Область техники, к которой относится изобретение
Изобретение относится к соединениям, которые являются модуляторами CCR2 рецептора. Соединения могут применяться в фармацевтических композициях, способах лечения заболеваний и нарушений, включающих патологическую активацию CCR2 рецепторов.
Предшествующий уровень техники
Хемокины, также известные как хемотаксические цитокины, представляют собой группу низкомолекулярных белков, которые вырабатываются широким рядом клеток и имеют разнообразную биологическую активность. Хемокины привлекают различные типы клеток иммунной системы, такие как макрофаги, Т-клетки, эозинофилы, базофилы и нейтрофилы, и заставляют их мигрировать из крови в различные лимфоидные и нелимфоидные ткани. Они опосредуют проникновение воспалительных клеток к сайтам воспаления и отвечают за начало и развитие многих воспалительных заболеваний (обзор см. в работах Schall, Cytokine, 3:165-183 (1991), Schall et al., Curr. Opin. Immunol., 6:865-873 (1994)).
Помимо стимулирования хемотаксиса, хемокины могут индуцировать другие изменения в реагирующих клетках, включая изменения формы клеток, экзоцитоз гранул, активизацию интегрина, образование биоактивных липидов (например, лейкотриенов), окислительный всплеск, связанный с активацией лейкоцитов, разрастание клеток, устойчивость к индуцированию апоптоза и ангиогенез. Таким образом, хемокины являются ранними триггерами воспалительного ответа, вызывающими высвобождение медиаторов воспаления, хемотаксис и просачивание к сайтам инфекции или воспаления. Они также являются стимуляторами множества клеточных процессов, имеющих важное физиологическое значение, а также патологические последствия.
Хемокины реализуют свое действие путем активации хемокиновых рецепторов, экспрессируемых реагирующими клетками. Хемокиновые рецепторы - это класс рецепторов, связанных с G-белком (GPCR), также известных как семь-трансмембранные рецепторы, находящихся на поверхности широкого ряда типов клеток, таких как лейкоциты, клетки эндотелия, клетки гладких мышц и опухолевые клетки.
Хемокины и хемокиновые рецепторы экспрессируются собственными клетками почек и инфильтратом клеток при почечном воспалении (Segerer et al., J. Am. Soc. Nephrol., 11:152-76 (2000); Morii et al., J. Diabetes Complications, 17:11-5 (2003); Lloyd et al. J. Exp. Med., 185:1371-80 (1997); Gonzalez-Cuadrado et al. Clin. Exp. Immuno,. 106:518-22 (1996); Eddy & Giachelli, Kidney Int., 47:1546-57 (1995); Diamond et al., Am. J. Physiol., 266:F926-33 (1994)). У человека CCR2 и лиганд MCP-1 принадлежат к белкам, экспрессируемым при почечном фиброзе, и коррелируют со степенью инфильтрации макрофагов в интерстициальную ткань (Yang et al., Zhonghua Yi Xue Za Zhi, 81:73-7 (2001); Stephan et al., J. Urol., 167:1497-502 (2002); Amann et al., Diabetes Care, 26:2421-5 (2003); Dai et al., Chin. Med. J. (Engl), 114:864-8 (2001)). В животных моделях почечного фиброза блокада CCR2 или MCP-1 приводит к заметному уменьшению тяжести воспаления почек (Kitagawa et al., Am. J. Pathol., 165:237-46 (2004); Wada et al., Am. J. Pathol., 165:237-46 (2004); Shimizu et al., J. Am. Soc. Nephrol., 14:1496-505 (2003)).
Кроме того CCR2 играет роль в развитии поликистозной болезни почек (PKD). Патология при ADPKD характеризуется почечными цистами, которые развиваются из канальцевых эпителиальных клеток и непрерывно увеличиваются на протяжении всей жизни пациента. Рост цист сопровождается присутствием большого числа инфильтрирующихся макрофагов, которые направляются в почки в ответ на травму. Таким образом, подавление иммунного рекрутирования в почки составляет терапевтическую стратегию при PKD (J Am Soc Nephrol. 2011 Oct;22(10):1809-14.)
Ревматоидный артрит представляет собой хроническое заболевание суставов, характеризующееся воспалением синовиальной оболочки, приводящим к разрушению хряща и кости. Хотя истинные причины данного заболевания неизвестны, считается, что макрофаги и Т-клетки типа Th-1 играют ключевую роль в начале и развитии данного хронического воспалительного процесса (Vervoordeldonk et al., Curr. Rheumatol. Rep., 4:208-17 (2002)).
MCP-1 находится среди нескольких хемокинов, включая MIP-1α и IL-8, идентифицированных в ревматоидной синовиальной оболочке (Villiger et al., J. Immunol., 149:722-7 (1992); Scaife et al., Rheumatology (Oxford), 43:1346-52 (2004); Shadidi et al., Scand. J. Immunol., 57:192-8 (2003); Taylor et al., Arthritis Rheum., 43:38-47 (2000); Tucci et al., Biomed. Sci. Instrum., 34:169-74 (1997)). Концентрация хемокиновых рецепторов CCR1, CCR2, CCR3 и CCR5 повышается в суставах мышей, страдающих артритом (Plater-Zyberk et al., Immunol. Lett., 57:117-20 (1997). Было показано, что блокировка активности MCP-1 с помощью CCR2 антагониста или антитела против MCP-1 эффективно уменьшает воспаление в суставах в экспериментальной модели ревматоидного артрита (Gong et al., J. Exp. Med., 186:131-7 (1997); Ogata et al., J. Pathol., 182:106-14 (1997)).
Опосредуемая хемокиновым рецептором инфильтрация макрофагов в жировые ткани также может вносить вклад в осложнения, причиной которых является ожирение - состояние избыточного накопления жира в организме. Ожирение обуславливает предрасположенность организма ко многим заболеваниям, таким как инсулиннезависимый диабет, гипертензия, инсульт и ишемическая болезнь сердца. При ожирении нарушается метаболическая и эндокринная функция жировой ткани, что приводит к повышенному высвобождению жирных кислот, гормонов и провоспалительных молекул. Макрофаги жировой ткани считаются ключевым источником провоспалительных цитокинов, включая ФНО-альфа, iNOS и IL-6 (Weisberg et al., J. Clin. Invest., 112:1796-808 (2003)). Поступление макрофагов в жировую ткань, по всей вероятности, опосредуется через MCP-1, вырабатываемый адипоцитами (Christiansen T, et al., Int J Obes (Lond). 2005 Jan; 29(1):146-50; Sartipy et al., Proc. Natl. Acad. Sci. U.S.A., 100:7265-70 (2003)).
Повышение уровня MCP-1 может вызывать дифференциацию адипоцитов и резистентность к инсулину, а также способствует патологиям, связанным с гиперинсулинемией и ожирением. MCP-1 сверхэкспрессирован в плазме крови мышей, страдающих ожирением, в сравнении с контрольной группой без ожирения, и основным источником является белая жировая ткань. Также было показано, что MCP-1 ускоряет заживление ран и оказывает прямой ангиогенный эффект на эпителиальные клетки, и может напрямую участвовать в перестройке жировой ткани при ожирении. (Sartipy P, Loskutoff DJ., Proc. Natl. Acad. Sci. U.S.A.,100:7265 (2003)).
Уровень MCP-1 в плазме крови заметно повышен у мышей, страдающих алиментарным ожирением (DIO), и была обнаружена сильная корреляция между уровнем MCP-1 в плазме крови и весом тела. Кроме того, повышение уровня MCP-1, вызванное диетой с высоким содержанием жира, вызывает изменения в CD11b положительных моноцитах у мышей с DIO. (Takahashi K, et al., J. Biol. Chem., 46654 (2003)).
Кроме того, считается, что хроническое воспаление в жировой ткани играет решающую роль в развитии инсулинoвой резистентности, связанной с ожирением (Xu H, et al., J Clin Invest. 2003 Dec;112(12):1821-30). Предполагают, что инсулинoвая резистентность, связанная с ожирением, представляет собой, по меньшей мере частично, хроническое воспалительное заболевание, начинающееся в жировых тканях. Многие специфические гены воспаления и макрофагов заметно активированы в белой жировой ткани в мышиной модели генетического ожирения и ожирения, вызванного диетой с высоким содержанием жира (DIO), и эта активация предшествует резкому росту уровня инсулина в крови.
Повышенная экспрессия моноцитарного CCR2 и моноцитарного хемоаттрактантного белка-1 была обнаружена у пациентов с сахарным диабетом (Biochemical and Biophysical Research Communications, 344(3):780-5 (2006)) в исследовании, включающем анализ пациентов, страдающих диабетом. Уровень MCP-1 в плазме крови и экспрессирование CCR2 на поверхности моноцитов у пациентов, страдающих диабетом, были значительно выше, чем у пациентов без диабета, и уровень MCP-1 в плазме крови коррелировал с HbA1c, триглицеридами, BMI, hs-CRP. Экспрессирование CD36 и CD68 на поверхности моноцитов было значительно увеличено у пациентов, страдающих диабетом, и меньше регулировался MCP-1 у диабетиков, увеличивая поглощение окисленных ЛПНП и создавая потенциал для трансформации ксантомных клеток. Повышенный уровень MCP-1 в крови и увеличенная экспрессия моноцитарных CCR2, CD36, CD68 коррелировали с плохим контролем глюкозы в крови и потенциально коррелируют с увеличением накопления моноцитов в стенках сосудов.
MCP-1 является потенциальным участником негативного противодействия жировой ткани и скелетных мышц (Bianco JJ, et al., Endocrinology, 2458 (2006)). MCP-1 может значительно снижать стимулируемое инсулином поглощение глюкозы и представляет собой ярко выраженный индуктор инсулиновой резистентности в клетках скелетных мышц человека. Жировая ткань представляет собой большой секреторный и эндокринно активный орган, вырабатывающий биологически активные белки, регулирующие энергетический обмен и чувствительность к инсулину.
CCR2 модулирует воспалительный и метаболический эффект при питании с высоким содержанием жира (Weisberg SP, et al., J. Clin. Invest., 115 (2006)). Генетический дефицит CCR2 снижал потребление пищи и замедлял развитие ожирения у мышей, находящихся на диете с высоким содержанием жира. У мышей с ожирением и примерно одинаковым отложением жира, дефицит CCR2 снижал содержание макрофагов и воспалительный профиль жировой ткани, повышал выработку адипонектина и улучшал гомеостаз глюкозы и чувствительность к инсулину. У животных, не страдающих ожирением, не отмечалось никакого влияния CCR2 генотипа на метаболизм. У мышей, находящихся на диете с высоким содержанием жира, CCR2 генотип влиял на питание, развитие ожирения и воспаление в жировой ткани. Было показано, что краткосрочный антагонизм подавлял накопление макрофагов в жировой ткани и инсулиновую резистентность.
Хемокины и хемокиновые рецепторы являются ключевыми регуляторами направленной миграции иммунных клеток. MCP-1 представляет собой сильный хемоаттрактант для моноцитов и Т-клеток; его выработка индуцируется при воспалительных состояниях, включая стимуляцию провоспалительных цитокинов и гипоксию. Взаимодействие между MCP-1 и CCR2 вызывает миграцию моноцитов, макрофагов, а также активированных Т-клеток и играет ключевую роль в патогенезе многих воспалительных заболеваний. Подавление функции CCR2 с помощью низкомолекулярных антагонистов, описанное в настоящем изобретении, представляет собой новый подход в лечении воспалительных нарушений.
Псориаз представляет собой хроническое воспалительное заболевание, характеризующееся гиперпролиферацией кератиноцитов и выраженной инфильтрацией лейкоцитов. Известно, что кератиноциты в участках кожи, пораженных псориазом, вырабатывают распространенный CCR2 лиганд MCP-1, особенно при стимулировании провоспалительными цитокинами, такими как ФНО-α (Vestergaard et al., Acta. Derm. Venereol., 84(5):353-8 (2004); Gillitzer et al., J. Invest. Dermatol., 101(2):127-31 (1993); Deleuran et al., J. Dermatol. Sci., 13(3):228-36 (1996)). Поскольку MCP-1 может вызывать миграцию к коже макрофагов и дендритных клеток, экспрессирующих CCR2, считается, что данная пара рецептор-лиганд играет важную роль в регуляции взаимодействия между пролиферирующими кератиноцитами и кожными макрофагами при развитии псориаза. Таким образом, низкомолекулярный антагонист мог бы применяться для лечения псориаза.
Помимо воспалительных заболеваний, хемокины и хемокиновые рецепторы вовлечены также в развитие раковых заболеваний (Broek et al., Br. J. Cancer, 88(6):855-62 (2003)). Опухолевые клетки стимулируют образование стромы, которая секретирует различные медиаторы, необходимые для роста опухоли, включая факторы роста, цитокины и протеазы. Известно, что уровень MCP-1 тесно связан с накоплением опухоль-связанных макрофагов, и прогностический анализ показывает, что высокий уровень экспрессии MCP-1 представляет собой важный индикатор раннего рецидива при раке груди (Ueno et al., Clin. Cancer Res., 6(8):3282-9 (2001)). Таким образом, низкомолекулярный антагонист хемокина мог бы снизить выработку рост-стимулирующих цитокинов путем блокирования накопления макрофагов в местах формирования опухоли.
CCR2 и его лиганд CCL2 также играют большую роль в создании микроокружения опухоли и регуляции направленного поступления в опухоль как благотворных, так и разрушительных иммунных клеток. В недавних клинических и преклинических публикациях была показана роль CCR2 в развитии различных солидных опухолей вследствие активации его выработки в трансформированных клетках или благодаря усиленному хемотаксису воспалительных моноцитов в опухоль, которые финально дифференцируются в миелоидные супрессорные клетки (MDSC) или макрофаги, ассоциированные с опухолью (TAM). Считают, что MDSC/TAM промотируют образование опухолей по следующим механизмам: (1) участвуют в создании общего иммуноподавляющего микроокружения, тем самым стимулируя рост опухолей, что ликвидирует цитотоксическую активность проникающих цитотоксических Т-клеток (Mitchem et al, Cancer Res 73(3): 1128-1141), (2) усиливают ангиогенез посредством выработки фактора роста эндотелия сосудов (VEGF) и других факторов роста (Murdoch et al, Nat Rev Cancer 8: 618-631), (3) предотвращают старение опухоли (Di Mitri et al, AOP, Nature, 2014), и (4) стимулируют формирование метастазов опухолей в удаленные органы (Qian et al, Nature 475: 222-227). Таким образом, низкомолекулярный антагонист может быть полезным для ограничения образования опухолей, усиления опухоль-специфичной иммунной активности и для ограничения формирования метастазов.
В настоящем тексте описаны соединения, являющиеся модуляторами CCR2 и преодолевающие некоторые ограничения, присущие ранее известным модуляторам CCR2.
Краткое описание изобретения
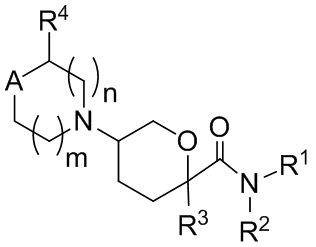
В одном аспекте, в настоящем изобретении описаны соединения, имеющее формулу:
 (I);
(I);
или их фармацевтически приемлемые соли, гидраты, стереоизомеры или ротамеры; где символы R1, R2, R3, R4, A и подстрочные индексы m и n имеют значения, описанные в Подробном описании изобретения.
Помимо описанных в настоящем тексте соединений, в настоящем изобретении описаны также фармацевтические композиции, содержащие одно или больше из этих соединений, а также способы применения этих соединений в методах терапии, в первую очередь направленных на лечение заболеваний, связанных с CCR2 сигнальной активностью.
В другом аспекте, в настоящем изобретении описаны методы диагностирования заболевания у пациента. В этих методах, описанные в настоящем тексте соединения вводят в меченом виде субъекту, затем проводят диагностическую визуализацию для определения наличия или отсутствия CCR2. В связанном с этим аспекте, метод диагностирования заболевания реализуют путем контакта образца ткани или крови с меченым соединением, как описано в настоящем тексте, и определяют наличие, отсутствие или количество CCR2 в исследуемом образце.
В другом аспекте, в настоящем изобретении описан способ модулирования работы хемокина, включающий контакт хемокинового рецептора с терапевтически эффективным количеством соединения или композиции по настоящему изобретению.
В другом аспекте, в настоящем изобретении описан способ лечения хемокин-опосредованного состояния или заболевания, включающий введение субъекту безопасного и эффективного количества соединения или композиции по настоящему изобретению.
В одном частном аспекте, настоящее изобретение касается способа лечения CCR2-опосредованного состояния или заболевания, включающего введение субъекту терапевтически эффективного количества соединения или фармацевтически приемлемой соли соединения, имеющего формулу I. В некоторых вариантах осуществления, CCR2-опосредованное состояние или заболевание представляет собой атеросклероз. В некоторых вариантах осуществления, CCR2-опосредованное состояние или заболевание представляет собой рестеноз. В некоторых вариантах осуществления, CCR2-опосредованное состояние или заболевание представляет собой множественный склероз. В некоторых вариантах осуществления, CCR2-опосредованное состояние или заболевание выбрано из группы, состоящей из воспалительной болезни кишечника, почечного фиброза, ревматоидного артрита, ожирения и инсулиннезависимого диабета. В некоторых вариантах осуществления, CCR2-опосредованное состояние или заболевание представляет собой диабет 2-го типа. В некоторых вариантах осуществления, CCR2-опосредованное состояние или заболевание выбрано из группы, состоящей из хронического обструктивного заболевания легких, идиопатического легочного фиброза и синдрома идиопатической пневмонии.
Помимо описанных в настоящем тексте соединений, в настоящем изобретении описаны также фармацевтические композиции, содержащие одно или больше из этих соединений, а также способы применения этих соединений в методах терапии, в первую очередь для лечения заболеваний, связанных с сигнальной активностью хемокинов.
Краткое описание чертежей
На фиг. 1A-1Y представлены структуры и активность репрезентативных соединений по настоящему изобретению. Соединения были получены как описано ниже, а также методами, описанными в примерах. Активность в анализе связывания охарактеризована следующим образом: +, 501 нM ≤ IC50 < 5000 нM; ++, 101 нM ≤ IC50 < 500 нM; и +++, 1 нM ≤ IC50 ≤ 100 нM.
На фиг. 2 приведена ORTEP структура фрагмента описанных в настоящем тексте соединений и показана стереохимия четвертичного центра (показан как несущий циклопропильную группу и идентифицирован как имеющий ‘S’ хиральность).
Подробное описание изобретения
Сокращения и определения
Термин "алкил", сам по себе и как часть другого заместителя, означает, если не указано иное, линейный или разветвленный углеводородный радикал, имеющий обозначенное число атомов углерода (например, C1-8 означает 1-8 атомов углерода). Примеры алкильных групп включают метил, этил, н-пропил, изопропил, н-бутил, трет-бутил, изобутил, втор-бутил, н-пентил, н-гексил, н-гептил, н-октил и т.п. Термин "алкенил" означает ненасыщенную алкильную группу, содержащую одну или больше двойных связей. Аналогично, термин «алкинил» означает ненасыщенную алкильную группу, содержащую одну или больше тройных связей. Примеры таких ненасыщенных алкильных групп включают винил, 2-пропенил, кротил, 2-изопентенил, 2-(бутадиенил), 2,4-пентадиенил, 3-(1,4-пентадиенил), этинил, 1- и 3-пропинил, 3-бутинил и их высшие гомологи и изомеры. Термин "циклоалкил" относится к углеводородным циклам, имеющим указанное число атомов в цикле (например, C3-6циклоалкил) и являющимся полностью насыщенными или имеющими не более одной двойной связи между вершинами цикла. "Циклоалкил" относится также к бициклическим и полициклическим углеводородным кольцам, таким как, например, бицикло[2.2.1]гептан, бицикло[2.2.2]октан и т.д. Термин "гетероциклоалкил" относится к циклоалкильной группе, содержащей 1-5 гетероатомов, выбранных из N, O, и S, где атомы азота и серы необязательно окислены, и атом(ы) азота необязательно кватернизован(ы). Гетероциклоалкил может представлять собой моноциклическую, бициклическую или полициклическую кольцевую систему. Неограничивающие примеры гетероциклоалкильных групп включают пирролидин, пиперидинил, имидазолидин, пиразолидин, бутиролактам, валеролактам, имидазолидинон, гидантоин, фталимид, пиперидин, 1,4-диоксан, морфолин, тиоморфолин, тиоморфолин-S-оксид, тиоморфолин-S,S-оксид, пиперазин, пиран, пиридон, 3-пирролин, тиопиран, пирон, тетрагидрофуран, тетрагидротиофен, хинуклидин и т.п.. Гетероциклоалкильная группа может быть присоединена к остальной части молекулы через атом углерода в цикле или гетероатом в цикле. В случае таких терминов, как циклоалкилалкил и гетероциклоалкилалкил, циклоалкильная или гетероциклоалкильная группа присоединена через алкильный или алкиленовый линкер к остальной части молекулы. Например, циклобутилметил - это циклобутильное кольцо, которое присоединено через метиленовый линкер к остальной части молекулы.
Термин "алкилен", сам по себе и как часть другого заместителя, означает двухвалентный радикал, являющийся производным алкана, примером которого может служить -CH2CH2CH2CH2-. В типичном случае, алкильная (или алкиленовая) группа содержит от 1 до 24 атомов углерода, при этом группы с 10 атомами углерода или меньше являются предпочтительными по настоящему изобретению. "Низший алкил" или "низший алкилен" представляет собой алкильную или алкиленовую группу с более короткой цепочкой, обычно содержащую четыре или меньше атомов углерода. Аналогично, «алкенилен» или «алкинилен» относится к ненасыщенным формам «алкилена», содержащим двойные или тройные связи, соответственно.
Термин “гетероалкил,” в отдельности или в комбинации с другим термином, означает, если не указано иное, устойчивый линейный или разветвленный или циклический углеводородный радикал или их комбинацию, состоящий из указанного числа атомов углерода и 1-3 гетероатомов, выбранных из группы, состоящей из O, N, Si и S, и где атомы азота и серы необязательно могут быть окислены, и гетероатом азота необязательно может быть кватернизован. Гетероатом(ы) O, N и S могут находиться в любом внутреннем положении гетероалкильной группы. Гетероатом Si может находиться в любом положении гетероалкильной группы, включая положение, по которому алкильная группа присоединена к остальной части молекулы. Примеры включают -CH2-CH2-O-CH3, -CH2-CH2-NH-CH3, -CH2-CH2-N(CH3)-CH3, -CH2-S-CH2-CH3, -CH2-CH2, -S(O)-CH3, -CH2-CH2-S(O)2-CH3, -CH=CH-O-CH3, -Si(CH3)3, -CH2-CH=N-OCH3 и -CH=CH-N(CH3)-CH3. До двух гетероатомов могут располагаться последовательно, как, например, в -CH2-NH-OCH3 и -CH2-O-Si(CH3)3. Сходным образом, термины “гетероалкенил” и “гетероалкинил”, в отдельности или в комбинации с другим термином, означают, если не указано иное, алкенильную группу или алкинильную группу, соответственно, которая содержит указанное число атомов углерода и от одного до трех гетероатомов, выбранных из группы, состоящей из O, N, Si и S, и где атомы азота и серы необязательно могут быть окислены, и гетероатом азота необязательно может быть кватернизован. Гетероатом(ы) O, N и S могут находиться в любом внутреннем положении гетероалкильной группы.
Термин “гетероалкилен”, в отдельности или как часть другого заместителя, означает двухвалентный радикал, насыщенный или ненасыщенный или полиненасыщенный, образованный из гетероалкила, например -CH2-CH2-S-CH2CH2-, -CH2-S-CH2-CH2-NH-CH2-, -O-CH2-CH=CH-, -CH2-CH=C(H)CH2-O-CH2- и -S-CH2-C≡C-. В случае гетероалкиленовых групп, гетероатомы могут также находиться в любом или в обоих терминальных положениях (например, алкиленокси, алкилендиокси, алкиленамино, алкилендиамино и т.п.).
Термины "алкокси," "алкиламино" и "алкилтио" (или тиоалкокси) применяются в их обычном смысле и относятся к алкильным группам, присоединенным к остальной части молекулы через атом кислорода, аминогруппу или атом серы, соответственно. Кроме того, для диалкиламино-групп, алкильные фрагменты могут быть одинаковыми или разными, а также могут объединяться с формированием 3-7-членного цикла с атомом азота, к которому они присоединены. Соответственно, группа, изображаемая как -NRaRb, включает пиперидинил, пирролидинил, морфолинил, азетидинил и т.п.
Термин "галоген" сам по себе или как часть другого заместителя означает, если не указано иное, атом фтора, хлора, брома или иода. Кроме того, такие термины как "галогеналкил," включают моногалогеналкил и полигалогеналкил. Например, термин "C1-4 галогеналкил" включает трифторметил, 2,2,2-трифторэтил, 4-хлорбутил, 3-бромпропил и т.п.
Термин "арил" означает, если не указано иное, полиненасыщенную, в типичном случае ароматическую углеводородную группу, которая может представлять собой один цикл или несколько циклов (до трех циклов), сопряженные или связанные ковалентно. Термин «гетероарил» относится к арильным группам (или циклам), содержащим от одного до пяти гетероатомов, выбранных из N, O, и S, где атомы азота и серы необязательно окислены, и атом(ы) азота необязательно кватернизован(ы). Гетероарильная группа может быть присоединена к остальной части молекулы через гетероатом. Неограничивающие примеры арильных групп включают фенил, нафтил и бифенил, в то время как неограничивающие примеры гетероарильных групп включают пиридил, пиридазинил, пиразинил, пиримидинил, триазинил, хинолинил, хиноксалинил, хиназолинил, циннолил, фталазинил, бензотриазинил, пуринил, бензоимидазолил, бензопиразолил, бензотриазолил, бензизоксазалил, изобензофурил, изоиндолил, индолизинил, бензотриазинил, тиенопиридинил, тиенопиримидинил, пиразолопиримидинил, имидазопиридины, бензотиаксолил, бензофуранил, бензотиенил, индолил, хинолил, изохинолил, изотиазолил, пиразолил, индазолил, птеридинил, имидазолил, триазолил, тетразолил, оксазолил, изоксазолил, тиадиазолил, пирролил, тиазолил, фурил, тиенил и т.п. Заместители в каждом из перечисленных выше арильных или гетероарильных циклических систем выбраны из группы приемлемых заместителей, описанных ниже.
Для краткости, термин "арил", когда применяется в комбинации с другими терминами (например, арилокси, арилтиокси, арилалкил), включает и арильные, и гетероарильные циклы, определение которым дано выше. Так, термин "арилалкил" включает такие радикалы, в которых арильная группа присоединена к алкильной группе, которая присоединена к остальной части молекулы (например, бензил, фенетил, пиридилметил и т.п.).
Указанные выше термины (например, "алкил", "арил" и "гетероарил") в некоторых вариантах осуществления включают как замещенные, так и незамещенные формы указанного радикала. Предпочтительные заместители для каждого типа радикала перечислены ниже. Для краткости, термины “арил” и “гетероарил” означают замещенные или незамещенные версии, описанные выше, в то время как термин "алкил" и соответствующие алифатические радикалы означают незамещенную версию, если специально не указано, что они замещенные.
Заместителями в алкильных радикалах (включая группы, которые часто именуются алкилен, алкенил, алкинил и циклоалкил) могут быть различные группы, выбранные из: -галоген, -OR’, -NR’R”, -SR’, -SiR’R”R”’, -OC(O)R’, -C(O)R’, -CO2R’, -CONR’R”, -OC(O)NR’R”, -NR”C(O)R’, -NR’-C(O)NR”R”’, -NR”C(O)2R’, -NH-C(NH2)=NH, -NR’C(NH2)=NH, -NH-C(NH2)=NR’, -S(O)R’, -S(O)2R’, -S(O)2NR’R”, -NR’S(O)2R”, -CN и -NO2, в количестве от нуля до (2 m’+1), где m’ это общее число атомов углерода в таком радикале. R’, R” и R”’ каждый независимо означают атом водорода, незамещенный C1-8 алкил, незамещенный гетероалкил, незамещенный арил, арил, замещенный 1-3 галогенами, незамещенный C1-8 алкил, C1-8 алкокси или C1-8 тиоалкокси группу, или незамещенные арил-C1-4 алкильные группы. Когда R’ и R” присоединены к одному и тому же атому азота, они могут объединяться с атомом азота с образованием 3-, 4-, 5-, 6- или 7-членного цикла. Например, -NR’R” включает 1-пирролидинил и 4-морфолинил. Термин “ацил”, когда применяется сам по себе или как часть другой группы, означает алкильный радикал, в котором два заместителя у атома углерода, ближайшего к точке присоединения данного радикала, заменены на заместитель =O (например, -C(O)CH3, -C(O)CH2CH2OR’ и т.п.).
Аналогично, заместители в арильных и гетероарильных группах варьируются и обычно выбраны из: -галоген, -OR’, -OC(O)R’, -NR’R”, -SR’, -R’, -CN, -NO2, -CO2R’, -CONR’R”, -C(O)R’, -OC(O)NR’R”, -NR”C(O)R’, -NR”C(O)2R’, -NR’-C(O)NR”R”’, -NH-C(NH2)=NH, -NR’C(NH2)=NH, -NH-C(NH2)=NR’, -S(O)R’, -S(O)2R’, -S(O)2NR’R”, -NR’S(O)2R”, -N3, перфтор(C1-C4)алкокси, и перфтор(C1-C4)алкил, в количестве от нуля до общего числа незанятых валентностей в ароматической циклической системе; и где R’, R” и R”’ независимо выбраны из атома водорода, C1-8 алкила, C3-6 циклоалкила, C2-8 алкенила, C2-8 алкинила, незамещенного арила и гетероарила, (незамещенный арил)-C1-4 алкила и незамещенный арилокси-C1-4 алкила. Другие подходящие заместители включают каждый из перечисленных выше заместителей для арила, присоединенных к атому в цикле алкиленовым мостиком из 1-4 атомов углерода.
Два из заместителей у соседних атомов арильного или гетероарильного кольца могут опционально быть замещены заместителем формулы -T-C(O)-(CH2)q-U-, где T и U независимо представляют собой -NH-, -O-, -CH2- или одинарную связь, и q представляет собой целое число от 0 до 2. Альтернативно, два из заместителей у соседних атомов арильного или гетероарильного кольца могут опционально быть замещены заместителем формулы -A-(CH2)r-B-, где A и B независимо представляют собой -CH2-, -O-, -NH-, -S-, -S(O)-, -S(O)2-, -S(O)2NR’- или одинарную связь, и r представляет собой целое число от 1 до 3. Одна из простых связей в новом цикле, образующемся таким образом, может опционально быть заменена на двойную связь. Альтернативно, два из заместителей у соседних атомов арильного или гетероарильного кольца могут опционально быть замещены заместителем формулы -(CH2)s-X-(CH2)t-, где s и t независимо представляют собой целые числа от 0 до 3, и X представляет собой -O-, -NR’-, -S-, -S(O)-, -S(O)2- или -S(O)2NR’-. Заместитель R’ в -NR’- и -S(O)2NR’- выбран из атома водорода или незамещенного C1-6 алкила.
При использовании в настоящем тексте, термин "гетероатом" включает кислород (O), азот (N), серу (S) и кремний (Si).
В случае описанных в настоящем тексте соединений, связь, идущая от заместителя (в типичном случае от группы R) к центру ароматического кольца (например, бензола, пиридина и т.п.) следует понимать как связь, осуществляющую соединение по любой доступной точке ароматического кольца. В некоторых вариантах осуществления, изображение включает также соединение с кольцом, которое сконденсировано с ароматическим кольцом. Например, связь, идущая к центру бензольного фрагмента индола, означает связь с любой доступной точкой шести- или пятичленного фрагмента индола.
Термин "фармацевтически приемлемые соли" включает соли действующих веществ, полученные с относительно нетоксичными кислотами или основаниями, в зависимости от конкретных заместителей в описанных в настоящем тексте соединениях. Когда соединения по настоящему изобретению содержат относительно кислые функциональные группы, можно получить основно-аддитивные соли путем взаимодействия нейтральной формы таких соединений с достаточным количество желаемого основания, даже без растворителя или в подходящем инертном растворителе. Примеры солей, являющихся производными фармацевтически приемлемых неорганических оснований, включают соли алюминия, аммония, кальция, меди, железа(II), железа (III), лития, магния, марганца, калия, натрия, цинка и т.д.. Соли, являющиеся производными фармацевтически приемлемых органических оснований, включают соли первичных, вторичных и третичных аминов, включая замещенные амины, циклические амины, природные амины и т.д., такие как аргинин, бетаин, кофеин, холин, N,N’-дибензилэтилендиамин, диэтиламин, 2-диэтиламиноэтанол, 2-диметиламиноэтанол, этаноламин, этилендиамин, N-этилморфолин, N-этилпиперидин, глюкамин, глюкозамин, гистидин, гидрабамин, изопропиламин, лизин, метилглюкамин, морфолин, пиперазин, пиперидин, полиаминовые смолы, прокаин, пурины, теобромин, тиэтиламин, триметиламин, трипропиламин, трометамин и т.п.. Когда соединения по настоящему изобретению содержат относительно основные функциональные группы, можно получить кислотно-аддитивные соли путем взаимодействия нейтральной формы таких соединений с достаточным количеством желаемой кислоты, без растворителя или в подходящем инертном растворителе. Примеры фармацевтически приемлемых кислотно-аддитивных солей включают соли с неорганическими кислотами, такими как хлористоводородная, бромистоводородная азотная, угольная, моногидроугольная, фосфорная, моногидрофосфорная, дигидрофосфорная, серная, моногидросерная, иодистоводородная или фосфористая кислота и т.п., а также соли с относительно нетоксичными органическим кислотами, такими как уксусная, пропионовая, изомасляная, малоновая, бензойная, янтарная, субериновая, фумаровая, миндальная, фталевая, бензолсульфоновая, пара-толуолсульфоновая, лимонная, винная, метансульфоновая и т.п.. Также охватываются соли с аминокислотами, такие как аргинаты и т.п., и соли таких органических кислот, как глюкуроновая или галактуроновая кислоты и т.п. (см, например, Berge, S.M., et al, “Pharmaceutical Salts”, Journal of Pharmaceutical Science, 1977, 66, 1-19). Некоторые частные соединения по настоящему изобретению содержат и основные, и кислотные функциональные группы, что позволяет таким соединениям образовывать как основно-аддитивные, так и кислотно-аддитивные соли.
Нейтральные формы соединений можно регенерировать путем взаимодействия соли с основанием или кислотой и выделения материнского соединения обычным способом. Материнская форма соединения отличается от различных солевых форм определенными физическими характеристиками, такими как растворимость в полярных растворителях, но во всем остальном соли эквивалентны материнским соединениям, в терминах настоящего изобретения.
Помимо солевых форм, в настоящем изобретении описаны соединения, представляющие собой пролекарственные формы. Пролекарства описанных в настоящем тексте соединений представляют собой соединения, которые легко претерпевают химические изменения в физиологических условиях, давая соединения по настоящему изобретению. Кроме того, пролекарства можно превратить в соединения по настоящему изобретению химическими или биохимическими методами в ex vivo условиях. Например, пролекарства можно медленно превратить в соединения по настоящему изобретению при помещении их в резервуар пластыря для чрезкожного введения с подходящим ферментативным или химическим реагентом.
Некоторые соединения по настоящему изобретению могут существовать в несольватированных формах, а также в сольватированных формах, включая гидратированные формы. В целом, сольватированные формы эквивалентны несольватированным формам, и все они охватываются настоящим изобретением. Некоторые соединения по настоящему изобретению могут существовать в нескольких кристаллических или аморфных формах. В целом, все физические формы эквивалентны для областей применения, охватываемых настоящим изобретением, и входят в объем настоящего изобретения.
Некоторые соединения по настоящему изобретению имеют асимметрические атомы углерода (оптические центры) или двойные связи; все рацематы, диастереомеры, геометрические изомеры, региоизомеры и индивидуальные изомеры (например, отдельные энантиомеры) входят в объем настоящего изобретения. Когда соединения описаны в настоящем тексте с определенной стереохимией (указано как R или S, или обозначено пунктирными связями или жирными линиями, обозначающими связь), такие соединения понимаются квалифицированными специалистами в данной области как практически не содержащие других изомеров (например, на 80%, 90%, 95%, 98%, 99% и до 100% свободные от другого изомера).
Соединения по настоящему изобретению могут также иметь неприродные соотношения изотопов по одному или больше атомов, составляющих эти соединения. Неприродные соотношения изотопов можно определить как находящиеся в диапазоне от природного количества до количества рассматриваемого атома равного 100%. Например, соединения могут быть радиоактивно мечены радиоактивными изотопами, такими как, например, тритий (3H), иод-125 (125I) или углерод-14 (14C), или нерадиоактивными изотопами, такими как дейтерий (2H) или углерод-13 (13C). Такие вариации изотопов могут открыть дополнительные области применения к описанным в других разделах настоящего описания. Например, изотопные модификации соединений по настоящему изобретению могут найти дополнительное применение, включая (но не ограничиваясь только ими) применение в качестве диагностических и/или визуализирующих реагентов, или в качестве цитотоксических/радиотоксических терапевтических средств. Кроме того, изотопные варианты соединений по настоящему изобретению могут иметь измененные фармакокинетические и фармакодинамические характеристики, которые могут вносить свой вклад в улучшение характеристик безопасности, переносимости или эффективности при лечении. Все изотопные вариации соединений по настоящему изобретению, радиоактивные и нерадиоактивные, входят в объем настоящего изобретения.
Общее
Настоящее изобретение касается соединений и их солей, композиций и способов, которые могут применяться в модулировании работы хемокинового рецептора, в частности работы CCR2. Модулирование активности хемокинового рецептора, при использовании в настоящем тексте в различных формах, охватывает антагонизм, агонизм, частичный антагонизм, обратный агонизм и/или частичный агонизм активности, связанной с конкретным хемокиновым рецептором, предпочтительно CCR2 рецептором. Соответственно, соединения по настоящему изобретению представляют собой соединения, которые модулируют по меньшей мере одну функцию или характеристику CCR2 млекопитающих, например человеческого CCR2 белка. Способность соединения модулировать функцию CCR2 можно продемонстрировать в анализе связывания (например, связывания лиганда или связывания агониста), анализе миграции, анализе сигнала (например, активация G-белка млекопитающих, индуцирование быстрого и короткого роста концентрации свободного цитозольного кальция), и/или анализе клеточной реакции (например, стимулирование хемотаксиса, экзоцитоза или высвобождения медиаторов воспаления лейкоцитами).
Соединения
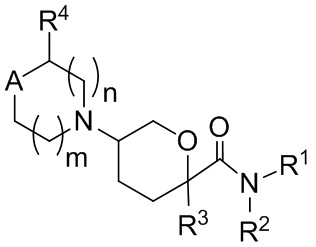
В одном аспекте, в настоящем изобретении описаны соединения, имеющие формулу I:
 (I);
(I);
или их фармацевтически приемлемая соль, гидрат, стереоизомер или ротамер; где
A представляет собой C(R5)(R6) или N(R5)
подстрочные индексы m и n каждый независимо представляют собой целые числа от 0 до 2, и m + n составляет ≤ 3;
R1 выбран из группы, состоящей из арила, арил-C1-4 алкила, гетероарила и гетероарил-C1-4 алкила, где гетероарильный фрагмент содержит 1-3 гетероатомов в качестве членов цикла, выбранных из N, O и S; и где арильные и гетероарильные группы или фрагменты необязательно замещены 1 - 5 заместителями Rx;
R2 выбран из группы, состоящей из H, C1-8 алкила, C3-8 циклоалкила, C3-8 циклоалкил-C1-4 алкила, арила, арил-C1-4 алкила, гетероарила и гетероарил-C1-4 алкила, где гетероарильный фрагмент содержит 1-3 гетероатомов в качестве членов цикла, выбранных из N, O и S; и где арильные и гетероарильные группы или фрагменты необязательно замещены 1 - 4 заместителями Rx;
или, необязательно, R1 и R2 объединены с атомом азота, к которому они присоединены, с образованием 6-11-членного моноциклического или конденсированного бициклического гетероциклического или гетероарильного кольца, где-NR1R2 необязательно дополнительно замещен 1 - 4 заместителями Rx;
R3 выбран из группы, состоящей из H, C1-8 алкила, C3-8 циклоалкила и C3-8 циклоалкил-C1-4 алкила, каждый из которых необязательно замещен 1-3 заместителями Ry;
R4 выбран из группы, состоящей из H, C1-8 алкила, необязательно замещенного 1 - 2 Ry, и -CO2H:
R5 выбран из группы, состоящей из C1-8 алкила, C1-8 алкокси-группы, C3-8 циклоалкила, C3-8 циклоалкилокси-группы, C3-8 циклоалкил-C1-4 алкила, C1-8 алкиламино-группы, ди-C1-8 алкиламино-группы, арила, арилокси-группы, ариламино-группы, арил-C1-4 алкила, гетероарила, гетероарилокси-группы, гетероариламино-группы и гетероарил-C1-4 алкила, каждый из которых необязательно замещен 1 - 5 заместителями Rz;
R6 выбран из группы, состоящей из H, F, OH, C1-8 алкила и C1-8 алкокси-группы, где C1-8 алкил и C1-8 алкокси группы необязательно замещены 1 - 3 заместителями Rz;
или, необязательно, R5 и R6 соединены с образованием спироциклического 5- или 6-членного циклоалкильного кольца, которое необязательно является ненасыщенным и имеет конденсированную арильную группу, которая необязательно замещена 1 - 4 заместителями Rz;
каждый Rx независимо выбран из группы, состоящей из галогена, -CN, -Rc, -CO2Ra, -CONRaRb, -C(O)Ra, -OC(O)NRaRb, -NRbC(O)Ra, -NRbC(O)2Rc, -NRa-C(O)NRaRb, -NRaC(O)NRaRb, -NRaRb, -ORa, -O-X1-ORa ,-O-X1-NRaRb, -O- X1-CO2Ra, -O-X1-CONRaRb, -X1-ORa, -X1-NRaRb,- X1-CO2Ra, -X1-CONRaRb, -SF5, -S(O)2NRaRb, и 5- или 6-членного арила или гетероарила, где каждый X1 представляет собой C1-4 алкилен; каждый Ra и Rb независимо выбран из атома водорода, C1-8 алкила и C1-8 галогеналкила, или в случае присоединения к одному и тому же атому азота могут объединяться с атомом азота, формируя 5- или 6-членное кольцо, содержащее в качестве членов цикла от 0 до 2 дополнительных гетероатомов, выбранных из N, O или S, и необязательно замещенное оксо-группой; каждый Rc независимо выбран из группы, состоящей из C1-8 алкила, C1-8 галогеналкила и C3-6 циклоалкила; и, необязательно, когда два заместителя Rx располагаются у соседних атомов, они объединены с образованием конденсированного 5- или 6-членного карбоциклического кольца, и где арильные или гетероарильные группы необязательно замещены 1-3 заместителями, выбранными из галогена, гидроксила, C1-4 алкила, C1-4 алкокси-группы, C1-4 галогеналкила и C1-4 галогеналкокси-группы;
каждый Ry независимо выбран из группы, состоящей из галогена, -CN, -Rf, -CO2Rd, -CONRdRe, -C(O)Rd, -OC(O)NRdRe, -NReC(O)Rd, -NReC(O)2Rf, -NRdC(O)NRdRe, -NRdC(O)NRdRe, -NRdRe, -ORd и -S(O)2NRdRe; где каждый Rd и Re независимо выбран из атома водорода, C1-8 алкила и C1-8 галогеналкила, или в случае присоединения к одному и тому же атому азота могут объединяться с атомом азота, формируя 5- или 6-членное кольцо, содержащее в качестве членов цикла от 0 до 2 дополнительных гетероатомов, выбранных из N, O или S; каждый Rf независимо выбран из группы, состоящей из C1-8 алкила, C1-8 галогеналкила и C3-6 циклоалкила;
каждый Rz независимо выбран из группы, состоящей из галогена, -CN, -Ri, -CO2Rg, -CONRgRh, -C(O)Rg, -OC(O)NRgRh, -NRhC(O)Rg, -NRhC(O)2Ri, -NRgC(O)NRgRh, -NRgRh, -ORg, -S(O)2NRgRh, -X1-Rj, -X1-NRgRh, -X1-CONRgRh, -X1-NRhC(O)Rg, -NHRj, -NHCH2Rj и тетразола; где каждый Rg и Rh независимо выбран из атома водорода, C1-8 алкила, C3-6 циклоалкила и C1-8 галогеналкила, или в случае присоединения к одному и тому же атому азота могут объединяться с атомом азота, формируя 5- или 6-членное кольцо, содержащее в качестве членов цикла от 0 до 2 дополнительных гетероатомов, выбранных из N, O или S, и необязательно замещенное одной или двумя оксо-группами; каждый Ri независимо выбран из группы, состоящей из C1-8 алкила, C1-8 галогеналкила и C3-6 циклоалкила; и каждый Rj выбран из группы, состоящей из C3-6 циклоалкила, пирролинила, пиперидинила, морфолинила, тетрагидрофуранила и тетрагидропиранила.
Следует понимать, что когда R1 и R2 объединены с атомом азота, к которому они присоединены, с образованием 6-11-членного моноциклического или конденсированного бициклического гетероциклического кольца, то данное 6-11-членное моноциклическое или конденсированное бициклическое гетероциклическое кольцо включает моноциклические гетероциклические кольца, сконденсированные с арильным или гетероарильным циклом.
В формуле I, заместитель R3, в одном варианте осуществления, выбран из группы, состоящей из H, метила, этила, пропила, изопропила, бутила, изобутила, втор-бутила, циклопропила, циклопропилметила, циклобутила и циклобутилметила.
В настоящем описании, квалифицированному специалисту в данной области будет понятно, что волнистая линия, пересекающая связь, означает точку присоединения данного заместителя или группы к остальной части молекулы.
Как отмечено выше, подстрочные индексы m и n представляют собой целые числа, выбранные из 0, 1 и 2, и m + n составляет ≤ 3. Когда подстрочный индекс равен 0, квалифицированному специалисту в данной области будет понятно, что имеется в виду циклическая структура с вершиной цикла A, но соседние вершины цикла по обе стороны скобок соединены связью. Соответственно, в настоящее изобретение входят структуры, где цикл, содержащий A в качестве вершины, включает:
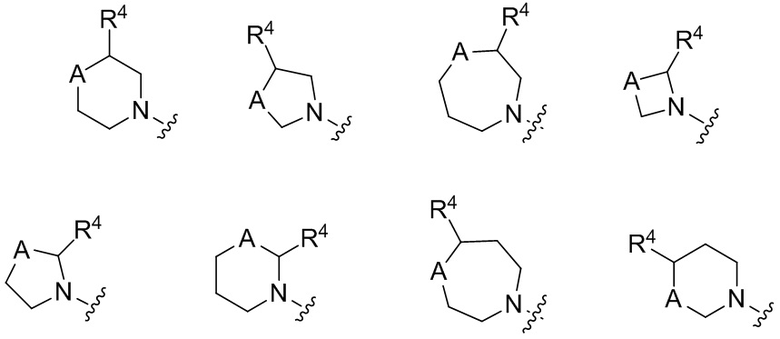 .
.
В одной частной группе вариантов осуществления, m и n оба равны 0. В другой частной группе вариантов осуществления, m и n оба равны 1. В другой частной группе вариантов осуществления, m равен 1, и n равен 0. В другой группе вариантов осуществления, m равен 1, и n равен 2.
В других частных вариантах осуществления, цикл, содержащий вершину A, представлен формулой, выбранной из:
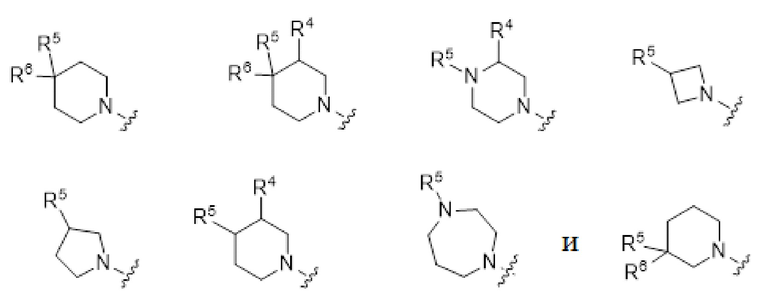 .
.
В одной подгруппе вариантов осуществления, соединения, имеющие формулу (I), представлены формулой:
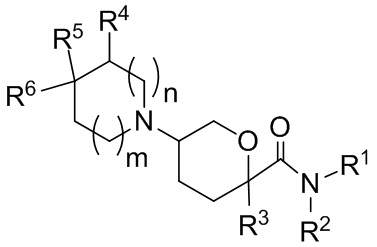 (Ia).
(Ia).
В рамках формулы (Ia), ряд частных вариантов осуществления может быть описан формулами Ia1, Ia2, Ia3, Ia4 и Ia5.
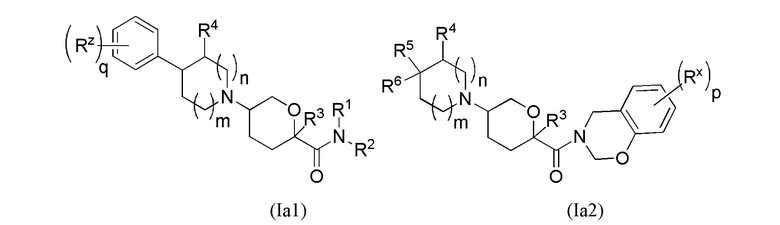
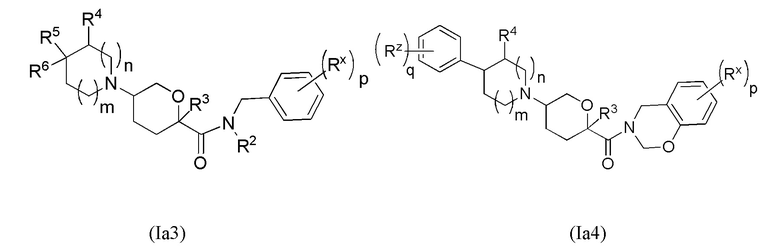
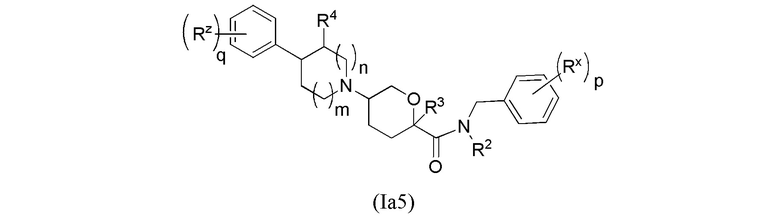
В каждой из формул Ia, Ia1, Ia2, Ia3, Ia4 и Ia5 указанные заместители (R1 - R6, Rx и Rz) и подстрочные индексы m и n имеют значения, указанные выше для формулы I. Подстрочные индексы p и q имеют следующие значения: для Ia1, Ia4 и Ia5, подстрочный индекс q представляет собой целое число от 0 до 5; для Ia2 и Ia4, подстрочный индекс p представляет собой целое число от 0 до 4; и для Ia3 и Ia5, подстрочный индекс p представляет собой целое число от 0 до 5.
В других частных вариантах осуществления, описанные в настоящем тексте соединения представлены формулами, выбранными из:
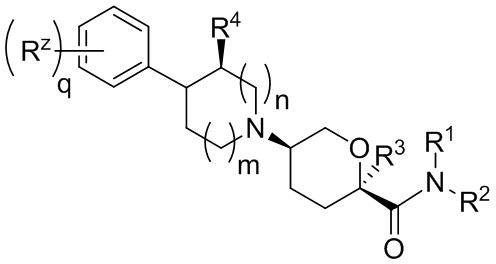
(Ia1’)
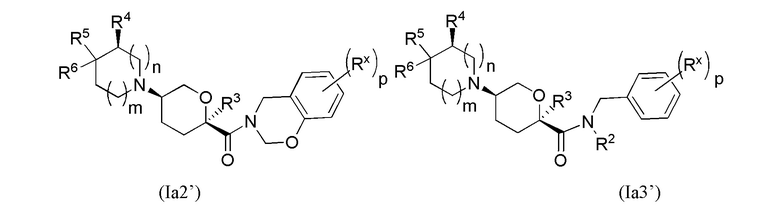
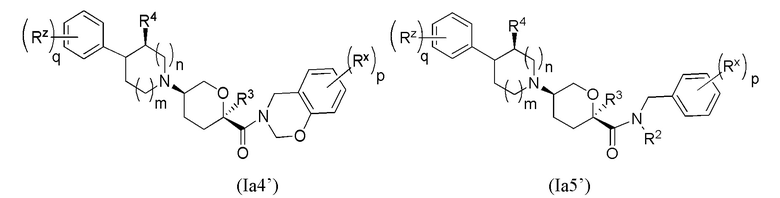
где каждое соединение практически не содержит других стереоизомеров, и где указанные заместители (R1 - R6, Rx и Rz) и подстрочные индексы m и n имеют значения, указанные выше для формулы I. Подстрочные индексы p и q имеют следующие значения: для Ia1’, Ia4’ и Ia5’, подстрочный индекс q представляет собой целое число от 0 до 5; для Ia2’ и Ia4’, подстрочный индекс p представляет собой целое число от 0 до 4; и для Ia3’ и Ia5’, подстрочный индекс p представляет собой целое число от 0 до 5.
В другой группе вариантов формулы I, A представляет собой C(R5)(R6), где R5 и R6 объединены с образованием кольца. Некоторые варианты осуществления приведены ниже:
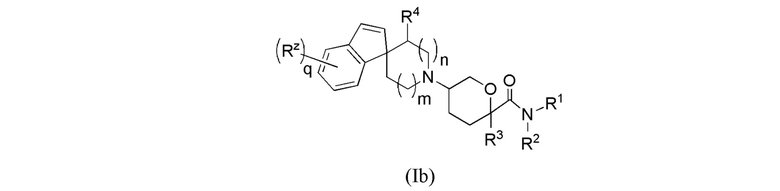
В каждой из формул Ib, Ib1 и Ib2 указанные заместители (R1 - R6, Rx и Rz) и подстрочные индексы m и n имеют значения, указанные выше для формулы I. Подстрочные индексы p и q имеют следующие значения: для Ib, Ib1 и Ib2, подстрочный индекс q представляет собой целое число от 0 до 5; для Ib1, подстрочный индекс p представляет собой целое число от 0 до 4; и для Ib2, подстрочный индекс p представляет собой целое число от 0 до 5.
В другой группе вариантов формулы I, A представляет собой NR5 (см.формулу Ic). Некоторые варианты осуществления приведены ниже:
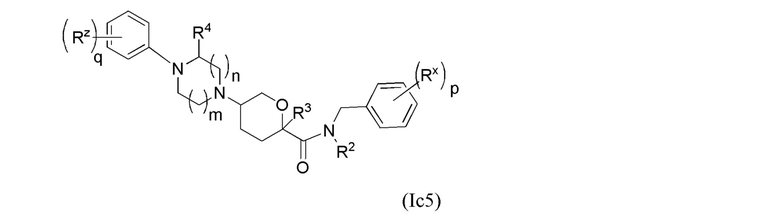
В каждой из формул Ic, Ic1, Ic2, Ic3, Ic4 и Ic5 указанные заместители (R1 - R6, Rx и Rz) и подстрочные индексы m и n имеют значения, указанные выше для формулы I. Подстрочные индексы p и q имеют следующие значения: для Ic1, Ic4 и Ic5, подстрочный индекс q представляет собой целое число от 0 до 5; для Ic2 и Ic4, подстрочный индекс p представляет собой целое число от 0 до 4; и для Ic3 и Ic5, подстрочный индекс p представляет собой целое число от 0 до 5.
В других частных вариантах осуществления, описанные в настоящем тексте соединения представлены формулами, выбранными из:
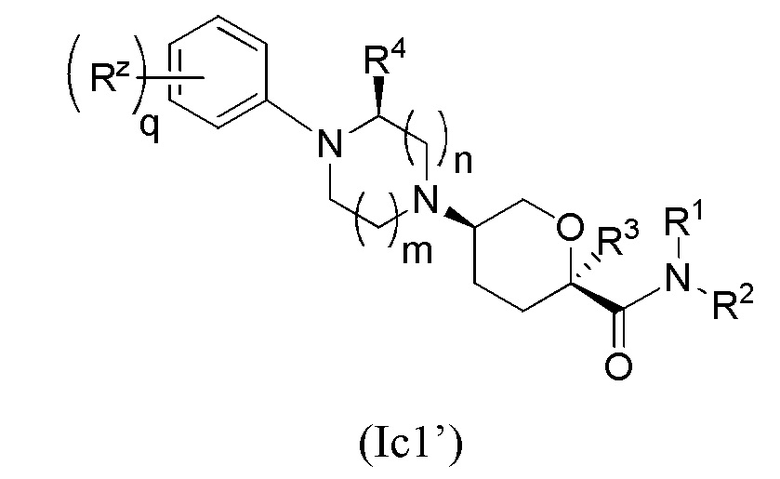

