Изобретение относится к фармацевтике, а именно к лекарственным средствам и препаратам, а также к способам их получения. Более конкретно изобретение касается парентеральных лекарственных средств, обладающих улучшенными фармакологическими показателями, в частности лучшей растворимостью в водных средах, улучшенной биодоступностью действующего вещества, пониженной токсичностью, пролонгированным действием и др.
Среди лекарственных средств существует много веществ, которые в силу своей химической природы обладают недостаточно хорошими фармакологическими показателями, например, имеют низкую растворимость в воде (липофильные соединения) и низкую биодоступность, проявляют значительную токсичность, быстро выводятся из организма, что приводит к необходимости частого приема или инъекций препарата для поддержания концентрации действующего вещества на терапевтическом уровне, и, как следствие, к увеличению вероятности проявления побочного действия и т.д. Все это в совокупности накладывает ряд ограничений на использование таких препаратов пациентами [Машковский М.Д. Лекарственные средства. М.: «Новая Волна», 2005, 1200 с.; Харкевич Д.А. Фармакология. М.: «Гэотар Медицина», 2006, 736 с.; Регистр лекарственных средств России. Энциклопедия лекарств. Издание 15-е. М.: «РЛС-2007», 2006. 1488 с.]. В этой связи разработка лекарственных средств, обладающих улучшенными фармакологическими показателями, позволяющих достичь более высокой эффективности при медикаментозном применении, является актуальной задачей.
Например, известно парентеральное лекарственное средство и способ получения такого средства в виде биоразлагаемой композиции, обладающей пролонгированным высвобождением биологически активного соединения [Патент РФ 2290950, 2007]. Это техническое решение связано с преодолением слишком быстрого выведение лекарства из организма. Лекарственное средство состоит из биоразлагаемого носителя, который представляет собой продукт высокотемпературной (120-220°С) поликонденсации многоатомного спирта (в частности, глицерина, пентаэритрита, маннита, сорбита) и алифатической альфа-гидроксикислоты (например, DL-молочной, гликолевой, альфа-гидроксимасляной и др.), при мольном соотношении сомономеров от 0,5:99,5 до 12:88; кроме того, композиция включает действующее вещество, выбранное из противовоспалительных нестероидных, противомикробных, противоопухолевых и других средств, а также жидкий пластификатор (трибутилцитрат, трибутирин, триэтилцитрат и др.), который вводится в массовом соотношении с носителем от 1:20 до 9:10. Способ получения данной композиции предполагает интенсивное перемешивание носителя, пластификатора и действующего вещества при температуре до 75°С с образованием однородной смеси. Такую смесь фасуют в ампулы с последующей стерилизацией гамма-излучением и хранят в холодильнике. Биотестирование на мышах лекарственного средства, содержащего в качестве действующего вещества цисплатин и полученного согласно данному техническому решению, показывает улучшение терапевтического эффекта (снижение темпа роста и массы опухоли), а также обнаруживает эффект пролонгированного высвобождения противоопухолевого агента. К недостаткам данного технического решения относятся нежелательные процессы, которые имеют место при высокой температуре на стадиях получения как полимерного носителя, так и конечной композиции, что значительно повышает опасность образования продуктов термодеструкции компонентов композиции, некоторые из которых могут оказаться токсичными для пользователей. Кроме того, препарат перед внутриопухолевым введением необходимо нагревать до 50°С, что делает его малопригодным в терапии человека.
