ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к соединениям, ингибирующим P2X пуринергический рецептор 3 (далее, P2X3 ингибиторы); в частности, настоящее изобретение относится к соединениям, которые являются производными аминохиназолина, способу получения указанных соединений, фармацевтическим композициям, включающим указанные соединения, и их терапевтическому применению.
Соединения по настоящему изобретению могут применяться в лечении множества расстройств, механизмы развития которых связаны с P2X3 рецепторами, таких как респираторные заболевания, включая кашель, астму, идиопатический легочный фиброз (ИЛФ) и хроническую обструктивную болезнь легких (ХОБЛ).
УРОВЕНЬ ТЕХНИКИ ИЗОБРЕТЕНИЯ
P2X рецепторы представляют собой ионные каналы клеточной поверхности, активируемые внеклеточным аденозин-5-трифосфатом (АТФ). Семейство P2X рецепторов представляет собой тримерные сборки, состоящие из субъединиц семи различных подтипов (P2X1-7), которые собираются в виде гомомерных и гетеромерных каналов. Все субъединицы имеют общую типологию, включающую внутриклеточные окончания, две трансмембранные спирали, образующие ионные каналы, и большой внеклеточный домен, содержащий сайт связывания АТФ. Гомомерные каналы P2X1, P2X2, P2X3, P2X4, P2X5 и P2X7 и гетеромерные каналы P2X2/3 и P2X1/5 были полностью охарактеризованы после гетерологичной экспрессии. P2X рецепторы широко распространены, и функциональные реакции наблюдаются в нейронах, нервной, эпителиальной, эндотелиальной, костной, мышечной и гемопоэтических тканях. На гладких мышцах P2X рецепторы реагируют на АТФ, высвобождаемый симпатическими двигательными нервами (например, при эякуляции). На сенсорных нервах они участвуют в инициировании афферентных сигналов в нескольких внутренних органах (например, мочевом пузыре, кишечнике) и играют ключевую роль в восприятии повреждающих ткани и воспалительных стимулов. Паракринные функции для передачи сигналов АТФ через P2X рецепторы весьма вероятны в нейрогипофизе, протоковых железах, эпителии дыхательных путей, почках, костах и кроветворных тканях (RA. North: Molecular Physiology of P2X Receptors; Physiol. Rev., Vol. 82, Oct. 2002). Все рецепторы P2X являются неселективными катионными каналами, проницаемыми для ионов Na+ и Ca+ и активируются АТФ; однако фармакология подтипов рецепторов изменяется в зависимости от чувствительности к АТФ и к антагонистам малых молекул (K. Kaczmarek-Hajek et al: Molecular и functional properties of P2X receptors - recent progress и persisting challenges; Purinergic Signalling 8:375-417, 2012).
У людей рецептор P2X3 был описан в сердце и спинном мозге на уровне мРНК и в DRG, кишечнике (нейроны межмышечного сплетения), мочевом пузыре (уротелий и субуротелий) и в зубной пульпе на уровне белка (Garcia-Guzman M. et al: Molecular characterization и pharmacological properties of the human P2X3 purinoceptor: Brain Res. Mol, Brain Res. 1997; 47(1-2):59-66).
Нейрофизиологическая роль P2X3 рецепторов в функции сенсорных нервов в дыхательных путях аналогична роли, опосредующей соматическую ноцицепцию (Undem B.J. и Nassenstein C.: Airway nerves и dyspnea associated with inflammatory airway disease, Respir. Physiol. Nerobiol. 167: 36-44, 2009). Это сходство привело к гипотезам, касающимся вовлеченности P2X3 рецепторов в симптомы дисфункции дыхательных путей, включая кашель и гиперактивность бронхов (Ford AP: In pursuit of P2X3 antagonists: novel therapeutics for chronic pain и и afferent sensitization, Purinergic signal 8 (suppl 1):3-26, 2012; North RA, Jarvis MF P2X Receptors as Drug Targets; Mol Pharmacol, 83:759-769, 2013). P2X3 субъединицы также локализованы во многих нейронах, особенно в DRG, узловых ганглиях, солитарном ядре и вкусовых рецепторах (Cheung K.K., Burnstock G.: Localization of P2X3 receptors и coexpression with P2X2 receptors during rat embryonic neurogenesis. J. Comp. Neurol. 443(4):368-382 2002).
P2X3 антагонисты были предложены для лечения диабетической невропатической боли (Guo J. et al.: Contributions of purinergic P2X3 receptors within the midbrain periaqueductal gray to diabetes-induced neuropathic pain, J. Physiol. Sci. Jan.; 65(1):99-104 2015).
P2X3 и P2X2/3 каналы играют важную роль в развитии суставной гипералгезии артритных суставов (Teixeira JM et al: P2X3 и P2X2/3Receptors Play a Crucial Role in Articular Hyperalgesia Development Through Inflammatory Mechanisms in the Knee Joint Experimental Synovitis, Mol Neurobiol Oct;54(8):6174-6186, 2017).
P2X3 являются потенциальной мишенью для терапевтического лечения боли в мочевом пузыре. Они также были предложены в качестве мишеней обезболивания для лечения боли в мочеточниках и для облегчения прохождения камня в мочеточнике (Canda A.E. et al: Physiology и pharmacology of the human ureter: basis for current и future treatments, Urol. Int. 78(4):289-98, 2007).
Сверхэкспрессия P2X3 влияет на низкую безрецидивную выживаемость у пациентов с гепатоцеллюлярной карциномой, что определяет P2X3 в качестве потенциальной терапевтической мишени (Maynard J.P. et al: P2X3 purinergic receptor overexpression is associated with poor recurrence-free survival in hepatocellular carcinoma patients Oncotarget Dec 1;6(38):41162-79, 2015).
Было высказано предположение, что P2X3 антагонисты могут улучшать восстановление эректильной функции (Li C.L. et al: Effects of intracavernous injection of P2X3 и NK1 receptor antagonists on erectile dysfunction induced by spinal cord transection in rats, Andrologia. Feb;47(1):25-9, 2015).
АТФ усиливает кашель, вызываемый лимонной кислотой и гистамином, это действие может быть ослаблено селективными P2X3 антагонистами (Kamei J. и Takahashi Y.: Involvement of ionotropic purinergic receptors in the histamine-induced enhancement of the cough reflex sensitivity in guinea pigs, Oct 10;547(1-3):160-4, 2006). У людей местная доставка АТФ вызывает кашель и бронхоспазм (Basoglu O.K. et al: Effects of aerosolized adenosine 5'-triphosphate vs adenosine 5'-monophosphate on dyspnea и airway caliber in healthy nonsmokers и patients with asthma, Chest. Oct;128(4):1905-9, 2005).
Терапевтическая перспективность P2X3 антагонистов для лечения хронического кашля была впервые выявлена Ford и Undem (Ford AP, Undem BJ: The therapeutic promise of АТР antagonism at P2X3 receptors in respiratory и urological disorders, Front Cell Neurosci, Dec 19;7:267, 2013). P2X3 экспрессируются афферентными нервами дыхательных путей и опосредуют гиперчувствительность кашлевого рефлекса, которая резко снижается при пероральном введении P2X3 антагониста AF-219 (Abdulqawi et al: P2X3 receptor antagonist (AF-219) in refractory chronic cough: a randomised, double-blind, placebo-controlled phase 2 study, Lancet 385, 1198-205, 2015).
АТФ является ключевым нейтотрансмиттером во вкусовой системе, действуя в основном через рецепторы гетеромильтиморов P2X2/3. Поэтому нарушение вкусовой функции может быть непроизвольным следствием пробного терапевтического лечения боли, хронического кашля и других состояний с использованием антагонистов пуринергических рецепторов P2X3 (Vandenbeuch A et al: Role of the ectonucleotidase NTPDase2 in taste bud function, Proc. Natl. Acad. Sci. USA, Sep 3;110(36):14789-94, 2013. Bo X. et al: Localization of АТР-gated P2X2 и P2X3 receptor immunoreactive nerves in rat taste buds, Neuroreport, 10(5):1107-11, 1999).
В качестве ингибиторов P2X3 и/или P2X2/3 в научной литературе были описаны различные соединения.
В WO2017058645 (Afferent Pharmaceuticals INC) раскрывается применение диаминопиримидиновых антагонистов P2X3/P2X2/3 для лечения расстройств, таких как кашель, хронический кашель и позыв к кашлю, включая кашель, связанный с респираторным заболеванием или нарушением, введением эффективного количества раскрытого соединения. Однако производные аминохиназолина не раскрываются.
В WO2017011729 (Patara Pharma LLC) описывается применение кромолина или его фармацевтически приемлемой соли и антагониста рецептора P2X3 и/или P2X2/3 в качестве противокашлевого средства для лечения легочных заболеваний и расстройств.
В WO2016091776, (Evotec AG) описываются 1,3-тиазол-2-ил-замещенные производные бензамида, которые ингибируют P2X3 рецептор, и фармацевтические композиции, содержащие указанные соединения, а также применение указанных соединений для лечения некоторых расстройств, включая респираторные заболевания.
В WO2016088838 (Shionogi) описываются производные пурина, обладающие новым антагонистическим действием в отношении P2X3 и/или P2X2/3 рецептора.
В WO2016084922 (Shionogi) описываются производные триазина, обладающие новым антагонистическим действием в отношении P2X3 и/или P2X2/3 рецептора.
WO2008123963 (Renovis) относится к конденсированным гетероциклическим соединениям класса тетрагидропиридо[4,3-д]пиримидинов и фармацевтическим композициям, включающим указанные соединения. Предоставлены также способы предотвращения и/или лечения некоторых расстройств, таких как нейродегенеративные расстройства, боль, астма, аутоиммунные расстройства, введением раскрытых соединений.
В WO2008130481 (Renovis) описываются 2-цианофенил-конденсированные гетероциклические соединения класса тетрагидропиридо[4,3-д]пиримидинов и фармацевтические композиции, включающие указанные соединения.
В WO2010033168 (Renovis) описываются серии бензамидов, замещенных фенилом или пиридилом, которые заявлены как применимые для лечения заболеваний, связанных с пуринергическими P2X рецепторами, точнее как антагонисты P2X3 рецептора и/или P2X2/3 рецептора. Однако в документе не раскрыты производные аминохиназолина.
WO2009110985 (Renovis) относится к фенил- и пиридил-замещенным бензамидным соединениям и фармацевтическим композициям, включающим указанные соединения, но не тиазол-замещенным бензамидам, что отличает указанные соединения от соединений по настоящему изобретению.
В WO2008000645 (Roche) раскрываются тетразол-замещенные ариламидные антагонисты P2X3 и/или P2X2/3 рецепторов, применимые для лечения урогенитальных заболеваний, состояний и расстройств, боли, желудочно-кишечных и респираторных заболеваний, состояний и расстройств.
Несмотря на достижения предшествующего уровня, все еще сохраняется потребность в новых аминохиназолиновых соединениях для лечения заболеваний, связанных с P2X3 рецепторами, во многих терапевтических областях, таких как, в частности, респираторные заболевания, предпочтительно в соединениях, обладающих селективным действием на P2X3 рецептор.
Следует подчеркнуть, что уровень техники не описывает и не предлагает производные аминохиназолина общей формулы (I) по настоящему изобретению, которые представляют собой решение вышеупомянутой потребности.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
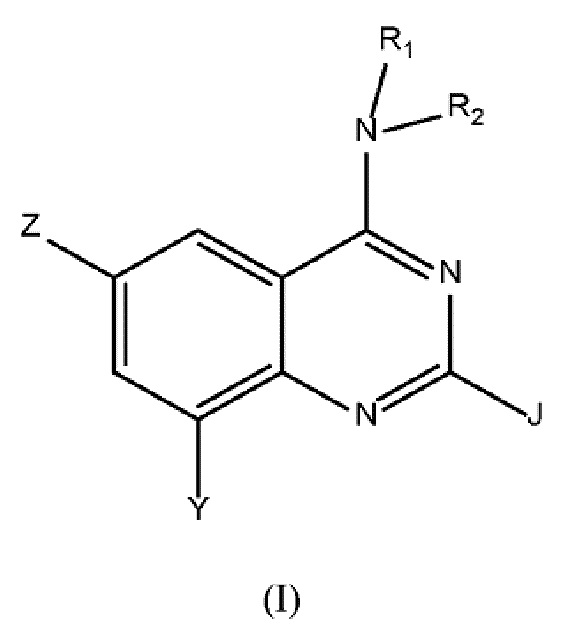
Настоящее изобретение относится к соединениям формулы (I)

где
Z выбран из группы, состоящей из (C3-C8)гетероциклоалкила, (RARB)N-, гетероарила, арила, где любой из указанных алкила, гетероарила, гетероциклоалкила и арила может быть необязательно замещенным одной или несколькими группами, выбранными из (C1-C3)алкила-, галогена, CN, (RARB)NC(O)-, (C1-C6)галогеналкила-, RAO-, (RARB)N(C1-C6)алкилена-, (C3-C7)циклоалкила-, RCSO2-,(RARB)N-;
R1 представляет собой Н или (C1-C4)алкил;
R2 выбран из группы, состоящей из (C1-C6)алкила-, гетероарил(C1-C4)алкила-, (C3-C8)гетероциклоалкил-(C1-C6)алкила-, гетероарил-(C1-C6)гидроксиалкила-, (C3-C8)гетероциклоалкила, (C3-C8)циклоалкил-(C1-C6)алкила-, арил-(C1-C4)алкила-, (RARB)N(C1-C6)алкилена-, (RARB)N(O)C(C1-C4)алкилена-, RAO(C1-C4)алкилена-, где любой из указанных алкила, алкилена, арила, гетероарила и гетероциклоалкила может быть необязательно замещенным одной или несколькими группами, выбранными из (C1-C3)алкила, RAO(C1-C4)алкилена-, (C1-C6)галогеналкила, галогена, оксо, RAO-, (C3-C8)гетероциклоалкил-(C1-C6)алкила-, гетероарила, (RARB)N-, -NHC(O)RC, -C(O)N(RARB), -SO2N(RARB), -O(C1-C4)алкилен-N(RARB), арила, необязательно замещенного галогеном, -ORC, арил-(C1-C4)алкила-, -C(O)RA;
RA и RB в каждом случае независимо представляют собой Н или выбраны из группы, состоящей из (C1-C4)алкила-, (C3-C8)циклоалкила, (C1-C6)галогеналкила, или
RA и RB вместе с атомом азота, к которому они присоединены, могут образовывать 5- или 6-членную насыщенную гетероциклическую моноциклическую кольцевую систему, необязательно содержащую дополнительный гетероатом, представляющий собой атом азота или кислорода, которая может быть необязательно замещенной одной или несколькими группами, выбранными из (C1-C4)алкила и оксогруппы;
RC в каждом случае представляет собой Н или выбран из группы, состоящей из (C1-C6)алкила, (RARB)N-, арил-(C1-C4)алкила-;
Y выбран из группы, состоящей из H, -ORD, RCSO2, галогена, -NHSO2RC, гетероарила, (C3-C8)гетероциклоалкила, где любой из указанных гетероарила и гетероциклоалкила может быть необязательно замещенным одной или несколькими группами, выбранными из (C1-C3)алкила и -C(O)N(RARB);
RD выбран из группы, состоящей из H, (C1-C6)алкила, (C3-C8)гетероциклоалкил-(C1-C6)алкила-, RCOC(O)(C1-C4)алкилена-, (RARB)N(C1-C6)алкилена-, (C3-C8)гетероциклоалкила, (C3-C8)циклоалкил-(C1-C6)алкила-, RCO(C1-C4)алкилена-, (RARB)N(O)C(C1-C4)алкилена-, где любой указанный гетероциклоалкил может быть необязательно замещенным одной или несколькими группами, выбранными из (C1-C3)алкила;
J представляет собой Н или выбран из группы, состоящей из (C1-C6)алкила, (RARB)N-, (C1-C6)галогеналкила, -ORC и галогена.
Во втором аспекте настоящее изобретение относится к фармацевтической композиции, включающей одно соединение формулы (I) или одну его фармацевтически приемлемую соль либо в комбинации с одним или несколькими другими активными ингредиентами - в смеси с одним или несколькими фармацевтически приемлемыми носителями или эксципиентами.
В третьем аспекте настоящее изобретение относится к соединению формулы (I) для применения в качестве лекарственного средства.
В дополнительном аспекте настоящее изобретение относится к применению соединения формулы (I) в лечении любого заболевания, в которое вовлечены рецепторы P2X3.
В другом дополнительном аспекте настоящее изобретение относится к соединению формулы (I) для применения в предотвращении и/или лечении респираторных заболеваний, включая кашель, подострый или хронический кашель, терапевтически резистентный кашель, идиопатический хронический кашель, поствирусный кашель, ятрогенный кашель, астму, идиопатический легочный фиброз (ИЛФ), хроническую обструктивную болезнь легких (ХОБЛ) и кашель, связанный с респираторными заболеваниями, такими как ХОБЛ, астма и бронхоспазм.
В еще одном дополнительном аспекте настоящее изобретение относится к соединению формулы Ib

где
R3 представляет собой ОН или галоген;
R4 представляет собой Н или OH;
R5 представляет собой галоген или -OMe;
R6 представляет собой галоген или Z;
Z принимает значения, определенные выше.
В еще одном дополнительном аспекте настоящее изобретение относится к применению соединения формулы (Ib) в качестве промежуточного соединения при получении соединений формулы (I).
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к соединению формулы (I)

где
Z выбран из группы, состоящей из (C3-C8)гетероциклоалкила, (RARB)N-, гетероарила, арила, где любой из указанных алкила, гетероарила, гетероциклоалкила и арила может быть необязательно замещенным одной или несколькими группами, выбранными из (C1-C3)алкила-, галогена, CN, (RARB)NC(O)-, (C1-C6)галогеналкила-, RAO-, (RARB)N(C1-C6)алкилена-, (C3-C7)циклоалкила-, RCSO2-, (RARB)N-;
R1 представляет собой Н или (C1-C4)алкил;
R2 выбран из группы, состоящей из (C1-C6)алкила-, гетероарил(C1-C4)алкила, (C3-C8)гетероциклоалкил-(C1-C6)алкила-, гетероарил-(C1-C6)гидроксиалкила-, (C3-C8)гетероциклоалкила, (C3-C8)циклоалкил-(C1-C6)алкила-, арил-(C1-C4)алкила-, (RARB)N(C1-C6)алкилена-, (RARB)N(O)C(C1-C4)алкилена-, RAO(C1-C4)алкилена-, где любой из указанных алкила, алкилена, арила, гетероарила и гетероциклоалкила может быть необязательно замещенным одной или несколькими группами, выбранными из (C1-C3)алкила, RAO(C1-C4)алкилена-, (C1-C6)галогеналкила, галогена, оксогруппы, RAO-, (C3-C8)гетероциклоалкил-(C1-C6)алкила-, гетероарила, (RARB)N-, -NHC(O)RC, -C(O)N(RARB), -SO2N(RARB), -O(C1-C4)алкилен-N(RARB), арила, необязательно замещенного галогеном, -ORC, арил-(C1-C4)алкила-, -C(O)RA;
RA и RB в каждом случае независимо представляют собой Н или выбраны из группы, состоящей из (C1-C4)алкила-, (C3-C8)циклоалкила-, (C1-C6) галогеналкила, или
RA и RB вместе с атомом азота, к которому они присоединены, могут образовывать 5- или 6-членную насыщенную гетероциклическую моноциклическую кольцевую систему, необязательно содержащую дополнительный гетероатом, представляющий собой атом азота или кислорода, которая может быть необязательно замещенной одной или несколькими группами, выбранными из (C1-C4)алкила и оксогруппы;
RC в каждом случае представляет собой Н или выбран из группы, состоящей из (C1-C6)алкила, (RARB)N-, арил-(C1-C4)алкила-;
Y выбран из группы, состоящей из H, -ORD, RCSO2, галогена, -NHSO2RC, гетероарила, (C3-C8)гетероциклоалкила, где любой из указанных гетероарила и гетероциклоалкила может быть необязательно замещенным одной или несколькими группами, выбранными из (C1-C3)алкила, -C(O)N(RARB);
RD в каждом случае выбран из группы, состоящей из H, (C1-C6)алкила, (C3-C8)гетероциклоалкил-(C1-C6)алкила-, RCOC(O)(C1-C4)алкилена-, (RARB)N(C1-C6)алкилена-, (C3-C8)гетероциклоалкила, (C3-C8)циклоалкил-(C1-C6)алкила-, RCO(C1-C4)алкилена-, (RARB)N(O)C(C1-C4)алкилена-, где любой указанный гетероциклоалкил может быть необязательно замещенным одной или несколькими группами, выбранными из (C1-C3)алкила;
J представляет собой Н или выбран из группы, состоящей из (C1-C6)алкила, (RARB)N-, (C1-C6)галогеналкила, -ORC и галогена.
ОПРЕДЕЛЕНИЯ
Термин «фармацевтически приемлемые соли», когда используется в настоящем описании, относится к производным соединений формулы (I), в которых исходное соединение является подходящим образом модифицированным превращением любой свободной кислотной или основной группы, если такая присутствует, в соответствующую аддитивную соль с любым основанием или кислотой, и которые обычно считаются фармацевтически приемлемыми.
Таким образом, подходящие примеры указанных солей могут включать аддитивные соли минеральных или органических кислот и основных остатков, таких как аминогруппы, а также аддитивные соли минеральных или органических оснований и кислотных остатков, таких как карбоксильные группы.
Катионы неорганических оснований, которые могут подходящим образом использоваться для получения солей, включают ионы щелочных или щелочно-земельных металлов, таких как калий, натрий, кальций или магний.
Соли, полученные взаимодействием основного соединения, выступающего в качестве основания, с неорганической или органической кислотой, включают, например, соли соляной кислоты, бромистоводородной кислоты, серной кислоты, фосфорной кислоты, метансульфоновой кислоты, камфорсульфоновой кислоты, уксусной кислоты, шавелевой кисоты, малеиновой кислоты, фумаровой кислоты, янтарной кислоты и лимонной кислоты.
Термин «галоген» или «атом галогена», когда используется в настоящем описании, включает атом фтора, хлора, брома и йода, предпочтительно атом хлора или фтора.
Термин «(Cx-Cy) алкил», где x и y представляют собой целые числа, относится к алкильному радикалу с прямой или разветвленной цепью, содержащему от x до y атомов углерода. Таким образом, когда, например, x представляет собой 1 и y представляет собой 6, указанный термин включает метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил и н-гексил.
Термин «(Cx-Cy) алкилен», где x и y представляют собой целые числа, когда используется в настоящем описании, относится к Cx-Cy алкильному радикалу, содержащему всего две ненасыщенные валентности, такому как двухвалентный метиленовый радикал.
Термин «(Cx-Cy) галогеналкил», где x и y представляют собой целые числа, относится к определенным выше группам «Cx-Cy алкил», в которых один или несколько атомов водорода замещены одним или несколькими атомами галогенов, которые могут быть одинаковыми или разными.
Таким образом, примеры указанных групп «(Cx-Cy) галогеналкил» могут включать галогенированные, полигалогенированные и полностью галогенированные алкильные группы, в которых все атомы водорода замещены атомами галогенов, например трифторметил, дифторметил или трифторэтил.
По аналогии, термин «(C1-C6) гидроксиалкил» или «(C1-C6)аминоалкил» относится к определенным выше группам «(C1-C6) алкил», в которых один или несколько атомов водорода замещены одним или несколькими гидроксильными (OH) или аминогруппами, соответственно. Примеры таких групп включают, соответственно, гидроксиметил, аминометил, диметиламинопропил и т.п.
В настоящем описании, если не указано иное, термин «аминоалкил» включает алкильные группы (т.е. группы «(C1-C6) алкил»), замещенные одной или несколькими аминогруппами (-NRARB). Таким образом, примером аминоалкила является моно-аминоалкильная группа, такая как RARBN-(C1-C6) алкил.
Что касается заместителей RA и RB, которые определены выше и далее, когда RA и RB вместе с атомом азота, к которому они присоединены, образуют 5-6-членный гетероциклический радикал, по меньшей мере еще один атом углерода кольца необязательно замещен по меньшей мере на один гетероатом (например, атом N, S или O) и/или может нести группы -оксозаместителя (=O). Разумеется, указанный гетероциклический радикал может быть необязательно дополнительно замещенным по любому доступному положению в кольце, а именно по атому углерода или по любому гетероатому, доступному для замещения. Замещение по атому углерода включает спиро-дизамещение, а также замещение по двум соседним атомам углерода с образованием в обоих случаях дополительного 5-6-членного гетероциклического кольца. Примерами указанных гетероциклических радикалов являются 1-пирролидинил, 1-пиперидинил, 1-пиперазинил, 4-метилпиперазинил, пиперазин-4-ил-2-он, 4-морфолинил, морфолинил-3-он, 1-(пиперазин-1-ил)этенон.
Термин «(Cx-Cy) циклоалкил», где х и y представляют собой целые числа, относится к насыщенным циклическим углеводородным группам, содержащим указанное число атомов углерода в кольце. Примеры таких групп включают циклопропил, циклобутил, циклопентил, циклогексил, циклогептил.
Термин «арил» относится к моноциклическим углеродным кольцевым системам, которые содержат в цикле 6 атомов, где кольцо является ароматическим. Примеры подходящих моноциклических арильных кольцевых систем включают, например, фенил.
Термин «гетероарил» относится к моно- или бициклическому ароматическому радикалу, содержащему один или несколько гетероатомов, выбранных из атомов S, N и O, и включает радикалы, содержащие два таких моноциклических кольца или одно такое моноциклическое кольцо и одно моноциклическое арильное кольцо, которые являются конденсированными через общую связь. Примерами подходящего 5-6-членного гетероарила являются тиенил, фурил, пирролил, имидазолил, тиазолил, изотиазолил, пиразолил, оксазолил, изоксазолил, изотиазолил, триазолил, тиадиазолил, оксадиазолил, пиридинил, пиридазинил, пиримидинил, пиразинил, тетразолил и триазинил.
Термин «гетероциклил» или «гетероциклический» относится к насыщенному моно-, би- или трициклическому неароматическому радикалу, содержащему один или несколько гетероатомов, выбранных из атомов S, N и O. В случае бициклических гетероциклических систем данный термин включает конденсированные спиро- и мостиковые бициклические системы.
Термин «(Cx-Cy) гетероциклоалкил», где х и y представляют собой целые числа, относится к насыщенным или частично ненасыщенным моноциклическим (Cx-Cy)циклоалкильным группам, в которых по меньшей мере один атом углерода в кольце замещен по меньшей мере на один гетероатом (например, атом N, S или O) или может нести группу -оксозаместителя (=O). Указанный гетероциклоалкил (т.e. гетероциклический радикал или гетероциклическая группа) может быть необязательно дополнительно замещенным по любому доступному положению в кольце, а именно по атому углерода или по любому гетероатому, доступному для замещения. Замещение по атому углерода включает спиро-дизамещение, а также замещение по двум соседним атомам углерода с образованием в обоих случаях дополнительного 5-6-членного гетероциклического кольца. Примерами (Cx-Cy) гетероциклоалкила являются пирролидинил, имидазолидинил, тиазолидинил, пиеразинил, пиперидинил, морфолинил, тиоморфолинил, дигидро- или тетрагидропиридинил, тетрагидротиофенил, азетидинил, оксетанил, тетрагидропиранил, пиранил, 2H- или 4H-пиранил, дигидро- или тетрагидрофуранил, дигидроизоксазолил, пирролидин-2-онил, дигидропирролил и т.п.
Конкретными примерами указанных гетероциклических радикалов являются тетрагидротиофен-1,1-диоксид, 3,3-дифторпирролидинил, 1-пирролидинил, 1-метил-2-пирролидинил, 1-пиперидинил, 1-пиперазинил, 4-морфолинил.
Термины «арилокси» и «арил(C1-C6)алкоксил» аналогично терминам «гетероарилoкси» и «гетероарил(C1-C6)алкоксил» относятся к присоединенным через кислородный мостик арильным или гетероарильным группам и присоединенным через цепочки арилалкоксильным или гетероарилалкоксильным группам. Примерами таких групп являются фенилокси, бензилокси и пиридинилокси, соответственно.
Термин «арил(C1-C6)алкил» относится к арильному кольцу, присоединенному к алкильным группам с прямой или разветвленной цепью, в которых количество атомов углерода составляет от 1 до 6, например фенилметилу (т.е. бензилу), фенилэтилу или фенилпропилу.
Термин «(Cz-Ck)гетероциклоалкил-(Cx-Cy)алкил», где z и k представляют собой целые числа, относится гетероциклическому кольцу, присоединенному к алкильным группам с прямой или разветвленной цепью, содержащим от x до y атомов углерода.
Аналогично, термин «гетероарил (Cx-Cy)алкил» или «арил (Cx-Cy)алкил» относится к гетероарильному или арильному кольцу, присоединенному к алкильным группам с прямой или разветвленной цепью, содержащим от x до y атомов углерода.
Термин «кольцевая система» относится к моно-, бициклическим или полициклическим кольцевым системам, которые могут быть насыщенными, частично ненасыщенными или ненасыщенными, таким как арил, (C3-C10) циклоалкил, (C3-C6)гетероциклоалкил или гетероарил.
Термины «группа», «радикал», «фрагмент» или «заместитель» являются синонимами и предназначены для обозначения функциональных групп или фрагментов молекул, которые могут присоединяться к связи или другим фрагментам или молекулам. Таким образом, например, термин «гетероциклический радикал» в настоящем описании относится к моно- или бициклическому насыщенному или частично насыщенному гетероциклический фрагменту (группе, радикалу), предпочтительно 4-11-членному моноциклическому радикалу, в котором по меньшей мере один дополнительный атом углерода необязательно замещен по меньшей мере на один дополнительный гетероатом, независимо выбранный из атомов N, S или O, и/или может нести оксогруппу (=O), причем указанный гетероциклический радикал необязательно дополнительно включает спиродизамещение, а также замещение по двум соседним или вицинальным атомам с образованием дополнительного 5-6-членного циклического или гетероциклического насыщенного, частично насыщенного или или ароматического кольца. Примерами указанных гетероциклических радикалов являются 1-пирролидинил, 1-пиперидинил, 1-пиперазинил, 4-морфолинил и т.п.
Тире («-»), которое не находится между двумя буквами или символами, означает точку присоединения заместителя. При графическом представлении точка присоединения циклической функциональной группы обозначается точкой («•»), расположенной на одном из доступных атомов кольца, где функциональная группа может присоединяться к связи или другому фрагменту молекул.
Оксо-фрагмент обозначается (O) как альтернатива другому обычному обозначению, например (=O). Таким образом, в настоящем описании в части, относящейся к общей формуле, карбонильная группа обычно представлена как -C(O)-, где группа в скобках является боковой группой, не включенной в цепь, а скобки используются, когда они способствуют устранению неоднозначности линейных формул; например, сульфонильная группа -SO2- также может быть представлена как -S(O)2- для устранения неоднозначности, связанной, например, с сульфинильной группой -S(O)O-.
Когда в соединениях формулы I присутствует основные аминогруппа или группа четвертичного аммония, физиологически приемлемые анионы могут быть выбраны из хлорида, бромида, йодида, трифторацетата, формиата, сульфата, фосфата, метансульфоната, нитрата, малеата, ацетата, цитрата, фумарата, тертрата, оксалата, сукцината, бензоата, п-толуолсульфоната, памоата и нафталиндисульфоната. Аналогично, в присутствии кислотных групп, таких как COOH группы, могут образовываться соли физиологически подходящих катионов, а также, например, соли, содержащие ионы щелочных или щелочно-земельных металлов.
Очевидно, что соединения формулы (I), когда содержат один или несколько стереогенных центров, могут существовать в виде оптических изомеров.
Когда в соединениях по настоящему изобретению присутствует по меньшей мере один стереогенный центр, они могут соответственно существовать в виде энантиомеров. Когда соединения по настоящему изобретению содержат два или более стереогенных центров, они могут дополнительно существовать в виде диастереоизомеров. Все такие одиночные энантиомеры, диастереоизомеры и их смеси в любом соотношении включены в объем настоящего изобретения. Абсолютная конфигурация (R) или (S) для углерода, являющегося стереогенным центром, присваивается в соответствии с правилами номенклатуры Кана-Ингольда-Прелога (Cahn-Ingold-Prelog), исходя из приоритетов групп.
Настоящее изобретение также относится к соответствующим дейтерированным производным соединений формулы (I).
Все предпочтительные группы или варианты осуществления соединений формулы I, описанные выше и далее, могут комбинироваться друг с другом и применяться также с соответствующими изменениями (mutatis mutandis).
В предпочтительном варианте осуществления настоящее изобретение относится к соединениям формулы (I), которая описана выше

где
Z выбран из группы, состоящей из гетероарила, арила, (RARB)N-, (C3-C8)гетероциклоалкила, где любой из указанных гетероарила, арила и гетероциклоалкила может быть необязательно замещенным одной или несколькими группами, выбранными из (C1-C3)алкила, галогена, CN, (RARB)NC(O)-;
R1 представляет собой Н или (C1-C4)алкил;
R2 выбран из группы, состоящей из гетероарил(C1-C4)алкила-, (RARB)N(O)C(C1-C4)алкилена-, где любой указанный гетероарил может быть необязательно замещенным одной или несколькими группами, выбранными из (C1-C3)алкила, галогена, (C1-C6)галогеналкила;
RA и RB в каждом случае независимо представляют собой H, (C1-C4)алкил- и (C3-C8)циклоалкил-, или RA и RB вместе с атомом азота, к которому они присоединены, могут образовывать 5- или 6-членную насыщенную гетероциклическую моноциклическую кольцевую систему, необязательно содержащую дополнительный гетероатом, представляющий собой атом кислорода или азота, которая может быть необязательно замещенной одной или несколькими группами, выбранными из оксогрупы, (C1-C4)алкила;
Y представляет собой H;
J представляет собой Н или выбран из группы, состоящей из (C1-C4)алкила, (RARB)N-, галогена, (C1-C6)галогеналкила.
В другом предпочтительном варианте осуществления настоящее изобретение относится к соединениям формулы (I), где
Z выбран из группы, состоящей из гетероарила и арила, где любой из указанных гетероарила и арила может быть необязательно замещенным одной или несколькими группами, выбранными из (C1-C3)алкила и галогена;
R1 представляет собой H;
R2 выбран из группы, состоящей из
(C3-C8)гетероциклоалкил-(C1-C6)алкила-, предпочтительно (пиперидинил)метила;
гетероарил(C1-C4)алкила-, предпочтительно (пиридинил)метила, (пиридинил)этила, (пиридазинил)метила, (пиридазинил)этил(пиримидинил)метила, (пиримидинил)этила, (оксадиазолил)этила, (тиадиазолил)этил-([1,2,4]триазоло[4,3-a]пиримидин-3-ил)метила, и
где любой из указанных алкила, гетероарила и гетероциклоалкила может быть необязательно замещенным одной или несколькими группами, выбранными из (C1-C3)алкила, (C1-C6)галогеналкила и -OH;
Y выбран из группы, состоящей из H и -ORD;
RD в каждом случае выбран из группы, состоящей из
(C1-C6)алкила, предпочтительно метила,
(C3-C8)гетероциклоалкил-(C1-C6)алкила-, предпочтительно (оксетанил)метила, (морфолинил)метила,
RCOC(O)(C1-C4)алкилена-, предпочтительно -CH2C(O)OH;
(C3-C8)гетероциклоалкила, предпочтительно тетрагидропиранила, пирролидинила, и
RCO(C1-C4)алкилена-, предпочтительно метоксиэтила;
J в каждом случае выбран из группы, состоящей из H и -ORC, предпочтительно представляет собой Н или -OH;
RC в каждом случае выбран из группы, состоящей из H и (C1-C6)алкила.
В соответствии с предпочтительным вариантом осуществления настоящее изобретение относится по меньшей мере к одному соединению из представленных в таблице 1 ниже и их фармацевтически приемлемым солям.
Таблица 1. Перечень предпочтительных соединений формулы (I)
В дополнительном предпочтительном варианте осуществления настоящее изобретение относится к соединению формулы (I), где
Z выбран из группы, состоящей из
гетероарила, предпочтительно пиримидинила, тиазолила, пиридинила, тиофенила,
арила, предпочтительно фенила,
(RARB)N-, где RA и RB вместе с атомом азота, к которому они присоединены, образуют 5- или 6-членную насыщенную гетероциклическую моноциклическую кольцевую систему, содержащую дополнительный гетероатом, представляющий собой атом кислорода или азота, причем указанный гетероциклический радикал является в свою очередь необязательно дополнительно замещенным одной или несколькими группами оксо, метила и фтора;
любой из указанных гетероарила и арила является дополнительно необязательно замещенным одной или несколькими группами, выбранными из
метила,
фтора,
RCSO2-, где RC выбран из группы, состоящей из фтора, -OH и
(RARB)N-, где RA и RB представляют собой H,
CN,
(RARB)NC(O)-, где RA и RB представляют собой H;
R1 представляет собой Н или метил;
R2 выбран из группы, состоящей из
гетероарил(C1-C4)алкила-, предпочтительно (пиридинил)метила, (пиридазил)метила, (пиримидинил)этила, (оксадиазолил)этила,
(RARB)N(O)C(C1-C4)алкилена-, предпочтительно RARB представляют собой H, циклопропил;
где любой указанный гетероарил может быть необязательно замещенным одной или несколькими группами, выбранными из метила, фтора и трифторметила;
Y представляет собой H;
J представляет собой Н или выбран из группы, состоящей из
галогена, предпочтительно хлора,
(C1-C4)алкила, предпочтительно метила,
(C1-C6)галогеналкила, предпочтительно трифторметила,
(RARB)N-, где RA и RB в каждом случае независимо представляют собой H, циклопропил и метил, или, альтернативно,
RA и RB вместе с атомом азота, к которому они присоединены, образуют 6-членную насыщенную гетероциклическую моноциклическую кольцевую систему, содержащую дополнительный гетероатом, который представляет собой атом кислорода.
В предпочтительном варианте осуществления настоящее изобретение относится к соединению формулы (I), которая определена выше,

где
Z выбран из группы, состоящей из гетероарила и арила, где любой из указанных гетероарила и арила может быть необязательно замещенным одной или несколькими группами, выбранными из (C1-C3)алкила, галогена, CN, (RARB)NC(O)-, (C1-C6)галогеналкила, RAO-, (RARB)N(C1-C6)алкилена-, (C3-C7)циклоалкила-, RCSO2-, (RARB)N-;
R1 представляет собой H или (C1-C6)алкил;
R2 выбран из группы, состоящей из (C1-C6)алкила, гетероарил(C1-C4)алкила-, (C3-C8)гетероциклоалкил-(C1-C6)алкила, гетероарил-(C1-C6)гидроксиалкила, арил-(C1-C4)алкила-, (C3-C8)гетероциклоалкила, (C3-C8)циклоалкил-(C1-C6)алкила-, (RARB)N(C1-C6)алкилена-, RAO(C1-C4)алкилена, где любой из указанных алкила, алкилена, арила, гетероарила, циклоалкила и гетероциклоалкила может быть необязательно замещенным одной или несколькими группами, выбранными из (C1-C3)алкила, RAO(C1-C4)алкилена, (C1-C6)галогеналкила, оксо, RAO-, (C3-C8)гетероциклоалкил-(C1-C6)алкила, гетероарила, арила, необязательно замещенного галогеном, RCO-, (RARB)N-, -NHC(O)RC,-C(O)N(RARB), галогена, -SO2N(RARB), -O(RAO(C1-C4)алкилен-N(RARB), арил-(C1-C4)алкила-, -C(O)RA;
RA и RB в каждом случае независимо представляют собой Н или выбраны из группы, состоящей из (C1-C4)алкила-, арила, (C1-C6) галогеналкила, или
RA и RB вместе с атомом азота, к которому они присоединены, могут образовывать 6-членную насыщенную гетероциклическую моноциклическую кольцевую систему, необязательно содержащую дополнительный гетероатом, представляющий собой атом азота или кислорода, которая может быть необязательно замещенной (C1-C4)алкилом- и оксо;
RC представляет собой Н или выбран из группы, состоящей из (C1-C6)алкила, (RARB)N-, арил-(C1-C4)алкила,
Y выбран из группы, состоящей из -ORD, RCSO2-, галогена, -NHSO2RC, гетероарила, (C3-C8)гетероциклоалкила, где любой из указанных гетероарила и гетероциклоалкила может быть необязательно замещенным одной или несколькими группами, выбранными из (C1-C3)алкила, -C(O)N(RARB);
J представляет собой Н или выбран из группы, состоящей из (C1-C6)алкила, -ORC;
RD представляет собой Н или (C1-C6)алкил.
В еще одном предпочтительном варианте осуществления настоящее изобретение относится к соединению формулы (I), которая определена выше,
где
Z выбран из группы, состоящей из гетероарила и арила, где любой из указанных гетероарила и арила может быть необязательно замещенным одной или несколькими группами, выбранными из (C1-C3)алкила, галогена, CN, (RARB)NC(O)-, (C1-C6)галогеналкила, RAO-, (RARB)N(C1-C6)алкилена-, (C3-C7)циклоалкила-, RCSO2-, (RARB)N-;
R1 представляет собой H;
R2 выбран из группы, состоящей из (C1-C6)алкила, гетероарил(C1-C4)алкила-, (C3-C8)гетероциклоалкил-(C1-C6)алкила, гетероарил-(C1-C6)гидроксиалкила, арил-(C1-C4)алкила-, (C3-C8)гетероциклоалкила, (C3-C8)циклоалкил-(C1-C6)алкила-, (RARB)N(C1-C6)алкилен-, RAO(C1-C4)алкилена,
где любой из указанных алкила, алкилена, арила, гетероарила, циклоалкила и гетероциклоалкила может быть необязательно замещенным одной или несколькими группами, выбранными из (C1-C3)алкила, RAO(C1-C4)алкилена-, (C1-C6)галогеналкила, оксо, RAO-, (C3-C8)гетероциклоалкил-(C1-C6)алкила, гетероарила, арила, необязательно замещенного галогеном, RCO-, (RARB)N-, -NHC(O)RC, -C(O)N(RARB), галогена -SO2N(RARB), -O(RAO(C1-C4)алкилен-N(RARB), арил-(C1-C4)алкила-, -C(O)RA,
RA и RB в каждом случае независимо представляют собой Н или выбраны из группы, состоящей из (C1-C4)алкила-, арила, (C1-C6)галогеналкила, или
RA и RB вместе с атомом азота, к которому они присоединены, могут образовывать 6-членную насыщенную гетероциклическую моноциклическую кольцевую систему, необязательно содержащую дополнительный гетероатом, представляющий собой атом азота или кислорода, которая может быть необязательно замещенной (C1-C4)алкилом- и оксогруппой;
RC представляет собой Н или выбран из группы, состоящей из (C1-C6)алкила, (RARB)N-, арил-(C1-C4)алкила-;
Y выбран из группы, состоящей из -ORD, RCSO2, галогена, -NHSO2RC, гетероарила, (C3-C8)гетероциклоалкила, где любой из указанных гетероарила и гетероциклоалкила может быть необязательно замещенным одной или несколькими группами, выбранными из (C1-C3)алкила, -C(O)N(RARB);
J представляет собой Н или выбран из группы, состоящей из (C1-C6)алкила, ORC;
RD представляет собой Н или (C1-C6)алкил.
В дополнительном предпочтительном варианте осуществления настоящее изобретение относится к соединению формулы (I), где
Z представляет собой Н или выбран из группы, состоящей из
(RARB)N-,
гетероарила, предпочтительно тиадиазолила, тиазолила, пиразолила, пиридазила, оксадиазолила, пиридинила, пиримидинила,
арила, предпочтительно фенила,
RAO-, где RA представляет собой H,
где каждый из указанных гетероарила и арила может быть необязательно замещенным одной или несколькими группами, выбранными из
метила,
галогена, предпочтительно фтора и хлора,
CN,
(RARB)NC(O)-, где RA и RB в каждом случае независимо представляют собой H или метил,
(C1-C6)галогеналкила, предпочтительно трифторметила и дифторметила,
RAO-, где RA представляет собой Н или выбран из метила, трифторметила и дифторметила,
(RARB)N(C1-C6)алкилена-, где RA и RB представляют собой метил,
циклопропила,
RCSO2-, где RC представляет собой метил,
(RARB)N-, где RA и RB независимо представляют собой H и метил;
R1 представляет собой H;
R2 выбран из группы, состоящей из
гетероарил(C1-C4)алкила-, предпочтительно ([1,2,4]триазоло[4,3-a]пиримидин-3-ил)метила, (триазолил)метила, (триазолил)этила, (имидазо[1,2-a]пиримидинил)метила, (пиримидинил)этила, (пиримидинил)метила, (пиразолил)метила, (пиридазинил)метила, (пиридазинил)этила (оксадиазолил)метила, (оксадиазолил)пропила, (пиридинил)метила, (пиридинил)этила, (оксадиазолил)этила,
(C3-C8)гетероциклоалкил-(C1-C6)алкила, предпочтительно (пиперидинил)метила, (тетразолил)метила, (морфолинил)этила,
гетероарил(C1-C6)гидроксиалкила-, предпочтительно (оксадиазолил)метанола,
(C3-C8)циклоалкил(C1-C6)алкила-, предпочтительно (циклопропил)метила,
арил-(C1-C4)алкила-, предпочтительно (фенил)метила,
(RARB)N(C1-C6)алкилена-, предпочтительно диметиламинобутила, диметиламинопропила,
где каждый из указанных арила, гетероарила, циклоалкила и гетероциклоалкила является необязательно дополнительно замещенным одной или несколькими группами, выбранными из
(C1-C3)алкила, предпочтительно метила и этила,
трифторметила,
оксо,
хлора,
RAO-, где RA выбран из группы, состоящей из трифторэтила, дифторэтила, метила и этила,
(C3-C8)гетероциклоалкил-(C1-C6)алкила-, предпочтительно (пиперидинил)метила,
(C3-C8)гетероциклоалкила, предпочтительно пиперазинила, необязательно дополнительно замещенного метилом,
гетероарила, предпочтительно пиридинила,
-NHC(O)RC, где RC представляет собой метил,
(RARB)N-, где RA и RB представляют собой метил,
RCO-, где RC представляет собой метил,
-C(O)N(RARB), где RA представляет собой H и RB представляет собой метил;
Y выбран из группы, состоящей из -ORD, RCSO2, галогена, -NHSO2RC, гетероарила, где любой такой гетероарил может быть необязательно замещенным одной или несколькими группами, выбранными из (C1-C3)алкила, -C(O)N(RARB);
RD представляет собой (C1-C6)алкил, предпочтительно метил;
J выбран из группы, состоящей из
(C1-C6)алкила, предпочтительно метила,
-ORC, где RC представляет собой Н или (C1-C6)алкил, предпочтительно метил.
В соответствии со всеми описанными выше конкретными вариантами осуществления настоящее изобретение относится по меньшей мере к одному соединению из представленных в таблице 2 ниже и их фармацевтически приемлемым солям.
Таблица 2. Перечень предпочтительных соединений формулы (I)

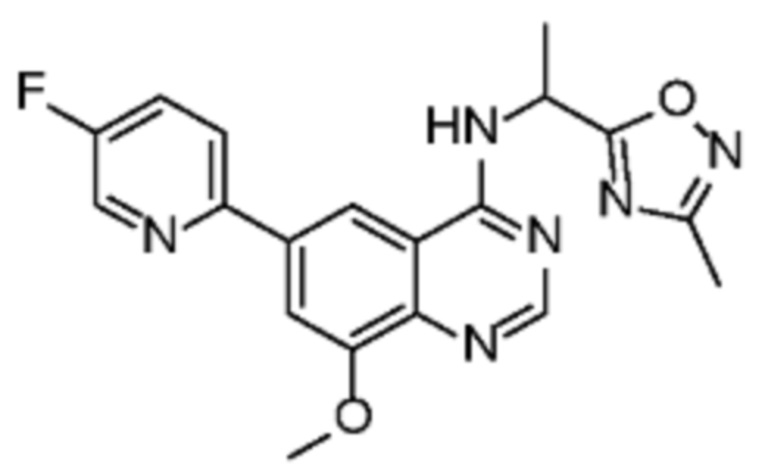

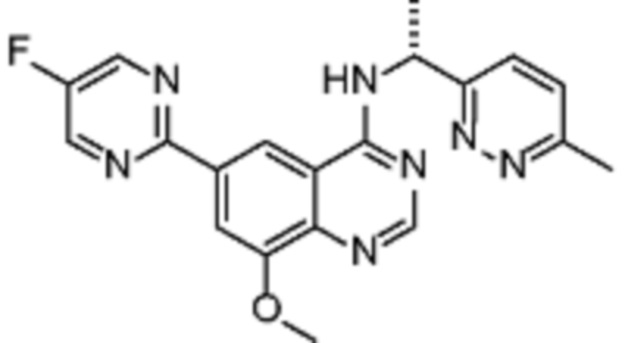


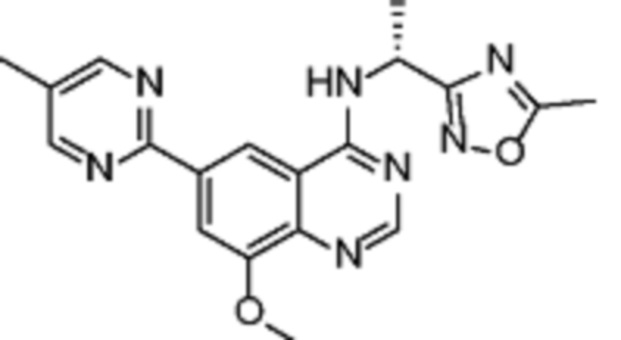
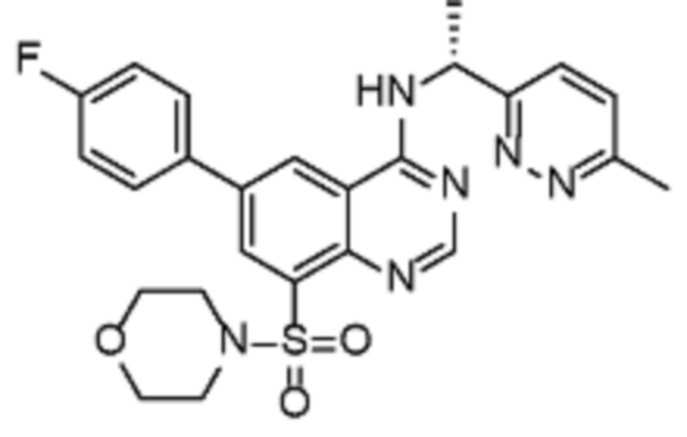
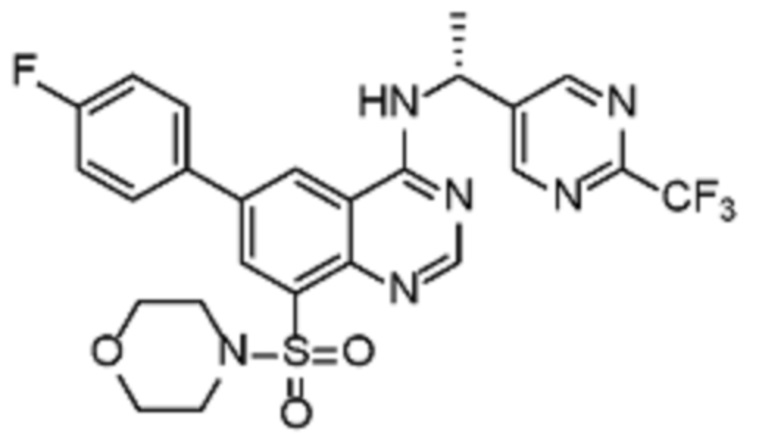
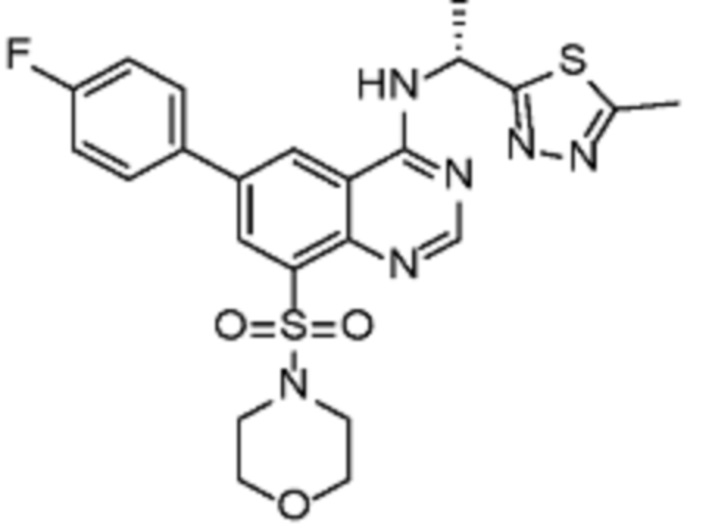

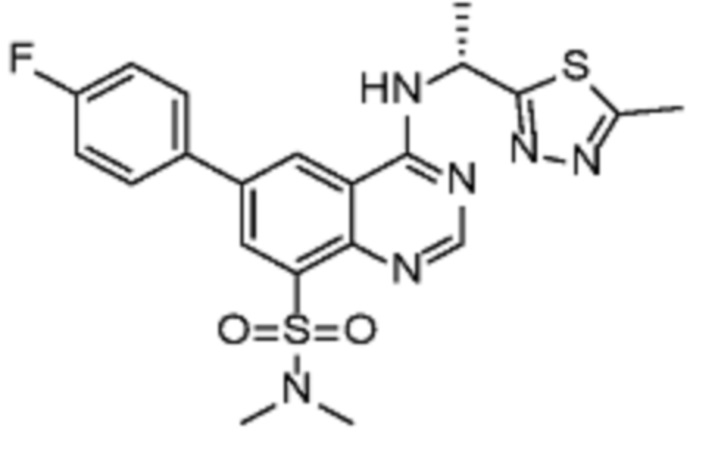



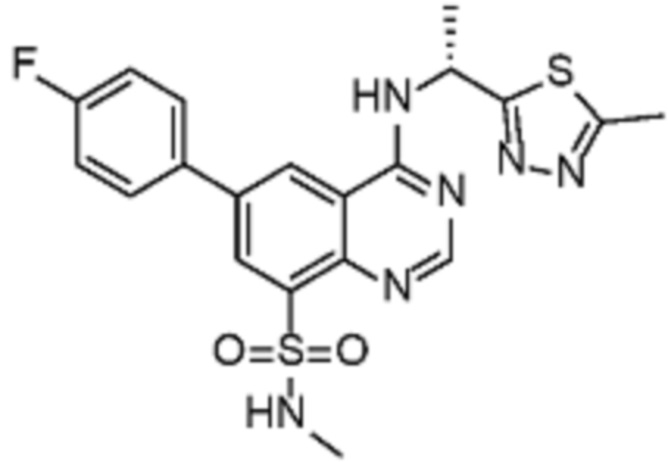
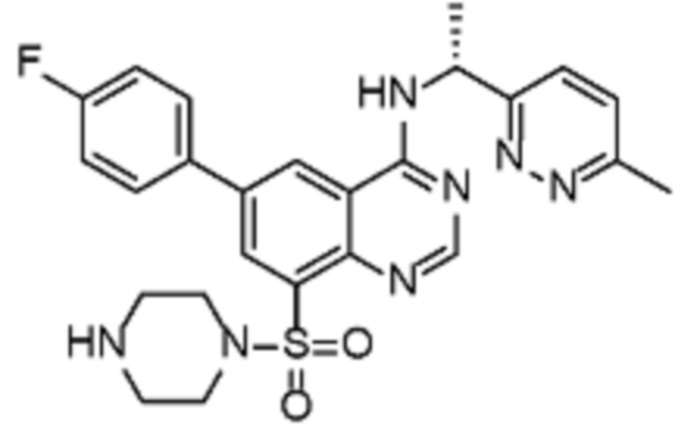
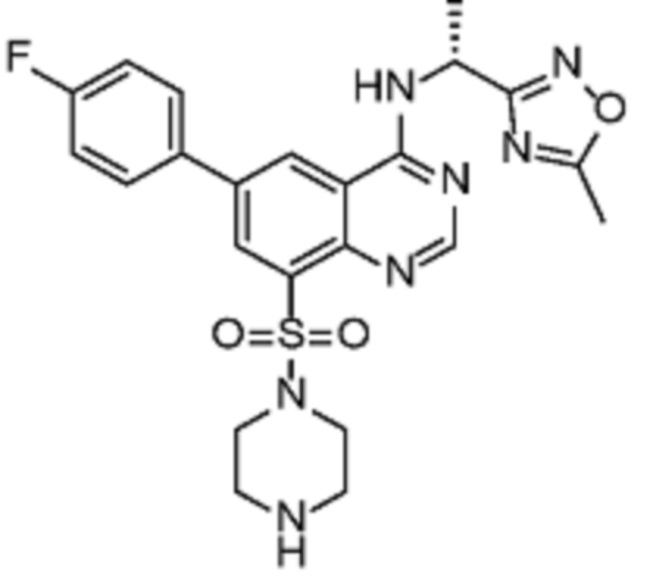

