Изобретение касается способа получения золотых катализаторов на носителе из пористого металлоксидного носителя и предшественника - золотохлористоводородной кислоты, в котором носитель вводится в контакт с предшественником - водным раствором золотохлористоводородной кислоты. Изобретение касается также золотого катализатора на металлоксидном носителе и его применения для окисления спиртов, альдегидов, полигидроксисоединений и углеводов.
Существует постоянная потребность в высокоактивных и стабильных катализаторах, которые могут быть использованными прежде всего для окисления органических соединений, таких как спирты, альдегиды, полигидроксисоединения и моно-, олиго- и полисахариды.
Известно применение палладиевых и платиновых катализаторов на носителях для окисления глюкозы. Однако оно в значительной степени ограничено вследствие незначительной селективности и скорости превращения. К тому же это ведет к относительно быстрому дезактивированию катализаторов ввиду блокирования поверхности катализатора вследствие адсорбции и/или в силу эффектов отравления.
Поэтому для промышленного производства продуктов окисления из углеводов приходится по-прежнему прибегать к ферментационным процессам, которые связаны с высокими затратами на оборудование и загрязнением сточными водами.
На этом основании должны быть разработаны новые типы катализаторов, которые обеспечивают эффективное каталитическое окисление, особенно углеводов, например, для получения альдоновых кислот с использованием кислорода в качестве окислителя, и, наряду с высокой активностью и селективностью, проявляют длительный срок службы.
Золотые катализаторы на носителе тоже известны. Они используются главным образом для окисления СО или пропена в газовой фазе и для селективного гидрирования. Золотые катализаторы на углеродном носителе могут также применяться для селективного окисления D-глюкозы в D-глюконовую кислоту в жидкой фазе. Из патента DE 10319917 А1 известны золотые катализаторы на носителе с распределенными в нанодисперсном состоянии частицами золота на углеродных или металлоксидных носителях. Они используются прежде всего для С1-селективного окисления глюкозы и других углеводов. Но активность этих катализаторов все же неудовлетворительна.
Известны способы получения золотых катализаторов путем импрегнирования носителей по «методу первоначального смачивания». Но в литературе такие способы импрегнирования описываются как непригодные для синтеза активных золотых катализаторов. Это обусловлено тем, что в этих способах, как правило, получаются слишком крупные частицы золота (больше чем 10 нм).
Поэтому технологическая проблема, лежащая в основе настоящего изобретения, в основном состоит в том, чтобы приготовить улучшенные золотые катализаторы на носителе и создать способ их получения, которые обладают улучшенной активностью, прежде всего при окислении органических соединений, таких как спирты, альдегиды и полигидроксисоединения.
Далее, в основе изобретения лежит проблема разработки способа селективного и эффективного окисления углеводов, в особенности для получения альдоновых кислот, которая преодолела бы недостатки современного состояния технологии.
Эта основополагающая технологическая проблема решается разработкой способа получения золотого катализатора на носителе из пористых металлоксидных носителей и золотохлористоводородной кислоты как предшественника, причем на стадии а) подготавливают металлоксидный носитель, на стадии b) носитель вводят в контакт с водным раствором золотохлористоводородной кислоты как предшественника. На стадии b) при этом получают импрегнированный предшественник катализатора, который на последующей стадии с) высушивают. Соответствующий изобретению способ в особенности отличается тем, что на стадии а) носитель готовится в сухом и преимущественно порошкообразном или гранулированном состоянии или в виде спрессованного порошкового блока, и на стадии b) объем водного раствора золотохлористоводородной кислоты как предшественника подбирают настолько максимально большим, чтобы он соответствовал объему пор носителя. Он может быть подобран и меньшим, но не большим, чем объем пор.
Если удельный объем пор носителя неизвестен, то предпочтительно измерить опытным путем объем водного раствора предшественника, который следует ввести в сухой носитель, для чего раствор предшественника постепенно прибавляется к сухому носителю до тех пор, пока носитель уже не сможет больше впитывать дальнейший объем раствора предшественника. Это прежде всего становится различимым по возникшему увлажнению материала носителя. Для каждого сорта металлоксидного носителя получается удельная поглотительная способность [в мл/г носителя катализатора], которая в первую очередь зависит от соотношения «поверхность/объем», от величины пор и от степени высушивания металлоксидного носителя. Под понятием «сухой» понимается, что пористый металлоксидный носитель в значительной степени не содержит никакой влаги в объеме пор, с тем, чтобы объем пор мог воспринять раствор предшественника.
В особенно предпочтительном варианте стадии от а) до с) последовательно выполняются многократно, то есть по меньшей мере дважды. В альтернативном варианте стадии b) и с) выполняются одновременно, то есть параллельно одна другой.
