Изобретение относится к соединениями, которые действуют на ноцицептин/ORL-1 рецепторную систему, а также на µ-опиоид рецепторную систему и которые отличаются, в частности, селективной эффективностью для лечения хронической боли (в частности, воспалительной боли, висцеральной боли, опухолевой боли, предпочтительно невропатической боли) без развития в тоже время четко выраженной эффективности в случае острой, ноцицептивной боли. Соединения в соответствии с изобретением представляют собой производные цис-тетрагидро-спиро(циклогексан-1,1'-пиридо[3,4-b]индол)-4-амина.
Хроническая боль может быть разделена на две большие группы. Патофизиологическая ноцицепторная боль запускается после тканевых повреждений путем возбуждения интактных ноцицепторов. Она включает, в частности, хроническую воспалительную боль. Боль, вызываемая механическими, метаболическими или воспалительными повреждениями самих нервов, с другой стороны, обозначается как невропатическая боль. Лечение хронической боли является основной медицинской сложной проблемой, поскольку, несмотря на наличие на рынке некоторых лекарственных средств, чрезвычайно эффективных в случае острой боли, они приводят во многих случаях к неудовлетворительному лечению боли в случае хронической и, в частности, невропатической боли.
Воспалительные процессы относятся к наиболее важным механизмам возникновения боли. Типичная воспалительная боль запускается посредством высвобождения брадикинина, гистамина и простагландинов с подкислением ткани и давлением эксудата на ноцицепторы. В результате этого зачастую происходит сенсибилизирующий эффект на центральную нервную систему, который сам по себе проявляется повышением спонтанной активности нейронов и более сильными ответными реакциями центральных нейронов (Coderre и др., Pain 1993, 52, 259-285). Эти изменения в ответном поведении центральных нейронов могут способствовать спонтанной боли и гипералгезии (повышенной болевой чувствительности на болевые раздражители), которые являются типичными для воспаленной ткани (Yaksh и др., PNAS 1999, 96, 7680-7686).
Показано, что нестероидные противовоспалительные средства (НСПВС), которые также имеют противовоспалительный компонент дополнительно к обезболивающему действию, в частности, эффективны для лечения воспалительной боли (Dickensen, A., International Congress and Symposium Series - Royal Society of Medicine (2000), 246, 47-54). Тем не менее, их применение для длительной терапии хронической боли ограничено, вследствие в отдельных случаях значительных нежелательных эффектов, таких как язвы желудочно-кишечного тракта или токсическое поражение почек. В случае интенсивной вплоть до чрезвычайно интенсивной воспалительной боли (например, в рамках хронического панкреатита), НСПВС, возможно, только незначительно уменьшают боль, но в связи с повышенных риском кровотечения, приводят к риску, который слишком высок. Последующей стадией является в общих случаях лечение с помощью µ-опиоидов, зависимость от наркотических средств широко распространена среди заинтересованных лиц (Vercauteren и др., Acta Anaesthesiologica Belgica 1994, 45, 99-105). Следовательно, существует острая потребность в соединениях, которые чрезвычайно эффективны в случае воспалительной боли и обладают уменьшенными возможностями вызывать зависимость.
Невропатическая боль развивается в тех случаях, когда периферические нервы повреждаются механическим, метаболическим или воспалительным образом. Параметры боли, которая возникает при этом, характеризуются главным образом посредством появления спонтанной боли, гипералгезии и аллодинии (боль, уже вызванная нетоксичными стимулами) (см. Baron, Clin. J. Pain 2000; 16 (2 Suppl), 12-20). Причины и характеристики, и следовательно, также потребности в лечении невропатической боли являются различными и изменяются. Она проявляется в результате повреждения или заболевания головного мозга, спинного мозга или периферических нервов. Возможными причинами являются операции (например, фантомная боль после ампутации), повреждения спинного мозга, удар, рассеянный склероз, злоупотребление алкоголем, или лекарственными средствами, или другими токсическими веществами, злокачественное новообразование и также метаболические заболевания, такие как диабет, подагра, почечная недостаточность или цирроз печени, а также инфекционные заболевания (в частности, опоясывающий герпес, болезнь Пфейфера, эрлихиоз, тиф, дифтерия, ВИЧ, сифилис или боррелиоз). Болевое ощущение имеет чрезвычайно разные признаки и симптомы (например, покалывающая, жгущая, стреляющая, электризующая или иррадиирущая боль), которая со временем может изменяться касательно количества и интенсивности.
Основная фармакологическая терапия невропатической боли включает трициклические антидепресанты и противосудорожные средства, которые используются в качестве монотерапии или также в комбинации с опиоидами. В большинстве случаев, такие лекарственные средства осуществляют только незначительное ослабление боли, в то время как освобождение от боли часто не достигается. Побочные эффекты, которые часто встречаются, не позволяют повысить дозировки лекарственных средств для достижения достаточного облегчения боли. В действительности, для удовлетворительного лечения невропатической боли часто требуется более высокая доза µ-опиоида, чем для лечения острой боли, вследствие этого побочные действия становятся еще более важными. Таким образом, в настоящее время невропатическая боль с трудом поддается лечению. Она облегчается только частично даже с помощью высоких доз опиоидов стадии-3 (Saudi Pharm. J. 2002, 10 (3), 73-85).
Опиоиды, которые используются для лечения невропатической боли, обычно также при этом эффективны по отношению к острой боли. До настоящего времени не представлялось возможным разделить лечение невропатической боли с одной стороны и острой боли с другой стороны. Следовательно, в зависимости от дозы опиоидов, подавляется любая болевая чувствительность, что может быть совершенно неблагоприятно. Острая боль имеет защитную функцию для организма, которая теряется, если чувствительность острой боли нарушена или подавлена. Следовательно, существует потребность в поддержании общей чувствительности боли, и при этом также контролирования невропатической боли.
Из уровня техники известны спироциклические циклогексановые производные, которые действуют на ноцицептин/ORL-1 и на µ-опиоид рецепторную систему. Эти соединения отличаются, в частности, чрезвычайно высокой структурной изменчивостью и пригодны, в частности, для лечения воспалительной и невропатической боли. В этой связи, можно привести ссылку, например, на полный документ WO 2004/043967, WO 2005/063769, WO 2005/066183 и WO 2006/108565.
Существует потребность в лекарственных средствах, которых эффективны для лечения хронической, в частности, невропатической, боли и которые в тоже время влияют в наименьшей возможной степени на ощущение острой боли. По мере возможности, такие лекарственные средства должны содержать такую небольшую дозу активного компонента, чтобы можно было обеспечить удовлетворительную терапию боли без проявления непереносимых побочных действий.
Задачей, лежащей в основе изобретения, является обеспечение новых соединений, которые пригодны в качестве лекарственных средств и обладают преимуществами по сравнению с уровнем техники.
Эта задача решается с помощью объектов формулы изобретения.
Изобретение относится к соединениям общей формулы (I)
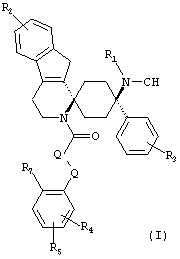
где
R1 представляет собой -H или СН3;
R2 представляет собой -H или -галоген;
R3 представляет собой -H или -галоген;
R4 представляет собой -H, -галоген или -OC1-3-алкил;
R5 представляет собой -H, -галоген или -OC1-3-алкил;
-Q1-Q2- образует группу -CH2- или -CR6=CH-; и
R6 и R7 либо оба одновременно представляют собой -H или вместе посредством мостика -S- образуют пятичленное кольцо;
в форме свободных оснований или физиологически приемлемых солей.
Неожиданно было обнаружено, что соединения в соответствии с изобретением действуют на ноцицептин/ORL-1 и на µ-опиоид рецепторную систему и чрезвычайно эффективны для лечения хронической боли, в частности невропатической боли, без подавления в то же время ощущения острой боли. Кроме того, эти соединения неожиданно проявляют - если это вообще имеет место - только очень незначительные опиоид-типичные побочные действия в болеутоляющем эффективном интервале дозирования.
Соединения в соответствии с изобретением проявляют очень высокую болеутоляющую эффективность для лечения хронической боли, в частности невропатической боли, предпочтительно после поли- или мононевропатических заболеваний.
Неожиданно было обнаружено, что соединения не оказывают влияния на нормальную ноцицепцию у здоровых животных или в здоровой ткани мононевропатических животных в дозах, которые приводят к практически полному устранению невропатической боли на моно- или полиневропатических моделях. Это обозначает, что соединения устраняют патологическое состояние (аллодинию или гипералгезию), но в то же время нарушают нормальную болевую чувствительность только очень незначительно - если это вообще имеет место. Следовательно, антиноцицептивное действие соединений является пренебрежимо малым.
Таким образом, соединения в соответствии с изобретением предоставляют возможность селективной эффективности по отношению к хронической боли, предпочтительно по отношению к невропатической боли, более предпочтительно по отношению к мононевропатической /невралгической или полиневропатической боли, еще более предпочтительно по отношению к боли в случае постгерпетической невралгии или в случае диабетической полиневропатии, предпочтительно с пренебрежимо малой антиноцицептивной эффективностью в случае острой боли. Эти необычные свойства соединений в соответствии с изобретением в совокупности является чрезвычайно важными для лечения боли.
Первый аспект изобретения относится к соединениям общей формулы (I)
где
R1 представляет собой -H или CH3; предпочтительно -CH3;
R2 представляет собой -H или -галоген; предпочтительно -H или -F; особенно предпочтительно -H;
R3 представляет собой -H или -галоген; предпочтительно -галоген; особенно предпочтительно -F;
R4 представляет собой -H, -галоген или -OC1-3-алкил; предпочтительно -H или -OCH3;
R5 представляет собой -H, -галоген или -OC1-3-алкил; предпочтительно -H или -OCH3;
-Q1-Q2- образует группу -CH2- или -CR6=CH-; и
R6 и R7 либо оба одновременно представляют собой -H или вместе посредством мостика -S- образуют пятичленное кольцо;
в форме свободных оснований или физиологически приемлемых солей.
Если -Q1-Q2- образует группу -CH2-, то соединения общей формулы (I) представляют собой амидные производные фенилуксусной кислоты.
Если -Q1-Q2- образует группу -CR6=CH-, то атом углерода той группы, с которой связан радикал R6, связан с атомом углерода карбонильной группы соединения общей формулы (I). В этом случае, R6 может представлять собой -H или ≠ -H. Если R6 представляет собой -H, то R7 также представляет собой -H. Если R6≠-H, то R6 и R7 вместе образуют, с помощью мостика -S-, пятичленное кольцо, таким образом, что тогда соединение общей формулы (I) представляет собой производное бензотиофена.
Соединения в соответствии с изобретением представляют собой выборку из соединений, раскрытых в WO 2004/043967, WO 2005/066183 и WO 2006/108565. Неожиданно было обнаружено, что спироамины в соответствии с изобретением, которые имеют цис конфигурацию в циклогексановом кольце по отношению к двум азотам (производные цис-тетрагидро-спиро(циклогексан-1,1'-пиридо[3,4-b]индол)-4-амина) обладают преимуществами по сравнению с другими гетероциклами.
Таким образом, цис-спироамиды в соответствии с изобретением, в отличие от других соединений в соответствии с WO 2004/043967, WO 2005/066183 и WO 2006/108565, проявляют на животной модели характерное действие по отношению к хронической, предпочтительно невропатической, боли, более предпочтительно боли в случае диабетической полиневропатии, без проявления существенного действия по отношению к острой боли в терапевтической дозе, необходимой для этого. В связи с тем, что различные побочные эффекты общепринятых обезболивающих средств связаны с механизмом действия по отношению к острой боли, то спироциклические цис-замещенные циклогексановые производные в соответствии с изобретением отличаются чрезвычайно благоприятным профилем побочных действий, в частности касательно опиоид-типических побочных действий.
Соединения в соответствии с изобретением предпочтительно являются ахиральными; основная структура общей формулы (I) не содержит хирального элемента (центра, оси или плоскости).
По отношению к спирокольцевой системе, соединения в соответствии с изобретением представляю собой изомеры, в которых схема замещения на спиро-циклогексановой кольцевой системе (не на индоле) также может быть обозначена цис/транс, Z/E или син/анти. "Цис-транс изомеры" представляют собой подгруппу стереоизомеров (конфигурационные изомеры).
В соединениях в соответствии с изобретением, два атома азота спироамина в каждом случае находятся в син-, или цис-, или Z-конфигурации относительно друг друга:
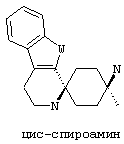
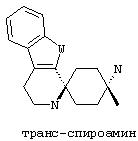
В предпочтительном варианте осуществления, избыток таким образом обозначенного цис-изомера составляет по меньшей мере 50%de, более предпочтительно по меньшей мере 75%de, еще более предпочтительно по меньшей мере 90%de, наиболее предпочтительно по меньшей мере 95%de и в особенности по меньшей мере 99%de.
Подходящие методы разделения изомеров (диастереоизомеров) известны специалисту в данной области техники. Примеры, которые могут быть упомянуты, включают колоночную хроматографию, препаративную ВЭЖХ и процессы кристаллизации. Процессы целенаправленного синтеза, в которых один изомер образуется в избытке, также в принципе известны специалисту в данной области техники.
Преимущества цис-изомера дополнительно в особенности неожиданно состоят в том, что в случае структурно родственных спироэфиров, обычно не цис-изомер, а транс-изомер обладает свойствами, которые являются благоприятными с фармакологической точки зрения (но которые иногда имеют другую природу по сравнению с преимуществами цис-спироаминов в соответствии с изобретением):
В предпочтительном варианте осуществления, соединения в соответствии с изобретением представлены в форме свободных оснований.
В другом предпочтительном варианте осуществления, соединения в соответствии с изобретением представлены в форме физиологически приемлемых солей.
Для целей описания, "соль" подразумевается как любая форма соединения, в которой оно приобретает ионную форму или заряжено и связано с противоионом (катионом или анионом) или находится в растворе. Термин также охватывает комплексы соединения с другими молекулами и ионами, в частности комплексы, которые связаны посредством ионных взаимодействий. Предпочтительные соли являются физиологически приемлемыми, в частности физиологически приемлемые соли с анионами или кислотами или также представляют собой соль, образованную с физиологически приемлемой кислотой.
Физиологически приемлемые соли с анионами или кислотами представляют собой соли данного конкретного соединения с неорганическими или органическими кислотами, которые являются физиологически приемлемыми - в особенности, когда применяются на людях и/или млекопитающих. Примеры физиологически приемлемых солей предпочтительных кислот представляют собой соли: соляной кислоты, бромистоводородной кислоты, серной кислоты, метансульфоновой кислоты, муравьиной кислоты, уксусной кислоты, щавелевой кислоты, янтарной кислоты, яблочной кислоты, винной кислоты, миндальной кислоты, фумаровой кислоты, молочной кислоты, лимонной кислоты, глутаминовой кислоты, сахариновой кислоты, монометилсебациновой кислоты, 5-оксо-пролина, гексан-1-сульфоновой кислоты, никотиновой кислоты, 2-, 3- или 4-аминобензойной кислоты, 2,4,6-триметил-бензойной кислоты, α-липоновой кислоты, ацетилглицина, ацетилсалициловой кислоты, гиппуровой кислоты и/или аспарагиновой кислоты. Особенно предпочтительным является гидрохлорид, цитрат и гемицитрат.
В предпочтительном варианте осуществления, соединение в соответствии с изобретением находится в форме свободного соединения или в форме физиологически приемлемой соли, но предпочтительно не в форме соли бензолсульфоновой кислоты, соли соляной кислоты или соли лимонной кислоты.
Для целей описания, "-галоген" предпочтительно обозначает -F, -Cl, -Br или -I, более предпочтительно -F или -Cl, в частности -F.
Для целей описания, "C1-3-алкил", в каждом случае независимо, является линейным или разветвленным, насыщенным или моно- или полиненасыщенным. Таким образом, "C1-3-алкил" включает ациклические насыщенные или ненасыщенные углеводородные радикалы, которые могут быть разветвленными или неразветвленными, то есть C1-3-алканилы, C1-3-алкенилы и C1-3-алкинилы.
Предпочтительными формами соединений общей формулы (I) являются соединения общей формулы (II), (III) или (IV):
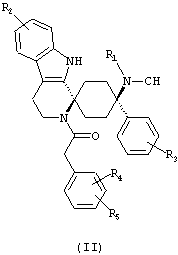
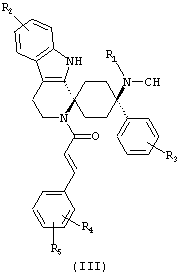
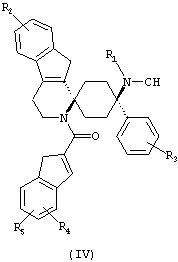
в форме свободных оснований или физиологически приемлемых солей.
Предпочтительно, R2 представляет собой -H и/или R3 представляет собой -F.
Предпочтительно, R4 и R5 либо оба представляют собой -H или оба -OCH3.
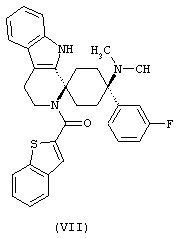
В особенно предпочтительном варианте осуществления, изобретение относится к соединениям, выбранным из группы, включающей соединения общих формул (V), (VI) и (VII)
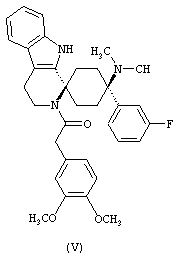
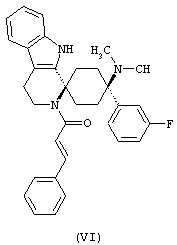
в форме свободных оснований или физиологически приемлемых солей.
Свободное основание соединения общей формулы (V) систематично может быть обозначено как 2',3',4',9'-тетрагидро-N,N-диметил-4-(3-фторфенил)-2'-(3,4-диметоксибензил)карбонил-спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол]-4-амин (цис-диастереоизомер) или также как 2-(3,4-диметоксифенил)-1-((1s,4s)-4-(диметиламино)-4-(3-фторфенил)-3',4'-дигидроспиро[циклогексан-1,1'-пиридо[3,4-b]индол]-2'(9'Н)-ил)этанон. Это соединение предпочтительно находится в форме свободного основания, в форме гидрохлорида, в форме цитрата или в форме гемицитрата.
Свободное основание соединения общей формулы (VI) систематично может быть обозначено как (Е)-2',3',4',9'-тетрагидро-N,N-диметил-4-(3-фторфенил)-2'-(2-фенилвинил)карбонил-спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол]-4-амин (цис-диастереоизомер) или также как (Е)-1-((1s,4s)-4-(диметиламино)-4-(3-фторфенил)-3',4,-дигидроспиро[циклогексан-1,1'-пиридо[3,4-b]индол]-2, (9'Н)-ил)-3-фенилпроп-2-ен-1-он. Это соединение предпочтительно находится в форме свободного основания, в форме гидрохлорида, в форме цитрата или в форме гемицитрата.
Свободное основание соединения общей формулы (VII) систематично может быть обозначено как 2',3',4',9'-тетрагидро-N,N-диметил-4-(3-фторфенил)-2'-(3,4-диметоксибензил)карбонил-спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол]-4-амин (цис-диастереоизомер) или также как бензо[b]тиофен-2-ил((1s,4s)-4-(диметиламино)-4-(3-фторфенил)-3',4,-дигидроспиро[циклогексан-1,1'-пиридо[3,4-b]индол]-2'(9'Н)-ил)метанон. Это соединение предпочтительно находится в форме свободного основания, в форме гидрохлорида, в форме цитрата или в форме гемицитрата.
Соединения, которые являются особенно предпочтительными в соответствии с изобретением, выбирают из группы, включающей
AMD-6цис
и их физиологически приемлемые соли и/или сольваты, в частности свободные основания, гидрохлориды, цитраты или гемицитраты.
Дальнейший аспект изобретения относится к соединениям в соответствии с изобретением в качестве лекарственных средств.
Дальнейший аспект изобретения относится к соединениям в соответствии с изобретением для применения для лечения невропатической и/или хронической боли, введение предпочтительно осуществляют два раза в сутки, один раз в сутки или менее часто, особенно предпочтительно не более чем один раз в сутки.
Изобретение также обеспечивает соединения в соответствии с изобретением для применения для лечения хронической боли. Предпочтительной является хроническая боль, выбранная из группы, включающей воспалительную боль, висцеральную боль, опухолевую боль и невропатическую боль. Невропатическая боль может быть мононевропатического/невралгического или полиневропатического происхождения.
Изобретение также обеспечивает соединения в соответствии с изобретением для применения для лечения боли в случае диабетической полиневропатии.
Изобретение также обеспечивает соединения в соответствии с изобретением для применения для лечения боли вследствие постгерпетической невралгии.
Соединения в соответствии с изобретением пригодны для лечения невропатической боли, предпочтительно мононевропатической/невралгической или полиневропатической боли. Боль представляет собой предпочтительно периферическую полиневропатическую боль или центральную полиневропатическую боль.
Полиневропатия или полиневропатическая боль предпочтительно является острой (вплоть до четырех недель), подострой (от четырех до восьми недель) или хронической (более чем восемь недель).
При полиневропатии, предпочтительно поражается двигательная, чувствительная, вегетативная, сенсомоторная или центральная нервная система. Симптомы предпочтительно распределены симметрично или асимметрично. Боль может быть слабой, умеренной, средней тяжести, тяжелой или очень тяжелой. Для оценки можно использовать шкалу невропатической боли (NPS) (см. B.S. Galer и др., Neurology 1997, 48, 332-8).
Примерами случаев периферической невропатической боли является диабетическая полиневропатия, постгерпетическая невралгия, радикулопатия, посттравматическая невралгия, полиневропатия, индуцированная химическими веществами, например, химиотерапией, фантомная боль конечностей, комплексный региональный синдром, ВИЧ-индуцированная сенсорная полиневропатия и алкогольная полиневропатия. Примерами случаев центральной полиневропатической боли является компрессионная миелопатия в результате стеноза суженных каналов, посттравматическая спинномозговая боль, боль вследствие удара, постишемическая миелопатия, индуцированная радиацией миелопатия, миелопатия, индуцированная рассеянным склерозом, и ВИЧ-индуцированная миелопатия.
