Область техники
Настоящее изобретение относится к новому типу соединений дигидроинден амида либо их фармацевтически приемлемым солям, их получению, фармацевтическим композициям, содержащим данные соединения, способам их применения для предотвращения или лечения заболеваний, связанных с ненормальной активностью протеин киназ, особенно в случае заболеваний, связанных с ненормальной активностью Abl, Bcr-Abl, c-Kit и PDGFR, а также их применению для производства медикаментов для предотвращения или лечения указанных заболеваний.
Предшествующий уровень техники
Протеинкиназы представляют собой ферменты, осуществляющие перенос фосфатной группы от нуклеозид трифосфата на конкретный сериновый, треониновый или тирозиновый остаток. Форфорилирование белка приводит к активации путей сигнальной трансдукции, играющих решающую роль в различных биологических процессах, включая рост клеток, метаболизм, дифференциацию и смерть. Известно, что нарушения сигналов, вызванные неправильной или ненормальной активностью протеинкиназ, связаны с большим числом болезней, включая рак, воспаления, аутоимунные заболевания, нарушения обмена веществ, инфекции, расстройства центральной нервной системы, сердечно-сосудистые заболевания и т.д. Поэтому протеинкиназы представляют собой привлекательный объект при разработке лекарств (Коген (Cohen, Nat Rev. Drug Discovery 2002, 1, 309)).
Гены abl и bcr представляют собой нормальные гены, расположенные на хромосомах 9 и 22 соответственно. Два гибридных гена образуются путем сегментного обмена между данными двумя генами: bcr-abl гена, расположенного на хромосоме 22q-n abl-bcr гена, расположенного на хромосоме 9q+. Белок 210kD (з210 BCr-Abl) кодируется bcr-abl геном хромосомы Philadelphia. Фрагмент Abl белка Bcr-Abl включает Abl тирозинкиназу, точно регулируемую в прототипе с-Abl, но постоянно активируемую белком слияния Bcr-Abl, что приводит к нарушению клеточного роста. Белок Bcr-Abl обнаружен у 95% пациентов с хронической гранулоцитной лейкемией (ХГЛ) и у 10-25% пациентов с острым лимфобластным лейкозом (ОЛЛ). Иматиниб, известный под торговым названием Gleevec, является ингибитором тирозинкиназы Bcr-Abl и был клинически одобрен в качестве эффективного состава для лечения ХГЛ (Друкер и др. Druker et al. N. Engl. J. Med. 2006, 355, 2408)). Однако, несмотря на постоянное лечение с использованием Иматиниба, у некоторых пациентов с ХГЛ наблюдаются рецидивы на заключительной фазе или на стадии бластного кризиса вследствие невосприимчивости к лекарству. Обоснованием невосприимчивости к лекарству на молекулярном уровне является возникновение мутантов, устойчивых к воздействию иматиниба в киназном домене Bcr-Abl белка. К настоящему моменту известно более 22 мутантов, среди которых наиболее распространенными являются M244V, G250E, Q252H, Y253H, Е255К, E255V, F311L, T351I, F317L, F359V, V379I, L387M, Н396Р, H396R и др. (Нарди и др. (Nardi, et al. Curr. Opin. Hematol. 2004, 11, 35)).
c-Kit (CD117, рецептор фактора стволовых клеток), кодируемый c-kit протоонкогеном, представляет собой рецептор фактора роста с тирозинкиназной активностью. Он может активироваться при связывании с фактором стволовых клеток (SCF). Мутации в c-kit приводят к постоянной активации функции c-Kit тирозинкиназы, которая далее приводит к активности тирозинкиназы независимо от лигандов, аутофосфорилированию c-Kit и нарушению клеточной пролиферации. Сверхэкспрессия и мутации c-Kit выявлены в большинстве гастроинтестинальных стромальных опухолях (ГИСО). Гастроинтестинальные стромальные опухоли представляют собой серию мезенхимальных опухолей, которые могут возникать из предшественников клеток тканей желудочно-кишечного тракта. Главным образом, они возникают у населения среднего и старшего возраста. Примерно 70% опухолей возникают в желудке, 20-30% опухолей - в тонком кишечнике и менее 10% - в пищеводе, ободочной и прямой кишке. Хорошо известно, что гастроинтестинальные стромальные опухоли являются устойчивыми к воздействиям классической химиотерапии, однако относительно эффективным является ингибирование c-Kit с помощью Иматиниба, поскольку предполагается, что c-Kit играют решающую роль в патогенезе данных болезней (Джоенсу и др. Joensuu et al. N. Engl. J. Med. 2001, 344, 1052). c-Kit сверхэкспрессируются и мутирует и при других различных видах рака, включая мастоцит, нейробластому, герминому, меланому, многоклеточный рак легкого, рак груди, оофорому и острую миелоидную лейкемию (см. Эдлинг и др. Edling et al. Int. J. Biochem. Cell Biol. 2007, 39, 1995; Lennartsson et al. Curr. Cancer Drug Targets, 2006, 6, 65).
Помимо участия в развитии раковых заболеваний, SCF/c-Kit также связаны с аутоимунными или воспалительными заболеваниями. SCF экспрессируются различными структурными и воспалительными клетками дыхательных путей. Большое количество путей активируется путем комбинации SCF и c-Kit, включая пути, использующие Фосфоинозитид-3-киназу (PI3), фосфолипазу С (PLC)-гамма, Src протеинкиназу, янус-киназу (JAK)/переносчики сигнала и активаторы транскрипции (STAT), а также митоген-активируемую протеинкиназу (MAP). Суппресия SCF/c-Kit пути может существенно снизить уровень гистамина, уменьшить пенентрацию тучных клеток и эозинофилов, снизить высвобождение интерлейкина (IL)-4 и повышенную биологическую активность дыхательных путей. Поэтому SCF/c-Kit является потенциальным объектом воздействия, контролирующим тучные клетки и эозинофилы, а также контролирующим активацию аутоимунных или воспалительных заболеваний, включая дерматиты, ревматоидный артрит, аллергический ринит, астму, анкилозирующий спондилит, псориаз и болезнь Крона (см. Ребер и др. Reber et al. Eur. J. Pharmacol. 2006, 533, 327; Paniagua et al. Nat. Clin. Prac. Rheum. 2007, 3, 190).
Рецепторы фактора роста тромбоцитов (PDGFR), такие как PDGFR-α и PDGFR-β, являются трансмембранными тирозинкиназными рецепторами, чьи лиганды образованы двумя цепями (PDGF-A), или двумя цепями В (PDGF-B), или гетеродимером одной цепи А и одной цепи В (PDGF-AB). Рецепторы фактора роста тромбоцитов претерпевают димеризацию при связывании лигандов, с последующей активацией тирозинкиназы и передачей сигнала по нисходящей. Изучения на животных in-vivo на PDGFs и PDGFRs показали, что PDGFR-α сигнализация играет роль в развитии гаструляции, мозговых и сердечных ганглионарных пластин, гонад, легких, кишечного тракта, кожи, центральной нервной системы и костей. Также известна роль сигнализирования PDGFR-β в ангиогенезе и раннем гемопоэзе. Сигнализация рецепторов фактора роста тромбоцитов также связана с другими различными заболеваниями. Аутокринная активация путей сигнализации факторов роста связана с некоторыми видами глиоматоза мелких кровеносных сосудов, миелопролиферативными синдромами, опухолями, множественной миеломы, а также саркомой, включая выбухающую дерматофибросаркому. Паракринное сигнализирование факторов роста обычно обнаруживается при эпителиальном раке. Он инициирует ингаляцию матрицы и может участвовать в эпителиально-мезенхимальном переходе и поэтому иметь влияние на развитие опухолей, анигиогенеза, инвазию и метастаз. Факторы роста тромбоцитов обуславливают органические паталогические изменения сосудистых заболеваний, таких как артероматоз, стеноз артерии, легочная гипертензия, болезни сетчатки, а также гепатофиброзы, включая легочный интестинальный фиброз, цирроз печени, склеродермию, гломерулосклероз и фиброз миокарда (см. Андраэ и др. (Andrae et al. Gene Dev. 2008, 22, 1276)). Поэтому сверэкспрессия PDGFR может помочь предотвратить и лечить вышеуказанные заболевания. Кроме того, сверхэкспрессия PDGFR может также помочь в лечении различных аутоимунных и воспалительных болезней, включая диабет, в частности диабет типа I, ревматоидный артит, псориаз, болезнь Крона и др. (Паниагуа и др. (Paniagua et al. Nat. Clin. Prac. Rheum. 2007, 3, 190; Louvet et al. Proc. Natl. Acad. Sci. USA, 2008, 105, 18895)).
Изобретение также относится к новому типу производных дигидроинденамида, которые могут ингибировать активность протеинкиназ, особенно одной или нескольких киназ, описанных выше. Данные соединения, поэтому, будут полезными при предотвращении или лечении заболеваний, связанных с нарушением или расстройством активности протеинкиназ, особенно в случае заболеваний, связанных с нарушением активности Abl, Bcr-Abl, c-Kit и PDGFR протеинкиназ.
Краткое описание изобретения
Изобретение представляет собой соединения Формулы I

или их фармацевтически приемлемые соли, либо пролекарства, где
R1 является насыщенной циклической аминогруппой, которая может необязательно быть замещена 1, 2, 3 или 4 R1a;
R1a представляет собой Н, галоген, цианогруппу, C1-6алкил, С1-6-гидроксиалкил, С1-6-галоалкил, C1-6-цианоалкил, ORa, SRa, NRbRc, NRbC(O)Rd, NRbS(O)2Rd, C(O)NRbRc, C(O)Rd, C(O)ORa, S(O)2Rd, C2-6алкенил, C2-6алкинил, арил, гетероарил, циклоалкил или гетероциклоалкил, где указанный C1-6алкил, С2-6алкенил, С2-6алкинил, арил, гетероарил, циклоалкил и гетероциклоалкил могут быть необязательно замещены 1, 2 или 3 группами, независимо выбранными из цианогруппы, галогена, ORa, SRa, NRbRc, NRb(CO)Rd, NRbS(O)2Rd, C(O)NRbRc, S(O)2NRbRc, C(O)Rd, C(O)ORa, S(O)2Rd, галоалкила, C1-6гидроксиалкила, С1-6цианоалкила, арила, гетероарила, циклоалкила и гетероциклоалкила. Иначе, две группы R1a вместе с присоединенными к ним атомами могут образовывать циклоалкил или гетероциклоалкил 3, 4, 5, 6 или 7-членного кольца и могут необязательно быть замещены 1, 2 или 3 группами, выбранными независимо из цианогруппы, галогена, ORa, SRa, NRbRc, NRb(CO)Rd, NRbS(O)2Rd, C(О)NRbRc, S(O)2NRbRc, C(O)Rd, C(O)ORa, S(O)2Rd, C1-6галогеналкила, C1-6гидроксиалкила, C1-6цианоалкила, C2.6 алкенила, C2-6алкинила, арила, гетероарила, циклоалкила и гетероциклоалкила;
R2 представляет собой Н, галоген, цианогруппу, ORa, SRa, NRbRc, C1-6алкил, С1-6гидроксиалкил, C1-6галогеналкил, C1-6цианоалкил, С2-6алкенил, С2-6алкинил; Иначе, две R2 группы вместе с присоединенными к ним атомами могут образовывать циклоалкил и гетероциклоалкил, представляющий собой 3, 4, 5, 6 или 7-членное кольцо, и могут быть необязательно замещены 1, 2 или 3 группами, выбранными независимо из цианогруппы, галогена, ORa, SRa, NRbRc, NRb(CO)Rd, NRbS(O)2Rd, C(O)NRbRc, S(O)2NRbRc, C(O)Rd, C(O)ORa, S(O)2Rd, C1-6галогеналкила, C1-6гидроксиалкила, C1-6цианоалкила, С2-6алкенила и С2-6алкинила;
R3 является H, галогеном, цианогруппой, ORa, SRa, NRbRc, С1-6алкилом, С1-6гидроксиалкилом, С1-6галогеналкилом, C1-6цианоалкилом, С2-6алкенилом, С2-6алкинилом, циклоалкилом или гетероциклоалкилом. Иначе, две R3 группы вместе с присоединенными к ним атомами могут образовывать циклоалкил и гетероциклоалкил 3, 4, 5, 6 или 7-членное кольцо и могут быть необязательно замещены 1, 2 или 3 группами, выбранными независимо из цианогруппы, галогена, ORa, SRa, NRbRc, C1-6алкила, C1-6галогеналкила, C1-6гидроксиалкила, C1-6цианоалкила, C2-6алкенила и С2-6алкинила;
W-X представляет собой амидную связь;
Y является гетероарилом, который может быть необязательно замещен на 1, 2 или 3 R4;
Z является гетероциклоалкилом или гетероарилом, который может быть необязательно замещен 1, 2 или 3 R5;
R4 и R5 независимо друг от друга выбирают из галогена, цианогруппы, ORa, SRa, NRbRc, С1-6алкила, C1-6гидроксиалкила, C1-6галогеналкила, C1-6цианоалкила, С2-6алкенила, С2-6алкинила, NRb(CO)Rd, C(O)NRbRc, NRbS(O)2Rd, S(O)2NRbRc, C(O)Rd, C(O)ORa, S(O)2Rd, циклоалкила, гетероциклоалкила, арила и гетероарила. Альтернативно, две R4 или две R5 группы вместе с присоединенными к ним атомами могут образовывать циклоалкил или гетероциклоалкил, представляющий собой 5, 6 или 7-членное кольцо, и могут необязательно быть замещены 1, 2 или 3 группами, независимо выбранными из галогена, цианогруппы, ORa, SRa, NRbRc, С1-6алкила, С1-6галогеналкила, гидроксиалкила, C1-6цианоалкила, С2-6алкенила и С2-6алкинила;
Ra, Rb, Rc и Rd независимо друг от друга выбирают из Н, C1-6алкила, C1-6галогеналкила, C1-6гидроксиалкила, С1-6цианоалкила, С2-6алкенила, С2-6алкинила, циклоалкила, гетероциклоалкила, арила и гетероарила; Альтернативно, Rb и Rc группы вместе с присоединенными к ним атомами азота могут образовывать гетероциклоалкил, представляющий собой 4, 5, 6 или 7-членное кольцо, и могут быть необязательно замещены 1, 2 или 3 группами, независимо выбранными из галогена, цианогруппы, C1-6алкила, С1-6галогеналкила, С1-6гидроксиалкила, С1-6цианоалкила, С2-6алкенила, С2-6алкинила, циклоалкила, гетероциклоалкила, арила и гетероарила;
n является целым числом от нуля до четырех; m является целым числом от нуля до двух.
Среди многих соединений Формулы I, а также их солей или пролекарств предпочтительными являются соединения по настоящему изобретению Формулы II