В предпочтительном варианте осуществления на стадии b) происходит контактирование металлоксидного носителя с золотохлористоводородной кислотой как предшественником путем прикапывания предшественника к носителю при перемешивании. В предпочтительном варианте предшественник набрызгивают на носитель, причем носитель преимущественно перемешивают. Во время перемешивания носитель предпочтительно высушивают с нанесенным предшественником (стадия с)). В одном из вариантов контактирование предшественника с носителем происходит в блоке для дражирования или в тарельчатом окомкователе, причем преимущественно прикапывается или набрызгивается и при необходимости одновременно высушивается. В еще одном варианте носитель находится в псевдоожиженном слое, и предшественник вводят в псевдоожиженный слой, преимущественно впрыскивают; при этом носитель преимущественно высушивают с нанесенным предшественником (стадия с)).
Преимущественно в качестве водного раствора золотохлористоводородной кислоты как предшественника вводится раствор золотохлористоводородной кислоты (HAuCl4) в водной кислоте, в особенности соляной кислоте, причем предпочтительно концентрация кислоты составляет от 0,1 моль/л до 12 моль/л, преимущественно от 1 моль/л до 4 моль/л, в особенности предпочтительно 2 моль/л. В особенно предпочтительном варианте исполнения значение рН готового раствора предшественника постоянно составляет 1 или ниже. Преимущественно или при необходимости - в зависимости от условий использования - раствор предшественника, употребляемый согласно изобретению, содержит по меньшей мере еще одну кислоту. Разумеется, вместо соляной кислоты могут быть использованы другие неорганические или органические кислоты.
В особенности предпочтительно для приготовления водного раствора предшественника отвешивать необходимое количество золотохлористоводородной кислоты непосредственно в водную кислоту и растворять. Предпочтительно для растворения золотохлористоводородной кислоты использовать водную кислоту преимущественно в концентрации от 0,1 моль/л до 12 моль/л, от 1 моль/л до 4 моль/л и особенно преимущественно 2 моль/л.
Измерения, проведенные с помощью просвечивающей электронной микроскопии (ТЕМ), показали, что полученные согласно изобретению катализаторы неожиданно обнаружили очень мелкие и активные частицы с величиной менее чем 10 нм, в особенности от 1 нм до 10 нм, преимущественно от 1 нм до 9 нм, особенно от 1 нм до 5 нм или даже от 1 нм до 2 нм. Авторам с помощью соответствующего изобретению способа впервые удалось по методу «первоначального смачивания» приготовить каталитически активные частицы золота с величинами явственно менее 10 нм. Эти научные данные являются неожиданными и противоречат описаниям в соответствующей технической литературе. Полученные золотые катализаторы проявляют до сих пор недостижимую активность, например, при преобразовании глюкозы. В особенности при использовании сильнокислого раствора предшественника (например, HCl с концентрацией 2 моль/л в качестве растворителя для золотохлористоводородной кислоты) и при применении пористого оксида алюминия, такого как Puralox KR-90, в качестве материала носителя удалось приготовление активнейшего золотого катализатора для окисления глюкозы. Катализатор, приготовленный согласно изобретению, проявляет активность до 2200  . В сравнении с известными для современного состояния технологии золотыми катализаторами это представляет собой повышение почти в четыре раза.
. В сравнении с известными для современного состояния технологии золотыми катализаторами это представляет собой повышение почти в четыре раза.
HAuCl4 в водном растворе нестабильна и гидролизуется. При этом в многочисленных последовательно переходящих друг в друга равновесиях происходит постепенный обмен атомов хлора на воду и гидроксильные группы: [AuCl4]-, [AuCl3(OH)]-, [AuCl2(OH)2]-, [AuCl2(OH)], [AuCl(OH)2], [Au(OH)3], [Au(OH)4]-. Эти равновесия являются зависящими от времени и от величины рН. С помощью в достаточной мере сниженного значения рН гидролиз может быть предотвращен.
Не вдаваясь в теорию, можно сказать, что в сильнокислом водном растворе (HCl с концентрацией 2 моль/л) преобладает тетрахлоридный комплекс [AuCl4]-. Присутствие этого комплекса неожиданно ведет к тому, что прежде всего при восстановлении предшественника катализатора очень мелкие частицы стабилизируются. В других менее кислых растворах, вероятно, это последовательно ведет к постепенному обмену ионов хлора на воду и гидроксид-ионы.
Предпочтительно в качестве носителя используются оксиды металлов, такие как оксид алюминия, оксид титана или оксид кремния. В особенности предпочтительно применение оксида алюминия (Al2O3), например Puralox SCFa-90 или Puralox KR-90.
Предпочтительно проводить высушивание на стадии с) при температурах, более высоких или равных комнатной температуре, преимущественно от 60°С до 200°С, особенно предпочтительно от 60°С до 100°С.
Предпочтительно в дополнительной стадии d), которая преимущественно проводится после стадии с), выполнять восстановление предшественника катализатора. Это делается преимущественно в токе водорода. Предпочтительно поток водорода имеет содержание водорода от 5 объемных процентов до 15 объемных процентов, преимущественно 10 объемных процентов. В зависимости от области использования поток водорода может при необходимости содержать по меньшей мере один инертный газ, такой как азот или благородный газ. В особенности предпочтительно, когда поток водорода состоит из газообразного водорода и по меньшей мере одного инертного газа. Альтернативно восстановление может быть проведено как жидкофазное восстановление известным путем с использованием подходящих восстановителей, таких как боргидрид натрия, соли муравьиной кислоты, углеводы, формальдегид или гидразин.
