Данное изобретение относится к группе новых солей соединений формулы
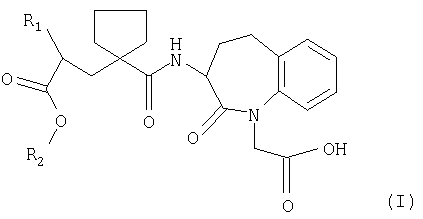
и их применению при получении фармацевтических соединений.
Бензазепины указанной выше формулы известны из ЕР 0733642, ЕР 0830863, WO 00/48601 и WO 01/03699. ЕР 0733642 относится к соединениям формулы (I) и их физиологически приемлемым солям, как таковым, и их применению при сердечной недостаточности. ЕР 0830863 WO 00/48601 и WO 01/03699 относятся к применению указанных выше соединений для улучшения желудочно-кишечного кровотока, при гипертензии, при лечении и профилактике поражений сердца, индуцированных адриамицином и аналогичными противоопухолевыми препаратами соответственно.
Предпочтительными бензазепинами являются соединения, где R1 представляет собой фенилэтильную группу или 1-нафтилэтильную группу, R2 и R3 оба представляют собой водород и где R4 представляет собой биолабильную эфирную группу. Подходящие группы, образующие биолабильные сложные эфиры, включают (C1-C6)-алкильные группы, фенил- или фенил- (C1-C6)-алкильные группы, которые необязательно замещены в фенильном кольце (C1-C6)-алкилом или (C2-C6)-алкиленовой цепью, связанной с двумя смежными атомами углерода, диоксоланилметильными группами, которые необязательно замещены в диоксолановом кольце (C1-C6)-алкильной или (C2-C6)-алканоилоксиметильными группами, оксиметильная группа которых необязательно замещена (C1-C6)-алкилом. Когда группа R4 представляет собой (C1-C6)-алкил, он является предпочтительно неразветвленной (C1-C4)-алкильной группой. В наиболее предпочтительных соединениях R4 представляет собой этил.
В ходе дальнейшего фармацевтического и клинического исследования обнаружено, что наиболее предпочтительные соединения, являясь твердыми пенами, имеют, таким образом, серьезный недостаток.
Для обеспечения воспроизводимой постоянной биодоступности активного ингредиента из твердой фармацевтической дозированной формы важно использовать гомогенную и воспроизводимую модификацию активного соединения. Следовательно, всегда существуют определенные сомнения относительно воспроизводимости и постоянства биодоступности соединений из материалов, которые в обычном состоянии не являются гомогенными, таких как твердая пена.
Также очевидно, что в промышленных масштабах очень тяжело выделить твердую пену. Кроме того, соединение мало растворимо в воде и, следовательно, очень трудно получать состав соединения, который может быть использован для внутривенного (в/в) введения. До данного изобретения внутривенный состав мог быть получен только из соответствующей дикислоты (смотри пример II в EP 0733642). Это означает, что для внутривенного состава должно быть использовано иное соединение, чем для перорального состава, нежелательного для фармацевтического соединения. В ходе дальнейших исследований обнаружено, что натриевая и калиевая соль моно-кислоты гораздо лучше растворимы в воде, но данные соли могут быть выделены только в виде твердой пены.
Задачей данного изобретения является обеспечение соли соединения общей формулы (I), которая должна отвечать следующим требованиям:
А) простой, в промышленном масштабе, способ выделения чистого твердого соединения кристаллизацией или осаждением;
Б) достаточно высокая растворимость в физиологической жидкости для получения внутривенного состава;
С) свойства твердого вещества, которые позволяют получать фармацевтическую композицию со стандартными вспомогательными веществами и на стандартном оборудовании;
Д) предпочтительно получение без существенных потерь хиральной и химической чистоты.
