Перекрестные ссылки на родственные заявки
[0001] Настоящая заявка испрашивает приоритет предварительных патентных заявок США № 62/537292, поданной 26 июля 2017, и 62/647492, поданной 23 марта 2018; содержание каждой из которых включено в настоящее изобретение с помощью ссылки во всей своей полноте.
Область техники настоящего изобретения
[0002] Настоящее изобретение относится к конкретным пролекарствам замещенных гетероциклических конденсированных гамма-карболинов, в свободной, твердой, фармацевтически приемлемой солевой и/или по существу чистой форме, как описано в настоящем изобретении, их фармацевтическим композициям, и способам применения в лечении заболеваний, включающих 5-HT2A рецепторный, серотониновый транспортерный (SERT), пути, включающие дофаминовые D1 и/или D2 рецепторные сигнальные системы, и/или μ-опиоидный рецептор, например, заболеваний и расстройств, таких как тревога, психоз, шизофрения, нарушения сна, сексуальные расстройства, мигрень, состояния, связанные с болью (включая головную боль, невропатическую боль и как сильный анальгетик), фибромиалгию, хроническую усталость, социальные фобии, желудочно-кишечные расстройства, такие как дисфункция моторики желудочно-кишечного тракта и ожирение; депрессия и поведенческие расстройства, такие как расстройства, связанные с психозом или болезнью Паркинсона; психоз, такой как шизофрения, связанный с депрессией; биполярное расстройство; наркомания, такая как опиатная зависимость и алкогольная зависимость, абстинентный наркотический синдром; обсессивно-компульсивное расстройство (OCD), обсессивно-компульсивное расстройство личности (OCPD) и родственные расстройства; и других психиатрических и неврологических заболеваний, а также к комбинациям с другими агентами. В некоторых вариантах осуществления, заболевания или расстройства могут включать терапевтически резистентную депрессию, кокаиновую зависимость и/или амфетаминовую зависимость, расстройство, связанное с употреблением опиоидов и симптомы отмены опиоидов.
УРОВЕНЬ ТЕХНИКИ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
[0003] Замещенные гетероциклические конденсированные гамма-карболины являются известными в качестве агонистов или антагонистов 5-HT2 рецепторов, в частности 5-HT2A и 5-HT2C рецепторов, в лечении расстройств центральной нервной системы. Данные соединения описаны в патенте США № 6548493; 7238690; 6552017; 6713471; 7183282; U.S. RE39680 и U.S. RE39679, в качестве новых соединений, пригодных для лечения расстройств, связанных с модуляцией 5-HT2A рецепторов, таких как ожирение, тревога, депрессия, психоз, шизофрения, расстройства сна, сексуальные расстройства, мигрень, заболевания, связанные с головной болью, социальные фобии, расстройства желудочно-кишечного тракта, такие как дисфункция моторики желудочно-кишечного тракта и ожирение. PCT/US08/03340 (WO 2008/112280) и его эквивалент в США US 2010/113781 и заявка США серийный No. 10/786935 (опубликованная как US 2004/209864) также описывают способы получения замещенных гетероциклических конденсированных гамма-карболинов и применение данных гамма-карболинов в качестве серотониновых агонистов и антагонистов, пригодных для борьбы и предотвращения расстройств центральной нервной системы, таких как аддиктивное поведение и расстройства сна.
[0004] Кроме того, WO/2009/145900 (и его эквивалент US 2011/071080) описывает применение конкретных замещенных гетероциклических конденсированных гамма-карболинов для лечения комбинации психоза и депрессивного расстройства, а также расстройства сна, депрессивных и/или поведенческих расстройств у пациентов с психозом или болезнью Паркинсона. В добавление к расстройствам, связанным с психозом и/или депрессией, настоящая патентная заявка описывает и заявляет применение данных соединений при низкой дозе для селективного антагонизма 5-HT2A рецепторов без воздействия или с минимальным воздействием на дофаминовые D2 рецепторы, посредством этого являясь пригодными для лечения расстройств сна без побочных эффектов, связанных с высокой загруженностью дофаминовых D2 путей или побочных эффектов других путей (например, GABAA рецепторов), связанных с общепринятыми седативно-гипнотическими агентами (например, бензодиазепинами), включая, но не ограничиваясь, развитие наркотической зависимости, мышечную гипотонию, слабость, головную боль, помутнение зрения, головокружение, тошноту, рвоту, эпигастральный дистресс, диарею, боли в суставах и боли в груди. WO 2009/114181 (и его эквивалент US 2011/112105) также описывает способы получения кристаллов солей присоединения толуолсульфокислоты данных замещенных гетероциклических конденсированных гамма-карболинов.
[0005] Кроме того, последние данные показывают, что приведенные выше замещенные гетероциклические конденсированные гамма-карболины могут действовать, частично, через антагонизм рецептора NMDA через передачу сигналов mTOR1, способом, подобным способу кетамина. Кетамин представляет собой селективный антагонист NMDA рецепторов. Кетамин действует через систему, которая не связана с обычными психогенными моноаминами (серотонин, норэпинефрин и дофамин), и это представляет собой главную причину его гораздо более быстрых эффектов. Кетамин непосредственно проявляет антагонизм по отношению к внесинаптическим глутаматергическим NMDA рецепторам, что также косвенно приводит к активации глутаматных рецепторов типа AMPA. Последующие эффекты включают пути нейротрофического фактора мозга (BDNF) и киназы mTORC1. Как и в случае с кетамином, последние данные свидетельствуют о том, что соединения, относящиеся к соединениям настоящего изобретения, усиливают как NMDA, так и AMPA-индуцированные токи в пирамидных нейронах медиальной префронтальной коры крыс посредством активации D1 рецепторов, и что это связано с усилением передачи mTORC1 сигналов.
[0006] Родственные заявки WO 2017/132408 и US 2017/319580 описывают новые оксо-метаболиты соединений, описанных в приведенных выше публикациях. Данные новые оксо-метаболиты сохраняют большую часть уникальной фармакологической активности исходных соединений, включая ингибирование серотониновых рецепторов, ингибирование SERT и модуляцию дофаминовых рецепторов. Однако неожиданно было обнаружено, что данные оксо-метаболиты также проявляют значительную активность относительно мю-опиатных рецепторов.
[0007] Обсессивно-компульсивное расстройство (OCD) и родственные расстройства, стали широко распространенными, и их трудно лечить. По оценкам, OCD поражает около 2,3% людей в определенный момент их жизни, и в течение указанного года, по оценкам, 1,2% людей во всем мире страдают от данного расстройства. Половина людей, страдающих от OCD, начинают проявлять симптомы в возрасте до 20 лет, что может серьезно повлиять на их способность получить адекватное и эффективное образование. Однако без эффективного лечения болезнь может длиться десятилетиями. Основой фармакологического лечения OCD являются селективные ингибиторы обратного захвата серотонина (SSRI). Вторая линия терапии представляет собой антипсихотические агенты, такие как кломипрамин, рисперидон, кветиапин и оланзапин. Значительное количество пациентов либо не реагируют на данные агенты, либо не могут справиться с побочными эффектами, вызванными данными агентами. Совсем недавно сообщалось, что опиоидный анальгетик, трамадол, может быть эффективным при лечении OCD. Опиаты действуют совершенно иначе, чем традиционные препараты для лечения OCD, поэтому они предлагают возможность лечения для людей, которые не могут принимать традиционные серотонинергические препараты или для которых эти агенты являются неэффективными. Однако сильные опиатные агенты могут вызывать привыкание, а их применение может быть противопоказано некоторым пациентам. Таким образом, сохраняется острая необходимость в новых способах лечения боли, OCD и других расстройств.
[0008] Расстройства, связанные с наркотической зависимостью, такие как расстройство, связанное с употреблением опиатов (OUD), представляют собой другую группу расстройств, которые трудно успешно лечить. Передозировка опиоидами уносит приблизительно 100 жизней в Соединенных Штатах каждый день, и увлечение опиоидами продолжает расти в Соединенных Штатах. Метадон, бупренорфин и налтрексон являются наиболее часто применяемыми способами лечения OUD. Метадон представляет собой агонист мю-опиоидного рецептора (MOP), бупренорфин представляет собой частичный агонист MOP, а налтрексон представляет собой антагонист MOP. Каждый из данных агентов имел ограниченный успех, и долгосрочная приверженность назначенным способам лечения OUD остается низкой. Кроме того, данные способы лечения часто усугубляют общие сопутствующие заболевания, связанные с OUD, такие как расстройства настроения и тревога, что еще больше увеличивает риск ремиссии. Резкое прекращение употребления опиоидов (то есть «завязка») также связано с серьезными побочными эффектами, включая дисфорию, депрессию и тревогу, и обычные лекарственные средства не решают данные проблемы и могут усугублять их. Таким образом, существует острая необходимость в улучшенных способах лечения OUD.
СУЩНОСТЬ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
[0009] Соединения формулы A и B, показанных ниже, представляют собой эффективные антагонисты серотонинового 5-HT2A рецептора и частичные агонисты мю-опиатного рецептора или агонисты смещенной активности. Данные соединения также взаимодействуют с дофаминовыми рецепторами, в частности дофаминовыми D1 рецепторами.

Также считают, что соединения формулы A и/или B, посредством их активности по отношению к рецепторам D1, могут также усиливать опосредованную NMDA и AMPA передачу сигналов через mTOR путь. Соединения формулы A и B и их аналоги являются пригодными для лечения или профилактики расстройств центральной нервной системы, но в данной области техники существует потребность в пролекарствах соединений формулы A и B, которые при введении пациенту могут обеспечить повышенные терапевтические концентрации или улучшение фармакокинетического распределения или динамики данных соединений. Настоящее изобретение удовлетворяет данную потребность обеспечением соединений формул I и II и других, которые являются пролекарствами соединений формул A и B и их аналогов. Благодаря своему полезному метаболическому и фармакокинетическому профилю, соединения настоящего изобретения являются особенно пригодными для формулирования в виде композиций пролонгированного действия или пролонгированного высвобождения, которые при введении пациенту могут обеспечить повышенные терапевтические концентрации соединений A и B и их аналогов в течение длительного периода времени.
[00010] В первом аспекте, настоящее изобретение относится к соединению (соединение I) формулы I:
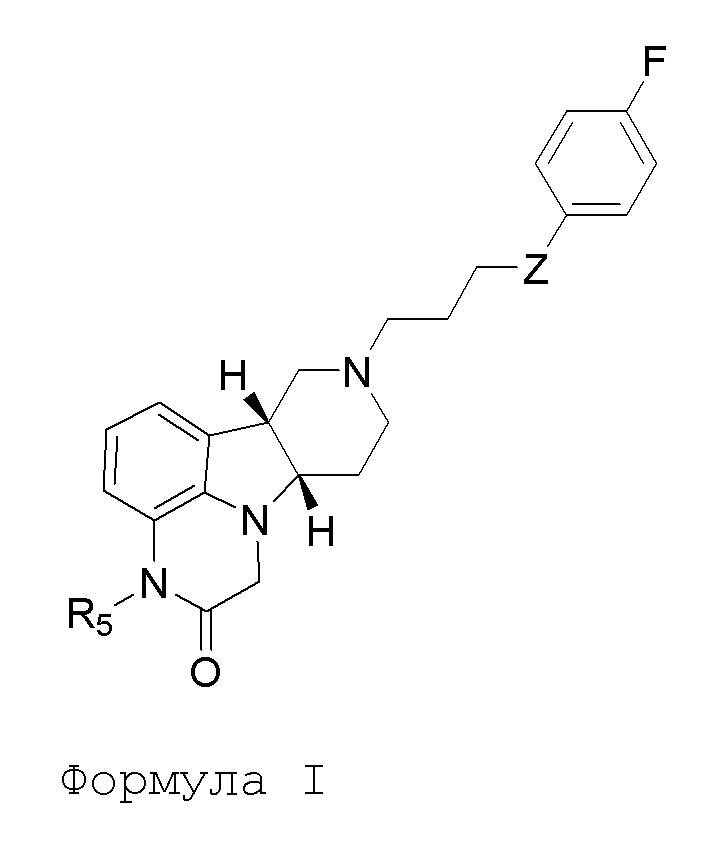
в которой:
R5 представляет собой -C(O)-O-C(Ra)(Rb)(Rc), -C(O)-O-CH2-O-C(Ra)(Rb)(Rc) или -C(R6)(R7)-O-C(O)-R8;
Z представляет собой O или -C(O)-;
R8 представляет собой -C(Ra)(Rb)(Rc), -O-C(Ra)(Rb)(Rc) или -N(Rd)(Re);
каждый Ra, Rb и Rc независимо выбран из H и C1-24алкила;
каждый Rd и Re независимо выбран из H и C1-24алкила;
каждый R6 и R7 независимо выбран из H, C1-6алкил, карбокси и C1-6алкоксикарбонила;
в свободной или солевой форме (например, фармацевтически приемлемой солевой форме), например, в выделенной или очищенной свободной или солевой форме (например, фармацевтически приемлемой солевой форме).
[00011] Настоящее изобретение относится к дополнительным примерным вариантам осуществления соединения формулы I, в свободной или солевой форме (например, фармацевтически приемлемой солевой форме), например, в выделенной или очищенной свободной или солевой форме (например, фармацевтически приемлемой солевой форме), включающим:
1.1 Соединение I, в котором Z представляет собой O;
1.2 Соединение I, в котором Z представляет собой -C(O);
1.3 Соединение I, 1.1, или 1.2, в котором R5 представляет собой -C(O)-O-C(Ra)(Rb)(Rc);
1.4 Соединение 1.3, в котором Ra представляет собой H, и каждый Rb и Rc независимо выбран из C1-24алкила, например, C1-20алкила, C5-20алкила, C9-18алкила, C10-16алкила или C11алкила, C12алкила, C13алкила, C14алкила, C15алкила или C16алкила;
1.5 Соединение 1.3, в котором Ra и Rb представляют собой H, и Rc представляет собой C1-24алкил, например, C1-20алкил, C5-20алкил, C9-18алкил, C10-16алкил или C11алкил, C12алкил, C13алкил, C14алкил, C15алкил или C16алкил;
1.6 Соединение 1.3, в котором каждый Ra, Rb и Rc независимо выбран из C1-24алкила, например, C1-20алкила, C5-20алкила, C9-18алкила, C10-16алкила или C11алкила, C12алкила, C13алкила, C14алкила, C15алкила или C16алкила;
1.7 Соединение 1.3, в котором каждый Ra, Rb и Rc представляет собой H;
1.8 Соединение 1.3, в котором Z представляет собой O, и Ra и Rb представляют собой H, и Rc представляет собой C10-14алкил (например, Rc представляет собой CH3(CH2)10 или CH3(CH2)14);
1.9 Соединение I, 1.1 или 1.2, в котором R5 представляет собой -C(O)-O-CH2-O-C(Ra)(Rb)(Rc);
1.10 Соединение 1.9, в котором Ra представляет собой H, и каждый Rb и Rc независимо выбран из C1-24алкила, например, C1-20алкила C5-20алкила, C9-18алкила, C10-16алкила или C11алкил, C12алкила, C13алкила, C14алкила, C15алкила или C16алкила;
1.11 Соединение 1.9, в котором Ra и Rb представляют собой H и Rc представляет собой C1-24алкил, например, C1-20алкил, C5-20алкил, C9-18алкил, C10-16алкил, или C11алкил, C12алкил, C13алкил, C14алкил, C15алкил или C16алкил;
1.12 Соединение 1.9, в котором каждый Ra, Rb и Rc независимо выбран из C1-24алкила, например, C1-20алкила, C5-20алкила, C9-18алкила, C10-16алкила или C11алкила, C12алкила, C13алкила, C14алкила, C15алкила или C16алкила;
1.13 Соединение 1.9, в котором каждый Ra, Rb и Rc представляет собой H;
1.14 Соединение I, 1.1, или 1.2, в котором R5 представляет собой -C(R6)(R7)-O-C(O)-R8, и R8 представляет собой -C(Ra)(Rb)(Rc);
1.15 Соединение I, 1.1, или 1.2, в котором R5 представляет собой -C(R6)(R7)-O-C(O)-R8, и R8 представляет собой -O-C(Ra)(Rb)(Rc);
1.16 Соединение 1.14 или 1.15, в котором Ra представляет собой H, и каждый Rb и Rc независимо выбран из C1-24алкила, например, C1-20алкила, C5-20алкила, C9-18алкила, C10-16алкила или C11алкила, C12алкила, C13алкила, C14алкила, C15алкила или C16алкила;
1.17 Соединение 1.14 или 1.15, в котором Ra и Rb представляют собой H, и Rc представляет собой C1-24алкил, например, C1-20алкил, C5-20алкил, C9-18алкил, C10-16алкил или C11алкил, C12алкил, C13алкил, C14алкил, C15алкил или C16алкил;
1.18 Соединение 1.14 или 1.15, в котором каждый Ra, Rb и Rc независимо выбран из C1-24алкила, например, C1-20алкила, C5-20алкила, C9-18алкила, C10-16алкила или C11алкила, C12алкила, C13алкила, C14алкила, C15алкила или C16алкила;
1.19 Соединение 1.14 или 1.15, в котором каждый Ra, Rb и Rc представляет собой H;
1.20 Соединение 1.14, в котором Z представляет собой O, и R6 представляет собой H, и R7 представляет собой C1-3алкил (например, R7 представляет собой метил или изопропил), и R8 представляет собой C10-14алкил (например, R8 представляет собой CH3(CH2)10 или CH3(CH2)14);
1.21 Соединение I, 1.1, или 1.2, в котором R5 представляет собой -C(R6)(R7)-O-C(O)-R8, и R8 представляет собой -N(Rd)(Re);
1.22 Соединение 1,21, в котором Rd представляет собой H, и Re независимо выбран из C1-24алкила, например, C1-20алкила, C5-20алкила, C9-18алкила, C10-16алкила или C11алкила, C12алкила, C13алкила, C14алкила, C15алкила или C16алкила;
1.23 Соединение 1.21, в котором каждый Rd и R4 независимо выбран из C1-24алкила, например, C1-20алкила, C5-20алкила, C9-18алкила, C10-16алкила или C11алкила, C12алкила, C13алкила, C14алкила, C15алкила или C16алкила;
1.24 Соединение 1,21, в котором каждый Rd и R4 представляет собой H;
1.25 Любое из соединений 1.14-1.24, в котором R6 представляет собой H, и R7 представляет собой H;
1.26 Любое из соединений 1.14-1.24, в котором R6 представляет собой C1-6алкил, и R7 представляет собой C1-6алкил;
1.27 Любое из соединений 1.14-1.24, в котором R6 представляет собой H, и R7 представляет собой C1-6алкил;
1.28 Любое из соединений 1.14-1.24, в котором R6 представляет собой H, и R7 представляет собой карбокси;
1.29 Любое из соединений 1.14-1.24, в котором R6 представляет собой H, и R7 представляет собой C1-6алкоксикарбонил, например, этоксикарбонил или метоксикарбонил;
1.30 Соединение I или любое из 1.1-1.29 в свободной форме;
1.31 Соединение I или любое из 1.1-1.29 в солевой форме, например, фармацевтически приемлемой солевой форме;
1.32 Соединение I или любое из 1.1-1.31 в по существу чистой диастереомерной форме (т.е., по существу свободной от других диастереомеров);
1.33 Соединение I или любое из 1.1-1.31, имеющее диастереомерный избыток более чем 70%, предпочтительно более чем 80%, более предпочтительно более чем 90% и самое предпочтительное более чем 95%.
1.34 Соединение I или любое из 1.1-1.33 в твердой форме.
в свободной или солевой форме (например, фармацевтически приемлемой солевой форме), например, в выделенной или очищенной свободной или солевой форме (например, фармацевтически приемлемой солевой форме).
[00012] Во втором аспекте, настоящее изобретение относится к соединению (соединение II) формулы II:
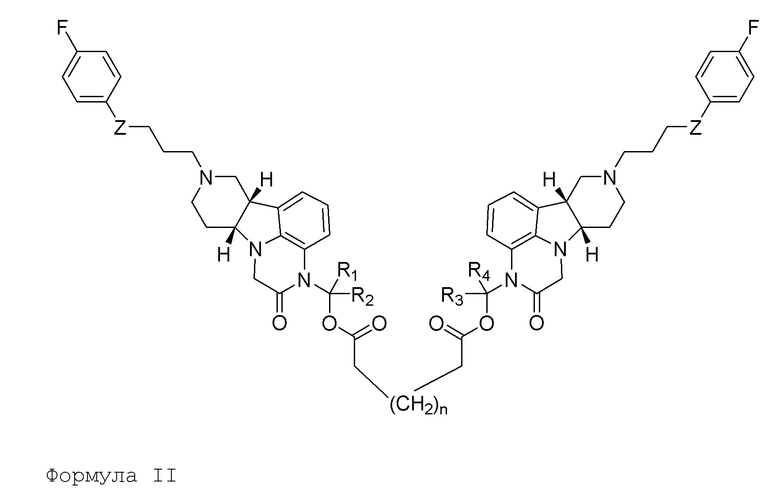
в которой:
Z представляет собой O, или -C(O)-;
каждый R1, R2, R3 и R4 независимо выбран из H и C1-6алкила;
n представляет собой целое от 1 до 23;
в свободной или солевой форме (например, фармацевтически приемлемой солевой форме), например, в выделенной или очищенной свободной или солевой форме (например, фармацевтически приемлемой солевой форме).
[00013] Настоящее изобретение относится к дополнительным примерным вариантам осуществления соединения формулы II, в свободной или солевой форме (например, фармацевтически приемлемой солевой форме), например, в выделенной или очищенной свободной или солевой форме (например, фармацевтически приемлемой солевой форме), включающим:
2.1 Соединение II, в котором Z представляет собой O;
2.2 Соединение II, в котором Z представляет собой -C(O);
2.3 Соединение II, 2.1 или 2.2, в котором R1 и R3 представляют собой H, и каждый R2 и R4 независимо выбран из C1-6алкила, например, C1-4алкила, C1-3алкила или C1-2алкила;
2.4 Соединение II, 2.1 или 2.2, в котором R1 и R2 представляют собой H, и каждый R3 и R4 независимо выбран из C1-6алкила, например, C1-4алкила, C1-3алкила или C1-2алкила;
2.5 Соединение II, 2.1 или 2.2, в котором каждый R1, R2, R3 и R4 независимо выбран из C1-6алкила, например, C1-4алкила, C1-3алкила или C1-2алкила;
2.6 Соединение II, 2.1 или 2.2, в котором каждый R1, R2, R3 и R4 представляет собой H;
2.7 Соединение II или любое из соединений 2.1-2.6, в котором n представляет собой целое от 1-21, например, 3-21 или 3-15 или 5-15 или 7-13 или 7-11;
2.8 Соединение II или любое из 2.1-2.7 в свободной форме;
2.9 Соединение II или любое из 2.1-2.7 в солевой форме, например, фармацевтически приемлемой солевой форме;
2.10 Соединение II или любое из 2.1-2.7 в твердой форме.
в свободной или солевой форме (например, фармацевтически приемлемой солевой форме), например, в выделенной или очищенной свободной или солевой форме (например, фармацевтически приемлемой солевой форме).
[00014] В третьем аспекте, настоящее изобретение относится к каждому из следующих соединений I или 1.1-1.34, соединения II или 2.1-2.10, (в настоящем изобретении далее вместе “соединения формул I-II и далее” или “соединения настоящего изобретения”) в фармацевтически приемлемой солевой форме (например, фармацевтически приемлемой солевой форме). Настоящее изобретение относится к дополнительным примерным вариантам осуществления соединений формул I-II и далее, включая:
5.1 Соединения формул I-II и далее, в которых соль представляет собой соль присоединения кислоты, выбранную из хлористоводородной, бромистоводородной, серной, сульфаминовой, фосфорной, азотной, уксусной, пропионовой, янтарной, гликолевой, стеариновой, молочной, яблочной, винной, лимонной, аскорбиновой, памовой, малеиновой, гидроксималеиновой, фенилуксусной, глутаминовой, бензойной, салициловой, сульфаниловой, 2-ацетоксибензойной, фумаровой, толуолсульфоновой, метансульфоновой, этандисульфоновой, щавелевой, изетионной и подобных;
5.2 Соединения формул I-II и далее, в которых соль представляет собой соль присоединения фумаровой кислоты;
5.3 Соединения формул I-II и далее, в которых соль представляет собой соль присоединения фосфорной кислоты;
5.4 Соединения формул I-II и далее, в которых соль представляет собой соль присоединения толуолсульфокислоты;
5.5 Любое из 5.1-5.4, в котором соль находится в твердой форме.
