Настоящее изобретение относится к новым поверхностно-активным веществам типа теломеров и к их использованию для получения метастабильных надмолекулярных систем. Эти стабильные надмолекулярные системы или наночастицы могут представлять собой липосомы или мицеллярные системы. Изобретение также относится к атипичным липосомам и наночастицам, полученным из данных поверхностно-активных веществ, и к их использованию в качестве векторов для активных ингредиентов, в особенности для терапевтически активных ингредиентов.
Некоторые амфифильные молекулы, природные фосфолипиды, обладают свойством ассоциироваться в воде с образованием метастабильных надмолекулярных образований сферической формы, называемых липосомами, которые содержат внутренний водный компартмент. Такие липосомы способны содержать терапевтически активные агенты внутри указанного внутреннего компартмента и могут, таким образом, быть использованными для транспортировки указанных активных агентов к клеткам-мишеням или тканям-мишеням. Исследование этих специфических векторов представляло собой предмет многочисленных публикаций, в которых были широко рассмотрены проблемы, а также потенциальные возможности (Barenholz, Curr. Opin. in Coll. And Int. Sci. 6 (2001) 66-77).
Однако использование липосом для транспортировки терапевтически активных ингредиентов имеет несколько основных недостатков.
Данные наноструктуры обычно обладают сравнительно низкой стабильностью с течением времени, так как они сливаются в среде, в которой они диспергированы, с образованием более крупных объектов, которые впоследствии быстро осаждаются. Это поведение в значительной степени ограничивает их стабильность и сохраняемый объем.
Биологическая стабильность данных векторов, т.е. время их удержания в кровотоке, непосредственно связана с их размером, который должен составлять менее 200 нм, для того, чтобы они могли достичь потенциальной мишени (Nagayasu et al., Adv. Drug Del. Rev. 40 (1999) 75-87). Однако кандидаты, наиболее эффективные с этой точки зрения, а именно однослойные липосомы небольших размеров, обладают недостатком, состоящим в том, что они имеют соотношение инкапсулированный медицинский продукт/липид меньшее по сравнению с однослойными липосомами больших размеров (Vernuri et al., Pharm. Acta Helv. 70 (1995) 95-111). Липиды, образующие указанные наноструктуры, обычно имеют высокую стоимость производства. Таким образом, низкая способность к инкапсулированию активных ингредиентов некоторых из этих липосом представляет собой экономическую проблему.
Наконец, необходима возможность обеспечить вектор, который может высвобождать свое содержимое постепенно и непрерывно. Такие свойства требуют использования высокоорганизованных и непроницаемых мембран, которые состоят из сложных и дорогостоящих липидных составов.
Несмотря на эти трудности, в настоящее время в продаже или на клинической фазе испытаний имеется определенное число фармацевтических препаратов на основе липосом. Наиболее существенное преимущество этих препаратов состоит в их превосходной переносимости по сравнению с использованием свободного активного ингредиента. В то же время их эффективность при эквивалентной дозе лишь незначительно выше. Это наблюдается в случае инкапсулированного в липосомы амфотерицина В (Ambisome®). Его инкапсулирование в липидных составах в значительной степени увеличивает его терапевтический индекс (Andres et al., Rev. Med. Interne, 22 (2001) 141-150).
Для того, чтобы уменьшить их быстрое удаление ретикулоэндотелиальной системой, были разработаны системы защиты липосом. Наиболее эффективная состоит в использовании фосфолипидов, замещенных полиэтиленгликолями, имеющими молекулярную массу в диапазоне от 1000 до 5000 в пропорции от 5 до 10% от общей смеси фосфолипидов. «Невидимые» липосомы, также называемые стелс-липосомами, образованные таким образом (продаваемые под торговым наименованием Stealth liposomes®), имеют большее время удержания в кровотоке по сравнению с традиционными липосомами (45 ч по сравнению с диапазоном от нескольких минут до нескольких часов). Увеличение времени циркуляции указанных липосом в крови способствует их аккумулированию в раковых тканях, которые особенно омываются (кровью), и их использование особенно пригодно для транспортировки противораковых составов (Gabizon et al., Cancer Res. 54 (1994) 987-992). В настоящее время на рынке представлены составы Stealth liposomes® на основе доксорубицина (Doxil®, Alza Corp.) для борьбы с синдромом Капоши. Однако определенные составы, состоящие из очень мелких (50 нм) липосом могут также рассматриваться как препараты длительной циркуляции. Примером этого является антираковый препарат даунорубицин (дауномицин), представленный на рынке NeXstar (DaunoXome®).
Для того, чтобы гарантировать механическую стабильность липосом как в ходе их хранения, так и при их использовании in vivo, можно представить себе несколько стратегий, включающих использование полимеров:
полимеризация поверхностно-активных веществ, составляющих мембрану липосомы, после ее образования (Bader et al., Adv. Polym. Sci. 64 (1985) 1-62 и Hotz et al., Adv. Mater. 10 (1998) 1387-1390);
взаимодействие амфифильных и неамфифильных ионных полимеров на поверхности внешней мембраны липосом (Hayashi et al., Biochem. Biophys. Acta, 1280 (1996) 120-126, Ishishara et al., Coll. And Surf., B: Biointerfaces 25 (2002) 325-333);
наконец, полимеризация гидрофильного мономера внутри внутренней водной полости липосомы представляет собой способ, который очень слабо изучен и был кратко описан Torchilin et al. (Makromol. Chem., Rapid communication, 8 (1987) 457-460). Его использовали в качестве инструмента для получения полимеризованного молекулярного отпечатка в патенте США (Perrot et al., патент США № 6217901, 17 апреля 2001).
Значительное ограничение, связанное с использованием полимеров для стабилизации липосом, представляет собой потенциальная токсичность, вызванная их аккумуляцией в лизосоме или собственной токсичностью неполимеризованного гидрофильного мономера (в случае акриламида). Для того, чтобы ограничить это явление, необходимо использовать полимеры с низкой молекулярной массой, которые более легко поддаются биологическому разрушению.
Использование мицелл, стабилизированных с использованием полимеризованных амфифильных соединений, комбинирующих гидрофильные и гидрофобные блоки, для транспортировки терапевтически активных агентов, которые в значительной степени нерастворимы в воде, представляло собой предмет многочисленных научных исследований (G.S. Kwon et al., Adv. Drug. Deliver. Rev., 16 (1995) 295-309, M. Jones et al., Eur. J. Pharm. Biopharm., 48 (1999) 101-111, V.P. Torchilin, J. Control. Release, 73 (2003) 137-172). Указанные векторные системы делают это возможным, в особенности для транспортировки и растворения определенного числа активных антираковых агентов, в особенности полициклических производных. Как общее правило, последние характеризуются очень низкой биологической доступностью при пероральном введении, а их внутривенные инъекции приводят из-за агрегации к эмболии кровеносных сосудов и к местной токсичности, связанной с твердыми отложениями (A.N. Lukyanov et al., Adv. Drug. Deliver. Rev., (2004) доступно в сети Интернет, Science Direct). Использование липосом, микроэмульсий или циклодекстринов представляет собой многообещающее решение, но все еще демонстрирующее слишком много ограничений, в частности слишком высокую вариабельность в растворении этих сравнительно нерастворимых активных ингредиентов, которое в значительной степени зависит от их структуры. Развитие малых полимерных мицеллярных систем, таким образом, представляет собой благоприятную альтернативу таким технологиям, в которых были заинтересованы заявители.
Благодаря особенно низкой ККМ (критической концентрации мицелообразования) полимерные поверхностно-активные вещества, составляющие указанные мицеллы, придают им в особенности высокую термодинамическую стабильность и чрезвычайно высокую способность к удержанию инкапсулированных активных агентов. Очень небольшой размер указанных наночастиц (менее чем 100 нм) придает им превосходную стабильность in vivo, а также пассивное нацеливание на в особенности хорошо снабжаемые (кровью) опухолевые участки.
Активное нацеливание указанных векторов можно осуществлять путем покрывания их поверхности нацеливающими молекулами, такими как антитела, пептиды, лектины, сахара, гормоны или специфические синтетические соединения.
В литературе упоминается большое число полимеров, которые являются амфифильными по своей природе. Они представляют собой обычно полимеры двухблочного типа, состоящие из различных гидрофильных и гидрофобных мономеров (M. Jones et al., Eur. J. Pharm. Biopharm., 48 (1999) 101-111, V.P. Torchilin, J. Control. Release, 73 (2001) 137-172). Также были исследованы другие амфифильные агенты, полученные из фосфолипидов и из полимеров полиэтиленгликоля и поливинилпирролидона (A.N. Lukyanov et al., Adv. Drug Deliver. Rev., (2004) доступно в Internet, Science Direct).
Первая цель настоящего изобретения состоит в развитии наноразмерных векторов, которые имеют низкую стоимость производства и имеют способность к транспортировке внутри своей внутренней полости большого семейства гидрофильных активных агентов. Наноразмерные векторы по изобретению позволяют инкапсулировать, удерживать и высвобождать вещества, которые можно дозировать. Заданные применения включают транспортировку активных ингредиентов, в особенности терапевтических активных ингредиентов, эпидермальную доставку косметических веществ и медицинскую диагностику; в частности транспортировку противораковых активных агентов, активных агентов для использования на основе вакцин, генетического материала, ферментов, гормонов, витаминов, сахаров, белков и пептидов, липидов или органических и неорганических молекул.
Эта цель достигнута посредством разработки и синтеза новых поверхностно-активных веществ, которые делают возможным получение наноразмерных векторов или липосом, которые имеют благоприятные свойства по сравнению с липосомами по предшествующему уровню техники.
Вторая цель настоящего изобретения состоит в разработке наноразмерных векторов, имеющих очень низкую стоимость производства и обладающих способностью транспортировать внутри своей гидрофобной полости или липидного слоя очень большое семейство гидрофобных активных агентов. Наноразмерные векторы по изобретению позволяют инкапсулировать, удерживать и высвобождать вещества, которые можно дозировать. Заданные применения включают транспортировку активных ингредиентов, в особенности терапевтических активных ингредиентов, эпидермальную доставку косметических веществ и медицинскую диагностику; в частности транспортировку противораковых активных агентов, активных агентов для использования на основе вакцин, генетического материала, ферментов, гормонов, витаминов, сахаров, белков и пептидов, липидов или органических и неорганических молекул.
Эта цель достигнута при помощи разработки и синтеза новых поверхностно-активных веществ теломерного типа, которые делают возможным получение мицелл и эллипсоидальных наночастиц или липосом, которые имеют благоприятные свойства по сравнению с полимерными мицеллами по предшествующему уровню техники.
Предметом настоящего изобретения являются соединения, имеющие формулу (I):
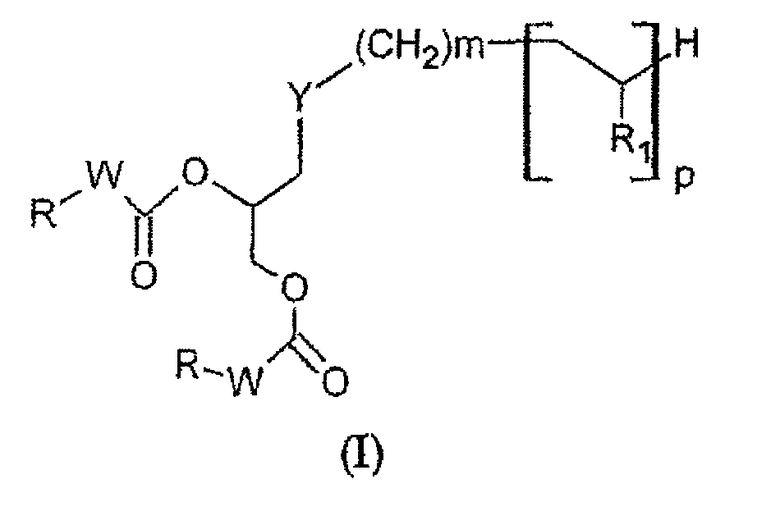
в которой:
• Y представляет собой атом серы или группу
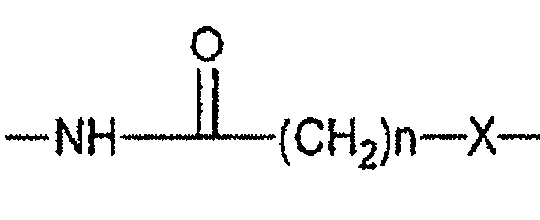
Причем Х выбирают из групп S и СН2, n представляет собой целое число, изменяющееся в диапазоне от 0 до 10, такое как, например, 0, 1, 2, 3, 4, 5 или 6;
• m представляет собой целое число, изменяющееся в диапазоне от 0 до 9, такое как, например, 0, 1, 2, 3, 4, 5 или 6;
и если Х=СН2, тогда 0 < m+n < 6;
• W представляет собой группу -NH- или группу -CH2-;
• р представляет собой целое число, изменяющееся в диапазоне от 0 до 50;
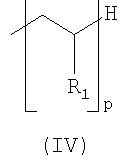
• R1 представляет собой группу, выбранную из следующих радикалов:
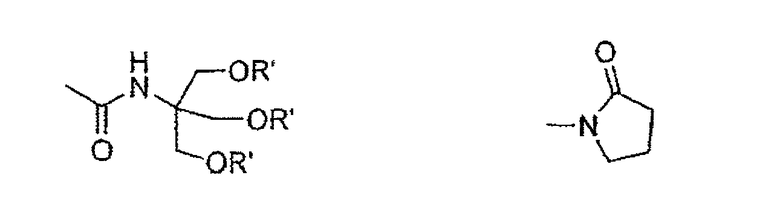
в которых R' представляет собой Н или гидрофильную группу, такую как, например, С4-С24 полигидроксилированное соединение на основе углеводорода; в частности, R' может быть выбрана из сахаров, таких как, например, галактоза, глюкоза, манноза или сиаловая кислота, связанных через их аномерный атом углерода;
• R представляет собой группу, выбранную из: С4-С24 радикалов на основе углеводорода; С4-С24 фторированных радикалов на основе углеводорода; С4-С24 тиоалкильных радикалов.
Группа R может быть, в частности, выбрана из следующих радикалов:
- тиооктильного радикала,
- С4-С24 радикалов на основе углеводорода, таких как н-бутил, трет-бутил, изобутил, н-пентил, изопентил, н-гексил, н-гептил, н-октил, н-нонил, н-децил, н-ундецил, н-додецил, н-тридецил, н-тетрадецил, н-пентадецил, н-гексадецил, н-гептадецил, н-октадецил или фитильный радикал
(CH3[CH(CH3)(CH2)3]3CH(CH3)CH2CH2)),
С4-С24 фторированных радикалов на основе углеводорода, таких как радикалы, отвечающие формуле -(СН2)t-(CF2)rF, в которой r и t оба представляют собой целые числа при условии 24 ≥ r+t ≥4, такие как, например:
-(CF2)4F; -(CF2)5F; -(CF2)6F; -(CF2)7F; -(CF2)8F; -(CF2)9F;
-(CF2)10F; -(CF2)11F; -(CF2)12F; -(CF2)13F; -(CF2)14F; -CH2-(CF2)3F;
-CH2-(CF2)4F; -CH2-(CF2)5F; -CH2-(CF2)6F; -CH2-(CF2)7F; -CH2-(CF2)8F; -CH2-(CF2)9F; -CH2-(CF2)10F; -CH2-(CF2)11F; -CH2-(CF2)12F; -CH2-(CF2)13F; -(CH2)2-(CF2)2F; -(CH2)2-(CF2)3F; -(CH2)2-(CF2)4F; -(CH2)2-(CF2)5F; -(CH2)2-(CF2)6F; -(CH2)2-(CF2)7F; -(CH2)2-(CF2)8F; -(CH2)2-(CF2)9F; -(CH2)2-(CF2)10F; -(CH2)2-(CF2)11F; -(CH2)2-(CF2)12F; -(CH2)3-(CF2)1F; -(CH2)13-(CF2)F; -(CH2)4-(CF2)6F; -(CH2)4-(CF2)8F;
-(CH2)4-(CF2)10F; -(CH2)10-(CF2)6F; -(CH2)10-(CF2)8F; -(CH2)10-(CF2)10F; и т.д.
Согласно первому предпочтительному варианту предметом настоящего изобретения являются соединения, имеющие формулу (IA):

в которой:
Х представляет собой атом S или группу -СН2-;
n представляет собой целое число, изменяющееся в диапазоне от 0 до 10, такое как, например, 0, 1, 2, 3, 4, 5 или 6;
m представляет собой целое число, изменяющееся в диапазоне от 0 до 9, такое как, например, 0, 1, 2, 3, 4, 5 или 6;
и если Х=СН2, тогда 0 < m+n < 6;
R представляет собой группу, выбранную из: С4-С24 радикалов на основе углеводорода; С4-С24 фторированных радикалов на основе углеводорода; С4-С24 тиоалкильных радикалов.
Предпочтительными цепями R являются цепи, которые способствуют тому, чтобы температура фазового перехода поверхностно-активного вещества формулы (I) была выше 37°С. В действительности, когда подобные поверхностно-активные вещества используют для получения липосом, такие поверхностно-активные вещества, которые имеют кристаллическую структуру при физиологической температуре, придают мембране липосомы бульшую жесткость и более высокую степень удержания растворенных веществ, инкапсулированных во внутреннем водном компартменте. Предпочтительно, R представляет собой цепь на основе углеводорода С12-С24 или цепь на основе фторированного углеводорода С8-С24.
