Настоящее изобретение относится к способу лечения рака у млекопитающего путем введения 4-хиназолинаминов в комбинации с другими противоопухолевыми соединениями. В частности, способ относится к способам лечения видов рака путем введения комбинации N-{3-хлор-4-[(3-фторбензил)окси]фенил}-6-[5-({[2-(метансульфонил)этил]амино}метил)-2-фурил]-4-хиназолинамина и его солей и сольватов вместе с дополнительными противоопухолевыми соединениями.
Эффективная химиотерапия для лечения рака является постоянной задачей в области онкологии. Обычно рак является результатом разрегулирования нормальных процессов, которые контролируют клеточное деление, дифференцировку и апоптическую гибель клеток. Апоптоз (запрограммированная гибель клеток) играет существенную роль в эмбриональном развитии и патогенезе различных заболеваний, таких как дегенеративные нейрональные заболевания, сердечно-сосудистые заболевания и рак. Одним из наиболее широко изучаемых путей, который вовлекает киназную регуляцию апоптоза, является передача клеточного сигнала от рецепторов факторов роста на клеточной поверхности к ядру (Crew and Erikson, Cell, 74:2115-17, 1993). В частности, передача клеточного сигнала от рецепторов факторов роста семейства erbВ.
Существует значительная кооперация среди семейства erbВ, которое регулирует клеточные эффекты, опосредованные этими рецепторами. Шесть различных лигандов, которые связываются с EGFR, включают в себя EGF (эпидермальный фактор роста), трансформирующий фактор роста, амфирегулин, гепарин-связывающий EGF, бетацеллюлин и эпирегулин (Alroy & Yarden, FEBS Letters, 410:83-86, 1997; Burden & Yarden, Neuron, 18: 847-855, 1997; Klapper et al., Proc Natl Acad Sci, 4994-5000, 1999). Херегулины, другой класс лигандов, связываются непосредственно с HER3 и/или HER4 (Holmes et al., Science, 256: 1205, 1992; Klapper et al., 1997, Oncogene, 14:2099-2109; Peles et al., Cell, 69:205, 1992). Связывание специфических лигандов вызывает гомо- или гетеродимеризацию рецепторов среди представителей семейства еrbВ (Carraway & Cantley, Cell, 78:5-8, 1994; Lemmon & Schlessinger, TrendsBiochemSci, 19:459-463, 1994). В отличие от других представителей рецепторов еrbВ, растворимый лиганд еще не идентифицирован для HER2, который, как оказалось, трансактивируется после гетеродимеризации. Гетеродимеризация рецептора еrbВ-2 с EGFR, HER3 или HER4 предпочтительнее, чем гомодимеризация (Klapper et al., 1999; Klapper et al., 1997). Димеризация рецепторов приводит к связыванию АТФ с каталитическим участком рецептора, активации рецепторной тирозинкиназы и аутофосфорилированию по С-концевым тирозиновым остаткам. Фосфорилированные тирозиновые остатки затем служат в качестве участков связывания с белками, такими как Grb2, Shc и фосфолипаза С, которые, в свою очередь, активируют нисходящие (downstream) сигнальные пути, включая Ras/MEK/Erk и PI3K/Akt пути, которые регулируют факторы транскрипции и другие белки, вовлеченные в биологические ответы, такие как пролиферация, клеточная подвижность, ангиогенез, клеточное выживание и дифференцировка (Alroy & Yarden, 1997; Burgering & Coffer, Nature, 376:599-602, 1995; Chan et al., AnnRevBiochem, 68:965-1014, 1999; Lewis et at., AdvCanRes, 74:49-139, 1998; Liu et al., Genes and Dev, 13:786-791, 1999; Muthuswamy et al., Mol & CellBio, 19,10:6845-6857, 1999; Riese & Stern, Bioessays, 20:41-48, 1998).
Разработано несколько методик, включающих моноклональные антитела (Маb), иммуноконъюгаты, вакцину против EGF и ингибиторы тирозинкиназ, которые нацелены на рецепторы семейства еrbВ и блокируют их активацию в раковых клетках (рассматриваются в (Sridhar et al., Lancet, 4,7:397-406, 2003)). Поскольку еrbВ2-содержащие гетеродимеры представляют собой наиболее стабильное и предпочтительное инициирующее событие для передачи сигнала, то подавление одновременно и еrbВ2, и EGFR является привлекательной терапевтической стратегией. Синтезирован ряд 6-тиазолилхиназолиновых двойных ингибиторов тирозинкиназ erbB-2/EGFR, которые обладают эффективностью в доклинических моделях рака (Cockerill et al., BiorgMedChemLett, 11:1401-1405, 2001; Rusnak et al., CanRes, 61:7196-7203, 2001a; Rusnak et al., MolCanTher, 1:85-94, 2001b). GW 572016 представляет собой 6-фуранилхиназолин, перорально активный, обратимый двойной киназный ингибитор как EGFR, так и еrbВ2 киназ (Rusnak et al., 2001b). В исследованиях человеческих ксенотрансплантатов GW572016 проявляет дозозависимое ингибирование киназ и селективно ингибирует опухолевые клетки, сверхэкспрессирующие EGFR или еrbВ2 (Rusnak et al., 2001b; Xia et al., Oncogene, 21:6255-6263, 2002).
Комбинированная терапия при лечении рака быстро становится скорее нормой, чем исключением. Онкологи непрерывно ищут противоопухолевые соединения, которые при использовании в комбинации обеспечивают более эффективное и/или улучшенное лечение индивидуума, страдающего от поражающего воздействия рака. Обычно успешная комбинированная терапия обеспечивает улучшенный и даже синергический эффект относительно монотерапии.
В ходе данной работы авторы настоящего изобретения обнаружили новые способы лечения рака, которые включают в себя введение N-{3-хлор-4-[(3-фторбензил)окси]фенил}-6-[5-({[2-(метансульфонил)этил]амино}метил)-2-фурил]-4-хиназолинамина (GW 572016), а также его солей и/или сольватов в комбинации с дополнительными противоопухолевыми соединениями.
Краткое изложение сущности изобретения
В первом аспекте настоящего изобретения предложен способ лечения рака молочной железы у млекопитающего, включающий введение указанному млекопитающему терапевтически эффективных количеств
1) соединения формулы (I'')
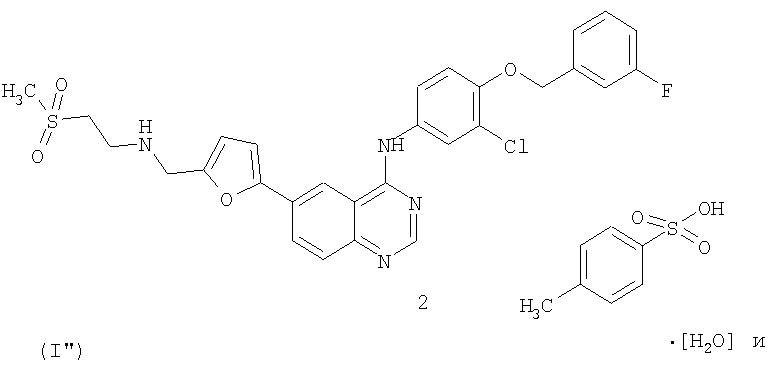
2) трастузумаба.
Во втором аспекте настоящего изобретения предложен способ лечения рака молочной железы у млекопитающего, включающий введение указанному млекопитающему терапевтически эффективных количеств
1) соединения формулы (I'')
2) летрозола.
В третьем аспекте настоящего изобретения предложен способ лечения рака молочной железы у млекопитающего, включающий введение указанному млекопитающему терапевтически эффективных количеств
1) соединения формулы (I'')
2) капецитабина.
В четвертом аспекте настоящего изобретения предложен способ лечения рака молочной железы у млекопитающего, включающий введение указанному млекопитающему терапевтически эффективных количеств
1) соединения формулы (I'')
2) тепотекана.
В пятом аспекте настоящего изобретения предложен способ лечения рака легких у млекопитающего, включающий введение указанному млекопитающему терапевтически эффективных количеств
1) соединения формулы (I'')
2) доцетаксела.
В шестом аспекте настоящего изобретения предложен способ лечения рака легких у млекопитающего, включающий введение указанному млекопитающему терапевтически эффективных количеств
1) соединения формулы (I'')
2)топотекана.
В седьмом аспекте настоящего изобретения предложен способ лечения колоректального рака у млекопитающего, включающий введение указанному млекопитающему терапевтически эффективных количеств
1) соединения формулы (I'')
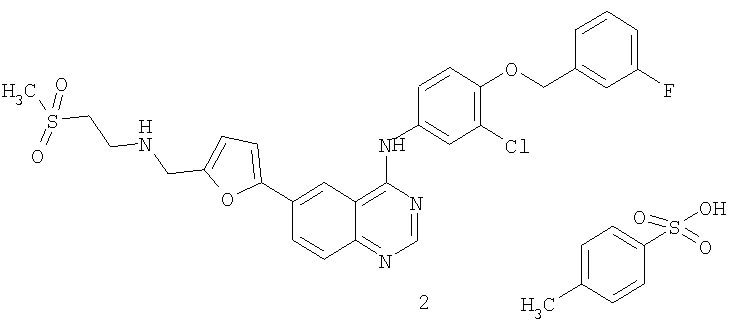

2) топотекана.
