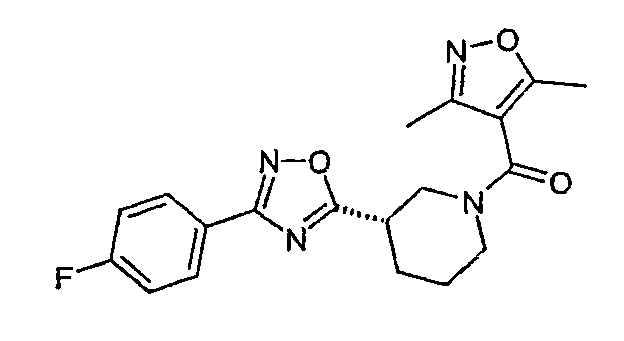
Соединение получают в соответствии с методикой, описанной в примере 13, используя 3,5-диметилизоксазол-4-карбонилхлорид в качестве выбранного ацилхлорида и гидрохлорид S-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидина (полученный в соответствии с методикой примера 12).
Выход: 20% (бесцветное масло); [α]D 20=+82° (c=0,88, CHCl3); ЖХМС (Tr): 9,10 мин (метод A); МС (ES+) m/z: 37H,1.
1H-ЯМР (CDCl3, 338 K, 300 МГц), δ (м.д.): 8,06 (дд, 2H); 7,15 (дд, 2H); 4,34 (уш.д, 1H); 3,88 (уш.д, 1H); 3,59 (дд, 1H); 3,34-3,17 (м, 2Н); 2,40 (с, 3Н); 2,33 (м, 1Н); 2,26 (с, 3Н); 2,12-1,89 (м, 2Н); 1,72-1,57 (м, 1Н).
Пример 18
{(S)-3-[3-(4-Фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}-[1,2,3]тиадиазол-4-илметанон
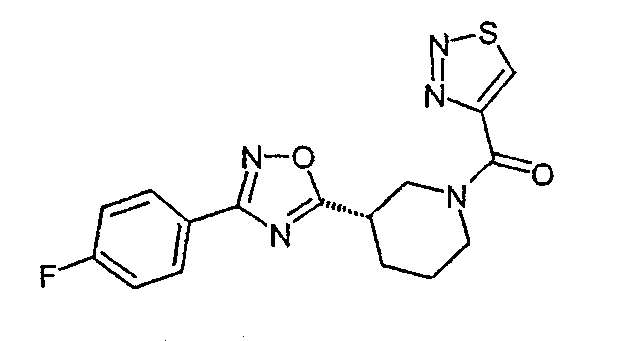
Соединение получают в соответствии с методикой, описанной в примере 13, используя 1,2,3-тиадиазол-4-карбонилхлорид в качестве выбранного ацилхлорида и гидрохлорид S-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидина (полученный в соответствии с методикой примера 12).
Выход: 44% (твердое вещество коричневатого цвета); т.пл.=90-91°С; [α]D 20=+104° (c=0,67, CHCl3); ЖХМС (Tr): 7,25 мин (метод A); МС (ES+) m/z: 36H,0.
1H-ЯМР (CDCl3, 343 K, 300 МГц), δ (м.д.): 9,09 (с, 1H); 8,04 (м, 2Н); 7,14 (дд, 2Н); 4,82 (уш.д, 1Н), 4,45 (уш.д, 1Н); 3,78 (уш.м, 1Н); 3,54-3,33 (м, 2Н), 2,45-2,33 (м, 1Н); 2,16-1,96 (м, 2Н); 1,91-1,76 (м, 1Н).
Пример 19
Бензотиазол-2-ил-{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон
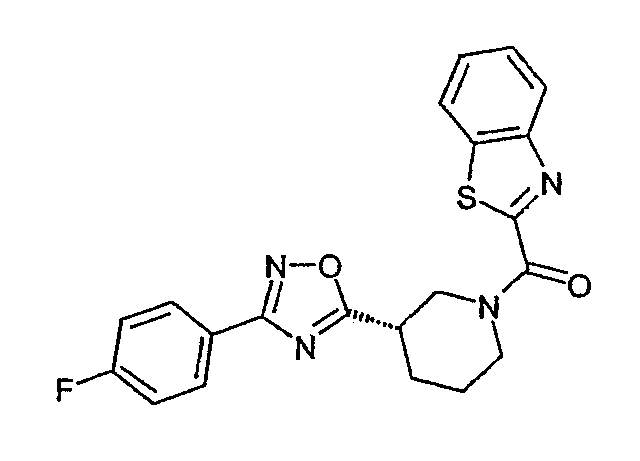
Соединение получают в соответствии с методикой, описанной в примере 13, используя 1,3-бензотиазол-2-карбонилхлорид в качестве выбранного ацилхлорида и гидрохлорид S-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидина (полученный в соответствии с методикой примера 12).
Выход: 70% (порошок бежевого цвета); т.пл.130-131°С; [α]D 20=+155° (c=0,8, CHCl3); ЖХМС (Tr): 8,11 мин (метод A); МС (ES+) m/z: 40H,1.
1H-ЯМР (CDCl3, 333 K, 300 МГц), δ (м.д.): 8,15-7,87 (м, 4Н); 7,58-7,41 (м, 2Н); 7,19-7,05 (дд, 2Н); 5,59-3,84 (уш.м, 2Н); 3,78-3,20 (м, 3Н); 2,47-2,30 (м, 1Н); 2,20-1,94 (м, 2Н); 1,92-1,73 (м, 1Н).
Пример 20
{(S)-3-[3-(4-Фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}-(5-метилизоксазол-3-ил)метанон
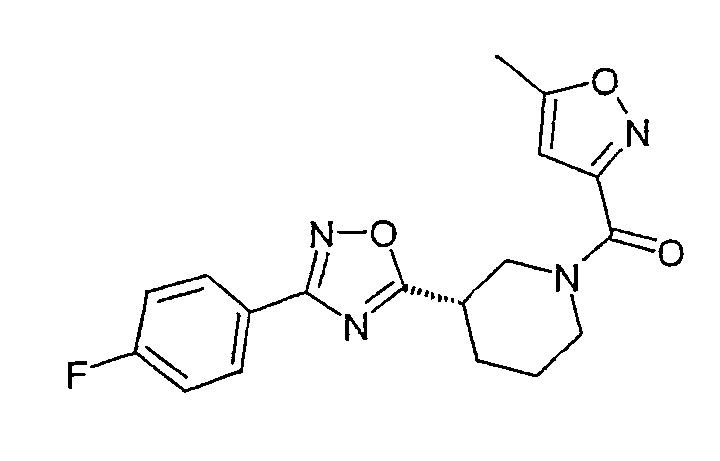
Соединение получают в соответствии с методикой, описанной в примере 13, используя 5-метилизоксазол-3-карбонилхлорид в качестве выбранного ацилхлорида и гидрохлорид S-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидина (полученный в соответствии с методикой примера 12).
Выход: 32% (бесцветное масло); [α]D 20=+112° (c=1,2, CHCl3); ЖХМС (Tr): 7,23 мин (метод A); МС (ES+) m/z: 357,1.
1H-ЯМР (CDCl3, 333 K, 300 МГц), δ (м.д.): 8,14-7,96 (м, 2H); 7,20-7,09 (м, 2H); 6,26 (с, 1H); 5,04-4,24 (уш.м, 2H); 3,97-3,58 (уш.м, 1H); 3,46-3,13 (м, 2H); 2,45 (с, 3H); 2,41-2,27 (м, 1H); 2,11-1,88 (м, 2H); 1,83-1,67 (м, 1H).
Пример 21
(1,5-Диметил-1H-пиразол-3-ил)-{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон
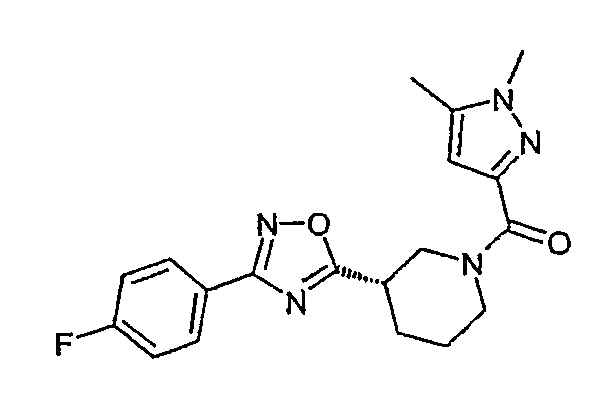
Соединение получают в соответствии с методикой, описанной в примере 13, используя 1,5-диметил-1H-пиразол-3-карбонилхлорид в качестве выбранного ацилхлорида и гидрохлорид S-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидина (полученный в соответствии с методикой примера 12).
Выход: 69% (твердое белое вещество); т.пл.=108-109°C; [α]D 20=+128° (c=0,9, CHCl3); ЖХМС (Tr): 7,00 мин (метод A); МС (ES+) m/z: 370,2.
1H-ЯМР (CDCl3, 338 K, 300 МГц), δ (м.д.): 8,12-8,02 (м, 2H); 7,20-7,09 (м, 2H); 6,42 (с, 1H); 5,06-4,95 (м, 1H); 4,70-4,54 (уш.м, 1H); 3,77 (с, 3H); 3,59-3,37 (уш.м, 1H); 3,36-3,03 (м, 2H), 2,40-2,24 (уш.м, 1H); 2,27 (с, 3H); 2,08-1,85 (м, 2H); 1,82-1,63 (м, 1H).
Пример 22
{(S)-3-[3-(4-Фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}-(4-трифторметилфенил)метанон
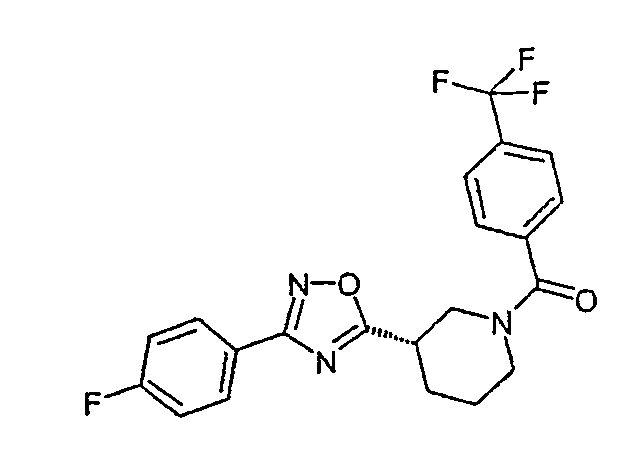
Соединение получают в соответствии с методикой, описанной в примере 13, используя 4-трифторметилбензоилхлорид в качестве выбранного ацилхлорида и гидрохлорид S-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидина (полученный в соответствии с методикой примера 12).
Выход: 54% (твердое белое вещество); т.пл.=109-110°C; [α]D 20=+95° (c=0,7, CHCl3); ЖХМС (Tr): 7,78 мин (метод A); МС (ES+) m/z: 42H,1.
1H-ЯМР (CDCl3, 333 K, 300 МГц), δ (м.д.): 8,11-8,0 (м, 2H); 7,67 (д, 2H); 7,52 (д, 2H); 7,20-7,09 (м, 2H); 4,58-4,17 (уш.м, 1H); 4,07-3,74 (уш.м 1H); 3,57 (дд, 1H); 3,38-3,17 (м, 2H); 2,40-2,25 (м, 1H); 2,15-1,85 (м, 2H); 1,78-1,58 (м, 1H).
Пример 23
4-{(S)-3-[3-(4-Фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-l-карбонил}бензонитрил
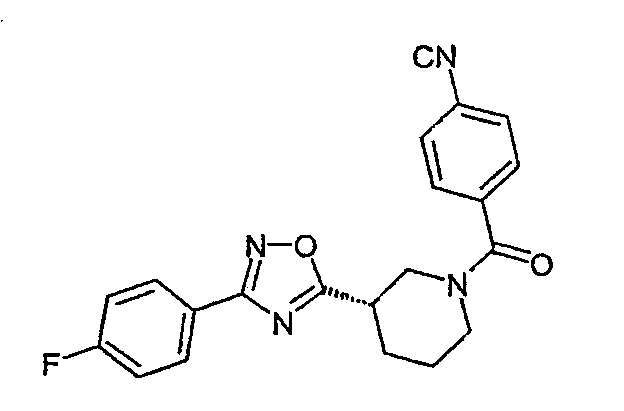
Соединение получают в соответствии с методикой, описанной в примере 13, используя 4-цианобензоилхлорид в качестве выбранного ацилхлорида и гидрохлорид S-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидина (полученный в соответствии с методикой примера 12).
Выход: 61% (твердое белое вещество); т.пл.=129-130°C; [α]D 20=+127° (c=1,1, CHCl3); ЖХМС (Tr): 7,25 мин (метод A); МС (ES+) m/z: 37H,1.
1H-ЯМР (CDCl3, 333 K, 300 МГц), δ (м.д.): 8,10-8,0 (м, 2H); 7,70 (дд, 2H); 7,51 (дд, 2H); 7,21-7,11 (м, 2H); 4,57-4,11 (уш.м, 1H); 4,05-3,73 (уш.м, 1H); 3,58 (дд, 1H); 3,40-3,17 (м, 2H); 2,40-2,26 (м, 1H); 2,16-1,85 (м, 2H); 1,78-1,58 (м, 1H).
Пример 24
{(S)-3-[3-(4-Фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}изоксазол-5-илметанон
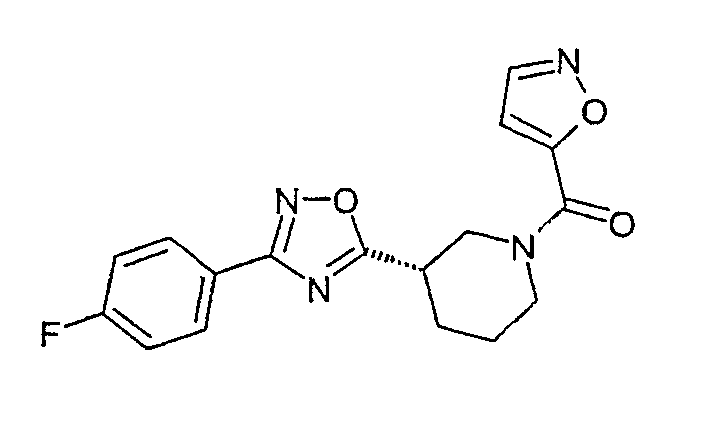
Соединение получают в соответствии с методикой, описанной в примере 13, используя изоксазол-5-карбонилхлорид в качестве выбранного ацилхлорида и гидрохлорид S-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидина (полученный в соответствии с методикой примера 12).
Выход: 50% (твердое белое вещество); т.пл.=93-94°C; [α]D 20=+127° (c=0,8, CHCl3); ЖХМС (Tr): 7,00 мин (метод A); МС (ES+) m/z: 34H,1.
1H-ЯМР (CDCl3, 333 K, 300 МГц), δ (м.д.): 8,28 (д, 1H); 8,10-8,0 (м, 2H); 7,20-7,09 (м, 2H); 6,75 (д, 1H); 4,87-4,27 (уш.м, 1H); 4,26-4,06 (м, 1H); 3,86-3,46 (уш.м, 1H); 3,46-3,20 (м, 2H); 2,46-2,27 (м, 1H); 2,18-1,88 (м, 2H); 1,86-1,67 (м, 1H).
Пример 25
(2,4-Дифторфенил)-{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон
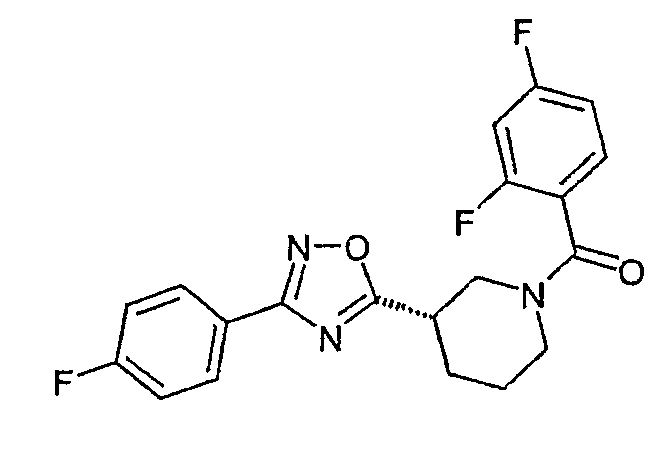
Соединение получают в соответствии с методикой, описанной в примере 13, используя 2,4-дифторбензоилхлорид в качестве выбранного ацилхлорида и гидрохлорид S-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидина (полученный в соответствии с методикой примера 12).
Выход: 59% (бесцветное масло); [α]D 20=+104° (c=1,1, CHCl3); ЖХМС (Tr): 7,51 мин (метод A); МС (ES+) m/z: 38H,1.
1H-ЯМР (CDCl3, 336 K, 300 МГц), δ (м.д.): 8,05 (дд, 2H); 7,38 (дд, 1H); 7,15 (дд, 2H); 6,98-6,80 (м, 2H); 5,13-3,72 (уш.м, 2H); 3,57-3,41 (м, 1H); 3,32-3,14 (м, 2H); 2,41-2,26 (м, 1H); 2,09-1,57 (м, 3H).
Пример 26
{(S)-3-[3-(4-Фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}-(3,4,5-трифторфенил)метанон
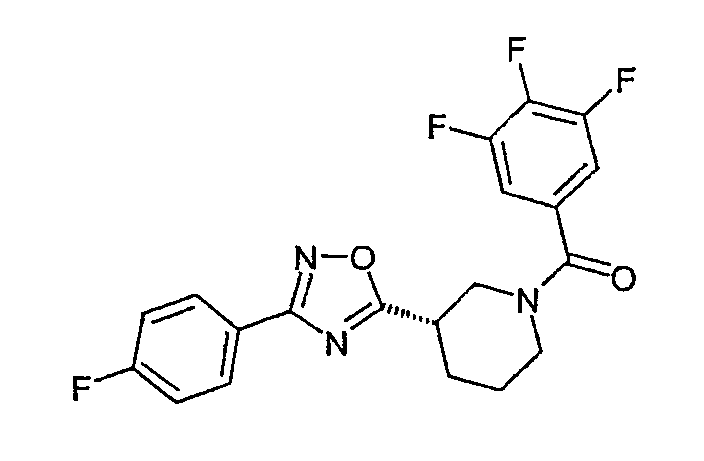
Соединение получают в соответствии с методикой, описанной в примере 13, используя 3,4,5-трифторбензоилхлорид в качестве выбранного ацилхлорида и гидрохлорид S-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидина (полученный в соответствии с методикой примера 12).
Выход: 63% (твердое белое вещество); т.пл.=139-140°C; [α]D 20=+81° (c=1,0, CHCl3); ЖХМС (Tr): 7,67 мин (метод A); МС (ES+) m/z: 406,1.
1H-ЯМР (CDCl3, 336 K, 300 МГц), δ (м.д.): 8,10-8,01 (м, 2H); 7,20-7,01 (м, 4H); 4,39-4,20 (м, 1H); 3,92-3,77 (м, 1H); 3,61 (дд, 1H); 3,42-3,18 (м, 2H); 2,38-2,25 (м, 1H); 2,15-1,85 (м, 2H); 1,77-1,59 (м, 1H).
Пример 27
(3-Хлор-4-фторфенил)-{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон
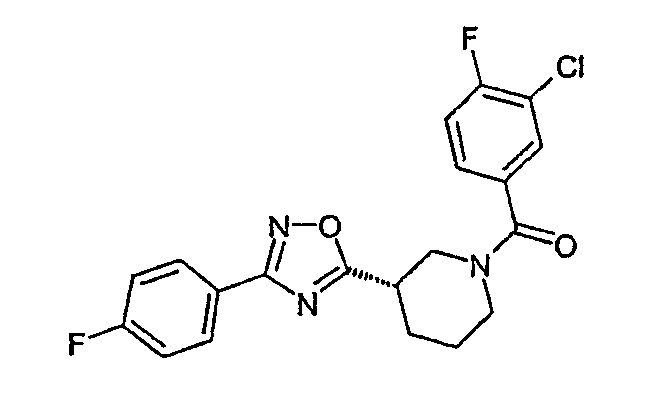
Соединение получают в соответствии с методикой, описанной в примере 13, используя 3-хлор-4-фторбензоилхлорид в качестве выбранного ацилхлорида и гидрохлорид S-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидина (полученный в соответствии с методикой примера 12).
Выход: 56% (твердое белое вещество); т.пл.=123-124°C; [α]D 20=+94° (c=1,1, CHCl3); ЖХМС (Tr): 8,00 мин (метод A); МС (ES+) m/z: 404,1.
lH-ЯМР (CDCl3, 338 K, 300 МГц), δ (м.д.): 8,12-8,00 (м, 2H); 7,51 (дд, 1H); 7,34-7,27 (м, 1H); 7,21-7,10 (м, 3H); 4,45-4,25 (м, 1H); 3,97-3,80 (м, 1H); 3,59 (дд, 1H); 3,40-3,18 (м, 2H); 2,39-2,26 (м, 1H); 2,12-1,86 (м, 2H); 1,77-1,58 (м, 1H).
Пример 28
{(S)-3-[3-(4-Фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}-(2-фенил-2H-пиразол-3-ил)метанон
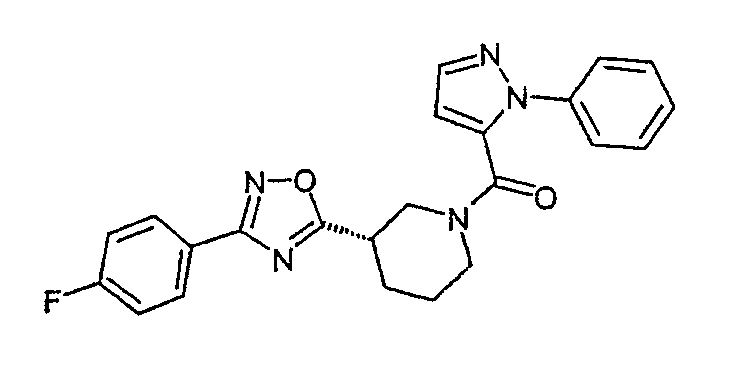
Соединение получают в соответствии с методикой, описанной в примере 13, используя 1-фенил-1H-пиразол-5-карбонилхлорид в качестве выбранного ацилхлорида и гидрохлорид S-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидина (полученный в соответствии с методикой примера 12)).
Выход: 34% (бесцветное масло); [α]D 20=+69° (c=0,5, CHCl3); ЖХМС (Tr): 7,19 мин (метод A); МС (ES+) m/z: 418,2.
1H-ЯМР (CDCl3, 338 K, 300 МГц), δ (м.д.): 8,10-7,98 (м, 2H); 7,68 (д, 1H); 7,59-7,52 (м, 2H); 7,48-7,31 (м, 3H); 7,19-7,09 (м, 2H); 6,54 (д, 1H); 5,02-4,03 (уш.м, 1H); 3,91-2,53 (уш.м, 4H); 2,42-1,68 (уш.м, 3H); 1,20-0,78 (уш.м, 1H).
Пример 29
{(S)-3-[3-(4-Фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}-(5-метил-2-фенил-2H-[1,2,3]триазол-4-ил)метанон
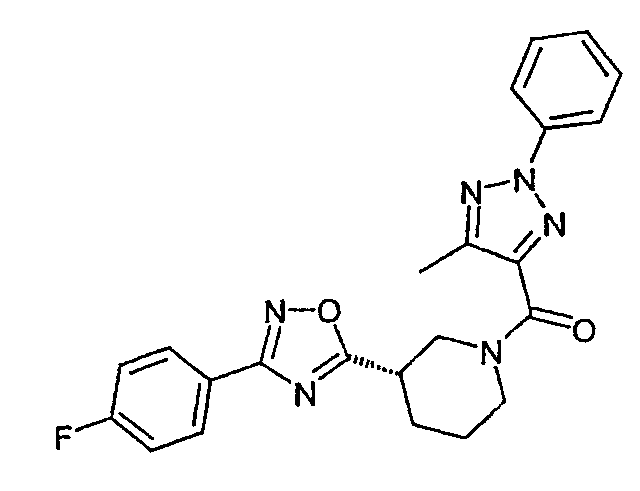
Соединение получают в соответствии с методикой, описанной в примере 13, используя 5-метил-2-фенил-2H-1,2,3-триазол-4-карбонилхлорид в качестве выбранного ацилхлорида и гидрохлорид S-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидина (полученный в соответствии с методикой примера 12).
Выход: 20% (не совсем белое масло); [α]D 20=+86° (c=0,3, CHCl3); ЖХМС (Tr): 8,20 мин (метод A); МС (ES+) m/z: 433,2.
1H-ЯМР (CDCl3, 338 K, 300 МГц), δ (м.д.): 8,15-7,95 (м, 4H); 7,55-7,28 (м, 3H); 7,22-7,07 (м, 2H); 5,05-4,75 (уш.м, 1H), 4,57-4,40 (уш.м, 1H); 3,83-3,65 (м, 1H); 3,56-3,15 (м, 2H); 2,54 (с, 3H); 2,47-2,29 (м, 1H); 2,23-1,66 (м, 3H).
