Данное изобретение относится к группе новых производных бензимидазолона и хиназолинона, которые являются агонистами рецепторов ORL1 (ноцицептина) человека. Изобретение также относится к получению названных соединений, к фармацевтическим композициям, содержащим фармакологически активное количество, по крайней мере, одного из названных производных имидазолона и хиназолинона в качестве активного ингредиента, а также к применению упомянутых фармацевтических композиций для лечения нарушений, в которые вовлечены рецепторы ORL1.
Изобретение относится к применению описываемого соединения для производства лекарственного средства, оказывающего благоприятное действие. Благоприятное действие раскрыто в описании или оно очевидно специалисту в данной области из спецификации и общих сведений в данной области. Изобретение также относится к применению соединения в соответствии с изобретением для производства лекарственного средства для лечения или предупреждения заболевания или состояния. Точнее, изобретение относится к новому применению для лечения заболевания или состояния, упоминаемого в описании или очевидного специалисту в данной области из спецификации и общих сведений в данной области. В воплощениях изобретения определенные соединения, представляемые в описании, используют для производства лекарственного средства.
Рецептор, «подобный опиоидному рецептору 1» (ORL1), был идентифицирован из библиотеки кДНК человека. Установлено, что названный «орфан-рецептор» имеет близкую гомологию с µ-, κ- и δ-опиоидными рецепторами (Mollereau et al., FEBS Lett., 341, 33-38, 1994; Bunzow et al., FEBS Lett., 347, 284-288, 1994). Несмотря на близкое сходство его последовательности и его структурное сходство с опиоидными рецепторами, лиганды классического опиоидного рецептора не взаимодействуют с рецепторами ORL1. В 1995 году нейропептид из 17 аминокислот был выделен из экстрактов мозга и, как впоследствии было показано, является природным лигандом рецептора ORL1, связанного с G-белком (Reinscheid et al., Science, 270, 792-794, 1995; Meunier et al., Nature, 377, 532-535, 1995). Полученный пептид назвали орфанин FQ или ноцицептин. Он не связывается с тремя традиционными опиоидными рецепторами. Эти данные послужили началом важных исследований функциональной роли рецептора ORL1 и новых лигандов для названного рецептора.
В результате исследований появились сотни публикаций, включая несколько обзоров (см., например, Grond et al., Anaesthesist, 51, 996-1005, 2002) и множество патентных заявок, в которых описывают как пептидные, так и непептидные лиганды. Описанные соединения в значительной степени различаются по эффективности в отношении рецепторов ORL1, а также по селективности (ORL1 против µ-опиатных рецепторов). Так как µ-опиатные рецепторы имеют широкое распространение в организме, отсутствие избирательности может привести к ряду нежелательных подобных опиатным побочных эффектов, таких как седативный эффект, угнетение дыхания, толерантность и зависимость (Drug News Perspect., 14, 335, 2001). Аналогично in vivo фармакодинамические и фармакокинетические свойства описываемых соединений очень изменчивы.
Ряд патентных заявок, связанных с ORL1, относятся к производным бензимидазолона: например, WO 98/54168, WO 99/36421, WO 00/006545, WO 00/08013, WO 01/39775 и US 20020128288. Из числа последних WO 01/39775 является наиболее близкой заявкой данному изобретению. Однако описанные в ней производные бензимидазолона, как оказалось, не соответствуют критериям, которые общепризнанно являются важными для применяемых терапевтических средств. Они характеризуются:
(1) наибольшей эффективностью (сродство к рецепторам ORL1 в диапазоне 166-1252 нМ);
(2) небольшой избирательностью в отношении µ-опиатных рецепторов (сродство в диапазоне 19-457 нМ);
(3) никаких доказательств относительно биодоступности после перорального введения, и
(4) никаких доказательств относительно доступности в ЦНС.
Наиболее мощным описанным агонистом ORL1 является Ro 64-6198. Соединение не содержит фрагмент бензимидазолона, но вместо этого имеет спиро-ядро (см.: EP0856514; Eur. J. Med. Chem., 35 (2000) 839-851 и Proc. Natl. Acad. Sci. USA., 2000, 97, 4938). Ro 64-6198 упоминают как мощное и избирательное соединение, легко пенетрирующее гематоэнцефалический барьер. Однако, несмотря на его благоприятные in vitro связывающие характеристики, профиль in vivo названного лиганда демонстрирует некоторые недостатки:
(1) он оказался менее эффективным на модели состояния страха, чем предполагали на основании данных in vitro;
(2) «терапевтическое окно» между желаемой эффективностью как агониста ORL1 и нежелательными опиатными побочными эффектами in vivo оказалось меньше, чем предполагали на основании данных in vitro.
Обсуждения производных бензимидазолона и Ro 64-6198, цитируемые выше, не указывают направления, как улучшить фармакологический профиль in vivo лучших описанных соединений. В обзоре, рассматривающем предмет обсуждения (“Characterisation of opiates, neuroleptics and synthetic analog at ORL1 and opioid receptors”, Eur. J. Pharmacol., 428, 29-36, 2001), Zaveri et al. заключают: «В отсутствие модели небольших молекул в активном центре, или кристаллической структуры рецептора ORL1, связанного с небольшой молекулой, необходимо быть крайне осторожным в оценке SAR среди различных классов лигандов рецептора ORL1, даже лигандов с очень близкими структурными свойствами».