Известны существенно более мягкие условия приготовления такого потенциального лекарственного средства, как стабилизированная форма динитрозильного комплекса железа (ДНКЖ) [Патент РФ 2291880, 2007]. Эта стабилизированная форма ДНКЖ обладает выраженным гипотензивным действием при парентеральном применении. Согласно данному техническому решению способ получения указанной формы заключается в том, что смешивают компоненты композиции (ДНКЖ 0,1-6,0 мас.%; полимерный стабилизатор 1-20 мас.%; вода до 100 мас.%), замораживают полученную композицию при -10…-196°С в течение 0,1-24 ч и высушивают лиофильно до остаточной влажности 0,1-2,0%. In vivo тестирование на крысах выявляет одинаковые по абсолютной величине эффекты снижения артериального давления как в случае свежеприготовленного ДНКЖ, который стабилен всего не более 30 мин при комнатной температуре, так и в случае стабилизированной полимерной формы ДНКЖ, которая хранилась при +25°С в течение двух месяцев, но при этом действие последней проявляется на протяжении более длительного (в 2-3 раза) времени, т.е. имеет место четко выраженный пролонгирующий эффект. Однако это известное техническое решение применимо только в отношении лекарственных веществ, хорошо растворимых в воде, т.е. является недостаточно универсальным. В случае же липофильных лекарственных субстанций таким путем нельзя получить соответствующую композицию, поскольку наблюдается либо коагуляция полимерного стабилизатора, либо неконтролируемое осаждение липофильного компонента.
Примером решения проблемы низкой растворимости липофильных лекарственных субстанций является известное лекарственное средство, содержащее плохо растворимое в воде действующее вещество (например, паклитаксел, тенипозид и др.) и солюбилизирующий агент, представляющий собой смесь (50:50 по объему) абсолютного этилового спирта и неполимерной добавки - полиоксиэтилированного касторового масла (так называемый Кремафор EL) [Патент США 5504102, 1996]. Это техническое решение позволяет получать достаточно стабильные растворы липофильных веществ, допустимые к парентеральному применению. К недостаткам данного лекарственного средства относится наличие в его составе Кремафора EL, который приводит к экстракции ди(2-гексил)фталата - пластификатора в поливинилхлоридных (ПВХ) изделиях, используемых в биомедицинском оборудовании. В результате вредный для человека ди(2-гексил)фталат солюбилизируется и попадает в организм пациентов. Кроме того, согласно данным Патента РФ 2291693 (2007), полиоксиэтилированное касторовое масло при парентеральном введении в организм человека может оказывать побочное действие, например, повышать кровяное давление, вызывать отдышку, приводить к нервному истощению и др.
Одним из определяющих фармакологических показателей лекарственных средств является их токсичность, причем особенно эта проблема актуальна в отношении противоопухолевых фармпрепаратов, многие из которых крайне токсичны и, наряду с раковыми, поражают и здоровые клетки. Так, например, известно парентеральное противоопухолевое лекарственное средство на основе вещества растительного происхождения - паклитаксела. Данное лекарственное средство представляет собой наночастицы, сформированные из альбумина сыворотки крови человека, с включенным в них паклитакселом [Патент США 5916596, 1999] при соотношении альбумин/фармакологический агент = 9:1 мас.ч. Такое лекарственное средство получают прибавлением раствора паклитаксела в метиленхлориде или хлороформе к водному раствору альбумина сыворотки крови человека с последующей гомогенизацией смеси при избыточном давлении. Далее органический растворитель из полученной эмульсии удаляют на роторном испарителе, а остаток замораживают и высушивают лиофильно. Биотестирование [Патент США 6537579, 2003] полученного таким образом лекарственного средства на мышах и крысах показывает существенное снижение токсичности (до 10 раз) по сравнению с лекарственным средством, известным под торговым названием "Тахоl" (на 6 мас.ч. паклитаксела приходится 527 мас.ч. Кремафора EL и 400 мас.ч. абсолютного этилового спирта).
Данное техническое решение [Патент США 5916596, 1999], как наиболее близкое к заявляемому по набору компонентов, включающему фармакологический агент, водорастворимый полимер и растворитель, выбрано нами за прототип.