Пример 30
(4-фтор-3-метилфенил)-{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон
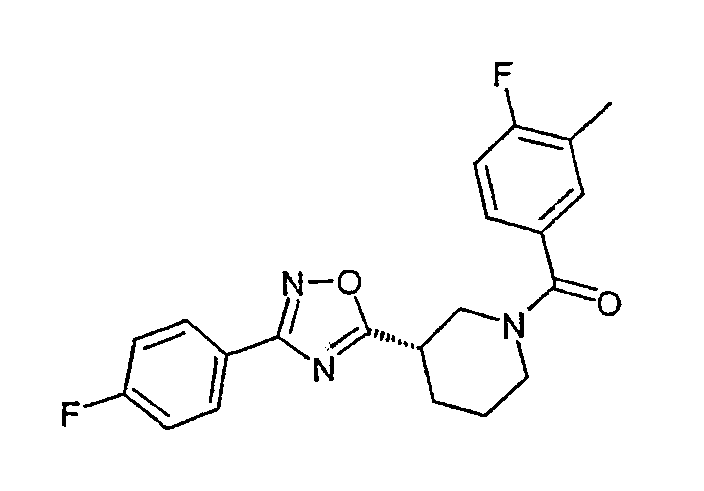
Соединение получают в соответствии с методикой, описанной в примере 13, используя 3-метил-4-фторбензоилхлорид в качестве выбранного ацилхлорида и гидрохлорид S-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидина (полученный в соответствии с методикой примера 12).
Выход: 52% (твердое белое вещество); т.пл.=106-107°C; [α]D 20=+99° (c=1,2, CHCl3); ЖХМС (Tr): 7,76 мин (метод A); МС (ES+) m/z: 384,2.
1H-ЯМР (CHCl3, 338 K, 300 МГц), δ (м.д.): 8,11-8,00 (м, 2H); 7,31-7,08 (м, 4H); 7,02 (дд, 1H); 4,49-4,32 (м, 1H); 4,06-3,91 (м, 1H); 3,51 (дд, 1H); 3,33-3,17 (м, 2H); 2,38-2,30 (м, 1H); 2,28 (с, 3H); 2,10-1,84 (м, 2H); 1,77-1,58 (м, 1H).
Пример 31
{(S)-3-[3-(4-Фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}-(5-пиридин-2-илтиофен-2-ил)метанон
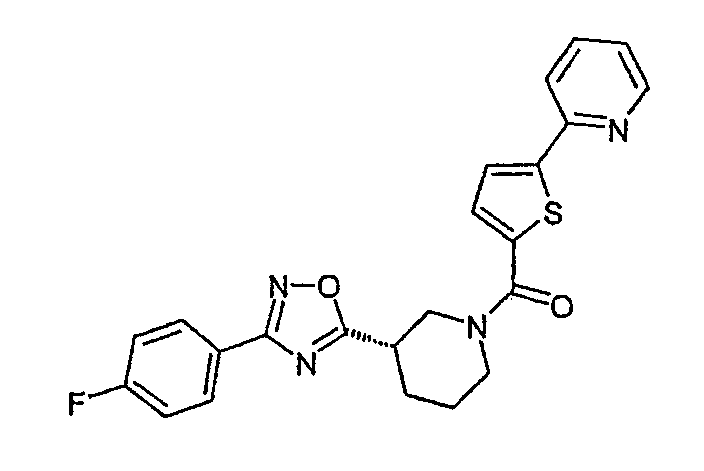
Соединение получают в соответствии с методикой, описанной в примере 13, используя 5-(2-пиридинил)-2-тиофенкарбонилхлорид в качестве выбранного ацилхлорида и гидрохлорид S-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидина (полученный в соответствии с методикой примера 12).
Выход: 13% (не совсем белый порошок); т.пл.=115-117°C; ЖХМС (Tr): 5,25 мин (метод A); МС (ES+) m/z: 435,2.
1H-ЯМР (CDCl3, 300 МГц), δ (м.д.): 8,59 (уш.д, 1H); 8,06 (дд, 2H); 7,73 (дд, 1H); 7,67 (д, 1H); 7,55 (д, 1H); 7,37 (д, 1H); 7,22 (м, 1H); 7,15 (дд, 2H); 4,69 (м, 1H); 4,32 (м, 1H); 3,57 (м, 1H); 3,36-3,24 (м, 2H); 2,37 (м, 1H); 2,09-1,89 (м, 2H); 1,83-1,67 (м, 1H).
Пример 32
{(S)-3-[3-(4-Фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}-(3-метил-тиофен-2-ил)метанон
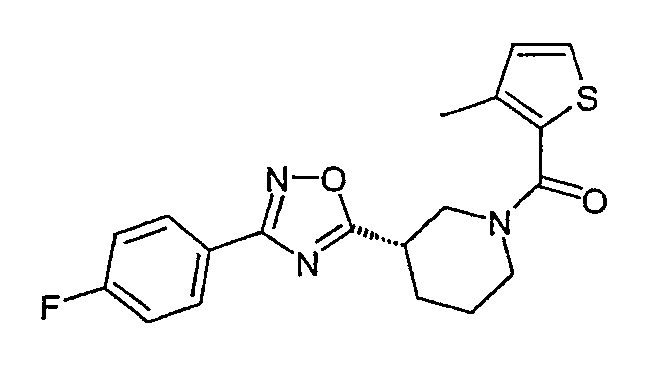
Соединение получают в соответствии с методикой, описанной в примере 13, используя 3-метил-тиофен-2-карбонилхлорид в качестве выбранного ацилхлорида и гидрохлорид S-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидина (полученный в соответствии с методикой примера 12).
Выход: 54% (твердое белое вещество); т.пл.=90-92°C; [α]D 20=+75° (c=0,95, CHCl3); ЖХМС (Tr): 5,58 мин (метод A); МС (ES+) m/z: 372,2.
1H-ЯМР (CDCl3, 300 МГц), δ (м.д.): 8,06 (дд, 2H); 7,27 (д, 1H); 7,15 (дд, 2H); 6,83 (д, 1H); 4,49 (м, 1H); 4,10 (м, 1H); 3,47 (дд, 1H); 3,30-3,15 (м, 2H); 2,33 (м, 1H); 2,27 (с, 3H); 2,04-1,85 (м, 2H); 1,80-1,62 (м, 1H).
Пример 33
{(S)-3-[3-(4-Фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}-(1-метил-1H-пиррол-2-ил)метанон
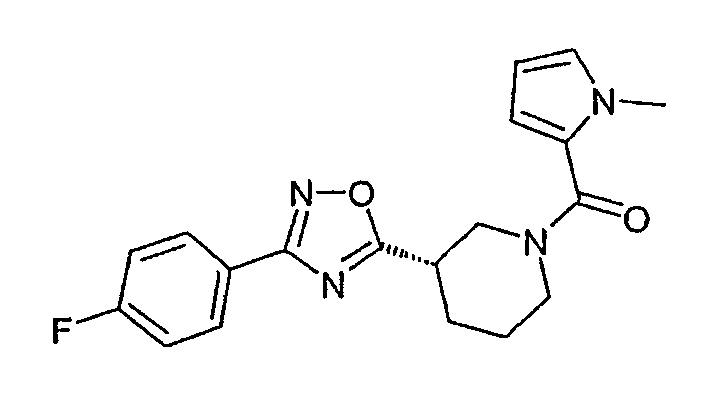
Соединение получают в соответствии с методикой, описанной в примере 13, используя 1-метил-1H-пиррол-2-карбонилхлорид в качестве выбранного ацилхлорида и гидрохлорид S-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидина (полученный в соответствии с методикой примера 12).
Выход: 64% (порошок бежевого цвета); т.пл.=84-85°C; [α]D 20=+101° (c=1,2, CHCl3); ЖХМС (Tr): 5,53 мин (метод A); МС (ES+) m/z: 355,2.
1H-ЯМР (CDCl3, 300 МГц), δ (м.д.): 8,06 (дд, 2H); 7,16 (дд, 2H); 6,70 (дд, 1H); 6,37 (дд, 1H); 6,09 (дд; 1H); 4,69 (м, 1H); 4,32 (м, 1H); 3,77 (с, 3H); 3,52 (дд, 1H); 3,24 (м, 2H); 3,34 (м, 1H); 2,08-1,86 (м, 2H); 1,77-1,61 (м, 1H).
Пример 34
Циклопентил-{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон
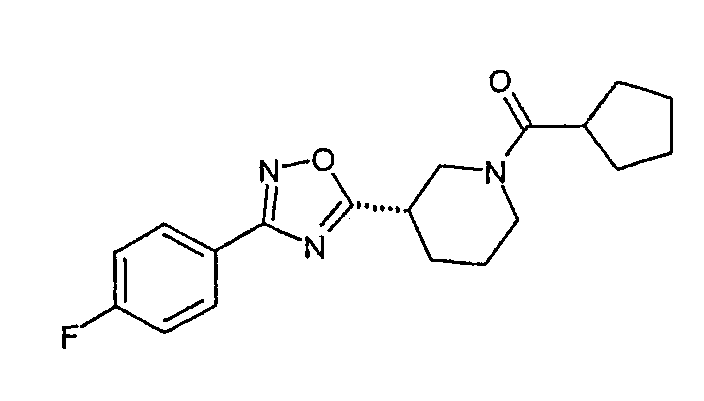
Соединение получают в соответствии с методикой, описанной в примере 13, используя циклопентанкарбонилхлорид в качестве выбранного ацилхлорида и гидрохлорид S-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидина (полученный в соответствии с методикой примера 12).
Выход: 91% (густое масло); [α]D 20=+95° (c=1,2, CHCl3); ЖХМС (Tr): 7,41 мин (метод A); МС (ES+) m/z: 344,2.
1H-ЯМР (CDCl3, 343 K, 300 МГц), δ (м.д.): 8,07 (дд, 2H); 7,15 (дд, 2H); 4,48 (м, 1H); 4,08 (м, 1H); 3,38 (м, 1H); 3,21-3,07 (м, 2H); 2,96 (м, 1H); 2,36-2,24 (м, 1H); 2,05-1,51 (м, 11H).
Пример 35
(3,4-Дифторфенил)-{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон
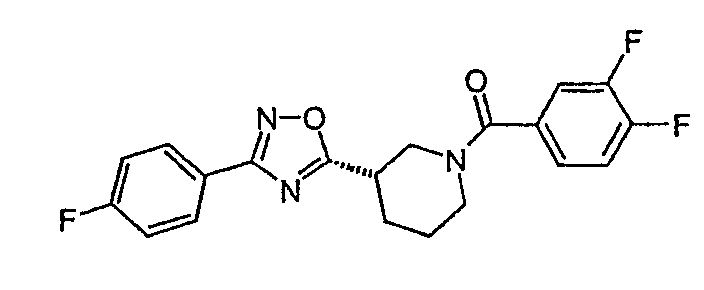
Соединение получают в соответствии с методикой, описанной в примере 13, используя 3,4-дифторбензоилхлорид в качестве выбранного ацилхлорида и гидрохлорид S-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидина (полученный в соответствии с методикой примера 12).
Выход: 59% (твердое белое вещество); т.пл.=120-121°C; [α]D 20=+105° (c=1,0, CHCl3); ЖХМС (Tr): 7,6 мин (метод A); МС (ES+) m/z: 388,0.
1H-ЯМР (CDCl3, 333 K, 300 МГц), δ (м.д.): 8,06 (дд, 2H); 7,28 (м, 1H); 7,22-7,11 (м, 4H); 4,36 (уш.д, 1H); 2,92 (уш.д, 1H); 3,57 (дд, 1H); 3,37-3,19 (м, 2H); 2,33 (м, 1H); 2,12-1,86 (м, 2H); 1,68 (м, 1H).
Пример 36
Бензотиазол-6-ил-{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон
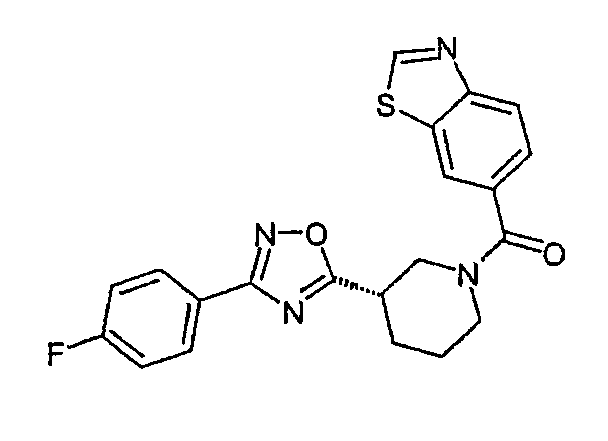
Смесь гидрохлорида S-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидина (100 мг, 0,35 ммоль, получение которого описано в примере 12), бензотиазол-6-карбоновой кислоты (70 мг, 0,38 ммоль), HOAT (72 мг, 0,52 ммоль), PS-DCC (ex Argonaut Technologies, 0,59 г, 0,70 ммоль, наполнение = 1,2 ммоль/г) и DIEA (90 мл, 0,52 ммоль) в сухом дихлорметане (6 мл) выдерживают в течение ночи на грохоте с вращением в одной плоскости (IKA Vibrax VXR). Смолу отфильтровывают и несколько раз промывают дихлорметаном; фильтрат промывают 1 н. HCl (10 мл × 2) и 5% (водный) K2CO3 (10 мл × 2), затем сушат над сульфатом натрия и упаривают при пониженном давлении. Сырой продукт очищают флэш-хроматографией (силикагель, элюент: ДХМ/MeOH 95/5), получая 50 мг бензотиазол-6-ил-{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-илпиперидин-1-ил}метанона.
Выход: 35% (белый порошок); т.пл.=63-64°C; [α]D 20=+105° (c=1,0, CHCl3); ЖХМС (Tr): 5,39 мин (метод A); МС (ES+) m/z: 409,1.
1H-ЯМР (CDCl3, 300 МГц), δ (м.д.): 9,08 (c, 1H); 8,17 (д, 1H); 8,07 (д, 1H); 8,05 (м, 2H); 7,57 (дд, 1H); 7,16 (дд, 2H); 5,00-3,71 (уш.м, 2H); 3,58 (м, 1H); 3,31 (м, 2H); 2,35 (м, 1H); 2,10-1,87 (м, 2H); 1,72 (м, 1H).
Пример 37
{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}тиазол-2-илметанон
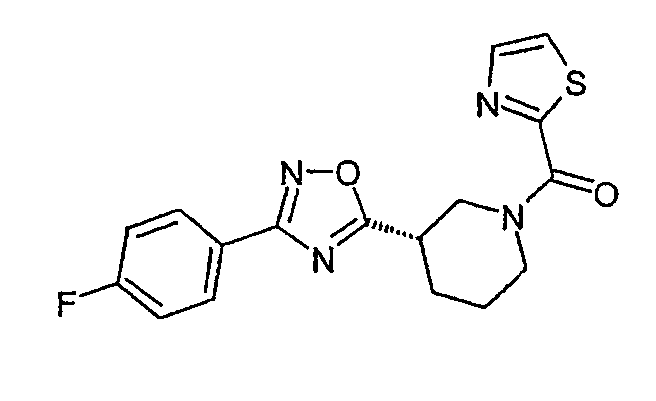
Соединение получают в соответствии с методикой, описанной в примере 36, используя 2-тиазолкарбоновую кислоту в качестве выбранной кислоты и гидрохлорид S-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидина (полученный в соответствии с методикой примера 12).
Выход: 55% (не совсем белый порошок); т.пл.=94-95°C; [α]D 20=+127° (c=0,9, CHCl3); ЖХМС (Tr): 5,54 мин (метод A); МС (ES+) m/z: 359,1.
1H-ЯМР (CDCl3, 300 МГц), δ (м.д.): 8,05 (уш.м, 2H); 7,89 (уш.м, 1H); 7,53 (уш.м, 1H); 7,15 (дд, 2H); 5,41, 4,94, 4,38, 4,04 и 3,44 (уш.м, 3H); 3,34 (уш.м, 2H); 2,36 (м, 1H); 2,13-1,92 (уш.м, 2H); 1,78 (м, 1H).
Пример 38
{(S)-3-[3-(4-Фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}-(4-метилтриазол-5-ил)метанон
Соединение получают в соответствии с методикой, описанной в примере 36, используя 4-метилтиазол-5-карбоновую кислоту в качестве кислоты и гидрохлорид S-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидина (полученный в соответствии с методикой примера 12).
Выход: 76% (порошок желтого цвета); т.пл.=122-124°C; [α]D 20=+101° (c=0,55, CHCl3); ЖХМС (Tr): 5,08 мин (метод A); МС (ES+) m/z: 373,1.
1H-ЯМР (CDCl3, 300 МГц), δ (м.д.): 8,75 (c, 1H); 8,06 (дд, 2H); 7,16 (дд, 2H); 4,42 (м, 1H); 3,97 (м, 1H); 3,56 (дд, 1H); 3,35-3,19 (м, 2H); 2,50 (c, 3H); 2,34 (м, 1H); 2,08-1,88 (м, 2H); 1,70 (м, 1H).
Пример 39
{(S)-3-[3-(4-Фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}-(6-морфолин-4-илпиридин-3-ил)метанон

Соединение получают в соответствии с методикой, описанной в примере 36, используя 6-морфолиноникотиновую кислоту в качестве выбранной кислоты и гидрохлорид S-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидина (полученный в соответствии с методикой примера 12).
Выход: 20% (не совсем белый порошок); т.пл.=112-114°C; [α]D 20=+111° (c=0,55,
CHCl3); ЖХМС (Tr): 4,96 мин (метод A); МС (ES+) m/z: 438,2.
1H-ЯМР (CDCl3, 300 МГц), δ (м.д.): 8,31 (д, 1H); 8,06 (дд, 2H); 7,65 (дд, 1H); 7,16 (дд, 2H); 6,63 (д, 1H); 4,51 (м, 1H); 4,11 (м, 1H); 3,81 (дд, 4H); 3,59 (дд, 4H); 3,50 (м, 1H); 3,24 (м, 2H); 2,35 (м, 1H); 2,05-1,86 (м, 2H); 1,69 (м, 1H).
Пример 40
{(S)-3-[3-(4-Фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}-(1Н-индол-5-ил)метанон
Соединение получают в соответствии с методикой, описанной в примере 36, используя индол-5-карбоновую кислоту в качестве выбранной кислоты и гидрохлорид S-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидина (полученный в соответствии с методикой примера 12).
Выход: 44% (твердое белое вещество); т.пл.=191-192°C; [α]D 20=+107° (c=0,85, CHCl3); ЖХМС (Tr): 5,44 мин (метод A); МС (ES+) m/z: 391,2.
1H-ЯМР (CDCl3, 333 K, 300 МГц), δ (м.д.): 8,19 (уш.c, 1H); 8,06 (дд, 2H); 7,75 (уш.c, 1H); 7,39 (д, 1H); 7,28 (дд, 1H); 7,24 (м, 1H); 7,14 (дд, 2H); 6,59 (м, 1H); 4,56 (м, 1H); 4,15 (м, 1H); 3,50 (дд, 1H); 3,33-3,18 (м, 2H); 2,34 (м, 1H); 2,07-1,85 (м, 2H); 1,73 (м, 1H).
Пример 41
2-(4-Фторфенил)-1-{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}этанон
Соединение получают в соответствии с методикой, описанной в примере 36, используя 4-фторфенилуксусную кислоту в качестве выбранной кислоты и гидрохлорид S-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидина (полученный в соответствии с методикой примера 12).
Выход: 37% (прозрачное масло); [α]D 20=+68° (c=0,6, CHCl3); ЖХМС (Tr): 5,58 мин (метод A); МС (ES+) m/z: 384,2.
1H-ЯМР (CDCl3, 333 K, 300 МГц), δ (м.д.): 8,06 (дд, 2H); 7,22 (дд, 2H); 7,16 (дд, 2H); 6,99 (дд, 2H); 4,65, 4,03, 3,47 и 3,00 (уш.м, 4H); 3,75 (c, 2H); 3,19 (ддд, 1H); 2,23 (м, 1H); 1,97 (м, 1H); 1,79 (м, 1H); 1,49 (м, 1H).
Пример 42
3-(4-Фторфенил)-1-{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}пропан-1-он

Соединение получают в соответствии с методикой, описанной в примере 36, используя 3-(4-фторфенил)пропионовую кислоту в качестве выбранной кислоты и гидрохлорид S-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидина (полученный в соответствии с методикой примера 12).
Выход: 50% (белый порошок); т.пл.=83-84°C; [α]D 2O=+80° (c=1,32, CHCl3); ЖХМС (Tr): 5,68 мин (метод A); МС (ES+) m/z: 398,4.
1H-ЯМР (CDCl3, 333 K, 300 МГц), δ (м.д.): 8,06 (дд, 2H); 7,17 (дд, 2H); 7,15 (дд, 2H); 6,95 (дд, 2H); 4,71, 3,93 и 3,44 (уш.м, 2H); 3,17 (м, 1H); 3,06 (уш.м, 1H); 2,98 (дд, 2H); 2,67 (дд, 2H); 2,26 (м, 1H); 1,97 (м, 1H); 1,83 (м, 1H); 1,55 (м, 1H).
Пример 43
{(S)-3-[3-(4-Фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}-изохинолин-3-илметанон
Соединение получают в соответствии с методикой, описанной в примере 36, используя изохинолин-3-карбоновую кислоту в качестве выбранной кислоты и гидрохлорид S-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидина (полученный в соответствии с методикой примера 12).
Выход: 56% (белое масло); [α]D 20=+150° (c=0,8, CHCl3); ЖХМС (Tr): 5,59 мин (метод A); МС (ES+) m/z: 403,2.
1H-ЯМР (CDCl3, 333 K, 300 МГц), δ (м.д.): 8,23 (д, 1H); 8,12 (д, 1H); 8,02 (уш.м, 2H); 7,84 (д, 1H); 7,76 (д, 1H); 7,74 (м, 1H); 7,59 (дд, 1H); 7,13 (уш.дд, 2H); 5,01, 4,51 и 4,18 (уш.м, 2H); 3,77-3,26 (уш.м, 3H); 2,39 (м, 1H); 2,18-1,78 (уш.м, 3H).
Пример 44
{(S)-3-[3-(4-Фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}хиноксалин-6-илметанон
Соединение получают в соответствии с методикой, описанной в примере 36, используя 6-хиноксалинкарбоновую кислоту в качестве выбранной кислоты и гидрохлорид S-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидина (полученный в соответствии с методикой примера 12). Чистый {(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}хиноксалин-6-илметанон получают после очистки продукта на силикагельном картридже (элюент: ДХМ/MeOH/NH4OH 98/2/0,2).
Выход: 83% (твердое белое вещество); [α]D 20=+120° (c=1,0, CHCl3); ЖХМС (Tr): 7,0 мин (метод A); МС (ES+) m/z: 404,0.
1H-ЯМР (CDCl3, 330 K, 300 МГц), δ (м.д.): 8,88 (c, 2H); 8,18 (дд, 2H); 8,06 (м, 2H); 7,82 (дд, 1H); 7,15 (дд, 2H); 4,47 (уш.м, 1H); 4,02 (уш.м, 1H); 3,65 (дд, 1H); 3,44-3,23 (м, 2H); 2,36 (м, 1H); 2,14-1,88 (м, 2H); 1,74 (м, 1H).
Пример 45
{(S)-3-[3-(4-Фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}-бензимидазол-6-илметанон
Соединение получают в соответствии с методикой, описанной в примере 36, используя бензимидазол-5-карбоновую кислоту в качестве выбранной кислоты и гидрохлорид S-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидина (полученный в соответствии с методикой примера 12). Чистый {(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}бензимидазол-6-илметанон получают после очистки продукта флэш-хроматографией (силикагель, элюент: ДХМ/MeOH/NH4OH 98/2/0,2).
Выход: 5% (твердое белое вещество); т.пл.=110-115°C; [α]D 20=+115° (c=1,0, CHCl3); ЖХМС (Tr): 6,28 мин (метод A); МС (ES+) m/z: 392,0.
1H-ЯМР (CDCl3, 300 МГц), δ (м.д.): 9,56 и 8,38 (c, 1H); 8,11-7,97 (м, 4H); 7,97-7,82 (м, 1H); 7,60-7,46 (м, 1H); 7,13 (м, 2H); 4,42 (уш.м, 1H); 3,97 (м, 1H); 3,59 (м, 1H); 3,31 (м, 2H); 2,34 (м, 1H); 2,11-1,84 (м, 2H); 1,72 (м, 1H).
Пример 46
(4-Фторфенил)-{(S)-3-[3-(2,4,6-трифторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон
46(A). трет-Бутиловый эфир S-3-[3-(2,4,6-трифторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-карбоновой кислоты
К раствору 2,4,6-трифторбензонитрила (1,5 г, 9,6 ммоль) в EtOH (5 мл) при комнатной температуре добавляют гидроксиламин (50% (масс.) водный раствор, 2,5 мл, 38 ммоль) и раствор кипятят с обратным холодильником при перемешивании в течение 1 часа. Растворитель удаляют при пониженном давлении, получая N-гидрокси-2,4,6-трифторбензамидин, который сразу используют в следующей стадии.
Смесь N-гидрокси-2,4,6-трифторбензамидин (9,6 ммоль), S-1-Boc-пиперидин-3-карбоновой кислоты (2,3 г, 9,6 ммоль), EDCI·HCl (2,87 г, 15 ммоль), HOBT (1,35 г, 9,6 ммоль) и DIEA (3,4 мл, 20 ммоль) в диоксане (10 мл) перемешивают в течение ночи при комнатной температуре в атмосфере азота. Добавляют еще одну порцию HOBT (1,35 г), EDCI·HCl (2,87 г) и DIEA (3,4 мл), после чего реакционную смесь нагревают до 60°C и выдерживают при данной температуре в течение 2 часов. Затем реакционную смесь кипятят с обратным холодильником в течение 18 часов и растворитель выпаривают при пониженном давлении. Остаток разбавляют водой (50 мл) и этилацетатом (50 мл), фазы разделяют и органический слой промывают последовательно водой (50 мл × 2) и 1 н. NaOH (50 мл × 2). Органический слой сушат над Na2SO4 и концентрируют при пониженном давлении. Очистка сырого продукта флэш-хроматографией (силикагель, элюент: ДХМ/гексан/MeOH 50/50/0,2) приводит к получению 0,7 г трет-бутилового эфира S-3-[3-(2,4,6-трифторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-карбоновой кислоты.
