Предметом настоящего изобретения являются новые N,O-сульфатированные гепарозаны, композиции, которые содержат эти N,O-сульфатированные гепарозаны, способ получения N,O-сульфатированных гепарозанов и фармацевтической композиции на их основе.
Известно, что гликозаминогликаны являются продуктами, которые могут быть получены путем извлечения из тканей животного происхождения. Некоторые из этих гликозаминогликанов обладают антикоагулирующими и антитромботическими свойствами, представляющими большой интерес. Типичными соединениями из этого семейства являются гепарин, его фрагменты и их производные, а также гепаринсульфат и дерматансульфат (хондроитинсульфат), которые, однако, имеют недостаток, обусловленный их происхождением и заключающийся в том, что они являются очень дорогостоящими.
В частности, известно, что дерматансульфат представляет собой семейство полимеров с переменной степенью полимеризации, образованных повторяющимися звеньями, состоящими из группы уроновой кислоты /идуронильной или глюкуронильной/ и из 4-ацетилсульфогалактозаминильной группы (H. W. Stuhlsatz "The Methodology of Connective Tissue Research", 1976, 137 146). Природный дерматансульфат имеет молекулярную массу, заключенную между 20000 и 40000 а. е. м. Это соединение представляет особый интерес в качестве антикоагулирующего и антитромботического средства (F. Fernandez et al. British Journal of Haematology, 1986, 64, 309 317).
С другой стороны, известно (I. Bjork, U. Zindahl "Molecular and Cellular Biochemistry, 1982, Dr. W Junk Publisher Pays Bas), что свертывание крови представляет собой сложный физиологический процесс, механизм которого может быть схематизирован и резюмирован следующим образом:
Некоторые стимулы, такие как контактная активация и тканевые факторы, дают начало последовательности активации для серии факторов свертывания, присутствующих в плазме крови, причем на приведенной выше схеме эти факторы обозначаются римскими цифрами, а наличие индекса "а" означает активированную форму /индекс "а" присутствует/ и неактивированную форму /индекс "а" отсутствует/ данного фактора свертывания.
Какова бы ни была природа стимула, конечные стадии являются идентичными: фактор Ха активирует фактор II /называемый также протромбином/, который в своей активированной форме /фактор IIа, называемый также тромбином/ вызывает частичный протеолиз растворимого фибриногена с высвобождением нерастворимого фибрина, т. е. основного компонента кровяного сгустка /тромба/.
В нормальных физиологических условиях белки-регуляторы, такие как антитромбин III /АТIII/ и гепариновый кофактор II /ГКII/, также присутствуют в плазме.
Антитромбин III проявляет ингибирующую активность по отношению к совокупности факторов свертывания, обозначенных звездочкой /*/ в приведенной выше схеме. Это ингибирование значительно усиливается в присутствии гепарина или синтетических олигосахаридов гепаринового типа /D. H. Atha et al. Biochemistry, 1985, 24, 6723 6729/.
Гепариновый кофактор II проявляет ингибирующую активность только по отношению к фактору IIа /тромбину/, который катализирует последнюю стадию свертывания. Эта активность значительно усиливается в присутствии гепарина или дерматансульфата (D. M. Tollefsen, J. Biol. Chem. /1983/, 258, 6713 6716).
Ингибирование фактора Ха или фактора IIа составляет примущественный способ для получения антикоагулирующей и антитромботической активности, потому что эти два фактора принимают участие на двух последних стадиях свертывания, которые являются независимыми от стимулирующего воздействия.
Чтобы обеспечить ингибирование только одного фактора IIa, имеется возможность, представляющая особый интерес, которая заключается в том, чтобы использовать специфичность гепаринового кофактора II и добиться усиления его ингибирующей активности. Дерматансульфат является известным продуктом, обладающим наиболее значительной усиливающей активностью этого типа.
Известно также, что образование основной гепариновой цепи происходит в две стадии. Сначала биосинтезируется гепарин, исходя из протеогликана - предшественника, у которого полисахаридная часть состоит из семейства полимеров с переменной степенью полимеризации, образованных повторяющимися β-Д-1,4-глюкоронил-a-N-ацетил-Д-1,4-глюкозамильными звеньями /дисахаридные звенья/ с молекулярной массой 379 а. е. м. Эта полисахаридная часть обычно называется N-ацетилгепарозан (J. Navia, Anal. Biochem. /1983/, 135, 134 - 140). Эта первая биосинтетическая стадия является единственным моментом, когда действительно можно говорить о "дисахаридном звене", так как вторая стадия биосинтеза будет изменять этот простой скелет значительным образом ("L'heparine, fabrication, Structure proprietes, analyses", J. P. Duclos, /1984/, p.p. 81 83, Masson Ed. France).
В самом деле, получившийся при биосинтезе природный гепарин является полисахаридом, образованным молекулами глюкуроновой кислоты и идуроновой кислоты /уроновыми кислотами/, сульфатированным, в случае необходимости, в положении 2 и связанным с молекулами глюкозамина, сульфатированного, в случае необходимости, в положении 6 и сульфатированного или ацетилированного на амине в положении 2.
Структура гепарина статически может быть изображена следующей формулой (i):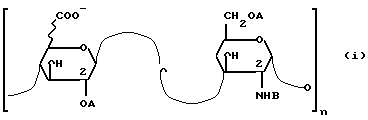
где А представляет собой Н и SO
Выражения "Н и SO
Кроме того, связь означает, что группа COO- в 20 30 приведенных выше дисахаридных звеньях имеет в некоторых случаях конфигурацию /ii/: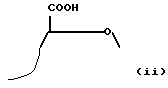
/речь идет о Д-глюкуроновой кислоте/, а в большинстве из n звеньев конфигурацию /iii/:
/речь идет о L-идуроновой кислоте/.
Следовательно, гепарин и фрагменты гепарина, такие, которые получаются в результате различных методов деполимеризации, являются макромолекулами, содержащими как звенья глюкуроновой кислоты, так и звенья идуроновой кислоты.
Некоторые способы деполимеризации позволяют получать фрагменты гепарина, имеющие молекулярный вес, заключенный между 2000 и 9000 а. е. м. и степень сульфатирования, превышающую по меньшей мере на 20% степень сульфатирования исходного гепарина. Такие "сверхсульфатированные" гепарины описаны в заявке на европейский патент ЕП-А-О 116 801, причем уроновые звенья у них имеют две упомянутые выше структуры /ii/ и /iii/.
Известно также, что некоторые бактерии рода Escherichia coli вырабатывают капсулярный полисахарид, называемый обычно антигеном К5, который представляет собой семейство полимеров, образованных повторяющимися β-D-1,4-глюкуронил-a-N-ацетил-Д-1,4-глюкозамильными звеньями (W. F. Vann et al. Eur. J. Biocem. 1981, 116, 359 364).
Этот полисахарид той же самой химической природы, что и полисахаридная часть протеогликана, предшественника гепарина, будет называться здесь N-ацетилгепарозаном. Этот продукт имеет молекулярную массу, заключенную между 105 и 2•106 а. е. м. а на уровне звеньев "уроновой кислоты" очень регулярную структуру, образованную исключительно Д-глюкуроновой кислотой (WF. Vann et al. Eur. J. Biochem. /1980/, 116, 359 364 и заявка на европейский патент ЕП-А-О N333243).
Заявка на европейский патент ЕП-А-О N333243 описывает О-сульфатированный полисахарид К5, а также некоторые из его фрагментов, образованных соответственно 4, 6 или 8 "сахарозными" звеньями, также О-сульфатированными. Эти соединения имеют антиангиогенную и антитуморальную активность при благоприятном соотношении этих активностей относительно антикоагулирующих свойств. Этот документ описывает также фрагменты N-ацетилгепарозана, образованные соответственно 4, 6, 8 или 10 "сахарозными" звеньями.
Заявка на европейский патент ЕП-А-О N333243 описывает также получение пентасахарида, имеющего структуру О-сульфатированного N-ацетилгепарозана, в результате полностью химического синтеза.
В настоящее время нами было обнаружено, что N, О-сульфатированные гепарозаны с переменным молекулярным весом, заключенным между 1500 и 15000 а. е. м. обладают антикоагулирующей активностью по отношению к гепариновому кофактору II и очень высокой анти-Ха активностью.
N, O-сульфатированные гепарозаны как предмет настоящего изобретения отличаются, следовательно, от других соединений, уже описанных в литературе, их оригинальной структурой, в частности их степенью сульфатирования /в том числе и сульфатирования на аминогруппе глюкозамина/ и их фармакологическими свойствами. Среди прочих фармакологических свойств они обладают антикоагулирующей активностью по отношению к гепариновому кофактору II /ГКII/, причем более сильной, чем аналогичная активность дерматансульфата, и они имеют фармакокинетические характеристики, представляющие большой интерес. Действительно, соединения по изобретению, которые являются продуктами, получаемыми в результате полухимического синтеза, обладают фармакологической активностью гликозаминогликанов, обычно применяемых в терапии, в частности фармакологической активностью гепарина, и могут использоваться для регулирования коагуляции и, в частности, могут найти свое применение в качестве антитромботических средств.
Предметом настоящего изобретения являются N, O-сульфатированные гепарозаны, образованные цепями или смесью цепей с молекулярной массой между 1500 и 15000 а. е. м. отличающиеся повторяющейся дисахаридной структурой со следующей формулой 1: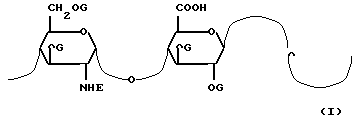
в которой: E представляет собой в 0 80% дисахаридных звеньев указанных N, O-сульфатированных гепарозанов ацетильную группу, а в оставшихся дисахаридных звеньях сульфатную группу и, в случае необходимости, атом водорода,
G представляет собой атом водорода и сульфатную группу и фармацевтически приемлемые соли указанных N, O-сульфатированных гепарозанов.
Степень сульфатирования N, O-сульфатированных гепарозанов, выраженная в виде отношения сульфат/карбоксил, составляет предпочтительно от 1,5 до 3,0.
Изобретение относится также к композиции N, O-сульфатированного гепарозана, содержащей по меньшей мере 70 мас. описанного выше N, O-сульфатированного гепарозана, который является предметом настоящего изобретения, а предпочтительно содержащей по меньшей мере 90 мас. N,O-сульфатированного гепарозана, который является предметом настоящего изобретения.
N,O-cульфатированные гепарозаны как предмет настоящего изобретения могут быть образованы идентичными полисахаридными цепями с хорошо определенной молекулярной массой, причем эта молекулярная масса находится в интервале 1500 15000 а. е. м. Они также могут быть образованы смесью цепей с переменными молекулярными массами, причем эти молекулярные массы заключены между 1500 и 15000 а. е. м. Дисперсия молекулярных масс этих цепей может быть более или менее значительной. Действительно, N, O-сульфатированные гепарозаны как предмет настоящего изобретения могут быть образованы цепями, имеющими между собой максимальное различие по молекулярной массе приблизительно 13500 а. е. м. или, напротив, только цепями, имеющими между собой различие по молекулярной массе приблизительно 300 а. е. м. что соответствует одному звену уроновой структуры /Д-глюкуроновая кислота или ее производные/ или глюкозаминовой структуры. Также является очевидным, что в зависимости от строения каждого N,O-гепарозана молекулярная масса цепей, имеющих либо наименьшую молекулярную массу, либо наибольшую молекулярную массу, может соответствовать любому значению, заключенному между 1500 и 15000 а. е. м.
Выражение "G представляет собой атом водорода или сульфатную группу", которое используется выше, показывает, что G в дисахаридном звене изображает для некоторых положений атом водорода, а для других оставшихся сульфатную группу. Аналогичным образом E изображает в некоторых дисахаридных звеньях ацильную группу, а в остальных из этих звеньев сульфатную группу или, в случае необходимости, атом водорода. Дисахаридные звенья N,O-сульфатированных гепарозанов не являются, следовательно, полностью идентичными.
Формула I изображает повторяющуюся дисахаридную структуру, образованную одним звеном глюкоамина и одним звеном Д-глюкуроновой кислоты. Указанные звенья могут инвертироваться /обращаться/, в частности, если считать, что дисахаридная структура с формулой I повторяется n раз, то звено-невосстановитель в цепях может являться, причем безразлично, либо глюкозаминовым звеном, таким как изображено в формуле I с гидроксигруппой в положении 4, причем это звено глюкозамина является сульфатированным или несульфатированным, либо Д-глюкуроновой кислотой, имеющей, в случае необходимости, двойную связь в положении С4 С5 и являющейся сульфатированной или несульфатированной. Звено-восстановитель может являться, причем безразлично, либо Д-глюкуроновой кислотой, такой как изображено в формуле I, замещенной водородом на аномерном кислороде, либо глюкозамином, либо 2:5-антидроманноструктурой, такой, которая получается вследствие деполимеризации под действием азотистой кислоты /2,5-ангидро-Д-манноза/ с последующим, в случае необходимости, окислением /2, 5-ангидро-Д-манноновая кислота/ или восстановлением /2,5-ангидро-Д-маннитол/.
Предпочтительными продуктами являются те, у которых два концевых звена, восстановитель и невосстановитель, цепей N,O-сульфатированных гепарозанов как предмета настоящего изобретения являются звеньями уроновой кислоты, сульфатированной или несульфатированной, глюкозамина, сульфатированного или несульфатированного, N-ацетилглюкозамина, сульфатированного или несульфатированного, или звеньями, имеющими 2,5-ангидроманноструктуру.
Предпочитают N, O-сульфатированные гепарозаны, у которых два концевых звена, восстановитель и невосстановитель, являются звеньями уроновой кислоты, сульфатированной или несульфатированной, глюкозамина, сульфатированного или несульфатированного, и N-ацетиглюкозамина, сульфатированного или несульфатированного.
