Настоящее изобретение относится к пероральной лекарственной форме с немедленным высвобождением активного вещества общей формулы
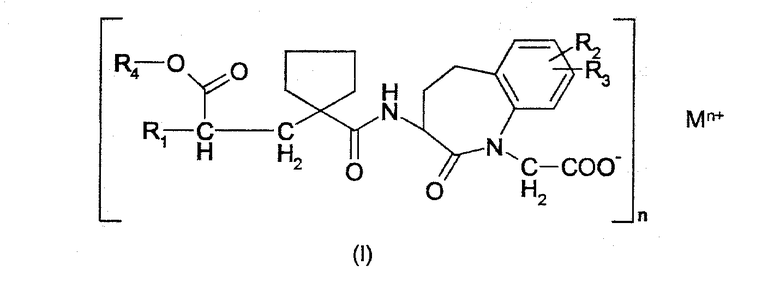
где:
R1 выбирают из группы, состоящей из (C1-C6)алкокси(C1-C6)алкила, который может быть замещен (C1-C6)алкокси, фенил-(C1-C6)-алкилом и фенилокси-(C1-C6)-алкилом, где фенильная группа может быть замещена (C1-C6)алкилом, (C1-C6)алкокси или галогеном и нафтил-(C1-C6)-алкилом,
R2 и R3 являются оба независимо водородом или галогеном,
R4 является группой, образующей биоразлагающийся сложный эфир,
M является водородом или ионом металла, предпочтительно - ионом двухвалентного металла,
n является 1, 2 или 3.
Различные активные вещества, включающие указанные выше соединения формулы (I), имеют очень малую растворимость в воде. Когда эти активные вещества вводят в организм, они часто имеют низкую биологическую доступность вследствие низкой растворимости в пищеварительном соке. Для решения этой проблемы были разработаны несколько способов, таких как микронизация, включение в циклодекстрины, использование инертных водорастворимых носителей, применение твердых дисперсий (WO 00/00179) или твердых растворов или нанокристаллических или аморфных форм активного вещества.
В WO 03/068266 описана пероральная лекарственная форма твердого раствора соединений формулы (I), имеющая повышенную биологическую доступность по сравнению с указанным активным веществом в традиционно изготавливаемой лекарственной форме. Хотя эта лекарственная форма характеризуется превосходной биологической доступностью, она имеет недостаток, заключающийся в том, что ее получают из расплавленной смеси, что приводит к некоторым ограничениям: ее следует приготавливать или в капсуле, или в таблетке с помощью метода экструзии из расплава. Кроме того, размер лекарственной формы будет слишком большим для более высоких доз.
Целью настоящего изобретения является разработка альтернативного варианта пероральной лекарственной формы соединения формулы I со значительным увеличением биологической доступности по сравнению с указанным активным веществом в традиционно получаемой лекарственной форме, которая является достаточно стабильной при практическом применении и которая также может быть использована для приготовления лекарственных форм с высоким содержанием активного вещества при приемлемом размере. Дополнительной целью настоящего изобретения является разработка лекарственной формы, которая может быть приготовлена с использованием стандартных методов и оборудования, чтобы не было необходимости в крупных капиталовложениях.
Эта цель может быть достигнута согласно настоящему изобретению за счет пероральной лекарственной формы с немедленным высвобождением активного вещества общей формулы
где:
R1 выбирают из группы, состоящей из (C1-C6)алкокси(C1-C6)алкила, который может быть замещен (C1-C6)алкокси, фенил-(C1-C6)-алкилом и фенилокси-(C1-C6)-алкилом, где фенильная группа может быть замещена (C1-C6)алкилом, (C1-C6)алкокси или галогеном и нафтил-(C1-C6)-алкилом,
R2 и R3 являются оба независимо водородом или галогеном,
R4 является группой, образующей биоразлагающийся сложный эфир,
M является водородом или ионом металла, предпочтительно - ионом двухвалентного металла,
n является 1, 2 или 3,
включающей:
a) указанное активное вещество в количестве до 65% от общей массы лекарственной формы;
b) по меньшей мере, 10 мас.% щелочного соединения или смеси щелочных соединений;
c) от 0,1 до 10 мас.% одного или более поверхностно-активных веществ, и
d) необязательно включает вспомогательные материалы в количестве от 1% до 45% от общей массы лекарственной формы.
M выбирают из группы, состоящей из Li+, Ca2+, Mg2+ и Zn2+ и предпочтительно - Ca2+. (C1-C6)-алкил является линейной или разветвленной алкильной группой, содержащей от 1 до 6 углеродных атомов. (C1-C6)-алкокси является линейной или разветвленной алкоксильной группой, содержащей от 1 до 6 углеродных атомов. Предпочтительно, R1 является фенилэтилом, R2 и R3 являются водородом и R4 является этилом.
Соединения общей формулы (I) описаны в EP0733642 и в WO 03/059939.
