ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Областью изобретения является приготовление сульфатированных полисахаридов с антикоагулянтной антитромботической активностью, начинающееся с полисахаридов микробиологического происхождения.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Природный гепарин представляет собой полимер, имеющий структуру гликозаминогликанов с различными молекулярными массами между 3000 Да и 30000 Да, состоящий из последовательности повторяющихся дисахаридных единиц, состоящих из уроновой кислоты (L-идуроновой или D-глукуроновой) и аминосахара (глюкозамина), связанными друг с другом β-1-4-связями. Уроновая кислота может быть сульфатирована во 2-м положении и глюкозамин может быть N-ацетилирован или N-сульфатирован и 6-O-сульфатирован. Кроме того, глюкозамин может также содержать сульфатную группу в 3-м положении.
Такие замещения являются существенными для образования участка связывания с высокой аффинностью к антитромбину (ATIII) и объясняют антикоагулянтную и антитромботическую активность полимера.
Гепарин является основным антикоагулянтным и антитромботическим агентом для терапевтического применения и вплоть до настоящего времени получаемым путем экстракции из органов животных. В попытке замены этого источника поступления и, следовательно, удовлетворения растущих потребностей в материале, вместе с тем исключающих любое случайное загрязнение со стороны инфекционных агентов, преимущественно вирусов или прионов, в последние годы были разработаны различные способы получения молекул с гепарино-подобной структурой, а также подобными характеристиками, начиная с полисахаридов N-ацетилгепарозанов, имеющих бактериальное происхождение, и таким образом, имеющихся в наличии без количественного ограничения.
Полисахарид N-ацетилгепарозан, выделенный из нескольких природных или рекомбинантных штаммов бактерий Escherichia Coli К5 или из Pasteurella multocida, имеет такую же первичную структуру природного предшественника гепарина, состоящую из повторяющейся последовательности D-глукуроновой кислоты и N-ацетилглюкозамина, соединенных друг с другом α-1-4-связями. Связь между дисахаридными единицами является, напротив, β-1-4.
Уроновая кислота может быть сульфатирована по двум различным положениям и глюкозамин может быть N-ацетилирован или N-сульфатирован и 6-O-сульфатирован. Кроме того, глюкозамин может содержать также сульфатную группу в 3-м положении.
Полисахарид N-ацетилгепарозан, выделенный из E.Coli К5 (Vann W.F., Schmidt М.А., Jann В., Jann К. (1981) в Eur. J. Biochem 116, 359-364), химически модифицировали как описано Lormeau et al. в патенте США N 5550116 и Casu et al. (Carb. Res 263-1994-271-284) или химическим и ферментативным способом в попытке получить продукты, наделенные биологической активностью, сопоставимой с биологической активностью экстрагируемого гепарина.
Кроме того, полуискусственные продукты должны подвергаться процессу деполимеризации для снижения молекулярной массы, что делает продукт более подходящим для различных терапевтических применений, в частности, это улучшает биодоступность и снижает риск кровотечения, связанного с их применением, и другие побочные эффекты.
Химические и ферментативные модификации бактериального полисахарида описываются, например, в патенте Италии N IТ1230785, где полисахарид К5 является N-деацетилированным и N-сульфатированным, кроме того, он подвергается ферментативной C5-эпимеризации глукуроновой кислоты. Такие преобразования сопровождаются другими преобразованиями ферментативного сульфатирования как на уроновой кислоте, так и на аминосахаре.
Патентная заявка WO 92/17509 описывает способ получения гепариноподобных продуктов, начинающийся с полисахарида К5, посредством N-дезацетилирования, N-сульфатирования и ферментативной C5-эпимеризации с последующим химическим O-сульфатированием и необязательно N-сульфатированием.
Патентная заявка WO 96/14425 и патент США N 5958899 описывают способ получения производных полисахарида К5, имеющих высокое содержание идуроновой кислоты, получаемых путем N-дезацетилирования, N-сульфатирования, ферментативной эпимеризации до идуроновой кислоты более чем 50% глукуроновой кислоты с использованием модифицированных буферов для получения предельной вязкости, с последующим сульфатированием по меньшей мере некоторых из свободных гидроксильных групп уроновой кислоты и глюкоэаминных групп.
Патентная заявка WO 97/433117 и патент США N 6162797 описывают приготовление производных К5 с высокими антикоагулянтными и антитромботическими активностями, полученных посредством N-деацетилирования, N-сульфатирования, ферментативной эпимеризации глукуроновой кислоты и O-суперсульфатированием и N-ресульфатированием.
Патентная заявка WO 98/42754, патент США №6197943 и Naggi A. et al. Carbohydrate Research 336 (2001) 283-290 описывают принцип получения сульфатированных гликозаминогликанов, в том числе производных полисахаридов К5, имеющих высокую антитромботическую активность in vitro, путем сольволитического десульфатирования суперсульфатированных предшественников и необязательного 6-O-ресульфатирования.
Патентные заявки WO 0172848 и WO 02/50125 описывают способ получения производных гликозаминогликанов из полисахарида К5, имеющего высокую антикоагулянтную и антитромботическую активность. Способ включает следующие стадии: а) N-деацетилирования, b) N-сульфатирования, с) ферментативной эпимеризации глукуроновой кислоты в идуроновую кислоту, d) суперсульфатирования, е) частичного химического десульфатирования, f) необязательного селективного 6-O-ресульфатирования. Способ характеризуется использованием фермента глюкуронил С5-эпимеразы в процессированной форме, в растворе или иммобилизованной. Кроме того, патентная заявка США N 09/732026 и Li et al. J. Blol. Chem, vol. 276, 213 (2001) 20069-20077 привели к открытию нового мышиного гена экспрессии фермента С5-эпимеразы, содержащего дополнительную последовательность на N-конце, которая делает возможным образование полных форм фермента, имеющих более высокое соотношение активность/стабильность относительно прежней.
КРАТКОЕ ИЗЛОЖЕНИЕ
Данное изобретение относится к способу получения сульфатированных гликозаминогликанов, получаемых из ацетил-N-гепарозана, который включает следующие стадии:
a) N-деацетилирования и N-сульфатирования полисахарида N-ацетилгепарозана, выделенного из природного или рекомбинантного бактериального источника,
b) ферментативной эпимеризации с помощью фермента глюкуронил С5-эпимеразы,
c) частичного O-сульфатирования в сочетании с частичным O-десульфатированием,
d) частичного 6-O-сульфатирования,
е) N-ресульфатирования
дополнительно включающему промежуточную контролируемую стадию деполимеризации, выполняемую альтернативно после стадии b), с) или d), и где такой способ характеризуется тем фактом, что частичное O-сульфатирование на стадии с) осуществляют с использованием молярного соотношения между сульфатирующим агентом и гидроксилами N-ацетилгепарозана, равного 5 или меньшего 5, более предпочтительно меньшего 2,5 или даже более предпочтительно меньшего 1,5, и тем фактом, что частичное 6-O-сульфатирование на стадии d) осуществляют с использованием молекулярного соотношения между сульфатирующим агентом и гидроксилами N-ацетилгепароэана, равного 2 или меньшего 2.
В соответствии с предпочтительным осуществлением промежуточную деполимеризацию выполняют после стадии эпимеризации b).
Как частичное O-сульфатирование, так и частичное 6-O-сульфатирование выполняют с помощью сульфатирующих агентов, выбранных из триэтиламина-SO3, триметиламина-SO3, пиридина-SO3, в апротонном полярном растворителе, предпочтительно не являющимся донором формильных групп, таком как тетраметиленсульфон, 2,4-диметилсульфолан, N,N,-диметилацетамид или N,N,-диэтилацетамид. Необязательно способ включает стадию аффинной селекции на матриксе, несущем антитромбин III или его фрагменты.
Изобретение также относится к сульфатированным гликозаминогликанам K5OS6OSNS-epi, полученным в соответствии с описанным способом, для фармацевтического применения. Эти продукты отличаются степенью 6-O-сульфатирования, превышающей 40%, и предпочтительно от 50% до 85%, очень близкой к значениям экстрагируемого гепарина, и наличием на редуцированном конце остатка безводного маннита, предпочтительно сульфатированного в положениях 1, 3 и 6. Они также отличаются степенью сульфатирования гидроксильной группы в положении 1 и 6 безводного маннита, равной 20% или превышающей 20%, и в соответствии с предпочтительным аспектом, полным отсутствием формильных групп на аминосахаре.
В соответствии с изобретением сульфатированные гликозаминогликаны демонстрируют биологическую активность против фактора Ха в плазме, превышающую биологическую активность биотехнологических гепаринов, полученных в соответствии с предшествующим уровнем техники, и соотношение между анти-Ха-активностью и анти-IIa-активностью, равной 1 или превышающей 1, сходное с экстрагируемыми гепаринами.
Продукты, получаемые в соответствии со способами изобретения:
a) обладают способностью высвобождать ингибитор тканевого фактора (TFPI) из клеток васкулярного эндотелиума, точно также как экстрагируемые гепарины или даже сильнее,
b) являются особенно устойчивыми к деградации гидролитическим ферментом, подобным гепариназе I,
с) обладают способностью ингибировать высвобождение протеаз тромбина и фактора Ха,
d) обнаруживают низкую аффинность для фактора PF4 (4-го тромбоцитарного фактора).
В соответствии с дальнейшим аспектом изобретение относится к биотехнологическим гепаринам (модифицированным N-ацетилгепарозанам), полученным в соответствии со способом изобретения, для терапевтического применения и к фармацевтическим композициям, содержащим такие продукты в виде активных компонентов. В соответствии с последующим аспектом изобретение рассматривает применение продуктов, полученных для приготовления лекарственных средств с антитромботическими и антикоагулянтными гепариноподобными активностями, для приготовления профибринолитических и антиагрегантных лекарственных средств и для приготовления лекарственных средств для профилактики и лечения тромбоэмболитических нарушений, вызванных врожденной или приобретенной нехваткой антитромбина III.
Дальнейший аспект изобретения относится к получению O-сульфатированных промежуточных продуктов K5OSNH2-epi и K5OS6OSNH2-epi, несущих свободную аминогруппу аминосахаров, предпочтительно свободную от формильных групп, причем такие промежуточные продукты могут быть выделены и использованы для получения N-сульфатированных и/или N-ацетилированных производных гепарозана.
ОПИСАНИЕ ФИГУР
Фигура 1. 1С-ЯМР спектр аномерной области полисахарида N-сульфата К5, эпимеризованного как описано в Примере 1.
Фигура 2. 1Н-ЯМР спектр продукта, полученного в Примере 1.
Фигура 3. 1Н-ЯМР спектр продукта, полученного в Примере 2.
Фигура 4. 13С-ЯМР спектр продукта, полученного в Примере 3.
Фигура 5. 13С-ЯМР спектр продукта, полученного в Примере 4.
Фигура 6. 13С-ЯМР спектр продукта, полученного в Примере 5.
Фигура 7. 13С-ЯМР спектр продукта, полученного в Примере 6.
Фигура 8. 13С-ЯМР-спектр продукта, полученного в Примере 7.
Фигура 9. 13С-ЯМР-спектр продукта, полученного в Примере 8.
Фигура 10. 13С-ЯМР-спектр продукта, полученного в Примере 9.
Фигура 11. 13С-ЯМР-спектр продукта, полученного в Примере 10.
Фигура 12. 13С-ЯМР-спектр продукта, полученного в Примере 11.
Фигура 13. 13С-ЯМР-спектр продукта, полученного в Примере 12.
Фигура 14. 13С-ЯМР-спектр продукта, полученного в Примере 13.
Фигура 15. 13С-ЯМР-спектр продукта, полученного в Примере 14.
Фигура 16. 13С-ЯМР-спектр продукта, полученного в Примере 15.
Фигура 17. 13С-ЯМР-спектр продукта, полученного в Примере 16.
Фигура 18. 1Н-ЯМР-спектр продукта, полученного в Примере 18.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В соответствии с главным аспектом настоящее изобретение относится к способу получения сульфатированных гликозаминогликанов, получаемых из N-ацетилгепарозана и названных с целью данного изобретения «биотехнологические гепарины», который включает следующие стадии:
а) N-деацетилирования и N-сульфатирования полисахарида N-ацетилгепарозана, выделенного из природного или рекомбинантного бактериального источника,
b) ферментативной эпимеризации с помощью фермента глюкуронил С5-эпимеразы,
c) частичного O-сульфатирования, в сочетании с частичным O-десульфатированием,
d) частичного селективного 6-O-сульфатирования,
e) N-ресульфатирования,
где этот способ дополнительно включает промежуточную стадию контролируемой деполимеризации, выполняемой альтернативно после стадии b), или с), или d), и где указанный способ характеризуется тем фактом, что O-сульфатирование является частичным, где на стадии с) этого достигают с использованием молярного соотношения между сульфатирующим агентом и гидроксильными группами субстрата (эпимеризованного N-ацетилгепарозана), равного 5 или меньшего 5, более предпочтительно, равного 2,5 или меньшего 2,5, или даже более предпочтительно, равного 1,5 или меньшего 1,5, и времени сульфатирования, меньшего 10 часов. Частичное 6-O-сульфатирование в соответствии со стадией d) получают с использованием молярного соотношения между сульфатирующим агентом и гидроксильными группами субстрата (эпимеризованного N-ацетилгепарозана), равного 2 или меньшего 2, или более предпочтительно, равного 1,5 или меньшего 1,5, и времени сульфатирования, меньшего 2 часов, или даже более предпочтительно, равного 90 минутам или меньшего 90 минут, или даже более предпочтительно, меньше чем 60 минут или равного 60 минутам при температуре, составляющей между 4°С и 30°С, предпочтительно между 10°С и 25°С.
Частичное O-сульфатирование в соответствии со стадией с) и частичное 6-O-сульфатирование в соответствии с этапом d) выполняют с использованием известных сульфатирующих агентов в апротонном полярном растворителе, предпочтительно не являющемся донором формильных групп, более предпочтительно выбранного из N,N,-диалкилацетамида (более предпочтительно N,N,-диметилацетамида или N,N,-диэтилацетамида) и сульфоланов (предпочтительно тетраметиленсульфона или 2,4-диметилсульфолана).
Использование органического растворителя, не являющегося донором формильных групп, связанное с условиями частичного сульфатирования, приводит к продуктам, отличающимся отсутствием формильных групп или их производных на аминосахаре, и к распределению сульфатных групп, сходному с распределением экстрагируемых гепаринов.