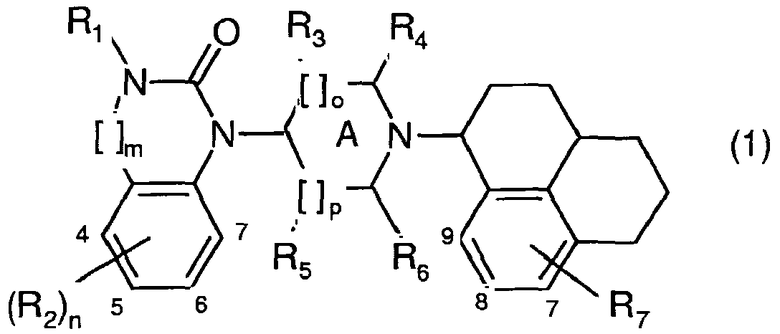
Неожиданно в настоящее время установлено, что в серии производных бензимидазолона и хиназолинона с новой комбинацией заместителей группа соединений, как было показано, имеет высокое сродство к рецепторам ORL1 человека. Кроме того, названные соединения, проявляющие хорошую избирательность в отношении рецепторов ORL1 относительно µ-опиатных рецепторов, являются легко биодоступными после перорального введения и проникают через гематоэнцефалический барьер. Фармакологический профиль in vitro и in vivo некоторых из названных соединений оказался лучше, чем профиль Ro 64-6198, в частности, относительно терапевтического окна между требуемой эффективностью как агониста ORL1 и нежелательными опиатными побочными эффектами in vivo. Изобретение относится к соединениям общей формулы (1):
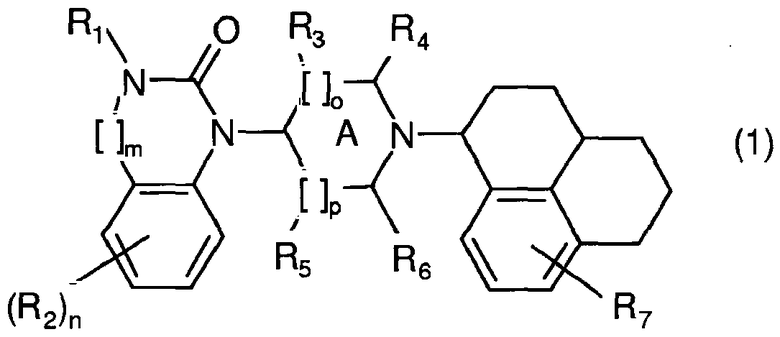
в которой:
R1 представляет собой Н, алкил(1-6С), алкил(1-3С)циклоалкил(3-6С), карбалкокси(2-7С) или ацил(2-7С),
[]m означает -(CH2)m-, в котором m равно 0 или 1,
R2 представляет собой галоген, CF3, алкил(1-6С), алкил(1-3С)циклоалкил(3-6С), фенил, амино, аминоалкил(1-3С), алкил(1-3С)амино, диалкил(1-3С)амино, циано, цианоалкил(1-3С), гидрокси, гидроксиалкил(1-3С), (1-3С)алкокси, OCF3, ацил(2-7С), трифторацетил, аминокарбоксил, (1-3С)алкилсульфонил или трифторметилсульфонил и n означает целое число 0-4 при условии, что, когда n равно 2, 3 или 4, заместители R2 могут быть одинаковыми или разными,
А представляет собой насыщенное или частично ненасыщенное кольцо,
[]о и []р представляют собой -(СН2)о- и -(СН2)р- соответственно при условии, что также возможно значение -СН-, когда А представляет собой частично ненасыщенное кольцо, и о и р независимо составляют 0, 1 или 2.
R3, R4, R5 и R6 независимо представляют собой водород, алкил(1-3С), алкил(1-3С)циклоалкил(3-6С), СН2ОН или (R3 и R5) или (R3 и R6), или (R4 и R5), или (R4 и R6) вместе могут образовывать алкиленовый мостик, содержащий 1-3 атома углерода, при условии, что, когда о равно 2, R3 означает водород, и когда р равно 2, R5 означает водород,
R7 представляет собой Н, галоген, CF3, алкил(1-6С), алкил(1-3С)циклоалкил(3-6С), амино, аминоалкил(1-3С), алкил(1-3С)амино, диалкил(1-3С)амино, гидрокси, гидроксиалкил(1-3С), (1-3С)алкокси, OCF3, ацил(2-7С), аминокарбоксил или (1-3С)алкилсульфонил,
и их фармакологически подходящим солям и пролекарствам.
Изобретение включает все соединения формулы (1), рацематы, смеси диастереомеров и индивидуальные стереоизомеры. Таким образом, соединения, в которых заместители на потенциально асимметрических атомах углерода имеют или R-конфигурацию, или S-конфигурацию, относятся к изобретению. Также пролекарства, то есть соединения, которые при введении человеку любым известным способом, превращаются в соединения формулы (1), относятся к изобретению. Пролекарства являются биообратимыми производными лекарственных молекул, используемых, чтобы преодолеть некоторые барьеры для действия родительской лекарственной молекулы. Упомянутые барьеры включают в себя растворимость, проницаемость, стабильность, предсистемный метаболизм и ограничения направленной доставки, не ограничиваясь перечисленным (J. Stella, “Prodrugs as therapeutics”, Expert Opin. Ther. Patents, 14(3), 277-280, 2004). В частности, это имеет отношение к соединениям с первичными или вторичными амино- или гидроксигруппами. Такие соединения могут взаимодействовать с органическими кислотами с образованием соединений формулы (1), в которых присутствует дополнительная группа, которую легко удаляют применением, например, амидина, енамина, основания Манниха, производного гидроксилметилена, производного О-(ацилоксиметилен карбамата), карбамата, сложного эфира, амида или енаминона. Пролекарство является неактивным соединением, которое при всасывании превращается в активную форму (Medical Chemistry: Principles and Practice, 1994, ISBN 0-85186-494-5, Ed.: F.D. King, p. 216).
Изобретение относится, в частности, к соединениям формулы (1), в которой: А означает насыщенное кольцо, R1 представляет собой водород, алкил(1-3С) или ацил(2-4С), R3, R4, R5 и R6 независимо представляют собой алкил(1-3С) или (R3 и R5), или (R3 и R6), или (R4 и R5), или (R4 и R6) вместе могут образовывать алкиленовый мостик, содержащий 1-3 атома углерода, при условии, что, когда о равно 2, R3 означает водород, и когда р равно 2, R5 означает водород, R7 представляет собой Н, галоген,
CF3, алкил(1-3С), амино, аминоалкил(1-3С), алкил(1-3С)амино, диалкил(1-3С)амино, гидрокси, (1-3С)алкокси или OCF3, и R2, m, n, o и p имеют значения, которые приведены выше.
Точнее, изобретение относится к соединениям формулы (1), в которой А означает насыщенное кольцо, m = 0, n = 0 или 1, o = 1, p = 1, R1 = H или ацетил, R2 представляет собой галоген, CF3, алкил(1-3С), амино, циано, OCH3 или OCF3, R3, R4, R5 и R6 независимо представляют собой водород или алкил(1-2С), или (R4 и R6) вместе могут образовывать алкиленовый мостик, содержащий 1-2 атома углерода, и R7 представляет собой Н, галоген, CF3, алкил(1-3С), амино, гидрокси или OCF3.
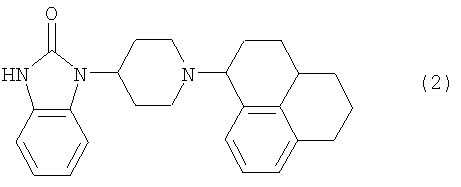
Более предпочтительным является соединение формулы (2) и его стериоизомеры. На нижеупомянутое соединение будет «пример 1».