Данная задача может быть выполнена получением солей металлов соединений общей формулы (I), как указывалось выше, где ион металла является ионом лития или ионом двухвалентного металла. Предпочтительными солями двухвалентных металлов являются соли кальция, магния и цинка. Наиболее предпочтительной является кальциевая соль. Неожиданно обнаружено, что данные соли, в противоположность солям натрия и калия, упомянутым в EP 0733642, имеют весьма желательные свойства, так как они могут быть выделены в твердой (аморфной) форме и растворяются в изотонической жидкости с pH 7,4 в количестве, по крайней мере в 10 раз превышающем количество соответствующей кислоты. Кроме того, соли двухвалентных металлов могут быть получены без рацемизации.
В следующем аспекте данного изобретения предлагается способ получения солей металлов, предпочтительно Li+ или солей двухвалентных Ca2+, Mg2+ или Zn2+.
Неожиданно было обнаружено, что соли лития и соли двухвалентных металлов соединения формулы I очень легко растворимы при комнатной температуре в слабополярных апротонных растворителях, таких как циклогексан, толуол, метил трет-бутиловый эфир и этилацетат.
Соли данного изобретения могут быть легко получены смешением гидроксида или подходящей соли требуемого металла с раствором или суспензией соединения формулы I в одном из вышеуказанных слабополярных апротонных растворителей. Альтернативно, когда гидроксид или соль требуемого металла недостаточно растворима для того, чтобы началось взаимодействие, может быть добавлено небольшое количество воды к раствору или суспензии в органическом растворителе и вода может быть удалена азеотропной отгонкой. В таком случае должен быть выбран неполярный апротонный растворитель, который образует азеотропную смесь с водой. Для металлов, которые имеют практически нерастворимый гидроксид, может быть добавлен металл в форме этилата (например, Mg(OEt)2) или в форме смешанного гидроксида/карбоната (3Zn(OH)2·2ZnCО3). Предпочтительным растворителем для вышеуказанного метода является метил трет-метиловый эфир или этилацетат. Если соль получена в растворе, то она может быть выделена первоначальным удалением воды, что может быть осуществлено с помощью азеотропной отгонки, с последующим смешением с осадителем. Осадителем является вторая жидкость, которую добавляют к раствору для уменьшения растворимости растворенного соединения, вызывая его осаждение/кристаллизацию и максимизацию выхода продукта. Для двух жидкостей (исходный растворитель и добавляемый осадитель) необходима полная смешиваемость одного с другим в любых соотношениях. Такой подход также используется для уменьшения растворимости неорганической соли в водном растворе добавлением смешиваемого с водой органического растворителя (ALFASSI Z. B. et al., AlChE J. 1984, 30, 874-6; MYDLARZ J. et al., J. Chem. Eng. Data 1989, 34, 124-6; MULLIN J. W. et al., Chem. Eng. Process. 1989, 26, 93-9). Примерами осадителя в рамках данного изобретения являются линейные углеводороды. Предпочтительными осадителями являются линейные C4-C10 углеводороды. Наиболее предпочтительным осадителем является н-гексан.
Поскольку почти во всех случаях соль выделяют в форме некристаллического осадка, который бывает гомогенным, иногда желательно проводить стадию настоящей кристаллизации для повышения чистоты активного соединения, которое должно отвечать жестким требованиям. Неожиданно было обнаружено, что соль S-α-метилбензиламина соединения формулы I наиболее подходит для очистки данных соединений, так как указанная соль является кристаллической и может быть легко перекристаллизована с высоким выходом из органических растворителей, предпочтительно спиртов, таких как этанол или изопропиловый спирт. Таким образом, данное изобретение также относится к солям S-α-метилбензиламина соединений формулы I. Указанные соли являются подходящими только в качестве промежуточных соединений в стадии очистки, поскольку S-α-метилбензиламин, как оказалось, слишком токсичен для применения.
Данные соли S-α-метилбензиламина соединения формулы I могут быть получены добавлением S-α-метилбензиламина к раствору соединения формулы I в этаноле или изопропаноле или другом подходящем спирте. Соль кристаллизуется из такого раствора при выстаивании и охлаждении (в зависимости от концентрации).