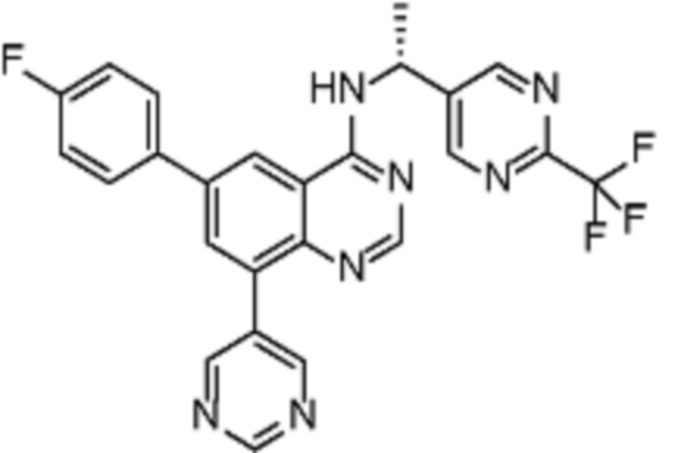
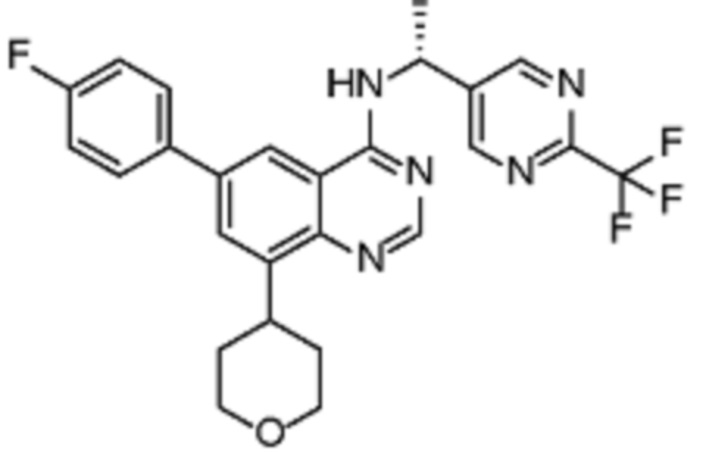
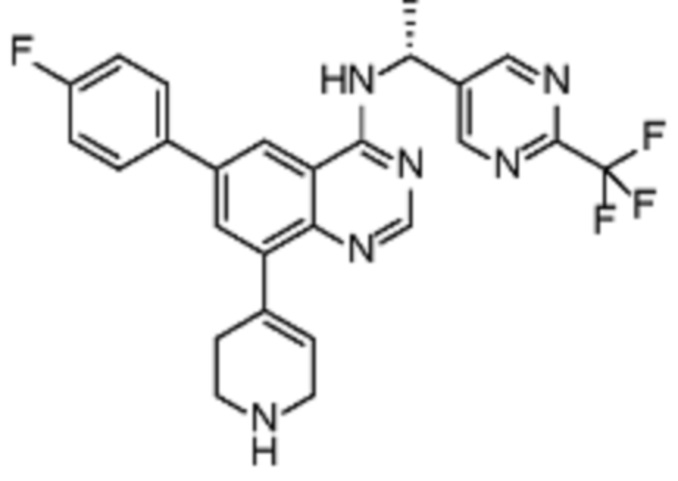
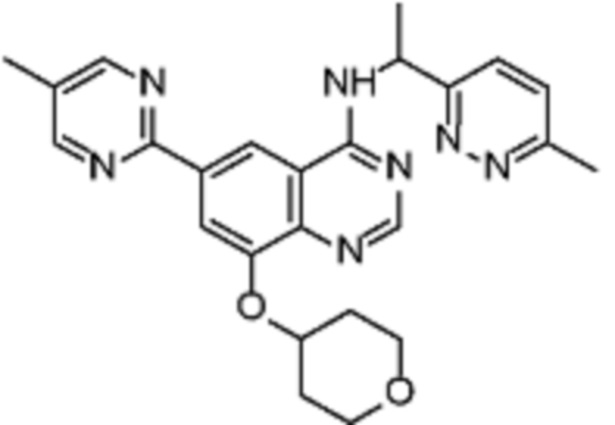


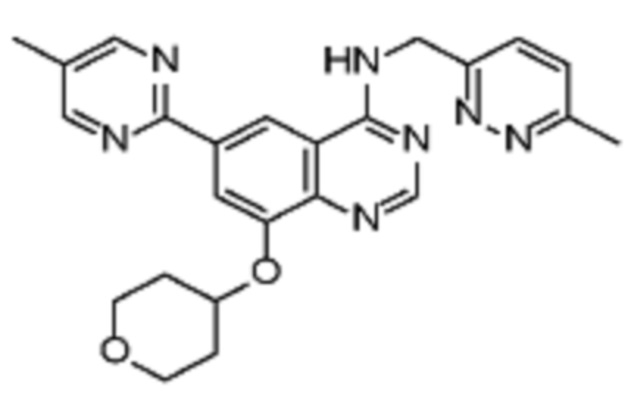
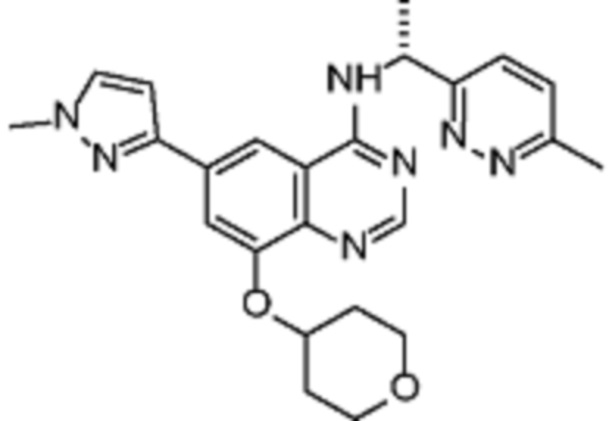
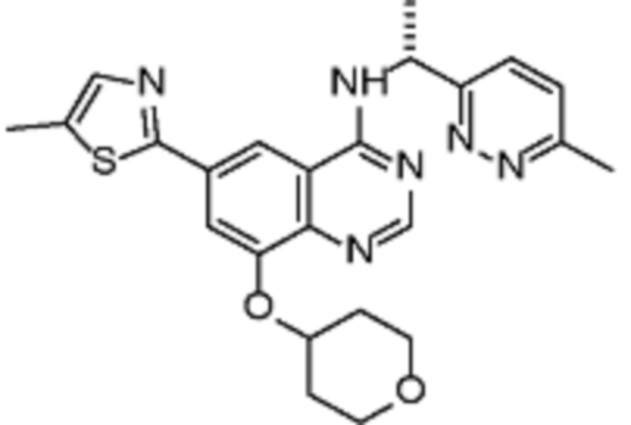

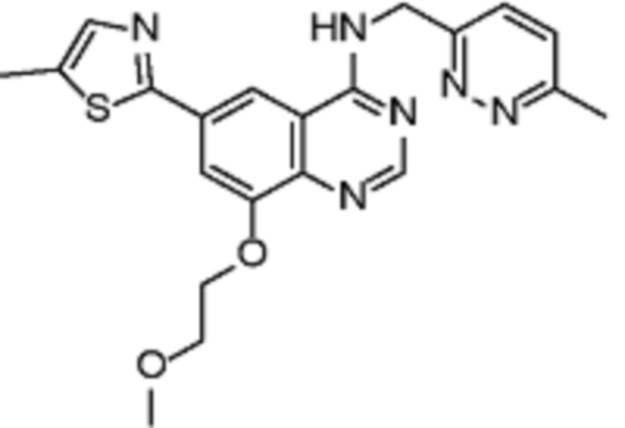

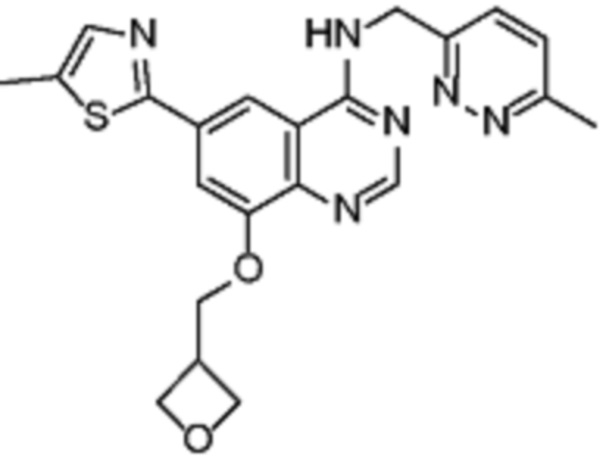
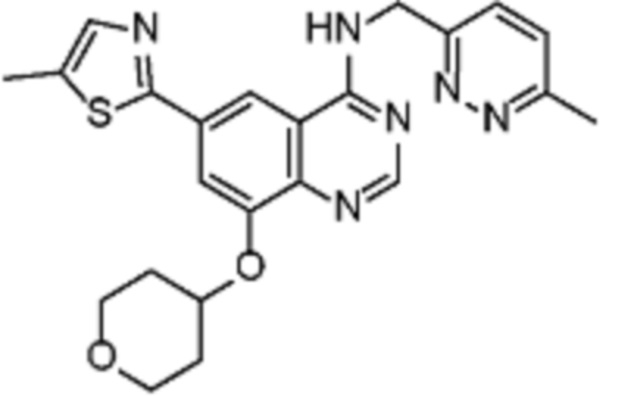
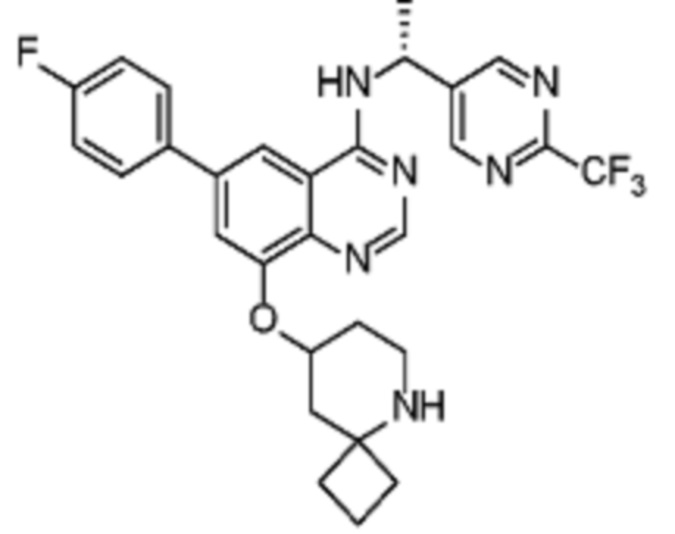


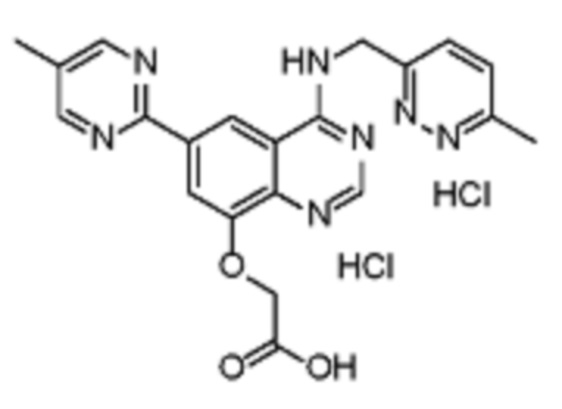
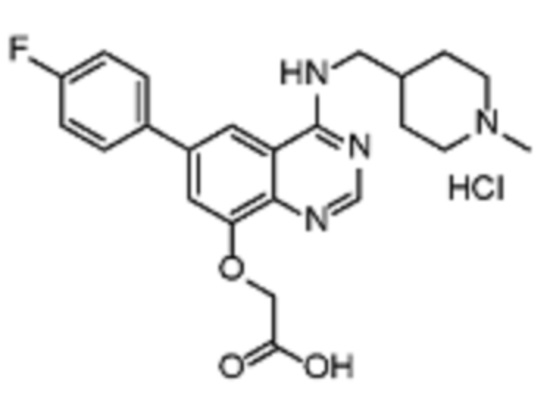
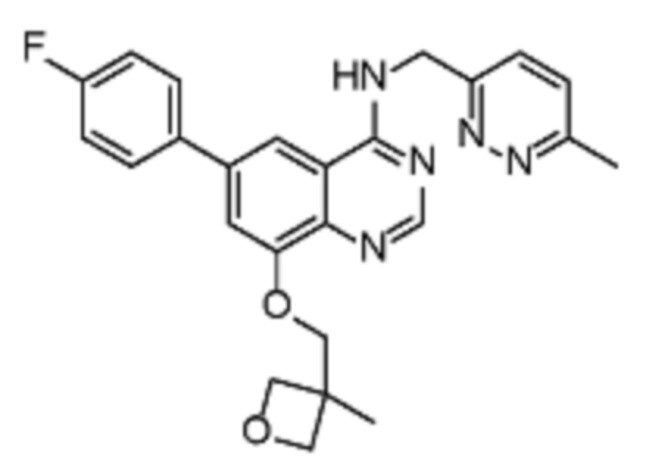
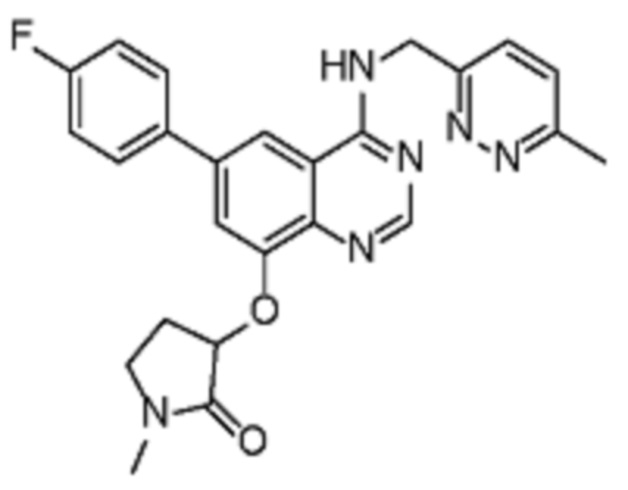
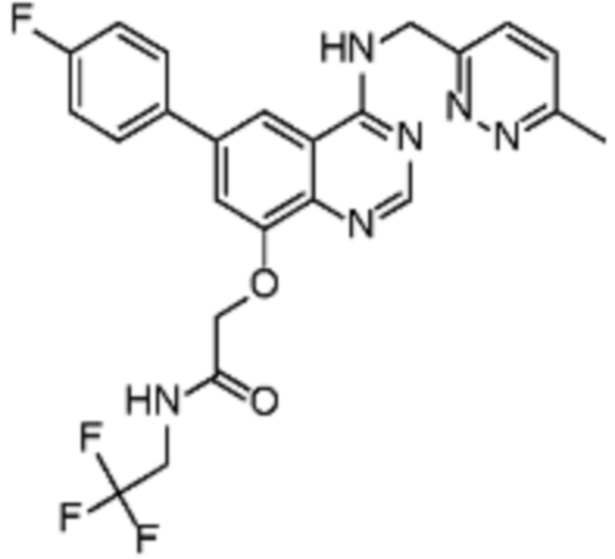
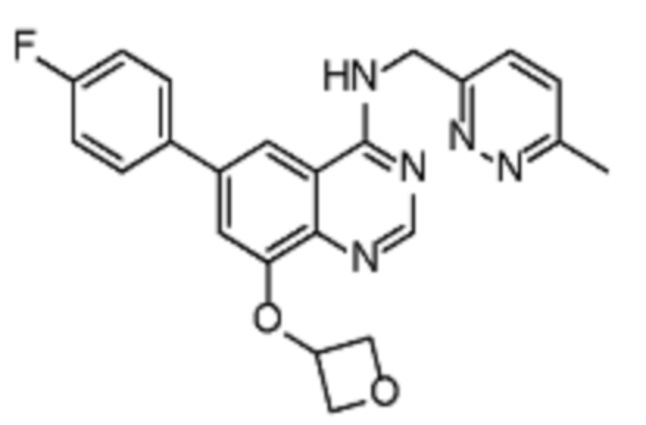



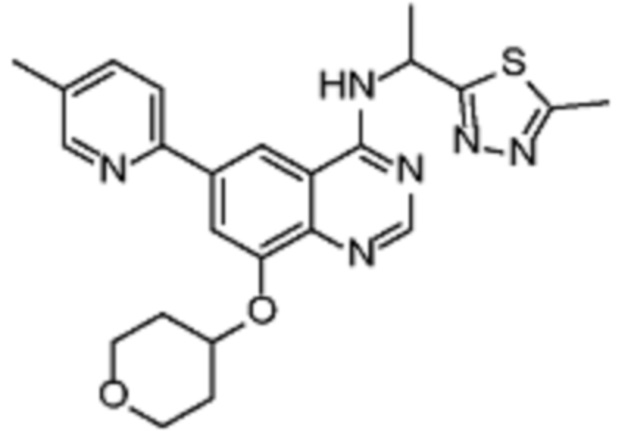
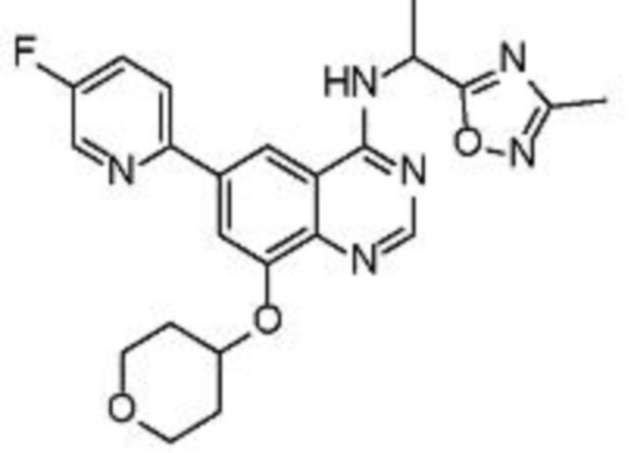

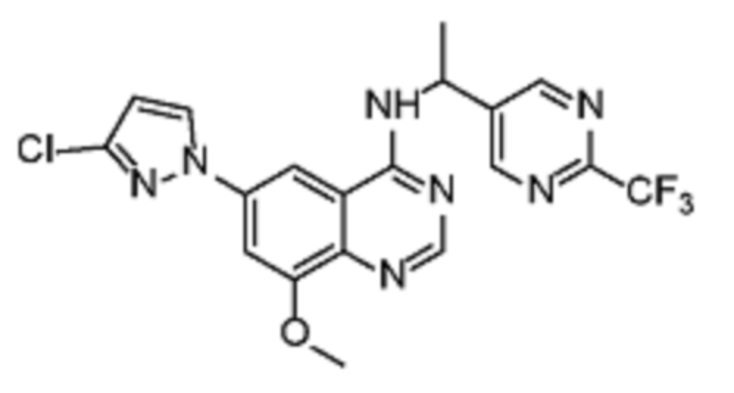







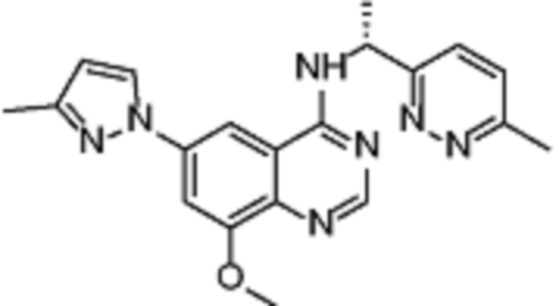
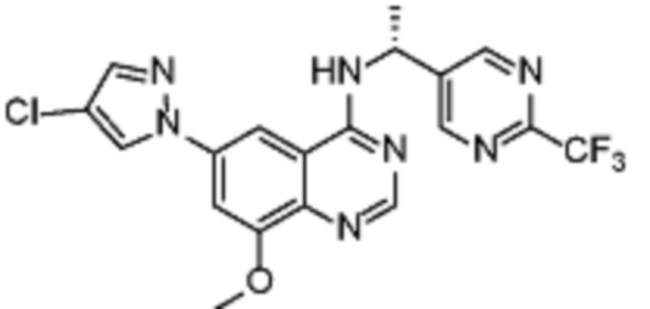


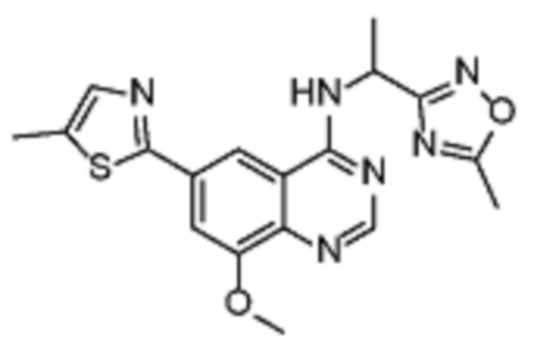




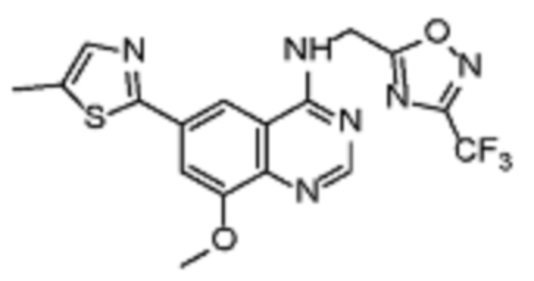
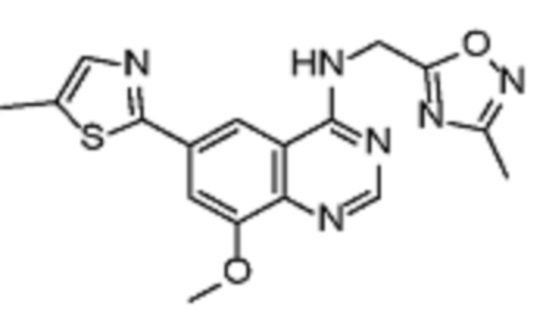


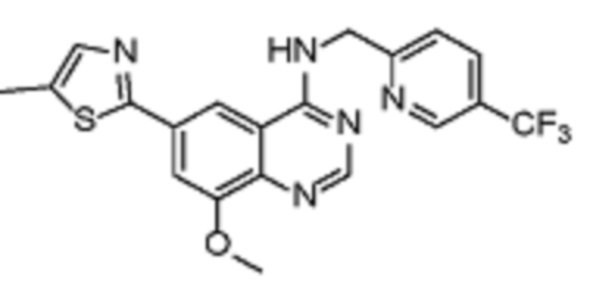
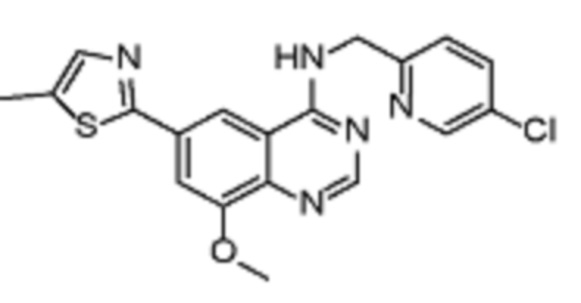

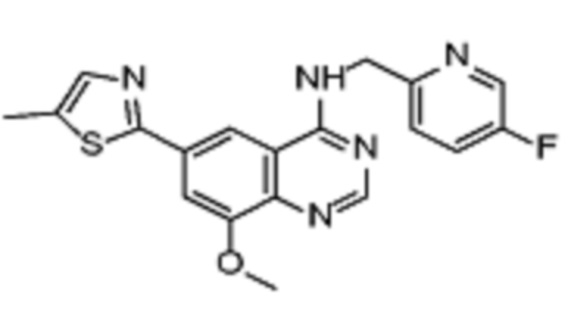

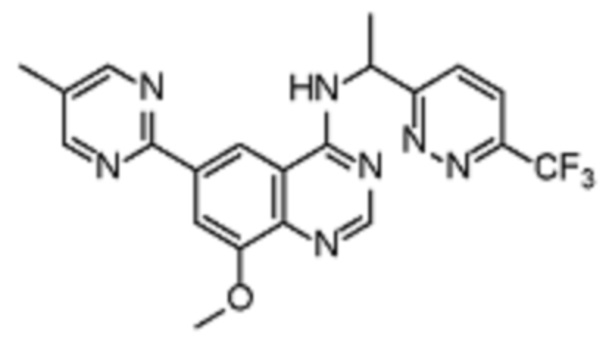



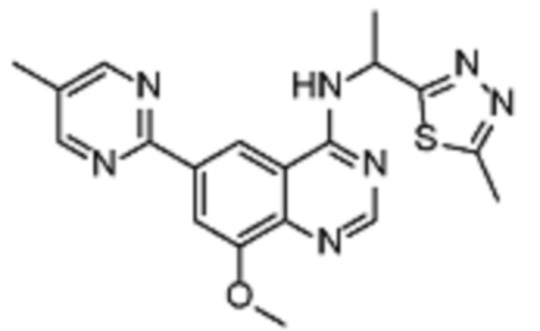
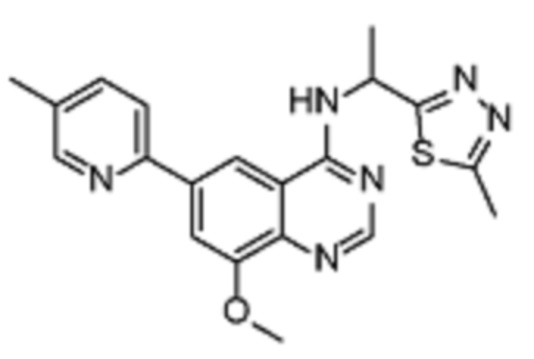



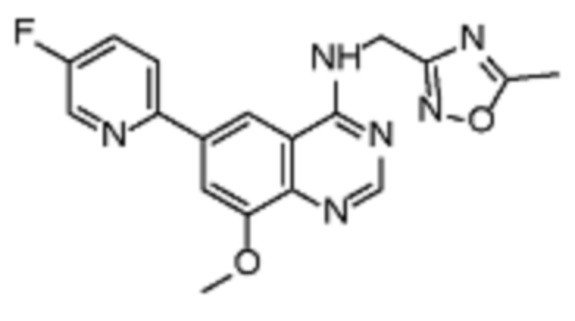
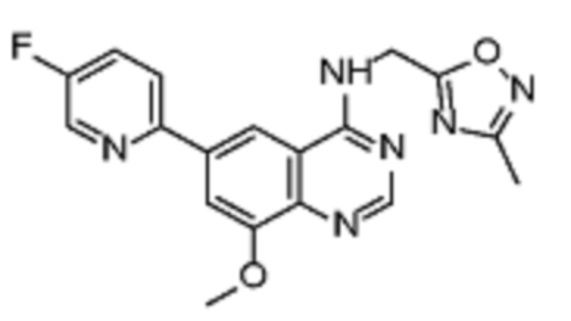
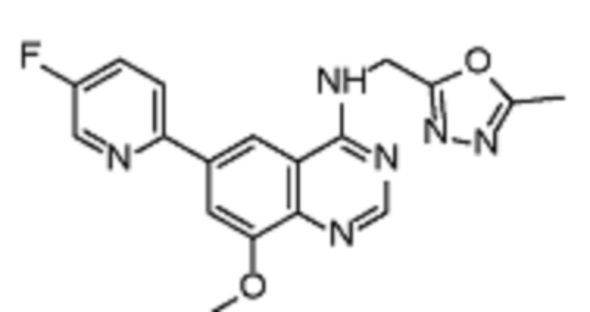
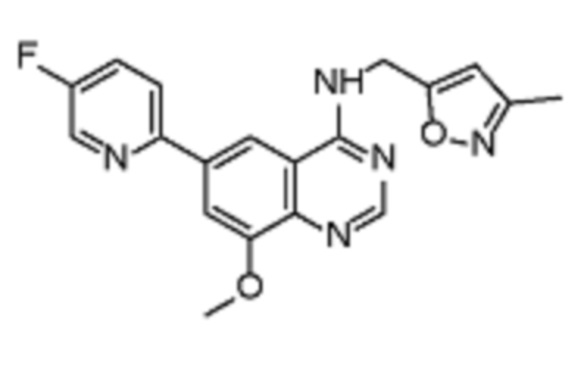
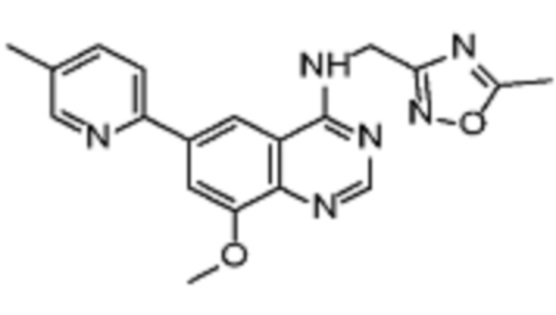



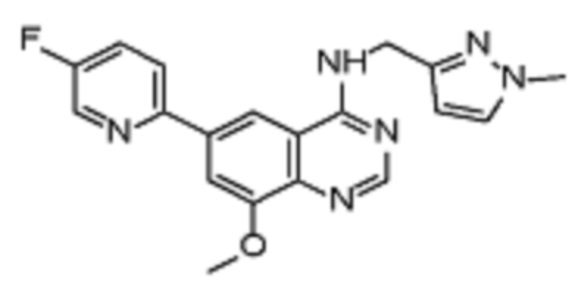





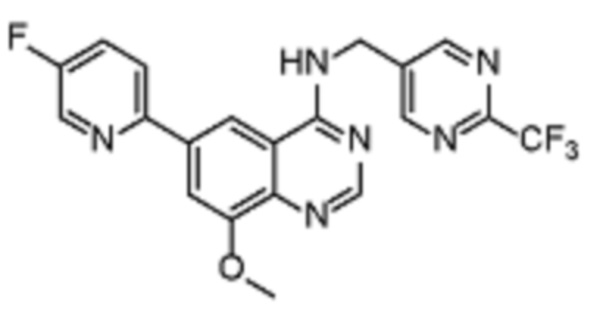
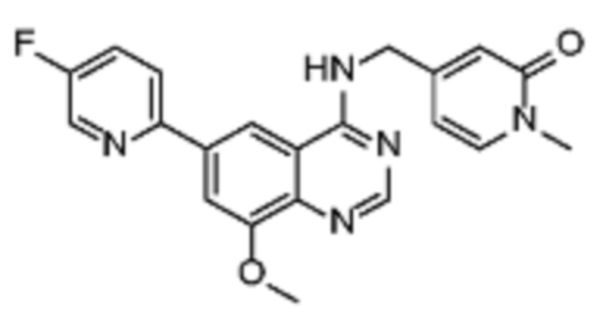
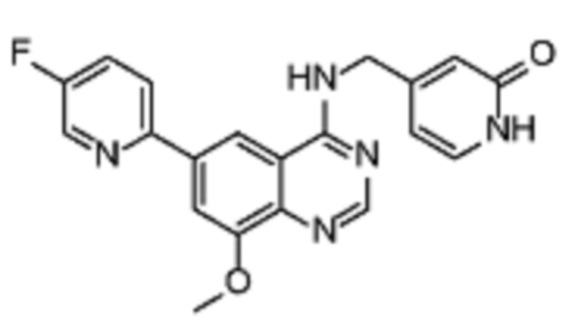
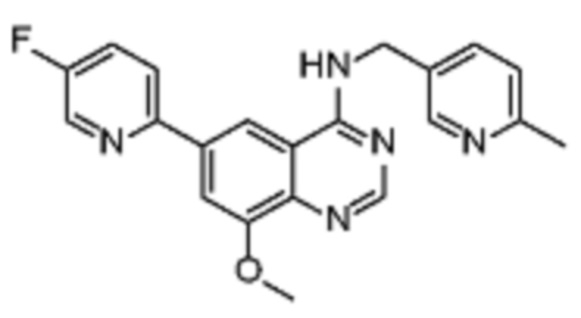


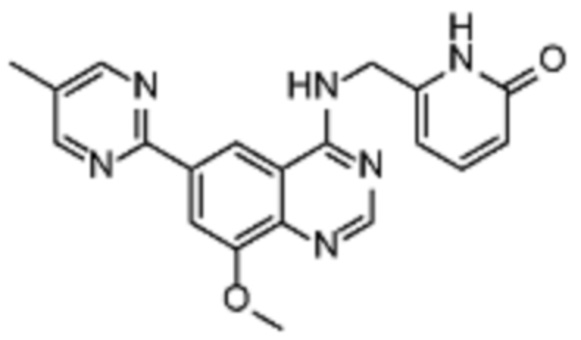

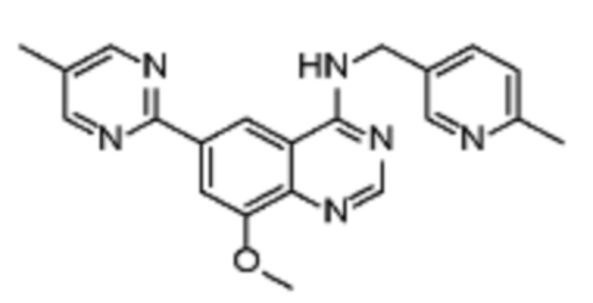
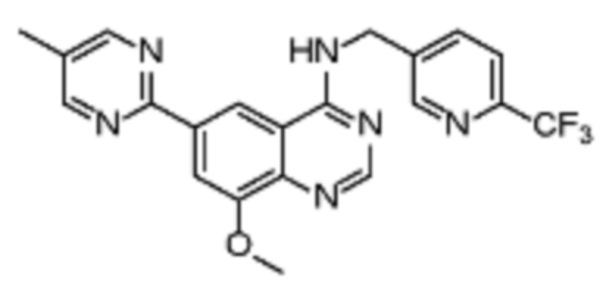









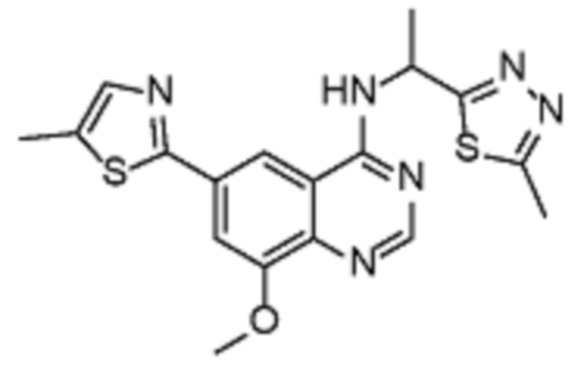

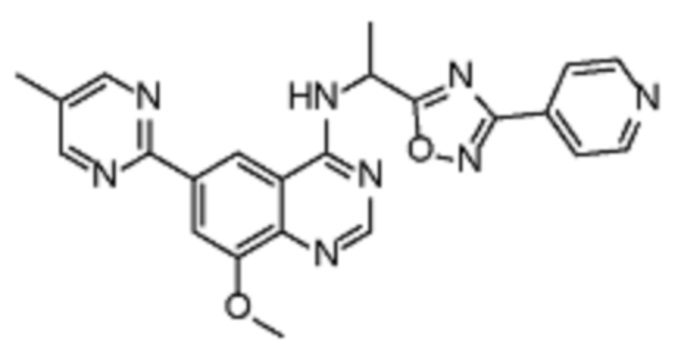
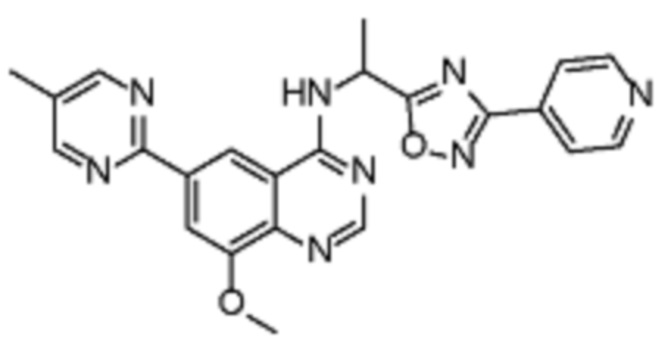


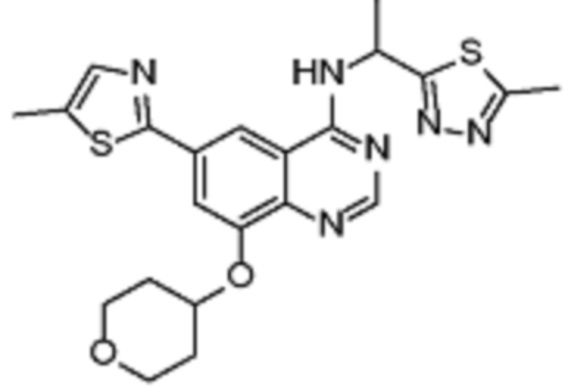
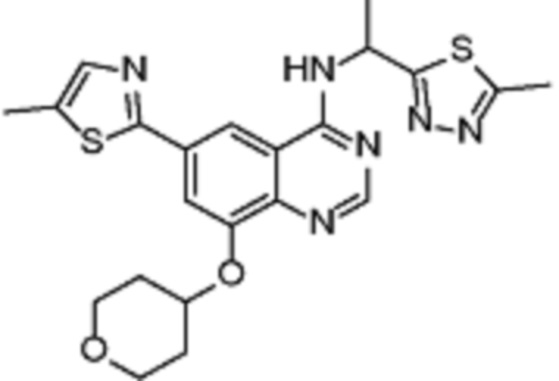


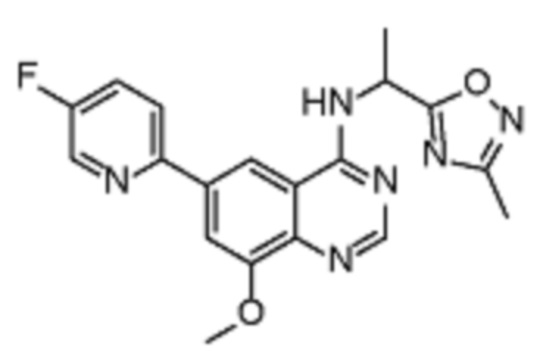

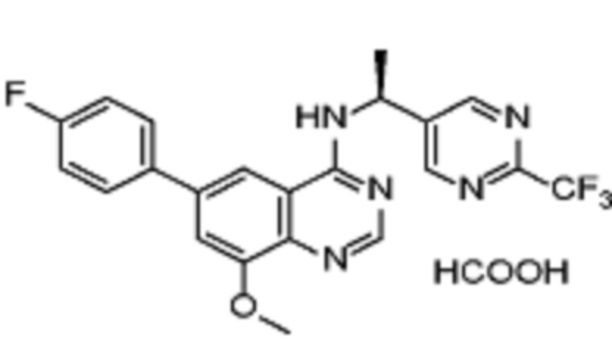
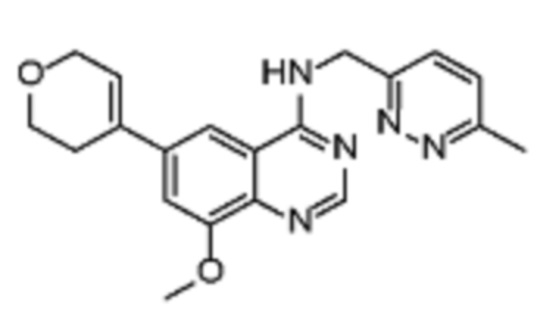

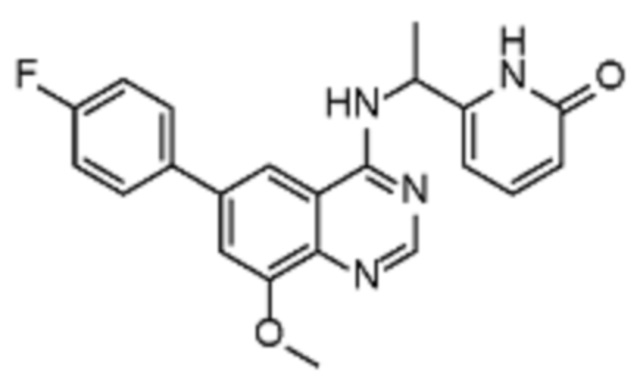

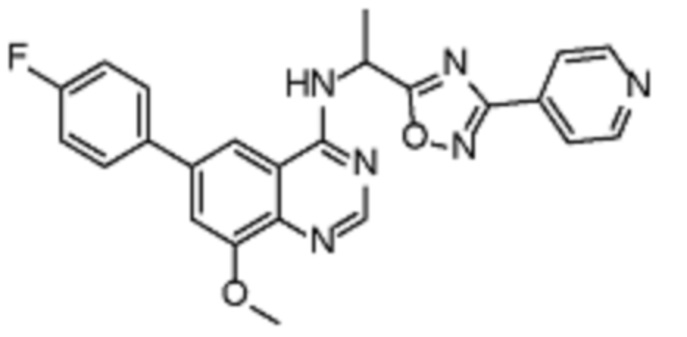
В дополнительном предпочтительном варианте осуществления настоящее изобретение относится к соединению формулы (I), где
Z представляет собой арил, где любой указанный арил может быть необязательно замещенным одной или несколькими группами, выбранными из (C1-C3)алкила, галогена, CN;
R1 представляет собой H;
R2 выбран из группы, состоящей из гетероарил(C1-C4)алкила-, (C3-C8)гетероциклоалкил-(C1-C6)алкила, (C3-C8)гетероциклоалкила, (C3-C8)циклоалкил-(C1-C6)алкила-, где любой из указанных алкила и гетероарила может быть необязательно замещенным одной или несколькими группами, выбранными из (C1-C3)алкила, (C1-C6)галогеналкила, оксогруппы, RAO-, арила, (RARB)N- и галогена;
RA и RB в каждом случае независимо представляют собой Н или выбраны из группы, состоящей из (C1-C4)алкила-, (C1-C6) галогеналкила;
Y выбран из группы, состоящей из -ORD, RCSO2, галогена, -NHSO2RC, гетероарила, гетероциклоалкила, где любой указанный гетероарил может быть необязательно замещенным одной или несколькими группами, выбранными из (C1-C3)алкила, -C(O)N(RARB);
J представляет собой Н или выбран из группы, состоящей из ORC;
RC представляет собой H или выбран из группы, состоящей из (C1-C6)алкила, (RARB)N-;
RD представляет собой Н или (C1-C6)алкил.
В соответствии с предпочтительным вариантом осуществления настоящее изобретение относится по меньшей мере к одному соединению, выбранному из представленных в таблице 3.
Таблица 3. Перечень предпочтительных соединений формулы (I)
В дополнительном предпочтительном варианте осуществления настоящее изобретение относится к соединению формулы (I), где
Z представляет собой гетероарил, где любой указанный гетероарил может быть необязательно замещенным одной или несколькими группами, выбранными из (C1-C3)алкила, галогена, CN, (C1-C6)галогеналкила;
R1 представляет собой H;
R2 выбран из группы, состоящей из гетероарил(C1-C4)алкила-, где любой из указанных алкила и гетероарила может быть необязательно замещенным одной или несколькими группами, выбранными из (C1-C3)алкила, (C1-C6)галогеналкила и -оксогруппы;
Y представляет собой -ORD;
J представляет собой H;
RD представляет собой (C1-C6)алкил.
В соответствии с предпочтительным вариантом осуществления настоящее изобретение относится по меньшей мере к одному соединению, выбранному из соединений, представленных в таблице 4.
Таблица 4. Перечень предпочтительных соединений формулы (I)
В дополнительном предпочтительном варианте осуществления настоящее изобретение относится к соединению формулы (I), определенной выше, где Y представляет собой -ORD, которое представлено формулой (Ia)

где
Z выбран из группы, состоящей из арила, где любой указанный арил может быть необязательно замещенным одной или несколькими группами, выбранными из галогенов;
R1 представляет собой H;
R2 выбран из группы, состоящей из гетероарил(C1-C4)алкила-, где любой указанный гетероарил может быть необязательно замещенным одной или несколькими группами, выбранными из (C1-C3)алкила, (C1-C6)галогеналкила;
RA и RB в каждом случае независимо представляют собой Н или выбраны из группы, состоящей из (C1-C4)алкила-;
RC в каждом случае представляет собой Н или выбран из группы, состоящей из (C1-C6)алкила;
RD выбран из группы, состоящей из H, (C1-C6)алкила, (C3-C8)гетероциклоалкил-(C1-C6)алкила-, RCOC(O)(C1-C4)алкилена-, (RARB)N(C1-C6)алкилена-, (C3-C8)гетероциклоалкила, RCO(C1-C4)алкилена-, (RARB)N(O)C(C1-C4)алкилена-, (C3-C8)циклоалкил-(C1-C6)алкила-, где любой указанный гетероциклоалкил может быть необязательно замещенным одной или несколькими группами, выбранными из (C1-C3)алкила;
J представляет собой H.
В дополнительном предпочтительном варианте осуществления настоящее изобретение относится к соединению формулы (Ia), где
Z представляет собой Н или выбран из группы, состоящей из
арила, предпочтительно фенила,
где каждый указанный арил может быть необязательно замещенным одной или несколькими группами, выбранными из
галогена, предпочтительно фтора;
R1 представляет собой H;
R2 выбран из группы, состоящей из
гетероарил(C1-C4)алкила-, предпочтительно (пиримидинил)этила, (пиридазинил)метила,
где каждый указанный гетероарил является необязательно дополнительно замещенным одной или несколькими группами, выбранными из
(C1-C3)алкила, предпочтительно метила,
трифторметила;
RD представляет собой Н или выбран из группы, состоящей из
(C1-C6)алкила, предпочтительно метила, пропила,
(C3-C8)гетероциклоалкил-(C1-C6)алкила-, предпочтительно (азетидинил)метила, (морфолинил)метила, (морфолинил)этила, (оксетанил)метила,
RCOC(O)(C1-C4)алкилена-, где RC выбран из группы, состоящей из H и этила,
(RARB)N(C1-C6)алкилена-, предпочтительно диметиламинопропила,
(C3-C8)гетероциклоалкила, предпочтительно тетрагидропиранила,
RCO(C1-C4)алкилена-, предпочтительно выбранного из группы, состоящей из метоксиэтила, пропанолила,
(RARB)N(O)C(C1-C4)алкилена-, предпочтительно диметилацетиламида, тетрагидрофуранила,
(C3-C8)циклоалкил-(C1-C6)алкила-, предпочтительно (циклопропил)метила,
где каждый указанный гетероциклоалкил может быть необязательно замещенным одной или несколькими группами, выбранными из метила, этила и пропила;
J представляет собой H.
В соответствии с конкретными вариантами осуществления настоящее изобретение относится по меньшей мере к одному соединению из представленных в таблице 5 ниже и их фармацевтически приемлемым солям
Таблица 5. Перечень предпочтительных соединений формулы (Ia)
Соединения формулы (I), включающие все представленные выше соединения или по меньшей мере одно из представленных выше соединений, обычно могут быть получены в соответствии с методиками, подробно описанными на схемах ниже, используя известные способы.
Схема 1

В одном варианте осуществления настоящего изобретения соеднение (IA) может быть получено в соответствии со схемой 1 из соединения (II). Соединение (II) было получено в соответствии с методикой, описанной в J.Med Chem., 2015, 58(8), 3548-3571.
Соединение (III) может быть получено из соединения (II) реакцией деоксиаминирования, которая опосредуется реагентами связывания, такими как PyBOP, с подходящим амином (реаг. 1).
Соединение (IA) может быть получено из соединения (III) реакцией кросс-сочетания с использованием металлического катализатора, такой как реакция Стилле (Stille) или реакция Сузуки (Suzuki), как описано в “Transition Metals for Organic Synthesis”, 2nd Ed, 1, 2004, с подходящим реагентом, таким как реагент 2 (реаг. 2).
Альтернативно, соединение (V) может быть получено из соединения (III) реакцией борилирования Мияуры (Miyaura) c использованием металлического катализатора.
Соединение (IA) может быть получено из соединения (V) реакцией кросс-сочетания с использованием металлического катализатора, такой как реакция Стилле или реакция Сузуки или аналогично, как описано в “Transition Metals for Organic Synthesis”, 2nd Ed, 1, 2004, с подходящим галогенорганическим соединением, таким как реагент 3 (реаг. 3).
В другом варианте осуществления соединение (IV) может быть получено исходя из соединения (II) реакцией кросс-сочетания с использованием металлического катализатора, такой как реакция Стилле или реакция Сузуки или аналогично, как описано в “Transition Metals for Organic Synthesis”, 2nd Ed, 1, 2004, с подходящим металлоорганическим реагентом, таким как реагент 2 (реаг. 2).
Соединение (IA) может быть получено из соединения (V) реакцией деоксиаминирования, которая опосредуется реагентами связывания, такими как PyBOP, с подходящим амином (реаг. 1).
Некоторые соединения (IA) могут содержать защитные группы гидроксильной или аминогруппы, которые затем удаляют в соответствии с известными методиками.
Схема 2

В другом вариате осуществления настоящего изобретения соединение (IB) может быть получено в соответствии со схемой 2 из соединений (VI).
Соединение (VII) может быть получено из соединения (VI) посредством реакции образования хиназолинового кольца, опосредуемой подходящими реагентами, такими как триэтилортоацетат или т.п.
Соединение (VIII) может быть получено из соединения (VII) реакцией кросс-сочетания с использованием металлического катализатора, такой как реакция Стилле или реакция Сузуки, или аналогичными реакциями с подходящими металлоорганическими реагентами (реаг.2), например борорганическими соединениями.
Соединения (IB) могут быть получены из соединения (VIII) реакцией деоксиаминирования, которая опосредуется реагентами связывания, такими как PyBOP, с подходящим амином (реаг. 1).
Некоторые соединения (IB) могут содержать защитные группы гидроксильной группы или аминогруппы, которые затем удаляются в соответствии с хорошо известными методиками.
Схема 3

В еще одном варианте осуществления настоящего изобретения соединение (IC) может быть получено из соединения (IX) в соответствии со схемой 3.
Соединение (X) может быть получено из соединения (IX) реакцией кросс-сочетания с использованием металлического катализатора, такой как реакция Стилле или реакция Сузуки или аналогично, с подходящими металлоорганическими реагентами (рeag.2), такими как, например, борорганические соединения.
Соединение (XI) может быть получено из соединения (X) галогенированием подходящими реагентами, такими как бром, NBS, NIS, йод, соли йодония или т.п.
Соединение (XII) может быть получено из соединения (XI) гидролизом в основной или кислотной среде.
Соединение (XIII) может быть получено из соединения (XII) реакцией образования хиназолинового кольца, которая опосредуется подходящими реагентами, такими как формамид или т.п.
Соединение (XIV) может быть получено из соединения (XIII) реакцией деоксиаминирования, которая опосредуется реагентами связывания, такими как PyBOP, в присутствии подходящего амина (реаг. 1).
Соединение (IC) может быть получено из соединения (XIV) реакцией кросс-сочетания с использованием металлического катализатора, такой как реакция Стилле или реакция Сузуки, или аналогичной с подходящими металлоорганическими реагентами (реаг. 6), такими как, например, борорганические соединения. Некоторые соединения (IC) могут содержать защитные группы гидроксильной группы или аминогруппы, которые затем удаляются в соответствии с хорошо известными методиками.
Соединение (IK) может быть получено из соединения (XIV) реакциями аминирования в присутствии подходящего реагента, такого как, например, метансульфонамид. Некоторые соединения (IK) могут содержать защитные группы гидроксильной группы или аминогруппы, которые затем удаляются в соответствии с хорошо известными методиками.
Схема 4

В еще одном варианте осуществления настоящего изобретения соединения (ID) могут быть получены в соответствии со схемой 4 из соединений (IA).
Соединение (XV) может быть получено из соединения (IA) реакцией деалкилирования, которая опосредуется сильными кислотами Льюиса, такими как BBr3 или т.п.
Соединения (ID) были получены из соединений (XV) алкилированием подходящими алкилирующими агентами (реаг. 4), такими как алкилхлориды, -бромиды, -йодиды, -мезилаты, -тозилаты или т.п.
Альтернативно, соединения (ID) могут быть получены из соединений (XV) и подходящего спирта реакцией Митсунобу или аналогичными, опосредуемыми, например, DEAD/PPh3, DIAD/PPh3 или CMT.
Некоторые соединения (ID) могут содержать защитные группы гидроксильной или аминогруппы, которые затем удаляют в соответствии с известными методиками.
Схема 5