Выход: 20%; ЖХМС (Tr): 10,10 мин (метод B); МС (ES+) m/z: 384,4.
1H-ЯМР (CDCl3, 300 МГц, 333 K), δ (м.д.): 6,80 (м, 2H); 4,29 (уш.д, 1H); 3,93 (ддд, 1H); 3,32 (дд, 1H); 3,19 (тт, 1H); 3,02 (ддд, 1H); 2,26 (м, 1H); 1,99-1,79 (м 2H); 1,70-1,56 (м, 1H); 1,47 (c, 9H),
46(B). Гидрохлорид S-3-[3-(2,4,6-Трифторфенил)-[1,2,4]оксадиазол-5-ил]пиперидина
трет-Бутиловый эфир S-3-[3-(2,4,6-трифторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-карбоновой кислоты (0,2 г, 0,52 ммоль) растворяют в дихлорметане (3 мл) и по каплям добавляют 5 мл HCl 4 н. (диоксановый раствор). Полученную смесь перемешивают при комнатной температуре в течение 2 часов. Растворитель выпаривают при пониженном давлении, получая 165 мг (выход: 100%) гидрохлорида S-3-[3-(2,4,6-трифторфенил)-[1,2,4]оксадиазол-5-ил]пиперидина в виде твердого белого вещества.
ЖХМС (Tr): 4,80 мин (метод B); МС (ES+) m/z: 284,4.
46(С). (4-Фторфенил)-{(S)-3-[3-(2,4,6-трифторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон
К суспензии гидрохлорида S-3-[3-(2,4,6-Трифторфенил)-[1,2,4]оксадиазол-5-ил]пиперидина (165 мг, 0,52 ммоль) в сухом дихлорметане (3 мл) по каплям при 0°С добавляют триэтиламин (154 мл, 1,09 ммоль) и 4-фторбензоилхлорид (62 мл, 0,52 ммоль). Реакционной смеси дают возможность нагреться до комнатной температуры и перемешивают в течение ночи в атмосфере азота. Затем раствор обрабатывают 0,5 н. HCl (5 мл) и фазы разделяют. Органический слой промывают последовательно 0,5 н. HCl (5 мл), 1 н. NaOH (5 мл) и водой (5 мл), затем сушат над Na2SO4 и упаривают при пониженном давлении. Сырой продукт очищают флэш-хроматографией (силикагель, элюент: гексан/этилацетат 8:2), получая 50 мг указанного в заголовке соединения. Дальнейшая очистка препаративной ВЭЖХ (колонка: SymmetryPrep Cl8, 7 мкМ, 19×300 мм; подвижная фаза A: вода/ацетонитрил/ТФУК 900/100/0,5, подвижная фаза B: вода/ацетонитрил/ТФУК 100/900/0,5, скорость истечения: 20 мл/мин) приводит к получению 25 мг (4-фторфенил)-{(S)-3-[3-(2,4,6-трифторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанона.
Выход: 12% (густое масло); ЖХМС (Tr): 7,0 мин (метод A); МС (ES+) m/z: 406,0.
1H-ЯМР (CDCl3, 333 K, 300 МГц), δ (м.д.): 7,45 (м, 2H); 7,18-7,00 (м, 2H); 6,90-6,75 (м, 2H); 4,40 (уш.д, 1H); 3,95 (уш.д, 1H); 3,52 (дд, 1H); 3,37-3,20 (м, 2H); 2,40-2,20 (м, 1H); 2,15-1,85 (м, 2H); 1,78-1,55 (м, 1H).
Пример 47
(4-Фторфенил)-[(S)-3-(3-пиридин-3-ил-[1,2,4]оксадиазол-5-ил)-пиперидин-1-ил]метанон
47(A). трет-Бутиловый эфир S-3-(3-пиридин-3-ил-[1,2,4]оксадиазол-5-ил)пиперидин-1-карбоновой кислоты
Соединение получают в соответствии с методикой, описанной в примере 46(A), исходя из 3-цианопиридина. Чистый трет-бутиловый эфир S-3-(3-пиридин-3-ил-[1,2,4]оксадиазол-5-ил)пиперидин-1-карбоновой кислоты получают после растирания продукта в смеси диэтилэфир/пентан 1:1 (Выход: 29%).
ЖХМС (Tr): 6,49 мин (метод A); МС (ES+) m/z: 331,1.
1H-ЯМР (CDCl3, 333 K, 300 МГц), δ (м.д.): 9,32 (д, 1H); 8,74 (дд, 1H); 8,37 (ддд, 1H); 7,43 (дд, 1H); 4,28 (уш.д, 1H); 3,93 (ддд, 1H); 3,34 (дд, 1H); 3,18 (тт, 1H); 3,05 (ддд, 1H); 2,27 (м, 1H); 2,00-1,81 (м, 2H); 1,68-1,57 (м, 1H).
47(B). Дигидрохлорид S-3-(5-пиперидин-3-ил-[1,2,4]оксадиазол-3-ил)пиридина
Соединение получают в соответствии с методикой, описанной в примере 46(B), исходя из трет-бутилового эфира S-3-(3-пиридин-3-ил-[1,2,4]оксадиазол-5-ил)пиперидин-1-карбоновой кислоты (выход: 100%).
ЖХМС (Tr): 1,78 мин (метод B); МС (ES+) m/z: 231,1.
47(С). (4-Фторфенил)-[(S)-3-(3-пиридин-3-ил-[1,2,4]оксадиазол-5-ил)пиперидин-1-ил]метанон
Соединение получают в соответствии с методикой, описанной в примере 46(C), исходя из дигидрохлорида S-3-(5-пиперидин-3-ил-[1,2,4]оксадиазол-3-ил)пиридина и используя 4-фторбензоилхлорид в качестве выбранного ацилхлорида. Чистый (4-фторфенил)-[(S)-3-(3-пиридин-3-ил-[1,2,4]оксадиазол-5-ил)пиперидин-1-ил]метанон получают после очистки сырого продукта флэш-хроматографией (силикагель, элюент: ДХМ/MeOH/NH4OH 99/1/0,1).
Выход: 98% (твердое клейкое вещество белого цвета); [α]D 20=+105° (c=1,05, CHCl3); ЖХМС (Tr): 5,8 мин (метод A); МС (ES+) m/z: 353,0.
1H-ЯМР (CDCl3, 333 K, 300 МГц), δ (м.д.): 9,30 (c, 1H); 8,8 (д, 1H); 8,3 (м, 1H); 7,55-7,35 (м, 3H); 7,20-7,0 (м, 2H); 4,50 (уш.д, 1H); 4,0 (уш.д, 1H); 3,50 (дд, 1H); 3,35-3,15 (м, 2H); 2,45 (м, 1H); 2,15-1,80 (м, 2H); 1,80-1,60 (м, 1H).
Пример 48
(4-Фторфенил)-[(S)-3-(3-пиридин-4-ил-[1,2,4]оксадиазол-5-ил)пиперидин-1-ил]метанон
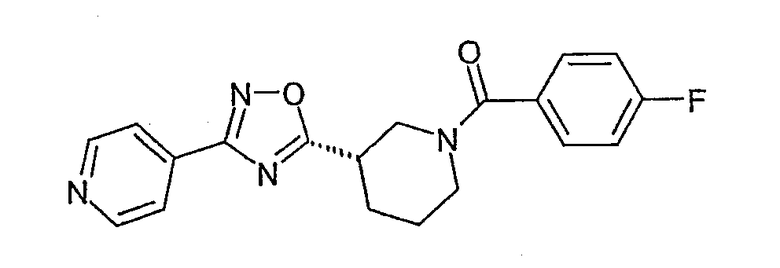
48(A). трет-Бутиловый эфир S-3-(3-пиридин-4-ил-[1,2,4]оксадиазол-5-ил)пиперидин-1-карбоновой кислоты
Соединение получают в соответствии с методикой, описанной в примере 46(A), исходя из 4-цианопиридина. Чистый трет-бутиловый эфир S-3-(3-пиридин-4-ил-[1,2,4]оксадиазол-5-ил)пиперидин-1-карбоновой кислоты получают после растирания сырого продукта в смеси гексан/диэтилэфир 1:1 (выход: 68%).
ЖХМС (Tr): 6,56 мин (метод A); МС (ES+) m/z: 331,1.
48(B). Дигидрохлорид S-4-(5-пиперидин-3-ил-[1,2,4]оксадиазол-3-ил)пиридина
Соединение получают в соответствии с методикой, описанной в примере 46(B), исходя из трет-бутилового эфира S-3-(3-пиридин-4-ил-[1,2,4]оксадиазол-5-ил)пиперидин-1-карбоновой кислоты (выход: 100%).
ЖХМС (Tr): 1,44 мин (метод B); МС (ES+) m/z: 231,1.
48(С). (4-Фторфенил)-[(S)-3-(3-пиридин-4-ил-[1,2,4]оксадиазол-5-ил)пиперидин-1-ил]метанон
Соединение получают в соответствии с методикой, описанной в примере 46(С), исходя из дигидрохлорида S-4(5-пиперидин-3-ил-[1,2,4]оксадиазол-3-ил)пиридина и используя 4-фторбензоилхлорид в качестве выбранного ацилхлорида. Чистый (4-фторфенил)-[(S)-3-(3-пиридин-4-ил-[1,2,4]оксадиазол-5-ил)пиперидин-1-ил]метанон получают после очистки сырого продукта флэш-хроматографией (2 последовательно соединенные колонки: силикагель, элюент: ДХМ/MeOH/NH4OH 99/1/0,1).
Выход: 38% (твердое белое вещество); т.пл.=113-115°C; [α]D 20=+112° (c=1,62, CHCl3); ЖХМС (Tr): 5,55 мин (метод A); МС (ES+) m/z: 353,0.
1H-ЯМР (CDCl3, 333 K, 300 МГц), δ (м.д.): 8,79 (м, 2H); 7,99 (м, 2H); 7,43 (дд, 2H); 7,10 (дд, 2H); 4,45 (уш.д, 1H); 3,97 (м, 1H); 3,54 (дд, 1H); 3,35-3,21 (м, 2H); 2,35 (м, 1H); 2,11-1,86 (м, 2H); 1,70 (м, 1H).
Пример 49
{(S)-3-[3-(2,4-Дифторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}-(4-фторфенил)метанон
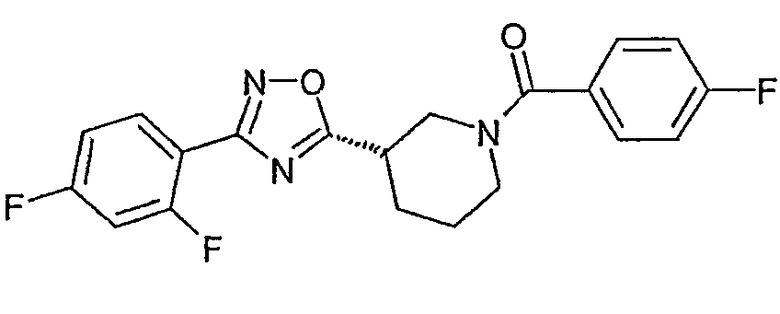
49(A). трет-Бутиловый эфир S-3-[3-(2,4-дифторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-карбоновой кислоты
К смеси 2,4-дифторбензонитрила (1,39 г, 10 ммоль) и DIEA (5,13 мл, 30 ммоль) в EtOH (15 мл) добавляют 2,12 г гидрохлорида гидроксиламина (30 ммоль) и реакционную смесь нагревают до 70°C и выдерживают при этой температуре в течение 48 часов. Половину объема растворителя удаляют при пониженном давлении. Смесь выливают в смесь ДХМ (100 мл) и воды (30 мл). Добавляют 2,5 мл 1 н. NaOH для достижения pH=9-10. Органическую фазу отделяют и водную фазу экстрагируют ДХМ. Органические слои объединяют, промывают, сушат над MgSO4, фильтруют и упаривают при пониженном давлении, получая N-гидрокси-2,4-дифторбензамидин, который используют в следующей стадии без дополнительной очистки.
Смесь N-гидрокси-2,4-дифторбензамидина (10 ммоль), S-1-Boc-пиперидин-3-карбоновой кислоты (2,3 г, 10 ммоль), EDCI·HCl (2,87 г, 15 ммоль), HOBT (1,35 г, 10 ммоль) и DIEA (3,4 мл, 20 ммоль) в диоксане (10 мл) перемешивают в течение ночи при комнатной температуре в атмосфере азота. Затем реакционную смесь кипятят с обратным холодильником в течение 18 часов и растворитель выпаривают при пониженном давлении. Остаток разбавляют водой (50 мл) и этилацетатом (50 мл), фазы разделяют и органический слой промывают последовательно водой (50 мл × 2) и 1 н. NaOH (50 мл × 2). Органический слой сушат над Na2SO4 и концентрируют при пониженном давлении. Очистка сырого продукта флэш-хроматографией (силикагель, элюент: ДХМ/гексан/MeOH 50/50/0,2) приводит к получению 2,4 г трет-бутилового эфира S-3-[3-(2,4-дифторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-карбоновой кислоты.
Выход: 66%; ЖХМС (Tr): 7,93 мин (метод A); МС (ES+) m/z: 366,2.
1H-ЯМР (CDCl3, 333 K, 300 МГц), δ (м.д.): 8,05 (м, 1H); 6,98 (м, 2H); 4,27 (уш.д, 1H); 3,92 (м, 1H); 3,31 (дд, 1H); 3,17 (тт, 1H); 3,03 (ддд, 1H); 2,25 (м, 1H); 1,95-1,78 (м, 2H); 1,77-1,53 (м, 1H); 1,46 (c, 9H).
49(B). Гидрохлорид S-3-[3-(2,4-дифторфенил)-[1,2,4]оксадиазол-5-ил]пиперидина
трет-Бутиловый эфир S-3-[3-(2,4-Дифторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-карбоновой кислоты (0,22 г, 0,6 ммоль) растворяют в дихлорметане (3 мл) и к полученному раствору по каплям добавляют 5 мл 4 н. HCl (раствор в диоксане). Полученную смесь перемешивают при комнатной температуре в течение 2 часов. Растворитель выпаривают при пониженном давлении, получая 180 мг (выход: 100%) гидрохлорида S-3-[3-(2,4-дифторфенил)-[1,2,4]оксадиазол-5-ил]пиперидина в виде твердого белого вещества. ЖХМС (Tr): 4,67 мин (метод B); МС (ES+) m/z: 266,2.
49(С). {(S)-3-[3-(2,4-Дифторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}-(4-фторфенил)метанон
К суспензии гидрохлорида S-3-[3-(2,4-дифторфенил)-[1,2,4]оксадиазол-5-ил]пиперидина (180 мг, 0,6 ммоль) в сухом дихлорметане (5 мл) по каплям при 0°С добавляют триэтиламин (180 мл, 1,26 ммоль) и 4-фторбензоилхлорид (71 мл, 0,6 ммоль). Реакционной смеси дают возможность нагреться до комнатной температуры и перемешивают ее в течение ночи в атмосфере азота. Затем раствор обрабатывают 0,5 н. HCl (5 мл) и фазы разделяют. Органический слой промывают последовательно 0,5 н. HCl (5 мл), 1 н. NaOH (5 мл) и водой (5 мл), затем сушат над Na2SO4 и упаривают при пониженном давлении. Сырой продукт очищают флэш-хроматографией (силикагель, элюент: ДХМ/гексан/MeOH 50/50/0,2), получая 50 мг указанного в заголовке соединения.
Выход: 86% (твердое клейкое вещество белого цвета); [α]D 20=+106° (c=1,05, CHCl3); ЖХМС (Tr): 7,13 мин (метод A); МС (ES+) m/z: 388,0.
1H-ЯМР (CDCl3, 333 K, 300 МГц), δ (м.д.): 8,00 (м, 1H); 7,50-7,35 (м, 2H); 7,15-6,90 (м, 4H); 4,4 (уш.д, 1H); 3,95 (уш.д, 1H); 3,50 (дд, 1H); 3,35-3,15 (м, 2H); 2,40-2,20 (м, 1H); 2,10-1,80 (м, 2H); 1,80-1,60 (м, 1H).
Пример 50
(4-Фторфенил)-[(S)-3-(3-нафталин-1-ил-[1,2,4]оксадиазол-5-ил)пиперидин-1-ил]метанон
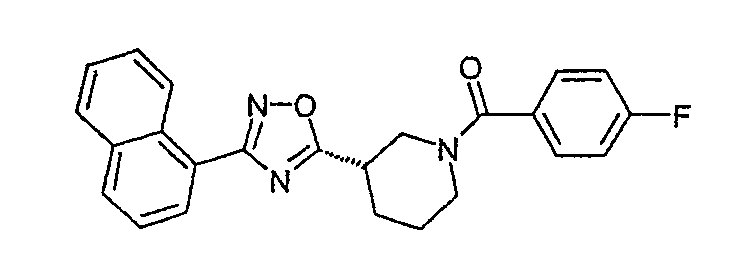
50(A). трет-Бутиловый эфир S-3-(3-нафталин-1-ил-[1,2,4]оксадиазол-5-ил)пиперидин-1-карбоновой кислоты
Соединение получают в соответствии с методикой, описанной в примере 49(A), исходя из трет-бутилового эфира 1-цианонафталина. Чистый трет-бутиловый эфир S-3-(3-нафталин-1-ил-[1,2,4]оксадиазол-5-ил)пиперидин-1-карбоновой кислоты получают после растирания сырого продукта в смеси диэтилэфир/гексан 1:1 и последовательно флэш-хроматографии (силикагель, элюент: ДХМ/MeOH 99,8/0,2) (выход: 66%).
ЖХМС (Tr): 8,64 мин (метод A); МС (ES+) m/z: 380,1.
50(B). Гидрохлорид S-3-(3-Нафталин-1-ил-[1,2,4]оксадиазол-5-ил)пиперидина
Соединение получают в соответствии с методикой, описанной в примере 49(B), исходя из трет-бутилового эфира S-3-(3-нафталин-1-ил-[1,2,4]оксадиазол-5-ил)пиперидин-1-карбоновой кислоты (выход: 100%).
ЖХМС (Tr): 5,42 мин (метод B); МС (ES+) m/z: 280,1.
50(С). (4-Фторфенил)-[(S)-3-(3-нафталин-1-ил-[1,2,4]оксадиазол-5-ил)пиперидин-1-ил]метанон
Соединение получают в соответствии с методикой, описанной в примере 49(С), исходя из гидрохлорида S-3-(3-нафталин-1-ил-[1,2,4]оксадиазол-5-ил)пиперидина и используя 4-фторбензоилхлорид в качестве выбранного ацилхлорида. Чистый (4-фторфенил)-[(S)-3-(3-нафталин-1-ил-[1,2,4]оксадиазол-5-ил)пиперидин-1-ил]метанон получают после очистки сырого продукта флэш-хроматографией (силикагель, элюент: ДХМ/гексан/MeOH 50/50/0,2).
Выход: 83% (твердое клейкое вещество белого цвета); [α]D 20=+88° (c=1,28, CHCl3); ЖХМС (Tr): 7,6 мин (метод A); МС (ES+) m/z: 402,1.
1H-ЯМР (CDCl3, 333 K, 300 МГц), δ (м.д.): 8,90 (д, 1H); 8,20 (д, 1H); 7,90 (дд, 2H); 7,60-7,40 (м, 5H); 7,15-7,00 (м, 2H); 4,50 (уш.д, 1H); 4,00 (уш.д, 1H); 3,60 (дд, 1H); 3,40-3,15 (м, 2H); 2,45-2,30 (м, 1H); 2,20-1,85 (м, 2H); 1,80-1,60 (м, 1H).
Пример 51
{(S)-3-[3-(2,6-Дифторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}-(4-фторфенил)метанон
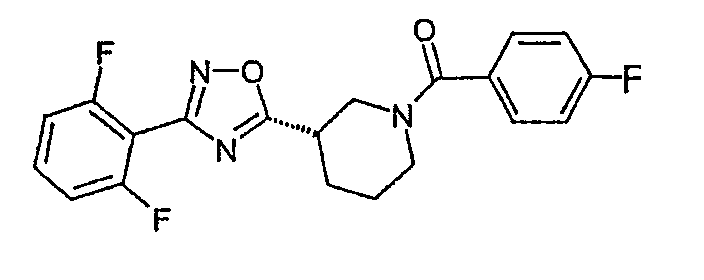
51(A). трет-Бутиловый эфир S-3-[3-(2,6-дифторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-карбоновой кислоты
Соединение получают в соответствии с методикой, описанной в примере 49(A), исходя из трет-бутилового эфира 2,6-дифторбензонитрила. S-3-[3-(2,6-дифторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-карбоновой кислоты получают после очистки продукта флэш-хроматографией (силикагель, элюент: ДХМ/MeOH 99,8/0,2) (Выход: 55%).
ЖХМС (Tr): 7,68 мин (Метод A); МС (ES+) m/z: 366,1.
1H-ЯМР (CDCl3, 333 K, 300 МГц), δ (м.д.): 7,44 (тт, 1H); 7,02 (дд, 2H); 4,29 (уш.д, 1H); 3,94 (ддд, 1H); 3,32 (дд, 1H); 3,19 (тт, 1H); 3,01 (ддд, 1H); 2,27 (м, 1H); 1,99-1,78 (м, 2H); 1,74-1,54 (м, 1H); 1,46 (c, 9H).
51(B). Гидрохлорид S-3-[3-(2,6-дифторфенил)-[1,2,4]оксадиазол-5-ил]пиперидина
Соединение получают в соответствии с методикой, описанной в примере 49(B), исходя из трет-бутилового эфира S-3-[3-(2,6-дифторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-карбоновой кислоты (выход: 100%).
ЖХМС (Tr): 4,24 мин (метод B); МС (ES+) m/z: 266,1.
51(С). {(S)-3-[3-(2,6-Дифторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}-(4-фторфенил)метанон
Соединение получают в соответствии с методикой, описанной в примере 49(С), исходя из гидрохлорида S-3-[3-(2,6-дифторфенил)-[1,2,4]оксадиазол-5-ил]пиперидина и используя 4-фторбензоилхлорид в качестве выбранного ацилхлорида. Чистый {(S)-3-[3-(2,6-дифторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}-(4-фторфенил)метанон получают после очистки сырого продукта флэш-хроматографией (2 последовательно соединенные колонки: силикагель, элюент: ДХМ/гексан/MeOH 50/50/0,2).
Выход: 60% (густое масло); [α]D 20=+97° (c=1,14, CHCl3); ЖХМС (Tr): 7,10 мин (метод A); МС (ES+) m/z: 388,1.
1H-ЯМР (CDCl3, 328 K, 300 МГц), δ (м.д.): 7,50-7,39 (м, 3H); 7,12-6,98 (м, 4H); 4,41 (уш.д, 1H); 3,99 (уш.д, 1H); 3,54 (дд, 1H); 3,35-3,21 (м, 2H); 2,35 (м, 1H); 2,11-1,87 (м, 2H); 1,75-1,60 (м, 1H).
Пример 52
(4-Фторфенил)-{(S)-3-[3-(2-метоксифенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон
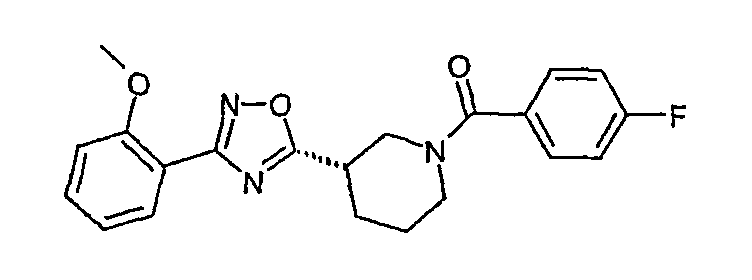
52(A). трет-Бутиловый эфир S-3-[3-(2-метоксифенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-карбоновой кислоты
Соединение получают в соответствии с методикой, описанной в примере 49(A), исходя из 2-метоксибензонитрила. Чистый трет-бутиловый эфир S-3-[3-(2-метоксифенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-карбоновой кислоты получают после очистки сырого продукта флэш-хроматографией (силикагель, элюент: гексан/этилацетат 8/2) (выход: 39%).
ЖХМС (Tr): 7,19 мин (метод A); МС (ES+) m/z: 360,1.
1H-ЯМР (CDCl3, 333 K, 300 МГц), δ (м.д.): 7,97 (дд, 1H); 7,45 (ддд, 1H); 7,06 (ддд, 1H); 7,04 (дд, 1H); 4,33 (уш.д, 1H); 3,97 (ддд, 1H); 3,95 (c, 3H); 3,28 (дд, 1H); 3,15 (тт, 1H); 2,98 (ддд, 1H); 2,27 (м, 1H); 1,98-1,79 (м, 2H); 1,69-1,53 (м, 1H); 1,47 (c, 9H).
52(B). Гидрохлорид S-3-[3-(2-метоксифенил)-[1,2,4]оксадиазол-5-ил]пиперидина
Соединение получают в соответствии с методикой, описанной в примере 49(B), исходя из трет-бутилового эфира S-3-[3-(2-метоксифенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-карбоновой кислоты (выход: 100%).