Предпочтительно выполняются одно или несколько следующих условий: X=S; n=2, m=1.
Предпочтительными соединениями формулы (IA) являются соединения, отвечающие формуле А, в которой R имеет то же определение, что и описано выше, n=2, X=S, m=1:

Формула А
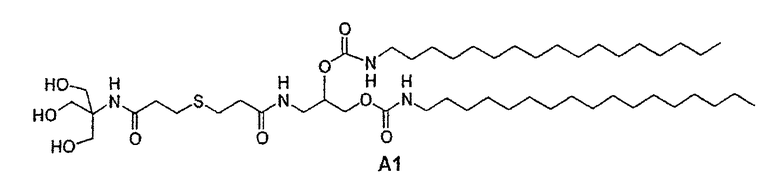
Среди этих соединений в особенности предпочтительным соединением является соединение А1, представленное ниже:
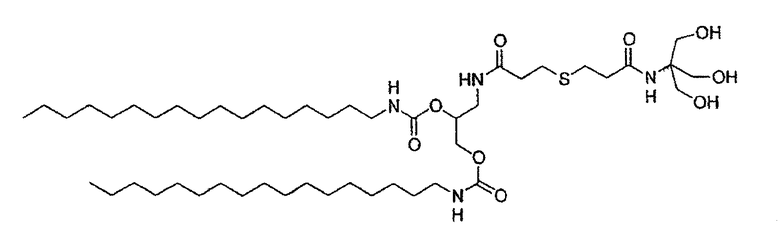
Соединение А1
Синтез молекул формулы (I) можно легко проводить при использовании традиционных способов органического синтеза. Несколько примеров синтеза проиллюстрировано в экспериментальной части.
Другой предмет изобретения состоит в использовании молекул формулы (I), преимущественно молекул формулы (IA) для получения липосом. Стенки липосом по предшествующему уровню техники в основном состоят из фосфолипидов.
Липосомы очень легко получают из поверхностно-активных веществ формулы (I), предпочтительно формулы (IA), на основе пленочного способа (Liposomes, a practical approach, R.R.C. New, Ed., Oxford University Press, New York, 1990). Этот способ может быть суммирован следующим образом.
Раствор поверхностно-активного вещества (I) или (IA), растворенного в метаноле или в хлороформе, медленно испаряют в круглодонной колбе для того, чтобы образовать тонкую пленку на стенке круглодонной колбы. Добавляют дистиллированную воду при 65°С, для того, чтобы повторно гидратировать пленку, с концентрацией 2,5 мг/мл. Полученный раствор затем подвергают обработке ультразвуком в течение 30 минут на ультразвуковой бане при температуре выше температуры фазового перехода диспергированного поверхностно-активного вещества, до тех пор, пока не будет получен голубоватый прозрачный раствор. В последней стадии также возможно заменить обработку ультразвуком повторной экструзией раствора через два поликарбонатных фильтра с размером пор 200 нм, установленных последовательно. Также можно представить себе двойную обработку ультразвуком и экструзией.
Для получения липосом по изобретению можно использовать другие традиционные способы получения липосом. В этом контексте в качестве ссылки можно указать S. Vernuri and C.T. Rhodes, Pharmaceutics Acta Helvetiae 70, (1995), 95-111.
Неожиданно наблюдается образование везикул удлиненной формы, такие везикулы носят название трубчатые везикулы из-за их размера, который составляет несколько десятков нанометров, и из-за их формы, которая напоминает трубу, закрытую с ее обеих концов.
Размер и механическую стабильность частиц, полученных в растворе, в зависимости от времени измеряли после фильтрации при помощи динамической дифракции света (High Performance Particle Sizer, Malvern). Природу полученных частиц исследовали при помощи просвечивающей электронной микроскопии после негативного окрашивания образца или после замораживания-скалывания (Фиг.4).
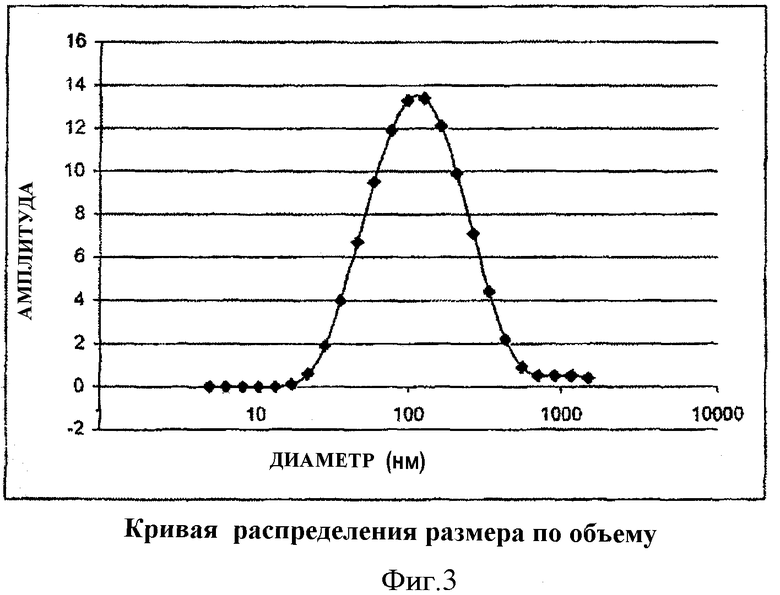
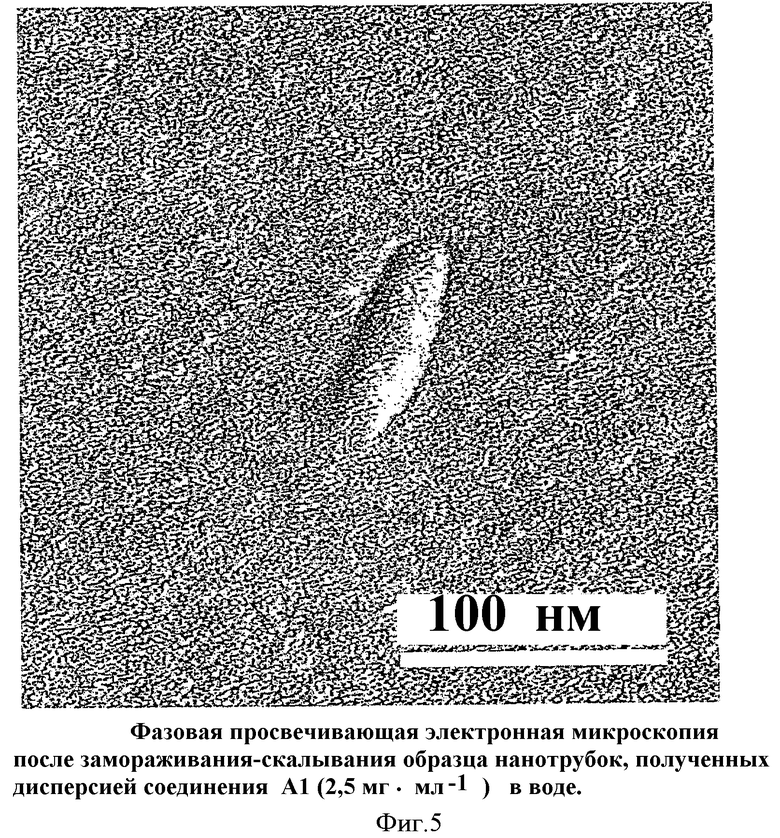
Измерения формы и размера, определенные путем рассматривания электронной микрофотографии, дали возможность продемонстрировать образование липосом удлиненной формы, называемых трубчатыми везикулами, замкнутыми с концов, среднее поперечное сечение которых составляет от 20 до 80 нм и средняя длина которых составляет от 200 до 500 нм (Фиг.3). Анализ способом замораживания-скалывания подтвердил образование указанных трубчатых везикул и их морфологические характеристики, а именно наличие внутренней водной полости, изолированной от внешней среды (Фиг.5).
Для заданного поверхностно-активного вещества формулы (IA) размер частиц является в значительной степени гомогенным: он изменяется в пределах диапазона ±10% от значения, предпочтительно ±5% вокруг центрального значения длины и диаметра.
Эти трубчатые везикулы имеют относительно высокую стабильность, поскольку не наблюдается изменения размера частиц после года хранения, тогда как липосомы, образованные из фосфатидил-холина яичного желтка, показывают изменение всего после лишь 5 дней хранения.
Доказательство наличия внутренней водной полости было получено опосредованно при помощи спектрофлуориметрических измерений кинетики инкапсулирования и высвобождения гидрофильного флуоресцентного зонда - карбоксифлуоресцеина. Измерения напрямую показывают более медленную кинетику высвобождения флуоресцентного зонда по сравнению с традиционным инкапсулированием в смеси фосфатидил-холина яичного желтка (Фиг.1).
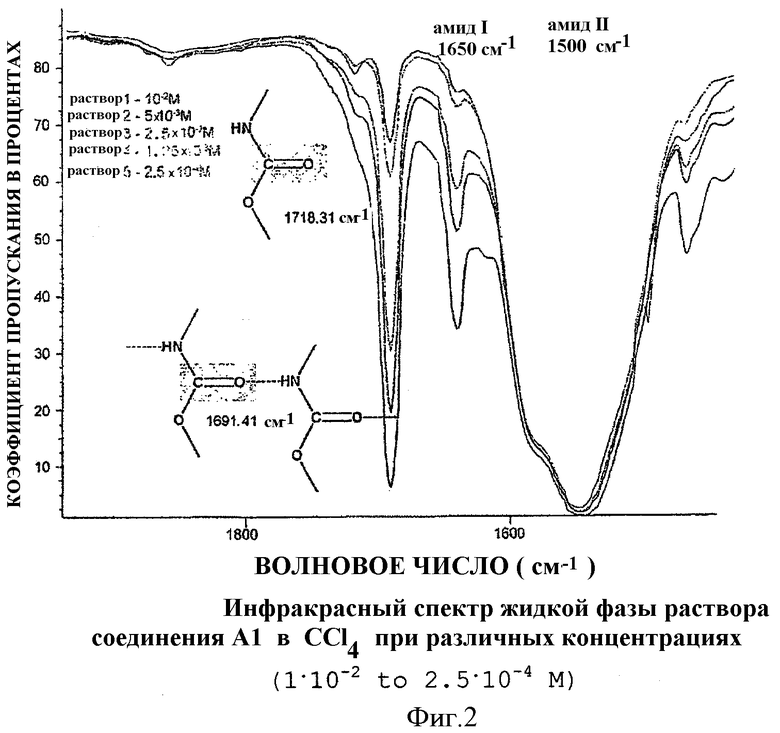
Заявители смогли продемонстрировать, что внутримолекулярные водородные связи между поверхностно-активными веществами, составляющими трубчатые везикулы, являются причиной их морфологии и их специфической стабильности. Действительно, спиртовые функции трис(гидроксиметил)аминометана, а также карбаматные функции обладают способностью генерировать сеть водородных связей между поверхностно-активными веществами, составляющими мембрану, таким образом стабилизируя трубчатые везикулы. Оказалось возможным продемонстрировать это при использовании инфракрасной спектроскопии жидкой фазы на основе преобразования Фурье (в CCl4). Увеличение интенсивности полосы при 1691 см-1, отвечающее карбонильным функциям, связанным с атомом водорода, фактически отмечается в спектре при увеличении концентрации поверхностно-активного вещества в растворе за счет его несвязанного гомолога (Фиг.2).
Здесь следует отметить, что замещение в поверхностно-активном веществе формулы (IA) карбаматной связи на сложно-эфирную связь между жирной цепочкой и звеном глицерина, как проиллюстрировано в структуре В, приводит к образованию в воде липосом традиционного строения, механическая стабильность которых составляет всего несколько часов, таким образом подтверждая выдвигаемую гипотезу, что трубчатые везикулы стабилизированы и организованы благодаря установлению специфических водородных связей (Фиг.6).
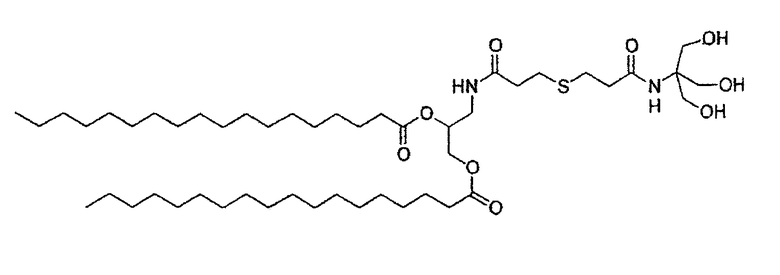
Структура В
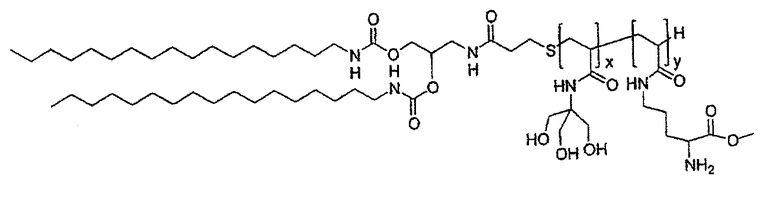
Предмет настоящего изобретения составляют также липосомы, или водная дисперсия везикул, отличающаяся тем, что они включают одно или несколько соединений формулы (I), преимущественно формулы (IA) в качестве компонента их стенок.
Эти липосомы демонстрируют оригинальные структурные характеристики, которые придают им неожиданные свойства, в частности улучшенную стабильность по сравнению с липосомами по предшествующему уровню техники. Липосомы по изобретению также демонстрируют способность к высвобождению активного ингредиента в течение длительного периода времени по сравнению с липосомами по предшествующему уровню техники.
Согласно второму предпочтительному варианту предмет настоящего изобретения представляет собой соединения, отвечающие формуле (IB):
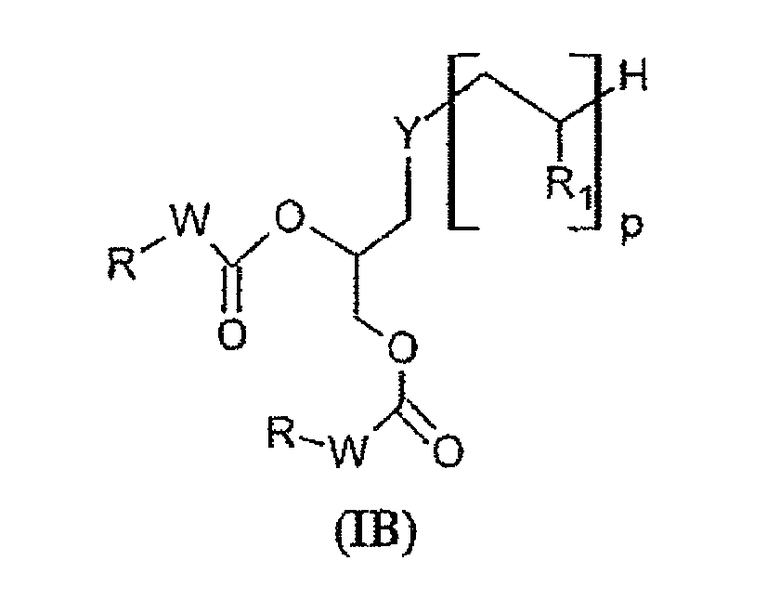
в которой:
Y представляет собой атом серы или группу -NH-CO-CH2CH2S-;
W представляет собой группу -NH- или группу -CH2-;
р представляет собой целое число, изменяющееся в диапазоне от 1 до 50;
R1 представляет собой группу, выбранную из следующих радикалов:

в которых R' представляет собой Н или гидрофильную группу, такую как, например, С4-С24 полигидроксилированное соединение на основе углеводорода. В частности, R' может быть выбрана из сахаров, таких как, например, галактоза, глюкоза, манноза или сиаловая кислота, связанных через их аномерный атом углерода;
R представляет собой группу, выбранную из: С4-С24 радикалов на основе углеводорода; С4-С24 фторированных радикалов на основе углеводорода; С4-С24 тиоалкильных радикалов.
Предпочтительными цепями R являются цепи, которые способствуют тому, чтобы критическая концентрация мицеллообразования (ККМ) поверхностно-активного вещества формулы (IB) составляла менее 10-5 М. В действительности, низкая ККМ придает наночастице  термодинамическую стабильность, а также способность к удержанию растворов, инкапсулированных во внутреннем гидрофобном компартменте. Предпочтительно, R представляет собой цепь на основе углеводорода С12-С24 или цепь на основе фторированного углеводорода С8-С24.
термодинамическую стабильность, а также способность к удержанию растворов, инкапсулированных во внутреннем гидрофобном компартменте. Предпочтительно, R представляет собой цепь на основе углеводорода С12-С24 или цепь на основе фторированного углеводорода С8-С24.
Синтез молекул формулы (IB) можно легко проводить с использованием традиционных способов органического синтеза. Несколько примеров синтеза проиллюстрировано в экспериментальной части.