В восьмом аспекте настоящего изобретения предложен способ лечения рака молочной железы у млекопитающего, включающий введение указанному млекопитающему терапевтически эффективных количеств
1) соединения формулы (I'')
2) соединения с антиэстрогенной активностью.
Подробное описание изобретения
Используемый в данном описании термин "опухоль" относится к аномальному росту клеток или ткани и, как подразумевается, включает в себя доброкачественные, то есть нераковые опухоли, и злокачественные, то есть раковые опухоли. Термин "опухолевый" означает или относится к опухоли.
Используемый в данном описании термин "эффективное количество" означает такое количество лекарственного средства или фармацевтического агента, которое вызовет биологический или лечебный ответ у ткани, системы, животного или человека, к которому стремится, например, исследователь или врач. Кроме того, термин "терапевтически эффективное количество" означает любое количество, которое при сравнении с действием на соответствующего субъекта, который не получал такого количества, приводит к улучшенному лечению, излечению, предупреждению или облегчению заболевания, расстройства или побочных эффектов, или снижению скорости развития заболевания или расстройства. Этот термин также включает в свои рамки количества, эффективные для усиления нормальной физиологической функции.
Как хорошо известно в данной области техники, виды рака или опухоли часто являются метастатическими, так как исходный (первичный) очаг злокачественного опухолевого роста распространяется на один или более анатомически обособленные участки. Ссылка на используемый в данном описании термин "опухоль" у субъекта включает в себя не только первичную опухоль, но также и метастатический опухолевый рост. Подобным же образом ссылка на термин "рак" и "лечение рака" включает в себя первичный и метастатический рак и лечение первичного рака, а также метастатических злокачественных участков.
"EGFR", также известный как "еrbВ-1", и "еrbВ-2", представляют собой протеин-тирозинкиназные трансмембранные рецепторы семейства еrbВ к факторам роста. Протеин-тирозинкиназы катализируют фосфорилирование специфических тирозильных остатков в различных белках, вовлеченных в регуляцию клеточного роста и дифференцировки (A.F.Wilks, Progress in Growth Factor Research, 1990, 2, 97-111; S.A.Courtneidge, Dev. Supp.I, 1993, 57-64; J.A. Cooper, Semin. Cell Biol., 1994, 5(6), 377-387; R.F. Paulson, Semin. Immunol., 1995, 7(4). 267-277; A.C. Chan, Curr. Opin. Immunol., 1996, 8(3), 394-401). Семейство ЕrbВ рецепторных тирозинкиназ I типа включает в себя еrbВ1 (также известный как рецептор эпидермального фактора роста (EGFR или HER1)), еrbВ2 (также известный как HER2), еrbВ3 и еrbВ4. Эти рецепторные тирозинкиназы широко экспрессируются в эпителиальных, мезенхимальных и нейрональных тканях, где они играют роль в регуляции клеточной пролиферации, выживания и дифференцировки (Sibilia and Wagner, Science, 269:234 (1995); Threadgill et al., Science, 269:230 (1995)). Повышенная экспрессия еrbВ2 или EGFR дикого типа или экспрессия конститутивно активированных рецепторов-мутантов трансформирует клетки in vitro (Di Fiore et al., 1987; DiMarco et al., Oncogene, 4:831 (1989); Hudziak et al., Proc. Natl. Acad. Sci. USA, 84:7159 (1987); Qian et al., Oncogene, 10:211 (1995)). Повышенная экспрессия еrbВ2 или EGFR коррелировала с худшим прогнозом болезни при некоторых видах рака молочной железы и разновидностях других злокачественных опухолей (Slamon et al., Science, 235: 177 (1987); Slamon et al., Science, 244:707 (1989); Bacus et al., Am. J. Clin. Path., 102:S13 (1994)).
Используемый в данном описании термин "сольват" относится к комплексному соединению различной стехиометрии, образованному при помощи растворенного вещества (в данном изобретении соединения формулы (I) или их соли) и растворителя. Такие пригодные для изобретения растворители могут не влиять на биологическую активность растворенного вещества. Примеры подходящих растворителей включают в себя, но не ограничиваются этим, воду, метанол, этанол и уксусную кислоту. Предпочтительно, используемый растворитель представляет собой фармацевтически приемлемый растворитель. Примеры подходящих фармацевтически приемлемых растворителей включают в себя, без ограничений, воду, этанол и уксусную кислоту. Наиболее предпочтительно используемый растворитель представляет собой воду.
Как изложено выше, настоящее изобретение направлено на способы лечения рака, включающие введение N-{3-хлор-4-[(3-фторбензил)окси]фенил}-6-[5-({[2-(метансульфонил)этил]амино}метил)-2-фурил]-4-хиназолинамина (GW 572016), а также его солей и/или сольватов в комбинации с другими противоопухолевыми соединениями.
Способы лечения рака, раскрытые в данном описании, включают введение соединения формулы (I).
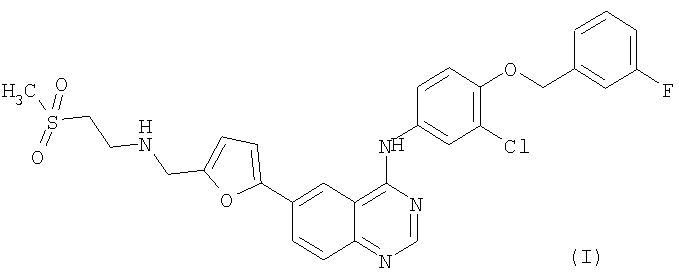
или его солей или сольватов.
В другом воплощении соединение является соединением формулы (I'), которое представляет собой дитозилатная соль соединения формулы (I) или его безводные или гидратированные формы. Дитозилатная соль соединения формулы (I) имеет химическое название дитозилат N-{3-хлор-4-[(3-фторбензил)окси]фенил}-6-[5-({[2-(метансульфонил)этил]амино}метил)-2-фурил]-4-хиназолинамина (GW 572016) и также известна как лапатиниб.
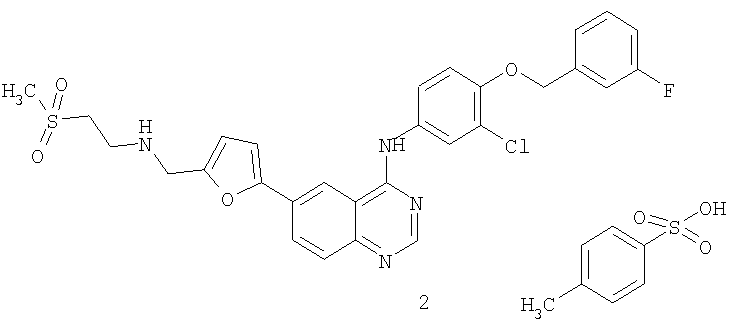
(I')
В одном воплощении соединение представляет собой безводную дитозилатную соль соединения формулы (I'). В другом воплощении соединение представляет собой соединение формулы (I''), которое является моногидратом дитозилатной соли соединения формулы (I').
Свободное основание, НСl соли и дитозилатные соли соединения формулы (I) могут быть получены согласно методикам вышеуказанной международной заявки на патент № РСТ/ЕР 99/00048, поданной 8 января 1999 г. и опубликованной как WО 99/35146 15 июля 1999 г., и международной заявки на патент № PCT/US 01/20706, поданной 28 июня 2001 г. и опубликованной как WO 02/02552 10 января 2002 г., и согласно соответствующим примерам, изложенным ниже. Одна такая методика получения дитозилатной соли соединения формулы (I) представлена далее на схеме 1.