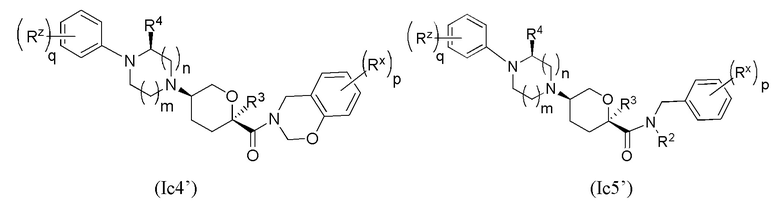
где каждое соединение практически не содержит других стереоизомеров, и где указанные заместители (R1 - R6, Rx и Rz) и подстрочные индексы m и n имеют значения, указанные выше для формулы I. Подстрочные индексы p и q имеют следующие значения: для Ic1’, Ic4’ и Ic5’, подстрочный индекс q представляет собой целое число от 0 до 5; для Ic2’ и Ic4’, подстрочный индекс p представляет собой целое число от 0 до 4; и для Ic3’ и Ic5’, подстрочный индекс p представляет собой целое число от 0 до 5.
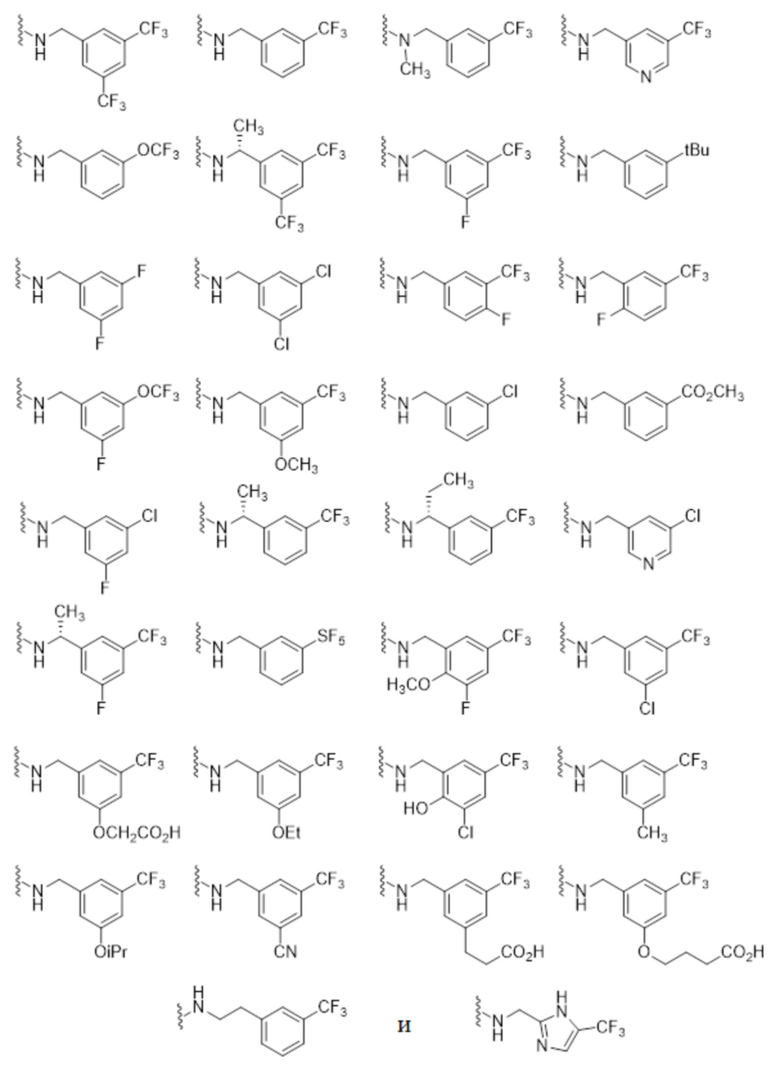
Другие частные варианты осуществления, соединения описаны для каждого из описанных выше I, Ia, Ia1, Ia1’, Ib, Ic, Ic1 и Ic1’, где -N(R1)(R2) выбран из:
 .
.
Другие частные варианты осуществления описаны для каждого из описанных выше I, Ia, Ia1, Ia1’, Ib, Ic, Ic1 и Ic1’, где -N(R1)(R2) выбран из:
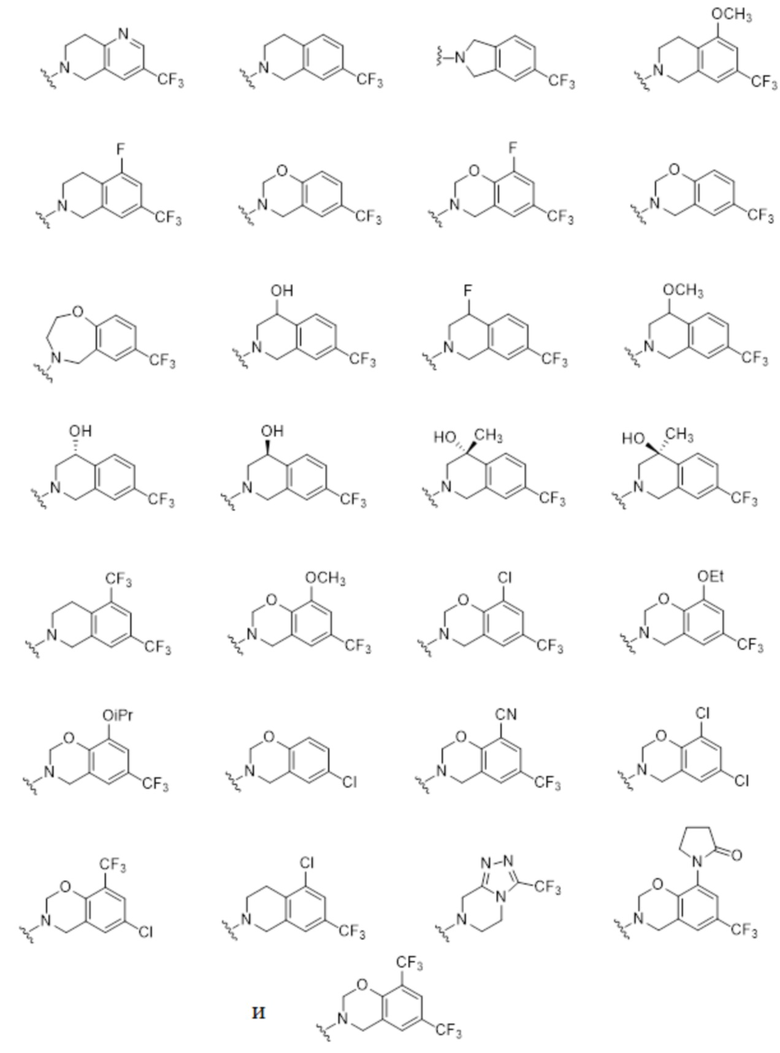 .
.
Другие частные варианты осуществления описаны для каждого из описанных выше I, Ia, Ia1, Ia1’, Ib, Ic, Ic1 и Ic1’, где -N(R1)(R2) выбран из:
 .
.
В некоторых вариантах осуществления описаны соединения, имеющие формулы I, Ia, Ia2, Ia3, Ia2’ и Ia3’, где A представляет собой C(R5)(R6) или показан в формуле как C(R5)(R6), где R5 выбран из арила, арилокси-группы, ариламино-группы, арил-C1-4 алкила, гетероарила, гетероарилокси-группы, гетероариламино-группы и гетероарил-C1-4 алкила, где арильные или гетероарильные группы или фрагменты выбраны из:
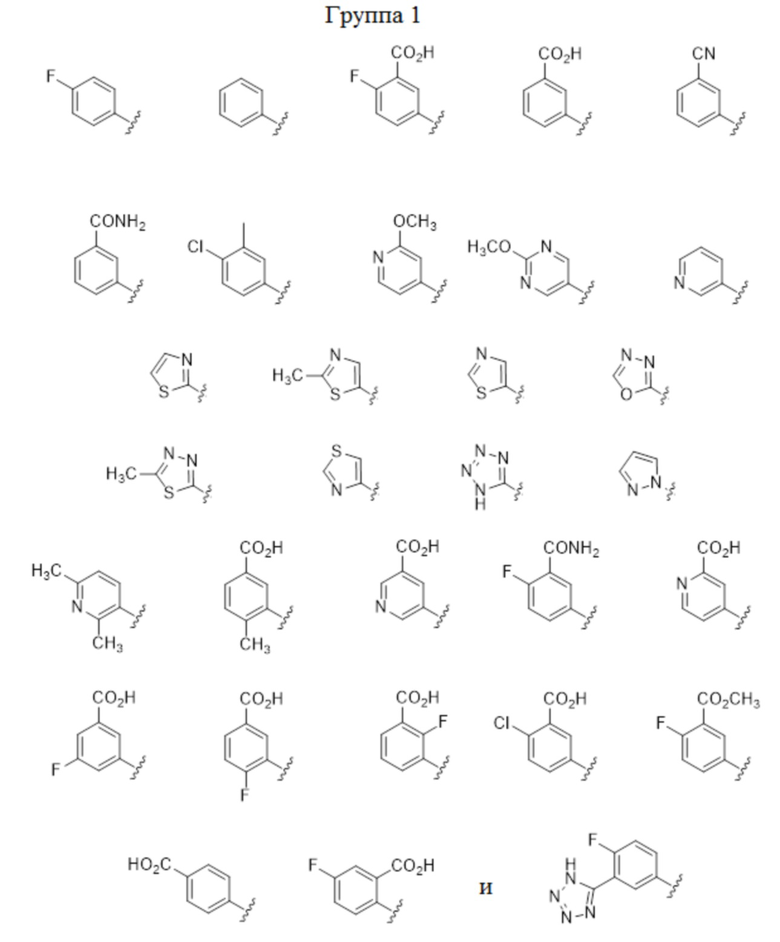 .
.
В некоторых частных вариантах осуществления описаны соединения, имеющие формулы I, Ia, Ia2, Ia3, Ia2’ и Ia3’, где A представляет собой C(R5)(R6) или показан в формуле как C(R5)(R6), где R5 выбран из арила, арилокси-группы, ариламино-группы и арил-C1-4 алкила, где арильная группа или фрагмент выбран из:
 .
.
В других частных вариантах осуществления, описаны соединения, имеющие формулы I, Ia, Ia2, Ia3, Ia2’ и Ia3’, где A представляет собой C(R5)(R6) или показан в формуле как C(R5)(R6), где R5 выбран из гетероарила, гетероарилокси-группы, гетероариламино-группы и гетероарил-C1-4 алкила, где гетероарильная группа или фрагмент выбран из:
 .
.
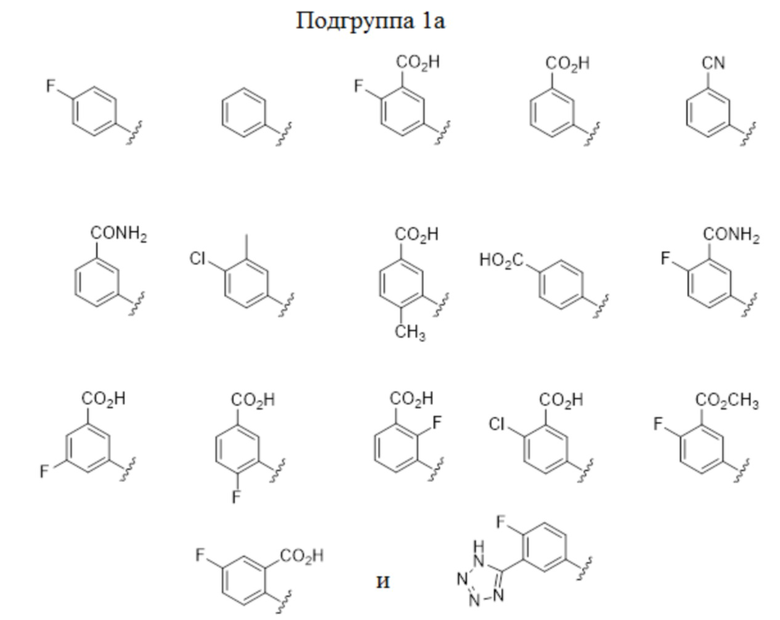
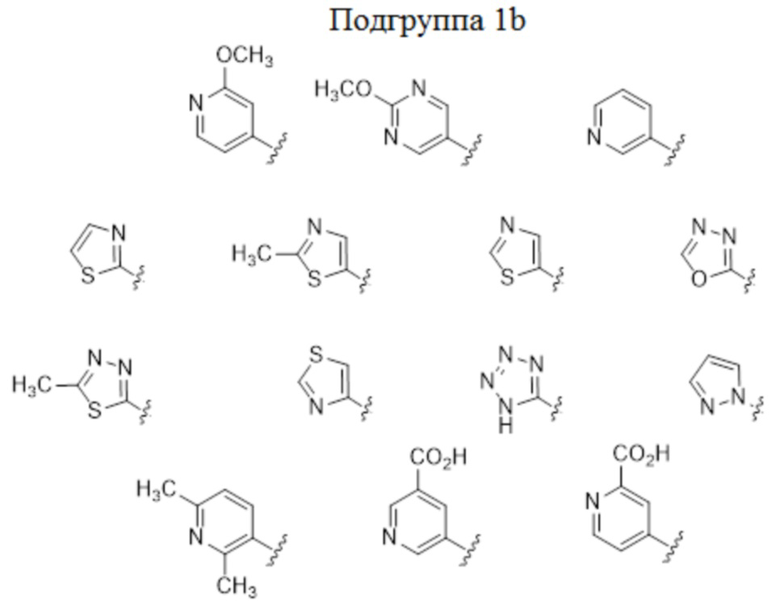
В некоторых вариантах осуществления, описаны соединения, имеющие формулы I, Ic, Ic2, Ic3, Ic2’ и Ic3’, где A представляет собой N(R5) или показан в формуле как N(R5), где R5 выбран из арила, арил-C1-4 алкила, гетероарила и гетероарил-C1-4 алкила, где арильные или гетероарильные группы или фрагменты выбраны из изображенной выше Группы 1. В некоторых частных вариантах осуществления, описаны соединения, имеющие формулы I, Ic, Ic2, Ic3, Ic2’ и Ic3’, где A представляет собой N(R5) или показан в формуле как N(R5), где R5 выбран из арила и арил-C1-4 алкила, где арильная группа или фрагмент выбраны из изображенной выше Подгруппы 1a. В других частных вариантах осуществления, описаны соединения, имеющие формулы I, Ic, Ic2, Ic3, Ic2’ и Ic3’, где A представляет собой N(R5) или показан в формуле как N(R5), где R5 выбран из гетероарила и гетероарил-C1-4 алкила, где гетероарильная группа или фрагмент выбраны из изображенной выше подгруппы 1b.
Получение соединений
Квалифицированным специалистам в данной области будет понятно, что имеются разные доступные способы синтеза молекул, представленных в Формуле изобретения. В целом, способы, которые могут применяться для синтеза соединений, представленных в Формуле изобретения, показаны на Схеме 1.
Схема 1