Если в предпочтительном варианте исполнения соответствующего изобретению способа стадии от а) до с), в особенности стадии b) и с), многократно проводятся последовательно, то предпочтительно предусмотреть, чтобы время от времени, преимущественно после каждого проведения стадий от а) до с), в особенности b) и с), выполнялось восстановление предшественника катализатора (стадия d)).
Предпочтительно проведение восстановления на стадии d) при температурах выше или равных 250°С. Согласно изобретению предпочтительно выполнение восстановления в течение от 10 мин до 300 мин, преимущественно от 80 до 120 мин.
Согласно изобретению также предусмотрено, что к носителю и/или водному раствору золотохлористоводородной кислоты как предшественника может быть добавлена по меньшей мере одна легирующая добавка. Она выбирается преимущественно из оксидов щелочных металлов, щелочноземельных металлов и редкоземельных металлов. Особенно предпочтительны добавки с натрием, калием, цезием, кальцием, церием и/или самарием. Предпочтительно вносить по меньшей мере одну легирующую добавку в количестве от 0,01 весового процента до 1 весового процента.
Дальнейший предмет настоящего изобретения также согласно этому заключается в применении золотохлористоводородной кислоты как предшественника, которая содержит раствор или суспензию золотохлористоводородной кислоты (HAuCl4) в растворителе или же соответственно состоит из них, причем растворителем является водная кислота в концентрации от 0,1 моль/л до 12 моль/л, преимущественно от 1 моль/л до 4 моль/л, особенно предпочтительно 2 моль/л. Предпочтительной кислотой является соляная кислота (HCl). Предпочтительно соляная кислота при необходимости присутствует в связи по меньшей мере с одной другой кислотой. Согласно изобретению эта золотохлористоводородная кислота как предшественник используется для получения золотых катализаторов на металлоксидных носителях преимущественно согласно вышеописанному способу.
Дальнейшим предметом настоящего изобретения также является золотой катализатор на металлоксидном носителе, который может быть получен вышеназванным способом или же соответственно приготовлен таковым. Соответствующий изобретению катализатор в особенности отличается тем, что средняя величина золотых частиц на носителе в основном составляет менее чем 10 нм, преимущественно менее или равна 5 нм, в особенности предпочтительно от 1 нм до 2 нм.
Предпочтительно соответствующий изобретению катализатор содержит золото в доле от 0,01 весового процента до 10 весовых процентов, преимущественно от 0,01 весового процента до 2 весовых процентов, в особенности преимущественно 0,3 весового процента.
Наконец, дальнейшим предметом настоящего изобретения является применение вышеназванного соответствующего изобретению катализатора для окисления органических выделенных соединений (эдуктов), которые в особенности избираются из спиртов, альдегидов и полигидроксисоединений. Согласно изобретению предпочтительно применять катализатор в гетерогенном катализе. То есть, катализатор присутствует как твердое вещество, тогда как окисляемые соединения находятся в жидкой фазе, например в виде водного раствора. Преимущественно используемый для окисления кислород затем в виде газа барботируется через жидкую фазу и благодаря интенсивному перемешиванию распределяется и растворяется в жидкой фазе. Катализатор преимущественно используется в форме порошка или гранулята. В дальнейшем предпочтительном варианте используются спрессованные порошковые блоки, например цилиндры, полые цилиндры, шарики или стержни.
В предпочтительном варианте исполнения готовится водный раствор окисляемого соединения или смеси эдуктов, который содержит этот эдукт в концентрации по меньшей мере около 10 ммол/л, преимущественно по меньшей мере около 100 ммол/л, 150 ммол/л, 200 ммол/л, 250 ммол/л, 1000 ммол/л или 1500 ммол/л. Затем водный раствор соединения добавляется к преимущественно порошкообразному катализатору согласно изобретению в количестве от около 10 мг/л до 10 г/л, причем предпочтительно на литр вносить около 1 г катализатора. Преимущественно соотношение между количеством окисляемого/окисляемых соединения(-ий) и количеством золота, нанесенного на металлоксидный носитель, составляет по меньшей мере около 300-400000, предпочтительно по меньшей мере 300, 500, 1000, 2000, 4000, 10000, 20000, 50000, 100000, 200000 или 400000.
Предпочтительно окисление соединения или смеси соединений проводится при значении рН от 7 до 11, преимущественно от 8 до 10. Предпочтительно температура поддерживается от 20°С до 140°С, от 40°С до 90°С и особенно предпочтительно от 40°С до 80°С. Давление составляет преимущественно от около 1 бар до около 25 бар. Предпочтительно кислород и/или воздух барботируется через водный раствор соединения, соединение, смесь или композицию со скоростью подачи газа от 100 мл/(мин × Lобъем реактора) до 10000 мл/(мин × Lобъем реактора), преимущественно 500 мл/(мин × Lобъем реактора).