Предметом настоящего изобретения является также способ получения композиции, содержащей от 70 до 100% N,O-сульфатированного гепарозана как предмета настоящего изобретения, отличающийся тем, что он включает последовательность следующих стадий:
стадия а: разведение посева Escherichia coli /K5/ для получения N-ацетилгепарозана,
стадия b: выделение и очистка образованного N-ацетилгепарозана для получения композиции, содержащей от 70% до 100% N-ацетилгепарозана, состоящего из цепей или из смеси цепей с молекулярной массой между 1500 и 15000 а. е. м. отличающихся повторяющейся дисахаридной структурой с формулой II:
стадия с: частичное диацетилирование этой N-ацетилгепарозановой композиции для получения композиции, содержащей от 70% до 100% гепарозана, состоящего из цепей или смеси цепей с молекулярной массой между 1500 и 15000 а. е. м. отличающихся повторяющейся дисахаридной структурой с формулой III: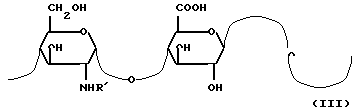
в которой R' представляет собой в 0 80% дисахаридных звеньях ацетильную группу, а в оставшихся дисахаридных звеньях атом водорода.
стадия d: либо частичное N,O-сульфатирование этой гепарозановой композиции,
либо частичное N,O-сульфатирование этой гепарозановой композиции с последующей стадией полного N-сульфатирования,
либо полное или частичное N-сульфатирование с последующей стадией полного или частичного О-сульфатирования, и, в случае необходимости, включает одну или несколько стадий разделения /фракционирования/ по молекулярным массам, осуществляемых по окончании стадий а, b, с или d.
Для получения композиции, содержащей от 70 до 100% N,O-сульфатированного гепарозана как предмета настоящего изобретения, в соответствии с изобретением предпочтительно используют в качестве посева (штамма) Escherichia coli /K5/ посев Escherichia coli SEBR 3282. Этот посев (штамм) является посевом (штаммом), производным от штамма Bi 8337-41 (010:К5:Н4) АТСС 23506 (описанного, в частности, D. S. Gupta et al. FEMS Microbiology Zetters, /1982/, 14, 75 78 и W. Vann, Eur. J. Biochem. /1981/, 116, 359 364).
Штамм Escherichia coli SEBR 3282 положительно отвечает на типовой тест специфичным фагом К5 в соответствии с методикой B. Kaiser et al (J. Clin. Microbiol. /1984/, 19, 2, 264 266). Следовательно, речь действительно идет о штамме Escherichia coli (K5). Этот штамм был представлен в CNCM Института Пастера, Париж, Франция, под N1-1013. Можно также использовать мутант, либо спонтанный, либо вызванный этим штаммом, а также другие соответствующие штаммы Escherichia coli /K5/, например, посев Bi 626-42 (012:KS:NM) ATCC 23508.
Используемая для культуры среда является предпочтительно средой, обогащенной азотсодержащим веществом, например средой, обогащенной дрожжевым экстрактом и гидролизатом казеина, это недорогое средство, содержащее значительное количество аминокислот.
Разведение посева (культивирование штамма) Escherichia coli /K5/ продолжается предпочтительно не менее двух часов после прекращения роста биомассы.
Выделение и очистка N-ацетилгепарозана для получения композиции, содержащей от 70 до 100% N-ацетилгепарозана (стадия В), состоящего из смеси цепей с молекулярной массой между 1500 и 15000 а. е. м. отличающихся повторяющейся дисахаридной структурой с формулой II, осуществляется при помощи способа, включающего по меньшей мере одну стадию осаждения и одну стадию ионообменной хроматографии. Эта стадия осуществляется при использовании предпочтительно колонки с Q-сефарозой или эквивалентной колонки. Осаждение осуществляется соответствующим органическим растворителем и, в частности, спиртом, предпочтительно этанолом. В ходе этого способа N-ацетилгепарозан может находиться в виде соли, предпочтительно в виде натриевой соли.
В качестве примера можно схематично изобразить предпочтительный способ выделения и очистки следующим образом:
стадия а1: осаждение этанолом,
стадия b1: диализ,
стадия с1: осаждение этанолом с последующей дегидратацией и сушкой,
стадия d1: очистка методом анионообменной хроматографии,
стадия e1: осаждение этанолом из элюата, полученного на стадии d1, дегидратация, сушка и измельчение (дробление).
Что касается стадий a1, b1 и c1, то порядок, в котором они осуществляются, имеет небольшое значение. Стадии a1 или c1 могут быть опущены.
На стадии e1 осаждение этанолом не является необходимым. Можно выделять N-ацетилгепарозан другими методами, как, например, выпариванием под вакуумом элюата, полученного на стадии d1.
Выделение и очистка N-ацетилгепарозана для получения композиции, содержащей от 70 до 100% N-ацетилгепарозана, состоящего из смеси цепей с молекулярной массой, заключенной между 1500 и 15000 а. е. м. могут также проводиться следующим образом:
стадия a'1: диализ,
стадия b'1: очистка в кислой среде, удаление нерастворимых примесей в водных растворах от pH 3,5 до pH 1,8,
стадия c'1: осаждение этанолом с последующими дегидратацией и сушкой,
стадия d'1: щелочной гидролиз и диализ,
стадия e'1: очистка методом анионообменной хроматографии,
стадия f'1: очистка при помощи эксклюзивной хроматографии.
Этот способ выделения и очистки также является предпочтительным способом по изобретению.
Щелочной гидролиз осуществляется при помощи раствора NaOH при температуре, заключенной между 30 и 80oC.
Применяя способы выделения и очистки, можно использовать в качестве исходного соединения либо суспензию, полученную по окончании разведения культуры, и в этом случае предварительная фильтрация является необходимой, либо продукт, который уже был подвергнут предварительной очистке, проведенной в соответствии со способом, который включает следующие стадии:
стадия a"1: центрифугирование суспензии, полученной по окончании разведения культуры,
стадия b"1: осуществление контакта всплывшей на поверхность фазы со щелочным раствором,
стадия c"1: предварительная фильтрация,
стадия d"1: концентрирование на мембране с определенным порогом отсекания, которая обеспечивает разделение по молекулярным массам,
стадия e"1: диализ.
Стадия e"1 не является необходимой.
В качестве щелочного раствора можно использовать 0,1 н раствор NaOH.
Предпочтительно проводят процесс предварительной очистки продукта, полученного по окончании разведения культуры, в соответствии со способом, описанным выше.
Разведение посевов Escherichia coli (K5) и способы выделения и очистки, а также стадия предварительной очистки, которые были упомянуты выше, позволят получить N-ацетилгепарозановые композиции, содержащие от 70 до 100% N-ацетилгепарозана, состоящего из смеси цепей с молекулярной массой между 1500 и 15000 а. е. м. отличающихся повторяющейся дисахаридной структурой с формулой II: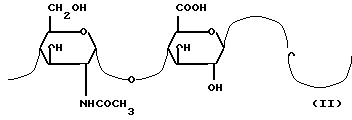
Действительно, благодаря описанным выше способам выделения и очистки и стадии предварительной очистки, оказывается возможным получать N-ацетилгепарозановые композиции, содержащие по меньшей мере от 80 до 100% N-ацетилгепарозана, состоящего из смеси цепей с молекулярной массой между 1500 и 15000 а. е. м.
N-ацетилгепарозаны, состоящие из смеси цепей с молекулярными массами, заключенными между 1500 и 15000 а. е. м. являются новыми продуктами и также составляют предмет настоящего изобретения.
Два концевых звена, восстановитель и невосстановитель, новых N-ацетилгепарозанов таких, которые получены в соответствии со способом, описанным выше, при использовании посева Escherichia coli SEBR 3282 или другого соответствующего посева, как указано выше, являются звеньями уроновой кислоты или N-ацетилглюкозамина.
В большинстве цепей концевое звено-невосстановитель является звеном уроновой кислоты с формулой (а):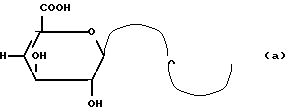
Возможное обогащение получаемой композиции N-ацетилгепарозаном, состоящим из смеси цепей с молекулярной массой между 1500 и 15000 а. е. м. имеющих повторяющуюся дисахаридную структуру с формулой II, может проводиться до различных степеней и, в частности, до выделения указанного N-ацетилгепарозана. Обогащение осуществляется путем использования классических методов разделения по молекулярным массам, таких как гель-проникающая хроматография и ультрафильтрация (A. A. Horner, Biochem. J. /1989/, 262, 953-958; G.Pyler et.al. I.Biol. Chem. /1988/, 263, 11, 5197-5201 и U. Lindahl et al. J. Biol. Chem. /1984/, 259, 20, 12368-12376). Можно также использовать метод этанольного разделения /заявка на европейский патент ЕП-А-О N287477/. Этот последний метод разделения имеет особую ценность среди других рассмотренных методов.
Описанные выше N-ацетилгепарозаны используются в качестве промежуточных соединений для получения N,O-сульфатированных гепарозанов как предмета настоящего изобретения, а также и для получения других производных.
Для получения N,O-сульфатированных гепарозанов как предмета настоящего изобретения можно также использовать другие известные N-ацетилгепарозаны, как, например, N-ацетилгепарозаны, состоящие в основном из полисахаридных цепей, которые все имеют одно и то же число дисахаридных звеньев, таких как описано в заявке на европейский патент ЕП-А-О N333243, и, в частности, состоящих из 6, 8 или 10 "сахарозных" звеньев, из N-ацетилгепарозановых фракций, состоящих из цепей, содержащих в среднем 10 моносахаридных звеньев (Gupta et al. FEMS Microbiology Letters /1983/, 16, 13 17) или из N-ацетилгепарозанов с высокой молекулярной массой, которые могут затем деполимеризоваться в соответствии с методами, используемыми для получения гепаринов с низким молекулярным весом.
Действительно, N, O-сульфатированные гепарозаны как предмет настоящего изобретения могут быть получены при помощи других способов, исходя из известного полисахарида (K5), имеющего высокую молекулярную массу. В этом случае стадия деполимеризации является необходимой.
Эта деполимеризация может проводиться перед деацетилированием N-ацетилгепарозана с высокой молекулярной массой, после деацетилирования, после N-сульфатирования или еще после N,O- сульфатирования.
Как было упомянуто выше, деполимеризация может осуществляться в соответствии с описанными в литературе методами для получения гепаринов с низким молекулярным весом, например, путем деполимеризации с периодатом, при помощи свободных радикалов, в результате бета-отщепления или при действии азотистой кислоты /заявки на патенты или патенты ЕП-О 040 144, ЕП-О 037 319, ЕП-О 121 067/. Эти методы приводятся в качестве примеров и не являются ограничивающими. Можно использовать любой другой метод деполимеризации гликозаминогликанов.
Когда N, O-сульфатированные гепарозаны синтезируют в результате деполимеризации, исходя из N-ацетилгепарозана с высокой молекулярной массой /предварительно деацетилированного и, в случае необходимости, N-сульфатированного/, подвергая его действию азотистой кислоты, то звено-восстановитель N, O-сульфатированных гепарозанов как предмета настоящего изобретения может иметь 2,5-ангидроманноструктуру и, в частности, структуру 2,5-ангидро-Д-маннозы. Деполимеризация под действием азотистой кислоты может проводиться после стадии N-деацетилирования или N-сульфатирования. Когда за этой стадией следует окисление или восстановление, то получают N,O-сульфатированные гепарозаны, имеющие в звене-восстановителе структуру соответственно 2,5-ангидро-Д-манноновой кислоты или 2,5-ангидро-Д-маннитола.
Частичное деацетилирование композиций, содержащих от 70 до 100% N-ацетилгепарозана, которое приводит к получению композиций, содержащих от 70 до 100% и даже от 80 до 100% гепарозана, состоящего из смеси цепей с молекулярной массой между 1500 и 15000 а. е. м. отличающихся повторяющейся дисахаридной структурой с формулой III, указанной выше, осуществляется в результате обработки при помощи деацетилирующего агента.
В качестве деацетилирующих агентов можно упомянуть пентасульфид фосфора, фторборат триэтилоксония, гидроксид натрия или гидразин, причем эти два последних агента имеют особую ценность. Можно также использовать сильные минеральные кислоты, такие как хлороводородная кислота, серная кислота и т. д. Продолжительность реакции зависит от выбранных экспериментальных условий и, в частности, от температуры и от концентрации деацетилирующего агента в реакционной среде.
Обогащение гепарозановой композиции гепарозаном, состоящим из цепей или из смеси цепей с молекулярной массой между 1500 и 15000 а. е. м. отличающихся повторяющейся дисахаридной структурой с формулой III, осуществляется в результате использования классических методов разделения по молекулярным массам, упомянутых выше /гель-проникающая хроматография, ультрафильтрация и этанольное разделение/. В этом случае получают композиции, содержащие от 90 до 100 мас. гепарозана, состоящего из смеси цепей с молекулярной массой между 1500 и 15000 а. е. м. имеющих повторяющуюся дисахаридную структуру с формулой III.
Для получения гепарозанов, состоящих из смеси цепей с молекулярной массой, заключенной между 1500 и 15000 а. е. м. можно также использовать N-ацетилгепарозаны с высоким молекулярным весом. Гепарозаны с высоким молекулярным весом, полученные после деацетилирования, могут деполимеризоваться при помощи методов, уже описанных при получении гепаринов с низким молекулярным весом и уже упомянутых в этой заявке. Продукты деполимеризации могут затем подвергаться фракционированию с целью получения предпочтительных гепарозанов по изобретению.
Гепарозаны, состоящие из смеси цепей с молекулярной массой между 1500 и 15000 а. е. м. отличающихся повторяющейся структурой с формулой III:
в которой R' представляет собой в 0 80% дисахаридных звеньев ацетильную группу, а в остальных дисахаридных звеньях атом водорода, являются новыми продуктами и в этом качестве также составляют часть изобретения.
Предпочтительными гепарозанами по изобретению являются гепарозаны, состоящие из смеси цепей с молекулярной массой между 1500 и 15000 а. е. м. отличающихся повторяющейся дисахаридной структурой с формулой III, в которой ацетильная группа /R'/ присутствует с содержанием, меньшим или равным 60%
Гепарозановые композиции, которые содержат по меньшей мере 70 мас. гепарозана, такого как описано выше, так-же составляют часть настоящего изобретения.