Предпочтительным соединением является кальциевая соль 1H-1-бензазепин-1-уксусной кислоты, 3-[[[1-[2-(этоксикарбонил)-4-фенилбутил]циклопентил]карбонил]амино]-2,3,4,5-тетрагидро-2-оксо-. Наиболее предпочтительным соединением является указанное соединение в 3S,2'R форме. Это соединение обозначается как соединение
S-Ca соответствующей кислоты (1H-1-бензазепин-1-уксусной кислоты, 3-[[[1-[2-(этоксикарбонил)-4-фенилбутил]циклопентил]-карбонил]амино]-2,3,4,5-тетрагидро-2-oxo-) обозначают как соединение S-H, и соответствующую соль S-α-метилбензиламина обозначают как соединение S-Mba.
Активное вещество формулы (I) обычно используют в количестве от 0,1 до 60% по массе, более предпочтительно - в количестве от 1 до 45% по массе, наиболее предпочтительно - в количестве от 10 до 45% по массе. Активное вещество может необязательно быть использовано в микронизированной форме.
Для облегчения понимания конкретных терминов, используемых в настоящей заявке, далее приводятся следующие определения.
Немедленное высвобождение обозначает высвобождение, по меньшей мере, 75% лекарственного средства в растворенной форме из лекарственной формы в течение 90 минут.
Достаточно устойчивый для практического применения означает приемлемую химическую и физическую стабильность в течение периода хранения, по меньшей мере, одного года в условиях окружающей среды, предпочтительно - по меньшей мере, в течение 2 лет, и еще более предпочтительно - по меньшей мере, в течение 3 лет, и наиболее предпочтительно, по меньшей мере, в течение 5 лет. Приемлемая химическая стабильность означает не более чем 5% разложения активного материала в течение периода хранения, предпочтительно - не более чем 3%, и наиболее предпочтительно - не более чем 1%. Приемлемая физическая стабильность означает отсутствие значительного изменения во внешнем виде, отсутствие разрушения таблетки при извлечении ее из блистерной упаковки в конце периода хранения и не более чем 20% изменение времени разрушения. Физическая стабильность является также приемлемой при растворении, по меньшей мере, 70% активного ингредиента в течение 60 минут в течение всего периода хранения.
Термин "микронизированный" относится к размеру частиц, где, в расчете на объем, более чем 95% частиц по размеру являются меньше чем 75 микрон.
Щелочное соединение выбирают из группы, состоящей из неорганических и органических щелочных соединений, таких как бикарбонат натрия, бикарбонат калия, карбонат натрия, карбонат калия, цитрат натрия, трис буфер, триэтаноламин, щелочные гидроксиды, такие как гидроксид натрия, гидроксид калия или гидроксид магния, щелочные фосфаты, такие как вторичный кислый фосфат калия и меглумин. Могут также быть использованы смеси этих щелочных соединений. Предпочтительными щелочными соединениями являются бикарбонат натрия, бикарбонат калия, карбонат натрия, карбонат калия и карбонат кальция. Наиболее предпочтительным щелочным соединением является бикарбонат натрия.
Щелочное соединение обычно используют в количестве, по меньшей мере, 10% от общей массы лекарственной формы. В случае применения карбоната его предпочтительно использовать в количестве 50% от общей массы лекарственной формы, более предпочтительно - в количестве, по меньшей мере, 55 мас.%, и наиболее предпочтительно - в количестве, по меньшей мере, 60 мас.%.
Предпочтительным поверхностно-активным ингредиентом являются гидрофильные поверхностно-активные вещества, и более предпочтительным - гидрофильное поверхностно-активное вещество, которое выбирают из группы, состоящей из неионных гидрофильных поверхностно-активных веществ и анионных гидрофильных поверхностно-активных веществ. Примерами неионных гидрофильных поверхностно-активных веществ являются эфиры полиоксиэтилена и сорбита, кромофоры и полоксамеры. Примерами анионных гидрофильных поверхностно-активных веществ являются лаурилсаркозинат натрия, докузат и фармацевтически приемлемые соли докузата. Может быть использована также смесь этих поверхностно-активных веществ. Более предпочтительными являются эфиры полиоксиэтилена и сорбита, лаурилсаркозинат натрия, докузат и фармацевтически приемлемые соли докузата. Еще более предпочтительными являются докузат кальция, докузат натрия и докузат калия. Наиболее предпочтительным поверхностно-активным ингредиентом является докузат натрия. Докузаты производятся в промышленности (например, фирмой Sigma Aldrich).
Докузаты обычно поставляют в виде кубиков со стороной около 1 см. Докузаты могут быть добавлены к сухой смеси ингредиентов после криогенного измельчения (то есть измельчения при низкой температуре, например, после охлаждения при помощи твердого диоксида углерода или жидкого азота) или в виде раствора, например, в дихлорметане, этилацетате или метил-трет-бутиловом эфире. В качестве альтернативного варианта докузат может быть соосажден с активным ингредиентом из органического раствора, содержащего как активный ингредиент, так и докузат, путем добавления антирастворителя, такого как гексан.