В соответствии со способом настоящего изобретения контролируемую деполимеризацию выполняют в качестве промежуточной стадии, которую вводят альтернативно после стадии b), с) или d), a не в финальной фазе как описано в известном уровне техники. Контролируемую деполимеризацию предпочтительно выполняют на эпимеризованном полисахариде N-сульфатгепарозане до или после стадии с) частичного O-сульфатирования. Контролируемая деполимеризация может быть выполнена физическими методами, в том числе обработкой гамма-лучами, или химическими способами, в том числе бета-гамма воздействием с азотистой кислотой или ее солями или обработкой солями йодной кислоты или воздействием свободными радикалами. В соответствии с предпочтительным аспектом деполимеризующим агентом является азотистая кислота, и полисахарид используют в количестве, составляющем от 1 до 100 мг соли/г полисахарида. Реакцию осуществляют при температуре, составляющей от 4 до 10°С. Более предпочтительно, контролируемую полимеризацию осуществляют в течение менее 30 минут в присутствии нитрита натрия и ее завершают прибавлением молярного избытка боргидрида натрия.
Промежуточная деполимеризация делает возможным получение низкомолекулярного продукта, предпочтительно с молекулярной массой, меньшей 15000 Да или равной 15000 Да, более предпочтительно составляющей от 3000 до 9000 Да, несущем остаток ангидроманнита на редуцированном конце, который обнаруживает, кроме того, сульфатирование гидроксила в 6-м положении, аналогично экстрагируемым гепаринам, сульфатирование гидроксилов в положении 1 и 3.
Однако способ является совместимым также с дальнейшей деполимеризацией, выполняемой в конце процесса. Заметили, что при проведении деполимеризации в промежуточной фазе конечные продукты имеют антикоагулянтную и антитромботическую активность и соотношение между анти-Ха и анти-IIа неожиданно выше, чем соотношение, обнаруженное в продуктах с такой же молекулярной массой, но полученных после деполимеризации, выполненной после стадий сульфатирования/десульфатирования и 6-O-сульфатирования, как продемонстрировано данными, представленными в Таблице 1.
В соответствии с предпочтительным способом осуществления изобретения полисахарид N-ацетилгепарозан предпочтительно получают из E.coli K5.
Способ может дополнительно и необязательно включать конечную стадию обогащения продуктов, полученных в результате стадий а)-е), состоящей из аффинной хроматографии на антитромбине III, как описано в Hook et al. FEBS Lett 1976, 66: 90-93.
N-деацетилирование и N-сульфатирование выполняют в соответствии со способами, известными в данной области, которые включают щелочной гидролиз, выполняемый при температуре, составляющей от 30 до 80°С, предпочтительно от 40 до 60°С, в течение периода времени, составляющего между 10 и 30 часами, предпочтительно между 15 и 20 часами, с последующей обработкой в течение периода времени вплоть до 12 часов при 20-65°С сульфатирующим агентом, предпочтительно пиридинсульфотриоксидом, в углекислом натрие.
Эпимеризацию на стадии b) выполняют с помощью природного или рекомбинантного фермента глюкоронил-С5-эпимеразы, предпочтительно в иммобилизованной форме.
Фермент является предпочтительно рекомбинантным ферментом, описанным в заявке WO 98/48006, или даже более предпочтительно ферментом, описанным в патенте США №09/732026, и предпочтительно экспрессированным и очищенным из клеток насекомых или из дрожжевых штаммов, таких как, например, Saccharomyces cerevisiae, Pichia methanolica, Hansenula polymorpha, Saccharomyces pombe, Kluyveromices lactis, Kluyveromices fragilis.
Иммобилизацию фермента предпочтительно выполняют на смолах CNBr Sepharose 4B (Pharmacia) или на полиметакриловых или на полистирольных смолах с эпоксидными группами или диоловыми группами, активированных с использованием CNBr, в буфере 100-300 мМ NaНСО3 или в 10-50 мМ фосфатном буфере при рН 7,0-8,3, более предпочтительно при рН 7,2-7,8, при температуре 4-25°С в течение 12-72 часов.
В соответствии с более предпочтительным аспектом реакцию эпимеризации проводят при температуре, не превышающей 35°С, предпочтительно при температуре, составляющей от 15 до 30°С, более предпочтительно, составляющей от 20 до 25°С.
Эпимеризацию выполняют в соответствии с известными способами, например, способом, описанным в заявке WO 01/72848, предпочтительно при температуре не выше, чем 35°С, более предпочтительно между 15 и 30°С, или даже более предпочтительно, между 20 и 25°С.
Буфером для эпимериэации предпочтительно является раствор HEPES (предпочтительно в концентрации 25 мМ) с рН, составляющим между 5,5-8,0, более предпочтительно между рН 6,5-7,0, и кроме того буфер содержит N-деацетилированный и N-сульфатированный полисахарид, 10-30 мМ ЭДТА, предпочтительно 15-25 мМ, CaCl2 (или альтернативно соли других двухвалентных катионов, например, Zn2+, Ва2+, Мg2+, Мn2+) в концентрации, составляющей между 70 и 150 мМ, более предпочтительно между 75-100 мМ. Раствор термостатируют при температуре, составляющей между 15° и 30°С (предпочтительно 20-25°С), предпочтительно рециркулируют при скорости потока 30-240 мл/час в течение периода времени между 1 и 24 часами. Колонка содержит предпочтительно от 1,2×107 до 3×1011 частей на миллион эквивалентов иммобилизованного фермента на инертном термостатированном носителе.
Приведенные выше определенные рабочие условия, в частности предварительно установленная температура, стабилизируют фермент C5-эпимеразу в течение тысячи часов, делая возможным заметное сохранение времени и реактивов для получения колонки для эпимеризации.
Частичное O-сульфатирование в способе (стадия с) выполняют путем использования известных сульфатирующих агентов, таких как триэтиламин-SO3, триметиламин-SO3, пиридин-SO3, в апротонном полярном растворителе, предпочтительно не являющимся донором формильных групп. Реакцию осуществляют с использованием молярного соотношения между сульфатирующим агентом и гидроксильными группами субстратов, равного 5 или меньшего 5, или предпочтительно равного 2,5 или меньшего 2,5, или более предпочтительно меньшего 1,5, в течение периода времени, равного 10 или меньшего 10 часов, более предпочтительно, равного 8 или меньшего 8 часов, предпочтительно составляющем от 1 до 6 часов, при температуре от 20°С до 70°С, предпочтительно от 30°С до 60°С.
Частичное O-сульфатирование с последующим частичным десульфатированием осуществляют путем воздействия с использованием десульфатирующего агента, такого как ДМСО, в метаноле в течение периода времени, составляющего от 10 до 240 минут, при температуре, составляющей от 45 до 90°С.
Каждая стадия способа может также включать осаждение и/или обессоливание промежуточного полисахарида в соответствии с известными методами.
Частичное 6-O-сульфатирование (стадия d) получают прибавлением сульфатирующего агента в молярном соотношении с гидроксильными группами субстратов, равным 2 или меньшем 2, или предпочтительно меньшем 1,5, в течение периода времени, равного 2 или меньшего 2 часов, или предпочтительно равного 90 или меньшего 90 минут, даже более предпочтительно между 4°С и 30°С, предпочтительно от 10°С и 25°С, в растворе с апротонным полярным растворителем, предпочтительно не являющимся донором формильных групп. В соответствии с альтернативным вариантом способа изобретения частичное 6-O-сульфатирование (стадия d) осуществляют после N-ресульфатирования, и стадии d) и е) выполняют в обратном порядке.
N-ресульфатирование (стадия е) предпочтительно осуществляют в карбонатном буфере прибавлением известного сульфатирующего агента, например, триэтиламина-SO3, триметиламина-SO3, пиридина-SO3.
В заключение необходимо отметить, что способ изобретения демонстрирует следующие инновационные элементы: частичное O-сульфатирование (O-сульфатирование и 6-O-сульфатирование), деполимеризацию, осуществляемую на промежуточной стадии, а не в качестве конечной стадии, и, кроме того, частичное O-сульфатирование и 6-O-сульфатирование осуществляют в апротонном полярном органическом растворителе, предпочтительно не являющимся донором формильных групп.
Производные N-ацетилгепарозана, полученные способом настоящего изобретения, обнаруживают характерную структуру и биологические отличия по отношению к биотехнологическим гепаринам, полученным в соответствии с хорошо известными в данной области способами.
С химической точки зрения полисахариды настоящего изобретения определяют как смесь полисахаридных цепей, представленных следующей общей формулой (I),
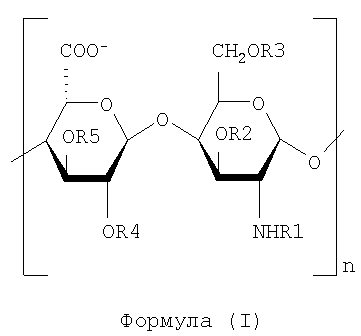
где n имеет пределы от 3 до 150, R1 может быть водородом, SO3-группой или ацетильной группой. R1 не представляет собой другие функциональные группы, такие как формильные группы. R2, R3, R4 и R5 могут быть водородом или SO3-группой, причем предпочтительно R1, R2, R3, R4, R5 являются замещенными как указано ниже:
- R1 от 85 до 97% SO3-группами, и/или от 3 до 15% ацетильными группами, и/или от 0 до 12% H+
- R2 от 15 до 60% SO3 -
- R3 SO3 - - группами до по меньшей мере 40%, предпочтительно от 50% до 85%
- по меньшей мере 20% единиц глукуроновой кислоты не являются сульфатированными в положениях R4 и R5.
В частности, деполимеризованные полисахариды в соответствии со способом изобретения демонстрируют на их редуцированном конце остаток ангидроманнита с одним или несколькими сульфатированными гидроксилами.
Это происходит при осуществлении деполимеризации в присутствии азотистой кислоты или ее производных, например, нитрита натрия, с последующим воздействием боргидридом натрия для получения соединений в соответствии с нижеследующей структурой (II),
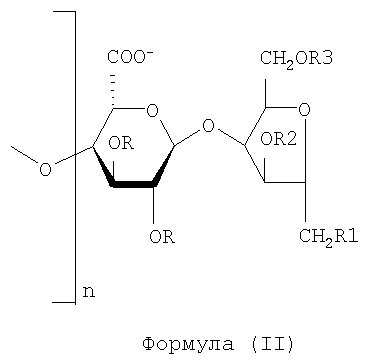
где R1, R2, R3 могут быть водородом или SO3 --группой и где предпочтительно:
- R1 колеблется от 0 до 100% SO3 -
- Р2 от 0 до 100% SO3 -
- R3 от 0 до 100% SO3 -
Предпочтительно R1 и R3 содержат от 20% до 85% SO3 -, и R2 - от 15 до 60% SO3 -.
Предпочтительными продуктами являются продукты с молекулярной массой, меньшей или равной 15000 Да, или предпочтительно составляющей от 1500 до 15000 Да, даже более предпочтительно от 3000 до 9000 Да.
Продукты, полученные в соответствии с изобретением, являются различными с точки зрения структуры по сравнению с продуктами предшествующего уровня техники, исходя из наличия множественных сигналов в области 13С ЯМР-спектра, составляющей от 79 до 89 импульсов в минуту, в частности от 80 до 86 импульсов в минуту, которые присутствуют в избытке по отношению к характерным сигналам ангидроманнита (см. фиг.10), которая показывает 13С ЯМР-спектр пробы при высоком разрешении, приготовленной в примере 9, и которые указывают на наличие сульфатированного в различной степени ангидроманнита, в частности, на гидроксилах в положении 1 и 6, в низкомолекулярных продуктах, приготовленных в соответствии со способом изобретения с использованием промежуточной деполимеризации.
Более конкретно, продукты, полученные в соответствии со способами изобретения, являются различными вследствие сульфатирования гидроксилов в положении 1 ангидроманнита, как показано усилением сигнала в области при 67-68 импульсов в минуту, и уменьшением исчезновения сигнала в области при 61-63 импульсов в минуту в спектре 13С ЯМР.
Различия обнаружены путем сравнения спектра на Фигуре 11 (соответствующего полисахариду, полученному как описано в примере 10) и спектра, показанного на Фигуре 9 (полисахарид, полученный как описано в примере 8). Эти различия являются более очевидными в форме двумерного ЯМР в соответствии со способом, описанным у Guerrini et al. Seminars in Thrombosis and Hermostasis, vol 27, 5, 473-482, 2001.
Главная характеристика этих участков является результатом частичного или полного сульфатирования имеющихся гидроксильных групп ангидроманнита, которые образуются на редуцированном конце полисахарида во время деполимеризации, при ее выполнении перед стадией сульфатирования и, в частности, сульфатированием гидроксилов в положении 1 и 6.
Дополнительной особенностью продуктов изобретения по отношению к продуктам и способу предшествующего уровня техники (например, особенностям, продемонстрированным в экспериментальном примере 6 и 7 относительно данной заявки) является отсутствие сигналов при 7-9,5 импульсов в минуту в спектре 1Н ЯМР и при 51 и 165 импульсов в минуту в спектре 13С ЯМР, что указывает на отсутствие химических групп, отличных от свободной аминогруппы или от ацетильной группы или от сульфатной группы, на глюкозамине во всех продуктах, полученных с низкой или высокой молекулярной массой.
И, наоборот, эти сигналы обычно присутствуют в продуктах, полученных способами, при которых осуществляют сульфатирование N-ацетилгепарозана в органических растворителях, донорах формильных групп, таких как N,N-диалкилформамиды.
Сульфатированные гликозаминогликаны, полученные в соответствии с данным изобретением, обнаруживают антикоагулянтную активность, измерянную в виде активности против фактора Ха в присутствии плазмы, выше чем антикоагулянтная активность биотехнологических гепаринов, полученных в соответствии с известными способами модификации. Кроме того, они обнаруживают соотношение между анти-Ха- и анти-IIа-активностями, равное или превышающее 1, очень сходное с соотношением экстрагируемых гепаринов.
В частности новые продукты демонстрируют:
a) ингибирующую фактор Ха активность выше, чем 50 МЕ/мг, более предпочтительно выше, чем 70 МЕ/мг в тестах, выполненных в присутствии плазмы крови человека. Активность против фактора Ха предпочтительно определяют как описано у Ten Gate H et al. Clin. Chem 3, 860-864 (1984) или в European Pharmacopoeia 1997 3rd edition. В присутствии плазмы эта биологическая активность является поразительно выше, чем активность биотехнологических гепаринов, полученных с использованием способов предшествующего уровня техники, и является сходной с активностью, определенной для экстрагируемых гепаринов с высокой или низкой молекулярной массой,
b) способность к активации TFPI (ингибитора пути тканевого фактора, описанного Bronze GJ Jr et al. Blood 71, 335-343, 1988), равную или превышающую способность к активации TFPI экстрагируемых гепаринов,
c) соотношение между анти-Ха- и анти-IIа-активностями, равное 1 или превышающее 1, при сопоставимой молекулярной массе. Более предпочтительно, соотношение является выше, чем 1,5.
d) устойчивость к перевариванию гепариназой I, равную или превышающую устойчивость экстрагируемых гепаринов,
e) способность к ингибированию высвобождения протеаз тромбина и фактора Ха,
f) низкую аффиность к фактору PF4 (фактор тромбоцитов-4).