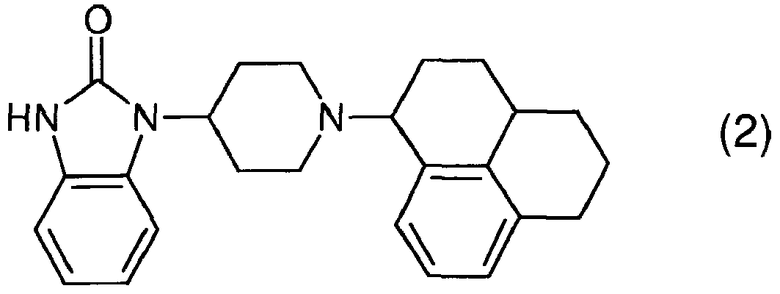
Соединения изобретения можно превращать в формы, подходящие для введения с помощью обычных способов, используя вспомогательные вещества и/или жидкие или твердые носители.
Фармацевтически подходящие соли можно получать, используя стандартные процедуры, хорошо известные в данной области, например, смешиванием соединения данного изобретения с подходящей солью. Подходящие аддитивные соли кислоты можно образовывать с неорганическими кислотами, такими как хлористоводородная кислота, или органическими кислотами, такими как фумаровая кислота.
Соединения изобретения общей формулы (1), а также их соли, имеют активность агониста ORL1. Их используют при лечении нарушений, в которые вовлечены рецепторы ORL1, или нарушений, которые лечат обработкой названных рецепторов. Например, используют при состояниях острой или хронической боли, нарушениях центральной нервной системы, особенно, но не ограничиваясь, для уменьшения симптомов состояния страха и стрессовых нарушений, депрессии, различных форм эпилепсии, «удара», нарушений, характеризующихся ухудшением познавательной способности и памяти, таких как болезнь Альцгеймера, болезнь Крейтцфельда-Якоба, болезнь Гентингтона, болезнь Паркинсона, нейрореабилитация (посттравматические повреждения мозга); острого повреждения мозга или спинного мозга, нарушений, связанных с веществами, включая нарушения, связанные с применением веществ (подобные зависимости и злоупотреблению), и нарушения, индуцированные веществом (подобные синдрому отмены вещества); нарушений приема пищи, подобных нервно-психической анорексии и нервно-психической булимии, ожирения; желудочно-кишечных нарушений, в частности синдрома воспаленной кишки, воспалительного заболевания кишечника (болезнь Крона) и язвенного колита, воспаления мочевых путей, почечных нарушений, характеризующихся дисбалансом удерживание/выделение воды или солевой экскреции; сердечно-сосудистых нарушений, таких как инфаркт миокарда, аритмия, артериальная гипертензия, тромбоз, анемия, атеросклероз, «грудная жаба»; кожных заболеваний, таких как крапивница, красная волчанка и зуд; глазных заболеваний, таких как глаукома; заболеваний органов дыхания, включающих в себя обструктивную болезнь легких, бронхит и кистозный фиброз; заболеваний иммунной системы и вирусных инфекций.
Соединения данного изобретения обычно вводят в виде фармацевтических композиций, которые представляют собой важные и новые воплощения изобретения вследствие присутствия в них соединений, точнее конкретных соединений, представляемых в описании. Типы фармацевтических композиций, которые можно использовать без ограничения, включают таблетки, жевательные таблетки, капсулы, растворы, растворы для парентерального введения, суппозитории, суспензии и другие типы, рассматриваемые в описании или очевидные специалисту в данной области из спецификации или общих сведений в данной области.
В воплощениях изобретения представляют фармацевтическую упаковку или набор, содержащий один или более контейнеров, заполненных одним или более ингредиентами фармацевтической композиции изобретения. К такому контейнеру(ам) могут быть присоединены описания, такие как инструкции для применения или уведомление в форме, предписанной Государственным агентством по регулированию производства, применению и безопасности фармацевтических продуктов, причем уведомление отражает одобрение агентства по производству, применению или безопасности при применении человеку и в ветеринарии.
ОБЩИЕ АСПЕКТЫ СИНТЕЗА
Соединения изобретения и их соли можно получать в соответствии с общим способом, представленным в общих чертах ниже в схеме 1:
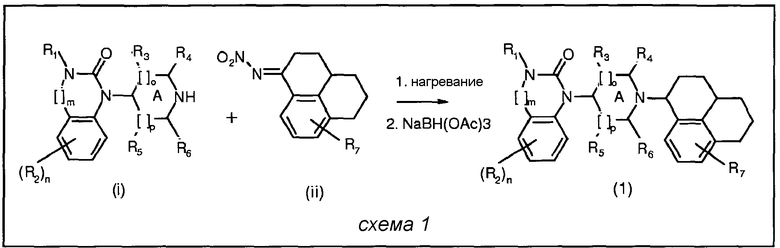
Исходные соединения для представленного общего способа получали следующим образом:
Бензимидазолоны [(i), когда m = 0] можно синтезировать по способам, описанным в J. Med. Chem., 30., 814-819, 1987 и WO 99/36421. Хиназолиноны [(i), когда m = 1] можно синтезировать в соответствии с Chem. Pharm. Bull., 33, 1116-1128, 1985. N-нитрооксимы (замещенного) 2,3,3а,4,5,6-гексагидро-1Н-фенален-1-она (ii) получали из соответствующих кетонов. Упомянутые кетоны в свою очередь получали из соответствующих (замещенных) тетралонов, как описано в Eur. J. Med. Chem. 35, 839-851, 2000.