или их фармацевтически приемлемые соли или пролекарства, где R1 представляет собой насыщенную циклическую аминогруппу, которую можно выбрать из пипиридинила, пиперазинила, пирролидинила, азетидинила и морфолинила, каждая из которых необязательно может быть замещена 1, 2, 3, или 4 R1a;
R1a представляет собой Н, галоген, цианогруппу, C1-6алкил, C1 _ 6гидроксиалкил, С1-6галогеналкил, С1-6цианоалкил, ORa, SRa, NRbRc, NRbC(O)Rd, NRbS(O)2Rd, C(O)NRbRc, C(O)Rd, C(O)ORa, S(O)2Rd, C2-6алкенил, C2-6алкинил, арил, гетероарил, циклоалкил или гетероциклоалкил, где указанные C1-6алкил, С2-6алкенил, С2-6алкинил, арил, гетероарил, циклоалкил и гетероциклоалкил могут быть необязательно замещены 1, 2 или 3 группами, выбранными независимо друг от друга из цианогруппы, галогена, ORa, SRa, NRbRc, NRb(CO)Rd, NRbS(O)2Rd, C(O)NRbRc, S(O)2NRbRc, C(O)Rd, C(O)ORa, S(O)2Rd, C1-6галогеналкила, C1-6гидроксиалкила, C1-6цианоалкила, арила, гетероарила, циклоалкила и гетероциклоалкила; Иначе, две R1a группы вместе с присоединенными к ним атомами могут образовывать циклоалкил или гетероциклоалкил 3, 4, 5, 6 или 7-членного кольца, а также могут быть необязательно замещены 1, 2 или 3 группами, выбранными независимо друг от друга из цианогруппы, галогена, ORa, SRa, NRbRc, NRb(CO)Rd, NRbS(O)2Rd, C(O)NRbRc, S(O)2NRbRc, C(O)Rd, C(O)ORa, S(O)2Rd, C1-6галогеналкила, C1-6гидроксиалкила, C1-6цианоалкила, С2-6алкенила, C2-6алкинила, арила, гетероарила, циклоалкила и гетероциклоалкила;
Y выбирают из пиридила, пиримидила, пиридазинила, пиразинила, триазинила, триазолила, изотиазолила, имидазолила, оксазолила, изоксазолила, триазолила или пиразолила, а также может быть необязательно замещен 1, 2, или 3 R4;
Z выбирают из пиридила, пиримидила, пиридазинила, пиразинила, триазинила, тиазолила, изотиазолила, имидазолила, оксазолила, изоксазолила, триазолила, пиразолила, азотистого оксазолила, пиридинола, пирроло-пиримидила, пиразоло-пиридила, пиразоло-пиримидила, хинолина, изохинолина, хиназолила, пиперазинила или морфолинила, а также могут быть необязательно замещены 1, 2 или 3 R5;
R4 и R5 выбирают независимо друг от друга из галогена, цианогруппы, ORa, SRa, NRbRc, C1-6алкила, C1-6гидроксиалкила, C1-6галогеналкила, C1-6цианоалкила, C2-6алкенила, С2-6алкинила, NRb(CO)Rd, C(O)NRbRc, NRbS(O)2Rd, S(O)2NRbRc, C(O)Rd, C(O)ORa, S(O)2Rd, циклоалкила, гетероциклоалкила, арила и гетероарила. Иначе, две R4 или R5 группы вместе с присоединенными к ним атомами могут образовывать циклоалкил или гетероциклоалкил, представляющий собой 5, 6 или 7-членное кольцо, а также могут быть необязательно замещены 1, 2 или 3 группами, выбранными независимо из галогена, цианогруппы, ORa, SRa, NRbRc, C1-6алкила, C1-6галогеналкила, C1-6гидроксиалкила, C1-6цианоалкила, C2-6алкенила и С2-6алкинила;
Ra, Rb, Rc и Rd независимо друг от друга выбирают из Н, C1-6алкила, C1-6галогеналкила, С1-6гидроксиалкила, C1-6цианоалкила, С2-6алкенила, C2-6алкинила, арила, гетероарила, циклоалкила и гетероциклоалкила. Альтернативно, Rb и Rc вместе с присоединенными к ним атомами азота могут образовывать 4, 5, 6 или 7-членное кольцо и могут быть необязательно замещены 1, 2 или 3 группами, выбранными независимо из галогена, цианогруппы, алкила, C1-6галогеналкила, C1-6гидроксиалкила, C1-6цианоалкила, С2-6алкенила, С2-6алкинила, арила, гетероарила, циклоалкила и гетероциклоалкила;
Среди многочисленных соединений Формулы I, а также их солей и пролекарств наиболее предпочтительными соединениями по настоящему изобретению являются соединения Формулы IIa

или их фармацевтически приемлемые соли или пролекарства, где R6 и R7 независимо друг от друга выбирают из Н, галогена, цианогруппы, C1-6алкила, C1-6гидроксиалкила, С1-6галогеналкила, C1-6цианоалкила, С2-6алкенила, С2-6алкинила. Иначе, R6 и R7 вместе с присоединенными к ним атомами образуют карбоцикл или гетероцикл, представляющий собой 5, 6 или 7-членного кольца, и могут быть необязательно замещены 1, 2 или 3 группами, выбранными независимо друг от друга из галогена, цианогруппы, ORa, SRa, NRbRc, С1-6алкила, С1-6гидроксиалкила, C1-6галогеналкила, C1-6цианоалкила, С2-6алкенила и С2-6алкинила;
R8 представляет собой Н, С1-6алкил, С2-6гидроксиалкил, С2-6галогеналкил, галогеналкил, C(O)NRbRc, C(O)Rd, C(O)ORa, S(O)2Rd, C3-6алкенил, С3-6алкинил, арил, гетероарил, циклоалкил и гетероциклоалкил. Причем указанные C1-6алкил, С3-6алкенил, С3-6алкинил, арил, гетероарил, циклоалкил и гетероциклоалкил могут быть необязательно замещены 1, 2 или 3 группами, выбранными независимо друг от друга из галогена, цианогруппы, ORa, SRa и NRbRc;
Y выбирают из пиридила, пиримидила, пиридазинила, пиразинила, триазинила, триазолила, изотиазолила, имидазолила, оксазолила, изоксазолила, триазолила или пиразолила, а также он может быть необязательно замещен 1, 2, или 3 R4;
Z выбирают из пиридила, пиримидила, пиридазинила, пиразинила, триазинила, тиазолила, изотиазолила, имидазолила, оксазолила, изоксазолила, триазолила, пиразолила, азотистого оксазолила, пириндола, пирроло-пиримидила, пиразоло-пиридила, пиразоло-пиримидила, хинолина, изохинолина, хиназолила, пиперазинила или морфолинила, а также может быть необязательно замещен 1, 2 или 3 R5;
R4 и R5 выбирают независимо друг от друга из галогена, цианогруппы, ORa, SRa, NRbRc, С1-6алкила, C1-6гидроксиалкила, C1-6галогеналкила, C1-6цианоалкила, С2-6алкенила, С2-6алкинила, NRb(CO)Rd, C(O)NRbRc, NRbS(O)2Rd, S(O)2NRbRc, C(O)Rd, C(O)ORa, S(O)2Rd, циклоалкила, гетероциклоалкила, арила и гетероарила. Иначе, две R4 или R5 группы вместе с присоединенными к ним атомами могут образовывать циклоалкил и гетероциклоалкил, представляющий собой 5, 6 или 7-членное кольцо, а также могут быть необязательно замещены 1, 2 или 3 группами, выбранными независимо из галогена, цианогруппы, ORa, SRa, NRbRc, С1-6алкила, C1-6галогеналкила, C1-6гидроксиалкила, C1-6цианоалкила, C2-6алкенила и С2-6алкинила;
Ra, Rb, Rc и Rd независимо друг от друга выбирают из Н, C1-6алкила, C1-6галогеналкила, C1-6гидроксиалкила, C1-6цианоалкила, С2-6алкенила, С2-6алкинила, арила, гетероарила, циклоалкила и гетероциклоалкила. Иначе, Rb и Rc вместе с присоединенными к ним атомами азота могут образовывать гетероциклоалкил, представляющий собой 4, 5, 6 или 7-членное кольцо, и могут быть необязательно замещены 1, 2 или 3 группами, выбранными независимо из галогена, цианогруппы, C1-6алкила, C1-6галогеналкила, гидроксиалкила, С1-6цианоалкила, С2-6алкенила, С2-6алкинила, арила, гетероарила, циклоалкила и гетероциклоалкила;
p представляет собой целое число от нуля до двух.
Среди множества соединений Формулы I, а также их солей и пролекарств, другими наиболее предпочтительными соединениями по настоящему изобретению являются вещества формулы