Способность к активированию TFPI из сосудистых эндотелиальных клеток усиливает антитромботическую и противовоспалительную активность этих продуктов и расширяет и улучшает терапевтические показания для тромбоза глубоких вен при хирургических процедурах, ишемических осложнений нестабильной стенокардии, инфаркта миокарда и случаев ишемии.
Способность к ингибированию образования протеазы, объединенная с повышенным образованием TFPI, делает возможным дальнейшее распространение терапевтических показаний этих продуктов для лечения сепсиса и его осложнений, таких как синдром диссеминированного внутрисосудистого свертывания (CID) и для лечения заболеваний, вызванных врожденным или приобретенным отсутствием антитромбина III.
Определение активности фактора TFPI после лечения с использованием продуктов настоящего изобретения выполняют, например, in vitro на клетках HUVEC в соответствии со способом, описанным Gory AM et al. Thromb. Haemostasis: 81: 589-593 (1999).
Производные N-ацетилгепарозана, полученные в соответствии с настоящим изобретением, (биотехнологические гепарины) являются особенно устойчивыми к деградации гидролитическими ферментами, такими как гепариназа I. Эта особенность вместе с возможностью получения продуктов с низкой молекулярной массой, которые сами по себе повышают биодоступность и снижают риск геморрагии, связанный с их применением, и коллатеральные эффекты по отношению к высокомолекулярным гепаринам, вместе с высокой активностью против фактора Ха и с низкой степенью сульфатирования, делает возможным их использование не только для парентерального, но также для перорального введения.
Как указано выше, продукт, полученный способом изобретения, обнаруживает способность ингибирования образования протеазы тромбина и фактора Ха.
Ингибирование образования протеаз предпочтительно выполняют в истощенной в отношении фибриноген плазме. Ингибирование образования как тромбина (фактора II), так и фактора Ха является предпочтительно измеряемым с использованием амидолитического метода как для внутренней, так и для наружной систем коагуляции.
В соответствии с обоими способами в обоих использованных системах, продукты, полученные по изобретению, демонстрируют сильную ингибирующую активность как для тромбина, так и для фактора Ха, и это свойство улучшает антитромбиновый профиль этих продуктов. Продукты, полученные в соответствии со способом изобретения, кроме того, являются наделенными низкой аффиностью к фактору PF4 (фактору тромбоцитов 4), которая может быть измерена в плазме в виде остаточной активности против фактора Ха после добавления фиксированного количества фактора PF4 в растворе, содержащем биотехнологические гепарины.
Остаточная активность против фактора Ха, определенная в виде процента относительно исходной активности, является выше, чем активность, полученная с экстрагируемыми гепаринами или с низкомолекулярными экстрагируемыми гепаринами, что указывает на более низкую аффинность к PF4.
Такая более низкая аффинность связывания улучшает клиническую характеристику продуктов, полученных в соответствии с настоящим изобретением, так как она снижает риск появления гепарин-индуцированной тромбоцитопении (HIT). Даже если предпочтительная молекулярная масса получаемого продукта является ниже чем 15000 Да, или более предпочтительно составляет от 3000 до 9000 Да, молекулярная масса продуктов > 15000 Да является легкодостижимой путем изменения условий деполимеризации, тем не менее поддерживающих их биологические свойства, например, высокую активность против фактора Ха, устойчивость к гепариназе и высвобождение фактора TFPI.
В заключение, биотехнологические гепарины, произведенные в соответствии с настоящим изобретением, обнаруживают следующие основные свойства:
- участок, охватывающий от 79 до 89 импульсов в минуту, или более точно, охватывающий от 80 до 86 импульсов в минуту 13С ЯМР, который отличается наличием избыточных многочисленных сигналов относительно характерных сигналов ангидроманнита и усилением сигнала при 67-68 импульсов в минуту и/или ослаблением сигнала или даже исчезновением при 61-62 импульсов в минуту у 13С ЯМР. Эти сигналы указывают на присутствие различно сульфатированного ангидроманнита, в частности сульфатированного в положении 1, 3 и 6 и даже более конкретно, сульфатированного по гидроксилу в положении 1, как подчеркнуто в сравнении между спектрами фигуры 11 и фигуры 9.
- предпочтительное отсутствие сигналов при 7-9,5 импульсах в минуту в спектре 1Н ЯМР и отсутствие сигнала при 51 и 165 импульсах в минуту в спектре 13С ЯМР, что указывает на отсутствие формильных групп.
- активность против фактора Ха (антикоагулянтную) в плазме, которая выше, чем антикоагулянтная активность биотехнологических гепаринов, полученных в соответствии со способами предшествующего уровня техники.
- соотношение анти-Ха-факторная активность/анти-IIa-факторная активность, которое равно или превышает таковое для биотехнологических гепаринов, полученных в соответствии со способами предшествующего уровня техники, предпочтительно выше или равно 1, или значительно более предпочтительно выше или равно 1,5.
- устойчивость к гепариназе, которая равна или выше чем у экстрагируемых гепаринов;
- способность к ингибированию образования тромбина и фактора Ха
- низкую аффинность к PF4.
Биологические активности вновь образованных биотехнологических гепаринов являются специфичными: в частности, соотношение между активностью против фактора Ха и активностью против фактора IIа равно 1 или выше чем 1, обычно соотношение в продуктах, полученных в соответствии с предшествующим уровнем техники, является ниже чем 1, что указывает на оптимальное соотношение между антитромботической и антикоагулянтной активностями, которое образуется из значениий АРТТ. Такое соотношение является сходным с соотношением экстрагируемых гепаринов.
Также оптимальным свойством по отношению к экстрагируемым гепаринам, оцениваемым из высоких значений HCII, является лучшее прямое ингибирование тромбина, что предполагает возможность применения продуктов изобретения при тромбоэмболитических и/или васкулярных нарушениях, вызываемых тромбином, и при приобретенном и при врожденном дефиците антитромбина III.
В соответствии с дальнейшим аспектом настоящее изобретение относится к применению продуктов, полученных в соответствии с описанными способами, в чистом виде или приготовленных в композиции с подходящими фармацевтически приемлемыми наполнителями или растворителями, для антикоагулянтного и антитромботического лечения или профилактики взамен экстрагируемых гепаринов и для получения лекарственных веществ с профибринолитической и антиагрегационной активностью.
Особенно подходящим является применение продуктов настоящего изобретения или композиций, содержащих такие продукты, в качестве активных ингредиентов для профилактики и лечения нестабильной стенокардии, инфаркта миокарда, тромбоза глубоких вен, легочной эмболии, случаев ишемии, а также для лечения сепсиса и для предотвращения его осложнений, например, синдрома диссеминированного внутрисосудистого свертывания (CID).
В соответствии с дальнейшим аспектом настоящее изобретение относится к применению продуктов изобретения для приготовления лекарственных средств для профилактики и лечения нестабильной стенокардии, артериальных тромбозов, атеросклероза и для лечения тромбоэмболитических заболеваний, вызванных врожденным или приобретенным дефицитом антитромбина III.
Продукты могут быть перенесены мицеллами, молекулами-носителями и т.д. и быть в результате особенно подходящими для перрорального применения помимо парентерального применения.
Следовательно, дополнительный аспект изобретения представляют фармацевтические композиции, содержащие в качестве активного ингредиента производные полисахаридов N-ацетилгепарозанов, полученных в соответствии со способами настоящего изобретения, в подходящих композициях как для перрорального, так и для парентерального применения.
В соответствии с дальнейшим аспектом настоящее изобретение относится к приготовлению O-сульфатированных промежуточных продуктов со свободной аминогруппой аминосахара и полностью утративших формильные группы и без какой бы то ни было антикоагулянтной активности, пригодные, например, при приготовлении конечных продуктов изобретения.
Особенно предпочтительным является промежуточный продукт KS-OSNH2-epi, полученный и выделенный в соответствии со способом, описанным в этом изобретении, который характеризуют как смесь полисахаридных цепей, представленных следующей общей формулой (III):
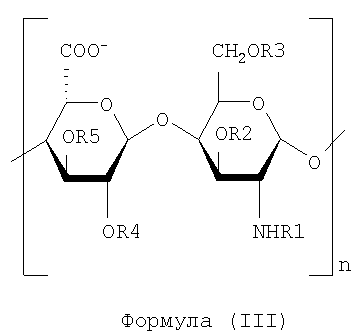
где n имеет пределы от 3 до 150, R1 может быть водородом или ацетильной группой со степенью ацетилирования, находящейся в пределах от 3% до 15%.
R1 не несет ни одну из функциональных групп, предпочтительно R1 не несет формильные группы: R2, R3, R4 и R5 могут быть водородом или SO3-группой, где диапазон сульфатирования предпочтительно составляет от 30% до 98%.
K5OSNH2-epi (1H ЯМР-спектр показан на фиг.16) имеет молекулярную массу, предпочтительно составляющую между 1500 и 15000 Да, или более предпочтительно от 3000 до 9000 Да и характеризуется антикоагулянтной активностью ниже чем 10 МЕ/мг, что определяют по активности против фактора Ха с помощью хромогенного метода (Coatest Heparin kit, Chromogenix).
K5OSNH2-epi является полезным, например, при приготовлении продуктов настоящего изобретения.
K5-OS6OSNH2-epi получают и он может быть выделен в соответствии с настоящим изобретением, его характеризуют как смесь полисахаридных цепей, представленных следующей общей формулой (IV):
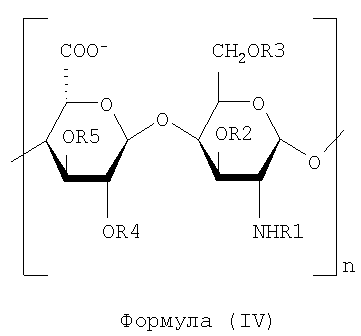
где n имеет пределы от 3 до 150, R1 может быть водородом или ацетильной группой, где степень ацетилирования составляет от 3% до 15%. R1 предпочтительно не несет другие функциональные группы. Предпочтительно R1 не несет формильные группы.
R2, R3, R4 и R5 могут быть водородом или SO3-группой, где R2, R3, R4, R5 являются предпочтительно замещенными как указано ниже:
- R2 от 15 до 60% SO3-
- R3 более чем 40%, предпочтительно от 50 до 85% SO3-
и по меньшей мере 20% единиц глукуроновой кислоты не являются сульфатированными в положениях R4 и R5.
Промежуточный продукт K5-OS6OSNH2-epi (13C ЯМР-спектр показан на фиг.15) имеет молекулярную массу, предпочтительно составляющую от 1500 до 15000 Да, или более предпочтительно от 3000 до 9000 Да. Промежуточный продукт K5-OS6OSNH2-epi отличается свободной аминогруппой на аминосахаре, несульфатирован и предпочтительно свободен от формильных групп. Он также отличается антикоагулянтной активностью, меньшей чем 10 МЕ/мг, что определяют измерением активности против фактора Ха с помощью хромогенного метода (Coatest heparin kit, Chromogenix). Промежуточный продукт K5-OS6OSNH2-epi является пригодным, например, для приготовления N-сульфатированных производных в соответствии с настоящим изобретением и не имеет какой бы то ни было антикоагулянтной активности.
В особенно предпочтительном варианте осуществления изобретения способ получения биогепаринов из бактериальных полисахаридов, например, из N-ацетилгепарозана (полисахарид Е.Coli K5) содержит следующие стадии:
Получение и очистка полисахаридов N-ацетилгепарозанов
Исходный материал является предпочтительно полисахаридом N-ацетилгепарозаном, представленным цепочкой дисахаридных единиц [-4)-GicA α 1-4 GIcNAc-(1-]n, образованными мономерами D-глукуроновой кислоты и N-ацетилглюкозамина, связанными β-1-4-связями. Полисахарид может быть получен, например, из природного штамма К5 Escherichia coli (штамм Вi 83337/41, серотип O10:К5:Н4) (в этом случае полисахарид является К5-полисахаридом) или из бактерии Pasteurella multocida тип D, или из их производных или мутантов, или рекомбинантных штаммов Escherichia coli, полученных, например, как описано у Finke A, et al. Journal of Bacteriology, 173 (13): 4088-4094, 1991, или у Drake CR, Roberts IS, Jann B, Jann К and Boulnois GJ. FEMS Microbiol Lett. 54 (1-3): 227-230, 1990.
Штамм Escherichia coli, подходящий для получения полисахарида К5, может быть получен также из общедоступных коллекций микроорганизмов, например, АТСС (Американская коллекция типовых культур-США) N АТСС 23506.
Штамм Multocida pasteurella тип D может быть получен из коллекции АТСС (АТСС N 12948).
Полисахарид N-ацетилгепарозан получают путем ферментации микроорганизмов и экстракции из культуральной жидкости. Очистку выполняют известными способами, такими как описаны, например, в патентной заявке WO 01/02597, в которой используют следующую культуральную жидкость: 2 г/л муки обезжиренной сои, 9,7 г/л K2HPO4, 2 г/л KH2PO4, 0,11 г/л MgCl2, 0,5 г/л лимоннокислого натрия, 1 г/л сульфата аммония, 2 г/л глюкозы (стерилизованной отдельно), вода очищенная до 1000 мл, рН 7,3.
Предварительную культуру предпочтительно инокулируют с помощью суспензии клеток Е.Coli Bi 8337/41 (O10:К5:Н4), полученной из косяка, хранящегося в триптическом соевом агаре. Предварительную культуру инкубируют при 37°С в течение 24 часов при перемешивании. На последующей стадии ферментер, содержащий упомянутую среду, инокулируют из расчета 0,1% с использованием вышеназванной предварительной культуры и проводят ферментацию в течение 18 часов при 37°С. В течение ферментации производят мониторинг рН, кислорода, остаточной глюкозы, произведенного К5-полисахарида и роста бактерий.
В конце ферментации доводят температуру до 80°С в течение 10 минут. Клетки отделяют от среды путем центрифугирования при 10000 об./мин и супернатант фильтруют через фильтрационные мембраны с пределом отсечения 1000-10000 Да до уменьшения объема приблизительно до 1/5. Затем К5-полисахарид осаждают добавлением 4 объемов ацетона и извлекают центрифугированием.