В еще одном варианте осуществления настоящего изобретения соединение (IE) может быть получено в соответствии со схемой 5 из соединений (XVI).
Соединение (XVII) может быть получено из соединения (XVI) реакцией деоксиаминирования, которая опосредуется реагентами связывания, такими как PyBOP или т.п., в присутствии подходящего амина (реаг. 1).
Соединения (IE) получают из соединений (XVII) реакцией кросс-сочетания с использованием металлического катализатора, такой как реакция Стилле, реакция Сузуки, или аналогичными с подходящими металлоорганическими реагентами (Reag. 2), такими как борорганические соединения.
Некоторые соединения (IE) могут содержать защитные группы гидроксильной группы или аминогруппы, которые затем удаляют в соответствии с хорошо известными методиками.
Схема 6
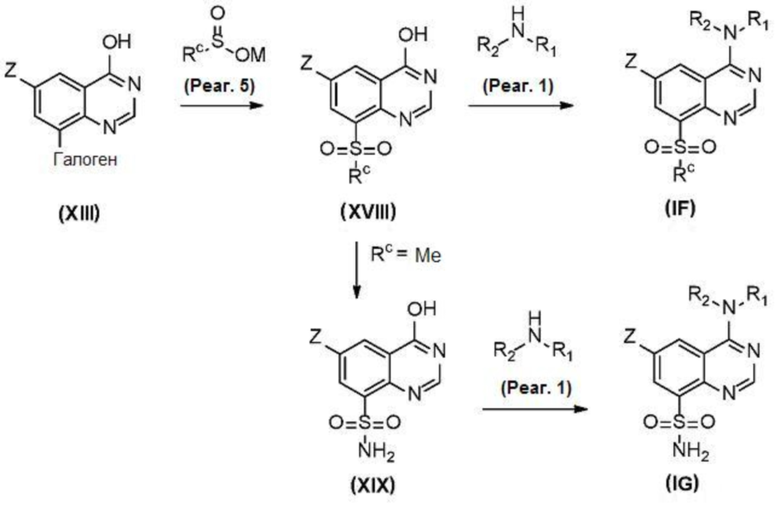
В еще одном варианте осуществления настоящего изобретения соединения (IF) и (IG) могут быть получены в соответствии со схемой 6 из соединений (XIII).
Соединение (XVIII) может быть получено из соединения (XIII) реакцией сульфинирования, катализируемой металлическим катализатором, с подходящим сульфинирующим агентом (реаг. 5), таким как, например, метансульфинат натрия.
Соединение (IF) может быть получено из соединения (XVIII) реакцией деоксиаминирования, которая опосредуется реагентами связывания, такими как PyBOP, в присутствии подходящего амина (реаг. 1). Некоторые соединения (IF) могут содержать защитные группы гидроксильной группы или аминогруппы, которые затем удаляются в соответствии с хорошо известными методиками.
В другом варианте осуществления настоящего изобретения соединение (XIX) может быть получено из соединения (XVIII) аминированием с трибутилборатом и (аминокси)сульфоновой кислотой, как описано в Tetr. Lett. 1994, 39, 7201.
Соединение (IG) может быть получено из соединения (XIX) реакцией деоксиаминирования, которая опосредуется такими реагентами, как PyBOP, в присутствии подходящего амина (реаг. 1).
Некоторые соединения (IG) могут содержать защитные группы гидроксильной группы или аминогруппы, которые затем удаляются в соответствии с хорошо известными методиками.
Схема 7
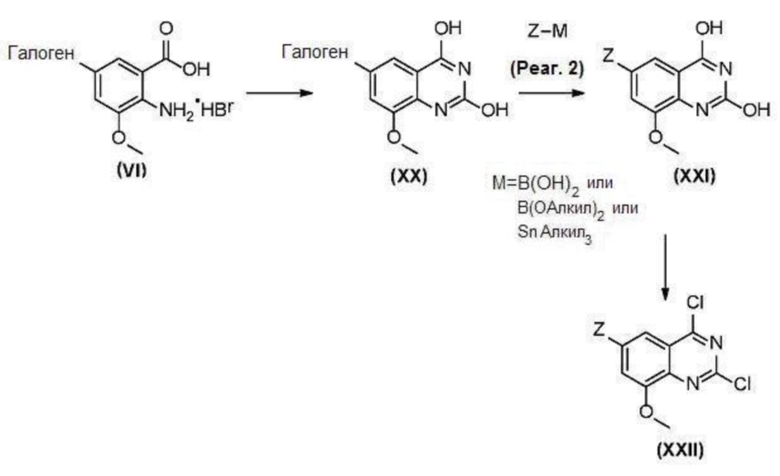

В другом варианте осуществления настоящего изобретения соединения (IH) и (IJ) могут быть получены в соответствии со схемой 7 из соединений (VI).
Соединение (XX) может быть получено из соединения (VI) реакцией образования хиназолинового кольца с использованием подходящих реагентов, таких как мочевина или т.п.
Соединение (XXI) может быть получено из соединения (XX) реакцией кросс-сочетания, катализируемой металлическый катализатором, такой как реакция Стилле или реакция Сузуки или аналогично, с подходящими металлоорганическими реагентами (реаг. 2), такими как, например, борорганические соединения.
Соединение (XXII) может быть получено из соединения (XXI) хлорированием с использованием подходящих реагентлв, таких как оксихлорид фосфора или т.п.
Соединение (XXIII) может быть получено из соединения (XXII) реакцией аминирования в присутствии подходящего амина (реаг. 1).
Соединение (IH) может быть получено из соединения (XXIII) гидролизом с использованием пододящего реагента, такого как, например, уксусная кислота.
Некоторые соединения (IH) могут содержать защитные группы гидроксильной группы или аминогруппы, которые затем удаляются в соответствии с хорошо известными методиками.
В другом варианте осуществления настоящего изобретения соединение (IJ) может быть получено из соединения (XXIII) взаимодействием с алкоксидами, такими как, например, метоксид натрия.
Некоторые соединения (IJ) могут содержать защитные группы гидроксильной группы или аминогруппы, которые затем удаляются в соответствии с хорошо известными методиками.
Схема 8

В еще одном варианте осуществления настоящее изобретения соединение (IK) может быть получено в соответствии со схемой 8 из соединений (XXIV).
Соединение (XXV) может быть получено из соединения (XXIV) реакцией аминирования в присутствии подходящего амина (рег. 6).
Соединение (XXVI) может быть получено из соединения (XXV) галогенированием подходящими реагентами, такими как бром, NBS, NIS, йод, соли йодония или т.п.
Соединение (XXVII) может быть получено из соединения (XXVI) реакцие образование хиназолинового кольца с подходящими реагентами, такими как формамид или т.п.
Соединение (XXVIII) может быть получено из соединения (XXVII) реакциями кросс-сочетания, такими как реакция Стилле, реакция Сузуки или аналогичные реакции, с подходящими металлоорганическими реагентами (реаг. 2), такими как, например, борорганические соединения. .
Cоединение (XXIII) может быть получено из соединения (XXII) реакцией аминирования в присутствии подходящего амина (реаг. 1).
Некоторые соединения (IL) могут содержать защитные группы гидроксильной группы или аминогруппы, которые удаляют в соответствии с хорошо известными методиками.
В конкретном аспекте настоящее изобретение относится к соединениям формулы (Ib)

где
R3 представляет собой ОН или галоген,
R4 представляет собой Н или OH;
R5 представляет собой галоген или -OMe;
R6 представляет собой галоген или Z;
Z принимает значения, определенные выше.
В дополнительном аспекте настоящее изобретение относится к применению соединений формулы (Ib) в качестве промежуточного соединения при получении соединений формулы (I), которая описана выше.
Соединения по настоящему изобретению обладают неожиданно выявленной эффективностью ингибирования P2X3 рецептора, и указанные соединения могут применяться для лечения респираторного заболевания.
В одном варианте осуществления было неожиданно обнаружено, что типичные соединения формулы (I) по настоящему изобретению эффективно и селективно ингибируют P2X3 рецептор, и указанные соединения могут применяться для лечения респираторного заболевания без неблагоприятного побочного эффекта, такого как потеря вкусовой реакции.
В предпочтительном варианте осуществления соединения формулы (I) являются селективными P2X3 антагонистами, где селективный P2X3 антагонист обладает по меньшей мере 10 кратной селективностью антагонизма в отношении гомомерного P2X3 рецептора по сравнению с антагонизмом в отношении гетеромерного P2X2/3 рецептора.
В дополнительном предпочтительном варианте осуществления селективный P2X3 антагонист обладает по меньшей мере 30-кратной селективностью в отношении гомомерного P2X3 рецептора по сравнению с антагонизмом в отношении гетеромерного P2X2/3 рецептора.
В дополнительном предпочтительном варианте осуществления селективный P2X3 антагонист обладает по меньшей мерее 50-кратной селективностью в отношении гомомерного P2X3 рецептора по сравнению с антагонизмом в отношении гетеромерного P2X2/3 рецептора.
Настоящее изобретение относится также к фармацевтической композиции, включающей только соединение формулы (I) или его фармацевтически приемлемую соль в смеси с одним или несколькими фармацевтически приемлемыми носителями либо комбинацию соединения формулы (I) или его фармацевтически приемлемой соли с одним или несколькими дополнительными активными ингредиентами в смеси с одним или несколькими фармацевтически приемлемыми носителями.
В одном аспекте настоящее изобретение относится к соединению формулы (I) по настоящему изобретению для применения в качестве лекарственного средства.
В дополнительном аспекте настоящее изобретение относится к применению соединения формулы (I) по настоящему изобретению или его фармацевтически приемлемой соли в производстве лекарственного средства для лечения расстройств, механизм развития которых связан с P2X3 рецептором, предпочтительно для лечения респираторных заболеваний.
Предпочтительно, настоящее изобретение относится к соединению формулы (I) для применения в предотвращении и/или лечении распираторных заболеваний, предпочтительно кашля, подострого или хронического кашля, терапевтически резистентного кашля, идиопатического хронического кашля, поствирусного кашля, ятрогенного кашля, астмы, идиопатического легочного фиброза (ИЛС), хронической обструктивной болезни легких (ХОБЛ) и кашля, связанного с респираторными заболеваниями, такими как ХОБЛ, астма и бронхоспазм.
Более предпочтительно, настоящее изобретение относится к соединениям формулы (I) для применения в предотвращении и/или лечении хронического кашля и кашля, связанного с респираторными заболеваниями, такими как ХОБЛ, астма и бронхоспазм.
Настоящее изобретение также относится к способу предотвращения и/или лечения расстройств, механизм развития которых связан с P2X3 рецепторами, причем указанный способ включает введение пациенту, нуждающемуся в таком лечении, терапевтически эффективного количества соединения по настоящему изобретению.
В частности, настоящее изобретение относится к способу предотвращения и/или лечения, где расстройство представляет собой кашель, подострый или хронический кашель, терапевтически резистентный кашель, идиопатический хронический кашель, поствирусный кашель, ятрогенный кашель, астму, идиопатический легочный фиброз (ИЛФ), хроническую обструктивную болезнь легких (ХОБЛ) и кашель, связанный с респираторными заболеваниями, такими как ХОБЛ, астма и бронхоспазм, где указанный способ включает введение подходящего количества соединения формулы (I) пациенту, нуждающемуся в этом.
В дополнительном предпочтительном варианте осуществления расстройство представляет собой хронический кашель.
Способы лечения по настоящему изобретению включают введение безопасного и эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли пациенту, нуждающемуся в этом. Термин «безопасное и эффективное количество», когда используется в настоящем описании в отношении соединения формулы (I) или его фармацевтически приемлемой соли или других фармацевтически активных ингредиентов, означает количество соединения, достаточное для лечения состояния пациента, но достаточно небольшое для использования без побочных эффектов, и тем не менее оно может быть в рабочем порядке определено специалистом в данной области. Соединения формулы (I) или их фармацевтически приемлемые соли могут вводиться один раз или в соответсвии со схемой лечения, где количество доз вводится с различными интервалами в течение заданного периода времени. Обычне суточные дозы могут изменяться в зависимости от конкретного выбранного способа введения.
Настоящее изобретение относится также к фармацевтическим композициям соединений формулы (I) в смеси с одним или несколькими фармацевтически приемлемыми носителями или эксципиентами, например носителями или эксципиентами, описанными в Remington’c Pharmaceutical Sciences Handbook, XVII Ed., Mack Pub., N.Y., U.S.A.
Введение соединений по настоящему изобретению и их фармацевтических композиций может осуществляться в соответствии с потребностями пациента, например перорально, назально, парентерально (подкожно, внутривенно, внутримышечно, интрастернально и инфузией) и ингаляцией.
Предпочтительно, соединения по настоящему изобретению могут вводиться перорально или ингаляцией.
Для введения соединений по настоящему изобретению могут использоваться различные твердые лекарственные формы, включая такие твердые формы как таблетки, желатиновые капсулы, капсуловидные таблетки, гранулы, пастилки и сыпучие порошки. Соединения по настоящему изобретению могут вводиться сами по себе или комбинироваться с рaзличными фармацевтически приемлемыми носителями, разбавителями (такими как сахароза, маннит, лактоза, крахмалы) и известными эксципиентами, включая суспендирующие агенты, солюбилизаторы, буферные агенты, связующие вещества, дезинтегрирующие ведествам консерванты, красители, вкусовые добавки, смазывающие вещества и т.п. Капсулы, таблетки и гели с замедленным высвобождением действующего вещества также являются преимущественными при введении соединений по настоящему изобретению.
Предпочтительно, соединения по настоящему изобретению вводятся в форме таблеток.
Различные жидкие лекарственные формы для перорального введения также могут использоваться для введения соединений по настоящему изобретению, включая водные и неводные растворы, эмульсии, суспензии, сиропы и эликсиры. Такие лекарственные формы также могут содержать подходящие известные инертные разбавители, такие как вода и подходящие известные эксципиенты, такие как консерванты, смаыивающие агенты, подстастители, вкусовые добавки, а также добавки для эмульгирования и/или суспендирования соединения по настоящему изобретению. Соединения по настоящему изобретению могут вводиться инъекцией, например внутривенно, в форме изотонического стерильного раствора.
Для лечения заболеваний дыхательных путей соединения по настоящему изобретению редпочтительно вводятся ингаляцией.
Препараты, которые могут вводиться ингаляцией, включают порошки, вводимые ингаляцией, дозируемые аэрозоли, содержащие пропеллент или препараты для введения ингаляцией, не содержащие пропеллент.
Для введения в форме сухого порошка могут применяться ингаляторы одной или множества доз, известные из предшествующего уровня техники. В этом случае порошком могут заполняться желатиновые, пластиковые или другие капсулы, картриджи, блистерные упаковки или резервуар.
Разбавитель или носитель, химически инертный по отношению к соединению по настоящему изобретению, например лактоза или любая другая добавка для увеличения массовой доли лекарственного средства, попадающего в верхние дыхательные пути, может добавляться к порошкообразным соединения по настоящему изобретению.
Аэрозоли для ингаляции, содержащие газообразный пропеллент, такой как гидрофторалканы, могут содержать соединения по настоящему изобретению в виде раствора или в диспергированной форме. Препараты, содержащие пропеллент, также могут содержать другие ингредиенты, такие как сорастворители, стабилизаторы и, необязательно, другие эксципиенты.
Препараты для ингаляции, свободные от пропеллента, включающие соединения по настоящему изобретению, могут быть в форме растворов или суспензий в водной, спиртовой или водноспиртовой среде, и они могут доставляться с помощью струйных или ультразвуковых небулайзеров, известных из предшествующего уровня, или небулайзеров мягкого тумана.
Предпочтительно, соединения по настоящему изобретению вводятся перорально.
Соединения по настоящему изобретению могут вводиться в форме единственного активного ингредиента или в комбинации с другими фармацевтически активными ингредиентами.
Предпочтительно, соединения по настоящему изобретению могут объединяться с терапевтическими лекарственными средствами или активными ингредиентами, применимыми для лечения заболеваний, которые связаны с P2X3 рецептором или опосредуются им.
Дозы соединений по настоящему изобретению зависят от различных факторов, включая, среди прочих, конкретное заболевание, подлежащее лечению, тяжесть симптомов, способ введения и т.п.
Настоящее изобретение также относится к устройству, содержающему фармацевтическую композицию, включающую соединение формулы (I) по настоящему изобретению, в форме ингалятора сухого порошка, содержащего разовую дозу или множество доз, или в форме дозирующего ингалятора.
Различные аспекты настоящего изобретения, описанные в настоящем документе, иллюстрируются представленными далее примерами, которые никоим образом не ограничивают настоящее изобретение.
Примеры экспериментов по тестированию, описанные в настоящем документе, служат для иллюстрации настоящего изобретения, но изобретение не ограничивается приведенными примерами.
ПРИМЕРЫ ПОЛУЧЕНИЯ ПРОМЕЖУТОЧНЫХ ПРОДУКТОВ И СОЕДИНЕНИЙ
Химические названия были получены с использованием программного обеспечения Dotmatics. В некоторых случаях вместо названий, сгенерированных программным обеспечением Dotmatics, используются общепринятые названия коммерчески доступных реагентов.
Все реагенты, синтез которых не описан в экспериментальной части, являются либо коммерчески доступными, либо известными соединениями или могут быть получены специалистами в данной области техники из известных соединений известными способами.
(R)-1-(2-(Трифторметил)пиримидин-5-ил)этанамин HCl, (R)-1-(6-метилпиридазин-3-ил)этан-1-амин HCl получают в соответстии с методикой, описанной в WO2016/091776.
Значения используемых аббревиатур
Et2O: диэтиловый эфир
Et3N: триэтиламин
TEA: триэтиламин
DCC: N, N'-дициклогексилкарбодиимид
PyBOP: гексафторфосфат (бензотриазол-1-илокси)трипирролидинофосфония
ДМФА: диметилформамид
EtOAc: этилацетат
КТ: комнатная температура
ТГФ: тетрагидрофуран
ДХМ: дихлорметан
MeOH: метиловый спирт
EtOH: этиловый спирт
ТФУК: трифторуксусная кислота
ЖХ-МС: жидкостная хроматография/масс-спектрометрия
ЖХВД: жидкостная хроматография высокого давления (ЖХВД)
ЖХСД: жидкостная хроматография среднего давления
СФХ: сверхкритическая флюидная хроматография
dppf: 1,1'-бис(дифенилфосфино)ферроцен
DIEA или DIPEA: N, N-диизопропилэтиламин
MeCN: ацетонитрил
MTBE: трет-бутилметиловый эфир
TBDMSCl: трет-бутил(хлор)диметилсилан
ДМСО: диметилсульфоксид
Boc2O: ди-трет-бутилдикарбонат
СВЭЖХ: сверхэффективная жидкостная хроматография.
Общее описание экспериментов и методов
Аналитические методы
Жидкостная хроматография-масс-спектрометрия
Метод 1
СВЭЖХ-МС выполняют на аппарате Waters Acquity I-Class с детектором Waters Diode Array, который соединенном с одноквадрупольным масс-спектрометром Waters SQD2, с использованием колонки Waters HSS C18 (1,8 мкм, 100 × 2,1 мм); 5% ацетонитрил/вода (с 0,1% муравьиной кислоты в каждом компоненте подвижной фазы) в течение 1,2 минуты с последующим с линейным градиентом 5-100% в течение 3,5 минуты и затем 100% в течение 1,5 минуты (F=0,5 мл/мин.).
Метод 2
СВЭЖХ-МС выполняют на аппарате Waters Acquity I-Class с детектором Waters Diode Array, который соединен с одноквадрупольным масс-спектрометром Waters SQD2, с использованием колонки Waters BEH Shield RP18 (1,7 мкм, 100 × 2,1 мм); 5% ацетонитрил/вода (с 10 мM бикарбоната аммония в каждом компоненте подвижной фазы) в течение 1,2 минуты с последующим линейным градиентом 5-100% в течение 3,5 минуты и затем - 100% в течение 1,5 минуты (F=0,5 мл/мин.).
Метод 3
СВЭЖХ-МС проводят на аппарате Waters DAD+Waters SQD2, соединеннном с одноквадрупольным СВЭЖХ-МС спектрометром, используя колонку Acquity UPLC BEH Shield RP18 (1,7 мкм 100 × 2,1 мм, картридж Plus guard); 5% ацетонитрил/вода (с 10 мM бикарбоната аммония в каждом компоненте подвижной фазы) в течение 0,4 минуты с последующим линейным градиентом 5-95% в течение 6,4 минуты и затем 95% в течение 1,2 минуты (F=0,4 мл/мин.).
Метод 4
СВЭЖХ-МС выполняют на аппарате Waters DAD+Waters SQD2 с одноквадрупольным СВЭЖХ-МС спектрометром с использованием колонки Acquity СВЭЖХ BEH Shield RP18 (1,7 мкм 100 × 2,1 мм, кардридж Plus guard); при заданной температуре колонки 5% ацетонитрила (степень чистоты - для анализа в дальней УФ области) с 0,1% (об./об.) муравьиная кислота/вода (высокой чистоты, полученной с помощью дополнительного блока PureLab Option) с 0,1% муравьиной кислотой в течение 0,4 минуты с последующим линейным градиентом 5-95% в течение 6,4 минуты, затем 95% в течение 1,2 минуты (F=0,4 мл/мин.).
Метод 5
Масс-спектрометер Aquity UPLC-QDa с обращенно-фазовой колонкой C18 (50 × 2,1 мм Acquity CSH, размер частиц 1,7 мкм, 40°C), элюирование с градиентом, A: 95/5 вода/ацетонитрил+0,05% муравьиной кислоты, B: 95/5 ацетонитрил/вода+0,05% муравьиной кислоты.
Градиент:
МС-детектирование, УФ детектор с фотодиодной матрицей (ДФМ)
МС метод ионизации: электрораспыление (положительный/отрицательный ион).
Метод 5A
Масс-спектрометр Aquity UPLC QDa с C18-обращенно-фазовой колонкой (50 × 2,1 мм Acquity CSH с размером частиц 1,7 мкм, 40 ºC), элюирование с градиентом - A: 95/5 вода/ацетонитрилe+0,05% муравьиной кислоты; B: 95/5 ацетонитрил/вода+0,05% муравьиной кислоты.
Градиент:
МС-детектирование, УФ ДФМ
МС метод ионизации: электрораспыление (положительный/отрицательный ион)
Метод 6
Масс-спектрометер Aquity UPLC-QDa с обращенно-фазовой колонкой C18 (50 × 2,1 мм Acquity BEH с размером частиц 1,7 мкм, 40°C); элюирование с градиентом, A: 95/5 вода/ацетонитрил+0,05% конц. раствора аммиака, B: 95/5 ацетонитрил/вода+0,05% конц. раствора аммиака.
Градиент:
МС-детектирование, УФ ДФМ
МС метод ионизации: электрораспыление (положительный/отрицательный ион).
Метод 7
Dionex UHPLC Ultimate 3000 с диодно-матричным детектором/Thermo Scientific MSQ Pluse в комплекте с колонкой aKinetex® 2,6 мкм XB-C18 (4,6 × 50 мм), 110A; 25°C, элюирование с градиентом, А: 0,1% об./об. раствор муравьиной кислоты в воде, B: 0,1% об./об. раствор муравьиной кислоты в ацетонитриле.
Градиент:
МС-детектирование, УФ ДФМ
МС метод ионизации: электрораспыление (положительный/отрицательный ион).
ЯМР
Анализ методом спектроскопии 1H ядерного магнитного резонанса (ЯМР) выполняют с использованием аппарата Bruker или Varian, работающего при 300 или 400 МГц, используя заявленный растворитель примерно при комнатной температуре, если не указано иное. Во всех случаях данные ЯМР согласуются с предполагаемыми структурами. Характеристические химические сдвиги (δ) даны в частях на миллион с использованием общепринятых аббревиатур для обозначения основных пиков: c - синглет; д - дублет; т - триплет; кв - квартет; дд - дублет дублетов; дт - дублет триплетов; м - мультиплет; уш. - уширенный.
Условия препаративной ЖХВД с обращенной фазой (ОФ-ЖХВД)
Очистку препаративной ЖХВД проводят в соответствии с ЖХВД с обращенной фазой, используя систему препаративной ЖХВД Waters Fractionlynx (2525 насос, 2996/2998, детектор ультрафиолетового и видимого диапазона (UV/VIS детектор), 2767 жидкостной манипулятор) или эквивалентной системой ЖХВД, такой как система Gilson Trilution UV directed. Жидкостной манипулятор Waters 2767 выступает в качестве автодозатора и отборника фракций. Для препаративной очистки соединений используют колонки Waters Sunfire OBD Phenomenex Luna Phenyl Hexyl, Waters Xbridge Phenyl (10 мкм, 19 × 150 мм) или Waters CSH Phenyl Hexyl (19 × 150, 5 мкм). Подходящие градиенты определенных концентраций подбирают на основе систем растворителей ацетонитрила и метанола в кислотных или основных условиях. Модификаторами, используемыми в кислотных/основных условиях, являются муравьиная кислота или трифторуксусная кислота (0,1% об./об.) и бикарбонат аммония (10 мM), соответственно. Очистку контролируют с помощью программного обеспечния Waters Fractionlynx software через мониторинг при 210-400 нм и инициируют пороговое значение сбора при 260 нм и, в случае использования Fractionlynx) при наличии целевого молекулярного иона, который выявляется в условиях API. Собранные фракции анализируют ЖХМС (системы Waters Acquity с Waters SQD).
Протокол разделения с помощью сверхкритической флюидной хроматографии (СФХ)
Диастереомерное разделение соединений проводят с помощью сверхкритической флюидной хроматографии (СФХ) с использованием препаративной СФХ системы Waters Thar Prep100 (насос P200 CO2, насос модификатора 2545, детектор УФ и видимого диапазона 2998, жидкостной манипулятор 2767 с многоуровнемым модулем инженции (Stacked Injection Module). Жидкостной манипулятор Waters 2767 выступает в качестве автодозатора и отборника фракций. Подходящие изократические методы подбирают на основе систем растворителей метанола, этанола или изопропанола в немодифицированных или основных условиях. Используемый стандартный метод СФХ модифицируют - CO2, 100 мл/мин., противодавление 120 бар, температура колонки 40°C. Модификатором, используемым в основных условиях, является диэтиламин (0,1% об./об.). Модификатором, используемым в кислотных условиях, является муравьиная кислота (0,1% об./об.) или трифторуксусеая кислота (0,1% об./об.). СФХ-очистку контролируют с помощью программного обеспечения Waters Fractionlynx через мониторинг при 210-400 нм и обычно инициируют пороговое значение сбора при 260 нм. Собранные фракции анализируют с помощью СФХ (системы Waters/Thar SFC c Waters SQD). Фрации, которые содержат целевой продукт, концентрируют вакуумным центрифугированием.
Условия анализа сверхкрической флюидной хроматографией-масс-спектрометрией
Метод 8
СФХ-МС проводят на системах Waters/Thar SFC с Waters SQD, используя колонку Lux Cellulose-3; 15% метиловый спирт/CO2 (с 0,1% диэтиламина), изократическое хроматографирование при 5 мл/мин., противодавлении 120 бар, температуре колонки 40°C.
Метод 9
СФХ-МС проводят на системах Waters/Thar SFC с Waters SQD, используя колонку Lux Cellulose-3, 20% метилый спирт/CO2 (с 0,1% диэтиламина); изократическое хроматографирование при 5 мл/мин., противодавлении 120 бар, температуре колонки 40°C.
Метод 10
СФХ-МС проводят на системах Waters/Thar SFC с Waters SQD, используя колонку Lux Cellulose-4, 55% этиловый спирт/CO2 (с 0,1% диэтиламина); изократическое хроматографирование при 5 мл/мин., противодавлении 120 бар, температуре колонки 40°C.
Метод 11
СФХ-МС проводят на системах Waters/Thar SFC с Waters SQD, используя колонку Lux Cellulose-4; 20% изопропиловый спирт/CO2 (с 0,1% диэтиламина), изократическое хроматографирование при 5 мл/мин., противодавлении 120 бар, температуре колонки 40°C.
Метод 12
СФХ-МС проводят на системах Waters/Thar SFC с Waters SQD, используя колонку Lux Cellulose-4; 30% изопропиловый сирт/CO2 (с 0,1% диэтиламина), изократическое хроматографирование при 5 мл/мин., противодавлении 120 бар, температуре колонки 40°C.
Метод 13
СФХ-МС проводят на системах Waters/Thar SFC с Waters SQD, используя колонку Lux Cellulose-4; 50% изопропиловый спирт/CO2 (с 0,1% диэтиламина), изократическое хроматографирование при 5 мл/мин., противодавлении 120 бар, температуре колонки 40°C.
Метод 14
СФХ-МС проводят на системах Waters/Thar с Waters SQD, используя колонку Lux Cellulose-4; 25% метиловый спирт/CO2 (с 0,1% диэтиламина), изократическое хроматографирование при 5 мл/мин., противодавлении 120 бар, температуре колонки 40°C.
Метод 15
СФХ-МС проводят на системах Waters/Thar SFC с Waters SQD, используя колонку YMC Amylose-C; 15% этиловый спирт/CO2 (с 0,1% диэтиламина), изократическое хроматографирование при 5 мл/мин., противодавлении 120 бар, температуре колонки 40°C.
Метод 16
СФХ-МС проводят на системах Waters/Thar SFC с Waters SQD, используя колонку YMC Amylose-C, 25% изопропиловый спирт/CO2 (с 0,1% диэтиламина), изократическое хроматографирование при 5 мл/мин., противодавлении 120 бар, температуре колонки 40°C.
Метод 17
СФХ-МС проводят на системах Waters/Thar SFC с Waters SQD, используя колонку YMC Amylose-C; 35% изопропиловый спирт/CO2 (с 0,1% диэтиламина), изократическое хроматографирование при 5 мл/мин., противодавлении 120 бар, температуре колонки 40°С.
Метод 18
СФХ-МС проводят на системах Waters/Thar SFC с Waters SQD, используя колонку YMC Amylose-C; 55% изопропиловый спирт/CO2 (с 0,1% диэтиламина), изократическое хроматографирование при 5 мл/мин., противодавлении 120 бар, температуре колонки 40°С.
Метод 19
СФХ-МС проводят на системах Waters/Thar SFC с Waters SQD, используя колонку YMC Amylose-C; 15% метиловый спирт/CO2 (с 0,1% диэтиламина), изократическое хроматографирование при 5 мл/мин., противодавлении 120 бар, температуре колонки 40°С.
Метод 20
СФХ-МС проводят на системах Waters/Thar SFC с Waters SQD, используя колонку YMC Amylose-C; 20% метиловый спирт/CO2 (с 0,1% диэтиламина), изократическое хроматографирование при 5 мл/мин., противодавлении 120 бар, температуре колонки 40°С.
Метод 21
СФХ-МС проводят на системах Waters/Thar SFC с Waters SQD, используя колонку YMC Cellulose-C; 15% изопропиловый спирт/CO2 (с 0,1% диэтиламина), изократическое хроматографирование при 5 мл/мин., противодавлении 120 бар, температуре колонки 40°С.
Метод 22
СФХ-МС проводят на системах Waters/Thar SFC с Waters SQD, используя колонку YMC Cellulose-C; 15% метиловый спирт/CO2 (с 0,1% диэтиламина), изократическое хроматографирование при 5 мл/мин., противодавлении 120 бар, температуре колонки 40°С.
Метод 23
СФХ-МС проводят на системах Waters/Thar SFC с Waters SQD, используя колонку YMC Cellulose-C; 25% метиловый спирт/CO2 (с 0,1% диэтиламина), изократическое хроматографирование при 5 мл/мин., противодавлении 120 бар, температуре колонки 40°С.
Метод 24
СФХ-МС проводят на системах Waters/Thar SFC с Waters SQD, используя колонку YMC Cellulose-SC; 55% изопропиловый спирт/CO2 (с 0,1% диэтиламина), изократическое хроматографирование при 5 мл/мин., противодавлении 120 бар, температуре колонки 40°С.
Метод 25
СФХ-МС проводят на системах Waters/Thar SFC с Waters SQD, используя колонку Lux Cellulose-3; 10% метиловый спирт/CO2 (с 0,1% диэтиламина), изократическое хроматографирование при 5 мл/мин., противодавлении 120 бар, температуре колонки 40°С.
Метод 26
СФХ-МС проводят на системах Waters/Thar SFC с Waters SQD, используя колонку Lux Cellulose-3; 25% метиловый спирт/CO2 (с 0,1% диэтиламина); изократическое хроматографирование при 5 мл/мин., противодавлении 120 бар, температуре колонки 40°С.
Метод 27
СФХ-МС 2проводят на системах Waters/Thar SFC с Waters SQD, используя колонку Lux Cellulose-3; 30% метиловый спирт/CO2 (с 0,1% диэтиламина), изократическое хроматографирование при 5 мл/мин., противодавлении 120 бар, температуре колонки 40°С.
Метод 28
СФХ-МС проводят на системах Waters/Thar SFC с Waters SQD, используя колонку Lux Cellulose-4; 40% изопропиловый спирт/CO2 (с 0,1% диэтиламина), изократическое хроматографирование при 5 мл/мин., противодавлении 120 бар, температуре колонки 40°С.
Метод 29
СФХ-МС проводят на системах Waters/Thar SFC с Waters SQD, используя колонку Lux Cellulose-4; 40% метиловый спирт/CO2 (с 0,1% диэтиламина), изократическое хроматографирование при 5 мл/мин., противодавлении 120 бар, температуре колонки 40°С.
Метод 30
СФХ-МС проводят на системах Waters/Thar SFC с Waters SQD, используя колонку Lux Cellulose-4; 50% метиловый спирт/CO2 (с 0,1% диэтиламина), изократическое хроматографирование при 5 мл/мин., противодавлении 120 бар, температуре колонки 40°С.
Метод 31
СФХ-МС проводят на системах Waters/Thar SFC с Waters SQD, используя колонку Lux Cellulose-4; 55% изопропиловый спирт/CO2 (с 0,1% диэтиламина), изократическое хроматографирование при 5 мл/мин., противодавлении 120 бар, температуре колонки 40°С.
Метод 32
СФХ-МС проводят на системах Waters/Thar SFC с Waters SQD, используя колонку Lux Cellulose-4; 55% метиловый спирт/CO2 (с 0,1% диэтиламина), изократическое хроматографирование при 5 мл/мин., противодавлении 120 бар, температуре колонки 40°С.
Метод 33
СФХ-МС проводят на системах Waters/Thar SFC с Waters SQD, используя колонку YMC Amylose-C; 20% этиловый спирт/CO2 (с 0,1% диэтиламина), изократическое хроматографирование при 5 мл/мин., противодавлении 120 бар, температуре колонки 40°С.
Метод 34
СФХ-МС проводят на системах Waters/Thar SFC с Waters SQD, используя колонку YMC Amylose-C; 30% изопропиловый спирт/CO2 (с 0,1% диэтиламина), изократическое хроматографирование при 5 мл/мин., противодавлении 120 бар, температуре колонки 40°С.
Метод 35
СФХ-МС проводят на системах Waters/Thar SFC с Waters SQD, используя колонку YMC Amylose-C; 30% метиловый спирт/CO2 (с 0,1% диэтиламина), изократическое хроматографирование при 5 мл/мин., противодавлении 120 бар, температуре колонки 40°С.
Метод 36
СФХ-МС проводят на системах Waters/Thar SFC с Waters SQD, используя колонку YMC Amylose-C; 40% метиловый спирт/CO2 (с 0,1% диэтиламина), изократическое хроматографирование при 5 мл/мин., противодавлении 120 бар, температуре колонки 40°С.
Метод 37
СФХ-МС проводят на системах Waters/Thar SFC с Waters SQD, используя колонку YMC Amylose-C; 55% метиловый спирт/CO2 (с 0,1% диэтиламина), изократическое хроматографирование при 5 мл/мин., противодавлении 120 бар, температуре колонки 40°С.
Метод 38
СФХ-МС проводят на системах Waters/Thar SFC с Waters SQD, используя колонку YMC Cellulose-C; 20% метиловый спирт/CO2 (с 0,1% диэтиламина), изократическое хроматографирование при 5 мл/мин., противодавлении 120 бар, температуре колонки 40°С.
Метод 39
СФХ-МС проводят на системах Waters/Thar SFC с Waters SQD, используя колонку YMC Cellulose-SC; 35% изопропиловый спирт/CO2 (с 0,1% диэтиламина), изократическое хроматографирование при 5 мл/мин., противодавлении 120 бар, температуре колонки 40°С.
Метод 40
СФХ-МС проводят на системах Waters/Thar SFC с Waters SQD, используя колонку YMC Cellulose-SC; 45% изопропиловый спирт/CO2 (с 0,1% диэтиламина), изократическое хроматографирование при 5 мл/мин., противодавлении 120 бар, температуре колонки 40°С.
ПРИМЕРЫ ПОЛУЧЕНИЯ ПРОМЕЖУТОЧНЫХ ПРОДУКТОВ И СОЕДИНЕНИЙ
Пример 1
6-(1-((6-(4-Фторфенил)-8-метоксихиназолин-4-ил)амино)этил)пиридин-2(1Н)-он

Стадия 1: Получение гидробромида 6-(1-аминоэтил)пиридин-2(1Н)-она
1-(6-Метоксипиридин-2-ил)этан-1-амин (250 мг, 1,64 ммоль) в гидробромиде (48% масс. в воде, 3,7 мл) нагревают до 80°C и выдерживают при указанной температуре в течение 24 часов. После этого смесь охлаждают до комнатной температуры и концентрируют в вакууме с получением указанного в заголовке соединения (360 мг, количественный выход) в виде твердого вещества коричневого цвета, которое используют в следующей стадии без дополнительной очистки.
¹H ЯМР (400 МГц, ДМСО): δ (8,39 уш. с, 3 H), 7,64 (дд, J=8,9, 7,1 Гц, 1 H), 6,64 (д, J=6,9 Гц, 1 H), 6,54 (д, J=8,8 Гц, 1 H), 4,38-4,30 (м, 1 H), 1,52 (д, J=6,8 Гц, 3 H). 1 NH/OH не обнаружены.
Стадия 2: Получение 6-(1-((6-(4-фторфенил)-8-метоксихиназолин-4-ил)амино)этил)пиридин-2(1Н)-она
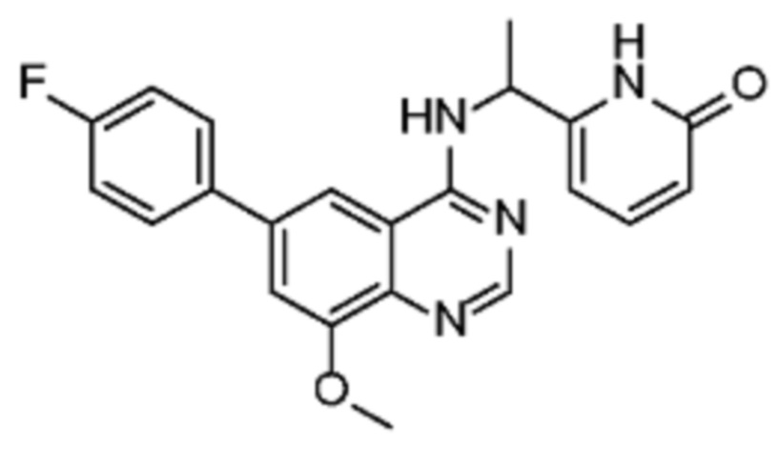
6-(4-Фторфенил)-8-метоксихиназолин-4-ол (150 мг, 0,56 ммоль), гексафторфосфат (бензотриазол-1-илокси)трипиррллидинофосфония (318 мг, 0,61 ммоль) и N, N-диизопропилэтиламин (0,48 мл, 2,78 ммоль) перемешивают в N, N-диметилформамидe (4 мл) при 40°C в течение 1 часа. К смеси добавляют гидробромид 6-(1-аминоэтил)пиридин-2(1Н)-она (146 мг, 0,67 ммоль) и перемешивают реакционную смесь при 40°C в течение 24 часов. Реакционную смесь охлаждают до комнатной температуры и добавляют этилацетат (10 мл) и воду (10 мл). Слои разделяют и водный слой экстрагируют этилацетатом(2 × 10 мл). Объединенные органические фракции промывают водой (2 × 10 мл), сушат над MgSO4, фильтруют и концентрируют в вакууме. Остаток очищают препаративной ЖХВД с получением указанного в заголовке соединения (35 мг, 16%) в виде твердого не совсем белого вещества.
¹H ЯМР (400 МГц, ДМСО): δ 11,73 (c, 1 H), 8,47 (c, 1 H), 8,43 (д, J=7,6 Гц, 1 H), 8,26 (д, J=1,8 Гц, 1 H), 8,02-7,97 (м, 2 H), 7,58 (д, J=1,5 Гц, 1 H), 7,47-7,38 (м, 3 H), 6,22 (д, J=9,1 Гц, 2 H), 5,43-5,35 (м, 1 H), 4,06 (c, 3 H), 1,64 (д, J=7,1 Гц, 3 H). ЖХМС (метод 4): [MH+]=391 при 3,01 мин.
Пример 2
6-(5-Фторпиридин-2-ил)-8-метокси-N-(1-(3-метил-1,2,4-оксадиазол-5-ил)этил)хиназолин-4-амин
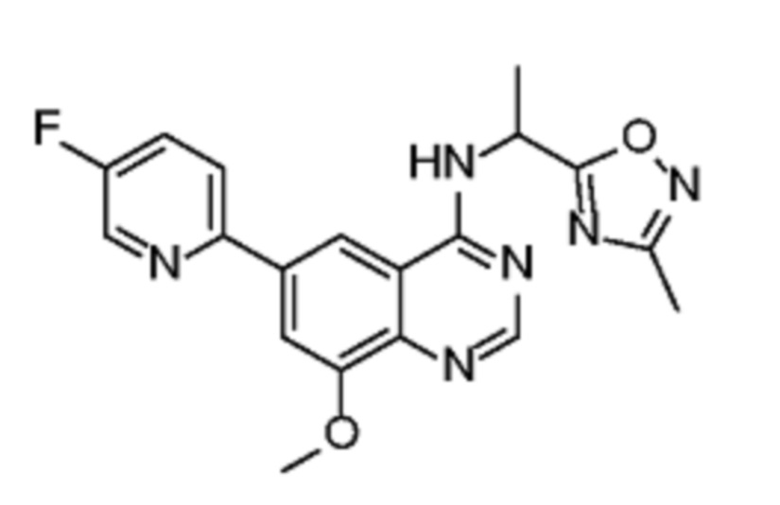
6-(5-Фторпиридин-2-ил)-8-метоксихиназолин-4-ол (140 мг, 0.42 ммоль), гексафторфосфат бензотриазол-1-илокситрипирролидинофосфония (294 ммоль, 0,56 ммоль) и N, N-диизопропилэтиламин (332 мг, 0,45 мл, 2,57 ммоль) растворяют в N, N-диметилформамидe (4 мл). Реакционную смесь нагревают до 40°C и выдерживают при указанной температуре в течение 2 часов, после чего добавляют (1-(3-метил-1,2,4-оксадиазол-5-ил)этан-1-амин (101 мг, 0,62 ммоль). Реакционную смесь перемешивают в течение дополнительных 24 часов и затем концентрируют в вакууме. Остаток очищают препаративной ЖХВД с получением указанного в заголовке соединения (12,1 мг, 6%) в виде твердого бесцветного вещества.
1H ЯМР (400 МГц, CDCl3): δ 8,53 (дд, J=2,8, 6,5 Гц, 1 H) 8,42 (c, 1 H), 7,89 (д, J=4,4 Гц, 1 H), 7,79 (м, 1 H), 7,61 (д, J=10,1 Гц, 1 H), 7,52 (м, 1 H), 7,16 (уш. с, 1 H), 5,86 (м, 1 H), 4,14 (c, 3 H), 2,45 (c, 3 H), 1,81 (д, J=6,6 Гц, 1 H). ЖХМС (метод 4): [MH+]=381 пр 3,76 мин.
Соединения примеров, представленные в таблице ниже, синтезуют в соответствии с методикой, описанной для получения 6-(5-фторпиридин-2-ил)-8-метокси-N-(1-(3-метил-1,2,4-оксадиазол-5-ил)этил)хиназолин-4-амина (пример 2).
структура
1H ЯМР
ЖХ-МС

ЖХМС (метод 3): [MH+]=382 при 2,81 мин.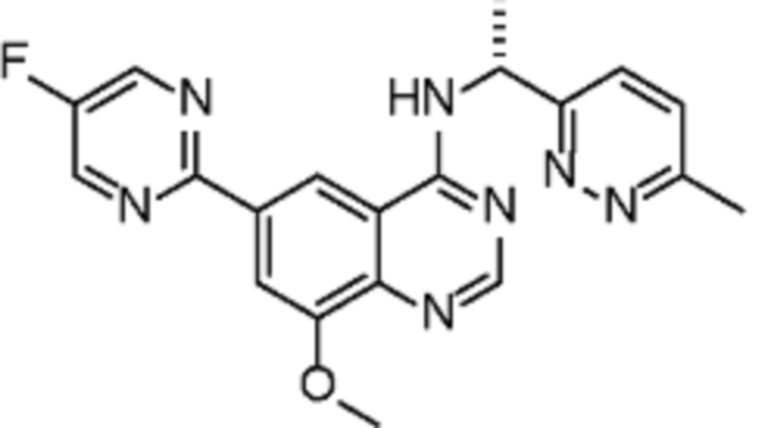
ЖХМС (метод 3): [MH+]=392 при 2,51 мин.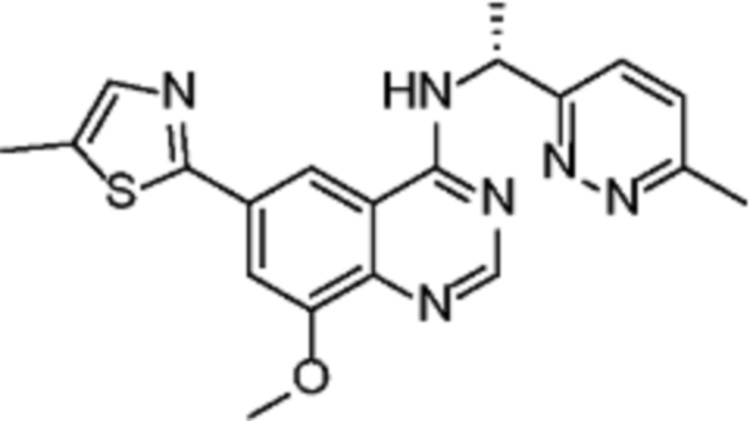
ЖХМС (метод 3): [MH+]=393 при 3,8 мин.
ЖХМС (метод 4): [MH+]=441 при 2,82 мин.
ЖХМС (метод 3): [MH+]=378 при 3,46 мин.
Промежуточное соединение 29
Получение 2-амино-3-(морфолиносульфонил)бензойной кислоты

К раствору морфолина (0,741 мл, 8,47 ммоль) в воде (50 мл) в течение 10 минут добавляют 3-(хлорсульфонил)-2-нитробензойную кислоту (0,75 г, 2,82 ммоль) и перемешивают в течение 30 минут, затем добавляют Pd/C 10% (50% вл.) (2,82 ммоль) и водную соляную кислоту (2N, в воде, 0,103 г, 2,82 ммоль) и перемешивают полученную реакционную смесь в течение ночи в атмосфере водорода (балон). Катализатор удаляют фильтрацией и растворитель удаляют при пониженном давлении. Сырой продукт очищают C18 флэш-хроматографией ((H2O/ACN)) 95:5+0,1% HCOOH}:{(ACN/H2O) 95:5+HCOOH 0,1%, элюирование с градиентом: от 100:0 до 0:100} с получением 2-амино-3-(морфолиносульфонил)бензойной кислоты (0,80 г, 2,79 ммоль, выход 99%) в виде твердого вещества коричневого цвета.
ЖХМС(метод 5): 0,75 мин., [M+H]+ 287,75
Соединения, представленные в таблице ниже, получают в соответствии с описанной выше методикой.
структура

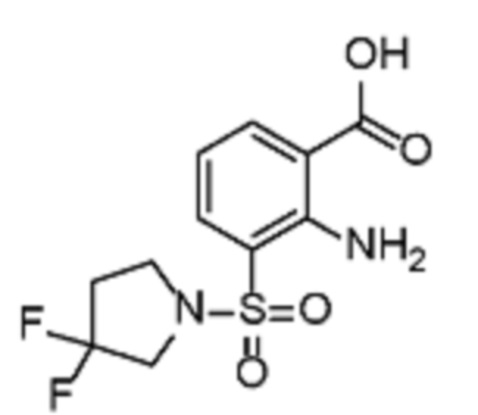


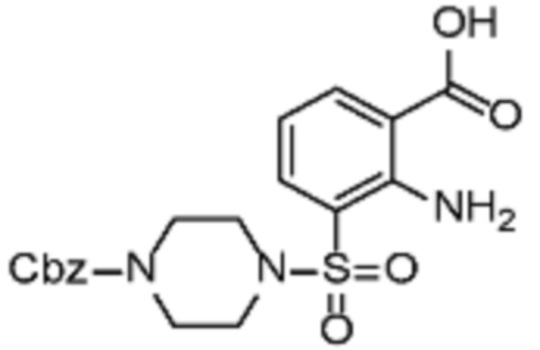
Промежуточное соединение 35
2-Амино-5-бром-3-(морфолиносульфонил)бензойная кислота

К раствору 2-амино-3-(морфолиносульфонил)бензойной кислоты (0,8 г, 2,79 ммоль) (промежуточное соединение 29) в ДМФА добавляют NBS (0,567 мл, 2,79 ммоль) и перемешивают полученный раствор в течение 30 мин. Затем расторитель удаляют и сырой продукт очищают C18 флэш-хроматографией {(H2O/ACN)) 95:5+0,1% HCOOH}:{(ACN/H2O) 95:5+HCOOH 0,1%, элюирование с градиентом: от 100:0 до 0:100} с получением 2-амино-5-бром-3-(морфолиносульфонил)бензойной кислоты (0,85 г, 2,328 ммоль, выход 83%) в виде твердого белого вещества.
ЖХМС(метод 5): 0,98 мин., 366,56 [M+H]+.
Соединения, представленные в таблице ниже, получают в соответствии с описанной выше методикой.
структура
ЖХ-МС





Промежуточное соединение 41
6-Бром-8-(морфолиносульфонил)хиназолин-4-ол

К раствору 2-амино-5-бром-3-(морфолиносульфонил)бензойной кислоты (600 мг, 1,643 ммоль) (промежуточное соединение 35) в формамиде (6 мл) добавляют этансульфоновую кислоту (0,6 мл, 1,643 ммоль), смесь нагревают до 120°C и выдерживают при указанной температуре в течение 48 часов. Реакционную смесь загружают непосредственно на С18-картридж и оищают обращенно-фазовой хроматографией ((H2O/ACN)) 95:5 +0,1% HCOOH}:{(ACN/H2O) 95:5+HCCOH 0,1%, элюирование с градиентом: от 100:0 до 0:100} с получением 6-бром-8-(морфолиносульфонил)хиназолин-4-ол (0,230 г, 0,615 ммоль, выход 37,4%) в виде твердого вещества коричневого цвета.
ЖХМС(метод 5): 0,72 мин., [M+H]+ 375,56.
Соединения, представленные в таблице ниже, получают в соответствии с описанной выше методикой.
структура
ЖХ-МС



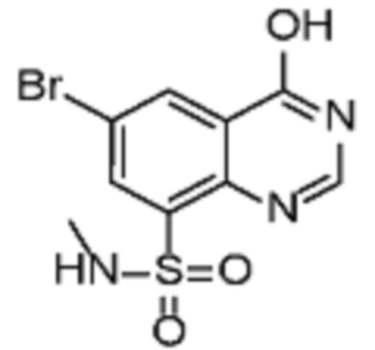
пиперазин-1-карбоксилат
Промежуточное соединение 47
6-(4-Фторфенил)-8-(морфолиносульфонил)хиназолин-4-ол
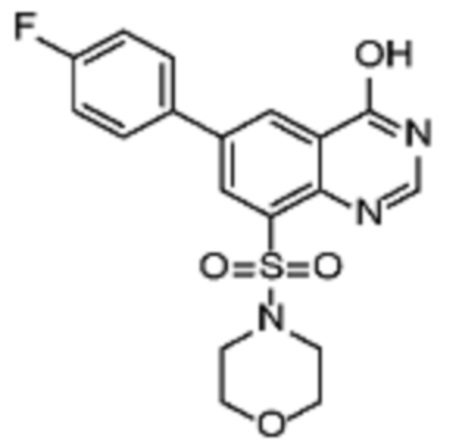
К суспензии 6-бром-8-(морфолиносульфонил)хиназолин-4-ол (250 мг, 0,668 ммоль) в диоксане (10 мл) добавляют воду (3 мл), 4-фторфенилбороновую кислоту (187 мг, 1,336 ммоль), K2CO3 (277 мг, 2,004 ммоль) и PdCl2(dppf)(56 мг, 0,07 ммоль) и перемешивают полученную реакционную смесь в течение ночи. Затем растворитель удаляют и сырой продукт очищают C18 флэш-хроматографией {(H2O/ACN) 95:5 +0,1% HCOOH}:{(ACN/H2O) 95:5+HCOOH 0,1%}, элюирование с градиентом: om 100:0 до 0:100, с получнием 6-(4-фторфенил)-8-(морфолиносульфонил)хиназолин-4-ола (204 мг, 0,524 ммоль, выход 78%) в виде твердого вещества коричневого цвета.
ЖХМС(метод 5): 0,87 мин., [M+H]+ 389,81
Соединения, представленные в таблице ниже, получают в соответствии с описанной выше методикой.
структура
ЖХ-МС
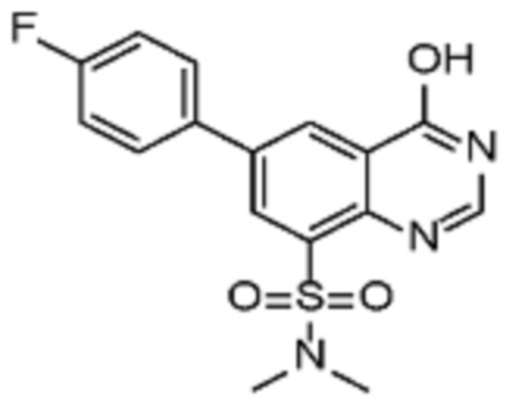
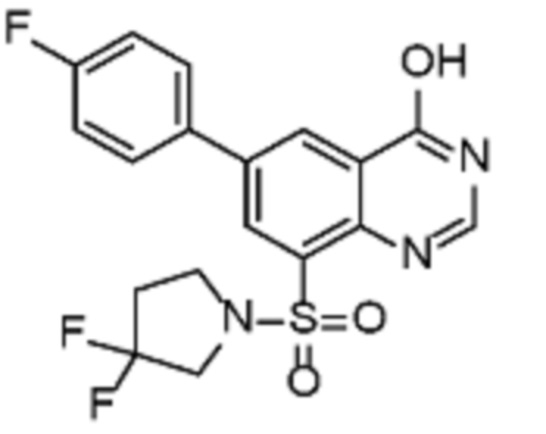



Пример 8
(R)-6-(4-Фторфенил)-N-(1-(6-метилпиридазин-3-ил)этил)-8-(морфолиносульфонил)хиназолин-4-амин
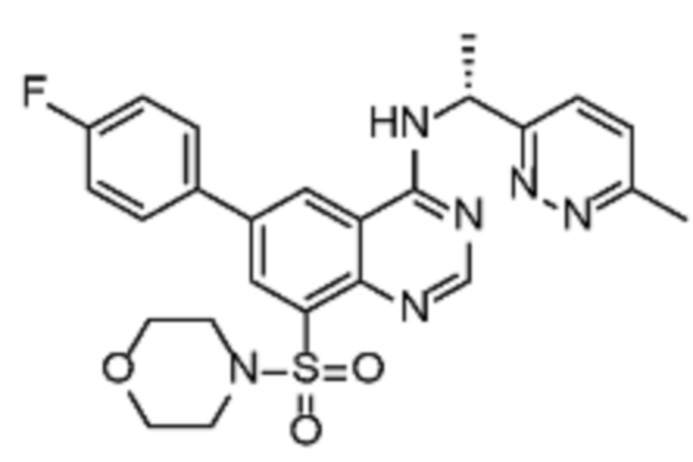
К раствору 6-(4-фторфенил)-8-(морфолиносульфонил)хиназолин-4-ол (60 мг, 0,154 ммоль) в сухом ДМФА (4 мл) добавляют PyBOP (96 мг, 0,185 ммоль) и DIPEA (0,059 мл, 0,339 ммоль) и перемешивают реакционную смесь в течение 30 минут. Затем добавляют дигидрохлорид ((R)-1-(6-метилпиридазин-3-ил)этан-1-амин (32,4 мг, 0,154 ммоль) и дополнительно перемешивают реакционную смесь в течение 30 минут. Затем растворитель удаляют и сырой продукт очищают C18 флэш-хроматографией (элюирование с градиентом: {(H2O/ACN) 95:5 +0,1% HCOOH}:{(ACN/H2O) 95:5+HCOOH 0,1%}, 100:0 до 0:100) с получением (R)-6-(4-фторфенил)-N-(1-(6-метилпиридазин-3-ил)этил)-8-(морфолиносульфонил)хиназолин-4-амина (10 мг, 0,020 ммоль, выход 12,76%) твердого вещества желтоватого цвета.
1H ЯМР (400 МГц, ДМСО-d6) δ м.д. 9,20 (c, 2 H), 9,01 (уш. д, J=6,58 Гц, 1 H), 8,95 (c, 1 H), 8,56 (c, 1 H), 8,54 (c, 1 H), 7,92 (уш. дд, J=8,22, 5,59 Гц, 2 H), 7,43 (уш. т, J=8,66 Гц, 2 H), 5,71 (уш. квин, J=6,80 Гц, 1 H), 4,33 (уш. т, J=4,82 Гц, 2 H), 3,52-3,61 (м, 4 H), 3,39-3,50 (м, 2 H), 1,75 (уш. д, J=7,02 Гц, 3 H).
ЖХМС(метод 5): 1,58 мин., 509,13 [M+H]+.
Соедиения, представленные в таблице ниже, получают в соответствии с методикой, описанной выше.
¹H ЯМР/ЖХ-МС