В качестве краткого обобщения, молекулу формулы (IB) синтезируют с группой R тиоалкильного типа, указанную молекулу используют в качестве агента-переносчика в реакции теломеризации в присутствии гидрофильного полимеризуемого реагента (типа трис- (гидроксиметил)акриламидометана или его производных, или винилпирролидона) и свободно-радикального инициатора, такого как α,α'-азобисбутиронитрил (AIBN) в растворе в метаноле, ТГФ (THF) или в ацетонитриле, доведенном до точки кипения. Изначальное соотношение полимеризуемого мономера и агента-переносчика делает возможным осуществление контроля степени полимеризации теломера и, таким образом, растворимости продукта. Последний получают осаждением из простого эфира.
Предпочтительными соединениями формулы (IB) являются соединения, в которых Y представляет собой S.
Другой предпочтительный вариант представляет собой такой, в котором р представляет собой целое число в диапазоне от 5 до 15.
Еще более преимущественно, предпочтение отдается соединениям, отвечающим формуле С ниже, в которой R определен так же, как указано выше, р представляет собой целое число в диапазоне от 5 до 15 и W=CH2:
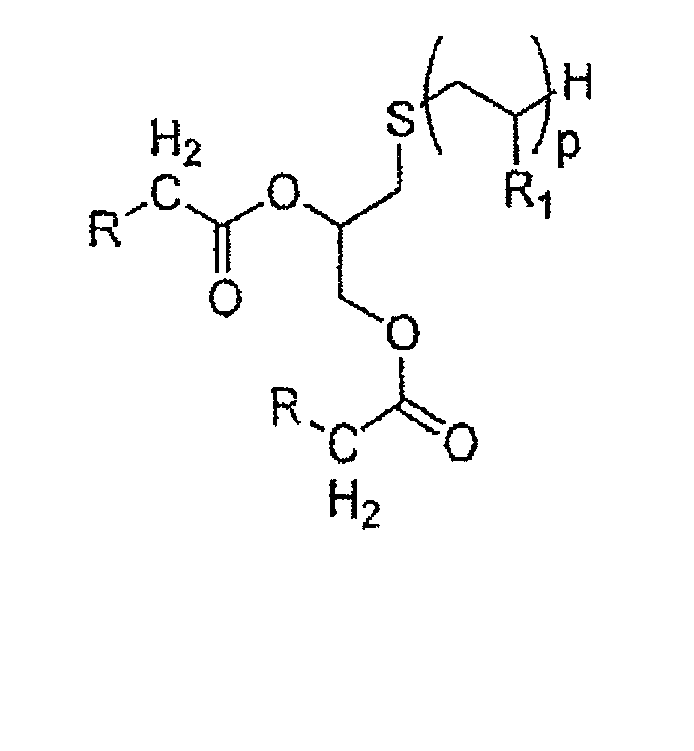
Соединение С
Среди указанных соединений в особенности предпочтительным соединением является С1, представленное ниже:

Соединение С1
Другим предметом изобретения является использование соединений формулы (I), преимущественно соединений формулы (IB), для получения наночастиц с гидрофобной полостью, а также полученные таким образом наночастицы. Наночастицы получают из поверхностно-активных веществ формулы (I) или (IB), чрезвычайно легко, при использовании пленочного способа, который хорошо известен специалистам в данной области техники и который описан в работе Liposomes, a practical approach, R.R.C. New, Ed., Oxford University Press, New York, 1990. Способ проводят, как описано выше для соединения формулы (IA).
Неожиданно, при использовании соединений с общей формулой (IB), в которых величина р составляет менее чем 15 и где R представляет собой цепь на основе углеводорода, включающую по крайней мере 12 атомов углерода, наблюдалось образование наночастиц удлиненной формы, называемых эллипсоидами из-за их формы, напоминающей форму зерен риса (Фиг.7а).
Исследование эллипсоидных частиц, полученных при использовании соединений структуры (IB)
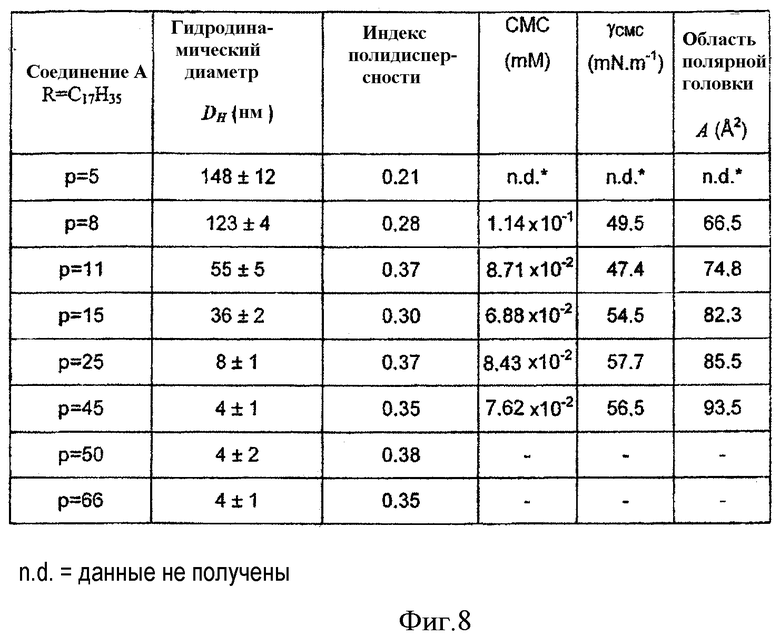
Действительно, при использовании производных соединений с общей формулой (IB), в которых р составляет в диапазоне от 5 до 15, наблюдалось образование оригинальных частиц (Фиг.8), форма которых напоминала зерно риса и размер которых уменьшался с увеличением р. В том случае, если р составляет более 15, размер полученных объектов составляет менее 10 нм и природа образуемых агрегатов в значительной степени является мицеллярной.
Размер и механическую стабильность с течением времени частиц, полученных в растворе, измеряли после фильтрации при помощи динамической дифракции света (High Performance Particle Sizer, Malvern). Природу полученных частиц исследовали при помощи просвечивающей электронной микроскопии (ТЕМ) после негативного окрашивания образца или после замораживания-скалывания. Критическую концентрацию агрегации этих поверхностно-активных веществ определяли тензиометрией или спектрофлуометрией при использовании способа флуоресцентной метки. Кроме того, способ тензиометрии по Wilhelmi сделал возможным определение максимальных поверхностных натяжений и площади полярной головки на границе раздела вода-воздух (Фиг.8). Эти соединения демонстрируют сравнительно низкие ККМ порядка 10-5 М. ККМ этих поверхностно-активных веществ фактически не изменяется как функция от р (Фиг.8). Для того, чтобы вызвать уменьшение или увеличение этой величины, необходимо соответственно увеличивать или уменьшать длину цепей на основе углеводородов. Температуру фазового перехода этих поверхностно-активных веществ определяли поляризационной спектрофлуриметрией и рассеянием света (Фиг.9). Температура фазового перехода фактически не изменяется для постоянных длин цепи при увеличении р (41°С<Tm<44°С).
Гидродинамический диаметр (D H) надмолекулярного образования изменяется обратно пропорционально изменению средней степени полимеризации теломеров: чем больше относительный объем полярного компонента, тем больше кривизна мембран (Фиг.8). Этот результат был подтвержден электронной микроскопией. За пределами р=20 все микрофотографии показывают мицеллярные растворы (Фиг.7b). С другой стороны, ниже этой величины они демонстрируют надмолекулярные образования, напоминающие продолговатые объекты в форме рисового зерна, которые не демонстрируют наличия внутренней водной полости при ТЕМ исследованиях после негативного окрашивания (Фиг.7а).
При использовании калибровки частиц оказывается, что для соединения структуры (С) с р=5 и R=C17H35 гидродинамический диаметр полученных частиц составляет 148 нм. Эти объекты демонстрируют чрезвычайно высокую стабильность с течением времени, которая увеличивается пропорционально величине р (от 2 недель для р=5 до нескольких месяцев для р=25).
Данные эллипсоидные частицы демонстрируют оригинальные структурные характеристики, и их средний гидродинамический диаметр можно легко изменять, варьируя р, т.е. число мономерных единиц, составляющих полимерную частицу. Частицы по изобретению также продемонстрировали способность инкапсулировать гидрофобные активные агенты. Это включение можно проводить при использовании технологических способов, хорошо известных специалистам в данной области техники. Например, инкапсулирование можно проводить растворением активного агента в предварительно полученном растворе эллипсоидов или мицелл, процедурой “масло в воде” или диализом. Терапевтические соединения, которые могут быть инкапсулированы, представляют собой все соединения, предпочтительно гидрофобные соединения, которые могут быть стабильно включены в эти мицеллярные или эллипсоидные образования. При использовании указанных объектов различные семейства слабогидрофильных или гидрофобных активных ингредиентов могут быть инкапсулированы или растворены, включая противораковые агенты, антибиотики, иммуномодуляторы, стероиды, противовоспалительные препараты или нуклеотиды. Могут также быть инкапсулированы или векторизированы гидрофильные соединения, способные образовывать комплекс с полярной частью наночастиц. Дозу активного агента, эффективно инкапсулированного в указанных наночастицах, определяют после фильтрации неинкапсулированного активного агента при использовании ВЭЖХ (HPLC), УФ (UV) или флуоресцентной спектрометрии, а также 1Н ЯМР.
В дополнение к соединению формулы (I) мицеллярные, эллипсоидальные или липосомные наночастицы по изобретению могут также предпочтительно содержать по крайней мере одно соединение, отвечающее формуле (II) ниже:
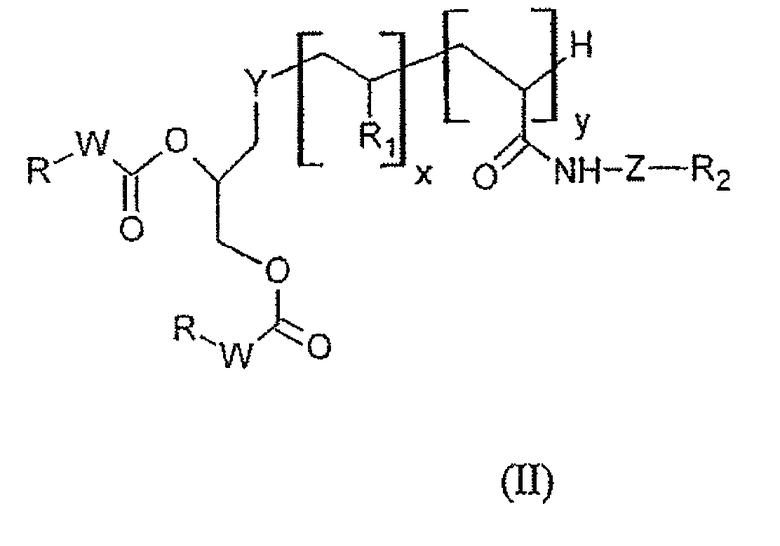
в которой:
- Y представляет собой атом серы или группу
-NH-CO-(CH2)n-X-, в которой Х представляет собой атом серы S или группу -CH2-, n представляет собой целое число в интервале от 0 до 10;
- W представляет собой группу -NH- или -CH2-;
- x представляет собой 0 или целое число в диапазоне от 1 до 30;
- y представляет собой 0 или целое число в диапазоне от 1 до 10;
- R1 представляет собой гидрофильную группу, выбранную из следующих радикалов:
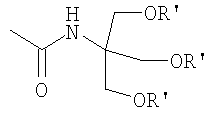
в которых R' представляет собой Н или гидрофильную группу, такую как, например, C4-С24 полигидроксилированное соединение на основе углеводорода; в особенности R' может быть выбрана из сахаров, таких как, например, галактоза, глюкоза, манноза или сиаловая кислота, связанных через их аномерный атом углерода;
- R2 представляет собой группу распознавания, которую выбирают в соответствии с клеточной мишенью; предпочтительно ее выбирают из групп, обладающих заметным сродством к биологической мишени активного ингредиента, транспортируемого в наночастице.
R2 может представлять собой сахарид по своей природе (для определения в качестве объекта специфических мембранных пектинов, которые находятся в специфических тканях и которые специфическим образом распознают либо галактозу - в случае печени, кости, определенных раковых опухолей; либо маннозу - в случае макрофагов, сердца; либо сиаловую кислоту - в случае эритроцитов, и т.д.), небольшие молекулы-эффекторы (небольшая молекула, концентрация которой регулирует активность молекулы определенного белка путем взаимодействия со специфическим участком связывания на молекуле белка и изменения его структуры (Айала Ф., Кайгер Дж. «Современная генетика». Том 3, стр.318)), быть гормональным по природе (как, например, стероиды), быть синтетическим по природе, как, например, Gleevek для определения в качестве объектов киназ, специфические антитела, фрагменты антител, антиген, биотин, который связывает определенные специфические белки, и, в более общем случае, представлять собой любой субстрат, для которого предшествующие исследования показали распознаваемую специфичность. Среди пептидов, которые могут быть использованы в настоящем изобретении, можно упомянуть, например, RGD-последовательность, известную благодаря своему сродству к αVβ3 интегринам.
Можно представить, что та же самая молекула формулы (II) содержит одну или несколько идентичных групп распознавания R2 или больше различных групп распознавания R2, которые дают возможность направлять частицы к нескольким отдельным биологическим мишеням.
- R группа подчиняется тем же правилам, что и определены выше для структуры соединения формулы (I).
Z представляет собой «ножку» (spacer arm), которая соединяет группу распознавания R2 с полимерной цепью. Z связана с R2 при помощи связи, которая может быть выбрана из функций
-О-СО-, -СО-NH-, -NH-CO-NH-, -NH-CO-O-, O-CO-O-, -O-, -CH=N- или -S- или за счет образования комплекса с атомом никеля (WoodleChikh et Lasical., Biochim. Biophys. Acta, 1113 (1992) 171-1999, Acta, 1567 (2002) 204-212). Последний может связываться, во-первых, с полигистидиновой меткой, присоединенной к агенту, определяющему объект, и, во-вторых, - с поликислотой типа NTA, присоединенной к полимерной цепи.
«Ножка» Z может состоять из белковой цепи. Последняя может быть присоединена к олигомерной цепи при помощи концевой аминогруппы боковой цепи или основной цепи. Эта «ножка» включает от 1 до 5 аминокислот, предпочтительно от 1 до 3 аминокислот.
Аминокислоты, составляющие «ножку» Z, выбирают из природных аминокислот, таких как аланин, аргинин, аспарагин, аспарагиновая кислота, цистеин, глутамин, глутаминовая кислота, глицин, гистидин, изолейцин, лейцин, лизин, метионин, фенилаланин, пролин, серин, треонин, триптофан, тирозин или валин, или синтетических аминокислот, таких как гидроксипролин, норлейцин, орнитин, цитруллин или циклогексилаланин. Эта «ножка» Z может состоять из тирозинового остатка, который дает возможность отслеживать вектор in vivo после введения метки с 125I или 131I.
В качестве группы Z можно также предположить использование Ω-аминокислот, таких как 3-аминопропионовая кислота и 4-аминомасляная кислота, а также этаноламина, 3-пропаноламина или диаминов формулы -NH-(CH2)r NH-, в которых r представляет собой целое число в диапазоне от 2 до 6.
Или Z-R2 группа представляет собой группу NTA, и, если происходит связывание за счет образования комплекса с атомом никеля, Z-R2 группа состоит из группы NTA формулы:
Согласно первому варианту изобретения, который касается липосом, полученных из молекул формулы (IA), предпочтительные соединения формулы (II) представляют собой соединения формулы (IIA) ниже:
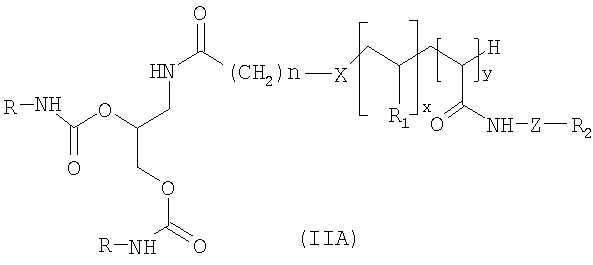
в которой X, n, z, y, R, R1 и R2 имеют те же значения, что и выше в формуле (II);
- предпочтительно x и y не представляют собой ноль одновременно;
- предпочтительно Х представляет собой S;
- предпочтительно n=2.
Согласно второму варианту изобретения, который касается липосом, полученных из молекул формулы (IB), предпочтительные соединения формулы (II) представляют собой соединения формулы (IIB) ниже:
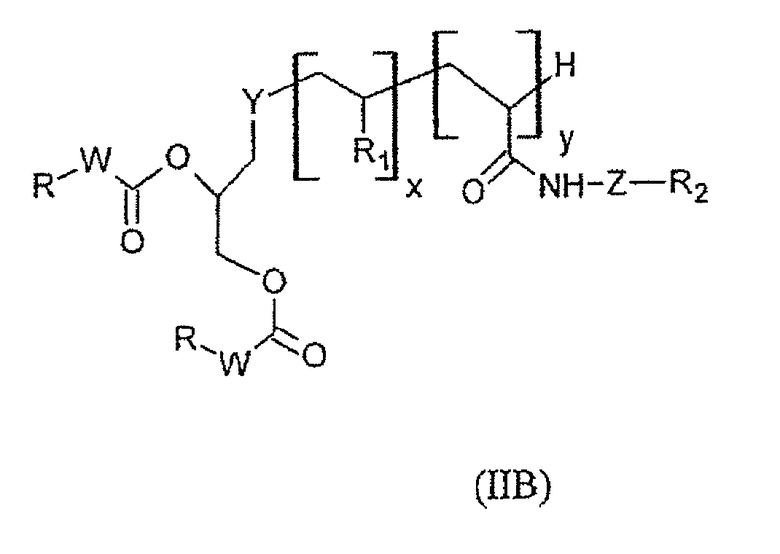
в которой:
- Y представляет собой атом серы или группу -NH-CO-CH2CH2S-;
- W, x, y, Z, R, R1 и R2 имеют те же значения, что и в формуле (II) выше.