Депротеинизацию осадка предпочтительно выполняют с использованием протеазы II типа из Aspergillus Orizae в буфере, содержащем 0,1 М NaCl и 0,15 М ЭДТА, рН 8, 0,5% додецилсульфат натрия (SDS), при 37°С в течение 90 минут.
Раствор подергают ультрафильтрации с использованием мембран с пределом отсечения 10000 Да и затем полисахарид осаждают с помощью ацетона. Чистота полисахарида обычно составляет около 80%, и чистоту определяют по меньшей мере одним из следующих аналитических методов: определением уроновых кислот (карбазольный метод), протонного МР и 13С ЯМР, УФ и/или по содержанию белка.
a) N-деацетилированио и N-сульфатирование полисахарида N-ацетилгепарозана из микроорганизмов
Часть, предпочтительно содержащую от 5 до 10 г очищенного К5-полисахарида, солюбилизируют в 200-2000 мл 2N гидроксида натрия и оставляют для прохождения реакции при 40-80°С до завершения деацетилирования (то есть 15-30 часов). Раствор доводят до нейтрального значения. Раствор, содержащий деацетилированный K5-полисахарид, выдерживают при 20-65°С, и добавляют в один прием 10-40 г углекислого натрия, а также 10-40 г сульфатирующего агента, выбранного из таких реагентов, как аддукты пиридинсульфотриоксида, триметиламинсульфотриоксид и т.д.
Сульфатирующий агент добавляют за период времени до 12 часов. В конце взаимодействия, если необходимо, раствор доводят до комнатной температуры и до значения рН, составляющего от 7,5 до 8.
Продукт очищают от солей известными способами, такими как, например, диафильтрацией с помощью спиральных мембран 1000 Да (препаративный картридж Millipore). Оставшийся продукт уменьшают в объеме до получения 10% концентрации полисахаридов. Концентрированный раствор, если необходимо, может быть высушен с помощью известных способов.
Отношение N-сульфатированного/N-ацетилу определяют с помощью 13С ЯМР.
b) Ферментативная эпимеризация с помощью глюкуронил С5-эпимеразы
Стадию С5-эпимеризации, которая заключается в эпимеризации части глукуроновой кислоты в идуроновую кислоту, выполняют с помощью фермента глюкуронил-С5-эпимеразы (называемую С5-эпимеразой) природного или рекомбинантного, или в растворе, или, предпочтительно, в иммобилизованной форме.
Для этой стадии используют рекомбинантный фермент С5-эпимеразу, как описано в заявке WO 98/48006. Предпочтительно, рекомбинантный фермент модифицирован, как описано в заявке на патент США №09/732026 и у Li et al. J. Biol. Chem, vol 276, 23, (2001) 20069-20077), и содержит дополнительную последовательность на его N-терминальном конце.
Рекомбинантный фермент предпочтительно экспрессируют и очищают из клеток насекомых или клеток дрожжей, предпочтительно принадлежащих к видам Saccharomyces cerevisiae, Pichia pastoris, Pichia methanolica, Hansenula polymorpha, Saccharomyces pombe, Kluyveromices lactis, Kluyveromices fragilis.
b.1) Иммобилизация С5-эпимеразы на смоле.
Рекомбинантный фермент может быть иммобилизован на различных инертных матрицах, таких как смолы, мембраны или стеклянные шарики, модифицированных с помощью функциональных групп известными способами, такими как цианобромид, глутаральдегид, карбодиимид, или позволяя ферменту взаимодействовать с ионообменной смолой, или позволяя ему абсорбироваться на мембранах.
В соответствии с предпочтительной реализацией фермент иммобилизуют на коммерческих смолах, таких как CNBr Sepharose 4B (Pharmacia) или на полистирольных или полиметакриловых смолах (Resindion Mitsubishi) с эпоксидными или диол-СNВr-активированными группами.
Особенно предпочтительной в соответствии с изобретением является иммобилизация фермента на полиметакриловой смоле с диольными группами, активированной CNBr, в буфере 100-300 мМ NaHCO3 при рН 7,0-8,3, предпочтительно рН 7,2-7,8, при температуре 4-25°С в течение 12-72 часов.
В соответствии с настоящим изобретением реакцию связывания фермента с инертным матриксом выполняют в присутствии N-деацетилированного N-сульфатированного К5-субстрата во избежание такого связывания, которое заключается в связывании активного сайта фермента с потерей активности.
Определение активности иммобилизованного фермента выполняют рециркуляцией через колонку, содержащую иммобилизованный фермент, определенного количества N-деацетилированного N-сульфатированного К5, теоретически превращаемое в cpm (число/минуту) иммобилизованного фермента, растворенного в буфере 25 мМ HEPES, 0,1 М КСl, 0,01% Triton Х-100 и 0,15 М ЭДТА при рН 7,4 при 37°С в течение 24 часов при потоке 0,5 мл/минуту. После очистки посредством метода DEAE-хроматографии и обессоливания на Sephadex G-10 продукт лиофилизируют и анализируют на содержание идуроновой кислоты методом протонного ЯМР в соответствии с заявкой WO 96/14425.
b.2) Эпимеризация с помощью иммобилизованного фермента
Реакция C5-эпимеризации может быть осуществлена, например, как описано в заявке WO 96/14425, в буфере для реакции при рН 7,4, содержащем предпочтительно 0,04 М HEPES или 0,05 М Tris, 0,4 M KCl, 0,06 M ЭДТА и Triton X-100 и одну или несколько добавок, таких как, в частности, глицерин или поливинилпирролидон.
Реакция может быть осуществлена как описано в заявке WO 01/72848, где используют раствор, содержащий 25 мМ HEPES, 50 мМ CaCl2, рН 7,4, при температуре 30-40°С.
Особенно предпочтительными являются температурные условия реакции и буфера, которые позволяют активности глюкуронил-С5-эпимеразы быть продолжительной и стабильной даже после иммобилизации на колонке.
20-1000 мл водного раствора, содержащего 0,001-10 г N-деацетилированного N-сульфатированного К5 и 10-30 мМ ЭДТА, предпочтительно 15-25 мМ, 25 мМ HEPES, CaCl2 в концентрации от 70 до 150 мМ (предпочтительно 75-100 мМ) при рН 5,5-8,0, предпочтительно рН 6,5-7,0 и термостатированного при температуре, составляющей от 15°С до 30°С (предпочтительно 20-25°С), предоставляют возможность циркуляции при потоке 30-240 мл/час в течение периода времени, составляющего от 1 до 24 часов, в колонке, содержащей от 1,2×107 и 3×1011 эквивалентов фермента, иммобилизованного на термостатированной инертной среде, при температуре, составляющей от 15°С до 30°С, предпочтительно от 20 до 25°С.
Вышеуказанные температурные и буферные условия повышают стабильность фермента на колонке в течение периода работы более чем 3000 часов и, следовательно, делают способ особенно выгодным. В конце реакции пробу очищают пропусканием через смолу DEAE или картридж Sartobind DEAE и осаждают добавлением 2 М NaCl и, наконец, обессоливают на смоле Sephadex G10 (Pharmacia) или очищают преципитацией 2 объемами этанола и пропусканием через смолу IR 120H+ для получения натриевой соли.
Продукт, полученный при указанных предпочтительных условиях, имеет степень эпимеризации, определенную способом протонного ЯМР, описанным в заявке WO 96/14425, по меньшей мере 50% (отношение идуроновой кислоты к суммарным уроновым кислотам).
Контролируемая деполимеризация.
Продукт, полученный на стадиях b), с) или d), подвергают контролируемой деполимеризации известными способами, такими как дезаминирование в присутствии азотистой кислоты, как описано в заявке WO 82/03627, или окислительным размыканием с периодатом натрия (ЕР 2874770), или воздействием свободными радикалами (ЕР 121067), или бета-элиминацией (ЕР 40144), или воздействием гамма-лучами (патент США №4987222) для получения фракций с молекулярной массой, предпочтительно составляющей от 1500 до 15000 Да, или даже более предпочтительно составляющей от 3000 до 9000 Да.
В соответствии с предпочтительным аспектом настоящего изобретения контролируемую деполимеризацию выполняют перед стадиями сульфатирования.
В частности, продукт, полученный в результате вышеупомянутых стадий, помещают в условия контролируемой деполимеризации в присутствии азотистой кислоты или в присутствии нитрита натрия. В этом случае используемое количество соли составляет от 1 до 100 мг на каждый грамм полисахарида, с последующим редуцированием избыточным боргидридом.
В соответствии со вторым предпочтительным вариантом осуществления пробу растворяют в 50-250 мл воды при 4°С для подкисления с помощью 1N соляной кислоты. Затем добавляют определенное количество нитрита натрия, составляющее от 5 до 500 мг, и реакцию продолжают в течение менее 60 минут, предпочтительно меньше чем 30 минут.
После распада избыточного боргидрида натрия продукт получают осаждением с использованием 3 объемов этанола и высушивают в вакуумной горячей сушке.
При осуществлении деполимеризации в конце способа она может быть выполнена как описано, например, в заявке WO 01/72848.
с) Частичное O-сульфатирование в сочетании с частичным O-десульфатированием.
Продукт, полученный в результате вышеупомянутых стадий, ресуспендируют в воде при концентрации 10%. Раствор охлаждают при 10°С и пропускают, поддерживая температуру 10°С, через катионообменную смолу IR-120 Н+. После пропускания раствора смолу промывают деионизированной водой до значения рН элюата, превышающего 6. Кислый раствор доводят до нейтрального значения прибавлением третичного амина или четвертичной аммониевой соли, например, 15% водного раствора гидроокиси тетрабутиламмония, получая подходящую аммониевую соль. Раствор может быть скоцентрирован до минимального объема и лиофилизирован.
Полученный продукт суспендируют в 10-1000 мл органического растворителя, состоящего предпочтительно из сульфолана или 2,4-диметилсульфолана. Альтернативно, органическим растворителем может быть диметилформамид (DMF), диметилсульфоксид (DMSO) или N,N,-диметилацетамид. Такой органический растворитель добавляют вместе с сульфатирующим агентом, таким как аддукт пиридин-SO3, в твердой форме или в виде раствора с теми же самыми растворителями, которые использовали выше.
Молярное соотношение между сульфатирующим агентом и полисахаридным субстратом (эпимеризованным N-сульфатированным К5), под которым подразумевают соотношение между сульфатирующим агентом и гидроксильными молями полисахаридного димера, поддерживают равным или меньшим 5, или более предпочтительно меньшим 2,5, или даже более предпочтительно меньшим 1,5.
Раствор поддерживают при температуре, составляющей от 20 до 70°С, предпочтительно составляющей от 30 до 60°С, в течение периода времени, меньшим 10 часов или равном 10 часам, более предпочтительно меньшим 8 часов или равном 8 часам, или даже более предпочтительно меньшим 6 часов или равном 6 часам. В конце реакции раствор в конечном счете охлаждают при комнатной температуре и доводят с помощью ацетона, насыщенного хлоридом натрия, до полного осаждения полисахарида.
Осадок отделяют от растворителя фильтрацией, растворяют в минимальном количестве деионизированной воды и доводят с помощью хлорида натрия до получения 0,2 М раствора. Раствор доводят до рН 7,5-8 добавлением 2N гидроксида натрия. Затем добавляют ацетон до полного осаждения. Осадок отделяют от растворителя фильтрацией. Полученное твердое вещество растворяют с помощью 10-100 мл деионизированной воды и очищают от остаточных солей посредством ультрафильтрации.
Аликвоту лиофилизируют для структурного анализа частично O-сульфатированного продукта с помощью 13С ЯМР и 1Н ЯМР.
Раствор, содержащий частично сульфатированный продукт, пропускают через катионообменную смолу IR-120 H+ или ее эквивалент.
Смолу промывают деионизированной водой до значения рН элюата, превышающего 6, и затем добавляют пиридин. Раствор концентрируют до минимального объема и лиофилизируют. Продукт обрабатывают 20-200 мл смеси DMSO/метанол (9/1 об./об.) и раствор хранят при 45-90°С в течение 10-420 минут. В конце раствор смешивают с 10-200 мл деионизированной воды и обрабатывают ацетоном, насыщенным хлоридом натрия, до полного осаждения.
Полученное твердое вещество очищают с помощью диафильтрации в соответствии с известными методами, и аликвоту лиофилизируют для структурного анализа 13С ЯМР.
d) Частичное 6-O-сульфатирование
Затем продукт с предыдущей стадии подвергают 6-O-сульфатированию. Содержание сульфатных групп в 6-O-положении определяют известными способами, например, ЯМР в соответствии с условиями, описанными Guerrini et al. Seminars in Thrombosis and Hemostasis, vol 27, 5, 473-482, 2001.
Полученный продукт ресуспендируют в воде при концентрации, составляющей от 5 до 10%, и хранят при комнатной температуре. Затем раствор пропускают через катионообменную смолу IR-120 H+ или ее эквивалент. После прохождения раствора смолу промывают деионизированной водой и доводят до нейтрального значения с помощью третичного амина или четвертичной аммониевой соли, таких как гидроокись тетрабутиламмония в водном растворе, получая подходящую аммониевую соль. Раствор концентрируют до минимального объема и лиофилизируют.
Полученный продукт суслендируют в 10-100 мл органического растворителя, содержащего диметилформамид, предпочтительно в сульфолане или в 2,4-диметилсульфолане или в N,N,-диметилацетамиде, и смешивают с сульфатирующим агентом, таким как аддукт пиридин-SO3, в твердой форме или в виде раствора с тем же самым растворителем.
Суспензию доводят до температуры, составляющей от 4°С до 30°С, предпочтительно от 10°С до 25°С, обрабатывают определенным количеством сульфатирующего агента, такого как аддукт пиридин-SO3, с использованием менее чем 2 эквивалентов по сравнению с гидроксильными группами, которые должны сульфатироваться, или даже более предпочтительно менее чем с 1,5 эквивалентами, в течение 10-90 минут и обрабатывают ацетоном, насыщенным хлоридом натрия, в количестве, необходимом для полного осаждения. Затем полученное твердое вещество очищают диафильтрацией в соответствии со способами, хорошо известными в данной области. Аликвоту лиофилизируют для структурного анализа методом 13С ЯМР.