Оказалось, что с соответствующими изобретению золотыми катализаторами при окислении альдоз достигается 100%-ная селективность для альдегидного положения. Соответствующие изобретению золотые катализаторы поэтому пригодны, кроме того, для селективного окисления углеводов. Под этим понимается в особенности окисление способной к окислению альдегидной группы при С1-углеродном атоме в углеводе до карбоксильной группы, в то время как спиртовые группы при других атомах углерода в углеводе не окисляются. Поэтому в результате получается преимущественно альдоновая кислота. Что касается соответствующих изобретению предпочтительно использованных углеводов, то речь преимущественно идет об альдозах, которые при С1-углеродном атоме содержат способную к окислению альдегидную группу, или о 2-кетозах, у которых при С1-углеродном атоме может быть введена способная к окислению альдегидная группа. Путем селективного окисления альдегидной группы получается альдоновая кислота. При селективном окислении смеси альдоз поэтому получается смесь различных альдоновых кислот.
Настоящее изобретение поэтому касается также применения соответствующих изобретению катализаторов для получения альдоновой кислоты или смеси различных альдоновых кислот путем селективного окисления одной или более альдоз со способной к окислению альдегидной группой.
Настоящее изобретение поэтому касается также применения для получения альдоновой кислоты или смеси различных альдоновых кислот с использованием одной или более 2-кетоз, причем 2-кетоза (кетозы) первоначально переводится(-ятся) в таутомерную(-ные) альдозную(-ные) форму (формы) со способной к окислению альдегидной группой и затем при использовании катализатора селективно окисляется/окисляются.
Согласно изобретению окисляемые углеводы включают как мономерные полигидроксиальдегиды или полигидроксикетоны, то есть моносахариды, их димеры и так далее до декамеров, то есть олигосахариды, такие как дисахариды, трисахариды и т.д., так и макромолекулярные полисахариды. Под «моносахаридами» в связи с настоящим изобретением понимаются соединения общей химической формулы
CnH2nОn, содержащие от 3 до 7 кислородных функциональных групп, причем природные моносахариды в основном являются гексозами и пентозами. Углеродная цепь моносахарида может быть неразветвленной или разветвленной. Под «олигосахаридами» понимаются соединения, которые получаются объединением от 2 до 10 моносахаридных молекул с выделением воды.
В особенности предпочтительно применение катализатора для селективного окисления углеводов, избранных из таких моносахаридов, как глюкоза, галактоза, манноза, ксилоза и рибоза, а также дисахаридных альдоз, таких как мальтоза, лактоза, целлобиоза и изомальтоза, а также дисахаридных 2-кетоз, таких как палатиноза, а также крахмальной патоки и мальтодекстринов, а также смесей этих углеводов.
При окислении глюкозы с использованием соответствующего изобретению способа получается глюконовая кислота в качестве продукта окисления. При окислении галактозы с использованием соответствующего изобретению способа получается галактоновая кислота в качестве продукта окисления.
В еще одном предпочтительном варианте исполнения окисляемым углеводом является олигосахарид, в особенности дисахарид. Окисляемый дисахарид преимущественно представляет собой дисахаридную альдозу, такую как мальтоза, лактоза, целлобиоза или изомальтоза. Согласно изобретению при селективном окислении мальтозы с использованием соответствующего изобретению способа получается мальтобионовая кислота в качестве продукта окисления. При использовании соответствующего изобретению способа при окислении лактозы получается лактобионовая кислота в качестве продукта окисления.
В дальнейшем предпочтительном варианте осуществления изобретения окисляемым олигосахаридом является дисахаридная кетоза. Что касается окисляемой дисахаридной кетозы, то речь преимущественно идет о палатинозе (изомальтулозе). Для окисления палатиноза согласно изобретению переводится в таутомерную альдозную форму, которая затем подвергается окислению.
В дальнейшем предпочтительном варианте исполнения изобретения окисляемым углеводом является мальтодекстрин. Мальтодекстрины представляют собой получаемые ферментативным разложением крахмала водорастворимые углеводы, в особенности эквивалентные декстрозы, с длиной цепи от 2 до 30, преимущественно от 5 до 20 фрагментов ангидроглюкозы с участием мальтозы. При селективном окислении мальтодекстрина с использованием соответствующего изобретению способа получается продукт окисления, который согласно изобретению содержит в составе, наряду с соответствующими олигосахаридными альдоновыми кислотами, также часть мальтобионовой кислоты и глюконовой кислоты.
В дальнейшем предпочтительном варианте исполнения окисляемым углеводом является крахмальная патока. Под крахмальной патокой понимается глюкозная патока, которая получается из крахмала и прежде всего существует в виде очищенного водного раствора, причем сухая масса, как правило, составляет по меньшей мере 70%.
В дальнейшем предпочтительном варианте исполнения окисляемым углеводом является фурфурол. Окисляемый фурфурол представляет собой преимущественно гидроксиметилфурфурол (HMF) или глюкозилоксиметилфурфурол (GMF).
Изобретение более подробно разъясняется нижеприведенными Примерами и Фиг.1-4, причем Примеры нельзя воспринимать как ограничивающие.