В предпочтительном варианте осуществления, невропатия, вызывающая невропатическую боль, связана с заболеванием, выбранным из группы, включающей сахарный диабет, васкулит, уремию, гипотиреоз, злоупотребление алкоголем, постгерпетическую невралгию, идиопатическую невропатию, хроническую воспалительную демиелинизирующую невропатию, многофокальную моторную невропатию, наследственную полиневропатию, синдром Гийена-Барре, интоксикацию [например, алкоголем, тяжелыми металлами {в частности Pb, Hg, As}, углеводородами, вследствие химиотерапии цитостатиками], порфириновую болезнь, инфекционные заболевания, злокачественные новообразования [например, миелома, амилоид, лейкоз, лимфома], злокачественную анемию, недостаточность витамина Е, синдром Рефсума, синдром Бессена - Корнцвейга, болезнь Фабри, васкулит и амилоидоз. Особенно предпочтительными являются диабетическая полиневропатия и постгерпетическая невралгия. Если заболевание представляет собой инфекционное заболевание, то его предпочтительно выбирают из группы, включающей мононуклеоз, эрлихиоз, тиф, дифтерию, проказу, ВИЧ, сифилис и боррелиоз.
Полиневропатическая боль предпочтительно представляет собой боль, вызванную полиневропатией в пределах значений ICD-10 (Международная статистическая классификация болезней и проблем, связанных со здоровьем (International Statistical Classification of Diseases and Related Health Problems)), Редакция ВОЗ, предпочтительно как указано в 2008).
Изобретение также обеспечивает соединения в соответствии с изобретением для применения для лечения состояний тревоги, стресса и синдромов, связанных со стрессом, депрессии, эпилепсии, болезни Альцгеймера, старческой деменции, общих когнитивных дисфункций, нарушений обучения и памяти (в качестве ноотропного), синдромов отмены, злоупотребления и/или зависимости от алкоголя, и/или наркотиков, и/или лекарственных средств, нарушений половых функций, сердечно-сосудистых заболеваний, гипотонии, гипертонии, шума в ушах, зуда, мигрени, тугоухости, недостаточной перистальтики кишечника, ухудшенного потребления пищи, анорексии, ожирения, опорно-двигательных нарушений, диареи, кахексии, недержания мочи, или в качестве миорелаксанта, противосудорожного средства или обезболивающего средства, или для совместного введения в случае лечения с помощью опиоидного анальгезирующего средства или с анестетиком, для диуреза или антинатрийуреза, анксиолизиса, для модуляции опорно-двигательной активности, для модуляции экспрессии нейромедиатора и лечения связанных с этим нейродегенеративных заболеваний, для лечения синдромов отмены и/или для уменьшения возможности вызывания привыкания опиоидами.
Изобретение также обеспечивает способ лечения, в частности одного из вышеуказанных показаний, млекопитающего, отличающегося от человека, или человека, нуждающегося в лечении хронической боли, предпочтительно невропатической боли, более предпочтительно боли в случае диабетической полиневропатии или постгерпетической невралгии, путем введения индивидуально терапевтически необходимой суточной дозы соединения в соответствии с изобретением, или лекарственной формы в соответствии с изобретением, в результате чего вместе с тем предпочтительно не осуществляется существенного подавления чувствительности отстрой ноцицепторной боли и/или не проявляются существенные опиоид-типичные побочные действия, в частности по существу не проявляются дыхательная недостаточность, и/или констипация, и/или задержка мочи, и/или тошнота, и/или рвота, и/или гипотония, и/или брадикардия, и/или привыкание, и/или зависимость, и/или эйфория, и/или депрессия, и/или седативный эффект, и/или головокружение.
Изобретение также обеспечивает способ лечения, в частности одного из вышеуказанных показаний, млекопитающего, отличающегося от человека, или человека, нуждающегося в лечении хронической боли, предпочтительно невропатической боли, более предпочтительно боли в случае диабетической полиневропатии или постгерпетической невралгии, путем введения суточной дозы X соединения в соответствии с изобретением, или лекарственной формы в соответствии с изобретением, в результате чего предпочтительно не осуществляется одновременного подавления чувствительности острой ноцицепторной боли и/или не проявляются существенные опиоид-типичные побочные действия, в частности по существу не проявляются дыхательная недостаточность, и/или констипация, и/или задержка мочи, и/или тошнота, и/или рвота, и/или гипотония, и/или брадикардия, и/или привыкание, и/или зависимость, и/или эйфория, и/или депрессия, и/или седативный эффект, и/или головокружение; где суточную дозу X выбирают из группы, включающей 0,001, 0,002, 0,003, 0,004, 0,005, 0,006, 0,007, 0,008, 0,009, 0,01, 0,02, 0,03, 0,04, 0,05, 0,06, 0,07, 0,08, 0,09, 0,1, 0,2, 0,3, 0,4, 0,5, 0,6, 0,7, 0,8, 0,9, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 мг.
Изобретение также обеспечивает соединения в соответствии с изобретением, имеющие аффинность к µ-опиоидному рецептору и к ORL-1 рецептору, которые
- значительно эффективны для лечения невропатической боли, предпочтительно у крыс, более предпочтительно в виде мононевропатической боли на модели в соответствии с Chung, и характеризуются полумаксимальной эффективной дозой 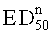
- по существу не являются значительно эффективными для лечения острой боли, предпочтительно у крыс, более предпочтительно в тесте отдергивания хвоста, в дозе, которая больше, чем 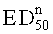
Таким образом, соединения в соответствии с изобретением, при введении в полумаксимальной эффективной дозе
В предпочтительном варианте осуществления, невропатическая боль представляет собой мононевропатическую или невралгическую боль, предпочтительно боль вследствие постгерпетической невралгии. В другом предпочтительном варианте осуществления, боль представляет собой полиневропатическую боль, предпочтительно боли в случае диабетической полиневропатии.
Предпочтительно, соединения в соответствии с изобретением по существу не являются значительно эффективными для лечения острой или ноцицептивной боли даже в дозе, которая больше, чем полумаксимальная эффективная доза
Полумаксимальная эффективная доза
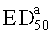
Подходящие методы изучения эффективности активного компонента для лечения невропатической боли и для определения полумаксимальной эффективной дозы
Например, определение можно осуществлять на животной модели (например, мышь или крыса), при помощи:
- мононевропатическую боль можно изучать в соответствии с Chung (S.H. Kim, J.M. Chung, Pain. 1992, 50(3), 355-63) или Bennett (G.J. Bennett, Y.K. Xie, Pain. 1988, 33(1), 87-107),
- боль в случае диабетической полиневропатии можно изучать с помощью стрептозотоцин (STZ)-индуцированного диабета (Е.К. Joseph, J.D. Levine, Neuroscience. 2003; 120(4):907-13), и
- острую боль можно изучать с помощью так называемого теста отдергивания хвоста (D'Amour и Smith, J. Pharm. Exp. Ther. 72, 1941, 74-9).
Определение предпочтительно осуществляют на животной модели, касательно эффективности по отношению к невропатической боли как эффективность по отношению к мононевропатической боли у крыс на модели в соответствии с Chung, и касательно эффективности по отношению к острой боли у крыс в тесте отдергивания хвоста, предпочтительно в каждом случае, как описано в экспериментальном разделе.
Таким образом, соединения в соответствии с изобретением предпочтительно имеют аффинность к µ-опиоидному рецептору и к ORL-1 рецептору, которые, у крыс,
- значительно эффективны для лечения мононевропатической боли на модели в соответствии с Chung и характеризуются с помощью полумаксимальной эффективной дозы
- по существу не являются значительно эффективными для лечения острой боли в тесте отдергивания хвоста в дозе, которая больше, чем
Оценку экспериментальных данных относительно статистически значимого различия между группами, получавшими дозу соединения, и контрольными группами, получавшими наполнитель, предпочтительно осуществляли с помощью дисперсионного анализа с повторными измерениями (повторные измерения ANOVA) и анализа post hoc (процедура множественных сравнений) в соответствии с Bonferroni, предпочтительно как описано в экспериментальном разделе. Уровень достоверности устанавливали на р<0,05. Размер групп обычно составлял n=10.
В принципе, сравнительное определение анальгезирующей эффективности по отношению к невропатической боли и острой, ноцицептивной боли, также можно осуществлять на людях, но это является менее предпочтительным, в частности, по этическим соображениям. В таком случае, исследование эффективности по отношению к невропатической боли, то есть у пациентов, страдающих от невропатической боли, может быть осуществлено в соответствии с Hansson Р, Backonja М, Bouhassira D. (2007). Usefulness and limitations of quantitative sensory testing: clinical and research application in neuropathic pain states. Pain. 129(3): 256-9. В таком случае исследование эффективности по отношению к острой боли можно осуществить в соответствии с Posner J, Telekes A, Crowley D, Phillipson R, Peck AW. (1985). Effects of an opiate on cold-induced pain and the CNS in healthy volunteers. Pain. 23(1):73-82.
Неожиданно было обнаружено, что соединения в соответствии с изобретением отличаются чрезвычайно благоприятным профилем побочных действий по сравнению с общепринятыми опиоидами стадии-3. Таким образом, даже при введении терапевтически эффективных доз, что является необходимым, в частности, для лечения невропатической боли, не наблюдается или имеют место главным образом только незначительно выраженные опиоид-типичные побочные действия, такие как, например, дыхательная недостаточность, констипация, задержка мочи, тошнота, рвота, гипотония, брадикардия, привыкание, зависимость, эйфория, депрессия, седативный эффект и головокружение. До настоящего времени, значительно уменьшенное проявление опиоид-типичных побочных действий дыхательная недостаточность, констипация, гипотония, брадикардия, нарушение способности координации движения (в качестве критерия побочных эффектов на центральную нервную систему), физическая и психическая зависимость были показаны экспериментально на животных моделях.
В предпочтительном варианте осуществления, соединения в соответствии с изобретением, при введении в полумаксимальной эффективной дозе
Подходящие методы для изучения индуцированной активным компонентом дыхательной недостаточности известны специалисту в данной области техники. Исследование предпочтительно осуществляют на модели определения газов крови у крыс в виде изменения артериального парциального давления O2 и CO2. Оценку экспериментальных данных относительно статистически значимого различия между группами, получавшими дозу соединения, и контрольными группами, получавшими наполнитель, предпочтительно осуществляли с помощью однофакторного дисперсионного анализа (однофакторный ANOVA), а также анализа post hoc в соответствии с Dunnett, предпочтительно как описано в экспериментальном разделе. Уровень достоверности устанавливали на p<0,05. Размер групп обычно составлял n=6. Для более подробного описания этой модели на животных, также ссылаемся на экспериментальный раздел.
В предпочтительном варианте осуществления, соединения в соответствии с изобретением, при введении в полумаксимальной эффективной дозе
Подходящие методы для изучения индуцированной активным компонентом констипации известны специалисту в данной области техники. Исследование предпочтительно осуществляют на модели прохождения активированного угля у мышей в качестве изменения скорости транзита пищи по желудочно-кишечному тракту. Оценку экспериментальных данных относительно статистически значимого различия между группами, получавшими дозу соединения, и контрольными группами, получавшими наполнитель, предпочтительно осуществляли с помощью однофакторного дисперсионного анализа (однофакторный ANOVA), а также анализа post hoc в соответствии с Dunnett, предпочтительно как описано в экспериментальном разделе. Уровень достоверности устанавливали на р<0,05. Размер групп обычно составлял n=10. Для более подробного описания этой модели на животных, также ссылаемся на экспериментальный раздел.
В предпочтительном варианте осуществления, соединения в соответствии с изобретением, при введении в полумаксимальной эффективной дозе
Подходящие методы для изучения индуцированной активным компонентом гипотонии известны специалисту в данной области техники. Исследование предпочтительно осуществляют на циркуляторной модели у бодрствующих кроликов с телеметрией в качестве изменения артериального давления (систолическое, диастолическое и среднее значение). Оценку экспериментальных данных относительно статистически значимого различия между группами, получавшими дозу соединения, и контрольными группами, получавшими наполнитель, предпочтительно осуществляли с помощью однофакторного дисперсионного анализа (однофакторный ANOVA), а также анализа post hoc в соответствии с Dunnett, предпочтительно как описано в экспериментальном разделе. Уровень достоверности устанавливали на р<0,05. Размер групп обычно составлял n=6. Для более подробного описания этой модели на животных, также ссылаемся на экспериментальный раздел.
В предпочтительном варианте осуществления, соединения в соответствии с изобретением, при введении в полумаксимальной эффективной дозе
Подходящие методы для изучения индуцированной активным компонентом брадикардии известны специалисту в данной области техники. Исследование предпочтительно осуществляют на циркуляторной модели у бодрствующих кроликов с телеметрией в качестве изменения частоты пульса. Оценку экспериментальных данных относительно статистически значимого различия между группами, получавшими дозу соединения, и контрольными группами, получавшими наполнитель, предпочтительно осуществляли с помощью однофакторного дисперсионного анализа (однофакторный ANOVA), а также анализа post hoc в соответствии с Dunnett, предпочтительно как описано в экспериментальном разделе. Уровень достоверности устанавливали на р<0,05. Размер групп обычно составлял n=6. Для более подробного описания этой модели на животных, также ссылаемся на экспериментальный раздел.
В предпочтительном варианте осуществления, соединения в соответствии с изобретением, при введении в полумаксимальной эффективной дозе
Подходящие методы для изучения индуцированной активным компонентом нарушение способности координации движения известны специалисту в данной области техники. Исследование предпочтительно осуществляют на модели RotaRod у мышей (в соответствии с Kuribara Н., Higuchi Y., Tadokoro S. (1977), Effects of central depressants on Rota-Rod and traction performance in mice. Japan. J. Pharmacol. 27, 117-126) в качестве изменения способности бегать на вращающемся барабане. Оценку экспериментальных данных относительно статистически значимого различия между группами, получавшими дозу соединения, и контрольными группами, получавшими наполнитель, предпочтительно осуществляли с помощью однофакторного дисперсионного анализа (однофакторный ANOVA), а также анализа post hoc в соответствии с Dunnett, предпочтительно как описано в экспериментальном разделе. Уровень достоверности устанавливали на р<0,05. Размер групп обычно составлял n=10. Для более подробного описания этой модели на животных, также ссылаемся на экспериментальный раздел.
В предпочтительном варианте осуществления, соединения в соответствии с изобретением, при введении в полумаксимальной эффективной дозе
Подходящие методы для изучения индуцированной активным компонентом физической зависимости известны специалисту в данной области техники. Исследование предпочтительно осуществляют на модели прыгания у мышей (аналогично Saelens JK, Arch Int Pharmacodyn 190: 213-218, 1971) в качестве индуцированной налоксоном абстиненции. Оценку экспериментальных данных относительно статистически значимого различия между группами, получавшими дозу соединения, и контрольными группами, получавшими наполнитель, предпочтительно осуществляли с помощью точного критерий Фишера для параметра "количество животных с абстинентным синдромом", а также с помощью критерия Крускала-Уоллиса для параметра "частота прыжков", предпочтительно как описано в экспериментальном разделе. Уровень достоверности устанавливали на р<0,05 в каждом случае. Размер групп обычно составлял n=12. Для более подробного описания этой модели на животных, также ссылаемся на экспериментальный раздел.
В предпочтительном варианте осуществления, соединения в соответствии с изобретением, при введении в полумаксимальной эффективной дозе
Подходящие методы для изучения индуцированной активным компонентом психической зависимости или привыкания известны специалисту в данной области техники. Исследование предпочтительно осуществляют с помощью предпочтения мест введения психоактивного вещества у крыс, предпочтительно как описано в Tzschentke, Т.М., Bruckmann, W. и Friderichs, F. (2002) Lack of sensitization during place conditioning у крыс is consistent with the low abuse potential of tramadol. Neuroscience Letters 329, 25-28. Оценку экспериментальных данных относительно статистически значимого различия в предпочтениях животных для активного компонента или наполнителя предпочтительно осуществляли с помощью парного t-критерия. Уровень достоверности устанавливали на р<0,05. Размер групп обычно составлял n=8. Для более подробного описания этой модели на животных, ссылаемся на описание метода в Tzschentke, Т.М., Bruckmann, W. и Friderichs, F. (2002) Neuroscience Letters 329, 25-28.
Соединения в соответствии с изобретением пригодны для лечения хронической боли, предпочтительно невропатической боли, более предпочтительно мононевропатической/невралгической или полиневропатической боли, еще более предпочтительно боли в случае постгерпетической невралгии или в случае диабетической полиневропатии.
Определения различных форм хронической боли известны специалисту в данной области техники. В этом связи можно привести ссылку, например, на Merskey Н., Bogduk N. Classification of chronic pain. Seattle: IASP Press 1994, Bennett G.J., Anesth Analg. 2003, 97, 619-20 и Backonja M.M., Anesth Analg. 2003, 97, 785-90.
Для целей описания, хроническая боль предпочтительно определяется как боль, которая существует в течение продолжительного периода (обычно по меньшей мере 3, 4, 5 или 6 месяцев) и продолжает существовать за пределами нормального времени излечения. Невропатическая боль предпочтительно определяется как боль или чувствительный феномен, которая вызывается поражением, заболеванием или дисфункцией центральной или периферической нервной системы. Для целей описания, острая боль предпочтительно определяется как неприятное чувствительное и эмоциональное восприятие, которое сопровождает острое или потенциальное повреждение ткани или описано в контексте такого повреждения (см. определения Международной ассоциации по изучению боли (International Association for the Study of Pain®) (IASP)).
Соединения в соответствии с изобретением имеют Ki значение на µ-опиоид рецепторе предпочтительно не более чем 1000 нМ, более предпочтительно не более чем 500 нМ, еще более предпочтительно 100 нМ, наиболее предпочтительно не более чем 50 нМ и, в особенности, не более чем 25 нМ.
Способы определения Ki значения на µ-опиоид рецепторе известны специалисту в данной области техники. Определение предпочтительно осуществляют в гомогенной партии в микротитровальных планшетах. Для этого, серийные разведения тестируемых соединений предпочтительно инкубировали в течение 90 минут при комнатной температуре с препаратом рецепторной мембраны (15-40 мкг белка на 250 мкл инкубируемой партии) СНО-K1 клеток, которые экспрессируют µ-опиат рецептора человека (RB-HOM препарат рецепторной мембраны от NEN, Zaventem, Belgium) в присутствии 1 нмоль/л радиоактивного лиганда [3H]-налоксон (NET719, NEN, Zaventem, Belgium) и 1 мг WGA-SPA шариков (SPA шарики агглютинина из проростков пшеницы от Amersham/Pharmacia, Freiburg, Germany), в общем объеме 250 мкл. Предпочтительно использовали в качестве инкубационного буфера 50 ммоль/л Трис-HCl, дополненной 0,05 мас.% азида натрия и 0,06 мас.% бычьего сывороточного альбумина. Для определения неспецифического связывания, дополнительно предпочтительно добавляли 25 мкмоль/л налоксона. После завершения 90-то минутного времени инкубирования, микротитровальные планшета предпочтительно центрифугировали в течение 20 минут при 1000 g и радиоактивность измеряли в р счетчике (Microbeta-Trilux, PerkinElmer Wallac, Freiburg, Germany). Определяли процент вытеснения радиоактивного лиганда из его связывания с µ-опиат рецептором человека при концентрации тестируемых веществ предпочтительно 1 мкмоль/л и выражали в виде процента ингибирования (% ингибирования) специфического связывания. На основе процента вытеснения различными концентрациями тестируемых соединений представляется возможным рассчитать ингибирующие концентрации IC50, которые вызывают 50% вытеснение радиоактивного лиганда. Их этого Ki значения для тестируемых веществ могут быть рассчитаны с помощью превращения согласно уравнению Ченга-Прусоффа.
Соединения в соответствии с изобретением имеют Ki значения для ORL1 рецептора предпочтительно не более чем 500 нМ, более предпочтительно не более чем 100 нМ, наиболее предпочтительно не более чем 50 нМ и, в особенности, не более чем 10 нМ.
Способы определения Ki значения для ORL1 рецептора известны специалисту в данной области техники. Определение предпочтительно осуществляют в анализе связывания с рецептором 3Н-ноцицептин/орфанин FQ с мембранами рекомбинантных CHO-ORL1 клеток. Эту систему тестирования предпочтительно осуществляют в соответствии с методом, предложенным Ardati и др. (Mol. Pharmacol., 51, 1997, р.816-824). Концентрация 3H-ноцицептин/орфанин FQ в этих тестах предпочтительно составляет 0,5 нМ. Исследования связывания предпочтительно осуществляют в каждом случае с 20 мкг мембранного белка на партию 200 мкл в 50 мМ Hepes, pH 7,4, 10 мМ MgCl2 и 1 мМ EDTA. Связывание с ORL1 рецептором предпочтительно определяют, используя в каждом случае 1 мг шариков WGA-SPA (Amersham-Pharmacia, Freiburg) путем инкубирования партии в течение одного часа при КТ и последующего измерения на сцинтилляционном счетчике Trilux (Wallac, Finland).
Изобретение также обеспечивает способ получения соединений в соответствии с изобретением. Подходящие способы синтеза соединений в соответствии с изобретением известны в принципе специалисту в данной области техники.