е) N-ресульфатирование
Раствор со стадии d), содержащий 6-O-сульфатированный полисахарид, доводят до 20-65°С и прибавляют 10-100 г карбоната натрия в виде однократной добавки и 10-100 г сульфатирующего агента, выбранного из имеющихся реагентов, таких как предпочтительно пиридинсульфотриоксид. Прибавление сульфатирующего агента выполняют в течение различного периода времени, вплоть до 12 часов. В конце реакции, если необходимо, раствор доводят до комнатной температуры и до значения рН, составляющего от 7,5 до 8, предпочтительно с использованием 2М гидроксида натрия.
Продукт очищают от солей известными способами, такими как, например, диафильтрация с использованием спиральной мембраны 1000 Да (т.е. Prepscale cartridge - Millipore). Процесс завершают при проводимости пермеата ниже 1000 мкСм, предпочтительно ниже 100 мкСм. Полученный продукт уменьшают в объеме фильтрацией до концентрации полисахаридов 10%. Аликвоту с концентрированного раствора лиофилизируют для структурного анализа 13С ЯМР.
Избирательное 6-O-сульфатирование и N-ресульфатирование, выполненное как описано в этой стадии, на продуктах, полученных на стадии е), может быть выполнено в различной последовательности, например, сначала N-ресульфатирование, а затем 6-O-сульфатирование, как описано в заявке WO 98/42754, без изменения биологических активностей конечного продукта.
Обогащение последовательностями, связывающими антитромбин III (необязательно).
Продукт, полученный как описано на предыдущей стадии, может быть необязательно дополнительно очищен хроматографией, например, на анионообменной хроматографической колонке, такой как, например, DEAE-колонки как описано у Lam L.H. et al. Bloch. And Biophysical Research Communication vol 69, 2, p.570-577, 1976. Альтернативно такой очистке или помимо нее продукт может быть подвергнут дополнительной аффинной хроматографии на колонках, содержащих целую или частичную последовательность антитромбина человека как описано, например, в Hook et al FEBS Lett 1976, 66: 90-93 или в патенте США N 4692435, или пептидные последовательности, имеющие высокую аффинность к гепарину, как описано в Liu S et al. Proc. Natl. Acad Sci USA 1980, 77: 6551-6555. Такое воздействие делает возможным отделение последующей элюцией солевым раствором NaCl, по меньшей мере фракции, способной связываться с твердой фазой.
Практически, 10-50 мг продукта, полученного со стадии N-ресульфатирования, наносят на аффинную колонку, на которой иммобилизовано 50-100 мг антитромбина III человека (Kedrion SpA, Lucca, Italia) в буфере 10 мМ Tris-HCl pH 7,4 и 0-0,15 M NaCl, 4°C. Затем колонку промывают по меньшей мере 3-мя объемами буфера 10 мМ Tris-HCl, pH 7,4.
Молекулы, связанные с высокой степенью аффинности к колонке, элюируют 10 мМ Tris-HCl pH 7,4, содержащим от 0,5 до 3 М NaCl.
Затем элюированный материал предпочтительно диафильтруют через спиральные мембраны с пределом отсечения 1000 Да для удаления солей и концентрируют лиофилизацией.
Аликвота может быть проанализирована карбазольным методом, ВЭЖХ, ЯМР и хромогенной реакцией для определения активности против фактора Ха.
Полученный материал демонстрирует более высокую активность против фактора Ха, улучшенную от 1,5 до 3 раз относительно активности исходного материала.
Такое же увеличение активности против фактора Ха получают при обработке таким же образом экстрагируемых гепаринов. Это является дополнительным знаком сходства биотехнологических гепаринов, полученных в соответствии с настоящим изобретением, с экстрагируемыми гепаринами в связывании с антитромбином III.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
ПРИМЕР 1. Получение биотехнологических гепаринов в соответствии со способом изобретения.
Придерживались следующих стадий:
a) начального получения полисахарида N-ацетилгепарозана из Esherichia coli K5,
b) N-деацетилирования/N-сульфатирования,
c) эпимеризации,
d) деполимеризации,
e) частичного O-сульфатирования/частичного О-десульфатирования,
f) частичного 6-O-сульфатирования,
g) N-ресульфатирования.
а) Получение полисахарида
Полисахарид N-ацетилгепарозан получали посредством ферментации штамма В1 8337/41 E.coli, серотип O10:k5:H4 (ATCC 23506), и последующей экстракции из культуральной среды и очистки в соответствии с описанием в патентной заявке WO 01/02597 с использованием следующей культуральной жидкости: 2 г/л обезжиренной соевой муки, 9,7 г/л К2НРO4, 2 г/л КН2PO4, 0,11 г/л MgCl2, 0,5 г/л лимоннокислого натрия, 1 г/л сульфата аммония, 2 г/л глюкозы (стерилизованной отдельно), очищенная вода до 1000 мл, рН 7,3.
Культуру инокулировали суспензией клеток, полученных из косяка, хранящегося в триптическом соевом агаре, при 37°С в течение 24 часов при перемешивании. Инокулят в ферментере, тип 5 (industrie Meccaniche di Bagnolo SpA), содержащий такую же, как указано, выше среду, засевали при 0,1% с помощью вышеуказанной предварительной культуры, и ферментацию проводили при температуре 37°С в течение 18 часов. В течение ферментации измеряли рН, кислород, остаточную глюкозу, произведенный полисахарид K5 и рост бактерии. В конце ферментации температуру поднимали до 80°С на 10 минут. Клетки отделяли от ростовой среды центрифугированием при 10000 об./мин и супернатант фильтровали с использованием фильтрационных мембран с пределом отсечения 10000 Да до уменьшения объема до 1/5. Полисахарид K5 осаждали добавлением 4-х объемов ацетона и в конечном счете выделяли центрифугированием.
Депротеинизацию полученного твердого вещества проводили протеазой II-го типа из Aspergillus Orizae в буфере 0,1 М NaCl и 0,15 М ЭДТА, при рН 8, содержащем SDS 0,5%, 37°С в течение 90 минут.
Полученный раствор подвергали ультрафильтрации с использованием мембран, имеющих номинальный уровень отсечения 10000 Да, и полисахариды осаждали с использованием ацетона. Чистоту полисахарида измеряли: определением уроновых кислот (карбазольным методом), протонным и углеродным 13 ЯМР, УФ и содержанием белка.
b) N-деацетилирование/N-сульфатирование
10 грамм продукта, полученного на стадии а), растворяли в 200 мл 2N гидроксида натрия и оставляли при 50°С в течение 18 часов. Раствор доводили до нейтрального рН с помощью 6N соляной кислоты. Получали N-деацетилированный полисахарид.
Раствор N-деацетилированного полисахарида выдерживали при 40°С и прибавляли 10 г углекислого натрия в виде отдельной добавки и 10 г аддукта пиридинсульфотриоксид в течение 10 минут. Полученный продукт, готовый N-деацетилированный N-сульфатированный полисахарид К5, очищали от солей диафильтрацией с использованием спиральной мембраны 1000 Да (препаративный картридж - Millipore). Процесс очистки завершали при проводимости пермеата ниже 100 мкСм.
Продукт доводили до 10% концентрации полисахарида с использованием такого же способа диафильтрации и затем лиофилизировали.
Соотношение N-сульфат/N-ацетил полученного продукта составляло 9,5/0,5 при определении посредством углеродного 13 ЯМР.
c) Эпимеризация
с-1) Иммобилизация С5-эпимеразы на смоле
5 мг рекомбинантной глюкуронил-С5-эпимеразы, полученной в соответствии с заявкой на патент США №09/732026 и Li et al J. Biol. Сhem., vol 276, 23, (2001) 20069-20077, растворяли в 200 мл буфера 0,25 М HEPES, рН 7,4, содержащего 0,1М KCl, 0,1% Triton X-100 и 15 мМ ЭДТА, 100 мг N-деацетилированного N-сульфатированного К5, полученного в соответствии с описанием на стадии b), прибавляли к раствору. Раствор подвергали диафильтрации через мембрану 30000 Да при 4°С до исчезновения N-деацетилированного N-сульфатированного К5 в диафильтрате. В растворе, задержанном мембраной, буфер был заменен диафильтрацией и замещением 200 мМ NaHCO3 при рН 7, и после концентрирования до 50 мл добавляли 50 мл смолы - CNBr-активированной Sepharose 4В, которую оставляли для взаимодействия в течение ночи при 4°С.
В конце взаимодействия определяли количество остаточного фермента в супернатанте с помощью метода Quantigold (Diversified Biotec) после декантации. В супернатанте фермент отсутствовал, что указывало на то, что с помощью описанного способа фермент был иммобилизован на 100%. Для заполнения сайтов смолы, оставшихся незанятыми, смолу промывали буфером 100 мМ Tris-HCl, при рН8.
Для измерения активности иммобилизованного фермента определенное количество иммобилизованного фермента, теоретически соответствующее 1,2×107 частей на миллион, наносили на колонку. В колонку, приготовленную таким образом, вносили 1 мг N-деацетилированного N-сульфатированного К5, полученного в соответствии со стадией b) и растворенного в буфере 25 мМ HEPES, 0,1 М KCl, 0,015 М ЭДТА, 0,01% Triton Х-100 при рН 7,4, предоставляя возможность его рециркуляции через указанную колонку при 37°С в течение ночи со скоростью потока 0,5 мл/минуту.
После очистки методом DEAE-хроматографии и обессоливания на Sephadex G-10 пробу лиофилизировали и анализировали на содержание идуроновой кислоты с помощью метода протонного ЯМР в соответствии с описанием в заявке на патент WO 96/14425.
с-2) Эпимеризация с помощью иммобилизованного фермента.
10 г N-деацетилированного N-сульфатированного полисахарида К5 растворяли в 600 мл буфере 25 мМ HEPES, рН 7,0, 15 мМ ЭДТА, содержащем 75 мМ CaCl2. Полученному раствору предоставляли возможность рециркуляции через 50 мл колонку, заполненную смолой, несущей иммобилизованный фермент.
Эту процедуру проводили при 28°С со скоростью потока 200 мл/час в течение 24 часов.
Полученный продукт очищали ультрафильтрацией и осаждали этанолом.
Преципитат перерастворяли в воде при концентрации 10%.
Полученный продукт демонстрировал 55% процентов эпимеризации, измеряемой с помощью 1Н ЯМР, как процентное соотношение идуроновой кислоты к общим уроновым кислотам (как показано на фиг.1).
d) Контролируемая деполимеризация.
Пробу, полученную со стадии с-2), подвергали контролируемой деградации с использованием нитрозной кислоты, как описано в заявке на патент WO 82/03627. В частности, 5 г пробы растворяли в 250 мл воды и доводили до 4°С с использованием термостатированной бани. рН доводили до рН 2,0 с помощью 1 N соляной кислоты при 4°С и затем добавляли 200 мг нитрита натрия. При необходимости рН доводили до 2 с использованием 1 N соляной кислоты и выдерживали при слабом перемешивании в течение 15 минут. Раствор нейтрализовали с помощью 1 N NaOH, охлаждали при 4°С.
Добавляли 250 мг боргидрида натрия, растворенного в 13 мл деионизированной воды, оставляя для взаимодействия в течение 4 часов. Раствор доводили до рН 5,0 с использованием 1 N соляной кислоты и оставляли на 10 минут для разрушения избытка боргидрида натрия, и затем раствор нейтрализовали с помощью 1 N NaOH. Продукт извлекали преципитацией 3-мя объемами этанола и затем высушивали в вакуумной горячей сушке. Полученный продукт обнаруживал молекулярную массу около 6000 Да.
е) Частичное O-сульфатирование/частичное O-десульфатирование
Продукт, образовавшийся на предыдущей стадии, ресуспендировали из расчета 10% концентрации в водном растворе. Раствор охлаждали при 10°С и пропускали при температуре 10° через катионообменную смолу IR-120+. После протекания этого раствора смолу промывали деионизированной водой до тех пор, пока рН элюата не становился выше 6. Затем кислый раствор нейтрализовали с использованием третичного амина или четвертичной аммониевой соли, такой как тетрабутиламмоний гидроксид в водном растворе при 15%, получая в результате аммониевую соль. Затем раствор концентрировали до минимального объема и лиофилизировали.
Образовавшийся в результате раствор ресуспендировали в 100 мл N,N,-диметилацетамида (DMA) и добавляли пиридин-SO3.
Затем добавляли определенное количество сульфатирующего агента в молярном соотношении 1,25 между сульфатирующим агентом и субстратом эпимеризованного N-сульфата К5 (в качестве молей гидроокисла).
Раствор выдерживали при 50°С в течение 360 минут. В конце реакции раствор охлаждали при комнатной температуре и добавляли ацетон, насыщенный хлоридом натрия, до полного осаждения.
Осадок отделяли от растворителя фильтрацией, растворяли в минимальном количестве деионизированной воды и добавляли хлорид натрия до получения 0,2 М раствора. Раствор доводили до рН 7,5 добавлением 2 N гидроксида натрия и добавляли ацетон для предоставления возможности преципитации. Затем преципитированный раствор отделяли от растворителя посредством фильтрации. Твердое вещество раствора, полученное таким образом, солюбилизировали посредством добавления 100 мл деионизированной воды и очищали от остаточных солей ультрафильтрацией.
Аликвоту лиофилизировали для структурного анализа частично O-сульфатированного продукта с помощью 13С ЯМР.
Раствору, содержащему частично сульфатированный продукт, предоставляли возможность протекания через катионообменную смолу IR-120+ или ее эквивалент. После пропускания этого раствора смолу промывали деионизированной водой до тех пор, пока постоянное значение рН не становилось выше 6. Кислый раствор доводили до нейтрального значения добавлением пиридина. Раствор концентрировали до минимального объема и лиофилизировали. Полученный продукт обрабатывали 100 мл смеси DMSO/метанол (9/1 об./об.) и полученный раствор выдерживали при 65°С в течение 240 минут.
Наконец, в раствор добавляли 200 мл деионизированной воды и затем обрабатывали ацетоном, насыщенным хлоридом натрия, в количестве, необходимом для полноты преципитации. Полученное твердое вещество очищали диафильтрацией в соответствии с известными способами и аликвоту лиофилизировали для структурного анализа 13С ЯМР.
f) частичное 6-O-сульфатирование
Затем продукт, полученный на стадии е), ресуспендировали в водный раствор при концентрации 10% и хранили при комнатной температуре. Раствор пропускали через катионообменную смолу IR-120+. Затем смолу промывали деионизированной водой и доводили до нейтрального значения посредством тетрабутиламмония гидроксида в водном растворе, получая аммониевую соль. Затем раствор концентрировали до минимального объема и лиофилизировали.
