Заявление о перекрестной ссылке
Данная заявка заявляет преимущества предварительной заявки США 60/553906, поданной 17 марта 2004. Для целей патентной практики Соединенных Штатов содержание указанной предварительной заявки приводится посредством ссылки.
Уровень техники
Данное изобретение относится к составам для полимеризации одного или нескольких мономеров или смесей мономеров, таких как этилен, и одного или нескольких сомономеров для формирования сополимерного продукта, имеющего уникальные физические свойства, к способу получения таких сополимеров и к полученным полимерным продуктам. В другом аспекте изобретение относится к способам применения этих полимеров в тех случаях, когда требуются уникальные сочетания физических свойств. Еще в одном аспекте изобретение относится к изделиям, полученным из этих полимеров. Полимеры по изобретению содержат два или более различных регионов или сегментов (блоков), в результате чего полимер обладает уникальными физическими свойствами. Такие мульти-блок-сополимеры и содержащие их полимерные смеси успешно применяются при получении твердых изделий, таких как отформованные изделия, пленки, листы и вспененные предметы, формованием, экструдированием или другими процессами, и применимы в качестве компонентов или ингредиентов в адгезивах, ламинатах, полимерных смесях и для других конечных целей. Полученные продукты применяются при производстве компонентов для автомобилей, таких как профили, бамперы и детали отделки; упаковочных материалов; изоляции электрических кабелей и для других целей.
Давно известно, что полимеры, содержащие структуру блочного типа, часто имеют превосходящие свойства по сравнению со статистическими сополимерами и смесями. Например, три-блок-сополимеры из стирола и бутадиена (SBS) и их гидрированные версии (SEBS) имеют превосходное сочетание теплостойкости и упругости. Другие блок-сополимеры также известны в технике. Как правило, блок-сополимеры, известные как термопластичные эластомеры (TPE), имеют желательные свойства, благодаря присутствию "мягких" или эластомерных блочных сегментов, соединяющих "жесткие" или кристаллизуемые или стекловидные блоки в том же полимере. При температурах вплоть до температуры плавления или температуры стеклования жестких сегментов полимеры демонстрируют эластомерный характер. При более высоких температурах полимеры становятся текучими, проявляющими термопластичное поведение. Известные способы получения блок-сополимеров включают анионную полимеризацию и регулируемую свободнорадикальную полимеризацию. К сожалению, указанные способы получения блок-сополимеров требуют последовательного присоединения мономеров и периодической технологии, и типы мономеров, которые могут быть успешно применены в таких способах, относительно ограничены. Например, при анионной полимеризации стирола и бутадиена для формирования блок-сополимера типа SBS каждая полимерная цепь требует стехиометрического количества инициатора, и полученные полимеры имеют чрезвычайно узкое молекулярномассовое распределение, Mw/Mn, предпочтительно от 1,0 до 1,3. Кроме того, анионные и свободнорадикальные процессы являются относительно медленными, результатом чего являются плохие экономические показатели процесса.
Было бы желательно получать блок-сополимеры каталитически, то есть в процессе, где более чем одну молекулу полимера получают на каждую молекулу катализатора или инициатора. Кроме того, было бы очень желательно получать блок-сополимеры из олефиновых мономеров, таких как этилен, пропилен и более высокие альфа-олефины, которые обычно являются неподходящими для применения в анионных или свободнорадикальных полимеризациях. В некоторых из этих полимеров очень желательно, чтобы некоторые или все блоки полимера содержали аморфные полимеры, такие как сополимер этилена и сомономера, особенно аморфные статистические сополимеры, содержащие этилен и α-олефин, имеющий 3, и особенно 4, или более атомов углерода. И наконец, было бы очень желательно, если бы была возможность применения непрерывного процесса производства блок-сополимеров.
Предшествующими исследователями установлено, что конкретные катализаторы гомогенной координационной полимеризации могут быть использованы для получения полимеров, имеющих структуру по существу "подобную блочной", путем подавления переноса цепи во время полимеризации, например, проведением процесса полимеризации в отсутствие агента переноса цепи и при достаточно низкой температуре, так что перенос цепи β-гидридным элиминированием или другими процессами переноса цепи по существу исключается. В таких условиях последовательное присоединение различных мономеров, как было сказано, имеет результатом образование полимеров, имеющих последовательности или сегменты различного мономерного содержания. Некоторые примеры таких каталитических составов и процессов рассматриваются Coates, Hustad and Reinartz в Angew. Chem. Int. Ed., 41, 2236-2257 (2002), а также в US-A-2003/0114623.
К сожалению, такие процессы требуют последовательного присоединения мономеров и имеют результатом получение только одной полимерной цепи на активный центр катализатора, который ограничивает производительность катализатора. К тому же требование относительно низких температур процесса увеличивает эксплуатационные затраты, делая такие процессы непригодными для коммерческой реализации. Более того, катализатор не может быть оптимизирован для формирования каждого соответствующего типа полимера, и поэтому полный процесс имеет результатом получение блоков или сегментов полимера менее чем максимальной эффективности и/или качества. Например, обычно неизбежно формирование конкретного количества преждевременно оборванного полимера, в результате чего образуются смеси, имеющие плохие полимерный свойства. Соответственно, при нормальных рабочих условиях для последовательно получаемых блок-сополимеров, имеющих Mw/Mn 1,5 или более, полученное распределение длин блоков является относительно негомогенным, не наиболее вероятным распределением. Наконец, последовательно получаемые блок-сополимеры необходимо получать в периодическом процессе, ограничивающем скорости и повышающем затраты по отношению к реакциям полимеризации, проводимым в непрерывном процессе.
По этим причинам было бы очень желательно предоставить способ получения олефиновых сополимеров в хорошо определенных блоках или сегментах в процессе с использованием катализаторов координационной полимеризации, способных работать при высоких каталитических эффективностях. В дополнение, было бы желательно предоставить способ и получаемые в результате блок-сополимеры или сегментированные сополимеры, где можно влиять на вставку концевых блоков или упорядочение блоков в полимере подходящим подбором условий процесса. Наконец, было бы желательно предоставить непрерывный процесс получения мульти-блок-сополимеров.
Применение некоторых алкильных соединений металлов и других соединений, таких как водород, в качестве агентов переноса цепи для прерывания роста цепи при полимеризации олефинов хорошо известно в технике. Кроме того, известно использование таких соединений, особенно алкиловых соединений алюминия, в качестве акцепторов или в качестве сокатализаторов при полимеризациях олефинов. В Macromolecules, 33, 9192-9199 (2000), применение конкретных триалкиловых соединений алюминия в качестве агентов переноса цепи в сочетании с конкретными двойными цирконоценовыми каталитическими составами имело результатом полипропиленовые смеси, содержащие небольшие количества полимерных фракций, содержащих как изотактические, так и атактические сегменты цепи. В Liu and Rytter, Macromolecular Rapid Comm., 22, 952-956 (2001), и Bruaseth and Rytter, Macromolecules, 36, 3026-3034 (2003), смеси этилена и 1-гексена полимеризовали подобным каталитическим составом, содержащим триметилалюминиевый агент переноса цепи. В последней ссылке авторы суммировали исследования предшествующего уровня техники следующим образом (некоторые цитаты пропущены):
"Смешивание двух металлоценов, имеющих известное поведение при полимеризации, может быть использовано для регулирования микроструктуры полимера. Было проведено несколько исследований полимеризации этена путем смешивания двух металлоценов. Общие наблюдения были такие, что путем объединения катализаторов, которые по отдельности дают полиэтен с различной Mw, может быть получен полиэтен с более широким и в некоторых случаях бимодальным MWD". [S]oares and Kim (J. Polym. Sci., Part A: Polym. Chem., 38, 1408-1432 (2000), разработан критерий для испытания бимодальности MWD полимеров, полученных с двойными односайтовыми катализаторами, как показано на примере сополимеризации этилен/1-гексен смесями Et(Ind)2ZrCl2/Cp2HfCl2 и Et(Ind)2ZrCl2/CGC (катализатор ограниченной геометрии) на носителе из диоксида кремния. Heiland and Kaminsky (Macromol. Chem., 193, 601-610 (1992)) исследовали смесь Et-(Ind)2ZrCl2 и гафниевого аналога в сополимеризации этена и 1-бутена.
Эти исследования не содержат какого-либо указания на взаимодействие между двумя различными сайтами, например, путем повторной адсорбции оборванной цепи на альтернативном сайте. Такие сообщения были опубликованы, однако, для полимеризации пропена. Chien et al. (J. Polym. Sci., Part A: Polym. Chem., 37, 2439-2445 (1999), Macromol., 30, 3447-3458 (1997)) исследована полимеризация пропена гомогенными бинарными цирконоценовыми катализаторами. Смесь изотактического полипропилена (i-PP), атактического полипропилена (а-PP) и стереоблочной фракции (i-PP-b-a-PP) была получена с бинарной системой, содержащей изоспецифический и аспецифический предшественник с боратом и TIBA в качестве сокатализатора. Путем применения бинарной смеси изоспецифического и синдиоспецифического цирконоценов была получена смесь изотактического полипропилена (i-PP), синдиотактического полипропилена (s-PP) и стереоблочной фракции (i-PP-b-s-PP). Механизм формирования стереоблочной фракции, вероятно, вовлекает обмен с растущими цепями между двумя различными каталитическими сайтами. Przybyla and Fink (Acta Polym., 50, 77-83 (1999) использовали два различных типа металлоценов (изоспецифический и синдиоспецифический) на носителе из того же диоксида кремния для полимеризации пропена. Они сообщали, что с конкретным типом носителя из диоксида кремния происходит перенос цепи между активными частицами в каталитической системе и образуется стереоблочный РР. Lieber and Brintzinger (Macromol. 3, 9192-9199 (2000)) предложили более подробное объяснение, как происходит перенос растущей полимерной цепи от одного типа металлоцена к другому. Они исследовали полимеризацию пропена каталитическими смесями из двух различных анса-цирконоценов. Различные катализаторы вначале исследовались индивидуально в отношении их тенденции к обмену алкил-полимерила с алкилалюминиевым активатором и затем по парам в отношении их способности производить полимеры со стереоблочной структурой. Они сообщали, что формирование стереоблок-полимеров смесью цирконоценовых катализаторов с различными стереоселективностями является случайным при эффективном обмене полимерилами между центрами Zr катализатора и центрами Al сокатализатора.
Brusath и Rytter затем раскрыли свои собственные наблюдения использования двойных цирконоценовых катализаторов для полимеризации смесей этилен/1-гексен и сообщили об эффектах влияния двухсайтового катализатора на активность полимеризации, внедрение сомономера и микроструктуру полимера при использовании метилалюмоксанового сокатализатора.
Анализ вышеупомянутых результатов показывает, что Rytter и сотрудники, вероятно, потерпели неудачу при попытке использовать сочетания катализатора, сокатализатора и третьих компонентов, которые были бы способны к повторной адсорбции полимерной цепи от агента переноса цепи на обоих активных каталитических сайтах, т.е. двусторонней повторной адсорбции. Несмотря на указание того, что обрыв цепи из-за присутствия триметилалюминия, вероятно, происходит, что касается полимера, образованного от катализатора, включающего минимальный сомономер, и после этого, вероятно, происходит обмен полимерилами с более открытым каталитическим сайтом с последующей возможной непрерывной полимеризацией, очевидность обратного перемещения полимерных лигандов представляется недостаточной в ссылке. Фактически, в более поздней информации, Rytter и др., Polymer, 45, 7853-7861 (2004), сообщалось, что никакого переноса цепи между каталитическими сайтами в действительности не было в более ранних экспериментах. Подобные полимеризации описаны в WO 98/34970.
В патентах США 6380341 и 6169151 сказано, что применение "дифференциального" металлоценового катализатора, то есть металлоцена, способного к относительно легкому превращению между двумя стереоизомерными формами, имеющими отличающиеся характеристики полимеризации, такие как отличающиеся константы сополимеризации, приводит к образованию олефиновых сополимеров, имеющих "блоковую" структуру. Неблагоприятно, что соответствующие стереоизомеры таких металлоценов обычно не имеют значительного различия по полимерообразующим свойствам и не способны к формированию как высоко кристаллических, так и аморфных сегментов блок-сополимера, например, из заданной мономерной смеси при фиксированных условиях реакции. Более того, так как относительный показатель двух "дифференциальных" форм катализатора не может быть изменен, нет возможности применения "дифференциальных" катализаторов для изменения состава полимерных блоков или соотношения соответствующих блоков. Наконец, в способах олефиновой блок-сополимеризации предшествующего уровня техники не было возможности легко контролировать упорядочение различных полимерных блоков и, особенно, контролировать характер завершающего блока или сегмента мульти-блок-сополимера. Для конкретных применений желательно получать полимеры, имеющие концевые блоки, которые являются высоко кристаллическими, которые функционализируются или более легко функционализируются или которые обладают другими отличающими их свойствами. Например, вероятно, что полимеры, где концевые сегменты или блоки являются кристаллическими или стекловидными, обладают улучшенным сопротивлением к истиранию и термическими свойствами, такими как предел прочности при растяжении, упругое восстановление и остаточная деформация при сжатии. Кроме того, полимеры, где блоки, имеющие аморфные свойства, являются внутренними или преимущественно соединенными между кристаллическими или стекловидными блоками, имеют усовершенствованные эластомерные свойства, такие как усовершенствованную силу сжатия и восстановление, особенно при повышенных температурах.
В JACS, 2004, 126, 10791-10712, Gibson и др. обсуждают эффекты "катализируемой живой полимеризации" на молекулярномассовое распределение. Авторы определяют катализируемую живую полимеризацию следующим образом:
"…если перенос цепи к алюминию устанавливает единственный механизм переноса и обмен растущей полимерной цепи между центрами переходного металла и алюминия является очень быстрым и обратимым, полимерные цепи будут казаться растущими на алюминиевых центрах. Это может быть затем обоснованно описанным как катализируемая реакция роста цепи на алюминии… Привлекательным проявлением этого типа реакции роста цепи является распределение по Poisson молекулярных масс продукта, как противопоставленное распределению по Schultz-Flory, которое возникает, когда перенос β-Н сопровождает распространение."
Авторы доложили результаты катализируемой живой гомополимеризации этилена с использованием содержащего железо катализатора в сочетаниях с ZnEt2, ZnMe2 или Zn(i-Pr)2. Гомолептические алкилы алюминия, бора, олова, лития, магния и свинца не вызывают катализируемого роста цепи. Применение GaMe3 в качестве сокатализатора имело результатом получение полимера, имеющего узкое молекулярномассовое распределение. Однако после анализа зависимого от времени распределения продукта авторы пришли к заключению, что эта реакция была "не просто катализируемой реакцией роста цепи". Ссылка терпит неудачу в раскрытии применения двух или более катализаторов в сочетании с челночным агентом цепи для получения мульти-блок-сополимеров. Подобные процессы с использованием единственных катализаторов описаны в патентах США 5210338, 5276220 и 6444876.
Более ранними разработчиками было заявлено о сформированных блок-сополимерах с использованием единственного катализатора типа Циглера-Натта в нескольких реакторах, соединенных последовательно, смотри, например, патенты США 3970719 и 4039632. Дополнительные процессы на основе катализатора Циглера-Натта и полимеры раскрыты в патентах США 4971936, 5089573, 5118767, 5118768, 5134209, 5229477, 5270276, 5270410, 5294581, 5543458, 5550194 и 5693713, а также в ЕР-А-470171 и ЕР-А-500530.
Несмотря на достижения предшествующих исследователей, в технике сохраняется потребность в способе полимеризации, который был бы пригоден для получения подобных блоковым сополимеров, особенно мульти-блок-сополимеров и в особенности линейных мульти-блок-сополимеров, с высоким выходом и селективностью. Более того, было бы желательно, если бы был предложен усовершенствованный способ получения мульти-блок-сополимеров, особенно линейных мульти-блок-сополимеров, из двух или более олефиновых мономеров, таких как этилен, и одного или нескольких сомономеров с использованием челночного агента. В дополнение было бы желательно предоставить такой усовершенствованный способ, который прогоден для получения мульти-блок-сополимеров, особенно линейных мульти-блок-сополимеров, имеющих относительно узкое молекулярномассовое распределение. Было бы дополнительно желательно предоставить способ получения сополимеров, имеющих более чем два сегмента или блока. Еще было бы желательно предоставить способ идентификации сочетаний катализаторов и челночных агентов цепи, способных давать такие мульти-блок-сополимеры. Еще дополнительно было бы желательно предоставить способ независимого контроля порядка различных полимерных блоков, особенно способ получения олефиновых блок-сополимеров, содержащих концевые блоки, имеющие высокую кристалличность и/или функциональность. И наконец, было бы желательно предоставить усовершенствованный способ получения каких-либо из вышеупомянутых желательных полимерных продуктов в непрерывном процессе без необходимого последовательного присоединения мономеров. Очень желательно, чтобы такой способ допускал независимый контроль количества и/или идентичности используемых челночного агента (агентов) и/или катализаторов.
Сущность изобретения
Согласно данному изобретению в настоящее время предложен состав для применения в полимеризации полимеризуемого присоединением мономера, предпочтительно двух или более полимеризуемых присоединением мономеров, особенно этилена и по меньшей мере одного сополимеризуемого сомономера для формирования сегментированного сополимера (мульти-блок-сополимера), указанный сополимер содержит два или более, предпочтительно три или более, сегментов или блоков, различных по одному или нескольким химическим или физическим свойствам, как дополнительно раскрыто здесь, состав содержит смесь или продукт реакции, полученный в результате объединения:
(А) первого катализатора полимеризации олефинов,
(В) второго катализатора полимеризации олефинов, способствующего получению полимеров, отличающихся по химическим или физическим свойствам от полимера, полученного с катализатором А в эквивалентных условиях полимеризации, и
(С) челночного агента цепи, и
предпочтительно смесь или продукт реакции, полученный в результате объединения:
(А) первого катализатора полимеризации олефинов, имеющего высокий показатель внедрения сомономера,
(В) второго катализатора полимеризации олефинов, имеющего показатель внедрения сомономера менее чем 95 процентов, предпочтительно менее чем 90 процентов, более предпочтительно менее чем 25 процентов и наиболее предпочтительно менее чем 10 процентов показателя внедрения сомономера катализатора А, и
(С) челночного агента цепи.
В другом варианте воплощения изобретения предложен способ подбора смеси катализаторов (А) и (В) и челночного агента цепи (С), способствующей получению мульти-блок-сополимеров по изобретению, особенно таких сополимеров, содержащих этилен в полимеризованной форме.
В дополнительном варианте воплощения данного изобретения предложен способ получения сегментированного сополимера, особенно такого сополимера, содержащего этилен и необязательно один или несколько полимеризуемых присоединением мономеров, иных чем этилен, указанный процесс содержит контактирование этилена и необязательно одного или нескольких полимеризуемых присоединением мономеров, иных чем этилен в условиях полимеризации присоединением с составом, содержащим:
смесь или продукт реакции, полученный в результате объединения:
(А) первого катализатора полимеризации олефинов, имеющего высокий показатель внедрения сомономера,
(В) второго катализатора полимеризации олефинов, имеющего показатель внедрения сомономера менее чем 90 процентов, предпочтительно менее чем 50 процентов, наиболее предпочтительно менее чем 5 процентов показателя внедрения сомономера катализатора А, и
(С) челночного агента цепи.
Предпочтительно указанный способ принимает форму непрерывного осуществляемого в растворе процесса формирования блок-сополимеров, особенно мульти-блок-сополимеров, предпочтительно линейных мульти-блок-сополимеров из двух или более мономеров, более конкретно этилена и С4-20 олефина или циклоолефина и наиболее предпочтительно этилена и С4-20 α-олефина, с использованием составных катализаторов, которые не способны к взаимопревращению. То есть катализаторы являются химически различными. В условиях непрерывно осуществляемой в растворе полимеризации процесс идеально подходит для полимеризации смесей мономеров при высоких превращениях мономеров. В таких условиях полимеризации челночное перемещение от челночного агента цепи к катализатору становится преимущественным по сравнению с ростом цепи, и мульти-блок-сополимеры, особенно линейные мульти-блок-сополимеры по изобретению, образуются с высокой эффективностью.
В другом варианте воплощения изобретения предложен сегментированный сополимер (мульти-блок-сополимер), особенно такой сополимер, содержащий этилен в полимеризованной форме, указанный сополимер содержит два или более, предпочтительно три или более сегментов, отличающихся по содержанию сомономера или плотности или по другому химическому или физическому свойству. Очень предпочтительно, если сополимер обладает молекулярномассовым распределением, Mw/Mn, менее чем 3,0, предпочтительно менее чем 2,8. Наиболее предпочтительно полимеры по изобретению являются мульти-блок-сополимерами этилена.
В еще одном варианте воплощения изобретения предложены функционализованные производные вышеупомянутых сегментированных или мульти-блок-сополимеров.
В еще дополнительном варианте воплощения данного изобретения предложена полимерная смесь, содержащая: (1) органический или неорганический полимер, предпочтительно гомополимер этилена или пропилена и/или сополимер этилена или пропилена и сополимеризуемого сомономера и (2) мульти-блок-сополимер по данному изобретению или полученный согласно способу по данному изобретению. В желательном варианте воплощения компонентом (1) является матричный полимер, содержащий полиэтилен высокой плотности или изотактический полипропилен, и компонентом (2) является эластомерный мульти-блок-сополимер. В предпочтительном варианте воплощения компонент (2) содержит окклюзии матричного полимера, образованные во время компаундирования компонентов (1) и (2).
Краткое описание чертежей
Фиг.1 является схематическим изображением процесса челночного перемещения полимерной цепи с вовлечением двух каталитических сайтов.
Фиг.2 показывает графики дельта DSC-CRYSTAF как функции DSC энтальпии плавления для примеров 1-19, сравнительных полимеров A-F и обычных сополимеров этилен/октен.
Фиг.3-27 являются DSC кривыми нагревания и соответствующими отчетами CRYSTAF для полимеров из примеров 1-19 и сравнительных полимеров A-F, включая распределение пиковых температур и интеграции фракций по массе для областей, соответствующих надлежащим пиковым температурам.
Фиг.28 является микрофотографией низкого разрешения, показывающей кристаллическую структуру различных сравнительных примеров, а также полимеров, полученных с применением изменяющихся количеств челночного агента цепи по изобретению.
Фиг.29 является микрофотографией высокого разрешения, показывающей морфологию различных сравнительного сополимера этилен/1-октен, а также трех мульти-блок-сополимеров, полученных согласно изобретению.
Фиг.30 изображает поведение при циклической 300-процентной деформации образцов, полученных из полимера из примера 17.
Фиг.31 изображает релаксацию напряжений сшитых волокон из полимера примера 11 и сравнительного G при 21°С и 40°С.
Фиг.32 и 33 являются графиками среднечисленной молекулярной массы полимера (Mn) как функции выхода для полимеризаций, проведенных в примерах 27 и 28 соответственно.
Фиг.34 является графиком пиковой температуры плавления против плотности для мульти-блок-сополимеров этилен/1-октен по изобретению (линия), а также для типичных традиционных сополимеров этилен/1-октен (кривая).
Фиг.35 является графиком динамического модуля упругости как функции температуры для сравнительных сополимеров этилен/1-октен и пропилен/этилен и для двух мульти-блок-сополимеров этилен/1-октен по изобретению, полученных с различными количествами челночного агента цепи.
Фиг.36-49 являются DSC кривыми нагревания и соответствующими отчетами CRYSTAF для полимеров из примеров 24-33 и сравнительных М-Р, соответственно, включая распределение пиковых температур и интеграции фракций по массе для областей, соответствующих надлежащим пиковым температурам.
Фиг.50 показывает графики дельта DSC-CRYSTAF как функции DSC энтальпии плавления для полимеров из примеров 24, 25, 29-33, сравнительных полимеров М-Р и обычных сополимеров этилен/октен.
Фиг. 51-53 являются микроскопическими изображениями атомного усиления (atomic force, вероятно наноразрешения) сделанных микротомом срезов образцов, отлитых под давлением пластинок изотактического полипропилена с модифицированной ударной вязкостью, соответствующими образцам а, b и d таблицы 13 соответственно.
Фиг.54 является графиком содержания октена фракций фракционированного TREF сополимера этилен/1-октен против температуры элюирования TREF фракции для полимера из примера 5 и сравнительных полимеров Е и F.
Фиг.55 является графиком содержания октена фракций фракционированного TREF сополимера этилен/1-октен против температуры элюирования TREF фракции для полимера из примера 5 и сравнительного F.
Подробное описание изобретения
Все ссылки на Периодическую таблицу элементов здесь будут относится к Периодической таблице элементов, опубликованной и охраняемой авторским правом CRC Press, Inc., 2003. Также любые ссылки на группу или группы должны быть группой или группами, отраженными в Периодической таблице элементов с использованием системы IUPAC для нумерации групп. Если не установлено иначе, подразумевается из контекста или привычно в технике, все части и проценты даны на основе массы. Для целей патентной практики Соединенных Штатов содержание какого-либо патента, патентной заявки или публикации, на которые ссылаются здесь, приобщаются к сему ссылкой во всей их полноте (или эквивалентная США их версия приобщается к сему ссылкой) особенно, что касается раскрытия технологий синтеза, определений (до такой степени не несовместимой с какими-либо определениями, приведенными здесь) и общеизвестности в технике.
Термин "содержащий" и его производные не предназначается для исключения присутствия какого-либо дополнительного компонента, стадии или процедуры, раскрыты они здесь или нет. Для того чтобы избежать какого-либо сомнения, все заявленные здесь составы, благодаря применению термина "содержащий", могут включать какую-либо дополнительную добавку, адъювант или соединение, полимерное оно или нет, если не установлено иначе. Напротив, термин "состоящий по существу из" исключает из сферы какого-либо последующего перечисления любой другой компонент, стадию или процедуру, за исключением тех, которые не являются существенными для работоспособности. Термин "состоящий из" исключает любой компонент, стадию или процедуру, конкретно не очерченные или не перечисленные. Термин "или", если не установлено иначе, относится к перечисленным членам отдельно, а также в любом сочетании.
Термин "полимер" включает и обычные гомополимеры, то есть, гомогенные полимеры, полученные из единственного мономера, и сополимеры (взаимозаменяемо упоминаемые здесь как интерполимеры), означающие полимеры, полученные путем взаимодействия по меньшей мере двух мономеров, или иным образом содержащие химически дифференцированные сегменты или блоки в них, даже если сформированы из единственного мономера. Более конкретно, термин "полиэтилен" включает гомополимеры этилена и сополимеры этилена и одного или нескольких С3-8 α-олефинов, в которых этилен составляет по меньшей мере 50 мол. процентов. Термин "кристаллический", если используется, относится к полимеру, который обладает точкой перехода первого порядка или кристаллической температурой плавления (Tm), которую определяют дифференциальной сканирующей калориметрией (DSC) или эквивалентной методикой. Термин может быть использован взаимозаменяемо с термином "полукристаллический". Термин "аморфный" относится к полимеру, у которого нет кристаллической температуры плавления, определяемой дифференциальной сканирующей калориметрией (DSC) или эквивалентной методикой.
Термин "мульти-блок-сополимер" или "сегментированный сополимер" относится к полимеру, содержащему два или более химически различных регионов или сегментов (упоминаемых как "блоки"), предпочтительно соединенных линейным образом, то есть к полимеру, содержащему химически дифференцированные звенья, которые соединены конец с концом по отношению к полимеризуемой этиленовой функциональности, скорее чем в виде подвесных или привитых. В предпочтительном варианте воплощения блоки отличаются по количеству или типу внедренного в них сомономера, плотности, степени кристалличности, размеру кристаллитов, приписываемому полимеру такого состава, типу или степени регулярности молекулярной структуры (изотактический или синдиотактический), регио-регулярности или регио-нерегулярности, количеству разветвлений, включая длинноцепочечное разветвление или гиперразветвление, гомогенности или другому химическому или физическому свойству. По сравнению с блок-сополимерами предшествующего уровня техники, включая сополимеры, полученные последовательным присоединением мономера, с дифференциальными катализаторами или методом анионной полимеризации, сополимеры по изобретению характеризуются уникальными распределениями и полидисперсностью полимера (PDI или Mw/Mn), распределением длин блоков и/или распределением количества блоков, благодаря, в предпочтительном варианте воплощения, эффекту челночного агента (агентов) в сочетании с составными катализаторами. Более конкретно, когда их получают в непрерывном процессе, полимеры желательно имеют PDI от 1,7 до 2,9, предпочтительно от 1,8 до 2,5, более предпочтительно от 1,8 до 2,2 и наиболее предпочтительно от 1,8 до 2,1. Когда их получают в периодическом или полупериодическом процессе, полимеры желательно имеют PDI от 1,0 до 2,9, предпочтительно от 1,3 до 2,5, более предпочтительно от 1,4 до 2,0 и наиболее предпочтительно от 1,4 до 1,8.
Термин "этиленовый мульти-блок-сополимер" означает мульти-блок-сополимер, содержащий этилен и один или несколько сополимеризуемых сомономеров, где этилен составляет множество полимеризованных мономерных звеньев по меньшей мере одного блока или сегмента в полимере, предпочтительно по меньшей мере 90 мол. процентов, более предпочтительно по меньшей мере 95 мол. процентов и наиболее предпочтительно по меньшей мере 98 мол. процентов указанного блока. На основе общей массы полимера этиленовые мульти-блок-сополимеры по данному изобретению предпочтительно имеют содержание этилена от 25 до 97 процентов, более предпочтительно от 40 до 96 процентов, еще более предпочтительно от 55 до 95 процентов и наиболее более предпочтительно от 65 до 85 процентов.
Так как соответствующие различимые сегменты или блоки, сформированные из двух или более мономеров, соединены в единые полимерные цепи, полимер не может быть полностью фракционирован с помощью стандартных методов селективной экстракции. Например, полимеры, содержащие регионы, которые являются относительно кристаллическими (сегменты высокой плотности), и регионы, которые являются относительно аморфными (сегменты низкой плотности), не могут быть селективно экстрагированы или фракционированы с помощью отличающихся растворителей. В предпочтительном варианте воплощения количество полимера, экстрагируемого с помощью диалкилэфирного или алканового растворителя, менее чем 10 процентов, предпочтительно менее чем 7 процентов, более предпочтительно менее чем 5 процентов и наиболее предпочтительно менее чем 2 процентов от общей массы полимера.
В дополнение, мульти-блок-сополимеры по изобретению желательно имеют PDI, соответствующий скорее распределению по Schultz-Flory, чем распределению по Poisson. Применение данного процесса полимеризации имеет результатом продукт, имеющий как полидисперсное распределение блоков, так и полидисперсное распределение размеров блоков. Конечным результатом этого является образование полимерных продуктов, имеющих улучшенные и отличающиеся физические свойства. Теоретические преимущества полидисперсного распределения блоков были ранее смоделированы и обсуждались в Potemkin, Physical Review E (1998) 57(6), p. 6902-6912, и Dobrynin, J. Chem. Phys. (1997) 107(21), p. 9234-9238.
В дополнительном варианте воплощения полимеры по изобретению, особенно полученные в реакторе непрерывного действия для полимеризации в растворе, обладают наиболее вероятным распределением длин блоков. Наиболее предпочтительными полимерами по изобретению являются мульти-блок-сополимеры, содержащие 4 или более блоков или сегментов, включая концевые блоки.
Следующая математическая обработка полученных полимеров основана на теоретически выведенных параметрах, которые представляются применимыми к полимерам по данному изобретению и демонстрируют, что, особенно в устойчивом состоянии в реакторе постоянного действия с хорошим перемешиванием, длины блоков конечного полимера, полученного с использованием 2 или более катализаторов, будут согласоваться с наиболее вероятным распределением, выведенным следующим образом, где рi означает вероятность распространения что касается последовательностей блоков от катализатора i. Теоретическая обработка основана на стандартных допущениях и методах, известных в технике, и используется в прогнозировании воздействий кинетики полимеризации на молекулярную архитектуру, включая применение выражений скорости реакций массового действия, на которые не влияют длины цепей или блоков. Такие методы ранее были раскрыты в W. H. Ray, J. Macromol. Sci., Rev. Macromol. Chem., C8, 1 (1972), и A. E. Hamielec and J. F. MacGregor, "Polymer Reaction Engineering", K. H. Reichert and W. Geisler, Eds., Hanser, Munich, 1983. В дополнение предполагается, что соседние последовательности, сформированные тем же катализатором, образуют единый блок. Для катализатора i фракция последовательностей с длиной n задана Xi[n], где n представляет целое число от 1 до бесконечности, представляющее число мономерных звеньев в блоке.
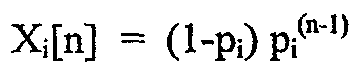
наиболее вероятное распределение длин блоков
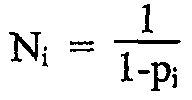
среднечисленная длина блока
Каждый катализатор имеет вероятность распространения (рi) и формирует сегмент полимера, имеющий уникальные среднюю длину блока и распределение. В наиболее предпочтительном варианте воплощения вероятность распространения определена как
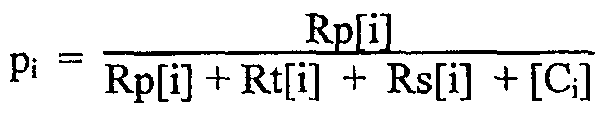
для каждого катализатора i={1, 2…}, где
Rp[i]=скорость расхода мономера катализатором i (моль/л),
Rt[i]=общая скорость переноса и обрывания цепи катализатором i (моль/л),
Rs[i]=скорость челночного перемещения находящегося в состоянии покоя полимера к другим катализаторам (моль/л) и
[Ci]=концентрация катализатора i (моль/л).
Находящиеся в состоянии покоя полимерные цепи относятся к полимерным цепям, которые присоединены к CSA.
Общий расход мономера или скорость распространения полимера, Rp[i], определяют, используя константу кажущейся скорости, kpi, умноженную на общую концентрацию мономера, [M], как следует:
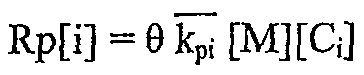
Общая скорость переноса цепи дана ниже, включая величины для переноса цепи к водороду (Н2), бета-гидридное элиминирование и перенос цепи к челночному агенту цепи (CSA). Время пребывания в реакторе задано θ, и каждая индексированная величина k является постоянной скорости.
Rt[i]=θkH2i[H2][Сi]+θkβi[Сi]+θkai[CSA][Ci]
Для двойной каталитической системы скорость челночного перемещения полимера между катализаторами 1 и 2 дана следующим образом:
Rs[1]=Rs[2]=θka1[CSA]θka2[C1][C2].
Если используют более 2 катализаторов, тогда приводят подобные члены и увеличивают сложность теоретического отношения для результата Rs[i], но окончательный вывод, что полученные распределения длин блоков являются наиболее вероятными, остается тем же.
Используемый здесь по отношению к химическому соединению, если конкретно не указано иначе, единственный термин включает все изомерные формы и наоборот (например, "гексан", включает все изомеры гексана отдельно или вместе). Термины "соединение" и "комплекс" используются здесь взаимозаменяемо по отношению к органическим, неорганическим и металлорганическим соединениям. Термин "атом" относится к наименьшей составляющей элемента, невзирая на ионное состояние, то есть независимо от того, несет он заряд или частичный заряд или нет или связан с другим атомом. Термин "гетероатом" относится к атому, иному чем углерод или водород. Предпочтительные гетероатомы включают F, Cl, Br, N, O, P, B, S, Si, Sb, Al, Sn, As, Se и Ge.
Термин "гидрокарбил" относится к одновалентным заместителям, содержащим атомы только водорода и углерода, включая разветвленные или неразветвленные, насыщенные или ненасыщенные, циклические, полициклические или нециклические разновидности. Примеры включают алкил-, циклоалкил-, алкенил-, алкадиенил-, циклоалкенил-, циклоалкадиенил-, арил- и алкинил- группы. "Замещенный гидрокарбил" относится к гидрокарбилгруппе, которая замещена одной или несколькими заместителями иными чем гидрокарбильные группы. Термин "содержащий гетероатом гидрокарбил" или "гетерогидрокарбил" относится к одновалентным группам, в которых по меньшей мере один атом, отличный от водорода или углерода, присутствует наряду с одним или несколькими атомами углерода и одним или несколькими атомами водорода. Термин "гетерокарбил" относится к группам, содержащим один или несколько атомов углерода и один или несколько гетероатомов и никаких атомов водорода. Связь между атомом углерода и каким-либо гетероатомом, а также связи между какими-либо двумя гетероатомами могут быть одинарными или множественными ковалентными связями или координационными или другими донорными связями. Таким образом, алкилгруппа, замещенная гетероциклоалкил-, арил- замещенный гетероциклоалкил- гетероарил-, алкил-замещенный гетероарил-, алкокси-, арилокси- дигидрокарбилборил-, дигидрокарбилфосфино-, дигидрокарбиламино-, тригидрокарбилсилил-, гидрокарбилтио- или гидрокарбилселеногруппой находится в сфере охвата термином гетероалкил. Примеры подходящих гетероалкилгрупп включают цианометил-, бензоилметил-, (2-пиридил)метил- и трифторметил-группы.
Используемый здесь термин "ароматический" относится к многоатомной циклической конъюгированной кольцевой системе, содержащей (4δ+2) π-электронов, где δ представляет собой целое число более чем или равное 1. Термин "конденсированный", используемый здесь по отношению к кольцевой системе, содержащей два или более многоатомных циклических колец, означает, что касается по меньшей мере двух ее колец, по меньшей мере одна пара соседних атомов включена в оба кольца. Термин "арил" относится к одновалентному ароматическому заместителю, который может быть единственным ароматическим кольцом или несколькими ароматическими кольцами, которые конденсированы вместе, связаны ковалентно или связаны с общей группой, такой как группа метилена или этилена. Примеры ароматического кольца (колец) включают фенил, нафтил, антраценил и бифенил, наряду с прочими.
"Замещенный арил" относится к арилгруппе, в которой один или несколько атомов водорода, связанные с каким-либо атомом углерода, замещены одной или несколькими функциональными группами, такими как алкил, замещенный алкил, циклоалкил, замещенный циклоалкил, гетероциклоалкил, замещенный гетероциклоалкил, галоген, галогеналкил (напр. CF3), гидрокси, амино, фосфидо, алкокси, амино, тио, нитро, и как насыщенные, так и ненасыщенные циклические углеводороды, которые конденсированы с ароматическим кольцом (кольцами), связаны ковалентно или связаны с общей группой, такой как группа метилена или этилена. Общей связующей группой может быть также карбонил, как в бензофеноне, или кислород, как в простом дифениловом эфире, или азот в дифениламине.
Термин, "показатель внедрения сомономера", относится к проценту coмономера, внедренного в coполимер, полученный в типичных условиях полимеризации этилен/coмономер в отсутствие других катализаторов полимеризации, идеально в условиях устойчивого состояния непрерывной полимеризации в растворе в углеводородном разбавителе при 100°C, давлении этилена 4,5 МПа (давление в реакторе), более чем 92 (более предпочтительно более чем 95) процентом превращении этилена и более чем 0,01 процентном превращении coмономера. Выбор комплексов металлов или каталитических составов, имеющих наибольшее различие в показателях внедрения coмономера, имеет результатом coполимеры из двух или более мономеров, имеющих наибольшее различие в свойствах блоков или сегментов, таких как плотность.
В некоторых случаях показатель внедрения сомономера может быть определен непосредственно, например, с помощью спектроскопического метода ЯМР. Часто, однако, какое-либо различие во внедрении coмономера следует определять опосредованно. Для полимеров, образованных из нескольких мономеров, это может быть осуществлено различными методами на основе реакционных способностей мономеров.
Для coполимеров, полученных с заданным катализатором, относительные количества coмономера и мономера в coполимере и, следовательно, состав coполимера определяют по относительным скоростям реакции coмономера и мономера. Математически молярное отношение coмономера к мономеру дано уравнением
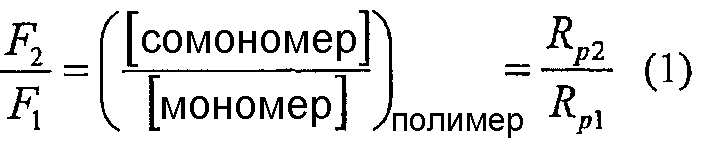
Здесь Rp2 и Rp1 означают скорости полимеризации сомономера и мономера, соответственно, и F2 и F1 означают молярные фракции каждого в сополимере. Так как F1+F2=1, мы можем преобразовать это уравнение в
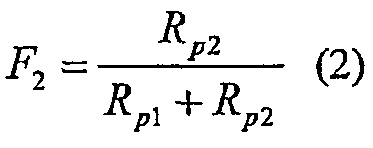
Индивидуальные скорости полимеризации сомономера и мономера обычно являются комплексными функциями температуры, катализатора и концентраций мономера/сомономера. На пределе, когда концентрация сомономера в реакционной среде падает до ноля, Rp2 падает до ноля, F2 становится равным нолю, и полимер состоит из чистого мономера. В лимитирующем случае отсутствия мономера в реакторе Rp1 становится равным нолю и F2 означает единицу (при условии, что сомономер может полимеризоваться один).
Для наиболее гомогенных катализаторов отношение сомономера к мономеру в реакторе в значительной степени определяет состав полимера, который определяют согласно или модели концевой сополимеризации, или модели предпоследней сополимеризации.
Для статистических сополимеров, в которых особенность последнего вставленного мономера диктует скорость, с которой вставляются последующие мономеры, используют модель концевой сополимеризации. В этой модели реакции вставки типа
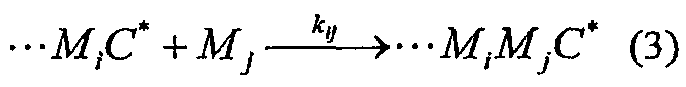
где С* представляет катализатор, Мi представляет мономер i, и kij означает константу скорости, имеющую уравнение скорости
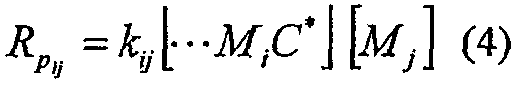
Молярную фракцию сомономера (i=2) в реакционной среде определяют по уравнению:
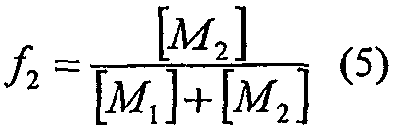
Упрощенное уравнение для состава сомономера может быть выведено, как раскрыто в George Odian, Principles of Polymerization, Second Edition, John Wiley and Sons, 1970, которое следует:
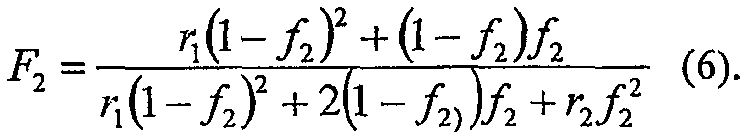
Из этого уравнения молярная фракция сомономера в полимере зависит только от молярной фракции сомономера в реакционной среде и двух зависимых от температуры констант сополимеризации, выраженных константами скорости вставки как
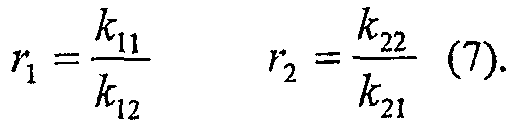
В качестве варианта в модели предпоследней coполимеризации индивидуальности последних двух мономеров, внедренных в растущую полимерную цепь, диктуют скорость последующего внедрения мономера. Реакции полимеризации проходят по типу

и индивидуальные уравнения скорости следующие:
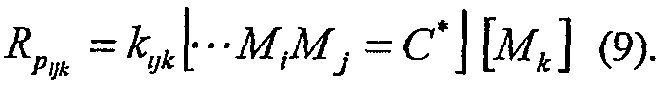
Содержание сомономера может быть рассчитано (снова, как раскрыто в George Odian, Supra.) как
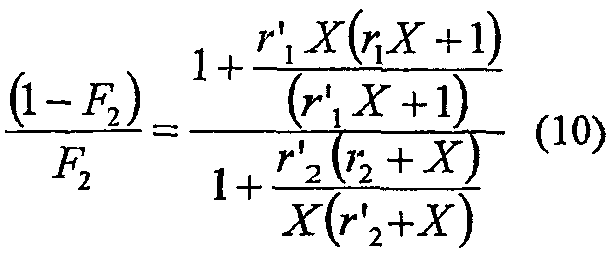
где Х определяют как
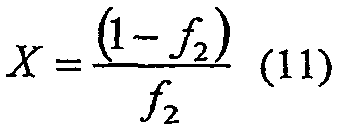
и константы сополимеризации определяют как
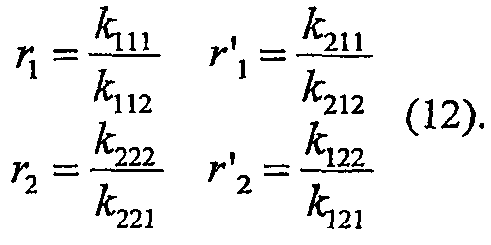
Также для этой модели полимерный состав является функцией только зависимых от температуры констант сополимеризации и молярной фракции сомономера в реакторе. Тоже самое также верно, когда может происходить обратное внедрение coмономера или мономера или в случае сополимеризации более чем двух мономеров.
Константы сополимеризации для применения в вышеупомянутых моделях могут быть прогнозированы с помощью хорошо известных теоретических методов или эмпирически выведены из действительных данных полимеризации. Подходящие теоретические методы раскрыты, например, в B. G. Kyle, Chemical and Process Thermodynamics, Third Addition, Prentice-Hall, 1999, и в Redlich-Kwong-Soave (RKS) Equation of State, Chemical Engineering Science, 1972, pp 1197-1203. Коммерчески доступные компьютерные программы могут быть использованы для облегчения выведения констант сополимеризации из экспериментально полученных данных. Одним примером такого программного обеспечения является Aspen Plus from Aspen Technology, Inc., Ten Canal Park, Cambridge, MA 02141-2201 USA.
На основе вышеупомянутых теоретических соображений данное изобретение может быть альтернативно описано как состав для применения в полимеризации двух или более полимеризуемых присоединением мономеров, особенно этилена и по меньшей мере одного сополимеризуемого сомономера, для формирования высокомолекулярного сегментированного сополимера (мульти-блок-сополимера), причем указанный сополимер содержит два или более, предпочтительно три или более сегментов или блоков, отличающихся по одному или нескольким химическим или физическим свойства, как дополнительно раскрыто здесь, состав содержит смесь или продукт реакции, полученный в результате объединения:
(A) первого катализатора полимеризации олефинов,
(B) второго катализатора полимеризации олефинов, способствующего получению полимеров, отличающихся по химическим или физическим свойствам от полимера, полученного с катализатором (A) при эквивалентных условиях полимеризации, и
(C) челночного агента цепи, и
где
r1 первого катализатора полимеризации олефинов (r1A) и
r1 второго катализатора полимеризации олефинов (r1В)
выбраны так, что отношение (r1A/r1В) в условиях полимеризации равно 0,5 или менее, предпочтительно 0,25 или менее, более предпочтительно 0,125 или менее, еще более предпочтительно 0,08 или менее, наиболее предпочтительно 0,04 или менее.
Дополнительно предложен процесс, предпочтительно осуществляемый в растворе процесс и наиболее предпочтительно непрерывный осуществляемый в растворе процесс для применения в полимеризации двух или более полимеризуемых присоединением мономеров, особенно этилена и по меньшей мере одного сополимеризуемого coмономера, для формирования высокомолекулярного сегментированного coполимера (мульти-блок-сополимера), указанный coполимер содержит два или более, предпочтительно три или более сегментов или блоков, отличающихся по одному или нескольким химическим или физическим свойствам, как дополнительно раскрыто здесь, процесс содержит стадии объединения двух или более полимеризуемых присоединением мономеров, особенно этилена и по меньшей мере одного сополимеризуемого coмономера, в условиях полимеризации с составом, содержащим смесь или продукт реакции, полученный в результате объединения:
(A) первого катализатора полимеризации олефинов,
(B) второго катализатора полимеризации олефинов, способствующего получению полимеров, отличающихся по химическим или физическим свойствам от полимера, полученного с катализатором (A) при эквивалентных условиях полимеризации, и
(C) челночного агента цепи, и
извлечения полимерного продукта, где
r1 первого катализатора полимеризации олефинов (r1A) и
r1 второго катализатора полимеризации олефинов (r1В)
выбраны так, что отношение (r1A/r1В) в условиях полимеризации равно 0,5 или менее, предпочтительно 0,25 или менее, более предпочтительно 0,125 или менее, еще более предпочтительно 0,08 или менее, наиболее предпочтительно 0,04 или менее.
Дополнительно предложен состав для применения в полимеризации двух или более полимеризуемых присоединением мономеров (называемых как мономер и сомономер(ы) соответственно), особенно этилена и по меньшей мере одного сополимеризуемого сомономера, до формирования высокомолекулярного сегментированного сополимера (мульти-блок-сополимера), указанный сополимер состоит из двух или более, предпочтительно из трех или более сегментов или блоков, отличающихся по одному или нескольким химическим или физическим свойствам, как дополнительно раскрыто здесь, состав содержит смесь или продукт реакции, полученный в результате объединения:
(А) первого катализатора полимеризации олефинов,
(В) второго катализатора полимеризации олефинов, способствующего получению полимеров, отличающихся по химическим или физическим свойствам от полимера, полученного с катализатором А в эквивалентных условиях полимеризации, и
(С) челночного агента цепи, где
содержание сомономера в молярных процентах сополимера, полученного с первым катализатором полимеризации олефинов (F1), и
содержание сомономера в молярных процентах сополимера, полученного со вторым катализатором полимеризации олефинов (F2),
подобраны так, что отношение (F1/F2) в условиях полимеризации равно 2 или более, предпочтительно 4 или более, более предпочтительно 10 или более, еще более предпочтительно 15 или более и наиболее предпочтительно 20 или более.
Дополнительно предложен способ, предпочтительно осуществляемый в растворе способ, более предпочтительно непрерывный осуществляемый в растворе способ для применения в полимеризации двух или более полимеризуемых присоединением мономеров (называемых как мономер и сомономер(ы) соответственно), особенно этилена и по меньшей мере одного сополимеризуемого сомономера, до формирования высокомолекулярного сегментированного сополимера (мульти-блок-сополимера), указанный сополимер состоит из двух или более, предпочтительно из трех или более сегментов или блоков, отличающихся по одному или нескольким химическим или физическим свойствам, как дополнительно раскрыто здесь, способ содержит стадии объединения в условиях полимеризации:
(А) первого катализатора полимеризации олефинов,
(В) второго катализатора полимеризации олефинов, способствующего получению полимеров, отличающихся по химическим или физическим свойствам от полимера, полученного с катализатором А в эквивалентных условиях полимеризации, и
(С) челночного агента цепи, где
содержание сомономера в молярных процентах сополимера, полученного с первым катализатором полимеризации олефинов (F1), и
содержание сомономера в молярных процентах сополимера, полученного со вторым катализатором полимеризации олефинов (F2),
выбраны так, что отношение (F1/F2) в условиях полимеризации равно 2 или более, предпочтительно 4 или более, более предпочтительно 10 или более, еще более предпочтительно 15 или более и наиболее предпочтительно 20 или более, в условиях полимеризации, и
извлечения полимерного продукта.
Мономеры
Подходящие мономеры для применения для получения полимеров по данному изобретению включают этилен и один или несколько полимеризуемых присоединением мономеров иных, чем этилен. Примеры подходящих coмономеров включают имеющие прямую цепь или разветвленные α-олефины из 3 до 30, предпочтительно 3 до 20 атомов углерода, такие как пропилен, 1-бутен, 1-пентен, 3-метил-1-бутен, 1-гексен, 4-метил-1-пентен, 3-метил-1-пентен, 1-октен, 1-децен, 1-додецен, 1-тетрадецен, 1-гексадецен, 1-октадецен и 1-эйкозен; циклоолефины из 3 до 30, предпочтительно 3 до 20 атомов углерода, такие как циклопентен, циклогептен, норборнен, 5-метил-2-норборнен, тетрациклододецен и 2-метил-1,4,5,8-диметано-1,2,3,4,4a,5,8,8a-октагидронафталин; ди- и полиолефины, такие как бутадиен, изопрен, 4-метил-1,3-пентадиен, 1,3-пентадиен, 1,4-пентадиен, 1,5-гексадиен, 1,4-гексадиен, 1,3-гексадиен, 1,3-октадиен, 1,4-октадиен, 1,5-октадиен, 1,6-октадиен, 1,7-октадиен, этилиденнорборнен, винилнорборнен, дициклопентадиен, 7-метил-1,6-октадиен, 4-этилиден-8-метил-1,7-нонадиен и 5,9-диметил-1,4,8-декатриен; ароматические винил-соединения, такие как моно или полиалкилстиролы (включая стирол, o-метилстирол, м-метилстирол, п-метилстирол, o,п-диметилстирол, o-этилстирол, м-этилстирол и п-этилстирол) и содержащие функциональную группу производные, такие как метоксистирол, этоксистирол, винилбензойная кислота, метилвинилбензоат, винилбензилацетат, гидроксистирол, o-хлорстирол, п-хлорстирол, дивинилбензол, 3-фенилпропен, 4-фенилпропен, a-метилстирол, винилхлорид, 1,2-дифторэтилен, 1,2-дихлорэтилен, тетрафторэтилен и 3,3,3-трифтор-1-пропен.
Челночные агенты цепи
Термин "челночный агент" относится к соединению или смеси соединений, используемой в составе по данному изобретению, которые способны вызывать обмен полимерила между по меньшей мере двумя активными каталитическими сайтами катализаторов, включенных в состав, в условиях полимеризации. То есть перенос фрагмента полимера происходит и к одному или нескольким активным каталитическим сайтам, и от них. В противоположность челночному агенту "агент переноса цепи" вызывает прекращение роста полимерной цепи и достигает однократного переноса растущего полимера от катализатора к агенту переноса. Предпочтительно челночный агент имеет отношение активностей RA-B/RB-A от 0,01 до 100, более предпочтительно от 0,1 до 10, наиболее предпочтительно от 0,5 до 2,0 и наиболее высоко предпочтительно от 0,8 до 1,2, где RA-B представляет скорость переноса полимерила от активного сайта катализатора A к активному сайту катализатора В посредством челночного агента, и RB-A означает скорость обратного переноса полимерила, т.е. скорость обмена, начинающегося от активного сайта катализатора В к активному сайту катализатора А посредством челночного агента. Желательно промежуточное соединение, образуемое между челночным агентом и цепью полимерила, является достаточно устойчивым, так что обрыв цепи является относительно редким. Желательно менее чем 90 процентов, предпочтительно менее чем 75 процентов, более предпочтительно менее чем 50 процентов и наиболее предпочтительно менее чем 10 процентов продуктов челнок-полимерил обрываются прежде достижения 3 отличающихся полимерных сегментов или блоков. В идеале, скорость челночного перемещения цепи (определяемая временем, необходимым для переноса полимерной цепи от сайта катализатора к челночному агенту цепи и затем обратно к сайту катализатора) эквивалентна скорости обрывания полимера или быстрее даже вплоть до 10 или даже в 100 раз быстрее чем скорость обрывания полимера. Это способствует образованию полимерного блока в тех же масштабах, что и распространению полимера.
Подбором различных сочетаний катализаторов, имеющих различные скорости внедрения сомономера, а также различные реакционные способности, и спариванием различных челночных агентов или смесей агентов с указанными сочетаниями катализаторов можно получать полимерные продукты, имеющие сегменты с различными плотностями или концентрациями сомономера, различными длинами блоков и различными числами таких сегментов или блоков в каждом сополимере. Например, если активность челночного агента низка по сравнению со скоростью каталитического распространения полимерной цепи одним или несколькими катализаторами, могут быть получены мульти-блок-сополимеры и полимерные смеси с увеличенной длиной блоков. И наоборот, если челночное перемещение является очень быстрым относительно распространения полимерной цепи, получают сополимер, имеющий более беспорядочную структуру цепи и более короткие длины блоков. Слишком быстрый челночный агент может давать мульти-блок-сополимер, имеющий по существу свойства статистического сополимера. Путем правильного подбора и каталитической смеси, и челночного агента могут быть получены относительно чистые блок-сополимеры, сополимеры, содержащие относительно большие полимерные сегменты или блоки, и/или смеси предшествующего с различными этиленовыми гомополимерами и/или сополимерами.
Подходящий состав, содержащий катализатор А, катализатор В и челночный агент цепи, может быть подобран для данного изобретения путем следующей многоступенчатой процедуры, специально адаптированной для дифференциации блоков на основе внедрения сомономера.
I. Один или несколько полимеризуемых присоединением, предпочтительно олефиновых, мономеров полимеризуют, используя смесь, содержащую потенциальный катализатор и потенциальный челночный агент цепи. Это испытание полимеризации желательно проводят, используя реактор периодического или полупериодического действия (т.е. без повторного снабжения катализатором или челночным агентом) предпочтительно с относительно постоянной концентрацией мономера, работающий в условиях полимеризации в растворе, обычно при использовании молярного отношения катализатора к челночному агенту цепи от 1:5 до 1:500. После образования подходящего количества полимера реакцию прекращают добавлением яда катализатора и измеряют свойства полимера (Mw, Mn и Mw/Mn или PDI).
II. Предыдущее испытание полимеризации и полимера повторяют при различных периодах времени реакции, обеспечивая серию полимеров, имеющих диапазон выходов и величин PDI.
III. Пары катализатор/челночный агент, демонстрирующие значительный перенос полимера как к челночному агенту, так и от него, характеризуются серией полимеров, где минимальный PDI менее чем 2,0, более предпочтительно менее чем 1,5 и наиболее предпочтительно менее чем 1,3. Более того, если происходит челночное перемещение цепи, Mn полимера будет увеличиваться, предпочтительно вначале линейно, когда превращение увеличивается. Наиболее предпочтительными парами катализатор/челночный агент являются пары, дающие Mn полимера как функцию превращения (или выхода полимера), соответствующую линии со статистической точностью (R2) более чем 0,95, предпочтительно более чем 0,99.
Стадии I-III затем проводят для одной или нескольких дополнительных пар потенциальных катализаторов и/или предполагаемых челночных агентов.
Затем подбирают подходящий состав, содержащий катализатор А, катализатор В и один или несколько челночных агентов цепи по изобретению так, что оба катализатора участвуют в челночном перемещении цепи одним или несколькими челночными агентами цепи, и катализатор А имеет более высокий показатель внедрения сомономера (или является иным образом способным к селективному формированию полимера) по сравнению с катализатором В в выбранных условиях реакции. Наиболее предпочтительно по меньшей мере один из челночных агентов цепи подвергается переносу полимера и в прямом, и в обратном направлениях (как установлено в предыдущем испытании) и с катализатором А, и с катализатором В. В дополнение, предпочтительно, что челночный агент цепи не уменьшает каталитическую активность (измеряемую по массе полимера, образовавшегося на массу катализатора за единицу времени) каждого катализатора (по сравнению с активностью в отсутствие челночного агента) более чем на 60 процентов, более предпочтительно такая каталитическая активность не уменьшается более чем на 20 процентов и наиболее предпочтительно каталитическая активность по меньшей мере одного из катализаторов увеличивается по сравнению с каталитической активностью в отсутствие челночного агента.
В качестве варианта возможно также выявлять желательные пары катализатор/челночный агент проведением ряда полимеризаций в стандартных условиях периодической реакции и измерением полученных среднечисленных молекулярных масс, PDI и выхода полимера или производительности. Подходящие челночные агенты характеризуются снижением полученной Mn без значительного расширения PDI или потери активности (уменьшение выхода или скорости).
Предшествующие испытания легко приспособляемы к высокопроизводительным методикам скрининга с использованием автоматизированных реакторов и аналитических датчиков и к формированию полимерных блоков, имеющих различные отличающиеся свойства. Например, некоторые потенциальные кандидаты в челночные агенты могут быть предварительно идентифицированы или синтезированы на месте путем объединения различных металлорганических соединений с различными источниками протонов и соединением или продуктом реакции, добавляемым в реакцию полимеризации с использованием каталитического состава полимеризации олефина. Отдельные полимеризации проводят при изменяемых молярных отношениях челночного агента к катализатору. В качестве минимального требования подходящими челночными агентами являются те, которые производят минимальный PDI менее чем 2,0 в экспериментах с изменяемым выходом, которые описаны выше, не оказывая значительного влияния на каталитическую активность, и предпочтительно улучшающие каталитическую активность, как описано выше.
Независимо от способа идентификации, априори, термин челночный агент имеет отношение к соединению, которое способствует получению идентифицированных в настоящее время мульти-блок-сополимеров или успешно применяется в условиях полимеризации, раскрытых здесь. Очень желательно, что мульти-блок-сополимеры, имеющие среднее число блоков или сегментов на среднюю цепь (которая определена как среднее число блоков различного состава, деленное на Mn полимера) более чем 3,0, более предпочтительно более чем 3,5, еще более предпочтительно более чем 4,0 и менее чем 25, предпочтительно менее чем 15, более предпочтительно менее чем 10,0, наиболее предпочтительно менее чем 8,0, формируют по изобретению.
Подходящие челночные агенты для применения здесь включают соединения или комплексы металлов групп 1, 2, 12 или 13, содержащие по меньшей мере одну C1-20 гидрокарбилгруппу, предпочтительно гидрокарбил-замещенные соединения алюминия, галлия или цинка, содержащие от 1 до 12 атомов углерода в каждой гидрокарбил-группе, и продукты их взаимодействия с источником протонов. Предпочтительными гидрокарбил-группами являются алкилгруппы, предпочтительно линейные или разветвленные C2-8 алкилгруппы. Наиболее предпочтительными челночными агентами для применения в данном изобретении являются соединения триалкилалюминия и диалкилцинка, особенно триэтилалюминий, три(изопропил)алюминий, три(изобутил)алюминий, три(н-гексил)алюминий, три(н-октил)алюминий, триэтилгаллий или диэтилцинк. Дополнительные подходящие челночные агенты включают продукт реакции или смесь, образованную объединением указанного металлорганического соединения, предпочтительно соединения три(C1-8)алкилалюминия или ди(C1-8)алкилцинка, особенно триэтилалюминия, три(изопропил)алюминия, три(изобутил)алюминия, три(н-гексил)алюминия, три(н-октил)алюминия или диэтилцинка, с менее чем стехиометрическим количеством (по отношению к числу гидрокарбил-групп) вторичного амина или гидроксильного соединения, особенно бис(триметилсилил)амина, трет-бутил(диметил)силоксана, 2-гидроксиметилпиридина, ди(н-пентил)амина, 2,6-ди(трет-бутил)фенола, этил(1-нафтил)амина, бис(2,3,6,7-дибензо-1-азациклогептанамина) или 2,6-дифенилфенола. Желательно, когда используют достаточно амино- или гидроксильного реагента, так что одна гидрокарбил-группа приходится на атом металла. Главными продуктами реакции указанных сочетаний, наиболее желательными для применения в данном изобретении в качестве челночных агентов, являются н-октилалюминий ди(бис(триметилсилил)амид), изопропилалюминий бис(диметил(трет-бутил)силоксид) и н-октилалюминий ди(пиридинил-2-метоксид), изобутилалюминий бис(диметил(трет-бутил)силоксан), изобутилалюминий бис(ди(триметилсилил)амид), н-октилалюминий ди(пиридин-2-метоксид), изобутилалюминий бис(ди(н-пентил)амид), н-октилалюминий бис(2,6-ди-трет-бутилфеноксид), н-октилалюминий ди(этил(1-нафтил)амид), этилалюминий бис(трет-бутилдиметилсилоксид), этилалюминий ди(бис(триметилсилил)амид), этилалюминий бис(2,3,6,7-дибензо-1-азациклогептанамид), н-октилалюминий бис(2,3,6,7-дибензо-1-азациклогептанамид), н-октиламмоний бис(диметил(трет-бутил)сульфоксид, этилцинк (2,6-дифенилфеноксид) и этилцинк(трет-бутоксид).
Специалист должен учитывать, что челночный агент подходящий для одного катализатора или сочетания катализаторов может не быть обязательно хорошим или даже удовлетворительным для применения с другим катализатором или сочетанием катализаторов. Некоторые потенциальные челночные агенты могут вредно влиять на характеристики одного или нескольких катализаторов и могут быть нежелательными для применения также и по этой причине. Соответственно, активность челночного агента цепи желательно приводят в равновесие с каталитической активностью катализаторов для достижения желательных свойств полимера. В некоторых вариантах воплощения изобретения наилучшие результаты могут быть достигнуты с применением челночных агентов, имеющих активность челночного перемещения цепи (которую измеряют по скорости переноса цепи), которая менее чем максимальная возможная скорость.