ЖХМС (метод 5): [MH+]=509,13 при 1,58 мин.
ЖХМС (метод 5): [MH+]=515,01 при 0,98 мин.
ЖХМС (метод 5): [MH+]=466,62 при 0,92 мин.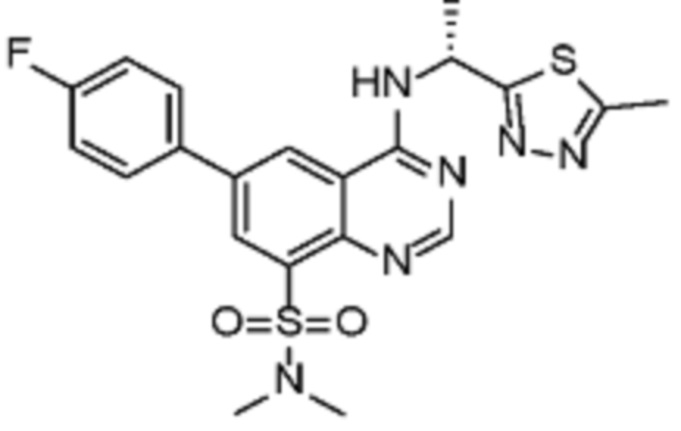
ЖХМС (метод 5): [MH+]=472.7 при 1,0 мин.
ЖХМС (метод 5): [MH+]=457,6 при 1,02 мин.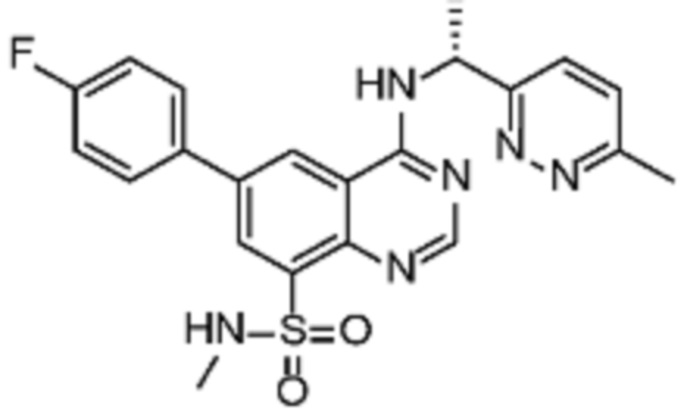
ЖХМС (метод 5A): [MH+]=452,92 при 1,61 мин.
ЖХМС (метод 5A): [MH+]=442,81 при 1,81 мин.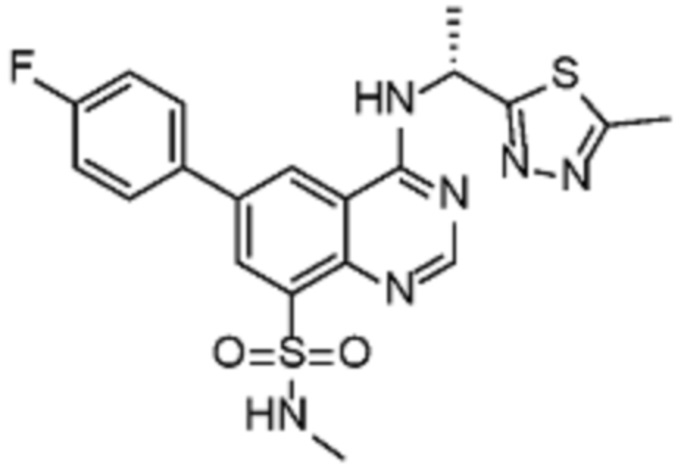
ЖХМС (метод ): [MH+]=458,02 при 1,02 мин.
Пример 22
(R)-6-(4-Фторфенил)-N-(1-(6-метилпиридазин-3-ил)этил)-8-(пиперазин-1-илсульфонилхиназолин-4-амин

К раствору бензил-4-((6-(4-фторфенил)-4-гидроксихиназолин-8-ил)сульфонил)пиперазин-1-карбоксилата (100 мг, 0,191 ммоль) (промежуточное соединение 52) в сухом ДМФА (5 мл) добавляют PyBOP (129 мг, 0,249 ммоль) и DIPEA (0,100 мл, 0,574 ммоль) и перемешивают реакционную смесь в течение 30 минут. Затем добавляют дигидрохлорид (R)-1-(6-метилпиридазин-3-ил)этан-1-амина (40,2 мг, 0,191 ммоль) и дополнительно перемешивают реакционную смесь в течение 30 минут. Растворитель удаляют и и снова растворяют сырой продукт в сухом ДХМ (5,00 мл) и добавляют трибромид бора (1 M в ДХМ, 1,914 мл, 1,914 ммоль) и перемешивают реакционную смесь в течение 1 часа. К смеси добавляют EtOH (2 мл), затем летучие компоненты удаляют и сырой продукт очищают флэш-хроматографией (С18, элюирование с градиентом: {(H2O/ACN) 95:5+0,1% HCOOH}:{(ACN/H2O) 95:5+HCOOH 0,1%}, от 100:0 до 0:100) с получением (R)-6-(4-фторфенил)-N-(1-(6-метилпиридазин-3-ил)этил)-8-(пиперазин-1-илсульфонилхиназолин-4-амина (7 мг, 0,014 ммоль, выход 7,21%) в виде твердого белого вещества.
1H ЯМР (400 МГц, ДМСО-d6) δ м.д. 9,25 (уш. д, J=6,58 Гц, 1 H), 9,08 (д, J=1,75 Гц, 1 H), 8,64 (уш. с, 2 H), 8,54 (д, J=1,75 Гц, 1 H), 8,50 (c, 1 H), 7,88-7,98 (м, 3 H), 7,76 (д, J=8,77 Гц, 1 H), 7,41 (т, J=8,77 Гц, 2 H), 5,76 (квин, J=6,96 Гц, 1 H), 3,46-3,58 (м, 4 H), 3,09 (уш. с, 4 H), 2,62 (c, 3 H), 1,71 (д, J=7,02 Гц, 3 H).
СВЭЖХ PRECLI-WI-0183 minuti rt, 4,29 мин., 508,18 m/z
Соединения, представленные в таблице ниже получают в соответствии с описанной выше методикой.
структура
¹H ЯМР/ЖХ-МС

ЖХМС (метод 5): [MH+]=458,02 при 1,02 мин.
Промежуточное соединение 20
(R)-6-(4-Фторфенил)-8-йод-N-(1-(2-(трифторметил)пиримидин-5-ил)этил)хиназолин-4-амин

Стадия 1: Получение метил-4-амино-4'-фтор-[1,1'-бифенил]-3-карбоксилата

Газообратный азот барботируют в течение 5 минут через смесь метил-2-амино-5-бромбензоата (2,00 г, 8,69 ммоль), 4-фторфенилбороновой кислоты, сложного пинаколового эфира (2,90 г, 12,04 ммоль), трехосновного фосфата калия (3,69 г, 17,39 ммоль), воды (3,5 мл) в N, N-диметилформамидe (10,5 мл), затем добавляют комплекс [1,1′-бис(дифенилфосфино)ферроцен]дихлорпалладия(II) с дихлорметаном (710 мг, 0,87 ммоль). Полученную смесь нагревают до 100°С и выдерживают при указанной температуре в течение 1,25 часов. После охлаждения до комнатной температуры реакционную смесь разбавляют водой (100 мл) и диэтиловым эфиром (100 мл) и отделяют органическую фазу. Водную фазу экстрагируют еще раз диэтиловым эфиром (100 мл) и затем этилацетатом (100 мл). Органические фазы объединяют, промывают водой (100 мл), сушат над MgSO4 и удаляют растворитель в вкууме. Остаток очищают хроматографией (силикагельл, элюирование с градиентом: 5-35% этилацетата в циклогексане) с получением указанного в заголовке соединения в виде твердого не совсем белого вещества (2,08 г, выход 97%).
1H ЯМР (400 МГц, CDCl3): δ 8,06 (д, J=2,3 Гц, 1 H), 7,50-7,44 (м, 3 H), 7,08 (дд, J=8,7, 8,7 Гц, 2 H), 6,74 (д, J=8,6 Гц, 1 H), 5,78 (c, 2 H), 3,90 (c, 3 H).
Стадия 2: Получение метил-4-амино-4'-фтор-5-йод-[1,1'-бифенил]-3-карбоксилата
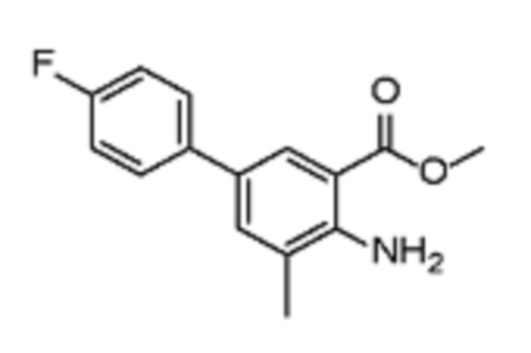
К раствору метил-4-амино-4'-фтор-[1,1'-бифенил]-3-карбоксилата (2,08 г, 8,48 ммоль) в дихлорметане (25 мл) добавляют тетрафторборат бис(пиридин)йодония (4,73 г, 12,72 ммоль) и ТФУК (2,1 мл, 27,42 ммоль). Полученную смесь перемешивают при комнатной температуре в течение 2 дней. ЖХВД анализ показывает 70% конверсию, после чего добавляют дополнительную порцию тетрафторбората бис(пиридин)йодония (1,25 г, 3,36 ммоль) и перемешивают смесь в течение дополнительеых 2,5 часов. Реакционную смесь разбавляют дихлорметаном (25 мл) и осторожно обрабатывают раствором NaHCO3 (7 г, 83 ммоль) в воде (100 мл). Водный слой отделяют и дополнительно экстрагируют дихлорметаном (2 × 25 мл). Органические фазы объединяют, 8% водным раствором тиосульфата натрия (100 мл), фильтруют через фриттовый фильтр и растворитель удаляют в вакууме. Остаток очищают хроматографией (силикагель, элюирование с градиентом: 0-25% этилацетата в циклогексане) с получением указанного в заголовке соединения в виде твердого не совсем белого вещества (2,71 г, 86%).
1H ЯМР (400 МГц, CDCl3): δ 8,10-8,07 (м, 1 H), 8,04-8,00 (м, 1 H), 7,46-7,41 (м, 2 H), 7,09 (дд, J=8,6, 8,6 Гц, 2 H), 6,48-6,37 (м, 2 H), 3,91 (c, 3 H).
Стадия 3: Получение 4-амино-4'-фтор-5-йод-[1,1'-бифенил]-3-карбоновой кислоты
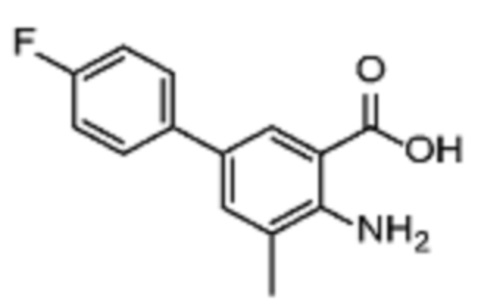
К раствору метил-4-амино-4'-фтор-5-йод-[1,1'-бифенил]-3-карбоксилата (2,59 г, 6,98 ммоль) в 1,4-диоксане (25 мл) и воде (5 мл) добавляют моногидрат гидроксида лития (1,75 г, 41,87 ммоль). Смесь перемешивают при комнатной температуре в течение 18 часов. Реакционную смесь разбавляют водой (100 мл) и диэтиловым эфиром (100 мл) и разделяют. Водную фазу подкисляют 1N HCl (45 мл) до pH=1 и экстрагируют дихлорметаном (3 × 50 мл). Органические фазы объединяют, фильтруют через гидрофобный фриттовый фильтр и растворитель удаляют в вакууме с получением указанного в заголовке соединения в виде твердого не совсем белого вещества (2,39 г, 96%).
¹H ЯМР (400 МГц, ДМСО): δ 13,17 (c, 1 H), 8,18 (д, J=2,0 Гц, 1 H), 8,09 (д, J=2,3 Гц, 1 H), 7,65 (дд, J=5,4, 8,5 Гц, 2 H), 7,27 (дд, J=8,8, 8,8 Гц, 2 H), 6,84 (c, 2 H).
Стадия 4: Получение 6-(4-фторфенил)-8-йодхиназолин-4(3H)-она

Раствор 4-амино-4'-фтор-5-йод-[1,1'-бифенил]-3-карбоновой кислоты (2,39 г, 6,69 ммоль) в формамидe (4 мл) нагревают до 130°С и выдерживают при указанной температуре в течение16 часов. После охлаждения до комнатной температуры реакционную смесь разбавляют водой (20 мл) и перемешивают в течение 20 минут, затем фильтруют. Твердый продукт промывают водой (3 × 5 мл), затем 10% MeOH в диэтиловом эфире (3 × 5 мл) с получением указанного в заголовке соединения в виде твердого не совсем белого вещества (2,14 г, 87%).
¹H ЯМР (400 МГц, ДМСО): δ 12,57 (c, 1 H), 8,66 (c, 1 H), 8,36 (c, 1 H), 8,28 (c, 1 H), 7,89 (дд, J=5,6, 7,8 Гц, 2 H), 7,38 (дд, J=8,6, 8,6 Гц, 2 H).
Стадия 5: Получение (R)-6-(4-фторфенил)-8-йод-N-(1-(2-(трифторметил)пиримидин-5-ил)этил)хиназолин-4-амина

К раствору 6-(4-фторфенил)-8-йодхиназолин-4-(3H)-она (1,14 г, 3,11 ммоль) в N, N-диметилформамидe (10 мл) осторожно добавляют гексафторфосфат (бензотриазол-1-илокси)трипирролидинофосфония (2,03 г, 1,25 ммоль) и ди-изопропилэтиламин (2,7 мл, 15,57 ммоль). Полученную смесь нагревают до 45ºС и выдерживают при указанной температуре в течение часа, затем добавляют гидрохлорид (R)-1-(2-(трифторметил)пиримидин-5-ил)этан-1-амина (0,96 г, 4,2 ммоль), поддерживая температуру смеси на уровне 45°С, и выдерживают при указанной температуре в течение 2 часов. После охлаждения до комнатной температуры смесь разбавляют этилацетатом (75 мл) и водой (175 мл). Органическую фазу промывают насыщенным раствором соли (2 × 20 мл), пропускают через гидрофобный фриттовый фильтр и растворитель удаляют в вакууме. Остаток очищают хроматографией (силикагель, элюирование с градиентом: 0-15% этилацетата в дихлорметане) с получением указанного в заголовке соединения в виде твердого не совсем белого вещества (1,27 г, 75%).
¹H ЯМР (400 МГц, ДМСО): δ 9,18 (c, 2 H), 8,88 (д, J=6,9 Гц, 1 H), 8,71 (д, J=1,8 Гц, 1 H), 8,67 (д, J=1,8 Гц, 1 H), 8,51 (c, 1 H), 7,95-7,90 (м, 2 H), 7,40 (дд, J=8,9, 8,9 Гц, 2 H), 5,73-5,67 (м, 1 H), 1,75 (д, J=7,2 Гц, 3 H).
Соединения, представленные в таблице ниже, получают в соответствии с описанной выше методикой, используя подходящий амин.
Пример 24
(R)-6-(4-Фторфенил)-8-(пиридазин-4-ил)-N-(1-(2-(трифторметил)пиримидин-5-ил)этил)хиназолин-4-амин
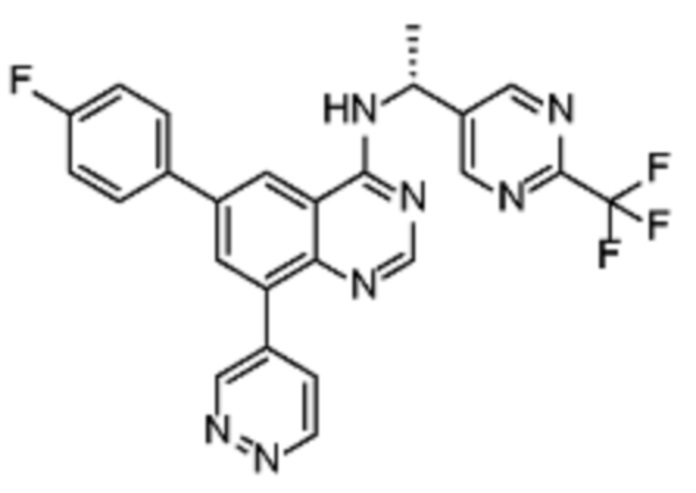
Газообразный азот барботируют в течение 10 минут через суспензию (R)-6-(4-фторфенил)-8-йод-N-(1-(2-(трифторметил)пиримидин-5-ил)этил)хиназолин-4-амина (50 мг, 0,093 ммоль), 4-(трибутилстаннил)пиридазина (44 мг, 0,12 ммоль, 1,3 экв.), тетракис(трифенилфосфин)палладия(0) (16 мг, 0,014 ммоль, 0,15 экв.) в 1,4-диоксане (1 мл). Дегазированную смесь переносят в предварительно нагретый до 90°C смеситель. Смесь перемешивают при 90°С в течение 17 часов. Полученный раствор охлаждают до комнатной температуры. Полученный остаток смешивают с насыщенным водным раствором фторида калия (10 мл) в течение 1 часа и экстрагируют этилацетатом (3 × 15 мл). Объединенные органические слои пропускают через гидрофобный фриттовый фильтр и концентрируют в вакууме. Остаток очищают с помощью SCX картриджа, промывают метанолом (10 мл), затем элюируют 10% об./об. 7 N аммония в метаноле. Полученный остаток очищают препаративной ЖХВД с получением указанного в заголовке соединения (6,0 мг, выход 13%) в виде твердого не совсем белого вещества.
¹H ЯМР (400 МГц, ДМСО): δ 9,65-9,63 (м, 1 H), 9,35 (дд, J=1,1, 5,3 Гц, 1 H), 9,21 (c, 2 H), 8,93 (д, J=6,9 Гц, 1 H), 8,84 (д, J=1,9 Гц, 1 H), 8,50 (c, 1 H), 8,36 (д, J=1,9 Гц, 1 H), 8,10 (дд, J=2,3, 5,3 Гц, 1 H), 8,07-8,02 (м, 2 H), 7,44 (дд, J=8,8, 8,8 Гц, 2 H), 5,72 (дд, J=7,0, 7,0 Гц, 1 H), 1,78 (д, J=7,0 Гц, 3 H). ЖХМС (метод 4): [MH+]=492 при 4,49 мин.
Пример 25
(R)-6-(4-Фторфенил)-8-(пиримидин-5-ил)-N-(1-(2-(трифторметил)пиримидин-5-ил)этил)хиназолин-4-амин
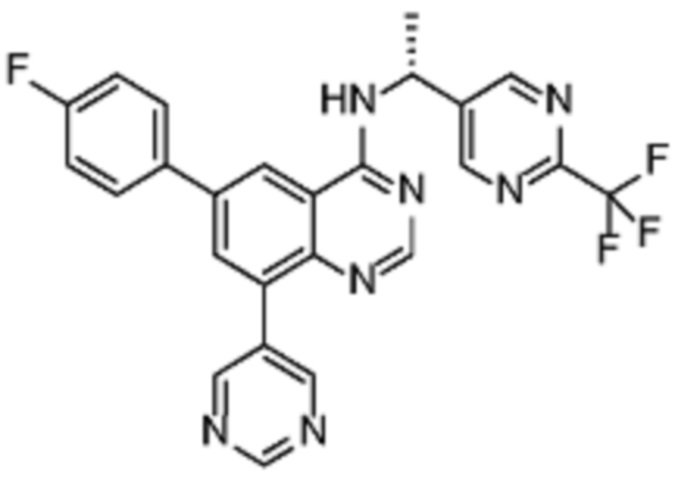
Газообразный азот барботируют в течение 10 минут через суспензию (R)-6-(4-фторфенил)-8-йод-N-(1-(2-(трифторметил)пиримидин-5-ил)этил)хиназолин-4-амина (100 мг, 0,18 ммоль), пиримидин-5-илбороновой кислоты (31 мг, 0,25 ммоль), фторида цезия (85 мг, 0,556 ммоль) в воде (0,5 мл) и N, N-диметилформамидe (2,0 мл), затем добавляют тетракис(трифенилфосфин)палладия(0) (21 мг, 0,018 ммоль). Полученную смесь нагревают до 95°С и выдерживают при указанной температуре в течение 18 часов. После этого реакционную смесь охлаждают до комнатной температуры и фильтруют через Celite, промывая этилацетатом (2 × 10 мл). Объединенные органические фракции распределяют с водой (15 мл), сушат с помощью картриджа фазоразделителя и концентрируют в вакууме. Полученный остаток очищают препаративной ЖХВД с получением указанного в заголовке соединения (49 мг, 55%) в виде твердого не совсем белого вещества.
¹H ЯМР (400 МГц, ДМСО): δ 9,22-9,18 (м, 5 H), 8,90 (д, J=6,9 Гц, 1 H), 8,80 (д, J=1,9 Гц, 1 H), 8,49 (c, 1 H), 8,34 (д, J=1,9 Гц, 1 H), 8,07-8,03 (м, 2 H), 7,43 (дд, J=8,9, 8,9 Гц, 2 H), 5,75-5,70 (м, 1 H), 1,78 (д, J=7,2 Гц, 3 H). ЖХМС (метод 3): [MH+]=492 при 4,88 мин.
Соединения, представленные в таблице ниже, получают в соответствии с методикой, описанной для получения (R)-6-(4-фторфенил)-8-(пиримидин-5-ил)-N-(1-(2-(трифторметил)пиримидин-5-ил)этил)хиназолин-4-амина.
структура
¹H ЯМР
ЖХ-МС

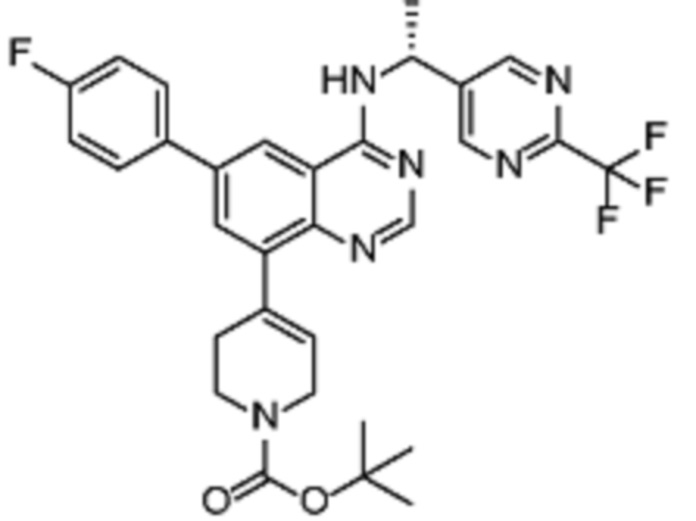
Пример 26
(R)-6-(4-Фторфенил)-8-(тетрагидро-2H-пиран-4-ил)-N-(1-(2-(трифторметил) пиримидин-5-ил)этил)хиназолин-4-амин

Смесь (R)-8-(3,6-дигидро-2H-пиран-4-ил)-6-(4-фторфенил)-N-(1-(2-(трифторметил)пиримидин-5-ил)этил)хиназолин-4-амина (46 мг, 0,093 ммоль) и 10% Pd/C (10 мг) в этаноле (15 мл) подвергают воздействию атмосферы водородй в течение 36 часов. После этого смесь пропускают через Celite и концентрируют в вакууме. Остаток очищают препаративной ЖХВД с получением указанного в заголовке соединения (14,2 мг, 30%) в виде твердого не совсем белого вещества.
¹H ЯМР (400 МГц, ДМСО): δ 9,18 (c, 2 H), 8,71 (д, J=6,9 Гц, 1 H), 8,54 (д, J=1,9 Гц, 1 H), 8,51 (c, 1 H), 7,97-7,92 (м, 3 H), 7,40 (дд, J=8,9, 8,9 Гц, 2 H), 5,72-5,66 (м, 1 H), 4,03-3,97 (м, 3 H), 3,60-3,52 (м, 2 H), 1,98-1,83 (м, 2 H), 1,78-1,71 (м, 4 H), 1,52-1,42 (м, 1 H). ЖХМС (метод 4): [MH+]=498 при 4,33 мин.
Пример 27
(R)-6-(4-Фторфенил)-8-(1,2,3,6-тетрагидропиридин-4-ил)-N-(1-(2-(трифторметил)пиримидин-5-ил)этил)хиназолин-4-амин
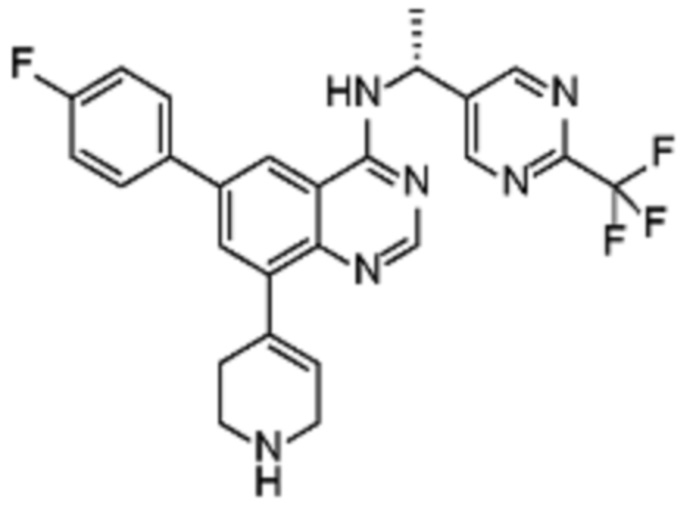
К раствору трет-бутил-(R)-4-(6-(4-фторфенил)-4-((1-(2-(трифторметил)пиримидин-5-ил)этил)амино)хиназолин-8-ил)-3,6-дигидропиридин-1(2H)-карбоксилата (промежуточное соединение 54) (120 мг, 0,20 ммоль) в метаноле (10,0 мл) добавляют 4N HCl раствор в 1,4-диоксане (1 мл, 4,04 ммоль). Смесь перемешивают при комнатной температуре в течение 18 часов. Реакционную смесь осторожно загружают на SCX картридж. Картридж промывают метанолом и фильтрат элюируют 7M раствором аммония в метаноле. Растворитель удаляют в вакууме с получением указанного в заголовке соединения (12,5 мг, 13%) в виде твердого не совсем белого вещества.
1H ЯМР (400 МГц, ДМСО): δ 9,18 (c, 2 H), 8,70 (д, J=6,9 Гц, 1 H), 8,58 (д, J=1,9 Гц, 1 H), 8,45 (c, 1 H), 7,92 (дд, J=5,4, 8,8 Гц, 2 H), 7,85 (д, J=1,9 Гц, 1 H), 7,39 (дд, J=8,9, 8,9 Гц, 2 H), 5,95 (c, 1 H), 5,71-5,66 (м, 1 H), 3,42 (д, J=2,5 Гц, 2 H), 2,96 (дд, J=5,5, 5,5 Гц, 2 H), 2,59-2,59 (м, 2 H), 1,75 (д, J=7,2 Гц, 3 H). NH не обнаружен. ЖХМС (метод 4): [MH+]=495 при 3,1 мин.
Промежуточное соединение 11
Гидробромид 2-амино-5-бром-3-метоксибензойной кислоты
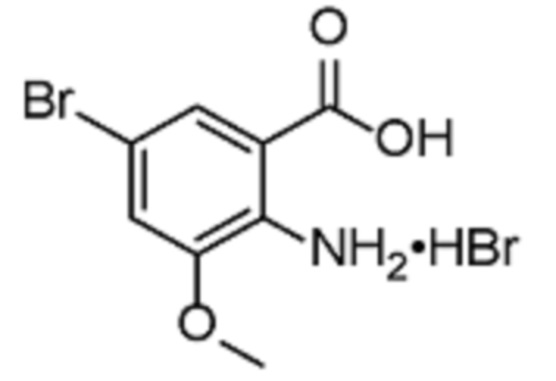
Раствор брома (6,0 г, 1,9 мл, 37,70 ммоль) в хлороформе (15 мл) по каплям в течение часа добавляют к суспензии 2-амино-3-метоксибензойной кислоты (6,0 г, 35,90 ммоль) в хлороформе (180 мл) при 0°C. Реакционную смесь перемешивают в течение дополнительных пяти часов и дают возможность смеси медленно нагреться до комнатной температуры. Растворитель удаляют в вакууме и остаток растирают с диэтиловым эфиром. Реакционную смесь фильтруют с получением указанного в заголовке соединения в виде твердого вещества бежевого цвета (11,3 г, выход 96%).
ЖХМС (метод 4): [MH+]=247 при 4,07 мин.
Промежуточное соединение 10
6-Бром-8-метоксихиназолин-4-ол

Раствор гидробромида 2-амино-5-бром-3-метоксибензойной кислоты (промежуточное соединение 11) (10,0 г, 30,60 ммоль) в формамидe (40 мл) нагревают до 165°С и выдерживают при указанной температуре в течение 18 часов. После охлаждения до комнатной температуры реакционную смесь разбавляют водой (100 мл), выливают в ледяную воду (400 мл) и фильтруют. Осадок промывают водой (200 мл) и диэтиловым эфиром (200 мл) с получением указанного в заголовке соединения в виде твердого вещества светло-коричневого цвета (5,9 г, 76%).
ЖХМС (метод 4): [MH+]=255 при 3,07 мин.
Промежуточное соединение 16
6-(4-Фторфенил)-8-метоксихиназолин-4-ол

Газообратный азот барботируют в течение 5 минут через смесь 6-бром-8-метоксихиназолин-4-ола (промежуточное соединение 10) (1,18 г, 4,63 ммоль), 4-фторфенилбороновой кислоты (710 мг, 5,09 ммоль) и карбоната цезия (5,73 г, 17,58 ммоль) в 1,4-диоксане (30 мл) и воде (7,5 мл), затем добавляют комплекс [1,1′-бис(дифенилфосфино)ферроцен]дихлорпалладия(II) с дихлорметаном (190 мг, 0,23 ммоль), реакционную смесь нагревают до 110°С и выдерживают при указанной температуре в течение 5 часов. После охлаждения до комнатной температуры смесь разбавляют водой (20 мл), фильтруют, твердый продукт промывают 10% метанолом в диэтиловом эфире, затем диэтиловым эфиром с получением указанного в заголовке соединения (1,0 г, 80%) в виде твердого вещества бежевого цвета.
ЖХМС (метод 5): [MH+]=271,1 при 0,81 мин.
Промежуточное соединение 17
6-(4-Фторфенил)-8-метокси-N-(1-(3-метил-1,2,4-оксадиазол-5-ил)этил)хиназолин-4-амин
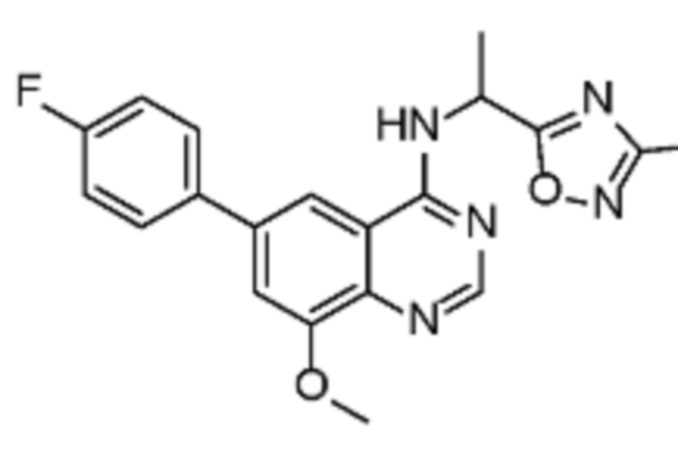
К раствору 6-(4-фторфенил)-8-метоксихиназолин-4-ола (промежуточное соединение 16) (100 мг, 0,37 ммоль) в N, N-диметилформамидe (2 мл) осторожно добавляют гексафторфосфат (бензотриазол-1-илокси)трипирролидинофосфония (212 мг, 0,41 ммоль) и ди-изопропилэтиламин (0,32 мл, 1,85 ммоль). Полученную смесь нагревают до 40ºC и перемешивают в течение 20 мин., добавляют 1-(3-метил-1,2,4-оксадиазол-5-ил)этан-1-амин (67 мг, 0,41 ммоль) и выдерживают смесь при 40°С в течение 18 часов. После охлаждения до комнатной температуры смесь разбавляют этилацетатом (50 мл) и водой (20 мл). Органическую фазу промывают насыщенным раствором соли (2 × 20 мл), пропускают через гидрофобный фриттовый фильтр и удаляют растворитель в вакууме. Остаток очищают препаративной ЖХВД с получением указанного в заголовке соединения (43 мг, 31%) в виде твердого белого вещества.
¹H ЯМР (400 МГц, ДМСО): δ 8,85 (д, J=6,8 Гц, 1 H), 8,49 (c, 1 H), 8,24 (c, 1 H), 7,99 (дд, J=5,6, 8,6 Гц, 2 H), 7,61 (c, 1 H), 7,44 (дд, J=8,8, 8,8 Гц, 2 H), 5,88-5,82 (м, 1 H), 4,08 (c, 3 H), 2,37 (c, 3 H), 1,78 (д, J=7,1 Гц, 3 H). ЖХМС (метод 4): [MH+]=375 при 3,29 мин.
Соединения, представленные в таблице ниже, синтезируют в соответствии с методикой, описанной для получения 6-(4-фторфенил)-8-метокси-N-(1-(3-метил-1,2,4-оксадиазол-5-ил)этил)хиназолин-4-амина.
Структура
1H ЯМР
ЖХ-МС
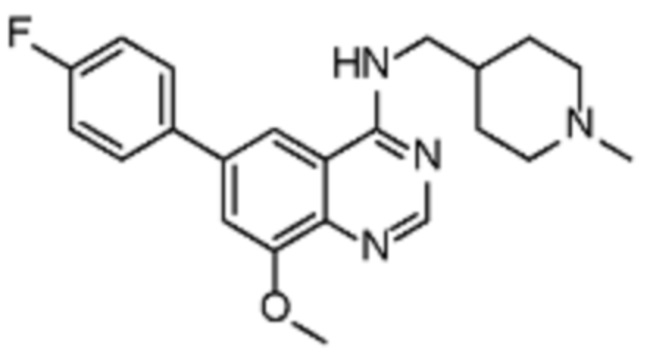
ЖХМС (метод 4): [MH+]=381 при 2,27 мин.
Промежуточное соединение 8
(R)-6-Бром-8-метокси-N-(1-(6-метилпиридазин-3-ил)этил)хиназолин-4-амин

К раствору 6-бром-8-метоксихиназолин-4-ола (промежуточное соединение 10) (65 мг, 0,27 ммоль) в N, N-диметилформамидe (1,5 мл) осторожно добавляют гексафторфосфат (бензотриазол-1-илокси)трипирролидинофосфония (139 мг, 0,27 ммоль) и ди-изопропилэтиламин (0,2 мл, 0,81 ммоль). Полученную смесь нагревают до 60°С и выдерживают при указанной температуре в течение часа, затем добавляют (R)-1-(6-метилпиридазин-3-ил)этан-1-амин (65 мг, 0,27 ммоль) и выдерживают при 60°С в течение 18 часов. После охлаждения до комнатной температуры реакционную смесь концентрируют непосредственно на силикагеле и очищают хроматографией (силикагель, элюирование с градиентом: 0-100% (10% MeOH в этилацетате) в этилацетате) с получением указанного в заголовке соединения в виде твердого вещества бежевого цвета (100 мг, количественный выход).
ЖХМС (метод 4): [MH+]=374 при 2,42 миг.
Промежуточные соединения, представленные в таблице ниже, синтезируют в соответствии с методикой, описанной для получения (R)-6-бром-8-метокси-N-(1-(6-метилпиридазин-3-ил)этил)хиназолин-4-амина (промежуточное соединение 8).
Структура
ЖХ-МС


Промежуточное соединение 5
6-(4-Фтор-3-метилфенил)-8-метокси-N-((6-метилпиридазин-3-ил)метил)хиназолин-4-амин
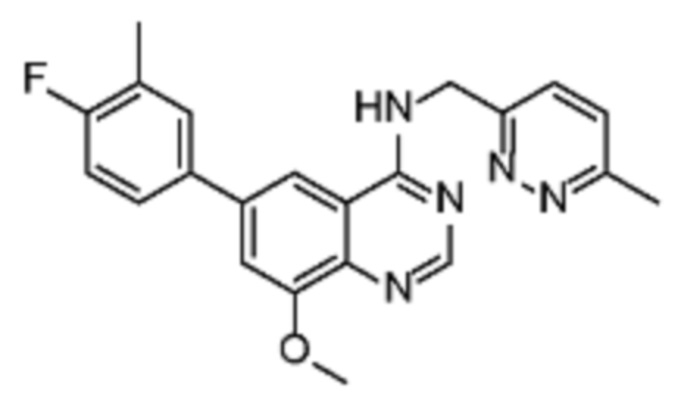
К раствору 6-бром-8-метокси-N-((6-метилпиридазин-3-ил)метил)хиназолин-4-амина (промежуточное соединение 9) (70 мг, 0,19 ммоль) в 1,4-диоксане (4,0 мл) добавляют 4-фтор-3-метилфенилбороновую кислоту (33 мг, 0,21 ммоль), тетракис(трифенилфосфин)палладий(0) (20 мг, 0,02 ммоль), карбонат калия (36 мг, 0,26 ммоль) и воду (0,5 мл). Полученную смесь нагревают до 95°С и выдерживают при указанной температуре в течение 18 часов. После охлаждения до комнатной температуры реакционную смесь фильтруют через Celite®. Осадок на Celite® промывают этилацетатом (2 × 20 мл). Объединенные органические фазы промывают насыщенным раствором соли (2 × 20 мл), фильтруют через гидрофобный фриттовый фильтр и растворитель удаляют в вакууме. Остаток очищают препаративной ЖХВД с получением указанного в заголовке соединения в виде твердого не совсем белого вещества (18 мг, 24%).
1Н ЯМР (400 МГц, ДМСО): δ 9,03 (дд, J=5,8, 5,8 Гц, 1H), 8,41 (c, 1H), 8,15 (д, J=1,8 Гц, 1H), 7,82 (дд, J=2,0, 7,4 Гц, 1H), 7,77-7,72 (м, 1H), 7,57-7,49 (м, 3H), 7,31 (дд, J=9,1, 9,1 Гц, 1H), 5,03 (д, J=5,8 Гц, 2H), 4,03 (c, 3H), 2,60 (c, 3H), 2,37 (д, J=1,8 Гц, 3H).
ЖХМС (метод 4): [MH+]=390 при 3,12 мин.
Соединения, представленные в таблице ниже, получают в соответствии с методикой, описанной для получения 6-(4-фтор-3-метилфенил)-8-метокси-N-((6-метилпиридазин-3-ил)метил)хиназолин-4-амина (промежуточное соединение 5).

ЖХМС (метод 3): [MH+]=394 при 3,79 мин.
PdCl2dppf используют в качестве катализатора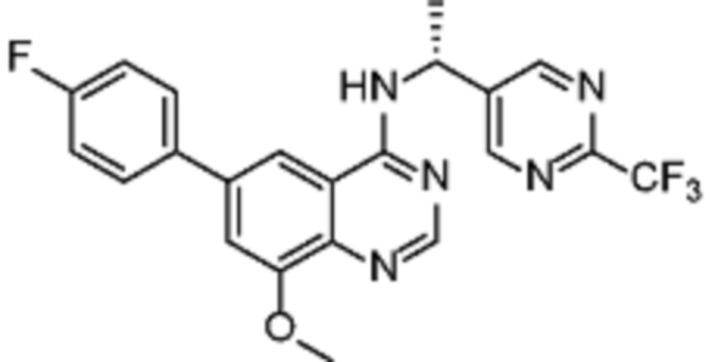
1H ЯМР (400 МГц, ДМСО-d6) δ м, д, 9,12 (c, 2 H), 8,55 (д, J=7,0 Гц, 1 H), 8,37 (c, 1 H), 8,14 (д, J=1,3 Гц, 1 H), 7,83-7,96 (м, 2 H), 7,48 (д, J=1,3 Гц, 1 H), 7,35 (т, J=8,8 Гц, 2 H), 5,50-5,60 (м, 1 H), 3,97 (c, 3 H), 1,69 (д, J=7,0 Гц, 3 H)
PdCl2dppf используют в качестве катализатора
1H ЯМР (400 МГц, ацетонитрил-d3) δ м.д. 8,43 (c, 1 H), 7,66-7,84 (м, 3 H), 7,53 (д, J=8,8 Гц, 1 H), 7,44 (д, J=1,3 Гц, 1 H), 7,39 (д, J=8,8 Гц, 1 H), 7,20-7,30 (м, 2 H), 7,00-7,15 (м, 1 H), 5,06 (c, 2 H), 4,04 (c, 3 H), 2,62 (c, 3 H)
Промежуточное соединение 14
8-Метокси-6-(5-метил-1,3,4-тиадиазол-2-ил)-N-((6-метилпиридазин-3-ил)метил)хиназолин-4-амин

Стадия 1: Получение 8-метокси-N-((6-метилпиридазин-3-ил)метил)-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)хиназолин-4-амина
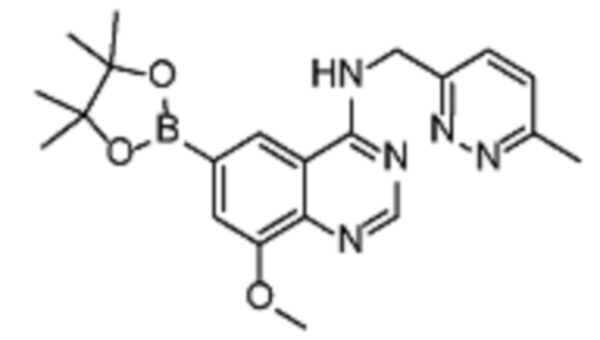
Газообратный азот барботируют в течение 5 минут через смесь 6-бром-8-метокси-N-((6-метилпиридазин-3-ил)метил)хиназолин-4-амина (промежуточное соединение 9) (250 мг, 0,69 ммоль), бис(пинаколато)дибора (194 мг, 0,76 ммоль), [1,1’-бис-(дифенилфосфино)ферроцен]дихлорпалладия(II) (25 мг, 0,03 ммоль) и ацетата калия (204 мг, 2,08 ммоль) в 1,4-диоксане (15,0 мл). Смесь нагревают до 90°С и выдерживают при указанной температуре в течение 18 часов. После охлаждения до комнатной температуры реакционную смесь фильтруют через Celite® и растворитель удаляют в вакууме. Остаток используют в следующей стадии без дополнительной очистки.
Стадия 2: Получение 8-метокси-6-(5-метил-1,3,4-тиадиазол-2-ил)-N-((6-метилпиридазин-3-ил)метил)хиназолин-4-амина
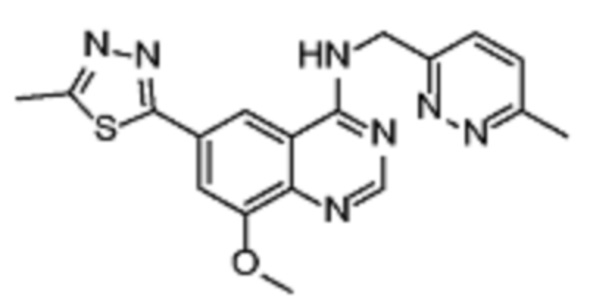
Газообратный азот барботируют в течение 5 минут через смесь 2-бром-5-метил-1,3,4-тиадиазола (34 мг, 0,19 ммоль), 8-метокси-N-((6-метилпиридазин-3-ил)метил)-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)хиназолин-4-амина (70 мг, 0,17 ммоль), карбоната калия (36 мг, 0,26 ммоль) и воды (0,5 мл) в 1,4-диоксане (4,0 мл), затем добавляют тетракис(трифенилфосфин)палладия(0) (20 мг, 0,02 ммоль). Полученную смесь нагревают до 95°С и выдерживают при указанной температуре в течение 16 часов. После охлаждения до комнатной температуры реакционную смесь фильтруют через Celite® и промывают этилацетатом (20 мл). Органические фазы объединяют, пропускают через гидрофобный фриттовый фильтр и растворитель удаляют в вакууме. Остаток очищают препаративной ЖХВД с получением указанного в заголовке соединения в виде твердого не совсем белого вещества (21,0 мг, 32%).
¹H ЯМР (400 МГц, ДМСО): δ 9,31 (дд, J=5,8, 5,8 Гц, 1 H), 8,47 (c, 1 H), 8,44 (д, J=1,7 Гц, 1 H), 7,80 (д, J=1,4 Гц, 1 H), 7,58 (д, J=8,7 Гц, 1 H), 7,51 (д, J=8,7 Гц, 1 H), 5,03 (д, J=5,8 Гц, 2 H), 4,03 (c, 3 H), 2,84 (c, 3 H), 2,60 (c, 3 H). ЖХМС (метод 3): [MH+]=380 при 2,13 мин.
Соединения, представленные в таблице ниже, получают в соответствии с методикой, описанной для получения 8-метокси-6-(5-метил-1,3,4-тиадиазол-2-ил)-N-((6-метилпиридазин-3-ил)метил)хиназолин-4-амина.
структура
¹H ЯМР
ЖХ-МС
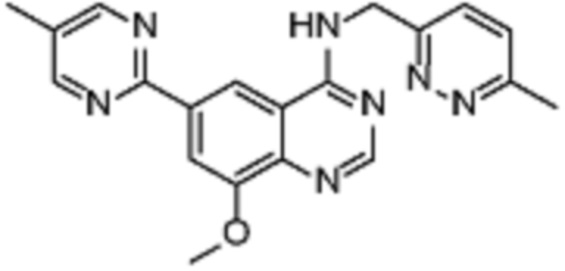
ЖХМС (метод 3): [MH+]=374 при 3,25 мин.
Промежуточное соединение 15a
8-Метокси-N-((6-метилпиридазин-3-ил)метил)-6-(5-метилтиазол-2-ил)хиназолин-4-амин

Стадия 1: Получение 6-(5,5-диметил-1,3,2-диоксаборинан-2-ил)-8-метокси-N-((6-метилпиридазин-3-ил)метил)хиназолин-4-амина

Газообратный азот барботируют в течение 5 минут через смесь 6-бром-8-метокси-N-((6-метилпиридазин-3-ил)метил)хиназолин-4-амина (промежуточное соединение 6) (100 мг, 0,28 ммоль), бис(неопентилгликолято)дибора (66 мг, 0,29 ммоль), комплекса [1,1’-bis(дтфенилфосфино)ферроцен]дихлорпалладия (II) с дихлорметаном (10 мг, 0,02 ммоль) и ацетата калия (54 мг, 0,55 ммоль) в 1,4-диоксане (3,0 мл). Смесь нагревают до 100°С и выдерживают при указанной температуре в течение3 часов. После охлаждения до комнатной температуры, смесь используют в следующей стадии в виде раствора в 1,4-диоксане без дополнительной очистки.
Стадия 2: Получение 8-метокси-N-((6-метилпиридазин-3-ил)метил)-6-(5-метилтиазол-2-ил)хиназолин-4-амина
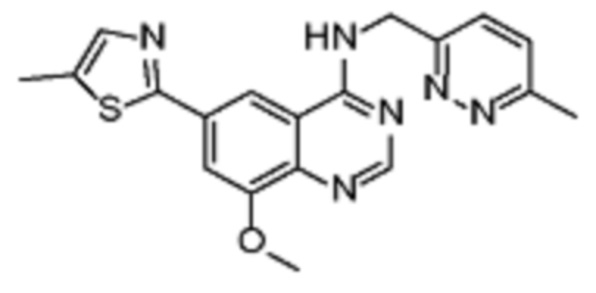
К раствору полученного выше 6-(5,5-диметил-1,3,2-диоксаборинан-2-ил)-8-метокси-N-((6-метилпиридазин-3-ил)метил)хиназолин-4-амина (100 мг, 0,28 ммоль) добавляют водный карбонат цезия (181 мг, 0,56 ммоль, 0,4 мл), 2-бром-5-метилтиазол (64 мг, 0,28 ммоль) и тетракис(трифенилфосфин)палладий(0) (20 мг, 0,02 ммоль). Полученную смесь нагревают до 95°С и выдерживают при указанной температуре в течение 16 часов. После охлаждения до комнатной температуры смесь фильтруют через Celite® и промывают осадок на фильтре этилацетатом (2 × 10 мл). Органические фазы промывают насыщенным водным каствором хлорида аммония (10 мл), пропускают через гидрофобный фриттовый фильтр и удаляют растворитель в вакууме. Остаток очищают препаративной ЖХВД с получением указанного в заголовке соединения в виде твердого не совсем белого вещества (25 мг, 24%).
1H ЯМР (400 МГц, ДМСО): δ 9,24 (дд, J=5,7, 5,7 Гц, 1 H), 8,43 (c, 1 H), 8,37 (д, J=1,5 Гц, 1 H), 7,75 (д, J=1,5 Гц, 1 H), 7,68 (д, J=1,1 Гц, 1 H), 7,57 (д, J=8,7 Гц, 1 H), 7,50 (д, J=8,7 Гц, 1 H), 5,02 (д, J=5,8 Гц, 2 H), 4,00 (c, 3 H), 2,60 (c, 3 H), 2,50 (c, 3 H). ЖХМС (метод 3): [MH+]=379 при 3,20 мин.
Промежуточное соединение 1
(R)-6-(4-Фторфенил)-4-((1-(2-(трифторметил)пиримидин-5-ил)этил)амино)-хиназолин-8-ол
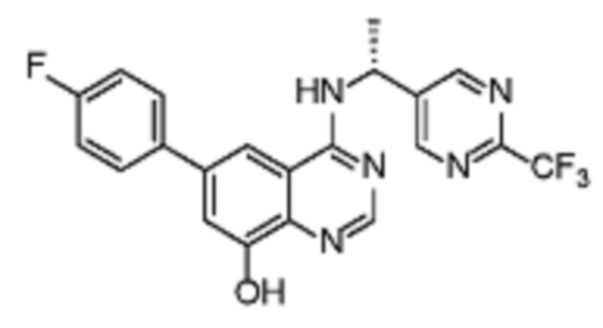
К раствору 6-(4-фторфенил)-8-метокси-N-[(1R)-1-[2-(трифторметил)-пиримидин-5-ил]этил]хиназолин-4-амина (промежуточное соединение 7a) (490 мг, 1,11 ммоль) в хлороформе (8 мл) при 0°C по каплям добавляют трибромид бора (0,32 мл, 3,32 ммоль). Реакционной смеси дают возможность нагреться до комнатной температуры, затем нагревают до 65°С и выдерживают при указанной температуре в течение 18 часов. После охлаждения до комнатной температуры реакционную смесь охлаждают на ледяной бане и гасят метанолом (2 мл). Растворитель удаляют в вакууме. Остаток разбавляют этилацетатом (50 мл) и промывают насыщенным водным раствором NaHCO3 (50 мл). Водный слой экстрагируют этилацетатом (2 × 20 мл). Органические фазы объединяют, пропускают через гидрофобный фриттовый фильтр и удаляют растворитель в вакууме с получением указанного в заголовке соединения в виде твердого вещества серого цвета (416 мг, выход 88%).
1H ЯМР (400 МГц, ДМСО): δ 9,18 (c, 2 H), 8,65 (д, J=7,0 Гц, 1 H), 8,46 (c, 1 H), 8,09 (c, 1 H), 7,86 (дд, J=5,5, 8,7 Гц, 2 H), 7,45 (д, J=1,3 Гц, 1 H), 7,37 (дд, J=8,8, 8,8 Гц, 2 H), 5,71-5,67 (м, 1 H), 1,75 (д, J=7,0 Гц, 3 H). OH не обнаружен.
Представленные далее промежуточные соединения получают в соответствии с описанной выше методикой, исходя из соединения, указанного в таблице.
ЖХ-МС
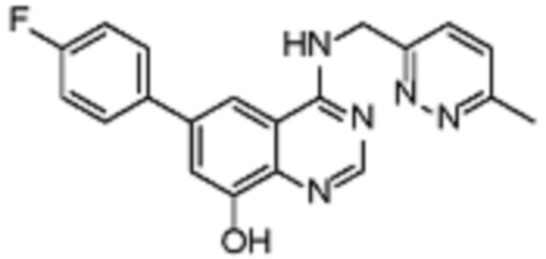

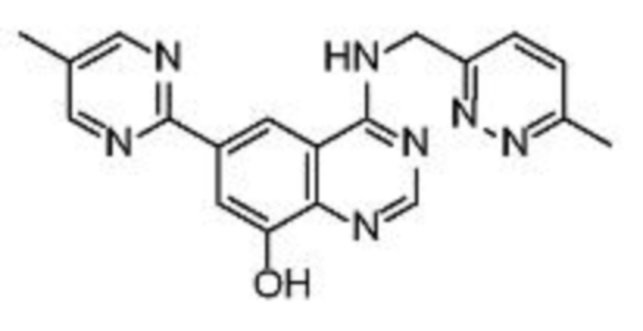
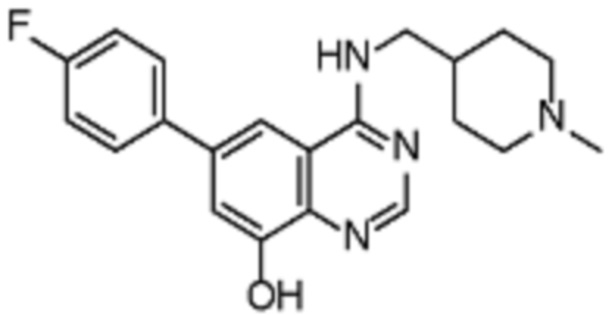
Промежуточное соединение 59
6-Бром-N-((6-метилпиридазин-3-ил)метил)-8-((тетрагидро-2H-пиран-4-ил)окси)хиназолин-4-амин

Стадия 1: Получение 6-бром-4-(((6-метилпиридазин-3-ил)метил)амино)хиназолин-8-ола