или их фармацевтически приемлемые соли или пролекарства, где R9 и R10 независимо друг от друга выбирают из Н, C1-6алкила, С2-6гидроксиалкила, С2-6галогеналкила, C1-6галогеналкила, C(O)NRbRc, C(O)Rd, C(O)ORa, S(O)2Rd, C3-6алкенила, С3-6алкинила, арила, гетероарила, циклоалкила и гетероциклоалкила. Причем указанный C1-6алкил, С3-6алкенил, С3-6алкинил, арил, гетероарил, циклоалкил и гетероциклоалкил независимо друг от друга могут быть замещены 1, 2 или 3 группами, независимо выбранными из галогена, цианогруппы, ORa, SRa и NRbR°. Иначе, R9 и R10 вместе с присоединенными к ним атомами могут образовывать циклоалкил или гетероциклоалкил, представляющий собой 5, 6 или 7-членное кольцо, и могут быть необязательно замещены 1, 2 или 3 группами, выбранными независимо из галогена, цианогруппы, ORa, SRa, NRbRc, С1-6алкила, C1-6гидроксиалкила, C1-6галогеналкила, C1-6цианоалкила, C2-6алкенила, С2-6алкинила, арила, гетероарила, циклоалкила и гетероциклоалкила;
R11 представляет собой Н, галоген, цианогруппу, ORa, SRa, NRbRc, C1-6алкил, C1-6гидроксиалкил, C1-6галогеналкил, C1-6цианоалкил, С2-6алкенил, С2-6алкинил;
Y выбирают из пиридила, пиримидила, пиридазинила, пиразинила, триазинила, тиазолила, изотиазолила, имидазолила, оксазолила, изоксазолила, триазолила или пиразолила, а также он может быть необязательно замещен 1, 2 или 3 R4;
Z выбирают из пиридила, пиримидила, пиридазинила, пиразинила, триазинила, тиазолила, изотиазолила, имидазолила, оксазолила, изоксазолила, триазолила, пиразолила, азотистого оксазолила, пиридинола, пирроло-пиримидила, пиразоло-пиридила, пиразоло-пиримидила, хинолина, изохинолина, хиназолила, пиперазинила или морфолинила, а также он может быть необязательно замещен 1, 2 или 3 R5;
R4 и R5 выбирают независимо друг от друга из галогена, цианогруппы, ORa, SRa, NRbRc, C1-6алкила, гидроксиалкила, C1-6галогеналкила, C1-6цианоалкила, C2-6алкенила, C2-6алкинила, NRb(CO)Rd, C(0)NRbRc, NRbS(O)2Rd, S(O)2NRbRc, C(O)Rd, C(O)ORa, S(O)2Rd, циклоалкила, гетероциклоалкила, арила и гетероарила. Иначе, две R4 или R5 группы вместе с присоединенными к ним атомами могут образовывать циклоалкил или гетероциклоалкил, представляющий собой 5, 6 или 7-членное кольцо, а также могут быть необязательно замещены 1, 2 или 3 группами, выбранными независимо из галогена, цианогруппы, ORa, SRa, NRbRc, С1-6алкила, C1-6галогеналкила, C1-6гидроксиалкила, C1-6цианоалкила, С2-6алкенила и С2-6алкинила;
Ra, Rb, Rc и Rd независимо друг от друга выбирают из Н, C1-6алкила, С1-6галогеналкила, C1-6гидроксиалкила, С1-6цианоалкила, С2-6алкенила, С2-6алкинила, арила, гетероарила, циклоалкила и гетероциклоалкила. Иначе, Rb и Rc вместе с присоединенными к ним атомами азота могут образовывать гетероциклоалкил, представляющий собой 4, 5, 6 или 7-членное кольцо, и могут быть необязательно замещены 1, 2 или 3 группами, выбранными независимо из галогена, цианогруппы, C1-6алкила, C1-6галогеналкила, С1-6гидроксиалкила, C1-6цианоалкила, С2-6алкенила, С2-6алкинила, арила, гетероарила, циклоалкила и гетероциклоалкила;
q представляет собой целое число от нуля до трех.
Другим объектом настоящего изобретения является способ регулирования активности протеинкиназ, причем указанный способ включает воздействие на упомянутую протеинкиназу указанных выше соединений или фармацевтически приемлемых солей или их пролекарств.
Предпочтительно, упомянутые протеинкиназы выбирают из Alb, Bcr-Abl, c-Kit и PDGFR. Также указанные протеинкиназы включают мутантные киназы, выбранные из мутантных Abl-киназы, Bcr-Abl киназы, c-Kit киназы и PDGFR киназы.
Другим объектом изобретения является использование упомянутых выше соединений или их фармацевтически приемлемых лекарств для производства медикаментов для лечения заболеваний или расстройств, связанных с активностью протеинкиназ или ненормальной пролиферацией клеток.
Еще одним объектом изобретения является способ лечения заболеваний или расстройств пациентов, связанных с активностью киназ, включающий введение эффективных доз вышеупомянутых соединений или их фармацевтически приемлемых солей либо пролекарств пациентам.
Подробное описание изобретения
Ниже будут описаны иллюстративные примеры. Однако данные воплощения являются исключительно демонстрационными и не ограничивают объем настоящего изобретения.
Следует применять следующие используемые в настоящем описании определения, если не указано иное.
«Галоген» включает фтор (F), хлор (Cl), бром (Br) и иод (I).
«Алкил» относится к линейным или разветвленным насыщенным углеводородным группам. Примеры алкилов включают С1-20алкилы, предпочтительно С1-6алкилы, такие как метил (Me), этил (Et), пропил (такие как н-пропил и изопропил), бутил (такие как н-бутил, изобутил и трет-бутил), амил (такие как н-амил, изоамил и неоамил), н-гексил и т.д. В каждой замещенной алкил- или алкил-замещенной группе, упомянутой выше, «алкил» имеет такое же значение, как описано выше.
«Гидроксиалкил» относится к алкилу, замещенному гидроксилом.
«Галогеналкил» относится к алкилу, замещенному одним или несколькими галогенами, таким как CH2F, CHF2, CF3, C2F5, CCl3 и т.д.
«Цианоалкил» или «цианозамещенный алкил» относится к алкилу, замещенному циано-группой.
«Алкенил» относится к алкилам, содержащим одну или несколько двойных связей углерод-углерод, таким как винил, пропенил, 1,3-бутадиенил, цис-бутенил, транс-бутенил и т.д.
«Алкинил» относится к алкилам, содержащим одну или несколько тройных связей углерод-углерод, таким как ацетиленил, пропинил и т.д.
«Циклоалкил» относится к неароматическому углеродному кольцу, включая циклоалкил, циклоалкенил и циклоалкинил. Циклоалкил может содержать мноциклическую или полициклическую кольцевую систему (такую как 2, 3 или 4 сконденсированных кольца), включая спироциклы. Циклоалкил может содержать 3-20 атомов углерода, а также как и 0, 1, 2 или 3 двойных связи и/или 0, 1 или 2 тройных связи. Циклоалкил также может включать одно кольцо или несколько конденсированных ароматических колец (т.е. с общей связью), например пентан, пентен, гексан и подобные замещенные производными бензола. Циклоалкил, содержащий одно или несколько конденсированных ароматических колец, может быть присоединен к другим группам через группу, входящую в состав ароматического кольца, или группу, не входящую в ароматическое кольцо. Примеры циклоалкилов включают циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклопентенил, циклогексенил, циклогексадиенил, циклогептатриенил, циклогексил, циклогептил, циклопентенил, циклогексенил, циклогексадиенил, циклогептатриенил, адамантил и т.д.
«Гетероциклоалкил» относится к неароматическому кольцу, где один или несколько атомов кольца являются гетероатомами, такими как N, О или S. Гетероциклоалкил может включать моноциклическую или полициклическую кольцевую систему (например, содержащую 2, 3 или 4 конденсированных кольца), включая спироциклы. Предпочтительные примеры гетероциклоалкилов включают, но не ограничиваются, азиридин, азетидин, тетрагидрофуран, тетрагидротиофен, пирролидин, оксазолидин, тиазолидин, имидазолидин, изоксазолидин, изотиазолидин, пиразолидин, морфолин, тиоморфолин, пиперазин, пиперидин и т.д. Гетероциклоалкил может также включать одно гетероциклическое кольцо или несколько конденсированных ароматических колец (т.е. с общей связью), например 2, 3-дигидробензофуран, 1,3-бензодиоксолан, бензо-1,4-диоксан, метилфталамид и нафталамид. Гетероциклоалкил, содержащий одно или несколько конденсированных ароматических колец, может быть присоединен к другим группам через группировку, входящую в ароматическое или неароматическое кольцо.
«Ароматическое кольцо» относится к моноциклическому или полициклическому (такому как 2, 3 или 4 конденсированных кольца) ароматическому углеводороду, такому как бензол, нафталин, антрацен, фенантрен и т.д.
«Гетеро-ароматическое кольцо» относится к ароматическим гетероциклам, включающим по меньшей мере один или несколько гетероатомов в кольце, таких как S, О или N. Гетероароматическое кольцо может содержать моноциклическую или полициклическую кольцевую систему (например, содержащую 2, 3 или 4 конденсированных кольца). Любой атом азота в гетероароматическом кольце может быть окислен с образованием оксида азота. Предпочтительные гетероароматические кольца включают, но не ограничиваются ими: пиридин, пиримидин, пиразин, пирадизин, триазин, фуран, тиофуран, имидазол, триазол, тетразол, тиазол, изотиазиол, 1,2,4-тиадиазол, пиррол, пиразол, оксазол, изоксазол, оксадиазол, бензофуран, бензотиофен, бензотиазол, индол, индазол, хинолин, изохинолин, пирин, карбазол, бензимидазол, пириндол, пирроло-пиримидин, пиразоло-пиридин, пиразоло-пиримидин и т.д.
«Необязательно» означает, что случай или ситуация, описываемые далее, могут иметь место или могут не иметь места. Упомянутое определение включает примеры случаев или ситуаций, описываемых в данном документе, когда они происходят или не происходят.
«Эффективная терапевтическая доза» относится к введению эффективного количества соединений формулы млекопитающему в случае необходимости подобного лечения. Эффективная терапевтическая доза может быть изменена, в зависимости от специфической активности медикамента, а также возраста, физиологического состояния, наличия других заболеваний и пищевого статуса пациента. Кроме того, определение применяемой эффективной терапевтической дозы зависит от другой возможной терапии, получаемой пациентом в данный промежуток времени.
«Лечение» означает любую терапию для лечения заболеваний млекопитающих, включая:
(i) предотвращение заболевания, т.е. предотвращение возникновения клинических симптомов заболевания;
(ii) подавление заболевания, т.е. предотвращение развития клинических симптомов заболевания, и/или
(iii) облегчение заболевания, т.е. достижение устранения клинических симптомов.
Во многих случаях соединение по настоящему изобретению может находится в виде кислой и/или основной соли вследствие наличия амино и/или карбоксильной группы или подобной.
«Соединение» в настоящем документе относится ко всем стереоизомерам, геометрическим изомерам, динамическим изомерам и изотопам.
Соединение по настоящему изобретению может быть ассиметричным, например, содержащим один или несколько стереоизомеров. Если не указано иное, включаются все стереоизомеры, такие как энантиомеры и диастереомеры. Соединение, содержащее асимметрически замещенный атом углерода, можно выделить в оптически активной чистой или рацемической форме. Оптически активную форму можно выделить из рацемической смеси или синтезировать, используя хиральные материалы или хиральные реагенты.
Соединения по настоящему изобретению также включают динамические изомеры. Динамические изомеры получают путем обмена между одинарной и прилегающей двойной связью, сопровождающееся миграцией протона.
Соединение по настоящему изобретению также включает конечное соединение или его промежуточное, включающее атомы изотопов. Атомы изотопов обладают таким же атомным номером, но различаются массовым числом. Например, изотопы водорода включают дейтерий и тритий.
Соединение по настоящему изобретению также включают фармацевтически приемлемые соли, получаемые путем конвертации основной группы исходного соединения в форму соли. Фармацевтически приемлемые соли включают, но не ограничиваются ими: неорганические или органические кислые соли основных групп, таких как цианамид. Фармацевтически приемлемые соли согласно данному документу можно синтезировать из исходного соединения, т.е. основная группа исходного соединения взаимодействует с 1-4 эквивалентами кислоты в системах растворителей. Подходящие соли перечислены в Remington's Pharmaceutical Science, 17th ed, Mark Publishing Company, Easton, Pa, 1985, p.1418 and Journal of Pharmaceutical Science, 66, 2 (1977).
Фармацевтически приемлемые соли присоединения кислоты можно получить из неорганических или органических солей. Соли, получаемые добавлением кислоты, можно получить из неорганических солей, включая соляную кислоту, бромоводородную кислоту, серную кислоту, азотную кислоту, фосфорную кислоту и т.д. Соли присоединения кислоты можно получить из органических кислот, включая уксусную кислоту, пропионовую кислоту, гликолевую кислоту, пировиноградную кислоту, щавелевую кислоту, яблочную кислоту, малоновую кислоту, янтарную кислоту, малеиновую кислоту, фумаровую кислоту, винную кислоту, лимонную кислоту, бензойную кислоту, коричную кислоту, миндальную кислоту, метансульфоновую кислоту, этилсульфоновую кислоту, толуол-п-сульфоновую кислоту, салициловую кислоту и т.д.
Используемый в настоящем изобретении термин «фармацевтически приемлемые носители» включает какие-либо и все растворители, дисперсные среды, покрытия, антибактериальные или противогрибковые агенты, изотонические агенты или средства, замедляющие абсорбцию, и подобные. Такие среды и агенты, используемые в фармацевтически активных соединениях, хорошо известны из уровня техники. Их использование в терапевтических композициях является предсказуемым, за исключением случаев, когда какие-либо обычные среды или агенты являются несовместимыми с активными веществами. Также в композиции можно включать дополнительные активные ингредиенты.
Описываемые в данном документе композиции предпочтительно готовить в виде единичной лекарственной формы. Термин «единичная лекарственная форма» относится к физически отдельной единице однократной дозы, которая подходит для введения человеку или другому млекопитающему. С целью достижения требующегося эффективного лечения, исходя из расположения, каждая единица содержит заранее определенное количество активных веществ, а также соответствующие подходящие фармацевтические эксципиенты (такие как таблетки, капсулы, ампулы). Соединения Формулы I являются эффективными в широком диапазоне доз и обычно вводятся в эффективном количестве. Предпочтительно, в отношении перорального введения, каждая единица дозы может содержать от 10 мг до 2 г соединения Формулы I, более предпочтительно от 10 до 700 мг; в то время как по отношению к парентеральному введению каждая единица дозы может содержать от 10 мг до 700 мг; более предпочтительно от 50 мг до 200 мг. Однако, следует подчеркнуть, что действительное вводимое количество соединений Формулы I определяется врачом, исходя из определенных условий, включая разновидность излечиваемой болезни, выбранный способ введения, конкретное соединение и его относительную активность, а также возраст, массу тела, отклик и тяжесть симптомов заболевания у пациента и т.п.
Для получения твердых композиций, таких как таблетки, основные активные компоненты смешивают с фармацевтическим эксципиентами (или носителями) с образованием твердой предварительной композиции, где содержится гомогенная смесь соединений по настоящему изобретению. Когда данную предварительную композицию называют гомогенной смесью, подразумевают, что активные компоненты равномерно распределены по всей композиции, что позволяет легко разделить композицию на единичные лекарственные формы с одинаковой эффективностью, такие как таблетки, пилюли или капсулы.
Таблетки или пилюли по настоящему изобретению могут быть покрытыми или сформированы в другие образцы, с целью получения лекарственной формы, обладающей преимуществом продления эффективности, или обеспечения защиты таблетки или пилюли от кислотной среды желудка. Например, таблетка или пилюля может включать компоненты внутренней и внешней дозы, где последняя существует в виде покрытия сверху. Указанные два типа компонентов могут отделяться энтеросолюбильным слоем, предотвращающим распад в желудке и позволяющим внутреннему компоненту целиком проникнуть в двенадцатиперстную кишку или замедленно высвобождаться. В качестве энеросолюбильного слоя или покрытия можно использовать разнообразные материалы, указанные материалы включают полимерные кислоты, а также смеси полимерных кислот и следующих материалов, таких как шеллак, гексадеканол и ацетатцеллюлозу.
Композиции, используемые для ингаляций и инсуффляций, включают фармацевтически приемлемые водные или органические растворители или растворы и суспензии смеси, а также порошки. Жидкие или твердые композиции могут содержать подходящие фармацевтические эксципиенты, упомянутые выше. Предпочтительно, данные композиции вводятся перорально или назально с целью достижения частичного или системного воздействия. Данные композиции в предпочтительных фармацевтически приемлемых растворителях можно распылять с помощью инертных газов: распыляемый раствор можно поместить непосредственно в устройство для распыления либо устройство распыления можно подсоединить к дыхательной маске или в импульсный агрегат для создания положительного давления. Композиции растворов, суспензий или порошков можно вводить с помощью устройства подходящим образом доставки лекарственных форм, предпочтительно пероральным или назальным.
В настоящем изобретении соединения и их фармацевтически приемлемые соли также включают формы сольватов или гидратов. В общем, формы сольватов или гидратов равноценны формам не-сольватов и не-гидратов и входят в объем настоящего изобретения. Некоторые соединения по настоящему изобретению могут вероятно существовать в виде поликристаллов или быть аморфными. Вкратце, все физические формы можно использовать равноценно, и все они входят в объем настоящего изобретения.
Настоящее изобретение также включает пролекарства соединений. Пролекарство представляет собой фармацевтическое вещество, полученное из исходного лекарства, и при попадании в организм посредством метаболизма переходит в исходное лекарство. Пролекарство можно получить путем введения одной или нескольких функциональных групп в исходное лекарство, которое высвободится при распаде замещающих групп in-vivo. Получение и использование пролекарств можно найти в документе Т.Хигучи и В.Стелла (Т.Higuchi and V.Stella), «Pro-drugs as Novel Delivery Systems", Vol.14 of the A.C.S. Symposium Series, and Bioreversible Carriers in Drug Design, ed. Edward B. Roche, American Pharmaceutial Association and Pergmon Press, 1987.
Настоящее изобретение также предлагает фармацевтические композиции, содержащие соединения формулы I или их фармацевтически приемлемые соли либо пролекарства и по меньшей мере один тип фармацевтически приемлемого носителя. Фармацевтические композиции по настоящему изобретению можно вводить перорально, инъекцией, спреем в виде ингаляций, подкожно, ректально, назально, вагинально, в брюшную полость, в виде внедрения или с помощью трансдермального пластыря и т.д.
С другой стороны, настоящее изобретение также предлагает способ регулирования активности протеинкиназы с помощью соединения Формулы I. Термин «регулирование киназной активности» в настоящем описании означает снижение активности протеинкиназы в некоторой степени при влиянии на киназу соединений дигдироинденамида по настоящему изобретению, по сравнению с активностью без применения соединений. Поэтому настоящее изобретение предлагает способ регулирования активности протеинкиназы путем воздействия на протеинкиназу соединений дигидроинденамида.
В частности, протеинкиназы, описанные в настоящем изобретении, представляют собой протеин-тирозинкиназы, включая Abl, Bcr-Abl, c-Kit и PDGFR.
Кроме того, протеинкиназы в настоящем изобретении включают также мутантные киназы, такие как Abl и Bcr-Abl киназы, мутантные c-Kit киназы и мутантные PDGFR киназы. Мутантные Abl и Bcr-Abl киназы включают, например, один или несколько следующих мутантов: M244V, G250E, Q252H, Y253F, Y253H, Е255К, E255V, F311L, T351I, F317L, М351Т, F359V, V379I, L387M, Н396Р, H396R и т.д.
С другой стороны, настоящее изобретение предлагает способ лечения заболеваний или расстройств, позволяющий регулировать активность протеинкиназы. Заболевания или расстройства, связанные с активностью протеинкиназы, включают рак, воспаления, аутоиммунные заболевания, расстройства метаболизма, инфекции, расстройства центральной нервной системы, сердечно-сосудистые заболевания и т.п.
Другим объектом настоящего изобретения являются соединения, описываемые в данном документе, которые можно использовать для лечения заболеваний или расстройств, связанных с абнормальной пролиферацией клеток, таких как рак, включая лейкемию, миелопролиферативный синдром, гематоз, гастроинтестинальная стромальная опухоль, рак толстой кишки, рак груди, рак желудка, оофорома, рак шейки матки, рак легкого, рак почек, рак простаты, рак мочевого пузыря, рак поджелудочной железы, нейробластома, опухоль тучных клеток, опухоль мозга, герминома, меланома, злокачественные образования, саркома, такая как выбухающая дерматофибросаркома, и т.п.
Другим объектом настоящего изобретения является возможность использования описываемых здесь соединений для лечения заболеваний, связанных с аутоиммунными расстройствами или воспалительными заболеваниями, включая диабеты, дерматит, ревматоидный артрит, аллергический ринит, астму, анкилозирующий спондилит, псориаз, болезнь Крона и т.д.
Другим объектом настоящего изобретения является возможность лечения сосудистых заболеваний, таких как атероматоз, стеноз кровеносного сосуда, легочная гипертензия, заболевания сетчатки, а также фиброзов, таких как легочный межуточный фиброз, гепатофиброз, цирроз печени, склеродермия, гломерулосклероз, фиброз миокарда и т.д.
Еще одним объектом настоящего изобретения являются способы получения соединений Формулы I. Соединения по настоящему изобретению можно получить с помощью следующих способов и процедур.

Промежуточное соединение формулы 1-5 можно получить согласно Схеме 1. 5-бромо-2,3-дигидроинден-1-он и CuCN кипятят в ДМФ с получением цианового интермедиата 1-1. Интермедиат 1-1 можно восстановить в промежуточное соединение 1-2, являющееся спиртом, взаимодействием с восстановителем, таким как борогидрид натрия, в растворителе, таком как метанол. Промежуточное соединение 1-2 может взаимодействовать с тионилхлоридом, что приведет к образованию хлор-группы, которую можно заместить циклической аминогруппой в присутствии триэтиламина или карбоната калия с получением интермедиата 1-4. Цианогруппу в интермедиате 1-4 можно подвергнуть гидролизу с образованием карбоновой кислоты, которую далее обрабатывают метанолом и тионилхлоридом с получением соединения 1-5. Два энантиомера соединения 1-4 или соединения 1-5 можно разделить хиральной высокоэффективной жидкостной хроматографией или кристаллизацией с использованием камфорсульфоновой кислоты.

В другом случае R1-замещенную 2,3-дигидроинденовую карбоновую кислоту (или ее эфир) можно получить способом, изображенным на Схеме 2. 5-бром-2,3-дигидроинден-1-он можно восстановить в промежуточное спиртовое соединение 2-1 взаимодействием с восстановителем, таким как борогидрид натрия, в растворителе типа метанола. После гидрокси-группу промежуточного соединения 2-1 можно перевести в хлоро-группу с помощью тионилхлорида, хлорид 2-2 можно заместить циклической аминогруппой, используя триэтиламин или карбонат калия в качестве основания, с получением интермедиата 2-3. Интермедиат 2-3 взаимодействует с СО в присутствии палладия, например диацетата палладия/1,3-бис-(фенилфосфин) пропана (дппп) или бис-(трифенилфосфин) палладий дихлорида (II) [(PPh3)2PdCl], в качестве катализатора с образованием промежуточного соединения формулы 2-4 в виде смеси двух энантиомеров. Если R в соединении 2-4 является Н, соединение 2-4 можно получить взаимодействием соединения 2-3 с бутиллитием, с последующим гашением диоксидом углерода. Соединение 2-3 и два энантиомера соединения 2-4 можно разделить хиральной высокоэффективной жидкостной хроматографией или способами с использованием хиральных кислот, таких как кристаллизация с помощью камфорсульфокислоты.

Конечные соединения формулы 3-4 можно получить, как показано на Схеме 3. Карбоновый эфир интермедиата 3-1 можно подвергнуть щелочному гидролизу, например в присутствии гидроксида натрия, с образованием карбоновой кислоты 3-2, которую далее конденсируют с производным анилина, получая конечное соединение формулы 3-4 с помощью связующего агента, такого как бензотриазол-1-илокситрис(диметиламино)фосфоний гексафторфосфат (БОФ) или O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфат (ГАТУ). Кроме того, карбоновую кислоту 3-2 можно обработать тионилхлоридом, получив хлорид кислоты 3-3, который далее взаимодействует с производным анилина, с образованием соединения формулы 3-4. Конечные соединения формулы 3-4 можно также получить реакцией между эфиром 3-1 и производным анилина в присутствии триалкилалюминия типа триметилалюминия или триэтилалюминия в качестве связующего агента.
Пример 1
Получение 1-(4-метилпиперазин-1-ил)-N-(4-метил-3-[(4-пиридин-3-илпиримидин-2-ил)амино]фенил)-2,3-дигидро-1H-инден-5-карбоксамида.