Схема 1
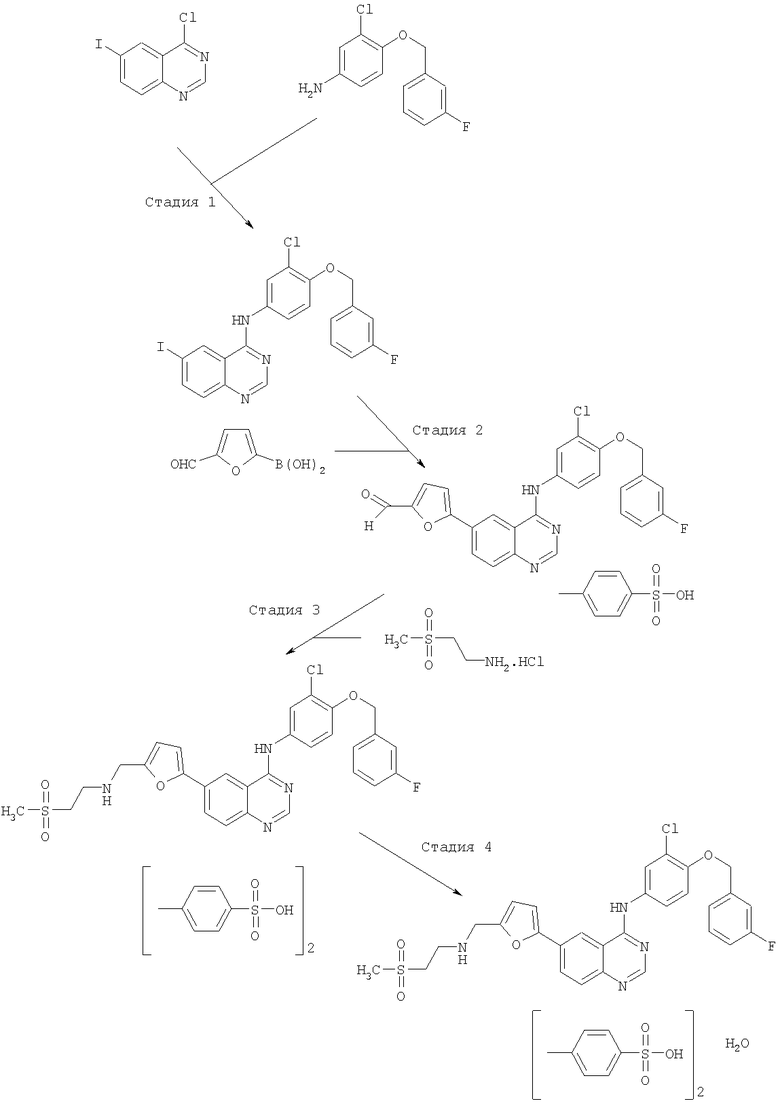
На схеме 1 получение дитозилатной соли соединения формулы (I) протекает в четыре стадии: Стадия 1: взаимодействие указанного бициклического соединения и амина с получением указанного производного иодхиназолина; Стадия 2: получение соответствующей альдегидной соли; Стадия 3: получение дитозилатной соли хиназолина; и Стадия 4: получение моногидрата дитозилатной соли.
Типично соли по настоящему изобретению представляют собой фармацевтически приемлемые соли. Соли, включенные в термин "фармацевтически приемлемые соли", относятся к нетоксичным солям соединений по этому изобретению. Соли соединений по настоящему изобретению могут включать в себя соли присоединения кислот, полученные по азоту заместителя в соединении по настоящему изобретению. Типичные соли включают в себя следующие соли: ацетат, бензолсульфонат, бензоат, бикарбонат, бисульфат, битартрат, борат, бромид, эдетат кальция, камсилат, карбонат, хлорид, клавуланат, цитрат, дигидрохлорид, эдетат, эдисилат, эстолат, эзилат, фумарат, глуцептат, глюконат, глутамат, гликолиларсанилат, гексилрезорцинат, гидрабамин, гидробромид, гидрохлорид, гидроксинафтоат, иодид, изетионат, лактат, лактобионат, лаурат, малат, малеат, манделат, мезилат, метилбромид, метилнитрат, метилсульфат, гидромалеат калия, мукат, напсилат, нитрат, N-метилглюкамин, оксалат, памоат (эмбонат), пальмитат, пантотенат, фосфат/дифосфат, полигалактуронат, салицилат калия, стеарат натрия, субацетат, сукцинат, таннат, тартрат, теоклат, тозилат, триэтиодид, триметиламмоний и валерат. Другие соли, которые не являются фармацевтически приемлемыми, могут быть полезны при получении соединений по этому изобретению, и они образуют дополнительный аспект изобретения.
В одном воплощении способ лечения рака представляет собой способ лечения рака молочной железы, при котором соединение формулы (I'') вводят с трастузумабом.
Трастузумаб представляет собой рекомбинантные человеческие моноклональные антитела, производные ДНК, которые селективно связываются с внеклеточным доменом HER2 (еrbВ2); трастузумаб имеется в продаже в виде лиофилизированного порошка для внутривенной инъекции под названием HERCEPTIN®. Трастузумаб назначают в виде одиночного агента для лечения пациентов с метастатическим раком молочной железы, при котором еrbВ2 подвергается сверхэкспрессии, которые ранее получали один или два химиотерапевтических режима.
В одном воплощении способ лечения рака представляет способ лечения рака молочной железы, при котором соединение формулы (I'') вводят с по меньшей мере одним соединением с антиэстрогенной активностью. Соединение с антиэстрогенной активностью может представлять собой антагонист эстрогеновых рецепторов или ингибитор синтеза эстрогенов. Типичные антагонисты эстрогеновых рецепторов включают в себя, но не ограничиваются этим, фулвестрант, тамоксифен и его метаболит 4-ОН-тамоксифен и торемифен. Характерные ингибиторы синтеза эстрогенов включают в себя ингибиторы ароматазы летрозол, анастрозол и экземестан.
Фулвестрант, 7-альфа-[9-(4,4,5,5-пентафторсульфинил)нонил]эстра-1,3,5-(10)-триен-3,17-бета-диол, имеется в продаже в виде инъекционного раствора под названием FASLODEX®. Фулвестрант показан для лечения гормонозависимого метастатического рака молочной железы у женщин в постменопаузе после антиэстрогенной терапии. Фулвестрант является антагонистом эстрогеновых рецепторов, который конкурентно связывается с эстрогеновым рецептором и подавляет белок ER (эстрогеновый рецептор) в клетках рака молочной железы человека.
Тамоксифен, 2-гидрокси-1,2,3-пропантрикарбоксилат (Z)2-[4-(1,2-дифенил-1-бутенил)фенокси]-N,N-диметилэтанамина (1:1), имеется в продаже в виде таблеток 10 или 20 мг под названием NOLVADEX®. Тамоксифен показан для лечения метастатического рака молочной железы у мужчин и женщин и в качестве адъювантной терапии при раке молочной железы. Тамоксифен является антагонистом эстрогеновых рецепторов, который конкурентно связывается с эстрогеновым рецептором.
Торемифен, цитрат 2-{p[(Z)-4-хлор-1,2-дифенил-1-бутенил]фенокси}-N,N-диметилэтиламина (1:1), имеется в продаже в виде таблеток 60 мг под названием FARESTON®. Торемифен показан для лечения опухолей, имеющих эстрогеновые рецепторы, и неизвестных опухолей при метастатическом раке молочной железы у женщин в постменопаузе. Торемифен является селективным модулятором эстрогеновых рецепторов, который связывается с эстрогеновым рецептором и может проявлять эстрогенную или антиэстрогенную активность в зависимости от продолжительности лечения, вида, пола, органа-мишени или выбранной конечной точки.
В другом воплощении способ лечения рака представляет собой способ лечения рака молочной железы, при котором соединение формулы (I'') вводят с летрозолом.
Летрозол представляет собой 4-4'-(1Н-1,2,4-триазол-1-илметилен)-дибензонитрил, который имеется в продаже в виде таблеток 2,5 мг под названием FEMARA®. Летрозол является перорально вводимым нестероидным ингибитором ароматазы. Конкретно, он является ингибитором синтеза эстрогенов, так как он ингибирует превращение андрогенов в эстрогены. Летрозол показан для лечения распространенного рака молочной железы у женщин в постменопаузе с прогрессированием заболевания после антиэстрогенной терапии.
Анастрозол представляет собой 1,3-бензолдиацетонитрил-α,α,α',α'-тетраметил-5-(1Н-1,2,4-триазол-1-илметил), который имеется в продаже в виде таблеток 1 мг под названием ARIMIDEX®. Анастрозол является перорально вводимым нестероидным ингибитором ароматазы. Конкретно, он является ингибитором синтеза эстрогенов, так как он ингибирует превращение андрогенов в эстрогены. Анастрозол показан для адъювантной терапии раннего рака молочной железы у женщин в постменопаузе.
Экземестан представляет собой 6-метиленандроста-1,4-диен-3,17-дион, который имеется в продаже в виде таблеток 25 мг под названием AROMASIN®. Экземестан является перорально вводимым стероидным ингибитором ароматазы. Конкретно, он является ингибитором синтеза эстрогенов, так как он ингибирует превращение андрогенов в эстрогены. Экземестан показан для лечения распространенного рака молочной железы у женщин в постменопаузе с прогрессированием заболевания после терапии тамоксифеном.
В одном воплощении способ лечения рака представляет собой способ лечения рака молочной железы, при котором соединение формулы (I'') вводят с капецитабином.
Капецитабин, 5'-дезокси-5-фтор-N-[(пентилокси)карбонил]-цитидин, имеется в продаже в виде таблеток 150 или 500 мг под названием XELODA®. Капецитабин является перорально вводимым пролекарством 5'-дезокси-5-фторуридином (5'-DFUR), которое превращается в 5-фторурацил in vivo. Капецитабин показан для лечения метастатического рака молочной железы, устойчивого к режиму лечения, включающему как паклитаксел, так и антрациклин.
В одном воплощении способ лечения рака представляет собой способ лечения рака молочной железы, при котором соединение формулы (I'') вводят с топотеканом.
Топотекан HCl, моногидрохлорид (S)-10-[(диметиламино)метил]-4-этил-4,9-дигидрокси-1Н-пирано[3',4',6,7]индолизино[1,2-b]хинолин-3,14-(4Н,12Н)-диона, имеется в продаже в виде инъекционного раствора под названием HYCAMTIN®. Топотекан является производным камптотецина, который связывается с комплексом топоизомераза I - ДНК и предотвращает повторное сшивание однонитевых разрывов, вызванных топоизомеразой I в ответ на напряжение скручивания молекулы ДНК. Топотекан показан для лечения второй линии метастатической карциномы яичника и мелкоклеточного рака легких. Ограничивающим дозу побочным эффектом топотекана HCl является миелосупрессия, первичная нейтропения.