Поверхностно-активное вещество обычно используют в количестве от 0,1% до 10% от общей массы лекарственной формы, предпочтительно - в количестве от 0,5 до 2,5%, более предпочтительно - в количестве от 0,8 до 1,5 мас.%, и наиболее предпочтительно - в количестве около 1,0 мас.%.
Предпочтительно, чтобы массовое отношение поверхностно-активного вещества к активному соединению было от 1:200 до 1:5, более предпочтительно - от 1:30 до 1:10, и наиболее предпочтительно - около 1:15. Предпочтительно, чтобы массовое отношение активного соединения к щелочному соединению составляло от 1:6 до 1:0,5, более предпочтительно - от 1:5 до 1:1,5, и наиболее предпочтительно - около 1:4. Предпочтительно, чтобы массовое отношение поверхностно-активного вещества к щелочному соединению составляло от 1:2000 до 1:5, более предпочтительно - от 1:100 до 1:10, и наиболее предпочтительно - около 1:60.
Лекарственная форма необязательно включает вспомогательные материалы в количестве до 45% от общей массы лекарственной формы и предпочтительно - от 1% до 45% от общей массы лекарственной формы. Примерами этих вспомогательных материалов являются:
a) связующие, такие как гуммиарабик, альгиновая кислота и ее соли, производные целлюлозы, метилцеллюлоза, гидроксиэтилцеллюлоза, гидроксипропилцеллюлоза, алюмосиликат магния, полиэтиленгликоль, камеди, полисахаридные кислоты, бентониты, гидроксипропилметилцеллюлоза, желатин, поливинилпирролидон, сополимер поливинилпирролидона/винилацетата, кросповидон, повидон, полиметакрилаты, гидроксипропилметилцеллюлоза, гидроксипропилцеллюлоза, крахмал, пептизированный крахмал, этилцеллюлоза, трагакантовая камедь, декстрин, микрокристаллическая целлюлоза, сахароза или глюкоза и другие подобные вещества.
b) разрыхлители, такие как крахмалы, пептизированный кукурузный крахмал, пептизированный крахмал, целлюлозы, сшитая карбоксиметилцеллюлоза, кросповидон, сшитый поливинилпирролидон, альгинатный комплекс кальция или натрия, глины, альгинаты, камеди или натрия крахмала гликолат и любые разрыхлители, используемые при изготовлении таблеток.
c) наполнители, такие как лактоза, карбонат кальция, фосфат кальция, двухосновный фосфат кальция, сульфат кальция, микрокристаллическая целлюлоза, порошок целлюлозы, декстроза, декстраты, декстран, крахмалы, пептизированный крахмал, сахароза, ксилит, лактит, маннит, сорбит, хлорид натрия, полиэтиленгликоль и другие подобные вещества.
d) стабилизаторы, такие как любые антиоксиданты, буферы или кислоты и другие подобные вещества.
e) смазывающие вещества, такие как стеарат магния, гидроксид кальция, тальк, коллоидный диоксид кремния, натрия стеарилфумарат, гидрированное растительное масло, стеариновая кислота, глицерилбехенат, стеараты магния, кальция и натрия, стеариновая кислота, тальк, воски, Stearowet, борная кислота, бензоат натрия, ацетат натрия, хлорид натрия, DL-лейцин, полиэтиленгликоли, олеат натрия или лаурилсульфат натрия и другие подобные вещества.
f) увлажняющие средства, такие как олеиновая кислота, моностеарат глицерина, моноолеат сорбита, монолаурат сорбита, триэтаноламинолеат, полиоксиэтиленсорбитанмоноолеат, полиоксиэтиленсорбитанмонолаурат, олеат натрия или лаурилсульфат натрия и другие подобные соединения,
g) разбавители, такие как лактоза, крахмал, маннит, сорбит, декстроза, микрокристаллическая целлюлоза, двухосновный фосфат кальция, разбавители на основе сахарозы, сахарная пудра, моногидрат одноосновного сульфата кальция, дигидрат сульфата кальция, тригидрат лактата кальция, декстраты, инозит, сухие вещества гидролизованных злаков, амилоза, порошкообразная целлюлоза, карбонат кальция, глицин или бентонит и другие подобные вещества,
h) вещества против слипания или вещества, способствующее скольжению, такие как тальк, кукурузный крахмал, DL-лейцин, лаурилсульфат натрия и стеараты магния, кальция или натрия и другие подобные вещества.
i) фармацевтически приемлемые носители, такие как гуммиарабик, желатин, коллоидный диоксид кремния, глицерофосфат кальция, лактат кальция, мальтодекстрин, глицерин, силикат магния, казеинат натрия, соевый лецитин, хлорид натрия, трикальцийфосфат, дикалийфосфат, стеароиллактилат натрия, каррагенан, моноглицерид, диглицерид или пептизированный крахмал и другие подобные вещества.
Предпочтительно, чтобы конечная лекарственная форма представляла собой гранулы, спрессованные таблетки или капсулы.