Предпочтительные пути синтеза описаны ниже:
Синтез кетоновых структурных единиц Е:
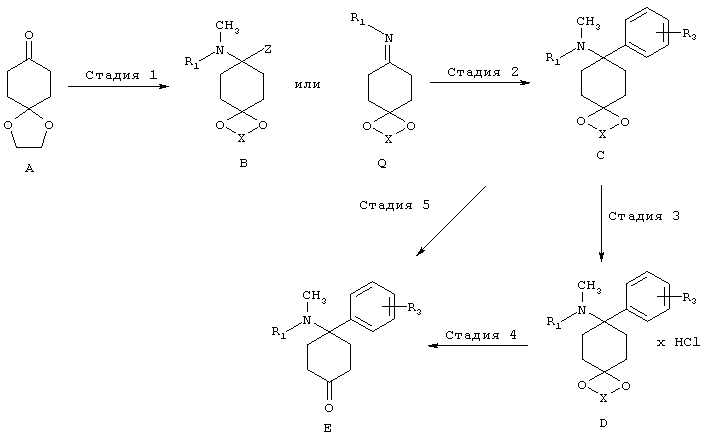
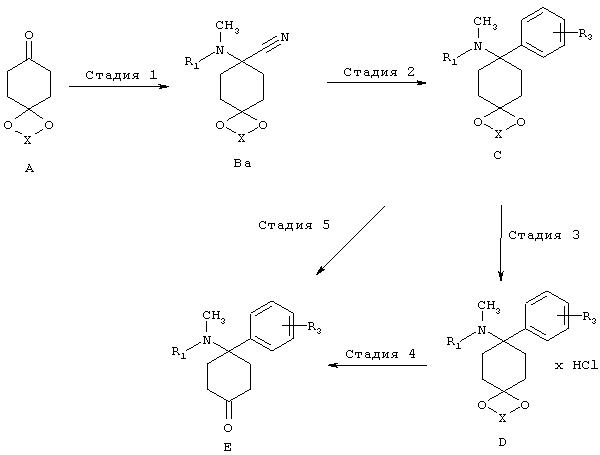
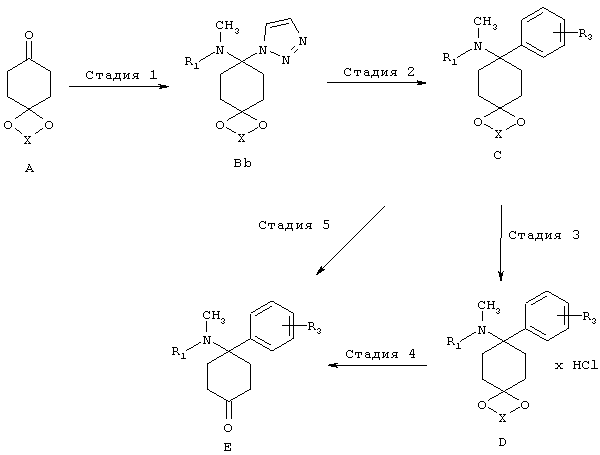
Стадия 1 (посредством B)
Структуры формулы В могут быть приготовлены путем реакции кетонов А с аминами и кислотными реагентами Z-H. Подходящие реагенты Z-H представляют собой, например, цианистый водород, 1,2,3-триазол, бензотриазол или пиразол. Особенно предпочтительным путем получения соединений структуры В является реакция кетонов с цианидами металлов и соответствующим амином в присутствии кислоты, предпочтительно в спирте, при температуре от -40 до 60°C, предпочтительно при комнатной температуре с цианидами щелочных металлов в метаноле. Дальнейшим особенно предпочтительным путем получения соединений структуры В является реакция кетонов с 1,2,3-триазолом и соответствующим амином в присутствии в условиях отвода воды, предпочтительно, используя отделитель воды, при повышенной температуре в инертном растворителе или используя молекулярное сито или другой осушающий реагент. Аналогичным образом, могут быть введены структуры, аналогичные В, используя бензотриазольные или пиразольные группы вместо триазольных групп.
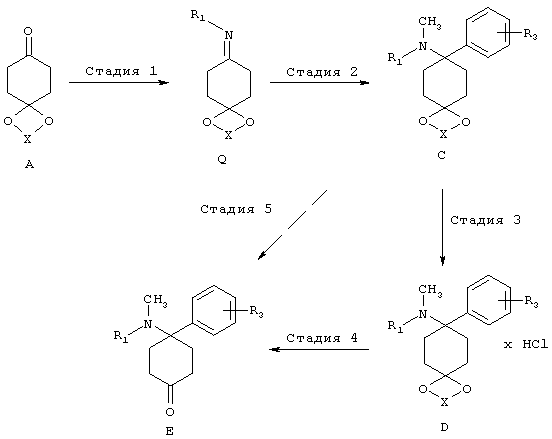
Стадия 1 (посредством Q)
Получение иминов общей формулы Q из кетонов А можно найти в общем уровнем техники.
Стадия 2 (посредством В)
В целом, ацетали С могут быть получены путем замещение подходящих уходящих групп Z в структурах формулы В. Подходящие уходящие группы представляют собой предпочтительно циано группы; 1,2,3-триазол-1-ильные группы. Дальнейшие уходящие группы представляют собой 1Н-бензо[d][1,2,3]триазол-1-ильные группы и пиразол-1-ильные группы (Katritzky и др., Synthesis 1989, 66-69). Особенно предпочтительным путем получения соединений структуры С является реакция аминонитрилов В (Z=CN) с соответствующими металлоорганическими соединениями, предпочтительно соединениями Гриньяра, предпочтительно в простых эфирах, предпочтительно при КТ. Металлоорганические соединения либо являются коммерчески доступными или могут быть получены в соответствии с общим уровнем техники.
Дальнейшим особенно предпочтительным путем получения соединений структуры С является реакция аминотриазолов В (Z = триазол) с соответствующими металлоорганическими соединениями, предпочтительно Соединения Гриньяра, предпочтительно в простых эфирах, предпочтительно при КТ. Металлоорганические соединения либо являются коммерчески доступными или могут быть получены в соответствии с общим уровнем техники.
Стадия 2 (посредством Q)
Аминоацетали C, имеющие не более одного заместителя на атоме азота, могут быть получены в соответствии с процессами, в принципе известными специалисту в данной области техники, путем добавления углеродных нуклеофилов к иминами Q, предпочтительно металлоорганических соединений в инертных растворителях, особенно предпочтительно с реагентами Гриньяра или литийорганических соединений, предпочтительно в простых эфирах, предпочтительно при температуре от 100 до КТ.
Стадия 4/5:
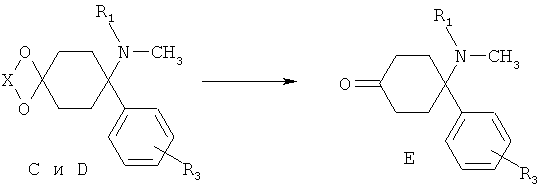
Соединения формулы Е могут быть выделены в свободной форме из соответствующих ацеталей С, или из их солей D, в соответствии с хорошо известным уровнем техники путем снятия защиты с помощью кислот. X выбирают из группы алкил, алкил/алкилиден/алкилиден, замещенный арилом или алкилом (насыщенный/ненасыщенный).
Получение С (R1≠-H) из Ca(R1=-H)
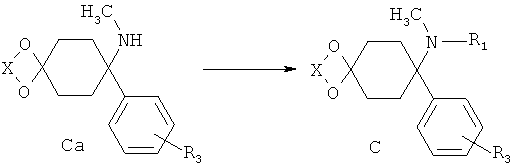
Аминоацетали Са, имеющие не более одного заместителя на атоме азота, могут быть превращены в соответствии с процессами, известными в принципе специалисту в данной области техники, например, путем восстановительного аминирования, в соответствующие аминоацетали С, имеющие один или два дополнительных заместителя на азоте.
Аминонитрильный путь, иминовый путь и триазольный путь
Необходимые кетоновые промежуточные соединения Е могут быть получены, например, в соответствии со следующими тремя различными путями: (1) аминонитрильный путь, (2) иминовый путь и (3) триазольный путь.
(1) Аминонитрильный путь:
В аминонитрильном пути синтезируют, как описано на последующей схеме синтеза, из кетонового предшественника А аминонитрил Ba, который превращают в структурные элементы С или D и дополнительно в Е, используя нуклеофил MR3. Этот путь синтеза уже был описан и использован в WO 2004/043967.
(2) Иминовый путь:
В иминовом пути синтезируют, как описано на последующей схеме, из кетонового предшественника А имин Q, который превращают в структурные элементы С и D и дополнительно в Е, используя нуклеофил MR3. Необходимые иминовые структурные элементы Q могут быть получены в соответствии с методом, известным специалисту в данной области техники (Layer, Chem. Rev., 1963, 8, 489-510). Для присоединения металлоорганических компонентов MR3 к имину Q используют процессы, известные из литературы (например, Maddox и др., J. Med. Chem., 1965, 8, 230-235. Kudzma и др., J. Med. Chem., 1989, 32, 2534-2542). Стадии 3, 4 и 5 осуществляют аналогично аминонитрильному пути.
(3) Триазольный путь:
В триазольном пути синтезируют, как описано на последующей схеме, из кетонового предшественника A триазол Bb, который превращают в структурные элементы C и D и дополнительно в E, используя нуклеофил MR3. Условия можно найти в указанных литературных ссылках: (a) Katritzky и др. Synthesis, 1992, 1295-1298. (b) Prashad, и др., Tetrahedron Lett. 2005, 46, 5455-5458.
Синтез спироаминов (AMN)
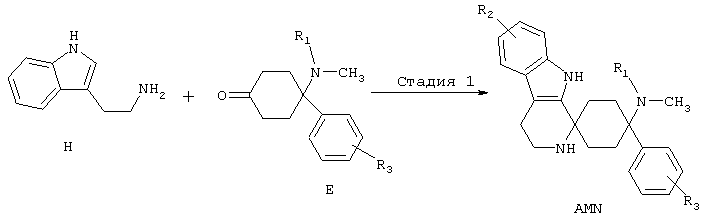
Триптамины типа Н могут реагировать в реакции тира Пиктета-Шпенглера с кетонами Е, с добавлением по меньшей мере реагента из группы кислот, кислотных ангидридов, сложных эфиров, солей, реагирующих со слабой кислотой или кислот Льюиса, с образованием продуктов формулы AMN.
Предпочтительно используют по меньшей мере один реагент из группы карбоновые кислоты, фосфорные кислоты или сульфоновые кислоты или их ангидриды, триалкилсилиловые эфиры карбоновых кислот, соли, реагирующие с кислотами, минеральные кислоты или кислоты Льюиса, выбранные из группы, включающей трифторид бора, хлорид индия (III), тетрахлорид титана, хлорид алюминия (III), или с добавлением по меньшей мере одной соли переходного металла, предпочтительно с добавлением по меньшей мере одного трифлата переходного металла (трифторметансульфоната переходного металла), особенно предпочтительно с добавлением по меньшей мере одного трифторметансульфоната переходного металла, выбиранного из группы, включающей трифторметансульфонат скандия (III), трифторметансульфонат иттербия (III) и трифторметансульфоната индия (III), необязательно с добавлением целита, со связанными с твердой фазой реактансами или реагентами, при повышенной или пониженной температуре, с или без микроволнового излучения, необязательно в подходящем растворителе или смеси растворителей, таких как, например, хлорированные или нехлорированные, в таком случае предпочтительно ароматические, углеводороды, ацетонитрил; в эфирных растворителях, предпочтительно в диэтиловом эфире или ТГФ; или в нитрометане, в подходящих случаях также в спиртах или воде. Особенно предпочтительным является применение пара-толуолсульфонат пиридиния, фосфорного ангидрида в присутствии целита, эфират трифторида бора, трифторуксусной кислоты, тетраизопропилового эфира ортотитановой кислотой совместно с трифторуксусной кислотой, трифторметансульфоновой кислоты триметилсилилового эфира, трифторметансульфоновой кислоты, метансульфоновой кислоты, трифторуксусной кислоты, уксусной кислоты, фосфорной кислоты, полифосфорной кислоты, полифосфатных эфиров, п-толуолсульфоновой кислоты, соляной кислоты HCl газ, серной кислоты совместно с ацетатным буфером, тетрахлорида олова.
Условия, указанные в последующих примерах, в свою очередь, предпочтительно используют.
Соединения общих формул Н и Е либо являются коммерчески доступными или их получение известно из уровня техники или может быть выведено из уровня техники с помощью способа, очевидного для специалиста в данной области техники. В этой связи особенно релевантными являются следующие ссылки: Jirkovsky и др., J. Heterocycl. Chem., 12, 1975, 937-940; Beck и др., J. Chem. Soc. Perkin 1, 1992, 813-822; Shinada и др., Tetrahedron Lett., 39, 1996, 7099-7102; Garden и др., Tetrahedron, 58, 2002, 8399-8412; Lednicer и др., J. Med. Chem., 23, 1980, 424-430; Bandini и др. J. Org. Chem. 67, 15; 2002, 5386-5389; Davis и др., J.Med.Chem. 35, 1, 1992, 177-184; Yamagishi и др., J.Med.Chem. 35, 11, 1992, 2085-2094; Gleave и др.; Bioorg.Med.Chem.Lett. 8, 10, 1998, 1231-1236; Sandmeyer, Helv.Chim.Acta; 2; 1919; 239; Katz и др.; J. Med. Chem. 31, 6, 1988; 1244-1250; Вас и др. Tetrahedron Lett. 1988, 29, 2819; Ma и др. J. Org. Chem. 2001, 66, 4525; Kato и др. J. Fluorine Chem. 99, 1, 1999, 5-8.
Синтез спироамидов (AMD)
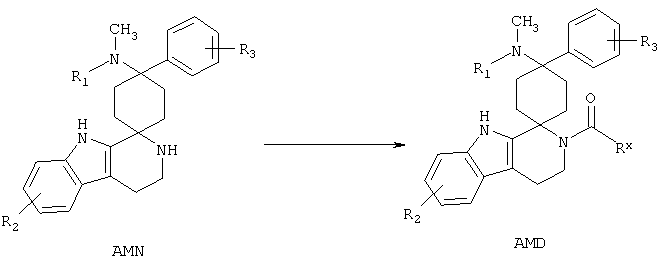
Соединения общей формулы AMN могут взаимодействовать с карбоновыми кислотами в по меньшей мере одном растворителе, предпочтительно выбранном из группы, включающей дихлорметан, ацетонитрил, диметилформамид, диэтиловый эфир, диоксан и тетрагидрофуран, с добавлением по меньшей мере одного связующего реагента, предпочтительно выбранного из группы, включающей карбонилдиимидазол (CDI), йодид 2-хлор-1-метилпиридиния (реагент Mukaiyama), N-(3-диметиламинопропил)-N'-этилкарбодиимид (EDCI), тетрафторборат O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония (TBTU), N,N'-дициклогексилкарбодиимид (DCC) и гексафторфосфат 1-бензотриазолилокси-трис-(диметиламино)-фосфония (ВОР), необязательно в присутствии по меньшей мере одного неорганического основания, предпочтительно выбранного из группы, включающей карбонат калия и карбонат цезия, или органического основания, предпочтительно выбранного из группы, включающей триэтиламин, диизопропилэтиламин и пиридин, и необязательно с добавлением 4-(диметиламино)пиридина или 1-гидроксибензотриазола, при температуре предпочтительно от 25°C до 150°C, необязательно с микроволновым облучением, получая соединения общей формулы AMD.
Соединения общей формулы AMN могут взаимодействовать с кислотными ангидридами и хлоридами карбоновых кислот в по меньшей мере одном растворителе, предпочтительно выбранном из группы, включающей дихлорметан, ацетонитрил, диметилформамид, диэтиловый эфир, диоксан и тетрагидрофуран, необязательно в присутствии по меньшей мере одного неорганического основания, предпочтительно выбранного из группы, включающей карбонат калия и карбонат цезия, или органического основания, предпочтительно выбранного из группы, включающей триэтиламин, диизопропилэтиламин и пиридин, и необязательно с добавлением 4-(диметиламино)пиридина или 1-гидроксибензотриазола, при температуре предпочтительно от 25°C до 150°C, необязательно с микроволновым облучением, получая соединения общей формулы AMD.
Касательно дальнейших подробностей синтеза соединений в соответствии с изобретением, в частности, относительно синтеза подходящих исходных структурных элементов, можно привести ссылку на WO 2004/043967, WO 2005/063769, WO 2005/066183, WO 2006/018184, WO 2006/108565, WO 2007/124903 и WO 2008/009416 полностью. Для специалиста в данной области техники понятно, что подходящие исходные структурные элементы для синтеза соединений в соответствии с изобретением могут быть получены аналогично схемам синтеза и примерам осуществления, раскрытым в настоящем изобретении.
Соединения в соответствии с изобретением действуют, например, на ORL1 и опиоид рецепторы, которые релевантны в связи с различными заболеваниями, таким образом, они пригодны в качестве активного компонента (лекарственного средства) в фармацевтической композиции.
Изобретение также обеспечивает фармацевтическую композицию, которая содержит физиологически приемлемый носитель и по меньшей мере одно соединение в соответствии с изобретением.
Предпочтительно, композиция в соответствии с изобретением является твердой, жидкой или пастообразной; и/или
- содержит соединение в соответствии с изобретением в количестве от 0,001 до 99 мас.%, предпочтительно от 1,0 до 70 мас.%, на основе общего веса композиции.
Фармацевтическая композиция в соответствии с изобретением необязательно может содержать подходящие добавки и/или вспомогательные вещества и/или необязательно дополнительные активные компоненты.
Примерами подходящих физиологически приемлемых носителей, добавок и/или вспомогательных веществ являются наполнители, растворители, разбавители, красители и/или связующие. Эти вещества известны специалисту в данной области техники (см. Н.Р. Fiedler, Lexikon der Hilfsstoffe fur Pharmazie, Kosmetik und angrenzende Gebiete, Editio Cantor Aulendoff).
Композиция в соответствии с изобретением содержит соединение в соответствии с изобретением в количестве предпочтительно от 0,001 до 99 мас.%, более предпочтительно от 0,1 до 90 мас.%, еще более предпочтительно от 0,5 до 80 мас.%, наиболее предпочтительно от 1,0 до 70 мас.% и, в особенности, от 2,5 до 60 мас.%, на основе общего веса композиции.
Композиция в соответствии с изобретением предпочтительно производится для системного, местного или локального введения, предпочтительно для перорального введения.
Изобретение также обеспечивает фармацевтическую лекарственную форму, которая содержит фармацевтическую композицию в соответствии с изобретением.
В предпочтительном варианте осуществления, лекарственная форма в соответствии с изобретением производится для введения два раза в сутки, для введения один раз в сутки или для введения менее часто, чем один раз в сутки, предпочтительно для введения не более, чем один раз в сутки.
Введение предпочтительно является системным, в частности пероральным.
В предпочтительном варианте осуществления, лекарственная форма в соответствии с изобретением содержит соединение в соответствии с изобретением в такой низкой дозе, которая не является существенно эффективной для лечения острой боли. Эта доза предпочтительно находится в диапазоне от 1,0 мкг до 10 мг, на основе молекулярного веса свободного основания.
Предпочтительно, доза составляет 0,001 мг±50%, 0,002 мг±50%, 0,003 мг±50%, 0,004 мг±50%, 0,005 мг±50%, 0,006 мг±50%, 0,007 мг±50%, 0,008 мг±50%, 0,009 мг±50%, 0,01 мг±50%, 0,02 мг±50%, 0,03 мг±50%, 0,04 мг±50%, 0,05 мг±50%, 0,06 мг±50%, 0,07 мг±50%, 0,08 мг±50%, 0,09 мг±50%, 0,1 мг±50%, 0,15 мг±50%, 0,2 мг±50%, 0,25 мг±50%, 0,3 мг±50%, 0,35 мг±50%, 0,4 мг±50%, 0,45 мг±50%, 0,5 мг±50%, 0,55 мг±50%, 0,6 мг±50%, 0,65 мг±50%, 0,7 мг±50%, 0,75 мг±50%, 0,8 мг±50%, 0,85 мг±50%, 0,9 мг±50%, 0,95 мг±50%, 1 мг±50%, 1,5 мг±50%, 2 мг±50%, 2,5 мг±50%, 3 мг±50%, 3,5 мг±50%, 4 мг±50%, 4,5 мг±50%, 5 мг±50%, 5,5 мг±50%, 6 мг±50%, 6,5 мг±50%, 7 мг±50%, 7,5 мг±50%, 8 мг±50%, 8,5 мг±50%, 9 мг±50%, 9,5 мг±50% или 10 мг±50%, на основе молекулярного веса свободного основания.
Более предпочтительно, доза составляет 0,001 мг±25%, 0,002 мг±25%, 0,003 мг±25%, 0,004 мг±25%, 0,005 мг±25%, 0,006 мг±25%, 0,007 мг±25%, 0,008 мг±25%, 0,009 мг±25%, 0,01 мг±25%, 0,02 мг±25%, 0,03 мг±25%, 0,04 мг±25%, 0,05 мг±25%, 0,06 мг±25%, 0,07 мг±25%, 0,08 мг±25%, 0,09 мг±25%, 0,1 мг±25%, 0,15 мг±25%, 0,2 мг±25%, 0,25 мг±25%, 0,3 мг±25%, 0,35 мг±25%, 0,4 мг±25%, 0,45 мг±25%, 0,5 мг±25%, 0,55 мг±25%, 0,6 мг±25%, 0,65 мг±25%, 0,7 мг±25%, 0,75 мг±25%, 0,8 мг±25%, 0,85 мг±25%, 0,9 мг±25%, 0,95 мг±25%, 1 мг±25%, 1,5 мг±25%, 2 мг±25%, 2,5 мг±25%, 3 мг±25%, 3,5 мг±25%, 4 мг±25%, 4,5 мг±25%, 5 мг±25%, 5,5 мг±25%, 6 мг±25%, 6,5 мг±25%, 7 мг±25%, 7,5 мг±25%, 8 мг±25%, 8,5 мг±25%, 9 мг±25%, 9,5 мг±25% или 10 мг±25%, на основе молекулярного веса свободного основания.
Особенно предпочтительно, доза составляет 0,001 мг, 0,002 мг, 0,003 мг, 0,004 мг, 0,005 мг, 0,006 мг, 0,007 мг, 0,008 мг, 0,009 мг, 0,01 мг, 0,02 мг, 0,03 мг, 0,04 мг, 0,05 мг, 0,06 мг, 0,07 мг, 0,08 мг, 0,09 мг, 0,1 мг, 0,15 мг, 0,2 мг, 0,25 мг, 0,3 мг, 0,35 мг, 0,4 мг, 0,45 мг, 0,5 мг, 0,55 мг, 0,6 мг, 0,65 мг, 0,7 мг, 0,75 мг, 0,8 мг, 0,85 мг, 0,9 мг, 0,95 мг, 1 мг, 1,5 мг, 2 мг, 2,5 мг, 3 мг, 3,5 мг, 4 мг, 4,5 мг, 5 мг, 5,5 мг, 6 мг, 6,5 мг, 7 мг, 7,5 мг, 8 мг, 8,5 мг, 9 мг, 9,5 мг или 10 мг, на основе молекулярного веса свободного основания.
В предпочтительном варианте осуществления, лекарственная форма в соответствии с изобретением содержит соединение в соответствии с изобретением в количестве 10 мкг±90%, более предпочтительно 10 мкг±75%, еще более предпочтительно 10 мкг±50%, наиболее предпочтительно 10 мкг±25%, и, в особенности, 10 мкг±10%, на основе молекулярного веса свободного основания.
В другом предпочтительном варианте осуществления, лекарственная форма в соответствии с изобретением содержит соединение в соответствии с изобретением в количестве 100 мкг±90%, более предпочтительно 100 мкг±75%, еще более предпочтительно 100 мкг±50%, наиболее предпочтительно 100 мкг±25%, и, в особенности, 100 мкг±10%, на основе молекулярного веса свободного основания.
В дальнейшем предпочтительном варианте осуществления, лекарственная форма в соответствии с изобретением содержит соединение в соответствии с изобретением в количестве 250 мкг+90%, более предпочтительно 250 мкг±75%, еще более предпочтительно 250 мкг±50%, наиболее предпочтительно 250 мкг±25%, и, в особенности, 250 мкг±10%, на основе молекулярного веса свободного основания.
В дальнейшем предпочтительном варианте осуществления, лекарственная форма в соответствии с изобретением содержит соединение в соответствии с изобретением в количестве 500 мкг±90%, более предпочтительно 500 мкг±75%, еще более предпочтительно 500 мкг±50%, наиболее предпочтительно 500 мкг+25%, и, в особенности, 500 мкг±10%, на основе молекулярного веса свободного основания.
В другом предпочтительном варианте осуществления, лекарственная форма в соответствии с изобретением содержит соединение в соответствии с изобретением в количестве 750 мкг±90%, более предпочтительно 750 мкг±75%, еще более предпочтительно 750 мкг±50%, наиболее предпочтительно 750 мкг±25%, и, в особенности, 750 мкг±10%, на основе молекулярного веса свободного основания.
В дальнейшем предпочтительном варианте осуществления, лекарственная форма в соответствии с изобретением содержит соединение в соответствии с изобретением в количестве 1000 мкг±90%, более предпочтительно 1000 мкг±75%, еще более предпочтительно 1000 мкг±50%, наиболее предпочтительно 1000 мкг±25%, и, в особенности, 1000 мкг±10%, на основе молекулярного веса свободного основания.
Лекарственная форма в соответствии с изобретением может быть введена, например, в виде жидкой дозированной форме в форме инъекционных растворов, каплей или соков, или в виде полутвердой дозированной формы в форме гранул, таблеток, пеллет, пленок, капсул, пластырей/наносимых распылением пластырей или аэрозолей. Выбор вспомогательных веществ и т.д. и их используемое количество зависит от того, будет ли вводиться лекарственная форма орально, перорально, парентерально, внутривенно, внутрибрюшинно, внутрикожно, внутримышечно, интраназально, буккально, ректально или местно, например, на кожу, слизистую оболочку или в глаза.
Формы введения в форме таблеток, драже, капсул, гранул, каплей, соков и сиропов пригодны для перорального введения, и растворы, суспензии, легко восстанавливаемые сухие препараты и также спреи пригодны для парентерального, местного и ингаляционного введения. Соединения в соответствии с изобретением в депо, в растворенной форме или в пластыре, необязательно с добавлением агентов, способствующих проникновению через кожу, представляют собой подходящие препараты для чрескожного введения.
Формы введения, которые могут вводиться перорально или чрескожно, могут высвобождать соединения в соответствии с изобретением замедленным образом. Соединения в соответствии с изобретением также могут вводиться в парентеральной депо форме с длительным высвобождением, такой как, например, импланты или имплантированные насосы. Другие дополнительные активные компоненты, известные специалисту в данной области техники, в принципе могут быть добавлены в формы введения в соответствии с изобретением.
В предпочтительном варианте осуществления, соединения в соответствии с изобретением высвобождаются из лекарственной формы незамедлительно (незамедлительное высвобождение, IR), то есть предпочтительно по меньшей мере 80% активного компонента, который изначально присутствует, высвобождается в условиях in vitro, предпочтительно в соответствии с Европейской Фармакопеей, через 20 минут.
Неожиданно было обнаружено, что соединения в соответствии с изобретением отличаются необычно долгим периодом полувыведения (t1/2) или фармакологической продолжительностью действия, таким образом, что сравнительно нечастого введения достаточно для достижения фармакологической эффективности, и соответственно купирования боли, которое продолжается сравнительно длительное время.
Формы введения с пролонгированным введением соединений в соответствии с изобретением для этого не являются абсолютно необходимыми; продолжительное действие наблюдается даже в случае быстрого высвобождения (IR) в связи с длительным периодом период полувыведения. IR свойство таких форм введения имеет дополнительное преимущество, состоящее в том, при наличии продолжительной эффективности, тем не менее, достигается быстрое поглощение активного компонента и, соответственно, быстрое начало фармакологической эффективности после первого введения. Таким образом, свойства IR форм введения комбинируются со свойствами PR форм введения (PR, пролонгированное высвобождение).
В предпочтительном варианте осуществления, лекарственная форма в соответствии с изобретением представляет собой лекарственную форму с быстрым высвобождением активного компонента (IR), которая содержит соединение в соответствии с изобретением, предпочтительно общей формулы (V) или (VI), в форме свободного основания или физиологически приемлемую соль, предпочтительно гидрохлорид, цитрат или гемицитрат, и изготавливают предпочтительно для перорального введения не более чем один раз в сутки, предпочтительно ровно один раз в сутки. В этой связи, "быстрое высвобождение активного компонента" обозначает, что в условиях in vitro, предпочтительно в соответствии с Европейской Фармакопеей, по меньшей мере 80% активного компонента, который изначально присутствует, высвобождается через 20 минут.
Количество соединений в соответствии с изобретением для введения пациенту изменяется в зависимости от веса пациента, типа введения, показания и тяжести заболевания. Обычно, вводят от 0,00005 до 50 мг/кг, предпочтительно от 0,001 до 0,5 мг/кг, более предпочтительно от 1 до 10 мкг/кг, по меньшей мере одного соединения в соответствии с изобретением.
Во всех вышеописанных вариантах осуществления форм введения в соответствии с изобретением особенно предпочтительным для лекарственной формы является содержание дополнительного активного соединения, наряду с по меньшей мере одним соединением в соответствии с изобретением.
ORL1 рецептор и µ-опиоид рецептор связаны, в частности, с появлением боли. Таким образом, соединения в соответствии с изобретением можно использовать для приготовления лекарственного средства для лечения хронической боли, предпочтительно невропатической боли, более предпочтительно мононевропатической/невралгической или полиневропатической боли, более предпочтительно боли в случае постгерпетической невралгии или в случае диабетической полиневропатии.
Последующие примеры служат для пояснения изобретения, но никоим образом не должны толковаться как ограничивающие.
В последующей номенклатуре стереохимии характерных соединений, "(Е)" относится к замещению в двойной связи, например, на производном коричной кислоты, и "цис" и "транс" относится к замещению на циклогексильном кольце.
Синтез индольных структурных элементов (H)
Структурный элемент H-1:
2-(1H-индол-3-ил)этанамин (H-1)
Коммерчески доступный во время синтеза от Aldrich.
Структурный элемент Н-2:
2-(5-Фтор-1H-индол-3-ил)этанамин (H-2)
Коммерчески доступный во время синтеза от Fluorochem.
Синтез кетоновых структурных элементов (E)
Структурный элемент E-1:
Диметил-(8-фенил-1,4-диоксаспиро[4,5]дец-8-ил)амин гидрохлорид (D-1)
Аминонитрил В-1 (21 г, 0,1 моль), растворенный в ТГФ (210 мл), добавляли в течение 15 минут, в атмосфере аргона и при охлаждении на льду, к 1.82М раствору хлорида фенилмагния в ТГФ (109 мл, 0,198 моль), и после этого перемешивали в течение 16 часов при комнатной температуре. Для обработки реакционной смеси, добавляли насыщенный раствор хлорида аммония (150 мл), при охлаждении на льду, и осуществляли экстракцию с помощью диэтилового эфира (3×100 мл). Органическую фазу экстрагировали путем встряхивания с водой (100 мл) и насыщенным раствором NaCl (100 мл) и концентрировали. Оставалось желтое масло (25,2 г). Неочищенный продукт растворяли в этил метил кетоне (280 мл), и добавляли ClSiMe3 (18,8 мл, 0,15 моль), при охлаждении на льду. После осуществления реакции в течение 6 часов, гидрохлорид D-1 может быть выделен в форме белого твердого вещества с выходом 35% (10,5 г).
4-Диметиламино-4-фенилциклогексанон (E-1)
Гидрохлорид D-1 (10,5 г, 35,2 ммоль) растворяли в 7,5 н. соляной кислоте (36 мл) и перемешивали в течение 96 часов при комнатной температуре. После завершения гидролиза, реакционную смесь экстрагировали с помощью диэтилового эфира (2×50 мл). При охлаждении на льду, водную фазу подщелачивали с помощью 5 н. раствора гидроксида натрия, экстрагировали с помощью дихлорметана (3×50 мл) и концентрировали. Таким образом может быть выделен кетон 6 в форме желтого твердого вещества, имеющего точку плавления 104-108°C, с выходом 97% (7,4 г).
Структурный элемент Е-2:
Вариант 1:
[8-(3-Фторфенил)-1,4-диоксаспиро[4,5]дец-8-ил]диметиламин гидрохлорид (D-2)
0,5М раствора бромида 3-фторфенилмагния в ТГФ (3, 750 мл, 375 ммоль) добавляли в течение 15 минут, в атмосфере аргона и при охлаждении на льду, к раствору аминонитрила В-1 (19,8 г, 94 ммоль) в ТГФ (100 мл), и после этого перемешивали в течение 16 часов при комнатной температуре. Для обработки реакционной смеси, добавляли насыщенный раствор хлорида аммония (150 мл) и воду (60 мл), при охлаждении на льду, и осуществляли экстракцию с помощью диэтилового эфира (3×100 мл). Органическую фазу экстрагировали путем встряхивания с водой (50 мл) и насыщенным раствором NaCl (50 мл) и концентрировали. Оставалось коричневое масло (26,5 г), которое, дополнительно к фенильному соединению 4, также содержало кеталь 2. Неочищенный продукт растворяли в этил метил кетоне (156 мл), и добавляли ClSiMe3 (17,8 мл, 141 ммоль), при охлаждении на льду. После осуществления реакции в течение 6 часов, гидрохлорид D-2 может быть выделен в форме белого твердого вещества, имеющего точку плавления 275-278°C, с выходом 55% (16,3 г).
Вариант 2:
[8-(3-Фтор-фенил)-1,4-диокса-спиро[4,5]дец-8-ил]-диметил-амин гидрохлорид (D-2)
Раствор 1-бром-3-фторбензола (5,00 г, 28,6 ммоль) в абс. эфире (15 мл) добавляли по каплям к суспензии магния (694 мг, 28,6 ммоль) в абс. эфире (10 мл) таким образом, что эфир кипел. После завершения добавления, перемешивали в течение 10 минут при КТ, после чего магний полностью растворялся. Реакционный раствор охлаждали на ледяной бане, и аминонитрил В-1 (3,00 г, 14,3 ммоль) в абс. ТГФ (30 мл) добавляли по каплям при 10°C. Партию перемешивали в течение ночи при комнатной температуре; к реакционной смеси добавляли 20% раствор NH4Cl (20 мл) и воду (30 мл), при охлаждении на льду, и осуществляли экстракцию с помощью простого эфира (3×50 мл). Органическую фазу промывали водой (50 мл) и затем насыщенным раствором NaCl (50 мл), высушивали над Na2SO4 и концентрировали в вакууме. Неочищенный продукт растворяли в этил метил кетоне (25 мл); добавляли ClSiMe3 (3,2 мл, 25 ммоль), при охлаждении на льду, и перемешивали в течение 5 часов при комнатной температуре. Полученный осадок отфильтровывали и высушивали в вакууме.
Выход D-2: 2,8 г (62%)
1Н-ЯМР (ДМСО-d6): 1,91 (8H, m); 2,54 (6Н, s); 3,91 (4Н, d); 7,37 (1Н, m); 7,61 (3Н, m).
Вариант 1:
4-Диметиламино-4-(3-фтор-фенил)-циклогексанон (Е-2)
Гидрохлорид D-2 (7,2 г, 22,75 ммоль) растворяли в воде (9,6 мл); добавляли концентрированную соляную кислоту (14 мл, 455 ммоль), и перемешивали в течение 4 дней при комнатной температуре. После завершения гидролиза, реакционную смесь экстрагировали с помощью диэтилового эфира (2×50 мл) и водную фазу подщелачивали с помощью 5 н. раствора гидроксида натрия, при охлаждении на льду, вследствие чего осаждался продукт. Кетон Е-2 может быть выделен в форме желтого твердого вещества, имеющего точку плавления 83-88°C, с выходом 50% (6,05 г).
Вариант 2:
4-Диметиламино-4-(3-фтор-фенил)-циклогексанон (Е-2)
Гидрохлорид D-2 (2,80 г, 8,86 ммоль) растворяли в воде (3,7 мл); добавляли концентрированную соляную кислоту (5,5 мл), и перемешивали в течение 4 дней при КТ. После завершения гидролиза, реакционную смесь экстрагировали с помощью простого эфира (2×10 мл), водный раствор подщелачивали с помощью 5 н. раствора гидроксида натрия, при охлаждении на льду, реакционную смесь экстрагировали с помощью дихлорметана (3×50 мл), и органическую фазу высушивали над сульфатом натрия и концентрировали в вакууме. Неочищенный продукт очищали путем флэш-хроматографии с CHCl3/МеОН (20:1).
Выход Е-2: 676 мг (32%), бесцветное твердое вещество Точка плавления: 62-67°C
1Н-ЯМР (ДМСО-d6): 2,02 (6Н, s); 2,12 (5Н, m); 2,45 (3Н, m); 7,24 (3Н, m); 7,43 (1Н, m).
Структурный элемент Е-3:
[8-(4-Фторфенил)-1,4-диоксаспиро[4,5]дец-8-ил]диметиламин гидрохлорид (D-3)
1М раствор бромида 4-фторфенилмагния в ТГФ (3, 125 мл, 125 ммоль) добавляли в течение 15 минут, в атмосфере аргона и при охлаждении на льду, к раствору аминонитрила В-1 (10,5 г, 50 ммоль) в ТГФ (150 мл), и после этого перемешивали в течение 16 часов при комнатной температуре. Для обработки реакционной смеси, добавляли насыщенный раствор хлорида аммония (37 мл) и воду (50 мл), при охлаждении на льду, и осуществляли экстракцию с помощью диэтилового эфира (3×100 мл). Органическую фазу экстрагировали путем встряхивания с водой (50 мл) и насыщенным раствором NaCl (50 мл) и концентрировали. Оставалось коричневое масло (12,55 г), которое содержало, дополнительно к фенильному соединению С-3, также кеталь В-1. Неочищенный продукт растворяли в этил метил кетоне (75 мл), и добавляли ClSiMe3 (9,5 мл, 75 ммоль), при охлаждении на льду. После осуществления реакции в течение 6 часов, гидрохлорид D-3 может быть выделен в форме белого твердого вещества с выходом 47% (7,48 г).
4-Диметиламино-4-(4-фторфенил)циклогексанон (Е-3)
Гидрохлорид D-3 (7,2 г, 22,75 ммоль) растворяли в воде (9,6 мл); добавляли концентрированную соляную кислоту (14 мл, 455 ммоль), и перемешивали в течение 4 дней при комнатной температуре. После завершения гидролиза, реакционную смесь экстрагировали с помощью диэтилового эфира (2×50 мл) и водную фазу подщелачивали с помощью 5 н. раствора гидроксида натрия, при охлаждении на льду, экстрагировали с помощью дихлорметана (3×50 мл) и концентрировали. Кетон Е-3 может быть выделен в форме желтого твердого вещества, имеющего точку плавления 128-133°C, с выходом 76% (4,05 г).
Структурный элемент Е-4:
Диметил-(8-тиофен-2-ил-1,4-диоксаспиро[4,5]дец-8-ил)амин гидрохлорид (D-4)
2-Йодтиофен (1, 22,9 г, 109 ммоль) растворяли, в атмосфере аргона, в ТГФ (80 мл), и 2М хлорид изопропилмагния (2, 35,7 мл, 72 ммоль) в ТГФ добавляли при 0°C в течение 30 минут. После осуществления реакции в течение 1 часа при 3-5°C, добавляли аминонитрил В-1 (10 г, 47,6 ммоль), растворенный в тетрагидрофуране (20 мл), и перемешивали в течение 20 часов при комнатной температуре. Обработку партии осуществляли путем добавления насыщенного раствора NH4Cl (85 мл) и экстрагирования с помощью диэтилового эфира (3×100 мл). Органическую фазу экстрагировали путем встряхивания с водой (50 мл) и насыщенным раствором NaCl (50 мл) и концентрировали. Представляется возможным получить темно-коричневое масло (21,3 г), которое содержало, дополнительно к желательному кеталю, аминонитрил В-1 и 2-йодтиофен. Неочищенный продукт растворяли в этил метил кетоне (140 мл), и добавляли ClSiMe3 (9,1 мл, 71,4 ммоль). После осуществления реакции в течение 6 часов, гидрохлорид D-4 выделяли в форме белого кристаллического соединения с выходом 60% (8,74 г).
4-Диметиламино-4-тиофен-2-илциклогексанон (Е-4)
Гидрохлорид D-4 (8,68 г, 28,6 ммоль) растворяли в 7,5 н. соляной кислоте (29 мл) и перемешивали в течение 48 часов при комнатной температуре. После завершения гидролиза, реакционную смесь экстрагировали с помощью диэтилового эфира (2×50 мл). Водную фазу подщелачивали с помощью 5 н. раствора гидроксида натрия, при охлаждении на льду, экстрагировали с помощью дихлорметана (3×50 мл) и концентрировали. Таким образом получали кетон Е-4 в форме желтого твердого вещества, имеющего точку плавления 108-110°C, с выходом 89% (5,66 г).
Структурный элемент Е-5:
N,N-Диметил-8-(тиофен-3-ил)-1,4-диоксаспиро[4,5]декан-8-амин (D-5)
3-Йодтиофен (1, 5 г, 23,8 ммоль) растворяли, в атмосфере аргона, в ТГФ (18 мл), и 2М изопропилмагния хлорид (2, 7,8 мл, 15,5 ммоль) в ТГФ добавляли в течение 8 минут при 0°C. После осуществления реакции в течение 1 часа при 3-5°C, добавляли аминонитрил В-1 (2, 16 г, 10,3 ммоль), растворенный в тетра гидрофура не (20 мл). После этого перемешивали в течение 20 часов при комнатной температуре. Обработку партии осуществляли путем добавления насыщенного раствора NH4Cl (20 мл) и экстрагирования с помощью диэтилового эфира (3×50 мл). Органическую фазу экстрагировали путем встряхивания с водой (20 мл) и насыщенным раствором NaCl (20 мл) и концентрировали. Получали светло-коричневое масло (3,95 г). Неочищенный продукт растворяли в этил метил кетоне (40 мл), и добавляли ClSiMe3 (1,95 мл, 15,5 ммоль). После осуществления реакции в течение 3 часов, желательный гидрохлорид может быть выделен в форме белого кристаллического соединения с выходом 60% (1,86 г) с точкой плавления 250-251°C.
4-(Диметиламино)-4-(тиофен-3-ил)циклогексанон (Е-5)
Гидрохлорид D-5 (1,8 г, 5,9 ммоль) растворяли в 7,5 н. соляной кислоте (7 мл) и перемешивали в течение 48 часов при комнатной температуре. После завершения гидролиза, реакционную смесь экстрагировали с помощью диэтилового эфира (2×30 мл); при охлаждении на льду, водную фазу подщелачивали с помощью 5 н. раствора гидроксида натрия, экстрагировали с помощью дихлорметана (3×30 мл) и концентрировали. Кетон Е-5 может быть выделен в форме желтого твердого вещества, имеющего точку плавления 147-150°C, с выходом 98% (1,27 г).
Синтез спироаминовых структурных элементов (AMNцис/AMNтранс)
Пример AMN-1цис:
2',3',4',9'-Тетрагидро-N,N-диметил-4-(фенил)-спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол]-4-амин (цис-диастереоизомер)
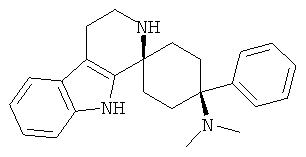
Примечание: В соответствии с этой процедурой, главным образом получали цис продукт AMN-1цис. Транс продукт AMN-1транс получали только в виде вторичного продукта или в неочищенной форме.
Кетон Е-1 (3,26 г, 15 ммоль) и триптамин Н-1 (2,4 г, 15 ммоль) растворяли в безводном МеОН (100 мл) с исключением кислорода. К этой смеси добавляли сульфат натрия (3 г). После осуществления реакции в течение 17 часов, растворитель отгоняли на роторном испарителе и остаток ресуспендировали в 1,2-дихлорэтане (100 мл). К реакционной смеси добавляли трифторуксусную кислоту (15 мл) и перемешивали в течение 1 часа при комнатной температуре. За осуществлением реакции наблюдали с помощью ТСХ. Для осуществления обработки, к партии добавляли H2O (40 мл) и значение pH доводили до 11 с помощью NaOH (5 моль/л). Осаждалось белое твердое вещество и отфильтровывали с отсасыванием над фриттой. Твердое вещество промывали с помощью H2O (3×5 мл) и высушивали. Оно представляло собой цис продукт AMN-1цис, который получали в форме белого твердого вещества, имеющего точку плавления 214-218°C, с выходом 4 г (74%). Маточную жидкость (водная фаза) экстрагировали с помощью 1,2-дихлорэтана (3×25 мл). Органическую фазу высушивали с помощью Na2SO4 и концентрировали. Твердый коричневый остаток перекристаллизовывали из МеОН (10 мл) и получали смесь цис-AMN-1цис и транс-AMN1-1транс спироамин (1:1). Смесь получали в форме белого твердого вещества с выходом 940 мг (17%).
1Н ЯМР (600 МГц, ДМСО-d6): 1,61 (m, 2 Н) 1,63 (m, 2 Н) 1,92 (s, 6Н) 2,12 (m, 2 Н) 2,39 (m, 2Н) 2,53 (t, J=5,36 Гц, 2Н) 2,99 (t, J=5,35 Гц, 2Н) 6,86 (m, 1Н) 6,91 (m, 1Н) 7,16 (d, J=7,52 Гц, 1Н) 7,28 (d, J=7,52 Гц, 1Н) 7,31 (m, 1Н) 7,43 (m, 4Н) 10,21 (s, 1Н)
Пример АММ-2цис:
2',3',4',9'-Тетрагидро-N,N-диметил-4-(3-фторфенил)-спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол]-4-амин(цис-диастереоизомер)
Кетон Е-2 (4,71 г, 20 ммоль) и триптамин Н-1 (3,2 г, 20 ммоль) растворяли в безводном МеОН (200 мл), в атмосфере аргона. После осуществления реакции в течение 24 часов, МеОН отгоняли и желтый, маслянистый остаток суспендировали в 1,2-дихлорэтане (200 мл). К реакционной смеси добавляли трифторуксусную кислоту (20 мл) и перемешивали в течение 2 часов при комнатной температуре. За осуществлением реакции наблюдали с помощью ТСХ. Для осуществления обработки, партию разводили с помощью H2O (100 мл) и значение pH доводили до 11 с помощью NaOH (5 моль/л). После добавления этил ацетата (50 мл), осаждалось белое твердое вещество при перемешивании и отфильтровывали с отсасыванием над фриттой. Твердое вещество промывали с помощью H2O (3×25 мл) и после этого высушивали. Это был цис-диастереоизомер AMN-2цис, который получали в форме белого твердого вещества, имеющего точку плавления 220-225°C, с выходом 5,5 г (73%).
1H ЯМР (600 МГц, ДМСО-d6): 1,61 (m, 2 Н) 1,62 (m, 2 Н) 1,93 (s, 6 Н) 2,11 (m, 2 Н) 2,38 (m, 2 Н) 2,53 (t, J=5,56 Гц, 2 Н) 2,99 (t, J=5,56 Гц, 2 Н) 6,87 (m, 1H) 6,92 (m, 1H) 7,14 (m, 1H) 7,17 (d, J=8,34 Гц, 1H) 7,20 (m, 1H) 7,25 (d, J=7,82 Гц, 1H) 7,28 (d, J=7,47 Гц, 1H) 7,47 (m, 1Н) 10,26 (s, 1H)
Пример AMN-2транс:
2',3',4',9'-Тетрагидро-N,N-диметил-4-(3-фторфенил)-спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол]-4-амин(транс-диастереоизомер)
Триптамин Н-1 (2,03 г, 12,7 ммоль) и кетон (Е-2, 3,0 г, 12,7 ммоль) растворяли в абс. метаноле (130 мл) и перемешивали в течение 16 часов при комнатной температуре, в атмосфере аргона. После этого реакционную смесь концентрировали. Остаток растворяли в абс. 1,2-дихлорэтане (130 мл); быстро добавляли трифторуксусную кислоту (12,7 мл), и перемешивали в течение 2 часов при комнатной температуре. При охлаждении на льду, добавляли воду (120 мл) и 5 н. раствор гидроксида натрия (40 мл) и перемешивали в течение 1 часа. Бесцветное твердое вещество, которое образовывалось таким образом, отделяли путем фильтрации и промывали с помощью 1,2-дихлорэтана (30 мл) и воды (4×25 мл). Цис-спироамин AMN-240C получали с выходом 77% (3,7 г) со следами транс-спироамина AMN-2транс. Фазы фильтрата разделяли.
Органическую фазу высушивали с помощью сульфата натрия и концентрировали, добавляли метанол (3 мл), и перемешивали в течение 1 часа при комнатной температуре. Осаждалось белое твердое вещество и отделяли путем фильтрации и промывали метанолом (4×3 мл). Транс-спироамин AMN-2транс получали с выходом 5% (250 мг) со следами цис-спироамина AMN-2400. После очистки путем хроматографии [силикагель 60 (20 г); метанол (200 мл)], получали транс-спироамин AMN-2транс (170 мг), имеющий точку плавления 296-299°C.
1Н ЯМР (600 МГц, ДМСО-d6): 1,55 (m, 2 Н) 1,62 (m, 2 Н) 1,88 (s, 6 Н) 2,26 (m, 2 Н) 2,43 (m, 2 Н) 2,55 (t, J=5,49 Гц, 2 Н) 2,96 (t, J=5,25 Гц, 2 Н) 6,91 (m, 1H) 6,99 (m, 1H) 7,08 (m, 1H) 7,14 (m, 1H) 7,20 (d, J=7,64 Гц, 1H) 7,32 (m, 2Н) 7,40 (m, 1H) 10,63 (s, 1H)
Пример AMN-3цис:
6'-Фтор-2',3',4',9'-тетрагидро-N,N-диметил-4-(3-фторфенил)спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол]-4-амин(цис-диастереоизомер)
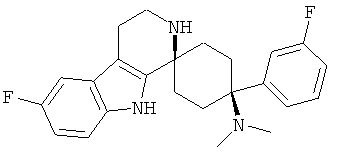
Кетон Е-2 (9,6 г, 41,2 ммоль) и фтортриптамин Н-2 (7,3 г, 41,2 ммоль) растворяли в этаноле (200 мл) и нагревали в течение 12 часов в колбе с обратным холодильником. После этого этанол отгоняли и неочищенный продукт суспендировали в 1,2-дихлорэтане (100 мл). К реакционной смеси добавляли трифторуксусную кислоту (90 мл) и перемешивали в течение 12 часов при комнатной температуре. За осуществлением реакции наблюдали с помощью ТСХ. Для осуществления обработки, партию подщелачивали с помощью 500 мл 1 н. раствора NaOH 0°C и после этого экстрагировали 3х с помощью 500 мл этил ацетата. Объединенные органические фазы высушивали над сульфатом магния и концентрировали при пониженном давлении. После добавления метанола (100 мл), осаждалось белое твердое вещество при перемешивании и отфильтровывали с отсасыванием над фриттой. Твердое вещество промывали метанолом (2×25 мл) и после этого высушивали. Это был цис-диастереоизомер AMN-34UC, который получали в форме белого твердого вещества с выходом 3,6 г (22%).
1Н ЯМР (ДМСО-d6, 400 МГц): δ 10,39 (s, 1Н), 7,44-7,49 (m, 1Н), 7,11-7,24 (m, 4Н), 7,00-7,04 (m, 1Н), 6,72-6,78 (m, 1Н), 2,95-2,98 (t, 2Н), 2,48-2,50 (m, 1Н), 2,36-2,39 (d, 2Н), 1,98-2,11 (m, 2Н), 1,91 (s, 6Н), 1,51-1,67 (m, 5Н) МС m/z (М+1): 396,4; Чистота (ВЭЖХ): 95,03%
Пример AMN-4цис:
6'-Фтор-2',3',4',9'-тетрагидро-N,N-диметил-4-(фенил)-спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол]-4-амин(цис-диастереоизомер)
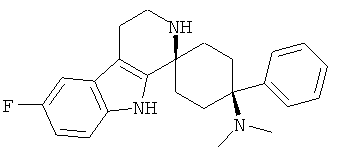
Кетон Е-1 (8,4 г, 47 ммоль) и фтортриптамин Н-2 (10,2 г, 47 ммоль) растворяли в этаноле (200 мл) и нагревали в течение 12 часов в колбе с обратным холодильником. После этого этанол отгоняли и неочищенный продукт суспендировали в 1,2-дихлорэтане (120 мл). К реакционной смеси добавляли трифторуксусную кислоту (100 мл) и перемешивали в течение 12 часов при комнатной температуре. За осуществлением реакции наблюдали с помощью ТСХ. Для осуществления обработки, партию подщелачивали с помощью 1 н. раствора NaOH при 0°C и после этого экстрагировали 3х с помощью 500 мл этил ацетата. Объединенные органические фазы высушивали над сульфатом магния и концентрировали при пониженном давлении. После добавления метанола (100 мл), осаждалось белое твердое вещество при перемешивании и отфильтровывали с отсасыванием над фриттой. Твердое вещество промывали метанолом (2×25 мл) и после этого высушивали. Это был цис-диастереоизомер AMN-4цис, который получали в форме белого твердого вещества с выходом 4 г (28%).
1H ЯМР (ДМСО-d6, 400 МГц): δ 10,36 (s, 1Н), 7,45-7,42 (t, 4Н), 7,32-7,29 (m, 1Н), 7,14-7,10 (m, 1Н), 7,03-7,00 (m, 1Н), 6,76-6,71 (m, 1Н), 2,99-2,96 (t, 2Н), 2,40-2,37 (d, 2Н), 2,13-2,04 (m, 2Н), 1,91 (s, 6Н), 1,88 (s, 1Н), 1,65-1,54 (m, 4Н), 1,23 (s, 1Н).
Пример AMN-5цис:
2',3',4',9'-Тетрагидро-N,N-диметил-4-(4-фторфенил)-спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол]-4-амин(цис-диастереоизомер)
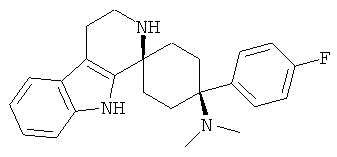
Кетон Е-3 (2800 мг, 11,90 ммоль) и триптамин (Н-1, 1910 мг, 11,90 ммоль) растворяли, в атмосфере аргона, в безводном метаноле (119 мл) и перемешивали в течение 18 часов. После этого метанол отгоняли в вакууме и остаток суспендировали в 1,2-дихлорэтане (119 мл). К реакционной смеси добавляли трифторуксусную кислоту (11,9 мл) и перемешивали в течение 2 часов при комнатной температуре. После этого реакционную смесь разводили с помощью 1,2-дихлорэтана (119 мл) и значение pH доводили до 11 с помощью 1 н. раствора гидроксида натрия, при охлаждении на льду. Образовывался светлый осадок. Смесь перемешивали в течение ночи при комнатной температуре. Осадок отфильтровывали с отсасыванием, промывали водой и высушивали в вакууме. Цис-диастереоизомер AMN-5цис (точка плавления 249-250°C, в некоторых случаях 225-230°C) может быть выделен с выходом 80% (3610 мг, 9,56 ммоль). Фазы разделяли. Органическую фазу высушивали с помощью сульфата натрия, фильтровали и летучие компоненты удаляли в вакууме. Светлый остаток (транс-диастереоизомер AMN-5транс) ресуспендировали в метаноле (5 мл) и перемешивали в течение 48 часов. Осадок отфильтровывали и высушивали в вакууме. Транс-диастереоизомер AMN-5транс (268-271°C) может быть выделен с выходом 6% (279 мг, 0,74 ммоль).
13С{1Н}-ЯМР (101 МГц, ДМСО-06) 6 част, на млн: 22,8 (1 С), 27,3 (2 С), 32,6 (2 С), 37,8 (2 С), 38,6 (1 С), 51,2 (1 С), 60,5 (1 С), 106,7 (1 С), 110,8 (1 С), 114,2 (2 С, d, J=21 Гц), 117,2 (1 С), 117,9 (1 С), 120,0 (1 С), 126,9 (1 С), 129,7 (2 С, d, J=8 Гц), 132,8 (1 С, d, J=3 Гц), 135,4 (1 С), 141,4 (1 С), 160,7 (1 С, d, J=242 Гц)
Синтез цис-спироамидных примеров (AMDцис)
Пример АМР-1цис:
(Е)-2',3',4',9'-Тетрагидро-N,N-диметил-4-фенил-2,-(2-фенилвинил)карбонил-спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол]-4-амин метансульфонат(1:1)(цис-диастереоизомер)
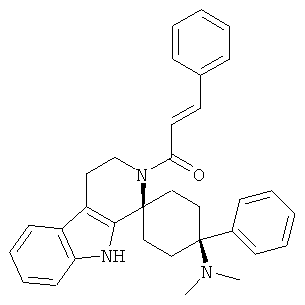
AMN-1400 растворяли в ТГФ (8 мл). После этого добавляли хлорид коричной кислоты (254 мг, 1,53 ммоль) и диизопропилэтиламин (216 мг, 1,67 ммоль), и перемешивали в течение 2 дней при КТ. После завершения реакции, твердое вещество отфильтровывали и к фильтрату добавляли насыщенный раствор Na2CO3. Водную фазу экстрагировали три раза каждый раз с помощью 10 мл этил ацетата. После этого органическую фазу высушивали над MgSO4 и концентрировали на роторном испарителе. Неочищенный продукт очищали путем колоночной хроматографии [силикагель 60; ДХМ/метанол (19: 1, 570 мл)]. Продукт получали с выходом 174 мг (26%). Для получения метансульфоната, только что полученный спироамид (174 мг, 0,355 ммоль) суспендировали в ДХМ (6 мл), и добавляли метансульфоновую кислоту (23,7 мкл, 0,355 ммоль) при КТ. После этого добавляли ацетон (0,8 мл), и добавляли достаточное количество диэтилового эфира для диспергирования мутности, которая появилась при встряхивании. Перемешивали дополнительно в течение 30 минут и полученное твердое вещество после этого отфильтровали с отсасыванием, с исключением воздуха, промывали с помощью диэтилового эфира и высушивали в течение 3 часов при 50°C под вакуумом с помощью насоса масла. Продукт AMD-1цис получали с выходом 159 мг (76%).
1Н ЯМР (600 МГц, ДМСО-d6) 1,65 (t, J=13,22 Гц, 2 Н) 2,20 (t, J=12,84 Гц, 2 Н) 2,51 (d, J=4,53 Гц, 9 Н) 2,87-3,16 (m, 4 Н) 4,13 (br. s., 2 Н) 6,92 (t, J=7,55 Гц, 1H) 6,99 (t, J=7,55 Гц, 1H) 7,20 (d, J=8,31 Гц, 1H) 7,31 (d, J=7,55 Гц, 1H) 7,36-7,51 (m, 5 Н) 7,56 - 7,69 (m, 3 Н) 7,74 (d, J=7,55 Гц, 2 Н) 7,82 (d, J=7,55 Гц, 2 Н) 9,62 (br, s, 1H)
Пример АМР-2цис
2',3',4',9'-Тетрагидро-N,N-диметил-4-(3-фтор-фенил)-2'-(4-хлорбензил)-карбонил-спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол]-4-амин (цис-диастереоизомер)
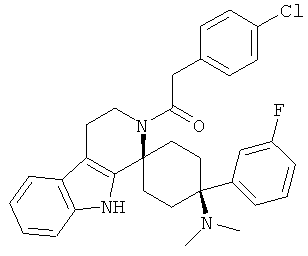
Спироамин (AMN-2цис; 396 мг, 1,05 ммоль) суспендировали в ДХМ (15 мл) в сосуде, пригодном для микроволн, и добавляли 2-(4-хлорфенил)ацетил хлорид (397 мг, 2,1 ммоль) и диизопропилэтиламин (269 мг, 2,1 ммоль). Реакционную смесь облучали в течение 10 минут при 120±С в микроволновом шкафу (Initiator Eight, Biotage). После завершения реакции (мониторинг с помощью ТСХ), реакционную смесь сначала фильтровали, добавляли диэтиловый эфир (15 мл), и снова фильтровали. Добавляли насыщенный раствор Na2CO3 (8 мл).
После разделения фаз, водную фазу снова промывали ДХМ. Объединенные органические фазы высушивали над MgSO4 и концентрировали на роторном испарителе. Неочищенный продукт очищали путем колоночной хроматографии [силикагель 60; ДХМ/метанол (19: 1)]. Продукт AMD-2цис получали с выходом 91 мг(16%).
1Н ЯМР (600 МГц, ДМСО-d6) d част, на млн. 1,55 (t, J=13,60 Гц, 2Н) 1,79 (t, J=12,84 Гц, 2Н) 1,92 (br. s., 6Н) 2,60-2,70 (m, 2Н) 2,73-2,87 (m, 2Н) 3,17 (d, J=5,29 Гц, 2Н) 3,89-4,01 (m, 4 Н) 6,90 (t, J=7,55 Гц, 1H) 6,97 (t, J=7,55 Гц, 1H) 7,08-7,16 (m, 1H) 7,18 (d, J=8,31 Гц, 1H) 7,21-7,30 (m, 3 Н) 7,34 (q, J=8,31 Гц, 4Н) 7,46 (q, J=7,30 Гц, 1H) 10,53 (s, 1H)
Пример АМР-3цис
2',3',4',9'-Тетрагидро-N,N-диметил-4-(3-фторфенил)-2'-(бензотиофен-2-ил)карбонил-спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол]-4-амин (цис-диастереоизомер)
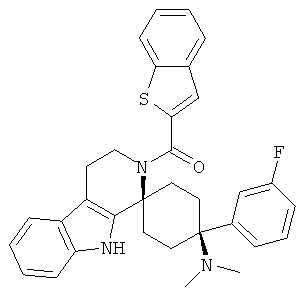
Спироамин (AMN-2цис; 264 мг, 0,7 ммоль) суспендировали в ДХМ (7 мл) в сосуде, пригодном для микроволн, и добавляли бензо[b]тиофен-2-карбонил хлорид (239 мг, 1,21 ммоль) и диизопропилэтиламин (180 мг, 1,4 ммоль). Реакционную смесь облучали в течение 10 минут при 100°C в микроволновом шкафу (Initiator Eight, Biotage). После завершения реакции (мониторинг с помощью ТСХ), реакционную смесь разводили с помощью ДХМ (15 мл) и фильтровали. Насыщенный раствор Na2CO3 (8 мл) добавляли к маточной жидкости. После разделения фаз, водную фазу еще два раза промывали ДХМ. Объединенные органические фазы высушивали над MgSO4 и концентрировали на роторном испарителе. Неочищенный продукт очищали путем колоночной хроматографии [силикагель 60; ДХМ/метанол (19:1)]. Продукт AMD-34UC получали с выходом 125 мг (33%).
1Н ЯМР (600 МГц, ДМСО-d6) d част, на млн. 1,63-1,79 (m, 2Н) 1,83-1,93 (m, 2Н) 1,95 (s, 6Н) 2,60 (d, J=13,60 Гц, 2Н) 2,65 (t, J=5,67 Гц, 2Н) 2,78-2,94 (m, 2Н) 4,08-4,22 (m, 2Н) 6,92 (t, J=7,55 Гц, 1H) 6,99 (t, J=7,55 Гц, 1H) 7,16 (t, J=8,31 Гц, 1H) 7,23 (d, J=8,31 Гц, 1H) 7,26-7,36 (m, 3Н) 7,44-7,54 (m, 3Н) 7,95 (s, 1H) 8,03 (d, J=7,55 Гц, 1H) 8,07 (d, J=8,31 Гц, 1H) 10,66 (s, 1H)
Пример АМР-4цис
2',3',4',9'-Тетрагидро-N,N-диметил-4-(3-фтор-фенил)-2'-(4-фторбензил)-карбонил-спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол]-4-амин метансульфонат(цис-диастереоизомер)
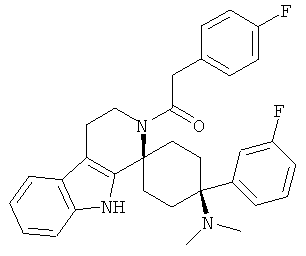
Спироамин (AMN-2цис; 600 мг, 1,59 ммоль) суспендировали в ДХМ (15 мл) в сосуде, пригодном для микроволн, и добавляли 2-(4-фторфенил)ацетил хлорид (548 мг, 3,18 ммоль) и диизопропилэтиламин (408 мг, 3,18 ммоль). Реакционную смесь облучали в течение 10 минут при 130°C в микроволновом шкафу (Initiator Eight, Biotage). После завершения реакции (мониторинг с помощью ТСХ), реакционную смесь сначала фильтровали, маточную жидкость разводили с помощью ДХМ (45 мл), и добавляли насыщенный раствор Na2CO3 (25 мл). После разделения фаз, органическую фазу снова промывали насыщенным раствором Na2CO3. Органическую фазу высушивали над MgSO4 и концентрировали на роторном испарителе. Неочищенный продукт очищали путем колоночной хроматографии [силикагель 60; ДХМ/метанол (4: 1)]. Продукт получали с выходом 150 мг (18%). Для получения метансульфоната, спироамид (150 мг, 0,29 ммоль) растворяли в ДХМ (1 мл), и добавляли метансульфоновую кислоту (18,9 мкл, 0,29 ммоль) при КТ. Смесь разводили с помощью диэтилового эфира, чтобы образовывалась регулируемая смесь. Твердое вещество отфильтровывали с отсасыванием, с исключением воздуха, промывали с помощью диэтилового эфира и высушивали при 50°C под вакуумом с помощью насоса масла. Продукт AMD-4цис получали с выходом 148 мг (83%).
1Н ЯМР (600 МГц, ДМСО-d6) d част, на млн. 1,58 (t, J=12,84 Гц, 2Н) 2,16 (t, J=12,09 Гц, 2Н) 2,31 (s, 3Н) 2,53-2,58 (m, 6Н) 2,58-2,68 (m, 2Н) 2,83-3,03 (m, 4Н) 3,98 (s, 2Н) 3,99-4,06 (m, 2Н) 6,92 (t, J=7,18 Гц, 1H) 6,99 (t, J=7,18 Гц, 1H) 7,14 (t, J=8,31 Гц, 2Н) 7,18 (d, J=8,31 Гц, 1H) 7,29 (d, J=7,55 Гц, 1H) 7,36 (t, J=6,42 Гц, 2 Н) 7,45 (t, J=7,93 Гц, 1H) 7,58-7,75 (m, 3 Н) 9,65 (br. s., 1H)
Пример АМР-5цис
(Е)-2',3',4',9'-Тетрагидро-N,N-диметил-4-(3-фторфенил)-2'-(2-фенилвинил)карбонил-спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол]-4-амин(цис-диастереоизомер)
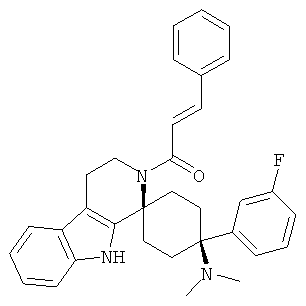
Спироамин (AMN-2цис; 378 мг, 1,0 ммоль) растворяли в безводном апротонном растворителе (6 мл); добавляли циннамоил хлорид (183 мг, 1,1 ммоль) и диизопропилэтиламин (155 мг, 1,2 ммоль), и перемешивали в течение ночи при КТ. После завершения реакции (мониторинг с помощью ТСХ), растворитель удаляли, остаток подвергали водной обработке, и осуществляли экстракцию с помощью галогенированного растворителя. Объединенные органические фазы высушивали над Na2S04 и концентрировали. Неочищенный продукт очищали путем колоночной хроматографии. При осуществлении концентрирования, твердое вещество осаждалось и отфильтровывали и после этого высушивали. Продукт получали с выходом 220 мг (43%).
1Н ЯМР (600 МГц, ДМСО-d6) d част, на млн. 1,63 (t, J=13,60 Гц, 2 Н) 1,84 (t, J=13,22 Гц, 2Н) 1,91 (s, 6Н) 2,54-2,63 (m, 2 Н) 2,65 (t, J=5,67 Гц, 2 Н) 2,82-3,02 (m, 2 Н) 3,17 (d, J=5,29 Гц, 2 Н) 4,00-4,22 (m, 2 Н) 6,90 (t, J=7,18 Гц, 1H) 6,97 (t, J=7,55 Гц, 1H) 7,11-7,18 (m, 1H) 7,20 (d, J=8,31 Гц, 1H) 7,23-7,33 (m, 3 Н) 7,35-7,54 (m, 5Н) 7,72 (d, J=6,80 Гц, 2Н) 10,59 (s, 1H)
Пример АМР-6цис:
(Е)-2',3',4',9'-Тетрагидро-N,N-диметил-4-(3-фторфенил)-2'-(2-фенилвинил)карбонил-спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол]-4-амин цитрат (цис-диастереоизомер)
Для получения соли, амид AMD-5цис (220 мг, 0,43 ммоль) растворяли в безводном апротонном растворителе (1,5 мл), и по мере возможности добавляли лимонную кислоту (83 мг, 0,43 ммоль), растворенную в незначительно полярном растворителе. Для осаждения продукта, неполярный растворитель добавляли по каплям. После этого твердое вещество отфильтровали с отсасыванием, с исключением воздуха, и высушивали при 50°C под вакуумом с помощью насоса масла. Продукт AMD-6цис получали с выходом 100 мг (33%).
Пример АМР-7цис
2',3',4',9'-Тетрагидро-N,N-диметил-4-(3-фторфенил)-2'-(3,4-диметоксибензил)карбонил-спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол]-4-амин (цис-диастереоизомер)
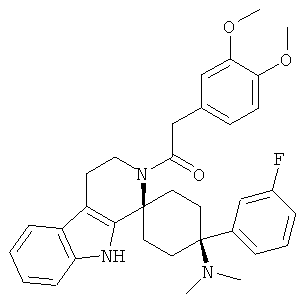
Спироамин (AMN-2цис; 200 мг, 0,54 ммоль) суспендировали в галогенированном растворителе (5 мл) в сосуде, пригодном для микроволн, и добавляли 2-(3,4-диметоксифенил)ацетил хлорид (230 мг, 1,1 ммоль) и диизопропилэтиламин (138 мг, 1,1 ммоль). Реакционную смесь облучали в течение 10 минут при 120°C в микроволновом шкафу (Initiator Eight, Biotage). После завершения реакции (мониторинг с помощью ТСХ), реакционную смесь сначала фильтровали и после этого к маточной жидкости добавляли раствор NaOH (5 н., 10 мл) добавляли. После разделения фаз, водную фазу экстрагировали три раза полярным апротонным растворителем (в каждом случае 5 мл). Объединенные органические фазы высушивали над MgSO4 и концентрировали. Неочищенный продукт очищали путем колоночной хроматографии. Продукт AMD-7цис получали с выходом 140 мг (47%).
1Н ЯМР (600 МГц, ДМСО-d6) d част, на млн. 1,54 (t, J=12,46 Гц, 2Н) 1,79 (t, J=13,22 Гц, 2Н) 1,85-1,96 (m, 6Н) 2,52-2,60 (m, 2Н) 2,62-2,72 (m, 2Н) 2,73-2,89 (m, 2Н) 3,74 (s, 3Н) 3,77 (s, 3Н) 3,82 (br. s., 2H) 3,90 (br. s., 2Н) 6,83-6,93 (m, 4Н) 6,97 (t, J=7,55 Гц, 1H) 7,13 (t, J=7,18 Гц, 1H) 7,19 (d, J=8,31 Гц, 1H) 7,20-7,32 (m, 3Н) 7,41-7,54 (m, 1H) 10,53 (s, 1H)
Пример АМР-8цис
(Е)-2',3',4',9'-Тетрагидро-N,N-диметил-6,-фтор-4-(3-фторфенил)-2'-(2-фенилвинил)карбонил-спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол]-4-амин (цис-диастереоизомер)
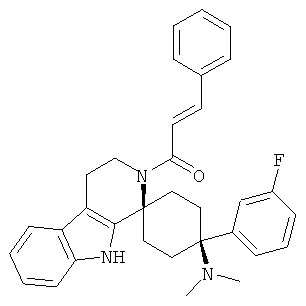
Суспензию спироамина AMN-3цис (0,197 г; 0,5 ммоль; 1 экв.) в 15 мл абс. ДХМ помещали в микроволновой сосуд. К этой суспензии последовательно добавляли этил-диизопропиламин (0,129 г; 1 ммоль; 2 экв.) и хлорид коричной кислоты (0,166 г; 1 ммоль; 2 экв.). Микроволновой сосуд закрывали и нагревали в течение 10 минут при 120°C в микроволновом шкафу (Initiator Eight, Biotage). Для осуществления обработки, к реакционной смеси добавляли 4 мл воды и 4 мл 1 н. раствора гидроксида натрия. Смесь перемешивали в течение 2 часов при КТ. После этого фазы разделяли и водную фазу экстрагировали 3× с помощью ДХМ. Объединенные органические фазы промывали водой и высушивали над сульфатом натрия. После удаления растворителя при пониженном давлении, остаток очищали путем колоночной хроматографии (силикагель; этил ацетат/циклогексан 1:2→1:0). Получали 0,087 г продукта AMD-8цис (33%).
ВЭЖХ/МС анализ: Rt=4,2 мин.; Чистота (УФ 200-400 нм) 97%; m/z=526,1
Пример AMD-9цис
2',3',4',9'-Тетрагидро-N,N-диметил-6,-фтор-4-(3-фторфенил)-2'-(бензил)карбонил-спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол]-4-амин (цис-диастереоизомер)
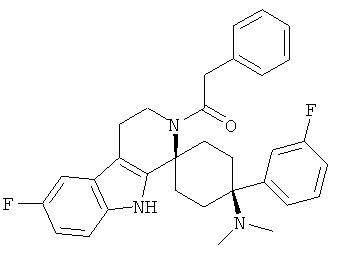
Суспензию спироамина AMN-3цис (0,25 г; 0,63 ммоль; 1 экв.) в 19 мл абс. ДХМ помещали в микроволновой сосуд. К этой суспензии последовательно добавляли этил-диизопропиламин (0,163 г; 1,26 ммоль; 2 экв.) и 2-фенилацетил хлорид (0,195 г; 1,26 ммоль; 2 экв.). Микроволновой сосуд закрывали и нагревали в течение 10 минут при 120±С в микроволновом шкафу (Initiator Eight, Biotage). Для осуществления обработки, к реакционной смеси добавляли 5 мл воды и 5 мл 1 н. раствора гидроксида натрия. Смесь перемешивали в течение 2 часов при КТ. После этого фазы разделяли и водную фазу экстрагировали 3× с помощью ДХМ. Объединенные органические фазы промывали водой и высушивали над сульфатом натрия. После удаления растворителя при пониженном давлении, остаток очищали путем колоночной хроматографии (силикагель; этил ацетат → этил ацетат/метанол 9:1). Получали 0,145 г продукта AMD-9цис (45%).
ВЭЖХ/МС анализ: Rt=3,9 мин.; Чистота (УФ 200-400 нм) 98%; m/z=514,1
Пример АМР-10цис
(Е)-2',3',4',9'-Тетрагидро-N,N-диметил-6'-фртор-4-фенил-2'-(2-фенилвинил)карбонил-спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол]-4-амин (цис-диастереоизомер)
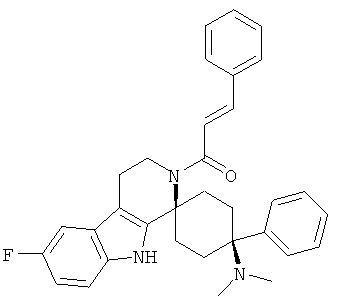
Раствор спироамина AMN-4цис (0,15 г; 0,397 ммоль; 1 экв.) в 9 мл абс. ТГФ добавляли в атмосфере азота при КТ к раствору хлорида коричной кислоты (0,198 г; 1,192 ммоль; 3 экв.) в 4,5 мл абс. ТГФ. После перемешивания в течение 1 часа при КТ, сначала 3 мл воды и, при охлаждении на льду, к мутному реакционному раствору добавляли 3 мл 1 н. раствора гидроксида натрия. Перемешивали в течение 1,5 часа. После удаления растворителя при пониженном давлении, полученное твердое вещество отфильтровывали и промывали водой. Неочищенный продукт очищали путем колоночной хроматографии (силикагель; этил ацетат). Получали 0,043 г продукта АМР-10цис (21%).
ВЭЖХ/МС анализ: Rt=4,2 мин.; Чистота (УФ 200-400 нм) 98%; m/z=508,2 Пример АМР-11цис
2',3',4',9'-Тетрагидро-N,N-диметил-4-(3-фторфенил)-2'-бензилкарбонил-спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол]-4-амин (цис-диастереоизомер)
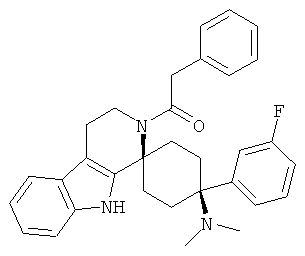
Цис-спироамин AMN-2цис (1,29 г, 3,4 ммоль) растворяли, с исключением кислорода, в абсолютном тетрагидрофуране (20 мл) и абсолютном дихлорметане (120 мл); добавляли основание Хьюнига (1,167 мл, 6,8 ммоль), и добавляли 2-фенилацетил хлорид (900 мкл, 6,8 ммоль) при комнатной температуре. После осуществления реакции в течение 30 минут, к смеси добавляли 5 н. раствор гидроксида натрия (100 мл), и перемешивали в течение 2 часов. Водную фазу отделяли и экстрагировали с помощью дихлорметана (3×10 мл). Объединенные органические фазы высушивали над Na2SO4 и после этого концентрировали. Неочищенный продукт выделяли и разделяли с помощью хроматографии [силикагель 60 (100 г); EtOAc (1000 мл)]. Цис-амид AMD-11цис получали в форме бесцветного твердого вещества с выходом 820 мг (49%) с точкой плавления 95-100°C.
13С-ЯМР (101 МГц, ДМСО-D6) δ част, на млн: 22,1, 29,1, 33,0, 38,0, 40,8, 43,1, 60,0, 60,3, 105,5, 111,1, 113,7, 113,2, 114,5, 114,7, 117,3, 118,4, 120,5, 123,8, 126,2, 126,5, 128,2, 129,0, 129,2, 129,3, 135,3, 136,5, 139,5, 140,6, 161,1, 163,5, 173,4
Пример AMD-12цис
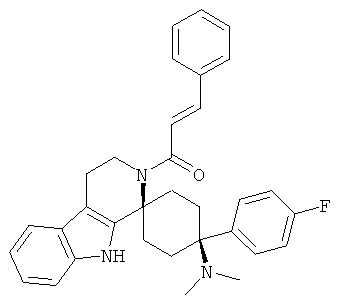
Основание Хьюнига (0,45 мл, 342 мг, 2,64 ммоль) и хлорид коричной кислоты (440 мг, 2,64 ммоль), растворенный в абсолютном дихлорметане (12 мл), добавляли по каплям последовательно в течение 10 минут, в атмосфере аргона, к суспензии цис-спироамина AMN-5цис (500 мг, 1,32 ммоль). Реакционную смесь перемешивали в течение 1 часа при комнатной температуре, и после этого добавляли воду (30 мл) и 1 н. раствор гидроксида натрия (5 мл) и перемешивали в течение 1,5 часа. После этого дихлорметан удаляли в вакууме. Светлое твердое вещество осаждалось и отделяли путем фильтрации и после этого промывали водой (3×30 мл). Таким образом полученный неочищенный продукт очищали путем хроматографии [силикагель 60 (70 г), этил ацетат/циклогексан 1:1 (500 мл), этил ацетат (1000 мл), этил ацетат/метанол 10:1 (330 мл), этил ацетат/метанол 4:1 (800 мл), метанол (300 мл)]. Для нанесения неочищенного продукта на колонку, необходимо растворить реакционный продукт в этил ацетате/циклогексане 1:1 с небольшим количеством тетрагидрофурана. Цис-амид AMD-124UC (точка плавления 145-155°C) получали в форме бесцветного твердого вещества с выходом 31% (204 мг, 0,40 ммоль).
13С{1Н}-ЯМР (101 МГц, ДМСО-D6) δ част, на млн: 22,5 (1 С), 29,3 (2 С), 32,6 (2 С), 37,8 (2 С), 41,3 (1 С), 59,5 (1 С), 60,3 (1 С, br), 105,4 (1 С), 111,1 (1 С), 114,3 (2 С, d, J=20 Гц), 117,3 (1 С), 118,4 (1 С), 120,5 (1 С), 123,1 (1 С), 126,6 (1 С), 127,9 (2 С), 128,7 (2 С), 129,3 (2 С), 129,8 (2 С, d, J=8 Гц), 132,4 (1 С, br), 135,1 (1 С), 135,4 (1 С), 139,4 (1 С), 140,4 (1 С), 160,9 (1 С, d, J=243 Гц), 170,3 (1 С) Синтез S транс-спироамидных сравнительных примеров (AMDтранс)
Пример AMD-3транс
2',3',4',9'-Тетрагидро-N,N-диметил-4-(3-фторфенил)-2'-(бензотиофен-2-ил)карбонил-спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол]-4-амин цитрат(1:1)(транс-диастереоизомер)
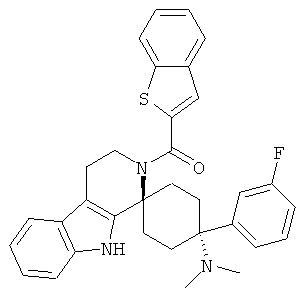
2',3',4',9'-Тетрагидро-N,N-диметил-4-(3-фторфенил)-2'-(бензотиофен-2-ил)карбонил-спиро[циклогексан-1,1'(1'H)-пиридо[3,4-b]индол]-4-амин (транс-диастереоизомер)
Хлорид бензо[b]тиофен-2-карбоновой кислоты (728 мг, 3,96 ммоль) растворяли, в атмосфере аргона, в абс. тетрагидрофуране (30 мл), и транс-спироамин AMN-2 транс (500 мг, 1,32 ммоль), растворенный в абс. тетрагидрофуране (60 мл), добавляли в течение 75 минут при комнатной температуре. Образовывался светлый осадок. После осуществления реакции в течение 2 часов, реакционную смесь разводили с помощью водой (15 мл); добавляли 1 н. раствор гидроксида натрия (15 мл), при охлаждении на льду, и перемешивали в течение 2,5 часов. Тетрагидрофуран удаляли в вакууме. Образовывалось твердое вещество и отделяли путем фильтрации и промывали водой (3×20 мл). Неочищенный продукт (587 мг) разделяли с помощью хроматографии [силикагель 60 (80 г); этил ацетат/циклогексан 1: 1 (1 л), этил ацетат/метанол 4: 1 (500 мл)]. Таким образом получали транс-амид в форме бесцветного твердого вещества с выходом 12% (82 мг) с точкой плавления 219-221°C.
13С-ЯМР (101 МГц, CDCI3) δ част, на млн: 22,4, 30,0, 30,9, 38,2, 46,4, 58,3, 59,5, 106,2, 111,0, 113,5, 113,7, 114,4, 114,7, 118,0, 119,1, 121,4, 122,5, 123,1, 124,7, 125,5, 125,8, 126,4, 128,7, 136,0, 138,7, 140,1, 140,4, 141,1, 142,1, 161,2, 163,7, 167,1
2',3',4',9'-Тетрагидро-N,N-диметил-4-(3-фторфенил)-2'-(бензотиофен-2-ил)карбонил-спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол]-4-амин цитрат (1:1) (транс-диастереоизомер; AMD-3транс)
Только что приготовленный транс-амид (82 мг, 0,152 ммоль) суспендировали при 80±С в этаноле (8 мл), и добавляли этанольный раствор (3 мл) лимонной кислоты (32 мг, 0,167 ммоль). При охлаждении до комнатной температуры, из прозрачного раствора осаждалось твердое вещество. Через 1,5 часа, смесь концентрировали до 2 мл, добавляли диэтиловый эфир (20 мл), и перемешивали в течение 20 минут. Бесцветное твердое вещество отделяли путем фильтрации и промывали с помощью диэтилового эфира (2×3 мл) (64 мг). Через 3 дня, дополнительное количество твердого вещества осаждалось из фильтрата при комнатной температуре и отфильтровывали с отсасыванием и промывали с помощью диэтилового эфира (2×2 мл) (35 мг). Две фракции объединяли. Таким образом получали транс-цитрат AMD-3транс с выходом 81% (89 мг) с точкой плавления 175-185°C.
Пример AMD-6транс
(Е)-2',3',4',9'-Тетрагидро-N,N-диметил-4-(3-фторфенил)-2'-(2-фенилвинил)карбонил-спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол]-4-амин цитрат (1:1) (транс-диастереоизомер)
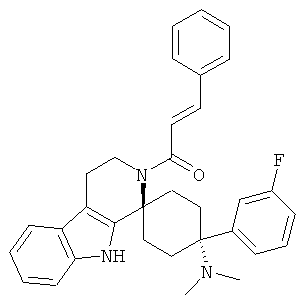
2',3',4',9'-Тетрагидро-N,N-диметил-4-(3-фторфенил)-2'-(2-фенилвинил)карбонил-спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол]-4-амин (транс-диастереоизомер)
Хлорид коричной кислоты (1,32 г, 7,92 ммоль) растворяли в атмосфере аргона в абс. тетрагидрофуране (30 мл), и неочищенный спироамин AMN-2цис (1,0 г, 2,64 ммоль, содержащий почти 10% транс-диастереоизомера AMN-2транс), растворенный в абс. тетрагидрофуране (60 мл), добавляли в течение 40 минут, при комнатной температуре. После осуществления реакции в течение 1 часа, к мутному реакционному раствору добавляли воду (20 мл) и, при охлаждении на льду, 1 н. раствор гидроксида натрия (20 мл), и перемешивали в течение 1,5 часа. Тетрагидрофуран удаляли в вакууме. Твердое вещество осаждалось и отделяли путем фильтрации и промывали водой (3×25 мл). Неочищенный продукт (1,16 г) разделяли с помощью хроматографии [силикагель 60 (200 г); этил ацетат/циклогексан 1: 1 (1,3 л), этил ацетат (1,6 л)]. Цис-амид получали в форме бесцветного твердого вещества с выходом 40% (540 мг) с точкой плавления 155-158°C. Транс-амид выделяли с выходом 7% (93 мг) с точкой плавления 151-155°C.
2',3',4',9'-Тетрагидро-N,N-диметил-4-(3-фторфенил)-2'-(2-фенилвинил)карбонил-спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол]-4-амин цитрат (1:1) (транс-диастереоизомер; AMD-6транс)
Только что приготовленный транс-амид (188 мг, 0,37 ммоль) растворяли при 80°C в этаноле (35 мл), и добавляли этанольный раствор (2 мл) лимонной кислоты (77 мг, 0,4 ммоль). Перемешивали в течение 2 часов при комнатной температуре, постепенно происходила кристаллизация. Смесь хранили в течение 1,5 часа при 5°C, и бесцветное твердое вещество отделяли путем фильтрации и промывали с помощью диэтилового эфира (3×3 мл) (146 мг). Фильтрат концентрировали и ресуспендировали в этаноле (1 мл), и добавляли диэтиловый эфир (20 мл). Через 16 часов, дополнительное количество бесцветной соли отделяли и промывали с помощью диэтилового эфира (2×2 мл) (36 мг). Две фракции объединяли и транс-цитрат AMD-6транс получали с выходом 71% (182 мг) с точкой плавления 161-164°C.
13С-ЯМР (101 МГц, ДМСО-D6) δ част, на млн: (транс диастереоизомер) 22,4, 29,2, 30,7, 37,9, 41,5, 43,1, 58,5, 59,6, 72,0, 105,5, 111,3, 113,2, 113,4, 113,5, 113,8, 117,3, 118,4, 120,5, 122,8, 123,1, 126,5, 127,7, 128,6, 129,1, 129,2, 135,0, 135,6, 139,8, 140,1, 160,7, 163,1, 169,9, 171,2, 175,2
Пример AMD-7транс
2',3',4',9'-Тетрагидро-N,N-диметил-4-(3-фторфенил)-2'-(3,4-диметоксибензил)карбонил-спиро[циклогексан-1,1'(1'Н)-пиридо[3,4-b]индол]-4-амин (транс-диастереоизомер)
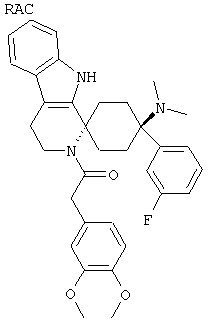
3,4-Диметоксифенилуксусную кислоту (1 г, 5,1 ммоль, 2,2 экв.) суспендировали в 25 мл абс. толуола, и добавляли тионил хлорид (0,84 мл, 11,6 ммоль, 5,0 экв.). Нагревали в течение 2 часов в колбе с обратным холодильником, и после этого растворитель удаляли. Остаток совместно перегоняли с абс. толуол (3×50 мл) и неочищенный продукт растворяли в дихлорметане (37 мл) и переносили в микроволновой сосуд. Добавляли спироамин AMN-2транс (0,875 мг, 2,32 ммоль) и основание Хьюнига (0,78 мл, 580 ммоль, 250 экв.), и микроволновой сосуд закрывали и нагревали в течение 20 минут при 120°C в микроволновом шкафу (Initiator Eight, Biotage). Для осуществления обработки, к реакционной смеси добавляли 17 мл воды и 17 мл 1 н. раствора гидроксида натрия. Эту смесь перемешивали в течение 2 часов при КТ. После этого фазы разделяли и водную фазу экстрагировали 3× с помощью дихлорметана. Объединенные органические фазы промывали водой и высушивали над сульфатом натрия. После удаления растворителя при пониженном давлении, остаток очищали путем колоночной хроматографии (силикагель; этил ацетат/н-гексан 2:1). Получали 0,236 г продукта AMD-7транс (18%).
ВЭЖХ/МС анализ: Rt=5,45 мин.; Чистота (УФ 200-400 нм)>99%; m/z=555,8
Синтез цис-спироэфирных сравнительных примеров (ЭФИРЦИС)
Пример ЭФИР-1цис
6'-Фтор-4',9'-дигидро-N,N-диметил-4-(3-тиенил)-спиро[циклогексан-1,1'(3'Н)-пирано[3,4-b]индол]-4-амин, метансульфонат (2:5) (цис-диастереоизомер)
Кетон Е-5 (446,6 мг, 2 ммоль) растворяли совместно с 5-фтортриптофолом (2, 394,4 мг, 2 ммоль) в абсолютном 1,2-дихлорэтане (30 мл). После этого к смеси добавляли метансульфоновую кислоту (0,13 мл, 2 ммоль), вследствие чего цвет реакционного раствора изменялся с красновато-коричневого на темно серый. Через 5 минут, начинало осаждаться светло-серое твердое вещество. Партию перемешивали в течение 20 часов при КТ. После этого метансульфонат цис-спироэфира отфильтровывали с отсасыванием и промывали с помощью 1,2-дихлорэтана (2×10 мл). Светло-серое твердое вещество получали с выходом 76% (733 мг) и с точкой плавления 143-145°C (ЭФИР-1цис). После этого к фильтрату добавляли 1 н. NaOH (30 мл), и перемешивали в течение 2 часов при КТ. Таким образом осаждали транс-спироэфир в форме бесцветного твердого вещества и получали, после фильтрации, с выходом 8% (58,5 мг).
1Н ЯМР (600 МГц, ДМСО-d6): 1,67 (m, 2 Н) 1,94 (m, 2 Н) 2,24 (m, 2 Н) 2,44 (s, 8 Н) 2,53 (s, 3 Н) 2,54 (s, 3 Н) 2,66 (t, J=5,27 Гц, 2 Н) 2,72 (m, 2 Н) 3,95 (t, J=5,28 Гц, 2 Н) 6,84 (m, 1H) 7,14 (m, 1H) 7,19 (dd, J=4,50 / 8,70 Гц, 1H) 7,47 (d, J=5,10 Гц, 1H) 7,83 (m, 1H) 8,07 (m, 1H) 9,67 (m, 1H) 10,80 (s, 1H)
Пример ЭФИР-2цис
4',9'-Дигидро-N,N-диметил-4-(2-тиенил)-спиро[циклогексан-1,1' (3'Н)-пирано[3,4-b]индол]-4-амин, метансульфонат (1:2) (цис-диастереоизомер)
Кетон Е-4 (223 мг, 1 ммоль) помещали совместно с триптофолом (2, 161 мг, 1 ммоль) в абсолютном дихлорметане (40 мл). После этого добавляли метансульфоновую кислоту (0,071 мл, 1,1 ммоль). Смесь перемешивали в течение 16 часов при КТ, вследствие чего осаждался метансульфонат спироэфира. Светло-серое твердое вещество (ЭФИР-2 чис) отфильтровывали с отсасыванием, промывали с помощью дихлорметана (2×10 мл) и получали с выходом 25% (117 мг) с точкой плавления 132±С. К фильтрату добавляли 1 н. NaOH (20 мл), и перемешивали в течение 16 часов при КТ. Органическую фазу отделяли и водную фазу экстрагировали с помощью дихлорметана (2×20 мл).
Органические фазы объединяли, высушивали и концентрировали, получали смесь веществ (274 мг) и разделяли с помощью хроматографии [силикагель G (20 г); этил ацетат/метанол 8:1]. Транс-спироэфир получали с выходом 54% (196 мг, точка плавления 235-238°C), и цис-спироэфир получали с выходом 10% (38 мг).
1Н ЯМР (600 МГц, ДМСО-d6)
1,82 (m, 2 Н) 1,98 (m, 2 Н) 2,33 (m, 2 Н) 2,36 (s, 6 Н) 2,60 (s, 3 Н) 2,61 (s, 3 Н) 2,53 (m, 2 Н) 2,70 (t, J=5,23 Гц, 2 Н) 3,96 (t, J=5,23 Гц, 2 Н) 6,94 (m, 1H) 7,00 (m, 1H) 7,21 (d, J=8,29 Гц, 1H) 7,34 (dd, J=3,74 / 5,28 Гц, 1H) 7,37 (d, J=7,37 Гц, 1H) 7,59 (d, J=2,76 Гц, 1H) 7,95 (d, J=5,32 Гц, 1H) 9,78 (m, 1H) 10,74 (s, 1H)
Оборудование и методы для ВЭЖХ-МС анализа: ВЭЖХ: Waters Alliance 2795 с PDA Waters 996; МС: ZQ 2000 MassLynx Single Quadrupol MC Детектор; Колонка: Waters Atlantis™ dC 18, 3 мкм, 2,1×30 мм; Температура колонки: 40°C, Элюент А: очищенная вода+0,1% муравьиная кислота; Элюент В: ацетонитрил (градиентная чистота)+0,1% муравьиная кислота; Градиент: от 0% В до 100% В в течение 8,8 мин, 100% В в течение 0,4 мин, от 100% В до 0% В в течение 0,01 мин, 0% В в течение 0,8 мин; Поток: 1,0 мл/мин; Ионизация: ES+, 25 В; Доводка: 100 мкл/мин. 70% метанол+0,2% муравьиная кислота; УФ: 200-400 нм.
Исследование фармакологических свойств характерных соединений
А) Сравнение анальгезирующей эффективности (в виде ED50 или % МРЕ при специфической тестируемой дозе) на модели острой боли (отдергивание хвоста, крысы/мыши) и на моделях мононевропатической боли (Chung, крысы; Bennett, крысы) или моделях полиневропатической боли (STZ полиневропатия, крысы).
Неожиданные фармакологические свойства соединений в соответствии с изобретением описаны главным образом путем сравнения друг с другом результатов для модели мононевропатической боли в соответствии с Chung у крыс и модели острой боли отдергивание хвоста у крыс. Посредством этого представляется возможным показать, что соединения в соответствии с изобретением не проявляют существенного антиноцицептивного действия на модели отдергивания хвоста у крыс при кратной дозе, которая имеет достоверную анальгезирующую эффективность на модели Chung (например,
Обезболивающий тест в тесте отдергивания хвоста у крыс
Тестируемые животные: Самки крыс Sprague Dawley (crl: CD (SD) беспородные; производитель: Charles River, Sulzfeld, Germany); вес тела: 130 - 190 г; животных содержали в стандартных клетках (клетки типа IV Makrolon, Ebeco, Castrop-Rauxel, Germany), заполненных в каждом случае не более чем 8 животными, с ритмом 12:12 часов свет/темнота и с едой и питьевой водой ad libitum.
Описание метода: Анальгезирующую эффективность тестируемых соединений исследовали в тесте поджога хвоста (отдергивание хвоста) у крыс в соответствии с методом D'Amour и Smith (J. Pharm. Exp. Ther. 72, 74 79 (1941)). Животных помещали поодиночке в специальные тестируемые клетки и основание хвоста подвергали воздействию сфокусированного теплового луча лампы (тип отдергивания хвоста 50/08/1.бс, Labtec, Dr. Hess). Интенсивность лампы устанавливали таким образом, чтобы время между включением лампы и неожиданным отрыванием хвоста (латентность отдергивания) у необработанных животных составлял 2,5-5 секунд. Перед введением тестируемого соединения, животных предварительно тестировали два раза в течение 30 минут и среднее значение этих измерений рассчитывали как среднее значение предварительного тестирования. Измерение боли в целом осуществляли через 5, 20, 40, 60, 90, 120, 180 и 240 минут после внутривенного введения тестируемого соединения или его наполнителя. Антиноцицептивное действие определяли как повышение латентности отдергивания в соответствии со следующей формулой: (% МРЕ)=[(T1-Т0)/(Т2-Т0)]×100, где: Т0= контрольный латентный период перед введением вещества, T1=латентный период после введения вещества, Т2=максимальная продолжительность воздействия горячего луча (12 секунд), МРЕ = максимально возможный эффект.
Для тестируемых соединений, имеющих антиноцицептивное действие, дозовую зависимость определяли путем введения 3-5 логарифмически возрастающих доз, которые включали пороговую дозу и максимально эффективную дозу. Полумаксимальную эффективную дозу (ED50) с соответствующими 95% доверительными интервалами определяли путем полулогарифмического регрессионного анализа во время максимального действия.
Статистическая оценка: Размер групп обычно составлял n=10. Дисперсионный анализ с повторными измерениями (повторные измерения ANOVA), а также анализ post hoc в соответствии с Bonferroni использовали для тестирования статистически значимого различия для %МРЕ данных между группами, получавшими дозу соединения, и контрольными группами, получавшими наполнитель. Уровень достоверности устанавливали на <0,05.
Отдергивание хвоста с уменьшенной интенсивностью горячего луча у крыс
Тестируемые животные: Самцов крыс Sprague-Dawley (производитель: Janvier, Le Genest St. Isle, France); вес тела: 200 - 250 г; животных содержали в стандартных клетках (клетки типа IV Makrolon, Ebeco, Castrop-Rauxel, Germany), заполненных в каждом случае не более, чем 5 животными, с ритмом 12:12 часов свет/темнота и с едой и питьевой водой ad libitum.
Описание метода: Модулирующую эффективность тестируемых веществ при остром вредном тепловом стимуле изучали в тесте горячего луча (отдергивание хвоста) у крыс в соответствии с методом D'Amour и Smith (J. Pharm. Exp. Ther. 72, 74 79 (1941)). Животных помещали поодиночке в специальные тестируемые компартменты и основание хвоста подвергали воздействию сфокусированного горячего луча измерительного устройства анальгезии (модель 2011, Rhema Labortechnik, Hofheim, Germany).
Интенсивность горячего луча устанавливали таким образом, чтобы время между включением лампы и неожиданным отрыванием хвоста (латентность отдергивания) у необработанных животных составляла приблизительно 12-13 секунд. Перед введением вещества в соответствии с изобретением, латентность отдергивания измеряли два раза с интервалом 5 минут и среднее значение определяли в виде контрольного латентного периода. Измерение латентности отдергивания хвоста осуществляли сначала через 10 минут после внутривенного введения тестируемого соединения или его наполнителя. После исчезновения антиноцицептивного эффекта (через 2-4 часа), измерения осуществляли с интервалами 30 минут вплоть до максимально 6,5 часов после введения вещества. Анти - или проноцицептивное действие определяли как повышение или понижение периода латентности отдергивания в соответствии со следующей формулой: (% МРЕ)=[(T1-Т0)/(Т2-Т0)]×100, где: Т0= контрольный латентный период перед введением вещества, T1= латентный период после введения вещества, Т2= максимальная продолжительность воздействия горячего луча (30 секунд), МРЕ= максимально возможный эффект. Для тестируемых соединений, имеющих антиноцицептивное действие, дозовую зависимость определяли путем введения 3-5 логарифмически возрастающих доз, которые включали пороговую дозу и максимально эффективную дозу. Полумаксимальную эффективную дозу (ED50) с соответствующими 95% доверительными интервалами определяли путем полулогарифмического регрессионного анализа во время максимального действия.
Статистическая оценка: Размер групп обычно составлял n=10. Дисперсионный анализ с повторными измерениями (повторные измерения ANOVA), а также анализ post hoc в соответствии с Bonferroni использовали для тестирования статистически значимого различия для % МРЕ данных между группами, получавшими дозу соединения, и контрольными группами, получавшими наполнитель. Уровень достоверности устанавливали на <0,05.
Обезболивающий тест в тесте отдергивания хвоста у мышей
Тестируемые животные: Самцы мышей NMRI (производитель: Charles River, Sulzfeld, Germany); вес тела: 20-25 г; животных содержали в стандартных клетках (клетки типа III Makrolon, Ebeco, Castrop-Rauxel, Germany) заполненных в каждом случае не более, чем 6 животными, с ритмом 12:12 часов свет/темнота и с едой и питьевой водой ad libitum.
Описание метода: Анальгезирующую эффективность тестируемого соединения исследовали в тесте горячего луча (отдергивание хвоста) у мышей в соответствии с методом D'Amour и Smith (J. Pharm. Exp. Ther. 72 74 79 (1941)). Животных помещали поодиночке в специальные исследовательские клетки и основание хвоста подвергали воздействию сфокусированного теплового луча электрической лампы (тип отдергивания хвоста type 55/12/10.fl Labtec, Dr. Hess). Интенсивность лампы устанавливали таким образом, чтобы время между включением лампы и неожиданным отрыванием хвоста (латентность отдергивания) у необработанных животных составляло 2,5-5 секунд. Перед введением тестируемого соединения, животных предварительно тестировали два раза в течение 30 минут и среднее значение этих измерений определяли как среднее значение предварительного тестирования. Измерение боли в целом осуществляли через 20, 40 и 60 минут после внутривенного введения тестируемого соединения или его наполнителя. Антиноцицептивное действие определяли как повышение периода латентности отдергивания в соответствии со следующей формулой: (% МРЕ)=[(T1-Т0)/(Т2-Т0)]×100, где: Т0= контрольный латентный период перед введением вещества, T1= латентный период после введения вещества, Т2= максимальная продолжительность воздействия горячего луча (12 секунд), МРЕ= максимально возможный эффект. Для тестируемых соединений, имеющих антиноцицептивное действие, дозовую зависимость определяли путем введения 3-5 логарифмически возрастающих доз, которые включали пороговую дозу и максимально эффективную дозу. Полумаксимальную эффективную дозу (ED50) с соответствующими 95% доверительными интервалами определяли путем полулогарифмического регрессионного анализа во время максимального действия.
Статистическая оценка: Размер групп обычно составлял n=10. Дисперсионный анализ с повторными измерениями (повторные измерения ANOVA), а также анализ post hoc в соответствии с Bonferroni использовали для тестирования статистически значимого различия для %МРЕ данных между группами, получавшими дозу соединения, и контрольными группами, получавшими наполнитель. Уровень достоверности устанавливали на <0,05.
Модель Chung: Мононевропатическая боль после лигатуры спинального нерва
Тестируемые животные: Самцов крыс Sprague Dawley (RjHan:SD беспородные; производитель: Janvier, Genest St. Isle, France), имеющих вес тела 140-160 г, содержали в стандартных клетках (клетки типа IV Makrolon, Ebeco, Castrop-Rauxel, Germany), заполненных в каждом случае не более, чем 8 животными, с ритмом 12:12 часов свет/темнота и с едой и питьевой водой ad libitum. Перед доставкой животных и операцией, соблюдали интервал одна неделя. После операции, животных тестировали несколько раз в течение периода 4-5 недель, соблюдая период отмывки по меньшей мере одна неделя.
Описание модели: При анестезии пентобарбиталом (Narcoren®, 60 мг/кг и/п, Merial GmbH, Hallbergmoos, Germany), в левых L5, L6 спинномозговых нервах удаляли часть паравертебральной мышцы и часть левого остистого отростка L5 поясничного позвонка. Спинномозговые нервы L5 и L6 осторожно выделяли и связывали плотной лигатурой (NC-шелк черный, USP 5/0, метрика 1, Braun Melsungen AG, Melsungen, Germany) (Kim и Chung 1992). После лигирования, мышцы и прилегающую ткань сшивали и рану закрывали с помощью металлических скоб. После восстановительного периода одна неделя, животных помещали в клетки с белым полом для измерения механической аллодинии. Порог отдергивания определяли на ипсилатеральной и/или контралатеральной задних лапах с помощью электронного филамента von Frey (Somedic АВ, Malmo, Sweden). Среднее значение для пяти стимуляций давало экспериментальную точку. Животных тестировали 30 минут перед и в различные промежутки времени после введения тестируемого вещества или раствора носителя. Данные представляли в виде % максимально возможного эффекта (% МРЕ) для предварительно тестируемых индивидуальных животных (=0% МРЕ) и тестируемых значений независимой плацебо контрольной группы (=100% МРЕ). Альтернативно, пороги отдергивания указывали в граммах. Для тестируемых соединений, имеющих обезболивающее действие, дозовую зависимость определяли путем введения 3-5 логарифмически возрастающих доз, которые включали пороговую дозу и максимально эффективную дозу. Полумаксимальную эффективную дозу (ED50) с соответствующими 95% доверительными интервалами определяли путем полулогарифмического регрессионного анализа во время максимального действия.
Статистическая оценка: Размер групп обычно составлял n=10. Дисперсионный анализ с повторными измерениями (повторные измерения ANOVA), а также анализ post hoc в соответствии с Bonferroni использовали для тестирования статистически значимого различия для %МРЕ данных между группами, получавшими дозу соединения, и контрольными группами, получавшими наполнитель. Уровень достоверности устанавливали на <0,05.
Ссылка: Kim, S.H. и Chung, J.M., An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat, Pain, 50 (1992) 355-363.
Модель Bennett: Мононевропатическая боль у крыс
Тестируемые животные: Самцы крыс Sprague Dawley (RjHan:SD беспородные; производитель: Janvier, Genest St. Isle, France), имеющих вес тела 140-160 г, содержали в стандартных клетках (клетки типа IV Makrolon, Ebeco, Castrop-Rauxel, Germany), заполненных в каждом случае не более, чем 8 животными, с ритмом 12:12 часов свет/темнота и с едой и питьевой водой ad libitum. Перед доставкой животных и операцией, соблюдали интервал одна неделя. После операции, животных тестировали несколько раз в течение периода 4 недели, соблюдая период отмывки по меньшей мере одна неделя.
Описание метода: Исследование эффективности невропатической боли осуществляли на модели Bennett (хроническая компрессия; Bennett и Xie, 1988, Pain 33: 87-107). Под анестезией наркореном, крысам накладывали четыре свободные лигатуры на правый седалищный нерв. У животных развивалась сверхчувствительность иннервированой лапы вследствие поврежденного нерва, которую измеряли количественно, после фазы восстановления в течение одной недели, приблизительно в течение четырех недель 4°C холодной металлической пластины (холодовая аллодиния). Животных наблюдали на пластине в течение 2 минут, и измеряли отдергивающих реакций поврежденной лапы.
Оценка и статистика: Исходя из предварительного значения перед введением вещества, действие вещества определяли в течение одного часа в четыре временные точки (например, 15, 30, 45, 60 минут после введения) и полученную площадь под фармакокинетической кривой (AUC) и ингибирование холодовой аллодинии в индивидуальных точках измерения выражали в виде процента действия относительно контрольного наполнителя (AUC) или исходного значения (индивидуальные точки измерения). Размер групп составлял n=10, достоверность анти-аллодинического действия (р<0,05) определяли с помощью дисперсионного анализа с повторными измерениями и анализа post hoc в соответствии с Bonferroni.
STZ модель: Полиневропатическая боль у крыс
Тестируемые животные: Самцы крыс Sprague Dawley (производитель: Janvier, Genest St. Isle, France); вес тела 140-160 г; животных содержали в стандартных клетках (клетки типа IV Makrolon, Ebeco, Castrop-Rauxel, Germany), заполненных в каждом случае не более чем 8 животных, с ритмом 12:12 часов свет/темнота и с едой и питьевой водой ad libitum.