В одном воплощении способ лечения рака представляет собой способ лечения рака легких, при котором соединение формулы (I'') вводят с доцетакселем. В одном воплощении рак легких представляет собой немелкоклеточный рак легких.
Доцетаксел, (2R,3S)-N-карбокси-3-фенилизосерина N-трет-бутиловый эфир, 13-эфир с 5β-20-эпокси-1,2α,4,7β,10β,13α-гексагидрокситакс-11-ен-9-он-4-ацетат-2-бензоатом, тригидрат, имеется в продаже в виде инъекционного раствора под названием TAXOTERE®. Доцетаксел показан для лечения рака молочной железы. Доцетаксел является полусинтетическим производным паклитаксела q.v., полученного с использованием природного предшественника, 10-деацетилбаккатина III, экстрагируемого из игл европейского тиса. Ограничивающей дозу токсичностью доцетаксела является нейтропения.
В одном воплощении способ лечения рака представляет собой способ лечения рака легких, при котором соединение формулы (I'') вводят с топотеканом. В одном воплощении рак легких представляет собой немелкоклеточный рак легких.
Топотекан является таким, как описано выше.
В одном воплощении способ лечения рака представляет собой способ лечения колоректального рака, при котором соединение формулы (I'') вводят с топотеканом.
Топотекан является таким, как описано выше.
В одном воплощении способ лечения рака представляет собой способ лечения рака молочной железы, при котором соединение формулы (I'') вводят с по меньшей мере одним ингибитором bcl-2.
Апоптоз или запрограммированная гибель клеток представляет собой механизм, посредством которого избыточные, нежелательные или поврежденные клетки удаляются из организма. Большая часть злокачественных процессов подвергается действию аберрантных апоптических путей, так как апоптоз блокируется или ингибируется, что приводит к улучшенному клеточному выживанию и, возможно, невосприимчивости к лечению. Bcl-2 является представителем семейства регуляторов апоптоза. Bcl-2 представляет собой супрессор апоптического пути и при сверхэкспрессии в раковых клетках может играть роль в стимуляции ракового развития и роста. По существу, предполагают, что ингибитор bcl-2 может быть эффективным при лечении рака (Sara et al., Current Med Chem, 11:1031-1040, 2004; Lie et al., CurrMed Chem - Anticancer Agents, 3:217-223, 2003). Одним известным ингибитором bcl-2 является НА14-1, который представляет собой этил-[2-амино-6-бром-4-(1-циано-2-этокси-2-оксоэтил)]-4Н-хромен-3-карбоксилат и который получают от Calbiochem из Сан-Диего, Калифорния.
Таким образом, виды комбинированной терапии согласно настоящему изобретению включают в себя введение соединение формулы (I''), а также применение по меньшей мере одного другого противоопухолевого агента. Такую комбинацию агентов можно вводить вместе или раздельно, и при раздельном введении оно может происходить одновременно или последовательно в любом порядке как близко, так и отдаленно по времени. Количества соединения формулы (I'') и другого фармацевтически активного агента (агентов) и относительные распределения введения во времени будут выбираться таким образом, чтобы достичь желаемого комбинированного терапевтического эффекта.
Также в настоящем изобретении рассматривают фармацевтические композиции, содержащие соединения формулы (I'') и по меньшей мере один противоопухолевый агент. Такие соединения формулы (I'') и по меньшей мере один противоопухолевый агент являются такими, как описано выше, и могут быть использованы в любых комбинациях, описанных выше, в способе лечения рака по настоящему изобретению.
Возможно, что для применения в способах лечения рака по настоящему изобретению терапевтически эффективные количества соединения формулы (I''), а также его солей или сольватов можно вводить не только в виде грубого химического вещества, но возможно в присутствии активного ингредиента в виде фармацевтической композиции. Таким образом, согласно изобретению дополнительно предложены фармацевтические композиции, которые можно вводить в способах лечения рака по настоящему изобретению. Фармацевтические композиции содержат терапевтически эффективные количества соединения формулы (I'') и его солей или сольватов и один или более чем один фармацевтически приемлемый носитель, разбавитель или эксципиент. Носитель (носители), разбавитель (разбавители) или эксципиент (эксципиенты) должны быть приемлемыми в смысле совместимости с другими ингредиентами лекарственного препарата и безвредности для его реципиента.
Фармацевтические препараты могут быть представлены в стандартных лекарственных формах, содержащих заданное количество активного ингредиента на стандартную дозу. Такая стандартная доза может содержать, например, от 0,5 мг до 1 г, предпочтительно от 1 мг до 700 мг, более предпочтительно от 5 мг до 100 мг соединения формулы (I) в зависимости от состояния, которое лечат, пути введения и возраста, массы и состояния пациента, или фармацевтические препараты могут быть представлены в стандартных лекарственных формах, содержащих заданное количество активного ингредиента на стандартную дозу. Предпочтительными стандартными лекарственными препаратами являются те, которые содержат суточную дозу или разделенную дозу, как изложено здесь ранее, или соответствующую часть этой дозы активного ингредиента. Кроме того, такие фармацевтические препараты могут быть приготовлены с помощью любого из способов, хорошо известных в области фармации.
Соединение формулы (I'') можно вводить любым подходящим путем. Подходящие пути введения включают в себя пероральный, ректальный, назальный, местный (включая трансбуккальный и сублингвальный), вагинальный и парентеральный (включая подкожный, внутримышечный, внутривенный, внутрикожный, интратекальный и эпидуральный). Следует понимать, что предпочтительный путь введения может варьироваться в зависимости, например, от состояния реципиента комбинации.
Фармацевтические препараты, предназначенные для перорального введения, могут быть представлены в виде отдельных форм, таких как капсулы или таблетки; порошки или гранулы; растворы или суспензии в водных или неводных жидкостях; съедобные пены или кремы; или жидкие эмульсии типа масло в воде или жидкие эмульсии типа вода в масле.
Например, для перорального введения в форме таблетки или капсулы активный лекарственный компонент можно объединять с пероральным, нетоксичным фармацевтически приемлемым инертным носителем, таким как этанол, глицерин, вода и тому подобное. Порошки приготавливают путем измельчения соединения до подходящего мелкого размера и смешения с измельченным аналогичным образом фармацевтическим носителем, таким как пищевой углевод, таким как, например, крахмал или маннит. Также могут присутствовать ароматизатор, консервант, диспергирующее вещество и краситель.
Капсулы приготавливают путем получения порошкообразной смеси, как описано выше, и наполнения сформированной желатиновой оболочки. Скользящие и смазывающие вещества, такие как коллоидный оксид кремния, тальк, стеарат магния, стеарат кальция или твердый полиэтиленгликоль, могут быть добавлены к порошкообразной смеси перед процедурой наполнения. Для улучшения доступности лекарства, когда капсулу проглатывают, также может быть добавлен разрыхлитель или солюбилизатор, такой как агар-агар, карбонат кальция или карбонат натрия.
Кроме того, когда желательно или необходимо, в смесь также могут быть включены подходящие связующие вещества, смазывающие вещества, разрыхлители и красители. Подходящие связующие вещества включают в себя крахмал, желатин, природные сахара, такие как глюкоза или бета-лактоза, кукурузные подсластители, природные и синтетические камеди, такие как гуммиарабик, трагакант или альгинат натрия, карбоксиметилцеллюлоза, полиэтиленгликоль, воски и тому подобное. Смазывающие вещества, используемые в этих лекарственных формах, включают в себя олеат натрия, стеарат натрия, стеарат магния, бензоат натрия, ацетат натрия, хлорид натрия и тому подобное. Разрыхлители включают в себя, без ограничения, крахмал, метилцеллюлозу, агар, бентонит, ксантановую камедь и тому подобное. Таблетки получают, например, путем приготовления порошкообразной смеси, гранулирования или агрегирования, добавления смазывающего вещества и разрыхлителя и прессования в таблетки. Порошкообразную смесь приготавливают путем смешивания соответствующим образом измельченного соединения с разбавителем или основанием, как описано выше, и возможно со связующим веществом, таким как карбоксиметилцеллюлоза, алигинат, желатин или поливинилпирролидон, замедлителем растворения, таким как парафин, ускорителем всасывания, таким как четвертичная соль, и/или поглощающим веществом, таким как бентонит, каолин или дикальций фосфат. Порошкообразную смесь можно гранулировать путем смачивания связующим веществом, таким как сироп, крахмальный клейстер, клейкое вещество акации или растворы целлюлозных или полимерных материалов, и продавливания через сито. В качестве альтернативы грануляции, порошкообразную смесь можно пропустить через таблетировочную машину, а полученные в результате не полностью сформированные агрегаты измельчить в гранулы. Гранулы можно смазывать для предотвращения прилипания к штампам, формирующим таблетки, путем добавления стеариновой кислоты, стеарата, талька или минерального масла. Смазанную смесь затем прессовали в таблетки. Соединения по настоящему изобретению также можно объединять с сыпучим инертным носителем и прессовать в таблетки непосредственно без проведения стадий гранулирования или агрегирования. Может быть предложено прозрачное или непрозрачное защитное покрытие, состоящее из изолирующего слоя шеллака, покрытие из сахара или полимерного материала и глянцевое покрытие из воска. К этим покрытиям могут быть добавлены красители, чтобы отличать различные стандартные дозировки.