ЖХМС (Tr): 4,40 мин (метод B); МС (ES+) m/z: 260,1.
52(С). (4-Фторфенил)-{(S)-3-[3-(2-метоксифенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил)метанон
Соединение получают в соответствии с методикой, описанной в примере 49(С), исходя из гидрохлорида S-3-[3-(2-метоксифенил)-[1,2,4]оксадиазол-5-ил]пиперидина и используя 4-фторбензоилхлорид в качестве выбранного ацилхлорида. Чистый (4-фторфенил)-{(S)-3-[3-(2-метоксифенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон получают после очистки сырого продукта флэш-хроматографией (силикагель, элюент: гексан/этилацетат 6/4).
Выход: 47% (клейкое твердое вещество); [α]D 20=+88° (c=0,98, CHCl3); ЖХМС (Tr): 7,33 мин (метод A); МС (ES+) m/z: 382,1.
1H-ЯМР (CDCl3, 333 K, 300 МГц), δ (м.д.): 7,93 (дд, 1H); 7,49-7,39 (м, 3H); 7,12-7,01 (м, 4H); 4,40 (уш.д, 1H); 4,00 (уш.д, 1H); 3,94 (c, 3H); 3,52 (дд, 1H); 3,25 (м, 2H); 2,34 (м, 1H); 2,09-1,86 (м, 2H); 1,68 (м, 1H).
Пример 53
(4-Фторфенил)-[(S)-3-(3-нафталин-2-ил-[1,2,4]оксадиазол-5-ил)-пиперидин-1-ил]метанон
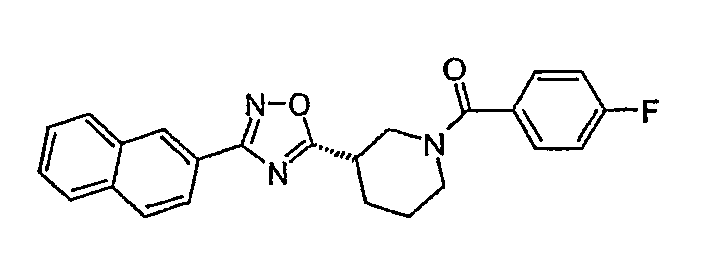
53(A). трет-Бутиловый эфир S-3-(3-нафталин-2-ил-[1,2,4]оксадиазол-5-ил)пиперидин-1-карбоновой кислоты
Соединение получают в соответствии с методикой, описанной в примере 49(A), исходя из 2-цианонафталина. Чистый трет-бутиловый эфир S-3-(3-нафталин-2-ил-[1,2,4-оксадиазол-5-ил)пиперидин-1-карбоновой кислоты получают после очистки сырого продукта флэш-хроматографией (силикагель, элюент: ДХМ/гексан/MeOH 50/50/0,2) (выход: 58%).
ЖХМС (Tr): 8,72 мин (метод A); МС (ES+) m/z: 380,1.
1H-ЯМР (CDCl3, 333 K, 300 МГц), δ (м.д.): 8,61 (уш.c, 1H); 8,14 (дд, 1H); 7,97-7,82 (м, 3H); 7,57-7,48 (м, 2H); 4,32 (уш.д, 1H); 3,95 (ддд, 1H); 3,36 (дд, 1H); 3,19 (тт, 1H); 3,05 (ддд, 1H); 2,29 (м, 1H); 2,02-1,82 (м, 2H); 1,71-1,58 (м, 1H).
53(B). Гидрохлорид S-3-(3-нафталин-2-ил-[1,2,4]оксадиазол-5-ил)пиперидина
Соединение получают в соответствии с методикой, описанной в примере 49(B), исходя из трет-бутилового эфира S-3-(3-нафталин-2-ил-[1,2,4]оксадиазол-5-ил)пиперидин-1-карбоновой кислоты (выход: 100%).
ЖХМС (Tr): 5,96 мин (метод B); МС (ES+) m/z: 280,1.
53(С). (4-Фторфенил)-[(S)-3-(3-нафталин-2-ил-[1,2,4]оксадиазол-5-ил)пиперидин-1-ил]метанон
Соединение получают в соответствии с методикой, описанной в примере 49(С), исходя из гидрохлорида S-3-(3-нафталин-2-ил-[1,2,4]оксадиазол-5-ил)пиперидина и используя 4-фторбензоилхлорид в качестве выбранного ацилхлорида. Чистый (4-фторфенил)-[(S)-3-(3-нафталин-2-ил-[1,2,4]оксадиазол-5-ил)-пиперидин-1-ил]метанон получают после очистки сырого продукта флэш-хроматографией (силикагель, элюент: гексан/этилацетат 8/2).
Выход: 14% (твердое белое вещество); т.пл.=142-143°C; [α]D 20=+123° (c=1,025, CHCl3); ЖХМС (Tr): 7,97 мин (метод A); МС (ES+) m/z: 402,1.
1H-ЯМР (CDCl3, 333 K, 300 МГц), δ (м.д.): 8,59 (уш.с, 1H); 8,12 (дд, 1H); 7,97-7,84 (м, 3H); 7,59-7,49 (м, 2H); 7,45 (дд, 2H); 7,10 (дд, 2H); 4,45 (уш.д, 1H); 4,01 (уш.д, 1H); 3,58 (дд, 1H); 3,29 (м, 2H); 2,38 (м, 1H); 2,14-1,89 (м, 2H); 1,78-1,65 (м, 1H).
Пример 54
(4-Фторфенил)-[(S)-3-(3-п-толил-[1,2,4]оксадиазол-5-ил)пиперидин-1-ил]метанон

54(A). трет-Бутиловый эфир S-3-(3-п-толил-[1,2,4]оксадиазол-5-ил)пиперидин-1-карбоновой кислоты
Соединение получают в соответствии с методикой, описанной в примере 49(A), исходя из 4-метилбензонитрила. Чистый трет-бутиловый эфир S-3-(3-п-толил-[1,2,4]оксадиазол-5-ил)пиперидин-1-карбоновой кислоты получают после растирания сырого продукта в смеси гексан/диэтилэфир 1/1 (Выход: 78%). ЖХМС (Tr): 11,0 мин (метод B); МС (ES+) m/z: 344,4.
1H-ЯМР (CDCl3, 333 K, 300 МГц), δ (м.д.): 7,96 (д, 2H); 7,26 (д, 2H); 4,28 (уш.д, 1H); 3,93 (ддд, 1H); 3,30 (дд, 1H); 3,13 (тт, 1H); 3,01 (ддд, 1H); 2,41 (c, 3H); 2,25 (м, 1H); 1,97-1,78 (м, 2H); 1,69-1,52 (м, 1H); 1,47 (c, 9H).
54(B). Гидрохлорид S-3-(3-п-толил-[1,2,4]оксадиазол-5-ил)пиперидина
Соединение получают в соответствии с методикой, описанной в примере 49(B), исходя из трет-бутилового эфира S-3-(3-п-толил-[1,2,4]оксадиазол-5-ил)пиперидин-1-карбоновой кислоты (выход: 100%).
ЖХМС (Tr): 5,3 мин (метод B); МС (ES+) m/z: 244,4.
54(С). (4-Фторфенил)-[(S)-3-(3-п-толил-[1,2,4]оксадиазол-5-ил)пиперидин-1-ил]метанон
Соединение получают в соответствии с методикой, описанной в примере 49(С), исходя из гидрохлорида S-3-(3-п-толил-[1,2,4]оксадиазол-5-ил)пиперидина и используя 4-фторбензоилхлорид в качестве выбранного ацилхлорида. Чистый (4-фторфенил)-[(S)-3-(3-п-толил-[1,2,4]оксадиазол-5-ил)пиперидин-1-ил]метанон получают после очистки сырого продукта флэш-хроматографией (силикагель, элюент: гексан/этилацетат 8/2).
Выход: 33% (густое масло); [α]D 20=+106° (c=1,0, CHCl3); ЖХМС (Tr): 9,5 мин (метод B); МС (ES+) m/z: 366,0.
1H-ЯМР (CDCl3, 333 K, 300 МГц), δ (м.д.): 7,94 (д, 2H); 7,43 (дд, 2H); 7,27 (д, 2H); 7,09 (дд, 2H); 4,40 (уш.д, 1H); 3,99 (уш.д, 1H); 3,52 (дд, 1H); 3,31-3,18 (м, 2H); 2,41 (c, 3H); 2,33 (м, 1H); 2,09-1,86 (м, 2H); 1,75-1,59 (м, 1H).
Пример 55
(4-Фторфенил)-{(S)-3-[3-(2-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон
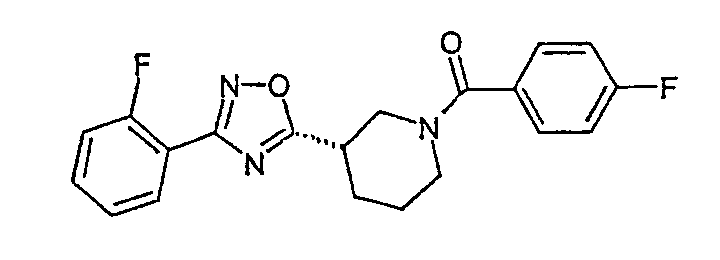
55(A). трет-Бутиловый эфир S-3-[3-(2-Фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-карбоновой кислоты
Соединение получают в соответствии с методикой, описанной в примере 49(A), исходя из трет-бутилового эфира 2-фторбензонитрила. Чистый трет-бутиловый эфир S-3-[3-(2-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-карбоновой кислоты получают после растирания сырого продукта в смеси гексан/диэтилэфир 1/1 (выход: 83%).
ЖХМС (Tr): 7,79 мин (метод A); МС (ES+) m/z: 348,1.
1H-ЯМР (CDCl3, 333 K, 300 МГц), δ (м.д.): 8,04 (ддд, 1H); 7,47 (м, 1H); 7,25 (ддд, 1H); 7,21 (м, 1H); 4,30 (уш.д, 1H); 3,94 (ддд, 1H); 3,32 (дд, 1H); 3,18 (тт, 1H); 3,02 (ддд, 1H); 2,26 (м, 1H); 1,99-1,79 (м, 2H); 1,69-1,53 (м, 1H); 1,47 (c, 9H).
55(B). Гидрохлорид S-3-[3-(2-Фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидина
Соединение получают в соответствии с методикой, описанной в примере 49(B), исходя из трет-бутилового эфира S-3-[3-(2-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-карбоновой кислоты (выход: 100%).
ЖХМС (Tr): 4,7 мин (метод B); МС (ES+) m/z: 248,1.
55(С). (4-Фторфенил)-{(S)-3-[3-(2-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон
Соединение получают в соответствии с методикой, описанной в примере 49(С), исходя из гидрохлорида S-3-[3-(2-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидина и используя 4-фторбензоилхлорид в качестве выбранного ацилхлорида. Чистый (4-Фторфенил)-{(S)-3-[3-(2-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон получают после очистки сырого продукта флэш-хроматографией (силикагель, элюент: гексан/этилацетат 8/2).
Выход: 22% (густое масло); [α]D 20=+102° (c=1,045, CHCl3); ЖХМС (Tr): 7,31 мин (метод A); МС (ES+) m/z: 370,1.
1H-ЯМР (CDCl3, 333 K, 300 МГц), δ (м.д.): 8,01 (ддд, 1H); 7,52-7,41 (м, 1H); 7,43 (дд, 1H); 7,29-7,18 (м, 2H); 7,09 (дд, 2H); 4,41 (уш.д, 1H); 3,99 (уш.д, 1H); 3,54 (дд, 1H); 3,27 (м, 2H); 2,34 (м, 1H); 2,10-1,87 (м, 2H); 1,76-1,61 (м, 1H).
Пример 56
(4-Фторфенил)-[(S)-3-(3-пиридин-2-ил-[1,2,4]оксадиазол-5-
ил)пиперидин-1-ил]метанон
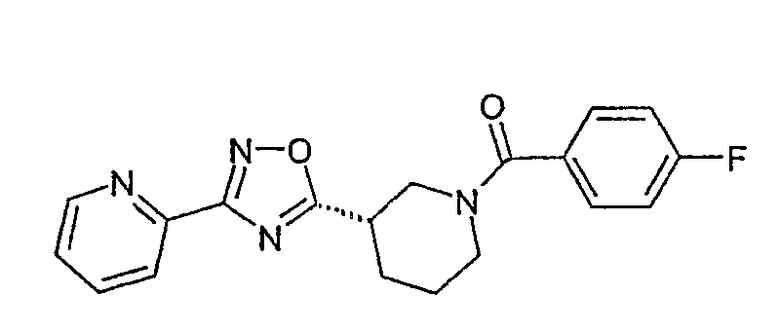
56(A). трет-Бутиловый эфир S-3-(3-пиридин-2-ил-[1,2,4]оксадиазол-5-ил)пиперидин-1-карбоновой кислоты
Соединение получают в соответствии с методикой, описанной в примере 49(A), исходя из 2-цианопиридина. Чистый трет-бутиловый эфир S-3-(3-пиридин-2-ил-[1,2,4]оксадиазол-5-ил)пиперидин-1-карбоновой кислоты получают после очистки сырого продукта флэш-хроматографией (силикагель, элюент: ДХМ/MeOH/NH4OH 98/2/0,2) (выход: 54%).
ЖХМС (Tr): 6,87 мин (метод A); МС (ES+) m/z: 331,2.
1H-ЯМР (CDCl3, 333 K, 300 МГц), δ (м.д.): 8,80 (ддд, 1H); 8,11 (ддд, 1H); 7,82 (ддд, 1H); 7,40 (ддд, 1H); 4,33 (уш.д, 1H); 3,98 (ддд, 1H); 3,33 (дд, 1H); 3,20 (тт, 1H); 2,99 (ддд, 1H); 2,28 (м, 1H); 2,03-1,79 (м, 2H); 1,69-1,54 (м, 1H), 1,48 (c, 9H).
56(B). Дигидрохлорид S-2-(5-Пиперидин-3-ил-[1,2,4]оксадиазол-3-ил)пиридина
Соединение получают в соответствии с методикой, описанной в примере 49(B), исходя из трет-бутилового эфира S-3-(3-пиридин-2-ил-[1,2,4]оксадиазол-5-ил)пиперидин-1-карбоновой кислоты (выход: 100%).
ЖХМС (Tr): 3,12 мин (метод B); МС (ES+) m/z: 231,2.
56(С). (4-Фторфенил)-[(S)-3-(3-пиридин-2-ил-[1,2,4]оксадиазол-5-ил)пиперидин-1-ил]метанон
Соединение получают в соответствии с методикой, описанной в примере 49(С), исходя из дигидрохлорида S-2-(5-пиперидин-3-ил-[1,2,4]оксадиазол-3-ил)пиридина и используя 4-фторбензоилхлорид в качестве выбранного ацилхлорида.
Выход: 78% (густое масло желтоватого цвета); [α]D 20=+103° (c=1,05, CHCl3); ЖХМС (Tr): 6,56 мин (метод A); МС (ES+) m/z: 353,0.
1H-ЯМР (CDCl3, 333 K, 300 МГц), δ (м.д.): 8,79 (м, 1H); 8,09 (ддд, 1H); 7,82 (ддд, 1H); 7,47-7,37 (м, 3H); 7,08 (дд, 2H); 4,44 (уш.д, 1H); 4,05 (уш.д, 1H); 3,54 (дд, 1H); 3,29 (тт, 1H); 3,21 (ддд, 1H); 2,36 (м, 1H); 2,13-1,86 (м, 2H); 1,76-1,61 (м, 1H).
Пример 57
S -(4-Фторфенил)-{3-[5-(4-фторфенил)-4-метил-4H-1,2,4-триазол-3-ил]пиперидин-1-ил}метанон
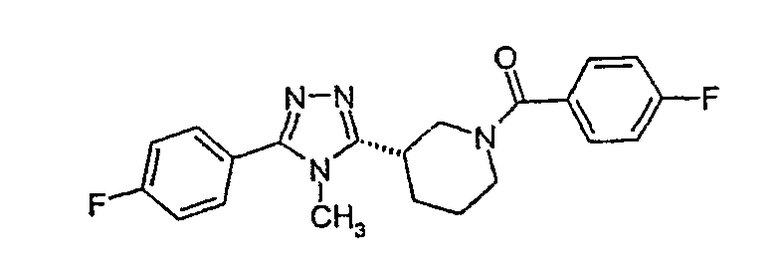
57(A). 1-(4-Фторбензоил)-(S)пиперидин-3-карбоновая кислота
К суспензии гидрохлорида (S)пиперидин-3-карбоновой кислоты (0,75 г, 4,53 ммоль) в сухом дихлорметане (30 мл ) по каплям при 0°С добавляют триэтиламин (1,97 мл, 14,0 ммоль) и 4-фторбензоилхлорид (543 мл, 4,53 ммоль). Полученный раствор перемешивают в течение ночи при комнатной температуре в атмосфере азота, добавляют 1 н. HCl (30 мл) и фазы разделяют. Органический слой промывают 1 н. HCl (30 мл), водой (30 мл), сушат над Na2SO4 и упаривают при пониженном давлении, получая 1,05 г 1-(4-фторбензоил)-(S)пиперидин-3-карбоновой кислоты в виде масла желтого цвета, которое используют на следующей стадии синтеза без дополнительной очистки. Выход: 92%; ЖХМС (Tr): 6,55 мин (метод B); МС (ES+) m/z: 252,3.
57(B). трет-Бутиловый эфир N'-[1-(4-фторбензоил)-(S)пиперидин-3-карбонил]гидразинкарбоновой кислоты
Смесь 1-(4-фторбензоил)-(S)-пиперидин-3-карбоновой кислоты (1,05 г, 4,17 ммоль), трет-бутилкарбазата (0,55 г, 4,17 ммоль), HOBT (0,562 г, 4,17 ммоль), EDCI·HCl (1,2 г, 6,25 ммоль) в сухом дихлорметане (8 мл) перемешивают в течение ночи при температуре окружающей среды в атмосфере азота. Затем добавляют 1 н. HCl (30 мл) и фазы разделяют. Органический слой промывают 1 н. HCl (30 мл), 1 н. NaOH (30 мл × 2), затем водой (30 мл). Выпаривание органического растворителя приводит к получению сырого масла желтого цвета, которое очищают флэш-хроматографией (силикагель, элюент: ДХМ/MeOH 70/1), получая 0,715 г трет-бутилового эфира N'-[1-(4-фторбензоил)-(S)пиперидин-3-карбонил]гидразинкарбоновой кислоты.
Выход: 47% (масло желтого цвета); ЖХМС (Tr): 6,40 мин (метод A); МС (ES+) m/z: 366,2.
1H-ЯМР (CDCl3, 333 K, 300 МГц), δ (м.д.): 8,04 (уш.с, 1H); 7,47-7,36 (м, 2H); 7,12-7,01 (м, 2H); 6,42 (уш.с, 1H); 4,12-3,90 (уш.м, 1H); 3,88-3,51 (уш.м, 2H); 3,34-3,09 (уш.м, 1H); 2,62-2,36 (уш.м, 1H); 2,18-1,84 (м, 2H); 1,81-1,64 (м, 1H); 1,47 (c, 9H); 1,52-1,43 (м, 1H).
57(С). Гидрохлорид гидразида 1-(4-фторбензоил)-(S)пиперидин-3-карбоновой кислоты
0,55 г (1,5 ммоль) трет-бутилового эфира N'-[1-(4-фторбензоил)-(S)пиперидин-3-карбонил]гидразинкарбоновой кислоты суспендируют в 5 мл дихлорметана и добавляют при 0°C 4 мл 1 н. HCl (раствор в диоксане). Раствору дают возможность нагреться до комнатной температуры и перемешивают его в течение 1 часа 30 минут. Растворитель выпаривают при пониженном давлении, получая 0,412 г гидразида 1-(4-фторбензоил)-(S)пиперидин-3-карбоновой кислоты в виде твердого белого, очень гигроскопичного вещества.
Выход: 91%; ЖХМС (Tr): 5,4 мин (метод B); МС (ES+) m/z: 266,2.
1H-ЯМР (CDCl3, 333 K + D2O, 300 МГц), δ (м.д.): 7,41 (дд, 2H); 7,08 (дд, 2H); 4,45 (м, 1H); 4,11 (м, 1H); 3,84 (м, 1H); 3,39 (дд, 1H); 3,13 (м, 1H); 2,39 (м, 1H); 1,95 (м, 2H); 1,85-1,75 (м, 1H).
57(D). 4-фтор-N-метилбензимидоилхлорид
Суспензию 4-фтор-N-метилбензамида (CAS: 701-49-5, 0,106 г, 0,69 ммоль) в тионилхлориде (202 мл, 2,78 ммоль) кипятят с обратным холодильником в течение 1 часа 30 минут. Растворитель удаляют при пониженном давлении, затем добавляют толуол и растворитель выпаривают при пониженном давлении, получая масло желтого цвета, которое сразу используют в следующей стадии.
57(E). S-(4-Фторфенил)-{3-[5-(4-фторфенил)-4-метил-4H-[1,2,4]триазол-3-ил]пиперидин-1-ил}метанон
К раствору 4-фтор-N-метилбензимидоилхлорида, полученному в соответствии с методикой, описанной в 58(D), в сухом толуоле (8 мл), в атмосфере азота добавляют гидрохлорид гидразина 1-(4-фторбензоил)-(S)пиперидин-3-карбоновой кислоты (0,21 г, 0,69 ммоль) и безводный триэтиламин (204 мл, 1,46 ммоль) и полученную смесь кипятят с обратным холодильником в течение 2 часов. Растворитель удаляют при пониженном давлении, остаток разбавляют дихлорметаном и промывают NaHCO3 (водный). Органический слой сушат над Na2SO4 и упаривают при пониженном давлении. Сырой продукт очищают флэш-хроматографией (2 последовательно соединенные колонки (первая колонка: силикагель, элюирование с градиентом: от ДХМ/MeOH 20:1 до ДХМ/MeOH 4:1; вторая колонка: силикагель, элюирование с градиентом: ацетон/этилацетат от 1:1 до 2:1), получая 20 мг S-(4-фторфенил)-{3-[5-(4-фторфенил)-4-метил-4H-[1,2,4]триазол-3-ил]пиперидин-1-ил}метанон.
Выход: 8% (твердое белое вещество); ЖХМС (Tr): 6,26 мин (метод A); МС (ES+) m/z: 383,1.
1H-ЯМР (CDCl3, 300 МГц), δ (м.д.): 7,65 (дд, 2H); 7,46 (дд, 2H); 7,20 (дд, 2H); 7,10 (дд, 2H); 4,46 (уш.м, 1H); 4,01 (уш.м, 1H); 3,66 (c, 3H); 3,39 (м, 1H); 3,19 (м, 1H); 2,98 (уш.м, 1H); 2,28-1,89 (м, 3H); 1,60 (м, 1H).
Пример 58
(4-Фторфенил)-{(S)-3-[5-(4-фторфенил)-[1,3,4]оксадиазол-2-ил]пиперидин-1-ил}метанон
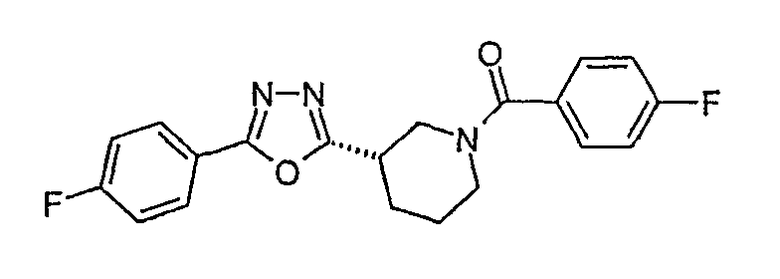
58(A). N'-[(S)-1-(4-фторбензоил)пиперидин-3-карбонил]гидразид 4-фторбензойной кислоты
Смесь гидрохлорида гидразида 1-(4-фторбензоил)-(S)-пиперидин-3-карбоновой кислоты, полученного в соответствии с методикой, описанной в примере 57(С) (2,97 г, 11,2 ммоль), 4-фторбензойной кислоты (1,68 г, 11,2 ммоль), HOBT (1,5 г, 11,2 ммоль), EDCI·HCl (3,2 г, 16,8 ммоль) и сухого триэтиламина (5,43 мл, 39,5 ммоль) в сухом дихлорметане (80 мл) перемешивают в течение ночи при температуре окружающей среды в атмосфере азота. Затем добавляют 1 н. HCl (80 мл) и фазы разделяют. Органический слой промывают 1 н. HCl (80 мл), 1 н. NaOH (80 мл × 2), затем водой (80 мл). Выпаривание органического растворителя приводит к получению сырого продукта в виде масла, которое очищают флэш-хроматографией (силикагель, элюент: ДХМ/MeOH/NH4OH 98/2/0,2). Соединение, полученное после колоночной хроматографии, снова очищают флэш-хроматографией (силикагель, элюент: ДХМ/MeOH/NH4OH 98/2/0,2), получая чистый N'-[(S)-1-(4-фторбензоил)пиперидин-3-карбонил]гидразид 4-фторбензойной кислоты в виде твердого белого вещества (250 мг).
Выход: 6%; ЖХМС (Tr): 5,88 мин (метод A); МС (ES+) m/z: 388,0.
1H-ЯМР (CDCl3, 333 K, 300 МГц), δ (м.д.): 8,91 (уш.с, 1H); 8,35 (уш.с, 1H); 7,83 (дд, 2H); 7,47 (дд, 2H); 7,13 (дд, 2H); 7,09 (дд, 2H); 4,03-3,76 (м, 3H); 3,32 (м, 1H); 2,61 (м, 1H); 2,22-1,93 (м, 2H); 1,77 (м, 1H); 1,55 (м, 1H).