i: DIEA, NaBH(OAc)3, ДХЭ
ii: Pd/C, H2, MeOH
iii: HATU, DIEA, ДМФА
iv: параформальдегид, TsOH, толуол, кипячение
Вариации показанных выше способов применяли для получения соединений по настоящему изобретению, некоторые из которых описаны в Примерах.
Семейство соединений, имеющих формулу I, представляющих особый интерес, состоит из соединений, их фармацевтически приемлемых солей, гидратов, стереоизомеров и ротамеров, приведенных на фиг. 1.
Фармацевтические композиции
Помимо описанных выше соединений, композиции для модулирования активности CCR2 у человека и животных в типичном случае содержат фармацевтический носитель или разбавитель.
Термин "композиция" при использовании в настоящем тексте охватывает продукт, содержащий указанные ингредиенты в указанных количествах, а также любой продукт, получающийся напрямую или косвенно при комбинации указанных ингредиентов в указанных количествах. Термин «фармацевтически приемлемый» означает, что носитель, разбавитель или вспомогательное вещество должны быть совместимы с другими ингредиентами в препарате и не наносить вреда пациенту, принимающему препарат.
Фармацевтические композиции для введения соединений по настоящему изобретению удобно выпускать в единичной лекарственной форме, и их можно приготовить любым из методов, хорошо известных в области фармацевтики и введения лекарственных средств. Все методы включают стадию соединения действующего вещества с носителем, который содержит один или несколько вспомогательных ингредиентов. В целом, фармацевтические композиции готовят путем однородного и равномерного смешивания действующего вещества с жидким носителем или тонко измельченным твердым носителем, или с обоими, и затем, при необходимости, формования продукта в желаемый препарат. В фармацевтическую композицию действующее вещество включают в количестве, достаточном для достижения желаемого эффекта при болезненном процессе или состоянии.
Фармацевтические композиции, содержащие действующее вещество, могут иметь форму, подходящую для перорального применения, например форму таблеток, пастилок, ромбовидных таблеток, водных или масляных суспензий, диспергируемых порошков или гранул, эмульсий и самоэмульгирующихся составов, как описано в заявке на патент США2002-0012680, твердых или мягких капсул, сиропов, эликсиров, растворов, буккальных пластырей, гелей для перорального применения, жевательной резинки, жевательных таблеток, шипучих порошков и шипучих таблеток. Композиции для перорального применения можно приготовить согласно любым методам, известным в области производства фармацевтических композиций, и такие композиции могут содержать одно или больше средств, выбранных из группы, состоящей из подсластителей, ароматизаторов, красителей, антиоксидантов и консервантов, для создания фармацевтически удачных и приятных на вид препаратов. Таблетки содержат действующее вещество в смеси с нетоксичными фармацевтически приемлемыми вспомогательными веществами, которые подходят для производства таблеток. Такими вспомогательными веществами могут быть, например, инертные разбавители, такие как целлюлоза, диоксид кремния, оксид алюминия, карбонат кальция, карбонат натрия, глюкоза, маннит, сорбит, лактоза, фосфат кальция или фосфат натрия, гранулирующие средства и разрыхлители, например кукурузный крахмал или альгиновая кислота; связующие средства, например поливинилпирролидон, целлюлоза, ПЭГ, крахмал, желатин или камедь акации, и лубриканты, например стеарат магния, стеариновая кислота или тальк. Таблетки могут не иметь покрытия или могут иметь нанесенное покрытие, которое растворяется в кишечнике или другим известным образом замедляет распад и всасывание в желудочно-кишечном тракте, тем самым обеспечивая продолжительное действие в течение длительного периода времени. Например, можно применять замедляющее вещество, такое как глицерил моностеарат или глицерил дистеарат. Также таблетки могут иметь покрытие, нанесенное по методике, описанной в патенте США 4,256,108; 4,166,452 и 4,265,874, с формированием осмотических терапевтических таблеток с замедленным высвобождением.
Препараты для перорального применения могут также иметь вид твердых желатиновых капсул, в которых действующее вещество смешано с инертным твердым разбавителем, например карбонатом кальция, фосфатом кальция или каолином, или вид мягких желатиновых капсул, в которых действующее вещество смешано с водной или масляной средой, например арахисовым маслом, жидким парафином или оливковым маслом. Кроме того, эмульсии могут быть приготовлены с несмешивающимся с водой ингредиентом, таким как масло, и стабилизированы поверхностно-активными веществами, такими как моно-диглицериды, ПЭГ-эфиры и т.п.
Водные суспензии содержат действующие вещества в смеси со вспомогательными веществами, подходящими для производства водных суспензий. Такими вспомогательными веществами являются суспендирующие средства, например натрия карбоксиметилцеллюлоза, метилцеллюлоза, гидрокси-пропилметилцеллюлоза, альгинат натрия, поливинилпирролидон, трагакантовая камедь и смола акации; диспергирующие и смачивающие средства, которые могут представлять собой природные фосфатиды, например лецитин, или продукты конденсации алкиленоксида с жирными кислотами, например полиоксиэтилен стеарат, продукты конденсации этиленоксида с длинноцепочечными алифатическими спиртами, например гептадекаэтиленоксицетанол, или продукты конденсации этиленоксида с частичными сложными эфирами, полученными из жирных кислот и гекситола, такие как полиоксиэтилен сорбитол моноолеат, или продукты конденсации этиленоксида с частичными сложными эфирами, полученными из жирных кислот и гекситол-ангидридами, например полиэтилен сорбитан моноолеат. Водные суспензии могут также содержать один или больше консервантов, например этил или н-пропил пара-гидроксибензоат, один или больше красителей, один или больше ароматизаторов, и один или больше подсластителей, таких как сахароза или сахарин.
Масляные суспензии можно приготовить суспендированием действующего вещества в растительном масле, например в арахисовом масле, оливковом масле, сезамовом масле или кокосовом масле, или в минеральном масле, таком как жидкий парафин. Масляные суспензии могут содержать загуститель, например пчелиный воск, твердый парафин или цетиловый спирт. Можно добавлять подсластители, такие как описанные выше, и ароматизаторы для получения приятного препарат для перорального приема. Такие композиции можно консервировать добавлением антиоксиданта, такого как аскорбиновая кислота.
Диспергируемые порошки и гранулы, подходящие для приготовления водных суспензий путем добавления воды, содержат действующее вещество в смеси с диспергирующим или смачивающим средством, супендирующим средством и одним или больше консервантами. Примерами подходящих диспергирующих и смачивающих средств могут являться вещества, уже упомянутые выше. Также могут присутствовать дополнительные вспомогательные вещества, например подсластители, ароматизаторы и красители.
Фармацевтические композиции по настоящему изобретению могут также иметь форму эмульсий типа масло-в-воде. Масляной фазой может служить растительное масло, например оливковое масло или арахисовое масло, или минеральное масло, например жидкий парафин, или их смесь. Подходящими эмульгаторами могут быть природные смолы, например смола акации или трагакантовая камедь, природные фосфатиды, например соевое масло, лецитин, и сложные эфиры или неполные сложные эфиры, полученные из жирных кислот и гекситол-ангидридов, например сорбитан моноолеат, и продукты конденсации указанных неполных сложных эфиров с этиленоксидом, например полиоксиэтилен сорбитан моноолеат. Эмульсии могут также содержать подсластители и ароматизаторы.
В сиропы и эликсиры можно добавлять подсластители, например глицерин, пропиленгликоль, сорбит и сахарозу. Такие препараты могут также содержать мягчитель, консервант, ароматизатор и/или краситель. Растворы для перорального приема можно готовить в комбинации с циклодекстрином, ПЭГ и поверхностно-активными веществами.
Фармацевтические композиции могут иметь форму стерильных инъецируемых водных или масляных суспензий. Такую суспензию можно готовить согласно методам из существующего уровня техники, применяя перечисленные выше подходящие диспергирующие или смачивающие средства, а также суспендирующие средства. Стерильный инъецируемый препарат может также представлять собой стерильный инъецируемый раствор или суспензию в нетоксичном парэнтерально-приемлемом разбавителе или растворителе, например, раствор в 1,3-бутандиоле. Среди подходящих носителей и растворителей, которые могут применяться, можно упомянуть воду, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, стерильные жирные масла широко применяются в качестве растворителя или суспендирующей среды. Для этой цели можно применять любую марку жирного масла, включая синтетические моно- и диглицериды. Кроме того, в препаратах для инъекций нашли применение жирные кислоты, такие как олеиновая кислота.
Соединения по настоящему изобретению можно также вводить в форме суппозиториев для ректального введения лекарственных препаратов. Такие композиции можно готовить смешиванием лекарственного средства с подходящим нераздражающим вспомогательным веществом, которое является твердым при обычных температурах, но жидким при ректальной температуре, и поэтому плавится в заднем проходе, высвобождая лекарственное средство. Такие вещества включают масло какао и полиэтиленгликоли. Кроме того, соединения можно вводить через глаза посредством растворов или мазей. Кроме того, можно осуществлять чрезкожное введение рассматриваемых соединений посредством ионофоретических пластырей и т.п.. Для местного нанесения применяют кремы, мази, гели, растворы или суспензии и т.д., содержащие соединения по настоящем изобретению. При использовании в настоящем тексте, местное нанесение включает применение жидкостей для промывания и полоскания для рта.
Соединения по настоящему изобретению можно также использовать в сочетании с носителем, который может представлять собой подходящий полимерный носитель, такой как, например, носитель для таргетированного лекарственного средства. Такие полимеры могут включать поливинилпирролидон, сополимер пирана, полигидрокси-пропил-метакриламид-фенол, полигидроксиэтил-аспартамид-фенол или полиэтиленоксид-полилизин, замещенный пальмитоильными остатками. Кроме того, соединения по настоящему изобретению можно использовать в сочетании с носителями, которые представляют собой биоразлагаемые полимеры, которые можно применять для достижения контролируемого высвобождения лекарственного средства, например с полимолочной кислотой, полигликолевой кислотой, сополимерами полимолочной и полигликолевой кислот, поли-эпсилон-капролактоном, полигидроксимасляной кислотой, полиортоэфирами, полиацеталями, полидигидропиранами, полицианоакрилатами и сшитыми или амфипатическими блок-сополимерами гидрогелей. Полимеры и полупропускающие полимерные матриксы можно формовать в изделия, такие как клапаны, стенты, сосуды, протезы и т.п.. В одном варианте осуществления настоящего изобретения, соединение по настоящему изобретению вводят в сочетание с полимером или полупропускающим полимерным матриксом, которому придана форма стента или стент-графта.
В некоторых вариантах осуществления описана фармацевтическая композиция, содержащая соединения по настоящему изобретению и дополнительно содержащая одно или больше дополнительных терапевтических соединений.
В некоторых вариантах осуществления, одно или больше дополнительных терапевтических соединений выбраны из одного или больше представителей из следующих: ингибитор Btk тирозинкиназы, ингибитор тирозинкиназного рецептора Erbb2; ингибитор тирозинкиназного рецептора Erbb4, ингибитор mTOR, ингибитор тимидилатсинтазы, ингибитор тирозинкиназного рецептора EGFR, антагонист эпидермального фактора роста, ингибитор Fyn тирозинкиназы, ингибитор Kit тирозинкиназы, ингибитор Lyn тирозинкиназы, модулятор рецептора NK-клеток, антагонист PDGF рецептора, ингибитор PARP, ингибитор поли-АДФ-рибоза-полимеразы, ингибитор поли-АДФ-рибоза-полимеразы 1, ингибитор поли-АДФ-рибоза-полимеразы 2, ингибитор поли-АДФ-рибоза-полимеразы 3, модулятор галактозилтрансферазы, ингибитор дигидропиримидин дегидрогеназы, ингибитор оротат фосфорибозилтрансферазы, модулятор теломеразы, ингибитор муцина 1, ингибитор муцина, агонист секретина, модулятор ФНО-связанного апоптоз-индуцирующего лиганда, стимулятор гена IL17, лиганд интерлейкина 17Е, агонист нейрокининового рецептора, ингибитор циклина G1, ингибитор контрольных точек, ингибитор PD-1, ингибитор PD-L1, ингибитор CTLA4, ингибитор топоизомеразы I, ингибитор протеинкиназы Alk-5, ингибитор лиганда фактора роста соединительной ткани, антагонист рецептора Notch-2, антагонист рецептора Notch-3, стимулятор гиалуронидазы, ингибитор протеинкиназы MEK-1; ингибитор протеинкиназы MEK-2, модулятор рецептора GM-CSF; модулятор лиганда ФНО-альфа, модулятор мезотелина, стимулятор аспарагиназы, стимулятор каспазы-3; стимулятор каспазы-9, ингибитор гена PKN3, ингибитор хеджехог-белка; антагонист рецептора белка Smoothened, ингибитор гена AKT1, ингибитор DHFR, стимулятор тимидинкиназы, модулятор CD29, модулятор фибронектина, лиганд интерлейкина-2, ингибитор серинпротеазы, стимулятор гена D40LG; стимулятор гена TNFSF9, ингибитор 2 оксоглутарат дегидрогеназы, антагонист рецептора трансформирующего фактора роста-бета II типа, ингибитор тирозинкиназного рецептора Erbb3, антагонист рецептора холецистокинина CCK2, модулятор белка опухоли Вильмса, модулятор Ras ГТФазы, ингибитор гистондеацетилазы, ингибитор циклинзависимой киназы 4, модулятор эстрогенового рецептора бета, ингибитор 4-1BB, ингибитор 4-1BBL, ингибитор PD-L2, ингибитор B7-H3, ингибитор B7-H4, ингибитор BTLA, ингибитор HVEM, ингибитор TIM3, ингибитор GAL9, ингибитор LAG3, ингибитор VISTA, ингибитор KIR, ингибитор 2B4, CD160, модулятор CD66e, антагонист рецептора ангиотензина II, ингибитор лиганда фактора роста соединительной ткани, ингибитор тирозинкиназы Jak1, ингибитор тирозинкиназы Jak2, двойной ингибитор тирозинкиназы Jak1/Jak2, стимулятор ангиотензин-превращающего фермента 2, антагонист рецептора гормона роста, ингибитор галектина-3, ингибитор натрий-глюкозного транспортера-2, антагонист эндотелина ET-A, антагонист минералокортикоидного рецептора, антагонист эндотелина ET-B, антагонист рецептора конечного продукта гликозилирования, лиганд адренокортикотропного гормона, агонист рецептора фарнезоида X, агонист сопряженного с G-белком рецептора желчной кислоты, ингибитор альдозоредуктазы, ингибитор ксантиноксидазы, агонист PPAR гамма, антагонист рецептора простаноида, антагонист рецептора FGF, антагонист рецептора PDGF, антагонист трансформирующего фактора роста, ингибитор MAP-киназы p38, антагонист рецептора VEGF-1, ингибитор протеинтирозинфосфатазы бета, стимулятор тирозинкиназного рецептора Tek, ингибитор PDE 5, антагонист минералокортикоидного рецептора, ингибитор ACE, ингибитор I-каппа B-киназы, стимулятор гена NFE2L2, ингибитор ядерного фактора каппа-В, ингибитор гена STAT3, ингибитор НАДФ-оксидазы 1, ингибитор НАДФ-оксидазы 4, ингибитор PDE 4, ингибитор ренина, ингибитор протеинкиназы MEKK-5, ингибитор мембранной медь-аминооксидазы, антагонист интегрина альфа-V/бета-3, усилитель чувствительности рецепторов к инсулину, модулятор калликреина 1, ингибитор циклооксигеназы 1 и стимулятор фенилаланин гидроксилазы.
В некоторых вариантах осуществления, одно или больше дополнительных терапевтических соединений выбраны из одного или больше представителей из следующих: бавитуксимаб, IMM-101, CAP1-6D, Rexin-G, генистеин, CVac, MM-D37K, PCI-27483, TG-01, моноцетиностат, LOAd-703, CPI-613, упамостат, CRS-207, NovaCaps, траметиниб, Atu-027, сонидегиб, GRASPA, трабедерсен, насторазепид, Vaccell, ореговомаб, истиратумаб, рефаметиниб, регорафениб, лапатиниб, селуметиниб, рукапариб, пелареореп, тарекстумаб, ПЭГ-илированная гиалуронидаза, варлитиниб, аглатимаген бесаденовек, GBS-01, GI-4000, WF-10, галунисертиб, афатиниб, RX-0201, FG-3019, пертузумаб, DCVax-direct, селинексор, глуфосфамид, вирулизин, иттрий (90Y) кливатузумаб тетраксетан, бривудин, нимотузумаб, альгенпантуцел-L, тегафур + гимерацил + отерацил калия + кальция фолинат, олапариб, ибрутиниб, пирарубицин, Rh-Apo2L, тертомотид, тегафур + гимерацил + отерацил калия, тегафур + гимерацил + отерацил калия, мазитиниб, Rexin-G, митомицин, эрлотиниб, адриамицин, дексаметазон, винкристин, циклофосфамид, фторурацил, топотекан, таксол, интерфероны, производные платины, таксан, паклитаксел, алкалоиды барвинка, винбластин, антрациклины, доксорубицин, эпиподофиллотоксины, этопозид, цисплатин, рапамицин, метотрексат, актиномицин D, доластатин 10, колхицин, эметин, триметрексат, метоприн, циклоспорин, даунорубицин, тенипозид, амфотерицин, алкилирующие агенты, хлорамбуцил, 5-фторурацил, камфотерицин, цисплатин, метронидазол, гливек, авастин, вектибикс, абалерикс, альдеслейкин, алемтузумаб, алитретиноин, аллопуринол, альтретамин, амифостин, анастрозол, триоксид мышьяка, аспарагиназа, азацитидин, AZD9291, BCG Live, бевакузимаб, фторурацил, бексаротен, блеомицин, бортезомиб, бусульфан, калустерон, капецитабин, камфотецин, карбоплатин, кармустин, целекоксиб, цетуксимаб, хлорамбуцил, кладрибин, клофарабин, циклофосфамид, цитарабин, дактиномицин, дарбепоэтин альфа, даунорубицин, денилейкин, дексразоксан, доцетаксел, доксорубицин (нейтральный), доксорубицин гидрохлорид, дромостанолон пропионат, эпирубицин, эпоэтин альфа, эстрамустин, этопозид фосфат, этопозид, эксеместан, филграстим, флоксуридин, флударабин, фулвестрант, гефитиниб, гемцитабин, гемтузумаб, госерелин ацетат, гистрелин ацетат, гидроксимочевина, ибритумомаб, идарубицие, ифосфамид, иматиниб мезилат, интерферон альфа-2a, интерферон альфа-2b, иринотекан, леналидомид, летрозол, лейковорин, левпролид ацетат, левамизол, ломустин, мегестрол ацетат, мелфалан, меркаптопурин, 6-MP, месна, метотрексат, метокссален, митомицин C, митотан, митоксантрон, нандролон, неларабин, нофетумомаб, опрелвекин, оксалиплатин, наб-паклитаксел, палифермин, памидронат, пегадемаза, пегаспаргаза, пегфилграстим, пеметрексед динатрия, пентостатин, пипоброман, пликамицин, порфимер натрия, прокарбазин, хинакрин, расбуриказа, ритуксимаб, роцилетиниб, сарграмостим, сорафениб, стрептозоцин, сунитиниб малеат, тальк, тамоксифен, темозоломид, тенипозид, VM-26, тестолактон, тиогуанин, 6-TG, тиотепа, топотекан, торемифен, тозитумомаб, трастузумаб, третиноин, ATRA, урамустин, валрубицин, винбластин, винкристин, винорелбин, золедронат, золедроновая кислота, пембролизумаб, ниволумаб, IBI-308, mDX-400, BGB-108, MEДИ-0680, SHR-1210, PF-06801591, PDR-001, GB-226, STI-1110, дурвалумаб, атезолизумаб, авелумаб, BMS-936559, ALN-PDL, TSR-042, KD-033, CA-170, STI-1014, FOLFIRINOX, KY-1003, олмесартан медоксомил, кандесартан, PBI-4050, барицитиниб, GSK-2586881, лозартан, дапаглифлозин пропандиол, пегвисомант, GR-MD-02, каналифлозин, ирбесартан, FG-3019, атрасентан, финеренон, спарсентан, босентан, дефибротид, фимасартан, азелирагон, пиридоксамин, кортикотропин, INT-767, эпалрестат, топироксостат, SER-150-DN, пирфенидон, VEGFR-1 mAb, AKB-9778, PF-489791, SHP-627, CS-3150, имидаприл, периндоприл, каптоприл, эналаприл, лизиноприл, зофеноприл, лизиноприл, хинаприл, беназеприл, трандолаприл, цилазаприл, фосиноприл, рамиприл, бардолоксон метил, ирбесартан + пропагерманий, GKT-831, MT-3995, TAK-648, TAK-272, GS-4997, DW-1029M, ASP-8232, VPI-2690B, DM-199, рейн, PHN-033, GLY-230, сапроптерин, сулодексид.
В некоторых вариантах осуществления, одно или больше дополнительных терапевтических соединений представляют собой ингибитор ангиотензин-превращающего фермента (ACE) или блокатор рецепторов ангиотензина II (ARB). В некоторых вариантах осуществления, одно или больше дополнительных терапевтических соединений представляют собой ингибитор ангиотензин-превращающего фермента (ACE). В некоторых вариантах осуществления, одно или больше дополнительных терапевтических соединений представляют собой блокатор рецепторов ангиотензина II (ARB). В некоторых вариантах осуществления, одно или больше дополнительных терапевтических соединений представляют собой олмесартан медоксомил, кандесартан, лозартан, ирбесартан, спарсентан, фимасартан, GSK-2586881, имидаприл, периндоприл, каптоприл, эналаприл, лизиноприл, зофеноприл, лизиноприл, хинаприл, беназеприл, трандолаприл, цилазаприл, фозиноприл или рамиприл.
В некоторых вариантах осуществления одно или больше дополнительных терапевтических соединений представляют собой FOLFIRINOX. В некоторых вариантах осуществления, одно или больше дополнительных терапевтических соединений представляют собой гемцитабин и паклитаксел. В некоторых вариантах осуществления, одно или больше дополнительных терапевтических соединений представляют собой гемцитабин и наб-паклитаксел.
Соединения, модулирующие активность CCR2
В настоящем изобретении описаны соединения, модулирующие активность по меньшей мере одного CCR2. Хемокиновые рецепторы представляют собой интегральные мембранные белки, которые взаимодействуют с внеклеточным лигандом, таким как хемокин, и опосредуют клеточный ответ на лиганд, например, хемотаксис, повышенная внутриклеточная концентрация ионов кальция и т.д.. Поэтому модулирование функции хемокинового рецептора, например нарушение взаимодействий хемокиновый рецептор-лиганд, модулирует опосредуемый хемокиновым рецептором ответ, и лечит или предотвращает патологическое состояние или заболевание, вызываемое хемокиновым рецептором. Модулирование функции хемокинового рецептора включает как стимулирование, так и подавление его функции. Тип осуществляемого модулирования зависит от характеристик соединения, являющегося, например, антагонистом или полным, частичным или обратным агонистом.
Без привязки к какой-либо конкретной теории, считают, что описанные в настоящем тексте соединения нарушают взаимодействие между хемокиновым рецептором и одним или больше когнатными лигандами. В частности, считают, что описанные в настоящем тексте соединения нарушают взаимодействие между CCR2 и CCR2 лигандом, таким как MCP-1. Соединения, охватываемые настоящим изобретением, включают (но не ограничиваются только ими) иллюстративные соединения, описанные в настоящем тексте, и их соли.
Например, подходящие соединения работают как потенциальные CCR2 антагонисты, и эта антагонистическая активность была дополнительно подтверждена в тестах на животных в отношении воспаления, одного из отличительных болезненных состояний для CCR2. Соответственно, описанные в настоящем тексте соединения могут применяться в фармацевтических композициях, способах лечения CCR2-опосредованных заболеваний, и в качестве контроля в тестах, направленных на идентификацию конкурентно-способных CCR2 антагонистов.
Способы лечения
Модулирование функции CCR2 рецептора
Соединения по настоящему изобретению могут применяться в качестве агонистов, (предпочтительно) антагонистов, частичных агонистов, обратных агонистов CCR2 рецепторов с различном контексте, как in vitro, так и in vivo. В одном варианте осуществления, соединения по настоящему изобретению представляют собой CCR2 антагонисты, которые могут применяться для ингибирования связывания лиганда CCR2 рецептора с CCR2 рецептором in vitro или in vivo. В целом, такие методы включают стадию контакта CCR2 рецептора с достаточным количеством одного или больше описанных в настоящем тексте модуляторов рецептора CCR2, в присутствии лиганда CCR2 рецептора в водном растворе и в условиях, подходящих для связывания лиганда с CCR2 рецептором. CCR2 рецептор может присутствовать в суспензии (например, в препарате изолированных мембран или клеток), в культивируемых или изолированных клетках, или в ткани или органе.
Предпочтительно, количество модулятора CCR2 рецептора, контактирующего с рецептором, должно быть достаточным для ингибирования связывания лиганда с CCR2 рецептором in vitro, при измерении, например, с помощью радиолигандного анализа, анализа притока кальция или анализа хемотаксиса, описанных в настоящем тексте.
В одном варианте осуществления настоящего изобретения, модуляторы CCR2 по настоящему изобретению применяют для модулирования, предпочтительно ингибирования, сигнал-преобразующей функции CCR2 рецептора, например, путем контакта одного или больше соединений по настоящему изобретению с CCR2 рецептором (in vitro или in vivo) в условиях, подходящих для связывания модулятора(-ов) с рецептором. Рецептор может находиться в растворе или суспензии, в препарате культивируемых или изолированных клеток, или в пациенте. Любое модулирование сигнал-преобразующей активности можно оценить посредством детектирования влияния на мобилизацию ионов кальция или посредством детектирования влияния на CCR2 рецептор-опосредованный клеточный хемотаксис. В целом, эффективное количество модулятора(-ов) СCR2 представляет собой количество, достаточное для модулирования сигнал-преобразующей активности CCR2 рецептора in vitro в ходе анализа мобилизации кальция, или CCR2 рецептор-опосредованного клеточного хемотаксиса в ходе анализа миграции.
Когда соединения по настоящему изобретению применяют для ингибирования CCR2 рецептор-опосредованного клеточного хемотаксиса, предпочтительно таксиса лейкоцитов, в ходе in vitro анализа хемотаксиса, такие методы включают контакт клеток крови (в частности, белых кровяных телец приматов, в особенности белых кровяных телец человека) с одним или больше соединениями по настоящему изобретению. Предпочтительно, концентрация достаточна для ингибирования хемотаксиса белых кровяных телец в ходе in vitro анализа хемотаксиса, так что уровни хемотаксиса, наблюдающиеся в контрольном эксперименте, существенно выше, как описано выше, чем уровни, наблюдающиеся в анализе при добавлении соединения по настоящему изобретению.
В другом варианте осуществления, соединения по настоящему изобретению могут также применяться для лечения пациентов, страдающих от состояний, реагирующих на модулирование CCR2 рецептора. При использовании в настоящем тексте, термин "лечить" или "лечение" включает как болезнь-модифицирующее лечение, так и симптоматическое лечение, которые оба могут быть профилактическим (т.е. проводящимся до появления симптомов, с целью предотвратить, задержать или уменьшить тяжесть симптомов) или терапевтическим (т.е. проводящимся после появления симптомов, с целью уменьшить степень тяжести и/или длительность симптомов). При использовании в настоящем тексте, состояние считают "реагирующим на модулирование CCR2 рецептора", если модулирование активности CCR2 рецептора приводит к уменьшению ненадлежащей активности CCR2 рецептора. При использовании в настоящем тексте, термин "пациенты" включает приматов (в особенности, людей), домашних животных-компаньонов (таких как собаки, кошки, лошади и т.п.) и домашний скот (такой как крупный рогатый скот, свиньи, овцы и т.п.), при применении описанных в настоящем тексте дозировок.
В некоторых вариантах осуществления, соединения по настоящему изобретению могут также применяться для лечения пациентов, страдающих воспалительным заболеванием или нарушением, сердечно-сосудистым или церебро-васкулярным нарушением, аутоиммунным заболеванием, раковым заболеванием или солидной опухолью. В некоторых вариантах осуществления, соединения по настоящему изобретению можно применять для лечения пациентов, страдающих заболеванием или нарушением, выбранным из группы, состоящей из следующих: диабетическая нефропатия, нефропатия, нейтропения, нейтрофилия, альбуминурия, диабетическая ретинопатия, фокально-сегментарный гломерулосклероз, гломерулосклероз, аллергия, фиброз, НАСГ (неалкогольный стеатогепатит), гемолитико-уремический синдром, атипичный гемолитико-уремический синдром (aHUS), C3-гломерулопатия, C3-гломерулонефрит, болезнь плотного осадка, мезангиопролиферативный гломерулонефрит, сепсис, септический шок, болезнь Альцгеймера, множественный склероз, инсульт, воспалительная болезнь кишечника, хроническая обструктивная болезнь легких, воспаление вследствие ожогов, травма легких, остеоартрит, атопический дерматит, хроническая уртикария, ишемически-реперфузионное повреждение, синдром острой дыхательной недостаточности, синдром системной воспалительной реакции, синдром полиорганной недостаточности, отторжение тканевого трансплантата, реакция «трансплантат против хозяина», сверхострое отторжение трансплантированных органов, инфаркт миокарда, тромбоз коронарных артерий, закупорка сосудов, постхирургическая реокклюзия сосудов, артериосклероз, травматическое повреждение центральной нервной системы, ишемическая болезнь сердца, ревматоидный артрит, системная красная волчанка, синдром Гийена-Барре, панкреатит, волчаночный нефрит, волчаночный гломерулонефрит, псориаз, болезнь Крона, васкулит, АНЦА-васкулит, синдром раздраженного кишечника, дерматомиозит, множественный склероз, бронхиальная астма, пемфигус, пемфигоид, склеродерма, миастения гравис, аутоиммунные гемолитические и тромбоцитопенические состояния, синдром Гудпасчера, иммуноваскулит, отторжение тканевого трансплантата, сверхострое отторжение трансплантированных органов, меланома, мелкоклеточная карцинома легких, немелкоклеточная карцинома легких, рак поджелудочной железы, рак молочной железы, рак мочевого пузыря, почечноклеточный рак, рак толстой и прямой кишки, гепатоцеллюлярная карцинома, плоскоклеточный рак головы и шеи, рак пищевода, рак яичника, рак предстательной железы, рак желудка, острый миелогенный лейкоз, лейкоз, патологические осложнения, связанные с группой заболеваний, состоящей из инсулинозависимого сахарного диабета, волчаночной нефропатии, нефрита Хеймана, мембранного нефрита, гломерулонефрита, ответа вследствие контактной чувствительности, и воспаление, вызванное контактом крови с искусственными поверхностями.
В некоторых вариантах осуществления, соединения по настоящему изобретению можно применять для лечения пациентов, страдающих от заболевания или нарушения, и вводить с одним или больше дополнительными терапевтическими соединениями. В некоторых вариантах осуществления, одно или больше дополнительных терапевтических соединений выбраны из одного или больше представителей из следующих: ингибитор Btk тирозинкиназы, ингибитор тирозинкиназного рецептора Erbb2; ингибитор тирозинкиназного рецептора Erbb4, ингибитор mTOR, ингибитор тимидилатсинтазы, ингибитор тирозинкиназного рецептора EGFR, антагонист эпидермального фактора роста, ингибитор Fyn тирозинкиназы, ингибитор Kit тирозинкиназы, ингибитор Lyn тирозинкиназы, модулятор рецептора NK-клеток, антагонист PDGF рецептора, ингибитор PARP, ингибитор поли-АДФ-рибоза-полимеразы, ингибитор поли-АДФ-рибоза-полимеразы 1, ингибитор поли-АДФ-рибоза-полимеразы 2, ингибитор поли-АДФ-рибоза-полимеразы 3, модулятор галактозилтрансферазы, ингибитор дигидропиримидин дегидрогеназы, ингибитор оротат фосфорибозилтрансферазы, модулятор теломеразы, ингибитор муцина 1, ингибитор муцина, агонист секретина, модулятор ФНО-связанного апоптоз-индуцирующего лиганда, стимулятор гена IL17, лиганд интерлейкина 17Е, агонист нейрокининового рецептора, ингибитор циклина G1, ингибитор контрольных точек, ингибитор PD-1, ингибитор PD-L1, ингибитор CTLA4, ингибитор топоизомеразы I, ингибитор протеинкиназы Alk-5, ингибитор лиганда фактора роста соединительной ткани, антагонист рецептора Notch-2, антагонист рецептора Notch-3, стимулятор гиалуронидазы, ингибитор протеинкиназы MEK-1; ингибитор протеинкиназы MEK-2, модулятор рецептора GM-CSF; модулятор лиганда ФНО-альфа, модулятор мезотелина, стимулятор аспарагиназы, стимулятор каспазы-3; стимулятор каспазы-9, ингибитор гена PKN3, ингибитор хеджехог-белка; антагонист рецептора белка Smoothened, ингибитор гена AKT1, ингибитор DHFR, стимулятор тимидинкиназы, модулятор CD29, модулятор фибронектина, лиганд интерлейкина-2, ингибитор серинпротеазы, стимулятор гена D40LG; стимулятор гена TNFSF9, ингибитор 2 оксоглутарат дегидрогеназы, антагонист рецептора трансформирующего фактора роста-бета II типа, ингибитор тирозинкиназного рецептора Erbb3, антагонист рецептора холецистокинина CCK2, модулятор белка опухоли Вильмса, модулятор Ras ГТФазы, ингибитор гистондеацетилазы, ингибитор циклинзависимой киназы 4, модулятор эстрогенового рецептора бета, ингибитор 4-1BB, ингибитор 4-1BBL, ингибитор PD-L2, ингибитор B7-H3, ингибитор B7-H4, ингибитор BTLA, ингибитор HVEM, ингибитор TIM3, ингибитор GAL9, ингибитор LAG3, ингибитор VISTA, ингибитор KIR, ингибитор 2B4, CD160, модулятор CD66e, антагонист рецептора ангиотензина II, ингибитор лиганда фактора роста соединительной ткани, ингибитор тирозинкиназы Jak1, ингибитор тирозинкиназы Jak2, двойной ингибитор тирозинкиназы Jak1/Jak2, стимулятор ангиотензин-превращающего фермента 2, антагонист рецептора гормона роста, ингибитор галектина-3, ингибитор натрий-глюкозного транспортера-2, антагонист эндотелина ET-A, антагонист минералокортикоидного рецептора, антагонист эндотелина ET-B, антагонист рецептора конечного продукта гликозилирования, лиганд адренокортикотропного гормона, агонист рецептора фарнезоида X, агонист сопряженного с G-белком рецептора желчной кислоты, ингибитор альдозоредуктазы, ингибитор ксантиноксидазы, агонист PPAR гамма, антагонист рецептора простаноида, антагонист рецептора FGF, антагонист рецептора PDGF, антагонист трансформирующего фактора роста, ингибитор MAP-киназы p38, антагонист рецептора VEGF-1, ингибитор протеинтирозинфосфатазы бета, стимулятор тирозинкиназного рецептора Tek, ингибитор PDE 5, антагонист минералокортикоидного рецептора, ингибитор ACE, ингибитор I-каппа B-киназы, стимулятор гена NFE2L2, ингибитор ядерного фактора каппа-В, ингибитор гена STAT3, ингибитор НАДФ-оксидазы 1, ингибитор НАДФ-оксидазы 4, ингибитор PDE 4, ингибитор ренина, ингибитор протеинкиназы MEKK-5, ингибитор мембранной медь-аминооксидазы, антагонист интегрина альфа-V/бета-3, усилитель чувствительности рецепторов к инсулину, модулятор калликреина 1, ингибитор циклооксигеназы 1 и стимулятор фенилаланин гидроксилазы.
В некоторых вариантах осуществления, одно или больше дополнительных терапевтических соединений выбраны из одного или больше представителей из следующих: бавитуксимаб, IMM-101, CAP1-6D, Rexin-G, генистеин, CVac, MM-D37K, PCI-27483, TG-01, моноцетиностат, LOAd-703, CPI-613, упамостат, CRS-207, NovaCaps, траметиниб, Atu-027, сонидегиб, GRASPA, трабедерсен, насторазепид, Vaccell, ореговомаб, истиратумаб, рефаметиниб, регорафениб, лапатиниб, селуметиниб, рукапариб, пелареореп, тарекстумаб, ПЭГ-илированная гиалуронидаза, варлитиниб, аглатимаген бесаденовек, GBS-01, GI-4000, WF-10, галунисертиб, афатиниб, RX-0201, FG-3019, пертузумаб, DCVax-direct, селинексор, глуфосфамид, вирулизин, иттрий (90Y) кливатузумаб тетраксетан, бривудин, нимотузумаб, альгенпантуцел-L, тегафур + гимерацил + отерацил калия + кальция фолинат, олапариб, ибрутиниб, пирарубицин, Rh-Apo2L, тертомотид, тегафур + гимерацил + отерацил калия, тегафур + гимерацил + отерацил калия, мазитиниб, Rexin-G, митомицин, эрлотиниб, адриамицин, дексаметазон, винкристин, циклофосфамид, фторурацил, топотекан, таксол, интерфероны, производные платины, таксан, паклитаксел, алкалоиды барвинка, винбластин, антрациклины, доксорубицин, эпиподофиллотоксины, этопозид, цисплатин, рапамицин, метотрексат, актиномицин D, доластатин 10, колхицин, эметин, триметрексат, метоприн, циклоспорин, даунорубицин, тенипозид, амфотерицин, алкилирующие агенты, хлорамбуцил, 5-фторурацил, камфотерицин, цисплатин, метронидазол, гливек, авастин, вектибикс, абалерикс, альдеслейкин, алемтузумаб, алитретиноин, аллопуринол, альтретамин, амифостин, анастрозол, триоксид мышьяка, аспарагиназа, азацитидин, AZD9291, BCG Live, бевакузимаб, фторурацил, бексаротен, блеомицин, бортезомиб, бусульфан, калустерон, капецитабин, камфотецин, карбоплатин, кармустин, целекоксиб, цетуксимаб, хлорамбуцил, кладрибин, клофарабин, циклофосфамид, цитарабин, дактиномицин, дарбепоэтин альфа, даунорубицин, денилейкин, дексразоксан, доцетаксел, доксорубицин (нейтральный), доксорубицин гидрохлорид, дромостанолон пропионат, эпирубицин, эпоэтин альфа, эстрамустин, этопозид фосфат, этопозид, эксеместан, филграстим, флоксуридин, флударабин, фулвестрант, гефитиниб, гемцитабин, гемтузумаб, госерелин ацетат, гистрелин ацетат, гидроксимочевина, ибритумомаб, идарубицие, ифосфамид, иматиниб мезилат, интерферон альфа-2a, интерферон альфа-2b, иринотекан, леналидомид, летрозол, лейковорин, левпролид ацетат, левамизол, ломустин, мегестрол ацетат, мелфалан, меркаптопурин, 6-MP, месна, метотрексат, метокссален, митомицин C, митотан, митоксантрон, нандролон, неларабин, нофетумомаб, опрелвекин, оксалиплатин, наб-паклитаксел, палифермин, памидронат, пегадемаза, пегаспаргаза, пегфилграстим, пеметрексед динатрия, пентостатин, пипоброман, пликамицин, порфимер натрия, прокарбазин, хинакрин, расбуриказа, ритуксимаб, роцилетиниб, сарграмостим, сорафениб, стрептозоцин, сунитиниб малеат, тальк, тамоксифен, темозоломид, тенипозид, VM-26, тестолактон, тиогуанин, 6-TG, тиотепа, топотекан, торемифен, тозитумомаб, трастузумаб, третиноин, ATRA, урамустин, валрубицин, винбластин, винкристин, винорелбин, золедронат, золедроновая кислота, пембролизумаб, ниволумаб, IBI-308, mDX-400, BGB-108, MEДИ-0680, SHR-1210, PF-06801591, PDR-001, GB-226, STI-1110, дурвалумаб, атезолизумаб, авелумаб, BMS-936559, ALN-PDL, TSR-042, KD-033, CA-170, STI-1014, FOLFIRINOX, KY-1003, олмесартан медоксомил, кандесартан, PBI-4050, барицитиниб, GSK-2586881, лозартан, дапаглифлозин пропандиол, пегвисомант, GR-MD-02, каналифлозин, ирбесартан, FG-3019, атрасентан, финеренон, спарсентан, босентан, дефибротид, фимасартан, азелирагон, пиридоксамин, кортикотропин, INT-767, эпалрестат, топироксостат, SER-150-DN, пирфенидон, VEGFR-1 mAb, AKB-9778, PF-489791, SHP-627, CS-3150, имидаприл, периндоприл, каптоприл, эналаприл, лизиноприл, зофеноприл, лизиноприл, хинаприл, беназеприл, трандолаприл, цилазаприл, фосиноприл, рамиприл, бардолоксон метил, ирбесартан + пропагерманий, GKT-831, MT-3995, TAK-648, TAK-272, GS-4997, DW-1029M, ASP-8232, VPI-2690B, DM-199, рейн, PHN-033, GLY-230, сапроптерин, сулодексид.
В некоторых вариантах осуществления одно или больше дополнительных терапевтических соединений представляют собой ингибитор ангиотензин-превращающего фермента (ACE) или блокатор рецепторов ангиотензина II (ARB). В некоторых вариантах осуществления, одно или больше дополнительных терапевтических соединений представляют собой ингибитор ангиотензин-превращающего фермента (ACE). В некоторых вариантах осуществления, одно или больше дополнительных терапевтических соединений представляют собой блокатор рецепторов ангиотензина II (ARB). В некоторых вариантах осуществления, одно или больше дополнительных терапевтических соединений представляют собой олмесартан медоксомил, кандесартан, лозартан, ирбесартан, спарсентан, фимасартан, GSK-2586881, имидаприл, периндоприл, каптоприл, эналаприл, лизиноприл, зофеноприл, лизиноприл, хинаприл, беназеприл, трандолаприл, цилазаприл, фозиноприл или рамиприл.
В некоторых вариантах осуществления одно или больше дополнительных терапевтических соединений представляют собой FOLFIRINOX. В некоторых вариантах осуществления, одно или больше дополнительных терапевтических соединений представляют собой гемцитабин и паклитаксел. В некоторых вариантах осуществления, одно или больше дополнительных терапевтических соединений представляют собой гемцитабин и наб-паклитаксел.
Состояния, которые можно лечить посредством модулирования CCR2:
Аутоиммунные нарушения - например, ревматоидный артрит, системная красная волчанка, синдром Гийена-Барре, панкреатит, волчаночный нефрит, волчаночный гломерулонефрит, псориаз, болезнь Крона, васкулит, синдром раздраженного кишечника, дерматомиозит, множественный склероз, бронхиальная астма, пемфигус, пемфигоид, склеродерма, миастения гравис, аутоиммунные гемолитические и тромбоцитопенические состояния, синдром Гудпасчера (и связанные с ним гломерулонефрит и легочное кровотечение), иммуноваскулит, отторжение тканевого трансплантата, сверхострое отторжение трансплантированных органов и т.п.
Воспалительные заболевания и связанные с ними состояния - например, нейтропения, сепсис, септический шок, болезнь Альцгеймера, множественный склероз, инсульт, воспалительная болезнь кишечника (IBD), воспаление вследствие сильных ожогов, повреждение легких, ишемически-реперфузионное повреждение, остеоартрит, а также синдром острой дыхательной недостаточности (ARDS), хроническое воспалительное заболевание легких (ХВЗЛ), синдром системной воспалительной реакции (SIRS), атопический дерматит, псориаз, хроническая уртикария и синдром полиорганной недостаточности (СПОН). Также в этот список входят патологические осложнения, связанные с инсулинозависимым сахарным диабетом (включая диабетическую ретинопатию), волчаночной нефропатией, нефритом Хеймана, мембранным нефритом и другими формами гломерулонефрита, ответа вследствие контактной чувствительности, и воспаление, вызванное контактом крови с искусственными поверхностями, которые могут вызывать активацию комплемента, что наблюдается, например, при искусственном кровообращении (например, во время гемодиализа или в аппарате для сердечно-легочной реанимации, например в связи с хирургической операцией на сосудах, такой как аортокоронарное шунтирование или замена сердечного клапана), или в связи с контактом с поверхностью другого искусственного сосуда или контейнера (например, вспомогательная желудочковая система, аппарат «искусственное сердце», системы переливания крови, пакеты для хранения крови, плазмафарез, тромбофарез и т.п.). Также в этот список входят заболевания, связанные с ишемически-реперфузионным повреждением, такие как заболевания, являющиеся следствием пересадки трансплантатов, включая трансплантаты солидных органов, и синдромы, такие как ишемически-реперфузионное повреждение, ишемический колит и сердечная ишемия. Соединения по настоящему изобретению можно также применять в лечении возрастной дегенерации макулы (Hageman et al, P.N.A.S,102: 7227-7232, 2005).
Сердечно-сосудистые и церебро-васкулярные нарушения - например, инфаркт миокарда, тромбоз коронарных артерий, окклюзия сосудов, постхирургическая реокклюзия сосудов, атеросклероз, травматическое повреждение центральной нервной системы и ишемическая болезнь сердца. В одном варианте осуществления, эффективное количество соединения по настоящему изобретению можно вводить пациенту, у которого имеется риск инфаркта миокарда или тромбоза (т.е. пациенту, у которого наблюдается один или больше известных факторов риска инфаркта миокарда или тромбоза, таких как (но не ограничиваясь только ими) ожирение, курение, высокое кровяное давление, гиперхолестеринемия, предыстория или генетическая история инфаркта миокарда или тромбоза), для снижения риска инфаркта миокарда или тромбоза).
Васкулит - васкулит характеризуется воспалением сосудов. Инфильтрация лейкоцитов приводит к разрушению стенок сосудов, и считается, что каскад комплемента играет важную роль в инициировании миграции лейкоцитов, наряду с повреждением в месте воспаления (Vasculitis, Second Edition, Edited by Ball and Bridges, Oxford University Press, pp 47-53, 2008). Соединения по настоящему изобретению можно применять для лечения лейкокластического васкулита, гранулематоза Вегенера, микроскопического полиангиита, синдрома Черджа-Стросса, болезни Шенлейна-Геноха, полиартеритис нодоза, быстропрогрессирующего гломерулонефрита, (RPGN), криоглобулинемии, гигантоклеточного артериита, болезни Бехчета и синдрома дуги аорты (TAK).
Рак - также играют большую роль в создании микроокружения опухоли и регуляции направленного поступления в опухоль как благотворных, так и разрушительных иммунных клеток. В недавних клинических и преклинических публикациях была показана роль CCR2 в развитии различных солидных опухолей, включая меланому, мелкоклеточную карциному легких, немелкоклеточную карциному легких, рак поджелудочной железы, рак молочной железы, рак мочевого пузыря, почечноклеточный рак, рак толстой и прямой кишки, гепатоцеллюлярную карциному, плоскоклеточный рак головы и шеи, рак пищевода, рак яичника, рак предстательной железы и рак желудка.
ВИЧ-инфекция, СПИД и вирусные инфекции - модуляторы CCR2 рецептора, описанные в настоящем тексте, можно применять для подавления ВИЧ-инфекции, замедления развития СПИД или уменьшения степени тяжести симптомов ВИЧ-инфекции, и СПИД, а также для лечения гепатита С.
Нейродегенеративные расстройства и родственные заболевания. В других аспектах описанные в настоящем тексте CCR2 антагонисты можно применять для лечения болезни Альцгеймера, множественного склероза и ухудшения когнитивной функции, связанного с операцией в условиях искусственного кровообращения и родственными процедурами.
В одном варианте осуществления настоящего изобретения, соединения по настоящему изобретению можно применять для лечения болезней, выбранных из группы, состоящей из сепсиса (и связанных с ним нарушений), ХОЗЛ, ревматоидного артрита, волчаночного нефрита и множественного склероза.
Описанные в настоящем тексте способы лечения включают, в целом, введение пациенту эффективного количества одного или больше соединений, описанных в настоящем тексте. Подходящие пациенты включают пациентов, страдающих от или подверженных (т.е. в рамках профилактического лечения) расстройству или заболеванию, указанному в настоящем тексте. Типичные пациенты для описанного в настоящем тексте лечения включают млекопитающих, в частности приматов, в особенности людей. Другие подходящие пациенты включают домашних животных-компаньонов, таких как собаки, кошки, лошади и т.п., или домашний скот, такой как крупный рогатый скот, свиньи, овцы и т.п.
В целом, описанные в настоящем тексте способы лечения включают введение пациенту эффективного количества одного или больше соединений, описанных в настоящем тексте. В предпочтительном варианте осуществления, соединение(я) по настоящему изобретению предпочтительно вводят пациенту (например, человеку) перорально или наружно. Эффективное количество может представлять собой количество, достаточное для модулирования активности CCR2 рецептора и/или количество, достаточное для уменьшения или облегчения симптомов, имеющихся у пациента. Предпочтительно, вводимое количество достаточно для создания в плазме крови концентрации соединения (или его активного метаболита, если соединение представляет собой пролекарство), достаточно высокого для обеспечения детектируемого ингибирования хемотаксиса белых кровяных телец in vitro. Режим введения может различаться в зависимости от применяемого соединения и конкретного состояния, подвергающегося лечению; для лечения большинства нарушений предпочтительна частота введения 4 раза в сутки или меньше. В целом, более предпочтителен режим введения 2 раза в сутки, в особенности предпочтительно введение 1 раз в сутки. Однако следует понимать, что конкретная дозировка и режим введения для каждого отдельного пациента будет зависеть от набора факторов, включая активность конкретного применяемого соединения, возраст, вес тела, общее состояние здоровья, пол, диета, время введения, способ введения, скорость экскреции, комбинацию с другими лекарствами (т.е. других лекарств, вводимых пациенту) и степени тяжести конкретного заболевания, подвергающегося терапии, а также от мнения лечащего врача. Обычно предпочтительным является применение минимальной дозы, достаточной для обеспечения эффективной терапии. Можно проводить мониторинг терапевтической эффективности с применением медицинских или ветеринарных критериев, подходящих для состояния, лечение или профилактика которого осуществляется.
В зависимости от заболевания и состояния субъекта, соединения и композиции по настоящему изобретению можно вводить перорально, парентерально (например, внутримышечной, внутрибрюшинной, внутривенной, интрацеребровентрикулярной, интрацистернальной инъекцией или инфузией, подкожной инъекцией, или в виде имплантата), ингаляционно, назально, вагинально, ректально, сублингвально или наружно, и их можно вводить в состав, по отдельности или совместно, подходящих дозированных форм, содержащих общеупотребимые нетоксичные фармацевтически приемлемые носители, вспомогательные вещества и разбавители, подходящие для каждого способа введения. Настоящее изобретение охватывает также введение соединений и композиций по настоящему изобретению в виде депо-препаратов.
Могут применяться уровни дозировки порядка от примерно 0,1 мг до примерно 140 мг на килограмм веса тела в сутки при лечении или профилактике состояний, включающих патогенную активность CCR2 (от примерно 0,5 мг до примерно 7 г на человека-пациента в сутки). Количество действующего вещества, которое можно комбинировать с носителями для приготовления однократной дозированной формы, будет варьироваться в зависимости от подвергающегося лечению субъекта и способа введения. Дозированные лекарственные формы обычно содержат от примерно 1 мг до примерно 500 мг действующего вещества. Для соединений, вводимых перорально, чрезкожно, внутривенно или подкожно, предпочтительно введение достаточного количества соединения для достижения концентрации в плазме крови от 5 нг (нанограмм)/мл до 10 мкг (микрограмм)/мл плазмы крови, более предпочтительно введение достаточного количества соединения для достижения концентрации в плазме крови от 20 нг/мл до 1 мкг/мл плазмы крови, наиболее предпочтительно введение достаточного количества соединения для достижения концентрации в плазме крови от 50 нг/мл до 200 нг/мл плазмы крови. Для прямого инъецирования в синовиальную оболочку (при лечении артрита), следует вводить достаточное количество соединения, чтобы достичь локальную концентрацию примерно 1 мМ.
Частота дозирования может также варьироваться в зависимости от применяемого соединения и конкретного излечиваемого заболевания. Однако, для лечения большинства нарушений предпочтителен режим дозирования 4 раза в сутки, три раза в сутки или меньше, а особенно предпочтителен режим дозирования один раз в сутки или 2 раза в сутки. Однако следует понимать, что конкретная дозировка для каждого отдельного пациента будет зависеть от набора факторов, включая активность конкретного применяемого соединения, возраста, веса тела, общего состояния здоровья, пола, диеты, времени введения, способа введения, скорости экскреции, комбинации лекарств (т.е. других лекарств, вводимых пациенту), степени тяжести конкретного заболевания, подвергающегося терапии, и от других факторов, включая мнение лечащего врача.
В некоторых вариантах осуществления, для лечения или профилактики состояний, требующих модулирование CCR2 рецептора, подходящий уровень дозировки обычно составляет примерно от 0,001 до 100 мг на килограмм веса тела пациента в сутки, и это количество можно вводить в виде однократной дозы или нескольких доз. Предпочтительно, уровень дозировки составляет от примерно 0,01 до примерно 25 мг/кг в сутки; более предпочтительно - от примерно 0,05 до примерно 10 мг/кг в сутки. Подходящий уровень дозировки может составлять примерно от 0,01 до 25 мг/кг в сутки, примерно от 0,05 до 10 мг/кг в сутки, или примерно 0,1 - 5 мг/кг в сутки. В указанном диапазоне дозировка может составлять от 0,005 до 0,05, от 0,05 до 0,5, от 0,5 до 5,0, или от 5,0 до 50 мг/кг в сутки. При пероральном введении, композиция предпочтительно имеет форму таблеток, содержащих от 1,0 до 1000 миллиграммов действующего вещества, в частности 1,0, 5,0, 10,0, 15,0, 20,0, 25,0, 50,0, 75,0, 100,0, 150,0, 200,0, 250,0, 300,0, 400,0, 500,0, 600,0, 750,0, 800,0, 900,0 и 1000,0 миллиграммов действующего вещества, с симптоматической регулировкой дозировки для пациента, подвергающегося лечению. Соединения можно вводить в режиме дозирования от 1 до 4 раз в сутки, предпочтительно один или два раза в сутки.
[0001] Однако следует понимать, что конкретная дозировка и частота дозирования для каждого отдельного пациента может варьироваться и зависит от набора факторов, включая активность конкретного применяемого соединения, метаболическую стабильность и длительность действия данного соединения, возраст, вес тела, наследственные признаки, общее состояние здоровья, пол, диету и время введения, скорость экскреции, комбинацию с другими лекарствами, степень тяжести конкретного состояния и терапии, которой подвергается субъект.
Комбинированная терапия - лекарственные средства для применения в комбинации с CCR2 соединениями
Фармакологические агенты, которые можно применять в комбинации с CCR2 антагонистами по настоящему изобретению, включают агенты, которые применяются для лечения атеросклероза, рестеноза, множественного склероза, фиброза легких, воспалительной болезни кишечника, ревматоидного артрита, реакции «трансплантат против хозяина», почечного фиброза, псориаза, отторжения трансплантата, ожирения, диабета, геиперхолестеринемии и рака.
При лечении рака могут применяться иммуномодуляторы, такие как описанные в настоящем изобретении, в комбинации с другими соединениями и реагентами, которые регулируют иммунный ответ или иммунное окружение опухоли. Это называют термином «иммуноонкология», и могут применяться разнообразные реагенты по отдельности или в комбинациях, включая малые молекулы, белки и пептиды, антитела, шпигельмеры, вирусы, олигорибонуклеотиды и другие методы доставки генетической информации, а также другие терапевтические средства, известные в данной области. Таким образом, композиции по настоящему изобретению можно применять в приготовлении иммуноонкологических препаратов.
В других вариантах осуществления, описанные в настоящем изобретении способы направлены на лечение аллергических заболеваний, при котором соединение или композицию по настоящему изобретению вводят отдельно или в комбинации со вторым терапевтическим агентом, где указанный второй терапевтический агент представляет собой антигистамин. В случае применения в комбинации, лечащий врач может вводить комбинацию соединения или композиции по настоящему изобретению и второго терапевтического агента. Также, соединение или композицию и второй терапевтический агент можно вводить последовательно, в любом порядке.
Соединения и композиции по настоящему изобретению можно комбинировать с другими соединениями и композициями, имеющими соответствующие свойства, для предотвращения и лечения целевого состояния или заболевания, таких как воспалительные состояния и заболевания, включая воспалительную болезнь кишечника, аллергические заболевания, псориаз, атопический дерматит и астму, и перечисленные выше патологии. Выбор подходящих средств для применения в составе комбинированной терапии может сделать квалифицированный специалист в данной области. Комбинация терапевтических средств может работать синерегетично, осуществляя лечение или профилактику различных нарушений. Применяя такой подход, можно достичь терапевтической эффективности с более низкими дозировками каждого агента, тем самым уменьшая риск возникновения нежелательных побочных эффектов.
Для лечения, предотвращения, ослабления, контроля или уменьшения роста солидных опухолей и метастазов, соединения по настоящему изобретению можно применять в комбинации со следующими: (1) стратегия противораковых вакцин, (2) модуляторы иммунных контрольных точек, такие как антагонистические антитела против ингибиторов иммунных контрольных точек (анти-PD1, анти-PD-L1, анти-CTLA4, анти-Tim3, анти-VISTA, анти-KIR) или агонистические антитела против иммунных акселераторов (анти-Lag3, анти-OX40, анти-ICOS, анти-4-1BB), (3) блокирующие или истощающие антитела против белков клеточной поверхности, обычно характеризующихся повышенным уровнем экспрессии в трансформированных клетках (CEACAM1, Syndecan-2, GRP78), (4) антиангиогенная терапия (анти-VEGF, анти-VEGFR, VEGFR низкомолекулярные ингибиторы), (5) антилимфоангиогенез (блокирующие антитела или ингибиторы против VEGF, FDF2, PDGF, а также их соответствующих рецепторов), (6) стандартная химиотерапия (Гемцитабин, Паклитаксел, Фолфоринокс), (7) лучевая терапия, (8) антагонисты других хемокинов (CCR1, CCR4, CCR6, CXCR4, CXCR2, CXCR7 низкомолекулярные ингибиторы, блокирующие антитела или истощающие антитела), (9) истощающие антитела против хемокинов, которые активируют вышеуказанные хемокиновые рецепторы, (10) ингибиторы, воздействующие на распространенные соматические мутации при раке, такие как ингибиторы, специфично воздействующие на следующие гены (BRAF, KRAS, NRAS, EGFR, CTNNB1, NOTCH1, PIK3CA, PTEN, APC, FLT3, IDH1, IDH2, KIT, TP53, JAK2).
Для лечения, предотвращения, ослабления, контроля или уменьшения риска воспаления, соединения по настоящему изобретению можно применять в комбинации с противовоспалительным или анальгетическим средством, таким как агонист опиатных рецепторов, ингибитором липоксигеназы, таким как ингибитор 5-липоксигеназы, ингибитором циклооксигеназы, таким как ингибитор циклооксигеназы-2, ингибитором интерлейкина, таким как ингибитор интерлейкина-1, антагонистом NMDA (N-метил-D-аспартата), ингибитором оксида азота или ингибитором синтеза оксида азота, нестероидным противовоспалительным средством или цитокин-подавляющим противовоспалительным средством, например с таким соединением, как ацетаминофен, аспирин, кодеин, биологические ФНО-секвестранты, фентанил, ибупрофен, индометацин, кеторолак, морфин, напроксен, фенацетин, пироксикам, стероидный анальгетик, суфентанил, сунлиндак, тенидап и т.п..
Сходным образом, соединения по настоящему изобретению можно вводить с болеутолящим средством; с потенциирующим средством, таким как кофеин, H2-антагонист, симетикон, гидроксид алюминия или магния; с противозастойным средством, таким как псевдоэфедрин; с противокашлевым средством, таким как кодеин; с диуретиком; с седативным или неседативным антигистаминным средством; с антагонистом очень позднего антигена (VLA-4); с иммунодепрессантом, таким как циклоспорин, такролимус, рапамиин, агонисты EDG рецептора или другие иммунодепрессанты FK-506 типа; со стероидом; с нестероидным противоастматическим средством, таким как β2-агонист, лейкотриеновый антагонист или ингибитор биосинтеза лейкотриена; с ингибитором фофодиэстеразы IV типа (PDE-IV); со средством, снижающим уровень холестерина, таким как ингибитор HMG-CoA редуктазы, секвестрант или ингибитор абсорбции холестерина; и с противодиабетическим средством, таким как инсулин, ингибиторы α-глюкозидазы или глитазоны.
Весовое соотношение соединения по настоящему изобретению и второго действующего вещества может варьироваться и зависит от эффективной дозы каждого ингредиента. Обычно применяют эффективную дозировку каждого ингредиента. Так, например, когда соединение по настоящему изобретению комбинируют с НПВС, весовое соотношение соединения по настоящему изобретению и НПВС обычно находится в диапазоне примерно от 1000:1 до 1:1000, предпочтительно от 200:1 до 1:200. Комбинации соединения по настоящему изобретению и других действующих веществ обычно также находятся в указанном диапазоне, но в каждом случае следует применять эффективную дозировку каждого действующего вещества.
В свете сказанного выше, в настоящем тексте описан ряд частных вариантов осуществления.
В одном варианте осуществления, в настоящем изобретении описан способ лечения CCR2-опосредованного состояния или заболевания, включающий введение субъекту безопасного и эффективного количества соединения или композиции по настоящему изобретению, где CCR2-опосредованное состояние или заболевание представляет собой атеросклероз.
В одном варианте осуществления, в настоящем изобретении описан способ лечения CCR2-опосредованного состояния или заболевания, включающий введение субъекту безопасного и эффективного количества соединения или композиции по настоящему изобретению, где CCR2-опосредованное состояние или заболевание представляет собой рестеноз.
В одном варианте осуществления, в настоящем изобретении описан способ лечения CCR2-опосредованного состояния или заболевания, включающий введение субъекту безопасного и эффективного количества соединения или композиции по настоящему изобретению, где CCR2-опосредованное состояние или заболевание представляет собой множественный склероз.
В одном варианте осуществления, в настоящем изобретении описан способ лечения CCR2-опосредованного состояния или заболевания, включающий введение субъекту безопасного и эффективного количества соединения или композиции по настоящему изобретению, где CCR2-опосредованное состояние или заболевание выбрано из группы, состоящей из воспалительной болезни кишечника, почечного фиброза, ревматоидного артрита, ожирения и инсулин-независимого диабета.
В одном варианте осуществления, в настоящем изобретении описан способ лечения CCR2-опосредованного состояния или заболевания, включающий введение субъекту безопасного и эффективного количества соединения или композиции по настоящему изобретению, где CCR2-опосредованное состояние или заболевание представляет собой диабет 2-го типа.
В одном варианте осуществления, в настоящем изобретении описан способ лечения CCR2-опосредованного состояния или заболевания, включающий введение субъекту безопасного и эффективного количества соединения или композиции по настоящему изобретению, где CCR2-опосредованное состояние или заболевание представляет собой диабетическую нефропатию.
В одном варианте осуществления, в настоящем изобретении описан способ лечения CCR2-опосредованного состояния или заболевания, включающий введение субъекту безопасного и эффективного количества соединения или композиции по настоящему изобретению, где CCR2-опосредованное состояние или заболевание представляет собой рак поджелудочной железы.
В одном варианте осуществления, в настоящем изобретении описан способ лечения CCR2-опосредованного состояния или заболевания, включающий введение субъекту безопасного и эффективного количества соединения или композиции по настоящему изобретению, где CCR2-опосредованное состояние или заболевание представляет собой рак или иммуноонкологические показания.
В одном варианте осуществления, в настоящем изобретении описан способ лечения CCR2-опосредованного состояния или заболевания, включающий введение субъекту безопасного и эффективного количества соединения или композиции по настоящему изобретению, где CCR2-опосредованное состояние или заболевание выбрано из группы, состоящей из хронического обструктивного заболевания легких, идиопатического легочного фиброза и синдрома идиопатической пневмонии.
В одном варианте осуществления, в настоящем изобретении описан способ лечения CCR2-опосредованного состояния или заболевания, включающий введение субъекту безопасного и эффективного количества соединения или композиции по настоящему изобретению, где введение осуществляют перорально, парэнтерально, ректально, чрезкожно, сублингвально, назально или наружно.
В одном варианте осуществления, в настоящем изобретении описан способ лечения CCR2-опосредованного состояния или заболевания, включающий введение субъекту безопасного и эффективного количества соединения или композиции по настоящему изобретению, где соединение вводят в комбинации с противовоспалительным или анальгетическим средством.
В одном варианте осуществления, в настоящем изобретении описан способ лечения CCR2-опосредованного состояния или заболевания, включающий введение субъекту безопасного и эффективного количества соединения или композиции по настоящему изобретению, где вводят также противовоспалительное или анальгетическое средство.
В одном варианте осуществления, в настоящем изобретении описан способ модулирования работы CCR2 в клетке, где работу CCR2 в клетке модулируют посредством контакта клетки с CCR2-модулирующим количеством соединения по настоящему изобретению.
В одном варианте осуществления, в настоящем изобретении описан способ лечения CCR2-опосредованного состояния или заболевания, включающий введение субъекту безопасного и эффективного количества соединения или композиции по настоящему изобретению, где заболевание выбрано из группы, состоящей из фиброза легких, отторжения трансплантата, реакции «трансплантат против хозяина» и рака.
В других вариантах осуществления, способы по настоящему изобретению направлены на лечение псориаза, где соединение или композиция по настоящему изобретению применяется в отдельности или в комбинации со вторым терапевтическим средством, таким как кортикостероид, лубрикант, кератолитик, производное витамина D3, PUVA и антралин.
В других вариантах осуществления, способы по настоящему изобретению направлены на лечение атопического дерматита, где соединение или композиция по настоящему изобретению применяется в отдельности или в комбинации со вторым терапевтическим средством, таким как лубрикант и кортикостероид.
В других вариантах осуществления, способы по настоящему изобретению направлены на лечение астмы, где соединение или композиция по настоящему изобретению применяется в отдельности или в комбинации со вторым терапевтическим средством, таким как β2-агонист и кортикостероид.
В другом аспекте настоящего изобретения, соединения по настоящему изобретению можно применять в различных нефармацевтических in vitro и in vivo целях. Например, соединения по настоящему изобретению можно метить и применять в качестве образцов для детектирования и локализации CCR2 рецептора (препараты клеток или образцы срезов тканей). Соединения по настоящему изобретению можно также применять в качестве положительного контроля в анализах активности CCR2 рецептора, т.е. в качестве стандартов для определения способности агента-кандидата связываться с CCR2 рецептором, или в качестве радиоактивных меток в позитрон-эмиссионной томографии (PET) или однофотонной эмиссионной компьютерной томографии (SPECT). Такие методы можно применять для характеристики CCR2 рецепторов в живых субъектах. Например, модулятор CCR2 рецептора можно пометить с помощью любой из ряда хорошо известных методик (например, введение радиоактивной радионуклидной метки, такой как тритий) и инкубировать с образцом в течение надлежащего времени инкубирования (например, определенного в ходе предварительного анализа временного профиля связывания). После инкубирования удаляют несвязанное соединение (например, промыванием), а связанное соединение детектируют с помощью любого метода, подходящего для применяемой метки (например, авторадиография или сцинтилляционные измерения в случае радиоактивно-меченых соединений; можно применять спектроскопические методы для детектирования люминисцентных групп и флуоресцентных групп). В качестве контроля можно аналогичным образом проанализировать такой же образец, содержащий меченое соединение и большее (например, в 10 раз большее) количество немеченого соединения. Большее количество детектируемой метки, остающейся в тестируемом образце, чем в контроле, указывает на присутствие CCR2 рецептора в образце. Детектирование, включая авторадиографию CCR2 рецептора (картирование рецепторов) в культивируемых клетках или образцах тканей можно осуществлять как описано Kuhar в разделах 8.1.1-8.1.9 в Current Protocols in Pharmacology (1998) John Wiley & Sons, New York.
Описанные в настоящем тексте соединения можно также применять во многих хорошо известных методах разделения клеток. Например, модуляторы можно связать с внутренней поверхностью планшета для тканевых культур или другой подложкой, для применения в качестве лигандов для иммобилизации и выделения CCR2 рецепторов (например, изоляции клеток, экспрессирующих рецептор) in vitro. В одном предпочтительном варианте применения, модулятор, связанный с флуоресцентным маркером, таким как флуоресцеин, вводят в контакт с клетками, которые затем анализируют (или выделяют) методом сортировки клеток с активированной флуоресценцией (FACS).
На фиг. 1, представлены структуры и активность репрезентативных соединений, описанных в настоящем тексте. Активность в анализе связывания охарактеризована следующим образом:
+, 501 нM ≤ IC50 < 5000 нM; ++, 101 нM ≤ IC50 < 500 нM; и +++, 1 нM ≤ IC50 ≤ 100 нM.
Примеры
Приведенные далее примеры служат для иллюстрирования, но не ограничения объема притязаний настоящего изобретения.
Реагенты и растворители, применение которых описано ниже, могут быть получены из коммерческих источников, таких как Aldrich Chemical Co. (Milwaukee, Wisconsin, USA). 1H-ЯМР спектры записывали на ЯМР-спектрометре Varian Mercury 400 МГц. Химические сдвиги пиков приведены относительно ТМС и представлены в следующем порядке: мультиплетность (с, синглет; д, дублет; т, триплет; кв, квадруплет; м, мультиплет) и число протонов. Результаты масс-спектрометрии приведены как соотношение массы к заряду, затем приведена относительная интенсивность каждого иона (в скобках). В примерах приведено единственное значение m/e для M+H (или, если указано, для M-H) иона, содержащего наиболее распространенные изотопы атомов. Распределение изотопов во всех случаях соответствует ожидаемой формуле. Анализ методом ионизации электроспреем (ESI) проводили на Hewlett-Packard MSD масс-спектрометре с электрораспылением, используя ВЭЖХ прибор HP1100 для введения образца. Обычно аналит растворяли в метаноле в концентрации 0,1 мг/мл, и 1 микролитр раствора вводили с растворителем в масс-спектрометр, который проводил сканирование в диапазоне от 100 до 1500 дальтон. Все соединения можно анализировать в режиме ESI с регистрацией положительно заряженных ионов, с применением в качестве растворителя смеси ацетонитрил/вода с 1% муравьиной кислоты. Приведенные ниже соединения можно также анализировать в режиме ESI с регистрацией отрицательно заряженных ионов, с применением в качестве растворителя 2 мM раствора NH4OAc в смеси ацетонитрил/вода.
В Примерах и тексте описания в настоящем изобретении применяются следующие аббревиатуры:
водн: водный; BBr3: трибромид бора; CH2Cl2 или ДХМ: дихлорметан; CH3CN: ацетонитрил; CH3OH или MeOH: метанол; DAST, (диэтиламино)серы трифторид; DavePhos, 2-дициклогексилфосфино-2′-(N,N-диметиламино)бифенил; 1,2-ДХЭ, 1,2-дихлорэтан; DIEA: N,N-диизопропилэтиламин; ДМФА: диметилформамид; DMP, периодинан Десс-Мартина; ДМСО: диметилсульфоксид; экв.: эквиваленты; Et3N: триэтиламин; Et2O: диэтиловый эфир; EtOH: этанол; EtONa, этоксид натрия; ч: час(ы); HATU, O-(7-азабензотиазол-1-ил)-N,N,N’,N’-тетраметилурония гексафторфосфат; HCl: хлороводород; H2O: вода; K2CO3: карбонат калия; KHSO4: бисульфат калия; LAH, литийалюминий гидрид; LDA, диизопропиламид лития; MgSO4: сульфат магния; мл: миллилитр; NaCl: хлорид натрия; NaH: гидрид натрия; NaHCO3: бикарбонат натрия; NaOEt: этоксид натрия; NaOH: гидроксид натрия; NaOMe: метоксид натрия; Na2SO4: сульфат натрия; NH4Cl: хлорид аммония; NMP: N-метил пирролидинон; pH: -log [H+]; POCl3, фосфорил трихлорид; PPTS, пиридиния п-толуолсульфонат; RP-HPLC, обращенно-фазовая высокоэффективная жидкостная хроматография; RT, комнатная температура; ТЭА, триэтиламин; ТФУК: трифторуксусная кислота; ТФУКA, трифторуксусный ангидрид; ТГФ: тетрагидрофуран; ТСХ: тонкослойная хроматография.
Соединения по настоящему изобретению можно синтезировать как описано ниже, используя различные реакции, известные квалифицированному специалисту в данной области. Квалифицированному специалисту в данной области будет также понятно, что можно применять альтернативные способы синтеза целевых соединений по настоящему изобретению, и что подходы, описанные в тексте настоящего документа, не являются исчерпывающими, но обеспечивают широко применимые и практичные пути синтеза описанных соединений.
Некоторые молекулы, заявленные в настоящем патенте, могут существовать в различных энантиомерных и диастереомерных формах, и в объем настоящего изобретения включены все такие варианты указанных соединений.
Подробное описание экспериментальных методик, применявшихся для синтеза ключевых соединений в настоящем тексте, дают молекулы, которые охарактеризованы идентифицирующими их физическими данными, а также связанными с ними структурными обозначениями.
Квалифицированному специалисту в данной области будет также понятно, что в ходе стандартных обработок реакционных смесей в органической химии часто применяются кислоты и основания. В ходе описанных в настоящем тексте экспериментальных методик иногда образуются соли материнских соединений, если они обладают кислотностью или основностью.
Примеры
Пример 1
Синтез [(2S,5S)-5-[4-(4-фторфенил)-1-пиперидил]-2-метил-тетрагидропиран-2-ил]-[3-(трифторметил)-7,8-дигидро-5H-1,6-нафтиридин-6-ил]метанона и [(2R,5R)-5-[4-(4-фторфенил)-1-пиперидил]-2-метил-тетрагидропиран-2-ил]-[3-(трифторметил)-7,8-дигидро-5H-1,6-нафтиридин-6-ил]метанона
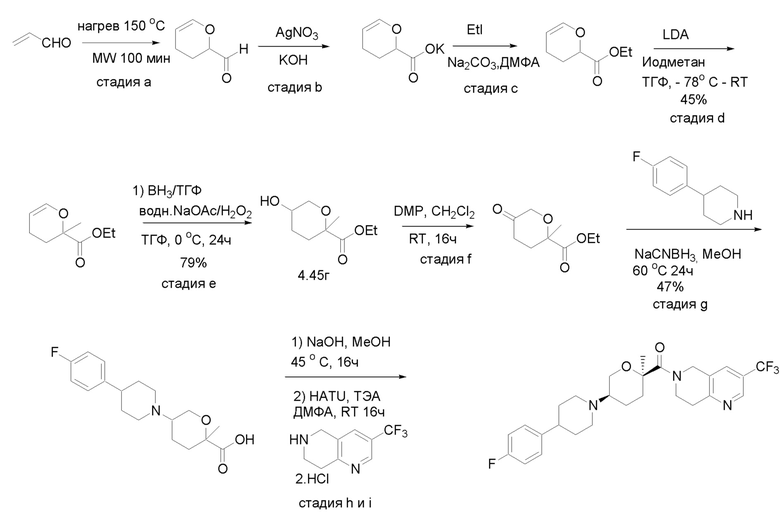
Стадия a. В микроволновую виалу помещали свежеперегнанный акролеин (40,0 г, 48,0 мл, 710 ммоль) и гидрохинон (50 мг). Полученную смесь нагревали 70 минут при 140°C, затем 30 минут при 150°C. После этого содержимое виалы фильтровали, промывали CH2Cl2 и упаривали при пониженном давлении. Полученный 3,4-дигидро-2H-пиран-2-карбоксальдегид (20,0 г, 50%) был достаточно чистым для непосредственного использования в следующей стадии.
Стадия b. Раствор AgNO3 (65 г, 383 ммоль) в воде (400 мл) добавляли в перемешиваемый раствор 3,4-дигидро-2H-пиран-2-карбальдегида (13 г, 116 ммоль) в этаноле (300 мл), после чего медленно добавляли раствор KOH (43 г, 766 ммоль) в воде (300 мл) в течение 1 часа. Полученную смесь фильтровали и упаривали. Остаток экстрагировали эфиром. Водный слой доводили до pH=3 добавлением 6н. раствора HCl и экстрагировали МТБЭ (2 x 200 мл). Органический слой упаривали и в остаток добавляли 1н. раствор NaOH до pH=12. Полученную смесь упаривали совместно с метанолом досуха, получая 3,4-дигидро-2H-пиран-2-карбоновую кислоту (8 г).
Стадия c. В перемешиваемую суспензию карбоновой кислоты, полученной как описано выше (8,0 г, 53,0 ммоль), в ДМФА (70 мл) при комнатной температуре добавляли Na2CO3 (5,0 г, 47,2 ммоль) и этилиодид (9,9 г, 63,6 ммоль). Реакционную смесь перемешивали в течение 6 часов при 110°C, затем охлаждали до комнатной температуры и разбавляли водой (100 мл). Полученный сырой продукт экстрагировали диэтиловым эфиром (4x100 мл) и объединенные органические слои промывали насыщенным водным раствором хлорида натрия, сушили над Na2SO4, фильтровали и упаривали при пониженном давлении, получая этил 3,4-дигидро-2H-пиран-2-карбоксилат (5 г, 60%) в виде желтого масла.
Стадия d. В охлажденный (-78°C) раствор этил 3,4-дигидро-2H-пиран-2-карбоксилата (10,4 г, 66,6 ммоль) в безводном ТГФ (150 мл) добавляли раствор LDA в ТГФ (2,0 M, 40 мл, 80,0 ммоль). Реакционную смесь перемешивали при -78°C в течение 1 часа, добавляли метилиодид (16,6 мл, 266,4 ммоль), перемешивали в течение 1 часа при -78°C и оставляли нагреваться до комнатной температуры. Реакцию гасили насыщенным водным раствором хлорида аммония и экстрагировали эфиром (2 x 100 мл). Органический экстракт промывали насыщенным водным раствором хлорида натрия, сушили над Na2SO4, фильтровали и упаривали в вакууме, получая этил-3,4-дигидро-2-метил-2H-пиран-2-карбоксилат (5,1 г, 45%) в виде бесцветного масла. 1H-ЯМР (400 МГц, CDCl3): δ 6,39 (дт, J = 1,9 Гц, 1H), 4,73 - 4,69 (м, 1H), 4,21 (кв, J = 7,5 Гц, 2H), 2,23 (дт, J = 1,9 Гц, 1H), 2,0-1,95 (м, 2H), 1,79-1,71 (м, 1H), 1,48 (с, 3H), 1,28 (т, J = 7,1 Гц, 3H).
Стадия e. В перемешиваемый раствор этил-3,4-дигидро-2-метил-2H-пиран-2-карбоксилата (5,1 г, 30,0 ммоль) в ТГФ (50 мл) при 0°C медленно добавляли BH3 в ТГФ (60 мл, 60,0 ммоль, 1M раствор). Реакционную смесь перемешивали при 0°C в течение 4 часов, и смесь выдерживали при 0°C в течение 16 часов. В полученную смесь затем добавляли NaOAc (7,98 г, 60,0 ммоль) и H2O2 (14,0 мл, 70,0 ммоль, 30% раствор). Реакционную смесь перемешивали при 0°C в течение 2 часов, затем оставляли нагреваться до комнатной температуры и гасили насыщенным Na2SO3 раствором. Полученную смесь экстрагировали этилацетатом (2 x 100 мл), и объединенные органические слои промывали насыщенным водным раствором хлорида натрия, сушили над Na2SO4, фильтровали и упаривали при пониженном давлении. Полученный сырой продукт очищали методом колоночной хроматографии на силикагеле, элюируя смесью этилацетат-гексан с градиентом от 10% до 60% этилацетат/гексан, получая этил тетрагидро-5-гидрокси-2-метил-2H-пиран-2-карбоксилат (4,43 г, 79%) в виде бесцветной вязкой жидкости.
Стадия f. В перемешиваемый раствор этил-тетрагидро-5-гидрокси-2-метил-2H-пиран-2-карбоксилата (4,4 г, 23,6 ммоль) в CH2Cl2 (150 мл) при 0°C добавляли периодинан Десс-Мартина (15,0 г, 35,4 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. После окончания реакции, смесь гасили насыщенным Na2S2O4 раствор, затем перемешивали в течение 30 минут, и результирующую смесь экстрагировали дихлорметаном (2 x 150 мл) и промывали водным раствором NaHCO3 и насыщенным водным раствором хлорида натрия, сушили над Na2SO4, фильтровали и упаривали при пониженном давлении. Полученный неочищенный остаток растирали в гексане и фильтровали, получая этил тетрагидро-2-метил-5-оксо-2H-пиран-2-карбоксилат (3,45 г).
Стадия g. В перемешиваемый раствор этил тетрагидро-2-метил-5-оксо-2H-пиран-2-карбоксилата (0,75 г, 4,07 ммоль), 4-(4-фторфенил)пиперидина (963 мг, 4,48 ммоль) в CH2Cl2 (15 мл) добавляли Na(OAc)3BH (27,8 г, 124,05 ммоль) при комнатной температуре. Реакционную смесь нагревали при 50°C в течение 16 часов. После окончания реакции, смесь гасили насыщенным раствором NaHCO3. Результирующую смесь экстрагировали дихлорметаном (2x150 мл) и промывали водным раствором NaHCO3 и насыщенным водным раствором хлорида натрия, сушили над Na2SO4, фильтровали и упаривали при пониженном давлении. Полученный сырой продукт очищали методом колоночной хроматографии на силикагеле, элюируя смесью этилацетат-гексан в градиенте от 10% до 60% этилацетат/гексан, получая этил-5-(4-(4-фторфенил)пиперидин-1-ил)-тетрагидро-2-метил-2H-пиран-2-карбоксилат (1,2 г, 47%). MS: (ES) m/z вычислено для C20H29FNO3 [M + H]+ 350,2, найдено 350,2.
Стадия h. В перемешиваемый раствор этил-5-(4-(4-фторфенил) пиперидин-1-ил)-тетрагидро-2-метил-2H-пиран-2-карбоксилата (1,2 г, 14,9 ммоль) в MeOH (5 мл) при комнатной температуре добавляли 4M раствор NaOH (3,0 мл). Реакционную смесь нагревали при 80°C в течение 16 часов. После окончания реакции, ее охлаждали до комнатной температуры и гасили 2н. раствором HCl (3 мл), после чего pH = 5. Водный раствор экстрагировали смесью CHCl3: iPrOH 3:1 (3 x 50 мл), и объединенные органические слои промывали насыщенным водным раствором хлорида натрия, сушили над Na2SO4, фильтровали и упаривали при пониженном давлении. Выделяли неочищенное соединение (1,05 г) в виде коричневого твердого вещества и использовали в следующей стадии без дополнительной очистки. MS: (ES) m/z вычислено для C18H24FNO3 [M + H]+ 321,2, найдено 321,2.
Стадия i. 5-(4-(4-Фторфенил)пиперидин-1-ил)-тетрагидро-2-метил-2H-пиран-2-карбоновую кислоту (150 мг, 0,467 ммоль), 3-(трифторметил)-5,6,7,8-тетрагидро-1,6-нафтиридин HCl (143 мг, 0,70 ммоль), HATU (266 мг, 0,70 ммоль) и диизопропилэтиламин (300 мг, 2,33 ммоль) смешивали в ДМФА (2,5 мл). Реакционную смесь перемешивали в течение 16 часов. После окончания реакции, смесь разбавляли водой, и полученный водный раствор экстрагировали этилацетатом (2x25 мл). Объединенные органические слои промывали насыщенным водным раствором хлорида натрия, сушили над Na2SO4, фильтровали и упаривали при пониженном давлении. Полученный сырой продукт очищали методом колоночной хроматографии на силикагеле, элюируя смесью этилацетат-гексан (20-100%), получая смесь диастереомеров, которую дополнительно очищали методом препаративной ТСХ (8:2 этилацетат:гексан) для разделения диастереоизомеров. Рацемический цис-изомер, [(2S,5S)-5-[4-(4-фторфенил)-1-пиперидил)]-2-метил-тетрагидропиран-2-ил]-[3-(трифторметил)-7,8-дигидро-5H-1,6-нафтиридин-6-ил]-метанон и [(2R,5R)-5-[4-(4-фторфенил)-1-пиперидил]-2-метил-тетрагидропиран-2-ил]-[3-(трифторметил)-7,8-дигидро-5H-1,6-нафтиридин-6-ил]метанон, (20 мг, 10%) выделяли в виде белого пушистого твердого вещества: 1H ЯМР (400 МГц, MeOH-d4) δ 8,71 (с, 1H), 8,04 (с, 1H), 7,23 (дд, J = 7,4, 5,5 Гц, 2H), 7,04 (дд, J = 8,6, 5,5 Гц, 2H), 5,35-5,25 (м, 1H), 5,05 (дд, J = 18,4, Гц, 1H), 4,72 - 4,62 (м, 1H), 4,40 - 4,35 (м, 1H), 4,30 - 4,22 (м, 1H), 4,15 - 3,90 (м, 1H), 3,70 - 3,55 (м, 2H), 3,50 - 3,40 (м, 2H), 3,28-3,05 (м, 4H), 2,95 - 2,80 (м, 1H), 2,75 - 2,60 (м, 1H), 2,28-2,20 (м, 1H), 2,15 - 2,05 (м, 2H), 1,95 - 1,75 (м, 3H), 1,53 (с, 3H). MS: (ES) m/z вычислено для C27H32F4N3O2 [M+H]+ 506,2, найдено 506,2.
Синтез (2S)-2-циклопропил-3,4-дигидропиран-2-карбоновой кислоты
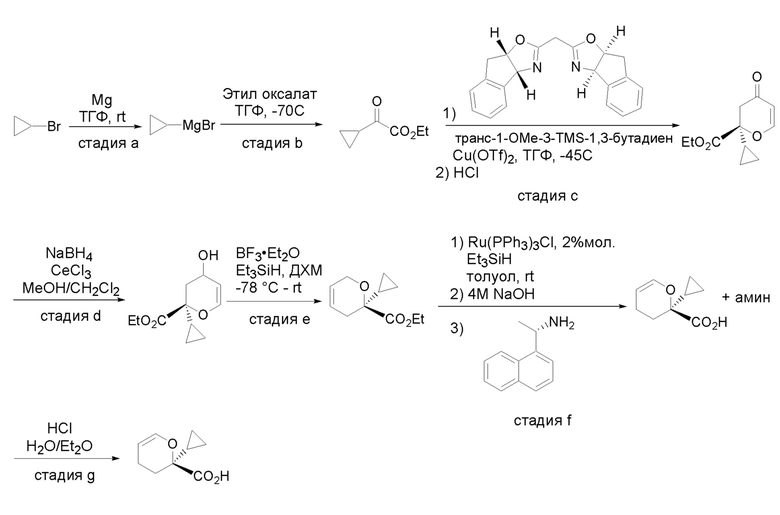
Стадия a. В 5-литровую колбу, оснащенную магнитной мешалкой, загружали мг стружку (106 г, 4,34 моль, 1,58 экв.) и безводный ТГФ (2,00 л) и продували током N2. мг активировали добавлением Br2 (1 мл, 19,5 ммоль) и перемешиванием при комнатной температуре в течение 15 мин. Раствор циклопропил бромида (500 г, 4,13 моль, 1,5 экв.) в безводном ТГФ (1,9 л) медленно добавляли в течение 2 часов. Реакционную смесь выдерживали в ледяной бане во время добавления, и скорость добавления контролировали таким образом, чтобы внутренняя температура была ниже 35°C. После добавления смесь перемешивали при комнатной температуре в течение 1,5 часов, затем охлаждали на ледяной бане в течение ночи. На следующий день реакционную смесь осторожно нагревали до 22°C, при этом растворялась большая часть осадка, и использовали в следующей стадии.
Стадия b. В 12-литровой колбе, оснащенной механической мешалкой (промывали 1 раз сухим ТГФ), растворяли диэтилоксалат (373 мл, 2,75 моль, 1 экв.) в ТГФ (1,00 л), и полученный раствор охлаждали в бане спирт/сухой лед. Раствор реактива Гриньяра с первой стадии переносили через тефлоновую канюлю в раствор оксалата в течение 3,5 часов, поддерживая внутреннюю температуру ниже -60°C. Колбу из-под реактива Гриньяра промывали ТГФ (100 мл), и промывочный раствор также переносили через тефлоновую канюлю в реакционную смесь. После добавления, реакционную смесь перемешивали еще 1,5 час, затем гасили добавлением водного раствора H2SO4 (3M, 740 мл) до значения pH ~2. Реакционную колбу вынимали из бани и оставляли нагреваться до комнатной температуры при перемешивании в течение ночи. В смесь добавляли насыщенный водный раствор хлорида натрия (600 мл), H2O (500 мл) и гексан (1,5 л). Полученную смесь интенсивно перемешивали и разделяли слои. Водный слой экстрагировали диэтиловым эфиром (2 x 400 мл), сушили над MgSO4, фильтровали и упаривали, получая коричневое масло. Это масло перегоняли в вакууме (~20 мм рт.ст.), получая желтое масло (318 г, 88% чистота согласно 1H ЯМР), которое использовали на следующей стадии.
Стадия c. 12-литровую 3-горлую колбу, оснащенную механической мешалкой и внутренним термометром, промывали безводным ТГФ (3 x) и загружали [3aR-[2(3'aR*, 8'aS*),3'aβ,8'aβ]]-(+)-2,2'-метиленбис[3a,8a-дигидро-8H-индено[1,2-d]оксазол] (49,56 г, 0,15 моль, 0,055 экв.), безводный ТГФ (6 л) и 3A молекулярные сита (500 г). Полученную смесь продували током N2 5 минут, затем добавляли Cu(OTf)2. Полученную смесь перемешивали в течение 1 часа, затем охлаждали в бане спирт/сухой лед. Когда внутренняя температура достигала -50°C, добавляли раствор кетона (467 г, 84% чистота, 2,76 моль, 1 экв.) в ТГФ (200 мл) в течение 15 мин. После добавления, реакционную смесь перемешивали в течение 45 мин, за это время внутренняя температура достигала -75°C. Добавляли раствор диена (510 г, 2,96 моль, 1,07 экв.) в ТГФ (200 мл) в течение 60 мин, поддерживая внутреннюю температуру ниже -67°C. Реакционную смесь затем перемешивали в течение 18 часов в бане с температурой -78°C. После этого колбу вынимали из бани, добавляли 1 M раствор HCl в насыщенном водном растворе хлорида натрия (3 л, 3,00 моль), и реакционную смесь оставляли перемешиваться на 2 часа. Добавляли гексан (1 л) и разделяли слои. Водный слой экстрагировали МТБЭ (750 мл), объединенные органические слои промывали насыщенным водным раствором хлорида натрия (750 мл), сушили над MgSO4 и фильтровали через целит. Фильтрат упаривали, получая темное коричневое масло (867 г). Это масло растворяли в ацетоне (1 л) и добавляли МТБЭ (2 л), в результате чего образовывался осадок. Полученную смесь фильтровали через целит и упаривали. Полученный продукт затем растворяли в MeCN (1 л) и промывали гексаном (3 x 400 мл). Объединенные гексановые фракции экстрагировали MeCN (200 мл) и объединенные MeCN-фракции упаривали, получая продукт в виде темно-коричневого масла (715 г, 86% ee). Энантиомерный избыток определяли методом ВЭЖХ, используя Pirkle Covalent (R,R) Whelk-01 5/100 Kromasil хиральную колонку (30:70 iPrOH:гексан в качестве элюента при 1 мл/мин, tR (мажорный) = 8,4 мин и tR (минорный) = 10,6 мин).
Стадия d. В 12-литровую 3-горлую колбу, оснащенную механической мешалкой и внутренним термометром, загружали CeCl3•7 H2O (514 г, 1,38 моль, 0,5 экв.) и MeOH (2,2 л). После растворения всех твердых компонентов смесь охлаждали до 0°C и добавляли неочищенный интермедиат 1 (714 г, 2,76 моль, 1 экв.)) в CH2Cl2 (2,2 л). Когда смесь достигала внутренней температуры 5°C, добавляли порциями твердый NaBH4 (124 г, 3,28 моль, 1,2 экв.) (~10г на порцию), поддерживая внутреннюю температуру ниже 16°C. После добавления баню удаляли. Медленно добавляли H2O (3 л), затем 1 M раствор лимонной кислоты (2,2 л). Слои разделяли, и водный слой экстрагировали дихлорметаном (3 x 500 мл). Объединенные органические слои промывали смесью 1:1 H2O:насыщ.р-р NaCl (600 мл), затем сушили над MgSO4, фильтровали и упаривали. Получали продукт в виде темного масла (598 г), содержащего 15 мол.% CH2Cl2, и выдерживали в морозильнике в течение ночи. Полученный продукт использовали в следующей стадии на следующий день без дополнительной очистки.
Стадия e. В 12-литровую 3-горлую колбу, оснащенную механической мешалкой и капельной воронкой, помещали продукт с предыдущей стадии (598 г, 92% чистота, 2,59 моль), растворенный в CH2Cl2 (4 л), и охлаждали в бане спирт/сухой лед. Когда внутренняя температура достигала -75°C, добавляли Et3SiH (497 мл, 3,11 моль, 1,2 экв.) и перемешивали в течение 5 минут, затем добавляли по каплям BF3•Et2O (384 мл, 3,11 моль, 1,2 экв.) в CH2Cl2 (768 мл) в течение 1,25 часа, поддерживая внутреннюю температуру ниже -68°C. Удаляли охлаждающую баню, и реакционную смесь перемешивали в течение 1,5 час, за это время внутренняя температура достигала 2 °C. Реакционную смесь разбавляли водой (1 л) и осторожно гасили насыщенным раствором NaHCO3 (2,5 л). Реакционную смесь перемешивали при комнатной температуре в течение 20 минут, затем органический слой отделяли, и водный слой экстрагировали дихлорметаном (500 мл). Объединенные органические слои сушили над MgSO4, фильтровали и упаривали, получая сырой продукт (558 г, >100%), который использовали в следующей стадии без очистки.
Стадия f. В 5-литровую 3-горлую колбу, оснащенную механической мешалкой, помещали неочищенное исходное вещество (2,59 моль), растворяли в толуоле (2,59 л) и пропускали N2 через раствор в течение 20 минут. Добавляли Et3SiH (414 мл, 2,59 моль), затем добавляли катализатор Уилкинсона (43,1 г, 47 ммоль, 0,018 экв.). Пропускали N2 через реакционный раствор в течение еще 5 минут, и смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь упаривали. Добавляли 4M раствор NaOH (1,13 л, 4,53 моль, 1,75 экв.) в смеси MeOH:H2O (1:1), и смесь перемешивали при 70 °C в течение 1 часа. Реакционную смесь упаривали в вакууме до ~50% от начального объема и нейтрализовывали до pH= 1-2 с помощью 2M раствора NaHSO4. Полученную темно-коричневую смесь экстрагировали этилацетатом (3 x 1,0 л). Объединенные органические слои сушили над MgSO4 и фильтровали. Полученный раствор разбавляли этилацетатом до общего объема 9,0 л. Полученный раствор интенсивно перемешивали, при этом добавляя (S)-1-(1-нафтил)этиламин (314 г, 1,83 моль, 0,7 экв.) в EtOH (150 мл). Немедленно выпадало в осадок белое твердое вещество, и смесь перемешивали в течение 2 часов, после чего фильтровали, промывая осадок этилацетатом (500 мл). Осадок сушили в вакууме, получая продукт в виде коричневатого твердого вещества (456 г, 88% ee). Добавляли 10 л MeCN и кипятили смесь 1 час. Реакционную смесь фильтровали, получая продукт в виде не совсем белого твердого вещества (428 г, 91% ee). Добавляли MeCN (6,8 л) и H2O (270 мл), и смесь кипятили в течение 9 часов. Смесь охлаждали до комнатной температуры. Отфильтровывали твердый осадок, промывали его ацетонитрилом. Сушили в вакуумном термошкафу, получая продукт в виде не совсем белого твердого вещества (352 г, >98% ee).
Стадия g. В полученную выше соль (46,0 г, 136 ммоль, 1,0 экв.) добавляли Et2O (500 мл) и 0,5 M водный раствор HCl (326 мл, 163 ммоль, 1,2 экв.). Полученную смесь перемешивали до полного растворения кислоты и затем разделяли слои. Водный слой экстрагировали диэтиловым эфиром (2 x 350 мл). Объединенные органические слои затем сушили над MgSO4, фильтровали, и упаривали, получая свободную кислоту, которую использовали дальше без очистки.
Синтез бензил (2S)-2-циклопропил-5-оксо-тетрагидропиран-2-карбоксилата
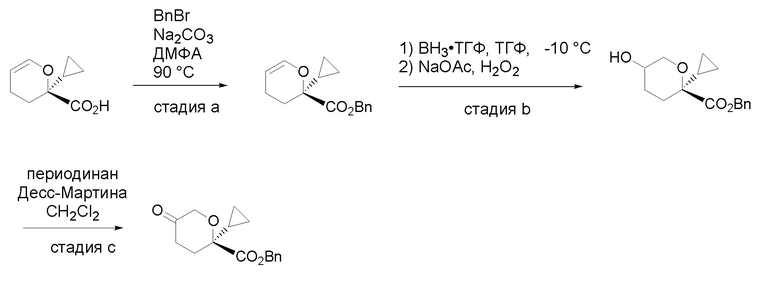
Стадия a. К (2S)-2-циклопропил-3,4-дигидро-2H-пиран-2-карбоновой кислоте (5,4 г, 32,1 ммоль, 1 экв.), растворенной в ДМФА (70 мл), добавляли Na2CO3 (5,1 г, 48,1 ммоль, 1,5 экв.). Перемешивали при 50 °C в течение 5 мин, затем добавляли бензилбромид (4,6 мл, 38,5 ммоль, 1,2 экв.), и реакционную смесь перемешивали при 90°C в течение 3 часов. Реакционную смесь охлаждали до комнатной температуры, затем разбавляли водой (150 мл) и экстрагировали диэтиловым эфиром (100 мл затем 2 x 50 мл). Объединенные органические слои сушили над MgSO4, фильтровали и упаривали, получая продукт в виде желтого масла (8,3 г).
Стадия b. (2S)-2-Циклопропил-3,4-дигидро-2H-пиран-2-карбоновой кислоты бензиловый эфир (8,3 г, 32 ммоль, 1 экв.) растворяли в ТГФ (100 мл) и охлаждали в бане со смесью насыщ.р-р NaCl/сухой лед. Добавляли BH3•ТГФ (1M раствор в ТГФ, 32 мл, 32 ммоль, 1 экв.) по каплям в течение 5 минут. Реакционную смесь выдерживали в холодильнике с температурой 8°C в течение ночи, затем возвращали в баню со смесью насыщ.р-р NaCl/сухой лед. Затем медленно добавляли раствор NaOAc (5,3 г, 64 ммоль, 2 экв.) в H2O (20 мл), и реакционную смесь перемешивали в течение 10 минут. Добавляли H2O2 (9,7 мл, 96 ммоль, 3 экв.) в один прием, охлаждающую баню удаляли, и реакционную смесь перемешивали в течение 3 часов. Реакционную смесь разбавляли насыщенным водным раствором хлорида натрия (50 мл) и отделяли органический слой. Водный слой экстрагировали диэтиловым эфиром (100 мл), и объединенные органические фазы сушили над MgSO4, фильтровали и упаривали. Остаток снова растворяли в Et2O, и белый осадок отделяли фильтрованием через целит. Фильтрат упаривали, получая желтое масло (8,8 г).
Стадия c. Масло, полученное на предыдущей стадии (99 г, 359 ммоль, 1 экв.), растворяли в CH2Cl2 (100 мл) и порциями добавляли периодинан Десс-Мартина (82,5 г, 430 ммоль, 1,5 экв.). После добавления реакционную смесь нагревали до кипения и перемешивали в течение 5,5 часов. Добавляли периодинан Десс-Мартина (15,2 г, 35,9 ммоль, 0,1 экв.), и смесь перемешивали в течение 1,5 часов при кипячении. Реакционную смесь охлаждали до комнатной температуры и перемешивали в течение ночи. Осторожно добавляли в реакционную смесь насыщенный водный раствор NaHCO3 (3 л), затем добавляли насыщенный водный раствор Na2S2O3 (100 мл), и смесь перемешивали в течение 1,5 часов. Затем смесь фильтровали через целит и отделяли органический слой. Водный слой экстрагировали дихлорметаном (2 x 300 мл), объединенные органические фазы сушили над MgSO4, фильтровали, и упаривали до одной трети общего объема. Добавляли циклогексан (500 мл) и упаривали до одной трети объема. Добавляли циклогексан (1 л) и целит (35 г), и смесь перемешивали при 40°C в течение 5 минут. Затем фильтровали смесь через целит, промывали циклогексаном и упаривали, получая темно-желтое масло (93,5 г). Часть полученного вещества (31,5 г) очищали хроматографией на силикагеле, применяя смесь 8:2 гексан:EtOAc в качестве элюента и получая сырой продукт (21 г, 67%).
Пример 2
Синтез 5-[1-[(3R,6S)-6-циклопропил-6-[[3-фтор-5-(трифторметил)фенил] метилкарбамоил]тетрагидропиран-3-ил]-4-пиперидил]-2-фторбензойной кислоты
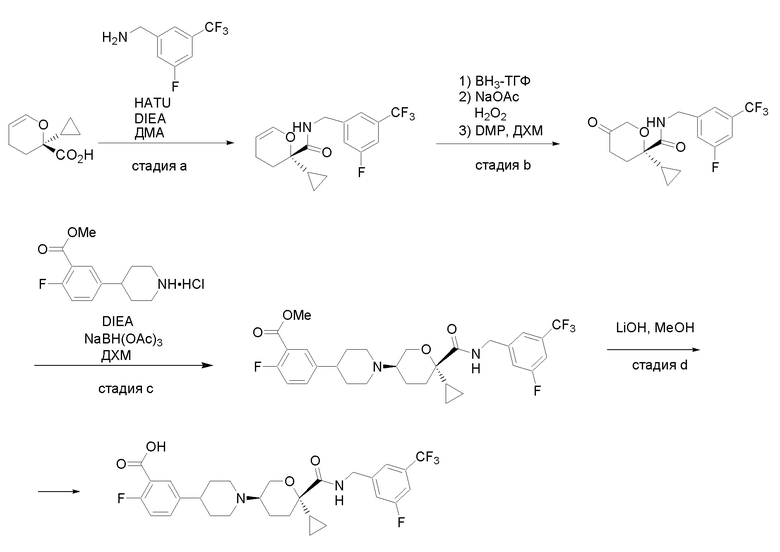
Стадия a. В смесь (2S)-2-циклопропил-3,4-дигидро-2H-2-карбоновой кислоты (730 мг, 4,3 ммоль), 3-фтор-5-трифторметил-бензиламина (1,0 г, 5,2 ммоль), диэтилизопропиламина (650 мг, 5 ммоль) в ДМФА (10 мл) добавляли HATU (2,2 г, 5,8 ммоль). Результирующий раствор перемешивали при комнатной температуре 3 часа. Реакцию гасили водой, экстрагировали этилацетатом и промывали насыщенным водным раствором хлорида натрия. Органический слой упаривали в вакууме, и остаток очищали методом флэш-хроматографии (от 2 до 25% этилацетата в гексане в качестве элюента), получая 1,2 г (2S)-2-циклопропил-N-(3-фтор-5-(трифторметил)бензил)-3,4-дигидро-2H-пиран-2-карбоксамида (84% выход) в виде светло-желтого сиропа и использовали его напрямую в следующей стадии. MS: (ES) m/z вычислено для C17H18F4NO2 [M + H]+ 344,1, найдено 344,1.
Стадия b. Полученный как описано выше амид (1,2 г, 3,5 ммоль) растворяли в ТГФ (10 мл), охлаждали на ледяной бане в атмосфере азота и добавляли боран в ТГФ (1M, 10 мл, 10 ммоль). Результирующую смесь перемешивали при 0°C 4 часа (мониторинг методом LC-MS) и медленно гасили раствором NaOAc тригидрата (2 г, 15 ммоль) в воде (6 мл), затем добавляли 30%-ный раствор H2O2 (2,5 мл). Результирующую смесь оставляли нагреваться до комнатной температуры и перемешивали в течение 1 часа. Полученную смесь разбавляли этилацетатом, промывали раствором NaHCO3 и насыщенным водным раствором хлорида натрия, сушили над MgSO4, фильтровали и упаривали в вакууме. Остаток растворяли в CH2Cl2 (50 мл) и добавляли периодинан Десс-Мартина (3,2 г, 7,5 ммоль). Полученную смесь перемешивали при комнатной температуре в течение ночи, гасили насыщенным раствором Na2S2O3 и упаривали при пониженном давлении. Остаток разбавляли этилацетатом, промывали раствором NaHCO3, 1н. раствором HCl, насыщенным водным раствором хлорида натрия и сушили над MgSO4, фильтровали и упаривали при пониженном давлении. Остаток очищали методом флэш-хроматографии (от 20 до 45% EtOAc в гексане в качестве элюента), получая 0,97 г (2S)-2-циклопропил-N-(3-фтор-5-(трифторметил)бензил)-5-оксотетрагидро-2H-пиран-2-карбоксамида (60% выход) в виде светло-желтого сиропа и использовали его напрямую в следующей стадии. MS: (ES) m/z вычислено для C17H18F4NO3 [M + H]+ 360,1, найдено 360,1.
Стадия c. Кетон, полученный как описано выше (140 мг, 0,39 ммоль), добавляли в смесь метил 2-фтор-5-(пиперидин-4-ил)бензоата гидрохлорида (200 мг, 0,73 ммоль) и N,N-диизопропилэтиламина (650 мг, 5 ммоль) в CH2Cl2 (10 мл), затем добавляли NaBH(OAc)3 (160 мг, 0,76 ммоль). Результирующую смесь перемешивали при комнатной температуре 1 час и затем в бане с температурой 45 °C в течение ночи. Реакционную смесь охлаждали до комнатной температуры, гасили насыщенным раствором NaHCO3, экстрагировали 10%-ным раствором MeOH в CH2Cl2 и сушили над MgSO4. После фильтрования и упаривания в вакууме, остаток очищали методом флэш-хроматографии (от 2 до 5% MeOH в CH2Cl2 в качестве элюента) и очищали методом препаративной ТСХ (75% EtOAc в гексане в качестве элюента), получая 30 мг метил 5-(1-((3R,6S)-6-циклопропил-6-(3-фтор-5-(трифторметил)бензилкарбамоил)тетрагидро-2H-пиран-3-ил)пиперидин-4-ил)-2-фторбензоата в виде желтой пены, и использовали его напрямую в следующей стадии. MS: (ES) m/z вычислено для C30H34F5N2O4 [M + H]+ 581,2, найдено 581,3.
Стадия d. Сложный эфир, полученный как описано выше (30 мг, 0,05 ммоль), растворяли в MeOH (3 мл) и воде (1 мл) и добавляли LiOH моногидрат (100 мг, 2,38 ммоль). Результирующую смесь перемешивали при комнатной температуре в течение ночи, разбавляли 2н. раствором HCl, экстрагировали 10%-ным MeOH в CH2Cl2 и сушили над MgSO4. После фильтрования и упаривания в вакууме, остаток очищали методом препаративной ТСХ (75% EtOAc в гексане в качестве элюента), затем методом обращенно-фазовой ВЭЖХ (C18 колонка, ацетонитрил-H2O с 0,1% ТФУК в качестве элюента), получая 25 мг целевого соединения в виде белого твердого вещества. 1H ЯМР (400 МГц, CD3OD) δ 8,84 (ушир, 1H), 7,83 (дд, J = 2,5, 6,9 Гц, 1H), 7,49 (дт, J = 2,1, 7,9 Гц, 2H), 7,36 (дд, J = 7,9, 9,3 Гц, 2 H), 7,19 (дд, J = 8,5, 10,6 Гц, 1H), 4,68 (дд, J = 7,1, 15,4 Гц, 1H), 4,36 (дд, J = 5,4, 15,4 Гц, 1H), 4,22-4,18 (м, 1H), 3,66-3,62 (м, 3H), 3,40-3,18 (м, 4H), 2,98-2,93 (м, 1H), 2,69-2,63 (м, 1H), 2,27-2,10 (м, 3H), 1,97-1,93 (м, 2H), 1,75-1,57 (м, 2H), 1,17-1,06 (м, 1H), 0,70-0,65 (м, 1H), 0,60-0,36 (м, 3H). MS: (ES) m/z вычислено для C29H32F5N2O4 [M + H]+ 567,2, найдено 567,3.
Пример 3
Синтез 3-[(3S,4R)-1-[(3R,6S)-6-циклопропил-6-[[3-фтор-5-(трифторметил)фенил] метилкарбамоил]тетрагидропиран-3-ил]-3-метил-4-пиперидил]бензойной кислоты и 3-[(3R,4S)-1-[(3R,6S)-6-циклопропил-6-[[3-фтор-5-(трифторметил)фенил] метилкарбамоил]тетрагидропиран-3-ил]-3-метил-4-пиперидил]бензойной кислоты
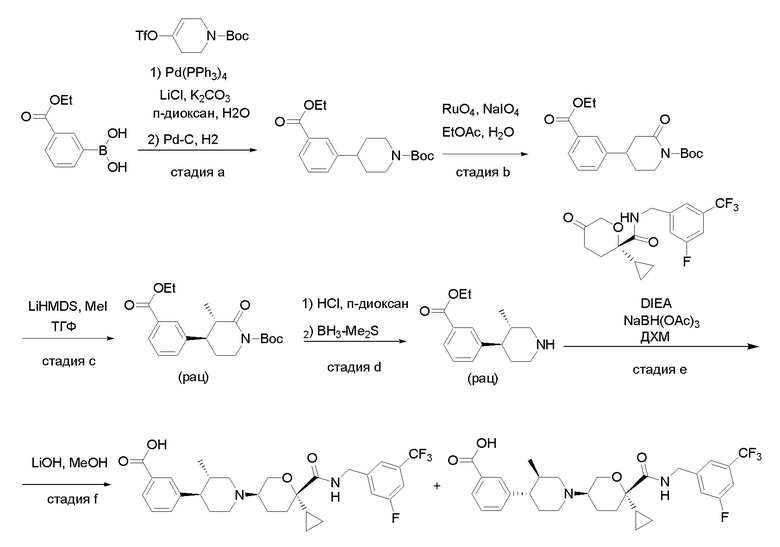
Стадия a. В смесь 3-(этоксикарбонил)фенилбороновой кислоты (4,0 г, 20,6 ммоль), трет-бутил 4-(трифторметилсульфонилокси)-5,6-дигидропиридин-1(2H)-карбоксилата (4,0 г, 12 ммоль), K2CO3 (4,2 г, 30 ммоль) и LiCl (4,3 г, 100 ммоль) в п-диоксане (75 мл) и воде (15 мл) добавляли Pd(PPh3)4 (600 мг, 0,5 ммоль). Результирующую смесь продували током N2 и затем нагревали до 100°C в атмосфере азота на 3 часа. После охлаждения до комнатной температуры, полученную смесь разбавляли этилацетатом и промывали насыщенным водным раствором хлорида натрия. Органический слой упаривали в вакууме, и остаток очищали методом флэш-хроматографии (от 2 до 25% этилацетата в гексане в качестве элюента), получая 2,4 г трет-бутил 4-(3-(этоксикарбонил)фенил)-5,6-дигидропиридин-1(2H)-карбоксилата (60% выход) в виде светло-желтого сиропа. Его растворяли в EtOH (40 мл), добавляли 10% Pd/C (400 мг) и гидрировали в течение ночи под шариком с водородом при комнатной температуре. Полученную смесь фильтровали через целит, и фильтрат упаривали в вакууме, получая 2,4 г трет-бутил 4-(3-(этоксикарбонил)фенил)пиперидин-1-карбоксилата в виде светло-желтого сиропа, который использовали напрямую в следующей стадии. MS: (ES) m/z вычислено для C19H28NO4 [M + H]+ 334,2, найдено 334,2.
Стадия b. Boc-пиперидин, полученный как описано выше (1,4 г, 4,2 ммоль), растворяли в EtOAc (70 мл) и медленно добавляли (за 30 минут) в раствор RuO4 (200 мг, 1,21 ммоль) и NaIO4 (5 г, 23,4 ммоль) в воде (20 мл). Полученную темную смесь перемешивали при комнатной температуре 3 часа. Реакцию гасили добавлением 10%-ного раствора Na2S2O3 (100 мл), смесь экстрагировали этилацетатом, промывали раствором NaHCO3, насыщенным водным раствором хлорида натрия и сушили над MgSO4. После фильтрования и упаривания в вакууме, остаток очищали методом флэш-хроматографии (от 5 до 25% EtOAc в гексане в качестве элюента), получая трет-бутил 4-(3-(этоксикарбонил)фенил)-2-оксопиперидин-1-карбоксилат (900 мг) в виде светло-желтого сиропа, который использовали напрямую в следующей стадии. MS: (ES) m/z вычислено для C19H25NNaO5 [M + Na]+ 370,2, найдено 370,1.
Стадия c. Boc-пиперидон, полученный как описано выше (900 мг, 2,6 ммоль), растворяли в ТГФ (5 мл) и по каплям добавляли в раствор LiHMDS (1M, 3 мл, 3 ммоль) в ТГФ в атмосфере азота при -78°C. Результирующий раствор перемешивали при -78°C в течение 30 минут, затем добавляли MeI (710 мг, 5 ммоль) и температуру медленно поднимали до -20°C в течение 3 часов. Реакцию гасили насыщенным водным раствором NH4Cl, смесь экстрагировали этилацетатом и промывали насыщенным водным раствором хлорида натрия. Органический слой упаривали в вакууме, и остаток очищали методом флэш-хроматографии (от 5 до 25% EtOAc в гексане в качестве элюента), получая (±)-(3S,4R)-трет-бутил 4-(3-(этоксикарбонил)фенил)-3-метил-2-оксопиперидин-1-карбоксилат (230 мг) в виде светло-желтого сиропа, который использовали напрямую в следующей стадии. MS: (ES) m/z вычислено для C20H27NNaO5 [M + Na]+ 384,2, найдено 384,1.
Стадия d. Boc-метилпиперидон, полученный как описано выше (230 мг, 0,63 ммоль), растворяли в CH2Cl2 (5 мл) и добавляли 4н. раствор HCl в диоксане (5 мл, 20 ммоль). После перемешивания при комнатной температуре в течение 2 часов, растворитель упаривали в вакууме, и остаток растворяли в ТГФ (5 мл). В полученный раствор, охлажденный на ледяной бане, медленно добавляли BH3•Me2S (2M, 1 мл). Результирующую смесь перемешивали при 0°C в течение 6 часов, затем оставляли при 4°C на 72 часа. Растворитель упаривали в вакууме, и остаток растворяли в EtOH (5 мл) с конц. HCl (0,1 мл, 12 ммоль). Полученную смесь перемешивали при 50°C в течение 30 минут. Органический слой упаривали в вакууме, и остаток очищали методом флэш-хроматографии (от 2 до 20% MeOH в CH2Cl2 с 2% NH4OH в качестве элюента), получая 110 мг of (±)-этил 3-((3S,4R)-3-метилпиперидин-4-ил)бензоата в виде светло-желтой пены, которую использовали напрямую в следующей стадии. MS: (ES) m/z вычислено для C15H22NO2 [M + H]+ 248,2, найдено 248,2.
Стадия e. Амин, полученный как описано выше (110 мг, 0,44 ммоль), растворяли в CH2Cl2 (10 мл) и добавляли N,N-диизопропилэтиламин (390 мг, 3 ммоль), (2S)-2-циклопропил-N-(3-фтор-5-(трифторметил)бензил)-5-оксотетрагидро-2H-пиран-2-карбоксамид (120 мг, 0,33 ммоль), и затем добавляли NaBH(OAc)3 (110 мг, 0,52 ммоль). Результирующую смесь перемешивали при комнатной температуре 1 час и затем при 45°C в течение ночи. Реакционную смесь охлаждали до комнатной температуры, гасили насыщенным водным раствором NaHCO3, экстрагировали 10%-ным раствором MeOH в CH2Cl2 и сушили над MgSO4. После фильтрования и упаривания в вакууме, остаток очищали методом флэш-хроматографии (от 2 до 5% MeOH в CH2Cl2 в качестве элюента) и препаративной ТСХ (60% EtOAc в гексане в качестве элюента), получая смесь (75 мг) этил 3-[(3S,4R)-1-[(3R,6S)-6-циклопропил-6-[[3-фтор-5-(трифторметил)фенил] метилкарбамоил]тетрагидропиран-3-ил]-3-метил-4-пиперидил]бензоата и этил 3-[(3R,4S)-1-[(3R,6S)-6-циклопропил-6-[[3-фтор-5-(трифторметил)фенил]метилкарбамоил] тетрагидропиран-3-ил]-3-метил-4-пиперидил]бензоата в виде желтой пены и использовали эту смесь напрямую в следующей стадии. MS: (ES) m/z вычислено для C32H39F4N2O4 [M + H]+ 591,3, найдено 591,2.
Стадия f. В раствор сложного эфира, полученного как описано выше (75 мг, 0,13 ммоль), в MeOH (5 мл) и воде (2 мл) добавляли LiOH моногидрат (210 мг, 5 ммоль). Результирующую смесь перемешивали при 80 °C в течение 2 часов, разбавляли 2н.раствором HCl, экстрагировали 10%-ным раствором MeOH в CH2Cl2 и сушили над MgSO4. После фильтрования и упаривания в вакууме, остаток очищали методом препаративной ТСХ (20% MeOH в EtOAc в качестве элюента) и затем обращенно-фазовой ВЭЖХ (C18 колонка, ацетонитрил-H2O с 0,1% ТФУК в качестве элюента), получая 30 мг целевого соединения (смесь диастереомеров) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, CD3OD) δ 8,87 (ушир, 1H), 7,85-7,91 (м, 2H), 7,27-7,51 (м, 5H), 4,68 (ддд, J = 4,0, 7,0, 15,6 Гц, 1H), 4,37 (дт, J = 4,6, 15,4 Гц, 1H), 4,26-4,20 (м, 1 H), 3,75-3,55 (м, 3H), 3,34-3,15 (м, 3H), 2,90 (кв, J = 11,9 Гц, 1H), 2,70-2,63 (м, 1H), 2,57-2,52 (м, 1H), 2,33-2,20 (м, 1H), 2,10-1,96 (м, 2H), 1,72-1,55 (м, 3H), 1,15-1,06 (м, 1 H), 0,77 (д, J = 8,0 Гц, 3H), 0,76-0,67 (м, 1H), 0,61-0,38 (м, 3H). MS: (ES) m/z вычислено для C30H35F4N2O4 [M + H]+ 563,3, найдено 563,3.
Пример 4
Синтез 5-[(2S)-4-[(3R,6S)-6-циклопропил-6-[[3-фтор-5-(трифторметил)фенил] метилкарбамоил]тетрагидропиран-3-ил]-2-метил-пиперазин-1-ил]-2-фтор-бензойной кислоты
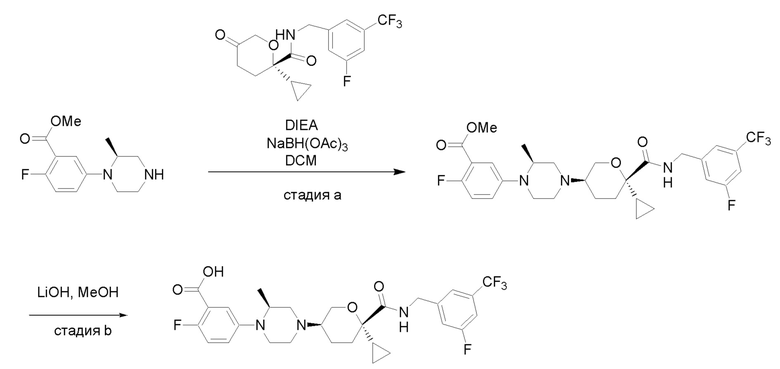
Стадия a. В смесь (S)-метил 2-фтор-5-(2-метилпиперазин-1-ил)бензоата ди-HCl соли (150 мг, 0,46) и N,N-диизопропилэтиламина (390 мг, 3 ммоль) в CH2Cl2 (10 мл) добавляли (2S)-2-циклопропил-N-(3-фтор-5-(трифторметил)бензил)-5-оксотетрагидро-2H-пиран-2-карбоксамид (150 мг, 0,42 ммоль) и NaBH(OAc)3 (211 мг, 1 ммоль). Результирующую смесь перемешивали при комнатной температуре 1 час и затем при 45°C в течение ночи. Реакционную смесь охлаждали до комнатной температуры, гасили насыщенным водным раствором NaHCO3, экстрагировали 10%-ным раствором MeOH в CH2Cl2 и сушили над MgSO4. После фильтрования и упаривания в вакууме, остаток очищали методом флэш-хроматографии (от 2 до 5% раствор MeOH в CH2Cl2 в качестве элюента) и затем препаративной ТСХ (50% EtOAc в гексане в качестве элюента), получая метил 5-[(2S)-4-[(3R,6S)-6-циклопропил-6-[[3-фтор-5-(трифторметил)фенил] метилкарбамоил]тетрагидропиран-3-ил]-2-метил-пиперазин-1-ил]-2-фтор-бензоат (60 мг) в виде желтой пены, которую использовали напрямую в следующей стадии. MS: (ES) m/z вычислено для C30H35F5N3O4 [M + H]+ 596,3, найдено 596,4.
Стадия b. В раствор сложного эфира, полученного как описано выше (60 мг, 0,10 ммоль), в MeOH (5 мл) и воде (2 мл) добавляли LiOH моногидрат (210 мг, 5 ммоль). Результирующую смесь перемешивали при 80°C в течение 2 часов, разбавляли 2н. раствором HCl, экстрагировали 10%-ным раствором MeOH в CH2Cl2 и сушили над MgSO4. После фильтрования и упаривания в вакууме, остаток очищали методом препаративной ТСХ (EtOAc в качестве элюента) и затем обращенно-фазовой ВЭЖХ (C18 колонка, ацетонитрил-H2O с 0,1% ТФУК в качестве элюента), получая 30 мг целевого соединения в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, CD3OD) δ 8,67 (ушир, 1H), 7,51-7,29 (м, 4H), 7,16-6,99 (м, 2H), 4,59 (дд, J = 6,8, 15,4 Гц, 1H), 4,43 (дд, J = 5,6, 15,4 Гц, 1H), 3,99 (ддд, J = 2,2, 4,3, 11,4 Гц, 1H), 3,54 (дт, J = 3,3, 6,3 Гц, 1H), 3,44-3,32 (м, 1H), 3,10-3,02 (м, 2H), 2,86-2,82 (м, 2H), 2,75-2,67 (м, 1H), 2,67-2,47 (м, 3H), 2,05-1,97 (м, 1H), 1,55 (дт, J = 3,6, 13,4 Гц, 1H), 1,44-1,26 (м, 1H), 1,15-1,02 (м, 1 H), 0,94 (д, J = 6 Гц, 3H), 0,94-0,83 (м, 1H), 0,77-0,66 (м, 1H), 0,61-0,36 (м, 3H). MS: (ES) m/z вычислено для C29H33F5N3O4 [M + H]+ 582,2, найдено 582,2.
Пример 5
Синтез 5-[(2S)-4-[(3R,6S)-6-циклопропил-6-[4-[3-(трифторметил)фенил]пиперазин-1-карбонил]тетрагидропиран-3-ил]-2-метил-пиперазин-1-ил]-2-фтор-бензойной кислоты
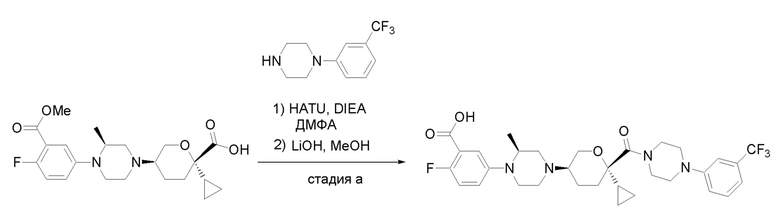
Стадия a. В смесь (2S,5R)-2-циклопропил-5-[(3S)-4-(4-фтор-3-метоксикарбонил-фенил)-3-метил-пиперазин-1-ил]тетрагидропиран-2-карбоновой кислоты (70 мг, 0,17 ммоль), 1-(3-(трифторметил)фенил)пиперазина (70 мг, 0,3 ммоль), N,N-диизопропилэтиламина (130 мг, 1 ммоль) в ДМФА (5 мл) добавляли HATU (110 мг, 0,29 ммоль). Результирующий раствор перемешивали при комнатной температуре 1 час. Реакцию гасили водой, экстрагировали этилацетатом и промывали насыщенным водным раствором хлорида натрия. Органический слой упаривали в вакууме, и остаток очищали методом флэш-хроматографии (от 10 до 35% этилацетата в гексане в качестве элюента), получая 85 мг промежуточного сложного эфира. Его растворяли в MeOH (5 мл) и воде (2 мл) и добавляли LiOH моногидрат (210 мг, 5 ммоль). Результирующую смесь перемешивали при комнатной температуре 2 часа, разбавляли 2н. раствором HCl, экстрагировали 10%-ным раствором MeOH в CH2Cl2 и сушили над MgSO4. После фильтрования и упаривания в вакууме, остаток очищали методом препаративной ТСХ (EtOAc в качестве элюента) и затем обращенно-фазовой ВЭЖХ (C18 колонка, ацетонитрил-H2O с 0,1% ТФУК в качестве элюента), получая 50 мг целевого соединения в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, CD3OD) δ 7,18-7,80 (м, 7H), 4,20-4,40 (м, 4H), 3,20-3,98 (м, 14H), 3,01-2,88 (м, 1H), 2,63 (д, J = 13,7 Гц, 1H), 2,35-2,25 (м, 1H), 1,85-1,78 (м, 1H), 1,56-1,48 (м, 1H), 1,30-1,20 (м, 1H), 1,04-0,94 (м, 3 H), 0,92-0,83 (м, 1H), 0,74-0,60 (м, 2H), 0,49-0,40 (м, 1H). MS: (ES) m/z вычислено для C32H39F4N4O4 [M + H]+ 619,3, найдено 619,2.
Синтез (2S,5R)-2-циклопропил-5-[(3S,4R)-4-(4-фторфенил)-3-метоксикарбонил-1-пиперидил]тетрагидропиран-2-карбоновой кислоты
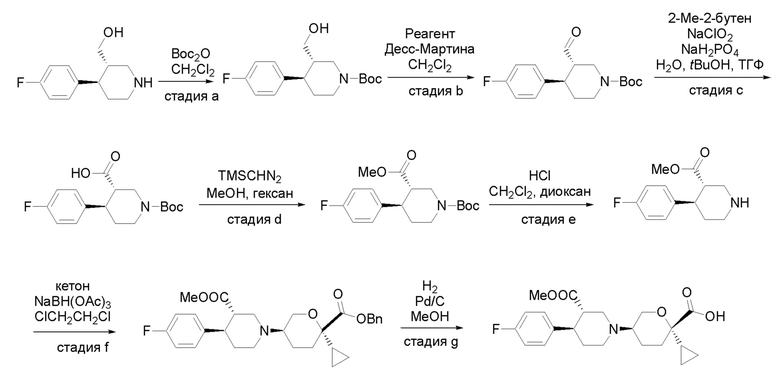
Стадия a. [(3S,4R)-4-(4-Фтор-фенил)-пиперидин-3-ил]-метанол (3,00 г, 14,3 ммоль, 1 экв.) суспендировали в CH2Cl2 (10 мл) и добавляли Boc ангидрид (3,23 г, 14,8 ммоль, 1,03 экв.). Смесь перемешивали в течение 2 часов, затем упаривали, получая сырой продукт, который использовали в следующей стадии без дополнительной очистки.
Стадия b. К сырому продукту, полученному на Стадии a (298 мг, 0,965 ммоль, 1 экв.), добавляли CH2Cl2 (2 мл) и периодинан Десс-Мартина (449 мг, 1,06 ммоль, 1,1 экв.). Реакционную смесь перемешивали в течение 1,5 часов при комнатной температуре. После этого смесь очищали хроматографией на силикагеле (гексан:EtOAc в качестве элюента), получая (3S,4R)-4-(4-фтор-фенил)-3-гидроксиметил-пиперидин-1-карбоновой кислоты трет-бутиловый эфир (167 мг, 56%).
Стадия c: В (3S,4R)-4-(4-Фтор-фенил)-3-гидроксиметил-пиперидин-1-карбоновой кислоты трет-бутиловый эфир (167 мг, 0,543 ммоль, 1 экв.) добавляли т-BuOH (3,5 мл) и 2-метил-2-бутен в ТГФ (2M, 2 мл, 4 ммоль, 7 экв.). Смесь охлаждали на ледяной бане и добавляли смесь NaClO2 (80%, 329 мг, 2,9 ммоль, 5 экв.) и NaH2PO4 (535 мг, 3,88 ммоль, 7 экв.) в H2O (1,8 мл). Полученную смесь перемешивали в течение 30 минут, затем упаривали. Остаток разделяли между EtOAc и H2O, слои разделяли и экстрагировали этилацетатом (3 x). Органическую фазу сушили над Na2SO4, фильтровали, и упаривали, получая сырой продукт (176 мг).
Стадия d. К сырому продукту, полученному на предыдущей стадии (4,17 г, 11 ммоль, 1 экв.), добавляли MeOH (15 мл), и полученный раствор охлаждали на ледяной бане. Добавляли триметилсилилдиазометан в гексане (2M, 10 мл, 20 ммоль, 1,8 экв.) до пожелтения раствора. Затем смесь упаривали и очищали хроматографией на силикагеле (гексан:EtOAc в качестве элюента), получая (3S,4R)-4-(4-Фтор-фенил)-пиперидин-1,3-дикарбоновой кислоты 1-трет-бутиловый эфир 3-метиловый эфир (3,69 г).
Стадия e. В (3S,4R)-4-(4-Фтор-фенил)-пиперидин-1,3-дикарбоновой кислоты 1-трет-бутиловый эфир 3-метиловый эфир (3,67 г, 10,9 ммоль, 1 экв.) добавляли CH2Cl2 (10 мл) и HCl в диоксане (4M, 10 мл, 40 ммоль, 3,7 экв.). Смесь перемешивали в течение 1,5 часов, затем упаривали. Остаток разбавляли дихлорметаном и промывали насыщенным водным раствором NaHCO3. Водный слой экстрагировали дихлорметаном (2 x), затем органическую фазу сушили над Na2SO4, фильтровали и упаривали, получая (3S,4R)-4-(4-Фтор-фенил)-пиперидин-3-карбоновой кислоты метиловый эфир (2,55 г).
Стадия f. В (3S,4R)-4-(4-фтор-фенил)-пиперидин-3-карбоновой кислоты метиловый эфир (2,55 г, 10,8 ммоль, 1,1 экв.) добавляли (2S)-2-Циклопропил-5-оксо-тетрагидро-пиран-2-карбоновой кислоты бензиловый эфир (2,73 г, 9,95 ммоль, 1 экв.) в дихлорэтане (30 мл), и смесь охлаждали на ледяной бане. Добавляли NaBH(OAc)3 (3,28 г, 15,5 ммоль, 1,5 экв.), и реакционную смесь затем перемешивали в течение ночи при комнатной температуре. Реакционную смесь разбавляли дихлорметаном и промывали насыщенным водным раствором NaHCO3. Водный слой экстрагировали дихлорметаном (2x), и органическую фазу затем сушили над Na2SO4, фильтровали и упаривали. Полученный сырой продукт очищали хроматографией на силикагеле (гексан:EtOAc в качестве элюента), получая целевой продукт (3,74 г) в виде цис/транс смеси.
Стадия g. В смесь со Стадии f (3,74 г, 7,54 ммоль) добавляли MeOH (25 мл) затем добавляли 10%-ный влажный Degussa type Pd/C (304 мг). Реакционную смесь перемешивали в атмосфере H2 3 часа. Реакционную смесь фильтровали через целит, промывая MeOH и CH2Cl2. Полученный раствор упаривали и очищали хроматографией на силикагеле (CH2Cl2:MeOH в качестве элюента), получая (2S,5R)-2-циклопропил-5-[(3S,4R)-4-(4-фторфенил)-3-метоксикарбонил-1-пиперидил]тетрагидропиран-2-карбоновую кислоту (1,318 г, 43%) в виде чистого изомера.
Пример 6
Синтез (3S,4R)-1-[(3R,6S)-6-циклопропил-6-[8-фтор-6-(трифторметил)-2,4-дигидро-1,3-бензоксазин-3-карбонил]тетрагидропиран-3-ил]-4-(4-фторфенил)пиперидин-3-карбоновой кислоты
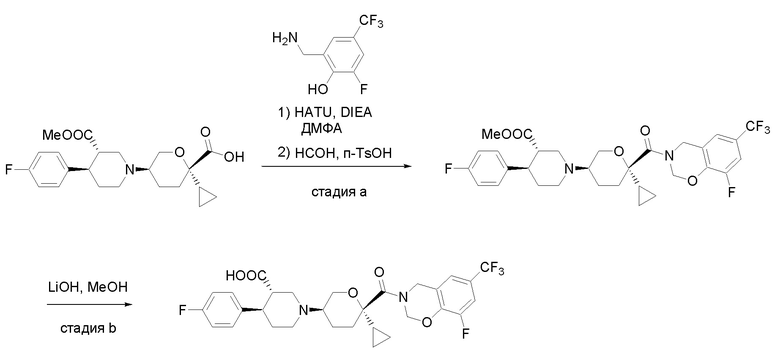
Стадия a: В смесь (2S,5R)-2-циклопропил-5-[(3S,4R)-4-(4-фторфенил)-3-метоксикарбонил-1-пиперидил]тетрагидропиран-2-карбоновой кислоты (100 мг, 0,25 ммоль), 2-(аминометил)-6-фтор-4-(трифторметил)фенола (80 мг, 0,38 ммоль) и N,N-диизопропилэтиламина (390 мг, 3 ммоль) в ДМФА (6 мл) добавляли HATU (150 мг, 0,39 ммоль). Результирующий раствор перемешивали при комнатной температуре 1 час. Реакцию гасили водой, экстрагировали этилацетатом и промывали насыщенным водным раствором хлорида натрия. Органический слой упаривали в вакууме, остаток растворяли в толуоле (10 мл) и добавляли параформальдегид (600 мг, 20 ммоль) и п-TsOH (150 мг, 0,87 ммоль). Результирующую смесь перемешивали при 100°C в течение 6 часов. После охлаждения до комнатной температуры, смесь разбавляли этилацетатом и промывали насыщенным водным раствором NaHCO3, насыщенным водным раствором хлорида натрия и сушили над MgSO4. После фильтрования и упаривания в вакууме, остаток очищали методом флэш-хроматографии (от 10% до 30% EtOAc в гексане в качестве элюента), получая метил (3S,4R)-1-[(3R,6S)-6-циклопропил-6-[8-фтор-6-(трифторметил)-2,4-дигидро-1,3-бензоксазин-3-карбонил]тетрагидропиран-3-ил]-4-(4-фторфенил) пиперидин-3-карбоксилат (80 мг) в виде желтой пены, которую использовали напрямую в следующей стадии. MS: (ES) m/z вычислено для C31H34F5N2O5 [M + H]+ 609,2, найдено 609,3.
Стадия b. Сложный эфир, полученный как описано выше (80 мг, 0,13 ммоль), растворяли в MeOH (5 мл) и воде (2 мл) и затем добавляли LiOH моногидрат (210 мг, 5 ммоль). Результирующую смесь перемешивали при 60 °C в течение 1 часа, разбавляли 2н. раствором HCl, экстрагировали 10%-ным раствором MeOH в CH2Cl2 и сушили над MgSO4. После фильтрования и упаривания в вакууме, остаток очищали методом препаративной ТСХ (EtOAc в качестве элюента) и затем обращенно-фазовой ВЭЖХ (C18 колонка, ацетонитрил-H2O с 0,1% ТФУК в качестве элюента), получая 50 мг целевого соединения в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, CD3OD) δ 7,40-7,36 (м, 2H), 7,32-7,20 (м, 2H), 7,04 (тд, J = 2,4, 9,1 Гц, 1H), 6,20 (ушир, 1H), 5,75 (ушир, 1H), 4,98-4,88 (м, 1H), 4,45-4,36 (м, 1H), 3,77-3,40 (м, 4H), 3,35- 2,98 (м, 8H), 2,65 (дт, J = 3,6, 13,7 Гц, 1H), 2,33-2,23 (м, 1H), 2,09-1,95 (м, 2H), 1,84-1,72 (м, 1H), 1,65-1,58 (м, 1H), 1,25-1,15 (м, 1H), 0,65-0,46 (м, 3H). MS: (ES) m/z вычислено для C30H32F5N2O5 [M + H]+ 595,2, найдено 595,3.
Синтез этил 3-[(3R,4S)-3-метил-4-пиперидил]бензоата
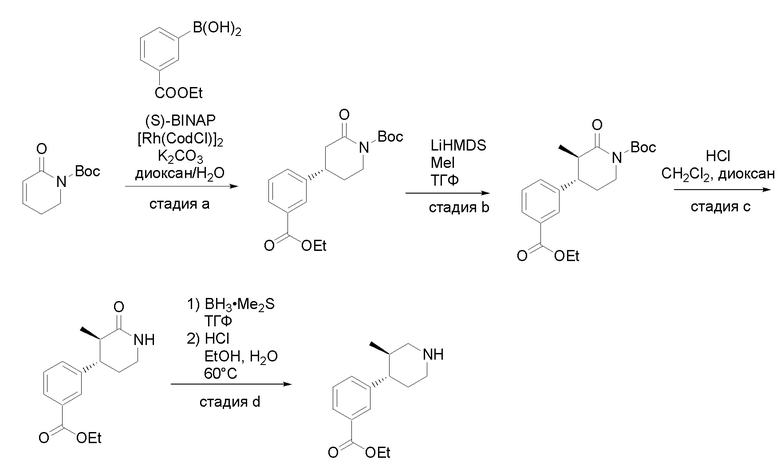
Стадия a. 3-Карбэтоксифенилбороновую кислоту (6,0 г, 31 ммоль, 2,2 экв.) растворяли в диоксане (50 мл) и охлаждали на ледяной бане. Добавляли K2CO3 (4,6 г, 33 ммоль, 2,4 экв.), затем H2O (5 мл). N2 барботировали через полученный раствор 2 минуты, затем добавляли (Rh(cod)Cl)2 (600 мг, 1,2 ммоль, 0,08 экв.) и (S)-BINAP (1,68 г, 2,56 ммоль, 0,18 экв.). N2 барботировали через полученный раствор еще 5 минут, затем медленно добавляли 6-оксо-3,6-дигидро-2H-пиридин-1-карбоновой кислоты трет-бутиловый эфир (2,70 г, 14 ммоль, 1 экв.). N2 барботировали через полученный раствор еще 5 минут, затем смесь оставляли нагреваться до комнатной температуры в течение 1 часа. Реакционную смесь нагревали до 45 °C на 1 час, затем охлаждали до комнатной температуры и перемешивали в течение ночи. Разбавляли реакционную смесь этилацетатом, промывали водой, затем насыщенным водным раствором хлорида натрия и упаривали. Остаток очищали хроматографией на силикагеле (от 5% до 20% EtOAc в гексане), получая (S)-4-(3-этоксикарбонил-фенил)-2-оксо-пиперидин-1-карбоновой кислоты трет-бутиловый эфир (3,5 г, 74%).
Стадия b. LiHMDS (1M, 13,4 мл, 13,4 ммоль, 1,3 экв.) добавляли в ТГФ (20 мл), и раствор охлаждали до -60 °C. Затем добавляли (S)-4-(3-этоксикарбонил-фенил)-2-оксо-пиперидин-1-карбоновой кислоты трет-бутиловый эфир (3,5 г, 10 ммоль, 1 экв.) в ТГФ в течение 10 минут, поддерживая внутреннюю температуру ниже -45°C. Реакционную смесь перемешивали в течение 45 минут в бане с температурой -78°C, затем добавляли MeI (2 мл). Реакционную смесь перемешивали в той же бане 3 часа, затем гасили добавлением NH4OH и нагревали до комнатной температуры. Полученную смесь экстрагировали этилацетатом (150 мл) и упаривали. Полученный остаток очищали хроматографией на силикагеле (от 5% до 15% EtOAc в гексане), получая (3R,4S)-4-(3-этоксикарбонил-фенил)-3-метил-2-оксо-пиперидин-1-карбоновой кислоты трет-бутиловый эфир (3,0 г, 83%).
Стадия c. (3R,4S)-4-(3-Этоксикарбонил-фенил)-3-метил-2-оксо-пиперидин-1-карбоновой кислоты трет-бутиловый эфир (3,0 г, 8,3 ммоль) растворяли в CH2Cl2 (20 мл) и добавляли HCl в диоксане (4н., 20 мл, 80 ммоль). Смесь оставляли перемешиваться при комнатной температуре на 4 часа, затем упаривали, получая продукт, который использовали напрямую в следующей стадии.
Стадия d. 3-((3R,4S)-3-Метил-2-оксо-пиперидин-4-ил)-бензойной кислоты этиловый эфир (8,3 ммоль, 1 экв.) растворяли в ТГФ (35 мл) и медленно добавляли BH3•SMe2 в ТГФ (2M, 12 мл, 24 ммоль, 3 экв.). Реакционную смесь оставляли в холодильнике при 4°C на 3 дня, затем раствор упаривали. Остаток растворяли в EtOH (35 мл) и добавляли концентрированную HCl (1,8 мл, 22 ммоль). Раствор перемешивали при 60°C в течение 2 часов и упаривали. Остаток очищали хроматографией на силикагеле (от 5% до 15% MeOH в CH2Cl2), получая этил 3-[(3R,4S)-3-метил-4-пиперидил]бензоат (1,2 г, 59%).
Пример 7
Синтез 3-[(3R,4S)-1-[(3R,6S)-6-циклопропил-6-[8-фтор-6-(трифторметил)-2,4-дигидро-1,3-бензоксазин-3-карбонил]тетрагидропиран-3-ил]-3-метил-4-пиперидил]бензойной кислоты
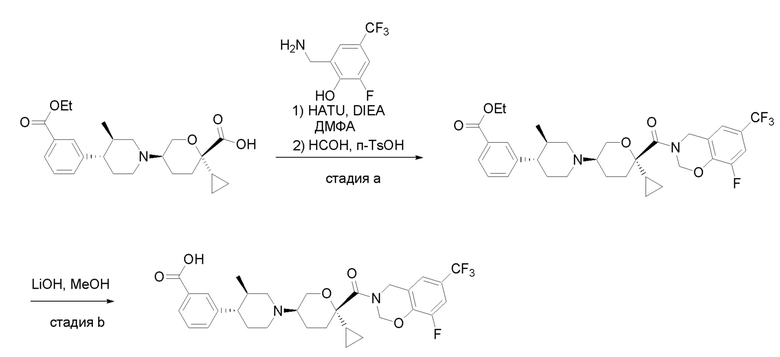
Стадия a. В смесь (2S,5R)-2-циклопропил-5-[(3R,4S)-4-(3-этоксикарбонилфенил)-3-метил-1-пиперидил]тетрагидропиран-2-карбоновой кислоты (85 мг, 0,21 ммоль), 2-(аминометил)-6-фтор-4-(трифторметил)фенола (60 мг, 0,29 ммоль), N,N-диизопропилэтиламина (390 мг, 3 ммоль) в ДМФА (6 мл) добавляли HATU (120 мг, 0,32 ммоль). Результирующий раствор перемешивали при комнатной температуре 1 час. Реакцию гасили водой (25 мл), экстрагировали этилацетатом (50 мл) и промывали насыщенным водным раствором хлорида натрия (25 мл). Органический слой упаривали в вакууме, остаток растворяли в толуоле (10 мл) и добавляли параформальдегид (600 мг, 20 ммоль) и п-TsOH (150 мг, 0,87 ммоль) при 100 °C в течение 6 часов. После охлаждения до комнатной температуры смесь разбавляли этилацетатом и промывали насыщенным водным раствором NaHCO3 (20 мл), насыщенным водным раствором хлорида натрия (20 мл) и сушили над MgSO4. После фильтрования и упаривания в вакууме, остаток очищали методом препаративной ТСХ (35% EtOAc в гексане в качестве элюента), получая этил 3-[(3R,4S)-1-[(3R,6S)-6-циклопропил-6-[8-фтор-6-(трифторметил)-2,4-дигидро-1,3-бензоксазин-3-карбонил]тетрагидропиран-3-ил]-3-метил-4-пиперидил]бензоат (15 мг) в виде желтой пены и использовали его напрямую в следующей стадии. MS: (ES) m/z вычислено для C33H39F4N2O5 [M + H]+ 619,3, найдено 619,3.
Стадия b. Сложный эфир, полученный как описано выше (15 мг, 0,024 ммоль), растворяли в MeOH (5 мл) и воде (2 мл), затем добавляли LiOH моногидрат (100 мг, 2,5 ммоль) при 60°C в течение 3 часов. Реакционную смесь разбавляли 2н. раствором HCl, экстрагировали 10%-ным раствором MeOH в CH2Cl2 и сушили над MgSO4. После фильтрования и упаривания в вакууме, остаток очищали методом препаративной ТСХ (EtOAc в качестве элюента) и затем методом обращенно-фазовой ВЭЖХ (C18 колонка, ацетонитрил-H2O с 0,1% ТФУК в качестве элюента), получая 7 мг целевого соединения в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, CD3OD) δ 7,94-7,82 (м, 2H), 7,48-7,37 (м, 4H), 6,20 (ушир, 1H), 5,75 (ушир, 1H), 4,98-4,90 (м, 1H), 4,45-4,35 (м, 1H), 3,70-3,38 (м, 4H), 3,35- 3,02 (м, 4H), 2,90-2,80 (м, 1H), 2,68-2,62 (м, 1H), 2,60-2,53 (м, 1H), 2,32-1,92 (м, 3H), 1,85-1,70 (м, 1H), 1,65-1,55 (м, 1H), 1,32-1,20 (м, 2H), 0,75 (д, J = 6,7 Гц, 3H), 0,65-0,46 (м, 3H). MS: (ES) m/z вычислено для C31H35F4N2O5 [M + H]+ 591,2, найдено 591,3.
Синтез 2-(аминометил)-6-хлор-4-(трифторметил)фенола
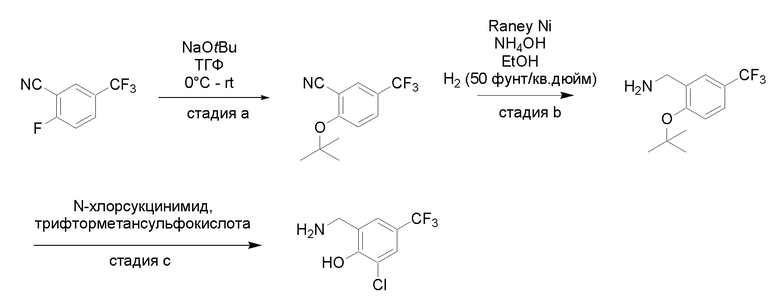
Стадия a. В раствор 2-фтор-5-(трифторметил)бензонитрила (50,0 г, 265 ммоль, 1 экв.) в ТГФ (1,0 л) добавляли NaOtBu (30,5 г, 317 ммоль, 1,2 экв.) на 10 минут при охлаждении на ледяной бане и оставляли при комнатной температуре в течение ночи. Полученную смесь затем упаривали, остаток разбавляли водой (400 мл) и экстрагировали гексаном (2 x 300 мл). Объединенные органические слои сушили над MgSO4, фильтровали и упаривали, получая желтое масло (61,7 г, 96%) которое затвердевало при стоянии. Его использовали в Стадии b.
Стадия b. 2-трет-Бутокси-5-трифторметил-бензонитрил (20,0 г, 82,3 ммоль) растворяли в EtOH (100 мл). Добавляли NH4OH (10 мл), затем добавляли суспензию никеля Ренея в воде (5 мл). Реакционную смесь встряхивали с H2 (50 фунт/кв.дюйм) в течение ночи. Полученную смесь фильтровали через целит и упаривали. Остаток растворяли в гексане и сушили над MgSO4, фильтровали и упаривали, получая желтое масло (19,8 г, 97%).
Стадия c. Трифторуксусную кислоту (2 мл) добавляли к 2-трет-бутокси-5-трифторметил-бензиламину (2,5 г, 10 ммоль, 1 экв.) при перемешивании. Осторожно добавляли в раствор триторметансульфокислоту (10 мл), затем порциями добавляли N-хлорсукцинимид (2,7 г, 20 ммоль, 2 экв.). Реакционную смесь перемешивали при комнатной температуре 2 часа, затем выливали в H2O и нейтрализовывали насыщенным водным раствором NaHCO3. Полученную смесь экстрагировали этилацетатом (2 x 25 мл), затем объединенные органические фазы сушили над MgSO4, фильтровали и упаривали. Полученный коричневый остаток растворяли в минимальном количестве MeOH, при этом образовывался серый осадок. Осадок отфильтровывали и промывали минимальным количеством MeOH, получая продукт в виде серого твердого вещества (850 мг, 37%).
Пример 8
Синтез 3-[(3R,4S)-1-[(3R,6S)-6-[8-хлор-6-(трифторметил)-2,4-дигидро-1,3-бензоксазин-3-карбонил]-6-циклопропил-тетрагидропиран-3-ил]-3-метил-4-пиперидил]бензойной кислоты
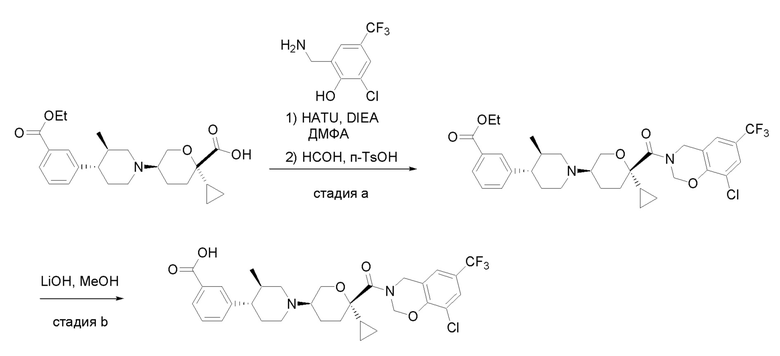
Стадия a. В смесь (2S,5R)-2-циклопропил-5-[(3R,4S)-4-(3-этоксикарбонилфенил)-3-метил-1-пиперидил]тетрагидропиран-2-карбоновой кислоты (80 мг, 0,19 ммоль), 2-(аминометил)-6-фтор-4-(трифторметил)фенола (50 мг, 0,22 ммоль), N,N-диизопропилэтиламина (260 мг, 2 ммоль) в ДМФА (6 мл) добавляли HATU (100 мг, 0,26 ммоль). Результирующий раствор перемешивали при комнатной температуре 1 час. Реакцию гасили водой, экстрагировали этилацетатом и промывали насыщенным водным раствором хлорида натрия. Органический слой упаривали в вакууме, остаток растворяли в толуоле (10 мл) и добавляли параформальдегид (600 мг, 20 ммоль) и п-TsOH (150 мг, 0,87 ммоль). Результирующую смесь перемешивали при 100°C в течение 6 часов. После охлаждения до комнатной температуры, смесь разбавляли этилацетатом (25 мл) и промывали насыщенным водным раствором NaHCO3 (15 мл), насыщенным водным раствором хлорида натрия (15 мл) и сушили над MgSO4. После фильтрования и упаривания в вакууме, остаток очищали методом препаративной ТСХ (35% EtOAc в гексане в качестве элюента), получая 50 мг этил 3-[(3R,4S)-1-[(3R,6S)-6-[8-хлор-6-(трифторметил)-2,4-дигидро-1,3-бензоксазин-3-карбонил]-6-циклопропил-тетрагидропиран-3-ил]-3-метил-4-пиперидил]бензоата в виде желтой пены, которую использовали напрямую в следующей стадии. MS: (ES) m/z вычислено для C33H39ClF3N2O5 [M + H]+ 635,2, найдено 635,2.
Стадия b. Сложный эфир, полученный как описано выше (50 мг, 0,079 ммоль), растворяли в MeOH (5 мл) и воде (2 мл), добавляли LiOH моногидрат (210 мг, 5 ммоль) при 60°C на 3 часа. Полученную смесь разбавляли 2н. раствором HCl, экстрагировали 10%-ным раствором MeOH в CH2Cl2 и сушили над MgSO4. После фильтрования и упаривания в вакууме, остаток очищали методом обращенно-фазовой ВЭЖХ (C18 колонка, ацетонитрил-H2O с 0,1% ТФУК в качестве элюента), получая 40 мг целевого соединения в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, CD3OD) δ 7,96-7,84 (м, 2H), 7,61-7,53 (м, 2H), 7,45 (д, J = 4,7 Гц, 2H), 6,17 (ушир, 1H), 5,88 (ушир, 1H), 4,98-4,90 (м, 1H), 4,45-4,35 (м, 1H), 3,70-3,37 (м, 4H), 3,35- 3,05 (м, 4H), 2,85 (т, J = 12,2 Гц, 1H), 2,65 (тд, J = 3,7, 13,9 Гц, 1H), 2,54 (дт, J = 4,8, 11,2 Гц, 1H), 2,34-2,26 (м, 1H), 2,16-1,92 (м, 2H), 1,85-1,70 (м, 1H), 1,65-1,55 (м, 1H), 1,32-1,20 (м, 2H), 0,75 (д, J = 6,7 Гц, 3H), 0,65-0,46 (м, 3H). MS: (ES) m/z вычислено для C31H35ClF3N2O5 [M + H]+ 607,2, найдено 607,4.
Пример 9
Синтез 5-[(3R,4S)-1-[(3R,6S)-6-циклопропил-6-[[3-фтор-5-(трифторметил)фенил] метилкарбамоил]тетрагидропиран-3-ил]-3-метил-4-пиперидил]-2-фтор-бензойной кислоты
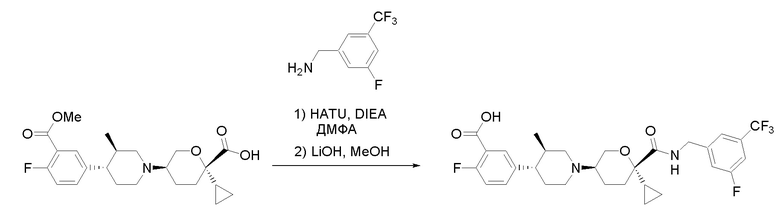
В смесь (2S,5R)-2-циклопропил-5-[(3R,4S)-4-(4-фтор-3-метоксикарбонил-фенил)-3-метил-1-пиперидил]тетрагидропиран-2-карбоновой кислоты (35 мг, 0,083 ммоль), 3-фтор-5-трифторметил-бензиламина (40 мг, 0,2 ммоль) и N,N-диизопропилэтиламина (130 мг, 1 ммоль) в ДМФА (5 мл) добавляли HATU (60 мг, 0,15 ммоль). Результирующий раствор перемешивали при комнатной температуре 1 час. Реакцию гасили водой (5 мл), экстрагировали этилацетатом (25 мл) и промывали насыщенным водным раствором хлорида натрия (25 мл). Органический слой упаривали в вакууме, и остаток очищали методом препаративной ТСХ (75% EtOAc в гексане в качестве элюента), получая 50 мг промежуточного сложного эфира. Его растворяли в MeOH (5 мл) и воде (2 мл) и затем добавляли LiOH моногидрат (210 мг, 5 ммоль). Результирующую смесь перемешивали при 60°C в течение 3 часов, разбавляли 2н. раствором HCl, экстрагировали 10%-ным раствором MeOH в CH2Cl2 и сушили над MgSO4. После фильтрования и упаривания в вакууме, остаток очищали методом обращенно-фазовой ВЭЖХ (C18 колонка, ацетонитрил-H2O с 0,1% ТФУК в качестве элюента), получая 50 мг целевого соединения в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, CD3OD) δ 8,87 (т, J = 6,3 Гц, 1H), 7,79 (дд, J = 2,4, 6,9 Гц, 1H), 7,53-7,32 (м, 3 H), 7,20 (дд, J = 8,5, 10,6 Гц, 1H), 4,98-4,90 (м, 1H), 4,66 (дд, J = 7,0, 15,6 Гц, 1H), 4,36 (дд, J = 5,4, 15,6 Гц, 1H), 4,23-4,15 (м, 1H), 3,69-3,52 (м, 3H), 3,44-3,22 (м, 3H), 3,22-3,10 (м, 1H), 2,87 (т, J = 12,0 Гц, 1H), 2,70-2,60 (м, 1H), 2,54 (дт, J = 3,9, 11,8 Гц, 1H), 2,30-2,20 (м, 1H), 2,10-1,86 (м, 3H), 1,75-1,57 (м, 2H), 1,17-1,05 (м, 1H), 0,78 (д, J = 6,5 Гц, 3H), 0,70-0,46 (м, 3H). MS: (ES) m/z вычислено для C30H34F5N2O4 [M + H]+ 581,2, найдено 581,4.
Пример 10
Синтез 3-[(3R,4S)-1-[(3R,6S)-6-циклопропил-6-[4-[3-фтор-5-(трифторметил)фенил] пиперазин-1-карбонил]тетрагидропиран-3-ил]-3-метил-4-пиперидил]бензойной кислоты
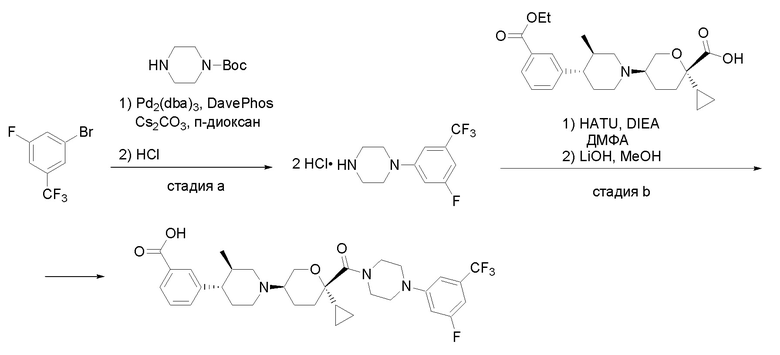
Стадия a. В смесь 1-бром-3-фтор-5-(трифторметил)бензола (1,0 г, 4,1 ммоль), трет-бутил пиперазин-1-карбоксилата (1,0 г, 5,4 ммоль) и Cs2CO3 (2,4 г, 7,4 ммоль) в п-диоксане (10 мл) добавляли DavePhos (120 мг, 0,3 ммоль). Результирующую смесь продували током азота и нагревали при 100°C в течение 13 часов. После охлаждения до комнатной температуры, реакционную смесь разбавляли этилацетатом и фильтровали через целит. Фильтрат упаривали в вакууме, и остаток очищали методом флэш-хроматографии (от 2 до 25% этилацетат в гексане в качестве элюента), получая 1,2 г промежуточного Boc-пиперазина. Его растворяли в CH2Cl2 (5 мл) и добавляли 4н. раствор HCl в диоксане (6 мл, 24 ммоль). После перемешивания при комнатной температуре в течение 3 часов, растворитель упаривали в вакууме, получая 1,2 г 1-(3-фтор-5-(трифторметил)фенил)пиперазин дигидрохлорида в виде желтого твердого вещества, которое использовали напрямую в следующей стадии. MS: (ES) m/z вычислено для C11H13F4N2 [M + H]+ 249,1, найдено 249,1.
Стадия b. В смесь пиперазин ди-HCl соли, полученной как описано выше (60 мг, 0,19 ммоль), (2S,5R)-2-циклопропил-5-[(3R,4S)-4-(3-этоксикарбонилфенил)-3-метил-1-пиперидил]тетрагидропиран-2-карбоновой кислоты (75 мг, 0,18 ммоль) и N,N-диизопропилэтиламина (130 мг, 1 ммоль) в ДМФА (5 мл) добавляли HATU (110 мг, 0,29 ммоль). Результирующий раствор перемешивали при комнатной температуре 1 час. Реакцию гасили водой, экстрагировали этилацетатом и промывали насыщенным водным раствором хлорида натрия. Органический слой упаривали в вакууме, и остаток очищали методом препаративной ТСХ (75% EtOAc в гексане в качестве элюента), получая 15 мг промежуточного сложного эфира. Его растворяли в MeOH (5 мл) и воде (2 мл), затем добавляли LiOH моногидрат (210 мг, 5 ммоль) при комнатной температуре на 2 часа, разбавляли 2н. раствором HCl, экстрагировали 10%-ным раствором MeOH в CH2Cl2 и сушили над MgSO4. После фильтрования и упаривания в вакууме, остаток очищали методом обращенно-фазовой ВЭЖХ (C18 колонка, ацетонитрил-H2O с 0,1% ТФУК в качестве элюента), получая 12 мг целевого соединения в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, CD3OD) δ 7,97-7,85 (м, 2H), 7,51-7,42 (м, 2H), 7,08-6,94 (м, 2H), 6,82 (д, J = 13,7 Гц, 1H), 4,38-4,30 (м, 2H), 4,30-4,14 (м, 4H), 3,90-3,55 (м, 5H), 3,48-3,05 (м, 4H), 2,92-2,82 (м, 1H), 2,66-2,49 (м, 2H), 2,27-1,98 (м, 4H), 1,85-1,72 (м, 1H), 1,60-1,50 (м, 1H), 1,25-1,18 (м, 1H), 0,95-0,82 (м, 1H), 0,78 (д, J = 6,5 Гц, 3H), 0,74-0,60 (м, 2H), 0,50-0,42 (м, 1H). MS: (ES) m/z вычислено для C33H40F4N3O4 [M + H]+ 618,3, найдено 618,3.
Пример 11
Синтез (2S,5R)-2-циклопропил-N-[[3-фтор-5-(трифторметил)фенил]метил]-5-[4-гидрокси-4-(3-пиридил)-1-пиперидил]тетрагидропиран-2-карбоксамида
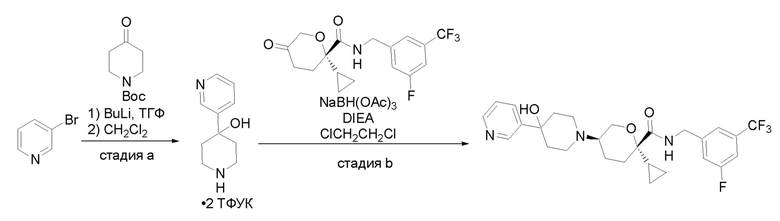
Стадия a. В охлажденный до -78 °C раствор 3-бромпиридина (1,58 г, 10 ммоль) в 20 мл ТГФ добавляли BuLi (1,6M, 6,25 мл, 10 ммоль). Результирующую смесь перемешивали при -78°C в течение 20 минут. Добавляли по каплям раствор 1-Boc-4-пиперидона (1,99 г, 10 ммоль) в ТГФ (10 мл). Реакционную смесь медленно нагревали до комнатной температуры в течение ночи. Гасили реакцию водным раствором NH4Cl (10 мл) и экстрагировали эфиром (100 мл). Объединенные органические слои промывали насыщенным водным раствором хлорида натрия и сушили над MgSO4. После упаривания, полученный сырой продукт очищали хроматографией на силикагеле (20% EtOA в гексане), получая бесцветное масло (1,5 г, 50%), которое растворяли в CH2Cl2 (30 мл) и охлаждали до 0°C. Добавляли 2,0 мл ТФУК, и результирующую смесь перемешивали при комнатной температуре 4 часа, затем упаривали при пониженном давлении, получая липкое масло (2,1 г, 95%), которое использовали в следующей стадии без дополнительной очистки.
Стадия b. ТФУК-соль амина (202 мг, 0,5 ммоль) растворяли в 1,2-дихлорэтане (2 мл) при комнатной температуре, затем добавляли iPr2NEt (130 мг, 1 ммоль). Результирующую смесь перемешивали при комнатной температуре 30 минут. Добавляли раствор кетона (104 мг, 0,3 ммоль) в 1,2-дихлорэтане (1,0 мл), затем NaBH(OAc)3 (318 мг, 1,5 ммоль). Результирующую смесь перемешивали при комнатной температуре в течение ночи. Реакцию гасили добавлением 5 мл водного раствора NaHCO3 и экстрагировали 30 мл CH2Cl2. Объединенные органические слои промывали насыщенным водным раствором хлорида натрия и сушили над MgSO4. После упаривания, полученный сырой продукт очищали методом препаративной обращенно-фазовой ВЭЖХ, получая целевой продукт (12 мг). 1H ЯМР (400 МГц, хлороформ-d) δ 8,75 (с, 1H), 7,37 (с, 1H), 7,22 (д, J = 8,7 Гц, 2H), 7,00 - 6,84 (м, 2H), 4,69 (дд, J = 15,6, 7,1 Гц, 1H), 4,42 (дд, J = 15,6, 5,6 Гц, 1H), 3,91 (ддд, J = 10,9, 4,4, 2,2 Гц, 1H), 3,35 (т, J = 10,7 Гц, 1H), 3,08 - 2,68 (м, 4H), 2,69 - 2,56 (м, 1H), 2,50 - 1,96 (м, 6H), 1,98 - 1,88 (м, 1H), 1,75-1,69 (м, 2H), 1,55 - 1,35 (м, 2H), 1,12 (тт, J = 8,5, 5,5 Гц, 1H), 0,61 - 0,35 (м, 4H). MS: (ES) m/z вычислено для C27H32F4N3O3 [M+1] 522,2, найдено 522,2.
Пример 12
Синтез 2-[3-[[[(2S,5R)-2-циклопропил-5-[4-(4-фторфенил)-1-пиперидил] тетрагидропиран-2-карбонил]амино]метил]-5-(трифторметил)фенокси]уксусной кислоты
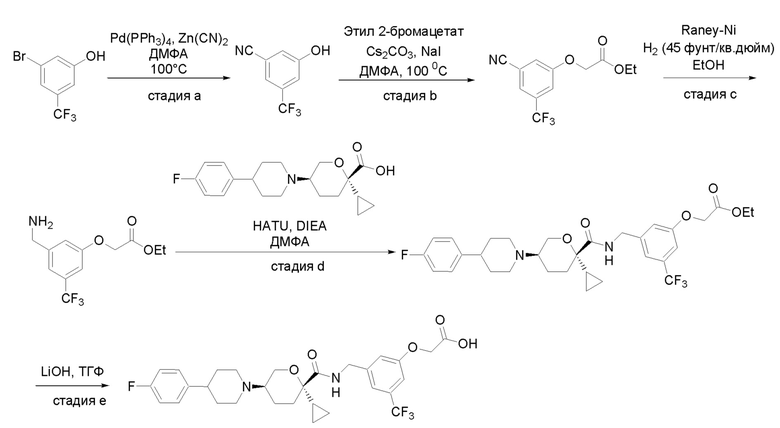
Стадия a. В раствор 3-бром-5-трифторметилфенола (7,2 г, 30 ммоль) в ДМФА (100 мл) добавляли Zn(CN)2 (3,51 г, 30 ммоль) и Pd(PPh3)4 (3,5 г, 6 ммоль). Результирующую смесь перемешивали при 100 °C в атмосфере азота в течение 4 часов. Добавляли EtOAc (250 мл), смесь промывали водой (2 x 50 мл) и насыщенным водным раствором хлорида натрия (100 мл), затем сушили над MgSO4 и упаривали при пониженном давлении. Остаток очищали хроматографией на силикагеле (20% EtOA в гексане), получая бесцветное масло (3,36 г, 60%).
Стадия b. В раствор 3-циано-5-трифторметилфенола (1,87 г, 10 ммоль) в ДМФА (20 мл) добавляли этил 2-бромацетат (2,5 г, 15 ммоль), Cs2CO3 (6,5 г, 20 ммоль) и NaI (1,5 г, 10 ммоль). Результирующую смесь перемешивали при 100°C в течение ночи, затем охлаждали до комнатной температуры. Добавляли EtOAc (250 мл), смесь промывали водой (2 x 50 мл) и насыщенным водным раствором хлорида натрия (100мл), после чего сушили над MgSO4 и упаривали при пониженном давлении. Остаток очищали хроматографией на силикагеле (10% EtOA в гексане), получая бесцветное масло (2,18 г, 80%).
Стадия c: В раствор полученного цианида (1,35 г, 5 ммоль) в EtOH (20 мл) добавляли суспензию никеля Ренея (1 мл) и NH4OH (2 мл). Полученную суспензию перемешивали при комнатной температуре при 45 фунт/кв.дюйм водорода в течение 4 часов. Реакционную смесь фильтровали через слой целита и упаривали при пониженном давлении, получая светло-голубое масло (1,1 г), которое использовали в следующей стадии без дополнительной очистки.
Стадия d: В раствор промежуточной (2S,5R)-2-циклопропил-5-[4-(4-фторфенил)-1-пиперидил]тетрагидропиран-2-карбоновой кислоты (52 мг, 0,149 ммоль) в ДМФА (2 мл) добавляли амин со Стадии c (100 мг), триэтиламин (0,5 мл) и HATU (190 мг). Результирующую смесь перемешивали при комнатной температуре в течение ночи. Добавляли EtOAc (20 мл), и смесь промывали насыщенным водным раствором NaHCO3 (2 x 5 мл) и насыщенным водным раствором хлорида натрия (5 мл). Объединенные органические слои сушили над MgSO4 и упаривали при пониженном давлении. Остаток очищали методом препаративной ТСХ (100% EtOAc), получая целевой продукт (35 мг).
Стадия e. Смесь сложного эфира (35 мг) в ТГФ (1 мл) и 1н. водном растворе LiOH (1 мл) перемешивали при комнатной температуре в течение ночи. Медленно добавляли 1н. водный раствор HCl, доводя pH смеси до 7,0, и затем смесь экстрагировали 2:1 смесью CHCl3:iPrOH (20 мл). Органическую фазу сушили над MgSO4 и упаривали при пониженном давлении. Остаток очищали методом обращенно-фазовой ВЭЖХ, получая целевой продукт (15 мг). 1H ЯМР (400 МГц, CD3OD) δ 7,51 (ушир.с, 1H), 7,40-7,30 (м, 2H), 7,24-7,18 (м, 2H), 7,02-6,95 (м, 2H), 4,55, 4,44 (ABкв., J = 15,4 Гц, 2H), 3,94 (ддд, J = 11,2, 4,4, 2,2 Гц, 1H), 3,39 (т, J = 11,0 Гц, 1H), 3,00-2,90 (м, 4H), 2,58-2,41 (м, 3H), 2,39-2,24 (м, 2H), 2,00-1,92 (м, 1H), 1,82-1,60 (м, 4H), 1,52 (тд, J = 13,5, 3,7 Гц, 1H), 1,40-1,26 (м, 1H), 1,10-1,02 (м, 1H), 0,70-0,64 (м, 1H), 0,57-0,49 (м, 1H), 0,46-0,30 (м, 3H). MS: (ES) m/z вычислено для C30H35F4N2O5 [M + H]+ 579,2, найдено 579,6.
Синтез (2S,5R)-2-циклопропил-5-[4-(4-фтор-3-метоксикарбонил-фенил)-1-пиперидил]тетрагидропиран-2-карбоновой кислоты
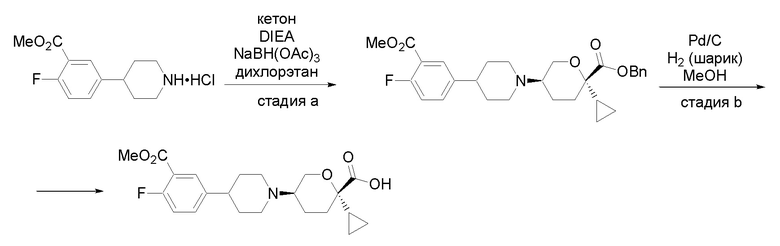
Стадия a: 2-Фтор-5-пиперидин-4-ил-бензойной кислоты метиловый эфир гидрохлорид (5,0 г, 18 ммоль, 1 экв.) и диизопропилэтиламин (3,2 мл, 18,2 ммоль, 1 экв.) в 1,2-дихлорэтане (100 мл) перемешивали при кипячении до гомогенности. Полученный раствор охлаждали до комнатной температуры и добавляли (2S)-2-циклопропил-5-оксо-тетрагидро-пиран-2-карбоновой кислоты бензиловый эфир (5,0 г, 18,2 ммоль, 1 экв.), затем добавляли NaBH(OAc)3 (7,7 г, 36,4 ммоль, 2 экв.). Смесь перемешивали при комнатной температуре 2 дня, затем нейтрализовывали насыщенным водным раствором NaHCO3. Органический слой отделяли, сушили над MgSO4, фильтровали и упаривали, получая сырой продукт.
Стадия b: К сырому продукту, полученному на Стадии a (18,2 ммоль), добавляли MeOH (100 мл) и 10% Pd/C (50% влажность, 7,7 г, 3,6 ммоль, 0,2 экв.). Смесь перемешивали под давлением H2 из воздушного шарика. После окончания, реакционную смесь фильтровали через целит и осадок на фильтре промывали смесью 1:1 MeOH:AcOH. Полученный раствор затем упаривали. Остаток разбавляли ацетоном. Образовавшийся белый осадок отфильтровывали, получая (2S,5R)-2-циклопропил-5-[4-(4-фтор-3-метоксикарбонил-фенил)-1-пиперидил]тетрагидропиран-2-карбоновую кислоту (1,1 г, 15%) в виде чистого цис диастереомера.
Синтез 5-хлор-7-(трифторметил)-1,2,3,4-тетрагидроизохинолина
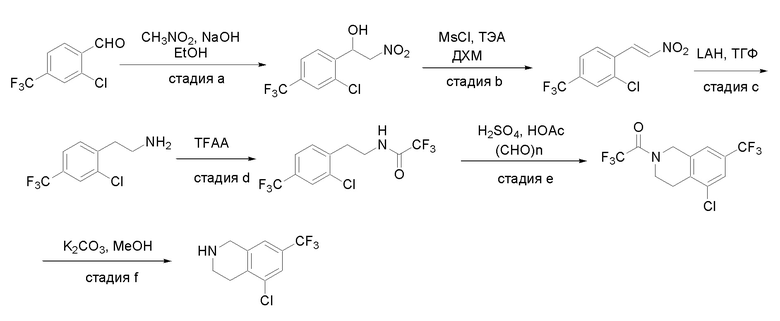
Стадия a. В охлажденный до 0 °C раствор 2-хлор-4-трифторметилбензальдегида (1,8 г, 9 ммоль) в EtOH (20 мл) добавляли CH3NO2 (610 мг, 10 ммоль). Добавляли по каплям водный раствор NaOH (10M, 1 мл), и результирующую смесь перемешивали при комнатной температуре 1 час. Реакцию гасили 1н. раствором HCl (10 мл) и экстрагировали этилацетатом (200 мл). Объединенные органические слои промывали водным раствором NaHCO3 и насыщенным водным раствором хлорида натрия, после чего сушили над MgSO4 и упаривали при пониженном давлении, получая бесцветное масло, которое использовали в следующей стадии без дополнительной очистки.
Стадия b. Полученное на предыдущей стадии масло растворяли в CH2Cl2 (20 мл), и раствор охлаждали до 0°C, затем добавляли триэтиламин (3,3 г, 30 ммоль). Добавляли по каплям MsCl (3,42 г, 30 ммоль), и результирующую смесь перемешивали при комнатной температуре 30 минут, после чего разбавляли дихлорметаном (200 мл). Объединенные органические слои промывали водным раствором NaHCO3 и насыщенным водным раствором хлорида натрия, затем сушили над MgSO4 и упаривали при пониженном давлении. Остаток очищали хроматографией на силикагеле (5% EtOAc в гексане), получая 1,35 г целевой продукт в виде желтого твердого вещества.
Стадия c: В охлажденный до 0°C раствор продукта с предыдущей стадии (625 мг, 2,5 ммоль) в ТГФ (20 мл) добавляли по каплям раствор LiAlH4 в ТГФ (1M, 8 мл). Результирующую смесь нагревали при 50°C в течение 5 часов. Полученную смесь охлаждали до 0°C и медленно гасили водой, затем фильтровали через слой целита и промывали МТБЭ. Фильтрат сушили и упаривали, получая бесцветное масло, которое использовали на следующей стадии.
Стадия d: Масло, полученное на Стадии c, растворяли в CH2Cl2 (20 мл), и полученный раствор охлаждали до 0°C. Добавляли по каплям трифторуксусный ангидрид (630 мг, 3 ммоль) и перемешивали при комнатной температуре 1 час. Полученную смесь разбавляли дихлорметаном (100 мл) и промывали водным раствором NaHCO3 и насыщенным водным раствором хлорида натрия, затем сушили над MgSO4 и упаривали при пониженном давлении. Остаток очищали хроматографией на силикагеле (10% EtOAc в гексане), получая 600 мг целевого продукта в виде белого твердого вещества.
Стадия e. В раствор ТФУК-защищенного амина, полученного на предыдущей стадии (1,0 г, 3,13 ммоль), в смеси H2SO4 (4 мл) и HOAc (8мл) добавляли параформальдегид (290 мг). Результирующую смесь перемешивали при комнатной температуре 72 часа, затем разбавляли этилацетатом (200 мл). Полученную смесь промывали водой и насыщенным водным раствором хлорида натрия, сушили над MgSO4, затем упаривали при пониженном давлении. Остаток очищали хроматографией на силикагеле (10% EtOAc в гексане), получая 300 мг целевого продукта в виде белого твердого вещества.
Стадия f. Твердый продукт со Стадии e растворяли в смеси MeOH (6 мл) и воды (2 мл), затем добавляли K2CO3 (300 мг). Результирующую смесь перемешивали при комнатной температуре в течение ночи. Добавляли 200 мл смеси CHCl3: iPrOH (2:1), органическую фазу отделяли и сушили над MgSO4, затем упаривали при пониженном давлении. Остаток очищали хроматографией на силикагеле (50% EtOAc в гексане), получая 190 мг целевого продукта в виде бесцветного масла.
Пример 13
Синтез 5-[(3R,4S)-1-[(3R,6S)-6-[5-хлор-7-(трифторметил)-3,4-дигидро-1H-изохинолин-2-карбонил]-6-циклопропил-тетрагидропиран-3-ил]-3-метил-4-пиперидил]-2-фтор-бензойной кислоты
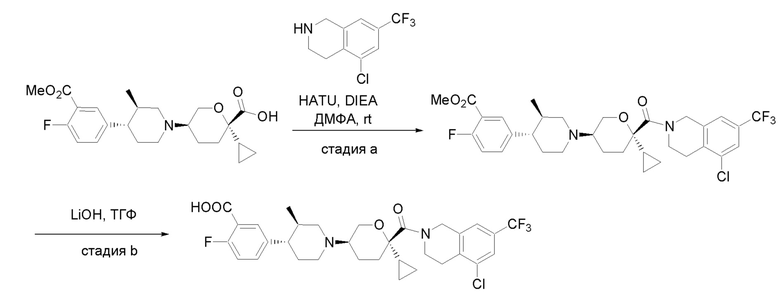
Стадия a. В раствор полученной промежуточной кислоты (52 мг, 0,149 ммоль) в ДМФА (2 мл) добавляли амин (100 мг), триэтиламин (0,5 мл) и HATU (190 мг). Результирующую смесь перемешивали при комнатной температуре в течение ночи. Добавляли EtOAc (20 мл), и смесь промывали насыщенным водным раствором NaHCO3 (2 x 5 мл) и насыщенным водным раствором хлорида натрия (5 мл). Объединенные органические слои сушили над MgSO4 и упаривали при пониженном давлении. Остаток очищали методом препаративной ТСХ (100% EtOAc), получая 25 мг целевого продукта.
Стадия b. В продукт со стадии a добавляли водный LiOH в ТГФ, как описано выше, получая финальный продукт (12 мг). 1H ЯМР (400 МГц, CD3OD) δ 7,43-7,40 (м, 1 H), 7,34-7,26 (м, 1 H), 7,20-7,14 (м, 1 H), 7,04-6,96 (м, 2 H), 5,49-5,16 (м, 1H), 4,85-4,52 (м, 1 H), 4,25-3,96 (м, 2 H), 3,30-3,18 (м, 1 H), 3,12-2,85 (м, 4 H), 2,59-2,38 (м, 3 H), 2,36-2,19 (м, 2 H), 2,09-1,97 (м, 1 H), 1,85-1,74 (м, 2 H), 1,73-1,58 (м, 2 H), 1,56-1,38 (м, 1H), 1,34-1,27 (м, 1 H), 1,23-0,97 (м, 1 H), 0,78 (д, J = 6,5 Гц, 3H), 0,76-0,20 (м, 4 H). MS: (ES) m/z вычислено для C32H36F4ClN2O4 [M + H]+ 623,2, найдено 623,1.
Пример 14
Синтез 3-[(3R, 4S)-1-[(3R,6S)-6-[5-хлор-7-(трифторметил)-3,4-дигидро-1H-изохинолин-2-карбонил]-6-циклопропил-тетрагидропиран-3-ил]-4-пиперидил]бензойной кислоты
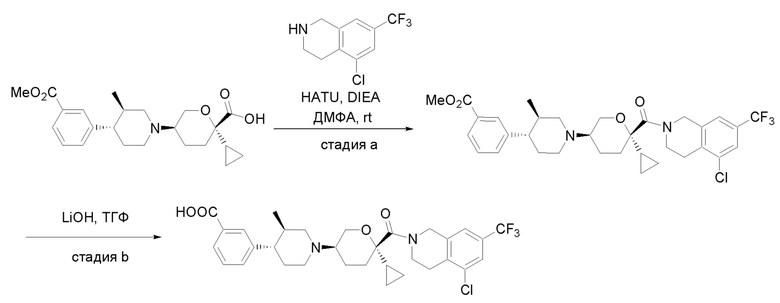
Стадия a. В раствор полученной промежуточной кислоты (52 мг, 0,149 ммоль) в ДМФА (2 мл) добавляли амин (100 мг), триэтиламин (0,5 мл) и HATU (190 мг). Результирующую смесь перемешивали при комнатной температуре в течение ночи. Добавляли EtOAc (20 мл), и смесь промывали насыщенным водным раствором NaHCO3 (2 x 5 мл) и насыщенным водным раствором хлорида натрия (5 мл). Объединенные органические слои сушили над MgSO4 и упаривали при пониженном давлении. Остаток очищали методом препаративной ТСХ (100% EtOAc), получая 25 мг целевого продукта.
Стадия b. В продукт со Стадии a добавляли водный LiOH в ТГФ, как описано выше, получая финальный продукт (12 мг). 1H ЯМР (400 МГц, CD3OD) δ 7,41-7,38 (м, 2 H), 7,34-7,26 (м, 1 H), 7,20-7,14 (м, 1 H), 7,04-6,96 (м, 2 H), 5,49-5,16 (м, 1H), 4,85-4,52 (м, 1 H), 4,25-3,96 (м, 2 H), 3,30-3,18 (м, 1 H), 3,12-2,85 (м, 4 H), 2,59-2,38 (м, 3 H), 2,36-2,19 (м, 2 H), 2,09-1,97 (м, 1 H), 1,85-1,74 (м, 2 H), 1,73-1,58 (м, 2 H), 1,56-1,38 (м, 1 H), 1,34-1,27 (м, 1 H), 1,23-0,97 (м, 1 H), 0,78 (д, J = 6,5 Гц, 3H), 0,76-0,20 (м, 4 H). MS: (ES) m/z вычислено для C32H37F3ClN2O4 [M + H]+ 605,2, найдено 605,1.
Пример 15
Синтез N-[[3,5-бис(трифторметил)фенил]метил]-5-спиро[инден-1,4'-пиперидин]-1'-ил-тетрагидропиран-2-карбоксамида
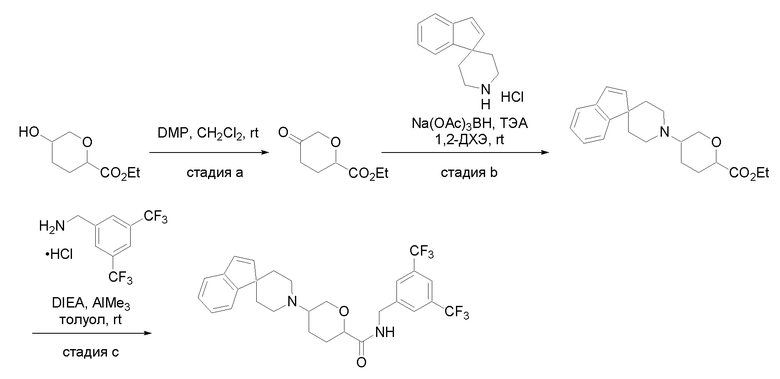
Стадия a. Этил 5-гидрокситетрагидропиран-2-карбоксилат (2,0 г, 11,5 ммоль) растворяли в безводном CH2Cl2 (80 мл) и добавляли периодинан Десс-Мартина (7,3 г, 17,2 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи, затем добавляли 10%-ный раствор Na2S2O3 (50 мл) и насыщенный раствор NaHCO3 (50 мл). Полученную смесь перемешивали в течение 15 минут, отделяли органический слой, сушили над MgSO4, фильтровали и упаривали. Остаток разбавляли эфиром, отфильтровывали белый осадок, и фильтрат упаривали, получая сырой продукт в виде желтого масла (1,5 г, 76%).
Стадия b. В суспензию 4-спироинден-пиперидин гидрохлорида (554 мг, 2,5 ммоль) в безводном 1,2-дихлорэтане (10 мл) добавляли триэтиламин (0,35 мл, 2,5 ммоль), и смесь перемешивали при комнатной температуре в течение 15 минут. Добавляли неочищенный кетон, полученный на стадии a (400 мг, 2,3 ммоль), затем твердый NaBH(OAc)3 (0,97 г, 4,6 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи, затем добавляли H2O (10 мл) и 2M раствор K2CO3 (10 мл). Органический слой отделяли и водную фазу дополнительно экстрагировали дихлорметаном (2 x 15 мл). Объединенные органические слои сушили над MgSO4, фильтровали и упаривали. Остаток очищали методом колоночной хроматографии (силикагель, CH2Cl2 → 10%-ный раствор MeOH в CH2Cl2), получая целевой продукт в виде смеси диастереомеров (460 мг, 59%). MS: (ES) m/z вычислено для C21H28NO3 [M + H]+ 342,2, найдено 342,1.
Стадия c. В суспензию 3,5-бис(трифторметил)фенил)-метанамин гидрохлорида (195,7 мг, 0,7 ммоль) в безводном толуоле (1 мл) добавляли N,N-диизопропилэтиламин (0,12 мл, 0,7 ммоль), и смесь перемешивали при комнатной температуре в течение 15 минут. Добавляли по каплям 2M раствор AlMe3 (0,7 мл, 1,4 ммоль, а через 10 минут добавляли сложный эфир, полученный на стадии b (120 мг, 0,35 ммоль) в безводном толуоле (2 мл). Полученную смесь перемешивали при комнатной температуре в течение ночи, затем добавляли H2O (5 мл) и тартрат калия (500 мг). Органический слой отделяли, и водную фазу экстрагировали диэтиловым эфиром. Объединенные органические слои сушили над MgSO4, фильтровали и упаривали. Остаток очищали методом колоночной хроматографии (силикагель, CH2Cl2 → 1:1 CH2Cl2:EtOAc) и затем методом C18 ВЭЖХ, получая продукт в виде смеси диастереомеров (75 мг, 33%). 1H ЯМР (400 МГц, CDCl3) δ 7,83-7,71 (м, 3 H), 7,41-7,27 (м, 4 H), 7,05 (т, J = 6,3 Гц, 1 H), 6,94-6,87 (м, 1 H), 6,74-6.66 (м, 1 H), 4,80-4,32 (м, 3 H), 4,21-3,92 (м, 1 H), 3,90-3,60 (м, 2 H), 3,27-2,89 (м, 6 H), 2,86-2,63 (м, 1 H), 2,58-2,38 (м, 1 H), 2,24-1,94 (м, 2 H), 1,72-1,47 (м, 2 H). MS: (ES) m/z вычислено для C28H29F6N2O2 [M + H]+ 539,2, найдено 539,2.
Пример 16
Синтез (2S,5R)-5-[4-(4-фторфенил)-1-пиперидил]-2-изопропил-N-[[5-(трифторметил)-3-пиридил]метил]тетрагидропиран-2-карбоксамида и (2R,5S)-5-[4-(4-фторфенил)-1-пиперидил]-2-изопропил-N-[[5-(трифторметил)-3-пиридил]метил]тетрагидропиран-2-карбоксамида
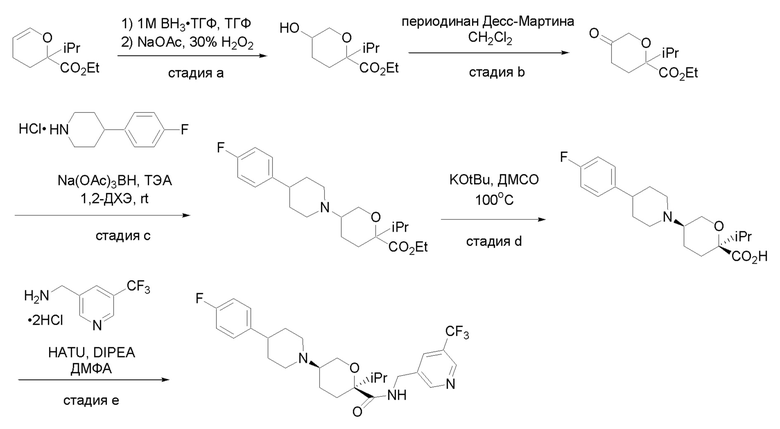
Стадия a. 2-Изопропил-3,4-дигидро-2H-пиран-2-карбоновой кислоты этиловый эфир (7,0 г, 35,0 ммоль) растворяли в безводном ТГФ (100 мл) и охлаждали до -10°C, затем добавляли по каплям 1M раствор BH3 в ТГФ (75 мл, 75,0 ммоль). Реакционную смесь выдерживали при 4 °C в течение ночи, затем охлаждали до -10°C и медленно добавляли раствор NaOAc (5,7 г, 70 ммоль) в H2O (15 мл). Через 10 минут добавляли 35%-ный H2O2 (10,2 мл, 105 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь разбавляли насыщенным водным раствором хлорида натрия (50 мл), и органический слой отделяли, сушили над MgSO4, фильтровали и упаривали. Остаток разбавляли эфиром (150 мл), отфильтровывали белый осадок и упаривали фильтрат, получая сырой продукт в виде масла (7,5 г, 98%).
Стадия b. Стадию окисления проводили аналогично примеру 15, получая желтое масло, 96%.
Стадия c. Стадию восстановительного аминирования проводили аналогично примеру 15, получая продукт в виде смеси диастереомеров (52%). MS: (ES) m/z вычислено для C22H33FNO3 [M + H]+ 378,2, найдено 378,1.
Стадия d. Продукт, полученный на стадии c (4,6 г, 12,2 ммоль), растворяли в ДМСО (25 мл) и добавляли KOtBu (3,4 г, 30,5 ммоль). Полученную смесь перемешивали при 100°C в течение 20 минут, затем охлаждали и разбавляли водой (100 мл), AcOH (10 мл) и экстрагировали этилацетатом (3 x 100 мл). Объединенные органические слои сушили над MgSO4, фильтровали и упаривали. Остаток разбавляли эфиром (50 мл), белый осадок продукта ((рац)-цис диастереомер) отфильтровывали и сушили в вакууме (600 мг, 14%). MS: (ES) m/z вычислено для C20H29FNO3 [M + H]+ 350,2, найдено 350,1.
Стадия e. В смесь кислоты, полученной на стадии d (40 мг, 0,115 ммоль), HATU (87 мг, 0,23 ммоль) в безводном ДМФА (1 мл) добавляли диизопропилэтиламин (0,1 мл, 0,575 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 15 минут, затем добавляли 5-трифторметил-пиридин-3-ил)-метиламин дигидрохлорид (29 мг, 0,116 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи, затем разбавляли водой (8 мл) и экстрагировали этилацетатом (2 x 5 мл). Объединенные органические слои сушили над MgSO4, фильтровали и упаривали. Остаток очищали методом препаративной ТСХ (силикагель, 2:8 гексан:EtOAc), получая целевой продукт в виде (рац)-цис диастереомера (10 мг, 17%). 1H ЯМР (400 МГц, CD3OD) δ 8,85-8,76 (м, 2 H), 8,14 (ушир.с, 1 H), 7,28-7,13 (м, 2 H), 7,06-6,90 (м, 2 H), 4,54, 4,49 (AB кв, J = 15,1 Гц, 2 H), 3,97 (ддд, J = 11,3, 4,6, 2,4 Гц, 1 H), 3,35 (т, J = 10,9 Гц, 1 H), 3,00-2,83 (м, 2 H), 2,57-2,21 (м, 5 H), 2,01-1,92 (м, 1 H), 1,88-1,58 (м, 5 H), 1,44 (тд, J = 13,2, 3,6 Гц, 1 H), 1,36-1,23 (м, 1 H), 0,91 (д, J = 6,9 Гц, 3 H), 0,83 (д, J = 6,9 Гц, 3 H). MS: (ES) m/z вычислено для C27H34F4N3O2 [M + H]+ 508,3, найдено 508,0.
Пример 17
Синтез (2S,5R)-2-циклопропил-5-[4-(4-фторфенил)-1-пиперидил]-N-[[3-фтор-5-(трифторметил)фенил]метил]тетрагидропиран-2-карбоксамида
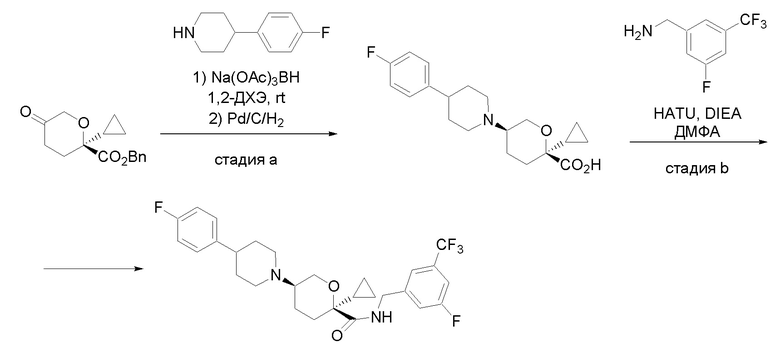
Стадия a. В раствор 4-(4-фторфенил)пиперидина (3,3 г, 13,9 ммоль) и бензил (2S)-2-циклопропил-5-оксо-тетрагидропиран-2-карбоксилата (4,0 г, 14,6 ммоль) в безводном 1,2-дихлорэтане (100 мл) добавляли твердый NaBH(OAc)3 (5,9 г, 27,8 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи, затем добавляли H2O (100 мл) и 2M K2CO3 (10 мл). Органический слой отделяли, и водную фазу экстрагировали дихлорметаном (2 x 50 мл). Объединенные органические слои сушили над MgSO4, фильтровали и упаривали. Остаток снова растворяли в MeOH (100 мл) и добавляли 10% Pd/C (50% влажность) (3 г, 1,4 ммоль) в атмосфере азота. Реакционную смесь интенсивно перемешивали в атмосфере H2 (шарик) в течение ночи, затем фильтровали через целит и упаривали. Остаток разбавляли ацетоном (15 мл) и Et2O (15 мл). Продукт в виде белого твердого вещества отфильтровывали, промывали эфиром (15 мл) и сушили в вакууме (850 мг, 15%). MS: (ES) m/z вычислено для C20H27FNO3 [M + H]+348,2, найдено 348,4.
Стадия b. Стадию сочетания с амином проводили аналогично примеру 16, получая желтоватое твердое вещество, 52%. 1H ЯМР (400 МГц, CD3OD) δ 7,51 (ушир.с, 1 H),7,40-7,30 (м, 2 H), 7,24-7,18 (м, 2 H), 7,02-6,95 (м, 2 H), 4,55, 4,44 (AB кв, J = 15,4 Гц, 2 H), 3,94 (ддд, J = 11,2, 4,4, 2,2 Гц, 1 H), 3,39 (т, J = 11,0 Гц, 1 H), 3,00-2,90 (м, 2 H), 2,58-2,41 (м, 3 H), 2,39-2,24 (м, 2 H), 2,00-1,92 (м, 1 H), 1,82-1,60 (м, 4 H), 1,52 (тд, J = 13,5, 3,7 Гц, 1 H), 1,40-1,26 (м, 1 H), 1,10-1,02 (м, 1 H), 0,70-0,64 (м, 1 H), 0,57-0,49 (м, 1 H), 0,46-0,30 (м, 2 H). MS: (ES) m/z вычислено для C28H32F5N2O2 [M + H]+ 523,2, найдено 523,0.
Пример 18
Синтез [(2S,5R)-2-циклопропил-5-[4-(4-фторфенил)-1-пиперидил]тетрагидропиран-2-ил]-[5-фтор-7-(трифторметил)-3,4-дигидро-1H-изохинолин-2-ил]метанона
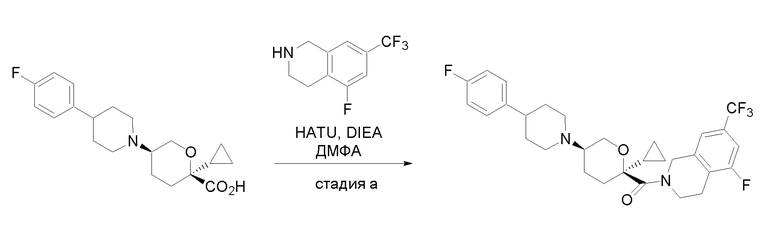
Стадия a. Указанное в заголовке соединение получали реакцией сочетания с получением амида аналогично примеру 16 (18%). 1H ЯМР (400 МГц, CD3OD) δ 7,38 (ушир.с, 1 H), 7,34-7,26 (м, 1 H), 7,25-7,14 (м, 2 H), 7,04-6,91 (м, 2 H), 5,49 (ушир.д, J = 17,4 Гц, 0,5 H), 5,16 (ушир.д, J = 17,3 Гц, 0,5 H), 4,85-4,52 (м, 1 H), 4,25-3,96 (м, 2 H), 3,30-3,18 (м, 1 H), 3,12-2,85 (м, 4 H), 2,59-2,38 (м, 3 H), 2,36-2,19 (м, 2 H), 2,09-1,97 (м, 1 H), 1,85-1,74 (м, 2 H), 1,73-1,58 (м, 2 H), 1,56-1,38 (м, 2 H), 1,34-1,27 (м, 1 H), 1,23-0,97 (м, 1 H), 0,76-0,20 (м, 4 H). MS: (ES) m/z вычислено для C30H34F5N2O2 [M + H]+ 549,3, найдено 549,0.
Пример 19
Синтез 5-[(2S)-4-[(3R,6S)-6-циклопропил-6-[6-(трифторметил)-2,4-дигидро-1,3-бензоксазин-3-карбонил]тетрагидропиран-3-ил]-2-метил-пиперазин-1-ил]-2-фтор-бензойной кислоты
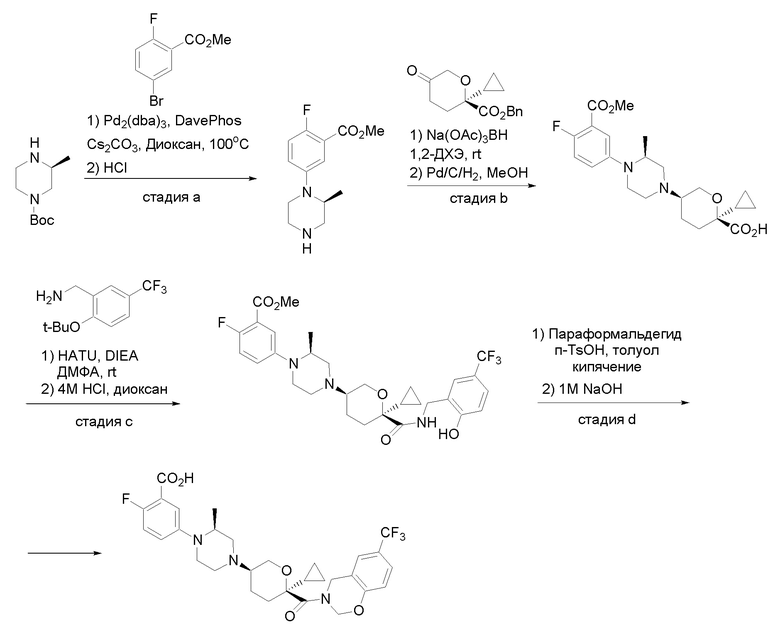
Стадия a. В раствор т-бутил (3S)-3-метилпиперазин-1-карбоксилата (10,0 г, 50,0 ммоль) и метил 5-бром-2-фтор-бензоата (11,6 г, 50 ммоль) в безводном дегазированном 1,4-диоксане (100 мл) добавляли безводный Cs2CO3 (24,4 г, 75,0 ммоль), затем DavePhos (1,2 г, 3,0 ммоль) и Pd2(dba)3 (2,3 г, 2,5 ммоль). Реакционную смесь перемешивали в атмосфере азота при 100°C в течение ночи, затем охлаждали до комнатной температуры, фильтровали через целит и упаривали. Остаток снова растворяли в EtOAc (200 мл), промывали 0,5M раствором AcOH (2 x 100 мл), сушили над MgSO4, фильтровали и упаривали. Сырой т-бутил (3S)-4-(4-фтор-3-метоксикарбонил-фенил)-3-метил-пиперазин-1-карбоксилат растворяли в 1,4-диоксане (40 мл) и 4M растворе HCl в 1,4-диоксане (40 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи, затем упаривали. Остаток разбавляли насыщенным раствором NaHCO3 (100 мл) и экстрагировали этилацетатом (2 x 100 мл). Объединенные органические слои сушили над MgSO4, фильтровали и упаривали. Полученный сырой продукт (5 г, 40%) использовали далее без дополнительной очистки.
Стадия b. Стадию восстановительного аминирования и синтез кислоты проводили аналогично примеру 17. Продукт очищали методом колоночной хроматографии (силикагель, CH2Cl2 → 9:1 CH2Cl2:MeOH), получая желтое твердое вещество (32%). MS: (ES) m/z вычислено для C22H30FN2O5 [M + H]+ 421,2, найдено 421,0.
Стадия c. В смесь кислоты, полученной на стадии b (150 мг, 0,36 ммоль), и HATU (205 мг, 0,54 ммоль) в безводном ДМФА (3 мл) добавляли диизопропилэтиламин (0,19 мл, 1,1 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 15 минут, затем добавляли 2-т-бутокси-5-(трифторметил)фенил]метанамин (99 мг, 0,4 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи, затем разбавляли водой (8 мл) и экстрагировали этилацетатом (2 x 5 мл). Объединенные органические слои сушили над MgSO4, фильтровали и упаривали. Остаток растворяли в 1,4-диоксане (2 мл) и 4M растворе HCl в 1,4-диоксане (2 мл) и перемешивали при комнатной температуре в течение 1часа, затем упаривали. Остаток разбавляли насыщенным раствором NaHCO3 (10 мл) и экстрагировали этилацетатом (3 x 5 мл). Объединенные органические слои сушили над MgSO4, фильтровали и упаривали. Полученный сырой продукт (170 мг, 80%) использовали далее без дополнительной очистки.
Стадия d. Смесь сырого фенола, полученная на стадии c (170 мг, 0,29 ммоль), параформальдегида (900 мг) и п-TsOH•H2O (300 мг) в толуоле (10 мл) перемешивали при кипячении (насадка Дина-Старка) 3 часа и затем упаривали. Остаток разбавляли насыщенным раствором NaHCO3 (10 мл) и экстрагировали этилацетатом (3 x 5 мл). Объединенные органические слои сушили над MgSO4, фильтровали и упаривали. Полученный сырой сложный эфир растворяли в MeOH (2 мл) и добавляли 4M раствор NaOH (0,5 мл, 1:1 MeOH:H2O). Полученную смесь перемешивали при комнатной температуре в течение 2часов и затем добавляли AcOH (0,5 мл). Реакционную смесь очищали методом C18 ВЭЖХ, получая продукт (52 мг, 22%). 1H ЯМР (400 МГц, CD3OD) δ 7,65 (с, 1 H), 7,54 (с, 1 H), 7,51-7,44 (м, 1 H), 7,35 (с, 1 H), 7,23-7,13 (м, 1 H), 7,04 (д, J = 8,6 Гц, 1 H), 6,27-6,09 (м, 1 H), 5,73-5,57 (м, 1 H), 4,87-4,76 (м, 1 H), 4,48-4,33 (м, 1 H), 3,71-3,34 (м, 4 H), 3,28-2,85 (м, 5 H), 2,64 (дт, J = 14,0, 3,7 Гц, 1 H), 2,37-2,24 (м, 1 H), 1,82-1,67 (м, 1 H), 1,58 (тд, J = 13,2, 3,6 Гц, 1 H), 1,35-1,14 (м, 2 H), 1,05-0,85 (м, 3 H), 0,72-0,36 (м, 4 H). MS: (ES) m/z вычислено для C30H34F4N3O5 [M + H]+ 592,2, найдено 592,0.
Пример 20
Синтез 5-[(2S)-4-[(3R,6S)-6-циклопропил-6-[8-фтор-6-(трифторметил)-2,4-дигидро-1,3-бензоксазин-3-карбонил]тетрагидропиран-3-ил]-2-метил-пиперазин-1-ил]-2-фтор-бензойной кислоты
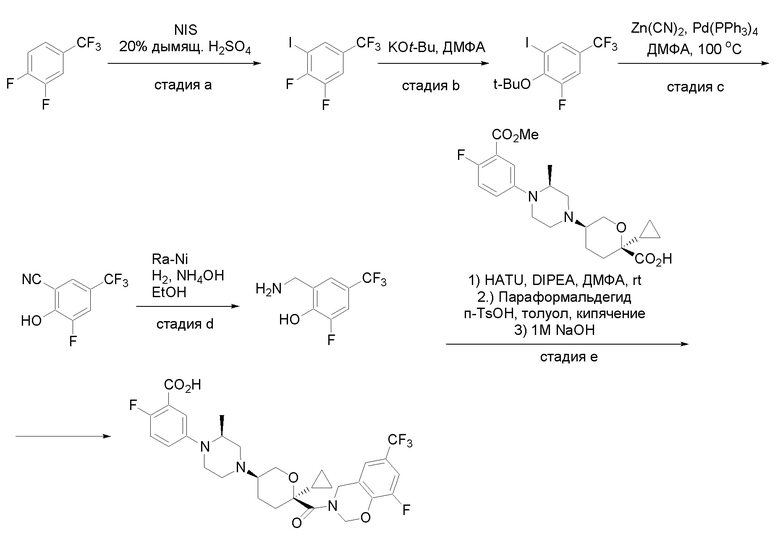
Стадия a. К 20% дымящей H2SO4 (100 мл), охлажденной до 0°C, добавляли 1,2-дифтор-4-(трифторметил)бензол (52,0 г, 285,7 ммоль), затем твердый NIS (70,7 г, 314,3 ммоль). Темно-коричневую густую смесь перемешивали при 0°C в течение 5 минут, затем медленно нагревали до комнатной температуры и перемешивали в течение 1 часа. Реакционную смесь выливали на лед и экстрагировали гексаном (2 x 300 мл). Объединенные органические слои промывали 10%-ным раствором Na2SO3, насыщенным раствором NaHCO3 и в конце насыщенным водным раствором хлорида натрия. Органический слой сушили над MgSO4, фильтровали и упаривали, получая сырой продукт в виде прозрачного масла (74 г, 84%).
Стадия b. К раствору 1,2-дифтор-3-иод-5-(трифторметил)бензола, полученного на стадии a (60,0 г, 194,8 ммоль) в безводном ДМФА (200 мл), охлажденному до 0°C, добавляли твердый KOtBu (24,0 г, 214,3 ммоль). Реакционную смесь перемешивали при 0°C в течение 30 мин, затем разбавляли водой (1 л) и экстрагировали гексаном (3 x 300 мл). Объединенные органические фазы сушили над MgSO4, фильтровали и упаривали. Остаток очищали фильтрованием через слой силикагеля и промывали 10%-ным EtOAc в гексане, получая желтое масло (63 г, 89%).
Стадия c. 2-т-Бутокси-1-фтор-3-иод-5-(трифторметил)бензол, полученный на стадии b (30,0 г, 82,9 ммоль), растворяли в безводном дегазированном ДМФА (200 мл), затем добавляли Zn(CN)2 (9,7 г, 82,9 ммоль) и Pd(PPh3)4 (4,7 г, 4,1 ммоль). Реакционную смесь перемешивали в атмосфере азота при 100 °C в течение ночи, разбавляли водой (1 л) и экстрагировали диэтиловым эфиром (2 x 300 мл). Объединенные органические слои промывали 1M раствором NaOH (2 x 80 мл) и затем водный слой нейтрализовывали 2M раствором HCl и экстрагировали эфиром (2 x 300 мл). Объединенные органические слои сушили над MgSO4, фильтровали и упаривали, получая сырой продукт в виде желтого масла (11 г, 65%), которое использовали далее без дополнительной очистки.
Стадия d. 3-Фтор-2-гидрокси-5-(трифторметил)бензонитрил, полученный на стадии c (11,0 г, 53,6 ммоль), растворяли в EtOH (50 мл) и добавляли 28%-ный NH4OH (5 мл) и суспензию никеля Ренея 2800 в H2O (3 мл). Реакционную смесь встряхивали в аппарате Парра в атмосфере H2 (50 фунт/кв.дюйм) в течение ночи, затем фильтровали через целит и упаривали. Полученный сырой продукт промывали эфиром, получая продукт в виде желтого твердого вещества (4,6 г, 41%). MS: (ES) m/z вычислено для C8H8F4NO [M + H]+ 210,1, найдено 210,2.
Стадия e. Целевое соединение получали аналогично предыдущим примерам (27%). 1H ЯМР (400 МГц, CD3OD) δ 7,69-7,60 (м, 1 H), 7,46-7,28 (м, 3 H), 7,18 (т, J = 9,5 Гц, 1 H), 6,40-6,14 (м, 1 H), 5,85-5,63 (м, 1 H), 5,11-4,92 (м, 1 H), 4,51-4,31 (м, 1 H), 3,71-3,36 (м, 4 H), 3,28-2,74 (м, 6 H), 2,65 (дт, J = 13,4, 3,4 Гц, 1 H), 2,40-2,24 (м, 1 H), 1,83-1,67 (м, 1 H), 1,59 (тд, J = 13,5, 3,8 Гц, 1 H), 1,32-1,11 (м, 1 H), 1,08-0,84 (м, 3 H), 0,77-0,27 (м, 4 H). MS: (ES) m/z вычислено для C30H33F5N3O5 [M + H]+ 610,2, найдено 610,0.
Пример 21
Синтез 5-[1-[(3R,6S)-6-циклопропил-6-[8-фтор-6-(трифторметил)-2,4-дигидро-1,3-бензоксазин-3-карбонил]тетрагидропиран-3-ил]-4-пиперидил]-2-фтор-бензойной кислоты
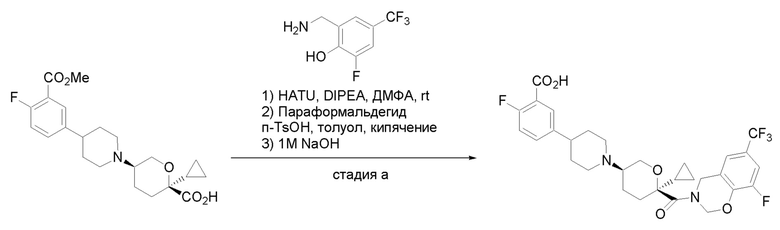
Стадия a. Целевое соединение получали аналогично предыдущим примерам (42%). 1H ЯМР (400 МГц, CD3OD) δ 7,80 (дд, J = 6,9, 2,5 Гц, 1 H), 7,53-7,43 (м, 1 H), 7,44-7,34 (м, 2 H), 7,17 (дд, J = 10,6, 8,5 Гц, 1 H), 6,25 (ушир.с, 1 H), 5,72 (ушир.с, 1 H), 5,12-4,91 (м, 2 H), 4,51-4,21 (м, 1 H), 3,75-3,51 (м, 3 H), 3,46-3,35 (м, 1 H), 3,23-3,08 (м, 2 H), 2,99-2,86 (м, 1 H), 2,70-2,56 (м, 1 H), 2,33-2,20 (м, 1 H), 2,20-2,06 (м, 2 H), 1,95-1,67 (м, 3 H), 1,68-1,53 (м, 1 H), 1,32-1,11 (м, 1 H), 0,79-0,22 (м, 4 H). MS: (ES) m/z вычислено для C30H32F5N2O5 [M + H]+ 595,2, найдено 595,0.
Пример 22
Синтез 5-[(2S)-4-[(3R,6S)-6-циклопропил-6-[4-фтор-7-(трифторметил)-3,4-дигидро-1H-изохинолин-2-карбонил]тетрагидропиран-3-ил]-2-метил-пиперазин-1-ил]-2-фтор-бензойной кислоты
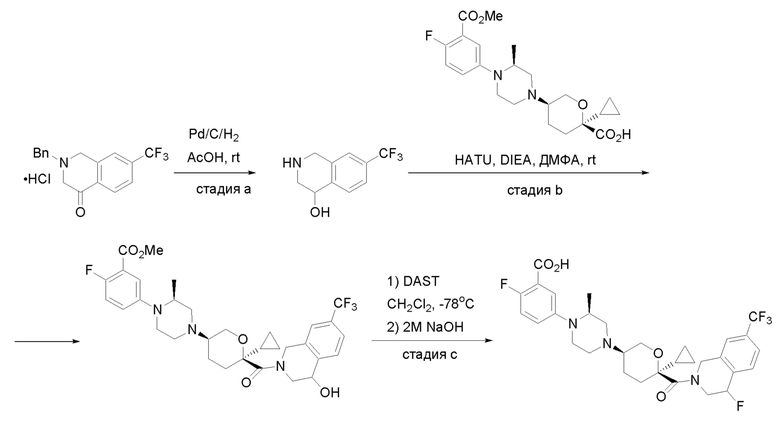
Стадия a. 2-Бензил-7-(трифторметил)-1,3-дигидроизохинолин-4-он (3,0 г, 8,8 ммоль) растворяли в AcOH (40 мл) и добавляли 10% Pd/C (50% влажность) (1,9 г, 0,9 ммоль) в атмосфере азота. Реакционную смесь интенсивно перемешивали в атмосфере H2 (шарик) в течение ночи, затем фильтровали через целит и упаривали. Остаток промывали CH3CN и Et2O, получая зеленоватое твердое вещество (2 г, 90%). MS: (ES) m/z вычислено для C10H11F3NO [M + H]+ 218,1, найдено 218,0.
Стадия b. Стадию сочетания с образованием амида проводили аналогично предыдущим примерам (53%). MS: (ES) m/z вычислено для C32H38F4N3O5 [M + H]+ 620,3, найдено 620,0.
Стадия c. Неочищенный спирт, полученный на стадии b (111 мг, 0,18 ммоль), растворяли в безводном CH2Cl2 (5 мл), охлаждали до -78°C, (добавляли диэтиламино)серы трифторид (94 мкл, 0,72 ммоль), и реакционную смесь перемешивали при -78°C в течение 1 часа. Добавляли MeOH (1 мл) и растворитель упаривали. Остаток растворяли в MeOH (1 мл) и добавляли 4M раствор NaOH (1 мл, 1:1 MeOH:H2O). Полученную смесь перемешивали при комнатной температуре в течение 1 часа и затем добавляли AcOH (0,5 мл). Реакционную смесь очищали методом C18 ВЭЖХ, получая целевой продукт (15 мг, 10%). 1H ЯМР (400 МГц, CD3OD) δ 7,73-7,63 (м, 4H), 7,42-7,29 (м, 1H), 7,22-7,12 (м, 1H), 5,94-5,58 (м, 2H), 5,52-5,25 (м, 1H), 4,50-4,22 (м, 1H), 3,80-3,36 (м, 3H), 3,26-2,80 (м, 7H), 2,68-2,56 (м, 2H), 2,35-2,20 (м, 1H), 1,86-1,46 (м, 2H), 1,35-0,30 (м, 8H). MS: (ES) m/z вычислено для C31H35F5N3O4 [M + H]+ 608,3, найдено 608,0.
Синтез метил 2-фтор-5-[[(3R)-пирролидин-3-ил]амино]бензоата
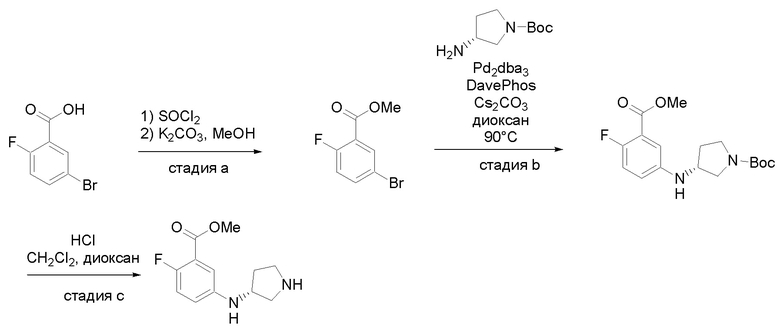
Стадия a. К 5-бром-2-фтор-бензойной кислоте (6,38 г, 29,1 ммоль, 1 экв.) добавляли тионил хлорид (10 мл, 290 ммоль, 10 экв.). Полученную смесь перемешивали при 80°C до завершения реакции, затем упаривали. Добавляли K2CO3 (10,87 г, 78,6 ммоль, 2,7 экв.), затем MeOH (30 мл). Полученный раствор перемешивали при комнатной температуре. Реакционную смесь фильтровали и осадок промывали метиленхлоридом. Органическую фазу затем промывали водой, сушили над MgSO4, фильтровали и упаривали, получая продукт (3,29 г, 14,1 ммоль, 49%).
Стадия b. К 5-бром-2-фтор-бензойной кислоты метиловому эфиру (652 мг, 2,80 ммоль, 1 экв.) добавляли Cs2CO3 (2,34 г, 7,18 ммоль, 2,5 экв.), затем добавляли 2-дициклогексилфосфино-2′-(N,N-диметиламино)бифенил (132 мг, 0,335 ммоль, 0,12 экв.), диоксан (6 мл) и (R)-3-Амино-пирролидин-1-карбоновой кислоты трет-бутиловый эфир (0,695 мл, 3,37 ммоль, 1,2 экв.). N2 барботировали через смесь в течение 5 минут, затем добавляли Pd2(dba)3 и снова барботировали через смесь N2 в течение 5 минут. Реакционную смесь затем нагревали при 90 °C в течение 2 дней. Смесь охлаждали до комнатной температуры, разбавляли дихлорметаном, промывали водой (3 x 20 мл), затем насыщенным водным раствором хлорида натрия и сушили над MgSO4, фильтровали и упаривали, получая сырой продукт. Его очищали хроматографией на силикагеле (гексан/EtOAc), получая целевой продукт (328 мг, 0,969 ммоль, 35%).
Стадия c. В (R)-3-(4-Фтор-3-метоксикарбонил-фениламино)-пирролидин-1-карбоновой кислоты трет-бутиловый эфир (328 мг, 0,969 ммоль) добавляли CH2Cl2 (1 мл), затем 4н. раствор HCl в диоксане (1 мл, 4 ммоль). Смесь перемешивали в течение ночи и затем упаривали. Остаток разделяли между насыщенным раствором NaHCO3 и EtOAc. Слои разделяли, водный слой экстрагировали 2:1 смесью CHCl3:iPrOH (5 x). Экстракты сушили над Na2SO4, фильтровали и упаривали, получая (R)-2-фтор-5-(пирролидин-3-иламино)-бензойной кислоты метиловый эфир (216 мг).
Пример 23
Синтез 5-[[(3R)-1-[(3R,6S)-6-циклопропил-6-[6-(трифторметил)-2,4-дигидро-1,3-бензоксазин-3-карбонил]тетрагидропиран-3-ил]пирролидин-3-ил]амино]-2-фтор-бензойной кислоты
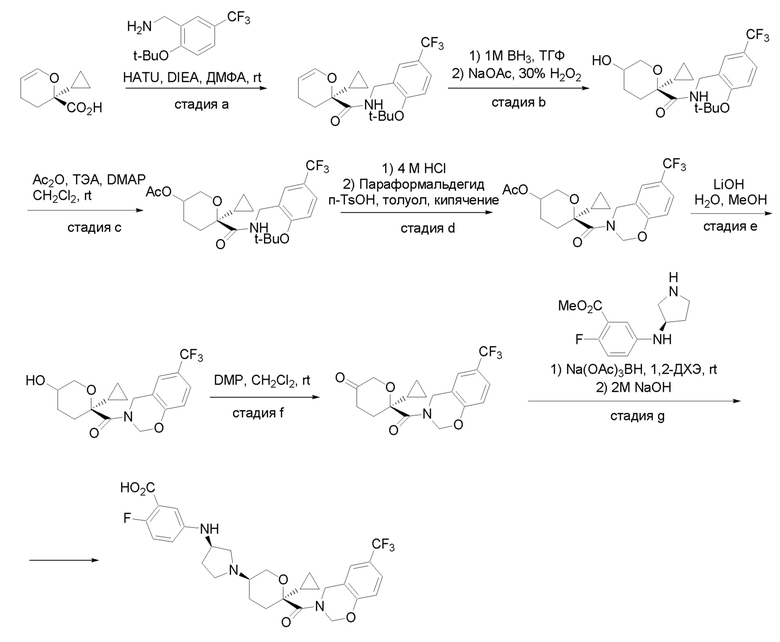
Стадия a. В смесь (2S)-2-циклопропил-3,4-дигидропиран-2-карбоновой кислоты (6,5 г, 38,9 ммоль) и HATU (16,3 г, 42,8 ммоль) в безводном ДМФА (80 мл) добавляли диизопропилэтиламин (13,5 мл, 77,8 ммоль), и смесь перемешивали при комнатной температуре в течение 15 минут, после чего добавляли раствор 2-т-бутокси-5-(трифторметил)фенил]метанамин (10,5 г, 42,8 ммоль) в безводном ДМФА (20 мл), и реакционную смесь перемешивали при комнатной температуре в течение 1 дня. Смесь разбавляли водой (600 мл) и экстрагировали EtOAc (3 x 200 мл). Объединенные органические слои промывали насыщенным водным раствором хлорида натрия (4 x 50 мл), сушили над MgSO4, фильтровали и упаривали. Полученный сырой продукт очищали методом колоночной хроматографии (силикагель, гексан → 9:1 гексан:EtOAc), получая белое твердое вещество (10,4 г, 67%).
Стадия b. Продукт, полученный на стадии a (10,0 г, 25,2 ммоль), растворяли в безводном ТГФ (100 мл) и охлаждали до -10 °C, затем добавляли по каплям 1M раствор BH3 в ТГФ (25,2 мл, 25,2 ммоль). Реакционную смесь выдерживали при 4 °C в течение ночи, затем охлаждали до -10°C и медленно добавляли раствор NaOAc (4,1 г, 50,4 ммоль) в H2O (15 мл). Через 10 минут добавляли 35%-ный H2O2 (7,6 мл, 75,6 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Смесь разбавляли насыщенным водным раствором хлорида натрия (50 мл), отделяли органический слой, сушили над MgSO4, фильтровали и упаривали. Остаток разбавляли эфиром (150 мл), белое твердое вещество отфильтровывали и фильтрат упаривали, получая сырой продукт в виде масла (10,5 г, колич.).
Стадия c. В продукт со Стадии b (10,4 г, 25,2 ммоль), добавляли триэтиламин (5,3 мл, 37,8 ммоль) и DMAP (0,3 г, 2,5 ммоль) в безводном CH2Cl2 (150 мл). Добавляли по каплям уксусный ангидрид (3,6 мл, 37,8 ммоль) при комнатной температуре, и смесь перемешивали в течение 1 дня. Реакционную смесь промывали водой (100 мл), затем насыщенным раствором NaHCO3 (100 мл), и органический слой сушили над MgSO4, фильтровали и упаривали. Полученный сырой продукт очищали методом колоночной хроматографии (силикагель, гексан → 7:3 гексан:EtOAc), получая вязкое желтое масло (8,0 г, 70%).
Стадия d. Продукт со Стадии c (8,0 г, 17,5 ммоль), растворяли в безводном 1,4-диоксане (10 мл) и 4M растворе HCl в 1,4-диоксане (30 мл); перемешивали при комнатной температуре в течение 1 час, затем упаривали. Остаток растворяли в толуоле (10 мл), затем добавляли параформальдегид (20 г) и п-TsOH•H2O (332 мг), реакционную смесь перемешивали при кипячении (насадка Дина-Старка) 2 часа, и затем упаривали. Остаток очищали методом колоночной хроматографии (силикагель, гексан → 7:3 гексан:EtOAc), получая вязкое желтое масло (6,2 г, 86%).
Стадия e. Продукт со Стадии d (6,2 г, 15,0 ммоль), растворяли в MeOH (70 мл) и добавляли раствор LiOH•H2O (3,1 г, 75 ммоль) в H2O (35 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, затем нейтрализовывали до pH = 7 добавлением 2M раствора AcOH и упаривали. Остаток разбавляли насыщенным водным раствором хлорида натрия (100 мл), экстрагировали дихлорметаном (3 x 50 мл), объединенные органические фазы сушили над MgSO4, фильтровали и упаривали, получая белое твердое вещество (5,3 г, 95%).
Стадия f. Стадию окисления проводили аналогично предыдущим примерам, получая желтое масло (колич.).
Стадия g. В раствор продукта по Стадии f (150 мг, 0,4 ммоль), и метил 2-фтор-5-[[(3R)-пирролидин-3-ил]амино]бензоата (98 мг, 0,4 ммоль) в безводном 1,2-дихлорэтане (5 мл) добавляли NaBH(OAc)3 (174 мг, 0,8 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение в течение ночи, затем промывали насыщенным раствором NaHCO3 (10 мл). Органический слой отделяли, сушили над MgSO4, фильтровали и упаривали. Остаток растворяли в MeOH (1 мл) и добавляли 4M раствор NaOH раствор (1 мл, 1:1 MeOH:H2O). Полученную смесь перемешивали при комнатной температуре в течение 1 часа, затем добавляли AcOH (0,5 мл). Реакционную смесь очищали методом C18 ВЭЖХ, получая продукт (39 мг, 12%). 1H ЯМР (400 МГц, CD3OD) δ 7,57-7,40 (м, 2H), 7,14-6,95 (м, 3H), 6,86-6,77 (м, 1H), 6,27-6,07 (м, 1H), 5,69-5,54 (м, 1H), 4,38-4,10 (м, 2H), 3,57-3,34 (м, 2H), 2,63-2,51 (м, 2H), 2,29-2,16 (м, 2H), 2,15-1,90 (м, 2H), 1,80-1,62 (м, 2H), 1,55 (тд, J = 13,3, 3,8 Гц, 2H), 1,30-1,09 (м, 2H), 0,72-0,36 (м, 5H). MS: (ES) m/z вычислено для C29H32F4N3O5 [M + H]+ 578,2, найдено 577,9.
Пример 24
Синтез 3-[(2S)-4-[(3R,6S)-6-циклопропил-6-[6-(трифторметил)-2,4-дигидро-1,3-бензоксазин-3-карбонил]тетрагидропиран-3-ил]-2-метил-пиперазин-1-ил]пропановой кислоты
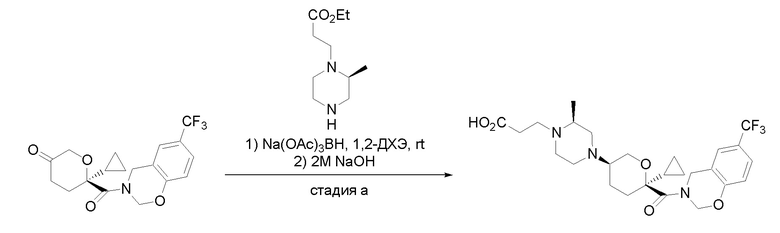
Стадия a. Целевое соединение получали аналогично предыдущим примерам, но применяя этил 3-[(2S)-2-метилпиперазин-1-ил]пропаноат (7,5%). 1H ЯМР (400 МГц, CD3OD) δ 7,56-7,43 (м, 2H), 7,05-6,97 (м, 1H), 6,22-6,06 (м, 1H), 5,80-5,61 (м, 1H), 4,16-3,98 (м, 1H), 3,60-3,39 (м, 2H), 3,27-2,87 (м, 8H), 2,79-2,57 (м, 4H), 2,57-2,41 (м, 2H), 2,05-1,91 (м, 1H), 1,54-1,39 (м, 2H), 1,32 (д, J = 6,4 Гц, 3H), 1,22-1,06 (м, 1H), 0,70-0,33 (м, 4H). MS: (ES) m/z вычислено для C26H35F3N3O5 [M + H]+ 526,3, найдено 526,0.
Пример 25
Синтез 5-[(2S)-4-[(3R,6S)-6-циклопропил-6-[8-этокси-6-(трифторметил)-2,4-дигидро-1,3-бензоксазин-3-карбонил]тетрагидропиран-3-ил]-2-метил-пиперазин-1-ил]-2-фтор-бензойной кислоты
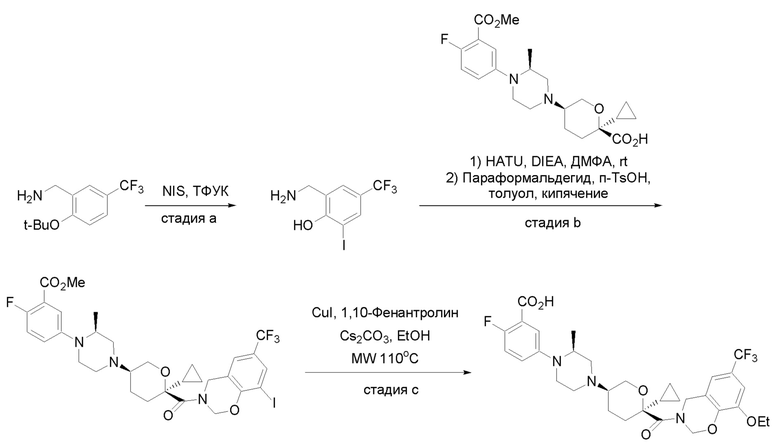
Стадия a. В раствор 2-трет-бутокси-5-(трифторметил)фенил]метанамина (4,0 г, 16,2 ммоль) в ТФУК (20 мл) добавляли N-иодсукцинимид (4,4 г, 19,4 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 30 минут. Смесь упаривали, затем остаток растворяли в EtOAc (250 мл) и промывали насыщенным раствором NaHCO3 (50 мл) и 10%-ным раствором Na2SO3 (50 мл). Органический слой сушили над MgSO4, фильтровали и упаривали, получая желтое твердое вещество (4,8 г, 93%).
Стадия b. Стадию b проводили аналогично предыдущим примерам, получая желтое твердое вещество (90%). MS: (ES) m/z вычислено для C31H35F4IN3O5 [M + H]+ 732,2, найдено 732,2.
Стадия c. Продукт со Стадии b (100 мг, 0,14 ммоль), 1,10-фенантролин (13 мг, 0,07 ммоль), CuI (13 мг, 0,07 ммоль) и Cs2CO3 (91 мг, 0,28 ммоль) помещали в микроволновую виалу и разбавляли безводным EtOH (3 мл). Реакцию проводили в микроволновом реакторе при 110°C в течение 1 часа, затем добавляли AcOH (1 мл), и реакционную смесь очищали методом C18 ВЭЖХ, получая продукт (12 мг, 10%). 1H ЯМР (400 МГц, CD3OD) δ 7,78-7,59 (м, 1H), 7,45-7,27 (м, 1H), 7,25-7,03 (м, 3H), 6,32-6,15 (м, 1H), 5,69-5,51 (м, 1H), 4,83-4,72 (м, 1H), 4,51-4,34 (м, 1H), 4,10 (кв, J = 7,0 Гц, 2H), 3,70-3,36 (м, 4H), 3,27-2,82 (м, 5H), 2,64 (дт, J = 13,7, 3,6 Гц, 1H), 2,37-2,25 (м, 1H), 1,83-1,66 (м, 1H), 1,59 (тд, J = 13,4, 3,7 Гц, 1H), 1,42 (т, J = 7,0 Гц, 3H), 1,39-1,14 (м, 2H), 1,07-0,85 (м, 3H), 0,73-0,37 (м, 4H). MS: (ES) m/z вычислено для C32H38F4N3O6 [M + H]+ 636,3, найдено 636,0.
Синтез метил 3-[(3R,4S)-3-метил-4-пиперидил]бензоата
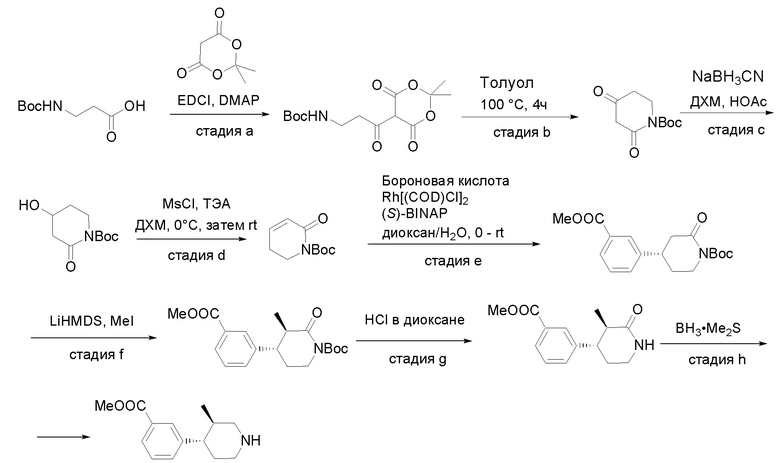
Стадия a. EDCI (191,7 г, 1,0 моль) добавляли в охлажденный на ледяной бане раствор 3-(трет-бутоксикарбониламино)пропановой кислоты (189,2 г, 1,0 моль), кислоты Мелдрума (144,1 г, 1,0 моль) и DMAP (135,0 г, 1,1 моль) в ДМФА (400 мл). Реакционную смесь оставляли перемешиваться при комнатной температуре в течение ночи. Реакцию гасили добавлением 200 мл H2O. Результирующую смесь добавляли по каплям в H2O (2,0 л) примеханическом перемешивании. Выпавший твердый осадок отфильтровывали и тщательно промывали водой до pH ~ 7, получая продукт (283 г, 90% выход). 1H ЯМР (300 МГц, CDCl3): δ 4,9 (ушир.с, 1H), 3,55 (кв, 2H), 3,45-3,25 (т, 3H), 1,75 (с, 6H), 1,4 (с, 9H).
Стадия b. Раствор трет-бутил-3-(2,2-диметил-4,6-диоксо-1,3-диоксан-5-ил)-3-оксопропилкарбамата (100 г, 317,46 ммоль) в толуоле (1500 мл) нагревали при 100 °C в течение 4 часов. Реакционную смесь охлаждали до комнатной температуры и упаривали при пониженном давлении, получая полученный сырой продукт. Очистка методом растирания в МТБЭ дала продукт (56 г, 83% выход). 1H ЯМР (300 МГц, CDCl3): δ 4,1 (т, 2H), 3,5 (с, 2H), 2,6 (т, 2H), 1,55 (с, 9H).
Стадия c. трет-Бутил 2,4-диоксопиперидин-1-карбоксилат (106,5 г, 500ммоль) растворяли в CH2Cl2 (1,0 л) и HOAc ( 60 мл). Результирующий раствор охлаждали до 0°C, затем порциями добавляли NaBH3CN (37,8 г, 600 ммоль). После перемешивания при комнатной температуре в течение ночи, реакционную смесь охлаждали до 0°C и медленно добавляли водный NH4OH, доводя pH до 9. Органическую фазу отделяли и промывали насыщенным водным раствором хлорида натрия (2 x 400 мл), затем сушили над MgSO4, фильтровали и упаривали, получая продукт в виде липкого масла.
Стадия d. Продукт со Стадии c, растворяли в CH2Cl2 (1,0 л), и полученный раствор охлаждали до 0°C. Добавляли триэтиламин (151,5 г, 1,5 моль), и результирующую смесь перемешивали при 0°C в течение 30 минут. Добавляли по каплям уксусный ангидрид (53,5 г, 525 ммоль). После перемешивания при комнатной температуре в течение ночи, добавляли воду (400 мл), и органический слой отделяли и промывали 5%-ным раствором KHSO4 (3 x 300 мл). Органическую фазу сушили над MgSO4, затем упаривали при пониженном давлении. Остаток пропускали через короткий слой SiO2 (500 г) и промывали 20%-ным раствором EtOA в гексане. Объединенные органические слои упаривали, получая продукт (69 г) в виде бесцветного масла, которое напрямую использовали в следующей стадии без дополнительной очистки.
Стадия e. Хлор(1,5-циклооктадиен)родий(I) димер (493 мг, 1 ммоль) и (S)-BINAP (1,56 г, 2,5 ммоль) добавляли в раствор 3-метоксикарбонилфенилбороновой кислоты (9,0 г, 50 ммоль) в диоксане (65 мл). Результирующую смесь перемешивали при комнатной температуре в атмосфере азота 1 час. Полученную смесь охлаждали до 0°C и добавляли H2O (10 мл), затем 6-оксо-3,6-дигидро-2H-пиридин-1-карбоновой кислоты трет-бутиловый эфир (9,3 г, 47,5 ммоль) в диоксане (5 мл) и триэтиламин (4,8 г, 47,5 ммоль). Результирующую смесь перемешивали при комнатной температуре в течение ночи в атмосфере азота. Полученную смесь разбавляли гептаном (65 мл), МТБЭ (17,5 мл) и водой (50 мл). Органическую фазу отделяли и промывали водой (50 мл). Водную фазу объединяли и экстрагировали смесью МТБЭ/гептан (1:2, 50 мл). Объединенные органические слои промывали насыщенным водным раствором хлорида натрия и сушили над MgSO4. Упариванием при пониженном давлении получали бледное твердое вещество, которое промывали смесью 10% EtOAc в гексане (100 мл), получая 10 г белого твердого вещества (соединение 5, 60% выход, 99% ee). Процент ee определяли методом ВЭЖХ с применением хиральной колонки (Regiscell 25см x 4,6мм, 5 микрон сферический) и 30% iPrOH в гексане в качестве элюента.
Стадия f. 4-(3-Метоксикарбонил-фенил)-2-оксо-пиперидин-1-карбоновой кислоты трет-бутиловый эфир (40 г, 120 ммоль) растворяли в безводном ТГФ (120 мл), и полученный раствор охлаждали до -78°C. Добавляли по каплям LiHMDS в ТГФ (1M, 122 мл, 122 ммоль), и результирующую смесь перемешивали при -78°C в течение 3 часов, затем добавляли по каплям MeI (33,84 г, 240 ммоль) при той же температуре. Полученную смесь медленно нагревали до комнатной температуры в течение ночи. Добавляли воду (100 мл), органический слой отделяли и промывали насыщенным водным раствором хлорида натрия и сушили. Упариванием при пониженном давлении получали коричневое твердое вещество, которое перекристаллизовывали из смеси 10% EtOAc в гексане (400 мл), получая целевой продукт (35 г, 84%) в виде бледно-желтого твердого вещества.
Стадия g. 4-(3-Метоксикарбонил-фенил)-3-метил-2-оксо-пиперидин-1-карбоновой кислоты трет-бутиловый эфир (10,4 г, 30 ммоль) добавляли порциями в раствор HCl в диоксане (4M, 20 мл, 80 ммоль). Результирующую смесь перемешивали при комнатной температуре в течение двух часов, затем упаривали при пониженном давлении, получая белое твердое вещество.
Стадия h. Продукт со Стадии g, растворяли в 30 мл безводного ТГФ. Результирующий раствор охлаждали до -78°C и добавляли по каплям раствор BH3•SMe2 в ТГФ (2M, 45 мл, 90 ммоль). После добавления, смесь перемешивали при комнатной температуре 48 часов, после чего охлаждали до -78°C. Осторожно гасили реакцию добавлением MeOH (10 мл). Добавляли концентрированную HCl (8 мл), и смесь перемешивали при 60°C в течение 2 часов. Удаляли MeOH при пониженном давлении и разбавляли остаток водой (20 мл). результирующую водную смесь экстрагировали смесью МТБЭ:гептан (1:2, 50 мл). Отделенный водный слой доводили до pH 9 добавлением водного раствора NH4OH, после чего экстрагировали смесью iPrOH:CHCl3 (1:2, 3 x 100 мл). Объединенные органические слои промывали насыщенным водным раствором хлорида натрия и сушили над MgSO4, затем упаривали при пониженном давлении. Остаток очищали хроматографией на силикагеле (10% MeOH в CH2Cl2, плюс 1% NH4OH), получая целевой продукт в виде бесцветного масла.
Синтез 2-(аминометил)-4,6-бис(трифторметил)фенола
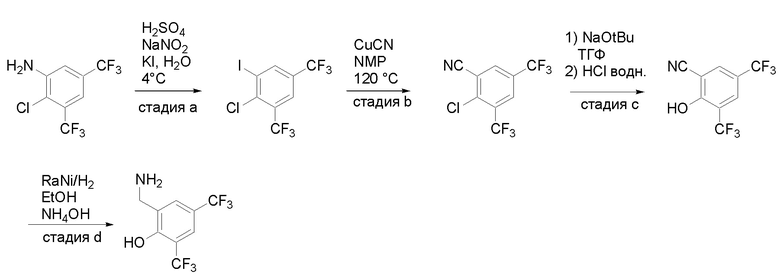
Стадия a. Смесь H2O (30 мл) и H2SO4 (30 мл) охлаждали на ледяной бане. Добавляли 2-Хлор-3,5-бис-трифторметиланилин (5,00 г, 19,0 ммоль, 1 экв.) в MeCN (30 мл) в течение 5 минут. Смесь перемешивали в течение 10 минут, затем добавляли по каплям NaNO2 (2,36 г, 34,2 ммоль, 1,8 экв.) в H2O (17 мл) в течение 5 минут, в это время внутренняя температура достигала 10°C. Смесь перемешивали 10 минут, затем выливали в охлажденный на льду раствор KI (11,0 г, 66,5 ммоль, 3,5 экв.) в H2O (30 мл). Смесь перемешивали в течение 2 часов, затем разбавляли CHCl3 (60 мл). Слои разделяли и водный слой экстрагировали CHCl3. Объединенные органические слои промывали насыщенным водным раствором NaHCO3 (2 x), затем насыщенным водным раствором Na2S2O3. Органическую фазу затем сушили над MgSO4, фильтровали и упаривали, получая продукт (6,58g, 93%) в виде светло-коричневого масла.
Стадия b. 2-Хлор-1-иод-3,5-бис-трифторметил-бензол (11,8 г, 31,5 ммоль, 1 экв.) растворяли в N-метилпирролидоне (32 мл) и добавляли CuCN (3,4 г, 38,2 ммоль, 1,2 экв.). Реакционную смесь нагревали при 120°C в течение 6 часов. Смесь охлаждали до комнатной температуры, затем разбавляли водой (100 мл) и гептаном (100 мл). Затем фильтровали смесь через целит и слои разделяли. Органический слой фильтровали через силикагель (10 г), элюируя гептаном (50 мл). Полученный раствор упаривали, получая продукт (6,6 г, 76%).
Стадия c. 2-Хлор-3,5-бис-трифторметил-бензонитрил (57,1 г, 209 ммоль, 1 экв.) растворяли в ТГФ (417 мл) и охлаждали на ледяной бане. Добавляли NaOtBu (24,0 г, 250 ммоль, 1,2 экв.) порциями в течение 10 минут, поддерживая внутреннюю температуру ниже 8°C. Через 30 минут реакцию оставляли нагреваться до комнатной температуры, затем перемешивали в течение 3 часов. Добавляли водный раствор HCl (4н., 627 мл, 2,51 моль, 12 экв.) и перемешивали при комнатной температуре в течение ночи. Реакционную смесь нагревали при 50°C в течение 2 часов. Полученный раствор упаривали, добавляли гептан (400 мл). Слои разделяли, органическую фазу сушили над MgSO4, фильтровали и упаривали. Полученное масло разбавляли гептаном до общего веса 160 г, затем охлаждали до -20°C. Смесь фильтровали, и твердую фазу промывали холодным гептаном (60 мл), получая целевой продукт (23,6 г, 44%).
Стадия d. 2-Гидрокси-3,5-бис-трифторметил-бензонитрил (2,79 г, 10,9 ммоль) растворяли в EtOH (50 мл) и NH4OH (5 мл). Добавляли суспензию никеля Ренея в воде (2,00 мл) и встряхивали реакционную смесь в атмосфере H2 (40 фунт/кв.дюйм) в течение дней. Реакционную смесь фильтровали, осадок промывали метанолом, и объединенный раствор упаривали. Остаток от упаривания растворяли в MeOH и добавляли HCl в Et2O (2M, 6 мл, 12 ммоль). Полученную смесь упаривали, затем растворяли в EtOAc. Промывали раствором NH4OH, затем упаривали. Полученный твердый продукт растирали в Et2O, получая 2-аминометил-4,6-бис-трифторметил-фенол (1,55 г, 55%).
Пример 26
Синтез 3-[(3R,4S)-1-[(3R,6S)-6-[6,8-бис(трифторметил)-2,4-дигидро-1,3-бензоксазин-3-карбонил]-6-циклопропил-тетрагидропиран-3-ил]-3-метил-4-пиперидил]бензойной кислоты