Эти гепарозаны и гепарозановые композиции являются промежуточными соединениями, пригодными для получения N,O-сульфатированных гепарозанов как предмета настоящего изобретения, но они также могут использоваться для получения других продуктов, например продуктов, которые подвергаются впоследствии эпимеризации на уровне звена Д-глюкуроновой кислоты.
Перед стадией частичного N,O-сульфатирования гепарозаны могут превращаться в соль органического основания или в соль четвертичного аммония. Для образования соли четвертичного аммония с гепарозанами используют предпочтительным образом тетрабутиламмоний.
Стадия частичного N,O-сульфатирования, осуществляемая в соответствии со способом, описанным во французском патенте N 2584728 от 10.11. 87 /соответствующем заявке ФР-85. 10787/, проводится в полярном апротонном растворителе, таком как диметилформамид, диметилсульфоксид, гексаметилфосфорамид или ацетонитрил, или в смеси этих растворителей при помощи, например, комплекса серного ангидрида /триоксида серы:SO3/ с органическим основанием, таким как триэтиламин, триметиламин или пиридин. Она может также проводиться при помощи хлорсульфоновой кислоты, растворенной в пиридине. Используют предпочтительно комплекс триоксид серы пиридин.
Можно также использовать другие агенты сульфатирования, в частности те, о которых сообщается в статье E. E. Gilbert в Chemichal Review, /1962/, 62, 549 589. Реакция N,O-сульфатирования осуществляется обычно при температуре, заключенной между 0o и 100oC, предпочтительно между 10o и 50oC, в течение 6 14 ч.
При осуществлении способа получения в конце реакции частичного N,O-сульфатирования композиция N,O-сульфатированного гепарозана, содержащая от 70 до 100% N, O-сульфатированного гепарозана как предмета настоящего изобретения, осаждается в результате прибавления хлорида натрия до получения 0,33 М раствора хлорида натрия, а затем адекватного количества этанола. Полученный осадок извлекается в 0,5 М растворе хлорида натрия. Затем этот раствор нейтрализуется. После прибавления адекватного количества этанола полученный осадок выделяется, извлекается при помощи высокоочищенной воды, отделяется от последней путем диализа, лиофилизуется и сушится.
Стадия N,O-сульфатирования, а также стадии очистки композиции N,O-сульфатированного гепарозана, описанные в предыдущем абзаце, могут повторяться один или несколько раз. Процесс очистки дается в качестве примера и не исключает эквивалентных способов.
За этой стадией N, O-сульфатирования предпочтительно следует стадия полного N-сульфатирования, которая осуществляется обычно в водном растворителе преимущественно со щелочным значением pH при помощи сульфатирующего агента, такого как комплекс серного ангидрида с органическим основанием, например, триметиламином.
По окончании стадии полного N-сульфатирования композиция N,O-сульфатированного гепарозана осаждается после прибавления хлорида натрия до получения 0,5 М раствора хлорида натрия и этанола. Полученный осадок затем снова переводится в раствор при помощи 0,5 М раствора хлорида натрия, снова осаждается этанолом, потом извлекается высокоочищенной водой, отделяется относительно последней путем диализа, лиофилизируется и сушится.
Обогащение композиции N,O-сульфатированного гепарозана N,O-сульфатированным гепарозаном осуществляется в результате использования классических методов разделения по молекулярным массам, которые уже упоминались. Осуществление этого обогащения представляет интерес.
N, O-сульфатированные гепарозаны как предмет настоящего изобретения и композиции N,O-сульфатированных гепарозанов, содержащие по меньшей мере 70% нового N,O-сульфатированного гепарозана как предмета настоящего изобретения, получаются преимущественно при помощи способа, включающего N-сульфатирование в качестве конечной реакции. Тогда Е для повторяющихся структур в формуле (I) представляет собой ацетильную группу и сульфатную группу.
Можно также получать N,O-сульфатированные гепарозаны как предмет настоящего изобретения, проводя сначала N-сульфатирование с последующим О-сульфатированием. Этот вариант способа используется предпочтительным образом.
N-сульфатирование осуществляется при помощи комплекса триоксида серы с органическим основанием, таким как триметиламин, триэтиламин или пиридин. Способ также может осуществляться при помощи хлорсульфоновой кислоты в растворе пиридина. Преимущественно используют комплекс триоксида серы с триэтиламином, а реакция N-сульфатирования проводится при температуре, заключенной между 20 и 80oC, в щелочной водной среде. По окончании реакции N-сульфатирования полученный таким образом продукт осаждается в результате прибавления адекватного количества этанола. Полученный осадок извлекается высокоочищенной водой, отделяется относительно последней путем диализа, лиофилизируется и сушится. Процесс очистки дается в качестве примера и не исключает эквивалентные способы. Стадии очистки могут повторяться несколько раз.
В соответствии со способом по изобретению перед стадией О-сульфатирования N-сульфатированный гепарозан превращается предпочтительно в соль органического основания или в соль четвертичного аммония. Для образования соли четвертичного аммония используют предпочтительным образом тетрабутиламмоний.
Реакция О-сульфатирования проводится в формамиде или в другом растворителе, химически эквивалентном, при помощи, например, комплекса триоксида серы с органическим основанием, таким как триметиламин, триэтиламин или пиридин. Используют предпочтительно комплекс триокиси серы -пиридин. Реакция О-сульфатирования обычно проводится при температуре, заключенной между 10oC и 50oC.
Затем N,O-сульфатированный гепарозан осаждается в результате прибавления к реакционной среде хлорида натрия до получения 0,33 М раствора NaCl, а потом адекватного количества этанола, как в случае N,O-сульфатирования. Затем приступают к очистке композиции N,O-сульфатированного гепарозана. Различные стадии были уже детальным образом описаны выше (осаждение этанолом, диализ и т.д.).
В соответствии с изобретением предпочтительный N,O-сульфатированный гепарозан содержит по меньшей мере 90 мас. цепей с молекулярной массой меньше 11000 а. е. м. Считается ценным, чтобы этот N,O-сульфатированный гепарозан содержал менее 0,2 мкмоль/мг аминогрупп (NH2).
Ацетильная группа предпочтительно присутствует с содержанием, меньшим или равным 60% и считается ценным, чтобы степень сульфатирования, выраженная как отношение сульфат/карбоксил, была бы заключена между 1,5 и 3,0.
Предпочтительными соединениями по изобретению являются N,O-сульфатированные гепарозаны, состоящие из цепей, имеющих среднюю молекулярную массу приблизительно от 400 до 700 а. е. м. и степень сульфатирования, выраженную как отношение сульфат/карбоксил, со значениями между 1, 7 и 3, а также N, O-сульфатированные гепарозаны, N-деацетилированные приблизительно на 80% (содержание присутствующих ацетильных групп составляет примерно 20%) и состоящие по меньшей мере на 70 мас. из цепей с молекулярными массами, заключенными между 5000 и 7000 а. е. м. которые имеют степень сульфатирования от 1,8 до 2,5, или еще N,O-сульфатированные гепарозаны, состоящие по меньшей мере на 70 мас. из цепей с молекулярными массами, заключенными между 10000 и 12000 а. е. м. которые N-деацетилированы приблизительно на 80% /содержание присутствующих ацетильных групп составляет примерно 20%/ и имеют степень сульфатирования от 1,8 до 2,5 или же еще N,O-сульфатированные гепарозаны, N-деацетилированные приблизительно на 40% /содержание присутствующих ацетильных групп составляет примерно 60%/ и состоящие по меньшей мере на 70 мас. из цепей с молекулярными массами, заключенными между 6000 и 8000 а. е. м. которые имеют степень сульфатирования от 2,0 до 2,8.
Предпочтительными N,O-сульфатированными гепарозанами по изобретению являются, в частности, следующие:
N,O-сульфатированные гепарозаны, N-деацетилированные приблизительно на 80% /содержание присутствующих ацетильных групп составляет примерно 20%/, которые состоят по меньшей мере на 80 мас. из цепей с молекулярными массами, заключенными между 2300 а. е. м. и 7200 а. е. м. и имеют степень сульфатирования от 1,8 до 2,5;
N,O-сульфатированные гепарозаны, N-деацетилированные приблизительно на 80% /содержание присутствующих ацетильных групп составляет примерно 20%/, которые состоят по меньшей мере на 80 мас. из цепей с молекулярными массами, заключенными между 3300 а. е. м. и 7700 а. е. м. и имеют степень сульфатирования от 1,8 до 2,5;
N,O-сульфатированные гепарозаны, N-деацетилированные приблизительно на 80% /содержание ацетильных групп составляет приблизительно 20%/, которые состоят по меньшей мере на 70 мас. из цепей с молекулярными массами, заключенными между 6900 а. е. м. и 13500 а. е. м, и имеют степень сульфатирования от 1,8 до 2,5;
N,O-сульфатированные гепарозаны, N-деацетилированные приблизительно на 40% /содержание присутствующих ацетильных групп составляет примерно 60%/, которые состоят по меньшей мере на 80 мас. из цепей, с молекулярными массами, заключенными между 4000 а. е. м. и 10300 а. е. м. и имеют степень сульфатирования от 2,0 до 2,8.
Под солями N,O-сульфатированных гепарозанов понимают все фармацевтически приемлемые соли. Эти соли получаются при помощи классических методов, описанных, в частности, для получения солей гепарина /патент США N4168377/.
Описанный выше способ получения N,O-сульфатированных гепарозанов, а также методы очистки позволяют получить N,O-сульфатированные гепарозаны в виде натриевой соли. Исходя из этих солей, можно получать, применяя методы, используемые для получения различных солей гепарина или гепарина в несолевой форме ("L'heparine, fabrication, structure, proprietes, analyses". J. P. Duclos, Masson Ed. France, /1984/, p.p. 81-83), либо другие соли N,O-сульфатированных гепарозанов, либо N, O-сульфатированные гепарозаны в несолевой форме.
N, O-сульфатированные гепарозаны как предмет настоящего изобретения обладают фармакологическими и биохимическими свойствами, которые представляют интерес и являются совершенно удивительными по отношению к известному уровню техники.
В частности, в отличие от сульфатированных продуктов антигена К5, описанных в европейском патенте ЕП-А-О N333243, которые проявляют антиангиогенную и антитуморальную активность с благоприятным соотношением этих активностей относительно антикоагулирующих свойств, N,O-сульфатированные гепарозаны по настоящему изобретению обладают хорошей активностью, регулирующей коагуляцию. Эта активность намного превосходит активность дерматансульфата по различным параметрам коагуляции, и она скорее может сравниться с активностью гепарина.
В частности, анти-IIа-активность, зависящая от АТШ или ГКП, для соединений, представляющих изобретение, была определена в соответствии с методиками, описанными Dupong D. et al. b Thrombosis and Haemostasis, /1988/, 60, 2, 236 239 для гепаринового кофактора ПI /ГКП/ и M. L. Larsen et al. b Thrombosis Research /1978/, 13, 2, 285 288 для антитромбина /АТШ/.
В обоих случаях тест заключается в измерении in vitro ингибирующего действия изучаемого соединения на очищенный тромбин /фактор IIа/ в присутствии очищенных ГКП и АТШ при определении амидолитической активности тромбина по отношению к хромогенному субстрату. Поскольку дерматансульфат, полученный в соответствии с методикой, описанной H. W. Stuhlsatz et al, в "The Methodology of Connective Tissue Research", /1976/, 137 146, обладает наибольшей анти-IIа-активностью, зависящей от ГКП, то он используется в качестве вещества для сравнения /стандарта/ в тесте по измерению этой активности, причем результат выражается в мг дерматансульфата /ДС/, эквивалентных по активности 1 мг изучаемого соединения (Экв мг ДС/мг).
Анти-Ха-активность /титр Yin-Wessler/ указанных соединений, представляющих изобретение, была измерена по методике, описанной E. T. Jin в J.Lab. Clin. Med. /1973/, 81, 2, 298 310, тогда как их общая антикоагулирующая активность была измерена в соответствии с тестом АРТТ, описанным R. R. Rroctoretal в Am. J. Clin. Path. /1961/, 36, 212 219.
Все тестированные соединения показали анти-IIa-активность, зависящую от ГКПI, явно превышающую аналогичную активность дерматансульфата. Анти-IIа-активность, зависящая от АТШ, и титр Jin-Wessler, хотя и меньшие аналогичных параметров для гепарина, оказались превышающими аналогичные параметры для дерматансульфата. Их титр по АРТТ от примерно 2 до примерно 20 раз превышает тот же параметр для дерматансульфата и может доходить до 60% от значения того же параметра для гепарина.
Следовательно, N,O-сульфатированные гепарозаны по изобретению проявляют специфичность и антикоагулирующую активность, представляющие особый интерес. Небольшая молекулярная масса этих соединений придает им, с другой стороны, фармакокинетические свойства, также представляющие значительный интерес.
N, O-сульфатированные гепарозаны по настоящему изобретению являются в значительной степени малотоксичными; их токсичность полностью совместима с их использованием в качестве медикаментов.
Таким образом, изобретение распространяется также на фармацевтические композиции, содержащие в качестве активного компонента N,O-сульфатированный гепарозан как предмет настоящего изобретения, одну из его солей или композицию N,O-сульфатированного гепарозана, содержащую по меньшей мере 70% этого N,O-сульфатированного гепарозана или одной из его солей, в комбинации с одним или несколькими фармацевтически приемлемыми носителями.
Эти фармацевтические композиции являются полезными, в частности, при лечении в качестве профилактики или для выздоровления от заболеваний стенки сосудов, таких как атеросклероз и артериосклероз, и при лечении состояний с гиперкоагулируемостью, наблюдаемых, например, после хирургических операций, с туморальными нарушениями или с нарушениями коагуляции, вызванными бактериальными, вирусными или ферментативными активаторами.
Дозировка может хорошо варьироваться в зависимости от возраста, веса и состояния здоровья пациента, от природы и серьезности заболевания, а также от пути введения.