58(B). (4-Фторфенил)-{3-[5-(4-фторфенил)-[1,3,4]оксадиазол-2-ил]пиперидин-1-ил}метанон
Смесь N'-[(S)-1-(4-фторбензоил)пиперидин-3-карбонил]гидразида 4-фторбензойной кислоты (100 мг, 0,26 ммоль), 4-толуонсульфонилхлорида (60 мг, 0,31 ммоль), 2-трет-бутилимино-2-диэтиламино-1,3-диметилпергидро-1,3,2-диазафосфора на твердом носителе (PS-BEMP, ex Fluka, 586 мг, 1,3 ммоль, наполнение 2,2 ммоль/г) в сухом тетрагидрофуране (6 мл) обрабатывают микроволнами в следующих условиях: цикл МВ: t=1 мин, P=100 Вт, время охлаждения = 2 мин; после 5 МВ циклов смолу отфильтровывают и повторно промывают дихлорметаном. Растворитель выпаривают при пониженном давлении, получая сырой продукт в виде твердого вещества, которое очищают колоночной флэш-хроматографией (силикагель, элюент: гексан/этилацетат 1:1). Указанное в заголовке соединение получают в виде твердого белого вещества (82 мг).
Выход: 85%; ЖХМС (Tr): 6,75 мин (метод A); МС (ES+) m/z: 370,1.
1H-ЯМР (CDCl3, 300 K, 300 МГц), δ (м.д.): 8,02 (м, 2H); 7,41 (м, 2H); 7,31-6,93 (м, 4H); 4,96-3,37 (м, 3H); 3,22 (м, 2H), 2,32 (м, 1H); 2,20-1,63 (м, 3H).
Пример 59
(2-Фторфенил)-{(S)-3-[2-(3,4-дифторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон
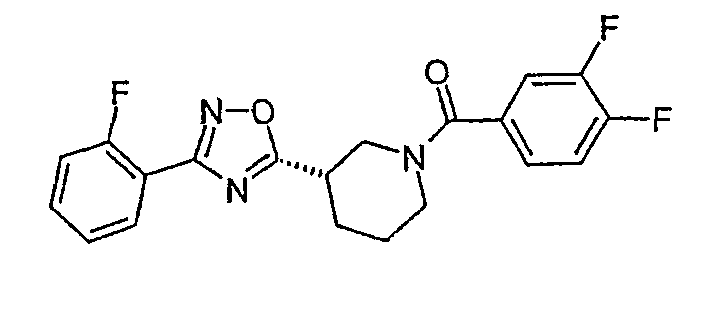
Соединение получают в соответствии с методикой, описанной в примере 49(С), исходя из гидрохлорида S-3-[3-(2-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидина и используя 3,4-дифторбензоилхлорид в качестве выбранного ацилхлорида. Чистый (2-Фторфенил)-{(S)-3-[2-(3,4-дифторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон получают после очистки сырого продукта флэш-хроматографией (силикагель, элюент:ДХМ//MeOH/NH4OH 99/1/0,1).
Выход: 61% (клейкое твердое вещество); т.пл.=115-119°C; [α]D 20=+92,2° (c=1,14,
CHCl3); ЖХМС (Tr): 7,25 мин (метод A); МС (ES+) m/z: 388,0.
1H-ЯМР (CDCl3, 333 K, 300 МГц), δ (м.д.): 8,01 (ддд, 1H); 7,48 (м, 1H); 7,33-7,15 (м, 5H); 4,37 (м, 1H); 3,93 (м, 1H); 3,59 (дд, 1H); 3,37-3,23 (м, 2H); 2,35 (м, 1H); 2,13-1,87 (м, 2H); 1,76-1,61 (м, 1H).
Пример 60
(4-Фторфенил)-{2-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]морфолин-4-ил}метанон
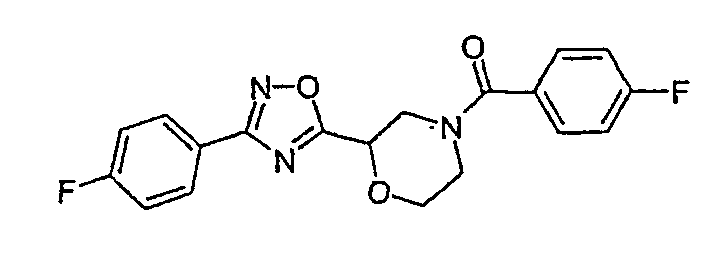
60(A). трет-Бутиловый эфир 2-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]морфолин-4-карбоновой кислоты
Смесь (R,S)-N-Boc-2-карбоксиморфолина (0,5 г, 2,16 ммоль), N-гидрокси-4-фторбензамидина (0,333 г, 2,16 ммоль), EDCI·HCl (0,621 г, 3,24 ммоль), HOBT (0,292 г, 2,16 ммоль) и безводного триэтиламина (605 мл, 4,32 ммоль) в диоксане (7 мл) перемешивают в течение ночи при комнатной температуре в атмосфере азота. После этого смесь кипятят с обратным холодильником в течение 4 часов и концентрируют при пониженном давлении. Остаток разбавляют этилацетатом (15 мл) и водой (15 мл) и фазы разделяют. Органический слой промывают 1 н. NaOH (15 мл) и раствором соли, сушат над сульфатом натрия и упаривают при пониженном давлении. Сырой продукт очищают флэш-хроматографией (силикагель, элюент: гексан/этилацетат 9/1), получая 325 мг (выход: 43%) трет-бутилового эфира 2-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]морфолин-4-карбоновой кислоты в виде белого масла.
ЖХМС (Tr): 7,51 мин (метод A); МС (ES+) m/z: 350,1.
1H-ЯМР (CDCl3, 300 МГц), δ (м.д.): 8,11 (дд, 2H); 7,16 (дд, 2H); 4,83 (дд, 1H); 4,27 (уш.м, 1H); 4,09 (м, 1H); 3,87 (м, 1H); 3,74 (ддд, 1H); 3,41 (уш.м, 1H); 3,23 (ддд, 1H); 1,48 (c, 9H).
60(B). Гидрохлорид 2-[3-(4-Фторфенил)-[1,2,4]оксадиазол-5-ил]морфолина
2-[3-(4-Фторфенил)-[1,2,4]оксадиазол-5-ил]морфолин-4-карбоновую кислоту (0,325 г, 0,93 ммоль) растворяют в дихлорметане (3 мл) и по каплям добавляют 5 мл 4 н. HCl (раствор в диоксане). Полученную смесь перемешивают при комнатной температуре в течение 2 часов. Растворитель выпаривают при пониженном давлении, получая 265 мг (выход: 100%) гидрохлорида 2-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]морфолина в виде твердого белого вещества.
ЖХМС (Tr): 5,68 мин (метод A); МС (ES+) m/z: 250,1.
1H-ЯМР (ДМСО, 300 МГц), δ (м.д.): 9,58 (уш.с, 1H); 8,09 (дд, 2H); 7,43 (дд, 2H); 5,38 (дд, 1H); 4,19-3,97 (м, 2H); 3,71 (дд, 1H); 3,45 (дд, 1H), 3,30-3,12 (м, 2H).
60(С). (4-Фторфенил)-{2-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]морфолин-4-ил}метанон
К суспензии гидрохлорида 2-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]морфолина (0,15 г, 0,53 ммоль) в безводном дихлорметане (6 мл) в атмосфере азота при 0°С последовательно добавляют триэтиламин (155 мл, 1,1 ммоль) и 4-фторбензоилхлорид (62 мл, 0,53 ммоль). Реакционной смеси дают возможность нагреться до комнатной температуры и перемешивают ее в течение ночи. К смеси добавляют 1 н. HCl (6 мл) и фазы разделяют. Органический слой промывают последовательно 1 н. HCl (6 мл), 1н NaOH (6 мл × 2) и водой, затем сушат над сульфатом натрия и упаривают при пониженном давлении. Сырой продукт очищают флэш-хроматографией (силикагель, элюент: гексан/этилацетат 7/3), получая 120 мг (4-фторфенил)-{2-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]морфолин-4-ил}метанона в виде твердого белого вещества.
Выход: 61%; т.пл.=116-117°C; ЖХМС (Tr): 7,33 мин (метод A); МС (ES+) m/z: 372,0.
1H-ЯМР (CDCl3, 300 МГц, 330 K), δ (м.д.): 8,08 (дд, 2H); 7,47 (дд, 2H); 7,16 (дд, 2H); 7,12 (дд, 2H); 4,90 (дд, 1H); 4,39 (уш.д,1H); 4,12 (ддд, 1H); 3,95 (уш.д, 1H); 3,79 (ддд, 1H); 3,71 (дд, 1H); 3,53 (ддд, 1H).
Пример 61
(4-Фторфенил)-{3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]-4-метилпиперазин-1-ил}метанон
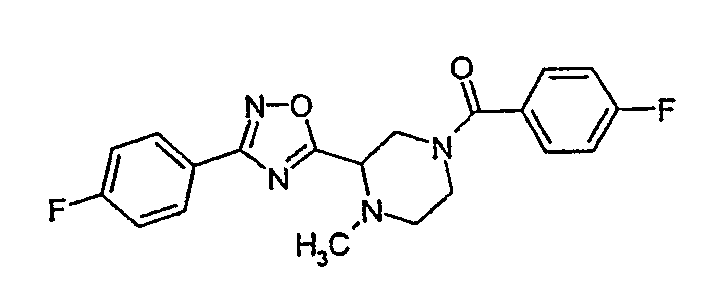
61(A). трет-Бутиловый эфир пиперазин-1,3-дикарбоновой кислоты
К раствору дигидрохлорида 2-пиперазинкарбоновой кислоты (1,0 г, 4,92 ммоль) в 20 мл смеси вода/диоксан 1:1 добавляют 6 н. NaOH добавляют для доведения pH до 11. После этого к смеси по каплям добавляют раствор BOC-ON® (1,34 г, 5,41 ммоль) в диоксане (5 мл) при сохранении во время добавления pH=11 и полученный раствор перемешивают в течение ночи при комнатной температуре. Добавляют еще 0,134 г BOC-ON® и реакционную смесь перемешивают в течение 2 часов. Растворитель выпаривают при пониженном давлении и остаток разбавляют смесью диэтилэфир/вода (60 мл). Фазы разделяют и pH водного слоя доводят до 7 медленным добавлением 1 н. HCl. Выпаривание воды при пониженном давлении приводит к получению указанного в заголовке соединения в виде твердого белого вещества, которое сушат в вакуумной печи при 50°C и используют в следующей стадии без дополнительной очистки.
ЖХМС (Tr): 3,3 мин (метод B); МС (ES+) m/z: 231,0.
61(B). трет-Бутиловый эфир 4-метилпиперазин-1,3-дикарбоновой кислоты
Сырой 1-трет-бутиловый эфир пиперазин-1,3-дикарбоновой кислоты (4,92 ммоль), полученный в соответствии с методикой примера 62(A), суспендируют в сухом ацетонитриле (30 мл) в атмосфере азота и к раствору добавляют формальдегид (37% водный раствор, 367 мл, 4,92 ммоль) и Na(OAc)3BH (2,3 г, 10,82 ммоль). Полученную смесь перемешивают при комнатной температуре в течение 3 часов, затем медленно добавляют насыщенный водный раствор NaHCO3 для доведения значения pH до 7. Смесь упаривают досуха при пониженном давлении, получая твердое вещество желтого цвета, которое используют в следующей стадии без дополнительной очистки. ЖХМС (Tr): 3,19 мин (метод B); МС (ES+) m/z: 245,0.
61(С). трет-Бутиловый эфир 3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]-4-метилпиперазин-1-карбоновой кислоты
Смесь N-гидрокси-4-фторбензамидина (0,758 г, 4,92 ммоль), 4-метилпиперазин-1,3-дикарбоновой кислоты (4,92 ммоль), полученной в соответствии с методикой примера 62(B), EDCI·HCl (1,41 г, 7,38 ммоль), HOBT (0,665 г, 4,92 ммоль) и безводного триэтиламина (1,38 мл, 9,84 ммоль) в диоксане (80 мл) перемешивают при комнатной температуре в атмосфере азота в течение выходных. Затем реакционную смесь кипятят с обратным холодильником в течение 7 часов и растворитель выпаривают при пониженном давлении. Остаток разбавляют водой (50 мл) и этилацетатом (50 мл), фазы разделяют и органический слой промывают последовательно водой (50 мл × 2) и 1 н. NaOH (50 мл × 2). Органический слой сушат над Na2SO4 и концентрируют при пониженном давлении, получая масло желтого цвета. Очистка сырого продукта флэш-хроматографией (силикагель, элюирование с градиентом: гексан/этилацетат от 8/2 до 7/3) приводит к получению 0,312 г трет-бутилового эфира 3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]-4-метилпиперазин-1-карбоновой кислоты в виде масла желтого цвета.
Общий выход трех стадий 22(A), 22(B) и 22(С): 18%. ЖХМС (Tr): 7,34 мин (метод B); МС (ES+) m/z: 363,1.
1H-ЯМР (CDCl3, 300 МГц, 328 K), δ (м.д.): 8,11 (дд, 2H); 7,16 (дд, 2H); 4,01-3,90 (м, 2H); 3,83-3,62 (м, 3H); 3,17 (м, 1H); 2,55 (м, 1H); 2,43 (c, 3H); 1,41 (c, 9H).
61(D). Дигидрохлорид 2-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]-1-метилпиперазина
4 н. HCl (3 мл раствора в диоксане) по каплям добавляют к раствору трет-бутилового эфира [3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]-4-метилпиперазин-1-карбоновой кислоты (0,312 г, 0,86 ммоль) в метаноле (8 мл) и раствор перемешивают в течение ночи при комнатной температуре. Выпаривание летучих компонентов при пониженном давлении приводит к получению дигидрохлорида 2-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]-1-метилпиперазина в виде твердого белого вещества (0,285 г).
Выход: 100%; ЖХМС (Tr): 5,63 мин (метод A); МС (ES+) m/z: 263,2.
1H-ЯМР (CDCl3, 300 МГц, 328 K), δ (м.д.): 8,11 (дд, 2H); 7,16 (дд, 2H); 4,01-3,90 (м, 2H); 3,83-3,62 (м, 3H); 3,17 (м, 1H); 2,55 (м, 1H); 2,43 (c, 3H); 1,41 (c, 9H).
61(E). (4-Фторфенил)-{3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]-4-метилпиперазин-1-ил}метанон
К суспензии дигидрохлорида 2-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]-1-метилпиперазина (0,285 г, 0,86 ммоль) в безводном дихлорметане (6 мл) в атмосфере азота при 0°С последовательно добавляют триэтиламин (374 мл, 2,7 ммоль) и 4-фторбензоилхлорид (102 мл, 0,86 ммоль). Реакционной смеси дают возможность нагреться до комнатной температуры и перемешивают смесь в течение 2 часов. Добавляют 1 н. NaOH (6 мл) и фазы разделяют. Органический слой промывают последовательно 1 н. NaOH (6 мл) и водой, затем сушат над сульфатом натрия и упаривают при пониженном давлении. Очистка сырого продукта флэш-хроматографией (силикагель, элюент: ДХМ/MeOH/NH4OH 99/1/0,1) приводит к получению (4-фторфенил)-{3-[3-(4-фторфенил)-[1,2,4-оксадиазол-5-ил]-4-метилпиперазин-1-ил}метанона (0,12 г).
Выход: 36%; ЖХМС (Tr): 6,36 мин (метод A); МС (ES+) m/z: 385,1.
1Н-ЯМР (CDCl3, 328 K, 300 МГц), δ (м.д.): 8,10 (дд, 2H); 7,40 (дд, 2H); 7,18 (дд, 2H); 7,06 (дд, 2H); 4,14-3,92 (м, 3H); 3,91-3,73 (м, 2H); 3,17 (м, 1H); 2,58 (м, 1H); 2,42 (c, 3H).
Пример 62
(4-Фторфенил)амид (S)-1-(4-Фторбензоил)пиперидин-3-карбоновой кислоты
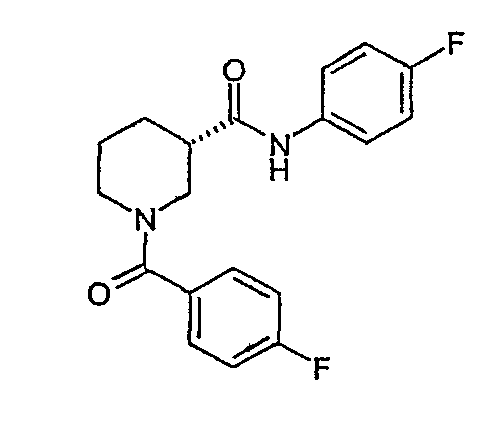
62(A). Трет-Бутиловый эфир (S)-3-(4-фторфенилкарбамоил)пиперидин-1-карбоновой кислоты
Смесь S-1-Boc-пиперидин-3-карбоновой кислоты (0,3 г, 1,30 ммоль), EDCI·HCl (0,376 г, 1,96 ммоль), HOBT (0,198 г, 1,30 ммоль) в сухом дихлорметане (6 мл) перемешивают при комнатной температуре в течение 1 часа в атмосфере азота. Затем добавляют 4-фторанилин (124 мл, 1,30 ммоль) и реакционную смесь перемешивают в течение ночи при температуре окружающей среды. Растворитель выпаривают при пониженном давлении и остаток разбавляют водой и этилацетатом. Фазы разделяют, органический слой промывают 2М Na2CO3 (водный), сушат над сульфатом натрия и концентрируют при пониженном давлении. Сырой продукт очищают фильтрацией через картридж с силикагелем (силикагель: 10 г, элюирование с градиентом: петролейный эфир/этилацетат от 9/1 до 8/2), получая 0,35 г трет-бутилового эфира (S)-3-(4-фторфенилкарбамоил)пиперидин-1-карбоновой кислоты.
Выход: 84%; ЖХМС (Tr): 7,08 мин (метод A); МС (ES+) m/z: 323,2.
1H-ЯМР (CDCl3, 300 МГц), δ (м.д.): 8,26 (уш.с, 1H); 7,53 (дд, 2H); 6,98 (дд, 2H); 3,82 (м, 1H); 3,67-3,46 (м, 2H); 3,23 (м, 1H); 2,49 (м, 1H); 2,06 (м, 1H); 1,89 (м, 1H); 1,63 (м, 1H); 1,50 (м, 1H); 1,46 (c, 9H).
62(B). Гидрохлорид (4-фторфенил)амида (S)-пиперидин-3-карбоновой кислоты
К охлажденному раствору трет-бутилового эфира (S)-3-(4-фторфенилкарбамоил)пиперидин-1-карбоновой кислоты (0,34 г, 1,05 ммоль) в дихлорметане (5 мл) по каплям добавляют 5,2 мл 4 н. HCl (раствор в диоксане) и раствор перемешивают при комнатной температуре в течение 45 минут. Растворитель выпаривают при пониженном давлении, получая указанное в заголовке соединение в виде твердого белого вещества (0,252 г).
Выход: 92%; ЖХМС (Tr): 5,39 мин (метод A); МС (ES+) m/z: 223,2.
1H-ЯМР (ДМСО, 300 МГц), δ (м.д.): 10,34 (c, 1H); 8,95 (уш.с, 2H); 7,63 (дд, 2H); 7,14 (дд, 2H); 3,18 (м, 2H); 3,02 (дд, 1H); 2,88 (м, 2H); 2,04 (м, 1H); 1,88-1,55 (м, 3H).
62(С). (4-Фторфенил)амид (S)-1-(4-Фторбензоил)пиперидин-3-карбоновой кислоты
4-фторбензоилхлорид (53 мл, 0,45 ммоль) добавляют при 0°C к раствору гидрохлорида (4-фторфенил)амида (S)-пиперидин-3-карбоновой кислоты (0,116 г, 0,45 ммоль) и триэтиламина (131 мл, 0,94 ммоль) в дихлорметане (3 мл) в атмосфере азота. Реакционную смесь перемешивают при комнатной температуре в течение 2,5 часов, растворитель выпаривают и остаток разбавляют водой и этилацетатом. Фазы разделяют и органический слой промывают 1 н. HCl (10 мл), 2М Na2CO3 (водный) (10 мл) и раствором соли (10 мл). Органический слой сушат над сульфатом натрия и концентрируют при пониженном давлении, получая 0,127 г (4-фторфенил)амида (S)-1-(4-фторбензоил)пиперидин-3-карбоновой кислоты в виде твердого белого вещества.
Выход: 82%; т.пл.=163-164°C; [α]D 20=+54,7° (c=0,995, CHCl3); ЖХМС (Tr): 6,68 мин (метод A); МС (ES+) m/z: 345,0.
1H-ЯМР (CDCl3, 333 K, 300 МГц), δ (м.д.): 8,28 (уш.с, 1H); 7,56 (дд, 2H); 7,41 (дд, 2H); 7,09 (дд, 2H); 7,01 (дд, 2H); 4,05 (уш.м, 1H); 3,89 (дд, 1H); 3,65 (уш.м, 1H); 3,40 (уш.м, 1H); 2,63 (м, 1H); 2,26 (м, 1H); 1,95 (м, 1H); 1,64 (м, 1H); 1,56 (м, 1H).
Пример 63
(4-Фторфенил)метиламид (S)-1-(4-фторбензоил)пиперидин-3-карбоновой кислоты
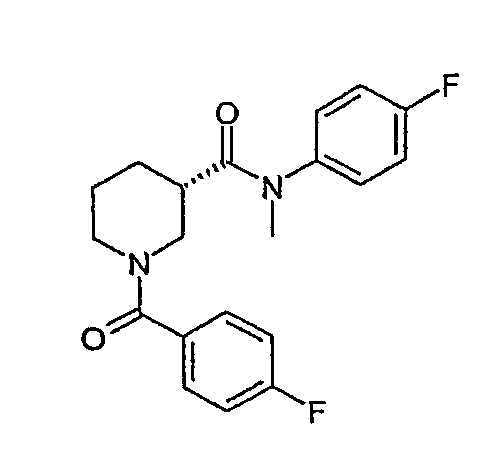
63(A). трет-Бутиловый эфир (S)-3-[(4-фторфенил)метилкарбамоил]пиперидин-1-карбоновой кислоты
Смесь S-1-Boc-пиперидин-3-карбоновой кислоты (0,3 г, 1,30 ммоль), EDCI·HCl (0,376 г, 1,96 ммоль), HOBT (0,198 г, 1,30 ммоль) в диоксане (4 мл) перемешивают при комнатной температуре в течение 1 часа в атмосфере азота. К смеси добавляют N-метил-4-фторанилин (164 мг, 1,30 ммоль), реакционную смесь нагревают до 80°C и выдерживают при этой температуре в течение 2 часов и затем перемешивают при комнатной температуре в течение 2 дней. Растворитель выпаривают при пониженном давлении и остаток разбавляют водой и этилацетатом. Фазы разделяют, органический слой промывают 2М Na2CO3 (водный), сушат над сульфатом натрия и концентрируют при пониженном давлении. Сырой продукт очищают фильтрацией через картридж с силикагелем (силикагель: 10 г, элюирование с градиентом: петролейный эфир/этилацетат: от 9/1 до 7/3), получая 0,209 г трет-бутилового эфира (S)-3-[(4-фторфенил)метилкарбамоил]пиперидин-1-карбоновой кислоты.
Выход: 47%; ЖХМС (Tr): 7,0 мин (метод A); МС (ES+) m/z: 337,2.
1H-ЯМР (CDCl3, 300 МГц), δ (м.д.): 7,22-7,07 (м, 4H); 4,00 (м, 2H); 3,22 (c, 3H); 2,89 (дд, 1H); 2,63 (м, 1H); 2,27 (м, 1H); 1,78-1,65 (м, 2H); 1,61-1,50 (м, 2H); 1,40 (c, 9H).
63(B). Гидрохлорид (4-фторфенил)метиламида (S)-пиперидин-3-карбоновой кислоты
К охлажденному раствору трет-бутилового эфира (S)-3-[(4-фторфенил)метилкарбамоил]пиперидин-1-карбоновой кислоты (0,205 г, 0,61 ммоль) в дихлорметане (4 мл) по каплям добавляют 3 мл 4 н. HCl (раствор в диоксане) и полученную смесь перемешивают при комнатной температуре в течение 1 часа. Растворитель выпаривают при пониженном давлении, получая указанное в заголовке соединение в виде твердого белого вещества (0,16 г).
Выход: 96%. ЖХМС (Tr): 5,37 мин (метод A); МС (ES+) m/z: 237,1.
1H-ЯМР (ДМСО + ТФУК, 333 K, 300 МГц), δ (м.д.): 8,98-8,45 (м, 2H); 7,43 (дд, 2H); 7,28 (дд, 2H); 3,16 (c, 3H); 3,16-2,67 (м, 5H); 1,80-1,34 (м, 4H).
63(С). (4-Фторфенил)метиламид (S)-1-(4-фторбензоил)пиперидин-3-карбоновой кислоты
4-фторбензоилхлорид (47 мл, 0,40 ммоль) добавляют при 0°C к раствору гидрохлорида (4-фторфенил)метиламида (S)пиперидин-3-карбоновой кислоты (0,11 г, 0,40 ммоль) и триэтиламина (112 мл, 0,80 ммоль) в дихлорметане (3 мл) в атмосфере азота. Реакционную смесь перемешивают при комнатной температуре в течение 2 часов, растворитель выпаривают и остаток разбавляют водой и этилацетатом. Фазы разделяют и органический слой промывают 1 н. HCl (10 мл), 1М Na2CO3 (водный) (10 мл) и раствором соли (10 мл). Органический слой сушат над сульфатом натрия и концентрируют при пониженном давлении, получая 0,121 г (4-фторфенил)метиламида (S)-1-(4-фторбензоил)пиперидин-3-карбоновой кислоты в виде клейкого твердого вещества.