Стадия a. В смесь 3-[(3R,4S)-3-метил-4-пиперидил]бензоата (4,5 г, 19,3 ммоль), бензил (2S)-2-циклопропил-5-оксо-тетрагидропиран-2-карбоксилата (5,8 ммоль, 21,2 ммоль) и триэтиламина (5,4 мл) в безводном 1,2-дихлорэтане (100 мл) добавляли Na(OAc)3BH (8,2 г, 38,6 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи, затем разбавляли водой (100 мл) и отделяли органический слой. Водный слой экстрагировали дихлорметаном (50 мл), и объединенные органические фазы сушили над MgSO4, фильтровали и упаривали. Остаток растворяли в MeOH (100 мл) и добавляли 10% Pd/C (50% влажность) (2,1 г, 1,0 ммоль) в атмосфере азота. Реакционную смесь интенсивно встряхивали в аппарате Парра по давлением водорода 55 фунт/кв.дюйм в течение ночи, затем фильтровали через целит и упаривали. Остаток разбавляли ацетоном (45 мл), продукт отфильтровывали, промывали ацетоном (5 мл) и Et2O (15 мл) и сушили в вакууме (2,4 г, 31%).
Стадия b. В смесь кислоты со Стадии a (2,2 г, 5,5 ммоль), и HATU (2,2 г, 5,8 ммоль) в безводном ДМФА (30 мл) добавляли диизопропилэтиламин (1,9 мл, 11,0 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 15 минут, затем добавляли 2-(аминометил)-4,6-бис(трифторметил)фенол (1,5 г, 5,8 ммоль) в безводном ДМФА (10 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи, затем разбавляли водой (300 мл) и экстрагировали этилацетатом (3 x 100 мл). Объединенные органические слои промывали насыщенным водным раствором хлорида натрия (4 x 50 мл), затем сушили над MgSO4, фильтровали и упаривали. Остаток растворяли в толуоле (300 мл) и добавляли параформальдегид (20 г), п-TsOH•H2O (2,1 г, 11 ммоль) и MsOH (0,7 мл, 11 ммоль). Реакционную смесь перемешивали при кипячении (насадка Дина-Старка) 5 часов, затем охлаждали до комнатной температуры, разбавляли насыщенным раствором NaHCO3 (200 мл) и экстрагировали этилацетатом (2 x 100 мл). Объединенные органические слои сушили над MgSO4, фильтровали и упаривали. Полученный сырой сложный эфир очищали методом колоночной хроматографии (силикагель, гексан → 1:1 гексан:EtOAc), получая белое твердое вещество (2,5 г, 70%). MS: (ES) m/z вычислено для C33H37F6N2O5 [M + H]+ 655,3, найдено 655,5.
Стадия c. Продукт со Стадии b (2,5 г, 3,8 ммоль), разбавляли 1M раствором NaOH (40 мл, 95:5; MeOH:H2O) и перемешивали при 40°C в течение 5 часов. Добавляли AcOH (2,3 мл, 40 ммоль) и смесь упаривали. Остаток разбавляли водой (200 мл) и экстрагировали дихлорметаном (2 x 100 мл). Объединенные органические слои сушили над MgSO4, фильтровали и добавляли 2M раствор HCl в Et2O (5,7 мл, 11,4 ммоль). Полученную смесь упаривали и остаток промывали эфиром (50 мл), получая желтоватое твердое вещество, которое представляло собой целевой продукт в виде HCl соли (2,15 г, 83%). 1H ЯМР (400 МГц, CD3OD) δ 7,97-7,83 (м, 3H), 7,81-7,73 (м, 1H), 7,51-7,41 (м, 2H), 6,21-5,89 (м, 2H), 5,19-4,92 (м, 2H), 4,45-4,32 (м, 1H), 3,71-3,52 (м, 3H), 3,48-3,35 (м, 1H), 3,24-3,09 (м, 1H), 2,85 (т, J = 12,0 Гц, 1H), 2,71-2,60 (м, 1H), 2,60-2,48 (м, 1H), 2,37-2,24 (м, 1H), 2,23-2,08 (м, 1H), 2,08-1,94 (м, 2H), 1,89-1,70 (м, 1H), 1,70-1,55 (м, 1H), 1,24-1,13 (м, 1H), 0,75 (д, J = 6,6 Гц, 3H), 0,72-0,28 (м, 4H). MS: (ES) m/z вычислено для C32H35F6N2O5 [M + H]+ 641,2, найдено 641,5.
Пример 27
Синтез 5-[(2S)-4-[(3R,6S)-6-[[3-хлор-5-(трифторметил)фенил]метилкарбамоил]-6-циклопропил-тетрагидропиран-3-ил]-2-метил-пиперазин-1-ил]-2-фтор-бензойной кислоты
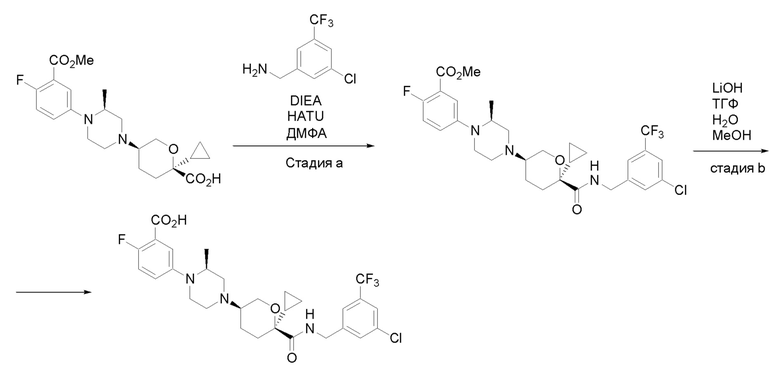
Стадия a. В виалу объемом 20 мл помещали (2S,5R)-2-циклопропил-5-[(3S)-4-(4-фтор-3-метоксикарбонил-фенил)-3-метил-пиперазин-1-ил]тетрагидропиран-2-карбоновую кислоту (39,7 мг, 0,094 ммоль), затем добавляли HATU (38,2 мг , 0,100 ммоль) и ДМФА (0,5 мл) при комнатной температуре . После добавления iPr2NEt (25 мкл, 0,144 ммоль), реакционную смесь перемешивали 1 минуту, затем добавляли 3-хлор-5-трифторметилбензиламин (18 мкл, 0,116 ммоль). Реакционную смесь перемешивали в течение ночи. Смесь разбавляли этилацетатом (20 мл) и водой (10 мл). Слои разделяли, и водную фазу экстрагировали этилацетатом (2 x 20 мл). После осушки безводным сульфатом натрия, реакционную смесь упаривали при пониженном давлении. Полученный продукт очищали методом флеш-хроматографии на силикагеле в градиенте от 15% до 33% этилацетата в гексане. Получали метил 5-[(2S)-4-[(3R,6S)-6-[[3-хлор-5-(трифторметил) фенил]метилкарбамоил]-6-циклопропил-тетрагидропиран-3-ил]-2-метил-пиперазин-1-ил]-2-фтор-бензоат с выходом 59% (34,2 мг).
Стадия b. Метил 5-[(2S)-4-[(3R,6S)-6-[[3-хлор-5-(трифторметил)фенил] метилкарбамоил]-6-циклопропил-тетрагидропиран-3-ил]-2-метил-пиперазин-1-ил]-2-фтор-бензоат (34,2 мг, 0,0559 ммоль) растворяли в ТГФ (0,25 мл) при комнатной температуре. Добавляли 1,5н. раствор гидроксида лития (60 мкл, 0,090 ммоль), затем метанол (60 мкл). Реакционную смесь перемешивали в течение ночи. Полученный сырой продукт очищали методом обращенно-фазовой ВЭЖХ, применяя градиент ацетонитрила в воде с 0,01% трифторметилуксусной кислоты (20% ~ 95%). После удаления растворителя получали 5-[(2S)-4-[(3R,6S)-6-[[3-хлор-5-(трифторметил)фенил]метилкарбамоил]-6-циклопропил-тетрагидропиран-3-ил]-2-метил-пиперазин-1-ил]-2-фтор-бензойную кислоту в виде бис-ТФУК соли (39,0 мг, 84% выход). 1H ЯМР (400 МГц, Метанол-d4) δ 8,85 (т, J = 6,3 Гц, 1H), 7,71 (ушир, 1H), 7,63 (д, J = 16 Гц, 1H), 7,61 (с, 1H), 7,40 (ушир, 1H), 7,22-7,17 (м, 1H), 4,84-4,90 (м, 1H), 4,70 (дд, J = 15,5, 7,1 Гц, 1H), 4,32 (дд, J = 15,6, 5,3 Гц, 1H), 4,24 (д, J = 11,3 Гц, 1H), 3,62 (т, J = 10,8 Гц, 1H), 3,00 - 3,62 (м, 8H), 2,74 - 2,58 (м, 1H), 2,32-2,27 (м, 1H), 1,79 - 1,52 (м, 2H), 1,22 - 1,06 (м, 1H), 0,99 (ушир, 3H), 0,71 (дт, J = 9,5, 4,6 Гц, 1H), 0,55 (дт, J = 9,8, 5,4 Гц, 1H), 0,47 (ддкв, J = 13,9, 9,0, 4,4 Гц, 1H). MS: (ES) m/z вычислено для C29H33ClF4N3O4 [M + H]+ 598,2, найдено 598,1
Пример 28
Синтез (3S,4R)-1-[(3R,6S)-6-циклопропил-6-[8-этокси-6-(трифторметил)-2,4-дигидро-1,3-бензоксазин-3-карбонил]тетрагидропиран-3-ил]-4-(4-фторфенил)пиперидин-3-карбоновой кислоты
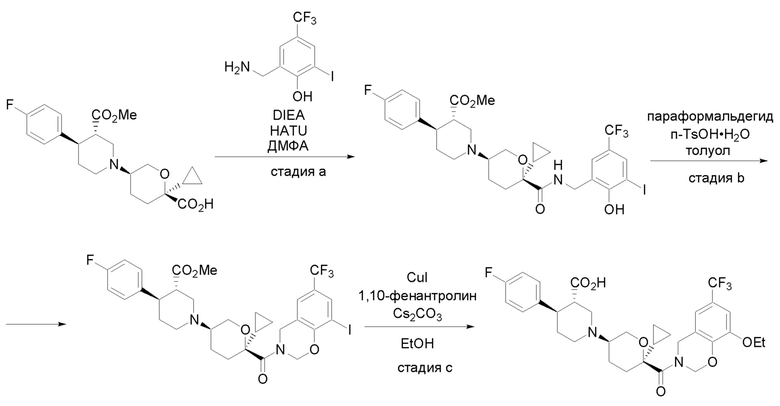
Стадия a. В виалу объемом 20 мл загружали (2S,5R)-2-циклопропил-5-[(3S,4R)-4-(4-фторфенил)-3-метоксикарбонил-1-пиперидил]тетрагидропиран-2-карбоновую кислоту (360 мг, 0,887 ммоль), HATU (356 мг, 0,936 ммоль) и ДМФА (3,5 мл) при комнатной температуре. Добавляли iPr2NEt (234 мкл, 1,34 мл), и реакционную смесь перемешивали в течение 15 минут. Часть реакционной смеси (1,56 мл) изымали для проведения другой реакции. В оставшуюся часть смеси добавляли 2-гидрокси-3-иод-5-трифторметилбензиламин (188 мг, 0,593 ммоль), и реакционную смесь перемешивали 5 часов. Добавляли еще 2-гидрокси-3-иод-5-трифторметилбензиламин (34,1 мг, 0,108 ммоль) для доведения реакции до конца. Когда все исходное соединение израсходовалось, реакционную смесь разбавляли водой (10 мл) и этилацетатом (25 мл). Слои разделяли, и водный слой экстрагировали этилацетатом (2 x 20 мл). Объединенные органические слои сушили над безводным сульфатом натрия, затем удаляли растворитель при пониженном давлении. Полученный сырой продукт использовали напрямую в следующей стадии.
Стадия b. Сырой продукт со Стадии a (~ 0,5 ммоль), растворяли в толуоле (5 мл). Добавляли п-толуолсульфокислоту моногидрат (195 мг, 1,03 ммоль), затем параформальдегид (~170 мг, 5,71 ммоль). Реакционную смесь перемешивали при 100°C. Сублимированный параформальдегид соскабливали со стенок колбы в реакционную смесь, в это же время добавляли еще параформальдегид (один шпатель каждые 10 - 20 минут). Этот процесс повторяли при нагревании при 100°C до достижения конверсии более 60% (примерно 5 часов). Реакционную смесь охлаждали и разбавляли этилацетатом (20 мл) и насыщенным водным раствором бикарбоната натрия (10 мл). Органический слой отделяли и экстрагировали этилацетатом (2 x 20 мл). Объединенные органические слои сушили над безводным сульфатом натрия. После удаления растворителя при пониженном давлении, продукт очищали методом хроматографии на силикагеле. Соединение элюировали градиентом от 17% до 50% этилацетата в гексане. Целевой продукт получали с выходом 69% (247 мг).
Стадия c. Продукт с предыдущей стадии (97,5 мг, 0,136 ммоль) помещали в виалу для проведения реакций в микроволновой печи, затем добавляли карбонат цезия (89,5 мг, 0,275 ммоль), 1,10-фенантролин (12,0 мг, 0,0666 ммоль), иодид меди (I) (12,9 мг, 0,0677 ммоль) и этанол (1 мл). Реакционную смесь нагревали в микроволновой печи 45 минут при 100°C. Добавляли воду (0,1 мл) и нагревали еще 10 минут при 100°C. После добавления 1н. раствора NaOH (0,1 мл), реакционную смесь разбавляли дихлорметаном, затем отфильтровывали нерастворенные частицы. Фильтрат упаривали и очищали методом препаративной ТСХ, применяя 17% метанола в этилацетате как элюент. Полученный продукт далее очищали методом обращенно-фазовой ВЭЖХ, получая (3S,4R)-1-[(3R,6S)-6-циклопропил-6-[8-этокси-6-(трифторметил)-2,4-дигидро-1,3-бензоксазин-3-карбонил]тетрагидропиран-3-ил]-4-(4-фторфенил)пиперидин-3-карбоновую кислоту (13,0 мг, 16% выход) в виде соли с ТФУК. 1H ЯМР (400 МГц, Метанол-d4) δ 7,23 - 7,28 (м, 2H), 7,02 - 7,15 (м, 4H), 6,24-6,21 (м, 1H), 5,63-5,59 (м, 1H) , 4,94-4,89 (м, 1H), 4,43-4,35 (м, 1H), 4,10 (кв, J = 7,0 Гц, 2H), 3,73 - 3,78 (м, 1H), 3,62 - 3,67 (м, 1H), 3,53 - 3,60 (м, 1H), 3,46 - 3,49 (м, 1H), 3,12 - 3,37 (м, 3H), 3,04 - 3,10 (м, 1H), 2,92 - 2,99 (м, 1H), 2,67-2,61 (м, 1H), 2,30-2,24 (м, 1H), 2,11-2,04 (м, 1H), 1,90 - 1,97 (м, 1H), 1,69 - 1,79 (м, 1H), 1,56-1,65 (м, 1H), 1,42 (т, J = 7,0 Гц, 3H), 1,20 - 1,26 (м, 1H), 0,59 - 0,70 (м, 3H), 0,44 - 0,49 (м, 1H). MS: (ES) m/z вычислено для C32H37F4N2O6 [M + H]+ 621,3, найдено 621,4.
Пример 29
Синтез 5-[(3R,4S)-1-[(3R,6S)-6-[6,8-бис(трифторметил)-2,4-дигидро-1,3-бензоксазин-3-карбонил]-6-циклопропил-тетрагидропиран-3-ил]-3-метил-4-пиперидил]-2-фторбензойной кислоты
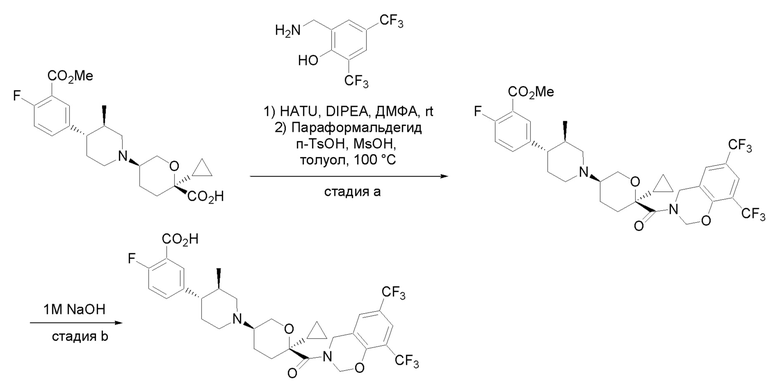
Стадия a. В смесь кислоты (2,2 г, 5,5 ммоль) и HATU (2,2 г, 5,8 ммоль) в безводном ДМФА (30 мл) добавляли диизопропилэтиламин (1,9 мл, 11,0 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 15 минут, затем добавляли 2-(аминометил)-4,6-бис(трифторметил)фенол (1,5 г, 5,8 ммоль) в безводном ДМФА (10 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи, затем разбавляли водой (300 мл) и экстрагировали этилацетатом (3 x 100 мл). Объединенные органические слои промывали насыщенным водным раствором хлорида натрия (4 x 50 мл), затем сушили над MgSO4, фильтровали и упаривали. Остаток растворяли в толуоле (300 мл) и параформальдегиде (20 г), добавляли п-TsOH x H2O (2,1 г, 11 ммоль) и MsOH (0,7 мл, 11 ммоль). Реакционную смесь перемешивали при кипячении (насадка Дина-Старка) 5 часов, затем охлаждали до комнатной температуры, разбавляли насыщенным раствором NaHCO3 (200 мл) и экстрагировали этилацетатом (2 x 100 мл). Объединенные органические слои сушили над MgSO4, фильтровали и упаривали. Полученный сырой сложный эфир очищали методом колоночной хроматографии (силикагель, гексан → 1:1 гексан:EtOAc), получая белое твердое вещество (2,5 г, 70%).
Стадия b. Продукт со Стадии a (2,5 г, 3,8 ммоль), разбавляли 1н. раствором NaOH (40 мл, 95:5; MeOH:H2O) и перемешивали при 40°C в течение 5 часов. Добавляли AcOH (2,3 мл, 40 ммоль) и смесь упаривали. Остаток разбавляли водой (200 мл) и экстрагировали дихлорметаном (2 x 100 мл). Объединенные органические слои сушили над MgSO4, фильтровали и добавляли 2M раствор HCl в Et2O (5,7 мл, 11,4 ммоль). Полученную смесь упаривали, и остаток промывали эфиром (50 мл), получая HCl соль целевого продукта в виде желтоватого твердого вещества (2,15 г, 83%). 1H ЯМР (400 МГц, CD3OD) δ 7,97-7,83 (м, 2H), 7,79-7,70 (м, 1H), 7,51-7,41 (м, 2H), 6,21-5,81 (м, 2H), 5,19-4,92 (м, 2H), 4,45-4,32 (м, 1H), 3,71-3,52 (м, 3H), 3,48-3,35 (м, 1H), 3,24-3,09 (м, 1H), 2,85 (т, J = 12,0 Гц, 1H), 2,71-2,60 (м, 1H), 2,60-2,48 (м, 1H), 2,37-2,24 (м, 1H), 2,23-2,08 (м, 1H), 2,08-1,94 (м, 2H), 1,89-1,70 (м, 1H), 1,70-1,55 (м, 1H), 1,24-1,13 (м, 1H), 0,75 (д, J = 6,6 Гц, 3H), 0,72-0,28 (м, 4H). MS: (ES) m/z вычислено для C32H34F7N2O5 [M + H]+ 659,2, найдено 658,7.
Биологические примеры
In vitro тесты
Реагенты
THP-1 клетки получали от American Type Culture Collection (Manassas, VA) и выращивали в RPMI среде для выращивания культур тканей, с добавлением 10% эмбриональной телячьей сыворотки (FCS) в увлажняемом инкубаторе с 5% CO2 при 37°C. Рекомбинантные моноцитарные хемотаксические белки MCP-1 получали от R&D Systems (Minneapolis, MN). 125I-меченый белок MCP-1 получали от Amersham (Piscataway, NJ). ChemoTX® микрокамеры для определения хемотаксиса покупали в Neuro Probe (Gaithersburg, MD). CyQUANT® набор для детекции пролиферации клеток покупали у Molecular Probes (Eugene, Oregon). Кальциевый индикаторный краситель Fluo-4 AM покупали у Molecular Devices (Mountain View, CA).
Анализ связывания лиганда
Анализ связывания лиганда можно применять для определения способности потенциальных CCR2 антагонистов блокировать взаимодействие между CCR2 и его лигандом MCP-1. CCR2-экспрессирующие клетки THP-1 центрифугировали и снова суспендировали в аналитическом буфере (20 мM HEPES pH 7,1, 140 мM NaCl, 1 мM CaCl2, 5 мM MgCl2, с 0,2% альбумина бычьей сыворотки) до концентрации 2,2 x 105 клеток/мл. Анализ связывания проводили следующим образом. Сначала добавляли 0,09 мл суспензии клеток (1 x 105 THP-1 клеток/лунка) в микропланшеты, содержащие соединения, с финальной концентрацией ~2-10 мкM каждого соединения для скрининга (или для оценки зависимости ответа от дозировки в рамках определения значения IC50). Затем добавляли 0,09 мл 125I-меченого MCP-1 (получен от Amersham; Piscataway, NJ), разведенного в аналитическом буфере до финальной концентрации ~50 пкM, что давало значение ~30000 импульсов в минуту на лунку, планшеты закрывали и инкубировали примерно 3 часа при 4°C на платформе со встряхиванием. Реакционные растворы фильтровали при отсасывании на GF/B стеклянных фильтрах, предварительно смоченных в 0,3%-ном растворе полиэтиленимина (PEI), на вакуумном харвестере клеток (Packard Instruments; Meriden, CT). Сцинтилляционную жидкость (50 мкл; Microscint 20, Packard Instruments) добавляли в каждую лунку, планшеты закрывали и измеряли радиоактивность в сцинтилляционном счетчике Top Count (Packard Instruments). Использовали контрольные лунки, содержащие либо только разбавитель (для общего числа импульсов), либо избыток MCP-1 (1 мкг/мл, для неспецифичного связывания) для вычисления процента полного ингибирования соединения. Использовали компьютерную программу Prism от GraphPad, Inc. (San Diego, Ca) для вычисления значений IC50. Значения IC50 представляют собой концентрации, необходимые для снижения связывания меченого MCP-1 с рецептором на 50%.
Анализ притока кальция
В анализе притока кальция замеряют увеличение внутриклеточной концентрации кальция после индуцированной лигандом активации рецептора, и его можно применять как вторичный анализ после первичного скрининга. Такой анализ можно проводить, например, на приборе FLIPR (Molecular Devices, Mountain View, CA). Для начала анализа, хемокин-экспрессирующие клетки (такие как клетки THP-1 для анализа CCR2) собирают центрифугированием суспензии клеток и заново суспендируют в концентрации 1,5 x 106 клеток/мл в HBSS (с 1% эмбриональной телячьей сыворотки). Затем клетки метят кальциевым индикаторным красителем Fluo-4 AM в течение 45 минут при 37°C при аккуратном встряхивании. После инкубирования клетки пеллетируют, промывают один раз HBSS и заново суспендируют в том же буфере с плотностью 1,6 x 106 клеток/мл. Сто микролитров меченых клеток смешивают с 10 мкл тестируемого соединения в необходимой концентрации в микропланшете. Добавляют хемокиновый белок (MCP-1 в финальной концентрации 0,1 нM для CCR2 анализа) для активирования рецептора. Степень ингибирования определяют путем сравнения сигналов кальция между клетками, обработанными и необработанными тестируемым соединением. Вычисление значений IC50 проводили с помощью нелинейного регрессионного анализа методом наименьших квадратов с применением Graphpad Prism (Graphpad Software, San Diego, CA).
Анализ миграции
Анализ миграции применяли для определения эффективности потенциальных антагонистов рецептора в блокировании миграции, опосредуемой хемокинами (такими как CCR2). Данный анализ стандартно проводили с применением микрокамерной системы ChemoTX® с поликарбонатной мембраной, имеющей размер пор 5 мкм. Для начала такого анализа, хемокин-экспрессирующие клетки (такие как THP-1 клетки для CCR2 анализа) собирали центрифугированием суспензии клеток при 1000 об/мин на центрифуге GS-6R Beckman. Пеллету клеток заново суспендировали в буфере для исследования хемотаксиса (HBSS с 0,1% BSA) в концентрации 10x106 клеток/мл для анализа CCR2. Нужные концентрации тестируемых соединений готовили из 10 мM стоковых растворов последовательными разбавлениями в буфере для исследования хемотаксиса. Равные объемы клеток и соединений смешивали и инкубировали при комнатной температуре в течение 15 минут. Затем 20 мкл смеси переносили на пористую мембрану миграционной микрокамеры, при этом 29 мкл хемокинового лиганда (0,1 нM хемокинового MCP-1 белка для CCR2 анализа) было помещено в нижнюю камеру. После инкубирования при 37°C (90 минут для CCR2), во время которого клетки мигрировали против градиента хемокина, анализ останавливали посредством удаления капель клеток с верха фильтра. Для количественной оценки клеток, мигрировавших через мембрану, добавляли 5 мкл 7X CyQUANT® раствора в каждую лунку в нижней камере и замеряли сигнал флуоресценции на флуоресцентном планшет-ридере Spectrafluor Plus (TECAN, Durham, NC). Степень ингибирования определяли посредством сравнения сигналов миграции между клетками, обработанными и необработанными тестируемым соединением. Вычисление значений IC50 проводили с помощью нелинейного регрессионного анализа методом наименьших квадратов с применением Graphpad Prism (Graphpad Software, San Diego, CA).
In vivo эффективность
Оценка соединений в крысиной модели артрита, индуцированного коллагеном
Проводили 17-дневное исследование артрита, индуцированного коллагеном II типа, для оценки действия соединений по настоящему изобретению на вызванную артритом клиническую припухлость голеностопного сустава. Крысиная модель артрита, индуцированного коллагеном, является экспериментальной моделью полиартрита, которая широко используется для предклинического тестирования противоревматических средств (см. Trentham et al., J. Exp. Med. 146(3):857-868 (1977), Bendele et al., Toxicologic Pathol. 27:134-142 (1999), Bendele et al., Arthritis Rheum. 42:498-506 (1999)). Отличительными чертами этой модели является надежное наступление и развитие сильного, легкоизмеримого многосуставного воспаления, выраженное разрушение хрящей вкупе с образованием паннуса, резорбция кости от легкой до средней степени тяжести, и пролиферация околохрящевой кости. Самок крыс линии Lewis (примерно 0,2 кг) усыпляли изофлураном и инъецировали им неполный адъювант Фрейнда, содержащий 2 мг/мл бычьего коллагена II типа, в основание хвоста и в два места на спине в день 0 и день 6 данного 17-дневного исследования. Соединения вводили ежедневно подкожной инъекцией со дня 9 до дня 17 в дозировке, например, 100 мг/кг и в объеме 1 мл/кг, в подходящем носителе. Измерение кавернометром диаметра голеностопного сустава проводили ежедневно, и уменьшение припухлости сустава принимали за меру эффективности.
Оценка соединений в животной модели человеческого язвенного колита
Мышиная модель, описанная Panwala и соавторами (Panwala, et al., J Immunol., 161(10):5733-44 (1998)), включает генетическое удаление мышиного гена множественной резистентности (MDR). Мыши с «выбитым» геном MDR (MDR-/-) подвержены развитию сильного спонтанного воспаления кишечника при содержании в специфических стерильных условиях. Воспаление кишечника, наблюдающееся у MDR-/- мышей, имеет патологическую картину, сходную с человеческой воспалительной болезнью кишечника (IBD), и определяется инфильтрацией Т-клеток Th1 типа в собственный слой толстого кишечника.
Другая мышиная модель IBD была описана Powrie с соавторами в работе Int Immunol., 5(11):1461-71 (1993), в которой субпопуляцию CD4+ T клеток (называемых CD45RB(high)) от иммунокомпетентных мышей очищали и адаптивно переносили иммунодефицитным мышам (таким как мыши C.B-17 scid). У животных с восстановленной популяцией CD45RBhighCD4+ T клеток развивалась летальная изнуряющая болезнь с сильными инфильтратами моноядерных клеток в кишечнике, патологически сходно с человеческой IBD.
Оценка соединений в животной модели человеческой болезни Крона
ФНО ARE(-/-) модель. Роль ФНО в болезни Крона у человека недавно была продемонстрирована успехом лечения с применением ФНО-альфа антител в работе Targan et al., N Engl J Med., 337(15):1029-35 (1997). У мышей с аномальной выработкой ФНО-альфа вследствие генетических изменений в гене ФНО (ARE-/-), развивается воспалительная болезнь кишечника по типу болезни Крона (см. Kontoyiannis et al., Immunity, 10(3):387-98 (1999)).
SAMP/yit модель. Эта модель описана в работе Kosiewicz и др. в J Clin Invest., 107(6):695-702 (2001). У линии мышей SAMP/Yit спонтанно развивается хроническое воспаление, локализованное в терминальном отделе подвздошной кишки. Результирующий илеит характеризуется массовой инфильтрацией активированных Т лимфоцитов в собственный слой и сильно напоминает человеческую болезнь Крона.
Оценка соединений в мышиной модели тиогликолят-индуцированного воспаления брюшины
Проводили 2-дневное исследование тиогликолят-индуцированного воспаления для оценки действия тестируемого соединения, соединения 5 (фиг. 1). Отличительными чертами этой модели является надежное наступление и развитие сильного, легкоизмеримого воспалительного клеточного инфильтрата. Для вызывания воспалительного перитонита у крыс линии Lewis, инъецировали Brewer-тиогликолят (1,0 мл, 4%-ный раствор в дистиллированной воде) интраперитонеально (i.p.). Перед этой инъекцией группа, которой вводили соединение, получала тестируемое соединение 4-хлор-3-трифторметил-N-[5-хлор-2-(2-метансульфонил-бензоил)-пиридин-3-ил]-бензолсульфонамид или носитель, а контрольная группа получала такой же объем PBS в форме интраперитонеальной инъекции. Через два дня проводили промывание брюшины ледяным PBS, содержащим 1 мM EDTA. Полученные клетки подсчитывали в устройстве для подсчета клеток (Coulter Counter; Coulter Pharmaceutical, Palo Alto, CA) и идентифицировали моноциты/макрофаги методом поточной цитометрии, используя их свойства светорассеяния.
Тестируемое соединение значительно и специфично подавляло число воспалительных макрофагов, появившихся после инъекции тиогликолята.
Оценка соединений в мышиной модели бактериальной инфекции
Проводили 1-дневное исследование инфекции стрептококковой пневмонии для оценки действия тестируемого соединения. В данной модели замеряли бактериальную инфекцию и ее распространение в животном после легочного инфицирования живой культурой бактерий, замер проводили по воспалительному клеточному инфильтрату и определению бактериальной нагрузки. Мышам линии C57/B6 вводили интраназально LD50 400 КОЕ в день 0. Группы подвергали обработке соединением или носителем за 1 день до бактериального инокулирования и дважды в день на протяжении исследования. Бактериальную нагрузку замеряли через 24 часа путем посева серийных разбавлений гомогенизированной ткани легких на агаровые пластинки и подсчета числа колоний.
Оценка соединений в мышиной модели карциномы легких
В данной области техники известно много животных опухолевых моделей, которые могут применяться, например, для оценки соединений. Например, в исследовании ксенотрансплантата карциномы легких, фрагменты опухоли A549 (30-40 мг) имплантировали в подкожное пространство голых мышей. Опухолям позволяли расти до размера примерно 150 мг (между 100 и 200 мг), в этот момент мышей вводили в настоящее исследование и начинали введение соединений. Мышам вводили исследуемое соединение или контроль (носитель). Мелфалан может быть включен в качестве положительного контроля (9мг/кг массы тела, интраперитонеальное введение, Q4Dx3). Опухоли замеряли два раза в неделю каверномером в двух измерениях и переводили в значение массы опухоли, используя формулу для сплющенного эллипсоида (a x b2/2), где a - это более длинный размер, и b - это более короткий размер, и принимая единицу плотности (1 мм3 = 1 мг). Вес тела также можно замерять дважды в неделю для оценки каких-либо побочных эффектов от введения соединения. Противоопухолевую активность оценивали по задержке роста опухоли в группе, которой вводили соединение, по сравнению с контрольной группой, получавшей только носитель.
Оценка соединений в мышиной модели глиобластомы
В данной области техники известно много животных опухолевых моделей, которые могут применяться, например, для оценки соединений. Например, в мышиной модели глиобластомы, 1x106 клеток U251MG имплантировали стереотактической инъекцией в мозг голых мышей. Через 20 дней опухоли облучали дозой 1-1 Грей радиации. После облучения мышам вводили (например, подкожно, интраперитонеально, перорально, парентерально или другим путем) соединение или контроль (носитель), и оставляли опухоли прогрессировать. Рост опухоли и/или смертность мониторили на протяжении всего исследования. Опухоли замеряли два раза в неделю каверномером в двух измерениях и переводили в значение массы опухоли, используя формулу для сплющенного эллипсоида (a x b2/2), где a - это более длинный размер, и b - это более короткий размер, и принимая единицу плотности (1 мм3 = 1 мг). Вес тела также можно замерять дважды в неделю для оценки каких-либо побочных эффектов от введения соединения. Противоопухолевую активность оценивали по задержке роста опухоли в группе, которой вводили соединение, по сравнению с контрольной группой, получавшей только носитель.
Оценка соединений в мышиной модели рака поджелудочной железы
В данной области техники известно много животных опухолевых моделей, которые могут применяться, например, для оценки соединений. Например, в мышиной модели рака поджелудочной железы, panco2 клетки (NCI) имплантировали ортотопически в поджелудочные железы мышей линии C57bl/6. Опухоли оставляли развиваться на три недели, затем животным вводили либо соединение 53 (фиг. 1), либо контроль (носитель) подкожной инъекцией один раз в сутки в течение 3 недель. Эффективность соединения оценивали по росту опухоли. У животных, которым вводили соединение 53 (фиг. 1), наблюдали значительно меньшие по размеру опухоли, чем у животных, которым вводили только носитель.
Оценка соединений в мышиной модели поликистозной болезни почек
В мышиной модели поликистозной болезни почек, у мышей Pkd1flox/flox:Pkhd1-Cre (Jackson Labs, ME) спонтанно развивается поликистозная болезнь почек вследствие генетически обусловленной почечно-селективной недостаточности мышиного ортолога человеческого гена PKD1, мутации в котором ответственны за наиболее распространенную и наиболее тяжелую форму ADPKD. Болезни позволяли развиваться, и в день 10 вводили соединение по настоящему изобретению или контроль (носитель) подкожной инъекцией один раз в сутки вплоть до дня 26. В этот момент животных умерщвляли и регистрировали массу почек, содержание мочевины в крови и число кист в почках. Эффективность оценивали как улучшение в этих показателях при добавлении соединения 53 (фиг. 1).
Следует понимать, что описанные в настоящем тексте примеры и варианты осуществления приведены исключительно в иллюстративных целях, и что квалифицированным специалистом в данной области могут быть предложены различные модификации и изменения в их свете, которые охватываются сутью настоящего изобретения и входят в объем его притязаний.
Все процитированные в настоящем тексте публикации, патенты и заявки на патенты включены в настоящий текст в полном объеме посредством ссылки.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОЛЕКАРСТВА СОПРЯЖЕННО-БИЦИКЛИЧЕСКИХ АНТАГОНИСТОВ C5aR | 2019 |
|
RU2794327C2 |
| ДИАРИЛ-ЗАМЕЩЕННЫЕ 6,5-СОПРЯЖЕННЫЕ ЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ КАК ИНГИБИТОРЫ C5aR | 2018 |
|
RU2796983C2 |
| 6-5 КОНДЕНСИРОВАННЫЕ КОЛЬЦА КАК ИНГИБИТОРЫ С5а | 2018 |
|
RU2780338C2 |
| ПРОИЗВОДНЫЕ ПИПЕРАЗИНИЛПИПЕРИДИНА В КАЧЕСТВЕ АНТАГОНИСТОВ ХЕМОКИНОВОГО РЕЦЕПТОРА | 2005 |
|
RU2369604C2 |
| СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ PD-L1 ЗАБОЛЕВАНИЙ | 2020 |
|
RU2838028C2 |
| АНТАГОНИСТЫ CXCR7 | 2013 |
|
RU2649004C2 |
| ДИАЗОЛЬНЫЕ ЛАКТАМЫ | 2013 |
|
RU2666730C2 |
| ТИОФЕНИЛЬНЫЕ И ПИРРОЛИЛЬНЫЕ АЗЕПИНЫ В КАЧЕСТВЕ ЛИГАНДОВ СЕРОТОНИНОВОГО 5-HT РЕЦЕПТОРА И ИХ ПРИМЕНЕНИЕ | 2007 |
|
RU2434872C2 |
| АМИДНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ АГОНИСТОВ GRP119 | 2014 |
|
RU2642429C2 |
| АНТАГОНИСТЫ ХЕМОКИНОВЫХ РЕЦЕПТОРОВ | 2013 |
|
RU2646762C2 |
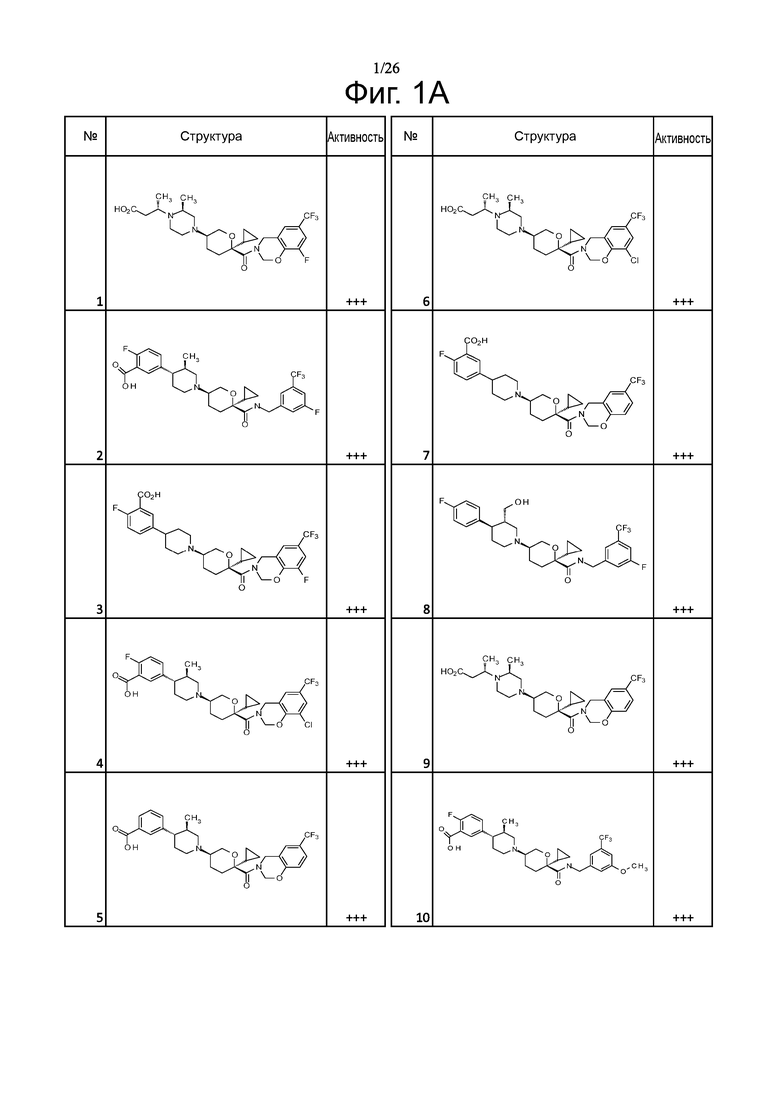
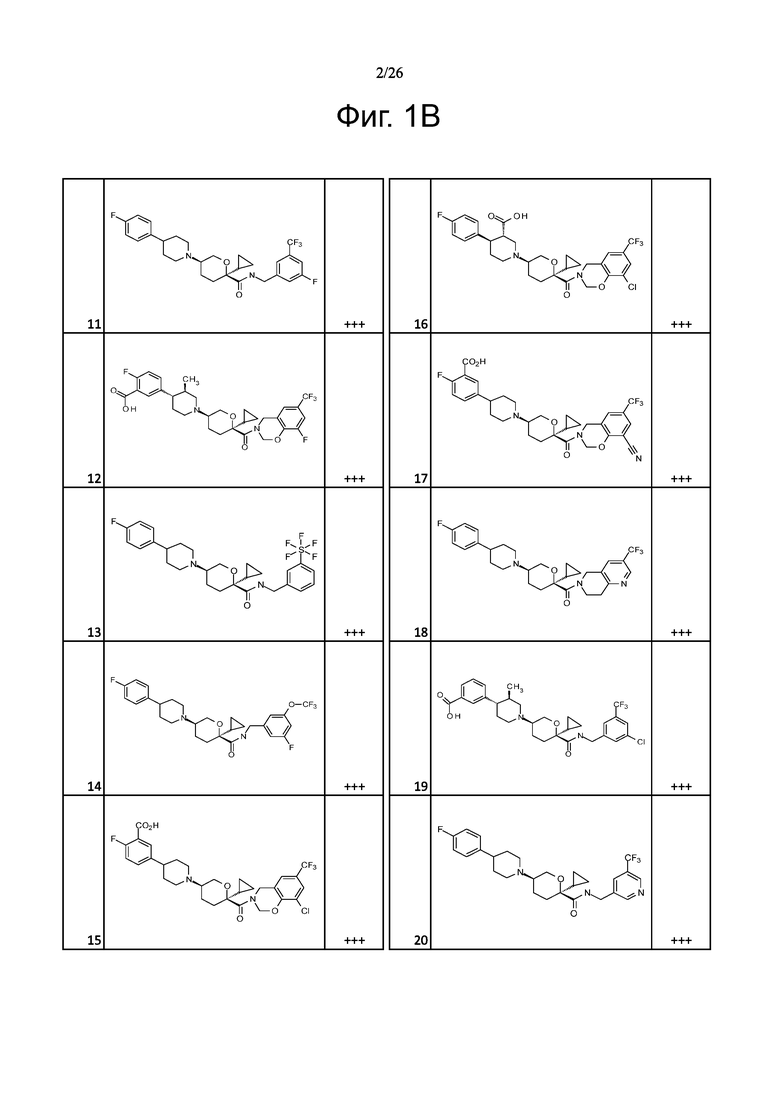
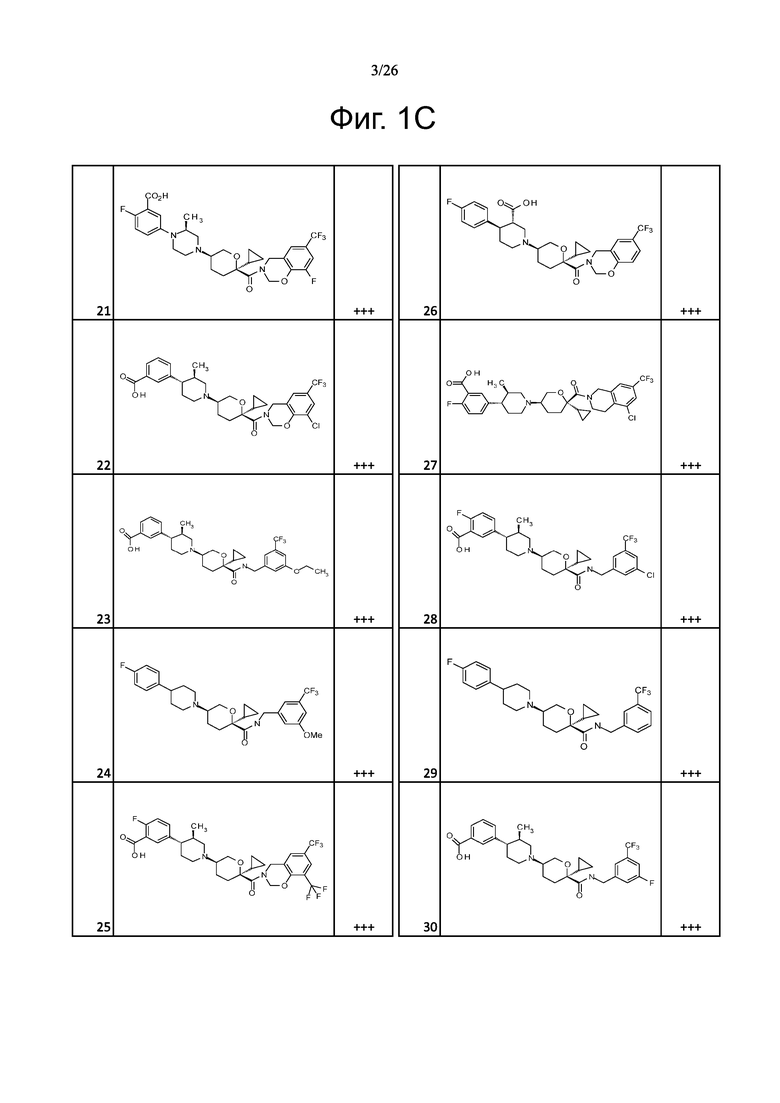
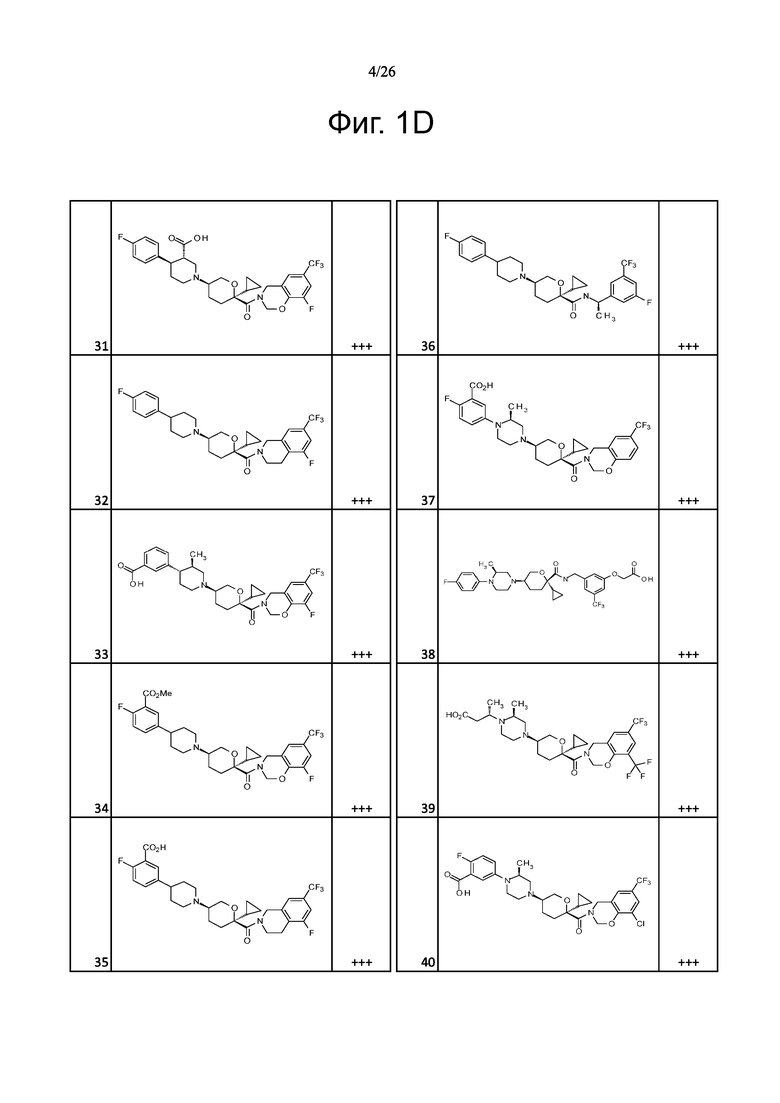
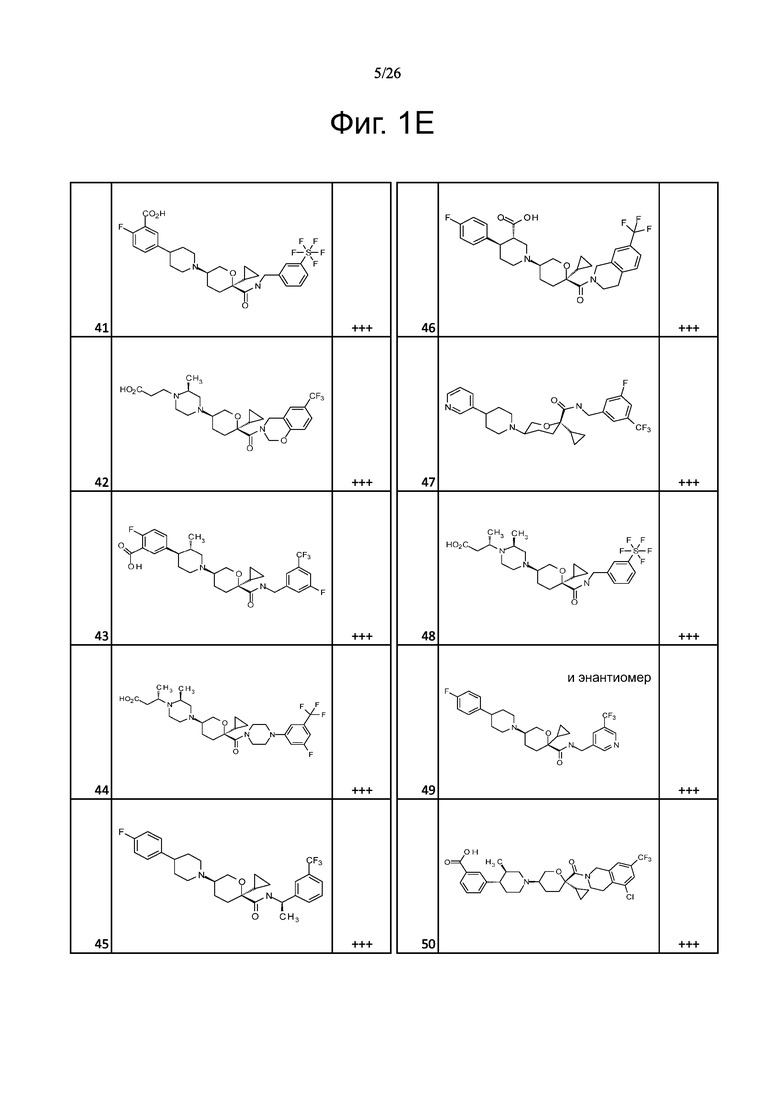
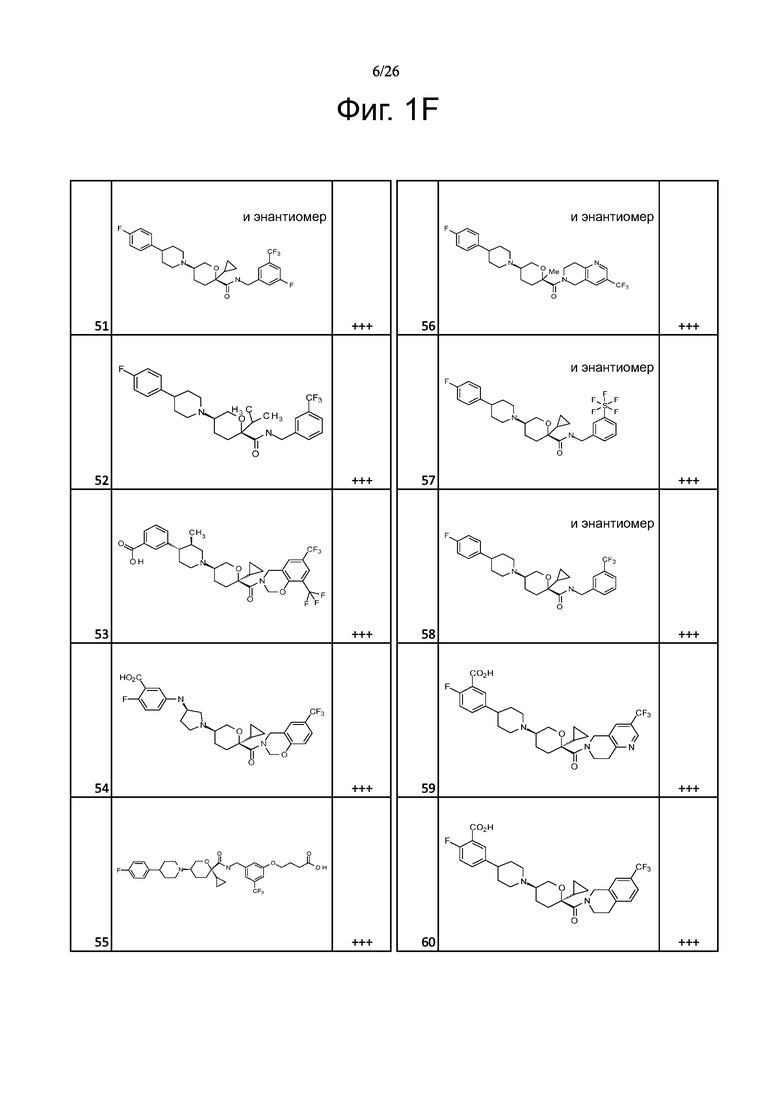
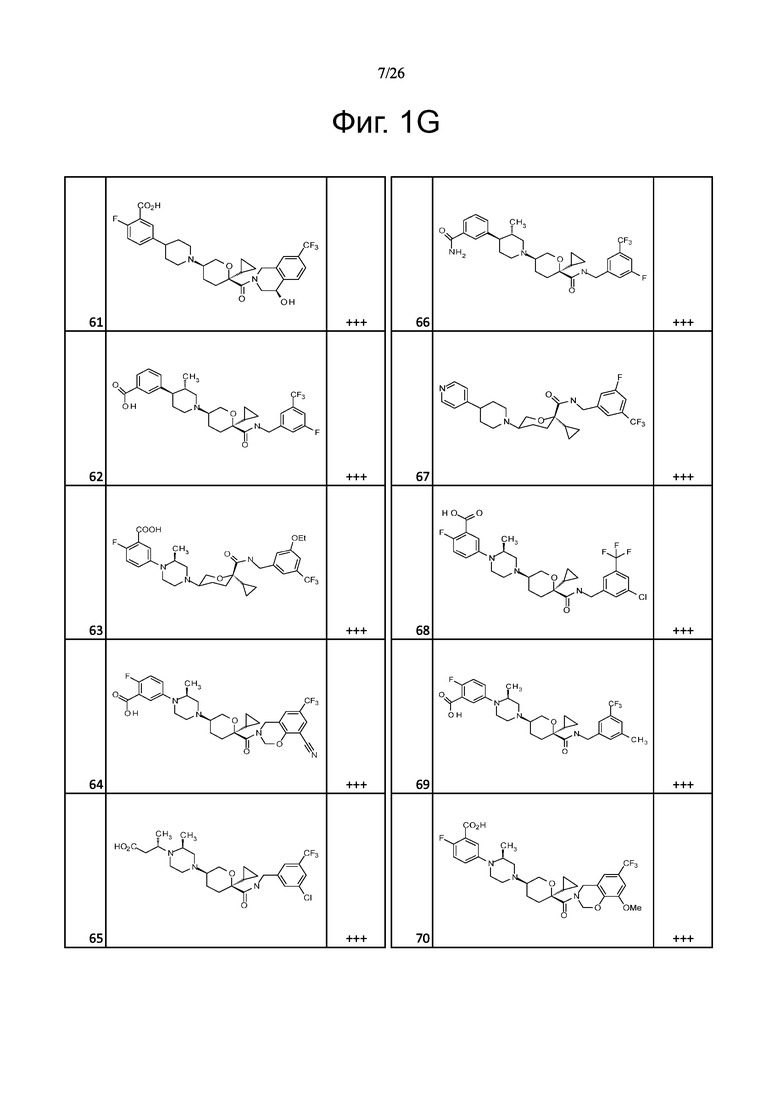
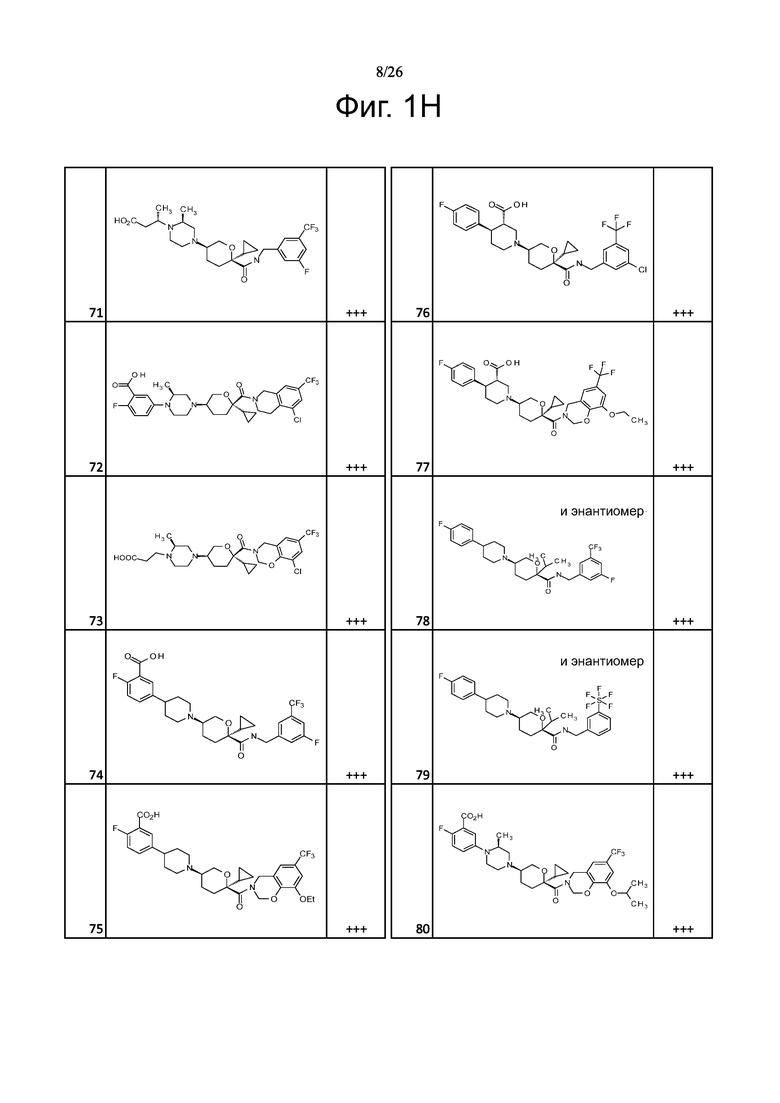
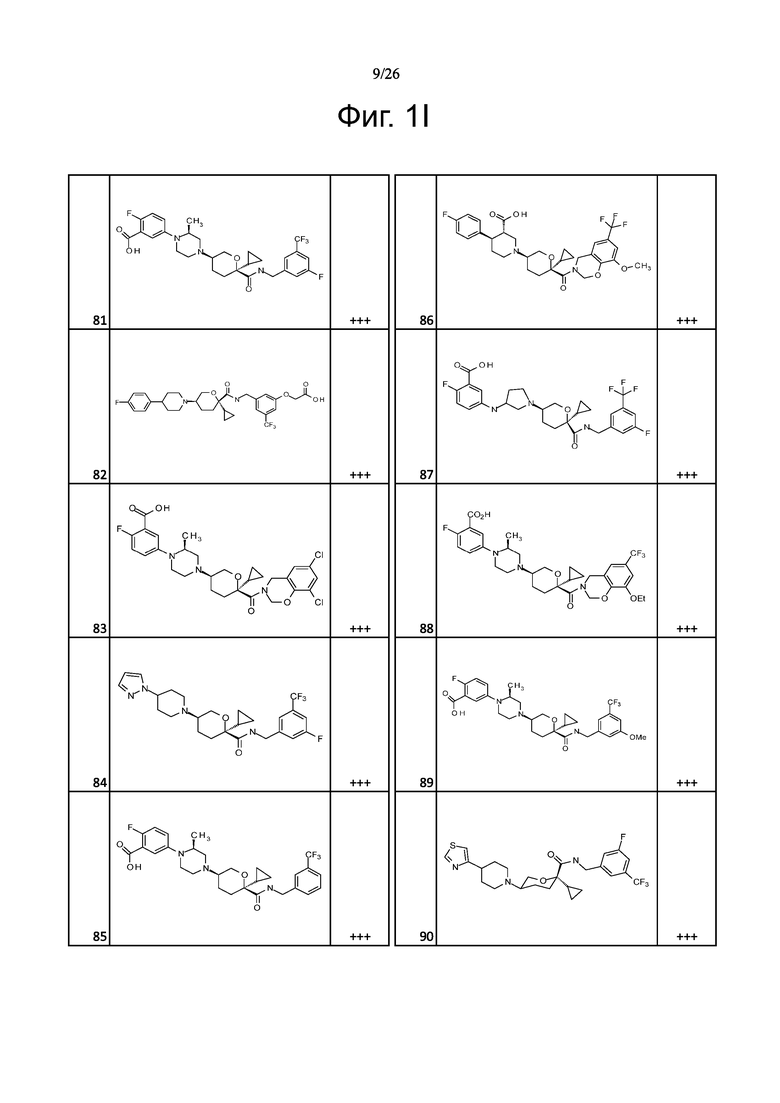
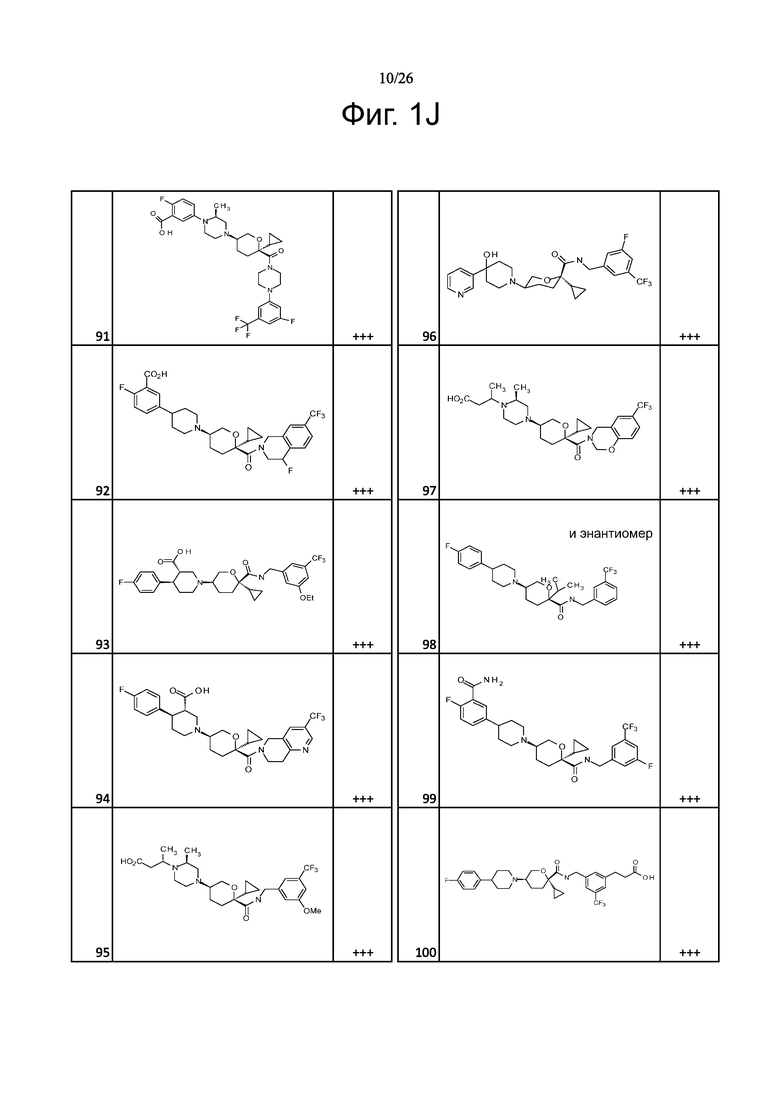
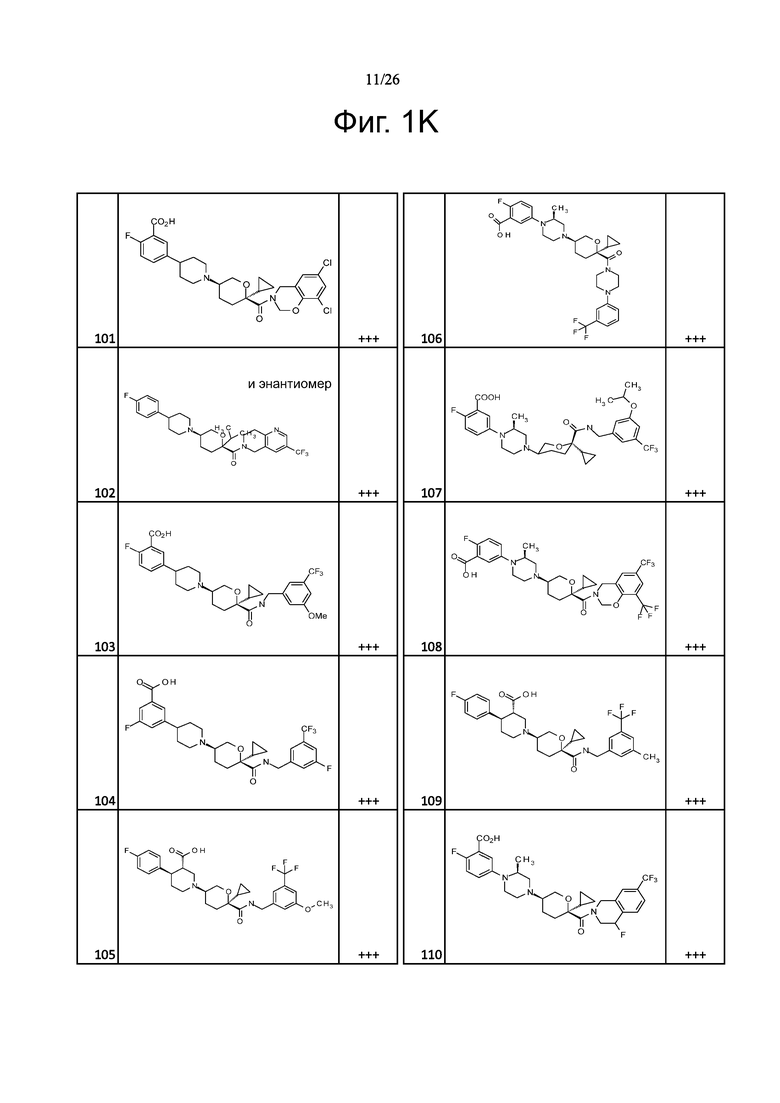
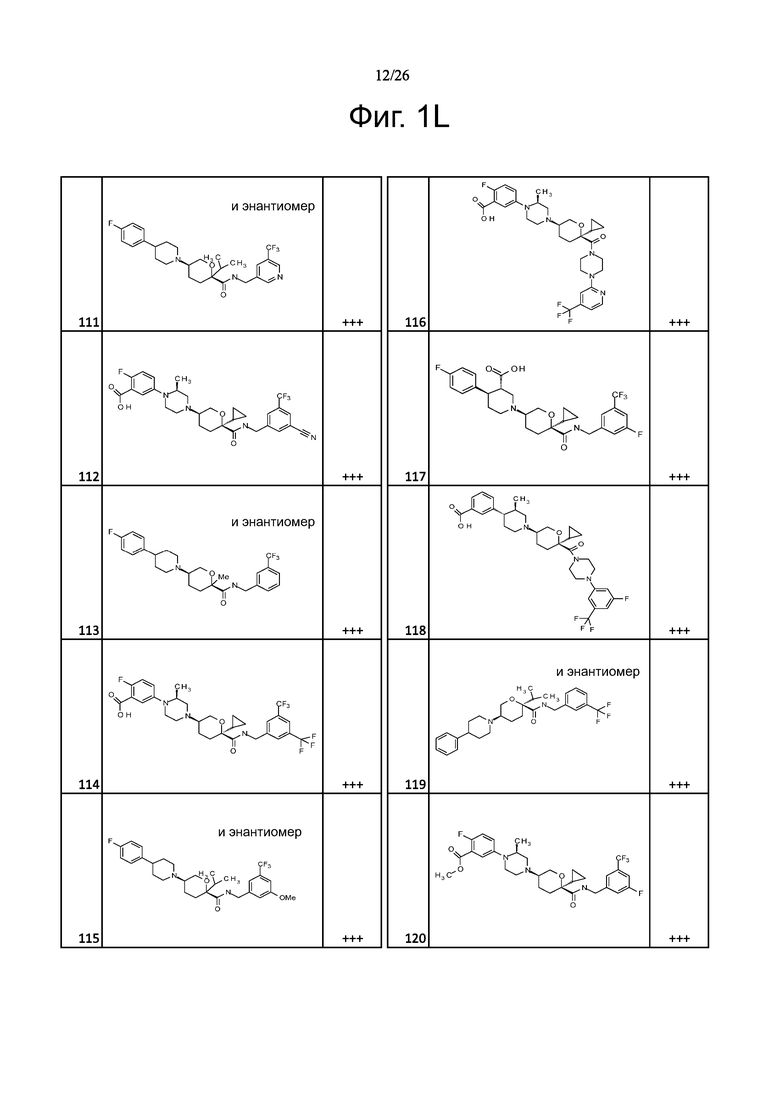
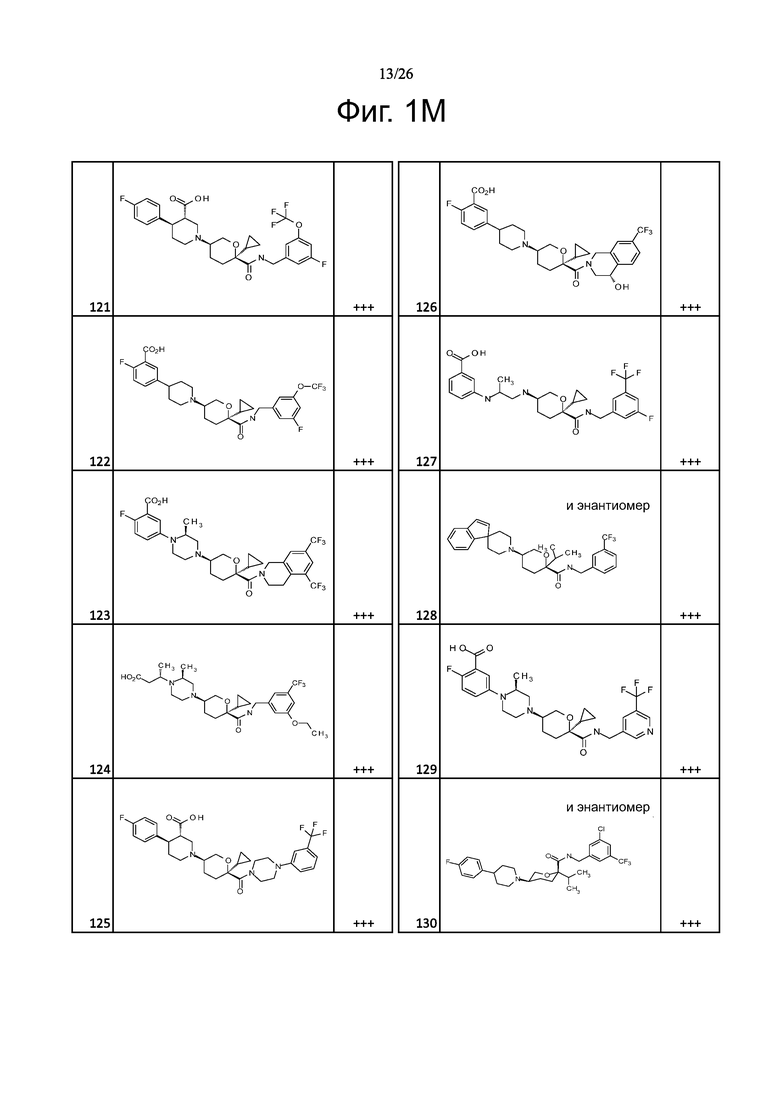
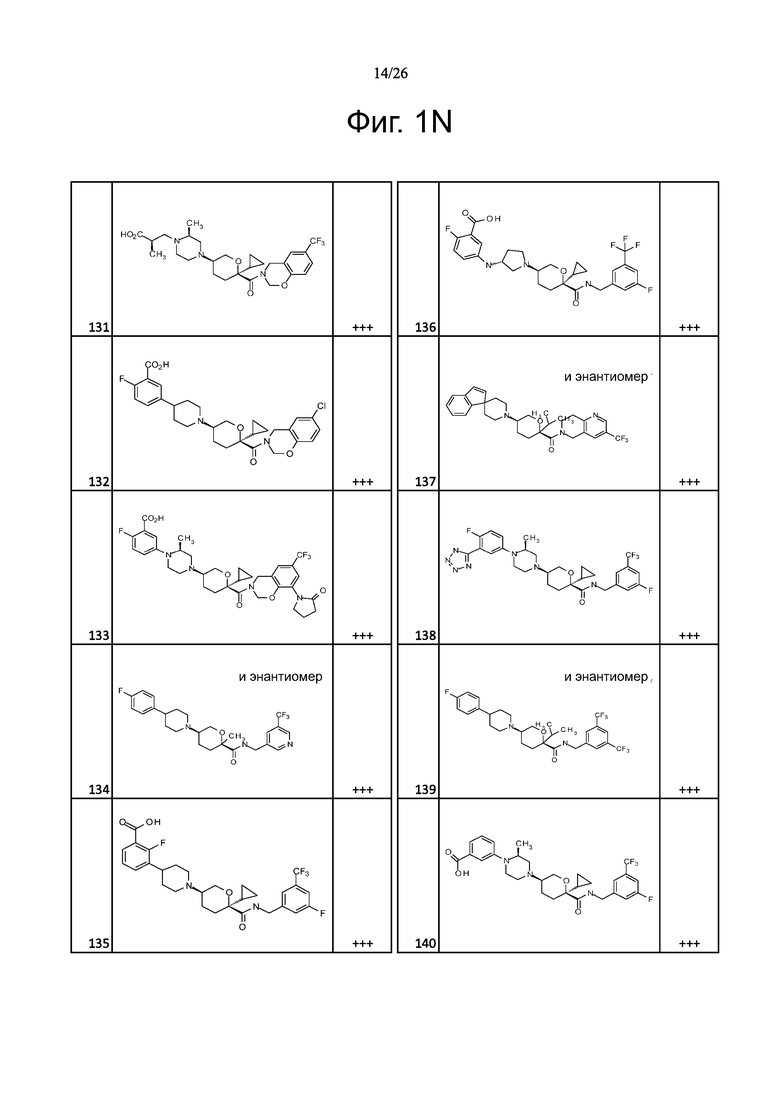
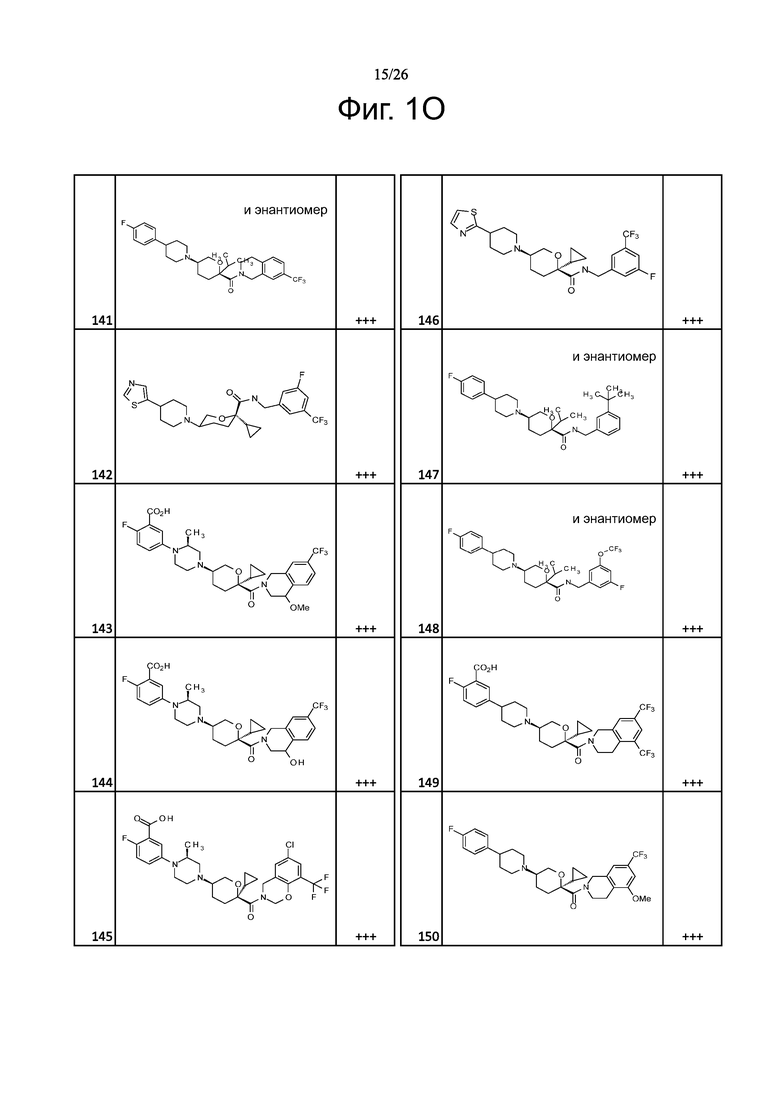
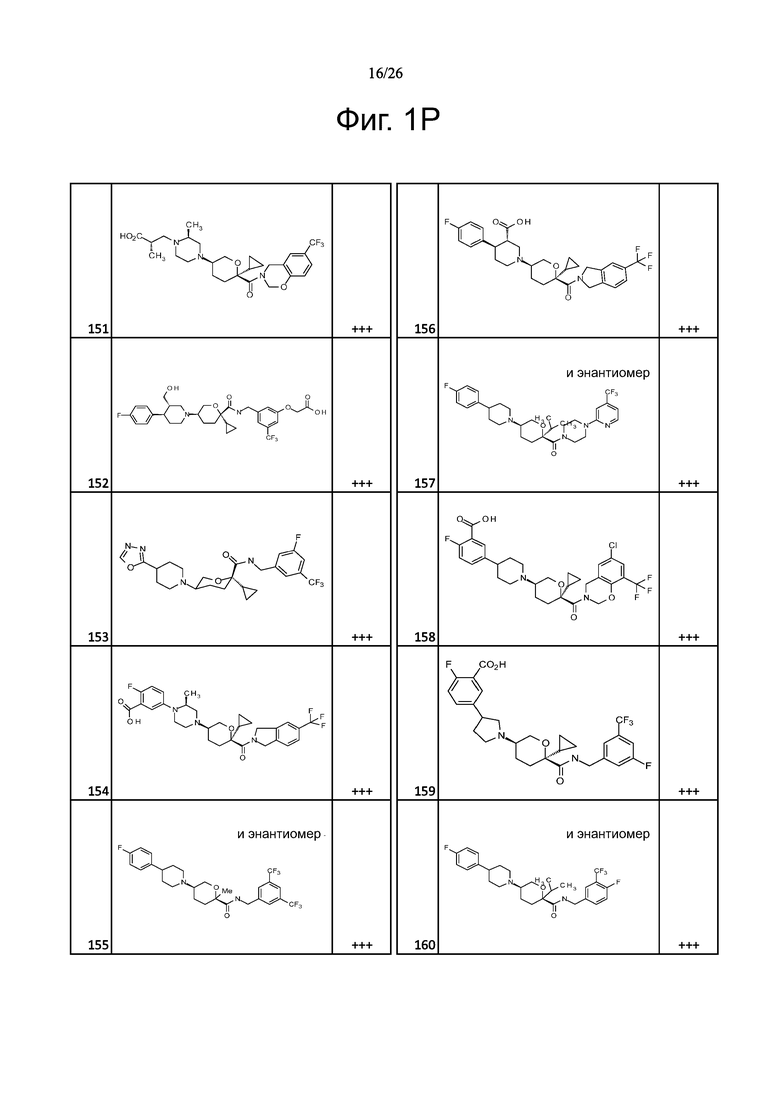
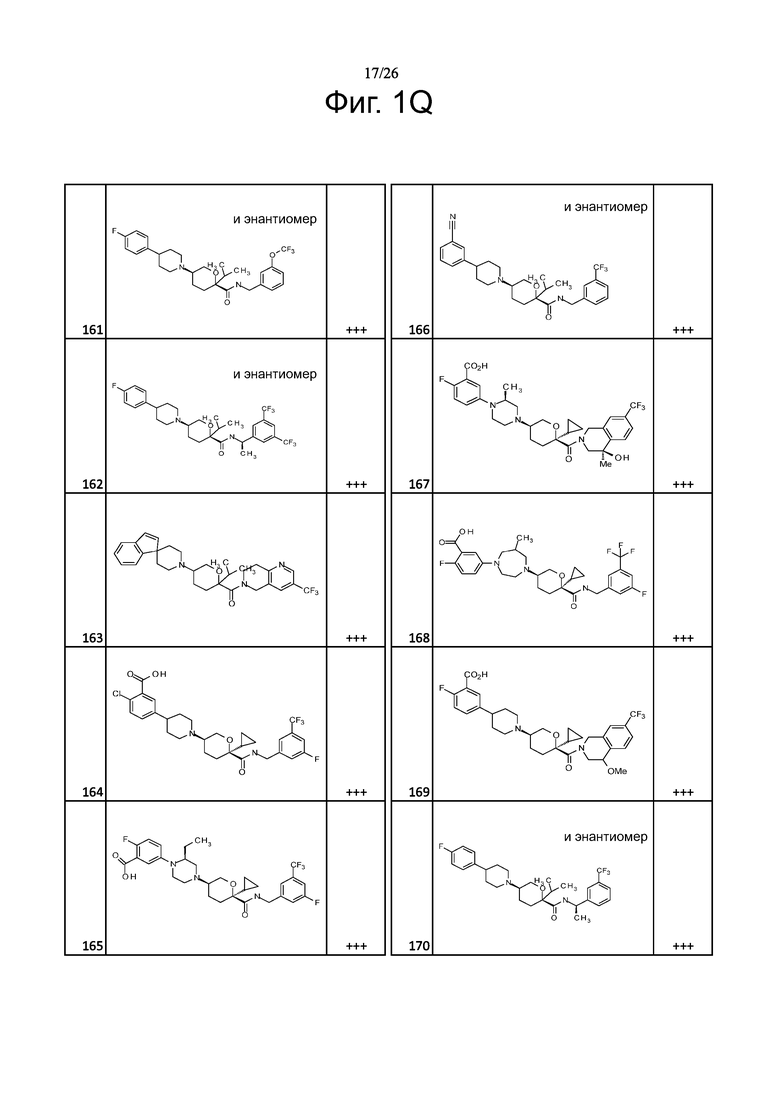