Предпочтительно, наночастицы (липосомы, трубчатые везикулы, мицеллы или эллипсоидальные частицы) по изобретению содержат от 1 до 5% одного или нескольких соединений формулы (II), которые делают возможным улучшение нацеливания этих наночастиц на их биологическую цель без ухудшения их структуры.
Указанные липидные теломеры формулы (II) обладают тем преимуществом, обусловленным их олигомерной гидрофильной частью, что они способны дистанцировать привитые агенты распознавания от поверхности трубчатых везикул, таким образом способствуя их распознаванию клетками-мишенями или тканями-мишенями. Другое преимущество, связанное с использованием указанных нацеливающих липидов (II), состоит в возможности увеличения числа единиц распознавания в одном соединении благодаря технологическому способу теломеризации. Факторы x и y фактически легко контролируемы и будут достаточно тесно зависеть от пропорции мономеров и от используемого в реакции телогенного агента.
Лигирование агентов распознавания можно проводить перед теломеризацией гидрофильной головки при условии совместимости с условиями реакции. Агенты распознавания можно также присоединять к олигомерной полярной головке после образования трубчатых везикул. Теломеризованную гидрофильную часть затем функционализируют группами, способными обеспечивать связывание с этими агентами распознавания. Различные технологические способы связывания, которые можно использовать, хорошо известны специалистам в данной области техники, и они, в частности, описаны в: Allen et al., Biochim. Biophys. Acta, 1237 (1995) 99-108; Sapra, Prog. Lipids Res., 42 (2003) 439-462, Hansen et al., Biochim. Biophys. Acta, 1239 (1995) 133-144.
Соединения формулы (II) составляют другой объект изобретения.
Соединения формулы (I) и формулы (II), описанные выше, могут быть обобщены в виде одной и той же формулы (III):

в которой R, W, m и Y определены так же, как и в формулах (I) и (II) выше, а R3 представляет собой группу, выбранную из
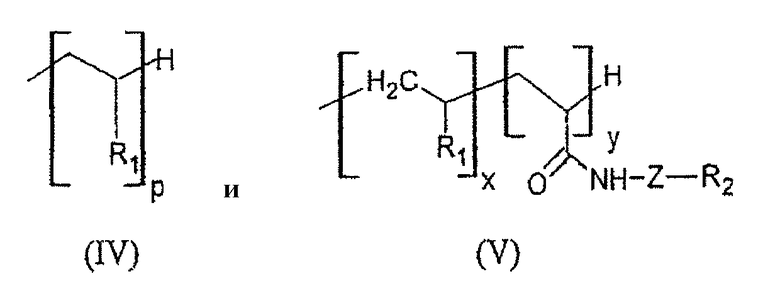
причем R1 имеет такое же определение, как и в формулах (I) и (II), р имеет такое же определение, как и в формуле (I), и x, y, Z и R2 имеют такое же определение, как и в формуле (II).
В частности, соединения формулы (IA) и формулы (IIA), описанные выше, могут быть обобщены в виде общей формулы (IIIA):
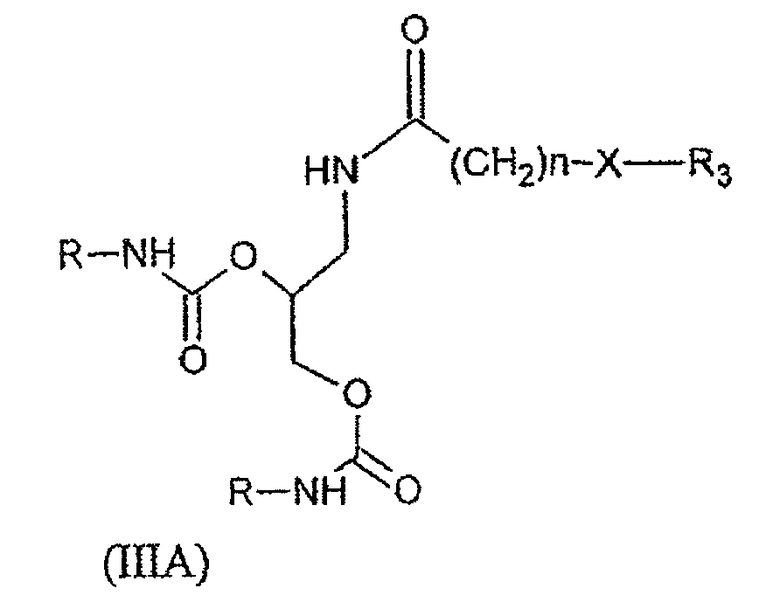
в которой X, n и R определены так же, как и в формулах (IA) и (IIA), и R3 представляет собой группу, выбранную из:
при этом m имеет такое же значение, как и в формуле (IA), R1, R2, Z, x и y имеют такие же значения, как и в формуле (IIA).
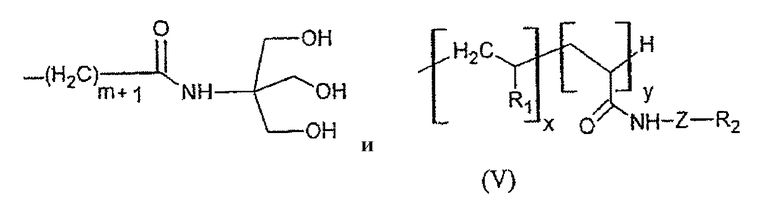
Соединения формулы (IB) и формулы (IIB), описанные выше, могут быть обобщены в виде одной и той же формулы (IIIB):
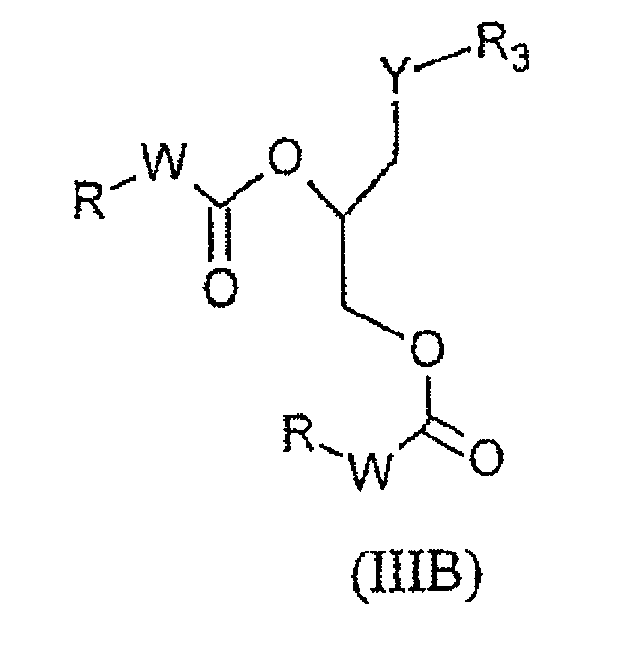
в которой R, W и Y имеют те же значения, что и в формулах (IB) и (IIB), и R3 представляет собой группу, выбранную из
причем R1 имеет такое же определение, как и в формулах (IB) и (IIB), р имеет такое же определение, как и в формуле (IB), и x, y, Z и R2 имеют такое же определение, как и в формуле (IIB).
Согласно предпочтительному варианту осуществления изобретения липосомы или трубчатые везикулы по изобретению получают из соединений формулы (IA) и необязательно (IIA) и стабилизируют на основе теломеризации или полимеризации мономера акрилового типа, содержащегося в их внутренней водной полости.
Для того, чтобы ограничить высвобождение растворенных веществ, инкапсулированных в указанных трубчатых везикулах, и увеличить их механическую стабильность, во внутренний водный компартмент трубчатых везикул можно вводить олигомерную или полимерную матрицу. Олигомерную матрицу получают после инкапсулирования образующих ее мономеров в трубчатых везикулах и удаления неинкапсулированных мономеров при помощи гель-проникающих способов разделения. Теломеризация, которая состоит в образовании полимера в присутствии агента переноса цепи, делает возможным получение небольших полимеров контролируемого размера. Низкая молекулярная масса этих полимеров способствует их удалению почками. Исключая аккумуляцию полимера в лизосоме, можно также избежать проблем токсичности.
Наночастицы, липосомы или трубчатые везикулы, включающие дополнительно к поверхностно-активным веществам формулы (IA) по крайней мере один олигомер или теломер, как описано выше, составляют другой предмет настоящего изобретения.
Олигомер или теломер состоит из ионогенных или неионогенных гидрофильных мономерных конструкционных блоков, выбранных из акриловой кислоты, метакриловой кислоты и метакриламидных производных и также акрилатных, метакрилатных, акриламидных и метакриламидных производных С1-С6 спиртов, С2-С12 полиспиртов, сахаров и аминокислот.
Данные сахара могут представлять собой:
- простые сахара, такие как глюкоза, рибоза, арабиноза, ксилоза, ликсоза, аллоза, альтроза, манноза, галактоза, фруктоза или талоза;
- дисахариды, такие как мальтоза, сахароза или лактоза.
Аминокислоты могут быть выбраны из природных аминокислот, таких как аланин, аргинин, аспарагин, аспарагиновая кислота, цистеин, глутамин, глутаминовая кислота, глицин, гистидин, изолейцин, лейцин, лизин, метионин, фенилаланин, пролин, серин, треонин, триптофан, тирозин или валин, или синтетических аминокислот, таких как гидроксипролин, норлейцин, орнитин, цитруллин или циклогексилаланин.
Среди коммерчески доступных мономеров, которые могут быть использованы в способе по изобретению, могут быть упомянуты в качестве примера трис(гидрокси)метилакриламидометан, натрийакрилат, гидроксиэтилакрилат, глюкозамоноакрилат, глюкоза-1-(N-метил)акриламид, глюкоза-2-акриламид, мальтоза-1-акриламид и моноакрилат сорбита.
Для того, чтобы модифицировать способность к высвобождению и к стабилизации теломера, в производстве теломера можно использовать водорастворимые сшивающие агенты, такие как глюкоза-1,2-диакриламид, сорбита диакрилат, сахарозы диакрилат, сахарозы ди(этилендиаминакриламид), канамицина тетраакриламид, канамицина диакриламид или другие сахара ди- или полифункционализированные акрилатами или акриламидами. В качестве общего правила могут быть использованы все гидрофильные соединения, способные присоединять по крайней мере две акрилатные или акриламидные группы. В качестве примера можно привести акрилатное производное трис(гидроксиметил)акриламидометана (соединение Е):
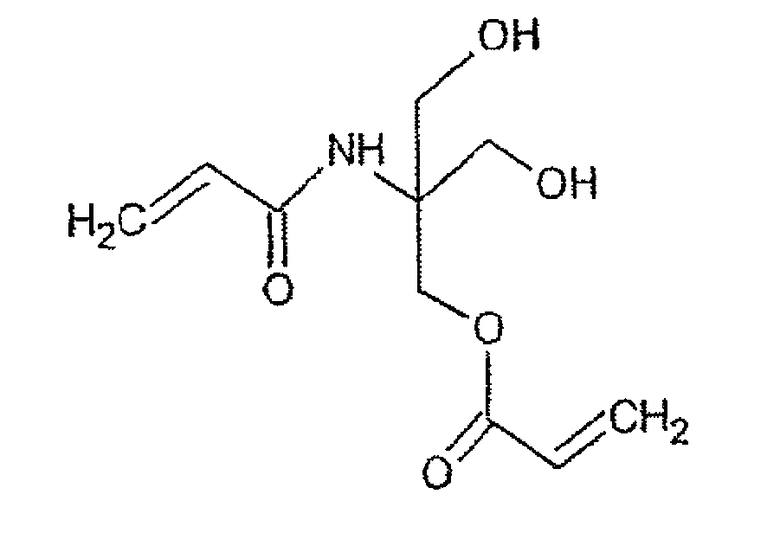
Соединение Е
Данные сшивающие агенты используются в соотношении в диапазоне от 1 до 5% по массе по отношению к массе мономера(ов).
Размер теломера контролируют за счет использования одного или нескольких гидрофильных или гидрофобных агентов переноса, которые вводят в мембрану или внутреннюю водную полость наночастиц и, в частности, трубчатых везикул. Агент переноса цепи может быть гидрофильным или гидрофобным, тиольного или фосфитного типа. Используемые агенты переноса цепи выбирают из гидрофильных тиолов, таких как тиолуксусная кислота, меркаптопропионовая кислота, тиоэтиленгликоль, цистамин или цистеин, или гидрофобных тиолов, таких как алкантиолы от С2 до С30, например соединение D (производное холестерина), для которого известна способность к интеграции в фосфолипидные мембраны. Синтез соединения D, в частности, описан в M. Wathier et al., Chem. Phys. Lipids (2002), 115, 17-37.
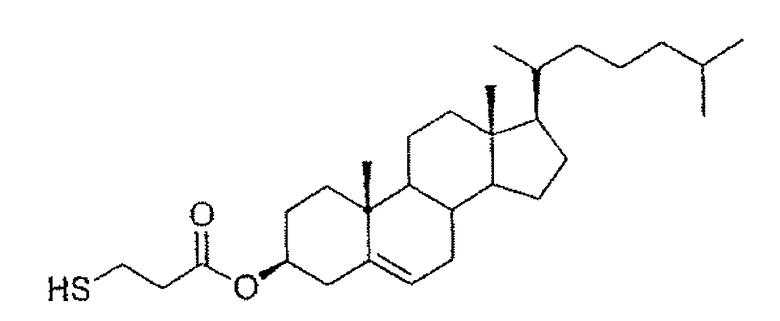
Соединение D
Агент переноса цепи может также быть выбран из ранее описанных лестничных тиолов. Со своей стороны, фосфиты могут быть гидрофильными, такими как диэтилфосфит, или гидрофобными, такими как диоктилфосфит, дидодецилфосфит или дигексадецилфосфит.
Для того, чтобы избежать разрушения мембраны трубчатых везикул, для гидрофильной структуры этих гидрофобных тиолов предпочтительно, чтобы она была сходной со структурой составляющих ее поверхностно-активных веществ. Общее звено этих телогенных агентов преимущественно состоит из аминоглицеринового звена, к которому привиты цепи жирных кислот посредством карбаматных связей согласно формуле (VI):
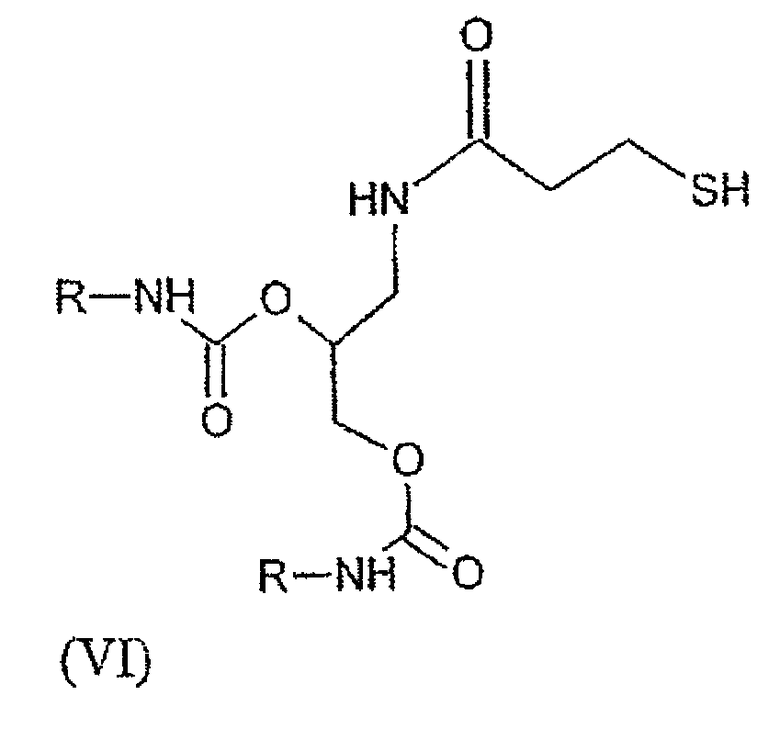
в которой R имеет то же значение, что и в формулах (I), (II) и (III) выше.
Для того, чтобы регулировать степень полимеризации, можно изменять долю агента переноса цепи, внедренного в мембрану, по отношению к количеству инкапсулированных мономеров. Отношение агент переноса цепи/липидное поверхностно-активное вещество (I) предпочтительно находится в диапазоне от 1 до 10% масс./масс.
Отношение агент переноса цепи/мономер может изменяться в диапазоне от 0,1 до 10% масс./масс.
Полимеризацию можно инициировать известным способом при помощи ультрафиолетового облучения или фотоинициаторов, окислительно-восстановительных инициирующих парных систем, нагреванием или термическими свободно-радикальными инициаторами и, в более общем случае, при использовании всех традиционных технологических способов, описанных в литературе (G. Odian, Principles of polymerization, 3. sup. rd. Ed, Wiley, New York, 1991).