Обычно, однако, предпочтительные челночные агенты обладают наивысшими скоростями переноса полимера, а также наивысшими эффективностями переноса (уменьшенным числом случаев обрыва цепи). Такие челночные агенты могут использоваться в уменьшенных концентрациях и все еще достигать желательной степени челночного перемещения. В дополнение, такие челночные агенты имеют результатом получение наиболее коротких возможных длин полимерных блоков. Очень желательно челночные агенты цепи с единственным сайтом обмена используют из-за того, что эффективная молекулярная масса полимера в реакторе снижается, тем самым уменьшая вязкость реакционной смеси и, следовательно, уменьшая эксплуатационные затраты.
Катализаторы
Подходящие для применения здесь катализаторы включают любое соединение или сочетание соединений, которые приспособлены для получения полимеров желательного состава или типа. Могут быть использованы и гетерогенные, и гомогенные катализаторы. Примеры гетерогенных катализаторов включают хорошо известные составы Циглера-Натта, особенно галогениды металлов группы 4 на носителе из галогенидов металлов группы 2 или смешанных галогенидов и алкоксидов и хорошо известные катализаторы на основе хрома или ванадия. Предпочтительно, однако, для облегчения использования и для получения полимерных сегментов узкого диапазона молекулярной массы в растворе, чтобы катализаторы для применения здесь были гомогенными катализаторами, содержащими относительно чистое металлорганическое соединение или комплекс металла, особенно соединения или комплексы на основе металлов, выбранных из групп 3-10 или ряда лантанидов Периодической таблицы элементов. Предпочтительно, чтобы какой-либо используемый здесь катализатор не оказывал значительного пагубного влияния на характеристики другого катализатора в условиях данной полимеризации. Желательно, чтобы никакой катализатор не терял активность более чем на 25 процентов, более предпочтительно более чем на 10 процентов в условиях данной полимеризации.
Комплексы металлов для применения здесь, имеющие высокий показатель внедрения сомономера (катализатор А) включают комплексы переходных металлов, выбранных из групп 3-15 Периодической таблицы элементов, содержащие один или несколько делокализованных π-связанных лигандов или лигандов поливалентного основания Льюиса. Примеры включают металлоцен, полуметаллоцен, имеющий ограниченную геометрию, и поливалентный пиридиламин или другие комплексы полихелатирующих оснований. Комплексы обычно изображают формулой: MKkXxZz или ее димером, где
М означает металл, выбранный из групп 3-15, предпочтительно 3-10, более предпочтительно 4-8 и наиболее предпочтительно группы 4 Периодической таблицы элементов;
К', независимо в каждом случае, представляет группу, содержащую делокализованные π-электроны или одну или несколько электронных пар, через которые К связана с М, указанная группа К содержит вплоть до 50 атомов, не считая атомы водорода, необязательно две или более групп К могут быть соединены вместе, образуя связанную мостиковой связью структуру, и еще необязательно одна или несколько групп К могут быть связаны с Z, X или и с Z, и с Х;
Х, независимо в каждом случае, представляет одновалентную анионную часть молекулы, имеющую вплоть до 40 неводородных атомов, необязательно одна или несколько групп Х могут быть связаны вместе, образуя двухвалентную или поливалентную анионную группу, и еще необязательно одна или несколько групп Х и одна или несколько групп Z могут быть связаны вместе, образуя тем самым часть молекулы, которая и ковалентно связана с М, и координирована с ним;
Z, независимо в каждом случае, представляет нейтральный донорный лиганд основания Льюиса из вплоть до 50 неводородных атомов, содержащий по меньшей мере одну неразделенную электронную пару, через которую Z координируется с М;
k означает целое число от 0 до 3;
х означает целое число от 1 до 4;
z означает целое число от 0 до 3 и
сумма k+x равна состоянию формального окисления М.
Подходящие комплексы металлов включают те, которые содержат от 1 до 3 π-связанных анионных или нейтральных лигандных групп, которые могут быть циклическими или нециклическими делокализованными π-связанными анионными лигандными группами. Примерами таких π-связанных групп являются конъюгированные или неконъюгированные, циклические или нециклические диеновые или диенилгруппы, аллилгруппы, группы боратабензола, группы фосфола и арена. Под термином "π-связанный" подразумевается, что лиганд-группа связана с переходным металлом путем распределения электронов от частично делокализованной π-связи.
Каждый атом в делокализованной π-связанной группе может быть независимо замещенным радикалом, выбранным из группы, состоящей из водорода, галогена, гидрокарбила, галогенгидрокарбила, замещенных гидрокарбилом гетероатомов, где гетероатом выбран из групп 14-16 Периодической таблицы элементов, и таких радикалов замещенных гидрокарбилом гетероатомов, дополнительно замещенных группой, содержащей гетероатом группы 15 или 16. Кроме того, два или более таких радикалов могут вместе образовывать конденсированную кольцевую систему, включая частично или полностью галогенированные конденсированные кольцевые системы, или они могут образовывать металлоцикл с металлом. Термин "гидрокарбил" охватывает C1-20 линейные разветвленные и циклические алкил-радикалы, C6-20 ароматические радикалы, C7-20 алкил-замещенные ароматические радикалы и C7-20 арил-замещенные алкил-радикалы. Подходящие гидрокарбил-замещенные гетератомные радикалы включают моно-, ди- и тризамещенные радикалы бора, кремния, германия, азота, фосфора или кислорода, где каждая из гидрокарбил-групп содержит от 1 до 20 атомов углерода. Примеры включают N,N-диметиламино, пирролидинил, триметилсилил, трет-бутилдиметилсилил, метил-ди(трет-бутил)силил, трифенилгермил и триметилгермил-группы. Примеры групп, содержащих гетероатом группы 15 или 16, включают амино-, фосфино-, алкокси- или алкилтиогруппы или их двухвалентные производные, например амид-, фосфид-, алкиленокси- или алкилентиогруппы, связанные с переходным металлом или металлом ряда лантанидов и связанные с гидрокарбилгруппой, π-связанной группой или гидрокарбил-замещенным гетератомом.
Примеры подходящих анионных делокализованных π-связанных групп включают циклопентадиенил, инденил, флуоренил, тетрагидроинденил, тетрагидрофлуоренил, октагидрофлуоренил, пентадиенил, циклогексадиенил, дигидроантраценил, гексагидроантраценил, декагидроантраценил-группы, группы фосфола и боратабензила, а также инертно замещенные их производные, особенно С1-10 гидрокарбил-замещенные или трис(С1-10 гидрокарбил)силил-замещенные их производные. Предпочтительными анионными делокализованными π-связанными группами являются циклопентадиенил, пентаметилциклопентадиенил, тетраметилциклопентадиенил, тетраметилсилилциклопентадиенил, инденил, 2,3-диметилинденил, флуоренил, 2-метилинденил, 2-метил-4-фенилинденил, тетрагидрофлуоренил, октагидрофлуоренил, 1-индаценил, 3-пирролидиноинден-1-ил, 3,4-(циклопента(l)фенантрен-1-ил и тетрагидроинденил.
Боратабензенил-лиганды являются анионными лигандами, которые являются борсодержащими аналогами бензола. Они известны из предшествующего уровня техники и описаны G. Herberich, et al., in Organometallics, 14,1, 471-480 (1995). Предпочтительные боратабензенил-лиганды соответствуют формуле:
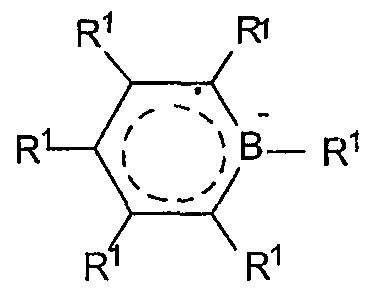
где R1 представляет инертный заместитель, предпочтительно выбранный из группы, состоящей из водорода, гидрокарбила, силила, галогена или гермила, указанный R1 имеет вплоть до 20 атомов, не считая водорода, и, необязательно, две соседние группы R1 могут быть соединены вместе. В комплексах, включающих двухвалентные производные таких делокализованных π-связанных групп, один их атом связан посредством ковалентной связи или ковалентно связанной двухвалентной группы с другим атомом комплекса, формируя таким образом связанную мостиковой связью систему.
Фосфолы являются анионными лигандами, которые представляют собой фосфорсодержащие аналоги циклопентадиенил-группы. Они известны из предшествующего уровня техники и описаны в WO 98/50392 и других источниках. Предпочтительные фосфол-лиганды соответствуют формуле:
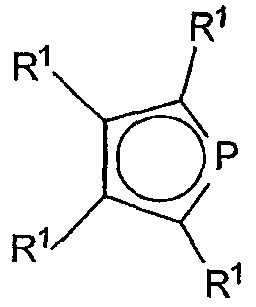
где R1 имеет указанные выше значения.
Предпочтительные комплексы переходных металлов для применения здесь соответствуют формуле: MKkXxZz или ее димеру, где
M представляет металл группы 4;
K представляет группу, содержащую делокализованные π-электроны, посредством которых K связана с M, указанная группа К содержит вплоть до 50 атомов, не считая атомы водорода, необязательно, две группы К могут быть соединены вместе, образуя связанную мостиковой связью структуру, и еще одна группа K может быть связана с X или Z;
X, в каждом случае, представляет одновалентную анионную группу, имеющую вплоть до 40 атомов, иных чем водород, необязательно, одна или несколько групп X и одна или несколько групп K связаны вместе с образованием металлоцикла, и еще, необязательно, одна или несколько групп X и одна или несколько групп Z связаны вместе, образуя таким образом группу, которая и ковалентно связана с М, и координирована с ним;
Z, независимо в каждом случае, представляет нейтральный донорный лиганд основания Льюиса из вплоть до 50 атомов, иных чем водород, содержащий по меньшей мере одну неразделенную электронную пару, посредством которой Z координирован с M;
k представляет целое число от 0 до 3,
x представляет целое число от 1 до 4,
z представляет целое число от 0 до 3 и
сумма, k+x, равна состоянию нормального окисления M.
Предпочтительные комплексы включают те, которые содержат одну или две группы К. Последние комплексы включают те, которые содержат мостиковую группу, связывающую две группы K. Предпочтительными мостиковыми группами являются группы, соответствующие формуле (ER'2)e, где E представляет кремний, германий, олово или углерод, R', независимо в каждом случае, представляет водород или группу, выбранную из силила, гидрокарбила, гидрокарбилoкси и их сочетаний, указанная группа R' имеет вплоть до 30 атомов углерода или кремния, и e означает 1-8. Предпочтительно R', независимо в каждом случае, представляет метил, этил, пропил, бензил, трет-бутил, фенил, метокси, этокси или фенокси.
Примерами комплексов, содержащих две группы К, являются соединения, соответствующие формуле:
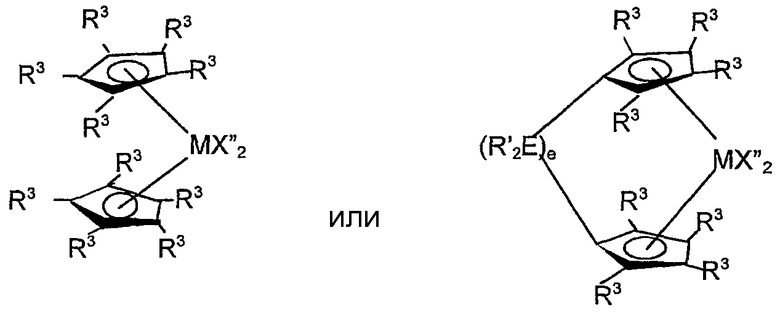
где M представляет титан, цирконий или гафний, предпочтительно цирконий или гафний, в состоянии нормального окисления +2 или +4;
R3, в каждом случае независимо, выбран из группы, состоящей из водорода, гидрокарбила, силила, гермила, циано, галогена и их сочетаний, указанная группа R3 имеет вплоть до 20 атомов, иных чем водород, или соседние группы R3 вместе образуют двухвалентное производное (то есть гидрокарбадиил, силадиил или гермадиил-группу), формируя тем самым конденсированную кольцевую систему, и
X", независимо в каждом случае, представляет анионную лиганд-группу из вплоть до 40 атомов, иных чем водород, или две группы X" вместе образуют двухвалентную анионную лиганд-группу из вплоть до 40 атомов, иных чем водород, или вместе представляют конъюгированный диен, имеющий от 4 дo 30 атомов, иных чем водород, связанный посредством делокализованных π-электронов с M, вследствие чего M находится в состоянии нормального окисления +2, и
R', E и e имеют указанные выше значения.
Примерами связанных мостиковой связью лигандов, содержащих две π-связанные группы, являются:
диметилбис(циклопентадиенил)силан,
диметилбис(тетраметилциклопентадиенил)силан,
диметилбис(2-этилциклопентадиен-1-ил)силан,
диметилбис(2-трет-бутилциклопентадиен-1-ил)силан,
2,2-бис(тетраметилциклопентадиенил)пропан,
диметилбис(инден-1-ил)силан,
диметилбис(тетрагидроинден-1-ил)силан,
диметилбис(флуорен-1-ил)силан,
диметилбис(тетрагидрофлуорен-1-ил)силан,
диметилбис(2-метил-4-фенилинден-1-ил)силан,
диметилбис(2-метилинден-1-ил)силан,
диметил(циклопентадиенил)(флуорен-1-ил)силан,
диметил(циклопентадиенил)(октагидрофлуорен-1-ил)силан,
диметил(циклопентадиенил)(тетрагидрофлуорен-1-ил)силан,
(1,1,2,2-тетраметил)-1,2-бис(циклопентадиенил)дисилан,
1,2-бис(циклопентадиенил)этан и
диметил(циклопентадиенил)-1-(флуорен-1-ил)метан.
Предпочтительные группы X" выбраны из групп гидрида, гидрокарбила, силила, гермила, галогенгидрокарбила, галогенсилила, силилгидрокарбила и аминогидрокарбила или две группы X" вместе образуют двухвалентное производное конъюгированного диена или еще вместе они могут образовывать нейтральный π-связанный конъюгированный диен. Наиболее предпочтительными группами X" являются C1-20 гидрокарбил-группы.
Примеры комплексов металлов указанной выше формулы, подходящие для применения в данном изобретении, включают:
бис(циклопентадиенил)цирконийдиметил,
бис(циклопентадиенил)цирконий дибензил,
бис(циклопентадиенил)цирконий метилбензил,
бис(циклопентадиенил)цирконий метилфенил,
бис(циклопентадиенил)цирконийдифенил,
бис(циклопентадиенил)титаналлил,
бис(циклопентадиенил)цирконийметилметоксид,
бис(циклопентадиенил)цирконийметилхлорид,
бис(пентаметилциклопентадиенил)цирконийдиметил,
бис(пентаметилциклопентадиенил)титандиметил,
бис(инденил)цирконийдиметил,
инденилфлуоренилцирконийдиметил,
бис(инденил)цирконийметил(2-(диметиламино)бензил),
бис(инденил)цирконийметилтриметилсилил,
бис(тетрагидроинденил)цирконийметилтриметилсилил,
бис(пентаметилциклопентадиенил)цирконийметилбензил,
бис(пентаметилциклопентадиенил)цирконийдибензил,
бис(пентаметилциклопентадиенил)цирконийметилметоксид,
бис(пентаметилциклопентадиенил)цирконийметилхлорид,
бис(метилэтилциклопентадиенил)цирконийдиметил,
бис(бутилциклопентадиенил)цирконийдибензил,
бис(трет-бутилциклопентадиенил)цирконийдиметил,
бис(этилтетраметилциклопентадиенил)цирконийдиметил,
бис(метилпропилциклопентадиенил)цирконийдибензил,
бис(триметилсилилциклопентадиенил)цирконийдибензил,
диметилсилилбис(циклопентадиенил)цирконийдихлорид,
диметилсилилбис(циклопентадиенил)цирконийдиметил,
диметилсилилбис(тетраметилциклопентадиенил)титан (III) аллил
диметилсилилбис(трет-бутилциклопентадиенил)цирконий дихлорид,
диметилсилилбис(н-бутилциклопентадиенил)цирконийдихлорид,
(диметилсилилбис(тетраметилциклопентадиенил)титан (III) 2-(диметиламино)бензил,
(диметилсилилбис(н-бутилциклопентадиенил)титан (III) 2-(диметиламино)бензил,
диметилсилилбис(инденил)цирконийдихлорид,
диметилсилилбис(инденил)цирконийдиметил,
диметилсилилбис(2-метилинденил)цирконийдиметил,
диметилсилилбис(2-метил-4-фенилинденил)цирконийдиметил,
диметилсилилбис(2-метилинденил)цирконий-1,4-дифенил-1,3-бутадиен,
диметилсилилбис(2-метил-4-фенилинденил)цирконий (II) 1,4-дифенил-1,3-бутадиен,
диметилсилилбис(4,5,6,7-тетрагидроинден-1-ил)цирконий дихлорид,
диметилсилилбис(4,5,6,7-тетрагидроинден-1-ил)цирконий диметил,
диметилсилилбис(тетрагидроинденил)цирконий(II) 1,4-дифенил-1,3-бутадиен,
диметилсилилбис(тетраметилциклопентадиенил)цирконий диметил
диметилсилилбис(флуоренил)цирконийдиметил,
диметилсилилбис(тетрагидрофлуоренил)цирконий бис(триметилсилил),
этиленбис(инденил)цирконийдихлорид,
этиленбис(инденил)цирконийдиметил,
этиленбис(4,5,6,7-тетрагидроинденил)цирконийдихлорид,
этиленбис(4,5,6,7-тетрагидроинденил)цирконийдиметил,
(изопропилиден)(циклопентадиенил)(флуоренил)цирконий дибензил и
диметилсилил(тетраметилциклопентадиенил)(флуоренил)цирконий диметил.
Дополнительный класс комплексов металлов, используемых в данном изобретении, соответствует предшествующей формуле: MKZZXX или ее димеру, где M, K, X, x и z имеют указанные выше значения и Z представляет заместитель из вплоть до 50 атомов иных чем водород, который вместе с K образует металлоцикл с M.
Предпочтительные заместители Z включают группы, содержащие вплоть до 30 атомов, иных чем водород, содержащие по меньшей мере один атом, такой как кислород, сера, бор или член группы 14 Периодической таблицы элементов, непосредственно присоединенный к K, и другой атом, выбранный из группы, состоящей из азота, фосфора, кислорода или серы, который ковалентно связан с M.
Более конкретно этот класс комплексов металлов группы 4, используемых согласно данному изобретению, включает "катализаторы ограниченной геометрии", соответствующие формуле:
где
M представляет титан или цирконий, предпочтительно титан, в состоянии нормального окисления +2, +3 или +4;
K1 представляет делокализованную π-связанную лиганд-группу, необязательно замещенную 1-5 группами R2,
R2, в каждом случае независимо, выбрана из группы, состоящей из водорода, гидрокарбила, силила, гермила, циано, галогена и их сочетаний, указанная группа R2 имеет вплоть до 20 атомов, иных чем водород, или соседние группы R2 вместе образуют двухвалентное производное (то есть гидрокарбадиил, силадиил или гермадиил), формируя тем самым конденсированную кольцевую систему,
каждый X представляет галоген, гидрокарбил, гидрокарбилокси или силил-группу, указанная группа имеет вплоть до 20 атомов, иных чем водород, или две группы Х вместе образуют нейтральный С5-30 конъюгированный диен или его двухвалентное производное;
x означает 1 или 2;
Y означает -O-, -S-, -NR'-, -PR'- и
X' представляет SiR'2, CR'2, SiR'2SiR'2, CR'2CR'2, CR'=CR', CR'2SiR'2 или GeR'2, где
R', независимо в каждом случае, представляет водород или группу, выбранную из силила, гидрокарбила, гидрокарбилокси и их сочетаний, указанный R' имеет вплоть до 30 атомов углерода или кремния.
Конкретные примеры указанных комплексов металлов ограниченной геометрии включают соединения, соответствующие формуле:
где
Ar представляет арил-группу из от 6 дo 30 атомов, не считая водород;
R4, независимо в каждом случае, представляет водород, Ar или группу, иную чем Ar, выбранную из таких групп, как гидрокарбил, тригидрокарбилсилил, тригидрокарбилгермил, галогенид, гидрокарбилокси, тригидрокарбилсилокси, бис(тригидрокарбилсилил)амино, ди(гидрокарбил)амино, гидрокарбадииламино, гидрокарбилимино, ди(гидрокарбил)фосфино, гидрокарбадиилфосфино, гидрокарбилсульфидо,
галоген-замещенный гидрокарбил,
гидрокарбилокси-замещенный гидрокарбил,
тригидрокарбилсилил-замещенный гидрокарбил, тригидрокарбилсилокси-замещенный гидрокарбил, бис(тригидрокарбилсилил)амино-замещенный гидрокарбил, ди(гидрокарбил)амино-замещенный гидрокарбил,
гидрокарбиленамино-замещенный гидрокарбил, ди(гидрокарбил)фосфино-замещенный гидрокарбил, гидрокарбиленфосфино-замещенный гидрокарбил или гидрокарбилсульфидо-замещенный гидрокарбил,
указанная группа R4 имеет вплоть до 40 атомов, не считая атомов водорода, и, необязательно, две соседние группы R4 могут быть соединены вместе, образуя полициклическую конденсированную кольцевую группу;
M представляет титан;
X' представляет SiR6 2, CR6 2, SiR6 2SiR6 2, CR6 2CR6 2, CR6=CR6, CR6 2SiR6 2, BR6, BR6L" или GeR6 2;
Y представляет -О-, -S-, -NR5-, -PR5-, -NR5 2 или -PR5 2;
R5, независимо в каждом случае, представляет гидрокарбил, тригидрокарбилсилил или тригидрокарбилсилилгидрокарбил, указанная группа R5 имеет вплоть до 20 атомов, иных чем водород, и, необязательно, две группы R5 или R5 вместе с Y или Z образуют кольцевую систему;
R6, независимо в каждом случае, представляет водород, или член, выбранный из гидрокарбила, гидрокарбилокси, силила, галогенированного алкила, галогенированного арила, -NR5 2 и их сочетаний, указанная группа R6 имеет вплоть до 20 атомов, иных чем водород, и, необязательно, две группы R6 или R6 вместе с Z образуют кольцевую систему;
Z представляет нейтральный диен или монодентатное или полидентатное основание Льюиса, необязательно связанные с R5, R6 или X;
X представляет водород, одновалентную анионную лиганд-группу, имеющую вплоть до 60 атомов, не считая водород, или две группы Х соединены вместе, формируя тем самым двухвалентную лиганд-группу;
x означает 1 или 2 и
z означает 0, 1 или 2.
Предпочтительными примерами указанных комплексов металлов являются замещенные при обоих положениях 3 и 4 циклопентадиенила или инденила группой Ar.
Примеры указанных комплексов металлов включают:
(3-фенилциклопентадиен-1-ил)диметил(трет-бутиламидо) силантитан дихлорид,
(3-фенилциклопентадиен-1-ил)диметил(трет-бутиламидо) силантитан диметил,
(3-фенилциклопентадиен-1-ил)диметил(трет-бутиламидо) силантитан (II) 1,3-дифенил-1,3-бутадиен;
(3-(пиррол-1-ил)циклопентадиен-1-ил)диметил(трет-бутиламидо)силантитан дихлорид,
(3-(пиррол-1-ил)циклопентадиен-1-ил)диметил(трет-бутиламидо)силантитан диметил,
(3-(пиррол-1-ил)циклопентадиен-1-ил))диметил(трет-бутиламидо)силантитан (II) 1,4-дифенил-1,3-бутадиен;
(3-(1-метилпиррол-3-ил)циклопентадиен-1-ил)диметил(трет-бутиламидо)силантитан дихлорид,
(3-(1-метилпиррол-3-ил)циклопентадиен-1-ил)диметил(трет-бутиламидо)силантитан диметил,
(3-(1-метилпиррол-3-ил)циклопентадиен-1-ил)диметил(трет-бутиламидо)силантитан (II) 1,4-дифенил-1,3-бутадиен;
(3,4-дифенилциклопентадиен-1-ил)диметил(трет-бутиламидо)силантитан дихлорид,
(3,4-дифенилциклопентадиен-1-ил)диметил(трет-бутиламидо)силантитан диметил,
(3,4-дифенилциклопентадиен-1-ил)диметил(трет-бутиламидо)силантитан (II) 1,3-пентадиен;
(3-(3-N,N-диметиламино)фенил)циклопентадиен-1-ил)диметил(трет-бутиламидо)силантитан дихлорид,
(3-(3-N,N-диметиламино)фенилциклопентадиен-1-ил)диметил (трет-бутиламидо)силантитан диметил,
(3-(3-N,N-диметиламино)фенилциклопентадиен-1-ил)диметил(трет-бутиламидо)силантитан (II) 1,4-дифенил-1,3-бутадиен;
(3-(4-метоксифенил)-4-метилциклопентадиен-1-ил)диметил(трет-бутиламидо)силантитан дихлорид,
(3-(4-метоксифенил)-4-фенилциклопентадиен-1-ил)диметил(трет-бутиламидо)силантитан диметил,
(3-(4-метоксифенил)-4-фенилциклопентадиен-1-ил)диметил(трет-бутиламидо)силантитан (II) 1,4-дифенил-1,3-бутадиен;
(3-фенил-4-метоксициклопентадиен-1-ил)диметил(трет-бутиламидо)силантитан дихлорид,
(3-фенил-4-метоксициклопентадиен-1-ил)диметил(трет-бутиламидо)силантитан диметил,
(3-фенил-4-метоксициклопентадиен-1-ил)диметил(трет-бутиламидо)силантитан (II) 1,4-дифенил-1,3-бутадиен;
(3-фенил-4-(N,N-диметиламино)циклопентадиен-1-ил)диметил(трет-бутиламидо)силантитан дихлорид,
(3-фенил-4-(N,N-диметиламино)циклопентадиен-1-ил)диметил(трет-бутиламидо)силантитан диметил,
(3-фенил-4-(N,N-диметиламино)циклопентадиен-1-ил)диметил(трет-бутиламидо)силантитан (II) 1,4-дифенил-1,3-бутадиен;
2-метил-(3,4-ди(4-метилфенил)циклопентадиен-1-ил)диметил(трет-бутиламидо)силантитан дихлорид,
2-метил-(3,4-ди(4-метилфенил)циклопентадиен-1-ил)диметил(трет-бутиламидо)силантитан диметил,
2-метил-(3,4-ди(4-метилфенил)циклопентадиен-1-ил)диметил(трет-бутиламидо)силантитан (II) 1,4-дифенил-1,3-бутадиен;
((2,3-дифенил)-4-(N,N-диметиламино)циклопентадиен-1-ил)диметил(трет-бутиламидо)силантитан дихлорид,
((2,3-дифенил)-4-(N,N-диметиламино)циклопентадиен-1-ил)диметил(трет-бутиламидо)силантитан диметил,
((2,3-дифенил)-4-(N,N-диметиламино)циклопентадиен-1-ил)диметил(трет-бутиламидо)силантитан (II) 1,4-дифенил-1,3-бутадиен;
(2,3,4-трифенил-5-метилциклопентадиен-1-ил)диметил(трет-бутиламидо)силантитан дихлорид,
(2,3,4-трифенил-5-метилциклопентадиен-1-ил)диметил(трет-бутиламидо)силантитан диметил,
(2,3,4-трифенил-5-метилциклопентадиен-1-ил)диметил(трет-бутиламидо)силантитан (II) 1,4-дифенил-1,3-бутадиен;
(3-фенил-4-метоксициклопентадиен-1-ил)диметил(трет-бутиламидо)силантитан дихлорид,
(3-фенил-4-метоксициклопентадиен-1-ил)диметил(трет-бутиламидо)силантитан диметил,
(3-фенил-4-метоксициклопентадиен-1-ил)диметил(трет-бутиламидо)силантитан (II) 1,4-дифенил-1,3-бутадиен;
(2,3-дифенил-4-(н-бутил)циклопентадиен-1-ил)диметил(трет-бутиламидо)силантитан дихлорид,
(2,3-дифенил-4-(н-бутил)циклопентадиен-1-ил)диметил(трет-бутиламидо)силантитан диметил,
(2,3-дифенил-4-(н-бутил)циклопентадиен-1-ил)диметил(трет-бутиламидо)силантитан (II) 1,4-дифенил-1,3-бутадиен;
(2,3,4,5-тетрафенилциклопентадиен-1-ил)диметил(трет-бутиламидо)силантитан дихлорид,
(2,3,4,5-тетрафенилциклопентадиен-1-ил)диметил(трет-бутиламидо)силантитан диметил и
(2,3,4,5-тетрафенилциклопентадиен-1-ил)диметил(трет-бутиламидо)силантитан (II) 1,4-дифенил-1,3-бутадиен.
Дополнительными примерами подходящих комплексов металлов для применения в качестве катализатора (A) здесь являются полициклические комплексы, соответствующие формуле:
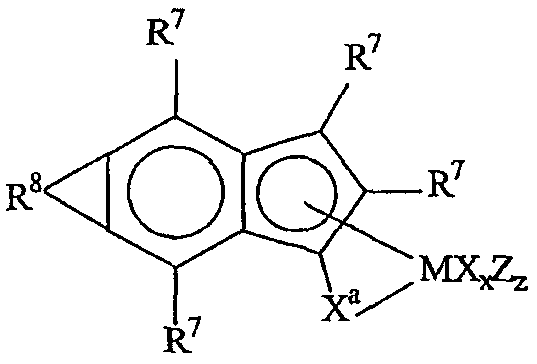
где M представляет титан в состоянии нормального окисления +2, +3 или +4;
R7, независимо в каждом случае, представляет гидрид, гидрокарбил, силил, гермил, галогенид, гидрокарбилокси, гидрокарбилсилокси, гидрокарбилсилиламино, ди(гидрокарбил)амино, гидрокарбиленамино, ди(гидрокарбил)фосфино, гидрокарбиленфосфино, гидрокарбилсульфидо, галоген-замещенный гидрокарбил, гидрокарбилокси-замещенный гидрокарбил, силил-замещенный гидрокарбил, гидрокарбилсилокси-замещенный гидрокарбил, гидрокарбилсилиламино-замещенный гидрокарбил, ди(гидрокарбил)амино-замещенный гидрокарбил, гидрокарбиленамино-замещенный гидрокарбил, ди(гидрокарбил)фосфино-замещенный гидрокарбил, гидрокарбиленфосфино-замещенный гидрокарбил или гидрокарбилсульфидо-замещенный гидрокарбил, указанная группа R7 имеет вплоть до 40 атомов, не считая водород, и, необязательно, две или более указанных групп могут вместе образовывать двухвалентное производное;
R8 представляет двухвалентную гидрокарбилен- или замещенную гидрокарбилен-группу, образующую конденсированную систему с остальной частью комплекса металла, указанная группа R8 содержит от 1 до 30 атомов, не считая водород;
Xa представляет двухвалентную группу или группу, содержащую одну σ-связь и нейтральную двухэлектронную пару, способную формировать координационно-ковалентную связь с M, указанная группа Xa содержит бор или член группы 14 Периодической таблицы элементов и также содержит азот, фосфор, серу или кислород;
X представляет одновалентную анионную лиганд-группу, имеющую вплоть до 60 атомов, единственную из класса лигандов, которые являются циклическими делокализованными π-связанными лиганд-группами, и, необязательно, две группы Х вместе образуют двухвалентную лиганд-группу;
Z, независимо в каждом случае, представляет нейтральное связывающее соединение, имеющее вплоть до 20 атомов;
x означает 0, 1 или 2 и
z означает ноль или 1.
Предпочтительными примерами таких комплексов являются 3-фенил-замещенные s-индеценил комплексы, соответствующие формуле:
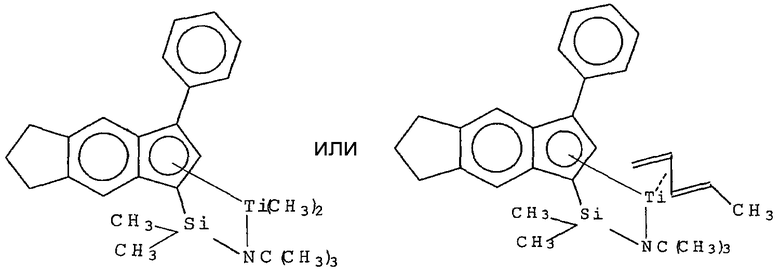
2,3-диметил-замещенные s-индеценил комплексы, соответствующие формулам:
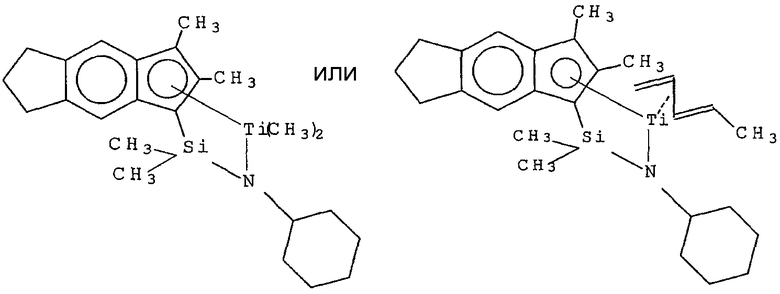
или 2-метил-замещенные s-индеценил комплексы, соответствующие формулам:
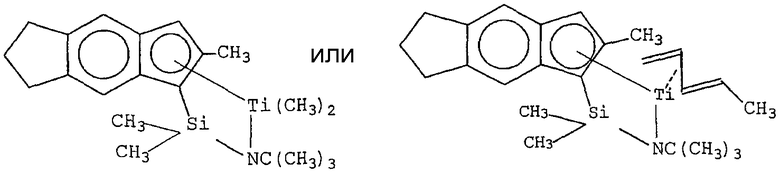
Дополнительными примерами комплексов металлов, которые успешно используются в качестве катализатора (A) согласно данному изобретению, включают комплексы формулы:
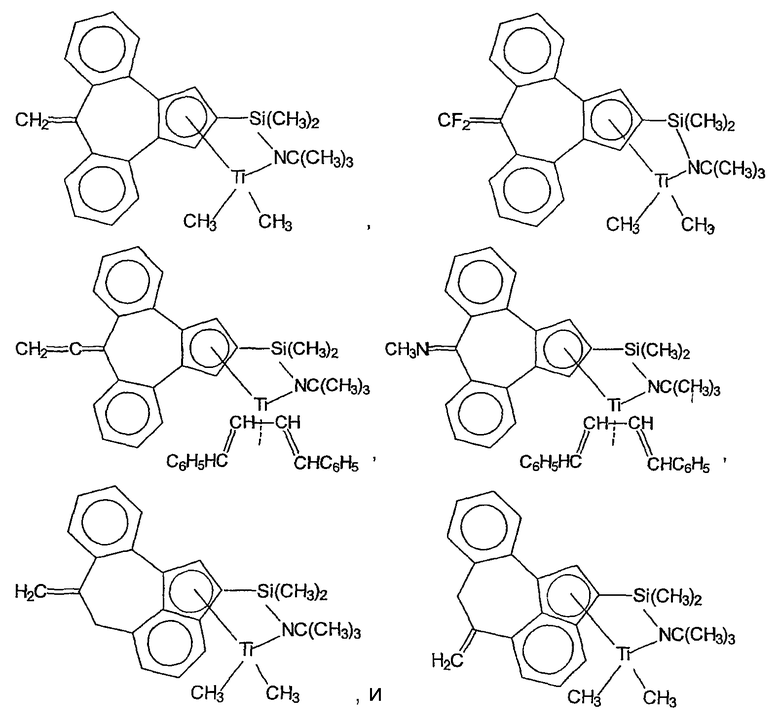
Конкретные комплексы металлов включают:
(8-метилен-1,8-дигидродибензо[e,h]азулен-1-ил)-N-(1,1-диметил этил)диметилсиланамид титан (II) 1,4-дифенил-1,3-бутадиен,
(8-метилен-1,8-дигидродибензо[e,h]азулен-1-ил)-N-(1,1-диметилэтил)диметилсиланамид титан (II) 1,3-пентадиен,
(8-метилен-1,8-дигидродибензо[e,h]азулен-1-ил)-N-(1,1-диметил этил)диметилсиланамид титан (III) 2-(N,N-диметиламино) бензил,
(8-метилен-l,8-дигидродибензо[e,h]азулен-1-ил)-N-(1,1-диметилэтил)диметилсиланамид титан (IV) дихлорид,
(8-метилен-1,8-дигидродибензо[e,h]азулен-1-ил)-N-(1,1-диметилэтил)диметилсиланамид титан (IV) диметил,
(8-метилен-1,8-дигидродибензо[e,h]азулен-1-ил)-N-(1,1-диметилэтил)диметилсиланамид титан (IV) дибензил,
(8-дифторметилен-1,8-дигидродибензо[e,h]азулен-1-ил)-N-(1,1-диметилэтил)диметилсиланамид титан (II) 1,4-дифенил-1,3-бутадиен,
(8-дифторметилен-1,8-дигидродибензо[e,h]азулен-1-ил)-N-(1,1-диметилэтил)диметилсиланамид титан (II) 1,3-пентадиен,
(8-дифторметилен-1,8-дигидродибензо[e,h]азулен-1-ил)-N-(1,1-диметилэтил)диметилсиланамид титан (III) 2-(N,N-диметиламино)бензил,
(8-дифторметилен-1,8-дигидродибензо[e,h]азулен-1-ил)-N-(1,1-диметилэтил)диметилсиланамид титан (IV) дихлорид,
(8-дифторметилен-1,8-дигидродибензо[e,h]азулен-1-ил)-N-(1,1-диметилэтил)диметилсиланамид титан (IV) диметил,
(8-дифторметилен-1,8-дигидродибензо[e,h]азулен-1-ил)-N-(1,1 -диметилэтил)диметилсиланамид титан (IV) дибензил,
(8-метилен-l,8-дигидродибензо[e,h]азулен-2-ил)-N-(1,1-диметилэтил)диметилсиланамид титан (II) 1,4-дифенил-1,3-бутадиен,
(8-метилен-1,8-дигидродибензо[e,h]азулен-2-ил)-N-(1,1-диметилэтил)диметилсиланамид титан (II) 1,3-пентадиен,
(8-метилен-1,8-дигидродибензо[e,h]азулен-2-ил)-N-(1,1-диметилэтил)диметилсиланамид титан (III) 2-(N,N-диметиламино)бензил,
(8-метилен-1,8-дигидродибензо[e,h]азулен-2-ил)-N-(1,1-диметилэтил)диметилсиланамид титан (IV) дихлорид,
(8-метилен-1,8-дигидродибензо[e,h]азулен-2-ил)-N-(1,1-диметилэтил)диметилсиланамид титан (IV) диметил,
(8-метилен-l,8-дигидродибензо[e,h]азулен-2-ил)-N-(1,1-диметилэтил)диметилсиланамид титан (IV) дибензил,
(8-дифторметилен-l,8-дигидродибензо[e,h]азулен-2-ил)-N-(1,1-диметилэтил)диметилсиланамид титан (II) 1,4-дифенил-1,3-бутадиен,
(8-дифторметилен-l,8-дигидродибензо[e,h]азулен-2-ил)-N-(1,1-диметилэтил)диметилсиланамид титан (II) 1,3-пентадиен,
(8-дифторметилен-1,8-дигидродибензо[e,h]азулен-2-ил)-N-(1,1-диметилэтил)диметилсиланамид титан (III) 2-(N,N-диметиламино)бензил,
(8-дифторметилен-l,8-дигидродибензо[e,h]азулен-2-ил)-N-(1,1-диметилэтил)диметилсиланамид титан (IV) дихлорид,
(8-дифторметилен-1,8-дигидродибензо[e,h]азулен-2-ил)-N-(1,1-диметилэтил)диметилсиланамид титан (IV) диметил,
(8-дифторметилен-1,8-дигидродибензо[e,h]азулен-2-ил)-N-(1,1-диметилэтил)диметилсиланамид титан (IV) дибензил и их смеси, особенно смеси позиционных изомеров.
Дополнительные пояснительные примеры комплексов металлов для применения согласно данному изобретению соответствуют формуле:
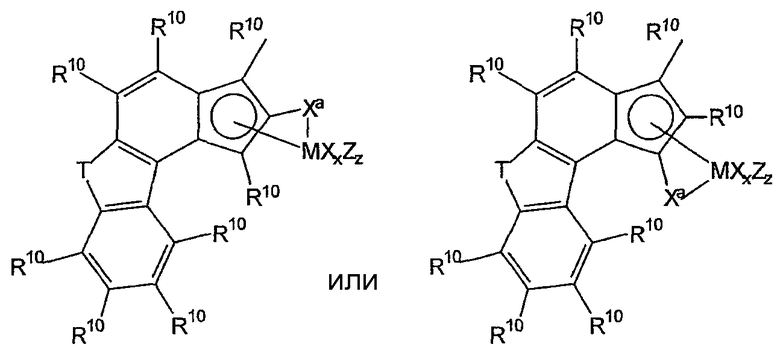
где M представляет титан в состоянии нормального окисления +2, +3 или +4;
T представляет -NR9- или -O-;
R9 представляет гидрокарбил, силил, гермил, дигидрокарбилборил или галогенгидрокарбил или вплоть до 10 атомов, не считая водород;
R10 независимо в каждом случае, представляет водород, гидрокарбил, тригидрокарбилсилил, тригидрокарбилсилилгидрокарбил, гермил, галогенид, гидрокарбилокси, гидрокарбилсилокси, гидрокарбилсилиламино, ди(гидрокарбил)амино, гидрокарбиленамино, ди(гидрокарбил)фосфино, гидрокарбиленфосфино, гидрокарбилсульфидо, галоген-замещенный гидрокарбил, гидрокарбилокси-замещенный гидрокарбил,
силил-замещенный гидрокарбил,
гидрокарбилсилокси-замещенный гидрокарбил, гидрокарбилсилиламино-замещенный гидрокарбил, ди(гидрокарбил)амино-замещенный гидрокарбил,
гидрокарбиленамино-замещенный гидрокарбил, ди(гидрокарбил)фосфино-замещенный гидрокарбил, гидрокарбиленфосфино-замещенный гидрокарбил или
гидрокарбилсульфидо-замещенный гидрокарбил, указанная группа R10 имеет вплоть до 40 атомов, не считая атомов водорода, и, необязательно, две или более указанных соседних групп R10 могут вместе образовывать двухвалентное производное, формируя тем самым насыщенное или ненасыщенное конденсированное кольцо;
Xa представляет двухвалентную группу, не имеющую делокализованных π-электронов, или такую группу, содержащую одну σ-связь и нейтральную двухэлектронную пару, способную формировать координационно-ковалентную связь с M, указанная группа Xа содержит бор или член группы 14 Периодической таблицы элементов и также содержит азот, фосфор, серу или кислород;
X представляет одновалентную анионную лиганд-группу, имеющую вплоть до 60 атомов, единственную из класса лигандов, которые являются циклическими лиганд-группами, связанными с M посредством делокализованных π-электронов, или две группы Х вместе представляют двухвалентную анионную лиганд-группу;
Z независимо в каждом случае представляет нейтральное связывающее соединение, имеющее вплоть до 20 атомов;
x означает 0, 1, 2, или 3 и
z означает 0 или 1.
Очень предпочтительно T означает =N(CH3), X представляет галоген или гидрокарбил, x означает 2, X' означает диметилсилан, z означает 0 и R10, в каждом случае, представляет водород, гидрокарбил, гидрокарбилокси, дигидрокарбиламино, гидрокарбиленамино, дигидрокарбиламино-замещенный гидрокарбил или гидрокарбиленамино-замещенный гидрокарбил из вплоть до 20 атомов, не считая водорода, и, необязательно, две группы R10 могут быть соединены вместе.
Пояснительные комплексы предшествующей формулы, которые могут быть использованы в практике данного изобретения, дополнительно включают следующие соединения:
(трет-бутиламидо)диметил[6,7]бензо[4,5:2',3'](1-метилизоиндол)-(3H)-инден-2-ил)силантитан (II) 1,4-дифенил-1,3-бутадиен,
(трет-бутиламидо)диметил[6,7]бензо[4,5:2',3'](1-метилизоиндол)-(3H)-инден-2-ил)силантитан (II) 1,3-пентадиен,
(трет-бутиламидо)диметил[6,7]бензо[4,5:2',3'](1-метилизоиндол)-(3H)-инден-2-ил)силантитан (III) 2-(N,N-диметиламино)бензил,
(трет-бутиламидо)диметил[6,7]бензо[4,5:2',3'](1-метилизоиндол)-(3H)-инден-2-ил)силантитан (IV) дихлорид,
(трет-бутиламидо)диметил[6,7]бензо[4,5:2',3'](1-метилизоиндол)-(3H)-инден-2-ил)силантитан (IV) диметил,
(трет-бутиламидо)диметил[6,7]бензо[4,5:2',3'](1-метилизоиндол)-(3H)-инден-2-ил)силантитан (IV) дибензил,
(трет-бутиламидо)диметил[6,7]бензо[4,5:2',3'](1-метилизоиндол)-(3H)-инден-2-ил)силантитан (IV) бис(триметилсилил),
(циклогексиламидо)диметил[6,7]бензо[4,5:2',3'](1-метилизоиндол)-(3H)-инден-2-ил)силантитан (II) 1,4-дифенил-1,3-бутадиен,
(циклогексиламидо)диметил[6,7]бензо[4,5:2',3'](1-метилизоиндол)-(3H)-инден-2-ил)силантитан (II) 1,3-пентадиен,
(циклогексиламидо)диметил[6,7]бензо[4,5:2',3'](1-метилизоиндол)-(3H)-инден-2-ил)силантитан (III) 2-(N,N-диметиламино)бензил,
(циклогексиламидо)диметил[6,7]бензо[4,5:2',3'](1-метилизоиндол)-(3H)-инден-2-ил)силантитан (IV) дихлорид,
(циклогексиламидо)диметил[6,7]бензо[4,5:2',3'](1-метилизоиндол)-(3H)-инден-2-ил)силантитан (IV) диметил,
(циклогексиламидо)диметил[6,7]бензо[4,5:2',3'](1-метилизоиндол)-(3H)-инден-2-ил)силантитан (IV) дибензил,
(циклогексиламидо)диметил[6,7]бензо[4,5:2',3'](1-метилизоиндол)-(3H)-инден-2-ил)силантитан (IV) бис(триметилсилил),
(трет-бутиламидо)ди(п-метилфенил)[6,7]бензо[4,5:2',3'](1-метилизоиндол)-(3H)-инден-2-ил)силантитан (II) 1,4-дифенил-1,3-бутадиен,
(трет-бутиламидо)ди(п-метилфенил)[6,7]бензо[4,5:2',3'](1-метилизоиндол)-(3H)-инден-2-ил)силантитан (II) 1,3-пентадиен,
(трет-бутиламидо)ди(п-метилфенил)[6,7]бензо[4,5:2',3'](1-метилизоиндол)-(3H)-инден-2-ил)силантитан (III) 2-(N,N-диметиламино)бензил,
(трет-бутиламидо)ди(п-метилфенил)[6,7]бензо[4,5:2',3'](1-метилизоиндол)-(3H)-инден-2-ил)силантитан (IV) дихлорид,
(трет-бутиламидо)ди(п-метилфенил)[6,7]бензо[4,5:2',3'](1-метилизоиндол)-(3H)-инден-2-ил)силантитан (IV) диметил,
(трет-бутиламидо)ди(п-метилфенил)[6,7]бензо[4,5:2',3'](1-метилизоиндол)-(3H)-инден-2-ил)силантитан (IV) дибензил,
(трет-бутиламидо)ди(п-метилфенил)[6,7]бензо[4,5:2',3'](1-метилизоиндол)-(3H)-инден-2-ил)силантитан (IV) бис(триметилсилил),
(циклогексиламидо)ди(п-метилфенил)[6,7]бензо[4,5:2',3'](1-метилизоиндол)-(3H)-инден-2-ил)силантитан (II) 1,4-дифенил-1,3-бутадиен,
(циклогексиламидо)ди(п-метилфенил)[6,7]бензо[4,5:2',3'](1-метилизоиндол)-(3H)-инден-2-ил)силантитан (II) 1,3-пентадиен,
(циклогексиламидо)ди(п-метилфенил)[6,7]бензо[4,5:2',3'](1-метилизоиндол)-(3H)-инден-2-ил)силантитан (III) 2-(N,N-диметиламино)бензил,
(циклогексиламидо)ди(п-метилфенил)[6,7]бензо[4,5:2',3'](1-метилизоиндол)-(3H)-инден-2-ил)силантитан (IV) дихлорид,
(циклогексиламидо)ди(п-метилфенил)[6,7]бензо[4,5:2',3'](1-метилизоиндол)-(3H)-инден-2-ил)силантитан (IV) диметил,
(циклогексиламидо)ди(п-метилфенил)[6,7]бензо[4,5:2',3'](1-метилизоиндол)-(3H)-инден-2-ил)силантитан (IV) дибензил и
(циклогексиламидо)ди(п-метилфенил)[6,7]бензо[4,5:2',3'](1-метилизоиндол)-(3H)-инден-2-ил)силантитан (IV) бис(триметилсилил).
Пояснительные комплексы металлов группы 4, которые могут быть использованы в практике данного изобретения, дополнительно включают:
(трет-бутиламидо)(1,1-диметил-2,3,4,9,10-η-1,4,5,6,7,8-гексагидронафталинил)диметилсилантитандиметил,
(трет-бутиламидо)(1,1,2,3-тетраметил-2,3,4,9,10-η-1,4,5,6,7,8-гексагидронафталинил)диметилсилантитандиметил,
(трет-бутиламидо)(тетраметил-η5-циклопентадиенил) диметилсилантитандибензил,
(трет-бутиламидо)(тетраметил-η5-циклопентадиенил) диметилсилантитандиметил,
(трет-бутиламидо)(тетраметил-η5-циклопентадиенил)-1,2-этандиилтитандиметил,
(трет-бутиламидо)(тетраметил-η5-инденил)диметилсилантитандиметил,
(трет-бутиламидо)(тетраметил-η5-циклопентадиенил) диметилсилантитан (III) 2-(диметиламино)бензил;
(трет-бутиламидо)(тетраметил-η5-циклопентадиенил) диметилсилантитан (III) аллил,
(трет-бутиламидо)(тетраметил-η5-циклопентадиенил) диметилсилантитан (III) 2,4-диметилпентадиенил,
(трет-бутиламидо)(тетраметил-η5-циклопентадиенил) диметилсилантитан (II) 1,4-дифенил-1,3-бутадиен,
(трет-бутиламидо)(тетраметил-η5-циклопентадиенил) диметилсилантитан (II) 1,3-пентадиен,
(трет-бутиламидо)(2-метилинденил)диметилсилантитан (II) 1,4-дифенил-1,3-бутадиен,
(трет-бутиламидо)(2-метилинденил)диметилсилантитан (II) 2,4-гексадиен,
(трет-бутиламидо)(2-метилинденил)диметилсилантитан (IV) 2,3-диметил-1,3-бутадиен,
(трет-бутиламидо)(2-метилинденил)диметилсилантитан (IV) изопрен,
(трет-бутиламидо)(2-метилинденил)диметилсилантитан (IV) 1,3-бутадиен,
(трет-бутиламидо)(2,3-диметилинденил)диметилсилантитан (IV) 2,3-диметил-1,3-бутадиен,
(трет-бутиламидо)(2,3-диметилинденил)диметилсилантитан (IV) изопрен,
(трет-бутиламидо)(2,3-диметилинденил)диметилсилантитан (IV) диметил,
(трет-бутиламидо)(2,3-диметилинденил)диметилсилантитан (IV) дибензил,
(трет-бутиламидо)(2,3-диметилинденил)диметилсилантитан (IV) 1,3-бутадиен,
(трет-бутиламидо)(2,3-диметилинденил)диметилсилантитан (II) 1,3-пентадиен,
(трет-бутиламидо)(2,3-диметилинденил)диметилсилантитан (II) 1,4-дифенил-1,3-бутадиен,
(трет-бутиламидо)(2-метилинденил)диметилсилантитан (II) 1,3-пентадиен,
(трет-бутиламидо)(2-метилинденил)диметилсилантитан (IV) диметил,
(трет-бутиламидо)(2-метилинденил)диметилсилантитан (IV) дибензил,
(трет-бутиламидо)(2-метил-4-фенилинденил)диметилсилантитан (II) 1,4-дифенил-1,3-бутадиен,
(трет-бутиламидо)(2-метил-4-фенилинденил)диметилсилантитан (II) 1,3-пентадиен,
(трет-бутиламидо)(2-метил-4-фенилинденил)диметилсилантитан (II) 2,4-гексадиен,
(трет-бутиламидо)(тетраметил-η5-циклопентадиенил)диметилсилантитан (IV) 1,3-бутадиен,
(трет-бутиламидо)(тетраметил-η5-циклопентадиенил) диметилсилантитан (IV) 2,3-диметил-1,3-бутадиен,
(трет-бутиламидо)(тетраметил-η5-циклопентадиенил) диметилсилантитан (IV) изопрен,
(трет-бутиламидо)(тетраметил-η5-циклопентадиенил)диметилсилантитан (II) 1,4-дибензил-1,3-бутадиен,
(трет-бутиламидо)(тетраметил-η5-циклопентадиенил)диметилсилантитан (II) 2,4-гексадиен,
(трет-бутиламидо)(тетраметил-η5-циклопентадиенил)диметил силантитан (II) 3-метил-1,3-пентадиен,
(трет-бутиламидо)(2,4-диметилпентадиен-3-ил)диметилсилантитандиметил,
(трет-бутиламидо)(6,6-диметилциклогексадиенил) диметилсилантитандиметил,
(трет-бутиламидо)(1,1-диметил-2,3,4,9,10-η-1,4,5,6,7,8-гексагидронафталин-4-ил)диметилсилантитандиметил,
(трет-бутиламидо)(1,1,2,3-тетраметил-2,3,4,9,10-η-1,4,5,6,7,8-гексагидронафталин-4-ил)диметилсилантитандиметил,
(трет-бутиламидо)(тетраметил-η5-циклопентадиенил)метилфенилсилантитан (IV) диметил,
(трет-бутиламидо)(тетраметил-η5-циклопентадиенил)метилфенилсилантитан (II) 1,4-дифенил-1,3-бутадиен,
1-(трет-бутиламидо)-2-(тетраметил-η5-циклопентадиенил)этандиилтитан (IV)диметил и
1-(трет-бутиламидо)-2-(тетраметил-η5-циклопентадиенил)этандиилтитан (II) 1,4-дифенил-1,3-бутадиен.
Другие делокализованные π-связанные комплексы, особенно комплексы, содержащие другие металлы группы 4, будут, конечно очевидны специалистам, и они раскрыты, наряду с другими источниками, в WO 03/78480, WO 03/78483, WO 02/92610, WO 02/02577, US 2003/0004286 и патентах США 6515155, 6555634, 6150297, 6034022, 6268444, 6015868, 5866704 и 5470993.
Дополнительными примерами комплексов металлов, которые успешно используются в качестве катализатора (A), являются комплексы поливалентных оснований Льюиса, такие как соединения, соответствующие формулам:
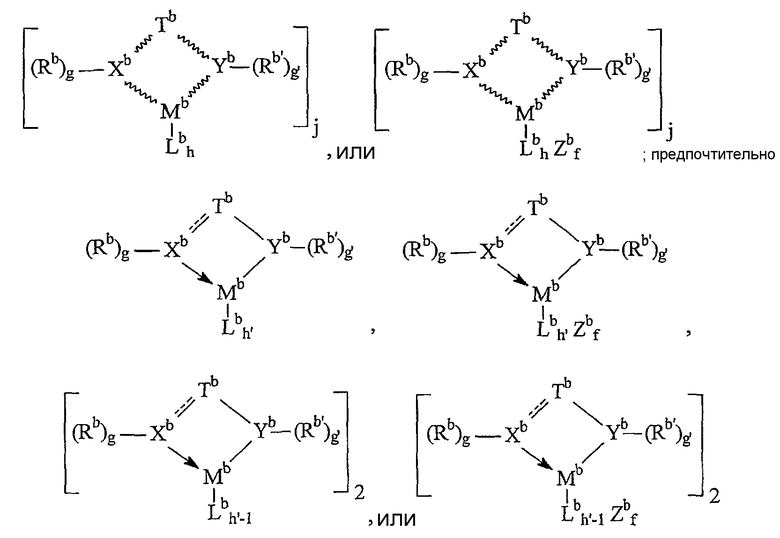
где Tb представляет мостиковую группу, предпочтительно содержащую 2 или более атомов, иных чем водород,
Xb и Yb, каждый независимо, выбраны из группы, состоящей из азота, серы, кислорода и фосфора, более предпочтительно оба Xb и Yb представляют азот,
Rb и Rb', независимо в каждом случае, представляют водород или C1-50 гидрокарбил-группы, необязательно содержащие один или несколько гетератомов, или их инертно замещенные производные. Неограничивающие примеры подходящих
Rb и Rb, групп включают алкил, алкенил, арил, аралкил, (поли)алкиларил и циклоалкил группы, а также их производные, замещенные азотом, фосфором, кислородом и галогеном. Конкретные примеры подходящих Rb и Rb' групп включают метил, этил, изопропил, октил, фенил, 2,6-диметилфенил, 2,6-ди(изопропил)фенил, 2,4,6-триметилфенил, пентафторфенил, 3,5-трифторметилфенил и бензил;
g означает 0 или 1;
Mb представляет металлический элемент, выбранный из групп 3-15 или ряда лантанидов Периодической таблицы элементов. Предпочтительно Mb представляет металл группы 3-13, более предпочтительно Mb представляет металл группы 4-10;
Lb представляет одновалентный, двухвалентный или трехвалентный анионный лиганд, содержащий от 1 до 50 атомов, не считая водород. Примеры подходящих групп Lb включают галогенид, гидрид, гидрокарбил, гидрокарбилокси, ди(гидрокарбил)амидо, гидрокарбиленамидо, ди(гидрокарбил)фосфидо, гидрокарбилсульфидо, гидрокарбилокси, три(гидрокарбилсилил)алкил и карбоксилаты. Более предпочтительными группами Lb являются С1-20 алкил, C7-20 аралкил и хлорид;
h представляет целое число от 1 до 6, предпочтительно от 1 до 4, более предпочтительно от 1 до 3, и j означает 1 или 2, с величиной h x j, выбранной, чтобы обеспечить баланс заряда;
Zb представляет нейтральную лиганд-группу, координированную с Mb, и содержит вплоть до 50 атомов, не считая водород. Предпочтительные группы Zb включают алифатические и ароматические амины, фосфины и простые эфиры, алкены, алкадиены и инертно замещенные их производные. Подходящие инертные заместители включают галоген, алкокси, арилокси, алкоксикарбонил, арилоксикарбонил, ди(гидрокарбил)амин, три(гидрокарбил)силильную и нитрильную группы. Предпочтительные группы Zb включают трифенилфосфин, тетрагидрофуран, пиридин и 1,4-дифенилбутадиен;
f представляет целое число от 1 до 3;
две или три группы из Tb, Rb и Rb могут быть соединены вместе с образованием структуры из одного или нескольких колец;
h′ представляет целое число от 1 до 6, предпочтительно от 1 до 4, более предпочтительно от 1 до 3;
зигзагообразная линия указывает какую-либо форму электронного взаимодействия, особенно координационные или ковалентные связи, включая множественные связи, стрелки означают координационные связи и пунктирные линии указывают необязательные двойные связи.
В одном варианте воплощения предпочтительно, что Rb имеет относительно низкую стерическую затрудненность, что касается Xb. В этом варианте воплощения наиболее предпочтительными группами Rb являются алкил-группы с линейной цепью, алкенил-группы с линейной цепью, алкил-группы с разветвленной цепью, где ближайшая точка разветвления по меньшей мере на 3 атома удалена от Xb, и их производные, замещенные галогеном, дигидрокарбиламино, алкокси или тригидрокарбилсилилом. Очень предпочтительными группами Rb в этом варианте воплощения являются алкил-группы с линейной цепью.
В то же время в этом варианте воплощения Rb' предпочтительно имеет относительно высокую стерическую затрудненность, что касается Yb. Неограничивающие примеры подходящих групп Rb' для этого варианта воплощения включают алкил или алкенил-группы, содержащие один или несколько вторичных или третичных углеродных центров, циклоалкил, арил, алкарил, алифатические или ароматические гетероциклические группы, органические или неорганические олигомерные, полимерные или циклические группы и их производные, замещенные галогеном, дигидрокарбиламино, алкокси или тригидрокарбилсилилом. Предпочтительные группы Rb' в этом варианте воплощения содержат от 3 дo 40, более предпочтительно от 3 дo 30 и наиболее предпочтительно от 4 дo 20 атомов, не считая водород, и являются разветвленными или циклическими.
Примерами предпочтительных групп Tb являются структуры, соответствующие следующим формулам:
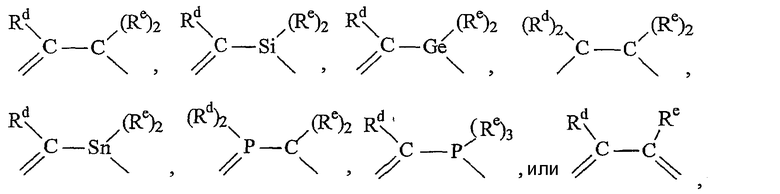
где
каждый Rd представляет С1-10 гидрокарбил группу, предпочтительно метил, этил, н-пропил, изопропил, трет-бутил, фенил, 2,6-диметилфенил, бензил или толил. Каждый Re представляет С1-10 гидрокарбил, предпочтительно метил, этил, н-пропил, изопропил, трет-бутил, фенил, 2,6-диметилфенил, бензил или толил. В дополнение, две или более групп Rd или Re или смеси групп Rd и Re могут вместе образовывать поливалентное производное гидрокарбил-группы, такое как 1,4-бутилен, 1,5-пентилен, или мультициклическую, с конденсированными кольцами поливалентную гидрокарбил- или гетерогидрокарбил-группу, такую как нафталин-1,8-диил.
Предпочтительные примеры предшествующих поливалентных комплексов оснований Льюиса включают:
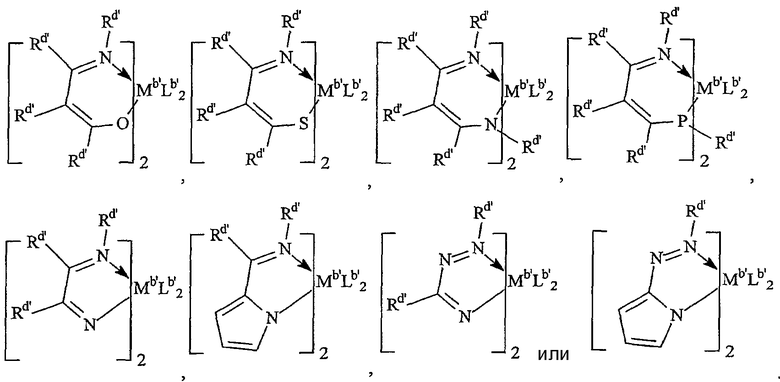
где Rd', в каждом случае, независимо выбран из группы, состоящей из водорода и С1-50 гидрокарбил-групп, необязательно содержащих один или несколько гетератомов, или их инертно замещенных производных, или еще, необязательно, две соседние группы Rd' групп могут вместе образовывать двухвалентную мостиковую группу;
d' означает 4;
Mb' представляет металл группы 4, предпочтительно титан или гафний, или металл группы 10, предпочтительно Ni или Pd;
Lb' представляет одновалентный лиганд из вплоть до 50 атомов, не считая водород, предпочтительно галогенид или гидрокарбил, или две группы Lb вместе представляют двухвалентную или нейтральную лиганд-группу, предпочтительно С2-50 гидрокарбилен, гидрокарбадиил или диен-группу.