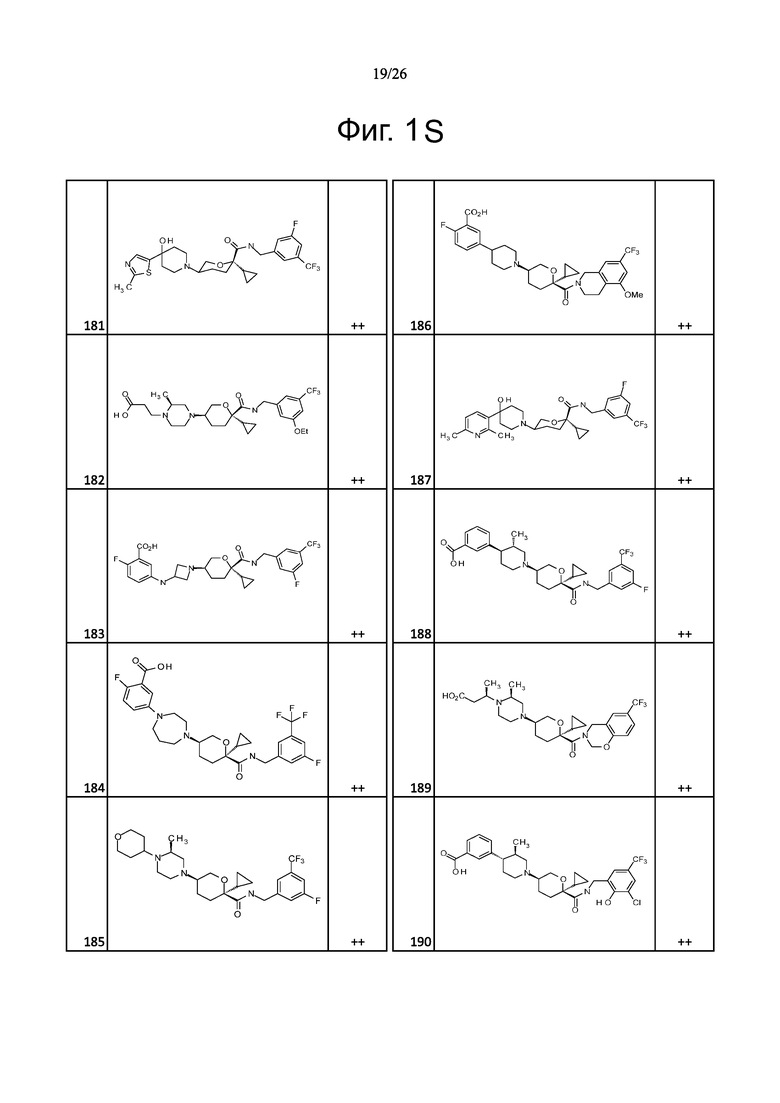
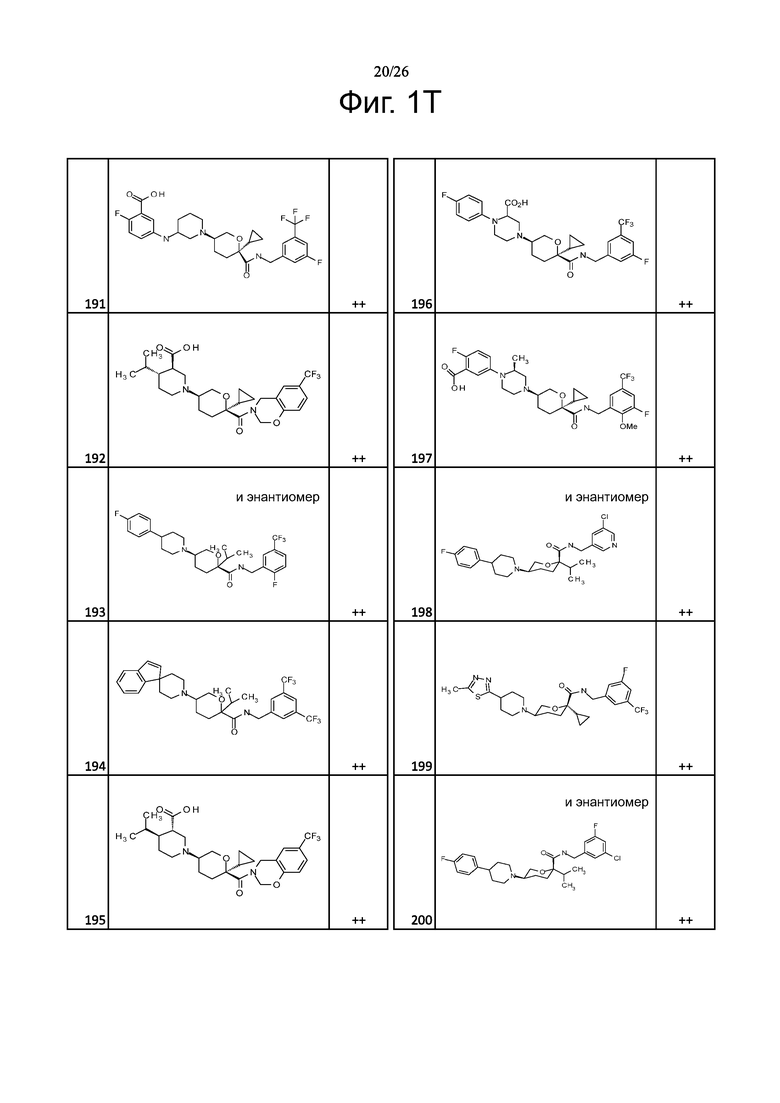
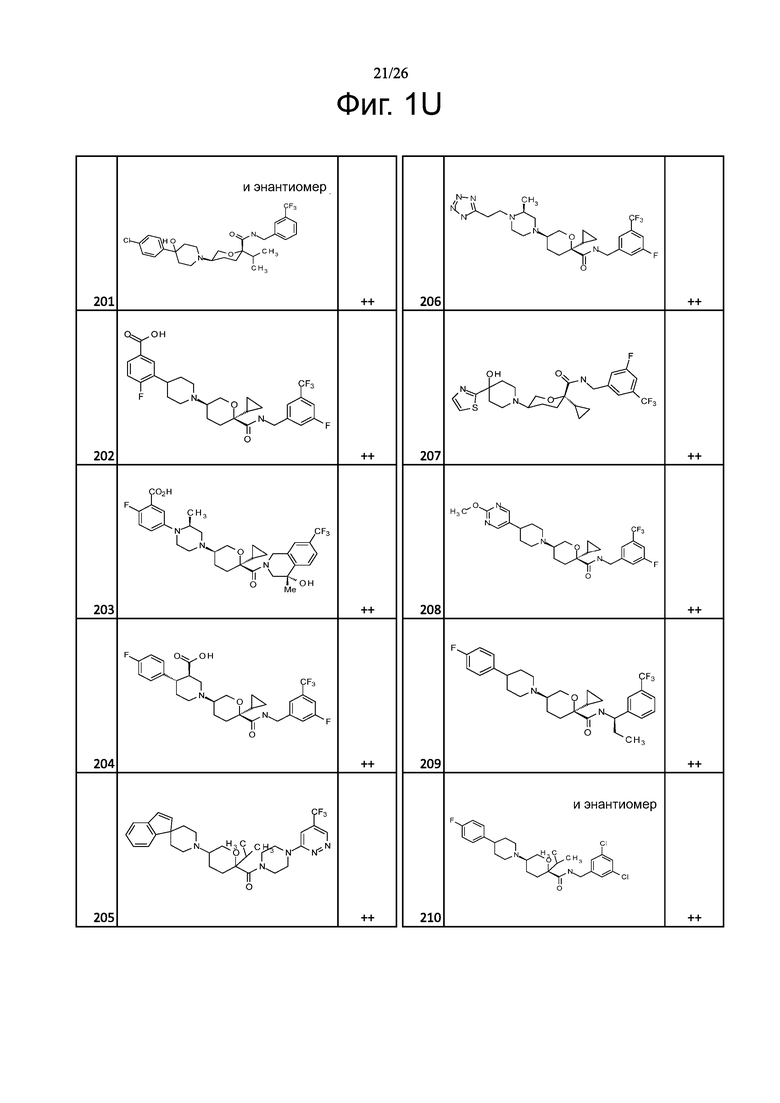
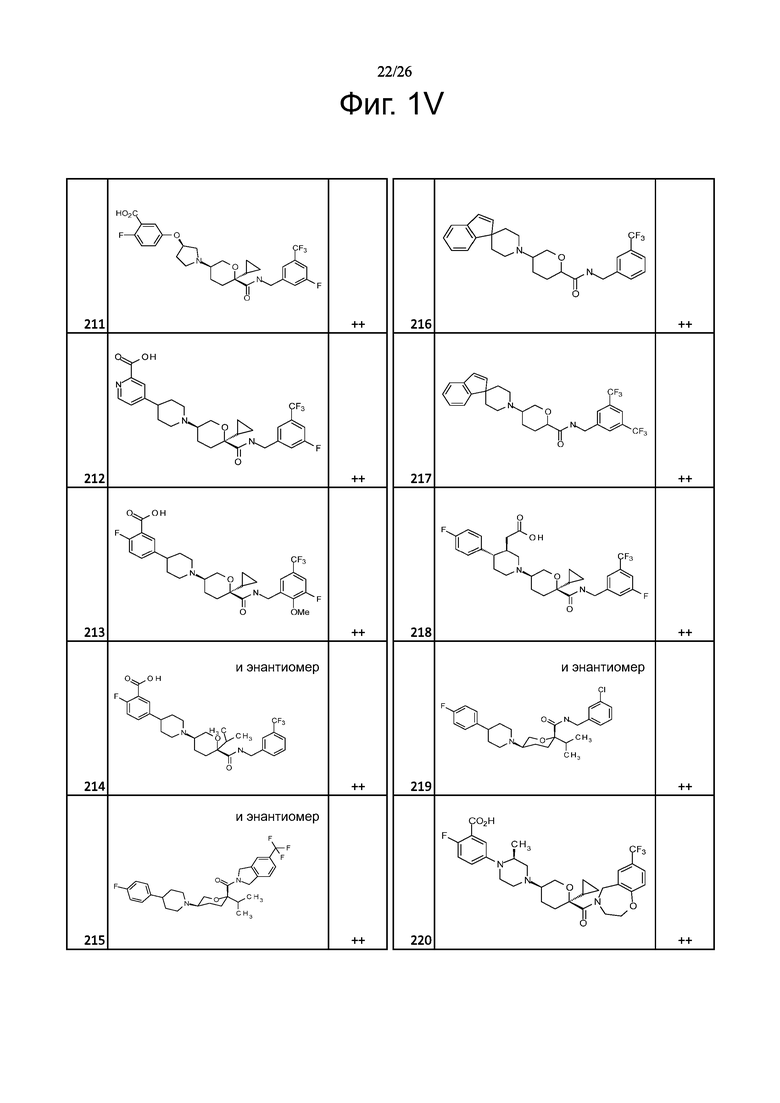
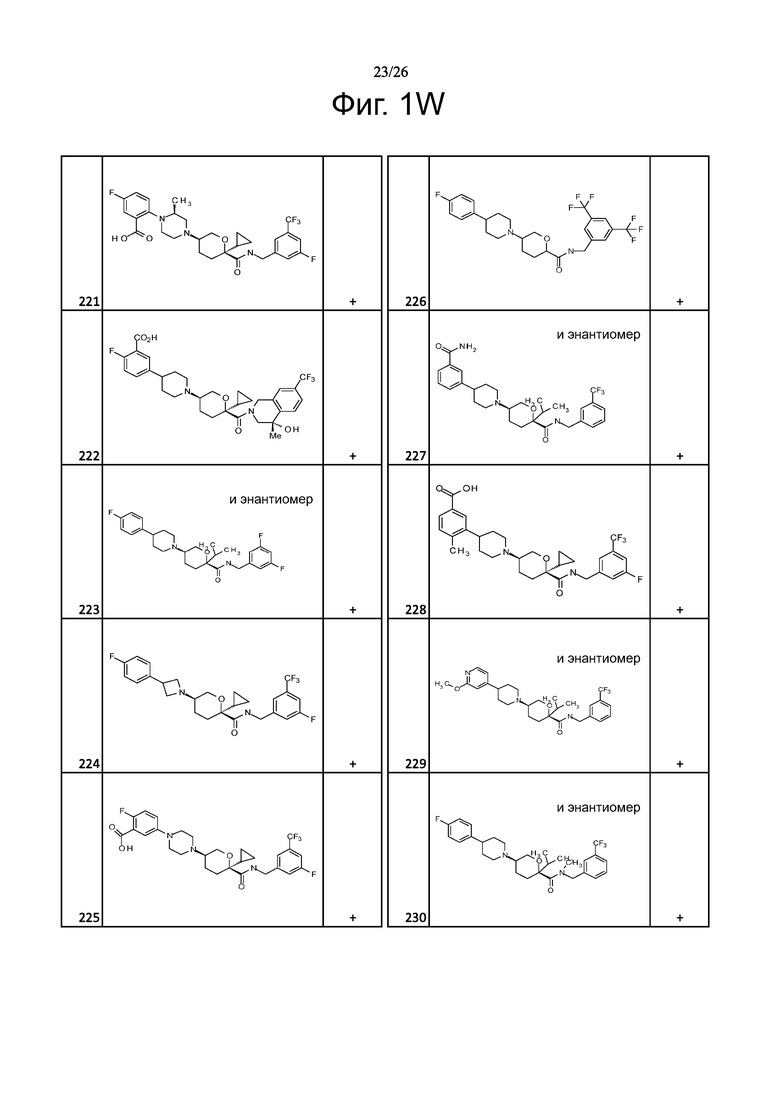