В конце полимеризации внутренняя водная полость трубчатых везикул содержит гидрофильный теломер, который необязательно имеет телогенную гидрофобную часть, интегрированную во внутреннюю мембрану ламеллы. Физико-химическое описание указанных стабилизированных трубчатых везикул представлено в экспериментальной части, и продемонстрированы их большая способность к высвобождению инкапсулированного растворенного вещества и их увеличенная механическая стабильность.
Заданные применения включают транспортировку активных ингредиентов, в особенности терапевтически активных ингредиентов, эпидермальную доставку косметических веществ и агентов для медицинской диагностики; в частности транспортировку противораковых активных агентов, вакцин, генетического материала, ферментов, гормонов, витаминов, сахаров, белков и пептидов, липидов или органических и неорганических молекул.
В контексте использования, связанного с вакцинами, эпитопы и пептиды могут быть интегрированы во внутреннюю водную матрицу или экспрессированы на поверхности трубчатых везикул при помощи системы ковалентного связывания, для того, чтобы улучшить иммунный ответ против эпитопов.
Таким образом, предметом настоящего изобретения также является любая композиция, в особенности любая терапевтическая, диагностическая, вакцинирующая или косметическая композиция, включающая по крайней мере один активный ингредиент в комбинации с наночастицей, липосомой, трубчатой везикулой, эллипсоидом или мицеллой, как описано выше, и, в особенности, любая композиция, включающая по крайней мере один активный ингредиент, инкапсулированный в липосому или трубчатую везикулу, эллипсоид или мицеллу, согласно настоящему изобретению.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
ЧЕРТЕЖИ
Фиг.1 иллюстрирует кинетику высвобождения карбоксифлуоресцеина, инкапсулированного в фосфатидилхолиновые липосомы (+) и трубчатые везикулы, состоящие из соединения А1 (•), измеряемую спектрофлуорометрией.
Фиг.2 представляет собой инфракрасный спектр жидкой фазы раствора соединения А1 в CCl4 при различных концентрациях (от 1×10-2 до 2,5×10-4 М).
Фиг.3 представляет собой кривую распределения размера на основе объема трубчатых везикул, измеренных при помощи электронной микроскопии.
Фиг.4 представляет собой фотографию, полученную при помощи фазовой просвечивающей электронной микроскопии после негативного окрашивания 2% уранилацетатом трубчатых везикул, полученных дисперсией соединения А1 (2,5 мг·мл-1) в воде.
Фиг.5 представляет собой фотографию, полученную при помощи фазовой просвечивающей электронной микроскопии после замораживания-скалывания образца трубчатых везикул, полученных дисперсией соединения А1 (2,5 мг·мл-1) в воде.
Фиг.6 представляет собой фотографию, полученную при помощи фазовой просвечивающей электронной микроскопии после негативного окрашивания 2% уранилацетатом образца трубчатых везикул, полученных дисперсией соединения структуры В (2,5 мг·мл -1) в воде.
Фиг.7 представляет собой микрофотографии, полученные фазовой просвечивающей электронной микроскопией (негативное окрашивание 20% уранилацетатом) водных дисперсий соединений формулы С с R=C17H35 и р=5 (a) и R=C17H35 и р=20 (b), и электронные микрофотографии после замораживания-скалывания соединения формулы С с R=C17H35 и р=8 (с).
Фиг.8 представляет собой таблицу, суммирующую результаты физико-химических исследований и исследования размера частиц водных дисперсий соединений структуры С с R=C17H35 и изменяющимся р.
Фиг.9 представляет температуры фазового перехода, измеренного при помощи светорассеяния, и повторяемость значений, измеренных при помощи спектрофлуорометрии.
Пример 1: синтез производного А1
Синтез производного А1 суммирован на схеме 1.
• Тритилмеркаптопропионовая кислота (1): коммерчески доступная молекула.
• рац-N-(2,3-дигидроксипропил)-3-(тритилмеркапто)пропионамид (2)
2 г 3-(тритилмеркапто)пропановой кислоты (5,75 ммоль), 0,523 г аминопропандиола (1 экв.) и 1,7 г N-(этоксикарбонил)-2-этокси-1,2-дигидрохинолина (EEDQ) (6,9 ммоль, 1,2 экв.) растворяли в 50 мл дихлорметана. Реакционную смесь кипятили с обратным холодильником в течение 16 часов. Неочищенную реакционную массу затем промывали насыщенным раствором бикарбоната натрия и затем нормальным раствором соляной кислоты, насыщенным хлоридом натрия, а затем высушивали над сульфатом натрия. После фильтрации через пористое стекло продукт очищали хроматографией на силикагеле, элюирование проводили с градиентом от чистого этилацетата до смеси этилацетат/метанол 9:1 (об./об.). Чистый продукт получали в форме белого порошка (2,12 г, выход: 87%).
Продукт также может быть получен в чистом виде, с идентичным выходом, при помощи кристаллизации неочищенной реакционной массы при комнатной температуре, из 8:2 (об.:об.) смеси этилацетат/метанол в течение 8 дней.
1Н ЯМР(/CDCl3): δ (м.д.) 7,40-7,25(15H, м, тритил-ароматика); 5,86(1Н, т, NH); 3,7(1Н, м, СНОН); 3,51(2Н, м, СН 2ОН); 3,31(2Н, м, CH 2NH); 3,09(2Н, м, ОН); 2,50(2Н, т, CH 2S); 2,04(2Н, м, SCH2CH 2CO).
13C ЯМР(/CHCl3): δ (м.д.) 172,9(CH2 CONH); 144,6(SCC фенил); 129,6(Cpara фенил); 128,0(Cortho фенил); 126,8 (Cmeta фенил); 70,9(CHOH); 66,9(SCPh3); 63,6(CH2OH); 42,1(NHCH2CH); 35,3(SCH2 CH2CO); 27,6(CH2S).
• рац-2,3-ди[N-(гептадецил)карбамоилоксипропил]-3-(тритилмеркапто)пропионамид (3)
2 г рац-N-(2,3-дигидроксипропил)-3-(тритил-меркапто)пропионамида (4,75 ммоль) и 2,8 г 1-изоцианатгептадекана (9,97 ммоль, 2,1 экв.) растворяли в 50 мл свежеперегнанного толуола при комнатной температуре и при барботировании аргона. Реакционную среду кипятили с обратным холодильником и добавляли к смеси на кончике шпателя 1,4-диазабицикло[2.2.2]октан (DABCO) (кат.). После 6 часов реакционную неочищенную смесь испаряли досуха и поглощали в минимальном количестве эфира, из которого продукт кристаллизовался при комнатной температуре. После фильтрации продукт получали в чистом виде в форме белого порошка (4,04 г, выход 86%).
1Н ЯМР -(CDCl3): δ (м.д.) 7,47-7,23(15H, м, тритил-ароматика); 6,02(1H, т, NH); 4,95-4,74(3Н, м, СНО, СН 2О); 4,18(2Н, м, NH); 3,44(2Н, т, CH 2NH); 3,12(4Н, кв, CH 2NH); 2,51(2Н, т, CH 2S); 2,06(2Н, т, SCH 2CH 2CO); 1,49(4Н, м, NHCH2CH 2), 1,28(54H, м, СН2 цепочки); 0,91(6Н, т, СН3).
13С ЯМР(CDCl3): δ (м.д.) 171,2(CH2 CONH); 156,1-155,9(OCONH); 144,7(SCC фенил); 129,6(Cpara фенил); 127,9(Cortho фенил); 126,7 (Cmeta фенил); 71,2(CHO); 66,8(SCPh3); 63,4(CH2O); 41,2(NCH2CH2); 40,0(NHCH2CH); 35,5(SCH2 CH2CO); 31,9(NCH2 CH2); 29,9-29,3(цепочки) 27,6(CH2S); 22,7(CH2CH3); 14,1(CH3).
• рац-2,3-ди[N-(гептадецил)карбамоилоксипропил]-3-меркаптопропионамид (4)
2 г соединения 3 (2,03 ммоль) и 0,236 г триэтилсилана (2,03 ммоль, 1 экв.) растворяли в минимальном количестве дихлорметана и охлаждали до 0°С при помощи ледяной бани. Добавляли при охлаждении прикапыванием 10% раствор трифторуксусной кислоты в дихлорметане с использованием делительной воронки. Сразу после завершения добавления среду нагревали до комнатной температуры и оставляли на 3 часа при перемешивании. После упаривания досуха, поглощения дихлорметаном и промывания насыщенным раствором хлорида натрия в дистиллированной воде и затем насыщенным раствором бикарбоната натрия, неочищенную реакционную массу упаривали досуха и поглощали этилацетатом, из которого она кристаллизовалась при охлаждении. Чистый продукт выделяли в форме белого порошка (1,3 г, выход 86%).
1Н ЯМР(CDCl3): δ (м.д.) 6,46(1H, т, NH); 4,97-4,95(3Н, м, СНО, СН 2О); 4,21(2Н, м, NH); 3,50(2Н, т, CH 2NH); 3,16(4Н, кв, CH 2NH); 2,81(2Н, кв, CH 2S); 2,51(2Н, т, SCH 2CH 2CO); 1,64(1Н, т, SH); 1,50(4Н, м, NHCH2CH 2), 1,27(54H, м, СН2 цепочки); 0,89(6Н, т, СН3).
13С ЯМР(CDCl3): δ (м.д.) 170,9(CH2 CONH); 156,1-156,0(OCONH); 71,3(CHO); 63,5(CH2O); 41,2(NCH2CH2); 40,4(NHCH2CH); 35,5(SCH2 CH2CO); 31,9(NCH2 CH2); 29,9-29,4(цепочки) 22,7(CH2CH3); 20,4(CH 2S); 14,1(CH3).
• Ацетилированное соединение А1
0,5 г соединения 4 (0,67 ммоль) и 1,01 г триацетилированного THAM (3,37 ммоль, 5 экв.) растворяли в 30 мл свежеперегнанного триэтиламина. Смесь доводили до 50°С при барботировании аргоном в течение 2 часов и затем охлаждали до комнатной температуры и испаряли досуха. Неочищенную реакционную массу поглощали этилацетатом так, чтобы сделать возможным промывание нормальным водным раствором соляной кислоты и затем насыщенным водным раствором бикарбоната натрия. Продукт затем очищали хроматографией на колонке с силикагелем, элюируемой чистым этилацетатом. После упаривания и высушивания продукт получали в чистом виде в форме белого порошка (0,33 г, выход: 46%)
1Н ЯМР(/CDCl3): δ (м.д.) 6,48(1H, т, CONHCH2); 6,37(1H, c, CONHC); 5,05-4,96(3Н, м, СНО, СН 2О); 4,45(6Н, c, CH2O); 4,22(2Н, м, NH); 3,48(2Н, т, CH 2NH); 3,17(4Н, кв, CH 2NH); 2,81(2Н, кв, CH 2S); 2,49(4Н, т, SCH 2CH 2CO); 2,10(9H, c, CH3); 1,44(4Н, м, NHCH2CH 2); 1,27(54H, м, СН2 цепочки); 0,90(6Н, т, СН3).
13С ЯМР(/CDCl3): δ (м.д.) 171,7-171,5(CH2 CONH); 170,6(ОСО); 156,2-156,0(OCNH); 71,2(CHO); 63,6(CH2O); 62,5(CH2O); 41,2(NCH2CH2); 40,2(NHCH2CH); 37,1-36,6(SCH2 CH2CO); 31,9(NCH2 CH2); 29,8-29,3(цепочки) 26,8(CH3); 22,6(CH2CH3); 20,8(CH 2S); 14,1(CH3).

Схема 1: Синтез производного А1
• Соединение А1
0,3 г ацетилированного производного (0,28 ммоль) растворяли в минимальном количестве метанола и затем добавляли на кончике шпателя метоксид натрия (катализатор). Смесь оставляли при комнатной температуре в течение 30 минут при перемешивании при поддержании рН в диапазоне от 8 до 9, если необходимо путем добавления метоксида натрия. Реакционную среду затем нейтрализовали путем добавления нескольких капель нормального водного раствора соляной кислоты. После упаривания досуха неочищенную реакционную смесь очищали хроматографией на силикагеле, элюирование проводили смесью 95:5 (об.:об.) этилацетат/метанол. Продукт получали в чистом виде в форме белого порошка (0,233 г, выход: 88%). Т. пл.: 129°С.
1Н ЯМР(CDCl3): δ (м.д.) 7,00(1H, c, CONHC); 6,72(1H, т, CONHCH2); 5,00(3H, м, CHO, CH 2O); 4,47(3Н, м, НО, СН 2О х3) 4,44(6Н, c, CH2O х3); 4,19(2Н, м, NH х2); 3,48(2Н, т, CH 2NH); 3,16(4Н, кв, CH 2NH х2); 2,85(2Н, кв, CH 2S); 2,65-2,50(4Н, т, SCH 2CH 2CO х2); 1,44(4Н, м, NHCH2CH 2 х2); 1,28(54H, м, СН2 цепочки); 0,91(6Н, т, СН3 х2).
13С ЯМР(CDCl3): δ (м.д.) 171,1(CH2 CONH); 156,2-156,0(OCONH); 71,2(CHO); 63,6(CH2O); 61,0(CH2OH); 41,2(NCH2CH2); 40,2(NHCH2CH); 37,1-36,6(SCH2 CH2CO); 31,9(NCH2 CH2); 29,8-29,3(цепочки) 22,6(CH2CH3); 20,41(CH 2S); 14,1(CH3).
Пример 2: Синтез сшивающего агента Е: 2-акрилоиламино-3-гидрокси-2-(гидроксиметил)пропилового сложного эфира акриловой кислоты
Соединение Е
• 5-акрилоиламино-2,2-диметил[1,3]диоксан-5-илметиловый сложный эфир акриловой кислоты
1 г (4,65 ммоль) ТНАМ-изопропилидена растворяли в минимальном количестве дихлорметана. Значение рН устанавливали и поддерживали равным 9 путем добавления нескольких капель триэтиламина и затем добавляли прикапыванием раствор, содержащий 0,378 мл акрилоилхлорида (4,65 ммоль, 1 экв.) в 4 мл дихлорметана. Реакцию отслеживали при помощи ТСХ (смесь 7:3 этилацетат/циклогексан) с исчезновением ТНАМ-изопропилидена (Rf:0,3). Среду затем нейтрализовали путем добавления муравьиной кислоты, испаряли досуха и очищали хроматографией на силикагеле при использовании градиента элюирования от 7:3 до 5:5 смеси этилацетат/циклогексан. Чистый продукт получали в форме желтого масла (1,1 г, выход 87%).
1Н ЯМР(CDCl3): δ (м.д.) 6,50(м, 1H, CHCONH); 6,43(д, 1H, CHC2b акриловый сложный эфир); 6,30(c, 1H, NH); 6,22(д, 1Н, CH 2b акриламид) 6,14(д, 1Н, CHCOO); 5,93(д, 1Н, CH 2a акриловый сложный эфир); 5,65(д, 1Н, CH 2a акриламид); 4,62(с, 2Н, СН2ОСО); 4,42(д, 2Н, СН2О); 3,80(д, 2Н, CH2O); 1,49(д, 6Н, CH3 х2);.
• 2-акрилоиламино-3-гидрокси-2-(гидроксиметил) пропиловый сложный эфир акриловой кислоты
1,1 г (4,08 ммоль) 5-акрилоиламино-2,2-диметил[1,3]диоксан-5-илметилового сложного эфира акриловой кислоты и 5,45 г Montomorillonite K10 смешивали в дихлорметане и оставляли при комнатной температуре в течение 4 дней при перемешивании. Эту смесь затем фильтровали через целит и промывали метанолом. Осадок на целитовом фильтре несколько раз поглощали метанолом при интенсивном перемешивании перед тем, как снова его отфильтровать. После упаривания досуха продукт получали в чистом виде в виде желтого масла (0,766 г, выход: 82%).
1Н ЯМР(CDCl3): δ (м.д.) 6,73(с, 1H, NH); 6,42(д, 1H, CH 2b акриловый сложный эфир); 6,21(д, 1Н, CH 2b акриламид) 6,14(д, 1Н, CHCOO); 5,88(д, 1Н, CH 2a акриловый сложный эфир); 5,69(д, 1Н, CH 2a акриламид); 4,70(м, 2Н, ОН х2); 4,39(д, 2Н, CH2O); 3,70(дд, 4Н, CH2OH).
Пример 3: Синтез целевого липидного теломера G
Этот синтез проиллюстрирован на схеме 2.
• Метиловый сложный эфир 5-акрилоиламино-2-трет-бутилоксикарбониламинопентановой кислоты (5)
4,00 г ацетатной соли L-(Boc)LysOMe (12,50 ммоль - 1 экв.) растворяли в 30 мл безводного дихлорметана. рН раствора доводили до 9 путем добавления DIEA и затем охлаждали до 0°С. 1,87 мл акрилоилхлорида (23 ммоль - 1,85 экв.) добавляли к реакционной смеси прикапыванием при поддержании рН раствора основным путем добавления DIEA. После перемешивания в течение 24 часов среду промывали водой и органическую фазу затем сушили над Na2SO4. Растворители испаряли при пониженном давлении и очистка флеш-хроматографией на силикагеле (элюент: смесь 8:2 этилацетат/циклогексан) позволила получить соединение 5 (3,67 г, выход: 76%) в форме полупрозрачного бесцветного масла. [α]D= + 7,5 (c, 1, CHCl3).