[00015] В четвертом аспекте, настоящее изобретение относится к фармацевтической композиции (фармацевтической композиции 6), содержащей соединение из любого из соединений I или 1.1-1.34, соединения II или 2.1-2.10, (вместе, соединений формул I-II и далее или соединений настоящего изобретения), например, в смеси с фармацевтически приемлемым разбавителем или носителем. Настоящее изобретение относится к дополнительным примерным вариантам осуществления фармацевтической композиции 6, включая:
6.1 Фармацевтическую композицию 6, содержащую соединение I или любое из 1.1-1.34;
6.2 Фармацевтическую композицию 6, содержащую соединение II или любое из 2.1-2.10;
6.3 Фармацевтическую композицию 6 или любую из 6.1-6.2, где соединение формулы I-II и далее находится в твердой форме;
6.4 Фармацевтическую композицию 6 или любую из 6.1-6.3, где соединение формул I-II и далее находится в фармацевтически приемлемой солевой форме (например, фармацевтически приемлемой солевой форме), как описано для соединений 5.1-5.5;
6.5 Фармацевтическую композицию 6 или любую из 6.1-6.4, где соединение формул I-II и далее находится в смеси с фармацевтически приемлемым разбавителем или носителем.
[00016] В следующем варианте осуществления, фармацевтические композиции настоящего изобретения предназначены для длительного или отсроченного высвобождения, например, депо, состав. В одном варианте осуществления, депо состав (депо состав 6.6) представляет собой фармацевтическую композицию любой из 6.1-6.5, предпочтительно в свободной или фармацевтически приемлемой солевой форме, и предпочтительно в смеси с фармацевтически приемлемым разбавителем или носителем, например, обеспечивая длительное или отсроченное высвобождение в виде инъецируемой депо формы.
[00017] В конкретном варианте осуществления, депо состав 6.6 содержит соединение из любых из соединения I или 1.1-1.34, или соединения II или 2.1-2.10, в свободной основной или фармацевтически приемлемой солевой форме, необязательно в кристаллической форме, где соединение измельчают или кристаллизуют до размера микрочастиц или наночастиц, например, частиц или кристаллов, имеющих размер частиц на основе объема (например, диаметр или Dv50) 0,5-100 микрон, например, например, 5-30 микрон, 10-20 микрон, 20-100 микрон, 20-50 микрон или 30-50 микрон. Данные частицы или кристаллы можно смешивать с подходящим фармацевтически приемлемым разбавителем или носителем, например, водой, получая депо состав для инъекции. Например, депо состав можно формулировать для внутримышечной или подкожной инъекции с дозой лекарственного средства, подходящей для 4-6 недельного лечения. В некоторых вариантах осуществления, частицы или кристаллы имеют площадь поверхности 0,1-5 м2/г, например, 0,5-3,3 м2/г или 0,8 1,2 м2/г.
[00018] В другом варианте осуществления, настоящее изобретение относится к фармацевтической композиции 6.7, которая представляет собой фармацевтическую композицию 6 или любую из 6.1-6.6, в которой соединение формул I-II и далее находится в полимерном матриксе. В одном варианте осуществления, соединение настоящего изобретения диспергируют или растворяют в полимерном матриксе. В следующем варианте осуществления, полимерный матрикс включает стандартные полимеры, применяемые в депо составах, такие как полимеры, выбранные из полиэфира гидроксижирной кислоты и ее производных, или полимера алкил альфа-цианоакрилата, полиалкиленоксалата, полиортоэфира, поликарбоната, полиорто-карбоната, поиламинокислота, эфира гиалуроновой кислоты и их смесей. В следующем варианте осуществления, полимер выбран из группы, состоящей из полилактида, поли d, l-лактида, полигликолида, PLGA 50:50, PLGA 85:15 и PLGA 90:10 полимера. В другом варианте осуществления, полимер выбран из поли(гликолевой кислоты), поли-D, L-молочной кислоты, поли-L-молочной кислоты, сополимеров перечисленных выше, поли(aлифатических карбоновых кислот), сополиоксалатов, поликапролактона, полидиоксанона, поли(ортокарбонатов), поли(ацеталей), поли(молочной кислоты-капролактона), полиртоэфиров, поли(гликолевой кислоты-капролактона), полиангидридов и природных полимеров, включая альбумин, казеин и воски, такие как, глицерин моно- и дистеарат, и подобных. В предпочтительном варианте осуществления, полимерный матрикс включает поли(d, l-лактид-со-гликолид).
[00019] (Фармацевтические) композиции 6 и 6.1-6.7 являются особенно пригодными для длительного или отсроченного высвобождения, где соединение настоящего изобретения высвобождается при разрушении полимерного матрикса. Данные композиции можно формулировать для контролируемого и/или замедленного высвобождения соединений настоящего изобретения (например, в виде депо композиции) в течение периода вплоть до 180 дней, например, от приблизительно 14 до приблизительно 30 до приблизительно 180 дней. Например, полимерный матрикс может разрушаться и высвобождать соединения настоящего изобретения в течение периода приблизительно 30, приблизительно 60 или приблизительно 90 дней. В другом примере, полимерный матрикс может разрушаться и высвобождать соединения настоящего изобретения в течение периода приблизительно 120 или приблизительно 180 дней.
[00020] В еще другом варианте осуществления, фармацевтические композиции настоящего изобретения, например, депо композицию настоящего изобретения, например, фармацевтическую композицию 6.6 или 6.7, формулируют для введения инъекцией.
[00021] В пятом аспекте, настоящее изобретение относится к соединениям формул I-II и далее, как описано в настоящем изобретении выше, в системе пероральной доставки с высвобождением, контролируемым осмотически (OROS), которая описана в WO 2000/35419 (US 2001/0036472) и EP 1539115 (публикация США No. 2009/0202631), содержание каждой из которых включено с помощью ссылки во всей своей полноте. Следовательно, в одном варианте осуществления седьмого аспекта, настоящее изобретение относится к фармацевтической композиции или устройству, включающему (a) желатиновую капсулу, содержащую соединение любой из формул I-II и далее в свободной или фармацевтически приемлемой солевой форме или фармацевтическую композицию настоящего изобретения, как описано в настоящем изобретении выше; (b) многослойную оболочку, наслоенную на желатиновую капсулу, содержащую, в направлении наружу от капсулы: (i) барьерный слой, (ii) вспениваемый слой и (iii) полупроницаемый слой; и (c) и отверстие, образованное или образуемое в оболочке (фармацевтическая композиция P.1).
[00022] В другом варианте осуществления, настоящее изобретение относится к фармацевтической композиции, содержащей желатиновую капсулу, содержащую жидкость, соединение формул I-II и далее в свободной или фармацевтически приемлемой солевой форме или фармацевтическую композицию настоящего изобретения, например, любую из фармацевтических композиций 6 или 6.1-6.7, причем желатиновая капсула окружена многослойной оболочкой, содержащей барьерный слой, контактирующий с внешней поверхностью желатиновой капсулы, вспениваемый слой, контактирующий с барьерным слоем, полупроницаемый слой, окружающий вспениваемый слой, и выходное отверстие, образованное или образуемое в оболочке (фармацевтическая композиция P.2).
[00023] В еще другом варианте осуществления седьмого аспекта, настоящее изобретение относится к композиции, включающей желатиновую капсулу, содержащую жидкость, соединение формул I-II и далее в свободной или фармацевтически приемлемой солевой форме или фармацевтическую композицию настоящего изобретения, например, любую из фармацевтических композиций 6 или 6.1-6.7, причем желатиновая капсула окружена многослойной оболочкой, содержащей барьерный слой, контактирующий с внешней поверхностью желатиновой капсулы, вспениваемый слой, контактирующий с барьерным слоем, полупроницаемый слой, окружающий вспениваемый слой, и выходное отверстие, образованное или образуемое в оболочке, где барьерный слой образует изолирующий слой между вспениваемым слоем и окружающей средой при выходном отверстии (фармацевтическая композиция P.3).
[00024] В еще другом варианте осуществления седьмого аспекта, настоящее изобретение относится к композиции, включающей желатиновую капсулу, содержащую жидкость, соединение формул I-II и далее в свободной или фармацевтически приемлемой солевой форме или фармацевтическую композицию настоящего изобретения, например, любую из фармацевтических композиций 6 или 6.1-6.7, причем желатиновая капсула окружена барьерным слоем, контактирующим с внешней поверхностью желатиновой капсулы, вспениваемым слоем, контактирующим с частью барьерного слоя, полупроницаемым слоем, окружающим, по меньшей мере, вспениваемый слой, и выходное отверстие, образованное или образуемое в лекарственной форме, распространяющееся от внешней поверхности желатиновой капсулы до окружающей среды применения (фармацевтическая композиция P.4). Вспениваемый слой можно образовывать в виде одной или более дискретных секций, таких как, например, две секции, расположенные на противоположных сторонах или концах желатиновой капсулы.
[00025] В конкретном варианте осуществления седьмого аспекта, соединение настоящего изобретения в системе пероральной доставки с высвобождением, контролируемым осмотически (т.е., в композиции P.1-P.4) находится в жидком составе, где состав может быть чистым, представлять собой жидкий активный агент, жидкий активный агент в растворе, суспензии, эмульсии или самоэмульгирующейся композиции или подобных.
[00026] Дополнительную информацию о составе системы пероральной доставки с высвобождением, контролируемым осмотически, включая характеристики желатиновой капсулы, барьерного слоя, вспениваемого слоя, полупроницаемого слоя и отверстия, можно найти в WO 2000/35419 и US 2001/0036472, содержание которых включено с помощью ссылки во всей своей полноте.
[00027] Другую систему пероральной доставки с высвобождением, контролируемым осмотически, для соединения формул I-II и далее или фармацевтической композиции настоящего изобретения можно найти в EP 1539115 (публикация США No. 2009/0202631), содержание которой включено с помощью ссылки во всей своей полноте. Следовательно, в другом варианте осуществления седьмого аспекта, настоящее изобретение относится к композиции или устройству, содержащему (a) два или более слоев, причем указанные два или более слоев содержат первый слой и второй слой, причем первый указанный слой содержит соединение формул I-II и далее, в свободной или фармацевтически приемлемой солевой форме, или фармацевтическую композицию, как описано в настоящем изобретении выше, причем второй указанный слой содержит полимер; (b) внешнюю оболочку, окружающую указанные два или более слоев; и (c) отверстие в указанной внешней оболочке (фармацевтическая композиция P.5).
[00028] В фармацевтической композиции P.5 предпочтительно применяют полупроницаемую мембрану, окружающую трехслойное ядро: в данных вариантах осуществления, первый слой называют первым слоем лекарственного средства, и он содержит низкие количества лекарственного средства (например, соединения формул I-II и далее) и осмотический агент, такой как соль, средний слой, называемый вторым слоем лекарственного средства, содержит большие количества лекарственного средства, вспомогательных веществ и не содержит соли; и третий слой, называемый слоем осмотического вещества, содержит осмотические агенты и не содержит лекарственного средства (фармацевтическая композиция P.6). По меньшей мере, одно отверстие просверливают через мембрану на конце первого слоя лекарственного средства таблетки в форме капсулы.
[00029] Фармацевтическая композиция P.5 или P.6 может содержать мембрану, образующую секцию, мембрану, окружающую внутренний защитный слой, по меньшей мере, одно выходное отверстие, образованное или образуемое в ней, и причем, по меньшей мере, часть мембраны является полупроницаемой; вспениваемый слой, расположенный в секции, удаленной от выходного отверстия и находящийся в гидравлическом сообщении с полупроницаемой частью мембраны; первый слой лекарственного средства, расположенный рядом с выходным отверстием; и второй слой лекарственного средства, расположенный в секции между первым слоем лекарственного средства и вспениваемым слоем, причем слои лекарственного средства содержат соединение настоящего изобретения в свободной или его фармацевтически приемлемой солевой форме (фармацевтическая композиция P.7). В зависимости от относительной вязкости первого слоя лекарственного средства и второго слоя лекарственного средства получают различные профили высвобождения. Является обязательным определить оптимальную вязкость для каждого слоя. В настоящем изобретении вязкость модулируют добавлением соли, хлорида натрия. Профиль доставки из ядра зависит от веса, состава и толщины каждого из слоев лекарственного средства.
[00030] В конкретном варианте осуществления, настоящее изобретение относится к фармацевтической композиции P.7, в которой первый слой лекарственного средства содержит соль, и второй слой лекарственного средства не содержит соли. Фармацевтическая композиция P.5-P.7 может необязательно содержать слой, способствующий течению, между мембраной и слоями лекарственного средства.
[00031] Фармацевтические композиции P.1-P.7 будут, в общем, называть составом системы пероральной доставки с высвобождением, контролируемым осмотически.
[00032] В восьмом аспекте, настоящее изобретение относится к способу (способ 1) лечения или профилактики расстройства центральной нервной системы, включающему введение нуждающемуся пациенту соединения формул I-II и далее или фармацевтической композиции 6 или 6.1-6.7 или P.1-P.7, например, способу 1, где вводимое соединение или композиция представляет собой:
1.1 Соединение I или любое из 1.1-1.34 в свободной или фармацевтически приемлемой солевой форме;
1.2 Соединение II или любое из 2.1-2.10 в свободной или фармацевтически приемлемой солевой форме;
1.3 Соединения варианта осуществления 5 или любого из 5.1-5.5;
1.4 Фармацевтическую композицию, как описано любой из фармацевтических композиций 6 и 6.1-6.7;
1.5 Депо композицию, как описано в депо композиции 6.6 или 6.7;
1.6 Фармацевтическую композицию P.1-P.7;
1.7 Состав системы пероральной доставки с высвобождением, контролируемым осмотически, как описано в настоящем изобретении выше;
[0033] В следующем варианте осуществления восьмого аспекта, настоящее изобретение относится к способу 1 или любому из способов 1.1-1.7, где способ дополнительно описан ниже:
1.8 Способ 1 или любой из способов 1.1-1.7, где расстройство центральной нервной системы представляет собой расстройство, выбранное из группы, состоящей из ожирения, тревоги (включая генерализованную тревогу, социальную тревогу и панические расстройства), депрессии (например, резистентной депрессии и MDD), психоза (включая психоз, связанный с деменцией, такой как галлюцинации при заболевании Паркинсона в прогрессирующей стадии или параноидальные идеи), шизофрении, расстройств сна (в частности расстройств сна, связанных с шизофренией и другими психиатрическими и неврологическими заболеваниями), сексуальных расстройств, мигрени, боли и состояний, связанных с болью, включая головную боль, идиопатическую боль, хроническую боль (такую как хроническая боль умеренной или средней тяжести, например, у пациентов, требующих 24 часовое лечение других заболеваний), невропатическую боль, зубную боль, фибромиалгию, хроническую усталость, агорафобию, социальные фобии, возбуждение при деменции (например, возбуждение при болезни Альцгеймера), возбуждение при аутизме и связанных аутистических расстройствах, расстройств желудочно-кишечного тракта, таких как дисфункция моторики желудочно-кишечного тракта, и деменции, например, деменции при болезни Альцгеймера или заболевании Паркинсона; поведенческих расстройств; наркомании, например, опиатной зависимости и/или алкогольной зависимости, и отказа от наркотиков или алкогольной зависимости (например, опиатной зависимости); передозировки опиатами; сопутствующих заболеваний, связанных с наркоманией, таких как депрессия, тревога и психоз; переедания; и обсессивно-компульсивного расстройства (OCD), обсессивно-компульсивного расстройства личности (OCPD) и родственных расстройств; или расстройства, связанного с применением опиатов (OUD);
1.9 Способ 1 или любому из способов 1.1-1.8, где расстройство центральной нервной системы представляет собой расстройство, включающее серотонин 5-HT2A, дофамин D1 и/или D2 рецепторную систему и/или пути транспортера обратного захвата серотонина (SERT), как аналогично описано в WO/2009/145900 и US 2011/071080, содержание которых включено в настоящее изобретение с помощью ссылки во всей своей полноте;
1.10 Способ 1 или любому из способов 1.1-1.9, где расстройство центральной нервной системы представляет собой расстройство, включающее μ-опиоидный рецептор;
1.11 Способ 1 или любому из способов 1.1-1.10, где расстройство центральной нервной системы представляет собой расстройство, выбранное из следующих: (i) психоза, например, шизофрении, у пациента, страдающего от депрессии; (2) депрессии у пациента, страдающего от психоза, например, шизофрении; (3) поведенческих расстройств, связанных с психозом и/или наркоманией, например, шизофренией или болезнью Паркинсона; (4) расстройств сна, связанных с психозом, например, шизофренией или болезнью Паркинсона; и (5) наркотической зависимости, расстройств, связанных с применением наркотических веществ, и/или расстройств, вызванных с наркотическими веществами, необязательно когда пациент страдает от остаточных симптомов тревоги или тревожного расстройства; и необязательно когда депрессия представляет собой терапевтически резистентную депрессию;
1.12 Способ 1 или любому из способов 1.1-1.11, где расстройство центральной нервной системы представляет собой психоз, например, шизофрению, и указанный пациент представляет собой пациента, страдающего от депрессии;
1.13 Способ 1 или любому из способов 1.1-1.12, где указанный пациент неспособен переносить побочные эффекты общепринятых антипсихотических лекарственных средств, например, хлорпромазина, галоперидола, дроперидола, флуфеназина, локсапина, молиндона мезоридазина, перфеназина, пимозида, прохлорперазина, промазина, тиоридазина, тиотиксена, трифлуоперазина, брекспипразола, карипразина, азенапина, луразидона, клозапина, арипипразола, оланзапина, кветиапина, рисперидона и зипразидона;
1.14 Способ 1 или любому из способов 1.1-1.13, где указанный пациент неспособен переносить побочные эффекты ненаркотических анальгетических средств и/или опиатных и опиоидных лекарственных средств или где применение опиатных лекарственных средств противопоказано указанному пациенту, например, из-за предшествующего злоупотребления наркотическими веществами или высокой вероятности злоупотребления наркотическими веществами, такими как опиатные и опиоидные лекарственные средства, включая, например, морфин, кодеин, тебаин, орипавин, дипропионат морфина, диникотинат морфина, дигидрокодеин, бупренорфин, эторфин, гидрокодон, гидроморфон, оксикодон, оксиморфон, фентанил, альфа-метилфентанил, алфентанил, трефантинил, брифентанил, ремифентанил, октфентанил, суфентанил, карфентанил, меперидин, продин, промедол, пропоксифен, декстропропоксифен, метадон, дифеноксилат, дезоцин, пентазоцин, феназоцин, буторфанол, налбуфин, лефорфанол, лефометорфан, трамадол, тапентадол, и анилеридин, или любые их комбинации.
1.15 Способ 1 или любому из способов 1.1-1.14, где указанный пациент неспособен переносить побочные эффекты общепринятых антипсихотических лекарственных средств, например, галоперидола, брекспиразола, карипразина, азенапина, луразидона, арипипразола, клозапина, оланзапина, кветиапина, рисперидона и зипразидона;
1.16 Способ 1 или любому из способов 1,1-1,15, где указанное расстройство представляет собой депрессию, и указанный пациент представляет собой пациента, страдающего от психоза, например, шизофрении, или болезни Паркинсона;
1.17 Способ 1 или любому из способов 1.1-1.15, где указанное расстройство представляет собой расстройство сна, и указанный пациент страдает от депрессии;
1.18 Способ 1 или любому из способов 1.1-1.15, где указанное одно или более расстройств представляет собой расстройство сна, и указанный пациент страдает от психоза, например, шизофрении;
1.19 Способ 1 или любому из способов 1.1-1.15, где указанное одно или более расстройств представляет собой расстройство сна, и указанный пациент страдает от болезни Паркинсона;
1.20 Способ 1 или любому из способов 1.1-1.15, где указанное одно или более расстройств представляет собой расстройство сна, и указанный пациент страдает от депрессии и психоза, например, шизофрении, или болезни Паркинсона.
1.21 Способ 1 или любой из 1.1-1.20, где указанный пациент страдает от наркомании, необязательно в сочетании с любым предшествующим заболеванием, например, где указанный пациент страдает от опиатной зависимости, кокаиновой зависимости, амфетаминовой зависимости и/или алкогольной зависимости, или от отказа от наркотиков или алкогольной зависимости (например, опиатной, кокаиновой или амфетаминовой зависимости), и необязательно где пациент страдает от сопутствующего заболевания, такого как тревога, депрессия или психоз, или остаточных симптомов тревоги или тревожного расстройства и/или измененного настроения (например, депрессии); дополнительно необязательно когда пациент страдает от опиатной передозировки;
1.22 Любой из приведенных выше способов, где эффективное количество составляет 1 мг-1000 мг, например, 2,5 мг-50 мг или для долгодействующего состава, 25 мг-1500 мг, например, 50 мг-500 мг, или 250 мг-1000 мг, или 250 мг-750 мг или 75 мг-300 мг;
1.23 Любой из приведенных выше способов, где эффективное количество составляет 1 мг-100 мг в день, например, 2,5 мг-50 мг в день;
1.24 Любой из приведенных выше способов, в котором заболевание, которое будут лечить, представляет собой дискинезию, например, у пациента, получающего дофаминергические лекарственные средства, например, лекарственные средства, выбранные из леводопы и вспомогательных веществ для леводопы (карбидопы, COMT ингибиторов, MAO-B ингибиторов), дофаминовых агонистов и антихолинергических лекарственных средств, например, леводопа;
1.25 Любой из приведенных выше способов, где пациент страдает от болезни Паркинсона.
[0034] Расстройства, связанные с применение наркотических веществ, и расстройства, вызванные наркотическими веществами, представляют собой две категории расстройств, связанных с наркотическими веществами, определенные пятым изданием DSM (the Diagnostic and Statistical Manual of Mental Disorders, DSM-V). Расстройство, связанное с применение наркотических веществ, представляет собой набор симптомов, являющихся результатом применения наркотического вещества, которое индивид продолжает принимать, несмотря на возникающие в результате проблемы. Расстройство, вызванное наркотическим веществом, представляет собой расстройство, вызванное применением самого наркотического вещества. Расстройства, вызванные наркотическими веществами, включают интоксикацию, абстиненцию, ментальные расстройства, вызванные наркотическими веществами, включая психоз, вызванный психоактивными веществами, биполярные и родственные расстройства, вызванные наркотическими веществами, депрессивные расстройства, вызванные наркотическими веществами, тревожные расстройства, вызванные наркотическими веществами, обсессивно-компульсивные и родственные расстройства, вызванные наркотическими веществами, расстройства сна, вызванные наркотическими веществами, сексуальные дисфункции, вызванные наркотическими веществами, бред, вызванный психоактивными веществами, и нейрокогнитивные расстройства, вызванные наркотическими веществами.
[0035] DSM-V включает критерии для классификации расстройства, связанного с применением наркотических веществ, как слабое, умеренное или тяжелое. В некоторых вариантах осуществления способов, описанных в настоящем изобретении, расстройство, связанное с применением наркотических веществ, выбрано из легкого расстройства, связанного с применением наркотических веществ, умеренного расстройства, связанного с применением наркотических веществ, или тяжелого расстройства, связанного с применением наркотических веществ. В некоторых вариантах осуществления, расстройство, связанное с применением наркотических веществ, представляет собой легкое расстройство, связанное с применением наркотических веществ. В некоторых вариантах осуществления, расстройство, связанное с применением наркотических веществ, представляет собой умеренное расстройство, связанное с применением наркотических веществ. В некоторых вариантах осуществления, расстройство, связанное с применением наркотических веществ, представляет собой тяжелое расстройство, связанное с применением наркотических веществ.