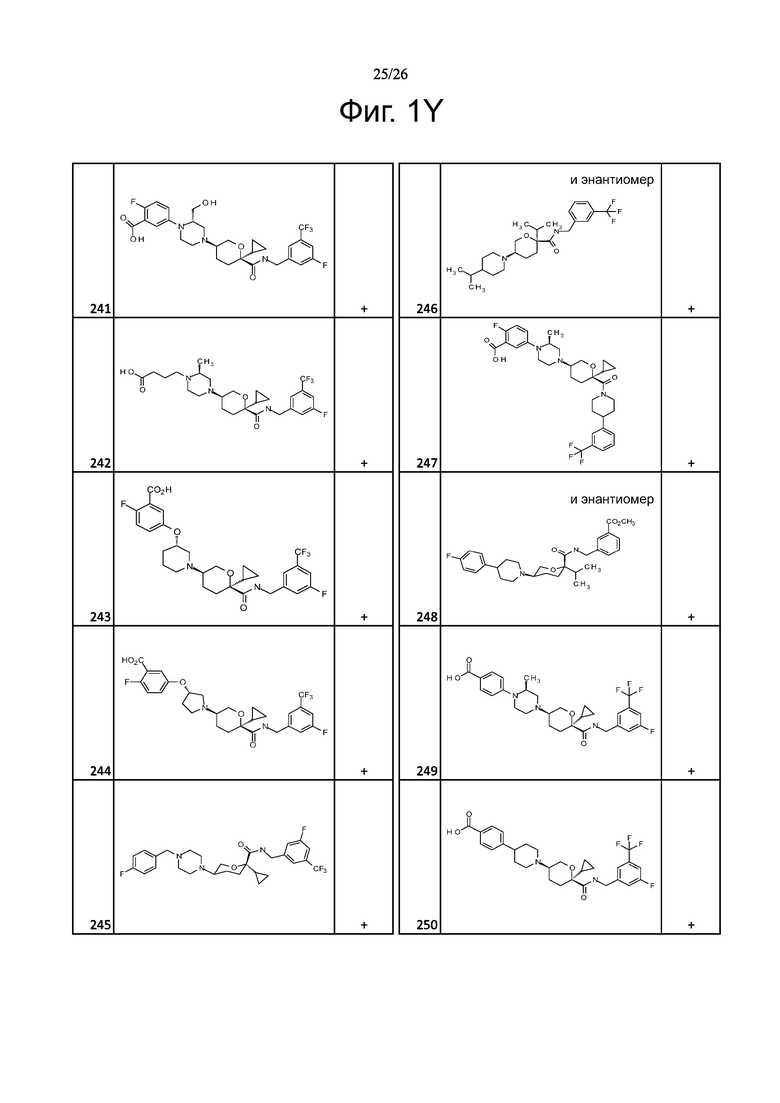
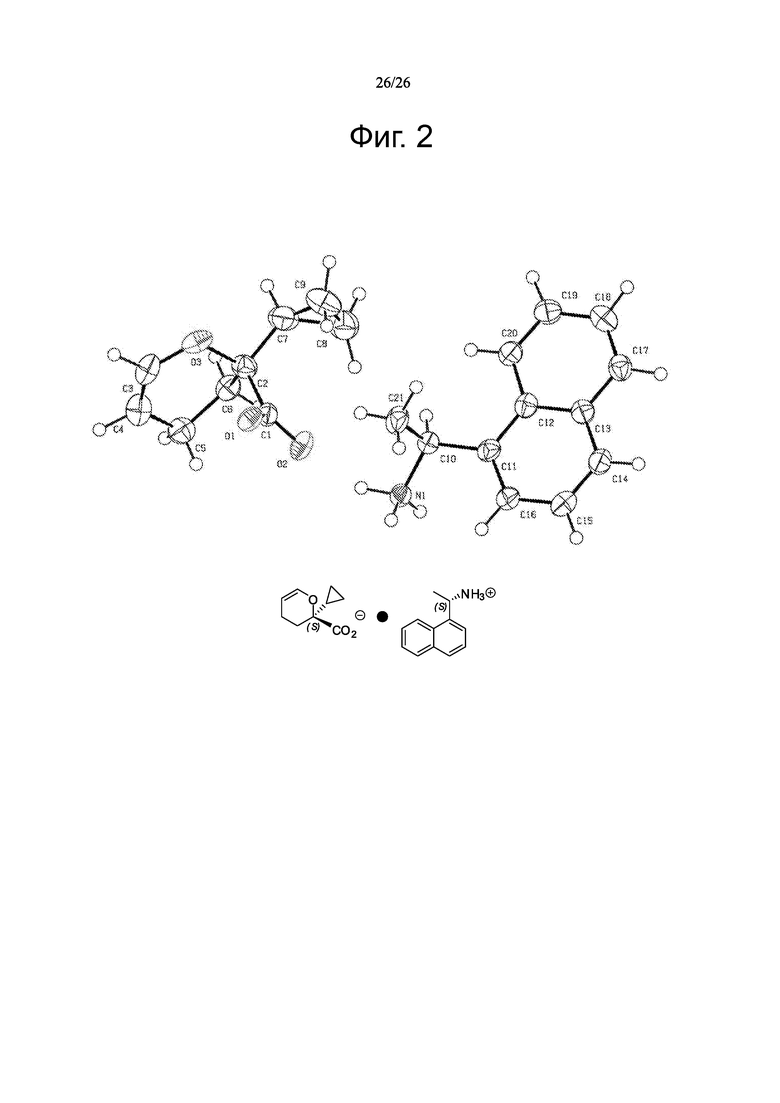
В настоящем изобретении описаны соединения общей формулы (I) или их фармацевтически приемлемые соли, являющиеся модуляторами CCR2 рецептора. Они могут применяться в фармацевтических композициях для лечения CCR2-опосредованного заболевания или нарушения, в способах лечения млекопитающего, страдающего от или склонного к заболеванию или нарушению, выбранного из группы, состоящей из почечного фиброза, поликистозной болезни почек, ревматоидного артрита, атопического дерматита, псориаза, нефропатии, рака поджелудочной железы и неалкогольного стеатогепатита (НС), а также в способах ингибирования CCR2 рецептор-опосредованного клеточного хемотаксиса, включающих контактирование белых кровяных телец млекопитающего с модулирующим CCR2 рецептором. 4 н. и 48 з.п. ф-лы, 2 ил., 29 пр.
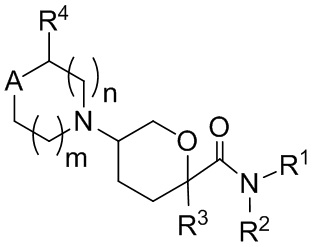 (I)
(I)
1. Соединение, имеющее формулу
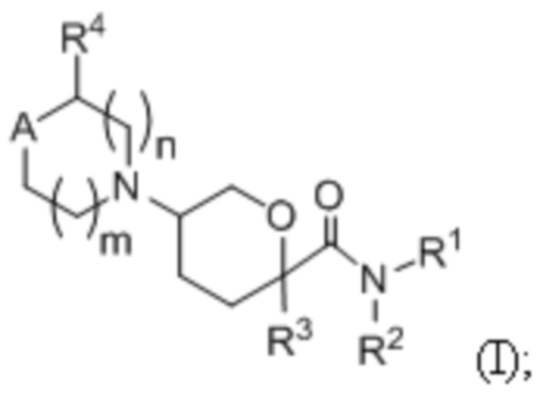
или его фармацевтически приемлемая соль; где
A представляет собой C(R5)(R6) или N(R5);
подстрочные индексы m и n, каждый, независимо представляют собой целые числа от 0 до 2, и m+n в сумме ≤ 3;
R1 выбран из группы, состоящей из фенила, пиридила, фенил-C1-4 алкила, пиридил-C1-4 алкила, имидазолил-C1-4 алкила; и где указанные группы или фрагменты фенила, пиридила и имидазолила необязательно замещены 1-3 Rx заместителями;
R2 выбран из группы, состоящей из H и C1-8 алкила;
или, необязательно, R1 и R2 объединены с атомом азота, к которому они присоединены, с образованием 6-11-членного моноциклического или конденсированного бициклического гетероциклического или гетероарильного кольца, где гетероциклическое или гетероарильное кольцо содержит в качестве членов цикла 1-4 гетероатома, выбранных из N и O, где -NR1R2 необязательно дополнительно замещен 1-3 заместителями Rx;
R3 выбран из группы, состоящей из H, C1-8 алкила, C3-8 циклоалкила;
R4 выбран из группы, состоящей из H, метила, необязательно замещенного 1-2 заместителями Ry, и -CO2H;
R5 выбран из группы, состоящей из C1-8 алкила, C1-8 алкокси-группы, фенила, фенил-C1-4 алкила, 5-6-членного гетероарила, содержащего в качестве членов цикла 1-3 гетероатома, выбранных из N, O и S, каждый из которых необязательно замещен 1-2 заместителями Rz;
R6 выбран из группы, состоящей из H и OH;
или, необязательно, R5 и R6 соединены с образованием спироциклического 5- или 6-членного циклоалкильного кольца, которое необязательно является ненасыщенным и имеет конденсированную арильную группу;
каждый Rx независимо выбран из группы, состоящей из галогена, -CN, -Rc, -CO2Ra, -NRaRb, -ORa,-O-X1-CO2Ra, -X1-CO2Ra, -SF5 и 6-членного арила или гетероарила, где гетероарильный фрагмент содержит в качестве членов цикла 1-2 атома азота, где каждый X1 представляет собой C1-4 алкилен; каждый Ra и Rb независимо выбран из атома водорода, C1-8 алкила и C1-8 галогеналкила, или, в случае присоединения к одному и тому же атому азота, могут объединяться с атомом азота, формируя 5- или 6-членное кольцо, содержащее в качестве членов цикла от 0 до 1 дополнительных гетероатомов, выбранных из O, и необязательно замещенное оксо-группой; каждый Rc независимо выбран из группы, состоящей из C1-8 алкила, и C1-8 галогеналкила, и где арильные или гетероарильные группы необязательно замещены 1-2 заместителями, выбранными из галогена, C1-4 алкила и C1-4 галогеналкила;
каждый Ry независимо выбран из группы, состоящей из -CO2Rd и -ORd; где каждый Rd и Re независимо выбран из атома водорода и C1-8 алкила;
каждый Rz независимо выбран из группы, состоящей из галогена, -CN, -CO2Rg, -CONRgRh, -ORg и тетразола; где каждый Rg и Rh независимо выбран из атома водорода и C1-8 алкила.
2. Соединение по п. 1, где R3 является представителем, выбранным из группы, состоящей из H, метила, этила, пропила, изопропила, бутила, изобутила, втор-бутила, циклопропила, циклопропилметила, циклобутила и циклобутилметила.
3. Соединение по п. 1, где m и n оба равны 0.
4. Соединение по п. 1, где m и n оба равны 1.
5. Соединение по п. 1, где m равен 1, и n равен 0.
6. Соединение по п. 1, где m равен 1, и n равен 2.
7. Соединение по любому из пп. 1-6, где цикл, содержащий вершину A, представлен формулой, выбранной из группы, состоящей из:
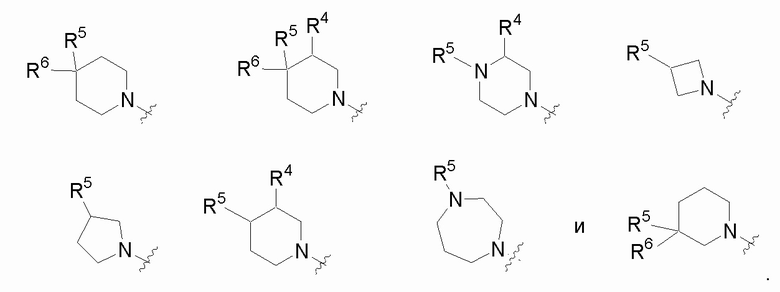
8. Соединение по п. 1, имеющее формулу:
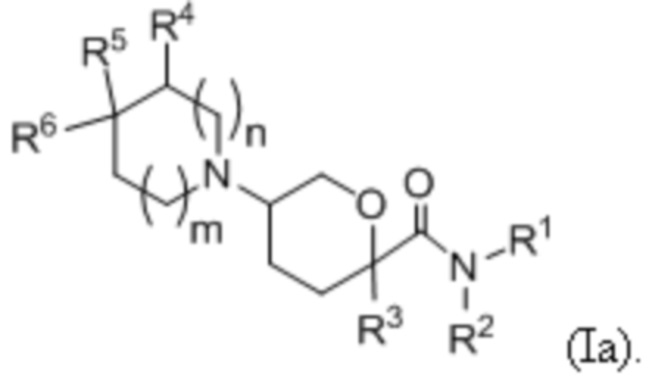
9. Соединение по п. 1 или 8, имеющее формулу:
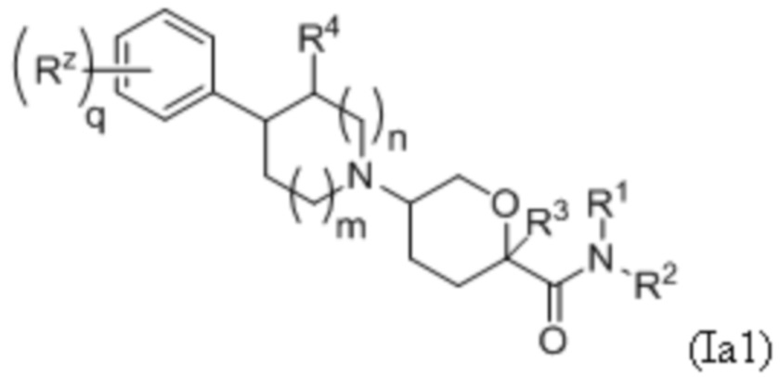
где подстрочный индекс q представляет собой целое число от 1 до 2.
10. Соединение по любому из пп. 1 или 8, имеющее формулу:
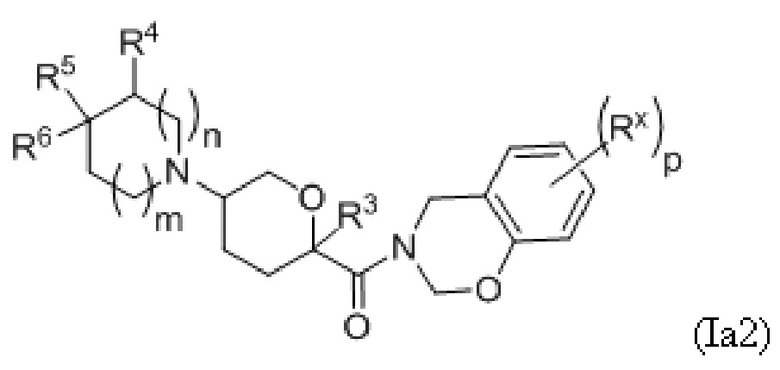
где подстрочный индекс p представляет собой целое число от 1 до 3.
11. Соединение по любому из пп. 1 или 8, имеющее формулу:

