Настоящее изобретение относится к группе гидронопольных производных, которые являются агонистами (веществами, обладающими сродством) к ORL1 (ноцицептиновым) рецепторам человека. Изобретение также относится к получению этих соединений, к фармацевтическим композициям, содержащим фармакологически активное количество, по меньшей мере, одного из этих новых производных гидронопола в качестве активного ингредиента, а также к применению этих фармацевтических композиций для лечения расстройств, связанных с ORL1 рецепторами.
′Opioid Receptor-Like 1′ (ORL1) рецептор идентифицировали из библиотеки кДНК человека. Было установлено, что этот 'сиротский рецептор' имеет близкую гомологию с µ-, κ- и δ-опиоидными рецепторами (Mollereau et al., FEBS Lett., 341, 33-38, 1994; Bunzow et al., FEBS Lett., 347, 284-288, 1994). Несмотря на близкое сходство его последовательности и структурное сходство с опиоидными рецепторами классические лиганды опиоидного рецептора не взаимодействуют с ORL1 рецепторами. В 1995 г. 17-аминокислотный нейропептид выделили из экстрактов (вытяжки) мозга и впоследствии показали, что он является природным лигандом G-протеин-связанного ORL1 рецептора (Reinscheid et al., Science, 270, 792-794, 1995; Meunier et al., Nature, 377, 532-535, 1995). Пептид назвали сиротой FQ или ноцицептином, и он не присоединяется к трем традиционным опиоидным рецепторам. Эти открытия дали начало фундаментальному исследованию функциональной роли ORL1 рецептора и новых лигандов для ORL1 рецептора. Это привело к нескольким сотням публикаций, включая несколько обзоров (смотри, например, Grond et al., Anaesthesist, 51, 996-1005, 2002), и к десяткам патентных заявок, описывающих как пептидные, так и непептидные лиганды, варьированные по силе и селективности (ORL-1 в сравнении с µ-опиатом). Поскольку рецепторы µ-опиата широко распределяются по всему телу, отсутствие селективности может привести к ряду нежелательных опиатоподобных побочных эффектов, например к седации, угнетению дыхания, запору, привыканию и зависимости (Drug News Perspect, 14, 335, 2001).
Производные 1,3,8-триазаспиро[4,5]декан-4-она описаны в патентной заявке JP-A-2000/169476, опубликованной 20 июня 2000 г.; в патентной заявке WO 01/07050 A1, опубликованной 1 февраля 2001 г., и в патентной заявке US 2003/0109539 A1, опубликованной 12 июня 2003 г. Однако ни в одной из заявок, приведенных выше, не указаны рецепторы µ-опиата. В патентной заявке EP 0 997 464 A1, опубликованной 3 мая 2000 г., указывают, что соединения 1,3,8-триазаспиро[4,5]деканона в качестве агонистов ORL1 рецептора обладают селективным сродством к ORL1 рецепторам, но конкретная информация о сродстве рецептора µ-опиата была ограничена утверждением того, что: «особенно предпочтительные соединения показали большее сродство к ORL-1 рецепторам, чем к мю-рецепторам (то есть, IC50 для ORL1-рецепторов/IC50 для мю-рецепторов были меньше, чем 1,0)». Более конкретной является патентная заявка US 2001/0041711, опубликованная 15 ноября 2001г. Эта патентная заявка описывает триазоспиро-соединения, имеющие сродство к рецептору ноцицептина. Соединения также были испытаны на рецепторах µ-, κ- и δ-опиата, но только за несколькими исключениями обнаружено, что они являются более сильнодействующими по отношению к рецепторам µ-опиата, чем к ORL1 рецепторам. Исключения были селективны по отношению к ORL1 рецепторам менее чем в два раза. Таким образом, ближайший известный уровень техники (прототип) не указывает, как создавать сильнодействующие (эффективные) ORL1 лиганды с точно выраженной селективностью по сравнению с рецепторами µ-опиата, то есть с селективностью, имеющей показатель, по меньшей мере, 10, не говоря уже о таких соединениях, которые также имеют хорошее бионакопление. Наконец, производные 1,3,8-триазаспиро[4,5]декан-4-она, замещенные гидроксиалкилом, полезные для лечения расстройств, опосредуемых (обусловленных) ORL1 рецептором, были опубликованы 18 марта 2004 г. в патентной заявке WO 2004/022558, зарегистрированной 5 сентября 2003г.
Неожиданно обнаружено, что в сериях гидронопольных производных группа соединений, как было показано, имеет очень высокое сродство к рецепторам ORL1 человека. Более того, эти соединения показывают хорошую селективность к рецепторам ORL1 относительно рецепторов µ-опиата и легко усвояемы после орального введения.
Изобретение относится к соединениям общей формулы (1)

в которой:
R1 представляет собой атом водорода, галоген, CF3, алкильную (1-6С) группу, циклоалкильную (3-6С) группу, фенильную группу, аминогруппу, алкил(1-3С)аминогруппу, диалкил(1-3С)аминогруппу, гидроксильную группу, гидроксиалкильную (1-3С) группу, (1-3С)алкоксигруппу, OCF3, карбоксильную группу, аминокарбонильную или (1-3С)алкилсульфонильную группу,
m является целым числом от 1 до 4, при условии, что, когда m равно 2, 3 или 4, заместители R1 могут быть одинаковыми или различными,
R2 представляет собой атом водорода, необязательно замещенную алкильную (1-6С) группу, циклоалкильную (3-6С) группу, -CH2OH, -CH2OCH3, карбоксильную группу, ацетильную группу, необязательно замещенную бензильную группу или группу Q следующей структуры (2):