Затем полученный продукт суспендировали в 100 мл DMA и в раствор DMA добавляли сульфатирующий агент пиридин-SO3. Раствор доводили до 10°С и обрабатывали частью сульфатирующего агента аддукта пиридин-SO3 в отношении 1,25 эквивалента сульфатирующего агента относительно гидроксила в течение 60 минут.
Раствор обрабатывали ацетоном, насыщенным хлоридом натрия, в количестве, необходимом для полной преципитации. Полученный твердый материал очищали посредством диафильтрации в соответствии с известным способом. Аликвоту лиофилизировали для структурного анализа 13С ЯМР.
g) N-ресульфатирование
Продукт растворяли в воде, доводили до температуры 40°С и добавляли одной добавкой 10 г углекислого натрия и 10 г пиридинсульфотриоксида в течение периода 10 минут.
В конце реакции, если необходимо, раствор приводили к комнатной температуре и затем, если необходимо, до рН ниже, чем 8,0 с использованием NaOH.
Затем продукт очищали от солей известными способами, такими как, например, диафильтрацией с использованием спиральной мембраны с пределом отсечения 1000 Да (препаративный картридж-Millipore). Процесс завершали при проводимости пермеата ниже чем 1000 мкСм, предпочтительно ниже, чем 100 мкСм. Задержанный продукт уменьшали в объеме вплоть до получения 10% концентрации полисахарида выполнением того же самого процесса фильтрации.
Спектр 1Н ЯМР показан на фиг.2.
Активность против фактора Ха полученного продукта, измеренная в плазме человека, составляла 140 МЕ/мг (см. таблицу 2) и соотношение между активность против фактора Хаю и анти-II активностью составляло 2,5.
ПРИМЕР 2. O-сульфатирование с использованием DMF
Пример номера 1 повторяли с изменением в том, что в стадии с) и f) частичное O-сульфатирование и частичное 6-O-сульфатирование выполняли с использованием диметилформамида (DMF) в качестве органического растворителя. Полученный продукт обнаруживал активность против фактора Ха в плазме 85,9 МЕ/мг (см. таблицу 2). ЯМР спектр представлен на фиг.3.
ПРИМЕР 3. Контролируемая деполимеризация в присутствии 50 мг/г субстрата нитрита натрия
Пример номера 1 повторяли с отличием в том, что контролируемую деполимеризацию выполняли с использованием 50 мг нитрита натрия на грамм полисахарида для получения молекулярной массы около 4200 Да.
Полученный продукт обнаруживал активность против фактора Ха в плазме 60,1 МЕ/мг (см. таблицу 2). 13С ЯМР спектр представлен на фиг.4.
ПРИМЕР 4. ПОЛУЧЕНИЕ биотехнологических гепаринов, имеющих молекулярную массу приблизительно 8000 Да (контролируемая деполимермзация с 20 мг/г субстрата нитрита натрия)
Пример номера 1 повторяли с отличием в том, на стадии а) выполняли контролируемую деполимеризацию с 20 мг нитрита натрия на г полисахарида для получения молекулярной массы около 8000 Да.
Полученный продукт обнаруживал активность против фактора Ха в плазме 150 МЕ/мг (см. таблицу 2). 13С ЯМР спектр представлен на фиг.5.
ПРИМЕР 5. ПОЛУЧЕНИЕ биотехнологических гепаринов, имеющих молекулярную массу около 20000 Да
Пример номер 5 выполняли в соответствии со следующими стадиями:
a) начального получения полисахарида N-ацетилгепарозана из Escherichia coli K5;
b) N-деацетилирования/N-сульфатирования;
c) эпимеризации;
d) частичного O-сульфатирования/частичного O-десульфатирования;
e) частичного 6-O-cульфaтиpoвaния/N-pecyльфaтиpoвaния,
где стадии а)-с) соответствуют каждой своей соответствующей стадии в примере 1 и где отсутствует деполимеримеризация (стадия d), стадия d) соответствует стадии е) в примере 1, а стадия е) соответствует стадии f) и g) в примере 1.
Полученный конечный продукт имел молекулярную массу около 20000 Да и являлся поддающимся деполимеризации. Активность против фактора Ха в плазме составляла 135 МЕ/мг (см. таблицу 2).
13С ЯМР спектр представлен на фиг.6.
ПРИМЕР 6. ПОЛУЧЕНИЕ биотехнологических гепаринов, имеющих молекулярную массу приблизительно 15000 Да (контролируемая деполимермзация с 5 мг/г субстрата нитрита натрия)
Пример 1 повторяли при тех же самых температурных и временных условиях, использованных на стадии d), но контролируемую деполимеризацию с нитритом натрия выполняли с использованием 5 мг нитрита натрия на г полисахарида для получения молекулярной массы около 15000 Да.
Полученный продукт демонстрировал активность против фактора Ха в плазме 180 МЕ/мг (см. таблицу 2); 13С ЯМР спектр представлен на фиг.7.
ПРИМЕР 7. Способ приготовления биотехнологических гепаринов в соответствии с известными способами.
В этом примере использовали условия способа O-суперсульфатирования и 6-O-сульфатирования, описанные в заявках WO 01/72848 и WO 02/50125. Вкратце, осуществление суперсульфатирования в диметилформамиде при 50°С в течение 18 часов, O-десульфатирования при 65°С в течение 150 минут и 6-O-сульфатирования в диметилформамиде при 0°С в течение 90 минут.
Конечный продукт демонстрировал молекулярную массу около 20000 Да и активность против фактора Ха в плазме 45 МЕ/мг (см. таблицу 2). 13С ЯМР спектр представлен на фиг.8.
ПРИМЕР 8. Получение биотехнологических гепаринов в соответствии с известными уровнями техники.
Этот пример выполняли с использованием тех же самых условий суперсульфатирования и 6-O-десульфатирования, описанных в заявках WO 01/72848 и WO 02/50125: вкратце, суперсульфатирование выполняли в диметилформамиде при 50°С в течение 18 часов, O-десульфатирование при 65°С в течение 150 минут и 6-O-сульфатирование в диметилформамиде при 0°С в течение 90 минут.
Кроме того, стадию деполимеризации выполняли в конце способа сульфатирования, как описано в заявках WO 01/72848 и WO 02/50125, в присутствии 40 мг/г субстрата нитрита натрия при 4°С в течение 15 минут.
Полученный конечный продукт имел молекулярную массу около 6000 Да и активность против фактора Ха в плазме 31,5 МЕ/мг. 13С ЯМР спектр представлен на фиг.9.
ПРИМЕР 9. Получение биотехнологических гепаринов: контроль условий деполимеризации и сульфатирования.
Пример 1 повторяли со следующими изменениями: контролируемую деполимеризацию (стадия d) в примере 1) выполняли с 40 мг нитрита натрия на г эпимеризованного K5NS при тех же самых временных и температурных условиях примера 1. Полученная молекулярная масса составляла около 6500 Да.
Частичное сульфатирование, (стадия е) примера 1, выполняли с молярным соотношением 5 между сульфатирующим агентом и субстратом эпимеризованного N-сульфатированного К5 и в течение периода 180 минут, в то время как O-десульфатирование выполняли в течение 60 минут.
Полученный продукт демонстрировал активность против фактора Ха в плазме 89 МЕ/мг (см. таблицу 2). 13С ЯМР спектр представлен на фиг.10.
ПРИМЕР 10. Получение биотехнологических гепаринов: контроль условий деполимеризации и сульфатирования.
Пример 1 повторяли и промежуточную деполимеризацию (стадия d) на N-полисахариде К5 выполняли при тех же самых временных и температурных условиях, описанных в примере 9, но с использованием 40 мг нитрита натрия на мг эпимеризованного K5NS для получения молекулярной массы около 6500 Да. Частичное сульфатирование, соответствующее (стадии е) в примере 1, выполняли с использованием молярного соотношениия 0,6 между сульфатирующим агентом и эпимеризованным субстратом N-сульфатированного К5 и в течение 8 часов времени инкубирования. O-десульфатирование выполняли в течение 30 минут.
Полученный продукт демонстрировал активность против фактора Ха в плазме 101 МЕ/мг (см. таблицу 2).
13С ЯМР спектр продукта, полученного в соответствии с этим примером, представлен на фиг.11.
ПРИМЕР 11. Отделение фракций полисахаридов, обладающих высокой аффинностью к антитромбину III, путем селекции на аффинной колонке.
Полисахарид получали в соответствии с примером 1. После стадии g) N-ресульфатирования продукт пропускали через аффинную колонку следующим образом: 20 мг продукта, полученного на стадии g) в примере 1, наносили на колонку со смолой CNBr-Sepharose 4B (Pharmacia), на которую предварительно иммобилизовали, в соответствии с известными способами, 100 мг антитромбина III человека (Kedrion SpA, Lucca, Italia) в буфере 10 мМ Tris-HCl при рН 7,4 и 0-0,15 М NaCl, при 4°С. После периода связывания в течение 60 минут колонку промывали по меньшей мере 3-мя объемами буфера 10 мМ Tris-HCl рН 7,4.
Молекулы, связанные с более высокой аффинностью с колонкой, элюировали добавлением градиента 10 мМ Tris-HCl рН 7,4, содержащем 2М NaCl.
Элюированный материал диафильтровали с помощью спиральной мембраны с пределом отсечения 1000 Да для освобождения от солей и концентрировали лиофилизацией.
Конечный продукт демонстрировал молекулярную массу 8500 Да и активность против фактора Ха в плазме 300 МЕ/мг (см. таблицу 2). 13С ЯМР спектр представлен на фиг.12.
ПРИМЕР 12. Получение биотехнологических гепаринов, имеющих низкую молекулярную массу около 6000 Да.
Пример 5 повторяли, но после стадии N-сульфатирования продукт деполимеризовали при тех же самых условиях, как описано для стадии d) примера 1.
Полученный конечный продукт имел молекулярную массу около 6000 Да и активность против фактора Ха в плазме 65 МЕ/мг (см. таблицу 2). 13С ЯМР спектр представлен на фиг.13.
ПРИМЕР 13. Получение биотехнологических гепаринов: контроль условий сульфатирования.
Пример 1 повторяли, изменяя O-сульфатирование (стадия е) примера 1, которое выполняли с молярным соотношением 5 между сульфатирующим агентом и эпимеризованным К5-сульфатированным субстратом в течение 8 часов при 50°С.
Полученный продукт демонстрировал активность против фактора Ха в плазме 75 МЕ/мг. 13С ЯМР спектр представлен на фиг.14.
ПРИМЕР 14. Получение промежуточного продукта K5-OS6OSNH2, epi (нересульфатированного промежуточного продукта биотехнологического гепарина).
Пример 1 повторяли без последней стадии ресульфатирования.
Конечный продукт имел молекулярную массу около 6000 Да и активность против фактора Ха 5 МЕ/мг (см. таблицу 2). 13С ЯМР спектр представлен на фиг.15.
ПРИМЕР 15. Получение промежуточного продукта K5-OS,NH2, epi (промежуточного продукта биотехнологического гепарина до стадий десульфатирования, 6-O-сульфатирования и N-ресульфатирования).
Повторяли пример 1 при изменении условий частичного O-сульфатирования (стадия е) примера 1, в то время как его выполняли с молярным соотношением 5 между сульфатирующим агентом и гидроксилами субстрата эпимеризованного К5 N-сульфата в течение 8 часов при 50°С и без последующих стадий десульфатирования, частичного O-сульфатирования и N-ресульфатирования.
Полученный продукт K5-OSNH2-epi имел молекулярную массу около 6000 Да, 95% сульфатирование гидроксилов и активность против фактора Ха 8 МЕ/мг (см. таблицу 2). Протонный 1Н ЯМР спектр показан на фиг.16.
ПРИМЕР 16. Получение биотехнологических гепаринов, с молекулярной массой около 6000 Да
Пример 1 выполняли со следующими стадиями:
a) начального получения полисахарида N-ацетилгепарозана из Escherichia coli K5,
b) N-деацетилирования/N-сульфатирования,
c) эпимеризации,
d) частичного O-сульфатирования/частичного O-десульфатирования,
e) контролируемой деполимеризации,
f) частичного 6-О-сульфатирования/N-ресульфатирования, где условия стадий а)-с) соответствуют условиям, описанным в соответствующих стадиях примера 1, и где стадии d) и е) осуществляют в обратном порядке по отношению к тем же самым стадиям примера 1.
Полученный конечный продукт имел молекулярную массу около 60000 Да и активность против фактора Ха в плазме 95 МЕ/мг (см. таблицу 2). Углеродный ЯМР спектр представлен на фиг.17.
ПРИМЕР 17. Определение биологической активности продуктов, полученных в соответствии с изобретением.
Измерение активности против фактора Ха: активность против фактора Ха оценивали в соответствии с хромогенным методом (Coatest Heparin kit, Chromogenix). Измерение выполняли в нормальной плазме человека с использованием таких реагентов, как хромогенный субстрат S2222 (Chromogenix), бычий фактор Ха (Chromogenix) и антитромбин III человека (Chromogenix). Реакцию осуществляли при 37°С в коагулометре ACL 9000 (International Laboratory) и считывание производили при 405 нм. Результаты показаны в таблице 2. Результаты, полученные в соответствии с известными способами (конкретно, продукты способов, полученные как описано в заявках WO 01/72848 и WO 02/50125), просуммированы в таблице 1, в которой сообщаются литературные данные для экстрагируемых гепаринов (Farced J et al. Exp Opin. Invest. Drugs, 1997, 6: 705-733). Активность против фактора Ха. Активность против фактора Ха оценивали в нормальной плазме человека в соответствии со следующей процедурой: 30 мкл 0,5 Ед/мл антитромбина III человека (Chromogenix) смешивали с 30 мкл раствора исследуемой пробы в различной концентрации и с 60 мкл бычьего тромбина до 5,3 нанокатал/мл (nKat/mI) (Chromogenix).
Раствор инкубировали в течение 70 секунд при 37°С, затем добавляли 60 мкл хромогенного субстрата S-2238 (Chromogenix). Реакцию регистрировали в течение 90 секунд со считыванием каждую секунду при 405 нм с использованием коагулометра ACL 9000 (International Laboratory). Результаты, полученные с известными продуктами (главным образом продуктами способов, описанных в заявках WO 01/72848 и WO 02/50125), просуммированы и в таблице 1.
Устойчивость к гепариназе. Устойчивость к гепариназе I определяли приготовлением низкомолекулярных и высокомолекулярных проб в буфере 20 мМ Tris-HCl, 50 мМ NaCl, 4 мМ CaCl; и 0,01% БСА при рН 7,5 в конечной концентрации 0,02%. Добавляли 20 единиц гепариназы I (Sigma, CAS номер 52227-76-6) к 100 мкл раствора, содержащего пробу, и реакционную смесь инкубировали при 25°С. Затем реакцию останавливали добавлением 50 мМ HCl с постоянными интервалами, каждые 10 минут, 1 час, 2 часа, 20 часов.