Стадия А: 1-оксо-2,3-дигидро-1Н-инден-5-карбонитрил

5-бромо-2,3-дигидро-1H-индин-1-он (21,1 г, 100 ммоль) и цианид меди (17.9 г 200 ммоль) смешали в 200 мл диметилформамида и перемешивали в течение ночи при 140°С. После того как раствор охладили до комнатной температуры, добавляли 500 мл этилацетата и осадок отфильтровали с использованием кизельгура. Осадок промывали несколько раз этилацетатом. Объединенный фильтрат дважды промывали 1 н. соляной кислотой и далее трижды солевым раствором, после сушили над безводным сульфатом магния, отфильтровывали и концентрировали. Неочищенный продукт очищали на силикагеле, элюируя смесью этилацетат/гексан (1:2), с получением 7,9 г требуемого соединения (50% выход). Данные масс-спектроскопии MS(M+1)=158,05.
Стадия В: 1-гидрокси-2,3-дигидро-1Н-инден-5-карбонитрил

1-оксо-2,3-дигидро-1H-инден-5-карбонитрил (7,85 г, 50 ммоль) растворяли в 50 мл метанола. К смеси постепенно добавляли боргидрид (2,3 г, 60 ммоль) в течение 30 мин. После 2 часов перемешивания раствор концентрировали. Остаток растворяли в этилацетате и полученный раствор дважды промывали бикарбонатом натрия, затем трижды солевым раствором и сушили над сульфатом магния, далее фильтровали и концентрировали, с получением 8 г желаемого соединения (100% выход). Данные масс-спектроскопии MS(M+1)=160,07.
Стадия С: 1-(4-метилпиперазин-1-ил)-2,3-дигидро-1Н-инден-5-карбонитрил

1-гидрокси-2,3-дигидро-1H-инден-5-карбонитрил (4,77 г, 30 ммоль) растворяли в 10 мл дихлорметана. При охлаждении льдом добавляли по каплям тионилхлорид (6,6 мл, 90 ммоль) в течение 15 минут. Раствор после перемешивания в течение 3 часов концентрировали. Остаток растворяли в этилацетате и полученный раствор трижды промывали охлажденным солевым раствором, сушили над безводным сульфатом магния и концентрировали с получением 1-хлор-2,3-дигидро-1H-инден-5-карбонитрила.
Полученный 1-хлор-2,3-дигидро-1Н-инден-5-карбонитрил растворяли в 80 мл ацетонитрила и далее добавили 1-метил пиперазин (6 г, 60 ммоль) и карбонат калия (4,14 г, 30 ммоль). После раствор перемешивали в течение ночи при 60°С, ацетонитрил удаляли концентрированием при пониженном давлении. Далее добавляли этилацетат. Полученный раствор трижды промывали солевым раствором, сушили над сульфатом магния, концентрировали и очищали на силикагеле, используя в качестве элюента 5% метанол/дихлорметан, получив, таким образом, 4,3 г целевого соединения (выход 60%). Данные масс-спектроскопии MS(M+1)=242,16.
Стадия D: Метил 1-(4-метилпиперазин-1-ил)-2,3-дигидро-1H-инден-5-карбоксилат

1-(4-метилпиперазин-1-ил)-2,3-дигидро-1Н-инден-5-карбонитрил (2,41 г, 10 ммоль) растворяли в 10 мл 2 н. раствора гидроксида натрия. Раствор перемешивали в течение ночи при 100°С и затем концентрировали. Полученный после вакуумной перегонки остаток растворяли в 30 мл метанола. По каплям при перемешивании в течение 1 часа добавляли тионилхлорид (3,3 мл). Смесь кипятили с обратным холодильником в течение ночи и затем концентрировали. Сперва добавляли воду, далее для перевода раствора в основной добавляли карбонат калия. Раствор трижды экстрагировали этилацетатом. Объединенные экстракты промывали солевым раствором, сушили над сульфатом магния, затем концентрировали. Далее выполняли очистку на силикагелевой колонке, используя в качестве элюента 5% метанол/дихлорметан, получив 2,1 г (выход 77%) целевого вещества. Данные масс-спектроскопии MS(M+1)=275,17.
Стадия Е: Получение 1-(4-метилпиперазин-1-ил)-N-(4-метил-3-[(4-пиридин-3-ил пиримидин-2-ил)амино]фенил)-2,3-дигидро-1H-инден-5-карбоксамидеамида

Метил 1-(4-метилпиперазин-1-ил)-2,3-дигидро-1Н-инден-5-карбоксилат (1,37 г, 5 ммоль) и 4-метил-N(3)-(4-пиридин-3-илпиримидин-2-ил)фенил-1,3-диамин (Сзакацс и др. (Szakacs et al). J. Med. Chem. 2005, 48:249) (1,66 г, 6 ммоль) растворяли в 30 мл толуола. Далее добавляли 2 М раствор триметилалюминия в толуоле (5 мл, 10 ммоль) и смесь перемешивали в течение ночи при 50°С. Реакция прошла не полностью. Далее добавляли другую порцию 2 М триметилалюминия в толуоле (3 мл, 6 ммоль). Смесь охлаждали во льду и перемешивали в течение ночи при 60°С. При перемешивании добавляли водный насыщенный раствор тартрата калия и натрия (50 мл). Раствор экстрагировали дихлорметаном (3×100 мл). Объединенные экстракты промывали бикарбонатом натрия (100 мл) и далее солевым раствором (2×100 мл), сушили над сульфатом магния, затем концентрировали. Очистку проводили на силикагелевой колонке, используя в качестве элюента 50% этилацетат/дихлорметаен/5-10% триэтиламина, с получением 1,5 г целевого соединения (выход 58%). Данные масс-спектроскопии MS(M+1)=520,27. 1H ЯМР (ДМСО (диметилсульфоксид)-d6, ppm): δ 10,10 (s, 1H); 9,20 (s, 1H); 8,95 (s, 1H); 8,62 (d, J=4,8 Гц, 1Н); 8,42 (d, J=4,8 Гц, 1Н); 8,40 (d, J=9,0 Гц, 1H); 8,00 (s, 1H); 7,75 (s, 1H); 7,72 (d, J=9,0 Гц, 1Н); 7,45 (dd, J=8,2 Гц, 4,8 Гц, 1Н); 7,40 (d, J=8,0 Гц, 1Н); 7,38 (d, J=4,8 Гц, 1Н); 7,28 (d, J=9,0 Гц, 1Н); 7,15 (d, J=9,0 Гц, 1Н); 4,26 (t, J=9,0 Гц, 1Н); 2,2-2,9 (m, 1Н); 2,15 (s, 3Н); 2,08 (s, 3Н); 2,0 (m, 2H).
Пример 2
Получение трет-бутил 4-{5-[({4-метил-3-[(4-пиридин-3-илпиримидин-2-ил)амино]фенил}амино)карбонил)-2,3-дигидро-1-H-инден-1-ил}пиперазин-1-карбоксилата
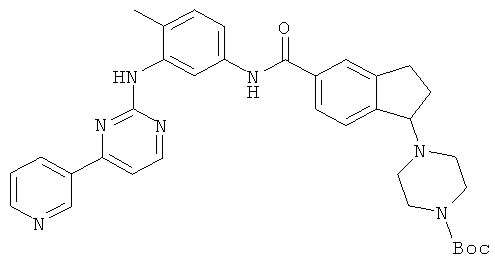
Стадия А: 5-бром-2,3-дигидро-1H-инден-1-ол

5-бром-2,3-дигидро-1H-инден-1-он (210 г, 1000 ммоль) суспендировали в 1 л метанола, далее постепенно в течение 1 часа добавляли боргидрид натрия (41,6 г, 1100 ммоль) при перемешивании. Растворитель упаривали при 50°С и пониженном давлении, после перемешивание продолжали еще в течение 1 часа. Далее добавляли этилацетат (1 л), затем добавляли насыщенный раствор бикарбоната натрия (500 мл). Перемешивание продолжали в течение некоторого времени, далее раствор переносили в делительную воронку, водную фазу удаляли. Органическую фазу дважды промывали насыщенным раствором бикарбоната натрия и дважды солевым раствором, затем сушили (сульфат магния) и, наконец, концентрировали, с получением 198 г (93%) целевого вещества.
Стадия В: 5-бром-1-хлор-2,3-дигидро-1Н-инден

5-бром-2,3-дигидро-1H-инден-1-ол (198 г, 934 ммоль) растворяли в 500 мл дихлорметана. При охлаждении льдом по каплям в течение 2 часов к раствору в дихлорметане добавляли тионилхлорид (275 мл, 3770 ммоль). Раствор концентрировали при 30°С и пониженном давлении и затем перемешивали в течение 2 часов при комнатной температуре. Остаток растворяли в этилацетате (1 л), и полученный раствор промывали ледяной водой (3×500 мл), затем солевым раствором (2×300 мл), сушили над сульфатом магния, затем концентрировали с получением 5-бром-1-хлор-2,3-дигидро-1H-индена.
Стадия С: трет-бутил 4-(5-бром-2,3-дигидро-1H-инден-1-ил) пиперазин-1-карбоксилат

5-бром-1-хлор-2,3-дигидро-1Н-инден (10 г, 43 ммоль) растворяли в 80 мл ацетонитрила, далее добавляли трет-бутил пиперазин-1-карбоксилат (9,7 г, 52 ммоль) и затем карбонат натрия (4,8 г, 45 ммоль). Смесь перемешивали в течение ночи при 60°С. Нерастворимый остаток отделяли фильтрацией, далее фильтрат концентрировали. Остаток отделяли на силикагелевой колонке, используя в качестве элюента этилацетат/гексан (1:2 до 1:1), с получением 12 г (выход 72%) целевого соединения. Данные масс-спектроскопии MS(M+1)=381,11, 383,11.
Стадия D: трет-бутил 4-[5-(этоксикарбонил)-2,3-дигидро-1H-инден-1-ил) пиперазин-1-карбоксилат

трет-бутил 4-(5-бром-2,3-дигидро-1Н-инден-1-ил) пиперазин-1-карбоксилат (11 г, 28,87 ммоль) растворяли в этаноле (50 мл), затем добавляли диметилсульфоксид (5 мл) и триэтиламин (5 мл). Систему вакуумировали и заполняли N2. Добавляли ацетат палладия (2 г) и 1,3-бис(дифенилфосфино)пропан (3 г). Систему вакуумировали и заполняли N2. Систему снова вакуумировали и перемешивали при 100°С в течение 24 часов с подключенными баллонами СО. После охлаждения до комнатной температуры смесь отфильтровывали с помощью кизельгура, который далее тщательно промывали этанолом. Фильтрат концентрировали. Остаток растворяли в этилацетате (500 мл), полученный раствор промывали солевым раствором (3×200 мл), сушили над сульфатом магния, концентрировали и, наконец, отделяли на силикагелевой колонке, используя в качестве элюента этилацетат/гексан (1:2 до 1:1), с получением 8,5 г (выход 79%) целевого соединения. Данные масс-спектроскопии MS(M+1)=375,22.
Стадия Е: 1-[4-(ВОС)пиперазин-1-ил]-2,3-дигидро-1H-инден-5-карбоновая кислота

Трет-бутил 4-[5-(этоксикарбонил)-2,3-дигидро-1H-инден-1-ил)пиперазин-1-карбоксилат (8 г, 21,36 ммоль) растворяли в 20 мл метанола, добавляли 30 мл гидроксида натрия (1 н.). Раствор перемешивали в течение ночи при комнатной температуре и еще два часа при 50°С, затем концентрировали. Остаток растворяли в воде (50 мл) и полученный раствор подкисляли до рН 5 с помощью 1 н. раствора HCl, далее экстрагировали этилацетатом (3×100 мл). Экстракты объединяли, сушили над сульфатом магния, далее концентрировали с получением целевого соединения. Данные масс-спектроскопии MS(M+1)=347,19.
Стадия F: трет-бутил 4-{5-[({4-метил-3-[(4-пиридин-3-илпиримидин-2-ил)амино]фенил}амино)карбонил]-2,3-дигидро-1Н-инден-1-ил}пиперазин-1-карбоксилат
1-[4-(ВОС)пиперазин-1-ил]-2,3-дигидро-1H-инден-5-карбоновую кислоту (7,4 г, 21,36 ммоль) и 4-метил-N(3)-[(4-пиридин-3-илпиримидин-2-ил)фенил-1,3-диамин (6,1 г, 22 ммоль) растворяли в 20 мл N,N-диметилформамида. Добавляли триэтиламин (8,9 мл, 64 ммоль) и O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфата (9,5 г, 25 ммоль). Раствор перемешивали в течение ночи при комнатной температуре, затем добавляли солевой раствор (100 мл) и этилацетат (200 мл). Водную фазу удаляли, слой этилацетата промывали солевым раствором (3×100 мл). Далее раствор сушили над сульфатом магния, концентрировали и, наконец, очищали на силикагелевой колонке, используя в качестве элюента метанол/метанхлорид (1:2 до 1:1), с получением 9,5 г (выход 73%) указанного соединения. Данные масс-спектроскопии MS(M+1)=606,31. 1H ЯМР (ДМСО-d6, ppm): δ 10,15 (s, 1H); 9,25 (s, 1H); 8,99 (s, 1H); 8,67 (d, J=4,8 Гц, 1H); 8,50 (d, J=5,2 Гц, 1H); 8,46 (d, J=8,4 Гц, 1H); 8,05 (s, 1H); 7,78 (s, 1H); 7,76 (d, J=8,0 Гц, 1Н); 7,50 (dd, J=8,0 Гц, 4,8 Гц, 1H); 7,46 (d, J=8,4 Гц, 1H); 7,41 (d, J=5,2 Гц, 1H); 7,38 (d, J=7,6 Гц, 1Н); 7,18 (d, J=8,8 Гц, 1H); 4,35 (t, J=7,2 Гц, 1H); 3,30 (m, 3Н); 3,05 (m, 1H); 2,08 (s, 2H); 2,42 (m, 2H); 2,30 (m, 2H); 2,20 (s, 3Н); 2,04 (m, 2H); 1,36 (s, 9H).
Пример 3
Получение N-(4-метил-3-[(4-пиридин-3-илпиримидин-2-ил)амино]фенил)-1-пиперазин-1-ил-2,3-дигидро-1Н-инден-5-карбоксамида

трет-бутил 4-{5-[({4-метил-3-[(4-пиридин-3-илпиримидин-2 ил)амино]фенил}амино)карбонил]-2,3-дигидро-1H-инден-1-ил}пиперазин-1 карбоксилат (2 г, 3,3 ммоль) растворяли в 4 н. растворе HCl в диоксане (10 мл). После перемешивания при комнатной температуре в течение 3 часов раствор концентрировали с получением твердого продукта. Продукт (100 мг) очищали высокоэффективной жидкостной хроматографией при рН=10 с получением целевого продукта. Данные масс-спектроскопии MS(M+1)=506,26. 1H ЯМР (ДМСО-d6, ppm): δ 10,08 (s, 1H); 9,20 (s, 1H); 8,93 (s, 1H); 8,62 (d, J=4,8 Гц, 1H); 8,44 (d, J=5,2 Гц, 1H); 8,40 (d, J=8,0 Гц, 1H); 8,00 (s, 1H); 7,72 (s, 1H); 7,70 (d, J=8,0 Гц, 1Н); 7,45 (dd, J=8,2 Гц, 4,8 Гц, 1Н); 7,41 (d, J=8,2 Гц, 1Н); 7,36 (d, J=5,2 Гц, 1Н); 7,31 (d, J=8,0 Гц, 1Н); 7,12 (d, J=8,8 Гц, 1Н); 4,22 (t, J=6,8 Гц, 1Н); 2,80 (m, 2H); 2,60 (m, 4H); 2,35 (m, 2H); 2,22 (m, 2H); 2,15 (s, 3H); 2,00 (m, 2H).
Пример 4
Получение 1-(4-этилпиперазин-1-ил)-N-(4-метил-3-[(4-пиридин-3-илпиримидин-2-ил)амино]фенил)-2,3-дигидро-1H-инден-5-карбоксамида