Однако прототип имеет ряд недостатков:
- применяются токсичные для внутренних органов человека (в частности, для печени) растворители - хлорорганические жидкости, хорошо сорбирующиеся различными полимерами. Используемый в способе-прототипе прием (эвакуирование на роторном испарителе) не обеспечивает полноту удаления метиленхлорида или хлороформа, особенно из адсорбционно-связанного полимером (альбумином) состояния;
- при эмульгировании органического раствора паклитаксела в водном растворе альбумина на границе раздела водной и органической фаз за счет неспецифических гидрофобных взаимодействий происходит денатурация белка, т.е. часть молекул альбумина теряет нативную конформацию, что нежелательным образом влияет на иммуногенность альбумина, а это, в свою очередь, может вызывать тяжелую аллергическую реакцию у пациентов с пониженным иммунитетом, например у раковых больных;
- способ получения конечного лекарственного средства требует сложного аппаратурного оформления процесса из-за необходимости работы при повышенном давлении на стадии эмульгирования;
- при реализации известного способа образуются вредные для окружающей среды отходы - отработанный хлорорганический растворитель.
Авторы данного изобретения предлагают новую фармацевтическую композицию для парентерального лекарственного средства и способ ее получения.
Ниже приведены определения и сокращения, использованные в описании изобретения.
Лекарственное средство - вещество или смесь веществ, предназначенное для лечения и предупреждения заболеваний.
Фармакологический агент - вещество, обладающее фармакологической активностью и используемое в качестве основного ингредиента лекарственного средства.
Нековалентный комплекс - комплекс, который образуется между молекулами веществ в подходящем растворителе за счет межмолекулярных взаимодействий нековалентной природы, а именно - водородного связывания, координационно-ионных, а также гидрофобных взаимодействий.
Остаточный растворитель - растворитель, который остается адсорбционно-связанным нековалентным комплексом после высушивания его раствора применяемым способом.
Фармакологический агент
I - ампициллин
II - блеомицин
III - дифенгидрамин
IV - добутамин
V - допамин
VI - доцетаксел
VII - ланреотид
VIII - октреотид
IX - паклитаксел
X - цефазолина натриевая соль
XI - этилметилгидроксипиридина сукцинат
XII - зидовудин
Водорастворимый полимер
А - альбумин сыворотки крови человека
В - 2-гидроксиэтилкрахмал
С - декстран
D - желатин
Е - поли(N-винилпирролидон)
F - смесь
Неполимерная добавка
α - (C10-C18)ацил-сорбит
β - олигооксиэтилен(C10-C18)ацил-сорбит
γ - (C10-C18)алкиловый эфир олигоэтиленгликоля
Растворитель
а - вода
б - диметилсульфоксид
в - N-метил-2-пирролидон
г - 2-пирролидон
д - этанол
** Метод высушивания:
ЛС - лиофильная сушка
PC - распылительная сушка
УП - упаривание в вакууме
*** Растворимость:
низк. - менее 1 г/л
сред. - 1-10 г/л
хор. - 10-100 г/л
выс. - более 100 г/л
**** в отн. ед. от стандарта (LD50)
н/о - не определяли
Технический результат настоящего изобретения заключается в создании новой фармацевтической композиции для получения парентерального лекарственного средства, обладающего набором улучшенных фармакологических показателей, таких как токсичность, продолжительность воздействия, биодоступность, и способа его получения.
Технический результат достигается тем, что фармацевтическая композиция для получения парентерального лекарственного средства включает фармакологический агент, водорастворимый полимер, неполимерную добавку и остаточный растворитель, где водорастворимый полимер выбран из группы: декстран, желатин, 2-гидроксиэтилкрахмал, поли(N-винилпирролидон), альбумин сыворотки крови человека или их смеси; неполимерная добавка выбрана из группы: (C10-C18)ацил-сорбит, олигооксиэтилен(C10-C18)ацил-сорбит, (C10-C18)алкиловый эфир олигоэтиленгликоля, причем фармакологический агент, водорастворимый полимер, неполимерная добавка и остаточный растворитель входят в состав фармацевтической композиции в виде нековалентного комплекса при следующем соотношении (мас.ч.): фармакологический агент 0,10-100, водорастворимый полимер 1-100, неполимерная добавка 0,001-100, остаточный растворитель 0,3-30.