Эта дозировка включает введение одной или нескольких доз примерно от 1 мг до 1 г в день, предпочтительно примерно от 5 мг до 500 мг в день, например, порядка 200 мг в день внутривенным путем или подкожным путем, при нерегулярных введениях или с регулярными интервалами, или с дневной дозой порядка от 200 мг до 1000 мг в день оральным путем.
Эти дозы, конечно, могут быть подобраны для любого пациента в зависимости от наблюдаемых результатов и анализов крови, проведенных предварительно.
Изобретение иллюстрируется приведенными ниже примерами.
Пример 1.
Получение N-ацетилгепарозана в основном с небольшой молекулярной массой /способ 1/.
1. Разведение посева бактерий Escherichia coli /K5/ и выделение фильтрата, содержащего N-ацетилгепарозан.
Засевают 400 мл среды В с составом, уточненным в приведенной ниже табл. 1, при помощи штамма Escherichia coli SEBR 3282 /хронированного в коллекцию штаммов Института Пастера, Париж, Франция, под N 1 1013/ и проводят инкубацию при перемешивании в течение 2 ч при 37oC.
Полученная предварительная культура переносится затем в ферментер объемом 18,5 л, содержащий 11 л среды А с составом, уточненным в приведенной ниже табл. 1, и проводят инкубацию в течение 4 ч при температуре 37oC и значении pH 7,4, причем парциальное давление кислорода поддерживается величиной 40 мм рт. ст. в результате регулирования подачи воздуха (до 20 л/мин) и перемешивания. Затем прибавляют глюкозу путем непрерывного введения стерильного раствора, содержащего 600 г/л глюкозы, с расходом 250 мл/ч в течение 8 ч.
Продолжают культивировать в тех же самых условиях температуры, pH и парциального давления кислорода в течение 10 ч после прекращения прибавления глюкозы. Наблюдение за оптической плотностью /ОП/ ( λ 600 нм) среды с культурой позволяет подтвердить, что отсутствует рост биомассы в течение последних 12 ч разведения культуры.
Затем охлаждают бульон для культуры до температуры 25oC, потом фильтруют его через мембрану с пористостью 0,22 мкм. Получают таким образом примерно 12 л фильтрата, содержащего N-ацетилгепарозан.
Таблица I.
Состав и получение среды A и среды B.
Среда A.
Среда А получается в результате объединения трех приведенных ниже стерильных растворов.
Раствор N 1.
В 700 мл высокоочищенной воды растворить в следующем порядке:
Комплексообразователь: N'-/трис-(гидроксиметил)-метил/-глицин (Трицин: поставляемый в торговлю фирмой Флука® 360 мг
FeSO4, 7H2O 280 мг
CaCl2, 2H2O 6,7 мг
MgCl2, 6H2O 1270 мг
K2SO4 500 мг
KCl 5000 мг
Гидролизат казеина /основной источник аминокислот /ГИКАЗЕШ/, поставляемый в торговлю фирмой Шеффилд® 25000 мг
Дрожжевой экстракт /поставляемый в торговлю фирмой Дифко®/ 18000 мг
Раствор олигоэлементов /см. ниже таблицу II/ 1 мл
Пеногаситель Структол I 673 /поставляемый в торговлю фирмой Шилл и Зейлахер® /: несколько капель пипеткой Пастера.
Установить значение pH 7,4 при помощи раствора КОН /d 1,38/ и довести до объема 850 мл при помощи высокоочищенной воды. Подвергнуть среду обработке в автоклаве в течение 45 мин при 120oC.
Раствор N 2.
Растворить 5 г K2HPO4 в примерно 40 мл высокоочищенной воды, затем довести до 50 мл тем же самым растворителем. Отфильтровать полученный раствор через фильтр с пористостью 0,2 мкм.
Раствор N 3.
Растворить 20,7 г глюкозы в адекватном количестве высокоочищенной воды и довести объем до 100 мл при помощи того же самого растворителя. Подвергнуть обработке в автоклаве при температуре 110oC в течение 30 мин.
Среда В.
Получение среды В является идентичным получению среды А с тем отличием, что следует прибавить, помимо прочего, 20 г буферного раствора со значением pH 7,2 /3-морфолинопропансульфокислота/ после прибавления пеногасителя.
Таблица II
Получение раствора олигоэлементов, используемого при получении среды А и среды В.
Растворить в 800 мл высокоочищенной воды в следующем порядке:
H3PO4 500 мг
Na2MgO4, 2H2O 1930 мг
CoCl2, 6H2O 11850 мг
CuSO4, 5H2O 25 мг
ZnSO4, 7H2O 2000 мг
AlCl3, 6H2O 2410 мг
Прибавить 100 мл хлороводородной кислоты с плотностью 1,19 и довести до объема 1000 мл при помощи высокоочищенной воды.
2. Выделение и очистка N-ацетилгепарозана в основном с небольшой молекулярной массой.
Стадия а. Этанольное осаждение.
Прибавляют к фильтрату примерно 48 л 95% (об/об) этанола и оставляют смесь для осаждения и декантирования при температуре 4oC в течение 8 ч. Удаляют всплывшую на поверхность фазу путем откачивания с последующим центрифугированием примерно в 1 л высокоочищенной воды.
Стадия b. Диализ.
В течение 24 ч проводят диализ раствора, полученного на предыдущей стадии, который помещается в устройство для выдавливания NOJAX 40, имеющее мембрану на основе целлюлозы с пористостью 24 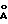 относительно высокоочищенной воды (1 объем раствора/ 6 объемов воды, возобновляются спустя 2 ч, 8 ч и 16 ч). Эта операция позволяет удалить небольшие молекулы, присутствующие в среде для культуры, такие как соли, сахароза, аминокислоты, олигонуклеотиды и олигопептиды.
относительно высокоочищенной воды (1 объем раствора/ 6 объемов воды, возобновляются спустя 2 ч, 8 ч и 16 ч). Эта операция позволяет удалить небольшие молекулы, присутствующие в среде для культуры, такие как соли, сахароза, аминокислоты, олигонуклеотиды и олигопептиды.
Стадия с. Осаждение, дегидратация и сушка.
К 1 объему подвергнутого диализу раствора прибавляют 0,5 М NaCl и 4 объема этанола. Дают образоваться осадку в течение 5 мин при комнатной температуре. Центрифугируют при 5000 g в течение 20 мин. Извлекают осадок после центрифугирования в этаноле, перемешивают полученную суспензию и оставляют в покое в течение 1 ч при комнатной температуре. Повторяют операции центрифугирования и перевода в суспензию. Снова центрифугируют при 5000 g в течение 20 мин. Сушат осадки, полученные после центрифугирования, в сушильном шкафу под вакуумом при 40oC в течение 24 ч.
Стадия d. Измельчение в порошок.
Сухие осадки центрифугирования измельчаются при помощи ступки в условиях отсутствия воды.
Стадия e. Анионообменная хроматография.
Извлекают измельченные осадки центрифугирования в буферном растворе, называемом буферным раствором Д, с составом 20 мМ Трис-НCl и pH 7,5 при расходе 100 мл/г. Полученный раствор хроматографируется на анионообменной колонке с сильным ионитом, имеющим матрицу из агарозы, сшитую при помощи четвертичного аммония /"О-сефароза с устойчивым потоком" фирмы Фармация® /, который предварительно приводился в равновесие с буферным раствором Д при соотношении 50 мл геля на 1 г порошка. Промывают гель достаточным количеством буферного раствора Д для возвращения к основной линии УФ-детектирования при 214 нм, а затем 25 мМ раствором пиперазина, pH которого установлен при значении 3,5. Проводят элюирование при помощи раствора с pH 3,5, имеющего следующий состав: 0,5 М NaCl и 25 мМ пиперазина. Элюат нейтрализуется при помощи 5 н раствора NaOH.
Стадия f. Осаждение, дегидратация, сушка и измельчение.
Повторяют операции, описанные выше на стадиях с и d, не прибавляя хлорида натрия.
N-ацетилгепарозан, полученный в результате стадии f, называется партия А.
Один из вариантов способа очистки заключается в последовательном осуществлении стадий а, с, d, e и f. Полученный таким образом N-ацетилгепарозан называется партия В.
3/ Описание N-ацетилгепарозана, полученного в результате различных стадий очистки
Спектр ядерного магнитного резонанса /ЯМР/.
Спектры ЯМР протона и углерода 13C сравнивают с аналогичными спектрами для N-ацетилгепарозана, описанного W. E. Vann (Eur. J. Biochem. /1981/, 116, 59 364).
Изучение спектров, полученных для N-ацетилгепарозана из партии A и из партии B, подтверждает химическую идентичность продукта с N-ацетилгепарозаном, описанным W. F. Vann. Речь идет о полимерных цепях, образованных повторяющимися структурами β-Д-1,4-глюкуронил-a-N-ацетил-Д-1,4-глюкозамила.
Определение молекулярно-массового распределения при помощи эксклюзивной хроматографии.
Молекулярно-массовое распределение определяется при помощи высокоэффективной жидкостной хромотографии /ВЭЖХ/ методом исключения в следующих условиях:
Колонка, наполненная шариками кремнезема с диаметром 10 мкм и с пористостью 250 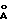 .
.
Элюент: 0,5 М водный раствор сульфата натрия.
Расход: 1 мл/мин.
УФ-детектирование с λ 205 нм.
Калибровка осуществляется при помощи набора олигосахаридов, производных гепарина, со следующими молекулярными массами: 1324, 1883, 2436, 3411, 3996, 4535, 4995, 5365, 6150, 6671, 7542, 8655, 10088, 11561, 12950, 14809, 17387 и 22674 а. е. м.
Принимая во внимание этот набор эталонов, учитывают только молекулярные массы между 934 а. е. м. и 56703 а. е. м. Принимают, что определяемая оптическая плотность пропорциональна количеству N-ацетилгепарозана. Однако точность метода экспоненциально уменьшается для высоких молекулярных масс и, в частности, превышающих 20000 а. е. м.
Весовая фракция, составляющая по меньшей мере 70% партии А, имеет молекулярную массу, заключенную между значениями 1700 и 8000 а. е. м.
Очень похожая хромотография получается при анализе партии В. Основной пик распределения находится примерно при 5000 а. е. м. Весовая фракция, составляющая по меньшей мере 70% партии В, имеет молекулярную массу, заключенную между 1500 и 8000 а. е. м.
Наблюдение за молекулярно-массовым распределением методом электрофореза на полиакриламидном геле.
Эти анализируемые образцы, а также метка окончания миграции, содержащая бромфеноловый синий, подвергаются электрофорезу в буферном растворе Трис-Бората, содержащем 15% полиакриламидного геля, полученного при полимеризации смеси 29/1 акриламида и N,N'-диметилен-бис-акриламида. Миграция осуществляется при токе 40 мА в течение примерно 4 ч на геле длиной 18 см до выхода метки окончания миграции. В этом случае гель окрашивается в синий алциановый цвет, а затем в серебряный цвет в соответствии с методом S. Pelkonen et al. (J. Bact. /1983/, 170, 6, 2646), специфичным для кислых полисахаридов.
Этот анализ методом электрофореза был проведен для частично очищенного продукта, полученного после стадии а, и для очищенного продукта /партия A или партия B/, полученного после конечной стадии, с целью проверить отсутствие значительного изменения молекулярно-массового распределения для N-ацетилгепарозана в ходе очистки.
Изучение профилей, полученных для частично очищенного продукта, выходящего после стадии а, и для очищенного продукта, выходящего после конечной стадии /партия A или партия B/, не выявляет значительных различий /наличие полос со сравнимой интенсивностью на одних и тех же участках миграции/. Следовательно, отсутствует значительное изменение молекулярно-массового распределения для N-ацетилгепарозана в ходе его очистки.
Количественный анализ уроновых кислот.
Количество уроновой кислоты на единицу массы очищенного продукта /партия A или партия B/, полученного после конечной стадии, определялось путем колориметрии в соответствии с методом, описанным T. Bitter (Analytical Biochemistry, /1962/, 4, 330-334). Этот метод количественного анализа основан на реакции гликозаминогликанов с карбазолом в кислой среде при нагревании, которая вызывает розовое окрашивание, пропорциональное количеству высвобождаемой уроновой кислоты.
Для партии А частично очищенный продукт, полученный после стадии d, и очищенный продукт, полученный после конечной стадии f, имеют содержание уроновой кислоты соответственно 1,3 и 2,1 мкмоль/мг.
Для партии В очищенный продукт после конечной стадии также имеет содержание уроновой кислоты величиной 2,1 мкмоль/мг.
Спектрофотометрия в ультрафиолетовой и видимой областях.
Очищенный продукт /партия A/ растворяется в высокоочищенной воде, а полученный раствор (С 1 мг/мл) помещается в оптическую кювету со слоем прохождения 1 см. Его спектр поглощения регистрируется между 200 и 600 нм.
Полученный спектр позволяет утверждать, в частности, на основании поглощения при 256 нм, что партия А содержит менее 1% АД N.
Количественный анализ всех протеинов.
Комплект "анализ протеинов", поставляемый в торговлю фирмой БИОРАД, используется для количественного анализа всех протеинов. Метод количественного анализа основан на том, что длина волны максимума поглощения для кислого раствора блестящей сини Coomassie g-250 смещается от 465 нм до 595 нм, когда протеины начинают в нем проявляться (Reisner et al. Anal. Biochem. /1975/, 64, 509).
Содержание всех протеинов в партии А является меньшим 1,5%
Количественный анализ свободных аминогрупп (NH2)
Этот количественный анализ был проведен в соответствии с методом, описанным Zensaku Josizawa et al. в Biochemica et Biophisica Acta. (1967), 141, 358 365.
Параметр содержания NH2 (выраженный в мкмоль/мг) является показателем количества деацетилированных b-Д-1,4-глюкуронил-a-N-ацетил-Д-1,4-глюкозаминильных звеньев и примесей, которые содержат свободную аминогруппу.
Партия А и партия В, каждая, имеют содержание NH2-групп величиной 0,05 мкмоль/мг. Отношение NH2/глюкуроновая кислота 0,054/2,1 является меньшим 2,5% Следовательно, на 100 b-Д-1,4-глюкуронил-a-N-ацетил-1,4-глюкозаминильных звеньев приходится менее 2,5 дисахаридных звеньев этого деацетилированного типа.