Каждую пробу анализировали как спектрофотометрическим измерением при 235 нм, так и ВЭЖХ гель-проникающей хроматографией для определения молекулярной массы. Полученные результаты представлены в таблице 3, из которой видно, что в то время, как экстрагируемые гепарины, используемые в исследовании, как высокомолекулярные (HMW), так и низкомолекулярные (Fraxiparina, Sanofi), в конечном счете разрушаются, как видно по снижению молекулярной массы, биотехнологические гепарины, полученные в соответствии с настоящим изобретением, являются стабильными (молекулярная масса является постоянной даже после 20-часовой обработки гепариназой I).
Активность TFPI. Определение активности на основе TFPI-фактора проводили in vitro на клетках HUVEC в соответствии со способом, описанным Gori AM. et al. Thromb. Haemostasis 1999:81:589-93, и сравнение проводили с коммерческим неразделенным гепарином (таблица 4).
Ингибирование синтеза протеаз. Определение ингибирования синтеза протеаз выполняли в плазме, истощенной в отношении фибриногена, в соответствии со способом, сообщенным Fareed et al. Path. Haem. Thromb. 2002: 32 (3): 56-65. После добавления исследуемых проб с различным разведением плазму активировали добавлением РТ (тромбопластина С) для активации собственной системы коагуляции или АРТТ (Dade Actin) для активации собственной системы коагуляции. Ингибирование как тромбина (фактора II), так и фактора Ха регистрировали с использованием коагулометра ACL 9000 (International Laboratory). Значения, полученные при разведении пробы 50 мкг/мл, представлены в таблице 5.
Аффинность к фактору PF4: Определение аффиности к фактору PF4 (4-му тромбоцитарному фактору) оценивали в плазме определением остаточной активности против фактора Ха после добавления фиксированного количества фактора PF4 в растворе, содержащем биотехнологические гепарины, полученные в соответствии с изобретением.
100 мкг плазмы, содержащей 0,8 МЕ/мл анти-Ха, добавляли до получения конечной концентрации PF4 10 мкг/мл. Остаточную активность против фактора Ха пробы измеряли с использованием Coatest heparin kit (Chromogenix) с использованием коагулометра ACL 9000 (International Laboratory). Остаточную активность против фактора Ха определяли как процент от исходной активности (таблица 6).
Измерение активированного частичного тромбопластинового времени (АРТТ): определение АРТТ выполняли с помощью пробы на коагуляцию с использованием коагулометра ACL 9000 (International Laboratory). Реакцию осуществляли при 37°С добавлением определенного количества цефалина (kit АРТТ International Laboratory cod 8468710) к пробе, разведенной должным образом, и последующим свертыванием после добавления хлорида кальция и итогового считывания при 660 нм.
Значения АРТТ приводятся в таблице 2 в виде процентного соотношения активности относительно первого международного стандарта низкомолекулярного гепарина 85/600.
Определение активности гепаринового кофактора II (HCII): определение HCII выполняли приготовлением реакционной смеси, содержащей 20 мкл HCII (Stago) 0,085 PEU/мл, 80 мкл раствора исследуемой пробы при различной концентрации, 50 мкл тромбина 0,18 Ед/мл (Boehringer) в 0,02 М Tris-буфере, рН 7,4, 0,15 М NaCl и 0,1% ПЭГ 6000. Раствор инкубировали в течение 60 секунд при 37°С, затем добавляли 50 мкл 1 мМ хромогенного субстрата Spectrozyme (American Diagnostic). Реакцию контролировали в течение 180 секунд с интервалами в 1 секунду при длине волны 405 нм на автоматическом коагулометре ACL 9000 (insrtumentation Laboratory).
Значения НСII приводятся в таблице 2 в виде процентного соотношения активности относительно первого международного стандарта низкомолекулярного гепарина 85/600.
В таблице 1 и 2 данные биологической активности биотехнологических гепаринов, полученных в соответствии с настоящим изобретением или в соответствии со способами, отличающимися O-суперсульфатированием независимо от использования растворителей, доноров формильных групп или не являющимися донорами формильных групп, таких как N,N,-диметилформамид, и отличающимися конечной стадией деполимеризации, или экстрагируемых гепаринов. Данные, в частности, показывают, что активность против фактора Ха, измеренная в плазме человека, является выше для продуктов, полученных в соответствии с данным способом, чем для продуктов предшествующего уровня техники, сравнимых по молекулярной массе.
В действительности, в продуктах, полученных в соответствии с настоящим изобретением, соотношение между биологической активностью, выраженной в виде активности против фактора Ха, и молекулярной массой является выше. Такое увеличение, по-видимому, обусловлено комбинированными особенностями способа:
- O-сульфатированием (O-сульфатирование и 6-O-сульфатирование), выполняемом в мягких условиях (смотри примеры 5 и 7 для сравнения):
- использованием апротонного полярного растворителя, не являющегося донором формильных групп, в течение стадий O-сульфатирования и 6-O-сульфатирования (смотри примеры 1 и 2 для сравнения);
- промежуточной стадией деполимеризации, которую выполняют перед стадией O-сульфатирования или перед стадией 6-O-сульфатирования, по сравнению с деполимеризацией, выполняемой в конце способа (смотри примеры 1 и 12 для сравнения).
Более высокое увеличение соотношения между биологической активностью, рассматриваемой как активность против фактора Ха в плазме, и молекулярной массой полученного продукта, а также соотношения между анти-Ха- и анти-IIa-активностями получают, когда O-сульфатирования (O-сульфатирование и 6-O-сульфатирование) являются частичными вместе с использованием полярного апротонного растворителя, не являющегося донором формильных групп, для O-сульфатирования и 6-O-сульфатирования, связанных с деполимеризацией, выполненной на промежуточной стадии как в примере 1 и в примере 6.
Однако способ является также совместимым с более сильным O-сульфатированием, связанным с частичным 6-O-сульфатированием, как описано в примере 13.
Более того, способ настоящего изобретения является также совместимым с конечной деполимеризацией, как описано в примере 12.
Дополнительный параметр, улучшенный в продуктах настоящего изобретения по сравнению с биотехнологическими гепаринами, полученными в соответствии с известными способами, является соотношение между анти-Ха-активностью и анти-IIa-активностью, свидетельствующее о зависимости между антитромботическими и антикоагулянтными свойствами, получающимися также из значения АРТТ, которое является сходным со значением экстрагируемых гепаринов.
У продуктов, полученных в соответствии с изобретением, такое соотношение является равным или превышающим 1, в то время как оно является ниже чем 1 у продуктов, полученных в соответствии со способами предшествующего уровня техники.
Другой характеристикой, показанной высокими значениями НСII, является более высокая способность прямого ингибирования тромбина по сравнению с экстрагируемыми гепаринами.
ПРИМЕР 18. Получение биотехнологических гепаринов с молекулярной массой около 6000 Да.
Пример 1 выполняли со следующими стадиями:
a) начального получения полисахарида N-ацетилгепарозана из Escherichia coli K5,
b) N-деацетилирования/N-сульфатирования,
c) эпимеризации,
d) частичного O-сульфатирования/частичного O-десульфатирования,
e) N-ресульфатирования,
f) контролируемой деполимеризации,
g) частичного 6-O-сульфатирования,
где условия стадий а)-с) соответствуют условиям, описанным в соответствующих стадиях примера 1, и где стадии d)-g) осуществляют в обратном порядке по отношению к тем же самым стадиям примера 1.
Полученный конечный продукт имел молекулярную массу около 60000 Да и активность против фактора Ха в плазме 92 МЕ/мг (см. таблицу 2). Протонный ЯМР спектр представлен на фиг.18.
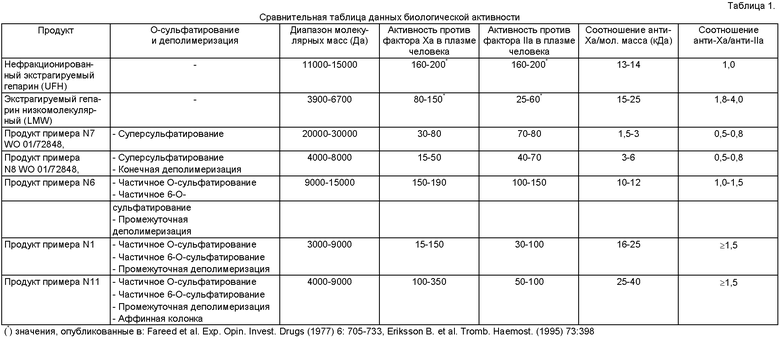
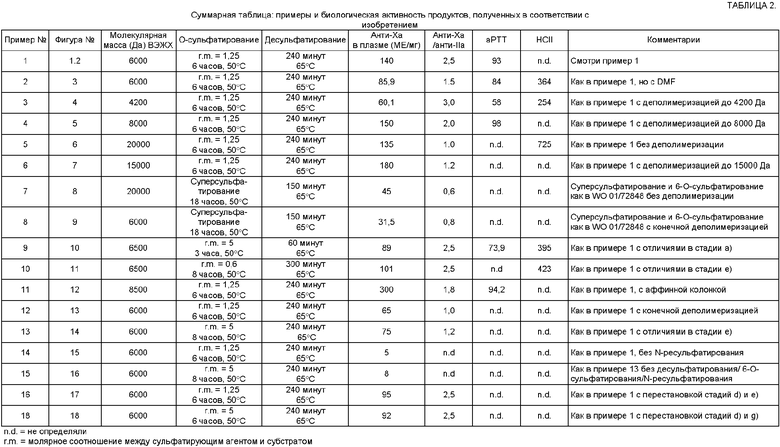
Сравнительный тост гидролиза гепариназы биотехнологических
Количественный анализ высвобождения TFPI-фактора из клето HUVEC In vitro
Количественный анализ ингибирования образования протез
Количественный анализ аффинности фактора PF4
| название | год | авторы | номер документа |
|---|---|---|---|
| ГЛИКОЗАМИНОГЛИКАНЫ, ПРОИЗВОДНЫЕ К5-ПОЛИСАХАРИДА, ОБЛАДАЮЩИЕ ВЫСОКОЙ АНТИКОАГУЛЯНТНОЙ И АНТИТРОМБОТИЧЕСКОЙ АКТИВНОСТЬЮ, И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2001 |
|
RU2283319C2 |
| ЭПИМЕРИЗОВАННЫЕ ПРОИЗВОДНЫЕ ПОЛИСАХАРИДА К5 С ВЫСОКОЙ СТЕПЕНЬЮ СУЛЬФАТИРОВАНИЯ | 2003 |
|
RU2333222C2 |
| ПОЛУЧЕНИЕ ПОЛИФУНКЦИОНАЛЬНЫХ СУЛЬФАТИРОВАННЫХ ПРОИЗВОДНЫХ НА ОСНОВЕ ПОРОШКОВОЙ ЦЕЛЛЮЛОЗЫ, ОБЛАДАЮЩИХ АНТИКОАГУЛЯНТНОЙ АКТИВНОСТЬЮ | 2009 |
|
RU2430729C2 |
| N,О-СУЛЬФАТИРОВАННЫЕ ГЕПАРОЗАНЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ АНТИТРОМБОТИЧЕСКОЙ АКТИВНОСТЬЮ | 1991 |
|
RU2099353C1 |
| ПОЛУСИНТЕТИЧЕСКИЕ СУЛЬФАМИНОГЕПАРОСАНСУЛЬФАТЫ, ИМЕЮЩИЕ ВЫСОКУЮ АНТИМЕТАСТАТИЧЕСКУЮ АКТИВНОСТЬ И ПОНИЖЕННЫЙ ГЕМОРРАГИЧЕСКИЙ РИСК | 1997 |
|
RU2176915C2 |
| СПОСОБ ПОЛУЧЕНИЯ СУЛЬФАТИРОВАННЫХ ПРОИЗВОДНЫХ АРАБИНОГАЛАКТАНА, ОБЛАДАЮЩИХ АНТИКОАГУЛЯНТНОЙ И ГИПОЛИПИДЕМИЧЕСКОЙ АКТИВНОСТЬЮ | 2007 |
|
RU2319707C1 |
| СПОСОБ ПОЛУЧЕНИЯ СУЛЬФАТИРОВАННЫХ ПРОИЗВОДНЫХ АРАБИНОГАЛАКТАНА, ОБЛАДАЮЩИХ АНТИКОАГУЛЯНТНОЙ И ГИПОЛИПИДЕМИЧЕСКОЙ АКТИВНОСТЬЮ | 2013 |
|
RU2532915C1 |
| Способ получения флуоресцентных производных арабиногалактана | 2021 |
|
RU2768707C1 |
| СПОСОБ СУЛЬФАТИРОВАНИЯ КАРБОКСИМЕТИЛЦЕЛЛЮЛОЗЫ | 2011 |
|
RU2471809C1 |
| СРЕДСТВО С АНТИТРОМБОТИЧЕСКОЙ АКТИВНОСТЬЮ | 2016 |
|
RU2627435C1 |
Способ получения сульфатированных гликозаминогликанов, получаемых из N-ацетилгепарозана, включает а) N-деацетилирование и N-сульфатирование полисахарида N-ацетилгепарозана, полученного из природных или рекомбинантных бактериальных штаммов, предпочтительно Е.coli K5, b) ферментативную эпимеризацию с помощью фермента глукоронил-С5-эпимеразы, с) частичное O-сульфатирование с последующим частичным O-десульфатированием, d) частичное 6-O-сульфатирование, е) N-сульфатирование и промежуточную стадию контролируемой деполимеризации, характеризующийся тем фактом, что оба O-сульфатирования (O-сульфатирование и 6-O-сульфатирование) являются частичными. Дополнительно изобретение относится к продуктам, полученным в соответствии со способом, которые демонстрируют соотношение между анти-Ха- и анти-IIa-активностями, равное 1 или превышающее 1, и к антитромботическим и антикоагулянтным фармацевтическим композициям, содержащим указанные продукты в сочетании с подходящими и фармацевтически приемлемыми наполнителями и/или растворителем. 8 н. и 53 з.п. ф-лы, 18 ил., 6 табл.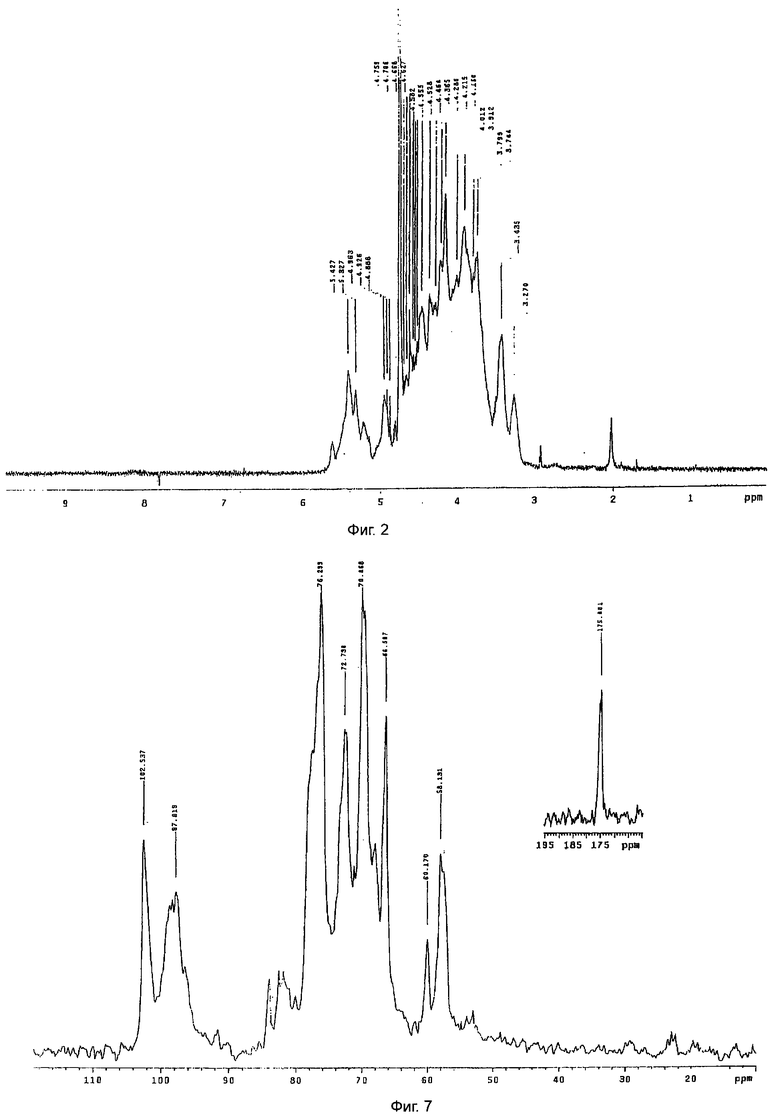
1. Способ получения сульфатов гликозаминогликанов, получаемых из N-ацетилгепарозана, включающий следующие стадии:
a) N-деацетилирования и N-сульфатирования полисахарида N-ацетилгепарозана, выделенного из природного или рекомбинантного штамма бактерий:
b) ферментативной эпимеризации с помощью фермента глюкоронил С5-эпимеразы;
c) частичного O-сульфатирования в сочетании с частичным O-десульфатированием;
d) частичного 6-O-сульфатирования;
e) N-ресульфатирования,
дополнительно включающий стадию контролируемой деполимеризации, альтернативно выполняемой после стадии b), с) или d), и где указанный способ характеризуется тем фактом, что частичное O-сульфатирование на стадии с) выполняют в течение менее чем 10 ч и с использованием молярного соотношения между сульфатирующим агентом и N-ацетилгепарозаном, равного 5 или меньшего 5, и тем фактом, что частичное 6-O-сульфатирование (стадия d) выполняют в течение периода времени, равного 2 ч или меньшего 2 ч, и с использованием молярного соотношения между сульфатирующим агентом и гидроксильными группами N-ацетилгепарозана, равного 2 или меньшего 2.