Технический результат достигается также тем, что способ получения фармацевтической композиции заключается в том, что готовят композицию смешиванием фармакологического агента, водорастворимого полимера и неполимерной добавки с растворителем в следующих соотношениях (мас.ч.):
причем растворитель представляет собой смесь воды с этанолом, или диметилсульфоксидом, или N-метил-2-пирролидоном, или 2-пирролидоном при их объемном соотношении от 99,9:0,1 до 50:50, с последующим высушиванием композиции до содержания остаточного растворителя 0,3-30 мас.ч.
Технический результат достигается также тем, что парентеральное лекарственное средство для лечения и предупреждения заболеваний нервной системы, или заболеваний сердечно-сосудистой системы, или онкологических заболеваний, или вирусных заболеваний, или инфекционных заболеваний, включает в свой состав вышеуказанную фармацевтическую композицию.
Выбор компонентов и количественного состава заявляемой фармацевтической композиции, а также параметров способа ее получения обусловлен следующими факторами.
1) Используемый растворитель должен являться допущенным к медицинскому применению и позволять готовить растворы компонентов композиции при получении нековалентного комплекса; предлагается использование в качестве такого растворителя смеси воды с этанолом, или диметилсульфоксидом, или N-метил-2-пирролидоном, или 2-пирролидоном при их объемном соотношении от
99,9:0,1 до 50:50.
2) Компоненты заявляемой фармацевтической композиции, т.е. фармакологический агент, водорастворимый полимер и неполимерная добавка, входят в ее состав в виде нековалентного комплекса, что обеспечивает пролонгирующий эффект в отношении действия фармакологического агента, химическая структура которого при этом не изменяется, но для диссоциации такого нековалентного комплекса требуется определенное дополнительное время, зависящее от индивидуальных свойств компонентов лекарственного средства. При приготовлении заявляемой фармацевтической композиции фармакологический агент, водорастворимый полимер и неполимерная добавка подбираются таким образом, чтобы в вышеуказанном общем растворителе они могли образовывать нековалентный комплекс за счет межмолекулярных взаимодействий нековалентной природы: водородного связывания, координационно-ионных, а также гидрофобных взаимодействий. Такой подход, с одной стороны, обеспечивает неизменность химической структуры фармакологического агента, поэтому не влияет на природу физиологической активности, проявляемую им, а с другой стороны, неполимерная добавка и водорастворимый полимер выполняют солюбилизирующую функцию, тем самым, наряду с пролонгирующим эффектом, обеспечивают лучшую растворимость в водной среде фармакологического агента, и, как следствие, повышенную биодоступность. В ряде случаев (см. примеры реализации заявляемого технического решения) наряду с вышеописанными эффектами наблюдается понижение токсичности препарата, т.е. в целом имеет место заметное комплексное (многофакторное) улучшение фармакологических показателей получаемого парентерального лекарственного средства по сравнению с аналогами и прототипом.
3) Выбор конкретных представителей фармакологического агента обусловлен сравнительно низкими фармакологическими показателями, например недостаточной растворимостью в водных средах, низкой биодоступностью, повышенной токсичностью, слишком быстрым выведением из организма и др. Однако в качестве фармакологического агента в заявляемом изобретении могут использоваться также и вещества с хорошими фармакологическими показателями, поскольку заявляемое техническое решение может обеспечить их дальнейшее улучшение. Предлагаемое изобретение предусматривает использование фармакологического агента, предназначенного для лечения и предупреждения заболеваний нервной системы, или заболеваний сердечно-сосудистой системы, или онкологических заболеваний, или вирусных заболеваний, или инфекционных заболеваний.