Комплексы поливалентного основания Льюиса для применения в данном изобретении особенно включают производные металла группы 4, особенно производные гафния замещенных гидрокарбиламином гетероарил-соединений, соответствующие формуле:
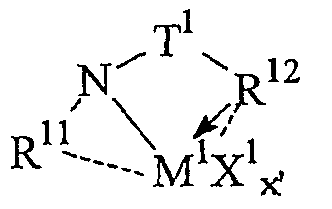
где R11 выбран из алкила, циклоалкила, гетероалкила, циклогетероалкила, арила и их инертно замещенных производных, содержащих от 1 до 30 атомов, не считая водород, или их двухвалентных производных;
T1 представляет двухвалентную мостиковую группу из от 1 до 41 атомов, иных чем водород, предпочтительно от 1 дo 20 атомов атомов, иных чем водород, и наиболее предпочтительно моно- или ди- замещенную С1-20 гидрокарбилом группу метилена или силана;
R12 представляет C5-20 гетероарил-группу, содержащую функциональность основания Льюиса, особенно пиридин-2-ил- или замещенный пиридин-2-ил, или ее двухвалентное производное;
M1 представляет металл группы 4, предпочтительно гафний;
X1 представляет анионный, нейтральный или дианионный лиганд;
x' представляет целое число от 0 до 5, указывающее число таких X1 групп, и
связи, необязательные связи и электронодонорные взаимодействия представлены линиями, пунктирными линиями и стрелками соответственно.
Предпочтительными комплексами являются те, где образование лиганда является результатом элиминирования водорода из аминогруппы и, необязательно, потери одной или нескольких дополнительных групп, особенно из R12. В дополнение, передача электронов от функциональной группы основания Льюиса, предпочтительно электронной пары, придает дополнительную стабильность металлическому центру. Предпочтительные комплексы металлов соответствуют формуле:
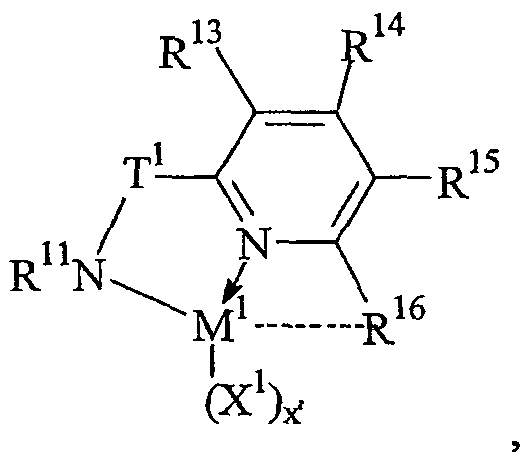
где
M1, X1, x', R11 и T1 имеют указанные выше значения,
R13, R14, R15 и R16 представляют водород, галоген или группы алкил, циклоалкил, гетероалкил, гетероциклоалкил, арил или силил из вплоть до 20 атомов, не считая водород, или соседние группы R13, R14, R15 или R16 групп могут быть соединены вместе, формируя тем самым производные из конденсированных колец, и
связи, необязательные связи и взаимодействия с передачей электронной пары представлены линиями, пунктирными линиями и стрелками соответственно.
Более предпочтительные примеры предшествующих комплексов металлов соответствуют формуле:
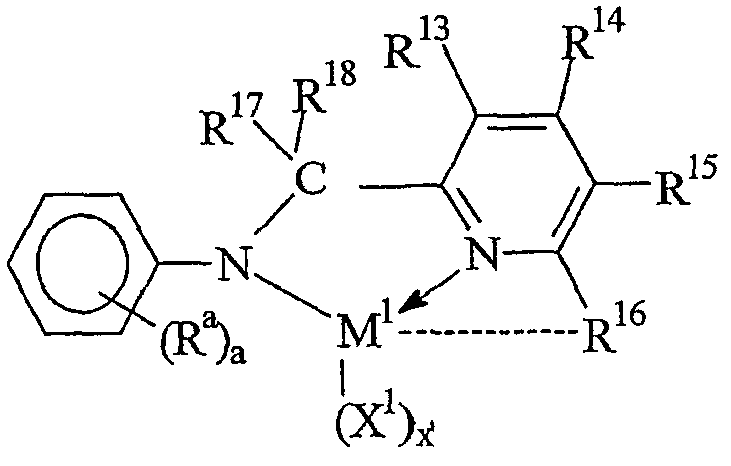
где
M1, X1 и x' имеют указанные выше значения,
R13, R14, R15 и R16 имеют указанные выше значения, предпочтительно R13, R14 и R15 представляют водород или С1-4 алкил, и R16 представляет C6-20 арил, наиболее предпочтительно нафталинил;
Ra, независимо в каждом случае, представляет С1-4 алкил и a означает 1-5, наиболее предпочтительно Ra в двух орто-положениях по отношению к азоту представляет изопропил или трет-бутил;
R17 и R18, независимо в каждом случае, представляют водород, галоген или C1-20 алкил или арил, наиболее предпочтительно один из R17 и R18 представляет водород и другой представляет C6-20 арил-группу, особенно 2-изопропил, фенил или конденсированную полициклическую арил-группу, наиболее предпочтительно антраценил, и
связи, необязательные связи и взаимодействия с передачей электронной пары представлены линиями, пунктирными линиями и стрелками соответственно.
Очень предпочтительные комплексы металлов для применения здесь в качестве катализатора (A) соответствуют формуле:
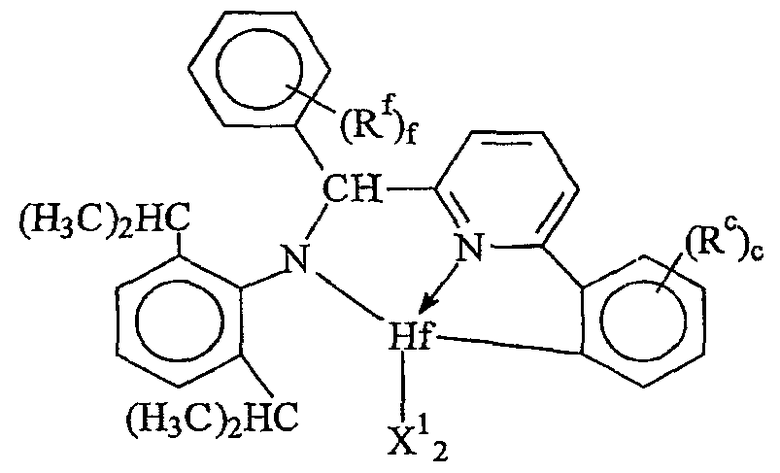
где X1, в каждом случае, представляет галогенид, N,N-диметиламидо или С1-4 алкил и предпочтительно, в каждом случае, X1 представляет метил;
Rf, независимо в каждом случае, представляет водород, галоген, С1-20 алкил или C6-20 арил, или две соседние группы Rf соединены вместе, формируя тем самым кольцо, и f означает 1-5 и
Rc, независимо в каждом случае, представляет водород, галоген, С1-20 алкил или C6-20 арил, или две соседние группы Rс соединены вместе, формируя тем самым кольцо, и с означает 1-5.
Наиболее предпочтительными примерами комплексов металлов для применения в качестве катализатора (A) согласно данному изобретению являются комплексы следующих формул:
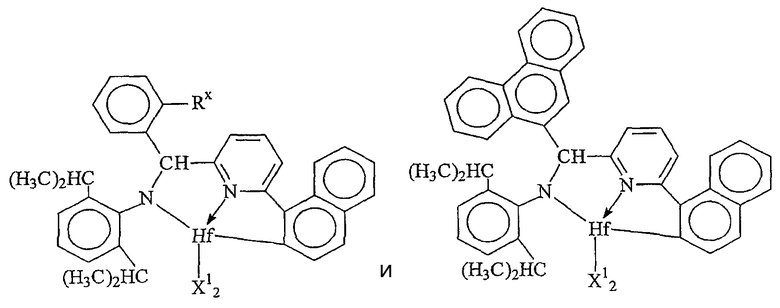
где Rх представляет С1-4 алкил или циклоалкил, предпочтительно метил, изопропил, трет-бутил или циклогексил, и
X1, в каждом случае, представляет галогенид, N,N-диметиламидо или С1-4 алкил, предпочтительно метил.
Примеры комплексов металлов, успешно используемых в качестве катализатора (A) согласно данному изобретению, включают:
[N-(2,6-ди(1-метилэтил)фенил)амидо)(o-толил)(α-нафталин-2-диил(6-пиридин-2-диил)метан)]гафний диметил;
[N-(2,6-ди(1-метилэтил)фенил)амидо)(o-толил)(α-нафталин-2-диил(6-пиридин-2-диил)метан)]гафний ди(N,N-диметиламидо);
[N-(2,6-ди(1-метилэтил)фенил)амидо)(o-толил)(α-нафталин-2-диил(6-пиридин-2-диил)метан)]гафний дихлорид;
[N-(2,6-ди(1-метилэтил)фенил)амидо)(2-изопропилфенил)(α-нафталин-2-диил(6-пиридин-2-диил)метан)]гафний диметил;
[N-(2,6-ди(1-метилэтил)фенил)амидо)(2-изопропилфенил)(α-нафталин-2-диил(6-пиридин-2-диил)метан)]гафний ди(N,N-диметиламидо);
[N-(2,6-ди(1-метилэтил)фенил)амидо)(2-изопропилфенил)(α-нафталин-2-диил(6-пиридин-2-диил)метан)]гафний дихлорид;
[N-(2,6-ди(1-метилэтил)фенил)амидо)(фенантрен-5-ил)(α-нафталин-2-диил(6-пиридин-2-диил)метан)]гафний диметил;
[N-(2,6-ди(1-метилэтил)фенил)амидо)(фенантрен-5-ил)(α-нафталин-2-диил(6-пиридин-2-диил)метан)]гафний ди(N,N-диметиламидо) и
[N-(2,6-ди(1-метилэтил)фенил)амидо)(фенантрен-5-ил)(α-нафталин-2-диил(6-пиридин-2-диил)метан)]гафний дихлорид.
В условиях реакции, используемых для получения комплексов металлов, используемых в данном изобретении, водород положения 2 группы α-нафталина, замещенной в положении 6 пиридин-2-ил-группы, подвергают элиминированию, уникально формируя таким образом комплексы металлов, где металл ковалентно связан и с полученной амидогруппой, и с положением 2 α-нафталинил-группы, а также стабилизирован координированием с атомом азота пиридинила через электронную пару атома азота.
Дополнительные подходящие комплексы металлов поливалентных оснований Льюиса для применения здесь включают соединения, соответствующие формуле:
где
R20 представляет ароматическую или инертно замещенную ароматическую группу, содержащую от 5 дo 20 атомов, не считая водород, или ее поливалентное производное;
T3 представляет группу гидрокарбилена или силана, имеющую от 1 до 20 атомов, не считая водород, или ее инертно замещенное производное;
M3 представляет металл группы 4, предпочтительно цирконий или гафний;
G представляет анионную, нейтральную или дианионную лиганд-группу; предпочтительно галогенид, гидрокарбил или дигидрокарбиламид, имеющую вплоть до 20 атомов, не считая водород;
g представляет число от 1 до 5, указывающее число таких G групп, и
связи и электронодонорные взаимодействия представлены линиями и стрелками соответственно.
Предпочтительно такие комплексы соответствуют формуле:
где
T3 представляет двухвалентную мостиковую группу из от 2 дo 20 атомов, не считая водород, предпочтительно замещенную или незамещенную группу C3-6 алкилена; и
Ar2, независимо в каждом случае, представляет группу арилена или алкил- или арил-замещенного арилена из от 6 дo 20 атомов, не считая водород;
M3 представляет металл группы 4, предпочтительно гафний или цирконий;
G, независимо в каждом случае, представляет анионную, нейтральную или дианионную лиганд-группу;
g представляет число от 1 до 5, указывающее число таких G групп, и
электронодонорные взаимодействия представлены стрелками.
Предпочтительные примеры комплексов металлов предшествующей формулы включают следующие соединения:
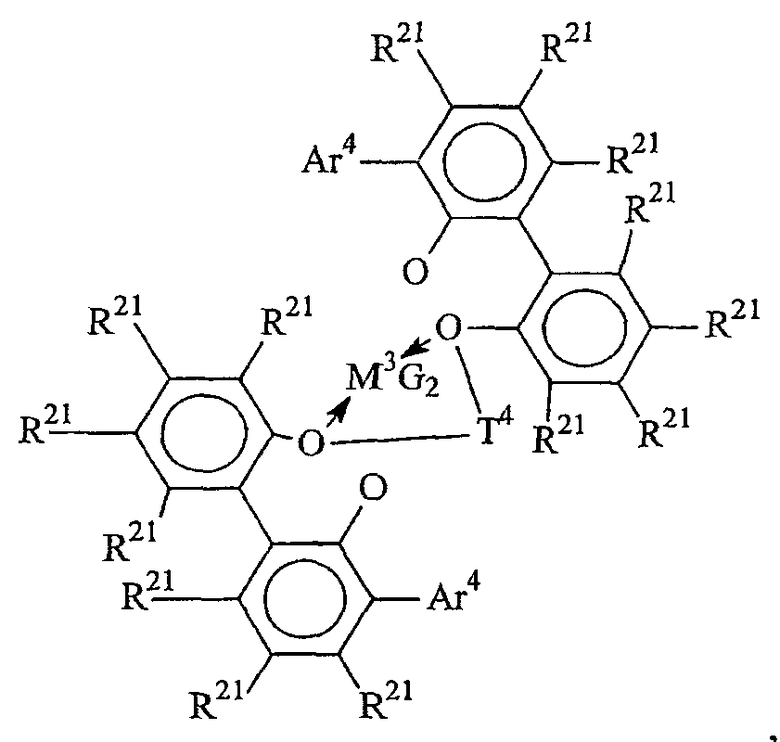
где M3 представляет Hf или Zr;
Ar4 представляет С6-20 арил или его инертно замещенное производное, особенно 3,5-ди(изопропил)фенил, 3,5-ди(изобутил)фенил, дибензо-1H-пиррол-1-ил или антрацен-5-ил и
T4, независимо в каждом случае, содержит группу С3-6 алкилена, группу С3-6 циклоалкилена или их инертно замещенные производные;
R21, независимо в каждом случае, представляет водород, галоген, гидрокарбил, тригидрокарбилсилил или тригидрокарбилсилилгидрокарбил из вплоть до 50 атомов, не считая водород, и
G, независимо в каждом случае, представляет галоген или группу гидрокарбил или тригидрокарбилсилил из вплоть до 20 атомов, не считая водород, или 2 группы G вместе представляют двухвалентное производное указанных гидрокарбила или тригидрокарбилсилила.
Особенно предпочтительными являются соединения формулы:
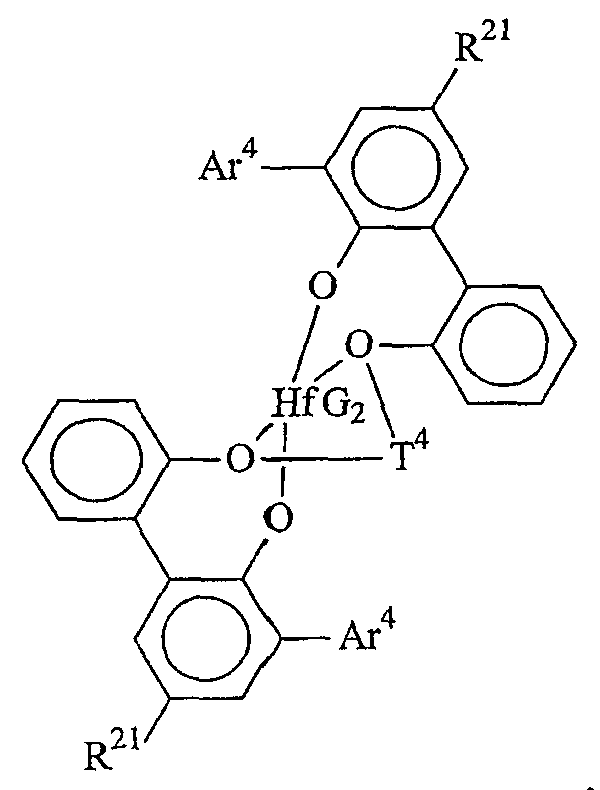
где Ar4 представляет 3,5-ди(изопропил)фенил, 3,5-ди(изобутил)фенил, дибензо-1H-пиррол-1-ил или антрацен-5-ил,
R21 представляет водород, галоген или С1-4 алкил, особенно метил,
T4 представляет пропан-1,3-диил или бутан-1,4-диил и
G представляет хлор, метил или бензил.
Наиболее предпочтительным комплексом металла предшествующей формулы является:
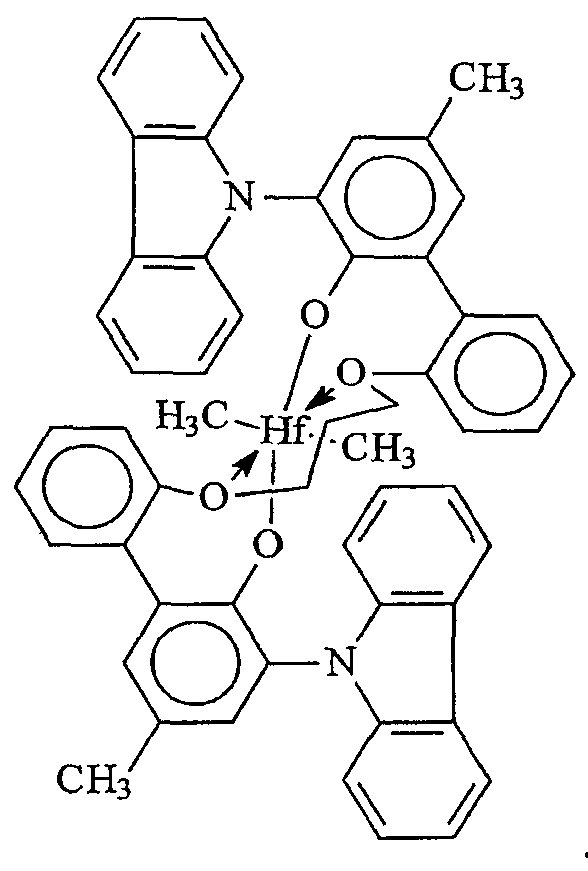
Указанные комплексы поливалентного основания Льюиса обычно получают стандартными процедурами металлирования и обмена лиганда с привлечением источника металла группы 4 и источника нейтрального полифункционального лиганда. В дополнение, комплексы могут быть также получены путем процесса элиминирования амида и гидрокарбилирования, исходя из тетраамида соответствующего металла группы 4 и агента гидрокарбилирования, такого как триметилалюминий. Другие технологии также могут быть использованы. Эти комплексы известны из, наряду с другими источниками, патентов США 6320005, 6103657, а также WO 02/38628, WO 03/40195 и США 04/0220050.
Катализаторы, имеющие высокие свойства внедрения сомономера, как также известно, повторно внедряют полученные на месте длинноцепочечные олефины, случайно образующиеся во время полимеризации через β-гидридное элиминирование и обрывание цепи растущего полимера или в результате другого процесса. Концентрация таких длинноцепочечных олефинов особенно увеличивается при использовании условий непрерывной полимеризации в растворе при высоких степенях превращения, особенно при степенях превращения этилена 95 процентов или более, более предпочтительно при степенях превращения этилена 97 процентов или более. В таких условиях небольшое, но поддающееся определению количество оканчивающегося олефином полимера может быть повторно внедрено в растущую полимерную цепь, что приводит к образованию длинноцепочечных ответвлений, то есть ветвей углерода длиной более чем могло бы быть результатом от другого сознательно добавленного сомономера. Более того, такие цепи отражают присутствие других сомономеров, представленных в реакционной смеси. То есть цепи могут также включать короткоцепочечное или длинноцепочечное разветвление, зависящее от состава сомономеров реакционной смеси. Длинноцепочечное разветвление олефиновых полимеров дополнительно описано в патентах США 5272236, 5278272 и 5665800. В одном аспекте изобретения степень длинноцепочечного разветвления в продукте заметно подавляют или элиминируют в целом применением челночных агентов цепи, чтобы заставить по существу все полимерные цепи оканчиваться челночным агентом цепи, а не образованием винил-групп, которые могут быть повторно внедрены с образованием длинноцепочечного ответвления. В этом варианте воплощения полученный полимерный блок является в высокой степени линейным, что приводит к благоприятным свойствам.
Альтернативно и более предпочтительно, разветвление, включая гиперразветвление, может быть индуцировано в конкретном сегменте данных мульти-блок-сополимеров применением специфических катализаторов, о которых известно, что они приводят к "гулянию цепи" в полученном полимере. Например, конкретные гомогенные связанные мостиковыми связями бис-инденил- или частично гидрированные бис-инденил-циркониевые катализаторы, раскрытые Kaminski, et al, J. Mol. Catal. A: Chemical. 102 (1995) 59-65; Zambelli, et al., Macromolecules. 1988, 21, 617-622, или Dias, et al, J. Mol. Catal. A: Chemical. 185 (2002) 57-64, могут быть могут быть использованы для получения разветвленных сополимеров из единственных мономеров, включая этилен. Катализаторы на основе высших переходных металлов, особенно никелевые и палладиевые катализаторы, как известно, приводят к гиперразветвленным полимерам (ответвления которых также разветвляются), как раскрыто в Brookhart, et al., J. Am. Chem. Soc., 1995, 117, … -6415.
В одном варианте воплощения изобретения наличие такого разветвления (длинноцепочечного разветвления, 1,3-присоединения или гиперразветвления) в полимерах по изобретению может быть ограничено только блоками или сегментами, являющимися результатом активности катализатора А. Соответственно, в одном варианте воплощения изобретения мульти-блок-сополимер, содержащий блоки или сегменты, различающиеся по наличию такого разветвления, в сочетании с другими сегментами или блоками, по существу лишенными такого разветвления (особенно высоко кристаллическими полимерными блоками с высокой плотностью или высокой степенью кристалличности), могут быть получены из реакционной смеси, содержащей единственный мономер, то есть без присоединения сознательно добавляемого сомономера. Очень предпочтительно, в отдельном варианте воплощения изобретения, мульти-блок-сополимер, содержащий чередующиеся неразветвленные этиленовые гомополимерные сегменты и разветвленные полиэтиленовые сегменты, особенно сегменты сополимера этилен/пропилен, может быть получен из первоначальной реакционной смеси, состоящей по существу из этилена в качестве полимеризуемого присоединением мономера. Наличие такого разветвления в мульти-блок-сополимерах по изобретению может быть выявлено по конкретным физическим свойствам полученных сополимеров, таким как уменьшенная дефектность поверхности во время экструзии расплава (уменьшенное растрескивание расплава), пониженная температура плавления, Tg, для аморфных сегментов по сравнению с неразветвленным полимерным сегментом и/или наличие последовательностей 1,3-присоединения или гиперразветвления, как определяют методом ЯМР. Количество указанных типов разветвления, имеющихся в полимерах по изобретению (как часть блоков или сегментов, содержащих его) обычно находится в пределах от 0,01 до 10 ответвлений на 1000 атомов углерода.
Подходящие соединения металлов для применения в качестве катализатора (В) включают предшествующие соединения металлов, упомянутые по отношению к катализатору (А), а также другие соединения металлов при условии, что в одном варианте воплощения изобретения они могут внедрять сомономер относительно слабо по сравнению с катализатором (А). Соответственно, в дополнение к ранее указанным комплексам металлов могут быть использованы следующие дополнительные комплексы металлов.
Производные групп 4-10, соответствующие формуле:
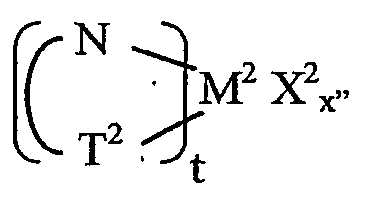
где
M2 представляет металл групп 4-10 Периодической таблицы элементов, предпочтительно металл группы 4, Ni(II) или Pd(II), наиболее предпочтительно цирконий;
T2 представляет группу, содержащую азот, кислород или фосфор;
X2 представляет галоген, гидрокарбил или гидрокарбилокси;
t означает один или два;
x" представляет число, выбранное, чтобы обеспечить баланс заряда,
и T2 и N связаны мостиковым лигандом.
Такие катализаторы ранее были раскрыты в J. Am. Chem. Soc., 118, 267-268 (1996), J. Am. Chem. Soc., 117, 6414-6415 (1995), и Organometallics, 16, 1514-1516 (1997), наряду с другими раскрытиями.
Предпочтительными примерами указанных комплексов металлов для применения в качестве катализатора (B) являются комплексы ароматических дииминов или ароматических диоксииминов с металлами группы 4, особенно с цирконием, соответствующие формуле:
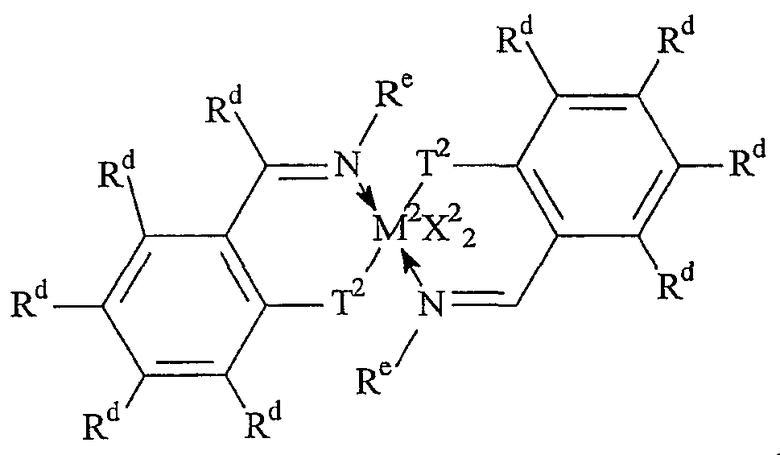
где
M2, T2 и Х2 имеют указанные выше значения,
Rd, независимо в каждом случае, представляет водород, галоген или Re и
Re, независимо в каждом случае, представляет C1-20 гидрокарбил или его производное, замещенное гетератомом, особенно F, N, S или P, более предпочтительно C1-10 гидрокарбил или его производное, замещенное F или N, наиболее предпочтительно алкил, диалкиламиноалкил, пирролил, пиперидинил, перфторфенил, циклоалкил, (поли)алкиларил или аралкил.
Наиболее предпочтительными примерами предшествующих комплексов металлов для применения в качестве катализатора (B) являются комплексы ароматического диоксиимина и циркония, соответствующие формуле:
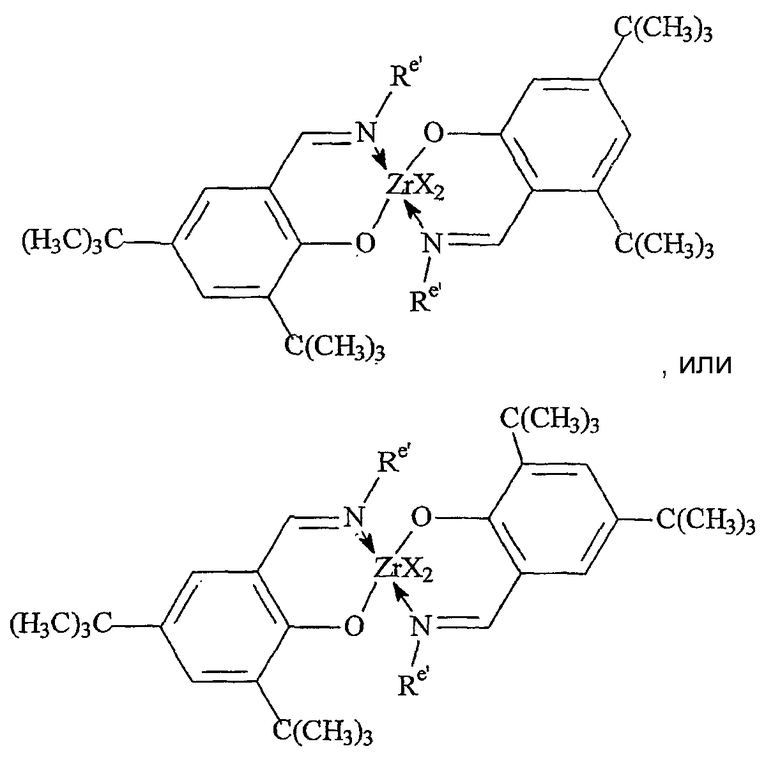
где
X2 имеет указанные выше значения, предпочтительно С1-10 гидрокарбил, наиболее предпочтительно метил или бензил, и
Re' представляет метил, изопропил, трет-бутил, циклопентил, циклогексил, 2-метилциклогексил, 2,4-диметилциклогексил, 2-пирролил, N-метил-2-пирролил, 2-пиперидинил, N-метил-2-пиперидинил, бензил, o-толил, 2,6-диметилфенил, перфторфенил, 2,6-ди(изопропил)фенил или 2,4,6-триметилфенил.
Указанные комплексы для применения в качестве катализатора (B), также включающие некоторые комплексы фосфинимина, раскрыты в EP-A-890581. Эти комплексы соответствуют формуле:
[(R f ) 3 -P=N] f M(K 2 )(R f ) 3-f ,
где
Rf представляет одновалентный лиганд или две группы Rf вместе представляют двухвалентный лиганд, предпочтительно Rf представляет водород или С1-4 алкил;
M представляет металл группы 4;
K2 представляет группу, содержащую делокализованные π-электроны, посредством которых группа K2 связана с M, указанная группа K2 содержит вплоть до 50 атомов, не считая атомы водорода, и
f означает 1 или 2.
Специалист должен учитывать, что в других вариантах воплощения изобретения критерием выбора сочетания катализатора (А) и (В) может быть любое другое отличающееся свойство полученных полимерных блоков, такое как сочетание на основе регулярности молекулярной структуры (изотактический/синдиотактический, изотактический/атактический или синдиотактический/атактический), содержание регио-ошибки или их сочетания.
Сокатализаторы
Каждый из катализаторов (A) и (B) на основе комплексов металлов (также взаимозаменяемо упоминаемые здесь как прокатализаторы) может быть активирован до образования активного каталитического состава путем объединения с сокатализатором, предпочтительно катионообразующим сокатализатором, сильной кислотой Льюиса или их сочетанием. В предпочтительном варианте воплощения челночный агент используют как для целей челночного перемещения цепи, так и в качестве компонента сокатализатора каталитического состава.
Комплексам металлов желательно придавать каталитическую активность объединением с катионообразующим сокатализатором, таким как те, которые были известны ранее в технике для применения с комплексами металлов группы 4 для полимеризации олефинов. Подходящие катионообразующие сокатализаторы для применения здесь включают нейтральные кислоты Льюиса, такие как С1-30 гидрокарбил-замещенные соединения металла группы 13, особенно соединения три(гидрокарбил)алюминия или три(гидрокарбил)бора и галогенированные (включая пергалогенированные) их производные, имеющие от 1 до 10 атомов углерода в каждой группе гидрокарбила или галогенированного гидрокарбила, более предпочтительно перфторированные три(арил)бор-соединения и наиболее предпочтительно трис(пентафтор-фенил)боран; неполимерные совместимые некоординирующие ионообразующие соединения (включая применение таких соединений в окислительных условиях), особенно применение солей аммония, фосфония, оксония, карбония, силилия или сульфония с совместимыми некоординирующими анионами или солей ферроцения, свинца или серебра с совместимыми некоординирующими анионами, и сочетания указанных катионообразующих сокатализаторов и технических приемов. Указанные активирующие сокатализаторы и активирующие технические приемы ранее были изучены в отношении различных комплексов металлов для полимеризации олефинов в следующих ссылках: EP-A-277003, US-A-5153157, US-A-5064802, US-A-5321106, US-A-5721185, US-A-5350723, US-A-5425872, US-A-5625087, US-A-5883204, US-A-5919983, US-A-5783512, WO 99/15534 и WO99/42467.
Сочетания нейтральных кислот Льюиса, особенно сочетание соединения триалкилалюминия, имеющего от 1 до 4 атомов углерода в каждой алкил-группе, и галогенированного соединения три(гидрокарбил)бора, имеющего от 1 до 20 атомов углерода в каждой гидрокарбил-группе, особенно трис(пентафторфенил)борана, дополнительные сочетания, таких смесей нейтральных кислот Льюиса с полимерным или олигомерным алюмоксаном и сочетания единственной нейтральной кислоты Льюиса, особенно трис(пентафторфенил)борана с полимерным или олигомерным алюмоксаном, могут быть использованы в качестве активирующих сокатализаторов. Предпочтительные молярные отношения комплекс металла:трис(пентафторфенил-боран:алюмоксан от 1:1:1 дo 1:5:20, более предпочтительно от 1:1:1,5 дo 1:5:10.
Подходящие ионообразующие соединения, применимые в качестве сокатализаторов в одном варианте воплощения данного изобретения, содержат катион, который является кислотой Бренстеда, способной передавать протон, и совместимый некоординирующий анион, A-. Используемый здесь термин "некоординирующий" означает анион или вещество, которое или не координирует с содержащим металл группы 4 комплексом предшественником и каталитическим производным, полученным из него, или который только слабо координируется с такими комплексами, оставаясь поэтому достаточно лабильным, чтобы быть замещенным нейтральным основанием Льюиса. Некоординирующий анион специфически относится к аниону, который, когда выступает в роли уравновешивающего заряд аниона в катионном металлическом комплексе, не переносит анионный заместитель или его фрагмент к указанному катиону, формируя тем самым нейтральные комплексы. "Совместимые анионы" - это анионы, которые не деградируют до нейтральности, когда первоначально сформированный комплекс разлагается, и не вмешиваются в желательную последующую полимеризацию или другие применения комплекса.
Предпочтительными анионами являются анионы, содержащие единственный координационный комплекс, содержащий несущее заряд металлическое или металлоидное ядро, такой анион способен уравновешивать заряд активных каталитических частиц (катиона металла), которые могут быть образованы при объединении двух компонентов. Кроме того, указанный анион должен быть достаточно лабильным, чтобы быть замещенным олефиновыми, диолефиновыми и ненасыщенными по типу ацетилена соединениями или другими нейтральными основаниями Льюиса, такими как простые эфиры или нитрилы. Подходящие металлы включают, но без ограничения указанным, алюминий, золото и платину. Подходящие металлоиды включают, но без ограничения указанным, бор, фосфор и кремний. Соединения, содержащие анионы, которые содержат координационные комплексы, содержащие единственный атом металла или металлоида, конечно, хорошо известны, и многие, в частности такие соединения, содержащие единственный атом бора в анионной части, коммерчески доступны.
Предпочтительно такие сокатализаторы могут быть представлены следующей общей формулой:
(L*-H) g + (A) g -
где
L* представляет нейтральное основание Льюиса,
(L*-H)+ представляет сопряженную с L* кислоту Бренстеда;
Ag' представляет некоординирующий совместимый анион, имеющий заряд g-, и
g представляет целое число от 1 до 3.
Более предпочтительно Ag- соответствует формуле:
[M'Q 4 ] -
где
M' представляет бор или алюминий в состоянии нормального окисления +3 и
Q, независимо в каждом случае, выбран из таких радикалов, как гидрид, диалкиламидо, галогенид, гидрокарбил, гидрокарбилоксид, галогензамещенный-гидрокарбил, галогензамещенный гидрокарбилокси и галоген-замещенный силилгидрокарбил (включая пергалогенированный гидрокарбил- пергалогенированный гидрокарбилокси- и пергалогенированный силилгидрокарбил-радикалы), указанный Q имеет вплоть до 20 атомов углерода при условии, что не более чем в одном случае Q галогенид. Примеры подходящих гидрокарбилоксидных групп Q раскрыты в US-A-5296433.
В более предпочтительном варианте воплощения d означает один, то есть противоион имеет единственный отрицательный заряд и представлен А-. Активирующие сокатализаторы, содержащие бор, которые особенно применимы при получении катализаторов по данному изобретению, могут быть представлены следующей общей формулой:
(L*-H) + (BQ 4 ) -
где
L* имеет указанные выше значения,
B представляет бор в нормальном состоянии окисления 3 и
Q представляет гидрокарбил-, гидрокарбилокси-, фторированную гидрокарбил-, фторированную гидрокарбилокси- или фторированную силилгидрокарбил-группу из вплоть до 20 атомов, иных чем водород при условии, что не более чем в одном случае Q гидрокарбил.
Предпочтительными солями основания Льюиса являются соли аммония, более предпочтительно соли триалкиламмония, содержащие одну или несколько С12-40 алкил-групп. Наиболее предпочтительно Q, в каждом случае, представляет фторированный арил, особенно пентафторфенил.
Пояснительными, но не ограничительными примерами соединений бора, которые могут быть использованы в качестве активирующего сокатализатора при получении усовершенствованных катализаторов по данному изобретению, являются тризамещенные соли аммония, такие как
триметиламмоний тетракис(пентафторфенил) борат,
триэтиламмоний тетракис(пентафторфенил) борат,
трипропиламмоний тетракис(пентафторфенил) борат,
три(н-бутил)аммоний тетракис(пентафторфенил) борат,
три(втор-бутил)аммоний тетракис(пентафторфенил) борат,
N,N-диметиланилиний тетракис(пентафторфенил) борат,
N,N-диметиланилиний н-бутилтрис(пентафторфенил) борат,
N,N-диметиланилиний бензилтрис(пентафторфенил) борат,
N,N-диметиланилиний тетракис(4-(трет-бутилдиметилсилил)-2,3,5,6-тетрафторфенил) борат,
N,N-диметиланилиний тетракис(4-(триизопропилсилил)-2,3,5,6-тетрафторфенил) борат,
N,N-диметиланилинийпентафторфенокситрис(пентафторфенил)борат
N,N-диэтиланилиний тетракис(пентафторфенил) борат,
N,N-диметил-2,4,6-триметиланилинийтетракис(пентафторфенил)борат,
диметилоктадециламмоний тетракис(пентафторфенил) борат,
метилдиоктадециламмоний тетракис(пентафторфенил) борат,
соли диалкиламмония, такие как
ди(изопропил)аммоний тетракис(пентафторфенил) борат,
метилоктадециламмоний тетракис(пентафторфенил) борат,
метилоктадодециламмоний тетракис(пентафторфенил) борат и
диоктадециламмоний тетракис(пентафторфенил) борат;
соли тризамещенного фосфония, такие как
трифенилфосфоний тетракис(пентафторфенил) борат,
метилдиоктадецилфосфоний тетракис(пентафторфенил) борат и
три(2,6-диметилфенил)фосфоний тетракис(пентафторфенил) борат;
соли дизамещенного оксония, такие как:
дифенилоксоний тетракис(пентафторфенил) борат,
ди(o-толил)оксоний тетракис(пентафторфенил) борат и
ди(октадецил)оксоний тетракис(пентафторфенил) борат;
соли дизамещенного сульфония, такие как:
ди(o-толил)сульфоний тетракис(пентафторфенил) борат и
метилоктадецилсульфоний тетракис(пентафторфенил) борат.
Предпочтительными катионами (L*-H)+ являются катионы метилдиоктадециламмония, катионы диметилоктадециламмония и катионы аммония, полученные из смесей триалкиламинов, содержащих одну или две С14-18 алкил-группы.
Другой подходящий ионообразующий активирующий сокатализатор содержит соль катионного окисляющего агента и некоординирующего совместимого аниона, представленную формулой:
(Ox h+ ) g (A g- ) h
где
Oxh+ представляет катионный окисляющий агент, имеющий заряд h+;
h представляет целое число от 1 до 3 и
Ag- и g имеют указанные выше значения.
Примеры катионных окисляющих агентов включают ферроцений, гидрокарбил-замещенный ферроцений, Ag+ или Pb+2. Предпочтительными вариантами воплощения Ag- являются такие анионы, определение которым дано ранее по отношению к содержащим кислоту Бренстеда активирующим сокатализаторам, особенно тетракис(пентафторфенил)борату.
Другой подходящий ионообразующий, активирующий сокатализатор содержит соединение, которое представляет соль иона карбения и некоординирующего совместимого аниона, представленную формулой:
[C] + A -
где
[C]+ представляет С1-20 ион карбения и
A- представляет некоординирующий совместимый анион, имеющий заряд -1. Предпочтительным ионом карбения является катион тритил, то есть трифенилметилий.
Дополнительный подходящий ионообразующий активирующий сокатализатор содержит соединение, которое представляет собой соль иона силилия и некоординирующего совместимого аниона, представленное формулой:
(Q 1 3 Si) + A -
где
Q1 представляет гидрокарбил и A- имеет указанные выше значения.
Предпочтительными активирующими сокатализаторами солями силилия являются триметилсилилий тетракиспентафторфенилборат, триэтилсилилий тетракиспентафторфенилборат и замещенные простым эфиром их аддукты. Соли силилия были ранее раскрыты в общих чертах в J. Chem Soc. Chem. Comm., 1993, 383-384, а также Lambert, J. B., et al., Organometallics. 1994, 13, 2430-2443. Применение указанных солей силилия в качестве активирующих сокатализаторов для катализаторов полимеризации присоединением раскрыто в US-A-5625087.
Некоторые комплексы спиртов, меркаптанов, силанолов и оксимов с трис(пентафторфенил)бораном также являются эффективными активаторами катализаторов и могут быть использованы согласно данному изобретению. Такие сокатализаторы раскрыты в US-A-5296433.
Подходящие активирующие сокатализаторы для применения здесь также включают полимерные или олигомерные алюмоксаны, особенно метилалюмоксан (MAO), модифицированный триизобутилалюминием метилалюмоксан (MMAO) или изобутилалюмоксан; модифицированные кислотой Льюиса алюмоксаны, особенно модифицированные пергалогенированным три(гидрокарбил)алюминием или пергалогенированным три(гидрокарбил)бором алюмоксаны, имеющие от 1 до 10 атомов углерода в каждой группе гидрокарбила или галогенированного гидрокарбила, и наиболее предпочтительно модифицированные трис(пентафторфенил)бораном алюмоксаны. Такие сокатализаторы раскрыты ранее в патентах США 6214760, 6160146, 6140521 и 6696379.
Класс сокатализаторов, содержащих некоординирующие анионы, упоминаемые как экспандированные анионы, дополнительно раскрытый в патенте США 6395671, может быть успешно использован для активации комплексов металлов по данному изобретению для полимеризации олефинов. Обычно такие сокатализаторы (иллюстрируемые сокатализаторами, имеющими анионы имидазолид, замещенный имидазолид, имидазолинид, замещенный имидазолинид, бензимидазолид или замещенный бензимидазолид) могут быть изображены следующим образом:
где
A*+ представляет катион, особенно катион, содержащий протон, и предпочтительно представляет катион тригидрокарбиламмония, содержащий одну или две С14-20 алкил-группы, особенно катион метил-ди(С10-40 алкил)аммония,
Q3, независимо в каждом случае, представляет водород или галоген, гидрокарбил, галогенкарбил, галогенгидрокарбил, силилгидрокарбил или силил, включая моно-, ди- и три(гидрокарбил)силил) группу из вплоть до 30 атомов, не считая водород, предпочтительно С1-20 алкил, и
Q2 представляет трис(пентафторфенил)боран или трис(пентафторфенил)алюман).
Примеры таких активаторов катализаторов включают соли тригидрокарбиламмония, особенно соли метилди(С14-20 алкил)аммония:
бис(трис(пентафторфенил)боран)имидазолид,
бис(трис(пентафторфенил)боран)-2-ундецилимидазолид,
бис(трис(пентафторфенил)боран)-2-гептадецилимидазолид,
бис(трис(пентафторфенил)боран)-4,5-бис(ундецил)имидазолид,
бис(трис(пентафторфенил)боран)-4,5-бис(гептадецил)имидазолид,
бис(трис(пентафторфенил)боран)имидазолинид,
бис(трис(пентафторфенил)боран)-2-ундецилимидазолинид,
бис(трис(пентафторфенил)боран)-2-гептадецилимидазолинид,
бис(трис(пентафторфенил)боран)-4,5-бис(ундецил)имидазолинид,
бис(трис(пентафторфенил)боран)-4,5-бис(гептадецил)имидазолинид,
бис(трис(пентафторфенил)боран)-5,6-диметилбензимидазолид,
бис(трис(пентафторфенил)боран)-5,6-бис(ундецил)бензимидазолид,
бис(трис(пентафторфенил)алюман)имидазолид,
бис(трис(пентафторфенил)алюман)-2-ундецилимидазолид,
бис(трис(пентафторфенил)алюман)-2-гептадецилимидазолид,
бис(трис(пентафторфенил)алюман)-4,5-бис(ундецил)имидазолид,
бис(трис(пентафторфенил)алюман)-4,5-бис(гептадецил)имидазолид,
бис(трис(пентафторфенил)алюман)имидазолинид,
бис(трис(пентафторфенил)алюман)-2-ундецилимидазолинид,
бис(трис(пентафторфенил)алюман)-2-гептадецилимидазолинид,
бис(трис(пентафторфенил)алюман)-4,5-бис(ундецил)имидазолинид,
бис(трис(пентафторфенил)алюман)-4,5-бис(гептадецил)имидазолинид,
бис(трис(пентафторфенил)алюман)-5,6-диметилбензимидазолид и
бис(трис(пентафторфенил)алюман)-5,6-бис(ундецил)бензимидазолид.
Другие активаторы включают те, которые описаны в публикации PCT WO 98/07515, такие как трис(2,2',2"-нонафторбифенил) фторалюминат. Сочетания активаторов также предусмотрены изобретением, например, алюмоксаны и ионизирующие активаторы в сочетаниях, смотри, например, EP-A-0 573120, публикации PCT WO 94/07928 и WO 95/14044 и патенты США 5153157 и 5453410. WO 98/09996 описывает активирование каталитических соединений перхлоратами, периодатами и иодатами, включая их гидраты. WO 99/18135 описывает применение органоборалюминиевых активаторов. WO 03/10171 раскрывает активаторы катализаторов, которые являются аддуктами кислот Бренстеда с кислотами Льюиса. Другие активаторы и способы активирования каталитического соединения описаны, например, в патентах США 5849852, 5859653, 5869723, EP-A-615981 и публикации PCT WO 98/32775. Все указанные активаторы катализаторов, а также любой другой известный активатор для катализаторов на основе комплексов переходных металлов может быть использован один или в сочетании согласно данному изобретению, однако, для достижения наилучших результатов лучше избегать содержащих алюмоксан сокатализаторов.
Используемое молярное отношение катализатор/сокатализатор предпочтительно находится в пределах от 1:10000 дo 100:1, более предпочтительно от 1:5000 дo 10:1, наиболее предпочтительно от 1:1000 дo 1:1. Алюмоксан, когда его применяют сам по себе в качестве активирующего сокатализатора, используют в большом количестве, обычно по меньшей мере 100-кратном количеству комплекса металла на молярной основе. Трис(пентафторфенил)боран, когда его применяют в качестве активирующего сокатализатора, используют в молярном отношении к комплексу металла от 0,5:1 дo 10:1, более предпочтительно от 1:1 дo 6:1, наиболее предпочтительно от 1:1 дo 5:1. Остальные активирующие сокатализаторы обычно используют приблизительно в эквимолярном количестве с комплексом металла.
Способ по изобретению с использованием катализатора А, катализатора В и челночного агента цепи С может быть дополнительно пояснен со ссылкой на фиг.1, где показан активированный каталитический сайт А, 10, который в условиях полимеризации формирует полимерную цепь 13, прикрепленную к активному каталитическому сайту, 12. Подобным образом активный каталитический сайт В, 20, производит дифференцированную полимерную цепь 23, прикрепленную к активному каталитическому сайту 22. Челночный агент С1 цепи, присоединенный к полимерной цепи, образованной активным катализатором В, 14, обменивает его полимерную цепь 23, на полимерную цепь 13, присоединенную к каталитическому сайту А. Дополнительный рост цепи в условиях полимеризации вызывает формирование мульти-блок-сополимера 18, присоединенного к активному каталитическому сайту А. Подобным образом челночный агент С2 цепи, прикрепленный к полимерной цепи, образованной активным каталитическим сайтом А, 24, обменивает его полимерную цепь, 13, на полимерную цепь 23, прикрепленную к каталитическому сайту В.
Дополнительный рост цепи в условиях полимеризации вызывает образование мульти-блок-сополимера 28, присоединенного к активному каталитическому сайту В. Растущие мульти-блок-сополимеры подвергаются повторному обмену между активным катализатором А и активным катализатором В посредством челночного агента С, результатом чего является формирование блока или сегмента с отличающимися свойствами, когда бы ни происходил обмен на противоположный активный каталитический сайт. Растущие полимерные цепи могут быть извлечены, пока они присоединены к челночному агенту цепи, и функционализованы, если желательно. В качестве варианта полученный полимер может быть извлечен отделением от активного каталитического сайта или челночного агента путем использования источника протонов или другого губительного агента.
Вероятно (без желания быть связанными таким предположением), что на состав из соответствующих сегментов или блоков и особенно на концевые сегменты полимерных цепей можно воздействовать через выбор условий процесса или других переменных процесса. В полимерах по изобретению характер концевых сегментов определяется относительными скоростями переноса или обрывания цепи для соответствующих катализаторов, а также относительными скоростями челночного перемещения цепи. Возможные механизмы обрывания цепи включают, но без ограничения указанным, элиминирование β-водорода, перенос β-водорода к мономеру, элиминирование β-метила и перенос цепи к водороду или другому обрывающему цепь реагенту, такому как органосилан или агент функционализирования цепи. Соответственно, когда используют низкую концентрацию челночного агента цепи, большинство концов полимерных цепей будут генерированы в реакторе полимеризации одним из указанных механизмов обрывания цепи, и относительные скорости обрывания цепей для катализаторов (А) и (В) будут определять преобладающую обрывающую цепь группу. То есть катализатор, имеющий наивысшую скорость обрывания цепи, будет создавать относительно больше концевых сегментов цепи в законченном полимере.
И наоборот, когда используют высокую концентрацию челночного агента цепи, большинство полимерных цепей внутри реактора и на выходе из зоны реакции присоединены к или связаны с челночным агентом цепи. В таких условиях реакции относительные скорости переноса цепи катализаторов полимеризации и относительная скорость челночного перемещения цепи двух катализаторов в основном определяют своеобразие завершающей цепь группы. Если катализатор (А) имеет более быструю скорость переноса цепи и/или челночного перемещения цепи, чем катализатор В, тогда большинство концевых сегментов цепи будут сегментами, создаваемыми катализатором (А).
При промежуточных концентрациях челночного агента цепи все три указанные фактора являются действенными в определении своеобразия концевого блока полимера. Предыдущая методология может быть расширена до анализа мульти-блок-сополимеров, имеющих более двух типов блоков, и для регулирования средних длин блоков и последовательностей блоков для указанных полимеров. Например, использование смеси катализаторов 1, 2 и 3 с челночным агентом цепи, для которого каждый тип катализатора создает отличающийся тип полимерного блока, дает линейный блок-сополимер с тремя различными типами блоков. Более того, если отношение скорости челночного перемещения к скорости распространения для трех катализаторов следует в порядке 1>2>3, тогда средняя длина блока для трех типов блоков будет следовать в порядке 3>2>1, и будет меньше случаев блоков 2-типа, смежных с блоками 3-типа, чем блоков 1-типа, смежных с блоками 2-типа.
Отсюда следует, что существует способ регулирования распределения длин блоков различных типов блоков. Например, путем подбора катализаторов 1, 2 и 3 (где 2 и 3 создают по существу тот же тип полимерного блока) и челночного агента цепи и скоростей, следующих в порядке 1>2>3, получают полимер, который будет иметь бимодальное распределение длин блоков, полученных от катализаторов 2 и 3.
Во время полимеризации реакционную смесь, содержащую один или несколько мономеров, приводят в контакт с активированным каталитическим составом согласно каким-либо подходящим условиям полимеризации. Процесс характеризуется применением повышенных температур и давлений. Водород может быть использован в качестве агента переноса цепи для контроля молекулярной массы согласно известным методикам, если желательно. Как и при других подобных полимеризациях очень желательно, чтобы используемые мономеры и растворители были достаточно высокой чистоты, чтобы не происходила дезактивация катализатора. Может быть использована любая подходящая технология очистки мономера, такая как удаление летучих примесей при пониженном давлении, контактирование с молекулярными ситами или оксидом алюминия с высокой удельной поверхностью или сочетание указанных процессов. Специалист будет учитывать, что отношение челночного агента цепи к одному или нескольким катализаторам и/или мономерам в процессе по данному изобретению может изменяться для того, чтобы получать полимеры, отличающиеся по одному или нескольким химическим или физическим свойствам.
В данном изобретении могут быть использованы носители, особенно в суспензионной или газофазной полимеризациях. Подходящие носители включают твердые измельченные имеющие высокую удельную поверхность оксиды металлов, оксиды металлоидов или их смеси (взаимозаменяемо упоминаемые здесь как неорганический оксид). Примеры включают тальк, оксид алюминия, оксид магния, оксид титана, диоксид циркония, Sn2O3, алюминосиликаты, боросиликаты, глины и их смеси. Подходящие носители предпочтительно имеют удельную поверхность, которую определяют порометрией азотом по методу БЭT, от 10 до 1000 м2/г и предпочтительно от 100 до 600 м2/г. Средний размер частиц обычно от 0,1 до 500 мкм, предпочтительно от 1 до 200 мкм, более предпочтительно от 10 до 100 мкм.
В одном варианте воплощения изобретения данный каталитический состав и необязательный носитель могут быть высушены при распылении или иным образом превращены в твердую порошкообразную форму, чтобы обеспечить состав, который легко транспортируется и которым легко манипулировать. Подходящие способы сушки при распылении содержащей жидкость суспензии хорошо известны в технике и успешно используются здесь. Предпочтительные технологии сушки при распылении каталитических составов для применения здесь описаны в патентах США 5648310 и 5672669.
Полимеризацию желательно проводят как непрерывную полимеризацию, предпочтительно непрерывную полимеризацию в растворе, в которой каталитические компоненты, челночный агент(ы), мономеры и необязательно растворитель, адъюванты, поглотители и вспомогательные добавки для полимеризации непрерывно подают в зону реакции и полимерный продукт непрерывно удаляют из нее. В сфере охвата терминов "непрерывный" и "непрерывно", которые используются в данном контексте, находятся такие процессы, в которых осуществляется периодическое добавление реагентов и удаление продуктов с такими малыми регулярными или нерегулярными интервалами, что во времени весь процесс является по существу непрерывным.
Каталитические составы могут быть полезно использованы в осуществляемом при высоком давлении в растворе, в суспензии или в газовой фазе процессе полимеризации. Для процесса полимеризации в растворе желательно использовать гомогенные дисперсии каталитических компонентов в жидком разбавителе, в котором полимер растворим в используемых условиях полимеризации. Один такой процесс с использованием чрезвычайно тонкого диоксида кремния или подобного диспергирующего агента для получения такой гомогенной каталитической дисперсии, где или комплекс металла, или сокатализатор является только слабо растворимым, раскрыт в US-A-5783512. Осуществляемый в растворе процесс получения новых полимеров по данному изобретению, особенно непрерывный, осуществляемый в растворе процесс предпочтительно проводят при температуре между 80°С и 250°С, более предпочтительно между 100°С и 210°С и наиболее предпочтительно между 110°С и 210°С. Осуществляемый при высоком давлении процесс обычно проводят при температурах от 100°С до 400°С и при давлениях выше 500 бар (50 МПа). Осуществляемый в суспензии процесс обычно использует инертный углеводородный разбавитель и температуры от 0°С вплоть до температуры чуть ниже температуры, при которой полученный полимер становится по существу растворимым в инертной среде полимеризации. Предпочтительные температуры при суспензионной полимеризации от 30°С, предпочтительно от 60°С вплоть до 115°С, предпочтительно вплоть до 100°С. Давления обычно находятся в пределах от атмосферного (100 кПа) до 500 фунтов на кв. дюйм (3,4 МПа).
Во всех указанных процессах предпочтительно используют условия непрерывной или по существу непрерывной полимеризации. Применение таких условий полимеризации, особенно непрерывных процессов полимеризации в растворе, с использованием двух или более разновидностей активных катализаторов полимеризации позволяет использовать повышенные температуры в реакторе, результатом чего является экономичное производство мульти-блок- или вегментированных сополимеров с высокими выходами и эффективностями. Могут быть использованы и условия гомогенной реакции, и условия реакции типа пробка-поток. Последние условия предпочтительны, когда желательно сужение к концу состава блока.
Оба каталитических состава (А) и (В) могут быть приготовлены в виде гомогенного состава добавлением необходимых комплексов металлов к растворителю, в котором будет проводиться полимеризация, или в разбавитель, совместимый с окончательной реакционной смесью. Желательный сокатализатор или активатор и челночный агент могут быть объединены с каталитическим составом предварительно, одновременно или после объединения с мономерами, которые должны быть полимеризованы, и каким-либо дополнительным реакционным разбавителем.
Все время отдельные ингредиенты, а также любой активный каталитический состав должны быть защищены от кислорода и влаги. Поэтому каталитические компоненты, челночный агент и активированные катализаторы следует готовить и хранить в атмосфере без кислорода и влаги, предпочтительно в сухом инертном газе, таком как азот.
Без ограничения каким-либо образом объема изобретения одним способом проведения процесса полимеризации является следующий. В реактор с мешалкой непрерывно вводят мономеры, которые должны быть полимеризованы, вместе с каким-либо растворителем или разбавителем. Реактор содержит жидкую фазу, состоящую по существу из мономеров вместе с каким-либо растворителем или разбавителем и растворенный полимер. Предпочтительные растворители включают С4-10 углеводороды или их смеси, особенно алканы, такие как гексан, или смеси алканов, а также один или несколько мономеров, используемых в полимеризации.
Катализаторы (А) и (В) наряду с сокатализатором и челночным агентом цепи непрерывно или периодически вводят в жидкую фазу реактора или в какую-либо ее рецикловую часть. Температуру и давление в реакторе можно регулировать, подбирая отношение растворитель/мономер, скорость добавления катализатора, а также с помощью охлаждающих или нагревающих змеевиков, рубашек или и того, и другого. Скорость полимеризации регулируют скоростью добавления катализатора. Содержание этилена в полимерном продукте определяется отношением этилена к сомономеру в реакторе, которое регулируют, манипулируя соответствующими скоростями подачи указанных компонентов в реактор. Молекулярную массу полимерного продукта контролируют, необязательно, путем регулирования других переменных полимеризации, таких как температура, концентрация мономера, или ранее упомянутым агентом переноса цепи, как это хорошо известно в технике. На выходе из реактора вытекающий поток приводят в контакт с агентом, уничтожающим катализатор, таким как вода, пар или спирт. Раствор полимера необязательно нагревают и полимерный продукт извлекают путем однократного испарения газообразных мономеров, а также остаточного растворителя или разбавителя при пониженном давлении и, если необходимо, проведения дополнительного удаления летучих компонентов в оборудовании, таком как экструдер с отводом газов. В непрерывном процессе среднее время пребывания катализатора и полимера в реакторе, как правило, от 5 минут до 8 часов и предпочтительно от 10 мин до 6 часов.
В качестве варианта указанная полимеризация может быть проведена в реакторе с циркуляцией непрерывного действия с градиентом мономера, катализатора или челночного агента, устанавливаемым между различными его зонами, или без градиента, с необязательным сопровождением раздельным добавлением катализаторов и/или агента переноса цепи и работающем при адиабатических или неадиабатических условиях полимеризации в растворе или при сочетании указанных реакторных условий. Примеры подходящих реакторов с циркуляцией и разнообразных подходящих технологических условий применительно к ним находятся в патентах США 5977251, 6319989 и 6683149.
Хотя и нежелательно, каталитический состав может быть также приготовлен и использован как гетерогенный катализатор путем адсорбирования необходимых компонентов на инертных неорганических или органических твердых частицах, как сообщалось ранее. В предпочтительном варианте воплощения гетерогенный катализатор получают совместным осаждением комплекса металла и продукта реакции инертного неорганического соединения и активатора, содержащего активный водород, особенно продукта реакции соединения три(С1-4 алкил)алюминия и аммониевой соли гидроксиарилтрис(пентафторфенил)бората, такой как аммониевая соль (4-гидрокси-3,5-ди-трет-бутилфенил)трис(пентафторфенил)бората. Когда каталитический состав приготовлен в гетерогенной форме или на носителе, его можно использовать в суспензионной или газофазной полимеризации. В качестве практического ограничения, суспензионная полимеризация имеет место в жидких разбавителях, в которых полимерный продукт по существу нерастворим. Предпочтительно разбавителем для суспензионной полимеризации является один или несколько углеводородов менее чем с 5 атомами углерода. Если желательно, насыщенные углеводороды, такие как этан, пропан или бутан, могут быть использованы как весь разбавитель или как часть его. Как с полимеризацией в растворе, α-олефиновый сомономер или смесь различных α-олефиновых сомономеров могут быть использованы как весь разбавитель или как часть его. Наиболее предпочтительно по меньшей мере большая часть разбавителя содержит α-олефиновый мономер или мономеры, которые должны быть полимеризованы.
Предпочтительно для применения в процессах газофазной полимеризации материал носителя и полученный катализатор имеют средний диаметр частиц от 20 до 200 мкм, более предпочтительно от 30 мкм до 150 мкм и наиболее предпочтительно от 50 мкм до 100 мкм. Предпочтительно для применения в процессах суспензионной полимеризации носитель имеет средний диаметр частиц от 1 мкм до 200 мкм, более предпочтительно от 5 мкм до 100 мкм и наиболее предпочтительно от 10 мкм до 80 мкм.
Подходящий для применения здесь процесс газофазной полимеризации по существу подобен известным процессам, используемым в промышленных масштабах для производства полипропилена, сополимеров этилен/α-олефин и других олефиновых полимеров. Используемый газофазный процесс может быть, например, того типа, в котором используют механически перемешиваемый слой или псевдоожиженный газом слой в качестве зоны реакции полимеризации. Предпочтительным является процесс, где реакцию полимеризации проводят в вертикальном цилиндрическом реакторе полимеризации, содержащем псевдоожиженный слой полимерных частиц на носителе или находящихся во взвешенном состоянии над перфорированной пластиной или решеткой для псевдоожижения потоком псевдоожижающего газа.
Газ, используемый для псевдоожижения слоя, содержит мономер или мономеры, которые должны быть полимеризованы, и также служит в качестве теплообменной среды для удаления тепла реакции из слоя. Горячие газы выходят из верха реактора обычно через зону транквилизации, известную также как зона снижения скорости, имеющую более широкий диаметр, чем псевдоожиженный слой, и где тонкие частицы, захваченные потоком газа, имеют благоприятную возможность вернутся под действием силы тяжести обратно в слой. Может быть также выгодно применять циклон для удаления ультратонких частиц из потока горячего газа. Газ затем обычно возвращают в слой через воздуходувку или компрессор и один или несколько теплообменников, чтобы отобрать тепло полимеризации от газа.
Предпочтительным способом охлаждения слоя в дополнение к охлаждению, обеспечиваемому охлажденным рецикловым газом, является подача летучей жидкости в слой, чтобы обеспечить испарительный охлаждающий эффект, что часто называют работой в режиме конденсации. Используемой в этом случае летучей жидкостью может быть, например, летучая инертная жидкость, например насыщенный углеводород, имеющий 3-8, предпочтительно 4-6 атомов углерода. В случае, когда мономер или сомономер сам по себе является летучей жидкостью или может быть конденсирован, чтобы обеспечить такую жидкость, его можно соответствующим образом вводить в слой, чтобы обеспечить испарительный охлаждающий эффект. Летучая жидкость испаряется в горячем псевдоожиженном слое с образованием газа, который смешивается с псевдоожижающим газом. Если летучей жидкостью является мономер или сомономер, он будет подвергаться некоторой полимеризации в слое. Испаренная жидкость затем выходит из реактора как часть горячего рециклового газа и поступает в компрессионную/теплообменную часть контура циркуляции. Рецикловый газ охлаждают в теплообменнике, и, если температура, до которой газ охлаждается, ниже точки росы, жидкость будет осаждаться из газа. Эту жидкость желательно возвращать непрерывно в псевдоожиженный слой. Возможно возвращать осажденную жидкость в слой в виде капель жидкости, переносимых в потоке рециклового газа. Этот тип процесса описан, например, в ЕР-89691, US 4543399, WO-94/25495 и US 5352749. Особенно предпочтительный способ возвращения жидкости в слой предусматривает отделение жидкости от потока рециклового газа и повторное впрыскивание этой жидкости непосредственно в слой, предпочтительно используя методику, которая создает тонкие капельки жидкости внутри слоя. Этот тип процесса описан в WO-94/28032.
Реакцию полимеризации, происходящую в псевдоожиженном слое, катализируют непрерывным или полунепрерывным добавлением каталитического состава по изобретению. Каталитический состав может быть подвергнут стадии предварительной полимеризации, например, путем полимеризации небольшого количества олефинового мономера в жидком инертном разбавителе, чтобы обеспечить каталитический композит, содержащий частицы катализатора на носителе, внедренные в частицы олефинового полимера также.