Выход: 84%; [α]D 20=+48,9° (c=1,020, CHCl3); ЖХМС (Tr): 6,61 мин (метод A); МС (ES+) m/z: 359,1.
1H-ЯМР (CDCl3, 333 K, 300 МГц), δ (м.д.): 7,28 (дд, 2H); 7,13-7,00 (м, 6H); 4,06 (уш.м, 2H); 3,20 (c, 3H); 3,17 (м, 1H); 2,89 (м, 1H); 2,40 (м, 1H); 1,94-1,66 (м, 3H); 1,28 (м, 1H).
Пример 64
(E)-3-(4-Фторфенил)-1-{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}пропенон
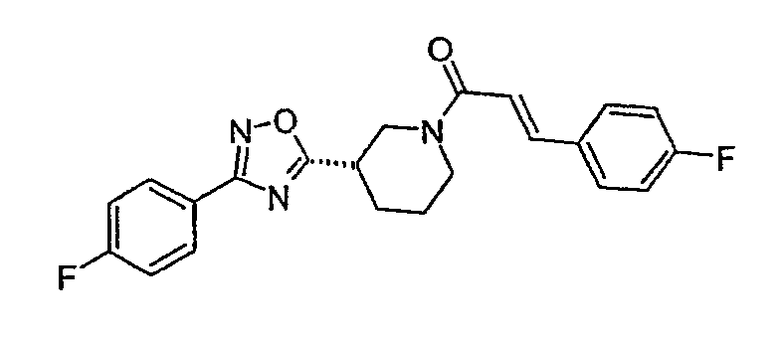
Соединение получают в соответствии с методикой, описанной в примере 36, используя 4-фторкоричную кислоту в качестве выбранной кислоты и гидрохлорид S-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидина (полученный в соответствии с методикой примера 12). Чистый (E)-3-(4-фторфенил)-1-{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}пропенон получают после растирания сырого продукта с диэтилэфиром.
Выход: 77% (твердое белое вещество); т.пл.=137-139°C; [α]D 20=+191,7° (c=1,49, CHCl3); ЖХМС (Tr): 7,62 мин (метод A); МС (ES+) m/z: 396,1.
1H-ЯМР (CDCl3, 300 МГц), δ (м.д.): 8,06 (дд, 2H); 7,63 (д, 1H); 7,50 (дд, 2H); 7,13 (дд, 2H); 7,06 (дд, 2H); 6,87 (д, 1H); 4,52 (м, 1H); 4,08 (ддд, 1H); 3,59 (м, 1H); 3,37 (ддд, 1H); 3,24 (м, 1H); 2,33 (м, 1H); 2,07 (м, 1H); 1,93 (м, 1H); 1,78-1,62 (м, 1H).
Пример 65
1-(4-{(S)-3-[3-(4-Фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-карбонил}пиперидин-1-ил)этанон
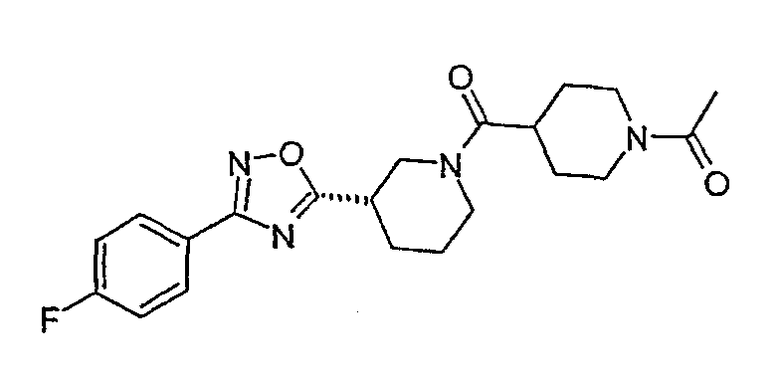
Соединение получают в соответствии с методикой, описанной в примере 36, используя 1-ацетилпиперидин-4-карбоновую кислоту в качестве выбранной кислоты и гидрохлорид S-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидина (полученный в соответствии с методикой примера 12). Чистый 1-(4-{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-карбонил}пиперидин-1-ил)этанон получают после очистки сырого продукта флэш-хроматографией (силикагель, элюент:
ДХМ/MeOH/NH4OH 98/2/0,2).
Выход: 57% (клейкое твердое вещество желтого цвета); [α]D 20=+88,3° (c=2,23, CHCl3); ЖХМС (Tr): 6,5 мин (метод A); МС (ES+) m/z: 401,2.
1H-ЯМР (CDCl3+D2O, 330 K, 300 МГц), δ (м.д.): 8,06 (дд, 2H); 7,15 (дд, 2H); 4,68-3,73 (м, 5H); 3,65-2,97 (м, 3H); 2,80 (м, 2H); 2,29 (м, 1H); 2,13-1,56 (м, 10H).
Пример 66
{(S)-3-[3-(4-Фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}тиофен-3-илметанон
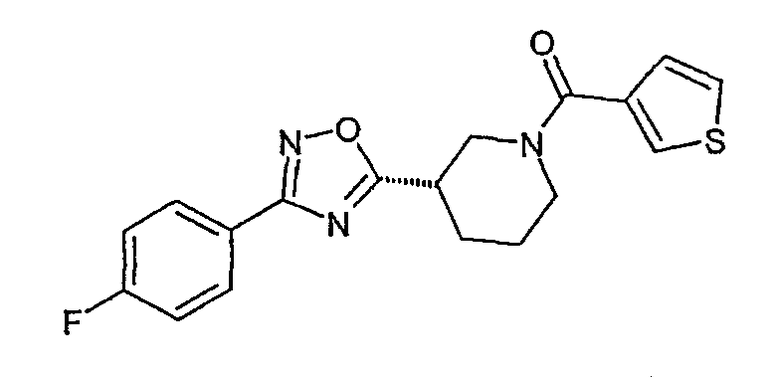
Соединение получают в соответствии с методикой, описанной в примере 36, используя тиофен-3-карбоновую кислоту в качестве выбранной кислоты и гидрохлорид S-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидина (полученный в соответствии с методикой примера 12). Чистый {(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}тиофен-3-илметанон получают после очистки сырого продукта флэш-хроматографией (силикагель, элюент: ДХМ/MeOH 99,5/0,5).
Выход: 57% (клейкое твердое вещество); [α]D 20=+79,8° (c=0,9, CHCl3); ЖХМС (Tr): 7,19 мин (метод A); МС (ES+) m/z: 358,1.
1H-ЯМР (CDCl3, 300 МГц), δ (м.д.): 8,07 (дд, 2H); 7,54 (дд, 1H); 7,34 (дд, 1H); 7,20 (дд, 1H); 7,15 (дд, 2H); 4,53 (м, 1H); 4,11 (м, 1H); 3,51 (дд, 1H); 3,32-3,19 (м, 2H); 2,35 (м, 1H); 2,09-1,87 (м, 2H); 1,77-1,61 (м, 1H).
Пример 67
{(S)-3-[3-(4-Фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}-(4-имидазол-1-илфенил)метанон
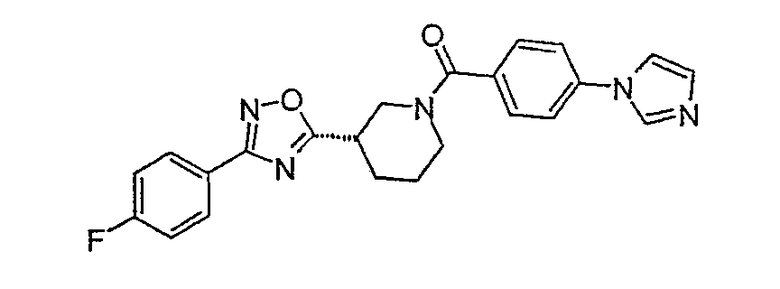
Соединение получают в соответствии с методикой, описанной в примере 36, используя 4-(1H-имидазол-1-ил)бензойную кислоту в качестве выбранной кислоты и гидрохлорид S-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидина (полученный в соответствии с методикой примера 12). Чистый {(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}-(4-имидазол-1-илфенил)метанон получают после очистки сырого продукта флэш-хроматографией (силикагель, элюент: ДХМ/MeOH/NH4OH 98,5/1,5/0,15).
Выход: 64% (твердое не совсем белое вещество); [α]D 20=+125,7° (c=1,707, CHCl3); ЖХМС (Tr): 6,66 мин (метод A); МС (ES+) m/z: 418,1.
1H-ЯМР (CDCl3, 300 МГц), δ (м.д.): 8,19 (уш.м, 1H); 8,07 (дд, 2H); 7,59 (д, 2H); 7,49 (д, 2H); 7,32 (м, 2H); 7,16 (дд, 2H); 4,42 (м, 1H); 3,99 (м, 1H); 3,59 (дд, 1H); 3,39-3,21 (м, 2H); 2,36 (м, 1H); 2,14-1,90 (м, 2H); 1,72 (м, 1H).
Пример 68
(4-Фторфенил)-[3-(5-фенилтетразол-2-ил)пиперидин-1-ил]метанон
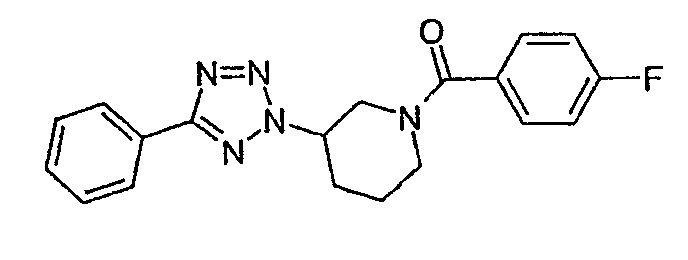
68(A). (4-Фторфенил)-(3-гидроксипиперидин-1-ил)метанон
Смесь 3-гидроксипиперидин (0,6 г, 5,93 ммоль), 4-фторбензойной кислоты (0,83 г, 5,93 ммоль), HOBT (0,8 г, 5,93 ммоль), EDCI·HCl (1,7 г, 8,9 ммоль) и сухого триэтиламина (1,66 мл, 11,86 ммоль) в сухом дихлорметане (30 мл) перемешивают в течение ночи при температуре окружающей среды в атмосфере азота. Затем добавляют 1 н. HCl (30 мл) и фазы разделяют. Органический слой промывают 1 н. HCl (30 мл), 1 н. NaOH (30 мл × 2), затем водой (30 мл). Выпаривание органического растворителя приводит к получению (4-фторфенил)-(3-гидроксипиперидин-1-ил)метанона в виде белого масла (0,7 г).
Выход: 53%; ЖХМС (Tr): 5,49 мин (метод A); МС (ES+) m/z: 224,1.
1H-ЯМР (CDCl3, 300 МГц), δ (м.д.): 7,43 (дд, 2H); 7,08 (дд, 2H); 3,83 (м, 1H); 3,73 (м, 1H); 3,56 (м, 1H); 3,43 (м, 2H); 1,99-1,79 (м, 2H); 1,74-1,42 (м, 2H).
68(B). (4-Фторфенил)-[3-(5-фенилтетразол-2-ил)пиперидин-1-ил]метанон
К раствору (4-фторфенил)-(3-гидроксипиперидин-1-ил)метанона (0,2 г, 0,89 ммоль) в сухом тетрагидрофуране (8 мл) в атмосфере азота одной порцией при 0°С добавляют трифенилфосфин (0,235 г, 0,89 ммоль). К реакционной смеси по каплям добавляют диизопропилазадикарбоксилат (DIAD, 175 мкл, 0,89 ммоль), поддерживая температуру реакционной смеси на уровне 0°C. Реакционной смеси дают возможность нагреться до комнатной температуры и смесь перемешивают при комнатной температуре в течение 24 часов. После этого при 0°С к смеси добавляют трифенилфосфин (0,118 г, 0,45 ммоль) и диизопропилазадикарбоксилат (DIAD, 87 мкл, 0,45 ммоль) и реакционную смесь перемешивают при комнатной температуре в течение ночи. Растворитель выпаривают при пониженном давлении, получая масло желтого цвета, которое очищают флэш-хроматографией (силикагель, элюент: гексан/этилацетат 6/4). Чистый (4-фторфенил)-[3-(5-фенилтетразол-2-ил)пиперидин-1-ил]метанон получают в виде густого масла (132 мг). Выход: 42%; ЖХМС (Tr): 7,04 мин (метод A); МС (ES+) m/z: 352,1.
1H-ЯМР (CDCl3, 300 МГц, 323 K), δ (м.д.): 8,12 (дд, 2H); 7,55-7,35 (м, 5H); 7,07 (дд, 2H); 4,90 (м, 1H); 4,50 (м, 1H); 4,33-3,69 (м, 3H); 3,36 (м, 1H); 2,57-2,28 (м, 1H); 2,14-1,96 (м, 1H); 1,84-1,66 (м, 1H).
Пример 69
(4-Фторфенил)-[(S)-3-(3-фенил-[1,2,4]оксадиазол-5-ил)пиперидин-1-ил]метанон
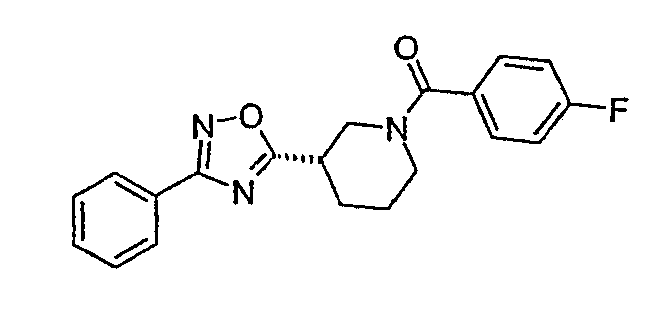
69(A). трет-Бутиловый эфир S-3-(3-фенил-[1,2,4]оксадиазол-5-ил)пиперидин-1-карбоновой кислоты
Соединение получают в соответствии с методикой, описанной в примере 46(A), исходя из бензонитрила. трет-Бутиловый эфир S-3-(3-фенил-[1,2,4]оксадиазол-5-ил)пиперидин-1-карбоновой кислоты получают в виде масла бежевого цвета, которое используют на следующей стадии без дополнительной очистки (выход: 85%).
ЖХМС (Tr): 7,83 мин (метод A); МС (ES+) m/z: 330,2.
69(B). Гидрохлорид S-3-(3-фенил-[1,2,4]оксадиазол-5-ил)пиперидина
Соединение получают в соответствии с методикой, описанной в примере 46(B), исходя из трет-бутилового эфира S-3-(3-фенил-[1,2,4]оксадиазол-5-ил)пиперидин-1-карбоновой кислоты (Выход: 100%).
69(С). (4-Фторфенил)-[(S)-3-(3-фенил-[1,2,4]оксадиазол-5-ил)пиперидин-1-ил]метанон
Соединение получают в соответствии с методикой, описанной в примере 46(С), исходя из гидрохлорида S-3-(3-фенил-[1,2,4]оксадиазол-5-ил)пиперидина и используя 4-фторбензоилхлорид в качестве выбранного ацилхлорида. Чистый (4-фторфенил)-[(S)-3-(3-фенил-[1,2,4]оксадиазол-5-ил)пиперидин-1-ил]метанон получают после очистки сырого продукта хроматографией на картридже из силикагеля (элюирование с градиентом: гексан/этилацетат от 8/2 до 6/4).
Выход: 60% (твердое белое вещество); т.пл.=116-118°C; [α]D 20=+99,3° (c=0,64, CHCl3); ЖХМС (Tr): 7,21 мин (метод A); МС (ES+) m/z: 352,2.
1H-ЯМР (CDCl3, 300 МГц, 323 K), δ (м.д.): 8,06 (м, 2H); 7,54-7,37 (м, 5H); 7,08 (м, 2H); 4,42 (м, 1H); 3,99 (м, 1H); 3,52 (дд, 1H); 3,26 (ддд, 2H); 2,34 (м, 1H); 2,12-1,59 (м, 3H).
Пример 70
(3,4-Дифторфенил)-[(S)-3-(3-фенил-[1,2,4]оксадиазол-5-ил)пиперидин-1-ил]метанон
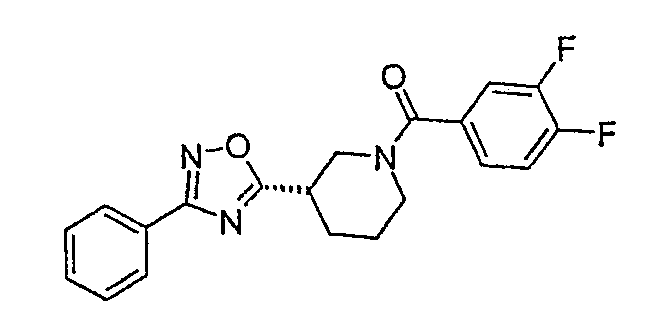
Соединение получают в соответствии с методикой, описанной в примере 46(С), исходя из гидрохлорида S-3-(3-фенил-[1,2,4]оксадиазол-5-ил)пиперидина и используя 3,4-дифторбензоилхлорид в качестве выбранного ацилхлорида. Чистый (3,4-дифторфенил)-[(S)-3-(3-фенил-[1,2,4]оксадиазол-5-ил)пиперидин-1-ил]метанон получают после очистки хроматографией с использованием силикагельного картриджа (элюирование с градиентом: гексан/этилацетат от 8/2 до 6/4).
Выход: 31% (твердое белое вещество); т.пл.=149-151°C; [α]D 20=+111,7° (c=0,55, CHCl3); ЖХМС (Tr): 7,33 мин (метод A); МС (ES+) m/z: 370,2.
1H-ЯМР (CDCl3, 300 МГц, 323 K), δ (м.д.): 8,06 (дд, 2H); 7,53-7,42 (м, 3H); 7,35-7,11 (м, 3H); 4,36 (м, 1H); 3,93 (м, 1H); 3,57 (дд, 1H); 3,40-3,16 (м, 2H); 2,33 (м, 1H); 2,14-1,56 (м, 3H).
Пример 71
(4-Фторфенил)-{(S)-3-[3-(4-нитрофенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон
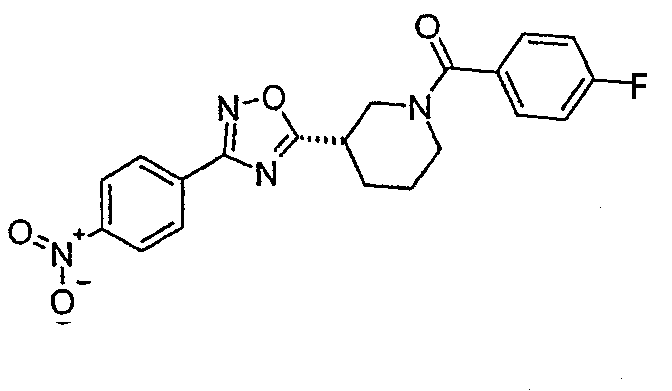
71(A). трет-Бутиловый эфир S-3-[3-(4-нитрофенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-карбоновой кислоты
Соединение получают в соответствии с методикой, описанной в примере 46(A), исходя из 4-нитробензонитрила. Чистый трет-бутиловый эфир S-3-[3-(4-нитрофенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-карбоновой кислоты получают в виде твердого вещества желтого цвета и используют в следующей стадии без дополнительной очистки (выход: 83%).
ЖХМС (Tr): 7,93 мин (метод A); МС (ES+) m/z: 375,1.
71(B). Гидрохлорид S-3-[3-(4-нитрофенил)-[1,2,4]оксадиазол-5-ил]пиперидина
Соединение получают в соответствии с методикой, описанной в примере 46(B), исходя из трет-бутилового эфира S-3-[3-(4-нитрофенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-карбоновой кислоты (выход: 100%).
71(С). (4-Фторфенил)-{(S)-3-[3-(4-нитрофенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон
Соединение получают в соответствии с методикой, описанной в примере 46(С), исходя из гидрохлорида S-3-[3-(4-нитрофенил)-[1,2,4]оксадиазол-5-ил]пиперидина и используя 4-фторбензоилхлорид в качестве выбранного ацилхлорида. Чистый (4-фторфенил)-{(S)-3-[3-(4-нитрофенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон получают после очистки сырого продукта хроматографией с использованием силикагельного картриджа (элюирование с градиентом: гексан/этилацетат от 8/2 до 6/4).
Выход: 48% (твердое вещество желтого цвета); т.пл.=162-164°C; [α]D 20=+111,5° (c=0,59, CHCl3); ЖХМС (Tr): 7,29 мин (метод A); МС (ES+) m/z: 397,1.
1H-ЯМР (CDCl3, 300 МГц, 323 K), δ (м.д.): 8,29 (дд, 4H); 7,43 (дд, 2H); 7,10 (дд, 2H); 4,47 (м, 1H); 3,98 (м, 1H); 3,54 (дд, 1H); 3,37-3,19 (м, 2H); 2,36 (м, 1H); 2,11-1,57 (м, 3H).
Пример 72
(3,4-Дифторфенил)-{(S)-3-[3-(4-нитрофенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон
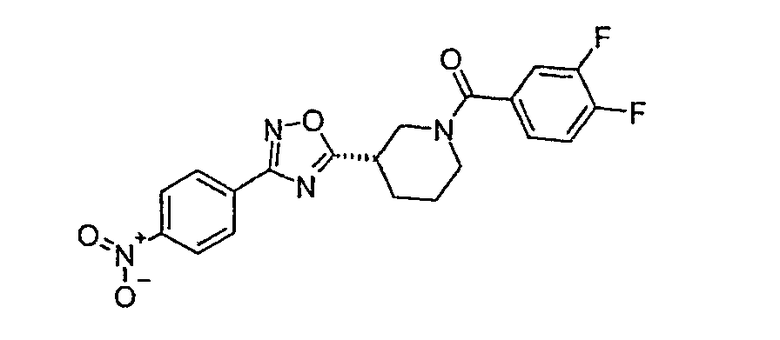
Соединение получают в соответствии с методикой, описанной в примере 46(С), исходя из гидрохлорида S-3-[3-(4-нитрофенил)-[1,2,4]оксадиазол-5-ил]пиперидина и используя 3,4-дифторбензоилхлорид в качестве выбранного ацилхлорида. Чистый (3,4-дифторфенил)-{(S)-3-[3-(4-нитрофенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон получают после очистки сырого продукта хроматографией с использованием силикагельного картриджа (элюирование с градиентом: гексан/этилацетат от 8/2 до 6/4).
Выход: 44% (твердое вещество желтого цвета); т.пл.=138-140°C; [α]D 20=+112,4° (c=0,50, CHCl3); ЖХМС (Tr): 7,39 мин (метод A); МС (ES+) m/z: 415,1.
1H-ЯМР (CDCl3, 300 МГц, 328 K), δ (м.д.): 8,38-8,19 (м, 4H); 7,35-7,10 (м, 3H); 4,41 (м, 1H); 3,92 (м, 1H); 3,58 (дд, 1H); 3,32 (м, 2H); 2,48-1,59 (м, 4H).
ФАРМАКОЛОГИЯ
Соединения, представленные в настоящем изобретении, являются положительными аллостерическими модуляторами mGluR5. Сами по себе данные соединения не проявляют связывания с ортостерическим сайтом распознавания глютамата и сами не активируют mGluR5. Но в присутствии соединений формулы I возрастает ответ mGluR5
на концентрацию глютамата или агониста mGluR5. Предполагается, что соединения формулы I оказывают воздействие на mGluR5 вследствие их способности расширять функцию рецептора.
Пример А
Изучение связывания mGluR5 на мембранном препарате мозга крысы
Активность соединений изобретения исследуют в соответствии с методикой радиолигандного связывания с использованием кортикальных мембран крысы и меченного радиоактивным тритием 2-метил-6-(фенилэтинил)пиридина ([3H]-MPEP) в качестве лиганда согласно способам, аналогичным описанным в публикациях: Gasparini et al. (2002) Bioorg. Med. Chem. Lett 12: 407-409; Anderson et al. (2002) J. Pharmacol. Exp. Ther. 303:1044-1051.
Мембранный препарат
Кортикальные слои иссекают из головного мозга крыс Sprague-Dawley (Charles River Laboratories, L'Arbresle, Франция) с массой тела 200-300 г. Ткани гомогенизируют в 10 объемах (об./мас.) ледяного 50 мМ HEPES-NaOH (рН 7,4) с использованием измельчителя Polytron (Kinematica AG, Luzern, Швейцария) и центрифугируют в течение 30 минут при 40000×g (4°С). Супернатант выгружают и пеллет промывают дважды повторным суспендированием в 10 объемах 50 мМ HEPES-NaOH. Затем мембраны собирают центрифугированием и промывают перед конечным суспендированием в 10 объемах 20 мМ HEPES-NaOH (рН 7,4). Концентрацию белка определяют c помощью метода Бредфорда (Bradford) (белковая проба Bio-Rad, Reinach, Швейцария) с использованием альбумина бычьей сыворотки в качестве стандарта.