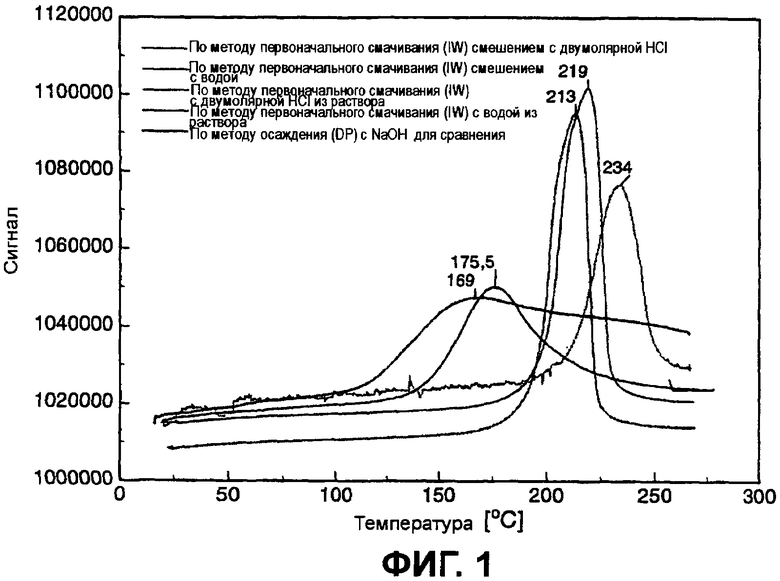
Фиг.1 показывает TPR-профиль (температурно-программированного восстановления) катализаторов.
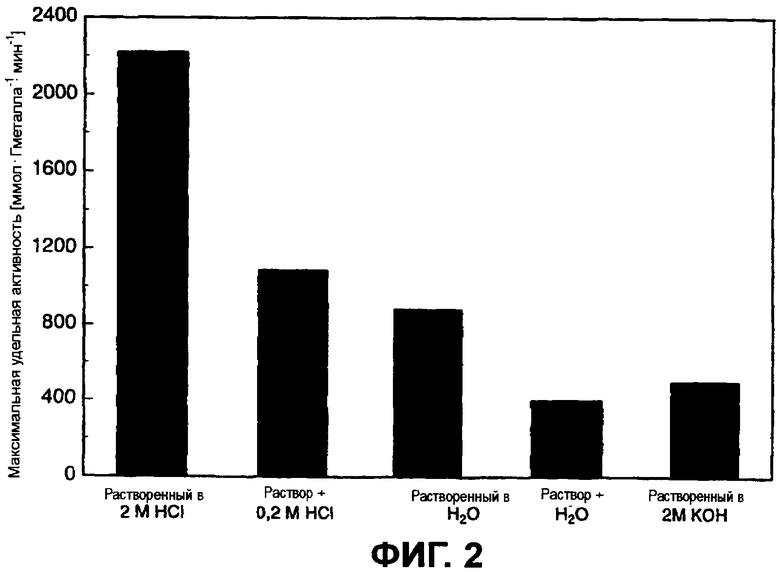
Фиг.2 показывает зависимость активности полученных катализаторов от использованного раствора предшественника.
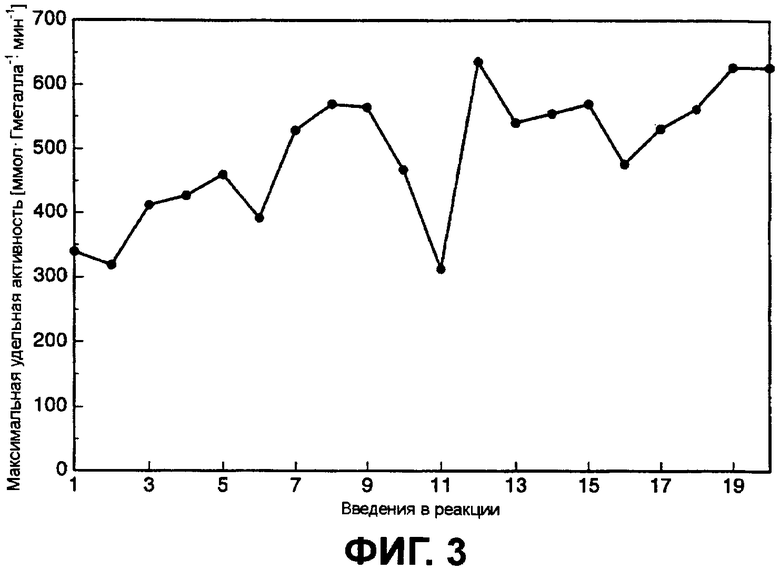
Фиг.3 показывает долговременную стабильность катализатора на протяжении 20 опытов «повторяющейся загрузки».