6-Бром-8-метокси-N-((6-метилпиридазин-3-ил)метил)хиназолин-4-амин (500 мг, 1,39 ммоль) суспендируют в хлороформе (10 мл) и охлаждают до 0°C. К смеси медленно добавляют трибромид бора (0,40 мл, 4,16 ммоль). Реакционную смесь перемешивают при 0°С и выдерживают при указанной температуре в течение 5 минут, затем нагревают до 70°С и выдерживают при указанной температуре в течение 16 часов. После этого смесь охлаждают до комнатной температуры и гасят метанолом. Растворители удаляют в вакууме. Остаток растирают с водой и сушат при пониженном давлении в течение 16 часов с получением указанного в заголовке соединения (480 мг, количественный выход) в виде твердого не совсем белого вещества.
ЖХМС (метод 4): [MH+]=346 при 2,07 мин.
Стадия 2: Получение 6-бром-N-((6-метилпиридазин-3-ил)метил)-8-((тетрагидро-2H-пиран-4-ил)окси)хиназолин-4-амина
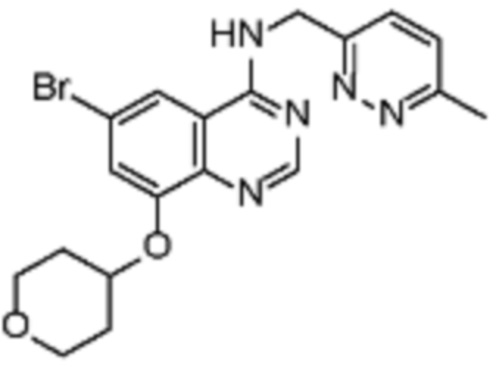
Смесь 6-бром-4-(((6-метилпиридазин-3-ил)метил)амино)хиназолин-8-ола (400 мг, 1,16 ммоль), тетрагидро-2H-пиран-4-илметансульфоната (458 мг, 2,54 ммоль) и карбоната цезия (1129 мг, 3,47 ммоль) в N, N-диметилформамидe (12 мл) нагревают до 50°С в атмосфере азота и выдерживают в указанных условиях в течение 16 часов. К реакционной смеси добавляют дополнительную аликвоту тетрагидро-2H-пиран-4-илметансульфоната (458 мг, 2,54 ммоль) и выдерживают реакционную смесь при 50°С в атмосфере азота в течение дополнительных 5 часов. Реакционную смесь охлаждают до комнатной температуры и затем распределяют между водой и этилацетатом. Две фазы разделяют и водную фазу further экстрагируют этилацетатом (4 × 10 мл). Объединенные органические слои were сушат над сульфатом магния, фильтруют и растворитель удаляют в вакууме. Остаток очищают обращенно-фазовой колоночной хроматографией (C18, элюирование с градиентом: 0-95% ацетонитрил (+ 0,1% гидроксида аммония) в воде (+ 0,1% гидроксида аммония) с получением указанного в заголовке соединения (110 мг, 22%) в виде твердого вещества коричневого цвета.
ЖХМС (метод 4): [MH+]=430 при 2,46 мин.
Промежуточные соединения, представленные в таблице ниже, получают в соответствии с методикой, описнной для получния 6-бром-N-((6-метилпиридазин-3-ил)метил)-8-((тетрагидро-2H-пиран-4-ил)окси)хиназолин-4-амина.
структура
¹H ЯМР
ЖХ-МС

ЖХМС (метод 4): [MH+]=444 при 2,61 мин.
Пример 28
Одиночный энантиомер 1 N-[1-(6-метилпиридазин-3-ил)этил]-6-(5-метилпиримидин-2-ил)-8-тетрагидропиран-4-илоксихиназолин-4-амина
Пример 29
Одиночный энантиомер 2 N-[1-(6-метилпиридазин-3-ил)этил]-6-(5-метилпиримидин-2-ил)-8-тетрагидропиран-4-илоксихиназолин-4-амина
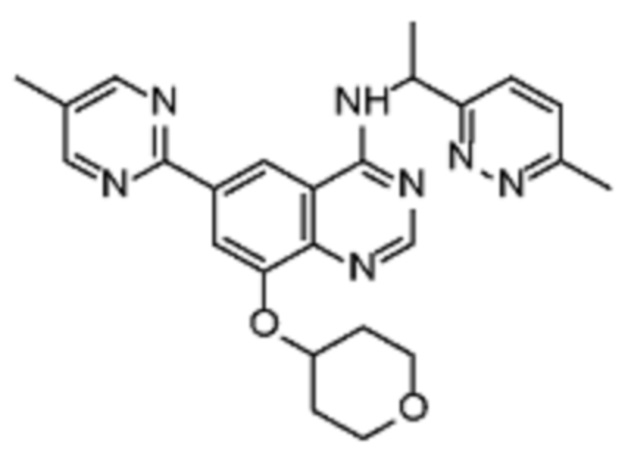
4-((1-(6-Метилпиридазин-3-ил)этил)амино)-6-(5-метилпиримидин-2-ил)хиназолин-8-ол (150 мг, 0,40 ммоль), тетрагидро-2H-пиран-4-ил метансульфонат (159 мг, 0,88 ммоль) и карбонат цезия (393 мг, 1,21 ммоль) растворяют в N, N-диметилформамидe (5 мл). Затем реакционную смесь нагревают до 50°С и выдерживают при указанной температуре в течение 16 часов. После этого смесь охлаждают до комнатной температуры, разбавляют водой (40 мл) и распределяют между водой и этилацетатом (50 мл). Фазы разделяют и водный слой дважды экстрагируют этилацетатом (2 × 40 мл). Объединенные органические слои сушат над гидрофобный фриттом и концентрируют в вакууме. Остаток очищают препаративной ЖХВД с получением указанного в заголовке соединения (50,6 мг, 27%) в виде твердого бесцветного вещества, которое представляет собой смесь двух энантиомеров. Одиночные изомеры получают очисткой рацемической описанной выше смеси хиральной препаративной СФХ.
Пример 28
Одиночный энантиомер 1 N-[1-(6-метилпиридазин-3-ил)этил]-6-(5-метилпиримидин-2-ил)-8-тетрагидропиран-4-илоксихиназолин-4-амина (16,9 мг, выход 66%):
1H ЯМР (400 МГц, CDCl3) δ 8,75 (c, 1 H), 8,68 (c, 2 H), 8,62 (д, J=2,9 Гц, 1 H), 8,28 (д, J=0,8 Гц, 1 H), 7,57 (д, J=5,4 Гц, 1 H), 7,45 (д, J=8,3 Гц, 1 H), 7,35 (д, J=8,3 Гц, 1 H), 5,84-5,76 (м, 1 H), 4,94-4,86 (м, 1 H), 4,15-4,08 (м, 2 H), 3,68-3,60 (м, 2 H), 2,75 (c, 3 H), 2,40 (c, 3 H), 2,25-2,16 (м, 2 H), 2,11-2,02 (м, 2 H), 1,76 (д, J=8,3 Гц, 3 H). ЖХМС (метод 4): [MH+]=394 при 2,81 мин. Хиральный анализ (метод 30): при 2,93 мин.
Пример 29
Одиночный энантиомер 2 N-[1-(6-метилпиридазин-3-ил)этил]-6-(5-метилпиримидин-2-ил)-8-тетрагидропиран-4-илоксихиназолин-4-амин (8,7 мг, 34%):
¹H ЯМР (400 МГц, CDCl3) δ 8,75 (c, 1 H), 8,68 (c, 2 H), 8,60 (д, J=2,9 Гц, 1 H), 8,28 (д, J=0,8 Гц, 1 H), 7,57 (д, J=5,4 Гц, 1 H), 7,45 (д, J=8,3 Гц, 1 H), 7,35 (д, J=8,3 Гц, 1 H), 5,84-5,76 (м, 1 H), 4,94-4,86 (м, 1 H), 4,15-4,08 (м, 2 H), 3,68-3,60 (м, 2 H), 2,75 (c, 3 H), 2,40 (c, 3 H), 2,25-2,16 (м, 2 H), 2,11-2,02 (м, 2 H), 1,76 (д, J=8,3 Гц, 3 H). ЖХМС (метод 3): [MH+]=394 при 3,68 мин. Хиральный анализ (метод 30): при 3,7 мин.
Пример 30
6-(5-Фтор-2-пиридил)-N-[(6-метилпиридазин-3-ил)метил]-8-тетрагидропиран-4-илоксихиназолин-4-амин
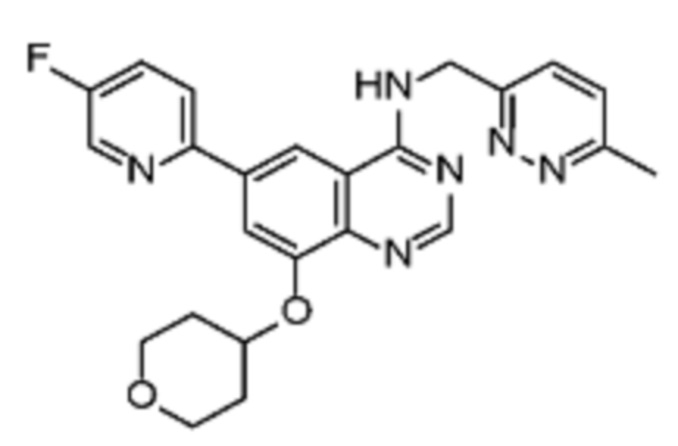
Газообразный азот барботируют в течение 5 минут через суспензию 6-бром-N-((6-метилпиридазин-3-ил)метил)-8-((тетрагидро-2H-пиран-4-ил)окси)хиназолин-4-амина (110 мг, 0,26 ммоль) и дихлорида бис(трифенилфосфин)палладия(II) (18 мг, 0,026 ммоль) в 1,4-диоксане (6 мл). К суспензии добавляют 5-фтор-2-(трибутилстаннил)пиридин (118 мг, 0,31 ммоль), смесь помещают в пробирку для микроволновой обработки и герметично закрывают. Реакционную смесь нагревают до 150°С и выдерживают при указанной температуре в течение 15 минут. После этого реакционную смесь разбавляют этилацетатом и фильтруют через слой Celite®. Раствворитель удаляют в вакууме и полученный остаток очищают препаративной ЖХВД с получением указанного в заголовке соединения (11,7 мг, 10%) в виде твердого бесцветного вещества.
¹H ЯМР (400 МГц, ДМСО): δ 9,14 (т, J=5,9 Гц, 1 H), 8,73 (д, J=2,9 Гц, 1 H), 8,60 (д, J=1,6 Гц, 1 H), 8,47 (c, 1 H), 8,25 (дд, J=4,3, 8,9 Гц, 1 H), 8,06 (д, J=1,5 Гц, 1 H), 7,96 (dt, J=3,0, 8,7 Гц, 1 H), 7,57 (д, J=8,7 Гц, 1 H), 7,50 (д, J=8,8 Гц, 1 H), 5,05 (д, J=5,7 Гц, 2 H), 4,98-4,90 (м, 1 H), 3,93 (td, J=4,6, 11,4 Гц, 2 H), 3,53 (ддд, J=2,7, 9,1, 11,6 Гц, 2 H), 2,60 (c, 3 H), 2,09-2,01 (м, 2 H), 1,78-1,68 (м, 2 H). ЖХМС (метод 4): [MH+]=447 при 2,69 мин.
Пример 31
N-[(6-Метилпиридазин-3-ил)метил]-6-(5-метилпиримидин-2-ил)-8-тетрагидропиран-4-илоксихиназолин-4-амин
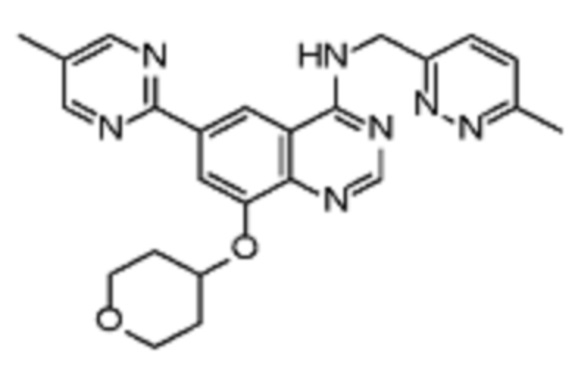
Газообразный азот барботируют в течение 5 минут через суспензию 6-бром-N-((6-метилпиридазин-3-ил)метил)-8-((тетрагидро-2H-пиран-4-ил)окси)хиназолин-4-амина (100 мг, 0,23 ммоль), бис(пинаколато)дибора (71 мг, 0,28 ммоль), комплекса [1,1’-бис(лифенилфосфино)ферроцен]дихллорпалладия(II) с дихлорметаном (9,5 мг, 0,012 ммоль) и ацетата калия (46 мг, 0,47 ммоль) в 1,4-диоксане (3 мл). Реакционную смесь нагревают до 100°С и выдерживают при указанной температуре в течение 4 часов. Реакционную смесь охлаждают до комнатной температуры и растворитель удаляют в вакууме. Остаток суспендируют в диоксане (3 мл) и воде (0,6 мл). К остатку добавляют 2-бром-5-метилпиримидин (39 мг, 0,22 ммоль), карбонат цезия (139 мг, 0,43 ммоль) и комлекс [1,1’-бис(дифенилфосфино)ферроцен]дихлорпалладия(II) с дихлорметаном (8,7 мг, 0,011 ммоль) и через полученную суспензию в течение 5 минут барботируют газообразный азот. После этого реакционную смесь нагревают до 100°С и выдерживают при указанной температуре в течение 16 часов. Реакционную смесь охлаждают до комнатной температуры, разбавляют метанолом и удаляют растворитель в вакууме. Полученный остаток очищают препаративной ЖХВД с получением указанного в заголовке соединения (15,8 мг, 15%) в виде твердого не совсем белого вещества.
¹H ЯМР (400 МГц, ДМСО): δ 9,31 (т, J=5,9 Гц, 1 H), 8,96 (д, J=1,5 Гц, 1 H), 8,84 (д, J=0,7 Гц, 2 H), 8,47 (c, 1 H), 8,28 (д, J=1,5 Гц, 1 H), 7,54 (д, J=8,6 Гц, 1 H), 7,49 (д, J=8,7 Гц, 1 H), 5,01 (д, J=5,7 Гц, 2 H), 4,95-4,88 (м, 1 H), 3,94 (td, J=4,6, 11,3 Гц, 2 H), 3,54 (ддд, J=2,7, 9,0, 11,6 Гц, 2 H), 2,60 (c, 3 H), 2,37 (c, 3 H), 2,09-2,02 (м, 2 H), 1,79-1,69 (м, 2 H). ЖХМС (метод 4): [MH+]=444 при 2,59 мин.
Пример 32
(R)-6-(1-Метил-1H-пиразол-3-ил)-N-(1-(6-метилпиридазин-3-ил)этил)-8-((тетрагидро-2H-пиран-4-ил)окси)хиназолин-4-амин
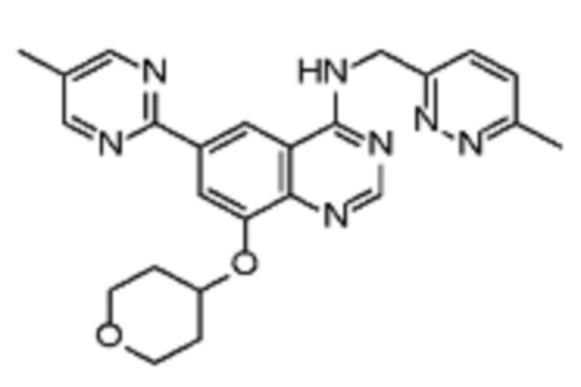
Газообразный азот барботируют в течение 10 минут через смесь (R)-6-бром-N-(1-(6-метилпиридазин-3-ил)этил)-8-((тетрагидро-2H-пиран-4-ил)окси)хиназолин-4-амина (120 мг, 0,270 ммоль), 1-метил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола (73 мг, 0.351 ммоль) и карбоната цезия (176 мг, 0,54 ммоль) в 1,4-диоксане (5 мл) и воде (0,75 мл). Затем добавляют [1,1′-бис(дифенилфосфино)ферроцен]дихлорпалладий (II) (20 мг, 0,027 ммоль), полученную реакционную смесь нагревают до 95°С и выдерживают при указанной температуре в течение 3 часов. После этого реакционную смесь охлаждают до комнатной температуры и фильтруют через Celite®. Смесь разбавляют водой (10 мл) и распределяют между водой и этилацетатом (15 мл). Фазы разделяют и водный слой экстрагируют этилацетатом (2 × 10 мл). Объединенные органические слои сушат над гидрофобным фриттом и концентрируют в вакууме. Остаток очищают препаративной ЖХВД с получением указанного в заголовке соединения (48 мг, 39%).
¹H ЯМР (400 МГц, ДМСО): δ 8,64 (д, J=8,4 Гц, 1 H), 8,41 (д, J=2,0 Гц, 1 H), 8,36 (c, 1 H), 7,83 (д, J=2,7 Гц, 1 H), 7,77 (д, J=1,4 Гц, 1 H), 7,62 (д, J=8,2 Гц, 1 H), 7,50 (д, J=7,5 Гц, 1 H), 6,91 (д, J=2,7 Гц, 1 H), 5,79-5,72 (м, 1 H), 4,92-4,84 (м, 1 H), 3,96 (c, 3 H), 3,92-3,88 (м, 2 H), 3,56-3,47 (м, 3 H), 2,60 (c, 3 H), 2,05-1,97 (м, 2 H), 1,73 (д, J=7,5 Гц, 3 H), 1,69-1,65 (м, 2 H). ЖХМС (метод 4): [MH+]=446 при 2,59 мин.
Пример 33
(R)-6-(5-Метилтиазол-2-ил)-N-(1-(пиридазин-3-ил)этил)-8-((тетрагидро-2H-пиран-4-ил)окси)хиназолин-4-амин
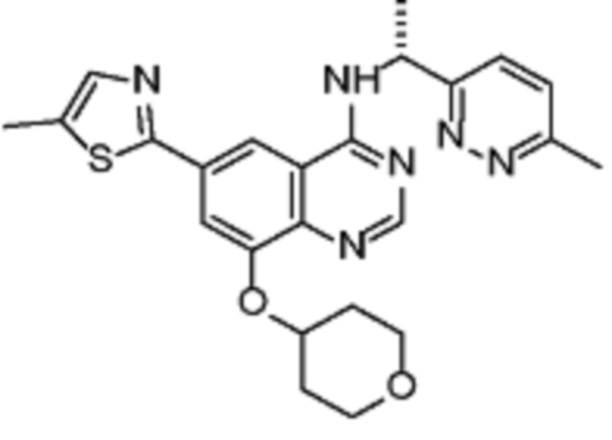
Стадия 1: Получение (R)-N-(1-(6-метилпиридазин-3-ил)этил)-8-((тетрагидро-2H-пиран-4-ил)окси)-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)хиназолин-4-амина

Газообразный азот барботируют в течение 10 минут через смесь (R)-6-бром-N-(1-(6-метилпиридазин-3-ил)этил)-8-((тетрагидро-2H-пиран-4-ил)окси)хиназолин-4-амина (200 мг, 0,45 ммоль), бис(пинаколато)дибора (137 мг, 0,54 ммоль) и ацетата калия (88 мг, 0,90 ммоль) в 1,4-диоксане (6 мл). Затем в реакционную смесь добавляют [1,1′- бис(дифенилфосфино)ферроцен]дихлорпалладий(II) (33 мг, 0,05 ммоль), полученную реакционную смесь нагревают до 90°С и выдерживают при указанной температуре в течение 2 часов. После этого реакционную смесь охлаждают до комнатной температуры и фильтруют через Celite®. Указанное в заголовке соединение используют непосредственно в виде раствора в 1,4-диоксане, предполагая количественный выход.
Стадия 2: Получение (R)-N-(1-(6-метилпиридазин-3-ил)этил)-6-(5-метилтиазол-2-ил)-8-((тетрагидро-2H-пиран-4-ил)окси)хиназолин-4-амина
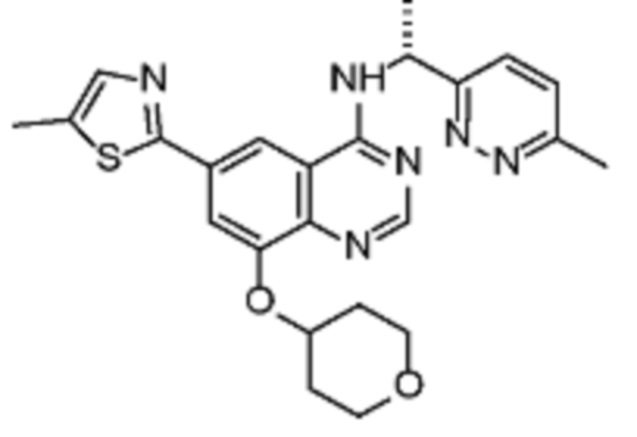
К (R)-N-(1-(6-метилпиридазин-3-ил)этил)-8-((тетрагидро-2H-пиран-4-ил)окси)-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)хиназолин-4-амину (6 мл, раствор в диоксане, 0,45 ммоль) добавляют 2-бром-5-метилтиазол (120 мг, 0,68 ммоль), карбонат цезия (291 мг, 0,90 ммоль) и воду (0,5 мл). Газообразный азот барботируют через реакционную смесь в течение 10 минут и затем добавляют тетракис(трифенилфосфин)палладий(0) (52 мг, 0,05 ммоль). После этого реакционную смесь нагревают до 90°С и выдерживают при указанной температуре в течение 16 часов. Затем реакционную смесь охлаждают до комнатной температуры, фильтруют через Celite® и концентрируют в вакууме. Остаток очищают колоночной хроматографией (силикагель, элюирование с градиентом: 0-60% этилацетата в циклогексане) с получением указанного в заголовке соединения (52 мг, 25%) в виде твердого не совсем белого вещества.
¹H ЯМР (400 МГц, ДМСО): δ 8,91 (д, J=7,4 Гц, 1 H), 8,53 (д, J=1,6 Гц, 1 H), 8,41 (c, 1 H), 7,81 (д, J=1,6 Гц, 1 H), 7,69 (д, J=1,1 Гц, 1 H), 7,63 (д, J=8,7 Гц, 1 H), 7,51 (д, J=8,8 Гц, 1 H), 5,80-5,71 (м, 1 H), 4,94-4,87 (м, 1 H), 3,96-3,88 (м, 2 H), 3,56-3,50 (м, 2 H), 2,59 (c, 3 H), 2,07-1,99 (м, 4 H), 1,72 (д, J=7,2 Гц, 6 H). ЖХМС (метод 4): [MH+]=463 при 2,86 мин.
Пример 34
4-[[4-[1-(6-метилпиридазин-3-ил)этиламино]-6-(5-метилпиримидин-2-ил)хиназолин-8-ил]оксиметил]тетрагидропиран-4-ол

(R)-4-((1-(6-Метилпиридазин-3-ил)этил)амино)-6-(5-метилпиримидин-2-ил)хиназолин-8-ол (35 мг, 0,094 ммоль), 1,6-диоксаспиро(2,5)октан (23,0 мг, 0,20 ммоль) и карбонат цезия (31,0 мг, 0,94 ммоль) суспендируют в N, N-диметилформамидe (0,6 мл). Реакционную смесь затем нагревают до 120°С и выдерживают при указанной температуре в течение 2 часов. После этого смесь охлаждают до комнатной температуры и фильтруют. Полученный фильтрат очищают препаративной ЖХВД с получением указанного в заголовке соединения (6 мг, 16%) в виде твердого вещества коричневого цвета.
1Н ЯМР (400 МГц, ДМСО): δ 9,08-9,00 (м, 2 H), 8,86 (c, 2 H), 8,45 (c, 1 H), 8,25 (c, 1 H), 7,63 (д, J=8,8 Гц, 1 H), 7,50 (д, J=8,7 Гц, 1 H), 5,82-5,76 (м, 1 H), 4,91 (c, 1 H), 4,01 (c, 2 H), 3,72 (д, J=7,0 Гц, 4 H), 2,60 (c, 3 H), 2,38 (c, 3 H), 1,97-1,87 (м, 2 H), 1,73 (д, J=7,0 Гц, 3 H), 1,57-1,50 (м, 2 H). ЖХМС (метод 4): [MH+]=488 при 2,70 мин.
Пример 35
8-(2-Метоксиэтокси)-N-((6-метилпиридазин-3-ил)метил)-6-(5-метилтиазол-2-ил)хиназолин-4-амин
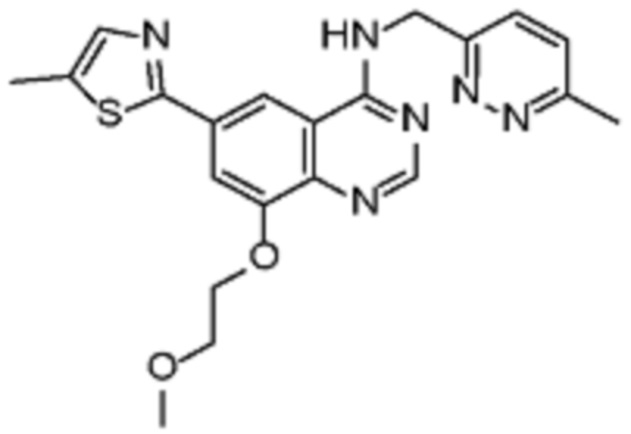
К раствору 4-(((6-метилпиридазин-3-ил)метил)амино)-6-(5-метилтиазол-2-ил)хиназолин-8-ола (100 мг, 0,27 ммоль) в N, N-диметилформамидe (2 мл) добавляют карбонат цезия (178 мг, 0,55 ммоль) с последующим добавлением 1-бром-2-метоксиэтана (55 мг, 0,39 ммоль). Реакционную смесь перемешивают при 100°С и выдерживают при указанной температуре в течение 16 часов. Смесь охлаждают до комнатной температуры, концентрируют в вакууме и остаток очищают препаративной ЖХВД с получением указанного в заголовке соединения (28,0 мг, 24%) в виде твердого бесцветного вещества.
1H ЯМР (400 МГц, ДМСО): δ 9,24 (дд, J=5,9, 5,9 Гц, 1 H), 8,45 (c, 1 H), 8,38 (д, J=1,6 Гц, 1 H), 7,76 (д, J=1,5 Гц, 1 H), 7,67 (д, J=1,3 Гц, 1 H), 7,57 (д, J=8,6 Гц, 1 H), 7,51 (д, J=8,7 Гц, 1 H), 5,02 (д, J=5,7 Гц, 2 H), 4,35-4,32 (м, 2 H), 3,81-3,77 (м, 2 H), 3,38 (c, 3 H), 2,60 (c, 3 H), 2,55 (д, J=1.6 Гц, 3 H). ЖХМС (метод 4): [MH+]=423 при 2,66 мин.
Соединения, представленные в таблице ниже, получают в соответствии с методикой, описанной для получния 8-(2-метоксиэтокси)-N-((6-метилпиридазин-3-ил)метил)-6-(5-метилтиазол-2-ил)хиназолин-4-амина.
структура
1H ЯМР
ЖХ-МС
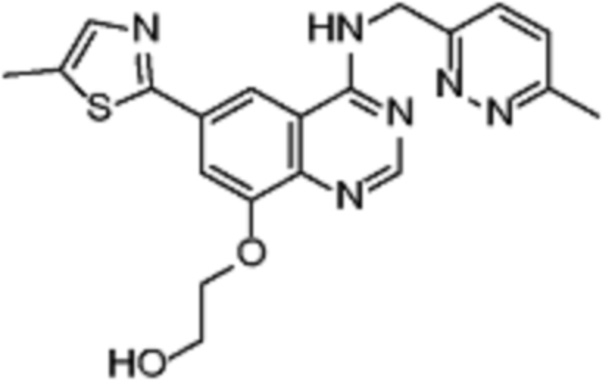
ЖХМС (метод 4): [MH+]=409 при 2,41 мин. 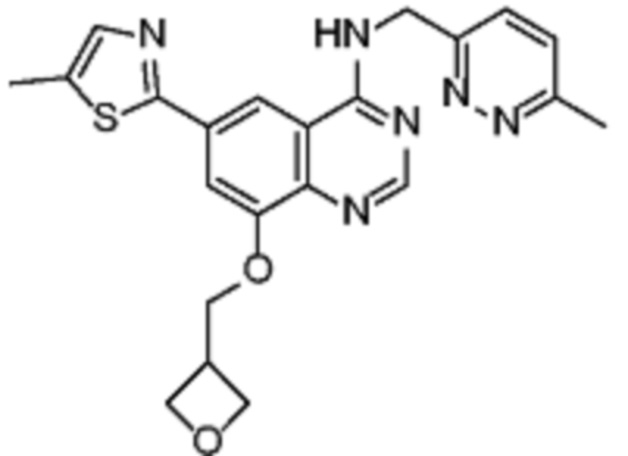
ЖХМС (метод 4): [MH+]=435 при 2,59 мин. 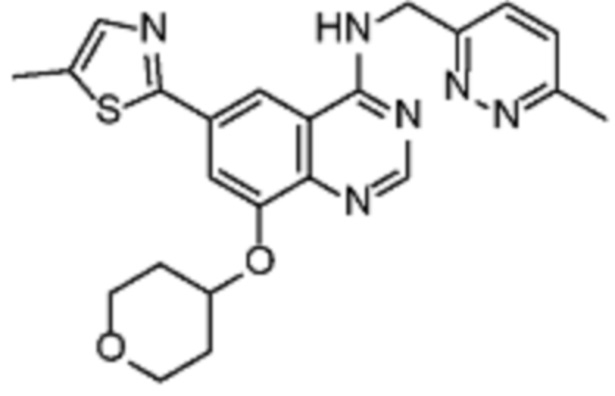
ЖХМС (метод 4): [MH+]=449 при 3,68 мин.
Cs2CO3
Пример 39
8-(5-Азаспиро[3.5]нонан-8-илокси)-6-(4-фторфенил)-N-[(1R)-1-[2-(трифторметил)пиримидин-5-ил]этил]хиназолин-4-амин
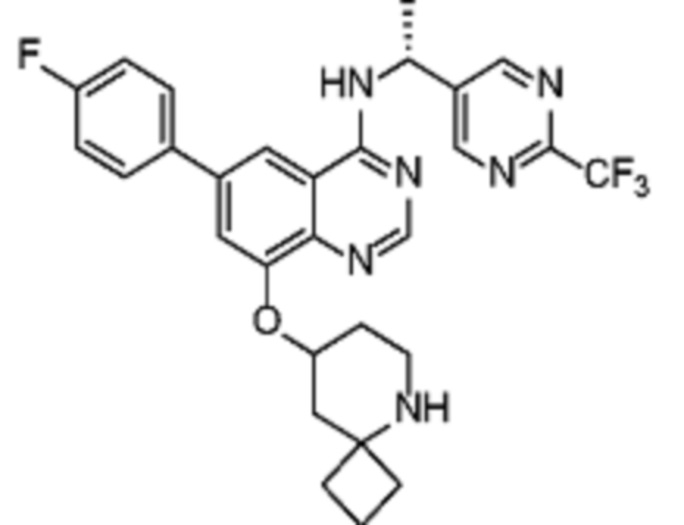
Стадия 1: Получение трет-бутил-8-((6-(4-фторфенил)-4-(((R)-1-(2-(трифторметил)пиримидин-5-ил)этил)амино)хиназолин-8-ил)окси)-5-азаспиро[3.5]нонан-5-карбоксилата

Газообратный азот барботируют в течение 5 минут через смесь (R)-6-(4-фторфенил)-4-((1-(2-(трифторметил)пиримидин-5-ил)этил)амино)хиназолин-8-ола (100 мг, 0,233 ммоль), трет-бутил-8-гидрокси-5-азаспиро[3.5]нонан-5-карбоксилата (62 мг, 0,256 ммоль) и цианометилтрибутилфосфорана (84 мг, 0,349 ммоль) в толуоле (3,0 мл). Смесь нагревают до 100°С и выдерживают при указанной температуре в течение 72 часов. После охлаждения до комнатной температуры растворитель удаляют в вакууме. Остаток очищают препаративной ЖХВД с получением указанного в заголовке соединения (41 мг, выход 27%) в виде твердого не совсем белого вещества.
ЖХМС (метод 3): [MH+]=653 при 6,17 мин.
Стадия 2: Получение 8-(5-азаспиро[3.5]нонан-8-илокси)-6-(4-фторфенил)-N-[(1R)-1-[2-(трифторметил)пиримидин-5-ил]этил]хиназолин-4-амина
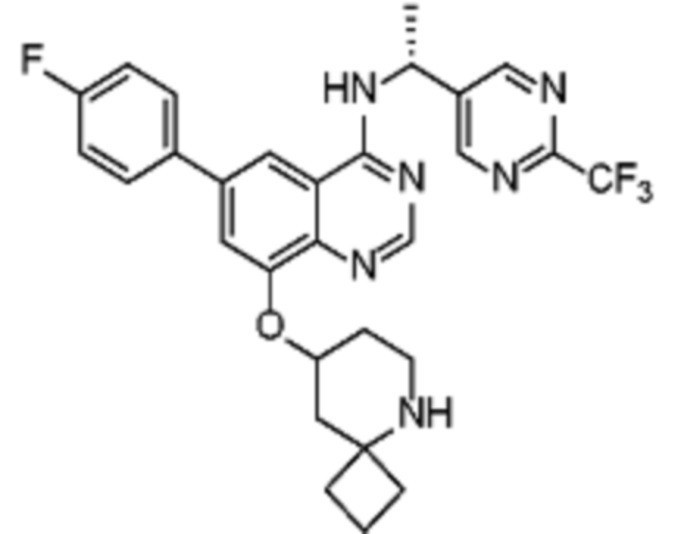
К раствору трет-бутил-8-((6-(4-фторфенил)-4-(((R)-1-(2-(трифторметил)пиримидин-5-ил)этил)амино)хиназолин-8-ил)окси)-5-азаспиро[3.5]нонан-5-карбоксилата (41 мг, 0,063 ммоль) в метаноле (1 мл) добавляют 4 N раствор HCl в 1,4-диоксане (1 мл). Полученную смесь перемешивают при комнатной температуре в течение 18 часов.
Реакционную смесь загружают непосредственно на SCX-картридж. Картридж промывают метанолом и фильтрат собирают, элюирование: 7M раствор аммиака в метаноле. Растворитель удаляют в вакууме и остаток очищают препаративной ЖХВД с получением указанного в заголовке соединения (11,0 мг, выход 31%) в виде твердого вещества бежевого цвета.
¹H ЯМР (400 МГц, ДМСО): δ 9,18 (c, 2 H), 8,68 (д, J=6,8 Гц, 1 H), 8,43 (c, 1 H), 8,25 (c, 1 H), 7,94-7,90 (м, 2 H), 7,63-7,61 (м, 1 H), 7,43-7,40 (м, 2 H), 5,71-5,64 (м, 1 H), 4,88-4,85 (м, 1 H), 3,05-3,03 (м, 1 H), 2,84-2,79 (м , 1 H), 2,34-2,31 (м, 1 H), 2,10-2,02 (м, 2 H), 1,80-1,73 (м, 2 H), 1,66-1,61 (м, 2 H), 1,47-1,41 (м, 4 H), 1,89 (д, J=7,2 Гц, 3 H). ЖХМС (метод 4): [MH+]=553 при 3,09 мин.
Соединение, представленное в таблице ниже, получают в соответствии с методикой, описанной для получния 8-(5-азаспиро[3.5]нонан-8-илокси)-6-(4-фторфенил)-N-[(1R)-1-[2-(трифторметил)пиримидин-5-ил]этил]хиназолин-4-амина.
структура
¹H ЯМР
ЖХ-МС

ЖХМС (метод 4): [MH+]=525 при 3,02 мин.
Пример 44
(R)-2-((6-(4-Фторфенил)-4-((1-(2-(трифторметил)пиримидин-5-ил)этил)амино)хиназолин-8-ил)окси)уксусная кислота
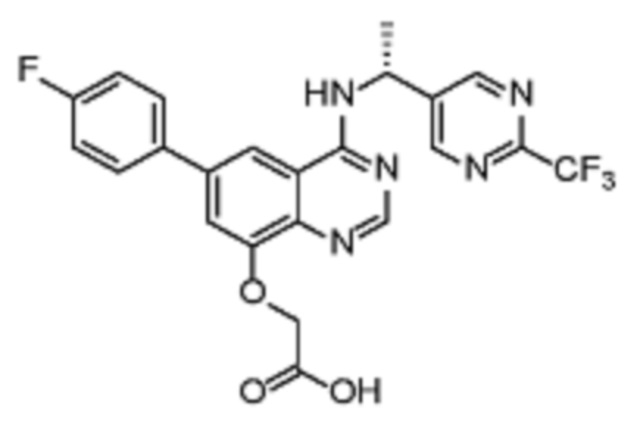
Стадия 1: трет-Бутил-(R)-2-((6-(4-фторфенил)-4-((1-(2-(трифторметил)пиримидин-5-ил)этил)амино)хиназолин-8-ил)окси)ацетат
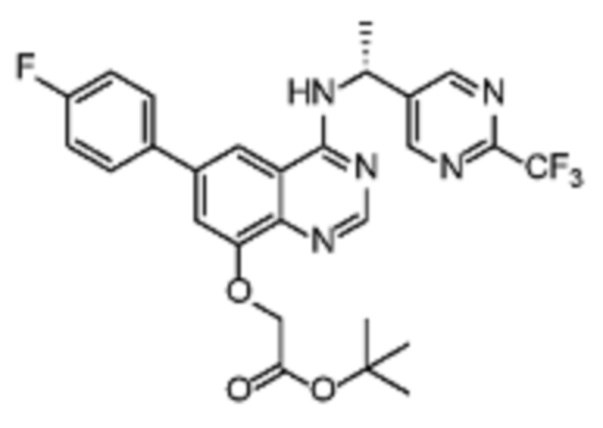
трет-Бутил-2-бромацетат (0,057 мл, 0,384 ммоль) добавляют при перемешивании к смеси (R)-6-(4-фторфенил)-4-((1-(2-(трифторметил)пиримидин-5-ил)этил)амино)хиназолин-8-ола (150 мг, 0,349 ммоль) (промежуточное соединение 1) и карбоната калия (155 мг, 1,122 ммоль) в ДМФА (3 мл). Смесь перемешивают при КТ в течение 1 часа, затем реакционную смесь разбавляют EtOAC и промывают насыщенным раствором соли (2 ×). Органический слой сушат над сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Очистка хроматографией (Biotage Isolera, 10 г KP-Sil картридж, элюирование с градиентом: от 0 до 50% EtOAc в дихлорметане в 15 CV) приводит к получению трет-бутил-(R)-2-((6-(4-фторфенил)-4-((1-(2-(трифторметил)пиримидин-5-ил)этил)амино)хиназолин-8-ил)окси)ацетата (149 мг, 0,274 ммоль, выход 78%) в виде белого порошка.
ЖХМС (метод 5 ): 0,97 мин., m/z 544,2 [M+H]+.

трет-Бутил-(R)-2-((6-(4-фторфенил)-4-((1-(2-(трифторметил)пиримидин-5-ил)этил)амино)хиназолин-8-ил)окси)ацетат (149 мг, 0,274 ммоль) растворяют в ТФУК (2 мл). Смесь перемешивают при КТ в течение 16 часов, затем летучие компоненты удаляют при пониженном давлении. Растирание в диэтиловом эфире приводит к получению (R)-2-((6-(4-фторфенил)-4-((1-(2-(трифторметил)пиримидин-5-ил)этил)амино)хиназолин-8-ил)окси)уксусной кислоты (134 мг, 0,275 ммоль, выход 100%) в виде порошка бледно-розового цвета.
ЖХМС (метод5 ): 0,81 мин., m/z 488,1 [M+H]+.
1H ЯМР (400 МГц, ДМСО-d6) δ м.д. 9,22 (уш. с, 1 H), 9,16 (c, 2 H), 8,55 (c, 1 H), 8,29 (c, 1 H), 7,87 (дд, J=8,66, 5,37 Гц, 2 H), 7,65 (уш. с, 1 H), 7,38 (т, J=8,77 Гц, 2 H), 5,75 (квин, J=6,63 Гц, 1 H), 5,07 (c, 2 H), 1,73 (д, J=7,02 Гц, 3 H).
Пример 55
Дигидрохлорид 2-((4-(((6-метилпиридазин-3-ил)метил)амино)-6-(5-метилпиримидин-2-ил)хиназолин-8-ил)окси)уксусной кислоты
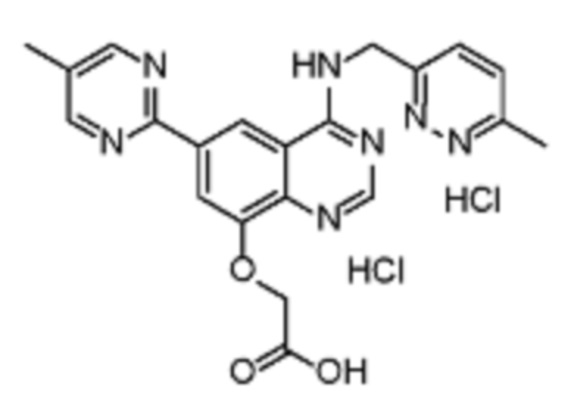
Стадия 1: трет-Бутил-2-((4-(((6-метилпиридазин-3-ил)метил)амино)-6-(5-метилпиримидин-2-ил)хиназолин-8-ил)окси)ацетат
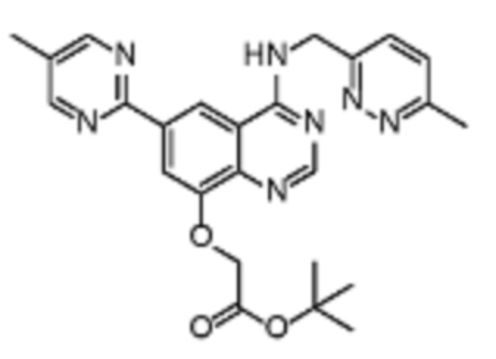
трет-Бутил-2-бромацетат (247 мкл, 1,669 ммоль) при перемешивании добавляют к смеси 4-(((6-метилпиридазин-3-ил)метил)амино)-6-(5-метилпиримидин-2-ил)хиназолин-8-ола (500 мг, 1,391 ммоль) (промежуточное соединение 57) и карбоната калия (456 мг, 3,30 ммоль) в смеси ДМФА/ТГФ (2:1, 9 мл). Смесь перемешивают при КТ в течение 16 часов. Реакцию гасят добавлением муравьиной кислоты (300 мкл, 7,95 ммоль) и удаляют при пониженном давлении летучие компоненты. Очистка ОФ-хроматографией (Biotage Isolera, 60 г C18 картридж, элюирование с градиентом: от 0 до 30% B в A; A: вода/ацетонитрил 95:5+0,1% HCOOH, B: ацетонитрил/вода 95:5+0,1% HCOOH) приводит к получению трет-бутил-2-((4-(((6-метилпиридазин-3-ил)метил)амино)-6-(5-метилпиримидин-2-ил)хиназолин-8-ил)окси)ацетата (451 мг, 0,952 ммоль, выход 68,5%) в виде воска оранжевого цвета.
ЖХМС (метод 5 ): 0,66 мин., 473,8 [M+H]+.
Стадия 2: Дигидрохлорид 2-((4-(((6-метилпиридазин-3-ил)метил)амино)-6-(5-метилпиримидин-2-ил)хиназолин-8-ил)окси)уксусной кислоты
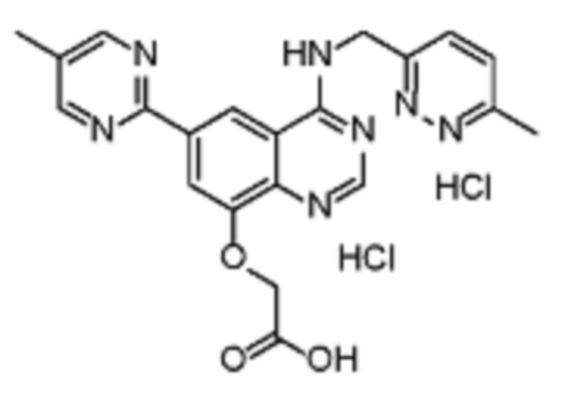
трет-Бутил-2-((4-(((6-метилпиридазин-3-ил)метил)амино)-6-(5-метилпиримидин-2-ил)хиназолин-8-ил)окси)ацетат перемешивают в 4N растворе HCl в 1,4-диоксане (20 мл, 80 ммоль) в течение 16 часов при КТ. Летучие компоненты удаляют при пониженном давлении с получением дигидрохлорида 2-((4-(((6-метилпиридазин-3-ил)метил)амино)-6-(5-метилпиримидин-2-ил)хиназолин-8-ил)окси)уксусной кислоты (552 мг, 1,126 ммоль, выход 81%) в виде порошка бледно-желтого цвета.
ЖХМС (метод 5 ): 0,45 мин., 418,0 [M+H]+.
1H ЯМР (400 МГц, ДМСО-d6) δ м.д. 11,20 (уш. т, J=5,48 Гц, 1 H), 9,18 (c, 1 H), 8,88 (c, 2 H), 8,80 (c, 1 H), 8,41 (c, 1 H), 7,77 (д, J=8,55 Гц, 1 H), 7,65 (д, J=8,55 Гц, 1 H), 5,16-5,28 (м, 4 H), 2,63 (c, 3 H), 2,38 (c, 3 H).
Пример 57
Гидрохлорид 2-((6-(4-Фторфенил)-4-(((1-метилпиперидин-4-ил)метил)амино)хиназолин-8-ил)окси)уксусной кислоты
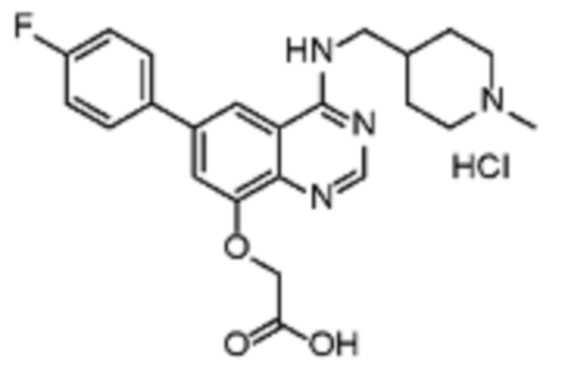
Стадия 1: трет-Бутил-(6-(4-фторфенил)-8-гидроксихиназолин-4-ил)((1-метилпиперидин-4-ил)метил)карбамат
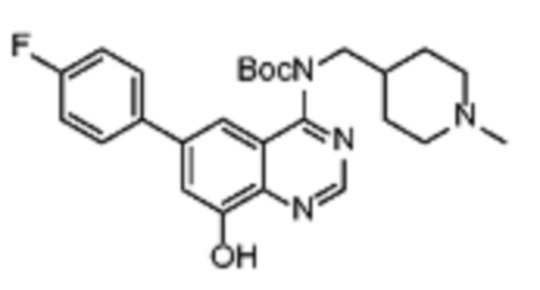
Раствор ди-трет-бутилдикарбоната (1,641 мл, 7,14 ммоль) в ТГФ (10 мл) добавляют при перемешивании к раствору 6-(4-фторфенил)-4-(((1-метилпиперидин-4-ил)метил)амино)хиназолин-8-ола (1,19 г, 3,25 ммоль) (промежуточное соединение 59) и DIPEA (1,244 мл, 7,14 ммоль) в ТГФ (20 мл). Затем добавляют 4-диметиламинопиридин (0,079 г, 0,649 ммоль) и смесь перемешивают в течение 16 часов при КТ. ЖХМС показывает полную конверсию в трет-бутил-(8-((трет-бутоксикарбонил)окси)-6-(4-фторфенил)хиназолин-4-ил)((1-метилпиперидин-4-ил)метил)карбамат. Летучие компоненты удаляют при пониженном давлении и остаток распределяют между EtOAc/ТГФ (1:1) и насыщенным водным раствором хлорида аммония. Слои разделяют и органический слой промывают насыщенным раствором соли, сушат над сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Сырой продукт снова растворяют в дихлорметане (20 мл) и обрабатывают 2,0 M диметиламином в ТГФ (4 мл). Смесь перемешивают при КТ в течение 4 часов, затем летучие компоненты удаляют при пониженном давлении. Очистка колоночной хроматографией (Biotage Isolera, 55 г NH картридж, элюирование с градиентом: от 0 до 30% EtOH в дихлорметане) приводит к получению трет-бутил-(6-(4-фторфенил)-8-гидроксихиназолин-4-ил)((1-метилпиперидин-4-ил)метил)карбамата (398 мг, 0,853 ммоль, выход 26,3%) в виде порошка оранжевого цвета.
ЖХМС (метод 5 ): 1,32 мин., 467,4 [M+H]+.
Стадия 2: Гидрохлорид 2-((6-(4-Фторфенил)-4-(((1-метилпиперидин-4-ил)метил)амино)хиназолин-8-ил)окси)уксусной кислоты
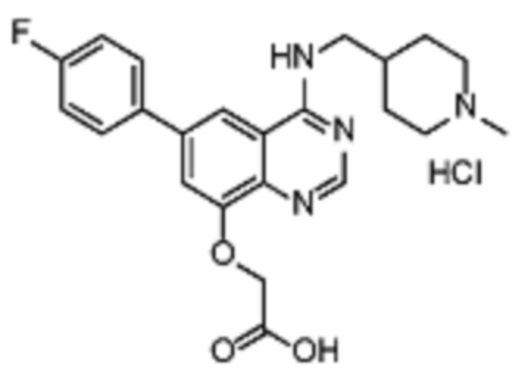
трет-Бутил-2-бромацетат (0,075 мл, 0,509 ммоль) добавляют при перемешивании к суспензии карбоната калия (147 мг, 1,061 ммоль) и трет-бутил-(6-(4-фторфенил)-8-гидроксихиназолин-4-ил)((1-метилпиперидин-4-ил)метил)карбамата (198 мг, 0,424 ммоль) в ДМФА (2 мл). Смесь перемешивают в течение 16 часов при КТ. Реакционную смесь разбавляют EtOAc и промывают насыщенным раствором соли (3 ×). Органический слой сушат над сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Очистка колоночной хроматографией (Biotage Isolera, 28 г NH картридж, элюирование с градиентом: от 0 до 20% AcOEt в дихлорметане) приводит к получению промежуточного трет-бутил-2-((4-((трет-бутоксикарбонил)((1-метилпиперидин-4-ил)метил)амино)-6-(4-фторфенил)хиназолин-8-ил)окси)ацетата. Продукт растворяют при КТ в 1,4-диоксане (3 мл), затем обрабатывают 4N раствором HCl в течение 18 часов при КТ. Летучие компоненты удаляют при пониженном давлении с получением гидрохлорида 2-((6-(4-фторфенил)-4-(((1-метилпиперидин-4-ил)метил)амино)хиназолин-8-ил)окси)уксусной кислоты (88,1 мг, 0,191 ммоль, выход 45,0%) в виде порошка бледно-желтого цвета.
ЖХМС (метод 5 ): 0,33 мин., 425,2 [M+H]+.
1H ЯМР (400 МГц, ДМСО-d6) δ м.д. 13,95-14,23 (уш. с, 1 H), 10,65-10,55 (уш. с, 1 H), 10,11-10,18 (м, 1 H), 8,72 (c, 1 H), 8,53 (c, 1 H), 7,95 (уш. дд, J=8,33, 5,48 Гц, 2 H), 7,87 (c, 1 H), 7,38 (т, J=8,77 Гц, 2 H), 5,19 (c, 2 H), 3,60-3,50 (м, 2 H), 3,17-3,34 (уш. с, 3H), 2,74-2,90 (м, 2 H), 2,67 (c, 3 H), 2,50-2,53 (м, 1 H), 1,97-2,05 (м, 1 H), 1,90 (уш. д, J=13,37 Гц, 2 H), 1,40-1,61 (м, 2 H).
Промежуточное соединение 2
6-(4-Фторфенил)-8-[(3-метил-1,2,4-оксадиазол-5-ил)метокси]-N-[(6-метилпиридазин-3-ил)метил]хиназолин-4-амин

Стадия 1: Получение (3-метил-1,2,4-оксадиазол-5-ил)метил метансульфоната

Раствор (3-метил-1,2,4-оксадиазол-5-ил)метанола (300 мг, 2,63 ммоль) и триэтиламина (1,1 мл, 7,89 ммоль) в ДХМ (6 мл) перемешивают при 0°С и выдерживают при указанной температуре в течение 5 минут. К смеси добавляют метансульфонилхлорид (0,41 мл, 5,26 ммоль) и перемешивают реакционную смесь при 0°С в течение 15 минут. Реакционную смесь затем нагревают до комнатной температуры и перемешивают в течение 16 часов. Реакционную смесь распределяют между ДХМ и водой и две фазы разделяют. Водную фазу экстрагируют ДХМ и объединенные органические фазы пропусают через бумагу для разделения фаз. Растворитель удаляют в вакууме с получением указанного в заголовке соединения в виде масла оранжевого цвета (290 мг, выход 57%).
¹H ЯМР (400 МГц, CDCl3): δ 4,66 (c, 2 H), 3,49 (c, 3 H), 2,43 (c, 3 H).
Стадия 2: Получение 6-(4-фторфенил)-8-[(3-метил-1,2,4-оксадиазол-5-ил)метокси]-N-[(6-метилпиридазин-3-ил)метил]хиназолин-4-амина