Фармацевтически приемлемые соли данного изобретения могут быть получены в соответствии со способами получения, известными на данном уровне техники. Могут быть использованы обычные лекарственные формы, такие как, например, таблетки, капсулы или суппозитории. Данные фармацевтические лекарственные формы могут быть получены с помощью известных способов, таких как прямое прессование, грануляция, экструзия, формование с использованием обычных твердых эксципиентов, таких как наполнители, например целлюлозы, лактозы и крахмалы, связующие вещества, например целлюлозы и поливинилпирролидон (пвп), дезинтегранты, например крахмалы и поперечно-сшитый поливинилпирролидон (пвп), агенты скольжения (глиданты), например коллоидный диоксид кремния, лубриканты, например магния стеарат или обычная жидкость и полутвердые эксципиенты, такие как полиэтиленгликоли, производные касторового масла, триглицериды и парафины. Кроме того, могут быть добавлены консерванты, например парабены, и эмульгаторы, например полисорбаты.
Фармацевтически приемлемые соли данного изобретения являются подходящими в качестве лекарственных препаратов для крупных млекопитающих, особенно для людей, для лечения сердечной недостаточности и для промотирования диуреза/натрийуреза, особенно для пациентов, страдающих от сердечной недостаточности, для улучшения желудочно-кишечного кровотока, при лечении гипертензии и при лечении и профилактики поражений сердца, вызванных адриамицином и аналогичными противоопухолевыми препаратами. С этой целью соединения данного изобретения могут быть использованы в медицинских формах, которые могут быть введены парентерально, особенно внутривенно, перорально или в виде суппозиториев. Используемые дозы могут варьировать индивидуально и в зависимости от природы состояния, которое подлежит лечению, конкретного применяемого вещества и способа введения. Медицинские формы с содержанием активного компонента от 1 до 800 мг на одну дозу в основном подходят для введения крупным млекопитающим, особенно людям.
Следующие примеры приведены с намерением дополнительно проиллюстрировать изобретение более детально и, следовательно, данные примеры в любом случае не подразумевают ограничения объема изобретения.
ПРИМЕРЫ.
Пример 1.
Основная методика получения солей металлов соединений формулы I.
Примерно 15 ммоль активного вещества в кислой форме растворяют или суспендируют в 40 мл слабо полярного апротонного растворителя. Добавляют раствор примерно 1,2 эквивалента реагента металла в воде или в этом же растворителе в качестве активного компонента. В некоторых случаях необходимо добавлять воду для того, чтобы началось взаимодействие. Воду удаляют азеотропной отгонкой. Когда реагент металла не представляет собой гидроксид или этилат, растворитель удаляют полностью, с последующим повторным растворением в 40-160 мл первоначального слабо полярного апротонного растворителя, с последующей фильтрацией для удаления непрореагировавшего реагента металла и необязательных образовавшихся других солей. Фильтрованный раствор добавляют к гексану и, когда образуется твердый продукт, его собирают на фильтре. При образовании смолы или масла большую часть растворителя декантируют и оставшийся растворитель выпаривают с получением твердой пены.
*Соединение II = 1H-1-бензазепин-1-уксусная кислота, 3-[[[1-[2-(этоксикарбонил)-4-(1-нафталенил)бутил]циклопентил]карбонил]амино]-2,3,4,5-тетрагидро-оксо-,[S-(R*,S*)]-.
Свойства полученных солей представлены в таблице 2. Содержание соединения было определено ВЭЖХ-методом (с использованием MACHEREY-NAGEL Nucleosil 100-5 C18-HD в качестве колонки, используя систему градиента начиная с 5% В и заканчивая 100% В, с фосфорно-кислым буфером с pH 5,1 в качестве элюента А и элюента В, который представляет собой ацетонитрил, смешанный с 10% элюента А). Содержание металла определяли с помощью комплексометрического титрования с раствором этилендиамин-тетра-уксусной кислоты для кальция и атомной эмиссионной спектроскопии (АЭС) для всех остальных металлов.