Жидкости для перорального введения, такие как растворы, сиропы и эликсиры, могут быть приготовлены в стандартной лекарственной форме, так чтобы данное количество содержало заданное количество соединения. Сиропы могут быть приготовлены путем растворения соединения в соответствующим образом ароматизированном водном растворе, тогда как эликсиры приготавливают с использованием нетоксичного спиртового носителя. Суспензии могут быть приготовлены в виде препарата путем диспергирования соединения в нетоксичном носителе. Также могут быть добавлены солюбилизаторы и эмульгаторы, такие как этоксилированные изостеариловые спирты и полиоксиэтиленовые эфиры сорбита, консерванты, ароматизирующие добавки, такие как масло перечной мяты или природные подсластители или сахарин или другие искусственные подсластители, и тому подобное.
Когда это целесообразно, стандартные лекарственные препараты для перорального введения могут быть микроинкапсулированы. Также может быть приготовлен препарат для пролонгированного или длительного высвобождения, например, путем покрытия или заключения гранулированного материала в полимеры, воск или тому подобное.
Агенты для применения согласно настоящему изобретению также могут быть введены в форме липосомальных систем доставки, таких как мелкие однослойные везикулы, крупные однослойные везикулы и многослойные везикулы. Липосомы могут быть образованы из разнообразных фосфолипидов, таких как холестерин, стеариламин или фосфатидилхолины.
Агенты для применения согласно настоящему изобретению также могут быть доставлены путем использования моноклональных антител в качестве индивидуальных носителей, с которыми связаны молекулы соединений. Соединения также можно связывать с растворимыми полимерами в качестве носителей для направленной доставки лекарственных средств. Такие полимеры могут включать в себя поливинилпирролидон, сополимер пирана, полигидроксипропилметакриламидфенол, полигидроксиэтиласпартамидфенол или полиэтиленоксидполилизин, замещенный остатками пальмитоила. Кроме того, соединения можно связывать с классом биодеградируемых полимеров, полезных для достижения контролируемого высвобождения лекарственного средства, например, полимолочной кислотой, полиэпсилонкапролактоном, полигидроксимасляной кислотой, полиортоэфирами, полиацеталями, полидигидропиранами, полицианоакрилатами и поперечно-сшитыми или амфипатическими блоксополимерами гидрогелей.
Фармацевтические препараты, предназначенные для трансдермального введения, могут быть представлены в виде отдельных пластырей, предназначенных для пребывания в тесном контакте с эпидермисом реципиента в течение длительного периода времени. Например, активный ингредиент может высвобождаться из пластыря посредством ионтофореза, как в общих чертах описано в Pharmaceutical Research, 3(6), 318 (1986).
Фармацевтические препараты, предназначенные для местного введения, могут быть приготовлены в виде мазей, кремов, суспензий, лосьонов, порошков, растворов, паст, гелей, спреев, аэрозолей и масел.
Для лечения глаза или других наружных тканей, например, ротовой полости и кожи, препараты предпочтительно используют в виде мази или крема для местного применения. При приготовлении в виде мази активный ингредиент может быть использован либо с парафиновой, либо с водорастворимой мазевой основой. Альтернативно, активный ингредиент может быть приготовлен в виде крема с кремовой основой типа масло-в-воде или основой типа вода-в-масле.
Фармацевтические препараты, предназначенные для местного введения в глаз, включают в себя глазные капли, в которых активный ингредиент растворен или суспендирован в подходящем носителе, особенно в водном растворителе.
Фармацевтические препараты, предназначенные для местного введения в ротовую полость, включают в себя леденцы, пастилки и раствор для полоскания полости рта.
Фармацевтические препараты, предназначенные для ректального введения, могут быть представлены в виде суппозиториев или в виде клизм.
Фармацевтические препараты, предназначенные для назального введения, в которых носитель представляет собой твердое вещество, включают в себя крупнозернистый порошок, имеющий размер частиц, например, в пределах от 20 до 500 микрон, который вводят посредством вдыхания через нос, то есть посредством быстрой ингаляции через носовой проход из порошкового контейнера, поднесенного близко к носу. Подходящие препараты, в которых носитель представляет собой жидкость для введения в виде назального спрея или в виде назальных капель, включают в себя водные или масляные растворы активного ингредиента.
Фармацевтические препараты, предназначенные для введения посредством ингаляции, включают в себя тонко измельченные порошки для ингаляции или туманы, которые могут быть получены посредством различных типов дозированных аэрозолей под давлением, небулайзеров или инсуффляторов.
Фармацевтические препараты, предназначенные для вагинального введения, могут быть представлены в виде пессариев, тампонов, кремов, гелей, паст, пен или препаратов в виде спреев.
Фармацевтические препараты, предназначенные для парентерального введения, включают в себя водные и неводные стерильные инъекционные растворы, которые могут содержать антиоксиданты, буферы, бактериостатики и растворенные вещества, которые делают препарат изотоническим по отношению к крови предполагаемого реципиента; и водные и неводные стерильные суспензии, которые могут включать в себя суспендирующие агенты и загустители. Препараты могут быть представлены в однодозовых или многодозовых контейнерах, например в запаянных ампулах и флаконах, и могут храниться в сублимированном (лиофилизированном) состоянии, требуя лишь добавления стерильного жидкого носителя, например стерильной воды для инъекций непосредственно перед применением. Приготовленные для немедленного приема инъекционные растворы и суспензии могут быть получены из стерильных порошков, гранул и таблеток.
Следует понимать, что кроме ингредиентов, подробно упомянутых выше, препараты могут включать в себя другие общепринятые в данной области техники агенты, принимая во внимание тип данного препарата, например препараты, подходящие для перорального введения, могут включать в себя ароматизаторы.
Как указано, млекопитающему вводят терапевтически эффективные количества конкретного соединения формулы (I). Типично, терапевтически эффективное количество одного из вводимых агентов по настоящему изобретению будет зависеть от ряда факторов, включая, например, возраст и массу млекопитающего, точно установленное состояние, требующее лечения, тяжесть состояния, природу препарата и путь введения. В конечном счете, терапевтически эффективное количество будет назначаться по усмотрению лечащего врача или ветеринара.
Типично, соединение формулы (I) будет назначаться в пределах от 0,1 до 100 мг/кг массы тела реципиента (млекопитающего) в сутки и более обычно в пределах от 1 до 10 мг/кг массы тела в сутки.
Следующие примеры предназначены только для иллюстрации, а не для ограничения объема изобретения каким-либо образом.
Примеры
Используемые в данном описании символы и условные обозначения, используемые в этих способах, схемах и примерах, соответствуют тем, которые используются в современной научной литературе, например, в журнале Американского химического общества (Journal of the American Chemical Society) или в журнале биологической химии (Journal of Biological Chemistry). Стандартные однобуквенные и трехбуквенные аббревиатуры обычно используют для обозначения аминокислотных остатков, которые, как принято, находятся в L-конфигурации, если не указано иначе. Если не указано иначе, все исходные материалы получали от торговых поставщиков и использовали без дополнительной очистки. В частности, в примерах и в описании могут быть использованы следующие аббревиатуры:
г (граммы);
мг (миллиграммы);
л (литры);
мл (миллилитры);
мкл (микролитры);
psi (фунты на квадратный дюйм);
М (молярный);
мМ (миллимолярный);
н (нормальный);
кг (килограмм);
в/в (внутривенный);
Гц (герц);
МГц (мегагерц);
моль (моли);
ммоль (миллимоли);
RT (комнатная температура);
мин (минуты);
ч (часы);
т.пл. (точка плавления);
ТСХ (тонкослойная хроматография);
Тr (время удерживания);
ОФ (обращенная фаза);
DCM (дихлорметан);
DCE (дихлорэтан);
DMF (N,N-диметилформамид);
НОАс (уксусная кислота);
TMSE (2-(триметилсилил)этил);
TMS (триметилсилил);
TIPS (триизопропилсилил);
TBS (трет-бутилдиметилсилил);
ВЭЖХ (высокоэффективная жидкостная хроматография);
THF (тетрагидрофуран);
DMSO (диметилсульфоксид);
EtOAc (этилацетат);
DME (1,2-диметоксиэтан);
EDTA (этилендиаминтетрауксусная кислота);
FBS эмбриональная бычья сыворотка
IMDM среда Дульбекко, модифицированная по методу Искова
IMS промышленные метилированные спирты
PBS фосфатно-солевой буфер
RPMI мемориальный институт Росвелл Парка
RIPA буфер *
* 150 мМ NaCl, 50 мМ трис-HCl, рН 7,5, 0,25% (мас./об.) - дезоксихолат, 1% NP-40, 5 мМ ортованадат натрия, 2 мМ фторид натрия и коктейль ингибиторов протеаз.
Если не указано иначе, все температуры выражены в °С (градусы Цельсия). Все реакции осуществляли в атмосфере инертного газа при комнатной температуре, если не указано иначе.