в которой:
[]n изображает символически -(CH2)n-, где n является целым числом от 0 до 7,
R3 представляет собой атом водорода или алкильную (1-3С) группу,
R4 представляет собой атом водорода, необязательно замещенную алкильную (1-6С) группу, насыщенное, ненасыщенное или частично насыщенное моно-, ди- или трициклическое необязательно замещенное кольцо, или алкильную (1-3С) группу, замещенную насыщенным, ненасыщенным или частично насыщенным необязательно замещенным пяти- или шестичленным кольцом, которое необязательно содержит один или более гетероатомов, или
(R3+R4) вместе с атомом азота, к которому они присоединены, представляют собой насыщенное, ненасыщенное или частично насыщенное моно-, ди- или трициклическое необязательно замещенное кольцо,
и к их фармакологически приемлемым солям и пролекарствам.
В описании заместителей аббревиатура 'алкильная (1-3С) группа' означает 'метильная группа, этильная группа, n-пропильная группа или изопропильная группа'. 'Необязательно замещенная' означает, что группа может быть дополнительно замещена или не замещена одной или более группами, выбранными из алкильной группы, алкенильной группы, алкинильной группы, арильной группы, фтора, хлора, брома, гидроксильной группы, алкилоксигруппы, алкенилоксигруппы, арилоксигруппы, ацилоксигруппы, аминогруппы, алкиламиногруппы, диалкиламиногруппы, ариламиногруппы, тиогруппы, алкилтиогруппы, арилтиогруппы, цианогруппы, оксогруппы, нитрогруппы, ацильной группы, амидогруппы, алкиламидогруппы, диалкиламидогруппы, карбоксильной группы, или два необязательных заместителя вместе с атомами углерода, к которому они присоединены, могут образовывать 5- или 6-членное ароматическое или неароматическое кольцо, содержащее 0, 1 или 2 гетероатома, выбранных из азота, кислорода или серы. В рамках контекста объяснения выражения 'необязательно замещенная', 'алкильная группа' означает С1-3-алкильная группа, 'алкенильная группа' означает С1-3-алкенильная группа, 'алкинильная группа' означает С1-3-алкинильная группа, 'ацильная группа' означает С1-3-ацильная группа и 'арильная группа' означает фурильная группа, тиенильная группа, пирролильная группа, оксазолильная группа, тиазолильная группа, имидазолильная группа, пиразолильная группа, изоксазолильная группа, изотиазолильная группа, пиридильная группа, пиридазинильная группа, пиримидинильная группа, пиразинильная группа, 1,3,5-триазинильная группа, фенильная группа, индазолильная группа, индолильная группа, индолизинильная группа, изоиндолильная группа, бензо[b]фуранильная группа, бензо[b]тиофенильная группа, бензимидазольная группа, бензтиазолильная группа, пуринильная группа, хинолинильная группа, изохинолильная группа, хинолильная группа, фталазинильная группа, хиназолинильная группа, хиноксалинильная группа, 1,8-нафтиридинильная группа, пиридинильная группа, нафтильная группа или азуленильная группа, предпочтительно фенильная группа, пиридильная группа или нафтильная группа. Необязательные заместители сами могут нести дополнительные необязательные заместители. Предпочтительные необязательные заместители включают С1-3-алкильную группу, такую как, например, метильная группа, этильная группа и трифторметильная группа, фтора, хлора, брома, гидроксильную группу, С1-3-алкилоксигруппу, такую как, например, метоксигруппа этоксигруппа и трифторметоксигруппа, и аминогруппу. 'Гетероатом' означает атом, такой как атом азота, атом кислорода или атом серы. 'Пяти- или шестичленные кольца' представляют собой, например: фурановые кольца, тиофеновые кольца, пиррольные кольца, оксазольные кольца, тиазольные кольца, имидазольные кольца, пиразольные кольца, изоксазольные кольца, изотиазольные, 1,2,3-оксадиазольные кольца, 1,2,3-триазольные кольца, 1,3,4-тиадиазольные кольца, пиридиновые кольца, пиридазиновые кольца, пиримидиновые кольца или пиразиновые кольца.
К изобретению относятся все соединения, имеющие формулу (1), рацематы, смеси диастереомеров и выделенные стереоизомеры. Таким образом, соединения, в которых заместители на потециально асимметричных атомах углерода находятся или в R-конфигурации, или в S-конфигурации, относятся к изобретению.
Пролекарства являются терапевтическими средствами (веществами), которые сами по себе являются неактивными, но превращаются в один или более активных метаболитов. Пролекарства являются биообратимыми производными молекул лекарственных веществ, используемыми для преодоления некоторых барьеров к (извлечению) полезности родительской молекулы лекарственного вещества. Эти барьеры включают в себя растворимость, проницаемость, стабильность, пресистематический метаболизм и ограничения по нацеливанию лекарства, но не ограничены этим (Medicinal Chemistry: Principles and Practice, 1994, ISBN 0-85186-494-5, Ed.: F. D. King, p. 215; J. Stella, “Prodrugs as therapeutics”, Expert Opin. Ther. Patents, 14(3), 277-280, 2004; P. Ettmayer et al., “Lessons learned from marketed and investigational prodrugs”, J.Med.Chem., 47, 2393-2404, 2004). Пролекарства, то есть соединения, которые при введении в человека любым известным способом подвергаются биотрансформации в соединения, имеющие формулу (1), относятся к изобретению. В частности, это относится к соединениям с первичными или вторичными аминогруппами или гидроксильными группами. Такие соединения могут быть подвергнуты реакции с органическими кислотами, давая соединения, имеющие формулу (1), в которой присутствует дополнительная группа, которая легко удаляется после введения, но которая не ограничивается, например, амидиновой группой, энаминовой группой, основанием Манниха, гидроксилметиленовым производным, O-(ацилоксиметиленкарбамат)-производным, карбаматной группой, сложноэфирной группой, амидогруппой или энаминоновой группой.
В частности изобретение относится к соединениям, имеющим формулу (1), в которой: R1 представляет собой атом водорода, галоген, CF3, алкильную (1-6С) группу, гидроксильную группу, (1-3С)алкоксигруппу или OCF3, m=1, и все другие радикалы имеют значения, приведенные выше.
В большей степени изобретение относится к соединениям, имеющим формулу (1), в которой: R1 представляет собой атом водорода, галоген, CF3, алкильную (1-6С) группу, гидроксильную группу, (1-3С)алкоксигруппу или OCF3, m=1, R2 представляет собой группу Q, имеющую общую формулу (2), и все другие радикалы имеют значения, приведенные выше.
В еще большей степени изобретение относится к соединениям, имеющим формулу (1), в которой: R1 представляет собой атом водорода, галоген, CF3, алкильную (1-6С) группу, гидроксильную группу, (1-3С)алкоксигруппу или группу OCF3, m=1, R2 представляет собой группу Q, имеющую общую формулу (2), R3 представляет собой метильную группу, R4 представляет собой алкильную (1-3С) группу, замещенную насыщенным, необязательно замещенным шестичленным кольцом, которое необязательно содержит один или более гетероатомов, и []n имеет значения, приведенные выше.
Наиболее предпочтительными соединениями изобретения являются соединения, имеющие формулу (1), в которой: R1 представляет собой атом водорода, галоген, CF3, алкильную (1-6С) группу, гидроксильную группу, (1-3С)алкоксигруппу или OCF3, m=1, R2 представляет собой группу Q, имеющую общую формулу (2), R3 представляет собой метильную группу, R4 представляет собой метиленовую группу, замещенную необязательно замещенным пиперидиновым кольцом, и []n имеет значения, приведенные выше.
Фармацевтически приемлемые соли могут быть получены путем использования стандартных способов, хорошо известных в данной области, например, смешением соединения настоящего изобретения с подходящей кислотой, например, неорганической кислотой, такой как соляная (хлористоводородная) кислота, или с органической кислотой.
Соединения настоящего изобретения общей формулы (1), а также их соли проявляют агонистическую активность к ORL1 рецепторам. Они являются полезными для лечения расстройств, связанных с ORL1 рецепторами, или, которые можно лечить посредством манипулирования этими рецепторами, особенно, но не ограничиваясь этим: состояний острой и хронической боли, нарушений обмена веществ, таких как нервная анорексия и нейрогенная булимия, ожирение; желудочно-кишечных расстройств, в частности, синдрома раздраженной толстой кишки, воспалительной болезни кишечника (гранулематозного энтерита и язвенного колита), поноса, запора, боли в животе, воспаления мочевого тракта, почечных расстройств, характеризующихся нарушениями баланса удержания/выделения воды или выделения соли; расстройств сердечно-сосудистой системы, таких как инфаркт миокарда, аритмия, повышенное кровяное давление, тромбоз, анемия, артериосклероз, стенокардия; офтальмологических расстройств, таких как глаукома; расстройств дыхательной системы, включающих хроническое обструктивное заболевание легких, бронхит и кистозный фиброз; заболеваний иммунной системы и вирусных инфекций.
Агонистические свойства соединений изобретения по отношению к ORL1 рецептору in vitro и in vivo определили, используя методики, приведенные ниже.
Сродство к ORL1 рецепторам человека
Сродство соединений к ORL1 рецепторам человека определили, используя реакцию связывания рецептора in vitro, описанный Ardati et al., Mol. Pharmacol., 51, 816, 1997. Кратко, препараты мембран были получены из клеток CHO (яичника китайского хомяка), в которых ORL1 рецептор человека стабильно выражен. Мембраны выдерживали с [3H]-ноцицептином в отсутствии или в присутствии тестовых соединений в различных концентрациях, разведенных в подходящем буфере. Неспецифическое связывание определили как связывание, остающееся в присутствии 10-6 М ноцицептина. Отделение связанной радиоактивности от свободной осуществили фильтрацией через стекловолоконные фильтры Паккарда GF/B с несколькими промываниями ледяным буфером, используя коллектор клеток Паккарда. Связанную радиоактивность измеряли сцинтилляционным счетчиком (Topcount, Packard), используя жидкий коктейль для сцинтилляционного счета (Microscint 0, Packard). Измеренную радиоактивность наносили на графике в зависимости от концентрации вытесняющего тестового соединения и кривые вытеснения рассчитывали логистической регрессией с четырьмя параметрами, получая в результате значения IC50, то есть значения IC50 для такой концентрации вытесняющего соединения, при которой вытеснено 50% радиолиганда. Значения сродства pK1 вычисляли c учетом поправок значений IC50 для концентрации радиолиганда и его сродства к ORL1 рецептору человека в соответствии с уравнением Ченга-Прусоффа:
pK1=-log(IC50/(1+S/Kd))
в котором IC50 представляет собой то же, что описано выше, S является концентрацией [3H]-ноцицептина, использованной в образце для анализа, выраженной в моль/л (обычно 0,2 нМ), и Kd является равновесной константой диссоциации [3H]-ноцицептина по отношению к ORL1 рецепторам человека (0,4 нМ).
Соединения изобретения имеют высокое сродство к ORL1 рецепторам в реакции связывания, описанной выше. Это свойство делает их полезными в лечении расстройств, связанных с ORL1 рецепторами, или которые можно лечить посредством манипулирования этими рецепторами.
Сродство к µ-опиатным рецепторам
Сродство соединений к µ-опиатным рецепторам определяли, используя реакцию связывания рецептора in vitro, описанную Wang et al., FEBS Letters, 338, 217, 1994. Кратко, препараты мембран получали из клеток CHO (яичника китайского хомяка), в которых µ-опиатный рецептор человека стабильно выражен, и выдерживали со специфическим лигандом µ-опиата [3H]-DAMGO (D-Ala2, N-Me-Phe4, глицинол5-Энкефалин) в отсутствии или в присутствии тестовых соединений в различных концентрациях, разведенных в подходящем буфере. Неспецифическое связывание определяли как связывание, остающееся в присутствии 10-6 М налоксона. Отделение связанной радиоактивности от свободной осуществляли таким же способом, как описано выше, и сродство соединений вычисляли подобным образом.
Соединения изобретения имеют низкое сродство к µ-опиатным рецепторам в реакции связывания, описанной выше. Таким образом, маловероятно, что они индуцируют нежелательные побочные эффекты, которые, как известно, имеют место с опиатами, такими как морфин.
Агонизм ORL1 рецептора in vitro
Активация G-протеин-связанного ORL1 рецептора ингибирует активность аденилатциклазы и снижает внутриклеточную концентрацию вторичного посланника (мессенджера) - циклоаденозинмонофосфата (cAMP). Активность соединений по отношению к ORL1 рецепторам измеряли, используя методику анализа, описанную Jenck et al., Proc. Natl. Acad. Sci. USA, 97, 4938-4943, 2000. Показано, что они являются сильнодействующими (эффективными) агонистами со значениями pEC50, соответствующими значениям их pKi.
Диарея, вызванная касторовым маслом, у мышей, находящихся в сознании
Показано, что пока есть пептид ноцицептин после подкожного введения, то соединения изобретения могут снижать диарею, вызванную касторовым маслом, у мышей. Поскольку периферически введенный пептид не проникает через гематоэнцефалический барьер указано, что снижение диареи, обусловленное ORL1, является периферически обусловленным.
Используемые животные: для этой модели диареи, вызванной касторовым маслом, использовали самцов мышей NMRI (Naval Medical Research Institute). Во всех экспериментах группа состояла из 10-12 животных.
Экспериментальные методики: в день эксперимента мыши получали или соединение, или носитель (с двухнедельными интервалами). По истечении 30 минут вводили орально касторовое масло (8 мл/кг массы тела) и животных помещали отдельно в клетках со свободным доступом к воде. Фекалии собирали по истечении 5 часов. В течение этого периода времени каждые 20 минут определяли визуальным осмотром качество фекалий. Бальная оценка диареи колебалась в диапазоне от 0 до 4, 0=нет выброса, 1=нормальный выброс (масса), 2=слабая диарея, 3=умеренная диарея, 4=сильная диарея. Таким образом, эта оценка по баллам отражает начало проявления и интенсивность диареи. В этих экспериментах определяли средний балл диареи и сухой вес фекалий.
Анализ данных: действие соединений приведено в виде относительных чисел (процент от контрольных значений). Исходные данные, регистрируемые в экспериментах, сравнивали с контрольными данными (без соединения) для одних и тех же животных посредством парных двусторонних критериев Стьюдента (t-tests) или с контрольной группой посредством непарного критерия Стьюдента (t-test). Значения p<0,05 принимали как статистически значимые.
Толстокишечный транзит (транзит по толстой кишке) у крыс, находящихся в сознании
Показано, что соединения изобретения не оказывают влияния на нормальный (регулярный) транзит по толстой кишке у крыс. Это также имело место в случае для пептида (Ноцицептина) после подкожного введения. Поскольку периферически введенный пептид не проникает через гематоэнцефалический барьер указано, что периферическая активация ORL1 рецептора не ухудшает нормальный желудочно-кишечный транзит. Напротив, периферическая активация µ-опиатного рецептора может сильно ухудшить транзит в этой модели. Таким образом, это исследование показывает селективность соединений изобретения по отношению к ORL1 рецептору.
Используемые животные: для экспериментов использовали самцов крыс Sprague Dawley. Во всех экспериментах группа состояла из 10-12 животных.
Экспериментальные методики: до проведения экспериментов крысам сделали постоянный титановый свищ слепой кишки под общей анестезией. Животным дали возможность восстановиться после хирургического вмешательства и приучили к кормлению в режиме свободного доступа для осуществления процесса поглощения еды в течение 3 часов в день. В день экспериментов после периода кормления через свищ в слепую кишку вводили вещество-маркер (2 мл суспензии, содержащей 80% сульфата бария) и животные получали или соединение, или носитель. Позже их помещали в метаболические клетки и собирали фекальные пеллеты ежечасно в течение 21 часа, используя автоматическую систему для сбора. В течение этого периода времени животные имели свободный доступ к воде. Содержание сульфата бария в фекалиях анализировали радиографически, а фекалии взвешивали. Зависимость содержания маркера в фекалиях от времени и количество фекалий позволили оценить среднее время удерживания сульфата бария, то есть время толстокишечного транзита. Были определены среднее время удерживания сульфата бария, содержащегося в пеллетах, и общая масса (выброс) фекалий.
Анализ данных: действие соединений приведено в виде относительных чисел (процент от контрольных значений). Исходные данные, регистрируемые в экспериментах, сравнивали с контрольными данными (без соединения) для одних и тех же животных посредством парных двусторонних критериев Стьюдента (t-tests). Значения p<0,05 принимали как статистически значимые. В модели толстокишечного транзита контрольные данные представляют собой среднее значение из двух контрольных экспериментов (до и после введения соединения, с недельными интервалами).
Висцеральная гиперчувствительность, вызванная уксусной кислотой, у крыс, находящихся в сознании
Показано, что соединения изобретения могут снижать висцеральную гиперчувствительность у крыс, пока имеется в наличии пептид Ноцицептин после подкожного введения. Поскольку периферически введенный пептид не проникает через гематоэнцефалический барьер указано, что снижение висцеральной гиперчувствительности, обусловленное ORL1, периферически обусловлено.
Используемые животные: взрослые самки крыс Sprague Dawley, вес тела: в диапазоне от 200 до 250 г. Группа состоит из 5-10 животных.
Экспериментальная методика: до проведения экспериментов животных подвергли голоданию в течение 24 часов со свободным доступом к воде. В толстую кишку (на 10 см, проксимальные к анальному отверстию) ввели уксусную кислоту (0,6%, 1,5 мл). По истечении 50 минут в дистальную толстую кишку ректально ввели резиновый шарик длиной 5 см (объемом 6-7 мл ) и закрепили путем связывания лентой с трубочкой (пробиркой), привязанной к хвосту крысы. Колоректальное растяжение осуществляли путем установления давления в шарике до 100 мбар (104 Па) в течение 10 минут. В течение этого периода времени путем визуального осмотра отслеживали число брюшных сужений (перетяжек). Продолжали эксперименты только на тех животных, которые реагировали на колоректальное растяжение более чем 10-ю брюшными перетяжками. Эти животные получали разовую дозу вещества или носителя и протокол колоректального растяжения повторяли в течение 30, 60, 90 и 120 минут после введения.
Анализ данных: результаты приведены как среднее значение ± статистическое отклонение. Число брюшных перетяжек, возникших в течение 30, 60, 90 и 120 минут после введения вещества или носителя, а также средние значения (30-120 мин) сравнили с предзначениями путем парных двусторонних критериев Стьюдента (t-тестов). Относительное количество брюшных перетяжек (% от предзначений), возникших в течение 30, 60, 90 и 120 минут в случае введения вещества и в случае введения носителя, и относительные средние значения (30-120 мин) для обоих случаев сравнивали между собой путем непарных двусторонних критериев Стьюдента (t-тестов). Значения p<0,05 были приняты как статистически значимые.
Агонизм ORL1 рецептора in vivo: отсутствие проникновения в центральную нервную систему (CNS)
Показано, что большинство соединений изобретения не проявляют активности при воздействии ультразвукового голосового сигнала, вызывающего стресс у взрослых особей (AUV), что описано Van der Poel et al., Psychopharmacology, 97, 147-148, 1989. Это показывает, что соединения не проникают через гематоэнцефалический барьер. В этом анализе пептид ноцицептин также является активным, но для того, чтобы продемонстрировать его действенность, необходимо его вводить непосредственно в мозг (внутримозговой (меж)желудочковой инъекцией).
ПРИМЕРЫ СИНТЕЗОВ ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ И КОНЕЧНЫХ ПРОДУКТОВ
(-)-транс-2-(6,6-Диметил-бицикло[3.1.1]гепт-2-ил)-этанол (5)

Миртанилбромид (2)
Трифенилфосфин (116 г, 0,44 моль) растворили в ацетонитриле (1 л) и охладили в ледяной бане в атмосфере N2. По каплям добавили бром (22,5 мл, 0,44 моль). Температуру экзотермической реакции поддерживали ниже 10°С. По завершении добавления ледяную баню удалили и медленно добавили (-)-транс-миртанол (1) (2,686 г, 0,44 моля), растворенный в ацетонитриле (250 мл). По завершении добавления светло-желтый раствор кипятили, используя прибор с насадкой Дина-Старка, в течение 3 часов. Во время реакции растворитель отгоняли в ловушку для воды и затем 20 раз удаляли (приблизительно 200 мл растворителя в итоге). Газохроматографический (GC) анализ показал полную конверсию исходного вещества. Смесь упарили досуха. Сырую (необработанную) смесь очистили на колонке с силикагелем (элюент: дихлорметан/диэтиловый эфир 1/1, объем/объем). Это дало 87,8 г бромида (2) (91%) в виде светло-желтого масла.
Миртанилцианид (3)
Миртанилбромид (2) (87,8 г, 0,41 моль) растворили в диметилформамиде (1 л). Добавили цианид натрия (40 г, 0,81 моль) и смесь перемешивали при кипячении с обратным холодильником в течение 5 часов. Газохроматографический анализ показал полную конверсию. Смесь разбавили водой (3 л) и экстрагировали трет-бутилметиловым эфиром (TBME, 3× 1,5 л). Органический слой промыли рассолом (насыщенным раствором NaCl), осушили над Na2SO4 и концентрировали досуха. Сырую смесь очистили на колонке с силикагелем (элюент: гептан/дихлорметан, 1/1, объем/объем) и получили 52,4 г (80%) цианида (3) в виде бесцветной жидкости.
Этиловый эфир (сложный) (4)
Этанол (500 мл) охладили на ледяной бане. Добавили по каплям серную кислоту (190 мл). Добавили цианид (3) (52,4 г, 0,32 моля), растворенный в этаноле (100 мл), и смесь перемешивали при кипячении с обратным холодильником в течение ночи. Газохроматографический анализ показал полное присоединение. Смесь охладили и добавили воду (1,5 л). Смесь экстрагировали трет-бутилметиловым эфиром (3×1,5 л). Органический слой промыли NaHCO3 (нас. 1 л), осушили над Na2SO4 и концентрировали. Выход: 54,2 г сложного эфира 5 (80%) в виде почти бесцветной жидкости. Сырой продукт (4) использовали в следующей реакции без очистки.
(-)-транс-2-(6,6-Диметил-бицикло[3.1.1]гепт-2-ил)-этанол (5)
К суспензии литийалюмогидрида (20 г, 0,52 моля) в тетрагидрофуране (1 л) добавили сложный эфир (4) (54,2 г, 0,26 моля), растворенный в тетрагидрофуране (500 мл). По завершении добавления смесь кипятили с обратным холодильником в течение 1 часа. Газохроматографический анализ показал полную конверсию исходного вещества. Смесь охладили на ледяной бане и осторожно добавили соляную кислоту (1М, 1 л). По завершении добавления смесь разбавили водой (1 л) и экстрагировали трет-бутилметиловым эфиром (3×1,5 л). Органический слой промыли рассолом (насыщенным раствором NaCl), осушили над Na2SO4 и концентрировали досуха. Сырую смесь подвергли очистке перегонкой Кугельрота (температура кипения 85°С, 3·10-2 мбар (3 Па)). Выход: 35,9 г соединения 1 (65%) в виде бесцветного масла.
(+)-транс-2-(6,6-Диметил-бицикло[3.1.1]гепт-2-ил)-этанол (10)