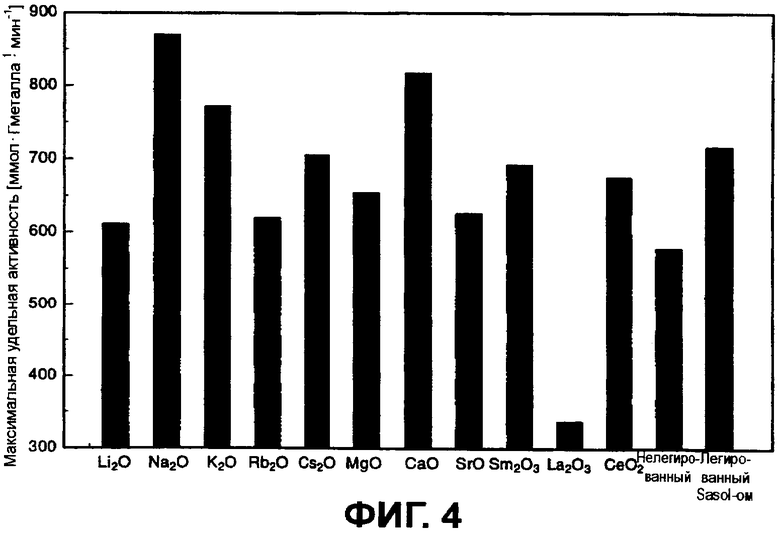
Фиг.4 показывает влияние различных промоторов (активаторов) на максимальную удельную активность золотых катализаторов, приготовленных согласно изобретению.
Пример 1: Приготовление катализатора
Материал носителя
В качестве материала носителя были использованы образцовые оксиды алюминия Puralox KR-90 и Puralox SCFa-90, легированная NaO особая форма (обе от фирмы Sasol). Они имеют следующие характеристики (таблица):
легированный NaO
Получение золотохлористоводородной кислоты как предшественника
Необходимое количество золотохлористоводородной кислоты в кристаллической форме (от фирмы Chempur, (50% Au)) растворяется в объеме растворителя, который максимально соответствует объему пор применяемого количества носителя.
Были приготовлены различные катализаторы, для которых предшественник HAuCl4 был растворен в соляной кислоте, воде и растворе едкого кали КОН. Кроме того, хранившийся в течение длительного времени водный раствор предшественника (25 г/л золота Au) был разбавлен соответственно водой и соляной кислотой. Были приготовлены следующие составы для золотохлористоводородной кислоты как предшественника:
1) навеска предшественника растворена в соляной кислоте концентрации 2 моль/л;
2) раствор предшественника, взятый из водного исходного раствора предшественника, разбавлен соляной кислотой концентрации 0,2 моль/л;
3) навеска предшественника растворена в воде;
4) раствор предшественника, взятый из водного исходного раствора предшественника, разбавлен водой;
5) навеска предшественника растворена в водном растворе КОН.
Чтобы получить катализаторы с различным содержанием золота, в каждую загрузку многократно вносили золотохлористоводородную кислоту, каждый раз с различными концентрациями, или же соответственно разбавляли. Требовалось получить золотые катализаторы с содержанием металла между 0,1 и 5%. На загрузку в каждом случае готовили 2 г золотого катализатора.
Пропитка металлоксидного носителя, метод первоначального смачивания
Растворы предшественника каждый раз в отдельных порциях при одновременном интенсивном перемешивании были постепенно добавлены по каплям к материалу носителя. Конец добавления определялся по возникающему увлажнению материала носителя, которое показывало насыщение объема пор и тем самым предел поглощающей способности носителя.
Высушивание, восстановление
Пропитанные предшественники катализатора были высушены в течение ночи в сушильном шкафу (при примерно 80°С) и затем в течение 3 ч подвергнуты восстановлению при 250°С в потоке газовой смеси азота и водорода (примерно 10% Н2). После этого охлаждение проводилось в потоке азота.
Результаты
а) Содержание золота
Для всех приготовленных золотых катализаторов прежде всего было определено содержание золота с помощью метода ICP-AES (индуктивно-связанная плазма - атомная эмиссионная спектроскопия). Были приготовлены золотые катализаторы с содержанием металла между 0,1 и 5%. Экспериментально определенные значения содержания золота были сравнены с теоретически рассчитанными.
Значения теоретически рассчитанного содержания золота прекрасно коррелировали с фактическим содержанием золота во всех загрузках. Удалось нанести золото на носитель без потерь.
b) Размер частиц
Снимки золотых катализаторов, полученные методом просвечивающей электронной микроскопии (ТЕМ), неожиданно показывают размеры частиц от 1 до максимально близкого к 10 нм.
с) Температура восстановления
Для всех катализаторов в каждом случае были сняты профили температурно-программируемого восстановления (TPR-профили). Фиг.1 показывает TPR-профили катализаторов. Из TPR-профилей можно определить явное различие в максимальных температурах. Наивысшую температуру восстановления при 234°С показывает катализатор, для которого навеска предшественника была внесена в сильнокислый раствор (здесь: соляная кислота HCl в концентрации 2 моль/л); самую низкую температуру показывает катализатор, для которого раствор предшественника был разбавлен водой. Из высокой температуры восстановления можно сделать вывод о сильной адсорбции золотого предшественника на носителе.