*Соединение II = 1H-1-бензазепин-1-уксусная кислота, 3-[[[1-[2-(этоксикарбонил)-4-(1-нафталенил)булил]циклопентил]карбонил]амино]-(2,3,4,5-тетрагидро-оксо-, [S-(R*,S*)]-.
Н.о.- не определено.
Исходя из данных таблицы 2 становится ясным, что соли Li, Ca, Mg и Zn, которые выделяют в виде твердого порошка растворимы в слабополярных апротонных растворителях. Примерами данных растворителей являются этилацетат, толуол, циклогексан и метил трет-бутиловый эфир. Данные соединения также растворимы в полярных апротонных растворителях, таких как ТГФ, ацетон, ацетонитрил, ДМФ и ДМСО. Установленное содержание металла в соли несколько выше, чем теоретическое содержание, но это нормально для данных типов методик и анализов. Во время образования соли с сильно основными одновалентными гидроокисями происходит деградация активного вещества, приводя к низкому содержанию соединения в конечной соли.
Пример 2.
Получение соли S-α-метилбензиламина 1H-1-бензазепин-1-уксусной кислоты, 3-[[[1-[(2R)-2-(этоксикарбонил)-4-фенилбутил]циклопентил]карбонил]амино]-2,3,4,5-тетрагидро-2-оксо-, (3S)-.
18 г 1H-1-бензазепин-1-уксусной кислоты, 3-[[[1-[(2R)-2-(этоксикарбонил)-4-фенилбутил]циклопентил]карбонил]амино]-2,3,4,5-тетрагидро-2-оксо-, (3S)- растворяют в 90 мл абсолютного этанола. 4,1 г S-α-метилбензиламина добавляют при 20-25°С. Спонтанно образующуюся кристаллическую суспензию нагревают до 40°С и перемешивают в течение одного часа. После охлаждения до 0-5°С и дополнительного перемешивания в течение 4 часов кристаллы собирают фильтрацией, промывают 40 мл охлажденного абсолютного этанола и высушивают при 45°С в вакуумной печи. 19 г соли S-α-метилбензиламина 1H-1-бензазепин-1-уксусной кислоты, 3-[[[1-[(2R)-2-(этоксикарбонил)-4-фенилбутил]циклопентил]карбонил]амино]-2,3,4,5-тетрагидро-2-оксо-, (3S)- получают в качестве первой порции.
Пример 3.
Получение кальциевой соли 1H-1-бензазепин-1-уксусной кислоты, 3-[[[1-[(2R)-2-(этоксикарбонил)-4-фенилбутил]циклопентил]карбонил]амино]-2,3,4,5-тетрагидро-2-оксо-, (3S)-.
К раствору 30 г соли S-α-метилбензиламина 1H-1-бензазепин-1-уксусной кислоты, 3-[[[1-[(2R)-2-(этоксикарбонил)-4-фенилбутил]циклопентил]карбонил]амино]-2,3,4,5-тетрагидро-2-оксо-, (3S)- в 120 мл метил трет-бутилового эфира (МТБЭ) добавляют 100 мл 1М водного раствора хлористоводородной кислоты, и полученную смесь перемешивают в течение 10 минут. Слои разделяют и органический слой промывают, по крайней мере, три раза 15 мл воды до тех пор, пока значение pH не достигнет значения выше 5. Добавляют 95% Ca(OH)2 в количестве 2 г, и смесь нагревают до 55°С с обратным холодильником. Когда после 30 минут количество суспензии незначительно уменьшится, добавляют 0,5 мл воды. Смесь кипятят с обратным холодильником в течение 2 часов и водоотделителем (ловушка Дина-Старка). После 2 часов дистиллят становится совершенно чистым, а реакционная смесь - слегка мутной. Смесь охлаждают до 30-35°С, добавляют в течение 30 минут посредством проточного фильтра к 240 мл гексана. Твердый продукт отделяют фильтрованием и промывают 50 мл гексана. После высушивания получают 25,6 г не совсем белого свободно сыпучего порошка.