Миртанилмезилат (7)
18,1 г (0,12 моля) (+)-транс-миртанола (6) добавили к раствору 18,5 мл мезилхлорида (2 экв., 0,24 моль, 27,5 г) и 49 мл пиридина (5 экв., 0,60 моля, 47,5 г) в 400 мл дихлорметана (DCM). Реакционную смесь перемешивали всю ночь при комнатной температуре. Добавили воду и реакционную смесь перемешивали в течение одного часа. Органический слой экстрагировали и водный слой экстрагировали еще два раза. Объединенные органические слои промыли (насыщенным NaHCO3, водой, рассолом (насыщенным раствором NaCl)), высушили (Na2SO4) и упарили в вакууме с получением 25,9 г (91%) мезилата (7) в виде бесцветного масла.
Миртанилцианид (8)
Миртанилмезилат (7) (25,9 г, 0,11 моля) растворили в диметилсульфоксиде (DMSO) (250 мл). Добавили цианид калия (4 экв., 29,2 г, 0,45 моля) и смесь перемешивали при 70°С в течение 2 дней. Газохроматографический анализ показал полную конверсию. Смесь разбавили водой (750 мл) и экстрагировали трет-бутилметиловым эфиром (3×300 мл). Органический слой промыли рассолом, высушили над Na2SO4 и концентрировали досуха, что дало 17,7 г (количественный выход) цианида (8) в виде бесцветного масла.
Этиловый эфир (сложный) (9)
Этанол (200 мл) охладили на ледяной бане. По каплям добавили серную кислоту (80 мл). Добавили цианид (8) (17,7 г, 0,11 моля), растворенный в этаноле (40 мл), и смесь перемешивали при кипячении с обратным холодильником в течение всей ночи. Газохроматографический анализ показал полное присоединение. Смесь охладили и добавили воду (1 л). Смесь экстрагировали трет-бутилметиловым эфиром (3×500 мл). Органический слой промыли NaHCO3 (нас., 500 мл), высушили над Na2SO4 и концентрировали. Выход: 20,4 г сложного эфира (9) (88%) в виде желтого масла. Сырой продукт (9) использовали в следующей реакции без очистки.
(+)-транс-Дигидронопол (10)
К суспензии литийалюмогидрида (7,4 г, 0,19 моля) в тетрагидрофуране (350 мл) добавили сложный эфир (9) (20,1 г, 0,09 моля), растворенный в тетрагидрофуране (200 мл). По завершении добавления смесь кипятили с обратным холодильником в течение 2 часов. Смесь охладили на ледяной бане и осторожно добавили соляную кислоту (1М, 1 л). По завершении добавления смесь разбавили водой (300 мл) и экстрагировали трет-бутилметиловым эфиром (3×500 мл). Органический слой промыли рассолом, осушили над Na2SO4 и концентрировали досуха. Сырую смесь подвергли очистке перегонкой Кугельрота (температура кипения 85°С, 8·10-2 миллибар (8 Па)). Выход: 9,2 г соединения (10) (61%) в виде бесцветного масла.
(-)-цис-2-(6,6-Диметил-бицикло[3.1.1]гепт-2-ил)-этанол (11)
Синтез цис-аналога: (+)-цис-2-(6,6-диметил-бицикло[3.1.1]гепт-2-ил)-этанола (18), с (-)-β-пиненом в качестве исходного вещества описан в J. Amer. Chem. Soc. 68, 638, 1946, и в патентах US 2,427,343, 2,427,344 и 2,427,345.

(+)-β-пинен (13)
В сухой стеклянной посуде трет-бутилоксид калия (KOt-Bu, 49,4 г, 0,44 моля) добавили к n-бутиллитию (176 мл, 2,5М в гексане). Суспензию охладили до -78°С. По каплям добавили (+)-α-пинен (12) (50 г, 0,37 моля). Реакционной смеси дали нагреться до комнатной температуры и затем перемешивали в течение 45 часов. Реакционную смесь охладили до -78°С и по каплям добавили B(OMe)3 (137 мл, 1,20 моля). Реакционной смеси дали нагреться до комнатной температуры (экзотермическая реакция!). По каплям добавили 10% HCl (водный, 250 мл) и реакционную смесь перемешивали в течение 1 часа. Слои разделили и водный слой экстрагировали гептаном (2×200 мл). Объединенные органические слои высушили над Na2SO4 и упарили досуха, что дало 36,7 г желтого масла. Сырой продукт подвергли очистке, используя перегонку Кугельрота (8-12 мбар (800-1200 Па), 50-60°С), что дало 36,6 г (0,27 моля, выход=73%, 88% чистоты) (+)-β-пинена (13) в виде бесцветного масла.
Миртанол (14)
(+)-β-пинен (13) (36,6 г,0,27 моля) растворили в тетрагидрофуране (100 мл) и охладили до 0°С. По каплям добавили BH3·диметилсульфид (BH3·DMS) в тетрагидрофуране (2 М, 47,3 мл). Реакционную смесь перемешивали в течение получаса. Добавили этанол (90 мл). Добавили 1 М NaOH (вод.) (95 мл). Реакционную смесь охладили до 0°С. По каплям добавили 33 мл 30% H2O2, при этом температуре не давали подниматься выше 35°С. Реакционную смесь кипятили с обратным холодильником в течение 1 часа и вылили в воду (1 л). Раствор экстрагировали трет-бутилметиловым эфиром. Объединенные органические слои промыли водой и рассолом, осушили над Na2SO4 и упарили досуха. Остающийся α-пинен отогнали, используя перегонку Кугельрота (8-9 мбар (800-900 Па), 50-60°С), получая 38,6 г (0,25 моля, выход=93%) (+)-цис-миртанола (14) в виде бесцветного масла.
Миртанилмезилат (15)
15,0 г (0,10 моля) (+)-цис-миртанола (14) добавили к раствору 15 мл мезилхлорида (экв., 0,20 моля) и 40 мл пиридина (5 экв., 0,50 моля) в 300 мл дихлорметана (DCM). Реакционную смесь перемешивали всю ночь при комнатной температуре. Добавили воду и реакционную смесь перемешивали в течение 1 часа. Органический слой экстрагировали и водный слой экстрагировали еще два раза. Объединенные органические слои промыли (насыщенным NaHCO3, водой, рассолом), осушили (Na2SO4) и упарили в вакууме с получением 21,6 г (выход=93%) мезилата (15) в виде бесцветного масла.
Миртанилцианид (16)
Миртанил мезилат (15) (21,6 г, 0,093 моля) растворили в диметилсульфоксиде (DMSO) (230 мл). Добавили цианид калия (4 экв., 24,2 г, 0,37 моля) и смесь перемешивали при 70°С в течение 8 дней. Газохроматографичекий анализ показал полную конверсию. Смесь разбавили водой и экстрагировали гептаном. Органический слой промыли рассолом, высушили над Na2SO4 и концентрировали досуха с получением 15,8 г (количественный выход) цианида (16) в виде бесцветного масла.
Этиловый эфир (сложный) (17)
Этанол (150 мл) охладили на ледяной бане. По каплям добавили серную кислоту (60 мл). Добавили цианид (16) (16 г), растворенный в этаноле (30 мл), и смесь перемешивали при кипячении с обратным холодильником всю ночь. Газохроматографичекий анализ показал полную конверсию. Смесь охладили и добавили воду (1 л). Смесь экстрагировали трет-бутилметиловым эфиром (3×500 мл). Органический слой промыли насыщенным NaHCO3 (водный, 500 мл), высушили над Na2SO4 и концентрировали досуха. Выход: 20,6 г сложного эфира (17) (количественный выход) в виде желтого масла. Сырой продукт (17) использовали в следующей реакции без очистки.
(+)-цис-Дигидронопол (18)
К суспензии литийалюмогидрида (8,3 г, 0,22 моля) в тетрагидрофуране (400 мл) добавили сложный эфир (17) (23,6 г, 0,11 моля), растворенный в тетрагидрофуране (200 мл). По завершении добавления смесь кипятили с обратным холодильником в течение 2 часов. Смесь охладили на ледяной бане и осторожно добавили соляную кислоту (1М, 1 л). По завершении добавления смесь разбавили водой (300 мл) и экстрагировали трет-бутилметиловым эфиром (3×500 мл). Органический слой промыли рассолом, осушили над Na2SO4 и концентрировали досуха, получая желтое масло (13,4 г). Сырую смесь подвергли очистке перегонкой Кугельрота (температура кипения 85°С, 8·10-2 мбар (8 Па)). Выход: 8,7 г соединения (18) (51 ммоль, y=47%) в виде бесцветного масла.
1-Мезил-2-(6,6-диметил-бицикло[3.1.1]гепт-2-ил)-этанол (20) (для всех стереоизомеров дигидронопола)

К суспензии 67 г (0,4 моля) (-)-цис-2-(6,6-Диметил-бицикло[3.1.1]гепт-2-ил)-этанола (19) в 300 мл дихлорметана при 0°С добавили 139 мл (1 моль) триэтиламина. К этой смеси добавили по каплям 55,2 г (0,48 моля) мезилхлорида в 100 мл дихлорметана. По истечении 5 часов при комнатной температуре реакция была завершена, и были добавлены 300 мл 1н. водного раствора HCl. После отделения водный слой промыли дихлорметаном дважды и объединенные органические слои промыли водой, осушили над сульфатом магния и концентрировали в вакууме, получая на выходе 91,6 г (0,37 моля, 91%) сырого оранжевого маслянистого продукта. Этот сырой материал использовали для следующей стадии без дополнительной очистки.
8-[2-(6,6-Диметил-бицикло[3.1.1]гепт-2-ил)-этил]-3-(3-метиламино-пропил)-1-фенил-1,3,8-триаза-спиро[4.5]декан-4-он (24)

8-[2-(6,6-Диметил-бицикло[3.1.1]гепт-2-ил)-этил]-1-фенил-
1,3,8-триаза-спиро[4.5]декан-4-он (22) [пример №1 в таблицах ниже]
Спиросоединение (21) (310 г, 1,34 моля) и (ди)гидронополмезилат (20) (371 г, 1,51 моля) растворили в метилэтилкетоне (MEK, 15 л). Добавили карбонат калия (735 г, 5,33 моля) и йодид натрия (226 г, 1,51 моля) и смесь кипятили с обратным холодильником в течение всей ночи. После охлаждения реакционной смеси растворитель выпарили. Твердый остаток сняли дихлорметаном (5 л) и потрясли с водой (4 л). Слои разделили, органический слой осушили над Na2SO4 и растворитель выпарили. Остающееся сухое вещество промыли диэтиловым эфиром (Et2O) (3 л) и отфильтровали. Фильтрат упарили и промыли Et2O (300 мл). Сухое вещество отфильтровали (466,3 г, 1,22 моля, 91%).
3-(3-Хлор-пропил)-8-[2-(6,6-диметил-бицикло[3.1.1]гепт-2-ил)-этил]-1-фенил-1,3,8-триаза-спиро[4.5]декан-4-он (23)
Тетрагидрофуран (1500 мл) охладили на водно-ледяной бане. Добавили спиросоединение (22) (150,8 г, 0,40 моля) и трет-бутоксид калия (49 г, 0,44 моля) и получающуюся в результате смесь перемешивали при 0°С в течение 30 минут. Смесь стала прозрачной. К раствору при 0°С добавили по каплям 1-бром-3-хлорпропан (43 мл, 0,44 моля) в тетрагидрофуране (150 мл). По завершении добавления прекращали охлаждение и раствор перемешивали при 50°С в течение 4 часов. После охлаждения смесь вылили в насыщенный KHSO4 (водный, 1000 мл) и разбавили этилацетатом (EtOAc) (500 мл). Слои разделили и водный слой экстрагировали EtOAc (3×750 мл). Объединенные органические слои промыли водой и рассолом (1×500 мл в каждом случае). После осушения над Na2SO4 растворитель выпарили с получением желтого масла (205,6 г, 0,45 моля, количественный выход).
8-[2-(6,6-Диметил-бицикло[3.1.1]гепт-2-ил)-этил]-3-(3-метиламино-пропил)-1-фенил-1,3,8-триаза-спиро[4.5]декан-4-он (24) [пример №13 в таблицах ниже]
Сырое спиросоединение (23) (162,8 г, 0,36 моля) растворили в растворе метиламина и этилового спирта (Fluka, 8M, 1154 мл, 9,23 моля). Добавили йодид натрия (2,16 г, 0,014 моля) и раствор перемешивали при 70°С под атмосферой N2 в течение 3 дней. После охлаждения реакционную смесь разбавили водой и этилацетатом (500 мл в каждом случае). Водный слой экстрагировали этилацетатом (3× 800 мл). Органический слой промыли рассолом (500 мл). После осушения над Na2SO4 упарили растворитель с получением желтого масла. Это масло очищали колоночной хроматографией (SiO2, CH2Cl2/MeOH 90:10, содержащий 1% 7N NH3/MeOH) c получением 30 г соединения (24) с чистотой 93% (согласно HPLC/MS), 96% (115 г, 0,25 моля, 70%).
Изменение типа замещения фенильного кольца в спирокаркасе 8-[2-(6,6-Диметил-бицикло[3.1.1]гепт-2-ил)-этил]-1-фенил-1,3,8-триаза-спиро[4.5]декан-4-она (22)