N-(4-метил-3-[(4-пиридин-3-илпиримидин-2-ил)амино]фенил)-1-пиперазин-1-ил-2,3-дигидро-1H-инден-5-карбоксамид тетрагидрохлорид (100 мг, 0,15 ммоль) растворяли в ДМФ (2 мл), далее добавляли триэтиламин (101 мг, 1 ммоль), а затем ацетальдегид (26 мг, 0.6 ммоль). После раствор перемешивали в течение 20 минут, далее добавляли триацетоксиборгидрид (128 мг, 0,6 ммоль). Полученный раствор перемешивали в течение ночи при комнатной температуре, затем очищали высокоэффективной жидкостной хроматографией при рН=10 с получением 50 мг (выход 63%) целевого соединения. Данные масс-спектроскопии MS(M+1)=523,29. 1H ЯМР (ДМСО-d6, ppm): δ 10,14 (s, 1Н); 9,25 (s, 1Н); 8,98 (s, 1Н); 8,67 (d, J=4,8 Гц, 1Н); 8,49 (d, J=5,2 Гц, 1Н); 8,46 (d, J=8,6 Гц, 1Н); 8,05 (s, 1Н); 7,77 (s, 1Н); 7,75 (d, J=8,8 Гц, 1Н); 7,50 (dd, J=8,0 Гц, 4,8 Гц, 1Н); 7,46 (d, J=8,2 Гц, 1Н); 7,41 (d, J=5,2 Гц, 1Н); 7,35 (d, J=7,6 Гц, 1Н); 7,17 (d, J=8,8 Гц, 1Н); 4,31 (t, J=6,8 Гц, 1Н); 2,2-3,0 (m, 12Н); 2,20 (s, 3H); 2, 03 (m, 2H); 0,95 (t, J=7,0 Гц, 3H).
Пример 5
Получение 1-(4-изопропилпиперазин-1ил)-N-(4-метил-3-[(4-пиридин-3-илпиримидин-2-ил)амино]фенил)-2,3-дигидро-1H-инден-5-карбоксамида

N-(4-метил-3-[(4-пиридин-3-илпиримидин-2-ил)амино]фенил)-1-пиперазин-1-ил-2,3-дигидро-1H-инден-5-карбоксамид тетрагидрохлорид (100 мг, 0,15 ммоль) растворяли в ДМФ (2 мл), далее добавляли триэтиламин (101 мг, 1 ммоль), затем ацетон (35 мг, 0,6 ммоль). После раствор перемешивали в течение 20 мин, затем добавляли триацетоксиборгидрид натрия (128 мг, 0,6 ммоль). Полученный раствор перемешивали в течение ночи при комнатной температуре и далее очищали высокоэффективной жидкостной хроматографией при рН=10 с получением 58 мг (выход 71%) указанного соединения. Данные масс-спектроскопии MS(M+1)=548.31. 1H ЯМР (ДМСО-d6, ppm): δ 10,14 (s, 1Н); 9,26 (s, 1Н); 8,98 (s, 1H); 8,67 (d, J=4,8 Гц, 1Н); 8,49 (d, J=4,8 Гц, 1Н); 8,46 (d, J=8,4 Гц); 8,05 (s, 1Н); 7,77 (s, 1Н); 7,74 (d, J=8,0 Гц, 1Н); 7,51 (dd, J=8,0 & 4,8 Гц, 4,8 Гц, 1Н); 7,46 (d, J=8,2 Гц, 1Н); 7,41 (d, J=5,2 Гц, 1Н); 7,35 (d, J=7,6 Гц, 1Н); 7,17 (d, J=8,4 Гц, 1Н); 4,30 (t, J=7,0 Гц, 1Н); 2,91 (m, 12Н); 2,81 (s, 3Н); 2,3-2,6 (m, 9Н); 2,02 (m, 2Н); 0,92 (t, J=6,4 Гц, 6Н).
Пример 6
Получение 1-[4-(2-гидроксиэтилпиперазин-1-ил)-N-(4-метил-3-[(4-пиридин-3-ил пиримидин-2-ил)амино]фенил)-2,3-дигидро-1H-инден-5-карбоксамида

N-(4-метил-3-[(4-пиридин-3-илпиримидин-2-ил)амино]фенил)-1-пиперазин-1-ил-2,3-дигидро-1H-инден-5-карбоксамид тетрагидрохлорид (100 мг, 0,15 ммоль) растворяли в ДМФ (2 мл), далее добавляли триэтиламин (101 мг, 1 ммоль) и {[трет-бутил (диметил)силил]оксо}ацетальдегид (100 мг, 0,6 ммоль). После раствор перемешивали в течение 20 минут, затем добавляли триацетоксиборгидрид натрия (128 мг, 0,6 ммоль). Полученный раствор перемешивали в течение ночи при комнатной температуре, далее очищали высокоэффективной жидкостной хроматографией. Высушенный продукт растворяли в 2 мл дихлорметана/2 мл трифторуксусной кислоты. Раствор концентрировали после перемешивания в течение ночи и очищали высокоэффективной жидкостной хроматографией при рН=10 с получением 38 мг (выход 46%) целевого соединения. Данные масс-спектроскопии MS(M+1)=550,29. 1H ЯМР (ДМСО-d6, ppm): δ 10,16 (s, 1H); 9,23 (s, 1H); 8,97 (s, 1H); 8,65 (d, J=4,4 Гц, 1H); 8,48 (d, J=6,0 Гц, 1H); 8,46 (d, J=4,8 Гц); 8,02 (s, 1H); 7,82 (m, 2Н); 7,51 (m, 1H); 7,40 (m, 2Н); 7,14 (d, J=8,4 Гц, 1H); 2,6-3,7 (m, 17Н); 2,16 (s, 3H).
Пример 7
Получение 1-[4-ацетилпиперазин-1-ил)-N-(4-метил-3-[(4-пиридин-3-илпиримидин-2-ил)амино]фенил)-2,3-дигидро-1H-инден-5-карбоксамида

N-(4-метил-3-[(4-пиридин-3-илпиримидин-2-ил)амино]фенил)-1-пиперазин-1-ил-2,3-дигидро-1H-инден-5-карбоксамид тетрагидрохлорид (100 мг, 0.15 ммоль) растворяли в ДМФ (2 мл), затем добавляли триэтиламин (101 мг, 1 ммоль) и ацетилхлорид (16 мг, 0,2 ммоль) при охлаждении в ледяной бане. После перемешивания в течение 20 минут полученный раствор очищали высокоэффективной жидкостной хроматографией при рН=10 с получением 45 мг (выход 55%) целевого соединения. Данные масс-спектроскопии MS(+1)=548,27. 1H ЯМР (ДМСО-d6, ppm): δ 10,15 (s, 1H); 9,26 (s, 1H); 8,99 (s, 1H); 8,66 (d, J=4,8 Гц, 1H); 8,49 (d, J=5,2 Гц, 1H); 8,45 (d, J=8,4 Гц); 8,05 (s, 1H); 7,79 (s, 1H); 7,76 (d, J=8,0 Гц, 1H); 7,50 (dd, J=8,0 & 4,8 Гц, 1H); 4,37 (t, J=7,0 Гц, 1H); 3,42 (m, 1H); 3,40 (m, 3Н); 2,91 (m, 1H); 2,83 (m, 1H); 2,2-2,5 (m, 4Н); 2,20 (s, 3Н); 2,06 (m, 2Н); 1,95 (s, 3H).
Пример 8
Получение N-[3-(4,5'-бипиримидин-2-иламино)-4-метилфенил]-1-(4-метилпиперазин-1-ил)-2,3-дигидро-1H-инден-5-карбоксамид

Стадия А: 5-ацетилпиримидин

5-бромпиримидин (3,18 г, 20 ммоль) растворяли в 50 мл тетрагидрофурана. При охлаждении до -78°С, по каплям при перемешивании добавляли 15 мл 1,6 М раствора н-бутиллития в гексане. После перемешивания раствора в течение 30 минут медленно добавляли раствор N-метоксил-N-метилацетамида (2,58 г, 25 ммоль) в растворе тетрагидрофурана (10 мл). Смесь перемешивали при -78°С в течение 1 часа, затем оставляли медленно нагреваться. Когда температура смеси достигала 0°С, добавляли водный раствор хлорида аммония. Полученный раствор трижды экстрагировали этилацетатом. Объединенные экстракты промывали солевым раствором, сушили над сульфатом магния, концентрировали при пониженном давлении, затем очищали на силикагелевой колонке, используя в качестве элюента 5% метанол/дихлорметана, с получением 1 г целевого вещества (выход 45%). Данные масс-спектроскопии MS(M+1)=123,05.
Стадия В: (2Е)-3-(Диметиламино)-1-пиримидин-5-илпроп-2-ен-1-он

5-Ацетилпиримидин (1 г, 8,2 ммоль) и N,N-диметилформамид диметилацеталь (1,3 г, 11 ммоль) растворяли в 20 мл изопропанола. Раствор перемешивали при 100°С в течение 24 часов, охлаждали до комнатной температуры и сконцентрировали при пониженном давлении. Далее к остатку добавляли этиловый эфир. После охлаждения в ледяной бане в течение двух часов осадок отфильтровывали, промывали холодный этиловым эфиром, сушили в вакууме с получением 1 г (выход 59%) целевого соединения. Данные масс-спектроскопии MS(M+1)=178,0.
Стадия С: N-(2-метил-5-нитрофенил)-4,5'-бипиримидин-2-амин

(2Е)-3-(диметиламино)-1-пиримидин-5-илпроп-2-ен-1-он (1 г, 5,6 ммоль) и М-(2-метил-5-нитрофенил)гуанидин нитрат (1,44 г, 5,6 ммоль) (Z. Szakacs et al. (Сзакацс и др.), J. Med. Chem. 2005, 48, 249) суспендировали в 20 мл изопропанола. Далее добавляли гидроксид натрия (0,28 г, 7 ммоль). Смесь перемешивали в течение ночи и охлаждали до комнатной температуры. Осадок отфильтровывали, промывали изопропанолом и диэтиловым эфиром. Фильтрат концентрировали при пониженном давлении, остаток растворяли в 15 мл изопропанола. Полученный раствор кипятили с обратным холодильником в течение ночи и охлаждали до комнатной температуры. Твердый остаток отфильтровывали, промывали изопропанолом и диэтиловым эфиром. Объединенный осадок промывали водой и диэтиловым эфиром, далее сушили в вакууме с получением 1,2 г (выход 70%) целевого соединения. Данные масс-спектроскопии MS(M+1)=309,10.
Стадия D: N(3)-4,5'-бипиримидин-2-ил-4-метилбензол-1,3-диамин

Дигидрат хлорида олова (3,6 г, 16 ммоль) растворяли в 10 мл концентрированной соляной кислоты. В раствор добавляли N-(2-метил-5-нитрофенил)-4,5'-бипиримидин 2-амин при интенсивном перемешивании. Смесь выливали в ледяную воду, после этого перемешивали в течение 2 часов. Далее нейтрализовали до рН>8 карбонатом натрия и экстрагировали 4 раза этилацетатом. Объединенные экстракты промывали солевым раствором, сушили над сульфатом магния и, наконец, концентрировали при пониженном давлении, с получением 0,7 г требуемого соединения. Данные масс-спектроскопии MS(M+1)=279,13.
Стадия Е: N-[3-(4,5'-бипиримидин-2-иламино)-4-метилфенил]-1-(4-метилпиперазин-1-ил)-2,3-дигидро-1H-инден-5-карбоксамид
Метил-1-(4-метилпиперазин-1-ил)-2,3-дигидро-1H-инден-5-карбоксилат (823 г, 3 ммоль) и N(3)-4,5'-бипиримидин-2-ил-4-метилбензол-1,3-диамин (973 г, 3,5 ммоль) суспендировали в 15 мл толуола, затем добавляли 2 М раствор триметилалюминия (3 мл, 6 ммоль). Смесь перемешивали в течение ночи при 50°С и снова добавили 2 М раствора триметилалюминия (2 мл, 4 ммоль). Раствор перемешивали в течение ночи при 60°С и затем оставляли охлаждаться в ледяной бане. При перемешивании добавляли насыщенный водный раствор тартрата калия и натрия. Полученный раствор экстрагировали дихлорметаном (3×100 мл). Объединенные экстракты промывали бикарбонатом натрия (100 мл) и солевым раствором (2×100 мл), сушили над сульфатом магния, концентрировали и очищали хроматографией на силикагелевой колонке, используя в качестве элюента 50% этилацетат/дихлорметан/5-10% триэтиламин, с получением 702 мг целевого соединения (выход 45%). Данные масс-спектроскопии MS(M+1)=527,27, 1Н ЯМР (ДМСО-d6, ppm): δ 10,10 (s, 1H); 9,46 (s, 2H); 9,28 (s, 1H); 9,08 (s, 1H); 8,50 (d, J=5,7 Гц, 1Н); 8,04 (s, 1H); 7,74 (s, 1H);7,70 (d, J=9,0 Гц, 1H); 7,46 (d, J=5,7 Гц, 1H); 7,42 (d, J=9,0 Гц, 1H); 7,32 (d, J=9,0 Гц, 1H); 7,15 (d, J=9,0 Гц, 1H); 4,25 (t, J=5,7 Гц, 1H); 2,2-2,9 (m, ЮН); 2,15 (s, 3H); 2,07 (s, 3H); 2,0 (m, 2H).
Пример 9
Получение 1-[(3S)-3-(диметиламино)пирролидин-1-ил]-N-{4-метил-3-[(4-пиридин-3-ил пиримидин-2-ил)амино]фенил}-2,3-дигидро-1H-инден-5-карбоксамида

Стадия А: (3S)-1-(5-бромо-2,3-дигидро-1H-инден-1-ил)-N,N-диметилпирролидин-3-амин
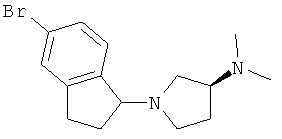
5-бром-1-хлор-2,3-дигидро-1H-инден (2,03 г, 8,76 ммоль) и (3S)-N,N-2,5-диметилпирролидин-3-амин (1 г, 8,76 ммоль) растворяли в 30 мл ацетонитрила, далее добавляли карбонат калия (1,81 г, 13,14 ммоль). Смесь перемешивали в течение ночи при 60°С, затем концентрировали. Остаток растворяли в этилацетате. Раствор промывали трижды солевым раствором, сушили над сульфатом магния, затем концентрировали. Далее проводили очистку колоночной хроматографией на силикагеле, в качестве элюента использовали этилацетат/дихлорметан/триэтиламин/метанол (10:10:1:1), с получением 1,3 г целевого соединения. Данные масс-спектроскопии MS(M+1)=309,0, 311,0.
Стадия В: Метил 1-[(35)-3-(N,N-диметиламино)пирролидин-1-ил]-2,3-дигидро-1H-инден-5-карбоксилат