1H-ЯМР: δ = 7,29 (1H, дд, J=2,2 и 8,1), 7,28 (1H, ддд, J=2,0, 6,6, 8,1), 7,25 (1H, дд, J=2,0 и 7,6), 7,19 (1H, ддд, J=2,2, 6,6, 7,6), 7,19 (2H, дддд, J=0,6, 1,7, 7,5, 7,8), 7,13 (1H, дд, J=1,3 и 7,5), 7,10 (2H, ддд, J=1,3, 2,1, 7,8), 4,39 (1H, д, J=16,9), 4,28 (1H, дд, J=8,1 и 11,7), 4,28 (1H, д, J=16,9), 4,07 (1H, дд, J=7,2 и 10,8), 4,01 (1H, дд, J=7,1 и 10,8), 3,33 (1H, ддд, J=8,0, 13,2, 13,7), 2,57 (1H, ддд, J=1,2, 7,1, 13,7), 2,52 (1H, дд, J=5,9 и 9,6), 2,49 (1H, дд, J=6,7 и 9,4), 2,31 (1H, дддд, J=3,3, 5,1, 9,2, 9,3), 2,29 (1H, дддд, J=7,1, 8,1, 13,1, 13,2), 2,03 (1H, дддд, J=1,2, 8,0, 11,7, 13,1), 2,0 (1H, дд, J=9,3 и 14,2), 1,82 (1H, дд, J=3,3 и 14,2), 1,82 (1H, ддд, J=5,9, 9,4, 13,6), 1,70 (1H, ддд, J=6,7, 9,6, 13,6), 2,02-1,42 (8H, м), 1,21 (3H, дд, J=7,1 и 7,2).
Пример 4.
Получение соли S-α-метилбензиламина соединения II = 1H-1-бензазепин-1-уксусной кислоты, 3-[[[1-[2-(этоксикарбонил)-4-(1-нафталенил)бутил]циклопентил]карбонил]амино]-2,3,4,5-тетрагидро-оксо-, [S-(R*,S*)]-.
21 г соединения II = 1H-1-бензазепин-1-уксусной кислоты, 3-[[[1-[2-(этоксикарбонил)-4-(1-нафталенил)бутил]циклопентил]карбонил]амино]-2,3,4,5-тетрагидро-оксо-, [S-(R*,S*)]- растворяют в 190 мл МТБЭ. Добавляют 45 мл этанола и 4,5 г S-α-метилбензиламина. После хранения в течение 4 дней при 4°С и перемешивания один раз в день кристаллы собирают фильтрацией, промывают 80 мл МТБЭ и сушат при 45°С в вакуумной печи. 19 г соли S-α-метилбензиламина соединения II = 1H-1-бензазепин-1-уксусная кислоты, 3-[[[1-[2-(этоксикарбонил)-4-(1-нафталенил)бутил]циклопентил]карбонил]амино]-2,3,4,5-тетрагидро-оксо-, [S-(R*,S*)]- получают в качестве первой порции.
Пример 5.
Получение кальциевой соли 1H-1-бензазепин-1-уксусной кислоты, 3-[[[1-[2-(этоксикарбонил)-4-(1-нафталенил)бутил]циклопентил]карбонил]амино]-2,3,4,5-тетрагидро-оксо-, [S-(R*,S*)]-.
К гетерогенной смеси 10 г соли S-α-метилбензиламина 1H-1-бензазепин-1-уксусной кислоты, 3-[[[1-[2-(этоксикарбонил)-4-(1-нафталенил)бутил]циклопентил]карбонил]амино]-2,3,4,5-тетрагидро-оксо-, [S-(R*,S*)]- в 80 мл метил-трет-бутилового эфира (МТБЭ) и 60 мл воды, добавляют в течение 15 минут при перемешивании 4,4 мл 36% водного раствора хлористоводородной кислоты, и полученную смесь перемешивают в течение 1,5 часов при комнатной температуре. Слои разделяют и органический слой промывают два раза 50 мл воды. Органический слой концентрируют до получения масла, добавляют 15 мл этилацетата, и полученный раствор снова концентрируют до получения масла. Масло опять растворяют в 80 мл этилацетата и добавляют 2 мл воды. Добавляют 95% Ca(OH)2 в количестве 0,56 г, и смесь кипятят с обратным холодильником в течение 4 часов водоотделителем (аппарат Дина-Старка). Раствор фильтруют и уменьшают его объем до 40 мл. Раствор охлаждают до 30-35°С, добавляют в течение 30 минут к 250 мл холодного гексана (10°С) и перемешивают в течение дополнительных 30 минут при 10°С. Твердый продукт отделяют фильтрованием и дважды промывают 10 мл гексана. После высушивания в вакууме (18 часов, 50°С, 120 мбар) получают 7,4 г свободно сыпучего порошка.