Опыты [ 3 H]-MPEP связывания
Мембраны оттаивают и снова суспендируют в связывающем буфере, содержащем 20 мм HEPES-NaOH, 3 мМ MgCl2, 3 мМ CaCl2, 100 мМ NaCl (рН 7,4). Сравнительные исследования проводят инкубированием в течение 1 часа при 4°С: 3 нМ [3H]-MPEP (39 Ci/ммоль, Tocris, Cookson Ltd, Bristol, Великобритания), 50 мкг мембран и соединения с концентрацией в интервале от 0,003 нМ до 30 мкМ в общем объеме 300 мкл. Неспецифическое связывание определяют с использованием 30 мкМ MPEP. Реакцию завершают быстрой фильтрацией через фильтровальные пластины из стекловолокна (96-луночные фильтровальные микропланшеты Unifilter GF/B, Perkin-Elmer, Schwerzenbach, Швейцария) с использованием 4×400 мкл ледяного буфера и клеточного харвестера (Filtermate, Perkin-Elmer, Downers Grove, США). Радиоактивность определяют жидкостной сцинтилляционной спектрометрией, используя планшет-ридер для считывания 96-луночных микропланшетов (TopCount, Perkin-Elmer, Downers Grove, США).
Анализ данных
Кривые ингибирования получают с использованием программного обеспечения Prism GraphPad (Graph Pad Software Inc, San Diego, США). IC50 определяют, по меньшей мере, по трем независимым экспериментам с помощью нелинейного регрессионного анализа кривых, построенных по 8 точкам концентрация-ответ.
Соединения настоящего изобретения обладают способностью ингибировать [3H]-MPEP связывание в кортикальных мембранах крыс с IC50 менее примерно 100 мкМ, менее примерно 30 мкМ и 10 мкМ, предпочтительно менее примерно 3 мкМ.
Пример В
При экспозиции ростовыми факторами (основной ростовой фактор фибробластов) культурированные астроциты крысы экспрессируют групповые I-Gq связанные mGluR транскрипты, а именно mGluR5, но не расщепленные варианты mGluR1, и следствием этого является функциональная экспрессия mGluR5 рецепторов. (Miller et al. (1995) J. Neurosci. 15:6103-9). Стимуляция mGluR5 рецепторов селективным агонистом CHPG и полная блокада гидролиза глутамат-индуцированного фосфоинозитида (PI) и последовательная внутриклеточная мобилизация кальция специфическим антагонистом в качестве МРЕР подверждает уникальную экспрессию mGluR5 рецепторов в этом препарате.
Данный препарат получают и применяют для оценки свойств соединений настоящего изобретения в отношении повышения мобилизации Са2+, индуцированной глютаматом, без проявления любой значительной активности при применении в отсутствие глютамата.
Первичная культура кортикальных астроцитов
Первичные глиальные культуры приготавливают из коры головного мозга эмбрионов крыс Sprague-Dawley возраста 15 дней с использованием модифицированных методов, описанных в публикациях: Mc Carthy and de Vellis (1980) J. Cell Biol. 85:890-902; Miller et al. (1995) J. Neurosci. 15(9):6103-9. Кору головного мозга иссекают и затем растирают в стерильном буфере, содержащем 5,36 мМ KCl, 0,44 мМ NaHCO3, 4,17 мМ KH2PO4, 137 мМ NaCl, 0,34 мМ NaH2PO4, 1 г/л глюкозы. Полученный клеточный гомогенат переносят в Т75 колбы, предварительно покрытые поли-D-лизином ((BIOCOAT, Becton Dickinson Biosciences, Erembodegem, Бельгия), в модифицированную по методу Дульбекко среду Игла (D-MEM GlutaMAX™ I, Invitrogen, Basel, Швейцария), сбуферированную 25 мМ HEPES и 22,7 мМ NaHCO3 и снабженную 4,5 г/л глюкозы, 1 мМ пирувата и 15% фетальной телячьей сыворотки (FCS, Invitrogen, Basel, Швейцария), пинициллином и стрептомицином, и инкубируют при 37°С с 5% СО2. Для последующего засевания содержание FCS снижают до 10%. По истечении 12 дней клетки помещают с помощью трипсинизации на 384-луночные микропланшеты, покрытые поли-D-лизином, при плотности 20000 клеток на лунку, в сбуферированную культуру, снабженную 5 нг/мл β-FGF (основной ростовой фактор фибробластов) (Invitrogen, Basel, Швейцария) и 10 нг/мл EGF (эпидермальный ростовой фактор) (Invitrogen, Basel, Switzerland).
Исследование Са 2+ мобилизации с использованием кортикальных астроцитов крысы
После инкубирования в течение 2 дней клетки промывают обычным буфером, содержащим 142 мМ NaCl, 6 мМ KCl, 1 мМ Mg2SO4, 1 мМ CaCl2, 20 мМ HEPES, 1 г/л глюкозы, 0,125 мМ сульфинпиразона (pH 7,4). Через 60 минут после добавления 4 мкМ Fluo-4 (TefLabs, Austin, TX) клетки промывают три раза 50 мкл PBS буфера и снова суспендируют в 45 мкл опытного буфера. Затем микропланшеты переносят в планшет-ридер Fluorometric Imaging Plate Reader (FLIPR, Molecular Devices, Sunnyvale, CA) для оценки внутриклеточного потока кальция. После мониторинга флуоресценции в течение 15 секунд для определения фонового уровня ДМСО растворы, содержащие представленные соединения настоящего изобретения в различных концентрациях, разбавленные опытным буфером (15 мкл 4× разбавления), добавляют на микропланшет с клетками в отсутствие или в присутствии 1 мкМ глютамата: используемая концентрация глютамата 1 мкМ, которая дает 20% от максимального ответа на глютамат (ЕС20) в таких экспериментальных условиях в соответствии с литературными данными, является концентрацией, используемой для обнаружения свойств положительных аллостерических модуляторов у соединений настоящего изобретения. Конечная ДМСО концентрация в опыте составляет 0,3%. После этого в каждом эксперименте отслеживают флуоресценцию как функцию времени в течение 3 минут и полученные данные анализируют с использованием Microsoft Excel и GraphPad Prism. Каждую точку данных определяют два раза.
Анализ данных
Кривые «концентрация-ответ» представленных соединений настоящего изобретения в присутствии ЕС20 глютамата получают с использованием программного обеспечения GraphPad Prism (Graph Pad Software Lac, San Diego, США). Кривые соответствуют логистическому уравнению с четырьмя параметрами (Y=Bottom + (Top-Bottom)/(l+10^((LogEC50-X)*HillSlope), позволяющему определять значения ЕС50. Каждую кривую получают с использованием троекратного эксперимента для каждой точки данных при 8 концентрациях.
Данные, представленные на фиг.1, показывают способность соединений примеров 12, 55 и 56 в концентрации 3 мкМ повышать стимулирование, вызванное 1 мкМ глютамата в первичной кортикальной mGluR5-экспрессирующей клеточной культуре. Соединения примеров 12, 55 и 56 не обладают статистически значимой агонистической активностью при испытании в отсутствие глютамата, как показывает сравнение со значением, полученным для буфера.
Каждая столбиковая диаграмма представляет среднее значение и среднеквадратическую ошибку точек, полученных в тройном эксперименте, и является репрезентативным показателем трех независимых экспериментов.
Данные, представленные на фиг.1, показывают повышение мобилизации Са2+, индуцированной 1 мкМ глютамата, в культуре астроцитов крыс в присутствии 3 мкМ соединений примеров 12, 55 и 56 настоящего изобретения.
Результаты, полученные в примере А и примере В, показывают, что соединения, описанные в настоящем изобретении, как таковые, не обладают действием на mGluR5. Вместе с тем, при добавлении соединений вместе с агонистом mGluR5, таким как глютамат или CHPG, эффект значительно возрастает по сравнению с эффектом применения одного агониста в такой же концентрации. Кроме того, соединения настоящего изобретения обладают способностью ингибировать связывание mGluR5 отрицательного аллостерического модулятора в кортикальном мембранном препарате крысы, свойством, описанным ранее для 3,3'-дифторбензальдазина (DFB), другого mGluR5 положительного аллостерического модулятора (O'Brien J.A. et al. (2003) Mol. Pharmacol. 64:731-40). Кроме того, DFB не способен ингибировать связывание [3Н]-хисквалата с ортостерическим глютаминовым сайтом (O'Brien J.A. et al. (2003) Mol. Pharmacol. 64:731-40). Эти данные показывают, что соединения настоящего изобретения являются положительным аллостерическим модулятором mGluR5 рецептора при испытании на природном препарате и не проявляют связывания с ортостерическим сайтом связывания рецептора.
Таким образом, ожидается, что положительные аллостерические модуляторы, предоставленные настоящим изобретением, повышают эффективность глютамата или mGluR5 агонистов при mGluR5 рецепторе. Поэтому предполагается, что положительные аллостерические модуляторы могут применяться для лечения различных неврологических и психиатрических расстройств, связанных с глютаматной дисфункцией, которая, как описано, подлежит лечению при таких заболеваниях, а также для лечения других расстройств, которые могут лечиться с помощью таких положительных аллостерических модуляторов.
Пример С
Модели шизофрении на животных
Фенциклидиновая (PCP) модель шизофрении: РСР-индуцированные повышения локомоторного движения представляют собой широко используемую модель шизофрении на животных. Данная модель основана на том факте, что фенциклидин вызывает шизофреноподобный синдром у людей, включая пациентов с повышенной двигательной активностью, нарушение когнитивной функции и ухудшение рабочей памяти (Steinpreis RE (1996) Behav Br Res. 74:45-55; Abi-Saab et al. (1998) Pharmacopsychiatry, 31:104-109). Далее было показано, что антипсихотические лекарственные средства, которые эффективны в лечении шизофрении, снижают активирующее локомоторное действие РСР (Gleason & Shannon (1997) Psychopharmacology, 129:79-84). Эти результаты показывают, что локомоторная активация, вызываемая РСР, является моделью, применимой для скрининга соединений, потенциально полезных для лечения шизофрении.
Амфетаминовая модель шизофрении: Амфетамин-индуцированные повышения локомоторного движения хорошо известны и широко применяются в качестве модели положительных симптомов шизофрении. Данная модель основана на том факте, что амфетамин повышает двигательную активность и может вызывать психотическое состояние у людей (Yui et al. (2000) Ann NY Acad Sci 914:1-12). Далее, хорошо известно, что амфетамин-индуцированные повышения локомоторной активности блокируются антипсихотическими лекарственными средствами, которые эффективны в лечении шизофрении (Arnt (1995) Eur J Pharmacol 283:55-62). Эти данные показывают, что локомоторная активация, вызываемая амфетамином, является моделью, применимой для скрининга соединений, потенциально полезных для лечения шизофрении.
Субъекты: Настоящие исследования проводят в соответствии с правилами содержания и использования животных Addex Pharmaceuticals и законами и директивами правительства Швейцарии по содержанию и использованию животных. Самцов мыши C57BL6/J (20-30 г) возраста 7 недель содержат группой при контролируемых температуре и влажности с 12 часовым циклом свет/темнота в течение, по меньшей мере, 7 дней перед использованием. Мышам обеспечен доступ к пище и воде по желанию за исключением времени проведения экспериментов по исследованию локомоторной активности.
Оценка локомоторной (двигательной) активности: Исследование действия соединений на РСР- или амфетамин-индуцированную локомоторную активацию проводят на мышах. Локомоторную активность мышей изучают, помещая их в белые пластиковые коробки площадью 35 см × 35 см с высотой стенки 40 см. Локомоторную активность (движение) контролируют с помощью системы видеонаблюдения (VideoTrack, Viewpoint, Champagne au Mont d'Or, Франция), которая записывает перемещения мышей. Мыши не боятся аппаратуры перед испытанием. В дни проведения испытаний опытные соединения (10, 30, 50 или 100 мг/кг (интраперитонеально)) или носитель вводят за 120 минут до инъекции РСР (5 мг/кг подкожно), амфетамина (3,0 мг/кг подкожно) или физиологического раствора. Мышей помещают в коробки для наблюдения за локомоторной активностью сразу после инъекции РСР, амфетамина или физиологического раствора, и их локомоторную активность, определяемую как пройденное ими расстояние, выраженное в сантиметрах (см), фиксируют в течение 60 минут.
Введение соединений: Соединения приготавливают в виде микросуспензии в стерильной воде (60% конечного объема) и Labrafil M1944 CS (абрикосовое масло - Gattefosse, Saint Priest, Франция) (40% конечного объема) и вводят в объеме 10 мл/кг. Мышам, которые в качестве лекарства получают носитель, вводят эквивалентный объем раствора носителя парентерально (i.p.) без добавления соединения. Гидрохлорид РСР (Sigma, Швейцария) растворяют в физиологическом растворе и вводят в дозе 5 мг/кг подкожно в объеме 10 мл/кг. Мышам, обработанным РСР, которые в качестве лекарства получают носитель, подкожно вводят равный объем физиологического раствора носителя. Сульфат D-амфетамина (Amino AG, Neuenhof, Швейцария) растворяют в физиологическом растворе и вводят в дозе 3,0 мг/кг подкожно в объеме 10 мл/кг. Мышам, обработанным D-амфетамином, которые в качестве лекарства получают носитель, подкожно вводят эквивалентный объем физиологического раствора носителя.
Статистический анализ: Статистический анализ выполняют с использованием программного обеспечения статистического анализа GraphPad PRISM (GraphPad, San Diego, CA, США). Данные анализируют с использованием анализа варианта в один проход (one-way analysis) (ANOVA) с последующими пост-хок Бонферрони-корректируемыми множественными сравнениями (post-hoc Bonferroni-corrected multiple comparisons), когда это подходит. Принимаемый уровень значимости р<0,05.
Действие соединений на РСР-индуцированную локомоторную активность мышей
Результаты, полученные в описанном выше эксперименте с использованием типичного соединения настоящего изобретения, представлены на фиг.2.
Данные, представленные на фиг.2, показывают, что типичное соединение настоящего изобретения значительно снижает возрастание двигательной активности, индуцированное PCP (f=13,39, df=(2,45), n=16/группа), в дозе 100 мг/кг и.п.
Действие соединений на амфетамин-индуцированную локомоторную активность мышей
Результаты, полученные в описанном выше эксперименте с использованием типичного соединения настоящего изобретения, представлены на фиг.3.
Данные, представленные на фиг.3, показывают, что типичное соединение настоящего изобретения снижает возрастание двигательной активности, индуцированное амфетамином (f=13,04, df=(4,82), n=8-33 мышей на группу), в дозах 50 и 100 мг/кг и.п.
Краткое обсуждение результатов испытания in vivo
Представленные выше данные показывают, что типичные соединения формулы I значительно снижают гиперлокомоторные эффекты РСР и амфетамина - двух широко распространенных моделей шизофрении на животных. Эти результаты подтвержают потенциал соединений формулы I в лечении шизофрении и родственных расстройств.
Соединения настоящего изобретения являются аллостерическими модуляторами mGluR5 рецепторов, они могут применяться для производства лекарственных средств, особенно для профилактики или лечения расстройств центральной нервной системы, а также других расстройств, модулируемых данным рецептором.
Соединения настоящего изобретения могут вводиться сами по себе или в сочетании с другими фармацевтическими лекарственными средствами, эффективными для лечения состояний, перечисленных выше.
Примеры препаратов
Типичными примерами составов препаратов соединений настоящего изобретения являются следующие композиции:
1) Таблетки
В данном примере соединение примера 12 может быть заменено любым соединением описанных примеров с 1 по 72, взятых в таком же количестве.
2) Суспензия:
Для перорального введения приготавливают водную суспензию, в которой каждый 1 миллилитр содержит от 1 до 5 мг одного из соединений описанных примеров, 50 мг натрийкарбоксиметилцеллюлозы, 1 мг бензоата натрия, 50 мг сорбита и воды - до 1 мл.
3) Препарат для инъекций
Композицию для парентерального введения приготавливают смешением 1,5% (мас.) активного ингредиента настоящего изобретения с 10% (об.) раствором пропиленгликоля и воды.
4) Мазь
В данном примере соединение примера 12 может быть заменено таким же количеством любого из соединений описанных примеров с 1 по 72.
Приемлемые изменения не должны рассматриваться отдельно от области настоящего изобретения. Таким образом, квалифицированному специалисту понятно, что описанное изобретение может видоизменяться без выделения этих изменений из области данного изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ТРИАЗОЛОПИРИДИНОВЫЕ СОЕДИНЕНИЯ-ИНГИБИТОРЫ JAK И СПОСОБЫ | 2009 |
|
RU2561104C2 |
| ТРИАЗОЛПИРИДИНОВЫЕ СОЕДИНЕНИЯ, ИНГИБИРУЮЩИЕ JAK, И СПОСОБЫ | 2009 |
|
RU2560153C2 |
| ПРОИЗВОДНЫЕ АМИНОХИНАЗОЛИНА В КАЧЕСТВЕ P2X3 ИНГИБИТОРОВ | 2020 |
|
RU2821714C2 |
| НОВЫЕ ЦИКЛИЧЕСКИЕ УГЛЕВОДОРОДНЫЕ СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ | 2008 |
|
RU2524949C2 |
| ПИРАЗОЛОПИРИМИДИНОВЫЕ СОЕДИНЕНИЯ-ИНГИБИТОРЫ JAK И СПОСОБЫ | 2009 |
|
RU2539568C2 |
| НОВЫЕ СОЕДИНЕНИЯ И КОМПОЗИЦИИ В КАЧЕСТВЕ ИНГИБИТОРОВ КАТЕПСИНА | 2004 |
|
RU2346943C2 |
| ПИРРОЛОПИРРОЛОВЫЕ КОМПОЗИЦИИ В КАЧЕСТВЕ АКТИВАТОРОВ ПИРУВАТКИНАЗЫ (PKR) | 2018 |
|
RU2736217C2 |
| АНТАГОНИСТЫ АРИЛСУЛЬФОНАМИДА CCR3 | 2010 |
|
RU2539591C2 |
| АЗАИНДОЛЬНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ФАКТОРА Xa | 2004 |
|
RU2330853C2 |
| БИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ ВЗАИМОДЕЙСТВИЯ/АКТИВАЦИИ PD1/PD-L1 | 2019 |
|
RU2777980C2 |
Изобретение относится к новым соединениям общей формулы I и их фармацевтически приемлемым солям. Соединения настоящего изобретения обладают свойствами положительного аллостерического модулятора mGluR5. В общей формуле I
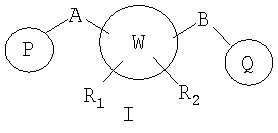
W представляет собой 6-членное гетероциклоалкильное кольцо с 1-2 гетероатомами, выбранными из N, О; R1 и R2 независимо представляют собой водород, C1-С6-алкил; Р и Q, каждый независимо, выбран из:
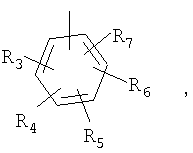
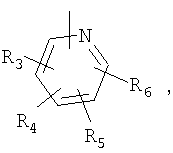

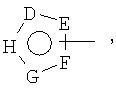
R3, R4, R5, R6 и R7 независимо представляют собой водород; галоген; -CN; нитро; C1-С6-алкил; С3-С6-циклоалкил; галоген-С1-С6-алкил; 5-6-членный гетероарил с 1-2 атомами N в качестве гетероатомов; 6-членный гетероцикл с 2 гетероатомами, представляющими собой N, О; фенил, необязательно замещенный галогеном; нафтил; -OR8; где необязательно два заместителя вместе с находящимися между ними атомами образуют 9-10-членное бициклическое арильное или гетероарильное кольцо с 1-2 гетероатомами, выбранными из N, S; R8 представляет собой водород, C1-С6-алкил; D, Е, F, G и Н независимо представляют собой
-C(R3)=, -O-, -N=, -N(R3)- или -S-; А представляет собой этинил, -C(=O)NR8- или группу формулы
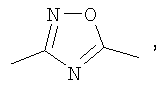
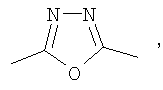
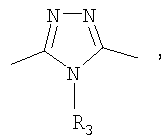

В представляет собой -С(=O)-С0-С2-алкил-, -С(=O)-С2-С6-алкенил-. Изобретение также относится к фармацевтической композиции на основе соединений изобретения. 2 н. и 18 з.п. ф-лы, 3 ил.
1. Соединение общей формулы I
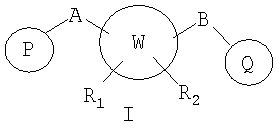
где W представляет собой 6-членное гетероциклоалкильное кольцо с 1-2 гетероатомами, выбранными из N, О;
R1 и R2 независимо представляют собой водород, C1-С6-алкил;
Р и Q, каждый независимо, выбран из
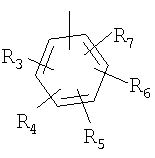 ,
, 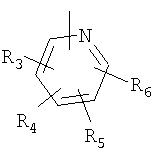 ,
, 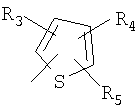 ,
, 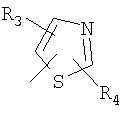 ,
, 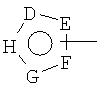
R3, R4, R5, R6 и R7 независимо представляют собой водород; галоген; -CN; нитро; C1-С6-алкил; С3-С6-циклоалкил; галоген-С1-С6-алкил; 5-6-членный гетероарил с 1-2 атомами N в качестве гетероатомов; 6-членный гетероцикл с 2-гетероатомами, представляющими собой N, О; фенил, необязательно замещенный галогеном; нафтил; -OR8; где необязательно два заместителя вместе с находящимися между ними атомами образуют 9-10-членное бициклическое арильное или гетероарильное кольцо с 1-2 гетероатомами, выбранными из N, S;
R8 представляет собой водород, C1-С6-алкил;
D, Е, Р, G и Н независимо представляют собой -С(R3)=, -O-, -N=, -N(R3)- или -S-;
А представляет собой этинил, -C(=O)NR8- или группу формулы
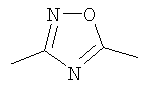
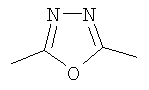

R3 принимает значения, определенные выше;
В представляет собой -С(=O)-С0-С2-алкил-, -С(=O)-С2-С6-алкенил-;
или фармацевтически приемлемые соли такого соединения,
за исключением следующих соединений:
4-(3-фенилоксадиазол-5-ил)-N-(4-бромфенил)аминокарбонилпиперидин;
N-бензоил-3-(3,4-диметоксифениламинокарбонил)пиперидин-4-он;
N-(3-цианофенилметилкарбонил)-4-(3-(2,3-дихлорфенил)пиразо-5-ил)пиперидин.
2. Соединение по п.1 формулы I-A
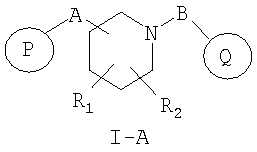
где R1 и R2 независимо представляют собой водород, C1-С6-алкил;
Р и Q, каждый независимо, выбран из
, , ,  ,
,
R3, R4, R5, R6 и R7 независимо представляют собой водород; галоген; -CN; нитро; C1-С6-алкил; С3-С6-циклоалкил; галоген-С1-С6-алкил; 5-6-членный гетероарил с 1-2 атомами N в качестве гетероатомов; 6-членный гетероцикл с 2 гетероатомами, представляющими собой N, О; фенил, необязательно замещенный галогеном; нафтил; -OR8; где необязательно два заместителя вместе с находящимися между ними атомами образуют 9-10-членное бициклическое арильное или гетероарильное кольцо с 1-2 гетероатомами, выбранными из N, S;
R8 представляет собой водород, C1-С6-алкил;
D, Е, F, G и Н независимо представляют собой -C(R3)=, -O-, -N=, -N(R3)- или -S-;
А представляет собой этинил, -C(=O)NR8- или группу формулы
R3 принимает значения, определенные выше;
В представляет собой -С(=O)-С0-С2-алкил-, -С(=O)-С2-С6-алкенил-;
или фармацевтически приемлемые соли такого соединения.
3. Соединение по п.1 или 2 формулы I-B
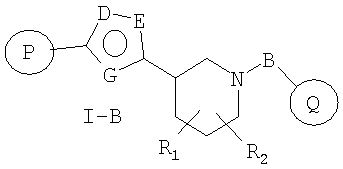
где R1 и R2 независимо представляют собой водород, C1-С6-алкил;
Р и Q, каждый независимо, выбран из:
, , , ,
R3, R4, R5, R6 и R7 независимо представляют собой водород; галоген; -CN; нитро; C1-С6-алкил; С3-С6-циклоалкил; галоген-С1-С6-алкил; 5-6-членный гетероарил с 1-2 атомами N в качестве гетероатомов; 6-членный гетероцикл с 2 гетероатомами, представляющими собой N, О; фенил, необязательно замещенный галогеном; нафтил; -OR8; где необязательно два заместителя вместе с находящимися между ними атомами образуют 9-10-членное бициклическое арильное или гетероарильное кольцо с 1-2 гетероатомами, выбранными из N, S;
R8 представляет собой водород, C1-С6-алкил;
D, Е, F, G и Н в Р и Q независимо представляют собой -C(R3)=, -O-, -N=, -N(R3)- или -S-;
D, Е и G в А независимо принимают значения, определенные для А в п.1;
В представляет собой -С(=O)-С0-С2-алкил-, -С(=O)-С2-С6-алкенил-;
или фармацевтически приемлемые соли такого соединения.