GW 572016 F представляет собой лапатиниб, химическое название которого - N-{3-хлор-4-[(3-фторбензил)окси]фенил}-6-[5-({[2-(метансульфонил)-этил]амино}метил)-2-фурил]-4-хиназолинамина дитозилата моногидрат.
Пример 1
Получение GW 572016 F
Стадия 1
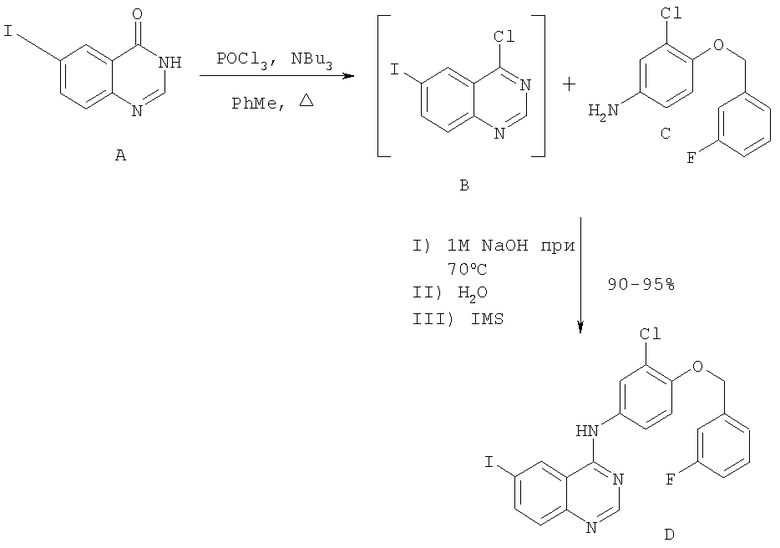
Перемешиваемый раствор 3Н-6-иодхиназолин-4-она (соединение А) в толуоле (5 об.ч.) обрабатывали три-н-бутиламином (1,2 экв.) при температуре от 20 до 25°С, затем нагревали до 90°С. Добавляли оксихлорид фосфора (1,1 экв.), реакционную смесь затем нагревали до температуры дефлегмации. Реакционную смесь охлаждали до 50°С и добавляли толуол (5 об.ч.). Соединение С (1,03 экв.) добавляли в виде твердого вещества, суспензию снова нагревали до 90°С и перемешивали в течение 1 часа. Суспензию переносили во второй сосуд; первый сосуд промывали толуолом (2 об.ч.) и объединяли с реакционной смесью. Реакционную смесь охлаждали до 70°С, и 1,0 М водный раствор гидроксида натрия (16 об.ч.) по каплям добавляли в течение 1 часа к перемешиваемой суспензии, поддерживая температуру содержимого между 68-72°С. Смесь перемешивали при 65-70°С в течение 1 часа и затем охлаждали до 20°С в течение 1 часа. Суспензию перемешивали при 20°С в течение 2 часов, продукт собирали фильтрацией и последовательно промывали водой (3×5 об.ч.) и этанолом (IMS, 2×5 об.ч.), затем сушили под вакуумом при 50-60°С.
Объемы приведены относительно количества используемого соединения А. Наблюдаемый диапазон выхода в процентах: от 90 до 95% в виде белых или желтых кристаллов.
Стадия 2
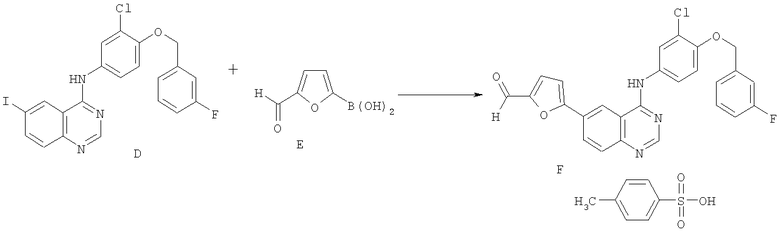
Смесь N-{3-хлор-4-[(3-фторбензил)окси]фенил}-6-иод-4-хиназолинамина - соединения D (1 мас.ч.), бороновой кислоты - соединения Е (0,37 мас.ч.; 1 экв.) и 10% палладия на угле (0,028 мас.ч.; 50% влажности) суспендировали в IMS (15 об.ч.). Полученную суспензию перемешивали в течение 5 минут, обрабатывали диизопропилэтиламином (0,39 об.ч.; 1,15 экв.) и затем нагревали до приблизительно 70°С в течение приблизительно 3 часов, когда реакцию завершали (определяли посредством ВЭЖХ-анализа). Смесь разбавляли тетрагидрофураном (THF; 15 об.ч.) и затем фильтровали в горячем состоянии для удаления катализатора. Сосуд промывали IMS (2 об.ч.).
Раствор пара-толуолсульфоновой кислоты моногидрата (1,5 мас.ч.; 4 экв.) в воде (1,5 об.ч.) добавляли в течение 5-10 минут к отфильтрованному раствору, который поддерживали при 65°С. После кристаллизации суспензию перемешивали при 60-65°С в течение 1 часа, охлаждали до приблизительно 25°С в течение 1 часа и перемешивали при этой температуре в течение еще 2 часов. Твердое вещество собирали фильтрацией, промывали IMS (3 об.ч.), затем сушили под вакуумом при приблизительно 50°С с получением соединения F в виде желто-оранжевого кристаллического твердого вещества (выделенного в виде этанольного сольвата, содержащего приблизительно 5% (мас./мас.) EtOH).
Стадия 3
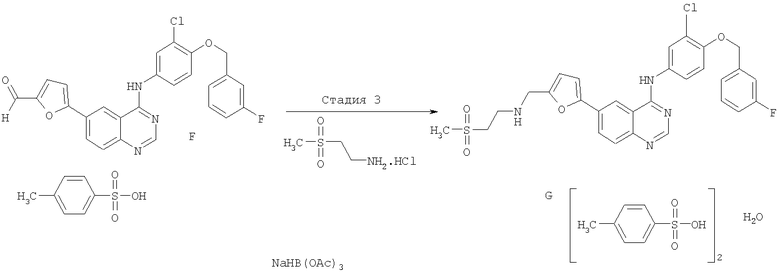
Соединение F (1 мас.ч.) и 2-(метилсульфонил)этиламина гидрохлорид (0,4 мас.ч.; 1,62 экв.) суспендировали в THF (10 об.ч.). Последовательно добавляли уксусную кислоту (0,354 об.ч.; 4 экв.) и диизопропилэтиламин (DIPEA; 1,08 об.ч.; 4,01 экв.). Полученный раствор перемешивали при 30-35°С в течение приблизительно 1 часа, затем охлаждали до приблизительно 22°С. Затем добавляли триацетоксиборгидрид натрия (0,66 мас.ч.; 2,01 экв.) в виде непрерывной подачи в течение приблизительно 15 минут (на этой стадии наблюдают некоторое вспенивание). Полученную смесь перемешивали при приблизительно 22°С в течение приблизительно 2 часов, затем отбирали образец для ВЭЖХ-анализа. Реакцию гасили путем добавления водного гидроксида натрия (25% мас./мас.; 3 об.ч.) с последующим добавлением воды (2 об.ч.) и перемешивали в течение приблизительно 30 минут (наблюдали некоторое вспенивание в начале добавления каустической соды).
Водную фазу затем отделяли, экстрагировали THF (2 об.ч.), и объединенные THF-экстракты затем дважды промывали 25% мас./об. водным раствором хлорида аммония (2×5 об.ч.)2. Готовили раствор пара-толуолсульфоновой кислоты моногидрата (p-TSA; 0,74 мас.ч.; 2,5 экв.) в воде (1 об.ч.)1, нагревали до приблизительно 60°С и добавляли затравку GW 572016 F (соединение G) (0,002 мас.ч.).
THF раствор свободного основания GW 572016 добавляли к раствору р-TSA в течение по меньшей мере 30 минут, поддерживая температуру реакционной массы 60±3°С. Полученную суспензию перемешивали при приблизительно 60°С в течение 1-2 часов, охлаждали до 20-25°С в течение часа и выдерживали при этой температуре в течение приблизительно 1 часа. Твердое вещество собирали фильтрацией, промывали смесью ТНF: вода, 95:5 (3×2 об.ч.), и сушили под вакуумом при приблизительно 35°С с получением GW 572016 F - соединения G в виде ярко-желтого кристаллического твердого вещества. Ожидаемый теоретический выход 80%, 117% мас./мас.
1 Минимальный реакционный объем приблизительно 1 об.ч.
2 Максимальный реакционный объем приблизительно 17 об.ч.
# Уточненный для анализа.
Стадия 4
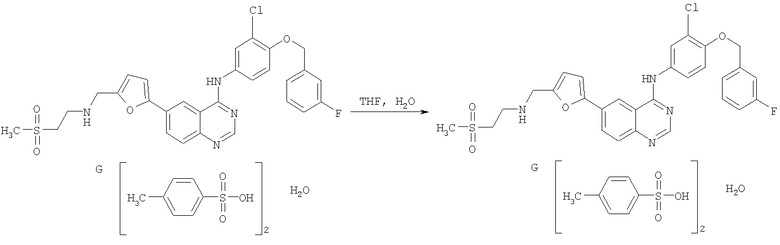
Суспензию N-{3-хлор-4-[(3-фторбензил)окси]фенил}-6-[5-({[2-(метансульфонил)этил]амино}метил)-2-фурил]-4-хиназолинамина дитозилата моногидрата - соединения G (1 мас.ч.) в тетрагидрофуране (THF; 14 об.ч.) и воде (6 об.ч.) нагревали до приблизительно 55°-60°С в течение 30 минут с получением раствора, который осветляли фильтрацией, и трубопроводы промывали в кристаллизационный сосуд смесью THF/вода (соотношение 7:3; 2 об.ч.). Полученный раствор нагревали до температуры дефлегмации, и тетрагидрофуран (9 об.ч.; 95% мас./мас. азеотропная смесь с водой) отгоняли при атмосферном давлении.