1H-ЯМР: δ = 7,99 (1H, широкий дублет, J=8), 7,88 (1H, дд, J=1,5 и 8), 7,73 (1H, широкий дублет, J=8), 7,56-7,44 (2H, м), 7,37 (1H, т, J=8), ˜7,36 (NH, д, J=8), 7,31 (1H, дд, J=1,5 и 8), 7,29 (1H, д, J=8), 7,24 (1H, тройной дублет, J=1,5, 8, 8), 7,21 (1H, дд, J=1,5 и 8), 7,13 (1H, тройной дублет, J=1,5, 8, 8), 4,48 (1H, д, J=16), 4,23 (1H, двойной триплет, J=8, 8, 12), 4,14-3,99 (3H, м), 3,56 (1H, тройной дублет, J=8, 13, 13), 3,02-2,96 (2H, м), 2,5-2,34(2H, м), 2,2-1,74 (8H, м), 1,6-1,24 (6H, м), 1,20 (3H, т, J=6).
Изобретение относится к соединениям общей формулы
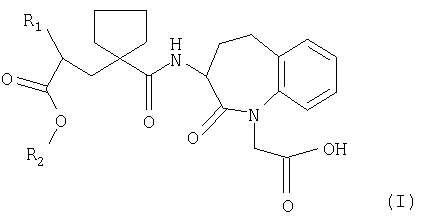
где R1 является фенил-(С1-С6)-алкильной группой или 1-нафтил-(С1-С6)-алкильной группой; R2 представляет собой биолабильную эфир образующую группу в виде фармацевтически приемлемой соли металла, где соль выбирают из группы, состоящей из соли лития, соли кальция, соли магния и цинка, а также к способу получения соединений и фармацевтической композиции, содержащей соли данного изобретения. Соединения используют при лечении заболеваний сердца или гипертензии, для улучшения желудочно-кишечного кровотока или при лечении и профилактике поражений сердца, индуцированных адриамицином и аналогичными противоопухолевыми препаратами. 9 н. и 6 з.п. ф-лы, 2 табл.
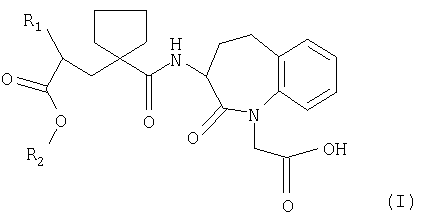
где R1 является фенил-(С1-С6)-алкильной группой или 1-нафтил-(С1-С6)-алкильной группой;
R2 представляет собой биолабильную эфиробразующую группу
в виде фармацевтически приемлемой соли металла, отличающееся тем, что соль выбирают из группы, состоящей из соли лития, соли кальция, соли магния и цинка.
Приоритет по пунктам:
| ПРОИЗВОДНЫЕ БЕНЗАЗЕПИН-, БЕНЗОКСАЗЕПИН- ИЛИ БЕНЗОТИАЗЕПИН-N-УКСУСНОЙ КИСЛОТЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ЛЕКАРСТВЕННОЕ СРЕДСТВО, ИХ СОДЕРЖАЩЕЕ | 1996 |
|
RU2159768C2 |
| ЕР 0830863 A1, 25.03.1998 | |||
| Радиоприемник | 1935 |
|
SU48601A1 |
| Машина для печатания ярлыков | 1956 |
|
SU103699A2 |