(3S)-1-(5-бром-2,3-дигидро-1Н-инден-1-ил)-N,N-2,5-диметилпирролидин-3-амин (1,3 г, 4,2 ммоль) растворяли в 30 мл метанола, 5 мл диметилсульфоксида и 7 мл триэтиламина. Реакционную колбу вакуумировали и заполняли N2. Далее добавляли ацетат палладия (0,24 г, 1 ммоль) и 1,3-бис(бифенилфосфино)пропан (0,5 г, 1,5 ммоль). Смесь перемешивали при 80°С в течение двух дней в присутствии СО. После охлаждения до комнатной температуры смесь фильтровали и концентрировали. Остаток растворяли в этилацетате. Полученный раствор промывали трижды солевым раствором, сушили над сульфатом магния, затем концентрировали. Далее выполняли очистку колоночной хроматографией на силикагеле, используя в качестве элюента этилацетат/дихлорметан/триэтиламин (10:10:1), с получением 0,7 г целевого соединения (выход 58%). Данные масс-спектроскопии MS(M+1)=289,1.
Стадия С: 1-[(3S)-3-(диметиламино)пирролидин-1-ил]-N-{4-метил-3-[(4-пиридин-3-ил пиримидин-2-ил)амино]фенил}-2,3-дигидроинден-5-карбоксамид
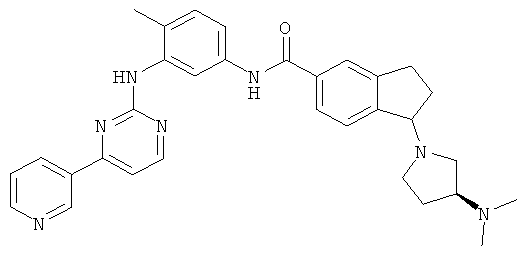
Метил 1-[(35)-3-(N,N-диметиламино)пирролидин-1-ил]-2,3-дигидроинден-5-карбоксилат (0,2 г, 0,69 ммоль) и 4-метил-М(3)-(4-пиридин-3-илпиримидин-2-ил)фенил-1,3-диамин (0,22 г, 0,8 ммоль) растворяли в 5 мл толуола. Далее добавляли 2 М раствор триметилалюминия в толуоле (1,3 мл, 2,6 ммоль). Смесь перемешивали при 60°С в течение 2 дней и далее охлаждали в ледяной бане. Затем добавляли тартрат калия и натрия в водном растворе (15 мл) и дихлорметан (50 мл). Органическую фазу отделили и водную экстрагировали дважды дихлорметаном. Объединенную органическую фазу промывали солевым раствором, сушили над сульфатом магния, затем концентрировали. Далее очищали высокоэффективной жидкостной хроматографией с получением 0,22 г (60% выхода) целевого вещества. Данные масс-спектроскопии MS(M+1)=534,29. 1Н ЯМР (CD3OD, ppm): δ 9,19 (s, 1H); 8,54 (d, J=5,2 Гц, 1H); 8,50 (d, J=8,4 Гц, 1H); 8,36 (d, J=5,2 Гц, 1H); 8,10 (s, 1H); 7,72 (s, 1H); 7,67 (d, J=8,4 Гц, 1H); 7,44 (d, J=5,2 Гц, 1H); 7,41 (d, J=8,4 Гц, 1H); 7,32 (d, J=8,4 Гц, 1H); 7,28 (d, J=5,2 Гц, 1H); 7,15 (d, J=8,4 Гц, 1H); 4,18 (m, 2H); 3,01 (m, 1H); 2,90 (m, 1H); 2,80 (m, 2H); 2,72 (m, 2H); 2,60 (m, 1H); 2,37 (m, 1H); 2,22 (s, 3Н); 2,16 (m, 1H); 2,14 (s, 6H); 1,95 (m, 1H); 1,62 (m, 1H).
Пример 10
Получение 1-[(3R)-3-(диметиламино)пирролидин-1-ил]-N-метил-3-[(4-пиридин-3-ил пиримидин-2-ил)амино]фенил}-2,3-дигидро-1H-инден-5-карбоксамида

Указанное соединение было получено способом, описанным в Примере 9. Данные масс-спектроскопии MS(M+1)=524,29, 1Н ЯМР (CD3OD, ppm): δ 9,19 (s, 1H); 8,54 (d, J=5,2 Гц, 1H); 8,50 (d, J=8,8 Гц, 1H); 8,36 (d, J=5,2 Гц, 1H); 8,10 (s, 1H); 7,72 (s, 1H); 7,6,7 (d, J=7,2 Гц, 1H); 7,44 (d, J=7,2 Гц, 1H); 7,41 (d, J=8,8 Гц, 1H); 7,32 (d, J=7,2 Гц, 1H); 7,28 (d, J=5,2 Гц, 1H); 7,15 (d, J=7,2 Гц, 1H); 4,18 (m, 1H); 3,02 (m, 1H); 2,95 (m, 1H); 2,85 (m, 2H); 2,75 (m, 2H); 2,65 (m, 1H), 2,39 (m, 1H); 2,24 (s, 3Н); 2,20 (m, 1H); 2,15 (s, 3Н); 1,98 (m, 1H); 1,65 (m, 1H).
Пример 11
Получение (1S)-1-(4-метилпиперазин-1-ил)-N-(4-метил-3-[(4-пиридин-3-илпиримидин-2-ил) амино]фенил)-2,3-дигидро-1H-инден-5-карбоксамида

Стадия А: 1-((15)-5-бром-2,3-дигидро-1H-инден-1-ил)-4-метилпиперазин

5-бром-1-хлор-2,3-дигидро-1H-инден (220 г, 950 ммоль) растворяли в ацетонитриле (1 л) и добавляли 1-метилпиперазин (150 г, 1500 ммоль) и карбонат калия (131 г, 950 ммоль). Смесь перемешивали в течение ночи при 60°С. Твердый осадок фильтровали, фильтрат концентрировали. Остаток растворяли в этилацетате (1 л) и полученный раствор промывали дважды гидроксидом натрия (2×300 мл), затем солевым раствором трижды (3×300 мл), сушили над сульфатом магния, концентрировали и очищали хроматографией на силикагелевой колонке, используя в качестве элюента 5% метанол/дихлорметан с получением 202 г продукта (выход 72%). Данные масс-спектроскопии MS(M+1)=295,07, 297,07.
Полученный продукт (202 г, 684,6 ммоль) растворяли в 2000 мл метанола и далее добавляли (1S)-(+)-10-камфорсульфоновую кислоту (318 г, 1369 ммоль), затем 4000 мл изопропанола. Раствор кипятили с обратным холодильником при нагревании в течение 10 минут, затем перемешивали в течение ночи при комнатной температуре. Осадок фильтровали. Когда жидкость перестала капать, осадок промывали изопропанолом и затем растворяли в 600 мл метанола. Далее добавляли изопропанол (1500 мл), раствор нагревали до кипения в течение 15 минут, и далее перемешивали в течение ночи при комнатной температуре. Осадок фильтровали. Когда жидкость перестала капать, осадок промывали изопропанолом и затем растворяли в 1 н. гидроксиде натрия (600 мл). Раствор перемешивали в течение 30 минут и затем трижды экстрагировали этилацетатом (3×300 мл). Объединенный экстракт промывали 1 н. гидроксидом натрия (300 мл) и солевым раствором (2×300 мл), сушили над сульфатом магния, затем концентрировали с получением 50 г целевого соединения. Хиральная чистота составила 99,7%, определенная хиральной высокоэффективной хроматографией. Рентгеновский монокристальный структурный анализ целевого соединения показал, что хиральный центр в 1 положении 2,3-дигидрпо-1H-индена находится в S конфигурации. Данные масс-спектроскопии MS(M+1)=295,07, 297,07.
Стадия В: Этил (1S)-1-(4-метилпиперазин-1-ил)-2,3-дигидро-1H-инден-5-карбоксилат

1-((1S)-5-бром-2,3-дигидро-1Н-инден-1-ил)-4-метилпиперазин (29.6 г, 100 ммоль) растворяли в 300 мл этанола, 30 мл ДМСО и 42 мл триэтиламина. Систему вакуумировали и заполняли N2. После добавляли ацетат палладия (2,4 г, 10 ммоль) и 1,3-бис(дифенилфосфино)пропан (3,3 г, 10 ммоль), систему вакуумировали и заполняли N2. После еще одного вакуумирования смесь перемешивали при 90°С в течение 2 дней в атмосфере СО. Далее охлаждали до комнатной температуры, раствор фильтровали с помощью кизельгура и затем концентрировали. Остаток растворяли в этилацетате (500 мл) и полученный раствор промывали солевым раствором (3×200 мл), сушили над сульфатом магния, концентрировали и затем отделяли хроматографией на силикагелевой колонке, используя в качестве элюента 50% этилацетат/45% дихлорметан/5% триэтиламин с получением 17,3 г целевого соединения (выход 60%). Данные масс-спектроскопии MS(M+1)=289,18.
Стадия С: (1S)-1-(4-метилпиперазин-1-ил)-N-(4-метил-3-[(4-пиридин-3-илпиримидин-2-ил)амино]фенил)-2,3-дигидро-1H-инден-5-карбоксамид

Этил (1S)-1-(4-метилпиперазин-1-ил)-2,3-дигидро-1H-инден-5-карбоксилат (7,2 г, 25 ммоль) и 4-метил-N(3)-(4-пиридин-3-илпиримидин-2-ил)фенил-1,3-диамин (8,3 г, 30 ммоль) растворяли в 150 мл толуола, далее добавляли 2 М раствор триметилалюминия в толуоле (20 мл, 40 ммоль). Полученный раствор перемешивали в течение ночи при 50°С, затем добавляли 20 мл 2 М раствора триметилалюминия в толуоле. После перемешивания при 60°С еще в течение 24 часов раствор охлаждали в ледяной бане и затем добавили водный раствор тартрата натрия и калия (200 мл), а затем дихлорметан (300 мл). Органическую фазу дважды экстрагировали дихлорметаном. Объединенные экстракты промывали дважды солевым раствором, сушили над сульфатом магния и затем концентрировали. Далее очищали колоночной хроматографией на силикагеле, в качестве элюента используя 50% этилацетат/дихлорметан/5-10% триэтиламин с получением 7,5 г (выход 58%) указанного соединения. MS(M+1)=520,27, 1Н ЯМР (ДМСС-d6, ppm): δ 10,10 (s, 1Н); 9,20 (s, 1Н); 8,95 (s, 1H); 8,66 (d, J=6,0 Гц, 1Н); 8,48 (d, J=6,0 10 Гц, 1Н); 8,43 (d, J=8,4 Гц, 1Н); 8,02 (s, 1Н); 7,77 (s, 1Н); 7,74 (d, J=8,4 Гц, 1Н); 7,48 (dd, 1Н); 7,42 (dd, 1H);7,40 (d, J=6,0 Гц, 1Н); 7,32 (d, J=8,4 Гц, 1Н); 7,18 (d, J=8,4 Гц, 1Н); 4,26 (t, J=8,4 Гц, 1Н); 2,2-3,0 (т, ЮН); 2,20 (s, 3Н); 2,12 (s, 3H); 2,02 (m, 2Н).
Пример 12
Получение (1R)-1-(4-метилпиперазин-1-ил)-N-(4-метил-3-[(4-пиридин-3-илпиримидин-2-ил)амино]фенил)-2,3-дигидро-1H-инден-5-карбоксамида

Стадия А: 1-((1S)-5-бром-2,3-дигидро-1H-инден-1-ил)-4-метилпиперазин

В стадии А примера 11 фильтрат метанол/изопропанол 1-(5-бром-2,3-дигидро-1H-инден-1-ил)-4-метил и (1S)-(+)-10-камфоросульфоновую кислоту концентрировали при пониженном давлении. Остаток растворяли в 1 л гидроксида натрия (1 н.). После перемешивания в течение 30 минут раствор экстрагировали этилацетатом (3×300 мл). Объединенные экстракты промывали 1 н. гидроксидом натрия (300 мл) и солевым раствором (3×300 мл), сушили над сульфатом магния и далее концентрировали с получением 140 г (474 ммоль) 1-(5-бром-2,3-дигидро-1H-инден-1-ил)-4-метилпиперазин, где доминирующим являлся R-энантиомер. Остаток растворяли в 1,4 л метанола, затем добавили (1R)-(-)-10-камфорсульфоновую кислоту (220 г, 948 ммоль) и 2,8 л изопропанола. Полученный раствор нагревали до кипения в течение 15 минут и далее перемешивали в течение ночи при комнатной температуре. Осадок фильтровали. Когда жидкость перестала капать, осадок промывали изопропанолом и растворяли в 600 мл метанола. После добавляли изопропанол (1500 мл), раствор нагревали до кипения в течение 15 минут и затем перемешивали в течение ночи при комнатной температуре. Осадок фильтровали. Далее после скалывания жидкости осадок промывали изопропанолом и затем растворяли в 800 мл гидроксида натрия (1 н.). Смесь перемешивали в течение 30 минут, затем экстрагировали трижды этилацетатом (3×300 мл). Объединенные экстракты промывали 1 н. раствором гидроксида натрия (500 мл) и солевым раствором (2×400 мл), сушили над сульфатом магния и затем концентрировали с получением 60 г желаемого соединения. Хиральная чистота составляла 99,8% по данным хиральной высокоэффективной жидкостной хроматографии. Данные масс-спектроскопии MS(M+1)=295,07, 297,07.
Стадия В: Этил (1R)-1-(4-метилпиперазин-1-ил)-2,3-дигидро-1H-инден-5-карбоксилат

Исходя из 1-((1R)-5-бром-2,3-дигидро-1H-инден-1-ил)-4-метилпиперазина, целевое соединение получали согласно способу, описанному на стадии В примера 11. Данные масс-спектроскопии MS(M+1)=289,18.
Стадия С: (1R)-1-(4-метилпиперазин-1-ил)-N-(4-метил-3-[(4-пиридин-3-илпиримидин-2-ил)амино] фенил)-2,3-дигидро-1H-инден-5-карбоксамид

Целевое соединение получали конденсацией этил (1R)-1-(4-метилпиперазин-1-ил)-2,3-дигидро-1H-инден-5-карбоксилата и 4-метил-N(3)-(4-пиридин-3-илпиримидин-2-ил) фенил-1,3-диамина согласно способу, описанному на стадии С примера 11. Данные масс-спектроскопии MS(M+1)=520.27. 1H ЯМР (ДМСО-d6, ppm): δ 10,15 (s, 1H); 9,22 (s, 1H); 8,98 (s, 1H); 8,64 (d, J=6,0 Гц, 1H); 8,46 (d, J=6,0 Гц, 1H); 8,42 (d, J=8,4 Гц, 1H); 8,02 (s, 1H); 7,75 (s, 1H); 7,72 (d, J=9,0 Гц, 1H); 7,50 (dd, 1H); 7,45 (dd, 1H);7,40 (d, J=5,4 Гц, 1H); 7,35 (d, J=8,4 Гц, 1H); 7,18 (d, J=9,0 Гц, 1H); 4,26 (t, J=6,0 Гц, 1Н); 2,2-3,0 (m, 10Н); 2,20 (s, 3Н); 2,12 (s, 3H); 2,3 (m, 2Н).
Пример 13
Получение (1S)-N-[3-(4,5'-бипиримидин-2-ил амино)-4-метилфенил]-1-(4-метилпиперазин-1-ил)-2,3-дигидро-1Н-инден-5-карбоксамида

Целевое соединение получали конденсацией этил (1S)-1-(4-метилпиперазин-1-ил)-2,3-дигидро-1H-инден-5-карбоксилата и N-(3)-4,5'-бипиримидин-2-ил-4-метилфенил-1,3-диамина по способу, описанному в стадии С примера 11. Данные масс-спектроскопии MS(M+1)=521,27, 1Н ЯМР (ДМСО-d6, ppm): δ 10,10 (s, 1Н); 9,40 (s, 2Н); 9,28 (s, 1Н); 9,08 (s, 1H); 8,50 (d, J=4,8 Гц, 1Н); 8,04 (s, 1Н); 7,74 (s, 1Н); 7,70 (d, J=9,0 Гц, 1Н); 7,46 (d, J=4,8 Гц, 1Н); 7,42 (d, J=7,8 Гц, 1Н); 7,32 (d, J=7,8 Гц, 1Н); 7,15 (d, J=9,0 Гц, 1Н); 4,25 (t, J=7,8 Гц, 1Н); 2,2-2,9 (m, 10Н); 2,15 (s, 3H); 2,07 (s, 3H); 2,0 (m, 2Н).
Пример 14
Получение (1R)-N-[3-(4,5'-бипиримидин-2-иламино)-4-метилфенил]-1-(4-метилпиперазин-1-ил)-2,3-дигидро-1Н-инден-5-карбоксамида

Указанное соединение получали конденсацией (1R)-1-(4-метилпиперазин-1-ил)-2,3-дигидроинден-5-карбоксилата и N-(3)-4,5'-бипиримидин-2-ил-4-метилфенил-1,3-диамин, согласно способу, описанному на стадии С примера 11. Данные масс-спектроскопии MS(M+1)=521,27, 1H ЯМР (ДМСО-d6, ppm): δ 10,10 (s, 1Н); 9,40 (s, 2Н); 9,28 (s, 1Н); 9,08 (s, 1Н); 8,50 (d, J=5,7 Гц, 1Н); 8,04 (s, 1Н); 7,74 (s, 1Н); 7,70 (d, J=8,4 Гц, 1Н); 7,46 (d, J=5,7 Гц, 1Н); 7,42 (d, J=8,4 Гц, 1Н); 7,32 (d, J=8,40 Гц, 1Н); 7,15 (d, J=8,4 Гц, 1Н); 4,25 (t, J=7,5 Гц, 1Н); 2,2-2,9 (m, 10Н); 2,15 (s, 3H); 2,07 (s, 3H); 2,0 (т, 2Н).
Пример 15
Получение (1S)-1-(4-метилпиперазин-1-ил)-N-(4-метил-3-[(4-пиридин-4-илпиримидин-2-ил) амино]фенил)-2,3-дигидро-1H-инден-5-карбоксамид
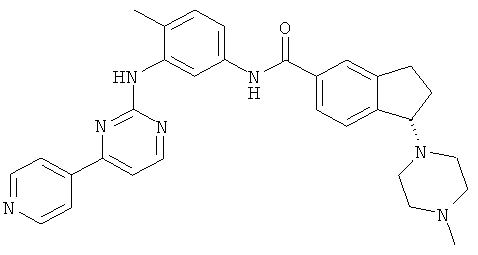
Стадия А: N-(2-Метил-5-нитрофенил)-4-пиридин-4-илпиримидин-2-амин