4. Соединение по п.1 или 2 формулы I-C
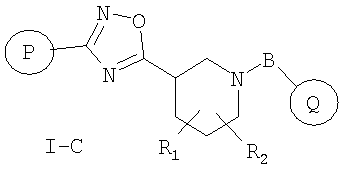
где R1 и R2 независимо представляют собой водород, C1-С6-алкил;
Р и Q, каждый независимо, выбран из
, , , ,
R3, R4, R5, R6 и R7 независимо представляют собой водород; галоген; -CN; нитро; C1-С6-алкил; С3-С6-циклоалкил; галоген-С1-С6-алкил; 5-6-членный гетероарил с 1-2 атомами N в качестве гетероатомов; 6-членный гетероцикл с 2 гетероатомами, представляющими собой N, О; фенил, необязательно замещенный галогеном; нафтил; -OR8; где необязательно два заместителя вместе с находящимися между ними атомами образуют 9-10-членное бициклическое арильное или гетероарильное кольцо с 1-2 гетероатомами, выбранными из N, S;
R8 представляет собой водород, C1-С6-алкил;
D, Е, F, G и Н независимо представляют собой -С(R3)=, -O-, -N=, -N(R3)- или -S-;
В представляет собой -С(=O)-С0-С2-алкил-, -С(=O)-С2-С6-алкенил-;
или фармацевтически приемлемые соли такого соединения.
5. Соединение по п.1 или 2 формулы I-D
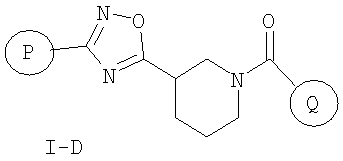
где Р и Q, каждый независимо, выбран из
, , , ,
R3, R4, R5, R6 и R7 независимо представляют собой водород; галоген; -CN; нитро; C1-С6-алкил; С3-С6-циклоалкил; галоген-С1-С6-алкил; 5-6-членный гетероарил с 1-2 атомами N в качестве гетероатомов; 6-членный гетероцикл с 2 гетероатомами, представляющими собой N, О; фенил, необязательно замещенный галогеном; нафтил; -OR8; где необязательно два заместителя вместе с находящимися между ними атомами образуют 9-10-членное бициклическое арильное или гетероарильное кольцо с 1-2 гетероатомами, выбранными из N, S;
R8 представляет собой водород, C1-С6-алкил;
D, Е, F, G и Н независимо представляют собой -С(R3)=, -O-, -N=, -N(R3)- или -S-;
или фармацевтически приемлемые соли такого соединения.
6. Соединение по п.1 или 2 формулы I-Е
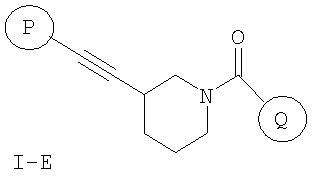
где Р и Q, каждый независимо, выбран из
, , , ,
R3, R4, R5, R6 и R7 независимо представляют собой водород; галоген; -CN; нитро; C1-С6-алкил; С3-С6-циклоалкил; галоген-С1-С6-алкил; 5-6-членный гетероарил с 1-2 атомами N в качестве гетероатомов; 6-членный гетероцикл с 2 гетероатомами, представляющими собой N, О; фенил, необязательно замещенный галогеном; нафтил; -OR8; где необязательно два заместителя вместе с находящимися между ними атомами образуют 9-10-членное бициклическое арильное или гетероарильное кольцо с 1-2 гетероатомами, выбранными из N, S;
R8 представляет собой водород, C1-С6-алкил;
D, Е, F, G и Н независимо представляют собой -C(R3)=, -O-, -N=, -N(R3)- или -S-;
или фармацевтически приемлемые соли такого соединения.
7. Соединение по п.1 или 2 формулы I-F
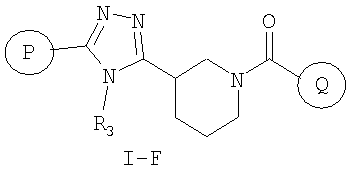
где Р и Q, каждый независимо, выбран из
, , , ,
R3, R4, R5, R6 и R7 независимо представляют собой водород; галоген; -CN; нитро; C1-С6-алкил; С3-С6-циклоалкил; галоген-С1-С6-алкил; 5-6-членный гетероарил с 1-2 атомами N в качестве гетероатомов; 6-членный гетероцикл с 2 гетероатомами, представляющими собой N, О; фенил, необязательно замещенный галогеном; нафтил; -OR8; где необязательно два заместителя вместе с находящимися между ними атомами образуют 9-10-членное бициклическое арильное или гетероарильное кольцо с 1-2 гетероатомами, выбранными из N, S;
R8 представляет собой водород, C1-С6-алкил;
D, Е, F, G и Н независимо представляют собой -С(R3)=, -O-, -N=, -N(R3)- или -S-;
или фармацевтически приемлемые соли такого соединения.
8. Соединение по п.1 формулы I-G
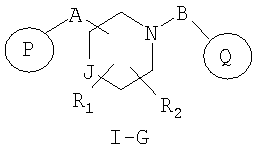
где R1 и R2 независимо представляют собой водород, C1-С6-алкил;
Р и Q, каждый независимо, выбран из
, , , ,
R3, R4, R5, R6 и R7 независимо представляют собой водород; галоген; -CN; нитро; C1-С6-алкил; С3-С6-циклоалкил; галоген-С1-С6-алкил; 5-6-членный гетероарил с 1-2 атомами N в качестве гетероатомов; 6-членный гетероцикл с 2 гетероатомами, представляющими собой N, О; фенил, необязательно замещенный галогеном; нафтил; -OR8; где необязательно два заместителя вместе с находящимися между ними атомами образуют 9-10-членное бициклическое арильное или гетероарильное кольцо с 1-2 гетероатомами, выбранными из N, S;
R8 представляет собой водород, C1-С6-алкил;
D, Е, F, G и Н независимо представляют собой -C(R3)=, -O-, -N=, -N(R3)- или -S-;
А представляет собой этинил, -C(=O)NR8- или группу формулы
R3 принимает значения, определенные выше;
В представляет собой -С(=O)-С0-С2-алкил-, -С(=O)-С2-С6-алкенил-;
J представляет собой -О- или -N(R11);
R11 представляет собой водород, C1-С6-алкил;
или фармацевтически приемлемые соли такого соединения.
9. Соединение по п.1 или 8 формулы I-H
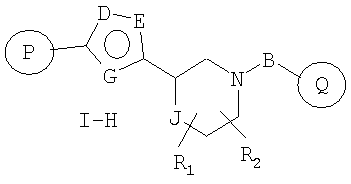
где R1 и R2 независимо представляют собой водород, C1-С6-алкил;
Р и Q, каждый независимо, выбран из
, , , ,
R3, R4, R5, R6 и R7 независимо представляют собой водород; галоген; -CN; нитро; C1-С6-алкил; С3-С6-циклоалкил; галоген-С1-С6-алкил; 5-6-членный гетероарил с 1-2 атомами N в качестве гетероатомов; 6-членный гетероцикл с 2 гетероатомами, представляющими собой N, О; фенил, необязательно замещенный галогеном; нафтил; -OR7; где необязательно два заместителя вместе с находящимися между ними атомами образуют 9-10-членное бициклическое арильное или гетероарильное кольцо с 1-2 гетероатомами, выбранными из N, S;
R8 представляет собой водород, C1-С6-алкил;
D, Е, F, G и Н в Р и Q независимо представляют собой -С(R3)=, -O-, -N=, -N(R3)- или -S-;
D, Е и G в А независимо принимают значения, определенные для А в п.1;
В представляет собой -С(=O)-С0-С2-алкил-, -С(=O)-С2-С6-алкенил-;
J представляет собой -О- или -N(R11)-;
R11 представляет собой водород, C1-C6-алкил;
или фармацевтически приемлемые соли такого соединения.
10. Соединение по п.1 или 8 формулы I-I
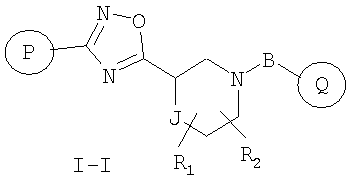
где R1 и R2 независимо представляют собой водород, C1-С6-алкил;
Р и Q, каждый независимо, выбран из
, , , ,
R3, R4, R5, R6 и R7 независимо представляют собой водород; галоген; -CN; нитро; C1-С6-алкил; С3-С6-циклоалкил; галоген-С1-С6-алкил; 5-6-членный гетероарил с 1-2 атомами N в качестве гетероатомов; 6-членный гетероцикл с 2 гетероатомами, представляющими собой N, О; фенил, необязательно замещенный галогеном; нафтил; -OR8; где необязательно два заместителя вместе с находящимися между ними атомами образуют 9-10-членное бициклическое арильное или гетероарильное кольцо с 1-2 гетероатомами, выбранными из N, S;
R8 представляет собой водород, C1-С6-алкил;
D, Е, F, G и Н независимо представляют собой -С(R3)=, -O-, -N=, -N(R3)- или -S-;
В представляет собой -С(=O)-С0-С2-алкил-, -С(=O)-С2-С6-алкенил-;
J представляет собой -О- или -N(R11)-;
R11 представляет собой водород, C1-С6-алкил;
или фармацевтически приемлемые соли такого соединения.
11. Соединение по п.1 или 8 формулы I-J
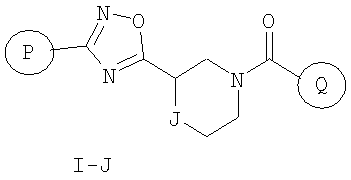
где Р и Q, каждый независимо, выбран из
, , , ,
R3, R4, R5, R6 и R7 независимо представляют собой водород; галоген; -CN; нитро; C1-С6-алкил; С3-С6-циклоалкил; галоген-С1-С6-алкил; 5-6-членный гетероарил с 1-2 атомами N в качестве гетероатомов; 6-членный гетероцикл с 2 гетероатомами, представляющими собой N, О; фенил, необязательно замещенный галогеном; нафтил; -OR8; где необязательно два заместителя вместе с находящимися между ними атомами образуют 9-10-членное бициклическое арильное или гетероарильное кольцо с 1-2 гетероатомами, выбранными из N, S;
R8 представляет собой водород, C1-С6-алкил;
D, Е, F, G и Н независимо представляют собой -С(R3)=, -O-, -N=, -N(R3)- или -S-;
J представляет собой -О- или -N(R11)-;
R11 представляет собой водород, C1-С6-алкил;
или фармацевтически приемлемые соли такого соединения.
12. Соединение по п.1 или 8 формулы I-K
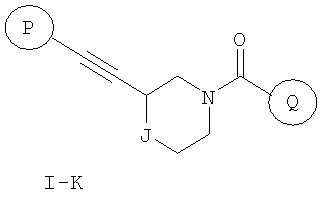
где Р и Q, каждый независимо, выбран из
, , , ,
где R3, R4, R5, R6 и R7 независимо представляют собой водород; галоген; -CN; нитро; C1-C6-алкил; С3-С6-циклоалкил; галоген-С1-С6-алкил; 5-6-членный гетероарил с 1-2 атомами N в качестве гетероатомов; 6-членный гетероцикл с 2 гетероатомами, представляющими собой N, О; фенил, необязательно замещенный галогеном; нафтил; -OR8; где необязательно два заместителя вместе с находящимися между ними атомами образуют 9-10-членное бициклическое арильное или гетероарильное кольцо с 1-2 гетероатомами, выбранными из N, S;
R8 представляет собой водород, C1-C6-алкил;
D, Е, F, G и Н независимо представляют собой -С(R3)=, -O-, -N=, -N(R3)- или -S-;
J представляет собой -О- или -N(R11)-;
R11 представляет собой водород, C1-С6-алкил;
или фармацевтически приемлемые соли такого соединения.
13. Соединение по п.1 или 8 формулы I-L
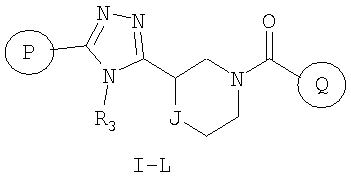
где Р и Q, каждый независимо, выбран из
, , , ,
R3, R4, R5, R6 и R7 независимо представляют собой водород; галоген; -CN; нитро; C1-C6-алкил; С3-С6-циклоалкил; галоген-С1-С6-алкил; 5-6-членный гетероарил с 1-2 атомами N в качестве гетероатомов; 6-членный гетероцикл с 2 гетероатомами, представляющими собой N, О; фенил, необязательно замещенный галогеном; нафтил; -OR8; где необязательно два заместителя вместе с находящимися между ними атомами образуют 9-10-членное бициклическое арильное или гетероарильное кольцо с 1-2 гетероатомами, выбранными из N, S;
R8 представляет собой водород, C1-С6-алкил;
D, Е, F, G и Н независимо представляют собой -С(R3)=, -O-, -N=, -N(R3)- или -S-;
J представляет собой -О- или -N(R11)-;
R11 представляет собой водород, C1-С6-алкил;
или фармацевтически приемлемые соли такого соединения.
14. Соединение по п.1 или 8 формулы I-М
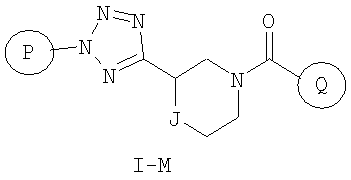
где Р и Q, каждый независимо выбран из
, , , ,
R3, R4, R5, R6 и R7 независимо представляют собой водород; галоген; -CN; нитро; C1-С6-алкил; С3-С6-циклоалкил; галоген-С1-С6-алкил; 5-6-членный гетероарил с 1-2 атомами N в качестве гетероатомов; 6-членный гетероцикл с 2 гетероатомами, представляющими собой N, О; фенил, необязательно замещенный галогеном; нафтил; -OR8; где необязательно два заместителя вместе с находящимися между ними атомами образуют 9-10-членное бициклическое арильное или гетероарильное кольцо с 1-2 гетероатомами, выбранными из N, S;
R8 представляет собой водород, C1-С6-алкил;
D, Е, F, G и Н независимо представляют собой -С(R3)=, -O-, -N=, -N(R3)- или -S-;
J представляет собой -О- или -N(R11)-;
R11 представляет собой водород, C1-С6-алкил;
или фармацевтически приемлемые соли такого соединения.
15. Соединение по п.1 или 8 формулы I-N
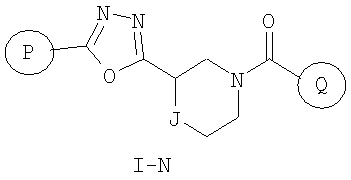
где Р и Q, каждый независимо, выбран из
, , , ,
R3, R4, R5, R6 и R7 независимо представляют собой водород; галоген; -CN; нитро; C1-C6-алкил; С3-С6-циклоалкил; галоген-С1-С6-алкил; 5-6-членный гетероарил с 1-2 атомами N в качестве гетероатомов; 6-членный гетероцикл с 2 гетероатомами, представляющими собой N, О; фенил, необязательно замещенный галогеном; нафтил; -OR8; где необязательно два заместителя вместе с находящимися между ними атомами образуют 9-10-членное бициклическое арильное или гетероарильное кольцо с 1-2 гетероатомами, выбранными из N, S;
R8 представляет собой водород, C1-С6-алкил;
D, Е, F, G и Н независимо представляют собой -C(R3)=, -O-, -N=, -N(R3)- или -S-;
J представляет собой -О- или -N(R11)-;
R11 представляет собой водород, C1-С6-алкил;
или фармацевтически приемлемые соли такого соединения.
16. Соединение по п.1, которое может существовать в виде оптических изомеров, где указанное соединение представляет собой рацемическую смесь или отдельные оптические изомеры.
17. Соединение по п.1, где указанные соединения выбраны из группы, включающей
(4-фторфенил)-[3-(4-фторфенилэтинил)пиперидин-1-ил]метанон;
(4-фторфенил)-{3-[5-(4-фторфенил)-4Н-[1,2,4]триазол-3-ил]пиперидин-1-ил}метанон;
(S)-(4-фторфенил)-{3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон;
(S)-(тиофен-2-ил)-{3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон;
{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}-(4-метил-2-пиразин-2-илтиазол-5-ил)метанон;
(2,4-дифторфенил)-{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон;
{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}-(3,4,5-трифторфенил)метанон;
{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}-(5-пиридин-2-илтиофен-2-ил)метанон;
циклопентил-{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон;
(3,4-дифторфенил)-{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон;
бензотиазол-6-ил-{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон;
(3,5-диметилизоксазол-4-ил)-{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон;
(4-фторфенил)-{(S)-3-[3-(2,4,6-трифторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон;
(4-фторфенил)-[(S)-3-(3-пиридин-3-ил-[1,2,4]оксадиазол-5-ил)пиперидин-1-ил]метанон;
(4-фторфенил)-[(S)-3-(3-пиридин-4-ил-[1,2,4]оксадиазол-5-ил)пиперидин-1-ил]метанон;
{(S)-3-[3-(2,4-дифторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}-(4-фторфенил)метанон;
(4-фторфенил)-[(S)-3-(3-п-толил-[1,2,4]оксадиазол-5-ил)пиперидин-1-ил]метанон;
(4-фторфенил)-{(S)-3-[3-(2-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон;
(4-фторфенил)-[(S)-3-(3-пиридин-2-ил-[1,2,4]оксадиазол-5-ил)пиперидин-1-ил]метанон;
(4-фторфенил)-{S-3-[5-(4-фторфенил)-[1,3,4]оксадиазол-2-ил]пиперидин-1-ил}метанон;
(2-фторфенил)-{(S)-3-[2-(3,4-дифторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон;
(4-фторфенил)-{2-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]морфолин-4-ил}метанон;
{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}тиофен-3-илметанон;
(4-фторфенил)-[3-(5-фенилтетразол-2-ил)пиперидин-1-ил]метанон;
(4-фторфенил)-[(S)-3-(3-фенил-[1,2,4]оксадиазол-5-ил)пиперидин-1-ил]метанон;
(3,4-дифторфенил)-[(S)-3-(3-фенил-[1,2,4]оксадиазол-5-ил)пиперидин-1-ил]метанон.
18. Соединение по п.1, где указанные соединения выбраны из группы, включающей
{3-[3-(4-метоксифенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}фенилметанон;
{3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}фенилметанон;
(4-фторфенил)-[3-(3-фенил-[1,2,4]оксадиазол-5-ил)пиперидин-1-ил]метанон;
(3-фторфенил)-[3-(3-фенил-[1,2,4]оксадиазол-5-ил)пиперидин-1-ил]метанон;
(4-фторфенил)-{3-[3-(3-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон;
(3-фторфенил)-{3-[3-(3-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон;
(4-фторфенил)-{3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон;
(3-фторфенил)-{3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон;
(R)-(4-фторфенил)-{3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон;
(4-фторфенил)-{3-[5-(4-фторфенил)-[1,2,4]оксадиазол-3-ил]пиперидин-1-ил)метанон;
(4-фторфенил)-{3-[5-(4-фторфенил)-4-метил-4Н-[1,2,4]триазол-3-ил]пиперидин-1-ил)метанон;
{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}-(2-фенилтиазол-4-ил)метанон;
{{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}-(2-метил-6-трифторметилпиридин-3-ил)метанон;
{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}-[1,2,3]тиадиазол-4-ил)метанон;
бензотиазол-2-ил-{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон;
{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}-(5-метилизоксазол-3-ил)метанон;
(1,5-диметил-1Н-пиразол-3-ил)-{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон;
{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}-(4-трифторметилфенил)метанон;
4-{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-карбонил}бензонитрил;
{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}изоксазол-5-илметанон;
(3-хлор-4-фторфенил)-{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил)метанон;
{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}-(2-фенил-2Н-пиразол-3-ил)метанон;
{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}-(5-метил-2-фенил-2Н-[1,2,3]триазол-4-ил)метанон;
(4-фтор-3-метилфенил)-{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон;
(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}-(3-метилтиофен-2-ил)метанон;
{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}-(1-метил-1Н-пиррол-2-ил)метанон;
{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}тиазол-2-илметанон;
{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}-(4-метилтиазол-5-ил)метанон;
{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}-(6-морфолин-4-илпиридин-3-ил)метанон;
{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}-(1Н-индол-5-ил)метанон;
2-(4-фторфенил)-1-{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил }этанон;
3-(4-фторфенил)-1-{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}пропан-1-он;
{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}изохинолин-3-илметанон;
{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}хиноксалин-6-илметанон;
{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}бензимидазол-6-илметанон;
(4-фторфенил)-[(S)-3-(3-нафталин-1-ил-[1,2,4]оксадиазол-5-ил)пиперидин-1-ил]метанон;
{(S)-3-[3-(2,6-дифторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}-(4-фторфенил)метанон;
(4-фторфенил)-{(S)-3-[3-(2-метоксифенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон;
(4-фторфенил)-[(S)-3-(3-нафталин-2-ил-[1,2,4]оксадиазол-5-ил)пиперидин-1-ил]метанон;
(4-фторфенил)-{3-[5-(4-фторфенил)-[1,2,4]оксадиазол-3-ил]пиперидин-1-ил}метанон;
(4-фторфенил)-{3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]-4-метилпиперазин-1-ил}метанон;
(4-фторфенил)амид(S)-1-(4-фторбензоил)пиперидин-3-карбоновой кислоты;
(4-фторфенил)метиламид(S)-1-(4-фторбензоил)пиперидин-3-карбоновой кислоты;
(Е)-3-(4-фторфенил)-1-{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}пропенон;
{(S)-3-[3-(4-фторфенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}-(4-имидазол-1-илфенил)метанон;
(4-фторфенил)-{(S)-3-[3-(4-нитрофенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон;
(3,4-дифторфенил)-{(S)-3-[3-(4-нитрофенил)-[1,2,4]оксадиазол-5-ил]пиперидин-1-ил}метанон.
19. Соединения по п.1, где указанные соединения выбраны из группы, включающей
(4-фторфенил)-{(S)-3-[5-(4-фторфенил)изоксазол-3-ил]пиперидин-1-ил}метанон;
(4-фторфенил)-{(S)-3-[5-(4-фторфенил)-1Н-имидазол-2-ил]пиперидин-1-ил}метанон;
(4-фторфенил)-{(S)-3-(4-(4-фторфенил)-1Н-имидазол-1-ил]пиперидин-1-ил}метанон;
(4-фторфенил)-{(S)-3-(4-(4-фторфенил)-1H-пиразол-1-ил]пиперидин-1-ил}метанон;
N-(1-(4-фторбензоил)пиперидин-3-ил)-4-фторбензамид;
(2-фторфенил)-{3-[2-(4-фторфенил)оксазол-5-ил]пиперидин-1-ил}метанон;
(2-фторфенил)-{3-[5-(4-фторфенил)оксазол-2-ил]пиперидин-1-ил}метанон;
(2-фторфенил)-{3-[5-(4-фторфенил)тиазол-2-ил]пиперидин-1-ил}метанон;
(2-фторфенил)-{3-[2-(4-фторфенил)тиазол-5-ил]пиперидин-1-ил}метанон;
(2-фторфенил)-{3-[5-(4-фторфенил)-[1,3,4]тиадиазол-2-ил]пиперидин-1-ил}метанон;
(2-фторфенил)-{3-[5-(4-фторфенил)-[1,2,4]оксадиазол-3-ил]пиперидин-1-ил}метанон;
(2-фторфенил)(3-(5-(4-фторфенил)изоксазол-3-ил)пиперидин-1-ил)метанон;
(2-фторфенил)(3-(5-(4-фторфенил)-1Н-имидазол-2-ил)пиперидин-1-ил)метанон;
(2-фторфенил)(3-(4-(4-фторфенил)-1Н-имидазол-1-ил)пиперидин-1-ил)метанон;
(2-фторфенил)(3-(4-(4-фторфенил)-1Н-пиразол-1-ил)пиперидин-1-ил)метанон;
N-(1-(4-фторбензоил)пиперидин-3-ил)-2-фторбензамид;
(2-фторфенил)-{3-[2-(3,4-дифторфенил)оксазол-5-ил]пиперидин-1-ил}метанон;
(2-фторфенил)-{3-[5-(3,4-дифторфенил)оксазол-2-ил]пиперидин-1-ил}метанон;
(2-фторфенил)-{3-[5-(3,4-дифторфенил)тиазол-2-ил]пиперидин-1-ил}метанон;
(2-фторфенил)-{3-[2-(3,4-дифторфенил)тиазол-5-ил]пиперидин-1-ил}метанон;
(2-фторфенил)-{3-[5-(3,4-дифторфенил)-[1,3,4]тиадиазол-2-ил]пиперидин-1-ил}метанон;
(2-фторфенил)-{3-[5-(3,4-дифторфенил)-[1,2,4]оксадиазол-3-ил]пиперидин-1-ил}метанон;
(2-фторфенил)(3-(5-(3,4-дифторфенил)изоксазол-3-ил)пиперидин-1-ил)метанон;
(2-фторфенил)(3-(5-(3,4-дифторфенил)-1Н-имидазол-2-ил)пиперидин-1-ил)метанон;
(2-фторфенил)(3-(4-(3,4-дифторфенил)-1Н-имидазол-1-ил)пиперидин-1-ил)метанон;
(2-фторфенил)(3-(4-(3,4-дифторфенил)-1Н-пиразол-1-ил)пиперидин-1-ил)метанон;
N-(1-(3,4-дифторбензоил)пиперидин-3-ил)-2-фторбензамид.
20. Фармацевтическая композиция, обладающая свойствами положительного аллостерического модулятора mGluR5, включающая терапевтически эффективное количество соединения по пп.1-19 и фармацевтически приемлемые носители и/или наполнители.
| Прибор для подсчета талонов | 1930 |
|
SU25768A1 |
| 0 |
|
SU154498A1 | |
| ПРОИЗВОДНЫЕ 5-ФЕНИЛ-3-(ПИПЕРИДИН-4-ИЛ)-1,3,4-ОКСАДИАЗОЛ-2(3Н)-ОНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И ЛЕКАРСТВЕННОЕ СРЕДСТВО | 1996 |
|
RU2167160C2 |
| US 3991064 A, 09.11.1976 | |||
| US 20030149049 A1, 07.08.2003 | |||
| WO 9902497 A2, 21.01.1999. | |||