1Н ЯМР(250 МГц, ДМСО-d6): δ 8,08(1H, т, J=5,6 Гц, NH-CO-О); 7,22(1H, д, J=7,7 Гц, NH-CO); 6,20(1Н, дд, Jцис=9,7 Гц и Jтранс=17,1 Гц, Нс); 6,05(1Н, дд, Jgem=2,7 Гц и Jтранс=17,1 Гц, Hb); 5,56(1Н, дд, Jgem=2,7 Гц и Jцис=9,7 Гц, На); 3,92(1Н, м, СН); 3,62(3Н, с, СН3-О); 3,10(2Н, кв, J=6,3 Гц, CH2-NH); 1,60(6Н, м, СН2 лизин), 1,38(9Н, с, СН3 Boc).
13С ЯМР(62,86 МГц CDCl3): δ 169,3, 161,3(CO); 136,8 (CIV аром.); 130,7(СН аром.); 128,6(CIV аром.); 128,6(СН аром.) м, 125,5(СН=N(O)), 72,2(CIV); 28,4(CH2-CO), 25,7(CH3 трет-бутил).
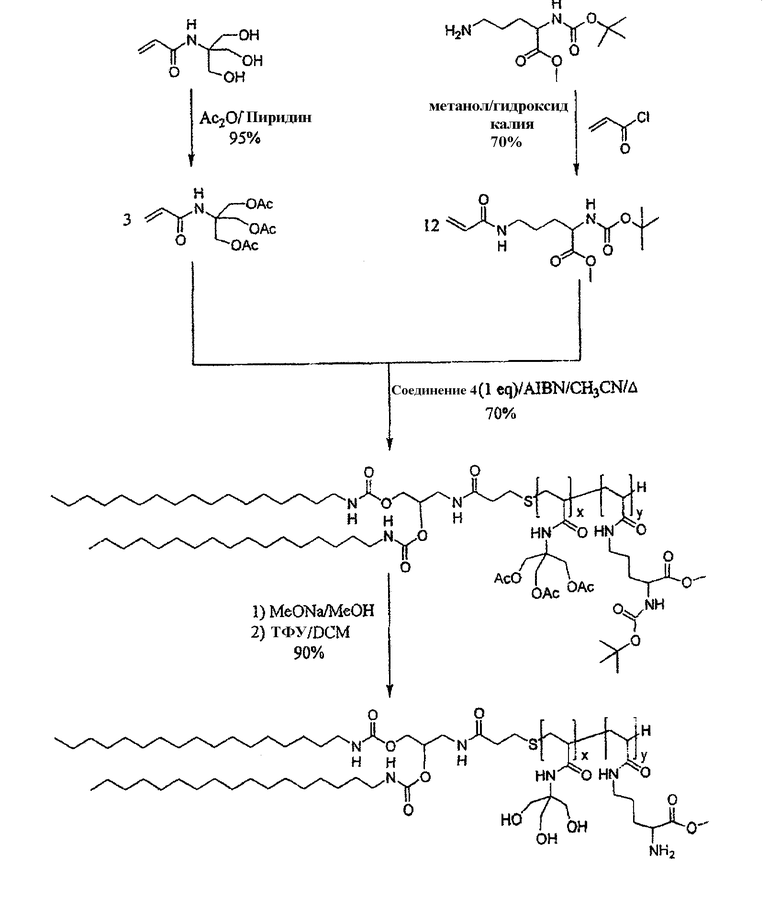
Схема синтеза целевого липидного теломера G
• Синтез теломеризованного липида G
0,767 г трис(ацетоксиметил)акриламидометанового мономера 6 (2,55 ммоль, 12 экв.) и 0,2 г мономера 5 (0,63 ммоль, 3 экв.) растворяли в 20 мл свежеперегнанного ацетонитрила в 100 мл двухгорлой круглодонной колбе, оснащенной сверху холодильником. Трис-(ацетоксиметил)акриламидометан 6 получали в соответствии с рекомендациями документа A Polidori et al., New. J. Chem. 1994, 18, 839-848. Реакционную смесь дегазировали под аргоном и кипятили с обратным холодильником. Добавляли 7 мг AIBN (4,29×10-2 ммоль, 0,2 экв.) и 0,157 г тиола 4 (0,21 ммоль, 1 экв.), растворенного в 5 мл свежеперегнанного и дегазированного ацетонитрила. Реакционную смесь кипятили с обратным холодильником в течение 4 ч, пока мономеры не были полностью израсходованы (определяли по ТСХ). Реакционную смесь концентрировали при пониженном давлении и неочищенную реакционную смесь фильтровали через сефадексовую колонку (1:1 смесь MeOH/CH2Cl2). Продукт затем растворяли в 100 мл метанола в присутствии каталитического количества метоксида натрия. После перемешивания в течение 5 ч реакционную среду нейтрализовали путем добавления кислотной смолы IRC 50. Смолу удаляли фильтрацией и растворитель удаляли при пониженном давлении. Продукт затем вводили в реакцию при охлаждении в кислой смеси ТФУ/CH2Cl2 (20%) в течение 3 ч.
Реакционную смесь затем концентрировали при пониженном давлении. Полученное масло поглощали несколько раз эфиром до тех пор, пока теломер не осаждался в форме белого порошка. Продукт растворяли в воде и лиофилизировали, пока не получали соединение G в форме белого порошка.
Среднюю степень полимеризации (DPn) и соотношение концентраций каждого мономера в макромолекуле (х и y) определяли при помощи 1Н ЯМР, сравнивая интегралы сигналов от метилов двух алкильных цепей при 1,1 м.д. с сигналами Tris(CH 2OH) при 4,3 м.д. и от лизина (CH 2NH) при 3,37 м.д. Заявители были способны определить следующие значения для x и y: x:30 и y:10.
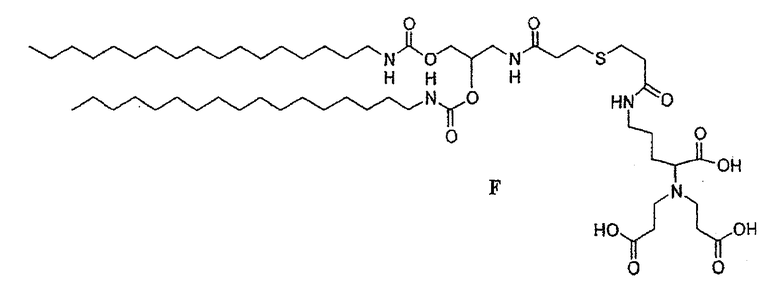
Пример 4: Синтез целевого липида F
• 5-Акрилоиламино-2-трет-бутоксикарбониламинопентановая кислота 7
2 г Вос лизина (8,13 ммоль) растворяли в 10 мл смеси 1:1 ацетонитрила - 2 н. гидроксида натрия. Реакционную среду охлаждали до 10оС. Добавляли прикапыванием акрилоилхлорид (1,1 г, 12,19 ммоль), растворенный в 10 мл ацетонитрила. Значение рН поддерживали равным 8 путем добавления 2 н. гидроксида натрия. В конце добавления реакционную смесь перемешивали при комнатной температуре в течение 2 ч и затем подкисляли 2 н. раствором HCl и экстрагировали этилацетатом (3×20 мл). Органическую фазу сушили и затем концентрировали при пониженном давлении. Конечный продукт получали в чистом виде в форме белого порошка путем перекристаллизации из этанола (1,9 г, 78%).
1Н ЯМР(250 МГц, ДМСО-d6): δ 8,02(1H, т, NH-CO-О); 7,18(1H, д, J=7,4 Гц, NH-CO); 6,25(1Н, дд, Jцис=9,4 Гц и Jтранс=17,1 Гц, Нс); 6,05(1Н, дд, Jgem=2,7 Гц и Jтранс=17,1 Гц, Hb); 5,6(1Н, дд, Jgem=2,7 Гц и Jцис=9,4 Гц, На); 3,92(1Н, м, СН); 3,10(2Н, кв, J=6,3 Гц, CH2-NH); 1,60(6Н, м, СН2 лизин), 1,38(9Н, с, СН3 Boc).
13С ЯМР(62,86 МГц ДМСО-d6): δ 169,3, 161,3(CO); 155,8 (СО уретан); 130,7(СН2=); 128,6(-СН=); 80,5(С tBu) 53,7(СН Lys); 40,3(CεLys); 33,2(CβLys); 29,8(СδLys); 28,4(CH2-CO); 25,7(CH3 трет-бутил).
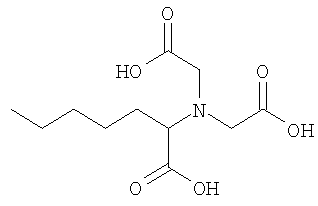
• 6-Акрилоиламино-2-[бис-(2-карбоксиэтил)амино]гексановая кислота 8
1,9 г соединения 7 (6,33 ммоль) растворяли в 20 мл смеси 1:1 трифторуксусная кислота - дихлорметан в 50 мл круглодонной колбе. После перемешивании в течение 2 ч растворитель испаряли при пониженном давлении и полученное масло поглощали несколько раз в хлороформе и испаряли, пока не был получен порошок. 1,94 г бромуксусной кислоты (12,66 ммоль) растворяли в 7 мл 2 н. гидроксида натрия в 25 мл круглодонной колбе. Раствор охлаждали до 0°С на ледяной бане и порошок, полученный после снятия защиты, растворенный в 11 мл водного 2 н. раствора гидроксида натрия, добавляли прикапыванием. После 2 ч реакции при комнатной температуре водный 2 н. раствор HCl добавляли, пока не был получен кислый рН. Продукт выпадал в виде осадка. Осадок отфильтровывали и фильтровали-сушили, и затем сушили при пониженном давлении. Продукт 8 получали в чистом виде в форме белого порошка после перекристаллизации из этанола (1,6 г, 57%).
1Н ЯМР(250 МГц, ДМСО-d6): δ 8,02(1H, т, NH-CO-О); 7,18(1H, д, J=7,4 Гц, NH-CO); 6,25(1Н, дд, Jцис=9,4 Гц и Jтранс=17,1 Гц, Нс); 6,05(1Н, дд, Jgem=2,7 Гц и Jтранс=17,1 Гц, Hb); 5,6(1Н, дд, Jgem=2,7 Гц и Jцис=9,4 Гц, На); 3,5(4Н, с, N-СН 2-COOH); 3,3(1Н, т, J=7,1 Гц, CH2-СН-N); 2,9(2Н, м, CH 2NH); 1-1,5(6Н, CH2CH2CH2).
13C ЯМР(62,86 МГц ДМСО-d6): δ 174,8, 173,6(COOH), 168,4 (CONH), 130,4(СН2=), 128,2(-СН=), 64,8(CH Lys), 53,8(N-СН 2-COOH), 40,3(CβLys); 30,5(CβLys); 29,8(СδLys).
• Синтез липида F
2,56 г соединения 4 (3,6 ммоль) и 1,6 г соединения 8 (3,6 ммоль) растворяли в 30 мл свежеперегнанного триэтиламина. Смесь доводили до 50°С при барботировании аргоном в течение 2 часов и затем охлаждали до комнатной температуры и испаряли досуха при пониженном давлении. Продукт получали в чистом виде в форме белого порошка после последовательной перекристаллизации из этилацетата (2,29 г, выход: 55%).
1Н ЯМР(250 МГц, ДМСО-d6): δ (м.д.) 7,12(1H, т, NH-CO-О); 6,48(1H, т, CONHCH2); 6,37(1Н, с, CONHC); 5,05-4,96(3Н, м, CHO, CH 2O); 4,45(6Н, c, CH2O); 4,22(2Н, м, NH); 3,48(6Н, c+т, N-CH2-СООН, CH 2NH); 3,25(1Н, т, J=7,1 Гц, СН2-СН-N); 3,17(2Н, м, CH 2NH); 2,81(4Н, т, CH 2S); 1,44(4H, м, NHCH2CH 2); 1,27(60H, м, CH2Lys, CH2 цепочки); 0,90(6Н, т, СН3).
13С ЯМР(250 МГц, ДМСО-d6): δ (м.д.) 171,7-171,5(COOH, CH2 CONH); 156,2-156,0(OCONH); 71,2(СНО); 64,5(СН Lys); 63,6(CH2O); 62,5(CH2O); 53,2(N-CH 2-COOH); 41,2(NCH2CH2); 40,2 40,1(CεLys, NHCH2CH); 37,1-36,6(SCH2 CH2CO); 31,9(CβLys); 29,8-29,3(цепочки и CδLys); 26,8(CH3); 22,6(CH2CH3); 20,8(CH2S); 14,1(CH3).
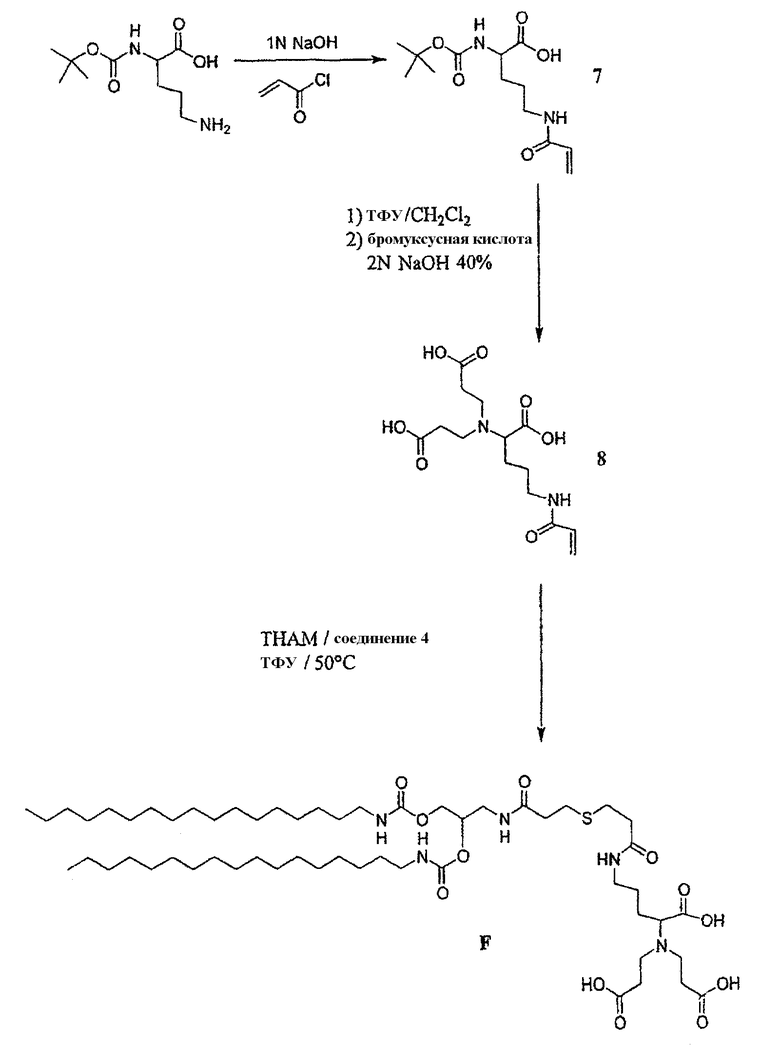
Схема синтеза целевого липида F
Пример 5: Получение трубчатых везикул из производного А1
Использованный материал
• Измерения размера трубчатых везикул
Распределение размера частиц измеряли при помощи фотонной корреляционной спектроскопии света образцов, растворенных в воде, при использовании устройства Malvern HPPS/NIBS, оснащенного 3 мВт He/Ne-лазером и 288-канальным коррелятором, оснащенным лавинными фотодиодами. Математическая обработка автокорреляционной кривой проводилась при помощи метода Контина (Contin).
• Дисперсия липида
Липиды А диспергировали в воде согласно пленочному способу в термостатируемой ультразвуковой бане Bransonic типа 2510Е (100 Вт, 42 Гц) выше температуры их фазового перехода в течение получаса.
• Образование дисперсии трубчатых везикул
Трубчатые везикулы получали, в первую очередь, путем растворения липида А1 в хлороформе. Раствор медленно концентрировали при пониженном давлении в 20-миллиметровой сердцевидной круглодонной колбе при использовании роторного испарителя. Полученную пленку затем сушили при пониженном давлении с использованием лопастного насоса. Липидную пленку затем повторно гидратировали дистиллированной водой при 65°С (на 10°С выше температуры фазового перехода липида) при концентрации 2,5 мг·мл-1. Смесь гомогенизировали вихревым перемешиванием в течение 5 минут и затем подвергали ультразвуковой обработке в течение 30 минут при 70°С. Полученный полупрозрачный голубоватый раствор фильтровали через 0,45 мкм фильтр и затем анализировали фотонным корреляционным спектрофотометром и трансмиссионной электронной микроскопией (фиг.3, 4 и 5).
При R = C17H35 (производное А1) температуру фазового перехода измеряли посредством определения флуоресцентной пробы, DPH, при использовании спектрофлуорометрии: Tm = 54°С.
Заявители получили трубчатые везикулы, средний гидродинамический диаметр которых, измеренный дифракцией света, составлял 132 нм (IP: 0,35).