В раствор добавляли затравку N-{3-хлор-4-[(3-фторбензил)окси]фенил}-6-[5-({[2-(метансульфонил)этил]амино}метил)-2-фурил]-4-хиназолинамина дитозилата моногидрата (0,002 мас.ч.). Как только кристаллизация начиналась, добавляли воду (6 об.ч.), поддерживая температуру реакции выше 55°С. Смесь охлаждали до 5°-15°С в течение приблизительно 2 часов. Твердое вещество собирали фильтрацией, промывали смесью тетрагидрофуран/вода (соотношение 3:7; 2 об.ч.), затем смесью тетрагидрофуран/вода (соотношение 19:1; 2 об.ч.) и сушили под вакуумом при 45°С с получением N-{3-хлор-4-[(3-фторбензил)окси]фенил}-6-[5-({[2-(метансульфонил)этил]амино}метил)-2-фурил]-4-хиназолинамина дитозилата моногидрата - соединения G в виде ярко-желтого кристаллического твердого вещества.
Пример 2
Лечение дозами лапатиниба и доцетаксела или топотекана
Клеточные линии получали из Американской Коллекции Типовых Культур. Клетки поддерживали во флаконах для выращивания тканевых культур в среде RPMI 1640 (Invitrogen #22400-089) с 10% эмбриональной бычьей сыворотки (FBS, HyClone # SH30071.03) и инкубировали при 37° Цельсия в атмосфере 5% СO2 до посева на планшет для определения IC50. Для определения IC50 клетки сеяли в подходящую среду в концентрации 5000 клеток на лунку в 96-луночный планшет для тканевых культур и снова помещали в термостат на ночь. Через приблизительно 24 часа после первоначального посева клетки подвергали воздействию GW 572016 в форме дитозилатной соли, только GW 572016F, только топотекана или доцетаксела, или GW 572016F и топотекана или тоцетаксела в комбинации. Клетки дозировали в 50% RPMI и 50% DMEM (среда Игла в модификации Дульбекко) с низким содержанием глюкозы, содержащей 5% FBS, 50 мкг/мл гентамицина и 0,3% DMSO. Все дозирование осуществляли последовательно и соотношение дозы каждого агента к GW 572016 F корректировали так, чтобы приблизительно отразить относительную эффективность каждого агента в каждой клеточной линии. В большинстве случаев агенты дозировали в одном фиксированном соотношении. В некоторых случаях данные также включают в себя значения Cl, полученные путем дозирования в формате матрицы. Включали значения Cl, полученные в результате дозирования на матрице, когда дозовое соотношение в обоих форматах дозирования составляло 1:1. Смотри формат дозирования ниже.
Через трое суток после воздействия соединения ростовую среду удаляли отсасыванием. Клеточную биомассу оценивали путем окрашивания клеток в 0,1 мл на лунку метиленовым синим (Sigma #M9140, 0,5% в смеси этанол: вода, 50:50) с последующей инкубацией при комнатной температуре в течение по меньшей мере 30 минут. Краситель отсасывали, и планшеты промывали путем погружения в деионизированную воду с последующей сушкой на воздухе. Краситель высвобождали из клеток добавлением 0,1 мл раствора для солюбилизации (1,0% N-лаурилсаркозин, натриевая соль, Sigma #1.5121 в PBS (фосфатно-солевом буфере)). Планшеты инкубировали при комнатной температуре в течение 40 минут. Оптическую плотность измеряли при 620 нм на устройстве Tecan Spectra для считывания микропланшетов. Процент ингибирования клеточного роста подсчитывали относительно необработанных контрольных лунок. Значения IC50 интерполировали, используя метод Левенберга (Levenberg) и Марквардта (Marquardt) (Mager, 1972) и уравнение:
y=Vmax*[1-(xn/(Kn+хn))], где "К" равно IC50.
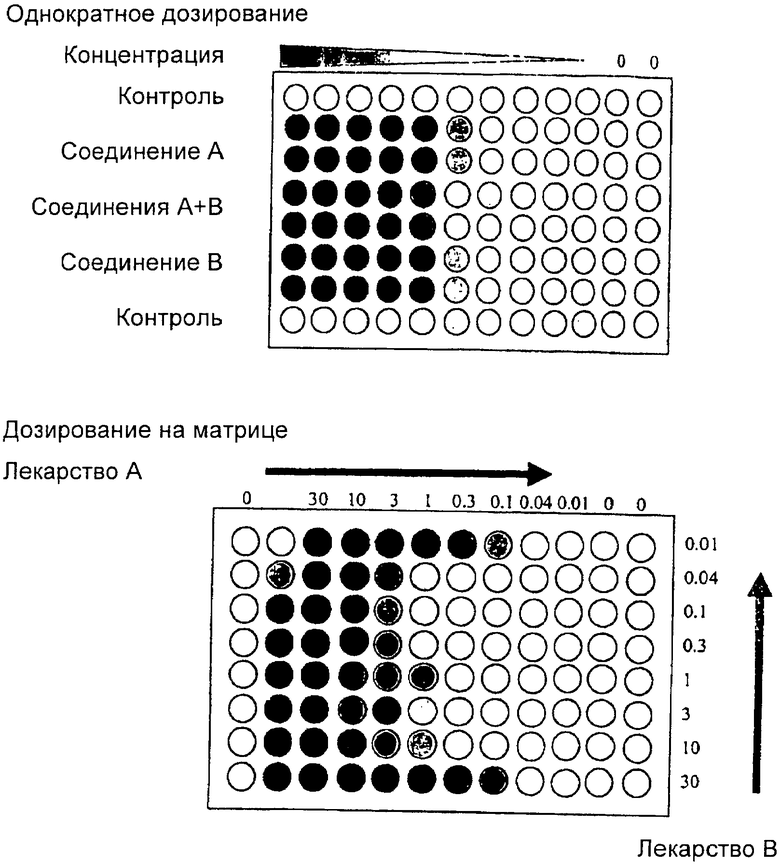
Значения IC50 получали для каждого агента отдельно и в комбинации. Значения IC50 включали в уравнение индекса комбинаций (Сl) по Сhоu и Talalay:
Da,comb/Da+Db,comb/Db, где Da и Db представляют собой значения IC50 каждого агента в отдельности. Da,comb представляет собой количество агента а в комбинации, где эффект составляет 50% ингибирования. Db,comb представляет собой количество агента b в комбинации, где эффект составляет 50% ингибирования. Если агенты дозируют в соотношении 1:1 друг к другу, то Da,comb=Db,comb. Значения более чем 1 означают антагонизм. Значения менее чем 1 означают синергизм. Можно предположить, что степень антагонизма или синергизма отражают путем отличия данного значения от 1,0, то есть 0,5 является более синергическим, чем 0,8, а 2,0 является более антагонистическим, чем 1,5. Важно отметить, что значение 1,0 означает предсказанную аддитивность. Возможно, комбинация дает больший ингибиторный эффект, чем каждый агент в отдельности, но тем не менее считается антагонистической. Это имеет место в тех случаях, когда величина комбинированного эффекта не больше, чем предсказано в математической модели. Другую аналитическую модель разрабатывают для сравнения комбинации с лучшим одиночным агентом. Модель Chou и Talalay также допускает, что индивидуальные агенты действуют независимо или по независимым путям и являются взаимно исключающими. Использование модели, которая допускает, что агенты работают с помощью такого же механизма, как GW 572016 F (взаимно не исключающие), увеличивает некоторые значения Cl, но не изменяет ранжирования агентов в этом наборе данных. Приведенная табл.1 включает в себя значения индекса комбинаций как для взаимно исключающего, так и для взаимно не исключающего определения Cl.
Пример 3
ВТ474 против комбинации лапатиниба и доцетаксела
Мышам с опухолями ВТ474 вводили только лапатиниб (200 и 100 мг/кг; один раз в сутки × 21 сутки, или за 2 суток или 1 сутки до введения доцетаксела) и в комбинации с доцетакселем (25 и 50 мг/кг внутрибрюшинно, один раз в неделю × 3 недели).
В обоих экспериментах высокоэффективным был только доцетаксел в дозе 50 мг/кг и в комбинации с лапатинибом. Однако в обоих экспериментах таксотер в дозе 25 и 50 мг/кг вызывал потерю массы после 3-недельного введения. В первом эксперименте смертей не было, а во втором эксперименте была одна смерть в группе из восьми мышей, получающих доцетаксел (25 мг/кг) и лапатиниб (200 мг/кг × 21 сутки). Все выжившие мыши быстро восстанавливали массу тела после завершения введения доз. Лечебные группы и результаты приведены в табл. 2.