где подстрочный индекс p представляет собой целое число от 1 до 3.
12. Соединение по любому из пп. 1, 8 или 10, имеющее формулу:
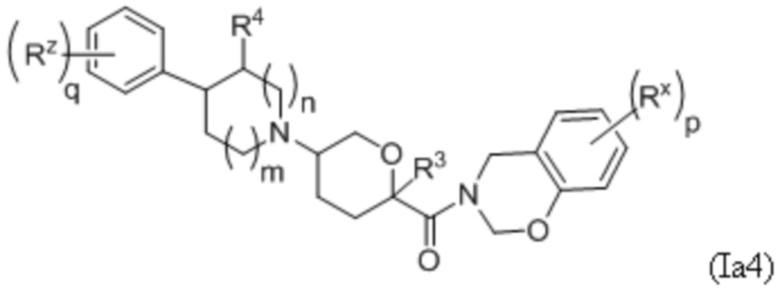
где подстрочный индекс q представляет собой целое число от 1 до 2, и подстрочный индекс p представляет собой целое число от 1 до 3.
13. Соединение по любому из пп. 1, 8, 9 или 11, имеющее формулу:
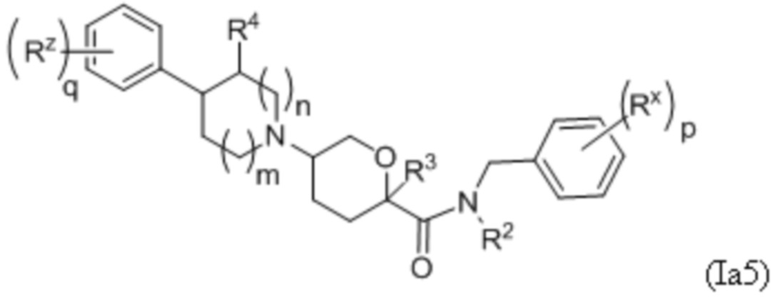
где подстрочный индекс q представляет собой целое число от 1 до 2, и подстрочный индекс p представляет собой целое число от 1 до 3.
14. Соединение по любому из пп. 1 или 8, имеющее формулу:
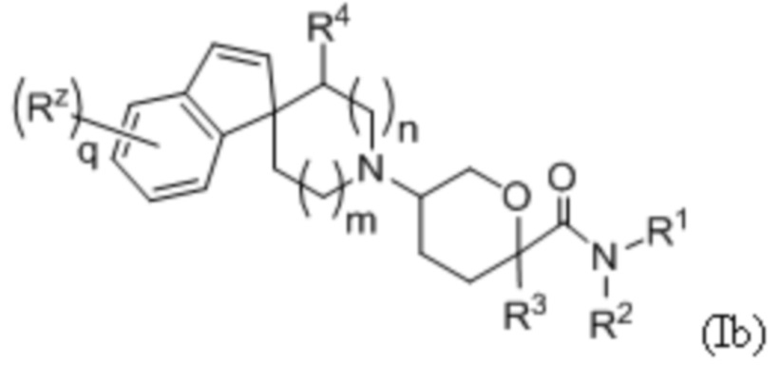
где подстрочный индекс q представляет собой целое число от 1 до 2.
15. Соединение по любому из пп. 1, 8 или 14, имеющее формулу:
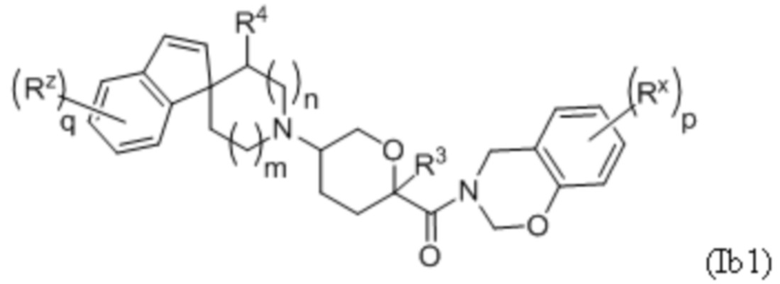
где подстрочный индекс q представляет собой целое число от 1 до 2, и подстрочный индекс p представляет собой целое число от 1 до 3.
16. Соединение по любому из пп. 1, 8 или 14, имеющее формулу:
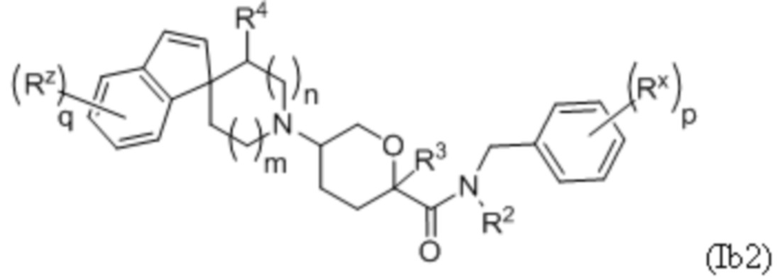
где подстрочный индекс q представляет собой целое число от 1 до 2, и подстрочный индекс p представляет собой целое число от 1 до 3.
17. Соединение по п. 1, имеющее формулу:
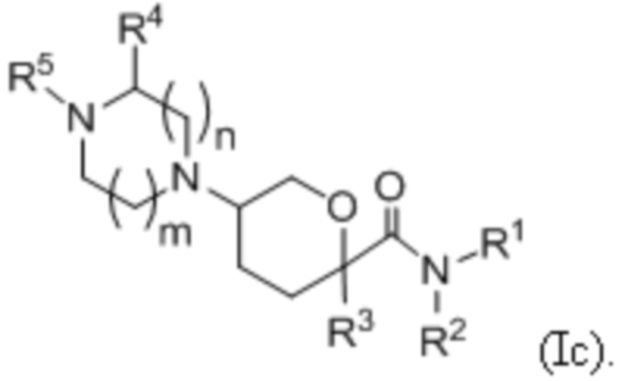
18. Соединение по любому из пп. 1 или 17, имеющее формулу:
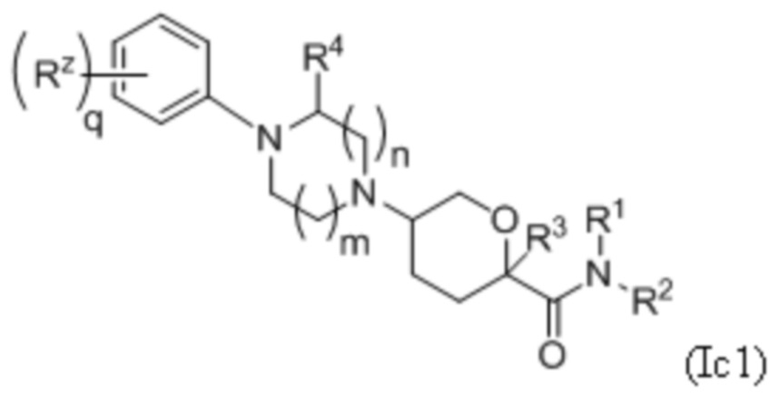
где подстрочный индекс q представляет собой целое число от 1 до 2.
19. Соединение по любому из пп. 1 или 17, имеющее формулу:
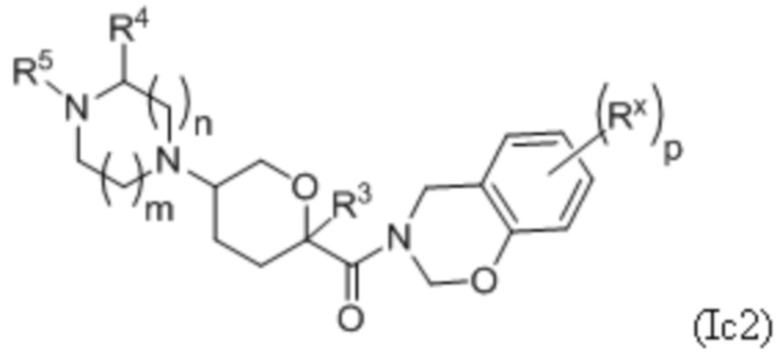
где p представляет собой целое число от 1 до 3.
20. Соединение по любому из пп. 1 или 17, имеющее формулу:
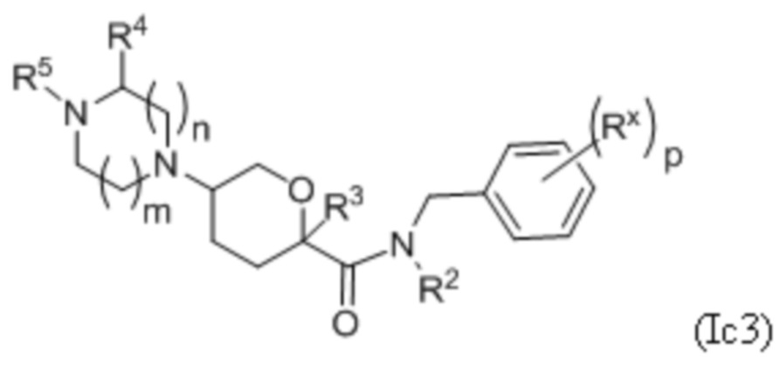
где подстрочный индекс p представляет собой целое число от 1 до 3.
21. Соединение по любому из пп. 1, 17 или 19, имеющее формулу:
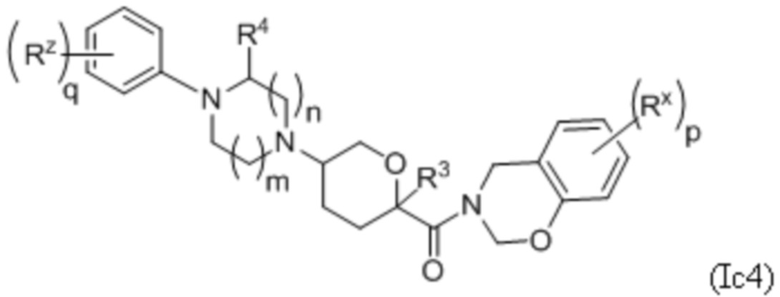
где подстрочный индекс q представляет собой целое число от 1 до 2, и подстрочный индекс p представляет собой целое число от 1 до 3.
22. Соединение по любому из пп. 1, 17 или 18, имеющее формулу:
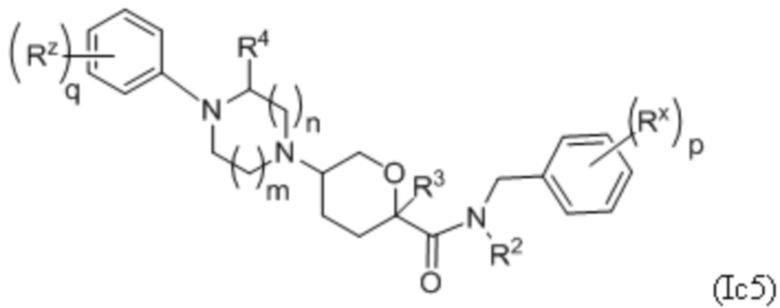
где подстрочный индекс q представляет собой целое число от 1 до 2, и подстрочный индекс p представляет собой целое число от 1 до 3.
23. Соединение по любому из пп. 1, 8 или 9, имеющее формулу:
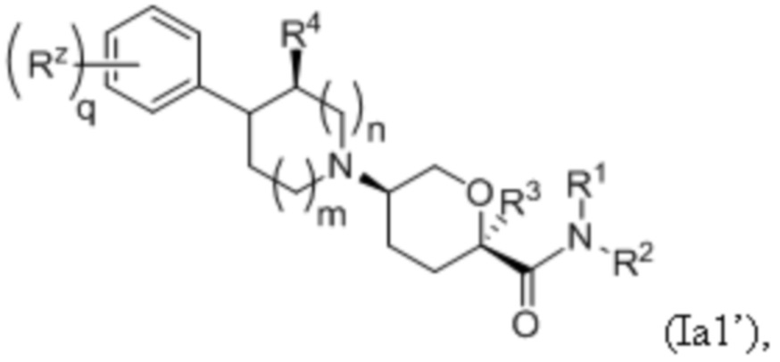
где подстрочный индекс q представляет собой целое число от 1 до 2, и где данное соединение практически не содержит других стереоизомеров.
24. Соединение по любому из пп. 1, 8 или 10, имеющее формулу:
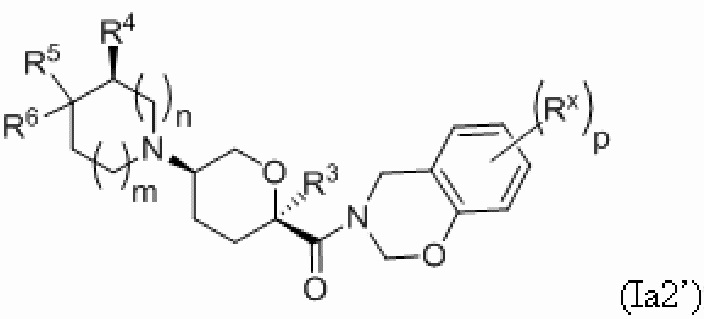
где подстрочный индекс p представляет собой целое число от 1 до 3, и где данное соединение практически не содержит других стереоизомеров.
25. Соединение по любому из пп. 1 или 8, имеющее формулу:
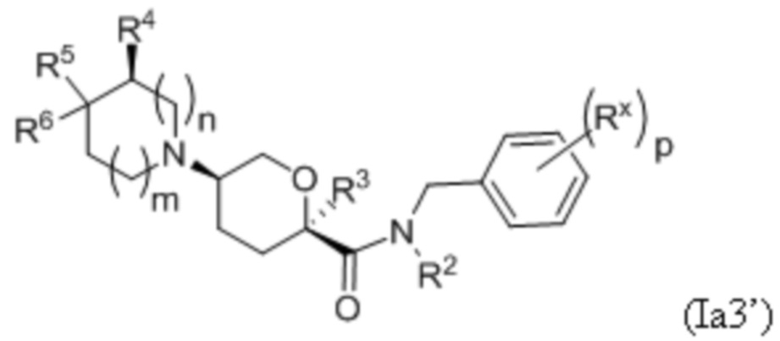
где подстрочный индекс p представляет собой целое число от 1 до 3, и где данное соединение практически не содержит других стереоизомеров.
26. Соединение по любому из пп. 1, 8 или 24, имеющее формулу:
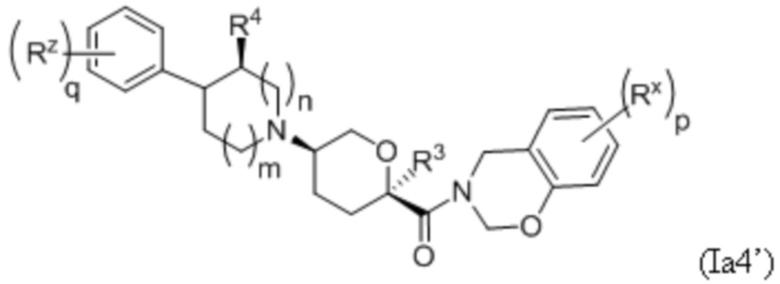
где подстрочный индекс q представляет собой целое число от 1 до 2, подстрочный индекс p представляет собой целое число от 1 до 3, и где данное соединение практически не содержит других стереоизомеров.
27. Соединение по любому из пп. 1, 8 или 25, имеющее формулу:
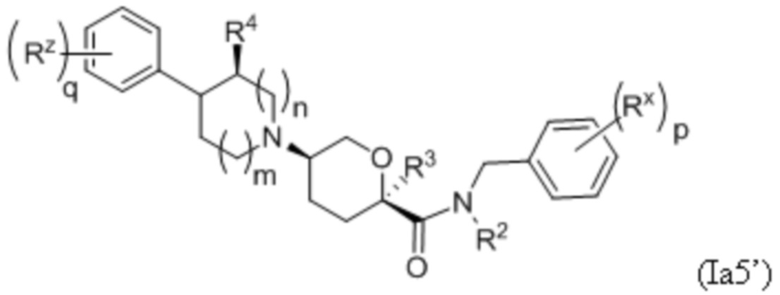
где подстрочный индекс q представляет собой целое число от 1 до 2, подстрочный индекс p представляет собой целое число от 1 до 3, и где данное соединение практически не содержит других стереоизомеров.
28. Соединение по любому из пп. 1, 17 или 18, имеющее формулу:
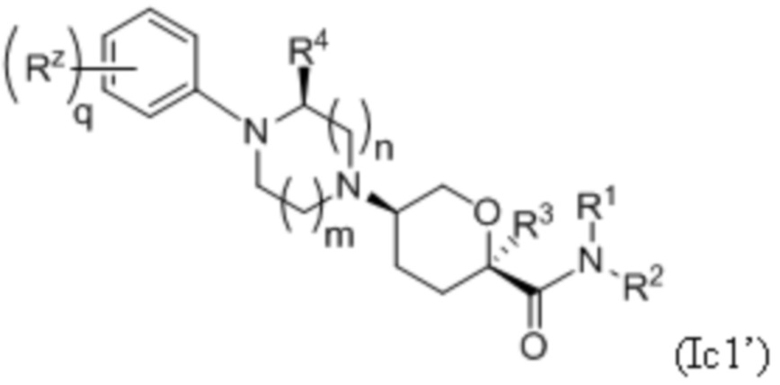
где данное соединение практически не содержит других стереоизомеров.
29. Соединение по любому из пп. 1, 8 или 19, имеющее формулу:
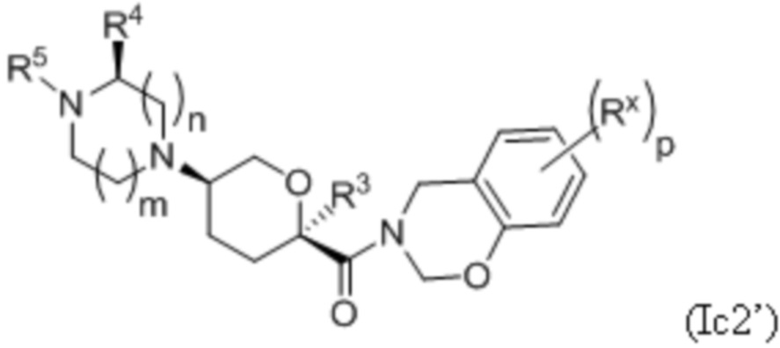
где подстрочный индекс p представляет собой целое число от 1 до 3, и где данное соединение практически не содержит других стереоизомеров.
30. Соединение по любому из пп. 1, 17 или 20, имеющее формулу:
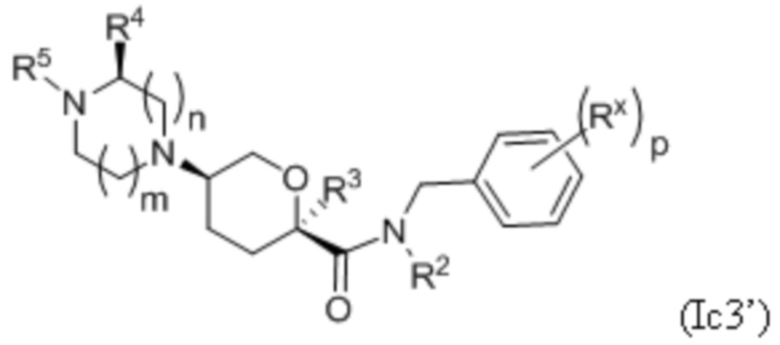
где подстрочный индекс p представляет собой целое число от 1 до 3, и где данное соединение практически не содержит других стереоизомеров.
31. Соединение по любому из пп. 1, 8, 19 или 29, имеющее формулу:
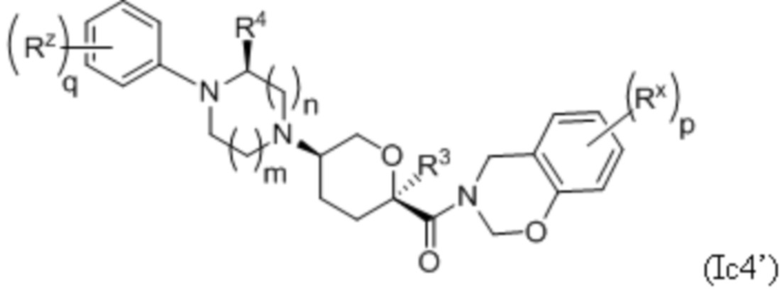
где подстрочный индекс q представляет собой целое число от 1 до 2, подстрочный индекс p представляет собой целое число от 1 до 3, и где данное соединение практически не содержит других стереоизомеров.
32. Соединение по любому из пп. 1, 17, 20 или 30, имеющее формулу:
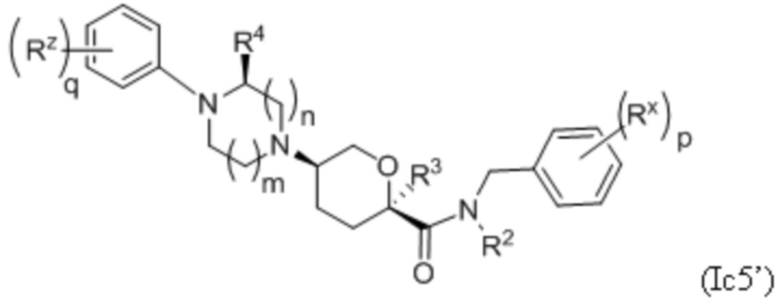
где подстрочный индекс q представляет собой целое число от 1 до 2, подстрочный индекс p представляет собой целое число от 1 до 3, и где данное соединение практически не содержит других стереоизомеров.
33. Соединение по любому из пп. 1-6, 8, 9, 14, 17, 18, 23 или 28, где -N(R1)(R2) выбран из группы, состоящей из:
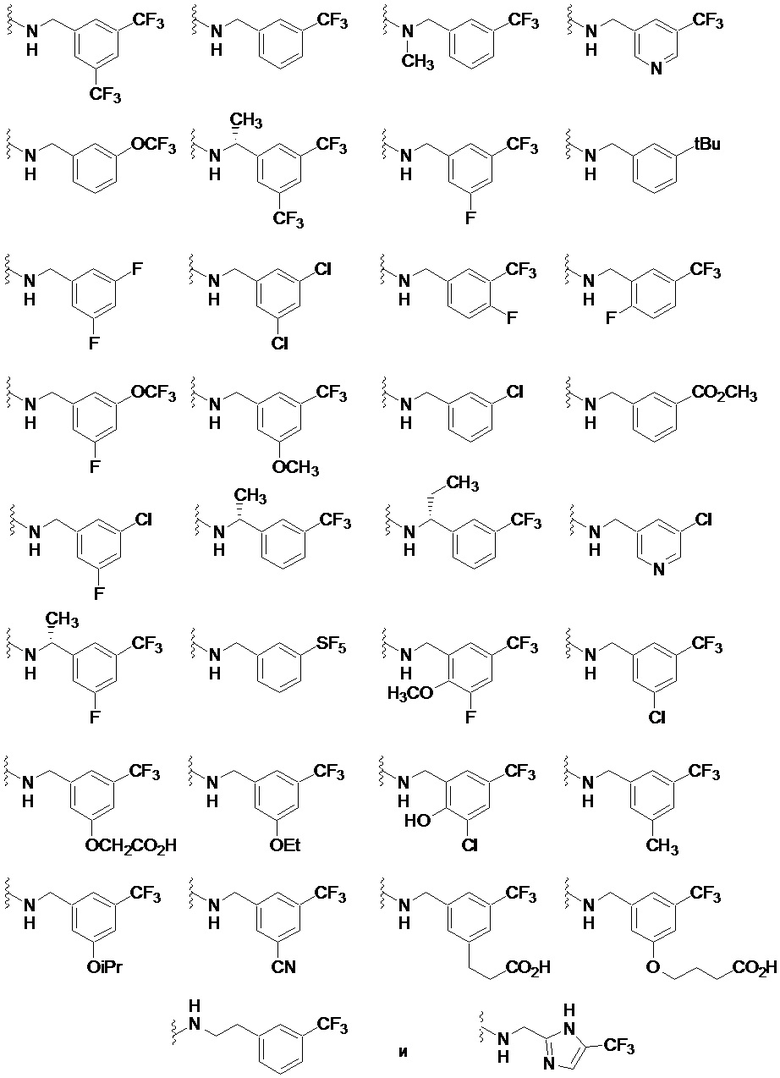 .
.
34. Соединение по любому из пп. 1-6, 8, 9, 14, 17, 18, 23 или 28, где -N(R1)(R2) выбран из группы, состоящей из:
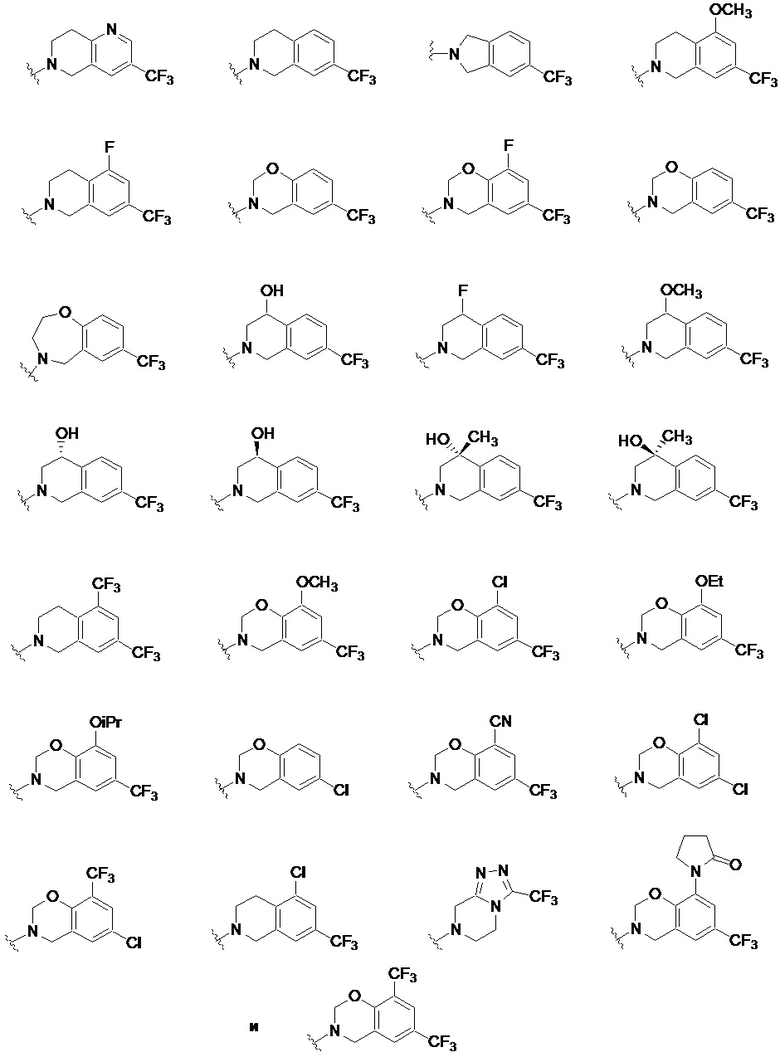 .
.
35. Соединение по любому из пп. 1-6, 8, 9, 14, 17, 18, 23 или 28, где -N(R1)(R2) выбран из группы, состоящей из:
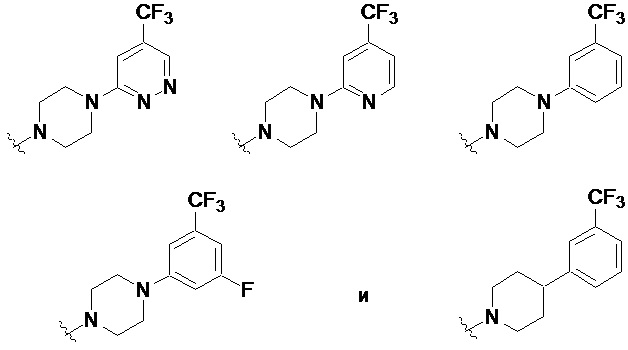 .
.
36. Соединение по любому из пп. 1-6, 8, 10, 11 или 24, 25, где A представляет собой C(R5)(R6), где R5 выбран из группы, состоящей из фенила, фенил-C1-4 алкила, 5-6-членного гетероарила, содержащего в качестве членов цикла 1-3 гетероатома, выбранных из N, O и S, где фенильные или 5-6-членные гетероарильные фрагменты выбраны из группы, состоящей из:

37. Соединение по п. 36, где R5 выбран из группы, состоящей из фенила, фенил-C1-4 алкила.
38. Соединение по п. 36, где R5 выбран из группы, состоящей из 5-6-членного гетероарила, содержащего в качестве членов цикла 1-3 гетероатома, выбранных из N, O и S.
39. Соединение по любому из пп. 1-6, 17, 19, 29 или 30, где A представляет собой N(R5), где R5 выбран из группы, состоящей из фенила, фенил-C1-4 алкила, 5-6-членного гетероарила, содержащего в качестве членов цикла 1-3 гетероатома, выбранных из N, O и S, где фенильные или 5-6-членные гетероарильные фрагменты выбраны из группы, состоящей из:
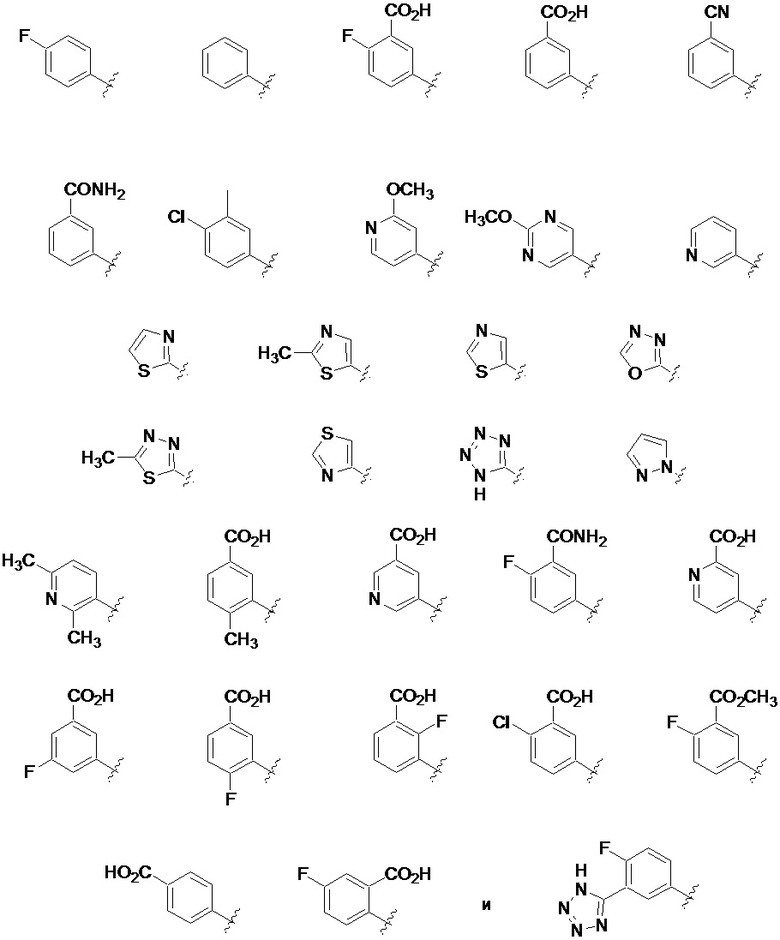 .
.
40. Соединение по п. 39, где R5 выбран из группы, состоящей из фенила, фенил-C1-4 алкила.
41. Соединение по п. 39, где R5 выбран из группы, состоящей из 5-6-членного гетероарила, содержащего в качестве членов цикла 1-3 гетероатома, выбранных из N, O и S.
42. Соединение по любому из пп. 1-6, 17, 19, 29 или 30, где A представляет собой N(R5), где R5 выбран из группы, состоящей из:
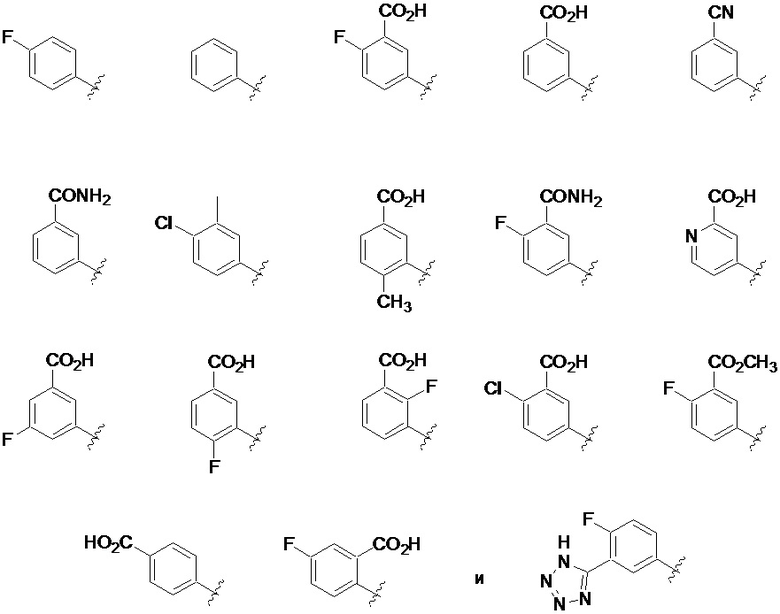 .
.
43. Соединение по п. 1, выбранное из группы, состоящей из:
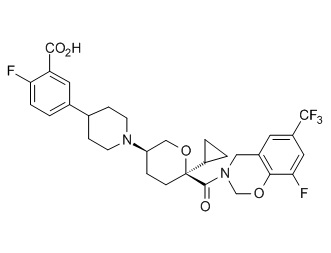

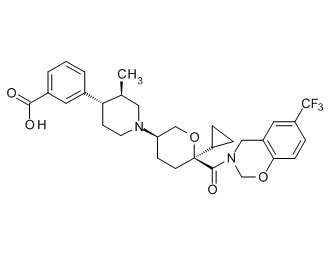
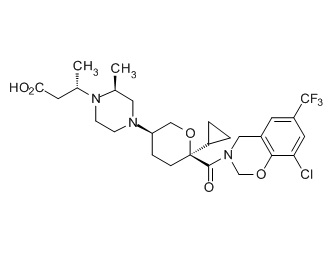
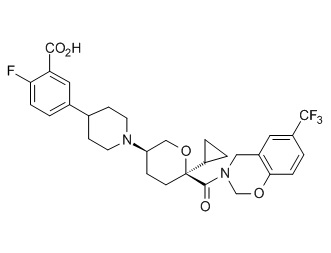
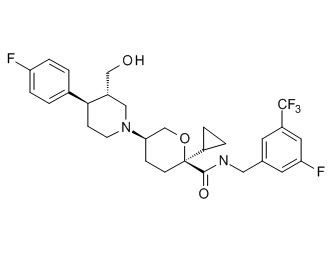
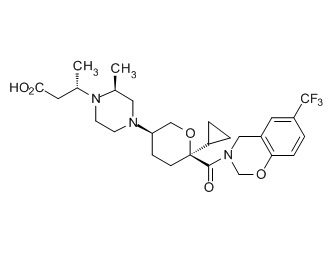
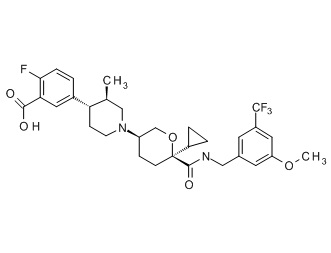
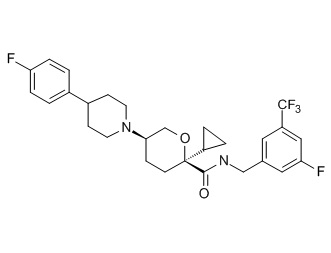