Описанная выше лекарственная форма может быть приготовлена с использованием традиционных методик получения и оборудования. Поэтому другим аспектом настоящего изобретения является разработка способа получения описанной выше лекарственной формы, включающего следующие стадии:
a) смешение активного вещества формулы I с щелочным соединением или смесью щелочных соединений и необязательно с одним или более вспомогательным материалом;
b) растворение поверхностно-активного вещества в растворителе, необязательно с одним или более вспомогательным материалом;
c) добавление раствора, включающего поверхностно-активное вещество в указанном растворителе, к смеси, содержащей активное вещество и щелочное соединение, и необязательно добавление одного или более вспомогательного материала;
d) сушка и просеивание полученных гранул и необязательно смешение с одним или более вспомогательным материалом;
e) необязательно прессование смеси в таблетки, необязательно с последующим нанесением покрытия или заполнением смесью капсул.
В другом варианте осуществления изобретения лекарственную форму получают способом, включающим следующие стадии:
a) растворение активного вещества формулы I в растворителе с получением первого раствора;
b) растворение поверхностно-активного вещества в растворителе с получением второго раствора;
c) смешение указанных первого и второго раствора;
d) соосаждение активного вещества и поверхностно-активного вещества из смешанного раствора путем добавления антирастворителя;
e) смешение соосажденной смеси, содержащей активное вещество и поверхностно-активное вещество, с щелочным соединением и необязательно с одним или более вспомогательным материалом;
f) сушка и просеивание полученных гранул и необязательно смешение с одним или более вспомогательным материалом;
g) необязательно прессование смеси в таблетки, необязательно с последующим нанесением покрытия или заполнением смесью капсул.
В еще одном варианте осуществления изобретения лекарственную форму получают способом, включающим следующие стадии:
a) смешение активного вещества формулы I с щелочным соединением или смесью щелочных соединений с одним или более поверхностно-активным веществом и необязательно с одним или более вспомогательным материалом;
b) уплотнение смеси в брикеты;
c) измельчение брикетов с образованием гранул;
d) смешение гранул с одним или более вспомогательным материалом;
e) необязательно прессование смеси в таблетки, необязательно с последующим нанесением покрытия или заполнением смесью капсул.
Когда этот способ получения сухой лекарственной формы, описанный в предыдущем параграфе, используют с докузатом в качестве поверхностно-активного вещества, докузат должен быть измельчен. Это измельчение может быть осуществлено с использованием метода криогенного измельчения, описанного выше в одном из параграфов.
Частью способа могут также быть различные стадии, такие как сушка, дробление, просеивание, смешение и расфасовка, но эти стадии не являются существенными признаками при получении лекарственной формы согласно настоящему изобретению.
Когда в качестве растворителя используют воду и содержание активного материала составляет выше чем 15% от общей массы лекарственной формы, предпочтительно использовать способ, включающий стадию уплотнения.
Подходящими растворителями для растворения поверхностно-активного вещества, используемыми в настоящем изобретении, являются, например, дихлорметан, этилацетат, метил-трет-бутиловый эфир и вода. Предпочтительным растворителем является вода, предпочтительно, с температурой от 50 до 95°C. Наиболее предпочтительно, чтобы в случае когда вода используется в качестве растворителя, температура составляла от 50 до 65°C.
Следующие примеры предназначены только для дополнительной более подробной иллюстрации изобретения, и поэтому предполагается, что эти примеры ни в коей мере не ограничивают объем изобретения.
ПРИМЕРЫ
Пример 1. Материалы и методы
Материалы
S-Ca может быть получен согласно методике, приведенной в примерах 2 и 3 патентной заявки WO 03/059939, используя в качестве исходного реагента кислоту, получаемую согласно примеру 2 патентной заявки EP 0733642.
Бикарбонат натрия может быть приобретен у фирмы Sigma Aldrich или Canton Labs, India.
Докузат натрия может быть приобретен у фирмы Sigma Aldrich.
Все другие вспомогательные материалы являются легко доступными выпускаемыми промышленностью материалами.
Методы
ИССЛЕДОВАНИЕ РАСТВОРИМОСТИ IN-VITRO
Растворяющая система
Растворимость таблеток определяют с помощью 900 мл 0,05 моль/л фосфатного буфера с pH 6,8 в качестве растворяющей среды, используя прибор для исследования 2 (мешалка) согласно фармакопее США (USP) при 50 об/мин.
Количество растворяющегося S-H определяют фильтрацией аликвот растворов и после их разбавления анализом с помощью УФ-поглощения при 240 нм. Для внешнего стандарта 17,0 мг соединения S-Ca растворяют в 50 мл метанола, 2,0 мл этого раствора с 2 мл растворяющей среды разбавляют до 25 мл метанолом.
Количество растворенного соединения S-H, выраженное в процентах относительно заданного количества, рассчитывают по уравнению 1:
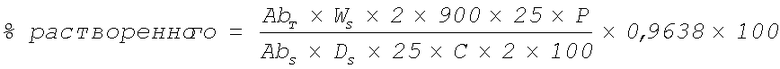
Уравнение 1 для вычисления количества растворенного соединения S-H.