Пример 2.
Получение N-ацетилгепарозана в основном с небольшой молекулярной массой (способ II).
1. Разведение посева бактерий Escherichia coli (K5) и выделение фильтрата, содержащего N-ацетилгепарозан, были осуществлены в соответствии с методикой, описанной в примере 1.
2. Выделение и очистка N-ацетилгепарозана в основном с небольшой молекулярной массой.
Стадия а. Диализ.
Подвергают диализу 375 мл фильтрата в соответствии со способом, описанным в примере 1 /2/. Выделение и очистка N-ацетилгепарозана в основном с небольшой молекулярной массой.
Стадия в/. После диализа получают приблизительно 1020 мл очищенного раствора.
Стадия b. Очистка в кислой среде.
К раствору, подвергнутому диализу, прибавляют адекватное количество 5 н раствора HCl для получения значения pH 3,5. Удаляют путем ценрифугирования полученный осадок, затем подкисляют раствор при помощи той же кислоты (5 н HCl) для получения значения pH 1,8. Может образоваться осадок, который удаляется путем ценрифугирования. Затем нейтрализуют раствор при помощи 5 н раствора NaOH.
Стадия с. Осаждение, дегидратация и сушка.
К нейтрализованному раствору прибавляют адекватное количество хлорида натрия, чтобы получить 0,5 М раствор NaOH, затем прибавляют 4 объема этанола. Дают образоваться осадку в течение 5 мин при комнатной температуре. Центрифугируют при 5000 g в течение 20 мин. Извлекают в этаноле осадки после центрифугирования, перемешивают полученную суспензию и оставляют в покое в течение 1 ч при комнатной температуре. Повторяют операции центрифугирования и перевода в суспензию. Снова центрифугируют при 5000 g в течение 20 мин. Сушат полученные осадки центрифугирования в сушильном шкафу под вакуумом при 40oC в течение 24 ч.
Стадия d. Щелочной гидролиз и диализ.
Продукт, полученный на предыдущей стадии после сушки, растворяется с образованием 2,5% (p/V) раствора в 0,25 н растворе NaOH. Выдерживают полученный таким образом раствор в течение 2 ч при температуре 50oC. Затем нейтрализуют при помощи 5н раствора HCl, а потом раствор, содержащий полисахарид, подвергают диализу в соответствии со способом, описанным в примере 1/2) Выделение и очистка N-ацетилгепарозана в основном с небольшой молекулярной массой, стадия b).
После диализа получают примерно 990 мл раствора.
Стадия е. Анионообменная хроматография.
К подвергнутому диализу раствору прибавляют адекватные количества пиперазина, ЭДТА и тритона Х-100 Пролабо®, чтобы получить соответственно концентрации 25 мМ по пиперазину, 2 мМ по ЭДТА и 0,2% (V/V) по тритону Х-100. Затем устанавливают значение pH 3,5 при помощи 5 н раствора HCl. Помещают этот раствор в колонку объемом 400 мл, содержащую Q-Сефарозу с устойчивым потоком, которая была приведена в равновесие с пиперазиновым буферным раствором, содержащим 25 мМ пиперазина, 2 мМ ЭДТА и 0,2 мМ тритона Х-100 (pH 3,5). Промывают пиперазиновым буферным раствором, элюируют 0,5 М раствором NaCl, затем осаждают при помощи 4 объемов этанола. Сушат под вакуумом при температуре 40oC. Получают таким образом примерно 9,85 г N-ацетилгепарозана.
Стадия f. Хроматография методом исключения.
Растворяют 4 г продукта, полученного на предыдущей стадии, в 60 мл буферного раствора с составом: 20 мМ трис-HCl с pH 7,5 и 1 М NaCl, затем пропускают через колонку объемом 200 мл с октил- Сефарозой® предварительно приведенной в равновесие с тем же самым буферным раствором. К незадержанной фракции прибавляют 4 объема этанола. Промывают образованный осадок и сушат его при температуре 40oC под вакуумом.
Получают таким образом 3,90 г N-ацетилгепарозана.
3) Описание N-ацетилгепарозана, полученного в результате различных стадий очистки.
Спектр ядерного магнитного резонанса (ЯМР).
Изучение спектров ЯМР протона и углерода 13С, полученных для этого N-ацетилгепарозана, подтверждает химическую идентичность продукта с N-ацетилгепарозаном, описанным W. F. Vann (Eur. J. Biochem. /1981/, 116, 59 - 364).
Определение молекулярно-массового распределения при помощи хроматографии методом исключения.
Молекулярно-массовое распределение определяется при помощи ВЭЖХ методом исключения в соответствии с методикой, используемой для определения молекулярно-массового распределения N-ацетилгепарозанов, описанных в примере 1. Весовая фракция, составляющая по меньшей мере 86% цепей, которые образуют партию по примеру 2, обладает молекулярной массой, заключенной между 1500 и 15000 а. е. м.
Количественный анализ уроновых кислот.
Полученный после стадии е N-ацетилгепарозан имеет содержание уроновых кислот величиной 1,94 мкмоль/мг.
Спектрофотометрия в ультрафиолетовой и видимой областях.
Полученный спектр позволяет утверждать, что синтезированный N-ацетилгепарозан содержит менее 1% АД N.
Количественный анализ всех протеинов.
Содержание всех протеинов в этой партии N-ацетилгепарозанов составляет менее 1%
Количественный анализ свободных аминогрупп (NH2).
Содержание NH2 групп составляет менее 0,1 мкмоль/мг.
Пример 3.
Получение N-ацетилгепарозана в основном с небольшой молекулярной массой (способ III).
1) Разведение посева Escherichia coli (K5).
Разведение посева Escherichia coli SEBR 3282 было проведено в соответствии с методикой, описанной в примере 1. Получают приблизительно 12 л культуры, содержащей N-ацетилгепарозан.
2) Предварительная очистка.
Стадия а. Центрифугирование.
По окончании разведения культуры ценрифугируют полученную суспензию (12 л) со скоростью 8000 оборотов/мин (т. е. между 1100 и 14000 g) в течение 20 мин.
Стадия b. Осуществление контакта с щелочным раствором.
После центрифугирования осадок удаляется, а всплывшая на поверхность фаза приводится в соприкосновение с 0,1 н раствором NaOH в течение примерно 1 ч.
Стадия с. Предварительная фильтрация.
Раствор, полученный на предыдущей стадии, подвергается предварительной фильтрации на фильтре 3М® серии 300 из полипропилена.
Стадия d. Концентрирование на мембране с определенным порогом отсекания.
Фильтрат, полученный на стадии с, концентрируют на фильтрующем элементе с полыми волокнами Амикон® рассчитанном на порог отсекания 1000 а. е. м. или эквивалентный. Получают таким образом раствор, обогащенный N-ацетилгепарозаном с небольшой молекулярной массой.
Стадия е. Диализ.
Раствор, обогащенный N-ацетилгепарозаном с небольшой молекулярной массой, подвергают диализу относительно высокоочищенной воды опять на системе Амикон® до очень высокого коэффициента разбавления (>10000).
3) Выделение и очистка N-ацетилгепарозана в основном с небольшой молекулярной массой.
Проводят процесс, как это указано в примере 1 /(2) Выделение и очистка N-ацетилгепарозана в основном с небольшой молекулярной массой, стадия с - стадия f/ и получают N-ацетилгепарозан, имеющий характеристики, подобные характеристикам для партии А, или, как это указано в примере 2 /2) Выделение и очистка N-ацетилгепарозана в основном с небольшой молекулярной массой, стадия а стадия f/.
N,O-сульфатированные гепарозаны
Пример 4.
Химическая модификация N-ацетилгепарозана в основном с небольшой молекулярной массой, полученного в примере 1.
1) Химическая модификация партии В.
Стадия а). Частичное деацетилирование.
Растворяют 500 мг партии В в 10 мл 1 н раствора NaOH. Доводят раствор до температуры 50oC и дают прореагировать при этой температуре в течение 8 ч при перемешивании. Затем нейтрализуют этот раствор при помощи 2 н раствора HCl, подвергают его диализу относительно высокоочищенной воды, затем лиофилизируют его.
Стадия b. Образование соли тетрабутиламмония.
Предыдущий лиофилизат извлекается в 20 мл высокоочищенной воды. Полученный раствор направляется на ионообменную колонку с ионитом на основе полистирола, сшитого дивинилбензолом (доуэкс 50 W фирмы Доу Кемисол® ), предварительно доведенную до кондиции в кислой среде, для того, чтобы регенерировать кислотную формулу продукта. Затем раствор смешивается с 0,4 мл 40%-ного раствора тетрабутиламмония. Проводят лиофилизацию.
Стадия с. Частичное N,O-сульфатирование.
Растворяют 421 мг соли, полученной на предыдущей стадии, в 35 мл диметилформамида и прибавляют 3,71 г комплекса триоксид серы-пиридин (поставляемого в торговлю фирмой Алдрих® в виде этанола S 755 6). Дают прореагировать при перемешивании в течение 6 ч при комнатной температуре. К одному объему реакционной среды прибавляют хлорид натрия до получения раствора с концентрацией 0,33 М по хлориду натрию, затем 2 объема этанола. Дают сформироваться осадку. Центрифугируют и удаляют всплывшую на поверхность фазу. Осадок после центрифугирования извлекается в 0,5 М раствора NaCl с последующей нейтрализацией. Затем прибавляют 2 объема этанола, дают сформироваться осадку, центрифугируют и извлекают осадок после центрифугирования при помощи высокоочищенной воды и лиофилизируют.
Повторяют совокупность операций, описанных в приведенном выше параграфе.
Полученный лиофилизат имеет следующие характеристики:
содержание уроновых кислот: 1,11 мкмоль/мг,
содержание свободных NH2-групп: 0,01 мкмоль/мг,
степень сульфатирования: 2,64 на дисахаридное звено.
Содержание уроновых кислот и NH2-групп определяется, как описано в примере 1.
Степень сульфатирования, называемая также отношением сульфат/карбоксил, есть среднее число сульфатных групп, приходящихся на одну карбоксильную группу; она определяется кондуктометрическим методом количественного анализа сульфатных групп и карбоксильных групп, который описан B. Casu et al. в Carbohydrate Research, (1975), 39, 168 176.
2) Химическая модификация партии А.
Партия А делится на три аликвотные части, называемые партия А1, партия А2 и партия А3.
Стадия а. Частичное деацетилирование и гель-фильтрация.
Партии А1, А2 и А3 были обработаны, как описано выше на стадии а) для партии В, с тем отличием, что только партия А1 подвергается диализу и лиофилизируется. Затем партии А1, А2 и А3 фракционируются в результате гель-фильтрации (называемой также гель-проникающей хроматографией или эксклюзивной хроматографией) при следующих условиях:
носитель в виде шариков с диаметром 25 75 мкм на основе аллилдекстрана, сшитого N, N'-метилен-бист-акриламидом (Сефакрил S 300 HR, поставляемый в торговлю фирмой Фармазия®),
используемый элюент: 0,4 М раствор NaCl.
Получают смеси фракций, которые соответствуют Кav от 0,46 до 1 для гепарозана, полученного из партии А1, от 0,43 до 1 для гепарозана, полученного из партии А2, и от 0,43 до 0,64 для гепарозана, полученного из партии А3. Эти фракции впоследствии называют "гель-фильтрационными гепарозанами".
Величина Кav является коэффициентом, обычно используемым в хроматографии методом исключения, и позволяет воспроизводить фракционирование в результате хроматографии методом исключения. Он определяется по формуле: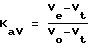
в которой:
Ve объем элюирования рассматриваемой фракции,
Vo объем исключения,
Vt полный объем геля.
Распределение по молекулярным массам для гепарозана, полученного из партии А1 сразу же после частичного диацетилирования, а также для гель-фильтрационных гепарозанов, полученных из партий А1, А2 и А3, определяется при помощи хроматографии методом исключения в соответствии с методикой, описанной в примере 1.
Некоторые результаты, выведенные на основании профилей элюирования, собраны в приведенной ниже таблице III, в которой PM0 представляет собой такую молекулярную массу, чтобы фракция 1% продукта имела бы молекулярную массу, превышающую PMо, PM1 представляет собой такую молекулярную массу, чтобы фракция 10 мас. продукта имела бы молекулярную массу, превышающую PM1, PM3 это такая молекулярная масса, чтобы фракция 10 мас. продукта имела бы молекулярную массу меньше PM3, PM2 представляет собой молекулярную массу, соответствующую максимуму абсорбции.
Установлено, что гель-фильтрация позволяет удалить неосновную фракцию гепарозана с высокой молекулярной массой.
Этот результат четко проявляется в табл. III, где установлено сильное уменьшение PMo вследствие операции гель-фильтрации. Гепарозан из каждой партии содержит по крайней мере 90 мас. цепей с молекулярными массами, меньшими 7000 а. е. м.
Гель-фильтрационные гепарозаны, происходящие из партий А1 и А2, затем обрабатываются, как описано в примере 1 /(2) Выделение и очистка N-ацетилгепарозана в основном с небольшой молекулярной массой, стадия с/.
Гель-фильтрационный гепарозан, происходящий из партии А3, не осаждается, а подвергается диализу относительно высокоочищенной воды.
Содержание уроновых кислот и свободных аминогрупп измеряется, как описано в примере 1.
Полученные результаты собраны в приведенной ниже табл. IV. Содержание остаточного ацетила было оценено при допущении, что имеется столько же глюкуронильных групп, сколько и глюкозаминильных групп.
Степень деацетилирования рассчитывалась как равная отношению содержания свободных аминогрупп к содержанию уроновых кислот.
Спектр ядерного магнитного резонанса.
Изучение спектра ядерного магнитного резонанса протонов, полученного для гель-фильтрационного гепарозана, происходящего из партии А3, позволило сделать вывод, что продукт обладает ожидаемой структурой.
Степень деацетилирования, рассчитанная путем интегрирования, оказалась равной 44% Это значение близко к значению, рассчитанному при использовании отношения содержания свободных аминогрупп к содержанию уроновых кислот (41% ).