[0036] Тревога и депрессия являются широко распространенными сопутствующими заболеваниями у пациентов, проходящих лечение от употребления наркотических веществ или злоупотребления наркотическими веществами. Стандартное лечение расстройства, связанного со злоупотреблением наркотическими веществами, представляет собой сочетание частичного опиоидного агониста бупренорфина с опиоидным антагонистом налоксоном, но ни один из данных лекарственных средств не оказывает какого-либо существенного влияния на тревогу или депрессию, таким образом, приводя к одному результату, заключающемуся в необходимости назначать третье лекарственное средство, такое как анксиолитическое средство бензодиазепинового класса или SSRI антидепрессант. Это усложняет схемы лечения и соблюдение пациентом режима лечения. Напротив, соединения настоящего изобретения обеспечивают опиатный антагонизм наряду с серотониновым антагонизмом и дофаминовой модуляцией. Это может привести к значительному улучшению лечения пациентов, применяющих или злоупотребляющих наркотическими веществами, сопровождающиеся тревогой и /или депрессией.
[0037] Соединения настоящего изобретения могут обладать анксиолитическими свойствами, уменьшающими потребность в лечении пациента анксиолитическим средством, когда указанные пациенты страдают от сопутствующей тревоги. Таким образом, в некоторых вариантах осуществления, настоящее изобретение относится к способу согласно способу 1 или любому из способов 1.1-1.25, в котором расстройство центральной нервной системы представляет собой наркотическую зависимость, расстройства, связанные с применением наркотических веществ, и/или расстройства, вызванные с наркотическими веществами, или расстройство, связанное со злоупотреблением наркотическими веществами, например, у пациента, страдающего от симптомов тревоги или которому поставлен диагноз тревоги в качестве сопутствующего расстройства, или в качестве остаточного расстройства, где способ не включает дополнительного введения анксиолитического агента, такого как бензодиазепин. Бензодиазепины представляют собой GABA-модулирующие соединения, включая соединения, обсуждаемые со ссылкой на способ 3.1 и 3.2 ниже.
[0038] В другом варианте осуществления восьмого аспекта, настоящее изобретение относится к способу 1 или любому из способов 1.1-1.7, где способ дополнительно описан ниже:
1.26 Способ 1 или любой из способов 1.1-1.25, где расстройство центральной нервной системы представляет собой расстройство, выбранное из обсессивно-компульсивного расстройства (OCD), обсессивно-компульсивного расстройства личности (OCPD), генерализованного тревожного расстройства, социального тревожного расстройства, панического расстройства, агорафобии, игровой зависимости, импульсивного расстройства пищевого поведения, телесного дисморфического расстройства, ипохондрии, патологического грумингового расстройства, клептомании, пиромании, синдрома нарушения внимания с гиперактивностью (ADHD), cиндрома дефицита внимания (ADD), расстройства контроля над побуждениями и родственных расстройств, и их комбинации.
1.27 Способ 1 или любой из способов 1.1-1.25, где расстройство центральной нервной системы выбрано из обсессивно-компульсивного расстройства (OCD), обсессивно-компульсивного расстройства личности (OCPD), социального тревожного расстройства, панического расстройства, агорафобии, игровой зависимости, импульсивного расстройства пищевого поведения, телесного дисморфического расстройства и расстройства контроля над побуждениями.
1.28 Способ 1 или любой из способов 1.1-1.25, где расстройство центральной нервной системы представляет собой обсессивно-компульсивное расстройство (OCD) или обсессивно-компульсивное расстройство личности (OCPD).
1.29 Любой приведенный выше способ, где указанный пациент не реагирует или не может переносить побочные эффекты терапии селективными ингибиторами обратного захвата серотонина (SSRI), такими как циталопрам, эсциталопрам, флуорксетин, флувоксамин, пароксетин и сертралин.
1.30 Любой приведенный выше способ, где указанный пациент не реагирует или не может переносить побочные эффекты терапии ингибиторами обратного захвата серотонина-норэпинефрина (SNRI), такими как венлафаксин, сибутрамин, дулоксетин, атомоксетин, десвенлафаксин, милнаципран и левомильнаципран.
1.31 Любой приведенный выше способ, где указанный пациент не реагирует или не может переносить побочные эффекты терапии антипсихотическим агентами, такими как кломипрамин, рисперидон, кветиапин и оланзапин.
1.32 Способ 1 или любой из способа 1.1-1.25, где расстройство центральной нервной системы представляет собой болевое расстройство, например, состояние, связанное с болью, такое как головная боль, идиопатическая боль, невропатическая боль, хроническая боль (например, хроническая боль умеренной или средней тяжести, например, у пациентов, нуждающихся в 24-часовом длительном лечении других заболеваний), фибромиалгия, зубная боль, травматическая боль или хроническая усталость.
1.33 Любой приведенный выше способ, где пациент не реагирует или не может переносить побочные эффекты ненаркотических анальгетических средств и/или опиатных и опиоидных лекарственных средств, или где применение опиатных лекарственных средств противопоказано для указанного пациента, например, из-за предшествующего злоупотребления наркотическими веществами или высокой вероятности злоупотребления наркотическими веществами, такими как опиатные и опиоидные лекарственные средства, включая, например, морфин, кодеин, тебаин, орипавин, дипропионат морфина, диникотинат морфина, дигидрокодеин, бупренорфин, эторфин, гидрокодон, гидроморфон, оксикодон, оксиморфон, фентанил, альфа-метилфентанил, алфентанил, трефантинил, брифентанил, ремифентанил, октфентанил, суфентанил, карфентанил, меперидин, продин, промедол, пропоксифен, декстропропоксифен, метадон, дифеноксилат, дезоцин, пентазоцин, феназоцин, буторфанол, налбуфин, лефорфанол, лефометорфан, трамадол, тапентадол и анилеридин или любые их комбинации;
1.34 Способ I или любой из способов 1.1-1.33, где расстройство или заболевание центральной нервной системы представляет собой наркоманию (например, опиатную зависимость (т.е., расстройство, связанное с употреблением опиоидов), кокаиновую зависимость, амфетаминовую зависимость и/или алкогольную зависимость) или отказ от наркотиков или алкогольную зависимость (например, опиатную, кокаиновую или амфетаминовую зависимость), и где пациент также страдает от сопутствующего заболевания, такого как тревога, депрессия или психоз; где пациент также необязательно страдает от опиатной передозировки;
1.35 Любой из приведенных выше способов, где эффективное количество составляет 1 мг-1000 мг, предпочтительно 2,5мг-50 мг, или для долгодействующего состава, 25 мг-1500 мг, например, 50 мг-500 мг, или 250 мг-1000 мг, или 250 мг-750 мг или 75 мг-300 мг;
1.36 Любой из приведенных выше способов, где эффективное количество составляет 1 мг-100 мг в день, предпочтительно 2,5 мг-50 мг в день.
[0039] В еще другом варианте осуществления, настоящее изобретение относится к любому из способов 1 или 1.1-1.36, как описано в настоящем изобретении выше, где расстройство представляет собой шизофрению или расстройство сна. В некоторых вариантах осуществления, указанная шизофрения связана с депрессией.
[0040] В еще другом варианте осуществления, настоящее изобретение относится к любому из способов 1.1-1.36, где депо композицию настоящего изобретения (например, депо композицию любой формулы 6.6-6.7) или (фармацевтическую) композицию 6 или 6.1-6.7, или фармацевтическую композицию P.1-P.7, вводят для контролируемого- и/или замедленного высвобождения соединений настоящего изобретения в течение периода от приблизительно 14 дней, от приблизительно 30 до приблизительно 180 дней, предпочтительно в течение периода приблизительно 30, приблизительно 60 или приблизительно 90 дней. Контролируемое- и/или замедленное высвобождение является особенно пригодным для обхождения досрочного прекращения терапии, в частности для терапии антипсихотическими лекарственными средствами, где неисполнение или несоблюдение режимов приема лекарственного средства представляет собой обычное явление.
[0041] В еще другом варианте осуществления, настоящее изобретение относится к любому способу 1 или 1.1-1.36, как описано в настоящем изобретении выше, где депо композицию настоящего изобретения вводят для контролируемого- и/или замедленного высвобождения соединений настоящего изобретения в течение некоторого периода времени.
[0042] В другом варианте осуществления восьмого аспекта, настоящее изобретение относится к способу 1 или любому из способов 1.1-1.36, например, любому способ лечения боли, где пациент страдает от расстройства желудочно-кишечного тракта и/или легочного расстройства. Традиционные опиоидные анальгетики страдают от двух основных побочных эффектов: желудочно-кишечные расстройства (включая тошноту, рвоту и запор) и угнетения дыхания. У 90-95% пациентов, принимающих опиоиды для длительной обезболивающей терапии, развивается серьезный запор, требующий длительного применения слабительных и/или клизм. Более сильные опиоиды, такие как морфин, оксикодон и гидроморфон, вызывают более тяжелые запоры, чем другие опиоиды. Угнетение дыхания представляет собой наиболее серьезный побочный эффект лечения опиоидами, поскольку оно создает риск смерти, особенно когда пациенты сочетают (преднамеренно или непреднамеренно) назначенные опиоидные анальгетики с другими легальными или нелегальными респираторными депрессантами (включая алкоголь). Пациенты, нуждающиеся в лечении боли, особенно в лечении хронической боли, подвергаются особому риску побочных эффектов, если они страдают от ранее имеющихся желудочно-кишечных или легочных расстройств. В отличие от традиционных опиоидных анальгетиков, соединения настоящего изобретения обеспечивают обезболивающий эффект без значительных побочных эффектов со стороны желудочно-кишечного тракта и без значительного угнетения дыхания. Следовательно, данные соединения обеспечат повышенную безопасность и эффективность для пациентов, нуждающихся в лечении боли, имеющих данные ранее существовавшие желудочно-кишечные и легочные расстройства. В следующих вариантах осуществления, соединение настоящего изобретения можно комбинироваться с традиционным опиатным агентом для обеспечения улучшенного контроля боли с эффектом снижения дозы по сравнению с традиционным опиатным агентом (и одновременно сниженным риском побочных эффектов).
[0043] Таким образом, в конкретных вариантах осуществления, настоящее изобретение дополнительно относится к:
1.37 Способу 1 или любому из 1.1-1.36, где пациент страдает от предшествующего или сопутствующего расстройства желудочно-кишечного тракта и/или легочного расстройства;
1.38 Способу 1.37, где предшествующее или сопутствующее расстройство выбрано из группы, состоящей из синдрома раздраженного кишечника, дисфункции тазовой диафрагмы, дивертикулита, воспалительного заболевания кишечника, рака толстой кишки или колоректального рака, целиакии, нецелиаковой чувствительности к глютену, астмы, хронического обструктивного заболевания легких (ХОБЛ), одышки, пневмонии, застойной сердечной недостаточности, интерстициальной болезни легких, пневмоторакса, бронхита, эмболии легкого и черепно-мозговой травмы (например, перелома грудины или ребер, ушиба межреберных мышц);
1.39 Способу 1.37 или 1.38, где расстройство центральной нервной системы представляет собой болевое расстройство, например, состояние, связанное с болью, такое как головная боль, идиопатическая боль, невропатическая боль, хроническая боль (например, хроническая боль от умеренной до умеренно тяжелой, например, у пациентов, нуждающихся в 24-часовом продолжительном лечении других заболеваний), фибромиалгия, зубная боль, травматическая боль или хроническая усталость;
1.40 Любого из способа 1 или 1.1-1.39, где расстройство центральной нервной системы представляет собой расстройство, связанное с применением опиатов, опиатную абстиненцию или опиатную зависимость, и где способ обеспечивает облегчение симптомов абстиненции (например, симптомов со стороны желудочно-кишечного тракта, таких как диарея, тревога, депрессия, боль, нарушение сна или любая их комбинация);
1.41 Любого из способа 1 или 1.1-1.40, где способ дополнительно включает сопутствующее введение другого опиатного или опиоидного агента, например, вводимого одновременно, отдельно или последовательно;
1.42 Способ 1,41, где дополнительный опиатный или опиоидный агент выбран из группы, состоящей из морфина, кодеина, тебаина, орипавина, дипропионата морфина, диникотината морфина, дигидрокодеина, бупренорфина, эторфина, гидрокодона, гидроморфона, оксикодона, оксиморфона, фентанила, альфа-метилфентанила, алфентанила, трефантинила, брифентанила, ремифентанила, октфентанила, суфентанила, карфентанила, меперидина, продина, промедола, пропоксифена, декстропропоксифена, метадона, дифеноксилата, дезоцина, пентазоцина, феназоцина, буторфанола, налбуфина, лефорфанола, лефометорфана, трамадола, тапентадола и анилеридина, или любых их комбинаций;
1.43 Любому из способа 1 или 1.1-1.42, где способ дополнительно включает совместное введение NMDA рецепторного антагониста, например, вводимого одновременно, отдельно или последовательно;
1.44 Способу 1,43, где NMDA рецепторный антагонист выбран из группы, состоящей из кетамина (например, S-кетамина и/или R-кетамина), гидроксиноркетамина, мемантина, декстрометорфана, декстроаллорфана, декстрорфана, амантадина и агматина, или любой их комбинации;
1.45 Любому из способов 1.37-1.44, где соединение представляет собой соединение формулы I, где Z представляет собой -O-.
[0044] В седьмом аспекте, настоящее изобретение относится к способу (способу 2) профилактики или лечения одного или более расстройств сна, включающему введение нуждающемуся пациенту соединения формул I-II и далее или фармацевтической композиции 6 или 6.1-6.7 или P.1-P.7, (способу 2) например, способу 2, где вводимое соединение или композиция представляет собой:
2.1 Соединение I или 1.1-1.34 в свободной или фармацевтически приемлемой солевой форме;
2.2 Соединение II или 2.1-2.10 в свободной или фармацевтически приемлемой солевой форме;
2.3 Соединение 5 или 5.1-5.5;
2.4 Фармацевтическую композицию, как определено любой из композиций 6 и 6.1-6.7;
2.5 Депо композиция, как определено в депо композиции 6.6 или 6.7;
2.6 Фармацевтическую композицию P.1-P.7;
2.7 Состава системы пероральной доставки с высвобождением, контролируемым осмотически, как описано в настоящем изобретении выше;
[0045] В следующем варианте осуществления седьмого аспекта, настоящее изобретение относится к способу 2 или 2.1-2.7, где расстройство сна включает бессонницу при поддержании сна, частое пробуждение, и ощущение неотдохнувшим при пробуждении; например:
2.8 Любой из приведенных выше способов, где расстройство сна представляет собой бессонницу при поддержании сна;
2.9 Любой из приведенных выше способов, где эффективное количество составляет 1 мг-5 мг, предпочтительно 2,5-5 мг в день;
2.10 Любой из приведенных выше способов, где эффективное количество составляет 2,5 мг или 5 мг в день;
2.11 Любой из приведенных выше способов, где расстройство сна присутствует у пациента, страдающего от или подверженного риску дискинезии, например, у пациента, получающего дофаминергические лекарственные средства, например, выбранные из леводопы и вспомогательных веществ для леводопы (карбидопа, COMT ингибиторов, MAO-B ингибиторов), дофаминовых агонистов и антихолинергических лекарственных средств, например, получающего леводопу;
2.12 Любой из приведенных выше способов, где пациент страдает от болезни Паркинсона.
[0046] В следующем варианте осуществления седьмого аспекта, настоящее изобретение относится к способу 2 или любому из 2.1-2.12, где расстройство сна включает бессонницу при поддержании сна, частое пробуждение и ощущение неотдохнувшим при пробуждении.
[0047] Соединения настоящего изобретения, фармацевтические композиции настоящего изобретения или депо композиции настоящего изобретения можно применять в комбинации со вторым терапевтическим агентом, особенно при более низких дозах, чем когда отдельные агенты применяют в качестве монотерапии, чтобы усилить терапевтическую активность комбинированных агентов, не вызывая нежелательных побочных эффектов, обычно возникающих при обычной монотерапии. Следовательно, соединения настоящего изобретения можно вводить одновременно, последовательно или продолжительно с другими антидепрессантными, антипсихотическими, другими гипотоническим агентами, и/или агентами, применяемыми для лечения болезни Паркинсона или поведенческих расстройств. В другом примере, побочные эффекты можно снижать или минимизировать введением соединения настоящего изобретения в комбинации с одним или более вторыми терапевтическими агентами в свободной или солевой форме (например, фармацевтически приемлемой солевой форме), где дозы (i) второго терапевтического агента (агентов) или (ii) или соединения настоящего изобретения и вторых терапевтических агентов, являются меньшими, чем если бы агенты/соединения вводили в виде монотерапии. В конкретном варианте осуществления, соединения настоящего изобретения являются пригодными для лечения дискинезии у пациента, получающего дофаминергические лекарственные средства, например, выбранные из леводопы и вспомогательных веществ для леводопы (карбидопа, COMT ингибиторов, MAO-B ингибиторов), дофаминовых агонистов и антихолинергических лекарственных средств, например, таких как применяемые в лечении болезни Паркинсона.
[0048] В некоторых следующих вариантах осуществления настоящего изобретения, фармацевтические композиции настоящего изобретения или депо композиции настоящего изобретения можно применять в комбинации со вторым терапевтическим агентом, особенно при более низких дозах, чем когда отдельные агенты применяют в качестве монотерапии для того, чтобы увеличить терапевтические активности комбинированных агентов, не вызывая нежелательных побочных эффектов, где второй терапевтический агент представляет собой опиатный антагонист или обратный агонист (например, налоксон). Соединения настоящего изобретения можно вводить одновременно, последовательно или продолжительно с данными опиатными антагонистами или опиатными обратными агонистами.
[0049] Следовательно, в восьмом аспекте, настоящее изобретение относится к способу 1 или любому из способов 1.1-1.45 или способу 2 или любому из 2.1-2.12, дополнительно включающему введение одного или более терапевтических агентов пациенту, где один или более терапевтических агентов выбраны из соединений, которые модулируют GABA активность (например, увеличивают активность и облегчают GABA передачу), GABA-B агониста, 5-HT рецепторного модулятора (например, 5-HT1A агониста, 5-HT2A антагониста, 5-HT2A обратного агониста и т.д.), агониста мелатонинового рецептора, модулятора ионных каналов (например, блокатора), серотонин-2 рецепторного антагониста/ингибитора обратного захвата (например, соединения, обладающего и 5-HT2 антагонизмом и ингибированием обратного захвата серотонина, т.е., SARI), антагониста орексинового рецептора, H3 агониста или антагониста, норадренергического агониста или антагониста, агониста галанина, CRH антагониста, гормона роста человека, агониста гормона роста, эстрогена, агониста эстрогена, нейрокинин-1 лекарственного средства, антидепрессанта, и опиатного агониста и/или частичного опиатного агониста (такого как агонист или частичный агонист мю-, каппа- или дельта-опиатного рецептора), или опиатного антагониста или обратного агониста (такого как антагонист или обратный агонист мю-, каппа- или дельта-опиатного рецептора), агониста ноцицептина, и антипсихотического агента, например, нестандартного антипсихотического агента, в свободной или фармацевтически приемлемой солевой форме (способ 1-A и 2-A соответственно; вместе “способ 3”). В следующих вариантах осуществления восьмого аспекта, настоящее изобретение относится к способу 1 или любому из способов 1.1-1.45, или способу 2 или любому из 2.1-2.12, дополнительно включающему введение пациенту одного или более терапевтических агентов, выбранных из приведенных выше и дополнительно выбранных из агонистов или частичных агонистов или антагонистов или обратных агонистов мю-опиатных, каппа-опиатных, дельта-опиатных и/или ноцицептиновых/oрфаниновых рецепторов. В следующих вариантах осуществления десятого аспекта, настоящее изобретение также обеспечивает способ 1 или любой из способов 1.1-45, или способ 2 или любой из 2.1-2.12, дополнительно включающий введение пациенту одного или более терапевтических агентов, выбранных из антагониста серотонинового HT6 рецептора, mGluR-2, -3 или -5 рецепторного агониста или антагониста (включая позитивные и негативные модуляторы и частичные агонисты).
[0050] В следующем варианте осуществления восьмого аспекта, настоящее изобретение относится к способу 3 (т.е., способу 1-A или 2-A), дополнительно включающему введение пациенту одного или более терапевтических агентов, как следует далее:
3.1 Способ 1-A или 2-A, где терапевтический агент (агенты) представляют собой соединения, которые модулируют GABA активностью (например, увеличивают активность и способствуют GABA передачи);
3.2 Способ 1-A или 2-A или 3.1, где GABA соединение выбрано из группы, состоящей из одного или более из доксепина, алпразолама, бромазепама, клобазама, клоназепама, клоразепата, диазепама, флунитразепама, флуразепама, лоразепама, мидазолама, нитразепама, оксазепама, темазепама, триазолама, индиплона, зопиклона, эзопиклона, залеплона, золпидема, габоксадола, вигабатрина, тиагабина, EVT 201 (Evotec Pharmaceuticals) и эстазолама;
3.3 Способ 1-A или 2-A, где терапевтический агент представляет собой дополнительный 5HT2a рецепторный антагонист;
3.4 Способ 1-A или 2-A или 3.3, где указанный дополнительный 5HT2a рецепторный антагонист выбран из одного или более из пимавансерина, кетансерина, рисперидона, эпливансерина, волинансерина (Sanofi-Aventis, France), прувансерина, MDL 100907 (Sanofi-Aventis, France), HY 10275 (Eli Lilly), APD 125 (Arena Pharmaceuticals, San Diego, CA) и AVE8488 (Sanofi-Aventis, France);
3.5 Способ 1-A или 2-A, где терапевтический агент представляет собой агонист мелатонинового рецептора;
3.6 Способ 1-A или 2-A или 3.5, где агонист мелатонинового рецептора выбран из группы, состоящей из одного или более из мелатонина, рамелтеона (ROZEREM®, Takeda Pharmaceuticals, Japan), VEC-162 (Vanda Pharmaceuticals, Rockville, MD), PD-6735 (фаза II исследований) и агомелатина;
3.7 Способ 1-A или 2-A, где терапевтический агент представляет собой блокатор ионных каналов;
3.8 Способ I-A или 2-A или 3.7, где указанный блокатор ионных каналов представляет собой один или более из ламотригина, габапентина и прегабалина.