2. Способ по п.1, в котором контролируемую промежуточную деполимеризацию выполняют после стадии b) эпимеризации.
3. Способ по п.1, в котором O-сульфатирование в соответствии со стадией с) выполняют с использованием молярного соотношения между сульфатирующим агентом и N-ацетилгепарозаном, меньшего 2,5.
4. Способ по п.3, в котором указанное молярное соотношение является равным или меньшим 1,5.
5. Способ по п.3, в котором частичное O-сульфатирование (стадия с) выполняют в течение периода времени, равного 6 ч или меньшего 6 ч.
6. Способ по п.1, в котором частичное 6-O-сульфатирование (стадия d) выполняют с использованием молярного соотношения между сульфатирующим агентом и гидроксильными группами N-ацетилгепарозана, меньшего 1,5 или равного 1,5.
7. Способ по п.6, в котором частичное 6-O-сульфатирование ((стадия d) способа) выполняют в течение периода времени, равного или меньшего 60 мин.
8. Способ по п.7, в котором сульфатирование выполняют в течение периода времени, равного или меньшего 30 мин.
9. Способ по п.1, в котором полисахарид N-ацетилгепарозан на стадии а) выделяют из Е.coli K5.
10. Способ по п.1, включающий дополнительную стадию f) аффинного выделения на матриксах со связанным антитромбином III или его фрагментами.
11. Способ по п.1, в котором частичное сульфатирование в соответствии со стадией с) и частичное 6-O-сульфатирование в соответствии со стадией d) выполняют с использованием сульфатирующего агента, выбранного из группы, состоящей из триэтиламина-SO3, триметиламина-SO3, пиридина-SO3, в апротонном растворителе.
12. Способ по п.11, в котором указанный апротонный полярный растворитель не является донором формильных групп.
13. Способ по п.12, в котором указанный апротонный полярный растворитель выбирают из тетраметиленсульфона, 2,4-диметилсульфолана, N,N-диметилацетамида и N,N-диэтилацетамида.
14. Способ по п.1, в котором частичное 6-O-сульфатирование (стадия d) выполняют при температуре, составляющей от 4 до 30°С.
15. Способ по п.14, в котором указанная температура охватывает от 10 до 25°C.
16. Способ по п.1, в котором промежуточную контролируемую деполимеризацию выполняют химическими или физическими способами.
17. Способ по п.16, в котором указанные физические способы включают воздействие гамма-лучами, и в котором указанные химические способы включают воздействие азотистой кислотой или ее солями, бета-элиминирование или воздействие йодной кислотой или свободными радикалами.
18. Способ по п.17, в котором деполимеризацию выполняют воздействием азотистой кислотой или ее солями.
19. Способ по п.18, в котором соотношение между азотистой кислотой или ее солями и сахаридом составляет от 1 до 100 мг соли на грамм полисахарида, и взаимодействие выполняют при температуре, составляющей от 4 до 10°С.
20. Способ по п.19, в котором контролируемую деполимеризацию выполняют в течение времени, меньшего 30 мин, и в присутствии азотистой кислоты или ее солей.
21. Способ по п.20, в котором солью азотистой кислоты является нитрит натрия.
22. Способ по п.16, в котором деполимеризацию завершают добавлением молярного избытка боргидрида.
23. Способ по п.11 для приготовления сульфатированных гликозаминогликанов, получаемых из N-ацетилгепарозана, включающий следующие стадии:
a) N-деацетилирования и N-сульфатирования полисахарида N-ацетилгепарозана, выделенного из природного или рекомбинантного штамма бактерий;
b) ферментативной эпимеризации с помощью фермента глюкуронил-С5-эпимеразы;
c) частичного O-сульфатирования в сочетании с частичным O-десульфатированием;
d) частичного 6-O-сульфатирования,
дополнительно включающий промежуточную стадию контролируемой деполимеризации, альтернативно выполняемой после стадии b), с) или d), где указанный способ характеризуется тем фактом, что частичное O-сульфатирование на стадии с) выполняют в течение периода времени, равного или меньшего 10 ч, и с использованием молярного соотношения между сульфатирующим агентом и N-ацетилгепарозаном, равного 5 или меньшего 5, и тем, что частичное 6-O-сульфатирование (стадия d) выполняют в течение периода времени, равного 2 ч или меньшего 2 ч, и с использованием молярного соотношения между сульфатирующим агентом и N-ацетилгепарозаном, равного 2 или меньшего 2, и тем, что O-сульфатирование на стадии с) и d) выполняют с сульфатирующим агентом, выбранным из триэтиламина-SO3, триметиламина-SO3, пиридина-SO3, в апротонном растворителе.
24. Способ по любому из пп.1-23, в котором реакцию С5-эпимеризации (стадия с) выполняют при температуре ниже 35°С, и в котором глюкоронил С5-эпимераза, или экстрагируемая, или рекомбинантная, является иммобилизованной на стационарной фазе.
25. Способ по п.24, в котором указанная рекомбинантная С5-эпимераза является мышиным ферментом, экспрессированном в клетках насекомых или в дрожжевых клетках.
26. Способ по п.24, в котором указанная температура составляет от 15 до 30°С.
27. Способ по п.26, в котором указанная температура составляет от 20 до 25°С.
28. Способ по п.24, в котором стационарной фазой является полистирольная или полиметакриловая смола с гепоксидными или диольными группами, активированными в присутствии CNBr, и указанную иммобилизацию С5-эпимеразы проводят в буфере, содержащем NaНСО3 в концентрации от 100 до 300 мМ, или в фосфатном буфере в концентрации от 10 до 50 мМ при рН от 7,0 до 8,3 при температуре, составляющей от 4 до 25°С, в течение периода времени, составляющего от 12 до 72 ч.
29. Способ по п.24, в котором эпимеризацию на стадии с) выполняют в буфере HEPES, содержащем ЭДТА от 10 до 30 мМ, CaCl2 от 70 до 150 мМ и с рН от 5,5 до 8,0.
30. Модифицированный N-ацетилгепарозан, получаемый способом в соответствии с п.9.
31. Модифицированный N-ацетилгепарозан по п.30 с молекулярной массой, равной 15000 Да или меньшей 15000 Да.
32. Модифицированный N-ацетилгепарозан по п.31 с молекулярной массой, составляющей от 3000 до 9000 Да.
33. Модифицированный N-ацетилгепарозан, получаемый способом в соответствии с п.12, в котором отсутствуют формильные группы в молекуле.
34. Модифицированный N-ацетилгепарозан по п.30, несущий на редуцированном конце остаток сульфатированного 2,5-ангидроманнита.
35. N-ацетилгепарозан по п.34, в котором гидроксильные группы в положении 1, 3 и 6 ангидроманнита являются частично сульфатированными.
36. N-ацетилгепарозан по п.35, в котором ангидроманнит является частично сульфатированным по гидроксилам в положении 1 и 6.
37. K5OS6OSNS-epi N-ацетилгепарозана по п.36, в котором степень сульфатирования гидроксильных групп в положении 1 составляет от 20 до 85%.
38. K5OS6OSNS-epi N-ацетилгепарозана по п.36, в котором степень сульфатирования гидроксильных групп в положении 6 глюкозамина превышает 40%.
39. K5OS6OSNS-epi N-ацетилгепарозана по п.38, в котором указанная степень сульфатирования составляет от 50 до 85%.
40. N-ацетилгепарозан по п.35, в котором степень сульфатирования гидроксильных групп в положении 3 глюкозамина составляет менее чем 60%.
41. K5OS6OSNH2-epi, получаемый способом по п.23.
42. K5OSNH2-epi, получаемый способом по п.23, стадий а)-с).
43. Модифицированный N-ацетилгепарозан по п.30, имеющий активность против фактора Ха, измеряемую в присутствии плазмы, равную 50 МЕ/мг или превышающую 50 МЕ/мг.
44. Модифицированный N-ацетилгепарозан по п.43, имеющий соотношение между активностями, ингибирующими фактор Ха и фактор Па, равное или превышающее 1,0.
45. Модифицированный N-ацетилгепарозан по п.43, имеющий активность активации TFPI, равную или превышающую активность экстрагируемых гепаринов.
46. Модифицированный N-аиетилгепарозан по п.43, имеющий устойчивость к гепариназе I выше, чем у экстрагируемых гепаринов.
47. Модифицированный N-ацетилгепарозан по п.43, ингибирующий образование протеаз тромбина и фактора Ха.
48. Модифицированный N-ацетилгепарозан по п.43, имеющий более низкую аффинность для фактора PF4 по сравнению с экстрагируемыми гепаринами.
49. Модифицированный N-ацетилгепарозан по п.43, имеющий более высокую активность HCI I, чем экстрагируемые гепарины.
50. Модифицированный N-ацетилгепарозан по п.43, имеющий в избытке множественные сигналы в 13С ЯМР спектре по сравнению с сигналами ангидроманнита в области, составляющей от 79 до 89 импульсов в минуту, и который не имеет сигналы при 51 и 165 импульсов в минуту и сигналы в области при импульсах в минуту 7-9,5 спектра 1Н ЯМР.
51. K5OS6OSN-epi, по п.50, имеющий 1Н ЯМР спектр, соответствующий фиг.2.
52. K5OS6OSN-epi, по п.50, имеющий 13С ЯМР спектр, соответствующий фиг.7.
53. Модифицированный N-ацетилгепарозан по п.30 или 43 для фармацевтического применения.
54. Применение продукта по п.53 для приготовления лекарственного препарата с гепариноподобной антитромботической и антикоагулянтной активностью.
55. Применение продукта по п.53 для приготовления лекарственного препарата с профибринолитической и антиагрегационной активностью.
56. Применение по п.54 для приготовления лекарственного препарата для профилактики и лечения нестабильной стенокардии, инфаркта миокарда, тромбоза глубоких вен, легочной эмболии и случаев ишемии.
57. Применение по п.54 для приготовления лекарственного препарата для лечения сепсиса и его осложнений, таких как синдром диссеминированного внутрисосудистого свертывания (CID).
58. Применение по п.55 для приготовления медицинского средства для профилактики и лечения нестабильной стенокардии, острого инфаркта миокарда, венозного и артериального тромбозов, легочной эмболии, случаев ишемии, атеросклероза.
59. Применение по п.54 или 55 для приготовления медицинского средства для профилактики и лечения тромбоэмболитических случаев вследствие врожденного или приобретенного дефицита антитромбина III.
60. Антитромботическая и антикоагулянтная фармацевтическая композиция, содержащая в качестве активного ингредиента любой из продуктов по п.30 или 43 в комбинации с подходящими наполнителями и/или растворителями.
61. Фармацевтическая композиция по п.60, по составу подходящая для перрорального введения.
| US 2003023079 A1, 30.01.2003 | |||
| US 6162797 A, 19.12.2000 | |||
| N,О-СУЛЬФАТИРОВАННЫЕ ГЕПАРОЗАНЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ АНТИТРОМБОТИЧЕСКОЙ АКТИВНОСТЬЮ | 1991 |
|
RU2099353C1 |