Стадия b. Частичное N,O-сульфатирование.
Гель-фильтрационные гепарозаны, происходящие из партий А1, А2 и А3, которые были подвергнуты деацетилированию, обрабатывают таким образом, как описано выше для партии В (стадия с частичное N,O-сульфатирование) с тем отличием, что не повторяют операции, описанные в первом параграфе.
В случае гепарозана, происходящего из партии А3, после первого осаждения осадок извлекается в высокоочищенной воде, подвергается диализу относительно высокоочищенной воды и оставляется в растворе.
Характеристики N,O-сульфатированных продуктов собраны в приведенной ниже табл. V.
Видно, что после реакции N, O-сульфатирования остаточное содержание NH2-групп (0,10 мкмоль/мг) превышает этот параметр для очищенных N-ацетилгепарозанов (0,05 мкмоль/мг). Следовательно, сульфатирование на атомах азота не является полным.
Стадия с. Полное N-сульфатирование.
Смешивают в объеме высокоочищенной воды величиной 20 мл, приходящемся на грамм используемого N,O-сульфатированного продукта, 1 мас. ч. N,O-сульфатированного продукта, 1 мас. ч. бикарбоната натрия, 1 мас. ч. комплекса триоксид серы-триметиламин и дают прореагировать при температуре 55oC в течение 20 ч при перемешивании. Затем разбавляют реакционную смесь (коэффициент разбавления 10), потом доводят удельную проводимость полученного раствора до удельной проводимости 0,5 М раствора хлорида натрия. Затем осуществляют осаждение в результате прибавления 2 объемов этанола, центрифугирование с последующим извлечением осадков ценрифугирования посредством 0,5 М раствора NaCl, потом второе осаждение в результате прибавления 2 объемов этанола. После извлечения в высокоочищенной воде и диализа относительно высокоочищенной воды продукты лиофилизируются и сушатся при 40oC под вакуумом.
Спектр ядерного магнитного резонанса.
Изучение спектра ЯМР 13C N,O-сульфатированного гепарозана, происходящего из партии А3, показывает, что для этого соединения спирт в положении 6 у глюкозаминильной группы полностью находится в виде сернокислого сложного эфира.
Некоторые характеристики полученных N,O-сульфатированных гепарозанов, определенные в соответствии с методиками, описанными выше, собраны в приведенной ниже табл. VI.
Установлено, что содержание остаточных NH2-групп (0,02 мкмоль/мг) является небольшим и меньшим, чем у очищенного N-ацетилгепарозана (0,05 мкмоль/мг для партии А и партии В), а также по сравнению с N,O-сульфатированным гепарозаном, полученным в результате частичного N,O-сульфатирования (уточняется в табл. V). Это доказывает полный характер реакций N-сульфатирования.
Молекулярно-массовое распределение продуктов, полученных после N-сульфатирования, анализируется при помощи методики, описанной в примере 1.
Некоторые результаты, выведенные из профилей элюирования, собраны в табл. VII. Определение величины PM0, PM1, PM2 и PM3 является идентичным определению, указанному для табл. III.
N,O-сульфатированный гепарозан из каждой партии содержит по меньшей мере 90 мас. цепей с молекулярными массами, меньшими 10000 а. е. м.
Действительно, партии А1, А2 и А3 содержат 80% цепей с молекулярными массами, заключенными соответственно между 2600 а. е. м. и 9000 а. е. м. 3400 и 10000 а. е. м. и 1800 и 7600 а. е. м.
Пример 5.
Химическая модификация N-ацетилгепарозана в основном с небольшой молекулярной массой, полученного в примере 2.
Синтез N, O-сульфатированных гепарозанов, полученных из продуктов, N-деацетилированных на 80%
Используемый N-ацетилгепарозан был получен в соответствии со способом, описанным в примере 2. Содержание уроновых кислот в этом продукте составляет 2,12 мкмоль/мг. Определение молекулярно-массового распределения было проведено при помощи хроматографии методом исключения в соответствии с методикой, описанной в примере 1. Фракция, составляющая по меньшей мере 87,5 мас. имеет цепи с молекулярными массами, заключенными между 1500 и 15000 а. е. м. Основной пик распределения находится примерно при 4900 а. е. м. Эта партия N-ацетилгепарозана называется партия С.
Стадия а. Частичное деацетилирование.
Растворяют 2,5 г партии С в 50 мл 2 н раствора NaOH. Доводят раствор до температуры 50oC и дают прореагировать при этой температуре в течение 8 ч при перемешивании. Устанавливают значение pH 8 при помощи 2 н раствора HCl, подвергают его диализу относительно высокоочищенной воды, затем лиофилизируют его.
Получают таким образом 1,96 г продукта.
Степень N-деацетилирования.
Количественный анализ свободных аминогрупп (NH2) показывает, что N-ацетилгепарозан был подвергнут N-деацетилированию на 80%
Стадия b. N-сульфатирование.
Предыдущий лиофилизат извлекают в 70 мл воды и прибавляют 2,5 г Na2CO3 и 2,5 г комплекса триоксид серы-триметиламин. Дают прореагировать в течение 20 ч при температуре 55oC.
Доводят удельную проводимость реакционной среды до удельной проводимости 0,5 М раствора NaCl в результате прибавления деминерализованной воды. Осаждают при помощи 4 объемов этанола. Центрифугируют. Снова растворяют осадок в 0,5 М раствора NaCl. Осаждают при помощи 4 объемов этанола. Центрифугируют.
Извлекают осадок после центрифугирования высокоочищенной водой, подвергают этот раствор диализу в соответствии со способом, который уже описан (пример 1), и лиофилизируют его.
Получают примерно 2 г продукта.
Стадия с. Образование соли тетрабутиламмония.
Лиофилизат, полученный на предыдущей стадии, превращается в соль тетрабутиламмония в соответствии со способом, описанным в примере 4 /1) Химическая модификация партии В, стадия b/.
Получают 2,7 г соли.
Стадия d. О-сульфатирование.
Растворяют 2,680 г соли, полученной на предыдущей стадии, в 196 мл формамида и прибавляют 11,27 г комплекса триоксид серы-пиридин. Дают прореагировать при перемешивании в течение 6 ч при температуре 30oC. Прибавляют 1 объем 2 М раствора NaCl на 5 объемов реакционной среды и устанавливают значение pH 7 при помощи раствора NaOH. Проводят повторное осаждение при помощи 2 объемов этанола, центрифугируют и повторно растворяют осадок в 0,5 М раствора NaCl. Затем прибавляют 2 объема этанола. Дают сформироваться осадку, центрифугируют и извлекают осадок после центрифугирования при помощи 80 мл высокоочищенной воды. Прибавляют 20 мл 2 М раствора NaCl, затем 4 объема этанола. Дают сформироваться осадку и центрифугируют.
Стадия е. Гель-фильтрация.
Продукт, полученный на стадии d, извлекается высокоочищенной водой, затем фракционируется в результате гель-фильтрации в соответствии с условиями, описанными в примере 4 /2) Химическая модификация партии А, стадия а/. Определяют молекулярные массы цепей, которые составляют композицию N, O-cульфатированного гепарозана, при помощи хроматографии методом исключения в соответствии со способом, описанным в примере 1. Группируют вместе с одной стороны фракции, содержащие цепи, которые имеют молекулярные массы, заключенные между 1500 и 12000 а. е. м. (раствор С(А)), а с другой стороны, фракции, содержащие N,O-сульфатированные гепарозаны, состоящие из цепей с молекулярными массами от 2000 до 30000 а. е. м. (раствор С(М)).
К раствору С(А) прибавляют 4 объема этанола. Дают сформироваться осадку; центрифугируют и извлекают осадок после ценрифугирования высокоочищенной водой. Подвергают диализу относительно высокоочищенной воды и лиофилизируют.
Получают 1,8 г продукта.
Полученный таким образом N,O-сульфатированный гепарозан называется партией С1.
некоторые характеристики этого N,O-сульфатированного гепарозана, определенные в соответствии с методиками, описанными выше, собраны в приведенной ниже табл. VIII.
Молекулярно-массовое распределение продуктов было оценено при помощи методики, описанной в примере 1. Некоторые результаты, выведенные на основании профиля элюирования, собраны в табл. IX.
Определение величин PMо, PM1, PM2 и PM3 является идентичным определению, указанному в табл. III.
N, O-сульфатированный гепарозан из партии С1 содержит 95 мас. цепей, заключенных между значениями 1500 и 15000 а. е. м. молекулярной массы.
Спектр ядерного магнитного резонанса (ЯМР).
Спектр ЯМР протона и углерода 13C получен для N,O-сульфатированного гепарозана из партии С1 (спектры зарегистрированы на приборе АМХ 500, растворитель Д2О).
Изучение спектров ЯМР протона и углерода 13C подтверждает ожидаемую структуру продукта. Речь идет, действительно, о N,O-сульфатированном гепарозане.
Изучение отношения интенсивностей протонов сахароз к протонам ацетильных групп спектра протонов приводит к предположению, что степень деацетилирования составляет 84% Это значение близко к значению, рассчитанному при использовании отношения содержания свободных аминогрупп к содержанию уроновых кислот (80%).
Спектр ЯМР углерода 13C подтверждает, в частности, что глюкозамин является почти полностью N-сульфатированным. Д-глюкуроновая кислота не является сульфатированной в положениях 2 и 3.
Стадия f. Гель-фильтрация.
К раствору С(М) прибавляют 4 объема этанола. Дают сформироваться осадку, ценрифугируют, извлекают осадок высокоочищенной водой, подвергают диализу и лиофилизируют.
Затем лиофилизат растворяется в 0,5 М растворе NaCl и фракционируется в результате гель-фильтрации в соответствии с условиями, определенными на стадии е.
Группируют вместе, с одной стороны, фракции, содержащие цепи, которые имеют молекулярные массы, заключенные между 1300 и 21000 а. е. м. (раствор С(B)), а с другой стороны, со значениями от 14000 и 37000 а.е.м. (раствор C(C)).
Эти два раствора обрабатываются, как указано выше для раствора С(А), и получают после лиофилизации из раствора С(В) партию С2, а из раствора (С(С))
партию С3.
Табл. X показывает некоторые результаты, выведенные на основании профилей элюирования, для продуктов из партии С2 и партии С3.
Определение величин PM0, PM1, PM2, PM3 является идентичным определению, указанному в табл. III.
N,O-сульфатированный гепарозан, соответствующий партии С2, содержит примерно 99 мас. цепей, молекулярная масса которых заключена между 1500 и 15000 а. е. м. a N,O-сульфатированный гепарозан, соответствующий партии С3, содержит примерно 73 мас. цепей, молекулярная масса которых заключена между 1500 и 15000 а. е. м.
Пример 6.
Химическая модификация N-ацетилгепарозана в основном с небольшой молекулярной массой, полученного по примеру 2. Синтез N,O-сульфатированного гепарозана, полученного из продукта, N-деацетилированного на 40% и имеющего степень сульфатирования 2,6.
N-ацетилгепарозан, используемый в качестве исходного вещества, был получен в соответствии со способом, описанным в примере 2. Содержание уроновых кислот в этом продукте составляет 1,96 мкмоль/мг. Эта партия называется партия Д.
Стадия а. Частичное деацетилирование.
Растворяют 3,7 г партии Д в 74 мл 1н NaOH и обрабатывают в соответствии со способом, описанным в примере 5 (стадия а). Деацетилирование осуществляется в атмосфере азота.
После лиофилизации получают 2,91 г продукта.
Степень N-деацетилирования.
Количественный анализ свободных аминогрупп (NH2) в продукте, полученном после лиофилизации, указывает, что N-ацетилгепарозан подвергается N-деацетилированию на 40%
Стадия b. N-сульфатирование.
Продукт, полученный на предыдущей стадии, был обработан при помощи 3,7 г комплекса триоксид серы-триметиламин в присутствии 3,7 г Na2CO3 и в соответствии со способом, описанным в примере 5 (стадия b).
Получают таким образом примерно 3 г N-сульфатированного гепарозана.
Стадия с. Образование соли тетрабутиламмония.
примерно 2 г N-сульфатированного гепарозана, полученного на предыдущей стадии, превращают в соответствии с методикой, описанной в примере 5 (стадия с), в соль тетрабутиламмония.
Получают 2,99 г лиофилизата.
Стадия d. О-сульфатирование.
Проводят реакцию 2,98 г соли, полученной на предыдущей стадии, с 14 г комплекса триоксид серы-триметиламин в соответствии со способом, описанным в примере 5 (стадия d), затем проводят осаждения и очистки, описанные в том же самом примере 5 (стадия d).
Получают 2,8 г продукта.
Стадия е. Гель-фильтрация.
Продукт, полученный на предыдущей стадии, фракционируется в результате гель-фильтрации при использовании методики и материалов, описанных в примере 4 /2) Химическая модификация партии А, стадия а/.
Молекулярно-массовое распределение фракций N,O-сульфатированного гепарозана определяется при помощи хроматографии методом исключения в соответствии с методикой, описанной в примере 1. Выделяют фракции, состоящие из N, O-сульфатированных гепарозанов, у которых большинство цепей имеют молекулярный вес, заключенный между 1500 и 15000 а. е. м.
Затем эти фракции концентрируются и подвергаются диализу относительно высокоочищенной воды. Потом проводят осаждение при добавлении 5 объемов этанола, центрифугируют, растворяют осадок в 0,5 М растворе NaCl и повторяют операцию осаждения.
Полученный таким образом очищенный продукт подвергается второму фракционированию при использовании протокола, упомянутого в начале этой стадии. Молекулярно-массовое распределение для фракций определяется при помощи хроматографии методом исключения в соответствии с уже описанной методикой. Собирают фракции, содержащие продукты, которые имеют молекулярные массы, заключенные между 4000 и 8000 а. е. м.
Эти фракции подвергают диализу, затем продукты осаждаются в результате прибавления этанола. Осадок извлекается и растворяется в 0,5 М растворе NaCl. Полученный таким образом раствор подвергается диализу относительно высокоочищенной воды и потом лиофилизируется.
Получают таким образом примерно 1 г N,O-сульфатированного гепарозана, называемого партия Д1.
Характеристики и молекулярно-массовое распределение этого N,O-сульфатированного гепарозана указываются соответственно в табл. XI и XII.
Определение величин PM0, PM1, PM2 и PM3 является идентичным определению, указанному в табл. III.
N,O-сульфатированный гепарозан из партии Д1 содержит 80 мас. цепей с молекулярными массами, заключенными между 4047 и 10305 а. е. м. и примерно 98,5 мас. этих цепей имеют молекулярную массу, заключенную между 1500 и 14600 а. е. м.
Спектр ядерного магнитного резонанса (ЯМР).
Спектры ЯМР протона и углерода 13С получены для N,O-сульфатированного гепарозана из партии Д1 (спектры зарегистрированы на приборе АМХ 500, растворитель Д2O).
Изучение спектров ЯМР протона и углерода 13С подтверждает ожидаемую структуру продукта. Речь идет, действительно, о N,O-сульфатированном гепарозане.
Спектр ЯМР углерода 13С подтверждает, в частности, что ни одна из аминогрупп глюкозаминового звена не существует в виде свободной аминогруппы (NH2). Глюкозамин является полностью сульфатированным в положении С6, напротив, все гидроксигруппы глюкуроновой кислоты не являются сульфатированными.
Пример 7.
Химическая модификация N-ацетилгепарозана в основном с небольшой молекулярной массой, полученного в примере 2.
Синтез N, O-сульфатированного гепарозана, полученного из продукта, N-деацетилированного на 40% и имеющего степень сульфатирования 3.
В качестве исходного вещества используют партию N-ацетилгепарозана, полученного в соответствии со способом, описанным в примере 2. Эта партия называется партия Е и имеет содержание уроновых кислот величиной 2 мкмоль/мг.
Молекулярно массовое распределение этого N-ацетилгепарозана было оценено при помощи методики, описанной в примере 1. Этот N-ацетилгепарозан содержит примерно 75 мас. цепей с молекулярными массами, заключенными между 1500 и 15000 а. е. м. а средняя молекулярная масса этих цепей составляет приблизительно 10900 а. е. м. Молекулярная масса основного компонента составляет 5135 а. е. м.
Стадия а. Частичное деацетилирование.
N-ацетилгепарозан был N-деацетилирован на 40% в соответствии с протоколом, упомянутым в примере 6 (стадия а). Для осуществления этого N-деацетилирования используют 2,5 г исходного вещества, которое растворяют в 50 мл 1 н NaOH. По окончании реакции pH реакционной среды доводится до 6 при помощи HCl, затем проводится концентрирование.
Степень N-деацетилирования.
Количественный анализ свободных аминогрупп (NH2) показывает, что N-ацетилгепарозан был подвергнут N-деацетилированию на 40%
Стадия b. Гель-фильтрация.
Используют колонку с Сефакрилом S 300 HR (5 см х 100 см), приведенную в равновесие с 0,5 М NaCl.
Молекулярно-массовое распределение гепарозана, содержащегося в различных фракциях, определяется при помощи хроматографии методом исключения. Группируют вместе фракции, содержащие цепи, имеющие молекулярную массу от 6000 до 20000 а. е. м. концентрируют на установке Ротавапор, осаждают при помощи 4 объемов этанола и сушат образовавшийся осадок.
Получают таким образом примерно 1 г продукта.
Стадия с. Образование соли тетрабутиламмония и частичное N,O-сульфатирование.
Для образования соли тетрабутил- аммония используют 300 мг продукта, полученного на предыдущей стадии, и применяют способ, описанный в примере 4 /1) Химическая модификация партии В, стадия b/. Получают 490 мг соли, которую растворяют в 30 мл формамида. Затем проводят частичное N,O-сульфатирование в соответствии со способом, описанным в примере 4 /1) Химическая модификация партии В, стадия с, первый параграф/.
Исходя из 490 мг соли, которую подвергают реакции с 2,25 г комплекса триоксид серы-пиридин, получают 406 мг N,O-сульфатированного гепарозана.
Стадия d. Полное N-сульфатирование и гель-фильтрация.
Растворяют 372 мг продукта, полученного на предыдущей стадии, в 15 мл 5% -ного раствора Na2CO3 и обрабатывают при помощи 372 мг комплекса триоксид серы-триметиламин в соответствии с методикой, описанной в примере 4 /2) Химическая модификация партии А, стадия с/.
Продукт, полученный после полного N-сульфатирования, затем фракционируется при использовании колонки с Сефакрилом S 300 HR (2,5 см х 100 см) в соответствии с уже описанным протоколом.
Фракции, имеющие молекулярную массу от 6000 до 20000 а. е. м. группируются вместе, концентрируются на установке Ротавапор, подвергаются экстенсивному диализу относительно холодной высокоочищенной воды и лиофилизируются.
Получают таким образом 260 мг N,O-сульфатированного гепарозана, называемого партия Е1.
Характеристики этого продукта собраны в табл. XIII.
Табл. XIV показывает молекулярно-массовое распределение цепей, которые образуют партию Е1.
Этот N,O-сульфатированный гепарозан содержит приблизительно 75 мас. цепей с молекулярными массами между 15000 и 150000 а. е. м. и приблизительно 65 мас. цепей, имеющих молекулярные массы между 7700 и 150000 а. е. м.
Определение величин PM0, PM1, PM2 и PM3 является идентичным определению, указанному в табл. III.
Пример 8.
Синтез двух партий N,O-сульфатированного гепарозана, полученных из продуктов, N-деацетилированных на 80% и имеющих степень сульфатирования 2,25 и 2,4.
Используемое исходное вещество (партия F) является партией N-ацетилгепарозана, полученного в соответствии с методикой, описанной в примере 2.
Определение молекулярно-массового распределения цепей, которые образуют это соединение, было осуществлено при помощи эксклюзивной хроматографии в соответствии с методикой, описанной в примере 1.
Изучение профиля элюирования позволяет установить, что партия F содержит 92 мас. цепей, молекулярная масса которых заключена между 1500 и 15000 а. е. м.
Основной пик находится примерно при 4800 а. е. м. Фракция партии F, составляющая по меньшей мере 80 мас. имеет молекулярную массу, заключенную между 2400 а. е. м. и 10000 а. е. м.
Содержание уроновых кислот в этом продукте составляет 2,4 мкмоль/мг.
Стадия а. Частичное деацетилирование.
Проводят процесс, как это описано в примере 5 (стадия а), используя 2,5 г партии F и 50 мл 2 н NaOH.
Получают 1,6 г продукта, N-деацетилированного на 80%
Степень N-деацетилирования в процентах была оценена при помощи количественного анализа свободных аминогрупп.
Стадии b и c. N-сульфатирование и образование соли тетрабутиламмония.
Эти две стадии являются идентичными стадиям b и с, описанным в примере 5.
Получают 4,7 г соли тетрабутиламмония.
Стадия d. O-сульфатирование.
Растворяют 2,9 г полученной выше соли в 290 мл формамида и прибавляют комплекс триоксид серы-пиридин.
Проводят процесс, как это описано на стадии d примера 5.
Осадок, полученный после второго центрифугирования, растворяют в высокоочищенной воде, затем подвергают диализу относительно высокоочищенной воды и лиофилизируют.
Получают таким образом 3,68 г продукта.
Стадия е. Гель-фильтрация.
Продукт, полученный на предыдущей стадии, растворяется в 0,5 М растворе NaCl, затем он помещается в колонку с Сефакрилом S 300 HR, приведенную в равновесие с 0,5 М NaCl. Эфлюент собирается при помощи коллектора фракций.
Фракции, имеющие молекулярную массу, заключенную между 1400 а. е. м. и 10000 а. е. м. группируются вместе и концентрируются на установке Ротавапор. Осаждают при помощи 4 объемов этанола, центрифугируют, извлекают осадок высокоочищенной водой, проводят экстенсивный диализ полученного таким образом раствора относительно высокоочищенной воды, лиофилизируют и сушат.
Получают таким образом 1,6 г N,O-сульфатированного гепарозана, называемого партия F1.
Фракции, соответствующие молекулярным массам, заключенным между 5000 а. е. м. и 35000 а. е. м. собираются, концентрируются на установке Ротавапор, а к полученному таким образом раствору прибавляют 4 объема этанола, центрифугируют, извлекают осадок в воде, проводят диализ полученного таким образом раствора относительно высокоочищенной воды и лиофилизируют. Лиофилизат подвергается новому фракционированию в соответствии с методикой, указанной в начале этой стадии.
Молекулярно-массовое распределение различных фракций определяется при помощи хроматографии методом исключения в соответствии с методикой, описанной в примере 1. Группируют вместе фракции, которые содержат N,O-сульфатированные гепарозаны, имеющие молекулярную массу, заключенную между 2000 и 26000 а. е. м. Осаждают при помощи 4 объемов этанола, центрифугируют и снова растворяют осадок в дистиллированной воде, проводят диализ и лиофилизируют. Получают таким образом 0,6 г N,O-сульфатированного гепарозана, называемого партия F2.
Характеристики партий F1 и F2 приводятся в табл. XV. Результаты, выведенные из профилей элюирования, собраны в табл. XVI.
Определение величин PM0, PM1, PM2 и PM3 является идентичным определению, указанному в табл. III.
N, O-сульфатированный гепарозан из партии F1 содержит приблизительно 99 мас. цепей, имеющих молекулярные массы, заключенные между 1500 и 15000 а. е. м. а аналогичный гепарозан из партии F2 содержит приблизительно 84,6 мас. цепей с молекулярными массами от 1500 до 15000 а. е. м. и приблизительно 70 мас. цепей с молекулярными массами от 6900 до 13500 а. е. м.
Спектр ядерного магнитного резонанса (ЯМР).
Спектры ЯМР протона и углерода 13C получены для N,O-сульфатированного гепарозана из партии F1 (спектры регистрируются на приборе АМХ 500, растворитель Д2O).
Изучение спектра протонов и, в частности, соотношения интенсивности протонов сахарозы к протонам деацетилированных групп приводит к предположению, что примерно 20% глюкозаминовых звеньев являются N-ацетилированными.
Изучение спектра 13C подтверждает, что глюкозаминовое звено является N-сульфатированным. Глюкуроновая кислота в большинстве дисахаридных структур не является О-сульфатированной в положениях 2 и 3.
Синтез А.
Получение N-ацетилгепарозана в основном с высокой молекулярной массой (способ 1).
1) Разведение посева Escherichia coli (K5) и выделение фильтрата, содержащего N-ацетилгепарозан.
Засевают 400 мл среды Д с составом, уточненным в приведенной ниже табл. XVII, при помощи посева Escherichia coli S EBR 3282 и проводят инкубацию при перемешивании в течение 2 ч при 37oC.
Полученная предварительная культура затем переносится в ферментер объемом 18,5 л, содержащий 11 л среды С с составом, также уточненным в приведенной ниже табл. XVII, и проводят инкубацию в течение 6 ч 30 мин при температуре 37oC и значении pH 7,2, причем парциальное давление кислорода поддерживается при значении 40 мм рт. ст. в результате регулирования подачи воздуха (до 20 л/мин) и перемешивания. Потом прибавляют глицерин, вводя непрерывно стерильный раствор, содержащий 500 г/л глицерина, с расходом 18 г/ч в течение 16 17 ч.
Продолжают разведение культуры при тех же самых условиях температуры, pH и парциального давления кислорода до почти полного потребления глицерина. Наблюдение за ОП (λ 600 нм) суспензии с культурой после окончания прибавления глицерина показывает, что достигается стационарное состояние или слабое разложение лизинами до прекращения разведения культуры в возрасте 28 - 30 ч в ферментере.
Затем охлаждают бульон с культурой до температуры 25oC, потом фильтруют его через мембрану с пористостью 0,22 мкм. Получают таким образом приблизительно 12 л фильтрата, содержащего N-ацетилгепарозан в основном с высокой молекулярной массой.
Таблица XVII.
Состав и получение среды С и среды Д.
Среда С.
В 900 мл высокоочищенной воды растворить в следующем порядке:
НТК (нитролотриуксусная кислота) 1000 мг
K2HPO4 790 мг
Глутаминовая кислота 11000 мг
MgCl2, 6H2O 500 мг
K2SO4 450 мг
FeSO4, 7H2O 18 мг
CaCl2, 2H2O 2 мг
NaCl 500 мг
KCl 5000 мг
Раствор олигоэлементов (см. табл. II, пример 1) 1 мг
Глицерин 10000 мг
Установить значение pH 7,2 при помощи концентрированного гидроксида калия с плотностью 1,38 и довести объем до 1000 мл при помощи высокоочищенной воды. Провести стерилизующую фильтрацию на мембране с пористостью 0,2 мкм.
Раствор глицерина.
Растворить 50 г глицерина в адекватном количестве высокоочищенной воды и довести объем до 1000 мл при помощи того же самого растворителя. Провести стерилизующую фильтрацию на мембране с пористостью 0,2 мкм.
Пеногаситель, используемый в ходе ферментации, является Структолом I 673 (фирмы Шилл и Зейлахер® ).
Среда D.
Получение среды D является идентичным получению среды С с тем отличием, что следует, помимо прочего, добавить буферный раствор (pH 7,2): 3-морфолинопропансульфокислота после прибавления пеногасителя.
2) Выделение и очистка N-ацетилгепарозана в основном с высокой молекулярной массой.
N-ацетилгепарозан в основном с высокой молекулярной массой был выделен и очищен в соответствии со способом, описанным в примере 2 /2) Выделение и очистка N-ацетилгепарозана в основном с небольшой молекулярной массой, стадия а/.
3) Описание полученного N-ацетилгепарозана.
Определение молекулярно-массового распределения при помощи хроматографии методом исключения.
Учитывая используемые в настоящее время стандарты, молекулярно-массовое распределение было оценено приблизительно.