Где:
Ab T = поглощение испытываемого препарата;
Ab S = поглощение стандартного препарата;
W S = масса взятого стандарта соединения S-Ca (в мг);
D S = разбавление для стандартного препарата;
P = содержание стандарта соединения S-Ca (в процентах);
C = заданное количество соединение S-H в каждой таблетке (то есть 150 мг или 300 мг);
0,9638 = коэффициент пересчета из соединения S-Ca в соединение S-H.
Пример 2. Получение лекарственной формы согласно настоящему изобретению путем грануляции в органическом растворителе
ПОЛУЧЕНИЕ ЛЕКАРСТВЕННОЙ ФОРМЫ
Количества S-Ca и бикарбоната натрия просеивают и смешивают. Докузат натрия растворяют в дихлорметане и гранулируют с помощью смесителя с получением хорошо перемешанной тестообразной массы. Гранулы сушат в центробежной сушилке. Высушенные гранулы просеивают, смешивают с мелкокристаллической целлюлозой, стеаратом магния, тальком и коллоидным диоксидом кремния и прессуют в таблетки. На таблетки наносят покрытие из Opadry, суспендированного в органических растворителях.
Композиция таблетки, содержащая 300 мг соединения S-H, полученная безводным способом
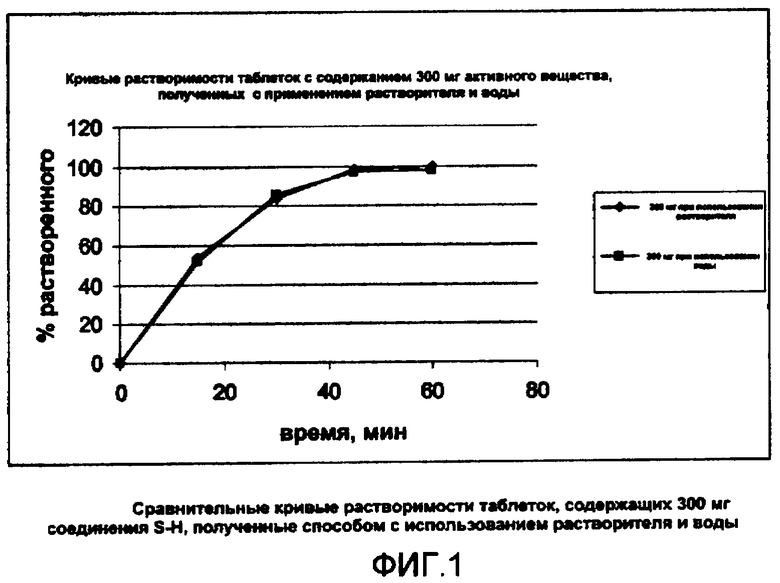
Кривые растворимости таблеток, полученных согласно этому примеру, приведены на фигуре 1 (обозначены символом ♦).
Пример 3. Получение лекарственной формы согласно настоящему изобретению с водной грануляцией и уплотнением
ПОЛУЧЕНИЕ ЛЕКАРСТВЕННОЙ ФОРМЫ
Смешивают около 33% от требуемого количества соединения S-Ca и требуемое количество бикарбоната натрия. Смесь увлажняют горячим водным раствором требуемого количества докузата натрия. Смесь гранулируют, гранулированный материал сушат и измельчают и оставшееся количество соединения S-Ca и около 50% от требуемых количеств натрия крахмала гликолата и стеарата магния смешивают с сухим гранулятом, уплотняют и измельчают. Оставшиеся количества натрия крахмала гликолата и стеарата магния и требуемые количества микрокристаллической целлюлозы, талька и коллоидного безводного диоксида кремния смешивают с гранулятом. Конечный гранулированный материал прессуют в таблетки. На таблетки наносят покрытия путем распыления на таблетки суспензии Opadry II в воде.
Композиция таблетки, содержащая 300 мг соединения S-Ca, полученная водной грануляцией и уплотнением
Avicel PH 101
Кривые растворимости, определенные методом, описанным в примере 1, таблеток, полученных согласно этому примеру, приведены на фигуре 1 (обозначены символом ■).
Пример 4. Получение лекарственной формы согласно настоящему изобретению с водной грануляцией
ПОЛУЧЕНИЕ ЛЕКАРСТВЕННОЙ ФОРМЫ
Смешивают требуемые количества S-Ca, бикарбоната натрия, около 50% количества натрия крахмала гликолата и около 54% количества микрокристаллической целлюлозы. Смесь увлажняют горячим водным раствором требуемого количества докузата натрия и Povidone K30. Смесь гранулируют, гранулированный материал сушат и измельчают.
Оставшиеся количества натрия крахмала гликолата и микрокристаллической целлюлозы и требуемые количества стеарата магния, талька и коллоидного безводного диоксида кремния смешивают с сухим гранулятом. Конечный гранулированный материал прессуют в таблетки. На таблетки наносят покрытия путем распыления на таблетки суспензии Opadry II в воде.