Пример 4
Клиническое исследование лапатиниба в комбинации с трастузумабом
В исследование включали пациентов с распространенным или метастатическим раком молочной железы, при котором наблюдается сверхэкспрессия белка еrbВ2 2+ или 3+, подтвержденная или иммуногистохимией и/или флуоресцентной гибридизацией in situ. Возрастающие дозовые уровни лапатиниба (750-1500 мг) вводили ежесуточно в течение 4 недель раз в квартал в комбинации с еженедельным стандартным введение доз трастузумаба (4 мг/кг ударная доза с последующими еженедельными инфузиями 2 мг/кг). Трех пациентов лечили каждым дозовым уровнем с увеличением до 6 в случае ограничивающей дозу токсичности. Получали ограниченные фармакокинетические образцы, чтобы определить какую-либо корреляцию между пиковыми концентрациями и токсичностью, связанной с лечением. Сообщали по отдельности об одном случае усталости 3 степени и одном случае тошноты 3 степени, ограничивающих дозу, при дозовом уровне 1500 мг/сутки. Диарея 1-2 степеней, анорексия, усталость и сыпь являются распространенными видами токсичности. Оценивание клинического ответа по критериям RECIST (критерии оценки ответа солидных опухолей на терапию) осуществляли на 8 неделе и затем каждые 8 недель.
26 пациентов лечили (группа 750-3; группа 1000-10; группа 1250-10; группа 1500-3). Средний возраст составлял 54 года (30-81). Выполняли семьдесят пять периодов лечения (4 недели = 1 период лечения): медиана 2. У двадцати пациентов оценивали ответ: 4 PR, продолжительность 1-4 месяца; 9 SD, продолжительность 1-5 месяцев и 7 PD, в пределах 1-6 месяцев.
После 152 периодов лечения оценка ответов: 5 PR, продолжительность 1,9; 2,6; 3,9; 5,0+ и 6,7+ месяца соответственно и 1 CR 7.7+ месяца.
CR - полный ответ, определенный как исчезновение целевых поражений;
PR - частичный ответ, определенный как уменьшение по меньшей мере на 30% целевых поражений;
SD - стабилизация заболевания, определяемая как отсутствие роста или некоторое уменьшение целевого поражения.
Пример 5
Клиническое исследование лапатиниба в комбинации с летрозолом
В исследование включали пациентов с распространенным раком молочной железы, положительным в отношении ER (рецепторы эстрогена) или PR (рецепторы прогестерона), или с другими опухолями (например, яичника, эндометрия), которые, вероятно, поддаются комбинированной терапии. Возрастающие дозы лапатиниба (1250-1500 мг) вводили в течение 4 недель раз в квартал в комбинации со стандартной дозой летрозола (2,5 мг/сутки). Трех пациентов лечили каждым дозовым уровнем с увеличением до 6 в случае ограничивающей дозу токсичности (DLT).
Семнадцать пациентов (17 женщин, средний возраст 50, в пределах 32-74 лет; медиана по шкале Карновского PS 90%) включали в исследование при 2 дозовых уровнях (группа 1250 мг - 4 пациента, группа 1500 мг - 13 пациентов). Выполняли тридцать три периода лечения (4 недели = 1 период лечения): медиана 2. Сообщали об одном случае DLT (диарея 3 степени) при дозовом уровне 1500 мг/сутки. Оптимально переносимый режим, как было определено, представлял собой 2,5 мг летрозола + 1500 мг/сутки лапатиниба. Диарея 1-2 степеней, тошнота, сыпь и усталость были распространенными видами негематологической токсичности. Из 16 оцениваемых пациентов у 3 пациентов классифицировали SD в течение ≥2 месяцев (рак молочной железы - 1 пациент, мочевого пузыря - 1 пациент, эндометрия - 1 пациент и шейки матки - 1 пациент), и у 4 пациентов классифицировали PD в пределах 2-4 месяцев.
Пример 6
Клиническое исследование лапатиниба в комбинации с капецитабином
Исследование 2 частей I фазы, объединяющее лапатиниб с капецитабином, осуществляли на 45 пациентах с распространенными солидными опухолями: (А) фаза увеличения дозы (24 пациента) и (В) фармакокинетическая фаза при оптимально переносимом режиме (OTR) (21 пациент): мужчины/женщины (23:22), средний возраст 57 лет (34-78), ECOG (Восточная объединенная онкологическая группа, Eastern Cooperative Oncology Group) (0/1/2:29/13/13), тяжело:легко переносящие предварительное лечение (23:22), типы опухолей (головы и шеи (H&N) (8), молочной железы (8), колоректальный (7), легких (6), другие (16)) и средний цикл 3 (1-9). Пациентов лечили в течение 14 суток капецитабином (С) (1500-2500 мг/м2) и ежесуточно лапатинибом (L) (1250-1500 мг) каждые 3 недели.
Виды ограничивающей дозу токсичности (DLT) представляли собой: мукозит 3 степени, усталость и анорексия - 1250 L/2000 С (n=1); сыпь 3 степени (n=1), диарея 3 степени (n=1) - 1500 L/2000 С и кровоточащий стоматит 2 степени (n=1), диарея 3 степени (n=1) - 1250 L/2500 С. Другие виды токсичности включали в себя стоматит (36%), тошноту/рвоту (30%), диарею (45%), неконъюгированную гипербилирубинемию (14%), усталость (19%), сыпь (38%) и ладонно-подошвенную эритродизестезию (hand foot syndrome) (29%). OTR представлял собой 1250 L/2000 С. Ответы (критерии RECIST) включали в себя 1 CR у женщины с воспалительным раком молочной железы, резистентным к трастузимабу и химиотерапии. Ее опухоль сверхэкспрессировала erbB2 (3+) с низким уровнем TS. Кроме того, наблюдали 4 PR (1 резистентный к эрлотинибу рак H&N; не поддающийся лечению таксаном рак H&N; рак молочной железы; рак желудка) и 6 SD >12 недель.
Пример 7
Комбинация лапатиниба с ингибитором Bcl-2
Исследовали эффект комбинации лапатиниба с ингибитором Bcl-2 (НА14-1) на рост клеток MCF-7 рака молочной железы человека, HER-2/neu-трансфицированных клеток линии MCF-7 и клеток линий MCF-7, резистентных к тамоксифену (ТАМ). Клеточный рост определяли с использованием МТТ-анализа (с использованием тетразолиевого красителя). К клеткам добавляли лапатиниб и НА14-1 в качестве монотерапии. Как лапатиниб (1-10 мкМ), так и НА14-1 (1-10 мкМ) давали дозозависимое ингибирование роста в каждой из 3 клеточных линий. Лечение комбинацией ингибитора EGFR/erbB-2, лапатиниба, и ингибитора Bcl-2, НА14-1, приводило к синергическому ингибированию роста всех 3 клеточных линий.
Настоящее изобретение относится к медицине, а именно к онкологии, и может быть использовано для лечения рака у млекопитающего. Способ по изобретению включает введение моногидрата дитозилатной соли N-{3-хлор-4-[(3-фторбензил)окси]фенил}-6-[5-({[2-(метансульфонил)этил]амино}метил)-2-фурил]-4-хиназолинамина (соединение 1) и капецитабина. Сочетание соединения 1 и капецитабина позволяет снизить риск прогрессирования заболевания за счет их синергического эффекта в отношении ингибирования роста раковых клеток. 2 табл.
Способ лечения рака молочной железы у млекопитающего, включающий введение указанному млекопитающему терапевтически эффективных количеств
1) соединения формулы (I'')
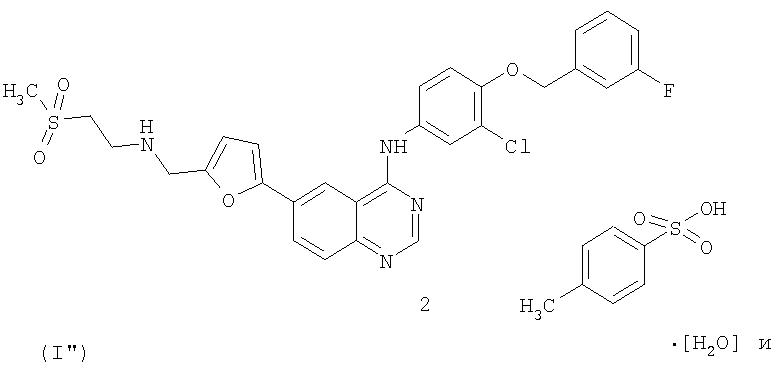
2) капецитабина.
| Прибор, замыкающий сигнальную цепь при повышении температуры | 1918 |
|
SU99A1 |
| СПОСОБ И КОМПОЗИЦИИ ДЛЯ ПЕРОРАЛЬНОГО ВВЕДЕНИЯ ТАКСАНОВ ПАЦИЕНТАМ | 1998 |
|
RU2205005C2 |
| РЛС | |||
| Энциклопедия лекарств, вып.11, 2004, подп | |||
| в печ | |||
| Видоизменение пишущей машины для тюркско-арабского шрифта | 1923 |
|
SU25A1 |
| HOWARD A | |||
| BURRIS, III | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |

