Область техники, к которой относится изобретение
Изобретение относится к области биотехнологии и, в частности, касается гликозаминогликанов, производных полисахарида К5, обладающих высокой антикоагулянтной и антитромботической активностью, полученных способом, включающим выделение полисахарида К5 из Escherichia Coli, Н-деацетилирование/N-сульфатирование, С-5 эпимеризацию, селективное O-десульфатирование, селективное 6-O-сульфатирование и N-сульфатирование, при этом указанную эпимеризацию осуществляют с использованием фермента глюкуроносил С-5 эпимеразы в растворенной или в иммобилизованной форме в присутствии специфических бивалентных катионов.
Уровень техники
Гликозаминогликаны - это биополимеры, получаемые путем промышленной экстракции различных органов животных, таких как слизистая кишечника, легкие и т.п. В зависимости от их структуры гликозаминогликаны подразделяются на гепарины, гепарансульфаты, дерматансульфаты, хондроитинсульфаты и ялуроновые кислоты. В частности, гепарин и гепарансульфат образованы повторением дисахаридных звеньев, состоящих из уроновой кислоты (L-идуроновая или D-глюкуроновая) и аминосахара (глюкозамин). Уроновая кислота может быть сульфатирована в положении 2, а глюкозамин по большей части бывает N-ацетилатирован (гепарансульфат) или N-сульфатирован (гепарин) и 6-O-сульфатирован. Кроме того, глюкозамин может также содержать сульфатную группу в положении 3.
Гепарин и гепарансульфат - это полидисперсные молекулы с молекулярным весом, варьирующим от 3000 до 30000 D. Гепарин, помимо таких известных свойств, как антикоагулянтная и антитромботическая активность, обладает также антилипемной, антипролиферативной, противовирусной, противоопухолевой активностями. Для того чтобы удовлетворить растущий спрос на сырье для этих новых областей терапии, необходим новый способ получения гепарина, альтернативный экстракции тканей животного происхождения. Природный биосинтез гепарина у млекопитающих и его особенности описаны в 1986 г. Линдалом и др.: Лейн Д. и Линдал Ю. (Ред.) «Гепарин: химические и биологические свойства; применение в лечебной практике» Эдварда Арнольда (Lane D. and Lindahl U. (Eds.) "Heparin-Chemical and Biological Properties; Clinical Applications", Edward Arnold, London, pp.159-190) и Линдал Ю., Файнголд Д.С. и Роден Л. (Lindahl U., Feingold D.S. and Roden L., (1986) TIBS, 11, 221-225).
Активность гепарина определяет последовательность пентасахарида - область, связывающая антитромбин III (ATIII) и называемая активным пентасахаридом; именно эта последовательность необходима для высокоаффинного связывания гепарина с ATIII. Эта последовательность содержит единственное звено глюкозамина, сульфатированного в положении 3, в других частях гепариновой молекулы оно не присутствует. Помимо механизма действия через ATIII, гепарин проявляет антикоагулянтную и антитромботическую активность путем активирования гепаринового кофактора II (HCII) с последующим селективным ингибированием тромбина. Известно, что минимальная последовательность, необходимая для активации HCII, представляет собой цепь из, по меньшей мере, 24 моносахаридов (Tollefsen D.M., Seminars in Thrombosis and Hemostasis 16, 66-70, 1990).
Как известно, К5 капсульный полисахарид, выделенный из Escherichia Coli и описанный Vann W.F., Schmidt M.A., Jann В., Jann К. (1981) в Eur. J. Biochem 116, 359-364, включает ту же последовательность предшественника гепарина и гепарансульфата (N-ацетил-гепаросан). Это соединение было химически модифицировано так, как это описано у Lormeau и др. в патенте США номер 5550116, а также у Casu и др. (Carb. Res 263-1994-271-284); химически и ферментативно - у Jann и др. (WO 92/17509) и у Casu и др., Carb. Letters 1, 107-114 (1994). В результате этих модификаций были получены вещества, обладающие в тестах in vitro антикоагулянтной биологической активностью, однако меньшей, чем у гепарина, полученного путем экстракции органов животных.
Сущность изобретения
Мы обнаружили новые гликозаминогликаны, производные К5-полисахарида Escherichia coli, обладающие молекулярным весом от 2000 до 30000 и содержащие от 25 до 50% по массе цепей, обладающих высоким сродством к ATIII, обладающие сильной антикоагулянтной и антитромботической активностью при соотношении активности HCII и активности анти-фактора Ха (antiXa) от 1.5 до 4 с преобладанием активности, ингибирующей тромбин.
Вышеуказанные гликозаминогликаны получены способом, включающим несколько этапов химической и ферментативной обработки; причем характерньм для последней является этап эпимеризации D-глюкуроновой кислоты до L-идуроновой кислоты с помощью глюкуроносил С-5 эпимеразы в растворенной или иммобилизованной форме в присутствии специфических двухвалентных катионов, при этом вышеуказанные ферменты выбирают из группы, состоящей из рекомбинантной глюкуроносил С-5 эпимеразы, глюкуроносил С-5 эпимеразы, выделенной из мышиной или крысиной мастоцитомы, и глюкуроносил С-5 эпимеразы, экстрагированной из печени крупного рогатого скота; а вышеуказанные двухвалентные катионы выбирают из группы, состоящей из Ва, Са, Mg и Mn.
Подробное описание изобретения
Данное изобретение относится к гликозаминогликанам, производным К5-полисахаридов Escherichia coli (далее называемых сокращенно К5), полученным способом, включающим следующие стадии:
a) Выделение К5-полисахарида из Escherichia coli
b) N-деацетилирование/N-сульфатирование
c) С-5 эпимеризация
d) Суперсульфатирование
e) Селективное O-десульфатирование
f) (Необязательно) селективное 6-O-сульфатирование
g) N-сульфатирование
Более подробно различные стадии этого способа рассмотрены ниже.
а) Выделение К5-полисахарида из Escherichia coli
E.coli выращивают в колбе Эриенмейера (Erienmeyer flask) согласно патенту М199А001465 с использованием среды состава:
рН 7.3
Среду стерилизуют при 120°С в течение 20 мин.
Раствор глюкозы готовят отдельно и стерилизуют при 120°С в течение 30 мин, а затем стерильно добавляют к среде.
В колбу Эриенмейера (Erienmeyer flask) добавляют суспензию клеток E.coli Bi 8337/41 (010:К5:Н4), выращенных на скошенном Triptic-соевом агаре (косячок); и инкубируют в течение 24 часов при 37°С при контролируемом перемешивании (160 оборотов/мин, пробег 6 см). Рост бактерий измеряют путем подсчета числа клеток с помощью микроскопа.
В 14 л ферментер Chemap-Braun, содержащий упомянутую выше среду, добавляют 0.1% культуры из вышеупомянутой колбы Эриенмейера (Erienmeyer flask) и проводят ферментацию в течение 18 часов при аэрации 1 vm (vm означает 1 объем воздуха на 1 объем жидкости в минуту) при перемешивании со скоростью 400 оборотов/мин (rpm ) при температуре 37°С. По окончании ферментации проводят определение рН, кислорода, остаточной глюкозы, синтезируемого полисахарида К5 и роста бактерий. В конце ферментации клетки инкубируют в течение 10 мин при 80°С.
Клетки отделяют от среды с помощью центрифугирования при 10000 оборот/мин, а супернатант подвергают ультрафильтрации с использованием модуля SS 316 (MST) с мембранами PES, обеспечивающими отсечение в 800 и 10000 D для уменьшения объема до 1/5. Полисахарид К5 осаждают добавлением 4 объемов ацетона при 4°С с последующей инкубацией в течение ночи при 4°С для осаждения. Затем К5 полисахарид отделяют или путем центрифугирования при 10000 оборот/мин в течение 20 мин., или путем фильтрации.
Депротеинизацию полученного осадка осуществляют с помощью протеазы типа II из Aspergillus Orizae в 0.1 М NaCl-буфере, содержащем 0.15 М EDTA, рН 8 и 0.5% SDS (10 милиграмм/литр фильтрата) при 37°С в течение 90 мин. Полученный раствор подвергают ультрафильтрации с использованием модуля SS 316 с мембранами, обеспечивающими отсечение 10000 D с двумя экстракциями 1 М NaCl и последующей промывкой до исчезновения поглощения в ультрафильтрате. Затем полисахариды К5 осаждают ацетоном; выход составляет 850 мг на литр фермента. Полученный препарат полисахарида характеризуют путем определения уроновых кислот (карбазольный метод), 1Н и 13С ЯМР, поглощения в УФ и содержания белка. Как правило, чистота превышает 80%.
Полученный полисахарид состоит из двух фракций, имеющих средний молекулярный вес 30000 и 5000 D соответственно, определенный с помощью HPLC с использованием двух Bio-sil SEC 250 (Bio Rad) - серийных колонках и Na2SO4 в качестве жидкой фазы со скоростью 0,5 мл/мин при комнатной температуре. Определение производят по отношению к стандартной кривой с использованием гепариновых фракций с известными молекулярными весами.
К 1% водному раствору очищенного полисахарида К5 добавляют тритон Х-100 до получения 5% раствора. Полученный раствор инкубируют в течение 2 часов при 55°С при перемешивании. Раствор нагревают до 75°С, и при последующем охлаждении до комнатной температуры в нем образуются две фазы. Верхнюю (органическую) фазу еще два раза подвергают термической обработке, при этом снова происходит образование двух фаз. Водную фазу, содержащую полисахарид, концентрируют при пониженном давлении и осаждают ацетоном или этанолом. Органическую фазу отбрасывают. Чистоту полученного образца определяют с помощью 1Н-ЯМР, она составляет 95%. Выход полисахарида составляет около 90%.
b) N-деацетилирование/N-сульфатирование
10 г очищенного полисахарида К5 растворяют в 100-2000 мл 2N-NaOH и инкубируют при 40-80°С до полного деацетилирования, как правило, не более 30 часов. Раствор оставляют при комнатной температуре и доводят рН до нейтрального с помощью 6N HCl. К раствору деацетилированного К5 добавляют 10-40 г карбоната натрия при температуре 20-65°С и 10-40 г сульфатирующего агента, выбираемого из числа легко доступных реагентов, таких как пиридин-SO3 аддукт, триметиламин-сульфотриоксид и др. Сульфатирующий агент добавляют в течение продолжительного времени, вплоть до 12 часов. В конце реакции, если это необходимо, температуру раствора понижают до комнатной, а рН доводят до 7.5-8 с помощью 5% раствора соляной кислоты.
Полученный продукт очищают от солей каким-либо из общеизвестных способов, например, диафильтрацией с помощью 1000 D спиральной мембраны (prepscale cartridge-Millipore). Диафильтрацию проводят до проводимости менее 1000 μS, предпочтительно менее 100 μS. Объем раствора уменьшают до получения 10% концентрации полисахарида, причем для концентрирования используют ту же систему фильтрации. При необходимости концентрированный раствор высушивают с помощью обычных методов. С помощью 13С-ЯМР было показано, что соотношение углерод N-сульфат/N-ацетил составляет от 10/0 до 7/3.
с) С-5 эпимеризация.
В соответствии с настоящим изобретением этап С-5 эпимеризации может быть выполнен с помощью фермента глюкуронил С-5 эпимеразы (далее С-5 эпимераза), используемой в растворенном или в иммобилизированном виде.
С-5 эпимеризадия с растворенным ферментом
От 1.2×107 до 1.2×1011 cpm (имп/мин) природного или рекомбинантного фермента С-5 эпимеразы, определенных в соответствии со способом, описанным Campbell P. и др., Analytical Biochemistry 131, 146-152 (1983), растворяют в 2-2000 мл 25 mM Hepes (N-2-гидроксиэтилпиперазин-N'-2-этансульфоновая кислота), рН от 5.5 до 7.4, содержащем 0.001-10 г N-деацетилированного, N-сульфатированного К5 и один или более ионов, выбранных из бария, кальция, магния, марганца с концентрацией от 10 до 60 mM. Реакцию проводят при температуре от 30 до 40°С, предпочтительно при 37°С, в течение 1-24 часов. В конце реакции фермент инактивируют при 100°С в течение 10 минут.
Продукт очищают на DEAE-сорбенте или с помощью DEAE картриджа Sartobind с элюцией 2М NaCl с последующим обессоливанием на Sephadex G-10, или осаждением 2 объемами этанола с последующей очисткой на IR 120 H+ для перевода его в натриевую соль. Соотношение идуроновой/глюкуроновой кислоты в полученном продукте составляет от 40:60 до 60:40, как было определено с помощью 1Н-ЯМР согласно WO96/14425.
С-5 эпимеризация с помощью иммобилизированного фермента
Фермент С-5 эпимеразу, природную или рекомбинантную, можно иммобилизовать на различных инертных носителях, например, на смолах, мембранах или стеклянных шариках, модифицированных различными функциональными группами, с использованием обычных способов иммобилизации для ферментов, например с помощью бромциана, глютардегида, карбодиимида или же с помощью ионообменных реакций фермента со смолой или путем осаждения фермента на мембране. В соответствии с настоящим изобретением иммобилизацию фермента на инертный носитель проводят в присутствии N-деацетилированного, N-сульфатированного субстрата К5 для предотвращения связывания по активному центру фермента с последующим уменьшением активности.
Активность иммобилизированного фермента определяют путем рециркуляции через колонку, содержащую иммобилизированный фермент, такого количества N-деацетилированного, N-сульфатированного К5, которое теоретически может быть ферментировано иммобилизированным ферментом в количестве cpm, растворенного в 25 mM Hepes, 0.1 М KCl, 0.01% Тритона X100 и 0.15 М EDTA при рН 7.4 и при 37°С в течение ночи со скоростью 0.5 мл/мин. После хроматографической очистки на DEAE и удаления солей на Sephadex G-10 продукт лиофилизируют и определяют содержание идуроновой кислоты методом 1Н-ЯМР. Соотношение идуроновой кислоты/глюкуроновой кислоты должно составить около 30:70.