Описание метода: Для индуцирования диабета, самцам крыс Sprague Dawley внутрибрюшинно инъецировали стрептозотоцин (STZ, 75 мг/кг). Диабетические крысы имели уровень глюкозы в крови по меньшей мере 17 мМ через одну неделю после инъекции STZ. Контрольным животным инъецировали раствор наполнителя. Определение порога механического ноцицептивного стимула (в граммах) осуществляли с помощью альгезиметра в испытании лапы под давлением в соответствии с Randall & Selitto (1957). В тесте, повышение давление стимула осуществляли на дорсальной поверхности задней лапы и записывали давление, которое немедленно приводило к рефлекторному отдергиванию лапы или издаванию звуков. Тесты проводили через три недели после индукции диабета. Порог механического ноцицептивного стимула измеряли перед и через 15, 30, 45 и 60 минут после введения вещества диабетическим животным и контрольным животным.
Ссылки: Randall LO, Selitto JJ. A method for measurement of analgesic activity on inflamed tissue. Arch. Int. Pharamcodyn. Arch. Int. Pharamcodyn. 1957; 111: 409-19
В) Сравнение болеутоляющего эффективного диапазона доз на модели мононевропатической боли (Chung, крысы) с диапазоном доз, при котором наблюдаются опиоид-типичные побочные действия.
Неожиданные фармакологические свойства соединений в соответствии с изобретением описаны главным образом путем сравнения друг с другом результатов, полученных на модели Chung у крыс (в качестве примера анальгезирующей эффективности по отношению к невропатической боли) и модели определения газов крови у крыс (в качестве примера дыхательной недостаточности как чрезвычайно тяжелого в настоящее время легко количественно определяемого опиоид-типичного побочного действия). Таким образом представляется возможным показать, что соединения в соответствии с изобретением не запускают существенную дыхательную недостаточность у крыс при введении в кратной дозе, которая обладает существенной анальгезирующей активностью на модели Chung (например,
Определение газов крови: Метод измерения артериального pCO2 и pO2 у крыс
Действие тестируемых веществ на дыхательную недостаточность изучали после в/в введения бодрствующим крысам с измерительными приборами. Тестируемым параметром являлось изменение парциального давления углекислого газа (pCO2) и парциального давления кислорода (pO2) в артериальной крови после введения вещества.
Тестируемые животные: Самцы крыс Sprague-Dawley (crl: CD (SD) беспородные; производитель: Charles River, Sulzfeld, Germany); вес: 250-275 г; животных содержали поодиночке в стандартных клетках (клетки типа II Makrolon, Ebeco, Castrop-Rauxel, Germany), с ритмом 12:12 часов свет/темнота и с едой и питьевой водой ad libitum.
Описание метода: По меньшей мере за 6 дней перед введением тестируемого вещества, РР катетер имплантировали в бедренную артерию и яремную вену крыс, при анестезии пентобарбиталом. Катетеры заполняли раствором гепарина (4000 ME) и закрывали проволочным мандреном. Введение тестируемого вещества или наполнителя осуществляли через венозный катетер. Перед введением вещества или наполнителя и в определенные точки времени после введения вещества или наполнителя, артериальный катетер в каждом случае открывали и продували приблизительно 500 мкл раствора гепарина. После этого около 100 мкл крови удаляли через катетер и отбирали с помощью гепаринизированного стеклянного капилляра. Катетер снова промывали раствором гепарина и снова закрывали. Артериальную кровь анализировали сразу с помощью устройства для определения газов крови (ABL 5, Radiometer GmbH, Willich, Germany). После минимального периода отмывки одна неделя, животные снова могут быть включены в тест.
Оценка теста и статистика: Устройство для определения газов крови автоматически определяет значения для pCO2 и pO2 крови в мм рт. ст. Действия веществ на парциальное давление рассчитывали в виде процента изменения относительно предварительных значений без вещества или наполнителя. Для статистической оценки, измеренные значения после введения вещества и одновременно измеренные значения после применения наполнителя сравнивали с помощью однофакторного дисперсионного анализа (однофакторный ANOVA) и анализа post hoc в соответствии с Dunnett. Уровень достоверности устанавливали на <0,05. Размер групп обычно составлял n=6.
Сердечнососудистые параметры: Способ измерения артериального давления и частота пульса у бодрствующих кроликов
Действие тестируемых веществ на сердечнососудистую систему изучали после в/в введения бодрствующим кроликам с телеметрией. Тестируемыми параметрами были изменение частоты пульса и артериального давления после введения вещества.
Тестируемые животные: Самки кроликов (New Zealand Whites; производитель: Charles River, Kisslegg, Germany); вес тела: около 3-5,5 кг; животных содержали поодиночке в специальных клетках для кроликов (W×D×Н=885×775×600 мм; Ebeco, Castrop-Rauxel, Germany), с ритмом 12:12 часов свет/темнота и с едой и питьевой водой ad libitum.
Приготовление теста: По меньшей мере за 21 день до начала эксперимента, блок телеметрической аппаратуры (TL11M2-D70-PCT от DSI, St. Paul, Minnesota, USA) для измерения артериального давления и электрокардиограммы (ЭКГ) имплантировали животным, при полной анестезии (изофлуран 2-3%). Таким образом катетер для измерения давления блока телеметрической аппаратуры вводили в A. femoralis и два бипотенциальных электрода фиксировали подкожно в области грудины или в области верхней левой грудной клетки. Передающий блок зашивали в кожный карман в области левого бока животных. Запись телеметрических сигналов осуществляли с помощью приемников типа RMC-1 (DSI). Для записи данных, хранения данных и обработки данных, использовали пакет программ Po-Ne-Mah (DSI).
Процедура теста: Введение вещества или наполнителя осуществляли через венозный катетер (V. auricularis). Перед введением вещества или наполнителя и в определенные точки времени после введения вещества или наполнителя, непосредственно определяли частоту пульса и артериальное давление (систолическое, диастолическое и среднее значение) с помощью калиброванной телеметрической системы и хранили в электронном виде. После минимального периода отмывки одна неделя, животных снова можно включать в тест.
Оценка теста и статистика: На основании измеренных значений артериального давления (в мм рт.ст.) и частоты пульса (в ударах в минуту) в определенные временные точки, в каждом случае определяли средние значения 10 последовательных сердечных ударов. Действие вещества на тестируемые параметры рассчитывали в виде процента изменения относительно предварительных значений без вещества или наполнителя. Для статистической оценки, измеренные значения после введения вещества и одновременно измеренные значения после введения наполнителя сравнивали с помощью однофакторного дисперсионного анализа (однофакторный ANOVA) и анализа post hoc в соответствии с Dunnett. Уровень достоверности устанавливали на <0,05. Размер групп обычно составлял n=6.
Тест прохождения активированного угля: Способ измерения скорости транзита пищи по желудочно-кишечному тракту у мышей
Тестируемые животные: Самцы мышей NMRI (производитель: Charles River, Sulzfeld, Germany), вес тела: 30-35 г; животных содержали в стандартных клетках (клетки типа IV Makrolon, Ebeco, Castrop-Rauxel, Germany), заполненных в каждом случае не более чем 18 животными, с ритмом 12:12 часов свет/темнота и с едой и питьевой водой ad libitum.
Описание теста: Перед тестом, животные голодали в течение 20-24 часов в клеточных вставках из проволочной сетки. Суспензию активированного угля (10% активированный уголь в 0,5% CMC раствора; вводимый объем: 0,1 мл/10 г вес тела) вводили животным перорально в качестве маркерного вещества прохождения через желудочно-кишечный тракт. После этого внутривенно вводили тестируемое вещество или раствор наполнителя. Через два часа после введения суспензии активированного угля, животных умертвляли посредством отравления CO2. После этого желудочно-кишечный тракт удаляли от желудка вплоть до и включая слепую кишку и растягивали на стеклянной пластинке, увлажненной 0,9% раствором NaCl. После этого измеряли расстояние пилорус-слепая кишка и расстояние, пройденное суспензией угля (самая дальняя точка).
Оценка теста: Для определения относительного ингибирования прохождения через желудочно-кишечный тракт, получали частное расстояния, пройденного суспензией угля (в см)/расстояние пилорус-слепая кишка (в см). Его указывали в виде % ингибирования. Для статистической оценки, измеренные значения после введения вещества и одновременно измеренные значения после введения наполнителя сравнивали с помощью однофакторного дисперсионного анализа (однофакторный ANOVA) и анализа post hoc в соответствии с Dunnett. Уровень достоверности устанавливали на<0,05. Размер групп обычно составлял n=10.
Rota-Rod тест: Метод изучения двигательной координации у мышей
Тестируемые животные: Самцы мышей CD-1 (производитель: Charles River, Sulzfeld, Germany), вес тела: 18-25 г; животных содержали в стандартных клетках (клетки типа IV Makrolon, Ebeco, Castrop-Rauxel, Germany), заполненных в каждом случае не более чем 18 животными, с ритмом 12:12 часов свет/темнота и с едой и питьевой водой ad libitum.
Описание метода: Относительно описания метода см.: Kuribara Н., Higuchi Y., Tadokoro S. (1977), Effects of central depressants on Rota-Rod and traction performance in mice. Japan. J. Pharmacol. 27, 117-126.
Статистическая оценка: Для статистической оценки, измеренные значения после введения вещества и одновременно измеренные значения после введения наполнителя сравнивали с помощью однофакторного дисперсионного анализа (однофакторный ANOVA) и анализа post hoc в соответствии с Dunnett.
Уровень достоверности устанавливали на <0,05. Размер групп обычно составлял n=10.
Тест прыжков: Метод изучения потенциала физической зависимости у мышей
Тестируемые животные: Самцы мышей NMRI (производитель: Charles River, Sulzfeld, Germany), вес тела: 20-24 г; животных содержали в стандартных клетках (клетки типа III Makrolon, Ebeco, Castrop-Rauxel, Germany), заполненных в каждом случае не более чем 6 животных, с ритмом 12:12 часов свет/темнота и с едой и питьевой водой ad libitum.
Описание метода: Тестируемые вещества вводили внутрибрюшинно в целом 7х в течение двух дней. 5 введений осуществляли в первый день в 9:00, 10:00, 11:00, 13:00 и 15:00 и во второй день в 9:00 и 11:00. Первые 3 введения вводили в возрастающих дозах (схема дозирования) и после этого в третьей части дозы. Синдром отмены вызывали налоксоном 30 мг/кг (и/п) через 2 часа после последнего введения вещества. Сразу после этого, животных помещали поодиночке в прозрачные коробки для наблюдения (высота 40 см, диаметр 15 см) и подсчитывали прыжковые реакции в течение периода времени 15 минут при 5-ти минутных интервалах. Морфин вводили сопутствующе в дозе в качестве сравнения /стандарта. Количественное определение абстиненции осуществляли с помощью количества прыжков с 0 до 10 минуты после введения налоксона. Определяли количество животных на группу с более чем 10 прыжками/10 минут и записывали в качестве "% положительных животных". Дополнительно, рассчитывали среднюю частоту прыжков на группу.
Статистическая оценка: Оценку экспериментальных данных относительно статистически значимого различия между группами, получавшими дозу соединения, и контрольными группами, получавшими наполнитель, предпочтительно осуществляли с помощью точного критерий Фишера для параметра "% положительных животных" и с помощью критерия Крускала-Уоллиса для параметра "частота прыжков", предпочтительно как описано в экспериментальном разделе. Уровень достоверности устанавливали на р<0,05 в каждом случае. Размер групп обычно составлял n=12.
Ссылка: Saelens JK, Arch Int Pharmacodyn 190: 213-218, 1971
Предпочтение мест введения психоактивного вещества: Метод изучения возможной индукции психической зависимости/привыкания у крыс
Описание метода: Относительно исследования предпочтения места см.: Tzschentke, Т.М., Bruckmann, W. и Friderichs, F. (2002) Lack of sensitization during place conditioning у крыс is consistent with the low abuse potential of tramadol. Neuroscience Letters 329, 25-28.
Статистическая оценка: Оценку экспериментальных данных относительно статистически значимого различия в предпочтениях животных для активного компонента или наполнителя предпочтительно осуществляли с помощью парного t-критерия. Уровень достоверности устанавливали на р<0,05. Размер групп обычно составлял n=8.
2) Факторы отличия рассчитывали в виде соотношения NOEL и среднего значения
система,
кролики
2) Факторы отличия рассчитывали в виде соотношения NOEL и среднего значения
Вывод: Примеры AMD-6цис и AMD-7цис выбраны для иллюстрации неожиданных фармакологических свойств соединений в соответствии с изобретением. Они представляют собой высокоаффинные лиганды к ORL1 рецептору и µ-опиоид рецептору, имеющие соотношение аффинности к ORL1 рецептору относительно аффинности к µ-опиоид рецептору приблизительно 5 или приблизительно 6. Примеры AMD-6цис и AMD-7цис свидетельствуют о том, что соединения в соответствии с изобретением имеют чрезвычайно высокую эффективность по отношению к невропатической боли (в данном случае:
2) NOEL (= Максимальная доза препарата, не приводящая к развитию наблюдаемых эффектов) обозначает верхнюю дозу без полученных экспериментальных данных (то есть дозу без достоверного эффекта)
Вывод: Соединения в соответствии с изобретением проявляют чрезвычайно хорошую эффективность по отношению к невропатической боли. Неожиданно, с другой стороны, не наблюдали существенного антиноцицептивного действия на модели острой боли даже в дозах, которые приблизительно в от 10 раз до более чем 100 раз выше за эффективные дозы на модели невропатии. Аналогичным образом, не наблюдали существенных опиоид-типичных побочных действий, неожиданно, на животных моделях для обнаружения побочных действий (например, определение газов крови, прохождение активированного угля через желудочно-кишечный тракт и RotaRod тест) для 10 раз до более чем в 300 раз более высоких дозах.
2) NOEL (= Максимальная доза препарата, не приводящая к развитию наблюдаемых эффектов) обозначает верхнюю дозу без полученных экспериментальных данных (то есть дозу без достоверного эффекта)
Вывод: Неожиданно, только цис-спироамины в соответствии с изобретением (в данном случае Примеры AMD-6цис и Пример AMN-2цис) проявляют очень хорошую эффективность по отношению к невропатической боли, и в то же время характеризуются отсутствием антиноцицептивного действия при острой боли. Аналогичным образом, не наблюдали существенных опиоид-типичных побочных действий на животных моделях для обнаружения побочных действий (как, например, в данном случае определение газов крови) в дозах, которые многократно выше. Транс-спироамины (в данном случае Сравнительный Пример AMD-6транс и Сравнительный Пример AMN-2транс), с другой стороны, не проявили отличий между дозами, которые эффективны по отношению к невропатической боли или по отношению к острой боли. Аналогичным образом, не было обнаружено отличий между дозами, при которых наблюдаются опиоид-типичные побочные действия (как, например, в данном случае определение газов крови). При суммарном сравнении, AMD-54MC и AMD-64MC проявили наибольшие отличия в максимально возможном анальгезирующем действии.
2) NOEL (= Максимальная доза препарата, не приводящая к развитию наблюдаемых эффектов) обозначает верхнюю дозу без полученных экспериментальных данных (то есть дозу без достоверного эффекта)
Вывод: Неожиданно, только цис-спироамины в соответствии с изобретением (в данном случае Пример AMN-2цис) проявляют очень хорошую эффективность по отношению к невропатической боли, и в то же время характеризуются отсутствием антиноцицептивного действия при острой боли. Аналогичным образом, не наблюдали существенных опиоид-типичных побочных действий на животных моделях для обнаружения побочных действий (как, например, в данном случае определение газов крови) в дозах, которые многократно выше. Цис-спироэфиры (в данном случае Сравнительный Пример Эфир-2цис и Сравнительный Пример Эфир-1цис), с другой стороны, не проявили заметных отличий между дозами, которые эффективны по отношению к невропатической боли или по отношению к острой боли.
2) NOEL (= Максимальная доза препарата, не приводящая к развитию наблюдаемых эффектов) обозначает наивысшую дозу без полученных экспериментальных данных (то есть дозу без достоверного эффекта)
Вывод: Сравнение AMD-5цис (свободное основание) и AMD-6цис (цитратная соль) показало отсутствие релевантных отличий фармакологических свойств основания и соли.
Ki [нМ]
ЕС50 [эфф.%]
| название | год | авторы | номер документа |
|---|---|---|---|
| СПИРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ЦИКЛОГЕКСАНА | 2008 |
|
RU2484092C2 |
| СПИРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ЦИКЛОГЕКСАНА | 2008 |
|
RU2470933C2 |
| ГИДРОКСИМЕТИЛЦИКЛОГЕКСИЛАМИНЫ | 2009 |
|
RU2514192C2 |
| ПРОИЗВОДНЫЕ СПИРО(5.5)УНДЕКАНА | 2009 |
|
RU2515895C2 |
| СПИРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ЦИКЛОГЕКСАНА СО СРОДСТВОМ К ORL1-РЕЦЕПТОРУ | 2004 |
|
RU2383544C2 |
| НОВЫЕ СПИРО[ИМИДАЗОЛИДИН-4,3'-ИНДОЛ]-2,2',5(1'Н)-ТРИОНЫ ДЛЯ ЛЕЧЕНИЯ СОСТОЯНИЙ, АССОЦИИРОВАННЫХ С ВАНИЛЛОИДНЫМ РЕЦЕПТОРОМ 1 | 2007 |
|
RU2421457C2 |
| ЗАМЕЩЕННЫЕ ЦИКЛОГЕКСИЛДИАМИНЫ | 2009 |
|
RU2526251C2 |
| СПИРОЦИКЛИЧЕСКИЕ АЗАИНДОЛЬНЫЕ ПРОИЗВОДНЫЕ | 2007 |
|
RU2468028C2 |
| ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ 1,2,3,4-ТЕТРАГИДРОИЗОХИНОЛИНА | 2005 |
|
RU2378257C2 |
| КОМБИНАЦИИ, СОДЕРЖАЩИЕ АЛЬФА-2-ДЕЛЬТА ЛИГАНДЫ И ИНГИБИТОРЫ ОБРАТНОГО ЗАХВАТА СЕРОТОНИНА/НОРАДРЕНАЛИНА | 2004 |
|
RU2320369C2 |
Изобретение относится к области органической химии, а именно к производным цис-тетрагидро-спиро(циклогексан-1,1'-пиридо[3,4-b]индол)-4-амина формулы (III) в форме свободных оснований или физиологически приемлемых солей, где R1 представляет собой Н или СН3; R2, R3, R4 и R5 представляют собой Н или галоген. Также изобретение относится к фармацевтической композиции на основе соединения формулы (III), обладающей активностью в отношении ORL1 рецептора и µ-опиоид рецептора. Технический результат: получены новые соединения, обладающие полезными биологическими свойствами. 2 н. и 6 з.п. ф-лы, 7 табл., 22 пр.
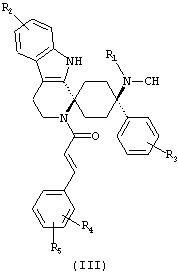
1. Соединение общей формулы (III)
где
R1 представляет собой -Н или -СН3;
R2 представляет собой -Н или -галоген;
R3 представляет собой -Н или -галоген;
R4 представляет собой -Н или -галоген и
R5 представляет собой -Н или -галоген;
в форме свободных оснований или физиологически приемлемых солей.
2. Соединение, как заявлено в п. 1, где R2 представляет собой -Н и/или R3 представляет собой -F.
3. Соединение, как заявлено в п. 1 или 2, которое выбирают из группы, включающей
- (Е)-1-((1s,4s)-4-(диметиламино)-4-фенил-3′,4′-дигидроспиро[циклогексан-1,1′-пиридо[3,4-b]индол]-2′(9′Н)-ил)-3-фенилпроп-2-ен-1-он;
- (Е)-1-((1s,4s)-4-(диметиламино)-4-(3-фторфенил)-3′,4′-дигидроспиро[циклогексан-1,1′-пиридо[3,4-b]индол]-2′(9′Н)-ил)-3-фенилпроп-2-ен-1-он;
- (Е)-1-((1s,4s)-4-(диметиламино)-6′-фтор-4-(3-фторфенил)-3′,4′-дигидроспиро[циклогексан-1,1′-пиридо[3,4-b]индол]-2′(9′Н)-ил)-3-фенилпроп-2-ен-1-он;
- (Е)-1-((1s,4s)-4-(диметиламино)-6′-фтор-4-фенил-3′,4′-дигидроспиро[циклогексан-1,1′-пиридо[3,4-b]индол]-2′(9′Н)-ил)-3-фенилпроп-2-ен-1-он и
- (Е)-1-((1s,4s)-4-(диметиламино)-4-(4-фторфенил)-3′,4′-дигидроспиро[циклогексан-1,1′-пиридо[3,4-b]индол]-2′(9′Н)-ил)-3-фенилпроп-2-ен-1-он;
в форме свободных оснований или физиологически приемлемых солей.
4. Соединение, как заявлено в п. 1 или 2, обладающее структурой
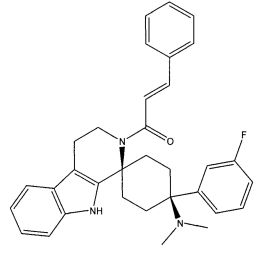
в форме свободного основания или физиологически приемлемых солей.
5. Соединение, как заявлено в п. 1 или 2, для применения в качестве лекарственного средства, обладающего активностью в отношении ORL1 рецептора и µ-опиоид рецептора.
6. Фармацевтическая композиция, обладающая активностью в отношении ORL1 рецептора и µ-опиоид рецептора, которая содержит физиологически приемлемый носитель и соединение в эффективном количестве, как заявлено в любом из пп. 1-4.
7. Соединение, как заявлено в п. 1 или 2, для применения для лечения невропатической и/или хронической боли.
8. Соединение, как заявлено в п. 7, где введение осуществляют не более чем один раз в сутки.
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| СПИРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ЦИКЛОГЕКСАНА | 2003 |
|
RU2354656C2 |