Пример 2: Каталитическое окисление глюкозы
Каталитическая эффективность катализаторов, приготовленных согласно Примеру 1, была испытана в жидкофазном окислении глюкозы в глюконовую кислоту.
Реакция была проведена в термостатированном стеклянном реакторе (объемом 500 мл) при 40°С. Барботирование выполнялось через стеклянный пористый фильтр со скоростью подачи кислорода 500 мл/мин. Начальная концентрация глюкозы составляла 100 ммол/л. Значение рН поддерживалось постоянным при рН 9 с помощью титратора (Titroline alpha, фирма Schott) и раствора едкого кали с концентрацией 2 моль/л. Поскольку для глюконовой кислоты речь идет о монокарбоновой кислоте, по израсходованным объемам щелочи при 100%-ной селективности можно непосредственно сделать вывод о полученном количестве кислоты. Дополнительный контроль выполнялся с помощью HPLC (высокоэффективной жидкостной хроматографии).
Результаты
а) Селективность
Приготовленные золотые катализаторы показали в этой реакции 100%-ную селективность для альдегидного положения в глюкозе.
b) Каталитическая активность
Конверсия во всех реакциях была полной (100%). Для сравнения катализаторов была выведена максимальная удельная активность.
Фиг.2 показывает зависимость активности приготовленных катализаторов от использованного раствора предшественника. Катализатор, для которого предшественник был растворен в сильнокислом растворе (здесь: соляная кислота HCl с концентрацией 2 моль/л), неожиданно показывает с большим отрывом наивысшую активность в 2200 ммолей глюкозы на грамм металла в минуту.
с) Долговременная стабильность
0,3%-ный Au-катализатор был вовлечен в 20 экспериментов «повторной загрузки» без потери активности. При исследовании долговременной стабильности оказалось, что катализаторы обладают отличной долговременной стабильностью. Вымывания золота не наблюдалось. Нарастание активности с увеличением числа опытов объясняется случаем сокращения лимитирования кислорода вследствие потерь катализатора.
Пример 3: Влияние добавок активаторов
В качестве добавок активаторов были использованы различные щелочные, щелочноземельные и редкоземельные металлы, и было изучено их влияние на активность золотых катализаторов, приготовленных согласно Примеру 1.
В качестве сравнения служили Puralox SCFA-90 («нелегированный» в Фиг.4) и легированный NaO Puralox SCFA-90 («легированный Sasol» в Фиг.4).
Результаты
Фиг.4 показывает влияние различных активаторов на максимальную удельную активность приготовленных согласно изобретению золотых катализаторов с содержанием золота 0,3 весового процента. Содержание активатора составляет в каждом случае 0,1 весового процента от общего количества катализатора. Почти каждый из внесенных активаторов проявил эффект повышения активности. Наибольший эффект повышения активности показали оксиды натрия, калия, цезия, кальция и церия, но не самария Sm2O3.
Сравнительный пример: Au/TiO 2 -катализатор с 0,5% Au
Приготовление
В качестве материала носителя был использован гидрат анатазного оксида титана TiO2 (Kronos, удельная поверхность SBET = 288 м2/г). К водной суспензии
50 г TiO2 в одном литре дистиллированной воды, которая была нагрета до 70°С и значение рН которой было установлено при 6,5 с помощью 0,2н. раствора NaOH, при постоянном значении рН в течение 3 ч при интенсивном перемешивании были добавлены 500 мг золотохлористоводородной кислоты (HAuCl4 × 3H2O) в 250 мл воды. Реакционная смесь перемешивается при 70°С в течение еще одного часа. После охлаждения до комнатной температуры добавляется раствор цитрата магния (2,318 г MgHC6H5O7 × 5H2O в 50 мл воды), величина рН которого перед этим была установлена при 6,5 с помощью 0,2н. раствора NaOH. После одночасового перемешивания твердое вещество отделяется центрифугированием, трижды промывается водой и затем высушивается в вакуумном сушильном шкафу при давлении <50 гПа в течение 17 ч при комнатной температуре и 4 ч при 50°С. Полученный предшественник легко растирается в порошок и нагревается на воздухе со скоростью подъема температуры 1 К/мин до 250°С и в течение 3 ч активируется при этой температуре.
Выход: 47,3 г
ТЕМ: доминируют частицы с диаметром менее чем 5 нм, единичные частицы с диаметром около 20 нм
ICP-АES-анализ: 0,45%
Окисление глюкозы
Катализатор, содержащий 0,5% Au, на TiO2-носителе (0,5% Au/TiO2) типа 149 (АСА) был использован для окисления глюкозы. Окисление глюкозы проводилось при следующих реакционных условиях: объем реакционной смеси (загрузка): 500 мл, количество катализатора: 1 г/л, начальная концентрация субстрата: 100 ммол/л, значение рН: 9; температура: 40°С, давление: 1 бар, скорость барботирования О2: 500 мл/мин, скорость перемешивания: 700 об/мин.
Результаты
Конверсия глюкозы: 100%
Максимальная удельная активность: 416 ммол глюкозы на грамм металла в минуту.