СПЕЦИАЛЬНЫЕ ПРИМЕРЫ СИНТЕЗА
Синтез соединения примера 1
Подробное описание синтеза соединения примера 1 представлено на схеме 2
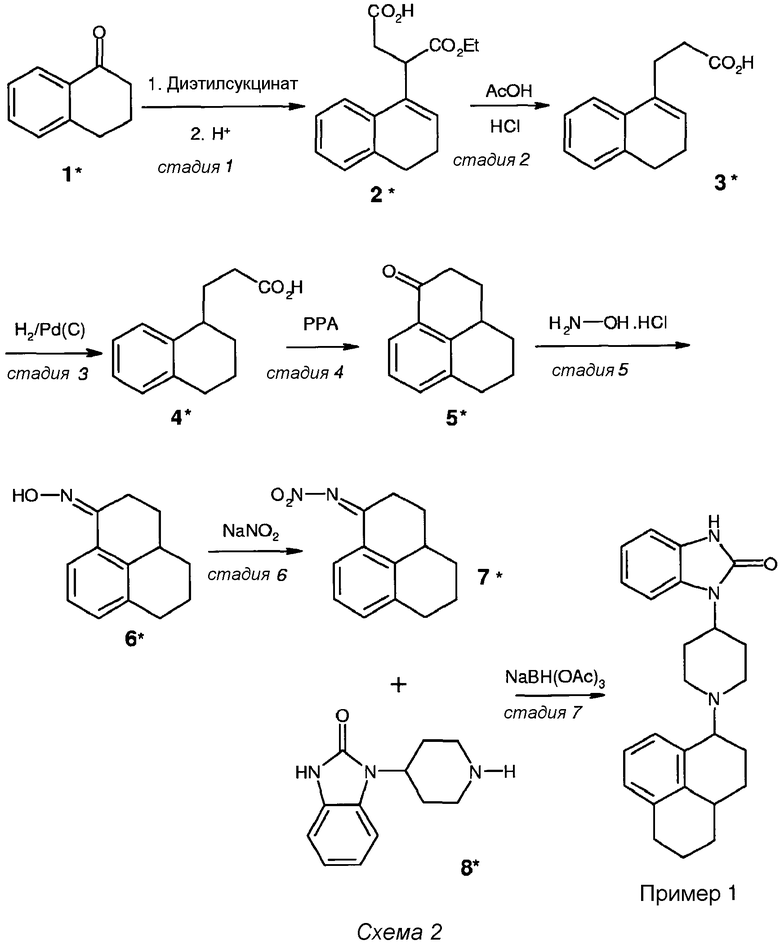
Первые четыре стадии схемы 2 проводили в соответствии с Eur. J. Med. Chem. 35, 839-851, 2000. Исходя из промежуточного соединения 5* (см. схему 2), синтез проводили следующим образом:
Стадия 5 (схема 2): Смесь 52 г (0,28 моль) 2,3,3а,4,5,6-гексагидро-1Н-фенален-1-она (соединение 5*), 28,1 г (0,40 моль) гидроксиламина·HCl и 55 г (0,40 моль) ацетата натрия·3Н2О в 500 мл 96% этанола перемешивали при 80°С в течение 4 часов и течение дополнительных 16 часов при комнатной температуре. Полученную смесь концентрировали в вакууме и добавляли 750 мл дихлорметана и 300 мл 5% водного раствора NaHCO3. Водный слой дважды промывали 100 мл дихлорметана, объединенные органические слои промывали 100 мл насыщенного солевого раствора, высушивали над MgSO4 и концентрировали в вакууме.
Стадия 6 (схема 2): 58,8 г (представляющие количественный выход) полученного не совсем белого твердого оксима (соединение 6*) суспендировали в 600 мл простого трет-бутилметилового эфира. При комнатной температуре к полученной суспензии добавляли 230 мл раствора 41,4 г (0,6 моль) нитрита натрия в воде с последующим добавлением 290 мл раствора 2 н. серной кислоты. После перемешивания при 40°С в течение 16 часов смесь охлаждали до комнатной температуры и добавляли 300 мл насыщенного водного раствора NaHCO3. Водный слой экстрагировали дважды 300 мл простого трет-бутилметилового эфира, и объединенные органические слои промывали 150 мл насыщенного солевого раствора, высушивали над MgSO4 и концентрировали в вакууме. Полученное коричневое масло очищали методом колоночной хроматографии (силикагель) с дихлорметаном в качестве элюента. Маслянистый продукт, полученный после концентрирования в вакууме, растирали с циклогексаном, и полученный осадок собирали фильтрованием и высушивали. Получали чистый NO2-имин (соединение 7*) (34 г, 0,148 моль, с выходом 52%) в виде не совсем белого твердого вещества с точкой плавления 64-69°С.
Стадия 7 (схема 2): смесь 6,51 г (30 ммоль) 4-(1-бензимидазолон)пиперидина (соединение 8*, ACROS), 6,9 г (30 ммоль) NO2-имина (соединение 7*) и 5,25 мл диизопропилэтиламина в 450 мл 1,2-дихлорэтана нагревали при 50°С и перемешивали в атмосфере N2 в течение 16 часов. После охлаждения до комнатной температуры добавляли 12,7 г (60 ммоль) NaBH(OAc)3 и полученную смесь перемешивали при комнатной температуре в атмосфере N2 в течение 24 часов. После концентрирования реакционной смеси в вакууме добавляли 500 мл дихлорметана и 500 мл водного 5% раствора NaHCO3 при перемешивании. Водный слой дважды промывали 100 мл дихлорметана, объединенные органические слои промывали 100 мл насыщенного солевого раствора, высушивали над MgSO4 и концентрировали в вакууме. Неочищенный продукт очищали методом колоночной хроматографии (силикагель) со смесью дихлорметан:метанол:аммиак (92:7,5:0,5) в качестве элюента. Чистый продукт получали концентрированием в вакууме (8,09 г, 21 ммоль, с 70% выходом, точка плавления 155-158°С). К 8,09 г (21 ммоль) чистого продукта добавляли 60 мл этанола и 2,44 г (21 ммоль) фумаровой кислоты. Концентрирование в вакууме полученного раствора давало фумарат примера 1 (10,53 г, 21 ммоль, количественный выход) в виде белой пены с М+ 358 m/z и точкой плавления 232-234°С.
Синтез соединения примера 2
Смесь 2,31 г (10 ммоль) 4-(1-хиназолинон)пиперидина и 2,3 г (10 ммоль) NO2-имина (соединение 7*) в 100 мл 1,2-дихлорэтана в атмосфере азота нагревали и перемешивали при 50°С в течение 7 часов и в течение дополнительных 16 часов при комнатной температуре. Впоследствии добавляли 4,2 г (20 ммоль) NaBH(OAc)3 и полученную смесь перемешивали при комнатной температуре в атмосфере N2 в течение 24 часов. После концентрирования реакционной смеси в вакууме 200 мл дихлорметана и 200 мл 5% водного раствора NaHCO3 добавляли при перемешивании. Водный слой дважды промывали 40 мл дихлорметана, объединенные органические слои промывали 40 мл насыщенного солевого раствора, высушивали над MgSO4 и концентрировали в вакууме. Неочищенный продукт очищали методом колоночной хроматографии (силикагель) со смесью дихлорметан:метанол:аммиак (94,5:5:0,5) в качестве элюента. Чистый продукт, полученный после концентрирования в вакууме (2,5 г, 6,2 ммоль), растворяли в 50 мл раствора HCl в этаноле. Концентрирование в вакууме давало хлорид соединения примера 2 (2,05 г, 4,7 ммоль, с выходом 47%) в виде белого аморфного твердого вещества с М+ 402 m/z и точкой плавления 172-180°С.