1-[2-(6,6-Диметил-бицикло[3.1.1]гепт-2-ил)-этил]-пиперидин-4-он (26):
Смесь 61,1 г (0,40 моля) пиперидонгидратгидрохлорида (25), 112,8 г (0,46 моля) дигидронополмезилата (20), 69,0 г (0,46 моля) NaI, 273 г (1,97 моля) K2CO3 и 4,3 л метилэтилкетона кипятили с обратным холодильником всю ночь. Смесь охладили до комнатной температуры и концентрировали в вакууме. Твердый остаток растворили в дихлорметане (1,5 л) и в воде (1,5 л) и слои разделили. Органический слой промыли водой (1 л) и осушили над Na2SO4. Слой концентрировали в вакууме с получением 113 г сырого продукта, который подвергли очистке колоночной хроматографией (SiO2, гептан:этилацетат, 6:1→1:1) и получили на выходе 77,7 г (0,31 моля, 78%) соединения (26) в виде оранжевого масла.
1-[2-(6,6-Диметил-бицикло[3.1.1]гепт-2-ил)-этил]-4-(3-фтор-фениламино)-пиперидин-4-карбонитрил (28а):
Раствор 20,0 г (80,2 ммоля) (26) и 8,4 мл (87 ммоля) 3-фторанилина (27а) в 65 мл уксусной кислоты охладили на холодной водяной бане. В течение 10 мин добавляли по каплям 10,7 мл (80,2 ммоля) триметилсилилцианида, поддерживая температуру ниже 40°С. Смесь перемешивали в течение 2 часов при комнатной температуре и вылили в смесь водного аммония (80 мл) и льда (80 г). Концентрированным NH3 pH довели до 10. Смесь экстрагировали хлороформом (3×200 мл). Объединенные органические слои осушили над Na2SO4 и концентрировали в вакууме, что дало 40,0 г сырого продукта, который подвергли очистке колоночной хроматографией (SiO2, гептан:этилацетат, 1:1) и получили 28,7 г (77,7 ммоля, 97%) соединения (28а). Сырой продукт также можно использовать в следующей стадии без очистки.
Амид 1-[2-(6,6-Диметил-бицикло[3.1.1]гепт-2-ил)-этил]-4-(3-фтор-фениламино)-пиперидин-4-карбоновой кислоты (29а):
Смесь 28,7 г (78 ммоля) соединения (28а), 135 мл муравьиной кислоты и 135 мл уксусного ангидрида перемешивали при комнатной температуре в течение 1 дня. Реакцию контролировали методами 1H-ЯМР-спектроскопии и масс-спектроскопии. По завершении реакции реакционную смесь вылили в лед с водой (800 мл). pH довели до 10 добавлением 33%-го NaOH (водного). Водный слой экстрагировали дихлорметаном (3×1 л). Объединенные органические слои осушили над Na2SO4 и концентрировали в вакууме. Твердый остаток растворили в 550 мл трет-бутилового спирта, 45 мл воды и 45 мл концентрированного водного аммония. При комнатной температуре добавили по каплям 90 мл 35%-го пероксида водорода. Смесь перемешивали всю ночь. Реакцию контролировали методом тонкослойной хроматографии (TLC). Добавили 900 мл воды и смесь экстрагировали дихлорметаном (3×500 мл). Объединенные органические слои осушили над Na2SO4 и концентрировали в вакууме с получением 29,3 г (76 ммоль, 98%) соединения (29а) в виде желтого сухого вещества, который использовали в следующей стадии без очистки.
8-[2-(6,6-Диметил-бицикло[3.1.1]гепт-2-ил)-этил]-1-(3-фтор-фенил)-1,3,8-триаза-спиро[4.5]декан-4-он (30а): [пример №9 в таблицах ниже]
Раствор 29,3 г (76 ммоль) соединения (29а) в 400 мл формамида нагревали в течение 2 часов при 200°С. Раствор поменял цвет из желтого в черный. Реакцию контролировали методом 1H-ЯМР-спектроскопии. По завершении реакции смесь охладили до комнатной температуры и вылили в лед с водой (800 г). Смесь экстрагировали дихлорметаном (6×1 л). Объединенные органические слои осушили над Na2SO4 и концентрировали в вакууме. Твердый остаток растворили в 1,2 л метанола и добавили порциями 4,3 г (114 ммоль) боргидрида натрия. Смесь перемешивали в течение 1 часа при комнатной температуре и в течение еще одного часа при 60°С. Реакционную смесь охладили до комнатной температуры и погасили 25 мл воды. Растворитель выпарили в вакууме. Твердый остаток растворили в 750 мл водного аммония и экстрагировали диметилхлорметаном (7×1,5 л). Объединенные органические слои осушили над Na2SO4 и концентрировали в вакууме с получением 24,8 г сырого продукта, который подвергли очистке колоночной хроматографией (SiO2, гептан:этилацетат, 1:1→1:3). Растирание элюированного продукта в порошок с диэтиловым эфиром дало 3,44 г соединения (30а) (8,6 ммоля, 11,3% в расчете на соединение (26)) в виде белого сухого вещества.
8-[2-(6,6-Диметил-бицикло[3.1.1]гепт-2-ил)-этил]-1-(3-метокси-фенил)-1,3,8-триаза-спиро[4.5]декан-4-он (30b): [пример №8 в таблицах ниже]
Последовательность стадий повторили, исходя из 68,0 г (0,27 моля) соединения (26); Соединение (30b) подвергли очистке колоночной хроматографией и растиранием в порошок с диэтиловым эфиром, что дало 13,9 г (34 ммоль, 12% выход в расчете на соединение (26)) в виде не совсем белого сухого вещества.
8-[2-(6,6-Диметил-бицикло[3.1.1]гепт-2-ил)-этил]-1-(3-хлор-фенил)-1,3,8-триаза-спиро[4.5]декан-4-он (30с): [пример №7 в таблицах ниже]
Последовательность стадий повторили, исходя из 67,4 г (0,27 моля) соединения (26); Соединение (30с) подвергли очистке колоночной хроматографией и растиранием в порошок с диэтиловым эфиром, что дало 7,42 г (17,8 ммоля, 6,6% выход в расчете на соединение (26)) в виде не совсем белого сухого вещества.
8-[2-(6,6-Диметил-бицикло[3.1.1]гепт-2-ил)-этил]-1-(3-трифторметил-фенил)-1,3,8-триаза-спиро[4.5]декан-4-он (30d): [пример №10 в таблицах ниже]
Для получения соединения (30d) выполнили ту же самую последовательность стадий, но вместо желательного продукта выделили соединение (29d). Поэтому последовательность стадий частично повторили. Соединение формилировали муравьиной кислотой и уксусным ангидридом, нагревали в формамиде и, в конце концов, восстановили боргидридом натрия. Сырой продукт очищали колоночной хроматографией (SiO2, этилацетат) и впоследствии растиранием в порошок с диэтиловым эфиром, что дало 7,39 г (6,6% общий выход в расчете на соединение (26)) в виде белого сухого вещества.
8-[2-(6,6-Диметил-бицикло[3.1.1]гепт-2-ил)-этил]-1-(4-фтор-фенил)-1,3,8-триаза-спиро[4.5]декан-4-он (30е): [пример №5 в таблицах ниже]
Последовательность стадий повторили, исходя из 45,0 г (0,18 моля) соединения (26); Соединение (30е) очищали колоночной хроматографией и растиранием в порошок с диэтиловым эфиром, что дало 9,82 г (24,5 ммоля, 13,6% выход в расчете на соединение (26)) в виде серого сухого вещества.
8-[2-(6,6-Диметил-бицикло[3.1.1]гепт-2-ил)-этил]-1-(4-метокси-фенил)-1,3,8-триаза-спиро[4.5]декан-4-он (30f): [пример №6 в таблицах ниже]
Последовательность стадий повторили, исходя из 45,0 г (0,18 моля) соединения (26); Соединение (30f) очищали колоночной хроматографией и растиранием в порошок с диэтиловым эфиром, что дало 8,94 г (21,7 ммоля, 12,1% выход в расчете на соединение (26)) в виде белого сухого вещества.