3.9 Способ 1-A или 2-A, где терапевтический агент представляет собой антагонист орексинового рецептора;
3.10 Способ 1-A или 2-A или 3.9, где антагонист орексинового рецептора выбран из группы, состоящей из орексина, 1,3-биарилмочевины, SB-334867-a (GlaxoSmithKline, UK), GW649868 (GlaxoSmithKline) и бензамидного производного;
3.11 Способ 1-A или 2-A, где терапевтический агент представляет собой серотонин-2 рецепторный антагонист/ингибитор обратного захвата(SARI);
3.12 Способ 1-A или 2-A или 3.11, где серотонин-2 рецепторный антагонист/ингибитор обратного захвата(SARI) выбран из группы, состоящей из одного или более Org 50081 (Organon-Netherlands), ритансерина, нефазодона, серзона и тразодона;
3.13 Способ 1-A или 2-A, где терапевтический агент представляет собой 5HTIa агонист;
3.14 Способ 1-A или 2-A или 3.13, где 5HTIa агонист выбран из группы, состоящей из одного или более из репинотана, саризотана, эптапирона, буспирона и MN-305 (MediciNova, San Diego, CA);
3.15 Способ 1-A или 2-A, где терапевтический агент представляет собой нейрокинин-1 лекарственное средство;
3.16 Способ 1-A или 2-A или 3.15, где нейрокининовое-1 лекарственное средство представляет собой казопитант (GlaxoSmithKline);
3.17 Способ 1-A или 2-A, где терапевтический агент представляет собой антипсихотический агент;
3.18 Способ 1-A или 2-A или 3.17, где антипсихотический агент выбран из группы, состоящей из хлорпромазина, галоперидола, дроперидола, флуфеназина, локсапина, мезоридазина, молиндона, перфеназина, пимозида, прохлорперазина промазина, тиоридазина, тиотиксена, трифлуоперазина, брекспиразола, карипразина, азенапина, луразидона, клозапина, арипипразола, оланзапина, кветиапина, рисперидона, зипразидона и палиперидона;
3.19 Способ 1-A или 2-A, где терапевтический агент представляет собой антидепрессант;
3.20 Способ 1-A или 2-A или 3.19, где антидепрессант выбран из амитриптилина, амоксапина, бупропиона, циталопрама, кломипрамина, дезипрамина, доксепина, дулоксетина, эсциталопрама, флуорксетина, флувоксамина, имипрамина, изокарбоксазида, мапротилина, миртазапина, нефазодона, нортриптилина, пароксетина, сульфата фенелзина, протриптилина, сертралина, транилципромина, тразодона, тримипрамина и венлафаксина;
3.21 Способ 1-A или 2-A, 3.17 или 3.18, где антипсихотический агент представляет собой нестандартный антипсихотический агент;
3.22 Способ 1-A или 2-A, или любой из 3.17-3.21, где нестандартный антипсихотический агент выбран из группы, состоящей из брекспиразола, карипразина, азенапина, луразидона, клозапина, арипипразола, оланзапина, кветиапина, рисперидона, зипразидона и палиперидона;
3.23 Способ 1-A или 2-A, где терапевтический агент выбран из любого из способов 3.1-3.22, например, выбран из группы, состоящей из модафинила, армодафинила, доксепина, алпразолама, бромазепама, клобазама, клоназепама, клоразепата, диазепама, флунитразепама, флуразепама, лоразепама, мидазолама, нитразепама, оксазепама, темазепама, триазолама, индиплона, зопиклона, эзопиклона, залеплона, золпидема, габоксадола, вигабатрина, тиагабина, EVT 201 (Evotec Pharmaceuticals), эстазолама, пимавансерина, кетансерина, рисперидона, эпливансерина, волинансерина (Sanofi-Aventis, France), прувансерина, MDL 100907 (Sanofi-Aventis, France), HY 10275 (Eli Lilly), APD 125 (Arena Pharmaceuticals, San Diego, CA), AVE8488 (Sanofi-Aventis, France), репинотана, саризотана, эптапирона, буспирона, MN-305 (MediciNova, San Diego, CA), мелатонина, рамелтеона (ROZEREM®, Takeda Pharmaceuticals, Japan), VEC-162 (Vanda Pharmaceuticals, Rockville, MD), PD-6735 (исследования II фазы), агомелатина, ламотригина, габапентина, прегабалина, орексина, 1,3-биарилмочевины, SB-334867-a (GlaxoSmithKline, UK), GW649868 (GlaxoSmithKline), бензамидного производного, Org 50081 (Organon-Netherlands), ритансерина, нефазодона, серзона, тразодона, касопитанта (GlaxoSmithKline), амитриптилина, амоксапина, бупропиона, циталопрама, кломипрамина, дезипрамина, доксепина, дулоксетина, эсциталопрама, флуорксетина, флувоксамина, имипрамина, изокарбоксазида, мапротилина, миртазапина, нефазодона, нортриптилина, пароксетина, сульфата фенелзина, протриптилина, сертралина, транилципромина, тразодона, тримипрамина, венлафаксина, хлорпромазина, галоперидола, дроперидола, флуфеназина, локсапина, мезоридазина, молиндона, перфеназина, пимозида, прохлорперазина, промазина, тиоридазина, тиотиксена, трифлуоперазина, брекспиразола, карипразина, азенапина, луразидона, клозапина, арипипразола, оланзапина, кветиапина, рисперидона, зипразидона и палиперидона;
3.24 Способ 1-A или 2-A, где терапевтический агент представляет собой H3 агонист;
3.25 Способ 1-A или 2-A, где терапевтический агент представляет собой H3 антагонист;
3.26 Способ 1-A или 2-A, где терапевтический агент представляет собой норадренергический агонист или антагонист;
3.27 Способ 1-A или 2-A, где терапевтический агент представляет собой агонист галанина;
3.28 Способ 1-A или 2-A, где терапевтический агент представляет собой CRH антагонист;
3.29 Способ 1-A или 2-A, где терапевтический агент представляет собой гормон роста человека;
3.30 Способ 1-A или 2-A, где терапевтический агент представляет собой агонист гормона роста;
3.31 Способ 1-A или 2-A, где терапевтический агент представляет собой эстроген;
3.32 Способ 1-A или 2-A, где терапевтический агент представляет собой агонист эстрогена;
3.33 Способ 1-A или 2-A, где терапевтический агент представляет собой нейрокининовое-1 лекарственное средство;
3.34 Способ 1-A или 2-A, где терапевтический агент комбинируют с соединениями формулы (I), и терапевтический агент представляет собой агент против болезни Паркинсона, такой как L-допа, ко-карелдопа, дуодопа, ставело, симметрел, бензтропин, бипериден, бромокриптин, энтакапон, перголид, прамипексол, проциклидин, ропинирол, селегилин и толкапон;
3.35 Способ 1-A или 2-A, где терапевтический агент представляет собой опиатный агонист или частичный опиатный агонист, например, мю-агонист или частичный агонист, или каппа-агонист или частичный агонист, включая смешенные агонисты/антагонисты (например, агент с частичной мю-агонистической активностью и каппа-антагонистической активностью);
3.36 Способ 3.35, где терапевтический агент представляет собой бупренорфин, необязательно, где указанный способ не включает совместную терапию анксиолитическим агентом, например, GABA соединением или бензодиазепином;
3.37 Способ 1-A или 2-A, где соединения формулы (I) можно применять для лечения расстройств сна, депрессии, психоза или любых их комбинаций, у пациентов, страдающих от перечисленных заболеваний и/или болезни Паркинсона;
3.38 Способ 1-A или 2-A, где заболевание выбрано из, по меньшей мере, одного или более из психоза, например, шизофрении, депрессии, поведенческих расстройств, расстройств сна (например, поддержания сна и/или засыпание) или любой комбинации данных расстройств;
3.39 Способ 1-A или 2-A, где терапевтический агент (агенты) представляет собой опиатный рецепторный антагонист или обратный агонист, например, полный опиатный антагонист, например, выбранный из налоксона, налтрексона, налмефена, метадона, налорфина, леваллорфана, самидорфана, налодеина, ципродима или норбиналторфимина;
3.40 Любой из приведенных выше способов, где расстройство представляет собой расстройство сна;
3.41 Любой из приведенных выше способов, где расстройство представляет собой расстройство сна, связанное с психозом, например, шизофренией или болезнью Паркинсона; в свободной или фармацевтически приемлемой солевой форме.
[0051] В девятом аспекте настоящего изобретения, комбинацию соединения настоящего изобретения и одного или более вторых терапевтических агентов, как определено в способах 1-A, 2-A или любом из способов 3 или 3.1-3.41, можно вводить пациенту в виде фармацевтической композиции или депо композиции, как описано в настоящем изобретении выше. Составы комбинации могут включать смеси комбинированных лекарственных средств, а также двух или более отдельных композиций лекарственных средств, где отдельные композиции можно, например, вводить пациенту совместно.
[0052] В конкретном варианте осуществления, способы 1-A, 2-A, 3 или 3.1-3.41 включают введение нуждающемуся пациенту соединения настоящего изобретения в комбинации с нестандартным антипсихотическим агентом, например, соединением, выбранным из брекспиразола, карипразина, азенапина, луразидона, клозапина, арипипразола, оланзапина, кветиапина, рисперидона, зипразидона, или палиперидона, в свободной или фармацевтически приемлемой солевой форме, например, где дозу нестандартного антипсихотического агента снижают и/или ослабляют побочные эффекты.
[0053] В другом варианте осуществления, способы 1-A, 2-A, 3 или 3.1-3.41 включают введение нуждающемуся пациенту, соединения настоящего изобретения в комбинации с антидепрессантом, например, амитриптилином, амоксапином, бупропионом, циталопрамом, кломипрамином, дезипрамином, доксепином, дулоксетином, эсциталопрамом, флуорксетином, флувоксамином, имипрамином, изокарбоксазидом, мапротилином, миртазапином, нефазодоном, нортриптилином, пароксетином, сульфатом фенелзина, протриптилином, сертралином, транилципромином, тразодоном, тримипрамином или венлафаксином, в свободной или фармацевтически приемлемой солевой форме. Альтернативно, антидепрессант можно применять в качестве вспомогательного лекарственного средства в добавление к соединению настоящего изобретения.
[0054] В еще другом варианте осуществления, способы 1-A, 2-A, 3 или 3.1-3.41 включает введение нуждающемуся пациенту, соединения настоящего изобретения в комбинации с соединением, которое модулирует GABA активность, например, соединением, выбранным из доксепина, алпразолама, бромазепама, клобазама, клоназепама, клоразепата, диазепама, флунитразепама, флуразепама, лоразепама, мидазолама, нитразепама, оксазепама, темазепама, триазолама, индиплона, зопиклона, эзопиклона, залеплона, золпидема, габоксадола, вигабатрина, тиагабина, EVT 201 (Evotec Фармацевтические средства), эстазолама или любых их комбинаций, в свободной или фармацевтически приемлемой солевой форме. В других вариантах осуществления, способы, описанные в настоящем изобретении, дополнительно не включают введение GABA соединения, бензодиазепина или любого другого анксиолитического агента.
[0055] В другом предпочтительном варианте осуществления, способы 1-A, 2-A, 3 или 3.1-3.41 включает введение нуждающемуся пациенту, соединения настоящего изобретения в комбинации с доксепином в свободной или фармацевтически приемлемой солевой форме. Дозы доксепина могут изменяться в любом диапазоне, известном специалисту в данной области техники. В одном примере, дозу 10 мг доксепина можно комбинировать с любой дозой соединения настоящего изобретения.
[0056] В другом варианте осуществления, способы 1-A, 2-A, 3 или 3.1-3.41 включает введение нуждающемуся пациенту соединения настоящего изобретения в комбинации с (включая в качестве части суточного режима дозирования) нестандартным стимулятором, например, модафинилом, адрафинилом или армодафинилом. Режим, вводящий соединение настоящего изобретения с данными лекарственными средствами, способствует более регулярному сну и избегает побочных эффектов, таких как психоз или мания, связанная с высокими уровнями данных лекарственных средств, например, при лечении биполярной депрессии, нарушения познавательных способностей, связанного с шизофренией, чрезмерной сонливости и усталости при состояниях, таких как болезнь Паркинсона и рак.
[0057] В другом варианте осуществления, способы 1-A, 2-A, 3 или 3.1-3.41 включает введение нуждающемуся пациенту соединения настоящего изобретения в комбинации (включая в качестве части суточного режима дозирования) с опиатным рецепторным антагонистом или обратным агонистом, например, полным опиатным антагонистом, например, выбранным из налоксона, налтрексона, налмефена, метадона, налорфина, леваллорфана, самидорфана, налодеина, ципродима или норбиналторфимина.
[0058] В некоторых из вышеизложенных вариантов осуществления, каждое из соединений формул I-II и далее; фармацевтических композиций 6 и 6.1-6.5; депо композиций 6.6 и 6.7; композиций P.1-P.7; способов 1 и 1.1-1.45; и способов 2 и 2.1-2.12; соединение настоящего изобретения по существу не содержит соединения формулы A и/или по существу не содержит соединения формулы B.
[0059] В десятом аспекте, настоящее изобретение относится к применению соединения, как определено далее:
11.1 Соединение I или 1.1-1.34 в свободной или фармацевтически приемлемой солевой форме;
11.2 Соединение II или 2.1-2.10 в свободной или фармацевтически приемлемой солевой форме;
11.3 Соединение 5 или 5.1-5.5;
11.4 Фармацевтическая композиция 6 и 6.1-6.7;
11.5 Фармацевтическая композиция P.1-P.7;
11.6 Состав системы пероральной доставки с высвобождением, контролируемым осмотически, как описано в настоящем изобретении выше;
(для получения лекарственного средства) для лечения или профилактики одного или более расстройств, как описано в настоящем изобретении выше, например, в любом из способа 1 или 1.1-1.45, любом из способа 2 и 2.1-2.12, и способа 3 или 3.3-3.41, или любых способах, описанных в одиннадцатом аспекте настоящего изобретения.
[0060] В одиннадцатом аспекте, настоящее изобретение относится к фармацевтической композиции, как описано в настоящем изобретении выше, например:
12.1 Фармацевтической композиции 6 и 6.1-6.7;
12.2 Фармацевтической композиции P.1-P.7;
12.3 Составу системы пероральной доставки с высвобождением, контролируемым осмотически, как описано в настоящем изобретении выше,
для применения в лечении или профилактике одного или более расстройств, как описано в настоящем изобретении выше, например, в любом из способов 1 и 1.1-1.33, способах 2 и 2.1-2.12, способах I-A, II-A, 3 или 3.1-3.41 или любых способах, описанных в девятом или десятом аспектах настоящего изобретения.
ПОДРОБНОЕ ОПИСАНИЕ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
[0061] Если не указано иное или не ясно из контекста, следующие термины, применяемые в настоящем изобретении, имеют следующие значения:
[0062] Соединения настоящего изобретения содержат биологически лабильные функциональные группы, находящиеся в соединениях так, что природная метаболическая активность будет удалять лабильные функциональные группы, давая в результате соединение формулы A и/или соединение формулы B. Как таковое, введение соединений настоящего изобретения нуждающемуся пациенту приводит в результате или к немедленному или к отсроченному высвобождению в тканях указанной персоны соединения формулы A и/или соединения формулы B. Ожидают, что соединения настоящего изобретения не обладают значительной фармакологической активностью сами по себе, но будут служить резервуаром фармакологически активных соединений формулы A и/или формулы B. Таким образом, соединения настоящего изобретения являются особенно пригодными для формулирования в виде долгодействующих инъецируемых препаратов (LAI) или “депо” фармацевтических композиций. Не будучи связанными теорией, инъецируемый “депо”, содержащий соединение настоящего изобретения, будет постепенно высвобождать в ткани тела указанное соединение, где в данных тканях указанное соединение будет постепенно подвергаться метаболизму, давая соединение формулы A и/или соединение формулы B. Данное депо составы можно дополнительно приспосабливать выбором подходящих компонентов для контроля скорости растворения и высвобождения соединений настоящего изобретения.
[0063] Показано, что соединения формулы A и B обладают рядом полезных фармацевтических свойств, каждое из которых, как ожидают, разделяется соединениями настоящего изобретения. Например, соединение формулы A и B обладает мощным 5-HT2A, D1 и Mu опиатным антагонизмом, вместе с умеренным D1, D2 и SERT антагонизмом. Более того, неожиданно обнаружили, что данные соединения могут функционировать как “смещенные” мю опиатные лиганды. Это значит, что когда соединения связываются с мю опиатными рецепторами, они могут функционировать как частичные мю агонисты через передачу сигнала, связанную с G-белком, но как мю антагонисты через сигнальную систему бета-аррестина. Это противоположно традиционным опиатным агонистам, морфину и фентанилу, которые имеют тенденцию сильно активировать как передачу сигналов, связанную с G-белком, так и сигнальную систему бета-аррестина. Считается, что активация сигнальной системы бета-аррестина данными лекарственными средствами опосредует желудочно-кишечную дисфункцию и угнетение дыхания, обычно опосредованные опиатными лекарственными средствами. Следовательно, ожидают, что соединения согласно настоящему изобретению, в частности соединения формулы I, приведут к облегчению боли с менее выраженными желудочно-кишечными и респираторными побочными эффектами, чем существующие опиатные анальгетики. Данный эффект был продемонстрирован в доклинических исследованиях и клинических исследованиях фазы II и фазы III для смещенного Mu агониста, оликеридина. Было показано, что олекиридин приводит к смещенному Mu агонизму через передачу сигналов, связанную с G-белком, со сниженной передачей сигналов посредством бета-аррестина по сравнению с морфином, и это было связано с его способностью вызывать анальгезию с уменьшенными респираторными побочными эффектами по сравнению с морфином. Более того, поскольку данные соединения являются антагонистами пути бета-аррестина, ожидают, что они будут пригодны в лечении опиатной передозировки, поскольку они будут ингибировать наиболее тяжелые побочные эффекты опиатов, при этом обеспечивая облегчение боли. Более того, данные соединения также обладают эффектом поддержания сна вследствие их серотонинергической активности. Поскольку многие люди, страдающие хронической болью, испытывают трудности со сном из-за боли, данные соединения могут помочь данным пациентам спать всю ночь благодаря синергетическому эффекту серотонинергической и опиатной рецепторной активности.
[0064] Таким образом, в определенных вариантах осуществления, соединения настоящего изобретения можно применять в способе лечения расстройства, связанного с применением опиатов (OUD), опиатной передозировкой или опиатной абстиненции, или отдельно, или в сочетании с опиатным антагонистом или обратным агонистом (например, налоксоном или налтрексоном). Ожидают, что соединения настоящего изобретения будут проявлять сильную способность смягчать дисфорию и сопутствующие психические расстройства, связанные с отменой наркотического средства (например, расстройства настроения и тревожные расстройства, нарушение сна), а также обеспечивать сильную анальгезию, но без побочных эффектов (например, эффектов на желудочно-кишечный тракт и угнетения дыхания) и аддиктивного потенциала, наблюдаемого с другими опиоидными лекарственными средствами (например, оксикодоном, метадоном или бупренорфином). Уникальный фармакологический профиль данных соединений также должен снижать риски неблагоприятных взаимодействий лекарственных средств (например, с алкоголем). Следовательно, данные соединения являются особенно подходящими для лечения расстройств, связанных с применением опиатов, и симптомов, связанных с опиатной абстиненцией. В добавление к непосредственному влиянию соединений на активность мю-рецепторов, влияние соединений на серотонинергические пути приводит к антидепрессантному действию, поддержанию сна и анксиолитическому действию. Поскольку депрессия и тревога являются ключевыми факторами, в первую очередь приводящими восприимчивых пациентов к употреблению опиоидов, соединения настоящего изобретения облегчат симптомы опиатной абстиненции и одновременно облегчат психиатрические сопутствующие заболевания, которые способствуют употреблению опиоидов-двусторонняя стратегия для снижения риска ремиссии. Поддержание сна, обеспечиваемое данными соединения, будет способствовать дальнейшему улучшению качества жизни пациентов, проходящих OUD лечение.
[0065] “Алкил”, как применяют в настоящем изобретении, представляет собой насыщенный или ненасыщенный углеводородный фрагмент, например, один-двадцать атомов углерода в длину, если не указано иначе; любой данный алкил может быть линейным или разветвленным (например, н-бутил или трет-бутил), предпочтительно линейным, если не указано иначе. Например, "C1-21 алкил" обозначает алкил, содержащий 1-21 атомов углерода. В одном варианте осуществления, алкил необязательно замещен одной или более гидрокси или C1-22алкокси (например, этокси) группами. В другом варианте осуществления, алкил содержит 1-21 атомов углерода, предпочтительно нормальную цепь и необязательно является насыщенным или ненасыщенным, например, в некоторых вариантах осуществления, когда R1 представляет собой алкильную цепь, содержащую 1-21 атомов углерода, предпочтительно 6-15 атомов углерода, 16-21 атомов углерода, например, так, что вместе с -C(O)-, с которым он соединен, например, при отщеплении от соединения формулы I, образует остаток природной или неприродной, насыщенной или ненасыщенной жирной кислоты.
[0066] Предполагается, что термин “фармацевтически приемлемый разбавитель или носитель” относится к разбавителям и носителям, которые являются пригодными в фармацевтических препаратах, и которые не содержат аллергенных, пирогенных или патогенных веществ, и веществ, которые, как известно, потенциально вызывают или способствуют заболеванию. Фармацевтически приемлемые разбавители или носители, таким образом, исключают телесные жидкости, такие как, например, кровь, моча, спинномозговая жидкость, слюна и подобные, а также их составляющие компоненты, такие как клетки крови и циркулирующие белки. Подходящие фармацевтически приемлемые разбавители и носители можно найти в любом из нескольких известных научных источников по фармацевтическим составам, например, Anderson, Philip O.; Knoben, James E.; Troutman, William G, eds., Handbook of Clinical Drug Data, Tenth Edition, McGraw-Hill, 2002; Pratt и Taylor, eds., Principles of Drug Action, Third Edition, Churchill Livingston, New York, 1990; Katzung, ed., Basic and Clinical Pharmacology, Ninth Edition, McGraw Hill, 20037ybg; Goodman and Gilman, eds., The Pharmacological Basis of Therapeutics, Tenth Edition, McGraw Hill, 2001; Remington's Pharmaceutical Sciences, 20th Ed., Lippincott Williams & Wilkins., 2000; и Martindale, The Extra Pharmacopoeia, Thirty-Second Edition (The Pharmaceutical Press, London, 1999); все из которых включены в настоящее изобретение с помощью ссылки во всей своей полноте.
[0067] Термины "очищенный," "в очищенной форме" или "в выделенной и очищенной форме" для соединения относятся к физическому состоянию указанного соединения после выделения из способа получения (например, из реакционной смеси), или природного источника или их комбинации. Таким образом, термин "очищенный," "в очищенной форме" или "в выделенной и очищенной форме" для соединения относится к физическому состоянию указанного соединения после его получения в результате способа очистки или способов, описанных в настоящем изобретении, или хорошо известных специалистам в данной области (например, хроматографии, перекристаллизации, LC-MS и LC-MS/MS способов и подобных), с достаточной степенью чистоты для того, чтобы охарактеризовать его стандартными аналитическими способами, описанными в настоящем изобретении или хорошо известными специалисту в данной области.
[0068] Если не указано иначе, соединения настоящего изобретения, например, соединение I или 1.1-1.34, соединение II или 2.1-2.10 (совместно, соединения формул I-II и далее) могут находиться в свободной форме или в виде соли, например, в виде солей присоединения кислоты. Соль присоединения кислоты соединения настоящего изобретения, которая является достаточно основной, например, соль присоединения кислоты, например, неорганической или органической кислоты, например, хлористоводородной, бромистоводородной, серной, фосфорной, уксусной, трифторуксусной, лимонной, малеиновой, толуолсульфоновой, пропионовой, янтарной, гликолевой, стеариновой, молочной, яблочной, винной, лимонной, аскорбиновой, памовой, гидроксималеиновой, фенилуксусной, глютаминовой, бензойной, салициловой, сульфаниловой, 2-ацетоксибензойной, фумаровой, толуолсульфоновой, метансульфоновой, этандисульфоновой, щавелевой, изетионовой кислота и подобных. Кроме того, соль соединения настоящего изобретения, которая является достаточно кислой, представляет собой соль щелочного металла, например, натриевую или калиевую соль, соль щелочноземельного металла, например, соль кальция или магния, аммониевую соль или соль с органическим основанием, который дает физиологически приемлемый катион, например, соль с метиламином, диметиламином, триметиламином, пиперидином, морфолином или трис(2-гидроксиэтил)амином. В конкретном варианте осуществления, соль соединения настоящего изобретения представляет собой соль присоединения толуолсульфокислоты. В другом конкретном варианте осуществления, соль соединения настоящего изобретения представляет собой соль присоединения фумаровой кислоты. В конкретном варианте осуществления, соль соединения настоящего изобретения представляет собой соль присоединения фосфорной кислоты.
[0069] Соединения настоящего изобретения предполагаются для применения в качестве фармацевтических средств, следовательно, фармацевтически приемлемые соли являются предпочтительными. Соли, которые не подходят для фармацевтического применения, могут быть пригодны, например, для выделения или очистки свободного соединения, и, следовательно, также включены в объем соединений настоящего изобретения.