Полимер получают непосредственно в пседоожиженном слое путем полимеризации мономера или смеси мономеров на псевдоожиженных частицах каталитического состава, каталитического состава на носителе или предварительно полимеризованного каталитического состава внутри слоя. Запуск реакции полимеризации осуществляют, используя слой предварительно сформированных полимерных частиц, которые предпочтительно подобны желательному полимеру, и кондиционируя слой сушкой инертным газом или азотом перед введением каталитического состава, мономеров и каких-либо других газов, которые желательно иметь в потоке рециклового газа, таких как разбавляющий газ, водородный агент переноса цепи или инертный газ, способный конденсироваться при работе в режиме газофазной конденсации. Полученный полимер выгружают непрерывно или полунепрерывно из псевдоожиженного слоя, когда желательно.
Газофазные процессы, наиболее подходящие для осуществления на практике данного изобретения, являются непрерывными процессами, которые предусматривают непрерывную подачу реагентов в зону реакции реактора и удаление продуктов из зоны реакции реактора, обеспечивая тем самым среду устойчивого состояния в зоне реакции реактора. Продукты легко извлекают, подвергая воздействию пониженного давления и, необязательно, повышенных температур (удаление летучих компонентов) согласно известным технологиям. Обычно псевдоожиженный слой газофазного процесса работает при температурах более чем 50°С, предпочтительно от 60°С до 110°С, более предпочтительно от 70°С до 110°С.
Примеры газофазных процессов, которые являются приспособляемыми для применения в процессе по данному изобретению, раскрыты в патентах США 4588790, 4543399, 5352749, 5436304, 5405922, 5462999, 5461123, 5453471, 5032562, 5028670, 5473028, 5106804, 5556238, 5541270, 5608019 и 5616661.
Как было упомянуто ранее, функционализованные производные мульти-блок-сополимеров также включены в данное изобретение. Примеры включают металлированные полимеры, где металл является остатком используемого катализатора или челночного агента цепи, а также дополнительные их производные, например продукт реакции металлированного полимера с источником кислорода и затем с водой с образованием полимера с концевыми гидроксильными группами. В другом варианте воплощения достаточное количество воздуха или другого гасящего агента добавляют, чтобы расщепить некоторые или все связи челночный агент-полимер, превращая тем самым по меньшей мере часть полимера в полимер с концевыми гидроксильными группами. Дополнительные примеры включают полимеры с концевыми олефиновыми группами, образованные β-гидридным элиминированием, и ненасыщенностью по типу этилена в полученном полимере.
В одном варианте воплощения изобретения мульти-блок-сополимер может быть функционализован малеинезацией (взаимодействием с малеиновым ангидридом или его эквивалентом), металлированием (таким как с алкиллитиевым реагентом, необязательно в присутствии основания Льюиса, особенно амина, такого как тетраметилэтилендиамин) или введением диена или защищенного олефина в процесс сополимеризации. После полимеризации с участием защищенного олефина защитная группа, например тригидрокарбилсилан, может быть удалена под воздействием более легко функционализируемого остатка. Методы функционализирования полимеров хорошо известны и раскрыты, например, в патенте США 5543458 и других источниках.
Так как существенная фракция полимерного продукта, покидающего реактор, оканчивается челночным агентом цепи, дальнейшее функционализирование является относительно легким. Металлированные разновидности полимера могут быть использованы в хорошо известных химических реакциях, таких как реакции подходящие для других соединений алкилалюминия, алкилгаллия, алкилцинка или соединений алкила с элементом группы 1, для формирования полимерных продуктов с амино-, гидрокси-, эпоксигруппами, группами кетона, сложного эфира, нитрила и других функционализованных по концам полимерных продуктов. Примеры подходящих методов реакций, которые могут быть адаптированы для применения здесь, описаны в Negishi, "Organometallics in Organic Synthesis", Vol. 1 and 2, (1980), и в других типовых публикациях по металлорганическому и органическому синтезу.
Полимерные продукты
Используя данный процесс, легко могут быть получены новые полимеры, особенно олефиновые сополимеры, включая мульти-блок-сополимеры одного или нескольких олефиновых мономеров. Очень желательно, полимеры являются сополимерами, содержащими в полимеризованной форме этилен и по меньшей мере один C3-20 α-олефиновый сомономер и, необязательно, один или несколько дополнительных coполимеризуемых сомономеров. Предпочтительными α-олефинами являются C3-8 α-олефины. Подходящие сомономеры выбраны из диолефинов, циклических олефинов и циклических диолефинов, галогенированных винил-соединений и винилиден- ароматических соединений. Более конкретно полимеры по изобретению включают следующие специфические воплощения.
В первом варианте воплощения изобретением является сополимер, имеющий по меньшей мере одну температуру плавления, Tm, в градусах Цельсия и плотность, d*, в граммах на кубический сантиметр, где числовые величины переменных соответствуют уравнению:
Tm>-2002,9+4538,5(d*)-2422,2(d*)2 ,
и где сополимер имеет Mw/Mn от 1,7 дo 3,5.
Во втором варианте воплощения изобретением является сополимер, имеющий по меньшей мере одну температуру плавления, Tm, в градусах Цельсия, и плотность, d*, в граммах на кубический сантиметр, где числовые величины переменных соответствуют уравнению:
Tm>-6288,1+13141(d*)-6720,3(d*)2.
В третьем варианте воплощения изобретением является сополимер, имеющий по меньшей мере одну температуру плавления, Tm, в градусах Цельсия, и плотность, d*, в граммах на кубический сантиметр, где числовые величины переменных соответствуют уравнению:
Tm≥858,91-1825,3(d*)+1112,8(d*)2.
В четвертом варианте воплощения изобретение относится к сополимеру, содержащему в полимеризованной форме этилен и С3-8 α-олефин, указанный сополимер имеет величину дельта (наивысший пик DSC минус наивысший пик CRYSTAF) более чем величина, y*, определяемая уравнением:
y*>-0,1299(ΔH)+62,81, предпочтительно уравнением:
y*>-0,1299(ΔH)+64,38 и более предпочтительно уравнением:
y*>-0,1299(ΔH)+65,95,
при теплоте плавления вплоть до 130 Дж/г, где пик CRYSTAF определяют, используя по меньшей мере 5 процентов совокупного полимера (то есть пик должен представлять по меньшей мере 5 процентов совокупного полимера), и если меньше чем 5 процентов полимера имеет идентифицируемый пик CRYSTAF, тогда температура CRYSTAF 30°C, и ΔH представляет числовую величину теплоты плавления в Дж/г. Еще более предпочтительно наивысший пик CRYSTAF содержит по меньшей мере 10 процентов совокупного полимера. Фиг.3-27 и 36-49 показывают кривые DSC и CRYSTAF для многих примеров по изобретению, а также пики многих сравнительных полимеров. Пики, используемые для расчета величины дельта, y*, идентифицируют на каждом чертеже наряду с интегрированной областью под кривой (показывающей процентную долю совокупного полимера). Фиг.2 и 50 показывают нанесенные на график данные для примеров по изобретению, а также для сравнительных примеров. Интегрированные площади пиков и пиковые температуры рассчитывают с помощью компьютеризованной графической программы, поставляемой производителем инструмента. Диагональная линия, показанная для статистических сравнительных этилен-октеновых полимеров, соответствует уравнению:
y*=-0,1299(ΔH)+62,81.
В пятом варианте воплощения изобретением является сополимер, имеющий предел прочности при растяжении выше 10 МПа, предпочтительно предел прочности при растяжении >11 МПа, более предпочтительно предел прочности при растяжении >13МПа и относительное удлинение при разрыве по меньшей мере 600 процентов, более предпочтительно по меньшей мере 700 процентов, очень предпочтительно по меньшей мере 800 процентов и наиболее предпочтительно по меньшей мере 900 процентов при скорости разделения ползуна 11 см/мин.
В шестом варианте воплощения изобретением является сополимер, имеющий величину дельта (пиковая температура наивысшего пика DSC (измеренная от базовой линии) минус пиковая температура наивысшего пика CRYSTAF (т.е. наивысшая числовая величина dW/dT)) более чем 48°C и теплоту плавления более чем или равную 130 Дж/г, где пик CRYSTAF определяют, используя по меньшей мере 5 процентов совокупного полимера (то есть пик должен представлять по меньшей мере 5 процентов совокупного полимера), и если менее чем 5 процентов полимера имеет идентифицируемый пик CRYSTAF, тогда температура CRYSTAF 30°C. Еще более предпочтительно наивысший пик CRYSTAF содержит по меньшей мере 10 процентов совокупного полимера. Фиг.3-27 и 36-49 показывают кривые DSC и CRYSTAF для многих примеров по изобретению, а также для многих сравнительных полимеров. Пики, используемые для расчета величины дельта, y*, идентифицируют на каждом чертеже наряду с интегрированной площадью под кривой (указывающей процентную долю совокупного полимера). На фиг.2 и 50 вертикальная линия иллюстрирует ΔH=130 Дж/г и горизонтальная линия иллюстрирует y*=48°C.
В седьмом варианте воплощения изобретением является сополимер, имеющий отношение динамических модулей упругости, G'(25°C)/G'(100°C), от 1 до 50, предпочтительно от 1 до 20, более предпочтительно от 1 до 10, и остаточную деформацию при сжатии при 70°C менее чем 80 процентов, предпочтительно менее чем 70 процентов, особенно менее чем 60 процентов, вплоть до остаточной деформации при сжатии 0 процентов.
В восьмом варианте воплощения изобретением является сополимер, имеющий теплоту плавления менее чем 85 Дж/г и силу слипания гранул, равную или менее чем 100 фунтов/фут2 (4800 Пa), предпочтительно равную или менее чем 50 фунтов/фут2 (2400 Па), особенно равную или менее чем 5 фунтов/футz (240 Пa) и настолько низкую как 0 фунтов/фут2 (0 Пa).
В девятом варианте воплощения изобретение относится к несшитому эластомерному сополимеру, содержащему в полимеризованной форме по меньшей мере 50 молярных процентов этилена, имеющему остаточную деформацию при сжатии при 70°C менее чем 80 процентов, предпочтительно менее чем 70 процентов, наиболее предпочтительно менее чем 60 процентов.
В десятом варианте воплощения изобретением является олефиновый сополимер, предпочтительно содержащий этилен и один или несколько coполимеризуемых сомономеров в полимеризованной форме, характеризующийся множественными блоками или сегментами из двух или более полимеризованных мономерных звеньев, различающихся по химическим или физическим свойствам (блокированный сополимер), наиболее предпочтительно мульти-блок-coполимер, указанный блок-сополимер имеет молекулярную фракцию, которая элюируется между 40°C и 130°C при фракционировании с использованием TREF, характеризующуюся тем, что указанная фракция имеет молярное содержание сомономера выше, предпочтительно по меньшей мере на 5 процентов выше, более предпочтительно по меньшей мере на 10 процентов выше, чем фракция сравнимого статистического этиленового сополимера, элюируемая между теми же температурами, где указанный сравнимый статистический этиленовый сополимер содержит тот же сомономер(ы) и имеет индекс расплава, плотность и молярное содержание сомономера (на основе всего полимера) в пределах 10 процентов от такой величины для блокированного сополимера. Предпочтительно Mw/Mn сравнимого сополимера также находится в пределах 10 процентов того же отношения для блокированного сополимера, и/или сравнимый сополимер имеет общее содержание сомономера в пределах 10 массовых процентов от такой величины для блокированного сополимера.
Содержание сомономера может быть измерено с применением любой подходящей методики, предпочтительны методы спектроскопии ядерного магнитного резонанса (ЯМР). Более того, для полимеров или смесей полимеров, имеющих относительно широкие TREF кривые, полимер, желательно, вначале фракционируют с применением TREF на фракции, каждая из которых имеет предел температуры элюирования 10°C или менее. То есть каждая элюируемая фракция имеет температурное окно сбора 10°C или менее. При использовании этой методики указанные блокированные сополимеры имеют по меньшей мере одну такую фракцию, имеющую более высокое молярное содержание сомономера, чем соответствующая фракция сравнимого сополимера.
Предпочтительно, для сополимеров этилена и 1-октена, блокированный сополимер имеет содержание сомономера фракции TREF, элюируемой между 40 и 130°C, более чем или равное количеству (-0,2013)T+20,07, более предпочтительно более чем или равное количеству (-0,2013)T+21,07, где T представляет числовую величину, измеренную в °С, пиковой температуры элюирования фракции TREF, которую сравнивают.
Фиг.54 графически изображает предшествующий вариант воплощения изобретения для блокированных сополимеров этилена и 1-октена, где график содержания сомономера против температуры элюирования TREF для различных сравнимых сополимеров этилен/1-октен (статистические coполимеры) подгоняют к линии, представляющей (-0,2013)T+20,07 (сплошная линия). Линия для уравнения (-0,2013)T+21,07 изображена пунктирной линией. Также изображены содержания сомономера для фракций нескольких блокированных сополимеров этилен/1-октен по изобретению (мульти-блок-coполимеры). Все фракции блокированного сополимера имеют значительно более высокое содержание 1-октена, чем любая линия при эквивалентных температурах элюирования. Это сказывается на характеристиках мульти-блок-coполимеров по изобретению и, как представляется, является следствием присутствия дифференцированных блоков в полимерных цепях, имеющих и кристаллический, и аморфный характер.
Фиг.55 графически отображает кривую TREF и содержание сомономеров полимерных фракций, например, 5 и сравнительной F. Пик, элюируемый от 40 дo 130°C, предпочтительно от 60°C дo 95°C для обоих полимеров, фракционируют на три части, элюируя каждую часть в температурном диапазоне менее чем 10°C. Действительные данные для примера 5 представлены треугольниками. Специалисту будет понятно, что соответствующая калибровочная кривая может быть построена для сополимеров, содержащих различные сомономеры, и линия, используемая в качестве сравнения, подогнана к величинам TREF, полученным от сравнительных сополимеров тех же мономеров, предпочтительно статистических coполимеров, полученных с использованием металлоцена или другого гомогенного каталитического состава. Блокированные сополимеры по данному изобретению характеризуются молярным содержанием сомономера более чем величина, определяемая из калибровочной кривой при той же температуре элюирования TREF, предпочтительно по меньшей мере на 5 процентов более, более предпочтительно по меньшей мере на 10 процентов более.
Для coполимеров этилена и α-олефина, полимеры по изобретению предпочтительно имеют PDI по меньшей мере 1,7, более предпочтительно по меньшей мере 2,0 и наиболее предпочтительно по меньшей мере 2,6, вплоть до максимальной величины 5,0, более предпочтительно вплоть до максимума 3,5 и особенно вплоть до максимума 2,7, теплоту плавления 80 Дж/г или менее, содержание этилена по меньшей мере 50 массовых процентов, температуру стеклования, Tg, менее чем -25°C, более предпочтительно менее чем -30°C, и/или одну и только одну Tm.
Полимеры могут быть дополнительно охарактеризованы глубиной пенетрации при термомеханическом анализе 1 мм при температуре по меньшей мере 90°C, а также модулем упругости при изгибе от 3 килофунт на квадратный дюйм (20 МПа) до 13 килофунт на квадратный дюйм (90 МПа). Альтернативно полимеры по изобретению могут иметь глубину пенетрации при термомеханическом анализе 1 мм при температуре по меньшей мере 104°C, а также модуль упругости при изгибе по меньшей мере 3 килофунт на квадратный дюйм (20 МПа). Сополимеры по изобретению могут дополнительно характеризоваться как имеющие остаточную деформацию при сжатии при 70°C менее чем 80 процентов, предпочтительно менее чем 70 процентов, наиболее предпочтительно менее чем 60 процентов. Полимеры по изобретению могут дополнительно характеризоваться как имеющие сопротивление истиранию (или объемную потерю) менее чем 90 мм3. Дополнительно полимеры по изобретению могут иметь такой динамический модуль упругости, G', один или в сочетании с другими свойствами, раскрытыми здесь, что log (G') более чем или равен 400 кPa, предпочтительно более чем или равен 1,0 МПа, при температуре 100°C. Более того, олефиновые полимеры по изобретению имеют относительно плоский график динамического модуля упругости как функции температуры в пределах 0 до 100°C (показано на фиг.35), что является характерным для блок-coполимеров, и до сих пор было неизвестно для всего олефинового coполимера, особенно coполимера этилена и одного или нескольких C3-8 алифатических α-олефинов. (Под термином "относительно плоский" в данном контексте подразумевается, что log G' (в Паскалях) уменьшается менее чем на один порядок величины между 50 и 100°C, предпочтительно между 0 и 100°C). Дополнительно, полимеры по изобретению могут иметь индекс расплава, I2, от 0,01 до 2000 г/10 мин, предпочтительно от 0,01 до 1000 г/10 мин, более предпочтительно от 0,01 до 500 г/10 мин и особенно от 0,01 до 100 г/10 мин. Полимеры по изобретению могут иметь молекулярные массы, Mw, от 1000 г/моль до 5000000 г/моль, предпочтительно от 1000 г/моль до 1000000, более предпочтительно от 10000 г/моль до 500000 г/моль и особенно от 10000 г/моль до 300000 г/моль. Плотность полимеров по изобретению может быть от 0,80 дo 0,99 г/см3 и предпочтительно для содержащих этилен полимеров от 0,85 г/см3 до 0,97 г/см3.
Полимеры по изобретению можно отличить от обычных статистических сополимеров, физических смесей полимеров и блок-сополимеров, полученных методами последовательного присоединения мономеров, с дифференциальными катализаторами, анионной или катионной живой полимеризации. В частности, по сравнению со статистическим сополимером из тех же мономеров и с тем же содержанием мономеров при эквивалентной кристалличности или модуле полимеры по изобретению имеют улучшенную (более высокую) теплостойкость, которую измеряют по температуре плавления, более высокую температуру пенетрации ТМА, более высокий предел прочности при растяжении при высокой температуре и/или более высокий динамический модуль упругости при скручивании при повышенной температуре, который определяют динамическим механическим анализом. По сравнению со статистическим сополимером, содержащим те же мономеры и с тем же содержанием мономеров, полимеры по изобретению имеют более низкую остаточную деформацию при сжатии, особенно при повышенных температурах, более низкую релаксацию напряжений, более высокое сопротивление ползучести, более высокую прочность на разрыв, более высокую устойчивость против слипания, более прочную сборку (setup) благодаря более высокой температуре кристаллизации (отверждения), более высокое восстановление (особенно при повышенных температурах), улучшенную износостойкость, более высокое усилие сжатия и улучшенное принятие масла и наполнителя.
Данные полимеры также обнаруживают уникальные взаимоотношения кристаллизации и распределения разветвлений. То есть данные полимеры имеют относительно большую разницу между самой высокой пиковой температурой, измеренной с помощью CRYSTAF и DSC как функцию тепла плавления, особенно по сравнению со статистическими сополимерами, содержащими те же мономеры и при том же уровне содержания мономеров, или с физическими смесями полимеров, такими как смесь полимера высокой плотности и сополимера более низкой плотности, при эквивалентной общей плотности. Предполагается, что это уникальное свойство полимеров по изобретению является результатом уникального распределения сомономера в блоках в главной цепи полимера. В частности, полимер желательно содержит чередующиеся блоки с отличающимся содержанием сомономера (включая блоки гомополимеров). Полимеры желательно имеют распределение по числу и/или размеру блока полимерных блоков, различающихся по плотности или содержанию сомономера, которое является распределением типа Schultz-Flory. В дополнение, полимеры по изобретению также имеют пиковую температуру плавления и профиль температуры кристаллизации, который уникально является независимым от морфологии плотность полимера/модуль. В предпочтительном варианте воплощения микрокристаллический порядок полимеров демонстрирует характерные сферолиты и пластинки, отличающие их от статистических или блок-сополимеров, даже при величинах PDI, которые менее чем 1,7 или даже менее чем 1,5 и ниже до менее чем 1,3. Уникальная кристаллическая морфология полимеров по изобретению, как предполагается, будет иметь результатом хорошие барьерные свойства, благодаря увеличенной извилистости кристаллической морфологии, что делает полимеры подходящими для применения для целей уплотнения и герметизации, таких как вкладыши в колпачки бутылок и пленки для упаковки продуктов, мяса и пищи. Фиг.28 представляет оптические микрофотографии низкого разрешения прессованных пленок, показывающие микрокристаллическую структуру трех мульти-блок-сополимеров по данному изобретению (все имеющие плотность около 0,88), но изготовленных с различными уровнями челночного агента цепи, показывающие изменяющуюся сферолитическую структуру, а также трех сравнительных полимеров, по существу линейного сополимера этилен/1-октен (сополимер Affinity™ с плотностью 0,875 г/см3, доступный от The Dow Chemical Company), линейного полиэтилена, имеющего плотность 0,94 г/см3, и полиэтиленовой смеси, полученной с двойными катализаторами в одном реакторе (внутриреакторная смесь). Фиг.29 представляет четыре полученные электронным сканирующим фотографированием микрофотографии высокого разрешения (масштаб 100 нм), три фотографии указанных образцов полимеров по изобретению, полученных с высоким, средним и низким уровнями содержания челночного агента цепи в реакторе, а также для сравнения микрофотографию по существу линейного сополимера этилен/1-октен (сополимер Affinity™ с плотностью 0,875 г/см3). Сравнение трех фотографий полимеров по изобретению в целом показывает уменьшение толщины и длины пластинок с увеличением уровней содержания челночного агента цепи.
Более того, полимеры по изобретению могут быть получены с использованием приемов воздействия на степень или уровень блочности. То есть, количество сомономера и длина каждого полимерного блока или сегмента могут быть изменены путем регулирования отношения и типа катализаторов и челночного агента, а также температуры полимеризации и других переменных полимеризации. Неожиданным полезным эффектом этого феномена является открытие того, что, когда степень блочности увеличивается, улучшаются оптические свойства, прочность на разрыв и свойства восстановления при высокой температуре полученного полимера. В частности, белесоватость уменьшается, тогда как прозрачность, прочность на разрыв и свойства восстановления при высокой температуре увеличиваются, когда среднее число блоков в полимере увеличивается. Путем подбора челночных агентов и сочетаний катализаторов, имеющих желательную способность переноса цепи (высокие скорости челночного перемещения с низкими уровнями обрывания цепей) другие формы обрывания полимера эффективно подавляются. Соответственно, если и наблюдается β-гидридная элиминация при полимеризации смесей сомономеров этилен/α-олефин по изобретению, то она незначительная, и полученные кристаллические блоки являются в высокой степени или по существу полностью, линейными, имеющими немного или не имеющими длинноцепочечного разветвления.
Другим неожиданным полезным эффектом изобретения является то, что избирательно могут быть получены полимеры, где концы цепей являются высококристаллическими. В эластомерных применениях уменьшение относительного количества полимера, который оканчивается аморфным блоком, уменьшает интермолекулярное разбавляющее действие на кристаллические области. Такой результат может быть достигнут подбором челночных агентов цепи и катализаторов, имеющих подходящую ответную реакцию на водород или другие агенты обрывания цепи. Конкретно, если катализатор, который создает высококристаллический полимер, является более чувствительным к обрыванию цепи (такому как путем применения водорода), чем катализатор, ответственный за образование менее кристаллического полимерного сегмента (такого как путем более высокого внедрения сомономера, формирования регио-ошибочного или атактического полимера), тогда высоко кристаллические полимерные сегменты будут преимущественно заполонять концевые части полимера. Не только полученные концевые группы кристаллические, но при обрывании цепи каталитический сайт, формирующий высококристаллический полимер, снова доступен для повторного инициирования формирования полимера. Первоначально сформированный полимер является, следовательно, другим высококристаллическим полимерным сегментом. Соответственно, оба конца полученного мульти-блок-сополимера преимущественно являются высоко кристаллическими.
Способность данных мульти-блок-сополимеров, полученных из этилена и сомономера, такого как 1-октен, сохранять свойства высокой температуры плавления, иллюстрируется ссылкой на фиг.34, которая представляет график кристаллической температуры плавления как функции плотности (содержания сомономера), при более низких плотностях кристаллические температуры плавления не снижаются значительно по сравнению с температурами мульти-блок-сополимеров более высокой плотности по изобретению (линия), тогда как традиционные статистические сополимеры обычно следуют хорошо известной кривой, отражающей потерю пиковой температуры кристаллического плавления, когда плотность уменьшается.
Другими очень желательными составами по данному изобретению являются эластомерные сополимеры этилена, С3-20 α-олефина, особенно пропилена, и необязательно одного или нескольких диеновых мономеров. Предпочтительные α-олефины для применения в этом варианте воплощения данного изобретения представлены формулой СН2=СНR*, где R* представляет линейную или разветвленную алкилгруппу из 1-12 атомов углерода. Примеры подходящих α-олефинов включают, но без ограничения перечисленными, пропилен, изобутилен, 1-бутен, 1-пентен, 1-гексен, 4-метил-1-пентен и 1-октен. Особенно предпочтительным α-олефином является пропилен. Полимеры на основе пропилена обычно упоминаются в технической литературе как полимеры ЕР или ЕРDM. Подходящие диены для применения для получения таких полимеров, особенно мульти-блок-полимеров типа ЕРDM, включают конъюгированные или неконъюгированные, с прямой или разветвленной цепью, циклические или полициклические диены, содержащие от 4 до 20 атомов углерода. Предпочтительные диены включают 1,4-пентадиен, 1,4-гексадиен, 5-этилиден-2-норборнен, дициклопентадиен, циклогексадиен и 5-бутилиден-2-норборнен. Особенно предпочтительным диеном является 5-этилиден-2-норборнен.
Так как содержащие диен полимеры содержат чередующиеся сегменты или блоки, содержащие большие или меньшие количества диена (включая его отсутствие) и α-олефина (включая его отсутствие), общее количество диена и α-олефина может быть уменьшено без потери последующих свойств полимера. То есть, так как диеновые и α-олефиновые мономеры преимущественно внедрены в один тип блока полимера скорее, чем равномерно или беспорядочно на протяжении всего полимера, они являются более эффективно используемыми, и впоследствии густота поперечных связей полимера может быть лучше контролируемой. Такие сшиваемые эластомеры и отвержденные продукты имеют полезные свойства, включая более высокий предел прочности при растяжении и улучшенное упругое восстановление.
Желательно полимеры по изобретению, полученные с двумя катализаторами, внедряющими различные количества сомономера, имеют массовое соотношение сформированных таким образом блоков от 95:5 до 5:95. Эластомерные полимеры желательно имеют содержание этилена от 20 до 90 процентов, содержание диена от 0,1 до 10 процентов и содержание α-олефина от 10 до 80 процентов на основе общей массы полимера. Дополнительно предпочтительно эластомерные мульти-блок-полимеры по данному варианту воплощения изобретения имеют содержание этилена от 60 до 90 процентов, содержание диена от 0,1 до 10 процентов и содержание α-олефина от 10 до 40 процентов на основе общей массы полимера. Предпочтительными полимерами являются высокомолекулярные полимеры, имеющие средневесовую молекулярную массу (Mw) от 10000 до около 2500000, предпочтительно от 20000 дo 500000, более предпочтительно от 20000 дo 350000, и полидисперсность менее чем 3,5, более предпочтительно менее чем 3,0, и вязкость по Муни (ML (1+4) 125°C) от 1 до 250.
Более предпочтительно такие полимеры имеют содержание этилена от 65 дo 75 процентов, содержание диена от 0 до 6 процентов и содержание α-олефина от 20 дo 35 процентов.
Полимер может быть маслонаполненным в количестве от 5 до около 75 процентов, предпочтительно от 10 дo 60 процентов, более предпочтительно от 20 дo 50 процентов, технологического масла от общей массы состава. Подходящие масла включают любые масла, которые обычно используют при производстве наполненных составов каучуков EPDM. Примеры включают и нафтеновые, и парафиновые масла, парафиновые масла предпочтительны.
Очень желательно состав отверждаемого каучука готовить введением одного или нескольких отвердителей наряду с традиционными ускорителями и другими вспомогательными веществами. Подходящими отвердителями являются отвердители на основе серы. Примеры подходящих отвердителей на основе серы включают, но без ограничения указанным, серу, тетраметилтиурам дисульфид (TMTD), дипентаметилентиурама тетрасульфид (DPTT), 2-меркаптобензотиазол (MВT), 2-меркаптобензотиазолата дисульфид (MBTS), цинк-2-меркаптобензотиазолат (ZMBT), цинк-диэтилдитиокарбаматцинк (ZDEC), цинк-дибутилдитиокарбамат (ZDBC), дипентаметилентиурама тетрасульфид (DPTT), N-трет-бутилбензотиазол-2-сульфаниламид (TBBS) и их смеси. Предпочтительная отверждающая система включает сочетание серы, MBT и TMTD. Желательно указанные компоненты использовать в количествах от 0,1 до 5 процентов, на основе общей массы состава.
Предпочтительный эластомерный состав по данному варианту воплощения изобретения может также включать сажу. Предпочтительно сажа присутствует в количестве от 10 дo 80 процентов, более предпочтительно от 20 дo 60 процентов, на основе общей массы состава.
Дополнительные компоненты данных составов, обычно используемые по данному изобретению, включают различные другие ингредиенты в количествах, которые не умаляют свойств полученного состава. Эти ингредиенты включают, но не ограничиваются указанными, активаторы, такие как оксид кальция или магния; жирные кислоты, такие как стеариновая кислота, и их соли; наполнители и усилители, такие как карбонат кальция или магния, диоксид кремния и силикаты алюминия; пластификаторы, такие как диалкиловые сложные эфиры дикарбоновых кислот; противостарители; мягчители; воски и пигменты.
Области применения и конечные применения
Полимеры по изобретению могут быть успешно использованы в разнообразных традиционных процессах получения термопластов для производства полезных изделий, включая объекты, содержащие по меньшей мере один пленочный слой, такие как однослойная пленка, или по меньшей мере один слой в многослойной пленке, полученной литьем, раздувом, каландрованием или процессами экструзионного покрытия, формованные изделия, такие как сформованные раздувом, инжекционным формованием или полученные центробежным формованием изделия; экструзионные изделия; волокна и тканые и нетканые материалы. Термопластичные составы, содержащие данные полимеры, включают смеси с другими натуральными или синтетическими полимерами, добавки, усиливающие агенты, добавки, усиливающие стойкость против воспламенения, антиоксиданты, стабилизаторы, красители, наполнители, агенты образования поперечных связей, порообразователи и пластификаторы. Особенно полезны многокомпонентные волокна, такие как волокна сердцевина/оболочка, имеющие внешний поверхностный слой, содержащий по меньшей мере частично один или несколько полимеров по изобретению.
Волокна могут быть получены из данных полимеров или смесей, такие как штапельные волокна, пакля, многокомпонентые, типа оболочка/сердцевина, скрученные и мононити. Подходящие процессы формирования волокон включают технологии прядения и раздува расплава, которые раскрыты в патентах США 4430563, 4663220, 4668566 и 4322027, штапельные волокна по гель-технологии, которые раскрыты в патенте США 4413110, тканые и нетканые материалы, которые раскрыты в патенте США 3485706, или структуры, изготовленные из таких волокон, включая смеси с другими волокнами, такими как полиэфирные, найлоновые или хлопковые, термоформованные изделия, экструдированные формы, включая профильные экструзионные изделия и изделия, полученные совместной экструзией, каландрованные изделия и полученные вытягиванием, скрученные или извитые нити или волокна. Описанные здесь новые полимеры применимы также для операций покрытия проводов и кабелей, а также для экструзии полотен в операциях вакуумного формования и получения формованных изделий, включая применение литьевого формования, процесса формования раздувом или процессы центробежного формования. Составы, содержащие олефиновые полимеры, могут быть также сформованы в законченные изделия, такие как упомянутые ранее, с использованием традиционных технологий переработки полиолефинов, которые хорошо известны специалистам в области переработки олефинов.
Дисперсии (как водные, так и неводные) также могут быть приготовлены с использованием данных полимеров или содержащих их составов. Пеноматериалы, содержащие полимеры по изобретению, также могут быть получены, как раскрыто в заявке РСТ 2004/027593, поданной 25 августа 2004. Полимеры могут быть также сшиты какими-либо известными средствами, такими как пероксид, электронный пучок, силан, азид или по другой технологии образования поперечных связей. Полимеры могут быть также химически модифицированы, например, привитой сополимеризацией (например, с использованием малеинового ангидрида (МАН), силанов или другого агента прививки), галогенированием, аминированием, сульфированием или другой химической модификацией.
Добавки или адъюванты могут быть включены в какой-либо состав, содержащий данные полимеры. Подходящие добавки включают наполнители, такие как органические или неорганические частицы, включая глины, тальк, диоксид титана, цеолиты, порошкообразные металлы, органические и неорганические волокна, включая углеродные волокна, волокна на основе нитрида кремния, стальную проволоку или сетку и найлоновые или полиэфирные ленты, частицы наноразмеров, глины и так далее; вещества для повышения клейкости, нефтяные мягчители, включая парафиновые и нафталиновые масла, и другие натуральные и синтетические полимеры, включая другие полимеры по изобретению.
Подходящие полимеры для смешивания с полимерами по изобретению включают термопластичные и нетермопластичные полимеры, включая натуральные и синтетические полимеры. Примеры полимеров для смешивания включают полипропилен (полипропилен с модифицированной ударной вязкостью, изотактический полипропилен, атактический полипропилен и статистические сополимеры этилен/пропилен), различные типы полиэтилена, включая полиэтилен высокого давления, свободнорадикальный LDPE, Ziegler-Natta LLDPE, металлоценовый полиэтилен (ПЭ), включая полученный в составном реакторе полиэтилен (смеси "в реакторе" из полиэтилена Ziegler-Natta и металлоценового полиэтилена, такие как продукты, раскрытые в патентах США 6545088, 6538070, 6566446,5844045, 5869575 и 6448341, сополимеры этилен-винилацетат (EVA), этилен/виниловый спирт, полистирол, полистирол с модифицированной ударной вязкостью, ABS, блок-сополимеры стирол/бутадиен и галогенированные их производные (SBS и SEBS) и термопластичные полиуретаны. Гомогенные полимеры, такие как олефиновые пластомеры и эластомеры, сополимеры на основе этилена и пропилена (например, полимеры, доступные под торговым обозначением VERSIFY™ от The Dow Chemical Company, и VISTAMAXX™, доступные от ExxonMobil, также могут быть применимы в качестве компонентов в смесях, содержащих полимеры по изобретению.
Подходящие конечные применения для указанных продуктов включают эластичные пленки и волокна, мягкие при контакте изделия, такие как рукоятки зубных щеток и рукоятки приборов, прокладки и профили, адгезивы (включая плавкие при нагревании адгезивы и эффективные под давлением адгезивы), обувь (включая обувные подошвы и стельки), детали и профили интерьера автомобилей, вспененные изделия (как с открытыми, так и с закрытыми ячейками), модификаторы ударной вязкости для других термопластичных полимеров, таких как полиэтилен высокой плотности, изотактический полипропилен или другие олефиновые полимеры, ткани с покрытием, шланги, трубопроводы, герметики, прокладки для колпачков, настилы и модификаторы индекса вязкости, известные также как модификаторы температуры текучести, для смазок.
В очень желательном варианте изобретения термопластичные составы, содержащие термопластичный матричный полимер, особенно изотактический полипропилен, и эластомерный мульти-блок-сополимер этилена и сополимеризуемого сомономера по изобретению, имеют уникальную способность формировать частицы типа сердцевина-оболочка, имеющие твердые кристаллические или полукристаллические блоки в форме ядра, окруженные мягкими или эластомерными блоками, формирующими "оболочку" вокруг окклюдированных доменов твердого полимера. Такие частицы формируются и диспергируются внутри матричного полимера под действием усилий, возникающих во время компаундирования или смешивания расплава. Такая очень желательная морфология, вероятно, является результатом уникальных физических свойств мульти-блок-сополимеров, которые дают возможность совместимым полимерным регионам, таким как матричные и эластомерные регионы с более высоким содержанием сомономера, мульти-блок-сополимера самосборки в расплаве, благодаря термодинамическим усилиям. Усилия сдвига во время компаундирования, вероятно, создают отдельные регионы матричного полимера, обогащенные эластомером. При отверждении такие регионы становятся окклюдированными эластомерными частицами, вставленными в полимерную матрицу.
Особенно желательными смесями являются термопластичные полиолефиновые смеси (ТРО), термопластичные эластомерные смеси (ТРЕ), термопластичные вулканизаты (TPV) и стироловые полимерные смеси. Смеси ТРЕ и TPV могут быть приготовлены объединением мульти-блок-сополимеров по изобретению, включая их функционализованные или ненасыщенные производные, с необязательным каучуком, включая обычные блок-сополимеры, особенно блок-сополимер SBS, и необязательно с образующим поперечные связи или вулканизирующим агентом. Смеси ТРО обычно получают смешиванием мульти-блок-сополимеров по изобретению с полиолефином и необязательно с образующим поперечные связи или вулканизирующим агентом. Указанные смеси могут быть использованы при получении формованного изделия и необязательно сшивании полученного формованного изделия. Подобная процедура с использованием различных компонентов ранее раскрыта в патенте США 6797779.
Подходящие традиционные блок-сополимеры для такого применения желательно имеют вязкость по Муни (ML 1+4 при 100°С) в пределах от 10 до 135, более предпочтительно от 25 до 100 и наиболее предпочтительно от 30 до 80. Подходящие полиолефины, главным образом, включают линейный полиэтилен или полиэтилен низкой плотности, полипропилен (включая атактический, изотактический, синдиотактический и его версии с модифицированной ударной вязкостью) и поли(4-метил-1-пентен). Подходящие стироловые полимеры включают полистирол, модифицированный каучуком полистирол (HIPS), сополимеры стирол/акрилонитрил (SAN), модифицированный каучуком SAN (ABS или AES) и сополимеры стирола и малеинового ангидрида.
Смеси могут быть приготовлены смешиванием или пластицированием соответствующих компонентов при температуре около или выше температуры плавления одного или обоих соединений. Для большинства мульти-блок-сополимеров эта температура может быть выше 130°С, наиболее обычно выше 145°С и наиболее предпочтительно выше 150°С. Может быть использовано типичное оборудование для перемешивания или пластицирования полимеров, которое позволяет достигать желательных температур и пластифицировать расплав смеси. Оно включает мельницы, месильные машины, экструдеры (как одночервячные, так и двухчервячные), смесители Бенбери, каландры и тому подобное. Последовательность смешивания и способ могут зависеть от конечного состава. Может быть использовано также сочетание смесителей Бенбери периодического действия и смесителей непрерывного действия, такое как смеситель Бенбери с последующим смесителем мельничного типа с последующим экструдером. Обычно состав ТРЕ или TPV будет иметь более высокую загрузку сшиваемого полимера (обычно это традиционный блок-сополимер, содержащий ненасыщенность) по сравнению с составами ТРО. Как правило, для составов ТРЕ или TPV массовое отношение блок-сополимера к мульти-блок-сополимеру может быть от около 90:10 до 10:90, более предпочтительно от 80:20 до 20:80 и наиболее предпочтительно от 75:25 до 25:75. Для применений ТРО массовое отношение мульти-блок-сополимера к полиолефину может быть от около 49:51 до около 5:95, более предпочтительно от 35:65 до около 10:90. Для применений модифицированного стиролового полимера массовое отношение мульти-блок-сополимера к полиолефину может быть от около 49:51 до около 5:95, более предпочтительно от 35:65 до около 10:90. Отношения могут быть изменены за счет изменения отношений вязкости различных компонентов. Есть заслуживающая внимания литература, поясняющая технологию изменения непрерывности фазы путем изменения отношений вязкости компонентов смеси, и специалист в этой области может проконсультироваться, если необходимо.
Составы смесей могут содержать технологические масла и технологические вспомогательные добавки. Технологические масла для каучука имеют конкретное обозначение по ASTM, и парафиновые, нафтеновые или ароматические технологические масла, все, являются подходящими для применения. Обычно используют от 0 до 150 частей, более предпочтительно от 0 до 100 частей и наиболее предпочтительно от 0 до 50 частей масла на 100 частей всего полимера. Более высокие количества масла могут приводить к усовершенствованию переработки полученного продукта при потерях в некоторых физических свойствах. Дополнительные технологические масла включают обычные воски, соли жирных кислот, такие как стеарат кальция или стеарат цинка, (поли)спирты, включая гликоли, простые эфиры (поли)спиртов, включая простые эфиры гликоля, сложные (поли)эфиры, включая сложные эфиры (поли)гликоля, и их солевые производные с металлами, особенно металлами группы 1 или 2 или с цинком.
Известно, что негидрированные каучуки, такие как содержащие полимеризованные формы бутадиена или изопрена, включая блок-сополимеры (здесь и далее - диеновые каучуки), имеют более низкую стойкость к УФ, озону и окислению, по сравнению с обыкновенно или высоко насыщенными каучуками. В применениях, таких как шины, изготовленные из составов, имеющих более высокие концентрации каучуков на основе диенов, как это известно, в состав вводят сажу для улучшения стойкости каучука, наряду с антиозоновыми добавками и антиоксидантами. Мульти-блок-сополимеры по данному изобретению, имеющие чрезвычайно низкие степени ненасыщенности, находят особое применение в качестве защитного поверхностного слоя (нанесенного как покрытие, совместно экструдированного или ламинированного) или стойкой против атмосферных воздействий пленки, приклеиваемой к изделиям, сформированным из традиционных полимерных составов, модифицированных диеновым эластомером.
Для традиционных применений TPO, TPV и TPE сажа является предпочтительной добавкой для УФ-абсорбции и стабилизирующих свойств. Примеры сажи включают ASTM N110, N121, N220, N231, N234, N242, N293, N299, S315, N326, N330, M332, N339, N343, N347, N351, N358, N375, N539, N550, N582, N630, N642, N650, N683, N754, N762, N765, N774, N787, N907, N908, N990 и N991. Указанные разновидности сажи имеют величины абсорбции иода в пределах от 9 дo 145 г/кг и средние объемы пор от 10 дo 150 см3/100 г. Как правило, используют сажи с размерами частиц, уменьшенными до такой степени, пока это позволяет критерий стоимости. Для многих таких применений данные мульти-блок-сополимеры и их смеси требуют немного или вообще не требуют сажи, давая тем самым возможность свободно планировать, вводить в состав альтернативные пигменты или нет. Разноцветные шины или шины под цвет автомобиля являются одной из возможностей.
Составы, включающие термопластичные смеси по данному изобретению, могут также содержать антиозонанты или антиоксиданты, которые известны химику по каучуку. Антиозонантами могут быть физические защитные средства, такие как парафиновые материалы, которые достигают поверхности и защищают деталь от кислорода или озона, или ими могут быть химические защитные средства, которые взаимодействуют с кислородом или озоном. Подходящие химические защитные средства включают модифицированные стиролом фенолы, бутилированный-октилированный фенол, бутилированный ди(диметилбензил) фенол, п-фенилендиамины, продукты реакции бутилирования п-крезола и дициклопентадиена (DCPD), полифенольные антиоксиданты, производные гидрохинона, хинолин, дифениленовые антиоксиданты, антиоксиданты на основе сложных тиоэфиров и их смеси. Некоторые примеры торговых наименований таких продуктов: антиоксидант Wingstay™ S, антиоксидант Polystay™ 100, антиоксидант Polystay™ 100 AZ, антиоксидант Polystay™ 200, антиоксидант Wingstay™ L, антиоксидант Wingstay™ LHLS, антиоксидант Wingstay™ K, антиоксидант Wingstay™ 29, антиоксидант Wingstay™ SN-1 и антиоксиданты Irganox™. В некоторых применениях используемые антиоксиданты и антиозонанты предпочтительно должны быть не оставляющими пятен и немигрирующими.
Для придания дополнительной устойчивости против УФ-излучения могут быть также использованы светостабилизаторы на основе затрудненных аминов (HALS) и УФ-поглотители. Подходящие примеры включают Tinuvin™ 123, Tinuvin™ 144, Tinuvin™ 622, Tinuvin™ 765, Tinuvin™ 770 и Tinuvin™ 780, доступные от Ciba Specialty Chemicals, и Chemisorb™ T944, доступный от Cytex Plastics, Houston TX, USA. Дополнительно вместе с соединением HALS в состав может быть введена кислота Льюиса для достижения превосходного качества поверхности, как раскрыто в патенте США 6051681.
Для некоторых составов может быть использован дополнительный процесс смешивания, чтобы предварительно диспергировать антиоксиданты, антиозонанты, сажу, УФ-поглотители и/или светостабилизаторы, чтобы получить маточную смесь и впоследствии готовить из нее полимерные смеси.
Подходящие сшивающие агенты (называемые также отвердителями или вулканизующими агентами) для использования здесь включают соединения на основе серы, на основе пероксида или на фенольной основе. Примеры указанных материалов имеются в технике, смотри патенты США: 3758643, 3806558, 5051478, 4104210, 4130535, 4202801, 4271049, 4340684, 4250273, 4927882, 4311628 и 5248729.
Когда используют отвердители на основе серы, могут быть также использованы ускорители и активаторы отверждения. Ускорители используют, чтобы регулировать время и/или температуру, необходимые для динамической вулканизации, и чтобы улучшать свойства получаемого сшитого изделия. В одном варианте воплощения используют единственный ускоритель или первичный ускоритель. Первичный ускоритель (ускорители) может быть использован в общих количествах в пределах от около 0,5 до около 4, предпочтительно от около 0,8 до около 1,5 частей на 100 частей каучука (phr) на основе общей массы состава. В другом варианте воплощения могут быть использованы сочетания первичного и вторичного ускорителей, причем вторичный ускоритель используют в меньших количествах, таких как от около 0,05 до около 3 частей на 100 частей каучука, для активации и улучшения свойств отвержденного изделия. Сочетания ускорителей обычно позволяют получать изделия, имеющие свойства несколько лучше чем у изделий, получаемых с использованием единственного ускорителя. В дополнение могут быть использованы ускорители замедленного действия, на которые не влияют нормальные температуры переработки, но которые, однако, дают удовлетворительное отверждение при обычных температурах вулканизации. Замедлители вулканизации также могут быть использованы. Подходящими типами ускорителей, которые могут быть использованы в данном изобретении, являются амины, дисульфиды, гуанидины, тиомочевины, тиазолы, тиурамы, сульфенамиды, дитиокарбаматы и ксантаты. Предпочтительно первичным ускорителем является сульфенамид. Если используют вторичный ускоритель, вторичным ускорителем предпочтительно является соединение гуанидина, дитиокарбамата или тиурама. Некоторые технологические добавки и активаторы отверждения, такие как стеариновая кислота и ZnO, также могут быть использованы. Когда используют отвердители на основе пероксида, совместно действующие активаторы или совместно действующие агенты могут быть использованы в сочетании с ними. Подходящие совместно действующие агенты включают триметилолпропан триакрилат (TMPTA), триметилолпропан триметакрилат (TMPTMA), триаллил цианурат (TAG), триаллил изоцианурат (TAIC), наряду с прочими. Применение пероксидных сшивающих агентов и необязательных совместно действующих агентов, используемых для частичной или полной динамической вулканизации, известно в технике и раскрыто, например, в публикации "Peroxode Vulcanization of Elastomer", Vol. 74, No 3, июль-август 2001.
Когда состав, содержащий мульти-блок-сополимер, является по меньшей мере частично сшитым, степень сшивки может быть измерена путем растворения состава в растворителе в течение заданного времени и расчета процентной доли геля или неэкстрагируемого компонента. Процентная доля геля обычно увеличивается с увеличением степени сшивки. Для отвержденных изделий по изобретению процентное содержание геля желательно находится в пределах от 5 до 100 процентов.
Мульти-блок-сополимеры по изобретению, а также их смеси лучше перерабатываются по сравнению с составами предшествующего уровня техники, благодаря, вероятно, более низкой вязкости расплава. Таким образом, состав или смесь демонстрирует улучшенный внешний вид, особенно в готовом формованном или экструдированном изделии. В то же время данные составы и их смеси уникально обладают усовершенствованными свойствами прочности расплава, которые позволяют с выгодой применять данные мульти-блок-сополимеры и их смеси, особенно смеси ТРО, в пеноматериалах и для термоформования, где прочность расплава в настоящее время неадекватна.
Термопластичные составы по изобретению могут также содержать органические или неорганические волокна или другие добавки, такие как крахмал, тальк, карбонат кальция, стекловолокна, полимерные волокна (включая найлоновые, вискозные, полиэфирные и полиарамидные), металлические волокна, чешуйки или частицы, вспучиваемые слоистые силикаты, фосфаты или карбонаты, такие как глины, слюда, оксид алюминия, алюминосиликаты или алюминофосфаты, углеродные частицы, углеродные волокна, наночастицы, включая нанотрубки, волластонит, графит, цеолиты и керамику, такую как карбид кремния, нитрид кремния или оксиды титана. Агенты на основе силана или другие связующие агенты также могут быть использованы для улучшения связывания наполнителей.
Термопластичные составы по изобретению, включая указанные смеси, могут быть переработаны обычными приемами формования, такими как инжекционное формование, экструзионное формование, термоформование, формование полых изделий заливкой и медленным вращением формы, многокомпонентное формование, формование с закладными деталями, формование раздувом и другие способы. Пленки, включая многослойные пленки, могут быть получены процессами литья или растяжки в раме, включая процессы раздува пленки.
Методы испытания
В предшествующем характеризующем раскрытии и в примерах, которые следуют далее, используются следующие аналитические методы:
Метод GPC для образцов 1-4 и A-C
Автоматизированный робот для обращения с жидкостями, снабженный нагреваемой иглой, отрегулированной на 160°C, используют для добавления достаточного количества 1,2,4-трихлорбензола, стабилизированного 300 м.д. Ионола (Ionol), к каждому высушенному образцу полимера, чтобы получить конечную концентрацию 30 мг/мл. Небольшой стеклянный стержень для перемешивания помещают внутрь каждой пробирки и образцы нагревают до 160°C в течение 2 часов на нагреваемом орбитальном вибраторе, вращающемся при 250 об/мин. Концентрированный раствор полимера затем разбавляют до 1 мг/мл, используя автоматизированный робот для обращения с жидкостями, снабженный нагреваемой иглой, отрегулированной на 160°C.
Систему Symyx Rapid GPC используют, чтобы определить данные молекулярной массы для каждого образца. Насос Gilson 350, отрегулированный на скорость потока 2,0 мл/мин, используют для перекачивания продутого гелием 1,2-дихлорбензола, стабилизированного 300 м.д. Ионола (Ionol), в качестве мобильной фазы через три колонки Plgel 10 микрометров (мкм) Mixed B 300 мм × 7,5 мм, размещенные последовательно и нагретые до 160°C. Детектор Polimer Labs ELS 1000 используют c испарителем, отрегулированным на 250°C, распылителем, отрегулированным на 165°C, и скорость потока азота устанавливают до 1,8 SLM при давлении 60-80 фунт на кв. дюйм (400-600 кПа) N2. Образцы полимера нагревают до 160°C и каждый образец впрыскивают в 250 мкл петлю с помощью робота для манипуляции с жидкостями и нагретой иглы. Применяют последовательный анализ образцов полимера, используя две переключаемые петли и перекрывающие впрыскивания. Данные образца собирают и анализируют с помощью программного обеспечения Symyx Epoch™. Пики вручную интегрируют и информацию о молекулярной массе представляют неисправленной против полистироловой стандартной калибровочной кривой.
Пик плавления DSC измеряют как максимум в скорости теплового потока (Вт/г) по отношению к прямой базовой линии на протяжении между -30°C и концом плавления. Теплоту плавления измеряют как площадь под кривой плавления между -30°C и концом плавления, используя линейную базовую линию.
Стандартный метод CRYSTAF
Распределения ответвлений определяют кристаллизационным аналитическим фракционированием (CRYSTAF), используя установку CRYSTAF 200, коммерчески доступную от PolymerChar, Valencia, Spain. Образцы растворяют в 1,2,4-трихлорбензоле при 160°C (0,66 мг/мл) в течение 1 ч и стабилизируют при 95°C в течение 45 минут. Температуры отбора образцов находятся в пределах от 95 дo 30°C при скорости охлаждения 0,2°C/мин. Инфракрасный детектор используют для измерения концентраций раствора полимера. Совокупную концентрацию растворимых веществ измеряют, когда полимер кристаллизуется, пока температура снижается. Аналитическая производная совокупного профиля отражает распределение короткоцепочечного разветвления полимера.
Пиковую температуру CRYSTAF и площадь идентифицируют модулем анализа пика, включенным в программное обеспечение CRYSTAF (Version 200l.b, PolymerChar, Valencia, Spain). Обычный порядок нахождения пика CRYSTAF устанавливает пиковую температуру как максимум в dW/dT и площадь между наибольшими положительными изгибами на любой стороне установленного пика на кривой производной. Для расчета кривой CRYSTAF предпочтительными технологичесими параметрами являются параметры с температурным пределом 70°C и с коэффициентами сглаживания выше температурного предела на 0,1 и ниже температурного предела на 0,3.
Стандартный метод DSC (исключая образцы 1-4 и A-C)
Результаты дифференциальной сканирующей калориметрии определяют, используя TAI модель Q1000 DSC, снабженную вспомогательным охлаждающим устройством RCS и автоматическим пробоотборником. Используют поток продувочного газа азота 50 мл/мин. Образец прессуют в тонкую пленку и расплавляют в прессе при около 175°С и затем охлаждают на воздухе до комнатной температуры (25°С). 3-10 мг материала вырезают в виде диска диаметром 6 мм, точно взвешивают, помещают в легкий алюминиевый поддон (ca 50 мг) и затем завертывают. Термическое поведение образца исследуют при следующем температурном профиле. Образец быстро нагревают до 180°С и выдерживают изотермически в течение 3 минут, чтобы удалить какую-либо предшествующую термическую историю. Образец затем охлаждают до -40°C при скорости охлаждения 10°C/мин и выдерживают при -40°C в течение 3 минут. Образец затем нагревают до 150 °C при скорости нагревания 10°C/мин. Кривые охлаждения и второго нагревания записывают.
Пик плавления DSC измеряют как максимум в скорости теплового потока (Вт/г) по отношению к линейной базовой линии на протяжении между -30°C и концом плавления. Теплоту плавления измеряют как площадь под кривой плавления между -30°C и концом плавления, используя линейную базовую линию.
Сопротивление истиранию
Сопротивление истиранию измеряют на формованных прессованием пластинках согласно ISO 4649. Дана средняя величина из 3 измерений. Пластинки для испытания имеют толщину 6,4 мм и сформованы прессованием с помощью горячего пресса (Carver Model #4095-4PR1001R). Гранулы помещают между листами политетрафторэтилена, нагревают при 190°С при 55 фунт на кв. дюйм (380 кПа) в течение 3 минут, затем при 1,3 МПа в течение 3 минут и затем при 2,6 МПа в течение 3 минут. Затем пластинки охлаждают в прессе проточной холодной водой при 1,3 МПа в течение 1 минуты и извлекают для испытания.
Метод GPC (исключая образцы 1-4 и A-C)
Гель-проникающая хроматографическая система состоит или из инструмента Polymer Laboratories Model PL-210 или Polymer Laboratories Model PL-220. Колонка и отделения карусели работают при 140°С. Используют три колонки Polymer Laboratories 10-micron Mixed-B. Растворителем является 1,2,4-трихлорбензол. Образцы готовят при концентрации 0,1 грамма полимера в 50 миллилитрах растворителя, содержащего 200 м.д. бутилированного гидрокситолуола (BHT). Образцы готовят путем легкого взбалтывания в течение 2 часов при 160°С. Используемый объем для впрыскивания 100 микролитров и скорость потока 1,0 мл/минута.
Калибрование набора колонок GPC осуществляют с 21 полистироловым стандартом узкого молекулярномассового распределения с молекулярными массами в пределах от 580 дo 8400000, стандарты подготовлены в виде 6 смесей "коктейлей" по меньшей мере с десятичным разрядом разделения между отдельными молекулярными массами. Стандарты приобретают у Polymer Laboratories (Shropshire, UK). Полистироловые стандарты готовят при 0,025 грамма в 50 миллилитрах растворителя для молекулярных масс, равных или более чем 1000000, и 0,05 грамма в 50 миллилитрах растворителя для молекулярных масс менее чем 1000000. Полистироловые стандарты растворяют при 80°C при спокойном взбалтывании в течение 30 минут. Узкие смеси стандартов запускают вначале и в порядке уменьшения компонента с наивышей молекулярной массой, чтобы свести к минимуму разложение. Пиковые молекулярные массы полистироловых стандартов преобразуют в молекулярные массы полиэтилена, используя следующее уравнение (которое описано в Williams and Ward, J. Polym. Sci. Polym. Let. 6, 621 (1968)):
M полиэтилен =0,431((M полистирол ).
Расчеты эквивалентной молекулярной массы полиэтилена осуществляют с помощью программного обеспечения Viscotek TriSEC Version 3,0.
Остаточная деформация при сжатии
Остаточную деформацию при сжатии измеряют согласно ASTM D 395. Образец готовят наложением круглых дисков диаметром 25,4 мм толщиной 3,2 мм, 2,0 мм и 0,25 мм, пока не будет достигнута общая толщина 12,7 мм. Диски вырубают из пластинок 12,7 см×12,7 см, сформованных прессованием горячим прессом при следующих условиях: давление ноль в течение 3 мин при 190°С, затем 86 МПа в течение 2 мин при 190°С, затем охлаждение внутри пресса холодной проточной водой при 86 МПа.
Плотность
Образцы для измерения плотности готовят согласно ASTM D 1928. Измерения проводят в течение одного часа прессования образца, используя ASTM D792, Метод B.
Модуль упругости при изгибе/секущий модуль/динамический модуль упругости
Образы формуют прессованием по ASTM D 1928. Модуль упругости при изгибе и 2 процентный секущий модуль измеряют согласно ASTM D-790. Динамический модуль упругости измеряют согласно ASTM D 5026-01 или по эквивалентной методике.
Оптические свойства
Пленки толщиной 0,4 мм формуют прессованием, используя горячий пресс (Carver Model #4095-4PR1001R). Гранулы помещают между листами политетрафторэтилена, нагревают при 190°С при 55 фунт на кв. дюйм (380 кПа) в течение 3 минут, затем при 1,3 МПа в течение 3 минут и затем при 2,6 МПа в течение 3 минут. Затем пленку охлаждают в прессе проточной холодной водой при 1,3 МПа в течение 1 минуты. Сформованные прессованием пленки используют для измерений оптических свойств, поведения при растяжении, восстановления и релаксации напряжений. Прозрачность измеряют с помощью нефелометра BYK Gardner Haze-gard, как оговорено в ASTM D 1746.
45° глянец измеряют с помощью гляриметра BYK Gardner Glossmeter Microgloss 45°, как описано в ASTM D-2457.
Внутреннюю белесоватость измеряют нефелометром BYK Gardner Haze-gard на основе процедуры А ASTM D 1003. Минеральные масла наносят на поверхность пленки, чтобы устранить поверхностные царапины.
Механические свойства (растяжимость, гистерезис и прочность)
Деформативность при растяжении по одной оси измеряют, используя образцы для испытания на растяжение ASTM D 1708. Образцы растягивают с помощью прибора Instron при 500% мин-1 при 21°C. Предел прочности при растяжении и относительное удлинение при разрыве сообщают как среднее от 5 образцов.
100% и 300% гистерезис определяют при циклическом приложении нагрузок до деформации 100% и 300%, используя образцы для испытания на растяжение ASTM D 1708 с прибором Instron™. Образец нагружают и освобождают от нагрузки при 267% мин-1 в течение 3 циклов при 21°С. Циклические эксперименты при 300% и 80°C проводят, используя камеру искусственного климата. В эксперименте при 80°С образцу дают возможность прийти в равновесное состояние в течение 45 минут при температуре испытания перед испытанием. При циклическом эксперименте с деформацией 300% при 21°С регистрируют усилие стягивания при деформации 150% при первом цикле без нагрузки. Процентное восстановление для всех экспериментов рассчитывают из первого цикла без нагрузки, используя деформацию, при которой нагрузка возвращалась к базовой линии. Процентное восстановление определяют как
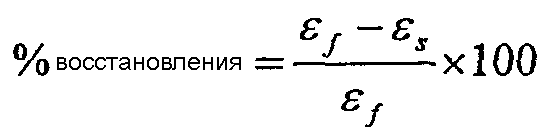
где εf представляет деформацию, взятую для циклической нагрузки, и εs представляет деформацию, когда нагрузка возвращается к базовой линии во время 1-го цикла снятия нагрузки.
Релаксацию напряжений измеряют при 50-процентной деформации и 37°C в течение 12 часов с помощью прибора Instron™, снабженного камерой искусственного климата. Геометрия шаблона 76 мм×25 мм×0,4 мм. После приведения в равновесное состояние при 37°C в течение 45 мин в камере искусственного климата, образец растягивают до деформации 50% при 333% мин-1. Напряжение регистрируют как функцию времени в течение 12 часов. Процентную релаксацию напряжений после 12 часов рассчитывают, используя формулу:
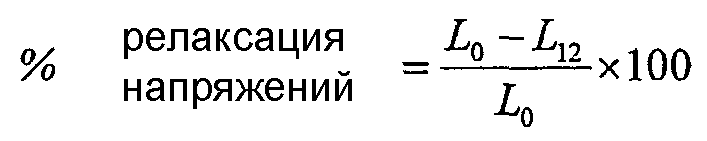
где L0 представляет нагрузку при деформации 50% при времени 0 и L12 представляет нагрузку при деформации 50% спустя 12 часов.
Эксперименты по разрыву образцов с надрезом при растяжении проводят на образцах, имеющих плотность 0,88 г/см3 или менее, используя прибор Instron™. Геометрия состоит из секции шаблона 76 мм×13 мм×0,4 мм с прорезанием надрезов 2 мм в образце до половины длины образца. Образец растягивают при 508 мм мин-1 при 21°С, пока он не разрывается. Энергию разрыва рассчитывают как площадь под кривой напряжение-удлинение вплоть до деформации при максимальной нагрузке. Сообщают среднее по меньшей мере от 3 образцов.
TMA
Термический механический анализ (температура пенетрации) проводят на дисках диаметром 30 мм и толщиной 3,3 мм, сформованных прессованием при 180°С и давлении формования 10 МПа в течение 5 минут с последующим быстрым охлаждением на воздухе. Используют инструмент TMA 7, брэнд доступный от Perkin-Elmer. При испытании зонд с кончиком радиусом 1,5 мм (P/N N519-0416) прикладывают к поверхности образцового диска с усилием 1Н. Температуру повышают при 5°C/мин от 25°C. Глубину проникновения зонда измеряют как функцию температуры. Эксперимент завершают, когда зонд проникает на 1 мм в образец.
DMA
Измерения при динамическом механическом анализе (DMA) проводят на дисках, сформованных прессованием на горячем прессе при 180°С при давлении 10 МПа в течение 6 минут и затем охлажденных водой в прессе при 90°C/мин. Испытание проводят с помощью управляемого ARES деформирующего вискозиметра (TA instruments), снабженного двойными консольными приспособлениями для испытания скручиванием.
Пластинку 1,5 мм прессуют и режут до бруска размерами 32×12 мм. Образец зажимают на обоих концах между приспособлениями, разделенными на 10 мм (зажимной интервал ΔL) и подвергают последовательному ступенчатому воздействию температур от -100°C дo 200°C (ступень 5°C). При каждой температуре измеряют модуль скручивания G' при круговой частоте 10 рад/с, амплитуду деформации поддерживают между 0,1 процента и 4 процентами для гарантии того, что вращающий момент достаточен и что измерение остается в линейном режиме.
Первоначальное статическое усилие 10 г поддерживают (режим автоматического напряжения), чтобы предотвратить провисание образца, когда происходит тепловое расширение. Как следствие, зажимной интервал ΔL увеличивается с температурой, особенно выше температуры плавления или размягчения образца полимера. Испытание прекращают при максимальной температуре или когда зазор между закреплениями достигает 65 мм.
Сила слипания гранул
Гранулы (150 г) загружают в полый цилиндр диаметром 2" (5 см), который изготовлен из двух половин, удерживаемых вместе шланговым зажимом. Нагрузку 2,75 фунта (1,25 кг) прилагают к гранулам в цилиндре при 45°С в течение 3 дней. Спустя 3 дня гранулы тесно объединяются в пробку цилиндрической формы. Пробку удаляют из формы и силу слипания гранул измеряют, создавая нагрузку давлением на цилиндр слипшихся гранул с помощью прибора Instron™, чтобы измерить усилие сжатия, необходимое для разрушения цилиндра на гранулы.