Трет-бутиловый сложный эфир 1-окса-6-аза-спиро[2.5]октан-6-карбоновой кислоты (32):
К раствору 44,9 г (0,225 моля) трет-бутилового сложного эфира 4-оксо-пиперидин-1-карбоновой кислоты (31) в 500 мл ацетонитрила последовательно добавили 59,5 г (0,27 моля) йодида триметилсульфоксония и 18,9 г (0,338 моля) тонко измельченного гидроксида калия. Реакционную смесь тщательно перемешивали в течение двух дней в атмосфере азота при комнатной температуре. После полной конверсии растворитель выпарили в вакууме и твердый остаток сняли дихлорметаном. Органический слой промыли водным раствором лимонной кислоты (6×), осушили над сульфатом натрия и концентрировали в вакууме. Этот сырой продукт использовали без дополнительной очистки в следующей стадии (43,5 г, 0,204 моля, 90,6% выход).
Трет-бутиловый сложный эфир 4-{[(3-{8-[2-(6,6-Диметил-бицикло[3.1.1]гепт-2-ил)-этил]-4-оксо-1-фенил-1,3,8-триаза-спиро[4.5]дек-3-ил}пропил)-метил-амино]-метил}-4-гидрокси-пиперидин-1-карбоновой кислоты (33): [пример №35 в таблицах ниже]
2,6 г (5,74 ммоля) соединения (24) поместили в колбу и разбавили 20 мл этанола. К этому раствору добавили 1,88 г (8,81 ммоля) эпоксида (32) и смесь кипятили с обратным холодильником до тех пор, пока метод тонкослойной хроматографии не показал полную конверсию. Для обработки реакционной смеси растворитель выпарили и твердый остаток сняли этилацетатом. После промывания водным раствором карбоната калия, сушки над сульфатом натрия и концентрирования в вакууме сырой материал очищали колоночной флэш хроматографией, что дало светло-желтое вязкое масло (3,51 г, 5,15 ммоля, 89,8% выход).
8-[2-(6,6-Диметил-бицикло[3.1.1]гепт-2-ил)-этил]-3-{3-[(4-гидрокси-пиперидин-4-илметил)-метил-амино]-пропил}-1-фенил-1,3,8-триаза-спиро[4.5]декан-4-он (34): [пример №37 в таблицах ниже]
Бутилоксикарбонильное производное (Boc-производное) (33) (2,79 г, 4,19 ммоля) растворили в тетрагидрофуране (25 мл). К этому раствору добавили 2 мл концентрированного водного раствора HCl и получающуюся в результате смесь перемешивали при комнатной температуре всю ночь. После того как метод тонкослойной хроматографии показал полную конверсию, при пониженном давлении удалили растворитель и получающийся в результате твердый остаток растворили в этилацетате. Промывание раствором карбоната калия, осушение органического слоя сульфатом натрия и концентрирование в вакууме обеспечили чистое указанное в подзаголовке соединение в виде светло-желтого вязкого масла (2,37 г, 3,8 ммоля, 90,7% выход).
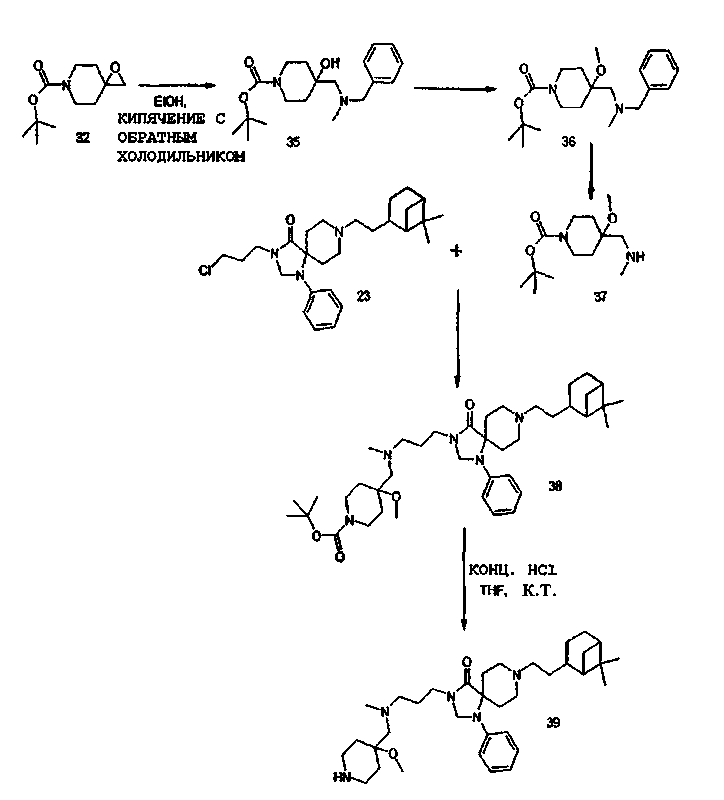
Трет-бутиловый сложный эфир 4-[(бензил-метил-амино)-метил]-4-гидрокси-пиперидин-1-карбоновой кислоты (35)
Эпоксид (32) (46 г, 216 ммоля) растворили в диоксане (300 мл). Добавили бензилметиламин (75 мл, 583 ммоль) и смесь перемешивали при кипячении с обратным холодильником в течение 90 часов. Метод тонкослойной хроматографии показал полную конверсию. Смесь упарили досуха. Избыток бензилметиламина удалили выпариванием в вакууме (0,05 мбар (5 Па), 80°С). Выход: 69,3 г аминоспирта (35) (96%) в виде оранжевого масла.
Трет-бутиловый сложный эфир 4-[(бензил-метил-амино)-метил]-4-метокси-пиперидин-1-карбоновой кислоты (36)
Спирт (35) (69,3 г, 200 ммоль) растворили в диметилформамиде (500 мл). В течение 30 мин добавляли 5-ю порциями NaH (9,2 г, 230 ммоль), промытый пентаном. По завершении добавления смесь перемешивали при температуре окружающей среды в течение 45 мин. За 1,5 мин добавили метилйодид (14,8 мл, 240 ммоль). Смесь перемешивали в течение 1,5 часов при температуре окружающей среды. Метод тонкослойной хроматографии показал почти 80-90% конверсию. Добавили дополнительные NaH (0,8 г, 20 ммоль) и метилйодид (1,2 мл, 20 ммоль) и смесь перемешивали в течение еще 2 часов при температуре окружающей среды. Избыток NaH разложили водой (100 мл) и смесь дополнительно разбавили водой (3,5 л). Смесь экстрагировали этилацетатом (2×1л, 500 мл). Органический слой промыли рассолом (1 л), осушили над Na2SO4 и концентрировали досуха. Следы диметилформамида удалили выпариванием в вакууме (0,4 мбар (40 Па), 80°С). Остающуюся смесь очистили на силикагеле (элюент: гептан/этилацетат, 4/1-3/1 объем/объем). Выход: 55,2 г амина (36) (80%) в виде светло-желтого масла.
Трет-бутиловый сложный эфир 4-метокси-4-метиламинометил-пиперидин-1-карбоновой кислоты (37)
Амин (36) (55 г, 158 ммоль) растворили в этилацетате (500 мл). Добавили палладиевый катализатор (Pd-C) (10%, влажный, 5 г) и смесь перемешивали в течение 23 часов в атмосфере водорода (1 бар (105 Па)). Метод тонкослойной хроматографии показал неполную конверсию. Добавили дополнительный Pd-C (2,5 г) и смесь перемешивали в атмосфере водорода (1 бар (105 Па)) в течение 110 часов. Метод ЯМР-спектроскопии показал полную конверсию. Смесь профильтровали через Целит, порцию Целита промыли этилацетатом и фильтрат выпарили досуха. Твердый остаток подвергли очистке перегонкой (высоковакуумной перегонкой из колбы в колбу, 0,04 мбар (4 Па), 130°С), что дало 35 г соединения (37) (86%) в виде бесцветного масла.
Синтез 8-[2-(6,6-Диметил-бицикло[3.1.1]гепт-2-ил)-этил]-3-{3-[(4-метокси-пиперидин-4-илметил)-метил-амино]-пропил}-1-фенил-1,3,8-триаза-спиро[4.5]декан-4-она (39) [пример №45 в таблицах ниже], исходя из хлорида (23) и амина (36), осуществили таким же способом, как описано ниже (общие методики).
Создание библиотеки с 8-[2-(6,6-Диметил-бицикло[3.1.1]гепт-2-ил)-этил]-3-(2-метиламино-этил}-1-фенил-1,3,8-триаза-спиро[4.5]декан-4-оном в качестве исходного вещества
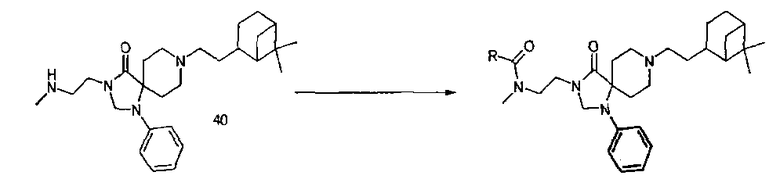
Библиотека амидов, способ I:
Производное N-метиламина (40) (1,832 г, 4,24 ммоля) растворили в 170 мл дихлорметана. Этот основной раствор использовали для получения амидов с участием различных растворов хлорангидридов следующим образом: 2 мл основного раствора (0,05 ммоля соединения (40)) обработали полимерсвязанным морфолином (0,162 ммоля). После перемешивания в течение 20 мин при комнатной температуре добавили соответствующий хлорангидрид (0,06 ммоля) в 2 мл дихлорметана и перемешивание продолжили в течение 1 дня при комнатной температуре. Реакцию контролировали методом тонкослойной хроматографии. Для того чтобы избавиться от остающихся хлорангидрида и производного N-метиламина, добавили соответственно полимерсвязанный трисамин и изоцианатный реагент (оба используют в качестве поглотителей). Вновь продолжили перемешивание при комнатной температуре и делали это в течение ночи, затем удалили полимеры фильтрацией. Фильтраты концентрировали при пониженном давлении. Используя этот протокол, синтезировали 69 соединений. Сродство каждого синтезированного амида к ORL1 рецептору человека измеряли по реакции связывания рецептора in vitro.
Библиотека амидов, способ II:
К 200 мкл исходных растворов основных растворов (0,25М в тетрагидрофуране) добавили 200 мкл исходных растворов хлорангидридов (0,25М в тетрагидрофуране), с последующим добавлением 50 мкл раствора триэтиламина (1,0М в тетрагидрофуране). После встряхивания в течение ночи (17 часов) при 30°С растворитель выпарили и сырые продукты сняли диметилсульфоксидом для анализа. (Примечание: нерастворимые реагенты добавили вручную). Используя этот протокол, синтезировали 26 соединений. Сродство каждого синтезированного амида к ORL1 рецептору человека измеряли по реакции связывания рецептора in vitro.

Библиотека Раскрытия Эпоксидов:
Производное N-метиламина (40) (1,316 г, 3,0 ммоль) растворили в 120 мл изопропилового спирта. Этот основной раствор использовали для получения аминоспиртов с участием различных растворов эпоксидов следующим образом: к 2 мл основного раствора (0,05 ммоля) соединения (40)) добавили раствор соответствующего эпоксида (0,075 ммоля) в 2 мл изопропилового спирта. Эту смесь нагрели до 80°С и грели в течение 2 дней. Для контроля за реакциями использовали метод тонкослойной хроматографии. Для обработки реакционной смеси добавили соответственно полимерсвязанный трисамин и изоцианатный реагент (оба используют в качестве поглотителей). Вновь продолжили перемешивание при комнатной температуре и делали это в течение 2 дней, и затем удалили полимеры простой фильтрацией. Фильтраты концентрировали при пониженном давлении. Используя этот протокол, синтезировали 27 соединений. Сродство каждого синтезированного аминоспирта к ORL1 рецептору человека измеряли по реакции связывания рецептора in vitro.

Библиотека карбамидов (мочевины):
К 200 мкл исходных растворов основных растворов (0,25М в тетрагидрофуране) добавили 200 мкл исходных растворов изоцианидов (0,25М) в тетрагидрофуране. Пробирки (пузырьки) закрыли и после встряхивания в течение ночи (17 часов) при 30°С растворитель выпарили и сырые продукты сняли диметилсульфоксидом для анализа. Используя этот протокол, синтезировали 71 соединение.
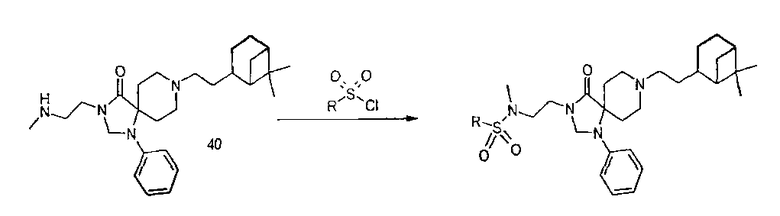
Библиотека сульфонамидов:
Исходные растворы приготовили из основных растворов (0,25М) в тетрагидрофуране и растворов сульфонилхлоридов (0,25М) в тетрагидрофуране. К 200 мкл основного раствора добавили 200 мкл раствора сульфонилхлорида с последующим добавлением 50 мкл 1,0М раствора диизопропилэтиламина (DIPEA) в тетрагидрофуране. Пробирки (пузырьки) закрыли и грели при 30°С в течение 16 часов. Продукты очищали катионообменной твердофазной экстракцией. Растворитель выпарили и сырые продукты сняли диметилсульфоксидом для анализа. Используя протокол, синтезировали 69 соединений.

Библиотека алкилирования:
Исходные растворы приготовили из основных растворов (0,25М) в диметилформамиде и растворов галогенидов (0,25М) в диметилформамиде. К 200 мкл основного раствора добавили 200 мкл раствора галогенида, содержащего 1 эквивалент KI, с последующим добавлением 50 мкл раствора диизопропилэтиламина (1,0 М). Пробирки (пузырьки) закрыли и грели в течение 17 часов. Конкретные модификации: альфагалогенкетоны при 30°С; другие при 60°С. Продукты подвергли очистке катионообменной твердофазной экстракцией. Растворитель выпарили и сырые продукты сняли диметилсульфоксидом для анализа. Используя протокол, синтезировали 61 соединение.

Библиотека Карбаматов:
К основному раствору (200 мкл, 0,25 М) в тетрагидрофуране добавили раствор диизопропилэтиламина (50 мкл, 2М) в тетрагидрофуране с последующим добавлением исходного раствора хлорформата (200 мкл, 0,25М) в тетрагидрофуране. Пробирки (пузырьки) закрыли и встряхивали в течение 24 часов при 30°С. Продукты очищали катионообменной твердофазной экстракцией. Растворитель выпарили и сырые продукты сняли диметилсульфоксидом для анализа. Используя этот протокол, синтезировали 21 соединение.