[0070] Соединения настоящего изобретения могут содержать один или более хиральных атомов углерода. Таким образом, соединения существуют в виде отдельной изомерной, например, энантиомерной или диастереомерной формы или в виде смеси индивидуальных форм, например, рацемических/диастереомерных смесей. Может присутствовать любой изомер, в котором асимметрический центр находится в (R)-, (S)- или (R, S)-конфигурации. Настоящее изобретение следует понимать как включающее отдельные оптически активные изомеры, а также их смеси (например, рацемические/диастереомерные смеси). Соответственно, соединения настоящего изобретения могут представлять собой рацемическую смесь или могут быть преимущественно, например, в чистой, или по существу чистой, изомерной форме, например, более чем 70% энантиомерный/диастереомерный избыток (“ee”), предпочтительно более чем 80% ee, более предпочтительно более чем 90% ee, самое предпочтительное более чем 95% ee. Очистку указанных изомеров и разделение указанных изомерных смесей можно осуществлять стандартными способами, известными в данной области техники (например, колоночной хроматографией, препаративной ТСХ, препаративной ВЭЖХ, псевдодвижущимся слоем и подобными).
[0071] Геометрические изомеры по природе заместителей вокруг двойной связи или кольца могут присутствовать в цис (Z) или транс (E) форме, и обе изомерные формы включены в объем настоящего изобретения.
[0072] Предполагается также, что соединения настоящего изобретения включают их стабильные и нестабильные изотопы. Стабильные изотопы представляют собой нерадиоактивные изотопы, которые содержат один дополнительный нейтрон по сравнению с распространенными нуклидами того же вида (т.е. элементом). Ожидается, что активность соединений, содержащих данные изотопы, будет сохранена, и данное соединение также будет пригодно для измерения фармакокинетических параметров неизотопных аналогов. Например, атом водорода в определенном положении в соединениях настоящего изобретения можно заменять дейтерием (стабильным изотопом, который не является радиоактивным). Примеры известных стабильных изотопов включают, но не ограничиваются, дейтерий, 13C, 15N, 18O. Альтернативно, нестабильные изотопы, которые являются радиоактивными изотопами, которые содержат дополнительные нейтроны по сравнению с распространенными нуклидами того же вида (т.е. элементом), например, 123I, 131I, 125I, 11C, 18F, могут заменять соответствующие распространенные варианты I, C и F. Еще один пример пригодного изотопа соединения настоящего изобретения представляет собой 11C изотоп. Данные радиоизотопы являются пригодными для радиоактивной визуализации и/или фармакокинетических исследований соединений настоящего изобретения. Кроме того, замена атомов с естественным распределением изотопов более тяжелыми изотопами может привести к желательному изменению фармакокинетических скоростей, когда данные замены осуществляют в метаболически лабильных положениях. Например, включение дейтерия (2H) вместо водорода может замедлить метаболическую деградацию, когда положение водорода представляет собой сайт ферментативной или метаболической активности.
[0073] Соединения настоящего изобретения можно включать в депо состав, например, диспергированием, растворением или инкапсулированием соединений настоящего изобретения в полимерный матрикс, как определено в любой из композиций 6 и 6.1-6.7, так что соединение будет непрерывно высвобождаться по мере разрушение полимера с течением времени. Высвобождение соединений настоящего изобретения из полимерного матрикса обеспечивает контролируемое и/или замедленное высвобождение соединений, например, из фармацевтической депо композиции, субъекту, например, теплокровному животному, такому как человек, которому вводили фармацевтический депо. Таким образом, фармацевтическое депо доставляет соединения настоящего изобретения субъекту в концентрациях, эффективных для лечения конкретного заболевания или состояния, в течение продолжительного периода времени, например, 14-180 дней, предпочтительно около 30, около 60 или около 90 дней.
[0074] Полимеры, пригодные для полимерного матрикса, в составе настоящего изобретения (например, депо композиции настоящего изобретения) могут включать полиэфир гидроксижирной кислоты и ее производных или других агентов, таких как полимолочная кислота, полигликолевая кислота, полицлимонная кислота, полияблочная кислота, поли-бета-гидроксимасляная кислота, полимер с раскрывающимся эпсилон-капролактоновым кольцом, сополимер молочной кислоты и гликолевой кислоты, сополимер 2-гидроксимасляной кислоты и гликолевой кислоты, сополимер полимолочной кислоты и полиэтиленгликоля или сополимер полигликолевой кислоты и полиэтиленгликоля) полимер алкил-альфа-цианоакрилата (например, поли(бутил-2-цианоакрилата)), полиалкиленоксалат (например, политриметиленоксалат или политетраметиленоксалат), полиортоэфир, поликарбонат (например, полиэтиленкарбонат или полиэтиленпропиленкарбонат), полиортокарбонат, полиаминокислота (например, поли-гамма-L-аланин, поли-гамма-бензил-L-глютаминовая кислота или поли-y-метил-L-глютаминовая кислота), эфир гиалуроновой кислоты и подобные, и можно применять один или более из данных полимеров.
[0075] Если полимеры представляют собой сополимеры, они могут быть любым из статистического, блочного и/или привитого сополимера. Когда вышеуказанные альфа-гидроксикарбоновые кислоты, гидроксидикарбоновые кислоты и гидрокситрикарбоновые кислоты обладают в своих молекулах оптической активностью, можно применять любой из D-изомеров, L-изомеров и/или DL-изомеров. Среди прочих, можно применять полимер альфа-гидроксикарбоновых кислот (полимер молочной кислоты и гликолевой кислоты), его сложные эфиры, сложные эфиры поли-альфа-цианоакриловой кислоты и т.д., и сополимер молочной кислоты и гликолевой кислоты (также называемый поли (лактид-альфа-гликолид) или поли(молочная-со-гликолевая кислота), и далее называемый PLGA) является предпочтительными. Таким образом, в одном аспекте полимер, применяемый для полимерных матриц, представляет собой PLGA. Термин PLGA включает полимеры молочной кислоты (также называемые полилактид, поли(молочная кислота) или PLA). Самое предпочтительное, полимер представляет собой биоразлагаемый поли(d, l-лактид-со-гликолидный) полимер.
[0076] В предпочтительном варианте осуществления, полимерный матрикс настоящего изобретения представляет собой биосовместимый и биоразлагаемый полимерный материал. Термин “биосовместимый” определяют как полимерный материал, который не является токсичным, не является канцерогенным и не вызывает значительного воспаления в тканях организма. Матричный материал должен быть биоразлагаемым, при этом полимерный материал должен разлагаться в результате процессов в теле до продуктов, легко утилизируемых организмом, и не должен накапливаться в организме. Продукты биоразложения также должны быть биосовместимыми с телом, так как полимерный матрикс является биосовместимым с телом. Конкретные пригодные примеры материалов полимерного матрикса включают поли(гликолевую кислоту), поли-D, L-молочную кислоту, поли-L-молочную кислоту, сополимеры приведенных выше, поли(алифатические карбоновые кислоты), сополиоксалаты, поликапролактон, полидиоксанон, поли(орто-карбонаты), поли(ацетали), поли(молочную кислоту-капролактон), полиортоэфиры, поли(гликолевую кислоту-капролактон), полиангидриды, и природные полимеры, включая альбумин, казеин и воски, такие как, глицерин моно- и дистеарат, и подобные. Предпочтительный полимер для применения на практике настоящего изобретения представляет собой dl(полилактид-со-гликолид). Предпочтительно, чтобы молярное отношение лактида к гликолиду в данном сополимере было в диапазоне приблизительно 75:25-50:50.
[0077] Пригодные PLGA полимеры могут иметь средневесовую молекулярную массу от приблизительно 5000 до 500000 дальтон, предпочтительно приблизительно 150000 дальтон. В зависимости от скорости разрушения, которую достигают, можно применять различную молекулярную массу полимеров. Для диффузионного механизма высвобождения лекарственного средства полимер должен оставаться неповрежденным до тех пор, пока все лекарственное средство не высвободится из полимерного матрицы и затем не разложится. Лекарственное средство также может высвобождаться из полимерного матрикса по мере того, как подвергается биоразрушению полимерное вспомогательное вещество.
[0078] PLGA можно получить любым общепринятым способом, или она может иметься в продаже. Например, PLGA можно получить полимеризацией с раскрытием кольца с подходящим катализатором из циклического лактида, гликолида и т.д. (смотри EP-0058481B2; Effects of polymerization variables on PLGA properties: molecular weight, composition and chain structure).
[0079] Считается, что PLGA является биоразлагаемым за счет разложения всей твердой полимерной композиции вследствие разрушения гидролизуемых и ферментативно расщепляемых сложноэфирных связей в биологических условиях (например, в присутствии воды и биологических ферментов, обнаруженных в тканях теплокровных животных, таких как человек), образуя молочную кислоту и гликолевую кислоту. Молочная и гликолевая кислота являются водорастворимыми, нетоксичными продуктами нормального метаболизма, которые могут дополнительно разлагаться с образованием углекислого газа и воды. Другими словами, считают, что PLGA разлагается посредством гидролиза ее сложноэфирных групп в присутствии воды, например, в организме теплокровного животного, такого как человек, с образованием молочной кислоты и гликолевой кислоты и созданием кислотного микроклимата. Молочная и гликолевая кислоты являются побочными продуктами различных метаболических путей в организме теплокровного животного, такого как человек, в нормальных физиологических условиях и, следовательно, хорошо переносятся и вызывают минимальную системную токсичность.
[0080] В другом варианте осуществления, полимерный матрикс, пригодный для настоящего изобретения, может включать звездообразный полимер, где структура полиэфира имеет звездообразную форму. Данные полиэстры содержат один полиольный остаток в качестве центрального фрагмента, окруженный цепями кислотных остатокв. Полиольный фрагмент может представлять собой, например, глюкозу или, например, маннитол. Данные эфиры являются известными и описаны в GB 2145422 и в патенте США No. 5538739, содержание которых включено с помощью ссылки.
[0081] Звездоообразные полимеры можно получить, применяя полигидрокси соединения, например, полиол, например, глюкозу или маннитол в качестве инициатора. Полиол содержит, по меньшей мере, 3 гидрокси группы и имеет молекулярный вес вплоть до приблизительно 20000 Дальтон, причем, по меньшей мере, 1, предпочтительно, по меньшей мере, 2, например, в качестве среднего 3 гидрокси группы полиола присутствуют в виде сложноэфирных групп, которые содержат полилактидные или со-полилактидные цепи. Разветвленные полиэстры, например, поли(d, l-лактид-со-гликолид) содержат центральный глюкозный фрагмент, содержащий лучи линейных полилактидных цепей.
[0082] Депо композиции настоящего изобретения (например, композиции 6 и 6.1-6.10 в полимерном матриксе), как описано в настоящем изобретении выше, могут содержать полимер в виде микрочастиц или наночастиц, или в жидкой форме, причем соединения настоящего изобретения диспергированы или инкапсулированы в нем. “Микрочастицы” означают твердые частицы, которые содержат соединения, или в растворе, или в твердой форме, где данное соединение диспергировано или растворено в полимере, который служит в качестве матрикса частиц. Путем соответствующего выбора полимерных материалов может быть получен состав микрочастиц, в котором полученные микрочастицы проявляют и свойства диффузионного высвобождения, и свойства биоразложения.
[0083] Когда полимер находится в виде микрочастиц, микрочастицы можно получить, применяя подходящий способ, такой как упаривание растворителя или способ экстракции растворителем. Например, в способе экстракции растворителем, соединения настоящего изобретения и полимер можно растворять в летучем органическом растворителе (например, кетоне, таком как ацетон, галогенированном углеводороде, таком как хлороформ или хлористый метилен, галогенированном ароматическом углеводороде, циклическом эфире, таком как диоксан, сложном эфире, такой как этилацетат, нитриле, таком как ацетонитрил, или спирте, таком как этанол) и диспергировать в водной фазе, содержащей подходящий стабилизатор эмульсии (например, поливиниловый спирт, PVA). Затем, органический растворитель упаривают, получая микрочастицы с инкапсулированными в них соединениями настоящего изобретения. В способе экстракции растворителем, соединения настоящего изобретения и полимер можно растворять в полярном растворителе (таком как ацетонитрил, дихлорметан, метанол, этилацетат или метилформиат), и затем диспергировать в водной фазе (такой как вода/PVA раствор). Эмульсию получают для обеспечения микрочастиц с инкапсулированными в них соединениями настоящего изобретения. Распылительная сушка представляет собой альтернативный способ получения микрочастиц.
[0084] Другой способ получения микрочастиц настоящего изобретения также описан в патенте США № 4389330 и патенте США № 4530840.
[0085] Микрочастицы настоящего изобретения можно получить любым способом, способным производить микрочастицы в диапазоне размеров, приемлемом для применения в инъецируемой композиции. Одним из предпочтительных способов получения является способ, который описан в патенте США № 4389330. В данном способе, жидкий активный агент растворяют или диспергируют в соответствующем растворителе. К среде, содержащей агент, добавляют полимерный матриксный материал в количестве относительно активного ингредиента, которое обеспечивает продукт, имеющий желаемую загрузку жидкого активного агента. Необязательно, все ингредиенты продукта из микрочастиц могут быть смешаны в среде растворителя вместе.
[0086] Растворители для соединений настоящего изобретения и материала полимерного матрикса, которые можно применять на практике настоящего изобретения, включают органические растворители, такие как ацетон; галогенированные углеводороды, такие как хлороформ, хлористый метилен и подобные; ароматические углеводородные соединения; галогенированные ароматические углеводородные соединения; циклические эфиры; спирты, такие как бензиловый спирт; этилацетат; и подобные. В одном варианте осуществления, растворителем для применения на практике настоящего изобретения может быть смесь бензилового спирта и этилацетата. Дополнительную информацию о получении микрочастиц, пригодных в настоящем изобретении, можно найти в патентной публикации США № 2008/0069885, содержание которой включено в настоящее исследование с помощью ссылок во всей своей полноте.
[0087] Количество соединений настоящего изобретения, включенных в микрочастицы, обычно находится в диапазоне от приблизительно 1% по весу до приблизительно 90% по весу, предпочтительно 30-50% по весу, более предпочтительно 35-40% по весу. % по весу обозначает части соединений настоящего изобретения на суммарный вес микрочастиц.
[0088] Фармацевтические депо композиции могут содержать фармацевтически приемлемый разбавитель или носитель, такой как смешиваемый с водой разбавитель или носитель.
[0089] Подробную информацию о составе системы пероральной доставки с высвобождением, контролируемым осмотически, можно найти в EP 1539115 (публикация США No. 2009/0202631) и WO 2000/35419 (US 2001/0036472), содержание каждой из которых включено с помощью ссылки во всей своей полноте.
[0090] "Терапевтически эффективное количество" представляет собой любое количество соединений настоящего изобретения (например, как содержится в фармацевтическом депо), которое, при введении субъекту, страдающему от заболевания или расстройства, эффективно вызывает уменьшение, ремиссию или регресс заболевания или расстройства в течение периода времени, как предназначено для лечения.
[0091] Дозировки, применяемые на практике настоящего изобретения, будут, конечно, варьироваться в зависимости, например, от конкретного заболевания или состояния, подлежащего лечению, конкретного применяемого соединения настоящего изобретения, способа введения и требуемой терапии. Если не указано иначе, количество соединения настоящего изобретения для введения (независимо от того, вводится ли оно в качестве свободного основания или в солевой формы) относится к количеству соединения настоящего изобретения в форме свободного основания (т.е. расчет количества основан на количестве свободного основания).
[0092] Соединения настоящего изобретения можно вводить любым удовлетворительным путем, включая перорально, парентерально (внутривенно, внутримышечно или подкожно) или трансдермально. В определенных вариантах осуществления, соединения настоящего изобретения, например, в виде депо состава предпочтительно вводили парентерально, например, инъекцией, например, внутримышечной или подкожной инъекцией.
[0093] В общем, удовлетворительные результаты для способа 1 и 1.1-1.45, способа 2 и 2.1-2.12 и способа 3 и 3.1-3.41 или применения соединений настоящего изобретения, как описано в настоящем изобретении выше, например, для лечения комбинации заболеваний, такой как комбинация, по меньшей мере, депрессии, психоза, например, (1) психоза, например, шизофрении, у пациента, страдающего от депрессии; (2) депрессии у пациента, страдающего от психоза, например, шизофрении; (3) поведенческих расстройств, связанных с психозом, например, шизофренией или болезнью Паркинсона; (4) расстройств сна, связанных с психозом, например, шизофренией или болезнью Паркинсона; и (5) наркотической зависимости, расстройств, связанных с применением наркотических веществ, и/или расстройств, вызванных наркотическими веществами, как указано выше, указаны для получения при пероральном введении в дозировках порядка приблизительно 1 мг-100 мг один раз в день, 2,5 мг-50 мг, например, 2,5 мг, 5 мг, l0 мг, 20 мг, 30 мг, 40 мг или 50 мг, один раз в день, путем перорального введения.
[0094] Удовлетворительные результаты для способа 2 или 2.1-2.12 или применения соединений настоящего изобретения, как описано в настоящем изобретении выше, например, для лечения отдельно расстройства сна указаны для получения при пероральном введении в дозировках порядка приблизительно 2,5 мг-5 мг, например, 2,5 мг, 3 мг, 4 мг или 5 мг, соединения настоящего изобретения в свободной или фармацевтически приемлемой солевой форме, один раз в день, предпочтительно пероральным введением.
[0095] Удовлетворительные результаты для способа 1-A или способа 2-A, или любого из 3.1-3.41 указаны для получения при меньше чем l00 мг, предпочтительно при меньше чем 50 мг, например, при меньше чем 40 мг, при меньше чем 30 мг, при меньше чем 20 мг, при меньше чем l0 мг, при меньше чем 5 мг, при меньше чем 2,5 мг, один раз в день. Удовлетворительные результаты для способа II-A или любого из 3.1-3.41 указаны для получения при меньше чем 5 мг, предпочтительно при меньше чем 2,5 мг.
[0096] Для лечения расстройств, описанных в настоящем изобретении, где депо композицию применяют для достижения большей продолжительности действия, дозы будут выше по сравнению с композицией с более коротким действием, например, более чем 1-100 мг, например, 25 мг, 50 мг, 100 мг, 500 мг, 1000 мг или более чем 1000 мг. Продолжительность действия соединений настоящего изобретения можно контролировать манипулированием полимерным составом, т.е. соотношением полимер:лекарственное средство, и размером микрочастиц. Когда композиция настоящего изобретения представляет собой депо композицию, предпочтительным является введение путем инъекции.
[0097] Фармацевтически приемлемые соли соединений настоящего изобретения можно получить из исходного соединения, которое содержит основную или кислотную группу общепринятыми химическими способами. Обычно, данные соли можно получить реакцией форм свободного основания данных соединений со стехиометрическим количеством соответствующей кислоты в воде или в органическом растворителе, или в смеси данных двух растворителей; обычно, предпочтительными являются неводные среды, такие как эфир, этилацетат, этанол, изопропанол или ацетонитрил. Дополнительные подробности получения данных солей, например, толуолсульфоновой соли в аморфной или кристаллической форме, можно найти в PCT/US0/03340 и/или предварительной заявке США № 61/036069 (каждый эквивалент US 2011/112105).
[0098] Фармацевтические композиции, содержащие соединения настоящего изобретения, можно получить, применяя общепринятые разбавители или вспомогательные вещества (один пример включает, но не ограничивается, кунжутное масло) и способы, известный в медицинской области техники. Таким образом, пероральные лекарственные формы могут включать таблетки, капсулы, растворы, суспензии и подобные.
[0099] Термин “совместно” при ссылке на терапевтическое применение обозначает введение двух или более активных ингредиентов пациенту как часть режима лечения заболевания или расстройства, независимо от того, назначаются ли два или более жидких активных агента в одно и то же или в разное время, или же одинаковыми или различными путями введения. Одновременное введение двух или более активных ингредиентов может происходить в разное время в один и тот же день, или в разные даты, или при разных частотах.
[00100] Термин “одновременно” при ссылке на терапевтическое применение обозначает введение двух или более активных ингредиентов в одно и то же время одним и тем же путем введения.
[00101] Термин “отдельно” при ссылке на терапевтическое применение обозначает введение двух или более активных ингредиентов в одно и то же время разными путями введения.
Способы получения соединений настоящего изобретения:
[0102] Соединение формулы A и способы его получения описаны в международной заявке PCT/US2017/15178, опубликованной как WO 2017/132408, и в US 2017/319580. Соединения настоящего изобретения, в которых Z представляет собой -O-, можно получить, например, реакцией соединения формулы A с подходящими алкилирующими и ацилирующими агентами, при необходимости, согласно схеме 1 ниже. Другие способы проведения аналогичных превращений являются известными специалисту в данной области техники.
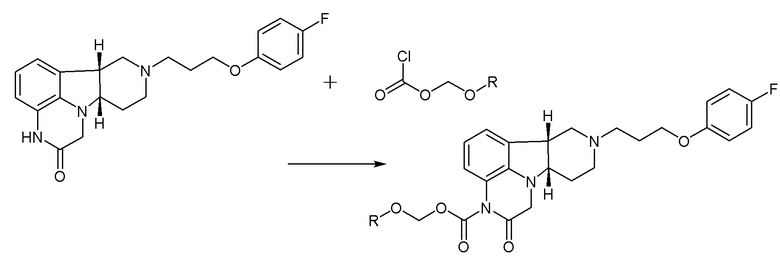
Схема 1
[0103] Другие соединения настоящего изобретения можно получить аналогичными способами, известными специалисту в данной области техники.
[0104] Выделение или очистку диастереомеров соединений настоящего изобретения можно осуществлять общепринятыми способами, известными в данной области техники, например, очисткой на колонке, препаративной тонкослойной хроматографией, препаративной ВЭЖХ, кристаллизацией, растиранием, псевдодвижущимся слоем и подобными.
[0105] Соли соединений настоящего изобретения можно получить аналогично способам, описанным в патенте США № 6548493; 7238690; 6552017; 6713471; 7183282; U.S. RE39680; U.S. RE39679; и WO 2009/114181 (US 2011/112105), содержание каждого из которых включено с помощью ссылки во всей своей полноте.
[0106] Диастереомеры полученных соединений можно разделить, например, ВЭЖХ, применяя CHIRALPAK® AY-H, 5 мкм, 30×250 мм при комнатной температуре и элюировать 10% этанол/90% гексан/0,1% диметилэтиламин. Пики можно детектировать при 230 нм, получая 98-99,9% ее диастереомера.
Пример 1: получение 4-((6bR,10aS)-2-оксо-2,3,6b,9,10,10a-гексагидро-1H,7H-пиридо[3',4':4,5]пирроло[1,2,3-de]хиноксалин-8-ил)-1-(4-фторфенил)-бутан-1-она
[0107] Этиловый эфир (6bR, 10aS)-2-оксо-2,3,6b,9,10,10a-гексагидро-1H,7H-пиридо[3',4':4,5]пирроло[1,2,3-de]хиноксалин-8-карбоновой кислоты (6,4 г, 21,2 ммоль) суспендировали в растворе уксусной кислоты HBr (64 мл, 33% вес/вес) при комнатной температуре. Смесь нагревали при 50°C в течение 16 ч. После охлаждения и обработки этилацетатом (300 мл), смесь фильтровали. Остаток на фильтре промывали этилацетатом (300 мл), и затем сушили в вакууме. Затем, полученную HBr соль суспедировали в метаноле (200 мл) и охлаждали сухим льдом в изопропаноле. При интенсивном перемешивании, раствор аммиака (10 мл, 7N в метаноле) медленно добавляли к суспензии, доводя pH смеси до 10. Полученную смесь сушили в вакууме без дополнительной очистки, получая неочищенный (6bR, 10aS)-2-оксо-2,3,6b,9,10,10a-гексагидро-1H,7H-пиридо[3',4':4,5]пирроло[1,2,3-de]хиноксалин (8,0 г), который применяли непосредственно в следующей стадии. MS (ESI) m/z 230,2 [M+H]+.