Измерения формы и размера, полученные на основе анализа электронных микрофотографий (фиг.4 и 5) дали возможность продемонстрировать образование трубчатых везикул, закрытых на своих концах, среднее поперечное сечение которых составляет 38 нм и средняя длина которых равна 247 нм. Анализ замораживанием-скалыванием подтвердил образование этих трубчатых везикул и их морфологические характеристики, т.е. наличие внутренней водной полости, изолированной от внешней среды.
Эти трубчатые везикулы имеют высокую стабильность, поскольку не наблюдается изменение размера частицы после одного года хранения, тогда как при тех же условиях липосомы, образованные из фосфатидилхолина яичного желтка, изменялись всего лишь через 5 дней.
Доказательства наличия водной внутренней полости проводились опосредованным образом путем спектрофлуорометрических измерений инкапсулирования и кинетики высвобождения гидрофильной флуоресцентной пробы, карбоксифлуоресцеина (фиг.1). Измерения непосредственным образом показывают более медленную кинетику высвобождения флуоресцентной пробы по сравнению с обычным инкапсулированием в смеси фосфатидилхолина яичного желтка.
Пример 6: Инкапсулирование карбоксифлуоресцеина в трубчатых везикулах, полученных из производного А1
Способность к инкапсулированию различных соединений определяли с использованием флуоресцентного зонда: 5 (6)-карбоксифлуоресцеина. Измеряли высвобождение данной флуоресцентной метки из везикул, полученных согласно стандартизированным способам.
Указанное исследование требует получения Tris буфера (15 мМ и 150 мМ NaCl) при рН 7,4, а также 120 мМ раствора карбоксифлуоресцеина, также при рН 7,4.
Исследованные соединения взвешивали (2,5 мг/мл) и растворяли в минимальном количестве метанола. Растворитель испаряли при пониженном давлении в роторном испарителе и полученную таким образом пленку сушили в потоке азота.
Добавляли 120 мМ раствора карбоксифлуоресцеина в Tris буфере, так чтобы получить дисперсию липидов, имеющую концентрацию, равную 2,5 мг/мл. Полученную суспензию перемешивали при использовании вихревой мешалки в течение 1 минуты и затем подвергали ультразвуковой обработке в ультразвуковой бане в течение 1 часа при 70°С.
Неинкапсулированную флуоресцентную пробу удаляли пропусканием через колонку Sephadex G25, предварительно уравновешенную Tris буфером. Собранную фракцию везикул немедленно исследовали спектрофлуорометрией.
Флуоресцентные измерения проводили при использовании спектрофлуориметра Jobin-Yvon (spectrofluoromax 2), оснащенного 150 Вт ксеноновой лампой. Все измерения проводили в кварцевой кювете, термостатированной при 25°С. Образцы анализировали при длине волны возбуждения 480 нм и испускаемой длине волны 530 нм в течение 4 часов.
Полосу пропускания фиксировали на 0,5 нм как для возбуждения, так и для испускания.
Для каждого измерения начальную интенсивность флуоресценции (F0) определяли через 30 минут после фильтрации через колонку. Высвобождение исследовали через 4 ч и измеряли интенсивность флуоресценции (Ft) через регулярные промежутки времени. Максимальной интенсивности флуоресценции (Fmax), которая соответствует 100% высвобождению, достигали после распада трубчатых везикул, достигаемого добавлением Triton Х100 (10% об./об.).
Процент высвобождения определяли на основе следующего уравнения (фиг.1):
%R=(F t -F o )/F max
Пример 7: Полимеризация мономера, инкапсулированного в трубчатых везикулах, полученных из липида А1.
Состав используемых растворов суммирован в Таблице 1.
• Получение пленки
20 мг липидов А1 растворяли в 2 мл раствора гидропероксида кумола в дихлорметане, который был свежеперегнанным и обезгаженным путем барботирования аргона (2,5×10-4 М) в сердцевидной круглодонной колбе.
Раствор испаряли досуха в роторном испарителе (температура бани не превышала 40оС) и пленку затем сушили при помощи лопастного насоса (1 час) и до использования помещали в инертную атмосферу. В случае инициирования дитионитом натрия пленку получали в дихлорметане, который был свежеперегнанным и обезгаженным путем барботирования аргона.
• Дисперсия
2 мл раствора мономера добавляли в дистиллированную воду (0,1 М), предварительно дезоксигенированную барботированием аргоном. Смесь перемешивали в течение 1 минуты и затем помещали на ультразвуковую баню на 60 минут при 70°С.
• Разделение-полимеризация
Полученную дисперсию загружали в колонку Sephadex G50 (2 см в диаметре с заполнением геля на высоту 10 см), предварительно уравновешенную 0,1 М NaCl буфером, который дезоксигенировали барботированием аргона в течение 30 минут.
2 мл голубоватой фракции, отвечающей элюату от 25 до 27 мл, собирали в круглодонную колбу. Затем добавляли 2 мл раствора метабисульфита натрия в NaCl буфере (2,5×10-4 М), так чтобы инициировать полимеризацию, которая протекает в инертной атмосфере при 37°С и в течение 1 (акриламид) и 3 (ТНАМ) часов.
В случае инициирования дитионитом натрия после отделения на колонке Sephadex добавляли 2 мл раствора инициатора в 0,1 М NaCl буфере (2,5×10-3 М).
Пример 8: Теломеризация мономера, инкапсулированного в трубчатые везикулы, полученные из липида А1
Состав используемых растворов суммирован в Таблице 2.
• Получение пленки
18 мг липида А1 и 2 мг телогенного соединения D растворяли в 2 мл раствора гидропероксида кумола в дихлорметане, который был свежеперегнанным и обезгаженным путем барботирования аргона (2,5×10-4 М) в сердцевидной круглодонной колбе.
Раствор испаряли досуха в роторном испарителе (температура бани не превышала 40оС) и пленку затем сушили при помощи лопастного насоса (1 час) и до использования помещали в инертную атмосферу. В случае инициирования дитионитом натрия пленку получали в дихлорметане, который был свежеперегнанным и обезгаженным путем барботирования аргона.
• Дисперсия
2 мл раствора мономера добавляли в дистиллированную воду (0,1 М), предварительно дезоксигенированную барботированием аргоном. Смесь перемешивали в течение 1 минуты и затем помещали на ультразвуковую баню на 60 минут при 70°С.
• Разделение-полимеризация
Полученную дисперсию загружали в колонку Sephadex G50 (2 см в диаметре с заполнением геля на высоту 10 см), предварительно уравновешенную 0,1 М NaCl буфером, который дезоксигенировали барботированием аргона в течение 30 минут.
2 мл голубоватой фракции, отвечающей элюату от 25 до 27 мл, собирали в круглодонную колбу. Затем добавляли 2 мл раствора метабисульфита натрия в NaCl буфере (2,5×10-4 М), так чтобы инициировать теломеризацию, которая протекает в инертной атмосфере при 37°С и в течение от 16 до 20 часов. В случае инициирования дитионитом натрия, после отделения на колонке Sephadex добавляли 2 мл раствора инициатора в 0,1 М NaCl буфере (2,5×10-3 М).
Пример 9: Синтез поверхностно-активного вещества общей формулы (IB)
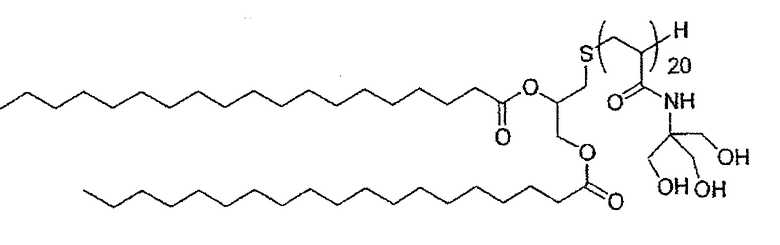
Соединение ТЕ 17-20
• рац-3-(тритилмеркапто)пропан-1,2-диол (Соединение 10)
10 г (92,6 ммоль) 3-меркапто-1,2-пропандиола и 13,54 мл (97,23 ммоль, 1,05 экв.) триэтиламина (ТЕА) растворяли в 100 мл ТГФ. 27,07 г (97,23 ммоль, 1,05 экв.) трифенилметилхлорида растворяли в 10 мл ТГФ и добавляли к смеси прикапыванием при температуре ниже 30°С. В конце добавления реакционную смесь оставляли на холоде при перемешивании и поддержании рН при 8-9 путем добавления ТЕА. Избыток трифенилметилхлорида удаляли путем добавления насыщенного раствора NaHCO3 перед испарением ТГФ при пониженном давлении. Неочищенный продукт поглощали CH2Cl2 перед тем как промыть нормальным раствором HCl и затем NaHCO3 и перед высушиванием над Na2SO4. Продукт окончательно очищали хроматографией на колонке с силикагелем, элюируя с градиентом (смесь циклогексан/ EtOAc от 7:3 до 1:1). Было получено 28,8 г чистого продукта в форме белого порошка. Rf продукта = 0,4 (ТСХ - 7:3 смесь EtOAC/ циклогексан). Выход: 89%. Т. пл.: 97-98°С.
1Н ЯМР(/CDCl3): δ(м.д.) 7,42-7,27(м, 15Н, ароматика тритила); 3,50(м, 3Н, СНОН, СН 2ОН); 2,80(м, 1Н, ОН); 2,50(т, 3Н, ОН, CH 2S).
13C ЯМР(CDCl3): δ(м.д.) 144,6(SCC фенил х3); 129,6(Сраra фенил х3); 128,0(Cortho фенил х6); 126,8(Cmeta фенил х6); 70,6(СНОН); 67,0(SCPh3); 65,5(CH2OH); 35,4(CH2S).
• рац-3-(тритилмеркапто)пропан-1,2-диилдигептадеканоат (соединение 11)
0,5 г (1,43 ммоль) соединения 10 и 0,523 г (4,28 ммоль, 3 экв.) диметиламинопиридина (DMAP) растворяли в 10 мл CH2Cl2 и затем охлаждали при использовании ледяной бани. Затем добавляли 0,952 г (3,14 ммоль, 2,2 экв.) стеароилхлорида, растворенного в 5 мл CH2Cl2 прикапыванием при использовании делительной воронки. После того, как добавление завершали, реакционную среду оставляли на 2 часа при перемешивании. Образованные соли DMAP удаляли фильтрацией и неочищенную реакционную массу затем испаряли досуха и в конце поглощали смесью MeOH/Et2O, из которой продукт кристализовали. Было получено 1,07 г чистого продукта в форме белого порошка. Rfпродукта = 0,3 (ТСХ - 7:3 смесь EtOAC/циклогексан). Выход: 85%. Т. пл.: 52-53°С.
1Н ЯМР(CDCl3): δ(м.д.) 7,30-7,15(м, 15Н, ароматика тритила); 5,16(квинт., 1/3Н, СНО); 4,78(квинт., 2/3Н, СНО); 4,1(дддд, 2Н, CH2O); 3,10(т, 2/3Н, CH 2S); 2,48(т, 4/3Н, CH 2S); 2,21(м, 4Н, CH 2CO х2); 1,30(м, 60Н, (СН2)15); 0,88(т, 6Н, СН 3)
13C ЯМР(/CDCl3): δ(м.д.) 172,2(OCOCH2 х2); 144,4(SCC фенил х3); 129,7(Cpara фенил х3); 128,2(Cortho фенил х6); 127,1(Cmeta фенил х6); 70,2(СНО); 67,4(SCPh3); 64,1(CH2O); 34,2(СН2СОО х2); 32,6(CH2S); 29,5((СН2)n х2); 24,9(CH2CH2COO х2); 22,7(СН2СН3 х2); 14,1(СН3 х2).
• рац-3-меркаптопропан-1,2-диил дигептадеканоат (соединение 12)
При использовании 0,5 г (0,56 ммоль) соединения 11 и 0,066 г (0,56 ммоль, 1 экв.) триэтилсилана после добавления 5% раствора ТФУ в CH2Cl2 и обычного промывания неочищенную реакционную массу испаряли досуха и очищали кристаллизацией из смеси MeOH/Et2O. Таким образом, выделяли 0,326 г чистого продукта в форме белого порошка. Rfпродукта = 0,5 (ТСХ - 7:3 смесь EtOAC/циклогексан). Выход: 90%. Т. пл.: 49-51°С.
1Н ЯМР(/CDCl3): δ(м.д.) 5,1(м, 1Н, СНО); 4,31(ддд, 2Н, CH2O); 3,10(т, 2/3Н, CH 2S); 2,73(т, 4/3Н, CH 2S); 2,30(м, 4Н, CH 2CO х2); 1,47(SH); 1,25(м, 60Н, (СН2)15 x2); 0,88(т, 6Н, СН 3 x2)
13C ЯМР(/CDCl3): δ(м.д.) 172,5(CH2OCO CHOCO); 71,8(CHO); 62,4(CH2O); 34,2(СН2СОО х2); 32,4(CH2SH); 29,5((СН2)n х2); 24,9(CH2CH2COO х2); 22,7(СН2СН3 х2); 14,1(СН3 х2).
Соединение ТЕ 17-20
1,09 г (6,25 ммоль, 5 экв.) трис(гидроксиметил)акриламидометана растворяли в 15 мл свежеперегнанного МеОН. Смесь перемешивали и барботировали аргоном и затем нагревали. Как только достигали температуры кипения, впрыскивали раствор, содержащий 0,04 г (0,25 ммоль, 0,2 экв.) AIBN и 0,8 г (1,25 ммоль) Соединения 12 в минимальном количестве свежеперегнанного ТГФ (приблизительно 1 мл), предварительно дегазированного потоком аргона. Реакцию отслеживали при помощи тонкослойной хроматографии) с исчезновением тиола (7:3 смесь этилацетат/циклогексан). После охлаждения до комнатной температуры неочищенную реакционную среду погружали в холодный Et2O при активном перемешивании, из которой осаждался теломер. Получали 1,2 г продукта в форме белого порошка. Выход: 70%.
1Н ЯМР(/ДМСО): δ (м.д.) 7,23(м, 19,8Н, NH xDPN); 4,78(м, 60Н, OH x3xDPn); 0,86(м, 6Н, СН3 x2).
| название | год | авторы | номер документа |
|---|---|---|---|
| АМИНОКИСЛОТЫ-ЛИПИДЫ | 2013 |
|
RU2670618C2 |
| НАЦЕЛИВАЮЩИЕ АМИНОКИСЛОТНЫЕ ЛИПИДЫ | 2013 |
|
RU2654210C2 |
| ПРОИЗВОДНЫЕ НА ОСНОВЕ ГИАЛУРОНОВОЙ КИСЛОТЫ, СПОСОБНЫЕ ОБРАЗОВЫВАТЬ ГИДРОГЕЛИ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ГИДРОГЕЛИ НА ОСНОВЕ УКАЗАННЫХ ПРОИЗВОДНЫХ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2013 |
|
RU2586931C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ АНТАГОНИСТЫ РЕЦЕПТОРА ВИТРОНЕКТИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ЛЕКАРСТВЕННОГО СРЕДСТВА И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, ИХ СОДЕРЖАЩИЕ | 2003 |
|
RU2412185C2 |
| СОДЕРЖАЩИЕ АЗОТ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ ИХ В КАЧЕСТВЕ АНТИБАКТЕРИАЛЬНЫХ ЛЕКАРСТВЕННЫХ СРЕДСТВ | 2008 |
|
RU2465276C2 |
| ПРОИЗВОДНЫЕ ИМИДАЗОЛОНОВ, СПОСОБ ПОЛУЧЕНИЯ И БИОЛОГИЧЕСКИЕ ПРИМЕНЕНИЯ | 2008 |
|
RU2491283C2 |
| 5-СУЛЬФАНИЛ-4Н-1,2,4-ТРИАЗОЛЬНЫЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ЛЕКАРСТВЕННОГО ПРЕПАРАТА | 2002 |
|
RU2367655C2 |
| СИСТЕМА ДОСТАВКИ НУКЛЕИНОВЫХ КИСЛОТ | 2002 |
|
RU2294192C2 |
| ФУНКЦИОНАЛИЗИРОВАННЫЕ ТОНКОПЛЕНОЧНЫЕ ПОЛИАМИДНЫЕ МЕМБРАНЫ | 2009 |
|
RU2519377C9 |
| КОМПЛЕКСЫ ТЕХНЕЦИЯ И РЕНИЯ С БИС(ГЕТЕРОАРИЛАМИ) И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2009 |
|
RU2539584C2 |





Настоящее изобретение относится к новым поверхностно-активным соединениям формулы III и их использованию для получения наночастиц, служащих в качестве векторов для биологически активных ингредиентов.


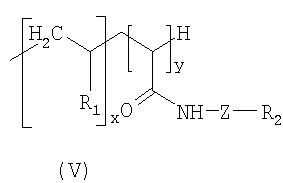
где R3 выбран из групп IV или V; Y - атом серы или -NH-CO-(CH2)n-X-; Х - атом серы или -СН2-; n и y целые числа от 0 до 10; R - углеводород, фторированный углеводород или тиоалкил; W - группа -NH- или -СН2-; p - целое число от 1 до 50; m - целое число от 0 до 9; и если Х=СН2, тогда 0<m+n<6; x - целое число от 0 до 30; R1 - выбран из следующих радикалов:
где R' представляет собой Н или полигидроксилированный углеводород; R2 - группа распознавания, обладающая сродством к биологической мишени; Z - спейсерная группа. Технический результат - разработка новых поверхностно-активных соединений и наночастиц на их основе. 4 н. и 18 з.п. ф-лы, 2 табл., 9 ил.