Индекс расплава
Индекс расплава, или I2, измеряют в соответствии с ASTM D 1238. Условие 190°C/2,16 кг. Индекс расплава или I10 также измеряют в соответствии с ASTM D 1238. Условие 190°C/10 кг.
ATREF
Анализ фракционирования элюированием при повышении температуры проводят согласно методу, описанному в патенте США 4798081. Состав, который должен быть проанализирован, растворяют в трихлорбензоле и позволяют ему кристаллизоваться в колонке, содержащей инертный носитель (дробь из нержавеющей стали), при медленном понижении температуры до 20°С при скорости охлаждения 0,1°C/мин. Колонку снабжают инфракрасным датчиком. Кривую хроматограммы ATREF затем создают при элюировании кристаллического полимерного образца из колонки путем медленного повышения температуры элюирующего растворителя (трихлорбензола) от 20 дo 120°C со скоростью 1,5°C/мин.
Фракционирование полимера путем TREF
Крупное фракционирование TREF проводят путем растворения 15-20 г полимера в 2 литрах 1,2,4-трихлорбензола (TCB) при перемешивании в течение 4 часов при 160°C. Раствор полимера вводят под избыточным давлением азота 15 фунтов на кв. дюйм (100 кПа) в стальную колонку 3 дюйма на 4 фута (7,6x12 см), набитую смесью 60:40 (об.:об.) сферических технического качества стеклянных шариков 30-40 меш (600-425 мкм) (доступных от Potters Industries, HC 30 Box 20, Brownwood, TX, 76801) и отрезков проволоки из нержавеющей стали диаметром 0,028" (0,7 мм) (доступной от Pellets, Inc. 63 Industrial Drive, North Tonawanda, NY, 14120). Колонку вставляют в терморегулируемую масляную рубашку, отрегулированную на 160°С. Колонку вначале охлаждают баллистически до 125°C, затем медленно охлаждают до 20°C при 0,04°C в минуту и выдерживают в течение одного часа. Свежий TCB вводят при около 65 мл/мин, тогда как температуру повышают при 0,167°C в минуту.
Порции приблизительно 2000 мл элюента из колонки препаративного TREF собирают в 16-позиционном нагреваемом коллекторе фракций. Полимер концентрируют в каждой фракции, используя ротационный испаритель, пока не останется около 50-100 мл раствора полимера. Концентрированным растворам дают отстояться в течение ночи перед добавлением избыточного метанола, фильтрованием и промыванием (прибл 300-500 мл метанола, включая конечное промывание). Стадию фильтрования осуществляют на 3-позиционной вакуумной станции фильтрования с использованием фильтровальной бумаги, покрытой 5,0 мкм политетрафторэтилена (доступной от Osmonics Inc., Cat# Z50WP04750). Отфильтрованные фракции сушат в течение ночи в вакуумной печи при 60°C и взвешивают на аналитических весах перед дополнительным испытанием.
I3 C ЯМР анализ
Образцы получают добавлением приблизительно 3 г смеси 50/50 тетрахлорэтан-d2/орто-дихлорбензол к образцу 0,4 г в 10 мм пробирке для ЯМР. Образцы растворяют и гомогенизируют нагреванием пробирки и ее содержимого до 150°С. Данные собирают с помощью спектрометра JEOL Eclipse™ 400 МГц или спектрометра Varian Unity Plus™ 400 МГц, соответствующие резонансной частоте 13C 100,5 МГц. Данные получают, используя 4000 переходных процессов на ряд данных с задержкой частоты повторения импульсов 6 секунд. Для достижения минимального отношения сигнал-шум для количественного анализа множественные ряды данных объединяют вместе. Ширина спектра 25000 Гц с минимальным размером ряда результатов обработки 32К. Образцы анализируют при 130°С в 10 мм датчике широкого диапазона. Внедрение сомономера определяют по методу триад Randall (Randal1, J.C.; JMS-Rev. Macromol. Chem. Phys., C29, 201-317 (1989).
Атомно-силовая микроскопия (AFM)
Срезы собирают от материала образца с помощью микротома Leica UCT™ с криокамерой FC, работающей при -80°C. Алмазный нож используют для рассечения всего материала образца на срезы толщиной 120 нм. Срезы помещают на поверхности только что расщепленной слюды и монтируют на стандартных металлических опорных дисках для образцов AFM с двойной углеродной лентой. Срезы исследуют с помощью DI NanoScope TV™ Multi-Mode AFM в режиме подключения к выводам с фазовым детектированием. Наночувствительные контакты используют во всех экспериментах.
Конкретные воплощения
Следующие конкретные воплощения изобретения и их сочетания являются особенно желательными и здесь разграничены, чтобы предоставить подробное раскрытие прилагаемой формулы изобретения.
1. Состав, содержащий смесь или продукт реакции, полученный в результате объединения:
(А) первого катализатора полимеризации олефинов,
(В) второго катализатора полимеризации олефинов, способствующего получению полимеров, отличающихся по химическим или физическим свойствам от полимера, полученного с катализатором (A) при эквивалентных условиях полимеризации, и
(С) челночного агента цепи.
1а. Состав, содержащий смесь или продукт реакции, полученный в результате объединения:
(А) первого катализатора полимеризации олефинов, имеющего высокий показатель внедрения сомономера,
(В) второго катализатора полимеризации олефинов, имеющего показатель внедрения сомономера менее чем 95 процентов, предпочтительно менее чем 90 процентов, более предпочтительно менее чем 25 процентов и наиболее предпочтительно менее чем 10 процентов от показателя внедрения сомономера катализатора (A), и
(А) челночного агента цепи.
2. Способ выбора смеси катализаторов (A) и (B) и челночного агента цепи (С) по п.1) или 1а), который способен давать мульти-блок-сополимер при контактировании олефинового мономера или смеси мономеров с указанной смесью в условиях полимеризации олефинов.
3. Способ получения мульти-блок-сополимера, содержащий контактирование одного или нескольких полимеризуемых присоединением мономеров в условиях полимеризации присоединением с составом, содержащим:
смесь или продукт реакции, полученные в результате объединения:
(A) первого катализатора полимеризации олефинов,
(B) второго катализатора полимеризации олефинов, способствующего получению полимеров, отличающихся по химическим или физическим свойствам от полимера, полученного с катализатором (A) в эквивалентных условиях полимеризации, и
(C) челночного агента цепи.
3a. Способ получения мульти-блок-сополимера, содержащий контактирование одного или нескольких полимеризуемых присоединением мономеров в условиях полимеризации присоединением с составом, содержащим:
смесь или продукт реакции, полученные в результате объединения:
(A) первого катализатора полимеризации олефинов, имеющего высокий показатель внедрения сомономера,
(B) второго катализатора полимеризации олефинов, имеющего показатель внедрения сомономера менее чем 90 процентов, предпочтительно менее чем 50 процентов, наиболее предпочтительно менее чем 5 процентов от показателя внедрения сомономера катализатора (A), и
(C) челночного агента цепи.
4. Мульти-блок-сополимер, содержащий в полимеризованной форме один или несколько полимеризуемых присоединением мономеров, указанный coполимер содержит два или более, предпочтительно три или более сегментов или блоков, различающихся по содержанию сомономера, кристалличности, регулярности молекулярной структуры, гомогенности, плотности, температуре плавления или температуре стеклования, предпочтительно указанный coполимер имеет молекулярномассовое распределение, Mw/Mn, менее чем 3,0, более предпочтительно менее чем 2,8.
4a. Мульти-блок-coполимер, содержащий в полимеризованной форме этилен и один или несколько coполимеризуемых сомономеров, указанный coполимер содержит два или более, предпочтительно три или более сегментов или блоков, различающихся по содержанию сомономера, кристалличности, регулярности молекулярной структуры, гомогенности, плотности, температуре плавления или температуре стеклования, предпочтительно указанный coполимер имеет молекулярномассовое распределение, Mw/Mn, менее чем 3,0, более предпочтительно менее чем 2,8.
5. Функционализованное производное мульти-блок coполимера по п.4.
6. Функционализованное производное мульти-блок coполимера по п.4a.
7. Олефиновый сополимер, имеющий по меньшей мере одну температуру плавления, Tm, в градусах Цельсия и плотность, d*, в граммах на кубический сантиметр, где числовые величины переменных соответствуют уравнению:
Tm>-2002,9+4538,5(d*)-2422,2(d*)2,
и где сополимер имеет Mw/Mn от 1,7 до 3,5.
8. Сополимер, содержащий в полимеризованной форме этилен и C3-8 α-олефин, имеющий по меньшей мере одну температуру плавления, Tm, в градусах Цельсия и плотность, d*, в граммах на кубический сантиметр, где числовые величины переменных соответствуют уравнению:
Tm>-2002,9+4538,5(d*)-2422,2(d*)2.
9. Мульти-блок-coполимер, содержащий в полимеризованной форме этилен и один или несколько coполимеризуемых сомономеров, имеющий по меньшей мере одну температуру плавления, Tm, в градусах Цельсия и плотность, d*, в граммах на кубический сантиметр, где числовые величины переменных соответствуют уравнению:
Tm>-2002,9+4538,5(d*)-2422,2(d*)2.
10. Олефиновый сополимер, имеющий Mw/Mn от 1,7 до 3,5,
величину дельта (наивысший пик DSC минус наивысший пик CRYSTAF) более чем величина y*, определяемая уравнением:
y*>-0,1299(ΔH)+62,81, предпочтительно уравнением:
y*>-0,1299(ΔH)+64,38 и более предпочтительно уравнением:
y*>-0,1299(ΔH)+65,95,
и теплоту плавления вплоть до 130 Дж/г,
где пик CRYSTAF определяют, используя по меньшей мере 5 процентов совокупного полимера, и если менее чем 5 процентов полимера имеет идентифицируемый пик CRYSTAF, тогда температура CRYSTAF 30°C, и ΔH представляет числовую величину теплоты плавления в Дж/г.
10a. Сополимер, содержащий в полимеризованной форме этилен и C3-8 α-олефин, указанный сополимер имеет величину дельта (наивысший пик DSC минус наивысший пик CRYSTAF) более чем величина y*, определяемая уравнением:
y*>-0,1299(ΔH)+62,81, предпочтительно уравнением:
y*>-0,1299(ΔH)+64,38 и более предпочтительно уравнением:
y*>-0,1299(ΔH)+65,95,
и теплоту плавления вплоть до 130 Дж/г,
где пик CRYSTAF определяют, используя по меньшей мере 5 процентов совокупного полимера, и если менее чем 5 процентов полимера имеет идентифицируемый пик CRYSTAF, тогда температура CRYSTAF 30°C, и ΔH представляет числовую величину теплоты плавления в Дж/г.
l0b. Мульти-блок-coполимер, имеющий величину дельта (наивысший пик DSC минус наивысший пик CRYSTAF) более чем величина y*, определяемая уравнением:
y*>-0,1299(ΔH)+62,81, предпочтительно уравнением:
y*>-0,1299(ΔH)+64,38 и более предпочтительно уравнением:
y*>-0,1299(ΔH)+65,95,
и теплоту плавления вплоть до 130 Дж/г,
где пик CRYSTAF определяют, используя по меньшей мере 5 процентов совокупного полимера, и если менее чем 5 процентов полимера имеет идентифицируемый пик CRYSTAF, тогда температура CRYSTAF 30°C, и ΔH представляет числовую величину теплоты плавления в Дж/г.
11. Олефиновый сополимер, имеющий предел прочности при растяжении выше 10 МПа, предпочтительно предел прочности при растяжении >11 МПа, более предпочтительно предел прочности при растяжении >13 МПа и относительное удлинение при разрыве по меньшей мере 600 процентов, более предпочтительно по меньшей мере 700 процентов, очень предпочтительно по меньшей мере 800 процентов и наиболее предпочтительно по меньшей мере 900 процентов при скорости разделения ползуна 11 см/минута.
11a. Мульти-блок-coполимер, содержащий в полимеризованной форме этилен и один или несколько coполимеризуемых сомономеров, имеющий предел прочности при растяжении выше 10 МПа, предпочтительно предел прочности при растяжении >11 МПа, более предпочтительно предел прочности при растяжении >13МПа и относительное удлинение при разрыве по меньшей мере 600 процентов, более предпочтительно по меньшей мере 700 процентов, очень предпочтительно по меньшей мере 800 процентов и наиболее предпочтительно по меньшей мере 900 процентов при скорости разделения ползуна 11 см/мин.
12. Олефиновый сополимер, имеющий величину дельта (наивысший пик DSC (измеренный от базовой линии) минус наивысший пик CRYSTAF) более чем 48°C и теплоту плавления более чем или равную 130 Дж/г, где пик CRYSTAF определяют, используя по меньшей мере 5 процентов совокупного полимера, и если менее чем 5 процентов полимера имеет идентифицируемый пик CRYSTAF, тогда температура CRYSTAF 30°C.
12a. Мульти-блок-coполимер, содержащий в полимеризованной форме этилен и один или более coполимеризуемых сомономеров, имеющий величину дельта (наивысший пик DSC (измеренный от базовой линии) минус наивысший пик CRYSTAF) более чем 45°C и теплоту плавления более чем или равную 130 Дж/г, где пик CRYSTAF определяют, используя по меньшей мере 5 процентов совокупного полимера, и если менее чем 5 процентов полимера имеет идентифицируемый пик CRYSTAF, тогда температура CRYSTAF 30°C.
13. Олефиновый сополимер, имеющий отношение динамических модулей упругости, G'(25°C)/G'(100°C), от 1 до 50, предпочтительно от 1 до 20, более предпочтительно от 1 до 10, и остаточную деформацию при сжатии при 70°C менее чем 80 процентов, предпочтительно менее чем 70 процентов, особенно менее чем 60 процентов, вплоть до остаточной деформации при сжатии 0 процентов.
13a. Мульти-блок-coполимер, содержащий в полимеризованной форме этилен и один или несколько coполимеризуемых сомономеров, имеющий отношение динамических модулей упругости, G'(25°C)/G'(100°C), от 1 до 50, предпочтительно от 1 до 20, более предпочтительно от 1 до 10, и остаточную деформацию при сжатии при 70°C менее чем 80 процентов, предпочтительно менее чем 70 процентов, особенно менее чем 60 процентов, вплоть до остаточной деформации при сжатии 0 процентов.
14. Олефиновый сополимер, имеющий теплоту плавления менее чем 85 Дж/г, предпочтительно менее чем 80 Дж/г, и силу слипания гранул, равную или менее чем 100 фунт/фут2 (4800 Пa), предпочтительно равную или менее чем 50 фунт/фут2 (2400 Пa), особенно равную или менее чем 5 фунт/фут2 (240 Пa) и настолько низкую как 0 фунт/фут2 (0 Пa).
14a. Мульти-блок-coполимер, содержащий в полимеризованной форме этилен и один или несколько coполимеризуемых сомономеров, имеющий теплоту плавления менее чем 85 Дж/г, предпочтительно менее чем 80 Дж/г, и силу слипания гранул равную или менее чем 100 фунтов/фут2 (4800 Пa), предпочтительно равную или менее чем 50 фунтов/фут2 (2400 Пa), особенно равную или менее чем 5 фунтов/фут2 (240 Пa), и настолько низкую как 0 фунтов/фут2 (0 Пa).
15. Несшитый эластомерный олефиновый сополимер, содержащий в полимеризованной форме по меньшей мере 50 молярных процентов этилена, имеющий остаточную деформацию при сжатии при 70°C менее чем 80 процентов, предпочтительно менее чем 70 процентов, наиболее предпочтительно менее чем 60 процентов.
15a. Несшитый эластомерный мульти-блок-coполимер, содержащий в полимеризованной форме по меньшей мере 50 молярных процентов этилена, имеющий остаточную деформацию при сжатии при 70°C менее чем 80 процентов, предпочтительно менее чем 70 процентов, наиболее предпочтительно менее чем 60 процентов.
16. Полимер по любому из пп.4-15, 4a, 5a, 10a-15a, l0b или получаемый способом по п.3 или 3a, имеющий единственную кристаллическую температуру плавления (Tm), которую измеряют DSC.
17. Полимер по любому из пп.4-15, 4a, 5a, 10a-15a, 10b или получаемый способом по п.3 или 3a, имеющий глубину пенетрации при термомеханическом анализе 1 мм при температуре по меньшей мере 90°C, предпочтительно при температуре по меньшей мере 100°C, и модуль упругости при изгибе от 3 килофунт на квадратный дюйм (20 МПа) дo 13 килофунт на квадратный дюйм (90 МПа).
18. Полимер по п.16, имеющий глубину пенетрации при термомеханическом анализе 1 мм при температуре по меньшей мере 90°C, предпочтительно при температуре по меньшей мере 100°C, и модуль упругости при изгибе от 3 килофунт на квадратный дюйм (20 МПа) дo 13 килофунт на квадратный дюйм (90 МПа).
19. Полимер по любому из пп.4-15, 4a, 5a, 10a-15a, 10b или получаемый способом по п.3 или 3a, имеющий объемную потерю при сопротивлении истиранию согласно ISO 4649 менее чем 90 мм3.
20. Полимер по п.16, имеющий объемную потерю при сопротивлении истиранию согласно ISO 4649 менее чем 90 мм3.
21. Полимер по п.17, имеющий объемную потерю при сопротивлении истиранию согласно ISO 4649 менее чем 90 мм3.
22. Полимер по п.18, имеющий объемную потерю при сопротивлении истиранию согласно ISO 4649 менее чем 90 мм3.
23. Полимер по любому из пп.4-15, 4a, 5a, 10a-15a, 10b или получаемый способом по п.3 или 3a, имеющий объемную потерю при сопротивлении истиранию согласно ISO 4649 менее чем 90 мм3 и имеющий динамический модуль упругости, G', такой, что log (G') более чем или равен 0,4 МПа, предпочтительно более чем или равен 1,0 МПа, при температуре 100°C.
24. Полимер по п.16, имеющий объемную потерю при сопротивлении истиранию согласно ISO 4649 менее чем 90 мм3 и имеющий динамический модуль упругости, G', такой, что log (G') более чем или равен 0,4 МПа, предпочтительно более чем или равен 1,0 МПа, при температуре 100°C.
25. Полимер по п.17, имеющий объемную потерю при сопротивлении истиранию согласно ISO 4649 менее чем 90 мм3 и имеющий динамический модуль упругости, G', такой, что log (G') более чем или равен 0,4 МПа, предпочтительно более чем или равен 1,0 МПа, при температуре 100°C.
26. Полимер по п.18, имеющий объемную потерю при сопротивлении истиранию согласно ISO 4649 менее чем 90 мм3 и имеющий динамический модуль упругости, G', такой, что log (G') более чем или равен 0,4 МПа, предпочтительно более чем или равен 1,0 МПа, при температуре 100°C.
27. Полимер по п.19, имеющий объемную потерю при сопротивлении истиранию согласно ISO 4649 менее чем 90 мм3 и имеющий динамический модуль упругости, G', такой, что log (G') более чем или равен 0,4 МПа, предпочтительно более чем или равен 1,0 МПа, при температуре 100°C.
28. Полимер по п.20, имеющий объемную потерю при сопротивлении истиранию согласно ISO 4649 менее чем 90 мм3 и имеющий динамический модуль упругости, G', такой, что log (G') более чем или равен 0,4 МПа, предпочтительно более чем или равен 1,0 МПа, при температуре 100°C.
29. Полимер по п.21, имеющий объемную потерю при сопротивлении истиранию согласно ISO 4649 менее чем 90 мм3 и имеющий динамический модуль упругости, G', такой, что log (G') более чем или равен 0,4 МПа, предпочтительно более чем или равен 1,0 МПа, при температуре 100°C.
30. Полимер по п.22, имеющий объемную потерю при сопротивлении истиранию согласно ISO 4649 менее чем 90 мм3 и имеющий динамический модуль упругости, G', такой, что log (G') более чем или равен 0,4 МПа, предпочтительно более чем или равен 1,0 МПа, при температуре 100°C.
31. Сшитое производное полимера по любому из пп.4-15, 4a, 5a, 10a-15a, 10b или получаемое способом по п.3 или 3a.
32. Сшитое производное полимера по п.16.
33. Сшитое производное полимера по п.17.
34. Сшитое производное полимера по п.18.
35. Сшитое производное полимера по п.19.
36. Сшитое производное полимера по п.20.
37. Сшитое производное полимера по п.21.
38. Сшитое производное полимера по п.22.
39. Сшитое производное полимера по п.23.
40. Сшитое производное полимера по п.24.
41. Сшитое производное полимера по п.25.
42. Сшитое производное полимера по п.26.
43. Сшитое производное полимера по п.27.
44. Сшитое производное полимера по п.28.
45. Сшитое производное полимера по п.29.
46. Сшитое производное полимера по п.30.
47. Полимер по любому из пп.4-15, 4a, 5a, 10a-15a, 10b или получаемый способом по п.3 или 3 a, или состав, содержащий его в форме пленки, по меньшей мере одного слоя многослойной пленки, по меньшей мере одного слоя ламинированного изделия, вспененного изделия, волокна, нетканого материала, сформованного литьем под давлением изделия, сформованного раздувом изделия, сформованного роторным прессованием изделия или адгезива.
48. Полимер по п.16 или состав, содержащий его в форме пленки, по меньшей мере одного слоя многослойной пленки, по меньшей мере одного слоя ламинированного изделия, вспененного изделия, волокна, нетканого материала, сформованного литьем под давлением изделия, сформованного раздувом изделия, сформованного роторным прессованием изделия или адгезива.
49. Полимер по п.17 или состав, содержащий его в форме пленки, по меньшей мере одного слоя многослойной пленки, по меньшей мере одного слоя ламинированного изделия, вспененного изделия, волокна, нетканого материала, сформованного литьем под давлением изделия, сформованного раздувом изделия, сформованного роторным прессованием изделия или адгезива.
50. Полимер по п.18 или состав, содержащий его в форме пленки, по меньшей мере одного слоя многослойной пленки, по меньшей мере одного слоя ламинированного изделия, вспененного изделия, волокна, нетканого материала, сформованного литьем под давлением изделия, сформованного раздувом изделия, сформованного роторным прессованием изделия или адгезива.
51. Полимер по п.19 или состав, содержащий его в форме пленки, по меньшей мере одного слоя многослойной пленки, по меньшей мере одного слоя ламинированного изделия, вспененного изделия, волокна, нетканого материала, сформованного литьем под давлением изделия, сформованного раздувом изделия, сформованного роторным прессованием изделия или адгезива.
52. Полимер по п.20 или состав, содержащий его в форме пленки, по меньшей мере одного слоя многослойной пленки, по меньшей мере одного слоя ламинированного изделия, вспененного изделия, волокна, нетканого материала, сформованного литьем под давлением изделия, сформованного раздувом изделия, сформованного роторным прессованием изделия или адгезива.
53. Полимер по п.21 или состав, содержащий его в форме пленки, по меньшей мере одного слоя многослойной пленки, по меньшей мере одного слоя ламинированного изделия, вспененного изделия, волокна, нетканого материала, сформованного литьем под давлением изделия, сформованного раздувом изделия, сформованного роторным прессованием изделия или адгезива.
54. Полимер по п.22 или состав, содержащий его в форме пленки, по меньшей мере одного слоя многослойной пленки, по меньшей мере одного слоя ламинированного изделия, вспененного изделия, волокна, нетканого материала, сформованного литьем под давлением изделия, сформованного раздувом изделия, сформованного роторным прессованием изделия или адгезива.
55. Полимер по п.23 или состав, содержащий его в форме пленки, по меньшей мере одного слоя многослойной пленки, по меньшей мере одного слоя ламинированного изделия, вспененного изделия, волокна, нетканого материала, сформованного литьем под давлением изделия, сформованного раздувом изделия, сформованного роторным прессованием изделия или адгезива.
56. Полимер по п.24 или состав, содержащий его в форме пленки, по меньшей мере одного слоя многослойной пленки, по меньшей мере одного слоя ламинированного изделия, вспененного изделия, волокна, нетканого материала, сформованного литьем под давлением изделия, сформованного раздувом изделия, сформованного роторным прессованием изделия или адгезива.
57. Полимер по п.25 или состав, содержащий его в форме пленки, по меньшей мере одного слоя многослойной пленки, по меньшей мере одного слоя ламинированного изделия, вспененного изделия, волокна, нетканого материала, сформованного литьем под давлением изделия, сформованного раздувом изделия, сформованного роторным прессованием изделия или адгезива.
58. Полимер по п.26 или состав, содержащий его в форме пленки, по меньшей мере одного слоя многослойной пленки, по меньшей мере одного слоя ламинированного изделия, вспененного изделия, волокна, нетканого материала, сформованного литьем под давлением изделия, сформованного раздувом изделия, сформованного роторным прессованием изделия или адгезива.
59. Полимер по п.27 или состав, содержащий его в форме пленки, по меньшей мере одного слоя многослойной пленки, по меньшей мере одного слоя ламинированного изделия, вспененного изделия, волокна, нетканого материала, сформованного литьем под давлением изделия, сформованного раздувом изделия, сформованного роторным прессованием изделия или адгезива.
60. Полимер по п.28 или состав, содержащий его в форме пленки, по меньшей мере одного слоя многослойной пленки, по меньшей мере одного слоя ламинированного изделия, вспененного изделия, волокна, нетканого материала, сформованного литьем под давлением изделия, сформованного раздувом изделия, сформованного роторным прессованием изделия или адгезива.
61. Полимер по п.29 или состав, содержащий его в форме пленки, по меньшей мере одного слоя многослойной пленки, по меньшей мере одного слоя ламинированного изделия, вспененного изделия, волокна, нетканого материала, сформованного литьем под давлением изделия, сформованного раздувом изделия, сформованного роторным прессованием изделия или адгезива.
62. Полимер по п.30 или состав, содержащий его в форме пленки, по меньшей мере одного слоя многослойной пленки, по меньшей мере одного слоя ламинированного изделия, вспененного изделия, волокна, нетканого материала, сформованного литьем под давлением изделия, сформованного раздувом изделия, сформованного роторным прессованием изделия или адгезива.
63. Полимер по п.31 или состав содержащий его в форме пленки, по меньшей мере одного слоя многослойной пленки, по меньшей мере одного слоя ламинированного изделия, вспененного изделия, волокна, нетканого материала, сформованного литьем под давлением изделия, сформованного раздувом изделия, сформованного роторным прессованием изделия или адгезива.
64. Полимер по п.32 или состав, содержащий его в форме пленки, по меньшей мере одного слоя многослойной пленки, по меньшей мере одного слоя ламинированного изделия, вспененного изделия, волокна, нетканого материала, сформованного литьем под давлением изделия, сформованного раздувом изделия, сформованного роторным прессованием изделия или адгезива.
65. Полимер по п.33 или состав, содержащий его в форме пленки, по меньшей мере одного слоя многослойной пленки, по меньшей мере одного слоя ламинированного изделия, вспененного изделия, волокна, нетканого материала, сформованного литьем под давлением изделия, сформованного раздувом изделия, сформованного роторным прессованием изделия или адгезива.
66. Полимер по п.34 или состав, содержащий его в форме пленки, по меньшей мере одного слоя многослойной пленки, по меньшей мере одного слоя ламинированного изделия, вспененного изделия, волокна, нетканого материала, сформованного литьем под давлением изделия, сформованного раздувом изделия, сформованного роторным прессованием изделия или адгезива.
67. Полимер по п.35 или состав, содержащий его в форме пленки, по меньшей мере одного слоя многослойной пленки, по меньшей мере одного слоя ламинированного изделия, вспененного изделия, волокна, нетканого материала, сформованного литьем под давлением изделия, сформованного раздувом изделия, сформованного роторным прессованием изделия или адгезива.
68. Полимер по п.36 или состав, содержащий его в форме пленки, по меньшей мере одного слоя многослойной пленки, по меньшей мере одного слоя ламинированного изделия, вспененного изделия, волокна, нетканого материала, сформованного литьем под давлением изделия, сформованного раздувом изделия, сформованного роторным прессованием изделия или адгезива.
69. Полимер по п.37 или состав, содержащий его в форме пленки, по меньшей мере одного слоя многослойной пленки, по меньшей мере одного слоя ламинированного изделия, вспененного изделия, волокна, нетканого материала, сформованного литьем под давлением изделия, сформованного раздувом изделия, сформованного роторным прессованием изделия или адгезива.
70. Полимер по п.38 или состав, содержащий его в форме пленки, по меньшей мере одного слоя многослойной пленки, по меньшей мере одного слоя ламинированного изделия, вспененного изделия, волокна, нетканого материала, сформованного литьем под давлением изделия, сформованного раздувом изделия, сформованного роторным прессованием изделия или адгезива.
71. Полимер по п.39 или состав, содержащий его в форме пленки, по меньшей мере одного слоя многослойной пленки, по меньшей мере одного слоя ламинированного изделия, вспененного изделия, волокна, нетканого материала, сформованного литьем под давлением изделия, сформованного раздувом изделия, сформованного роторным прессованием изделия или адгезива.
72. Полимер по п.40 или состав, содержащий его в форме пленки, по меньшей мере одного слоя многослойной пленки, по меньшей мере одного слоя ламинированного изделия, вспененного изделия, волокна, нетканого материала, сформованного литьем под давлением изделия, сформованного раздувом изделия, сформованного роторным прессованием изделия или адгезива.
73. Полимер по п.41 или состав, содержащий его в форме пленки, по меньшей мере одного слоя многослойной пленки, по меньшей мере одного слоя ламинированного изделия, вспененного изделия, волокна, нетканого материала, сформованного литьем под давлением изделия, сформованного раздувом изделия, сформованного роторным прессованием изделия или адгезива.
74. Полимер по п.42 или состав, содержащий его в форме пленки, по меньшей мере одного слоя многослойной пленки, по меньшей мере одного слоя ламинированного изделия, вспененного изделия, волокна, нетканого материала, сформованного литьем под давлением изделия, сформованного раздувом изделия, сформованного роторным прессованием изделия или адгезива.
75. Полимер по п.43 или состав, содержащий его в форме пленки, по меньшей мере одного слоя многослойной пленки, по меньшей мере одного слоя ламинированного изделия, вспененного изделия, волокна, нетканого материала, сформованного литьем под давлением изделия, сформованного раздувом изделия, сформованного роторным прессованием изделия или адгезива.
76. Полимер по п.44 или состав, содержащий его в форме пленки, по меньшей мере одного слоя многослойной пленки, по меньшей мере одного слоя ламинированного изделия, вспененного изделия, волокна, нетканого материала, сформованного литьем под давлением изделия, сформованного раздувом изделия, сформованного роторным прессованием изделия или адгезива.
77. Полимер по п.45 или состав, содержащий его в форме пленки, по меньшей мере одного слоя многослойной пленки, по меньшей мере одного слоя ламинированного изделия, вспененного изделия, волокна, нетканого материала, сформованного литьем под давлением изделия, сформованного раздувом изделия, сформованного роторным прессованием изделия или адгезива.
78. Полимер по п.46 или состав, содержащий его в форме пленки, по меньшей мере одного слоя многослойной пленки, по меньшей мере одного слоя ламинированного изделия, вспененного изделия, волокна, нетканого материала, сформованного литьем под давлением изделия, сформованного раздувом изделия, сформованного роторным прессованием изделия или адгезива.
79. Состав по п.1 или 1a, где челночным агентом является соединение тригидрокарбилалюминия или дигидрокарбилцинка, содержащее от 1 до 12 атомов углерода в каждой гидрокарбилгруппе.
80. Состав по п.79, где челночным агентом является триэтилалюминий или диэтилцинк.
81. Состав по п.1 или 1a, где катализатор (A) содержит комплекс, содержащий переходный металл, выбранный из групп 4-8 Периодической таблицы элементов, и один или несколько делокализованных, π-связанных лигандов или лигандов поливалентного основания Льюиса.
82. Состав по п.81, где катализатор (A) соответствует формуле:
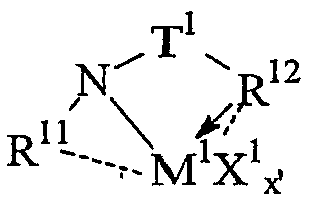
где
R11 выбран из алкила, циклоалкила, гетероалкила, циклогетероалкила, арила и инертно замещенных их производных, содержащих от 1 до 30 атомов, не считая водород, или их двухвалентных производных;
T1 представляет двухвалентную мостиковую группу из от 1 до 41 атомов, иных чем водород, предпочтительно от 1 до 20 атомов, иных чем водород, и наиболее предпочтительно моно- или ди-С1-20 гидрокарбил-замещенную группу метилена или силана,
R12 представляет C5-20 гетероарилгруппу, содержащую функциональность основания Льюиса, особенно пиридин-2-ил- или замещенную пиридин-2-ил-группу или ее двухвалентное производное;
M1 представляет металл группы 4, предпочтительно гафний;
X1 представляет анионную, нейтральную или дианионную лиганд-группу;
x' означает число от 0 до 5, указывающее число таких X1 групп, и
связи, необязательные связи и электронодонорные взаимодействия представлены линиями, пунктирными линиями и стрелками соответственно.
83. Состав по п.82, где катализатор (B) отвечает формуле:
где
M2 представляет металл групп 4-10 Периодической таблицы элементов;
T2 означает группу, содержащую азот, кислород или фосфор;
X2 представляет галоген, гидрокарбил или гидрокарбилокси;
t означает один или два;
x" означает целое число, выбранное, чтобы обеспечить баланс заряда,
и T2 и N связаны мостиковым лигандом.
84. Способ по п.3 или 3a, который является непрерывным процессом.
85. Способ по п.84, который является осуществляемым в растворе процессом.
86. Способ по п.85, где этилен и один или несколько coполимеризуемых сомономеров полимеризуют.
87. Способ по п.86, где превращение этилена в реакторе составляет по меньшей мере 95 процентов.
88. Способ по п.84, где катализатор (A) соответствует формуле:
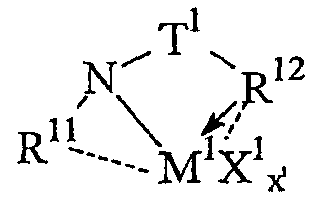
где
R11 выбран из алкила, циклоалкила, гетероалкила, циклогетероалкила, арила и инертно замещенных их производных, содержащих от 1 до 30 атомов, не считая водород, или их двухвалентных производных;
T1 представляет двухвалентную мостиковую группу из от 1 до 41 атомов, иных чем водород, предпочтительно от 1 до 20 атомов, иных чем водород, и наиболее предпочтительно моно- или ди-С1-20 гидрокарбил-замещенную группу метилена или силана,
R12 представляет C5-20 гетероарилгруппу, содержащую функциональность основания Льюиса, особенно пиридин-2-ил- или замещенную пиридин-2-ил-группу или ее двухвалентное производное;
M1 представляет металл группы 4, предпочтительно гафний;
X1 представляет анионную, нейтральную или дианионную лиганд-группу;
x' означает число от 0 до 5, указывающее число таких X1 групп, и
связи, необязательные связи и электронодонорные взаимодействия представлены линиями, пунктирными линиями и стрелками соответственно.
89. Способ по п.88, где катализатор (B) соответствует формуле:
где
M2 представляет металл групп 4-10 Периодической таблицы элементов;
T2 означает группу, содержащую азот, кислород или фосфор;
X2 представляет галоген, гидрокарбил или гидрокарбилокси;
t означает один или два;
x" означает целое число, выбранное, чтобы обеспечить баланс заряда,
и T2 и N связаны мостиковым лигандом.
90. Мульти-блок-coполимер, содержащий в полимеризованной форме этилен и coполимеризуемый сомономер.
91. Олефиновый полимер, имеющий относительно плоский график динамического модуля упругости, характеризующийся тем, что log G' (в Паскалях) уменьшается менее чем на один порядок величины между 50 и 100°C.
92. Способ по п.3 или 3a, в котором отношение челночного агента цепи к одному или нескольким катализаторам и/или мономерам изменяют, чтобы получать полимеры, различающиеся по одному или нескольким химическим или физическим свойствам.
93. Полимерная смесь, содержащая: (1) органический или неорганический полимер, предпочтительно гомополимер этилена или пропилена и/или coполимер этилена и coполимеризуемого сомономера, и (2) полимер по любому из пп.4-15, 4a, 5a, 10a-15a, 10b или получаемый способом по п.3 или 3a.
94. Полимерная смесь по п.93, где компонент (1) является органическим термопластичным полимером.
95. Полимерная смесь по п.94 где компонент (1) является пропиленовым гомополимером.
96. Полимерная смесь по п.95, где компонент (1) является высоко изотактическим полипропиленом.
97. Полимерная смесь по п.93, где компонент (2) является эластомерным coполимером этилена и одного или нескольких coполимеризуемых сомономеров.
98. Полимерная смесь по п.94, где компонент (2) является эластомерным coполимером этилена и одного или нескольких coполимеризуемых сомономеров.
99. Полимерная смесь по п.95 где компонент (2) является эластомерным coполимером, этилена и одного или нескольких coполимеризуемых сомономеров.
100. Полимерная смесь по п.96, где компонент (2) является эластомерным coполимером этилена и одного или нескольких coполимеризуемых сомономеров.
101. Полимерная смесь по п.93, где компонент (2) является эластомерным coполимером этилена и одного или нескольких coполимеризуемых сомономеров в форме частиц, содержащих окклюзии компонента (1).
102. Полимерная смесь по п.94, где компонент (2) является эластомерным coполимером этилена и одного или нескольких coполимеризуемых сомономеров в форме частиц, содержащих окклюзии компонента (1).
103. Полимерная смесь по п.95, где компонент (2) является эластомерным coполимером этилена и одного или нескольких coполимеризуемых сомономеров в форме частиц, содержащих окклюзии компонента (1).
104. Полимерная смесь по п.96, где компонент (2) является эластомерным coполимером этилена и одного или нескольких coполимеризуемых сомономеров в форме частиц, содержащих окклюзии компонента (1).
105. Полимерная смесь по п.93, где компонент (2) является эластомерным coполимером этилена и одного или нескольких coполимеризуемых сомономеров в форме частиц, содержащих окклюзии компонента (1), там указанные окклюзии сформированы при смешивании расплава компонентов (1) и (2).
106. Полимерная смесь по п.94, где компонент (2) является эластомерным coполимером этилена и одного или нескольких сополимеризуемых сомономеров в форме частиц, содержащих окклюзии компонента (1), там указанные окклюзии сформированы при смешивании расплава компонентов (1) и (2).
107. Полимерная смесь по п.95, где компонент (2) является эластомерным coполимером этилена и одного или нескольких coполимеризуемых сомономеров в форме частиц, содержащих окклюзии компонента (1), там указанные окклюзии сформированы при смешивании расплава компонентов (1) и (2).
108. Полимерная смесь по п.96, где компонент (2) является эластомерным coполимером этилена и одного или нескольких coполимеризуемых сомономеров в форме частиц, содержащих окклюзии компонента (1), там указанные окклюзии сформированы при смешивании расплава компонентов (1) и (2).
109. Способ получения полимерной смеси, содержащей: (1) органический или неорганический термопластичный полимер, предпочтительно гомополимер этилена или пропилена и/или a coполимер этилена и coполимеризуемого сомономера, и (2) эластомерный полимер в форме частиц, содержащих окклюзии компонента (1), там указанный способ содержит смешивание в расплаве компонентов (1) и (2) в условиях сдвига так, чтобы сформировать окклюзии компонента (1) в диспергированных частицах компонента (2).
110. Способ по п.109, где компонент (1) является изотактическим полипропиленом.
111. Способ по п.110, где компонент (2) является coполимером этилена и coполимеризуемого сомономера.
Специалист будет принимать во внимание, что раскрытое здесь изобретение может быть осуществлено на практике без какого-либо компонента, стадии или ингредиента, которые не были конкретно раскрыты.
Примеры
Следующие примеры представлены в качестве дополнительного пояснения изобретения и не должны быть истолкованы как ограничение. Термин "в течение ночи", если используется, относится к времени приблизительно 16-18 часов, термин "комнатная температура", относится к температуре 20-25°C и термин "смешанные алканы" относится к коммерчески получаемой смеси C6-9 алифатических углеводородов, доступной под торговым обозначением Isopar E® от Exxon Mobil Chemicals Inc. В случае, когда наименование соединения здесь не согласуется с его структурным изображением, структурное изображение будет контролем. Синтез всех комплексов металлов и подготовку всех отсеивающих экспериментов проводят в атмосфере сухого азота, используя технику сухой камеры. Все используемые растворители имеют чистоту для ЖХВР, и их сушат перед использованием.
MMAO относится к модифицированному метилалюмоксану, модифицированному триизобутилалюминием метилалюмоксану, доступному коммерчески от Akzo-Noble Corporation.
Катализатором (A1) является [N-(2,6-ди(1-метилэтил)фенил)амидо)(2-изопропилфенил)(α-нафталин-2-диил(6-пиридин-2-диил)метан)]гафний диметил, полученный согласно методам из WO 03/40195, 2003US0204017, USSN 10/429024, поданных 2 мая 2003, и WO 04/24740.
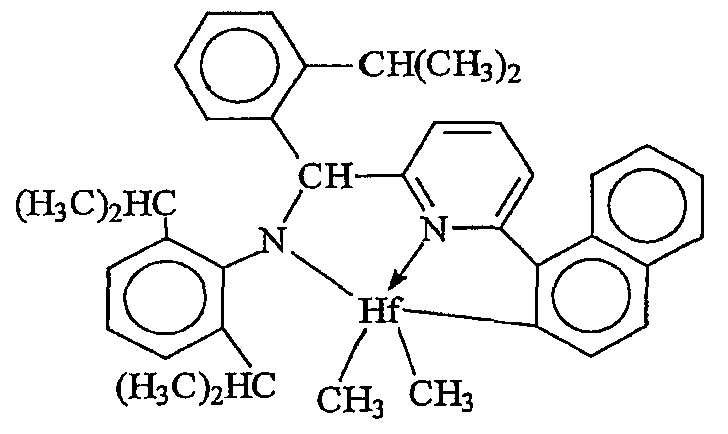
Катализатором (А2) является [N-(2,6-ди(1-метилэтил)фенил)амидо)(2-метилфенил)(1,2-фенилен-(6-пиридин-2-диил)метан)]гафний диметил, полученный согласно методам из WO 03/40195, 2003US0204017, USSN 10/429024, поданных 2 мая 2003, и WO 04/24740.
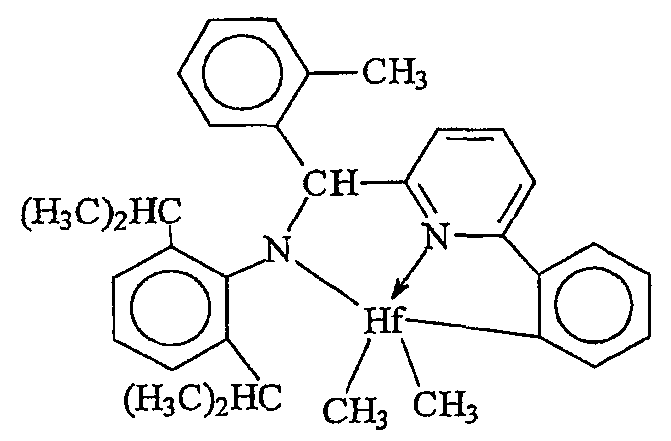
Катализатором (А3) является бис[N,N'''-(2,4,6-три(метилфенил)амидо)этилендиамин]гафний дибензил.
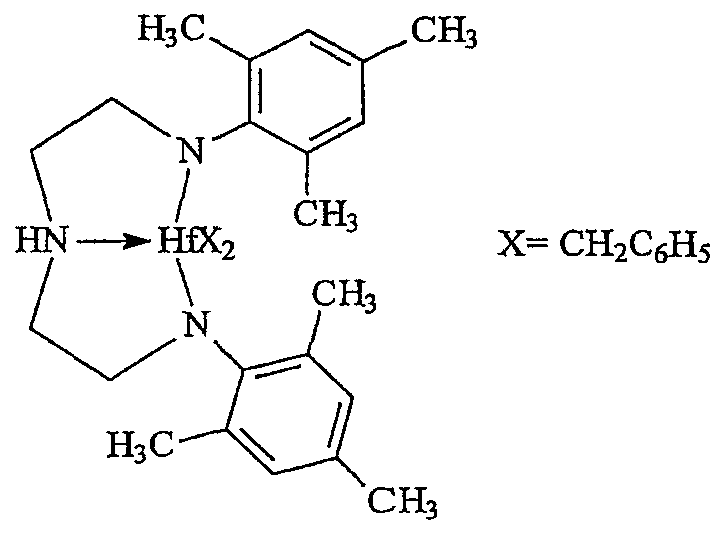
Катализатором (A4) является бис((2-оксоил-3-(дибензо-1H-пиррол-1-ил)-5-(метил)фенил)-2-феноксиметил)циклогексан-1,2-диил цирконий (IV) дибензил, полученный по существу согласно методам из US-A-2004/0010103.
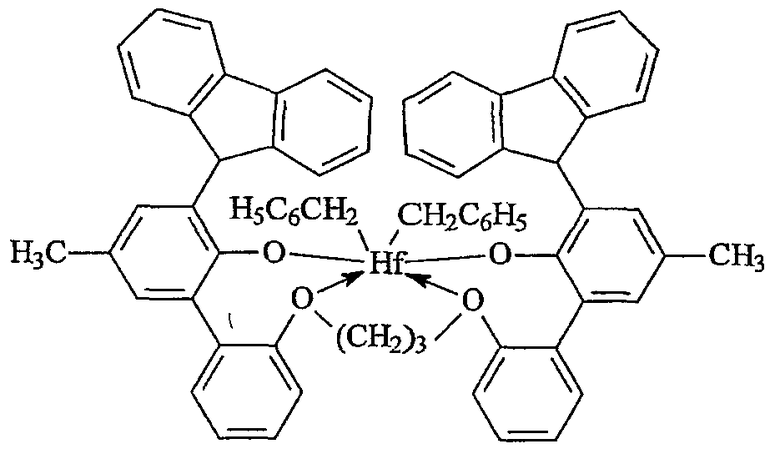
Катализатором (Bl) является 1,2-бис-(3,5-ди-трет-бутилфенилен)(1-(N-(1-метилэтил)имино)метил)(2-оксоил)цирконий дибензил.
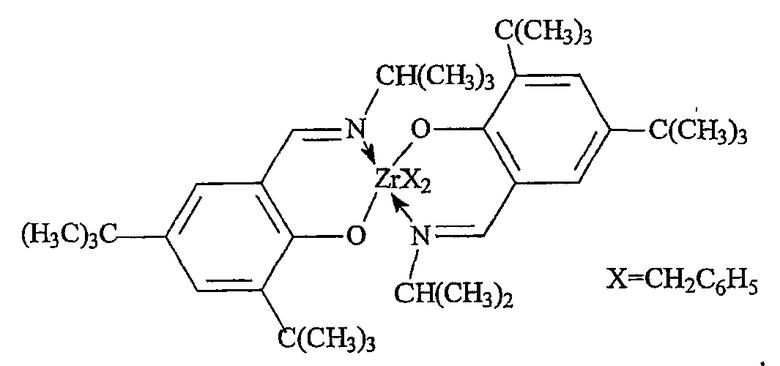
Катализатор (Bl) получают следующим образом.
a) Получение (1-метилэтил)(2-гидрокси-3,5-ди(трет-бутил) фенил)метилимина
3,5-Ди-трет-бутилсалицилальдегид (3,00 г) добавляют к 10 мл изопропиламина. Раствор быстро становится ярко-желтым. После перемешивания при температуре окружающей среды в течение 3 часов летучие вещества удаляют в вакууме, получая ярко-желтое кристаллическое твердое вещество (выход 97 процентов).
b) Получение 1,2-бис-(3,5-ди-трет-бутилфенилен)(1-(N-(1-метилэтил)имино)метил)(2-оксоил) цирконий дибензила
Раствор (1-метилэтил)(2-гидрокси-3,5-ди(трет-бутил)фенил) имина (605 мг, 2,2 ммоль) в 5 мл толуола медленно добавляют к раствору Zr(CH2Ph)4 (500 мг, 1,1 ммоль) в 50 мл толуола. Полученный темно-желтый раствор перемешивают в течение 30 мин. Растворитель удаляют при пониженном давлении, получая желательный продукт в виде красно-коричневого твердого вещества.
Катализатором (B2) является 1,2-бис-(3,5-ди-трет-бутилфенилен)(1-(N-(2-метилциклогексил)имино)метил)(2-оксоил) цирконий дибензил
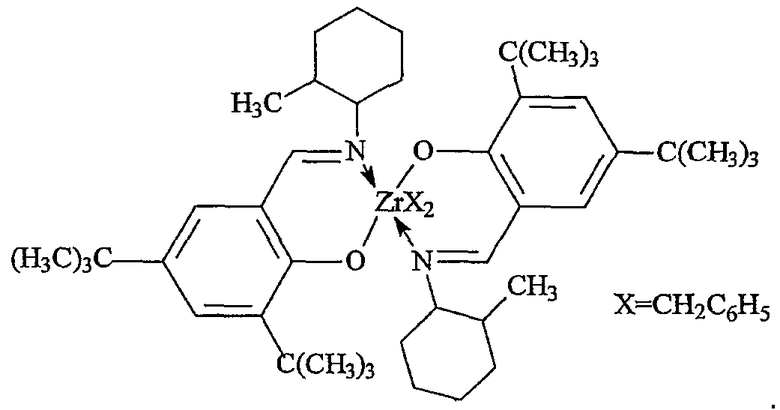
Катализатор (B2) получают следующим образом.
a) Получение (1-(2-метилциклогексил)этил)(2-оксоил-3,5-ди(трет-бутил)фенил)имина
2-Метилциклогексиламин (8,44 мл, 64,0 ммоль) растворяют в метаноле (90 мл) и добавляют ди-трет-бутилсалицилальдегид (10,00 г, 42,67 ммоль). Реакционную смесь перемешивают в течение трех часов и затем охлаждают до -25°C в течение 12 ч. Полученный желтый твердый осадок собирают фильтрованием, промывают холодным метанолом (2×15 мл) и затем сушат при пониженном давлении. Выход 11,17 г желтого твердого вещества. 1H ЯМР согласуется с желательным продуктом как смесью изомеров.
b) Получение бис-(1-(2-метилциклогексил)этил)(2-оксоил-3,5-ди(трет-бутил)фенил)имино)цирконий дибензила
Раствор (1-(2-метилциклогексил)этил)(2-оксоил-3,5-ди(трет-бутил)фенил)имина (7,63 г, 23,2 ммоль) в 200 мл толуола медленно добавляют к раствору Zr(CH2Ph)4 (5,28 г, 11,6 ммоль) в 600 мл толуола. Полученный темно-желтый раствор перемешивают в течение 1 часа при 25°C. Раствор разбавляют дополнительно 680 мл толуола, чтобы получить раствор, имеющий концентрацию 0,00783 M.
Катализатором (C1) является (трет-бутиламидо)диметил(3-N-пирролил-1,2,3,3a,7a-η-инден-1-ил)силантитан диметил, полученный по существу согласно методам из патента США 6268444:
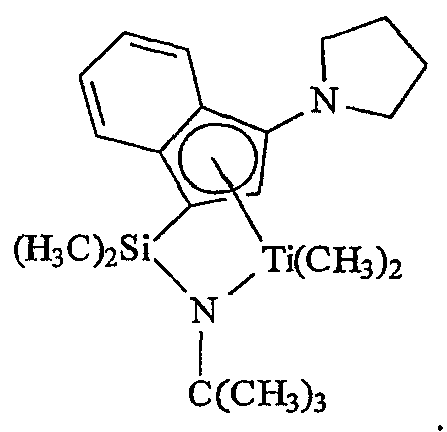
Катализатором (С2) является (трет-бутиламидо)ди(4-метилфенил)(2-метил-1,2,3,3a,7a-η-инден-1-ил)силантитан диметил, полученный по существу согласно методам из US-A-2003/004286:
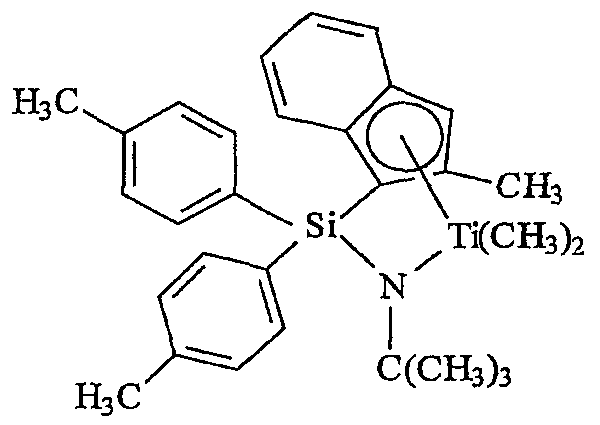
Катализатором (C3) является (трет-бутиламидо)ди(4-метилфенил)(2-метил-1,2,3,3a,8a-η-s-индацен-1-ил)силантитан диметил, полученный по существу согласно методам из US-A-2003/004286:
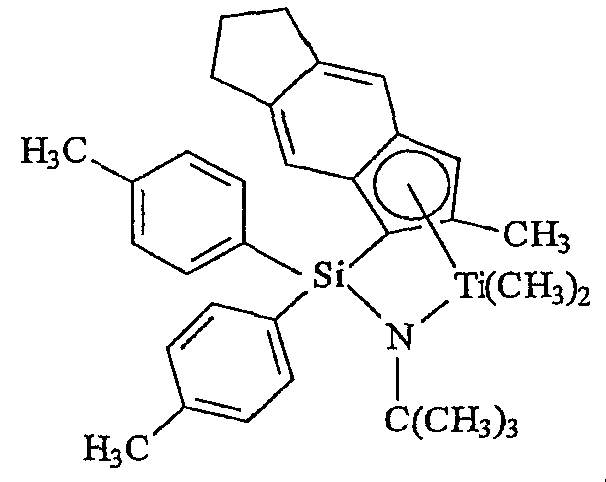
Катализатором (D1) является бис(диметилдисилоксан)(инден-1-ил)цирконий дихлорид, доступный от Sigma-Aldrich:
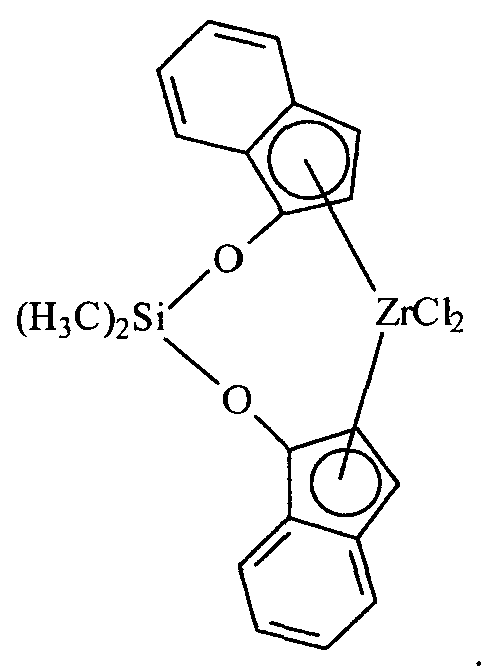
Сокатализатор 1
Смесь солей метилди(С14-18 алкил)аммония тетракис(пентафторфенил)бората (здесь и далее борат армииния), получаемая взаимодействием длинноцепочечного триалкиламина (Armeen™ M2HT, доступного от Akzo-Nobe1, Inc.), HCl и Li[B(C6F5)4], по существу как раскрыто в патенте США 5919983, пример 2.
Сокатализатор 2
Смешанная соль С14-18 алкилдиметиламмония и бис(трис(пентафторфенил)алюман)-2-ундецилимидазолида, полученная согласно патенту США 6395671, пример 16.
Челночные агенты
Используемые челночные агенты включают диэтилцинк (DEZ, SA1), ди(изобутил)цинк (SA2),
ди(н-гексил)цинк (SA3), триэтилалюминий (TEA, SA4), триоктилалюминий (SA5), триэтилгаллий (SA6),
изобутилалюминий бис(диметил(трет-бутил)силоксан) (SA7), изобутилалюминий бис(ди(триметилсилил)амид) (SA8),
н-октилалюминий ди(пиридин-2-метоксид) (SA9),
бис(н-октадецил)изобутилалюминий (SA10),
изобутилалюминий бис(ди(н-пентил)амид) (SA11),
н-октилалюминийбис(2,6-ди-трет-бутилфеноксид) (SA12),
н-октилалюминий ди(этил(1-нафтил)амид) (SA13),
этилалюминийбис(трет-бутилдиметилсилоксид) (SA14),
этилалюминий ди(бис(триметилсилил)амид) (SA15),
этилалюминий бис(2,3,6,7-дибензо-1-азациклогептанамид) (SA16),
н-октилалюминий бис(2,3,6,7-дибензо-1-азациклогептанеамид) (SA17),
н-октилалюминий бис(диметил(трет-бутил)силоксид (SA18),
этилцинк (2,6-дифенилфеноксид) (SA19) и
этилцинк (трет-бутоксид) (SA20).
Примеры 1-4. Сравнительные примеры A-C
Общие условия высокопроизводительной параллельной полимеризации
Полимеризации проводят, используя высокопроизводительный реактор параллельной полимеризации (PPR), доступный от Symyx technologies, Inc. и эксплуатируемый по существу согласно патентам США 6248540, 6030917, 6362309, 6306658 и 6316663. Сoполимеризации этилена проводят при 130°C и 200 фунт на кв. дюйм (1,4 МПа) с этиленом по требованию с использованием 1,2 эквивалента сокатализатора 1 на основе всего используемого катализатора (1,1 эквивалента, когда присутствует MMAO). Ряд полимеризаций проводят в параллельном работающем под давлением реакторе (PPR), составленном из 48 отдельных реакторных ячеек в наборе 6×8, которые снабжены предварительно взвешенными стеклянными пробирками. Рабочий объем каждой реакторной ячейки 6000 мкл. В каждой ячейке контролируют температуру и давление и обеспечивают перемешивание отдельными перемешивающими лопастями. Мономерный газ и гасящий газ подают непосредственно в установку PPR и регулируют автоматическими клапанами. Жидкие реагенты роботизировано добавляют в каждую реакторную ячейку шприцами, и растворителем в резервуаре являются смешанные алканы. Порядок добавления: растворитель из смешанных алканов (4 мл), сомономер этилена и 1-октена (1 мл), сокатализатор 1 или смесь сокатализатор 1/MMAO, челночный агент и катализатор или смесь катализаторов. Когда используют смесь сокатализатора 1 и MMAO или смесь двух катализаторов, реагенты предварительно смешивают в небольшом сосуде перед добавлением в реактор. Когда реагент не включают в эксперимент, указанный порядок добавления выстраивают иным образом. Полимеризации проводят приблизительно 1-2 минуты, пока не будут достигнуты заданные уровни потребления этилена. После гашения реакции СО реакторы охлаждают и стеклянные пробирки выгружают. Пробирки переносят в установку центрифуга/вакуумная сушилка и сушат в течение 12 часов при 60°С. Пробирки, содержащие высушенный полимер, взвешивают, и разность между этим весом и весом тары представляет выход полимера. Результаты сведены в таблицу 1. В таблице 1 и где-либо еще в заявке сравнительные примеры обозначены звездочкой (*).
Примеры 1-4 демонстрируют синтез линейных блок-сополимеров по данному изобретению, о чем свидетельствует формирование имеющего очень узкое MWD по существу мономодального coполимера, когда присутствует DEZ, и бимодального имеющего широкое молекулярномассовое распределение продукта (смеси раздельно полученных полимеров) в отсутствие DEZ. Благодаря тому факту, что катализатор (A1), как известно, внедряет больше октена чем катализатор (B1), различные блоки или сегменты полученных coполимеров по изобретению являются различимыми на основе разветвления или плотности.
Можно видеть, что полимеры, полученные по изобретению, имеют относительно узкую полидисперсность (Mw/Mn) и большее содержание блок-сополимера (тримера, тетрамера или более высокого) чем полимеры, полученные в отсутствие челночного агента.
Дальнейшие данные, характеризующие полимеры в таблице 1, определены со ссылкой на чертежи. Более конкретно результаты DSC и ATREFF показаны далее.
Кривая DSC на фиг.3 для полимера из примера 1 показывает температуру плавления (Tm) 115,7°C с теплотой плавления 158,1 Дж/г. Соответствующая кривая CRYSTAF показывает наивысший пик при 34,5°C с площадью пика 52,9 процента. Разность между DSC Tm и Tcrystaf равна 81,2°C.
Кривая DSC на фиг.4 для полимера из примера 2 показывает пик с температурой плавления (Tm) 109,7°C с теплотой плавления 214,0 Дж/г. Соответствующая кривая CRYSTAF показывает наивысший пик при 46,2°C с площадью пика 57,0 процентов. Разность между DSC Tm и Tcrystaf равна 63,5°C.
Кривая DSC на фиг.5 для полимера из примера 3 показывает пик с температурой плавления (Tm) 120,7°C с теплотой плавления 160,1 Дж/г. Соответствующая кривая CRYSTAF показывает наивысший пик при 66,1°C с площадью пика 71,8 процента. Разность между DSC Tm и Tcrystaf равна 54,6°C.
Кривая DSC на фиг.6 для полимера из примера 4 показывает пик с температурой плавления (Tm) 104,5°C с теплотой плавления 170,7 Дж/г. Соответствующая кривая CRYSTAF показывает наивысший пик при 30°C с площадью пика 18,2 процента. Разность между DSC Tm и Tcrystaf равна 74,5°C.
Кривая DSC на фиг.22 (сравнительный пример A) показывает температуру плавления (Tm) 90,0°C с теплотой плавления 86,7 Дж/г. Соответствующая кривая CRYSTAF показывает наивысший пик при 48,5°C с площадью пика 29,4 процента. Обе эти величины согласуются с полимером, который является низким по плотности. Разность между DSC Tm и Tcrystaf равна 41,8°C.
Кривая DSC на фиг.23 (Сравнительный пример B) показывает температуру плавления (Tm) 129,8°C с теплотой плавления 237,0 Дж/г. Соответствующая кривая CRYSTAF показывает наивысший пик при 82,4°C с площадью пика 83,7 процентов. Обе эти величины согласуются с полимером, который является высоким по плотности. Разность между DSC Tm и Tcrystaf равна 47,4°C.
Кривая DSC на фиг.24 (Сравнительный пример C) показывает температуру плавления (Tm) 125,3°C с теплотой плавления 143,0 Дж/г. Соответствующая кривая CRYSTAF показывает наивысший пик при 81,8°C с площадью пика 34,7 процента, а также более низкий кристаллический пик при 52,4°C. Разделение на два пика согласуется с присутствием высоко кристаллического и низко кристаллического полимера. Разность между DSC Tm и Tcrystaf равна 43,5°C.
Примеры 5-19. Сравнительные примеры D-F. Непрерывная полимеризация в растворе. Катализатор Al/B2+DEZ
Непрерывные полимеризации в растворах проводят в управляемом компьютером реакторе-автоклаве, снабженном внутренней мешалкой. Очищенный растворитель из смеси алканов (Isopar™ E, доступный от ExxonMobil, Inc.), этилен при 2,70 фунт/час (1,22 кг/час), 1-октен и водород (когда используют) подают в реактор емкостью 3,8 л, снабженный рубашкой для регулирования температуры и внутренней термопарой. Подачу растворителя в реактор измеряют массовым расходомером. Диафрагменный насос переменной производительности регулирует скорость потока растворителя и давление в реакторе. При разгрузке насоса боковой поток отбирают, чтобы обеспечить приливные потоки для линий впрыскивания катализатора и сокатализатора 1 и реакторной мешалки. Эти потоки измеряют массовыми расходомерами Micro-Motion и регулируют распределительными клапанами или регулировкой вручную игольчатых клапанов. Остальной растворитель объединяют с 1-октеном, этиленом и водородом (когда используют) и подают в реактор. Массовый расходомер используют для доставки водорода в реактор, когда необходимо. Температуру раствора растворитель/мономер регулируют, используя теплообменник перед входом в реактор. Этот поток поступает в донную часть реактора. Растворы каталитических компонентов дозируют с помощью насосов и массовых расходомеров, объединяют с проталкивающим катализатор растворителем и вводят в донную часть реактора. Реактор работает при заполнении жидкостью под давлением 500 фунтов на кв. дюйм (3,45 МПа) при энергичном перемешивании. Продукт удаляют через выпускные линии на верху реактора. Все выпускные линии из реактора снабжены паровым спутником и изолированы. Полимеризацию прекращают добавлением небольшого количества воды в выпускную линию наряду с какими-либо стабилизаторами или другими добавками и пропусканием смеси через статический смеситель. Продуктовый поток затем нагревают, пропуская через теплообменник перед удалением летучих примесей. Полимерный продукт извлекают путем экструзии с помощью экструдера с отводом газов и охлаждаемого водой гранулятора. Подробности процесса и результаты находятся в таблице 2. Выборочные свойства полимера представлены в таблице 3.