Целевое соединение получали реакцией конденсации между (2Е)-3-(диметиламино)-1-пиридин-4-илпроп-2-ен-1-оном и N-(2-метил-5-нитрофенил)гуанидин нитратом, согласно способу, описанному на Стадии С примера 8. Данные масс-спектроскопии MS(M+1)=308,11.
Стадия В: 4-метил-N(3)-(4-пиридин-4-илпиримидин-2-ил)бензол-1,3-диамин

Указанное соединение получили восстановлением N-(2-метил-5-нитрофенил)-4-пиридин-4-илпиримидин-2-амина согласно способу, описанному на Стадии D примера 8. Данные масс-спектроскопии MS(M+1)=278,13.
Стадия С: (1S)-1-(4-метилпиперазин-1-ил)-N-(4-метил-3-[(4-пиридин-4-илпиримидин-2-ил)амино]фенил)-2,3-дигидро-1Н-инден-5-карбоксамид

Целевое соединение получали реакцией конденсации между этил (1S)-1-(4-метилпиперазин-1-ил)-2,3-дигидро-1Н-инден-5-карбоксилатом и 4-метил-N(3)-(4-пиридин-4-илпиримидин-2-ил)бензол-1,3-диамином согласно способу, описанному на стадии С примера 11. Данные масс-спектроскопии MS(M+1)=520,27, 1H ЯМР (ДМСО-d6, ppm): δ 10,14 (s, 1H); 9,04 (s, 1H); 8,07 (d, J=4,4 Гц, 2H); 8,55 (d, и=4,8 Гц, 1H); 8,06 (s, 1H); 8,04 (d, J=4,4 Гц, 2Н); 7,78 (s, 1H); 7,75 (d, J=8,8H4, 1H); 7,45 (d, J=7,6 Гц, 1H); 7,44 (d, J=4,8 Гц, 1H); 7,35 (d, J=7,6 Гц, 1H); 7,18 (d, J=8,8 Гц, 1H); 4,31 (t, J=7,2 Гц, 1H); 2,0-3,0 (m, 10Н); 2,19 (s, 3H); 2,12 (s, 3H); 2,04 (m, 2H).
Пример 16
Получение (1S)-1-(4-метилпиперазин-1-ил)-N-(4-метил-3-[(4-пиридин-3-илпиримидин-2-ил)амино]фенил)-2,3-дигидро-1Н-инден-5-карбоксамид сульфата

Стадия А: (18)-1-(4-метилпиперазин-1-ил)-2,3-дигидроинден-5-карбоновая кислота

В защитной атмосфере N2, 1-((1S)-5-бром-2,3-дигидро-1Н-инден-1-ил)-4-метилпиперазин (660 г, 2,235 моль) и ТГФ (3,3 л) добавляли в 10 л трехгорлую колбу, затем раствор перемешивали до полного растворения. Температуру смеси понижали до -78°С в жидкой азотно-ацетоновой бане. н-Бутиллитий (н-BuLi) (2,5 М в растворе гексана) (1072 мл, 2,682 моль, 1,2-кратный) по каплям добавляли к раствору при температуре -78°С ~ -82°С. После перемешивали в течение 10 минут, когда анализ ЖХ-МС (Жидкостная хроматография-масс-спектрометрия) показал завершение реакции исходных веществ, осторожно добавляли сухой лед (170 г, 3,86 моль, 1,73-избыток). Раствор далее перемешивали в течение 10 минут при температуре -60°С ~ -75°С. После завершения реакции холодную баню удаляли и добавляли водный раствор 2 н. HCl для регулировки величины рН до значения рН=2. Большую часть воды упаривали на роторном испарителе. Далее смесь сушили в течение ночи при 50°С ~ 60°С в вакуумной сушильной печи с получением целевого соединения (1289 г, действительный продукт 583 г обеспечивает 100% выход). Данный сырой продукт непосредственно использовали в следующей стадии.
Стадия В: (1S)-1-(4-метилпиперазин-1-ил)-2,3-дигидро-инден-5-карбонил хлорид гидрохлорид

SOCl2 (2,5 л) добавляли в 5 л трехгорлую колбу. (1S)-1-(4-метилпиперазин-1-ил)-2,3-дигидро-1H-инден-5-карбоновую кислоту (1289 г, действительное содержание 583 г, эквивалентно 2,235 моль) добавляли партиями в течение 1 часа. Раствор нагревали при кипячении в течение ночи, затем охлаждали до комнатной температуры. Большую часть SOCl2 удаляли с помощью роторного испарителя. После добавления этилацетата (1,5 л) раствор охлаждали до 0°С, фильтровали с подсосом с получением белого осадка, который затем сушили в вакууме с получением целевого соединения (около 1325 г, выход 100%).
Стадия С: (1S)-1-(4-метилпиперазин-1-ил)-N-(4-метил-3-[(4-пиридин-3-илпиримидин-2-ил)амино]фенил)-2,3-дигидро-1Н-инден-5-карбоксамид

N-(5-амино-2-метилфенил)-4-(3-пиридил)-2-аминопиримидин (681 г, 2,46 моль, 1,1-кратный) растворяли в пиридине (3 л). (1S)-1-(4-метилпиперазин-1-ил)-2,3-дигидро-1H-инден-5-карбонил хлорид гидрохлорид (1325 г, действительное максимальное содержание 626 г, эквивалентно 2,235 моль, 1-кратный) медленно добавляли в течение 300 минут при перемешивании. Раствор очень нагревался, поскольку реакция протекала весьма бурно, однако охлаждение не требовалось. После раствор перемешивали в течение ночи при комнатной температуре, раствор далее при перемешивании добавляли к водному раствору 2 н. гидроксида натрия (2 л), сразу же добавляли дихлорметана (2 л). После перемешивания в течение некоторого времени раствор переносили в 5 литровую делительную воронку и отделяли слой дихлорметана. Водную фазу экстрагировали дихлорметаном (2×500 мл). Экстракты объединяли, сушили над безводным сульфатом магния, затем концентрировали. Остаток растворяли в дихлорметане и затем очищали колоночной хроматографией на силикагеле, в качестве элюента используя дихлорметан/5% метанол/1% триэтиламин. Фракции, содержащие нужный продукт, объединяли и концентрировали при пониженном давлении. Остаток растворяли в 1 л теплого этилацетата, и после перемешивания стали выпадать кристаллы. Осадок фильтровали и сушили в вакууме при 50°C с получением целевого продукта (617 г, 53% общий выход трех стадий). Данные масс-спектроскопии MS(M+1)=520,27.
Стадия D: (1S)-1-(4-метилпиперазин-1-ил)-N-(4-метил-3-[(4-пиридин-3-илпиримидин-2-ил)амино]фенил)-2,3-дигидро-1Н-инден-5-карбоксамид сульфат