4) В предлагаемом изобретении выбор водорастворимого полимера и неполимерной добавки обусловлен, с одной стороны, их химической природой, предопределяющей возможность образования нековалентного комплекса с конкретным фармакологическим агентом, а с другой стороны, их доступностью, безвредностью при парентеральном применении, сохранением свойств без изменения при длительном хранении и т.д. Заявляемое техническое решение предусматривает использование в качестве водорастворимого полимера таких природных и синтетических соединений, как декстран, желатин, 2-гидроксиэтилкрахмал, поли(N-винилпирролидон), альбумин сыворотки крови человека и их смесей, а в качестве неполимерной добавки - (C10-C18)ацил-сорбита, олигооксиэтилен(C10-C18)ацил-сорбита, (C10-C18)алкилового эфира олигоэтиленгликоля.
5) Количественный состав заявляемой фармацевтической композиции найден экспериментально и определялся как необходимая терапевтическая доза для конкретного фармакологического агента, так и технологическими особенностями реализации заявляемого способа, например, растворимостью компонентов композиции при приготовлении нековалентного комплекса «фармакологический агент -водорастворимый полимер - неполимерная добавка». Согласно предлагаемому техническому решению растворитель входит в состав в количестве 10-200 мас.ч., а остальные компоненты в количестве: 0,10-100 мас.ч. (фармакологический агент), 1-100 мас.ч. (водорастворимый полимер) и 0,001-100 мас.ч. (неполимерная добавка).
б) При реализации способа получения фармацевтической композиции для парентерального лекарственного средства, обладающего улучшенными фармакологическими показателями, сначала готовится композиция заявляемого состава, которая затем высушивается. Сушку осуществляют известными методами, например, упариванием, распылительной сушкой или лиофильной сушкой, что определяется свойствами (в частности, термостойкостью) конкретного фармакологического агента, а также летучестью используемого растворителя и, наконец, экономическими параметрами выбранного метода сушки. Заявляемый диапазон содержания остаточного растворителя в получаемом парентеральном лекарственном средстве определен экспериментально и находится в пределах 0,3-30 мас.ч. Получение препарата с меньшим чем 0,3 мас.ч. содержанием остаточного растворителя в принципе возможно, но это требует длительного вакуумирования и часто при повышенной температуре, что ведет к неоправданным избыточным энергозатратам. С другой стороны, более высокое чем 30 мас.ч. содержание остаточного растворителя в получаемом согласно предлагаемому изобретению лекарственном средстве нежелательно из-за опасности развития микрофлоры, а также размокания лекарственного средства в случае повышенной гигроскопичности его ингредиентов.
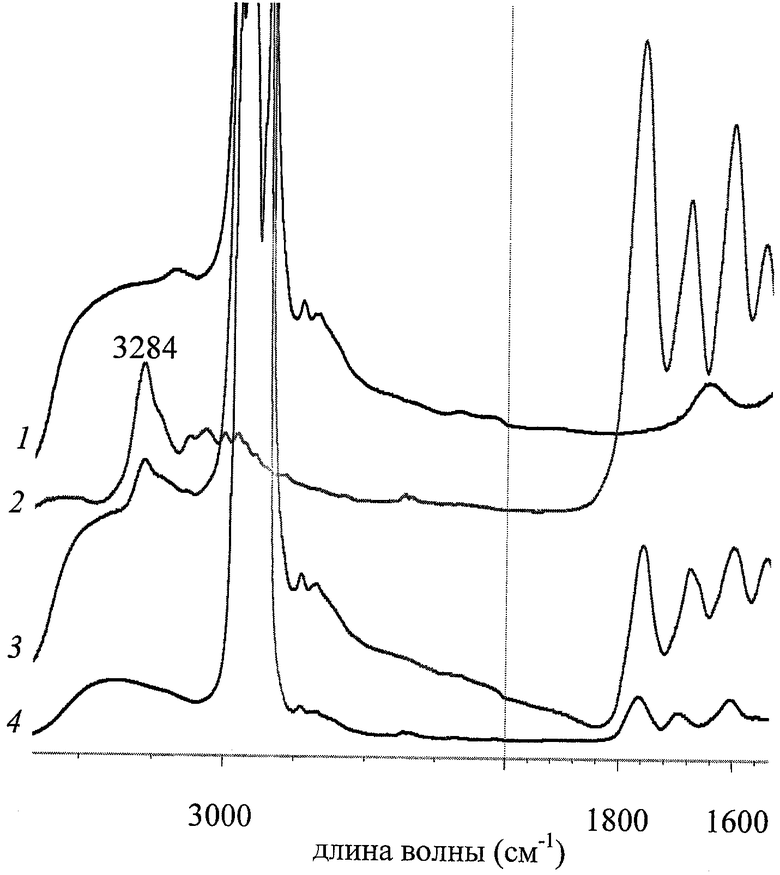
Изобретение поясняется чертежом: на чертеже представлены ИК спектры цефазолина натриевой соли (2), декстрана (1), механической смеси цефазолина натриевой соли, декстрана и додецил-сорбита (3), а также нековалентного комплекса «цефазолина натриевая соль - декстран - додецил-сорбита» (4).