[0108] Неочищенный (6bR, 10aS)-2-оксо-2,3,6b,9,10,10a-гексагидро-1H,7H-пиридо[3',4':4,5]пирроло[1,2,3-de]хиноксалин (1,4 г) растворяли в DMF (14 мл), и затем последовательно добавляли KI (2,15 г) и 4-хлор-4'-фторбутирофенон (2 мл). Смесь дегазировали аргоном, с последующим добавлением N, N-диизопропилэтиламина (DIPEA, 2 мл). Смесь нагревали при 78°C в течение 2 ч. После охлаждения, растворители удаляли при пониженном давлении. Темно-коричневый остаток суспендировали в дихлорметане (100 мл), и затем экстрагировали водой (30 мл). Органический слой отделяли и сушили над K2CO3. После фильтрации, растворители удаляли при пониженном давлении. Полученный неочищенный продукт очищали колоночной хроматографией на силикагеле, элюбируя 0-10% метанола в этилацетате, содержащем 0,1% 7N аммиака в метаноле, получая 4-((6bR,10aS)-2-оксо-2,3,6b,9,10,10a-гексагидро-1H,7H-пиридо[3',4':4,5]пирроло[1,2,3-de]хиноксалин-8-ил)-1-(4-фтор-фенил)бутан-1-он в виде светло-желтого твердого остатка (767 мг). 1H ЯМР (500 МГц, DMSO-d6) δ 10,3 (с, 1H), 8,1-8,0 (м, 2H), 7,3 (дд, J=8,86 Гц, 2H), 6,8 (д, J=7,25 Гц, 1H), 6,6 (дд, J=7,55 Гц, 1H), 6,6 (д, J=7,74 Гц, 1H), 3,8 (д, J=14,49 Гц, 1H), 3,3-3,3 (м, 1H), 3,2-3,2 (м, 1H), 3,1-3,0 (м, 1H), 3,0 (т, J=6,88 Гц, 2H), 2,8-2,8 (м, 1H), 2,6-2,5 (м, 1H), 2,3-2,2 (м, 2H), 2,1-2,0 (м, 1H), 1,9-1,8 (м, 1H), 1,8 (т, J=6,99 Гц, 2H), 1,6 (т, J=11,25 Гц, 2H). MS (ESI) m/z 394,2 [M+H]+.
Пример 2: получение (6bR,10aS)-8-(3-(4-фторфенокси)пропил)-6b,7,8,9,10,10a-гексагидро-1H-пиридо[3',4':4,5]пирроло[1,2,3-de]хиноксалин-2(3H)-она
[0109] Смесь (6bR,10aS)-6b,7,8,9,10,10a-гексагидро-1H-пиридо[3',4':4,5]пирроло[1,2,3-de]хиноксалин-2(3H)-она (100 мг, 0,436 ммоль), 1-(3-хлорпропокси)-4-фторбензола (100 мкл, 0,65 ммоль) и KI (144 мг, 0,87 ммоль) в DMF (2 мл) дегазировали аргоном в течение 3 минут и добавляли DIPEA (150 мкл, 0,87 ммоль). Полученную в результате смесь нагревали до 78°C и перемешивали при данной температуре в течение 2 ч. Смесь охлаждали до комнатной температуры, и затем фильтровали. Остаток на фильтре очищали колоночной хроматографией на силикагеле, применяя градиент 0-100% этилацетата в смеси метанол/7N NH3 в метаноле (1:0,1 об/об) в качестве элюента, получая частично очищенный продукт, который дополнительно очищали полупрепаративной ВЭЖХ системой, применяя градиент 0-60% ацетонитрила в воде, содержащей 0,1% муравьиную кислоту, в течение 16 минут, получая указанный в заголовке продукта в виде твердого вещества (50 мг, выход 30%). MS (ESI) m/z 406,2 [M+1]+. 1H ЯМР (500 МГц, DMSO-d6) δ 10,3 (с, 1H), 7,2-7,1 (м, 2H), 7,0-6,9 (м, 2H), 6,8 (дд, J=1,03, 7,25 Гц, 1H), 6,6 (т, J=7,55 Гц, 1H), 6,6 (дд, J=1,07, 7,79 Гц, 1H), 4,0 (т, J=6,35 Гц, 2H), 3,8 (д, J=14,74 Гц, 1H), 3,3-3,2 (м, 3H), 2,9 (дд, J=6,35, 11,13 Гц, 1H), 2,7-2,6 (м, 1H), 2,5-2,3 (м, 2H), 2,1 (т, J=11,66 Гц, 1H), 2,0 (д, J=14,50 Гц, 1H), 1,9-1,8 (м, 3H), 1,7 (т, J=11,04 Гц, 1H).
Пример 3: Определение функциональных свойств клеточных и ядерных рецепторов
[0110] Определение функциональных свойств клеточных и ядерных рецепторов проводили на соединении примера 1 согласно способу Wang, J.B. et al. (1994), FEBS Lett., 338:217-222. Соединение тестировали при нескольких концентрациях, определяя IC50 или EC50. Клеточные агонистические эффекты рассчитывали в виде процента контрольной реакции на известный эталонный агонист для каждой мишени, и клеточный антагонистический эффект рассчитывали в виде ингибирования в процентах реакции контрольного эталонного агониста для каждой мишени.
[0111] Следующий тест проводили, определяя эффект соединения примера 1 на μ (MOP) (h) рецептор:
(Рецептор)
(агонистический эффект)
MOP рецептор
(CHO клетки)
(0,3 мкМ DAMGO
для контроля)
37°C
(антагонистический эффект)
MOP рецептор
(CHO клетки)
(20 нМ)
37°C
[0112] Что касается антагонистов, наблюдаемые константы диссоциации (KB) рассчитывали, применяя модифицированное уравнение Ченга-Прусоффа:
где A=концентрация эталонного агониста в тесте, и EC50A=EC50 величина эталонного агониста.
[0113] Было обнаружено, что соединение примера 1 обладает μ (MOP) (h) (антагонистическим эффектом) с IC50 1,3×10-6M; и KB 1,4×10-7M.
[0114] Результаты по агонистической активности выражали в виде процента реакции контрольного агониста:
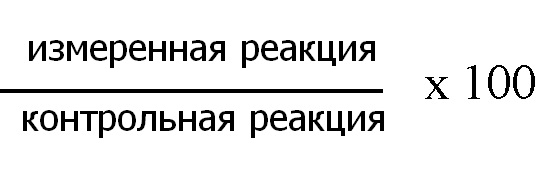
и антагонистическую активность в виде ингибирования в процентах максимальной реакции контрольного агониста:
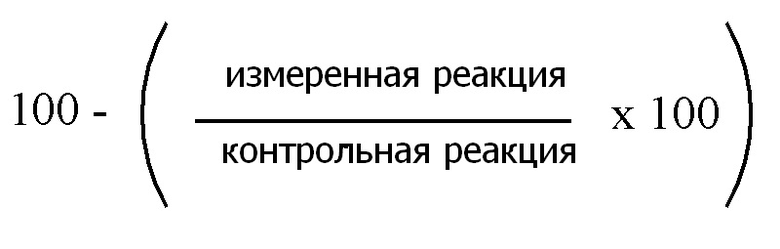
Полученной в присутствии соединения примера 1.
[0115] Величину EC50 (концентрация, вызывающая полумаксимальный ответ) и величину IC50 (концентрация, вызывающая полумаксимальное ингибирование реакции контрольного агониста) определяли нелинейным регрессионным анализом кривых зависимости реакции от концентрации, полученных со средними значениями повторения, применяя аппроксимацию кривой уравнением Хилла:
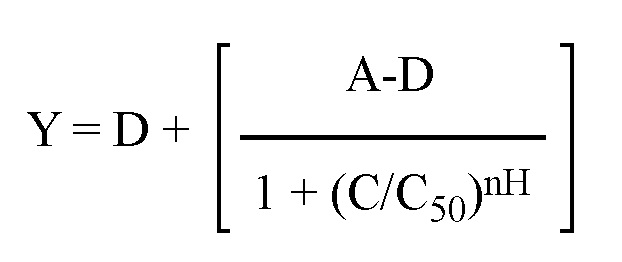
где Y=реакция, A=левая асимптота кривой, D=правая асимптота кривой, C=концентрация соединения, и C50=EC50 или IC50, и nH=коэффициент наклона. Анализ проводили, применяя программное обеспечение, разработанное самостоятельно, и проверяли сравнением с данными, полученными коммерческим программным обеспечением SigmaPlot ® 4.0 для Windows ® (© 1997 SPSS Inc.).
Пример 4: профиль рецепторного связывания
[0116] Рецепторное связывание определяли для соединений примера 1 и 2. Применяли следующие литературные способы, каждый из которых включен в настоящее изобретение с помощью ссылки во всей своей полноте: 5-HT2A: Bryant, H.U. et al. (1996), Life Sci., 15:1259-1268; D2: Hall, D.A. and Strange, P.G. (1997), Brit. J. Pharmacol., 121:731-736; D1: Zhou, Q.Y. et al. (1990), Nature, 347:76-80; SERT: Park, Y.M. et al. (1999), Anal. Biochem., 269:94-104; Mu opiate receptor: Wang, J.B. et al. (1994), FEBS Lett., 338:217-222.
[0117] В общем, результаты выражали в виде процента контрольного специфического связывания:
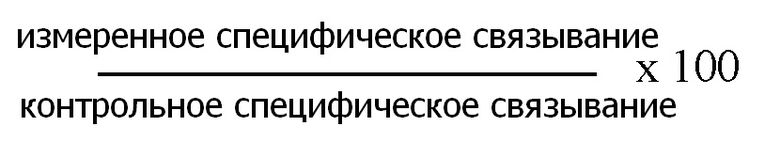
И в виде ингибирования в процентах контрольного специфического связывания:
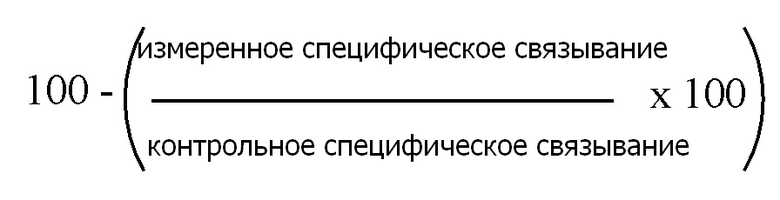
Полученного в присутствии испытуемых соединений.
[0118] Величины IC50 (концентрация, вызывающая полумаксимальное ингибирование контрольного специфического связывания) и коэффициенты Хилла (nH) определяли нелинейным регрессионным анализом конкурентных кривых, полученных со средними значениями повторения, применяя аппроксимацию кривой уравнением Хилла:
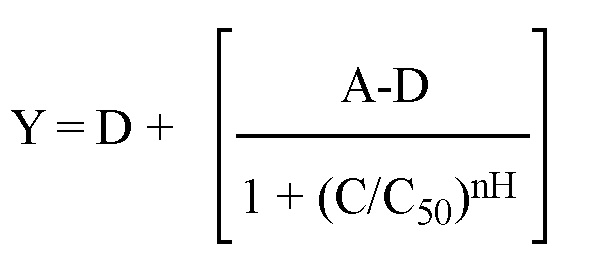
где Y=специфическое связывание, A=левая асимптота кривой, D=правая асимптота кривой, C=концентрация соединения, C50=IC50, и nH=коэффициент наклона. Данный анализ проводили, применяя программное обеспечение, разработанное самостоятельно, и проверяли сравнением с данными, полученными коммерческим программным обеспечением SigmaPlot® 4.0 для Windows® (© 1997 SPSS Inc.). Константы ингибирования (Ki) рассчитывали, применяя уравнение Ченга-Прусоффа:
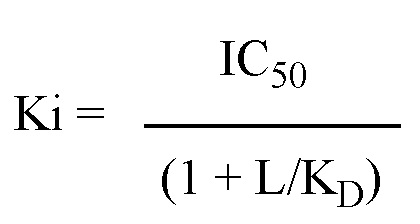
где L=концентрация радиолиганда в данном тесте, и KD=сродство радиолиганда к рецептору. График Скэтчарда применяли для определения KD.
[0119] Получены следующие результаты по рецепторному сродству:
Пример 5: модель судорожного движения головой, вызванного DOI, на мышах
[0120] R-(-)-2,5-диметокси-4-йодамфетамин (DOI) представляет собой агонист семейства серотониновых 5-HT2 рецепторов. При введении мышам он вызывает поведенческий профиль, связанный с частыми судорожными движениями головой. Частота этих судорожных движений головой в течение заданного периода времени может быть взята в качестве оценки агонизма 5-HT2 рецепторов в мозге. И наоборот, данный поведенческий анализ можно применять для определения 5-HT2 рецепторного антагонизма в мозге введением DOI с или без антагониста и регистрацией снижения вызванных DOI судорожных движений головой после введения антагониста.
[0121] Способ Darmani et al., Pharmacol Biochem Behav. (1990) 36:901-906 (содержание которого включено с помощью ссылки во всей своей полноте) применяют с некоторыми модификациями. (±)-DOI HCl вводили подкожно, и мышей немедленно помещали в обычную пластиковую клетку. Количество судорожных движений головой подсчитывали в течение 6 минут, начиная с 1 минуты после введения DOI. Испытуемое соединение вводили перорально за 0,5 ч до введения DOI. Результаты рассчитывали как EC50 для уменьшения вызванных DOI судорожных движений головой. Результаты показаны в следующей таблице:
[0122]
Результаты показывают, что соединения примера 1 и 2 эффективно блокируют судорожные движения головой, вызванные DOI, сопоставимо с in-vitro 5-HT2A результатами, показанными в примере 4.
Пример 6: Тест на подергивание хвостом на мышах
[0123] Тест на подергивание хвостом на мышах представляет собой меру анальгезии, показываемую порогом болевого рефлекса у мышей с задержкой. Самцов мышей CD-1 располагали хвостами под сфокусированным лучом высокоинтенсивного инфракрасного источника тепла, что приводило к нагреванию хвоста. Животное могло отвести свой хвост от источника тепла в любое время, когда ему становится неудобно. Записывали продолжительность времени (задержку) между включением нагревательного прибора и отдергиванием мышью хвоста с пути источника тепла. Введение морфина приводило к анельгезии, и это вызывало задержку реакции мыши на тепло (увеличенная задержка). Предварительное введение антагониста морфинового рецептора (MOR), т.е., налоксона (NAL), обращает эффект и приводит к нормальному времени ожидания. Данный тест применяют как функциональный анализ для определения антагонизма мю-опиатных рецепторов.
Пример 6a: антагонизм анальгезии, вызванной морфином, соединениями примеров 1 и 2
[0124] Десять самцов мышей CD-1 (в возрасте приблизительно 8 недель) распределяли в каждую из 5 групп обработки. Группы обрабатывали следующим способом: группа (1) [отрицательный контроль]: вводили 0,25% метилцеллюлозную среду перорально за 60 минут перед тестом на подергивание хвостом, и среду на основе соляного раствора за 30 минут до теста на подергивание хвостом; группа (2) [положительный контроль]: вводили 0,25% метилцеллюлозую среду перорально, за 60 мин до теста и 5 мг/кг морфина в соляном растворе за 30 мин до теста; группа (3) [положительный контроль]: вводили 3 мг/кг налоксона в соляном растворе за 50 минут до теста и 5 мг/кг морфина в соляном растворе за 30 мин до теста; группы (4)-(6): вводили 0,1 мг/кг, 0,3 мг/кг или 1 мг/кг испытуемого соединения в 0,25% метилцеллюлозной среде перорально за 60 мин до теста и 5 мг/кг морфина за 30 мин до теста. Эксперимент повторяли для соединений примера 1 и примера 2. Результаты показаны в следующей таблице в виде средней задержки в секундах:
среда/среда
[0125] Результаты показывают, что оба соединения примера 1 и примера 2 проявляют зависимую от дозы блокаду вызванной морфином мю-опиатной рецепторной активности.
Пример 6b: анальгезия соединением примера 2, ингибированная налоксоном
[0126] Во втором исследовании, применяя тест на подергивание хвостом на мышах, как определено выше, соединение примера 2 дополнительно сравнивали при дозах 1,0 мг/кг, 3,0 мг/кг и 10 мг/кг относительно морфина при 5 мг/кг с и без предварительной дозировки налоксона при 3 мг/кг (интраперитонеально). В группах с предобработкой, налоксон вводили за 20 минут до теста на подергивание хвостом. В группах без предобработки, соляной раствор вводили за 20 минут до теста на подергивание хвостом. В каждой группе, среду, морфин или соединение примера 2 вводили за 30 минут до теста на подергивание хвостом. Результаты показаны в таблице ниже в виде средней задержки в секундах:
[0127] Обнаружено, что введение соединения примера 2 при всех дозах значительно увеличивало задержку подергивания хвостом, и что данный эффект ослабляется предобработкой налоксоном. Данный результат демонстрирует зависящий от дозы анальгетический эффект, вызванный соединением примера 2, и дополнительно предполагает, что данный эффект опосредован мю-опиоидным рецепторным агонизмом.
Пример 6c: временная динамика для анальгезии, соединение примера 2
[0128] Тест на подергивание хвостом, как определено выше, повторяли для определения временной динамики анальгезии, являющейся результатом введения соединения примера 2. Мышам вводили подкожно или (1) среду за 30 минут до теста, (2) 5 мг/кг морфина за 30 минут до теста, или (3)-(7) 1 мг/кг соединения примера 3 за 30 минут, 2 часа, 4 часа, 8 часов или 24 часа до теста. Результаты показаны в таблице ниже в виде средней задержки в секундах:
[0129] Результаты показывают, что соединение примера 2 вызывает эффективную анальгезию при введении за 30 минут или 2 часа до теста на подергивание хвостом (ANOVA, P<0,001 относительно среды). При введении за 4 часа, 8 часов или 24 часа до теста на подергивание хвостом, соединение примера 2 при 1 мг/кг не вызывало анальгетический эффект, значительно отличающийся от контроля со средой. Таким образом, соединение примера 2 не вызывало пролонгированной анальгезии, что означает, что оно будет иметь более низкий потенциал злоупотребления и меньший риск взаимодействия лекарств по сравнению с другими опиатными анальгетиками.
Пример 6d: Анальгезия после длительного введения соединения примера 2
[0130] Тест на подергивание хвостом, описанный выше, повторяли, применяя модель тестирования, в которой животные подвергались 14-дневному длительному режиму обработки, с последующей кратковременной обработкой за 30 минут до теста на подергивание хвостом. Мышей разделяли по трем широким группам с шестью подгруппами из 10 мышей каждая. Три группы получали длительную обработку (A) средой, (B) соединением примера 2 при 0,3 мг/кг или (C) соединением примера 2 при 3,0 мг/кг. Каждая подгруппа дополнительно получала в качестве кратковременной обработки (1) среду или (2)-(6) соединение примера 2 при 0,01, 0,03, 0,1, 0,3 или 1,0 мг/кг. Все введения осуществляли подкожно. Результаты показаны в таблице ниже в виде средней задержки до подергивания хвостом в секундах:
[0131] Обнаружено, что 0,1, 0,3 и 1,0 мг/кг кратковременная обработка соединением примера 2 вызывала статистически значимый дозозависимый анальгетический эффект по сравнению с группой с кратковременной обработкой средой. Это верно для каждой из длительных группа (A), (B) и (C). По сравнению с предобработкой средой, предобработка соединением примера 2 при 0,3 мг/кг или 3,0 мг/кг обычно показывала статистически значимое снижение задержки подергивания хвостом при сравнении с теми же подгруппами с кратковременной обработкой. Данные результаты показывают, что в то время как некоторая толерантность к анальгетическому эффекту соединения примера 2 наступает после 14 дней длительной обработки, полученная анальгезия остается эффективной, несмотря на длительную предобработку.
Пример 7: ЦНС фосфопротеиновый профиль
[0132] Комплексное исследование молекулярного фосфорилирования также проводили для исследования профиля центральной нервной системы (ЦНС) соединений примера 1 и примера 2. Степень фосфорилирования белков для отобранных ключевых белков центральной нервной системы измеряли на прилежащих ядрах мышей. Исследуемые белки включали ERK1, ERK2, Glu1, NR2B и TH (тирозин идроксилаза), и соединения примера 1 и 3 сравнивали с антипсихотическими агентами рисперидоном и галоперидолом.
[0133] Мышей обрабатывали соединением примера 1 или 2 при 3 мг/кг или галоперидолом при 2 мг/кг. Мышей умерщвляли через 30 минут-2 часа после инъекции сфокусированным микроволновым облучением черепа, которое сохраняло фосфопротеин мозга, как он существует на момент смерти. Затем, прилежащие ядра отсекали от каждого мозга, разрезали слоями и замораживали в жидком азоте. Образцы дополнительно получали для анализа фосфопротеина SDS-PAGE электрофореза, с последующим специфическим для фосфопротеина иммуноблоттингом, как определено в Zhu H, et al., Brain Res. 2010 Jun 25; 1342:11-23. Фосфорилирование в каждом сайте определяли количественно, нормализовали к общему уровню белка (нефосфорилированного) и выражали в процентах от уровня фосфорилирования у контрольных мышей, обработанных средой.
[0134] Результаты показывают, что соединение ни примера 1, ни примера 2, не имело значительного эффекта на фосфорилирование тирозингидроксилазы по Ser40 через 30 минут или 60 минут, в отличие от галоперидола, который вызывал более чем 400% увеличение, и рисперидона, который вызывал более чем 500% увеличение TH фосфорилирования. Это демонстрирует, что соединения настоящего изобретения не нарушают дофаминовый метаболизм.
[0135] Результаты дополнительно демонстрируют, что ни соединение примера 1 ни соединение примера 2 не имело значительного эффекта на NR2B фосфорилирование по Tyr1472 через 30-60 минут. Соединения вызывали небольшое увеличение GluR1 фосфорилирования по Ser845, и небольшое снижение ERK2 фосфорилирования по Thr183 и Tyr185. Фосфорилирование белков по различным сайтам в конкретных белках, как известно, связано с различными активностями клетки, такими как белковый транспорт, активность ионных каналов, сила синаптической передачи сигналов и изменения в экспрессии генов. Показано, что фосфорилирование Tyr1472 в NMDA глютаматном рецепторе является важным для поддержания нейропатической боли. Фосфорилирование Ser845 GluR1 глютаматного рецептора AMPA типа связано с несколькими аспектами усиления синаптической передачи и усиления синаптической локализации рецептора для поддержки долгосрочной потенциации, связанной с когнитивными способностями. Также сообщалось, что фосфорилирование данного остатка приводит к увеличению вероятности открытия каналов. Фосфорилирование ERK2 киназы, члена MAP киназного каскада, по остаткам T183 и Y185 необходимо для полной активации данной киназы, ERK2 участвует во многих аспектах клеточной физиологии, включая рост клеток, выживание и регуляцию транскрипции. Сообщалось, что данная киназа является важной для синаптогенеза и когнитивной функции.
Пример 8: Исследование с «закапыванием шариков» на мышах (OCD модель)
[0136] Тест с «закапыванием шариков» применяли для измерения повторяющегося и связанного с тревогой поведения на грызунах. Он основан на наблюдении, что крысы и мыши будут закапывать вредные или безвредные объекты в своих настилах, и его применяли в качестве модели на животных для измерения эффекта фармакологических вмешательств в лечение повторяющихся расстройств поведения, таких как OCD.
[0137] Вначале, мышей разделяли на четыре группы обработки: (1) среда отрицательный контроль, (2) 0,3 мг/кг соединения примера 2, (3) 1,5 мг/кг соединения примера 2, и (4) 20 мг/кг MPEP (2-метил-6-(фенилэтинил)пиридина), положительный контроль. MPEP представляет собой селективный mGluR5 глютаматный рецепторный антагонист. Мышам в группах (2) и (3) вводили перорально соединение примера 2 при указанной дозе в 0,5% метилцеллюлозной водной среде за 30 минут до теста. Мышам в группах (1) вводили перорально среду, и мышам в группе (4) вводили интраперитонеальную инъекцию MPEP непосредственно перед началом теста.