(1S)-1-(4-метилпиперазин-1-ил)-N-(4-метил-3-[(4-пиридин-3-илпиримидин-2-ил)амино]фенил)-2,3-дигидро-1H-инден-5-карбоксамид (248 г, 0,477 моль) растворяли в этаноле (4,76 л). После перемешивания в течение 20 минут раствор фильтровали на фильтре с отсосом. Фильтрат добавляли в трехгорлую 20 л колбу. Медленно через воронку добавляли раствор серной кислоты (46,74 г, 0,477 моль, 1-кратный), разбавленный этанолом (532 мл) при перемешивании с получением желтоватой суспензии. Затем добавляли этанол (9,5 л). Смесь кипятили с обратным холодильником в течение 2 часов до тех пор, пока суспензия не приобретала молочно-белый цвет. Суспензию охлаждали до комнатной температуры, фильтровали на фильтре с подсосом, затем сушили с получением целевого продукта (183 г, 57,5%). рН фильтрата переводили в основной (рН≈11) водным раствором NaOH, затем экстрагировали дихлорметаном (4×200 мл). Экстракт сушили и концентрировали с получением (1S)-1-(4-метилпиперазин-1-ил)-N-(4-метил-3-[(4-пиридин-3-илпиримидин-2-ил)амино]фенил)-2,3-дигидро-1H-инден-5-карбоксамида (98 г, 39,5%). Температура плавления указанного продукта: 187~189°С. Элементарный анализ C31H36N7O7S1.5, вычисленное значение: С 55,84; Н 5,44; N 14,71; экспериментальное значение: С 55,72; Н 5,70; N 14,40.
Регулирование активности протеинкиназы и ингибирование пролиферации клеток соединениями по изобретению можно выявить с помощью способов, описанных ниже.
Пример А: Исследование ферментативной активности Abl, c-Kit и PDGFR киназы
Активность соединений по настоящему изобретению Abl, c-Kit и PDGFR киназ исследовали Тестом сдвига подвижности (ТСП, mobility shift assay). АТФ концентрация измерялась для каждой киназы в Км, т.е. Abl Km ATP=12 мкМ, с-Kit Km АТР=87 мкМ, PDGFR Km ATP=38 мкМ.
Материалы: Abl (приобретена у компании Carna, Lot No. 06CBS-2988C); с-Kit (приобретена у компании BPS, Cat. No. 40250, Lot No. 1003); PDGFR (приобретена у компании BPS, Cat. No. 40263, Lot No. 1001); ДМСО (приобретена у компании Sigma, Cat. No. D2650, Lot No. 474382); 96-луночный культуральный планшет (приобретена у компании Corning, Cat. No. 3365, Lot No. 22008026); 384- луночный культуральный планшет (приобретен у компании Corning, Cat. No. 3573, Lot No. 12608008); Staurosporine (приобретен у компании Sigma, Cat. No. S4400-1MG, Lot No. 046K4080).
Способы:
1. Получение киназного буфера и останавливающего буфера;
(1) Киназный буфер: 62,5 мМ HEPES, рН 7,5; 0,001875% Brij-35; 12,5 мМ MgCl2; 2,5 мМ DTT (дитиотреитол);
(2) Останавливающий буфер: 100 мМ HEPES, рН 7,5; 0,015% Brij-35; 0,2% покровное вещество №3; 50 мМ ЭДТА;
(3) Получение киназного раствора: киназный раствор получали растворением киназ в киназном буфере, описанном выше. По отношению к c-Kit киназе предварительную активационную обработку нужно выполнять следующим образом: 700 нМ c-Kit, 2 vM ATP, 4 мМ DTT и 10 мМ MgCl2 растворяли в киназном буфере. После инкубирования при 28°С в течение 15 минут раствор добавляли к киназному буферу;
(4) Получение полипептидного раствора: Полипептид FAM и АТФ растворяли в киназном буфере;
(5) Киназный раствор переносили в культуральный планшет. Полученные концентрации АТФ в Abl, c-Kit и PDGFR составляли 0,45 нМ, 12 нМ, 8 нМ соответственно;
(6) Полипептидный раствор переносили в культуральный планшет.Полученные концентрации АТФ в Abl, c-Kit и PDGFR окружении составляли 12 мкМ, 87 мкМ и 38 мкМ соответственно. Полученные концентрации MgCl2 во всех случаях составляли 10 мМ;
(7) Смесь в каждой лунке планшета инкубировали при 28°С в течение одного часа для Abl, 40 минут для c-Kit и 5 часов для PDGFE. Далее для прекращения реакции добавляли останавливающий буфер;
(8) Данные, собранные с помощью оборудования Caliper? далее обработали с помощью программного обеспечения XLift для расчета величин IC50.
Требуемые концентрации (IC50, нМ) каждого соединения по настоящему изобретению, приводящие к 50% ингибированию, приведены в Таблице 1. При этом для сравнения приведены величины IC50, приводящие к ингибированию данных трех киназ в аналогичных экспериментальных условиях в случае Иматиниба. В качестве положительного контроля в настоящем исследовании использовали стауроспорин.
Как показано в таблице 1, соединения по изобретению обладают очень высокой ингибирующей активностью в отношении Abl, c-Kit и PDGFR: величины IC50 в диапазоне от 2,2 нМ до 2044 нМ при ингибировании Abl, величины IC50 в диапазоне от 8,5 нМ до 2260 нМ при ингибировании c-Kit и величины IC50 в диапазоне от 9,6 нМ до 233 нМ в случае ингибирования PDGFR. За исключением примеров 2, 7, 12 и 14, соединения по изобретению обладают большей активностью по сравнению с Иматинибом при ингибировании трех киназ данного типа.
Пример В: Исследование киназной активности мутантов Abl и c-Kit
Ингибиторная активность соединений по настоящему изобретению мутантов Abl, c-Kit и PDGFR киназ определяли в исследовании меченных изотопом фосфора АТФ (33Р-АТФ).
1. Используя свежеприготовленный реакционный буфер, получали раствор субстрата: 20 mM HEPES, рН 7,5, 10 мМ MgCl2, 1 мМ ЕГТА, 0,02% Brij 35, 0,02 мг/мл BSA, 0,1 мМ Na3VO4, 2 мМ DTT, 1% ДМСО. Для c-Kit и c-Kit (V654A), дополнительно, в буфер добавили 2 мМ MnCl2;
2. К вышеуказанному раствору субстрата добавляли требуемый кофермент;
3. Добавляли киназы и аккуратно перемешивали;
4. Исследуемые соединения растворяли в ДМСО, далее добавляли вышеуказанный киназный расттвор с помощью акустического метода (Есhо550, диапазон нанолитры) и инкубировали в течение 20 минут;
5. Для инициирования реакции к указанному выше реакционному раствору добавляли 33Р-АТР;
6. Смесь инкубировали в течение 2 часов при комнатной температуре;
7. Активность киназ определяли фильтрацией-комбинацией;
8. Данные обрабатывали в Excel и вычитали контрольные данные. Для получения величин IC50 строили кривую с помощью программного обеспечения GraphPad Prism.
Величины IС50 соединения по примеру 11, которое может ингибировать Abl и 7 мутантов, а также c-Kit и 5 мутантов, перечислены в Таблице 2. Также, для сравнения в Таблице 2 также перечислены величины IC50 для Нилотиниб, ингибирующего те же мутанты в аналогичных экспериментальных условиях. В качестве положительного контроля в настоящем исследовании использовали стауроспорин.
Как показано в Таблице 2, соединения по примеру 11 обладают большей ингибирующей способностью при ингибировании мутантов Abl и c-Kit по сравнению с Нилотинибом. Нилотиниб (торговое название Tasigna) обладает хорошим эффектом при лечении пациентов, страдающих от лейкемии с невосприимчивостью к Иматинибу (торговое название Gleevec). Поскольку соединение по примеру 11 обладает большим эффектом при ингибировании мутантов Иматиниба в тесте по сравнению Нилотинибом, соединения по изобретению позволят достичь лучших результатов при лечении пациентов, страдающих от лейкемии с невосприимчивостью к Иматинибу. Мутанты c-Kit широко распространены в гастроинтестинальных стромальных опухолях, при мастоците и острой миелоидной лейкемии. Как показано в Таблице 2, соединение по примеру 11 обладает хорошим эффектом при ингибировании всех типов c-Kit мутантов. Поэтому соединение по изобретению можно применять для лечения гастроинтестинальных стромальных опухолях, при мастоците и острой миелоидной лейкемии.
Пример С: Исследование К562 клеток
Ингибиторная активность соединений по изобретению по отношению к росту клеток хронической миелоидной лейкемии К562 изучали люминесцентным способом CellTiter-Glo.
Материалы: К562 клеточный штамм (получен от АТСС, Cat. No. CCL-243, Lot No. 50644810); IMDM (получен от Invitrogen, Cat. No. 12440-053); фетальная бычья сыворотка (получен от Invitrogen, Cat. No. 10099141, Lot No. 613866);
ДМСО (получен от Sigma, Cat. No. D2650, Lot No. 077k2357); 96-луночный культуральный планшет получен от Coming, Cat. No. 3903); 15 мл пробирки для центрифугирования (получены от Greiner, Cat. No. 0703115, Lot No. 2012-01);
набор анализа жизнеспособности клеток (CellTiter-Glo) (заказан у Promega, Cat. No. G7571, Lot No. 256984); стауроспорин (заказан у Sigma, Cat. No. S4400-1MG, Lot No. 046K4080).
Методики:
1. Посев клеточных культур
(1) Получение полной среды: Полная среда состояла из тщательно перемешанной смеси 90% IMDM и 10% фетальной сыворотки;
(2) Отобрали клеточный штамм с хорошим характером роста;
(3) Клеточную суспензию переносили в пробирки для центрифугирования с использованием пипетки и далее центрифугировали при скорости 800-1000 об/мин в течение 3-5 минут;
(4) Надосадочную жидкость в пробирке удалили пипеткой;
(5) В пробирки добавили точный объем среды и далее клетки ресуспендировали, осторожно набирая раствор в пипетку и выпуская обратно;
(6) Клетки пересчитывали с помощью камеры для подсчета клеток крови;
(7) Отрегулировали концентрацию клеточной суспензии до 4х104 клеток/мл;
(8) Клеточную суспензию добавляли в 96-луночный культуральный планшет, по 100 мкл в лунку, т.е. 4000 клеток в лунку. Планшет инкубировали в течение ночи в атмосфере CO2 в инкубаторе.
2. Получение и добавление соединения
(1) Соединения растворяли в ДМСО и затем разбавляли с получением 10 разных концентраций;
(2) 0,5 мкл раствора соединения переносили в культуральный планшет;
(3) Культуральный планшет инкубировали при 37°С в инкубаторе в течение 72 часов.
3. Тест и анализ
(1) Инвертированным микроскопом изучали клеточную морфологию;
(2) В каждую лунку добавили 100 мкл реагента анализа жизнеспособности клеток;
(3) Планшет стряхивали в течение 2 минут в шейкере, добиваясь клеточного лизиса;
(4) Для стабилизации сигнала люминесценции планшет выдерживали при комнатной температуре в течение 10 минут;
(5) К нижней части планшета присоединяли белую мембрану, планшет измеряли с помощью оборудования Flexstation 3 (люминесценция, время интегрирования 500 мс);
(6) Результаты регистрировали и анализировали.
Требуемые концентрации (IC50, нМ) соединений по примерам 3, 11, 12, 13 и 15 по настоящему изобретению, приводящие к 50% ингибированию, показаны в Таблице 3. Кроме того, для сравнения в Таблице 3 приведены IC50 величины для Иматиниба, ингибирующего рост клеток К562 в аналогичных экспериментальных условиях. В качестве положительного контроля в настоящем исследовании использовали стауроспорин.
Как видно из Таблицы 3, соединения по примерам 3,11,12,13и15 обладают очень высокой ингибирующей активностью в отношении роста клеток хронической миелоидной лейкемии K562. За исключением примера 12, концентрации (IC50, нМ) соединений по примеру 3, 11, 13 и 15, приводящие к 50% ингибированию скорости роста клеток К562 существенно ниже концентраций Иматиниба (p≤0,05). Соединение по примеру 12 являлось оптическим энантиомером соединения примера 11. Хотя его ингибирующая активность на рост клеток К562 была в 65 раз ниже соединения по примеру 11, оно было более эффективным по сравнению с Иматинибом. Можно предположить, что соединения по настоящему изобретению можно использовать для эффективного лечения хронической миелоидной лейкемии.
Пример D: Исследование клеточных штаммов К562, KU812, MEG-01, Kasumi-1 и Sup-B15
В настоящем изобретении также тестировали ингибиторную активность соединений по настоящему изобретению в отношении роста клеток хронической миелоидной лейкемии К562, KU812, MEG-01, клеток острой миелоидой лейкемии Kasumi-1 и клеток острой лимфатической лейкемии Sup-B15.
Материалы: Спектрофотометр SpectraMAX Plus для измерения микропланшетов, модель 3011 (поставляется Molecular Devices Corp, California, USA); инкубатор CO2 с водяной рубашкой (поставляется Therma, USA); инвертированный микроскоп Chongguang XDS-1B (Chongqing Optical & Electrical Instrument Co.,Ltd., Chongqing, China); порошковый водный реагент CellTiter 96® MTS (поставляется Promega, Cat. No. G1112); феназин метасульфат (ФМС) (поставляется Sigma, Product No. P9625); RPMI1640 (поставляется GIBCO, USA, Cat. No. 31800-022); IMDM (поставляется GIBCO, USA, Cat. No. 12200-036); фетальная телячья сыворотка (FBS) (поставляется GIBCO, USA, Cat. No.FCS100).
Способы:
1. Получение исследуемого раствора
(1) Получение раствора ФМС: ФМС растворили в фосфатно-солевой буфер Дюльбекко (DPBS), получив концентрацию 0,92 г/мл. Далее раствор фильтровали и помещали в стерильный и светонепроницаемый контейнер;
(2) Получение раствора MTS: а) 21 мл DPBS добавляли в светонепроницаемый контейнер; б) 42 мг порошка MTS взвешивали и далее добавляли к DPBS; в) перечисленные выше компоненты перемешивали с помощью электромагнитной мешалки до полного растворения порошка; г) измеряли величину рН. Предпочтительно уровень рН должен находиться в диапазоне от 6,0 до 6,5. Если рН будет выше 6,5, его следует довести до 6,5 с помощью 1 н. HCl; д) раствор фильтровали и помещали в стерильный светонепроницаемый контейнер;
(3) Получение смеси MTS/ФМС: а) 2 мл раствора переносили в пробирку; б) 100 мкл раствора ФМС добавляли в пробирку; с) пробирку аккуратно встряхивали, чтобы перемешать раствор.
2. Посев клеточных культур
(1) Подсчет клеток выполняли с помощью камеры подсчета клеток крови после роста клеток до определенного размера;
(2) Концентрацию клеток установливали равной до 2,78×104 клеток/мл средой RPMI1640, содержащей 10% FBS (K562, KU812, MEG-01 или Kasumi-1 клетки) или среду IMDM, содержащую 0,05 мМ 2-меркаптоэтанола и 20% ФБС (Sup-B15 клетки);
(3) 180 мкл клеточной суспензии добавляли в каждую лунку 96-луночного культурального планшета с получением конечной плотности клеток 5×103 на лунку.
3. Получение и добавление соединений
(1) Исследуемые соединения растворяли в ДМСО и затем разбавляли с получением 10 разных концентраций;
(2) 20 мкл раствора каждой концентрации переносили в каждую лунку, уже содержащую клеточную суспензию (по 3 лунки для каждой концентрации);
(3) Планшеты инкубировали в течение 72 часов при 37°С, 5% СО2 и 95% влажности.
4. Тестирование и анализ
(1) 40 мкл раствора MTS/ФМС переносили с помощью пипетки в каждую лунку, содержащую 200 мкл среды, с получением общего раствора 240 мкл на каждую ячейку;
(2) Планшет инкубировали в течение 1-4 часов при 37°С, 5% CO2 и 95% влажности;
(3) Регистрировали абсорбцию при длине волны 490 нм с помощью SpectraMax Plus;
(4) IC50 величины рассчитывали с помощью 5 версии программного обеспечения GraphPad Prism.
В Таблице 4 приведены концентрации соединения по ПРИМЕРУ 16 (IC50, нМ), требуемые для 50% ингибирования К562, KU812, MEG-01, Kasumi-1 и Sup-815 клеточных штаммов. Кроме того, для сравнения также приведена ингибирующая активность Иматиниба и Нилотиниба в аналогичных экспериментальных условиях. В качестве положительного контроля использовали стауроспорин.
Как видно из Таблицы 4, соединение по примеру 16 проявляет очень высокую ингибирующую активность по отношению к росту клеточных штаммов хронической миелоидной лейкемии К562, KU812 и MEG-01, клеточных штаммов острой миелоидной лейкемии Kasumi-1, а также клеточных штаммов острой лимфатической лейкемии Sup-B15. Величины IC50 для данного соединения варьировались от 0,024 нМ до 39,6 нМ. Кроме того, активность соединения по примеру 16 при ингибировании роста данных клеточных штаммов была гораздо выше Иматиниба или Нилотиниба. Данные результаты позволяют предположить, что соединения по настоящему изобретению можно использовать для эффективного лечения хронической миелоидной лейкемии, острой миелоидной лейкемии и острой лимфатической лейкемии.
Несмотря на то, что данное изобретение описано вместе с иллюстративными воплощениями, специалистам данной области техники будут понятны возможные различные модификации и отклонения, не приводящие к изменению объема и целей настоящего изобретения. Следует понимать, что изобретение не ограничено иллюстративными примерами, приведенными в данном документе.
| название | год | авторы | номер документа |
|---|---|---|---|
| БИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ ВЗАИМОДЕЙСТВИЯ/АКТИВАЦИИ PD1/PD-L1 | 2019 |
|
RU2777980C2 |
| КОНДЕНСИРОВАННЫЕ ТРИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛОВ В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2689777C1 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛОПИРИДИНА В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ | 2015 |
|
RU2733400C2 |
| Новое соединение в качестве ингибитора протеинкиназы и содержащая его фармацевтическая композиция | 2019 |
|
RU2785734C1 |
| КОНДЕНСИРОВАННЫЕ ТРИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ИМИДАЗОЛА В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2679914C9 |
| КОНДЕНСИРОВАННЫЕ ТРИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ИМИДАЗОЛА В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2700004C1 |
| НОВЫЕ АНТИГЕЛЬМИНТНЫЕ СОЕДИНЕНИЯ | 2019 |
|
RU2828007C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ PRMT5 | 2018 |
|
RU2797822C2 |
| СОЕДИНЕНИЯ И КОМПОЗИЦИИ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗЫ | 2006 |
|
RU2383545C2 |
| Соединения пиразола, их фармацевтические композиции и их применение | 2019 |
|
RU2770835C1 |
Изобретение относится к новым дигидроинденамидам, выбранным из соединений, соответствующих общей формуле II, или их фармацевтически приемлемым солям. Соединения ингибируют активность протеинкиназ, выбранных из Abl, c-Kit и PDGFR, и могут найти применение для лечения заболеваний, связанных с нарушением активности указанных протеинкиназ, например лейкемии и других видов рака. В общей Формуле II

R1 представляет собой пиперазинил, который может быть необязательно замещен одним R1a; R1a представляет собой H, CH3, C(O)Rd или C(O)ORa; Y представляет собой пиримидил; Z представляет собой пиридил или пиримидил; Ra представляет собой трет-бутил и Rd представляет собой CH3. Указанные соединения представляют собой трет-бутил-4-{5-[({(4-метил-3-[(4-пиридин-3-илпиримидин-2-ил)амино]фенил}амино)карбонил)-2,3-дигидро-1H-инден-1-ил}пиперазин-1-карбоксилат; N-(4-метил-3-[(4-пиридин-3-илпиримидин-2-ил)амино]фенил)-1-пиперазин-1-ил-2,3-дигидро-1H-инден-5-карбоксамид; 1-[4-ацетилпиперазин-1-ил)-N-(4-метил-3-[(4-пиридин-3-илпиримидин-2-ил)амино]фенил)-2,3-дигидро-1H-инден-5-карбоксамид; (1R)-1-(4-метилпиперазин-1-ил)-N-(4-метил-3-[(4-пиридин-3-илпиримидин-2-ил)амино]фенил)-2,3-дигидро-1H-инден-5-карбоксамид; (1S)-N-[3-(4,5′-бипиримидин-2-иламино)-4-метилфенил]-1-(4-метилпиперазин-1-ил)-2,3-дигидро-1H-инден-5-карбоксамид; (1R)-N-[3-(4,5′-бипиримидин-2-иламино)-4-метилфенил]-1-(4-метилпиперазин-1-ил)-2,3-дигидро-1H-инден-5-карбоксамид; (1S)-1-(4-метилпиперазин-1-ил)-N-(4-метил-3-[(4-пиридин-4-илпиримидин-2-ил)амино]фенил)-2,3-дигидро-1H-инден-5-карбоксамид и (1S)-1-(4-метилпиперазин-1-ил)-N-(4-метил-3-[(4-пиридин-3-илпиримидин-2-ил)амино]фенил)-2,3-дигидро-1H-инден-5-карбоксамидсульфат. 4 н.п. ф-лы, 4 табл., 16 пр.
1. Соединение Формулы II
или его фармацевтически приемлемая соль, где
R1 представляет собой пиперазинил, который может быть необязательно замещен одним R1a;
R1a представляет собой H, CH3, C(O)Rd или C(O)ORa;
Y представляет собой пиримидил;
Z представляет собой пиридил или пиримидил;
Ra представляет собой трет-бутил и Rd представляет собой CH3 и
где указанное соединение выбрано из:
трет-бутил-4-{5-[({(4-метил-3-[(4-пиридин-3-илпиримидин-2-ил)амино]фенил}амино)карбонил)-2,3-дигидро-1H-инден-1-ил}пиперазин-1-карбоксилата;
N-(4-метил-3-[(4-пиридин-3-илпиримидин-2-ил)амино]фенил)-1-пиперазин-1-ил-2,3-дигидро-1H-инден-5-карбоксамида;
1-[4-ацетилпиперазин-1-ил)-N-(4-метил-3-[(4-пиридин-3-илпиримидин-2-ил)амино]фенил)-2,3-дигидро-1H-инден-5-карбоксамида;
(1R)-1-(4-метилпиперазин-1-ил)-N-(4-метил-3-[(4-пиридин-3-илпиримидин-2-ил)амино]фенил)-2,3-дигидро-1H-инден-5-карбоксамида;
(1S)-N-[3-(4,5′-бипиримидин-2-иламино)-4-метилфенил]-1-(4-метилпиперазин-1-ил)-2,3-дигидро-1H-инден-5-карбоксамида;
(1R)-N-[3-(4,5′-бипиримидин-2-иламино)-4-метилфенил]-1-(4-метилпиперазин-1-ил)-2,3-дигидро-1H-инден-5-карбоксамида;
(1S)-1-(4-метилпиперазин-1-ил)-N-(4-метил-3-[(4-пиридин-4-илпиримидин-2-ил)амино]фенил)-2,3-дигидро-1H-инден-5-карбоксамида и
(1S)-1-(4-метилпиперазин-1-ил)-N-(4-метил-3-[(4-пиридин-3-илпиримидин-2-ил)амино]фенил)-2,3-дигидро-1H-инден-5-карбоксамидсульфата.
2. Фармацевтическая композиция, обладающая свойствами ингибитора активности протеинкиназ, выбранных из Abl, c-Kit и PDGFR, где указанная фармацевтическая композиция содержит терапевтически эффективное количество соединения или его фармацевтически приемлемой соли по п.1, а также по меньшей мере один фармацевтически приемлемый носитель.
3. Способ ингибирования активности протеинкиназы, выбранной из Abl, c-Kit и PDGFR, где указанный способ включает воздействие соединения или его фармацевтически приемлемой соли по п.1 на указанную протеинкиназу.
4. Применение соединения или его фармацевтически приемлемой соли по п.1 при производстве лекарственных препаратов для лечения расстройств или заболеваний, где указанные заболевания или расстройства связаны с активностью протеинкиназы, выбранной из Abl, c-Kit и PDGFR.
| WO2005113494 (A2), 01.12.2005 | |||
| CN1944398A,11.04.2007 | |||
| ИНГИБИТОРЫ ТИРОЗИНКИНАЗ | 2003 |
|
RU2348627C2 |
| ИНГИБИТОРЫ ТИРОЗИНКИНАЗ | 2008 |
|
RU2445309C2 |
| АЦИЛИРОВАННЫЕ ИНДАНИЛАМИНЫ И ИХ ИСПОЛЬЗОВАНИЕ В КАЧЕСТВЕ ФАРМАЦЕВТИЧЕСКИХ СРЕДСТВ | 2002 |
|
RU2339614C2 |
| АЦИЛИРОВАННЫЕ ГЕТЕРОАРИЛКОНДЕНСИРОВАННЫЕ ЦИКЛОАЛКЕНИЛАМИНЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ФАРМАЦЕВТИЧЕСКИХ СРЕДСТВ | 2003 |
|
RU2338743C2 |