Ниже приводится пример реализации заявляемого изобретения, который иллюстрирует, но не ограничивает его объем.
Пример 1
В 10 мас.ч. растворителя, содержащего 99,9 об. ч. воды и 0,1 об. ч. этанола, растворяют 0,5 мас.ч. цефазолина натриевой соли и 1 мас.ч. декстрана. Далее к полученному раствору добавляют 0,001 мас.ч. додецил-сорбита (α(C12) - в обозначении таблицы). Этот раствор замораживают и высушивают сублимационно (лиофильная сушка) до содержания остаточного растворителя ~0,3 мас.ч. (определено титрованием по методу К. Фишера). ИК-спектроскопические исследования (см.чертеж) подтверждают образование нековалентного комплекса «цефазолин - декстран - додецил-сорбит» за счет водородных связей: если спектр механической смеси (кривая 3) содержит все характерные полосы поглощения, в частности четкую полосу при 3284 см-1 валентных колебаний связей N-H амидных группировок цефазолина натриевой соли (кривая 2), то в ИК-спектре соответствующего нековалентного комплекса (кривая 4) эта полоса в явном виде не присутствует, а имеется «плечо» широкой полосы при 3300-3400 см-1, отвечающей колебаниям водородно-связанных N-H-фрагментов.
Метод аналитического ультрацентрифугирования показывает, что гидродинамический радиус частиц парентерального лекарственного средства в воде не превышает 100 нм.
Определение фармакологических показателей полученного парентерального лекарственного средства проводят на мышах весом около 30 г. В качестве контрольного образца используют чистый фармакологический агент. Количество животных в каждой группе 8 особей. Препараты вводят внутрибрюшинно в физиологическом растворе в дозе (в расчете на фармакологический агент) 20 мг/кг. Образцы крови животных отбирают через 5, 15, 30, 60, 120 и 240 минут и проводят пробоподготовку по стандартной методике. Концентрацию действующего вещества в пробах определяют с помощью хроматомасс-спектрометрии. Анализ полученных результатов in vivo тестирования парентерального лекарственного средства, приготовленного согласно заявляемому техническому решению, показывает, что по сравнению с контрольным образцом обнаруживается повышение биодоступности (на 20%) и увеличение (в 1,9 раза) времени полувыведения (Т1/2) фармакологического агента, т.е. детектируется существенный эффект пролонгации. Таким образом, в данном случае (пример 1.2) заявляемое лекарственное средство обладает выраженными улучшенными фармакологическими показателями, обусловленными совокупностью признаков предлагаемого технического решения, а именно тем, что фармакологический агент входит в состав лекарственного средства в виде нековалентного комплекса с водорастворимым полимером и неполимерной добавкой.
Предлагаемое изобретение имеет следующие преимущества по сравнению с аналогами и прототипом.