Композиция таблетки, содержащей 150 мг S-Ca, полученная водной грануляцией
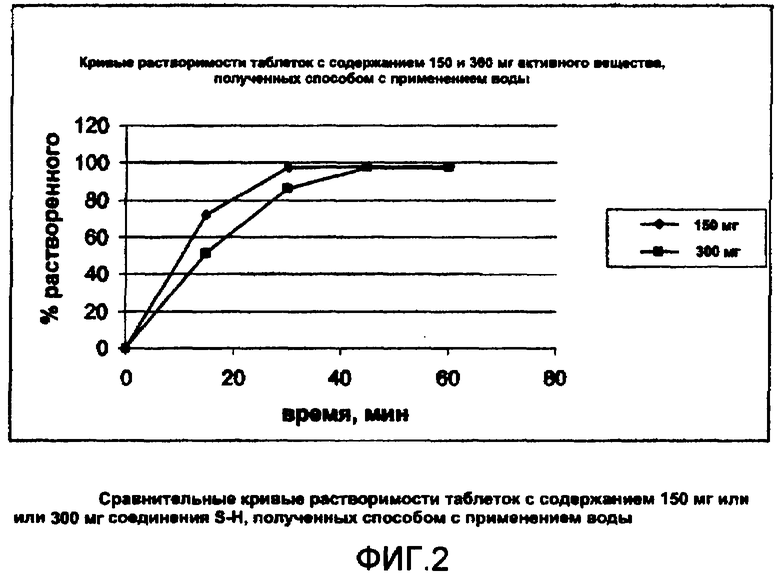
Кривые растворимости таблеток, полученных согласно примерам 3 и 4, приведены на фигуре 2 (символ ♦ для таблеток 150 мг и символ ■ для таблеток 300 мг).
Пример 5. Получение стандартной лекарственной формы, содержащей 400 мг S-Ca
ПОЛУЧЕНИЕ ЛЕКАРСТВЕННОЙ ФОРМЫ
Требуемое количество S-Ca уплотняют в сухом виде с помощью катка, измельчают и просеивают. Требуемые количества микрокристаллической целлюлозы, сшитого поливинилпирролидона и стеарилфумарата натрия смешивают с уплотненным порошком. Конечный гранулированный материал прессуют в таблетки. На таблетки наносят покрытия путем распыления на таблетки суспензии Opadry II в воде.
Композиция таблетки, содержащая 400 мг соединения S-Ca, полученная без поверхностно-активного вещества и щелочного материала
Avicel PH 101
Пример 6. Исследование биологической доступности
В открытом, рандомизированном, пересекающемся исследовании биологическую доступность разовой дозы у здоровых добровольцев в форме предпочтительной лекарственной формы сравнивали с лекарственной формой, полученной без карбоната и без поверхностно-активного вещества. Пероральный раствор используют в качестве контроля. Соединение S-Ca используют в качестве лекарственного вещества. Пациентам вводили следующие лекарственные формы:
- раствор, содержащий количество активного материала, соответствующее 200 мг соединения S-H, в цитратном буфере;
- лекарственную форму таблетки, полученной согласно примеру 1, с композицией, приведенной в таблице 1;
- лекарственную форму таблетки, полученной согласно примеру 5.
Данные по биологической доступности, нормализованной к дозе и вычисленной как отношение к лекарственной форме в виде раствора, приведены в таблице 5 и показаны на фигуре 3.
Биологическая доступность, измеренная как нормализованное по дозе отношение AUC (площади под фармакокинетической кривой) к лекарственной форме в цитратном буфере
| название | год | авторы | номер документа |
|---|---|---|---|
| ПЕРОРАЛЬНАЯ КОМПОЗИЦИЯ ТВЕРДОГО РАСТВОРА МАЛОРАСТВОРИМОГО В ВОДЕ АКТИВНОГО ВЕЩЕСТВА | 2003 |
|
RU2314811C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, ВКЛЮЧАЮЩИЕ МОДУЛЯТОР S1Р | 2007 |
|
RU2487703C2 |
| ТВЕРДОЕ ЛЕКАРСТВЕННОЕ СРЕДСТВО С КОНТРОЛИРУЕМЫМ ВЫСВОБОЖДЕНИЕМ | 2006 |
|
RU2467740C2 |
| ТВЕРДЫЕ СОЛИ БЕНЗАЗЕПИНОВЫХ СОЕДИНЕНИЙ И ИХ ПРИМЕНЕНИЕ ПРИ ПОЛУЧЕНИИ ФАРМАЦЕВТИЧЕСКИХ СОЕДИНЕНИЙ | 2003 |
|
RU2303041C2 |
| НЕПОСРЕДСТВЕННО ПРЕССУЕМАЯ МАТРИЦА ДЛЯ РЕГУЛИРУЕМОГО ВЫСВОБОЖДЕНИЯ ОДНОКРАТНЫХ ЕЖЕДНЕВНЫХ ДОЗ КЛАРИТРОМИЦИНА | 2000 |
|
RU2237480C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2011 |
|
RU2589842C2 |
| ТВЕРДЫЙ ПРЕПАРАТ С КОНТРОЛИРУЕМЫМ ВЫСВОБОЖДЕНИЕМ | 2006 |
|
RU2496480C2 |
| АЛКИЛТИОПИРИМИДИНЫ В КАЧЕСТВЕ АНТАГОНИСТОВ CRTH2 | 2008 |
|
RU2491280C2 |
| КОМПОЗИЦИИ И СПОСОБЫ ЛЕЧЕНИЯ СИНДРОМА РАЗДРАЖЕННОГО КИШЕЧНИКА | 2017 |
|
RU2751770C2 |
| КОМПОЗИЦИИ И СПОСОБЫ ДЛЯ ЛЕЧЕНИЯ РАКА | 2017 |
|
RU2770081C2 |

Настоящее изобретение относится к пероральной лекарственной форме с немедленным высвобождением производных бензазепин-1-уксусной кислоты, которая включает указанное активное вещество в количестве до 65% от общей массы лекарственной формы; 10 мас.% щелочного соединения или смеси щелочных соединений; от 0,1 до 10% одного или более поверхностно-активных веществ и необязательно включает вспомогательные материалы в количестве от 1% до 45% от общей массы лекарственной формы. Изобретение относится также к способом получения указанной лекарственной формы. Изобретение позволяет получить стабильные при применении лекарственные формы с повышенной биологической доступностью активного вещества. 4 н. и 17 з.п. ф-лы, 3 ил., 5 табл.