N-ацетилгепарозан, полученный для этого синтеза, состоит из цепей с молекулярными массами, заключенными между 20000 а. е. м. и 50000 а. е. м. а средняя молекулярная масса находится приблизительно при 100000 200000 а. е. м.
Количественный анализ уроновых кислот.
Содержание уроновых кислот в очищенном продукте, полученном на выходе конечной стадии, составляет величину 2,2 мкмоль/м.
Спектрофотометрия в ультрафиолетовой и видимой областях.
Полученный спектр позволяет утверждать, что эта партия содержит менее 0,5 АД N.
Количественный анализ всех протеинов.
Содержание всех протеинов является меньшим 0,5%
Количественный анализ свободных аминогрупп NH2.
Содержание NH2-групп является меньшим 0,1 мкмоль/мг.
Синтез В.
Получение N-ацетилгепарозана в основном с большой молекулярной массой (способ II).
1) Разведение посева Escherichia coli (K5).
Разведение посева Escherichia coli SEBR 3282 было осуществлено в соответствии с методикой, описанной в синтезе А. Получают приблизительно 12 л культуры, содержащей N-ацетилгепарозан в основном с высокой молекулярной массой.
2) Предварительная очистка.
Проводят процесс, как это описано в примере 3 /2) Предварительная очистка/, применяя на стадии d фильтрующий элемент с полыми волокнами Амикон® с порогом отсечения 30000 а. е. м. или эквивалентный.
3) Выделение и очистка N-ацетилгепарозана в основном с высокой молекулярной массой.
Проводят процесс, как это указано в примере 3 /3) Выделение и очистка N-ацетилгепарозана в основном с небольшой молекулярной массой/.
Синтез С.
Химическая модификация N-ацетилгепарозана в основном с высокой молекулярной массой, полученного по синтезу А.
В качестве исходного вещества для различных химических модификаций используют N-ацетилгепарозан, называемый партия G1, который получается в соответствии со способом, описанным в синтезе А.
Содержание уроновых кислот в этом продукте составляет 2,41 мкмоль/мг.
Стадия а. Частичное деацетилирование.
Растворяют 1,9 г партии G1 в 38,5 мл 2н NaOH. Доводят раствор до температуры 50oC и дают прореагировать при этой температуре в течение 8 ч в атмосфере азота.
Затем устанавливают значение pH 8,25 путем прибавления 2 н HCl. Проводят диализ относительно высокоочищенной воды, затем лиофилизируют.
Получают таким образом 1,6 г продукта.
Степень N-деацетилирования.
Количественный анализ свободных аминогрупп (NH2) показывает, что N-ацетилгепарозан был подвергнут N-деацетилированию на 80%
Стадия b. N-сульфатирование.
Растворяют 1,3 г продукта, полученного на предыдущей стадии, в 57 мл высокоочищенной воды, прибавляют 1,9 г Na2CO3 и 1,9 г комплекса триоксид серы-триметиламин и доводят до температуры 55oC в течение 20 ч. Затем доводят удельную проводимость раствора до удельной проводимости 0,5 М раствора NaCl путем прибавления деминерализованной воды и осаждают при помощи 4 объемов этанола. Центрифугируют, извлекают осадок 0,5 М раствором NaCl, осаждают при помощи 4 объемов этанола и центрифугируют.
Растворяют осадок в высокоочищенной воде и проводят диализ высокоочищенной водой в соответствии со способом, описанным в примере 1, затем лиофилизируют.
Получают таким образом 1,809 г N-сульфатированного гепарозана.
Стадия с. Образование соли тетрабутиламмония.
Растворяют 800 мг продукта, полученного на предыдущей стадии, в 100 мл воды. Помещают этот раствор в ионообменную колонку с Доуэкс 50W8 и проводят процесс, как это описано в примере 4 /1) Химическая модификация партии В, стадия b/.
После лиофилизации получают примерно 1,3 г соли.
Стадия d. О-сульфатирование.
Растворяют полученную выше соль в 80 мл формамида, прибавляют 5,6 г комплекса триоксид серы-пиридин. Дают прореагировать в течение 6 ч при температуре 30oC, затем прибавляют 16 мл 2 М NaCl. Доводят до значения pH 7 и осаждают при помощи 2 объемов этанола. Извлекают осадок 0,5 М раствором NaCl, снова его осаждают при помощи 2 объемов этанола, проводят диализ относительно высокоочищенной воды и концентрируют на установке Ротавапор.
Стадия е. Гель-фильтрация.
Концентрированный раствор, полученный на предыдущей стадии, фракционируется путем гель-фильтрации при использовании колонки с Сефакрилом S 300 HR и с применением в качестве элюента 0,5 М раствора NaCl.
Выделяют фракции, которые соответствуют молекулярным массам, заключенным между 10000 и 500000 а. е. м.
Прибавляют 2 объема этанола, затем концентрируют. Потом доводят удельную проводимость до удельной проводимости 0,5 М раствора NaCl, прибавляя воду.
Повторяют фракционирование путем гель-фильтрации и собирают фракции, содержащие N,O-сульфатированный гепарозан, состоящий из цепей, имеющих средние молекулярные массы от 100000 до 200000 а. е. м. а также фракции, содержащие N,O-сульфатированный гепарозан, состоящий из цепей, имеющих средние молекулярные массы приблизительно 50000 а. е. м. и приблизительно 12000 а. е. м.
К каждой из трех этих фракций прибавляют 5 объемов этанола, подвергают их диализу относительно высокоочищенной воды и подвергают полученные после диализа растворы лиофилизации.
Получают таким образом 3 партии N,O-сульфатированных гепарозанов:
партия G1 представляет собой N,O-сульфатированный гепарозан, состоящий из цепей, имеющих среднюю молекулярную массу от 100000 до 200000 а. е. м. выделяют 0,14 г этой партии;
партия G2 представляет собой N,O-сульфатированный гепарозан, состоящий из цепей, имеющих среднюю молекулярную массу приблизительно 50000 а. е. м. выделяют 0,350 г этого N,O-сульфатированного гепарозана;
партия G3 представляет собой N,O-сульфатированный гепарозан, состоящий из цепей, имеющих среднюю молекулярную массу 12000 а. е. м. выделяют приблизительно 0,100 г этой последней партии.
Характеристики трех партий N,O-сульфатированного гепарозана, описанных в этом примере, собраны в табл. XVIII.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ПОЛИСАХАРИДОВ С ВЫСОКОЙ АНТИТРОМБОТИЧЕСКОЙ АКТИВНОСТЬЮ В ПЛАЗМЕ | 2004 |
|
RU2361881C2 |
| Способ получения о-ацилированных глюкозаминогликанов | 1989 |
|
SU1831487A3 |
| ЭПИМЕРИЗОВАННЫЕ ПРОИЗВОДНЫЕ ПОЛИСАХАРИДА К5 С ВЫСОКОЙ СТЕПЕНЬЮ СУЛЬФАТИРОВАНИЯ | 2003 |
|
RU2333222C2 |
| ГЛИКОЗАМИНОГЛИКАНЫ, ПРОИЗВОДНЫЕ К5-ПОЛИСАХАРИДА, ОБЛАДАЮЩИЕ ВЫСОКОЙ АНТИКОАГУЛЯНТНОЙ И АНТИТРОМБОТИЧЕСКОЙ АКТИВНОСТЬЮ, И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2001 |
|
RU2283319C2 |
| БИОСИНТЕТИЧЕСКИЙ ГЕПАРИН | 2017 |
|
RU2760706C2 |
| НЕАНТИКОАГУЛЯНТНЫЕ ГЛИКОЗАМИНОГЛИКАНЫ, СОДЕРЖАЩИЕ ПОВТОРЯЮЩИЕСЯ ДИСАХАРИДНЫЕ ЗВЕНЬЯ, И ИХ МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ | 2012 |
|
RU2617764C2 |
| СПОСОБ ПОЛУЧЕНИЯ СУЛЬФАТИРОВАННОГО ПРОИЗВОДНОГО ГАЛАКТАНА | 1990 |
|
RU2028381C1 |
| СПОСОБ ПОЛУЧЕНИЯ СУЛЬФАТИРОВАННОГО ХИТОЗАНА | 1992 |
|
RU2048474C1 |
| СПОСОБ ПОПЕРЕЧНОГО СШИВАНИЯ КАРБОКСИЛИРОВАННЫХ ПОЛИСАХАРИДОВ | 1999 |
|
RU2230073C2 |
| Способ очистки хондроитин сульфата | 2015 |
|
RU2693262C2 |

Использование: в медицине в качестве антитромботических средств. Сущность изобретения: продукты: N, O-сульфатированные гепарозаны, состоящие из цепей или смесей цепей с молекулярной массой между 1500 и 15000 а. е. м., характеризующиеся повторяющейся дисахаридной структурой ф-лы I, указанной в описании, где Е - ацетильная группа в количестве 0 - 60% от дисахаридных звеньев N,O-сульфатированных гепарозанов, остальные дисахаридные звенья содержат сульфатную группу и, в случае необходимости, атом водорода, G - водород или сульфатная группа, причем степень сульфатирования гепарозанов, выраженная как отношение сульфат/карбоксил, заключена между 1,9 - 3, или их фармацевтически приемлемые соли. Способ получения I включает последовательность следующих стадий: а) культивирование штамма бактерий Escherichia coli /KS/CNCM N l-1013, b), выделение из посева и очистка N-ацетилгепарозана с получением композиции, содержащей от 70 до 100% гепарозана, с) частичное деацетилирование полученной композиции, содержащей от 70 до 100% гепарозана, d) частичное или полное N,O-сульфатирование полученной композиции. 4 с. и 24 з. п. ф-лы, 18 табл.
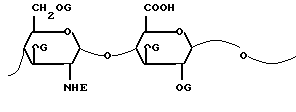
в которой E ацетильная группа в количестве 0 60% от дисахаридных звеньев N, O-сульфатированных гепарозанов, остальные дисахаридные звенья содержат сульфатную группу и, в случае необходимости, атом водорода;
G атом водорода или сульфатная группа,
причем степень сульфатирования гепарозанов, выраженная как отношение сульфат/карбоксил, заключена между 1,9 3, или их фармацевтически приемлемые соли.
6. N,O-сульфатированные гепарозаны по п.1, состоящие из по меньшей мере 70 мас. цепей с мол.м. между 10000 и 12000 а.е.м. степень сульфатирования составляет 1,9 2,5 и содержание ацетильных групп меньше или равно 20%
7. N,O-сульфатированные гепарозаны по п.1, которые состоят из по меньшей мере 70 мас. цепей с мол.м. между 6000 и 8000 а.е.м. степень сульфатирования составляет 2,0 2,8 и содержание ацетильных групп меньше или равно 60%
8. N,O-сульфатированные гепарозаны по п.1, которые состоят из по меньшей мере 80 мас. цепей с мол.м. между 2300 и 7200 а.е.м. степень сульфатирования составляет 1,9 2,5 и содержание ацетильных групп меньше или равно 20%
9. N,O-сульфатированные гепарозаны по п.1, которые состоят из по меньшей мере 80 мас. цепей с мол.м. между 3300 и 7700 а.е.м. степень сульфатирования составляет 1,9 2,5 и содержание ацетильных групп меньше или равно 20%
10. N,O-сульфатированные гепарозаны по п.1, которые состоят из по меньшей мере 70 мас. цепей с мол.м. между 6900 и 13500 а.е.м. степень сульфатирования составляет 1,9 2,5 и содержание ацетильных групп меньше или равно 20%
11. N,O-сульфатированные гепарозаны по п.1, которые состоят из по меньшей мере 80 мас. цепей с мол.м. заключенными между 4000 и 10300 а.е.м. степень сульфатирования составляет 2,0 2,8 и содержание ацетильных групп меньше или равно 60%
12. Способ получения N,O-сульфатированных гипарозанов по п.1, отличающийся тем, что он включает последовательность следующих стадий: стадия a: культивирование штамма бактерий Escherichia coli (K5) CNCM N 1-1013; стадия b: выделение из посева и очистка N-ацетилгепарозана с получением композиции, содержащей 70 100% N-ацетилгепарозана, состоящего из цепей или из смеси цепей с мол.м. между 1500 15000 а.е.м. характеризующихся повторяющейся дисахаридной структурой с формулой II
стадия c: частичное деацетилирование полученной композиции для получения композиции, содержащей 70 100% гепарозана, состоящего из цепей или из смеси цепей с мол. м. между 1500 и 15000 а.е.м. характеризующихся повторяющейся дисахаридной структурой с формулой III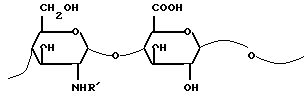
в которой R' представляет ацетильную группу в количестве 0 60% от дисахаридных звеньев, а в остальных дисахаридных звеньях атом водорода;
стадия d: либо частичное N,0-сульфатирование полученной гепарозановой композиции, либо частичное N, 0-сульфатирование полученной композиции с последующей стадией полного N-сульфатирования, либо полное или частичное N-сульфатирование с последующей стадией полного или частичного 0-сульфатирования,
и включает, в случае необходимости, одну или несколько стадий разделения по молекулярным массам, осуществляемым по окончании стадий a, b, c или d.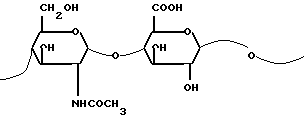
причем оба концевых звена, восстановитель и невосстановитель указанного N-ацетилгепарозана являются звеьями уроновых кислот или N-ацетилглюкозамина.
27. Гепарозаны, состоящие из смеси цепей с мол.м. между 1500 и 15000 а. е.м. характеризующиеся повторяющейся дисахаридной структурой формулы III
в которой R'- ацетильная группа в 0 60% дисахаридных звеньев, в остальных дисахаридных звеньях R' атом водорода.
| EP, заявка, 0214879, кл | |||
| Топка с несколькими решетками для твердого топлива | 1918 |
|
SU8A1 |
| EP, заявка, 0116801, кл | |||
| Топка с несколькими решетками для твердого топлива | 1918 |
|
SU8A1 |
| EP, заявка, 0209924, кл | |||
| Топка с несколькими решетками для твердого топлива | 1918 |
|
SU8A1 |