Библиотека трет-карбамидов (трет-мочевины):
Методика проведения опыта:
Эта методика подчинена реакции карбамоилхлорида и 2×75 вторичных аминов. Все пробирки (пузырьки) и колбы следует сушить при 100°С в вакууме. Все растворители следует сушить (молекулярными ситами в случае CH2Cl2 и K2CO3 в случае CH3CN).
Стадия 1
9,2 ммоля основного раствора растворили в 92 мл дихлорэтана (молекулярные сита 4Å) и получили 0,1 М раствор. К этому раствору добавили 5,68 мл диизопропилэтиламина (3,5 экв.). Смесь охладили до 0°С (ледяная баня) и сразу же добавили раствор 2,728 г (4,6 ммоля) трифосгена в 36,8 мл дихлорэтана. Ледяную баню удалили и смесь перемешивали в течение 30 минут. Реакцию контролировали методом тонкослойной хроматографии и методами жидкостной хроматографии и масс-спектроскопии. Реакционную смесь концентрировали при пониженном давлении в течение 1 часа при 40°С и 20 мбар (2000 Па). Сырой продукт растворили в 36,8 мл CH3CN (осушенный на K2CO3) и добавили 1,92 мл диизопропилэтиламина, получая 0,25 М раствор карбамоилхлорида (B).
Стадия 2
К 200 мкл раствора (0,25М) вторичных аминов в CH3CN добавили 200 мкл раствора (0,25М) карбамоилхлорида (В) в CH3CN с последующим добавлением 1 эквивалента диизопропиламина. Пробирки (пузырьки) закрыли и в течение 17 часов подвергали встряхиванию при 30°С. Реакционную смесь концентрировали, растворили в этилацетате и промыли 5%-ным раствором NaHCO3. Растворитель выпарили и сырой продукт сняли диметилсульфоксидом для анализа. Используя этот протокол синтезировали 49 соединений.
СИНТЕЗ ИНДИВИДУАЛЬНЫХ СОЕДИНЕНИЙ
Синтоны, используемые для получения описанных примеров:
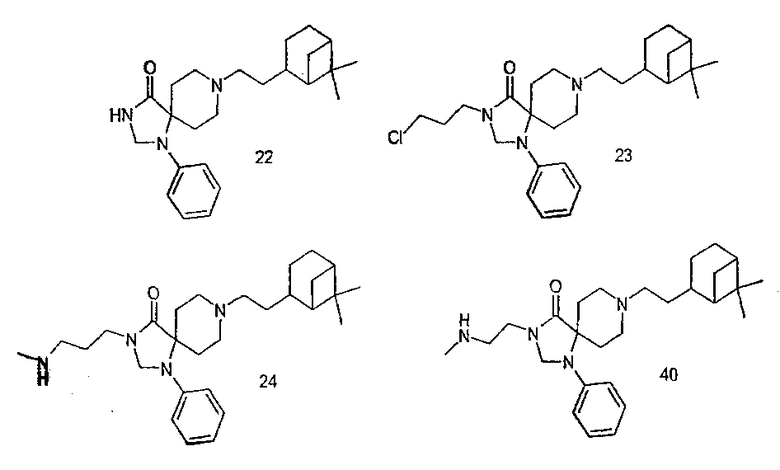
Реакции алкилирования с соединениями (24)/(40): общая методика:
Производное метиламина растворили в тетрагидрофуране и добавили 1,1 эквивалента диизопропилэтиламина. К этой смеси добавили соответствующий алкилирующий реагент (1 эквивалент) и раствор нагрели и кипятили с обратным холодильником, реакцию контролировали методом тонкослойной хроматографии. После полной конверсии раствор концентрировали и твердый остаток сняли водным раствором карбоната натрия. Водный слой экстрагировали несколько раз дихлорметаном. Объединенные слои сушили над сульфатом натрия, концентрировали и сырой продукт очищали колоночной хроматографией (SiO2, этилацетат или CH2Cl2/MeOH в качестве элюентов).
3-{3-[(2,4-Дифтор-бензил)-метил-амино]-пропил}-8-[2-(6,6-диметил-бицикло[3.1.1]гепт-2-ил)-этил]-1-фенил-1,3,8-триаза-спиро[4.5]декан-4-он
выход: 52% [пример №23 в таблицах ниже]
8-[2-(6,6-Диметил-бицикло[3.1.1]гепт-2-ил)-этил]-3-[3-(метил-пиридин-4-илметил-амино)-пропил]-1-фенил-1,3,8-триаза-спиро[4.5]декан-4-он
выход: 32% [пример №225 в таблицах ниже]
8-[2-(6,6-Диметил-бицикло[3.1.1]гепт-2-ил)-этил]-3-[3-(метил-пиридин-3-илметил-амино)-пропил]-1-фенил-1,3,8-триаза-спиро[4.5]декан-4-он
выход: 15% [пример №26 в таблицах ниже]
вариант:
8-[2-(6,6-Диметил-бицикло[3.1.1]гепт-2-ил)-этил]-3-[3-(метил-пиридин-2-ил-амино)-пропил]-1-фенил-1,3,8-триаза-спиро[4.5]декан-4-он
1,7 г производного метиламина (соединение 24) растворили в 4 мл 2-фторпиридина и кипятили с обратным холодильником при 150°С. После полной конверсии реакционную смесь вылили в воду и водный слой экстрагировали этилацетатом несколько раз. Объединенные органические слои сушили над сульфатом натрия, концентрировали в вакууме и очищали колоночной хроматографией (SiO2, этилацетат).
Выход: 60% [пример №83 в таблицах ниже]
Реакции раскрытия эпоксидов с соединением (24)/(40):
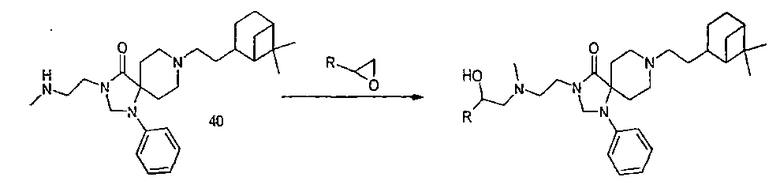
Общая методика:
Производное метиламина растворили в EtOH/H2O (объем/объем=10/1,2 ммоль/мл). После добавления эпоксида (1,5 экв.) смесь нагрели до кипения (с обратным холодильником), реакцию контролировали методом тонкослойной хроматографии. После полной конверсии раствор концентрировали и твердый остаток сняли водным раствором карбоната калия. Водный слой экстрагировали несколько раз этилацетатом. Объединенные органические слои сушили над сульфатом натрия, концентрировали и сырой продукт дополнительно подвергли очистке колоночной хроматографией (SiO2, CH2Cl2/MeOH в качестве элюентов).
3-{3-[(2,3-Дигидрокси-пропил)-метил-амино]-пропил}-8-[2-(6,6-диметил-бицикло[3.1.1]гепт-2-ил)-этил]-1-фенил-1,3,8-триаза-спиро[4.5]декан-4-он выход: 31% [пример №92 в таблицах ниже]
3-{2-[(2,3-Дигидрокси-пропил)-метил-амино]-этил}-8-[2-(6,6-диметил-бицикло[3.1.1]гепт-2-ил)-этил]-1-фенил-1,3,8-триаза-спиро[4.5]декан-4-он выход: 46% [пример №178 в таблицах ниже]
8-[2-(6,6-Диметил-бицикло[3.1.1]гепт-2-ил)-этил]-3-{3-[(2-гидрокси-циклогексил)-метил-амино]-прпил}-1-фенил-1,3,8-триаза-спиро[4,5]декан-4-он выход: 80% [пример №29 в таблицах ниже] (прим. при синтезе в качестве дополнительного основания использовали карбонат калия (2,5 экв.))
8-[2-(6,6-Диметил-бицикло[3.1.1]гепт-2-ил)-этил]-3-{3-[(2-гидрокси-3-морфолин-4-ил-пропил)-метил-амино]-пропил}-1-фенил-1,3,8-триаза-спиро[4.5]декан-4-он выход: 65% [пример №93 в таблицах ниже]
Реакции замещения с соединением (23):
Как правило, реакции замещения проводят в апротонном полярном растворителе (например, в ацетонитриле, диметилсульфоксиде или N-диметилформамиде) следующим образом:
Исходное вещество растворили в соответствующем растворителе. 0,1 экв. йодида натрия и 2 экв. основания (например, карбоната калия или диизопропилэтиламина) поместили в реакционную колбу и затем к этому раствору добавили соответствующий амин (2-4 экв.). Реакционную смесь нагрели и контролировали реакцию методом тонкослойной хроматографии. После стандартной водной обработки реакционной смеси твердый остаток дополнительно очищали колоночной хроматографией (SiO2, CH2Cl2/MeOH в качестве элюентов).

Получение оптически чистых исходных веществ в соответствии с литературными методиками [J.Prakt.Chem.329, 235 (1987)]
2-Метиламино-циклогексанол
Оксид циклогексана (147 г, 1,5 моля) растворили в 8М растворе метиламина в этаноле (750 мл) и перемешивали при 40°С в течение 16 часов. Реакционную смесь концентрировали в вакууме, что дало рацемический 2-метиламиноциклогексанол в виде светло-коричневого масла (195 г, 100%). Согласно данным газовой хроматографии этот продукт был не менее 99% чистоты и его использовали без дополнительной очистки.
Соль (1R,2R)-2-метиламино-циклогексанол-(R)-миндальной кислоты и (1S,2S)-2-метиламино-циклогексанол-(S)-миндальная кислота
Рацемический 2-метиламино-циклогексанол (195 г, макс. 1,5 моля) и (R)-(-)-миндальную кислоту (228 г, 1,5 моля) добавили к 2-пропанолу (1,2 л) и нагрели до температуры кипения (с обратным холодильником). Раствору дали медленно остыть до комнатной температуры и перемешивали всю ночь. Образовавшийся осадок собрали фильтрацией, промыли 2-бутаноном и сушили на воздухе, что дало белое сухое вещество (160 г, энантиомерный избыток (e.e.)=92%). Маточный раствор концентрировали, что дало коричневое масло (275 г), которое затвердевает при стоянии. Белое сухое вещество грели в течение 10 мин при температуре кипения с обратным холодильником в 2-бутаноне (1,7 л). Смеси дали остыть до комнатной температуры при перемешивании и в течение 16 часов перемешивали при комнатной температуре. Осадок собрали фильтрацией и сушили на воздухе, что дало соль (1R,2R)-2-метиламино-циклогексанол-(R)-миндальной кислоты (151,5 г, 538 ммоль, 36%) в виде белого сухого вещества с e.e. 99%.
Первый маточный раствор (275 г, 0,98 моля) добавили к раствору NaOH (200 г, 5 моль) в воде (800 мл) и рассолу (насыщенному раствору NaCl) (800 мл). Добавили дихлорметан (400 мл) и после перемешивания в течение 15 мин слои разделили. Водный слой вновь экстрагировали дихлорметаном (3×400 мл). Объединенные дихлорметаном органические слои сушили (Na2SO4) и концентрировали, что дало коричневое масло (118,5 г, 93,5% выход). Это масло очищали перегонкой Кугельрота (p=0,3 мбар (30 Па), T=70-80°С), что дало (1S, 2S)-2-метиламино-циклогексанол (105 г, 813 ммоль, 83% выход) с e.e. 66%. Этот обогащенный материал растворили в 2-пропаноле (700 мл). Добавили (S)-(+)-миндальную кислоту (124 г, 815 ммоль) и смесь нагрели до температуры кипения (с обратным холодильником). Получающемуся в результате раствору дали медленно остыть до комнатной температуры и при этой температуре перемешивали в течение 16 часов. Образовавшийся осадок собрали фильтрацией, промыли 2-бутаноном и сушили на воздухе, что дало белое сухое вещество (172 г). Эту первую соль нагрели и кипятили с обратным холодильником в течение 15 мин в 2-бутаноне (2,0 л). Смеси (получили непрозрачный раствор) дали медленно остыть до комнатной температуры и перемешивали в течение 16 часов. Осадок собрали фильтрацией и сушили, что дало соль (1S, 2S)-2-метиламино-циклогексанол-(S)-миндальной кислоты (160 г, 569 ммоль, 38%) в виде белого сухого вещества с e.e.>99%.
(1R,2R)-(-)-2-Метиламино-циклогексанол [Для определения взаимосвязи конфигурации и вращения смотри J.Pract.Chem. 329, 235 (1987), и Tetrahedron Asymm., 10, 4619 (1999)]
NaOH (108 г, 2,69 моля) растворили в воде (350 мл). Добавили рассол (400 мл) и охладили до комнатной температуры. Добавили дихлорметан (300 мл) и соль (1R, 2R)-2-метиламино-циклогексанол-(R)-миндальной кислоты (151,5 г, 538 ммоль) и в течение 10 мин смесь тщательно перемешивали. Слои разделили и водный слой вновь экстрагировали дихлорметаном (3×200 мл) (разделение слоев является процедурой, требующей времени). Объединенные дихлорметаном органические слои сушили (Na2SO4) и концентрировали, что дало масло (67,7 г, 97% выход). Это масло объединили с двумя другими порциями: 2,3 г и 6,4 г (обе с e.e.=99+%) и очистили перегонкой Кугельрота (p=0,5 мбар (50 Па), T=80-90°С), что дало бесцветное масло (72,0 г, 92%) с 94% чистотой согласно данным газовой хроматографии. Это масло вновь подвергли перегонке Кугельрота (p=0,3 мбар (30 Па)), что дало продукт, содержащий фракции: Фракция 1; T=40-60°С: 11,6 г с 89% чистотой согласно данным газовой хроматографии, Фракция 2; Т=60-65°С: 60,1 г GI0302-1 в виде бесцветного масла с 99% чистотой и e.e. 99,5%. [α]670=-51,5 (c=0,14, метанол).
(1S,2S)-(+)-2-Метиламино-циклогексанол [Для определения взаимосвязи конфигурации и вращения смотри J.Pract.Chem. 329, 235 (1987), и Tetrahedron Asymm., 10, 4619 (1999)]
NaOH (108 г, 2,69 моля) растворили в воде (400 мл). Добавили рассол (400 мл) и охладили до комнатной температуры. Добавили хлороформ (300 мл) и соль (1S,2S)-2-метиламино-циклогексанол-(S)-миндальной кислоты (160 г, 569 ммоль) и в течение 5 мин смесь тщательно перемешивали. Слои разделили и водный слой вновь экстрагировали хлороформом (3×350 мл). Объединенные хлороформом слои сушили (Na2SO4) и концентрировали, что дало масло (68 г, 93% выход). Это масло очищали перегонкой Кугельрота (p=0,1 мбар (10 Па)), что дало две фракции, содержащие продукт:
Фракция 1: Т=40-55°С: 17,5 г в виде бесцветного масла с 99% чистотой согласно данным газовой хроматографии,
Фракция 2: Т=55-60°С: 48,8 г в виде бесцветного масла с 99,9% чистотой согласно данным газовой хроматографии.
Обе фракции объединили и получили 66,2 г (512 ммоль, 90%) в виде бесцветного масла с e.e. 99,9% [α]670=+53,6 (c=0,14, метанол).
8-[2-(6,6-Диметил-бицикло[3.1.1]гепт-2-ил)-этил]-3-{3R-[(2R-гидрокси-циклогексил)-метил-амино]-пропил}-1-фенил-1,3,8-триаза-спиро[4.5]декан-4-он [пример №30 в таблицах ниже]
2,5 г бициклического хлорсодержащего соединения (23) растворили в 5 мл ацетонитрила. К этому раствору последовательно добавили 1,5 г (2 экв.) карбоната калия, 90 мг (0,1 экв.) йодида натрия, 780 мг (1R,2R)-(-)-2-метиламино-циклогексанола и, в заключение, 3 капли воды. Эту смесь нагрели и кипятили с обратным холодильником в течение 8 часов. Для обработки реакционную смесь разбавили этилацетатом и промыли сначала водной лимонной кислотой и потом водным раствором гидрокарбоната натрия. Органический слой сушили над сульфатом натрия, концентрировали и твердый остаток дополнительно очищали колоночной хроматографией (SiO2, CH2Cl2/MeOH в качестве элюента), что дало 1,75 г (58%) светло-желтого масла.
8-[2-(6,6-Диметил-бицикло[3.1.1]гепт-2-ил)-этил]-3-{3S-[(2S-гидрокси-циклогексил)-метил-амино]-пропил}-1-фенил-1,3,8-триаза-спиро[4.5]декан-4-он [пример №31 в таблицах ниже]
Соединение получали в соответствии с протоколом, приведенным выше для стереоизомера. Выход: 48% в виде светло-желтого масла.
Реакции Манниха с бороновыми кислотами
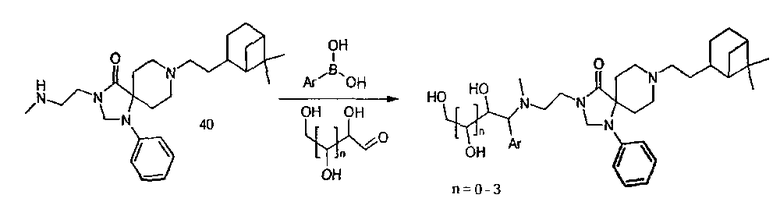
Следующий экспериментальный протокол использовали для синтеза нескольких соединений аналогичным способом, соответственно исходя из соединения (40) и соединения (24).
8-[2-(6,6-Диметил-бицикло[3.1.1]гепт-2-ил)-этил]-3-{3-[(1-фуран-2-ил-2,3,4,5-тетрагидрокси-пентил)-метил-амино]-пропил}-1-фенил-1,3,8-триаза-спиро[4.5]декан-4-он [пример №52 в таблицах ниже]
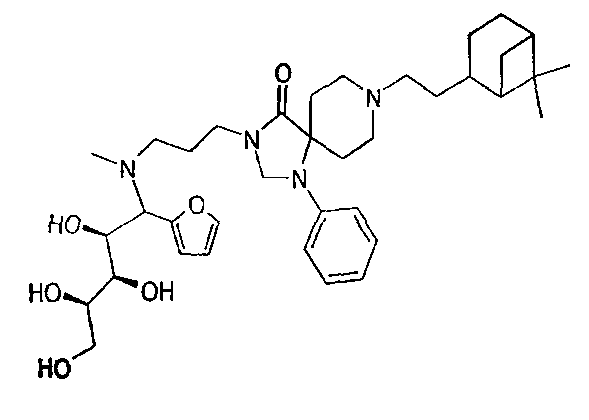
Раствор амина (24) (2,66 г, 5,876 ммоля) и 2-фуранил-борной кислоты (920 мг, 8,22 ммоля) в 30 мл EtOH и 0,5 мл H2O нагрели до 40°С при тщательном перемешивании. К этому раствору маленькими порциями в течение 15 мин при 40°С добавили 1,06 г (7,05 ммоля) D-(+)-ксилозы.
По истечении 2,5 часов реакционную смесь вылили в водный раствор NaHCO3 и водный слой экстрагировали 3 раза дихлорметаном. Объединенные органические слои концентрировали и сняли этилацетатом. Этот органический слой промыли несколько раз водной лимонной кислотой (10%). Объединенные водные слои затем нейтрализовали водным раствором NaHCO3 и нейтральный водный слой экстрагировали CH2Cl2. Объединенные органические экстракты сушили над сульфатом натрия и концентрировали в вакууме, что обеспечило сырое желтое масло, которое очищали колоночной хроматографией (SiO2, этилацетат/метанол: 20/1-10/1). Очистка дала на выходе названное в подзаголовке соединение в виде аморфного белого сухого вещества (1,40 г, 2,14 ммоля, 37%).
Альтернативная методика обработки реакционной смеси: После полной конверсии (контроль методом тонкослойной хроматографии) реакционную смесь охладили до комнатной температуры, добавили 1 мл трифторуксусной кислоты и остающийся раствор перемешивали в течение 10 мин при комнатной температуре. После концентрирования в вакууме проводили дополнительную очистку сырого продукта колоночной хроматографией.
Используя такую же методику, как описана выше, синтезировали следующие образцы:
8-[2-(6,6-Диметил-бицикло[3.1.1]гепт-2-ил)-этил]-3-{3-[метил-(2,3,4,5-тетрагидрокси-1-тиофен-2-ил-пентил)-амино]-пропил}-1-фенил-1,3,8-триаза-спиро[4.5]декан-4-он [пример № 59 в таблицах ниже] выход 80% (D-ксилоза и 2-тиофенил-бороновая кислота в качестве исходных веществ)
8-[2-(6,6-Диметил-бицикло[3.1.1]гепт-2-ил)-этил]-3-{2-[метил-(2,3,4,5-тетрагидрокси-1-тиофен-2-ил-пентил)-амино]-этил}-1-фенил-1,3,8-триаза-спиро[4.5]декан-4-он [пример № 58 в таблицах ниже] выход 13% (D-ксилоза и 2-тиофенил-бороновая кислота в качестве исходных веществ)
8-[2-(6,6-Диметил-бицикло[3.1.1]гепт-2-ил)-этил]-3-{2-[(1-фуран-2-ил-2,3,4,5,6-пентагидрокси-гексил)-метил-амино]-этил}-1-фенил-1,3,8-триаза-спиро[4.5]декан-4-он [пример № 55 в таблицах ниже] выход: 75% (D-глюкоза и 2-фуранил-бороновая кислота в качестве исходных веществ)
8-[2-(6,6-Диметил-бицикло[3.1.1]гепт-2-ил)-этил]-3-{2-[(1-фуран-2-ил-2,3,4,5-тетрагидрокси-пентил)-метил-амино]-этил}-1-фенил-1,3,8-триаза-спиро[4.5]декан-4-он [пример № 53 в таблицах ниже] выход: 42% (D-ксилоза и 2-фуранил-бороновая кислота в качестве исходных веществ)
8-[2-(6,6-Диметил-бицикло[3.1.1]гепт-2-ил)-этил]-3-{2-[(1-фуран-2-ил-2,3,4,5-тетрагидрокси-пентил)-метил-амино]-этил}-1-фенил-1,3,8-триаза-спиро[4.5]декан-4-он [пример № 52 в таблицах ниже] выход: 60% (L-ксилоза и 2-фуранил-бороновая кислота в качестве исходных веществ)
8-[2-(6,6-Диметил-бицикло[3.1.1]гепт-2-ил)-этил]-3-{2-[(1-фуран-2-ил-2,3-дигидрокси-пропил)-метил-амино]-этил}-1-фенил-1,3,8-триаза-спиро[4.5]декан-4-он [пример № 49 в таблицах ниже] выход: 66% (D,L-глицериновый альдегид и 2-фуранил-бороновая кислота в качестве исходных веществ)
3-{2-[(2,3-Дигидрокси-1-тиофен-3-ил-пропил)-метил-амино]-этил}-8-[2-(6,6-диметил-бицикло[3.1.1]гепт-2-ил)-этил]-1-фенил-1,3,8-триаза-спиро[4.5]декан-4-он [пример № 47 в таблицах ниже] выход: 39% (D,L-глицеринальдегид и 3-тиофенил-бороновая кислота в качестве исходных веществ)
8-[2-(6,6-Диметил-бицикло[3.1.1]гепт-2-ил)-этил]-3-{2-[метил-(2,3,4,5-тетрагидрокси-1-тиофен-3-ил-пентил)-амино]-этил}-1-фенил-1,3,8-триаза-спиро[4.5]декан-4-он [пример № 48 в таблицах ниже] выход: 28% (L-арабиноза и 3-тиофенил-бороновая кислота в качестве исходных веществ)
8-[2-(6,6-Диметил-бицикло[3.1.1]гепт-2-ил)-этил]-3-{2-[метил-(2,3,4,5-тетрагидрокси-1-тиофен-3-ил-пентил)-амино]-этил}-1-фенил-1,3,8-триаза-спиро[4.5]декан-4-он [пример № 55 в таблицах ниже] выход: 21% (L-ксилоза и 3-тиофенил-бороновая кислота в качестве исходных веществ)
8-[2-(6,6-Диметил-бицикло[3.1.1]гепт-2-ил)-этил]-3-{2-[метил-(2,3,4,5,6-пентагидрокси-1-тиофен-3-ил-гексил)-амино]-этил}-1-фенил-1,3,8-триаза-спиро[4.5]декан-4-он [пример № 54 в таблицах ниже] выход: 16% (D-глюкоза и 3-тиофенил-бороновая кислота в качестве исходных веществ)
Далее изобретение проиллюстрировано с помощью следующих конкретных примеров (предназначенных только для того, чтобы дополнительно проиллюстрировать изобретение более детально, и, следовательно, так или иначе не рассматриваемых как ограничение объема изобретения), перечисленных в таблице ниже и представленных общими формулами (1) и (2):