[0138] Тест проводили в прямоугольных клетках с 4-5 см настилом из древесного щепа в комнате с опущенными шторами и закрытой дверью, чтобы свести к минимуму отвлекающие факторы. Пятнадцать прозрачных мраморных шариков равномерно распределяли поверху настила в три ряда по пять мраморных шариков. Одну мышь помещали в каждую клетку. Мышь и клетку оставляли в покое на 30 минут. В конце теста мышь вынимали, и подсчитывается количество мраморных шариков, закопанных, по меньшей мере, на 2/3 их глубины. Результаты показаны в следующей таблице:
[0139] Результаты показывают, что по сравнению с контролем, имеется статистически значимое снижение «закапывания шариков» для мышей, обработанных 0,3 мг/кг соединения примера 2 (p<0,01) и 1,5 мг/кг соединения примера 2 (p<0,001). Кроме того, есть данные о четкой зависимости доза-реакция. Результаты подтверждают полезность соединения примера 2 в показаниях для лечения OCD.
Пример 9: анализ Мю-опиатной рецепторной активности
[0140] Соединения примера 1 и 2 тестировали в CHO-K1 клетках, экспрессирующих hOP3 (человеческий мю-опиатный рецептор μ1 подтип), применяя цАМФ набор для анализа на основе HTRF (цАМФ Dynamic2 набор для анализа от Cisbio, # 62AM4PEB). Замороженные клетки размораживали в 37°C водяной бане и суспендировали в 10 мл F-12 среде Хэма, содержащей 10% FBS. Клетки извлекали центрифугированием и суспендировали в буфере для анализа (5 нМ KCl, 1,25 мМ MgSO4, 124 мМ NaCl, 25 мМ HEPES, 13,3 мМ глюкозы, 1,25 мМ KH2PO4, 1,45 мМ CaCl2, 0,5 г/л BSA без протеазы, дополненном 1 мМ IBMX). Бупренорфин, мю-опиатный рецепторный частичный агонист, и налоксон, мю-опиатный рецепторный антагонист, и DAMGO, синтетический опиоидный пептидный полный агонист, применяли в качестве контролей.
[0141] Для анализа на агонизм, 12 мкл клеточной суспензии (2500 клеток/лунка) смешивали с 6 мкл форксолина (10 мкм конечная концентрация для анализа), и 6 мкл испытуемого соединения при увеличивающихся концентрациях смешивали в лунках 384-луночного белого планшета, и планшеты выдерживали в течение 30 минут при комнатной температуре. После добавления буфера для лизиса и одного часа дополнительного инкубирования, измеряли концентрации цАМФ согласно инструкции набора. Все данные анализа определяли в трех экземплярах. Аппроксимацию кривой проводили, применяя XLfit программное обеспечение (IDBS), и EC50 величины определяли, применяя 4-параметрическую экспоненциальную аппроксимацию. Тест на агонизм определяет способность испытуемого соединения ингибировать стимулированное форскалином накопление цАМФ.
[0142] Для анализа на антагонизм, 12 мкл клеточной суспензии (2500 клеток/лунка) смешивали с 6 мкл испытуемого соединения при увеличивающихся концентрациях, и смешивали в лунках 384-луночного белого планшета, и планшет инкубировали в течение 10 минут при комнатной температуре. Добавляли 6 мкл смеси DAMGO (D-Ala2-N-MePhe4-Gly-олэнкефалин, 10 нМ конечная концентрация для анализа) и форксолина (10 мкм конечная концентрация для анализа), и планшеты выдерживали в течение 30 минут при комнатной температуре. После добавления буфера для лизиса и одного часа дополнительного ингибирования, измеряли концентрации цАМФ согласно инструкции набора. Все данные анализа определяли в трех экземплярах. Аппроксимацию кривой проводили, применяя XLfit программное обеспечение (IDBS), и EC50 величины определяли, применяя 4-параметрическую экспоненциальную аппроксимацию. Наблюдаемую константу диссоциации (KB) рассчитывали, применяя модифицированное уравнение Ченга-Прусоффа. Тест на антагонизм измеряет способность испытуемого соединения обращать ингибирование стимулированного форскалином накопления цАМФ, вызванного DAMGO.
[0143] Результаты показаны в таблице ниже. Результаты показывают, что соединение примера 2 представляет собой слабый антагонист Mu рецептора, показывая гораздо большую IC50 по сравнению с налоксоном, и оно показывает умеренно высокое сродство, но частичный агонист, показывая только приблизительно 22% агонистическую активность относительно DAMGO (по сравнению с до приблизительно 79% активности для бупренорфина относительно DAMGO). Также показано, что соединение примера 1 обладает умеренно сильной частичной агонистической активностью.
[0144] Бупренорфин представляет собой лекарственное средство, применяемое для лечения хронической боли и опиатной абстиненции, но он страдает от той проблемы, что пользователи могут стать зависимыми из-за его высокой частичной агонистической активности. Чтобы компенсировать это, применяют коммерческую комбинацию бупренорфина с налоксоном (продаваемую как субоксон). Не будучи связанными теорией, считают, что соединения настоящего изобретения, которые являются более слабыми частичными мю агонистами, чем бупренорфин, с некоторой умеренной антагонистической активностью, позволят пациенту более эффективно лечиться от боли и/или опиатной абстиненции с более низким риском зависимости.
[0145] В дополнительном родственном исследовании, применяя сигнальный путь, блокирующий рекомбинантный человеческий MOP-бета, обнаружено, что соединение примера 2 не стимулирует сигнальную систему бета-аррестина через MOP рецептор при концентрациях вплоть до 10 мкМ, но что оно представляет собой антагонист с IC50 0,189 мкМ. Напротив, полный опиоидный агонист Met-энкефалин стимулирует сигнальную систему бета-аррестина с EC50 0,08 мкм.
Пример 10: исследование зависимости/переносимости на крысах
[0146] Соединение примера 2 оценивали при повторяющемся (28 дня) ежедневном подкожном введении самцам крыс Спрег-Доули, отслеживая влияние лекарственного средства при дозировании и определяя, есть ли фармакологическая толерантность. Кроме того, поведенческие, физические и физиологические признаки у крыс отслеживали после внезапного прекращения повторяющегося дозирования, чтобы определить, вызывает ли соединение физическую зависимость при отмене. Кроме того, фармакокинетическое исследование проводится параллельно с исследованием толерантности и зависимости, чтобы определить уровни лекарственного средства в плазме при конкретных дозах, применяемых в исследовании толерантности и зависимости. Морфин применяли в качестве положительного контроля для обеспечения достоверности модели и в качестве эталонного средства для сравнения из аналогичного фармакологического класса.
[0147] Соединение примера 2 оценивали при двух дозах, 0,3 и 3 мг/кг, вводимых подкожно четыре раза в день. Было обнаружено, что повторяющееся введение дает пиковые концентрации в плазме 15-38 нг/мл (среднее, n=3) для 0,3 мг/кг дозы, и 70-90 нг/мл (среднее, n=3) для 3 мг/кг дозы. Пиковая концентрация достигалась через 30 минут-1,5 часа после введения со сравнимыми результатами, полученными в первый, 14 и 28 день введения.
[0148] При обеих дозах примера 2 обнаруживали, что не наблюдается значительного влияния на массу тела животного, потребление пищи и воды или температуру тела в течение фазы приема или отмены. Преобладающими поведенческими и физическими эффектами, вызванными повторяющимся введением при 0,3 мг/кг, являются сгорбленная поза, хвост Штрауба и пилоэрекция во время фазы дозирования. При более высокой дозе основными наблюдаемыми поведенческими и физическими признаками являются сгорбленная поза, подавленное поведение, хвост Штрауба, биение хвостов и пилоэрекция.
[0149] Аналогичный профиль поведенческих и физических признаков наблюдали после внезапного прекращения введения соединения на 28 день исследования. В то время как вставание на задние лапы и повышение тонуса тела не наблюдали во время фазы введения дозы при 0,3 мг/кг, было обнаружено, что данные параматры значительно увеличиваются во время фазы отмены. При более высокой дозе умеренное вставание на задние лапы наблюдали во время фазы введения дозы, но во время фазы отмены вставание на задние лапы является более выраженным, и наблюдается повышенный тонус тела.
[0150] В качестве положительного контроля, морфин дозировали при 30 мг/кг перорально дважды в день. Данный режим дозирования, как и ожидалось, связан с изменениями массы тела, потребления пищи и воды, ректальной температуры и клиническими признаками, соответствующими развитию толерантности и абстиненции, вызванной отменой. Масса тела значительно увеличилась по сравнению с контрольной группой, получавшей среду, в дни 2 и 3, в то время как она значительно уменьшилась с 5 дня. Морфин значительно уменьшал потребление пищи в дни 1-9. После этого обычно наблюдалось снижение потребления пищи по сравнению с контрольной группой, но оно существенно не отличалось от контроля в дни 9, 13, 14, 16, 18, 21, 22 и 25. Данные эффекты на массу тела и потребление пищи демонстрируют толерантность к эффекту морфина.
[0151] Также обнаружено, что потребление воды группой, получавшей морфин, значительно ниже, чем у контрольной группы на 25 из 28 дней во время фазы введения дозы. Температура тела также обычно является меньшей, чем у контрольной группы во время фазы введения дозы, особенно на 20, 21 и 27 день. Наблюдали, что преобладающими поведенческими эффектами, вызываемыми морфином во время фазы введения дозы, являются хвост Штрауба, прыжки, рытье, повышение тонуса тела, повышение двигательной активности, взрывные движения и экзофтальм.
[0152] Более того, наблюдали, что воздержание от введения морфина на 28 день приводит в результате к первоначальному дополнительному снижению потребления пищи, с последующей рецидивирующей булимией, со значительно повышенным потреблением пищи на 33 день относительно контрольной группы. К 35 дню потребление пищи возвращалось к контрольным уровням. Аналогично, у крыс, которые ранее получали морфин, также наблюдалось первоначальное снижение потребления воды на 29 день, с последующей рецидивирующей чрезмерной жаждой (потребление воды возвращалось к контрольным уровням к 31 дню). Кроме того, статистически значимое снижение температуры тела в прямой кишке наблюдали во время дозирования, но температура тела возвращалась к контрольным уровням во время фазы отмены.
[0153] Более того, новые поведенческие и физические признаки наблюдали во время фазы отмены морфина, и это свидетельствует о наличии зависимости. Данные признаки включают в себя пилоэрекцию, атаксию/походку вразвалку, отряхивание мокрой собаки и сдавленный живот. Другие аномальные поведения, наблюдаемые во время фазы приема, постепенно исчезали во время фазы отмены. К 35 дню вставание на задние лапы было единственным поведенческим признаком или физическим признаком, наблюдаемым с высокой частотой встречаемости у крыс, которые ранее получали морфин.
[0154] Таким образом, показано, что повторяющееся введение морфина дает явные признаки толерантности и зависимости в данном исследовании, при этом изменения массы тела, потребления пищи и воды, ректальной температуры и клинических признаков соответствуют развитию толерантности и зависимости, вызванной отменой. Это демонстрирует достоверность способа исследования в выявлении физиологических изменений при введении и отмене дозирования.
[0155] Напротив, повторяющееся введение соединения примера 2 при 0,3 и 3 мг/кг четыре раза в день не вызывало толерантности при подкожном дозировании в течение 28 дней. Более того, при отмене сходный, но уменьшающийся профиль поведенческих и физических признаков наблюдали при самой высокой дозе, которую не считали клинически значимой. Таким образом, было обнаружено, что соединение примера 2 в общем не вызывает синдрома физической зависимости при прекращении дозирования.
Пример 11: исследование оксикодон-зависимой отмены на мышах
[0156] Оксикодон вводили самцам мышей C57BL/6J в течение 8 дней в режиме с повышением дозы 9, 17,8, 23,7 и 33 мг/кг два раза в день (7 часов между инъекциями) в 1-2, 3-4, 5-6 и 7-8 дни, соответственно. На утро девятого дня мышам вводили подкожно соединение примера 3 при 0,3, 1 или 3 мг/кг. За этим через 30 минут следовала инъекция среды или инъекция 3 мг/кг налоксона. Другая группа мышей служила в качестве отрицательных контролей, и вместо оксикодона данным мышам вводили соляной раствор в 1-8 дни. В 9 день данным мышам вводили или среду (с последующим налоксоном, как выше) или соединение примера 2 при 3 мг/кг, подкожно (с последующим налоксоном, как выше).
[0157] На 9 день, сразу после инъекции налоксона (или среды), мышей по отдельности помещали в прозрачные пластиковые клетки и непрерывно наблюдали в течение тридцати минут. Мышей контролировали на наличие общих соматических признаков опиатной абстиненции, включая прыжки, отряхивание мокрой собаки, тремор лап, движение в обратную сторону, птоз и диарею. Все данные виды поведения регистрировали как новые случаи, когда они были разделены, по меньшей мере, одной секундой или когда их прерывало нормальное поведение. Вес тела животного также регистрировали непосредственно до и через 30 минут после введения налоксона (или среды). Данные анализировали с помощью ANOVA с последующим тестом Тьюки для множественных сравнений, при необходимости. Уровень значимости устанавливали при р <0,05.
[0158] Результаты показаны в таблице ниже:
(2) на 9 день, с последующим
(3) через 30 минут
[0159] Общее количество признаков включает тремор лап, прыжки и отряхивание мокрой собаки. У мышей, получавших оксикодон, обнаружено, что налоксон вызывает значительное число общих признаков, тремор лап, прыжки и изменение массы тела (p ≤ 0,0001 для каждой). При всех испытуемых дозах соединение примера 2 приводило к значительному снижению общего количества признаков и тремора лап. Кроме того, при 3,0 мг/кг соединение также вызывало значительное снижение количества прыжков и снижало потерю массы тела.
[0160] Данные результаты показывают, что соединение примера 2 дозозависимо ослабляет признаки и симптомы опиатной абстиненции после внезапной отмены введения опиата у зависимых от опиата крыс.
Пример 12: Тест с введением в лапу формалина (модель воспалительной боли)
[0161] Введение под плантарный апоневроз химических раздражителей, таких как формалин, вызывает немедленную боль и дискомфорт у мышей, с последующим воспалением. Подкожная инъекция 2,5% раствора формалина (37% по массе водного формальдегида, разбавленного соляным раствором) в заднюю лапу приводит к двухфазному ответу: острому болевому ответу и отсроченному воспалительному ответу. Таким образом, данная модель на животных предоставляет информацию как об острой боли, так и подострой/тонической боли у одного и того же животного.
[0162] Мышей С57 сначала приучали в камере наблюдения. За 30 минут до введения формалина мышам подкожно вводили среду или 5 мг/кг морфина (в соляном растворе), вводимого подкожно, или соединение примера 2 (в 45% вес/об водном цикодекстрине), вводимого подкожно, при 0,3, 1,0 или 3,0 мг/кг. Кроме того, другой набор мышей обрабатывали контрольной средой или соединением примера 2 по 3,0 мг/кг, пероральным введением, а не подкожным введением.
[0163] Затем, мышам делали подкожную инъекцию в подошвенную поверхность левой задней лапы 20 мкл 2,5% раствора формалина. В течение следующих 40 минут записывали общее время, потраченное на облизывание или укусы обработанной задней лапы. Первые 10 минут представляют острый ноцицептический ответ, в то время как последующие 30 минут представляют отсроченный воспалительный ответ. С интервалом в одну минуту поведение каждого животного оценивали, применяя «среднюю поведенческую оценку», которая оценивает по шкале от 0 до 4:
0: нет реакции, животное спит
1: животное легко ходит на обработанной лапе, например, на цыпочках
2: животное поднимает обработанную лапу
3: животное встряхивает обработанную лапу
4: животное облизывает и кусает обработанную лапу
Данные анализировали ANOVA, с последующим апостериорным сравнения с тестами Фишера, где это уместно. Значимость устанавливали на p<0,05.
[0164] Результаты показаны в таблице ниже.
мин
мин
[0165] Результаты показывают значительный эффект обработки в течение как периода реакции ранней фазы (0-10 мин), так и поздней фазы (11-40 мин). Апостериорное сравнение показывает, что, по сравнению с обработкой средой, подкожная инъекция морфина или соединения примера 2 (при 3 мг/кг) значительно снижает балл по шкале болевого поведения, вызванного инъекцией формалина, а также значительно снижает продолжительность облизывания. Апостериорное сравнение также показывает, что подкожная инъекция морфина или соединения примера 2 (при 3 мг/кг), а также соединения примера 2 перорально (при 3 мг/кг), значительно снижает продолжительность времени, потраченного на облизывание. Хотя средняя оценка болевого поведения также снижалась, применяя 1,0 мг/кг соединения подкожного и 3,0 мг/кг перорально, данные эффекты не были статистически значимыми в данном исследовании. Продолжительность облизывания была аналогичным образом уменьшена, применяя 1,0 мг/кг соединения примера 2 подкожно, но результат не был статистически значимым в данном исследовании. Также было установлено, что ни одна из мышей в исследовании не претерпела значительных изменений массы тела ни в одной из исследуемых групп.
Пример 13: самовведение на крысах, зависимых от героина
[0166] Исследование проводили для определения, вводят ли крысы, зависимые от героина, самостоятельно соединение примера 2, и обнаружили, что они этого не делают, еще раз подчеркивая не вызывающую привыкание природу соединений настоящего изобретения.
[0167] Исследование проводилось в три этапа. На первом этапе крыс сначала обучали нажимать рычаг для получения еды, а затем их снабжали внутривенным яремным катетером и обучали самостоятельному введению героина. В ответ на сигнал (зажигание света в клетке), три нажатия животным рычага приводило к однократной инъекции героина через катетер. Героин предоставляли при начальной дозе 0,05 мг/кг/инъекция, а позже увеличивали до 0,015 мг/кг/инъекция. Затем данную натренированную реакцию подавляли заменой героина на соляной раствор. На втором этапе соляной раствор заменяли раствором соединения примера 2 при одной из четырех доз: 0,0003 мг/кг/инъекция, 0,001 мг/кг/инъекция, 0,003 мг/кг/инъекция и 0,010 мг/кг/инъекция. Каждой отдельной крысе вводили одну или две разные дозы соединения возрастающим способом. Затем, данную реакцию подавляли инъекцией соляного раствора, с последующей третьей фазой, которая повторяла употребление героина в дозе 0,015 мг/кг/инъекция. Цель третьего этапа заключалась в демонстрации того, что крысы все еще проявляют аддиктивное поведение к героину в конце исследования. Результаты исследования показаны в таблице ниже:
[0168] Результаты показывают, что есть статистически значимое усиление нажатия рычага крысами при приеме героина, но значимой разницы при введении физиологического раствора или соединения примера 2 не было. Таким образом, результаты предполагают, что крысы не становятся зависимыми от соединения примера 2.
Пример 14: Фармакокинетические данные на животных
[0169] Применяя стандартные способы, фармакокинетический профиль соединения примера 2 исследовали на нескольких животных.
Пример 14a: ФК исследования на крысах
[0170] В первом исследовании, крысам вводили соединение примера 2 внутривенным болюсом (внутривенно) при 1 мг/кг в 45% Trapposol среде или перорально (перорально) при 10 мг/кг в 0,5% CMC среде (N=3 для каждой группы). Во втором исследовании, крысам вводили соединение примера 2 при 10 мг/кг перорально или 3 мг/кг подкожно (подкожно), каждый в 45% Trapposol среде (N=6 для каждой группы). Концентрации в плазме лекарственного средства измеряли в моменты времени от 0 до 48 часа после дозирования. Репрезентативные результаты представлены ниже (* показывает концентрацию в плазме ниже величины, измеряемой количественно):
Пример 14b: ФК исследования на мышах
[0171] Аналогичное исследование на мышах проводили, применяя 10 мг/кг пероральное введение соединения примера 2, и получали следующие результаты: Tmax=0,25 часа; Cmax=279 нг/мл; AUC (0-4ч)=759 нг-ч/мл; соотношение в крови и плазме (0,25-4 ч) колеблется от 3,7 до 6,6. Исследование также проводили при дозе 0,1 мг/кг подкожно. Репрезентативные результаты показаны в таблице ниже:
(нг/мл)
(нг-ч/мл)
[0172] Вместе данные результаты показывают, что соединение примера 2 хорошо поглощается и распределяется в мозге и тканях и сохраняется с достаточно длительным периодом полувыведения, позволяющим вводить терапевтические дозы один раз в день.
Пример 15: функционирование желудочно-кишечного тракта
[0173] Эффект соединения примера 2 на моторику желудочно-кишечного тракта у крыс проверяли мониторингом скорости кишечного транзита болюса с активированным углем. Крыс обрабатывали (1) водной карбоксиметилцеллюлозной средой, (2) морфином (5 мг/кг, подкожно) или (3) соединением примера 3 (при 0,3, 1,0 или 3,0 мг/кг, подкожно) за 30 минут до перорального болюса 15% водного активированного угля. Измеренным результатом является степень подвижности, рассчитанная как расстояние, пройденное углем, в виде доли от полной длины кишечника животного. Результаты показаны в таблице ниже:
[0174] Данные результаты показывают, что соединение примера 2 не обладает значительным эффектом на моторику желудочно-кишечного тракта при дозах вплоть до 3 мг/кг. Напротив и, как и ожидалось, морфин приводил в результате к приблизительно 50% снижению моторики желудочно-кишечного тракта.
[0175] В следующем эксперименте, крыс предварительно обрабатывали за 60 минут до болюса с углем средой, морфином (5 мг/кг) или соединением примера 2 (3 мг/кг), каждый подкожно, с последующей обработкой морфином (5 мг/кг), морфином плюс соединение примера 2 (0,3 мг/кг или 3 мг/кг) или только соединением примера 2 (3 мг/кг). Результаты показаны в таблице ниже. Для групп 2 и 3, морфин вводили вначале, с последующей немедленной инъекцией соединения примера 2:
[0176] Результаты показывают, что соединение примера 2 обращает ингибирование моторики желудочно-кишечного тракта, вызванное морфином при введении или одновременно или последовательно перед морфином, причем блокада эффекта морфина является большей, когда применяют предобработку.
[0177] Не будучи связанными теорией, предполагают, что данные различия обусловлены тем, что соединение примера 2 действует как смещенный MOP лиганд, и его неспособностью активировать пути сигнальной системы бета-аррестина далее в каскаде, которые, как было показано, опосредуют опиатно-связанные побочные эффекты, в том числе запор и угнетение дыхания.
Пример 16: функция легких
[0178] Эффект соединения примера 2 на функцию легких у крыс оценивали отслеживанием частоты дыхания, дыхательного объема и минутного объема у крыс после подкожного введения соединения примера 2 при 0,3, 1,0 и 3,0 мг/кг, по сравнению с контролем со средой. Измерения проводили через 0, 15, 60, 120 и 240 минут после введения соединения. Обнаружено, что нет никаких существенных различий между группой со средой и любой из тестируемых групп в любой момент времени. Результаты показаны ниже для 60 минут, что типично для полученных результатов:
Пример 17: получение 1-(Ацилокси)алкильных производных
[0179] Серию соединений согласно формуле I, в которой Z представляет собой -O- и в которых R5 представляет собой -C(R6)(R7)-O-C(O)-R8, и в которых R6, R7 и R8 являются следующими, получали, исходя из соединения 14a, являющегося предшественником:
[0180] Каждое соединение получали реакцией соединения 17a, (6bR,10aS)-8-(3-(4-фторфенокси)пропил)-6b,7,8,9,10,10a-гексагидро-1H-пиридо[3',4':4,5]пирроло[1,2,3-de]хиноксалин-2(3H)-она, с подходящим хлорметилалкилатом 17x (R8CO2C(R6)(R7)Cl). Каждый из требуемых хлорметилалкилатов получали из соответствующих хлорангидрдиов кислот, R8COCl.