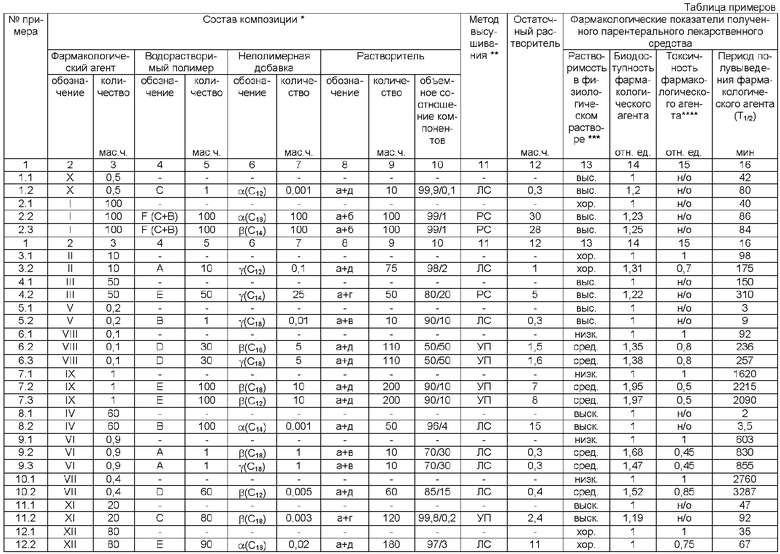
1) Заявляемое техническое решение характеризуется высокой степенью универсальности в отношении возможностей получения парентеральных лекарственных средств с улучшенными фармакологическими показателями на основе самых разнообразных фармакологических агентов, предназначенных для лечения и предупреждения заболеваний нервной системы (номера примеров в таблице 4.2, 11.2) или заболеваний сердечно-сосудистой системы (5.2, 8.2), или онкологических заболеваний (3.2, 6.2, 6.3, 7.2, 7.3, 9.2, 9.3, 10.2), или вирусных заболеваний (12.2), или инфекционных заболеваний (1.2, 2.2, 2.3). В частности, данные столбцов 13-16 таблицы примеров показывают существенное улучшение фармакологических показателей у представителей подобных лекарственных средств, что подтверждает универсальность предлагаемого изобретения.
2) Достигаемый эффект улучшения фармакологических показателей заявляемого парентерального лекарственного средства имеет синергический характер, т.е., как правило, наблюдается улучшение не какого-то одного конкретного фармакологического показателя, а целого набора таких показателей (номера примеров в таблице 2.2, 2.3, 3.2, 6.2, 6.3, 7.2, 7.3, 9.2, 9.3, 10.2, 12.2). Помимо основной функции, определяющей роль водорастворимого полимера в заявляемом техническом решении (комплексное улучшение фармакологических показателей), этот компонент нековалентного комплекса может одновременно выполнять еще и такие дополнительные функции как, например, связывание и выведение из организма нежелательных веществ типа органических токсикантов с помощью поли(N-винилпирролидона), альбумина, 2-гидроксиэтилкрахмала или декстрана.
3) Одно из принципиальных преимуществ предлагаемого изобретения относится к значительному повышению растворимости в воде липофильных фармакологических агентов (номера примеров в таблице 2.2, 2.3, 6.2, 6.3, 7.2, 7.3, 9.2, 9.3,10.2), что повышает их биодоступность и в результате позволяет снизить применяемую терапевтическую дозу, а это особенно важно для веществ с выраженной токсичностью, в частности противоопухолевых средств.
4) При реализации предлагаемого технического решения исключается применение токсичных хлорсодержащих органических растворителей. Более того, после приготовления раствора компонентов композиции этот раствор затем высушивают, поэтому небольшое количество растворителя, остающегося адсорбционно-связанным в получаемом препарате, не является опасным и тем более токсичными для пациентов, принимающих такое лекарственное средство, что подтверждается данными биотестирования, а именно понижением токсичности заявляемых лекарственных средств по сравнению с фармакологическим агентом (номера примеров в таблице 3.2, 6.2, 6.3, 7.2, 7.3, 9.2, 9.3, 10.2, 12.2).