Там, где в таблице ниже в колонке под заголовком «стереохимия» приведено название соединения (например, (-)-цис-гидронопол или D-ксилоза), это означает, что соединение по существу использовали в конечной стадии реакции.
роксипиперидин
рокси-пропил
разин-1-ил)-пропил
рокси-пропил
рокси-пропил
ацетил
азол-4-ил)-карбоксил
зол-4-карбоксил
мидил
(1)* R2 = (2-метокси-этоксиметил)
(2)* R2 = 4-гидрокси-пиперидин-4-ил-метил
(3)* R2= 1-бензил-пиперидин-4-ил-метил
(4)* R2 = пиперидин-4-ил-метил
(5)* R2 = 4-бензил-морфолин-2- ил-метил
(6)* R2 = ацетил
(7)* R2 = 3-аминобензил
Аналитические данные примеров из вышеприведенной таблицы приведены в таблице ниже. Подробности методов анализа перечисленных в таблице, а именно: "BASIS", "STANDARD", "CURVE 4", "AMAP 2" и "AMAP3" пояснены ниже.
лярный вес
ления
ния
АНАЛИТИЧЕСКИЕ МЕТОДЫ (ГАЗОВАЯ ХРОМАТОГРАФИЯ-МАСС СПЕКТРОСКОПИЯ)
ОСНОВНОЙ МЕТОД
Градиентный режим: 0,00=изократический (с постоянным составом элюента), 1,00=линейный
(мин)
(мл/мин)
СТАНДАРТНЫЙ МЕТОД
Подвижная фаза для жидкостной хроматографии Waters Alliance 2790
Длина хода поршня дегазатора (OnStroke Length) автоматическая
Колонка для жидкостной хроматографии Waters Alliance 2790
Быстрое уравновешивание в жидкостной хроматографии Waters Alliance 2790
Ввод/вывод (I/O) Waters Alliance 2790
Переключатель (Switch) 1: нет изменений; переключатель 2: нет изменений; переключатель 3: нет изменений; переключатель 4: нет изменений
Установка аналогового выхода (Analog Output Setting): скорость потока
Градиентный режим (программа) жидкостной хроматографии Waters Alliance 2790
Градиентный режим включает 6 точек входа:
(кривая)
Начальная длина волны (нм) 225,00
Конечная длина волны (нм) 260,00
Разрешение (нм) 1,2
Частота выборки (спектр/секунда) 1,000
Характеристика Фильтра 1
Длительность экспозиции (мин) Автоматическая интерполяция 656
Время останова для сбора данных (YesAcquisition) (мин) 10,75
Waters996 усилитель-распределитель импульсов (PDA) Аналоговый канал 1
Электролюминесцентная спектроскопия Фотолюминесценция (ELS PL ELS) 1000 (Темп. 80°C)
МЕТОД CURVE 4
Жидкостная хроматография Waters Alliance 2790
Колонка для жидкостной хроматографии Waters Alliance 2790
Положение колонки Колонка 1 время уравновешивания (мин) 0,00
Температура колонки (°C) 20
Предел температуры колонки (°C) 20
Waters Alliance 2790 Жидкостной Хроматограф (Быстрое уравновешивание )
Поток системного пути за пределами колонки (мл/мин) 0,00
Время системы (мин) 0,00
Время восстановления равновесия (мин) 0,00
Объем форколонки (мкл) 0,00
Ввод/вывод (I/O) Waters Alliance 2790
Переключатель (Switch) 1: нет изменений; переключатель 2: нет изменений; переключатель 3: нет изменений; переключатель 4: нет изменений
Установка аналогового выхода (Analog Output Setting): скорость потока
Градиентный режим (программа) жидкостной хроматографии Waters Alliance 2790
Градиентный режим включает 6 точек входа:
(мл/мин)
Начальная длина волны (нм) 205,00
Конечная длина волны (нм) 350,00
Разрешение (нм) 1,2
Частота выборки (спектр/секунда) 1,000
Характеристика Фильтра 1
Длительность воздействия (мин) Автоматическая интерполяция 656
Время останова для сбора данных (YesAcquisition) (мин) 10,75
Waters996 Усилитель-распределитель импульсов (PDA) Аналоговый канал 1
Электролюминесцентная спектроскопия Фотолюминесценция (ELS PL ELS) 1000 (Темп. 80°C)
МЕТОД AMAP 2
Система для Жидкостной Хроматографии и Масс-спектроскопии состоит из 2 микронасосов (Perkin Elmer серии 200). Насосы присоединены друг к другу Т-образным смесителем объемом 50 мкл. Смеситель присоединен к автоматическому прибору для взятия проб (Gilson 215).
Метод жидкостной хроматографии:
A=100% воды с 0,025% HCOOH and 10 ммоль NH4HCOO pH=±3
B=100% ацетонитрила с 0,025% HCOOH
Автоматический пробоотборник имеет петлю для впрыскивания объемом 2 мкл. Автоматический пробоотборник присоединен к колонке (Phenomenex Luna C18(2) 30·4,6 мм) с частицами размером 3 мкм. Колонку термостатируют при 40°C в термошкафу для колонки (Perkin Elmer серии 200). Колонка присоединена к УФ-фотометру (ABI 785), применяемому для биосистем, с проточной кюветой объемом 2,7 мкл. Длину волны устанавливают 254 нм. УФ-фотометр присоединен к масс-спектрометру (Sciex API 150EX). Масс-спектрометр имеет следующие параметры:
Диапазон сканирования: 150-900 атомных единиц массы
Полярность: положительная
Режим сканирования: профильный (по сечению)
Разрешение Q1: единица
Длина шага: 0,10 атомных единиц массы
Время одного сканирования: 0,500 сек
Распылитель (NEB): 10
Газовая заслонка (CUR): 10
Источник йонов (IS): 5200 вольт
Температура (TEM): 325°C
Дефлектор (DF): 30 вольт
Потенциал фокусировки (FP): 225 вольт
Потенциал на входе (EP): 10 вольт
Детектор светорассеяния присоединен к Sciex API 150. Детектор светорассеяния представляет собой детектор Sedere Sedex 55, функционирующий при 50°C и давлении N2 3 бар (3·105 Па). Вся система контролируется компьютером (Dell optiplex GX400), функционирующим в системе Windows NT.
МЕТОД AMAP 3
Идентичен методу AMAP 2, за исключением метода жидкостной хроматографии (LC), причем последний характеризуется следующими параметрами:
Время
(мкл/мин)
ПРИМЕРЫ ПРИГОТОВЛЕНИЯ КОМПОЗИЦИИ, ВКЛЮЧАЮЩЕЙ СОЕДИНЕНИЕ, ИСПОЛЬЗУЕМОЕ В ИССЛЕДОВАНИЯХ НА ЖИВОТНЫХ
Для орального (per oral) введения желательное количество (вплоть до 20 мкмоль) сухого вещества Примера 1 добавили к 1 мл 1%-ной (вес/объем) метилгидроксиэтил-целлюлозы и 0,1%-ному (вес/объем) полоксамеру (poloxamer) в воде. Соединение суспендировали встряхиванием и перемешиванием в течение 10 минут.
Для подкожного (s.c.) введения желательное количество (вплоть до 15 мкмоль) сухого вещества Примера 1 растворили или суспендировали в 1 мл физиологического раствора.
Фармакологические данные
58 (p.o.)
44 (p.o.)
| название | год | авторы | номер документа |
|---|---|---|---|
| Производные гуанина | 2020 |
|
RU2750867C1 |
| Противогрибковый полусинтетический полиеновый антибиотик, его водорастворимые соли и фармацевтические композиции на их основе | 2018 |
|
RU2688658C1 |
| ИНГИБИТОРЫ MASP-2 И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2019 |
|
RU2832708C2 |
| Способ получения противогрибкового полусинтетического полиенового антибиотика | 2020 |
|
RU2751333C1 |
| КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ЦВИТТЕРИОННЫЕ АЛКИЛАЛКАНОИЛАМИДЫ И/ИЛИ АЛКИЛАЛКАНОАТЫ | 2016 |
|
RU2725999C2 |
| ПИТАТЕЛЬНАЯ КОМПОЗИЦИЯ И ДЕТСКАЯ СМЕСЬ ДЛЯ СТИМУЛЯЦИИ МИЕЛИНИЗАЦИИ ГОЛОВНОГО МОЗГА | 2016 |
|
RU2766208C2 |
| ПИТАТЕЛЬНЫЕ КОМПОЗИЦИИ И ДЕТСКИЕ СМЕСИ, СПОСОБСТВУЮЩИЕ ПЕРВИЧНОЙ МИЕЛИНИЗАЦИИ | 2016 |
|
RU2766014C2 |
| ПИТАТЕЛЬНАЯ КОМПОЗИЦИЯ И ДЕТСКАЯ СМЕСЬ ДЛЯ СТИМУЛИРОВАНИЯ ПЕРВИЧНОЙ МИЕЛИНИЗАЦИИ | 2016 |
|
RU2766598C2 |
| КОМПОЗИЦИИ И ИХ ПРИМЕНЕНИЕ | 2016 |
|
RU2793427C2 |
| ПИТАТЕЛЬНАЯ КОМПОЗИЦИЯ И ДЕТСКАЯ СМЕСЬ ДЛЯ СТИМУЛИРОВАНИЯ МИЕЛИНИЗАЦИИ ГОЛОВНОГО МОЗГА | 2016 |
|
RU2795441C2 |
Изобретение относится к новым соединениям общей формулы (1)