(3-Метил-1,2,4-оксадиазол-5-ил)метилметансульфонат (176 мг, 0,46 ммоль), 6-(4-фторфенил)-4-(((6-метилпиридазин-3-ил)метил)амино)хиназолин-8-ол (150 мг, 0,23 ммоль) и карбонат цезия (223 мг, 0,69 ммоль) растворяют в ДМФА (4 мл). Реакционную смесь нагревают до 55°С и выдерживают при указанной температуре в течение 16 часов. Реакционную смесь охлаждают до комнатной температуры и затем распределяют между ДХМ и водой. Две фазы разделяют и водную фазу экстрагируют ДХМ (×2). Объединенные органические фазы пропусают через бумагу для разделения фаз и концентрируют в вакууме. Очистка препаративной ЖХВД приводит к получению указанного в заголовке соединения (78,1 мг, 75%) в виде твердого не совсем белого вещества.
¹H ЯМР (400 МГц, ДМСО): δ 9,13 (т, J=5,9 Гц, 1 H), 8,45 (c, 1 H), 8,30 (д, J=1,5 Гц, 1 H), 7,95-7,90 (м, 2 H), 7,75 (д, J=1,6 Гц, 1 H), 7,59-7,49 (м, 2 H), 7,43-7,38 (м, 2 H), 5,80 (c, 2 H), 5,04 (д, J=5,9 Гц, 2 H), 2,61-2,60 (м, 3 H), 2,39 (c, 3 H). ЖХМС (метод 3): [MH+]=458 при 4,29 мин.
Соединения, представленные далее, получают в соответствии с описанными выше методиками получения промежуточного соединения 2, исходя из реагентов, указанных в таблице.
¹H ЯМР
ЖХ-МС
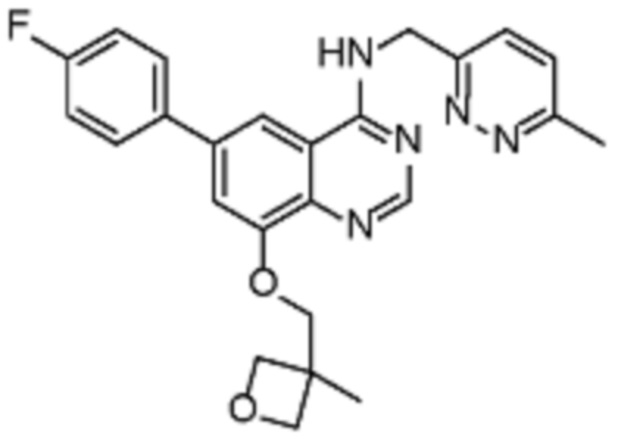
ЖХМС (метод 3): [MH+]=446 при 3,9 мин.
ЖХМС (метод 4): [MH+]=459 при 2,93 мин.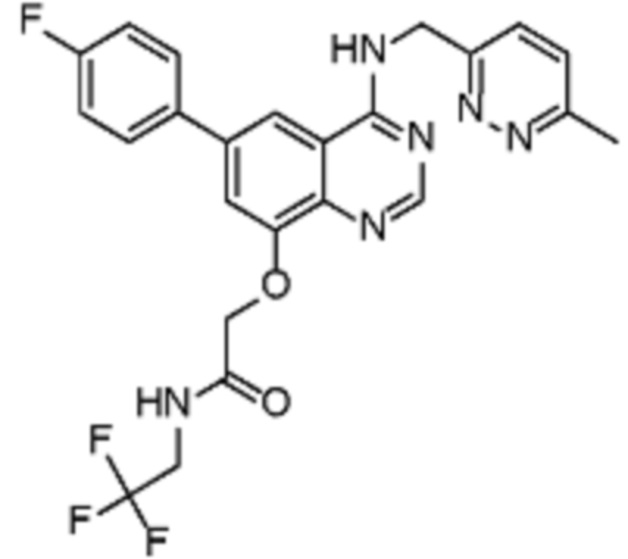
ЖХМС (метод 4): [MH+]=501 при 3,22 мин.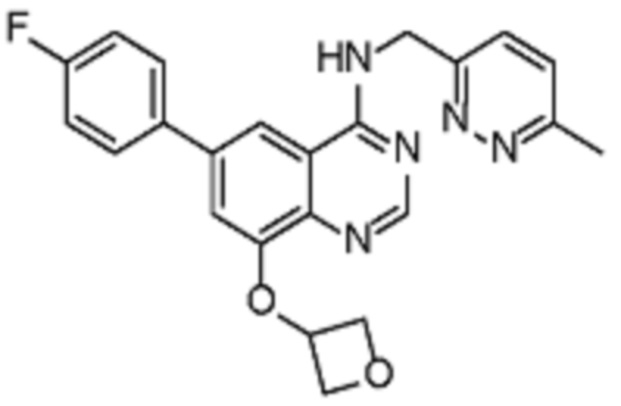
ЖХМС (метод 4): [MH+]=418 при 2,69 мин.
Пример 81
N-[1-(5-Метил-1,3,4-тиадиазол-2-ил)этил]-6-(5-метилтиазол-2-ил)-8-тетрагидропиран-4-илоксихиназолин-4-амин
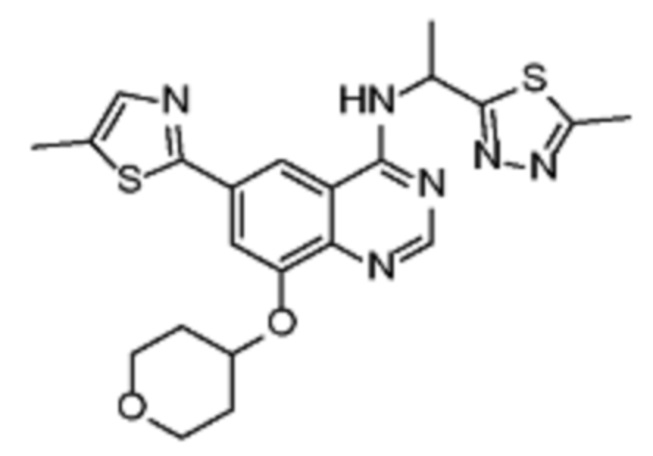
Стадия 1: Получение метил-2-нитро-3-((тетрагидро-2H-пиран-4-ил)окси)бензоата

Метил-3-гидрокси-2-нитробензоат (2000 мг, 10,14 ммоль), тетрагидропиран-4-илметансульфонат (3600 мг, 19,97 ммоль) и карбонат калия (6900 мг, 49,92 ммоль) суспендируют в ацетонитриле (40 мл), реакционную смесь нагревают до 82°С и выдерживают при указанной температуре в течение 16 часов в атмосфере азота. Реакционную смесь охлаждают до комнатной температуры и фильтруют. Фильтрат концентрируют при пониженном давлении, растворяют в ацетонитриле и фильтруют. Фильтрат концентрируют при пониженном давлении и очищают флэш-хроматографией (элюирование: 45% этилацетат в циклогексане) с получением указанного в заголовке соединения в виде прозрачного бесцветного масла (1500 мг, выход 52%).
ЖХМС (метод 3): [M+NH4]+= 299 при 4,04 мин.
Стадия 2: Получение метил-2-амино-3-((тетрагидро-2H-пиран-4-ил)окси)бензоата
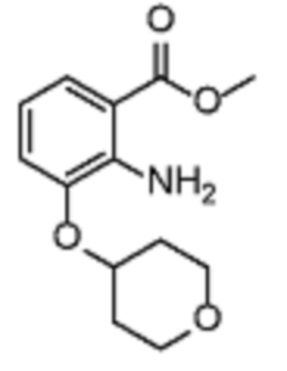
К раствору метил-2-нитро-3-((тетрагидро-2H-пиран-4-ил)окси)бензоата (2200 мг, 7,82 ммоль) в этаноле (22 мл) и метанола (5 мл) добавляют 1-метил-1,4-циклогексадиен (4,4 мл, 39,11 ммоль) . Реакционную смесь дегазируют азотом в течение 20 минут. Добавляют палладий на угле (10%, 832 мг, 0,782 ммоль и реакционную смесь нагревают до 80°С в атмосфере азота в течение 16 часов. Реакционную смесь охлаждают до комнатной температуры и фильтруют. Фильтрат концентрируют в вакууме и остаток очищают колоночной флэш-хроматографией (силикагель, элюирование: 25% этилацетата в циклогексане) с получением указанного в заголовке соединения (822 мг, 42%) в виде твердого не совсем белого вещества.
ЖХМС (метод 4): [MH+]=252 при 4,27 мин.
Стадия 3: Получение метил-2-амино-5-бром-3-((тетрагидро-2H-пиран-4-ил)окси)бензоата
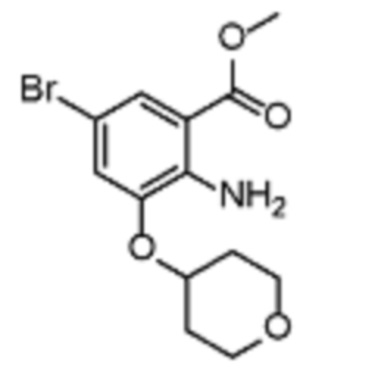
Метил-2-амино-3-((тетрагидро-2H-пиран-4-ил)окси)бензоат (886 мг,3,53 ммоль) растворяют в хлороформе (10 мл) и перемешивают при 5°С в течение 5 минут. К смеси по каплям добавляют бром (0,19 мл, 3,70 ммоль) в хлороформе (6 мл), при этом цвет изчезает между каплями. Реакционную смесь перемешивают при 5°С в течение 1 часа. Реакционную смесь гасят тиосульфатом натрия (3 мл) и смесь перемешивают в течение 10 минут. Добавляют насыщенный водный раствор гидрокарбоната натрия и перемешивают реакционную смесь в течение 5 минут. Две фазы разделяют и водную фазу экстрагируют ДХМ (× 2). Объединенные органические фазы сушат и концентрируют в вакууме с получением указанного в заголовке соединения (1106 мг, выход 95%).
¹H ЯМР (400 МГц, CDCl3): δ 7,62 (д, J=2,3 Гц, 1 H), 6,94 (д, J=2,3 Гц, 1 H), 6,03-5,99 (м, 2 H), 4,51-4,43 (м, 1 H), 4,01-3,95 (м, 2 H), 3,87 (c, 3 H), 3,63-3,56 (м, 2 H), 2,10-2,02 (м, 2 H), 1,86-1,77 (м, 2 H).
Стадия 4: Получение 6-бром-8-((тетрагидро-2H-пиран-4-ил)окси)хиназолин-4-ола

Метил-2-амино-5-бром-3-((тетрагидро-2H-пиран-4-ил)окси)бензоат (1106 мг, 3,35 ммоль) растворяют в ТГФ (5 мл), метаноле (5 мл) и воде (5 мл). Добавляют гидроксид лития (176 мг, 4,19 ммоль) и перемешивают полученную реакционную смесь при комнатной температуре в течение 18 часов. Реакционную смесь концентрируют в вакууме. Остаток перемешивают в формамидe (4 мл) при 140°С в течение 16 часов. Реакционную смесь охлаждают до комнатной температуры, затем разбавляют водой и фильтруют. Твердый продукт промывают водой (× 2), затем смесью диэтиловый эфир/метанол (9:1) (× 2) и наконец эфиром с получением указанного в заголовке соединения (1045 мг, выход 96%).
ЖХМС (метод 4): [MH+]=326 при 3,12 мин.
Стадия 5: Получение 6-(5-метилтиазол-2-ил)-8-((тетрагидро-2H-пиран-4-ил)окси)хиназолин-4-ола

6-Бром-8-((тетрагидро-2H-пиран-4-ил)окси)хиназолин-4-ол (348 мг, 1,07 ммоль), ацетат калия (210 мг, 2,14 ммоль) и бис(пинаколато)дибор (326 мг, 1,29 ммоль) дегазируют в диоксане (13 мл). Добавляют комплекс [1,1′-бис(дифенилфосфино)ферроцен]дихлорпалладия(II) с дихлорметаном (44 мг, 0,054 ммоль), реакционную смесь нагревают до 100°С и выдерживают при указанной температуре в течение 20 часов. Реакционной смеси дают возможность охладиться до комнатной температуры и к смеси добавляют 2-бром-5-метилтиазол (258 мг, 1,45 ммоль), карбонат цезия (698 мг, 2,14 ммоль) и воду (4 мл). Реакционную смесь дегазируют азотом и затем добавляют тетракис(трифенилфосфин)палладий(0) (37 мг, 0,032 ммоль). Реакционную смесь нагревают до 100°С и выдерживают при указанной температуре в течение 23 часов. После этого реакционную смесь охлаждают до комнатной температуры и распределяют между EtOAc и водой. Две фазы разделяют и экстрагируют водную фазу EtOAc (× 1) и ДХМ (× 1). Органические фазы сушат над MgSO4, объединяют и концентрируют в вакууме. Остаток очищают колоночной флэш-хроматографией (силикагель, элюирование с градиентом: 0-100% этилацетат/EtOH (3:1) в циклогексане) с получением указанного в заголовке соединения (176 мг, 20%).
ЖХМС (метод 4): [MH+]=344 при 3,21 мин.
Стадия 6: Получение N-[1-(5-метил-1,3,4-тиадиазол-2-ил)этил]-6-(5-метилтиазол-2-ил)-8-тетрагидропиран-4-илоксихиназолин-4-амина

Смесь 6-(5-метилтиазол-2-ил)-8-((тетрагидро-2H-пиран-4-ил)окси)хиназолин-4-ола (176 мг, 0,513 ммоль, чистота 40%), оксихлорида фосфора(V) (0,062 мл, 0,67 ммоль), DIPEA (0,45 мл, 2,56 ммоль) в толуоле (5 мл) нагревают до 90°С в атмосфере азота и выдерживают в указанных условиях в течение 2 часов. Реакционную смесь охлаждают до комнатной температуры и концентрируют в вакууме. Остаток распределяют между между водой и дихлорметаном. Объединенные органические слои промывают насыщенным раствором гидрокарбоната натрия, пропускают через гидрофобный фриттовый фильтр и концентрируют в вакууме. Остаток (80 мг) растворяют в хлороформе (0,5 мл) и добавляют 1-(5-метил-1,3,4-тиадиазол-2-ил)этан-1-амин (37 мг, 0,26 ммоль). Реакционную смесь нагревают в герметичной пробирке в атмосфере азота до 70°С и выдерживают в казанных условиях в течение 18 часов. После этого полученную смесь охлаждают до комнатной температуры. Реакционную смесь разбавляют охлажденной водой и перемешивают в течение 10 минут. Смесь распределяют между этилацетатом и водой. Две фазы разделяют и водную фазу дополнительно экстрагируют этилацетатом (× 2). Объединенные органические фазы пропусают через бумагу для разделения фаз и растворитель удаляют в вакууме. Остаток очищают препаративной ЖХВД с получением указанного в заголовке соединения (27,5 мг, выход 29%) в виде твердого не совсем белого вещества.
1H ЯМР (400 МГц, ДМСО): δ 9,09-9,08 (м, 1 H), 8,55 (c, 1 H), 8,44 (д, J=1,3 Гц, 1 H), 7,84 (c, 1 H), 7,69 (д, J=1,1 Гц, 1 H), 6,01-5,96 (м, 1 H), 4,97-4,90 (м, 1 H), 3,95-3,91 (м, 2 H), 3,58-3,51 (м, 2 H), 2,67 (c, 3 H), 2,55 (c, 3 H), 2,09-2,02 (м, 2 H), 1,81 (д, J=7,0 Гц, 3 H), 1,78-1,67 (м, 2 H). ЖХМС (метод 4): [MH+]=469 при 2,94 мин.
Соединение, представленное в таблице ниже, получают в соответствии с методикой, описнной для получния N-[1-(5-метил-1,3,4-тиадиазол-2-ил)этил]-6-(5-метилтиазол-2-ил)-8-тетрагидропиран-4-илоксихиназолин-4-амина.
структура
¹H ЯМР
ЖХ-МС

ЖХМС (метод 3): [MH+]=451 при 3,79 мин.
Соединения, представленные в таблице ниже, получают в виде одиночных изомеров очисткой с помощью хиральной препаративной СФХ соответствующей рацемической смеси, которую получают в соответствии с методикой, описанной для получения N-[1-(5-метил-1,3,4-тиадиазол-2-ил)этил]-6-(5-метилтиазол-2-ил)-8-тетрагидропиран-4-илоксихиназолин-4-амина.
структура
¹H ЯМР
ЖХ-МС
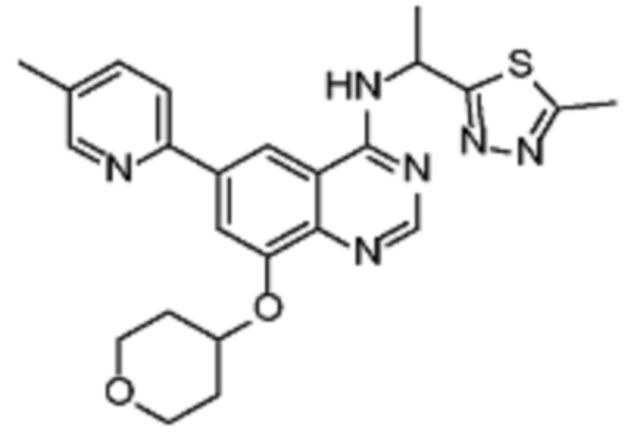
ЖХМС (метод 4): [MH+]=[MH+]=463 при 2,9 мин. Хиральный анализ (метод 8) при 2,20 мин.
ЖХМС (метод 4): [MH+]=463 при 2,9 мин. Хиральный анализ (метод 8) при 3,98 мин.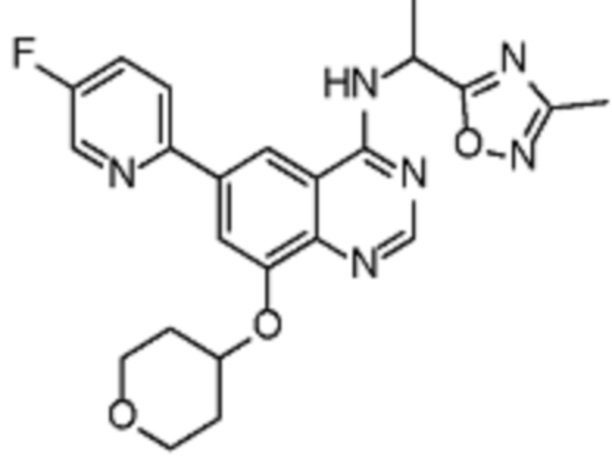
ЖХМС (метод 3): [MH+]=451 при 4,39 мин. Хиральный анализ (метод 40) при 3,49 мин.
Пример 94
6-(4-Фторфенил)-8-(2-метилпиразол-3-ил)-N-[(6-метилпиридазин-3-ил)метил]хиназолин-4-амин
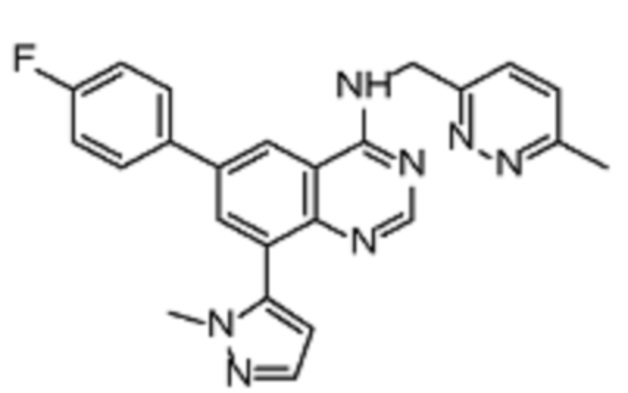
Стадия 1: Получение 6-(4-фторфенил)-8-йод-4-((2-(триметилсилил)этокси)метокси)хиназолина
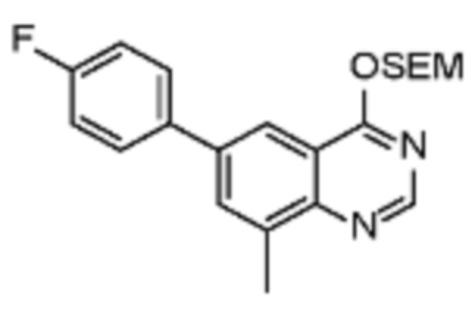
6-(4-Фторфенил)-8-йодхиназолин-4-ол (2,0 г, 5,46 ммоль) суспендируют в ДМФА (50 мл) и охлаждают до 0°С. Порциями добавляют гидрид натрия (60% в минеральном масле, 262 мг, 6,55 ммоль) и перемешивают реакционную смесь при 0°С в течение 30 минут. К смеси добавляют при 0°С 2-(триметилсилил)этоксиметилхлорид (1,5 мл, 8,19 ммоль) и реакционной смеси дают возможность нагреться до комнатной температуры в течение 16 часов. Реакционную смесь гасят водой и экстрагируют EtOAc (× 3). Объединенные органические фазы пропусают через бумагу для разделения фаз и растворитель удаляют в вакууме. Остаток очищают колоночной хроматографией (силикагель, элюирование с градиентом: 0-100% ДХМ в циклогексане, затем 0-100% этилацетата в ДХМ) с получением указанного в заголовке соединения (1,2 г, 44%) в виде твердого белого вещества.
ЖХМС (метод 3): [MH+]=497 при 6,43 мин.
Стадия 2: Получение 6-(4-фторфенил)-8-(1-метил-1H-пиразол-5-ил)-4-((2-триметилсилил)этокси)метокси)хиназолина
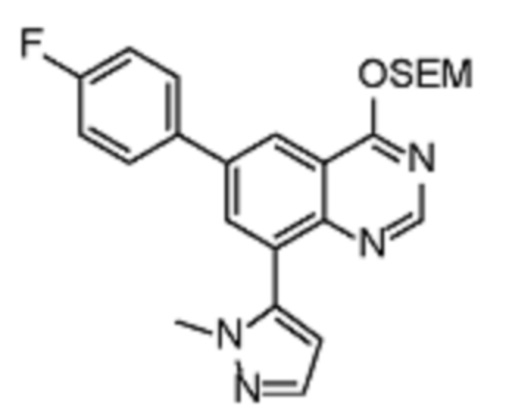
К 6-(4-фторфенил)-8-йод-4-((2-(триметилсилил)этокси)метокси)хиназолину (500 мг, 1,01 ммоль) добавляют сложный пинаколовый эфир 1-(метил-1H-пиразол)-5-бороновой кислоты (210 мг, 1,01 ммоль), карбонат цезия (1313 мг, 4,03 ммоль), тетракис(трифенилфосфин)палладий(0) (175 мг, 0,15 ммоль), диоксан (4 мл) и воду (2 мл). Реакционную смесь нагревают до 105°С и выдерживают при указанной температуре в течение 20 часов. К смеси добавляют дополнительные аликваты сложного пинаколового эфира 1-(метил-1H-пиразол-5-бороновой кислоты (210 мг, 1,01 ммоль) и тетракис(трифенилфосфин)палладия(0) (175 мг, 0,15 ммоль), реакционную смесь нагревают до 105°С и выдерживают при указанной температуре в течение 24 часов. Реакционную смесь охлаждают до комнатной температуры и распределяют между ДХМ и водой. Две фазы разделяют и водную фазу экстрагируют ДХМ. Объединенные органические фазы пропусают через бумагу для разделения фаз и растворитель удаляют в вакууме. Очистка колоночной хроматографией (силикагель, элюирование с градиентом: 0-50% этилацетата в циклогексане) приводит к получению указанного в заголовке соединения (388 мг, 85%) в виде масла желтого цвета.
¹H ЯМР (400 МГц, CDCl3): δ 8,60 (д, J=2,0 Гц, 1 H), 8,16 (c, 1 H), 7,96 (д, J=2,0 Гц, 1 H), 7,70-7,61 (м, 3 H), 7,18 (т, J=8,5 Гц, 2 H), 6,41 (д, J=1,8 Гц, 1 H), 5,46 (c, 2 H), 3,78 (c, 3 H), 3,70 (т, J=8,2 Гц, 2 H), 0,97 (т, J=8,3 Гц, 2 H), 0,00 (c, 9 H).
Стадия 3: Получение 6-(4-фторфенил)-8-(1-метил-1H-пиразол-5-ил)хиназолин-4-ола

Раствор 6-(4-фторфенил)-8-(1-метил-1H-пиразол-5-ил)-4-((2-триметилсилил)этокси)метокси)хиназолина (388 мг, 0,86 ммоль) в ДХМ (7,5 мл) обрабатывают трифторуксусной кислотой (2,5 мл) и реакционную смесь перемешивают при комнатной температуре в течение 3 часов. Растворители удаляют при пониженном давлении и остаток гасят насыщенным раствором NaHCO3 и водой. Полученный твердый продукт собирают фильтрацией и сушат в вакууме с получением указанного в заголовке соединения (185 мг, выход 67%) в виде твердого вещества розового цвета.
ЖХМС (метод 4): [MH+]=321 при 3,80 мин.
Стадия 4: Получение 6-(4-фторфенил)-8-(2-метилпиразол-3-ил)-N-[(6-метилпиридазин-3-ил)метил]хиназолин-4-амина
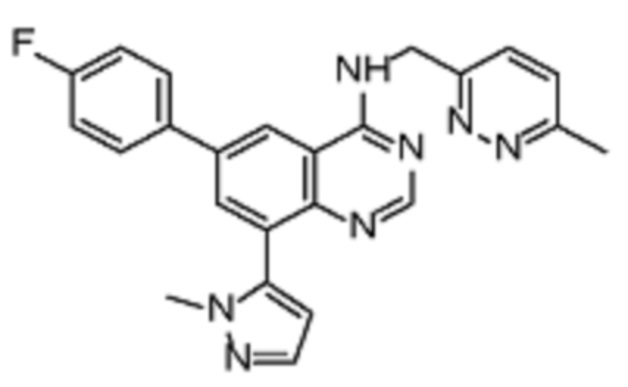
6-(4-Фторфенил)-8-(1-метил-1H-пиразол-5-ил)хиназолин-4-ол (100 мг, 0,31 ммоль) суспендируют в тионилхлориде (0,46 мл, 6,24 ммоль) и ДМФА (0,0002 мл, 0,003 ммоль). Реакционную смесь нагревают до 95°С, выдерживают при указанной температуре в течение 30 минут и затем охлаждают до комнатной температуры. Растворитель удаляют в вакууме. Полученный остаток суспендируют в диоксане (3,0 мл) и после этого добавляют DIPEA (272 мкл, 1,56 ммоль) и дигидрохлорид (6-метилпиридазин-3-ил)метанамина (92 мг, 0,47 ммоль). Реакционную смесь нагревают до 95°С, выдерживают при указанной температуре в течение 20 часов и затем охлаждают до комнатной температуры. Реакционную смесь распределяют между водой и ДХМ. Две фазы разделяют и водную фазу экстрагируют ДХМ. Объединенные органические фазы пропусают через бумагу для разделения фаз и растворитель удаляют в вакууме. Остаток очищают препаративной ЖХВД с получением указанного в заголовке соединения (12 мг, 9%) в виде твердого вещества коричневого цвета.
1H ЯМР (400 МГц, ДМСО): δ 9,34 (т, J=5,7 Гц, 1 H), 8,80 (д, J=2,0 Гц, 1 H), 8,47-8,46 (м, 1 H), 8,12 (д, J=2,0 Гц, 1 H), 8,00-7,95 (м, 2 H), 7,63-7,60 (м, 1 H), 7,52 (дд, J=3,4, 5,3 Гц, 2 H), 7,45-7,37 (м, 2 H), 6,46-6,45 (м, 1 H), 5,09-5,05 (м, 2 H), 3,66 (c, 3 H), 2,61 (c, 3 H).
ЖХМС (метод 3): [MH+]=426,5 при 4,3 мин.
Пример 95
6-(3-Хлор-1H-пиразол-1-ил)-8-метокси-N-(1-(2-(трифторметил)пиримидин-5-ил)этил)хиназолин-4-амин
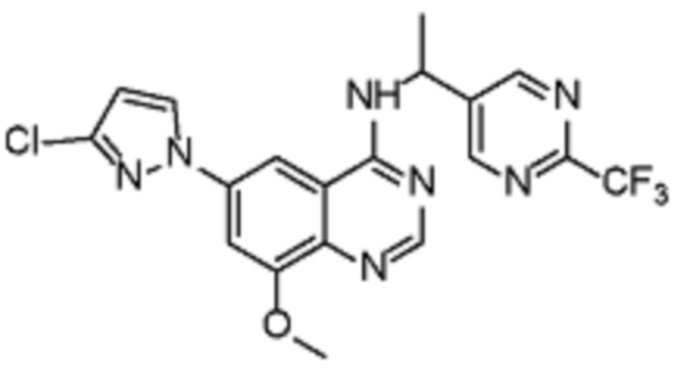
Газообразный азот барботируют через смесь 6-бром-8-метокси-N-[(1R)-1-[2-(трифторметил)пиримидин-5-ил]этил]хиназолин-4 амина (200 мг, 0,47 ммоль), бромида меди (I) (3,3 мг, 0,02 ммоль), 3-хлор-1H-пиразола (57 мг, 0,56 ммоль) и карбоната цезия (304 мг, 0,93 ммоль). Добавляют 1-(пиридин-2-ил)пропан-2-он (6,3 мг, 0,05 ммоль) в N, N-диметилформамидe (1,0 мл и переносят реакционную смесь в предварительно нагретый смеситель. Смесь нагревают до 120°С и выдерживают при указанной температуре в течение 16 часов. Реакционную смесь охлаждают, фильтруют через Celite® и промывают осадок на фильтре этилацетатом (2 × 5 мл). Остаток очищают колоночной хроматографией (силикагель, элюирование с градиентом: 0-7% метанола в дихлорметане), затем препаративной ЖХВД с получением указанного в заголовке соединения (30,0 мг, выход 14%) в виде твердого не совсем белого вещества.
¹H ЯМР (400 МГц, ДМСО): δ 9,17 (c, 2 H), 8,93 (c, 1 H), 8,63 (д, J=7,0 Гц, 1 H), 8,42 (c, 1 H), 8,33 (д, J=2,0 Гц, 1 H), 8,01 (c, 1 H), 7,70 (д, J=2,1 Гц, 1 H), 5,70-5,64 (м, 1 H), 4,02 (c, 3 H), 1,74 (д, J=7,2 Гц, 3 H). ЖХМС (метод 4): [MH+]=450,2 при 3,60 мин.
Соединения, представленные в таблице ниже, получают в соответствии с методикой, описанной для получения 6-(3-хлор-1H-пиразол-1-ил)-8-метокси-N-(1-(2-(трифторметил)пиримидин-5-ил)этил)хиназолин-4-амина.
структура
¹H ЯМР
ЖХ-МС

ЖХМС (метод 4): [MH+]=397 при 2,92 мин.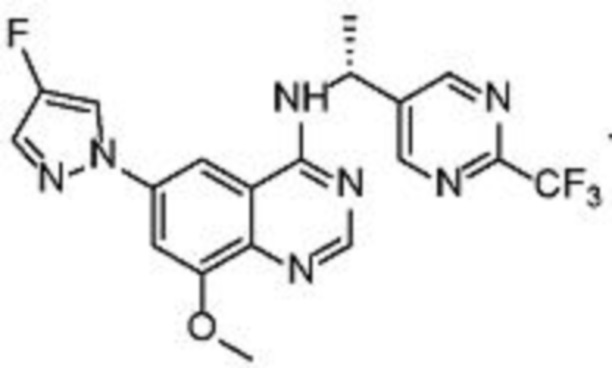
ЖХМС (метод 4): [MH+]=434 при 3,38 мин.
ЖХМС (метод 3): [MH+]=380 при 3,70 мин.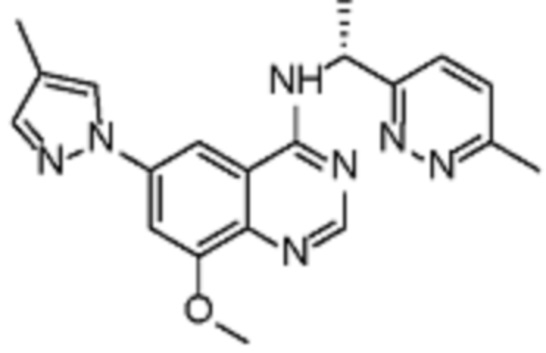
ЖХМС (метод 3): [MH+]=376 при 3,73 мин.
ЖХМС (метод 3): [MH+]=396 при 4,00 мин.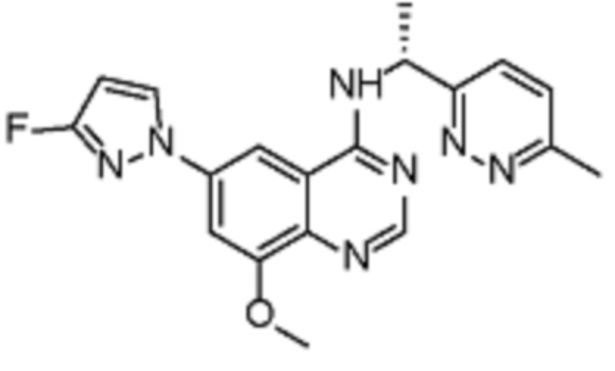
ЖХМС (метод 3): [MH+]=379 при 3,73 мин.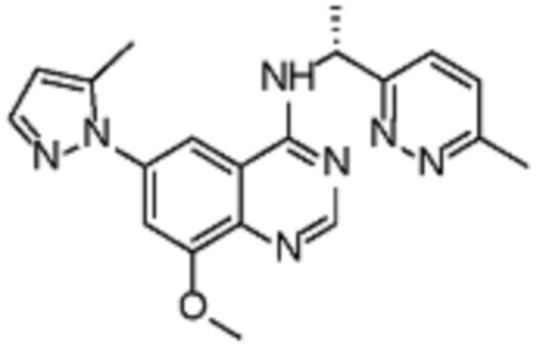
ЖХМС (метод 3): [MH+]=376 при 2,41 мин.
ЖХМС (метод 3): [MH+]=376 при 2,60 мин.
ЖХМС (метод 4): [MH+]=450 при 4,55 мин.
Промежуточное соединение 23
6-Бром-8-метокси-4-((2-(триметилсилил)этокси)метокси)хиназолин

6-Бром-8-метоксихиназолин-4-ол (200 мг, 0,78 ммоль) растворяют в N, N-диметилформамидe (10 мл) и реакционную смесь охлаждают до 0°С. К полученной смеси небольшими порциями добавляют гидрид натрия (60% дисперсия в минеральном масле, 38 мг, 0,94 ммоль) и перемешивают реакционную смесь в течение 30 мин. К смеси по каплям добавляют (2-хлорметоксиэтил)триметилсилан (0,21 мл, 1,18 ммоль). Реакционную смесь перемешивают при 0°С в течение 1 часа и затем дают возможность смеси нагреться до комнатной температуры. Реакционную смесь гасят водой (5 мл) и распределяют между водой и этилацетатом (15 мл). Фазы разделяют и водный слой промывают этилацетат (2 × 10 мл). Объединенные органические фазы сушат (MgSO4), фильтруют и концентрируют в вакууме. Сырой продукт очищают колоночной хроматографией (силикагель, элюирование с градиентом: 0-40% этилацетата в циклогексане) с получением указанного в заголовке соединения (181 мг, 60%) в виде твердого бесцветного вещества.
1H ЯМР (400 МГц, CDCl3): δ 8,17 (c, 1 H), 8,04 (д, J=2,0 Гц, 1 H), 7,3 (д, J=2,0 Гц, 1 H), 5,43 (c, 2 H), 4,02 (c, 3 H), 3,66 (м, J=4,1 Гц, 2 H), 0,95 (м, J=4,1 Гц, 2 H)), 0,02 (c, 9 H).
ЖХМС (метод 3): [MH+]=385 при 5,24 мин.
Промежуточное соединение 24
8-Метокси-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-4-((2-(триметилсилил)этокси)метокси)хиназолин

Газообразный азот барботируют в течение 10 минут через смесь 6-бром-8-метокси-4-((2-(триметилсилил)этокси)метокси)хиназолина (190 мг, 0,49 ммоль), бис(пинаколато)дибора (150 мг, 0,59 ммоль) и ацетата калия (97 мг, 0,99 ммоль) в 1,4-диоксане (5 мл), затем добавляют [1,1′-бис(дифенилфосфино)ферроцен]дихлорпалладий(II) (36 мг, 0,05 ммоль). Реакционную смесь нагревают до 95°С и выдерживают при указанной температуре в течение 2 часов. После этого реакционную смесь охлаждают до комнатной температуры и фильтруют через Celite®. Указанное соединение используют непосредственно в виде раствора в 1,4-диоксане, предполагая количественный выход.
Промежуточное соединение 25
6-(5-Фторпиридин-2-ил)-8-метокси-4-((2-(триметилсилил)этокси)метокси)хиназолин
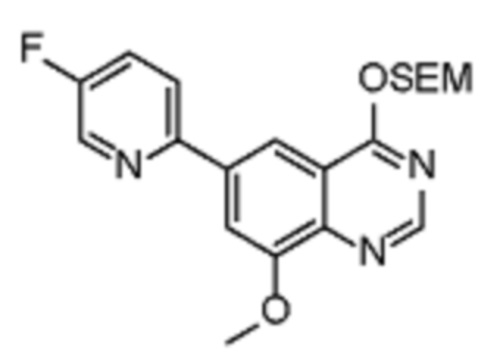
К 8-метокси-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-4-((2-(триметилсилил)этокси)метокси)хиназолину (330 мг, 0,78 ммоль) добавляют 2-бром-5-фторпиридин (137 мг, 0,78 ммоль), карбонат цезия (508 мг, 1,56 ммоль), 1,4-диоксан (8 мл) и воду (1 мл). Газообразный азот барботируют через реакционную смесь в течение 10 минут, после чего добавляют тетракис(трифенилфосфин)палладий(0) (90 мг, 0,078 ммоль). Затем реакционную смесь нагревают до 90°С и выдерживают при указанной температуре в течение 16 часов. После этого реакционную смесь охлаждают до комнатной температуры, фильтруют через Celite® и концентрируют в вакууме. Остаток очищают колоночной хроматографией (силикагель, элюирование с градиентом: 0-60% этилацетата в циклогексане) с получением указанного в заголовке соединения (226 мг, 72%) в виде твердого вещества бледно-желтого цвета.
1H ЯМР (400 МГц, ДМСО): δ 8,76 (д, J=2,4 Гц, 1 H), 8,46 (c, 1 H), 8,42 (д, J=1,7 Гц, 1 H), 8,30 (дд, J=9,1 Гц, 4,4 Гц, 1 H), 8,06 (д, J=1,7 Гц, 1 H), 7,91 (дт, J=2,4, 9,1Гц, 1 H), 5,44 (c, 2 H), 4,06 (c, 3 H), 3,64 (т, J=8,1Гц, 2 H), 0,97 (т, J=8,1 Гц, 2 H), 0,01 (c, 9 H).
ЖХМС (метод 3): [MH+]=402 при 5,43 мин.
Промежуточное соединение, представленное в таблице ниже, синтезируют в соответствии с методикой, описанной для получения 6-(5-фторпиридин-2-ил)-8-метокси-4-((2-(триметилсилил)этокси)метокси)хиназолина.
1H ЯМР
ЖХ-МС
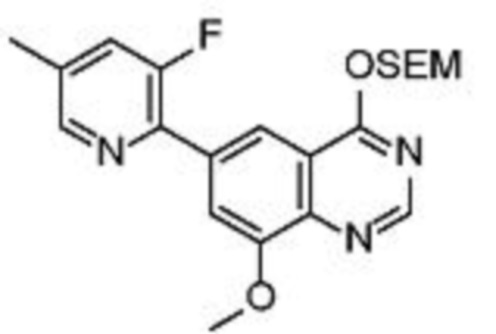
Промежуточное соединение 26
6-(5-Фторпиридин-2-ил)-8-метоксихиназолин-4-ол
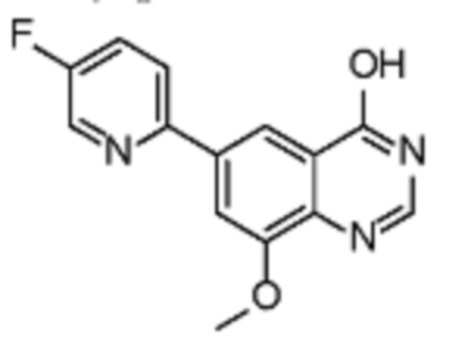
6-(5-Фторпиридин-2-ил)-8-метокси-4-((2-(триметилсилил)этокси)метокси)хиназолин (225 мг, 0,56 ммоль) растворяют в дихлорметане (5 мл) и трифторуксусной кислоте (0,21 мл, 2,81 ммоль). Реакционную смесь перемешивают в течение 16 часов. Реакционную смесь концентрируют в вакууме с получением указанного в заголовке соединения (142 мг, 93%) в виде твердого вещества оранжевого цвета.
¹H ЯМР (400 МГц, ДМСО): δ 8,76 (д, J=2.4 Гц, 1 H), 8,39 (д, J=1,7 Гц, 1 H), 8,29 (дд, J=9,0 Гц, 4,6 Гц, 1 H), 8,13 (c, 1 H), 8,05 (д, J=1,7 Гц, 1 H), 7,92 (дт, J=2,4, 9,1 Гц, 1 H), 5,69 (уш. с, 1 H), 4,06 (c, 3 H). ЖХМС (метод 3): [MH+]=272 при 3,08 мин.
Промежуточное соединение, представленное в таблице ниже, синтезируют в соответствии с методикой, описанной для получения 6-(5-фторпиридин-2-ил)-8-метоксихиназолин-4-ола (промежуточное соединение 26).
1H ЯМР
ЖХ-МС

Промежуточное соединение 61
4-Хлор-8-метокси-6-(5-метилпиримидин-2-ил)хиназолин
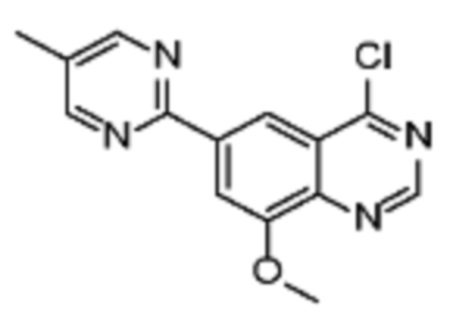
8-Метокси-6-(5-метилпиримидин-2-ил)хиназолин-4-ол (526 мг, 2,00 ммоль) (промежуточное соединение 27) и N, N-диизопропилэтиламин (760 мг, 5,9 ммоль) растворяют в толуоле (12 мл), нагревают до 120°С и выдерживают при указанной температуре в течение 20 минут. К раствору добавляют оксихлорид фосфора (331 мг, 2,2 ммоль) и перемешивают реакционную смесь в течение 2 часов. Реакционную смесь охлаждают до комнатной температуры и концентрируют в вакууме. Полученный остаток растирают с ацетонитрилом с получением указанного в заголовке соединения (284 мг, 50%) в виде твердого не совсем белого вещества.
¹H ЯМР (400 МГц, CDCl3): δ 9,08 (c, 1 H), 8,96 (c, 1 H), 8,73 (c, 2 H), 8,44 (c, 1 H), 4,24 (c, 3 H), 2,42 (c, 3 H).
Промежуточное соединение 28
8-Метокси-6-(1-метил-1H-пиразол-3-ил)хиназолин-4-ол

Газообразный азот барботируют в течение 10 минут через смесь 6-бром-8-метоксихиназолин-4-ола (613 мг, 2,4 ммоль), 1-метил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола (500 мг, 2,4 ммоль) и карбоната цезия (1570 мг, 4,8 ммоль) в 1,4-диоксане (25 мл) и воде (4 мл). Затем добавляют [1,1′-бис(дифенилфосфино)ферроцен]дихлорпалладий(II) (36 мг, 0,05 ммоль), полученную реакционную смесь нагревают до 95°С и выдерживают при указанной температуре в течение 3 часов. После этого реакционную смесь охлаждают до комнатной температуры и концентрируют в вакууме до трети объема. Затем добавляют воду (40 мл) и образовавшийся осадок собируют фильтрацией с получением указанного в заголовке соединения в виде твердого бесцветного вещества (449 мг, 73%)
1H ЯМР (400 МГц, ДМСО) δ 12,28 (c, 1 H), 8,06 (c, 1 H), 8,02 (c, 1 H), 7,80 (c, 1 H), 7,73 (c, 1 H), 6,86 (c, 1 H), 3,99 (c, 3 H,), 3,92 (c, 3 H). ЖХМС (метод 3): [MH+]=257 при 2,77 мин..
Пример 105
(8-Метокси-N-(1-(5-метил-1,3,4-оксадиазол-2-ил)этил)-6-(1-метил-1H-пиразол-3-ил)хиназолин-4-амин

Стадия 1: Получение 4-хлор-8-метокси-6-(1-метил-1H-пиразол-3-ил)хиназолин

8-Метокси-6-(1-метил-1H-пиразол-3-ил)хиназолин-4-ол (250 мг, 0,976 ммоль) и N, N-диизопропилэтиламин (151,0 мг, 1,17 ммоль) растворяют в толуоле (10 мл), полученный раствор нагревают 120°С и выдерживают при указанной температуре в течение 20 минут. К смеси добавляют оксихлорид фосфара (179 мг, 0,11 мл, 1,17 ммоль) и реакционную смесь перемешивают в течение 4 часов. Затем реакционную смесь охлаждают и концентрируют в вакууме с получением твердого вещества бледно-розового цвета (256 мг). Сырой остаток используют в следующей стадии без дополнительной очистки.
Стадия 2: Получение (8-метокси-N-(1-(5-метил-1,3,4-оксадиазол-2-ил)этил)-6-(1-метил-1H-пиразол-3-ил)хиназолин-4-амиан
4-Хлор-8-метокси-6-(1-метил-1H-пиразол-3-ил)хиназолин (150 мг, 0,546 ммоль) и N, N-диизопропилэтиламин (352 мг, 2,73 ммоль) растворяют в 1,4-диоксанe (5 мл). К раствору добавляют 1-(5-метил-1,3,4-оксадиазол-2-ил)этан-1-амин (139 мг, 1,09 ммоль) и перемешивают реакционную смесь при 50°С в течение 5 дней. Реакционную смесь охлаждают и концентрируют в вакууме. Остаток очищают препаративной ЖХВД с получением указанного в заголовке соединения в виде твердого не совсем белого вещества (11 мг, выход 6,3%).
1H ЯМР (400 МГц, ДМСО): δ 8,59 (д, J=7,8 Гц, 1 H), 8,41 (c, 1 H), 8,29 (д, J=1,3 Гц, 1 H), 7,83 (д, J=2,3 Гц, 1 H), 7,69 (д, J=1,0 Гц, 1 H), 6,89 (д, J=2,3 Гц, 1 H), 5,78-5,73 (м, 1 H), 3,98 (c, 3 H), 3,95 (c, 3 H), 2,58 (c, 3 H), 1,68 (д, J=7,2 Гц, 3 H). ЖХМС (метод 4): [MH+]=366 при 2,64 мин.
Соединения, представленные в таблице ниже, получают в соответствии с методикой, описнной для получния 8-метокси-N-(1-(5-метил-1,3,4-оксадиазол-2-ил)этил)-6-(1-метил-1H-пиразол-3-ил)хиназолин-4-амина.
структура
1H ЯМР
ЖХ-МС
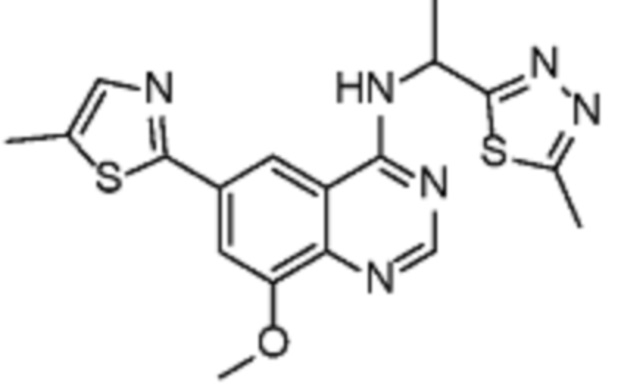
ЖХМС (метод 4): [MH+]=399 при 2,80 мин.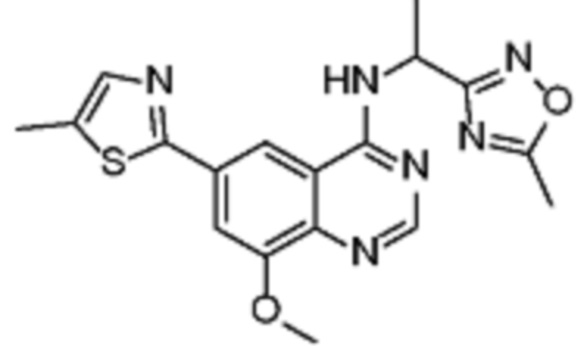
ЖХМС (метод 4): [MH+]=383 при 3,37 мин. 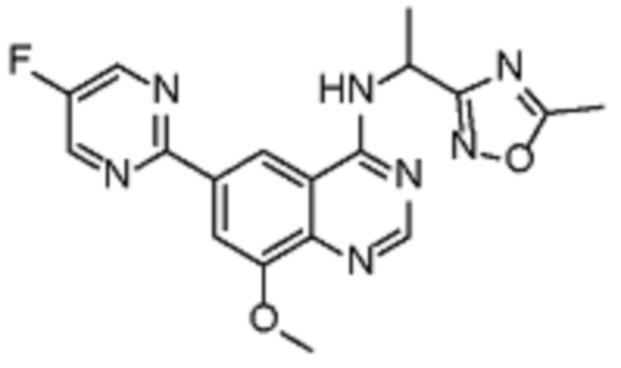
ЖХМС (метод 3): [MH+]=382 при 2,76 мин.
ЖХМС (метод 3): [MH+]=398 при 2,64 мин.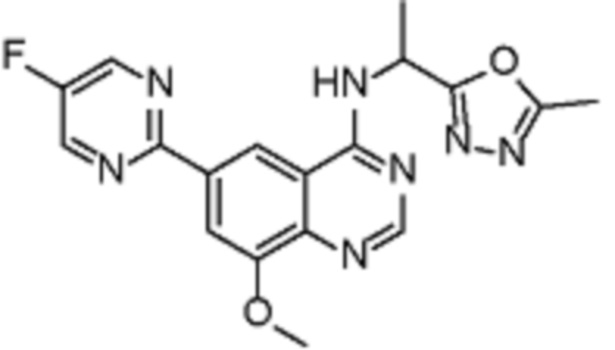
ЖХМС (метод 4): [MH+]=381 при 3,37 мин.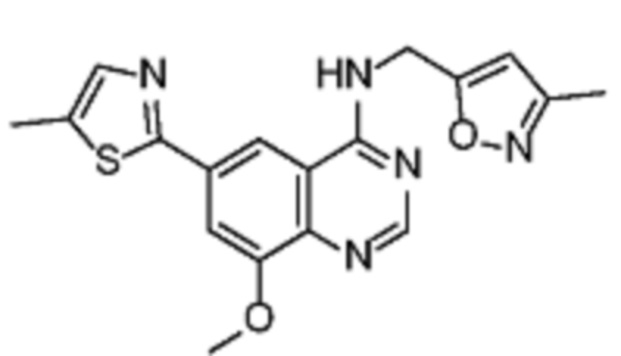
ЖХМС (метод 4): [MH+]=368 при 2,95 мин.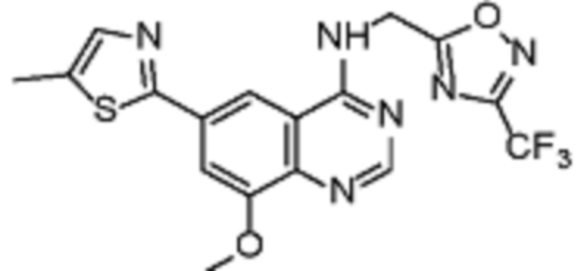
ЖХМС (метод 4): [MH+]=424 при 3,48 мин.
ЖХМС (метод 4): [MH+]=369 при 2,70 мин.
ЖХМС (метод 3): [MH+]=385 при 3,66 мин.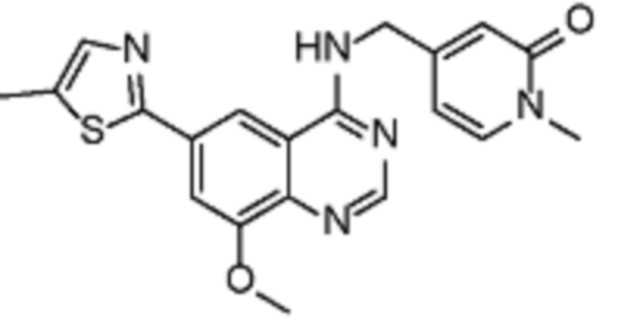
ЖХМС (метод 4): [MH+]=394 при 4,49 мин. 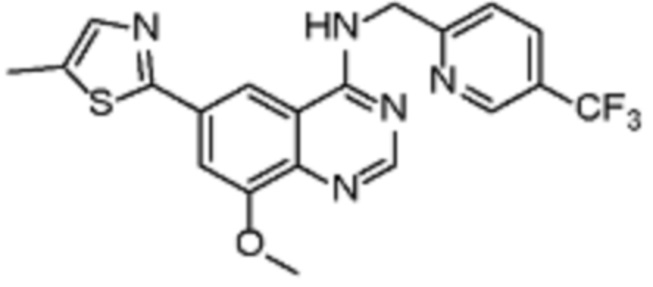
ЖХМС (метод 4): [MH+]=432 при 3,40 мин. 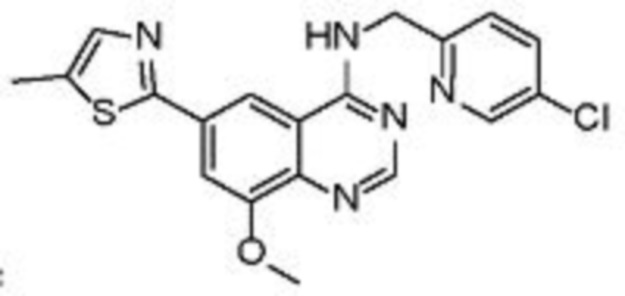
ЖХМС (метод 4): [MH+]=398 при 3,20 мин. 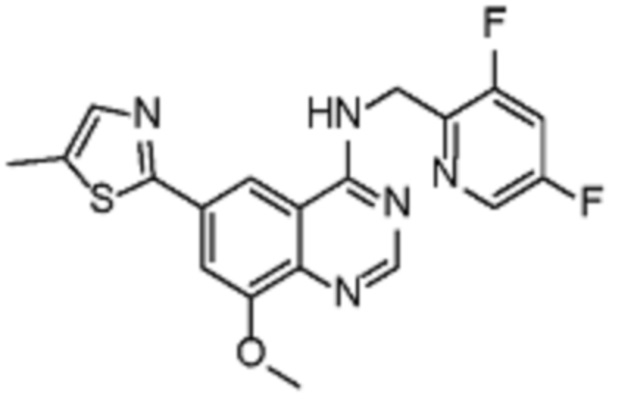
ЖХМС (метод 4): [MH+]=400 при 3,11 мин. 
ЖХМС (метод 4): [MH+]=382 при 2,99 мин.
ЖХМС (метод 3): [MH+]=444 при 5,04 мин.
Пример 120
Одиночный энантиомер 1 8-метокси-6-(5-метилпиримидин-2-ил)-N-[1-[6-(трифторметил)пиридазин-3-ил]этил]хиназолин-4-амина
Пример 121
Одиночный энантиомер 2 8-метокси-6-(5-метилпиримидин-2-ил)-N-[1-[6-(трифторметил)пиридазин-3-ил]этил]хиназолин-4-амина
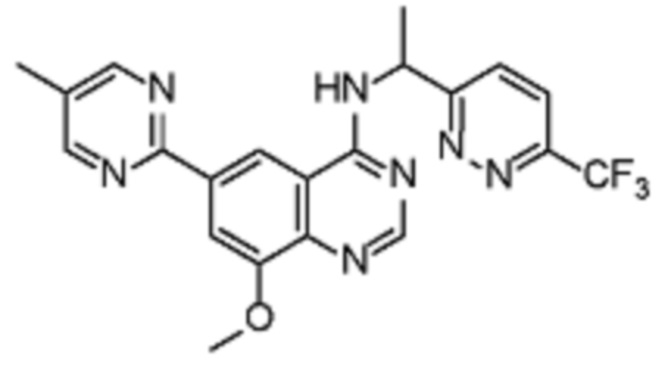
Смесь 6-(5-метилпиримидин-2-ил)-8-метоксихиназолин-4-ола (промежуточное соединение 27) (74 мг, 0.276 ммоль), оксихлорида фосфора(V) (0,034 мл, 0,36 ммоль) и DIPEA (0,24 мл, 1,38 ммоль) в толуоле (4 мл) нагревают до 90°С в атмосфере азота и выдерживают в указанных условиях в течение 2 часов. Реакционную смесь охлаждают до комнатной температуры и концентрируют в вакууме. Остаток распределяют между водой и дихлорметаном. Объединенные органические слои промывают насыщенным раствором гидрокарбоната натрия, пропускаяют гидрофобный фриттовый фильтр и концентрируют в вакууме. Остаток (88 мг, 0,275 ммоль) растворяют в хлороформе (0,5 мл) и к полученному раствор добавляют 1-(2-(трифторметил)пиримидин-5-ил)этан-1-амин (105 мг, 0,55 ммоль). Реакционную смесь нагревают до 70°С в герметичной пробирке в атмосфере азота и выдерживают в указанных условия в течение 18 часов. После этого полученную смесь охлаждают до комнатной температуры. Смесь разбавляют охлажденной водой и перемешивают в течение 10 минут, затем добавляют этилацетат и воду. Две фазы разделяют и водную фазу дополнительно экстрагируют этилацетатом (2 × 50 мл). Объединенные органические фазы пропусают через бумагу для разделения фаз и растворитель удаляют в вакууме с получением твердого не совсем белого вещества (69,2 мг, 57%). Остаток очищают препаративной хиральной СФХ с получением указанного в заголовке соединения в виде твердого не совсем белого вещества.
Пример 120
Одиночный энантиомер 1 8-метокси-6-(5-метилпиримидин-2-ил)-N-[1-[6-(трифторметил)пиридазин-3-ил]этил]хиназолин-4-амина : 43,8 мг, 63%
¹H ЯМР (400 МГц, ДМСО): δ 9,13-9,07 (м, 2 H), 8,86 (c, 2 H), 8,40 (c, 1 H), 8,24-8,19 (м, 2 H), 8,07 (д, J=8,9 Гц, 1 H), 5,93-5,88 (м, 1 H), 4,02 (c, 3 H), 2,39 (c, 3 H), 1,80 (д, J=7,2 Гц, 3 H). ЖХМС (метод 4): [MH+]=442 при 3,95 мин. Хиральный анализ (метод 36) при 0,73 мин.
Пример 121
Одиночный энантиомер 2 8-метокси-6-(5-метилпиримидин-2-ил)-N-[1-[6-(трифторметил)пиридазин-3-ил]этил]хиназолин-4-амиа: 8,0 мг, 11%.
1H ЯМР (400 МГц, ДМСО): δ 9,13-9,07 (м, 2 H), 8,87 (c, 2 H), 8,40 (c, 1 H), 8,24-8,19 (м, 2 H), 8,07 (д, J=8,8 Гц, 1 H), 5,93-5,88 (м, 1 H), 4,02 (c, 3 H), 2,39 (c, 3 H), 1,80 (д, J=7,2 Гц, 3 H). ЖХМС (метод 4): [MH+]=442 при 3,95 мин. Хиральный анализ (метод 36) при 1,54 мин.
Соединения, представленные в таблице ниже, получают в виде одиночных изомеров в соответствии с методикой, описанной для получения 8-метокси-6-(5-метилпиримидин-2-ил)-N-[1-[6-(трифторметил)пиридазин-3-ил]этил]хиназолин-4-амина.
структура
¹H ЯМР
ЖХ-МС