Синтез соединения примера 13
Подробное описание синтеза соединения примера 13 представлено на схеме 3:
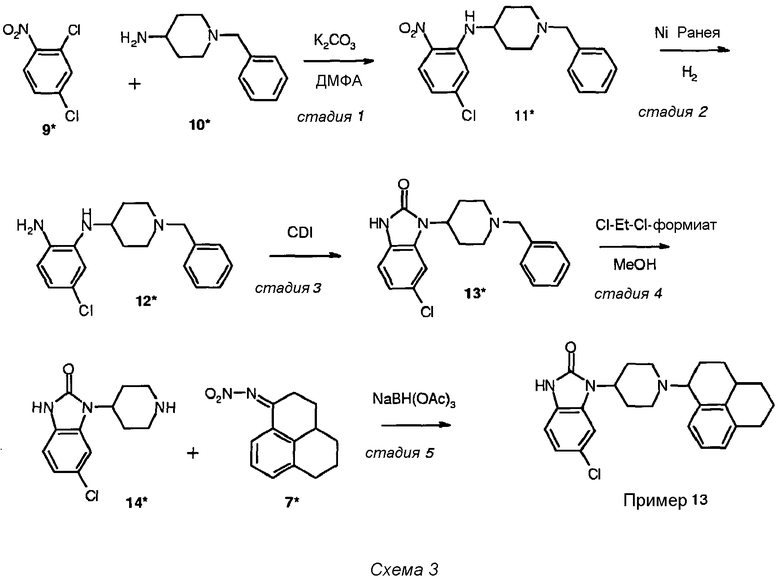
Стадия 1 (схема 3): Раствор 3,84 г (20 ммоль) 2,4-дихлорнитробензола (соединение 9*, Aldrich), 4,1 мл (20 ммоль) 4-амино-1-бензилпиперидина (соединение 10*, Aldrich), 4,46 г (32 ммоль) K2CO3 в 50 мл диметилформамида перемешивали при 95°С в атмосфере N2 в течение 18 часов. После охлаждения до комнатной температуры реакционную смесь вливали в смесь вода (150 мл)-дихлорметан (250 мл). Водный слой дважды экстрагировали 50 мл дихлорметана, и объединенные органические слои дважды промывали 50 мл воды, высушивали над MgSO4 и концентрировали в вакууме. Полученный неочищенный продукт очищали методом колоночной хроматографии (силикагель) со смесью дихлорметан:метанол (97:3) в качестве элюента. После концентрирования в вакууме получали чистый продукт в виде желтого маслянистого вещества (4,8 г, 13,8 ммоль, с выходом 69%).
Стадия 2 (схема 3): Порцию Ni Ранея (Aldrich R 2800 [7440-02-0], ~500 мг) дважды промывали 10 мл 96% этанола и впоследствии в атмосфере N2 добавляли к раствору 4,8 г (13,8 ммоль) соединения 11* в 200 мл 96% этанола. Раствор гидратировали при комнатной температуре и давлении 1 атмосфера в течение 2,5 часов. Смесь впоследствии фильтровали через Hyflo, промывали 300 мл 96% этанола и фильтрат концентрировали в вакууме, чтобы получить количественный выход соединения 12* в виде окрашенного маслянистого вещества (4,36 г, 13,8 ммоль, с 100% выходом).
Стадия 3 (схема 3): К раствору 4,36 г (13,8 ммоль) соединения 12*, продукта с предыдущей стадии, в 200 мл ацетонитрила, перемешиваемого при комнатной температуре в атмосфере азота, добавляли 3,36 г (20,7 ммоль) 1,1'-карбонилдиимидазола (CDI, ACROS). Осадок, который начал образовываться через 10 минут и образование которого возрастало вплоть до 3 часов, собирали фильтрованием, промывали ацетонитрилом (200 мл) и высушивали в вакууме, получая почти чистое соединение 13* (3,30 г, 9,7 ммоль, с 70% выходом).
Стадия 4 (схема 3): К суспензии 3,30 г (9,7 ммоль) соединения 13* в 90 мл 1,2-дихлорэтана, перемешанного в атмосфере N2 и охлажденного до 0°С, по каплям добавляли порцию 1-хлорэтил-хлорформиата (1,17 мл, 10,7 ммоль). После перемешивания при 0°С в течение 30 минут и при 80°С в течение 90 минут смесь охлаждали снова до 0°С и по каплям добавляли вторую порцию 1-хлорэтил-хлорформиата (1,17 мл, 10,7 ммоль). Смесь перемешивали еще раз при 0°С в течение 30 минут и при 80°С в течение 16 часов. После охлаждения до комнатной температуры смесь концентрировали в вакууме и к остатку добавляли 75 мл метанола. Полученный раствор перемешивали при 65°С в течение 1 часа и концентрировали в вакууме. После добавления 75 мл дихлорметана к полученному коричневому полутвердому веществу оно затвердевало при перемешивании в течение 1 часа. Осадок собирали фильтрованием, промывали 100 мл дихлорметана и высушивали. Полученный неочищенный продукт очищали методом колоночной хроматографии (силикагель) со смесью дихлорметан-метанол-аммиак (92:7,5:0,5) в качестве элюента. После концентрирования в вакууме, получали соединение 14* в виде белого твердого вещества (1,61 г, 6,4 ммоль, с выходом 66%).
Стадия 5 (схема 3): Смесь 1,61 г (6,4 ммоль) соединения 14* и 1,47 г (6,4 ммоль) NO2-имина (соединение 7*) в 200 мл 1,2-дихлорэтана в атмосфере N2 нагревали и перемешивали при 50°С в течение 16 часов. После охлаждения до комнатной температуры добавляли 2,76 г (13 ммоль) NaBH(OAc)3 и полученную смесь перемешивали при комнатной температуре в атмосфере N2 в течение 24 часов. Слегка окрашенный раствор вливали в смесь 300 мл дихлорметана, 100 мл воды и 50 мл водного 5% раствора NaHCO3. Водный слой дважды промывали 70 мл дихлорметана, объединенные органические слои высушивали над MgSO4 и концентрировали в вакууме. Неочищенный продукт очищали методом колоночной хроматографии (силикагель) со смесью дихлорметан:метанол:аммиак (92:7,5:0,5) в качестве элюента. Очищенный продукт концентрировали в вакууме и он загустевал при последующем одновременном выпаривании с ацетонитрилом. После перемешивания в 100 мл простого диизопропилового эфира осадок собирали фильтрованием и высушивали, получая соединение примера 13 (1,35 г, 3,2 ммоль, с выходом 50%) в виде слегка окрашенного чистого твердого вещества с М+ 422 m/z и точкой плавления 185-188°С.
С помощью описанных и сравнимых способов синтезировали следующие специфические примеры. Они предназначены для дальнейшей иллюстрации изобретения более подробно и поэтому не предназначены для ограничения сферы действия изобретения каким-либо образом. Информация по структуре данных соединений, все из которых изображают общей формулой (1), представлена в таблице ниже.
ФАРМАКОЛОГИЧЕСКИЕ ИССЛЕДОВАНИЯ
In vitro и in vivo исследовали свойства соединений изобретения как агонистов рецептора ORL1, а также их µ-опиатную активность (отсутствие ее), используя способы, описанные ниже.