Полученные полимеры испытывают путем DSC и ATREFF, как с предыдущими примерами. Результаты следующие.
Кривая DSC на фиг.7 (полимер примера 5) показывает пик с температурой плавления (Tm) 119,6°C с теплотой плавления 60,0 Дж/г. Соответствующая кривая CRYSTAF показывает наивысший пик при 47,6°C с площадью пика 59,5 процента. Дельта между DSC Tm и Tcrystaf равна 72,0°C.
Кривая DSC на фиг.8 (полимер примера 6) показывает пик с температурой плавления (Tm) 115,2°C с теплотой плавления 60,4 Дж/г. Соответствующая кривая CRYSTAF показывает наивысший пик при 44,2°C с площадью пика 62,7 процента. Дельта между DSC Tm и Tcrystaf равна 71,0°C.
Кривая DSC на фиг.9 (полимер примера 7) показывает пик с температурой плавления 121,3°C с теплотой плавления 69,1 Дж/г. Соответствующая кривая CRYSTAF показывает наивысший пик при 49,2°C с площадью пика 29,4 процента. Дельта между DSC Tm и Tcrystaf равна 72,1°C.
Кривая DSC на фиг.10 (полимер примера 8) показывает пик с температурой плавления (Tm) 123,5°C с теплотой плавления 67,9 Дж/г. Соответствующая кривая CRYSTAF показывает наивысший пик при 80,1°C с площадью пика 12,7 процента. Дельта между DSC Tm и Tcrystaf равна 43,4°C.
Кривая DSC на фиг.11 (полимер примера 9) показывает пик с температурой плавления (Tm) 124,6°C с теплотой плавления 73,5 Дж/г. Соответствующая кривая CRYSTAF показывает наивысший пик при 80,8°C с площадью пика 16,0 процентов. Дельта между DSC Tm и Tcrystaf равна 43,8°C.
Кривая DSC на фиг.12 (полимер примера 10) показывает пик с температурой плавления (Tm) 115,6°C с теплотой плавления 60,7 Дж/г. Соответствующая кривая CRYSTAF показывает наивысший пик при 40,9°C с площадью пика 52,4 процента. Дельта между DSC Tm и Tcrystaf равна 74,7°C.
Кривая DSC на фиг.13 (полимер примера 11) показывает пик с температурой плавления (Tm) 113,6°C с теплотой плавления 70,4 Дж/г. Соответствующая кривая CRYSTAF показывает наивысший пик при 39,6°C с площадью пика 25,2 процента. Дельта между DSC Tm и Tcrystaf равна 74,1°C.
Кривая DSC на фиг.14 (полимер примера 12) показывает пик с температурой плавления (Tm) 113,2°C с теплотой плавления 48,9 Дж/г. Соответствующая кривая CRYSTAF не показывает пика, равного или выше 30°C. (Tcrystaf для целей дополнительного расчета устанавливают поэтому при 30°C). Дельта между DSC Tm и Tcrystaf равна 83,2°C.
Кривая DSC на фиг.15 (полимер примера 13) показывает пик с температурой плавления (Tm) 114,4°C с теплотой плавления 49,4 Дж/г. Соответствующая кривая CRYSTAF показывает наивысший пик при 33,8°C с площадью пика 7,7 процента. Дельта между DSC Tm и Tciystaf равна 84,4°C.
Кривая DSC на фиг.16 (полимер примера 14) показывает пик с температурой плавления (Tm) 120,8°C с теплотой плавления 127,9 Дж/г. Соответствующая кривая CRYSTAF показывает наивысший пик при 72,9°C с площадью пика 92,2 процента. Дельта между DSC Tm и Tciystaf равна 47,9°C.
Кривая DSC на фиг.17 (полимер примера 15) показывает пик с температурой плавления (Tm) 114,3°C с теплотой плавления 36,2 Дж/г. Соответствующая кривая CRYSTAF показывает наивысший пик при 32,3°C с площадью пика 9,8 процента. Дельта между DSC Tm и Tcrystaf равна 82,0°C.
Кривая DSC на фиг.18 (полимер примера 16) показывает пик с температурой плавления (Tm) 116,6°C с теплотой плавления 44,9 Дж/г. Соответствующая кривая CRYSTAF показывает наивысший пик при 48,0°C с площадью пика 65,0 процентов. Дельта между DSC Tm и Tcrystaf равна 68,6°C.
Кривая DSC на фиг.19 (полимер примера 17) показывает пик с температурой плавления (Tm) 116,0°C с теплотой плавления 47,0 Дж/г. Соответствующая кривая CRYSTAF показывает наивысший пик при 43,1°C с площадью пика 56,8 процента. Дельта между DSC Tm и Tcrystaf равна 72,9°C.
Кривая DSC на фиг.20 (полимер примера 18) показывает пик с температурой плавления (Tm) 120,5°C с теплотой плавления 141,8 Дж/г. Соответствующая кривая CRYSTAF показывает наивысший пик при 70,0°C с площадью пика 94,0 процентов. Дельта между DSC Tm и Tcrystaf равна 50,5 °C.
Кривая DSC на фиг.21 (полимер примера 19) показывает пик с температурой плавления (Tm) 124,8°C с теплотой плавления 174,8 Дж/г. Соответствующая кривая CRYSTAF показывает наивысший пик при 79,9°C с площадью пика 87,9 процента. Дельта между DSC Tm и Tcrystaf равна 45,0°C.
Кривая DSC на фиг.25 (сравнительный пример D) показывает пик с температурой плавления (Tm) 37,3°C с теплотой плавления 31,6 Дж/г. Соответствующая кривая CRYSTAF не показывает пика, равного и выше 30°C. Обе эти величины согласуются с полимером, который имеет низкую плотность. Дельта между DSC Tm и Tcrystaf равна 7,3°C.
Кривая DSC на фиг.26 (сравнительный пример E) показывает пик с температурой плавления (Tm) 124,0°C с теплотой плавления 179,3 Дж/г. Соответствующая кривая CRYSTAF показывает наивысший пик при 79,3°C с площадью пика 94,6 процента. Обе эти величины согласуются с полимером, который имеет высокую плотность. Дельта между DSC Tm и Tcrystaf равна 44,6°C.
Кривая DSC на фиг.27 (сравнительный пример F) показывает пик с температурой плавления (Tm) 124,8°C с теплотой плавления 90,4 Дж/г. Соответствующая кривая CRYSTAF показывает наивысший пик при 77,6°C с площадью пика 19,5 процента. Разъединение между двумя пиками согласуется с присутствием и высококристаллического, и низкокристаллического полимера. Дельта между DSC Tm и Tcrystaf равна 47,2°C.
Испытание физических свойств
Образцы полимеров оценивают по физическим свойствам, таким как свойства высокотемпературной стойкости, что подтверждают температурным испытанием ТМА, сила слипания гранул, высокотемпературное восстановление, высокотемпературная остаточная деформация при сжатии и отношение динамических модулей упругости, G'(25°C)/G'(100°C). Некоторые коммерчески доступные полимеры включают в испытания: сравнительный пример G* представляет по существу линейный coполимер этилен/l-октен (AFFINITY™ KC8852G, доступный от The Dow Chemical Company), сравнительный пример H* представляет эластомерный по существу линейный coполимер этилен/1-октен (AFFINITY™ EG8100, доступный от The Dow Chemical Company), Сравнительный пример I представляет по существу линейный coполимер этилен/1-октен (Affinity PL 1840, доступный от The Dow Chemical Company), сравнительный пример J представляет гидрированный триблок-coполимер стирол/бутадиен/стирол (Kraton™ G1652, доступный от KRATON Polymers), сравнительный пример K* представляет термопластичный вулканизат (TPV, полиолефиновая смесь, содержащая диспергированный в ней сшитый эластомер). Результаты представлены в таблице 4.
В таблице 4 сравнительный пример F* (который является физической смесью двух полимеров, полученной одновременными полимеризациями с использованием катализаторов А1 и В1) имеет температуру пенетрации глубиной 1 мм 70°С, тогда как примеры 5-9 имеют температуру пенетрации глубиной 1 мм 100°С или выше. Кроме того, примеры 10-19 все имеют температуру пенетрации глубиной 1 мм более чем 85°С, причем большинство имеют температуру ТМА 1 мм более чем 90°С и даже более чем 100°С. Это показывает, что новые полимеры имеют улучшенную стабильность размеров и при более высоких температурах по сравнению с физической смесью. Сравнительный пример J* (коммерческий SEBS) имеет хорошую температуру ТМА 1 мм около 107°С, но имеет очень плохую (при высокой температуре 70°С) остаточную деформацию после сжатия около 100 процентов и он также терпит неудачу по восстановлению (образец разрушен) во время высокотемпературного (80°С) восстановления после 300-процентного напряжения. Таким образом, примеры полимеров имеют уникальное сочетание свойств, недоступное даже для некоторых коммерчески доступных высокоэффективных термопластичных эластомеров.
Подобным образом таблица 4 показывает низкое (хорошее) отношение динамического модуля упругости, G'(25°С)/G'(100°С), равное 6 или менее для полимеров по изобретению, тогда как физическая смесь (сравнительный пример F*) имеет отношение динамического модуля упругости 9 и статистический сополимер этилен/октен (сравнительный пример G) подобной плотности имеет отношение динамического модуля упругости величиной на порядок больше (89). Желательно, чтобы отношение динамического модуля упругости полимера было бы настолько близким к 1, насколько возможно. Такие полимеры будут относительно не подвержены действию температуры, и готовые изделия из таких полимеров можно было бы успешно использовать в широком температурном диапазоне. Эта особенность отношения динамического модуля упругости и независимости от температуры особенно полезны в областях применения эластомеров, таких как составы эффективных под давлением адгезивов.
Данные в таблице 4 демонстрируют, что полимеры по изобретению имеют улучшенные характеристики по прочности слипания гранул. В частности, пример 5 имеет прочность слипания гранул 0 МПа, что означает их свободную сыпучесть в условиях испытания в сравнении со сравнительными примерами F* и G*, которые обнаруживают значительное слипание. Прочность слипания важна, так как бестарная перевозка полимеров, имеющих большую силу слипания, может иметь результатом комкование и слеживание продукта при хранении или транспортировке, что приводит к затруднению обращения с ним.
Остаточная деформация при сжатии при высокой температуре (70°С) для полимеров по изобретению является, в основном, хорошей, имеющей значение обычно менее чем около 80 процентов, предпочтительно менее чем около 70 процентов и особенно предпочтительно менее чем около 60 процентов. В противоположность этому сравнительные примеры F, G, H и J, все имеют остаточную деформацию при сжатии при 70°С 100 процентов (максимально возможная величина, указывающая на отсутствие восстановления). Хорошая высокотемпературная остаточная деформация при сжатии (низкие численные величины) особенно необходима в таких применениях, как прокладки, оконные профили, о-кольцевые прокладки и тому подобное.
2. измерен при 38°С в течение 12 часов
Таблица 5 показывает результаты испытаний механических свойств для новых полимеров, а также различных сравнительных полимеров при температурах окружающей среды. Можно видеть, что данные полимеры имеют очень хорошее сопротивление истиранию при испытании в соответствии с ISO 4649, обычно показывая потерю объема менее чем около 90 мм3, предпочтительно менее чем около 80 мм3 и особенно предпочтительно менее чем около 50 мм3. В этом испытании более высокие числа показывают более высокую потерю объема и, следовательно, более низкую стойкость к истиранию.
Прочность на разрыв, которую измеряют прочностью на разрыв при испытании при растяжении образцов с надрезом полимеров по изобретению, обычно равна 1000 мДж или выше, как показано в таблице 5. Прочность на разрыв полимеров по изобретению может быть такой высокой как 3000 мДж или даже такой высокой как 5000 мДж. Сравнительные примеры обычно имеют прочность на разрыв не выше чем 750 мДж.
Таблица 5 также показывает, что полимеры по изобретению имеют улучшенное напряжение при сжатии при 150-процентной деформации (продемонстрировано более высокими величинами напряжения при сжатии), чем некоторые сравнительные примеры. Сравнительные примеры F, G и Н имеют величину напряжения при сжатии при 150-процентной деформации 400 кПа или менее, тогда как полимеры по изобретению имеют величины напряжения при сжатии при 150-процентной деформации 500 кПа (пример 11) до такой высокой как около 1100 кПа (пример 17). Полимеры, имеющие более высокие величины напряжения при сжатии при 150-процентной деформации были бы очень полезны для упругих применений, таких как эластичные волокна и ткани, особенно нетканые материалы. Другие применения включают подгузники, гигиенические принадлежности и медицинские корсажи предметов одежды, такие как небольшие детали и эластичные повязки.
Таблица 5 также показывает, что релаксация напряжений (при 50-процентной деформации) также является усовершенствованной (меньшей) для полимеров по изобретению по сравнению, например, со сравнительным примером G. Более низкая релаксация напряжений означает, что полимер сохраняет свою силу лучше в применениях, таких как подгузники и другие предметы одежды, где желательно сохранение упругих свойств в течение продолжительных периодов времени при температурах тела.
Оптическое испытание
Оптические свойства, представленные в таблице 6, даны на основе формованных прессованием пленок по существу без ориентирования. Оптические свойства полимеров могут изменяться в широких пределах, благодаря изменению в размерах кристаллов, являющегося результатом изменения количества челночного агента цепи, используемого при полимеризации.
Экстракции мульти-блок-сополимеров
Проводят исследования экстракции полимеров из примеров 5,7 и сравнительного примера Е. В экспериментах образец полимера взвешивают в стеклянном фриттованном наконечнике для экстракции и вставляют в экстрактор типа Kumagawa. Экстрактор с образцом продувают азотом и в круглодонную колбу на 500 мл загружают 350 мл простого диэтилового эфира. Колбу затем вставляют в экстрактор. Простой эфир нагревают при перемешивании. Отмечают время, когда простой эфир начинает конденсироваться на наконечнике, и дают возможность происходить экстракции в атмосфере азота в течение 24 часов. В это время нагревание прекращают и раствору дают возможность остывать. Остающийся в экстракторе простой эфир возвращают в колбу. Простой эфир в колбе выпаривают в вакууме при температуре окружающей среды и полученные твердые вещества продувают до сухости азотом. Остаток переносят во взвешенную колбу, используя последовательные смывы гексаном. Объединенные смывы гексана затем выпаривают с другой продувкой азотом и остаток сушат в вакууме в течение ночи при 40°С. Какой-либо остающийся в экстракторе простой эфир продувают до сухости азотом.
Во вторую чистую круглодонную колбу загружают 350 мл гексана и затем присоединяют ее к экстрактору. Гексан нагревают до кипения с возвращением флегмы при перемешивании и поддерживают при кипении с возвращением флегмы в течение 24 часов после того, как заметят начало конденсации на наконечнике. Нагревание затем прекращают и колбе позволяют остыть. Какой-либо гексан, остающийся в экстракторе, переносят обратно в колбу. Гексан удаляют выпариванием в вакууме при температуре окружающей среды и какой-либо остаток, остающийся в колбе переносят во взвешенную колбу, используя последовательные смывы гексаном. Гексан в колбе выпаривают продувкой азотом и остаток сушат в вакууме в течение ночи при 40°С.
Образец полимера, остающийся в наконечнике, после экстракций переносят из наконечника во взвешенную колбу и сушат в вакууме в течение ночи при 40°С. Результаты приведены в таблице 7.
Изготовление и испытание изделия
Волокна
Образцы полимера из примера 11, примера 17 и сравнительного примера G формуют в комплексный пучок из 24 волокон с круглыми поперечными сечениями на линии формования волокна (Fourn), снабженной двадцатью четырьмя 25х1 мм многоканальными мундштуками, температура прядильной головки 260°С, температура расплава 302°С и скорость мотальной машины 70 м/мин. Другие условия формования перечислены в таблице 8. Титр полученного пучка приблизительно 95-100 денье (г/9000 м).
Волокна сшивают пропусканием шесть раз через машину сшивки электронным пучком, работающую при дозе электронного пучка 32 KGy/проход, давая общую дозу 192 KGy. Между проходами волокна охлаждают до -10°С.
Поведение при растяжении полученных несшитых и сшитых волокон измеряют согласно BISTA Test Methods for Bare Elastic Yarns, Chapter 6: Tensile Properties using Option C clamps and Option A test speed. Предел прочности и удлинение при разрыве сообщаются в среднем из 5 повторов. Поведение восстановления сшитых волокон также измеряют, используя BISTA Test Methods for Bare Elastic Yarns, Chapter 7: Viscoelastic Properties. Procedure A, где волокно циклически нагружают до 300-процентной деформации. Долговременную деформацию в процентах рассчитывают на начало 6-го цикла, как указано в методе испытания. Результаты циклического поведения при 300-процентной деформации для волокон, полученных из полимера примера 17, показаны на фиг.30.
Релаксацию напряжений сшитых волокон измеряют от 10-процентной деформации при переменных температурах 21°С и 40°С. В эксперименте 13 петель волокон пучка с длиной окружности 324 мм устанавливают на машине для испытания Instron 2 крючками, получая в результате измерительную базу 162 мм. Образец растягивают до 10-процентной деформации при скорости 100-процентное удлинение/минута при 21°С и затем выдерживают 10 минут. Затем следует термическая обработка: 10 минут при 40°С в водяной бане, 10 минут при 21°С на воздухе, 10 минут при 40°С в водяной бане и 10 минут при 21°С на воздухе. Время для переноса образца между водяной баней и камерой воздушного охлаждения 6 секунд. Во время всего процесса нагрузку отслеживают. Процентное изменение нагрузки от нагрузки при 35 минутах и нагрузки при 45 минутах рассчитывают, используя формулу:
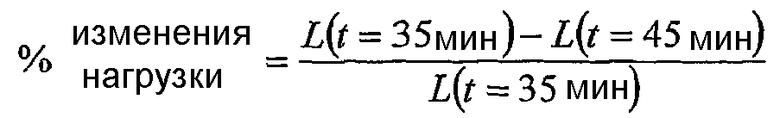
где L(t=35 мин) и L(t=45 мин) представляют нагрузки при 35 минутах и 45 минутах, соответствующие средним периодам воздействия по меньшей мере 40°С водяной бани и 21°С воздуха, соответственно. Результаты показаны на фиг.31. Свойства волокон также сведены в таблицу 9.
Денье)
денье)
В результате сшивки и волокон, полученных в примере 11, и сравнительных волокон G их прочность увеличилась с некоторой потерей в удлинении. Оба примера показали сходную долговременную деформацию приблизительно 135 процентов. На фиг.31 пример 11 показывает более низкую релаксацию напряжений, чем сравнительный пример G, а также является менее чувствительным к температуре. Процентное изменение нагрузки между 40°С (35 мин) и 21°С (35 мин) приведено в таблице 9. Волокно, полученное в примере 11, обнаруживает только 4-процентное изменение нагрузки, тогда как волокно сравнительного примера G показывает 25-процентное изменение. Низкая чувствительность к температуре при релаксации напряжений важна для обеспечения продолжительной сохранности бобин волокна. Высокая чувствительность к температуре при релаксации напряжений может приводить к порче бобин во время хранения в помещении без климат-контроля, так как волокно попеременно релаксирует и сжимается из-за температурных колебаний. Это может приводить к таким проблемам, как плохое поведение при размотке волокна и разрывы волокна при последующей переработке волокна.
Пены
Образцы полимеров (пример 5 и коммерчески доступного сополимера этилен/винилацетат Elvax™ 460, содержащего 18 процентов ацетата и имеющего индекс расплава 2, доступного от DuPont Inc., сравнительного примера L) компаундируют в расплаве с порообразователем на основе азида (AZ130, азодикарбонамидный порообразователь, доступный от Uniroyal, Inc.,), оксидом цинка, стеариновой кислотой и сшивающим агентом на основе пероксида (ди-трет-бутил-перокси-изопропил-бензол пероксид, 40 процентов активного вещества на носителе из диоксида кремния, пероксид Perkadox™ 1440, доступный от Azo Nobel, Inc.,), формуют прессованием в пластинки и дают возможность увеличиться в объеме.
Условия компаундирования: валковая мельница @ 130°С, 10 мин.
Условия формования и вспенивания. Листы из валковой мельницы предварительно нагревают до 90°С в печи в течение 15 минут, затем подают в форму, предварительно нагретую до 180°С, прессуют (механический затвор) и отверждают при этой температуре в течение 10 минут. После удаления образцам дают возможность увеличиваться в объеме. Подробный состав (части по массе) приведен в таблице 10.
Испытание свойств на полученных вспененных стренгах проводят следующим образом.
Плотность пены измеряют согласно ASTM 792, сопротивление истиранию измеряют согласно ISO 4649, усадку измеряют при комнатной температуре после воздействия на образец 70°С в течение 40 минут, согласно SATRA PM70, остаточную деформацию при сжатии измеряют при комнатной температуре после 1,5 и 24 часов воздействия на образец температуры 50°С в течение 6 часов согласно ISO 815, твердость А по Шору измеряют согласно ISO 868, сопротивление раздиру измеряют согласно стандартам SATRA ТМ65 и предел прочности при растяжении и удлинение измеряют согласно DIN 53504. Результаты представлены в таблице 11.
Результаты таблицы 11 показывают, что термические и механические свойства сшитой пены, полученной из примера 7, лучше свойств полученной подобным образом пены из сравнительного примера L. В частности, пена, полученная из примера 7, имеет более низкую усадку, более низкую остаточную деформацию при сжатии и более высокое сопротивление раздиру и удлинение, чем сравнительная пена. Эти свойства делают полимеры по изобретению хорошо подходящими для применения во многих областях применения с высокими эксплуатационными качествами, таких как обувные подошвы, настилы и конструкционные материалы.
Сшитые электронным пучком пленки
Сформованные прессованием пленки толщиной 0,4 мм сшивают в атмосфере азота с помощью устройства для сшивки облучением электронным пучком (Sterigenics, San Diego). Суммарную дозу электронного пучка 22,4 Мрад дают последовательно за 7 проходов через электронный пучок при 3,2 Мрад на проход. Все образцы показали уровень геля между 75 и 90 процентами, как измерено согласно ASTM D-2765. Механические свойства облученных пленок по существу не затронуты сшивкой. Хотя примеры по изобретению и сравнительные примеры обнаруживают сходные окончательные свойства, примеры по изобретению показывают более высокие процентное восстановление, напряжение сжатия и релаксацию напряжений, чем сравнительные примеры. Результаты представлены в таблице 12.
Модификация ударной вязкости полипропилена
Серию смесей изотактического полипропилена с модифицированной ударной вязкостью, содержащего 20 процентов по массе эластомера этилен/октен, получают в устройстве для приготовления смесей Haake, снабженном двухчервячным 18 мм экструдером Leistritz (L/D=30), двухшнековым питателем K-TRON K2VT20, двумя охлаждающими сборниками с циркуляцией охлажденной воды и чоппером c 4 лезвиями для резки стренг Berlyn PEL-2. Во всех смесях использовали полипропилен РР-314-02ZhPP, доступный от The Dow Chemical Co., имеющий MFR 2 дг/мин, измеренную согласно ASTM D1238 (230°С, 2,16 кг).
Устройство для циркуляции воды присоединено к рубашке приемного отверстия экструдера и установлено на 20°С, чтобы удерживать полимер от расплавления и закупоривания питающего отверстия. Температурные зоны экструдера установлены на 120, 165, 190, 230 и 230°С, соответственно. Экструзионная головка установлена на 230°С. Перед экструзией крышку, снабженную линией азота, помещают на верх питающего бункера. Область перехода от разгрузочного отверстия питателя к конусу питающего отверстия экструдера уплотняют тяжелой алюминиевой фольгой. Экструдер предварительно нагревают, калибруют и прогоняют пустым в течение нескольких минут, пропуская азот через систему, чтобы выдуть из нее кислород. Трехкилограммовые образцы для смешивания при расплавлении готовят смешиванием вручную объединенных компонентов в мешке из пластика перед экструзией.
Из полимерных образцов литьем под давлением формуют бруски для испытания и испытывают бруски с надрезом на ударную вязкость по Изоду при 23°С согласно ASTM D-256 и на модуль упругости при изгибе согласно ASTM D-790. Условия литьевого формования следующие. Образцы формуют литьем под давлением при температуре расплава 243°С, время заполнения 6,7 с при давлении 3400 фунтов на кв. дюйм (23 МПа), время удерживания 12 с при давлении 3400 фунтов на кв. дюйм (23 МПа) и общее время цикла 28 секунд. Отдельные подробности и результаты даны в таблице 13.
2. ENGAGE™ VP8770: 0,885 г/см3, 1 г/10 мин (I2), доступный от The Dow Chemical Co.
Результаты таблицы 13 показывают, что мульти-блок-сополимеры по изобретению являются высоко эффективными в качестве модификаторов ударной вязкости, когда их смешивают с изотактическим полипропиленом. Удивительно, что образец, изготовленный с полимером из примера 5, полученный с более высоким отношением челночный агент цепи/суммарный катализатор, результатом чего явилось увеличенное число блоков на полимерную молекулу (более "монолитный" полимер), показывает даже более низкий модуль и ударную вязкость, чем образец b, который является компаундированным с полимером примера 8, который является менее "монолитным" полимером. Это наблюдение указывает на то, что степень блочности, которая регулируется количеством челночного агента цепи, в мульти-блок-сополимерах по изобретению может сильно затрагивать баланс жесткость/ударовязкость полимерных смесей.
Дополнительные факты различия в свойствах полимерных смесей очевидны из сравнения фиг.51-53, которые являются микроскопическими изображениями атомного усиления, полученных на микротоме окрашенных тетраоксидом осмия срезов образцов сформованных литьем под давлением пластинок b, a и d, соответственно. На микрофотографиях темные области представляют эластомер сополимера этилен/октен, тогда как более светлые области представляют полипропиленовую гомополимерную матрицу. Из микрофотографий можно видеть, что мульти-блок-сополимеры, полученные с низкими отношениями CSA к катализатору (сополимеры низкой "блочности"), неожиданно образуют морфологию типа сердцевина-оболочка в смесях (фиг.51). Мульти-блок-сополимеры с высокими отношениями CSA (фиг.52) обнаруживают домены, по-видимому, твердого эластомера подобные по виду результатам, полученным с использованием традиционных этилен/октеновых модификаторов ударной вязкости (фиг.53).
Преимущества обладания уникальной морфологией, показанные на фиг.51 (морфология окклюдированного каучука), включают превосходный баланс жесткость/ударовязкость, высокую эффективность ударопрочности (уменьшенное количество каучука для достижения заданной ударовязкости) и более высокое сопротивление истиранию (более низкую склонность к стрессовому побелению). Более того, коэффициент преломления эластомера легко изменить регулированием количества присутствующих окклюзий. Это дает возможность сравнять коэффициенты преломления эластомера и матричного полимера, результатом являются смеси, обнаруживающие улучшенный баланс оптической прозрачности, жесткости, ударовязкости и сопротивления истиранию. Кроме того, такие смеси (то есть смеси, содержащие мульти-блок-сополимеры более низкой блочности) обнаруживают более высокую температуру термического разложения, улучшенную морфологическую стабильность (сохранение полимерных свойств после множества стадий обработки). Ранее такие свойства были достижимы только в смеси, содержащей дополнительные компоненты, такие как трехкомпонентые смеси эластомера, полиэтилена высокой плотности и изотактического полипропилена.
Получение образцов пленок раздувом
Образцы мульти-блок-сополимера (пример 14) и традиционного сополимера этилен/октен (сравнительный пример I) формуют в однослойные пленки на лабораторной линии раздува пленок. Полимерные образцы расплавляют в экструдере, пропускают через кольцевую головку, расширяют воздухом, охлаждают и разрезают на ориентированные по двум направлениям пленки. Условия формирования пленок представлены в таблице 14.
Образцы полученных пленок испытывают на нормированное сопротивление раздиру пленки в поперечном направлении (CD) и в направлении выхода из машины (MD) согласно ASTM D1922, свойства слипания согласно ASTM D3354-96 и коэффициент трения (COF) согласно ASTM D1894-01. Результаты находятся в таблице 15.
мин
МD
Пленка, полученная из полимера примера 15, показывает более высокую прочность на раздир (CD и MD), чем пленка, полученная из полимера сравнительного примера I. Дополнительно, она обнаруживает более сбалансированную прочность на раздир (меньшее отношение CD/MD), чем сравнительная пленка. И прочность слипания, и COF для пленки, изготовленной из примера 14, ниже чем в сравнительном примере I. Такое сочетание свойств пленки указывает на то, что пленки, изготовленные из мульти-блок-сополимеров по изобретению, имеют более высокое сопротивление раздиру и более высокое сопротивление слипанию, чем пленки, изготовленные из традиционных сополимеров этилен/октен.
Приготовление маслонаполненных полимерных смесей
Компаундированные смеси готовят при 190°C в предварительно нагретом смесителе Haake Rheomix™ 600 объемом 69 мл. Роторы вращаются при приводной скорости 50 об/мин, когда полимер добавляют и перерабатывают в расплав. Путем отслеживания вращающего момента смесителя контролируют плавление. Как только плавление полимера завершится, в расплавленный полимер добавляют шприцем парафиновое масло (RENOIL™ 625, доступное от Renkert Oil, Inc.). Как только добавление масла завершится, затвор опускают на расплав и перемешивание продолжают в течение 15 минут. Общая масса масла и полимера равна 55 граммам. Роторы затем останавливают, чан открывают и полученную смесь удаляют, расплющивают и охлаждают в прессе.
Смешанные и несмешанные полимеры формуют прессованием в пластинки 5"×5"×0,125"(125×125×3 мм) на прессе для ламинирования при следующих условиях:
1) 3 минуты без давления при 190°C,
2) 2 минуты при усилии затвора 30000 фунтов (133 кН) при 190°C и затем
3) 3 минуты при 25°C при усилии затвора 30000 фунтов (13 кН).
Полученные пластинки испытывают на твердость по Шору А ручным твердомером и на термостойкость (TMA). Результаты по твердости представлены средней величиной из 5 измерений при длительностях 1 и 5 секунд в случайных точках на поверхности пластинки. Результаты представлены в таблице 16.
Результаты таблицы 16 показывают, что полимер по изобретению имеет твердость по Шору А, подобную твердости сравнительного полимера, но показывает приблизительно на 40°С более высокую температуру TMA. Удивительно, что наполненный 30 массовыми процентами масла полимер имеет твердость по Шору А, подобную наполненному 40 массовыми процентами масла сравнительному полимеру, но имеет более чем на 30°С более высокую температуру TMA. Этот результат показывает, что полимер примера 17 обнаруживает более высокое принятие масла и более хорошее сохранение термических и механических свойств, таких как термостойкость, которую измеряют по температуре TMA, и предел прочности на разрыв по сравнению со сравнительным полимером H*. Такое сочетание низкой твердости и высокой температуры ТМА полезно во многих областях применения мягких эластомеров, таких как мягкие при прикосновении формованные изделия и эффективные под давлением адгезивы.
Пример 20. Способ выбора пары катализатор А/челночный агент
Ряд сополимеризаций этилен/1-октен проводят, используя отличающиеся молярные отношения катализатор/челночный агент и превращения мономеров. Сoкатализатором, используемым во всех полимеризациях, является сoкатализатор 2. Полученные полимеры подвергают измерению молекулярной массы (Mw и Mn), используя GPC. Показатель полидисперстности (PDI=Mw/Mn) рассчитывают для каждого полимера. Результаты сведены в таблицу 17 и графически представлены на фиг.32. На фиг.32 линия является статистическим соответствием данным с величиной R2 0,961.
i) 6-мл реакционный сосуд, содержащий вставку в виде стеклянного пузырька, загружают смешанными алканами (2,70 мл) и затем создают давление до 110 фунтов на кв. дюйм (0,77 МПа) этиленом. Октен (100 мкл) и затем смесь coкатализатора (4,2 мМ в толуоле, 0,100 мл, 420 нмоль) и диэтилцинка (10 мкмоль) добавляют шприцем. Катализатор (A) (3,5 мМ в толуоле, 0,100 мл, 350 нмоль) добавляют шприцем. Спустя 15 секунд, реакцию гасят добавлением CO. Стеклянную вставку удаляют и летучие компоненты удаляют в вакууме. Выход полимера=0,0938 г. Mw=14560; Mn=8267; PDI=1,76.
ii) 6-мл реакционный сосуд, содержащий вставку в виде стеклянного пузырька, загружают смешанными алканами (2,70 мл) и затем создают давление до 110 фунтов на кв. дюйм (0,77 МПа) этиленом. Октен (100 мкл) и затем смесь coкатализатора (4,2 мМ в толуоле, 0,100 мл, 420 нмоль) и диэтилцинка (10 мкмоль) добавляют шприцем. Катализатор (A) (3,5 мМ в толуоле, 0,100 мл, 350 нмоль) добавляют шприцем. Через 30 секунд реакцию гасят добавлением CO. Стеклянную вставку удаляют и летучие компоненты удаляют в вакууме. Выход полимера=0,1173 г. Mw=16677; Mn=9774; PDI=1,71.
iii) 6-мл реакционный сосуд, содержащий вставку в виде стеклянного пузырька, загружают смешанными алканами (2,70 мл) и затем создают давление до 110 фунтов на кв. дюйм (0,77 МПа) этиленом. Октен (100 мкл) и затем смесь coкатализатора (4,2 мМ в толуоле, 0,100 мл, 420 нмоль) и диэтилцинка (10 мкмоль) добавляют шприцем. Катализатор (A) (3,5 мМ в толуоле, 0,100 мл, 350 нмоль) добавляют шприцем. Через 51 секунду реакцию гасят добавлением CO. Стеклянную вставку удаляют и летучие компоненты удаляют в вакууме. Выход полимера=0,1360 г. Mw=20557; Mn=12773; PDI=1,61.
iv) 6-мл реакционный сосуд, содержащий вставку в виде стеклянного пузырька, загружают смешанными алканами (2,70 мл) и затем создают давление до 110 фунтов на кв. дюйм (0,77 МПа) этиленом. Октен (100 мкл) и затем смесь coкатализатора (4,2 мМ в толуоле, 0,100 мл, 420 нмоль) и диэтилцинка (10 мкмоль) добавляют шприцем. Катализатор (A) (3,5 мМ в толуоле, 0,100 мл, 350 нмоль) добавляют шприцем. Через 98 секунд реакцию гасят добавлением CO. Стеклянную вставку удаляют и летучие компоненты удаляют в вакууме. Выход полимера=0,1748 г. Mw=26379; Mn=13161; PDI=2,00.
v) 6-мл реакционный сосуд, содержащий вставку в виде стеклянного пузырька, загружают смешанными алканами (2,70 мл) и затем создают давление до 110 фунтов на кв. дюйм (0,77 МПа) этиленом. Октен (100 мкл) и затем смесь coкатализатора (4,2 мМ в толуоле, 0,100 мл, 420 нмоль) и диэтилцинка (10 мкмоль) добавляют шприцем. Катализатор (A) (3,5 мМ в толуоле, 0,100 мл, 350 нмоль) добавляют шприцем. Через 291 секунду реакцию гасят добавлением CO. Стеклянную вставку удаляют и летучие компоненты удаляют в вакууме. Выход полимера=0,2191 г. Mw=33777; Mn=18201; PDI=1,86.
vi) 6-мл реакционный сосуд, содержащий вставку в виде стеклянного пузырька, загружают смешанными алканами (2,70 мл) и затем создают давление до 110 фунтов на кв. дюйм (0,77 МПа) этиленом. Октен (100 мкл) и затем смесь coкатализатора (4,2 мМ в толуоле, 0,100 мл, 420 нмоль) и диэтилцинка (10 мкмоль) добавляют шприцем. Катализатор (A) (3,5 мМ в толуоле, 0,100 мл, 350 нмоль) добавляют шприцем. Через 1201 секунду реакцию гасят добавлением CO. Стеклянную вставку удаляют и летучие компоненты удаляют в вакууме. Выход полимера=0,2681 г. Mw=46539; Mn=24426; PDI=1,91.
Эти результаты показывают, что механизм челночного перемещения цепи (и прямой, и обратный обмен полимерила) между катализатором (A) и челночным агентом цепи, а именно диэтилцинком, имеет место во время полимеризации, благодаря тому факту, что Mn полученного полимера линейно увеличивается с выходом полимера, тогда как PDI остается менее чем или равным величине до двух для всех полимеризаций.
Пример 21. Способ выбора пары катализатор B2/челночный агент
Ряд полимеризаций этилен/1-октен проводят, используя различные молярные отношения катализатор/челночный агент и превращения мономера с coкатализатором 2. Измеряют молекулярную массу (Mw и Mn) полученных полимеров, используя GPC. Показатель полидисперстности (PDI=Mw/Mn) рассчитывают для каждого полимера. Результаты представлены в таблице 18 и графически изображены на фиг.33. На фиг.33 линия является статистическим соответствием данным с величиной R2 0,995.
i) 6-мл реакционный сосуд, содержащий вставку в виде стеклянного пузырька, загружают смешанными алканами (2,334 мл) и затем создают давление до 110 фунтов на кв. дюйм (0,77 МПа) этиленом. Октен (200 мкл) и затем смесь coкатализатора (1,8 мМ в толуоле, 0,233 мл, 419 нмоль) и диэтилцинка (10 мкмоль) добавляют шприцем. Катализатор (B2) (1,5 мМ в толуоле, 0,233 мл, 350 нмоль) добавляют шприцем. Через 18 секунд реакцию гасят добавлением CO. Стеклянную вставку удаляют и летучие компоненты удаляют в вакууме. Выход полимера=0,0542 г. Mw=7626; Mn=5281; PDI=1,44.
ii) 6-мл реакционный сосуд, содержащий вставку в виде стеклянного пузырька, загружают смешанными алканами (2,334 мл) и затем создают давление до 110 фунтов на кв. дюйм (0,77 МПа) этиленом. Октен (200 мкл) и затем смесь coкатализатора (1,8 мМ в толуоле, 0,233 мл, 419 нмоль) и диэтилцинка (10 мкмоль) добавляют шприцем. Катализатор (B2) (1,5 мМ в толуоле, 0,233 мл, 350 нмоль) добавляют шприцем. Через 39 секунд реакцию гасят добавлением CO. Стеклянную вставку удаляют и летучие компоненты удаляют в вакууме. Выход полимера=0,0769 г. Mw=10501; Mn=7523; PDI=1,40.
iii) 6-мл реакционный сосуд, содержащий вставку в виде стеклянного пузырька, загружают смешанными алканами (2,334 мл) и затем создают давление до 110 фунтов на кв. дюйм (0,77 МПа) этиленом. Октен (200 мкл) и затем смесь coкатализатора (1,8 мМ в толуоле, 0,233 мл, 419 нмоль) и диэтилцинка (10 мкмоль) добавляют шприцем. Катализатор (B2) (1,5 мМ в толуоле, 0,233 мл, 350 нмоль) добавляют шприцем. Через 59 секунд реакцию гасят добавлением CO. Стеклянную вставку удаляют и летучие компоненты удаляют в вакууме. Выход полимера=0,1071 г. Mw= 15840; Mn=10971; PDI=1,44.
iv) 6-мл реакционный сосуд, содержащий вставку в виде стеклянного пузырька, загружают смешанными алканами (2,334 мл) и затем создают давление до 110 фунтов на кв. дюйм (0,77 МПа) этиленом. Октен (200 мкл) и затем смесь coкатализатора (1,8 мМ в толуоле, 0,233 мл, 419 нмоль) и диэтилцинка (10 мкмоль) добавляют шприцем. Катализатор (B2) (1,5 мМ в толуоле, 0,233 мл, 350 нмоль) добавляют шприцем. Через 103 секунды реакцию гасят добавлением CO. Стеклянную вставку удаляют и летучие компоненты удаляют в вакууме. Выход полимера=0,1365 г. Mw=21664; Mn=12577; PDI=1,72.
v) 6-мл реакционный сосуд, содержащий вставку в виде стеклянного пузырька, загружают смешанными алканами (2,334 мл) и затем создают давление до 110 фунтов на кв. дюйм (0,77 МПа) этиленом. Октен (200 мкл) и затем смесь coкатализатора (1,8 мМ в толуоле, 0,233 мл, 419 нмоль) и диэтилцинка (10 мкмоль) добавляют шприцем. Катализатор (B2) (1,5 мМ в толуоле, 0,233 мл, 350 нмоль) добавляют шприцем. Через 173 секунды реакцию гасят добавлением CO. Стеклянную вставку удаляют и летучие компоненты удаляют в вакууме. Выход полимера=0,1829 г. Mw=25221; Mn=16245; PDI=1,55.
vi) 6-мл реакционный сосуд, содержащий вставку в виде стеклянного пузырька, загружают смешанными алканами (2,334 мл) и затем создают давление до 110 фунтов на кв. дюйм (0,77 МПа) этиленом. Октен (200 мкл) и затем смесь coкатализатора (1,8 мМ в толуоле, 0,233 мл, 419 нмоль) и диэтилцинка (10 мкмоль) добавляют шприцем. Катализатор (B2) (1,5 мМ в толуоле, 0,233 мл, 350 нмоль) добавляют шприцем. Через 282 секунды реакцию гасят добавлением CO. Стеклянную вставку удаляют и летучие компоненты удаляют в вакууме. Выход полимера=0,2566 г. Mw=35012; Mn=23376; PDI=1,50.
Эти результаты показывают, что механизм челночного перемещения цепи (и прямой, и обратный обмен полимерила) между катализатором (В2) и челночным агентом цепи диэтилцинком имеет место во время полимеризации, благодаря тому факту, что Mn полученного полимера линейно увеличивается с выходом полимера, тогда как PDI остается менее чем два и обычно менее чем 1,5 для всех полимеризаций.
Пример 22. Комбинаторный скрининг пар катализатор/челночный агент
Условия реакции примеров 1-4 по существу повторяют, используя различные катализаторы, coкатализатор 1 и потенциальные челночные агенты. Свыше 500 реакций проводят. Полученные сополимеры этилен/1-октен испытывают на Mn и PDI и скорость производства полимера сравнивают со скоростями, полученными от контроля с использованием ММАО вместо челночного агента. Наилучшие составы затем выбирают на основе наибольшего уменьшения молекулярной массы (Mn), наибольшего уменьшения PDI и наибольшего уменьшения (или действительного увеличения) скорости полимеризации. Выбранные сочетания, показывающие наилучшие результаты (упорядоченные по уменьшению Mn), представлены в таблице 19.
По данным таблицы 19 подходящие сочетания катализатора и челночного агента могут быть выбраны. Следует подчеркнуть, что предпочтительные сочетания катализатора и челночного агента в различных вариантах воплощения могут быть выбраны на основе желательной цели, такой как максимальное уменьшение Mn или усовершенствование скорости производства в сочетании с более умеренным уменьшением Mn. Дополнительно, указанные результаты основаны на единственном сочетании катализатор/челночный агент, тогда как на практике влияние, если это имеет место, присутствия одного или нескольких дополнительных катализаторов и применения условий непрерывной полимеризации следует учитывать при выборе сочетания катализаторов и челночного агента (агентов).
Пример 23. Формирование функционализованного мульти-блок-сополимера
В реактор на 1 л загружают 600 мл сухого дезоксигенированного гексана и 40 ммоль диэтилцинка и нагревают до 100°C в атмосфере азота. В реакторе создают давление до 10 фунтов на кв. дюйм (70 кПа) этиленом. Смесь 10 мкмоль катализатора (Al), 10 мкмоль катализатора (Bl) и 50 микромоль ММAO затем впрыскивают в реактор и этилен подают по потребности, чтобы поддерживать давление 10 фунтов на кв. дюйм (70 кПа) в течение 40 минут. Реактор затем вентилируют, охлаждают до температуры окружающей среды и продувают азотом в течение 20 минут. При энергичном продувании азотом поток воздуха вводят в донную часть реактора в течение 1 часа и полученную суспензию перемешивают еще час. Суспензию продукта реакции затем удаляют из реактора, перемешивают с водой и сушат, получая 25,5 г полимера. Анализ GPC показывает Mw=1271, Mn=1018, Mw/Mn=1,25. Анализ 1H ЯМР показывает 27-процентное превращение возможных оканчивающихся цинком концов цепей.
Примеры 24-28. Сополимеризация этилен/1-бутен
Непрерывные полимеризации в растворах проводят, следуя процедуре, описанной выше для примеров 5-19 со следующими исключениями: используемым сомономером во всех примерах является 1-бутен, а для примеров 25 смесь DEZ и MAO (молярное отношение 99:1) используют в качестве челночного агента цепи (CSA). Подробности процесса и результаты приведены в таблице 19. Можно видеть, что смесь челночных агентов цепи имеет результатом приблизительно 40-процентное улучшение эффективности при получении по существу подобных продуктов (плотность=0,88, I2=2). Свойства выбранных полимеров представлены в таблицах 21-24. Термические свойства полимера следующие.
Кривая DSC на фиг.36 для полимера примера 24 показывает пик с температурой плавления 114,9°С с теплотой плавления 44,1 Дж/г. Соответствующая кривая CRYSTAF показывает наивысший пик при 42,6°С с площадью пика 48,4 процента. Разность между DSC Tm и Tcrystaf равна 72,3°С.
Кривая DSC на фиг.37 для полимера примера 25 показывает пик с температурой плавления 114,5°С с теплотой плавления 41,5 Дж/г. Соответствующая кривая CRYSTAF показывает наивысший пик при 41,0°С с площадью пика 24,2 процента. Разность между DSC Tm и Tcrystaf равна 73,5°C.
Кривая DSC на фиг.38 для полимера примера 26 показывает пик с температурой плавления 116,7°С с теплотой плавления 45,7 Дж/г. Соответствующая кривая CRYSTAF показывает наивысший пик при 40,2°С с площадью пика 6,1 процента. Разность между DSC Tm Tcrystaf равна 76,5°C.
Кривая DSC на фиг.39 для полимера примера 27 показывает пик с температурой плавления 118,4°С с теплотой плавления 47,1 Дж/г. Соответствующая кривая CRYSTAF показывает наивысший пик при 40,2°С с площадью пика 6,1 процента. Разность между DSC Tm и Tcrystaf равна 79,8°С.
Кривая DSC на фиг.40 для полимера примера 28 показывает пик с температурой плавления 121,3°С с теплотой плавления 143,4 Дж/г. Соответствующая кривая CRYSTAF показывает наивысший пик при 74,4°С с площадью пика 96.6 процент. Разность между DSC Tm и Tcrystaf равна 46,9°C.
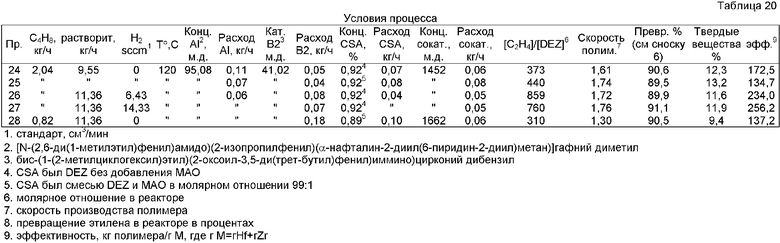
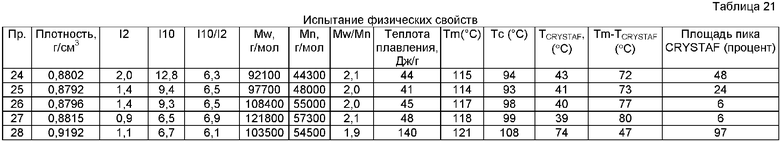
Примеры 29-33, сравнительные примеры М-Р
Условия реакции примеров 1-4 по существу повторяют, чтобы получить сополимеры этилена и разных алифатических сомономеров (1-гексен, 1-октен, 1-декан, 1,5-гексадиен и 4-метил-1-пентен). Используемым челночным агентом цепи является триоктилалюминий (SAS). МАО является замещающим CSA для сравнительных примеров М-Р. Подробности процесса отражены в таблице 25. Свойства полимеров приведены в таблице 26.
Термические свойства полученных полимеров следующие.
Кривая DSC на фиг.41 для полимера примера 29 показывает пик с температурой плавления 121,6°С с теплотой плавления 138,7 Дж/г. Соответствующая кривая CRYSTAF показывает наивысший пик при 61,0°С с площадью пика 17,8 процента. Разность между DSC Tm и Tcrystaf равна 60,6°C.
Кривая DSC на фиг.42 для полимера примера 30 показывает пик с температурой плавления 123,3°С с теплотой плавления 146,3 Дж/г. Соответствующая кривая CRYSTAF показывает наивысший пик при 50,6°С с площадью пика 25,4 процента. Разность между DSC Tm и Tcrystaf равна 72,7°С.
Кривая DSC на фиг.43 для полимера примера 31 показывает пик с температурой плавления 120,7°С с теплотой плавления 160,3 Дж/г. Соответствующая кривая CRYSTAF показывает наивысший пик при 52,3°С с площадью пика 95,1 процента. Разность между DSC Tm и Tcrystaf равна 68,4°С.
Кривая DSC на фиг.44 для полимера примера 32 показывает пик с температурой плавления 122,9°С с теплотой плавления 183,2 Дж/г. Соответствующая кривая CRYSTAF показывает наивысший пик при 64,1°С с площадью пика 95,2 процента. Разность между DSC Tm и Tcrystaf равна 58,7°С.
Кривая DSC на фиг.45 для полимера примера 33 показывает пик с температурой плавления 120,8°С с теплотой плавления 177,9 Дж/г. Соответствующая кривая CRYSTAF показывает наивысший пик при 64,1°С с площадью пика 95,7 процента. Разность между DSC Tm и Tcrystaf равна 56,7°С.
Кривая DSC на фиг.46 для полимера сравнительного примера M* показывает пик с температурой плавления 121,9°С с теплотой плавления 112,3 Дж/г. Соответствующая кривая CRYSTAF показывает наивысший пик при 78,9°С с площадью пика 36,1 процента. Разность между DSC Tm и Tcrystaf равна 43,0°С.
Кривая DSC на фиг.47 для полимера сравнительного примера N* показывает пик с температурой плавления 121,7°С с теплотой плавления 85,5 Дж/г. Соответствующая кривая CRYSTAF показывает наивысший пик при 30,0°С с площадью пика 69,7 процента. Разность между DSC Tm и Tcrystaf равна 91,7°С. Однако следует отметить, что Mw/Mn для этого сравнительного примера равно 15 и значительно больше чем в примерах по изобретению.
Кривая DSC на фиг.48 для полимера сравнительного примера O* показывает пик с температурой плавления 122,6°С с теплотой плавления 134,9 Дж/г. Соответствующая кривая CRYSTAF показывает наивысший пик при 81,1°С с площадью пика 40,4 процента. Разность между DSC Tm и Tcrystaf равна 41,5°С.
Кривая DSC на фиг.49 для полимера сравнительного примера P* показывает пик с температурой плавления 121,9°C с теплотой плавления 148,2 Дж/г. Соответствующая кривая CRYSTAF показывает наивысший пик при 82,8°С с площадью пика 33,3 процента. Разность между DSC Tm и Tcrystaf равна 39,1°С.
Фиг.50 представляет график разности пиковых температур DSC Tm - ТCRYSTAF как функцию энтальпии плавления DSC для примеров 24, 25, 29-33, сравнительных полимеров M-P и коммерчески полученных сополимеров этилен/октен.
Примеры 34-36, сравнительные примеры Q-S
Условия реакции примеров 1-4 по существу повторяют, чтобы получить сополимеры этилена и разных ароматических и циклоалифатических сомономеров (стирол, циклопентен и бицикло[2,2,1]гепт-2-ен (норборнен)). Используемым челночным агентом цепи является диэтилцинк (SA1). ММАО является замещающим CSA для сравнительных примеров Q-S. Подробности полимеризации приведены в таблице 27. Свойства полимеров указаны в таблице 28.