5) Предлагаемый способ получения фармацевтической композиции, которая входит в состав парентерального лекарственного средства, обладающего улучшенными фармакологическими показателями, в отличие от аналогов и прототипа предусматривает использование стандартного для фармацевтического производства оборудования, т.е. характеризуется хорошей технологичностью. Также заявляемое изобретение основано на применении очень мягких режимов всех технологических стадий, что обеспечивает сохранение физиологической активности действующего вещества в составе получаемого лекарственного средства.
Фармацевтическая композиция для получения парентерального лекарственного средства включает фармакологический агент, водорастворимый полимер, неполимерную добавку и растворитель. Водорастворимый полимер выбран из группы: декстран, желатин, 2-гидроксиэтилкрахмал, поли(N-винилпирролидон), альбумин сыворотки крови человека или их смеси. Неполимерная добавка выбрана из группы: (С10-С18)ацил-сорбит, олигооксиэтилен(С10-C18)ацил-сорбит, (С10-С18)алкиловый эфир олигоэтиленгликоля. Растворитель представляет собой смесь воды с этанолом, или диметилсульфоксидом, или N-метил-2-пирролидоном, или 2-пирролидоном. Фармакологический агент, указанные водорастворимый полимер, неполимерная добавка и остаточный растворитель входят в состав средства в виде нековалентного комплекса. Изобретение обеспечивает повышенную биодоступность фармакологических агентов и позволяет снизить применяемую терапевтическую дозу, что особенно важно для веществ с выраженной токсичностью. 2 н.п. ф-лы, 1 ил., 1 табл.
1. Фармацевтическая композиция для получения парентерального лекарственного средства, включающая фармакологический агент, водорастворимый полимер, неполимерную добавку и остаточный растворитель, отличающаяся тем, что водорастворимый полимер выбран из группы: декстран, желатин, 2-гидроксиэтилкрахмал, поли(N-винилпирролидон), альбумин сыворотки крови человека или их смеси; неполимерная добавка выбрана из группы: (С10-С18)ацил-сорбит, олигооксиэтилен-(С10-С18)ацил-сорбит, (С10-С18)алкиловый эфир олигоэтиленгликоля, причем фармакологический агент, водорастворимый полимер, неполимерная добавка и остаточный растворитель входят в состав средства в виде нековалентного комплекса при следующем соотношении, мас.ч.: фармакологический агент 0,10-100, водорастворимый полимер 1-100, неполимерная добавка 0,001-100, остаточный растворитель 0,3-30.
2. Способ получения фармацевтической композиции по п.1, заключающийся в том, что готовят композицию смешиванием фармакологического агента, водорастворимого полимера и неполимерной добавки с растворителем в следующих соотношениях, мас.ч.:
причем растворитель представляет собой смесь воды с этанолом, или диметилсульфоксидом, или N-метил-2-пирролидоном, или 2-пирролидоном при их объемном соотношении от 99,9:0,1 до 50:50 с последующим высушиванием композиции до содержания остаточного растворителя 0,3-30 мас.ч.
| US 5916596 А, 29.06.1999 | |||
| ЖЕВАТЕЛЬНАЯ РЕЗИНКА С ПОКРЫТИЕМ, СОДЕРЖАЩАЯ НИКОТИН, СПОСОБ ЕЕ ПОЛУЧЕНИЯ И ЕЕ ПРИМЕНЕНИЕ | 2002 |
|
RU2291688C2 |
| КОМПОЗИЦИИ, СОДЕРЖАЩИЕ КОМПЛЕКСЫ ВКЛЮЧЕНИЯ | 2001 |
|
RU2288921C2 |
| ФАРМАЦЕВТИЧЕСКИЙ СОСТАВ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И СПОСОБ УВЕЛИЧЕНИЯ БИОЛОГИЧЕСКОЙ ДОСТУПНОСТИ АКТИВНОГО ВЕЩЕСТВА С ИСПОЛЬЗОВАНИЕМ ДАННОГО СОСТАВА | 1991 |
|
RU2104715C1 |