Сродство в отношении рецепторов ORL1 человека
Сродство соединений к рецепторам ORL1 человека определяли, используя исследование связывания рецептора in vitro, описанное Ardati et al., Mol. Pharmacol., 51, 816, 1997. Кратко, препараты мембран получали из клеток CHO, в которых рецептор ORL1 человека стабильно экспрессировался. Мембраны инкубировали с [3H]-ноцицептином в отсутствие или в присутствии тестируемого соединения при различных концентрациях, разбавленного подходящим буфером. Никакого специфического связывания не наблюдали, когда связывание происходило в присутствии 10-6 М ноцицептина. Отделение связанной радиоактивности от свободной производили фильтрованием через фильтры из стеклянных волокон Packard GF/B с несколькими промывками охлажденным льдом буфером, используя клеточный харвестер Packard. Связанную радиоактивность определяли на сцинтилляционном счетчике (Topcount, Packard), используя сцинтилляционный коктейль (Microscint O, Packard). Определяемую радиоактивность вычерчивали в виде кривой против концентрации замещающего тестируемого соединения, и кривые замещения рассчитывали по методу логистической регрессии по четырем параметрам, дающему в результате величины IC50, то есть такой концентрации тестируемого соединения, при которой 50% радиолиганда замещается. Величину сродства pKi рассчитывали посредством коррекции значений IC50 для концентрации радиолиганда и его сродства относительно рецептора ORL1 человека по уравнению Ченга-Прусоффа:
PKi = -log(IC50/(1 + S/Kd))
в котором IC50 является таким, как описывают выше, S означает концентрацию [3H]-ноцицептина, используемую в исследовании, выраженную в моль/л (обычно 0,2 нМ), и Kd является константой диссоциации равновесия [3H]-ноцицептина для рецепторов ORL1 человека (0,4 нМ).
Соединения изобретения имеют высокое сродство к рецепторам ORL1 по данным описанного выше исследования связывания. Это свойство делает соединения полезными для лечения нарушений, в которые вовлечены рецепторы ORL1 или которые можно лечить коррекцией названных рецепторов.
Сродство к µ-опиатным рецепторам человека
Сродство соединений к µ-опиатным рецепторам определяли, используя исследование связывания рецептора in vitro, описанного Childers et al, Eur. J. Pharm. 55, 11, 1979. Кратко, препараты мембран получали из клеток CHO, в которых стабильно экспрессировался µ-опиатный рецептор человека и мембраны инкубировали с [3H]-налоксоном в отсутствие или в присутствии тестируемых соединений в диапазоне концентраций от 10 мкМ со снижением до 0,1 нМ, разбавленных подходящим буфером. Никакого специфического связывания не наблюдали, когда связывание происходило в присутствии 10-7 М тартрата леваллорфана. Отделение связанной радиоактивности от свободной производили, как описано выше, и сродство соединений рассчитывали подобным образом, используя концентрацию (S) 1 нМ [3H]-налоксона, и при значении Kd 1,3 нМ.
Большинство соединений изобретения имели низкое сродство к µ-опиатным рецепторам при описанном выше исследовании связывания. Таким образом, маловероятно, что они будут вызывать нежелательные побочные эффекты, которые, как известно, имеют место с опиатами, подобными морфину.
In vitro ORL1-рецепторный агонизм
Активация рецептора ORL1, связанного с белком G, ингибирует активность аденилатциклазы и снижает внутриклеточную концентрацию вторичного мессенджера цАМФ. Используя способ, который описан Jenck et al., Proc. Natl. Acad. Sci. USA, 97, 4938-4043, 2000, определяли активность соединений на рецепторах ORL1. Продемонстрировано, что соединения являются сильными агонистами.
In vivo ORL1-рецепторный агонизм
При внутрибрюшинном и/или пероральном введении соединений изобретения было показано, что они высокоактивны при кондиционированной ультразвуковой дистресс-вокализации (CUDV), процедуре, описанной Van der Poel et al., Psychopharmacology, 97, 147-148, 1989. Это демонстрирует не только, что соединения имеют хорошую биодоступность после перорального введения, но что они также проходят через гематоэнцефалический барьер. Пептидный ноцицептин также был активен в упомянутом исследовании, но для того чтобы продемонстрировать его действие, оказалось необходимым, вводить ноцицептин непосредственно в мозг (с помощью внутримозговой-желудочковой инъекции).
Агонист ORL1 индуцирует снижение кровяного давления
С интервалами в 5 минут крысам, анестезированным пентобарбиталом натрия 80 мг/кг, внутрибрюшинно вводили возрастающие внутривенные дозы агониста ORL1, приводя к снижению кровяного давления. Описанное снижение выражали как соотношение ED80 (средняя эффективная доза):доза, приводящая к 20% снижению кровяного давления по сравнению с контролем.
В экспериментах по взаимодействию однократную внутривенную дозу 2 мг/кг опиатного антагониста налоксона или 1 мг/кг избирательного антагониста ORL1 J-113397 вводили за 10 минут до первой дозы агониста. Указанная доза J-113397 оказалась способной полностью противодействовать действию ноцицептина. Эта доза налоксона, как известно, предотвращает индуцированное морфином снижение кровяного давления, но не оказывает влияние на индуцированное ноцицептином снижение кровяного давления.
Действие, индуцированное агонистом ORL1 и морфином, на потребление пищи
Последние исследования показали, что потребление пищи можно фармакологически регулировать лигандами опиатных рецепторов. Sanger и McCarthy (Increased food and water intake produced in rats by opiate receptor agonist. Psychopharmacology, 74(3): 217-220, 1981) показали, что системное введение морфина приводит к увеличению потребления пищи, действию, которое является противоположным действию неизбирательного опиатного антагониста налоксона. Кроме того, Ciccocioppo et al. (Reversal of stress- and CRF-induced anorexia in rats by the synthetic nociceptin/orphanin FQ receptor agonist, Ro 64-6198. Psychopharmacology, 161(2): 113-119, 2002) сообщил, что системное введение агониста ORL1 Ro 64-6198 также повышает потребление пищи крысами.
Самцов крыс Wistar размещали поодиночке и предоставляли свободный доступ к пище и воде. Однократную дозу наполнителя Ro 64-6198 (1, 3, 6, 10 мг/кг), соединения примера 1 (0,3, 1, 3, 6, 10 мг/кг) или морфина (1, 3, 10 мг/кг) вводили внутрибрюшинно, и пищу убирали за пределы досягаемости животными. Через 15 минут после введения взвешенное количество пищи (5-6 шариков = 25-30 грамм) повторно вносили в клетку животного. Затем пищу вновь взвешивали через 60 и 120 минут. Всех животных повторно использовали во время 4 отдельных экспериментов с минимальными интервалами времени 5 дней между экспериментами. Предпринимали предосторожности, чтобы гарантировать, чтобы никакой посторонний шум не индуцировал любой дополнительный стресс для животного. В тех экспериментах, в которых опиатный антагонист налоксон (1, 3, 10 мг/кг) или антагонист ORL1 J113397 (3, 10, 30 мг/кг) вводили до введения агониста (Ro 64-6198, 6 мг/кг; пример 1, 10 мг/кг или морфин, 2 мг/кг), антагонист вводили внутрибрюшинно за 30 минут до введения агониста. Во всех экспериментальных группах присутствовало минимум шесть животных на группу.