ЖХМС (метод 3): [MH+]=378 при 3,93 мин. Хиральный анализ (метод 12) при 2,72 мин.
ЖХМС (метод 4): [MH+]=378 при 2,8 мин. Хиральный анализ (метод 12) при 3,73 мин.
ЖХМС (метод 3): [MH+]=394 при 3,32 мин. Хиральный анализ (метод 39) при 1,56 мин.
ЖХМС (метод 3): [MH+]=394 при 3,32 мин. Хиральный анализ (метод 39) при 2,50 мин.
ЖХМС (метод 4): [MH+]=393 при 2,76 мин. Хиральный анализ (метод 9) при 1,43 мин.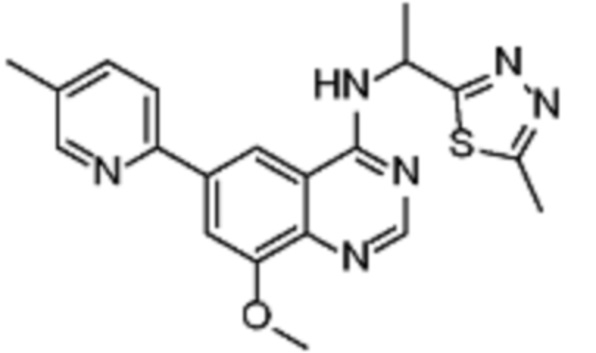
ЖХМС (метод 4): [MH+]=393 при 2,76 мин. Хиральный анализ (метод 12 при 2,66 мин.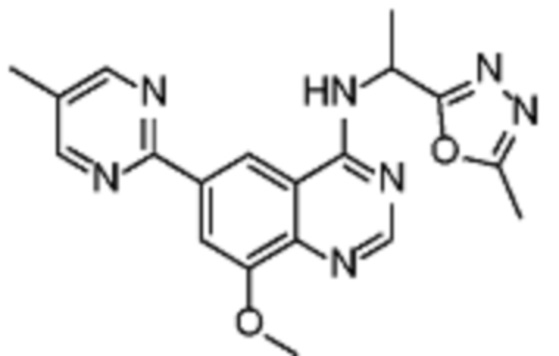
ЖХМС (метод 4): [MH+]=378 при 2,53 мин. Хиральный анализ (метод 13) при 1,49 мин.
ЖХМС (метод 4): [MH+]=378 при 2,53 мин. Хиральный анализ (метод 13) при 1,83 мин.
Промежуточное соединение 21
Получение 4-хлор-6-(5-фторпиридин-2-ил)-8-метоксихиназолина

8-Метокси-6-(5-фторпиридин-2-ил)хиназолин-4-ол (750 мг, 2,81 ммоль) суспендируют в тионилхлориде (4,1 мл, 56,12 ммоль) и добавляют ДМФА (0,0005 мл, 0,006 ммоль). Реакционную смесь нагревают до 95°С и выдерживают при указанной температуре в течение 4 часов. Реакционную смесь охлаждают до комнатной температуры и растворитель удаляют в вакууме с получением 4-хлор-6-(5-фторпиридин-2-ил)-8-метоксихиназолина (800 мг, 98%). Сырой остаток переносят непосредственно в следующую стадию без дополнительной очистки.
Пример 132
6-(5-Фторпиридин-2-ил)-8-метокси-N-((5-метил-1,2,4-оксадиазол-3-ил)метил)хиназолин-4-амин
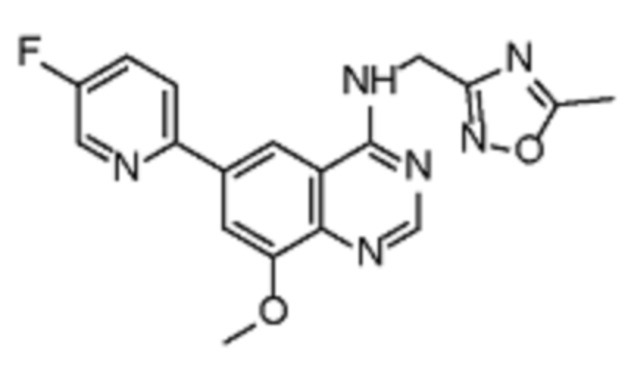
К суспензии 4-хлор-6-(5-фторпиридин-2-ил)-8-метоксихиназолина (150 мг, 0,51 ммоль) в диоксане (5 мл) добавляют DIPEA (0,45 мл, 2,59 ммоль) с последующим добавлением гидрохлорида (1,3,4-оксадиазол-2-ил)метанамина (105 мг, 0,77 ммоль). Реакционную смесь нагревают до 100°С и выдерживают при указанной температуре в течение 20 часов, затем дают возможность охладиться до комнатной температуры и растворитель удаляют в вакууме. Остаток переносят в ДХМ (25 мл), затем перемешивают в течение 30 мин., фильтруют через картридж разделения фаз и растворитель удаляют в вакууме. Остаток очищают препаративной ЖХВД с получением указанного в заголовке соединения (17,9 мг, 9%) в виде твердого не совсем белого вещества.
¹H ЯМР (400 МГц, ДМСО): δ 9,08 (т, J=5,6 Гц, 1 H), 8,59 (д, J=2,1 Гц, 1 H), 8,52 (д, J=1,5 Гц, 1 H), 8,49 (c, 1 H), 8,10 (д, J=8,1 Гц, 1 H), 8,04 (д, J=1,5 Гц, 1 H), 7,82 (дд, J=1,5, 8,2 Гц, 1 H), 4,99 (д, J=5,5 Гц, 2 H), 4,03-4,02 (м, 3 H), 2,48 (c, 3 H).
ЖХМС (метод 4): [MH+]=367 при 2,67 мин.
Соединения, представленные в таблице ниже, получают в соответствии с методикой, описанной для получения 6-(5-фторпиридин-2-ил)-8-метокси-N-((5-метил-1,2,4-оксадиазол-3-ил)метил)хиназолин-4-амина.
структура
¹H ЯМР
ЖХ-МС

ЖХМС (метод 4): [MH+]=367 при 2,71 мин.
ЖХМС (метод 4): [MH+]=367 при 2,49 мин.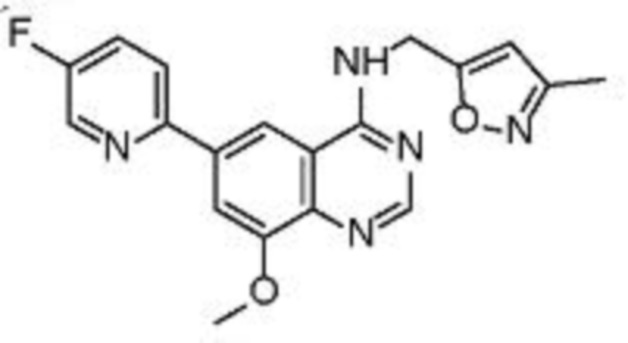
ЖХМС (метод 4): [MH+]=366 при 2,85 мин. 
ЖХМС (метод 3): [MH+]=363 при 3,36 мин.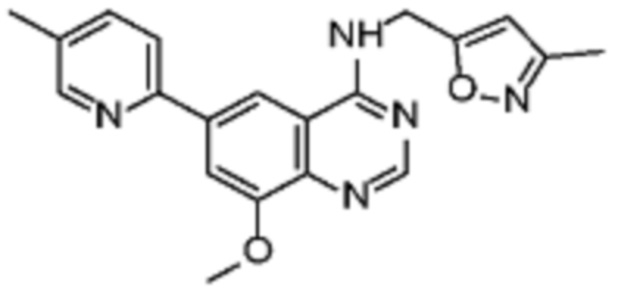
ЖХМС (метод 3): [MH+]=362,4 при 3,59 мин.
ЖХМС (метод 3): [MH+]=366 при 4,12 мин. 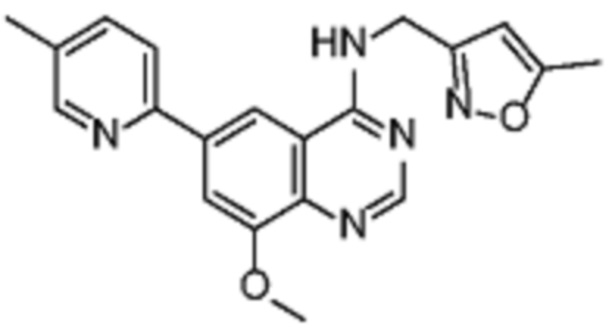
ЖХМС (метод 4): [MH+]=362 при 2,94 мин. 
ЖХМС (метод 3): [MH+]=365 при 3,33 мин.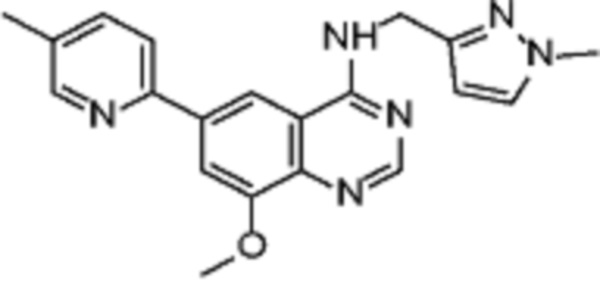
ЖХМС (метод 3): [MH+]=361 при 3,9 мин.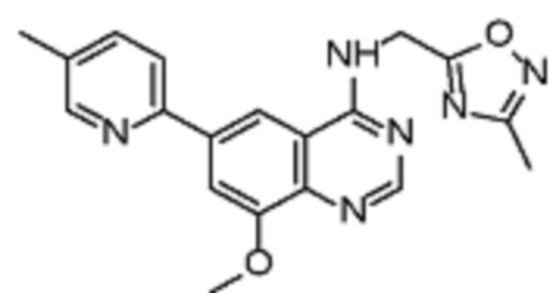
ЖХМС (метод 4): [MH+]=363 при 2,7 мин.
ЖХМС (метод 4): [MH+]=363 при 2,47 мин.
ЖХМС (метод 4): [MH+]=418 при 3,19 мин.
ЖХМС (метод 4): [MH+]=429 при 3,09 мин.
ЖХМС (метод 3): [MH+]=431 при 4,42 мин.
ЖХМС (метод 4): [MH+]=392 при 2,49 мин.
ЖХМС (метод 4): [MH+]=378 при 2,47 мин.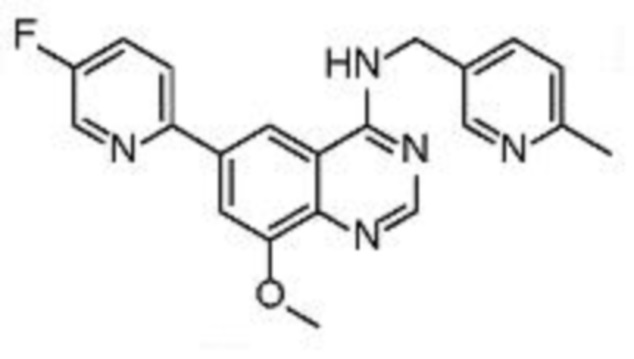
ЖХМС (метод 3): [MH+]=376 при 3,56 мин.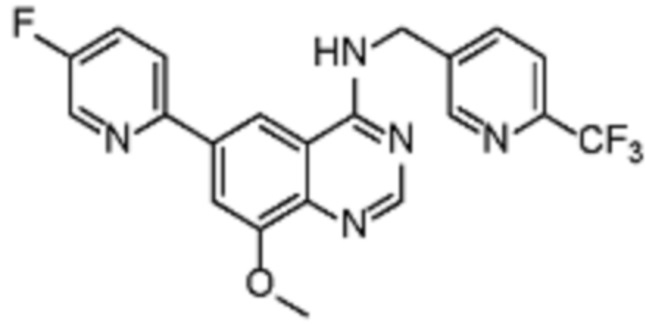
ЖХМС (метод 4): [MH+]=430 при 3,36 мин.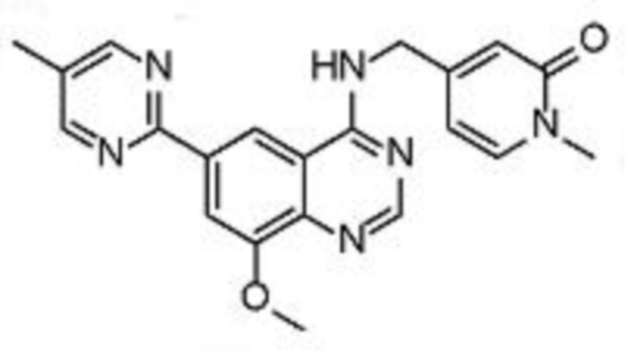
ЖХМС (метод 3): [MH+]=389 при 3,21 мин.
ЖХМС (метод 3): [MH+]=375 при 3,22 мин.
ЖХМС (метод 4): [MH+]=380 при 2,49 мин.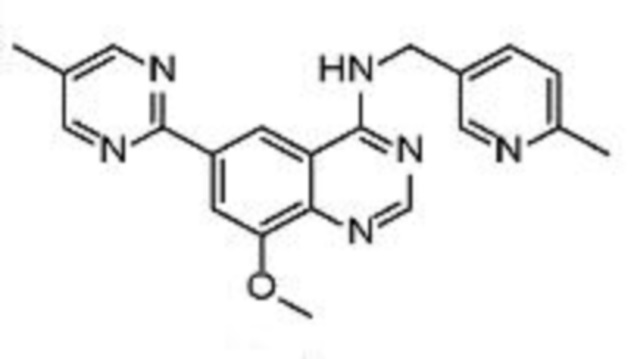
ЖХМС (метод 4): [MH+]=373 при 1,99 мин.
ЖХМС (метод 4): [MH+]=427 при 3,19 мин.
Пример 160
2-((8-Метокси-6-(5-метилпиримидин-2-ил)хиназолин-4-ил)амино)-1-морфолиноэтан-1-он

Стадия 1: Получение метил-(8-метокси-6-(5-метилпиримидин-2-ил)хиназолин-4-ил)глицината

К суспензии сырого 4-хлор-8-метокси-6-(5-метилпиримидин-2-ил)хиназолина (500 мг, 1,86 ммоль) в диоксане (5 мл) добавляют DIPEA (1,6 мл, 9,32 ммоль) и сложный метиловый эфир глицина (382 мг, 4,29 ммоль). Смесь нагревают до 95°С и выдерживают при указанной температуре в течение 18 часов. Реакционную смесь охлаждают до комнатной температуры и концентрируют в вакууме. Полученный остаток очищают колоночной флэш-хроматографией (силикагель, элюирование: 20% этилацетата в метаноле) с получением указанного в заголовке соединения (552 мг, выход 87%) в виде твердого вещества бледно-желтого цвета.
ЖХМС (метод 3): [MH+]=340 при 2,47 мин.
Стадия 2: Получение Метил N-(трет-бутоксикарбонил)-N-(8-метокси-6-(5-метилпиримидин-2-ил)хиназолин-4-ил)глицината

К раствору метил-(8-метокси-6-(5-метилпиримидин-2-ил)хиназолин-4-ил)глицината (550 мг, 1,62 ммоль) в дихлорметане (25 мл) добавляют DMAP (20 мг, 0,16 ммоль), триэтиламин (0,56 мл, 4,05 ммоль) и ди-трет-бутилдикарбонат (707 мг, 3,24 ммоль). Смесь перемешивают при комнатной температуре в течение 3 часов. После этого смесь концентрируют в вакууме и остаток очищают колоночной хроматографией (силикагель, элюирование: 10% метанол/этилацетат) с получением указанного в заголовке соединения (689 мг, 96%) в виде твердого вещества желтого цвета.
ЖХМС (метод 3): [MH+]=440 при 4,71 мин.
Стадия 3: Получение N-(трет-бутоксикаронил)-N-(8-метокси-6-(5-метилпиримидин-2-ил)хиназолин-4-ил)глицина
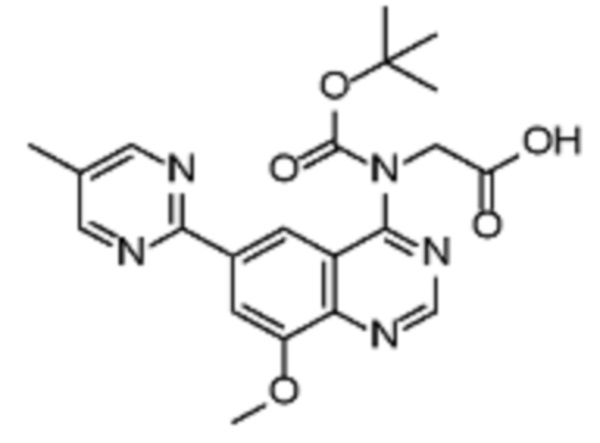
К суспензии метил-N-(трет-бутоксикарбонил)-N-(8-метокси-6-(5-метилпиримидин-2-ил)хиназолин-4-ил)глицината (680 мг, 1,55 ммоль) в смеси ТГФ (20 мл) и метанола (20 мл) добавляют 1 M аводный раствор гидроксида лития (3,9 мл, 2,5 ммоль). Смесь перемешивают при комнатной температуре в течение 5 часов. После этого реакционную смесь разбавляют водой (20 мл). Смесь подкисляют до pH 7, используя 1 M водный раствор соляной кислоты. Водную смесь концентрируют в вакууме и остаток очищают ОФ-хроматографией (элюирование с градиентом: 0-100% ACN/вода (NH4HCO3 модификатор)) с получением указанного в заголовке соединения в виде твердого не совсем белого вещества (498 мг, 75%).
ЖХМС (метод 4): [MH+]=426 при 3,98 мин.
Стадия 4: Получение трет-бутил-(8-метокси-6-(5-метилпиримидин-2-ил)хиназолин-4-ил)(2-морфолино-2-оксоэтил)карбамата

К раствору N-(трет-бутоксикарбонил)-N-(8-метокси-6-(5-метилпиримидин-2-ил)хиназолин-4-ил)глицина (50 мг, 0,12 ммоль) в ДМФА (2 мл) добавляют триэтиламин (0,033 мл, 0,24 ммоль), HATU (58 мг, 0,15 ммоль) и морфолин (0,015 мл, 0,18 ммоль). Смесь перемешивают при комнатной температуре в течение 18 часов. После этого смесь концентрируют в вакууме и остаток очищают обращено-фазовой хроматографией (элюирование с градиентом: 0-100% ACN/вода (NH4HCO3 модификатор) с получением указанного в заголовке соединения в виде твердого не совсем белого вещества (38 мг, 65%).
ЖХМС (метод 4): [MH+]=495 при 3,97 мин.
Стадия 5: Получение 2-((8-метокси-6-(5-метилпиримидин-2-ил)хиназолин-4-ил)амино)-1-морфолиноэтан-1-она

К раствору трет-бутил-(8-метокси-6-(5-метилпиримидин-2-ил)хиназолин-4-ил)(2-морфолино-2-оксоэтил)карбамата (28 мг, 0,057 ммоль) в диоксане (2 мл) добавляют 4 M раствор соляной кислоты в диоксане (2 мл). Смесь перемешивают при комнатной температуре в течение 2 часов. После этого смесь концентрируют в вакууме. Остаток очищают препаративной обращенно-фазовой ЖХВД с получением указанного в заголовке соединения в виде твердого бесцветного вещества (13 мг, 58%).
1H ЯМР (400 МГц, ДМСО): δ 8,85 (c, 3 H), 8,69 (дд, J=5,6, 5,6 Гц, 1 H), 8,46 (c, 1 H), 8,21 (д, J=1,4 Гц, 1 H), 4,41 (д, J=5,6 Гц, 2 H), 4,02 (c, 3 H), 3,69-3,64 (м, 2 H), 3,63-3,57 (м, 4 H), 3,49 (дд, J=4,6, 4,6 Гц, 2 H), 2,38 (c, 3 H). ЖХМС (метод 4): [MH+]=395 при 2,33 мин.
Пример 156
8-Метокси-N-((3-метил-1,2,4-оксадиазол-5-ил)метил)-6-(5-метилпиримидин-2-ил)хиназолин-4-амин
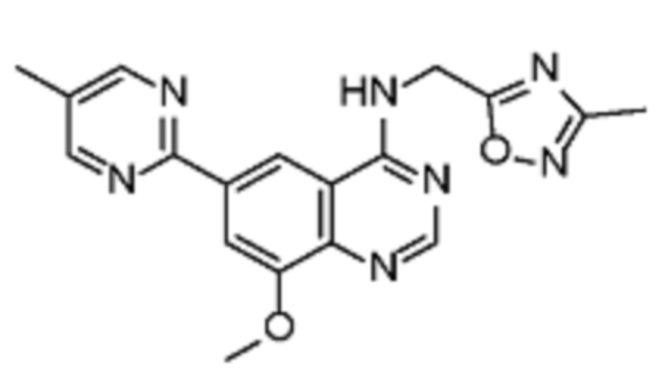
К раствору N-(трет-бутоксикарбонил)-N-(8-метокси-6-(5-метилпиримидин-2-ил)хиназолин-4-ил)глицина (100 мг, 0,24 ммоль) в ДМФА (3 мл) добавляют 1-этил-3-(3-диметиламинопропил)карбодиимид (59 мг, 0,31 ммоль) и перемешивают полученную смесь при комнатной температуре в течение 10 минут. К смеси добавляют N-гидроксиацетимидамид (26 мг, 0,35 ммоль), полученную смесь нагревают до 120°С и выдерживают при указанной температуре в течение 45 минут. После этого смесь концентрируют в вакууме. Остаток растворяют 1,4-диоксане (2 мл) и обрабатывают 4 M HCl в 1,4-диоксане (5 мл). Полученную смесь перемешивают при комнатной температуре в течение дополнительного 1 часа и затем концентрируют в вакууме. Остаток очищают препаративной ЖХВД с получением указанного в заголовке соединения (18,5 мг, 12% для двух втадий) в виде твердого вещества светло-коричневого цвета.
¹H ЯМР (400 МГц, ДМСО): δ 9,36 (т, J=5,6 Гц, 1 H), 8,90-8,85 (м, 3 H), 8,48-8,47 (м, 1 H), 8,24 (д, J=1,4 Гц, 1 H), 4,98 (д, J=5,6 Гц, 2 H), 4,04-4,03 (м, 3 H), 2,38 (c, 3 H), 2,31 (c, 3 H).
ЖХМС (метод 4): [MH+]=364 при 2,63 мин.
Пример 157
6-(3-Фтор-5-метил-2-пиридил)-8-метокси-N-[(1R)-1-(5-метил-1,2,4-оксадиазол-3-ил)этил]хиназолин-4-амин
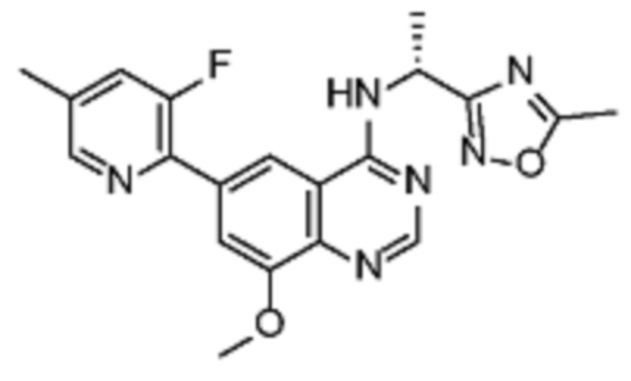
Раствор 6-(3-фтор-5-метилпиридин-2-ил)-8-метокси-4-((2-(триметилсилил)этокси)метокси)хиназолина (700 мг, 1,68 ммоль) в ДХМ (20 мл) обрабатывают трифторуксусной кислотой (2,6 мл) и перемешивают реакционную смесь при комнатной температуре в течение 19 часов. Растворители удаляют при пониженном давлении и остаток гасят насыщенным раствором NaHCO3 и водой. Обращующийся твердый осадок собирают фильтрацией и сушат в вакууме с получением 6-(3-фтор-5-метилпиридин-2-ил)-8-метоксихиназолин-4-ола (470 мг) в виде твердого не совсем белого вещества. Часть осадка (100 мг, 0,35 ммоль) суспендируют в тионилхлориде (0,51 мл, 7,01 ммоль) и ДМФА (0,005 мл). Реакционную смесь нагревают до 90°С и выдерживают при указанной температуре в течение 3 часов, затем охлаждают до комнатной температуры. Растворитель удаляют в вакууме. Остаток растворяют в ДХМ и промывают насыщенным раствором NaHCO3, водой, затем сушат и удаляют растворитель в вакууме. Остаток суспендируют в диоксане (2,0 мл) и DIPEA (0,30 мл, 1,75 ммоль) и добавляют гидрохлорид [(R)-1-(5-метил-1,2,4-оксадиазол-3-ил)этан-1-амина (79 мг, 0,525 ммоль). Реакционную смесь нагревают до 55°С и выдерживают при указанной температуре в течение 4 дней, затем охлаждают до комнатной температуры. Реакционную смесь распределяют между этилацетатом и водой. Две фазы разделяют, водную фазу экстрагируют этилацетатом. Объединенные органические фазы концентрируют в вакууме и остаток очищают препаративной ЖХВД с получением указанного в заголовке соединения (38,1 мг, 27%) в виде твердого белого вещества.
1H ЯМР (400 МГц, ДМСО): δ 8,80 (д, J=7,5 Гц, 1 H), 8,48 (д, J=3,4 Гц, 2 H), 8,43 (c, 1 H), 7,79 (квд, J=2,5, 7,0 Гц, 1 H), 7,70 (c, 1 H), 5,77-5,73 (м, 1 H), 3,98 (c, 3 H), 2,57 (c, 3 H), 2,44 (c, 3 H), 1,66 (д, J=7,2 Гц, 3 H). ЖХМС (метод 4): [MH+]=395 при 3,03 мин. Хиральный анализ (метод 35) при 1,75 мин.
Пример 158
6-(5-Фтор-2-пиридил)-8-метокси-N-[(1R)-1-(1-метилпиразол-3-ил)этил]хиназолин-4-амин

Стадия 1: Получение (R)-2-метил-N-((1-метил-1H-пиразол-3-ил)метилene)propane-2-сульфинамида

К раствору 1-метил-1H-пиразол-3-карбальдегида (500 мг, 4,54 ммоль) в ДХМ (5,0 мл) добавляют сульфат меди (1667 мг, 10,4 ммоль) и (R)-(+)-2-метил-2-пропансульфинамид (605 мг, 4,99 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 72 часов в атмосфере азота. Полученную смесь фильтруют через слой Celite® и промывают твердый осадок ДХМ. Фильтрат концентрируют при пониженном давлении, растворяют в ацетонитриле и фильтруют. Фильтрат концентрируют при пониженном давлении и очищают флэш-хроматографией (элюирование с градиентом: 0-50% этилацетата в циклогексане) с получением указанного в заголовке соединения (955 мг, 99%) в виде твердого белого вещества.
¹H ЯМР (400 МГц, CDCl3): δ 8,63 (c, 1 H), 7,40 (д, J=2,3 Гц, 1 H), 6,77 (д, J=2,5 Гц, 1 H), 3,99 (c, 3 H), 1,25-1,25 (м, 9 H).
Стадия 2: Получение (R)-2-метил-N-((R)-1-(1-метил-1H-пиразол-3-ил)этил)пропан-2-сульфинамида

Раствор (R)-2-метил-N-((1-метил-1H-пиразол-3-ил)метилен)пропан-2-сульфинамида (500 мг, 2,34 ммоль) в ДХМ (10,0 мл) охлаждают до -60°С. К смеси медленно добавляют бромид метилмагния в диэтиловом эфире (3,0 M, 1,0 мл, 2,93 ммоль). Реакционную смесь перемешивают при -60°С в течение 1 часа, затем медленно нагревают до комнатной температуры. После этого реакционную смесь снова охлаждают до -60°С и медленно добавляют еще одну аликвоту бромида метилмагния в диэтиловом эфире (3,0M, 0,5 мл, 1,47 ммоль). Реакционную смесь нагревают до комнатной температуры в течение ночи. Реакцию гасят насыщенным водным раствором NH4Cl и две фазы разделяют. Водную фазу экстрагируют ДХМ (2 × 50 мл) и объединенные органические фазы пропускают через бумагу для разделения фаз. Растворитель удаляют в вакууме. Остаток очищают колоночной флэш-хроматографией (силикагель, элюирование с градиентом: 0-10% метанола в ДХМ) с получением указанного в заголовке соединения (206 мг, 38%) в виде бесцветного масла.
ЖХМС (метод 3): [MH+]=230 при 3,41 мин.
Стадия 3: Получение гидрохлорида (R)-1-(1-метил-1H-пиразол-3-ил)этан-1-амина

К раствору (R)-2-метил-N-((R)-1-(1-метил-1H-пиразол-3-ил)этил)пропан-2-сульфонамида (206 мг, 0,90 ммоль) в диоксане (2,2 мл) добавляют раствор соляной кислоты в диоксане (4,0M, 2,2 мл, 8,98 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 18 часов. Растворитель удаляют в вакууме. Остаток растирают с этилацетатом с получением указанного в заголовке соединения (160 мг, количественный выход) в виде твердого белого вещества.
ЖХМС (метод 3): [MH+]=126 при 1,88 мин.
Стадия 4: Получение 6-(5-фтор-2-пиридил)-8-метокси-N-[(1R)-1-(1-метилпиразол-3-ил)этил]хиназолин-4-амина

К суспензии 4-хлор-6-(5-фторпиридин-2-ил)-8-метоксихиназолина (106 мг, 0,37 ммоль) в диоксане (3,0 мл) добавляют DIPEA (0,32 мл, 1,84 ммоль) с последующим добавлением гидрохлорида (R)-1-(1-метил-1H-пиразол-3-ил)этан-1-амина (89 мг, 0,55 ммоль). Реакционную смесь нагревают до 95°С и выдерживают при указанной температуре в течение 20 часов, затем смеси дают возможность охладиться до комнатной температуры. Реакционную смесь распределяют между ДХМ и водой и фазы разделяют. Водную фазу промывают ДХМ (2 × 50 мл) и затем воду удаляют в вакууме. Остаток очищают препаративной ЖХВД с получением указанного в заголовке соединения (5,25 мг, 4%) в виде твердого не совсем белого вещества.
1H ЯМР (400 МГц, ДМСО): δ 8,72 (д, J=3,0 Гц, 1 H), 8,56 (д, J=1,6 Гц, 1 H), 8,51 (д, J=8,3 Гц, 1 H), 8,46 (c, 1 H), 8,31 (дд, J=4,3, 9,2 Гц, 1 H), 7,99 (д, J=1,5 Гц, 1 H), 7,98-7,92 (м, 1 H), 7,60 (д, J=2,1 Гц, 1 H), 6,20 (д, J=2,1 Гц, 1 H), 5,79-5,71 (м, 1 H), 4,00 (c, 3 H), 3,81-3,81 (м, 3 H), 1,62 (д, J=6,9 Гц, 3 H). ЖХМС (метод 4): [MH+]=379 при 3,00 мин. Хиральный анализ (метод 29) при 2,4 мин.
Соеднения, представленные в таблице ниже, получают в соответствии с методикой, описанной для получения 6-(5-фтор-2-пиридил)-8-метокси-N-[(1R)-1-(1-метилпиразол-3-ил)этил]хиназолин-4-амина.
структура
¹H ЯМР
ЖХ-МС

ЖХМС (метод 4): [MH+]=375 при 2,84 мин. Хиральный анализ (метод 29) при 3,1 мин.
Пример 161
6-(4-Фторфенил)-N-[(5-фтор-2-пиридил)метил]-8-метоксихиназолин-4-амин
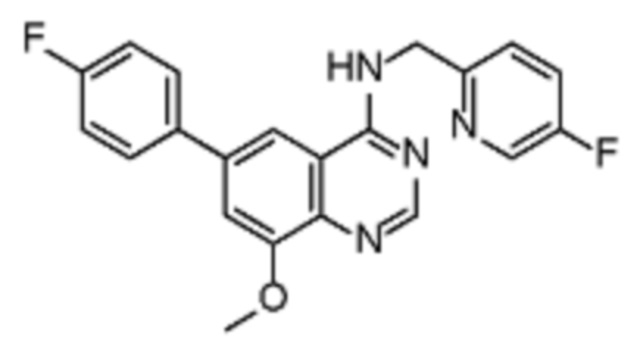
Стадия 1: Получение гидрохлорида 6-(4-фторфенил)-8-метоксихиназолин-4-амина
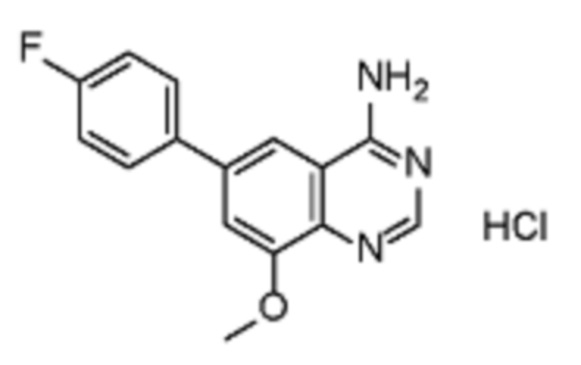
Раствор 4-хлор-6-(4-фторфенил)-8-метоксихиназолина (100 мг, 0,346 ммоль) в 7M растворе аммиака в метаноле (5 мл, 35,0 ммоль) перемешивают при комнатной температуре в течение 18 часов в герметичной пробирке. Реакционную смесь упаривают досуха с получением указанного в заголовке соединения (105 мг, количественный выход) в виде твердого вещества бежевого цвета, которое переносят в следующую стадию без дополнительной очистки.
Стадия 2: Получение 6-(4-фторфенил)-N-[(5-фтор-2-пиридил)метил]-8-метоксихиназолин-4-амина
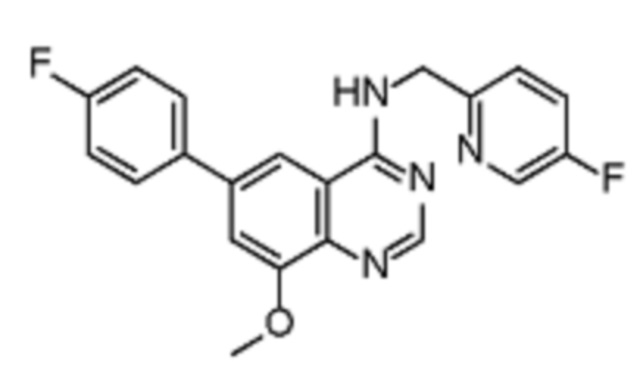
В пробирку для микроволновой обработки загружают гидрохлорид 6-(4-фторфенил)-8-метоксихиназолин-4-амина (40 мг, 0,132 ммоль), 5-фторпиколинальдегид (19,0 мг, 0,149 ммоль), этанол (1 мл), уксусную кислоту (0,05 мл) и триацетоксиборгидрид натрия (47 мг, 0,223 ммоль). Полученную смесь подвергают микроволновой обработке в течение 30 минут при 170°C. После охлаждения до комнатной температуры реакционную смесь концентрируют в вакууме. Остаток очищают препаративной ЖХВД с получением указанного в заголовке соединения (1,5 мг, 3%) в виде твердого не совсем белого вещества.
1H ЯМР (400 МГц, ДМСО): δ 9,04-9,01 (м, 1 H), 8,57 (д, J=3,2 Гц, 1 H), 8,45 (c, 1 H), 8,22 (д, J=1,6 Гц, 1 H), 8,01-7,95 (м, 2 H), 7,72-7,68 (м, 1 H), 7,58 (д, J=1,6 Гц, 1 H), 7,49-7,47 (м, 1 H), 7,45-7,39 (м, 2 H), 4,92 (д, J=5,6 Гц, 2 H), 4,06 (c, 3 H). ЖХМС (метод 3): [MH+]=379 при 4,5 мин.
Пример 188
6-(3,6-Дигидро-2H-пиран-4-ил)-8-метокси-N-((6-метилпиридазин-3-ил)метил)хиназолин-4-амин

К смеси 6-бром-8-метокси-N-((6-метилпиридазин-3-ил)метил)хиназолин-4-амина (промежуточное соединение 9) (150 мг, 0,41 ммоль), сложного пиноколового эфира 3,6-дигидро-2H-пиран-4-уксусной кислоты (175 мг, 0,834 ммоль) и дихлорида бис(трифенилфосфин)палладия(II) (15 мг, 0,02 ммоль) в 1,2-диметоксиэтане (5 мл) добавляют карбонат цезия (271 мг, 0,83 ммоль) в воде (0,5 мл). Реакционную смесь перемешивают в атмосфере азота при 100°С в течение 16 часов. После этого реакционную смесь фильтруют через слой Celite® и промывают этилацетатом (10 мл). Растворитель удаляют в вакууме и полученный остаток очищают препаративной ЖХВД с получением указанного в заголовке соединения (38 мг, 22%) в виде твердого бесцветного вещества.
1H ЯМР (400 МГц, ДМСО): δ 8,96 (дд, J=5,8, 5,8 Гц, 1 H), 8,38 (c, 1 H), 7,86 (д, J=1,3 Гц, 1 H), 7,55-7,48 (м, 2 H), 7,39 (д, J=1,4 Гц, 1 H), 6,51 (c, 1 H), 5,01 (д, J=5,9 Гц, 2 H), 4,32-4,30 (м, 2 H), 3,96 (c, 3 H), 3,90 (дд, J=5,5, 5,5 Гц, 2 H), 2,65-2,59 (м, 5 H). ЖХМС (метод 3): [MH+]=364 при 3,27 мин.
Соединения, представленные в таблице ниже, получают в виде одиночных изомеров с помощью очистки хиральной препаративной СФХ соответствующей рацемической смеси, описанной выше.
структура
¹H ЯМР
ЖХ-МС

ЖХМС (метод 3): [MH+]=377 при 4,17 мин. Хиральный анализ (метод 28) при 2,01 мин.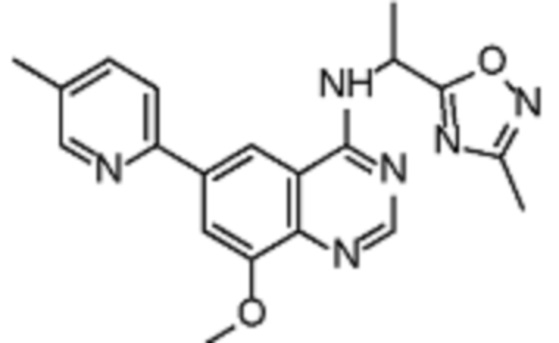
ЖХМС (метод 3): [MH+]=377 при 4,2 мин. Хиральный анализ (метод 28) при 2,83 мин.
ЖХМС (метод 4): [MH+]=399 при 2,80 мин. Хиральный анализ (метод 20) при 1,10 мин.
ЖХМС (метод 4): [MH+]=399 при 2,80 мин. Хиральный анализ (метод 20) при 3,16 мин.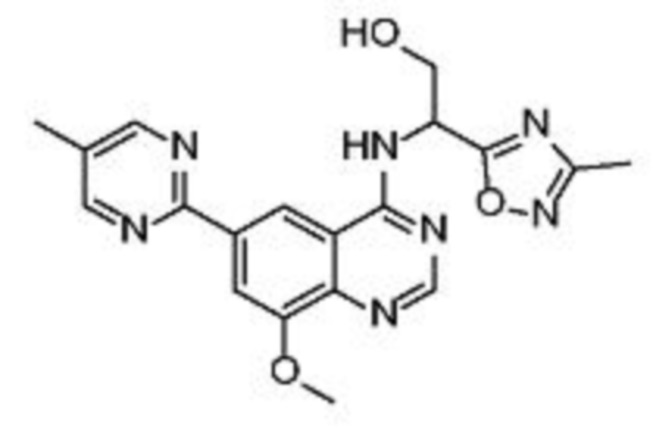
ЖХМС (метод 4): [MH+]=394 при 2,61 мин. Хиральный анализ (метод 28) при 4,13 мин.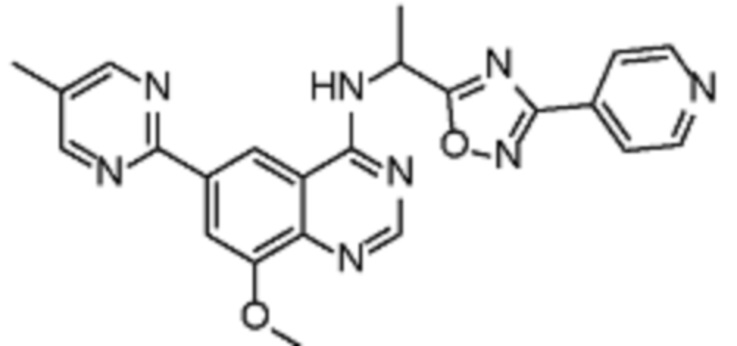
ЖХМС (метод 4): [MH+]=441 при 2,78 мин. Хиральный анализ (метод 13) при 2,23 мин.
ЖХМС (метод 4): [MH+]=441 при 2,81 мин. Хиральный анализ (метод 13) при 2,84 мин.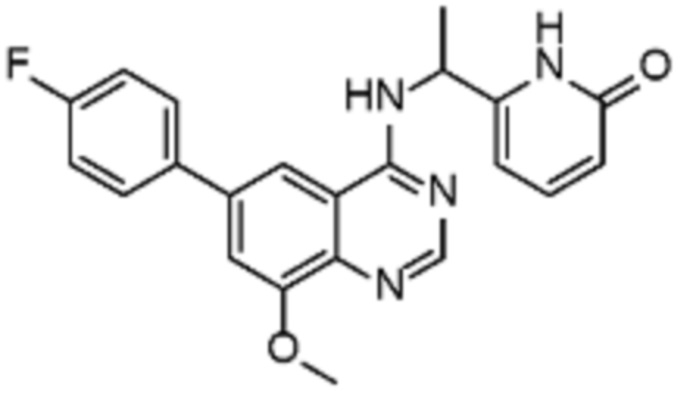
ЖХМС (метод 4): [MH+]=391 при 2,95 мин. Хиральный анализ (метод 34) при 2,99 мин.
ЖХМС (метод 4): [MH+]=391 при 2,94 мин. Хиральный анализ (метод 34) при 4,43 мин.
ЖХМС (метод 4): [MH+]=443 при 3,28 мин. Хиральный анализ (метод 17) при 2,24 мин.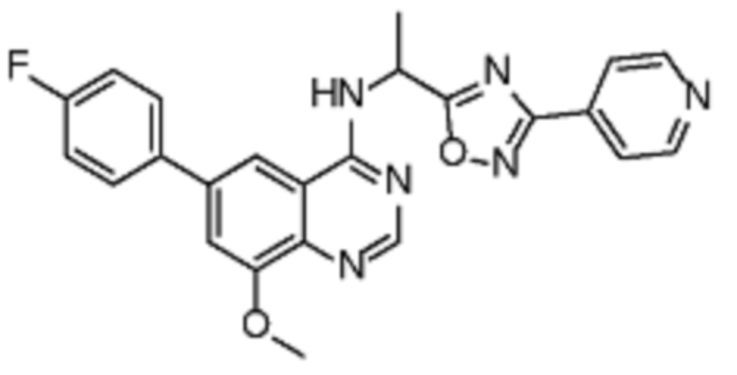
ЖХМС (метод 4): [MH+]=443 при 3,29 мин. Хиральный анализ (метод 17) при 3,41 мин.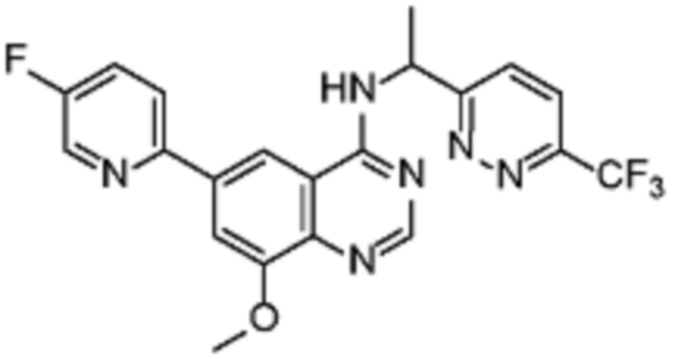
ЖХМС (метод 4): [MH+]=445 при 3,42 мин. Хиральный анализ (метод 35) при 1,35 мин.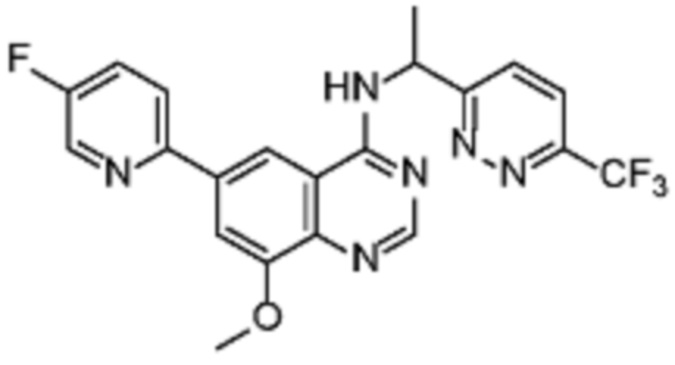
ЖХМС (метод 4): [MH+]=445 при 3,43 мин. Хиральный анализ (метод 35) при 2,10 мин.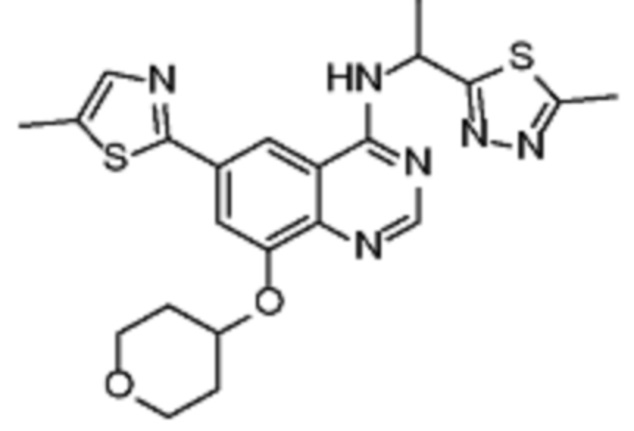
ЖХМС (метод 3): [MH+]=469 при 3,69 мин. Хиральный анализ (метод 9) при 1,51 мин.
ЖХМС (метод 3): [MH+]=469 при 3,7 мин. Хиральный анализ (метод 9) при 3,52 мин.
ЖХМС (метод 4): [MH+]=397 при 2,75 мин. Хиральный анализ (метод 32) при 1,36 мин.
ЖХМС (метод 4): [MH+]=397 при 2,75 мин. Хиральный анализ (метод 32) при 2 мин.
ЖХМС (метод 4): [MH+]=382 при 2,86 мин. Хиральный анализ (метод 28) при 1,51 мин.
ЖХМС (метод 3): [MH+]=381 при 2,90 мин. Хиральный анализ (метод 28) при 2,07 мин.
ФАРМАКОЛОГИЧЕСКАЯ АКТИВНОСТЬ СОЕДИНЕНИЙ ПО НАСТОЯЩЕМУ ИЗОБРЕТЕНИЮ
Электрофизиологический анализ in vitro в отношении P2X3
Клетки, экспрессирующие P2X3 рецепторы, выращивают в соответствии со стандартной практикой и выдерживают при 37°C в атмосфере СО2 с 5% влажностью. Клетки высевают в T175 колбы за 2 дня до анализа и диссоциируют из колб с использованием TrypLE при достижении 80-90% слияния. Диссоциированные клетки снова суспердируют в среде, не содержащей сыворотку, при плотности клеток 3×106 клеток/мл и загружают в автоматизированную пэтч-клемп систему Sophion Qube. Буфер внеклеточного анализа содержит 145 мM NaCl, 4 мM KCl, 2 мM CaCl2, 1 мM MgCl2, 10 мM HEPES и 10 мM глюкозы при pH 7,4. Раствор внутриклеточного анализа содержит 140 мM CsF, 10 мM NaCl, 10 мM EGTA, 10 мM HEPES при pH 7,2. Исходные растворы агонистов получают в H2O и разбавляют омывающим раствором перед использованием. Концентрация исходных растворов всех антагонистов в ДМСО составляет 10 мM, и исходные растворы разбавляют перед использованием. Все эксперименты проводят методом точечной фиксации потенциала в конфигурации «целая клетка» при комнатной температуре, причем 384 отдельных клетки одновременно фиксируют при потенциале -60 мВ на аппарати Sophion Qube. Два исходных ответа определяют с использованием α,β-MeАТФ (800 нM) с последующим применения агонистов, которые вымывают буфером внеклеточного анализа, содержащим 0,5 Ед./мл апиразы. После второго применения агониста антагонист инокулируют в отсутствие α,β-MeАТФ в течение 10 минут. После предварительной инкубации антагониста совместно вводят 800 нM α,β-MeАТФ и антагонист для определения ингибиторного действия антагониста. Одну концентрацию антагониста оценивают в отношении одной клетки, при этом на другие клетки на 384-регистрирующей подложке наносят антагонист в других концентрациях. Контрольную амплитуду тока P2X3 получают из пиковой амплитуды тока второго ответа агониста перед преинкубацией с антагонистом. Пиковую амплитуду тока P2X3 в присутствии антагониста используют для вычисления ингибиторного действия антагониста в каждой концентрации в соответствии со следующим уравнением:
Ингибирование P2X3, выраженное в процентах,=((контрольная пиковая амплитуда P2X3 - пиковая амплитуда антагониста P2X3)/контрольная пиковая амплитуда P2X3)*100
Кривые «концентрация-ответ» строят из результатов исытаний 10 различных концентраций, причем каждую концентрацию антагониста тестируют по меньшей мере на двух отдельных клетках. Концентрацию антагони, при которой P2X3 ток ингибируется на 50% (IC50), определяют аппроксимацией данных по следующему уравнению:
Y=a + [(b-a)/(1+10^((log c-x)d)]
где ‘a’ представляет собой минимальный ответ, ‘b’ представляет собой максимальный ответ, ‘c’ представляет собой IC50, ‘d’ представляет собой угловой коэффициент Хилла.
Результаты, полученные для отдельных соединений и представленные в таблице 6 ниже, выражены в виде диапазона активности.
Таблица 6
где эффективность ингибиторной активности соединений в отношении P2X3 характеризуется в соответствии со следующими критериями:
+++: pIC50 hP2X3> 6,5
++: 6,5 <pIC50 hP2X3> 5,5
+: 5,5 <pIC50 hP2X3>4,5
Электрофизиологический анализ in vitro в отношении P2X2/3
Типичные соединения по настоящему изобртению также тестируют в отношении P2X2/3 рецептора.
Для анализа активности в отношении P2X2/3 рецептора используют протокол анализа, описанный для P2X3 рецептора, с двумя модификациями: 1) в качестве агониста используют 10 мкM АТФ; и 2) среднюю амплитуду тока оределяют через семь секунд после применения агониста.
Результаты, представленные в таблице 7, показывают, что типичные соединения по настоящему изобретению являются селективными агонистами P2X3.
Таблица 7
где эффективность ингибиторной активности соединений в отношении изоформ P2X3 или P2X2/3 характеризуется в соответствии со следующими критериями:
+++: pIC50 hP2X3 или hP2X2/3 > 6,5
++: 6,5 < pIC50 hP2X3 или hP2X2/3 > 5,5
+: 5,5 < pIC50 hP2X3 или hP2X2/3 >4,5
Сравнительный пример A
6-(4-Фторфенил)-4-[(6-метил-3-пиридил)метокси]пиридо[2,3-d]пиримидин

Стадия 1: Синтез 6-бром-4-((6-метилпиридин-3-ил)метокси)пиридо[2,3-д]пиримидина

6-Бромпиридо[2,3-d]пиримидин-4(3H)-он (202 мг, 0,89 ммоль) (промежуточное соединение 1), 5-гидроксиметил-2-метилпиридин (110 мг, 0,89 ммоль) и трифенилфосфин (328 мг, 1,25 ммоль) перемешивают в сухом ТГФ (7 мл), к полученной смеси по каплям добавляют раствор диизопропилазодикарбоксилата (229 мкл, 1,16 ммоль) в сухом ТГФ (3 мл) и перемешивают получнную смесь при комнатной температуре в течение 6 часов. Реакционную смесь фильтруют и осадок на фильтре промывают смесью (2:1) ДХМ/MeOH (20 мл). Фильтраты объединяют и удаляют растворитель в вакууме. Остаток очищают хроматографией (силикагель, элюирование с градиентом: 0-100% EtOAc в ДХМ) с получением 6-бром-4-((6-метилпиридин-3-ил)метокси)пиридо[2,3-d]пиримидина (164 мг, выход 55%).
1H ЯМР (400 МГц, ДМСО): δ 9,08 (д, J=2,6 Гц, 1 H), 8,89 (c, 1 H), 8,67 (д, J=2,6 Гц, 1 H), 8,55 (д, J=2,1 Гц, 1 H), 7,72 (дд, J=2,4, 8,0 Гц, 1 H), 7,24 (д, J=8,0 Гц, 1 H), 5,20 (c, 2 H), 2,44 (c, 3 H).
Стадия 2: Синтез 6-(4-фторфенил)-4-[(6-метил-3-пиридил)метокси]пиридо[2,3-d]пиримидина