20-1000 мл 25 mM раствора HEPES, pH от 6 до 7.4, содержащего один или несколько ионов, выбранных из бария, кальция, магния, марганца, с концентрацией от 10 до 60 mM, а также 0.001-10 г N-деацетилированного N-сульфатированного К5, при температуре от 30 до 40°С подвергают рециркуляции со скоростью 30-160 мл/час от 1 до 24 часов в колонке, содержащей от 1.2×107 до 3×1011 cpm-эквивалентов иммобилизированного фермента на инертном носителе. После окончания реакции продукт очищают с помощью способов, которые рассмотрены в разделе эпимеризации в растворе. Соотношение идуроновой и глюкуроновой кислот в полученном продукте составляет от 40:60 до 60:40.
d) Суперсульфатирование
10% раствор эпимеризованного продукта, полученного на стадии (с), охлаждают до 10°С, а затем пропускают через IR-120 Н* или эквивалентную катионообменную смолу (35-100 мл). Хроматографию проводят при температуре 10°С. После нанесения раствора колонку промывают деионизированной водой до pH элюата не более 6 (около 3 объемов деионизированной воды). Кислый раствор доводят до нейтрального pH с помощью третичного или четвертичного амина, как, например, тетрабутиламмониум гидроксид (15% водный раствор), при этом образуется соответствующая соль аммония. Раствор концентрируют до минимального объема и модифицируют. Полученный продукт суспендируют в 20-500 мл DMF или DMSO и добавляют 15-300 г сульфатирующего агента, такого как пиридин-SO3-аддукт в твердом виде или в растворе DMF или DMSO. Раствор инкубируют при 20-70°С, предпочтительно при 40-60°С, в течение 2-24 часов.
По окончании реакции раствор охлаждают до комнатной температуры и продукт осаждают ацетоном, насыщенным хлоридом натрия. Осадок отделяют от растворителя с помощью фильтрации, растворяют в минимальном объеме деионизированной воды (например, 100 мл) и добавляют хлорид натрия до концентрации 0.2 М раствора. рН раствора доводят до 7.5-8 с помощью 2N NaOH и добавляют ацетон для осаждения. Осадок отделяют от растворителя фильтрацией. Полученное твердое вещество растворяют в 100 мл деионизированной воды и очищают от оставшихся солей ультрафильтрацией, как это описано на стадии (b). Аликвоты лиофилизируют для структурного анализа суперсульфатированного продукта методом 13С-ЯМР.
Содержание сульфатов на дисахарид в полученном продукте составляет 2.0-3.5, что было определено согласно Casu и др., Carbohyd. Res. Vol.39, рр.168-176 (1975). Положение 6 аминосахара сульфатировано на 80-95%, а положение 2 вообще не сульфатировано. Другие сульфатные группы присутствуют в положении 3 аминосахара и в положении 2 и 3 уроновой кислоты.
е) Селективное O-десульфатирование
Раствор продукта, полученного на стадии (d), хроматографируют на IR-120 Н+ или эквивалентной катионноообменной смоле (35-100 мл). После нанесения раствора смолу промывают деионизированной водой до рН элюата более 6 (около 3 объемов деионизированной воды). Кислый раствор нейтрализуют добавлением пиридина. Раствор концентрируют до минимального объема и модифицируют. К продукту добавляют 20-2000 мл раствора DMSO/метанол (9/1 объем/объем) и полученный раствор инкубируют при 45-90°С в течение 1-8 часов. В конце к раствору добавляют 10-200 мл деионизированной воды, а затем осаждают ацетоном, насыщенным хлоридом натрия.
При селективном O-десульфатировании сначала удаляются сульфатные группы из положения 6 аминосахара, затем из положений 3 и 2 уроновой кислоты и, наконец, - из положения 3 аминосахара. Получаемое твердое вещество очищают диафильтрацией, как это описано для стадии (b). Аликвоты лиофилизируют для проведения анализа с помощью 13С-ЯМР. Если из ЯМР-анализа следует, что содержание сульфатов в положении 6 аминосахара превышает 60% (содержание сульфата вычисляют согласно Casu и др. Arzneimittel-forschiung Drug Research 33-1, 135-142 (1983)), следует переходить непосредственно к стадии (g). В противном случае следует переходить к стадии (f).
f) Селективная 6-O-сульфатирование (необязательно)
Для получения третичной или четвертичной соли раствор, содержащий продукт, полученный на стадии (е) обрабатывают согласно стадии (d), однако все операции проводят при 20-25°С.
Аммонийную соль суспендируют в 20-500 мл DMF до образования взвеси. Суспензию охлаждают до 0°С и обрабатывают сульфатирующим агентом, таким как пиридин-SO3, аддукт, в количестве, рассчитанном как функция процентного содержания сульфата в положении 6 аминосахара, который должен быть восстановлен с учетом минимального 60% содержания 6-O-сульфата, вычисленного как описано выше. Количество сульфатирующего агента составляет, как правило, от двух до десяти эквивалентов, принимая во внимание гидроксильные функции по отношению к сульфату. Сульфатирующий агент добавляют в один прием или же в несколько приемов, но не более чем за 20 минут. Сульфатирующий агент можно добавлять в виде порошка или же растворенным в небольшом объеме DMF. Раствор инкубируют при 0-5°С в течение 0.5-3 часов. Для осаждения продукта к раствору добавляют ацетон, насыщенный хлоридом натрия. Осажденное твердое вещество очищают диафильтрацией, как это описано на стадии (b). Аликвоты лиофилизируют для проведения структурного анализа методом 13С-ЯМР. Если содержание 6-O-сульфата, определенное с помощью ЯМР, оказывается ниже 60%, стадию повторяют.
g) N-сульфатирование
N-сульфатирование продукта, полученного на стадии (f) или на стадии (е), проводят аналогично стадии (b). Полученный продукт характеризуют с помощью 1Н-ЯМР и 13С-ЯМР. С помощью биологических тестов было показано, что гликозаминогликаны согласно изобретению обладают активностями antiXa, APTT, HCII, Anti IIa и сродством к ATIII.
Получаемый продукт можно фракционировать с помощью хроматографии или же путем ультрафильтрации, получая при этом фракции с низким молекулярным весом от 2000 до 8000 D и с высоким молекулярным весом от 25000 до 30000 D, или же продукт можно подвергнуть контролируемой деполимеризации. С помощью известных методов, таких как, например, деаминирование азотистой кислотой, как это описано в WO8203627.
Сравнение биологических активностей гликозаминогликанов согласно изобретению (IN-2018 UF и IN-2018 LMW), UF-Гепарина (4-й междунар. Стандарт) и LMW -Гепарина (1-й междунар. Стандарт) приведены в Табл.1
UF
Ссылки
1. Thomas D.P. et al., Thrombosis and Haemostasis 45, 214-(1981) against the IV heparin international standard.
2. Andersson et al. Thrombosis Res. 9, 575 (1976) against the IV heparin international standard.
3. Опыт выполняется путем смешивания 20 мл HCII (Stago) 0.05 PEU/мл, растворенных в воде, с 80 мкл растворенного образца с различными концентрациями и с 50 мкл тромбина (0.18 U/ml- Boehringer) в 0.02 М ТРИС-буфере, рН 7.4, содержащем 0.15 М NaCI и 0.1% PEG-6000. Раствор инкубируют в течение 60 с при 37°С, а затем к нему добавляют 50 мкл 1 mM хромогенного субстрата Spectrozyme (American Diagnostic). В течение 180 с каждую секунду записывается поглощение при 405 nm с помощью автоматического коагулометра ACL-7000 (IL).
4. Опыт выполняется путем смешивания 30 мкл раствора 0.5 U/мл ATIII (Chromogenix), растворенного в 0.1 М ТРИС-буфера, рН 7.4, с 30 мкл раствора образца с различными концентрациями и с 60 мкл тромбина (5.3 nKat/Mn-Chromogenix) в 0.1 М рН 7.4 ТРИС-буфере. Раствор инкубируют в течение 70 с при 37°С, а затем добавляют 60 мкл 0.5 mM хромогенного субстрата S-2238 (Chromogenix) в воде. Реакцию контролируют в течение 90 с, записывая каждую секунду поглощение при 405 nm с помощью автоматического коагулометра ACL-7000 (IL).
5. Harenberg and De Vries, J. Chromatography 261, 287-292 (1983).
6. Hook M. et al. FEBS Letters 66, 90-93 (1976).
Как следует из Табл.1, продукт, полученный согласно изобретению, обладает активностью, сопоставимой с активностью гепарина, полученного путем экстракции в отношении фактора Ха (1), и пониженной в отношении общей (2) антикоагулянтной активности, в то же время активность в отношении ингибирования тромбина (3, 4) оказалась у продукта согласно изобретению значительно выше. Приведенные результаты показывают, что продукт согласно изобретению обладает большей антитромботической активностью и меньшими побочными эффектами типа кровотечения по сравнению с гепарином, полученным путем экстракции.
Благодаря своим свойствам гликозаминогликаны согласно настоящему изобретению можно использовать в чистом виде или в комбинации с фармацевтически приемлемыми носителями или разбавителями при антикоагулянтной и антитромботической терапии (The Pharmacological Basis of Therapeutics, 3rd, Edit. Louis S. Goodman & Alfred Gilman, p.1448).
Следовательно, настоящее изобретение также включает композиции, содержащие эффективное количество гликозаминогликанов, согласно изобретению в комбинации с фармацевтически приемлемыми носителями или разбавителями.
Настоящее изобретение также относится к способу терапии, включающему введение эффективного количества, составляющего от 1 до 100 мг/день, гликозаминогликанов согласно изобретению для антикоагулятного и антитромботического лечения.
Сведения, подтверждающие возможность осуществления изобретения
ПРИМЕР 1
Пример 1 выполняют поэтапно в следующей последовательности:
а) 10 г полисахарида К5, полученного путем ферментации, как описано в патенте М199А001465, и имеющего чистоту 80% (фиг.2), растворяли в деионизированной воде до концентрации 1%. Затем добавляли Тритон Х-100 до концентрации 5%. Раствор инкубировали при 55°С при помешивании в течение 2 часов. Раствор нагревали до 75°С и инкубировали при этой температуре до образования однородной взвеси. Затем раствор быстро охлаждали до комнатной температуры. При охлаждении образовывались две фазы. Верхнюю (органическую) фазу подвергали повторной тепловой обработке, как описано выше, еще 2 раза. Водяную фазу, содержащую полисахарид К5, концентрировали при пониженном давлении до 1/10 объема, после чего осаждали ацетоном или этанолом. Органическую фазу отбрасывали. Выделенный продукт представлял собой полисахарид К5 с чистотой около 90%, как было определено с помощью 1Н-ЯМР-спектрометра (фиг.3) по отношению к спектру внутреннего стандарта (фиг.1).
b) Продукт, полученный на стадии (а), растворяли в 1000 мл 2N NaON и инкубировали при температуре 60°С в течение 18 часов. Раствор охлаждали до комнатной температуры и рН доводили до нейтрального с помощью 6N соляной кислоты. Таким образом получали N-деацетилированный полисахарид К5.
При 40°С к раствору, содержащему N-деацетилированный К5, добавляли 10 г карбоната натрия и 10 г пиридин-сульфотриоксида аддукта в течение 10 минут. В конце реакции раствор охлаждали до комнатной температуры, а затем доводили рН до 7,5-8 раствором соляной кислоты. Полученный продукт, представляющий собой N-деацетилированный N-сульфатированный полисахарид К5, очищали от солей с помощью диафильтрации с использованием спиральной мембраны 1000D (prepscale cartridge-Millipore). Диафильтрацию проводили до электропроводности растворенного вещества ниже 100 мкСм. Отфильтрованный полисахарид доводили до концентрации 10% с помощью аналогичной системы диафильтрации, а затем лиофилизировали. Соотношение N-сульфат/N-ацетил в полученном веществе составляло 9,5/0,5, как было определено методом 13С- ЯМР (фиг.4).
c) 1-Иммобилизация С-5 эпимеразы
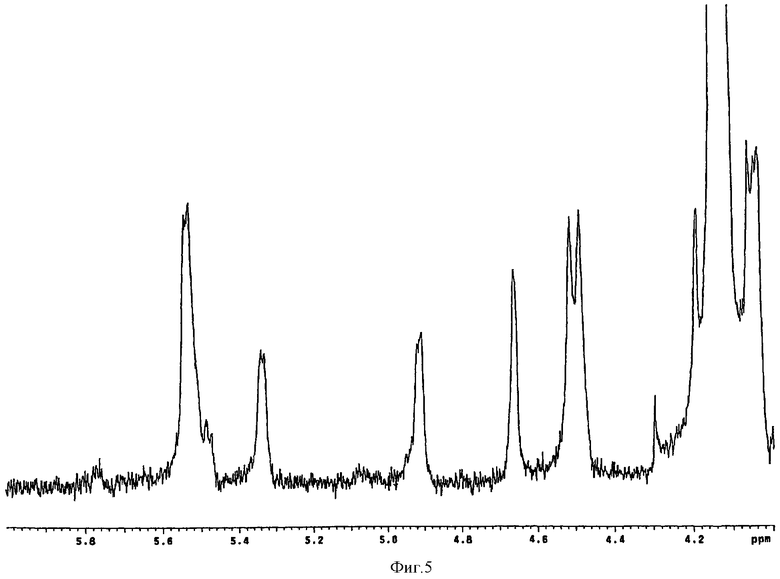
5 мг рекомбинантной эпимеразы С-5, полученной согласно WO98/48006 (соответствует примерно 1.2×10 cpm), растворяли в 200 мл 0,25 М Hepes, рН 7,4, содержащего 0.1 М KCl, 0.1% Тритон Х-100 и 15 мМ EDTA, добавляли 100 мг N-деацитилированного N-сульфатированного К5, полученного согласно стадии (b). Раствор подвергали диафильтрации на мембране 30000 D при температуре 4°С до исчезновения N-деацитилированного N-сульфатированного К5 в диафильтрате. С помощью диафильтрации проводили замену буфера на 200 мМ NaHCO3 с рН 7. После концентрирования до 50 мл добавляли 50 мл активированной CNBr Sepharose 4В и инкубировали при 4°С в течение ночи. По завершении реакции суспензию центрифугировали и определяли количество остаточного фермента в супернатанте методом Quantigold (Diversified Biotec). Оказалось, что в супернатанте фермент отсутствует, что подтверждает, что вышеописанный метод позволяет иммобилизовать фермент на 100%. Для блокирования оставшихся активных групп сорбент промывали 100 мМ ТРИС-HCl, рН 8. Для определения активности иммобилизованного фермента сорбент помещали в лабораторную колонку в количестве, теоретически соответствующем 1.2×107 cpm. Затем 1 мг N-деацетилированного N-сульфатированного К5, полученного согласно стадии (b), растворенного в 25 мМ Hepes, 0,1 М KCl, 0,015 М EDTA, 0,01% Тритон Х-100, рН 7,4, рециркулировали в указанной колонке со скоростью 0,5 мл/мин при 37°С в течение ночи. Полученный продукт хроматографировали на DEAE сорбенте, обессоливали на Sephadex G10, лиофилизировали и определяли содержание идуроновой кислоты с помощью 1H-ЯМР, согласно WO96/14425. Соотношение идуроновая/глюкоуроновая кислота составляло 30/70 (фиг.5).
2-Эпимеризация
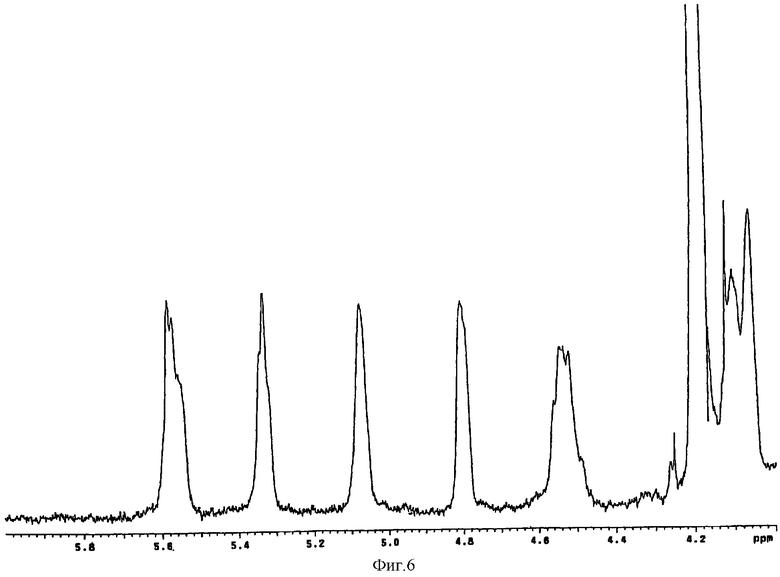
10 г N-деацетилированного N-сульфатированного полисахарида К5 растворяли в 600 мл 25 мМ Hepes, рН 6,5, содержащего 50 мМ CaCl2. Полученный раствор рециркулировали через 50-мл лабораторную колонку, содержащую иммобилизованный фермент. Рециркуляцию проводили в течение 24 часов со скоростью 200 мл/час при температуре 37°С. Полученный продукт очищали с помощью ультрафильтрации и осаждали этанолом. Осадок повторно растворяли в воде до концентрации 10%. В результате получали эпимеризованный продукт с соотношением идуроновая/глюкуроновая кислота 48/52 по сравнению с соотношением 0/100 в исходном продукте. Процентную величину эпимеризации вычисляли с помощью 1Н-ЯМР (фиг.6). Выход продукта, вычисленный по определению содержания уроновых кислот по отношению к стандарту с помощью карбазольного метода (Bitter and Muir Anal. Biochem. 39, 88-92-1971), составлял 90%.
d) Раствор, содержащий эпимеризованный продукт с концентрацией 10%, полученный на стадии (с), охлаждали до 10°С, а затем хроматографировали на IR-120 Н+ (50 мл) при температуре 10°С. После нанесения колонку промывали тремя объемами деионизированной воды. рН растворенного вещества был выше 6. Кислый раствор нейтрализовали 15% водным раствором гидроксида тетрабутиламмония. Полученный раствор концентрировали до 1/10 объема в вакууме на роторном испарителе при 40°С, а затем лиофилизировали. Продукт суспендировали в 200 мл DMF и добавляли 150 г пиридин-SO3 аддукта, растворенного в 200 мл DMF. Раствор инкубировали в течение 18 часов при 45°С. По завершении реакции раствор охлаждали до комнатной температуры и добавляли 1200 мл ацетона, насыщенного хлоридом натрия. Осадок отделяли от растворителя с помощью фильтрации, растворяли в 100 мл деионизированной воды и добавляли хлорид натрия до 0,2 М. С помощью 2 N гидроксида натрия рН раствора доводили до 7,5-8 и добавляли 300 мл ацетона. Осадок отфильтровывали. Полученное твердое вещество растворяли в 100 мл деионизированной воды и очищали от остаточных солей с помощью диафильтрации аналогично стадии (b). Результаты 13С-ЯМР анализа лиофилизированного образца суперсульфатированного продукта представлены на фиг.7.
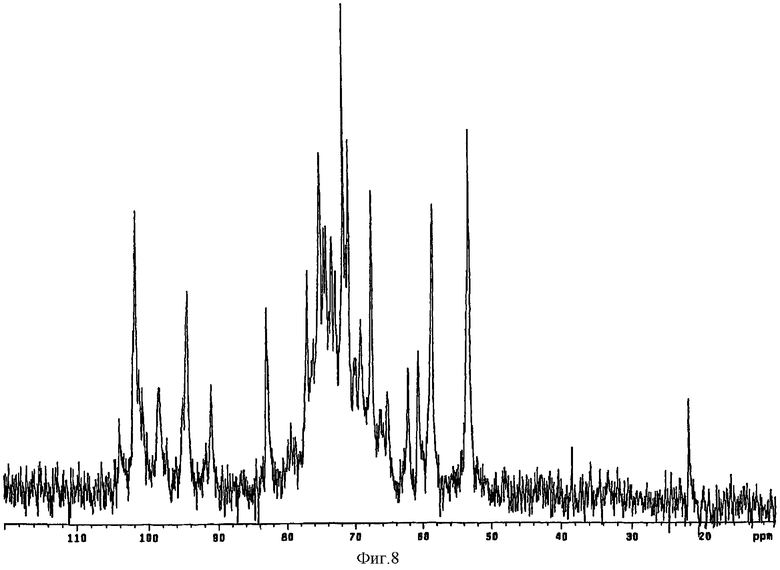
e) Раствор, содержащий продукт, полученный на стадии (d), хроматографировали на IR-120 H+ (50 мл). После нанесения колонку промывали тремя объемами деионизированной воды. рН растворенного вещества был выше 6. Кислый раствор нейтрализовали пиридином. Полученный в результате раствор концентрировали до 1/10 объема в вакууме на испарителе при 40°С, а затем лиофилизировали. К полученному продукту в форме пиридиновой соли добавляли в 500 мл раствора DMSO/метанол (9/1 V/V). Раствор инкубировали в течение 3,5 часов при 60°С, добавляли 50 мл деионизированной воды и 1650 мл ацетона, насыщенного хлоридом натрия. Полученное твердое вещество очищали с помощью диафильтрации согласно стадии (b) с получением 10% раствора. Данные 13С-ЯМР анализа лиофилизированного образца приведены на фиг.8. Как следует из фиг.8, содержание сульфатов в положении 6 аминосахара составляло 35%.
f) Раствор, содержащий продукт, полученный на стадии (е), хроматографировали на IR-120 Н* (50 мл). После нанесения колонку промывали тремя объемами деионизированной воды. рН растворенного вещества был выше 6. Кислый раствор затем нейтрализовали 15% водным раствором гидроксида тетрабутиламмония. Полученный раствор концентрировали до 1/10 объема в вакууме на роторном испарителе при температуре 40°С и лиофилизировали. Полученный продукт в форме соли тетрабутиламмония суспендировали в 200 мл DMF. Суспензию охлаждали до 0°С и добавляли 100 мл пиридин-SO3 аддукта в DMF. Сульфатирующий реагент добавляли однократно. Раствор инкубировали при 0°С в течение 1,5 часов, а затем добавляли в 750 мл ацетона, насыщенного хлоридом натрия. Полученное твердое вещество очищали с помощью диафильтрации согласно стадии (b).
g) Раствор, полученный на стадии (f), подвергали N-сульфатированию
Данные 13С-ЯМР анализа лиофилизированного образца показаны на фиг.9.
Химико-физические и биологические свойства полученного продукта приведены в Табл.2, строка 3, в сравнении с международным стандартом на гепарин IV и международным стандартом на гепарин I с низким молекулярным весом.
ПРИМЕР 2
Продукт получали согласно способу, описанному в Примере 1, с тем отличием, что на стадии (с) использовали иммобилизованную С-5 эпимеразу, выделенную из мастоцитомы мыши, согласно Jacobsson et al., J. Biol. Chem. 254, 2975-2982 (1979), в буфере, содержащем 40 мМ CaCl2, рН 7,4. Соотношение идуроновая/глюкуроновая кислот в полученном продукте составляло 59,5:40,5. Свойства продукта приведены в Табл.2, строка 4.
ПРИМЕР 3
Продукт получали согласно способу, описанному в Примере 1, с тем отличием, что на стадии (с) использовали иммобилизованную С-5 эпимеразу, выделенную из печени крупного рогатого скота, согласно WO96/14425, при этом рН буфера для реакции составлял рН 7,4, а время реакции составляло 32 часа. Время реакции на стадии (е) составляло 4 часа. Соотношение идуроновая/глюкуроновая кислот в полученном продукте составляло 55,4:44,6. Свойства продукта приведены в Табл.2, строка 5.
ПРИМЕР 4
Продукт получали согласно способу, описанному в Примере 1, с тем отличием, что на стадии (с) использовали рекомбинантную С-5 эпимеразу в растворе, а для эпимеризации использовали 10 г N-деацетилированного N-сульфатированного К5, растворенного в 1000 мл 25 мМ Hepes, рН 6,5, содержащего 50 мМ CaCl2. К этому раствору добавляли эквивалент 1,5×1011 cpm рекомбинантного фермента, описанного в Примере 1. Раствор инкубировали при температуре 37°С в течение 24 часов. Фермент инактивировали при температуре 100°С в течение 10 мин и фильтровали через фильтр 0,45 μ до получения чистого раствора, содержащего продукт. Полученный продукт очищали методом диафильтрации и осаждали этанолом или ацетоном. Осадок растворяли в воде до концентрации, равной 10%, и обрабатывали аналогично Примеру 1, однако время реакции на стадии (е) составило 2 часа. Соотношение идуроновая/глюкуроновая кислот в полученном продукте составляло 56:44. Свойства продукта приведены в Табл.2, строка 6.
ПРИМЕР 5
Продукт получали согласно способу, описанному в Примере 4, с использованием на стадии (с) фермента из мастоцитомы мыши, аналогично Примеру 2, в буферном растворе с рН 7,4, содержащем 40 мМ BaCl2. Реакцию проводили в течение 18 часов. Кроме этого, на стадии (е) время реакции составляло 3 часа. Соотношение идуроновая/глюкуроновая кислот в полученном продукте составляло 40,1:59,9. Свойства продукта приведены в Табл.2, строка 7.
ПРИМЕР 6
Продукт получали согласно способу, описанному в Примере 4, с использованием на стадии (с) С-5 эпимеразы из печени крупного рогатого скота, аналогично Примеру 3, в буферном растворе, содержащем 12,5 мМ MnCl2. Реакцию проводили в течение 14 часов. Кроме этого на стадии (е) время реакции составляло 4 часа. Соотношение идуроновая/глюкуроновая кислот в полученном продукте составляло 44,3:55,7. Свойства продукта приведены в Табл.2, строка 8.
ПРИМЕР 7
Продукт получали согласно способу, описанному в Примере 4, с использованием на стадии (с) буферного раствора с рН 7,4, содержащего 37,5 мМ MgCl2. Реакцию проводили в течение 16 часов. Кроме этого на стадии (е) время реакции составляло 4 часа. Соотношение идуроновая/глюкуроновая кислот в полученном продукте составляло 47,5:52,5. Свойства продукта приведены в Табл.2, строка 9.
ПРИМЕР 8
Продукт получали согласно способу, описанному в Примере 3, с использованием на стадии (с) буферного раствора с рН 7,0, содержащего 10 мМ MgCl2 и 5 мМ MnCl2. Реакцию проводили в течение 24 часов. Кроме этого на стадии (е) время реакции составляло 3 часа. Соотношение идуроновая/глюкуроновая кислот в полученном продукте составляло 44,8:55,2. Свойства продукта приведены в Табл.2, строка 10.
ПРИМЕР 9
Продукт получали согласно способу, описанному в Примере 6, с использованием на стадии (с) буферного раствора с рН 7,4, содержащего 10 мМ MgCl2, 5 мМ CaCl2, 10 мМ MnCl2. Реакцию проводили в течение 24 часов. Кроме этого на стадии (е) время реакции составляло 3 часа. Соотношение идуроновая/глюкуроновая кислот в полученному продукте составляло 52:48. Свойства продукта приведены в Табл.2, строка 11.
ПРИМЕР 10
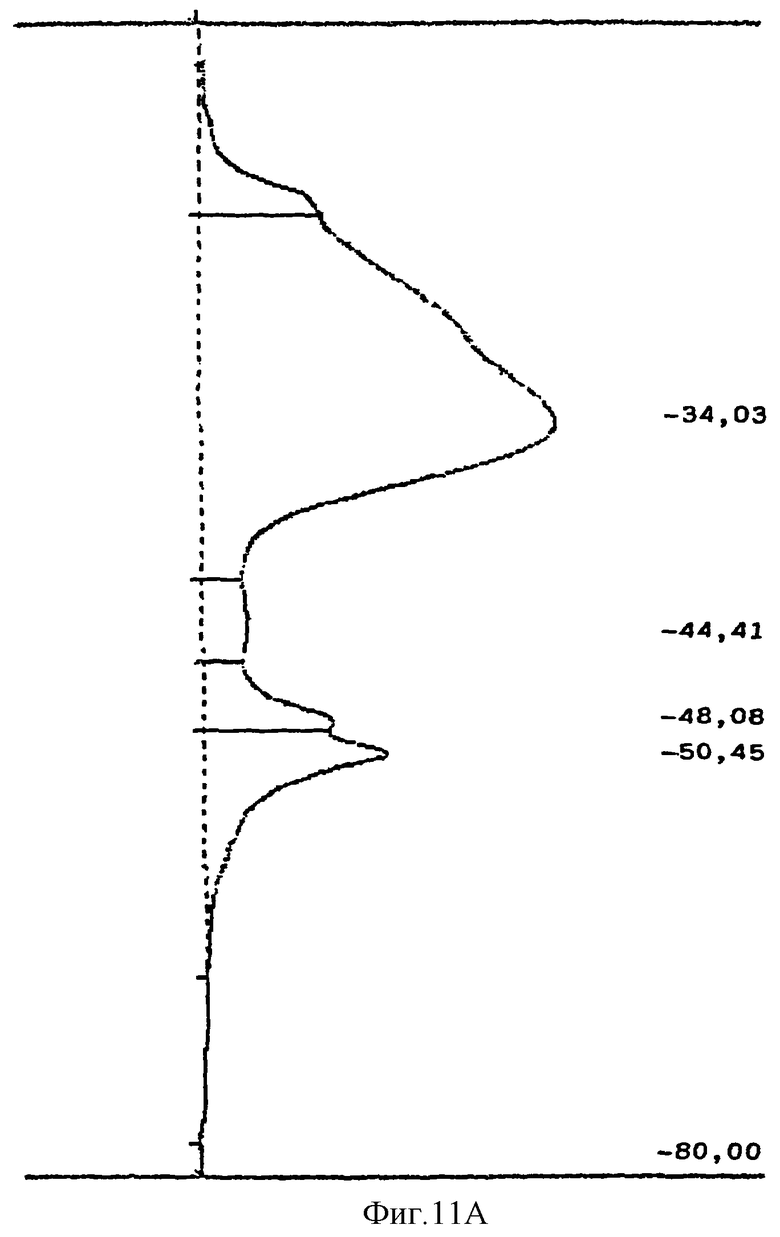
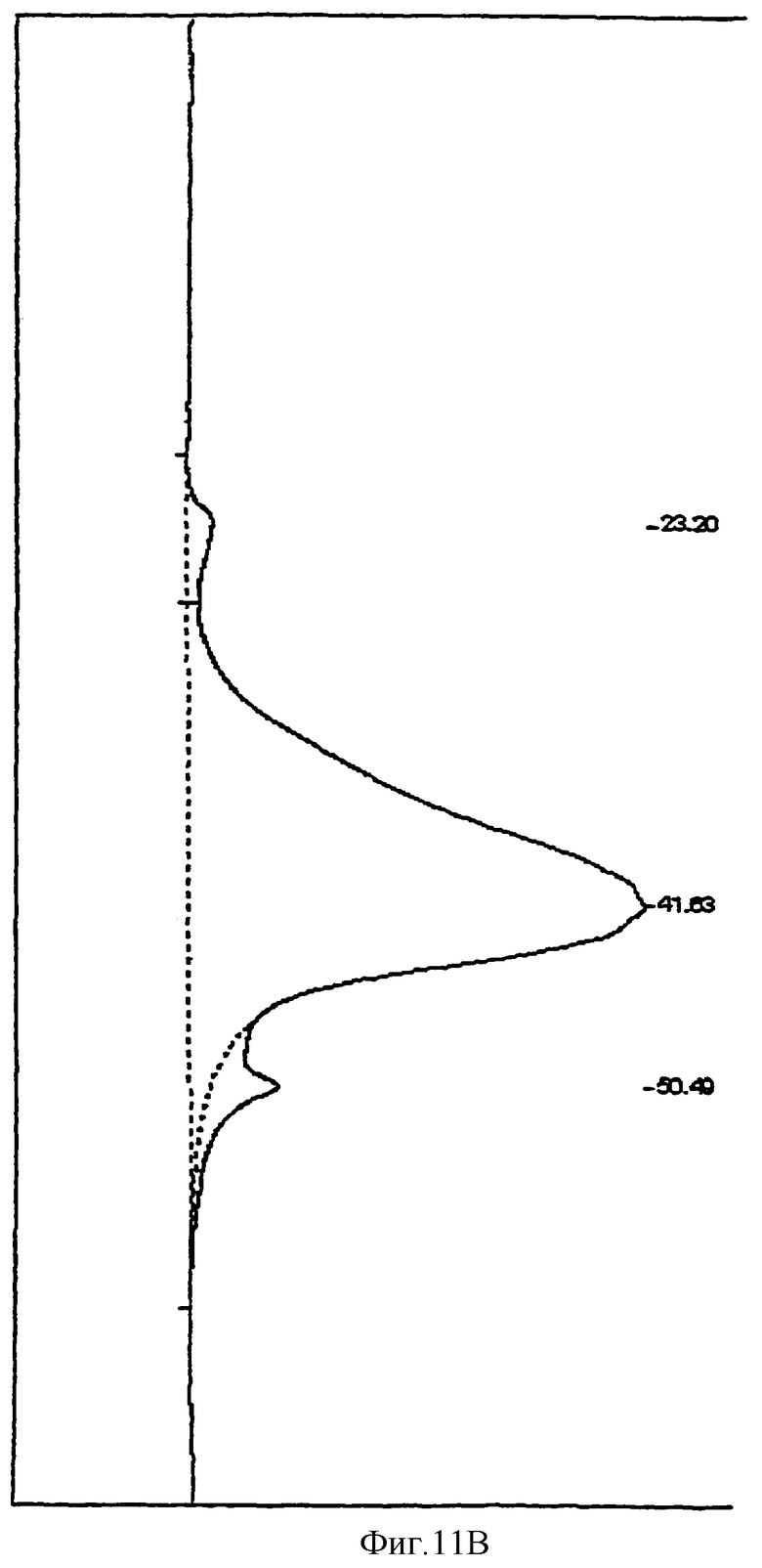
Продукт, полученный в Примере 3, с молекулярной массой, определенной согласно Harenberg and De Vries, J. Chromatography 261, 287-292 (1983) (фиг.10), разделяли методом гель-фильтрации. Для этого 1 г продукта растворяли в 20 мл 1 М буферного раствора, содержащего NaCl, и наносили на колонку, содержащую 1000 мл катионита Sephacryl HR S-400 (Amersham-Pharmacia). Элюрацию проводили 2000 мл 1М буферного раствора, содержащего NaCl, и собирали фракции объемом 50 мл с помощью коллектора фракций (Gilson). Содержание продукта во фракциях определяли с помощью карбазольного метода (Bitter and Muir, Anal. Biochem. 39, 88-92-1971). Фракции объединяли в группы А и В с высоким и с низким молекулярным весом соответственно. С помощью вакуумного испарителя объединенные фракции упаривали до концентрации 10% и обессоливали на колонке, содержащей 500 мл Sephadex G-10 (Amersham-Pharmacia). Обессоленные продукты лиофилизировали и получали объединенные фракции А и В (фиг.11А и фиг.11В). Свойства полученных продуктов приведены в Табл.2, строки 12 и 13.