ПРЕПАРАТЫ СОЕДИНЕНИЙ, КОТОРЫЕ ИСПОЛЬЗОВАЛИ В ИССЛЕДОВАНИЯХ НА ЖИВОТНЫХ
Приготовление соединения примера 1
Для перорального (р.о.) введения: к требуемому количеству (0,5-15 мг) твердого соединения примера 1 в стеклянной пробирке добавляли несколько стеклянных гранул и твердое вещество измельчали при интенсивном перемешивании в течение 2 минут. После добавления 1 мл раствора 1% метилцеллюлозы в воде соединение суспендировали при интенсивном перемешивания в течение 10 минут. Что касается концентраций до и выше 1 мг/мл, оставшиеся частицы в суспензии еще суспендировали, используя ультразвуковую камеру.
Для внутрибрюшинного (i.p.) введения: к требуемому количеству (0,5-15 мг) твердого соединения примера 1 в стеклянной пробирке добавляли несколько стеклянных гранул, и твердое вещество измельчали при интенсивном перемешивании в течение 2 минут. После добавления 1 мл раствора 1% метилцеллюлозы и 5% маннита в воде соединение суспендировали при интенсивном перемешивании в течение 10 минут. Наконец, рН приводили к 7.
Приготовление соединения примера 2
Для перорального (р.о.) введения: к требуемому количеству (0,5-15 мг) твердого соединения примера 2 в стеклянной пробирке добавляли несколько стеклянных гранул, и твердое вещество измельчали при интенсивном перемешивании в течение 2 минут. После добавления 1 мл раствора 1% метилцеллюлозы в воде соединение суспендировали при интенсивном перемешивании в течение 10 минут. РН приводили к 7 добавлением нескольких капель водной NaOH (0,1 н.). Оставшиеся в суспензии частицы еще суспендировали, используя ультразвуковую камеру.
Для внутрибрюшинного (i.p.) введения: препараты получали по способу, аналогичному способу, использованному для р.о. введения с использованием 1% метилцеллюлозы и 5% маннитом вместо 1% метилцеллюлозы в воде.
ФАРМАКОЛОГИЧЕСКИЕ ДАННЫЕ
функциональный ORL1-агонизм
*CUDV = кондиционированная ультразвуковая дистресс-вокализация; i.p. = внутрибрюшинно; р.о. = per os (перорально).
Из данных, представленных в таблице выше, очевидно, что соединение примера 1 является в десять раз более мощным, чем Ro 64-6198, при введении внутрибрюшинным способом и в три раза более мощным при пероральном введении.
Вышеприведенные данные показывают, что снижающему кровяное давление действию соединения примера 1 (диапазон 10-3000 мкг/кг) противодействовал J-113397 (смещение в зависимости доза-ответ от 356 до 1138 мкг/кг) и действие налоксона он не предотвращал (никакого смещения в зависимости доза-ответ 356 и 334 мкг/кг). Действие Ro 64-6198 (диапазон 10-1000 мкг/кг) предотвращалось J-113397 (смещение в зависимости доза-ответ от 84 до 264 мкг/кг) и действие двух наивысших доз налоксона оказалось также чувствительным (смещение в зависимости доза-ответ от 84 до 141 мкг/кг). Очевидно, Ro 64-6198 имеет µ-опиатный компонент.
Морфин (1,25, 2,5, 5 и 10 мг/кг), Ro 64-6198 (1, 3 и 10 мг/кг, внутрибрюшинно) и соединения примера 1 (0,3, 1, 3, 6, 10 мг/кг, внутрибрюшинно), все приводили к зависимому от дозы увеличению потребления пищи, которое во всех случаях было значительным по сравнению с группой, обработанной наполнителем. Системное введение опиатного антагониста налоксона (3 или 30 мг/кг, внутрибрюшинно) одного и антагониста ORL1 J-113397 (30 мг/кг, внутрибрюшинно) одного не оказывало влияние на потребление пищи.
Ro 64-6198 или соединением примера 1
Увеличение потребления пищи после введения морфина предотвращали в присутствии антагониста опиатного рецептора налоксона. Увеличение потребления пищи, ассоциированное с соединением примера 1, полностью предотвращали антагонистом ORL1 J-113397, но не антагонистом опиатного рецептора налоксоном. J-113397 не предотвращал (статистически достоверно) увеличение потребления пищи, связанное с Ro 64-6198, тогда как предварительная обработка налоксоном приводила к значительному изменению индукции потребления пищи посредством Ro 64-6198.
Представленные данные позволяют предположить, что увеличение в потреблении пищи, связанное с соединением примера 1, опосредовано агонизмом к рецепторам ORL1 и неопиатным рецепторам. Увеличение потребления пищи, ассоциированное с Ro 64-6198, частично было предотвращено налоксоном, но не J-113397, указывая, что в Ro 64-6198 имеется опиатный компонент, который не присутствует в соединении примера 1. В заключение, соединение примера 1 действует как более избирательный агонист ORL1 по сравнению с Ro 64-6198.