в которой: R1 представляет собой атом водорода, галоген, CF3, (1-3С)алкоксигруппу, m является целым числом от 1 до 4, при условии, что, когда m равно 2, 3 или 4, заместители R1 могут быть или одинаковыми или различными, R2 представляет собой атом водорода, алкильную (1-6С) группу, необязательно замещенную алкоксигруппой, циклоалкильную (3-6С) группу, -СН2ОН, -СН2ОСН3, ацетильную группу, бензильную группу, необязательно замещенную аминогруппой, или группу Q следующей структуры (2):
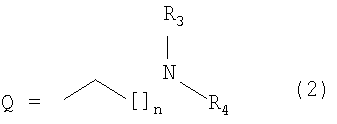
в которой: []n символически изображает -(СН2)n-, где n является целым числом от 0 до 7, R3 представляет собой атом водорода или алкильную (1-3С) группу, R4 представляет собой атом водорода, алкильную (1-6С) группу, необязательно замещенную одной или более группами, выбранными из алкильной группы, арильной группы, фтора, хлора, брома, гидроксильной группы, алкилоксигруппы, арилоксигруппы, ацилоксигруппы, аминогруппы, алкиламиногруппы, диалкиламиногруппы, ариламиногруппы, тиогруппы, алкилтиогруппы, арилтиогруппы, цианогруппы, оксогруппы, нитрогруппы, ацильной группы, амидогруппы, алкиламидогруппы, диалкиламидогруппы, карбоксильной группы, насыщенное, ненасыщенное или частично насыщенное моно- или дициклическое 5-10-членное кольцо, необязательно замещенное одной или более группами, выбранными из алкильной группы, арильной группы, фтора, хлора, брома, гидроксильной группы, алкилоксигруппы, арилоксигруппы, ацилоксигруппы, аминогруппы, алкиламиногруппы, диалкиламиногруппы, ариламиногруппы, тиогруппы, алкилтиогруппы, арилтиогруппы, цианогруппы, оксогруппы, нитрогруппы, ацильной группы, амидогруппы, алкиламидогруппы, диалкиламидогруппы, карбоксильной группы, или алкильную (1-3С) группу, замещенную насыщенным, ненасыщенным или частично насыщенным пяти- или шестичленным кольцом, которое необязательно содержит один или более гетероатомов, таких как атом азота, атом кислорода или атом серы, необязательно замещенным одной или более группами, выбранными из алкильной группы, арильной группы, фтора, хлора, брома, гидроксильной группы, алкилоксигруппы, арилоксигруппы, ацилоксигруппы, аминогруппы, алкиламиногруппы, диалкиламиногруппы, ариламиногруппы, тиогруппы, алкилтиогруппы, арилтиогруппы, цианогруппы, оксогруппы, нитрогруппы, ацильной группы, амидогруппы, алкиламидогруппы, диалкиламидогруппы, карбоксильной группы, или (R3+R4) вместе с атомом азота, к которому они присоединены, представляют собой насыщенное, ненасыщенное или частично насыщенное моно- или дициклическое пяти- или шестичленное кольцо, которое необязательно содержит один или более гетероатомов, таких как атом азота, атом кислорода или атом серы, необязательно замещенным одной или более группами, выбранными из алкильной группы, арильной группы, фтора, хлора, брома, гидроксильной группы, алкилоксигруппы, арилоксигруппы, ацилоксигруппы, аминогруппы, алкиламиногруппы, диалкиламиногруппы, ариламиногруппы, тиогруппы, алкилтиогруппы, арилтиогруппы, цианогруппы, оксогруппы, нитрогруппы, ацильной группы, амидогруппы, алкиламидогруппы, диалкиламидогруппы, карбоксильной группы, и ко всем стереоизомерам, а также к фармакологически приемлемым солям. Изобретение также относится к фармацевтическим композициям и к применению соединений. Технический результат - получение новых биологически активных соединений, проявляющих агонистическую активность к ORL1 рецепторам. 3 н. и 6 з.п. ф-лы, 3 табл.
1. Соединения общей формулы (1)
в которой R1 представляет собой атом водорода, галоген, CF3, (1-3С)алкоксигруппу, m является целым числом от 1 до 4, при условии, что, когда m равно 2, 3 или 4, заместители R1 могут быть или одинаковыми, или различными, R2 представляет собой атом водорода, алкильную (1-6С) группу, необязательно замещенную алкоксигруппой, циклоалкильную (3-6С) группу, -СН2ОН, -СН2ОСН3, ацетильную группу, бензильную группу, необязательно замещенную аминогруппой, или группу Q следующей структуры (2):
в которой []n символически изображает -(CH2)n-, где n является целым числом от 0 до 7,
R3 представляет собой атом водорода или алкильную (1-3С)группу,
R4 представляет собой атом водорода, алкильную (1-6С) группу, необязательно замещенную одной или более группами, выбранными из алкильной группы, арильной группы, фтора, хлора, брома, гидроксильной группы, алкилоксигруппы, арилоксигруппы, ацилоксигруппы, аминогруппы, алкиламиногруппы, диалкиламиногруппы, ариламиногруппы, тиогруппы, алкилтиогруппы, арилтиогруппы, цианогруппы, оксогруппы, нитрогруппы, ацильной группы, амидогруппы, алкиламидогруппы, диалкиламидогруппы, карбоксильной группы, насыщенное, ненасыщенное или частично насыщенное моно- или дициклическое 5-10-членное кольцо, необязательно замещенное одной или более группами, выбранными из алкильной группы, арильной группы, фтора, хлора, брома, гидроксильной группы, алкилоксигруппы, арилоксигруппы, ацилоксигруппы, аминогруппы, алкиламиногруппы, диалкиламиногруппы, ариламиногруппы, тиогруппы, алкилтиогруппы, арилтиогруппы, цианогруппы, оксогруппы, нитрогруппы, ацильной группы, амидогруппы, алкиламидогруппы, диалкиламидогруппы, карбоксильной группы, или алкильную (1-3С) группу, замещенную насыщенным, ненасыщенным или частично насыщенным пяти- или шестичленным кольцом, которое необязательно содержит один или более гетероатомов, таких как атом азота, атом кислорода или атом серы, необязательно замещенным одной или более группами, выбранными из алкильной группы, арильной группы, фтора, хлора, брома, гидроксильной группы, алкилоксигруппы, арилоксигруппы, ацилоксигруппы, аминогруппы, алкиламиногруппы, диалкиламиногруппы, ариламиногруппы, тиогруппы, алкилтиогруппы, арилтиогруппы, цианогруппы, оксогруппы, нитрогруппы, ацильной группы, амидогруппы, алкиламидогруппы, диалкиламидогруппы, карбоксильной группы, или
(R3+R4) вместе с атомом азота, к которому они присоединены, представляют собой насыщенное, ненасыщенное или частично насыщенное моно- или дициклическое пяти- или шестичленное кольцо, которое необязательно содержит один или более гетероатомов, таких как атом азота, атом кислорода или атом серы, необязательно замещенным одной или более группами, выбранными из алкильной группы, арильной группы, фтора, хлора, брома, гидроксильной группы, алкилоксигруппы, арилоксигруппы, ацилоксигруппы, аминогруппы, алкиламиногруппы, диалкиламиногруппы, ариламиногруппы, тиогруппы, алкилтиогруппы, арилтиогруппы, цианогруппы, оксогруппы, нитрогруппы, ацильной группы, амидогруппы, алкиламидогруппы, диалкиламидогруппы, карбоксильной группы, и все стереоизомеры, а также фармакологически приемлемые соли.
2. Соединения по п.1 общей формулы (1), в которой R1 представляет собой атом водорода, галоген, CF3, (1-3С)алкоксигруппу, m=1, и все другие радикалы имеют значения, приведенные в п.1.
3. Соединения по п.2 общей формулы (1), в которой R2 представляет собой группу Q, имеющую общую формулу (2), и все другие радикалы имеют значения, приведенные в п.2.
4. Соединения по п.3 общей формулы (1), в которой R3 представляет собой метильную группу, R4 представляет собой алкильную (1-3С) группу, замещенную насыщенным, необязательно замещенным шестичленным кольцом, которое необязательно содержит один или более гетероатомов, таких как атом азота, атом кислорода, атом серы, и все другие радикалы имеют значения, приведенные в п.3.
5. Соединения по п.4 общей формулы (1), в которой R4 представляет собой метиленовую группу, замещенную необязательно замещенным пиперидиновым кольцом, и все другие радикалы имеют значения, приведенные в п.4.
6. Соединения по любому из пп.1-5, включая все их стереоизомеры, фармацевтически приемлемые соли и пролекарства для применения в медицине в качестве агонистов ORL1 рецептора.
7. Фармацевтические композиции, проявляющие агонистическую активность к ORL1 рецепторам, содержащие фармакологически активное количество, по меньшей мере, одного из соединений, по любому из пп.1-5 в качестве активного ингредиента.
8. Применение соединения по любому из пп.1-5 для получения фармацевтической композиции для лечения состояний острой и хронической боли, нарушений обмена веществ, ожирения; желудочно-кишечных расстройств, воспаления мочевого тракта, почечных расстройств, характеризующихся нарушениями баланса удержания/выделения воды или выделения соли, расстройств сердечно-сосудистой системы, офтальмологических расстройств, расстройств дыхательной системы, заболеваний иммунной системы и вирусных инфекций.
9. Применение по п.8, отличающееся тем, что указанными нарушениями обмена веществ являются нервная анорексия и нейрогенная булимия; что указанными желудочно-кишечными расстройствами являются синдром раздраженной толстой кишки, воспалительное заболевание кишечника (гранулематозный энтерит и язвенный колит), диарея, запор, боль в животе; что указанными расстройствами сердечно-сосудистой системы являются инфаркт миокарда, аритмия, повышенное кровяное давление, тромбоз, анемия, артериосклероз, стенокардия; что указанным офтальмологическим расстройством является глаукома; что указанными расстройствами дыхательной системы являются хроническое обструктивное заболевание легких, бронхит и кистозный фиброз.
| Перекатываемый затвор для водоемов | 1922 |
|
SU2001A1 |
| Способ и приспособление для нагревания хлебопекарных камер | 1923 |
|
SU2003A1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| JP 2000169476 A, 20.06.2000 | |||
| EP 0997464 A, 03.05.2000 | |||
| RU 2002101935 A, 10.08.2003. | |||