Селективность по глюконовой кислоте >99,5%
Изобретение касается способа получения золотых катализаторов на носителе из пористого металлоксидного носителя и золотохлористоводородной кислоты как предшественника, катализатора и его применения для окисления спиртов, альдегидов, полигидроксисоединений и углеводов. Описан способ получения золотого катализатора на носителе, включающий стадии: а) приготовление носителя в сухом виде, b) контактирование носителя с водным раствором предшественника - золотохлористоводородной кислоты, причем объем раствора настолько максимально большой, как объем пор носителя, с тем, чтобы получить импрегнированный предшественник катализатора, и с) высушивание импрегнированного предшественника катализатора, в котором водный раствор предшественника - золотохлористоводородной кислоты - представляет собой раствор HAuCl4 и водной соляной кислоте с концентрацией кислоты от 0,1 моль/л до 12 моль/л. Описан катализатор, полученный этим способом, его применение для окисления органических соединений, в том числе для получения альдоновых кислот. Технический эффект - повышение активности и селективности катализатора. 5 н. и 11 з.п. ф-лы, 1 табл., 4 ил.
1. Способ получения золотого катализатора на носителе для окисления углеводов из пористого металлоксидного носителя и предшественника золотохлористоводородной кислоты, включающий стадии:
a) приготовление носителя в сухом виде,
b) контактирование носителя с водным раствором предшественника - золотохлористоводородной кислоты, причем объем раствора настолько максимально большой, как объем пор носителя, с тем, чтобы получить импрегнированный предшественник катализатора, и
c) высушивание импрегнированного предшественника катализатора, в котором водный раствор предшественника - золотохлористоводородной кислоты представляет собой раствор HAuCl4 в водной соляной кислоте с концентрацией кислоты от 0,1 до 12 моль/л.
2. Способ по п.1, в котором на стадии b) дополнительно водный раствор предшественника - золотохлористоводородной кислоты постепенно и только в таком количестве прибавляют к сухому носителю, пока носитель уже не может больше впитать последующий объем раствора.
3. Способ по п.1 или 2, в котором на дополнительной стадии d) восстановление предшественника катализатора проводят в токе водорода при температурах выше или равных 250°С.
4. Способ по п.3, в котором на стадии d) восстановление проводят в течение от 10 до 300 мин.
5. Способ по п.3, в котором на стадии d) поток водорода содержит водород в количестве от 5 до 15 об.% и при необходимости инертный газ.
6. Способ по п.1 или 2, в котором на дополнительной стадии d) восстановление предшественника катализатора проводят как жидкофазное восстановление.
7. Способ по п.1, в котором на стадии с) высушивание проводят при температурах от 60 до 200°С.
8. Способ по п.1, в котором выбранный металлоксидный носитель представляет собой оксид алюминия, оксид кремния и оксид титана.
9. Способ по п.1, в котором к носителю и/или раствору предшественника дополнительно вводят легирующие добавки, выбранные из оксидов щелочных металлов, щелочноземельных металлов и редкоземельных металлов.
10. Применение предшественника золотохлористоводородной кислоты в виде раствора или суспензии HAuCl4, в водной соляной кислоте с концентрацией кислоты от 0,1 до 12 моль/л для получения золотого катализатора на металлоксидном носителе для селективного окисления углеводов.
11. Золотой катализатор на металлоксидном носителе для селективного окисления углеводов, получаемый способом по одному из пп.1-9.
12. Катализатор по п.11, отличающийся тем, что средняя величина частиц золота на носителе составляет менее чем 10 нм.
13. Катализатор по п.11 или 12, отличающийся тем, что доля золота составляет от 0,01 до 10 вес.%.
14. Применение катализатора по одному из пп.11-13 для окисления органических соединений, выбираемых из спиртов, альдегидов и полигидроксисоединений.
15. Применение по п.14 для селективного окисления углеводов, выбираемых из глюкозы, галактозы, маннозы, ксилозы и рибозы; мальтозы, лактозы, целлобиозы и изомальтозы; палатинозы; крахмальной патоки; мальтодекстринов, а также их смесей.
16. Применение катализатора по одному из пп.11-13 для получения альдоновых кислот путем окисления.
| WO 2005003072 A1, 13.01.2005 | |||
| СПОСОБ И УСТРОЙСТВО ДЛЯ ПОЛУЧЕНИЯ МОНОКАРБОНОВЫХ КИСЛОТ ИЗ УГЛЕВОДОВ, ПРОИЗВОДНЫХ УГЛЕВОДОВ ИЛИ ПЕРВИЧНЫХ СПИРТОВ | 1994 |
|
RU2129541C1 |
| DE 10319917 A1, 09.12.2004 | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| ПЛАСТИНЧАТЫЙ СВЕРХПРОВОДЯЩИЙ ПРОВОД, СПОСОБ ЕГО ИЗГОТОВЛЕНИЯ И СВЕРХПРОВОДНИКОВЫЙ УЗЕЛ ПРОВОДА | 2006 |
|
RU2408956C2 |
| JP 2005154302 A, 16.06.2005. | |||