Газообразный азот барботируют через смесь 6-бром-4-((6-метилпиридин-3-ил)метокси)пиридо[2,3-d]пиримидина (84 мг, 0,254 ммоль), сложного пинаколового эфира 4-фторфенилбороновой кислоты (76 мг, 0,342 ммоль) и фторида цезия (116 мг, 0,761 ммоль) в ДМФА (1 мл) и воде (0,3 мл). Спустя 5 минут к смеси добавляют тетракис(трифенилфосфин)палладий(0) (29 мг, 0,025 ммоль), полученную смесь нагревают до 95°C и выдерживают при указанной температуре в течение 16 часов. Реакционную смесь разбавляют водой (6 мл) и EtOAc (3 мл). Водную фазу экстрагируют EtOAc (2 × 10 мл). Объединенные органические фазы пропускают через гидрофобный фриттовый фильтр, объединяют и удаляют растворитель в вакууме. Очистка препаративной обращено-фазовой ЖХВД приводит к получению 6-(4-фторфенил)-4-[(6-метил-3-пиридил)метокси]пиридо[2,3-d]пиримидина (52 мг, 59%) в форме 0,5 экв. формиатной соли.
1H ЯМР (400 МГц, ДМСО): δ 9,35 (д, J=2,8 Гц, 1 H), 8,92 (c, 1 H), 8,73 (д, J=2,5 Гц, 1 H), 8,61 (д, J=1,5 Гц, 1 H), 8,32 (c, 0,5 H), 7,97 (дд, J=5,3, 8,6 Гц, 2 H), 7,77 (дд, J=2,1, 8,0 Гц, 1 H), 7,42 (дд, J=8,8, 8,8 Гц, 2 H), 7,29 (д, J=8,1 Гц, 1 H), 5,27 (c, 2 H), 2,49 (c, 3 H).
ЖХМС (метод 4): [MH+]=347 при 2,82 мин.
Соединение, представленное в таблице ниже, получают в соответствии с методикой, описанной для получения 6-(4-фторфенил)-4-[(6-метил-3-пиридил)метокси]пиридо[2,3-d]пиримидина.
структура
1H ЯМР
ЖХ-МС
6-(5-Метилпиридин-2-ил)-4-((6- метилпиридин-3-ил)метокси)пиридо[2,3- d]пиримидин
ЖХМС (метод 3): [MH+]=344 при 3,31 мин.
Активность соединений сравнительных примеров A и B в отношении P2X3 тестируют in vitro с помощью электрофизиологического анализа, описанного выше.
Результаты, полученные для отдельных соединений, представлены в таблице 8 ниже и выражены в виде диапазона активности.
Таблица 10
Неактивное соеднение: pIC50 hP2X3<4,5.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ПИРИДОПИРИМИДИНОВ В КАЧЕСТВЕ ИНГИБИТОРОВ P2X | 2020 |
|
RU2833037C2 |
| ПРОИЗВОДНЫЕ ПИРИМИДИН-2,4-ДИАМИНА ДЛЯ ЛЕЧЕНИЯ РАКА | 2013 |
|
RU2672916C2 |
| ИНГИБИТОРНЫЕ СОЕДИНЕНИЯ | 2013 |
|
RU2673079C2 |
| ПРОИЗВОДНЫЕ 3-(5-ХЛОР-2-ОКСОБЕНЗО[D]ОКСАЗОЛ-3(2Н)-ИЛ)ПРОПАНОВОЙ КИСЛОТЫ В КАЧЕСТВЕ КМО ИНГИБИТОРОВ | 2014 |
|
RU2693016C1 |
| ИНГИБИТОРЫ КАНАЛОВ С ТРАНЗИТОРНЫМ ПОТЕНЦИАЛОМ НА ОСНОВЕ ОКСАДИАЗОЛОВ | 2019 |
|
RU2818244C2 |
| СОЕДИНЕНИЯ 2,3-ДИГИДРОХИНАЗОЛИНА В КАЧЕСТВЕ ИНГИБИТОРОВ NaV1.8 | 2020 |
|
RU2833870C2 |
| ПРОИЗВОДНЫЕ ОКСАДИАЗОЛАМИНА В КАЧЕСТВЕ ИНГИБИТОРА ГИСТОНДЕАЦЕТИЛАЗЫ 6 И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2016 |
|
RU2695133C1 |
| ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ КАРБОНУКЛЕОЗИДА, ПРИМЕНЯЕМЫЕ В КАЧЕСТВЕ ПРОТИВОРАКОВЫХ АГЕНТОВ | 2017 |
|
RU2712944C1 |
| ПРОИЗВОДНЫЕ 3-(6-ХЛОР-3-ОКСО-3,4-ДИГИДРО-(2H)-1,4-БЕНЗОКСАЗИН-4-ИЛ)ПРОПИОНОВОЙ КИСЛОТЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ KMO | 2016 |
|
RU2746001C2 |
| ПИРРОЛОПИРИМИДИНЫ В КАЧЕСТВЕ ПОТЕНЦИАТОРОВ МВТР | 2017 |
|
RU2757457C2 |
Настоящее изобретение относится к соединениям формулы I, где Z выбран из группы, состоящей из (C3-C8)гетероциклоалкила, гетероарила, фенила, где любой из указанных гетероарила, гетероциклоалкила и фенила может быть необязательно замещенным одной или несколькими группами, выбранными из (C1-C3)алкила-, галогена, (C1-C6)галогеналкила-; R1 представляет собой Н или (C1-C4)алкила; R2 выбран из группы, состоящей из гетероарил(C1-C4)алкила-, (C3-C8)гетероциклоалкил-(C1-C6)алкила-, гетероарил-(C1-C6)гидроксиалкила, (RARB)N(O)C(C1-C4)алкилена-, где любой из указанных алкила, алкилена, гетероарила и гетероциклоалкила может быть необязательно замещенным одной или несколькими группами, выбранными из (C1-C3)алкила, (C1-C6)галогеналкила, галогена, оксогруппы, гетероарила; RA и RB в каждом случае независимо представляют собой Н или выбраны из группы, состоящей из (C1-C4)алкила-, (C1-C6)галогеналкила, или RA и RB вместе с атомом азота, к которому они присоединены, могут образовывать 5- или 6-членную насыщенную гетероциклическую моноциклическую кольцевую систему, необязательно содержащую дополнительный гетероатом, представляющий собой атом азота или кислорода, которая может быть необязательно замещенной одной или несколькими группами, выбранными из (C1-C4)алкила и оксогруппы; Y выбран из группы, состоящей из -ORD, RCSO2, гетероарила, (C3-C8)гетероциклоалкила, где любой из указанных гетероарила и гетероциклоалкила может быть необязательно замещенным одной или несколькими группами, выбранными из (C1-C3)алкила; RC в каждом случае представляет собой Н или выбран из группы, состоящей из (C1-C6)алкила, (RARB)N-; RD в каждом случае выбран из группы, состоящей из H, (C1-C6)алкила, (C3-C8)гетероциклоалкил-(C1-C6)алкила-, RCOC(O)(C1-C4)алкилена-, (C3-C8)гетероциклоалкила, RCO(C1-C4)алкилена-, (RARB)N(O)C(C1-C4)алкилена, где любой указанный гетероциклоалкил может быть необязательно замещенным одной или несколькими группами, выбранными из (C1-C3)алкила-; J представляет собой Н. Также предложены фармацевтическая композиция, соединение формулы Ib и его применение в качестве промежуточного соединения при получении соединений формулы (I). Соединения по настоящему изобретению способны ингибировать P2X пуринорецептор 3 и могут применяться в лечении множества расстройств, механизм развития которых связан с P2X3 рецепторами, таких как респираторные заболевания, включая кашель, астму, идиопатический легочный фиброз (ИЛФ) и хроническую обструктивную болезнь легких (ХОБЛ). 5 н. и 13 з.п. ф-лы, 10 табл., 190 пр.
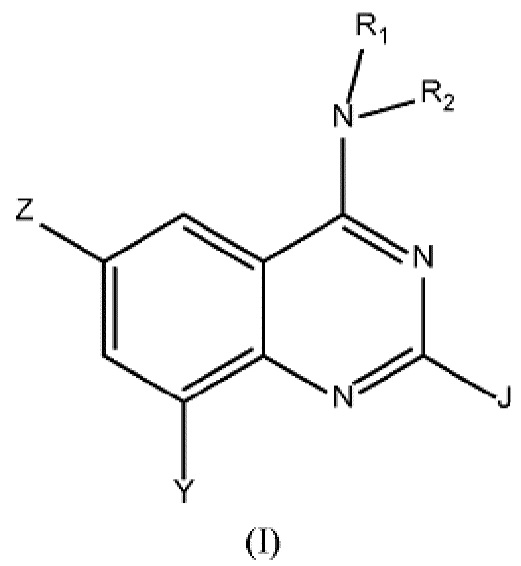
1. Соединение формулы (I)
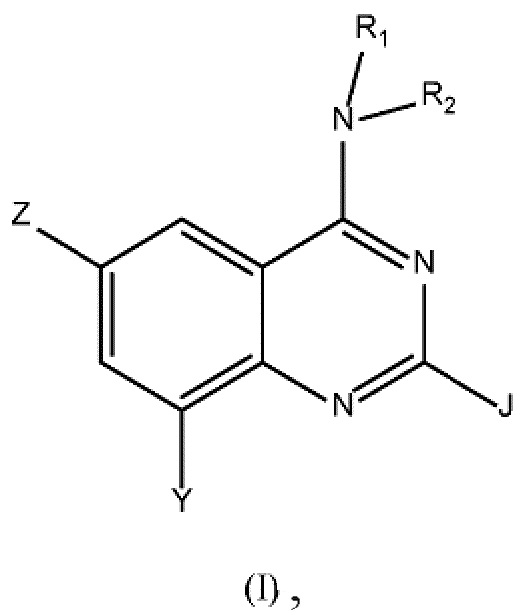
где Z выбран из группы, состоящей из (C3-C8)гетероциклоалкила, гетероарила, фенила, где любой из указанных гетероарила, гетероциклоалкила и фенила может быть необязательно замещенным одной или несколькими группами, выбранными из (C1-C3)алкила-, галогена, (C1-C6)галогеналкила-;
R1 представляет собой Н или (C1-C4)алкил;
R2 выбран из группы, состоящей из гетероарил(C1-C4)алкила-, (C3-C8)гетероциклоалкил-(C1-C6)алкила-, гетероарил-(C1-C6)гидроксиалкила, (RARB)N(O)C(C1-C4)алкилена-, где любой из указанных алкила, алкилена, гетероарила и гетероциклоалкила может быть необязательно замещенным одной или несколькими группами, выбранными из (C1-C3)алкила, (C1-C6)галогеналкила, галогена, оксогруппы, гетероарила;
RA и RB в каждом случае независимо представляют собой Н или выбраны из группы, состоящей из (C1-C4)алкила-, (C1-C6)галогеналкила, или
RA и RB вместе с атомом азота, к которому они присоединены, могут образовывать 5- или 6-членную насыщенную гетероциклическую моноциклическую кольцевую систему, необязательно содержащую дополнительный гетероатом, представляющий собой атом азота или кислорода, которая может быть необязательно замещенной одной или несколькими группами, выбранными из (C1-C4)алкила и оксогруппы;
Y выбран из группы, состоящей из -ORD, RCSO2, гетероарила, (C3-C8)гетероциклоалкила, где любой из указанных гетероарила и гетероциклоалкила может быть необязательно замещенным одной или несколькими группами, выбранными из (C1-C3)алкила;
RC в каждом случае представляет собой Н или выбран из группы, состоящей из (C1-C6)алкила, (RARB)N-;
RD в каждом случае выбран из группы, состоящей из H, (C1-C6)алкила, (C3-C8)гетероциклоалкил-(C1-C6)алкила-, RCOC(O)(C1-C4)алкилена-, (C3-C8)гетероциклоалкила, RCO(C1-C4)алкилена-, (RARB)N(O)C(C1-C4)алкилена, где любой указанный гетероциклоалкил может быть необязательно замещенным одной или несколькими группами, выбранными из (C1-C3)алкила-;
J представляет собой Н.
2. Соединение формулы I по п. 1, выбранное из группы, состоящей из следующих соединений:
6-(1-((6-(4-фторфенил)-8-метоксихиназолин-4-ил)амино)этил)пиридин-2(1Н)-он,
6-(5-фторпиридин-2-ил)-8-метокси-N-(1-(3-метил-1,2,4-оксадиазол-5-ил)этил)хиназолин-4-амин.
6-(5-фторпиримидин-2-ил)-8-метокси-N-(1-(3-метил-1,2,4-оксадиазол-5-ил)этил)хиназолин-4-амин,
((R)-6-(5-фторпиримидин-2-ил)-8-метокси-N-(1-(6-метилпиридазин-3-ил)этил)хиназолин-4-амин,
(R)-8-метокси-N-(1-(6-метилпиридазин-3-ил)этил)-6-(5-метилтиазол-2-ил)хиназолин-4-амин,
8-метокси-N-[(1R)-1-(5-метил-1,2,4-оксадиазол-3-ил)этил]-6-(5-метилпиримидин-2-ил)хиназолин-4-амин,
одиночный энантиомер N-[1-(6-метилпиридазин-3-ил)этил]-6-(5-метилпиримидин-2-ил)-8-тетрагидропиран-4-илоксихиназолин-4-амина,
6-(5-фтор-2-пиридил)-N-[(6-метилпиридазин-3-ил)метил]-8-тетрагидропиран-4-илоксихиназолин-4-амин,
N-[(6-метилпиридазин-3-ил)метил]-6-(5-метилпиримидин-2-ил)-8-тетрагидропиран-4-илоксихиназолин-4-амин,
(R)-6-(1-метил-1H-пиразол-3-ил)-N-(1-(6-метилпиридазин-3-ил)этил)-8-((тетрагидро-2H-пиран-4-ил)окси)хиназолин-4-амин,
(R)-6-(5-метилтиазол-2-ил)-N-(1-(пиридазин-3-ил)этил)-8-((тетрагидро-2H-пиран-4-ил)окси)хиназолин-4-амин,
8-(2-метоксиэтокси)-N-((6-метилпиридазин-3-ил)метил)-6-(5-метилтиазол-2-ил)хиназолин-4-амин,
2-((4-(((6-метилпиридазин-3-ил)метил)амино)-6-(5-метилтиазол-2-ил)хиназолин-8-ил)окси)этан-1-ол,
N-((6-метилпиридазин-3-ил)метил)-6-(5-метилтиазол-2-ил)-8-(оксетан-3-илметокси)хиназолин-4-амин,
N-[(6-метилпиридазин-3-ил)метил]-6-(5-метилтиазол-2-ил)-8-тетрагидропиран-4-илоксихиназолин-4-амин,
(R)-2-((6-(4-фторфенил)-4-((1-(2-(трифторметил)пиримидин-5-ил)этил)амино)хиназолин-8-ил)окси)уксусная кислота,
дигидрохлорид 2-((4-(((6-метилпиридазин-3-ил)метил)амино)-6-(5-метилпиримидин-2-ил)хиназолин-8-ил)окси)уксусной кислоты,
гидрохлорид 2-((6-(4-фторфенил)-4-(((1-метилпиперидин-4-ил)метил)амино)хиназолин-8-ил)окси)уксусной кислоты,
6-(4-фторфенил)-N-((6-метилпиридазин-3-ил)метил)-8-(оксетан-3-илокси)хиназолин-4-амин,
N-[1-(5-метил-1,3,4-тиадиазол-2-ил)этил]-6-(5-метилтиазол-2-ил)-8-тетрагидропиран-4-илоксихиназолин-4-амин,
6-(5-фтор-2-пиридил)-N-[(1R)-1-(5-метил-1,2,4-оксадиазол-3-ил)этил]-8-тетрагидропиран-4-илоксихиназолин-4-амин,
одиночный энантиомер 6-(5-метил-2-пиридил)-N-[1-(5-метил-1,3,4-тиадиазол-2-ил)этил]-8-тетрагидропиран-4-илоксихиназолин-4-амина,
одиночный энантиомер 6-(5-фтор-2-пиридил)-N-[1-(3-метил-1,2,4-оксадиазол-5-ил)этил]-8-тетрагидропиран-4-илоксихиназолин-4-амина,
6-(3-хлор-1H-пиразол-1-ил)-8-метокси-N-(1-(2-(трифторметил)пиримидин-5-ил)этил)хиназолин-4-амин,
6-(3-хлор-1H-пиразол-1-ил)-8-метокси-N-(1-(6-метилпиридазин-3-ил)этил)хиназолин-4-амин,
(R)-6-(4-фтор-1H-пиразол-1-ил)-8-метокси-N-(1-(2-(трифторметил)пиримидин-5-ил)этил)хиназолин-4-амин,
(R)-6-(4-фтор-1H-пиразол-1-ил)-8-метокси-N-(1-(6-метилпиридазин-3-ил)этил)хиназолин-4-амин,
(R)-8-метокси-6-(4-метил-1H-пиразол-1-ил)-N-(1-(6-метилпиридазин-3-ил)этил)хиназолин-4-амин,
(R)-6-(4-хлор-1H-пиразол-1-ил)-8-метокси-N-(1-(6-метилпиридазин-3-ил)этил)хиназолин-4-амин,
(R)-6-(3-фтор-1H-пиразол-1-ил)-8-метокси-N-(1-(6-метилпиридазин-3-ил)этил)хиназолин-4-амин,
(R)-8-метокси-6-(5-метил-1H-пиразол-1-ил)-N-(1-(6-метилпиридазин-3-ил)этил)хиназолин-4-амин,
(R)-8-метокси-6-(3-метил-1H-пиразол-1-ил)-N-(1-(6-метилпиридазин-3-ил)этил)хиназолин-4-амин,
(R)-6-(4-хлор-1H-пиразол-1-ил)-8-метокси-N-(1-(2-(трифторметил)пиримидин-5-ил)этил)хиназолин-4-амин,
(8-метокси-N-(1-(5-метил-1,3,4-оксадиазол-2-ил)этил)-6-(1-метил-1H-пиразол-3-ил)хиназолин-4-амин,
8-метокси-N-(1-(5-метил-1,3,4-тиадиазол-2-ил)этил)-6-(5-метилтиазол-2-ил)хиназолин-4-амин,
8-метокси-N-(1-(5-метил-1,2,4-оксадиазол-3-ил)этил)-6-(5-метилтиазол-2-ил)хиназолин-4-амин,
6-(5-фторпиримидин-2-ил)-8-метокси-N-(1-(5-метил-1,2,4-оксадиазол-3-ил)этил)хиназолин-4-амин,
6-(5-фторпиримидин-2-ил)-8-метокси-N-(1-(5-метил-1,3,4-тиадиазол-2-ил)этил)хиназолин-4-амин,
6-(5-фторпиримидин-2-ил)-8-метокси-N-(1-(5-метил-1,3,4-оксадиазол-2-ил)этил)хиназолин-4-амин,
8-метокси-N-[(3-метилизоксазол-5-ил)метил]-6-(5-метилтиазол-2-ил)хиназолин-4-амин,
8-метокси-6-(5-метилтиазол-2-ил)-N-[[3-(трифторметил)-1,2,4-оксадиазол-5-ил]метил]хиназолин-4-амин,
8-метокси-N-[(3-метил-1,2,4-оксадиазол-5-ил)метил]-6-(5-метилтиазол-2-ил)хиназолин-4-амин,
8-метокси-N-((5-метил-1,3,4-тиадиазол-2-ил)метил)-6-(5-метилтиазол-2-ил)хиназолин-4-амин,
4-(((8-метокси-6-(5-метилтиазол-2-ил)хиназолин-4-ил)амино)метил)-1-метилпиридин-2(1Н)-он,
8-метокси-6-(5-метилтиазол-2-ил)-N-((5-(трифторметил)пиридин-2-ил)метил)хиназолин-4-амин,
N-((5-хлорпиридин-2-ил)метил)-8-метокси-6-(5-метилтиазол-2-ил)хиназолин-4-амин,
N-((3,5-дифторпиридин-2-ил)метил)-8-метокси-6-(5-метилтиазол-2-ил)хиназолин-4-амин,
N-((5-фторпиридин-2-ил)метил)-8-метокси-6-(5-метилтиазол-2-ил)хиназолин-4-амин,
одиночный энантиомер 8-метокси-6-(5-метилпиримидин-2-ил)-N-[1-[6-(трифторметил)пиридазин-3-ил]этил]хиназолин-4-амина,
одиночный энантиомер 8-метокси-N-[1-(3-метил-1,2,4-оксадиазол-5-ил)этил]-6-(5-метилпиримидин-2-ил)хиназолин-4-амина,
одиночный энантиомер 8-метокси-6-(5-метилпиримидин-2-ил)-N-[1-(5-метил-1,3,4-тиадиазол-2-ил)этил]хиназолин-4-амина,
одиночный энантиомер 8-метокси-6-(5-метил-2-пиридил)-N-[1-(5-метил-1,3,4-тиадиазол-2-ил)этил]хиназолин-4-амина,
одиночный энантиомер 8-метокси-N-[(1R)-1-(5-метил-1,3,4-оксадиазол-2-ил)этил]-6-(5-метилпиримидин-2-ил)хиназолин-4-амина,
6-(5-фторпиридин-2-ил)-8-метокси-N-((5-метил-1,2,4-оксадиазол-3-ил)метил)хиназолин-4-амин,
6-(5-фторпиридин-2-ил)-8-метокси-N-((3-метил-1,2,4-оксадиазол-5-ил)метил)хиназолин-4-амин,
6-(5-фторпиридин-2-ил)-8-метокси-N-((5-метил-1,3,4-оксадиазол-2-ил)метил)хиназолин-4-амин,
6-(5-фторпиридин-2-ил)-8-метокси-N-((3-метилизоксазол-5-ил)метил)хиназолин-4-амин,
8-метокси-N-((5-метил-1,2,4-оксадиазол-3-ил)метил)-6-(5-метилпиридин-2-ил)хиназолин-4-амин,
8-метокси-N-((3-метилизоксазол-5-ил)метил)-6-(5-метилпиридин-2-ил)хиназолин-4-амин,
6-(5-фтор-2-пиридил)-8-метокси-N-[(5-метилизоксазол-3-ил)метил]хиназолин-4-амин,
8-метокси-N-[(5-метилизоксазол-3-ил)метил]-6-(5-метил-2-пиридил)хиназолин-4-амин,
6-(5-фтор-2-пиридил)-8-метокси-N-[(1-метилпиразол-3-ил)метил]хиназолин-4-амин,
8-метокси-N-[(1-метилпиразол-3-ил)метил]-6-(5-метил-2-пиридил)хиназолин-4-амин,
8-метокси-N-[(3-метил-1,2,4-оксадиазол-5-ил)метил]-6-(5-метил-2-пиридил)хиназолин-4-амин,
8-метокси-N-[(5-метил-1,3,4-оксадиазол-2-ил)метил]-6-(5-метил-2-пиридил)хиназолин-4-амин,
8-метокси-6-(5-метилпиримидин-2-ил)-N-[[3-(трифторметил)-1,2,4-оксадиазол-5-ил]метил]хиназолин-4-амин,
8-метокси-6-(5-метилпиримидин-2-ил)-N-[[2-(трифторметил)пиримидин-5-ил]метил]хиназолин-4-амин,
6-(5-фторпиридин-2-ил)-8-метокси-N-((2-(трифторметил)пиримидин-5-ил)метил)хиназолин-4-амин,
4-(((6-(5-фторпиридин-2-ил)-8-метоксихиназолин-4-ил)амино)метил)-1-метилпиридин-2(1Н)-он,
4-(((6-(5-фторпиридин-2-ил)-8-метоксихиназолин-4-ил)амино)метил)пиридин-2(1Н)-он,
6-(5-фторпиридин-2-ил)-8-метокси-N-((6-метилпиридин-3-ил)метил)хиназолин-4-амин,
6-(5-фторпиридин-2-ил)-8-метокси-N-((6-(трифторметил)пиридин-3-ил)метил)хиназолин-4-аминамин,
4-(((8-метокси-6-(5-метилпиримидин-2-ил)хиназолин-4-ил)амино)метил)-1-метилпиридин-2(1Н)-он,
6-(((8-метокси-6-(5-метилпиримидин-2-ил)хиназолин-4-ил)амино)метил)пиридин-2(1Н)-он,
8-метокси-N-((5-метил-1,3,4-тиадиазол-2-ил)метил)-6-(5-метилпиримидин-2-ил)хиназолин-4-амин,
8-метокси-N-((6-метилпиридин-3-ил)метил)-6-(5-метилпиримидин-2-ил)хиназолин-4-амин,
6-(5-фторпиридин-2-ил)-8-метокси-N-((6-(трифторметил)пиридин-3-ил)метил)хиназолин-4-амин,
8-метокси-N-((3-метил-1,2,4-оксадиазол-5-ил)метил)-6-(5-метилпиримидин-2-ил)хиназолин-4-амин,
6-(3-фтор-5-метил-2-пиридил)-8-метокси-N-[(1R)-1-(5-метил-1,2,4-оксадиазол-3-ил)этил] хиназолин-4-амин,
6-(5-фтор-2-пиридил)-8-метокси-N-[(1R)-1-(1-метилпиразол-3-ил)этил]хиназолин-4-амин,
8-метокси-N-[(1R)-1-(1-метилпиразол-3-ил)этил]-6-(5-метил-2-пиридил)хиназолин-4-амин,
6-(4-фторфенил)-N-[(5-фтор-2-пиридил)метил]-8-метоксихиназолин-4-амин,
одиночный энантиомер 8-метокси-N-[1-(3-метил-1,2,4-оксадиазол-5-ил)этил]-6-(5-метил-2-пиридил)хиназолин-4-амина,
одиночный энантиомер 8-метокси-N-(1-(5-метил-1,3,4-тиадиазол-2-ил)этил)-6-(5-метилтиазол-2-ил)хиназолин-4-амина,
одиночный энантиомер 2-((8-метокси-6-(5-метилпиримидин-2-ил)хиназолин-4-ил)амино)-2-(3-метил-1,2,4-оксадиазол-5-ил)этан-1-ола,
одиночный энантиомер 6-(5-фтор-2-пиридил)-8-метокси-N-[1-[6-(трифторметил)пиридазин-3-ил]этил]хиназолин-4-амина,
одиночный энантиомер N-[1-(5-метил-1,3,4-тиадиазол-2-ил)этил]-6-(5-метилтиазол-2-ил)-8-тетрагидропиран-4-илоксихиназолин-4-амина,
одиночный энантиомер 6-(5-фторпиридин-2-ил)-8-метокси-N-(1-(5-метил-1,3,4-тиадиазол-2-ил)этил)хиназолин-4-амина,
одиночный энантиомер 6-(5-фторпиридин-2-ил)-8-метокси-N-(1-(3-метил-1,2,4-оксадиазол-5-ил)этил)хиназолин-4-амина,
формиат 6-(4-фторфенил)-8-метокси-N-[(1S)-1-[2-(трифторметил)пиримидин-5-ил]этил]хиназолин-4-амина,
одиночный энантиомер 6-(1-((6-(4-фторфенил)-8-метоксихиназолин-4-ил)амино)этил)пиридин-2(1Н)-она.
3. Соединение формулы I по п. 1
где Z выбран из группы, состоящей из гетероарила и фенила, где любой из указанных гетероарила и фенила может быть необязательно замещенным одной или несколькими группами, выбранными из (C1-C3)алкила, галогена, (C1-C6)галогеналкила;
R1 представляет собой Н или (C1-C6)алкил;
R2 выбран из группы, состоящей из гетероарил(C1-C4)алкила-, (C3-C8)гетероциклоалкил-(C1-C6)алкила, гетероарил-(C1-C6)гидроксиалкила, где любой из указанных алкила, гетероарила и гетероциклоалкила может быть необязательно замещенным одной или несколькими группами, выбранными из (C1-C3)алкила, (C1-C6)галогеналкила, оксогруппы, гетероарила;
RA и RB в каждом случае независимо представляют собой Н или выбраны из группы, состоящей из (C1-C4)алкила-, (C1-C6)галогеналкила, или
RA и RB вместе с атомом азота, к которому они присоединены, могут образовывать 6-членную насыщенную гетероциклическую моноциклическую кольцевую систему, необязательно содержащую дополнительный гетероатом, представляющий собой атом азота или кислорода, которая может быть необязательно замещенной (C1-C4)алкилом- и оксогруппой;
RC представляет собой Н или выбран из группы, состоящей из (C1-C6)алкила, (RARB)N-;
Y выбран из группы, состоящей из -ORD, RCSO2-, гетероарила, (C3-C8)гетероциклоалкила, где любой из указанных гетероарила и гетероциклоалкила может быть необязательно замещенным одной или несколькими группами, выбранными из (C1-C3)алкила;
J представляет собой Н;
RD представляет собой Н или (C1-C6)алкил.
4. Соединение формулы I по п. 1,
где Z выбран из группы, состоящей из гетероарила и фенила, где любой из указанных гетероарила и фенила может быть необязательно замещенным одной или несколькими группами, выбранными из (C1-C3)алкила и галогена;
R1 представляет собой Н;
R2 выбран из группы, состоящей из гетероарил(C1-C4)алкила-, (C3-C8)гетероциклоалкил-(C1-C6)алкила, гетероарил-(C1-C6)гидроксиалкила, где любой из указанных алкила, гетероарила и гетероциклоалкила может быть необязательно замещенным одной или несколькими группами, выбранными из (C1-C3)алкила, (C1-C6)галогеналкила, оксогруппы, гетероарила;
RA и RB в каждом случае независимо представляют собой Н или выбраны из группы, состоящей из (C1-C4)алкила-, (C1-C6)галогеналкила, или
RA и RB вместе с атомом азота, к которому они присоединены, могут образовывать 6-членную насыщенную моноциклическую гетероциклическую кольцевую систему, необязательно содержащую дополнительный гетероатом, представляющий собой атом азота или кислорода, которая может быть необязательно замещенной (C1-C4)алкилом- и оксогруппой;
RC представляет собой Н или выбран из группы, состоящей из (C1-C6)алкила, (RARB)N-;
Y выбран из группы, состоящей из -ORD, RCSO2-, гетероарила, (C3-C8)гетероциклоалкила, где любой из указанных гетероарила и гетероциклоалкила может быть необязательно замещенным одной или несколькими группами, выбранными из (C1-C3)алкила;
J представляет собой Н;
RD представляет собой Н или (C1-C6)алкил.
5. Соединение формулы I по пп. 1, 3 и 4, выбранное из группы, состоящей из следующих соединений:
6-(1-((6-(4-фторфенил)-8-метоксихиназолин-4-ил)амино)этил)пиридин-2(1Н)-он,
6-(5-фторпиридин-2-ил)-8-метокси-N-(1-(3-метил-1,2,4-оксадиазол-5-ил)этил)хиназолин-4-амин,
6-(5-фторпиримидин-2-ил)-8-метокси-N-(1-(3-метил-1,2,4-оксадиазол-5-ил)этил)хиназолин-4-амин,
((R)-6-(5-фторпиримидин-2-ил)-8-метокси-N-(1-(6-метилпиридазин-3-ил)этил)хиназолин-4-амин,
(R)-8-метокси-N-(1-(6-метилпиридазин-3-ил)этил)-6-(5-метилтиазол-2-ил)хиназолин-4-амин,
8-метокси-6-(5-метилпиримидин-2-ил)-N-(1-(3-(пиридин-4-ил)-1,2,4-оксадиазол-5-ил)этил)хиназолин-4-амин,
8-метокси-N-[(1R)-1-(5-метил-1,2,4-оксадиазол-3-ил)этил]-6-(5-метилпиримидин-2-ил)хиназолин-4-амин,
(R)-6-(4-фторфенил)-N-(1-(6-метилпиридазин-3-ил)этил)-8-(морфолиносульфонил)хиназолин-4-амин,
((R)-6-(4-фторфенил)-8-(морфолиносульфонил)-N-(1-(2-(трифторметил)пиримидин-5-ил)этил)хиназолин-4-амин,
(рац)-6-(4-фторфенил)-N-(1-(5-метил-1,3,4-тиадиазол-2-ил)этил)-8-(морфолиносульфонил)хиназолин-4-амин,
(R)-6-(4-фторфенил)-N,N-диметил-4-((1-(6-метилпиридазин-3-ил)этил)амино)хиназолин-8-сульфонамид,
6-(4-фторфенил)-N,N-диметил-4-((1-(5-метил-1,3,4-тиадиазол-2-ил)этил)амино)хиназолин-8-сульфонамид,
6-(4-фторфенил)-N,N-диметил-4-((1-(5-метил-1,2,4-оксадиазол-3-ил)этил)амино)хиназолин-8-сульфонамид,
(R)-6-(4-фторфенил)-N-метил-4-((1-(6-метилпиридазин-3-ил)этил)амино)хиназолин-8-сульфонамид,
(R)-6-(4-фторфенил)-N-метил-4-((1-(5-метил-1,2,4-оксадиазол-3-ил)этил)амино)хиназолин-8-сульфонамид,
(6-(4-фторфенил)-N-метил-4-((1-(5-метил-1,3,4-тиадиазол-2-ил)этил)амино)хиназолин-8-сульфонамид,
(R)-6-(4-фторфенил)-N-(1-(6-метилпиридазин-3-ил)этил)-8-(пиперазин-1-илсульфонил)хиназолин-4-амин,
(R)-6-(4-фторфенил)-N-(1-(5-метил-1,2,4-оксадиазол-3-ил)этил)-8-(пиперазин-1-илсульфонил)хиназолин-4-амин,
(R)-6-(4-фторфенил)-8-(пиридазин-4-ил)-N-(1-(2-(трифторметил)пиримидин-5-ил)этил)хиназолин-4-амин,
(R)-6-(4-фторфенил)-8-(пиримидин-5-ил)-N-(1-(2-(трифторметил)пиримидин-5-ил)этил)хиназолин-4-амин,
(R)-6-(4-фторфенил)-8-(тетрагидро-2H-пиран-4-ил)-N-(1-(2-(трифторметил)пиримидин-5-ил)этил)хиназолин-4-амин,
(R)-6-(4-фторфенил)-8-(1,2,3,6-тетрагидропиридин-4-ил)-N-(1-(2-(трифторметил)пиримидин-5-ил)этил)хиназолин-4-амин,
одиночный энантиомер N-[1-(6-метилпиридазин-3-ил)этил]-6-(5-метилпиримидин-2-ил)-8-тетрагидропиран-4-илоксихиназолин-4-амина,
6-(5-фтор-2-пиридил)-N-[(6-метилпиридазин-3-ил)метил]-8-тетрагидропиран-4-илоксихиназолин-4-амин,
N-[(6-метилпиридазин-3-ил)метил]-6-(5-метилпиримидин-2-ил)-8-тетрагидропиран-4-илоксихиназолин-4-амин,
(R)-6-(1-метил-1H-пиразол-3-ил)-N-(1-(6-метилпиридазин-3-ил)этил)-8-((тетрагидро-2H-пиран-4-ил)окси)хиназолин-4-амин,
(R)-6-(5-метилтиазол-2-ил)-N-(1-(пиридазин-3-ил)этил)-8-((тетрагидро-2H-пиран-4-ил)окси)хиназолин-4-амин,
4-[[4-[1-(6-метилпиридазин-3-ил)этиламино]-6-(5-метилпиримидин-2-ил)хиназолин-8-ил]оксиметил]тетрагидропиран-4-ол,
8-(2-метоксиэтокси)-N-((6-метилпиридазин-3-ил)метил)-6-(5-метилтиазол-2-ил)хиназолин-4-амин,
2-((4-(((6-метилпиридазин-3-ил)метил)амино)-6-(5-метилтиазол-2-ил)хиназолин-8-ил)окси)этан-1-ол,
N-((6-метилпиридазин-3-ил)метил)-6-(5-метилтиазол-2-ил)-8-(оксетан-3-илметилокси)хиназолин-4-амин,
N-[(6-метилпиридазин-3-ил)метил]-6-(5-метилтиазол-2-ил)-8-тетрагидропиран-4-илоксихиназолин-4-амин,
8-(5-азаспиро[3.5]нонан-8-илокси)-6-(4-фторфенил)-N-[(1R)-1-[2-(трифторметил)пиримидин-5-ил]этил]хиназолин-4-амин,
8-((2-азабицикло[2.2.1]гептан-5-ил)окси)-6-(4-фторфенил)-N-((R)-1-(2-(трифторметил)пиримидин-5-ил)этил)хиназолин-4-амин,
(R)-2-((6-(4-фторфенил)-4-((1-(2-(трифторметил)пиримидин-5-ил)этил)амино)хиназолин-8-ил)окси)уксусная кислота,
дигидрохлорид 2-((4-(((6-метилпиридазин-3-ил)метил)амино)-6-(5-метилпиримидин-2-ил)хиназолин-8-ил)окси)уксусной кислоты,
гидрохлорид 2-((6-(4-фторфенил)-4-(((1-метилпиперидин-4-ил)метил)амино)хиназолин-8-ил)окси)уксусной кислоты,
6-(4-фторфенил)-8-[(3-метилоксетан-3-ил)метокси]-N-[(6-метилпиридазин-3-ил)метил]хиназолин-4-амин,
3-[6-(4-фторфенил)-4-[(6-метилпиридазин-3-ил)метиламино]хиназолин-8-ил]окси-1-метилпирролидин-2-он,
2-[6-(4-фторфенил)-4-[(6-метилпиридазин-3-ил)метиламино]хиназолин-8-ил]окси-N-(2,2,2-трифторэтил)ацетамид,
6-(4-фторфенил)-N-((6-метилпиридазин-3-ил)метил)-8-(оксетан-3-илокси)хиназолин-4-амин,
N-[1-(5-метил-1,3,4-тиадиазол-2-ил)этил]-6-(5-метилтиазол-2-ил)-8-тетрагидропиран-4-илоксихиназолин-4-амин,
6-(5-фтор-2-пиридил)-N-[(1R)-1-(5-метил-1,2,4-оксадиазол-3-ил)этил]-8-тетрагидропиран-4-илоксихиназолин-4-амин,
одиночный энантиомер 6-(5-метил-2-пиридил)-N-[1-(5-метил-1,3,4-тиадиазол-2-ил)этил]-8-тетрагидропиран-4-илоксихиназолин-4-амина,
одиночный энантиомер 6-(5-фтор-2-пиридил)-N-[1-(3-метил-1,2,4-оксадиазол-5-ил)этил]-8-тетрагидропиран-4-илоксихиназолин-4-амина,
6-(4-фторфенил)-8-(2-метилпиразол-3-ил)-N-[(6-метилпиридазин-3-ил)метил]хиназолин-4-амин,
6-(3-хлор-1H-пиразол-1-ил)-8-метокси-N-(1-(2-(трифторметил)пиримидин-5-ил)этил)хиназолин-4-амин,
6-(3-хлор-1H-пиразол-1-ил)-8-метокси-N-(1-(6-метилпиридазин-3-ил)этил)хиназолин-4-амин,
(R)-6-(4-фтор-1H-пиразол-1-ил)-8-метокси-N-(1-(2-(трифторметил)пиримидин-5-ил)этил)хиназолин-4-амин,
(R)-6-(4-фтор-1H-пиразол-1-ил)-8-метокси-N-(1-(6-метилпиридазин-3-ил)этил)хиназолин-4-амин,
(R)-8-метокси-6-(4-метил-1H-пиразол-1-ил)-N-(1-(6-метилпиридазин-3-ил)этил)хиназолин-4-амин,
(R)-6-(4-хлор-1H-пиразол-1-ил)-8-метокси-N-(1-(6-метилпиридазин-3-ил)этил)хиназолин-4-амин,
(R)-6-(3-фтор-1H-пиразол-1-ил)-8-метокси-N-(1-(6-метилпиридазин-3-ил)этил)хиназолин-4-амин,
(R)-8-метокси-6-(5-метил-1H-пиразол-1-ил)-N-(1-(6-метилпиридазин-3-ил)этил)хиназолин-4-амин,
(R)-8-метокси-6-(3-метил-1H-пиразол-1-ил)-N-(1-(6-метилпиридазин-3-ил)этил)хиназолин-4-амин,
(R)-6-(4-хлор-1H-пиразол-1-ил)-8-метокси-N-(1-(2-(трифторметил)пиримидин-5-ил)этил)хиназолин-4-амин,
(8-метокси-N-(1-(5-метил-1,3,4-оксадиазол-2-ил)этил)-6-(1-метил-1H-пиразол-3-ил)хиназолин-4-амин,
8-метокси-N-(1-(5-метил-1,3,4-тиадиазол-2-ил)этил)-6-(5-метилтиазол-2-ил)хиназолин-4-амин,
8-метокси-N-(1-(5-метил-1,2,4-оксадиазол-3-ил)этил)-6-(5-метилтиазол-2-ил)хиназолин-4-амин,
6-(5-фторпиримидин-2-ил)-8-метокси-N-(1-(5-метил-1,2,4-оксадиазол-3-ил)этил)хиназолин-4-амин,
6-(5-фторпиримидин-2-ил)-8-метокси-N-(1-(5-метил-1,3,4-тиадиазол-2-ил)этил)хиназолин-4-амин,
6-(5-фторпиримидин-2-ил)-8-метокси-N-(1-(5-метил-1,3,4-оксадиазол-2-ил)этил)хиназолин-4-амин,
8-метокси-N-[(3-метилизоксазол-5-ил)метил]-6-(5-метилтиазол-2-ил)хиназолин-4-амин,
8-метокси-6-(5-метилтиазол-2-ил)-N-[[3-(трифторметил)-1,2,4-оксадиазол-5-ил]метил]хиназолин-4-амин,
8-метокси-N-[(3-метил-1,2,4-оксадиазол-5-ил)метил]-6-(5-метилтиазол-2-ил)хиназолин-4-амин,
8-метокси-N-((5-метил-1,3,4-тиадиазол-2-ил)метил)-6-(5-метилтиазол-2-ил)хиназолин-4-амин,
4-(((8-метокси-6-(5-метилтиазол-2-ил)хиназолин-4-ил)амино)метил)-1-метилпиридин-2(1Н)-он,
8-метокси-6-(5-метилтиазол-2-ил)-N-((5-(трифторметил)пиридин-2-ил)метил)хиназолин-4-амин,
N-((5-хлорпиридин-2-ил)метил)-8-метокси-6-(5-метилтиазол-2-ил)хиназолин-4-амин,
N-((3,5-дифторпиридин-2-ил)метил)-8-метокси-6-(5-метилтиазол-2-ил)хиназолин-4-амин,
N-((5-фторпиридин-2-ил)метил)-8-метокси-6-(5-метилтиазол-2-ил)хиназолин-4-амин,
одиночный энантиомер 8-метокси-6-(5-метилпиримидин-2-ил)-N-[1-[6-(трифторметил)пиридазин-3-ил]этил]хиназолин-4-амина,
одиночный энантиомер 8-метокси-N-[1-(3-метил-1,2,4-оксадиазол-5-ил)этил]-6-(5-метилпиримидин-2-ил)хиназолин-4-амина,
одиночный энантиомер 8-метокси-6-(5-метилпиримидин-2-ил)-N-[1-(5-метил-1,3,4-тиадиазол-2-ил)этил]хиназолин-4-амина,
одиночный энантиомер 8-метокси-6-(5-метил-2-пиридил)-N-[1-(5-метил-1,3,4-тиадиазол-2-ил)этил]хиназолин-4-амина,
одиночный энантиомер 8-метокси-N-[(1R)-1-(5-метил-1,3,4-оксадиазол-2-ил)этил]-6-(5-метилпиримидин-2-ил)хиназолин-4-амина,
6-(5-фторпиридин-2-ил)-8-метокси-N-((5-метил-1,2,4-оксадиазол-3-ил)метил)хиназолин-4-амин,
6-(5-фторпиридин-2-ил)-8-метокси-N-((3-метил-1,2,4-оксадиазол-5-ил)метил)хиназолин-4-амин,
6-(5-фторпиридин-2-ил)-8-метокси-N-((5-метил-1,3,4-оксадиазол-2-ил)метил)хиназолин-4-амин,
6-(5-фторпиридин-2-ил)-8-метокси-N-((3-метилизоксазол-5-ил)метил)хиназолин-4-амин,
8-метокси-N-((5-метил-1,2,4-оксадиазол-3-ил)метил)-6-(5-метилпиридин-2-ил)хиназолин-4-амин,
8-метокси-N-((3-метилизоксазол-5-ил)метил)-6-(5-метилпиридин-2-ил)хиназолин-4-амин,
6-(5-фтор-2-пиридил)-8-метокси-N-[(5-метилизоксазол-3-ил)метил]хиназолин-4-амин,
8-метокси-N-[(5-метилизоксазол-3-ил)метил]-6-(5-метил-2-пиридил)хиназолин-4-амин,
6-(5-фтор-2-пиридил)-8-метокси-N-[(1-метилпиразол-3-ил)метил]хиназолин-4-амин,
8-метокси-N-[(1-метилпиразол-3-ил)метил]-6-(5-метил-2-пиридил)хиназолин-4-амин,
8-метокси-N-[(3-метил-1,2,4-оксадиазол-5-ил)метил]-6-(5-метил-2-пиридил)хиназолин-4-амин,
8-метокси-N-[(5-метил-1,3,4-оксадиазол-2-ил)метил]-6-(5-метил-2-пиридил)хиназолин-4-амин,
8-метокси-6-(5-метилпиримидин-2-ил)-N-[[3-(трифторметил)-1,2,4-оксадиазол-5-ил]метил]хиназолин-4-амин,
8-метокси-6-(5-метилпиримидин-2-ил)-N-[[2-(трифторметил)пиримидин-5-ил]метил]хиназолин-4-амин,
6-(5-фторпиридин-2-ил)-8-метокси-N-((2-(трифторметил)пиримидин-5-ил)метил)хиназолин-4-амин,
4-(((6-(5-фторпиридин-2-ил)-8-метоксихиназолин-4-ил)амино)метил)-1-метилпиридин-2(1Н)-он,
4-(((6-(5-фторпиридин-2-ил)-8-метоксихиназолин-4-ил)амино)метил)пиридин-2(1Н)-он,
6-(5-фторпиридин-2-ил)-8-метокси-N-((6-метилпиридин-3-ил)метил)хиназолин-4-амин,
6-(5-фторпиридин-2-ил)-8-метокси-N-((6-(трифторметил)пиридин-3-ил)метил)хиназолин-4-амин,
4-(((8-метокси-6-(5-метилпиримидин-2-ил)хиназолин-4-ил)амино)метил)-1-метилпиридин-2(1Н)-он,
6-(((8-метокси-6-(5-метилпиримидин-2-ил)хиназолин-4-ил)амино)метил)пиридин-2(1Н)-он,
8-метокси-N-((5-метил-1,3,4-тиадиазол-2-ил)метил)-6-(5-метилпиримидин-2-ил)хиназолин-4-амин,
8-метокси-N-((6-метилпиридин-3-ил)метил)-6-(5-метилпиримидин-2-ил)хиназолин-4-амин,
6-(5-фторпиридин-2-ил)-8-метокси-N-((6-(трифторметил)пиридин-3-ил)метил)хиназолин-4-амин,
8-метокси-N-((3-метил-1,2,4-оксадиазол-5-ил)метил)-6-(5-метилпиримидин-2-ил)хиназолин-4-амин,
6-(3-фтор-5-метил-2-пиридил)-8-метокси-N-[(1R)-1-(5-метил-1,2,4-оксадиазол-3-ил)этил]хиназолин-4-амин,
6-(5-фтор-2-пиридил)-8-метокси-N-[(1R)-1-(1-метилпиразол-3-ил)этил]хиназолин-4-амин,
8-метокси-N-[(1R)-1-(1-метилпиразол-3-ил)этил]-6-(5-метил-2-пиридил)хиназолин-4-амин,
2-((8-метокси-6-(5-метилпиримидин-2-ил)хиназолин-4-ил)амино)-1-морфолиноэтан-1-он,
6-(4-фторфенил)-N-[(5-фтор-2-пиридил)метил]-8-метоксихиназолин-4-амин,
одиночный энантиомер 8-метокси-N-[1-(3-метил-1,2,4-оксадиазол-5-ил)этил]-6-(5-метил-2-пиридил)хиназолин-4-амина,
одиночный энантиомер 8-метокси-N-(1-(5-метил-1,3,4-тиадиазол-2-ил)этил)-6-(5-метилтиазол-2-ил)хиназолин-4-амина,
одиночный энантиомер 2-((8-метокси-6-(5-метилпиримидин-2-ил)хиназолин-4-ил)амино)-2-(3-метил-1,2,4-оксадиазол-5-ил)этан-1-ола,
одиночный энантиомер 8-метокси-6-(5-метилпиримидин-2-ил)-N-(1-(3-(пиридин-4-ил)-1,2,4-оксадиазол-5-ил)этил)хиназолин-4-амина,
одиночный энантиомер 6-(5-фтор-2-пиридил)-8-метокси-N-[1-[6-(трифторметил)пиридазин-3-ил]этил]хиназолин-4-амина,
одиночный энантиомер N-[1-(5-метил-1,3,4-тиадиазол-2-ил)этил]-6-(5-метилтиазол-2-ил)-8-тетрагидропиран-4-илоксихиназолин-4-амина,
одиночный энантиомер 6-(5-фторпиридин-2-ил)-8-метокси-N-(1-(5-метил-1,3,4-тиадиазол-2-ил)этил)хиназолин-4-амина,
одиночный энантиомер 6-(5-фторпиридин-2-ил)-8-метокси-N-(1-(3-метил-1,2,4-оксадиазол-5-ил)этил)хиназолин-4-амина,
формиат 6-(4-фторфенил)-8-метокси-N-[(1S)-1-[2-(трифторметил)пиримидин-5-ил]этил]хиназолин-4-амина,
6-(3,6-дигидро-2H-пиран-4-ил)-8-метокси-N-((6-метилпиридазин-3-ил)метил)хиназолин-4-амин,
одиночный энантиомер 6-(1-((6-(4-фторфенил)-8-метоксихиназолин-4-ил)амино)этил)пиридин-2(1Н)-она,
одиночный энантиомер 6-(4-фторфенил)-8-метокси-N-(1-(3-(пиридин-4-ил)-1,2,4-оксадиазол-5-ил)этил)хиназолин-4-амина.
6. Соединение формулы (I) по п. 1, где Y представляет собой -ORD, представленное формулой (Ia)

где Z выбран из группы, состоящей из фенила, где любой указанный фенил может быть необязательно замещенным одной или несколькими группами, выбранными из галогена;
R1 представляет собой Н;
R2 выбран из группы, состоящей из гетероарил(C1-C4)алкила-, где любой указанный гетероарил может быть необязательно замещенным одной или несколькими группами, выбранными из (C1-C3)алкила, (C1-C6)галогеналкила;
RA и RB в каждом случае независимо представляют собой Н или выбраны из группы, состоящей из (C1-C4)алкила-;
RC в каждом случае представляет собой Н или выбран из группы, состоящей из (C1-C6)алкила;
RD выбран из группы, состоящей из H, (C1-C6)алкила, (C3-C8)гетероциклоалкил-(C1-C6)алкила-, RCOC(O)(C1-C4)алкилена-, , (C3-C8)гетероциклоалкила, RCO(C1-C4)алкилена-, (RARB)N(O)C(C1-C4)алкилена, где любой указанный гетероциклоалкил может быть необязательно замещенным одной или несколькими группами, выбранными из (C1-C3)алкила;
J представляет собой H.
7. Соединение формулы I по п. 6, выбранное из следующих соединений:
(R)-2-((6-(4-фторфенил)-4-((1-(2-(трифторметил)пиримидин-5-ил)этил)амино)хиназолин-8-ил)окси)уксусная кислота,
6-(4-фторфенил)-8-[(3-метилоксетан-3-ил)метокси]-N-[(6-метилпиридазин-3-ил)метил]хиназолин-4-амин,
3-[6-(4-фторфенил)-4-[(6-метилпиридазин-3-ил)метиламино]хиназолин-8-ил]окси-1-метилпирролидин-2-он,
6-(4-фторфенил)-N-((6-метилпиридазин-3-ил)метил)-8-(оксетан-3-илокси)хиназолин-4-амин,
одиночный энантиомер 6-(1-((6-(4-фторфенил)-8-метоксихиназолин-4-ил)амино)этил)пиридин-2(1Н)-она.
8. Фармацевтическая композиция, ингибирующая активность P2X пуринергического рецептора 3, включающая только соединение по пп. 1-7 или его фармацевтически приемлемую соль в смеси с одним или несколькими фармацевтически приемлемыми носителями или эксципиентами либо комбинацию соединения по пп. 1-7 или его фармацевтически приемлемой соли с одним или несколькими дополнительными активными ингредиентами в смеси с одним или несколькими фармацевтически приемлемыми носителями или эксципиентами.
9. Фармацевтическая композиция по п. 8 для перорального введения.
10. Фармацевтическая композиция по пп. 8 и 9 для применения в качестве лекарственного средства.
11. Фармацевтическая композиция по пп. 8 и 9 для применения в лечении любого заболевания, в которое вовлечены рецепторы P2X3.
12. Фармацевтическая композиция по пп. 8 и 9 для применения в предотвращении и/или лечении респираторных заболеваний, включая кашель, подострый или хронический кашель, терапевтически резистентный кашель, идиопатический хронический кашель, поствирусный кашель, ятрогенный кашель, астму, идиопатический легочный фиброз (ИЛФ), хроническую обструктивную болезнь легких (ХОБЛ) и кашель, связанный с респираторными заболеваниями, такими как ХОБЛ, астма и бронхоспазм.
13. Фармацевтическая композиция по п. 12 для применения в лечении хронического кашля.
14. Соединение формулы Ib

где R3 представляет собой OH или галоген,
R4 представляет собой Н;
R5 представляет собой -OMe;
R6 представляет собой Z;
Z принимает значения, определенные выше.
15. Применение соединений формулы (Ib) по п. 14 в качестве промежуточного соединения при получении соединений формулы (I) по п. 1.
16. Применение соединения по любому одному из пп. 1-7 для лечения любого заболевания, в которое вовлечены рецепторы P2X3.
17. Применение по п. 16, где заболевание выбрано из группы, состоящей из респираторных заболеваний, включая кашель, подострый или хронический кашель, терапевтически резистентный кашель, идиопатический хронический кашель, поствирусный кашель, ятрогенный кашель, астму, идиопатический легочный фиброз (ИЛФ), хроническую обструктивную болезнь легких (ХОБЛ) и кашель, связанный с респираторными заболеваниями, такими как ХОБЛ, астма и бронхоспазм.
18. Применение по п. 16 для лечения хронического кашля.
| WO 2016053794 A1, 07.04.2016 | |||
| PAN Y | |||
| et al, Pharmacophore and 3D-QSAR Characterization of 6-Arylquinazolin-4-amines as Cdc2-like Kinase 4 (Clk4) and Dual Specificity Tyrosine-phosphorylation-regulated Kinase 1A (Dyrk1A) Inhibitors, Journal of Chemical Information and Modeling, 2013, v | |||
| Веникодробильный станок | 1921 |
|
SU53A1 |
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| Катодный генератор | 1924 |
|
SU938A1 |
| FINLAY H.J | |||
| et al, Discovery of | |||