ПРИМЕР 11
Продукт, полученный в Примере 4, подвергали контролируемому расщеплению азотистой кислотой согласно WO 8203627. Для этого 5 г образца растворяли в 250 мл воды и охлаждали до 4°С. рН доводили до 2,0 с помощью 1 N соляной кислоты, охлажденной до 4°С, после чего быстро добавляли 10 мл 1% раствора нитрита натрия. При необходимости рН вновь доводили до 2,0 с помощью 1 N соляной кислоты и инкубировали в течение 15 мин при слабом перемешивании. Раствор нейтрализовали 1 N NaOH, охлажденным до 4°С. Затем добавляли 250 мг борогидрида натрия, растворенного в 13 мл деионизированной воды, и раствор инкубировали 4 часа. Затем рН раствора доводили до 5,0 с помощью 1 N соляной кислоты, инкубировали в течение 10 минут для нейтрализации избытка борогидрида натрия и нейтрализовали 1 N NaOH. Продукт осаждали тремя объемами этанола и высушивали в вакуумной печи. Свойства полученного продукта приведены в Табл.2, строка 14.
(4th int. STD)
(1st int.Std)
Ссылки (1)-(6) определены выше для Табл.1. Из результатов Таблицы 2 следует, что продукт, полученный способом, согласно изобретению обладает активностью, сравнимой с экстрактивным гепарином, в отношении к Ха-фактору (1), пониженной общей (2) антикоагулянтной активностью и повышенной активностью в отношении ингибирования тромбина (3, 4). Таким образом, комбинация свойств полученного продукта определяет его повышенную антитромботическую активность при уменьшении побочных эффектов, таких как кровотечение, наблюдаемое для экстрактивного гепарина.
ПРИМЕР 12
Два образца Q93B/159 и Q93B/160 были получены согласно примеру 3 из предшествующего уровня техники WO 9743317.
Свойства этих двух продуктов показаны в таблице 3 и сравнены со значением для продуктов, полученных согласно предыдущим примерам.

Как видно из таблицы 3, значения HCII/AntiXa соотношения для известных производных полисахарида Q93B/159 и Q93B/160 (1,13 и 1,34) ниже диапазона значений, приведенного в пункте 1 формулы изобретения (1,5-4,0). Другие производные полисахарида, раскрытые в WO 9743317 и проверенные аналогичным образом, также имели значения HCII/AntiXa соотношения ниже 1,5. Это последовательное и существенное различие в значении величины HCII/AntiXa соотношения доказывает, что производные, известные из WO 9743317, отличаются от производных полисахаридов, заявленных в пункте 1 формулы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ПОЛИСАХАРИДОВ С ВЫСОКОЙ АНТИТРОМБОТИЧЕСКОЙ АКТИВНОСТЬЮ В ПЛАЗМЕ | 2004 |
|
RU2361881C2 |
| ЭПИМЕРИЗОВАННЫЕ ПРОИЗВОДНЫЕ ПОЛИСАХАРИДА К5 С ВЫСОКОЙ СТЕПЕНЬЮ СУЛЬФАТИРОВАНИЯ | 2003 |
|
RU2333222C2 |
| ПРОИЗВОДНЫЕ СУЛЬФАТИРОВАННЫХ ПОЛИСАХАРИДОВ, ИХ СПОСОБ ПОЛУЧЕНИЯ, МОДИФИКАЦИЯ И ПРИМЕНЕНИЕ | 2016 |
|
RU2708327C2 |
| N,О-СУЛЬФАТИРОВАННЫЕ ГЕПАРОЗАНЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ АНТИТРОМБОТИЧЕСКОЙ АКТИВНОСТЬЮ | 1991 |
|
RU2099353C1 |
| НЕАНТИКОАГУЛЯНТНЫЕ ГЛИКОЗАМИНОГЛИКАНЫ, СОДЕРЖАЩИЕ ПОВТОРЯЮЩИЕСЯ ДИСАХАРИДНЫЕ ЗВЕНЬЯ, И ИХ МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ | 2012 |
|
RU2617764C2 |
| СПОСОБ ПРОДУКЦИИ СУЛЬФАТИРОВАННОГО ПОЛИСАХАРИДА И СПОСОБ ПРОДУКЦИИ PAPS | 2021 |
|
RU2811941C1 |
| БИОСИНТЕТИЧЕСКИЙ ГЕПАРИН | 2017 |
|
RU2760706C2 |
| СПОСОБ ПОЛУЧЕНИЯ НИЗКОМОЛЕКУЛЯРНОГО ГЕПАРИНА - НАДРОПАРИНА КАЛЬЦИЯ | 2024 |
|
RU2837407C1 |
| ОПОСРЕДУЮЩИЕ ТРАНСПОРТ КОЛЛОИДНЫЕ ЛЕКАРСТВЕННЫЕ СОЕДИНЕНИЯ | 2009 |
|
RU2519225C2 |
| Способ получения о-ацилированных глюкозаминогликанов | 1989 |
|
SU1831487A3 |






Описано N-деацетилированное N-сульфатированное производное полисахарида К5, эпимеризованное, по меньшей мере, до 40% содержания L-идуроновой кислоты по отношению к общему содержанию уроновых кислот, имеющее молекулярный вес от 2000 до 30000 Да, содержащее от 25 до 50% по весу цепей, обладающих высоким сродством к ATIII, и обладающее антиокоагулянтной и антитромботической активностью с соотношением вышеуказанных активностей HCII/antiXa от 1,5 до 4. Также описан способ получения производного полисахарида К5, включающий выделение полисахарида К5 из Escherichia Coli, N-деацетилирование и N-сульфатирование, С-5 эпимеризацию D-глюкуроновой кислоты до L-идуроновой кислоты, суперсульфатирование, селективное O-десульфатирование, селективное 6-O-сульфатирование и N-сульфатирование, причем С-5 эпимеризацию производят с использованием фермента глюкуроносил С-5 эпимеразы в растворенной или в иммобилизованной форме в присутствии бивалентных катионов, выбранных из группы, включающей Ва, Са, Mg и Mn. 4 н. и 8 з.п. ф-лы, 3 табл., 11 ил.
| WO 9743317 А, 20.11.1997 | |||
| WO 9614425 А, 17.05.1996 | |||
| N,О-СУЛЬФАТИРОВАННЫЕ ГЕПАРОЗАНЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ АНТИТРОМБОТИЧЕСКОЙ АКТИВНОСТЬЮ | 1991 |
|
RU2099353C1 |