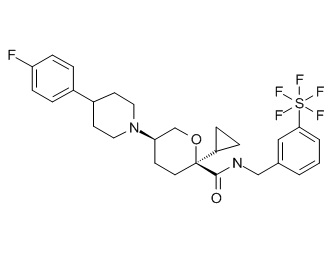
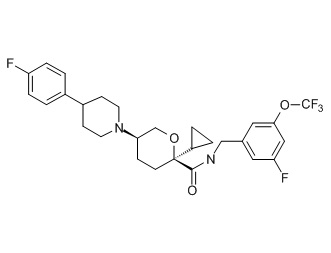
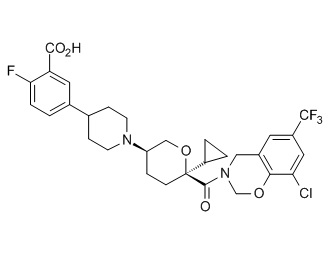
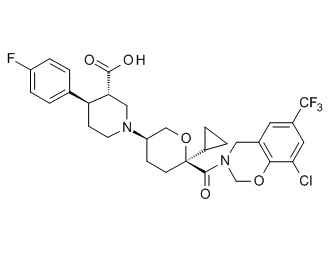
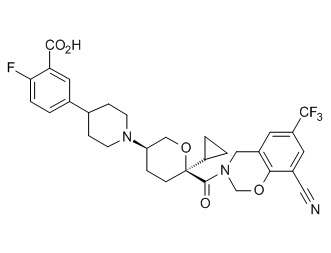
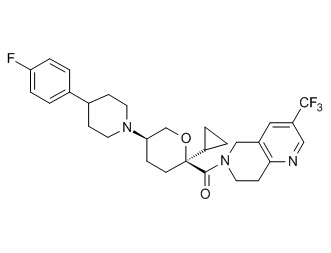
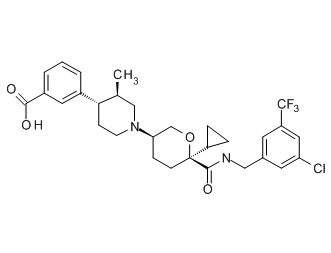
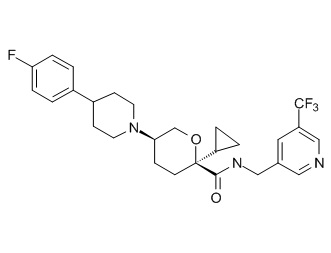
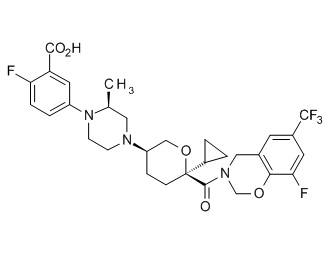
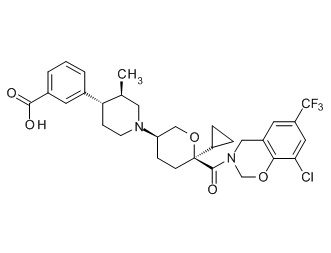
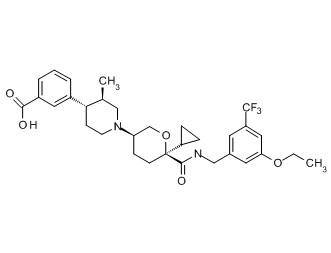
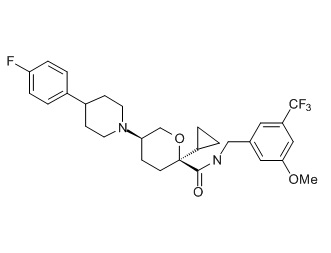
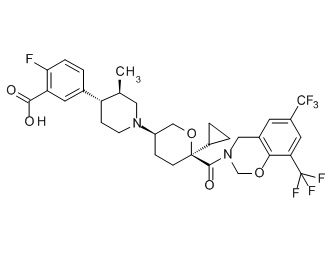
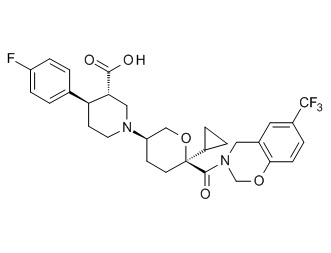
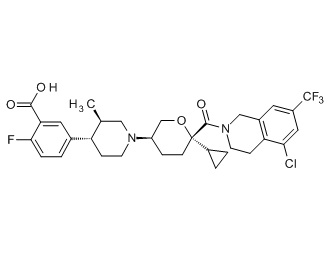
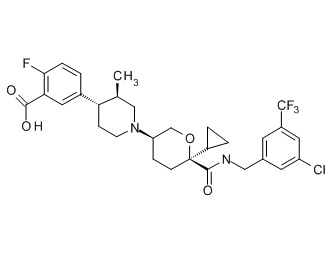
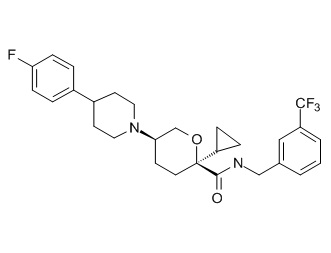
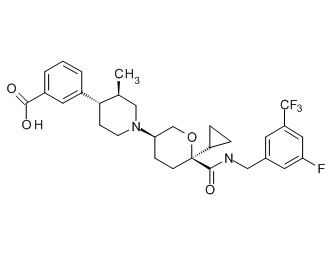
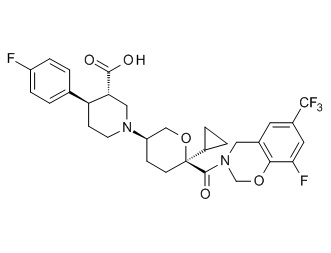
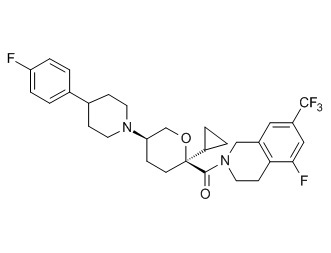
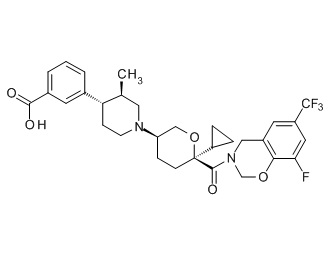
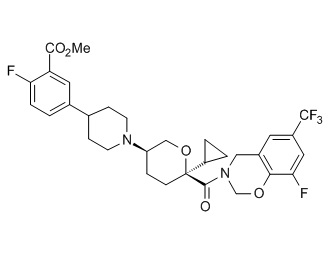
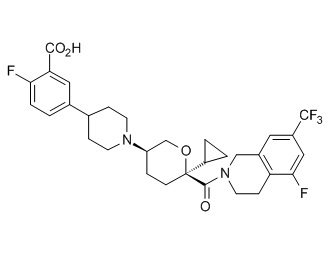
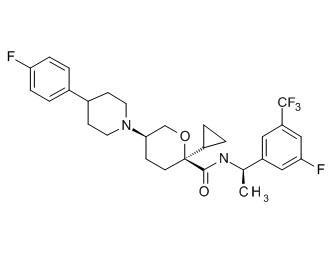
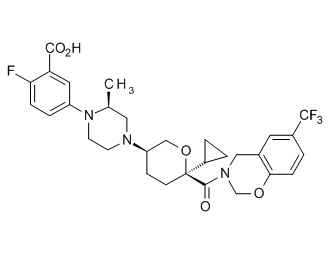

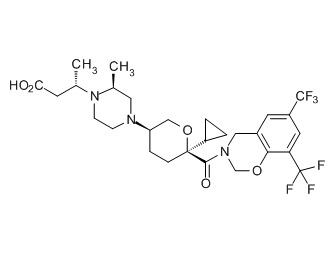
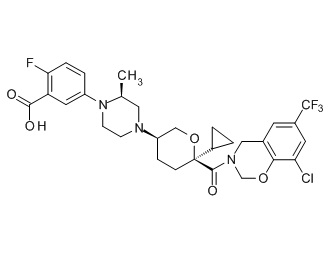
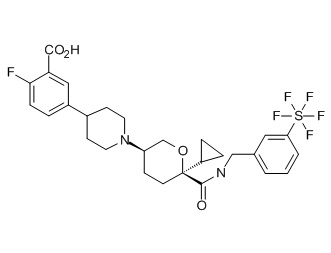
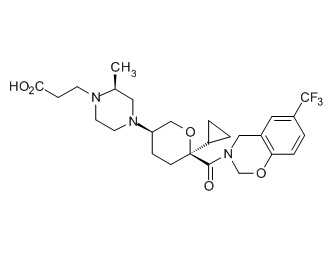
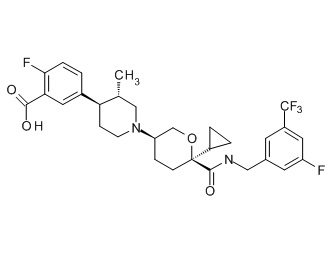
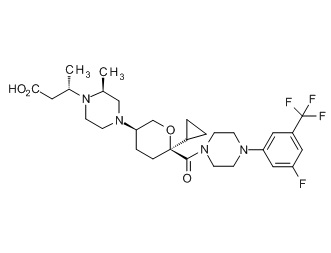
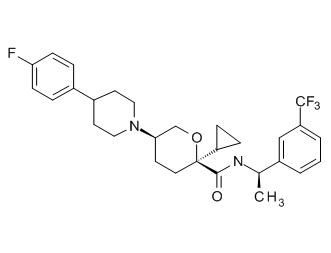
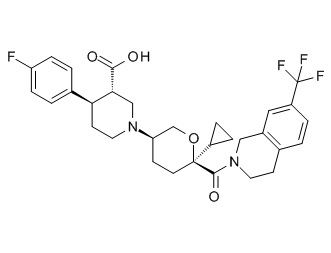
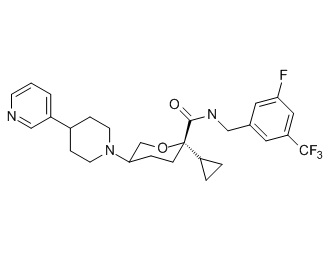
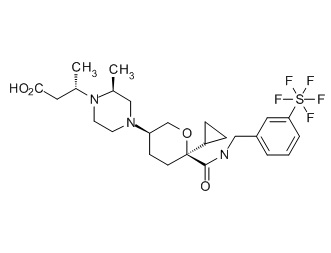
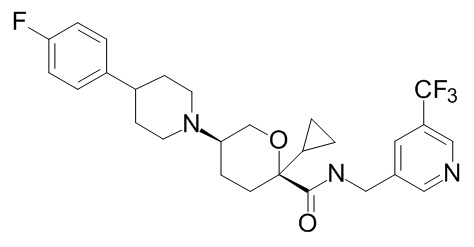
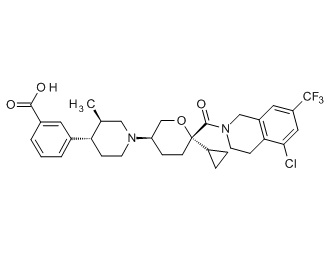
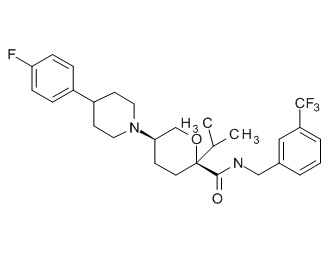
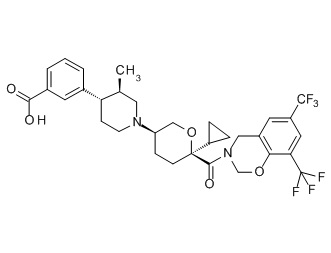
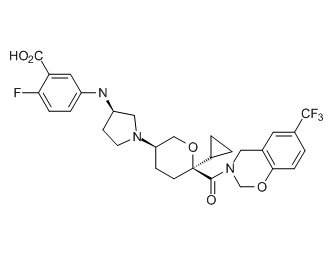
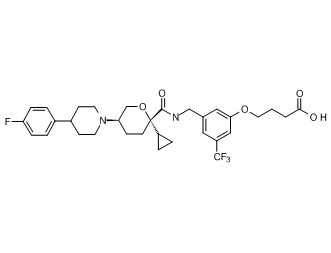
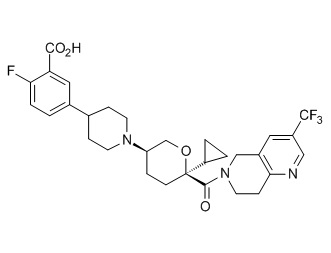
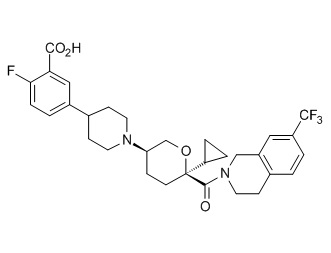
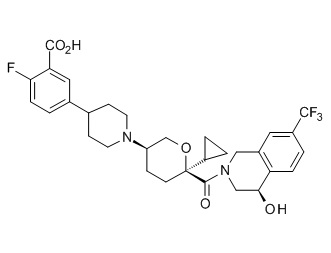
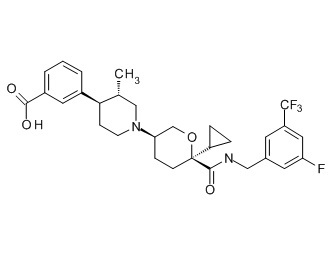
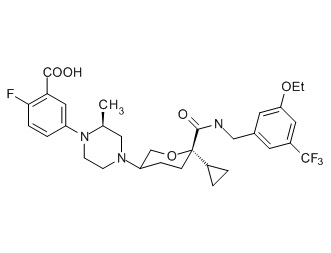
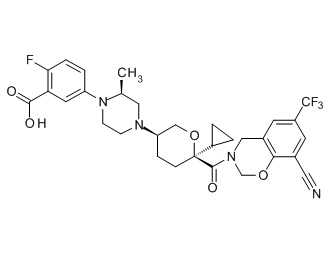
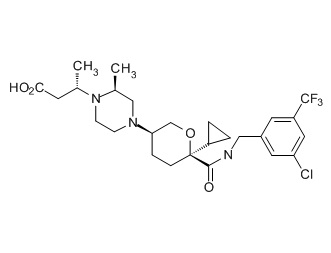
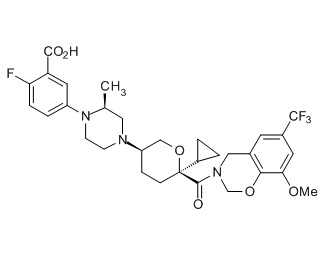
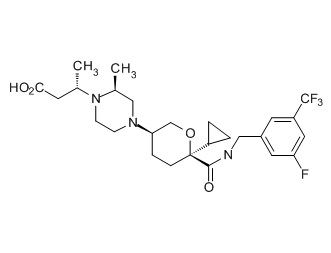
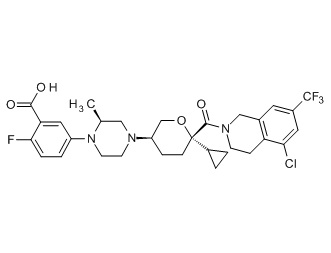
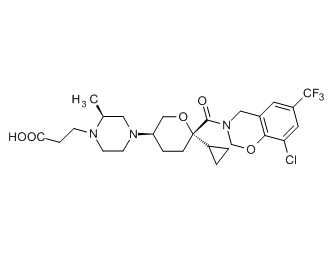

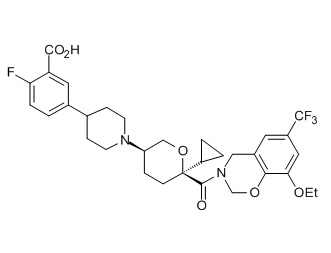
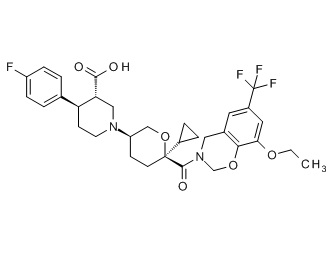
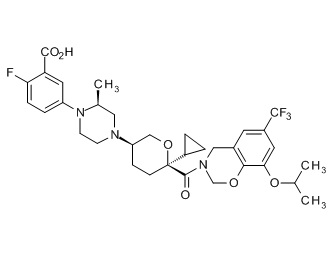
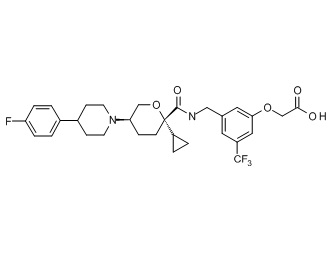
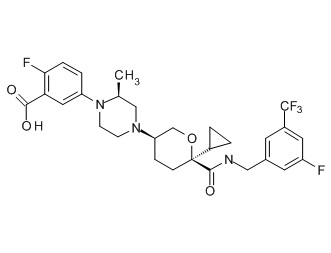
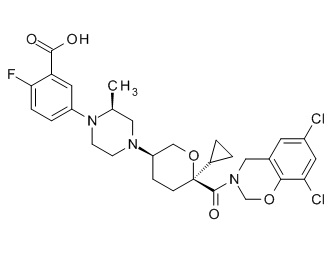
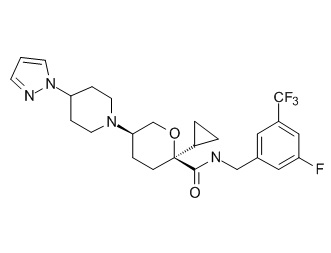
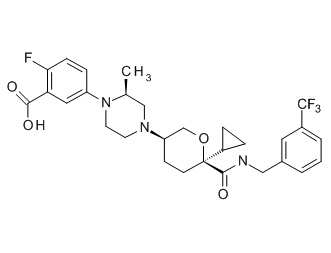
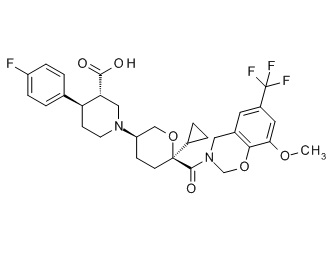
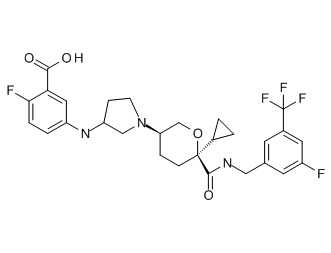
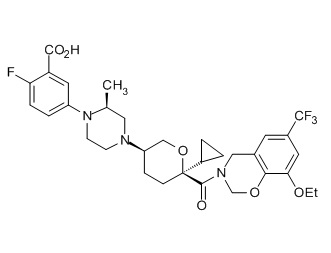
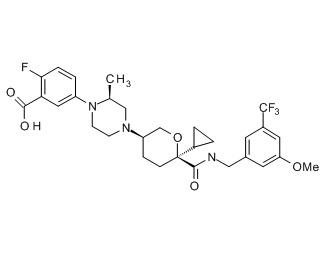
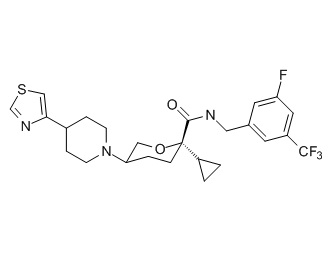
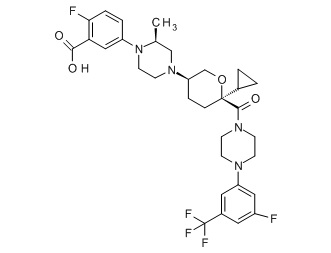
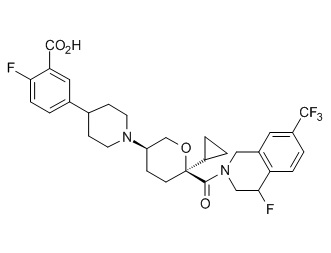
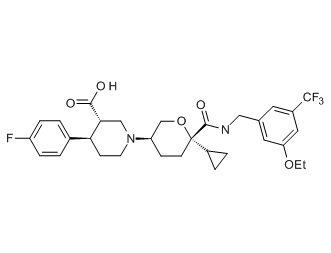
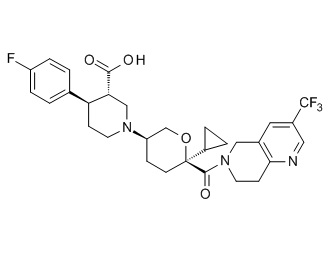
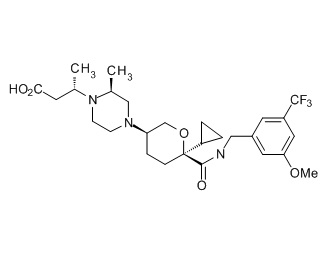
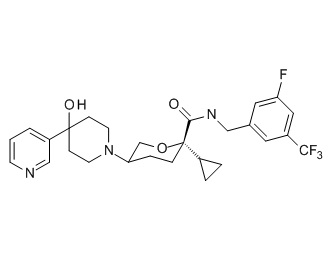
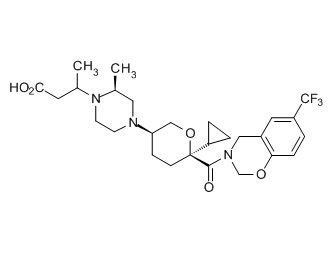
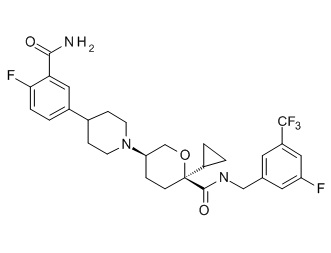
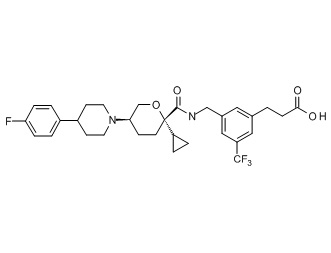
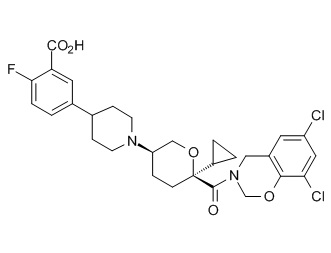
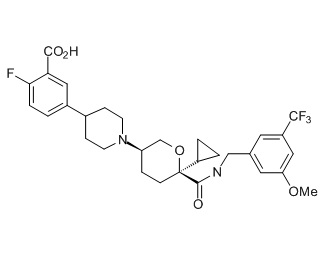
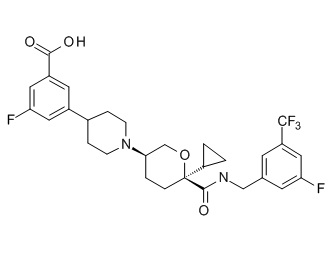
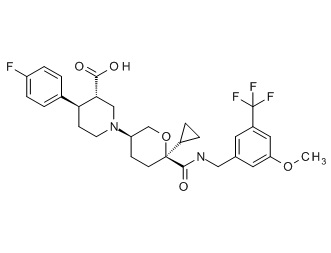
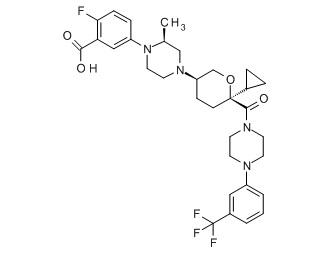
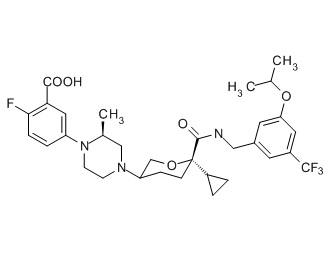
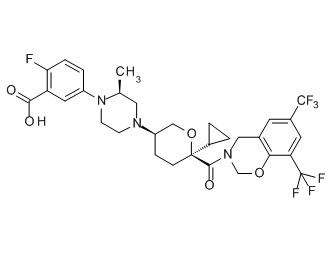
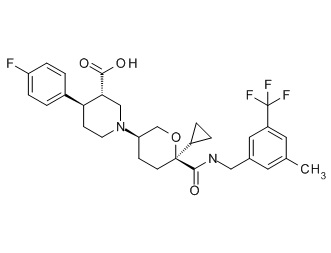
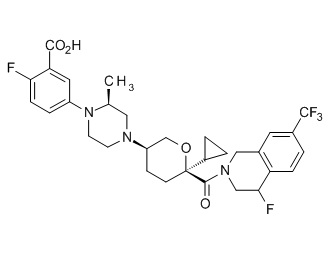
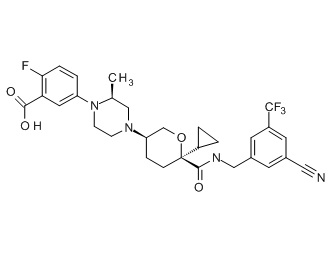
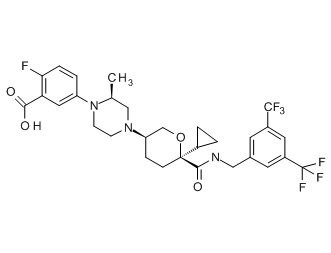
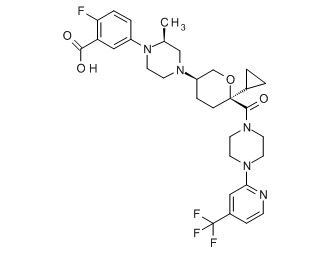
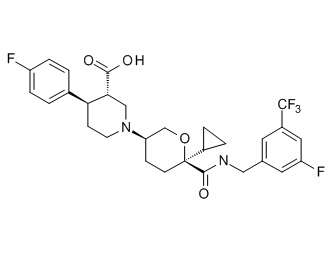
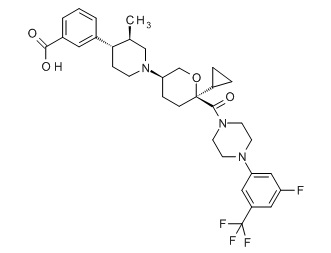
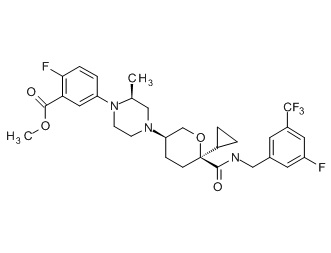
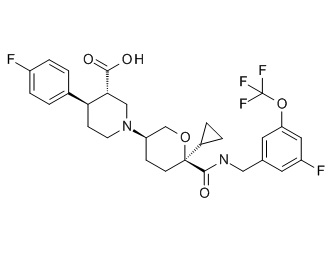
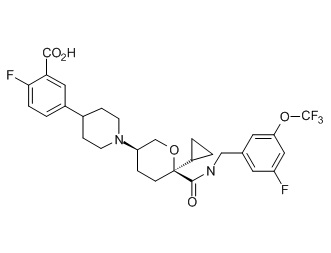
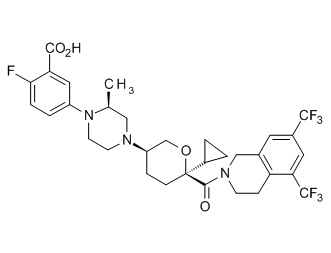
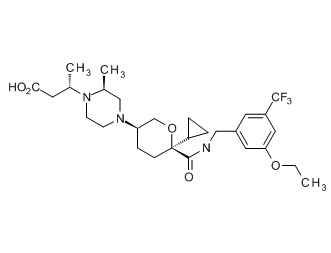
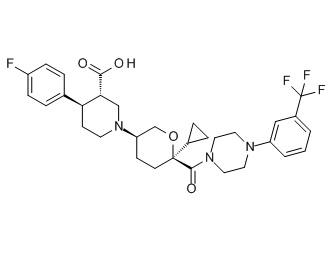
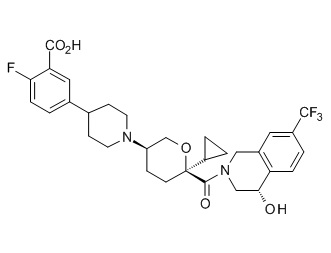
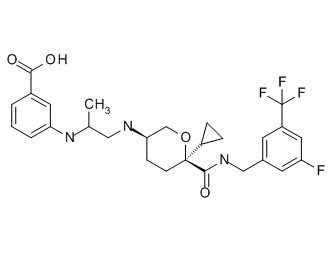
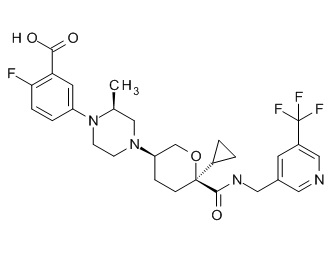
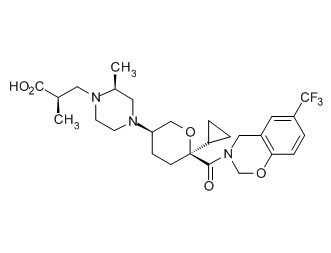
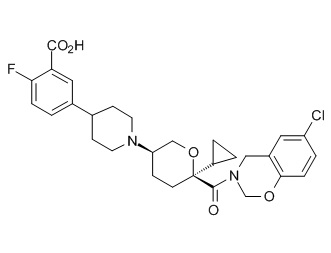
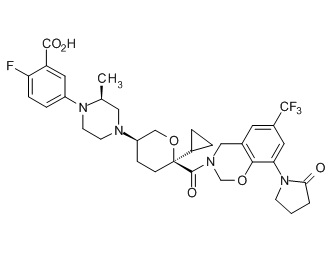
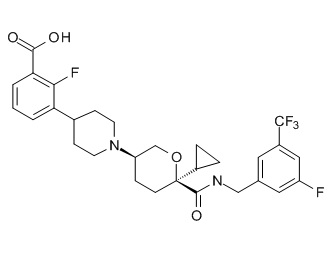
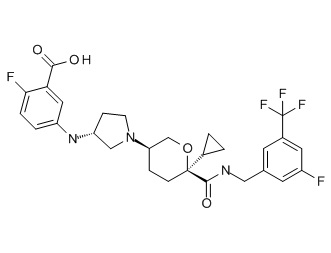
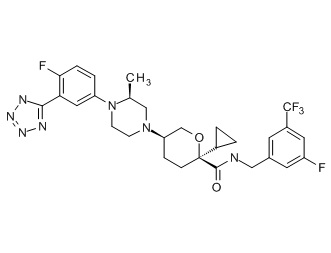
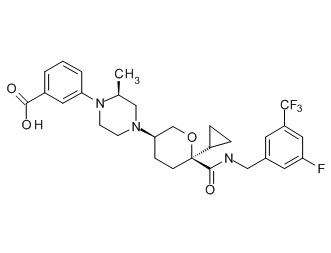
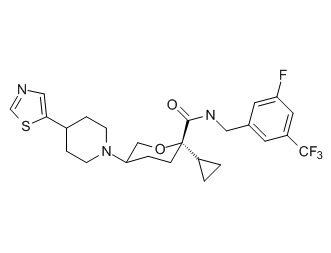
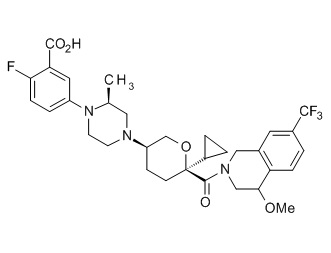
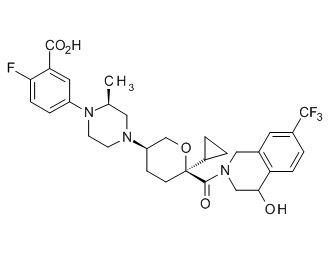
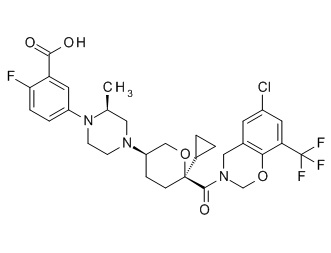
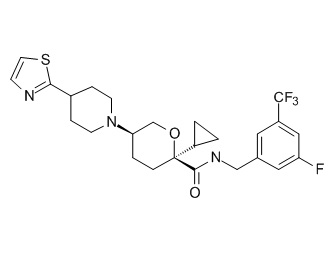
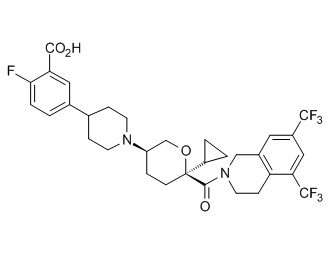
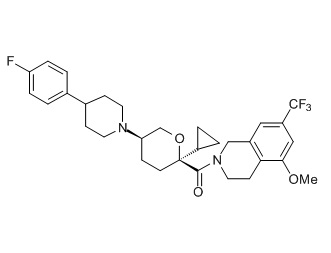
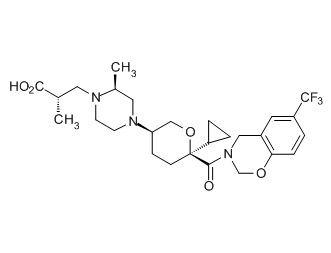
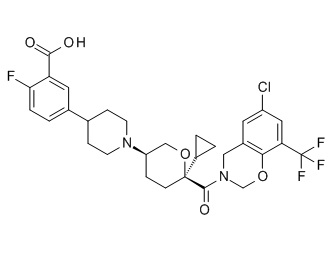
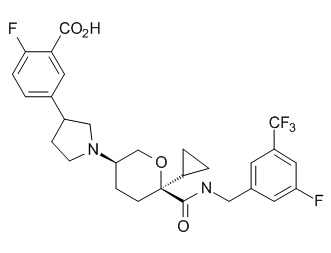
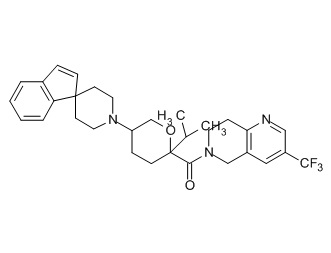

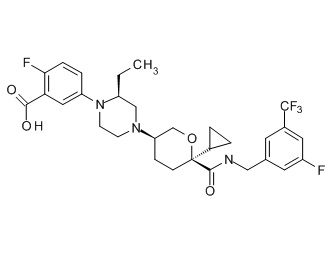
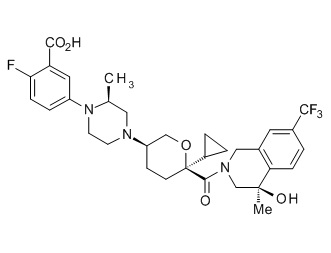
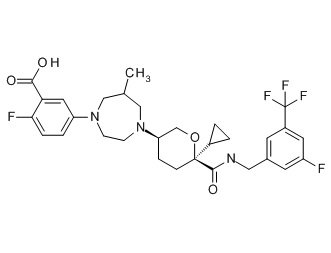
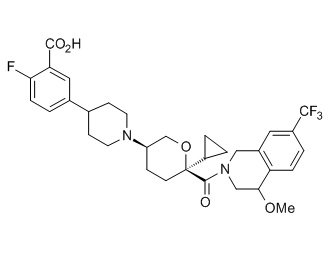
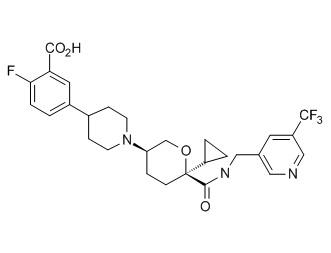
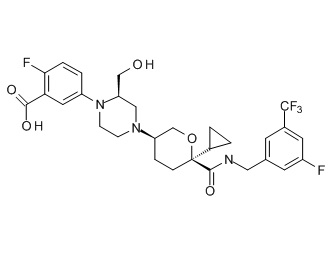
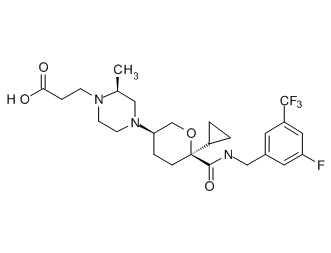

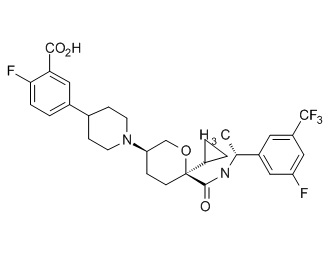
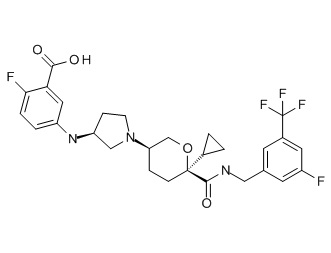
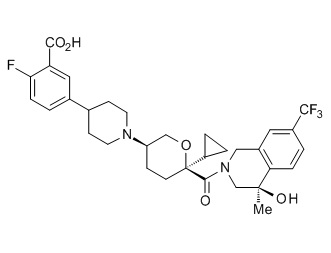
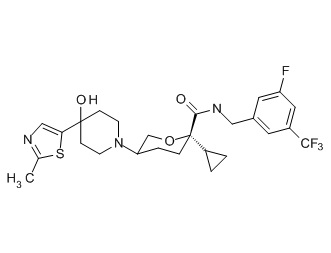
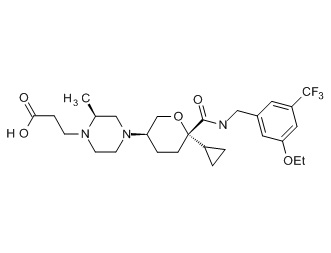
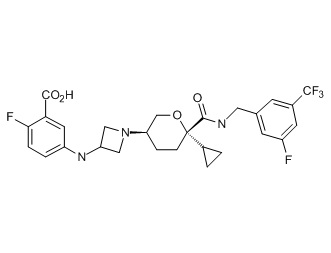
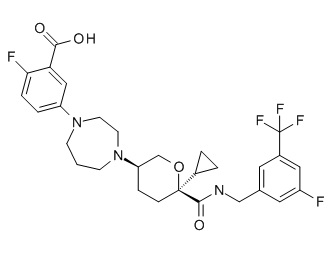
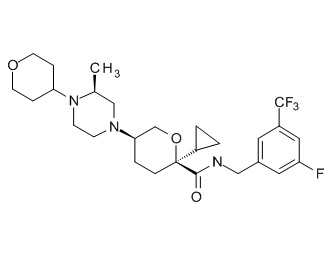

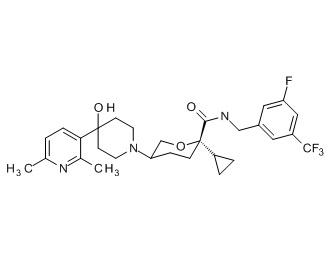
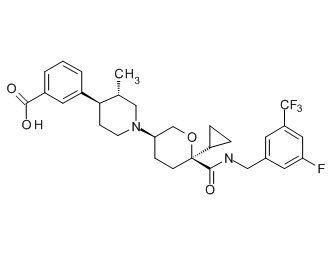
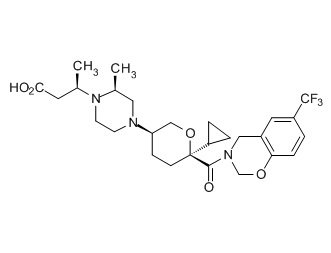
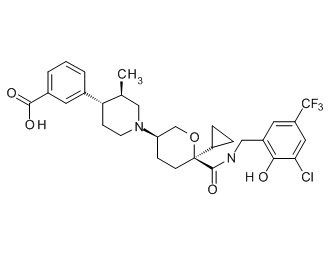
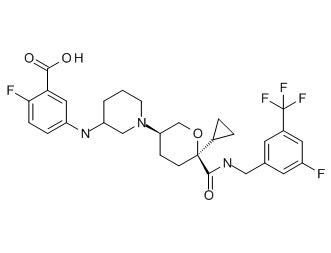
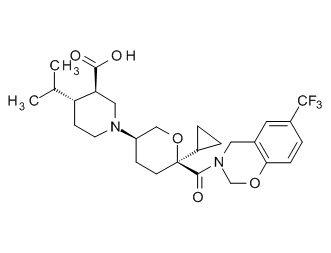
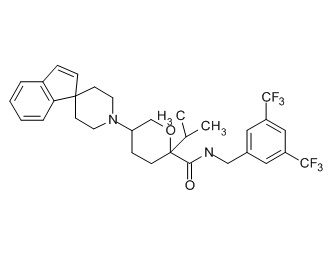
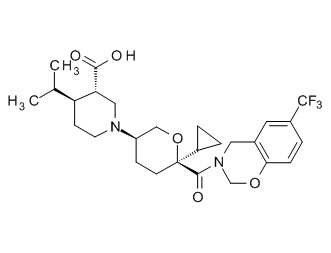
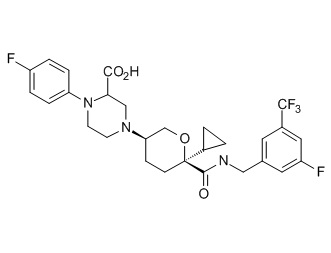
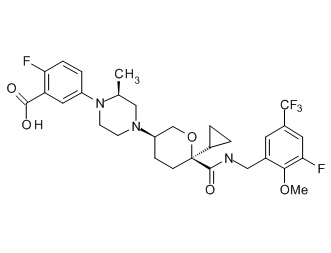
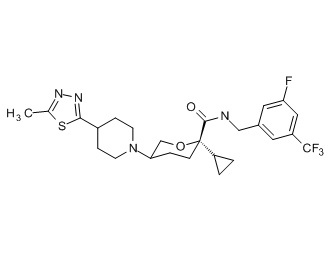
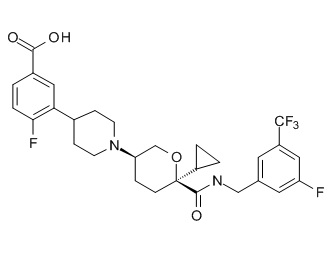
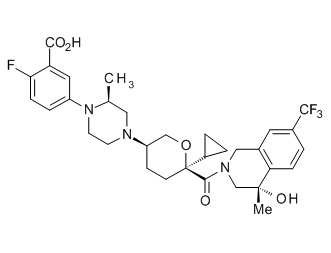
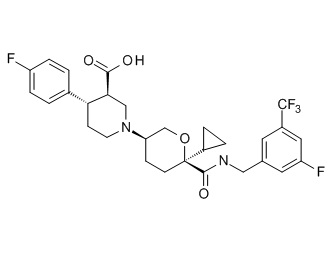
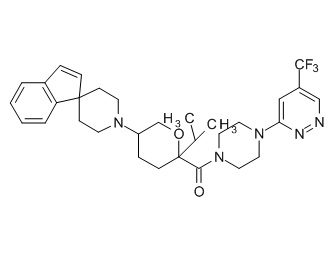
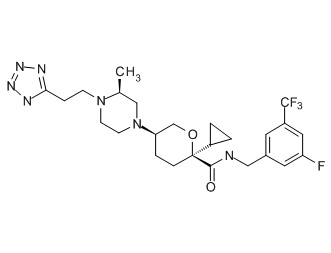
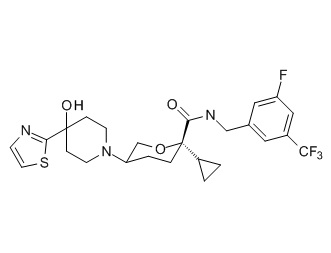
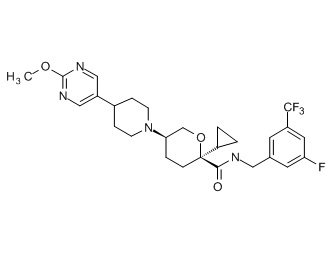
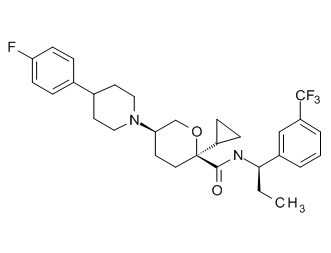
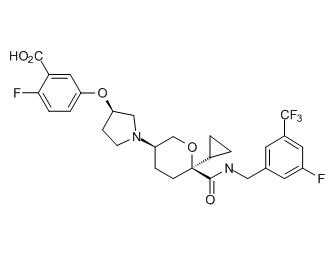
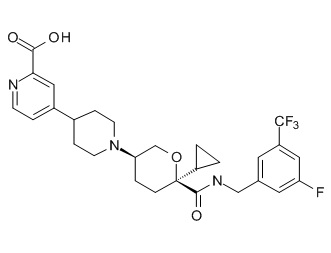
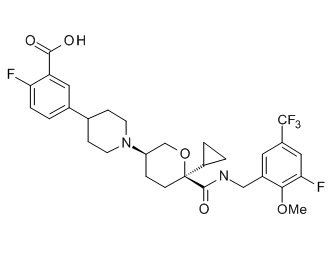
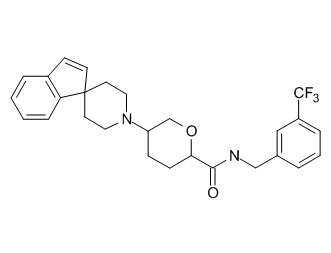
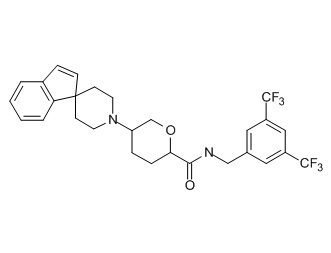
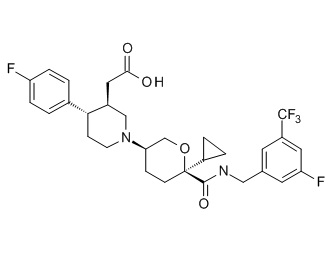
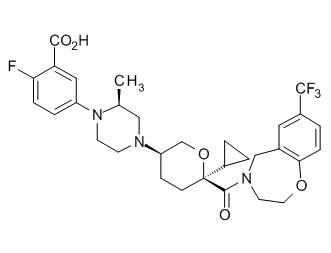
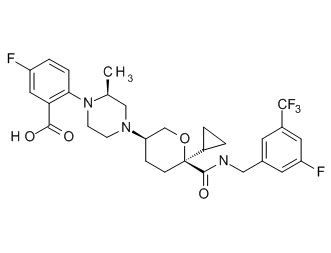
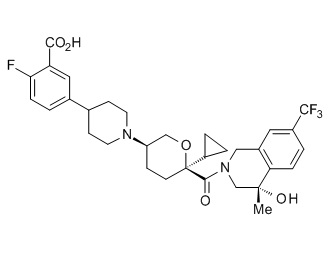

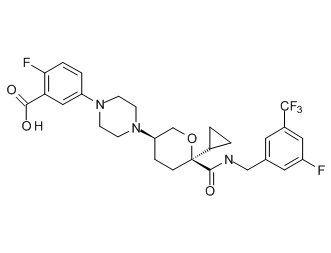
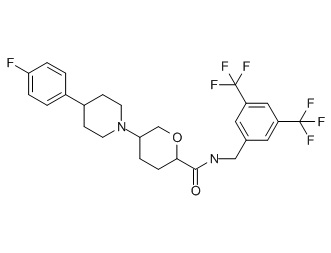


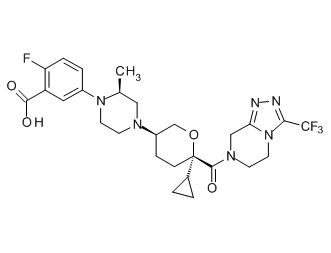
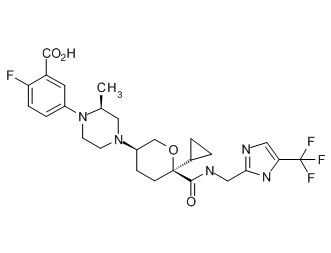
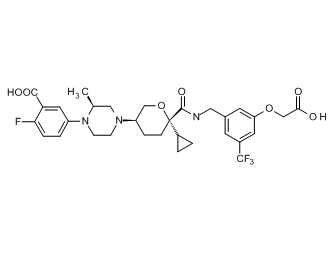
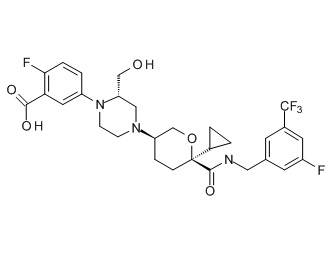
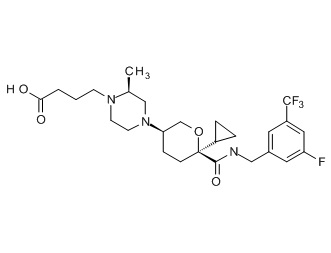
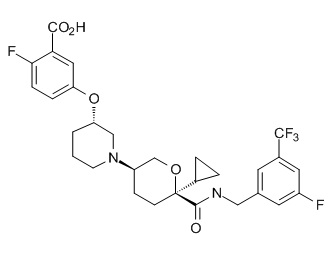
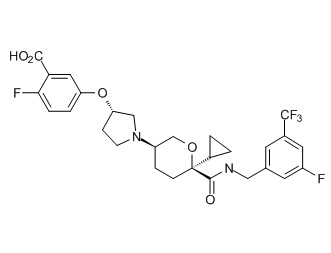
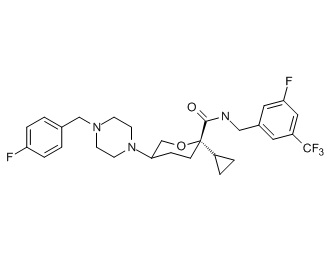
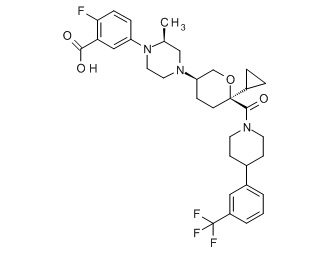
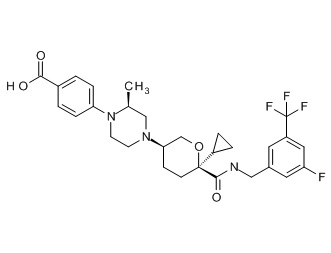
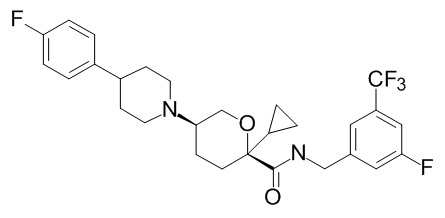

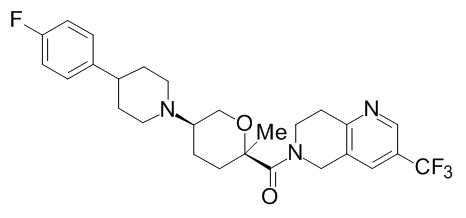
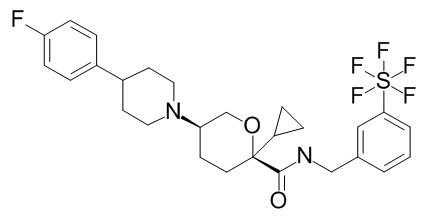

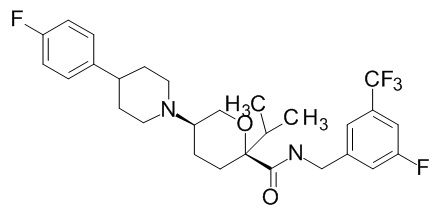
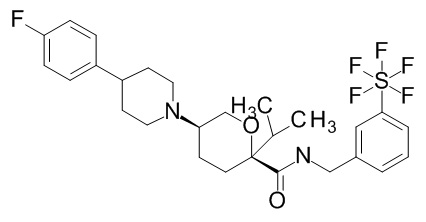
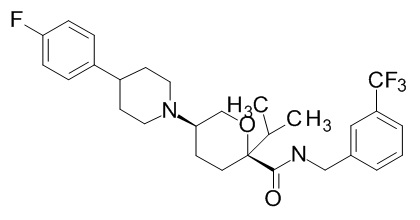

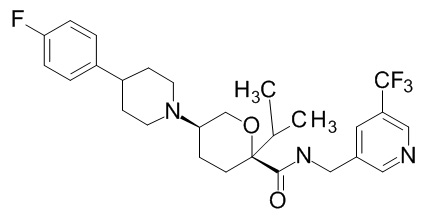
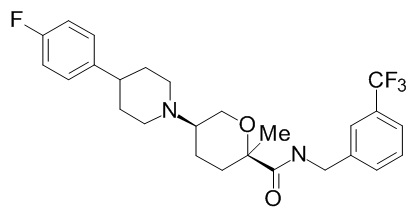
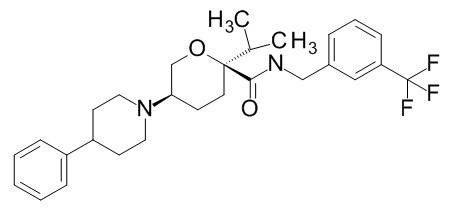
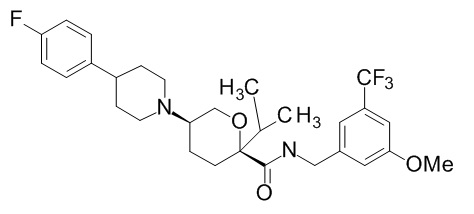
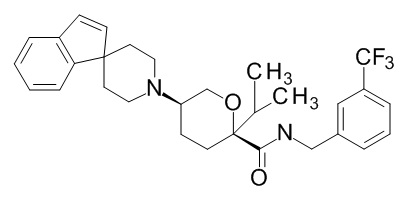
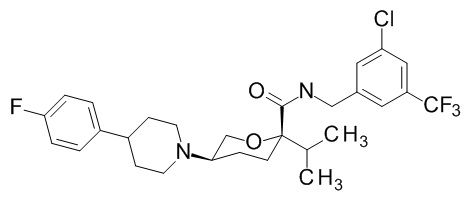
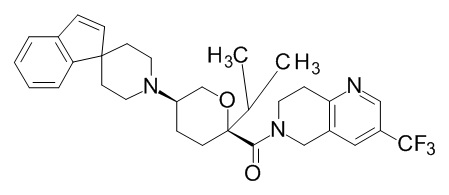
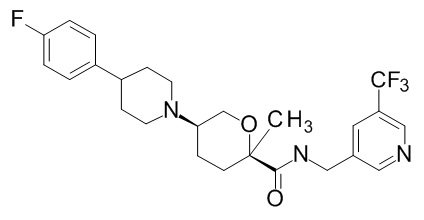
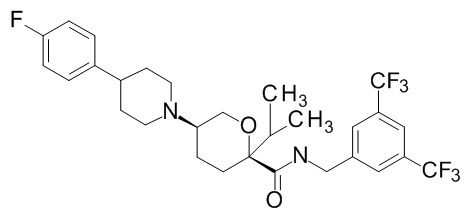
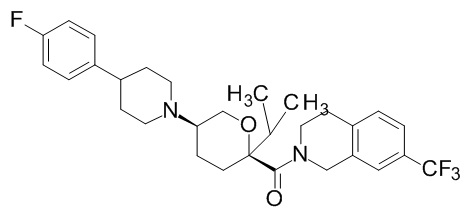

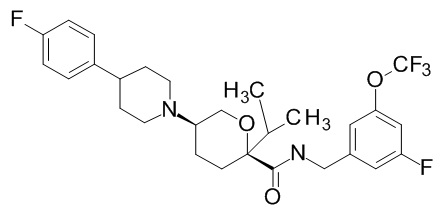
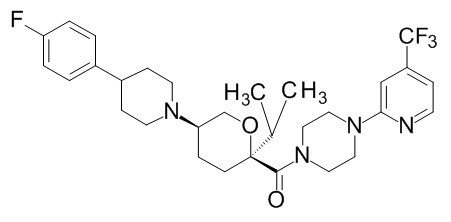
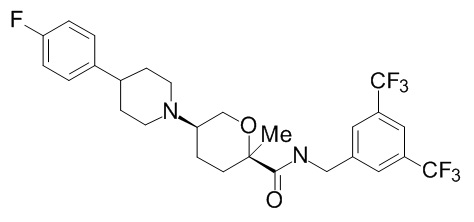
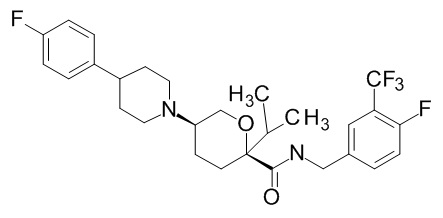
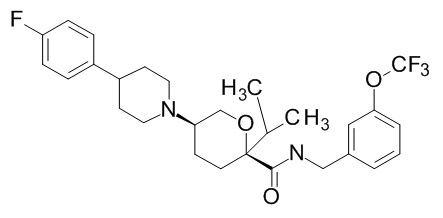
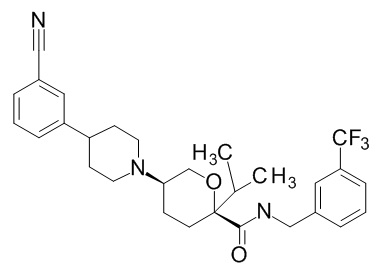
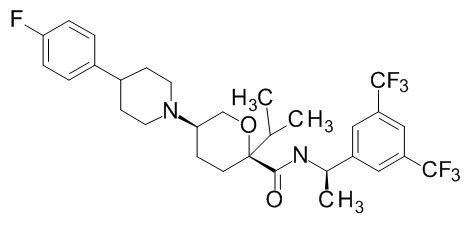
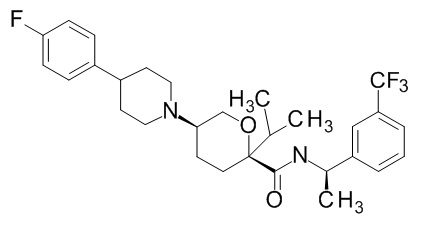
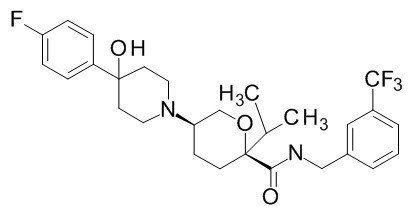
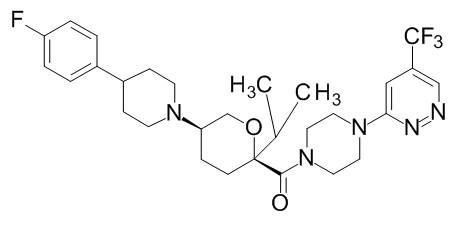
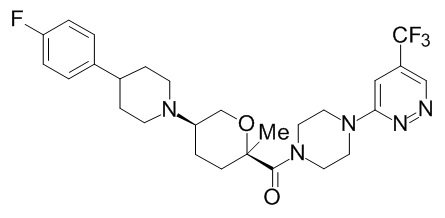
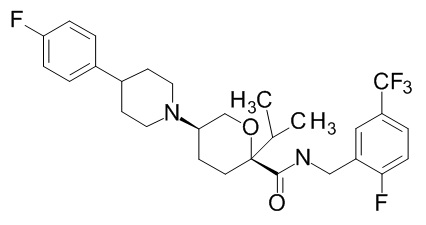
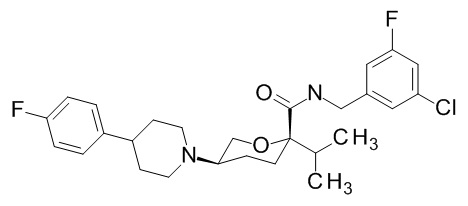
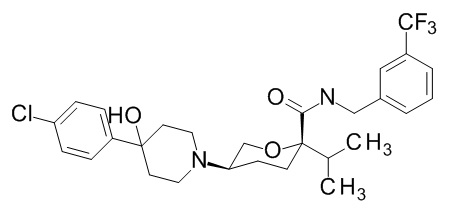
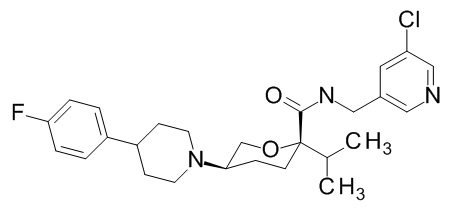
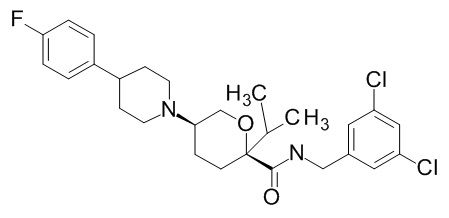
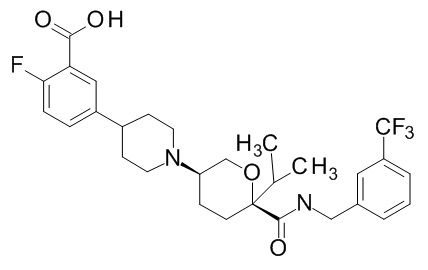

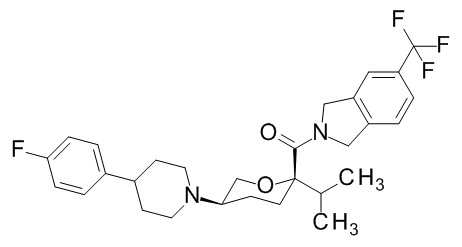
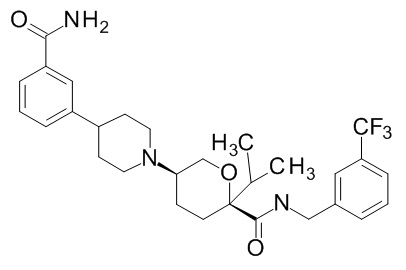
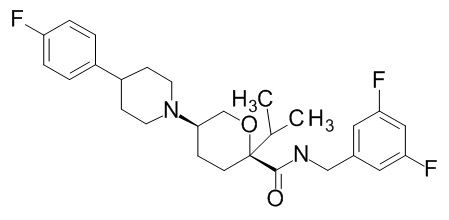
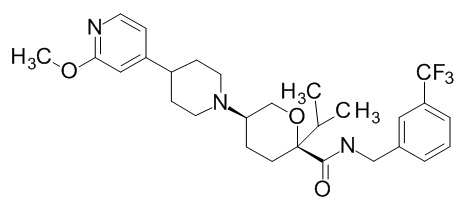
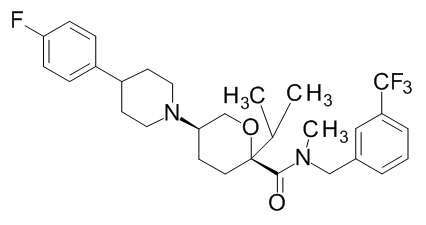
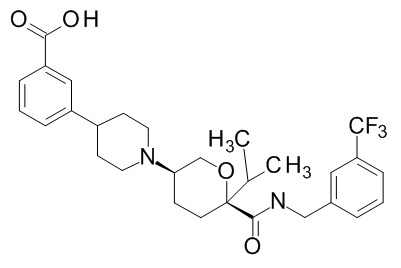
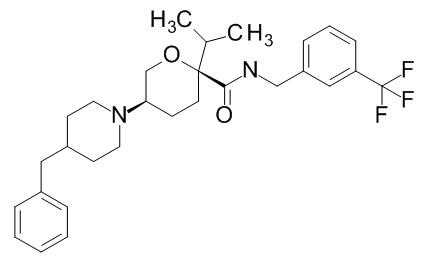
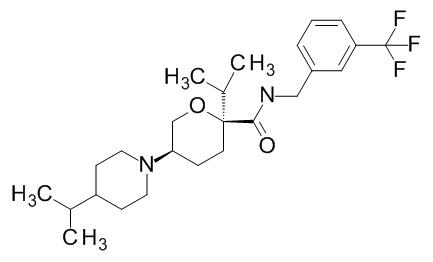 и
и 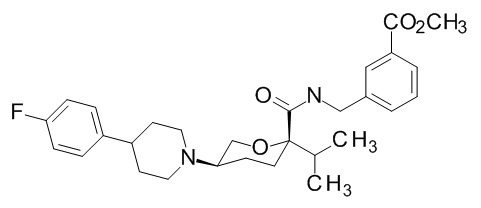
или его фармацевтически приемлемая соль.
44. Фармацевтическая композиция для лечения CCR2-опосредованного заболевания или нарушения, содержащая фармацевтически приемлемый носитель и соединение по любому из пп. 1-43.
45. Способ лечения млекопитающего, страдающего от или склонного к заболеванию или нарушению, выбранного из группы, состоящей из почечного фиброза, поликистозной болезни почек, ревматоидного артрита, атопического дерматита, псориаза, нефропатии, рака поджелудочной железы и неалкогольного стеатогепатита (НС), включающий введение млекопитающему эффективного количества соединения по любому из пп. 1-43 или фармацевтической композиции по п. 44.
46. Способ ингибирования CCR2 рецептор-опосредованного клеточного хемотаксиса, включающий контактирование белых кровяных телец млекопитающего с модулирующим CCR2 рецептор количеством соединения по любому из пп. 1-43 или фармацевтической композиции по п. 44.
47. Способ по п. 45, где указанная нефропатия представляет собой диабетическую нефропатию.
48. Способ по п. 45, где указанное заболевание или нарушение представляет собой рак поджелудочной железы.
49. Способ по п. 45, где указанное заболевание или нарушение представляет собой неалкогольный стеатогепатит (НС).
50. Соединение по п. 1, имеющее формулу
 ,
,
или его фармацевтически приемлемая соль.
51. Соединение по п. 1, имеющее формулу
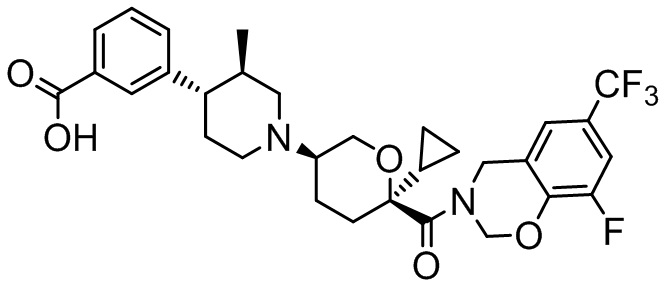
или его фармацевтически приемлемая соль.
52. Соединение по п. 1, имеющее формулу
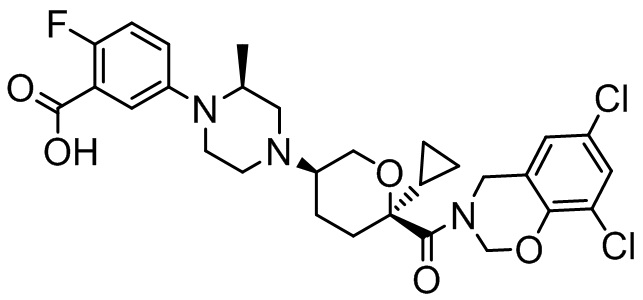
или его фармацевтически приемлемая соль.
| WO 2005080371 A1, 01.09.2005 | |||
| WO 2003093266 A1, 13.11.2003 | |||
| WO 2004092124 A2, 28.10.2004 | |||
| WO 2005067502 A2, 28.07.2005 | |||
| Изложница с суживающимся книзу сечением и с вертикально перемещающимся днищем | 1924 |
|
SU2012A1 |
| US 6949546 B2, 27.09.2005 | |||
| WO 2003092586 A2, 13.11.2003 | |||
| Пастеризатор для молока | 1928 |
|
SU14960A1 |