| название | год | авторы | номер документа |
|---|---|---|---|
| БИЦИКЛО-3.1.1-ГЕПТАН-ЗАМЕЩЕННЫЕ БЕНЗИМИДАЗОЛОН- И ХИНАЗОЛИНОН-ПРОИЗВОДНЫЕ АГОНИСТЫ ORL1 РЕЦЕПТОРОВ ЧЕЛОВЕКА | 2004 |
|
RU2357964C2 |
| СПИРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ЦИКЛОГЕКСАНА СО СРОДСТВОМ К ORL1-РЕЦЕПТОРУ | 2004 |
|
RU2383544C2 |
| СПИРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ЦИКЛОГЕКСАНА | 2008 |
|
RU2470933C2 |
| ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ | 2018 |
|
RU2777366C2 |
| ГИДРОНОПОЛЬНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ АГОНИСТОВ ПО ОТНОШЕНИЮ К ORL1 РЕЦЕПТОРАМ ЧЕЛОВЕКА | 2004 |
|
RU2351600C2 |
| ПРОИЗВОДНЫЕ ЦИС-ТЕТРАГИДРО-СПИРО(ЦИКЛОГЕСАН-1,1'-ПИРИДО[3,4-в]ИНДОЛ)-4-АМИНА, ПОЛЕЗНЫЕ ПРИ ЛЕЧЕНИИ НЕВРОПАТИЧЕСКОЙ И/ИЛИ ХРОНИЧЕСКОЙ БОЛИ | 2011 |
|
RU2592283C2 |
| ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ | 2018 |
|
RU2780002C2 |
| ПРОИЗВОДНЫЕ СПИРО(5.5)УНДЕКАНА | 2009 |
|
RU2515895C2 |
| ГИДРОКСИМЕТИЛЦИКЛОГЕКСИЛАМИНЫ | 2009 |
|
RU2514192C2 |
| ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ 4-АМИНОЦИКЛОГЕКСАНА | 2009 |
|
RU2525236C2 |
Изобретение относится к новым соединениям общей формулы (1), их оптически активным стереоизомерам, а также фармакологически пригодным солям, обладающим свойствами ORL1 и µ-опиатных рецепторов. Соединения могут быть использованы для приготовления лекарственного средства для лечения нарушений и заболеваний, таких как нарушения пищевого поведения, артериальная гипертензия. В общей формуле (1)
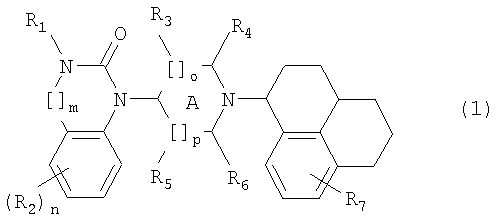
R1 представляет собой Н, алкил(1-6С), []m означает -(СН2)m-, в котором m равно 0 или 1, R2 представляет собой галоген, СF3, алкил(1-6С), фенил, циано, цианоалкил(1-3С), гидрокси, (1-3С)алкокси, ОСF3, ацил(2-7С), трифторацетил, (1-3С)алкилсульфонил или трифторметилсульфонил, и n означает целое число 0-4 при условии, что, когда n равно 2, 3 или 4, заместители R2 могут быть одинаковыми или разными, А означает насыщенное кольцо,
[]о и []р представляют собой -(СН2)о- и -(СH2)р, и о и р независимо соответствуют 0, 1 или 2, R3, R4, R5 и R6 независимо представляют собой водород, алкил(1-3С), или (R4 и R6) вместе могут образовывать алкиленовый мостик, содержащий 1-3 атома углерода, при условии, что, когда о равно 2, R3 означает водород, и когда р равно 2, R5 означает водород, R7 представляет собой Н, галоген, алкил(1-6С). Изобретение также относится к фармацевтической композиции, промежуточным соединениям (3) для получения соединений формулы (1). 5 н. и 3 з.п. ф-лы, 3 схемы, 1 табл.
1. Соединения общей формулы (1)
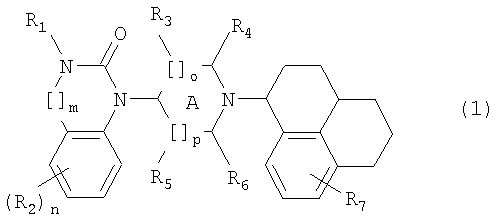
в которой R1 представляет собой Н, алкил(1-6С),
[]m означает -(СН2)m-, в котором m равно 0 или 1,
R2 представляет собой галоген, СF3, алкил(1-6С), фенил, циано, цианоалкил(1-3С), гидрокси, (1-3С)алкокси, ОСF3, ацил(2-7С), трифторацетил, (1-3С)алкилсульфонил или трифторметилсульфонил, и n означает целое число 0-4, при условии, что, когда n равно 2, 3 или 4, заместители R2 могут быть одинаковыми или разными,
А означает насыщенное кольцо,
[]о и []р представляют собой -(СH2)о- и -(СH2)р, и о и р независимо соответствуют 0, 1 или 2,
R3, R4, R5 и R6 независимо представляют собой водород, алкил(1-3С), или (R4 и R6) вместе могут образовывать алкиленовый мостик, содержащий 1-3 атома углерода, при условии, что, когда о равно 2, R3 означает водород, и когда р равно 2, R5 означает водород,
R7 представляет собой Н, галоген, алкил(1-6С),
или их оптически активные стереоизомеры, а также фармакологически пригодные соли.
2. Соединения по п.1 общей формулы (1), в которой А означает насыщенное кольцо, R1 представляет собой водород, алкил(1-3С),
R3, R4, R5 и R6 независимо представляют собой водород или алкил(1-3С), или (R4 и R6) вместе могут образовывать алкиленовый мостик, содержащий 1-3 атома углерода, при условии, что, когда о равно 2, R3 означает водород, и когда р равно 2, R5 означает водород, R7 представляет собой Н, галоген, алкил(1-3С), и R2, m, n, о и р имеют значения, которые приведены выше.
3. Соединения по п.1 общей формулы (1), в которой А означает насыщенное кольцо, m=0, n=0 или 1, о=1, р=1, R1=Н, R2 представляет собой галоген, СF3, алкил(1-3С), циано, ОСН3 или ОСF3, R3, R4, R5 и R6 независимо представляют собой водород или алкил(1-2С), или (R4 и R6) вместе могут образовывать алкиленовый мостик, содержащий 1-2 атома углерода, и R7 представляет собой Н, галоген, алкил(1-3С).
4. Соединение по п.1 формулы (2) и его стереоизомеры:
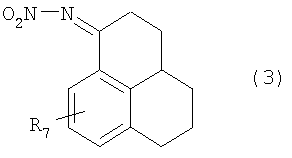
5. Соединения общей формулы (3):
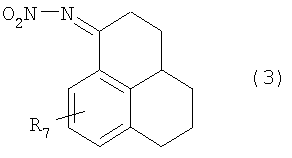
в которой R7 имеет значение, которое приведено в п.1.
6. Фармацевтические композиции, обладающие свойствами ORL1 и µ-опиатных рецепторов, содержащие фармакологически активное количество, по крайней мере, одного из соединений по любому из пп.1-4 в качестве активного ингредиента.
7. Применение соединения по любому из пп.1-4 для приготовления фармацевтической композиции для лечения нарушений, опосредованных действием рецепторов ORL1.
8. Применение по п.7, где названные состояния и нарушения выбраны из нарушений пищевого поведения, артериальной гипертензии.
| ЗАМЕЩЕННЫЕ АМИНОСОЕДИНЕНИЯ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1998 |
|
RU2197474C2 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| ZAVERI N | |||
| et al | |||
| "Characterizattion of Opiates, Neuroleptics, and Synthetic Fnalogs at ORL1 and Opioid Receptors", EUROPEN JOURNAL OF PHARMACOLODGY, 2001, 428, p.29-36. | |||