1. Пероральная лекарственная форма с немедленным высвобождением активного соединения общей формулы
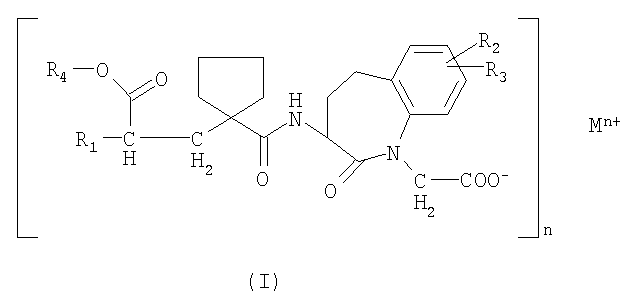
где R1 выбирают из группы, состоящей из (С1-С6)алкокси(С1-С6)алкила,
который может быть замещен (С1-С6)алкокси, фенил-(С1-С6)-алкилом и фенилокси-(С1-С6)-алкилом, где фенильная группа может быть замещена (С1-С6)алкилом, (С1-С6)алкокси или галогеном, и нафтил-(С1-С6)-алкилом,
R2 и R3 являются оба независимо водородом или галогеном,
R4 является группой, образующей биоразлагающийся сложный эфир,
М является водородом или ионом металла, предпочтительно ионом двухвалентного металла,
n является 1, 2 или 3;
включающая
а) указанное активное вещество в количестве до 65% от общей массы лекарственной формы;
b) по меньшей мере, 10 мас.% щелочного соединения или смеси щелочных соединений;
c) от 0,1 до 10 мас.% одного или более поверхностно-активных веществ и,
d) необязательно, включает вспомогательные материалы в количестве от 1 до 45% от общей массы лекарственной формы.
2. Пероральная лекарственная форма с немедленным высвобождением активного вещества по п.1, в которой щелочное соединение выбирают из группы, состоящей из неорганических и органических щелочных соединений, таких как бикарбонат натрия, бикарбонат калия, карбонат натрия, карбонат калия, цитрат натрия, трисбуфер, триэтаноламин, щелочные гидроксиды, такие как гидроксид натрия, гидроксид калия или гидроксид магния, щелочные фосфаты, такие как вторичный кислый фосфат калия, и меглумин или смесей этих щелочных соединений.
3. Пероральная лекарственная форма с немедленным высвобождением активного вещества по п.1 или 2, в которой поверхностно-активным веществом является гидрофильное поверхностно-активное вещество.
4. Пероральная лекарственная форма с немедленным высвобождением активного вещества по п.3, в которой гидрофильное поверхностно-активное вещество выбирают из группы, включающей кремофоры, полоксамеры, сложные эфиры полиоксиэтилена и сорбита, докузат и фармацевтически приемлемые соли докузата или их смеси.
5. Пероральная лекарственная форма с немедленным высвобождением активного вещества по п.4, в которой поверхностно-активное вещество выбирают из группы, состоящей из докузата натрия, докузата калия, докузата кальция.
6. Пероральная лекарственная форма с немедленным высвобождением активного вещества по пп.1 и 2, в которой М является Са2+.
7. Пероральная лекарственная форма с немедленным высвобождением активного вещества по пп.1 и 2, в которой массовое отношение поверхностно-активного вещества к активному соединению составляет от 1:200 до 1:5.
8. Пероральная лекарственная форма с немедленным высвобождением активного вещества по пп.1 и 2, в которой массовое отношение активного соединения к щелочному соединению составляет от 1:6 до 1:0,5.
9. Пероральная лекарственная форма с немедленным высвобождением активного вещества по пп.1 и 2, отличающаяся тем, что количество щелочного соединения составляет более 55 мас.%, предпочтительно более 60 мас.%.