| название | год | авторы | номер документа |
|---|---|---|---|
| СОСТАВ КАТАЛИЗАТОРА, СОДЕРЖАЩИЙ ЧЕЛНОЧНЫЙ АГЕНТ, ДЛЯ ФОРМИРОВАНИЯ МУЛЬТИ-БЛОК-СОПОЛИМЕРА ВЫСШЕГО ОЛЕФИНА | 2005 |
|
RU2375381C2 |
| КОМПОЗИЦИЯ КАТАЛИЗАТОРА, СОДЕРЖАЩАЯ АГЕНТ ЧЕЛНОЧНОГО ПЕРЕНОСА ЦЕПИ ДЛЯ ОБРАЗОВАНИЯ СОПОЛИМЕРА ЭТИЛЕНА | 2005 |
|
RU2359979C2 |
| БЛОК-СОПОЛИМЕРЫ ЭТИЛЕНА/α-ОЛЕФИНОВ | 2006 |
|
RU2409595C2 |
| ВОЛОКНА, ИЗГОТОВЛЕННЫЕ ИЗ СОПОЛИМЕРОВ ПРОПИЛЕНА/α-ОЛЕФИНОВ | 2006 |
|
RU2404299C2 |
| ТЕРМОРЕАКТИВНЫЕ ЭЛАСТОМЕРЫ | 1995 |
|
RU2159779C2 |
| ПОЛИОЛЕФИНОВАЯ СОПОЛИМЕРНАЯ КОМПОЗИЦИЯ, СПОСОБ ПОЛИМЕРИЗАЦИИ (ВАРИАНТЫ), МЕТАЛЛОЦЕНОВЫЙ КАТАЛИЗАТОР, ПЛЕНКА НА ОСНОВЕ КОМПОЗИЦИИ И СМЕСЬ ДВУХ ИЛИ БОЛЕЕ ПОЛИМЕРНЫХ КОМПОНЕНТОВ | 1997 |
|
RU2190632C2 |
| МЕТАЛЛОКОМПЛЕКСЫ, СОДЕРЖАЩИЕ ЗАМЕЩЕННЫЙ ИНДЕНИЛ, И СПОСОБ ПОЛИМЕРИЗАЦИИ ОЛЕФИНОВ | 1996 |
|
RU2175325C2 |
| КОМПЛЕКСЫ МЕТАЛЛОВ, СОДЕРЖАЩИЕ ЛИГАНДЫ 3-АРИЛЗАМЕЩЕННОГО ИНДЕНИЛА, КАТАЛИЗАТОР ПОЛИМЕРИЗАЦИИ ОЛЕФИНОВ И СПОСОБ ПОЛИМЕРИЗАЦИИ | 1997 |
|
RU2186073C2 |
| ПОЛИМЕРИЗАЦИЯ ОЛЕФИНОВ ИЗ РАСТВОРА | 1997 |
|
RU2190627C2 |
| ПОЛИМЕРНЫЕ СМЕСИ ИЗ ИНТЕРПОЛИМЕРОВ ЭТИЛЕН/α-ОЛЕФИН С УЛУЧШЕННОЙ СОВМЕСТИМОСТЬЮ | 2006 |
|
RU2408622C2 |
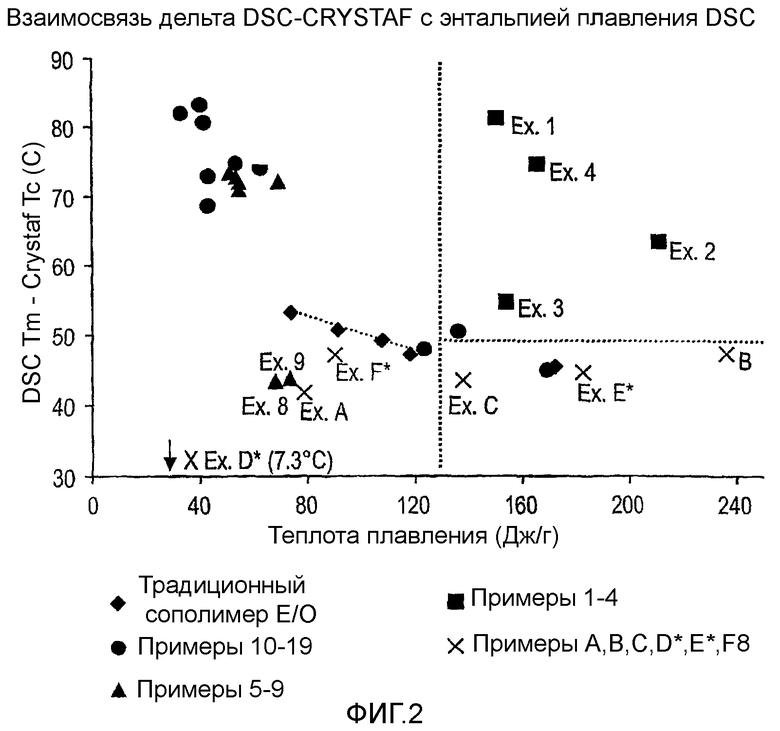
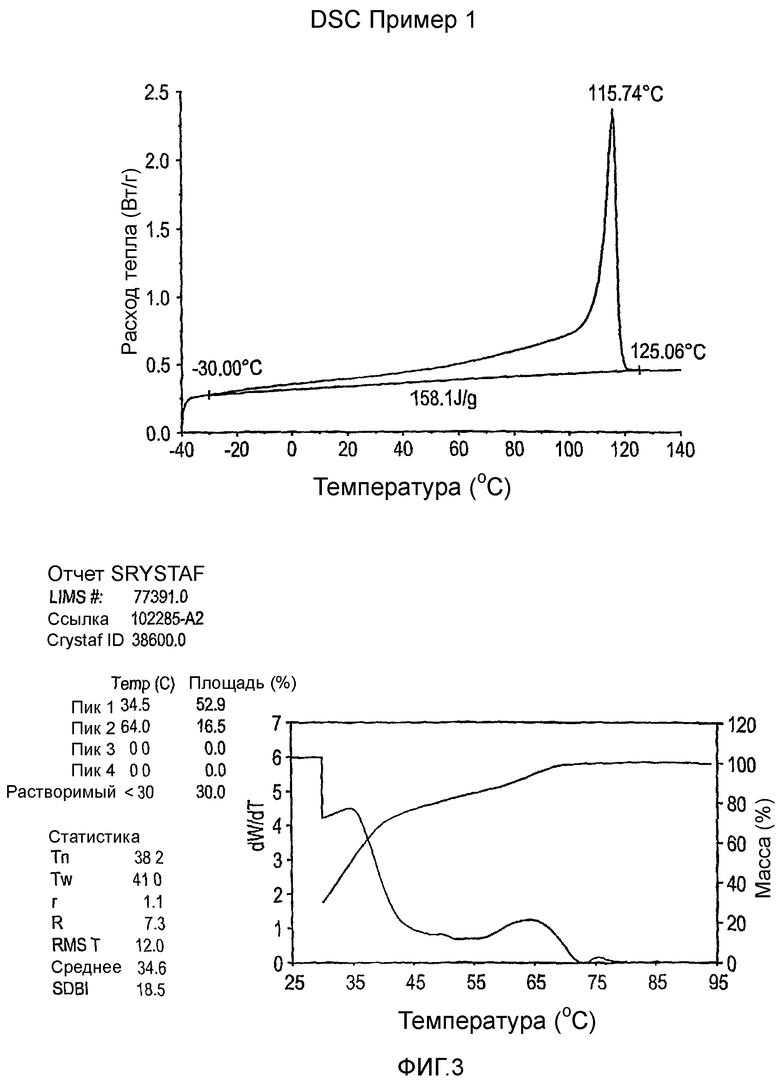
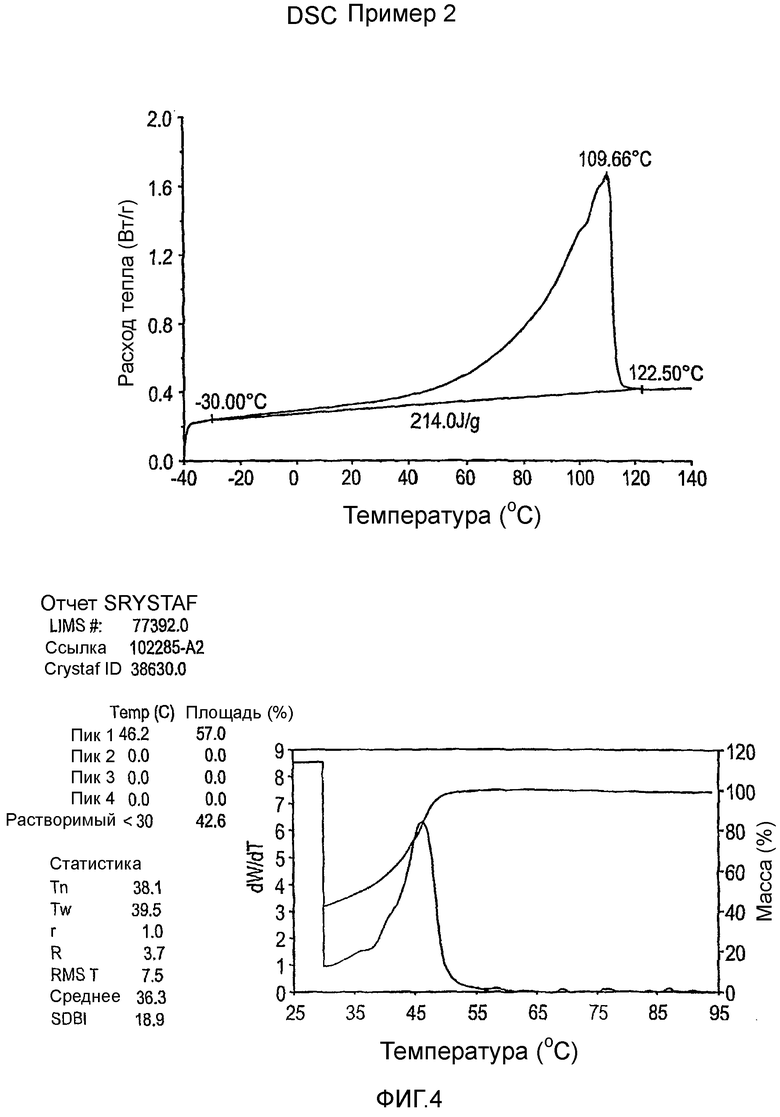
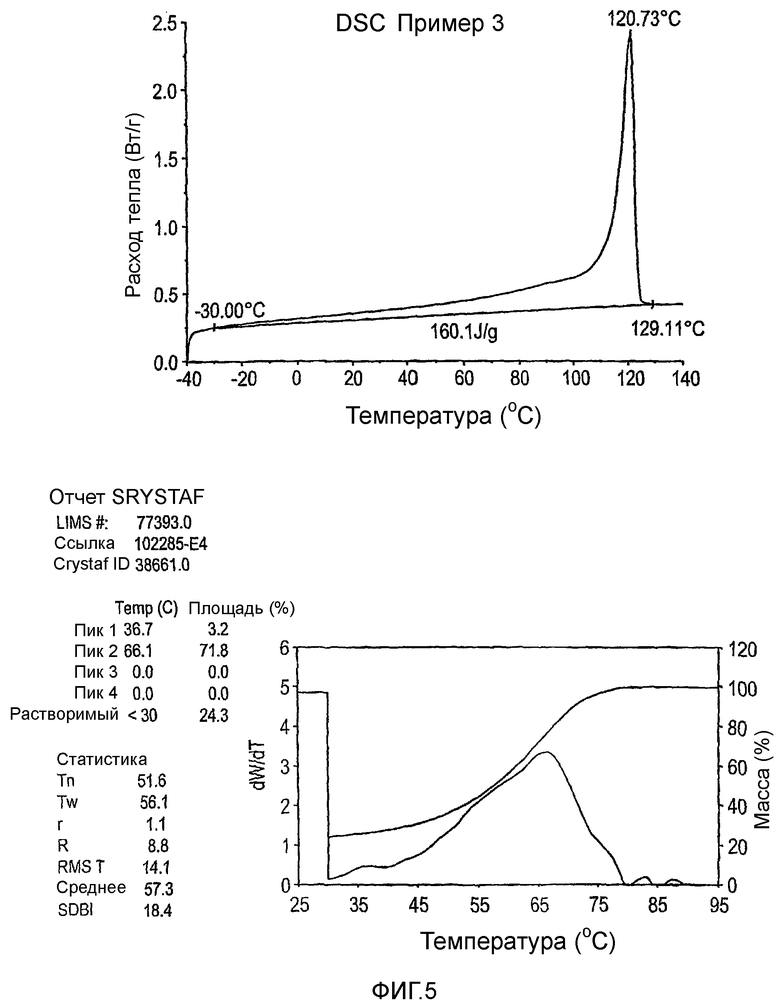
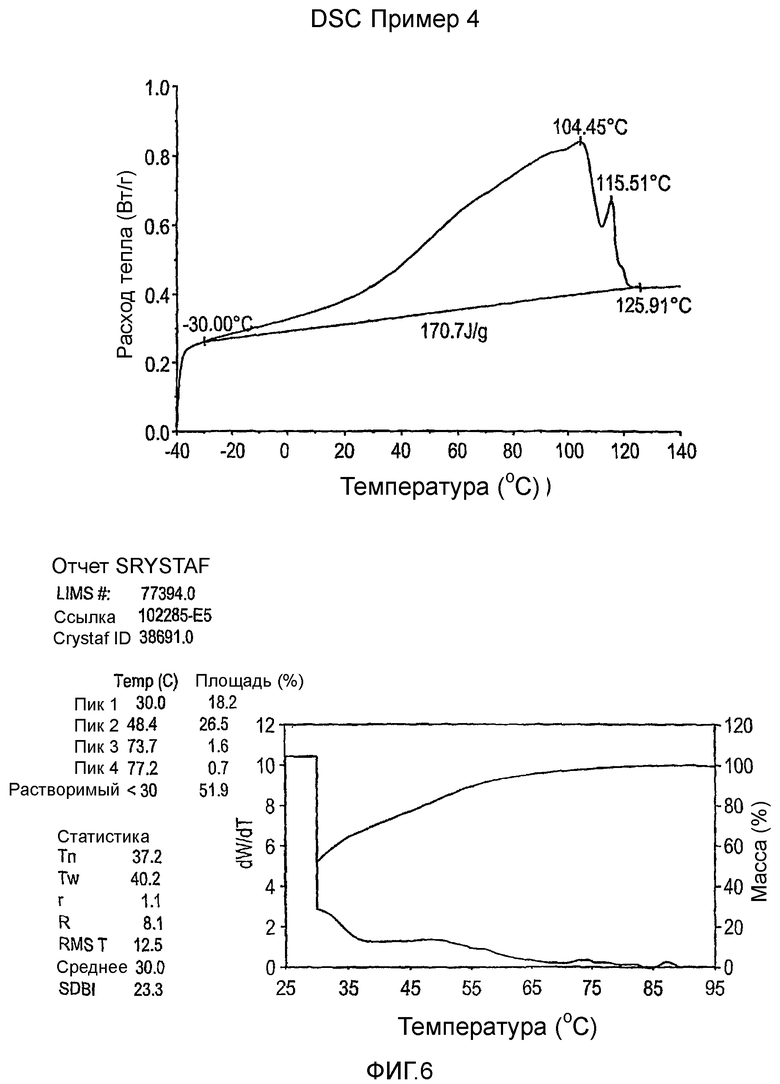
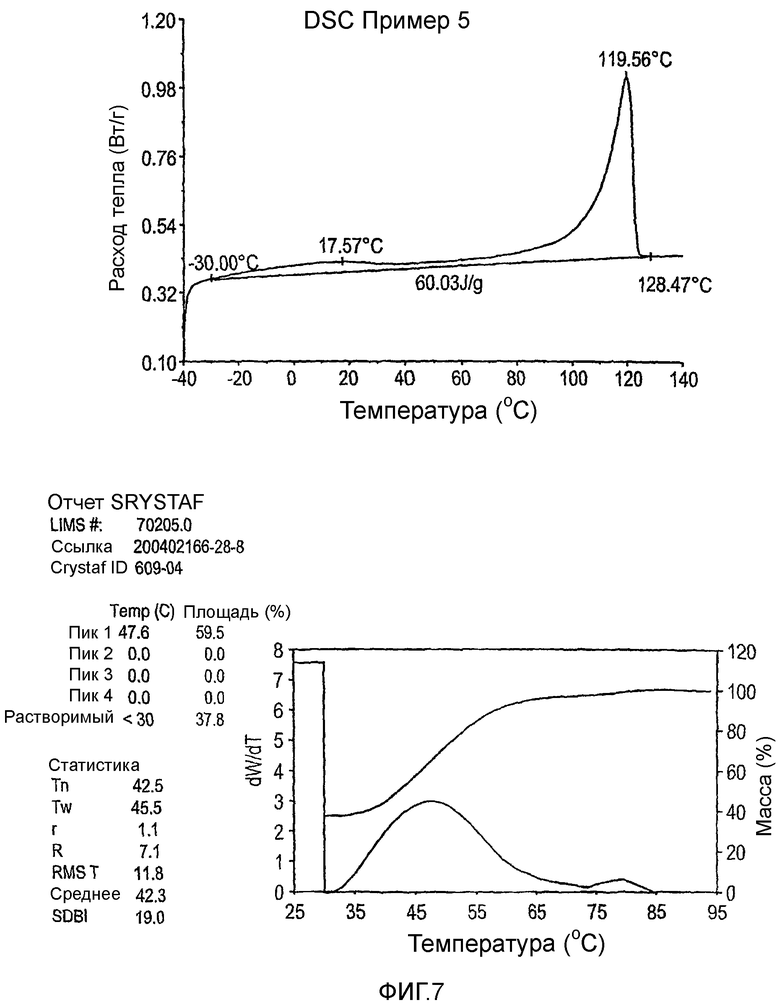
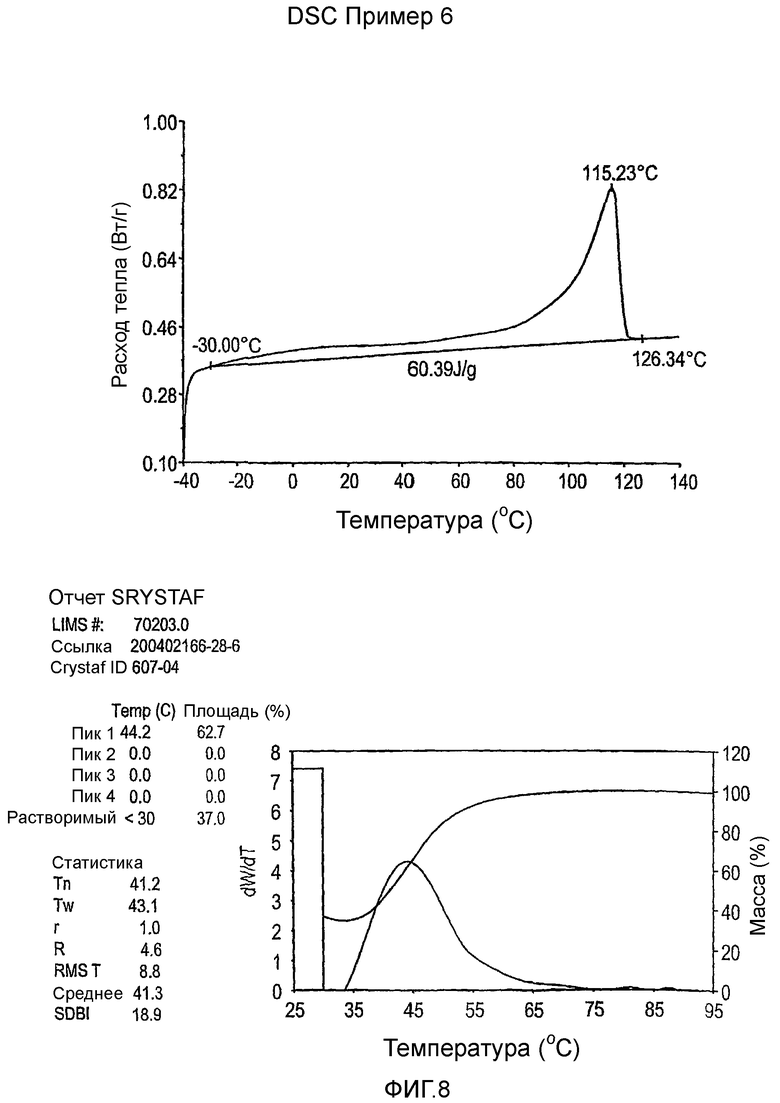
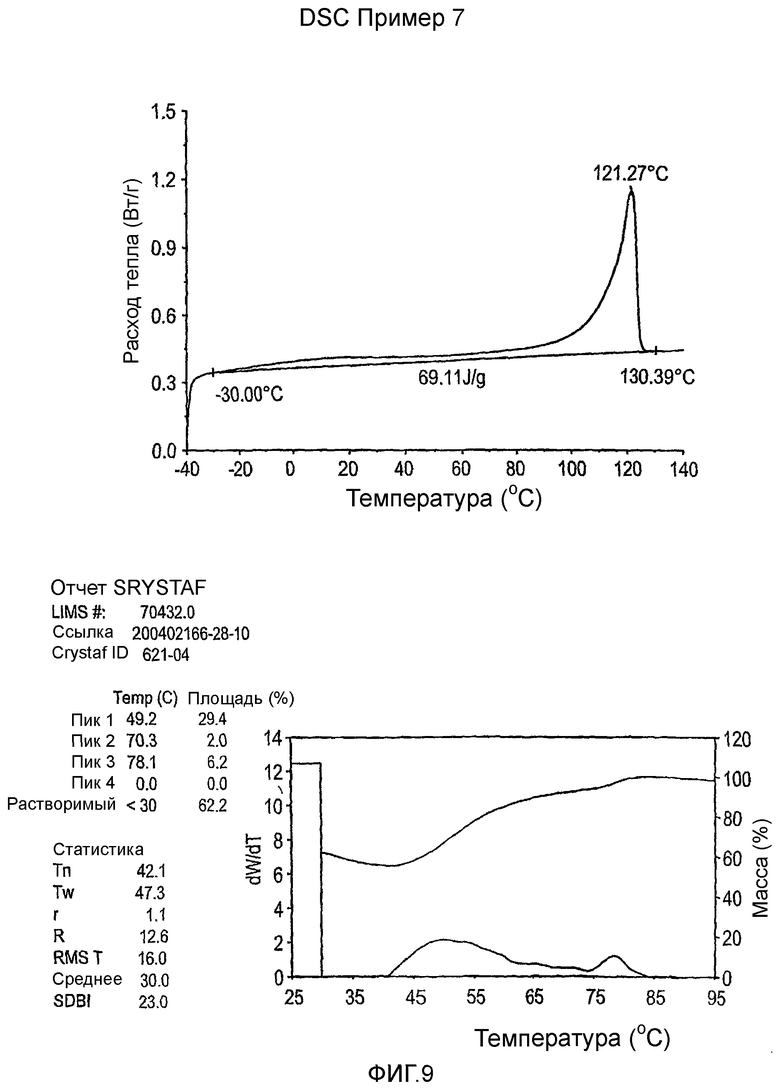
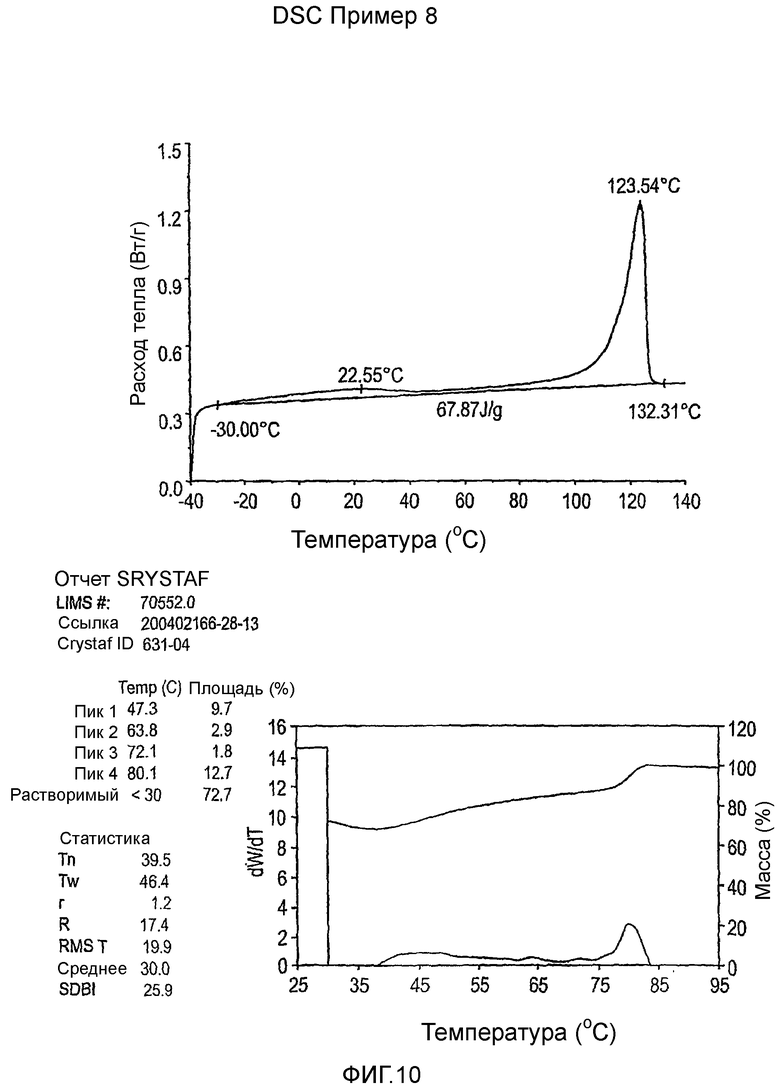
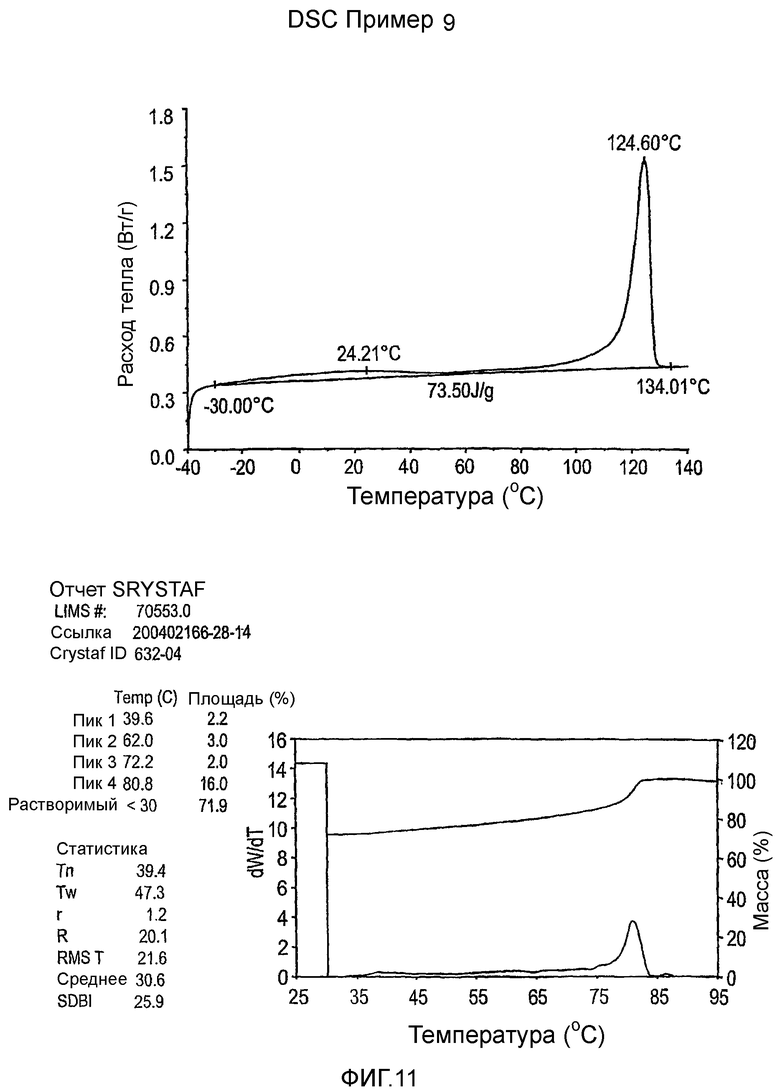
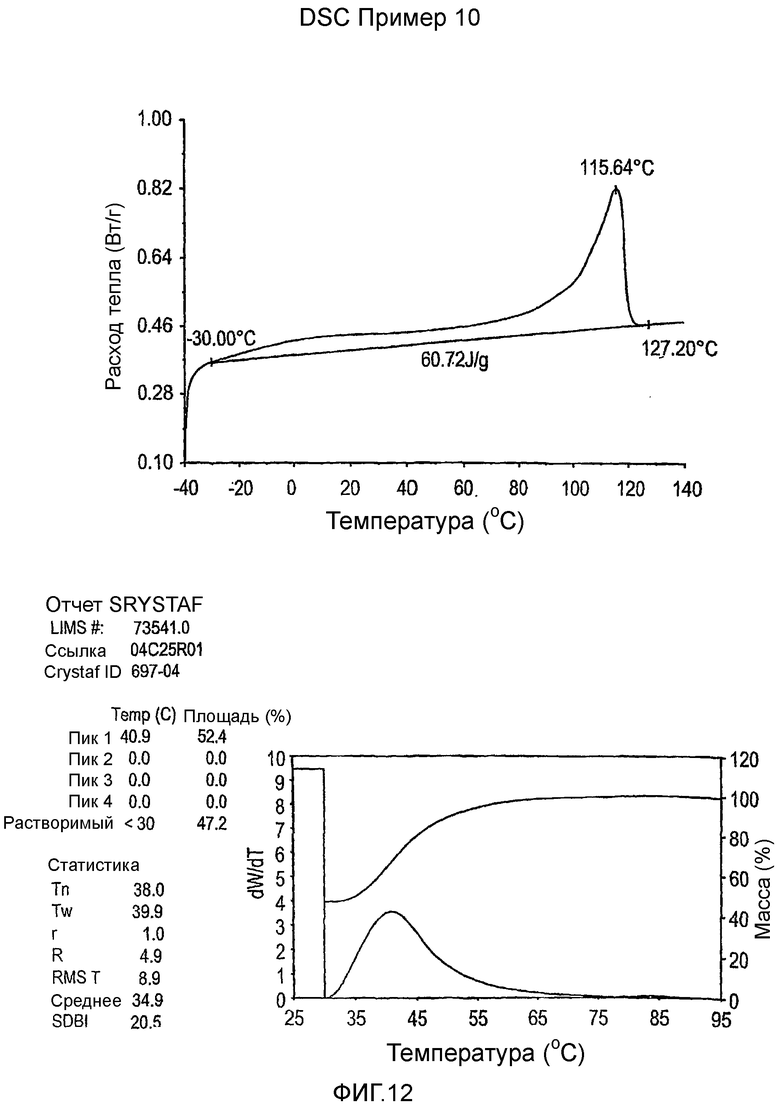
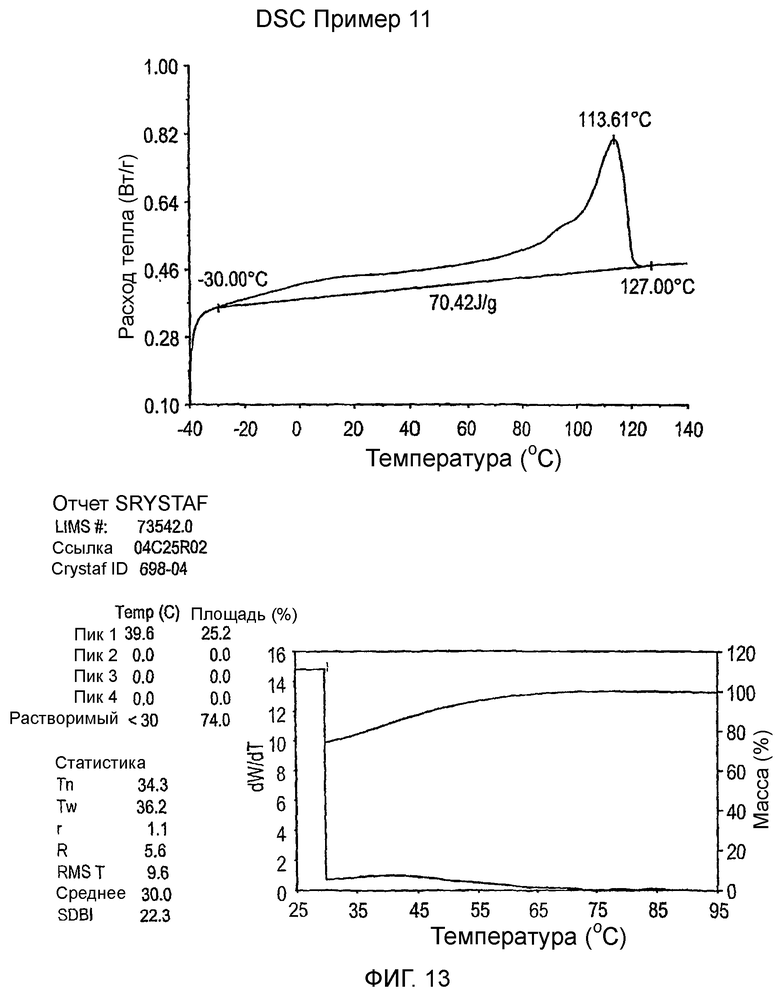
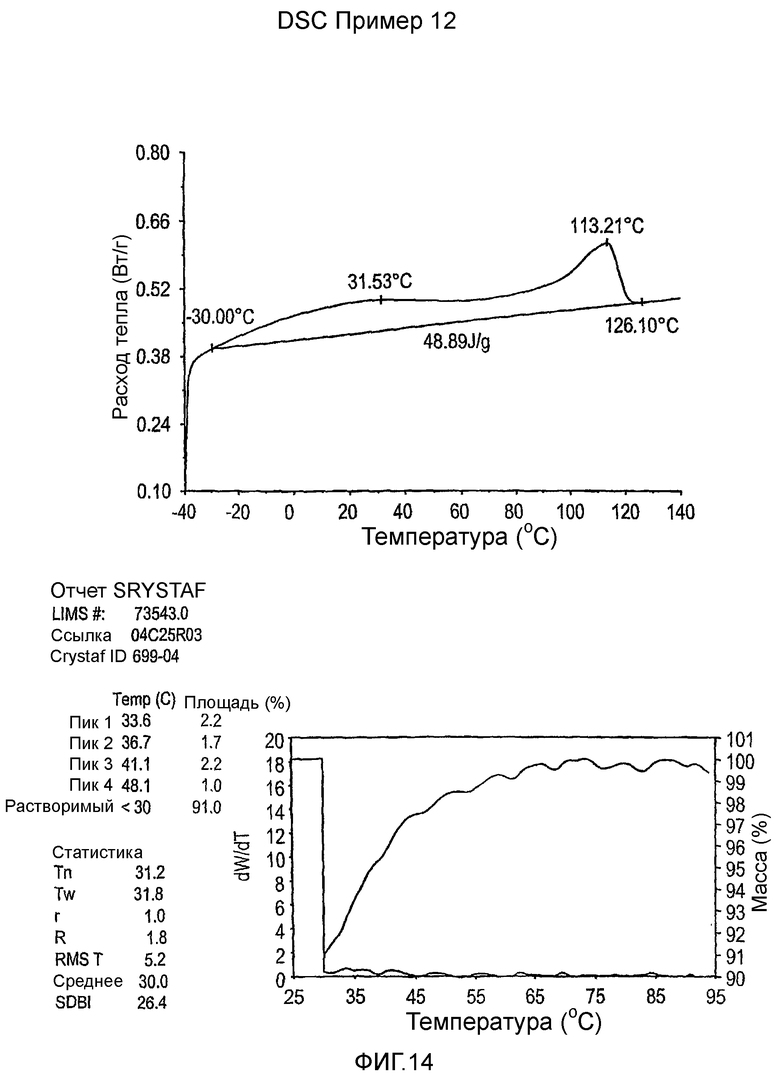
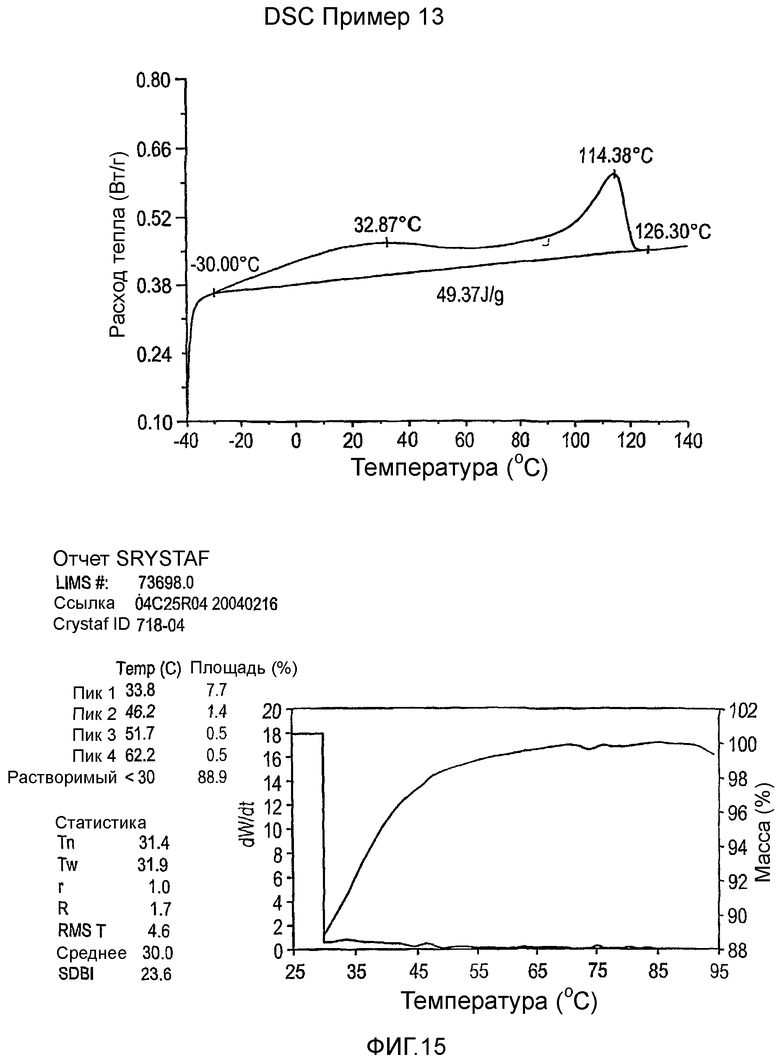
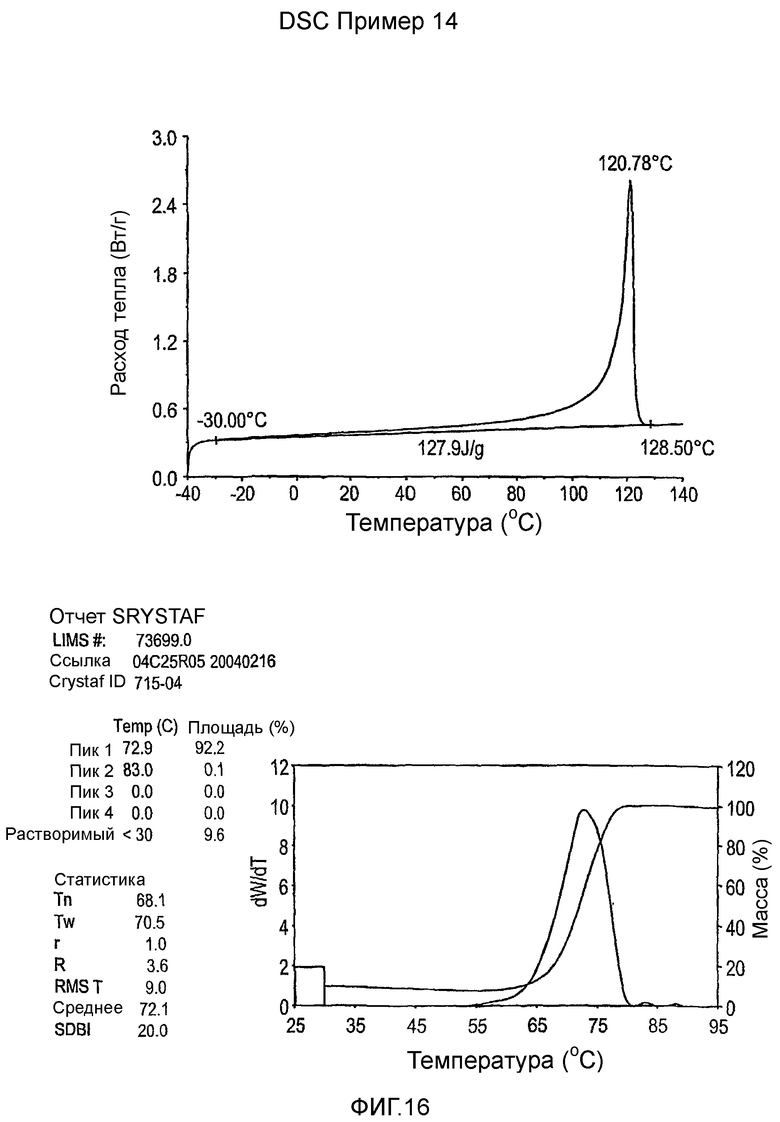
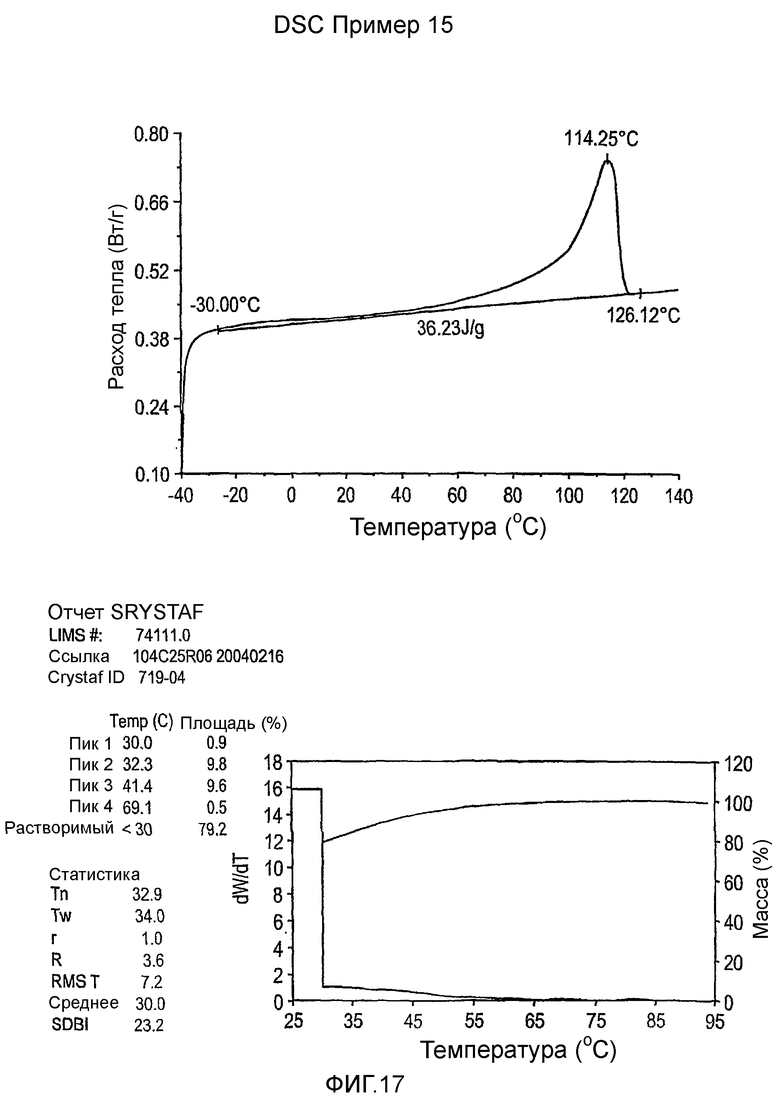
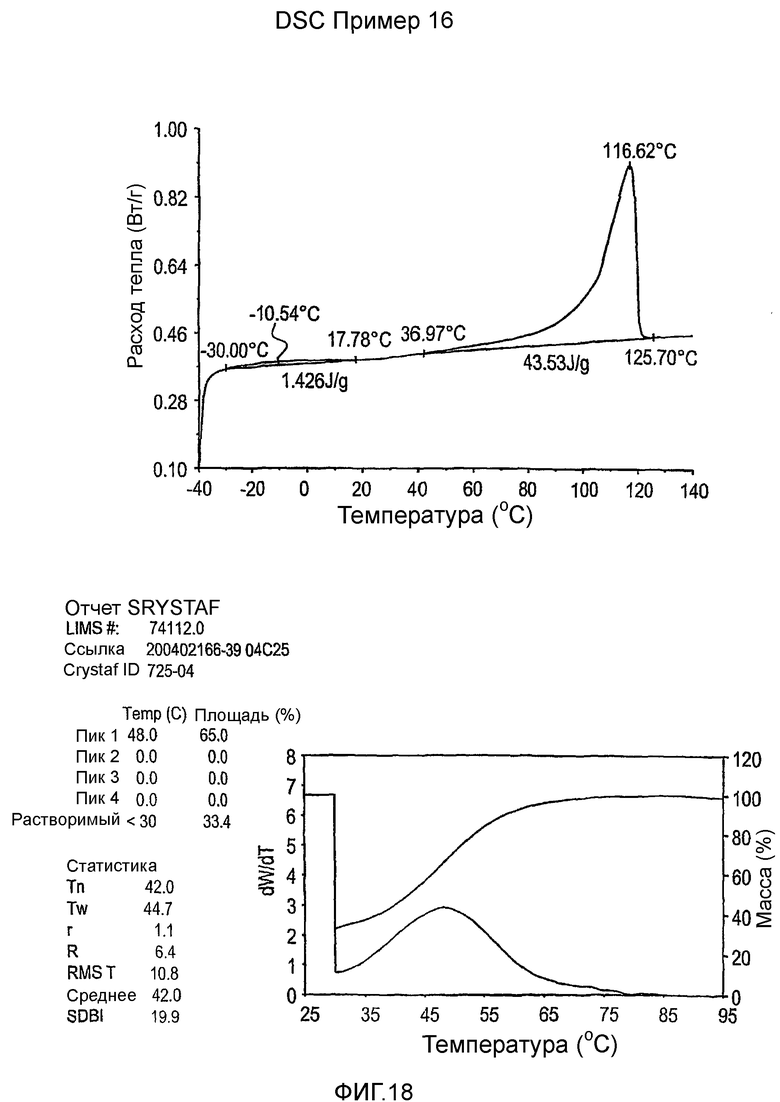
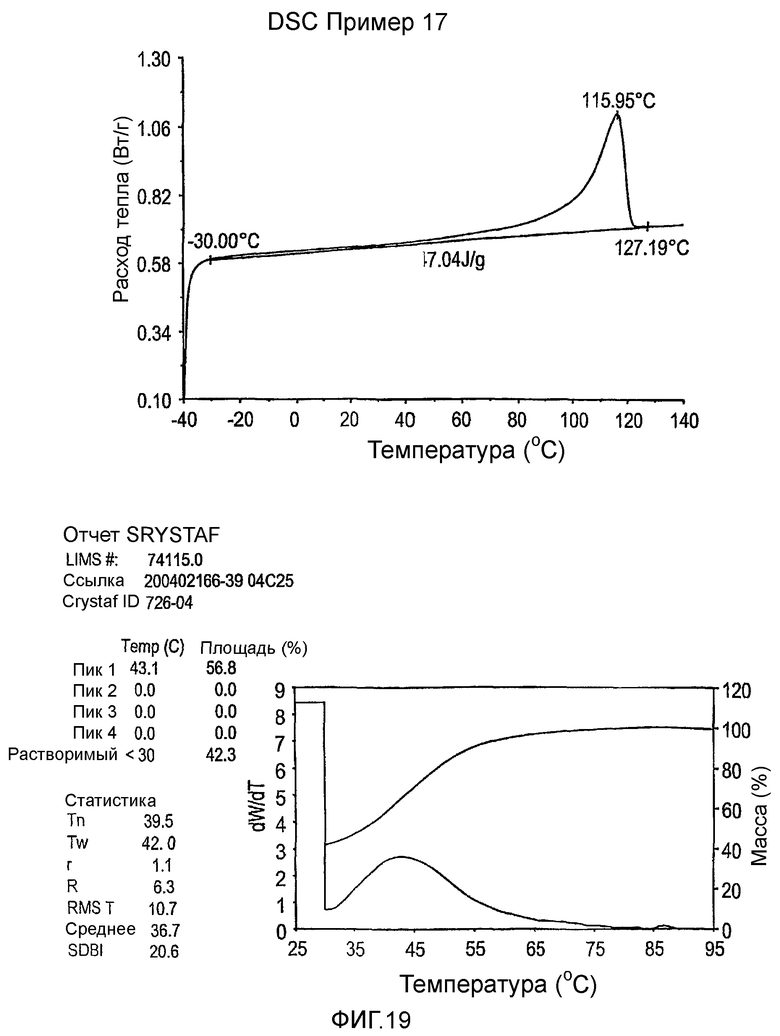
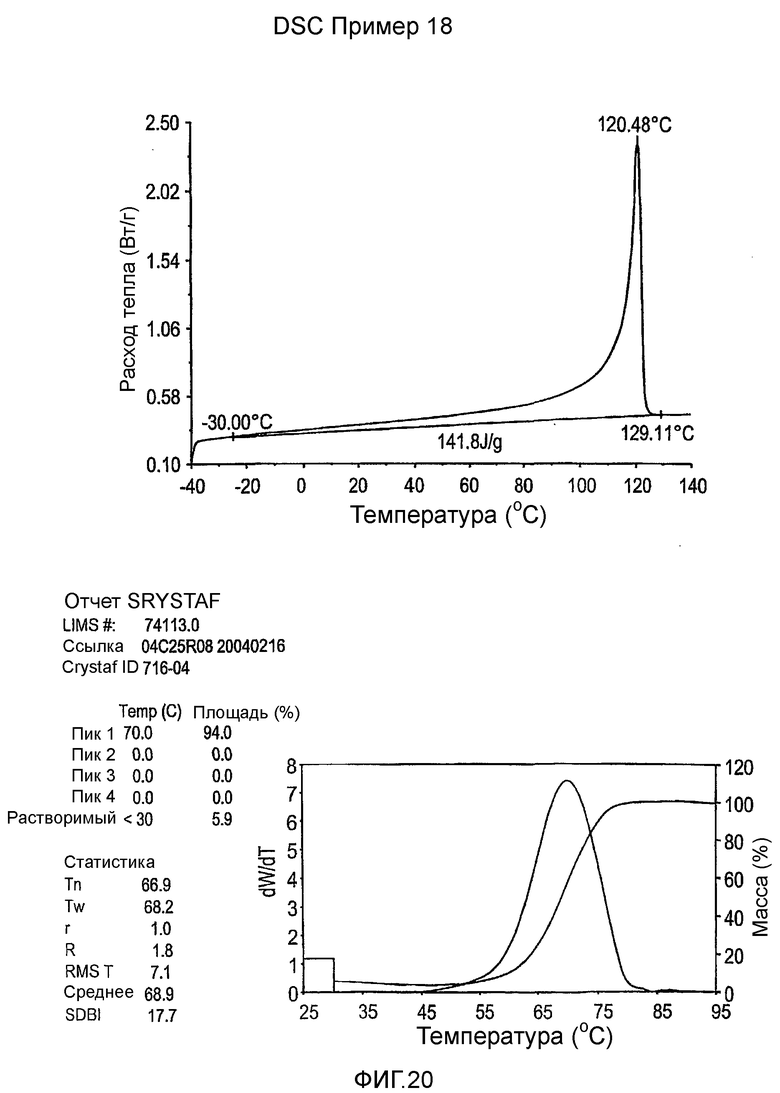
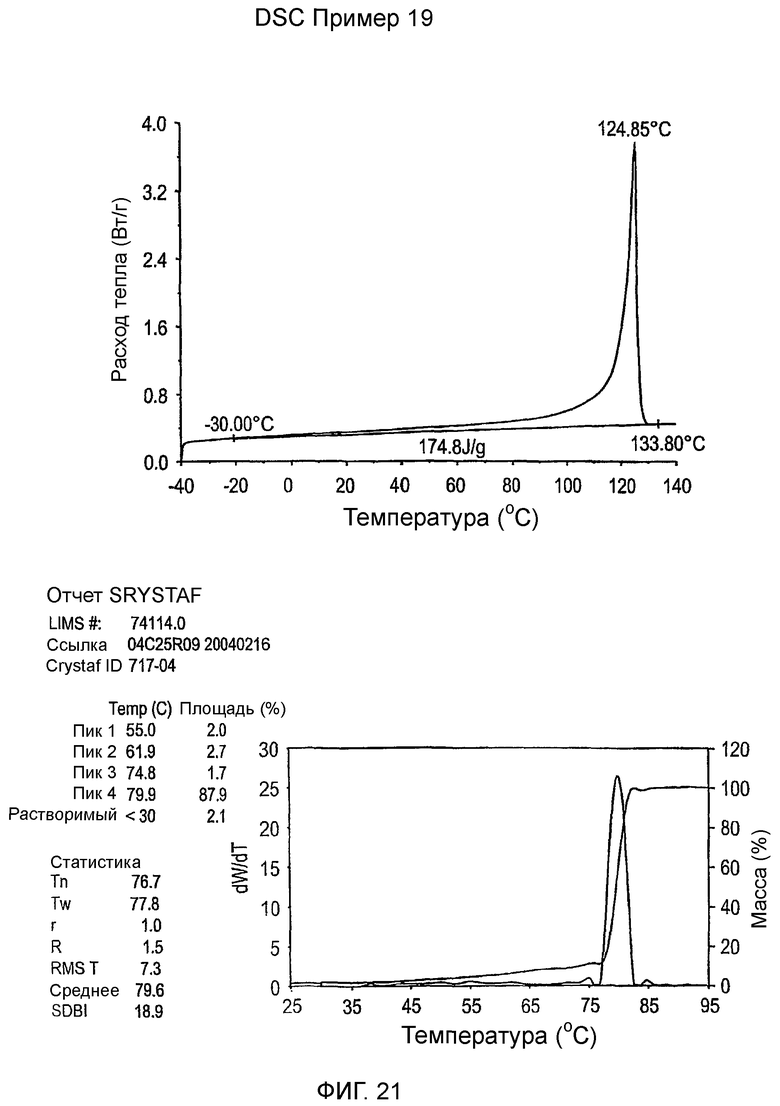
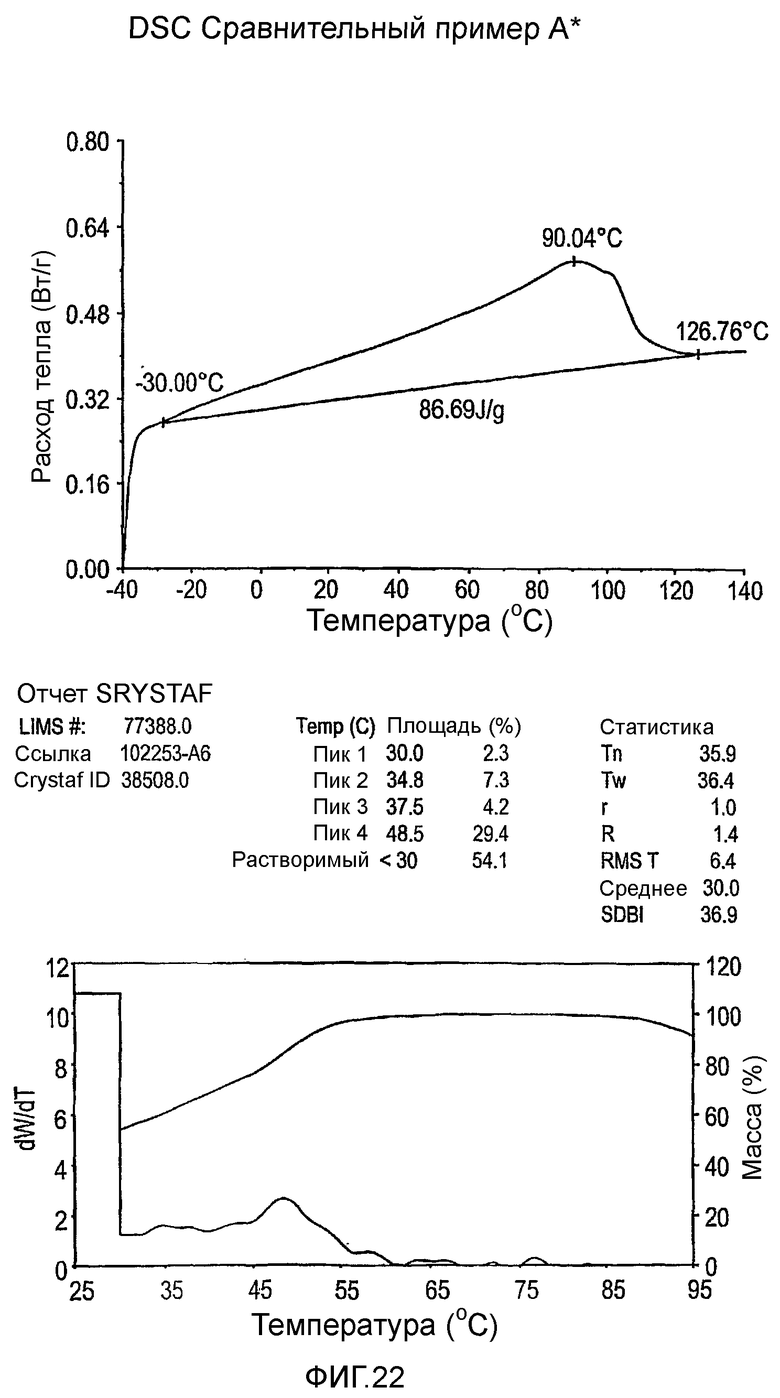
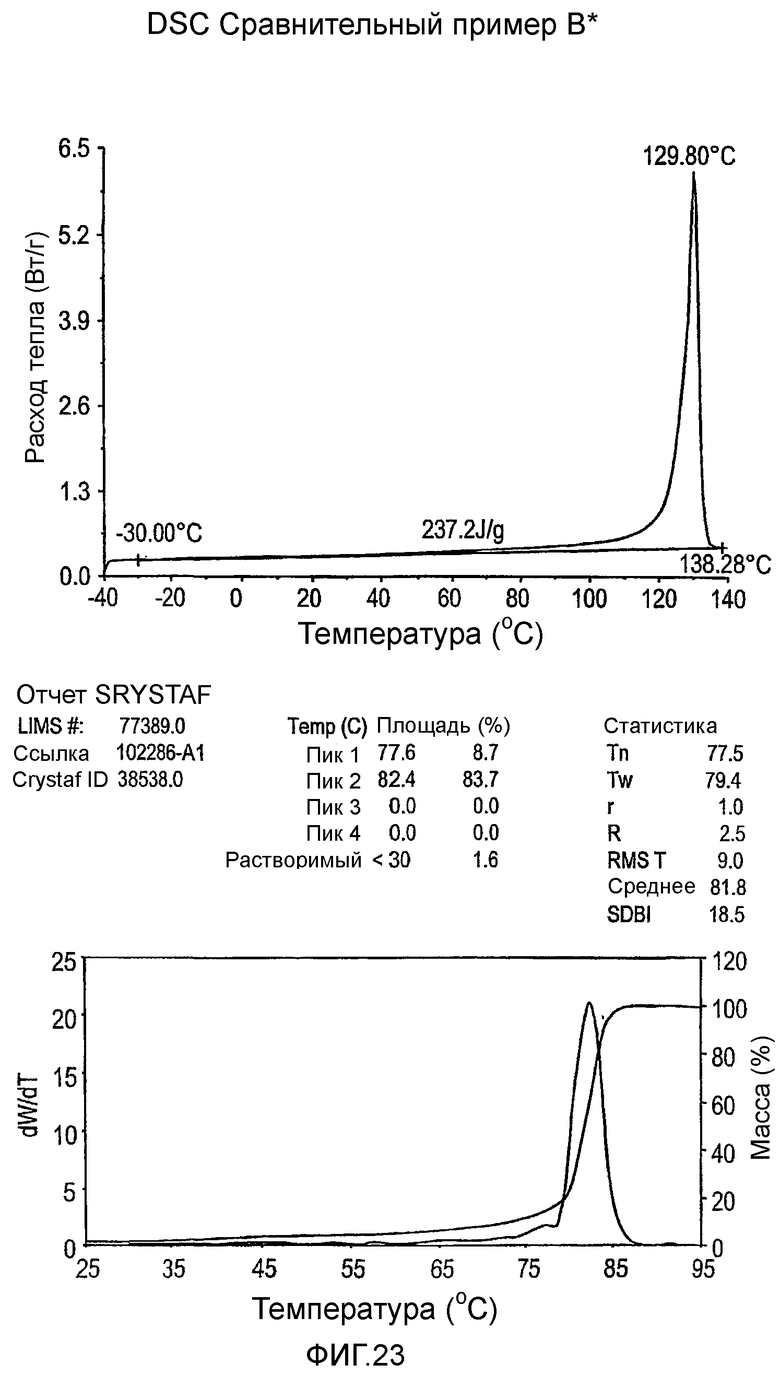
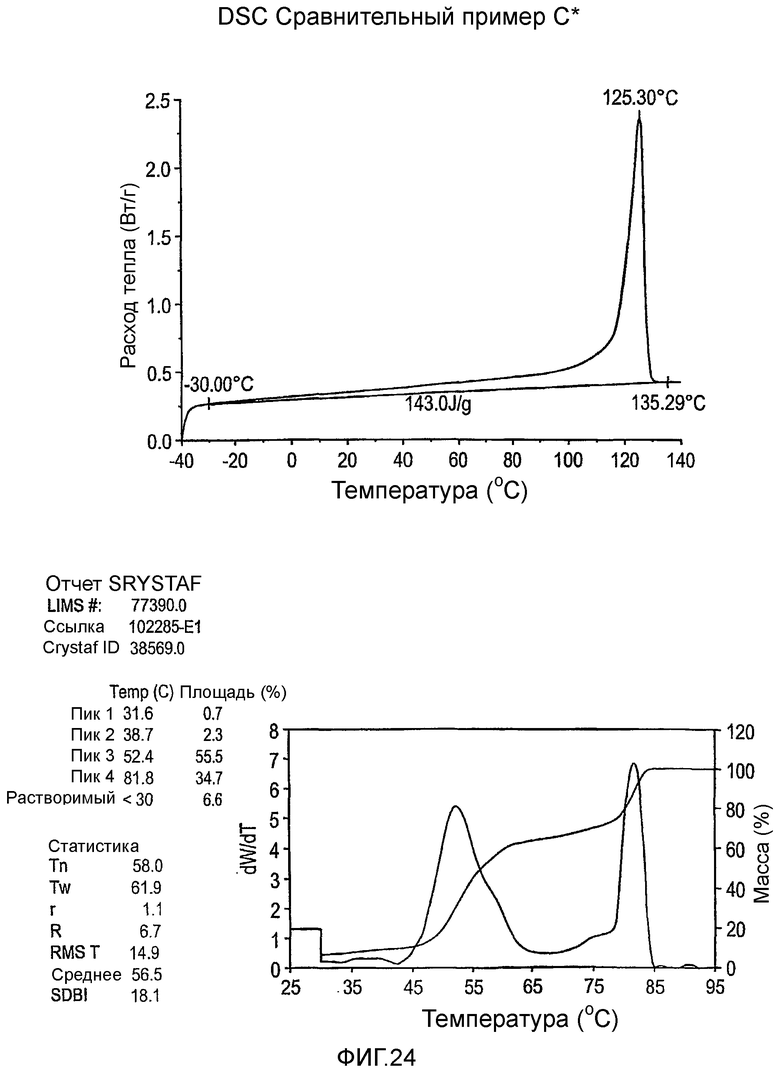
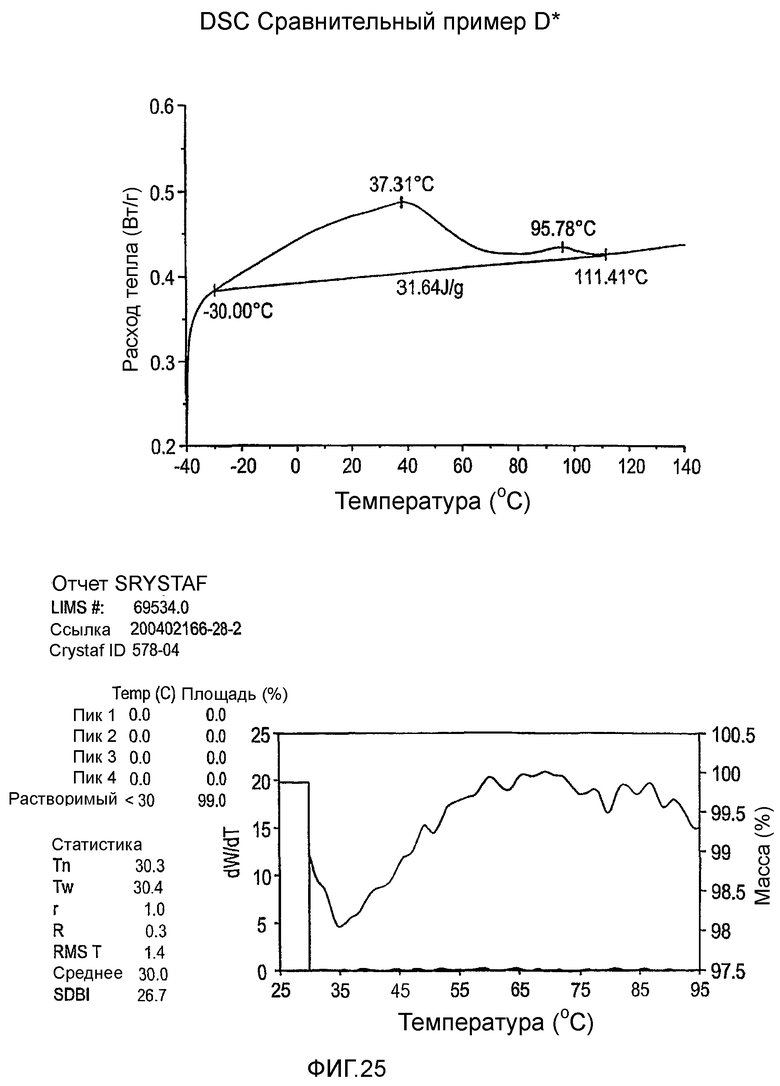
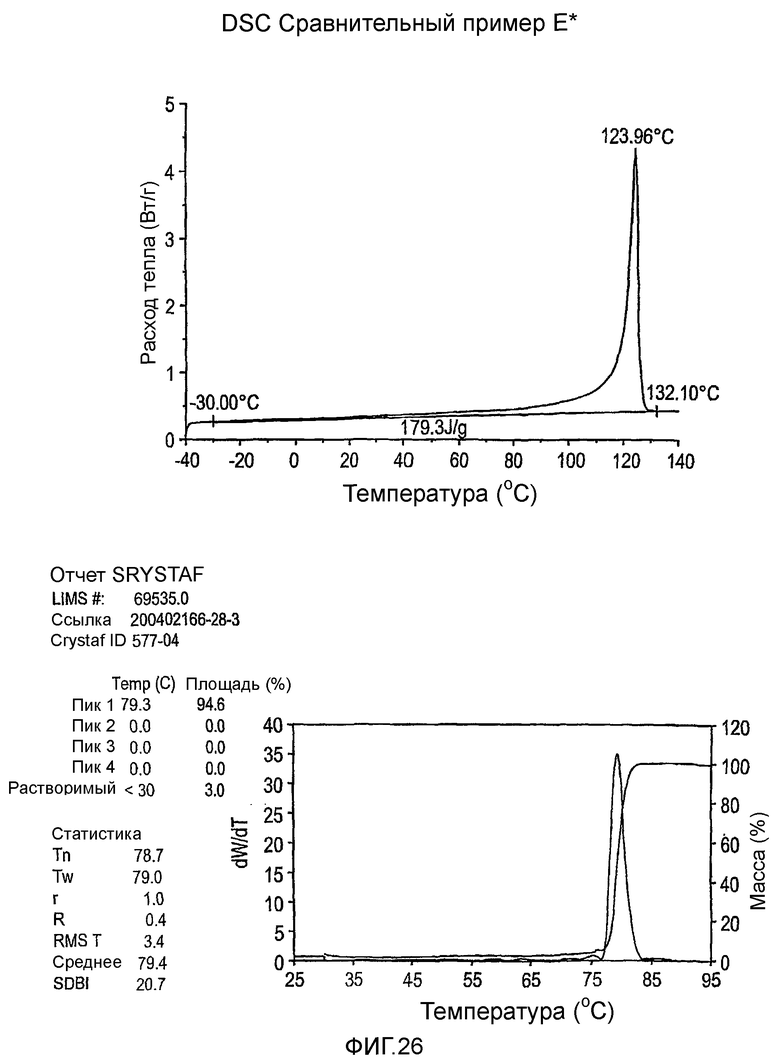
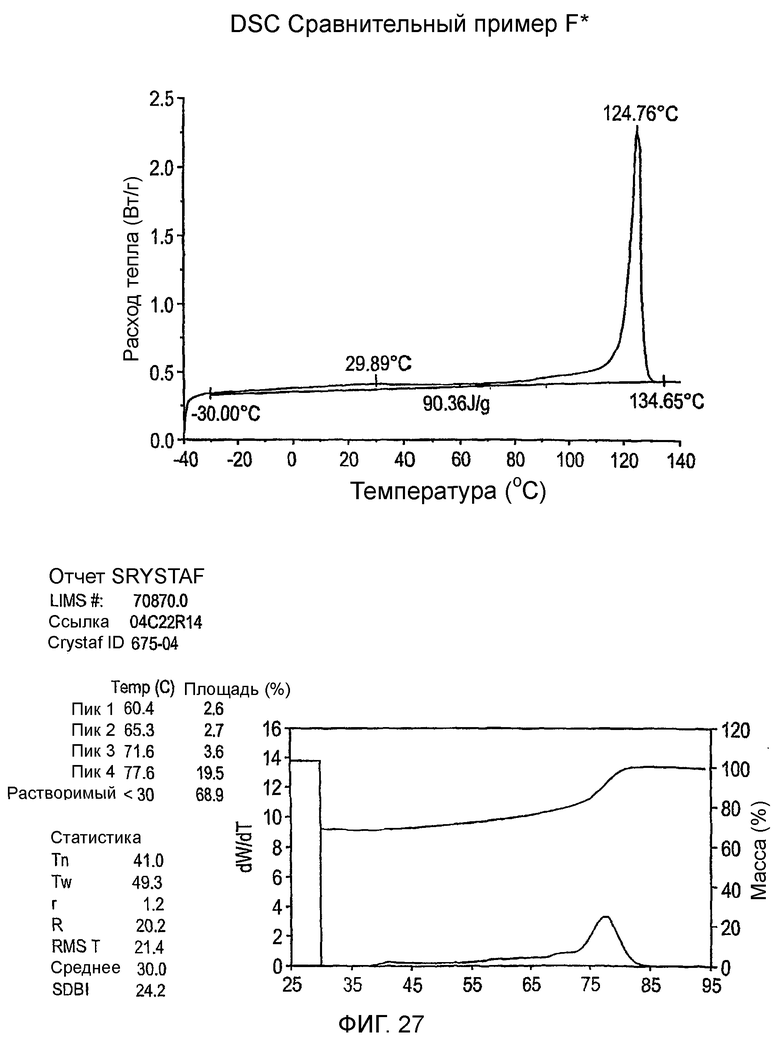

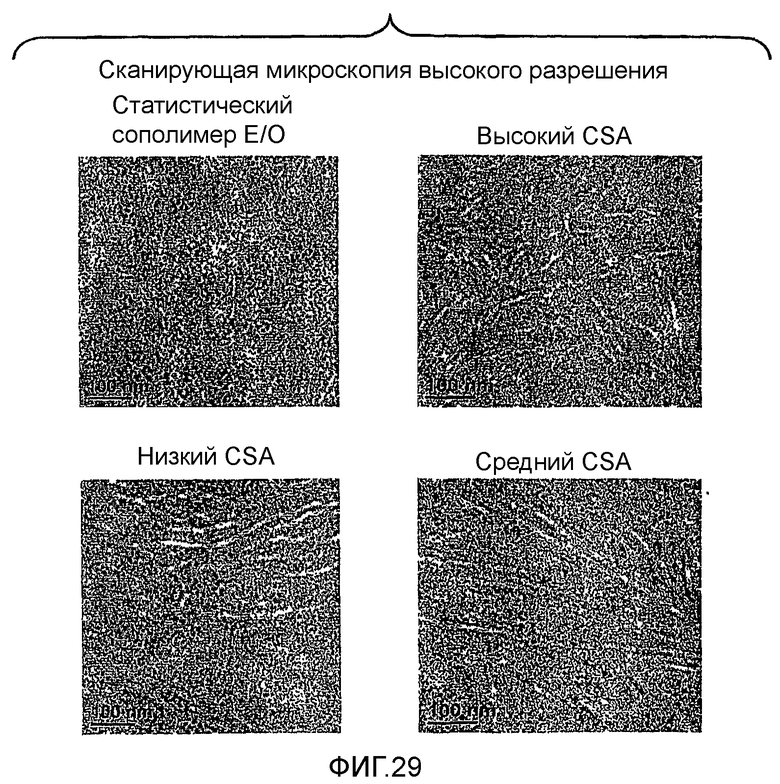
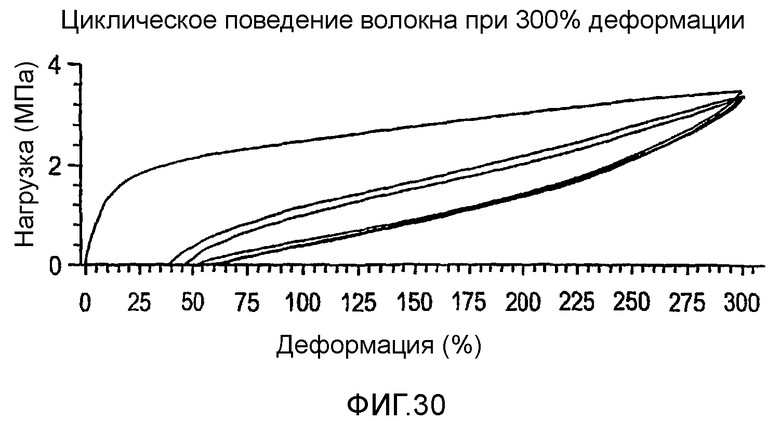
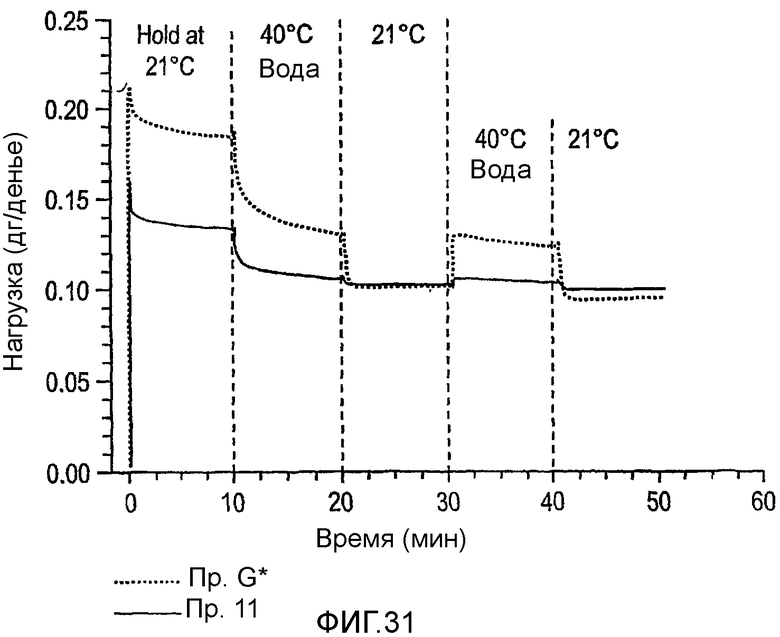
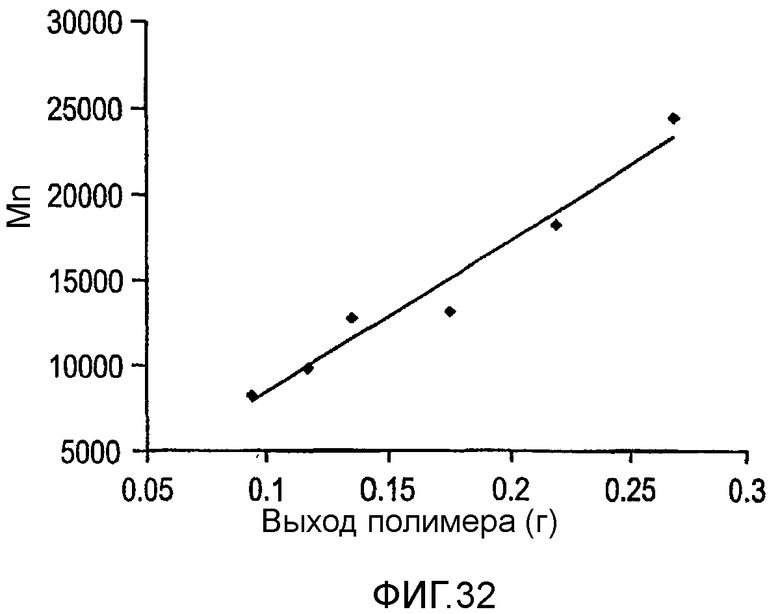
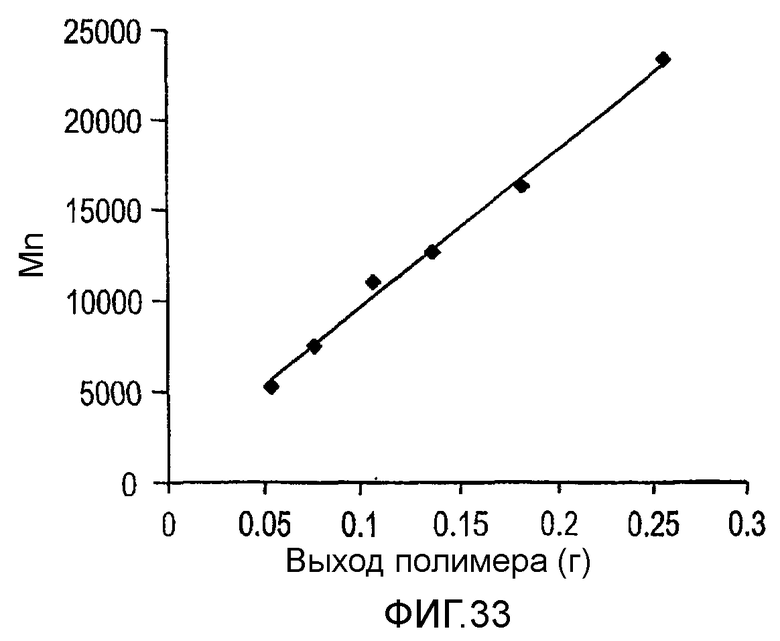
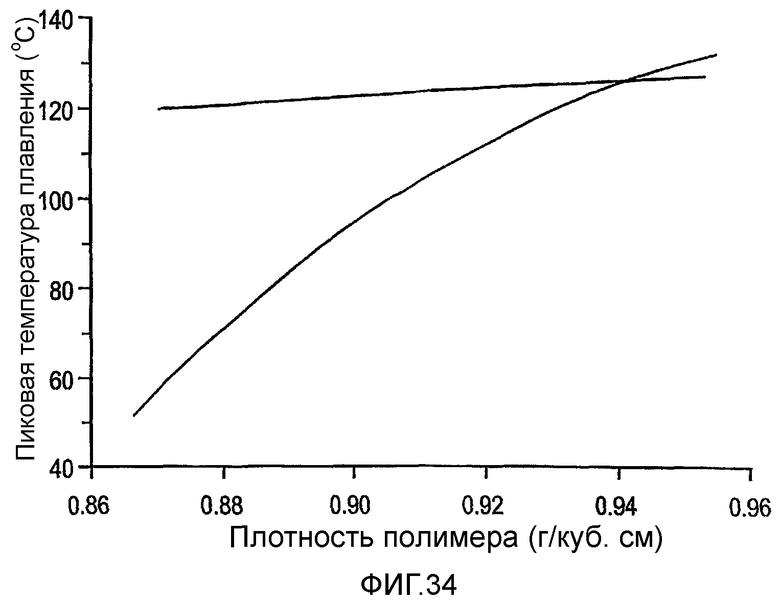
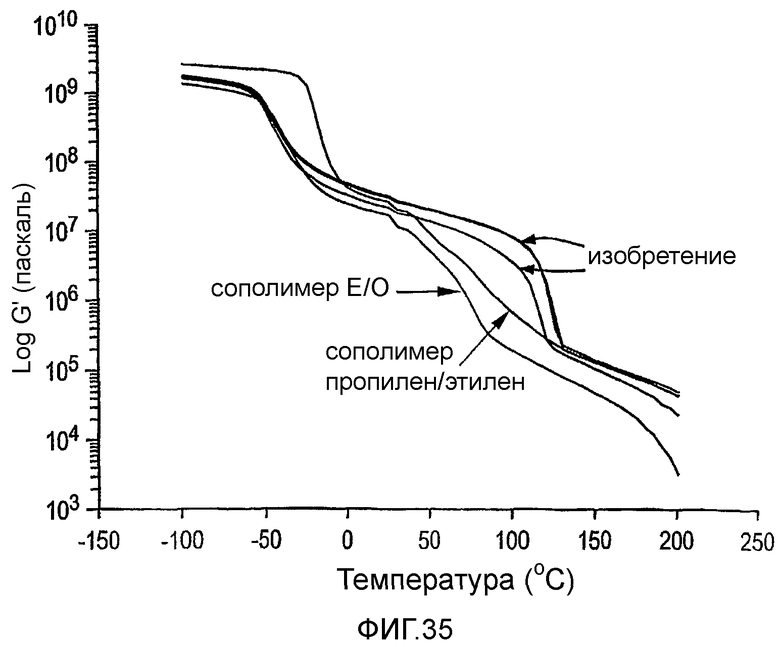
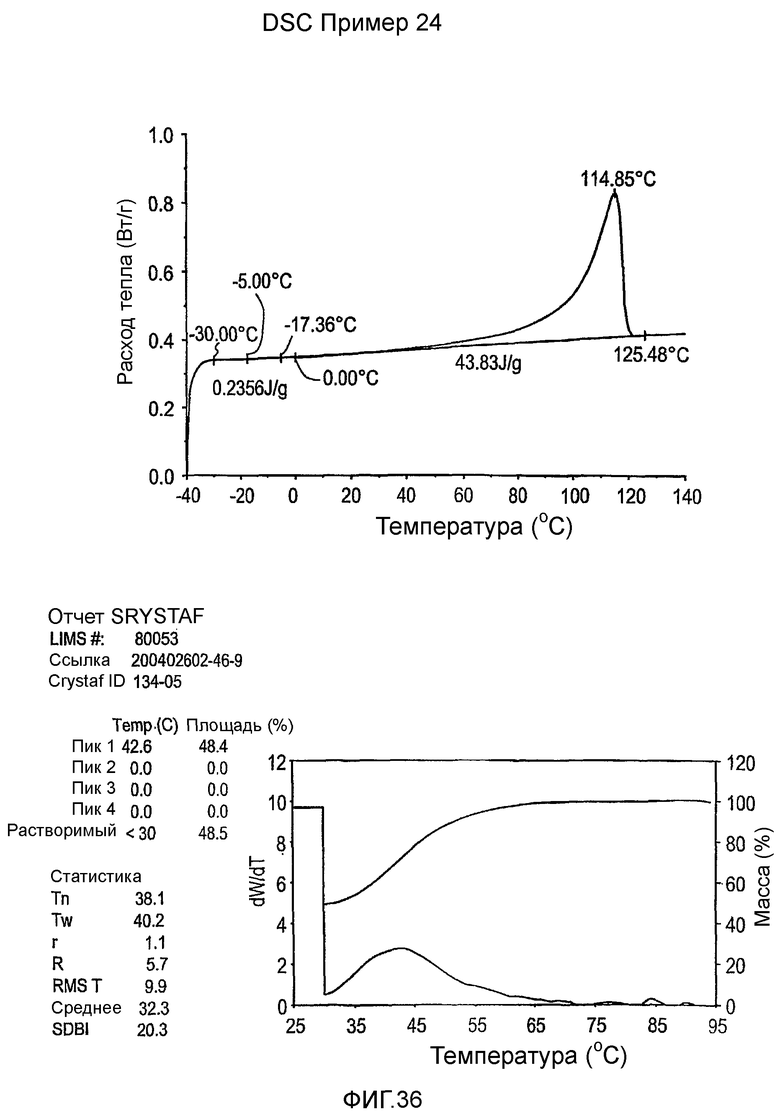
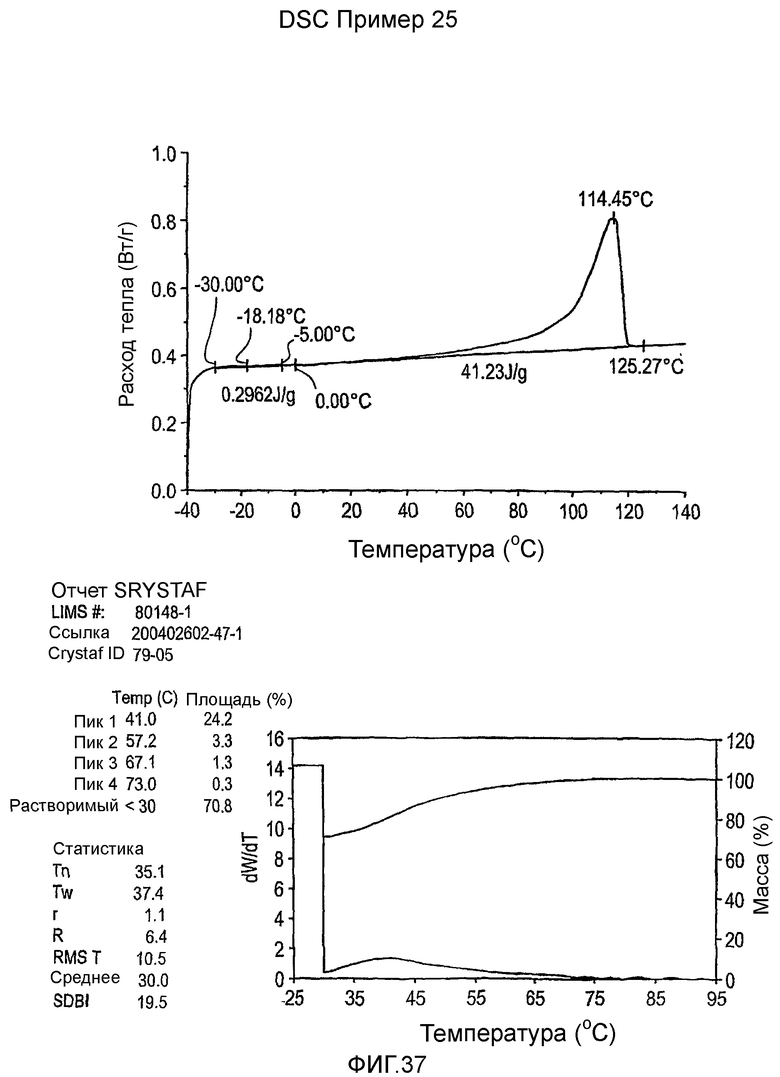
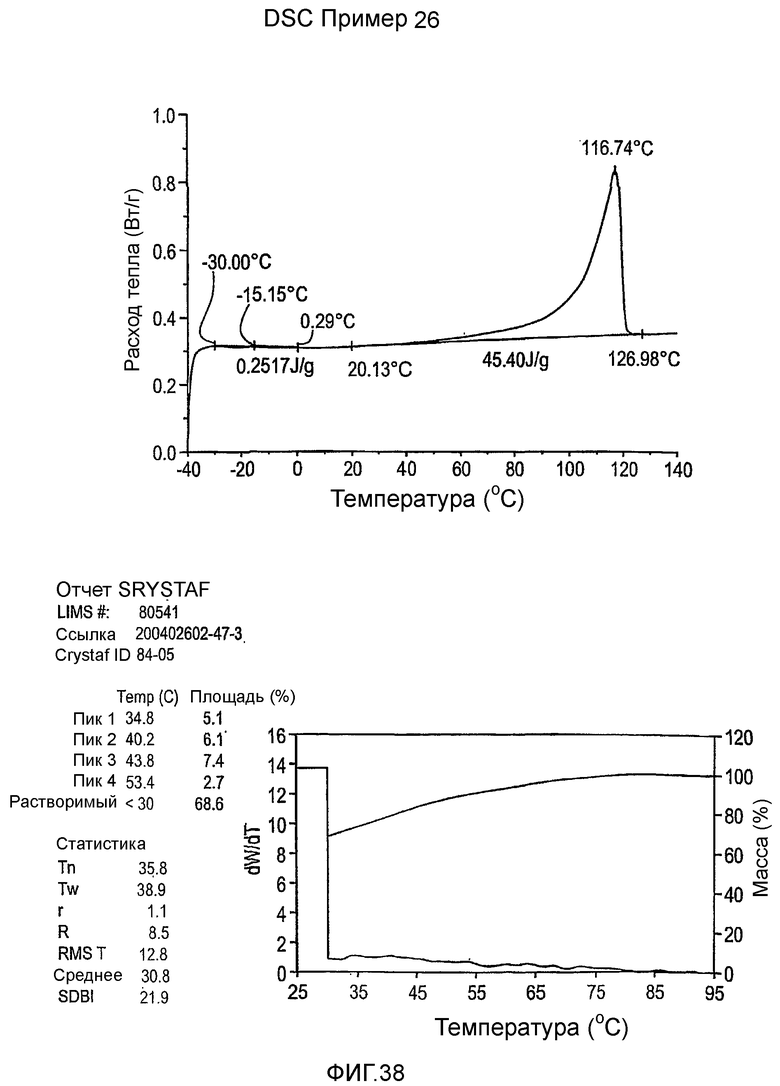
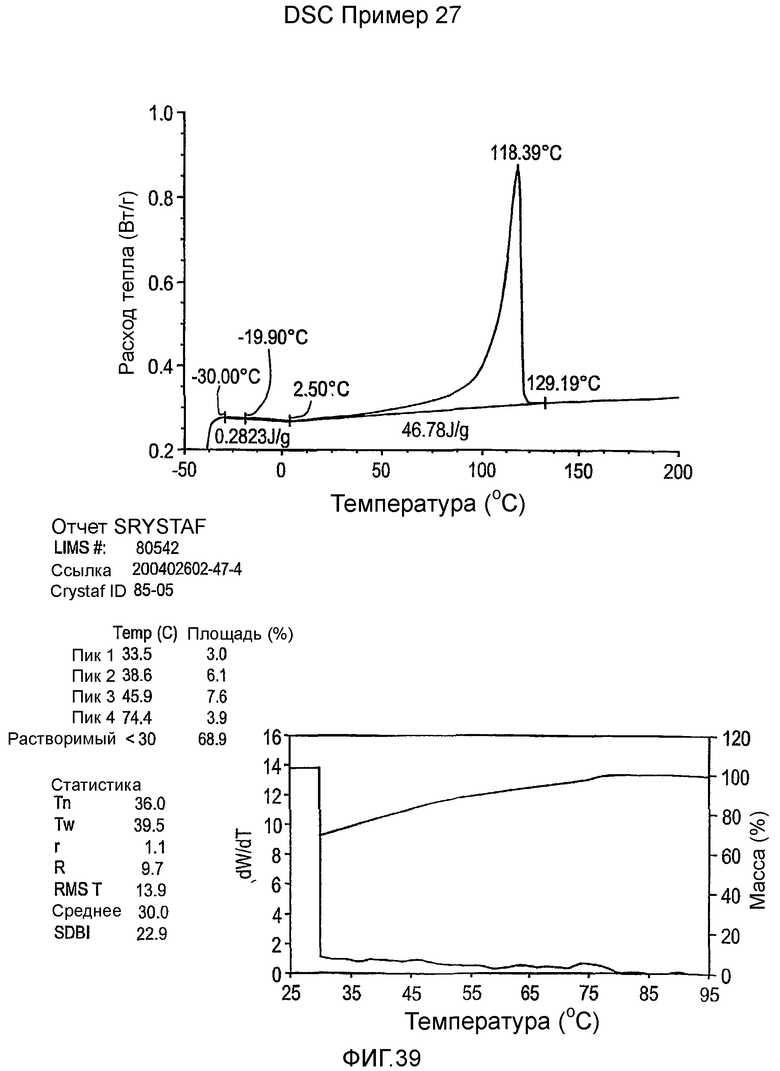
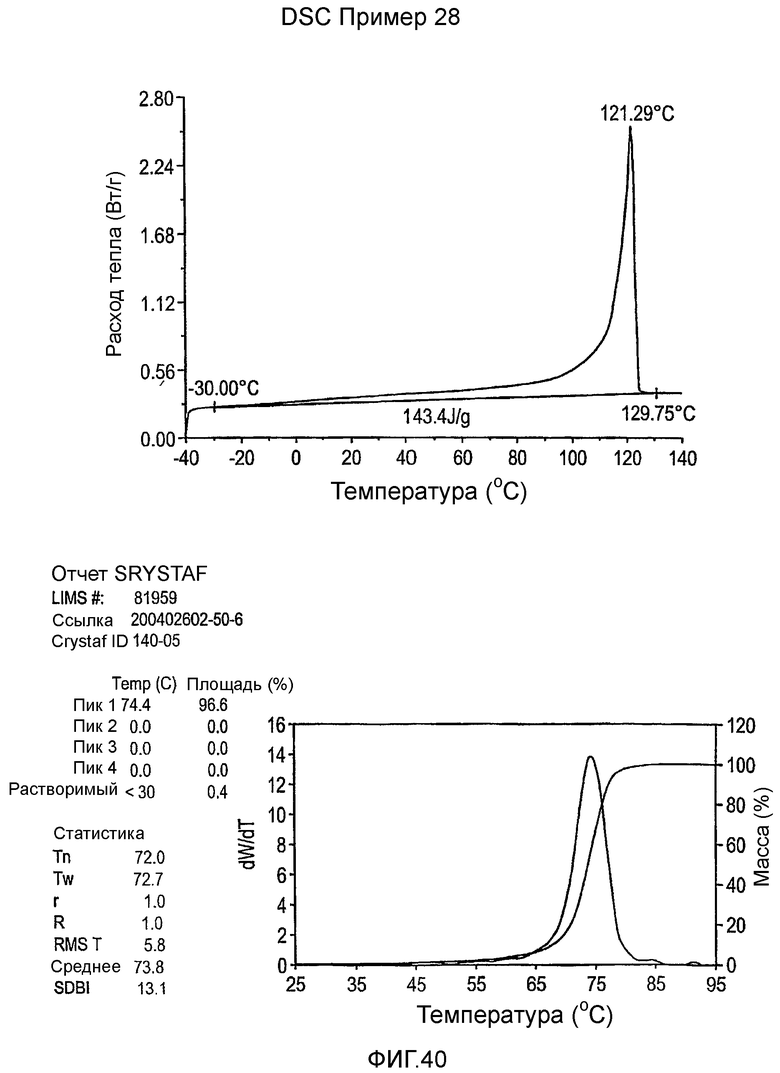
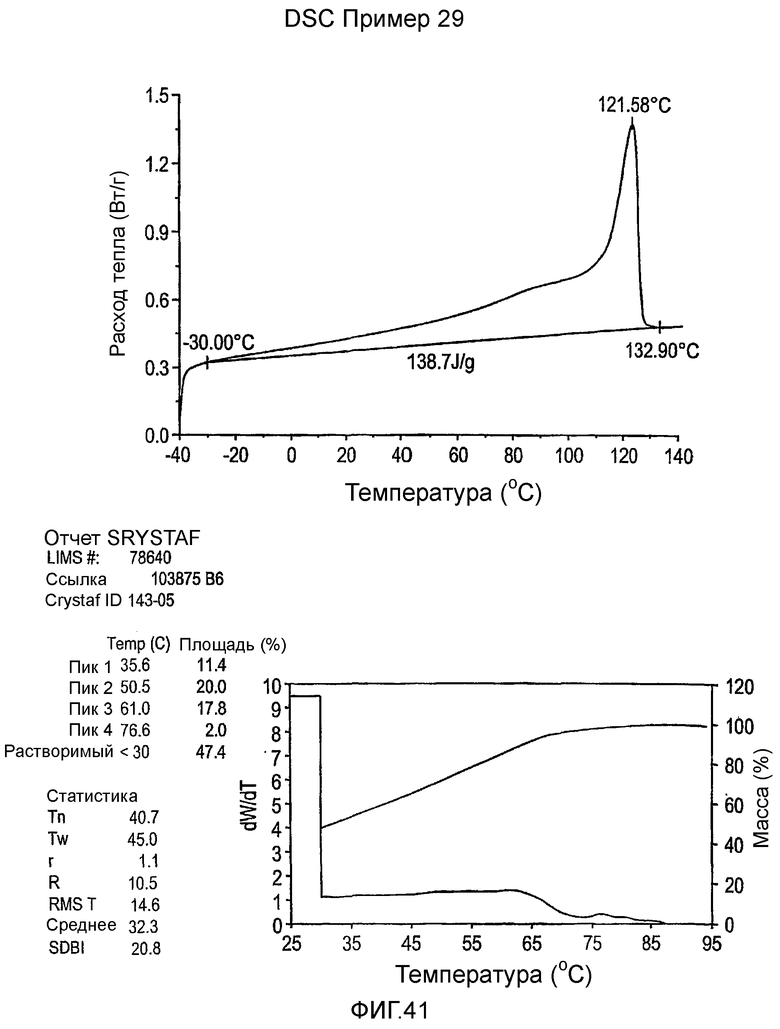
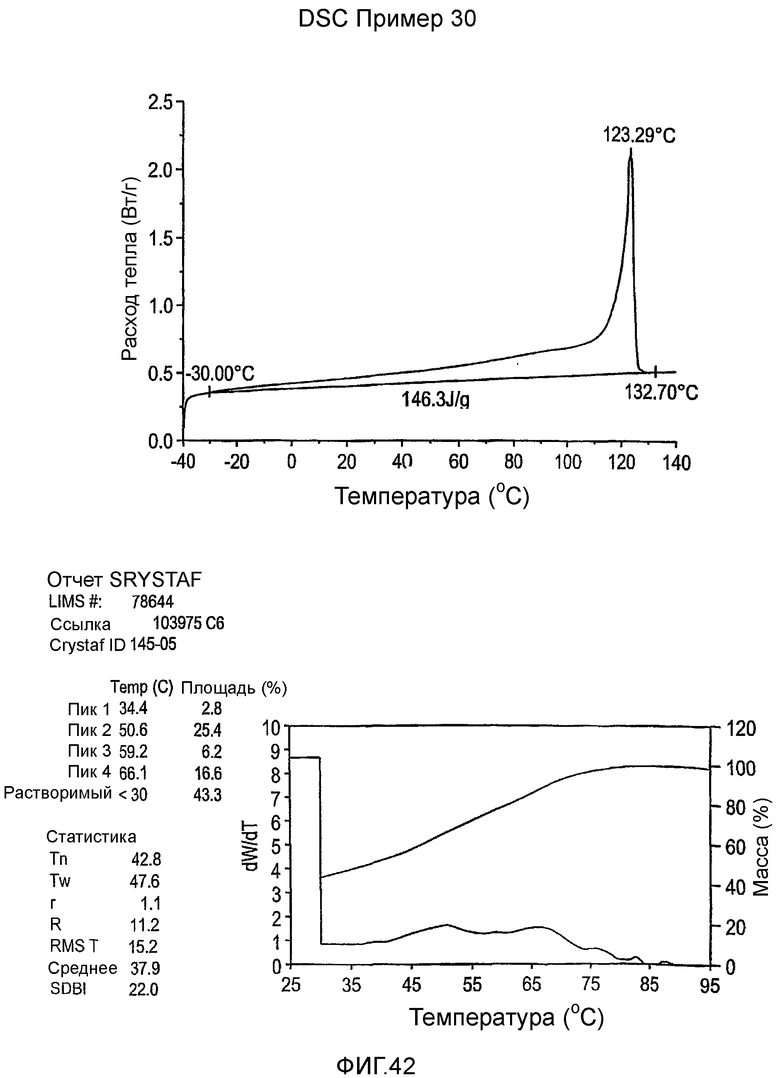
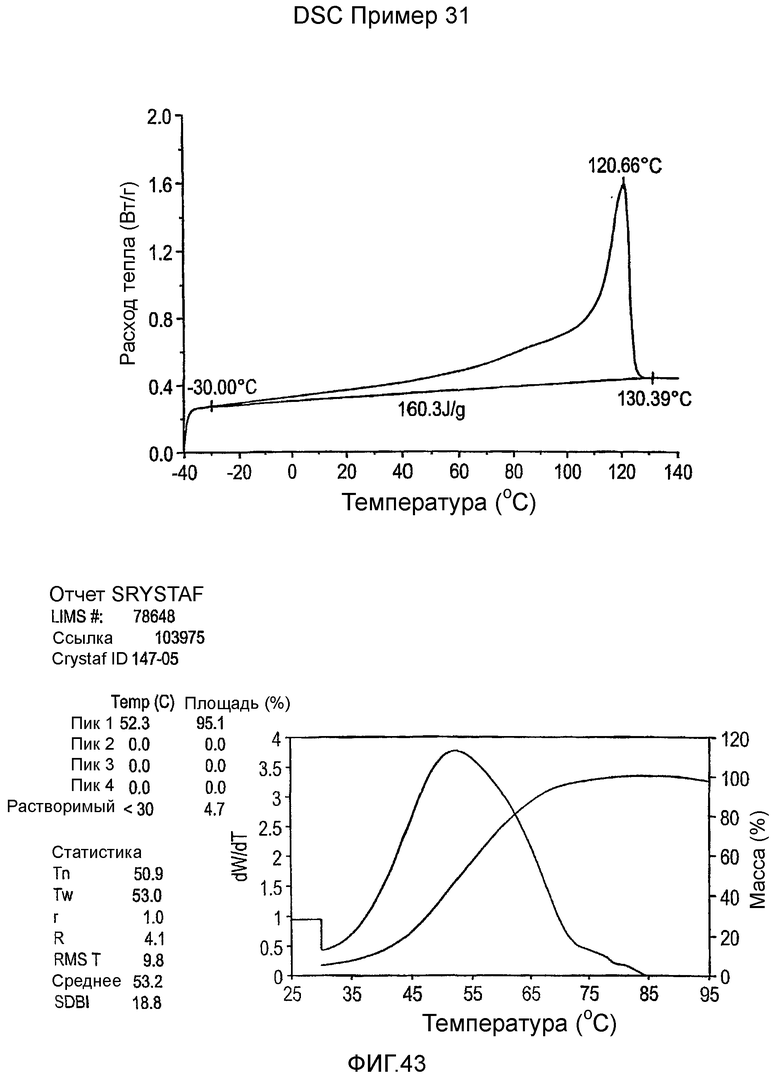
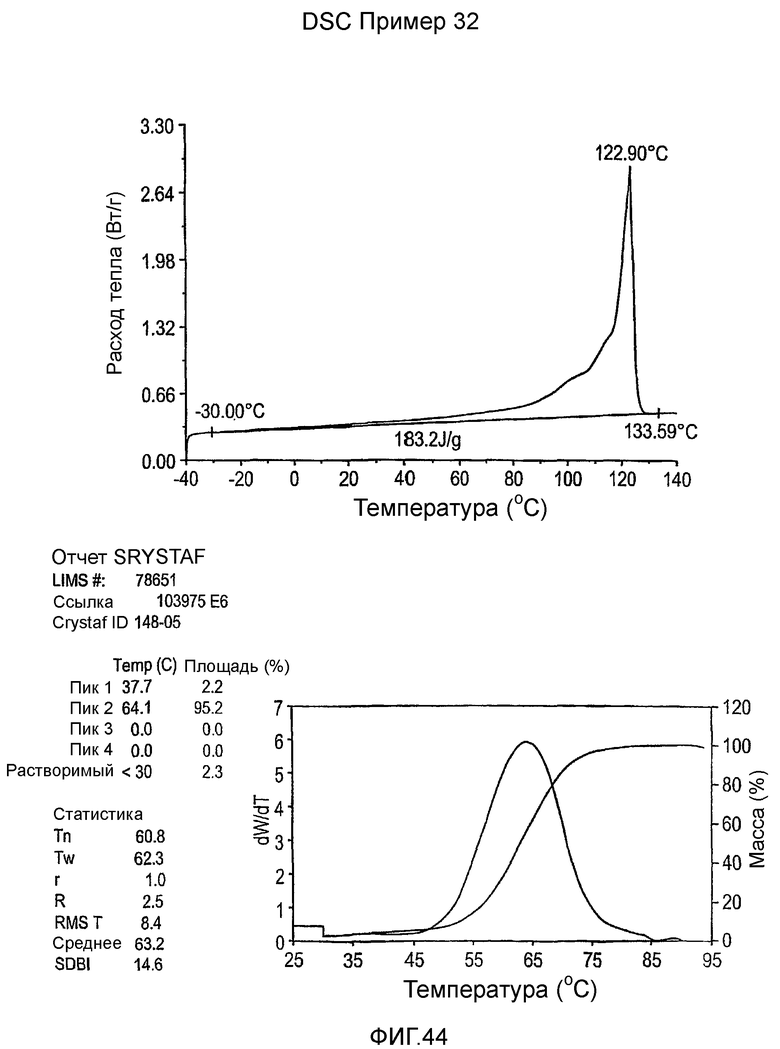
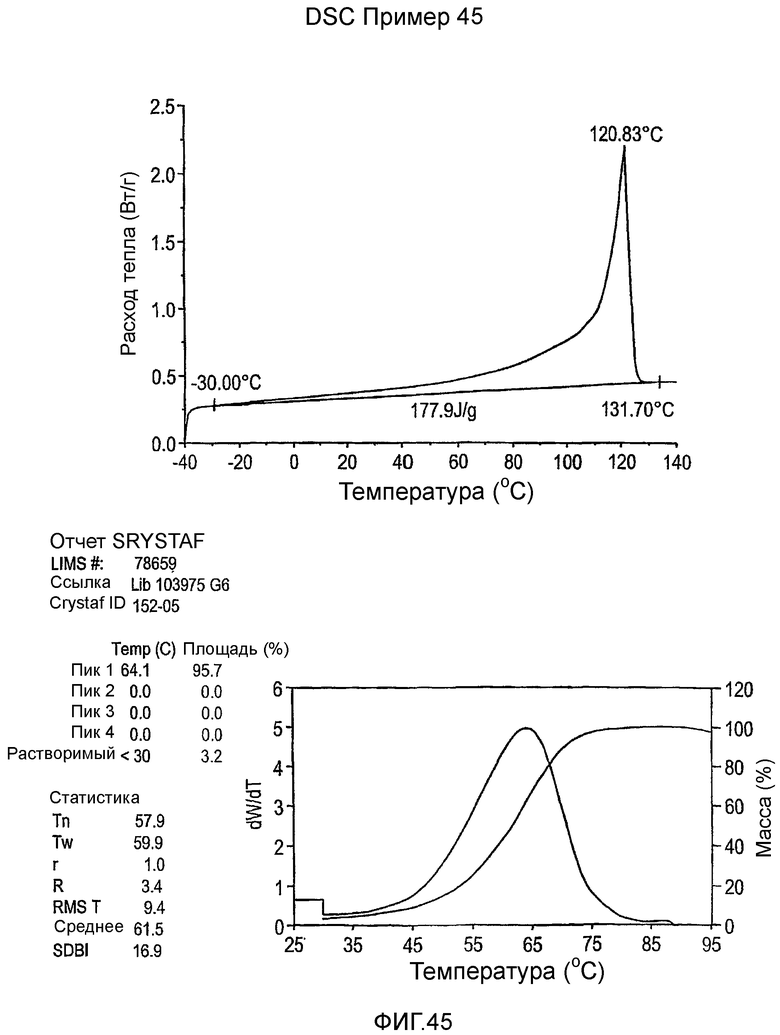
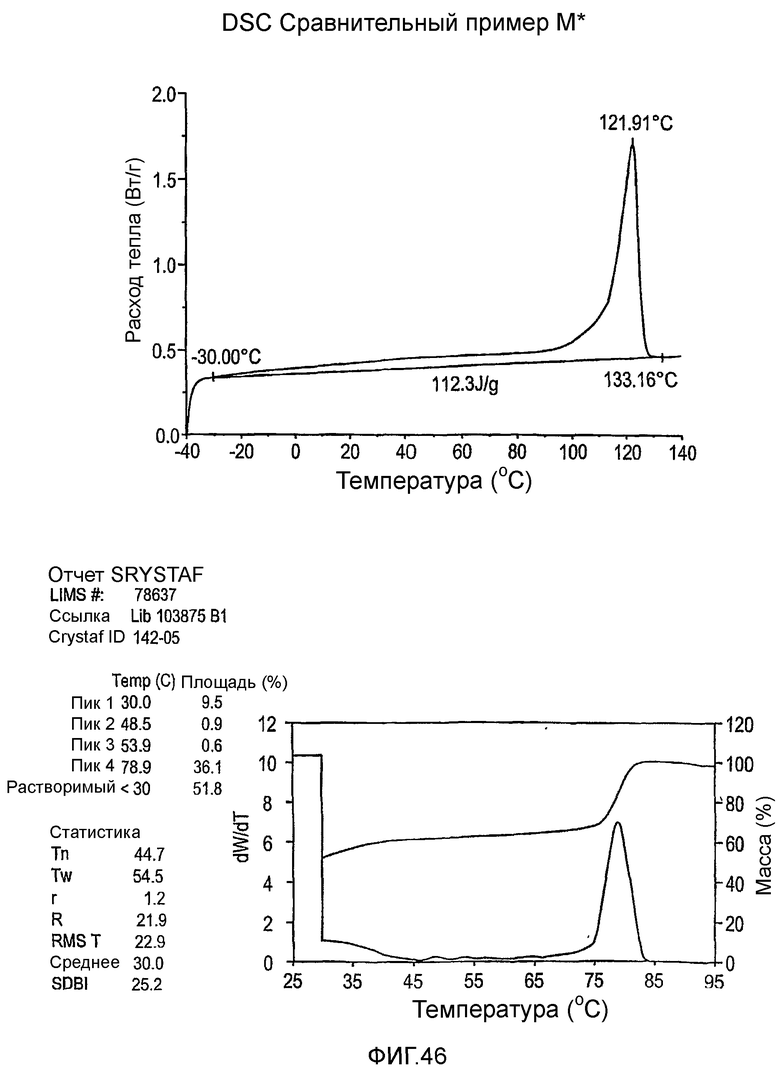
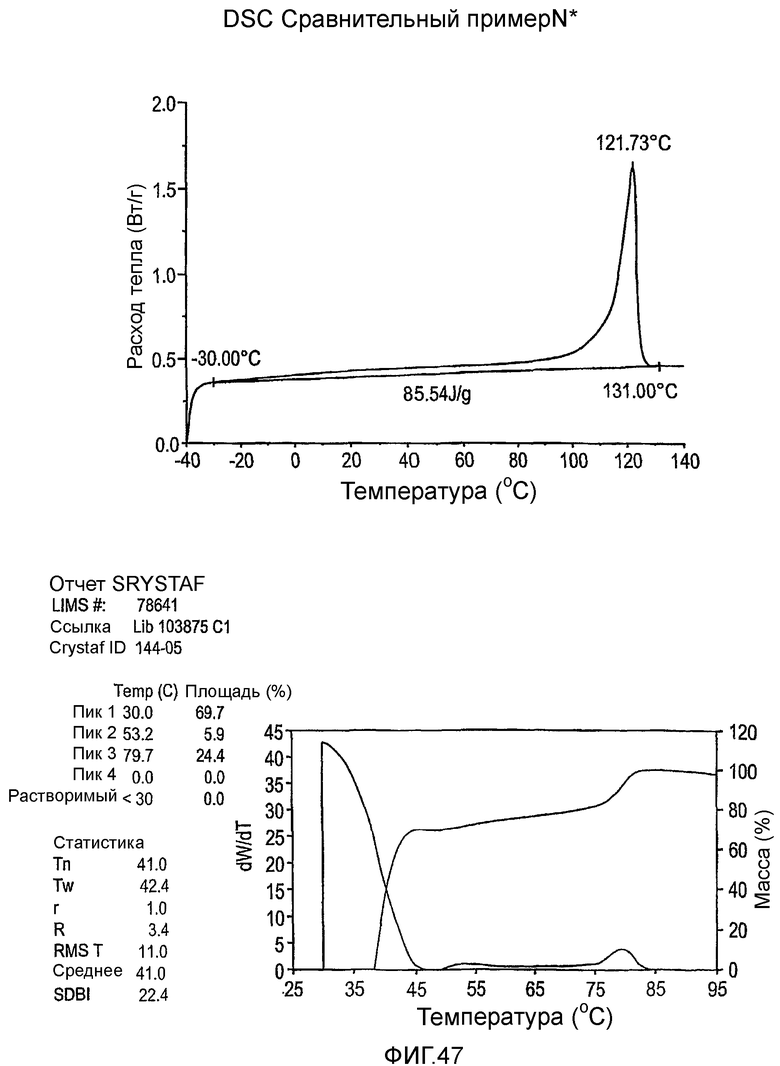
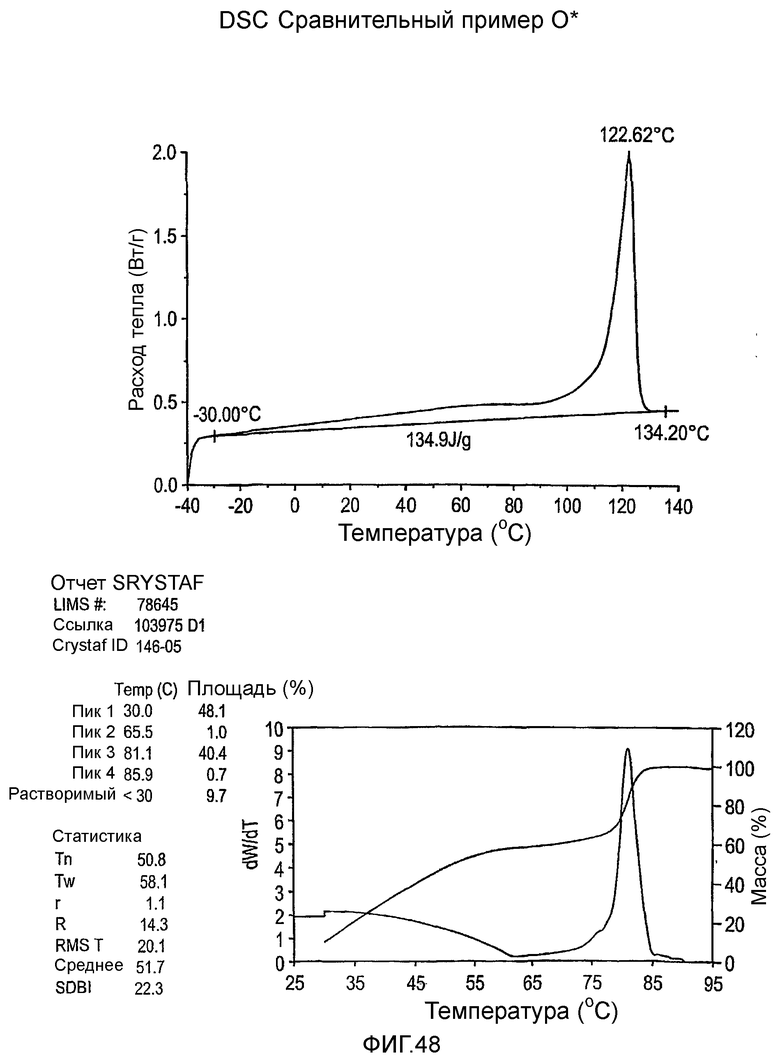
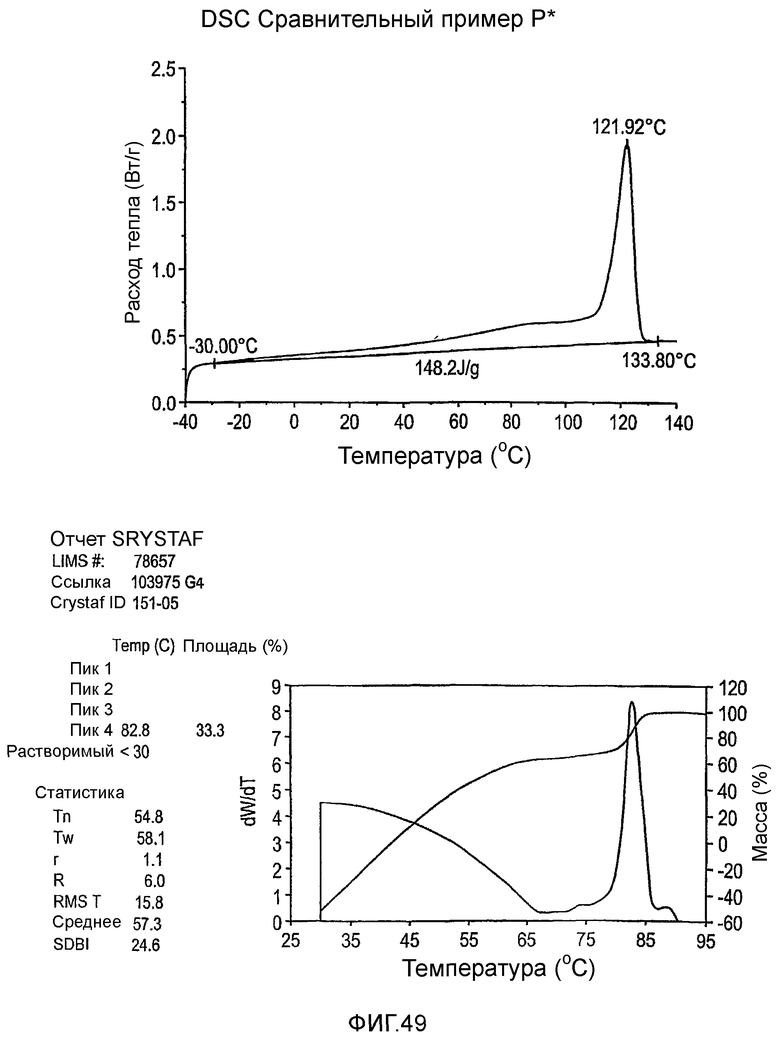
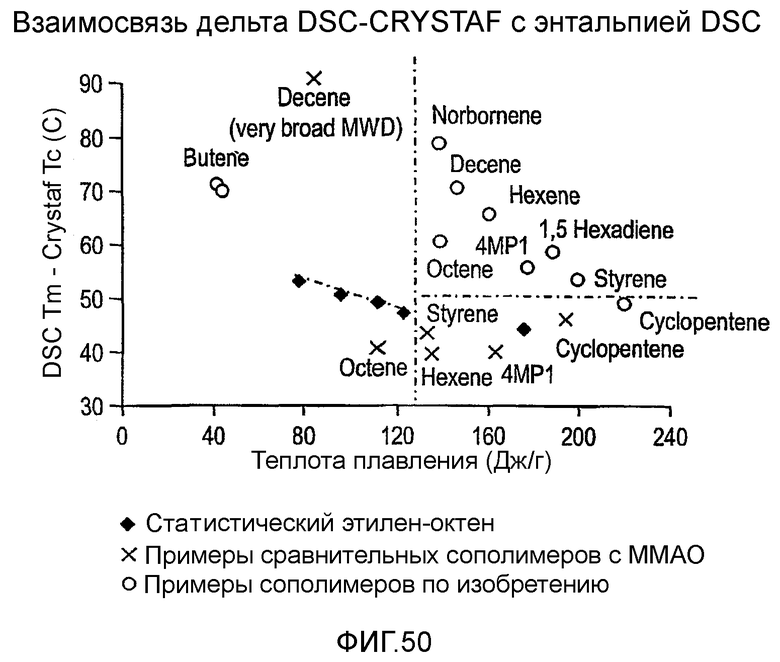
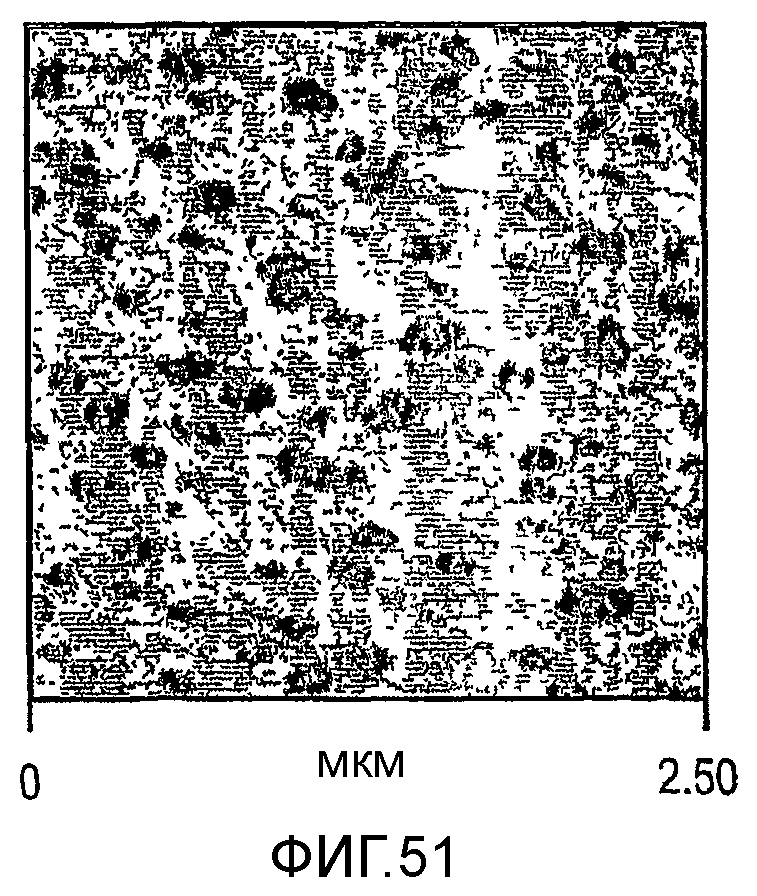
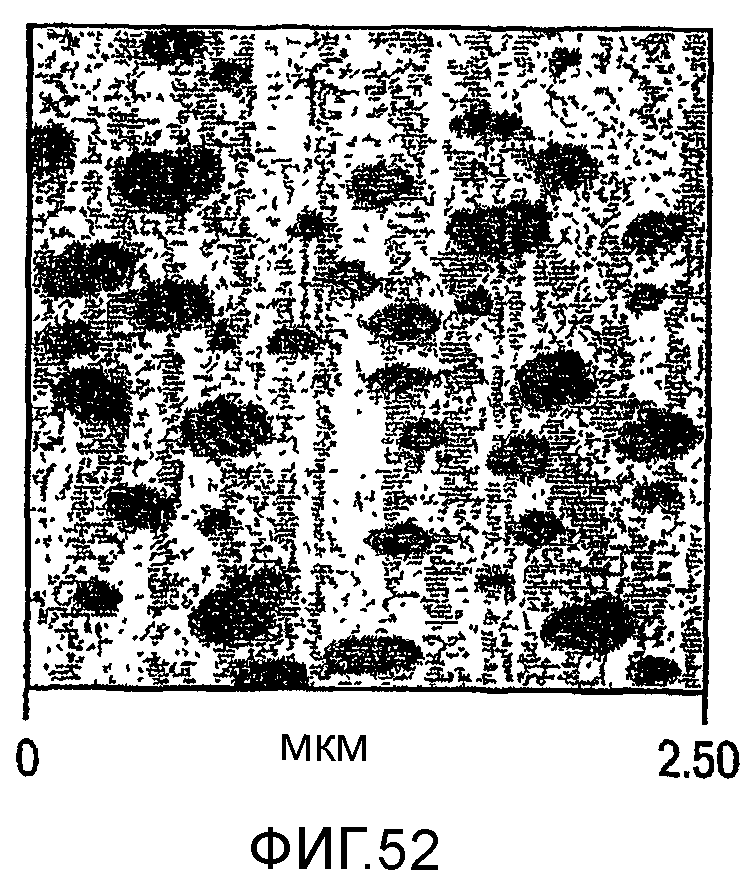
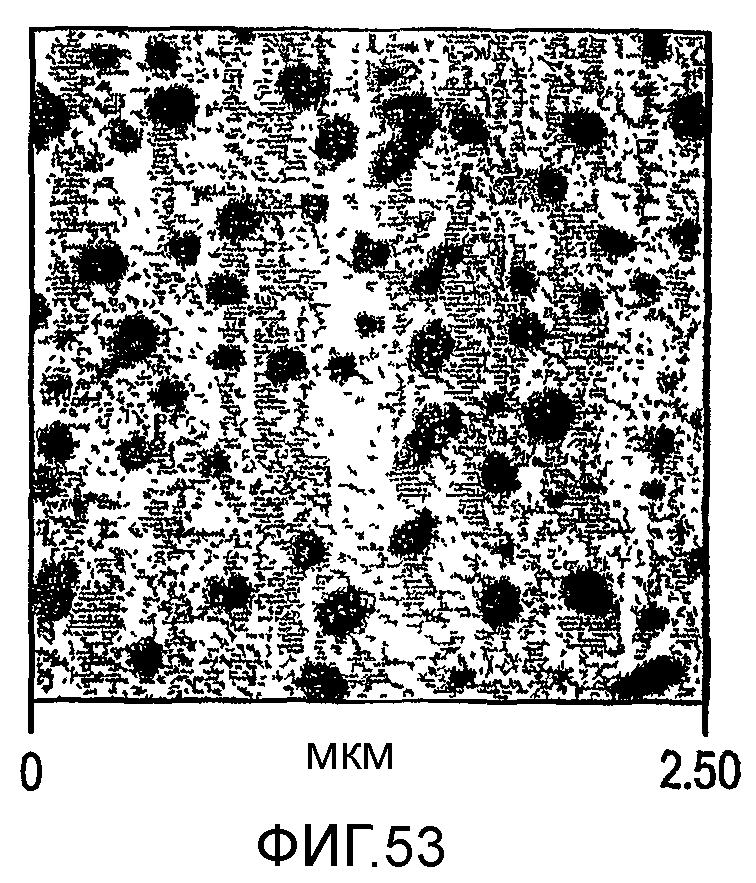
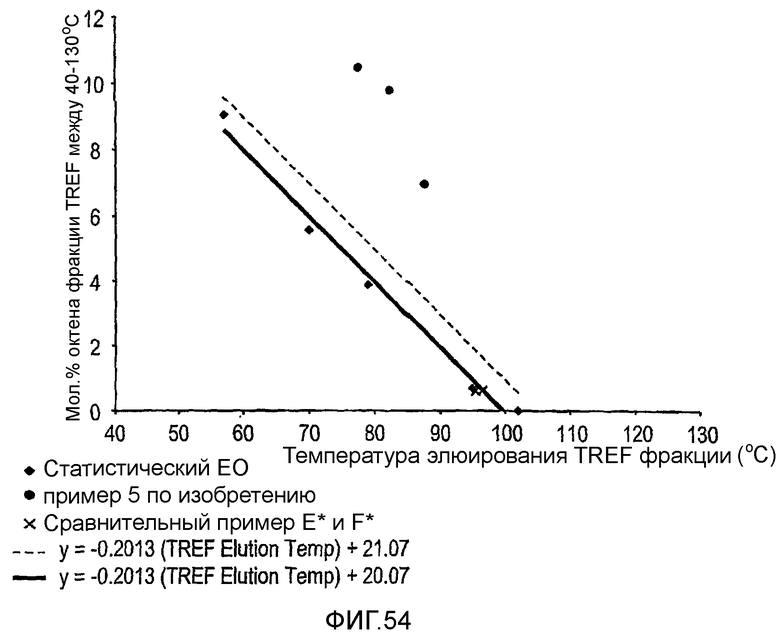
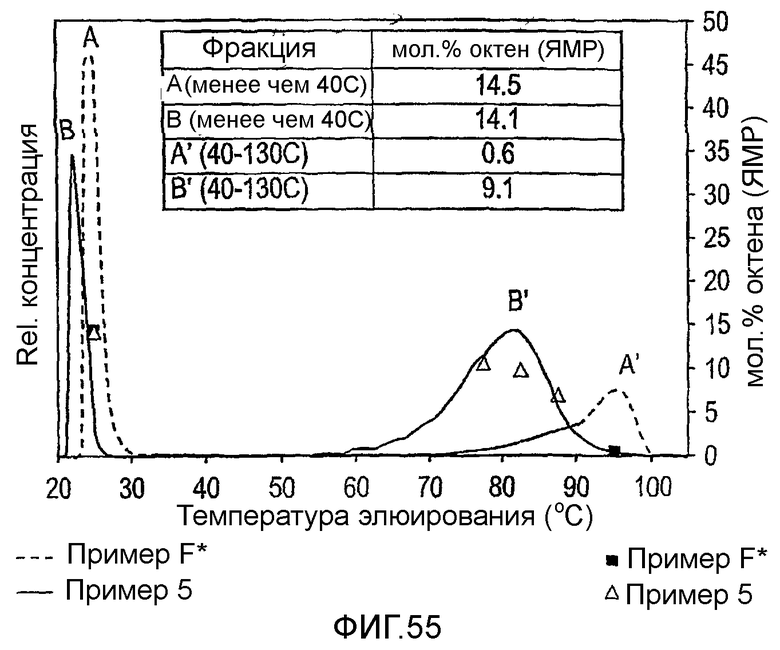
Изобретение относится к составу для применения при получении этилен-α-олефиновых мульти-блок-сополимеров, обладающих одной кристаллической температурой плавления Тm, определенной методом дифференциальной сканирующей калориметрии (DSC). Указанный состав содержит смесь или продукт реакции, полученный объединением: (А) первого катализатора полимеризации олефинов, (В) второго катализатора полимеризации олефинов, способствующего получению полимеров, отличающихся по химическим или физическим свойствам от полимера, полученного с катализатором (А) при эквивалентных условиях полимеризации, и (С) челночного агента цепи, причем в условиях полимеризации олефинов челночный агент цепи переносит фрагменты полимера между активными сайтами катализаторов (А) и (В). Технический результат - получение мульти-блок-сополимеров с высоким выходом и селективностью. 16 н. и 11 з.п. ф-лы, 28 табл., 55 ил.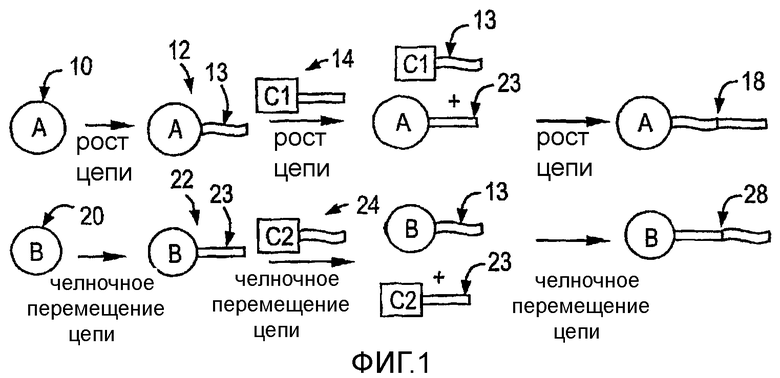
1. Состав для применения при получении этилен-α-олефиновых мульти-блок-сополимеров, обладающих одной кристаллической температурой плавления Тm, определенной методом дифференциальной сканирующей калориметрии (DSC), указанный состав содержит смесь или продукт реакции, полученный объединением:
(A) первого катализатора полимеризации олефинов,
(B) второго катализатора полимеризации олефинов, способствующего получению полимеров, отличающихся по химическим или физическим свойствам от полимера, полученного с катализатором (А) при эквивалентных условиях полимеризации, и
(C) челночного агента цепи, причем в условиях полимеризации олефинов челночный агент цепи переносит фрагменты полимера между активными сайтами катализаторов (А) и (В).
2. Состав по п.1, где катализатор (В) имеет показатель внедрения сомономера менее, чем показатель внедрения сомономера катализатора (А).
3. Состав по п.1, где челночным агентом является соединение алюминия, цинка или галлия, содержащее по меньшей мере один гидрокарбильный заместитель, имеющий от 1 до 12 атомов углерода.
4. Каталитический состав по п.3, где челночным агентом является соединение диалкилцинка.
5. Состав по п.1, где катализатор (А) содержит комплекс, содержащий переходный металл, выбранный из групп 4-8 Периодической таблицы элементов, и один или несколько делокализованных π-связанных лигандов или поливалентных лигандов основания Льюиса.
6. Состав по п.5, где катализатор (А) отвечает формуле:
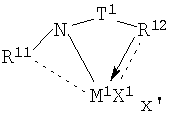
где R11 выбран из алкила, циклоалкила, гетероалкила, циклогетероалкила, арила и их инертно замещенных производных, содержащих от 1 до 30 атомов, не считая водород, или их двухвалентных производных;
Т1 представляет двухвалентную мостиковую группу из от 1 до 41 атомов, отличных от водорода, и
R12 представляет C5-20 гетероарилгруппу, содержащую функциональность основания Льюиса;