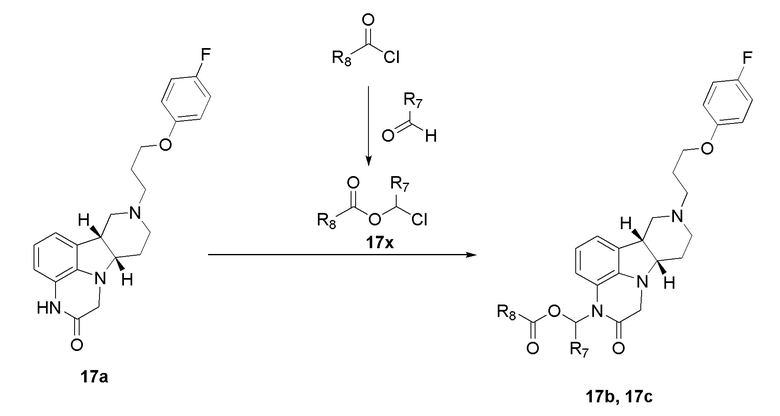
[0181] Например, хлорметилдодеканоат (CH3(CH2)10-C(O)O-CH2Cl) получали следующим способом. К перемешиваемой суспензии хлорида цинка(II) (294 мг, 2,16 ммоль) и параформальдегида (842 мг, 28,08 ммоль) в безводном ацетонитриле (4 мл) при 0°C добавляли лаурилхлорид (5,0 мл, 21,6ммоль) по каплям под аргоном. Суспензию перемешивали при 0°C в течение 10 мин и при комнатной температуре в течение 10 мин, и затем нагревали при 70°C в течение 24 ч. Затем, реакционную смесь охлаждали до комнатной температуры и выливали в дихлорметан (100 мл). Полученную в результате суспензию фильтровали под вакуумом и промывали дихлорметаном (2×10 мл). К объединенному фильтрату добавляли насыщенный бикарбонат натрия (100 мл), и смесь перемешивали при комнатной температуре в течение 1,5 ч. Дихлорметановую фазу отделяли и промывали насыщенным бикарбонатом натрия (2×60 мл), и упаривали досуха при пониженном давлении. Полученный в результате остаток дополнительно сушили при высоком вакууме, получая хлорметилдодеканоат в виде светло-оранжевого масла (4,523 г, 84% выход). Данный неочищенный продукт непосредственно применяли в следующей реакции без дополнительной очистки. Хлорметилдодеканоат применяли для получения соединения 17b. Для получения соединения 17с применяли хлорметилпальмитат, который получают из пальмитоилхлорида и параформальдегида. Аналогичные соединения 17x можно получить аналогичным способом, применяя другие хлорангидриды карбоновых кислот, таких как октаноилхлорид или стеарилхлорид, наряду с другими альдегидами, такими как ацетальдегид, пропионовый альдегид или изобутиральдегид.
[0182] Соединение 17b. Суспензию йодида калия (131 мг, 0,786 ммоль), карбоната калия (181 мг, 1,31 ммоль), N, N-диизопропилэтиламина (137 мкл, 0,786ммоль), N, N-диметилпиридин-4-амина (64 мг, 0,524ммоль), хлорметилдодеканоата (522 мг, 2,10ммоль) и 17a (212 мг, 0,556ммоль) в безводном DMF (1,5 мл) барботировали аргоном в течение 5 мин. Полученную в результате суспензию нагревали при 125°C микроволнами в течение 5 ч, и растворитель удаляли при пониженном давлении. Полученный неочищенный продукт очищали колоночной хроматографией на силикагеле, применяя градиент 0-100% этилацетата в гексане в качестве элюента, получая ((6bR,10aS)-8-(3-(4-фторфенокси)пропил)-2-оксо-6b,7,8,9,10,10a-гексагидро-1H-пиридо[3',4':4,5]пирроло[1,2,3-de]хиноксалин-3(2H)-ил)метилдодеканоат (17b) в виде светло-оранжевого твердого остатка (150 мг, 45% выход). MS (ESI) m/z 594,4089 [M+H]+. 1H ЯМР (500 МГц, хлороформ-d) δ 6,96 (дд, J=9,1, 8,2 Гц, 2H), 6,90 (с, 1H), 6,82 (д, J=7,0 Гц, 4H), 6,16 (д, J=10,5 Гц, 1H), 5,70 (д, J=10,5 Гц, 1H), 4,05 (д, J=14,5 Гц, 1H), 3,99 (д, J=6,6 Гц, 2H), 3,42 (д, J=14,4 Гц, 1H), 3,38-3,25 (м, 1H), 2,91 (д, J=28,2 Гц, 1H), 2,74 (с, 1H), 2,50 (с, 2H), 2,34 (т, J=7,5 Гц, 2H), 2,28 (т, J=7,9 Гц, 1H), 1,96 (с, 3H), 1,86 (с, 1H), 1,70-1,59 (м, 2H), 1,54 (с, 1H), 1,35-1,16 (м, 17H), 0,88 (т, J=6,9 Гц, 3H).
[0183] Соединение 17x получали аналогичным способом, применяя подходящие галогениды 14x. Например, соединение 17c получали, применяя хлорметилпальмитат. MS (ESI) m/z 650,4348 [M+H]+. 1H ЯМР (500 МГц, хлороформ-d) δ 6,96 (дд, J=9,1, 8,2 Гц, 2H), 6,89 (дд, J=5,5, 2,8 Гц, 1H), 6,86-6,78 (м, 4H), 6,16 (д, J=10,5 Гц, 1H), 5,70 (д, J=10,5 Гц, 1H), 4,05 (д, J=14,5 Гц, 1H), 3,97 (т, J=6,3 Гц, 2H), 3,41 (д, J=14,5 Гц, 1H), 3,32 (д, J=5,6 Гц, 2H), 2,97-2,89 (м, 1H), 2,74 (д, J=11,3 Гц, 1H), 2,57-2,42 (м, 2H), 2,34 (т, J=7,5 Гц, 2H), 2,30-2,20 (м, 1H), 2,00-1,91 (м, 3H), 1,85 (т, J=11,0 Гц, 1H), 1,62 (p, J=7,4 Гц, 2H), 1,53 (с, 2H), 1,35-1,18 (м, 23H), 0,88 (т, J=6,9 Гц, 3H).
Пример 18: получение алкоксикарбонильных (карбаматных) производных
[0184] Получали серию соединений согласно формуле I, в которой Z представляет собой -O- и в которой R5 представляет собой -C(O)-O-C(Ra)(Rb)(Rc), и в которой Ra, Rb и Rc являются следующими:
[0185] Каждое соединение получали реакцией соединения 17a с подходящим алкоксикарбонилхлоридом, RcCH2O(CO)Cl:
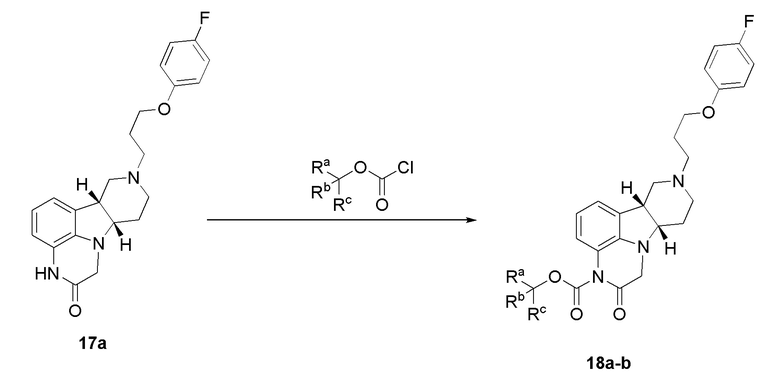
[0186] Соединение 18b. Смесь N, N-диизопропилэтиламина (219 мкл, 1,572 ммоль), N, N-диметилпиридин-4-амина (64 мг, 0,524 ммоль), гексадецилоксикарбонилхлорида (320 мг, 1,05 ммоль) и 17a (200 мг, 0,524 ммоль) в безводном DMF (3 мл) барботировали аргоном в течение 5 мин. Смесь перемешивали при комнатной температуре в течение 5 ч, и растворитель удаляли при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле, применяя градиент 0-100% этилацетата в гексане в качестве элюента, получая заявленное в заголовке соединение в виде светло-оранжевого твердого остатка (197 мг, 58% выход). MS (ESI) m/z 650,4970 [M+H]+. 1H ЯМР (500 МГц, хлороформ-d) δ 7,04 (д, J=8,2 Гц, 1H), 6,96 (дд, J=9,2, 8,2 Гц, 2H), 6,91 (д, J=7,3 Гц, 1H), 6,86-6,76 (м, 3H), 4,40 (т, J=6,7 Гц, 2H), 4,04-3,94 (м, 3H), 3,35 (д, J=14,1 Гц, 2H), 3,26 (д, J=5,3 Гц, 1H), 2,92 (с, 1H), 2,74 (с, 1H), 2,50 (с, 2H), 2,24 (д, J=14,8 Гц, 1H), 2,00-1,86 (м, 4H), 1,83-1,74 (м, 2H), 1,48-1,38 (м, 2H), 1,26 (с, 25H), 0,88 (т, J=6,9 Гц, 3H).
[0187] Соединение 18a. Соединение 18a получали аналогичным способом, применяя додеканоксикарбонилхлорид. MS (ESI) m/z 594,4180 [M+H]+. 1H ЯМР (500 МГц, хлороформ-d) δ 7,06 (дд, J=8,2, 0,9 Гц, 1H), 6,99-6,91 (м, 3H), 6,86-6,76 (м, 3H), 4,41 (т, J=6,7 Гц, 2H), 4,00-3,96 (м, 2H), 3,53 (с, 1H), 3,36 (д, J=14,0 Гц, 1H), 3,29 (д, J=4,0 Гц, 1H), 3,12 (с, 1H), 2,96 (с, 1H), 2,70 (д, J=11,9 Гц, 2H), 2,48 (с, 1H), 2,19 (с, 1H), 2,13-2,02 (м, 2H), 2,03-1,94 (м, 1H), 1,83-1,72 (м, 2H), 1,47-1,39 (м, 2H), 1,27 (д, J=6,3 Гц, 18H), 0,88 (т, J=6,9 Гц, 3H).
Пример 19: Фармакокинетический анализ соединений 17b и 18a
[0188] ФК исследования на крысах проводили по существу согласно тому же протоколу, как применяли в примере 14a. Крысам вводили подкожно дозу соединения примера 2, соединения 17b или соединения 18a при 30 мг/кг, в среде, состоящей из 3% CMC, 0,1% Tween 20 в нормальном соляном растворе (0,9% NaCl) (N=4 для каждой группы). Образцы плазмы отбирали через 2, 5, 15 и 30 минут и через 1, 2, 6, 8, 12, 24, 48, 72, 96, 120, 144, 168. 240, 336, 504 и 720 часов. Образцы анализировали на концентрацию введенного соединения (пример 2, соединение 17b или 18a), а также на соединение примера 2 (которое представляет собой гидролитический метаболит соединений 17b и 18a). Репрезентативные результаты показаны в таблице ниже (* показывает уровень аналита ниже измеряемого количественно уровня):
Данные результаты показывают, что соединения настоящего изобретения подвергаются эффективному метаболическому гидролизу, давая терапевтические концентрации в плазме активной лекарственной молекулы, соединения примера 2. Более того, результаты показывают, что данные соединения подвергаются постепенному in vivo гидролизу, который значительно продлевает временные рамки, в течение которых обнаруживают измеримые концентрации в плазме соединения примера 2 (по сравнению с результатами, показанными в примере 14a). Например, когда соединение примера 2 вводили крысам подкожно при терапевтически эффективной дозе 3 мг/кг, концентрация в плазме лекарственного средства, как наблюдалось, падала до приблизительного исходного уровня через 24 часа после дозирования (смотри пример 14a). Напротив, когда пролекарственное соединение 17b вводили при дозе 30 мг/кг, значительные концентрации в плазме соединения примера 2 получали вплоть до 336 часов после.
| название | год | авторы | номер документа |
|---|---|---|---|
| ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ | 2018 |
|
RU2780002C2 |
| ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ | 2017 |
|
RU2743513C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ ИЛИ ПРОФИЛАКТИКИ РАССТРОЙСТВА ЦЕНТРАЛЬНОЙ НЕРВНОЙ СИСТЕМЫ | 2017 |
|
RU2776800C2 |
| ЭНАНТИОМЕРНО ЧИСТЫЙ ОПИОИДНЫЙ ДИАРИЛМЕТИЛПИПЕРАЗИН И СПОСОБЫ ЕГО ПРИМЕНЕНИЯ | 2002 |
|
RU2309153C2 |
| ПРИМЕНЕНИЕ СИГМА-ЛИГАНДОВ ПРИ ИНДУЦИРОВАННОЙ ОПИОИДАМИ ГИПЕРАЛГЕЗИИ | 2011 |
|
RU2589899C2 |
| НЕЙРОГЕНЕЗ, ОПОСРЕДОВАННЫЙ ПРОИЗВОДНЫМ 4-АЦИЛАМИНОРИРИДИНА | 2007 |
|
RU2451512C2 |
| ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ | 2017 |
|
RU2733975C2 |
| СИНТЕТИЧЕСКИЕ ПЕПТИДНЫЕ АМИДЫ | 2007 |
|
RU2500685C2 |
| ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ | 2019 |
|
RU2809023C2 |
| ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ | 2011 |
|
RU2591194C2 |
Изобретение относится к соединениям формулы I в свободной или фармацевтически приемлемой солевой форме, а также к их фармацевтическим композициям и способам применения в лечении заболеваний. Технический результат: получены новые пролекарства замещенных гетероциклических конденсированных гамма-карболинов формулы I, которые могут быть применимы для лечения или профилактики расстройства центральной нервной системы, где расстройство представляет собой расстройство, включающее серотониновый 5-HT2A, транспортер обратного захвата серотонина (SERT), дофаминовый D1 и/или D2 путь и/или μ-опиоидный рецептор. 4 н. и 11 з.п. ф-лы, 19 табл., 19 пр.
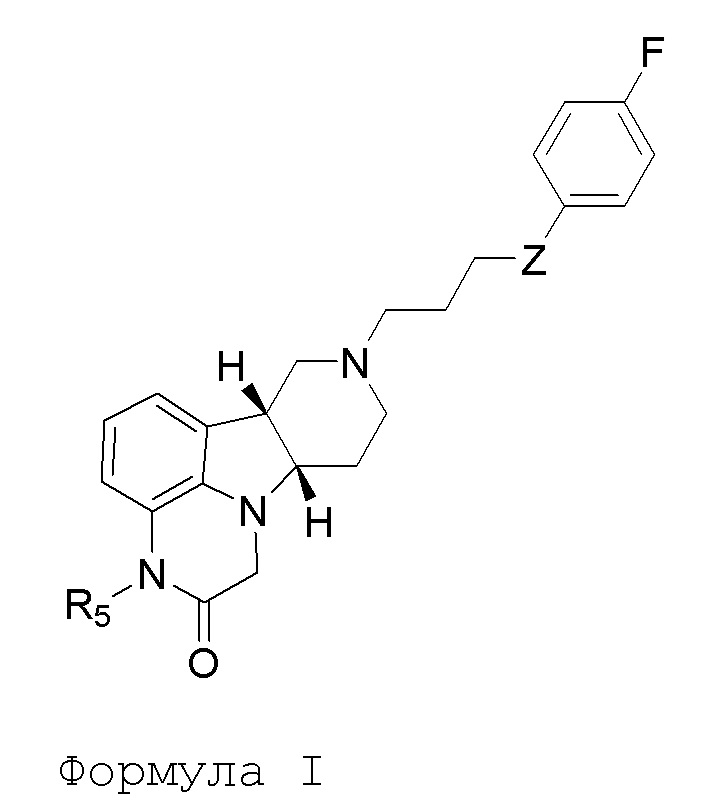
1. Соединение формулы I:
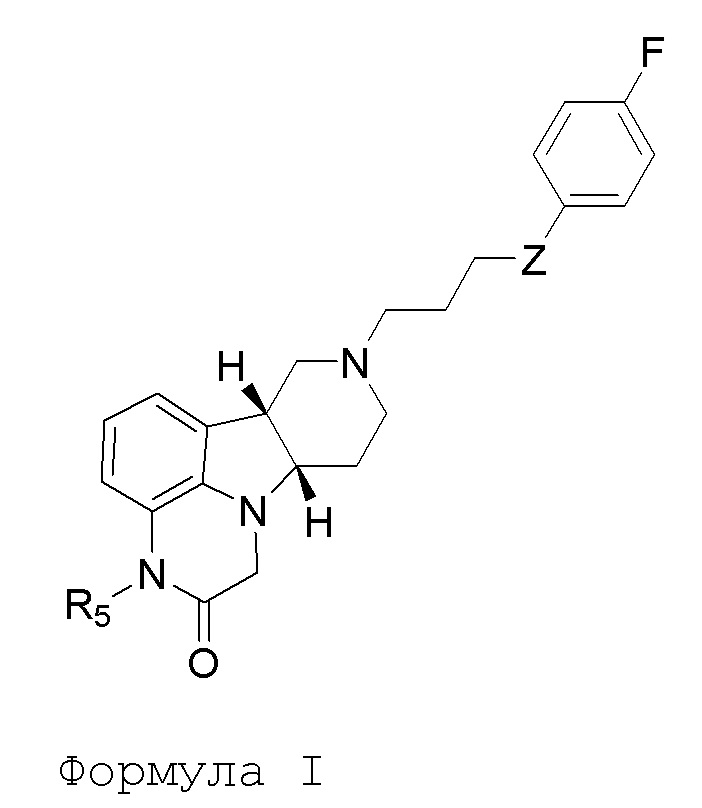
в которой:
R5 представляет собой -C(O)-O-C(Ra)(Rb)(Rc) или -C(R6)(R7)-O-C(O)-R8;
Z представляет собой O;
R8 представляет собой -C(Ra)(Rb)(Rc);
каждый Ra, Rb и Rc независимо выбран из H и C1-24алкила;
каждый R6 и R7 представляет собой H;
в свободной или фармацевтически приемлемой солевой форме.
2. Соединение по п. 1, в котором R5 представляет собой -C(O)-O-C(Ra)(Rb)(Rc).
3. Соединение по п. 1, в котором R5 представляет собой -C(R6)(R7)-O-C(O)-R8, и R8 представляет собой -C(Ra)(Rb)(Rc).
4. Соединение по любому из пп. 1-3 в форме фармацевтически приемлемой соли.
5. Фармацевтическая композиция для лечения расстройства, включающего серотониновый 5-HT2A, транспортер обратного захвата серотонина (SERT), дофаминовый D1 и/или D2 путь и/или μ-опиоидный рецептор, содержащая эффективное количество соединения по любому из пп. 1-4, в свободной или фармацевтически приемлемой солевой форме, в смеси с фармацевтически приемлемым разбавителем или носителем;
где расстройство выбрано из группы, состоящей из тревоги, депрессии, психоза, шизофрении, расстройств сна, сексуальных расстройств, боли и состояний, связанных с болью, социальных фобий, возбуждения при деменции, деменции при болезни Альцгеймера или болезни Паркинсона; поведенческих расстройств; опиатной зависимости, кокаиновой зависимости; отказа от опиатной зависимости; опиатной передозировки; сопутствующих состояний, связанных с наркоманией; обсессивно-компульсивного расстройства (OCD), обсессивно-компульсивного расстройства личности (OCPD), синдрома нарушения внимания с гиперактивностью (ADHD), синдрома дефицита внимания (ADD), расстройства контроля над побуждениями, и их комбинации.
6. Фармацевтическая композиция по п. 5, где расстройство выбрано из шизофрении, тревоги, депрессии, головной боли, идиопатической боли, невропатической боли, хронической боли, фибромиалгии, наркомании, отказа от наркотиков и опиатной передозировки.
7. Фармацевтическая композиция по п. 5 или 6, где композицию формулируют в виде долгодействующего инъецируемого препарата, например, для внутримышечной или подкожной инъекции.
8. Фармацевтическая композиция по п. 7, где композиция содержит соединение в свободной или фармацевтически приемлемой солевой форме или в кристаллической форме, где указанное соединение получают в виде частиц или кристаллов, имеющих объемный размер частиц 0,5-100 микрон, например 5-30 микрон, 10-20 микрон, 20-100 микрон, 20-50 микрон или 30-50 микрон; и необязательно, где частицы имеют площадь поверхности 0,1-5 м2/г, например 0,5-3 м2/г или 0,8-1,2 м2/г.
9. Способ лечения или профилактики расстройства центральной нервной системы, где расстройство представляет собой расстройство, включающее серотониновый 5-HT2A, транспортер обратного захвата серотонина (SERT), дофаминовый D1 и/или D2 путь и/или µ-опиоидный рецептор, включающий введение нуждающемуся пациенту эффективного количества соединения по любому из пп. 1-4 в свободной или фармацевтически приемлемой солевой форме, или фармацевтической композиции по любому из пп. 5-8,
где расстройство выбрано из группы, состоящей из тревоги, депрессии, психоза, шизофрении, расстройств сна, сексуальных расстройств, боли и состояний, связанных с болью, социальных фобий, возбуждения при деменции, деменции при болезни Альцгеймера или болезни Паркинсона; поведенческих расстройств; опиатной зависимости; отказа от опиатной зависимости; опиатной передозировки; сопутствующих состояний, связанных с наркоманией; обсессивно-компульсивного расстройства (OCD), обсессивно-компульсивного расстройства личности (OCPD), синдрома нарушения внимания с гиперактивностью (ADHD), синдрома дефицита внимания (ADD), расстройство контроля над побуждениями, и их комбинации.
10. Способ по п. 9, где указанное расстройство центральной нервной системы выбрано из группы, состоящей из депрессии, шизофрении, боли и состояний, связанных с болью, опиатной зависимости, отказа от опиатной зависимости и опиатной передозировки.
11. Способ по п. 9, где указанное расстройство центральной нервной системы выбрано из следующих: (i) психоза у пациента, страдающего от депрессии; (2) депрессии у пациента, страдающего от психоза; (3) поведенческих расстройств, связанных с психозом; (4) расстройств сна, связанных с психозом; и (5) наркотической зависимости, расстройств, связанных с применением наркотических веществ, и/или расстройств, вызванных наркотическими веществами.
12. Способ по п. 9, где указанное расстройство центральной нервной системы представляет собой расстройство, выбранное из обсессивно-компульсивного расстройства (OCD), обсессивно-компульсивного расстройства личности (OCPD), генерализованного тревожного расстройства, социального тревожного расстройства, панического расстройства, синдрома нарушения внимания с гиперактивностью (ADHD), cиндрома дефицита внимания (ADD), расстройства контроля над побуждениями, и их комбинации.
13. Способ по п. 9, где расстройство центральной нервной системы выбрано из головной боли, идиопатической боли, невропатической боли, хронической боли, фибромиалгии, опиатной зависимости, отказа от опиатной зависимости и опиатной передозировки.
14. Применение соединения по любому из пп. 1-4 в свободной или фармацевтически приемлемой солевой форме, или фармацевтической композиции по любому из пп. 5-8 в свободной или фармацевтически приемлемой солевой форме, для получения лекарственного средства для лечения или профилактики расстройств центральной нервной системы, где расстройство представляет собой расстройство, включающее серотониновый 5-HT2A, транспортер обратного захвата серотонина (SERT), дофаминовый D1 и/или D2 путь и/или μ-опиоидный рецептор;
где расстройство выбрано из группы, состоящей из тревоги, депрессии, психоза, шизофрении, расстройств сна, сексуальных расстройств, боли и состояний, связанных с болью, социальных фобий, возбуждения при деменции, деменции при болезни Альцгеймера или болезни Паркинсона; поведенческих расстройств; опиатной зависимости; отказа от опиатной зависимости; опиатной передозировки; сопутствующих состояний, связанных с наркоманией; обсессивно-компульсивного расстройства (OCD), обсессивно-компульсивного расстройства личности (OCPD), синдрома нарушения внимания с гиперактивностью (ADHD), синдрома дефицита внимания (ADD), расстройства контроля над побуждениями, и их комбинации.
15. Применение по п. 14, где расстройство выбрано из шизофрении, тревоги, депрессии, головной боли, идиопатической боли, невропатической боли, хронической боли, фибромиалгии, опиатной зависимости, отказа от опиатной зависимости и опиатной передозировки.
| WO2014145192A1, 18.09.2014 | |||
| ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ | 2011 |
|
RU2591194C2 |