10. Пероральная лекарственная форма с немедленным высвобождением активного вещества по пп.1 и 2, отличающаяся тем, что щелочным соединением является бикарбонат натрия.
11. Пероральная лекарственная форма с немедленным высвобождением активного вещества по пп.1 и 2, отличающаяся тем, что поверхностно-активным ингредиентом является докузат натрия.
12. Пероральная лекарственная форма с немедленным высвобождением активного вещества по пп.1 и 2, отличающаяся тем, что указанным активным веществом является кальциевая соль, 3-[[[1-[2-(этоксикарбонил)-4-фенилбутил]циклопентил]карбонил]амино]-2,3,4,5-тетрагидро-2-оксо-1H-1-бензазепин-1-уксусной кислоты, предпочтительно в 3S, 2'R форме.
13. Пероральная лекарственная форма с немедленным высвобождением активного вещества по п.8, отличающаяся тем, что указанным активным веществом является кальциевая соль, 3-[[[1-[2-(этоксикарбонил)-4-фенилбутил]циклопентил]карбонил]амино]-2,3,4,5-тетрагидро-2-оксо-1Н-1-бензазепин-1-уксусной кислоты, предпочтительно в 3S, 2'R форме.
14. Пероральная лекарственная форма с немедленным высвобождением активного вещества по п.9, отличающаяся тем, что указанным активным веществом является кальциевая соль 3-[[[1-[2-(этоксикарбонил)-4-фенилбутил]циклопентил]карбонил]амино]-2,3,4,5-тетрагидро-2-оксо-1Н-1-бензазепин-1-уксусной кислоты предпочтительно в 3S, 2'R форме.
15. Пероральная лекарственная форма с немедленным высвобождением активного вещества по п.10, отличающаяся тем, что указанным активным веществом является кальциевая соль 3-[[[1-[2-(этоксикарбонил)-4-фенилбутил]циклопентил]карбонил]амино]-2,3,4,5-тетрагидро-2-оксо-1H-1-бензазепин-1-уксусной кислоты предпочтительно в 3S, 2'R форме.
16. Пероральная лекарственная форма с немедленным высвобождением активного вещества по п.11, отличающаяся тем, что указанным активным веществом является кальциевая соль 3-[[[1-[2-(этоксикарбонил)-4-фенилбутил]циклопентил]карбонил]амино]-2,3,4,5-тетрагидро-2-оксо-1Н-1-бензазепин-1-уксусной кислоты предпочтительно в 3S, 2'R форме.
17. Пероральная лекарственная форма с немедленным высвобождением активного вещества по п.1 в виде гранул, прессованных таблеток или капсул.
18. Способ получения лекарственной формы по пп.1-17, включающий следующие стадии:
a) смешение активного вещества формулы I со щелочным соединением или смесью щелочных соединений и необязательно с одним или более вспомогательными материалами;
b) растворение поверхностно-активного вещества в растворителе, необязательно с одним или более вспомогательными материалами;
c) добавление раствора, включающего поверхностно-активное вещество в указанном растворителе, к смеси, содержащей активное вещество и щелочное соединение, и необязательно добавление одного или более вспомогательных материалов;
d) сушка и просеивание полученных гранул и необязательно смешение с одним или более вспомогательными материалами;
e) необязательно прессование смеси в таблетки, необязательно с последующим нанесением покрытия или заполнением смесью капсул.
19. Способ получения лекарственной формы по пп.1-17, включающий следующие стадии:
a) растворение активного вещества формулы I в растворителе с получением первого раствора;
b) растворение поверхностно-активного вещества в растворителе с получением второго раствора;
c) смешение указанных первого и второго растворов;
d) соосаждение активного вещества и поверхностно-активного вещества из смешанного раствора путем добавления антирастворителя;
e) смешение соосажденной смеси, содержащей активное вещество и поверхностно-активное вещество, со щелочным соединением и необязательно с одним или более вспомогательными материалами;
f) сушка и просеивание полученных гранул и необязательно смешение с одним или более вспомогательными материалами;
g) необязательно прессование смеси в таблетки, необязательно с последующим нанесением покрытия или заполнением смесью капсул.
20. Способ получения лекарственной формы по пп.1-17, включающий следующие стадии:
a) смешение активного вещества формулы I с щелочным соединением или смесью щелочных соединений с одним или более поверхностно-активными веществами и необязательно с одним или более вспомогательными материалами;
b) уплотнение смеси в брикеты;
c) измельчение брикетов с образованием гранул;
d) смешение гранул с одним или более вспомогательными материалами;
e) необязательно прессование смеси в таблетки, необязательно с последующим нанесением покрытия или заполнением смесью капсул.
21. Способ по п.20, в котором поверхностно-активным веществом является докузат и в котором добавляемый докузат подвергают измельчению с использованием метода криогенного размола перед стадией смешения.
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| WO 03068266 A1, 21.08.2003 | |||
| ЕР 0830863 A1, 25.03.1998 | |||
| Способ лечения ишемической болезни сердца | 1977 |
|
SU733642A1 |