М1 представляет металл группы 4;
X1 представляет анионную, нейтральную или дианионную лиганд-группу;
х' означает число от 0 до 5, указывающее число таких X1 групп, и связи, необязательные связи и электронодонорные взаимодействия представлены линиями, пунктирными линиями и стрелками соответственно.
7. Состав по п.6, где катализатор (В) отвечает формуле:
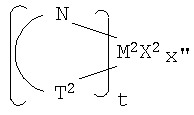
где М2 представляет металл групп 4-10 Периодической таблицы элементов;
Т2 означает группу, содержащую азот, кислород или фосфор;
Х2 представляет галоген, гидрокарбил или гидрокарбилокси;
t означает один или два;
х'' означает целое число, выбранное, чтобы обеспечить баланс заряда,
и Т2 и N связаны мостиковым лигандом.
8. Способ получения этилен- α-олефинового мульти-блок-сополимера, имеющего одну кристаллическую температуру плавления Тm, определенную методом DSC, указанный способ включает в себя контактирование одного или нескольких полимеризуемых присоединением мономеров в условиях полимеризации присоединением с составом по п.1.
9. Этилен- α-олефиновый мульти-блок-сополимер, получаемый способом по п.8, содержащий два или более сегментов или блоков, отличающихся по содержанию сомономера, кристалличности, плотности, температуре плавления или температуре стеклования.
10. Этилен- α-олефиновый мульти-блок-сополимер, получаемый способом по п.8, имеющий температуру плавления, Тm, в градусах Цельсия, и плотность, d*, в граммах на кубический сантиметр, где числовые величины переменных соответствуют уравнению:
Tm>-2002,9+4538,5(d*)-2422,2(d*)2 и где сополимер имеет Мw/Мn от 1,7 до 3,5.
11. Этилен- α-олефиновый мульти-блок-сополимер, получаемый способом по п.8, имеющий Mw/Mn от 1,7 до 3,5, величину дельта (наивысший пик DSC минус наивысший пик CRYSTAF) более чем величина у*, определяемая по уравнению:
у*>-0,1299(ΔН)+62,81,
и теплоту плавления вплоть до 130 Дж/г,
где пик CRYSTAF определяют, используя по меньшей мере 5% совокупного полимера, и, если менее чем 5% полимера имеет идентифицируемый пик CRYSTAF, тогда температура CRYSTAF 30°C и ΔН является численной величиной тепла плавления в Дж/г.
12. Этилен- α-олефиновый мульти-блок-сополимер, получаемый способом по п.8, имеющий предел прочности на разрыв выше 10 МПа и удлинение при разрыве по меньшей мере 600% при скорости разделения ползуна 11 см/мин.
13. Этилен- α-олефиновый мульти-блок-сополимер, получаемый способом по п.8, имеющий величину дельта (наивысший пик DSC (измеренный от базовой линии) минус наивысший пик CRYSTAF) более чем 48°С и теплоту плавления более чем или равную 130 Дж/г, где пик CRYSTAF определяют, используя по меньшей мере 5% совокупного полимера, и, если менее чем 5% полимера имеет идентифицируемый пик CRYSTAF, тогда температура CRYSTAF 30°C.
14. Этилен- α-олефиновый мульти-блок-сополимер, получаемый способом по п.8, имеющий отношение динамических модулей упругости, G′(25°C)/G′(100°C) от 1 до 50 и остаточную деформацию при сжатии при 70°С менее чем 80%.
15. Этилен- α-олефиновый мульти-блок-сополимер, получаемый способом по п.8, имеющий теплоту плавления менее чем 85 Дж/г и силу слипания гранул, равную или менее чем 100 фунт/фут2 (4800 Па).
16. Несшитый эластомерный этилен- α-олефиновый мульти-блок-сополимер, получаемый способом по п.8, содержащий в полимеризованной форме по меньшей мере 50 мол.% этилена, имеющий остаточную деформацию при сжатии 70°С менее чем 80%.
17. Этилен- α-олефиновый мульти-блок-сополимер, получаемый способом по п.8, содержащий единственную кристаллическую температуру плавления (Тm), которую измеряют DSC.
18. Этилен- α-олефиновый мульти-блок-сополимер, получаемый способом по п.8, имеющий глубину пенетрации по термомеханическому анализу 1 мм при температуре по меньшей мере 90°С и модуль упругости при изгибе от 3 кфунтов на кв. дюйм (20 МПа) до 13 кфунтов на кв. дюйм (90 МПа).
19. Этилен- α-олефиновый мульти-блок-сополимер по п.18, имеющий глубину пенетрации по термомеханическому анализу 1 мм при температуре по меньшей мере 90°С и модуль упругости при изгибе от 3 кфунтов на кв. дюйм (20 МПа) до 13 кфунтов на кв. дюйм (90 МПа).
20. Этилен- α-олефиновый мульти-блок-сополимер, получаемый способом по п.8, имеющий объемную потерю при сопротивлении истиранию согласно ISO 4649 менее чем 90 мм3.
21. Этилен- α-олефиновый мульти-блок-сополимер по п.18, имеющий объемную потерю при сопротивлении истиранию согласно ISO 4649 менее чем 90 мм3.
22. Этилен- α-олефиновый мульти-блок-сополимер, получаемый способом по п.8, имеющий объемную потерю при сопротивлении истиранию согласно ISO 4649 менее чем 90 мм3 и имеющий динамический модуль упругости, G', такой, что log (G') более чем или равен 0,4 МПа при температуре 100°С.
23. Этилен- α-олефиновый мульти-блок-сополимер по п.18, имеющий объемную потерю при сопротивлении истиранию согласно ISO 4649 менее чем 90 мм3 и имеющий динамический модуль упругости, G', такой, что log (G') более чем или равен 0,4 МПа при температуре 100°С.
24. Этилен- α-олефиновый мульти-блок-сополимер по п.20, имеющий динамический модуль упругости, G', такой, что log (G') более чем или равен 1,0 МПа, при температуре 100°С.
25. Этилен- α-олефиновый мульти-блок-сополимер по п.21, имеющий динамический модуль упругости, G', такой, что log (G') более чем или равен 1,0 МПа, при температуре 100°С.
26. Сшитое производное мульти-блок-сополимера по любому из пп.9-25 или получаемое способом по п.8.
27. Этилен- α-олефиновый мульти-блок-сополимер по любому из пп.9-25 или получаемый способом по п.8, или содержащий его состав, в виде пленки, по меньшей мере одного слоя многослойной пленки, по меньшей мере одного слоя ламинированного изделия, вспененного изделия, волокна, нетканого материала, сформованного литьем под давлением изделия, сформованного раздувом изделия, полученного центробежным формованием изделия или адгезива.
| PRZYBLA, FINK: "Two different on the same silica supported metallocene catalysts, activated by various trialkilaluminums", ACTA POLYMERICA, vol.50, 21.04.1999, pages 77-83 | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| LIEBER SUSANNA ET AL: "Propene polymerization with catalyst mixtures containing different ansa-circonocenes: chain transfer to alkilaluminum | |||