1. Соединение формулы (III):
в которой R3 представляет собой группу, выбранную из:
 и
и 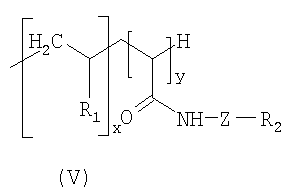
Y представляет собой атом серы или группу -NH-CO-(CH2)n-X-, X представляет собой атом серы S или группу -CH2-, n представляет собой целое число в диапазоне от 0 до 10;
R представляет собой группу, выбранную из радикалов на основе С4-С24-углеводорода; радикалов на основе фторированного С4-С24-углеводорода; С4-С24-тиоалкильных радикалов;
W представляет собой группу -NH- или -СН2-;
p представляет собой целое число в диапазоне от 1 до 50;
m представляет собой целое число, изменяющееся в диапазоне от 0 до 9;
и если Х=СН2, тогда 0<m+n<6;
х представляет собой целое число в диапазоне от 0 до 30;
y представляет собой 0 или целое число в диапазоне от 1 до 10;
R1 представляет собой группу, выбранную из следующих радикалов:
в которых R' представляет собой Н или С4-С24-полигидроксилированное соединение на основе углеводорода;
R2 представляет собой группу распознавания, обладающую сродством к биологической мишени и выбранную из: сахаридов, гормонов, синтетических молекул, антител, фрагментов антитела, биотина, пептидов, небольших молекул-эффекторов, которые делают возможным взаимодействие с рецепторами клеточной поверхности, и, если происходит связывание за счет образования комплекса с атомом никеля, Z-R2 группа состоит из группы NTA формулы:
Z представляет собой спейсерную «ножку» (spacer arm); Z связана с R2 при помощи связи, которая может быть выбрана из функций -O-СО-, -CO-NH-, -NH-CO-NH-, -NH-CO-O-, O-СО-O-, -O-, -CH=N- или -S- или за счет образования комплекса с атомом никеля; Z может быть выбрана из белковой цепи, Q-аминокислот, этаноламина, 3-пропаноламина и диаминов формулы -NH-(CH2)p'NH-, в которых р' представляет собой целое число в диапазоне от 2 до 6, или группа -Z-R2 представляет собой группу NTA формулы:
или соединение формулы III представляет собой соединение формулы F:
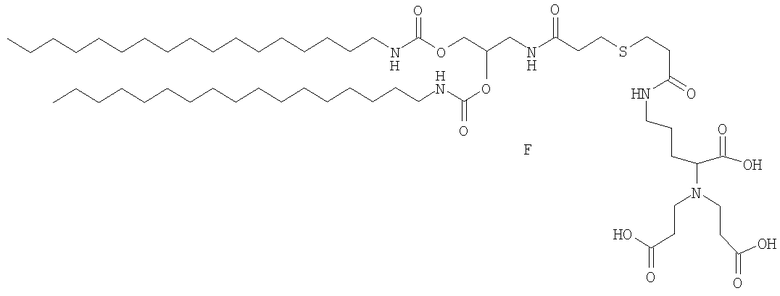 .
.
2. Соединение по п.1, отличающееся тем, что группу R выбирают из следующих радикалов:
тиооктильного радикала,
н-бутила, трет-бутила, изобутила, н-пентила, изопентила, н-гексила, н-гептила, н-октила, н-нонила, н-децила, н-ундецила, н-додецила, н-тридецила, н-тетрадецила, н-пентадецила, н-гексадецила, н-гептадецила, н-октадецила или фитильного радикала (СН3[СН(СН3)(СН2)3]3СН(СН3)СН2СН2),
радикалов на основе фторированного углеводорода, отвечающих формуле -(CH2)t(CF2)rF, в которой г и t оба представляют собой целые числа при условии: 14≥r+t≥4.
3. Соединение по п.1, отвечающее формуле (I):
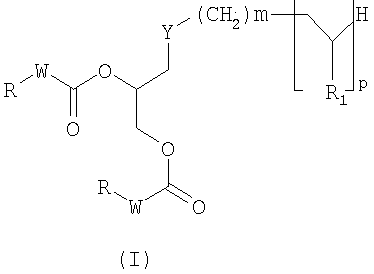
в которой:
Y представляет собой атом серы или группу
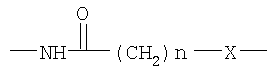
причем Х выбирают из групп S и СН2, n представляет собой целое число, изменяющееся в диапазоне от 0 до 10;
m представляет собой целое число, изменяющееся в диапазоне от 0 до 9; и если Х=СН2, тогда 0<m+n<6;
W представляет собой группу -NH- или группу -СН2-;
p представляет собой целое число, изменяющееся в диапазоне от 1 до 50;
R1 представляет собой группу, выбранную из следующих радикалов:
В которых R' представляет собой Н или С4-С24-полигидроксилированное соединение на основе углеводорода;
R представляет собой группу, выбранную из: радикалов на основе С4-С-24-углеводорода; радикалов на основе фторированного С4-С24-углеводорода; С4-С24-тиоалкильных радикалов.
4. Соединение по п.3, отличающееся тем, что оно отвечает формуле (IA):
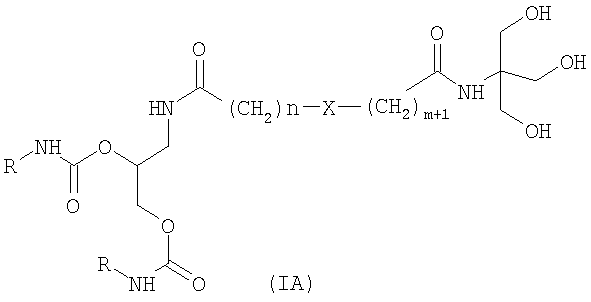
в которой:
Х представляет собой атом S или группу -СН2-;
n представляет собой целое число, изменяющееся в диапазоне от 0 до 10;
m представляет собой целое число, изменяющееся в диапазоне от 0 до 9;
если Х=СН2, тогда 0<m+n<6;
R представляет собой группу, выбранную из: радикалов на основе С4-С24-углеводорода; радикалов на основе фторированного С4-С24-углеводорода; С4-С24-тиоалкильных радикалов.
5. Соединение по п.4, отличающееся тем, что R выбрана так, чтобы соединение (IA) имело температуру фазового перехода выше 37°С.
6. Соединение по п.4 или 5, отличающееся тем, что оно отвечает формуле А:
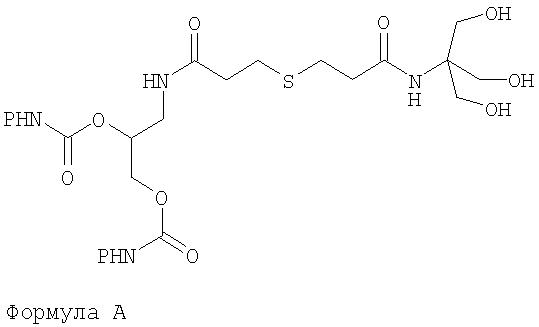
где R представляет собой группу, выбранную из: радикалов на основе С4-С24-углеводорода; радикалов на основе фторированного С4-С24-углеводорода; С4-С24-тиоалкильных радикалов.
7. Соединение по п.6, отличающееся тем, что оно отвечает формуле А1:
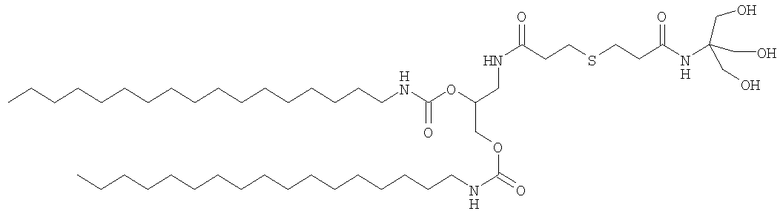
Соединение А1
8. Соединение по п.3, отвечающее формуле (IB):
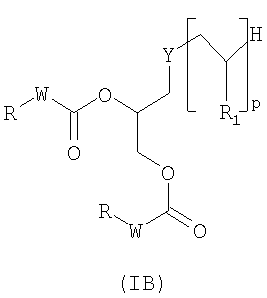
в которой:
Y представляет собой атом серы или группу -NH-CO-CH2CH2S-;
W представляет собой группу -NH- или группу -СН2-;
p представляет собой целое число, изменяющееся в диапазоне от 1 до 50;
R1 представляет собой группу, выбранную из следующих радикалов:
в которых R' представляет собой Н или С4-С24-полигидроксилированное соединение на основе углеводорода;
R представляет собой группу, выбранную из: радикалов на основе С4-С24-углеводорода; радикалов на основе фторированного С4-С24-углеводорода; С4-С24-тиоалкильных радикалов.
9. Соединение по п.8, отличающееся тем, что R выбрана так, чтобы соединение (IB) имело критическую мицеллярную концентрацию менее чем 10-5 М.
10. Соединение по п.8, отличающееся тем, что оно удовлетворяет одному или нескольким из указанных ниже условий:
p представляет собой целое число в диапазоне от 1 до 15;
Y представляет собой S.
11. Соединение по любому из пп.8-10, отличающееся тем, что оно отвечает формуле С, в которой p представляет собой число в диапазоне от 5 до 15:
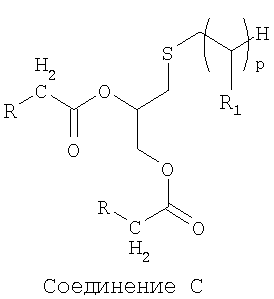
где R представляет собой группу, выбранную из: радикалов на основе С4-С24-углеводорода; радикалов на основе фторированного С4-С24-углеводорода; С4-С24-тиоалкильных радикалов.
12. Соединение по п.11, отличающееся тем, что оно отвечает формуле С1:
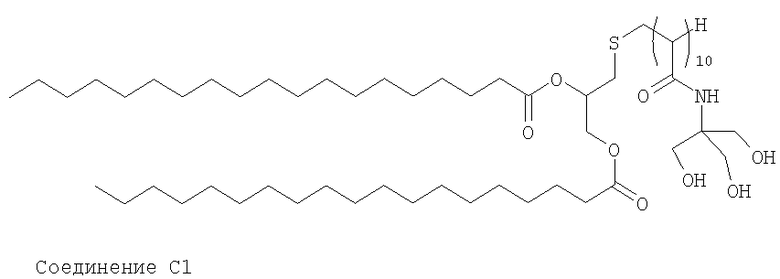
13. Соединение по п.1, отличающееся тем, что оно отвечает формуле (II):
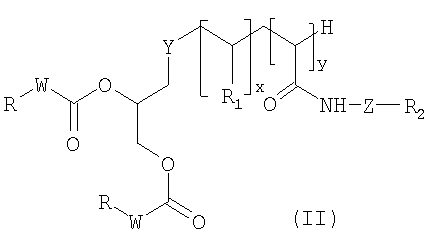
в которой:
Y представляет собой атом серы или группу -NH-CO-(CH2)n-X-, в которой Х представляет собой атом серы S или группу -СН2-, n представляет собой целое число в диапазоне от 0 до 10;
R представляет собой группу, выбранную из радикалов на основе С4-С24-углеводорода; радикалов на основе фторированного С4-С24-углеводорода; С4-С24-тиоалкильных радикалов;
W представляет собой группу -NH- или -СН2-;
x представляет собой целое число в диапазоне от 0 до 30;
y представляет собой 0 или целое число в диапазоне от 1 до 10;
R1 представляет собой группу, выбранную из следующих радикалов:
в которых R' представляет собой Н или С4-С24-полигидроксилированное соединение на основе углеводорода;
R2 представляет собой группу распознавания, обладающую сродством к биологической мишени и выбранную из сахаридов, гормонов, синтетических молекул, антител, фрагментов антитела, биотина, пептидов, небольших молекул-эффекторов, которые делают возможным взаимодействие с рецепторами клеточной поверхности, и, если происходит связывание за счет образования комплекса с атомом никеля, Z-Rz группа состоит из группы NTA формулы:
;
Z представляет собой спейсерную «ножку»; Z связана с R2 при помощи связи, которая может быть выбрана из функций -O-СО-, -CO-NH-, -NH-CO-NH-, -NH-CO-O-, O-СО-O-, -O-, -CH-N- или -S- или за счет образования комплекса с атомом никеля;
Z может быть выбрана из белковой цепи, Ω-аминокислот, этаноламина, 3-пропаноламина и диаминов формулы -NH-(CH2)p' NH-, в которых p' представляет собой целое число в диапазоне от 2 до 6, или группа -Z-R2 представляет собой группу NTA формулы:
14. Соединение по п.13, отличающееся тем, что оно отвечает формуле (IIА):
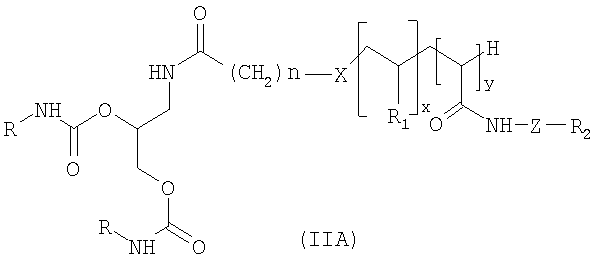
где R, R1, Z, R2, x, y, n определены в п.13.
15. Соединение по п.14, отличающееся тем, что выполняется одно или несколько из следующих условий:
X=S,
n=2
R1 выбирают из следующих радикалов:
в которых R' представляет собой Н или С4-С24-полигидроксилированное соединение на основе углеводорода;
R выбирают из следующих радикалов:
тиооктильного радикала,
н-бутила, трет-бутила, изобутила, н-пентила, изопентила, н-гексила, н-гептила, н-октила, н-нонила, н-децила, н-ундецила, н-додецила, н-тридецила, н-тетрадецила или фитильного радикала (СН3[СН(СН3)(СН2)3]3СН(СН3)СН2СН2),
фторированных радикалов на основе углеводорода, отвечающих формуле -(CH2)t-(CF2)rF, в которой г и t представляют собой два целых числа при условии: 14≥r+t≥4,
R2 выбирают из: сахаридов, гормонов, синтетических молекул, антител, фрагментов антитела, биотина, пептидов, небольших молекул-эффекторов, которые делают возможным взаимодействие с рецепторами клеточной поверхности, и, если происходит связывание за счет образования комплекса с атомом никеля, Z-R2 группа состоит из группы NTA формулы:
.
16. Соединение по п.1, отличающееся тем, что оно отвечает формуле F:
Формула F
17. Соединение по п.13, отличающееся тем, что оно отвечает формуле (IIB):
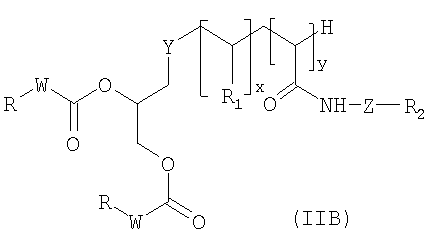
в которой:
Y представляет собой атом серы или группу -NH-CO-CH2CHzS-;
R, R1, R2, N, Z, х, у определены в п.13.
18. Наночастицы, представляющие собой трубчатые везикулы, для транспортировки терапевтически активных ингредиентов, в которых составной частью их стенок являются одно или несколько соединений формулы (I) по любому из пп.3-12.
19. Наночастицы по п.18, отличающиеся тем, что они также включают от 1 до 5% одного или нескольких соединений формулы (II) по любому из пп.13-17 в качестве составляющей части стенок трубчатых везикул.
20. Наночастицы по любому из пп.18 и 19, отличающиеся тем, что они также содержат теломер или полимер, образованный во внутренней полости трубчатых везикул из мономера акрилового типа.
21. Комбинация наночастиц по любому из пп.18-20 с соединением, выбранным из: терапевтически активных ингредиентов, косметических веществ, диагностических агентов и вакцин.
22. Терапевтическая, диагностическая, вакцинная или косметическая композиция для транспортировки терапевтически активных ингредиентов, включающая по меньшей мере один активный ингредиент в комбинации с наночастицами по любому из пп.18-20.
| СПОСОБ ПОЛУЧЕНИЯ ЛИПОСОМ | 1994 |
|
RU2071765C1 |
| US2003017976 А1, 23.01.2003 | |||
| FUMIO ITOH ET ALL, Chem | |||
| Pharm | |||
| Bull., 1998, 46(2), 255-273 | |||
| TSUNEAKI HIDA ET ALL, 1995, vol | |||
| Приспособление для автоматической односторонней разгрузки железнодорожных платформ | 1921 |
|
SU48A1 |
| MUNEAKI KURIMURA ET ALL, Chem | |||
| Pharm | |||
| Bull., 1993, 41(11), 1965-1970 | |||
| MUNEAKI KURIMURA ET ALL, Chem | |||
| Pharm | |||
| Bull., 1991, 39(10), 2590-2596 | |||
| MUNEAKI KURIMURA ET ALL, Chem | |||
| Pharm | |||
| Bull., 1990, 38(4), 1110-1112. | |||

